Настоящее изобретение относится к твердым пероральным лекарственным формам, содержащим липосомы, причем указанные липосомы содержат конъюгаты проникающих в клетку пептидов (СРР) и соединение, выбранное из липида и жирной кислоты, где указанные липосомы содержатся в капсуле, покрытой энтеросолюбильной оболочкой, или таблетке, покрытой энтеросолюбильной оболочкой. Настоящее изобретение также относится к применению указанных твердых пероральных лекарственных форм для пероральной доставки терапевтических и диагностических средств.
Пероральная доставка лекарственного средства считается наиболее подходящим способом применения, в частности, для лечения хронических заболеваний, которые требуют длительного и многократного введения лекарственного средства. Пероральный путь обеспечивает высокую безопасность лекарств и широко применяется среди пациентов благодаря его удобству. Кроме того, поскольку стерильность не требуется для пероральных лекарственных форм, затраты на производство, хранение и распространение снижаются, что может способствовать улучшению здравоохранения в странах третьего мира. Подсчитано, что 90% всех продаваемых лекарственных форм предназначены для перорального применения.
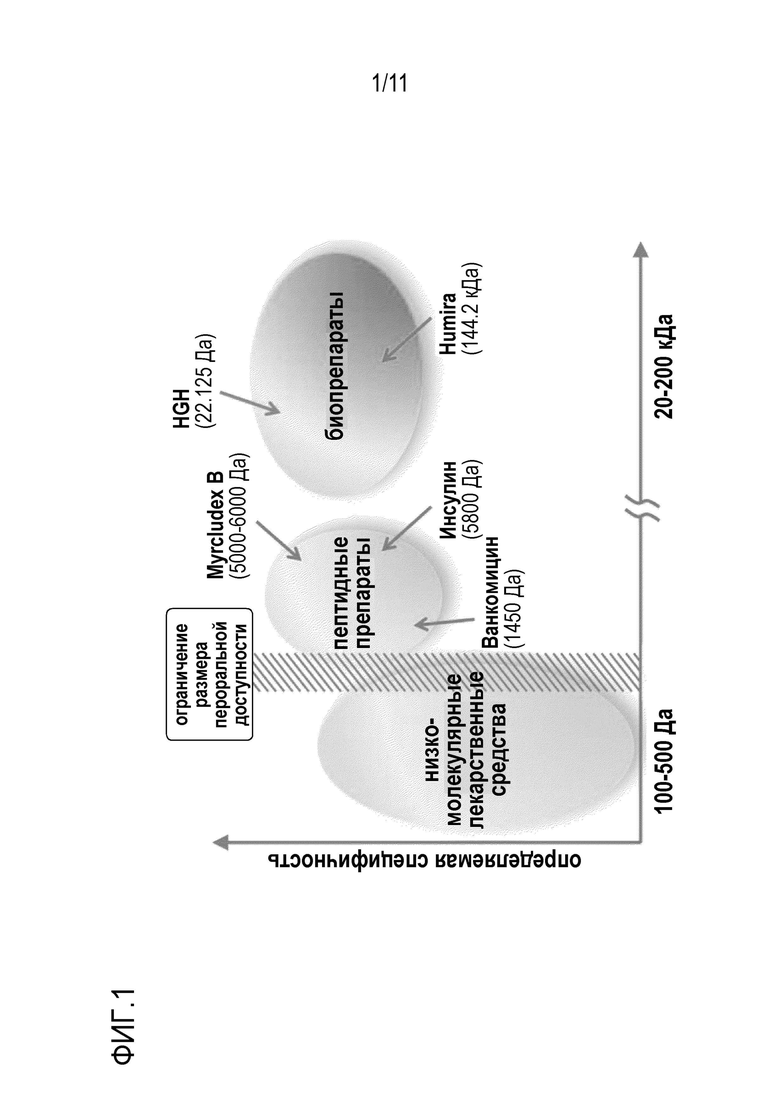
Количество макромолекулярных лекарственных средств, например, пептидов, белков и антител, присутствующих на фармацевтическом рынке, неуклонно растет (фиг. 1). Однако ограничивающим фактором для этих перспективных лекарственных средств является плохая биодоступность при пероральном введении. Из-за своих размеров антитела обладают особенно плохой биодоступность при пероральном введении. В частности, многие макромолекулярные препараты демонстрируют как очень плохую стабильность в кислых условиях в желудке после перорального введения, так и плохую абсорбцию через желудочно-кишечный барьер. Таким образом, такие лекарственные средства должны вводиться подкожно или внутривенно, что увеличивает необходимые медицинские усилия и приводит к увеличению затрат, снижению комплаентности пациента и увеличению риска осложнений.
Для преодоления этой проблемы, в последние годы были опробованы различные подходы к улучшению биодоступности, включая твердые липидные наночастицы, нано- или микроэмульсии и липосомы. Однако обычные липосомальные составы не были очень убедительными из-за их нестабильности в желудочно-кишечном тракте (ЖКТ). Кроме того, были разработаны некоторые липосомальные системы-носители, содержащие тетраэфирные липиды, которые демонстрируют значительно повышенную стабильность во внутрижелудочной среде. Однако тетраэфирные липиды могут быть выделены только из архей и только в лабораторном масштабе, что требует огромных усилий и очень высоких затрат. Соответственно, тетраэфирные липиды доступны только в очень ограниченных количествах. Кроме того, соответствующие липосомальные составы пока доступны только в виде суспензий, демонстрирующих плохую стабильность при хранении и комплаентность.
Соответственно, технической задачей, лежащей в основе настоящего изобретения, является создание твердых дозированных лекарственных форм для перорального введения, обеспечивающих улучшенную биодоступность при пероральном введении макромолекулярных лекарственных средств. Решение вышеуказанной технической задачи достигается с помощью вариантов осуществления, охарактеризованных в формуле изобретения и изложенных в настоящем описании.
В частности, в первом аспекте настоящее изобретение относится к твердой пероральной лекарственной форме, включающей липосомы, причем указанные липосомы включают конъюгат
(а) по меньшей мере одного типа проникающего в клетки пептида (CPP), и
(b) соединения, выбранного из липида и жирной кислоты;
где указанный конъюгат является частью двойного липидного слоя липосомы;
где лекарственная форма представляет собой гастрорезистентную твердую пероральную лекарственную форму. Предпочтительно, указанные липосомы содержатся в капсуле, покрытой энтеросолюбильной оболочкой, или таблетке, покрытой энтеросолюбильной оболочкой.
Используемый в настоящем описании термин «твердая пероральная лекарственная форма» относится к твердой лекарственной форме для перорального введения, то есть к капсуле, таблетке, гранулам или пеллетам.
“Гастрорезистентная” означает, что лекарственная форма по существу не высвобождает свое активное средство - такое как терапевтическое или диагностическое средство - в кислой среде желудка субъекта. Лекарственные формы с энтеросолюбильным покрытием являются гастрорезистентными.
Кроме того, термин «липосома», как используется в настоящем описании, относится к искусственно полученным везикулам, состоящим из липидных бислоев. Липосомы могут использоваться для доставки средств, благодаря их уникальному свойству инкапсулировать область водного раствора внутри липофильной мембраны. Липофильные соединения могут растворяться в липидном бислое, и таким образом липосомы могут нести как липофильные, так и гидрофильные соединения. Чтобы доставить молекулы к участкам действия, липидный бислой может сливаться с другими бислоями, такими как клеточные мембраны, доставляя таким образом содержимое липосомы. При получении липосом в растворе средства указанное средство может быть доставлено во внутреннюю полость липосомы.
Липосомы, используемые в композициях по настоящему изобретению, конкретно не ограничиваются конкретными липидами. В частности, липиды, используемые для образования указанных липосом, могут представлять собой любые подходящие липиды, известные в данной области. Эти липиды включают, но не ограничиваются ими, холестерин или его производные, фосфолипиды, лизофосфолипиды или их комбинации. Соответственно, в предпочтительном варианте осуществления указанные липосомы включают один или несколько липидов, выбранных из группы, состоящей из холестерина и его производных, фосфолипидов, лизофосфолипидов и их комбинаций. Предпочтительными производными холестерина в контексте настоящего изобретения являются стероиды и соединения, имеющие основную структуру стероидной молекулы. Предпочтительно указанные липосомы включают фосфолипиды, где указанные фосфолипиды могут быть синтетическими, полусинтетическими или природными фосфолипидами или их комбинациями. В общем, подходящие липиды выбраны из группы, состоящей из фосфатидилхолинов, фосфатидилэтаноламинов, фосфатидилинозитов, фосфатидилсеринов, цефалинов, фосфатидилглицеринов, лизофосфолипидов и их комбинаций. В конкретном варианте осуществления настоящего изобретения липосомы включают яичный фосфатидилхолин (E-PC; лецитин) и холестерин, предпочтительно в количестве примерно 85-95 моль% E-PC, более предпочтительно примерно 89 моль% E-PC и примерно 5-10 моль% холестерина. В одном варианте осуществления липосомы включают яичный фосфатидилхолин (E-PC; лецитин) и холестерин в количестве примерно 85-95 моль% E-PC, более предпочтительно примерно 89 моль% E-PC и примерно 5-10 моль% холестерина, где относительные количества относятся к соответствующему содержанию в бислое липосом.
Липосомы, которые следует использовать согласно настоящему изобретению, а также твердые пероральные лекарственные формы как таковые, могут дополнительно включать любые другие подходящие средства, известные в данной области, такие как, например, ингибиторы ферментов, усилители проницаемости или другие липофильные или гидрофильные вещества или их комбинации, которые можно использовать для стабилизации липосом или для изменения свойств липосом. Необязательные стабилизирующие средства включают тетраэфирные липиды. Такие липофильные или гидрофильные вещества конкретно не ограничены и известны в данной области. Они включают, например, витамин Е, жирные кислоты, воски и моно-, ди- и триглицериды и их смеси. Кроме того, могут быть добавлены вещества, повышающие биодоступность включенных активных средств, такие как ингибиторы ферментов, модуляторы плотных контактов, хелатирующие агенты или их смеси.
Конъюгаты, содержащиеся в липосомах по настоящему изобретению, представляют собой конъюгаты по меньшей мере одного типа проникающего в клетки пептида (CPP), и соединения, выбранного из липида и жирной кислоты. Соединение, с которым конъюгированы CPP, предпочтительно представляет собой подходящий липид, как определено выше, например, липид, выбранный из группы, состоящей из холестерина и его производных, фосфолипидов, лизофосфолипидов и их комбинаций, где фосфолипиды являются предпочтительными, и где указанные липиды могут быть модифицированными и/или активированными липидами. Примерами соединений в этом аспекте являются 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин-N-[4-(p-малеимидометил)циклогексанкарбоксамид] (натриевая соль) или 1,2-диолеоил-sn-глицеро-3-фосфоэтанол-амин-N-[4-(p-малеимидометил)циклогексан-карбоксамид] (натриевая соль). Предпочтительно, СРР конъюгированы с указанным соединением через линкер, такой как линкер, выбранный из группы, состоящей из бифункциональных PEG-линкеров, известных в данной области. CPP могут быть мономерными или димеризованными. В этом контексте мономерные CPP могут быть ковалентно конъюгированы с фосфолипидом через линкер, или димеризованные CPP, где возможны гомо- и гетеродимеры, ковалентно конъюгированы с фосфолипидом через линкер. Димеризация CPP может быть осуществлена любым способом, известным в данной области. В конкретном варианте осуществления СРР димеризуют с помощью трипептида KAK. Однако прикрепление также возможно без линкера, например, при использовании модифицированных и/или активированных липидов, таких как, например, фосфолипиды с малеимид-модифицированной головной группой.
Подходящие фосфолипиды для ковалентного конъюгирования CPP особенно не ограничены определенными фосфолипидами. В частности, фосфолипиды, используемые для ковалентного конъюгирования CPP, могут быть любыми подходящими фосфолипидами, известными в данной области техники, где указанные фосфолипиды могут быть синтетическими, полусинтетическими или природными фосфолипидами или их комбинациями. Обычно подходящие фосфолипиды могут быть выбраны из группы, состоящей из фосфатидилхолинов, фосфатидилэтаноламинов, фосфатидилинозитов, фосфатидилсеринов, цефалинов, фосфатидилглицеринов, лизофосфолипидов и их комбинаций. Конкретным фосфолипидом в этом аспекте является яичный фосфатидилхолин (E-PC; лецитин). Кроме того, подходящие фосфолипиды включают PEG-модифицированные версии вышеуказанных фосфолипидов, например, DSPE-PEG(2000) малеимид (1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин-N-[малеимид(полиэтиленгликоль)-2000] (аммониевая соль)).
Подходящие линкеры для ковалентного конъюгирования CPP с фосфолипидами конкретно не ограничены и известны в данной области. Они включают, например, бифункциональные PEG-линкеры в целом; например, SM(PEG)24 (пегилированный длинноцепочечный сшивающий агент SMCC). Подходящими типичными линкерами являются SMCC (сукцинимидил-4-(N-малеимидометил)циклогексан-1-карбоксилат)-линкер и линкер 6-малеимидогексановой кислоты. Длина PEG-фрагмента в таких линкерах влияет на эффективность инкапсуляции лекарственных средств, которые могут быть включены в липосомы, содержащиеся в твердых пероральных лекарственных формах по настоящему изобретению. Соответственно, указанный PEG-фрагмент предпочтительно имеет длину 8-50 отдельных PEG-единиц. Кроме того, способы ковалентного конъюгирования CPP с фосфолипидами через линкеры конкретно не ограничены и известны в данной области техники.
В предпочтительных вариантах осуществления липосомы, присутствующие в твердых пероральных лекарственных формах по настоящему изобретению, не включают каких-либо тетраэфирных липидов (TEL), которые представляют собой специфические липиды, полученные из археи, например, экстремофильный архей Sulfolobus acidocaldarius.
Согласно настоящему изобретению твердые пероральные лекарственные формы могут дополнительно включать по меньшей мере один фармацевтически приемлемый эксципиент и/или по меньшей мере один ингибитор протеазы и/или по меньшей мере один ингибитор липазы в твердой пероральной лекарственной форме, такой как капсула, покрытая энтеросолюбильной оболочкой, или таблетка, покрытая энтеросолюбильной оболочкой. Они могут присутствовать во внутренней полости липосом, в липидном двойном слое липосом (например, образуя часть двойного слоя путем ковалентного или нековалентного присоединения) или снаружи липосом (например, в других частях лекарственной формы). Предпочтительно, указанный, по меньшей мере, один фармацевтически приемлемый эксципиент выбран из группы, состоящей из сорбитан моностеарата, трипальмитина, цетилпальмитата, альгината, этилолеата, C8 триглицеридов, C10 триглицеридов, целлюлозы, дисахаридов, моносахаридов, олигосахаридов, стеарата магния, кукурузного крахмала, лимонной кислоты, винной кислоты, кислых солей аминокислот и их комбинаций. Кроме того, указанный, по меньшей мере, один ингибитор протеазы предпочтительно выбран из группы, состоящей из апротинина, ингибитора трипсина соевых бобов, бацитрацина, гликохолата натрия, бестатина, лейпептина, цистатина, камостат мезилата и их комбинаций. Кроме того, указанный, по меньшей мере, один ингибитор липазы предпочтительно выбран из группы, состоящей из орлистата, липстатина, хитина, хитозана, сапонина, флавоноидного гликозида, полифенола, эбелактона A и B, эстерастина, валилактона, панклицина, проантоцианидина, вибралактона и их комбинаций.
CPP, которые можно использовать в связи с настоящим изобретением, конкретно не ограничены и известны в данной области техники. Предпочтительно, CPP представляют собой CPP, показывающие положительный суммарный заряд. Кроме того, CPP по настоящему изобретению могут быть линейными или циклизованными CPP, причем особенно предпочтительными являются циклизованные CPP. Термины «циклизованный пептид», «циклический пептид» и «циклопептид» используются в настоящем описании как синонимы.
Более предпочтительно, CPP выбраны из группы, состоящей из
- пенетратина (такого как SEQ ID NO:1; RQIKIWFQNRRMKWKK), полученного из Drosophila melanogaster,
- TAT (трансактиватор транскрипции)-пептида (такого как SEQ ID NO:2; CGRKKKRRQRRRPPQC), полученного из HIV-1,
- MAP (модель амфипатического пептида) (такого как SEQ ID NO:3; GALFLGFLGAAGSTMGAWSQPKSKRKV), который представляет собой искусственный пептид,
- R9 (такого как SEQ ID NO:4; RRRRRRRRR), который представляет собой искусственный пептид,
- pVEC (такого как SEQ ID NO:5; LLIILRRRIRKQAHAHSK-амид), который представляет собой СРР, полученный из мышиного сосудистого эндотелиального кадгерина,
- транспортана (такого как SEQ ID NO:6; GWTLNSAGYLLGKINLKALAALAKISIL-amide), который получен из человеческого нейропептида галанина, и
- MPG (такого как SEQ ID NO:7; GALFLGFLGAAGSTMGAWSQPKSKRKV), который получен из HIV,
их комбинаций и их димеров. В варианте осуществления CPP представляет собой циклизованный R9.
В этом контексте все вышеперечисленные пептиды могут присутствовать в линейной или в циклизованной форме, причем циклизованная форма является предпочтительной. Кроме того, CPP могут состоять из L-аминокислот, D-аминокислот или их смесей, причем для линейных CPP предпочтительными являются D-аминокислоты.
В отличие от линейных CPP, проникающие в клетку циклопептиды являются менее чувствительными к гидролизу пептидазами, т.е., как было показано, они являются ферментативно более стабильными.
Используемый в настоящем описании термин «циклизованный пептид» не следует истолковывать как относящийся к пептиду, имеющему только одну кольцевую систему, т.е., настоящее изобретение не ограничивается моноциклическими пептидами. Соответственно, настоящее изобретение также относится к циклопептидам, в которых две или более кольцевых систем ковалентно связаны друг с другом. Кроме того, циклопептиды могут также включать аминокислоты, которые не являются частью кольцевой системы. Таким образом, пептидные боковые цепи могут присутствовать в циклопептидах. Предпочтительно циклопептиды представляют собой моноциклические пептиды и более предпочтительно моноциклические пептиды, не имеющие пептидные боковые цепи.
В варианте осуществления циклопептиды представляют собой положительно заряженные. Таким образом, увеличивается поглощение соответствующих липосом слизистой оболочкой. Например, циклопептиды, как определено выше, могут включать в основном фрагменты лизина и/или аргинина, которые имеют изоэлектрические точки около 9,5 и 11, соответственно. Благодаря своей дополнительной аминогруппе эти две аминокислоты являются положительно заряженными в нейтральных и даже в слабоосновных условиях. Соответственно, циклопептид, в основном включающий фрагменты указанных двух специфических аминокислот, также является положительно заряженным в нейтральных и слабоосновных условиях. В настоящем описании термин «в основном включающий» означает, что по меньшей мере 50%, предпочтительно по меньшей мере 60%, более предпочтительно по меньшей мере 70% и особенно предпочтительно по меньшей мере 80% аминокислот, образующих молекулу циклопептида, представляют собой фрагменты лизина и/или аргинина. Таким образом, гарантируется, что циклопептиды имеют положительный заряд в нейтральных и слабоосновных условиях, то есть имеют изоэлектрическую точку более 7. Следовательно, в конкретном варианте осуществления настоящего изобретения циклопептиды указанных выше липосом, входящих в состав твердых пероральных лекарственных форм по настоящему изобретению, имеют изоэлектрическую точку более 7,0, предпочтительно более 7,5, более предпочтительно более чем 8,0 и особенно предпочтительно более 8,5. В этом контексте изоэлектрическая точка циклопептида является средним арифметическим изоэлектрических точек аминокислот, образующих циклопептид.
В конкретном варианте осуществления настоящего изобретения циклопептиды включают от 2 до 19, предпочтительно от 3 до 16, более предпочтительно от 4 до 14 и особенно предпочтительно от 6 до 12 фрагментов аргинина, а также один фрагмент, выбранный из группы, состоящей из тирозина, треонина, серина и цистеина. Например, циклопептиды могут включать девять фрагментов аргинина и один фрагмент цистеина в кольцевой системе и называются как производное циклического цистеина R9 (такое как SEQ ID NO:8; RRRRRRRRRC). Другим предпочтительным примером является циклопептид, включающий девять фрагментов аргинина и один фрагмент лизина в кольцевой системе, который называется как производное R9K (SEQ ID NO:9; RRRRRRRRRK).
Аминокислоты, образующие циклопептиды, не ограничиваются протеиногенными аминокислотами. В настоящем описании аминокислоты могут быть выбраны из любых аминокислот, известных в данной области техники, и могут включать соответствующий D-энантиомер, L-энантиомер или любую их смесь. Здесь аминокислоты могут быть дополнительно функционализированы таким образом, чтобы ковалентно присоединяться к модифицированным биоразлагаемым полимерным цепям.
Согласно настоящему изобретению CPP-конъюгаты являются частью двойного липидного слоя липосомы. В этом контексте термин «являющийся частью двойного липидного слоя липосомы» предназначен для обозначения того факта, что указанные конъюгаты интегрированы в указанный двойной липидный слой через свою липидную и/или жирнокислотную часть, т.е. указанная липидная и/или жирнокислотная часть содержится в двойном липидном слое, тогда как CPP-часть конъюгата присутствует на поверхности липосомы.
Предпочтительно липосомы, содержащиеся в твердых пероральных лекарственных формах по настоящему изобретению, включают указанные СРР в количестве 0,05-5 моль% или 0,05-3 моль%, предпочтительно 0,05-2 моль% и более предпочтительно 0,1-1 моль% из расчета на общее количество липидов и/или жирных кислот. В случае мономерных СРР, они предпочтительно содержатся в количестве 0,05-2 моль%, предпочтительно 0,1-1 моль%, из расчета на общее количество липидов и/или жирных кислот. В случае димеризованных СРР, они предпочтительно содержатся в количестве 0,05-0,5 моль%, предпочтительно 0,1 моль%, из расчета на общее количество липидов и/или жирных кислот. Было обнаружено, что коэффициент полидисперсности липосом увеличивается с увеличением количества CPP в случае высоких пропорций CPP. Если количество CPP слишком велико, липосомы не образуются из-за высокого положительного заряда CPP, который приведет к высоким силам отталкивания.
Липиды и/или жирные кислоты, используемые для получения липосом, также могут быть присоединены к самонаводящимся структурам, таким как пептидные последовательности, антитела, рецептор лиганды, поверхностно-активные вещества и/или их комбинации.
В предпочтительном варианте осуществления настоящего изобретения липосомы, содержащиеся в твердых пероральных лекарственных формах по настоящему изобретению, лиофилизированы. Средства для лиофилизации липосом конкретно не ограничены и известны в данной области. Преимущественно липосомы могут быть лиофилизированы, например, с использованием 300-500 мМ сахарозы в качестве лиопротектора. Лиофилизация, как определено выше, обеспечивает длительное хранение твердых пероральных лекарственных форм по настоящему изобретению.
В предпочтительном варианте осуществления липосомы, содержащиеся в твердых пероральных лекарственных формах по настоящему изобретению, показывают Z-среднее значение, измеренное посредством динамического рассеяния света после разбавления в водной среде, самое большее 350 нм и коэффициент полидисперсности (PDI) самое большее 0,3, где Z-среднее от 100 до 200 нм и коэффициент полидисперсности от 0,1 до 0,3, предпочтительно около 0,2, является особенно предпочтительным.
Способы получения липосом особо не ограничены и известны в данной области. Они включают, например, гомогенизацию высокого давления, экструзию, введение этанола и двойное асимметричное центрифугирование (DAC).
В предпочтительных вариантах осуществления твердые пероральные лекарственные формы по настоящему изобретению могут дополнительно включать, по меньшей мере, одно терапевтическое средство и/или, по меньшей мере, одно диагностическое средство в лекарственной форме, такой как капсула, покрытая энтеросолюбильной оболочкой, или таблетка, покрытая энтеросолюбильной оболочкой.
Соответствующие терапевтические средства конкретно не ограничены и включают любые средства, для которых пероральная доставка может представлять интерес. Они включают, например, макромолекулы, такие как пептидные лекарственные средства (например, ванкомицин, глатирамер ацетат, Myrcludex B, октреотид, все виды инсулина и лираглутид, а также другие аналоги GLP (глюкагоноподобного пептида), такие как эксенатид, ликсисенатид, албиглутид, дулаглутид, таспоглютид и семаглутид) и белки или антитела (например, этанерцепт; Пэгфилграстим; адалимумаб, инфликсимаб, ритуксимаб, эпоэтин альфа, тратузумаб, ранибизумаб, бета-интерферон, омализумаб). Другие примеры включают фармацевтически активные средства, выбранные из группы, состоящей из гормона роста человека, гормона, высвобождающего гормон роста, пептида, высвобождающего гормон роста, интерферонов, колониестимулирующих факторов, интерлейкинов, фактора активации макрофагов, пептида макрофагов, В-клеточного фактора, Т-клеточного фактора, белка А, ингибитора аллергии, гликопротеинов некроза клеток, иммунотоксина, лимфотоксина, фактора некроза опухолей, супрессоров опухолей, фактора роста метастаз, альфа-1-антитрипсина, альбумина и его полипептидных фрагментов, аполипопротеин-Е, эритропоэтина, фактора VII, фактора VIII, фактора IX, активирующего плазминоген фактора, урокиназы, стрептокиназы, белка С, С-реактивного белка, ингибитора ренина, ингибитора коллагеназы, супероксиддисмутазы, тромбоцитарного фактора роста, эпидермального фактора роста, остеогенного фактора роста, костного морфогенетического белка, кальцитонина, инсулина, атриопептина, хрящевого индуцирующего фактора, фактора роста соединительной ткани, фолликулостимулирующего гормона, лютеинизирующего гормона, гормона, стимулирующего высвобождение лютеинизирующего гормона, факторов роста нервов, паратиреоидного гормона, релаксина, секретина, соматомедина, инсулиноподобного фактора роста, адренокортикального гормона, глюкагона, холецистокинина, панкреатического полипептида, гастрин-высвобождающего пептида, кортикотропин-рилизинг-фактора, тиреостимулирующего гормона, моноклональных или поликлональных антител против различных вирусов, бактерий или токсинов, вакцинных антигенов из вирусов, октреотида, циклоспорина, рифампицина, лопинавира, ритонавира, ванкомицина, телаванцина, оритаванцина, далбаванцина, бисфосфонатов, итраконазола, даназола, паклитаксела, циклоспорина, напроксена, капсаицина, альбутерол сульфата, тербуталин сульфата, дифенгидрамин гидрохлорида, хлорфенирамин малеата, лоратидин гидрохлорида, фексофенадин гидрохлорида, фенилбутазона, нифедипина, карбамазепина, напроксена, циклоспорина, бетаметазона, даназола, дексаметазона, преднизона, гидрокортизона, 17 бета-эстрадиола, кетоконазола, мефенамовой кислоты, беклометазона, алпразолама, мидазолама, миконазола, ибупрофена, кетопрофена, преднизолона, метилпреднизона, фенитоина, тестостерона, флунизолида, дифлунизала, будесонида, флутиказона, инсулина, глюкагоноподобного пептида, C-пептида, эритропоэтина, кальцитонина, лютеинизирующего гормона, пролактина, адренокортикотропного гормона, лейпролида, интерферона альфа-2b, интерферона бета-Ia, сарграмостима, альдеслейкина, интерферона альфа-2а, ингибитора интерферона альфа-n3альфа-протеиназы, этидроната, нафарелина, хорионического гонадотропина, простагландина E2, эпопростенола, акарбоза, метформина, десмопрессина, циклодекстрина, антибиотиков, противогрибковых препаратов, стероидов, противораковых лекарственных средств, анальгетиков, противовоспалительных средств, антигельминтиков, антиаритмических средств, пенициллинов, антикоагулянтов, антидепрессантов, противодиабетических средств, противоэпилептических средств, антигистаминных средств, антигипертензивных средств, антимускариновых средств, противомикобактериальнох средств, противоопухолевых средств, ЦНС-активных средств, иммунодепрессантов, антитиреоидных средств, противовирусных средств, анксиолитических седативных средств, снотворных, нейролептиков, вяжущих средств, бета-адреноблокаторов, препаратов и заменителей крови, кардиоинотропных средств, контрастных средств, кортикостероидов, средств от кашля, отхаркивающих средств, муколитиков, диуретиков, дофаминергических средств, антипаркинсонических средств, кровоостанавливающих средств, иммунологических средств, регуляторов липида, мышечных релаксантов, парасимпатомиметиков, кальцитонина паращитовидной железы, простагландинов, радиофармацевтических средств, половых гормонов, стероидов, антиаллергических средств, стимуляторов, аноретиков, симпатомиметиков, тиреоидных препаратов, вазидилататоров, ксантинов, гепаринов, терапевтических олигонуклеотидов, соматостатинов и их аналогов, и их фармакологически приемлемых органических и неорганических солей или комплексов металлов.
Кроме того, соответствующие диагностические средства конкретно не ограничены и включают любые средства, для которых пероральная доставка может представлять интерес.
Указанные выше средства могут присутствовать в твердых пероральных лекарственных формах по настоящему изобретению, заключенных в липосомы, т.е. во внутреннюю полость указанных липосом, например, когда указанные средства являются гидрофильными, или интегрированы в липосомальную мембрану, например, когда указанные средства являются липофильными. В этом контексте инкапсуляция терапевтических и/или диагностических средств зависит от гидрофильности указанных средств и способа получения липосом.
В предпочтительном варианте осуществления содержание терапевтического и/или диагностического средства в твердых пероральных лекарственных формах в соответствии с настоящим изобретением составляет более 0 и не более 50% (масс./масс.) по отношению к твердой пероральной лекарственной форме, предпочтительно от 5 до 20% (масс./масс.).
Энтеросолюбильные покрытия, т.е., покрытия, которые являются устойчивыми к желудочному соку, для использования в контексте настоящего изобретения конкретно не ограничены и известны в данной области. Типичное покрытие содержит 3% (масс./масс.) воды, 1,25% (масс./масс.) триацетина, 82,75% (масс./масс.) изопропанола и 13% (масс./масс.) Eudragit L 100. В этом контексте Eudragit L 100 представляет собой анионный сополимер метакриловой кислоты и этилакрилата в соотношении кислота:сложный эфир 1:1.
Твердые пероральные лекарственные формы по настоящему изобретению могут быть использованы в медицине. Лекарственные формы по настоящему изобретению являются особенно полезными для лечения хронических заболеваний, которые требуют регулярного введения терапевтических средств. Предпочтительно указанные твердые пероральные лекарственные формы предназначены для применения при лечении сепсиса, диабета, ревматизма, акромегалии, всех видов гепатита, всех видов рака или анемии. Кроме того, лекарственные формы могут быть использованы для лечения аутоиммунных заболеваний, заболеваний, которые требуют введения иммунодепрессантов, инфекционных заболеваний, заболеваний, которые требуют введения гормонов роста, энзимодефицитных заболеваний или нейродегенеративных заболеваний (т.е. болезни Альцгеймера, болезни Паркинсона).
В связанном аспекте настоящее изобретение относится к применению твердой пероральной лекарственной формы по настоящему изобретению для пероральной доставки по меньшей мере одного терапевтического средства и/или по меньшей мере одного диагностического средства. Изобретение дополнительно включает способ лечения субъекта, нуждающегося в этом, включающий
- введение субъекту количества твердой пероральной лекарственной формы по настоящему изобретению, достаточного для достижения желаемого терапевтического эффекта при пероральной доставке.
Субъектом может быть млекопитающее, такое как человек.
В этом контексте термин «пероральная доставка» относится к доставке одного или нескольких средств путем перорального введения указанных лекарственных форм.
В этом аспекте все соответствующие ограничения и определения, предоставленные для других аспектов настоящего изобретения, используются аналогичным образом. В частности, твердые пероральные лекарственные формы, терапевтические средства и диагностические средства являются такими, как определено выше.
В дополнительном связанном аспекте настоящее изобретение относится к способу доставки по меньшей мере одного терапевтического средства и/или по меньшей мере одного диагностического средства субъекту, включающему стадию введения, предпочтительно перорального введения, твердой пероральной лекарственной формы по настоящему изобретению указанному субъекту.
В этом аспекте все соответствующие ограничения и определения, предоставленные для других аспектов настоящего изобретения, используются аналогичным образом. В частности, твердые пероральные лекарственные формы, терапевтические средства и диагностические средства являются такими, как определено выше.
Используемый в настоящем изобретении термин «примерно» предназначен для использования в качестве модификатора ±10% от указанного значения. Например, термин «примерно 5%» предназначен для охвата диапазона от 4,5 до 5,5%.
Термины «включающий/включает», «состоящий из/состоит из» и «состоящий по существу из/состоит по существу из» используются в настоящем документе взаимозаменяемо, то есть каждый из указанных терминов может быть явно заменен на один из двух других терминов.
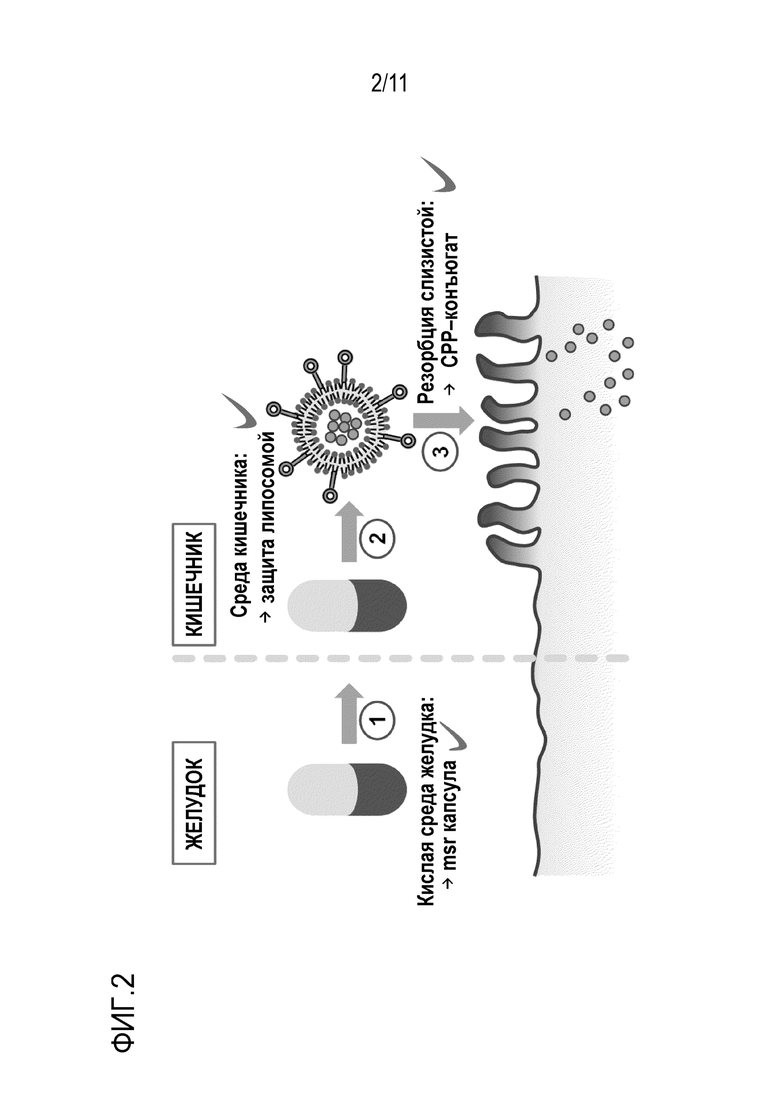
Настоящее изобретение преимущественно предоставляет, помимо прочего, твердые пероральные лекарственные формы, включающие СРР-связанные липосомы для перорального введения, причем указанные липосомы, например, содержатся в капсуле, покрытой энтеросолюбильной оболочкой, или таблетке, покрытой энтеросолюбильной оболочкой. CPP значительно увеличивают резорбцию липосом на слизистой оболочке кишечника, тогда как кишечнорастворимая лекарственная форма обеспечивает прохождение через желудок (фиг. 2). Таким образом, достигается значительное увеличение пероральной биодоступности лекарственных средств, содержащихся в указанных липосомах. Это обеспечивает возможность перорального введения широкого спектра макромолекулярных лекарств, что, в свою очередь, повышает комплаентность. Кроме того, лиофилизация липосом увеличивает стабильность при хранении соответствующих лекарственных форм.
Краткое описание фигур
На фигурах показано:
Фигура 1: В предшествующем уровне техники только лекарственные средства с молекулярной массой <500 Да можно было вводить перорально. Большинство лекарственных средств с более высокой молекулярной массой, например, пептиды и биологические препараты, должны вводиться парентерально.
Фигура 2: Липосомы, покрытые CPP, вводят перорально. Следовательно, липосомы лиофилизируют и переводят в твердую лекарственную форму. Прохождение через желудок обеспечивается энтеросолюбильным покрытием лекарственной формы. Кроме того, резорбция липосом на слизистой оболочке кишечника значительно повышается благодаря CPP.
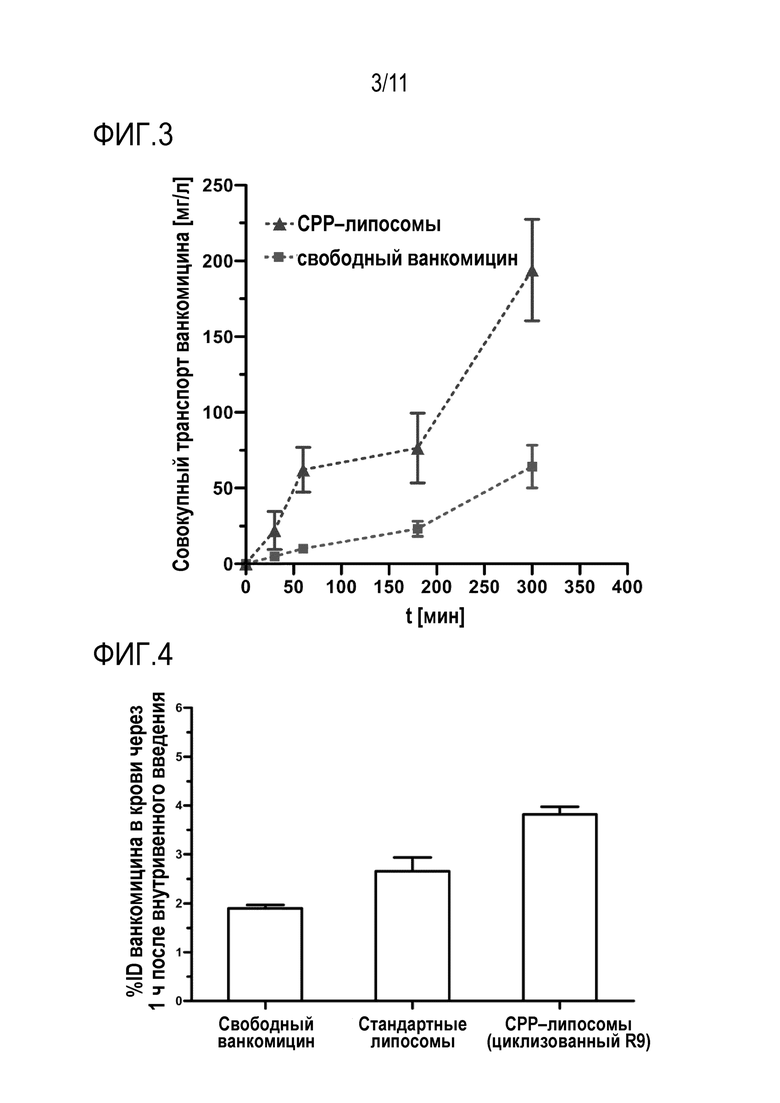
Фигура 3: Исследования в камере Уссинга липосом, покрытых циклическим R9 CPP по сравнению со свободным ванкомицином.
Фигура 4: Уровни ванкомицина в крови после перорального применения свободного ванкомицина и ванкомицина, включенного в липосомы, содержащие циклический R9 СРР, у крыс линии Wistar.
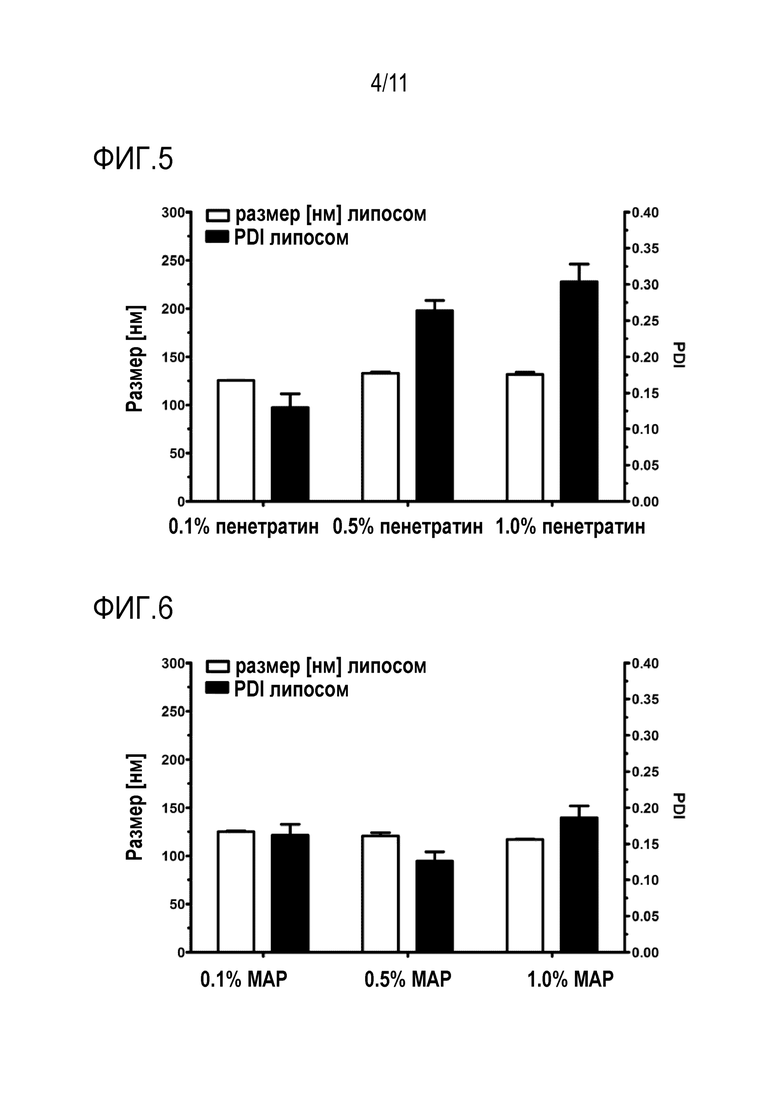
Фигура 5: Размер и PDI липосом, содержащих 0,1-1 моль% пенетратина CPP.
Фигура 6: Размер и PDI липосом, содержащих 0,1-1 моль% CPP MAP.
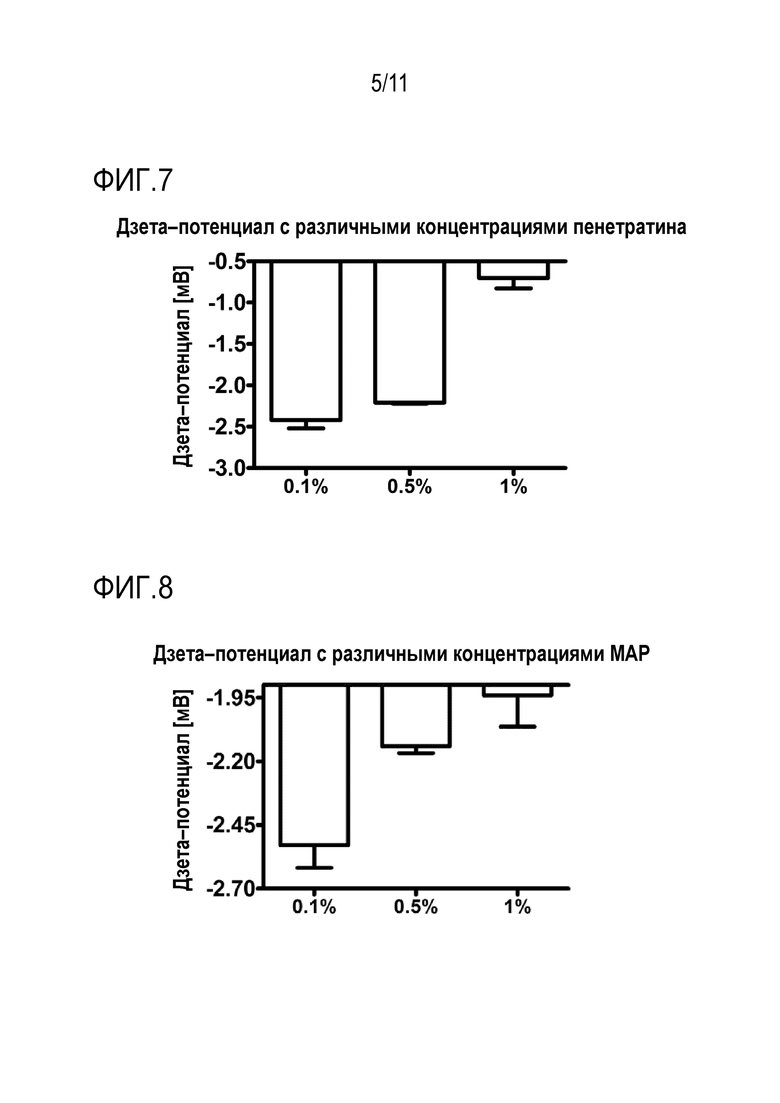
Фигура 7: Дзетапотенциалы липосом, содержащих 0,1-1 моль% пенетратина CPP.
Фигура 8: Дзетапотенциалы липосом, содержащих 0,1-1 моль% CPP MAP.
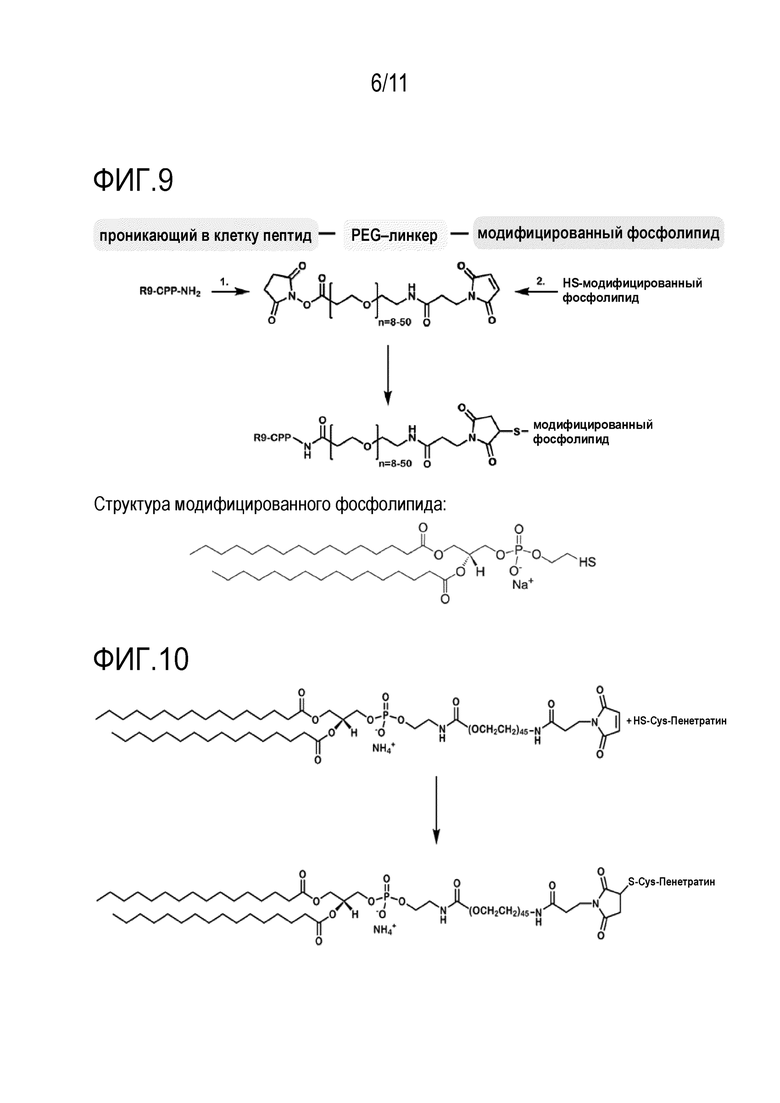
Фигура 9: Синтез предпочтительных, иллюстративных конъюгатов.
Фигура 10: Синтез предпочтительных, иллюстративных конъюгатов.
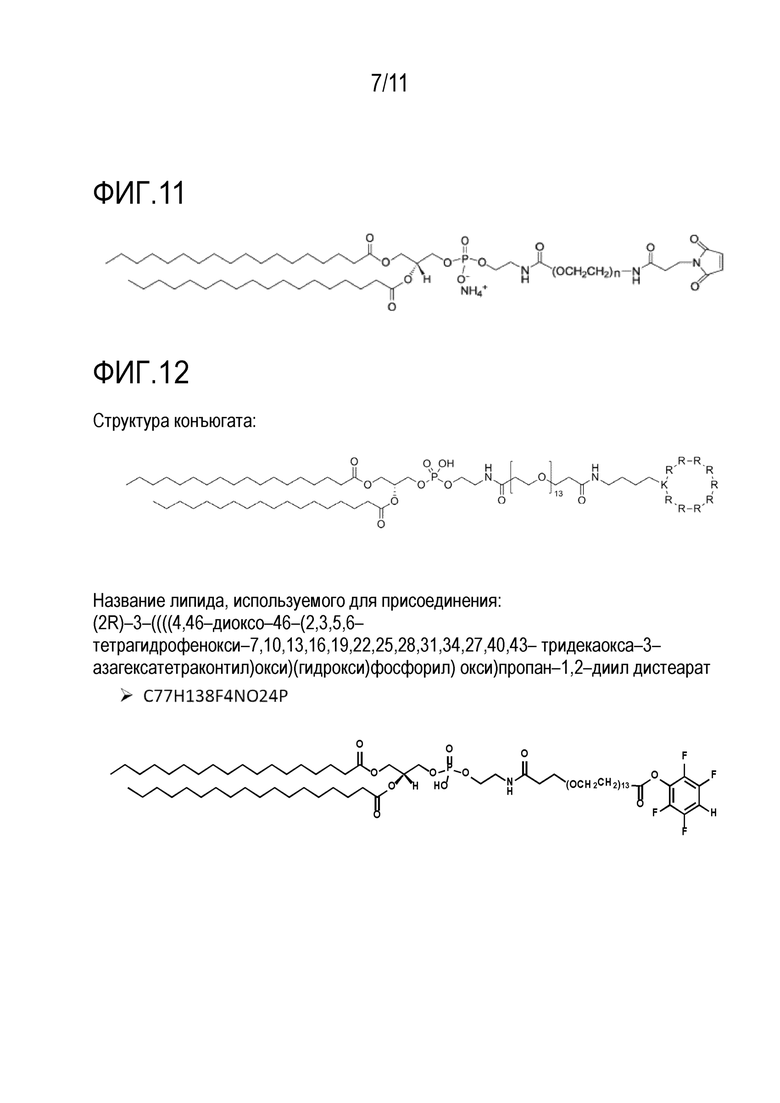
Фигура 11: Структура модифицированного фосфолипида, используемого для получения конъюгата.
Фигура 12: Пример конъюгата, использованного в исследованиях на животных, и модифицированного фосфолипида.
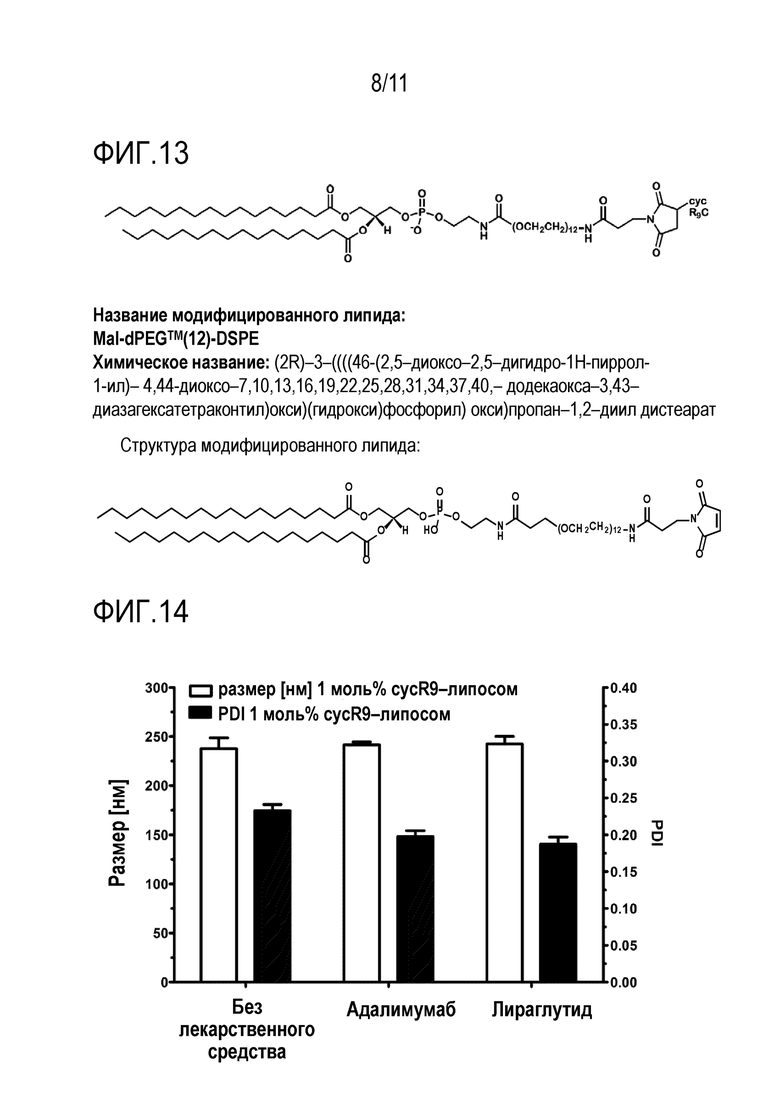
Фигура 13: Дополнительный пример конъюгата и модифицированного фосфолипида.
Фигура 14: PDI и размер разных липосом.
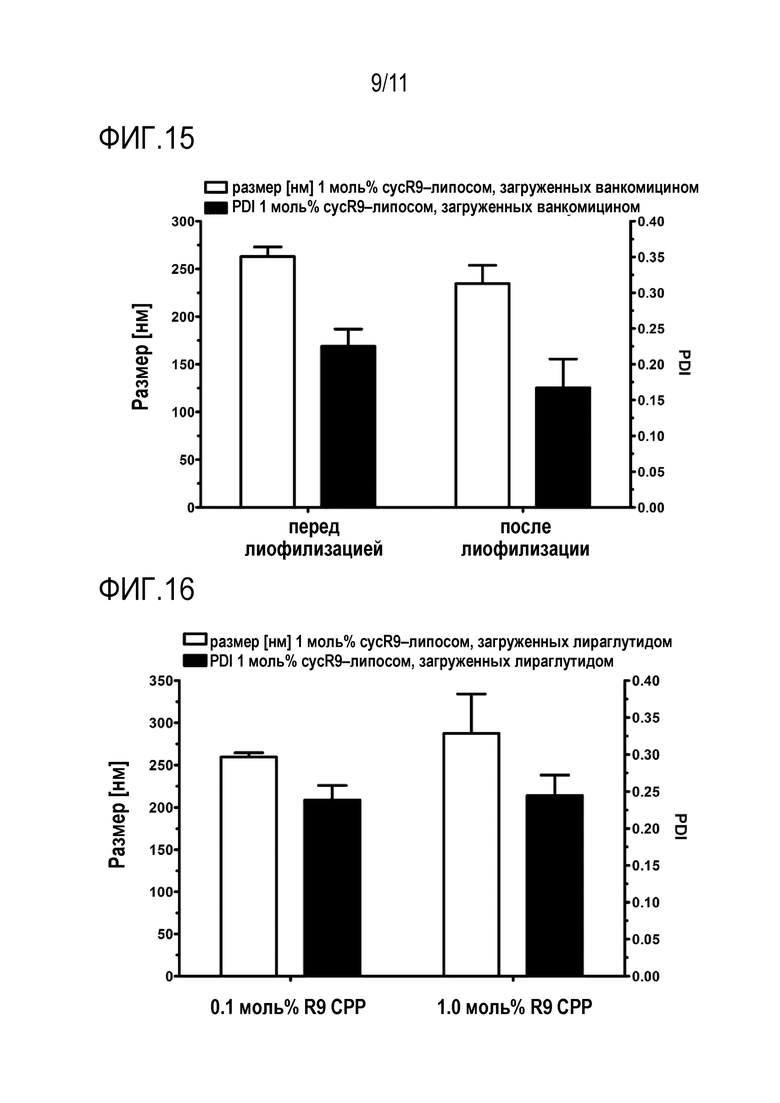
Фигура 15: PDI и размер ванкомицин-нагруженных липосом после лиофилизации.
Фигура 16: PDI и размер лираглутид-нагруженных липосом после лиофилизации.
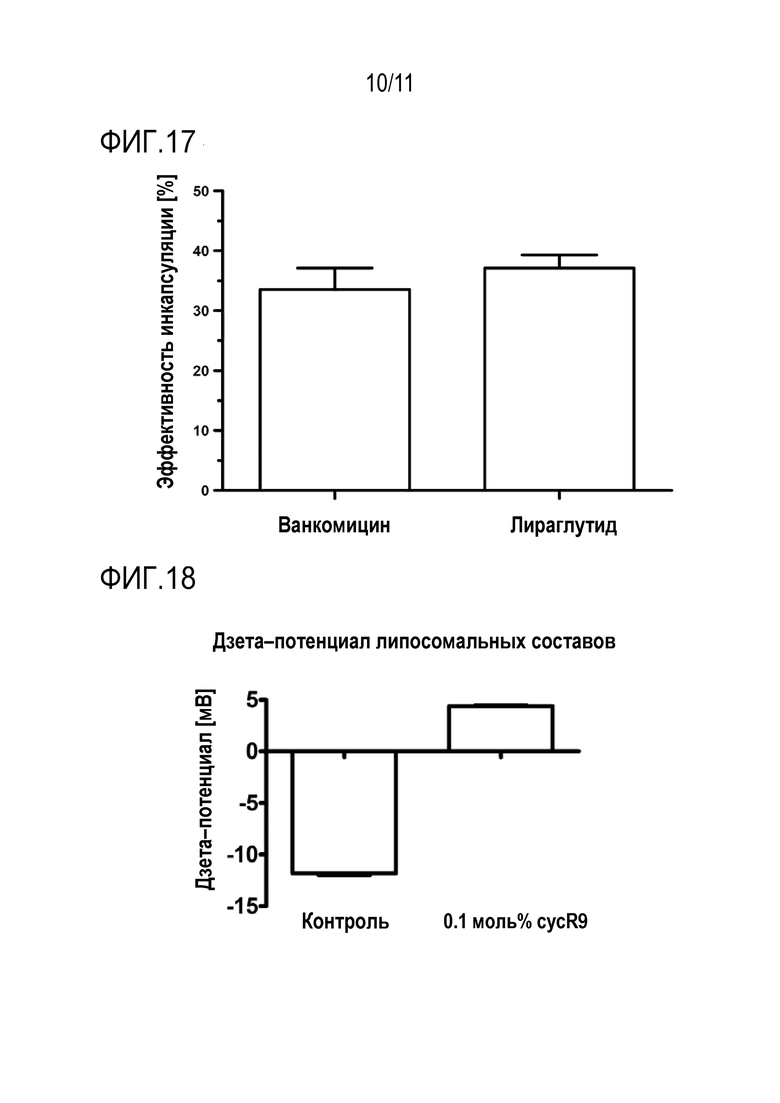
Фигура 17: Эффективность инкапсуляции для ванкомицина и лираглутида.
Фигура 18: Дзетапотенциал липосомальных составов, полученных по примеру 7.
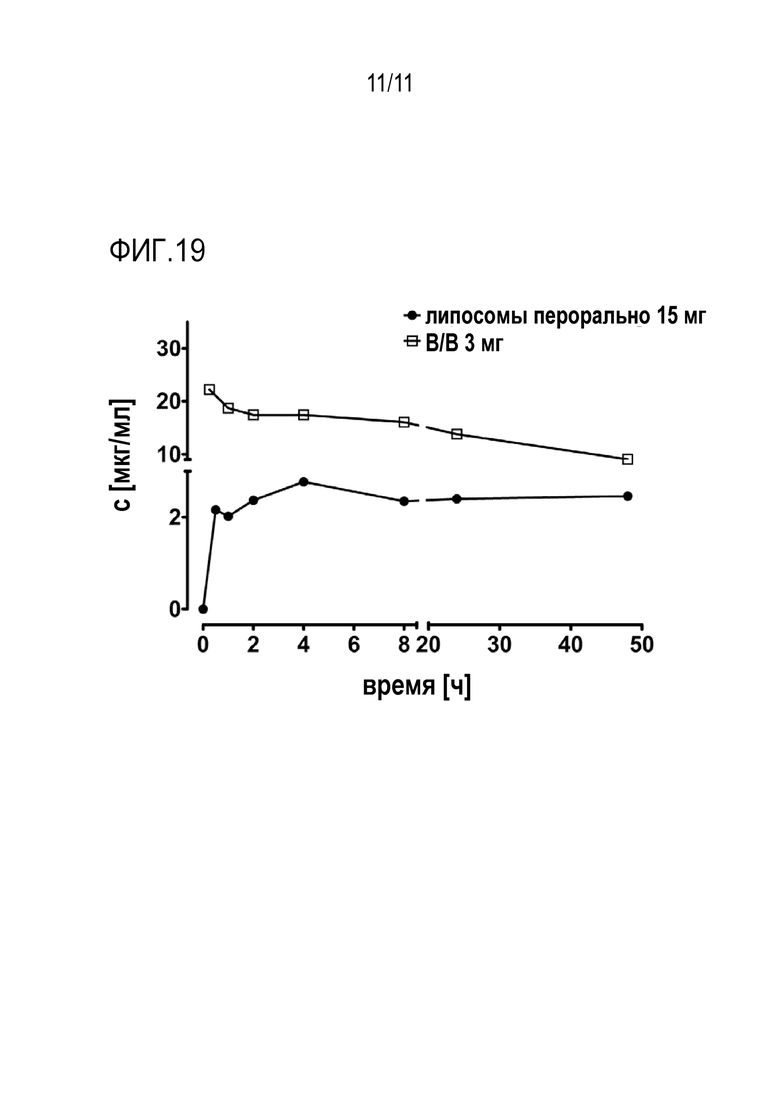
Фигура 19: Сравнение значений AUC для адалимумаба при внутривенном и пероральном введении у собак.
Примеры
Настоящее изобретение будет дополнительно проиллюстрировано в следующих примерах, не ограничиваясь ими.
1. Получение липосом
Липосомы получали пленочным методом с последующим двойным асимметричным центрифугированием с использованием SpeedMixer™. Хлороформ/метанол 9:1 (об./об.) использовали в качестве растворителя. Органический растворитель выпаривали потоком азота. Полученную липидную пленку сушили в течение 1 ч в вакуумной камере. После этого добавляли 20 мг стеклянных шариков. Липосомы получали путем быстрого перемешивания при 3000 об/мин в двойной асимметричной центрифуге с использованием специального держателя для флаконов. Было выполнено три анализа, и были добавлены разные количества PBS, как показано в следующей таблице.
2. Получение конъюгатов
a. Конъюгаты примеров, показанных на фиг. 3 и 4
На фиг. 9 показан синтез предпочтительных, иллюстративных конъюгатов, в частности тех, которые используются в примерах, показанных на фиг. 3 и 4. Линкер, используемый в этом примере, представлял собой SM(PEG)8, пегилированный длинноцепочечный SMCC кросслинкер и сукцинимидил([N-малеимидопропионамидо]-этиленгликоль)сложный эфир, соответственно. R9-CPP обозначает либо линейный, либо циклический нона-аргинин пептид. В случае циклического он циклизуется через лизин (R9K) и связывается с боковой цепью функциональную аминогруппу этого лизина.
Для связывания циклического CPP с бифункциональным PEG-линкером в качестве первой стадии циклизованный CPP связывали дополнительным лизином с линкером (1.). Для этой реакции использовали избыток CPP. На второй стадии этот промежуточный продукт связывали с модифицированным тиолом фосфолипидом (2.). Модифицированный фосфолипид представлял собой 1,2-дипальмитоил-sn-глицеро-3-фосфотиоэтанол, натриевую соль.
b. Конъюгаты примеров, показанных на фиг. 5-8
На фиг. 10 показан синтез предпочтительных, иллюстративных конъюгатов, в частности тех, которые используются в примерах, показанных на фиг.5-8.
Модифицированный цистеином пенетратин связывали с фосфолипидом, модифицированным в головной группе, что показано на фиг. 11. Его химическое название представляет собой 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин-N-[малеимид(полиэтиленгликоль)-2000], аммониевая соль.
c. Конъюгат, используемый в исследованиях на животных
На фигуре 12 показан конъюгат, который использовали в липосомах для исследований на животных, и модифицированный фосфолипид, используемый для получения конъюгата.
d. Дополнительный конъюгат
Получали дополнительный конъюгат, включающий циклический R9C. Полученный конъюгат и модифицированный липид, использованный для его получения, показаны на фиг. 13.
3. Определение уровней в крови
Уровни ванкомицина в крови оценивали у крыс линии Wistar после перорального введения лекарственного средства. В исследовании на животных сравнивали пероральную биодоступность липосом, включающих проникающие в клетку пептиды (СРР), конъюгированные, как показано на фиг.9, со свободным ванкомицином после перорального применения. Стандартные липосомы служили контролем и состояли из лецитина и холестерина. Уровни ванкомицина в крови определяли иммуноанализом. За 6 ч до перорального применения корм крыс убирали. Для перорального применения крыс подвергали действию наркоза с помощью изофлурана, и пероральное введение осуществляли с помощью желудочного зонда. Через 1 ч после перорального введения крыс умерщвляли и определяли уровни ванкомицина в крови. Результаты показаны на фиг. 4.
Испытание на животных показали значительное увеличение пероральной доступности ванкомицина при использовании СРР-липосом по сравнению со свободным пептидом и стандартными липосомами (фиг. 4).
4. Испытание в камере Уссинга
Для испытания в камере Уссинга использовали липосомальный состав, включающий 1 мол. % циклического R9-производного, показанного на фиг.9, и сравнивали со свободным пептидом (ванкомицином). Липосомы получали в фосфатном буфере с pH=6,8, как описано выше в примере 1. Для этого эксперимента тонкую кишку умерщвленных крыс (Sprague Dawley) промывали NaCl и затем фиксировали в камере Уссинга (поверхность 0,64 см2; Firma NaviCyte, MA1 66-00XX). После этого камеры заполняли инкубационной средой (буферный раствор Кребса-Рингера) и добавляли эквивалентные количества ванкомицина в липосомальном составе и свободный пептид. Испытание проводили в течение 5 часов. В определенные моменты времени отбирали пробы из акцепторной камеры и заменяли буфером Кребса-Рингера. Определяли совокупное количество ванкомицина в образцах. Результаты показаны на фиг. 3.
Исследования в камере Уссинга показали сильное увеличение транспорта ванкомицина через слизистый барьер для CPP-липосом в отличие от свободного пептида. Следовательно можно предположить, что CPP-липосомы сильно усиливают транспорт веществ через слизистые барьеры (фиг. 3).
5. Размер частиц и PDI
Размер частиц и PDI всех липосомальных составов определяли при комнатной температуре с использованием Zetasizer Nano ZS от Malvern™ (Malvern Instruments Ltd., Worcestershire, United Kingdom). Размер и PDI измеряли после разбавления до концентрации липидов 0,076 мг/мл с помощью 10 мМ фосфатного буфера с pH 7,4 с использованием автоматического режима.
На фиг. 5 показан размер и PDI липосом, содержащих 0,1-1 моль% CPP пенетратина. Размер липосом остается почти постоянным, в то время как PDI показывает увеличение при использовании более высоких количеств пенетратина.
На фиг. 6 показаны размер и PDI липосом, содержащих 0,1-1 моль% CPP MAP. Количество CPP, включенного в липосомы, не влияло на размер липосом и PDI.
6. Дзета-потенциал
Дзета-потенциал всех липосомальных составов определяли при комнатной температуре с использованием Zetasizer Nano ZS от Malvern™ (Malvern Instruments Ltd., Worcestershire, United Kingdom). Дзета-потенциал определяли после разбавления до концентрации липидов 0,95 мг/мл 50 мМ фосфатным буфером с pH 7,4. Параметры по умолчанию для автоматического режима Zetasizer Nano ZS от Malvern™ (Malvern Instruments Ltd., Worcestershire, United Kingdom) были следующими: количество измерений=3; продолжительность анализа=10 с; количество анализов=10; время установления равновесия=60 с; индекс рефракции растворителя 1,330; индекса рефракции полистирольной кюветы 1,590; вязкость=0,8872 мПа с; температура=25°С; диэлектрическая постоянная=78,5 Ф/м; режим обратного рассеяния (173°); автоматический выбор напряжения; уравнение Смолуховского.
На фиг. 7 сравниваются дзета-потенциалы липосом, содержащих 0,1-1 мол.% CPP-пенетратина. Чем выше количество CPP, тем выше был дзета-потенциал из-за положительного заряда CPP.
На фиг. 8 показаны дзета-потенциалы липосом, содержащих 0,1-1 моль% CPP MAP. Чем выше количество CPP, тем выше дзета-потенциал. Обычно желательно, чтобы дзета-потенциал находился в диапазоне от -5 до 10 мВ, причем предпочтительным является значение от -3 до 7 мВ. По возможности, дзета-потенциал должен быть положительным. Однако, если дзета-потенциал слишком высок, липосомы не будут стабильными.
7. Получение конъюгата для исследований на животных (адалимумаб у собак)
Пептидный синтез циклизованного R9 проводили на хлор-(2'-хлор)тритил полистирольной смоле с емкостью 0,89 ммоль/г. Загрузку осуществляли с 0,8 ммоль Fmoc-Lys(Boc)-OH на грамм смолы в DCM с 3 эквивалентами DIPEA в течение 2 часов. Избыточные 2-хлортритильные функции гасили путем загрузки MeOH со смесью DCM/MeOH/DIPEA 17:2:1. Девять последовательных стадий сочетания Fmoc-Arg(Pbf)-OH с последующим в DMF с избытком 7 эквивалентов аминокислоты; 6,6 эквивалентов HBTU и 4 эквивалента DIPEA. Fmoc-группы удаляли обработкой 20% пиперидином в DMF. В промежутке между стадиями смолу тщательно промывали DMF.
Расщепление пептида, защищенного боковой цепью, достигали смесью DCM/трифторэтанол/уксусная кислота в соотношении 7:2:1. Раствор для расщепления трижды выпаривали вместе с толуолом на ротационном испарителе. Пептид, защищенный боковой цепью, растворяли в DMF в концентрации 3 мг/мл. Циклизацию проводили с 4 эквивалентами PyAOP и DIPEA при комнатной температуре в течение ночи. После прекращения реакции с водой раствор концентрировали до пятидесятой-сотой исходного объема и защищенный боковой цепью циклопептид осаждали, выливая в холодный трет-бутилметиловый эфир.
Осажденный защищенный циклопептид сушили и затем снимали защиту смесью 5% дитиоэтанола в TFA. После осаждения диэтиловым эфиром циклопептид очищали.
Полученный пептид затем использовали для получения конъюгатов и липосом, как описано ранее. Липосомы нагружали адалимумабом и лираглутидом, соответственно. Также получали контрольные липосомы без препарата.
Размер и PDI липосом показаны на фиг. 14.
Липосомы лиофилизировали. PDI и размер ванкомицин-нагруженных липосом после лиофилизации показаны на фиг. 15. PDI и размер ванкомицин-нагруженных липосом после лиофилизации показаны на фиг. 16. Лиофилизация не оказала существенного влияния на размер или PDI.
Дзета-потенциал этих липосомальных составов показан на фиг. 18. В связи с высоким положительным зарядом CPP-конъюгата, cycR9-CPP-липосомы демонстрируют положительный дзета-потенциал в отличие от контрольных липосом.
8. Эффективность инкапсуляции
Эффективность инкапсуляции всех пептидных модельных веществ определяли с помощью ВЭЖХ с обращенной фазой (Agilent 1100 Series) с использованием колонки C18 (Chromolith® Performance RP-18e, 100-3 мм) с использованием линейного градиента 0,1% TFA в воде (элюент А) до 0,1% TFA в ацетонитриле (элюент B) в течение 5 мин (скорость потока 2 мл/мин; поглощение УФ-излучения λ=214 нм). После процесса быстрого перемешивания липосомы разделяли на две части по 100 мкл каждая. Часть 1 использовали для расчета значения 100%, полученного разрушением липосом путем добавления 50 мкл 1% Triton™ X-100, и определения площади под кривой (AUC) модельного вещества с помощью ВЭЖХ. Часть 2 очищали гель-фильтрационной хроматографией Sephadex G-25 (колонки NAP™-5) и определяли количественно так же, как в части 1.
Эффективность инкапсуляции E(%) рассчитывали с использованием следующего уравнения:
E(%)=([AUC]модельного вещества части2/[AUC] модельного вещества части1) ×100%
где [AUC] модельного вещества части2 представляет собой концентрацию модельного вещества в очищенной липосомальной фракции, а [AUC] модельного вещества части1 представляет собой концентрацию модельного вещества в липосомальной суспензии
На фиг.17 сравнение результатов для обоих модельных веществ.
9. Исследования на животных
Исследование PK с однократным введением перорального адалимумаба (Humira™) в сравнении с Адалимумабом после внутривенного введения. В качестве животных использовали 2 собак породы бигль. Пероральная доза адалимумаба составляла 15 мг, инкапсулированных в липосомы, содержащие 1 моль% конъюгата cycR9, описанного ранее. Липосомы лиофилизировали и затем помещали в капсулу с энтеросолюбильным покрытием. Внутривенная доза составила 3 мг. Временные точки отбора образцов крови для перорального введения были: 0 (до введения дозы), 0:15, 0:30, 1:00, 2:00, 4:00, 8:00, 24:00, 48:00, 96:00 часов после введения. Временные точки отбора образцов крови для внутривенного введения были: 0 (до введения дозы), 0:15, 0:30, 1:00, 2:00, 4:00, 8:00, 24:00, 48:00, 96:00 часов после введения. Все образцы крови анализировали с помощью ELISA (IDKmonitor® Adalimumab drug level ELISA, Immundiagnostik AG, Bensheim).
Результаты проиллюстрированы на фиг. 19. В результате сравнения значений AUC можно получить биодоступность адалимумаба 3,55% после перорального введения. Эти результаты демонстрируют огромный потенциал этого липосомального состава для пероральной доставки макромолекулярных средств. Насколько известно заявителям, о такой высокой биодоступности для перорального введения антител ранее не сообщалось.
--->
Список последовательностей
<110> Universität Heidelberg
<120> ЛИПОСОМАЛЬНЫЕ КОМПОЗИЦИИ И СОДЕРЖАЩИЕ ИХ ТВЕРДЫЕ
ПЕРОРАЛЬНЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ
<130> UH0001P-WO
<160> 8
<170> PatentIn version 3.5
<210> 1
<211> 16
<212> БЕЛОК
<213> Drosophila melanogaster
<400> 1
Arg Gln Ile Lys Ile Trp Phe Gln Asn Arg Arg Met Lys Trp Lys Lys
1 5 10 15
<210> 2
<211> 16
<212> БЕЛОК
<213> Вирус иммунодефицита человека типа 1
<400> 2
Cys Gly Arg Lys Lys Lys Arg Arg Gln Arg Arg Arg Pro Pro Gln Cys
1 5 10 15
<210> 3
<211> 27
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> искусственно сконструированный проникающий в клетку пептид
<400> 3
Gly Ala Leu Phe Leu Gly Phe Leu Gly Ala Ala Gly Ser Thr Met Gly
1 5 10 15
Ala Trp Ser Gln Pro Lys Ser Lys Arg Lys Val
20 25
<210> 4
<211> 9
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> искусственно сконструированный проникающий в клетку пептид
<400> 4
Arg Arg Arg Arg Arg Arg Arg Arg Arg
1 5
<210> 5
<211> 18
<212> БЕЛОК
<213> Mus musculus
<220>
<221> MISC_FEATURE
<222> (18)..(18)
<223> K-амид
<400> 5
Leu Leu Ile Ile Leu Arg Arg Arg Ile Arg Lys Gln Ala His Ala His
1 5 10 15
Ser Lys
<210> 6
<211> 28
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (28)..(28)
<223> L-амид
<400> 6
Gly Trp Thr Leu Asn Ser Ala Gly Tyr Leu Leu Gly Lys Ile Asn Leu
1 5 10 15
Lys Ala Leu Ala Ala Leu Ala Lys Ile Ser Ile Leu
20 25
<210> 7
<211> 27
<212> БЕЛОК
<213> Вирус иммунодефицита человека
<400> 7
Gly Ala Leu Phe Leu Gly Phe Leu Gly Ala Ala Gly Ser Thr Met Gly
1 5 10 15
Ala Trp Ser Gln Pro Lys Ser Lys Arg Lys Val
20 25
<210> 8
<211> 10
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> искусственно сконструированный проникающий в клетку пептид
<400> 8
Arg Arg Arg Arg Arg Arg Arg Arg Arg Cys
1 5 10
<210> 9
<211> 10
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> искусственно сконструированный проникающий в клетку пептид
<400> 9
Arg Arg Arg Arg Arg Arg Arg Arg Arg Lys
1 5 10
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИПОСОМЫ, СОДЕРЖАЩИЕ ПРОНИКАЮЩИЕ В КЛЕТКИ ПЕПТИДЫ И ТЕТРАЭФИРНЫЕ ЛИПИДЫ, ДЛЯ ПЕРОРАЛЬНОЙ ДОСТАВКИ МАКРОМОЛЕКУЛ | 2016 |
|
RU2743431C2 |
| ЛИПОСОМНЫЕ КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2015 |
|
RU2757110C2 |
| УСОВЕРШЕНСТВОВАННЫЕ ЛИПОСОМЫ И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2482837C2 |
| СОЕДИНЕНИЯ ДЛЯ НАЦЕЛЕННОЙ ДОСТАВКИ ЛЕКАРСТВЕННОГО СРЕДСТВА И УСИЛЕНИЯ АКТИВНОСТИ siPHK | 2012 |
|
RU2769872C2 |
| НАЦЕЛИВАЮЩИЕ АМИНОКИСЛОТНЫЕ ЛИПИДЫ | 2013 |
|
RU2654210C2 |
| ЛИПОСОМНЫЕ КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2005 |
|
RU2574926C2 |
| СОЕДИНЕНИЯ ДЛЯ НАЦЕЛЕННОЙ ДОСТАВКИ ЛЕКАРСТВЕННОГО СРЕДСТВА И УСИЛЕНИЯ АКТИВНОСТИ siPHK | 2012 |
|
RU2632888C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЛИПИДЫ, КОТОРЫЕ НЕСУТ ПОЛЯРНЫЙ И НЕПОЛЯРНЫЙ ФРАГМЕНТ | 2002 |
|
RU2294738C2 |
| ОДНОРАЗОВАЯ СИСТЕМА ДЛЯ СТЕРИЛЬНОГО ПОЛУЧЕНИЯ ЧАСТИЦ ИЗ ЛИПИДОВ И НУКЛЕИНОВЫХ КИСЛОТ | 2012 |
|
RU2642640C2 |
| Пероральная система доставки вещества белковой природы (варианты), защитная оболочка системы доставки (варианты) | 2014 |
|
RU2665367C2 |
Изобретение может быть использовано в фармацевтике и относится к твердой пероральной лекарственной форме, содержащей липосомы, которые включают конъюгат по меньшей мере одного типа проникающего в клетки пептида (CPP) и соединения, выбранного из липида и жирной кислоты. Указанный конъюгат является частью двойного липидного слоя липосомы. CPP-часть конъюгата присутствует на поверхности липосомы и СРР выбран из группы, состоящей из линейного или циклизованного пенетратина (SEQ ID NO:1; RQIKIWFQNRRMKWKK), линейного или циклизованного TAT (трансактиватор транскрипции)-пептида (SEQ ID NO:2; CGRKKKRRQRRRPPQC), линейного или циклизованного R9 (SEQ ID NO:4; RRRRRRRRR) или циклизованного СPP, содержащего по меньшей мере 50% фрагментов лизина и/или аргинина. Лекарственная форма представляет собой гастрорезистентную твердую пероральную лекарственную форму, дополнительно включающую по меньшей мере одно пептидное лекарственное средство, белок или антитело в твердой пероральной лекарственной форме. Также изобретение относится к применению твердой пероральной лекарственной формы для пероральной доставки пептидного лекарственного средства, белка или антитела. Технический результат заключается в создании твердых дозированных лекарственных форм, обеспечивающих улучшенную биодоступность при пероральном введении макромолекулярных лекарственных средств. 2 н. и 9 з.п. ф-лы, 1 табл., 19 ил., 9 пр.
1. Твердая пероральная лекарственная форма, включающая липосомы, причем указанные липосомы включают конъюгат
(a) по меньшей мере одного типа проникающего в клетки пептида (CPP) и
(b) соединения, выбранного из липида и жирной кислоты;
где указанный конъюгат является частью двойного липидного слоя липосомы;
где CPP–часть конъюгата присутствует на поверхности липосомы и СРР выбран из группы, состоящей из линейного или циклизованного пенетратина (такого как SEQ ID NO:1; RQIKIWFQNRRMKWKK), линейного или циклизованного TAT (трансактиватор транскрипции)–пептида (такого как SEQ ID NO:2; CGRKKKRRQRRRPPQC), линейного или циклизованного R9 (такого как SEQ ID NO:4; RRRRRRRRR), или циклизованного СPP, содержащего по меньшей мере 50% фрагментов лизина и/или аргинина;
где лекарственная форма представляет собой гастрорезистентную твердую пероральную лекарственную форму, дополнительно включающую по меньшей мере одно пептидное лекарственное средство, белок или антитело в твердой пероральной лекарственной форме.
2. Твердая пероральная лекарственная форма по п.1, где проникающий в клетки пептид (CPP) представляет собой циклизованный СPP, содержащий от 3 до 16 фрагментов аргинина.
3. Твердая пероральная лекарственная форма по п.1 или 2, где указанные липосомы содержатся в капсуле, покрытой энтеросолюбильной оболочкой, или таблетке, покрытой энтеросолюбильной оболочкой.
4. Твердая пероральная лекарственная форма по одному или нескольким из пп.1–3, дополнительно включающая по меньшей мере один фармацевтически приемлемый эксципиент, и/или по меньшей мере один ингибитор протеазы, и/или по меньшей мере один ингибитор липазы в твердой пероральной лекарственной форме.
5. Твердая пероральная лекарственная форма по одному или нескольким из пп.1–4, где указанный по меньшей мере один тип СРР представляет собой циклизованный CPP.
6. Твердая пероральная лекарственная форма по одному или нескольким из пп. 1–5, где указанные липосомы включают указанный по меньшей мере один тип СРР в количестве 0,05–5 моль % из расчета на общее количество липидов и/или жирных кислот.
7. Твердая пероральная лекарственная форма по одному или нескольким из пп. 1–6, где соединение, с которым указанный по меньшей мере один тип СРР конъюгирован, выбрано из группы, состоящей из холестерина и соединений, имеющих основную структуру стероидной молекулы, фосфолипидов, лизофосфолипидов и их комбинаций.
8. Твердая пероральная лекарственная форма по одному или нескольким из пп. 1–7, где указанный по меньшей мере один тип CPP конъюгирован с указанным соединением через бифункциональный PEG–линкер.
9. Твердая пероральная лекарственная форма по одному или нескольким из пп. 1–8, где указанные липосомы лиофилизированы.
10. Твердая пероральная лекарственная форма по одному или нескольким из пп. 1–9 для терапевтического и/или диагностического применения в медицине с целью пероральной доставки по меньшей мере одного пептидного лекарственного средства, белка или антитела.
11. Применение твердой пероральной лекарственной формы по любому из пп.1–10 для пероральной доставки по меньшей мере одного пептидного лекарственного средства, белка или антитела.
| Silvia F | |||
| Pantze et al.: "Matrix liposomes: A solid liposomal formulation for oral administration: Solid liposomes for oral administration" European journal of lipid science and technology, 2014, vol.116, no.9, pp.1145-1154 | |||
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Zhang Dongdong et al.: "Cell-penetrating peptides as noninvasive transmembrane vectors for | |||

