Область техники, к которой относится изобретение
В настоящем изобретении описаны соединения и фармацевтические композиции, содержащие одно или больше из указанных соединений или их фармацевтически приемлемых солей, которые эффективно подавляют связывание или работу хемокинов с CCR9 хемокиновым рецептором. В качестве антагонистов или модуляторов CCR9 хемокинового рецептора, указанные соединения и композиции могут найти применение в лечении различных иммунных нарушений, патологических состояний и заболеваний.
Предшествующий уровень техники
Хемокины, также известные как хемотактические цитокины, представляют собой группу белков с низким молекулярным весом, которые высвобождаются широким рядом клеток и обладают различной биологической активностью. Хемокины привлекают различные типы клеток иммунной системы, такие как макрофаги, Т-клетки, эозинофилы, базофилы и нейтрофилы, и заставляют их мигрировать из крови в различные лимфоидные и нелимфоидные ткани. Они обеспечивают проникновение воспалительных клеток к сайтам воспаления и отвечают за инициацию и развитие многих воспалительных заболеваний (см. обзор в работе Schall, Cytokine, 3:165-183 (1991), Schall et al., Curr. Opin. lmmunol., 6:865-873 (1994)).
Помимо стимулирования хемотаксиса, хемокины могут вызывать другие изменения в реагирующих клетках, включая изменение формы клеток, экзоцитоз гранул, повышение содержания интегрина, образование биоактивных липидов (например, лейкотриенов), респираторный взрыв, связанный с активацией лейкоцитов, пролиферацию клеток, устойчивость к индуцированию апоптоза и ангиогенез. Таким образом, хемокины являются ранними инициирующими факторами воспалительного ответа, вызывающими высвобождение медиаторов воспаления, хемотаксис и проникновение крови из сосудов к сайтам инфицирования или воспаления. Они также стимулируют множество клеточных процессов, имеющих важную физиологическую роль и патологические последствия.
Хемокины осуществляют свое действие путем активации хемокиновых рецепторов, экспрессируемых клетками, способными к реакции. Хемокиновые рецепторы представляют собой класс рецепторов, связанных с G-белком, известных также как рецепторы с семью трансмембранными петлями, которые находятся на поверхности разнообразных типов клеток, таких как лейкоциты, клетки эндотелия, клетки гладкой мускулатуры и опухолевые клетки.
Хемокины и хемокиновые рецепторы экспрессируются собственными клетками почек и инфильтрующимися клетками во время воспаления почек (Segerer et al., J. Am. Soc. Nephrol., 11:152-76 (2000); Morii et al., J. Diabetes Complications, 17:11-5 (2003); Lloyd et al. J. Exp. Med., 185:1371-80 (1997); Gonzalez-Cuadrado et al. Clin. Exp. Immunol., 106:518-22 (1996); Eddy & Giachelli, Kidney Int., 47:1546-57 (1995); Diamond et al., Am. J. Physiol., 266:F926-33 (1994)).
Инфильтрацию T-лимфоцитов (Т-клеток) в тонкий кишечник и толстый кишечник связывают с патогенезом целиакии, пищевой аллергии, ревматоидного артрита, воспалительных заболеваний кишечника (IBD), которые включают болезнь Крона и язвенный колит. Блокирование поступления соответствующих Т-клеток в кишечник может дать эффективный способ лечения воспалительных заболеваний кишечника человека. Недавно было обнаружено, что хемокиновый рецептор-9 (CCR(9)) экспрессирован на Т-клетках, базирующихся в кишечнике, в периферической крови, и его уровень повышен у пациентов с воспалением тонкого кишечника, таким как болезнь Крона и целиакия. Единственный идентифицированный на сегодняшний день лиганд CCR(9), TECK (хемокин, экспрессируемый тимусом) экспрессирован в тонком и толстом кишечнике, и считается, что данная пара лиганд-рецептор играет важнейшую роль в развитии воспалительных заболеваний кишечника. В частности, данная пара обуславливает миграцию в кишечник воспалительных клеток, вызывающих заболевания. См., например, Zaballos et al., J. Immunol., 162(10):5671-5675 (1999); Kunkel et al., J. Exp. Med., 192(5):761-768 (2000); Papadakis et al., J. Immunol., 165(9):5069-5076 (2000); Papadakis et al., Gastroenterology, 121(2):246-254 (2001); Campbell et al., J. Exp. Med., 195(1):135-141 (2002); Wurbel et al., Blood, 98(9):2626-2632 (2001); и Uehara et al., J. Immunol, 168(6):2811-2819 (2002); Rivera-Nieves et al., Gastroenterology, 2006 Nov;131(5):1518-29; и Kontoyiannis et al., J. Exp. Med., Vol. 196, Number 12, Dec. 16, 2002. Кроме того, было показано, что лимфоциты, несущие CCR(9), опосредуют патогенез филяриоза (лимфатическое филяриозное заболевание), а подавление CCR(9) коррелирует с уменьшением патологии в таких патологических состояниях. См., например, Babu et al., Journal of Infectious Diseases, 191: 1018-26, 2005.
Идентификация соединений, которые модулируют работу CCR(9), представляет собой привлекательное новое семейство терапевтических средств для лечения воспалительных и других патологических состояний и заболеваний, связанных с активацией CCR(9), таких как воспалительное заболевание кишечника.
Краткое описание изобретения
Настоящее изобретение касается соединений и их фармацевтически приемлемых солей, композиций и способов, которые могут применяться для модулирования работы CCR(9). Указанные соединения и их соли, композиции и способы, описанные в настоящем тексте, могут применяться в лечении или предотвращении опосредуемых хемокинами патологических состояний или заболеваний, включая некоторые воспалительные и иммунорегуляторные нарушения и заболевания.
Было показано, что соединения по настоящему изобретению модулируют CCR(9), как проиллюстрировано в примерах.
В одном аспекте, соединения по настоящему изобретению могут быть представлены формулой (I):
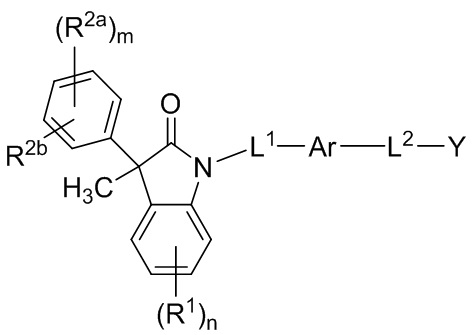 (I)
(I)
или их фармацевтически приемлемые соли, где
Ar представляет собой 5-10-членное ароматическое или гетероароматическое кольцо, необязательно замещенное 1-3 заместителями R3;
L1 выбран из группы, состоящей из одинарной связи, C1-6 алкилена и C1-6 гетероалкилена,
L2 выбран из группы, состоящей из одинарной связи, C1-6 алкилена и C1-6 гетероалкилена,
Y представляет собой CO2H или биоизостер карбоновой кислоты;
каждый R1 и каждый R2a независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила, C1-6 галогеналкоксигруппы, C3-6 циклоалкила и C2-6 алкенила, где алкильные, циклоалкильные и алкенильные фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, OH, CN, C1-3 алкила, C1-3 галогеналкила и C1-3 алкоксигруппы;
R2b выбран из группы, состоящей из H, галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C3-6 циклоалкила и C2-6 алкенила, где алкильные, циклоалкильные и алкенильные фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, CN, C1-3 алкила, C1-3 галогеналкила и C1-3 алкоксигруппы;
или, необязательно, один R2a и R2b, когда они присоединены к соседним атомам фенильного цикла, могут быть объединены с образованием 5- или 6-членного гетероциклоалкильного кольца, содержащего один или два атома в цикле, независимо выбранных из O, N и S, где указанное гетероциклоалкильное кольцо необязательно замещено 1-3 заместителями, выбранными из атома фтора и C1-3 алкила;
каждый R3 независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила, C1-6 галогеналкоксигруппы, C3-6 циклоалкила, и C2-6 алкенила;
подстрочный индекс m представляет собой целое число от 0 до 4; и
подстрочный индекс n представляет собой целое число от 0 до 3.
В другом аспекте, в настоящем изобретении описаны композиции, которые могут применяться для модулирования активности хемокинов. В одном варианте осуществления, композиция по настоящему изобретению содержит соединение по настоящему изобретению и фармацевтически приемлемый носитель или вспомогательное вещество.
В другом аспекте, в настоящем изобретении описаны способы модулирования работы хемокинов в клетке, включающие контакт клетки с терапевтически эффективным количеством соединения или композиции по настоящему изобретению.
В другом аспекте, в настоящем изобретении описаны способы модулирования работы хемокинов, включающие контакт хемокинового рецептора с терапевтически эффективным количеством соединения или композиции по настоящему изобретению.
В другом аспекте, в настоящем изобретении описаны способы лечения опосредуемого хемокинами патологического состояния или заболевания, включающие введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению. Введение может быть пероральным, парентеральным, ректальным, чрескожным, сублингвальным, назальным или местным. В некоторых аспектах, соединение можно вводить в комбинации с противовоспалительным средством или анальгетиком.
Помимо описанных в настоящем тексте соединений, в настоящем изобретении описаны также фармацевтические композиции, содержащие одно или больше из указанных соединений, а также способы применения указанных соединений в терапевтических способах, в первую очередь для лечения заболеваний, связанных с сигнальной активностью хемокинов. Опосредуемое CCR(9) заболевание или патологическое состояние может представлять собой воспалительное заболевание кишечника, аллергическое заболевание, псориаз, атопический дерматит, астму, фиброзные заболевания, отторжение трансплантата, реакцию отторжения трансплантата, синдром Шегрена, иммуноопосредованные пищевые аллергии, аутоиммунные заболевания, целиакию, ревматоидный артрит, тимому, рак вилочковой железы, лейкоз, солидную опухоль, или острый лимфобластный лейкоз, меланому, первичный склерозирующий холангит, гепатит и воспалительную болезнь печени, послеоперационную непроходимость кишечника, болезнь Крона или язвенный колит.
Краткое описание чертежей
Неприменимо
Подробное описание изобретения
Общее
Настоящее изобретение касается соединений и их солей, композиций и способов, которые могут применяться для модулирования работы хемокинового рецептора, в особенности работы CCR(9). Модулирование активности хемокинового рецептора, которое используется в настоящем тексте в различных формах, охватывает антагонизм, агонизм, частичный антагонизм, обратный агонизм и/или частичный агонизм активности, связанной с определенным хемокиновым рецептором, предпочтительно CCR(9) рецептором. Соответственно, соединения по настоящему изобретению представляют собой соединения, которые модулируют по меньшей мере одну функцию или характеристику CCR(9) млекопитающих, например, человеческого белка CCR(9). Способность указанных соединений модулировать работу CCR(9) можно продемонстрировать в анализе связывания (например, анализ связывания с лигандом или связывания с агонистом), анализе хемотаксиса (исследование миграции), анализе сигнала (например, активация G-белка млекопитающих, возникновение быстрого и транзиторного повышения концентрации свободного кальция в цитозоле), и/или анализе клеточного ответа (например, стимулирование хемотаксиса, экзоцитоза или высвобождения медиаторов воспаления лейкоцитами).
Сокращения и определения
Термин "алкил", сам по себе и как часть другого заместителя, означает, если не указано иное, линейный или разветвленный углеводородный радикал, имеющий обозначенное число атомов углерода (например, C1-8 означает 1-8 атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п. Термин "алкенил" означает ненасыщенную алкильную группу, содержащую одну или больше двойных связей. Аналогично, термин «алкинил» означает ненасыщенную алкильную группу, содержащую одну или больше тройных связей. Примеры таких ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры. Термин "циклоалкил" относится к углеводородным циклам, имеющим указанное число атомов в цикле (например, C3-6циклоалкил) и являющимся полностью насыщенными или имеющими не более одной двойной связи между вершинами цикла. "Циклоалкил" относится также к бициклическим и полициклическим углеводородным кольцам, таким как, например, бицикло[2.2.1]гептан, бицикло[2.2.2]октан и т.д. Термин "гетероциклоалкан” или “гетероциклоалкил" относится к циклоалкильной группе, содержащей 1-5 гетероатомов, выбранных из N, O, и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероциклоалкан может представлять собой моноциклическую, бициклическую или полициклическую кольцевую систему. Неограничивающие примеры гетероциклоалкановых групп включают пирролидин, имидазолидин, пиразолидин, бутиролактам, валеролактам, имидазолидинон, гидантоин, диоксолан, фталимид, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-оксид, пиперазин, пиран, пиридон, 3-пирролин, тиопиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин и т.п. Гетероциклоалкановая группа может быть присоединена к остальной части молекулы через атом углерода в цикле или гетероатом в цикле.
Термин "алкилен" в отдельности или как часть другого заместителя означает двухвалентный радикал, являющийся производным алкана, в качестве примера можно привести -CH2CH2CH2CH2-. В типичном случае алкильная (или алкиленовая) группа содержит от 1 до 24 атомов углерода, предпочтительными по настоящему изобретению являются группы, содержащие 10 или меньше атомов углерода. «Низший алкил» или «низший алкилен» представляет собой короткоцепочечную алкильную или алкиленовую группу, обычно содержащую четыре или меньше атомов углерода. Аналогично, «алкенилен» или «алкинилен» означает ненасыщенные формы «алкилена», содержащие двойные или тройные связи, соответственно. Термин "гетероалкилен" означает алкиленовую группу, в которой один или два атома углерода заменены на N, O или S.
При использовании в настоящем тексте, волнистая линия " ", пересекающая простую, двойную или тройную связь в любой изображенной в настоящем тексте химической структуре, означает точку присоединения простой, двойной или тройной связи к остальной части молекулы.
", пересекающая простую, двойную или тройную связь в любой изображенной в настоящем тексте химической структуре, означает точку присоединения простой, двойной или тройной связи к остальной части молекулы.
Термины "алкокси," "алкиламино" и "алкилтио" (или тиоалкокси) применяются в их обычном смысле и относятся к алкильным группам, присоединенным к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Кроме того, для диалкиламиногрупп, алкильные фрагменты могут быть одинаковыми или разными, а также могут объединяться с формированием 3-7-членного цикла с атомом азота, к которому они присоединены. Соответственно, группа, изображаемая как -NRaRb, включает пиперидинил, пирролидинил, морфолинил, азетидинил и т.п.
Термин "ди-(C1-4 алкил)амино-C1-4 алкил" относится к аминогруппе, несущей две C1-4 алкильные группы, которые могут быть одинаковыми или разными (например, метил, этил, пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил), и которая присоединена к остальной части молекулы через C1-4 алкильную группу (1-4-углеродная алкиленовая связывающая группа). Примеры ди-(C1-4 алкил)амино-C1-4 алкильных групп включают диметиламинометил, 2-(этил(метил)амино)этил, 3-(диметиламино)бутил и т.п.
Термин "галоген" сам по себе или как часть другого заместителя означает, если не указано иное, атом фтора, хлора, брома или иода. Кроме того, такие термины как "галогеналкил" и "галогеналкокси" включают моногалоген- и полигалоген- версии алкила и алкоксигруппы, соответственно. Например, термин "C1-4 галогеналкил" включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п.
Термин "арил" или “ароматическое кольцо” означает, если не указано иное, полиненасыщенную, в типичном случае ароматическую, углеводородную группу, которая может представлять собой один цикл или несколько циклов (до трех циклов), сопряженные или связанные ковалентно. Аналогично, термин «гетероарил» или “гетероароматическое кольцо” означает арильные группы (или циклы), содержащие от одного до пяти гетероатомов, выбранных из N, O и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероарильная группа или гетероароматическое кольцо могут быть присоединены к остальной части молекулы через гетероатом. Неограничивающие примеры арильных групп включают фенил, нафтил и бифенил, а неограничивающие примеры гетероарильных групп включают пиридил, пиридазинил, пиразинил, пиримидинил, триазинил, хинолинил, хиноксалинил, хиназолинил, циннолил, фталазинил, бензотриазинил, пуринил, бензоимидазолил, бензопиразолил, бензотриазолил, бензизоксазалил, изобензофурил, изоиндолил, индолизинил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиразолопиримидинил, имидазопиридины, бензотиаксолил, бензофуранил, бензотиенил, индолил, хинолил, изохинолил, изотиазолил, пиразолил, индазолил, птеридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, тиадиазолил, пирролил, тиазолил, фурил, тиенил и т.п. Заместители в каждой из перечисленных выше арильных или гетероарильных циклических системах выбраны из группы приемлемых заместителей, описанных ниже.
Термин "арилалкил" включает радикалы, в которых арильная группа присоединена к алкильной группе (например, бензил, фенетил и т.п.). Аналогично, термин "гетероарил-алкил" включает радикалы, в которых гетероарильная группа присоединена к алкильной группе (например, пиридилметил, тиазолилэтил и т.п.).
При использовании в настоящем тексте, термин "гетероатом" включает в себя кислород (О), азот (N), серу (S) и кремний (Si).
Термин "биоизостер карбоновой кислоты" относится к группе, имеющей полярный и/или кислотный характер, которая работает как замена карбоновой кислоты. Различные биоизостеры известны для карбоновых кислот, включая гидроксамовые кислоты, эфиры гидроксамовых кислот, фосфоновые кислоты, фосфиновые кислоты, сульфоновые кислоты, сульфиновые кислоты, сульфамиды, ацилсульфамиды, ацилмочевины, сульфонилмочевины, циклопентан-1,2-дионы, замещенные фенолы и гетероциклические биоизостеры, приведенные ниже:
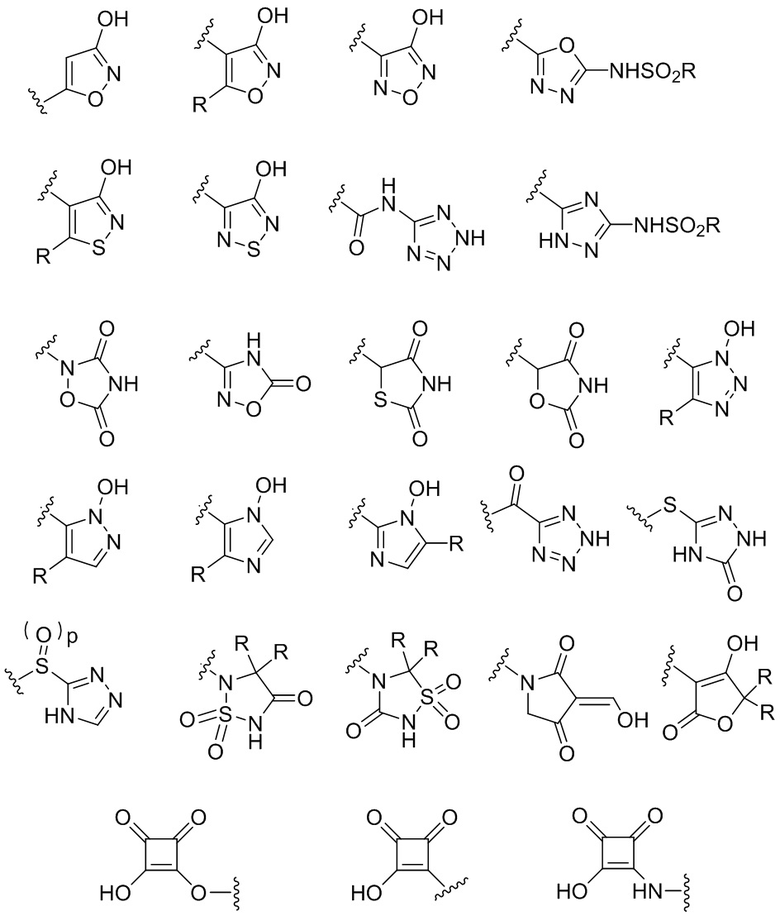
где p равен 0, 1 или 2, и где каждая группа R независимо выбрана из группы, состоящей из H, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гидроксиалкила, C1-C6 алкоксигруппы или C1-C4 алкил-O-C1-C4 алкила.
Другими примерами биоизостеров карбоновых кислот являются тетразолил или тетразолонил, где тетразолил или тетразолонил необязательно замещены C1-6 алкилом, C1-6 галогеналкилом, C1-6 гидроксиалкилом, -C1-6 алкоксигруппой или C1-4алкил-O-C1-4алкилом. Другие примеры биоизостеров карбоновых кислот описаны в статье Journal of Medicinal Chemistry, 2016, 59, 3183-3203, которая включена в настоящий текст посредством ссылки.
Термин "фармацевтически приемлемые соли" включает соли веществ, полученные с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей в описанных в настоящем тексте соединениях. Когда соединения по настоящему изобретению содержат относительно кислые функциональные группы, можно получить основно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количество желаемого основания, даже без растворителя или в подходящем инертном растворителе. Примеры солей, являющихся производными фармацевтически приемлемых неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа(II), железа (III), лития, магния, марганца, калия, натрия, цинка и т.д. Соли, являющиеся производными фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, природные амины и т.д., такие как аргинин, бетаин, кофеин, холин, N,N’-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, можно получить кислотно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количеством желаемой кислоты, без растворителя или в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли с неорганическими кислотами, такими как хлористоводородная, бромистоводородная, азотная, угольная, моногидроугольная, фосфорная, моногидрофосфорная, дигидрофосфорная, серная, моногидросерная, иодистоводородная или фосфористая кислота и т.п., а также соли с относительно нетоксичными органическими кислотами, такими как уксусная, пропионовая, изомасляная, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, пара-толуолсульфоновая, лимонная, винная, метансульфоновая и т.п. Также охватываются соли с аминокислотами, такие как аргинаты и т.п., и соли таких органических кислот, как глюкуроновая или галактуроновая кислоты и т.п. (см, например, Berge, S.M., et al., “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19). Некоторые частные соединения по настоящему изобретению содержат и основные, и кислотные функциональные группы, что позволяет таким соединениям образовывать как основно-аддитивные, так и кислотно-аддитивные соли.
Нейтральные формы соединений можно регенерировать путем взаимодействия соли с основанием или кислотой и выделения материнского соединения обычным способом. Материнская форма соединения отличается от различных солевых форм определенными физическими характеристиками, такими как растворимость в полярных растворителях, но во всем остальном соли эквивалентны материнским соединениям, в терминах настоящего изобретения.
Помимо солевых форм, в настоящем изобретении описаны соединения в форме пролекарства. Пролекарства описанных в настоящем тексте соединений представляют собой соединения, которые легко претерпевают химические изменения в физиологических условиях, давая соединения по настоящему изобретению. Кроме того, пролекарства могут превращаться в соединения по настоящему изобретению химическими или биохимическими методами в условиях in vivo. Например, пролекарства могут медленно превращаться в соединения по настоящему изобретению при помещении в резервуар чрескожного пластыря с подходящим ферментативным или химическим реагентом.
Некоторые соединения по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. В целом, сольватированные формы эквивалентны несольватированным формам, и все они охватываются настоящим изобретением. Некоторые соединения по настоящему изобретению могут существовать в нескольких кристаллических или аморфных формах. В целом, все физические формы эквивалентны для областей применения, охватываемых настоящим изобретением, и входят в объем настоящего изобретения.
Некоторые соединения по настоящему изобретению имеют асимметрические атомы углерода (оптические центры) или двойные связи; все рацематы, диастереомеры, геометрические изомеры, региоизомеры и индивидуальные изомеры (например, отдельные энантиомеры) входят в объем настоящего изобретения. Когда показана стерохимическая конфигурация, это означает соединение, в котором присутствует один из изомеров, и оно практически не содержит другого изомера. “Практически не содержит” другого изомера означает соотношение этих двух изомеров по меньшей мере 80/20, более предпочтительно 90/10 или 95/5 или больше. В некоторых вариантах осуществления, один из изомеров присутствует в количестве по меньшей мере 99%.
Соединения по настоящему изобретению могут также иметь неприродные соотношения изотопов по одному или больше атомов, составляющих эти соединения. Неприродные соотношения изотопов можно определить как находящиеся в диапазоне от природного количества до количества рассматриваемого атома равного 100%. Например, соединения могут быть радиоактивно мечены радиоактивными изотопами, такими как, например, тритий (3H), иод-125 (125I) или углерод-14 (14C), или нерадиоактивными изотопами, такими как дейтерий (2H) или углерод-13 (13C). Такие вариации изотопов могут открыть дополнительные области применения к описанным в других разделах настоящего описания. Например, изотопные модификации соединений по настоящему изобретению могут найти дополнительное применение, включая (но не ограничиваясь только ими) применение в качестве диагностических и/или визуализирующих реагентов, или в качестве цитотоксических/радиотоксических терапевтических средств. Кроме того, изотопные варианты соединений по настоящему изобретению могут иметь измененные фармакокинетические и фармакодинамические характеристики, которые могут вносить свой вклад в улучшение характеристик безопасности, переносимости или эффективности при лечении. Все изотопные вариации соединений по настоящему изобретению, радиоактивные и нерадиоактивные, входят в объем настоящего изобретения. Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может обеспечить определенные терапевтические преимущества вследствие большей метаболической стабильности. Например, может возрастать время полужизни in vivo, или можно уменьшить требования к дозировке.
Соединения по настоящему изобретению, имеющие формулу I, могут существовать в различных изомерных формах. В настоящем тексте, термины цис или транс используются в их обычном для химии значении, т.е. они означают взаимное расположение заместителей относительно определенной плоскости, например, двойной связи или циклической системы, такой как система декалинового типа или гидрохинолонового типа: в цис-изомере заместители находятся по одну сторону от плоскости, в транс-изомере заместители находятся по разные стороны от плоскости. Кроме того, настоящее изобретение охватывает различные конформеры, а также различные ротамеры. Конформеры представляют собой конформационные изомеры, которые могут различаться вследствие вращения вокруг одной или больше σ-связей. Ротамеры представляют собой конформеры, которые различаются вращением вокруг только одной σ-связи.
Соединения
В настоящем изобретении описаны соединения, которые модулируют активность CCR(9). Хемокиновые рецепторы представляют собой интегрированные в мембрану белки, которые взаимодействуют с внеклеточным лигандом, таким как хемокин, и обуславливают клеточный ответ на лиганд, например, хемотаксис, повышение концентрации ионов кальция в клетке и т.д. Поэтому модулирование работы хемокинового рецептора, например, нарушение взаимодействия хемокинового рецептора с лигандом, будет модулировать ответ, обуславливаемый хемокиновым рецептором, и позволит лечить или предотвращать патологическое состояние или заболевание, опосредуемое хемокиновым рецептором. Модулирование работы хемокинового рецептора включает как усиление, так и подавление работы. Тип осуществленного модулирования зависит от характеристик соединения, например, это может быть антагонист или полный, частичный или обратный агонист.
Например, соединения по настоящему изобретению работают как сильные CCR(9) антагонисты, и эта антагонистическая активность была подтверждена в тестах воспаления на животных, одного из характерных болезненных состояний для CCR(9). Соответственно, описанные в настоящем изобретении соединения могут применяться в фармацевтических композициях, способах лечения CCR(9)-опосредуемых заболеваний, и как контроль в тестах, направленных на выявление конкурентных антагонистов CCR(9).
В настоящем изобретении описаны соединения, имеющие формулу (I):
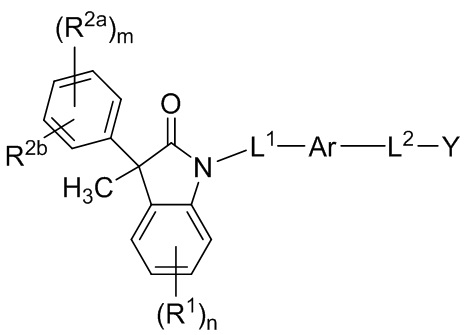 (I)
(I)
или их фармацевтически приемлемые соли, где
Ar представляет собой 5-10-членный ароматическое или гетероароматическое кольцо, необязательно замещенное 1-3 заместителями R3;
L1 выбран из группы, состоящей из одинарной связи, C1-6 алкилена и C1-6 гетероалкилена,
L2 выбран из группы, состоящей из одинарной связи, C1-6 алкилена и C1-6 гетероалкилена,
Y представляет собой CO2H или биоизостер карбоновой кислоты;
каждый R1 и каждый R2a независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила, C1-6 галогеналкоксигруппы, C3-6 циклоалкила и C2-6 алкенила, где алкильные, циклоалкильные и алкенильные фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, OH, CN, C1-3 алкила, C1-3 галогеналкила и C1-3 алкоксигруппы;
R2b выбран из группы, состоящей из H, галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C3-6 циклоалкила и C2-6 алкенила, где алкильные, циклоалкильные и алкенильные фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, CN, C1-3 алкила, C1-3 галогеналкила и C1-3 алкоксигруппы;
или, необязательно, один R2a и R2b, когда они присоединены к соседним атомам фенильного цикла, могут быть объединены с образованием 5- или 6-членного гетероциклоалкильного кольца, содержащего один или два атома в цикле, независимо выбранных из O, N и S, где указанное гетероциклоалкильное кольцо необязательно замещено 1-3 заместителями, выбранными из атома фтора и C1-3 алкила;
каждый R3 независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила, C1-6 галогеналкоксигруппы, C3-6 циклоалкила и C2-6 алкенила;
подстрочный индекс m представляет собой целое число от 0 до 4; и
подстрочный индекс n представляет собой целое число от 0 до 3.
В некоторых вариантах осуществления, Y выбран из группы, состоящей из:
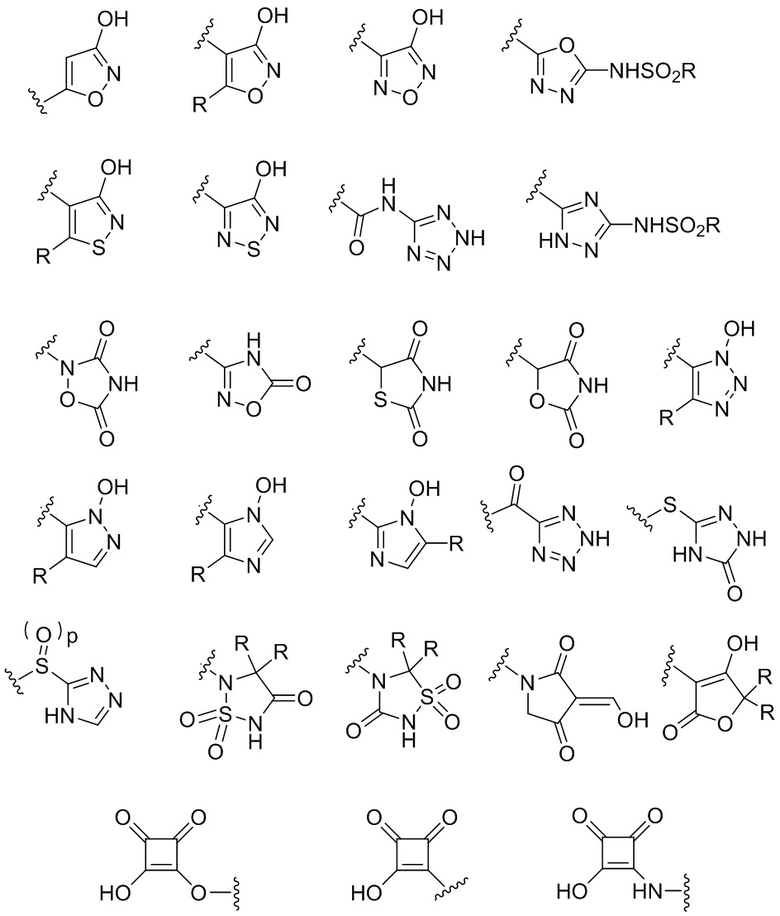
тетразолила и тетразолонила, где тетразолил или тетразолонил необязательно замещены заместителем R,
где p равен 0, 1 или 2, и где каждая группа R независимо выбрана из группы, состоящей из H, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гидроксиалкила, C1-C6 алкоксигруппы или C1-C4 алкил-O-C1-C4 алкила.
В некоторых вариантах осуществления, Y выбран из группы, состоящей из: тетразолила и тетразолонила, где тетразолил или тетразолонил необязательно замещены C1-6 алкилом, C1-6 галогеналкилом, C1-6 гидроксиалкилом, -C1-6 алкоксигруппой или C1-4алкил-O-C1-4алкилом.
В некоторых вариантах осуществления, описано соединение, имеющее формулу, выбранную из группы, состоящей из:
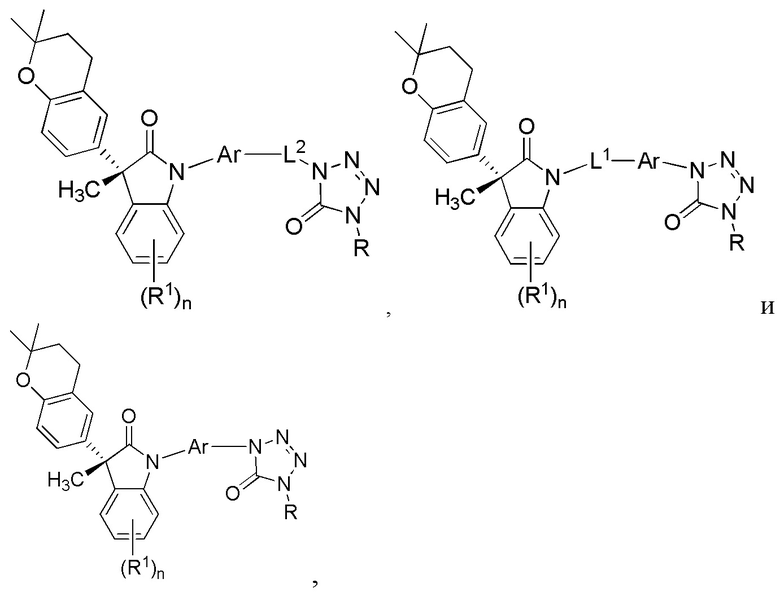
или его фармацевтически приемлемые соли, где указанное соединение практически не содержит других изомеров.
В некоторых вариантах осуществления, описано соединение, имеющее формулу:
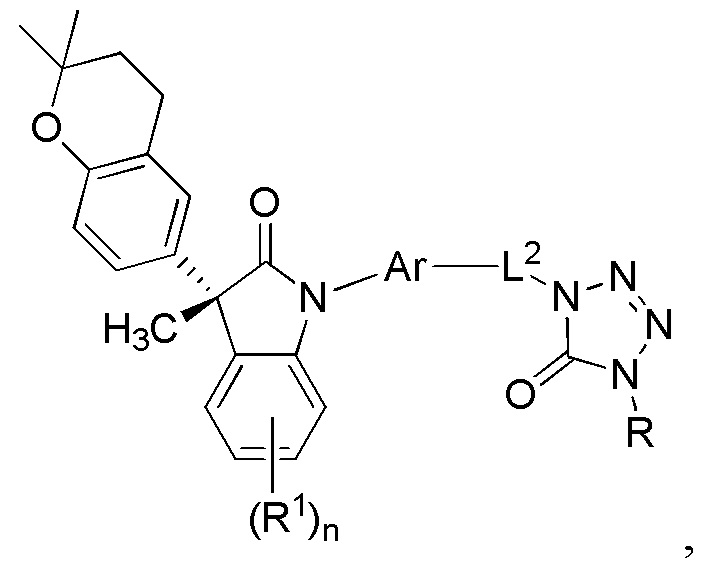
или его фармацевтически приемлемые соли, где L2 представляет собой C1-3 алкилен и где указанное соединение практически не содержит других изомеров.
В некоторых вариантах осуществления, описано соединение, имеющее формулу (I’):
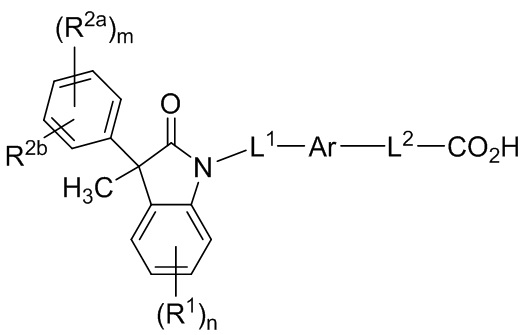 (I’)
(I’)
или его фармацевтически приемлемые соли, где
Ar представляет собой 5-10-членный ароматическое или гетероароматическое кольцо, необязательно замещенное 1-3 заместителями R3;
L1 выбран из группы, состоящей из одинарной связи, C1-6 алкилена и C1-6 гетероалкилена,
L2 выбран из группы, состоящей из одинарной связи, C1-6 алкилена и C1-6 гетероалкилена,
каждый R1 и каждый R2a независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила, C1-6 галогеналкоксигруппы, C3-6 циклоалкила и C2-6 алкенила, где алкильный, циклоалкильный и алкенильный фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, CN, C1-3 алкила, C1-3 галогеналкила и C1-3 алкоксигруппы;
R2b выбран из группы, состоящей из H, галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C3-6 циклоалкила и C2-6 алкенила, алкильный, циклоалкильный и алкенильный фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, CN, C1-3 алкила, C1-3 галогеналкила и C1-3 алкоксигруппы;
или, необязательно, один R2a и R2b, когда они присоединены к соседним атомам фенильного цикла, могут быть объединены с образованием 5- или 6-членного циклогетероалкильного кольца, содержащего один или два атома в цикле, независимо выбранных из O, N и S, где указанное циклогетероалкильное кольцо необязательно замещено 1-3 заместителями, выбранными из атома фтора и C1-3 алкила;
каждый R3 независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила, C1-6 галогеналкоксигруппы, C3-6 циклоалкила и C2-6 алкенила;
подстрочный индекс m представляет собой целое число от 0 до 4; и
подстрочный индекс n представляет собой целое число от 0 до 3.
В одной группе вариантов осуществления для каждой из формул (I) и (I’), Ar выбран из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3.
В некоторых частных вариантах формулы (I) и (I’), L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-. В других частных вариантах формулы (I), L2 выбран из группы, состоящей из одинарной связи, -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2- и -CH2CH2CH2-.
В некоторых частных вариантах формулы (I) и (I’), n равен 1 или 2. В других частных вариантах формулы (I) и (I’), m равен 1, 2 или 3.
В других вариантах осуществления, описаны подходящие соединения, имеющие формулу (Ia):
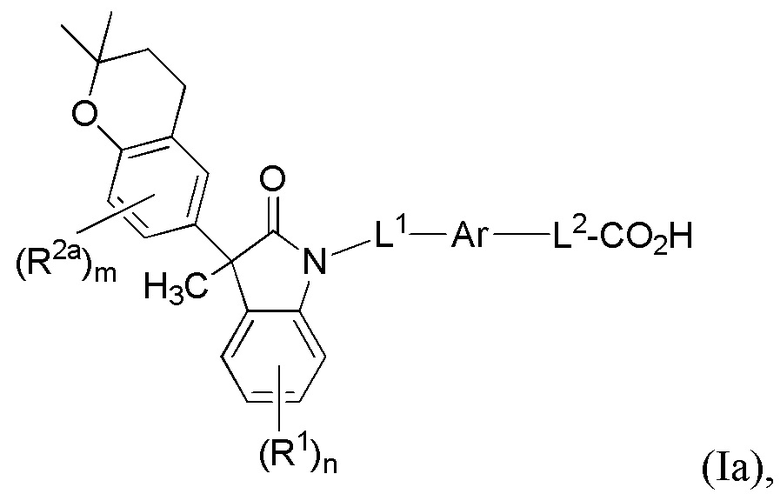
или их фармацевтически приемлемые соли.
В одной группе частных вариантов осуществления, Ar выбран из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3.
В другой группе частных вариантов осуществления, L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-. В другой группе частных вариантов осуществления, L2 выбран из группы, состоящей из одинарной связи, -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2- и -CH2CH2CH2-.
В других частных вариантах, подходящие соединения выбраны из:
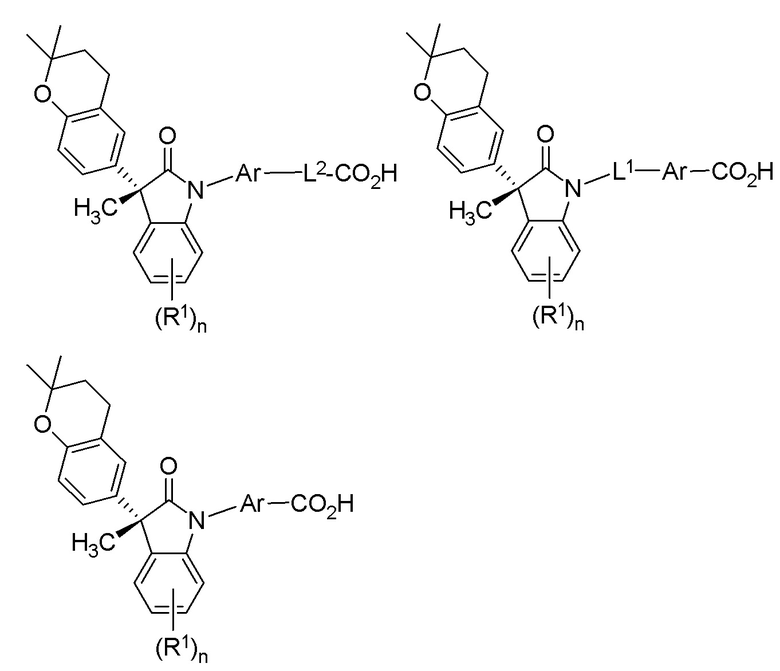
(Ia1) (Ia2) и (Ia3)
или их фармацевтически приемлемые солей, где указанное соединение практически не содержит других изомеров.
В формуле (Ia1), (Ia2) и (Ia3), частными вариантами осуществления являются такие, где Ar выбран из группы, состоящей из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3. В других вариантах осуществления, Ar выбран из группы, состоящей из 1,3-фенилена и 1,4-фенилена, каждый из которых необязательно замещен 1-2 заместителями R3. В некоторых вариантах осуществления, относящихся к формулам (Ia1), (Ia2) и (Ia3), L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-. В других вариантах формул (Ia1), (Ia2) и (Ia3), L2 выбран из группы, состоящей из -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-,-CH2- и -CH2CH2CH2-. В других вариантах формул (Ia1), (Ia2) и (Ia3), R1 выбран из группы, состоящей из галогена, цианогруппы, C1-3 алкила, C1-3 алкоксигруппы, C1-3 галогеналкила, C1-3 галогеналкоксигруппы, C3-5 циклоалкила и C2-3 алкенила. В других вариантах формул (Ia1), (Ia2) и (Ia3), R1 выбран из группы, состоящей из атома хлора, метила, цианогруппы, этила, циклопропила, трифторметила и трифторметоксигруппы.
В других частных вариантах, подходящие соединения выбраны из:
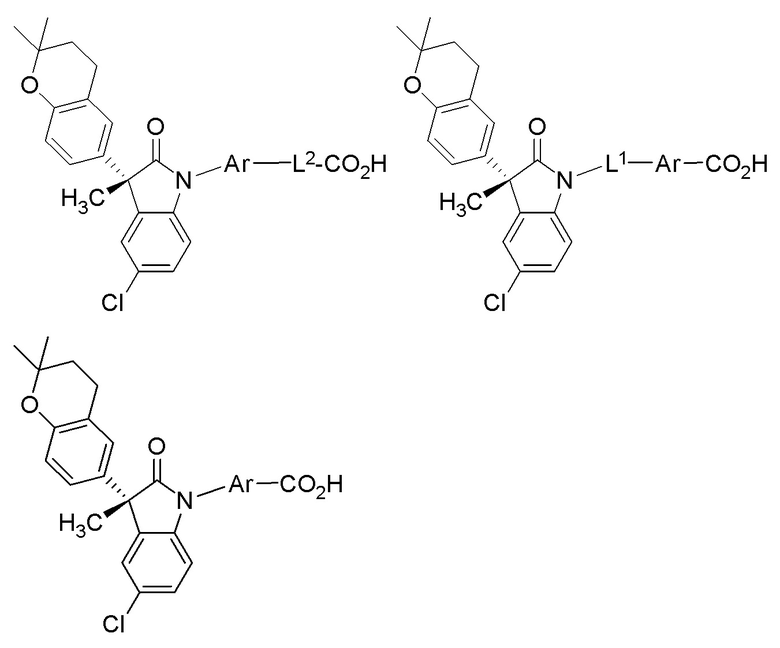
(Ia1’) (Ia2’) и (Ia3’)
или их фармацевтически приемлемых солей, где указанное соединение практически не содержит других изомеров.
В других вариантах осуществления, описаны подходящие соединения, имеющие формулу (Ib):
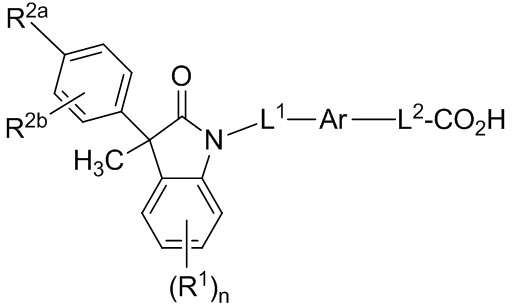
(Ib)
или их фармацевтически приемлемые соли.
В некоторых вариантах формулы (Ib), R2b представляет собой атом водорода. В других вариантах формулы (Ib), Ar выбран из группы, состоящей из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3. В других вариантах формулы (Ib), L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-. В других вариантах формулы (Ib), L2 выбран из группы, состоящей из одинарной связи, -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2- и -CH2CH2CH2-.
В других частных вариантах, подходящие соединения выбраны из:
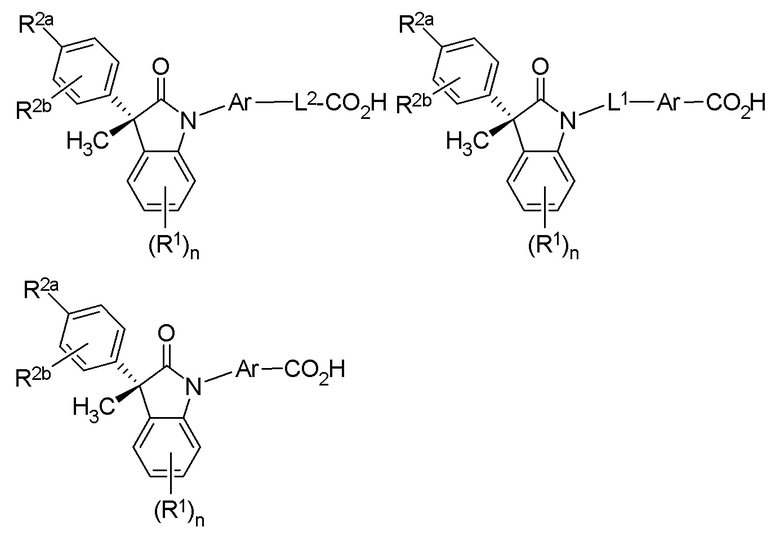
(Ib1) (Ib2) и (Ib3)
или их фармацевтически приемлемых солей, где указанное соединение практически не содержит других изомеров.
В формуле (Ib1), (Ib2) и (Ib3) частными вариантами осуществления являются такие, где Ar выбран из группы, состоящей из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3. В некоторых вариантах осуществления, относящихся к формулам (Ib1), (Ib2) и (Ib3), Ar выбран из группы, состоящей из 1,3-фенилена и 1,4-фенилена, каждый из которых необязательно замещен 1-2 заместителями R3. В других вариантах формул (Ib1), (Ib2) и (Ib3), R3 выбран из группы, состоящей из CH3, CH2CH3, CH2CH2CH3, CH(CH3)2 и CH2OH. В других вариантах формул (Ib1), (Ib2) и (Ib3), L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-. В других вариантах формул (Ib1), (Ib2) и (Ib3), L2 выбран из группы, состоящей из -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2- и -CH2CH2CH2-. В других вариантах формул (Ib1), (Ib2) и (Ib3), R1 выбран из группы, состоящей из галогена, цианогруппы, C1-3 алкила, C1-3 алкоксигруппы, C1-3 галогеналкила, C1-3 галогеналкоксигруппы, C3-5 циклоалкила и C2-3 алкенила, или R1 выбран из группы, состоящей из атома хлора, метила, цианогруппы, этила, циклопропила, трифторметила и трифторметоксигруппы.
В других частных вариантах, подходящие соединения выбраны из:
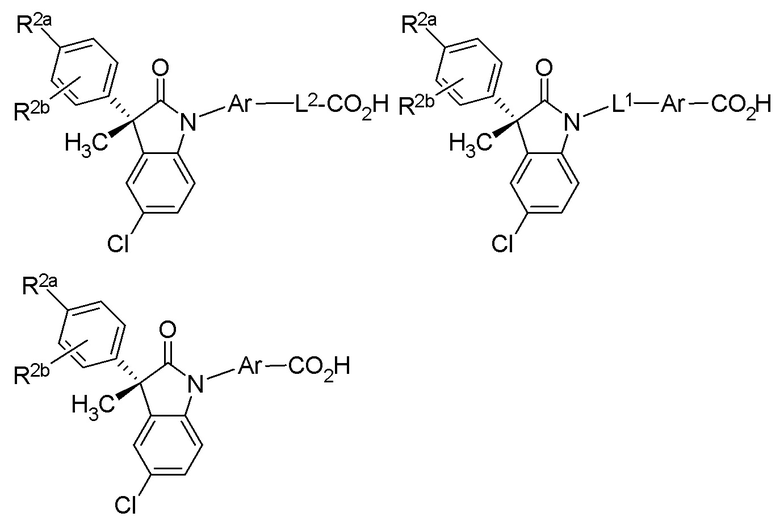
(Ib1’) (Ib2’) и (Ib3’)
или их фармацевтически приемлемых солей, где указанное соединение практически не содержит других изомеров.
Получение соединений
Описанное в настоящем изобретении соединение можно получить согласно приведенной ниже общей схеме. Исходя из подходящего замещенного эфира фенилуксусной кислоты, реакция с замещенным галогеннитробензолом в присутствии основания, с последующим добавлением метилиодида, дает скелет 2-оксоиндольного цикла, имеющего четвертичный центр в альфа-положении к карбоксильной группе. Расщепление изомеров, с последующим восстановлением нитрогруппы и циклизацией, дает замещенный 2-оксоиндол. Реакция по индольному атому азота с присоединением замещенной Ar группы или линкера (L1), содержащего присоединенную замещенную Ar группу, дает изображенные целевые соединения. Квалифицированному специалисту в данной области будет понятно, что могут быть применены модификации относительно общего направления, изображенного ниже на схеме, с получением различных соединений, имеющих формулу (I).
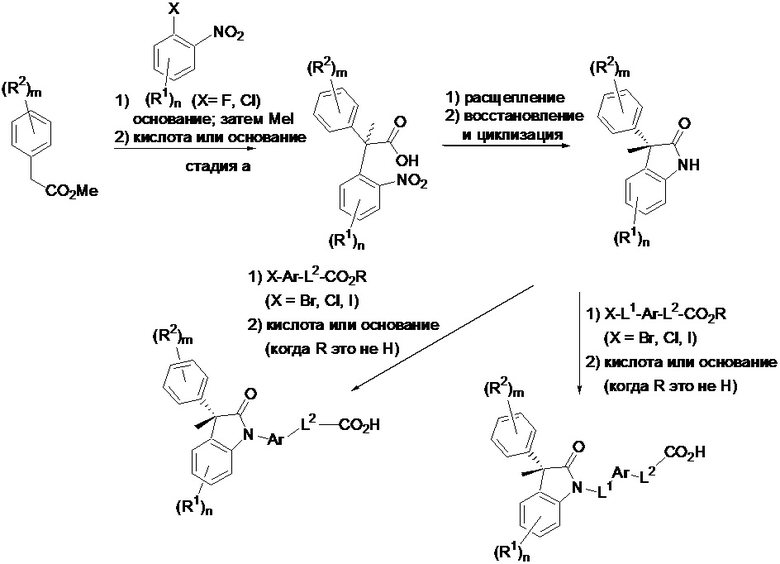
Композиции, модулирующие активность хемокинов
В другом аспекте, в настоящей заявке описаны композиции, которые модулируют активность хемокинов, в частности активность CCR(9). В целом, композиции для модулирования активности хемокинового рецептора у людей и животных содержат фармацевтически приемлемое вспомогательное вещество или разбавитель и соединение, имеющее любую из формул I, I’, Ia, Ib, Ia1, Ia2, Ia3, Ia1’, Ia2’, Ia3’, Ib1, Ib2, Ib3, Ib1’, Ib2’ и Ib3’.
Термин “композиция” при использовании в настоящем тексте охватывает продукт, содержащий указанные ингредиенты в указанных количествах, а также любой продукт, который получается, напрямую или опосредованно, при комбинировании указанных ингредиентов в указанных количествах. Под термином “фармацевтически приемлемый” понимается носитель, разбавитель или вспомогательное вещество, которое должно быть совместимым с другими ингредиентами препарата и не наносить вреда принимающему его пациенту.
Фармацевтические композиции для введения соединений по настоящему изобретению можно выпускать в виде дозированных лекарственных форм, и их можно готовить любым из способов, известных в фармакологии. Все способы включают стадию объединения действующего вещества с носителем, который содержит один или несколько вспомогательных ингредиентов. В целом, фармацевтические композиции получают путем однородного и тщательного смешивания действующего вещества с жидким носителем или тонко измельченным твердым носителем, или с обоими, и затем, при необходимости, придание продукту формы желаемого препарата. В фармацевтической композиции действующее вещество присутствует в количестве, достаточном для оказания целевого эффекта на болезнь или на патологическое состояние.
Фармацевтические композиции, содержащие действующее вещество, могут иметь форму, подходящую для перорального применения, например, они могут быть в виде таблеток, саше, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий и самоэмульгирующихся препаратов, как описано в Заявке на Патент США №6,451,339, твердых или мягких капсул, или сиропов или эликсиров. Композиции, предназначенные для перорального приема, можно готовить любыми способами, известными в области производства фармацевтических композиций. Такие композиции могут содержать один или больше агентов, выбранных из подсластителей, ароматизаторов, красителей и консервантов, для создания фармацевтически привлекательных и приятных на вкус препаратов. Таблетки содержат действующее вещество в смеси с другими нетоксичными фармацевтически приемлемыми вспомогательными веществами, подходящими для производства таблеток. Такими вспомогательными веществами могут быть, например, инертные разбавители, такие как целлюлоза, диоксид углерода, оксид алюминия, карбонат кальция, карбонат натрия, глюкоза, маннит, сорбит, лактоза, фосфат кальция или фосфат натрия; гранулирующие агенты и агенты, ускоряющие распад таблеток, например, кукурузный крахмал или альгиновую кислоту; связующие агенты, например, поливинилпирролидон, целлюлозу, крахмал, желатин или смолу акации, и лубриканты, например, стеарат магния, стеариновую кислоту или тальк. Таблетки могут не иметь покрытия или они могут быть покрыты кишечнорастворимой оболочкой, или иметь покрытие, нанесенное каким-либо другим известным способом, с целью замедления распадения и всасывания в желудочно-кишечном тракте, тем самым обеспечивая пролонгированное действие в течение более длительного времени. Например, можно применять такие замедляющие распадение материалы как глицерил моностеарат или глицерил дистеарат. На таблетки можно также наносить покрытие способами, описанными в патентах США № 4 256 108; 4 166 452 и 4 265 874, с образованием осмотических таблеток с замедленным высвобождением.
Препараты, предназначенные для перорального приема, могут также иметь вид твердых желатиновых капсул, где действующее вещество смешано с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где действующее вещество смешано с водной или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом. Кроме того, можно готовить эмульсии с нерастворяющимися в воде ингредиентами, такими как масла, и стабилизировать их поверхностно-активными веществами, такими как моно-диглицериды, сложные эфиры ПЭГ и т.п.
Водные суспензии содержат действующие вещества в смеси со вспомогательными веществами, подходящими для производства водных суспензий. Такие вспомогательные вещества представляют собой суспендирующие агенты, например, натрия карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую камедь, и смолу акации; диспергирующие или увлажняющие агенты могут представлять собой природные фосфатиды, например, лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например, полиоксиэтилен стеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например, гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и гекситола, такие как полиоксиэтилен сорбитмоноолеат, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и ангидридами гекситола, например, полиэтилен сорбитанмоноолеат. Водные суспензии могут также содержать один или больше консервантов, например, этил или н-пропил парагидроксибензоат, один или больше красителей, один или больше ароматизаторов, и один или больше подсластителей, таких как сахароза или сахарин.
Масляные суспензии можно получать путем суспендирования действующего вещества в растительном масле, например, в арахисовом масле, оливковом масле, сезамовом масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загустители, например, пчелиный воск, твердый парафин или цетиловые спирты. Можно добавлять подсластители, такие как перечисленные выше, и красители, для получения приятной на вкус композиции для перорального приема. Такие композиции можно стабилизировать добавлением антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии при добавлении воды, содержат действующее вещество в смеси с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или больше консервантами. Примеры диспергирующих или увлажняющих агентов, суспендирующих агентов приведены выше. Могут также присутствовать дополнительные вспомогательные вещества, например, подсластители, ароматизаторы и красители.
Фармацевтические композиции по настоящему изобретению могут также иметь форму эмульсий типа масло-в-воде. Масляной фазой может служить растительное масло, например, оливковое масло или арахисовое масло, или минеральное масло, например, жидкий парафин или их смесь. Подходящие эмульгаторы могут представлять собой природные смолы, например, смолу акации или трагакантовую камедь, природные фосфатиды, например, соевое масло, лецитин, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гекситола, например, сорбитан моноолеат, и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например, полиоксиэтилен сорбитанмоноолеат. Эмульсия может также содержать подсластители и ароматизаторы.
В состав сиропов и эликсиров могут входить подсластители, например, глицерин, пропиленгликоль, сорбит или сахароза. Такие препараты могут также содержать средства, уменьшающие раздражение, консервант, ароматизаторы и красители. Растворы для перорального приема можно готовить в комбинации с, например, циклодекстрином, ПЭГ и поверхностно-активными веществами.
Фармацевтические композиции могут иметь форму стерильных инъецируемых водных или масляных суспензий. Такую суспензию можно готовить по известным в данной области методикам, с применением подходящих диспергаторов или увлажняющих агентов и суспендирующих агентов, которые были указаны выше. Стерильные инъецируемые препараты могут также представлять собой стерильные инъецируемые растворы или суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например, могут иметь форму раствора в 1,3-бутандиоле. Среди подходящих носителей и растворителей, которые можно использовать, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные масла часто применяют в качестве растворителя или суспендирующей среды. Для этой цели можно использовать любое безвкусное нелетучее масло, включая синтетические моно- и диглицериды. Кроме того, в приготовлении инъецируемых препаратов находят применение жирные кислоты, такие как олеиновая кислота.
Описанные в настоящем тексте соединения можно также вводить в форме суппозиториев для ректального введения лекарственного средства. Такие композиции можно готовить путем смешивания лекарственного средства с подходящим нераздражающим вспомогательным веществом, которое твердое при комнатной температуре, но переходит в жидкое состояние при температуре тела, и поэтому плавится в прямой кишке с высвобождением лекарственного средства. Такими материалами являются масло какао и полиэтиленгликоли. Кроме того, соединения можно вводить в виде глазных препаратов как капли или мази. Кроме того, можно осуществлять чрескожное введение рассматриваемых соединений посредством ионофорезных пластырей и т.п.
Для местного нанесения применяют кремы, мази, гели, растворы или суспензии, содержащие соединения по настоящему изобретению. В контексте настоящего изобретения, местное нанесение включает также применение полосканий и растворов для рта.
Фармацевтические композиции и способы по настоящему изобретению могут дополнительно включать другие терапевтически активные соединения, как отмечено в настоящем тексте, такие как применяющиеся в лечении указанных выше патологических состояний.
В одном варианте осуществления, в настоящем изобретении описана композиция, состоящая из фармацевтически приемлемого носителя и соединения по настоящему изобретению.
Способы лечения
В зависимости от подвергающегося лечения заболевания и состояния пациента, соединения и композиции по настоящему изобретению можно вводить перорально, парентерально (например, внутримышечно, интраперитонеально, внутривенно, интрацеребрально, путем интрацистернальной инъекции или инфузии, путем подкожной инъекции или с помощью импланта), путем ингаляции, назально, вагинально, ректально, сублингвально или наружно, и их можно вводить в состав препаратов, по отдельности или совместно, в состав подходящих дозированных лекарственных форм, содержащих обычно используемые нетоксичные фармацевтически приемлемые носители, вспомогательные вещества и носители, подходящие для каждого способа введения. Настоящее изобретение также охватывает введение соединений и композиций по настоящему изобретению в виде депонируемых препаратов.
В лечении или предотвращении патологических состояний, которые требуют модулирования хемокинового рецептора, подходящий диапазон дозировок обычно составляет примерно от 0.01 до 100 мг на кг веса тела пациента, которые можно вводить в виде одной или нескольких доз. Предпочтительно, диапазон дозировок составляет примерно от 0.01 до 25 мг/кг в сутки; более предпочтительно примерно от 0.05 до 10 мг/кг в сутки. Подходящий диапазон дозировок может составлять примерно от 0.01 до 25 мг/кг в сутки, примерно от 0.05 до 10 мг/кг в сутки, или примерно от 0.1 до 5 мг/кг в сутки. Внутри указанного диапазона, дозировка может составлять от 0.005 до 0.05, от 0.05 до 0.5, от 0.5 до 5.0, или от 5.0 до 50 мг/кг в сутки. При пероральном введении, композиции предпочтительно вводят в виде таблеток, содержащих от 1.0 до 1000 миллиграммов действующего вещества, в частности 1.0, 5.0, 10.0, 15.0, 20.0, 25.0, 50.0, 75.0, 100.0, 150.0, 200.0, 250.0, 300.0, 400.0, 500.0, 600.0, 750.0, 800.0, 900.0 и 1000.0 миллиграммов действующего вещества, с симптоматическим регулированием дозировки для пациента, подвергающегося лечению. Соединения можно вводить в режиме от 1 до 4 раз в сутки, предпочтительно один или два раза в сутки.
Однако следует понимать, что конкретная дозировка и частота введения для каждого конкретного пациента может варьироваться и зависит от разных факторов, включая активность конкретного применяемого соединения, метаболическую устойчивость и длительность действия этого соединения, возраст, вес тела, наследственность, общее состояние здоровья, пол, диету, способ и время введения, скорость экскреции, применяемые в комбинации лекарств, тяжесть конкретного патологического состояния и терапию, применяемую к конкретному пациенту.
В некоторых вариантах осуществления, соединения по настоящему изобретению вводят как часть комбинированной терапии. Например, некоторое количество химиотерапевтического средства или облучения применяется к пациенту до, после или в комбинации с соединениями по настоящему изобретению. В некоторых вариантах осуществления, количество является субтерапевтическим, когда химиотерапевтическое средство или облучение применяется в отдельности. Квалифицированным специалистам в данной области будет понятно, что «комбинации» могут означать комбинации в лечении (т.е. два или больше лекарственных средств можно вводить в виде смеси, или по меньшей мере одновременно, или по меньшей мере вводить пациенту в разное время, но так, чтобы они оба находились в кровотоке пациента в одно и то же время). Кроме того, композиции по настоящему изобретению можно вводить до или после второго терапевтического воздействия, например, до или после применения химиотерапии или облучения.
В других вариантах осуществления, описанные в настоящем изобретении способы направлены на лечение аллергических заболеваний, где соединение или композицию по настоящему изобретению вводят отдельно или в комбинации со вторым терапевтическим средством, где указанное второе терапевтическое средство представляет собой антигистаминный или противовоспалительный препарат. При применении в комбинации, лечащий врач может вводить комбинацию соединения или композиции по настоящему изобретению и второго терапевтического средства. Также, описанное соединение или композицию и второе терапевтическое средство можно вводить последовательно, в любом порядке.
Соединения и композиции по настоящему изобретению можно комбинировать с другими соединениями и композициями, имеющими родственное применение для предотвращения и лечения интересующего патологического состояния или заболевания, такого как воспалительные состояния и заболевания, включая воспалительное заболевание кишечника (включая болезнь Крона и язвенный колит), аллергические заболевания, псориаз, атопический дерматит и астму, и перечисленные выше патологии. Выбор подходящих средств для применения в комбинированной терапии может сделать квалифицированный специалист в данной области. Комбинация терапевтических средств может работать синергетически, осуществляя лечение или профилактику различных заболеваний. Применяя такой подход, можно достичь терапевтической эффективности с более низкими дозировками каждого средства, тем самым снижая потенциал возникновения нежелательных побочных эффектов.
При лечении, профилактике, смягчении симптомов, борьбе или снижении риска воспаления, соединения по настоящему изобретению можно применять в комбинации с противовоспалительным или анальгетическим средством, таким как опиатный агонист, ингибитор липоксигеназы, такой как ингибитор 5-липоксигеназы, ингибитор циклооксигеназы, такой как ингибитор 2-циклооксигеназы, ингибитор интерлейкина, такой как ингибитор интерлейкина-1, NMDA антагонист, ингибитор окиси азота или ингибитор синтеза окиси азота, аминосалицилаты, кортикостероиды и другие иммунодепрессанты, нестероидные противовоспалительные средства или цитокин-подавляющие противовоспалительные средства, например, с таким соединением, как ацетаминофен, аспирин, кодеин, биологические ФНО-секвестранты, биологические агенты, воздействующие на α4β7, ингибиторы ACE2, ингибиторы протеинкиназы С, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацетин, пироксикам, стероидный анальгетик, суфентанил, сулиндак, тенидап и т.д.
Сходным образом, соединения по настоящему изобретению можно вводить с болеутоляющим средством; потенцирующим средством, таким как кофеин, H2-антагонист, симетикон, гидроксид алюминия или магния; противозастойным средством, таким как псевдоэфедрин; противокашлевым средством, таким как кодеин; диуретиком; седативным или неседативным антигистаминным средством; антагонистом VLA-4; иммунодепрессантом, таким как циклоспорин, такролимус, рапамицин, антагонисты рецептора EDG или другие иммунодепрессанты типа FK-506; стероидом; нестероидным противоастматическим средством, таким как β2-агонист, лейкотриеновый антагонист или ингибитор биосинтеза лейкотриена; ингибитором фосфодиэстеразы типа IV (PDE-IV); агентом, понижающим уровень холестерина, таким как ингибитор HMG-CoA редуктазы, секвестрант или ингибитор всасывания холестерина; и противодиабетическим средством, таким как инсулин, ингибиторы α-глюкозидазы или глитазоны.
Весовое соотношение соединения по настоящему изобретению и второго действующего вещества может варьироваться и зависит от эффективной дозировки каждого ингредиента. В целом, применяют эффективную дозу каждого ингредиента. Так, например, когда соединение по настоящему изобретению комбинируют с НПВС, весовое соотношение соединения по настоящему изобретению и НПВС обычно находится в диапазоне от примерно 1000:1 до примерно 1:1000, предпочтительно от примерно 200:1 до примерно 1:200. Комбинации соединений по настоящему изобретению и других действующих веществ обычно находятся в указанном выше диапазоне, но в каждом случае следует применять эффективную дозу каждого активного ингредиента.
Способы лечения или предотвращения CCR(9)-опосредованных патологических состояний или заболеваний
В другом аспекте, в настоящем изобретении описаны способы лечения или предотвращения CCR(9)-опосредованного патологического состояния или заболевания посредством введения субъекту, страдающему таким патологическим состоянием или заболеванием, терапевтически эффективного количества любого соединения, имеющего формулу I, I’, Ia, Ib, Ia1, Ia2, Ia3, Ia1’, Ia2’, Ia3’, Ib1, Ib2, Ib3, Ib1’, Ib2’ или Ib3’. Соединения для применения в данных способах включают соединения, имеющие формулу I, I’, Ia, Ib, Ia1, Ia2, Ia3, Ia1’, Ia2’, Ia3’, Ib1, Ib2, Ib3, Ib1’, Ib2’ и Ib3’, соединения, описанные в вариантах осуществления, соединения, приведенные ниже в примерах, и соединения, для которых в настоящем тексте приведены конкретные структуры. В настоящем тексте, термин "субъект" включает животных, таких как млекопитающие, включая (но не ограничиваясь только ими) приматов (например, людей), коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей и т.п. В предпочтительных вариантах осуществления, субъектом является человек.
В настоящем тексте, выражение "CCR(9)-опосредованное патологическое состояние или заболевание" и родственные ему выражения и термины означают патологическое состояние или заболевание, характеризующееся ненадлежащей, т.е. ниже или выше нормальной, функциональной активностью CCR(9). Ненадлежащая функциональная активность CCR(9) может являться следствием экспрессирования CCR(9) в клетках, которые в норме не экспрессируют CCR(9), повышенного экспрессирования CCR(9) (что приводит, например, к воспалительным и иммунорегулируемым нарушениям и заболеваниям) или пониженного экспрессирования CCR(9). Ненадлежащая функциональная активность CCR(9) может также являться следствием секретирования TECK клетками, которые в норме не секретируют TECK, повышенного экспрессирования TECK (что приводит, например, к воспалительным и иммунорегулируемым нарушениям и заболеваниям) или пониженного экспрессирования TECK. CCR(9)-опосредованное патологическое состояние или заболевание может полностью или частично быть опосредовано ненадлежащей функциональной активностью CCR(9). Однако, CCR(9)-опосредованное патологическое состояние или заболевание представляет собой такое патологическое состояние или заболевание, при котором модулирование CCR(9) оказывает определенный эффект на основное патологическое состояние или заболевание (например, CCR(9) антагонист приводит к некоторому улучшению самочувствия пациента, по меньшей мере у некоторых пациентов).
Термин "терапевтически эффективное количество" означает количество обсуждаемого соединения, которое обеспечивает биологический или медицинский ответ клетки, ткани, системы или животного, такого как человек, которого добивается исследователь, ветеринар, врач или другой человек, осуществляющий лечение.
Заболевания и патологические состояния, связанные с воспалением, иммунными нарушениями, инфекцией и раковыми заболеваниями, можно лечить или предотвращать соединениями по настоящему изобретению, композициями и способами. В одной группе вариантов осуществления, заболевания или патологические состояния, включая хронические заболевания, у людей или других животных, можно лечить ингибиторами функции CCR(9). Такие заболевания или патологические состояния включают: (1) аллергические заболевания, такие как системный анафилактический ответ или гиперчувствительность, аллергия на лекарственные средства, аллергия на укусы насекомых и пищевая аллергия, (2) воспалительные заболевания кишечника, такие как болезнь Крона, язвенный колит, микроскопический колит, илеит и энтерит, и послеоперационная кишечная непроходимость, (3) вагинит, (4) псориаз и воспалительные дерматозы, такие как дерматит, экзема, атопический дерматит, аллергический контактный дерматит, крапивница и кожный зуд, (5) васкулит, (6) спондилоартропатия, (7) склеродерма, (8) астма и аллергические заболевания дыхательных путей, такие как аллергическая астма, аллергический ринит, гиперчувствительность легких и т.п., (9) аутоиммунные заболевания, такие как фибромиалгия, анкилозирующий спондилоартрит, ювенильный ревматоидный артрит, болезнь Стилла, ювенильный ревматоидный полиартрит, ювенильный ревматоидный артрит, ревматическая полимиалгия, ревматоидный артрит, псориатический артрит, остеоартрит, ревматоидный полиартрит, рассеянный склероз, системная красная волчанка, диабет I типа, диабет II типа, гломерулонефрит и т.п., (10) отторжение трансплантата (включая отторжение аллотрансплантата), (11) болезнь «трансплантат против хозяина» (включая и острую, и хроническую), (12) другие заболевания, при которых необходимо подавить нежелательный воспалительный ответ, такие как атеросклероз, миозит, нейродегенеративные заболевания (например, болезнь Альцгеймера), энцефалит, менингит, гепатит, нефрит, сепсис, саркоидоз, аллергический конъюнктивит, отит, хроническая обструктивная болезнь легких, синусит, синдром Бехчета и подагра, (13) иммуноопосредованные пищевые аллергии, такие как целиакия, (14) фиброз легких и другие фиброзные заболевания, (15) синдром раздраженного кишечника, (16) первичный склерозирующий холиангит, (17) рак (включая первичный и метастатический), (18) связанные с бактериями синдромы, такие как гемоколит и гемолитический уремический синдром, (19) меланома, (20) первичный склерозирующий холангит, (21) послеоперационная непроходимость кишечника, (22) гепатит и воспалительные болезни печени, (23) синдром Шегрена.
В другой группе вариантов осуществления, заболевания или патологические состояния можно лечить модуляторами и агонистами CCR(9). Примеры заболеваний, которые можно лечить посредством модулирования работы CCR(9), включают раковые заболевания, сердечно-сосудистые заболевания, заболевания, в которых играют роль ангиогенез или образование новых сосудов (новообразования, ретинопатия и дегенерация желтого пятна), инфекционные заболевания (вирусные инфекции, например, ВИЧ-инфекция, и бактериальные инфекции) и иммуносупрессивные заболевания, такие как патологические состояния при пересадке органов и кожи. Термин «патологические состояния при пересадке органов» включает патологические состояния при пересадке костного мозга и солидных органов (например, почки, печени, легкого, сердца, поджелудочной железы или их комбинаций).
Предпочтительно, описанные в настоящем изобретении способы касаются лечения заболеваний или патологических состояний, выбранных из воспалительного заболевания кишечника, включая болезнь Крона и язвенный колит, аллергических заболеваний, псориаза, атопического дерматита и астмы, аутоиммунных заболеваний, таких как ревматоидный артрит, и иммуноопосредованной пищевой аллергии, такой как целиакия.
В других вариантах осуществления, описанные в настоящем изобретении способы касаются лечения псориаза, где соединение или композиция по настоящему изобретению применяется в отдельности или в комбинации со вторым терапевтическим средством, таким как кортикостероид, лубрикант, кератолитическое средство, производное витамина D3, ПУВА и антралин.
В других вариантах осуществления, описанные в настоящем изобретении способы касаются лечения атопического дерматита с применением соединения или композиции по настоящему изобретению в отдельности или в комбинации со вторым терапевтическим средством, таким как лубрикант и кортикостероид.
В других вариантах осуществления, описанные в настоящем изобретении способы касаются лечения астмы с применением соединения или композиции по настоящему изобретению в отдельности или в комбинации со вторым терапевтическим средством, таким как β2-агонист и кортикостероид.
Наборы и упаковки
Термины “набор” и “фармацевтический набор” относятся к продаваемому набору или упаковке, содержащим, в одном или больше подходящих контейнерах, одну или больше фармацевтических композиций и инструкции по их применению. В одном варианте осуществления, описаны наборы, содержащие соединение, имеющее формулу I, I’, Ia, Ib, Ia1, Ia2, Ia3, Ia1’, Ia2’, Ia3’, Ib1, Ib2, Ib3, Ib1’, Ib2’ или Ib3’, или его фармацевтически приемлемые соли, и инструкции по их применению. В одном варианте осуществления, описаны наборы, содержащие соединение, имеющее формулу I, I’, Ia, Ib, Ia1, Ia2, Ia3, Ia1’, Ia2’, Ia3’, Ib1, Ib2, Ib3, Ib1’, Ib2’ или Ib3’, или его фармацевтически приемлемые соли, в комбинации с одним или больше (например, одним, двумя, тремя, одним или двумя, или от одного до трех) дополнительными терапевтическими средствами, и инструкции по их применению.
В одном варианте осуществления, соединения по настоящему изобретению вводят в состав дозированных форм для однократного приема, которые упакованы в единую упаковку. Единая упаковка включает (но не ограничивается только ими) флакон, флакон с защитой от детей, ампулу и тубу. В одном варианте осуществления, соединения по настоящему изобретению и, необязательно, дополнительные терапевтические средства, вводят в состав дозированных форм для однократного приема, и каждая однократная дозированная форма индивидуально упакована в отдельную упаковку. Такие индивидуально упакованные однократные дозированные формы могут содержать фармацевтическую композицию в любой форме, включая (но не ограничиваясь только ими) жидкую форму, твердую форму, порошковую форму, форму гранулята, шипучего порошка или таблетки, твердых или мягких капсул, эмульсий, суспензий, сиропа, суппозиториев, таблетки, пастилок, таблеток для рассасывания, раствора, буккального пластыря, тонкой пленки, орального геля, жевательной таблетки, жевательной резинки и одноразовых шприцов. Такие индивидуально упакованные однократные дозированные формы можно комбинировать в упаковке, изготовленной из одного или больше из следующих материалов: бумага, тонкий картон, картон, металлическая фольга и пластиковая фольга, например, блистерная упаковка. Одну или больше дозированных форм можно вводить один или несколько раз в сутки. Одну или больше дозированных форм можно вводить три раза в сутки. Одну или больше дозированных форм можно вводить два раза в сутки. Одну или больше дозированных форм можно вводить в первый день, и одну или больше дозированных форм можно вводить в последующие дни.
Комбинированная терапия
Соединения по настоящему изобретению могут выпускаться в отдельности или в комбинации с одним или больше другими лекарственными средствами. Примеры терапевтических средств, которые можно комбинировать с соединением или композицией по настоящему изобретению, для введения по отдельности или в составе одной фармацевтической композиции, включают (но не ограничиваются только ими) модуляторы CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10, CCR11, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7, CX3CR1, ChemR23, C5aR, C5a и C5, или любую их комбинацию. В некоторых вариантах осуществления, модулятор является антагонистом.
Примеры терапевтических средств, которые можно комбинировать с соединением или композицией по настоящему изобретению, для введения по отдельности или в составе одной фармацевтической композиции, включают (но не ограничиваются только ими) CCX354, CCX9588, CCX140, CCX872, CCX598, CCX6239, CCX9664, CCX2553, CCX 2991, CCX282, CCX025, CCX507, CCX430, CCX765, CCX224, CCX662, CCX650, CCX832, CCX168, CCX168-M1, бразикумаб, будезонид, устекинумаб, эверолимус, глатирамер ацетат, натализумаб, этанерцепт, микофенолят мофетил, бродалумаб, каннабидиол, форалумаб, тралокинумаб, тамибаротен, месалазин, голимумаб, тедуглутид, инфликсимаб, ропивакаин, филготиниб, этролизумаб, SHP-647, элафибранор, ABC-294640, окрелизумаб, тофацитиниб, цертолизумаб пегол, адалимумаб, сарграмостим, абатацепт, кларитромицин, GSK-2982772, упадацитиниб, эдасалонексент, секукинумаб, ванкомицин, ведолизумаб, талидомид, ритуксимаб, катридекаког, RBX-2660, ампион, нитазоксанид, финголимод, тоцилизумаб, росиптор ацетат, AST-120, ризанкизумаб, телотристат этипрат, леналидомид, аликафорсен, тосуфлоксацин, интерферон бета-1a, E-6011, KAG-308, дексаметазон натрия фосфат, озанимод, доципарстат кобитолимод, месалазин, PUR-0110, апремиласт, месалазин, валганцикловир, такролимус, монгерсен, реместемцел-L, GS-5745, E-6011, E-6007, каротеграст метил, пиклиденозон, PF-06480605, балсалазид, пимекролимус, месалазин, рекомбинантный интерферон бета-1a, налтрексон, адалимумаб, амиселимод, брилацидин, басиликсимаб, этрасимод, LP-02, розиглитазон, плеканатид, лаквинимод, рифабутин + кларитромицин + клофазимин, инфликсимаб, тилдакизумаб, омега-3-карбоновые кислоты, TOP-1288, пефицитиниб, рифамицин, рифаксимин, JNJ-64304500, ASP-3291, DLX-105, зилеутон, 99mTc меченый аннексин V-128, ALT-836, биферонекс, клотримазол, гивиностат, Trichuris suis ova, INV-103, K(D)PT, BI-655064, глепаглутид, LYC-30937 EC, TRX-318, LY-3074828, ларазотид ацетат, IBP-9414, клазакизумаб, месалазин, эклометазон дипропионат, NN-8828, олокизумаб, бертилимумаб, мидисмазе, KRP-203, преднизолон, PF-06687234, STNM-01, KHK-4083, FE-999301, DLX-105, VB-201, DNVX-078, рифаксимин, Clostridium butyricum MIYAIRI 588, OPS-2071, сотрастаурин, абрилумаб, QBECO, анакинра, FFP-104, GLPG-1205, долканатид, PDA-002, молграмостим, месалазин, метронидазол, перепозиционированный налтрексон, вателизумаб, зукапсаицин, циклоспорин, опрелвекин, прулифлоксацин, рекомбинантный человеческий лактоферрин, алеквел, SAN-300, STP-206, GLPG-0974, P-28-GST, N-6022, ФНО-альфа киноид, ETX-201, низкомолекулярный гепарин, ETX-201, GED-0507-34-Levo, метенкефалин ацетат + тридекактид ацетат, HMPL-004, SB-012, TRK-170, бета-1,3/1,6-глюкан, месаламин + N-ацетилцистеин, 99mTc-сулесомаб, обсалазин, месалазин бациллы лихениформис, балсалазид натрий, пропионил-L-карнитин, Clostridium butyricum, беклометазон дипропионат, ацеманнан и SPD-480, или любая их комбинация.
Примеры терапевтических средств, которые можно комбинировать с соединением или композицией по настоящему изобретению, для введения по отдельности или в составе одной фармацевтической композиции, включают (но не ограничиваются только ими) IL-23 антагонист, агонист глюкокортикоида, IL-6 агонист, IL-12 антагонист, ингибитор mTOR комплекс 1, mTOR ингибитор, ингибитор адгезии молекулы к клетке, антагонист интегрина альфа-4/бета-1, ФНО-антагонист, связывающийся с ФНО агент, модулятор ФНО-рецептора II типа, ингибитор инозин монофосфат дегидрогеназы, ингибитор биосинтеза PurH пурина, антагонист рецептора интерлейкина 17A, модулятор каннабиноидного CB1 рецептора, модулятор каннабиноидного CB2 рецептора, модулятор каннабиноидного рецептора, CD3 антагонист, IL-13 антагонист, агонист рецептора ретиноевой кислоты альфа, агонист рецептора ретиноевой кислоты бета, агонист ретиноидного рецептора, ингибитор циклооксигеназы, ингибитор лиганда ФНО-альфа, агонист глюкагоноподобного пептида 2, ингибитор натриевых каналов, ингибитор Jak1 тирозинкиназы, антагонист интегрина альфа-4/бета-7, антагонист интегрина альфа-E, антагонист интегрина бета-7, модулятор иммуноглобулина G2, ингибитор MAdCAM, усилитель чувствительности рецепторов к инсулину, агонист PPAR альфа, агонист PPAR дельта, модулятор коллагена, ингибитор дигидрокерамид дельта 4 десатуразы, ингибитор сфингозин киназы 1, ингибитор сфингозин киназы 2, ингибитор aB-лимфоцит антиген CD20, ингибитор JAK тирозинкиназы, ингибитор Jak3 тирозинкиназы, CSF-1 агонист, агонист GM-CSF рецептора, стимулятор цитотоксического T-лимфоцит ассоциированного белка-4, ингибитор T-клеточного поверхностного гликопротеина CD28, ингибитор RIP-1 киназы, ингибитор ядерного фактора каппа-В, IL-17 антагонист, ингибитор пептидогликан-распознающего белка, антагонист интегрина альфа-4/бета-7, ингибитор B-лимфоцит связанного антигена CD20, агонист фактора XIII, ингибитор антигена-1 стволовых клеток, антагонист каннабиноидного рецептора; модулятор сфингозин-1-фосфатного рецептора-1, IL-6 антагонист, модулятор рецептора IL-6, стимулятор SH2 домена инозитол фосфатазы 1, ингибитор триптофан 5-гидроксилазы, ингибитор гена ICAM1, ингибитор ДНК-гиразы, ингибитор топоизомеразы IV, лиганд интерферон-бета, ингибитор лиганда фракталкина, агонист EP4 простаноидного рецептора, агонист сфингозин-1-фосфатного рецептора-1, модулятор сфингозин-1-фосфатного рецептора-1, модулятор сфингозин-1-фосфатного рецептора-5, ингибитор катепсина G, ингибитор каскада реакций комплемента, ингибитор эластазы, агонист гепарина, антагонист L-селектина, антагонист P-селектина, ингибитор ядерного фактора каппа В, TLR-9 агонист, модулятор интерлейкина-1 бета лиганда, PDE 4 ингибитор, ингибитор ДНК-полимеразы, SMAD-7 ингибитор, ингибитор TGF бета-1, ингибитор металлопротеазы-9, ингибитор лиганда фракталкина, антагонист интегрина, агонист аденозинового A3 рецептора, ингибитор лиганда фактора некроза опухоли 15, IL-10 антагонист, IL-2 антагонист, IL-4 антагонист, антагонист рецептора интерферона гамма, лиганд интерферон бета, антагонист опиоидного рецептора, ингибитор альфа-субъединицы IL-2 рецептора, стимулятор сфингозин 1 фосфат фосфатазы 1, усилитель чувствительности рецепторов к инсулину, агонист PPAR гамма, агонист натрийуретического рецептора типа C, ингибитор ацилтрансферазы, антагонист аполипопротеина C3, ингибитор адаптерной молекулы crk, IL-8 антагонист, ингибитор лиганда интерлейкин-1 бета, ингибитор Src тирозинкиназы, ингибитор Syk тирозинкиназы, ингибитор ДНК РНК полимеразы, ингибитор РНК полимеразы, агонист меланокортина, ингибитор 5-липоксигеназы, ингибитор тканевого фактора, лиганд интерферон бета, модулятор брадикининового рецептора, ингибитор гистон деацетилазы, агонист P2X7 пуринергического рецептора, стимулятор митохондриального белка теплового шока с весом 10 кДа, антагонист рецептора лиганда CD40, агонист глюкагоноподобного пептида 2, модулятор F1F0 АТФ синтазы, CD3 антагонист, ингибитор зонулина, ингибитор циклооксигеназы, модулятор липоксигеназы, IL-21 антагонист, антагонист CCR3 хемокина, ингибитор лиганда эотаксина, модулятор супероксид-дисмутазы, агонист сфингозин-1-фосфатного рецептора-1, модулятор CD29, лиганд интерлейкин-10, ингибитор гена CHST15, ингибитор OX40 лиганда, модулятор рецептора IL-6, ингибитор ядерного фактора каппа В, модулятор рецептора онкостатина M, ингибитор STAT, ингибитор STAT-3, TLR-2 антагонист, TLR-4 антагонист, ингибитор РНК полимеразы, ингибитор протеинкиназы C альфа, ингибитор протеинкиназы C бета, ингибитор протеинкиназы С дельта, ингибитор протеинкиназы C эпсилон, ингибитор протеинкиназы C эта, ингибитор протеинкиназы C тета, антагонист IL-1 рецептора типа I, ингибитор CD40 лиганда, антагонист рецептора CD40 лиганда, антагонист сопряженного с G-белком рецептора 84, агонист рецептора гуанилат циклазы, CD49b антагонист, агонист ваниллоидного рецептора VR1, ингибитор кальциневрина, IL-11 агонист, агонист PDGF рецептора, ингибитор ДНК-гиразы, стимулятор лактоферрина, антагонист интегрина альфа-1/бета-1, антагонист рецептора 2 свободных жирных кислот, ингибитор алкогольдегидрогеназы 5, ингибитор глутатион редуктазы, антагонист рецептора интерферона гамма, низкомолекулярный гепарин, агонист PPAR гамма, агонист рецептора ACTH, адренокортикотропный гормон, агонист рецептора опиоидного фактора роста, IL-6 антагонист, модулятор лиганда интерлейкин-1 бета, ингибитор ядерного фактора каппа В; ингибитор фактора транскрипции GATA 3, ингибитор ядерного фактора каппа В, ингибитор оксидоредуктазы, глюкокортикоидный агонист, агонист рецептора интерферона гамма, или любая их комбинация.
Примеры
Приведенные ниже примеры представлены для иллюстрации, но не для ограничения настоящего изобретения.
Реагенты и растворители, использовавшиеся в примерах, можно получить из коммерческих источников, таких как Aldrich Chemical Co. (Milwaukee, Wisconsin, USA). 1H-ЯМР спектры записывали на ЯМР спектрометре Varian Mercury с рабочей частотой 400 МГц. Значения пиков приведены относительно ТМС и записаны в следующем порядке: мультиплетность (с, синглет; д, дублет; т, триплет; кв, квадруплет; м, мультиплет) и число протонов. Результаты масс-спектрометрии приведены в виде соотношения массы к заряду, затем указана относительная интенсивность каждого иона (в скобках). В таблицах приведено одно значение m/e для иона M+H (или, если указано, M-H), содержащего наиболее распространенные изотопы атомов. Во всех случаях изотопное распределение соответствует ожидаемой формуле. Масс-спектральный анализ методом ионизации электроспрея (ESI) проводили на масс-спектрометре Hewlett-Packard MSD с применением ВЭЖХ HP1100, оснащенного Agilent Zorbax SB-C18, 2.1X50 мм, 5 мкм колонкой для ввода образца. Обычно аналит растворяли в метаноле в концентрации 0.1 мг/мл, и 1 микролитр раствора вводили с растворителем в масс-спектрометр, со сканированием в диапазоне от 100 до 1500 дальтон. Все соединения можно анализировать в режиме регистрации ESI положительно заряженных ионов, используя в качестве растворителя смесь ацетонитрил/вода с 1% муравьиной кислоты. Описанные ниже соединения можно также анализировать в режиме регистрации ESI отрицательно заряженных ионов, используя в качестве растворителя 2 мМ раствор NH4OAc в смеси ацетонитрил/вода.
Следующие сокращения применяются в примерах и в тексте настоящего описания:
ВЭЖХ, высокоэффективная жидкостная хроматография; ДМФА, Диметилформамид; ТФУК, Трифторуксусная кислота; ТГФ, тетрагидрофуран; EtOAc, Этилацетат; BOC2O, ди-трет-бутил дикарбонат или BOC ангидрид; DIPEA, диизопропилэтиламин; HBTU, O-(бензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфат; dppf, 1,1'-бис(дифенилфосфино)ферроцен; Pd2(dba)3, трис(дибензилиденацетон)дипалладий(0); DMP, диметилфталат; Me, метил; Et, этил; ДХМ, дихлорметан; rt, комнатная температура.
Соединения по настоящему изобретению можно синтезировать как описано ниже, используя разнообразные реакции, известные квалифицированному специалисту в данной области. Квалифицированному специалисту будет также понятно, что могут применяться альтернативные способы синтеза целевых соединений по настоящему изобретению, и что описанные в тексте настоящей заявки подходы не являются исчерпывающими, но дают широко применимые и практичные подходы к синтезу интересующих соединений.
Некоторые молекулы, описанные в настоящем патенте, могут существовать в различных энантиомерных и диастереомерных формах, и все такие варианты указанных соединений входят в объем настоящего изобретения.
Подробное описание экспериментальных методик, применяющихся для синтеза ключевых соединений в настоящем тексте, приводят к молекулам, которые описаны посредством идентифицирующих их физических параметров, а также структурных изображений.
Квалифицированному специалисту будет также понятно, что в стандартной обработке реакционных смесей в органической химии часто применяются кислоты и основания. В ходе описанных в настоящем патенте экспериментальных методик иногда образуются соли материнских соединений, если эти соединения обладают необходимой кислотностью или основностью.
Пример 1. Синтез (3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-индолин-2-она
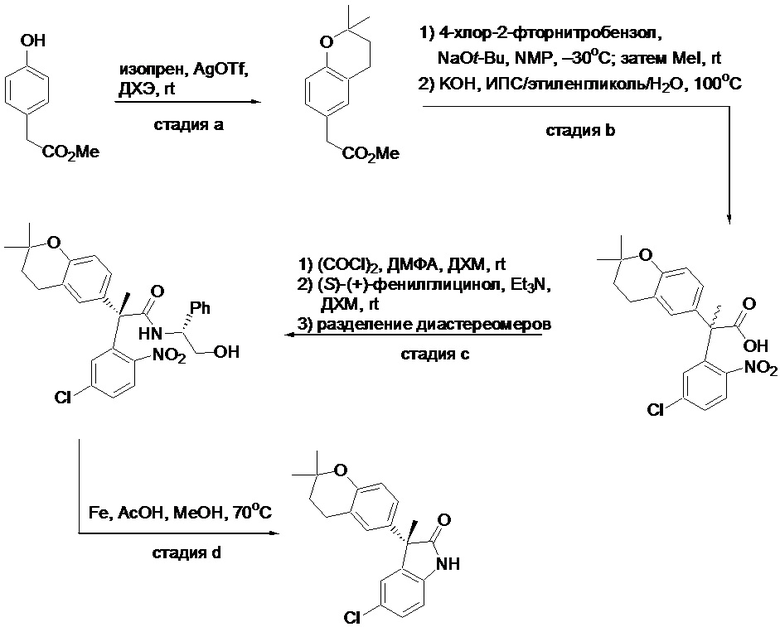
a) Раствор трифлата серебра (0.385 г, 1.50 ммоль) в 1,2-дихлорэтане (1.0 л) нагревали при 90°C в течение 3 часов с обратным холодильником в атмосфере азота. Полученную мутную смесь охлаждали до комнатной температуры, после чего добавляли метил 4-гидроксифенилацетат (24.9 г, 150 ммоль). Изопрен (15.3 г, 225 ммоль) в 1,2-дихлорэтане (100 мл) добавляли по каплям в течение 10 минут, и смесь перемешивали при комнатной температуре в течение 2 час. Раствор упаривали, разбавляли этилацетатом (150 мл) и промывали насыщенным водным раствором NaHCO3 (2 × 100 мл) и 1M раствором NaHSO4 (2 × 100 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом флэш-хроматографии (1–10% EtOAc в гексане) дала метил 2-(2,2-диметилхроман-6-ил)ацетат.
b) В охлажденный (–30°C) раствор трет-бутоксида натрия (93.0 г, 963 ммоль) в безводном N-метилпирролидоне (500 мл) в атмосфере азота медленно добавляли метил 2-(2,2-диметилхроман-6-ил)ацетат (80.8 г, 344 ммоль), затем 4-хлор-2-фторнитробензол (62.1 г, 354 ммоль) и еще N-метилпирролидон (150 мл). После перемешивания при –30 °C в течение 1 часа, метилиодид (42.8 мл, 688 ммоль) добавляли шприцом, и смесь перемешивали еще 20 мин. Реакцию гасили добавлением 3M HCl (600 мл), и смесь нагревали до комнатной температуры. Реакционную смесь экстрагировали метил-трет-бутиловым эфиром (1 × 500 мл), сушили над Na2SO4, фильтровали через слой силикагеля и упаривали в вакууме, получая коричневое твердое вещество. Полученный сырой продукт разбавляли добавлением раствора изопропилового спирта (520 мл), воды (260 мл) и этиленгликоля (340 г), и затем охлаждали до 0°C. Добавляли гидроксид калия (130 г, 2.32 моль), и раствор нагревали при 100°C в течение 4 часов. После окончания реакции, смесь снова охлаждали до 0°C, подкисляли до pH 3 водным 3M раствором HCl и экстрагировали этилацетатом (2 × 300 мл). Органические слои сушили над Na2SO4, фильтровали через слой силикагеля и упаривали в вакууме, получая 2-(5-хлор-2-нитро-фенил)-2-(2,2-диметилхроман-6-ил)пропионовую кислоту.
c) Оксалилхлорид (2.10 мл, 24.0 ммоль) и диметилформамид (0.10 мл) последовательно добавляли в перемешиваемый раствор 2-(5-хлор-2-нитро-фенил)-2-(2,2-диметилхроман-6-ил)пропионовой кислоты (7.21 г, 18.5 ммоль) в дихлорметане (100 мл) при комнатной температуре. Через 1.5 часа реакционную смесь упаривали и снова растворяли в дихлорметане (100 мл). Добавляли триэтиламин (7.77 мл, 55.2 ммоль) и (S)-(+)-фенилглицинол (2.54 г, 18.5 ммоль), и смесь перемешивали при комнатной температуре до завершения реакции (1 час). Смесь упаривали, разбавляли этилацетатом (300 мл) и промывали водным 1M раствором HCl (1 × 200 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом флэш-хроматографии (1–8% ТГФ в ДХМ) разделяла диастереомеры 2-(5-хлор-2-нитро-фенил)-2-(2,2-диметилхроман-6-ил)-N-(2-гидрокси-1-фенил-этил)пропанамида. Элюирующийся вторым диастереомер вводили в последующие реакции.
d) В раствор 2-(5-хлор-2-нитро-фенил)-2-(2,2-диметилхроман-6-ил)-N-(2-гидрокси-1-фенил-этил)пропанамида (3.95 г, 7.76 ммоль) и уксусной кислоты (4 мл) в метаноле (80 мл) добавляли порошок железа (4.0 г, 71.7 ммоль), и реакционную смесь нагревали при 70°C в течение 2 часов. После охлаждения до комнатной температуры, смесь разбавляли этилацетатом (150 мл) и промывали 1M раствором HCl (1 × 100 мл) и водой (1 × 100 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом флэш-хроматографии (0–40% EtOAc в гексане) дала (3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-индолин-2-он.
Пример 2. Альтернативный синтез (3R)- 5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-индолин-2-она
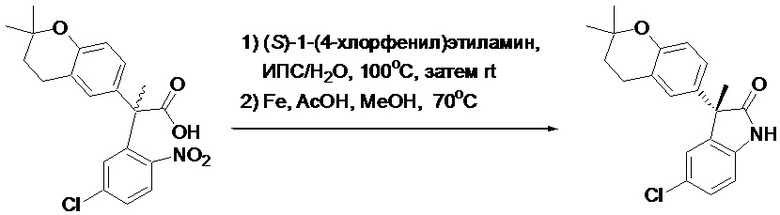
Раствор 2-(5-хлор-2-нитро-фенил)-2-(2,2-диметилхроман-6-ил)пропионовой кислоты (37.3 г, 95.6 ммоль) и (S)-1-(4-хлорфенил)этиламина в изопропиловом спирте (150 мл) и воде (50 мл) нагревали при 100°C до растворения всей твердой фазы. Раствор затем оставляли медленно охлаждаться до комнатной температуры и оставляли в спокойном состоянии на ночь. Выпавшую из раствора соль отфильтровывали и промывали смесью 2:1 ИПС-H2O (180 мл), получая чистое кристаллическое вещество (17.0 г, 33%, er >100:1 в свободной форме). Раствор кристаллической (S)-1-(4-хлорфенил)этиламиновой соли 2-(5-хлор-2-нитро-фенил)-2-(2,2-диметилхроман-6-ил)пропионовой кислоты (546 мг, 1.0 ммоль), порошка железа (224 мг, 4.0 ммоль) и уксусной кислоты (480 мг, 8.0 ммоль) в метаноле (5.0 мл) нагревали при 70°C в течение 1 часа. После охлаждения до комнатной температуры, смесь разбавляли этилацетатом (50 мл) и промывали 1M раствором HCl (1 × 50 мл) и водой (1 × 50 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка методом флеш-хроматографии (0–40% EtOAc в гексане) дала (3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-индолин-2-он.
Пример 3. Синтез 3-[(3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-2-оксо-индолин-1-ил]бензойной кислоты

В раствор (3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-индолин-2-она (45 мг, 0.13 ммоль), трет-бутил-3-иодбензоата (80 мг, 0.27 ммоль), транс-N,N’-диметилциклогексан-1,2-диамина (10 мг, 0.07 ммоль) и карбоната калия (70 мг, 0.51 ммоль) в диоксане (2.5 мл) добавляли иодид меди (10 мг, 0.053 ммоль). Смесь продували азотом и нагревали до 100°C. Через 1 час смесь охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл). Органический слой промывали 1 M раствором HCl (1 × 20 мл), водой (1 × 20 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме. Полученный сырой продукт разбавляли добавлением дихлорметана (1 мл) и трифторуксусной кислоты (2 мл) и перемешивали при комнатной температуре в течение 5 часов. Смесь упаривали и очищали методом обращенно-фазной ВЭЖХ (C18 колонка, ацетонитрил–H2O с 0.1% ТФУК в качестве элюента), получая указанное в заголовке соединение. 1H ЯМР (400 МГц, Хлороформ-d) δ 8.22 – 8.09 (м, 2H), 7.69 (дддд, J = 8.0, 1.8, 1.3, 0.4 Гц, 1H), 7.66 – 7.58 (м, 1H), 7.26 – 7.17 (м, 2H), 7.06 (д, J = 2.3 Гц, 1H), 6.99 (ддд, J = 8.5, 2.5, 0.6 Гц, 1H), 6.88 – 6.81 (м, 1H), 6.73 (д, J = 8.6 Гц, 1H), 2.76 (т, J = 6.7 Гц, 2H), 1.87 (с, 3H), 1.78 (т, J = 6.7 Гц, 2H), 1.32 (д, J = 1.5 Гц, 6H); МС: (ES) m/z вычислено для C27H25ClNO4 [M + H]+ 462.1, найдено 462.5.
Пример 4. Синтез 4-[[(3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-2-оксо-индолин-1-ил]метил]бензойной кислоты
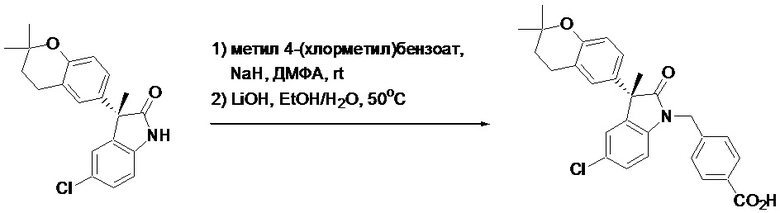
В охлажденный (0°C) раствор (3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-индолин-2-она (45 мг, 0.13 ммоль) в безводном диметилформамиде (0.80 мл) в атмосфере азота добавляли гидрид натрия (20 мг, 60%-ная суспензия в минеральном масле, 0.50 ммоль). После перемешивания при 0°C в течение 10 минут, раствор оставляли нагреваться до комнатной температуры и добавляли метил 4-(хлорметил)бензоат (25 мг, 0.14 ммоль). Смесь оставляли перемешиваться на 30 минут при комнатной температуре, после чего реакцию осторожно гасили добавлением 1 M раствора HCl (25 мл) и экстрагировали этилацетатом (50 мл). Органический слой промывали водой (1 × 20 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме. Полученный сырой продукт снова растворяли в смеси этанола (2 мл) и воды (1 мл). Добавляли моногидрат гидроксида лития (100 мг, 2.4 ммоль), и смесь перемешивали при 50°C в течение 30 мин. После охлаждения до комнатной температуры, реакцию гасили добавлением 1 M раствора HCl (25 мл) и экстрагировали этилацетатом (50 мл). Органический слой промывали водой (1 × 20 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом обращенно-фазной ВЭЖХ (C18 колонка, ацетонитрил–H2O с 0.1% ТФУК в качестве элюента) дала указанное в заголовке соединение. 1H ЯМР (400 МГц, Хлороформ-d) δ 8.05 (д, J = 8.2 Гц, 2H), 7.34 (д, J = 8.1 Гц, 2H), 7.19 – 7.12 (м, 2H), 6.99 (д, J = 2.4 Гц, 1H), 6.91 (дд, J = 8.6, 2.5 Гц, 1H), 6.72 (д, J = 8.5 Гц, 1H), 6.67 – 6.60 (м, 1H), 5.09 – 4.89 (м, 2H), 2.74 (т, J = 6.8 Гц, 2H), 1.81 (с, 3H), 1.78 (т, J = 6.7 Гц, 2H), 1.32 (с, 6H); МС: (ES) m/z вычислено для C28H27ClNO4 [M + H]+ 476.1, найдено 476.2.
Пример 5. Синтез 3-[(3R)-3-(4-трет-бутилфенил)-5-хлор-3-метил-2-оксо-индолин-1-ил]бензойной кислоты
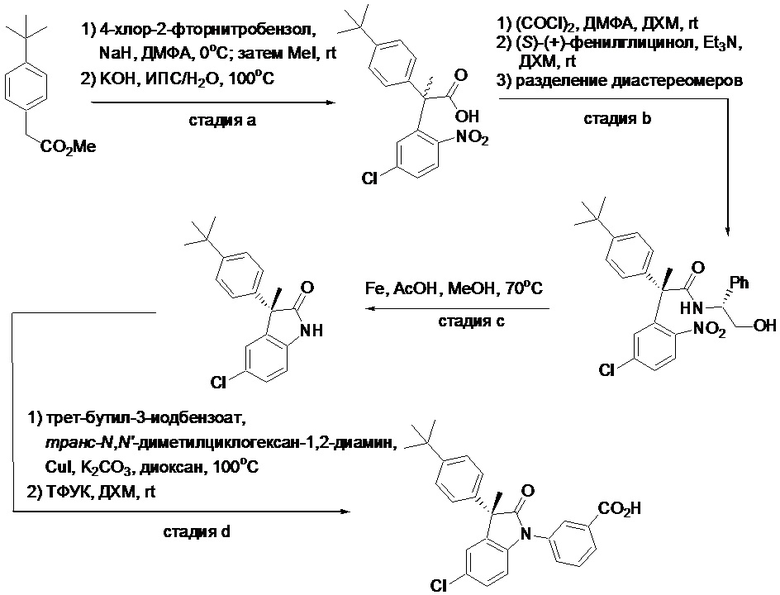
a) В охлажденный (0°C) раствор гидрида натрия (1.30 г, 39.9 ммоль) в безводном диметилформамиде (5 мл) в атмосфере азота медленно добавляли метил п-трет-бутилфенилацетат (1.70 г, 8.22 ммоль) в диметилформамиде (5 мл), и смесь перемешивали при 0°C в течение 30 минут. Затем 4-хлор-2-фторнитробензол (1.60 г, 9.05 ммоль) в диметилформамиде (3 мл) добавляли по каплям в течение 10 мин. После 1.5 часов перемешивания при 0°C, добавляли метилиодид (0.51 мл, 16.4 ммоль), и смесь оставляли нагреваться до комнатной температуры и перемешивали еще 3 часа. Реакцию гасили добавлением насыщенного водного раствора NH4Cl (50 мл). Смесь экстрагировали этилацетатом (2 × 50 мл), органические слои сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка методом флеш-хроматографии (0–20% EtOAc в гексане) дала метил 2-(4-трет-бутилфенил)-2-(3-хлорфенил)пропионат. Полученное соединение растворяли в изопропиловом спирте (40 мл) и воде (20 мл) с гидроксидом калия (2.3 г, 41.1 ммоль), и смесь нагревали при 100°C в течение 2 час. Смесь охлаждали до комнатной температуры, подкисляли до pH 3 1М раствором HCl и экстрагировали этилацетатом (2 × 50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом флеш-хроматографии (0–30% EtOAc в гексане) дала 2-(4-трет-бутилфенил)-2-(3-хлорфенил)пропионовую кислоту.
b) Оксалилхлорид (0.57 мл, 6.6 ммоль) и диметилформамид (4 капли) последовательно добавляли в перемешиваемый раствор 2-(4-трет-бутилфенил)-2-(3-хлорфенил)пропионовой кислоты (2.11 г, 5.5 ммоль) в дихлорметане (35 мл) при комнатной температуре. Через 2 часа реакционную смесь упаривали и снова растворяли в дихлорметане (30 мл). Добавляли триэтиламин (2.3 мл, 16.5 ммоль) и (S)-(+)-фенилглицинол (750 мг, 5.5 ммоль), и смесь перемешивали при комнатной температуре в течение 1 час. Смесь упаривали, разбавляли этилацетатом (100 мл) и промывали водным 1 M раствором HCl (1 × 50 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом флэш-хроматографии (1–15% EtOAc в ДХМ) разделяла диастереомеры (2R)-2-(4-трет-бутилфенил)-2-(3-хлорфенил)-N-[(1R)-2-гидрокси-1-фенил-этил)]пропанамида. Элюирующийся вторым диастереомер вводили в последующие реакции.
c) В раствор (2R)-2-(4-трет-бутилфенил)-2-(3-хлорфенил)-N-[(1R)-2-гидрокси-1-фенил-этил)]пропанамида (250 мг, 0.52 ммоль) и уксусной кислоты (0.22 мл) в метаноле (2.6 мл) добавляли порошок железа (87 мг, 1.56 ммоль), и реакционную смесь нагревали до 70°C в течение 2 час. После охлаждения до комнатной температуры, смесь разбавляли этилацетатом (20 мл) и промывали 1 M раствором HCl (1 × 10 мл) и водой (1 × 10 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом флеш-хроматографии (0–40% EtOAc в гексане) дала (3R)-3-(4-трет-бутилфенил)-5-хлор-3-метил-индолин-2-он.
d) В раствор (3R)-3-(4-трет-бутилфенил)-5-хлор-3-метил-индолин-2-она (34 мг, 0.11 ммоль), трет-бутил-3-иодбензоата (65 мг, 0.22 ммоль), транс-N,N’-диметилциклогексан-1,2-диамина (10 мг, 0.07 ммоль) и карбоната калия (70 мг, 0.51 ммоль) в диоксане (2.5 мл) добавляли иодид меди (10 мг, 0.053 ммоль). Смесь продували азотом и нагревали до 100°C. Через 1 час смесь охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл). Органический слой промывали 1 M раствором HCl (1 × 20 мл), водой (1 × 20 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме. Полученный сырой продукт разбавляли добавлением дихлорметана (1 мл) и трифторуксусной кислоты (2 мл) и перемешивали при комнатной температуре в течение 5 час. Смесь упаривали и очищали методом обращенно-фазной ВЭЖХ (C18 колонка, ацетонитрил–H2O с 0.1% ТФУК в качестве элюента), получая указанное в заголовке соединение. 1H ЯМР (400 МГц, Хлороформ-d) δ 8.18 – 8.10 (м, 2H), 7.72 – 7.59 (м, 2H), 7.40 – 7.35 (м, 2H), 7.29 (д, J = 2.1 Гц, 1H), 7.28 – 7.26 (м, 1H), 7.24 (д, J = 7.7 Гц, 2H), 6.87 – 6.81 (м, 1H), 1.91 (с, 3H), 1.30 (с, 9H); МС: (ES) m/z вычислено для C26H25ClNO3 [M + H]+ 434.1, найдено 434.2.
Пример 6. Синтез 5-[(3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-2-оксо-индолин-1-ил]-2-метокси-бензойной кислоты

В раствор (3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метилиндолин-2-она (60 мг, 0.18 ммоль), метил 5-иод-2-метоксибензоата (80 мг, 0.27 ммоль), транс-N,N’-диметилциклогексан-1,2-диамина (10 мг, 0.07 ммоль) и карбоната калия (102 мг, 0.73 ммоль) в диоксане (3.0 мл) добавляли иодид меди (20 мг, 0.10 ммоль). Смесь продували азотом и нагревали до 100°C. Через 1 час смесь охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл). Органический слой промывали 1 M раствором HCl (1 × 20 мл), водой (1 × 20 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме. Полученный сырой продукт снова растворяли в смеси этанола (2 мл) и воды (1 мл). Добавляли моногидрат гидроксида лития (100 мг, 2.4 ммоль), и смесь перемешивали при 50°C в течение 30 мин. После охлаждения до комнатной температуры, реакцию гасили добавлением 1 M раствора HCl (25 мл) и экстрагировали этилацетатом (50 мл). Органический слой промывали водой (1 × 20 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом обращенно-фазной ВЭЖХ (C18 колонка, ацетонитрил–H2O с 0.1% ТФУК в качестве элюента) дала указанное в заголовке соединение. 1H ЯМР (400 МГц, Хлороформ-d) δ 8.21 (д, J = 2.7 Гц, 1H), 7.66 (дд, J = 8.9, 2.7 Гц, 1H), 7.29 – 7.16 (м, 3 H), 7.01 (д, J = 2.4 Гц, 1H), 6.96 (дд, J = 8.6, 2.5 Гц, 1H), 6.78 (дд, J = 8.1, 0.7 Гц, 1H), 6.73 (д, J = 8.6 Гц, 1H), 4.13 (с, 3H), 2.75 (т, J = 6.7 Гц, 2H), 1.85 (с, 3H), 1.78 (т, J = 6.7 Гц, 2H), 1.32 (с, 6H); МС: (ES) m/z вычислено для C28H27ClNO5 [M + H]+ 492.2, найдено 492.3.
Пример 7. Синтез 5-[(3R)-3-(2,2-диметилхроман-6-ил)-3,5-диметил-2-оксо-индолин-1-ил]-2-метил-бензойной кислоты

В раствор (3R)-3-(2,2-диметилхроман-6-ил)-3,5-диметилиндолин-2-она (32 мг, 0.10 ммоль), метил 5-иод-2-метилбензоата (36 мг, 0.13 ммоль), транс-N,N’-диметилциклогексан-1,2-диамина (10 мг, 0.07 ммоль) и карбоната калия(28 мг, 0.20 ммоль) в диоксане (3.0 мл) добавляли иодид меди (6.0 мг, 0.03 ммоль). Смесь продували азотом и нагревали до 100°C. Через 1 час смесь охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл). Органический слой промывали 1 M раствором HCl (1 × 20 мл), водой (1 × 20 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме. Полученный сырой продукт снова растворяли в смеси этанола (2 мл) и воды (1 мл). Добавляли моногидрат гидроксида лития (100 мг, 2.4 ммоль), и смесь перемешивали при 50°C в течение 30 мин. После охлаждения до комнатной температуры, реакцию гасили добавлением 1 M раствора HCl (25 мл) и экстрагировали этилацетатом (50 мл). Органический слой промывали водой (1 × 20 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом обращенно-фазной ВЭЖХ (C18 колонка, ацетонитрил–H2O с 0.1% ТФУК в качестве элюента) дала указанное в заголовке соединение. 1H ЯМР (400 МГц, Хлороформ-d) δ 8.19 (т, J = 1.9 Гц, 1H), 8.13 – 8.06 (м, 1H), 7.76 – 7.69 (м, 1H), 7.61 (т, J = 7.9 Гц, 1H), 7.14 – 7.03 (м, 3H), 7.03 – 6.97 (м, 1H), 6.86 – 6.77 (м, 1H), 6.71 (д, J = 8.6 Гц, 1H), 2.75 (т, J = 6.8 Гц, 2H), 2.36 (д, J = 0.9 Гц, 3H), 1.85 (с, 3H), 1.78 (т, J = 6.7 Гц, 2H), 1.31 (д, J = 1.8 Гц, 6H); МС: (ES) m/z вычислено для C29H30NO4 [M + H]+ 456.6, найдено 456.0.
Пример 8. Синтез 2-[4-[(3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-2-оксо-индолин-1-ил]фенил]уксусной кислоты

В раствор (3R)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-индолин-2-она (60 мг, 0.18 ммоль), 4-иодфенилуксусной кислоты (92 мг, 0.35 ммоль), транс-N,N’-диметилциклогексан-1,2-диамина (10 мг, 0.07 ммоль) и карбоната калия (97 мг, 0.70 ммоль) в диметилформамиде (3.0 мл) добавляли иодид меди (20 мг, 0.10 ммоль). Смесь продували азотом и нагревали до 110°C. Через 1 час смесь охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл). Органический слой промывали 1 M раствором HCl (1 × 20 мл), водой (1 × 20 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме. Очистка полученного сырого продукта методом обращенно-фазной ВЭЖХ (C18 колонка, ацетонитрил–H2O с 0.1% ТФУК в качестве элюента) дала указанное в заголовке соединение. 1H ЯМР (400 МГц, Хлороформ-d) δ 7.49 – 7.41 (м, 2H), 7.41 – 7.34 (м, 2H), 7.20 (д.кв, J = 4.3, 2.1 Гц, 2H), 7.04 (д, J = 2.3 Гц, 1H), 6.96 (ддд, J = 8.6, 1.9, 1.2 Гц, 1H), 6.85 (д, J = 8.9 Гц, 1H), 6.71 (д, J = 8.5 Гц, 1H), 3.71 (с, 2H), 2.74 (т, J = 6.7 Гц, 2H), 1.84 (с, 3H), 1.78 (т, J = 6.7 Гц, 2H), 1.31 (д, J = 1.8 Гц, 6H); МС: (ES) m/z вычислено для C28H27ClNO4 [M + H]+ 476.2, найдено 476.2.
Пример 9. Синтез (S)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-1-(4-((5-оксо-4,5-дигидро-1H-тетразол-1-ил)метил)фенил)индолин-2-она
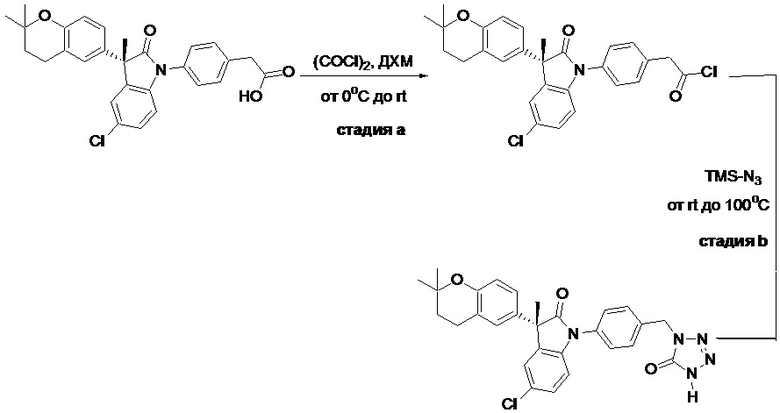
Стадия a. В раствор (S)-2-(4-(5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-2-оксоиндолин-1-ил)фенил)уксусной кислоты (190.4 мг, 0.40 ммоль) в дихлорметане (1.6 мл) при 0oC в атмосфере азота добавляли по каплям раствор оксалилхлорида (52 мкл, 0.60 ммоль) в дихлорметане (0.3 мл). Реакционную смесь перемешивали при 0oC 5 минут и затем нагревали до комнатной температуры и перемешивали 4 часа. Все растворители удаляли в вакууме и добавляли в остаток дихлорметан (2 мл). Смесь упаривали в вакууме, и данный процесс повторяли еще раз, получая ацилхлоридный продукт, который напрямую использовали в следующей стадии.
Стадия b. В ацилхлорид, полученный на предыдущей стадии, добавляли азидотриметилсилан (0.32 мл, 2.4 ммоль) при комнатной температуре (выделение газа!). Смесь нагревали до 100oC в атмосфере азота и перемешивали 37 часов. Реакционную смесь охлаждали до комнатной температуры и упаривали в вакууме. Добавляли к остатку воду и дихлорметан. Органический слой отделяли, сушили и упаривали. Полученный сырой продукт очищали методом хроматографии на силикагеле (50% этил ацетат/гексан), получая целевой продукт. 1H ЯМР (400 МГц, хлороформ-d) δ 7.58 – 7.51 (м, 2H), 7.47 – 7.41 (м, 2H), 7.22 – 7.16 (м, 2H), 7.03 (д, J = 2.4 Гц, 1H), 6.96 (дд, J = 8.6, 2.5 Гц, 1H), 6.85 – 6.80 (м, 1H), 6.70 (д, J = 8.6 Гц, 1H), 5.16 (с, 2H), 2.73 (т, J = 6.7 Гц, 2H), 1.83 (с, 3H), 1.77 (т, J = 6.7 Гц, 2H), 1.30 (д, J = 2.0 Гц, 6H). МС: (ES) m/z вычислено для C28H26ClN5O3 [M + H]+ 516.2, найдено 516.5.
Пример 10. Синтез (S)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-1-(4-((4-метил-5-оксо-4,5-дигидро-1H-тетразол-1-ил)метил)фенил)индолин-2-она
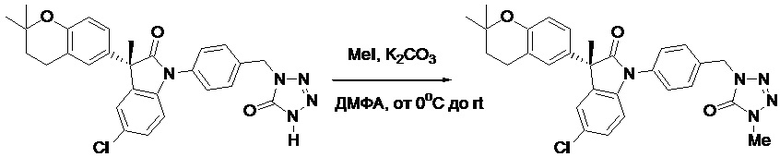
В смесь (S)-5-хлор-3-(2,2-диметилхроман-6-ил)-3-метил-1-(4-((5-оксо-4,5-дигидро-1H-тетразол-1-ил)метил)фенил)индолин-2-она (204 мг, 0.395 ммоль) и иодметана (49 мкл, 0.791 ммоль) в ДМФА (1.0 мл) при 0oC добавляли K2CO3 (138 мг, 1.0 ммоль). Реакционную смесь перемешивали при 0oC в течение 20 минут и затем при комнатной температуре 14 часов. Смесь выливали в воду (10 мл) и дихлорметан (3 мл). Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенные органические слои сушили и упаривали. Полученный сырой продукт очищали методом хроматографии на силикагеле (50% этил ацетат/гексан), получая целевой продукт. 1H ЯМР (400 МГц, хлороформ-d) δ 7.58 – 7.52 (м, 2H), 7.44 – 7.39 (м, 2H), 7.21 – 7.16 (м, 2H), 7.03 (д, J = 2.4 Гц, 1H), 6.98 – 6.93 (м, 1H), 6.85 – 6.80 (м, 1H), 6.70 (д, J = 8.6 Гц, 1H), 5.13 (с, 2H), 3.61 (с, 3H), 2.73 (т, J = 6.7 Гц, 2H), 1.82 (с, 3H), 1.76 (т, J = 6.7 Гц, 2H), 1.30 (д, J = 1.8 Гц, 6H). МС: (ES) m/z вычислено для C29H28ClN5O3 [M + H]+ 530.2, найдено 530.5.
Соединения, полученные аналогично описанным выше способам и протестированные методом анализа хемотаксиса в плазме крови, приведены ниже в таблице. A2 вычисляли как описано, и активность представлена в Таблице 1 в виде:
+, 20000 нM ≥ A2 ≥ 500 нM; ++, 500 нM > A2 ≥ 100 нM; +++, 100 нM > A2.
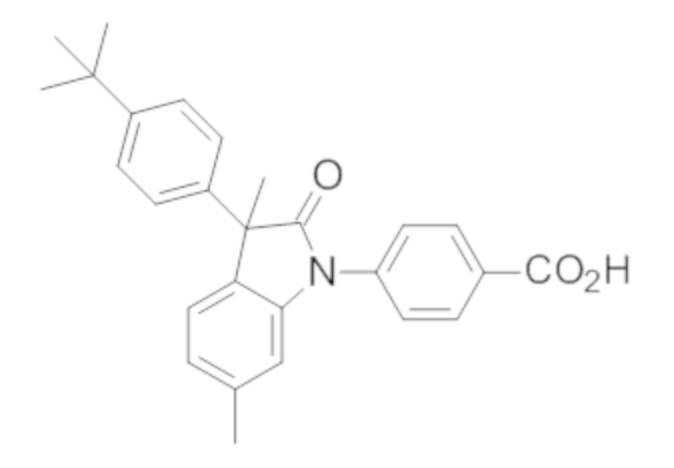
1.001
[M + H]+ 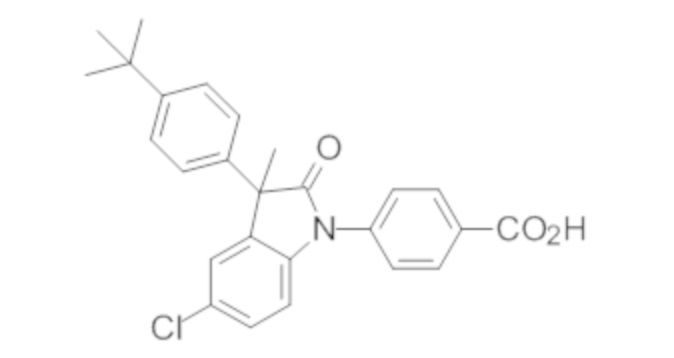
1.002
[M + H]+ 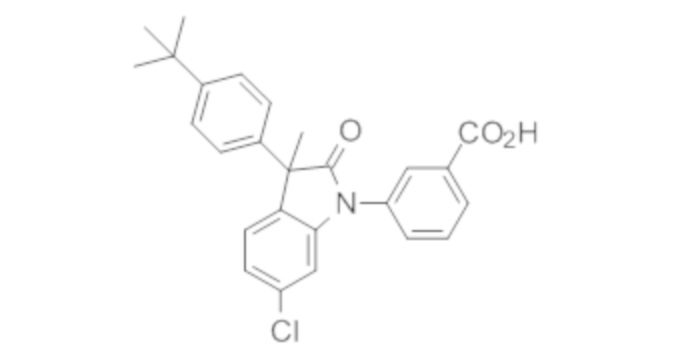
1.003
[M + H]+ 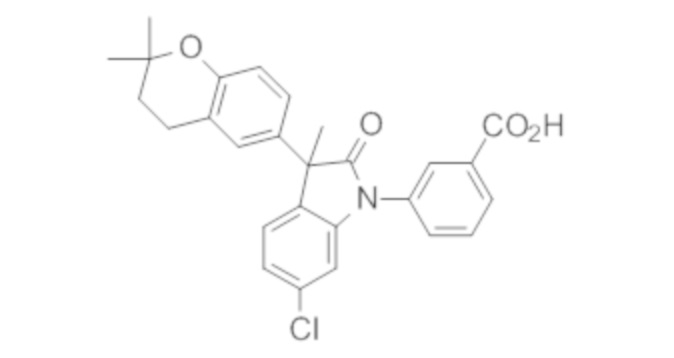
1.004
[M + H]+ 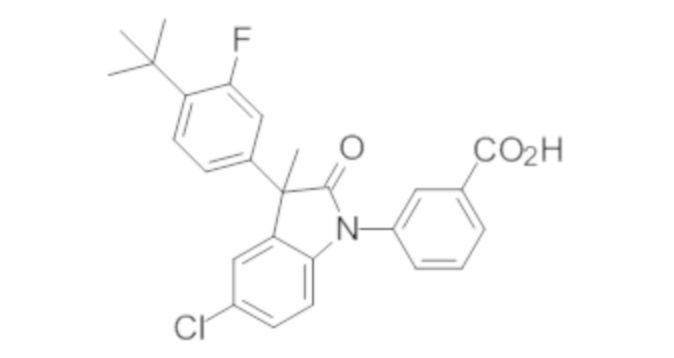
1.005
[M + Na]+ 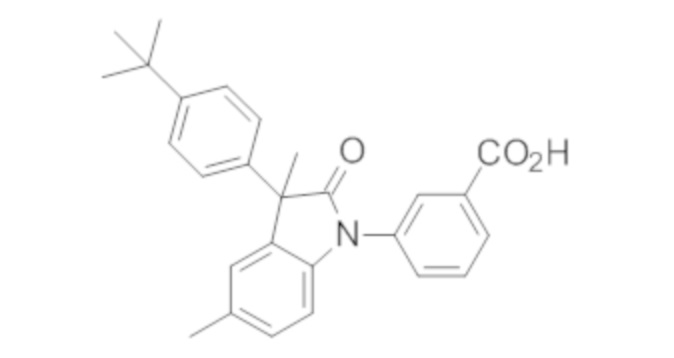
1.006
[M + H]+ 
1.007
[M + H]+ 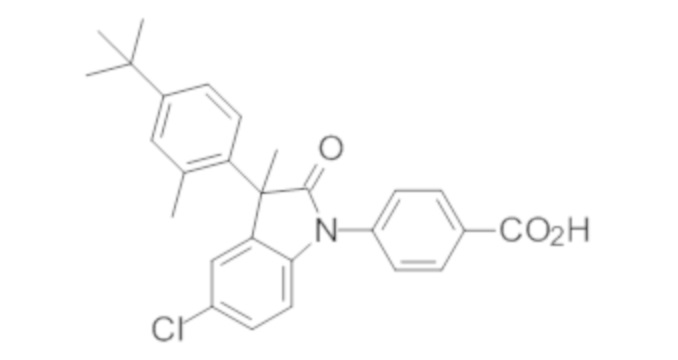
1.008
[M + H]+ 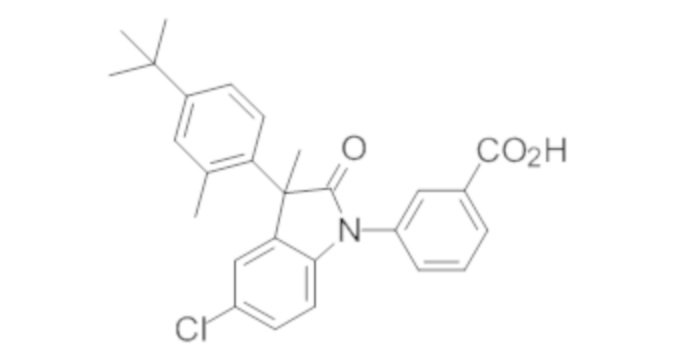
1.009
[M + H]+ 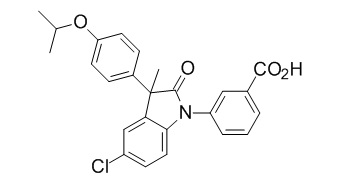
1.010
[M + H]+ 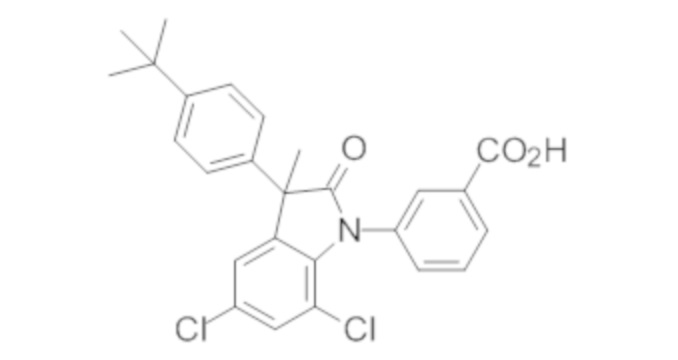
1.011
[M + H]+ 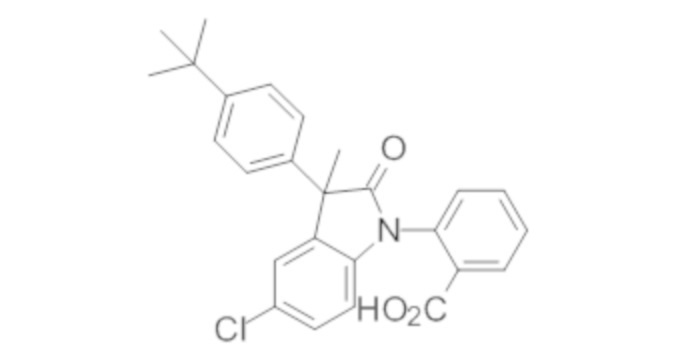
1.012
[M + H]+ 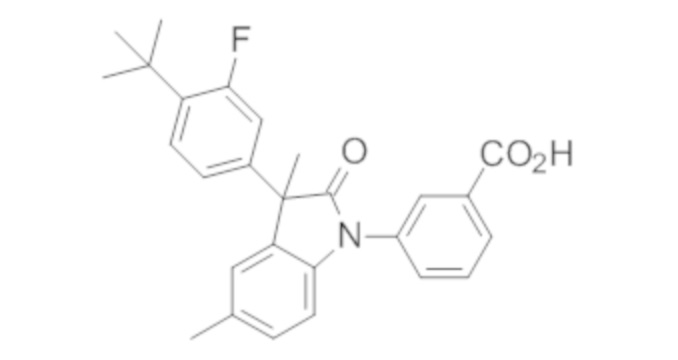
1.013
[M + H]+ 
1.014
[M + H]+ 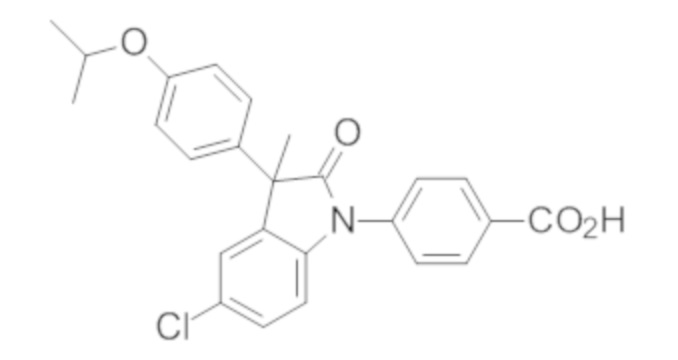
1.015
[M + H]+ 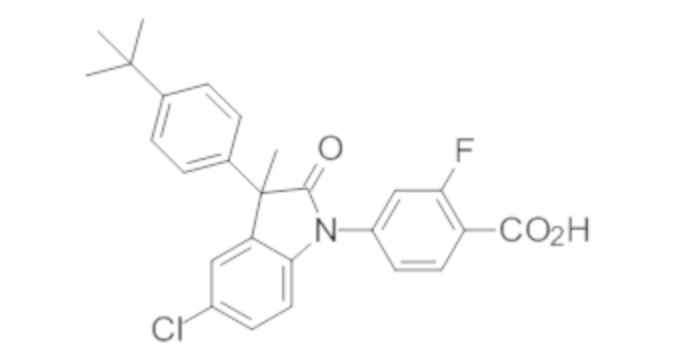
1.016
[M + H]+ 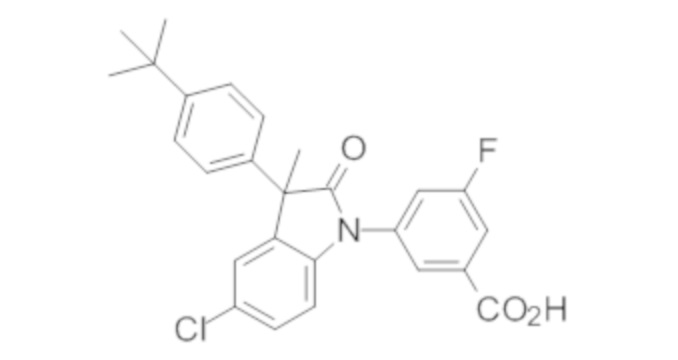
1.017
[M + H]+ 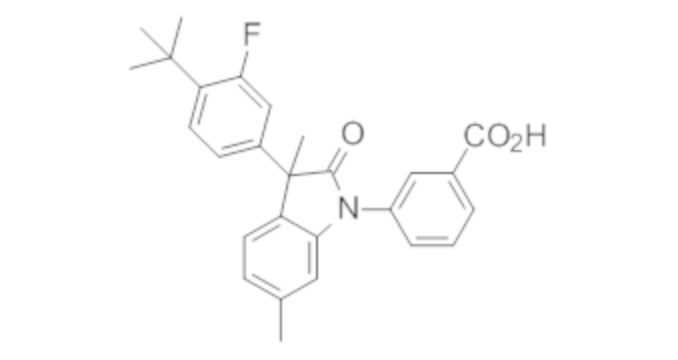
1.018
[M + H]+ 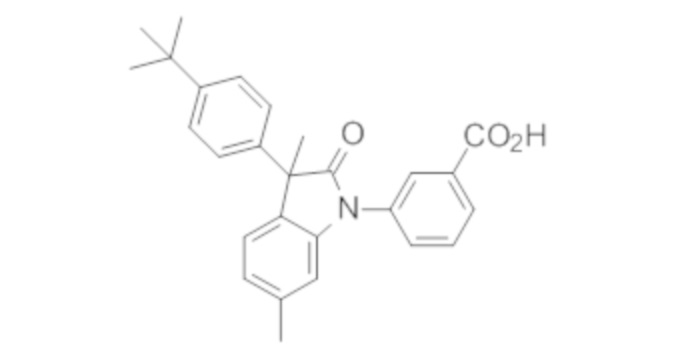
1.019
[M + H]+ 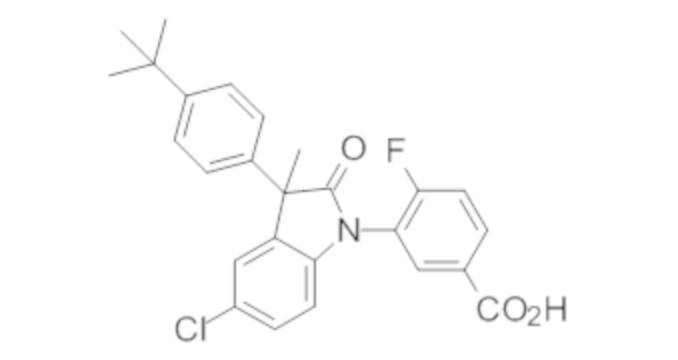
1.020
[M + H]+ 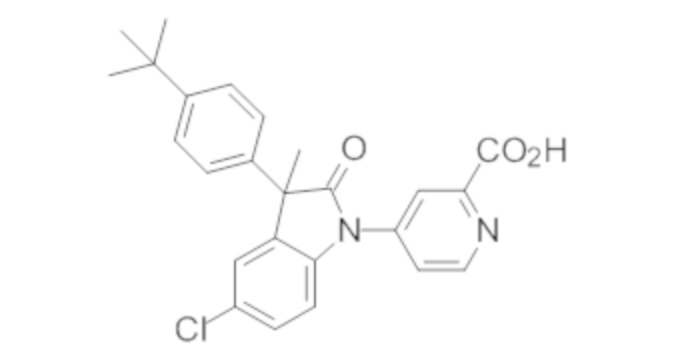
1.021
[M + H]+ 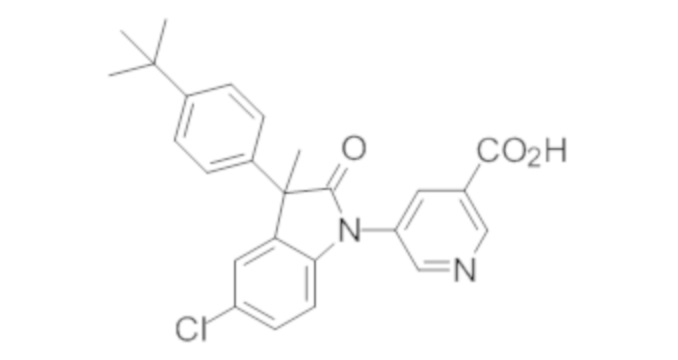
1.022
[M + H]+ 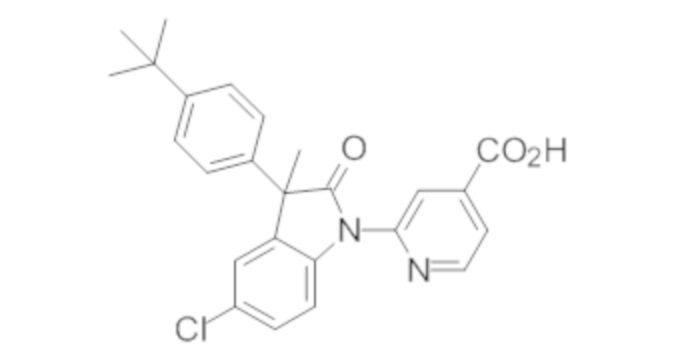
1.023
[M + H]+ 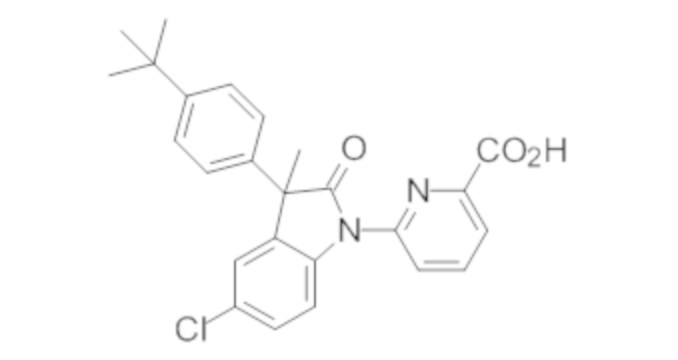
1.024
[M + Na]+ 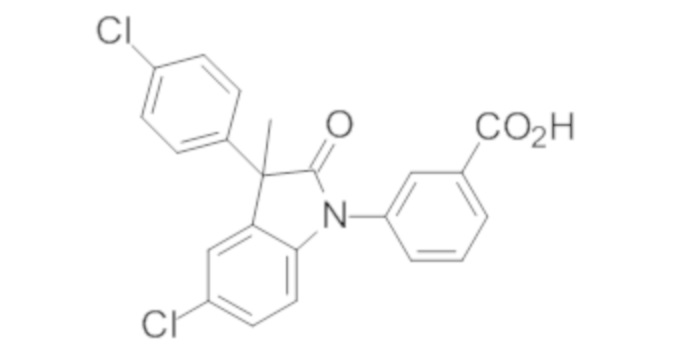
1.025
[M + Na]+ 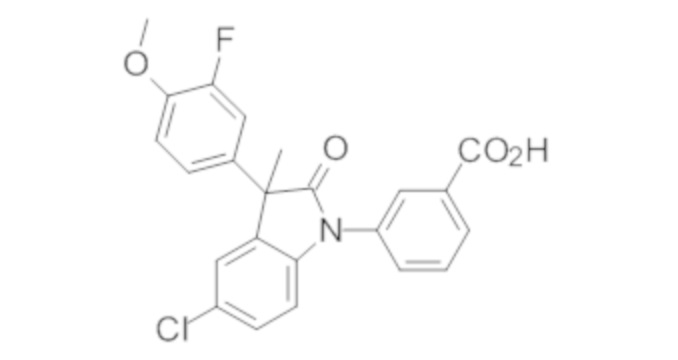
1.026
[M + H]+ 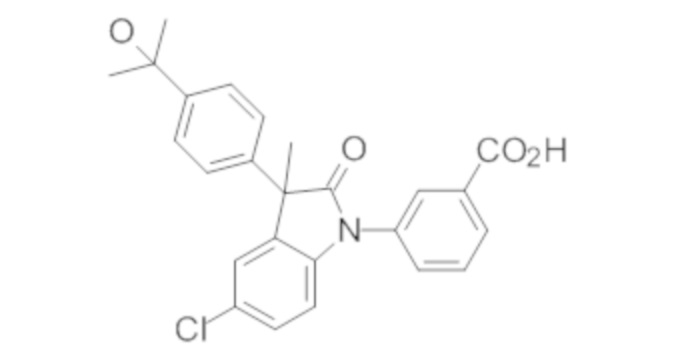
1.027
[M + Na]+ 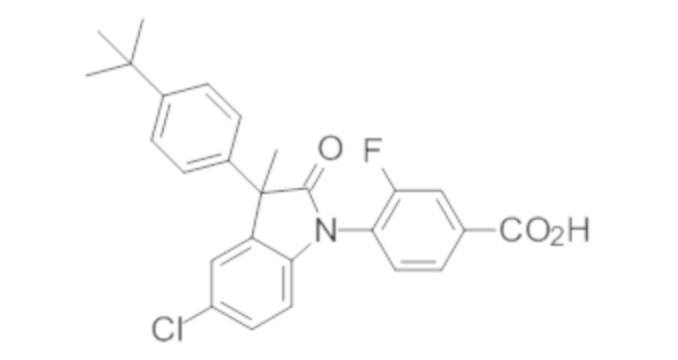
1.028
[M + H]+ 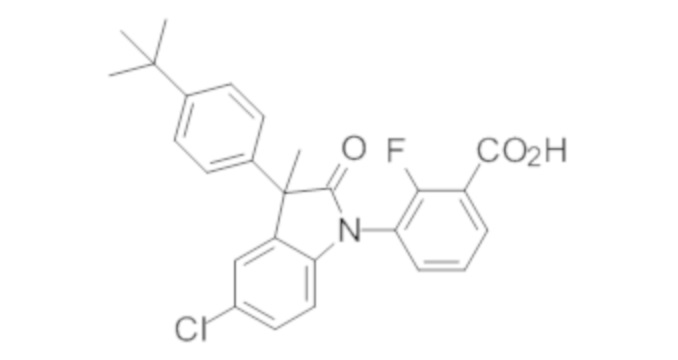
1.029
[M + H]+ 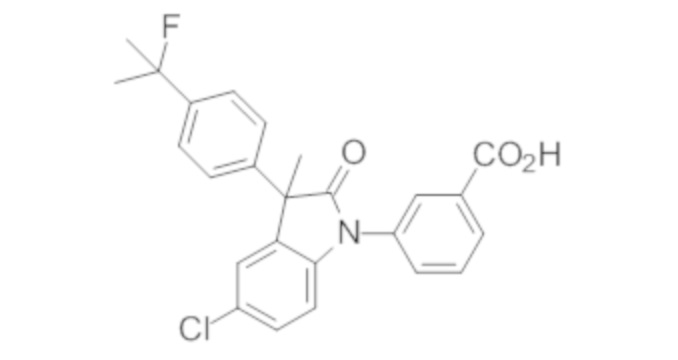
1.030
[M + Na]+ 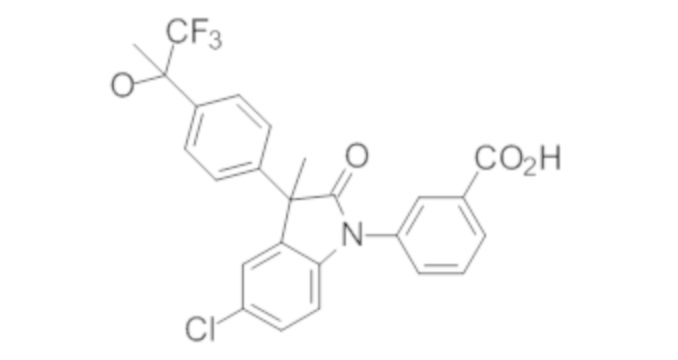
1.031
[M + Na]+ 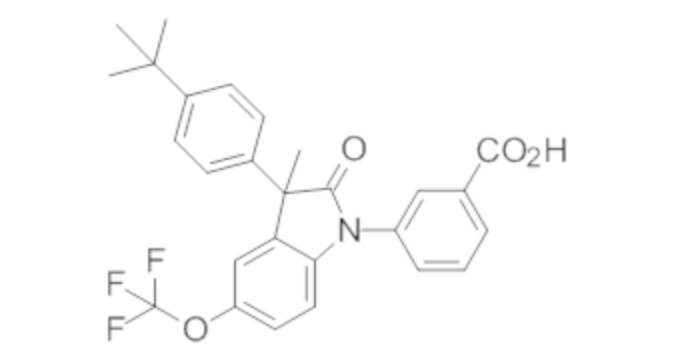
1.032
[M + H]+ 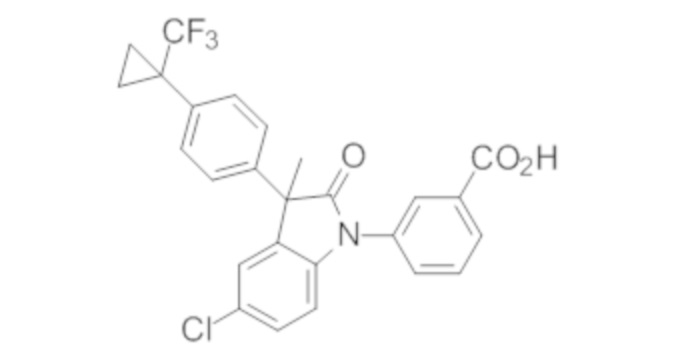
1.033
[M + H]+ 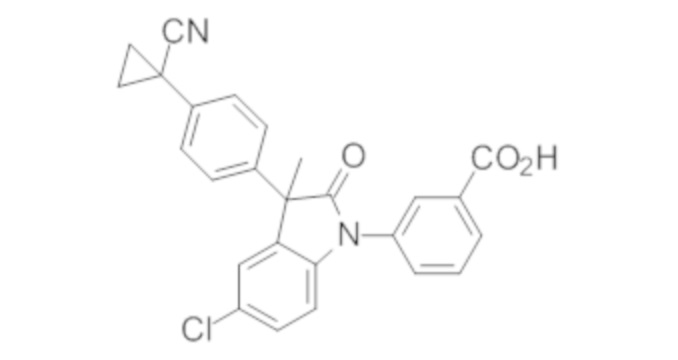
1.034
[M + H]+ 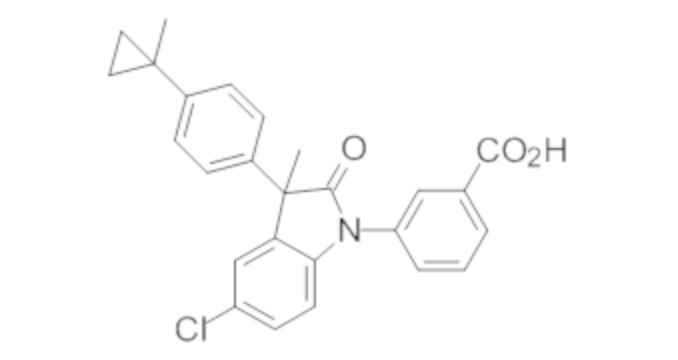
1.035
[M + H]+ 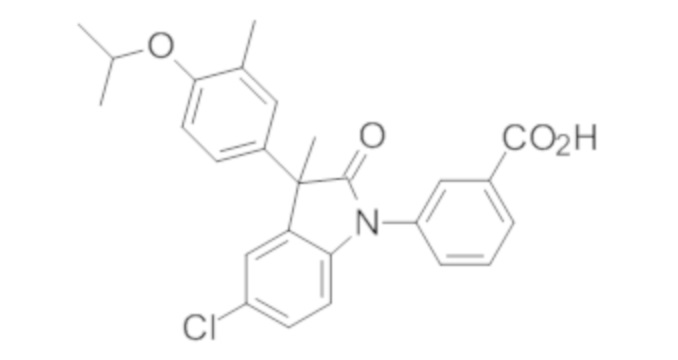
1.036
[M + H]+ 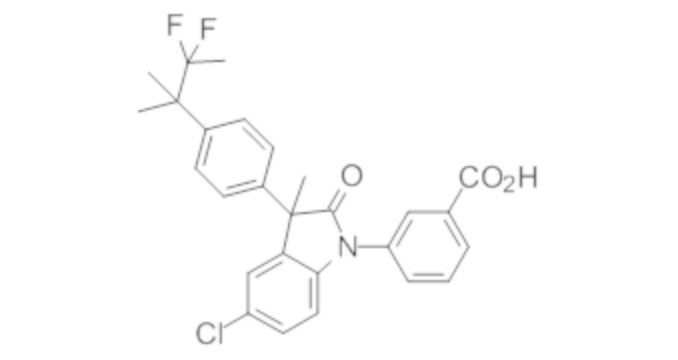
1.037
[M + H]+ 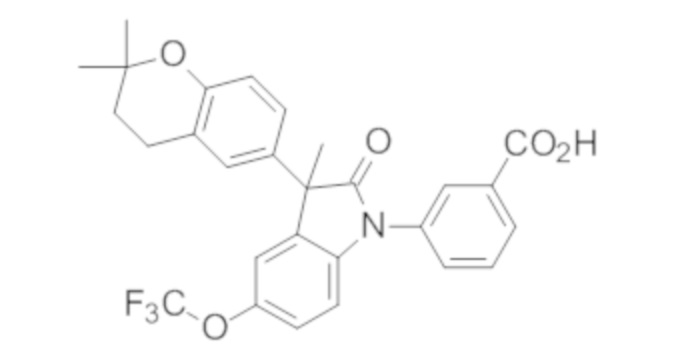
1.038
[M + H]+ 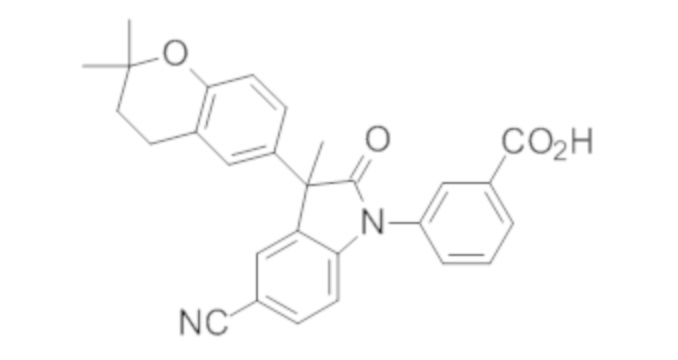
1.039
[M + H]+ 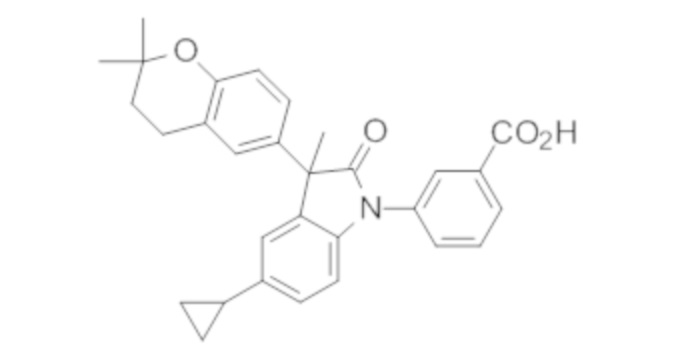
1.040
[M + H]+ 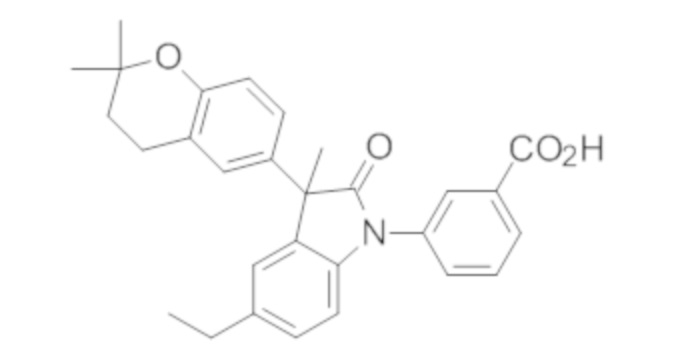 1.041
1.041
[M + H]+ 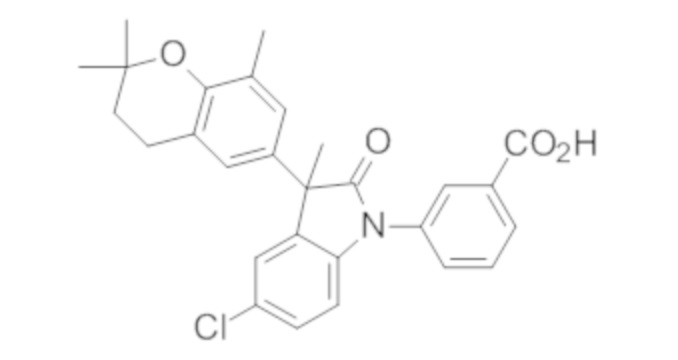
1.042
[M + H]+ 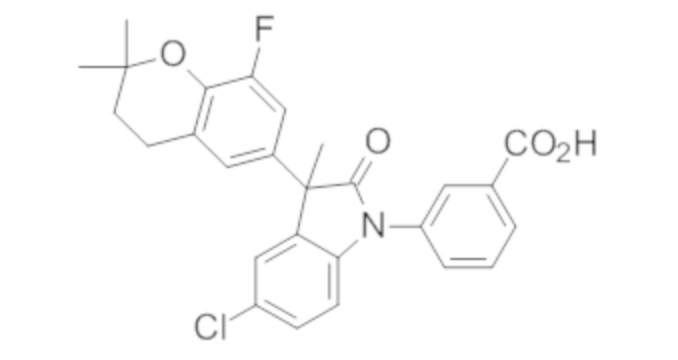
1.043
[M + H]+ 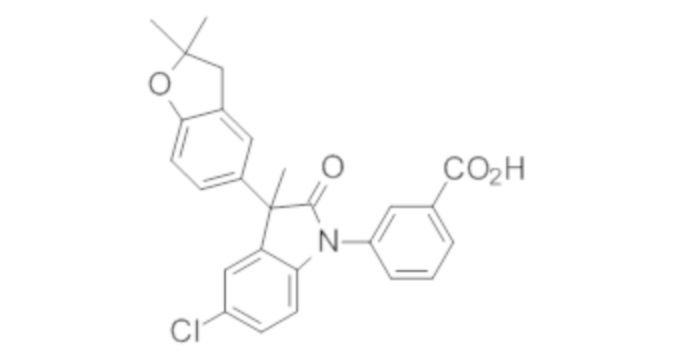
1.044
[M + H]+ 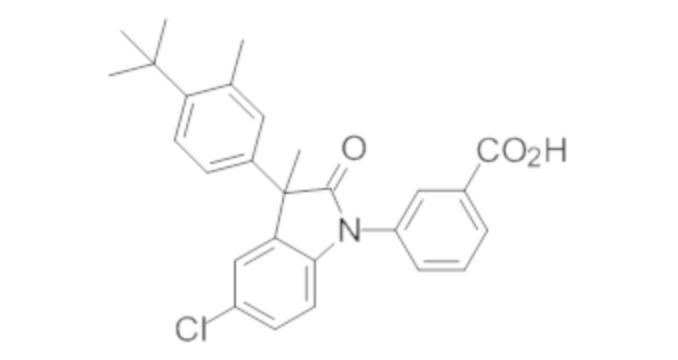 1.045
1.045
[M + Na]+ 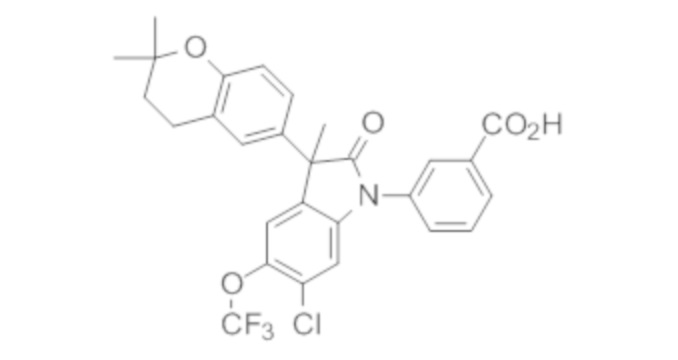
1.046
[M + H]+ 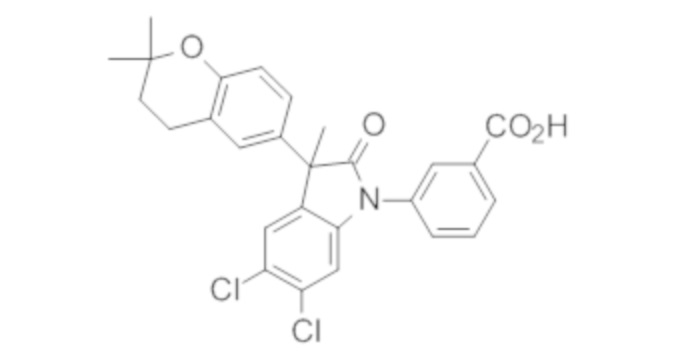
1.047
[M + Na]+ 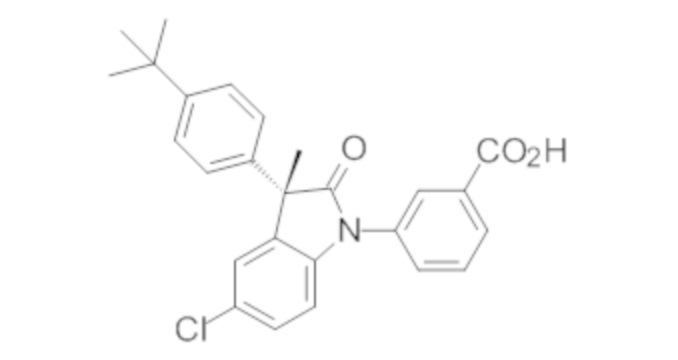
1.048
[M + H]+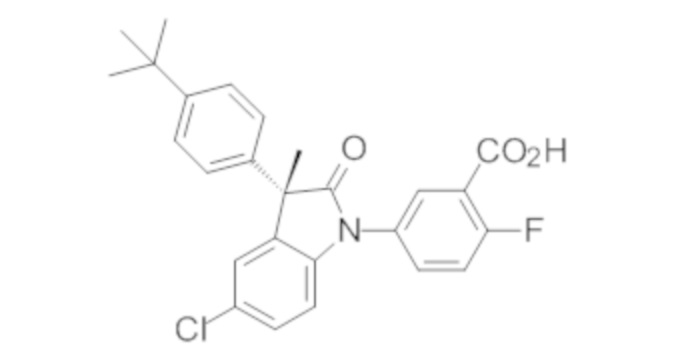
1.049
[M + H]+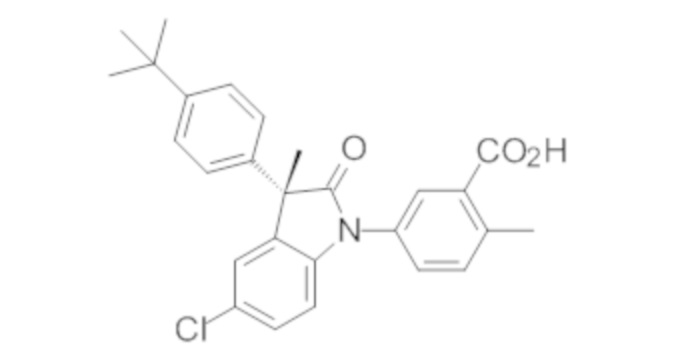
1.050
[M + H]+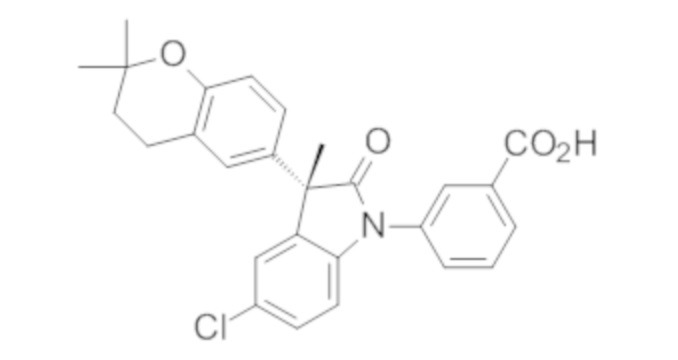
1.051
[M + H]+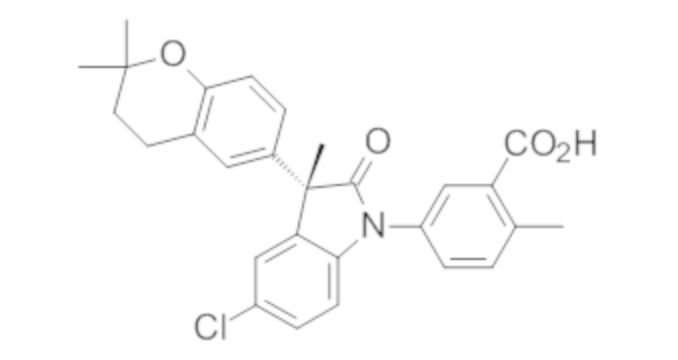
1.052
[M + H]+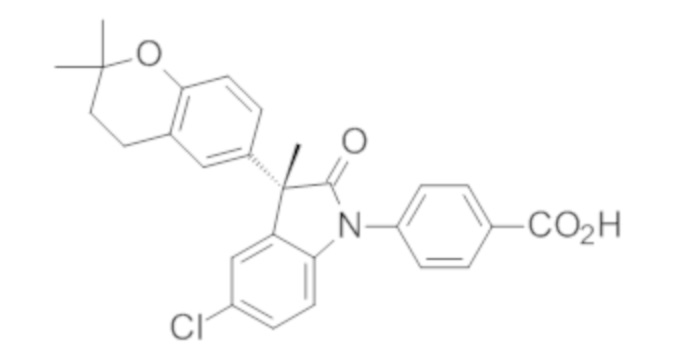
1.053
[M + H]+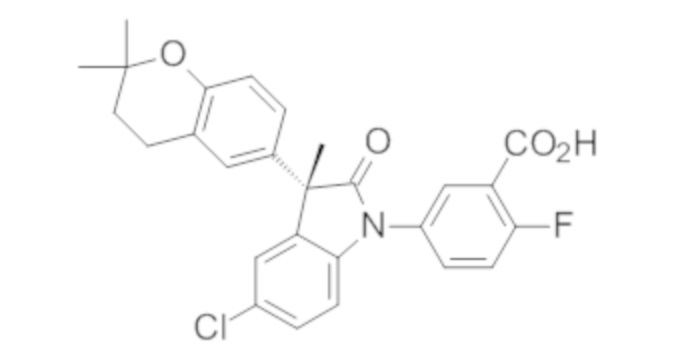
1.054
[M + H]+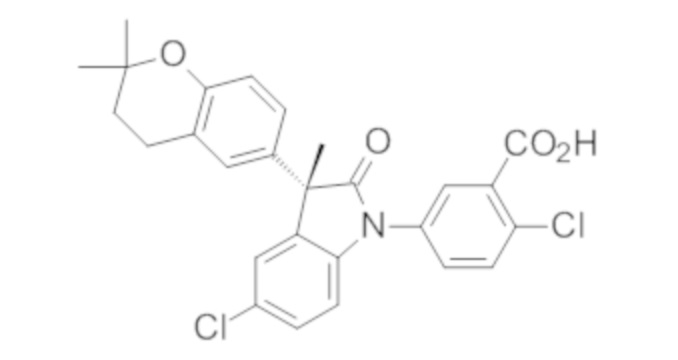
1.055
[M + H]+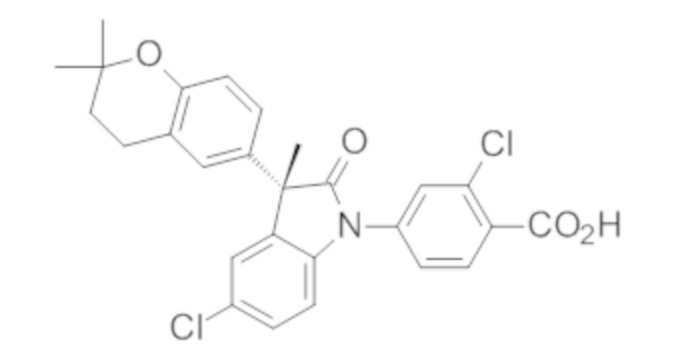
1.056
[M + H]+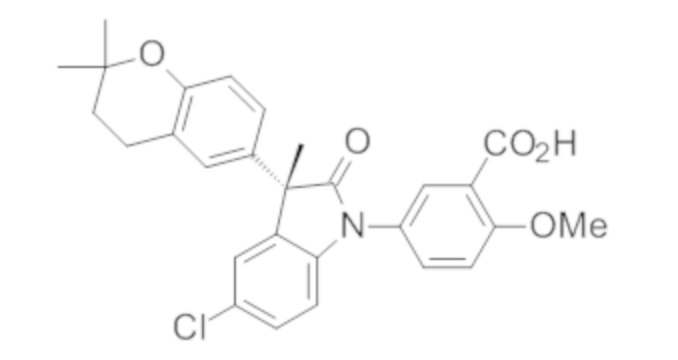
1.057
[M + H]+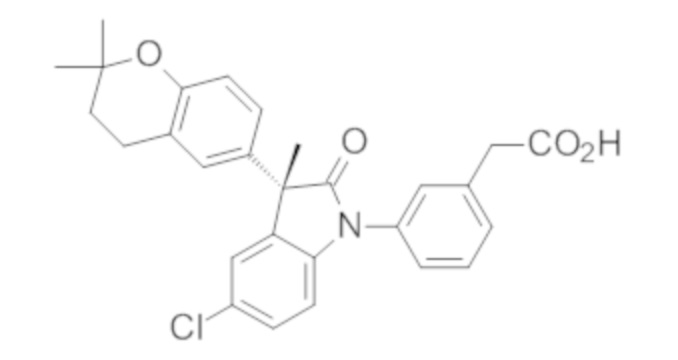
1.058
[M + H]+ 1.059
1.059
[M + H]+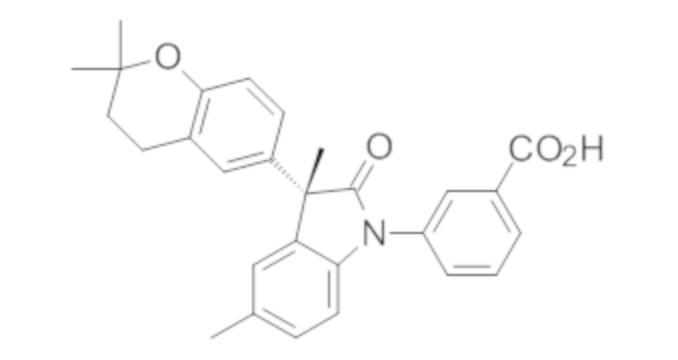
1.060
[M + H]+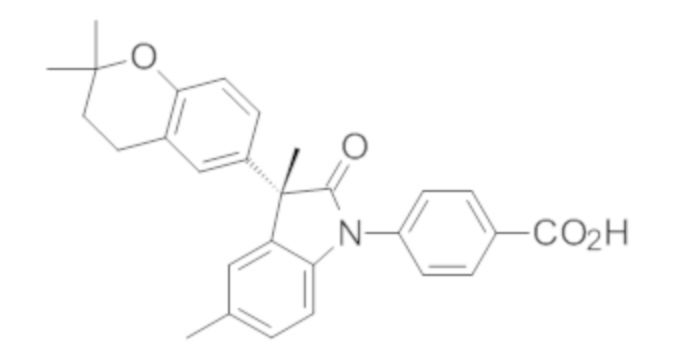
1.061
[M + H]+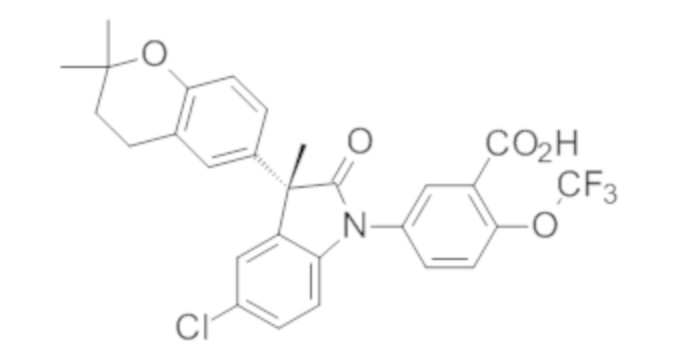
1.062
[M + H]+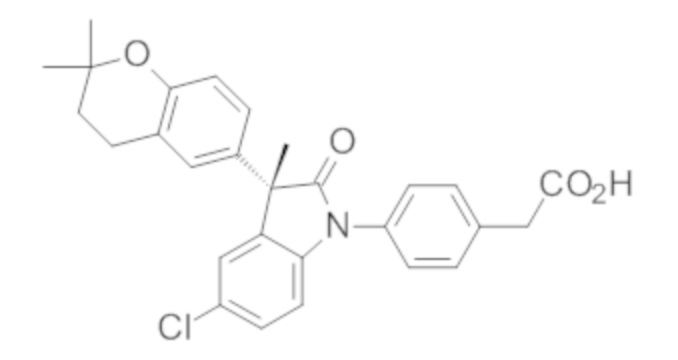
1.063
[M + H]+ 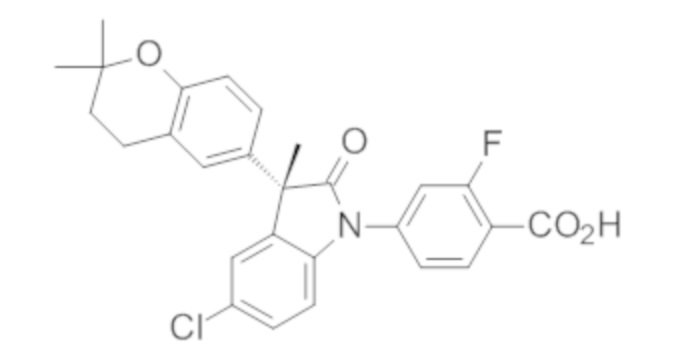
1.064
[M + H]+ 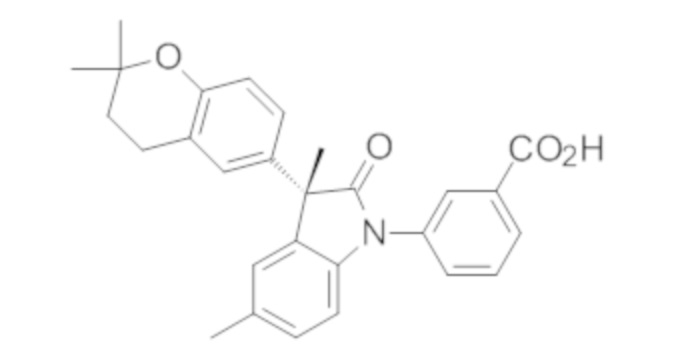
1.065
[M + H]+ 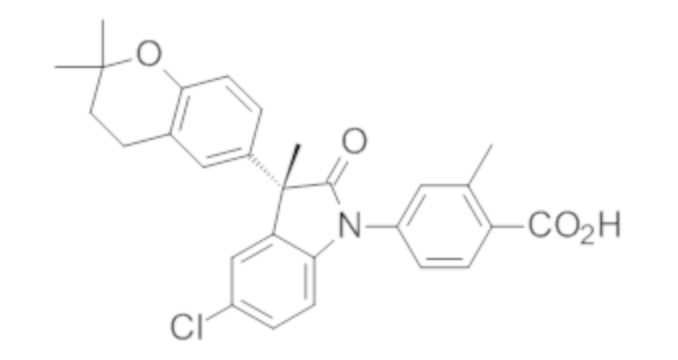
1.066
[M + H]+ 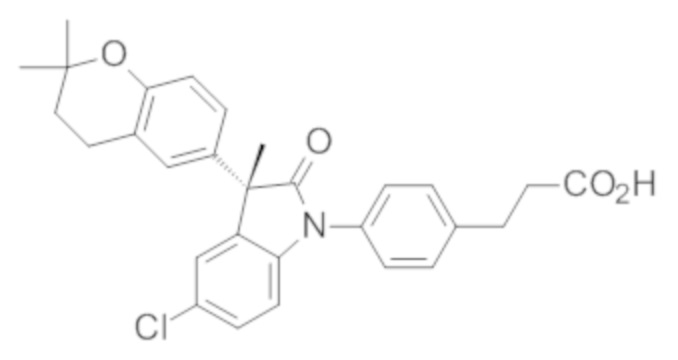
1.067
[M + H]+ 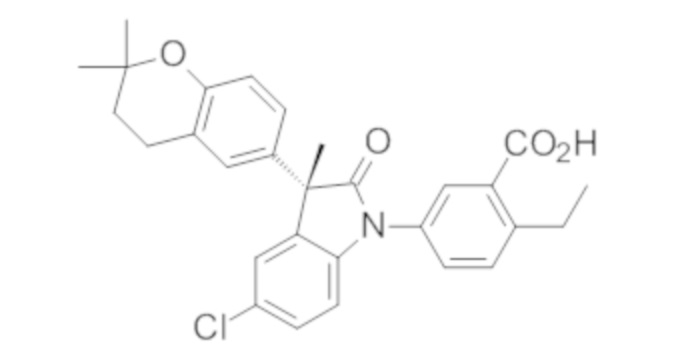
1.068
[M + H]+ 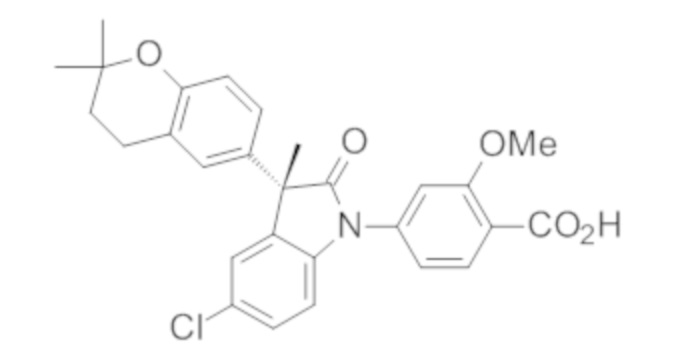
1.069
[M + H]+ 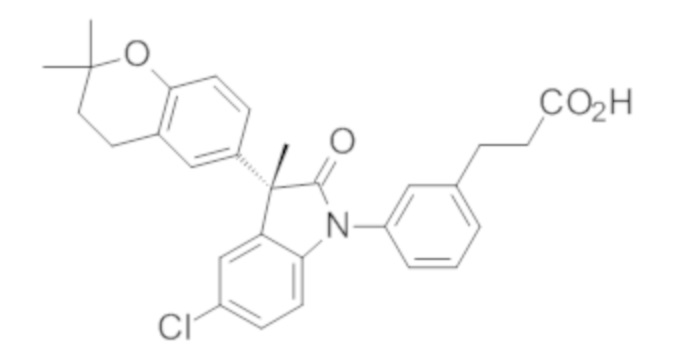
1.070
[M + H]+ 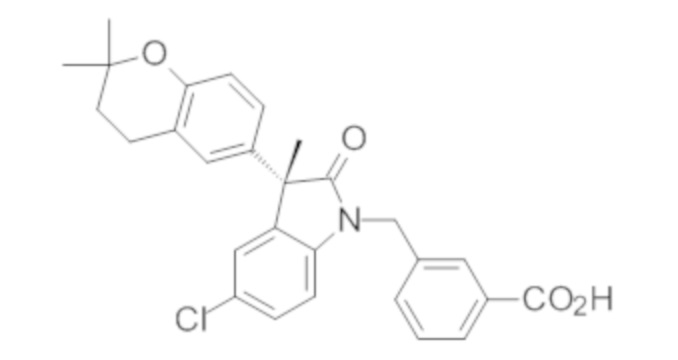
1.071
[M + H]+ 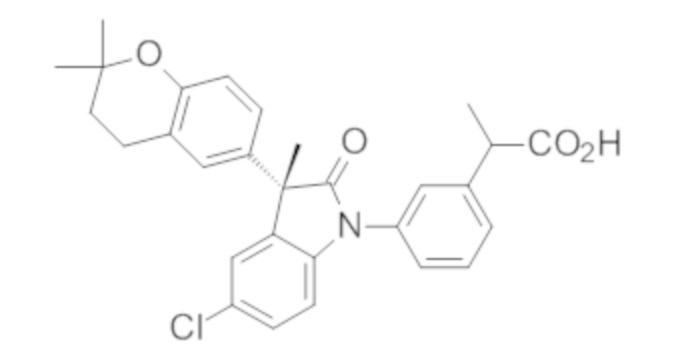
1.072
[M + H]+ 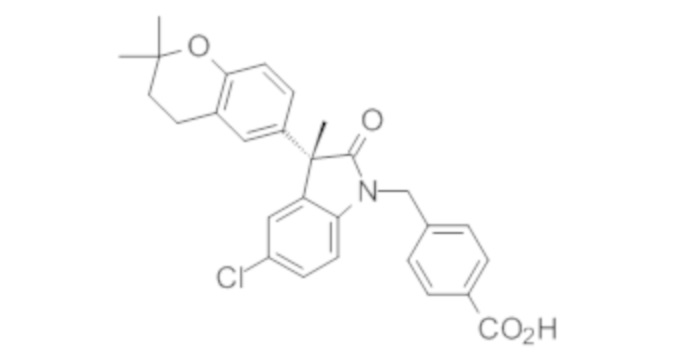
1.073
[M + H]+ 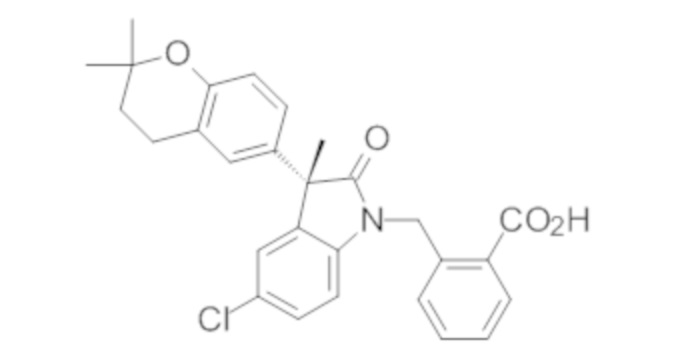
1.074
[M + H]+ 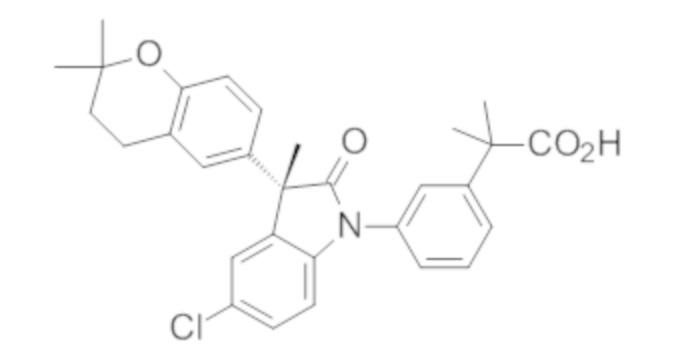
1.075
[M + H]+ 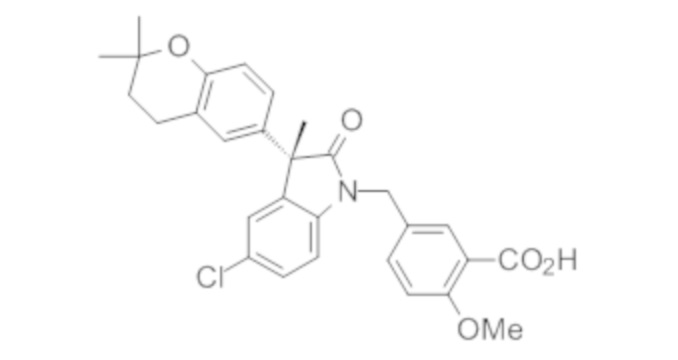
1.076
[M + Na]+ 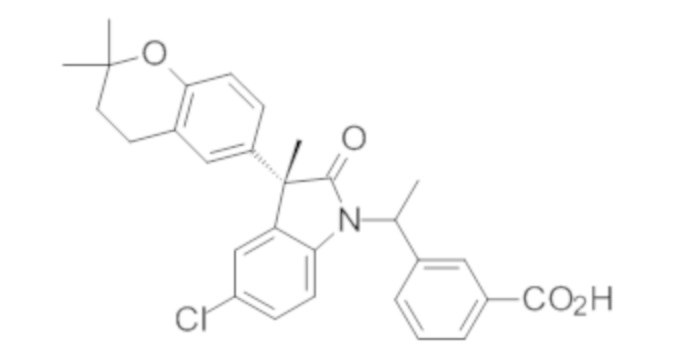
1.077
[M + Na]+ 
1.078
[M + Na]+ 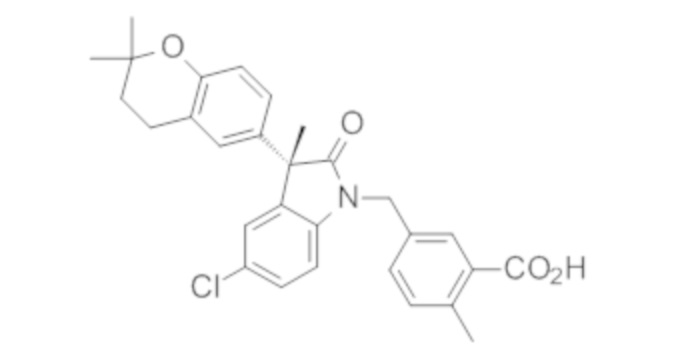
1.079
[M + Na]+ 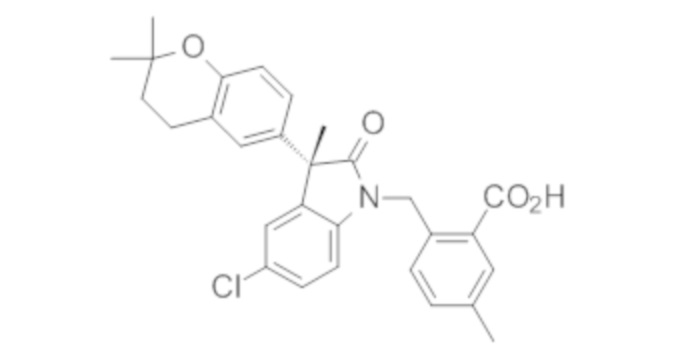
1.080
[M + H]+ 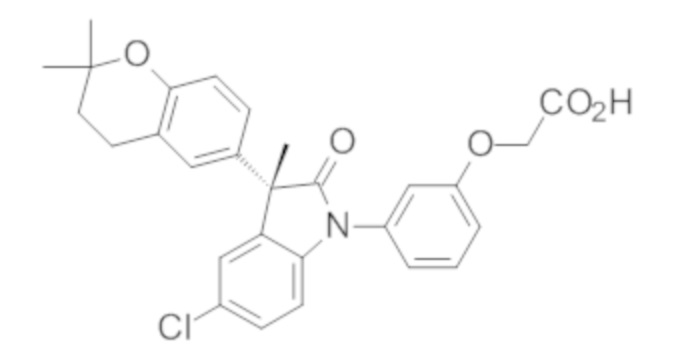
1.081
[M + H]+ 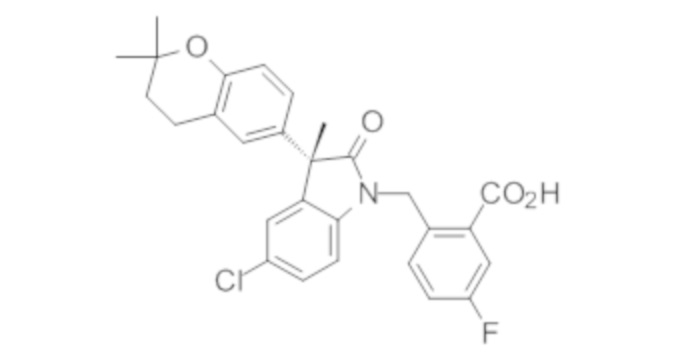
1.082
[M + H]+ 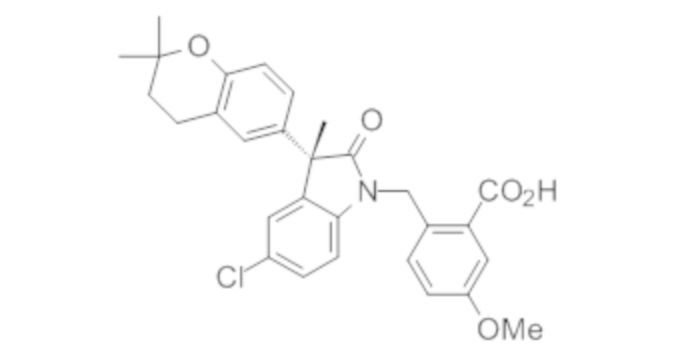
1.083
[M + H]+ 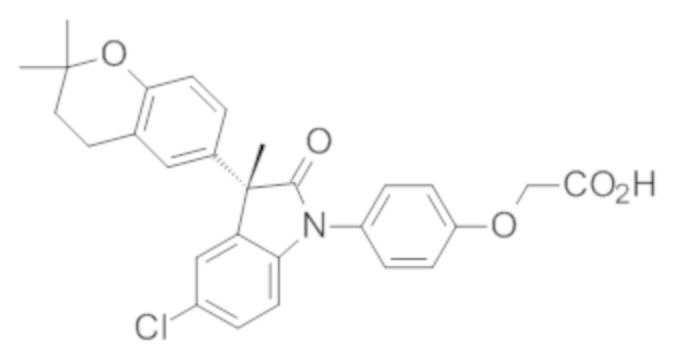
1.084
[M + H]+ 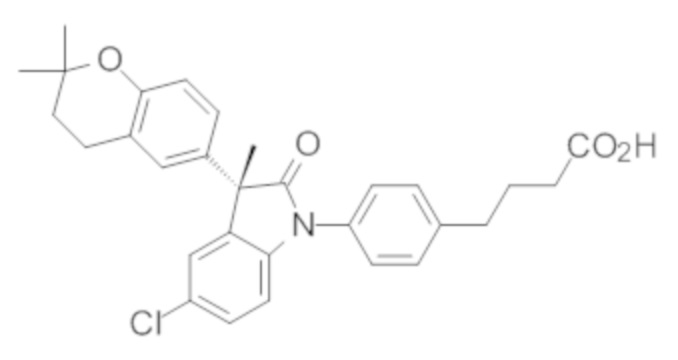
1.085
[M + H]+ 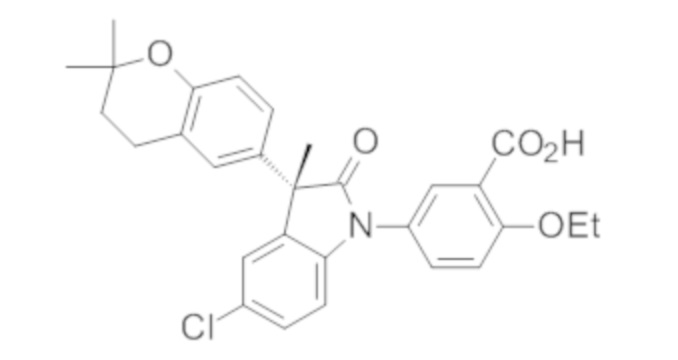
1.086
[M + H]+ 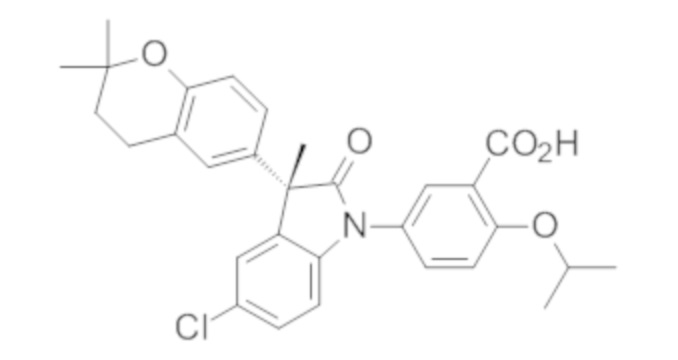
1.087
[M + H]+ 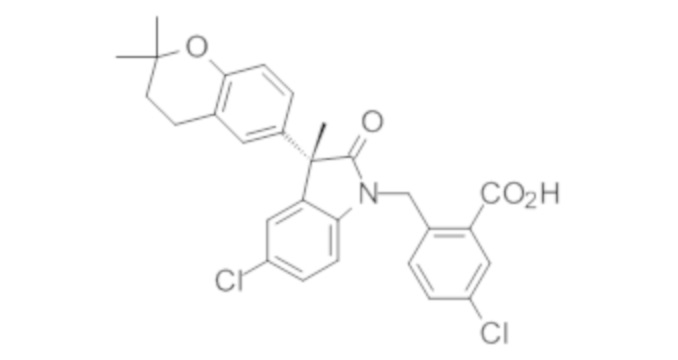
1.088
[M + H]+ 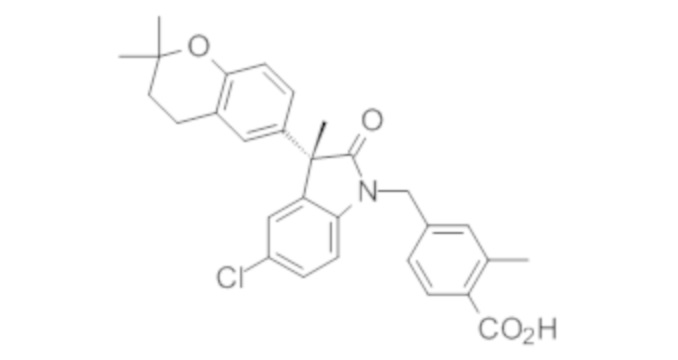
1.089
[M + H]+ 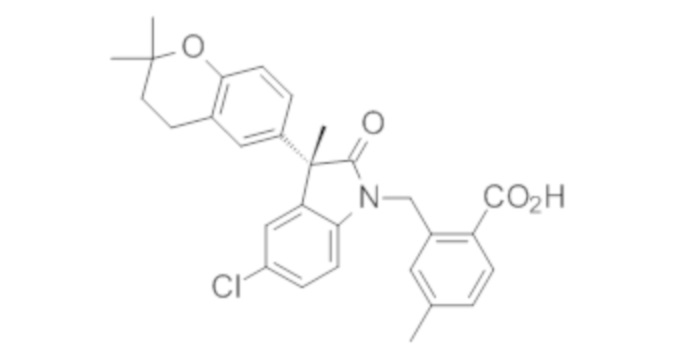
1.090
[M + H]+ 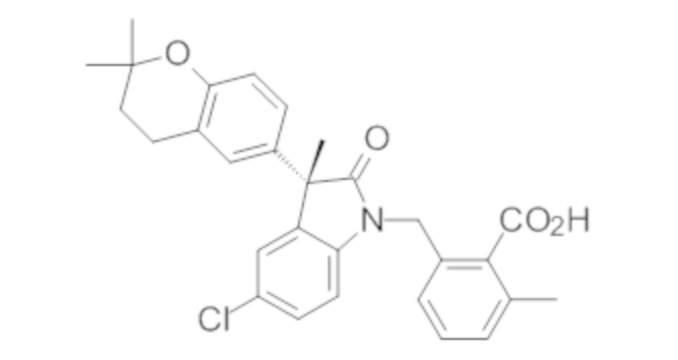
1.091
[M + Na]+ 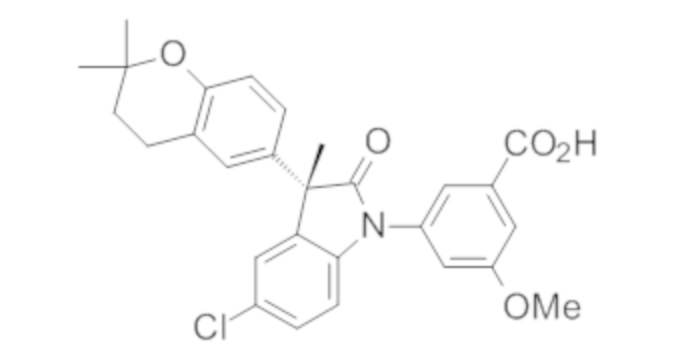
1.092
[M + H]+ 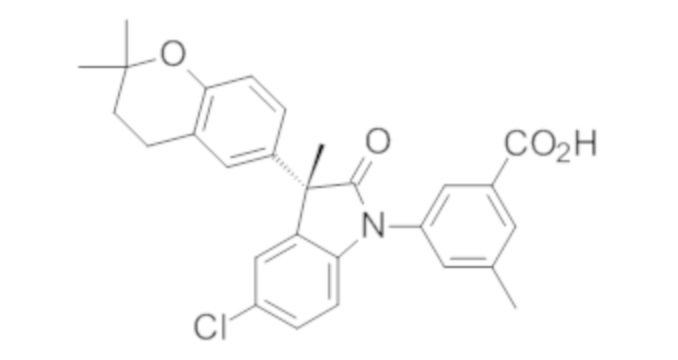
1.093
[M + H]+ 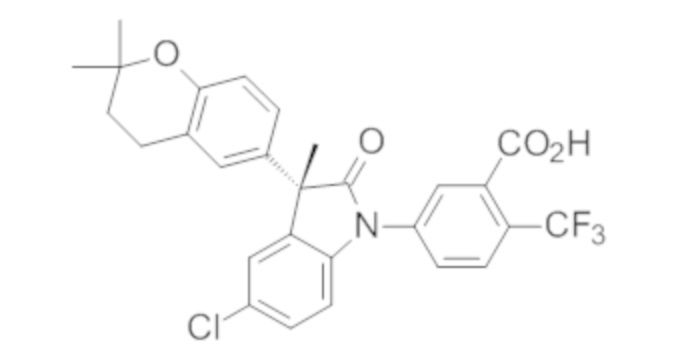
1.094
[M + H]+ 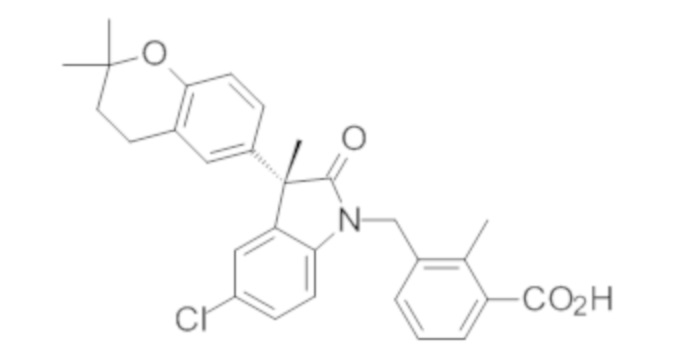
1.095
[M + H]+ 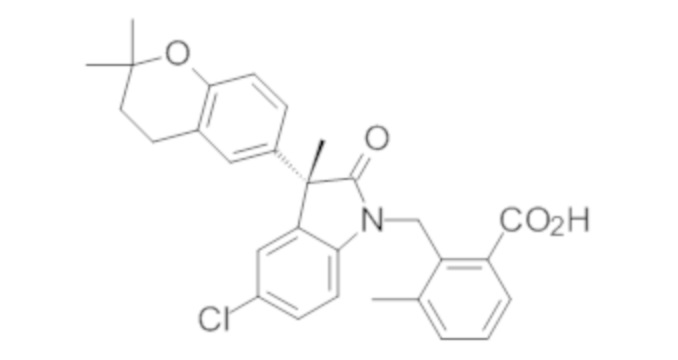
1.096
[M + H]+ 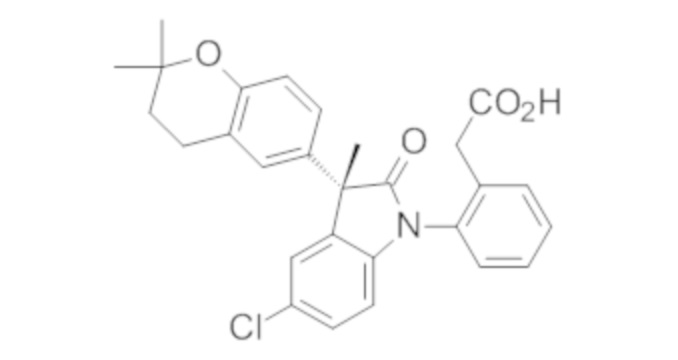 1.097
1.097
[M + H]+ 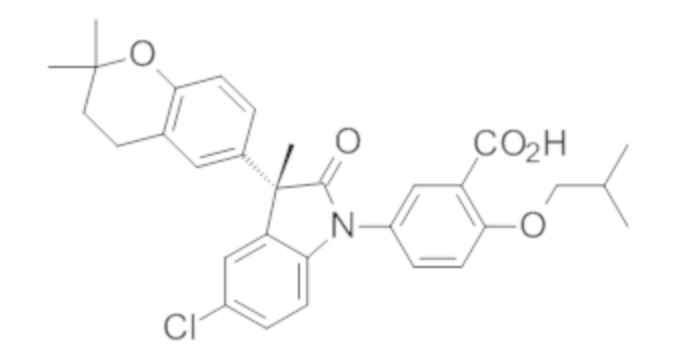
1.098
[M + H]+ 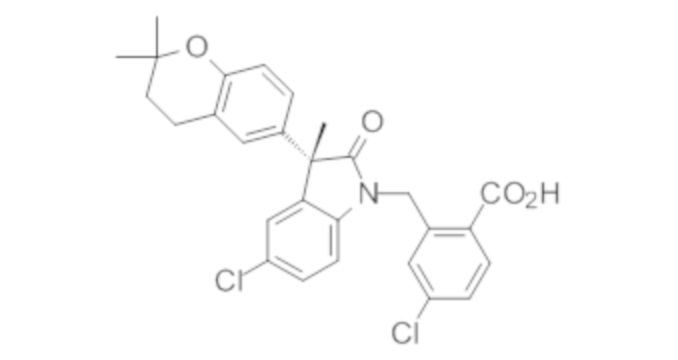
1.099
[M + Na]+ 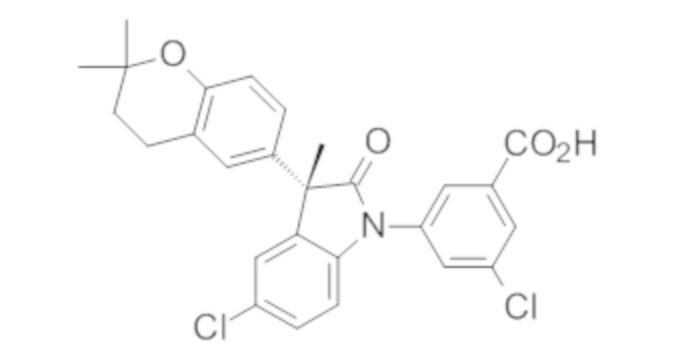
1.100
[M + H]+ 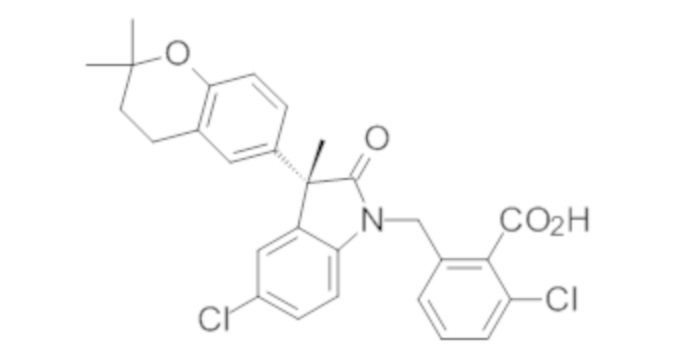
1.101
[M + Na]+ 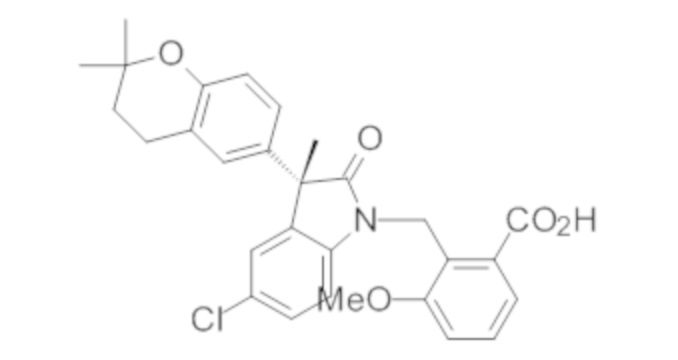
1.102
[M + H]+ 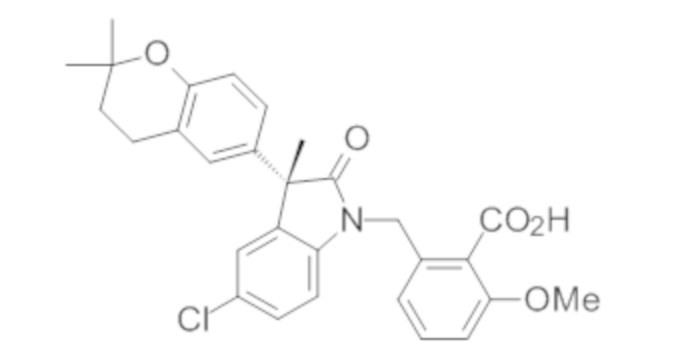
1.103
[M + H]+ 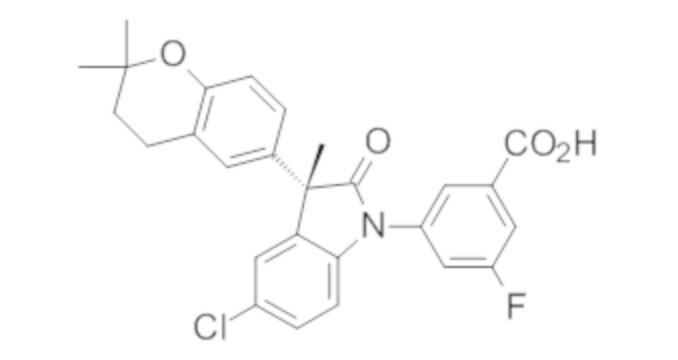
1.104
[M + H]+ 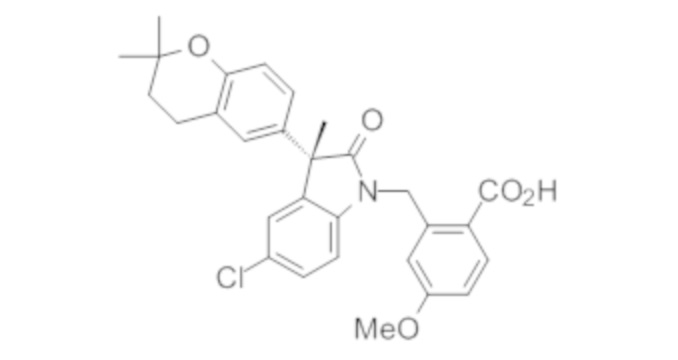
1.105
[M + H]+ 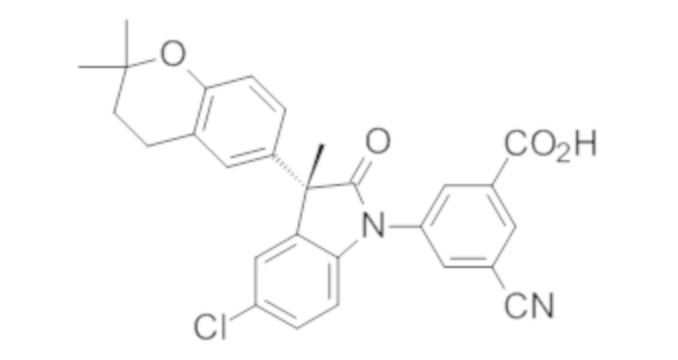
1.106
[M + H]+ 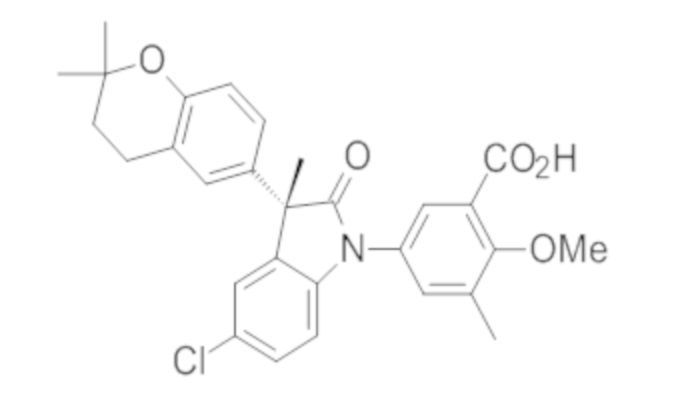
1.107
[M + H]+ 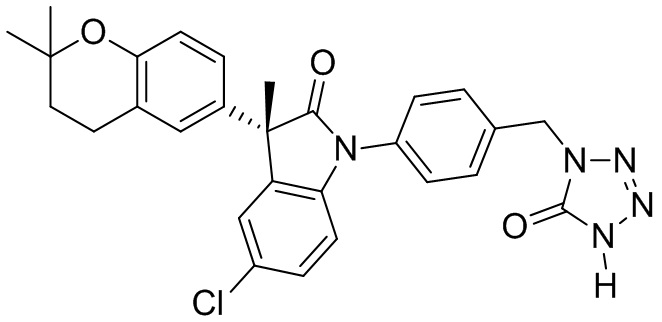
1.108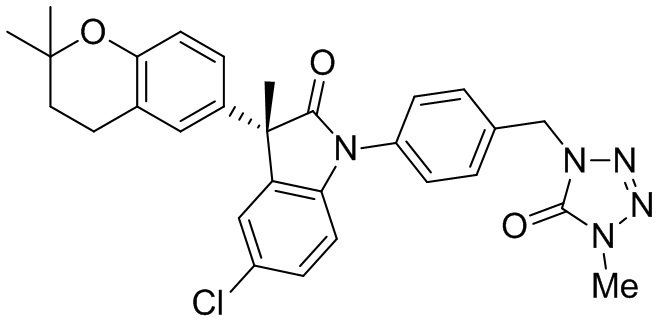
1.109
Биологические примеры
Определение эффективности модуляторов хемокинов
Примеры in vitro тестов - Реагенты
MOLT-4 клетки получали от Американской коллекции клеточных культур (American Type Culture Collection (Manassas, VA)) и выращивали в культуральной среде RPMI с добавлением 10% эмбриональной телячьей сыворотки (FCS) в увлаженном инкубаторе с 5% CO2 при 37°C. Рекомбинантные человеческие хемокиновые белки TECK получали от R&D Systems (Minneapolis, MN). Микрокамеры для тестов хемотаксиса ChemoTX™ покупали у Neuro Probe (Gaithersburg, MD). Наборы для исследования пролиферации клеток CyQUANT™ покупали у Molecular Probes (Eugene, Oregon). Кальциевый индикаторный краситель Fluo-4 AM покупали у Molecular Devices (Mountain View, CA).
In vitro тесты
Можно применять разные тесты для исследования описанных в настоящем тексте соединений, включая тесты передачи сигнала, тесты хемотаксиса (анализ миграции), анализ связывания с лигандом и другие тесты клеточного ответа. Тесты передачи сигнала хемокиновым рецептором можно применять для оценки способности соединения, такого как потенциальный CCR(9) антагонист, блокировать CCR(9) лиганд-(например, TECK)-индуцированный сигнал. Блокирование такого сигнала может использоваться в лечении различных заболеваний, таких как воспалительные заболевания кишечника, аллергическое заболевание, псориаз, атопический дерматит, астма, фиброзные заболевания, отторжение трансплантата, иммуноопосредованные пищевые аллергии, аутоиммунные заболевания, целиакия, ревматоидный артрит, тимома, рак вилочковой железы, лейкоз, солидная опухоль, острый лимфобластный лейкоз, меланома, первичный склерозирующий холангит, гепатит или послеоперационная непроходимость кишечника.
Тест хемотаксиса можно также применять для определения функции рецептора и оценки описанных в настоящем изобретении соединений. Эти тесты основаны на функциональной миграции клеток in vitro или in vivo, вызываемой неким агентом, и их можно применять для оценки связывания лигандов, ингибиторов или агонистов и/или их влияния на хемотаксис. В данной области техники известны разные тесты хемотаксиса, и любой подходящий анализ можно применять для тестирования соединений по настоящему изобретению. Примеры подходящих тестов включают описанные в PCT/US97/15915; Springer et al., WO 94/20142; Berman et al., Immunol. Invest., 17:625-677 (1988); и Kavanaugh et al., J. Immunol., 146:4149-4156 (1991)).
Оценка тестируемого модулятора в тесте хемотаксиса в сыворотке крови
Тест хемотаксиса в сыворотке крови использовали для определения эффективности потенциальных антагонистов рецепторов в блокировании миграции, осуществляемой при посредстве хемокиновых рецепторов, таких как CCR(9). Данный анализ проводили с использованием микрокамерной системы ChemoTX® с поликарбонатной мембраной, имеющей поры размером 5 мкм. MOLT-4 клетки собирали центрифугированием при 400 x g при комнатной температуре, затем суспендировали в концентрации 50 миллионов на мл в сыворотке крови человека, содержащей 50 мM HEPES (финальный pH 7.2). Затем тестируемое соединение или эквивалентный объем растворителя (ДМСО) добавляли в смесь клеток и сыворотки, с финальной концентрацией ДМСО 0.125% (об/об), и полученную смесь инкубировали при 37° C в течение 1 часа. Отдельно разбавляли рекомбинантный человеческий TECK буфером для теста хемотаксиса (HBSS + 0.1% BSA), обычно покрывая диапазон от 0.1 нM до 500 нM, после чего 29 мкл разбавленного хемокина помещали в нижние лунки планшета ChemoTX®. На планшет клали поликарбонатную мембрану с размером пор 5 мкм, и 20 мкл смеси клетки/соединение помещали на каждую лунку мембраны. Планшеты инкубировали при 37°C в течение 90 минут, после чего удаляли поликарбонатную мембрану и добавляли в нижние лунки 5 мкл ДНК-интеркалирующего агента CyQUANT (Invitrogen, Carlsbad, CA). Интенсивность флуоресценции, соответствующую числу мигрировавших клеток, замеряли с помощью планшета-ридера Spectrafluor Plus (TECAN, San Jose, CA).
Значения A2 вычисляли по приведенному ниже уравнению, сравнивая эффективность тестируемого соединения с эффективностью контрольного образца, содержащего только ДМСО, при экви-активных концентрациях хемокина:
Log(A2)= log[лек.средство(M)] - log[(A’/A)-1]
где A означает активность агониста в отсутствие антагониста, а A’ означает активность агониста в присутствии антагониста при данной концентрации лек. средства ([лек. средство(M)]).
Модель in vivo эффективности для человеческого IBD (воспалительного заболевания кишечника)
Инфильтрацию T клеток в тонкий кишечник и толстый кишечник связывают с патогенезом воспалительных заболеваний кишечника человека, которые включают целиакию, болезнь Крона и язвенный колит. Считается, что блокирование поступления релевантных Т клеток в кишечник является эффективным подходом к лечению IBD человека. CCR(9) экспрессирован в T клетках, мигрирующих в кишечник, в периферической крови, и уровень CCR(9) повышен у пациентов с воспалением тонкого кишечника, таким как болезнь Крона и целиакия. CCR(9) лиганд TECK экспрессируется в тонком кишечнике. Поэтому считается, что данная пара лиганд-рецептор играет роль в развитии IBD вследствие участия в миграции Т клеток в кишечник. Существует несколько животных моделей, и они могут применяться для оценки интересующих соединений, таких как потенциальные CCR(9) антагонисты, по способности воздействовать на такую миграцию Т клеток и/или патологическое состояние или заболевание, что может сделать возможным предсказание эффективности антагонистов у людей.
Животные модели с патологией, сходной с человеческим язвенным колитом
Мышиная модель, описанная в работе Panwala и соавторов (Panwala et al., J Immunol., 161(10):5733-44 (1998)), включает генетическое удаление мышиного гена множественной лекарственной устойчивости (MDR). Мыши с выключенным геном MDR (MDR-/-) подвержены развитию тяжелого спонтанного воспаления кишечника при содержании в специфических беспатогенных условиях. Воспаление кишечника, наблюдаемое у MDR-/- мышей, характеризуется патологией, сходной с человеческим воспалительным заболеванием кишечника (IBD), и определяется инфильтрацией Т клеток типа Th1 в собственную пластинку слизистой оболочки толстого кишечника.
Другая мышиная модель была описана в работе Davidson et al., J Exp Med., 184(1):241-51(1986). В этой модели удаляли мышиный ген IL-10, и мыши испытывали дефицит выработки интерлейкина 10 (IL-10-/-). У этих мышей развивалось хроническое воспалительное заболевание кишечника (IBD), преимущественно в толстом кишечнике, которое имеет такие же гистопатологические особенности, как человеческий IBD.
Другая мышиная модель IBD была описана в работе Powrie et al., Int. Immunol., 5(11):1461-71 (1993), где подсет T клеток CD4+ (именуемых CD45RB(high)) от иммунокомпетентных мышей очищали и адоптивно переносили иммунодефицитным мышам (таким как мыши C.B-17 scid). У животных с восстановленной популяцией T клеток CD45RBhighCD4+ развивалась летальная изнуряющая болезнь с сильной инфильтрацией моноядерных клеток в кишечнике, патологически сходная с человеческим IBD.
ФНО ARE(-/-) модель.
Роль ФНО в болезни Крона у человека была недавно продемонстрирована посредством успешного лечения с использованием антител к ФНО-альфа в работе Targan et al., N. Engl. J Med., 337(15):1029-35 (1997). У мышей с аномальной выработкой ФНО альфа из-за генетического изменения в гене TNF (ARE-/-) развивались воспалительные болезни кишечника (см. Kontoyiannis et al., Immunity, 10(3):387-98 (1999)).
Примеры тестов на эффективность in vivo
Оценка испытуемого модулятора в модели CCR(9)-зависимого трафика T-клеток.
Суспензии клеток готовили из селезенки и лимфатических узлов мышей линии OT-I Tg CD45.1. Суммарно 15 X 106 клеток (около 3 х 106 CD8 T клеток) инъецировали совпадающим по полу конгенным мышам линии CD45.2 C57BL/6n (возраст 8-10 недель). Через 24 часа животных иммунизировали принудительным пероральным введением 25мг белка овальбумина (Sigma-Aldrich, St. Louis, MO) + 10мкг холерного токсина (Calbiochem, San Diego, CA). CCR(9) антагонист – соединение 1.063 (Таблица 1) – вводили перед пероральным введением овальбумина, тайминг введения диктовался фармакокинетикой в организме мышей. Через пять дней после иммунизации животных усыпляли и извлекали тонкий кишечник. Удаляли Пейеровы бляшки и, после промывки фосфатно-солевым буферным раствором, вскрывали желудок на влажном квадратном куске ткани Optima (Allegiance Healthcare). Слизистую оболочку соскребали скальпелем и затем растворяли при перемешивании в 50 мл среды, содержащей 10% сыворотки новорожденных телят и DTT (1 мM), в течение 15 минут при комнатной температуре. После центрифугирования, полученные пеллеты заново растворяли в фосфатно-солевом буферном растворе, содержащем 10% сыворотки новорожденных телят, перемешивали на вихревой мешалке 3 минуты и быстро пропускали через колонку со стекловатой (1.6 г, упаковано в 20-миллитровый шприц; Fisher Scientific). Внутриэпителиальные лимфоциты тонкого кишечника дополнительно очищали в градиенте фиколл-пак и окрашивали с помощью mAbs для анализа методом проточной цитометрии. Детектирование и количественное определение перенесенных OT-1 Tg CD45.1 T клеток осуществляли методом проточной цитометрии. В данной модели, добавление соединения по настоящему изобретению приводило к значительному снижению частоты миграции OT-1 Tg CD45.1 T клеток в тонкий кишечник в ответ на антиген.
Оценка испытуемого модулятора в модели подавления распространения ВИЧ
В гуманизированных мышах BLT (bone marrow/liver/thymus, т.е. с пересаженным костным мозом, печенью и тимусом), SCID мышам с диабетом без ожирения (NOD) (у которых отсутствуют эндогенные T и B клетки) хирургически имплантируют эмбриональные органоиды тимуса и печени, как в SCID-hu системе. Мышей затем подвергают сублетальным дозам облучения и пересаживают им аутологичные CD34+ стволовые клетки, полученные из эмбриональной печени, которые располагаются в костном мозге мышей, эффективно принимающем трансплантируемый человеческий костный мозг, что приводит к наличию в периферической крови ряда человеческих клеток, включая зрелые T и B лимфоциты, моноциты, макрофаги и дендритные клетки, которые все демонстрируют интенсивную инфильтрацию в органы и ткани, включая печень, легкие и желудочно-кишечный тракт. После трансплантации, мышам вводят соединение по настоящему изобретению для подавления миграции человеческих клеток в желудочно-кишечный тракт, главный источник взаимодействия Т клеток с ВИЧ. Эффективность соединений оценивают по снижению вирусной нагрузки в крови, определяемой стандартными методами.
Оценка испытуемого модулятора в модели артрита
Проводили 17-дневное исследование коллаген-индуцированного артрита II типа для оценки влияния модулятора на вызванное артритом клиническое распухание голеностопного сустава. Коллаген-индуцированный артрит у крыс является экспериментальной моделью полиартрита, которая широко применяется для доклинических испытаний многочисленных противоревматических препаратов (см. Trentham et al., J. Exp. Med. 146(3):857-868 (1977), Bendele et al., Toxicologic Pathol. 27:134-142 (1999), Bendele et al., Arthritis. Rheum. 42:498-506 (1999)). Отличительными чертами данной модели являются надежное возникновение и развитие устойчивого, легко измеримого многосуставного воспаления, выраженное разрушение хряща вкупе с образованием паннуса, резорбция костной ткани (от слабой до умеренной) и разрастание околохрящевой кости.
Самок крыс линии Льюис (вес примерно 0.2 килограмма) усыпляли изофлураном и инъекционно вводили им неполный адъювант Фрейнда, содержащий 2 мг/мл бычьего коллагена II типа, в основание хвоста и в два сайта на спине в дни 0 и 6 данного 17-дневного исследования. Тестируемый модулятор вводили ежедневно посредством подкожной инъекции со дня 9 по день 17 в дозировке 100 мг/кг и в объеме 1 мл/кг в носителе (24.5 % Cremaphore EL, 24.5% масло, 1% бензиловый спирт и 50% дистиллированная вода). Измерение диаметра голеностопного сустава штангенциркулем проводили ежедневно, и уменьшение распухания голеностопного сустава принимали за критерий эффективности.
Оценка испытуемого модулятора в мышиной модели астмы
В данном примере описана методика определения эффективности антагонистов в лечении астмы. Животную модель астмы можно получить путем сенсибилизации грызунов к экспериментальному антигену (например, овальбумину) стандартной иммунизацией и последующим введением того же антигена в легкие грызунов посредством распыления аэрозоля. Три группы грызунов, по 10 грызунов в группе, подвергали активной сенсибилизации в День 0 путем однократной интраперитонеальной инъекции 100мкг овальбумина в фосфатно-солевом буферном растворе (PBS) с адъювантом, например, гидроксидом алюминия. Через 11 дней после сенсибилизации, животных помещали в камеру из плексигласа и подвергали воздействию аэрозоля с овальбумином (1%) в течение 30 минут с помощью ультразвукового генератора аэрозоля (De Vilbliss). Одной группе мышей дополнительно вводили PBS и Tween 0.5% интраперитонеально после первой сенсибилизации и с различным режимом дозирования после этого, вплоть до воздействия аэрозоля с овальбумином. Вторая серия состояла из групп мышей, которым вводили различные дозировки CCR4 антагониста (интраперитонеально, внутривенно, подкожно, внутримышечно, перорально или любым другим способом) при первой сенсибилизации и с различным режимом дозирования после этого, вплоть до воздействия аэрозоля с овальбумином. Третья серия мышей, выступающая в роли положительного контроля, состояла из групп, которым вводили мышиный IL-10 интраперитонеально, или антитела к IL4 интраперитонеально, или антитела к IL5 интраперитонеально, при первой сенсибилизации и с различным режимом дозирования после этого, вплоть до воздействия аэрозоля с овальбумином. Затем проводили исследование животных в различные моменты времени после воздействия аэрозоля с овальбумином на предмет работы легких, наличия инфильтратов клеток в бронхоальвеолярных выделениях, гистологического исследования легких и измерения титра овальбумин-специфичного иммуноглобулина Е в плазме крови.
Приведенное выше подробное описание является иллюстративным, а не ограничивающим, и следует понимать, что суть и объем настоящего изобретения в полной мере определяется формулой изобретения, включающей все эквиваленты.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕТРАЗОЛ-СОДЕРЖАЩИЕ ИНГИБИТОРЫ РЕГУЛИРУЮЩЕЙ АПОПТОТИЧЕСКИЕ СИГНАЛЫ КИНАЗЫ 1 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2807545C2 |
| ГЕТЕРОАРИЛ-БИФЕНИЛ АМИНЫ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2837843C1 |
| ПРОИЗВОДНЫЕ 2-((4-(4-(БЕНЗИЛОКСИ)ПИРИМИДИН-2-ИЛ)ПИПЕРАЗИН-1-ИЛ)МЕТИЛ)-1Н-БЕНЗО[d]ИМИДАЗОЛА В КАЧЕСТВЕ НИЗКОМОЛЕКУЛЯРНЫХ АГОНИСТОВ РЕЦЕПТОРА ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-1 | 2024 |
|
RU2838323C1 |
| Четвертичные аммониевые соединения на основе 3-гидроксипиридина, обладающие антибактериальной активностью | 2021 |
|
RU2778507C1 |
| ПРОИЗВОДНЫЕ 1-ТИО-D-ГЛЮЦИТОЛА | 2006 |
|
RU2387649C2 |
| ТЕТРАГИДРОПИРАНИЛЦИКЛОПЕНТИЛТЕТРАГИДРОПИРИДОПИРИДИНОВЫЕ МОДУЛЯТОРЫ АКТИВНОСТИ РЕЦЕПТОРА ХЕМОКИНА | 2003 |
|
RU2285004C2 |
| 5-5 КОНДЕНСИРОВАННЫЕ КОЛЬЦА КАК ИНГИБИТОРЫ С5а | 2018 |
|
RU2780322C2 |
| БИЦИКЛИЧЕСКОЕ ГЕТЕРОАРОМАТИЧЕСКОЕ КОЛЬЦЕВОЕ ПРОИЗВОДНОЕ | 2019 |
|
RU2798838C2 |
| ФЕНИЛКАРБАМАТЫ И ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТА ГИДРОЛАЗЫ АМИДОВ ЖИРНЫХ КИСЛОТ (FAAH) И МОДУЛЯТОРОВ D3 ДОПАМИНОВОГО РЕЦЕПТОРА (D3DR) | 2014 |
|
RU2693457C2 |
| АНТАГОНИСТ CASR | 2004 |
|
RU2315036C2 |
Изобретение относится к оксиндольным соединениям формулы (I), где значения R1, R2a, R2b, L1, L2, Ar, Y определены в формуле изобретения, которые могут применяться для лечения CCR(9)-опосредованных патологических состояний или заболеваний. 3 н. и 40 з.п. ф-лы, 1 табл., 10 пр.
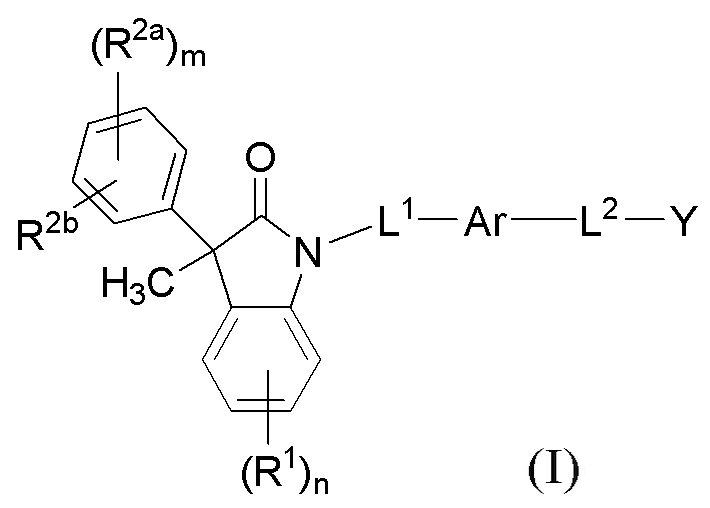
1. Соединение, имеющее формулу (I)
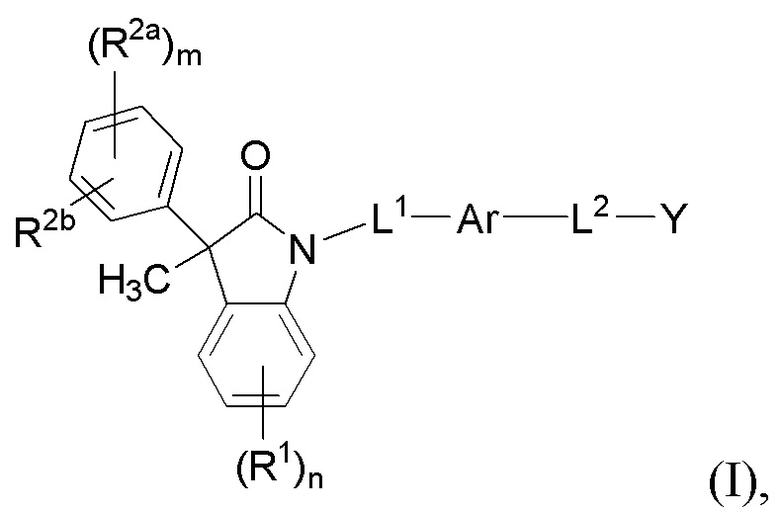
или его фармацевтически приемлемые соли, где
Ar представляет собой 5-10-членное ароматическое или гетероароматическое кольцо, содержащее от 1 до 5 гетероатомов в качестве вершин цикла, выбранных из N, О и S, необязательно замещенное 1-3 заместителями R3;
L1 выбран из группы, состоящей из одинарной связи и C1-6 алкилена,
L2 выбран из группы, состоящей из одинарной связи, C1-6 алкилена и C1-6 гетероалкилена, в котором один атом углерода алкиленовой группы заменен на O,
Y представляет собой CO2H или выбран из группы, состоящей из тетразолила и тетразолонила, где тетразолил и тетразолонил необязательно замещены C1-6 алкилом или C1-6 галоалкилом;
каждый R1 и каждый R2a независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила, C1-6 галогеналкоксигруппы, и C3-6 циклоалкила, где алкильные и циклоалкильные фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, OH, CN, C1-3 алкила и C1-3 галогеналкила;
R2b выбран из группы, состоящей из H, галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы и C3-6 циклоалкила, где алкильные, и циклоалкильные фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, CN, C1-3 алкила и C1-3 галогеналкила;
или, необязательно, один R2a и R2b, когда они присоединены к соседним атомам фенильного цикла, могут быть объединены с образованием 5- или 6-членного гетероциклоалкильного кольца, содержащего один или два атома в цикле, независимо выбранных из O, N и S, где указанное гетероциклоалкильное кольцо необязательно замещено 1-3 заместителями, выбранными из атома фтора и C1-3 алкила;
каждый R3 независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила и C1-6 галогеналкоксигруппы;
подстрочный индекс m представляет собой целое число от 0 до 4; и
подстрочный индекс n представляет собой целое число от 0 до 3.
2. Соединение по п. 1, имеющее формулу (I’)
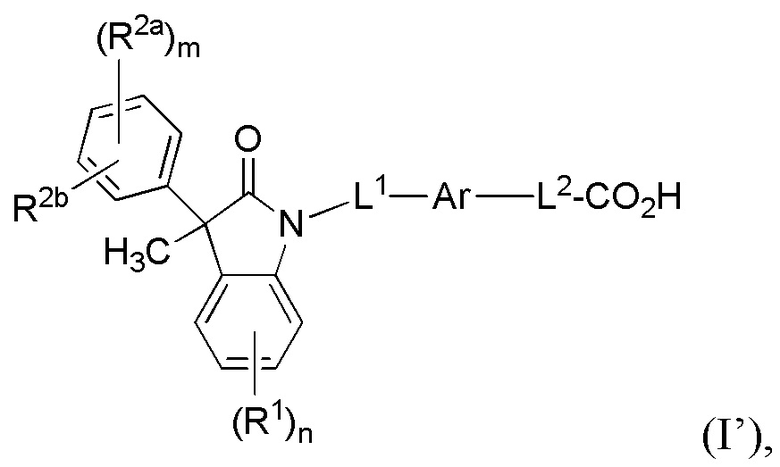
или его фармацевтически приемлемые соли, где
Ar представляет собой 5-10-членное ароматическое или гетероароматическое кольцо, содержащее от 1 до 5 гетероатомов в качестве вершин цикла, выбранных из N, О и S, необязательно замещенное 1-3 заместителями R3;
L1 выбран из группы, состоящей из одинарной связи и C1-6 алкилена,
L2 выбран из группы, состоящей из одинарной связи, C1-6 алкилена и C1-6 гетероалкилена, в котором один атом углерода алкиленовой группы заменен на O,
каждый R1 и каждый R2a независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила, C1-6 галогеналкоксигруппы и C3-6 циклоалкила, где алкильные и циклоалкильные фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, CN, C1-3 алкила и C1-3 галогеналкила;
R2b выбран из группы, состоящей из H, галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы и C3-6 циклоалкила, где алкильные и циклоалкильные фрагменты необязательно замещены 1-3 заместителями, выбранными из атома фтора, CN, C1-3 алкила и C1-3 галогеналкила;
или, необязательно, один R2a и R2b, когда они присоединены к соседним атомам фенильного цикла, могут быть объединены с образованием 5- или 6-членного гетероциклоалкильного кольца, содержащего один или два атома в цикле, независимо выбранных из O, N и S, где указанное гетероциклоалкильное кольцо необязательно замещено 1-3 заместителями, выбранными из атома фтора и C1-3 алкила;
каждый R3 независимо выбран из группы, состоящей из галогена, цианогруппы, C1-6 алкила, C1-6 алкоксигруппы, C1-6 галогеналкила и C1-6 галогеналкоксигруппы;
подстрочный индекс m представляет собой целое число от 0 до 4; и
подстрочный индекс n представляет собой целое число от 0 до 3.
3. Соединение по п. 1, имеющее формулу, выбранную из группы, состоящей из:
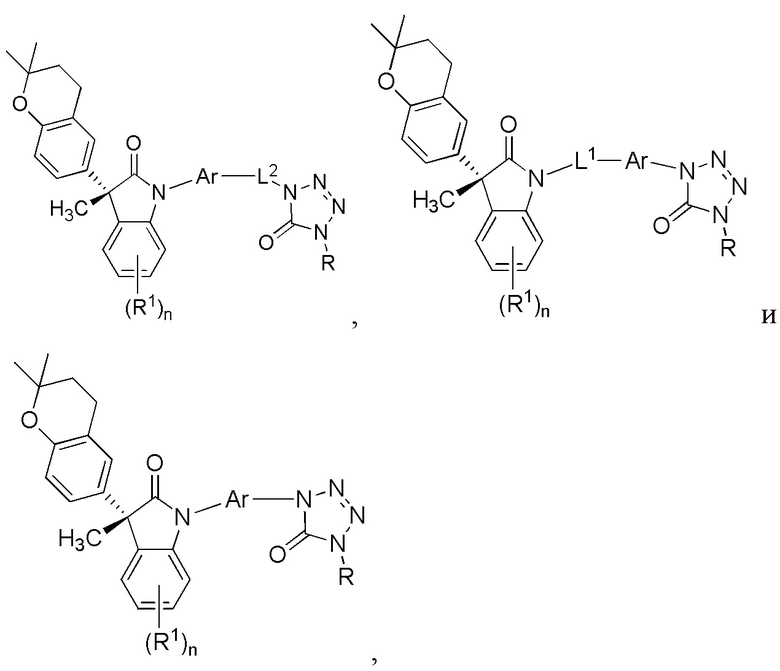
или его фармацевтически приемлемые соли, где указанное соединение практически не содержит других изомеров.
4. Соединение по любому из пп. 1 - 3, имеющее формулу
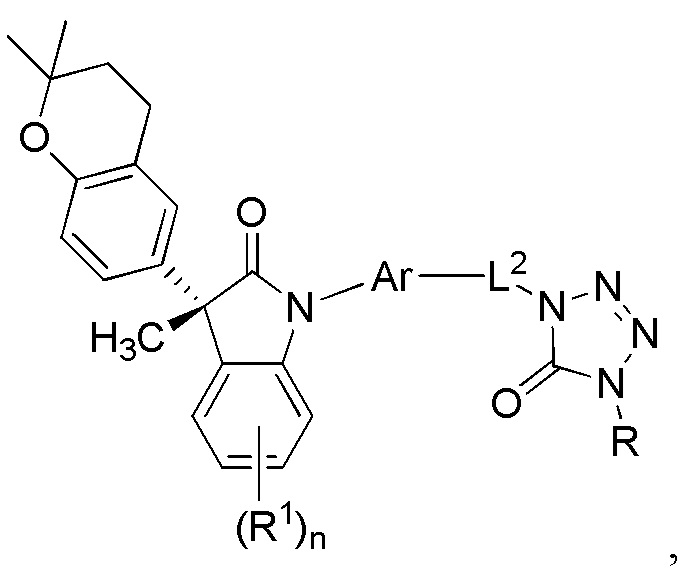
или его фармацевтически приемлемые соли, где L2 представляет собой C1-3 алкилен и где указанное соединение практически не содержит других изомеров.
5. Соединение по любому из пп. 1 - 4, где Ar выбран из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3.
6. Соединение по любому из пп. 1 - 3, где L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-.
7. Соединение по любому из пп. 1, 2, где L2 выбран из группы, состоящей из одинарной связи, -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2- и -CH2CH2CH2-.
8. Соединение по любому из пп. 1 - 7, где n равен 1 или 2.
9. Соединение по любому из пп. 1 - 8, где m равен 1, 2 или 3.
10. Соединение по п. 1, имеющее формулу
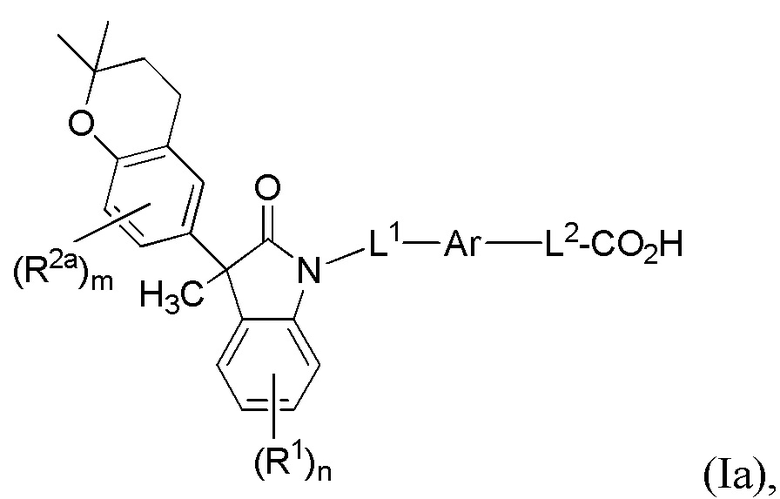
или его фармацевтически приемлемые соли.
11. Соединение по п. 10, где Ar выбран из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3.
12. Соединение по п. 10 или 11, где L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-.
13. Соединение по любому из пп. 10 - 12, где L2 выбран из группы, состоящей из одинарной связи, -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2- и -CH2CH2CH2-.
14. Соединение по п. 10, имеющее формулу, выбранную из группы, состоящей из:
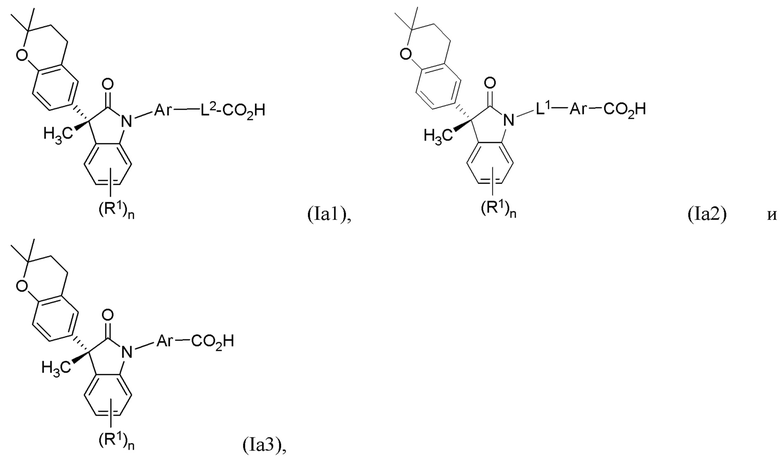
или его фармацевтически приемлемые соли, где указанное соединение практически не содержит других изомеров.
15. Соединение по п. 14, где Ar выбран из группы, состоящей из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3.
16. Соединение по п. 14 или 15, где Ar выбран из группы, состоящей из 1,3-фенилена и 1,4-фенилена, каждый из которых необязательно замещен 1-2 заместителями R3.
17. Соединение по любому из пп. 14 - 16, где L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-.
18. Соединение по любому из пп. 14 - 17, где L2 выбран из группы, состоящей из -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2- и -CH2CH2CH2-.
19. Соединение по любому из пп. 14 - 18, где R1 выбран из группы, состоящей из галогена, цианогруппы, C1-3 алкила, C1-3 алкоксигруппы, C1-3 галогеналкила, C1-3 галогеналкоксигруппы и C3-5 циклоалкила.
20. Соединение по любому из пп. 14 - 18, где R1 выбран из группы, состоящей из хлора, метила, цианогруппы, этила, циклопропила, трифторметила и трифторметоксигруппы.
21. Соединение по любому из пп. 14 - 20, имеющее формулу, выбранную из группы, состоящей из:
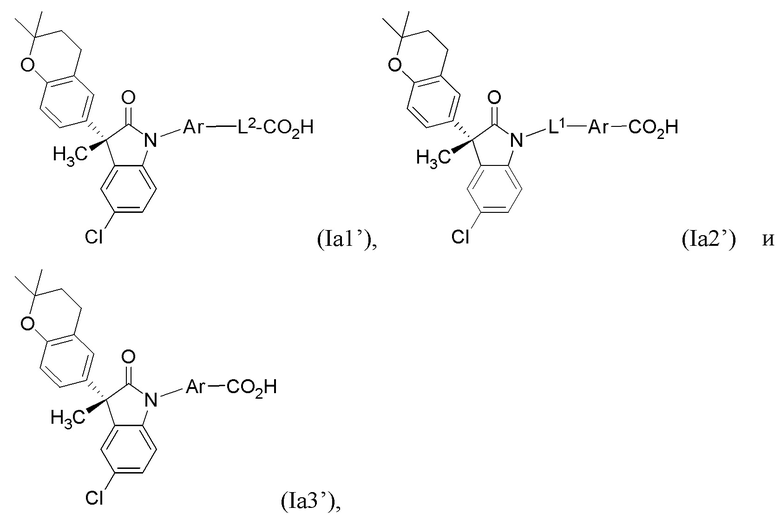
или его фармацевтически приемлемые соли, где указанное соединение практически не содержит других изомеров.
22. Соединение по п. 1, имеющее формулу
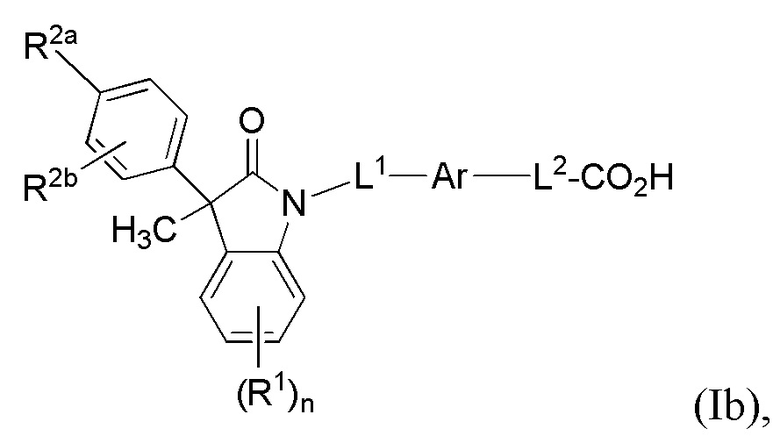
или его фармацевтически приемлемые соли.
23. Соединение по п. 22, где R2b представляет собой атом водорода.
24. Соединение по п. 22 или 23, где Ar выбран из группы, состоящей из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3.
25. Соединение по любому из пп. 22 - 24, где L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-.
26. Соединение по любому из пп. 22 - 25, где L2 выбран из группы, состоящей из одинарной связи, -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2- и -CH2CH2CH2-.
27. Соединение по любому из пп. 22 - 26, имеющее формулу, выбранную из группы, состоящей из:
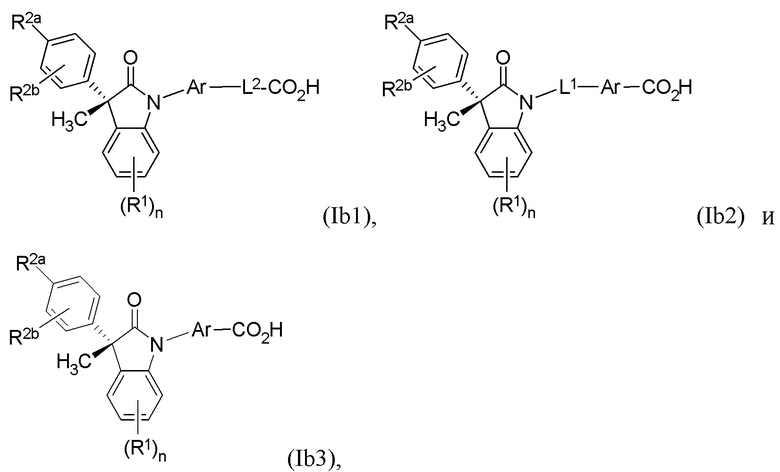
или его фармацевтически приемлемые соли, где указанное соединение практически не содержит других изомеров.
28. Соединение по п. 27, где Ar выбран из группы, состоящей из бензола, пиридина и хинолина, каждый из которых необязательно замещен 1-2 заместителями R3.
29. Соединение по п. 27 или 28, где Ar выбран из группы, состоящей из 1,3-фенилена и 1,4-фенилена, каждый из которых необязательно замещен 1-2 заместителями R3.
30. Соединение по любому из пп. 27 - 29, где R3 выбран из группы, состоящей из CH3, CH2CH3, CH2CH2CH3 и CH(CH3)2.
31. Соединение по любому из пп. 27 - 30, где L1 выбран из группы, состоящей из одинарной связи, -CH2- и -CH(CH3)-.
32. Соединение по любому из пп. 27 - 31, где L2 выбран из группы, состоящей из -O-CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2- и -CH2CH2CH2-.
33. Соединение по любому из пп. 27 - 32, где R1 выбран из группы, состоящей из галогена, цианогруппы, C1-3 алкила, C1-3 алкоксигруппы, C1-3 галогеналкила, C1-3 галогеналкоксигруппы и C3-5 циклоалкила.
34. Соединение по любому из пп. 27 - 32, где R1 выбран из группы, состоящей из хлора, метила, цианогруппы, этила, циклопропила, трифторметила и трифторметоксигруппы.
35. Соединение по п. 27, имеющее формулу, выбранную из группы, состоящей из:
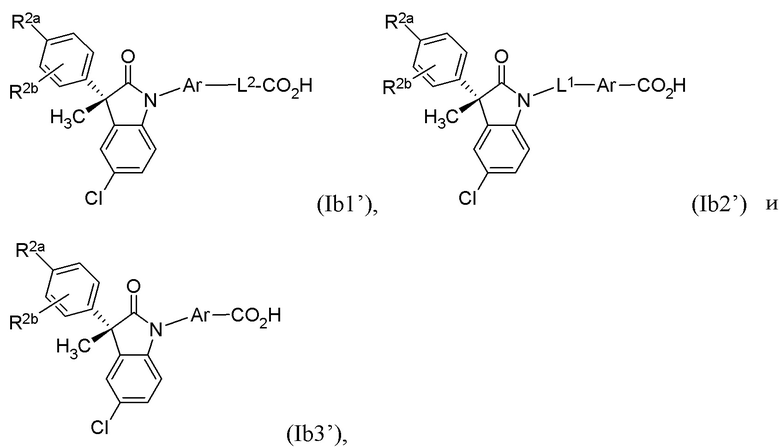
или его фармацевтически приемлемые соли, где указанное соединение практически не содержит других изомеров.
36. Соединение по п. 1 или его фармацевтически приемлемые соли, выбранное из группы, состоящей из:
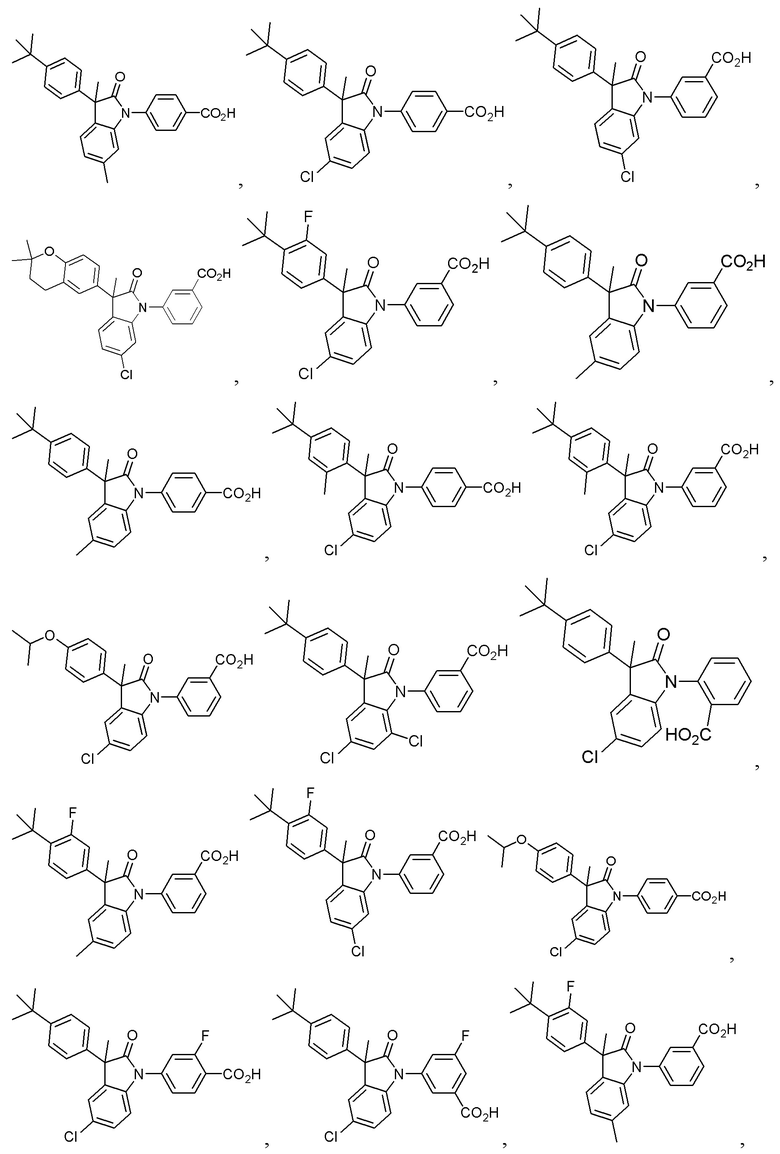

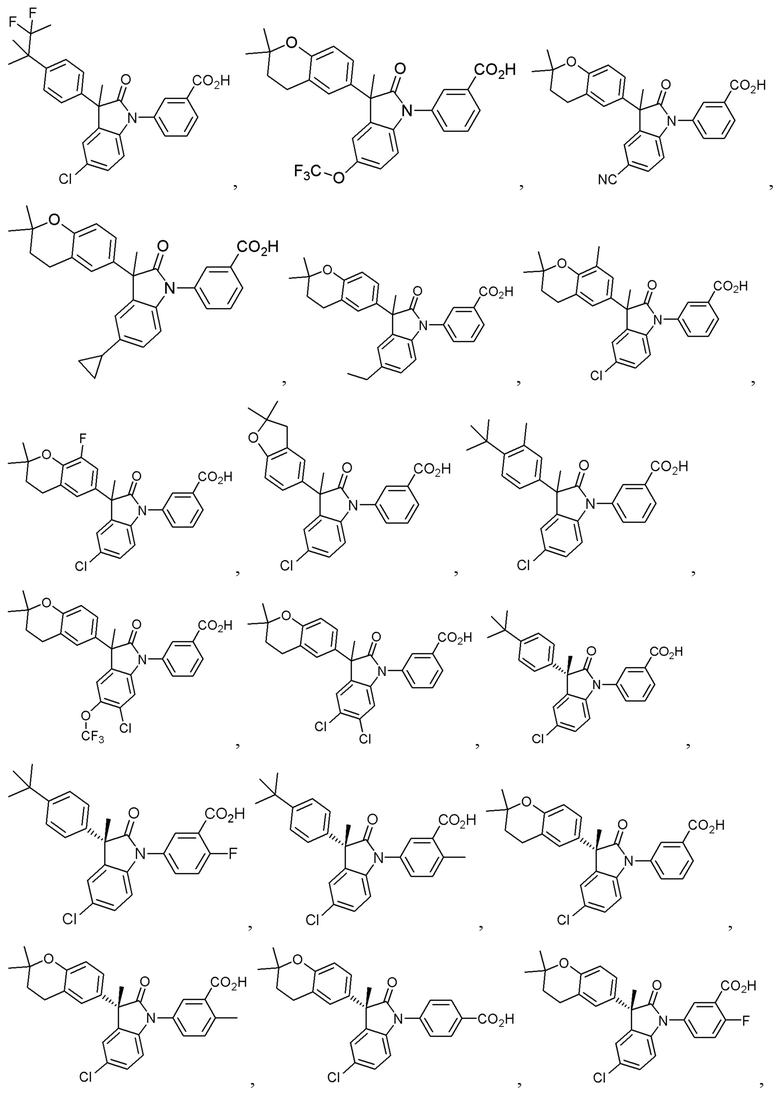
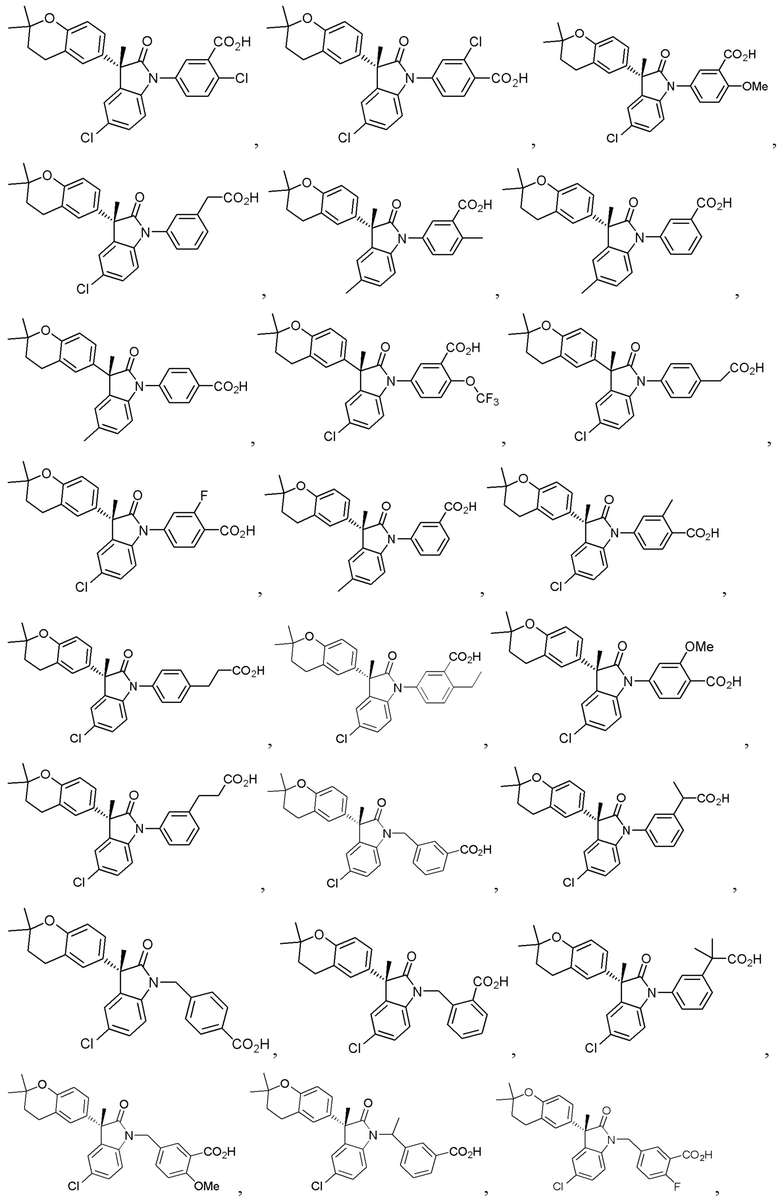
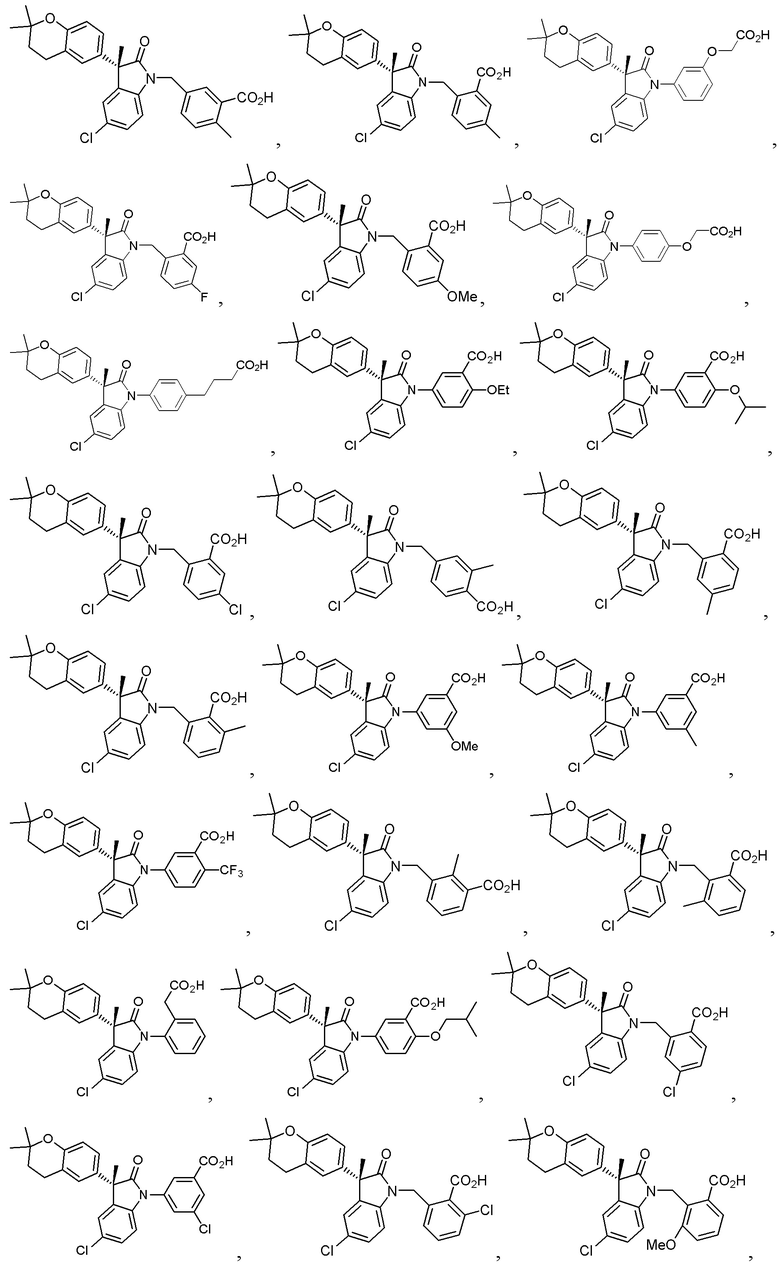
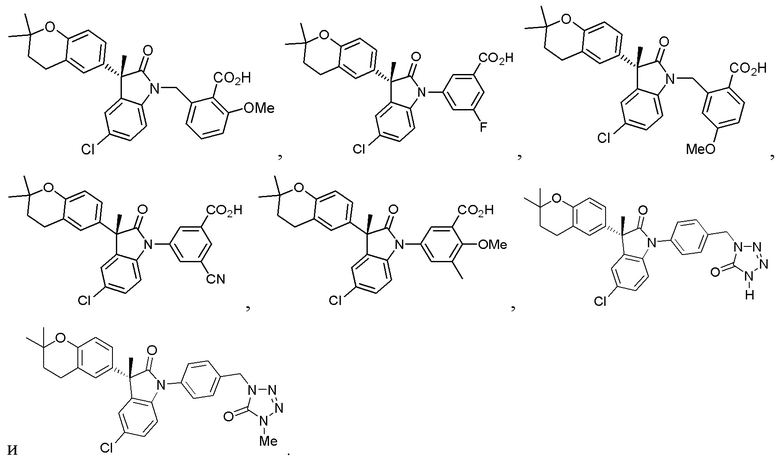
37. Соединение по п. 1 или его фармацевтически приемлемая соль, имеющее формулу
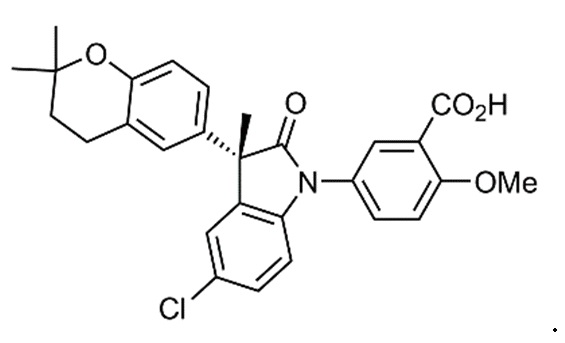
38. Соединение по п. 1 или его фармацевтически приемлемая соль, имеющее формулу
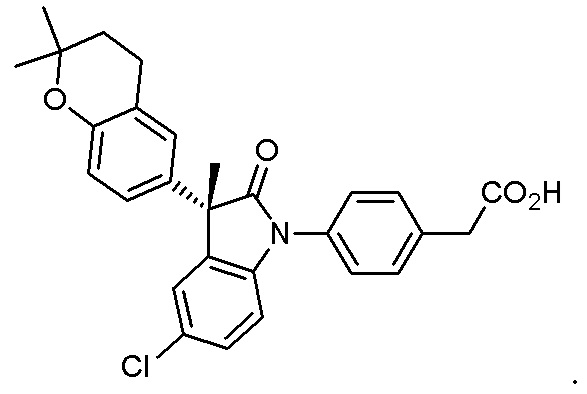
39. Соединение по п. 1 или его фармацевтически приемлемая соль, имеющее формулу
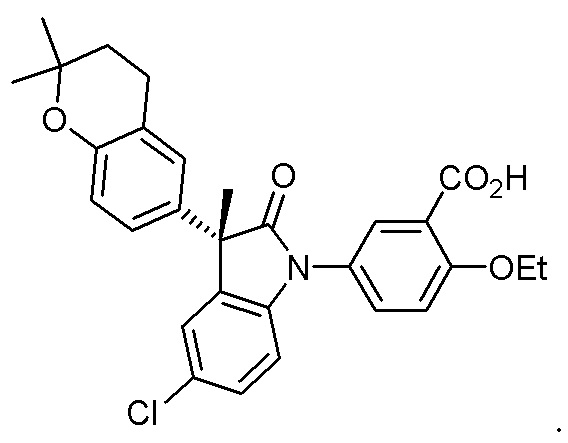
40. Фармацевтическая композиция для лечения CCR(9)-опосредованного заболевания или нарушения, содержащая фармацевтически приемлемое вспомогательное вещество и по меньшей мере одно соединение по любому из пп. 1 - 39 или его фармацевтически приемлемую соль.
41. Способ лечения CCR(9)-опосредованного заболевания или патологического состояния, выбранного из группы, состоящей из воспалительных заболеваний кишечника, болезни Крона и язвенного колита.
42. Способ по п. 41, где указанное заболевание или патологическое состояние выбрано из группы, состоящей из воспалительных заболеваний кишечника.
43. Способ по п. 41, где указанное заболевание или патологическое состояние выбрано из группы, состоящей из болезни Крона и язвенного колита.
| US 20130225580 A1, 29.08.2013 | |||
| US 20040171654 A1, 02.09.2004 | |||
| US 20130102604 A1, 25.04.2013 | |||
| US 20150291623 A1, 15.10.2015 | |||
| US 20070203184 A1, 30.08.2007 | |||
| WO 2008087278 A2, 24.07.2008 | |||
| ПРОИЗВОДНЫЕ N-СУЛЬФОНИЛ-2-ОКСО-ИНДОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИМЕЮЩАЯ АГЕНТ, АКТИВНЫЙ В ОТНОШЕНИИ РЕЦЕПТОРОВ ВАЗОПРЕССИНА И/ИЛИ ОЦИТОЦИНА | 1993 |
|
RU2135469C1 |

