ОБЛАСТЬ ТЕХНИКИ
[001] Настоящее изобретение относится к новым соединениям и способам их получения в области химии нуклеотидов. В частности, настоящее изобретение относится к нуклеотидам и олигонуклеотидам, содержащим модифицированную фосфатную группу, и способу их получения. Настоящее изобретение может применяться в цитологических исследованиях, в диагностике ДНК- или РНК-содержащих патогенов, в генной терапии, а также в терапии различных заболеваний бактериальной и вирусной природы, в том числе, COVID-19.
УРОВЕНЬ ТЕХНИКИ
[002] Производные нуклеиновых кислот (НК), такие как синтетические олигонуклеотиды, модифицированные различными дополнительными функциональными группами, широко используются как инструменты исследования в различных областях молекулярной биологии, биотехнологии и медицины. Одним из наиболее перспективных направлений можно считать использование олигонуклеотидов в качестве терапевтических агентов - более 11 препаратов уже одобрено организацией FDA (Food & Drug Administration, США) и более 150 находятся на различных стадиях клинических испытаний. Например, к применению в клинике одобрены такие олигонуклеотидные препараты, как подавляющий ангиогенез аптамер Macugene (Pegaptanib sodium), препараты Exondys 51 (eteplirsen) и Vyondys 53 (golodirsen) для лечения мышечной дистрофии Дюшенна, препарат для лечения спинальной мышечной атрофии Spinraza (nusinersen) и др (https://www.fda.gov/drugs/development-approval-process-drugs/drug-approvals-and-atabases).
[003] Многие олигонуклеотидные препараты направлены на лечение заболеваний, вызванных мутациями в одном или нескольких генах пациента. Соответственно, такие препараты направлены на корректировку экспрессии генов, ответственных за развитие заболевания.
[004] Олигонуклеотиды способны ингибировать транскрипцию, трансляцию или модулировать активность продукта целевого гена. Ингибирование транскрипции осуществляется путем связывания ДНК триплекс-образующими олигонуклеотидами [1], в том числе пептидными нуклеиновыми кислотами (PNA) [2]. Ингибирование трансляции, или антисмысловой (антисенс) механизм осуществляется путем блокирования трасляции со специфической мРНК [3]. Модуляция активности белка, кодируемого целевым геном, происходит за счет связывания олигонуклеотида с самим белком или с низкомолекулярным кофактором, например, в случае аптамеров [4].
[005] Большинство известных олигонуклеотидов действуют по антисмысловому механизму, связываясь со специфической мРНК в клетке, однако принцип и условия ингибирования трансляции отличаются для разных типов олигонуклеотидов. Например, малые интерферирующие РНК (siRNA) вызывают каталитическое расщепление специфической матричной РНК (мРНК) путем формирования НК-белкового комплекса RISC, обладающего нуклеазной активностью. Таким образом, взаимодействие siRNA с целевой мРНК приводит к деградации последней, предотвращая трансляцию мРНК на рибосомах в кодируемый ею белок [5].
[006] НК-энзимы - каталитические нуклеиновые кислоты, представляющие собой олигонуклеотиды с характерной вторичной структурой, также вызывают расщепление мРНК. Однако будучи сами катализаторами гидролитического расщепления РНК, НК-энзимы не нуждаются в клеточных белках [6].
[007] Большинство антисмысловых аналогов олигонуклеотидов связываются с мРНК и подавляют трансляцию, действуя по принципу стерического блокирования [7]. К их числу относятся многие аналоги олигонуклеотидов с модифицированным остатком рибозы: 2'-фтор [8], 2'-O-метильные [9], 2'-O-β-метоксиэтильные (2'-МОЕ) [10], LNA [11]. Аналоги с незаряженной межнуклеотидной фосфатной группой: метилфосфонаты [12], фосфотриэфиры [13] и фосфорамиды [14] также действуют по принципу стерического блокирования. Аналогичный механизм осуществляется и в случае производных НК где модифицированными являются одновременно остатки рибозы и фосфатной группы, например пептидные нуклеиновые кислоты (PNA) [15] и морфолиновые олигонуклеотиды (РМО) [16].
[008] Олигонуклеотидные препараты также разрабатываются и применяются для лечения болезней, вызванных такими вирусами, как цитомегаловирус человека (HCMV), ВИЧ (HIV-1), гепатит В (HBV), гепатит С (HСV), вирус Эбола, респираторно-синцитиальный вирус (РСВ), коронавирус SARS-CoV, вызывающий тяжелый острый респираторный синдром (ТОРС) и др. [17-24].
[009] Разработка терапии с использованием антисмысловых олигонуклеотидов является перспективным направлением борьбы с коронавирусом SARS-CoV-2, вызывающим COVID-19. В частности, разрабатываются антисмысловые олигонуклеотиды, комплементарные элементу FSE (frameshift stimulation element) - высококонсервативному участку генома SARS-CoV-2 [25]. Применение олигонуклеотидов для борьбы с коронавирусом SARS-CoV-2 особенно актуально ввиду сложности получения эффективной вакцины из-за высокой скорости мутирования генов, кодирующих белки оболочки вируса.
[0010] Независимо от механизма действия, соединения, пригодные для создания лекарственных препаратов, в частности олигонуклеотиды, должны обладать терапевтическим потенциалом. Под терапевтическим потенциалом понимается целый спектр полезных свойств необходимых для достижения выраженного терапевтического эффекта. В частности, терапевтические олигонуклеотиды должны обладать следующими свойствами:
[0011] а) Улучшенное проникновение в клетки, предпочтительно в отсутствие трансфекционных агентов;
[0012] Улучшенное проникновение обеспечивает достижение биологической мишени и необходимость применения более низких дозировок при применении соответствующих препаратов.
[0013] б) Низкая токсичность для клеток;
Низкая токсичность позволяет применять соответствующие препараты в широком диапазоне концентраций с достижением лучших терапевтических эффектов, без вредных последствий для организма.
[0014] в) Высокая стабильность в биологических средах;
[0015] Высокая стабильность позволяет препарату дольше находиться в организме, достигая нужного терапевтического эффекта при минимальном количестве введений препарата. Кроме того, высокая стабильность в биологических средах обеспечивает возможность разнообразного способа введения препарата.
[0016] г) Способностью формировать прочные и специфические комплексы с биологической мишенью;
[0017] Способность формировать прочные комплексы с биологической мишенью обеспечивает необходимый уровень необратимости процесса воздействия для достижения терапевтического эффекта. При этом специфичность образования соответствующих комплексов обеспечивает отсутствие воздействия на другие биологические мишени, минимизируя побочное воздействие на организм.
[0018] д) Доступностью получения.
[0019] Доступным способом получения соединений можно считать такой способ, который будет совместим с существующими и широко применяемыми протоколами синтеза в различных областях техники.
[0020] Существуют технологические аспекты создания соответствующих классов соединений. Например, возможность доведения олигонуклеотидного препарата до действующего лекарства будет сильно лимитирована при использовании не гибкого, дорогостоящего или слишком специфичного, т.е. малосовместимого с широкоприменяемыми, способа получения. Например, в настоящее время амидофосфитный метод является наиболее эффективным и широко используемым способом синтеза олигонуклеотидов. Протоколы амидофосфитного метода с использованием автоматических ДНК/РНК-синтезаторов известны специалистам в области химии нуклеотидов. Являясь востребованной и быстро развивающейся областью, химия автоматического синтеза нуклеиновых кислот и их производных постоянно пополняется новыми, в том числе коммерчески доступными компонентами для олигонуклеотидного синтеза, таких как различные нуклеозидные и ненуклеозидные мономеры, различные модификаторы, в частности флуоресцентные метки и гасители флуоресценции, а также различные защитные группы.
[0021] Таким образом, при получении олигонуклеотида, обладающего терапевтическим потенциалом, совместимость способа получения с существующими синтетическими протоколами амидофосфитной химии является одним из наиболее критических факторов конкурентного преимущества при создании олигонуклеотидных препаратов.
[0022] Обладание даже одним из перечисленных полезных свойств позволяет рассматривать его как соединение, обладающее терапевтическим потенциалом. Чем большее количество перечисленных полезных свойств сочетает в себе определенный класс соединений, и чем выше степень проявления этих свойств, тем выше терапевтический потенциал данного класса соединений и тем более он перспективен для создания терапевтических препаратов.
[0023] Природные олигонуклеотиды хоть и способны образовывать избирательные комплементарные комплексы с биологической мишенью, не являясь при этом токсичным классом соединений для организма, не обладают достаточной стабильностью в биологических средах и плохо проникают в клетки. Для улучшения перечисленных параметров, в состав олигонуклеотидов вводятся различные химические модификации. Введение различных химических модификаций в состав олигонуклеотида, в частности, позволяет повысить эффективность его проникновения через клеточную мембрану, устойчивость к ферментативному гидролизу, устойчивость в большом диапазоне рН, специфичность и стабильность комплекса, образуемого с комплементарным участком целевой нуклеиновой кислоты, при этом сохраняя низкий уровень токсичности для организма [26].
[0024] В структуре олигонуклеотида присутствует несколько положений для модификации - азотистые основания, остаток рибозы, межнуклеотидная фосфатная группа.
[0025] Модификация по межнуклеотидной фосфатной группе выгодно отличаются от остальных позиций для введения неприродных химических групп. Введение модификаций через фосфатную группу остова незначительно влияет на фундаментальное свойство олигонуклеотидов - способностью формировать прочные и специфические комплексы с биологической мишенью. При этом введение различных групп способно наделить создаваемое соединение новыми свойствами в широком диапазоне.
[0026] Известны различные варианты модификаций межнуклеотидных фосфатных групп, например, метилфосфонаты [27], тиофосфаты [28,29], дитиофосфаты [30], боранофосфаты [31], (WO1991008213A1, опубл. 13.06.1991; МПК A61K 31/69, A61K 31/70, A61K 31/7135, A61P 29/00, A61P 3/06, A61P 35/00, C07H 21/00, C07H 21/04, C07H 23/00, C12N 15/113, C12Q 1/68), амидофосфаты, фосфорилгуанидины и другие.
[0027] Метилфосфонатные олигонуклеотиды обладают высокой устойчивостью к ферментативному гидролизу нуклеазами, а также несколько увеличенной степенью образования комплементарного комплекса. В то же время, метилфосфонатные олигонуклеотиды являются химически нестойкими и легко подвергаются щелочному гидролизу. Кроме того, для получения метилфосфонатных олигонуклеотидов применяются способы получения отличные от амидофосфитного синтеза, что снижает эффективность их получения и лишает производителей возможности использования широкого спектра коммерчески доступных мономеров и модификаторов. Необходимость использования специальных мономеров значительно снижает перспективы метилфосфонатных олигонуклеотидов как платформы для создания терапевтических препаратов [32, 33].
[0028] Боранофосфатные олигонуклеотиды обладают повышенной ферментативной и химической стабильностью. В отличие от метилфосфонатных модификаций боранофосфатные модификации не приводят к исчезновению отрицательного заряда на фосфатной группе. В результате метилфосфонатные олигонуклеотиды способны рекрутировать РНКазу Н для расщепления гибридного ДНК/РНК комплекса. Такой механизм воздействия на целевую мишень, в отличие от простого стерического блокирования, позволяет работать терапевтическому агенту в каталитическом режиме. Однако образуемые с такими модифицированными олигонуклеотидами комплементарные комплексы менее стабильны по сравнению с природными олигонуклеотидами. Кроме того, способы получения боранофосфатных олигонуклеотидов также несовместимы с амидофоситным синтезом, и также требуют получения набора специальных мономеров [34, 35].
[0029] Амидофосфатные олигонуклеотиды содержат N-замещенную амино группу вместо атома кислорода в составе фосфатной группы. За счет нейтрализации отрицательного заряда фосфатной группы, амидофосфатные олигонуклеотиды более устойчивы к действию нуклеаз. Заместители при аминогруппе могут быть источником введения разнообразных функциональных групп в состав создаваемого олигонуклеотида. Однако основным недостатком амидофосфатов, ограничивающим их применение, является подверженность кислотному гидролизу вследствие протонирования аминогруппы [36-38].
[0030] Тиофосфатные олигонуклеотиды - один из немногих классов соединений, нашедший применение в области создания терапевтических НК. Тиофосфатные олигонуклеотиды стабильны к действию клеточных нуклеаз, их синтез совместим с протоколами твердофазного амидофосфитного синтеза. Однако данный тип модификаций не подразумевает никакого разнообразия вводимых групп, кроме как замены атома кислорода на атом серы. Таким образом, изменение свойств в рамках данного класса может быть достигнуто лишь варьированием количества и места введения тиофосфатных звеньев. Также тиофосфатные олигонуклеотиды обладают относительно высокой токсичностью и несколько пониженной способностью образовывать комплексы с НК, по сравнению немодифицированными олигонуклеотидами. Вероятно, именно эти недостатки сдерживают широкое применение данного класса соединений в качестве терапевтических препаратов [39].
[0031] Дитиофосфатные олигонуклеотиды способны активировать РНКазу Н, хотя и с меньшей эффективностью, чем тиофосфатные олигонуклеотиды. При этом дитиофосфатные олигонуклеотидыеще более устойчивы к действию нуклеаз. Однако из-за повышенного содержания серы дитиофосфатные олигонуклеотиды менее специфичны в ингибировании трансляции засчет более сильного связывания с белками. Кроме того, их химический синтез по сравнению с тиофосфатами более сложен.
[0032] К модифицированным по фосфатной группе олигонуклеотидам можно также отнести морфолиновые олигонуклеотиды, у которых изменен весь рибозо-фосфатный остов на морфолино-фосфордиамидный. Такой остов крайне далек от природного, что исключает возможность взаимодействия морфолиновых олигонуклеотидов с любыми НК-зависимыми ферментами, в том числе и с РНКазой Н. Морфолиновые олигонуклеотиды стабильны к действию нуклеаз, а также способны образовывать достаточно прочные комплементарные комплексы с НК. Однако данный класс соединений содержит лишь один представитель, что делает крайне затруднительным варьирование свойств морфолиновых олигонуклеотидов. Кроме того, получение морфолиновых олигонуклеотидов требует специализированного оборудования, мономеров и реактивов. Используемые способы получения морфолиновых олигонуклеотидов несовместимы со стандартным амидофосфитным синтезом [16, 40, 41].
[0033] Стоит отметить что все выше перечисленные модификации, кроме тиофосфатов и морфолиновых олигонуклеотидов, несмотря на давнюю известность не нашли применения в области создания терапевтических олигонуклеотидов. Часто основной причиной этому служит именно низкий уровень сочетаемости способов их получения со стандартным амидофосфитным методом синтеза.
[0034] Недавно открытый класс фосфорилгуанидиновых олигонуклеотидов также является классом соединений, содержащих модификацию по межнуклеотидной фосфатной группе (RU2708237C2, опубл. 05.12.2019; МПК: A61K 31/712, A61P 31/12, C07F 9/24, C07H 19/10, C07H 19/20). В данном случае это остаток замещенного или незамещенного гуанидина, что делает получаемую фосфорилгуанидиновую группу электронейтральной. Представители данного класса олигонуклеотидов стабильны к действию клеточных нуклеаз и устойчивы в широком диапозоне рH. Также при введение достаточно большого количества таких модификаций в состав создаваемого олигонуклеотида, предпочтительно по всем межнуклеотидным фосфатным группам, можно добиться незначительного улучшения проникновения в клетки. Однако при введении большого количества модификаций в состав олигонуклеотида, его остов оказывается сильно отличным от природного рибозо-фосфатного остова. В результате такие модифицированные олигонуклеотиды теряют способность к взаимодействию с НК-зависимыми ферментами, в частности с РНКазой Н, что сильно ограничивает доступные механизмы терапевтического действия данного класса соединений.
[0035] Таким образом, существует необходимость в создании соединений, обладающих высоким терапевтическим потенциалом, получаемых доступным легко масштабируемым способом.
ТЕРМИНЫ И ОПРЕДЕЛЕНИЯ
[0036] Алкил - разветвленный или неразветвленный циклический или ациклический заместитель на основе насыщенных углеводородов со свободной валентностью на атоме углерода. Например, C1-4алкильные группы включают в себя, в том числе, метильный, этильный, н-пропильный, изопропильный или трет-бутильный радикалы.
[0037] Алкенил - разветвленный или неразветвленный ациклический заместитель со свободной валентностью на атоме углерода на основе ненасыщенных углеводородов, содержащих, по крайней мере, одну углерод-углеродную двойную связь, который может содержать или не содержать углерод-углеродных тройных связей.
[0038] Алкинил - разветвленный или неразветвленный ациклический заместитель со свободной валентностью на атоме углерода на основе ненасыщенных углеводородов, содержащих, по крайней мере, одну углерод-углеродную тройную связь. При этом алкинил может содержать или не содержать двойные углерод-углеродные связи.
[0039] Арил - понимается ароматическая или гетероароматическая органическая группа со свободной валентностью на атоме углерода или, в некоторых вариантах реализации, на гетероатоме. Примеры ароматических групп могут включать в себя, в том числе, фенил и нафтил (1-нафтил или 2-нафтил). Арильные группы могут быть моноциклическими или полициклическими.
[0040] Защитная группа - это химическая группа, которая используется для временного блокирования реакционного сайта в органическом соединении и может удаляться в определенных условиях.
[0041] Линкер - ненуклеотидная химическая группа, которая может соединять соседние нуклеотиды в олигонуклеотиде (межнуклеотидный линкер); или соединять нуклеотид или его аналог с другой ненуклеотидной группой; или соединять олигонуклеотид или его аналог с полимерным носителем; или соединять нуклеозид или его аналог с полимерным носителем.
[0042] Нуклеозид - химическое соединение, содержащее остаток сахара и остаток гетероциклического основания. Примеры нуклеозидов могут включать, в том числе, рибозу, 2-дезоксирибозу, арабинозу и т.п. Примеры гетероциклических оснований могут включать в себя, в том числе, тимин, урацил, цитозин, аденин, гуанин, пурин, гипоксантин, ксантин, 2-аминопурин, 2,6-диаминопурин, 5-метилцитозин. 5-фторурацил, 5-хлорурацил. 5-бромурацил. 5-иодурацил. 5-трифторметилурацил. 5-фторцитозин. 5-хлорцитозин. 5-бромцитозин. 5-иодцитозин. 2-тиоурацил, 4-тиоурацил, 2-тиотимин, 4-тиотимин, 5-пропинилурацил. 5-пропинилцитозин, 7-деазааденин, 7-деазагуанин, 7-деаза-8-азааденин, 7-деаза-8-азагуанин, изоцитозин, изогуанин и т.п.
[0043] Под термином «нуклеозид» также может подразумеваться защищенный нуклеозид, аналог нуклеозида и защищенный аналог нуклеозида.
[0044] Аналог нуклеозида - используется для обозначения модифицированного нуклеозида, в котором остаток сахара заменён иной циклической или ациклической структурой. Примеры аналогов нуклеозидов, в которых остаток сахара заменён иной циклической структурой, могут включать в себя, в том числе, мономеры морфолиноолигонуклеотидов (РМО) и трицикло-ДНК. Примеры аналогов нуклеозидов, в которых остаток сахара заменён иной ациклической структурой, могут включать в себя, в том числе, мономеры пептидных нуклеиновых кислот (РNA) и глицериновых нуклеиновых кислот (GNA). Также, термин «аналог нуклеозида» используется для обозначения нуклеозида, содержащего химическую модификацию, например, заместитель в остатке сахара и/или в гетероциклическом основании. Примеры таких аналогов нуклеозидов могут включать в себя, в том числе, 2'-замещённые 2'-дезоксинуклеозиды, такие как 2'-амино и 2'-фтор, и рибонуклеозиды, такие как 2'-О-метил, 2'-О-аллил, 2'-О-β-метоксиэтилрибонуклеозиды, «замкнутые» нуклеозиды (LNA) и т.п.
[0045] В том числе аналоги нуклеозидов могут включать в себя аналоги, в которых остаток сахара заменён морфолиновым кольцом, как показано на формуле ниже:
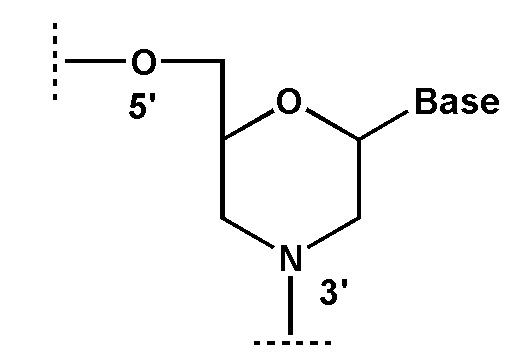
[0046] где Base - гетероциклическое основание.
[0047] В структурах данного типа обозначения 3' и 5' применяются по аналогии с природными нуклеозидами. В частности, на показанной выше структуре гидроксиметильный заместитель в морфолиновом кольце соответствует 5'-концу, а третья валентность атома азота - 3'-концу нуклеозида.
[0048] Защищенный нуклеозид - нуклеозид, содержащий одну или более защитных групп. Защищенным может быть и аналог нуклеозида. Например, DMTr-нуклеозид содержит DMTr защитную группу на 5'-конце.
[0049] Нуклеотид - химическое соединение, содержащее нуклеозид и хотя бы одну фосфатную группу, присоединенную к нему ковалентной связью. Примером ковалентной связи независимо и без ограничений является эфирная связь между 3'-, 2'- или 5'-гидроксильной группой нуклеозида и фосфатной группой.
[0050] Под термином «нуклеотид» также могут подразумеваться аналог нуклеотида.
[0051] Аналог нуклеотида - химическое соединение, содержащее аналог нуклеозида и хотя бы одну фосфатную группу, присоединенную к нему ковалентной связью. Примеры аналогов нуклеотидов с замененным остатком сахара могут включать, не ограничиваясь этим, 2'-замещенные 2'-дезоксинуклеотиды, такие как 2'-амино и 2'-фтор, и рибонуклеотиды, такие как 2'-О-метил, 2'-O-аллил, 2'-O-β-метоксиэтилрибонуклеотиды, «замкнутые» нуклеотиды (LNA), морфолино-нуклеотиды, трицикло-дезоксирибонуклеотиды, гликоль-нуклеотиды.
[0052] Олигонуклеотид - химическое соединение, состоящее из двух или более нуклеотидов, соединенных между собой в полимерную цепь. Олигонуклеотид может представлять собой фрагмент ДНК или РНК. Олигонуклеотиды могут быть одноцепочечными или двуцепочечными, т.е. содержать две цепи с высокой степенью комплементарности. При этом любая из цепей или обе могут быть модифицированы согласно настоящему изобретению. Ключевым признаком олигонуклеотида является способность образовывать устойчивые дуплексы с комплементарными участками НК и их производных за счет нековалентной связи. Примером подобной нековалентной связи может служить водородная связь.
[0053] Под термином «олигонуклеотид» также может подразумеваться аналог олигонуклеотида или модифицированный олигонуклеотид, содержащий модификацию, не охватываемую настоящим изобретением.
[0054] Аналог олигонуклеотида - вариант олигонуклеотида, который включает не менее одного аналога нуклеотида, и при этом общее количество нуклеотидов и/или аналогов нуклеотидов составляет два или более. При этом аналог олигонуклеотида может полностью состоять из аналогов нуклеотидов. При этом аналог олигонуклеотида может включать по меньшей мере одну фосфатную группу, которая может быть модифицирована согласно настоящему изобретению.
[0055] Аналоги олигонуклеотидов могут содержать, например, химические группировки на 3'- и/или 5'-конце олигонуклеотида (например, остатка 3'-«инвертированного» нуклеозида), остатки высокомолекулярного соединения низкой иммуногенности (например, полиэтиленгликоля ), низкомолекулярные соединения (например, холестерин), пептиды (например, пептиды, облегчающие проникновение в клетки), фосфатные группы с модификациями, не охватываемыми настоящим изобретением (например, тиофосфатную группу). Аналоги олигонуклеотидов также могут содержать модифицированные гетероциклические основания. Например, химическая модификация гетероциклических оснований может включать в себя, в том числе, замещение по С-5 пиримидинового нуклеотида, замещение по С-7 7-деазапуринового нуклеотида, замещение по экзоциклической аминогруппе, введение остатков 4-тиоурацила, 5-бром- и/или 5-иодурацила и пр. Аналоги олигонуклеотидов также могут содержать модифицированные остатки сахара. Например, модификация остатка сахара может включать в себя введение 2'-аминонуклеотида, 2'-фторнуклеотида, 2'-O-метилрибонуклеотида, 2'-O-аллилрибонуклеотида, 2'-O-β-метоксиэтилрибонуклеотида, «замкнутого» нуклеотида (locked nucleic acid, LNA) и/или трицикло-ДНК нуклеотида. Кроме того, в аналогах олигонуклеотидов связи между центральным атомом фосфора в фосфатной группе могут осуществляться, в том числе, через атом кислорода (обычный фосфат), атом азота (N3'-P5' фосфорамид) или атом серы (3'-тиофосфат); соответственно, 3'- и/или 5'-конец нуклеозида может оканчиваться, в том числе, гидроксильной группой, как в природном нуклеозиде, 3'-аминогруппой (N3'-Р5' фосфорамид) или 3'-меркаптогруппой (3'-тиофосфат). Примеры аналогов олигонуклеотидов также могут включать, в том числе, тиофосфаты (PS), селенофосфаты, дитиофосфаты, фосфорамиды, боранофосфаты, фосфордиамидные производные морфолиноолигонуклеотидов (РМО), трицикло-ДНК, фосфорилгуанидиновые олигонуклеотиды (PGO) и пептидные нуклеиновые кислоты (PNA). В PNA фосфатные группы заменены пептидными связями. Однако существуют варианты PNA, включающие фосфатные группы, которые могут быть модифицированы согласно настоящему изобретению.
[0056] Защищенный олигонуклеотид - олигонуклеотид, содержащий одну или более защитных групп.
[0057] Фосфатная группа - остаток фосфорной кислоты H3PO4, в котором один или более атомов водорода замещены органическим радикалом с получением, соответственно, фосфомоноэфира, фосфодиэфира или фосфотриэфира. Под термином фосфатная группа также может подразумеваться модифицированная фосфатная группа.
[0058] Модифицированная фосфатная группа - фосфатная группа, в которой любой из атомов кислорода замещен любой химической группой. Примерами заместителей могут быть, в том числе, атомы серы, селена, иминогруппа (-NHR), остаток борана (-ВН3-), остаток замещенного или незамещенного гуанидина. Предпочтительными примерами модифицированной фосфатной группы являются тиофосфатная группа, фосфорамидная группа, фосфорилгуанидиновая группа.
[0059] Финальное деблокирование - удаление защитных групп и отщепление олигонуклеотида или его аналога от твердофазного носителя (в случае, когда способ получения олигонуклеотида или его аналога реализуется в твердофазном варианте).
[0060] Терапевтический олигонуклеотид - олигонуклеотид или аналог олигонуклеотида, обладающий терапевтическим потенциалом, длиной 5-1000 нуклеотидов, используемый в составе лекарства при терапии рака, геномных нарушений и инфекционных заболеваний различной природы, например, в качестве ASO для ингибирования трансляции (siRNA, микроРНК), для модуляции сплайсинга за счет пропуска экзона, для коррекции генотипа с помощью метода CRISPR/Cas, а также в качестве аптамеров для специфичного ингибирования белков-мишеней или направленного транспорта лекарств.
[0061]
СОКРАЩЕНИЯ И УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
[0062] ВИЧ - вирус иммунодефицита человека
[0063] ДНК - дезоксирибонуклеиновая кислота
[0064] НК - нуклеиновая кислота (ДНК или РНК)
[0065] офВЭЖХ - обращённо-фазовая высокоэффективная жидкостная хроматография ВЭЖХ (RP-HPLC)
[0066] РНК - рибонуклеиновая кислота
[0067] ASO - antisense oligonucleotide, антисмысловой олигонуклеотид
[0068] DMF - диметилформамид
[0069] DMTr - 4,4'-диметокситритильная группа
[0070] FAM - 6-карбоксифлуоресцеин
[0071] FBS - Fetal Bovine Serum, фетальная бычья сыворотка
[0072] HEK293T - Human Embryonic Kidney 293, клеточная линия, полученная из эмбриональных почек человека
[0073] HepG2 - клеточная линия, полученная из гепатоцеллюлярной карциномы человека
[0074] IMDM - Iscove's Modified Dulbecco's Medium, среда для культивации клеток млекопитающих; модификация среды DMEM (Dulbecco′s Modified Eagle′s Medium), содержащая селенит натрия, добавочные аминокислоты и витамины, пируват натрия, HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) и нитрат калия вместо нитрата железа.
[0075] LNA - locked nucleic acid, «замкнутая нуклеиновая кислота»
[0076] МЕ - Международная Единица, единица измерения дозы вещества, основанная на его биологической активности. Количества вещества в 1 МЕ для разных классов веществ разные. Единицы действия, ЕД, чаще всего совпадают с МЕ.
[0077] МЕМ - Minimum Essential Medium, среда Игла; среда для культивирования клеточных культур, содержит буферный раствор для поддержания оптимального рН 7,4, глюкозу, аминокислоты, витамины и другие вещества.
[0078] mQ - деионизированная вода, вода I типа (сверхчистая вода)
[0079] Opti-MEM - среда для культивирования клеточных культур; является модификацией минимальной среды MEM, рекомендованной для трасфекции.
[0080] PBS - Phosphate buffered saline, изотонический натрий-фосфатный буфер, представляющий собой водный раствор солей, содержащий хлорид натрия, гидрофосфат натрия, хлорид калия и дигидрофосфат калия. Осмолярность и концентрации ионов в растворе приблизительно соответствуют концентрациям в теле человека.
[0081] РG - protective group, защитная группа
[0082] РМО - phosphorodiamidate morpholino oligomer, олигонуклеотиды на основе морфолинов.
[0083] PNA - peptide nucleic acid. Термин «пептидные нуклеиновые кислоты» обычно относят к аналогам олигонуклеотидов, в которых, в том числе, фосфатные группы заменены пептидными связями. Однако, термин «пептидные нуклеиновые кислоты» может также включать в себя соединения, которые содержат модифицированные фосфатные группы, являющиеся предметом настоящего изобретения. Таким образом, следует считать, что такого рода соединения также могут быть охвачены данным изобретением.
[0084] siRNA - small interfering RNA, малые интерферирующие РНК
[0085] T98G - клеточная линия, полученная из глиобластомы человека
[0086] THF - тетрагидрофуран
[0087] Ts - тозил
[0088] * - в Примерах обозначает положение заявленной модификации в составе олигонуклеотида
[0089] d - в Примерах обозначает положение додециламидофосфатной группы в составе олигонуклеотида.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0090] Задачей настоящего изобретения является создание соединений, обладающих терапевтическим потенциалом, а также разработка доступного способа их получения.
[0091] Данная задача решается заявляемым изобретением за счет достижения такого технического результата, как создание соединений, в том числе олигонуклеотидов, которые могут обладать одним или несколькими из следующих свойств:
[0092] а) улучшенным проникновением в клетки, предпочтительно в отсутствие трансфекционных агентов;
[0093] б) низкой токсичностью для клеток;
[0094] в) высокой химической и ферментативной устойчивостью;
[0095] г) способностью формировать прочные и специфические комплексы с биологической мишенью.
[0096] Также важной частью технического результата является возможность получения заявляемых соединений доступным способом (д), т.е способом совместимым с существующими широко применяемыми методами синтеза.
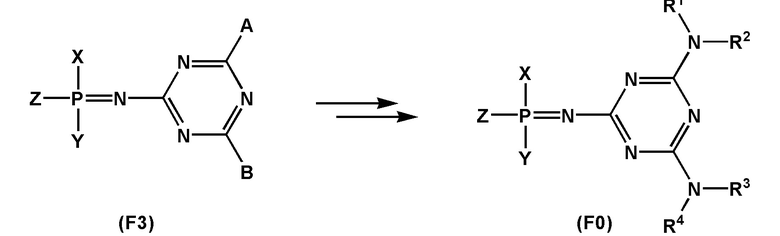
[0097] Заявленный технический результат достигается за счет того, что заявленное изобретение включает соединения, соответствующие формуле F0:
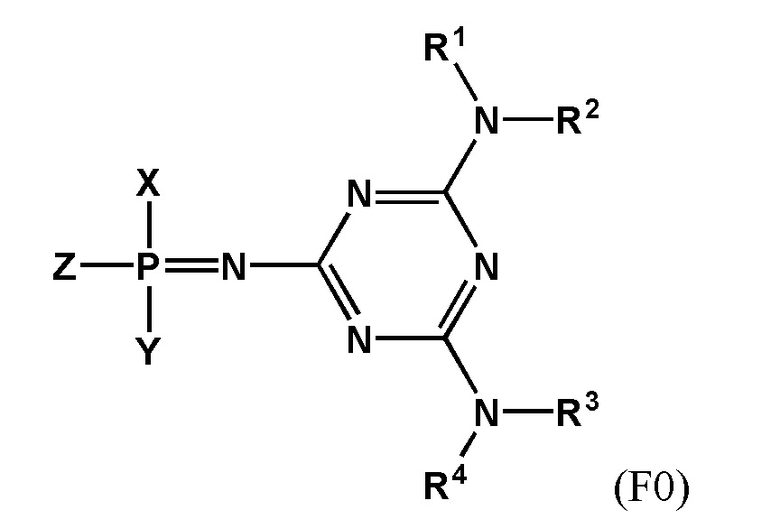
[0098] В котором заместитель Z выбран из группы: -OН, -SН, -SeН, -NНRN, -O-PG, -S-PG, -Se-PG или -N(PG)RN.
[0099] В одном варианте реализации Х выбран из группы, состоящей из 5'-O–конца нуклеозида или олигонуклеотида, а Y выбран из группы, состоящей из 3'-O–конца нуклеозида или олигонуклеотида, –H, –OH, –SH, –NHRN, –O-PG или –S-PG, линкера, монофосфата или дифосфата.
[00100] В другом варианте реализации Y выбран из группы, состоящей из 5'-O-конца нуклеозида или олигонуклеотида, а X выбран из группы, состоящей из 3'-O-конца нуклеозида или олигонуклеотида, -H, -OH, -SH, -NHRN, -O-PG или -S-PG, линкера, монофосфата или дифосфата.
[00101] Заместители R1, R2, R3, R4 выбраны из ряда –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать различные липофильные и/или катионные группы.
[00102] Используя различные заместители R1, R2, R3 и R4 в составе триазиновой группы, можно широко варьировать липофильность и заряд заявленных соединений. Таким образом можно модулировать параметры биораспеределения в организме, в частности, способность проникать в определенный тип клеток. Это может применяться при создании терапевтических препаратов направленного действия.
[00103] Остаток триазиновой группы придает химическую и ферментативную устойчивость заявленным соединениям. Кроме того, фосфатная группа, модифицированная согласно настоящему изобретению, является электронейтральной, что повышает способность заявленных соединений проникать через клеточную мембрану. Эти свойства характерны для всех заявленных соединений независимо от заместителей, входящих в состав триазинового остатка.
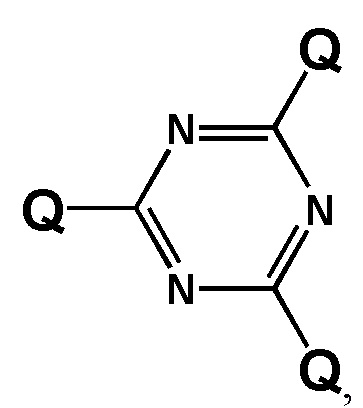
[00104] Предметом настоящего изобретения может являться олигонуклеотид, включающий по меньшей мере одну модифицированную фосфатную группу, соответствующую формуле Fx:
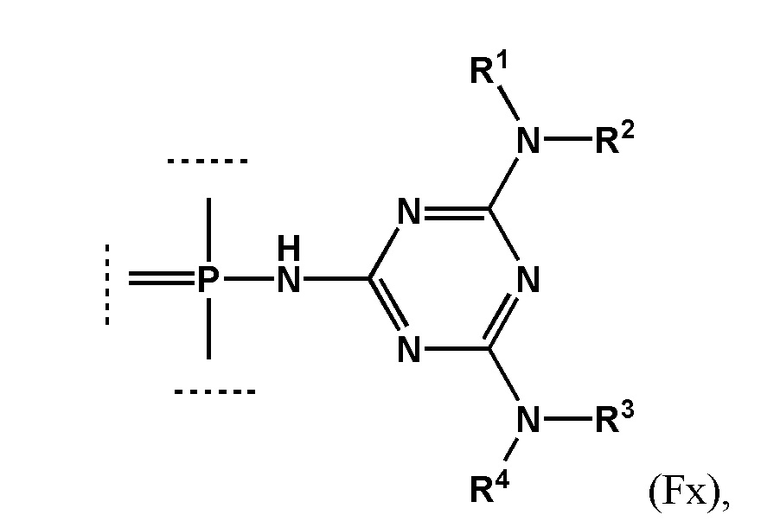
где - показывает место присоединения соответствующих олигонуклеотиду заместителей.
[00105] Наличие даже одной заявленной модификации может улучшать терапевтический потенциал создаваемого соединения, в частности, способность олигонуклеотида проникать через клеточные мембраны. Это позволяет создавать модифицированные олигонуклеотиды с желаемой эффективностью и специфичностью проникновения в клетки при минимальной степени модифицированности олигонуклеотида. Минимизация количества модификаций заметно упрощает процесс получения заявленного олигонуклеотида, и получаемый остов соединения сохраняет способность к взаимодействию с клеточными ферментами. Не менее важно, что заявленная модификация, не ослабляет способность модифицированных олигонуклеотидов образовывать специфические комплексы с комплементарными участками НК или их производными.
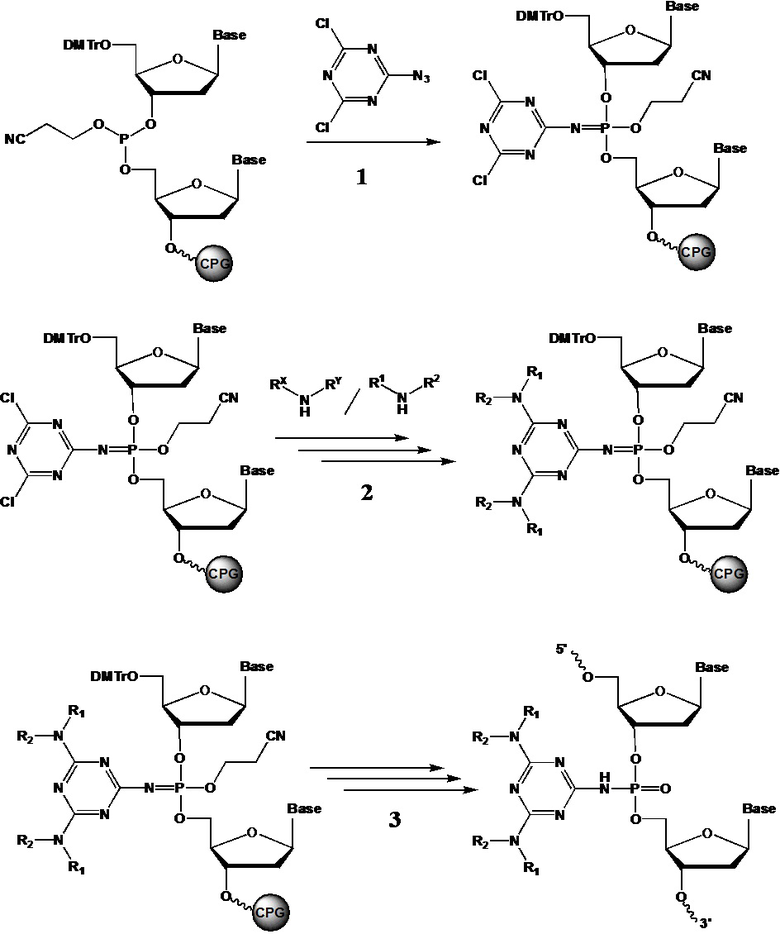
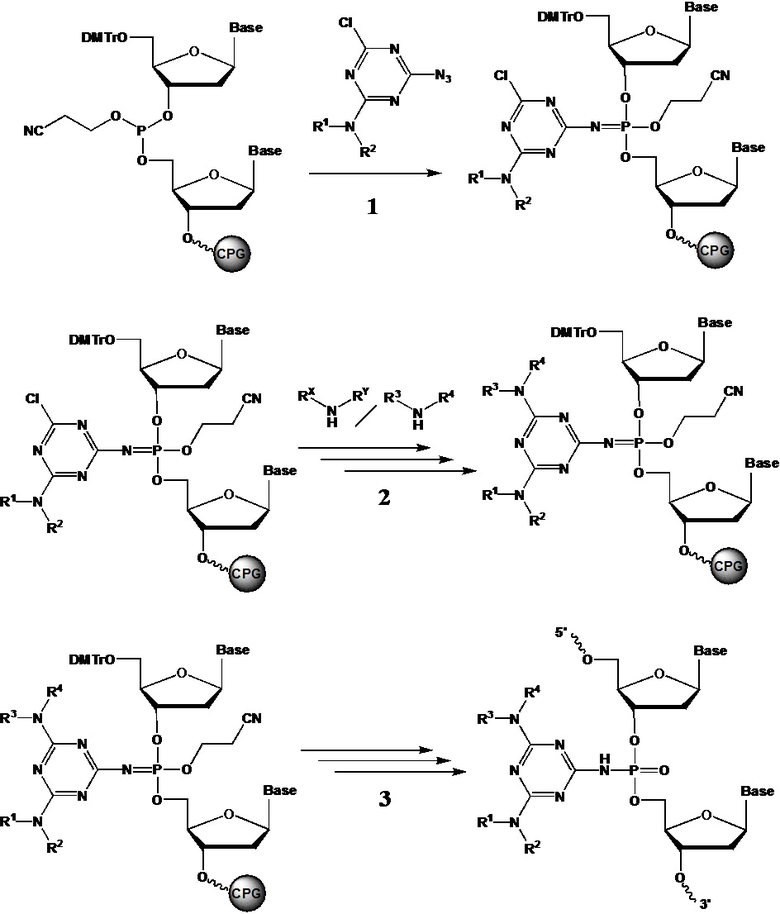
[00106] Предметом настоящего изобретения также является способ получения соединения, соответствующего формуле (F0). Способ заключается во взаимодействии производного трехвалентного фосфора формулы (F1) с азидотриазином формулы (F2) с получением соединения формулы (F3), c последующим обработкой аминами HNR1R2, HNR3R4 или HNRXRY. При этом заместители RX и RY будут преобразованы в заместители R1, R2, R3, R4 с использованием известных в области техники реакций превращения соответствующих реакционных групп, входящих в состав RX и RY.
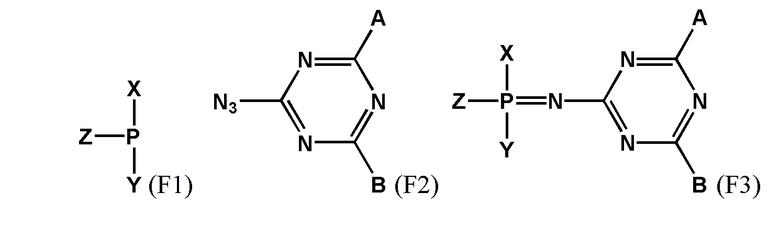
[00107] Заместители X, Y, Z определены так же, как они определены в формуле (F0).
[00108] A и B могут быть независимо выбраны из ряда –NR1R2, –NR3R4, –NRXRY, –Q.
[00109] Заместители R1, R2, R3, R4, R как они описаны в п. 1определены так же, как они определены в формуле (F0).
[00110] Q - это группа, способная вступать в реакции замещения. Q выбирается из ряда: –OR, –OC(O)R, –OS(O)2R, –CN, –Cl, –Br, –I, –F, –N3. Группа Q может быть замещена на –NR1R2, –NR3R4, –NRXRY в реакции с соответствующим амином HNR1R2, HNR3R4, HNRXRY.Заместители RX, RY выбраны из ряда, содержащего –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2– и/или –N(CH2CH2)2N–, а также оканчиваться группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2))2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3.
[00111] При этом R представляет собой заместитель, выбранный из ряда –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2– и/или –N(CH2CH2)2N– и могут оканчиваться группами –NRN2, –ORN, –SRN, –OC(O)RN, –NHC(O)RN, –C(O)ORN, –C(O)NHRN, –N=C(N(R2))2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3. При этом заместители R1, R2, R3, R4 определены так же, как они определены в формуле (F0).
[00112] В одном из вариантов реализации способа A = –NR1R2, B = –NR3R4. В этом случае получение соединения, соответствующего формуле (F0), не требует дополнительных химических преобразований.
[00113] В другом варианте реализации способа A = B = –Q. Еще в одном варианте реализации способа A = –NR1R2, B = –Q. В обоих случаях, когда один или оба заместителя A и B представляют собой –Q, группу Q замещают на –NR1R2, –NR3R4, –NRXRY в реакции с соответствующим амином HNR1R2, HNR3R4, HNRXRY. Замещение может проводиться как в составе соединения формулы (F2), так и в составе соединения формулы (F3).
[00114] Заместители RX и RY содержат реакционно способные центры, которые дополнительно подвергают химическому превращению или последовательности химических превращений, приводя структуры заместителей RX и RY к структурам R1, R2, R3, R4. Причем преобразование RX, RY в R1, R2, R3 или R4 может проводиться как в составе соединения формулы (F2), так и в составе соединения формулы (F3).
[00115] Заместители R1, R2, R3, R4, RX и RY могут широко определяться использованием соответствующих коммерческих и отдельно синтезированных аминов HNR1R2, HNR3R4 и HNRXRY, что обеспечивает гибкость выбранного способа получения и вариативность получаемого класса соединений с общей формулой (F0). При этом RX и RY могут быть преобразованы в R1, R2, R3 или R4 в несколько последовательных этапов. Такая многоэтапность преобразований RX и RY также обеспечивает возможность получения широко вариативного ряда заявленных соединений без усложнения способа.
[00116] В одном из воплощений способа, относящему к получению олигонуклеотида, производное трехвалентного фосфора - это Н-фосфонатное звено, полученное согласно Н-фосфонатному методу олигонуклеотидного синтеза, или фосфитное звено, полученного согласно амидофосфитному методу олигонуклеотидного синтеза.
[00117] При этом способ получения модифицированных олигонуклеотидов согласно настоящему изобретению может быть реализован в жидкофазном варианте, когда все реагенты во всех реакциях находятся в растворе. В предпочтительном варианте реализации заявленный способ осуществляется в твердофазном варианте, где синтезируемый олигонуклеотид иммобилизован на твердофазном носителе. В качестве твердофазного носителя может быть использован полимерный носитель.
[00118] В предпочтительном варианте реализации конденсация всех амидофосфитных звеньев производится в автоматическом режиме с помощью ДНК-синтезатора. При этом триазиновая группа может быть введена в любую межнуклеотидую фосфатную группу синтезируемого олигонуклеотида.
[00119] В рамках настоящего изобретения модифицированными могут быть одна и более межнуклеотидных фосфатных групп в составе олигонуклеотида. В одном из вариантов реализации модифицированными могут быть все межнуклеотидные фосфатные группы в составе олигонуклеотида. Однако в предпочтительном варианте реализации олигонуклеотид содержит одну или две модифицированных межнуклеотидных фосфатных групп.
[00120] Одним из достоинств заявленного способа является возможность его совмещения со стандартным протоколом амидофосфатного синтеза, что заметно упрощает и удешевляет получение модифицированных олигонуклеотидов.
[00121] Не менее важным достоинством заявленного способа является большая вариативность получаемых соединений, которая обеспечивается иерархической последовательностью этапов получения целевого соединения.
[00122] Настоящее изобретение может применяться в цитологических исследованиях, в молекулярной диагностике, в области создания терапевтических препаратов, направленных на лечение рака, геномных нарушений, различных заболеваний бактериальной и вирусной природы, в том числе, COVID-19. В частности, олигонуклеотиды, модифицированные согласно настоящему изобретению, могут быть использованы в таких методиках, как, например, альтернативный сплайсинг, антисмысловая микроРНК- и siRNA-терапия, использование аптамеров, редактирование генома с помощью CRISPR/Cas и другие.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
[00123] На Фиг. 1 приведены примеры хроматографического анализа (офВЭЖХ) получаемых соединений.
[00124] На Фиг. 2 приведены примеры масс-спектрометрического анализа (ESI MS) получаемых соединений.
[00125] На Фиг. 3 показаны результаты исследования химической устойчивости модифицированного олигонуклеотида в щелочных условиях.
[00126] На Фиг. 4 показаны результаты исследования химической устойчивости модифицированных олигонуклеотидов в кислой среде.
[00127] На Фиг. 5 показаны результаты исследования ферментативной устойчивости триазиниламидофосфатных модифицированных олигонуклеотидов.
[00128] На Фиг. 6 представлены результаты исследования эффективности проникновения модифицированного олигонуклеотида в культивируемые клетки человека HEK293T, T98G.
[00129] На Фиг. 7 показаны результаты лазерной конфокальной микроскопии клеток человека, трансфицированных модифицированным олигонуклеотидом.
[00130] На Фиг. 8 представлены результаты исследования эффективности проникновения в клетки человека олигонуклеотидов, содержащих додецильные остатки в составе модификаций с различными остовами.
[00131] На Фиг 9 приведены результаты исследования эффективности проникновения в клетки человека HEPG2 модифицированных олигорибонуклеотидов.
[00132] На Фиг. 10 представлены результаты исследования цитотоксичности модифицированных олигонуклеотидов.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[00133] В приведенном ниже подробном описании реализации изобретения приведены многочисленные детали реализации, призванные обеспечить отчетливое понимание настоящего изобретения. Однако, квалифицированному в предметной области специалисту, очевидно, каким образом можно использовать настоящее изобретение, как с данными деталями реализации, так и без них. В других случаях хорошо известные методы, процедуры и компоненты не описаны подробно, чтобы не затруднять излишне понимание особенностей настоящего изобретения.
[00134] Кроме того, из приведенного изложения ясно, что изобретение не ограничивается приведенной реализацией. Многочисленные возможные модификации, изменения, вариации и замены, сохраняющие суть и форму настоящего изобретения, очевидны для квалифицированных в предметной области специалистов.
[00135] Настоящее изобретение относится к соединениям, соответствующим формуле F0:
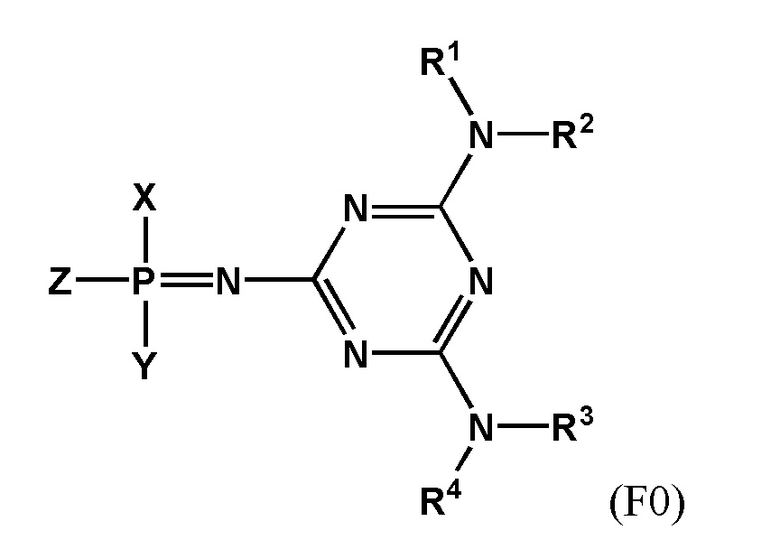
[00136] В котором заместитель Z выбран из группы: –OН, –SН, –SeН, –NНRN, –O-PG, –S-PG, –Se-PG или –N(PG)RN.
[00137] В одном варианте реализации Х выбран из группы, состоящей из 5'-O–конца нуклеозида или олигонуклеотида, а Y выбран из группы, состоящей из 3'-O–конца нуклеозида или олигонуклеотида, –H, –OH, –SH, –NHRN, –O-PG или –S-PG, линкера, монофосфата или дифосфата.
[00138] В другом варианте реализации Y выбран из группы, состоящей из 5'-O–конца нуклеозида или олигонуклеотида, а X выбран из группы, состоящей из 3'-O–конца нуклеозида или олигонуклеотида, –H, –OH, –SH, –NHRN, –O-PG или –S-PG, линкера, монофосфата или дифосфата.
[00139] Примеры защитных групп (PG) могут включать, в том числе, ацетильную (Ас), бензоильную (Bz), изобутирильную (Ibu), m-бутилфеноксиацетильную (Tac), левулинильную (Lev), метильную (Me), β-цианэтильную (СЕ), аллильную (All), о-хлорфенильную (o-ClPh), 4,4'-диметокситритильную (DMTr), 4-метокситритильную (MMTr), m-бутилдиметилсилильную (TBDMS), триизопропилсилилоксиметильную (ТОМ) и другие группы.
[00140] Примеры линкеров могут включать, в том числе, сукцинильный, дигликолильный, оксалильный, гидрохинон-O,O'-диацетильный (Q-линкер), фталоильный, 4,5-дихлорфталоильный, малонильный, глутарильный, диизопропилсилильный, 1,1,3,3-тетраизопропилдисилоксан-1,3-диильный, BHQ-линкер, аминолинкер и другие линкеры.
[00141] Заместители R1, R2, R3, R4 выбраны из ряда –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2–, –N(CH2CH2)2N– и/или  , а также оканчиваться группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2)2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3,
, а также оканчиваться группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2)2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3,
 или
или 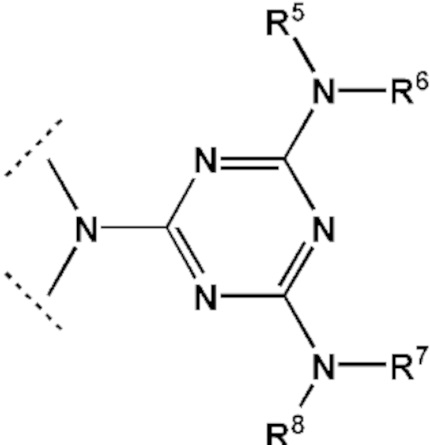 ,
,
[00142] Заместители R5, R6, R7, R8 выбраны из ряда, содержащего –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2– и/или –N(CH2CH2)2N–, а также оканчиваться группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2))2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3.
[00143] При этом R представляет собой заместитель, выбранный из ряда –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2– и/или –N(CH2CH2)2N– и могут оканчиваться группами –NRN2, –ORN, –SRN, –OC(O)RN, –NHC(O)RN, –C(O)ORN, –C(O)NHRN, –N=C(N(R2))2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3.
[00144] При этом PG - это защитная группа, а RN представляет собой –H или –C1-4алкил.
[00145] В некоторых вариантах воплощения заместители R1 и R2; R3 и R4; R5 и R6; R7 и R8 совместно со связанным с ними атомом могут формировать 5-8 членный гетероцикличный заместитель, выбранный из группы, состоящей N-пирролидинила, N-пиперидинила, N-азепанила, N-азоканила или N-пиперазинила.
[00146] В заявленных соединениях вводимый остаток триазиновой группы придает им химическую и ферментативную устойчивость. Кроме того, фосфатная группа, модифицированная согласно настоящему изобретению, является электронейтральной, в отличие от отрицательно заряженной немодифицированной фосфатной группы, что повышает способность заявленного соединения проникать через клеточную мембрану. Эти свойства характерны для всего класса описываемых соединений независимо от заместителей, входящих в состав триазинового остатка.
[00147] Используя различные заместители R1, R2, R3 и R4 в составе триазиновой группы, можно широко варьировать липофильность и заряд получаемого соединения, и соответственно модулировать способность заявленного соединения к биораспеределению в организме, в частности, проникать в клетки в зависимости от состава клеточной мембраны определенного типа клеток. Это может применяться при создании терапевтических препаратов, которые должны специфично проникать только в определенный тип клеток.
[00148] Важно отметить, что наличие даже одной заявленной модификации может улучшать терапевтический потенциал создаваемого соединения, в частности, способность олигонуклеотида проникать через клеточные мембраны. Это позволяет создавать модифицированные олигонуклеотиды с желаемой эффективностью и специфичностью проникновения в клетки при минимальной степени модифицированности олигонуклеотида. Минимизация количества модификаций заметно упрощает процесс получения заявленного олигонуклеотида.
[00149] Кроме того, минимизация количества модификаций, в предпочтительном варианте реализации единичная модификация, локализованная в 3-конце или 5 -конце олигонуклеотида, минимально меняет общую структуру рибозо-фосфатного остова олигонуклеотида. Это сохраняет возможность взаимодействия модифицированного олигонуклеотида с НК-ферментами, в частности с РНКазой Н.
[00150] Не менее важно, что заявленная модификация незначительно сказывается на способности модифицированных олигонуклеотидов образовывать специфические комплексы с комплементарными участками НК-мишеней. Это объясняется тем что в комплементарном комплексе триазиновая группа и присоединенные к ней заместители, при образовании комплексов с комплементарными участками ДНК или РНК оказываются экспонированными наружу от двойной спирали и тем самым не оказывают влияния на комплементарное взаимодействие азотистых оснований внутри нее.
[00151] В зависимости от заместителей модифицированная фосфатная группа может быть хиральной. В случае, если стереохимическая конфигурация не обозначена, структура включает в себя как Rp, так и Sp конфигурацию, как раздельно, таки и в виде смеси: например, рацемической смеси (рацемата). Например, структура:
включает в себя следующие структуры, как показано ниже:
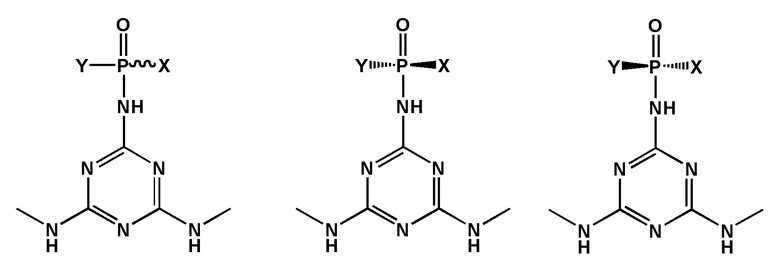
[00152] Заявленные соединения также могут включать в себя более одного хирального центра. В таком случае следует считать, что структура охватывает все возможные энантиомеры и диастереомеры.
[00153] В частном случае, когда заместитель Z представляет собой -OH, формула F0 может быть записана в виде двух таутомерных форм:
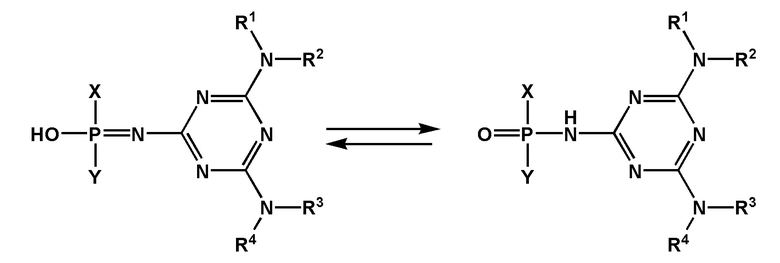
[00154] Предметом настоящего изобретения может быть олигонуклеотид, содержащий хотя бы одну модифицированную фосфатную группу соответствующую формуле (Fx):
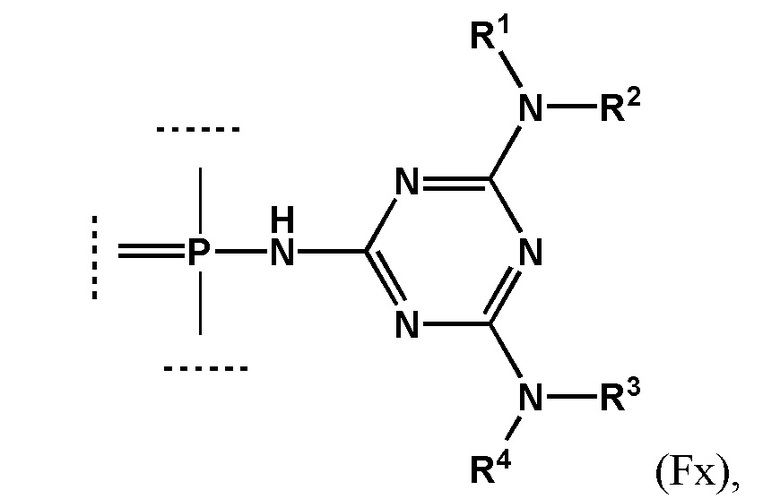 (Fx),
(Fx),
где - показывает место присоединения соответствующих олигонуклеотиду заместителей, а R1, R2, R3 и R4 определяются так же, как в формуле (F0).
[00155] Формула (Fx) отражает структуру олигонуклеотида, модифицированного согласно настоящему изобретению после этапа финального деблокирования.
[00156] Олигонуклеотиды согласно настоящему изобретению могут состоять из любого количества нуклеотидов, не менее 2. Например, олигонуклеотид может иметь минимальную длину в 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 или 40 нуклеотидов. Опционально, олигонуклеотид может иметь максимальную длину в 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, или 500 нуклеотидов, хотя и более длинные олигонуклеотиды могут быть использованы в отдельных применениях. Исключительно для примера, может быть получен олигонуклеотид, состоящий из 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 или 100 нуклеотидов.
[00157] В олигонуклеотидах, являющихся предметом настоящего изобретения, один или несколько, например, 2, 3, 4, 5, 6, 7, 8, 9,10 или более нуклеотидов, или же все нуклеотиды могут содержать фосфатную группу, модифицированную в соответствии с настоящим изобретением. В предпочтительно варианте реализации олигонуклеотид содержит не более двух заявленных модификаций. В еще более предпочтительном варианте реализации олигонуклеотид содержит одну заявленную модификацию.
[00158] Олигонуклеотиды длиной от 5 до 500 нуклеотидов, модифицированные согласно настоящему изобретению, могут применяться при терапии рака, геномных нарушений и инфекционных заболеваний, например, в качестве ASO для ингибирования трансляции (siRNA, микроРНК), для модуляции сплайсинга за счет пропуска экзона, для коррекции генотипа с помощью метода CRISPR/Cas, а также в качестве аптамеров для специфичного ингибирования белков-мишеней или направленного транспорта лекарств и в других применениях. Длина модифицированного олигонуклеотида 5-500 нуклеотидов, с одной стороны, обеспечивает специфичность комплекса, который формирует олигонуклеотид с комплементарным участком НК. С другой стороны, указанная длина позволяет получать модифицированный олигонуклеотид, используя автоматический ДНК-синтезатор.
[00159] Олигонуклеотиды в рамках настоящего изобретения могут быть получены и выделены в чистом виде.
[00160] Часть предпочтительных соединений, охватываемых настоящим изобретением, соответствуют формулам, приведённым ниже:
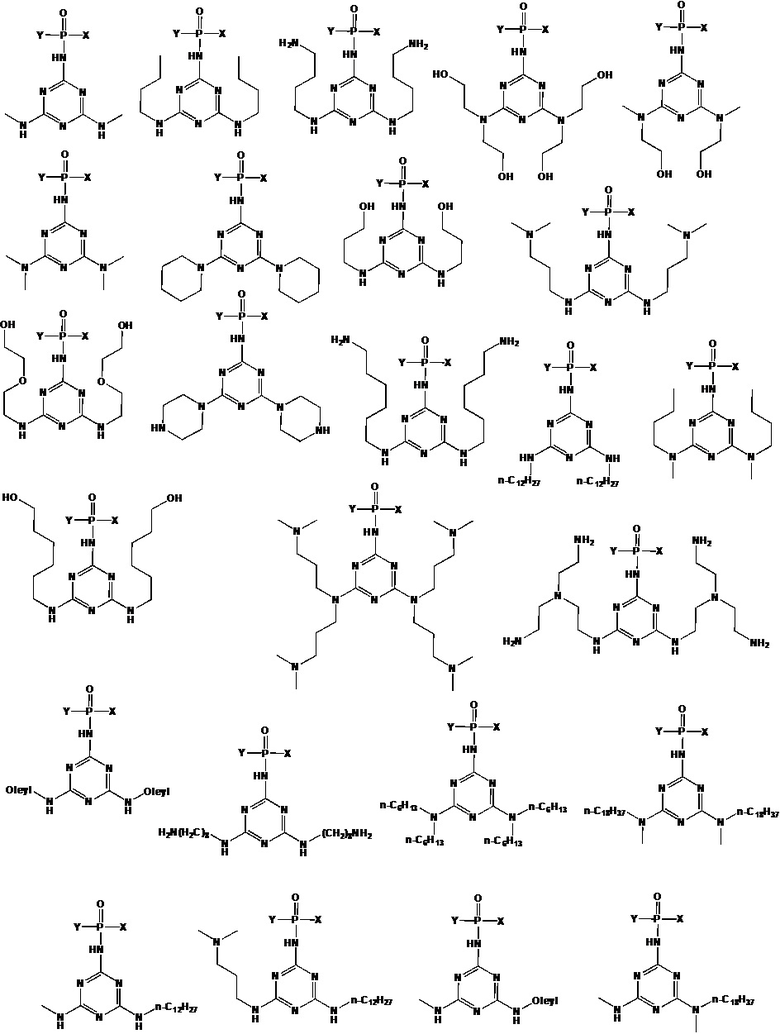
СПОСОБ ПОЛУЧЕНИЯ ЗАЯВЛЕННОГО СОЕДИНЕНИЯ
[00161] Способ получения заявленных соединений формулы (F0) заключается во взаимодействии производного трехвалентного фосфора формулы (F1) с азидотриазином формулы (F2) с получением соединения формулы (F3), как показано на схеме.
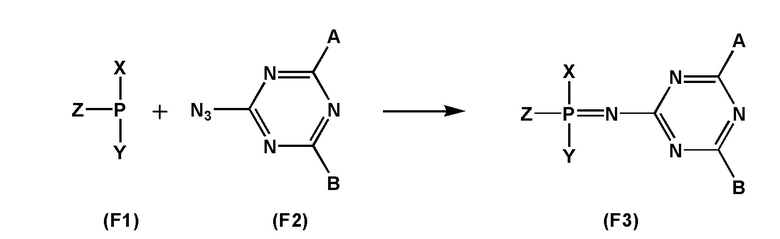
[00162] Заместители X, Y, Z определены так же, как они определены в формуле (F0).
[00163] A и B могут быть независимо выбраны из ряда –NR1R2, –NR3R4, –NRXRY, –Q.
[00164] Заместители R1, R2, R3, R4 определены так же, как они определены в формуле (F0).
[00165] Q - это группа, способная вступать в реакции замещения. Q может представлять собой, например, –OR; –OC(O)R; –OS(O)2R; –CN; –Cl; –Br; –I; –F; –N3. В рамках заявленного способа группу
[00166] Q замещают на –NR1R2, –NR3R4, –NRXRY в реакции с соответствующим амином HNR1R2, HNR3R4, HNRXRY. Причем замещение может производиться как в составе используемого азидотриазина (F2), так и в составе соединения (F3).
[00167] Использование азидотриазина (F2) для окисления производного трехвалентного фосфора (F1) обеспечивает сразу несколько технических преимуществ. Во-первых, ввиду акцепторной природы триазиновой группы, азидотриазины являются гораздо более реакционно способными соединениями, чем азиды с более электроно-донорными заместителями, такими как, например, алкил-азиды. Во-вторых, структурные особенности используемого азидотриазина позволяют вводить сразу две функциональные группы (А и B) в состав получаемого соединения в пределах одной фосфатной группы, что в большинстве случаев позволяет наделять получаемое соединение желаемым свойством используя лишь единичную модификацию.
[00168] RX и RY представляют собой заместители, которые содержат в своем составе липофильные и/или катионные группы, а кроме того содержат реакционноспособные центры, которые будут дополнительно подвергнуты химическому превращению иди последовательности химических превращений, приводя структуры заместителей к структурам R1, R2, R3или R4. Причем преобразование RX, RY в R1, R2, R3 или R4 может проводиться как в составе соединения (F2), так и в составе соединения (F3).
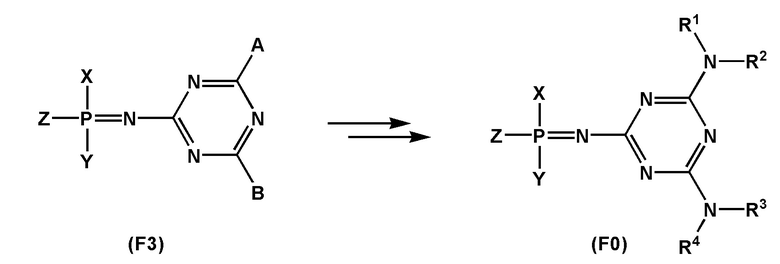
[00169] Заместители R1, R2, R3, R4, RX и RY могут широко определяться использованием соответствующих коммерческих и отдельно синтезированных аминов HNR1R2, HNR3R4 и HNRXRY, что обеспечивает гибкость заявленного способа получения и разнообразие представителей получаемого класса соединений с общей формулой (F0).
Заместители RX, RY выбраны из ряда, содержащего –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2– и/или –N(CH2CH2)2N–, а также оканчиваться группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2))2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3.
[00170] В частных вариантах реализации, RX, RY либо R1, R2, R3, R4 определяются аминами, приведенными ниже:
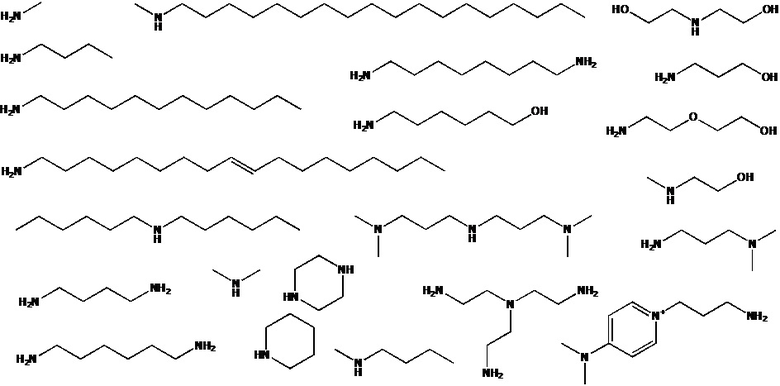
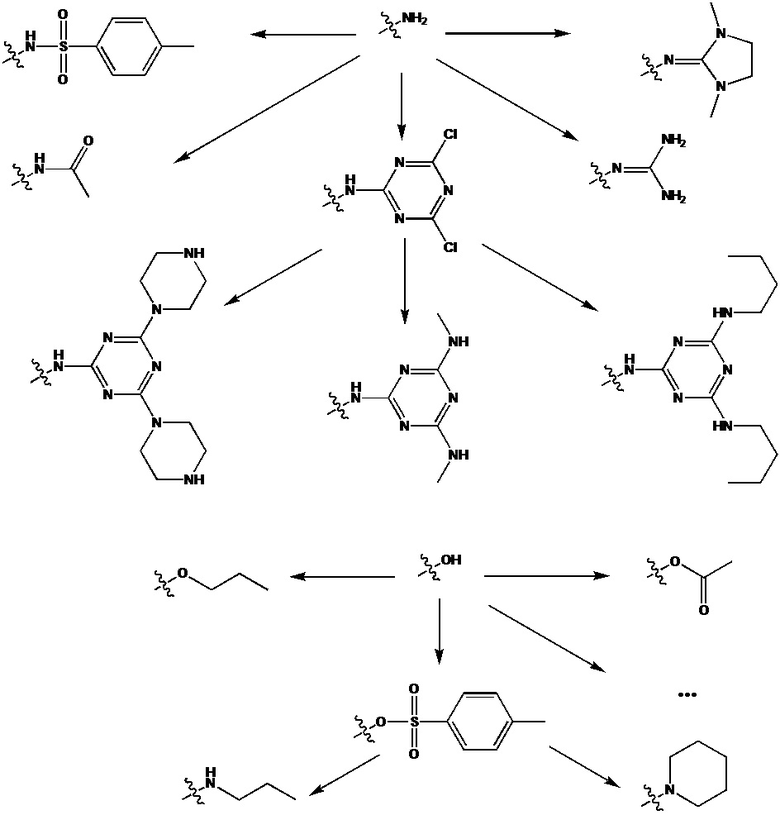
[00171] Присутствующие функциональные группы, такие как –NH2, –OH, –NH–, в составе заместителей RХ, RY могут быть преобразованы в другие функциональные группы с использованием простых реакций органического синтеза, известных специалистам предметной области.
[00172] В частности, если RX, RY содержат в своем составе группу –NH2, то эта группа может быть преобразована, например, в –NRX12 в реакции с QRX1; или в –NHRX1 в реакции с TsORX1; или в –NHC(O)RX1 в реакции с QC(O)RX1; или в –NHS(O)2RX1 в реакции с QS(O)2RX1; или в –N=C(N(RN2))2 в реакции с [QC+(NRN2)2]E-; или в 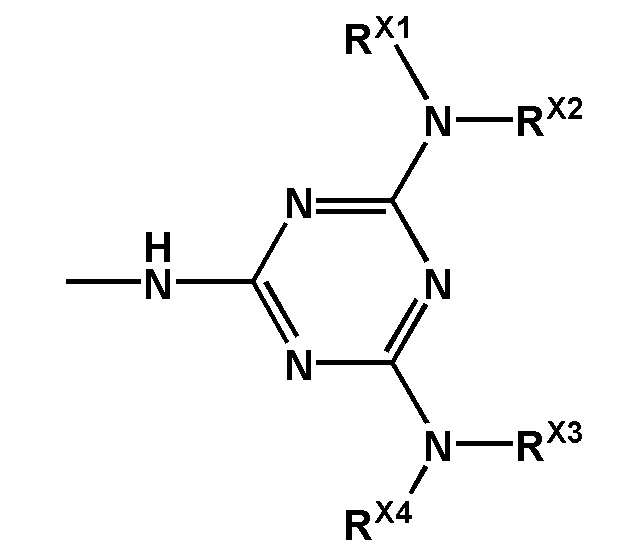
в последовательно проведенных реакциях с
 либо
либо 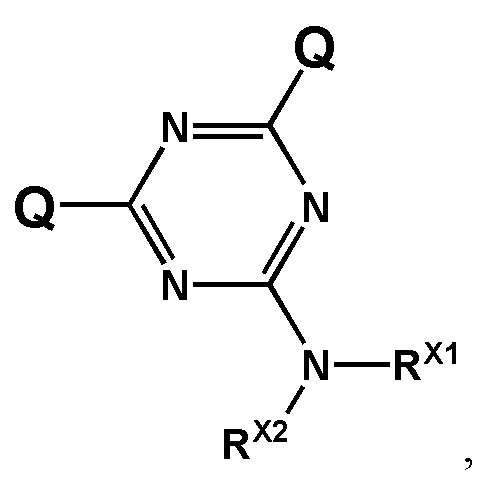 либо
либо 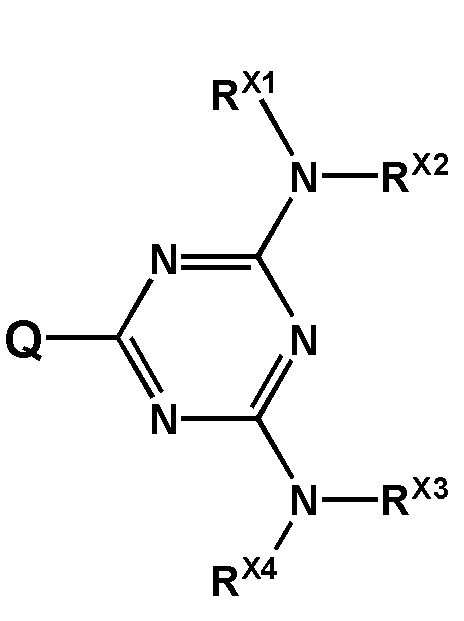 и аминами NHRX1RX2 и NHRX3RX4.
и аминами NHRX1RX2 и NHRX3RX4.
[00173] Если RX, RY содержат в своем составе группу –OH, то эта группа может быть преобразована, например, в –ORX1 в реакции с RX1; или в –NHRX1 в последовательно проведенных реакциях с QTs и амином NH2RX1; или в нуклеотид или олигонуклеотид после продолжения автоматического синтеза, где группа –OH является точкой роста олигонуклеотидной цепи.
[00174] При этом RX1, RX2, RX3, RX4 выбираются из ряда, содержащего –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2– и/или –N(CH2CH2)2N–, а также оканчиваться группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2))2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3.
[00175] Если RX1, RX2, RX3, RX4 содержат группу –NH2, то эта группа может быть преобразована в –NR2 в реакции с QR; или в –NHR в реакции с TsOR; или в –NHC(O)R в реакции с QC(O)R; или в –NHS(O)2R в реакции с QS(O)2R; или в –N=C(N(RN2))2 в реакции с [QC+(NRN2)2]E-; или в 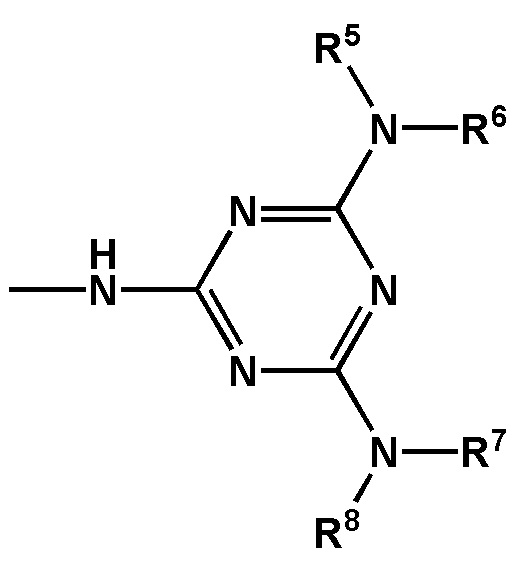 в последовательно проведенных реакциях с
в последовательно проведенных реакциях с
[00176]  , либо
, либо , либо
, либо 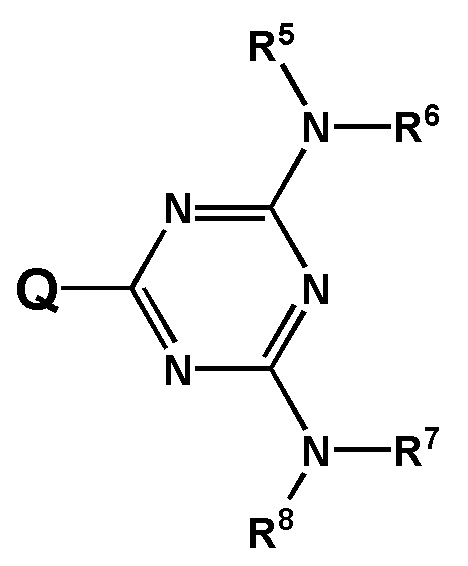 и аминами NHR5R6 и NHR7R8.
и аминами NHR5R6 и NHR7R8.
[00177] Если RX1, RX2, RX3, RX4 оканчиваются на –OH, то эта терминальная группа может быть преобразована в –OR в реакции с QR; или в –NHR в последовательно проведенных реакциях с QTs и амином NH2R; или в нуклеотид или олигонуклеотидную последовательность после продолжения автоматического синтеза.
[00178] При этом заместители R5, R6, R7, R8, R, RN как они описаны в (F0).
[00179] При этом E- представляет собой анионную составную часть, выбранную из группы, содержащей I-, Br-, Cl-, сукцинимид ((CH2)2(CO)2N-), CCl3-, CBr3-, CI3-, CHI2-, трифторметансульфонат (CF3SO3-), пара-толуолсульфонат (C7H7SO3-), дихлорфосфат (PO2Cl2-), перхлорат (ClO4-), тетрафторборат (BF4-), тетрафенилборат (BPh4-), или гексафторфосфат (PF6-).
[00180] В частных вариантах реализации в случае наличия в RX, RY первичных или вторичных аминогрупп (–NH2, –NH–) возможно получение модифицированных групп, оканчивающихся на остатки, выбираемых из ряда тозил, ацетил, остаток незамещенного или замещенного гуанидинина, остаток 4,6-дихлоро-1,3,5-триазина. В случае выбора 4,6-дихлоро-1,3,5-триазина как концевого остатка возможна его дальнейшая модификация с использованием аминов NHRX1RX2, выбираемых из ряда метиламина, бутиламина, пиперазина и др.
[00181] В частных вариантах реализации в случае наличия в RX, RY гидроксигрупп –ОН данные группы могут быть преобразованы в другие производные в реакции с соответствующими нуклеофильными реагентами. В частности гидроксигруппы могут быть преобразовнаы в отсатки простого эфира, сложного эфира, являться точкой роста новой олигонуклеотидной последовательности. В случае замещения остатком тозила возможна его дальнейшая модификация с использованием аминов NHRX1RX2 с получением соответствующих остатков вторичных и первичных аминов.
[00182] Некоторые примеры химических преобразований групп –NH2 и –OH представлены ниже:
[00183] Олигонуклеотид, содержащий хотя бы одну модифицированную фосфатную группу, соответствующую формуле (Fx), также может быть получен в рамках заявленного способа, с использованием стандартных протоколов автоматического твердофазного амидофосфитного синтеза.
[00184] Амидофосфитный метод является наиболее эффективным и широко используемым способом синтеза олигонуклеотидов. Протоколы амидофосфитного синтеза известны специалистам в области получения олигонуклеотидов и в основном осуществляются в твердофазном варианте с использованием автоматического ДНК-синтезатора. Вкратце, DMTr-нуклеозид, иммобилизованный на полимерном носителе, подвергается детритилированию, а затем конденсации с соответствующим образом активированным нуклеозид-амидофосфитом с образованием фосфит-триэфира. За этим обычно следует «кэппинг», т.е. ацилирование непрореагировавших гидроксильных групп нуклеозидов, иммобилизованных на полимерном носителе, после чего фосфит-триэфир окисляется в фосфотриэфир при помощи соответствующего окислителя. Данный цикл повторяют до окончания наращивания целевой олигонуклеотидной последовательности.
[00185] Способ получения модифицированных олигонуклеотидов согласно настоящему изобретению, включает следующие этапы:
[00186] 1) синтез олигонуклеотидной цепи проводится на ДНК синтезаторе по стандартному амидофосфитному протоколу вплоть до этапа конденсации звена, чья фосфатная часть будет модифицирована согласно настоящему изобретению; После конденсации соответствующего мономера, синтез останавливают после образования фосфит-триэфира (F1), до стадий «кэппинга» и окисления. Образовавшийся на этапе конденсации фосфит-триэфир (F1) обрабатывают азидотриазином (F2) при температуре от 5°С до 65°С, предпочтительно от 14°С до 29°С, еще более предпочтительно от 20°С до 25°С, с образованием соединения (F3);
[00187] 2) при необходимости проводят обработку безводным раствором амина с замещением А и/или В на остатки –NR1R2, –NR3R4, либо –NRXRY в составе соединения (F3) и проводят дополнительные химические превращения функциональных групп RX и RY до получения структур R1, R2, R3 и R4;
[00188] 3) продолжают синтез олигонуклеотида по амидофосфитному протоколу вплоть до следующего этапа конденсации звена, чья фосфатная часть будет модифицирована согласно настоящему изобретению и повторяют этапы 1, 2; либо продолжают синтез до получения полноразмерного олигонуклеотида. Проводят финальное деблокирование синтезированного олигонуклеотида c получением целевой структуры согласно формуле (Fx).
[00189] В том случае, когда А и В в составе азидотриазина (F2) представляют собой –NR1R2 и –NR3R4, этап 2) опускают.
[00190] В одном варианте реализации А и В в составе азидотриазина (F2) могут представлять собой атомы хлора. 2-азидо-4,6-дихлоро-1,3,5-триазин может быть получен в одну стадию из коммерчески доступного цианурхлорида, как показано в Примере 1.
[00191] В другом варианте реализации азидотриазин (F2) представляет собой 2-азидо-4-алкиламино-6-хлоро-1,3,5-триазин, который может быть получен из цианурхлорида и соответствующего амина, как показано в Примерах 2-4.
[00192] В другом варианте реализации азидотриазин (F2) представляет собой 2-азидо-4,6-диалкиламино-1,3,5-триазин, который может быть получен из цианурхлорида и соответствующих аминов, как показано в Примере 5.
[00193] Описанный способ получения модифицированного олигонуклеотида, может быть реализован в жидкофазном варианте, когда все реагенты во всех реакциях находятся в растворе и не связаны с твердофазными носителями. Однако предпочтительно заявленный способ реализуется в твердофазном варианте, когда синтезируемый олигонуклеотид связан с твердофазным носителем.
[00194] В качестве твердофазного носителя может быть использован полимерный носитель. Примеры полимерных носителей могут включать, в том числе, стекло с контролируемым размером пор (CPG), полистирольные смолы, TentaGel®, TSK Gel®Toyopearl®, поливиниловый спирт, ацетат целлюлозы и т.п.
[00195] Проведение дополнительных обработок олигонуклеотида на твердой фазе обеспечивает такие технологические преимущества, как простота отделения продукта обработки от непрореагировавшего раствора реагента, малые затраты модифицирующих агентов, высокая эффективность проводимых реакций.
[00196] В предпочтительном варианте реализации конденсация всех амидофосфитных мономеров производится в автоматическом режиме с помощью автоматического ДНК-синтезатора. При этом триазиновая группа может быть введена в любую межнуклеотидую фосфатную группу синтезируемого олигонуклеотида.
[00197] Одним из главных достоинств заявленного способа является возможность введения в состав олигонуклеотида большого разнообразия комбинаций органических радикалов R1, R2, R3 R4 в составе даже одной триазинамидофосфатной модификации. При этом заявленный способ предусматривает первоначальное получение общего предшественника - соединения (F3) - c возможностью дальнейшей последовательной сборки целевых органических радикалов R1, R2, R3 R4, путем обработки соединения (F3) различными реагентами, как описано выше. Такая иерархичная сборка финальной структуры (F0) позволяет получать широкий спектр представителей заявленного класса соединений доступным способом, особым преимуществом которого является совместимость со стандартным протоколом автоматического твердофазного амидофосфитного метода синтеза олигонуклеотидов. Кроме того благодаря химической устойчивости как в щелочных, так и кислотных средах получаемой амидотриазиновой связи данного типа модификации в структуре (Fx), получаемые модифицированные олигонуклеотиды стабильны в широком диапозоне значений pH.
[00198] Широкое разнообразие олигонуклеотидов, получаемых в рамках заявленного способа, проиллюстрировано Примерами с 6 по 43.
ОПИСАНИЕ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
[00199] В настоящих материалах заявки представлено предпочтительное раскрытие осуществления заявленного технического решения, которое не должно использоваться как ограничивающее иные, частные воплощения его реализации, которые не выходят за рамки испрашиваемого объема правовой охраны и являются очевидными для специалистов в соответствующей области техники.
[00200] Далее термин «олигонуклеотид» будет применяться к олигодезоксирибонуклеотидам, если специально не указано что-либо другое.
ОБЩИЕ МЕТОДЫ
[00201] Модифицированные олигонуклеотиды, были синтезированы на автоматическом ДНК-синтезаторе ASM-800 (Биоcсет, Россия) согласно амидофосфитному протоколу [42] в масштабе 0,2 мкмоль с использованием стандартных реакторов объемом 25 мкл.
[00202] Получение фосфит-триэфира для введения в реакцию Штаудингера с азидотриазином проводили следующим образом. Реактор, содержащий 10 мг полимерного носителя на основе пористого стекла (500 Å) с иммобилизованным нуклеозидом (загрузка нуклеозида 40 мкмоль/г) помещали в ДНК синтезатор и запускали протокол автоматизированного твердофазного синтеза олигонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Проводили синтез олигонуклеотидной цепи вплоть до звена, чья фосфатная часть будет модифицирована триазиновым остатком. После стадии конденсации с получением фосфит-триэфира, синтез останавливали, реактор вынимали из синтезатора, подсушив на водоструйном насосе после промывки сухим ацетонитрилом, полимерный носитель с иммобилизованным фосфит-триэфиром переносили из реактора в пластиковую пробирку для дальнейшей реакции с соответствующим азидотриазином и, при необходимости, дополнительных обработок.
[00203] Введение остатка 6-карбоксифлуоресцеина в виде флуоресцеинового амидофосфитного мономера осуществлялось по специальному протоколу с концентрацией флуоресцеинового амидофосфитного мономера 0,1 М, суммарным объемом флуоресцеинового амидофосфитного мономера 70 мкл, временем подачи флуоресцеинового амидофосфитного мономера 30 минут. Во всех последующих примерах остаток 6-карбоксифлуоресцеина в составе олигонуклеотида обозначается как [FAM].
[00204] Для всех реакций использовали пластиковые пробирки объемом 1,5 мл с завинчивающейся крышкой. После твердофазного синтеза полимерный носитель из реактора переносили в пластиковую пробирку и проводили этап финального деблокирования концентрированным водным раствором метиламина. После финального деблокирования супернатант упаривали досуха в вакууме на установке SpeedVac. К остатку прибавляли 200 мкл деионизированной воды (mQ) и отделяли полимерный носитель центрифугированием.
[00205] Модифицированные олигонуклеотиды выделяли с помощью обращенно-фазовой высокоэффективной жидкостной хроматографии (офВЭЖХ). Выделение проводили на хроматографе Agilent 1200 (США) с колонкой orbax SB-C18 (5 мкм) 4,6 × 150 мм в градиенте ацетонитрила в 20 мМ ацетате триэтиламмония, рН 7 от 0 до 90%, в течение 30 мин при скорости потока 1.5 мл/мин. Фракции, содержащие целевой продукт, упаривали в вакууме на установке SpeedVac. Затем олигонуклеотиды высаживали добавлением 1 мл раствора 1 M LiClO4 в ацетоне, осадок промывали ацетоном и сушили на воздухе 20 минут. Для контроля чистоты модифицированных олигонуклеотидов использовали денатурирующий гель-электрофорез в полиакриламидном геле (ПААГ) с 15% или 20% метилен-бис-акриламида с визуализацией полосок путём окрашивания раствором красителя Stains-All. Молекулярные массы модифицированных олигонуклеотидов определяли при помощи масс-спектроскопии MALDI-TOF или ESI в варианте регистрации положительных или отрицательных ионов.
ВАРИАНТЫ РЕАЛИЗАЦИИ СПОСОБА ПОЛУЧЕНИЯ МОДИФИЦИРОВАННЫХ ОЛИГОНУКЛЕОТИДОВ
[00206] Вариант 1. Введение остатка 4,6-диалкиламино-1,3,5-триазино-2-амидофосфата с использованием 2-азидо-4,6-дихлоро-1,3,5-триазина.
[00207] 1) Синтез олигонуклеотидной цепи вплоть до звена, чья фосфатная часть будет модифицирована триазиновым остатком проводили на ДНК синтезаторе, как описано в общих методах. Далее фосфит-триэфир, иммобилизованный на твердофазном носителе переносили в пробирку и добавляли 200 мкл 0,03-1 М раствора 2-азидо-4,6-дихлоро-1,3,5-триазина, задували аргоном и встряхивали на шейкере в течение 15 минут при комнатной температуре. Суспензию центрифугировали на 13400 об/мин в течение 30 сек, отбирали супернатант, трижды промывали полимерный носитель 200 мкл сухого ацетонитрила. 2) В пластиковую пробирку с полимерным носителем добавляли 200 мкл раствора (0.1 - 3M) амина, содержащего желаемые функциональные остатки, либо функциональные остатки для дальнейшей модификации, такие как -OH, -NH2, -NH- и т.д. В последнем случае проводили последовательные химические превращения путем обработок различными растворами для дополнительной модификации, такие как реакции с тозилхлоридом, хлорангидридами, аминами, гуанидирующими реагентми и др.; (см. раздел ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ). Между обработками растворами суспензию центрифугировали на 13400 об/мин в течение 30 сек, отбирали супернатант, трижды промывали полимерный носитель 200 мкл сухого ацетонитрила. 3) Носитель с иммобилизованным модифицированным олигонуклеотидом переносили в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез по амидофосфитному протоколу. После завершения олигонуклеотидного синтеза проводили финальное деблокирование концентрированным водным раствором метиламина.
[00208] Вариант 2. Введение остатка 4,6-диалкиламино-1,3,5-триазино-2-амидофосфата с использованием 2-азидо-4-алкиламино-6-хлоро-1,3,5-триазинов.
[00209] 1) Синтез олигонуклеотидной цепи вплоть до звена, чья фосфатная часть будет модифицирована триазиновым остатком, проводили на ДНК синтезаторе, как описано в общих методах. Далее фосфит-триэфир, иммобилизованный на твердофазном носителе переносили в пробирку и добавляли 200 мкл 0,1 - 3 М раствора 2-азидо-4-алкиламино-6-хлоро-1,3,5-триазина в безводном ацетонитриле или толуоле, задували аргоном и встряхивали на шейкере в течение 1 - 3 часов при комнатной температуре. Суспензию центрифугировали на 13400 об/мин в течение 30 сек, отбирали супернатант, трижды промывали полимерный носитель 200 мкл сухого ацетонитрила.
[00210] 2) В пластиковую пробирку с полимерным носителем добавляли 200 мкл раствора (0.1 - 3M) амина, содержащего желаемые функциональные остатки, либо функциональные остатки для дальнейшей модификации, такие как -OH, -NH2, -NH- и т.д. В последнем случае проводили последовательные химические превращения путем обработок различными растворами для дополнительной модификации, такие как реакции с тозилхлоридом, хлорангидридами, аминами, гуанидирующими реагентми и др.; (см. раздел ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ). Между обработками растворами суспензию центрифугировали на 13400 об/мин в течение 30 сек, отбирали супернатант, трижды промывали полимерный носитель 200 мкл сухого ацетонитрила.
[00211] 3) Носитель с иммобилизованным модифицированным олигонуклеотидом переносили в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез по амидофосфитному протоколу. После завершения олигонуклеотидного синтеза проводили финальное деблокирование концентрированным водным раствором метиламина.
[00212] Вариант 3. Введение остатка 4,6-диалкиламино-1,3,5-триазино-2-амидофосфата с использованием 2-азидо-4,6-диалкиламино-1,3,5-триазинов.1) Синтез олигонуклеотидной цепи вплоть до звена, чья фосфатная часть будет модифицирована триазиновым остатком проводили на ДНК синтезаторе, описано в общих методах. Далее фосфит-триэфир, иммобилизованный на твердофазном носителе переносили в пробирку и добавляли 200 мкл 0,5 М растворов 2-азидо-4,6-диалкиламино-1,3,5-триазина в DMF, задували аргоном и встряхивали на шейкере в течение 1 - 3 часов при 65°С. Суспензию центрифугировали на 13400 об/мин в течение 30 сек, отбирали супернатант, трижды промывали полимерный носитель 200 мкл сухого ацетонитрила.2) Носитель с иммобилизованным модифицированным олигонуклеотидом переносили в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез по β-цианэтильному фосфитамидному протоколу. После завершения олигонуклеотидного синтеза проводили финальное деблокирование концентрированным водным раствором метиламина.
ПРИМЕРЫ
[00213] В настоящих материалах заявки представлено предпочтительное раскрытие осуществления заявленного технического решения, которое не должно использоваться как ограничивающее иные, частные воплощения его реализации, которые не выходят за рамки испрашиваемого объема правовой охраны и являются очевидными для специалистов в соответствующей области техники.
[00214] Примеры хроматографического и масс-спектрометрического анализа получаемых соединений представлены на рисунках 1 и 2.
[00215] Получение азидо-триазинов
Пример 1. Получение 2-азидо-4,6-дихлоро-1,3,5-триазина
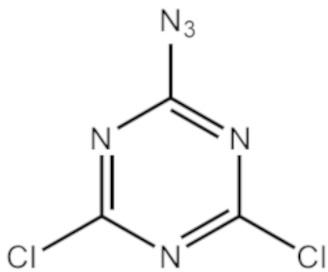
[00216] К раствору цианурхлорида (3,0 г, 16,3 ммоль) в 40 мл ацетона добавляли 1 эквивалент азида натрия (1,06 г, 16,3 ммоль), перемешивали на магнитной мешалке в течение 2 часов при комнатной температуре. Ацетон упаривали, осадок растворяли в хлористом метилене. Вещество очищали от избытка хлорида натрия в делительной воронке, последовательно четыре раза промывали 20 мл концентрированного раствора NaCl. Органический слой высушивали над Na2SO4, упаривали досуха, образовавшийся осадок сушили под вакуумом. Получали 2,36 г (76%) продукта в виде белого осадка. Дополнительную очистку вещества проводили методом колоночной хроматографии с использованием 50 г силикагеля в смеси хлористый метилен/гексан с градиентом хлористого метилена от 0 до 25%. Хроматографические фракции, содержащие продукт, упаривали досуха, сушили в вакууме. Получали 0,616 г (19,8%) чистого продукта.
Пример 2. Получение 2-азидо-4-додециламино-6-хлоро-1,3,5-триазина
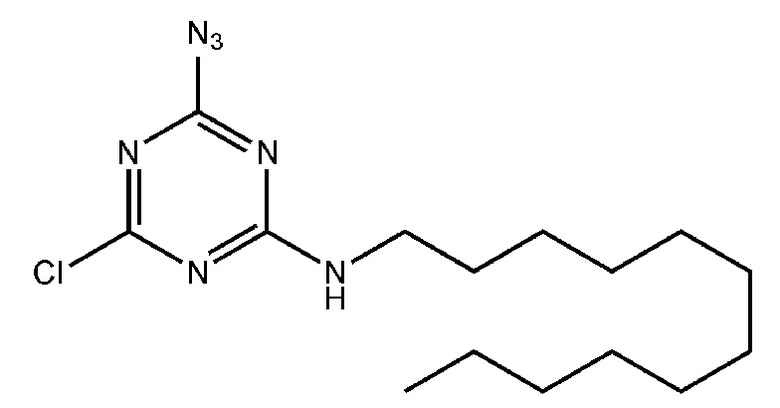
[00217] К раствору цианурхлорида (0,7 г, 3,8 ммоль) в 25 мл хлороформа добавляли 1 эквивалент додециламина (0,7 г, 3,8 ммоль) и 10 мл 10% (масс.) раствора NaOH, перемешивали на магнитной мешалке в течение cуток при комнатной температуре. Вещество очищали от избытка хлорида натрия в делительной воронке, трижды промывали 15 мл концентрированного раствора NaCl. Органический слой высушивали над Na2SO4, упаривали досуха, образовавшийся осадок сушили под вакуумом. Дополнительную очистку вещества проводили методом перекристаллизации в смеси метанол/хлороформ 5:1 при 0°С. Смесь для перекристаллизации отделяли от осадка, сушили в вакууме. Получали 0,872 г (68,7%) продукта в виде белого осадка 2-додециламино-4,6-дихлоро-1,3,5-триазина.
[00218] К раствору 2-додециламино-4,6-дихлоро-1,3,5-триазина (0,822 г, 2,47 ммоль) в 40 мл ацетона добавляли 1,25 эквивалента азида натрия (0,2 г, 3,09 ммоль), перемешивали на магнитной мешалке в течение 5 часов при комнатной температуре. Ацетон упаривали, осадок растворяли в хлористом метилене. Вещество очищали от избытка хлорида натрия в делительной воронке, четыре раза промывали 15 мл концентрированного раствора NaCl. Органический слой высушивали над Na2SO4, упаривали досуха, образовавшийся осадок сушили под вакуумом. Получали 0,693 г (82,5 %) продукта в виде белого осадка. Дополнительную очистку вещества проводили методом колоночной хроматографии с использованием 40 г силикагеля в смеси этилацетат/гексан с градиентом этилацетата от 0 до 10%. Хроматографические фракции, содержащие продукт, упаривали досуха, сушили в вакууме. Получали 0,168 г (20%) чистого продукта в виде белого осадка 2-азидо-4-додециламино-6-хлоро-1,3,5-триазина.
[00219] Для приготовления 0,1 М раствора растворяли 6,8 мг 2-азидо-4-додециламино-6-хлоро-1,3,5-триазина в 200 мкл сухого толуола, встряхивали на шейкере до растворения осадка, задували аргоном. Раствор готовили непосредственно перед проведением реакции.
Пример 3. Получение 2-азидо-4-олеиламино-6-хлоро-1,3,5-триазина
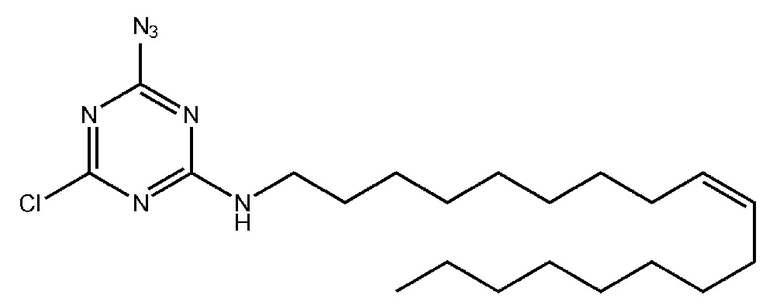
[00220] К раствору цианурхлорида (0,7 г, 3,8 ммоль) в 20 мл хлороформа добавляли 1 эквивалент олеиламина (1,25 мл, 3,8 ммоль) и 10 мл 10% (масс.) раствора NaOH, перемешивали на магнитной мешалке в течение cуток при комнатной температуре. Вещество очищали от избытка хлорида натрия в делительной воронке, трижды промывали 15 мл концентрированного раствора NaCl. Органический слой высушивали над Na2SO4, упаривали досуха, образовавшийся осадок сушили под вакуумом. Получали 1,498 г (95%) розового маслообразного продукта. Дополнительную очистку вещества проводили методом колоночной хроматографии с использованием 40 г силикагеля в смеси этилацетат/гексан с градиентом этилацетата от 0 до 7,5%. Хроматографические фракции, содержащие продукт, упаривали досуха, сушили в вакууме. Получали 0,953 г (60%) чистого маслообразного продукта - 2-олеиламино-4,6-дихлоро-1,3,5-триазина.
[00221] К раствору 2-олеиламино-4,6-дихлоро-1,3,5-триазина (0,881 г, 2,12 ммоль) в 30 мл ацетона добавляли 1,25 эквивалента азида натрия (0,173 г, 2,65 ммоль), перемешивали на магнитной мешалке в течение 6 часов при комнатной температуре. Ацетон упаривали, осадок растворяли в хлористом метилене. Вещество очищали от избытка хлорида натрия в делительной воронке, четыре раза промывали 15 мл концентрированного раствора NaCl. Органический слой высушивали над Na2SO4, упаривали досуха, образовавшийся осадок сушили под вакуумом. Получали 0,787 г (87,9 %) желтого маслообразного продукта. Дополнительную очистку вещества проводили методом колоночной хроматографии с использованием 40 г силикагеля в смеси этилацетат/гексан с градиентом этилацетата от 0 до 10%. Хроматографические фракции, содержащие продукт, упаривали досуха, сушили в вакууме. Получали 0,438 г (49%) чистого маслообразного продукта - 2-азидо-4-олеиламино-6-хлоро-1,3,5-триазина.
[00222] Для приготовления 0,1 М раствора растворяли 8,4 мг 2-азидо-4-олеиламино-6-хлоро-1,3,5-триазина в 200 мкл сухого ацетонитрила, встряхивали на шейкере до растворения осадка, сушили над ситами в течение суток при комнатной температуре, задували аргоном. Раствор готовили непосредственно перед проведением реакции.
Пример 4. Получение 2-азидо-4-(N-метил-N-октадецил)амино-6-хлоро-1,3,5-триазина
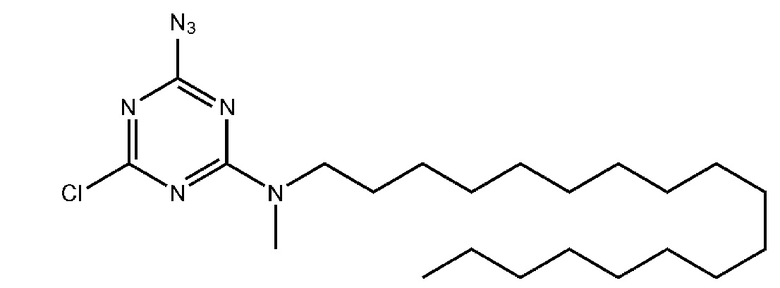
[00223] К раствору цианурхлорида (0,25 г, 1,36 ммоль) в 20 мл хлороформа добавляли 1 эквивалент (N-метил-N-октадецил)амина (0,38 г, 1,36 ммоль) и 10 мл 10% (масс.) раствора NaOH, перемешивали на магнитной мешалке в течение cуток при комнатной температуре. Вещество очищали от избытка хлорида натрия в делительной воронке, трижды промывали 15 мл концентрированного раствора NaCl. Органический слой высушивали над Na2SO4, упаривали досуха, образовавшийся осадок сушили под вакуумом. Получали 0,472 г (80%) чистого продукта в виде белого осадка 2-(N-метил-N-октадецил)амино-4,6-дихлоро-1,3,5-триазина.
[00224] К раствору 2-(N-метил-N-октадецил)амино-4,6-дихлоро-1,3,5-триазина (0,428 г, 0,99 ммоль) в 30 мл ацетона добавляли 1,2 эквивалента азида натрия (0,079 г, 1,19 ммоль), перемешивали на магнитной мешалке в течение 6 часов при комнатной температуре. Ацетон упаривали, осадок растворяли в хлористом метилене. Вещество очищали от избытка хлорида натрия в делительной воронке, четыре раза промывали 15 мл концентрированного раствора NaCl. Органический слой высушивали над Na2SO4, упаривали досуха, образовавшийся осадок сушили под вакуумом. Получали 0,351 г (80,5 %) продукта в виде белого осадка. Дополнительную очистку вещества проводили методом колоночной хроматографии с использованием 35 г силикагеля в смеси этилацетат/гексан с градиентом этилацетата от 0 до 4%. Хроматографические фракции, содержащие продукт, упаривали досуха, сушили в вакууме. Получали 0,306 г (49%) чистого продукта в виде белого осадка 2-азидо-4-(N-метил-N-октадецил)амино-6-хлоро-1,3,5-триазина.
[00225] Для приготовления 0,1 М раствора растворяли 8,8 мг 2-азидо-4-(N-метил-N-октадецил)амино-6-хлоро-1,3,5-триазина в 200 мкл сухого толуола, встряхивали на шейкере до растворения осадка, задували аргоном. Раствор готовили непосредственно перед проведением реакции.
Пример 5. Получение 2-азидо-4,6-дибутиламино-1,3,5-триазина
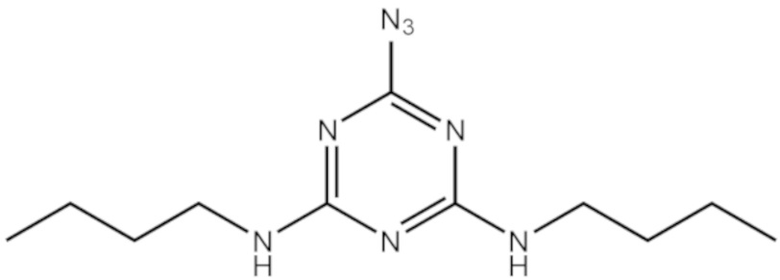
[00226] К раствору 2-азидо-4,6-дихлоро-1,3,5-триазина (0,25 г, 1,3 ммоль) в 20 мл THF добавляли 4 эквивалента бутиламина (0,52 мл, 5,2 ммоль), перемешивали на магнитной мешалке в течение суток при комнатной температуре. Отфильтровывали осадок гидрохлорида бутиламина на стеклянном пористом фильтре (16 пор), оставшийся раствор упаривали досуха, образовавшийся осадок сушили под вакуумом. Получали 0,34 г (98,6%) чистого продукта в виде белого осадка 2-азидо-4,6-дибутиламино-1,3,5-триазина.
[00227] Для приготовления 0,5 М раствора растворяли 26,4 мг 2-азидо-4,6-дибутиламино-1,3,5-триазина в 200 мкл сухого DMF, встряхивали на шейкере до растворения осадка, задували аргоном. Раствор готовили непосредственно перед проведением реакции.
[00228] Получение модифицированных олигонуклеотидов
Пример 6. Получение модифицированных олигонуклеотидов по варианту 1 с введением остатков метиламина
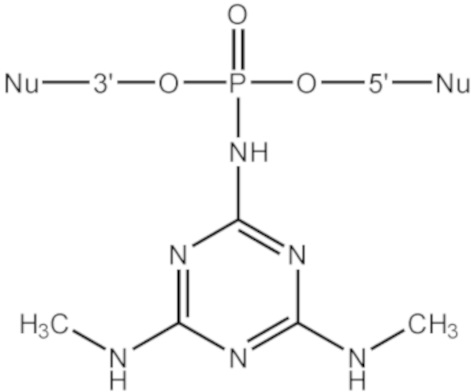
[00229] В данном примере получали модифицированные олигонуклеотиды 5'-d(T*TTTT), 5'-d(T*TTTTTTTTT), 5'-d(T*TTTTT), 5'-d(TT*TTTTT), 5'-d(T*T*TTTTTTTT).
[00230] Модифицированные олигонуклеотиды получали по варианту 1, где пункт 2) пропускали.
[00231] Молекулярные массы:
5'-d(T*TTTT): расчетная [M] 1595,19, полученная [M+H] 1595,41.
5'-d(TT*TTTTT): расчетная [M] 2202,0, полученная [M-H] 2203,0.
Пример 7. Синтез модифицированных олигонуклеотидов по варианту 1 с обработкой раствором пиперидина
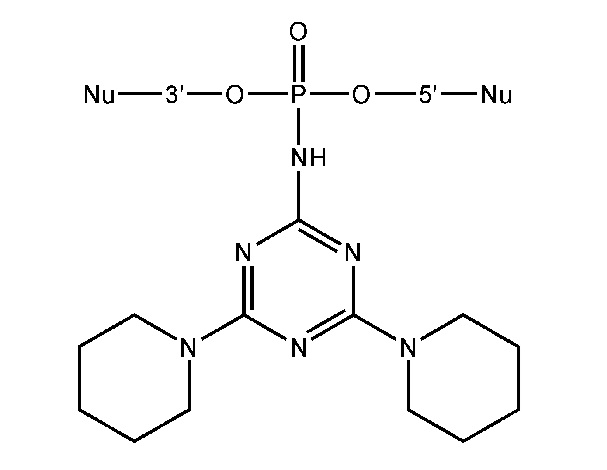
[00233] В данном примере получали модифицированные олигонуклеотиды 5'-d(T*TTTT), 5'-d(T*TTTTTTTTT), 5'-d(T*TTTT*T).
[00233] Модифицированные олигонуклеотиды получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) пиперидин в сухом ацетонитриле, 1 час, 25°С.
[00234] Молекулярная масса:
5'-d(T*TTTT*T): расчетная [M] 2250,0, полученная [M-H] 2250,8.
Пример 8. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором (N-метил)бутиламина
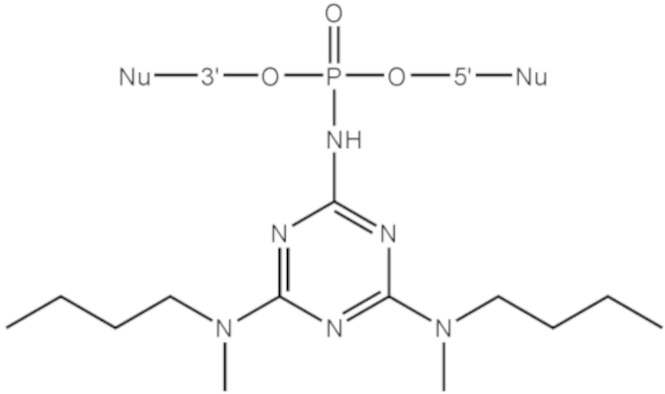
[00235] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 50% (об.) (N-метил)бутиламин в сухом ацетонитриле, 30 минут, 25°С.
[00236] Молекулярная масса:
5'-d(T*TTTTT): расчетная [M] 2011,6, полученная [M-H] 2010,6.
Пример 9. Синтез модифицированных олигонуклеотидов по варианту 1 с обработкой раствором додециламина
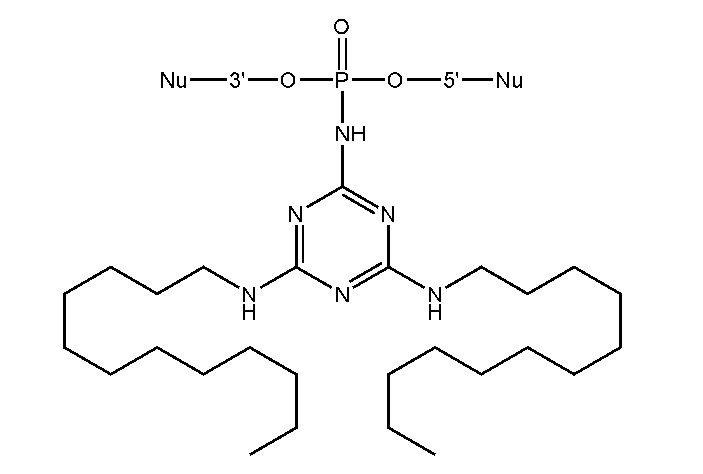
[00237] В данном примере получали модифицированные олигонуклеотиды 5'-d(T*TTTT), 5'-d(TTTT*T), 5'-d(CTGACTATGAAGTAT*T), 5'-[FAM]-d(CTGACTATGAAGTAT*T), 5'-[FAM]-d(CTGACTATGAAGTAT*T*T), 5'-d(TTTTTTTTT*T), 5'-d(AATACTTCATAGTCAGT*T).
[00238] Модифицированные олигонуклеотиды получали по варианту 1, где последовательные обработки в пункте 2) - 3 М додециламин в сухом пиридине, 30 минут, 55°С.
[00239] Молекулярные массы:
5'-d(T*TTTT): расчетная [M] 1903,8, полученная [M-H] 1902,6.
5'-d(AATACTTCATAGTCAGT*T): расчетная [M] 5917,4, полученная [M-H] 5916,4.
5'-[FAM]-d(CTGACTATGAAGTAT*T): расчетная [M] 5876,8, полученная [M-H] 5876,4.
Пример 10. Синтез модифицированных олигонуклеотидов по варианту 1 с обработкой раствором бутиламина
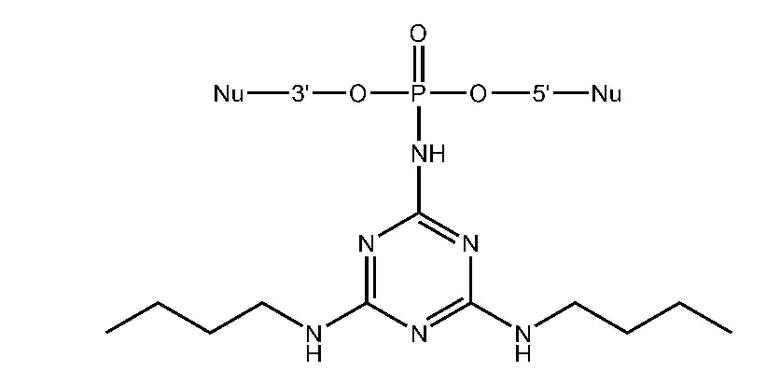
[00240] В данном примере получали модифицированные олигонуклеотиды 5'-d(CTGACTATGAAGTAT*T), 5'-[FAM]-d(CTGACTATGAAGTAT*T), 5'-d(TTTTTTTTT*T), 5'-d(T*T*TTTTTTTT).
[00241] Модифицированные олигонуклеотиды получали по варианту 1, где последовательные обработки в пункте 2) - 20% (об.) бутиламин в сухом ацетонитриле, 1 час, 40°С.
[00242] Молекулярная масса:
5'-[FAM]-d(CTGACTATGAAGTAT*T): расчетная [M] 5652,5, полученная [M-H] 5652,1.
Пример 11. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором олеиламина
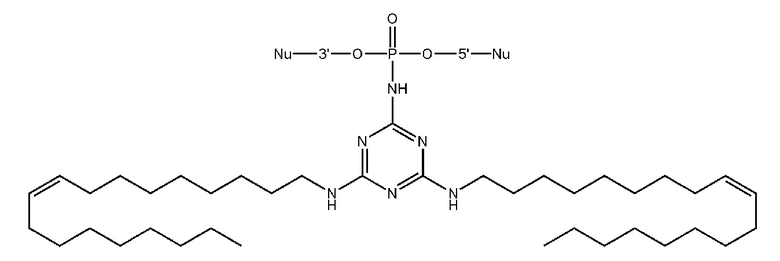
[00243] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 2 М олеиламин в сухом пиридине, 30 минут, 55°С.
[00244] Молекулярная масса:
5'-d(T*TTTT): расчетная [M] 2068,1, полученная [M-H] 2066,1.
Пример 12. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором (N-метил-N-октадецил)амина
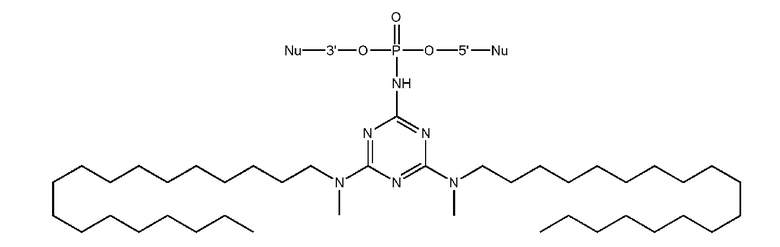
[00245] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 2 М N-метил-N-октадециламин в сухом пиридине, 30 минут, 55°С.
[00246] Молекулярная масса:
5'-d(T*TTTT): расчетная [M] 2100,0, полученная [M-H] 2099,1.
Пример 13. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором диметиламина
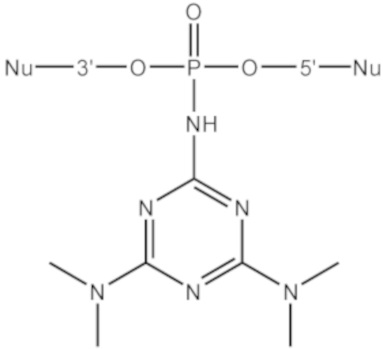
[00247] В данном примере получали модифицированный олигонуклеотид 5'-d(T*TTTTT) получали по варианту 1, где пункт 2 пропускали; олигонуклеотид деблокировали с твердофазного носителя с использованием конц. (ок. 40%) водн. раствора диметиламина при 55°С.
[00248] Молекулярная масса:
5'-d(T*TTTTT): расчетная [M] 1927,4, полученная [M-H] 1926,4.
Пример 14. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором дигексиламина
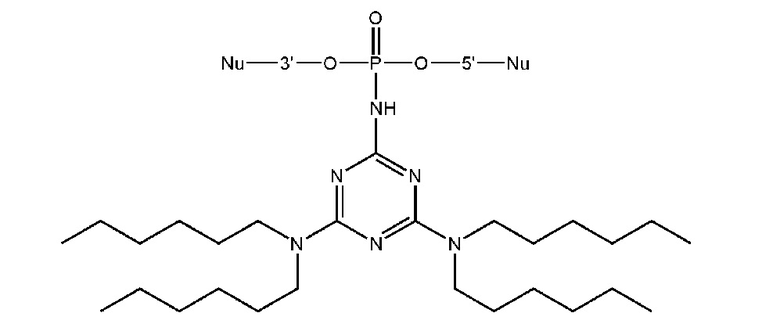
[00249] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) дигексиламин в сухом ацетонитриле, 1 час, 25°С.
[00250] Молекулярная масса:
5'-d(T*TTTT): расчетная [M] 1903,8, полученная [M-H] 1902,7.
Пример 15. Синтез модифицированных олигонуклеотидов по варианту 1 с обработкой раствором (N,N-диметиламино)пропиламина
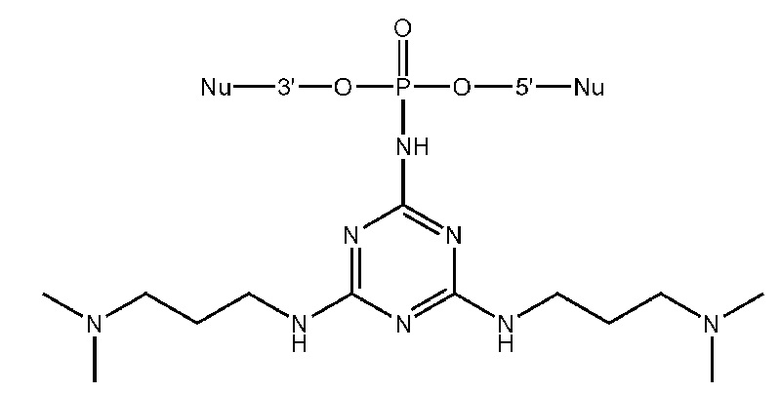
[00251] В данном примере получали модифицированные олигонуклеотиды 5'-d(T*TTTT), 5'-d(CTGACTATGAAGTAT*T), 5'-[FAM]-d(CTGACTATGAAGTAT*T), 5'-d(TTTTTTTTT*T), 5'-d(T*TTTTTTTTT), 5'-d(TTTTTTTT*T*T)
[00252] Модифицированные олигонуклеотиды получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) (N,N-диметиламино)пропиламин в сухом ацетонитриле, 1 час, 25°С.
[00253] Молекулярная масса:
5'-d(T*TTTT): расчетная [M] 1737,43, полученная [M+H] 1737,64.
Пример 16. Синтез модифицированных олигонуклеотидов по варианту 1 с обработкой раствором 3,3'-иминобис(N,N-диметил-пропиламина)
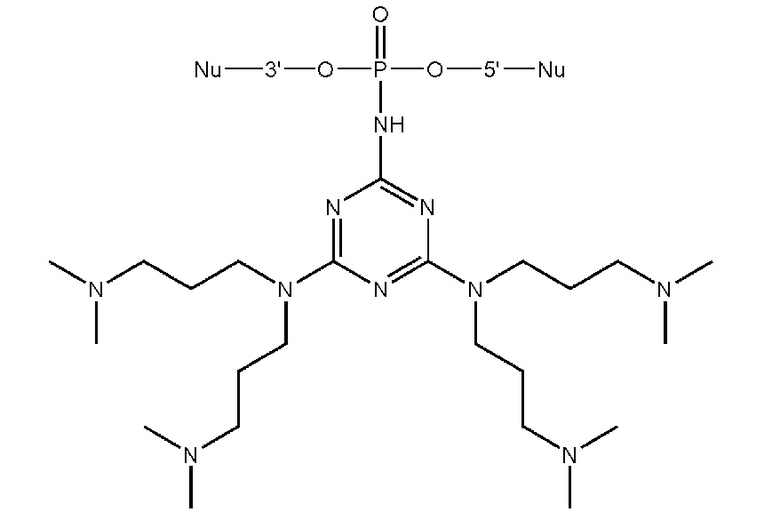
[00254] В данном примере получали модифицированные олигонуклеотиды 5'-d(T*TTTT), 5'-d(TTTT*T), 5'-d(T*T), 5'-d(CTGACTATGAAGTAT*T), 5'-[FAM]-d(CTGACTATGAAGTAT*T), 5'-d(T*TTTTTTTTT), 5'-d(TTTTTTTTT*T).
[00255] Модифицированные олигонуклеотиды в данном варианте получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) 3,3'-иминобис(N,N-диметил-пропиламин) в сухом ацетонитриле, 1 час, 25°С.
[00256] Молекулярные массы:
5'-d(TTTT*T): расчетная [M] 1907,7, полученная [M-H] 1906,6.
5'-d(T*T): расчетная [M] 995,1, полученная [M+H] 994,1.
Пример 17. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором 1,6-диаминогексана
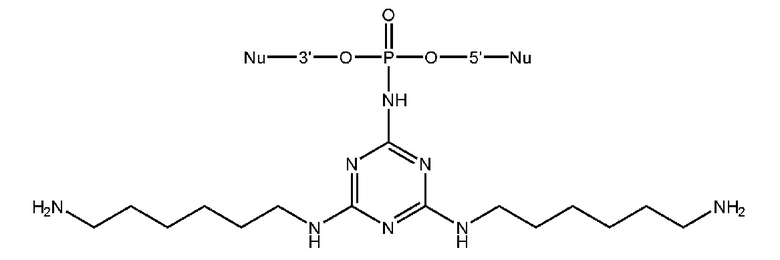
[00257] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 2 М 1,6-диаминогексан в сухом ацетонитриле, 1 час, 55°С.
[00258] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 3286,5, полученная [M-H] 3285,8.
Пример 18. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором 1,4-диаминобутана
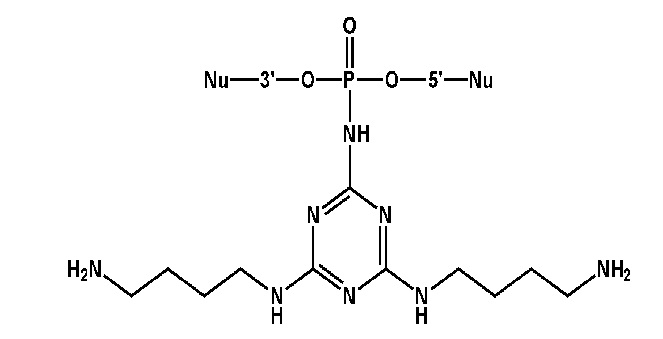
[00259] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) 1,4-диаминобутан в сухом ацетонитриле, 1 час, 55°С.
[00260] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 3230,3, полученная [M-H] 3229,7.
Пример 19. Синтез модифицированных олигонуклеотидов по варианту 1 с обработкой раствором трис-(2-аминоэтил)амина
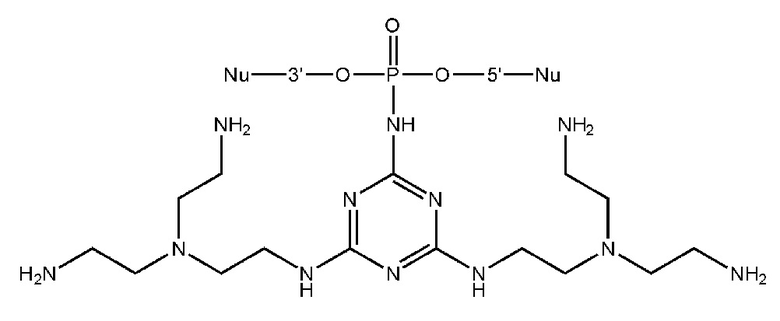
[00261] В данном примере модифицированные олигонуклеотиды 5'-d(T*TTTT), 5'-d(T*T), 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) трис-(2-аминоэтил)амин в сухом ацетонитриле, 1 час, 25°С.
[00262] Молекулярные массы:
5'-d(T*TTTTTTTTT): расчетная [M] 3346,5, полученная [M-H] 3345,0.
5'-d(T*TTTT): расчетная [M] 1825,5, полученная [M-H] 1824,2.
Пример 20. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором пиперазина
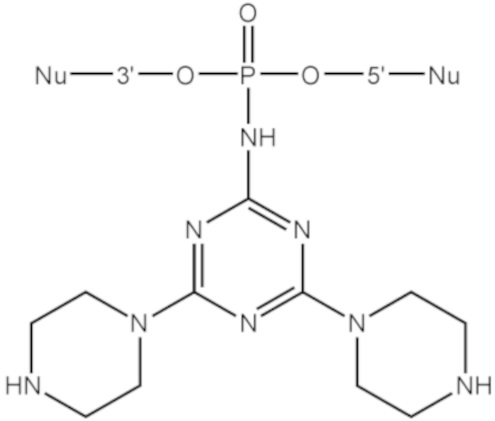
[00263] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 2 М пиперазин в сухом хлороформе, 2 часа, 25°С.
[00264] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 3226,3, полученная [M-H] 3225,7.
Пример 21. Синтез модифицированного олигонуклеотида по варианту 1 с последовательными обработками растворами трис-(2-аминоэтил)амина и гуанидина и S-метилмочевины
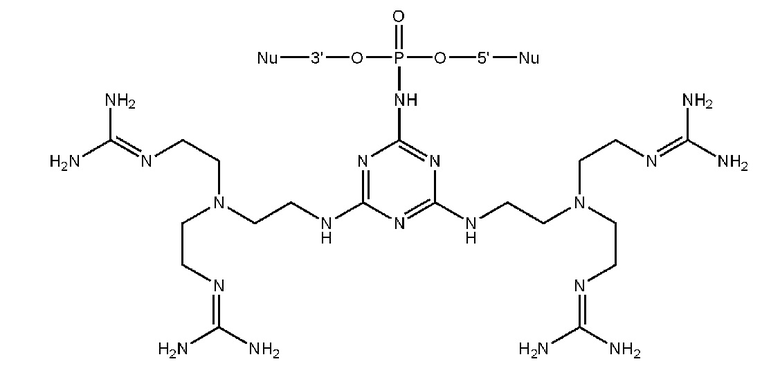
[00265] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) трис-(2-аминоэтил)амин в сухом ацетонитриле, 1 час, 25°С → 10 мг S-метилмочевина/100 мкл сухой ацетонитрил/50 мкл 25 мМ NaHCO3, сутки, 55°С.
[00266] Молекулярная масса:
5'-d(T*TTTTT): расчетная [M] 2297,9, полученная [M-H] 2297,0.
Пример 22. Синтез модифицированых олигонуклеотидов по варианту 1 с последовательными обработками растворами трис-(2-аминоэтил)амина и диметилимидазолидиний хлорид гексафторфосфата
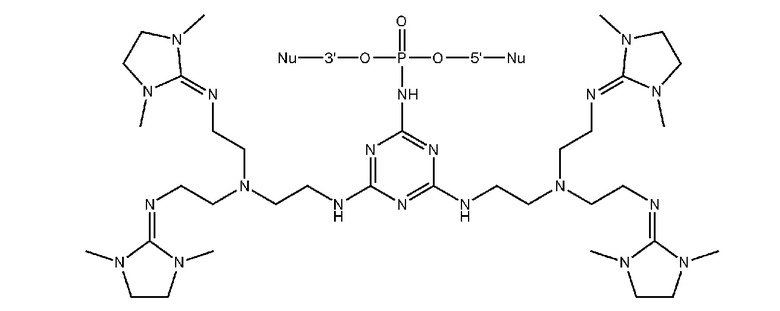
[00267] В данном примере получали модифицированные олигонуклеотиды 5'-d(T*TTTT), 5'-d(T*TTTTTTTTT), 5'-[FAM]-d(T*TTTTTTTTT), 5'-d(T*T).
[00268] Модифицированные олигонуклеотиды в данном варианте получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) трис-(2-аминоэтил)амин в сухом ацетонитриле, 1 час, комнатная температура → 10 мг диметилимидазолидиний хлорид гексафторфосфат/200 мкл сухой ацетонитрил/7 мкл DIPEA, 30 минут, 55°С.
[00269] Молекулярная масса:
5'-d(T*T): расчетная [M] 1297,5, полученная [M+H] 1296,0.
Пример 23. Синтез модифицированных олигонуклеотидов по варианту 1 с последовательными обработками растворами трис-(2-аминоэтил)амина и уксусного ангидрида
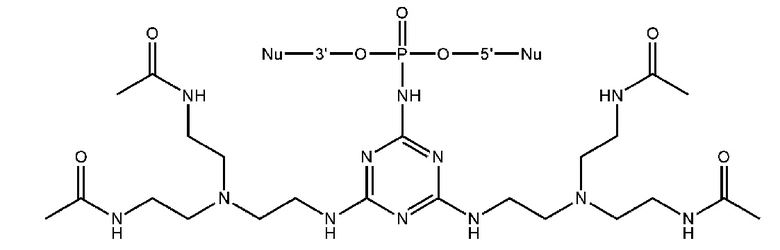
[00270] В данном примере получали модифицированные олигонуклеотиды 5'-d(T*TTTT), 5'-d(T*TTTTTTTTT), 5'-[FAM]-d(T*TTTTTTTTT).
[00271] Модифицированные олигонуклеотиды в данном варианте получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) трис-(2-аминоэтил)амин в сухом ацетонитриле, 1 час, 25°С → стандартные растворы и условия стадии кэпирования в автоматическом твердофазном амидофосфитном синтезе.
[00272] Молекулярные массы:
5'-d(T*TTTTTTTTT): расчетная [M] 3514,6, полученная [M-H] 3513,3.
5'-d(T*TTTT): расчетная [M] 1993,7, полученная [M-H] 1992,4.
Пример 24. Синтез модифицированного олигонуклеотида по варианту 1 с последовательными обработками растворами пиперазина и тозилхлорида
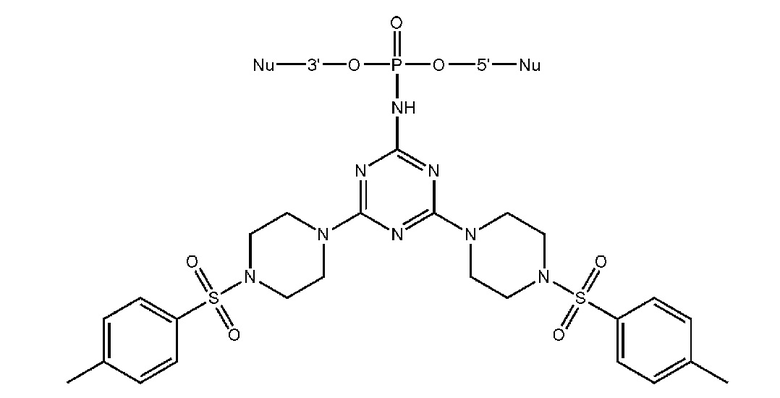
[00273] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 2 М пиперазин в сухом хлороформе, 2 часа, 25°С → 2 М тозилхлорид в сухом ацетонитриле с добавлением 2 М DIPEA в сухом ацетонитриле, 1 час, 25°С.
[00274] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 3534,6, полученная [M-H] 3533,4.
Пример 25. Синтез модифицированного олигонуклеотида по варианту 1 с последовательными обработками растворами 1,6-диаминогексана и тозилхлорида
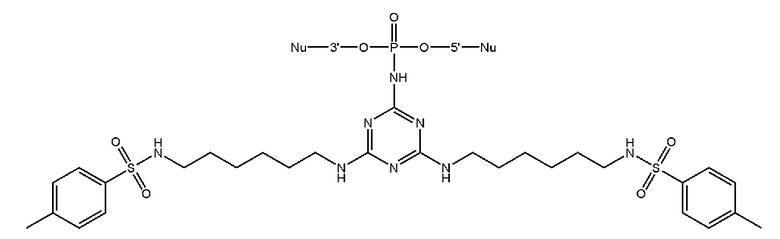
[00275] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 2 М 1,6-диаминогексан в сухом ацетонитриле, 1 час, 55°С → 2 М тозилхлорид в сухом ацетонитриле с добавлением 2 М DIPEA в сухом ацетонитриле, 1 час, 25°С.
[00276] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 3594,8, полученная [M-H] 3593,4.
Пример 26. Синтез модифицированного олигонуклеотида по варианту 1 с последовательными обработками растворами пиперазина, цианурхлорида и метиламина
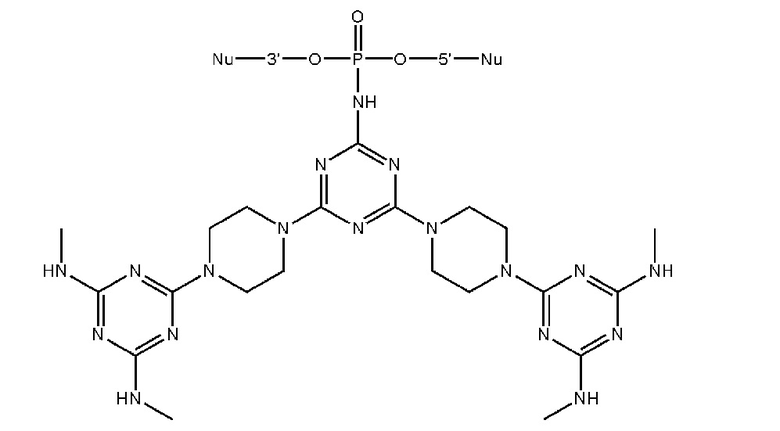
[00277] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 2 М пиперазин в сухом хлороформе, 2 часа, 25°С → 1.5 М цианурхлорид в сухом ацетонитриле с добавлением 1.5 М DIPEA в сухом ацетонитриле, 15 минут, 25°С.
[00278] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 3498,0, полученная [M-H] 3498,9.
Пример 27. Синтез модифицированного олигонуклеотида по варианту 1 с последовательными обработками растворами пиперазина, цианурхлорида и бутиламина
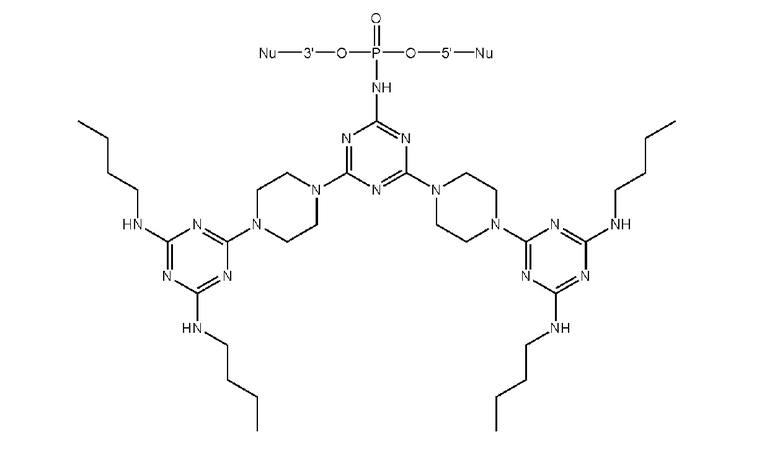
[00279] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 2 М пиперазин в сухом хлороформе, 2 часа, 25°С → 1.5 М цианурхлорид в сухом ацетонитриле с добавлением 1.5 М DIPEA в сухом ацетонитриле, 15 минут, 25°С → 10% (об.) бутиламин в сухом ацетонитриле, 1 час, 55°С.
[00280] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 3668,9, полученная [M-H] 3667,2.
Пример 28. Синтез модифицированного олигонуклеотида по варианту 1 с последовательными обработками растворами пиперазина, цианурхлорида, пиперазина и тозилхлорида
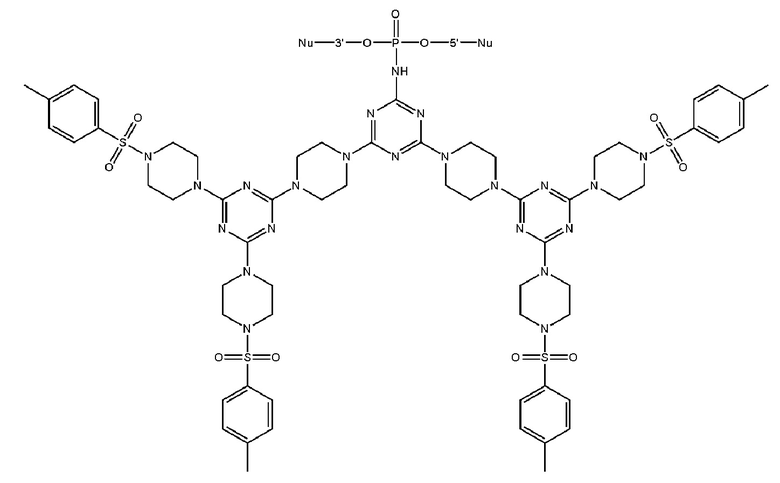
[00281] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 2 М пиперазин в сухом хлороформе, 2 часа, 25°С → 1.5 М цианурхлорид в сухом ацетонитриле с добавлением 1.5 М DIPEA в сухом ацетонитриле, 15 минут, 25°С → 2 М пиперазин в сухом хлороформе, 15 минут, 25°С → 2 М тозилхлорид в сухом ацетонитриле с добавлением 2 М DIPEA в сухом ацетонитриле, 1 час, 25°С.
[00282] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 4337,7, полученная [M-H] 4336,0.
Пример 29. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором (N-метил)аминоэтанола
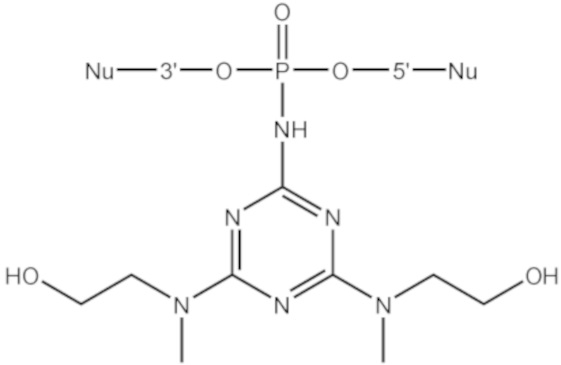
[00283] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 10% 2-(N-метил)аминоэтанол в сухом ацетонитриле, 1 час, 25°С.
[00284] Молекулярная масса:
5'-d(T*TTTTT): расчетная [M] 1987,5, полученная [M-H] 1986,6.
Пример 30. Синтез модифицированных олигонуклеотидов по варианту 1 с обработкой раствором диэтаноламина
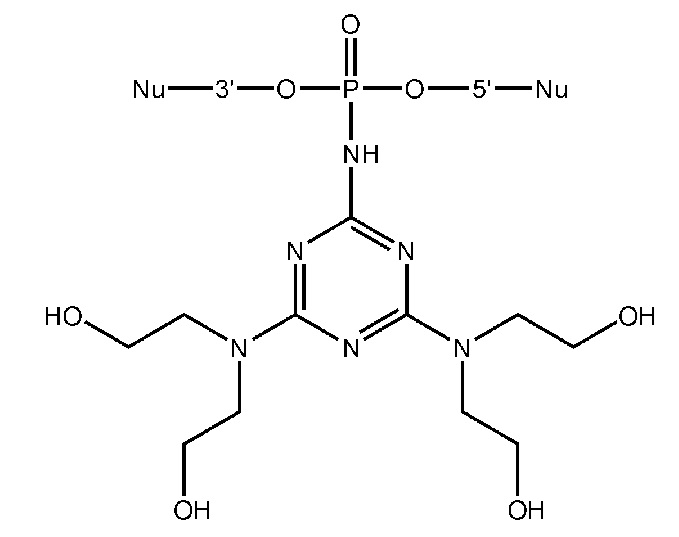
[00285] В данном примере модифицированные олигонуклеотиды 5'-d(T*TTTTTTTTT), 5'-d(T*TTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 5% (об.) диэтаноламин в смеси ацетон:ацетонитрил 1:1, 1 час, 55°С.
[00286] Молекулярные массы:
5'-d(T*TTTTT): расчетная [M] 2047,5, полученная [M-H] 2046,4.
5'-d(T*TTTTTTTTT): расчетная [M] 3264,3, полученная [M-H] 3262,8.
Пример 31. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором 3-аминопропанола
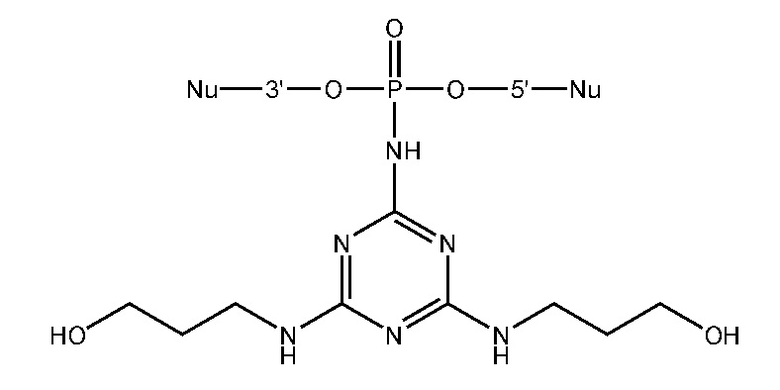
[00287] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) – 1.5 М 3-аминопропанол-1 в сухом ацетонитриле, 1 час, 55°С.
[00288] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 3204,3, полученная [M-H] 3203,6.
Пример 32. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором 6-аминогексанола
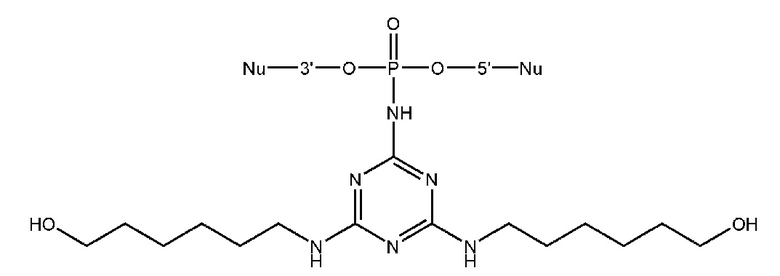
[00289] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 1.5 М 6-аминогексанол-1 в сухом ацетонитриле, 1 час, 55°С.
[00290] Молекулярная масса:
5'-d(T*TTTTTTTTT): расчетная [M] 3288,4, полученная [M-H] 3287,7.
Пример 33. Синтез модифицированного олигонуклеотида по варианту 1 с обработкой раствором 2-(2-аминоэтокси)этанола
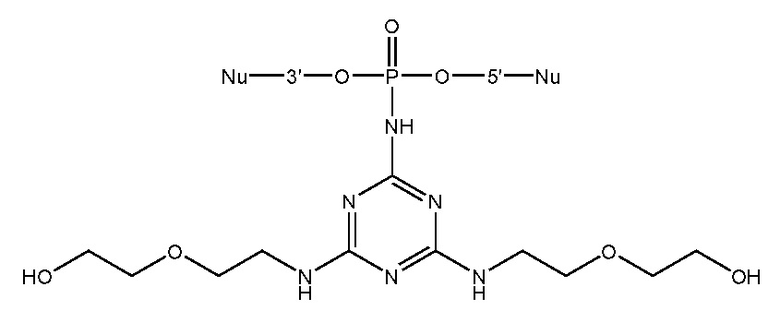
[00291] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 62,5% (об.) 2-(2-аминоэтокси)этанол в сухом ацетонитриле, 1 час, 25°С.
[00292] Молекулярная масса:
5'-d(T*TTTTT): расчетная [M] 2047,5, полученная [M-H] 2046,4.
Пример 34. Синтез модифицированных олигонуклеотидов по варианту 1 с последовательными обработками растворами 3-аминопропанола и пиперидина
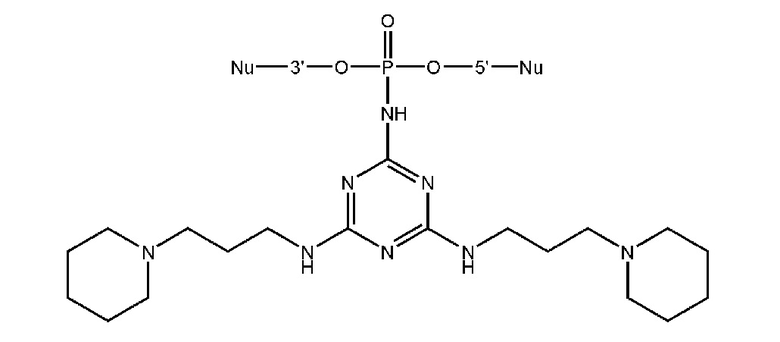
[00293] В данном примере модифицированные олигонуклеотиды 5'-d(T*TTTTTTTTT), 5'-d(T*T) получали по варианту 1, где последовательные обработки в пункте 2) - 1.5 М 3-аминопропанол-1 в сухом ацетонитриле, 1 час, 55°C → 2 М тозилхлорид в сухом ацетонитриле с добавлением 2 М DIPEA в сухом ацетонитриле, 1 час, 25°С → 10% (об.) пиперидин в сухом ацетонитриле, сутки, 40°С.
[00294] Молекулярная масса:
5'-d(T*T): расчетная [M] 905,0, полученная [M+H] 905,4.
Пример 35. Синтез модифицированного олигонуклеотида по варианту 1 с последовательными обработками растворами 3-аминопропанола и амидофосфитных мономеров тимидилатных звеньев
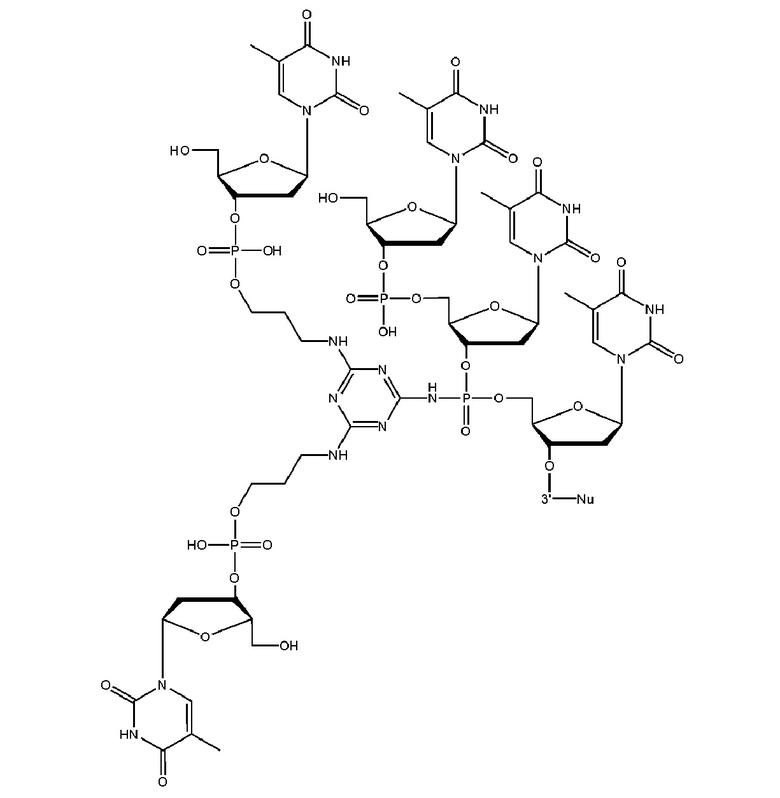
[00295] В данном примере модифицированный олигонуклеотид 5'-d(TT(T)2*TTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) – 1.5 М 3-аминопропанол-1 в сухом ацетонитриле, 1 час, 55°С → конденсация тимидилатного звена по стандартному протоколу твердофазного фосфорамидитного синтеза.
[00296] Молекулярная масса:
5'-d(TT(T)2*TTTTTT): расчетная [M] 3204,3, полученная [M-H] 3203,6.
Пример 36. Синтез модифицированного олигонуклеотида по варианту 1 с последовательными обработками растворами 6-аминогексанола и амидофосфитных мономеров тимидилатных звеньев
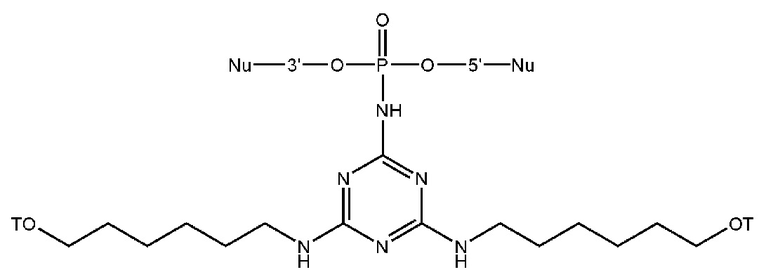
[00297] В данном примере модифицированный олигонуклеотид 5'-d(ТT(T)2*TTTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 1.5 М 6-аминогексанол-1 в сухом ацетонитриле, 1 час, 55°С → конденсация тимидилатного звена по стандартному протоколу твердофазного фосфорамидитного синтеза.
[00298] Молекулярная масса:
5'-d(ТT(T)2*TTTTTT): расчетная [M] 3288,4, полученная [M-H] 3287,7.
Пример 37. Синтез модифицированного олигонуклеотида по варианту 1 с последовательными обработками растворами 2-(2-аминоэтокси)этанола и амидофосфитных мономеров тимидилатных звеньев
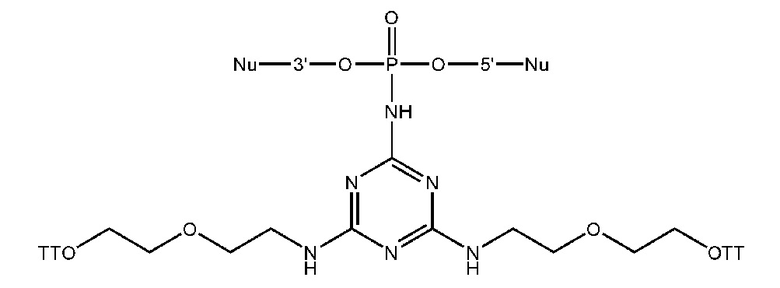
[00299] В данном примере модифицированный олигонуклеотид 5'-d(TTT(T2)2*TTTTT) получали по варианту 1, где последовательные обработки в пункте 2) - 62,5% (об.) 2-(2-аминоэтокси)этанол в сухом ацетонитриле, 1 час, 25°С → конденсация двух тимидилатных звеньев по стандартному протоколу твердофазного фосфорамидитного синтеза.
[00300] Молекулярная масса:
5'-d(T*TTTTT): расчетная [M] 3872,7, полученная [M-H] 3871,2.
Пример 38. Синтез модифицированных олигонуклеотидов по варианту 2 с использованием 2-азидо-4-додециламино-6-хлоро-1,3,5-триазина с обработкой раствором метиламина
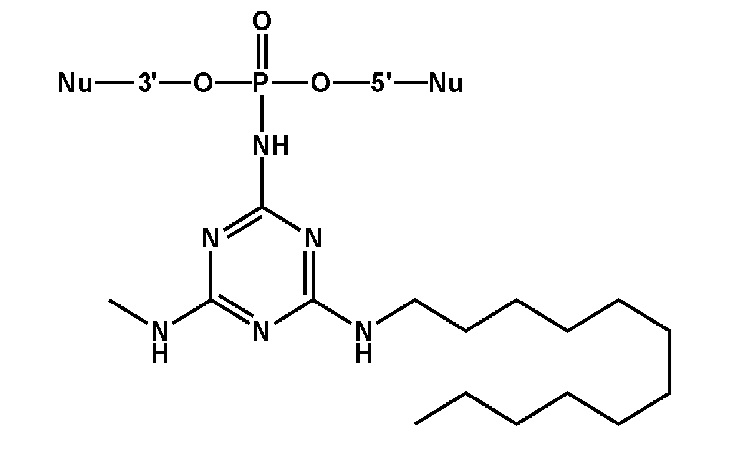
[00301] В данном примере модифицированные олигонуклеотиды 5'-d(T*TTTT), 5'-d(TTTT*TTTT) получали по варианту 2 с использованием 0,1 М раствора 2-азидо-4-додециламино-6-хлоро-1,3,5-триазина в толуоле в пункте 1), пункт 2) пропускали.
[00302] Молекулярная масса:
5'-d(T*TTTT): расчетная [M] 1749,48, полученная [M+H] 1749,57.
Пример 39. Синтез модифицированного олигонуклеотида по варианту 2 с использованием 2-азидо-4-додециламино-6-хлоро-1,3,5-триазина с обработкой раствором (N,N-диметиламино)пропиламина
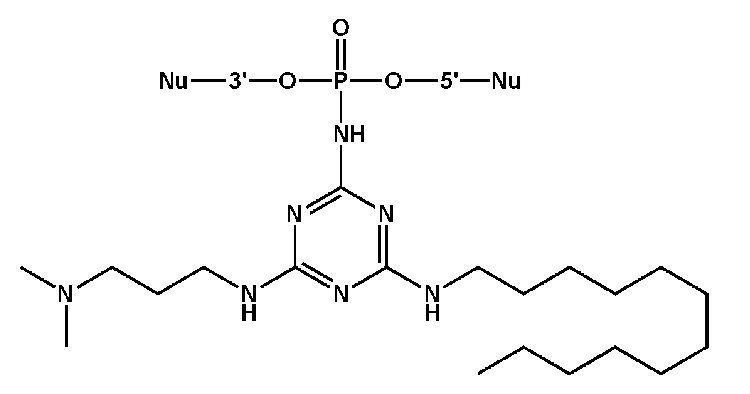
[00303] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTT) получали по варианту 2 с использованием 0,1 М раствора 2-азидо-4-додециламино-6-хлоро-1,3,5-триазина в толуоле в пункте 1), последовательные обработки в пункте 2) - 10% (об.) (N,N-диметиламино)пропиламин в сухом ацетонитриле, 1 час, 25°С.
[00304] Молекулярная масса:
5'-d(T*TTTT): расчетная [M] 1820,61, полученная [M+H] 1820,59.
Пример 40. Синтез модифицированного олигонуклеотида по варианту 2 с использованием 2-азидо-4-олеиламино-6-хлоро-1,3,5-триазина с обработкой раствором метиламина
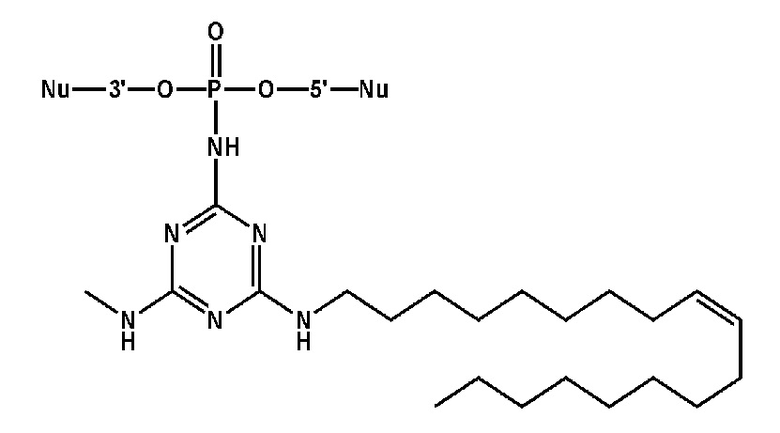
[00305] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTT) получали по варианту 2 с использованием 0,1 М раствора 2-азидо-4-олеиламино-6-хлоро-1,3,5-триазина в толуоле в пункте 1), пункт 2) пропускали.
[00306] Молекулярная масса:
5'-d(T*TTTT): расчетная [M] 1831,65, полученная [M+H] 1831,30.
Пример 41. Синтез модифицированного олигонуклеотида по варианту 2 с использованием 2-азидо-4-(N-метил-N-октадецил)амино-6-хлоро-1,3,5-триазина с обработкой раствором метиламина
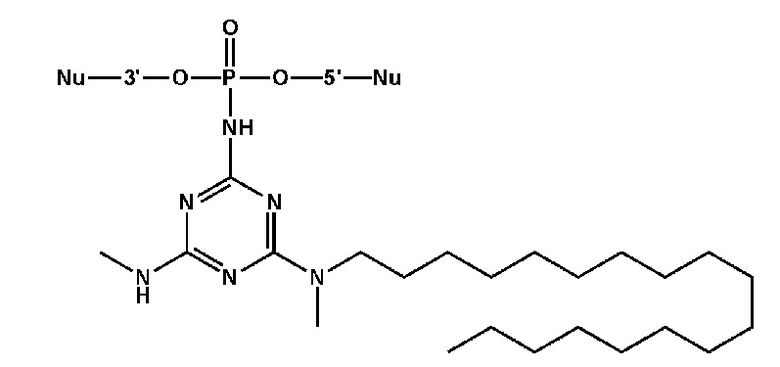
[00307] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTT) получали по варианту 2 с использованием 0,1 М раствора 2-азидо-4-(N-метил-N-октадецил)амино-6-хлоро-1,3,5-триазина в толуоле в пункте 1), пункт 2) пропускали.
[00308] Молекулярная масса:
5'-d(T*TTTT): расчетная [M] 1847,67, полученная [M+H] 1847,59.
Пример 42. Синтез модифицированного олигонуклеотида варианту 3 с использованием 2-азидо-4,6-дибутиламино-1,3,5-триазина
[00309] В данном примере модифицированный олигонуклеотид 5'-d(T*TTTT) получали по варианту 3 с использованием 0,5 М раствор 2-азидо-4-додециламино-6-хлоро-1,3,5-триазина в DMF в пункте 1).
[00310] Молекулярная масса:
5'-d(T*TTTT): расчетная [M] 1679,35, полученная [M+H] 1679,53.
Пример 43. Синтез модифицированных олигорибонуклеотидов по варианту 1 с обработкой раствором пиперидина
[00311] В данном примере модифицированные олигонуклеотиды 5'-AmCmGmUm*Um, 5'-UmUmUmUm*[BHQ], 5'-AmCmGmUm*[NH2]; m в этом примере обозначает 2'-O-метильные нуклеотиды в составе последовательности; [BHQ] и [NH2] обозначают полимерные носители CPG с остатками гасителя флуоресценции Black Hole Quencher™ и гексиламина соответственно.
[00312] Модифицированные олигонуклеотиды получали по варианту 1, где последовательные обработки в пункте 2) - 10% (об.) пиперидин в сухом ацетонитриле, 1 час, 25°С.
[00313] Молекулярные массы:
5'-AmCmGmUm*Um: расчетная [M] 1844,5, полученная [M-H] 1843,5.
5'-UmUmUmUm*[BHQ]: расчетная [M] 2017,7, полученная [M-H] 2016,5.
5'-AmCmGmUm*[NH2]: расчетная [M] 1703,4, полученная [M-H] 1702,5.
Исследование терапевтического потенциала модифицированных олигонуклеотидов
Пример 44. Исследование химической устойчивости олигонуклеотидов, содержащих триазиниламидофосфатную модификацию
[00314] Олигонуклеотид O2, 5-d(T*TTTТTTTTТ), где * - триазиниламидофосфатное звено с бутильными остатками, был синтезирован, как описано в примере 10. Для исследования устойчивости получаемого олигонуклеотида финальное деблокирование проводилось концентрированным водным раствором метиламина в течение 30 мин при 65°С. Из профиля хроматографии на Фиг. 3 видно, что в реакционной смеси отсутствуют какие-либо пики, свидетельствующие о деградации получаемого целевого продукта.
[00315] Для исследования устойчивости олигонуклеотида О2 в кислых условиях к раствору олигонуклеотида добавляли концентрированный раствор соляной кислоты до концентрации 0,1 М. После 30, 90, 180 и 780 минут отбирали равные по объему аликвоты реакционной смеси и нейтрализовали добавлением избытка концентрированного водного раствора аммиака. Затем аликвоты упаривали и проводили аналитическую офВЭЖХ для оценки степени кислотного гидролиза модифицированного олигонуклеотида. Параллельно проводили аналогичный эксперимент с олигонуклеотидом Bz 5-d(TbTTTТTTTTТ), где b -бензиламидофосфатное звено.
[00316] Результаты экспериментов, представленные на Фиг. 4, показывают, что количество олигонуклеотида, содержащего бензиламидофосфатную модификацию (Bz), уже через три часа снизилось более чем на 60%, а после 13 часов более чем на 80%, относительно исходного количества. В то же время количество олигонуклеотида, содержащего триазиниламидофосфатную модификацию (O2) не менялось на протяжении всего времени эксперимента. Это говорит о специфической устойчивости триазиниламидофосфатной модификации.
[00317] Таким образом, было показано, что олигонуклеотиды, содержащие модифицированную фосфатную группу соответствующую формуле (Fх) обладают высокой устойчивостью как в щелочных, так и в кислотных условиях.
Пример 45. Проверка устойчивости модифицированных олигонуклеотидов к действию нуклеаз цельноклеточных экстрактов клеток HEK293T и T98G
[00318] Для исследования ферментативной устойчивости использовали олигонуклеотид 5'-[FAM]-CTGACTATGAAGTAT*T-3', где * - положение триазиниламидофосфатного звена, содержащего бутильные остатки.
[00319] Для сравнения использовали контрольный немодифицированный олигонуклеотид 5'-[FAM]-CTGACTATGAAGTATT-3'.
[00320] Реакционные смеси содержали 1 мг/мл белков экстракта культивируемых клеток человека T98G или HEK293T, 0.1 мкМ одного из указанных олигонуклеотидов и буферные компоненты 10 мМ MgCl2, 50 мМ трис-HCl (pH=8.0), 50 мМ NaCl. Реакции проводили в течение 7.5 и 15 минут для каждого экстракта при 37°С. Реакции останавливали введением ЭДТА до конечной концентрации 20 мМ. После этого аликвоты анализировали с помощью электрофореза в ПААГ в денатурирующих условиях [43].
[00321] Результаты эксперимента представлены на Фиг. 5. На панели А показана электрофореграмма, полученная для немодифицированного олигонуклеотида, на панели В - для модифицированного олигонуклеотида. На каждой панели приняты следующие обозначения: 1 - контрольный раствор олигонуклеотида; 2 - HEK293T, 7,5 мин; 3 - HEK293T, 15 мин; 4 - T98G, 7,5 мин; 5 - T98G, 15 мин.
[00322] Как видно из полученных экспериментальных данных введение даже одной триазиниламидофосфатной модификации значительно увеличивает ферментативную устойчивость модифицированного олигонуклеотида в обоих клеточных экстрактах.
[00323] Таким образом, было показано, что олигонуклеотиды, содержащие модифицированную фосфатную группу соответствующую формуле (Fх) обладают высокой устойчивостью в биологических средах.
Пример 46. Исследование методом проточной цитофлуорометрии эффективности трансфекции клеток HEK293T и T98G олигонуклеотидом OSN4F
[00324] Для исследования эффективности проникновения в клетки использовали олигонуклеотид 5'-[FAM]-CTGACTATGAAGTAT*T-3' (OSN4F), где * - положение триазиниламидофосфатного звена, несущего два додецильных остатка, и контрольный немодифицированный олигонуклеотид 5'-[FAM]-CTGACTATGAAGTATT-3' (OSNF).
[00325] Культивируемые клетки человека HEK293T и T98G высеивали в лунки 24-луночного планшета в концентрации 20×104 кл/лун (HEK293T) или 12×104 кл/лун (T98G) в 500 мкл/лун среды IMDM, содержащей 10% FBS и 1%-ный раствор антибиотиков-антимикотика (10 мг/мл стрептомицина, 10000 ед/мл пенициллина и 25 мг/мл амофтерицина (IMP Biomedicals, Германия) (далее полная среда) и инкубировали в течение 18 ч для прикрепления клеток. Далее, клеточную среду заменяли на 200 мкл/лун среды IMDM в отсутствие сыворотки и антибиотиков. Олигонуклеотиды (OSNF, OSN4F) растворяли в среде Opti-MEM, и добавляли к клеткам до финальной концентрации 1 или 5 мкМ (по 50 мкл/лун). В качестве положительного контроля использовали трансфекцию клеток контрольным олигонуклеотидом OSNF с помощью коммерчески доступного трансфектанта Lipofectamine 2000 (Thermo Fisher Scientific, США). Трансфекцию выполняли согласно протоколу фирмы-изготовителя. Клетки инкубировали в течение 4 ч в стандартных условиях. После инкубации клетки снимали с культурального пластика с помощью 2%-ного трипсина (MP Biomedicals, США), ресуспендировали в полной среде IMDM, центрифугировали при 200 g в течение 5 мин, промывали PBS и фиксировали в 2%-ном растворе формальдегида в PBS (10 мин, комнатная температура). Клетки анализировали на проточном цитометре NovoCyte 3000 (ACEA Biosciences, США). Все экспериментальные точки выполняли в трех повторах для статистического анализа. Эффективность проникновения олигонуклеотидов характеризовали двумя показателями - процентом флуоресцентных клеток в популяции и средней интенсивностью флуоресценции клеток в образце, которые измеряли с помощью проточной цитометрии через 4 ч после трансфекции. Результаты эксперимента представлены на Фиг. 6. Гистограммы А и В показывают процент флуоресцентных клеток, гистограммы Б и Г - среднюю интенсивность флуоресценции. RFU – относительные флуоресцентные единицы, LF – Lipofectamine 2000. Данные представлены как среднее±стандартное отклонение.
[00326] Из представленных данных видно, что олигонуклеотид OSN4F в отсутствие дополнительных трансфекционных агентов эффективно проникает в клетки HEK293T как с точки зрения количества трансфицированных клеток, так и с точки зрения интенсивности флуоресценции. При этом эффективность проникновения OSN4F была сопоставима с эффективностью проникновения контрольного олигонуклеотида OSNF в присутствии коммерческого трансфекционного агента Lipofectamine 2000.
[00327] По гистограммам В и Г видно, что в отсутствие дополнительных трансфектантов модифицированный олигонуклеотид OSN4F проникает в клетки T98G эффективнее контрольного олигонуклеотида. Однако можно заметить, что для клеток глиобластомы T98G эффективность проникновения олигонуклеотидов кснижена по сравнению с клетками почек HEK293T, при этом сохранялась дозозависимая закономерность увеличения эффективности трансфекции. Одной из причин отличия эффективности проникновения олигонуклеотида являются особенности состава клеточных мембран HEK293T и T98G, обусловленные различием типов клеток.
[00328] Таким образом, модифицированный олигонуклеотид OSN4F способен эффективно проникать в культивируемые клетки человека HEK293T и T98G в отсутствие дополнительных трансфецирующих агентов. При этом эффективность его проникновения гораздо выше, чем у контрольного немодифицированного олигонуклеотида. Это говорит о положительном влиянии указанной модификации на способность к проникновению в клетки человека.
[00329] Важно, что эффективность проникновения модифицированного олигонуклеотида OSN4F без дополнительных трансфецирующих агентов сопоставима с эффективностью проникновения контрольного немодифицированного олигонуклеотида с помощью широко используемого трансфецирующего агента Lipofectamine2000.
[00330] Различия в эффективности проникновения модифицированного олигонуклеотида OSN4F в различные типы клеток говорят об избирательности проникновения, которая может быть обусловлена особенностями состава клеточных мембран. Такая избирательность вместе с зависимостью эффективности проникновения от дозы олигонуклеотида предоставляет возможности для создания высокоспецифических терапевтических препаратов.
Пример 46а. Данные конфокальной микроскопии по проникновению модифицированного олигонуклеотида в клетки человека
[00331] В данном исследовании использовали олигонуклеотид 5'-[FAM]-CTGACTATGAAGTAT*T-3' (OSN4F), где * - положение триазиниламидофосфатного звена, несущего два додецильных остатка.
[00332] Культивируемые клетки человека HEK293T высеивали на покровные стекла, помещенные в лунки 24-луночного планшета, в концентрации 6×104 кл/лун в полной среде IMDM и инкубировали при 37°С в атмосфере 5%-ного СО2 в течение 12-18 ч для прикрепления клеток. Далее, среду заменяли на IMDM в отсутствие сыворотки и антибиотика, содержащую OSN4F в концентрации 1 мкМ. Клетки инкубировали в присутствии олигонуклеотида при 37°С в атмосфере 5%-ного СО2 в течение 4 ч. После инкубации покровные стекла с клетками помещали на предметные стекла в каплю среды ProLong Glass Antifade Mounting Medium (ThermoFisher Scientific, США), содержащей NucBlue для окраски клеточных ядер, и инкубировали стекла в горизонтальном положении в темноте при комнатной температуре в течение 12 ч для полимеризации среды.
[00333] Внутриклеточную локализацию олигонуклеотида OSN4F исследовали с помощью конфокального микроскопа LSM710 (Zeiss, Германия), используя объектив plan-apochromat 63×/1.40 Oil DIC M27 (Zeiss, Германия) и два канала - синий и зеленый. Флуоресценция в синем канале при длине волны возбуждающего лазера 405 нм соответствовала NucBlue (окраска клеточных ядер); зеленый канал при длине волны возбуждающего лазера 488 нм соответствовал флуоресценции олигонуклеотида OSN4F, меченого остатком 6-карбоксифлуоресцеина.
[00334] На Фиг. 7 показана типичная серия микрофотографий клеток HEK293T, трансфецированных модифицированным олигонуклеотидом OSN4F. На левой фотографии видны ядра клеток, по центру - OSN4F (центр), справа - наложение каналов.
[00335] Данные конфокальной микроскопии показывают, что модифицированный олигонуклеотид OSN4F эффективно проникает в клетки человека, не налипая на клеточную мембрану, а локализуясь в цитоплазме и различных компартментах клетки. Это обеспечивает возможность модифицированным олигонуклеотидам достигать широкого круга разнообразных внутриклеточных молекулярных мишеней.
Пример 47. Сравнение эффективности проникновения в клетки человека олигонуклеотидов, содержащих липофильные остатки в составе различных остовов модификации
[00336] Исследование проводили на культивируемых клетках человека HEK293T. Работа с клетками и условия трансфекции осуществлялась как в примере 45.
[00337] Для исследования влияния остова модификации на эффективность проникновения использовали следующие модифицированные олигонуклеотиды: OSN4F - c триазиниламидофосфатной модификацией; PGO - с фосфорилгуанидиновой модификацией; PN - c двумя амидофосфатными модификациями; и ODF - c двумя ненуклеотидными звеньями. При этом все олигонуклеотиды содержали по два додецильных остатка.
OSN4F 5 - [FAM]CTGACTATGAAGTAT*T -3
PGO 5 - [FAM]CTGACTATGAAGTAT*T -3
PN 5 - [FAM]GGTAGCAAGTCGAGA*C*T -3
ODF 5 - [FAM]CTGACTATGAAGTAT[D][D]T -3
[00339] Результаты эксперимента представлены на Фиг. 8. Процент флуоресцентных клеток (слева) и среднюю интенсивность флуоресценции (справа) измеряли с помощью проточной цитометрии через 4 ч после трансфекции. RFU - относительные флуоресцентные единицы, LF - Lipofectamine 2000. Данные представлены как среднее±стандартное отклонение.
[00339] Из представленных данных видно, что уже при концентрации 1 мкМ олигонуклеотид OSN4F эффективно проникает в клетки. При этом частота его проникновения значительно выше, чем у всех остальных использованных олигонуклеотидов. Если посмотреть на интенсивность флуоресценции, то очевидно, что олигонуклеотид OSN4F проникает в клетки в больших количествах, чем остальные олигонуклеотиды.
[00340] Важно отметить, что все исследованные в данном примере олигонуклеотиды несли в своем составе одинаковое количество липофильных додецильных остатков. Это означает, что именно триазиниламидофосфатный остов, определяющий принадлежность к классу патентуемых соединений, обеспечивает улучшенное проникновение в клетки.
Пример 48. Исследование эффективности проникновения в клетки человека модифицированных олигорибонуклеотидов
[00341] Сравнение эффективности проникновения модифицированных олигорибонуклеотидов (ON1, ON2 и ON3) проводили на клеточной линии гепатоцеллюлярной карциномы человека (HepG2). Клетки культивировали в 24-луночном планшете в среде МЕМ, содержащей 10% FBS, 1 мМ пирувата натрия и смесь антибиотиком 100 ЕД/мл (пенецилин/стрептомицин). По достижении 70% конфлюэнтности среду удаляли, а монослой промывали натрий-фосфатным буфером для удаления следов сыворотки, далее к культуре клеток приливали среду OptiMEM, содержащию один из конъюгатов в концентрации 1 или 2,5, или 5 мкМ. Клетки в присутствии конъюгата олигорибонуклеотида инкубировали 12 часов в СО2-инкубаторе. После чего среда менялась на МЕМ, далее клетки инкубировали в СО2-инкубаторе еще 2 часа с целью полной деградации налипшего на поверхность клеток конъюгата. Эффективность проникновения в клетки олигорибонуклеотидов оценивали с помощью проточной цитофлуорометрии, для чего клетки перед анализом отмывали натрий-фосфатным буфером и открепляли от поверхности планшета с помощью раствора Трипсин-ЭДТА по стандартному протоколу. Анализ проводили в среде DME не содержащей красителя феноловый красный.
ON1 5'- rGrArGrCrCrCrCrCrUrArCrArGrGrGrCrUrCrUrUT*T-FAM -3ꞌ
ON2 5'- rGrArGrCrCrCrCrCrUrArCrArGrGrGrCrUrCrUrUT*T-FAM -3ꞌ
ON3 5'- rGrArGrCrCrCrCrCrUrArCrArGrGrGrCrUrCrUrUT*T-FAM -3ꞌ
ON1 (*) =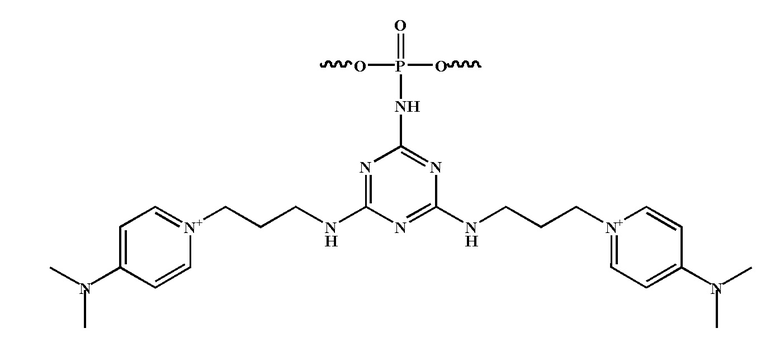
ON2 (*) = 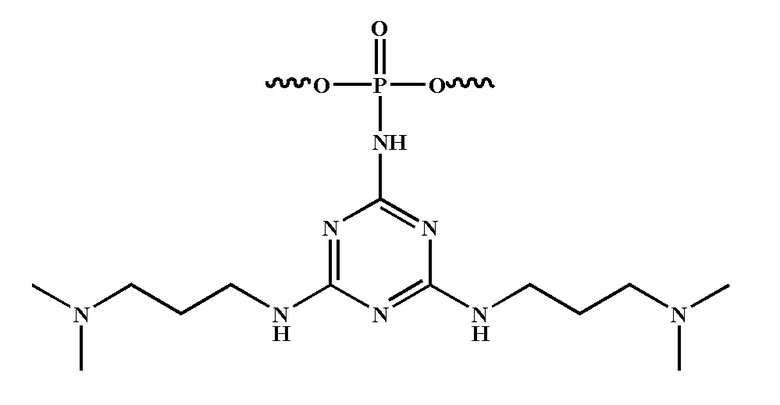
ON3 (*) =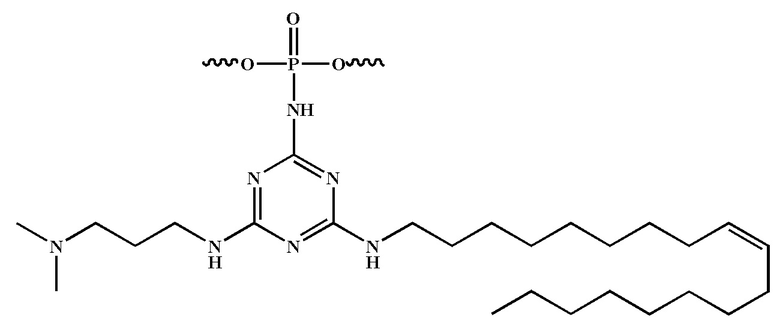
[00342] Результаты эксперимента представлены на Фиг. 9. Видно, что все исследованные в данном примере модифицированные олигорибонуклеотиды способны проникать в клетки человека. Причем эффективность проникновения в клетки у модифицированных олигорибонуклеотидов значительно выше, чем в контроле.
[00343] Таким образом, проведенный анализ показал, что различные представители класса патентуемых соединений обладают таким свойством как улучшенное проникновение в клетки человека в отсутствие трансфекционных агентов.
Пример 49. Исследование эффективности образования комплементарных комплексов с участием модифицированных олигонуклеотидов
[00344] Эффективность образования комплементарных комплексов исследовали для модифицированных олигонуклеотидов, содержащих триазиниламидофосфатную группу с остатками бутила (OSN1F) и додециламина (OSN4F, ISN4), а также немодифицированные олигонуклеотиды (OSNF, ISN).
[00345] Олигонуклеотиды OSN1F, OSN4F и OSNF смешивали с комплементарным олигонуклеотидом ISN. Олигонуклеотидов OSN1F и OSNF смешивали с модифицированным комплементарным олигонуклеотидом ISN4. Смешивание проводили в фосфатном буфере PBS при концентрации олигонуклеотидов 2,5 мкМ. Результаты экспериментов по измерению температуры плавления получаемых комплементарных комлексов представлены в таблице:
комплекс
плавления (ºС)
[00346] Видно, что во всех случаях модифицированные олигонуклеотиды способны эффективно связываться с коплементарными олигонуклеотидами. При этом температура плавления образуемых комплементарных комплексов лишь незначительно отличается от температуры плавления комплементарных комплесков, образованных олигонуклеотидами, не содержащими триазиновую модификацию (OSNF/ISN).
[00347] Приведенные данные показывают, что вне зависимости от размеров заместителей при триазиновой группе, в комплементарном комплексе триазиновая группа при образовании комплексов с комплементарными участками оказывается экспонированной наружу от двойной спирали и тем самым не оказывает влияния на комплементарное взаимодействие азотистых оснований внутри нее.
[00348] Это говорит о том, что фосфатная группа согласно формуле (Fx) не оказывают существенного влияния на способность олигонуклеотидов формировать прочные и избирательные комплементарные комплексы с биологической мишенью.
Пример 50. Исследование цитотоксичности модифицированных олигонуклеотидов.
[00349] Цитотоксичность олигонуклеотида OSN4F исследовали на клеточных культурах HEK293T и T98G в режиме реального времени с помощью прибора xCELLigence (ACEA Biosciences, США) в течение 24 ч. Клетки высевали на 16-луночные Е-планшеты в плотности 105 кл/лун в 150 мкл/лун полной среды IMDM и инкубировали в стандартных условиях в течение 20 ч для прикрепления клеток ко дну планшета. После этого среду заменяли на 150 мкл/лун среды IMDM, содержащей 10% FBS и 0.5, 1, 2.5, 5, 10, 20, 50 мкМ олигонуклеотида OSN4F. Клетки инкубировали в течение 24 ч в стандартных условиях. Значения клеточных индексов измеряли каждые 30 мин. Дозозависимые кривые выживаемости клеток строили с помощью программного обеспечения MS Excel для временной точки 24 ч после добавления олигонуклеотида к клеткам. Значения IC50 рассчитывали как концентрацию олигонуклеотида, необходимого для снижения клеточного индекса на 50% по сравнению с контрольными клетками, инкубированными в отсутствие олигонуклеотида.
[00350] Результаты эксперимента представлены на Фиг. 10. Видно, что олигонуклеотид OSN4F является нетоксичным в отношении клеток HEK293T и T98G (IC50 201.8 мкМ и 385.8 мкМ, соответственно). Рассчитанные значения IC50 значительно превышают типовые концентрации терапевтических олигонуклеотидов, (1-5 мкМ).
[00351] Экспериментальные данные показывают, что олигонуклеотид, модифицированный согласно настоящему изобретению, обладает низкой токсичностью для клеток человека.
Список цитированной литературы
1. Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB. The triple helix: 50 years later, the outcome. Nucleic Acids Res. 2008;36:5123-5138.
2. Nielsen PE. Sequence-selective targeting of duplex DNA by peptide nucleic acids. Curr Opin Mol Ther. 2010;12:184-191.
3. Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A. 1978;75:280-284.
4. Lee JF, Stovall GM, Ellington AD. Aptamer therapeutics advance. Curr Opin Chem Biol. 2006;10:282-9.
5. Crooke ST. Progress in antisense technology: The end of the beginning. Methods Enzymol. 2000;313:3-45.
6. Xu ZJ, Yang LF, Sun LQ, Cao Y. Use of DNAzymes for cancer research and therapy. Chinese Sci Bull. 2012;57:3404-3408.
7. Pérez B, Rodríguez-Pascau L, Vilageliu L, Grinberg D, Ugarte M, Desviat LR. Present and future of antisense therapy for splicing modulation in inherited metabolic disease. J Inherit Metab Dis. 2010;33:397-403.
8. Ruckman J, Green LS, Beeson J, Waugh S, Gillette WL, Henninger DD, et al. 2'-fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165): Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J Biol Chem. 1998;273:20556-67.
9. Lamond AI, Sproat BS. Antisense oligonucleotides made of 2'-O-alkylRNA: their properties and applications in RNA biochemistry. FEBS Lett. 1993;325:123-7.
10. Chi KN, Eisenhauer E, Fazli L, Jones EC, Goldenberg SL, Powers J, et al. A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2′-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J Natl Cancer Inst. 2005;97:1287-96.
11. Kaur H, Babu BR, Maiti S. Perspective on chemistry and therapeutic applications of locked nucleic acid (LNA). Chem Rev. 2007;107:4672-97.
12. Jäger A, Engels J. Synthesis of deoxynucleoside methylphosphonates via a phosphonamidite approach. Tetrahedron Lett. 1984;25:1437-40.
13. Hau JF, Asseline U, Thuong NT. Octathymidylates involving alternating neopentylphosphothionotriester-phosphodiester linkages with controlled stereochemistry at the modified P-center. Tetrahedron Lett. 1991;32:2497-8.
14. Jäger A, Levy MJ, Hecht SM. Oligonucleotide N-Alkylphosphoramidates: Synthesis and Binding to Polynucleotides. Biochemistry. 1988;27:7237-7246.
15. Egholm M, Buchardt O, Nielsen PE, Berg RH. Peptide Nucleic Acids (PNA). Oligonucleotide Analogues with an Achiral Peptide Backbone. J Am Chem Soc. 1992;114:1895-1897.
16. Summerton J, Weller D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187-95.
17. Spurgers KB, Sharkey CM, Warfield KL, Bavari S. Oligonucleotide antiviral therapeutics: Antisense and RNA interference for highly pathogenic RNA viruses. Antiviral Res. 2008;78:26-36.
18. Berkhout B, Haasnoot J. Nucleic acids-based therapeutics in the battle against pathogenic viruses. Handb Exp Pharmacol. 2009;189:243-63.
19. Ahn DG, Lee W, Choi JK, Kim SJ, Plant EP, Almazán F, et al. Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Res. 2011;91:1-10.
20. Lee CH, Kim JH, Lee SW. Prospects for nucleic acid-based therapeutics against hepatitis C virus. World J Gastroenterol. 2013;19:8949-62.
21. H. F. Rosenberg, J. B. Domachowske. Inflammatory Responses to Respiratory Syncytial Virus (RSV) Infection and the Development of Immunomodulatory Pharmacotherapeutics. Curr Med Chem. 2012;19:1424-31.
22. Choi JH, Croyle MA. Emerging Targets and Novel Approaches to Ebola Virus Prophylaxis and Treatment. BioDrugs [Internet]. 2013;27:565-583. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3624763/pdf/nihms412728.pdf
23. Maepa MB, Roelofse I, Ely A, Arbuthnot P. Progress and prospects of anti-HBV gene therapy development. Int J Mol Sci. 2015;16:17589-610.
24. Asha K, Kumar P, Sanicas M, Meseko C, Khanna M, Kumar B. Advancements in Nucleic Acid Based Therapeutics against Respiratory Viral Infections. J Clin Med. 2018;8:6.
25. Zhang K, Zheludev IN, Hagey RJ, Teng M, Wu P, Haslecker R, et al. Cryo-electron Microscopy and Exploratory Antisense Targeting of the 28-kDa Frameshift Stimulation Element from the SARS-CoV-2 RNA Genome. bioRxiv [Internet]. 2020; Available from: https://doi.org/10.1101/2020.07.18.209270
26. Sharad S. Antisense Therapy: An Overview. Intech [Internet]. 2016. p. 1-11. Available from: https://www.intechopen.com/books/advanced-biometric-technologies/liveness-detection-in-biometrics
27. Miller PS, Yano J, Yano E, Carroll C, Jayaraman K, Ts'o POP. Nonionic Nucleic Acid Analogues. Synthesis and Characterization of Dideoxyribonucleoside Methylphosphonates. Biochemistry. 1979;18:5134-43.
28. Stec WJ, Zon G, Egan W, Stec B. Automated Solid-Phase Synthesis, Separation, and Stereochemistry of Phosphorothioate Analogues of Oligodeoxyribonucleotides. J Am Chem Soc. 1984;106:6077-6079.
29. Burgers PMJ, Eckstein F. Diastereomers of S'-O-Adenosyl S'-O-Uridyl Phosphorothioate: Chemical Synthesis and Enzymatic Properties. Biochemistry. 1979;18:592-596.
30. Seeberger PH, Caruthers MH, Yau E. 2'-Deoxynucleoside Dithiophosphates: Synthesis and Biological Studies. J Am Chem Soc. 1995;117:1472-1478.
31. Hall AHS, Wan J, Spesock A, Sergueeva Z, Shaw BR, Alexander KA. High potency silencing by single-stranded boranophosphate siRNA. Nucleic Acids Res. 2006;34:2773-81.
32. Hogrefe RI, Vaghefi MM, Reynolds MA, Young KM, Arnold LJ. Deprotection of methylphosphonate oligonucleotides using a novel one-pot procedure. Nucleic Acids Res. 1993;21:2031-2038.
33. Reynolds MA, Hogrefe RI, Jaeger JA, Schwartz DA, Riley TA, Marvin WB, et al. Synthesis and thermodynamics of oligonucleotides containing chirally pure R(p) methylphosphonate linkages. Nucleic Acids Res. 1996;24:4584-4591.
34. Rait V, Sergueev D, Summers J, He K, Huang F, Krzyzanowska B, et al. Boranophosphate nucleic acids - A versatile DNA backbone. Nucleosides and Nucleotides. 1999;18:1379-80.
35. Hall AHS, Wan J, Shaughnessy EE, Shaw BR, Alexander KA. RNA interference using boranophosphate siRNAs: Structure-activity relationships. Nucleic Acids Res. 2004;32:5991-6000.
36. Letsinger RL, Singman CN, Histand G, Salunkhe M. Cationic oligonucleotides. J Am Chem Soc. 1988;110:4470-1.
37. Laurent A, Naval M, Debart F, Vasseur JJ, Rayner B. Chiral and steric effects in the efficient binding of α-anomeric deoxyoligonucleoside N-alkylphosphoramidates to ssDNA and RNA. Nucleic Acids Res. 1999;27:4151-4159.
38. Maier MA, Guzaev AP, Manoharan M. Synthesis of chimeric oligonucleotides containing phosphodiester, phosphorothioate, and phosphoramidate linkages. Org Lett. 2000;2:1819-22.
39. Aboul-Fadl T. Antisense Oligonucleotides: The State of the Art. Curr Med Chem. 2005;12:2193-214.
40. Alter J, Lou F, Rabinowitz A, Yin HF, Rosenfeld J, Wilton SD, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175-7.
41. Wu B, Moulton HM, Iversen PL, Jiang J, Li J, Li J, et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci U S A. 2008;105:14814-9.
42. Sinha ND, Biernat J, Mcmanus J, Köster H. Polymer support oligonucleotide synthesis XVIII: Use of β-cyanoethyi-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product. Nucleic Acids Res. 1984;12:4539-4557.
43. Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning : a laboratory manual. 2nd edition. Cold Spring Harb ,New York. 1989.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДИФИЦИРОВАННЫЕ ОЛИГОНУКЛЕОТИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2708237C2 |
| МОДИФИЦИРОВАННЫЕ ОЛИГОНУКЛЕОТИДЫ, АКТИВИРУЮЩИЕ РНКазу Н | 2017 |
|
RU2740501C2 |
| Фосфорамидные азольные олигонуклеотиды, способ синтеза фосфорамидных азольных олигонуклеотидов и способ матричного ферментативного синтеза ДНК с их использованием | 2023 |
|
RU2823099C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ХИМИЧЕСКОГО РАСЩЕПЛЕНИЯ И СНЯТИЯ ЗАЩИТЫ ДЛЯ СВЯЗАННЫХ С ПОВЕРХНОСТЬЮ ОЛИГОНУКЛЕОТИДОВ | 2019 |
|
RU2766688C2 |
| Алкинсодержащий амидофосфит для функционализации синтетических олигонуклеотидов и способ его получения | 2021 |
|
RU2781226C1 |
| ЛИГАНДНЫЕ СОЕДИНЕНИЯ, КОНЪЮГАТЫ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2819894C1 |
| АНАЛОГИ L-РИБО-ЗНК | 2000 |
|
RU2258708C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДОФОСФИТНОГО МОНОМЕРА АХИРАЛЬНОЙ НЕНУКЛЕОТИДНОЙ ВСТАВКИ ДЛЯ МОДИФИКАЦИИ ОЛИГОНУКЛЕОТИДОВ | 2011 |
|
RU2460721C1 |
| ОЛИГОДЕЗОКСИРИБОНУКЛЕОТИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2117011C1 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ НУКЛЕОЗИДЫ И ОЛИГОМЕРЫ, ПОЛУЧЕННЫЕ ИЗ НИХ | 2017 |
|
RU2824141C2 |
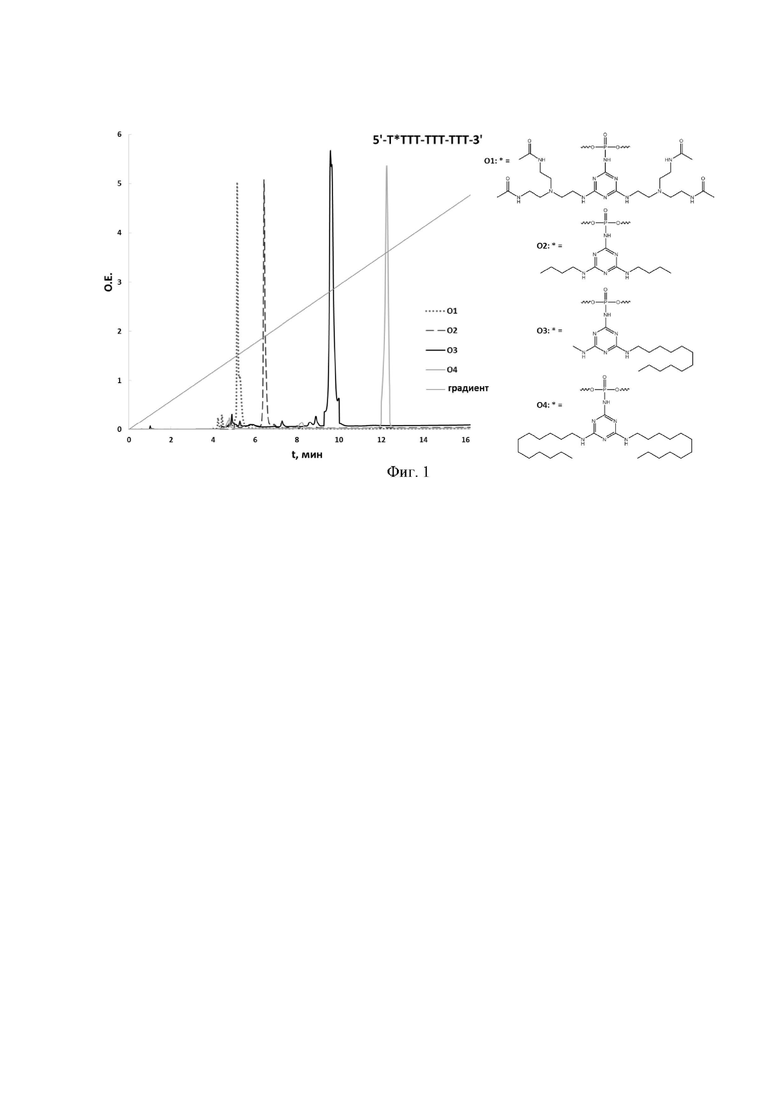
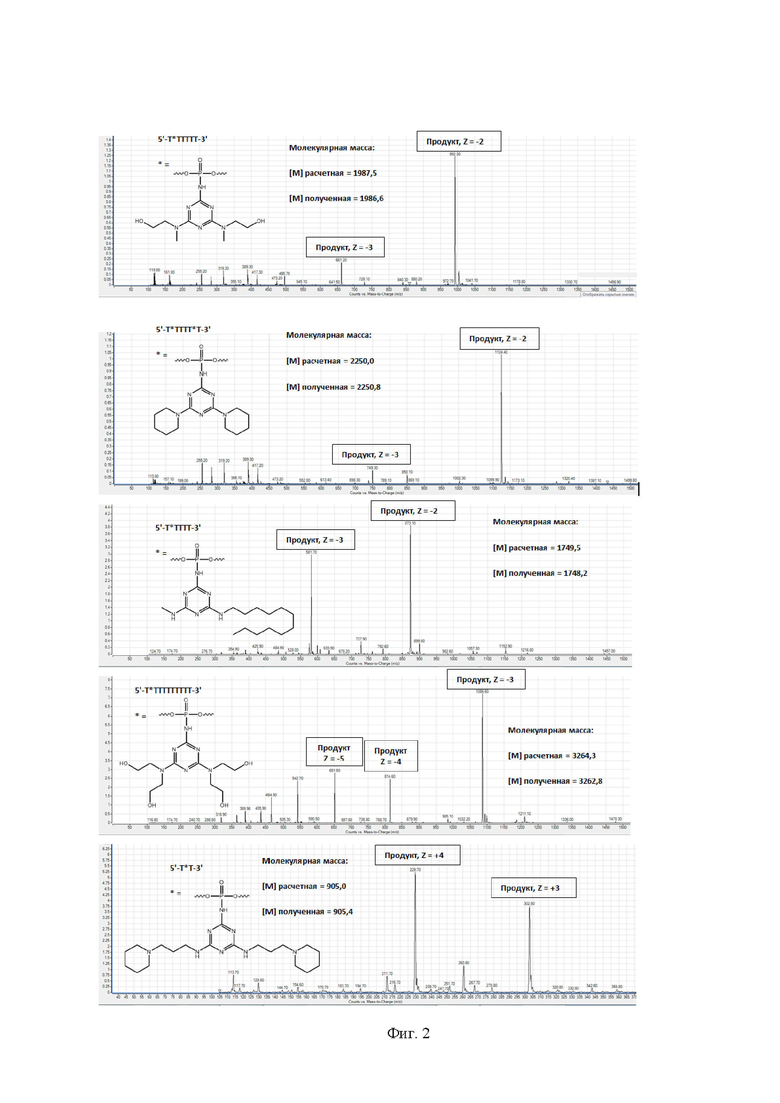
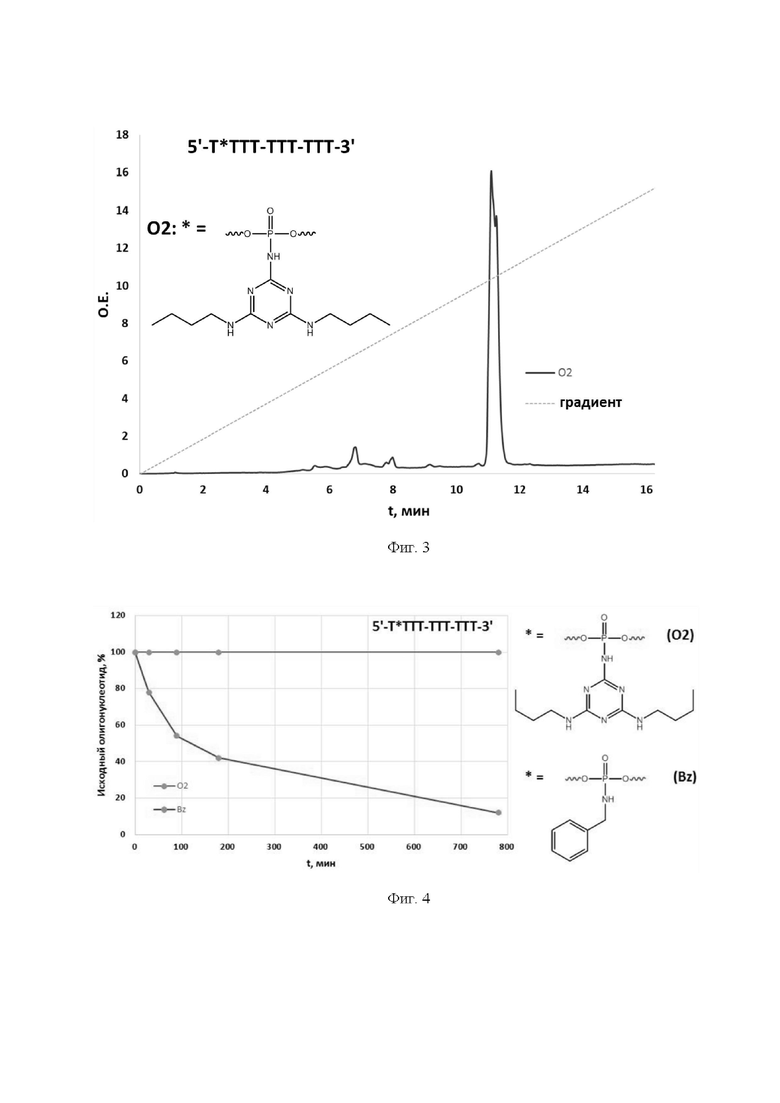
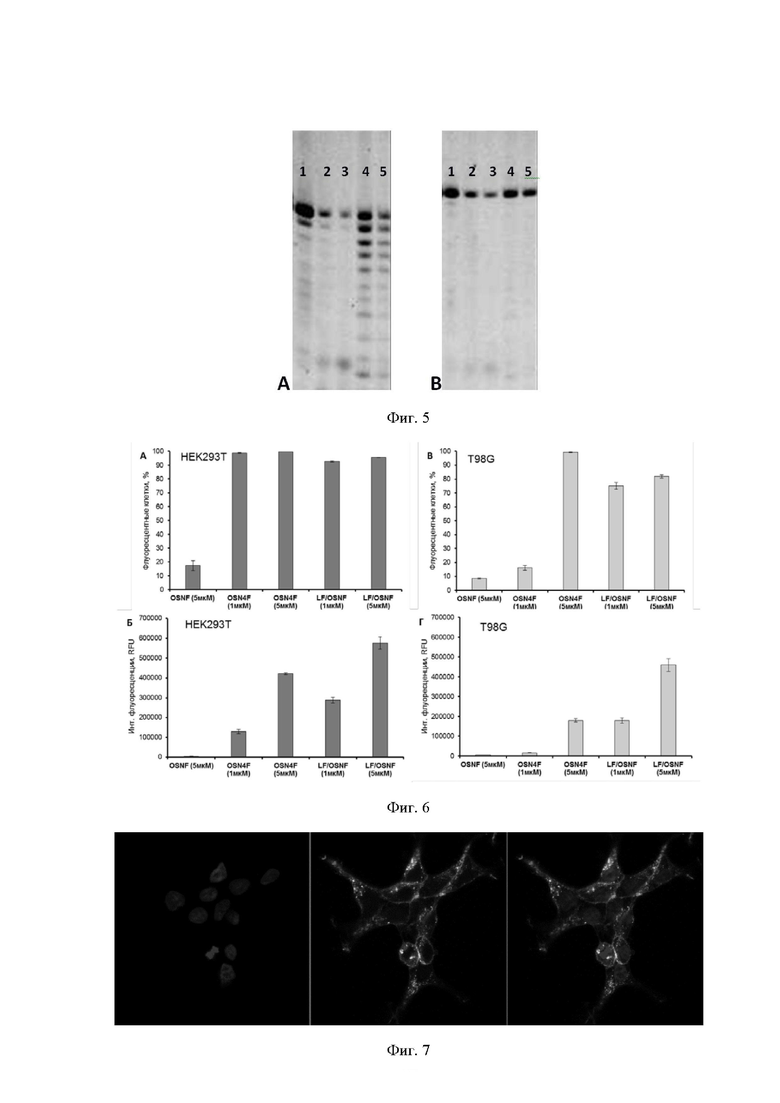
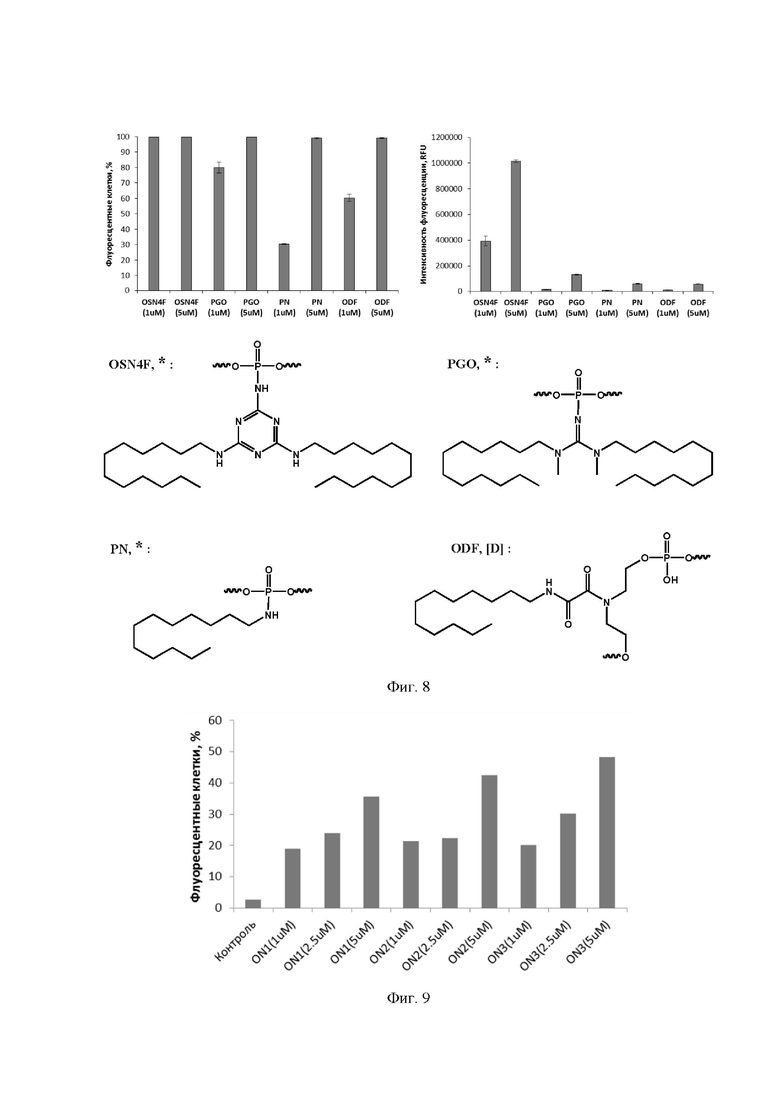
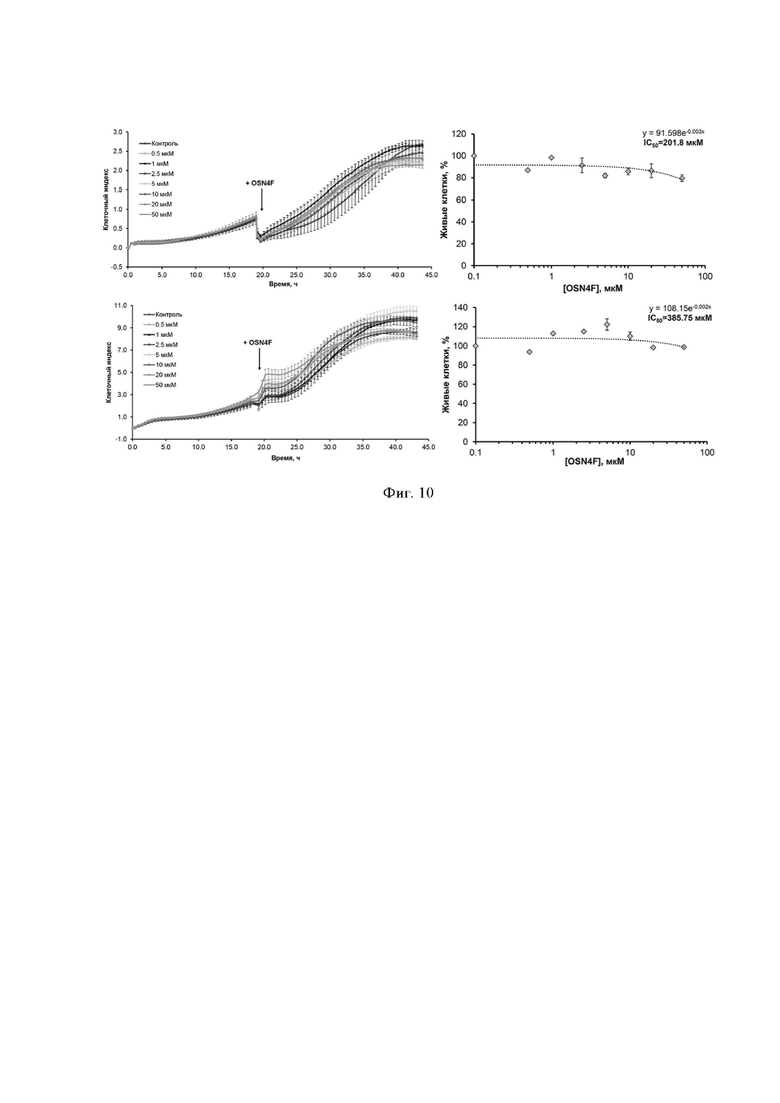
Изобретение относится к соединению, отвечающему формуле F0, в котором заместитель Z выбран из группы: –OН, –SН, –SeН, –NНRN, –O-PG, –S-PG, –Se-PG или –N(PG)RN; заместители Х и Y выбраны из группы, состоящей из 5'-O–конца нуклеозида или олигонуклеотида, 3'-O–конца нуклеозида или олигонуклеотида, ,–H, –OH, –SH, –NHRN, –O-PG или –S-PG, монофосфата, дифосфата, линкера; при этом либо Х, либо Y представляет собой 5'-O–конец нуклеозида или олигонуклеотида или 3'-O–конец нуклеозида или олигонуклеотида; заместители R1, R2, R3,R4 выбраны из ряда –H, –C1-18алкил,–C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2–, –N(CH2CH2)2N– и/или структуры (а), связанные с двумя атомами углерода основной цепи заместителя, а также оканчиваться группами–NR2, –OR, –SR, –OC(O)R, –NHC(O)R,–C(O)OR,–C(O)NHR, –N=C(N(R2)2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3, (в) или (с); при этом PG — это защитная группа, химически инертная в ходе автоматического синтеза, отщепляемая на этапе финального деблокирования олигонуклеотидов, при этом RN представляет собой –H или –C1-4алкил; при этом пары R1 и R2; R3 и R4; R5 и R6; R7 и R8 совместно со связанным с ними атомом азота необязательно формируют 5-8-членный гетероцикличный заместитель, выбранный из N-пирролидинила, N-пиперидинила, N-азепанила, N-азоканила или N-пиперазинила. Изобретение относится к способам получения соединений по изобретению формулы F0. Способ получения осуществляют путем взаимодействия производного трехвалентного фосфора формулы (F1) с азидотриазином формулы (F2 с последующим получением соединения формулы (F0). Способ получения также осуществляют путем взаимодействия производного трехвалентного фосфора формулы (F1) с азидотриазином формулы (F3) с получением соединения формулы (F4) с последующими превращениями в соединение формулы (F0). Технический результат – соединения для применения в качестве исследовательского средства, invitro и invivo, действующего через образование комплементарного комплекса с целевой нуклеиновой кислотой. 4 н. и 13 з.п. ф-лы, 10 ил., 1 табл., 49 пр.
1. Соединение, отвечающее формуле F0
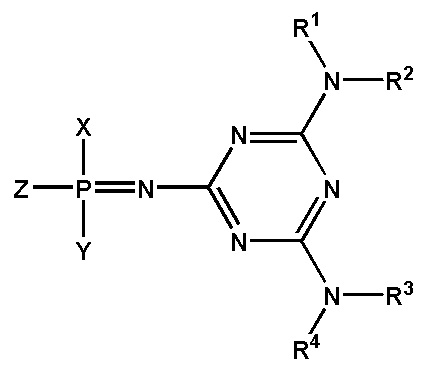 (F0),
(F0),
в котором заместитель Z выбран из группы: –OН, –SН, –SeН, –NНRN, –O-PG, –S-PG, –Se-PG или –N(PG)RN;
заместители Х и Y выбраны из группы, состоящей из 5'-O–конца нуклеозида или олигонуклеотида, 3'-O–конца нуклеозида или олигонуклеотида, ,–H, –OH, –SH, –NHRN, –O-PG или –S-PG, монофосфата, дифосфата, линкера;
при этом либо Х, либо Y представляет собой 5'-O–конец нуклеозида или олигонуклеотида или 3'-O–конец нуклеозида или олигонуклеотида;
заместители R1, R2, R3,R4 выбраны из ряда –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2–, –N(CH2CH2)2N– и/или 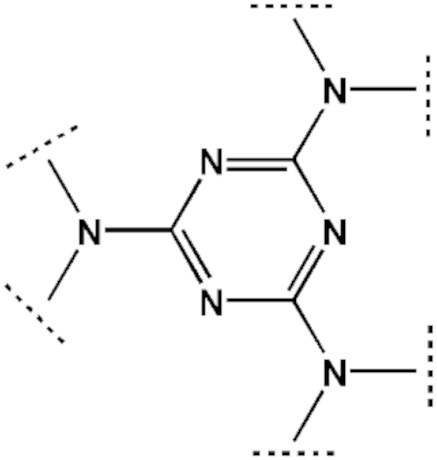 , связанные с двумя атомами углерода основной цепи заместителя,
, связанные с двумя атомами углерода основной цепи заместителя,
а также оканчиваться группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R,–C(O)OR,–C(O)NHR, –N=C(N(R2)2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3,
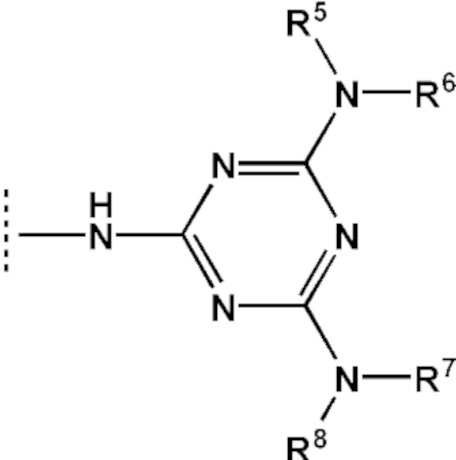 или
или 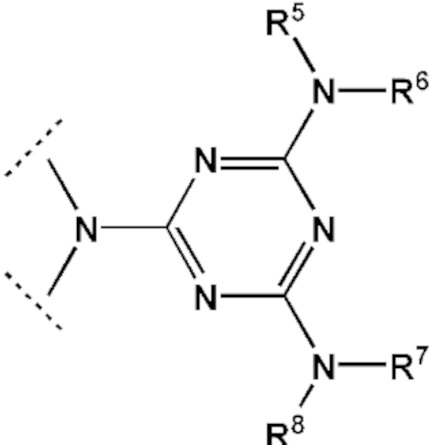 ;
;
при этом заместители R5, R6, R7, R8 выбраны из ряда, содержащего –H, –C1-18алкил,–C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, -NHC(O)–, –NHS(O)2–, –N(CH2CH2)2N–, связанные с двумя атомами углерода основной цепи заместителя,
а также оканчиваться группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2))2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3;
при этом R представляет собой заместитель, выбранный из ряда –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2–, –N(CH2CH2)2N–, связанные с двумя атомами углерода основной цепи заместителя,
и могут оканчиваться группами –NRN2, –ORN, –SRN, –OC(O)RN, –NHC(O)RN, –C(O)ORN, –C(O)NHRN, –N=C(N(RN2))2, –S(O)RN, –S(O)2RN, –S(O)2NRN2, –CN, –Cl, –Br, –I, –F, –N3;
при этом PG — это защитная группа, химически инертная в ходе автоматического синтеза, отщепляемая на этапе финального деблокирования олигонуклеотидов,
при этом RN представляет собой –H или –C1-4алкил;
при этом пары R1 и R2; R3 и R4; R5 и R6; R7 и R8 совместно со связанным с ними атомом азота необязательно формируют 5-8-членный гетероцикличный заместитель, выбранный из N-пирролидинила, N-пиперидинила, N-азепанила, N-азоканила или N-пиперазинила.
2. Соединение по п. 1, в котором Х выбран из группы, состоящей из 5'-O–конца нуклеозида или олигонуклеотида, а Y выбран из группы, состоящей из 3'-O–конца нуклеозида или олигонуклеотида, –H, –OH, –SH, –NHRN, –O-PG или –S-PG, линкера, монофосфата или дифосфата.
3. Соединение по п. 1, в котором Y выбран из группы, состоящей из 5'-O–конца нуклеозида или олигонуклеотида, а X выбран из группы, состоящей из 3'-O–конца нуклеозида или олигонуклеотида, –H, –OH, –SH, –NHRN, –O-PG или –S-PG, линкера, монофосфата или дифосфата.
4. Соединение по п. 1, в котором заместители R1, R2, R3,R4 выбраны из ряда –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые включают группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2–, –N(CH2CH2)2N– и/или , связанные с двумя атомами углерода основной цепи заместителя.
5. Соединение по п. 1, в котором заместители R1, R2, R3,R4 выбраны из ряда –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые оканчиваются группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2)2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3,
или .
6. Химическое соединение по п. 5, в котором заместители R5, R6, R7, R8 выбраны из ряда, содержащего –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые включают группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2–, –N(CH2CH2)2N–, связанные с двумя атомами углерода основной цепи заместителя.
7. Химическое соединение по п. 5, в котором заместители R5, R6, R7, R8выбраны из ряда, содержащего –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые оканчиваются группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2))2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3.
8. Соединение, по п. 1, в котором R1 = R3,R2 = R4.
9. Соединение, по п. 8, в котором R1 = R3 = –H или –CH3.
10. Соединение, по п. 9, в котором R2 = R4 = –C1-18алкил или –C1-18алкенил.
11. Соединение, по п. 1, представляющее собой олигонуклеотид.
12. Способ получения соединения по п. 1, заключающийся во взаимодействии производного трехвалентного фосфора формулы (F1) с азидотриазином формулы (F2) с последующим получением соединения формулы (F0)
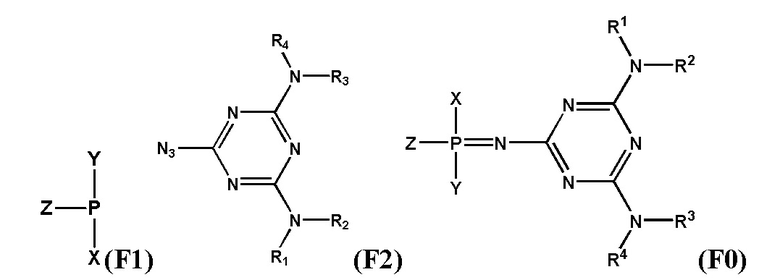
13. Способ получения соединения по п. 1, заключающийся во взаимодействии производного трехвалентного фосфора формулы (F1) с азидотриазином формулы (F3) с получением соединения формулы (F4) с последующими превращениями в соединение формулы (F0)
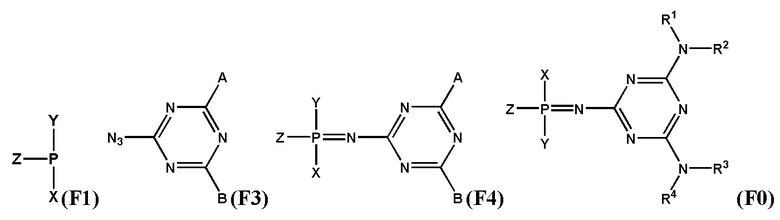
при этом группа A может быть выбрана из ряда –NR1R2, –NR3R4 , –NRXRY, –Q,
а группа B может быть выбрана из ряда –NRXRY, –Q,
где заместители RX,RY преобразуют в заместители R1, R2, R3,R4 с использованием реакций превращения соответствующих реакционных групп, входящих в состав RX,RY,
причем заместители RX,RY выбирают из ряда, содержащего –H, –C1-18алкил, –C2-18aлкенил, –C2-18aлкинил и –C6-18aрил, которые могут включать группы –NH–, –N<, –О–, –NHC(O)–, –NHS(O)2– и/или –N(CH2CH2)2N–, а также оканчиваться группами –NR2, –OR, –SR, –OC(O)R, –NHC(O)R, –C(O)OR, –C(O)NHR, –N=C(N(R2))2, –S(O)R, –S(O)2R, –S(O)2NR2, –CN, –Cl, –Br, –I, –F, –N3;
при этом Q представляет собой группу, которую замещают на –NR1R2, –NR3R4, –NRXRY в реакции с соответствующим амином HNR1R2, HNR3R4, HNRXRY,
при этом Q выбирают из ряда: –OR, –OC(O)R, –OS(O)2R, –CN, –Cl, –Br, –I, –F, –N3.
14. Способ по п. 12 или 13, где производное трехвалентного фосфора это Н-фосфонатное звено, полученное согласно Н-фосфонатному методу олигонуклеотидного синтеза, или фосфитное звено, полученного согласно амидофосфитному методу олигонуклеотидного синтеза.
15. Способ по п. 13, где A = B = –Q.
16. Способ по п. 15, где Q = Cl.
17. Применение соединения по п. 1 в качестве исследовательского средства, in vitro и in vivo, действующего через образование комплементарного комплекса с целевой нуклеиновой кислотой.
| A | |||
| B | |||
| Borkovec et al., "Insect Chemosterilants | |||
| III | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| A | |||
| B | |||
| Borkovec et al., "Insect Chemosterilants | |||
| V | |||
| Derivatives of Melamine" Journal of Medicinal Chemistry, 10(3), 1967, 457-461 | |||
| М | |||
| И | |||
| Буковский и др | |||
| "Фосфазотриазины | |||
| III | |||
| Окислительное иминирование соединений | |||
