Хемокины представляют собой семейство небольших (70-120 аминокислот), противовоспалительных цитокинов с мощным хемотактическим действием. Хемокины представляют собой хемотактические цитокины, которые выделяются широким спектром клеток для привлечения различных клеток, таких как моноциты, макрофаги, Т-клетки, эозинофилы, базофилы и нейтрофилы к местам воспаления (описано в Schall, Cytokine, 3, 165-183 (991) и Murphy, Rev. Immun., 12, 593-633 (1994)). Эти молекулы изначально были определены четырьмя сохраненными цистеинами и делятся на два подсемейства на основе расположения первой цистеиновой пары. В семействе СХС-хемокинов, которое включает IL-8, GROα, NAP-2 и IP-10, эти два цистеина разделены одной аминокислотой, в то время как в семействе СС-хемокинов, которое включает RANYES, MCP-1, MCP-2, MCP-3, MIP-1α, MIP-1β и эотаксин, эти два остатка являются соседними.
Хемокины секретируются широким спектром клеток и связываются со специфическими рецепторами, сопряженными с G-белком (GPCR) (описано в Horuk, Trends Pharm. Sci., 15, 159-165 (1994)), присутствующими в лейкоцитах и других клетках. Такие рецепторы хемокина образуют подсемейство GPCR, которое в настоящее время состоит из пятнадцати охарактеризованных членов и некоторого количества отдельных членов. В отличие от рецепторов для разнородных хемоаттрактантов, таких как С5а, fMLP, PAF и LTB4, рецепторы хемокина более селективно экспрессируются на подмножестве лейкоцитов. Таким образом, образование специфических хемокинов обеспечивается механизм для пополнения определенных подмножеств лейкоцитов.
При связывании со своими родственными лигандами, рецепторы хемокина преобразовывают внутриклеточный сигнал через связанный с ним трехмерный G белок, что приводит к быстрому увеличению концентрации внутриклеточного кальция. Существует, по крайней мере, семь рецепторов хемокина человека, которые связываются или реагируют на β-хемокины по следующей характерной схеме: CCR-1 (или "CKR-1" или "CC-CKR-1") [MIP-1α, MIP-1β, MCP-3, RANTES] (Ben-Barruch, et al., J. Biol. Chem., 270, 22123-22128 (1995); Beote, et al., Cell, 72, 415-425 (1993)); CCR-2A и CCR-2B (или "CKR-2A"/"CKR-2A" или "CC-CKR-2A"/"CC-CKR-2A") [MCP-1, MCP-2, MCP-3, MCP-4]; CCR-3 (или "CKR-3" или "CC-CKR-3") [Эотаксин, Эотаксин 2, RANTES, MCP-2, MCP-3] (Rollins, et al., Blood, 90, 908-928 (1997)); CCR-4 (или "CKR-4" или "CC-CKR-4") [MIP-1α, RANTES, MCP-1] (Rollins, et al., Blood, 90, 908-928 (1997)); CCR-5 (или "CKR-5" или "CC-CKR-5") [MIP-1α, RANTES, MIP-1β] (Sanson, et al., Biochemistry, 35, 3362-3367 (1996)); и антиген группы крови Даффи [RANTES, MCP-1] (Chaudhun, et al., J. Biol. Chem., 269, 7835-7838 (1994)). β-хемокины включают эотаксин, MIP («воспалительный белок макрофага»), МСР («белок хемоаттрактант моноцита») и RANTES («регулируемый при активации, экспрессированный и секретированный нормальными Т») среди прочих цитокинов.
Рецепторы хемокина, такие как CCR-1, CCR-2, CCR-2A, CCR-2B, CCR-3, CCR-4, CCR-5, CXCR-3, CXCR-4, считаются важными медиаторами воспалительных и иммунорегуляторных заболеваний и расстройств, включая астму, ринит и аллергические заболевания, а также аутоиммунных патологий, таких как ревматоидный артрит и атеросклероз. Считается, что люди, которые являются гомозиготными для делеции 32-основной пары в гене CCR-5, менее восприимчивы к ревматоидному артриту (Gomez, et al., Arthritis & Rheumatism, 42, 989-992 (1999)). Обзор роли эозинофилов в аллергическом воспалении представлен Kita, H., et al., в J. Exp. Med. 183, 2421-2426 (1996). Общий обзор роли хемокинов в аллергическом воспалении представлен Lugster, A.D., в New England J. Med., 338(7), 426-445 (1998). Подмножество хемокинов представляет собой мощные хемоаттрактанты для моноцитов и макрофагов. Наиболее охарактеризованными из них является МСР-1 (белок хемоаттрактант моноцита-1), первичным рецептором которого является CCR2. МСР-1 вырабатывается во многих типах клеток в ответ на воспалительную стимуляцию у различных видов, включая грызунов и человека, и стимулирует хемотаксис в моноцитах и подмножестве лимфоцитов. В частности, вырабатывание МСР-1 соотносится с инфильтрацией моноцитов и макрофагов в место воспаления. Делеция либо МСР-1, либо CCR2 гомогенной рекомбинацией у мышей дает значительное ослабление пополнения моноцитов в ответ на инъекцию тиогликолята и инфекцию Listeria monocytogenes (Lu et al., J. Exp. Med., 187, 601-608 (1998); Kurihara et al., J. Exp. Med., 186, 1757-1762 (1997); Boring et al., J. Clin. Invest., 100, 2552-2561 (1997); Kuziel et al., Proc. Natl. Acad. Sci., 12053-12058 (1997)). Более того, эти животные показывают пониженную инфильтрацию моноцитов в гранулематозные повреждения, вызванные инъекцией шистосомала или микобактериальных антигенов (Boring et al., J. Clin. Invest., 100, 2552-2561 (1997); Warmington et al., Am. J. Path., 154, 1407-1416 (1999)). Эти данные подтверждают, что вызванная МСР-1 активация CCR2 играет основную роль в пополнении моноцитов в местах воспаления, и что антагонизм этой активности даст значительное подавление иммунного ответа с получением терапевтической пользы при иммуновоспалительных и аутоиммунных заболеваниях. Следовательно, агенты, которые модулируют рецепторы хемокина, такие как рецептор CCR-2, будут полезны при таких расстройствах и заболеваниях. Кроме того, пополнение моноцитов в воспаленных повреждениях стенок сосудов является основным компонентом патогенеза и образования атерогенных бляшек. МСР-1 вырабатывается и секретируется эндотелиальными клетками и клетками интимальных гладких мышц после повреждения стенки сосуда при гиперхолестеринемических состояниях. Моноциты, пополняющиеся в месте повреждения, инфильтруются в стенку сосуда и дифференцируются в пенистые клетки в ответ на выделение МСР-1. Некоторые группы продемонстрировали, что размер повреждения аорты, содержание макрофагов и некроз ослаблены у МСР-1 -/- или CCR2-/- мышей, обратно скрещенных с АРО-Е -/-, LDL-R -/- или Apo B трансгенными мышами, получающими пищу с высоким уровнем жира (Boring et al., Nature, 394, 894-897 (1998); Gosling et al., J. Clin Invest., 103, 773-778 (1999)). Таким образом, антагонисты CCR2 могут ингибировать образование атеросклеротических повреждений и патологического развития посредством уменьшения пополнения моноцитов и дифференциации в стенку артерии.
Данное изобретение представляет соединения, которые являются модуляторами активности рецептора хемокина, и полезны для профилактики или лечения определенных воспалительных и иммунорегуляторных расстройств и заболеваний, аллергических заболеваний, атопических состояний, включая аллергический ринит, дерматит, конъюнктивит и астму, а также аутоиммунных патологий, таких как ревматоидный артрит и атеросклероз. Данное изобретение также представляет фармацевтические композиции, содержащие такие соединения, и применение данных соединений и композиций для профилактики и лечения таких заболеваний, в которые вовлечены рецепторы хемокина.
Данное изобретение относится к соединениям формулы I

где R3 является кислородом или отсутствует;
R8 выбирают из:
(а) водорода,
(b) С1-3алкила, который является незамещенным или замещен 1-6 атомами фтора,
(с) -О-С1-3алкила,
(d) фтора и
(е) гидрокси;
и их фармацевтически приемлемым солям и отдельным диастереомерам.
В одном из вариантов данного изобретения R3 отсутствует. В одном из вариантов данного изобретения R3 является водородом.
В данном изобретении предпочтительно, когда R8 выбирают из:
(a) водорода,
(b) трифторметила,
(c) метила,
(d) метокси,
(e) этокси,
(f) этила,
(g) фтора и
(h) гидрокси.
Примеры соединений в соответствии с данным изобретением включают те соединения, которые представлены в примерах и их фармацевтически приемлемые соли и отдельные диастереомеры.
Соединения в соответствии с данным изобретением имеют, по крайней мере, два асимметричных центра в положениях 1 и 3 циклопентильного кольца. Дополнительные асимметричные центры могут присутствовать в зависимости от природы различных заместителей в молекуле. Каждый такой асимметричный центр будет независимо давать два оптических изомера, и подразумевается, что все возможные оптические изомеры и диастереомеры в смесях и как чистые или частично очищенные соединения включены в объем данного изобретения. Независимый синтез диастереомеров и энантиомеров или их хроматографическое разделение могут быть проведены методами, известными в данной области техники, соответствующей модификацией методики, описанной ниже. Их абсолютная стереохимия может быть определена рентгеновской кристаллографией кристаллических продуктов или кристаллических промежуточных соединений, которые получают, при необходимости, с реагентом, содержащим асимметрический центр известной абсолютной конфигурации.
Как понятно специалистам в данной области техники, С1-3алкил включает группу, имеющую 1, 2 или 3 атома углерода в линейном или разветвленном расположении, так, что С1-3алкил включает метил, этил, н-пропил и изопропил.
Фраза «фармацевтически приемлемые» относится к соединениям, материалам, композициям и/или дозированным формам, которые, с точки зрения медицины, подходят для применения в контакте с тканями человека и животных без излишней токсичности, повреждения, аллергической реакции или других проблем или осложнений, и имеют разумное соотношение польза/риск. В данном описании термин «фармацевтически приемлемые соли» относится к производным, в которых исходное соединение модифицируют превращением в его кислотную или основную соль. Примеры фармацевтически приемлемых солей включают, но не ограничены ими, минеральные или органические кислотные соли основных остатков, таких как амины; щелочные или органические соли кислых остатков, таких как карбоновые кислоты; и подобные. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммониевые соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Например, такие обычные не токсичные соли включают соли, полученные из неорганических кислот, таких как хлористоводородная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и подобные; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфанилиновая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изетионовая и подобные.
Фармацевтически приемлемые соли в соответствии с данным изобретением могут быть получены из исходных соединений, которые содержат основную или кислотную группу, обычными химическими методами. Обычно такие соли могут быть получены взаимодействием свободной кислой или основной формы указанных соединений со стехиометрическим количеством соответствующего основания или кислоты в воде или органическом растворителе, или в смеси обоих; предпочтительно в неводной среде, такой как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил. Подходящие соли описаны, например, в Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, 1985, стр. 1418.
Определенные соединения в соответствии с данным изобретением включают соединения, которые выбирают из группы, включающей: соединения, указанные в заголовках примеров; и их фармацевтически приемлемые соли и отдельные диастереомеры.
Соединения в соответствии с данным изобретением полезны в способе модулирования активности рецептора хемокина у пациента, при необходимости такой модуляции, где способ включает введение эффективного количества соединения. Данное изобретение предназначено для применения следующих соединений в качестве модуляторов активности рецептора хемокина. В частности, эти соединения полезны в качестве модуляторов рецепторов хемокина, в частности, CCR-2.
Применение соединений в соответствии с данным изобретением в качестве модуляторов активности рецептора хемокина может быть продемонстрировано методом, известным в данной области техники, таким как исследования связывания хемокина, описанные Van Riper, et al., в J. Exp. Med., 177, 851-865 (1993), которые могут быть легко адаптированы для измерения связывания CCR-2.
Сродство рецептора в исследовании на связывание CCR-2 определяют измерением ингибирования 125I-MCP-1 к эндогенному рецептору CCR-2 в различных клетках, включая моноциты, клетки ТНР-1, или после гетерологической экспрессии клонированного рецептора в эукариотные клетки. Клетки суспендируют в связывающем буфере (50 мМ HEPES, рН 7,2, 5 мМ MgCl2, 1 мМ CaCl2 и 0,50% BSA) и добавляют к тестируемому соединению или ДМСО и 125I-MCP-1 при комнатной температуре в течение 1 часа для связывания. Затем клетки собирают на фильтры GBF, промывают 25 мМ HEPES буфером, содержащим 500 мМ NaCl, и рассчитывают клетки, связанные с 125I-MCP-1.
В хемотаксическом исследовании, хемотаксис проводят с применением РВМС, обедненного Т-клетками из цельной венозной крови или лейкофорезной крови, и очищают центрифугированием Фиколл-Гипак с последующим розеттингом с обработанными нейраминидазом эритроцитами овец. После выделения клетки промывают HBSS, содержащим 0,1 мг/мл BSA и суспендируют при 1×107 клеток/мл. Клетки метят в темноте флуоресцентом с 2 мкМ Calcien-AM (молекулярные пробы) в течение 30 минут при температуре 37°C. Меченые клетки дважды промывают и суспендируют при 5×106 клеток/мл в RPMI 1640 с L-глутамином (без красного фенола), содержащим 0,1 мг/мл BSA. МСР-1 (Petrotech) при 10 нг/мл разводят в той же среде или только среду добавляют на дно лунок (27 мкл). В верхнюю часть фильтра добавляют моноциты (150000 клеток) (30 мкл) с последующим 15 минутным предварительным инкубированием с ДМСО или с различными концентрациями тестируемого соединения. В равных концентрациях тестируемое соединение или ДМСО добавляют на дно лунок для предотвращения разбавления диффузией. После 60 мин инкубации при температуре 37°C, 5% СО2, фильтр удаляют и верхнюю часть промывают HBSS, содержащим 0,1 мг/мл BSA для удаления клеток, которые не перешли на фильтр. Спонтанную миграцию (хемокинез) определяют в отсутствие хемоаттрактанта.
В частности, соединения из представленных ниже примеров обладают активностью при связывании с CCR-2 рецептором в указанном выше исследовании, обычно с IC50 примерно ниже 1 мкМ. Такие результаты показывают активность, присущую соединениям при применении их в качестве модуляторов активности рецептора хемокина.
Рецепторы хемокина млекопитающих представляют собой мишень для вмешательства или стимулирования функции эозинофила и/или лимфоцитов у млекопитающих, таких как человек. Соединения, которые ингибируют или стимулируют функцию рецептора хемокина, особенно полезны для модулирования функции эозинофила и/или лимфоцита для терапевтических целей. Следовательно, соединения, которые ингибируют или стимулируют функцию рецептора хемокина, могут быть полезны для лечения, профилактики, улучшения, контроля или снижения риска широкого спектра воспалительных и иммунорегуляторных расстройств и заболеваний, аллергических заболеваний, атопических состояний, включая аллергический ринит, дерматит, конъюнктивит и астму, а также аутоиммунных патологий, таких как ревматоидный артрит и атеросклероз. Например, соединение мгновенного действия, которое ингибирует одну или более функций рецептора хемокина млекопитающего (например, рецептора хемокина человека), может вводиться для ингибирования (например, снижения или профилактики) воспаления. В результате ингибируется один или более воспалительных процессов, таких как эмиграция лейкоцитов, хемотаксис, экзоцитоз (например, ферментов, гистамина) или выделение медиатора воспаления.
В дополнение к приматам, таким как человек, метод в соответствии с данным изобретением может применяться для лечения различных других млекопитающих. Например, можно лечить млекопитающих, включающих, но не ограниченных ими, коров, овец, коз, лошадей, собак, кошек, морских свинок, крыс или других коровьих, овечьих, лошадиных, собачьих, кошачьих, крысиных или мышиных видов. Однако метод также может применяться к другим видам, таким как пернатые (например, куры).
Заболевания и состояния, связанные с воспалением и инфекцией, могут лечиться с применением соединений в соответствии с данным изобретением. В предпочтительном варианте заболеванием или состоянием является такое, в котором действие лимфоцитов ингибируется или стимулируется для модулирования воспалительной реакции.
Заболевания и состояния человека и других видов, которые могут лечиться ингибиторами функции рецептора хемокина, включают, но не ограничены ими: воспалительные или аллергические заболевания и состояния, включая респираторные аллергические заболевания, такие как астма, особенно бронхиальная астма, аллергический ринит, гиперчувствительность легких, гиперчувствительный пульмонит, эозинофильная пневмония (например, синдром Леффлера, хроническая эозинофильная пневмония), гиперчувствительность замедленного типа, промежуточная болезнь легких (ПБЛ) (например, идиопатический фиброз легких или ПБЛ, связанная с ревматоидным артритом, системная красная волчанка, анкилозирующий спондилит, системный склероз, синдром Съоргена, полимиозит или дерматомиозит); системная анафилаксия или реакция гиперчувствительности, аллергии к лекарственным средствам (например, к пенициллину, цефалоспоринам), аллергия к укусам насекомых; аутоиммунные заболевания, такие как ревматоидный артрит, псориазный артрит, рассеянный склероз, системная красная волчанка, бульбоспинальный паралич, детский начальный диабет; гломерулонефрит, аутоиммунный тироидит, болезнь Беккета; отторжение трансплантата (например, при трансплантации), включая отторжение аллотрансплантата или реакцию «трансплантат против хозяина»; воспалительные болезни кишечника, такие как болезнь Крона и язвенный колит; спондилоартропатии; склеродерма; псориаз (включая псориаз, медиированный Т-клетками) и воспалительные кожные болезни, такие как дерматит, экзема, атопический дерматит, аллергический контактный дерматит, крапивница; васкулит (например, некротизированный, кожный и гиперчувствительный васкулит); эозинофильный миозит, эозинофильный фасцит; рак кожи или органов с инфильтрацией лейкоцитов. Другие заболевания или состояния, при которых могут лечиться нежелательные ингибируемые воспалительные реакции, включают, но не ограничены ими, реперфузионные повреждения, атеросклероз, определенные гематологические злокачественные образования, вызванная цитокином токсическая реакция (например, септический шок, эндотоксический шок), полимиозит, дерматомиозит.
Заболевания или состояния людей или других видов, которые могут лечиться модуляторами действия рецептора хемокина, включают, но не ограничены ими: иммунодепрессия, такая как у пациентов с синдромом иммунодефицита, таким как СПИД или другие вирусные инфекции, у пациентов, принимающих лучевое лечение, химиотерапию, терапию для аутоиммунных заболеваний или лекарственную терапию (например, терапию кортикостероидами), которые вызывают иммунодепрессию; иммунодепрессия вследствие врожденного дефицита функции рецептора или других причин; и инфекционные заболевания, такие как паразитические заболевания, включая, но, не ограничиваясь ими, гельминтные инфекции, такие как нематоды (круглые черви), (Trichuriasis, Enterobiasis, Ascariasis, Hookworm, Strongylodiasis, Trichinosis, Filariasis), трематоды (плоские черви) (Schistosomiasis, Clonorchiasis), цестоды (ленточные черви) (Echinococcosis, Taeniasis saginata, Cysticercosis), висцеральные черви, мигрени от висцеральных личинок (например, Toxocara), эозинофильные гастроэнтериты (например, Anisaki sp., Phocanema sp.) и мигрени от кожных личинок (Ancylostona brazilience, Ancylostoma caninum). Кроме того, лечение указанных выше воспалительных, аллергических и аутоиммунных заболеваний может проводиться для промоторов функции рецептора хемокина, если они обеспечивают доставку достаточного количества соединения для прекращения экспрессии рецептора в клетках через индуцирование интернализации рецептора хемокина, или доставку соединения способом, который придает неправильное направление миграции клеток.
Соединения в соответствии с данным изобретением полезны для лечения, профилактики, улучшения, контроля или снижения риска широкого спектра воспалительных и иммунорегуляторных расстройств и заболеваний, аллергических состояний, атопических состояний, а также аутоиммунных патологий. В определенном варианте, данное изобретение относится к применению соединений в соответствии с данным изобретением для лечения, профилактики, улучшения, контроля или снижения риска аутоиммунных заболеваний, таких как ревматоидный артрит или псориазный артрит.
В другом варианте, данное изобретение может применяться для оценки предполагаемых специфических агонистов или антагонистов рецепторов хемокина, включая CCR-2. Следовательно, данное изобретение относится к применению таких соединений для получения и проведения скриннинговых исследований соединений, которые модулируют активность рецепторов хемокина. Например, соединения в соответствии с данным изобретением полезны для выделения мутантов рецептора, которые являются превосходными инструментами для скрининга более мощных соединений. Более того, соединения в соответствии с данным изобретением полезны для установления или определения места связывания других соединений с рецепторами хемокина, например, посредством конкурентного ингибирования. Соединения в соответствии с данным изобретением также полезны для оценки предполагаемых специфических модуляторов рецепторов хемокина, включая CCR-2. Как признано в данной области техники, всесторонней оценке специфических агонистов и антагонистов указанных выше рецепторов хемокина препятствует отсутствие доступности не-пептидных (метаболически устойчивых) соединений с высоким сродством связывания к таким рецепторам. Таким образом, соединения в соответствии с данным изобретением являются коммерческими продуктами, которые могут продаваться в этих целях.
Данное изобретение также относится к способу получения медикамента для модулирования активности рецептора хемокина у человека и животных, включающему объединение соединения в соответствии с данным изобретением с фармацевтическим носителем или разбавителем.
Данное изобретение также относится к применению данных соединений для лечения, профилактики, улучшения, контроля или снижения риска инфекции ретровирусом, в частности, вирусом герпеса или вирусом иммунодефицита человека (ВИЧ), или лечения и задержки наступления последующих патологических состояний, таких как СПИД. Лечение СПИД или профилактика или лечение инфекции ВИЧ определено как включающее, но не ограниченное ими, лечение широкого спектра состояний ВИЧ-инфекций: СПИД, ССК (СПИД-связанный комплекс), симптоматический и бессимптомный, и острый или потенциальный контакт с ВИЧ. Например, соединения в соответствии с данным изобретением полезны для лечения инфекции ВИЧ после предполагаемого контакта с ВИЧ через, например, переливание крови, трансплантации органов, обмена жидкостями тела, укусов, случайных уколов иглой или контакта с кровью пациента во время хирургических операций.
В предпочтительном варианте данного изобретения соединение в соответствии с данным изобретением может применяться в способе ингибирования связывания хемокина с рецептором хемокина, таким как CCR-2 клетки мишени, который включает взаимодействие клетки мишени с количеством соединения, которое является эффективными для ингибирования связывания хемокина с рецептором хемокина.
Объектом лечения в указанном выше способе является млекопитающее, предпочтительно человек, мужского или женского пола, для которого желательно модулирование активности рецептора хемокина. «Модулирование» в данном контексте подразумевает антагонизм, агонизм, частичный антагонизм, обратный агонизм и/или частичный агонизм. В предпочтительном варианте данного изобретения модулирование относится к антагонизму в отношении активности рецептора хемокина. Термин «терапевтически эффективное количество» означает такое количество соединения в соответствии с данным изобретением, которое вызывает биологическую или медицинскую реакцию ткани, системы, животного или человека, которая ожидается исследователем, ветеринаром, врачом или другим клиницистом.
Термин «композиция», используемый в данном описании, относится к продукту, содержащему определенные ингредиенты в определенных количествах, а также к любому продукту, который получают, прямо или опосредованно, из сочетания определенных ингредиентов в определенных количествах. Под «фармацевтически приемлемым» понимают носитель, разбавитель или эксципиент, который должен быть совместим с другими ингредиентами препаративной формы, и не наносить вреда пациенту. Под терминами «введение» и/или «вводимое» соединение понимают обеспечение соединением в соответствии с данным изобретением пациента при необходимости лечения. В данном случае термин «лечение» означает как лечение, так и профилактику или профилактическую терапию указанных выше состояний.
Комбинированная терапия для модулирования активности рецептора хемокина для лечения, профилактики, улучшения, контроля или снижения риска воспалительных и иммунорегуляторных расстройств и заболеваний, включая астму и аллергические болезни, а также аутоиммуные патологии, такие как ревматоидный артрит и атеросклероз, и патологии, указанные выше, иллюстрируется сочетанием соединений в соответствии с данным изобретением и других соединений, которые известны для таких целей.
Например, при лечении, профилактике, улучшении, контроле или снижении риска воспаления, соединение данного изобретения может применяться в сочетании с противовоспалительными или аналгезирующими агентами, такими как агонист опиата, ингибитор липоксигеназы, такой как ингибитор 5-липоксигеназы, ингибитор циклооксигеназы, такой как ингибитор циклооксигеназы-2, ингибитор интерлейкина, такой как ингибитор интерлейкина-1, антагонист NMDA, ингибитор оксида азота или ингибитор синтеза оксида азота, нестероидное противовоспалительное средство или цитокин-подавляющее противовоспалительное средство, например, с соединением, таким как ацетаминофен, аспирин, кодеин, эмбрел, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацетин, пироксикам, стероидный аналгетик, суфентанил, санлиндак, тенидап и подобные. Также, соединения в соответствии с данным изобретением могут вводиться с обезболивающими; потенцирующими средствами, такими как кофеин, антагонистом Н2, симетиконом, гидроксидом алюминия или магния; противозастойным средством, таким как фенилефрин, фенилпропаноламин, псевдофедрин, оксиметазолин, эфинефрин, нафазолин, ксилометанолин, пропилгексендрин или лево-дезокси-эфедрин; средством против кашля, таким как кодеин, гидрокодон, карамифен, карбетапентан или декстраметофан; мочегонным; и седативным или неседативным антигистамином.
Также соединения в соответствии с данным изобретением могут применяться в сочетании с другими лекарственными средствами, которые применяются для лечения/профилактики/подавления или уменьшения интенсивности симптомов заболеваний или состояний, для которых полезны соединения в соответствии с данным изобретением. Такие другие лекарственные средства могут вводиться способом и в количестве, применяемом обычно, одновременно или последовательно с соединением в соответствии с данным изобретением. Если соединение в соответствии с данным изобретением применяют одновременно с одним или несколькими лекарственными средствами, предпочтительно применять фармацевтическую композицию, содержащую такие другие лекарственные средства в дополнение к соединению в соответствии с данным изобретением. Следовательно, фармацевтические композиции в соответствии с данным изобретением представляют собой такие композиции, которые также содержат один или несколько других активных ингредиентов, в дополнение к соединению в соответствии с данным изобретением.
Примеры других активных ингредиентов, которые могут объединяться с соединением в соответствии с данным изобретением, и вводиться отдельно или в той же фармацевтической композиции, включают, но не ограничены ими: (а) антагонисты VLA-4, такие как описано в US 5510332, WO 95/15973, WO 96/01644, WO 96/06108, WO 96/20216, WO 96/22966, WO 96/31206, WO 96/40781, WO 97/03094, WO 97/02289, WO 98/42656, WO 98/53814, WO 98/53817, WO 98/53818, WO 98/54207 и WO 98/58902, (b) стероиды, такие как беклометазон, метилпреднизолон, бета-метазон, преднизолон, дексаметазон и гидрокортизон; (с) иммунодепрессанты, такие как циклоспорин, такролимус, рапамицин и другие иммунодепрессанты типа FK-506; (d) антигистамины (антагонисты Н1-гистамина), такие как бромфенирамин, хлорфенирамин, дексхлорфенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипелленамин, гидроксизин, метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин, фенирамин, пириламин, астемизол, терфенадин, лоратадин, дезлоратадин, цетиризин, фексофенадин, дезкарбоэтоксилоратадин и подобные; (е) нестероидные противоастматические средства, такие как β2-агонисты (тербуталин, метапротеренол, фенотерол, изоэтарин, альбутерол, битолтерол и пирбутерол), теофиллин, кромолин натрия, атропин, ипратропия бромид, антагонисты лейкотриена (зафирлукаст, монтелукаст, пранлукаст, иралукаст, побилукаст, SKB-106, 203), ингибиторы биосинтеза лейкотриена (зилеутон, BAY-1005); (f) нестероидные противовоспалительные средства (NSAID), такие как производные пропионовой кислоты (альминопрофен, беноксапрофен, буклоксовая кислота, капрофен, фенбуфен, фенопрофен, флупрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен, пранопрофен, супрофен, тиарофеновая кислота и тиоксапрофен), производные уксусной кислоты (индометацин, ацеметацин, алклофенак, клиданак, диклофенак, фенклофенак, фенклозовая кислота, фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, зулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенаминовой кислоты (флуфенаминовая кислота, меклофенаминовая кислота, мефенаминовая кислота, нифлуминовая кислота и толфенаминовая кислота), производные бифенилкарбоновой кислоты (дифлунизал и флуфенизал), оксикамы (изоксикам, пироксикам, судоксикам и теноксикам), салицилаты (ацетилсалициловая кислота, сульфазалазин) и пиразолоны (апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (g) ингибиторы циклооксигеназы-2 (СОХ-2); (h) ингибиторы фосфодиэстеразы типа IV (PDE-IV); (i) другие антагонисты рецепторов хемокина, особенно CCR-1, CCR-2, CCR-3, CXCR-3 и CCR-5; (j) агенты, понижающие холестерин, такие как ингибиторы HMG-CoA редуктазы (ловастатин, симвастатин и правастатин, флувастатин, аторвастатин, розувастатин и другие статины), секвестраты (холестирамин и колестипол), ингибиторы абсорбции холестерина (эзитимиб), производные никотиновой и фенофибровой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (k) противодиабетические агенты, такие как инсулин, сульфонилмочевины, бигуаниды (метформин), ингибиторы α-глюкозидазы (акарбоза) и глитазоны (троглитазон и пиоглитазон); (l) препараты интерферона-бета (интерферон-бета-1α, интерферон-бета-1β); (m) другие соединения, такие как 5-аминосалициловая кислота и ее пролекарства, антиметаболиты, такие как азатиоприн и 6-меркаптопурин и цитотоксические химиотерапевтические агенты для лечения раковых заболеваний.
Массовое соотношение соединения в соответствии с данным изобретением ко второму активному ингредиенту может варьироваться и зависит от эффективной дозы каждого ингредиента. В общем, применяется эффективная доза каждого ингредиента. Таким образом, например, если соединение в соответствии с данным изобретением объединяют с NSAID, массовое соотношение соединения в соответствии с данным изобретением к NSAID обычно варьируется от около 1000:1 до около 1:1000, предпочтительно от около 200:1 до 1:200. Сочетания соединения в соответствии с данным изобретением и других активных ингредиентов обычно также находится в указанных выше пределах, но в каждом случае должна применяться эффективная доза каждого ингредиента. В таких комбинациях соединение в соответствии с данным изобретением и другой активный ингредиент могут вводиться отдельно или в сочетании. Кроме того, введение одного элемента может осуществляться перед, во время или после введения другого агента (агентов).
Соединения в соответствии с данным изобретением могут вводиться пероральным, парентеральным путями (например, внутримышечным, внутрибрюшинным, внутривенным, интрецеребровентрикулярным путями, интрацистернальными инъекциями или инфузиями, подкожными инъекциями или имплантатами), ингаляцией, интраназальным, ректальным, вагинальным, подъязычным или местными путями введения и могут быть сформулированы, по одному или вместе, в подходящие стандартные лекарственные формы, содержащие обычные, нетоксичные, фармацевтически приемлемые носители, адъюванты и наполнители, подходящие для каждого пути ведения. Кроме лечения теплокровных животных, таких как мыши, крысы, лошади, скот, овцы, собаки, кошки, обезьяны и т.д., соединения в соответствии с данным изобретением являются эффектными для лечения людей.
Фармацевтические композиции для введения соединений в соответствии с данным изобретением могут быть в удобных стандартных лекарственных формах и могут быть получены любым методом, хорошо известным в области фармацевтики. Все методы включают стадию объединения активного ингредиента с носителем, который состоит из одного или нескольких дополнительных ингредиентов. В общем, фармацевтические композиции получают однородным и тщательным соединением активного ингредиента с жидким носителем или тонкоизмельченным твердым носителем, или обоими, и затем, при необходимости, формованием продукта в желательную лекарственную форму. В фармацевтической композиции активное соединение содержится в количестве, достаточном для получения желаемого эффекта в имеющемся процессе или состоянии заболевания. В данном контексте термин "композиция" относится к продукту, содержащему определенные ингредиенты в определенных количествах, а также к любому продукту, который получают прямо или опосредованно, из сочетания определенных ингредиентов в определенных количествах.
Фармацевтические композиции, содержащие активный ингредиент, могут быть в форме, подходящей для перорального применения, например, в виде таблеток, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, жестких или мягких капсул, или сиропов или эликсиров. Композиции, предназначенные для перорального применения, могут быть получены любым методом, известным в области получения фармацевтических композиций, и такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из подсластителей, вкусовых агентов, красителей и консервантов, для получения фармацевтически превосходных и вкусных композиций. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые подходят для получения таблеток. Эти эксципиенты могут быть, например, инертными разбавителями, такими как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующими и разрыхляющими агентами, например, кукурузным крахмалом или альгиновой кислотой; связующими агентами, например, крахмалом, желатином или акацией, и смазывающими агентами, например, стеаратом магния, стеариновой кислотой или тальком. Таблетки могут не иметь оболочки или могут быть покрыты известными методами для того, чтобы отложить разложение и абсорбцию в желудочно-кишечном тракте и, тем самым, обеспечить пролонгированное действие в течение более длительного периода. Например, в качестве замедляющего вещества может применяться такое как глицерилмоностеарат или глицерилдистеарат. Они также могут быть покрыты с применением методик, описанных в патентах США 4256108, 4166452 и 4265874, с получением осмотических терапевтических таблеток для контролируемого высвобождения.
Композиции для перорального применения могут быть представлены в твердых желатиновых капсулах, в которых активные ингредиенты смешаны с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в мягких желатиновых капсулах, в которых активные ингредиенты смешаны с водной или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом. Водные суспензии содержат активные материалы в смеси с эксципиентом, подходящими для получения водных суспензий. Такие эксципиенты включают суспендирующие агенты, например, натрийкарбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую смолу и аравийскую камедь; диспергирующие или смачивающие агенты представляют собой природные фосфатиды, например, лецитин или продукты конденсации алкиленоксида и жирных кислот, например, полиоксиэтиленстеарат, или продукты конденсации этиленоксида и длинноцепных алифатических спиртов, например, гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида и частичных эфиров, полученых из жирных кислот и гекситола, такие как полиоксиэтиленсорбитолмоноолеат, или продукты конденсации этиленоксида и частичных эфиров, полученных из жирных кислот и ангидридов гекситола, например, полиэтиленсорбитанмоноолеат. Водные суспензии также могут содержать один или более консервант, например, этил или н-пропил, н-гидроксибензоат, один или более краситель, один или более вкусовой агент, один или более подсластитель, такой как сахароза или сахарин. Масляные суспензии могут быть получены суспендированием активного ингредиента в растительном масле, например, арахисовом масле, оливковом масле, конопляном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загущающий агент, например, пчелиный воск, твердый парафин или цетиловый спирт. Для получения приятных пероральных композиций могут быть добавлены подсластители, такие, как описано выше, и вкусовые агенты. Эти композиции могут сохраняться с помощью добавления антиокислителя, такого как аскорбиновая кислота. Диспергируемые порошки и гранулы, подходящие для получения водной суспензии добавлением воды, содержат активный ингредиент в смеси с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или более консервантами. Подходящие диспергирующие или увлажняющие агенты и суспендирующие агенты представлены теми, которые указаны выше. Также могут присутствовать дополнительные наполнители, такие как подсластители, вкусовые агенты и красители. Фармацевтические композиции в соответствии с данным изобретением также могут быть в виде эмульсий масло в воде. Масляная фаза может представлять собой растительное масло, например, оливковое масло или арахисовое масло, или минеральное масло, например, жидкий парафин или их смесь. Подходящие эмульгирующие агенты могут быть природными смолами, например, аравийской камедью или трагакантом, природными фосфатидами, например, соевыми бобами, лецитином и сложными эфирами или частичными эфирами, полученными из жирных кислот и ангидридов гекситола, например, моноолеатом сорбитана, и продуктами конденсации указанных частичных эфиров и этиленоксида, например, полиоксиэтиленсорбитанмоноолеатом. Эмульсии также могут содержать подсластители и вкусовые агенты, например, глицерин, пропиленгликоль, сорбит или сахарозу. Такие композиции также могут содержать болеутоляющие средства, консерванты и вкусовые агенты и красители.
Фармацевтические композиции могут быть в виде стерильных водных или масляных суспензий для инъекций. Эти суспензии могут быть получены в соответствии с методами, известными в данной области, с применением подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, которые указаны выше. Стерильные лекарственные формы для инъекций также могут быть стерильными растворами или суспензиями для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в растворе 1,3-бутандиола. Среди приемлемых носителей и растворителей, которые могут применяться, можно отметить воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды могут применяться стерильные, жирные масла. Для этой цели может применяться любое мягкое жирное масло, включая синтетические моно- или диглицериды. Кроме того, для получения форм для инъекций могут применяться жирные кислоты, такие как олеиновая кислота. Соединения в соответствии с данным изобретением также могут вводиться в виде суппозиториев для ректального введения лекарственного средства. Такие композиции могут быть получены смешиванием лекарственного средства с подходящим не раздражающим эксципиентом, который является твердым при обычных температурах, но жидким при ректальной температуре и, следовательно, плавится в прямой кишке для высвобождения лекарственного средства. Такими материалами являются кокосовое масло и полиэтиленгликоли. Для местного применения используют кремы, мази, желе, растворы или суспензии, и т.д., содержащие соединения в соответствии с данным изобретением. (Для целей данного изобретения композиции для местного применения включают полоскания для рта и полоскания для горла).
Фармацевтические композиции и способ в соответствии с данным изобретением могут также включать другие терапевтически активные соединения, как указано выше, которые обычно применяют для лечения указанных патологических состояний. При лечении, профилактике, улучшении, контроле и снижении риска состояний, которые требуют модулирования рецептора хемокина, подходящая дозировка обычно составляет от около 0,01 до 500 мг на килограмма массы тела пациента в день и может вводиться одной или несколькими дозами. Предпочтительно, уровень дозирования составляет от около 0,1 до 250 мг/кг в день, более предпочтительно от около 0,5 до около 100 мг/кг в день. Подходящие дозы составляют от около 0,01 до 250 мг/кг в день, от около 0,05 до 100 мг/кг в день, от около 0,1 до 50 мг/кг в день. В этом интервале доза может составлять 0,05-0,5, 0,5-5 или 5-50 мг/кг в день. Для перорального введения композиции предпочтительно имеют форму таблеток, содержащих 1,0-1000 миллиграмм активного ингредиента, предпочтительно 2,0-500, более предпочтительно 3,0-200, особенно 1, 5, 10, 15, 20, 25, 30, 50, 75, 100, 125, 150, 175, 200, 250, 300, 400, 500, 600, 750, 800, 900 и 1000 миллиграмм активного ингредиента для доведения дозы в зависимости от подвергаемого лечению пациента. Соединения могут вводиться в режиме от 1 до 4 раз в день, предпочтительно один или два раза в день.
Должно быть понятно, что определенный уровень дозирования и частота дозирования для любого конкретного пациента может варьироваться и зависит от множества факторов, включая активность определенного применяемого соединения, метаболическую стойкость и продолжительность действия данного соединения, возраста, массы тела, общего состояния здоровья, пола, диеты, способа и времени введения, скорости высвобождения, комбинации лекарственного средства, тяжести определенного состояния и принимаемой пациенту терапии.
Несколько способов получения соединения в соответствии с данным изобретением проиллюстрированы в представленных ниже примерах. Соединения в соответствии с данным изобретением могут быть получены модификацией методик, описанных в примерах. Исходные вещества получают известными методами или как показано в примерах. Представленные ниже примеры даны для иллюстрации и не ограничивают раскрываемое изобретение. Ниже представлены примеры методов получения соединений, используемых в представленных ниже примерах, или которые могут быть заместителями соединений, представленных в примерах, которые могут не быть коммерчески доступными.
Концентрирование растворов обычно осуществляют на роторном испарителе при пониженном давлении. Флэш-хроматографию проводят на силикагеле (230-400 меш). ЖХСД - жидкостную хроматографию среднего давления, проводят в неподвижной фазе силикагеля, если не указано иначе. ЯМР спектр получают в растворе CDCl3, если не указано иначе. Константы взаимодействия (J) даны в герцах (Гц). Аббревиатуры: диэтиловый эфир (эфир), триэтиламин (ТЭА), N,N-диизопропилэтиламин (ДИЭА), насыщенный водный (нас.), комнатная температура (кт), часы (ч), минуты (мин).
В некоторых случаях порядок проведения представленных ниже реакций может быть изменен, чтобы способствовать реакции или избежать нежелательных продуктов реакции. Примеры представлены только для иллюстрации и не ограничивают раскрываемое изобретение.
Ниже представлены примеры методик получения соединений, используемых в представленных ниже примерах, или которые могут быть заместителями соединений, представленных в примерах, которые могут не быть коммерчески доступными.
Промежуточное соединение 1

Промежуточное соединение 1 получают по методике, описанной в J. Am. Chem. Soc., 1991, 113, 2079-2089.
Промежуточное соединение 2

К раствору тетрагидро-4Н-пиран-4-она (5,0 г, 50 ммоль) и гексаметилфосфорамида (8,70 мл) в тетрагидрофуране (150 мл) медленно добавляют раствор диизопропиламида лития (31,25 мл, 3М раствор) в 125 мл тетрагидрофурана при температуре -78°C. Реакционную смесь перемешивают в течение 5 мин и затем добавляют этилиодид (16,0 мл, 200 ммоль). Смесь постепенно нагревают до температуры 0°C в течение более 2 ч. Реакционную смесь гасят насыщенным раствором NH4Cl и затем экстрагируют простым эфиром (4×100 мл). Эфирный слой промывают насыщенным раствором соли, сушат (безводный сульфат магния), концентрируют и очищают флэш-хроматографией на колонке, элюируя смесью гексан/этилацетат (4:1) с получением промежуточного соединения 2 (1,20 г, 20%).
Промежуточное соединение 3

Стадия А

К смеси 5,6-дигидро-4-метокси-2Н-пирана (10,0 г, 87,5 ммоль) в метаноле (200 мл) при температуре 0°C по каплям добавляют раствор 3-хлорпероксибензойной кислоты (30,2 г, 175 ммоль) в метаноле (50 мл) через дополнительную воронку. Полученный раствор перемешивают в течение 5 ч, давая ему нагреваться до комнатной температуры. Метанол удаляют при пониженном давлении с получением белого твердого вещества. Вещество растворяют в 500 мл дихлорметана и охлаждают до температуры 0°C. К энергично перемешиваемой смеси порциями добавляют избыток твердого гидроксида кальция (50-60 г). После перемешивания в течение еще 30 мин смесь фильтруют через слой целита и фильтрат выпаривают при пониженном давлении с получением 11,62 г (82%) желаемого продукта в виде прозрачного масла.
1H ЯМР (500 МГц, CDCl3) δ 3,88-3,80 (м, 2H), 3,73-3,68 (м, 2H), 3,54-3,48 (м, 1H), 3,28 (с, 3H), 3,27 (с, 3H), 2,00-1,93 (м, 1H), 1,82-1,77 (м, 1H).
Стадия В

К охлажденному (0°C) раствору продукта со стадии А, промежуточного соединения 3 (9,40 г, 58,0 ммоль) в тетрагидрофуране (200 мл) в атмосфере азота медленно добавляют NaH (2,32 г, 58,0 ммоль) и полученную суспензию перемешивают в течение 1 ч при температуре 0°C. Затем к суспензии через шприц добавляют иодметан (7,22 мл, 116 ммоль) и полученную смесь перемешивают в течение ночи, давая ей нагреваться до комнатной температуры. Реакцию гасят насыщенным раствором хлорида аммония (200 мл) и органический слой затем удаляют, используя делительную воронку. Водный слой экстрагируют простым эфиром (3×150 мл) и все органические слои объединяют, сушат над безводным сульфатом натрия, фильтруют и выпаривают в вакууме. Очистку проводят флэш-хроматографией на колонке, используя градиентное элюирование смесью 10-60% простой эфир/гексан с получением 8,46 г (83%) желаемого продукта в виде прозрачного масла.
1H ЯМР (500 МГц, CDCl3) δ 3,98 (дд, J=2,5, 12,4 Гц, 1H), 3,77 (ддд, J=3,5, 7,1, 10,8 Гц, 1H), 3,57 (дд, J=1,4, 12,4 Гц, 1H), 3,50 (дд, J=2,5, 11,7 Гц, 1H), 3,46 (с, 3H), 3,25 (с, 3H), 3,22 (с, 3H), 3,22-3,20 (м, 1H), 1,96 (ддд, J=4,7, 11,8, 16,5 Гц, 1H), 1,75 (уш. дд,J=1,7, 14,2 Гц, 1H).
Стадия С

Раствор продукта со стадии В, промежуточного соединения 3 (3,0 г, 17,04 ммоль) в смеси тетрагидрофуран/вода (60 мл/10 мл) обрабатывают концентрированной хлористоводородной кислотой (6 мл) и полученный раствор перемешивают при комнатной температуре в течение 1 ч. Смесь концентрируют в вакууме для удаления тетрагидрофурана и затем водный слой экстрагируют простым эфиром (6×50 мл). Органические слои объединяют, сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении с получением промежуточного соединения 24 (1,75 г, 79%) в виде прозрачного масла.
1H ЯМР (500 МГц, CDCl3) δ 4,23 (ддд, J=1,2, 11,4, 12,4 Гц, 1H), 4,15-4,09 (м, 1H), 3,82 (дд, J=5,95, 8,7 Гц, 1H), 3,74 (ддд, J=5,5, 8,5, 13,6 Гц, 1H), 3,56 (дд, J=8,8, 11,3 Гц, 1H), 3,50 (с, 3H), 2,61 (app дд, J=5,0, 8,9 Гц, 2H).
Промежуточное соединение 4
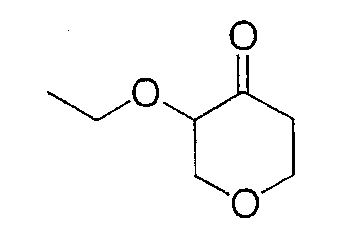
Это промежуточное соединение получают аналогично методике получения промежуточного соединения 3, за исключением того, что иодметан заменяют иодэтаном. Очистка ЖХСД (градиентное элюирование 0-40% этилацетат/гексан) дает 693 мг (66%) конечного соединения в виде прозрачного масла.
Промежуточное соединение 5

К смеси 5,6-дигидро-4-метокси-2Н-пирана (0,5 г, 4 ммоль) в смеси ацетонитрил/вода (15 мл, 1:1) при комнатной температуре добавляют бис(тетрафторборат) 1-(хлорметил)-4-фтор-1,4-диазониабицикло[2.2.2]октана (1,5 г, 4,4 ммоль, SELECTFLUOR™) одной порцией и полученную реакционную смесь перемешивают при комнатной температуре до завершения реакции. Затем добавляют твердый NaCl и реакционную смесь экстрагируют простым эфиром (4×50 мл). Эфирный слой сушат (безводный сульфат магния) и концентрируют с получением 0,34 г (65%) указанного в заголовке соединения, которое не требует дальнейшей очистки.
1H ЯМР (500 МГц, CDCl3) δ 4,95 (м, 1H), 4,4-4,21 (м, 2H), 3,72-3,65 (м, 2H), 2,75 (м, 2H).
Промежуточное соединение 6

Стадия А
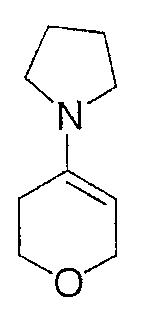
Смесь тетрагидро-4Н-пиран-4-она (10,0 г, 100 ммоль) и пирролидина (11 г, 150 ммоль) перемешивают при комнатной температуре в течение 1 ч. Избыток пирролидина удаляют в вакууме и остаток сушат в течение ночи в высоком вакууме. Энамин получают в виде желтой жидкости (14,7 г), которую используют на следующей стадии без дальнейшей очистки.
Стадия В

Энамин, полученный на стадии А, промежуточное соединение 6 (1,54 г, 10 ммоль) и 4-N,N-диметилпиридин (1,22 г) обрабатывают N,N-диметилформамидом (25 мл). Смесь охлаждают до температуры 0°C и добавляют твердый трифторметансульфонат 5-(трифторметил)дибензотиофения (4,0 г, 10 ммоль). Полученную смесь перемешивают при температуре 0°C в течение 1 ч, затем гасят 30 мл концентрированной водной HCl. Полученную смесь перемешивают в течение 2 ч и затем экстрагируют простым эфиром (4×70 мл). Объединенные эфирные слои промывают водой (50 мл) и насыщенным раствором соли (50 мл), сушат над Na2SO4, фильтруют и выпаривают. Остаток очищают на силикагеле (элюент: 10% эфир/гексан) с получением двух компонентов. Более полярный компонент (200 мг) является желаемым продуктом. 1Н-ЯМР показал, что продукт может существовать в форме полукеталя.
1H ЯМР (500 МГц, CDCl3) δ 4,43-3,38 (м, 5H), 3,24, 3,18 (сс, 3H) 2,52 (м, 1H), 1,82 (м, 1H).
Менее полярный продукт (100 мг) подтверждают как альфа-альфа'-дитрифторметилтетрагидро-4Н-пиран-4-он.
1H ЯМР (500 МГц, CDCl3) δ 4,59 (дд, 2H), 3,24, 3,80 (т, J=11,3 Гц, 2H), 3,42 (м, 2H).
Промежуточное соединение 7

Стадия А

К раствору 5-трифторметил-2-пиридиналя (51 г, 310 ммоль) и ацетата натрия (26,2 г, 319 ммоль) в ледяной уксусной кислоте (200 мл) добавляют бром (16,7 мл, 325 ммоль) и полученную смесь нагревают до температуры 80°C в течение 2,5 ч. Реакционную смесь охлаждают до комнатной температуры и затем выпаривают при пониженном давлении. Остаток нейтрализуют насыщенным NaHCO3 раствором и экстрагируют этилацетатом (3×200 мл). Органические слои объединяют, сушат над MgSO4, фильтруют и выпаривают в вакууме с получением 74,45 г (98%) неочищенного продукта.
1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, J=2,6 Гц, 1H), 7,89 (м, 1H).
Стадия В

В атмосфере азота замещенный пиридин, описанный на стадии А, промежуточное соединение 7 (48,8 г, 202 ммоль) добавляют небольшими порциями к суспензии NaH (8,9 г, 220 ммоль) в безводном тетрагидрофуране (500 мл). После завершения добавления промежуточного соединения реакционную смесь охлаждают до температуры -78°C и обрабатывают трет-бутиллитием (260 мл, 444 ммоль), который добавляют по каплям через шприц. После перемешивания в течение 5 мин медленно добавляют N,N-диметилформамид (50 мл, 707 ммоль) для поддержания температуры раствора ниже -50°C. Полученную смесь затем перемешивают в течение 10 ч, давая ей нагреваться до комнатной температуры. Смесь гасят 2н. HCl и затем разбавляют этилацетатом (1000 мл). Органический слой отделяют, промывают насыщенным раствором соли, сушат над MgSO4 и выпаривают в вакууме. Желаемый продукт осаждают из этилацетата и гексана и фильтруют с получением светло-коричневого твердого вещества (28,55 г, 74%).
1H ЯМР (500 МГц, CD3OD) δ 10,13 (с, 1H), 8,21 (с, 2H).
Стадия С
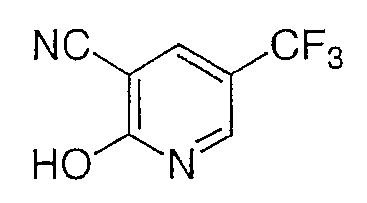
Смесь промежуточного соединения со стадии В, промежуточного соединения 7 (18 г, 95 ммоль), формиата натрия (7,1 г, 110 ммоль), гидрохлорида гидроксиламина (7,3 г, 110 ммоль) и муравьиной кислоты (150 мл) перемешивают при комнатной температуре в течение 2 ч и затем нагревают до температуры кипения с обратным холодильником в течение ночи. Реакционную смесь охлаждают и дают выстаиваться при комнатной температуре в течение 7 дней. Реакционную смесь выливают в воду и экстрагируют этилацетатом (3×). Объединенные органические слои промывают водой (2×), насыщенным NaHCO3 и насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме с получением желаемого продукта в виде коричневого порошка (17,84 г, 90%).
1H ЯМР (400 МГц, CD3OD) δ 8,37 (д, J=2,7 Гц, 1H), 8,19 (кв, J=0,7 Гц, 0,3 Гц, 1H).
Стадия D

К смеси оксихлорида фосфора (13.4 мл, 144 ммоль) и хинолина (8,7 мл, 73 ммоль) добавляют продукт со стадии С, промежуточное соединение 8 (24,6 г, 131 ммоль) и полученную смесь нагревают до температуры кипения с обратным холодильником в течение 3 ч. Реакционную смесь охлаждают до температуры 100°C перед медленным добавлением воды (70 мл). Затем смесь охлаждают до комнатной температуры и осторожно нейтрализуют насыщенным NaHCO3 раствором. Водный слой экстрагируют этилацетатом (3×) и органические слои объединяют, сушат над MgSO4, фильтруют и выпаривают в вакууме. Неочищенный продукт очищают флэш-хроматографией с получением (23,5 г, 87%) желаемого соединения.
1H ЯМР (500 МГц, CDCl3) δ 8,88 (д, J=2,0 Гц, 1H), 8,26 (д, J=2,5 Гц, 1H).
Стадия Е

К суспензии NaH (7,8 г, 200 ммоль) в тетрагидрофуране (100 мл) в атмосфере азота по каплям добавляют раствор трет-бутилметилмалоната (20 мл, 120 ммоль) в безводном тетрагидрофуране (100 мл) через шприц. Реакционную смесь перемешивают в течение 0,5 ч, затем медленно добавляют раствор промежуточного соединении, полученного на стадии D, промежуточное соединение 8 (20,1 г, 97,6 ммоль) в тетрагидрофуране (200 мл), через шприц. Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем гасят насыщенным раствором NH4Cl. Органический слой отделяют и водный слой экстрагируют этилацетатом (3×). Объединенные органические слои промывают водой (3×), сушат над Na2SO4, фильтруют и выпаривают в вакууме. Флэш-хроматография дает 31,76 г (95%) чистого желаемого продукта.
1H ЯМР (500 МГц, CDCl3) δ 9,03 (д, J=1,5 Гц, 1H), 8,25 (д, J=2,0 Гц, 1H), 5,25 (с, 1H), 3,86 (с, 3H), 1,52 (с, 9H).
Стадия F

Суспензию Ni Ренея (1 г) и продукта со стадии Е, промежуточное соединение 7 (18,2 г, 52,9 ммоль) в этаноле (130 мл) помещают на аппарат Парра и гидрируют при 40 ф/д2 Н2 в течение ночи. Суспензию фильтруют через целит и фильтрат выпаривают в вакууме с получением 16,35 г (98%) неочищенного продукта.
1H ЯМР (500 МГц, CDCl3) δ 8,83 (с, 1H), 7,89 (с, 1H), 7,82 (с, 1H), 4,83 (д, J=16 Гц, 1H), 4,72 (с, 1H), 4,49 (д, J=16 Гц, 1H), 1,45 (с, 9H).
Стадия G

К смеси продукта со стадии F, промежуточного соединения 7 (16 г, 51 ммоль) в дихлорметане (60 мл) добавляют ТФУК (30 мл) и полученную смесь перемешивают при комнатной температуре в течение 0,5 ч. Раствор выпаривают при пониженном давлении и остаток растворяют в дихлорметане. Смесь нейтрализуют медленным добавлением раствора насыщенного бикарбоната натрия и органический слой удаляют. Водный слой экстрагируют дихлорметаном (4×) и объединенные органические слои сушат над Na2SO4, фильтруют и выпаривают в вакууме с получением 10,42 г (95%) желаемого продукта.
1H ЯМР (500 МГц, CDCl3) δ 8,81 (с, 1H), 7,78 (с, 1H), 7,30 (с, 1H), 4,63 (с, 2H), 3,90 (с, 2H).
Стадия Н
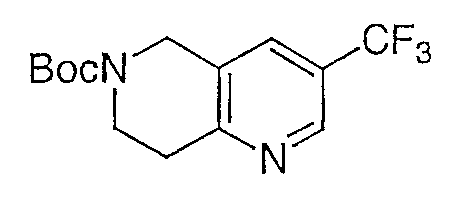
К раствору продукта со стадии G, промежуточного соединения 7 (18,0 г, 83,3 ммоль) в тетрагидрофуране (50 мл) добавляют 1,0 М борана в тетрагидрофуране (417 мл, 420 ммоль) и полученный раствор перемешивают при комнатной температуре в течение ночи. Раствор выпаривают при пониженном давлении и остаток обрабатывают раствором 1% HCl/метанол. Полученную смесь нагревают до температуры 50°C в течение ночи для разложения комплекса борана. Обработку кислым метанолом повторяют дважды для того, чтобы удостовериться в удалении комплекса борана. Раствор такого неочищенного продукта (83,3 ммоль, предположительно 100% превращение) и диизопропилэтиламина (43 мл, 250 ммоль) в дихлорметане обрабатывают ди-трет-бутилдикарбонатом (35,4 г, 167 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи. Раствор промывают насыщенным водным раствором бикарбоната натрия, водой и насыщенным раствором соли. Водные слои объединяют и опять промывают дихлорметаном (2×). Затем объединенные органические слои сушат над Na2SO4, фильтруют и выпаривают досуха. Неочищенный продукт очищают флэш-хроматографией и ЖХСД с получением (11,89 г, 47%) в виде желтого твердого вещества.
1H ЯМР (500 МГц, CDCl3) δ 8,69 (с, 1H), 7,66 (с, 1H), 4,67 (с, 2H), 3,79 (т, J=6,0 Гц, 2H), 3,08 (т, J=5,5 Гц, 2H), 1,51 (с, 9H).
Стадия I
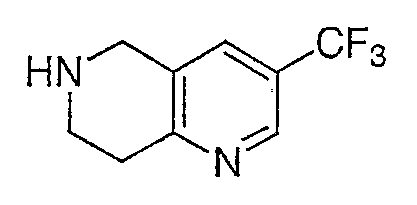
Продукт, описанный на стадии Н, промежуточное соединение 8 (11,89 г) обрабатывают раствором 4н. HCl в диоксане. Раствор перемешивают при комнатной температуре в течение 2 ч и затем выпаривают в вакууме с получением промежуточного соединения 8 (10,85 г, 99%) в виде желтого порошка. ЖХ-МС для C9H10F3N2 рассчитано 202,07, найдено [M+H]+ 203,0.
Промежуточное соединение 8

Методика А
Стадия А

Смесь (1S)-(+)-2-азабицикло[2.2.1]гепт-5-ен-3-она (10,3 г, 94,4 ммоль) в этилацетате (200 мл) и 10% Pd/C (0,5 г) гидрируют при комнатной температуре. Через 24 часа реакционную смесь фильтруют и выпаривают, оставляя менее 10,4 г (100%) продукта, который помещают в 250 мл метанола и HCl (12 М, 6 мл). Полученную смесь перемешивают при комнатной температуре до тех пор, пока реакция не завершится (72 ч). Выпаривание метанола с последующей сушкой в высоком вакууме дает указанное в заголовке соединение в виде не совсем белого твердого вещества (16,0 г, 96%).
1H ЯМР (500 МГц, D2О): δ 3,70 (с, 3H), 3,01 (м, 1H), 2,38 (м, 1H), 2,16-1,73 (м, 6H).
Стадия В

К суспензии промежуточного соединения со стадии А (10,2 г, 56,8 ммоль) в сухом дихлорметане (200 мл) добавляют бензофенонимин (10,2 г, 56,8 ммоль) при комнатной температуре и полученную смесь перемешивают в течение 24 ч. Реакционную смесь фильтруют и фильтрат выпаривают с получением желтого масла, которое растирают с простым эфиром (100 мл), фильтруют и выпаривают. Эту операцию повторяют дважды, чтобы удостовериться, что продукт не содержит примеси хлорида аммония. Полученное масло тщательно сушат в вакууме с получением указанного в заголовке соединения (18,03 г, >100%) и оно не требует дальнейшей очистки.
1H ЯМР (500 МГц, CDCl3): δ 7,5-7,18 (м, 10H), 3,75 (м, 1H), 3,7 (с, 3H), 2,78 (м, 1H), 2,26-1,71 (м, 6H).
Стадия С

К раствору диизопропиламида лития (полученного из диизопропиламина (7,7 г, 76 ммоль) и н-бутиллития (30,4 мл, 2,5 М в гексане, 76 ммоль) в тетрагидрофуране (120 мл) при температуре -78°C добавляют простой эфир со стадии В (18,0 г, 56,8 ммоль). Полученный раствор красного цвета перемешивают в течение 20 мин, затем гасят 2-иодпропаном (14,9 мг, 88 ммоль). Реакционную смесь постепенно нагревают в течение 3 часов до температуры 0°C и такую температуру поддерживают в течение еще 3 часов. Реакцию гасят водой и экстрагируют этилацетатом. Органический слой промывают водой, насыщенным раствором соли, сушат (безводный сульфат магния) и концентрируют с получением масла. К раствору неочищенного основания Шиффа (20,0 г) в тетрагидрофуране (100 мл) добавляют HCl (5,0 мл, 12 М). Полученную реакционную смесь перемешивают при комнатной температуре в течение 3 ч. После удаления всех летучих веществ гидрохлоридную соль помещают в дихлорметан (250 мл), добавляют насыщенный раствор бикарбоната натрия (250 мл) и ди-трет-бутилдикарбонат (26,0 г, 1,4 экв.). Полученную смесь отделяют и промывают водой, насыщенным раствором соли, сушат (безводный сульфат магния) и концентрируют с получением масла. Очистка флэш-хроматографией на колонке (элюент:гексан/этилацетат 19:1) дает желаемый продукт (4,91 г, 30%).
1H ЯМР (500 МГц, CDCl3): 4,79 (уш., 1H), 4,01 (м, 1H), 3,71 (с, 3H), 2,18-1,60 (м, 6H), 1,44 (с, 9H), 0,87 (д, J=6,9 Гц, 3H), 0,86 (д, J=6,9 Гц, 3H).
Стадия D
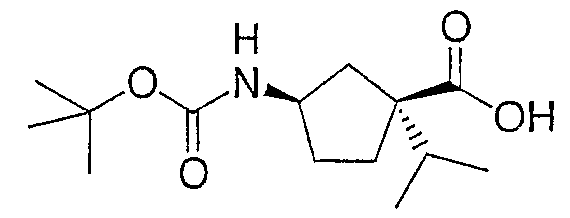
К раствору сложного эфира со стадии С (4,91 г, 17,2 ммоль) в метаноле (100 мл) добавляют раствор LiOH (3,6 г, 85 ммоль) в воде (20 мл) и тетрагидрофуране (10 мл). Полученную смесь нагревают до температуры 80°C до завершения реакции (18 ч). Метанол удаляют в вакууме и неочищенный продукт помещают в смесь вода/этилацетат (200 мл, 1:4) и охлаждают до температуры 0°C. Кислотность смеси доводят до рН 6. Слой этилацетата отделяют, промывают водой, насыщенным раствором соли, сушат (безводный сульфат магния) и концентрируют с получением масла. Очистка флэш-хроматографией на колонке (элюент:гексан/этилацетат 1:1 + 2% AcOH) дает промежуточное соединение 8 (3,9 г, 84%).
1H ЯМР (500 МГц, CDCl3): 11,36 (уш., 1H), 6,49 (уш., 1H), 4,83 (м, 1H), 3,71 (с, 3H), 2,30-1,55 (м, 6H), 1,46 (с, 9H), 0,94 (д, J=6,9 Гц, 3H), 0,933 (д, J=6,9 Гц, 3H).
Методика В
Стадия А

Коммерчески доступную (1R,4S)-4-аминоциклопент-2-ен-1-карбоновую кислоту превращают в гидрохлорид метилового эфира классическим методом.
Стадия В

К суспензии амина со стадии А (6,31 г, 35,5 ммоль) в ацетоне (40 мл), воде (20 мл) порциями добавляют твердый NaHCO3 (6,6 г, 78 ммоль). Через 5 мин добавляют раствор ди-трет-бутилдикарбоната (8,5 г, 39 ммоль) в ацетоне (60 мл) и реакционную смесь перемешивают при комнатной температуре. Через 3 ч ацетон удаляют в вакууме и остаток разделяют между простым эфиром (500 мл) и насыщенным водным раствором NaHCO3 (120 мл). Эфирный слой дополнительно промывают водным раствором NaHCO3 (1×100 мл), насыщенным раствором соли (1×100 мл), сушат над безводным Na2SO4, концентрируют и очищают флэш-хроматографией (15% этилацетат/гексан) с получением продукта (7,25 г, 85%).
Стадия С

К раствору бис(триметилсилил)амида лития (10,4 г, 62,1 ммоль) в тетрагидрофуране (100 мл) добавляют раствор промежуточного соединения со стадии В (6,71 г, 27,8 ммоль) в тетрагидрофуране (10 мл) в течение 10 мин при температуре -78°C. Полученный раствор перемешивают при температуре -78°C в течение 30 мин, затем добавляют одной порцией изопропилиодид (3,3 мл, 33 ммоль). Реакционную смесь нагревают до температуры -25°C и эту температуру поддерживают в течение ночи. Затем реакцию гасят насыщенным водным раствором NH4Cl (250 мл). Органический слой отделяют и водный слой далее экстрагируют диэтиловым эфиром (3×100 мл). Объединенные органические слои затем промывают насыщенным раствором соли (1×100 мл), сушат над безводным Na2SO4, фильтруют, концентрируют и очищают флэш-хроматографией (5-10% этилацетат/гексан) с получением продукта (5,66 г, 72%) в виде прозрачного масла (цис/транс=4,3/1).
1H ЯМР (500 МГц, CDCl3) цис-изомер: δ 5,79 (с, 2H), 4,75 (м, 1H), 3,72 (с, 3H), 2,28-2,20 (м, 2H), 2,0 (дд, J=15, 4 Гц, 1H), 1,45 (с, 9H), 0,85 (д, J=6,6 Гц, 3H), 0,81 (д, J=7 Гц, 3H).
Стадия D

К раствору продукта со стадии С (1,6 г, 5,7 ммоль) в тетрагидрофуране (50 мл), метаноле (50 мл) и воде (10 мл) добавляют моногидрат LiOH (400 мг) и реакционную смесь нагревают до температуры кипения с обратным холодильником до тех пор, пока ТСХ не покажет завершение реакции. Органические растворители удаляют в вакууме и водный слой промывают простым эфиром (1×) и затем медленно подкисляют концентрированной HCl до достижения рН 4. Полученную суспензию экстрагируют CH2Cl2 (3×). Объединенные органические слои сушат над безводным MgSO4, фильтруют и концентрируют с получением продукта в виде смеси двух цис/транс-изомеров (1,5 г) в виде пенящегося твердого желтого вещества. Это твердое вещество растворяют в этилацетате (2 мл) при нагревании и разбавляют гексаном (50 мл) с получением прозрачного раствора. Раствор охлаждают до комнатной температуры в течение 1 ч и затем выдерживают при температуре -25°C в холодильнике в течение ночи. Транс-изомер кристаллизуется вместе с некоторым количеством желаемого цис-изомера (500 мг всего). Маточный раствор собирают и концентрируют с получением указанного в заголовке соединения (1 г, 66%, только цис-изомер).
1H ЯМР (500 МГц, CDCl3) цис-изомер: δ 5,80 (м, 2H), 4,80 (м, 11-1), 2,40-2,20 (м, 2H), 2,15-2,0 (м, 1H), 1,5 (м, 9H), 1,0-0,8 (м, 3H).
Стадия Е

К раствору продукта со стадии D (1 г) в этаноле (30 мл) добавляют 10% Pd/C (100 мг) и полученную смесь перемешивают в аппарате Парра при давлении 50 ф Н2 в течение ночи. Смесь фильтруют через целит и концентрируют в вакууме с получением указанного в заголовке соединения (1 г, 99%).
1H ЯМР (500 МГц, CDCl3): 11,36 (уш., 1H), 6,49 (уш., 1H), 4,83 (м, 1H), 3,71 (с, 3H), 2,30-1,55 (м, 6H), 1,46 (с, 9H), 0,94 (д, J=6,9 Гц, 3H), 0,933 (д, J=6,9 Гц, 3H).
Промежуточное соединение 9

Стадия А

Промежуточное соединение 8 (4,6 г, 16 ммоль) и промежуточное соединение 11 (4,0 г, 14 ммоль) сначала сушат азеотропной перегонкой с толуолом (3×50 мл) и помещают в высокий вакуум на 30 мин. В атмосфере азота последовательно добавляют 4-диметиламинопиридин (1,08 г, 8,60 ммоль), безводный дихлорметан (40 мл) и диизопропилэтиламин (7,0 мл, 40 ммоль). После превращения промежуточного соединения 8 в раствор добавляют гексафторфосфат бром-трис-пирролидинофосфония (6,80 г, 14,3 ммоль), с немедленным добавлением еще диизопропилэтиламина (7,0 мл, 40 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем гасят насыщенным NaHCO3. Водный слой опять промывают дихлорметаном (3×50 мл) и органические слои объединяют, сушат над Na2SO4, фильтруют и выпаривают в вакууме. Неочищенный продукт очищают флэш-хроматографией на колонке (ступенчатый градиент 0-60% этилацетат/гексан) с получением продукта (4,80 г, 74%) в виде желтой пены.
1H ЯМР (500 МГц, CDCl3) δ 8,72 (с, 1H), 7,70 (с, 1H), 4,88 (уш.д, J=17,0 Гц, 1H), 4,78 (д, J=17,6 Гц, 1H), 4,04-3,84 (м, 2H), 3,52 (уш.с, 1H), 3,12 (уш.т, J=5,6 Гц, 1H), 2,32-2,06 (м, 3H), 1,98-1,70 (м, 4H), 1,64-1,54 (м, 1H), 1,44 (с, 9H), 0,92-0,82 (м, 6H). ЖХ-МС для C23H32F3N3О рассчитано 4555,24, найдено [M+H]+ 456,2.
Стадия В

Продукт со стадии В, промежуточное соединение 19 (1,2 г, 2,6 ммоль) растворяют с 4н. HCl в диоксане (50 мл) и полученный раствор перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь выпаривают в вакууме с получением продукта (904 мг, 97%) в виде белого порошка. ЖХ-МС для C18H24F3N3О рассчитано 355,20, найдено [M+H]+ 356,2.
Промежуточное соединение 10

Стадия А

К раствору продукта, описанного на стадии А, промежуточного соединения 19 (2,0 г, 4,4 ммоль) в дихлорметане (80 мл) добавляют 3-хлорпероксибензойную кислоту (2,11 г, 8,83 ммоль) и полученный раствор перемешивают в течение ночи при комнатной температуре. Смесь охлаждают до температуры 0°C и, при энергичном перемешивании, порциями добавляют твердый гидроксид кальция (около 6 г). Суспензию перемешивают в течение еще 30 мин, затем фильтруют через целит для удаления всех твердых веществ. Фильтрат выпаривают в вакууме и остаток очищают ЖХСД (градиентное элюирование 40-100% этилацетат/гексан) с получением 1,32 г (64%) желаемого соединения.
1H ЯМР (500 МГц, CDCl3) δ 8,46 (с, 1H), 7,28 (с, 1H), 4,88 (уш.д, J=17,2 Гц, 1H), 4,78 (д, J=17,7 Гц, 1H), 4,05-3,84 (м, 2H), 3,12 (уш.с, 1H), 2,34-2,06 (м, 3H), 1,88-1,70 (м, 4H), 1,62-1,54 (м, 1H), 1,43 (с, 9H), 0,90-0,85 (м, 6H). ЖХ-МС для C23H32F3N3О5 рассчитано 471,20, найдено [M+H]+ 472,2.
Стадия В

Продукт со стадии В, промежуточное соединение 20 (1,32 г, 2,82 ммоль) растворяют в 4н. HCl в диоксане (50 мл) и полученный раствор перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь выпаривают в вакууме с получением продукта (1,10 г, 98%) в виде белого порошка. ЖХ-МС для C18H24F3N3О2 рассчитано 371,20, найдено [M+H]+ 372,2.
Пример 1
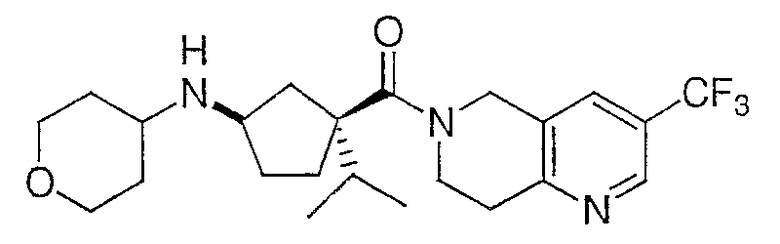
Раствор промежуточного соединения 9 (980 мг, 2,08 ммоль), тетрагидро-4Н-пиран-4-она (320 мг, 3,13 ммоль), диизопропилэтиламина (1,10 мл, 6,24 ммоль) и дробленых молекулярных сит (4Е, 500 мг) в дихлорметане (50 мл) обрабатывают триацетоксиборгидридом натрия (2,20 г, 10,4 ммоль) и перемешивают при комнатной температуре в течение ночи. Реакцию гасят насыщенным раствором бикарбоната натрия (50 мл) и разбавляют 25 мл дихлорметана. Органический слой отделяют и водный слой промывают дихлорметаном (2×25 мл). Органические слои объединяют, сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают ВЭЖХ с обращенной фазой с получением соединения Примера 1 (915 мг, 86,0%). ЖХ-МС для C23H31F3N3О2 рассчитано 439,24, найдено [M+H]+ 440,2.
Пример 2
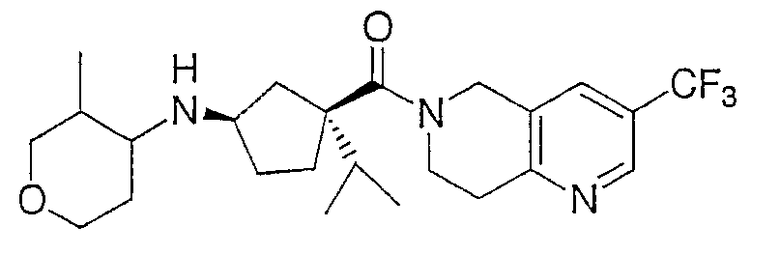
Раствор промежуточного соединения 9 (304 мг, 0,712 ммоль), промежуточного соединения 1 (160 мг, 1,42 ммоль), диизопропилэтиламина (370 мкл, 2,14 ммоль) и дробленых молекулярных сит (4Е, 150 мг) в дихлорметане (25 мл) обрабатывают триацетоксиборгидридом натрия (755 мг, 3,56 ммоль) и перемешивают при комнатной температуре в течение ночи. Реакцию гасят насыщенным раствором бикарбоната натрия (25 мл) и разбавляют 25 мл дихлорметана. Органический слой отделяют и водный слой промывают дихлорметаном (2×20 мл). Органические слои объединяют, сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Остаток очищают препаративной ТСХ (элюент: 0,5% NH4OH/5% метанол/94,5% CH2Cl2) с получением 239 мг (74%) конечного продукта в виде смеси диастереомеров. Цис и транс рацематы относительно пиранового кольца разделяют ВЭЖХ на колонке с Preparative ChiralCel OD (элюент: 5% этанол/95% гексан). Цис рацемат далее отделяют на колонке с Preparative ChiralCel AD (элюент: 5% этанол/95% гексан). ЖХ-МС для C24H35F3N3О2 рассчитано 453,26, найдено [M+H]+ 454,30.
Пример 3

Продукт получают по методике примера 2 за исключение того, что промежуточное соединение 1 заменяют промежуточным соединением 2. Очистка препаративной ТСХ (элюент: 0,5% NH4OH/5% метанол/94,5% CH2Cl2) дает 203 мг (92%) в виде смесь четырех диастереомеров. Отдельные изомеры получают очисткой ВЭЖХ на колонке с Preparative ChiralCel OD, элюируя 5% этанол/95% гексаном при скорости потока 9 мл/мин. ЖХ-МС для C25H36F3N3О2 рассчитано 467,28, найдено [M+H]+ 468,3 для всех 4 изомеров.
Пример 4
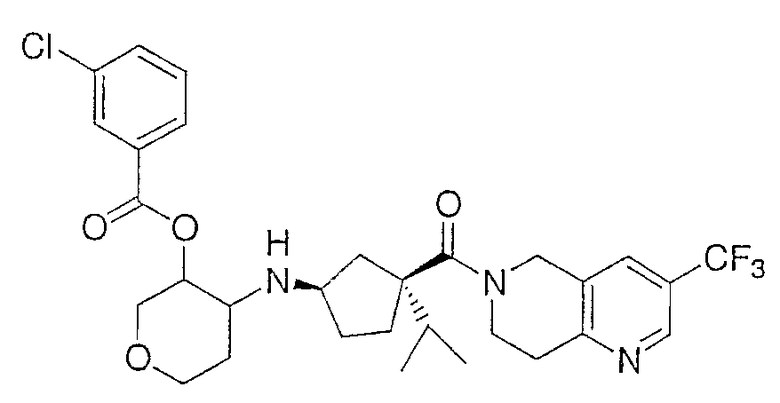
Продукт получают по методике примера 2 за исключением того, что промежуточное соединение 1 заменяют промежуточным соединением 5. Очистка дает 312 мг (88%) продукта в виде смеси четырех диастереомеров. ЖХ-МС для C30H36ClF3N3О4 рассчитано 593,23, найдено [M+H]+ 594,3.
Пример 5

К раствору продукта, описанного в примере 4 (286 мг, 0,482 ммоль) в метаноле добавляют 0,5 М раствор метоксида натрия в метаноле (1,2 мл, 0,58 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 2 ч. После завершения реакции смесь выпаривают в вакууме и очищают препаративной ТСХ (элюент: 1,0% NH4OH/10% метанол/89% CH2Cl2) с получением соединения примера 21 (201 мг, 91,6%) в виде смеси четырех диастереомеров. ЖХ-МС для C23H33F3N3О3 рассчитано 455,24, найдено [M+H]+ 456,25.
Пример 6

Раствор промежуточного соединения 9 (500 мг, 1,17 ммоль), промежуточного соединения 3 (458 мг, 3,51 ммоль), диизопропилэтиламина (407 мкл, 2,34 ммоль) и дробленых молекулярных сит (4Е, 250 мг) в дихлорметане (25 мл) обрабатывают триацетоксиборгидридом натрия (1,24 г, 5,85 ммоль) и перемешивают при комнатной температуре в течение ночи. Реакцию гасят насыщенным раствором бикарбоната натрия (25 мл) и разбавляют 25 мл дихлорметана. Органический слой отделяют и водный слой промывают дихлорметаном (2×20 мл). Органические слои объединяют, сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Остаток очищают препаративной ТСХ (элюент: 1,0% NH4OH/10% метанол/89% CH2Cl2) с получением 210 мг (86%) конечного продукта в виде смеси четырех диастереомеров. Отдельные изомеры получают ВЭЖХ на колонке с Preparative ChiralCel OD, элюируя 20% этанолом и 80% гексаном при скорости потока 9 мл/мин. ЖХ-МС для C24H34F3N3О3 рассчитано 469,21, найдено [M+H]+ 470,2 для всех 4 изомеров.
3-й изомер из колонки ChiralCel OD: 1H ЯМР (500 МГц, CDCl3) δ 8,72 (с, 1H), 7,69 (с, 1H), 4,87 (уш.д, J=17,2 Гц, 1H), 4,75 (д, J=17,4 Гц, 1H), 4,12 (дд, J=3,1, 12,4 Гц, 1H), 3,99-3,86 (м, 3H), 3,47-3,39 (м, 1H), 3,41 (с, наложение, 3H), 3,35-3,30 (м, 2H), 3,20-3,08 (м, 3H), 2,87-2,80 (м, 1H), 2,62-2,54 (м, 1H), 2,16-2,02 (м, 2H), 1,95 (уш.с, 1H), 1,88-1,81 (м, 1H), 1,78-1,57 (м, 6H), 1,41-1,32 (м, 1H), 0,96 (д, J=6,7 Гц, 3H), 0,84 (д, J=6,6 Гц, 3H).
4-й изомер из колонки ChiralCel OD: 1H ЯМР (500 МГц, CDCl3) δ 8,72 (с, 1H), 7,69 (с, 1H), 4,87 (уш.д, J=17,6 Гц, 1H), 4,75 (д, J=17,5 Гц, 1H), 4,10 (дд, J=3,1, 12,3 Гц, 1H), 3,99-3,88 (м, 3H), 3,46-3,39 (м, 1H), 3,41 (с, наложение, 3H), 3,35-3,30 (м, 2H), 3,17-3,09 (м, 3H), 2,86-2,80 (м, 1H), 2,64-2,55 (м, 1H), 2,16-2,10 (м, 1H), 2,05 (уш.с, 1H), 1,95-1,82 (м, 2H), 1,76-1,55 (м, 6H), 1,33-1,24 (м, 1H), 0,95 (д, J=6,7 Гц, 3H), 0,83 (д, J=6,6 Гц, 3H).
Пример 7
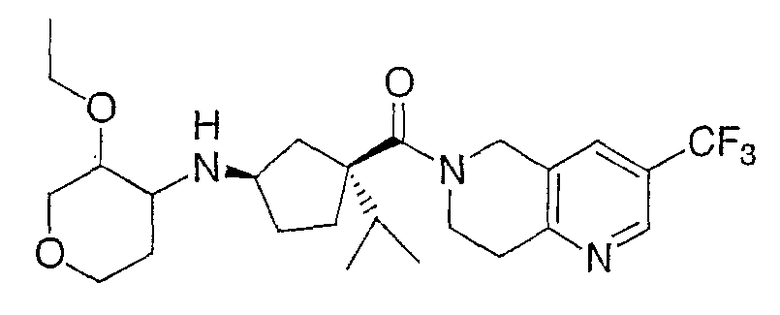
Продукт получают по методике примера 2 за исключением того, что промежуточное соединение 1 заменяют промежуточным соединением 4. Отдельные изомеры получают очисткой ВЭЖХ на колонке с Preparative ChiralCel OD, элюируя 15% этанолом и 85% гексаном при скорости потока 9 мл/мин. ЖХ-МС для С25Н36F3N3О3 рассчитано 483,23, найдено [М+Н]+ 484,2 для всех 4 изомеров.
ЖХМС: 2,7 мин, 484,3 ЯМР (СО3OD): м.д. 8,82 (с, 1Н), 8,23 (с, 1Н), 4,94 (с, 2Н), 4,86 (Н2O), 4,24-4,4 (м, 1Н), 3,9-4,4 (м, 3Н), 3,8 (м, 1Н), 3,7 (с, 1Н), 3,6 (м, 2Н), 3,46 (м, 2Н), 3,4 (м, 1Н), 3,3 (МеОН), 3,2 (м, 3Н), 2,4 (м, 1Н), 2,2-2,4 (м, 4Н), 2,90 (м, 1Н), 1,2 (м, 4Н), 0,9 (с, 6Н)
Пример 8

Продукт получают по методике примера 2 за исключением того, что промежуточное соединение 1 заменяют промежуточным соединением 5. ЖХ-МС для С24Н31F3N3O2 рассчитано 457,23, найдено [М+Н]+ 458,2 для всех 4 изомеров.
Пример 9

Продукт получают по методике примера 2 за исключением того, что промежуточное соединение 1 заменяют промежуточным соединением 6. Отдельные изомеры получают очисткой ВЭЖХ на колонке с Preparative ChiralCel OD, элюируя 5% этанолом и 95% гексаном при скорости потока 9 мл/мин. ЖХ-МС для С24Н31F6N3O2 рассчитано 507,23, найдено [М+Н]+ 508,2 для всех 4 изомеров.
Пример 10

Раствор промежуточного соединения 10 (641 мг, 1,60 ммоль), тетрагидро-4Н-пиран-4-она (220 мг, 2,24 ммоль), молекулярных сит (4Е, 320 мг) в дихлорметане (20 мл) обрабатывают триацетоксиборгидридом натрия (1,70 г, 8,00 ммоль) и перемешивают при комнатной температуре не более 5 ч. Реакцию гасят насыщенным раствором бикарбоната натрия (50 мл) и разбавляют 30 мл дихлорметана. Органический слой отделяют и водный слой промывают дихлорметаном (2×30 мл). Органические слои объединяют, сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают препаративной ТСХ (элюент: 0,75% NH4OH/7,5% метанол/91,75% CH2Cl2) с получением 626 мг (86%) конечного продукта.
1H ЯМР (500 МГц, CDCl3) δ 8,45, (с, 3H), 7,25 (с, 1H), 4,88 (уш.д, J=17,4 Гц, 1H), 4,77 (д, J=17,6 Гц, 1H), 4,00-3,85 (м, 4H), 3,41 (app т, J=11,7 Гц, 2H), 3,22,(p, J=6,8 Гц, 1H), 3,13-3,07 (м, 2H), 2,82-2,74 (м, 1H), 2,54-2,47 (м, 1H), 2,14 (дд, J=6,8, 12,8 Гц, 1H), 2,07-2,00 (м, 1H), 1,94-1,86 (м, 2H), 1,84-1,77 (м, 3H), 1,65-1,57 (м, 2H), 1,46-1,26 (м, 3H), 0,93 (д, J=6,8 Гц, 3H), 0,83 (д, J=6,8 Гц, 3H). ЖХ-МС для C23H32F3N3О3 рассчитано 455,24, найдено [M+H]+ 456,20.
Пример 11
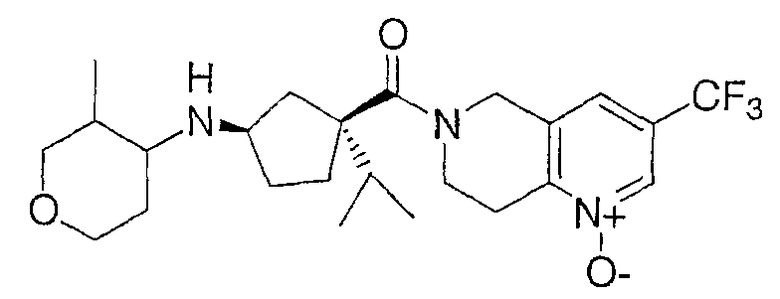
Продукт получают по методике примера 10 за исключением того, что тетрагидро-4Н-пиран-4-он заменяют промежуточным соединением 1. Отдельные изомеры получают очисткой ВЭЖХ на колонке с Preparative ChiralCel OD, элюируя 7% этанолом и 93% гексаном при скорости потока 9 мл/мин. ЖХ-МС для C24H34F3N3О3 рассчитано 469,24, найдено [M+H]+ 470,20 для всех 4 изомеров.
Пример 12

Продукт получают по методике примера 10 за исключением того, что тетрагидро-4Н-пиран-4-он заменяют промежуточным соединением 2. Отдельные изомеры получают очисткой ВЭЖХ на колонке с Preparative ChiralCel OD, элюируя 5% этанолом и 95% гексаном при скорости потока 9 мл/мин. ЖХ-МС для C25H36F3N3О3 рассчитано 483,24, найдено [M+H]+ 484,20 для всех 4 изомеров.
Пример 13

Продукт получают по методике примера 10 за исключением того, что тетрагидро-4Н-пиран-4-он заменяют промежуточным соединением 3. Отдельные изомеры получают очисткой ВЭЖХ на колонке с Preparative ChiralCel OD, элюируя 21% этанолом и 79% гексаном при скорости потока 9 мл/мин. ЖХ-МС для C24H34F3N3О4 рассчитано 485,25, найдено [M+H]+ 486,30 для всех 4 изомеров.
Хотя данное соединение описано и проиллюстрировано конкретными вариантами, специалисту в данной области техники будет понятно, что различные адаптации, изменения, модификации, замещения, удаления или добавления могут быть сделаны не выходя за рамки и объем данного изобретения. Например, могут применяться эффективные дозы, отличные от определенных доз, указанных выше, вследствие изменения реакции лечимого млекопитающего по любому из показаний соединением, указанным выше. Также наблюдаемые фармакологические реакции могут изменяться согласно и в зависимости от определенного выбранного активного соединения, или присутствующих фармацевтических носителей, а также типа лекарственной формы и способа введения, и такие ожидаемые вариации или различия в результатах рассматриваются в объектах и практике данного изобретения. Поэтому подразумевается, что данное изобретение определено представленной ниже формулой изобретения, и пункты указанной формулы изобретения трактуются так широко, насколько это разумно.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕНЗОКСАЗИНИЛ-АМИДОЦИКЛОПЕНТИЛ-ГЕТЕРОЦИКЛИЧЕСКИЕ МОДУЛЯТОРЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2301802C2 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| ЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДЫ ДЛЯ ИНГИБИРОВАНИЯ ГАММА-СЕКРЕТАЗЫ | 2004 |
|
RU2334743C2 |
| ЗАМЕЩЕННЫЕ МОРФОЛИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПРОТИВОДЕЙСТВИЯ ВЕЩЕСТВУ Р ИЛИ БЛОКИРОВАНИЯ РЕЦЕПТОРОВ НЕЙРОКИНИНА-1 | 1995 |
|
RU2170233C2 |
| ДИАРИЛ-5,6-КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЗАЩИТЫ ОТ ВОЗДЕЙСТВИЯ ЛЕЙКОТРИЕНОВ | 1993 |
|
RU2154065C2 |
| ПРОИЗВОДНЫЕ СУЛЬФОНАМИДА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ БЛОКИРОВАНИЯ ДЕЙСТВИЯ ФИБРИНОГЕНА И СПОСОБ ЛЕЧЕНИЯ ТРОМБОЗОВ У МЛЕКОПИТАЮЩИХ | 1992 |
|
RU2116296C1 |
| АНТИДИАБЕТИЧЕСКИЕ ОКСАЗОЛИДИНДИОНЫ | 2005 |
|
RU2355682C2 |
| ПРОИЗВОДНЫЕ ФЕНОКСИФЕНИЛУКСУСНОЙ КИСЛОТЫ | 1994 |
|
RU2139273C1 |
| ПРОИЗВОДНЫЕ ДИАЗЕПИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ АРИТМИИ | 1994 |
|
RU2155587C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ СНИЖЕНИЯ КОЛИЧЕСТВА ТАХИКИНИНОВ ПРИ ЛЕЧЕНИИ ИЛИ ПРОФИЛАКТИКЕ ФИЗИОЛОГИЧЕСКИХ СОСТОЯНИЙ, АССОЦИИРУЕМЫХ С ИЗБЫТКОМ ТАХИКИНИНОВ | 1994 |
|
RU2131426C1 |
Изобретение относится к тетрагидропиранилциклопентилтетрагидропиридопиридинам формулы I: 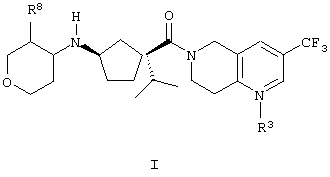
где R3 является кислородом или отсутствует; R8 выбирают из: (а) водорода, (b) C1-3алкила, который является незамещенным или замещен 1-6 атомами фтора, (с) -O-C1-3алкила, (d) фтора и (е) гидрокси; и их фармацевтически приемлемым солям и отдельным диастереомерам. Эти соединения являются модуляторами активности рецептора хемокина. Изобретение также относятся к фармацевтической композиции на основе соединений формулы I, способу модулирования активности рецептора хемокина у животных и человека и способу получения лекарственного средства. Технический результат - получение новых модуляторов активности рецептора хемокина. 5 н. и 3 з.п. ф-лы.

где
R3 является кислородом или отсутствует;
R8 выбирают из
(a) водорода,
(b) C1-3алкила, который является незамещенным или замещен 1-6 атомами фтора,
(c) -O-С1-3алкила,
(d) фтора и
(e) гидрокси;
и их фармацевтически приемлемые соли и отдельные диастереомеры.
(a) водорода,
(b) трифторметила,
(c) метила,
(d) метокси,
(e) этокси,
(f) этила,
(g) фтора и
(h) гидрокси.



и их фармацевтически приемлемые соли и отдельные диастереомеры.
| US 20020012664 A1, 31.01.2002 | |||
| ПРОИЗВОДНЫЕ 1,8-БЕНЗО(B)НАФТИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2047613C1 |
| ПРОИЗВОДНЫЕ 8-АРИЛ-1,7-НАФТИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 1997 |
|
RU2174123C2 |

