Настоящее изобретение относится к композиции. Кроме того, настоящее изобретение относится к соединению и липосоме и к применению композиции, соединения или липосомы в терапии, в частности в генной терапии (прежде всего для введения гена).
Одним из аспектов генной терапии является введение чужеродной нуклеиновой кислоты (такой как ДНК) в клетки таким образом, чтобы экспрессируемый протеин мог обладать требуемой терапевтической функцией1a.
Примеры терапии такого типа включают встраивание генов ТК, TSG или ILG с целью лечения рака; встраивание гена CFTR с целью лечения фиброзно-кистозной дегенерации; встраивание генов NGF, ТН или LDL с целью лечения нейродегенеративных и сердечно-сосудистых заболеваний; встраивание антагониста гена IL-1 с целью лечения ревматоидного артрита; встраивание антигенов ВИЧ и гена ТК. с целью лечения СПИДа и инфекций, вызываемых цитомегаловирусом (ЦМВ); встраивание антигенов и цитокинов, действующих в качестве вакцин; и встраивание β-глобина с целью лечения связанных с гемоглобинопатией состояний, таких как талассемия.
Многие современные методы генной терапии основаны на применении аденовирусных векторов, таких как Ad3 или Ad5, или других генных векторов.
Однако при их применении возникают серьезные проблемы2a. Это обусловливает необходимость разработки менее опасных невирусных методов переноса генов3a.
Невирусные векторы хорошо известны в данной области. В сущности, для невирусной генной терапии требуется вектор, который является миметиком вируса, но не является патогенным. С учетом того факта, что нуклеиновая кислота является отрицательно заряженной, было разработано два типа катионных невирусных векторов (которые оба служат для конденсации нуклеиновой кислоты), а именно липосомы и полимеры. Образовавшиеся при этом их комплексы с ДНК (липоплексы и полиплексы соответственно) все еще являются катионными, что облегчает эндоцитоз (поглощение клеткой) на анионной клеточной поверхности. После интернализации комплекс может подвергаться трем указанным на фиг.1 превращениям.
Обладающая большим потенциалом невирусная система переноса предусматривает применение катионных липосом4а. Так, катионные липосомы, которые обычно состоят из нейтрального фосфолипида и катионного липида, применяли для переноса в клетку ДНК4а, мРНК5а, антисмысловых олигонуклеотидов,6a протеинов7а и лекарственных средств8a. Многочисленные катионные липосомы имеются в продаже4a, 9a, в последнее время был синтезирован также целый ряд новых катионных липидов10a. Эффективность этих липосом доказана как в опытах in vitro4a, так и in vivo11a.
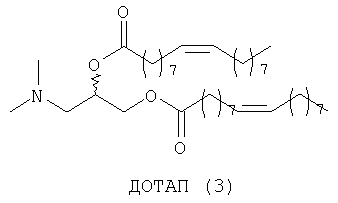
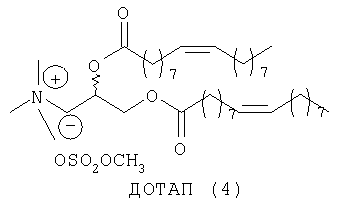
Липосомы получают путем молекулярной самосборки липидов. Катионные липосомы часто приготавливают в виде смеси как катионных липидов, так и нейтральных «хелперных» липидов. DOPC (ДОФХ, диолеилфосфатидилхолин) (1) и DOPE (ДОФЭ, диолеил-L-α-фосфатидилэтаноламин) (2) являются примерами двух таких нейтральных хелперных липидов, которые так называют из-за того, что они могут повышать способность к транфекции катионных липосом и могут помогать катионным липидам образовывать липосомы. Кроме того, 2 является наиболее широко распространенным из всех хелперных липидов и для него неоспоримо доказана способность повышать трансфекцию (введение и экспрессию гена) липоплексов, в состав которых он входит, что связано с его специфичными фузогенными свойствами2б. В противоположность этому DODAP (ДОДАП, диолеил-3-N,N-диметиламинопропан-1,2-диол) (3) (при рН 4,0)2в и DOTAP (ДОТАП, диолеил-3-N,N,N-триметиламмониопропан-1,2-диол (4) являются катионными липидами. Их структуры связывают ДНК в результате электростатических взаимодействий.
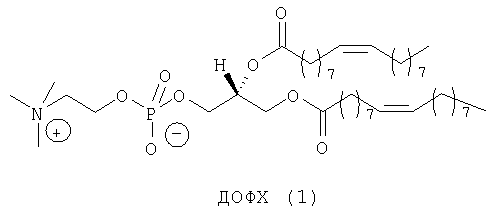
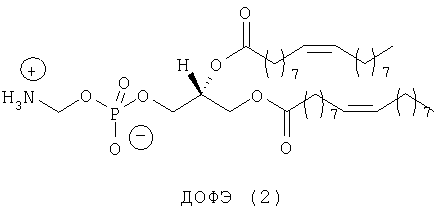
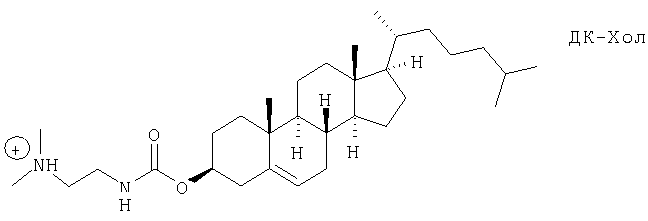
Одна из наиболее широко применяемых систем катионных липосом состоит из смеси нейтрального фосфолипида диолеилфосфатидинилэтаноламина (обычно обозначаемого «ДОФЭ») и катионного липида, 3β-[N-(N',N'-диметиламиноэтан)карбамоил]холестерина (обычно обозначаемого «ДК-Хол»)12a.
Ценность липосом в качестве агентов для введения лекарственных средств в сочетании с различными композициями уже продемонстрирована на стадии клинических испытаний6б, эти липосомы позволяют осуществлять эффективное инкапсулирование подлежащей введению молекулы лекарственного средства в их водных внутренних областях. Катионные липосомы электростатически взаимодействуют с плазмидной ДНК, но сами по себе не обладают способностью инкапсулировать нуклеиновую кислоту. Образовавшиеся в результате частицы, обозначенные липоплексами (ЛП), эффективно осуществляют трансфекцию in vitro.
Несмотря на эффективность известных катионных липосом, все еще сохраняется потребность в оптимизации эффективности переноса генов катионными липосомами, применяемыми для генной терапии человека 10а. С учетом практически полной расшифровки генома человека применение генов для терапевтических целей, т.е. для генной терапии, должно привести к революции в медицине. В этом контексте повышается внимание исследователей к невирусному введению генов, как более безопасной альтернативе для применения на человеке, даже если оно пока остается менее эффективным, чем основанные на применении вирусов технологии.
В этой области, особенно в последнее десятилетие, начали применять сложные макромолекулярные конструкции, включая многие элементы существующих технологий (вирусные протеины или пептиды, липосомы, полимеры, стратегии направленного переноса и маскирующие свойства).
В WO 01/48233 предложена система, основанная на триплексе, состоящем из вирусного корового протеина Mu, плазмидной ДНК и катионной липосомы (LMD). Эта базисная технология позволила получить хорошие результаты in vitro и многообещающие результаты in vivo. Но так же как и другие невирусные технологии, эта система требует дальнейшего усовершенствования для внедрения в терапию in vivo.
Это подразумевает достижение более высокого уровня стабильности частиц в биологических жидкостях (сыворотка, слизистая легкого) наряду с сохранением способности к эффективной трансфекции.
Это требование является одним из препятствий для внедрения всех существующих технологий. Современные стабильные композиции[1, 2] обеспечивают невысокий уровень трансфекции, а наиболее эффективные в настоящее время агенты для трансфекции имеют очень значительные ограничения с точки зрения применения, связанные с их нестабильностью[3-7].
После введения (в кровь в случае системного применения или в слизистую оболочку в случае местного нанесения на легкое) заряженные комплексы подвергаются воздействию соли и биологических макромолекул, что приводит к выраженной коллоидной агрегации и адсорбции биологически активных элементов (опсонины) на их поверхностях[8-7]. Эти носители генов подвергаются резким изменениям, которые могут включать осаждение, связывание протеинов, что приводит к элиминации частиц макрофагами, и к нарушению поверхностей, приводящих к их деструкции. Наиболее широко применяемая стабилизированная композиция включает привитые к поверхности цепи полиэтиленгликоля (ПЭГ)[12, 13]. ПЭГ представляет собой нетоксичный нейтральный полиэфир, который имеет большой исключенный объем для большинства макромолекул. К сожалению, композиции, характеризующиеся необходимым уровнем стабилизации, теряют их способность переносить ген; вероятно, из-за их пониженной неспецифической аффинности в отношении клеток или из-за потери необходимых свойств в отношении деструкции эндосом[14, 15].
Альтернативный подход, направленный на преодоление деструктивного действия биологических жидкостей на липоплексы, предусматривает попытку имитировать естественное состояние и нанесение на поверхность липидных бислоев с полисахаридами[16, 17].
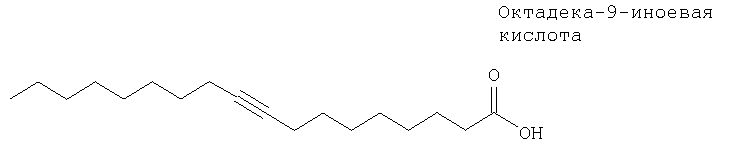
В 1991 г. были опубликованы данные о том, что октадека-9-иноевая кислота (6) обладает способностью связывать ДНК8б. Для октадека-9-иноевой кислоты характерна кажущаяся константа скорости диссоциации для ДНК 1,8 мМ; она ингибирует опосредуемое топоизомеразой I связывание ДНК с фильтром, но не ингибирует опосредуемую ДНК-топоизомеразой I релаксацию суперскрученной плазмидной ДНК. Кроме того, жирная кислота является слабым ингибитором ДНК-полимеразы α.
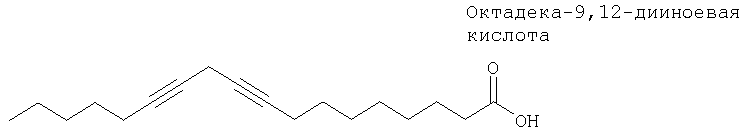
Липооксигеназа тромбоцитов (LOX) может избирательно ингибироваться ацетиленовыми жирными кислотами. Например, октадека-9,12-дииноевая кислота необратимо инактивирует Fe(III)-LOX9б.
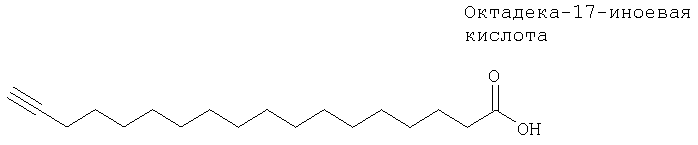
Цитохром Р4504А4 (ЦИТ4А4) представляет собой связанный с легким цитохром Р 450, который участвует в метаболизме простагландинов и арахидоновой кислоты с образованием их ω-гидроксилированных продуктов. Простагландины играют важную роль в регуляции репродуктивной, сосудистой и воспалительной систем. Установлено, что октадека-17-иноевая кислота является эффективным ингибитором10б обеспечивающего связывание с субстратом «кармана» ЦИТ4А4.
Октадека-5-иноевая кислота (таририновая кислота) ингибирует вылупление яиц С.tomontosicollis11б.
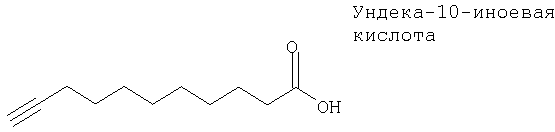
Ундека-10-иноевая кислота, которая является ингибитором цитохрома Р4504А1, ингибирует специфичную для этаноламина реакцию замены фосфолипидного основания в микросомах печени крыс12б.
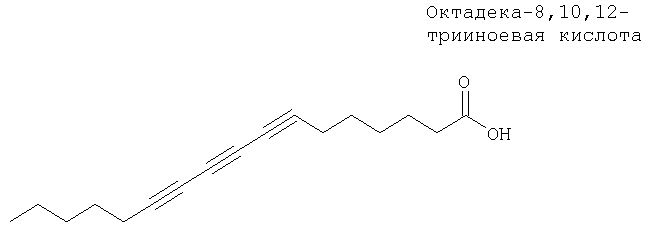
Установлено, что целый ряд ацетиленовых жирных кислот, например октадека-8,10,12-трииноевая кислота, являются сильными ингибиторами фермента циклооксигеназы и слабыми ингибиторами 5-липоксигеназы13б.
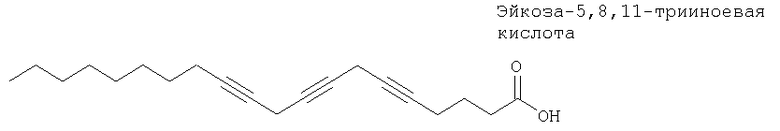
Ацетиленовая жирная кислота, такая как эйкоза-5,8,11-трииноевая кислота, ингибирует глутатион-S-трансферазу14б печени млекопитающих.
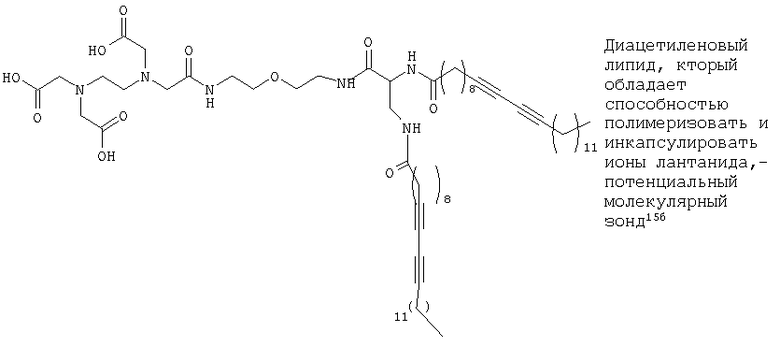
Липосомы, содержащие флуоресцентную метку и/или хелатирующие липиды металлы, находят применение в качестве зондов в области клеточной биологии, например, для оценки перемещения инкапсулированной ДНК при использовании в невирусной генной терапии. Были получены конъюгированные диацетиленовые липиды с использованием головных групп этилендиаминтетрауксусной кислоты (ЭДТК), которые обладают способностью хелатировать ионы лантанидов15б. Эти липиды (каждый с двумя конъюгированными группами ацетиленовой жирной кислоты (например, диацетиленовой жирной кислоты) были успешно включены в липосомы, и затем им давали пройти полимеризацию. Исследования, проведенные с использованием флуоресцентной метки, позволили установить, что диацетиленовые функциональные группы (неполимеризованный липид) и конъюгированные алкены (после полимеризации) можно применять в качестве активаторов ионов лантанидов.
Установлено, что диацетиленовые фосфолипиды подвергаются полимериации после включения в липосомы и обработки УФ-светом16б. Такая система является более стабильной, чем неполимеризованные липиды, и ее можно применять для систем с медленным высвобождением лекарственного средства.

Junichi с соавторами описали (WO 95/03035) применение полимеризованных липосом с повышенной стабильностью для перорального введения лекарственных средств. Фармацевтические соединения, предназначенные для перорального введения, можно капсулировать в полимеризованные липосомы для их введения в тонкую кишку. Образующие липосомы фосфолипиды полимеризуются через двойную связь, которую несут олефиновые и ацетиленовые фосфолипиды. Такая полимеризация повышает прочность, приводя к меньшей текучести липосом. По этой причине полимеризация фосфолипидов может найти ограниченное применение в невирусной генной терапии, если процесс полимеризации не будет обратимым. С одной стороны, липосомы должны сохранять стабильность в крови, но с другой стороны, они должны быть нестабильными внутри клетки; текучесть (и как следствие сродство к НII-фазе), вероятно, имеет решающее значение для повышения трансфекции in vitro.
Установлено, что при полимеризации внутри липосом диацетиленовые фосфатидилхолины не образуют тромбы in vitro. Этот аспект стабильности может являться результатом того, что полимеризованные фосфатидилхолины не могут участвовать в коагуляции18б.
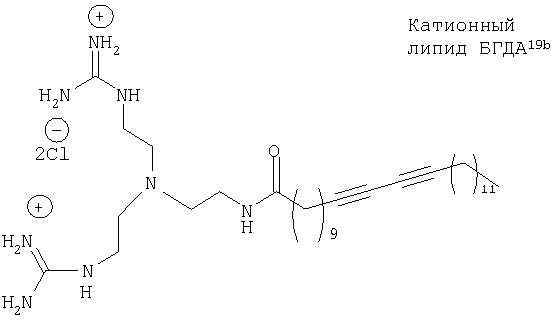
В 2001 г. было установлено, что катионный липид бисгуанидийдиацетилен (БГДА) обладает высокой эффективностью при использовании для трансфекции in vitro, когда он включен в композицию в виде катионных липосом в сочетании с ДОФЭ19б. Присутствие диацетиленовой функциональной группы является предпосылкой для полимеризации и, следовательно, новой ступенью для трансфекции генов. Этому может способствовать дальнейшее исследование взаимосвязи между структурой и активностью комплексов липид/ДНК путем оценки воздействий способных к полимеризации доменов.
Настоящее изобретение направлено на решение проблем, существующих в данной области.
Первым объектом настоящего изобретения является композиция, содержащая (I) соединение, представляющее собой липид, который содержит по меньшей мере один неполярный фрагмент и полярный фрагмент, где неполярный фрагмент представлен формулой X-Y-Z-, где Х обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С, Y обозначает О или СН2 и Z обозначает необязательную гидрокарбильную группу, в которой полярный фрагмент представлен формулой -[Т]mПГГ, где [Т]m обозначает необязательную группу, выбранную из ряда, включающего С(O), NH, NR1, NHC(O), C(O)NH, NR1C(O), C(O)NR1 и СН2, где R1 обозначает гидрокарбильную группу, где ПГГ обозначает полярную головную группу и где m обозначает количество неполярных фрагментов, (II) терапевтический агент.
Другим объектом настоящего изобретения является липосома, содержащая соединение, представляющее собой липид, который содержит по меньшей мере один неполярный фрагмент и полярный фрагмент, где неполярный фрагмент представлен формулой X-Y-Z-, где Х обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С, Y обозначает О или СН2 и Z обозначает необязательную гидрокарбильную группу, в которой полярный фрагмент представлен формулой -[Т]mПГГ, где [Т]m обозначает необязательную группу, выбранную из ряда, включающего С(O), NH, NR1, NHC(O), C(O)NH, NR1C(O), C(O)NR1 и СН2, где R1 обозначает гидрокарбильную группу, где ПГГ обозначает полярную головную группу и где m обозначает количество неполярных фрагментов; где соединение не представляет собой производное диолеилфосфатидилхолина ДО(4-ин)ФХ, ДО(9-ин)ФХ и ДО(14-ин)ФХ.
Следующим объектом настоящего изобретения является соединение, представляющее собой липид, который содержит по меньшей мере один неполярный фрагмент и полярный фрагмент, где неполярный фрагмент представлен формулой X-Y-Z-, где Х обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С, Y обозначает О или CH2 и Z обозначает необязательную гидрокарбильную группу, в которой полярный фрагмент представлен формулой -[Т]mПГГ, где [Т]m обозначает необязательную группу, выбранную из ряда, включающего С(O), NH, NR1, NHC(O), C(O)NH, NR1C(O), C(O)NR1 и СН2, где R1 обозначает гидрокарбильную группу, где ПГГ обозначает полярную головную группу и где m обозначает количество неполярных фрагментов; где соединение не представляет собой ДО(4-ин)ФХ, ДО(9-ин)ФХ и ДО(14-ин)ФХ.
Еще одним объектом настоящего изобретения является применение соединения, представляющего собой липид, для приготовления лекарственного средства, предназначенного для лечения генетического нарушения или состояния или заболевания, где соединение представляет собой липид, который содержит по меньшей мере один неполярный фрагмент и полярный фрагмент, где неполярный фрагмент представлен формулой X-Y-Z-, где Х обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С, Y обозначает О или СН2 и Z обозначает необязательную гидрокарбильную группу, в которой полярный фрагмент представлен формулой -[Т]mПГГ, где [Т]m обозначает необязательную группу, выбранную из ряда, включающего С(O), NH, NR1, NHC(O), C(O)NH, NR1C(O), C(O)NR1 и СН2, где R1 обозначает гидрокарбильную группу, где ПГГ обозначает полярную головную группу и где m обозначает количество неполярных фрагментов.
Еще одним объектом настоящего изобретения является соединение, композиция или липосома, предлагаемые в настоящем изобретении, предназначенные для применения в терапии.
Следующим объектом настоящего изобретения является применение соединения, композиции или липосомы для приготовления лекарственного средства, предназначенного для лечения генетического нарушения, или состояния, или заболевания.
Еще одним объектом настоящего изобретения является катионная липосома, образованная с использованием соединения, предлагаемого в настоящем изобретении, или соединения, полученного с помощью способа, предлагаемого в настоящем изобретении.
Следующим объектом настоящего изобретения является способ получения катионной липосомы, который предусматривает получение катионной липосомы с использованием соединения, предлагаемого в настоящем изобретении, или соединения, полученного с помощью способа, предлагаемого в настоящем изобретении.
Еще одним объектом настоящего изобретения является катионная липосома, предлагаемая в настоящем изобретении, или катионная липосома, полученная с помощью способа, предлагаемого в настоящем изобретении, которая предназначена для применения в терапии.
Другим объектом настоящего изобретения является применение катионной липосомы, предлагаемой в настоящем изобретении, или катионной липосомы, полученной с помощью способа, предлагаемого в настоящем изобретении, для приготовления лекарственного средства, предназначенного для лечения генетического нарушения, или состояния, или заболевания.
Следующим объектом настоящего изобретения является комбинация, содержащая нуклеотидную последовательность и один или несколько следующих ингредиентов: соединение, предлагаемое в настоящем изобретении, соединение, полученное способом, предлагаемым в настоящем изобретении, липосома, предлагаемая в настоящем изобретении, или липосома, полученная способом, предлагаемым в настоящем изобретении, композиция, предлагаемая в настоящем изобретении, или композиция, полученная способом, предлагаемым в настоящем изобретении.
Еще одним объектом изобретения является комбинация, предлагаемая в настоящем изобретении, предназначенная для применения в терапии.
Следующим объектом настоящего изобретения является применение комбинации, предлагаемой в настоящем изобретении, для приготовления лекарственного средства, предназначенного для лечения генетического нарушения, или состояния, или заболевания.
Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение, предлагаемое в настоящем изобретении, соединение, полученное способом, предлагаемым в настоящем изобретении, композицию, предлагаемую в настоящем изобретении, или композицию, полученную способом, предлагаемым в настоящем изобретении, в смеси с фармацевтическим и необязательно в смеси с фармацевтически приемлемым разбавителем, носителем или эксципиентом.
И еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая катионную липосому, предлагаемую в настоящем изобретении, катионную липосому, полученную способом, предлагаемым в настоящем изобретении, в смеси с фармацевтическим средством и необязательно в смеси с фармацевтически приемлемым разбавителем, носителем или эксципиентом.
Некоторые другие объекты изобретения приведены в прилагаемой формуле изобретения.
При создании изобретения были изучены такие вопросы, как усиление эндосомолиза (например, на основе данных о падении значения рН по мере созревания эндосомы с образованием лизосомы), направленный специфический для клетки перенос (липопептиды), конденсация ДНК, направленный перенос в ядро и, наконец, улучшение стабильности in vivo с сохранением фузогенности липоплекса.
Вероятно, имеющим решающее значение преимуществом соединения или композиции, предлагаемой в настоящем изобретении, является то, что их можно применять для получения катионной липосомы, которую можно использовать в генной терапии, в частности для введения нуклеиновых кислот (включая гены и антисмысловые ДНК/РНК) в клетки (in vitro, in vivo и ex vivo) для достижения терапевтической эффективности.
Агенты для генной терапии, как правило, вводят внутривенно. Парадоксально, что катионные липосомы должны обладать стабильностью в крови, но это свойство должно сочетаться с их нестабильностью внутри клетки, что позволяет обеспечивать выделение введенной нуклеиновой кислоты. Компоненты сыворотки крови снижают биологическую активность известных к настоящему времени катионных липосом, что может привести к клиренсу или перемещению терапевтической нуклеиновой кислоты и к плохим результатам in vivo. При создании изобретения было обнаружено, что активность in vitro в отношении трансфекции ацетиленовых соединений, предлагаемых в настоящем изобретении, сопоставима с активностью ДОФЭ. Это позволяет предположить, что тройная связь углерод-углерод более устойчива к окислению, чем двойные связи, и это может также повышать прочность мембран. Ацетиленовые соединения могут способствовать повышению стабильности в сыворотке крови, что может обеспечивать получение более хороших результатов in vivo.
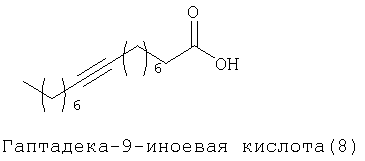
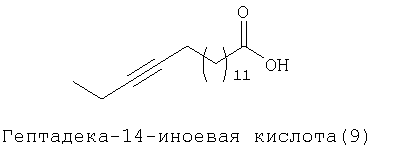
При создании изобретения был изучен широкий спектр соединений, подпадающих под объем изобретения и были получены хорошие результаты. В частности, были изучены соответствующие аналоги ДОФХ, ДОФЭ, ДОДАП и ДОТАП, каждый с тремя моноацетиленовыми жирными кислотами, несущими 18 атомов углерода (С 18) (соединения 5, 6 и 7) и двумя моноацетиленовыми жирными кислотами, несущими 17 атомов углерода (С 17) (соединения 8 и 9).
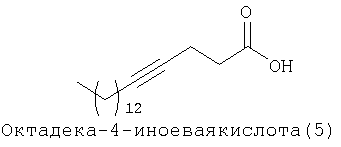
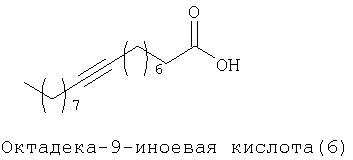
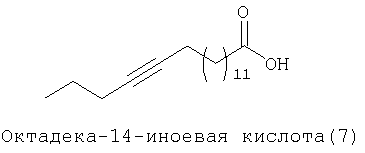
Был осуществлен синтез пяти моноацетиленовых аналогов нескольких липидов, а именно ДОФХ (1), ДОФЭ (2), ДОДАП (3) и ДОТАП (4). Эти липиды должны обладать улучшенной способностью к межмолекулярной упаковке, тем самым снижая текучесть липосомных структур и повышая время полужизни липосом в кровотоке in vivo.
Присутствие тройной связи С≡С, вероятно, дает преимущество, поскольку она в меньшей степени подвержена окислению, чем двойная связь С=С20б, и в целом обладает меньшей чувствительностью к действию электрофильных агентов21б. Кроме того, двойная связь более чувствительна к цис-/транс-изомеризации, в то время как тройная связь не подвержена такой изомеризации. Кислые условия и УФ-свет могут ускорить этот процесс. Весьма вероятно, что находящиеся в цис-положении двойные связи в ДОФЭ имеют решающее значение для эффективности ДОФЭ в отношении трансфекции, обусловливая способность содержащих ДОФЭ липосом формировать НII-фазу и в результате обусловливая фузогенность (см. ниже). Изомеризация с образованием более стабильной транс-формы может привести к снижению или даже к полной потере способности к трансфекции21в.
Липиды с двойными связями в цис-положении, такие как ДОФЭ, придают текучесть мембранам, в состав которых они входят. Ацетиленовые аналоги, такие как производное диолеилфосфатид-9-инилэтаноламина ДО(9-ин)ФЭ, вероятно, обусловливают большую прочность и, как следствие, повышенную стабильность in vivo состоящих из них липосом.
Rürup с соавторами установили, что температура фазового перехода (Tm) слоистого геля в жидкий кристалл (Lβ/Lα) 9-инового аналога ДОФХ (ДО(9-ин)ФХ, (33) примерно на 15° выше (-3,4°С), чем в случае стандартного ДОФЭ (-18°С). Кроме того, указанные исследователи установили, что если тройную связь сдвинуть в любом направлении от середины цепи жирного ацила, то значение Tm возрастет (фиг.11), как это происходит в случае аналогов с олефиновой (двойной) связью.
Таким образом, соответствующие аналоги ДОФЭ могут обладать одинаковым поведением не только в отношении перехода Lβ/Lα, но также в отношении фазового перехода Lα/НII. Температура фазового перехода (Тh) слоистого жидкого кристалла в инвертированный гексагональный кристалл (Lα/НII) составляет 10°С для стандартного ДОФЭ21г. Вследствие его сродства к НII-фазе ДОФЭ рассматривают как фузогенный липид, поскольку, когда мембраны расплавляются, структурные промежуточные продукты аналогичны тем, которые участвуют в фазовых переходах бислоя (Lα) в НII-фазу. Вероятно, эта фузогенная способность придает содержащим ДОФЭ липосомам эффективность в отношении трансфекции благодаря слиянию мембран липосом и эндосом, что способствует выделению плазмидной ДНК в цитоплазму.
Из-за низкого значения Тh невозможно приготавливать липосомы на основе ДОФЭ при физиологических условиях, т.е. при 37°С (или в действительности при 25°С) и при рН 7,0. С учетом того, что у ДО(9-ин)ФХ Tm примерно на 15°С выше, чем у ДОФХ, Tm ДО(9-ин)ФЭ может быть примерно на 15°С выше, чем у ДОФЭ. Если эта ситуация имеет место, то, следовательно, это может облегчать приготовление при комнатной температуре (КТ) катионных липосом, содержащих моноацетилированные аналоги липидов, предлагаемых в изобретении, таких как ДОФЭ, а не сами липиды. В конце концов, это может привести к снижению положительных зарядов катионных LMD, приводя к получению частиц для трансфекции, которые являются более стабильными не только из-за лучшей межмолекулярной упаковки вследствие наличия более жестких бислойных структур, но и более стабильными из-за того, что частицы являются более нейтральными. При применении невирусной генной терапии in vivo возникает целый ряд проблем, одной из них является наличие многочисленных отрицательно заряженных компонентов в крови. Они могут «метить» катионные LMD в качестве ингредиентов, предназначенных для деструкции посредством электростатических взаимодействий, или в более простом варианте могут замещать анионный комплекс mu-ДНК. С использованием ацетиленовых аналогов, предлагаемых в настоящем изобретении, можно создавать большее количество анионных векторов, которые являются более стабильными с точки зрения агрегации и деструкции.
Подробное описание изобретения
В контексте настоящего описания понятие «ацетиленовый гидрокарбил» обозначает группу, содержащую по меньшей мере С и Н, которая имеет по меньшей мере одну связь -С≡С- и необязательно может содержать один или несколько других приемлемых заместителей. Примерами таких заместителей могут являться галоген, алкокси-, нитро-, углеводородная группа, N-ацильная группа, циклическая группа и т.д. Помимо того, что заместители могут представлять собой циклическую группу, циклическую группу может образовывать комбинация заместителей. Если гидрокарбильная группа содержит более одного атома С, то не является обязательным, чтобы эти атомы углерода были связаны друг с другом. Например, по меньшей мере два из атомов углерода могут быть связаны через пригодный элемент или группу. Таким образом, гидрокарбильная группа может содержать гетероатомы. Приемлемые гетероатомы хорошо известны специалистам в данной области и включают, например, серу, азот и кислород.
Специалисту в данной области должно быть очевидно, что фраза «X обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С» обозначает, что каждый Х содержит одну и только одну связь С≡С.
Понятие «гидрокарбильная группа» в контексте настоящего описания обозначает группу, которая содержит по меньшей мере С и Н и которая не обязательно может содержать один или несколько других приемлемых заместителей. Примерами таких заместителей могут являться галоген, алкокси-, нитро-, углеводородная группа, N-ацильная группа, циклическая группа и т.д. Помимо того, что заместители могут представлять собой циклическую группу, циклическую группу может образовывать комбинация заместителей. Если гидрокарбильная группа содержит более одного атома С, то не является обязательным, чтобы эти атомы углерода были связаны друг с другом. Например, по меньшей мере два из атомов углерода могут быть связаны через пригодный элемент или группу. Таким образом, гидрокарбильная группа может содержать гетероатомы. Приемлемые гетероатомы хорошо известны специалистам в данной области и включают, например, серу, азот и кислород.
Согласно предпочтительному варианту осуществления изобретения гидрокарбильная группа представляет собой углеводородную группу.
В контексте настоящего описания понятие «углеводород» обозначает любую алкильную группу, алкенильную группу, алкинильную группу, ацильную группу, где эти группы могут быть линейными, разветвленными или циклическими, или арильную группу. Понятие «углеводород» включает также эти же группы, но необязательно замещенные. Если углеводород имеет разветвленную структуру и несет заместитель(и), то замещение может быть либо в углеводородном скелете, либо в боковой цепи; в другом варианте замещение может быть и в углеводородном скелете, и в боковой цепи.
В широком смысле объектом настоящего изобретения является композиция, содержащая (I) соединение, представляющее собой липид, который содержит по меньшей мере один неполярный фрагмент и полярный фрагмент, где неполярный фрагмент представлен формулой X-Y-Z-, где Х обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С, Y обозначает О или СН2 и Z обозначает необязательную гидрокарбильную группу, в которой полярный фрагмент представлен формулой -[Т]mПГГ, где [Т]m обозначает необязательную группу, выбранную из ряда, включающего С(O), NH, NR1, NHC(O), C(O)NH, NR1C(O), C(O)NR1 и СН2, где R1 обозначает гидрокарбильную группу, где ПГГ обозначает полярную головную группу и где m обозначает количество неполярных фрагментов, (II) терапевтический агент.
Другим объектом настоящего изобретения в широком смысле является липосома, содержащая соединение, представляющее собой липид, который содержит по меньшей мере один неполярный фрагмент и полярный фрагмент, где неполярный фрагмент представлен формулой X-Y-Z-, где Х обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С, Y обозначает О или СН2 и Z обозначает необязательную гидрокарбильную группу, в которой полярный фрагмент представлен формулой -[Т]mПГГ, где [Т]m обозначает необязательную группу, выбранную из ряда, включающего С(O), NH, NR1, NHC(O), C(O)NH, NR1C(O), C(O)NR1 и СН2, где R1 обозначает гидрокарбильную группу, где ПГГ обозначает полярную головную группу и где m обозначает количество неполярных фрагментов; где соединение не представляет собой ДО(4-ин)ФХ, ДО(9-ин)ФХ и ДО(14-ин)ФХ.
Следующим объектом настоящего изобретения в широком смысле является соединение, представляющее собой липид, который содержит по меньшей мере один неполярный фрагмент и полярный фрагмент, где неполярный фрагмент представлен формулой X-Y-Z-, где Х обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С, Y обозначает О или СН2 и Z обозначает необязательную гидрокарбильную группу, в которой полярный фрагмент представлен формулой -[Т]mПГГ, где [Т]m обозначает необязательную группу, выбранную из ряда, включающего С(O), NH, NR1, NHC(O), C(O)NH, NR1C(O), C(O)NR1 и СН2, где R1 обозначает гидрокарбильную группу, где ПГГ обозначает полярную головную группу и где m обозначает количество неполярных фрагментов; где соединение не представляет собой ДО(4-ин)ФХ, ДО(9-ин)ФХ, ДО(14-ин)ФХ, ДО(4-ин)ФЭ и ДО(14-ин)ФЭ.
И еще одним объектом настоящего изобретения в широком смысле является применения соединения, представляющего собой липид, для приготовления лекарственного средства, предназначенного для лечения генетического нарушения, или состояния, или заболевания, где соединение представляет собой липид, который содержит по меньшей мере один неполярный фрагмент и полярный фрагмент, где неполярный фрагмент представлен формулой X-Y-Z-, где Х обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С, Y обозначает О или СН2 и Z обозначает необязательную гидрокарбильную группу, в которой полярный фрагмент представлен формулой -[Т]mПГГ, где [Т]m обозначает необязательную группу, выбранную из ряда, включающего С(O), NH, NR1, NHC(O), C(O)NH, NR1C(O), C(O)NR1 и CH2, где R1 обозначает гидрокарбильную группу, где ПГГ обозначает полярную головную группу и где m обозначает количество неполярных фрагментов.
Согласно некоторым предпочтительным вариантам осуществления приведенных выше объектов изобретения в широком смысле по меньшей мере один Х обозначает ацетиленовую гидрокарбильную группу, содержащую одну связь С≡С.
Согласно одному из предпочтительных вариантов осуществления изобретения ацетиленовая гидрокарбильная группа представляет собой алкинильную группу.
Ниже изобретение проиллюстрировано на примерах, не ограничивающих его объем.
Предпочтительные варианты осуществления изобретения
Соединение может представлять собой анионный липид.
Предпочтительно соединение представляет собой нейтральный липид.
Предпочтительно соединение представляет собой катионный липид.
Полярный фрагмент
Полярная головная группа (ПГГ)
Специалисту в данной области должно быть очевидно, что полярная головная группа может быть получена из приемлемого липида. Под понятием «липид» понимают соединение, основой которой являются жирные кислоты или близко родственные соединения, такие как соответствующий спирт или сфингозиновое основание.
Согласно одному из вариантов осуществления полярную головную группу получают из фосфолипидов, церамидов, триацилглицеринов, лизофосфолипидов, фосфатидилсеринов, глицеринов, спиртов, соединений, содержащих алкоксигруппу, моноацилглицеринов, ганглиозидов, сфингомиелинов, цереброзидов, фосфатидилхолинов, фосфатидилэтаноламинов, фосфатидилинозитов (ФИ), диацилглицеринов, фосфатидиновых кислот, глицероуглеводов, полиспиртов и фосфатидилглицеринов.
Согласно одному из вариантов осуществления полярную головную группу получают из фосфолипидов, церамидов, триацилглицеринов, лизофосфолипидов и фосфатидилсеринов.
Предпочтительно полярную головную группу получают из фосфолипида.
Предпочтительно фосфалипид представляет собой нейтральный или анионный фосфолипид.
Согласно одному из предпочтительных вариантов осуществления изобретения фосфолипид выбирают из фосфатидилхолина (ФХ) и фосфатидилэтаноламина (ФЭ), такого как диолеил-L-α-фосфатидилэтаноламин (ДОФЭ).
Предпочтительно полярную головную группу получают из 3-N,N-диметиламинопропан-1,2-диола (ДАП) или 3-N,N,N-триметиламмониопропан-1,2-диола(ТАП).
Согласно одному из объектов полярная головная группа (ПГГ) может представлять собой группу -W-линкер-ГГ, где W выбирают из ряда, включающего СН2, О, NR1 и S, где R1 обозначает Н или гидрокарбильную группу, где линкер представляет собой необязательную линкерную группу и ГГ обозначает головную группу.
Головная группа (ГГ) может быть полярной или неполярной. Когда ГГ обозначает неполярную группу, то ей может быть придана полярность с помощью группы -C(O)W-линкер-. Такие полярные группы подпадают под настоящее определение при условии, что группа -С(O)-линкер-ГГ является полярной и ГГ является полярной, если присоединена к группе -С(O)W-линкер-.
Согласно одному из объектов головная группа (ГГ) может представлять собой алкильную группу. Предпочтительно алкил содержит по меньшей мере 5 атомов углерода, например, представляет собой С5-С100алкильную группу, C5-С80алкильную группу, С5-С60алкильную группу, С5-С50алкильную группу, C5-С40алкильную группу, С5-С30алкильную группу или С5-С20алкильную группу.
Согласно одному из объектов головную группу (ГГ) получают из фосфолипидов, церамидов, триацилглицеринов, лизофосфолипидов, фосфатидилсеринов, глицеринов, спиртов, соединений, содержащих алкоксигруппу, моноацилглицеринов, ганглиозидов, сфингомиелинов, цереброзидов, фосфатидилхолинов, фосфатидилэтаноламинов, фосфатидилинозитов (ФИ), диацилглицеринов, фосфатидиновых кислот, глицероуглеводов, полиспиртов и фосфатидилглицеринов.
Согласно одному из предпочтительных вариантов осуществления изобретения головная группа имеет формулу
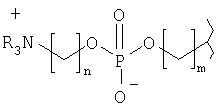
или формулу
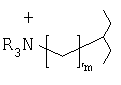
где R независимо друг от друга выбирают из ряда, включающего Н и гидрокарбил, m обозначает 1-10 и n обозначает 1-10.
Предпочтительно R выбирают из ряда, включающего Н и С1-С6алкил, более предпочтительно Н и C1-С3алкил, более предпочтительно Н и метил.
Предпочтительно m обозначает 1-5, более предпочтительно 1, 2 или 3.
Предпочтительно n обозначает 1-5, более предпочтительно 1, 2 или 3.
Линкер
Линкерная группа -W-линкер-ГГ может представлять собой любую приемлемую группу. Как правило, линкерная группа представляет собой гидрокарбильную группу.
Понятие «гидрокарбильная группа» в контексте настоящего описания обозначает группу, которая содержит по меньшей мере С и Н и которая не обязательно может содержать один или несколько других приемлемых заместителей. Примерами таких заместителей могут являться галоген, алкокси-, нитро-, алкильная группа, циклическая группа и т.д. Помимо того, что заместители могут представлять собой циклическую группу, циклическую группу может образовывать комбинация заместителей. Если гидрокарбильная группа содержит более одного атома С, то не является обязательным, чтобы эти атомы углерода были связаны друг с другом. Например, по меньшей мере два из атомов углерода могут быть связаны через пригодный элемент или группу. Таким образом, гидрокарбильная группа может содержать гетероатомы. Приемлемые гетероатомы хорошо известны специалистам в данной области и включают, например, серу, азот и кислород. Примером гидрокарбильной группы, который не ограничивает объем изобретения, является ацильная группа.
Как правило, гидрокарбильная группа представляет собой углеводородную группу. В контексте настоящего описания понятие «углеводород» обозначает любую алкильную группу, алкенильную группу, алкинильную группу, где эти группы могут быть линейными, разветвленными или циклическими, или арильную группу. Понятие «углеводород» включает также эти же группы, но необязательно замещенные. Если углеводород имеет разветвленную структуру и несет заместитель(и), то замещение может быть либо в углеводородном скелете, либо в боковой цепи; в другом варианте замещение может быть и в углеводородном скелете и в боковой цепи.
Согласно одному из предпочтительных объектов необязательная линкерная группа может не присутствовать. Согласно одному из предпочтительных объектов все необязательные линкерные группы отсутствуют.
Если одна, несколько или все необязательные линкерные группы отсутствуют, то группу/соединение, из которых получают полярную головную группу, как правило, выбирают из одной или нескольких -ОН-групп. Это позволяет получать простую эфирную связь между неполярным и полярным фрагментами.
Как должно быть очевидно специалисту в данной области, когда необязательный линкер присутствует, то две или большее количество W-групп может быть связано или не связно с одним и тем же атомом линкера. Согласно некоторым объектам две или большее количество W-групп может быть связано с различными атомами линкера.
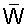
В группе -W-линкер-ГГ W выбирают из ряда, включающего СН2, О, NR1 и S, где R1 обозначает Н или гидрокарбильную группу.
Согласно одному из предпочтительных объектов группа W обозначает О или NR1.
R1 предпочтительно обозначает Н или углеводородную группу.
R1 предпочтительно обозначает Н, С1-С30-, C1-C25-, С1-С20-, C1-C15-, С1-С10-, C1-C5- или С5-С15гидрокарбильную группу.
R1 предпочтительно обозначает Н, C1-С30-, C1-C25-, С1-С20-, C1-C15-, C1-С10-, C1-C5- или С5-С15углеводородную группу.
R1 предпочтительно обозначает Н, необязательно замещенную C1-С30-, С1-С25-, C1-C20-, C1-C15-, C1-C10-, C1-C5- или С5-С15алкильную группу.
R1 предпочтительно обозначает Н, незамещенную C1-С30-, C1-C25-, C1-C20-, C1-C15-, C1-С10-, C1-C5- или С5-С15алкильную группу.
Неполярный фрагмент
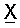
Как указано выше, Х обозначает гидрокарбильную цепь. Под понятием «гидрокарбильная цепь» понимают линейную гидрокарбильную группу.
В приведенных ниже определениях под длиной цепи понимают наибольшую длину цепи, непосредственно связанной с атомами в фрагменте X. Должно быть очевидно, что цепь не включает атомы циклических заместителей или заместителей концевого атома углерода.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из ряда, включающего необязательно замещенный алкил, необязательно замещенный алкенил и необязательно замещенный алкинил.
Согласно одному из предпочтительных объектов ацетиленовая гидрокарбильная группа содержит от 3 до 30 атомов углерода, например от 10 до 25 атомов углерода, от 15 до 20 атомов углерода, 15, 16, 17 или 18 атомов углерода.
Предпочтительно ацетиленовую гидрокарбильную группу получают из жирной кислоты, выбранной из ряда, включающего следующие кислоты:
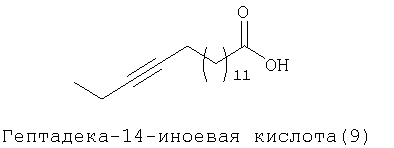
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из необязательно замещенных С6-С24алкинильных групп.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из необязательно замещенных алкинильных групп с длиной цепи 6-24 атома.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из необязательно замещенных алкинильных групп с длиной цепи 10-18 атомов.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из необязательно замещенных алкинильных групп с длиной цепи 16 или 17 атомов.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из незамещенных алкинильных групп.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из незамещенных С6-С24алкинильных групп.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из незамещенных алкинильных групп с длиной цепи 6-24 атома.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из незамещенных С10-С18алкинильных групп.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из незамещенных алкинильных групп с длиной цепи 10-18 атомов.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из незамещенных C16- или С17алкинильных групп.
Согласно одному из предпочтительных объектов Х обозначает группу, выбранную из незамещенных алкинильных групп с длиной цепи 16 или 17 атомов.
Согласно одному из предпочтительных объектов Х обозначает углеводородную цепь. Под «углеводородной цепью» понимают линейную углеводородную группу.
Когда Х содержит одну или несколько двойных связей, предпочтительно по меньшей мере одна из них, более предпочтительно каждая, находится в цис-конфигурации.
Согласно одному из предпочтительных объектов связь С≡С ацетиленовой углеводородной группы расположена на расстоянии 2-15 атомов углерода от концевого атома ацетиленовой гидрокарбильной группы.
Согласно одному из предпочтительных объектов связь С≡С ацетиленовой углеводородной группы расположена на расстоянии 2 атомов углерода от концевого атома ацетиленовой гидрокарбильной группы.
Согласно одному из предпочтительных объектов связь С≡С ацетиленовой углеводородной группы расположена на расстоянии 3 атомов углерода от концевого атома ацетиленовой гидрокарбильной группы.
Согласно одному из предпочтительных объектов связь С≡С ацетиленовой углеводородной группы расположена на расстоянии 7 атомов углерода от концевого атома ацетиленовой гидрокарбильной группы.
Согласно одному из предпочтительных объектов связь С≡С ацетиленовой углеводородной группы расположена на расстоянии 13 атомов углерода от концевого атома ацетиленовой гидрокарбильной группы.
Y
Как указано выше, Y обозначает О или СН2.
Согласно одному из предпочтительных объектов Y обозначает CH2.
Согласно одному из предпочтительных объектов, когда Y обозначает СН2, то цепь X-Y-Z содержит четное количество атомов углерода. Следует понимать, что под длиной цепи X-Y-Z понимают наиболее длинную цепь непосредственно связанных атомов в фрагменте X-Y-Z. Должно быть очевидно, что цепь не должна включать атомы циклических заместителей или заместителей концевого атома углерода.
Согласно одному из предпочтительных объектов X-Y-Z содержит четное количество атомов.
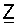
Как указано выше, Z обозначает необязательную гидрокарбильную группу.
Согласно одному из объектов Z обозначает алкильную группу.
Согласно одному из объектов Z обозначает C1-С10-, предпочтительно C1-С6-, предпочтительно С1-С3 алкильную группу. Предпочтительно Z обозначает -СН2-.
Соединения
Согласно одному из объектов соединение имеет формулу
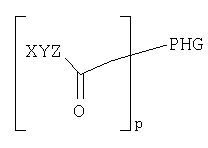
где р обозначает по меньшей мере 1, например, 1-10000, 1-1000, 1-100, 1-50, 1-20, 1-10, предпочтительно 1-5, предпочтительно 1, 2 или 3, и где каждый W, X, Y и Z выбирают независимо друг от друга.
Примеры приемлемых соединений, из которых получают полярную головную группу для данных значений р, приведены ниже
Согласно одному из объектов соединение имеет формулу
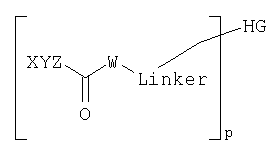
где р обозначает число от 1 до 10, предпочтительно от 1 до 5, предпочтительно 1, 2 или 3, и где каждый W, X, Y и Z выбирают независимо друг от друга.
Согласно одному из объектов соединение имеет формулу
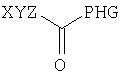
Согласно одному из объектов соединение имеет формулу
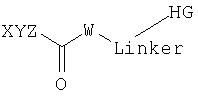
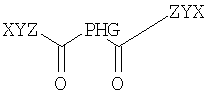
Предпочтительно соединение содержит по меньшей мере два неполярных фрагмента, где каждый фрагмент независимо друг от друга выбирают из неполярных фрагментов формулы X-Y-Z.
Согласно одному из предпочтительных объектов соединение имеет формулу
где каждый W, X, Y и Z выбирают независимо друг от друга.
Согласно одному из предпочтительных объектов соединение имеет формулу
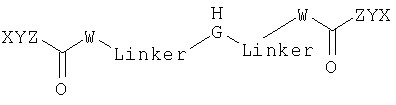
где каждый W, X, Y и Z выбирают независимо друг от друга.
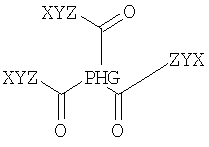
Согласно одному из объектов соединение содержит по меньшей мере 3 неполярных фрагмента, где каждый фрагмент независимо друг от друга выбирают из неполярных фрагментов формулы X-Y-Z-.
Согласно одному из предпочтительных объектов соединение имеет формулу
где каждый W, X, Y и Z выбирают независимо друг от друга.
Согласно одному из предпочтительных объектов соединение имеет формулу
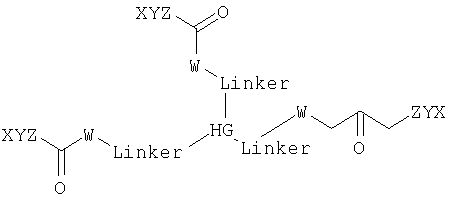
где каждый W, X, Y и Z выбирают независимо друг от друга.
Предпочтительным объектом настоящего изобретения является соединение формулы
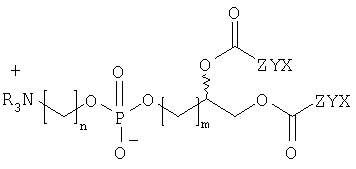
где R независимо друг от друга выбирают из Н и гидрокарбила, n обозначает число от 1 до 10, m обозначает число от 1 до 10. Настоящее изобретение относится также к соединению
- в смеси или в сочетании с нуклеотидной последовательностью;
- для применения в терапии;
- для применения с целью приготовления лекарственного средства, предназначенного для лечения генетического нарушения, или состояния, или заболевания;
- к полученной на его основе липосоме;
- к способу получения катионной липосомы, предусматривающему получение катионной липосомы из соединения;
- к катионной липосоме и ее применению;
- к фармацевтической композиции, содержащей соединение в смеси с фрмацевтическим средством и необязательно в смеси с фармацевтически приемлемым разбавителем, носителем или эксципиентом;
- в форме S- или R-изомера, предпочтительно в форме R-изомера.
Предпочтительно - ZYX обозначает группу формулы СрН2р-3, где р обозначает от 3 до 30, предпочтительно от 10 до 25, предпочтительно от 15 до 20, предпочтительно 15, 16, 17 или 18 атомов углерода.
Ниже представлены другие наиболее предпочтительные объекты настоящего изобретения. Объектом изобретения может являться соединение формулы
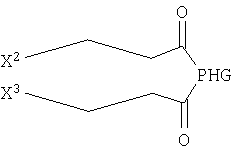
где X2 и Х3 независимо друг от друга выбирают из незамещенного С10-С18алкинила;
соединение формулы
где X2 и X3 независимо друг от друга выбирают из незамещенного С14алкинила и незамещенного С15алкинила;
соединение формулы
где Х2 и Х3 независимо друг от друга выбирают из СН3(СН2)12С≡С-, СН3СН2СН2С≡С(СН2)10-, СН3(СН2)7С≡С(СН2)5-, СН3(СН2)6С≡С(СН2)5- и СН3СН2С≡С(СН2)10-;
соединение формулы
где Х2 и Х3 независимо друг от друга выбирают из незамещенного С10-С18алкинила, где полярную головную группу получают из полярной головной группы фосфолипида;
соединение формулы
где X2 и Х3 независимо друг от друга выбирают из незамещенного С14алкинила и незамещенного С15алкинила, где полярную головную группу получают из полярной головной группы фосфолипида;
соединение формулы
где Х2 и Х3 независимо друг от друга выбирают из СН3(СН2)12С≡С-, СН3СН2СН2С≡С(СН2)10-, СН3(СН2)7С≡С(СН2)5-, СН3(СН2)6С≡С(СН2)5- и СН3СН2С≡С(СН2)10-, где полярную головную группу получают из полярной головной группы фосфолипида;
соединение формулы
где X2 и Х3 независимо друг от друга выбирают из незамещенного С10-С18алкинила, где полярную головную группу получают из полярной головной группы липида, выбранного из ряда, включающего фосфатидилхолин (ФХ), фосфатидилэтаноламин (ФЭ), 3-N,N-диметиламинопропан-1,2-диол (ДАП) и 3-N,N,N-триметиламмониопропан-1,2-диол (ТАП);
соединение формулы
где Х2 и X3 независимо друг от друга выбирают из незамещенного С14алкинила и незамещенного С15алкинила, где полярную головную группу получают из полярной головной группы липида, выбранного из ряда, включающего фосфатидилхолин (ФХ), фосфатидилэтаноламин (ФЭ), 3-N,N-диметиламинопропан-1,2-диол (ДАП) и 3-N,N,N-триметиламмониопропан-1,2-диол (ТАП);
соединение формулы
где Х2 и Х3 независимо друг от друга выбирают из СН3(СН2)12С≡С-, СН3СН2СН2С≡С(СН2)10-, СН3(СН2)7С≡С(СН2)5-, СН3(СН2)6С≡С(СН2)5- и СН3СН2С≡С(СН2)10-; где полярную головную группу получают из полярной головной группы липида, выбранного из ряда, включающего фосфатидилхолин (ФХ), фосфатидилэтаноламин (ФЭ), 3-N,N-диметиламинопропан-1,2-диол (ДАП) и 3-N,N,N-триметиламмониопропан-1,2-диол (ТАП).
Дополнительные объекты изобретения
Предпочтительным терапевтическим агентом композиции является нуклеотидная последовательность. Предпочтительно соединение находится в смеси или в сочетании с нуклеотидной последовательностью.
Нуклеотидная последовательность может представлять собой часть экспрессионный системы или всю такую систему, которую можно применять в терапии, например генной терапии.
Согласно предпочтительному объекту соединение по настоящему изобретению находится в смеси с конденсированным комплексом полипептид/нуклеиновая кислота, что позволяет получать невирусный вектор для введения нуклеиновой кислоты. Конденсированный комплекс полипептид/нуклеиновая кислота предпочтительно включает комплексы, описанные в WO 01/48233. В WO 01/48233 описаны невирусные векторы, предназначенные для введения нуклеиновой кислоты, которые содержат конденсированный комплекс полипептид/нуклеиновая кислота и катионный липид, причем комплекс включает (а) представляющую интерес нуклеотидную последовательность (NOI); и (б) один или несколько вирусных упаковочных полипептидов для нуклеиновой кислоты или их производных, где полипептиды или их производные (I) обладают способностью к связыванию с NOI; и (II) обладают способностью конденсировать NOI; причем NOI является гетерологичной по отношению к полипептиду. Предпочтительно полипептиды или их производные обладают способностью связываться с входящей в комплекс нуклеиновой кислотой. Предпочтительно полипептиды или их производные обладают способностью конденсировать входящую в комплекс нуклеиновую кислоту. Предпочтительно входящая в комплекс нуклеиновая кислота является гетерологичной относительно полипептидов или их производных.
Соединение по настоящему изобретению можно применять для частичной или полной замены катионного липида, описанного в WO 01/48233.
Таким образом, предпочтительным объектом настоящего изобретения является невирусный вектор для введения нуклеиновой кислоты, содержащий конденсированный комплекс полипептид/нуклеиновая кислота, катионный липид и соединение, предлагаемое в настоящем изобретении, причем комплекс включает
(а) представляющую интерес нуклеотидную последовательность (NOI); и
(б) один или несколько вирусных упаковочных полипептидов для нуклеиновой кислоты или их производных, где полипептиды или их производные (I) обладают способностью к связыванию с NOI; и (II) обладают способностью конденсировать NOI; и где NOI является гетерологичной по отношению к полипептиду;
невирусный вектор для введения нуклеиновой кислоты, содержащий конденсированный комплекс полипептид/нуклеиновая кислота и соединение, предлагаемое в настоящем изобретении, причем комплекс включает (а) нуклеиновую кислоту, которая имеет представляющую интерес последовательность (NOI); и (б) один или несколько вирусных упаковочных полипептидов для нуклеиновой кислоты или их производных, где полипептиды или их производные (I) обладают способностью к связыванию с NOI; и (II) обладают способностью конденсировать NOI; и где NOI является гетерологичной по отношению к полипептиду.
Соединение, предлагаемое в настоящем изобретении, можно объединять с липосомой или получать на его основе мицеллярную форму, способствующую введению.
Согласно еще одному объекту изобретения на основе соединения, предлагаемого в настоящем изобретении, можно получать кохелатные векторы, предназначенные для введения. Кохелатные векторы для введения представляют собой новую технологическую основу для перорального введения лекарственных средств. Кохелаты представляют собой фосфолипид-катионные преципитаты, состоящие из простых, встречающихся в естественных условиях продуктов, например фосфатидилсерина и кальция. Кохелаты представляют собой систему, размер которой может быть близок к нанометрам, которую можно капсулировать с использованием гидрофобных, амфифильных, отрицательно или положительно заряженных фрагментов.
Согласно одному из объектов соединение, предлагаемое в настоящем изобретении, находится в выделенной форме или очищенной форме.
Например, соединение может иметь форму или чистоту, которые отличаются от формы или чистоты в биологической системе, например, in vivo.
На основе соединений, предлагаемых в настоящем изобретении, можно получать фармацевтическую композицию, которая содержит соединение, предлагаемое в изобретении, необязательно в смеси с фармацевтически приемлемым носителем, разбавителем, эксципиентом или адъювантом.
Фармацевтическая композиция
Настоящее изобретение относится также к фармацевтической композиции, содержащей терапевтически эффективное количество агента, предлагаемого в изобретении, и фармацевтически приемлемые носитель, разбавитель или эксципиенты (включая их комбинации).
Такая композиция представляет собой композицию, содержащую или состоящую из терапевтически эффективного количества фармацевтически активного агента. Предпочтительно она включает фармацевтически приемлемые носитель, разбавитель или эксципиенты (включая их комбинации). Приемлемые носители или разбавители, которые используют в терапевтических целях, хорошо известны в фармацевтике и описаны, например, в Remington's Pharmaceutical Sciences, Mack Publishing Co. (A.R.Gennaro (ред.), 1985). Выбор фармацевтического носителя, эксципипента или разбавителя должен зависеть от предполагаемого пути введения и соответствовать стандартной фармацевтической практике. Фармацевтические композиции могут содержать в качестве носителя, эксципиента разбавителя, или в дополнение к ним, любой(ые) приемлемый(ые) носитель(и), замасливатель(и), суспендирующий(ие) агент(ы), агент(ы) для нанесения покрытия, солюбилизатор(ы).
Может потребоваться вводить указанную фармацевтическую композицию в стерильной форме. Она может находиться в виде стандартной дозы и, как правило, в запечатанном контейнере. Можно применять широкое разнообразие стандартных доз.
Согласно настоящему изобретению фармацевтические композиции могут включать один или несколько из следующих ингредиентов: консерванты, солюбилизаторы, стабилизаторы, смачивающие вещества, эмульгаторы, подслащивающие вещества, красители, корригенты, одоранты, соли (соединения, предлагаемые в настоящем изобретении, сами могут иметь форму фармацевтически приемлемой соли), буферы, агенты для нанесения покрытия, антиоксиданты, суспендирующие агенты, адъюванты, эксципиенты и разбавители. Примеры консервантов включают бензоат натрия, сорбиновую кислоту и эфиры пара-гидроксибензойной кислоты.
Помимо соединений, предлагаемых в настоящем изобретении, они могут содержать другие терапевтически активные агенты. Если применяют два или большее количество терапевтических агентов, то их можно вводить по отдельности (например, в различные моменты времени и/или с использованием различных путей введения), поэтому не требуется, чтобы они всегда присутствовали в одной композиции. Такая совместная терапия подпадает под объем настоящего изобретения.
Путь введения
Фармацевтическую композицию, предлагаемую в настоящем изобретении, можно адаптировать для введения любым приемлемым путем. Например, ее можно вводить перорально (включая щечный или подъязычный), ректальный, назальный, местный (включая щечный, подъязычный или трансдермальный), вагинальный или парентеральный (включая подкожный, внутримышечный, внутривенный или интрадермальный) пути. Такую композицию можно приготавливать с помощью любого метода, известного в данной области, например, смешением одного или нескольких действующих веществ с приемлемым носителем.
Для введения фармацевтических композиций, предлагаемых в настоящем изобретении, в зависимости от требуемого пути введения можно применять различные системы, предназначенные для введения лекарственных средств. Системы для введения лекарственных средств описаны, например, у Langer (Science, 249, 1991, сс.1527-1533) и у Illum и Davis (Current Opinions in Biotechnology, 2, 1991, сс.254-259). Ниже более подробно будут описаны различные пути введения лекарственных средств.
Агенты, предлагаемые в настоящем изобретении, можно вводить индивидуально, но, как правило, их вводят в виде фармацевтической композиции, например, когда агент смешивают с приемлемым фармацевтическим эксципиентом, разбавителем или носителем, выбранным в зависимости от требуемого пути введения и в соответствии со стандартной фармацевтической практикой.
Например, агент можно вводить (например, перорально или местно) в форме таблеток, капсул, капель, эликсиров, растворов или суспензий, которые могут содержать корригены или краситель и которые можно применять для немедленного, замедленного, модифицированного, непрерывного, периодического или контролируемого введения.
Таблетки могут содержать эксципиенты, такие как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, вторичный кислый фосфат кальция и глицин, разрыхлители, такие как крахмал (предпочтительно кукурузный, картофельный или маниоковый крахмал), натрий гликолят крахмала, кроскармелоза натрия и некоторые комплексы с силикатами, и связующие вещества, применяемые при грануляции, такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (ГПМЦ), гидроксипропилцеллюлоза (ГПЦ), сахароза, желатин и аравийская камедь. Кроме того, могут быть включены замасливатели, такие как стеарат магния, стеариновая кислота, глицерилбегенат и тальк.
Твердые композиции аналогичного типа можно применять также в качестве наполнителей желатиновых капсул. Предпочтительными эксципиентами для этой цели являются лактоза, крахмал, целлюлоза, молочный сахар или высокомолекулярные полиэтиленгликоли. При применении в виде водных суспензий и/или эликсиров агент можно объединять с различными подслащивающими веществами или корригентами, окрашивающими агентами или красителями, эмульгирующими и/или суспендирующими агентами и с разбавителями, такими как вода, этанол, пропиленгликоль и глицерин и их комбинациями.
Пути введения (доставки) включают (но не ограничиваясь ими) один или несколько следующих путей: пероральный (например, в виде таблетки, капсулы или раствора для приема внутрь), местный, через слизистую оболочку (например, в виде назального спрея или аэрозоля для ингаляции), назальный, парентеральный (например, с помощью инъецируемой формы), желудочно-кишечный, интраспинальный, внутрибрюшинный, внутримышечный, внутривенный, внутриматочный, внутриглазной, внутрикожный, внутричерепной, внутритрахеальный, внутривлагалищный, интрацеребровентрикулярный, интрацеребральный, подкожный, глазной (включая введение в стекловидное тело или в глазную полость), чрескожный, ректальный, щечный, через пенис, вагинальный, эпидуральный, подъязычный.
Следует понимать, что не является необходимым вводить все агенты одинаковым путем. Аналогично этому, если композиция содержит более одного действующего вещества, то эти компоненты можно вводить различными путями.
Если агент, предлагаемый в настоящем изобретении, вводят парентерально, то примерами такого введения агента являются один или несколько таких путей, как внутривенный, внутриартериальный, внутрибрюшинный, подоболочечный, внутрижелудочковый, внутриматочный, интрастернальный, внутричерепной, внутримышечный или подкожный; и/или с помощью инфузии.
Пероральное введение
Фармацевтические композиции, предназначенные для перорального введения, можно применять в виде капсул или таблеток; в виде порошков или гранул; в виде растворов, сиропов или суспензий (в водный или неводных жидкостях); в виде съедобной пены или в виде взбитых масс; или в виде эмульсий. Таблетки или твердые желатиновые капсулы могут содержать лактозу, кукурузный крахмал или его производные, стеариновую кислоту или ее соли. Мягкие желатиновые капсулы могут содержать растительные масла, воски, жиры, полутвердые или жидкие полиолы и т.д. Растворы и сиропы могут содержать воду, полиолы и сахара. Для приготовления суспензий можно использовать масла (например, растительные масла) с получением суспензий типа масло/вода или вода/масло. На действующее вещество, предназначенное для перорального введения, можно наносить покрытие с помощью материала, который замедляет разрушение и/или абсорбцию действующего вещества в желудочно-кишечном тракте, или путем смешения с таким материалом (например, можно применять глицерилмоностеарат или глицерилдистеарат). Указанное замедленное высвобождение действующего вещества может происходить в течение многих часов, и при необходимости действующее вещество можно защищать от разложения в желудке. Фармацевтические композиции для перорального введения можно приготавливать с целью облегчения высвобождения действующего вещества в определенной области желудочно-кишечного тракта, что связано с конкретным значением рН или ферментативной средой.
(II) Чрескожное введение
Фармацевтические композиции, предназначенные для чрескожного введения, можно применять в виде дискретных бляшек, которые служат для поддерживания тесного контакта с эпидермисом реципиента в течение длительного периода времени. Например, действующее вещество может выходить из бляшки в результате ионофореза (ионофорез описан в Pharmaceutical Research, 3 (6), 1986, с.318).
(III) Местное применение
В альтернативном варианте агент, предлагаемый в настоящем изобретении, можно вводить в форме суппозитория или пессария или его можно наносить местно в форме геля, гидрогеля, лосьона, раствора, крема, мази или пылящего порошка. Агент, предлагаемый в настоящем изобретении, можно применять также накожно или чрескожно, например, с помощью кожной бляшки. Их можно вводить также легочным или ректальным путями. Их можно применять также глазным путем. Для введения в глаз соединения можно приготавливать в виде микросуспензий в изотоническом стерильном физиологическом растворе с определенным значением рН или предпочтительно в виде растворов в изотоническом стерильном физиологическом растворе с определенным значением рН необязательно в сочетании с консервантом, таким как бензилалконийхлорид. В другом варианте соединение можно применять в форме мази, например вазелиновой мази.
Для местного нанесения на кожу можно приготавливать приемлемую мазь, содержащую соединение, суспендированное или растворенное, например, смешанное с одним или несколькими из следующих ингредиентов: минеральное масло, вазелиновое масло, белый вазелин, пропиленгликоль, полиоксиэтиленполиоксипропиленовое производное, эмульгирующий воск и вода. В другом варианте на его основе можно приготавливать приемлемый лосьон или крем путем суспендирования или растворения, например, в смеси одного или нескольких следующих ингредиентов: минеральное масло, сорбитанмоностеарат, полиэтиленгликоль, жидкий парафин, полисорбат 60, цетиловые эфиры воска, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и вода.
(IV) Ректальное введение
Фармацевтические композиции, предназначенные для ректального введения, можно применять в виде суппозиториев или клизм.
(V) Назальное введение
Фармацевтические композиции, предназначенные для назального введения, могут включать твердые носители, например порошки (предпочтительно размер частиц которых составляет 20-500 мкм). Порошки можно вводить таким образом, чтобы их можно было вдыхать через нос, т.е. путем быстрой ингаляции через нос из контейнера для порошка, который держат близко к носу. В другом варианте композиции для назального введения могут включать жидкие носители, например, представлять собой назальные спреи или назальные капли. Композиции могут включать водные или масляные растворы действующего вещества.
Композиции для введения путем ингаляции могут поставляться в специально приспособленных для этой цели устройствах, например, таких как находящиеся под давлением аэрозольные баллоны, распылители или инсуффляторы. Эти устройства можно конструировать таким образом, чтобы они обеспечивали введение предварительно определенных доз действующего вещества.
(VI) Вагинальное введение
Фармацевтические композиции, предназначенные для вагинального введения, могут иметь форму пессариев, тампонов, кремов, гелей, паст, пены или спреев.
(VII) Парентеральное введение
Если агент, предлагаемый в изобретении, вводят парентерально, то примерами такого введения агента являются один или несколько таких путей, как внутривенный, внутриартериальный, внутрибрюшинный, подоболочечный, внутрижелудочковый, внутриматочный, интрастернальный, внутричерепной, внутримышечный или подкожный; и/или с помощью инфузии.
При парентеральном введении агент предпочтительно применять в форме стерильного водного раствора, который может содержать другие вещества, например соли или глюкозу, в количестве, достаточном для получения изотоничного крови раствора. Водные растворы при необходимости можно соответствующим образом забуферивать (предпочтительно до рН 3-9). Приготовление приемлемых парентеральных композиций в стерильных условиях легко осуществлять с помощью стандартных фармацевтических методов, хорошо известных специалистам в данной области.
Чрескожный
Понятие «чрескожный» относится к введению соединения путем прохождения через кожу в кровоток.
Через слизистую оболочку (трансмукозальный)
Понятие «трансмукозальный» относится к введению соединения через слизистую ткань в кровоток.
Трансуретральный или внутриматочный
Понятия «трансуретральный» или «внутриматочный» относятся к введению лекарственного средства в матку таким образом, что лекарственное средство контактирует и проходит через стенку матки и входит в кровоток.
Увеличение проницаемости или ускорение проникновения
Понятия «увеличение проницаемости» или «ускорение проникновения» относятся к повышению проницаемости кожи или слизистой ткани для выбранного фармакологически активного вещества, в результате чего скорость, с которой соединение проникает через кожу или слизистую ткань, повышается. Вещества, увеличивающие проницаемость, могут включать, например, диметиосульфоксид (ДМСО), диметилформамид (ДМФ), N,N-диметилацетамид (ДМА), децилметилсульфоксид (CIOMSO), полиэтиленгликольмонолаурат (ПЭГМЛ), глицерилмонолаурат, лецитин, 1-замещенные азациклогептаноны, в частности 1-N-додецилциклазациклогептаноны (которые поступают в продажу под товарным знаком Azone ТМ, фирма Nelson Research & Development Co., Инвин, шт.Калифорния), спирты и т.п.
Носители или наполнители
Понятия «носители» или «наполнители» относится к носителям, пригодным для введения соединения, и включают любой такой носитель, известный в данной области, например любую жидкость, гель, растворитель, жидкий разбавитель, солюбилизатор или т.п., которые являются нетоксичными и которые не оказывают вредного воздействия ни на один из компонентов композиции.
Примеры фармацевтически приемлемых носителей включают, например, воду, соляные растворы, спирт, силикон, воски, вазелин, растительные масла, полиэтиленгликоли, пропиленгликоль, сахара, желатин, лактозу, амилозу, стеарат магния, тальк, поверхностно-активные вещества, кремниевую кислоту, вязкий парафин, парфюмерное масло, моноглицериды и диглицериды жирных кислот, петролейные эфиры жирных кислот, гидроксиметилцеллюлозу, поливинилпирролидон и т.п.
Эпидермальное введение лекарственного средства (трансферсомы)
Трансферсомы («несущие тельца») представляют собой сложный комплекс, наиболее часто пузырчатые, би- или многокомпонентные агрегаты, которые обладают способностью пересекать барьеры и переносить продукт от места нанесения к мишени. Трансферсомы поступают в продажу от фирмы IDEA Corporation, Мюнхен, Германия, и TRANSFERSOME является товарным знаком компании. Технологию чрескожного введения лекарственного средства, основанную на использовании трансферсом, можно применять для контролируемого и неинвазивного введения широкого спектра крупных молекул, а также для улучшенного введения небольших молекул, включая антагонисты метаболических ферментов и/или лекарственных средств, предлагаемых в настоящем изобретении.
Трансферсомы можно оптимизировать с целью достижения очень высокой гибкости и саморегуляции мембран. В результате этого они могут деформироваться и получают способность эффективно пересекать микропористый барьер, даже если доступные проходы существенного меньше среднего размера агрегата. Формы в виде трансферсом, как правило, состоят из амфипатических соединений, суспендированных в водном растворе, необязательно содержат биологически совместимые поверхностно-активные вещества. Пузырчатые трансферсомы состоят из липидного бислоя, окружающего водное ядро, и дополнительно содержат по меньшей мере один компонент, который обладает способностью размягчать мембрану. Таким образом, бислой трансферсомы является более гибким, чем даже метастабильная липосомная мембрана. Так, пузырьки трансферсомы легко изменяют форму путем местного регулирования связанного с окружающей средой стресса.
Кожа является одним из лучших биологических барьеров. Ее самая поверхностная часть, роговой слой, толщина которого составляет менее 10% от толщины кожи, на 80% определяет проницаемость кожного барьера. Этот защищающий организм слой состоит из перекрывающихся, рыхлых корнеоцитов, организованных в колончатые кластеры, закрытых состоящими из нескольких пластин липидными слоями, которые ковалентно связаны с клеточными мембранами и очень плотно упакованы. Как правило, среднее количество и уровень упорядоченности межклеточных липидных пластин возрастает по мере приближения к поверхности кожи. Это сопровождается непрерывным, но нелинейным снижением содержания воды вблизи поверхности. Несмотря на это, пик проницаемости кожного барьера локализован во внутренней области рогового слоя, в котором межклеточные липидные покрытия уже сформированы, но еще не подвергнуты риску, связанному с отделением клеток кожи.
Проникновение агрегатов трансферсом через кожу зависит от гибкости, гидрофильности и способности сохранять целостность пузырьков, при этом форма агрегата претерпевает существенные изменения. Когда суспензию пузырьков трансферсом помещают на поверхность кожи, вода испаряется из относительно сухой поверхности кожи и пузырьки начинают высыхать. Благодаря выраженной полярности большинства ингредиентов трансферсом, большому количеству гидрофильных групп на мембране, способствующих мягкости мембраны, пузырьки присоединяются к областям с высоким содержанием воды в узких щелях между примыкающими клетками в кожном барьере, делая кожу проницаемой для носителя. Этот механизм в сочетании с очень большой способностью пузырьков к деформации позволяет агрегатам трансферсом временно открывать тонкие «трещины», через которые вода обычно испаряется из кожи. Так образуются каналы между клетками кожи, которые на два порядка шире, чем исходные поры. Через такие новые активированные проходы могут проходить пузырьки, обладающие достаточной способностью к деформации, что поддерживает их целостность, но изменяет их форму, приспосабливая ее к каналу. Проходя по образовавшимся «виртуальным путям» или «виртуальным каналам» в роговом слое, трансферсомы достигают областей с высоким содержанием воды в более глубоких слоях кожи. Там пузырьки перераспределяются. Поскольку трансферсомы являются слишком большими для того, чтобы локально проникнуть в кровеносных сосуд, они обходят капиллярное русло и достигают подкожной ткани, где они накапливаются.
Хотя небольшие молекулы, которые пересекают роговой слой кожи (stratum corneum), как правило, выходят из кожи через кровоток, введение лекарственных средств с помощью пузырьков трансферсом позволяет осуществлять накапливание лекарственного средства глубоко под кожей. Из-за большого размера пузырьки медленно выходят из кожи и связанное с ними лекарственное средство может накапливаться в области-мишени. Следовательно, опосредуемое трансферсомой введение тестируемых лекарственных средств имеет тенденцию сдвигать область распределения лекарственного средства к более глубоко расположенной ткани под местом нанесения.
Гематоэнцефалический барьер (ТЭБ)
Фармацевтические композиции могут быть созданы так, чтобы они преодолевали гематоэнцефалический барьер (ГЭБ). Например, можно выбрать носитель, такой как жирная кислота, инозит или холестерин, который обладает способностью проникать через ГЭБ. Носитель может представлять собой вещество, которое проникает в мозг с помощью специфической транспортной системы в эндотелиальных клетках мозга, такой как инсулиноподобный фактор I или II. Носитель может быть связан с действующим веществом или может содержать/находиться в смеси с действующим веществом. Для прохождения ГЭБ можно применять липосомы. В WO 91/04014 описана основанная на липосомах система введения, в которой действующее вещество может быть инкапсулировано/погружено и в которой молекулы, в норме проходящие через ГЭБ (например, инсулин или инсулиноподобный фактор I или II), находятся на наружной поверхности липосомы. Основанные при применении липосом системы введения обсуждаются также в патенте США 4704355.
Введение с помощью полимеров/терапевтические агенты
Агенты можно также вводить, связывая их с полимерами. Терапевтические системы, основанные на использовании полимеров, как предполагается, являются эффективными системами введения, и они, как правило, содержат один или несколько подлежащих введению агентов, связанных с полимерной молекулой, которая действует в качестве носителя. При этом агенты расположены в полимерном каркасе и переносятся к клетке-мишени вместе с полимером.
Агенты могут быть связаны, слиты, смешаны, объединены или каким-либо иным образом соединены с полимером. Связь и т.д. между агентом и полимером может быть постоянной или временной и может включать ковалентные или нековалентные взаимодействия (в том числе ионные взаимодействия, гидрофобные силы, взаимодействия Ван-дер-Ваальса) и т.д. Точный механизм связи не имеет решающего значения в том случае, если агент достигает клетки-мишени практически вместе с полимером. Для простоты в контексте настоящего описания «агент, связанный с полимерным носителем» обозначают как «конъюгат полимер-агент».
Можно применять любой пригодный полимер, например натуральный или синтетический полимер, предпочтительно полимер-носитель представляет собой синтетический полимер, такой как ПЭГ. Более предпочтительно полимер-носитель представляет собой биологически инертную молекулу. Конкретными примерами полимеров являются полиэтиленгликоль (ПЭГ), сополимеры N-(2-гидроксипропил)метакриламида (ГПМА), дендримеры полиамидоамина (ПАМАМ), ОЭМА (оксиэтилметакрилат), линейные полиамидоаминные полимеры и т.д. Для присоединения агента к полимеру можно применять любой приемлемый линкер. Предпочтительно линкер представляет собой биологически разлагаемый линкер. Применение биологически разлагаемых линкеров позволяет контролировать высвобождение агента во внеклеточную или внутриклеточную среду. Высокомолекулярные макромолекулы не могут проходить в клетки путем пассивной диффузии и вместо этого поглощаются в виде окруженных мембраной пузырьков. Находясь внутри пузырька, внутриклеточные ферменты могут оказывать воздействие на конъюгат полимер-агент, приводя к высвобождению агента. Контролируемое внутриклеточное высвобождение позволяет избегать токсических побочных воздействий, связанных с многими лекарственными средствами.
Кроме того, агенты можно конъюгировать, соединять и т.д. с помощью методов, хорошо известных в данной области, с любым пригодным полимером, и вводить их. Агенты могут, в частности, включать любые молекулы, обозначенные как «вторичные агенты», такие как полипептиды, нуклеиновые кислоты, макромолекулы и т.д., как описано в приведенном ниже разделе. В частности, как указано, агент может представлять собой пролекарство.
Возможность осуществлять выбор исходного полимера позволяет конструировать конъюгаты полимер-агент с требуемыми свойствами. Молекулярную массу полимера (и следовательно, конъюгата полимер-агент), а также его заряд и характеристики гидрофобности можно точно подбирать. Преимуществами, связанными с применением конъюгатов полимер-агент, является экономичность их получения, стабильность (более длительный срок годности при хранении) и снижение иммуногенности и побочных воздействий. Кроме того, конъюгаты полимер-агент особенно важны для направленного переноса в опухолевые клетки из-за явления повышенной проницаемости и сохранения (ПРС), в результате чего растущие опухоли становятся более «проницаемыми» для находящихся в кровотоке макромолекул и крупных частиц, позволяя им легко входить во внутреннюю область опухоли. Для них также характерны повышенное накопление и низкая токсичность (как правило, 10-20% от токсичности агента в свободном состоянии). Применение сверхразветвленных дендримеров, например дендримеров ПАМАМ, особенно целесообразно, поскольку они позволяют получать монодисперстные композиции и также обеспечивают гибкость сайтов присоединения (во внутренней или внешней области дендримера). Конъюгаты полимер-агент чувствительны к значениям рН, например, конъюгаты, содержащие полиаминдоаминовые полимеры, можно целенаправленно изменять в зависимости от конкретной внутриклеточной среды. Это позволяет обеспечивать высвобождение лекарственного средства только тогда, когда связанный с полимером терапевтический агент находится в среде с определенным значением рН или диапазоном значений рН, т.е. внутри конкретного внутриклеточного компартмента. Конъюгаты полимер-агент дополнительно могут содержать ингредиенты, обеспечивающие направленный перенос, такие как иммуноглобулин или антитело, которые направляют конъюгат полимер-агент к определенным тканям, органам или клеткам, содержащим мишень, например конкретный антиген. Другие обеспечивающие направленный перенос ингредиенты приведены в настоящем описании и хорошо известны в данной области.
Конкретные примеры конъюгатов полимер-агент включают «Smancs», который представляет собой конъюгат, содержащий сополимер стирола и малеинового ангидрида и противоопухолевый агент неокарзиностатин, и конъюгат, содержащий ПЭГ (полиэтиленгликоль) и L-аспарагиназу, предназначенный для лечения лейкоза; РК1 (конъюгат сополимера ГПМА и противоракового лекарственного средства доксорубицина); РК2 (аналогичен РК1, но содержит также галактозную группу для направленного переноса к первичной и вторичной опухоли печени); конъюгат сополимера ГПМА и противоракового агента каптотецина; конъюгат сополимера ГПМА и противоракового агента паклитаксела; сополимер ГПМА-платинат и т.д. Любые из этих конъюгатов полимер-агент можно применять для совместного введения в трансгенные клетки по настоящему изобретению.
Уровни доз
Как правило, лечащий врач может определять эффективную дозу, которая является наиболее приемлемой для каждого пациента. Конкретный уровень доз и частота их введения может варьироваться для каждого пациента в зависимости от различных факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия этого соединения, возраст, вес тела, общее состояние здоровья, пол, диету, путь и продолжительность обработки, скорость выделения, комбинацию лекарственных средств, серьезность конкретного состояния и индивидуальные особенности лечения, которому ранее подвергался пациент. Агент и/или фармацевтическую композицию по настоящему изобретению можно вводить по схеме от 1 до 10 раз в день, например один или два раза в день.
Для перорального и парентерального введения человеку суточную дозу агента можно применять в виде однократной или разделенных доз.
В зависимости от потребности агент можно вводить в дозе от 0,01 до 30 мг/кг веса тела, например от 0,1 до 10 мг/кг, более предпочтительно от 0,1 до 1 мг/кг веса тела. Должно быть очевидно, что указанные дозы даны в качестве примера усредненных доз. Естественно, что в отдельных случаях они могут быть выше или ниже указанных диапазонов доз.
Терапевтически эффективное количество
Понятие «терапевтически эффективное количество» относится к количеству терапевтического агента, которое является эффективным для конкретной цели. Поскольку потребности каждого пациента могут варьироваться, определение оптимальных диапазонов эффективных количеств каждого из аддуктов оксида азота известно специалистам в данной области. Как правило, схему приема лекарственного средства для лечения состояния с помощью соединений и/или композиций, предлагаемых в настоящем изобретении, выбирают в зависимости от различных факторов, таких как тип, возраст, вес, пол, диета и медицинское состояние пациента, серьезность дисфункции, путь введения, фармакологические особенности, такие как активность, эффективность, фармакокинетические и токсикологические профили конкретного применяемого соединения, используемая система для введения лекарственного средства и от того, является ли соединение компонентом лекарственной комбинации, и специалист в данной области может ее регулировать. Таким образом, фактически применяемая схема приема лекарственного средства может варьироваться в широких пределах и может отличаться от изложенной в описании предпочтительной схемы приема лекарственного средства.
Индивидуум
В контексте настоящего описания понятие «индивидуум» относится к позвоночным животным, в частности представителям млекопитающих. Понятие включает (но не ограничиваясь ими) домашних животных, применяемых для спорта животных, приматов и человека.
Фармацевтические комбинации
Как правило, агент можно использовать в сочетании с одним или несколькими фармацевтически активными агентами. Иногда другой агент представляет собой вспомогательный агент.
Пациент
Понятие «пациент» относится к животным, предпочтительно млекопитающим, более предпочтительно к человеку.
Фармацевтически приемлемая соль
Агент может иметь форму и/или его можно вводить в виде фармацевтически приемлемой соли, например кислотно-аддитивной соли или соли присоединения основания, или ее сольвата, включая гидрат. Обзор приемлемых солей см. у Berge и др., J. Pharm. Sci., 66, 1977, сс.1-19.
Как правило, фармацевтически приемлемую соль можно легко получать с помощью соответствующей кислоты или основания. Соль можно осаждать из раствора и собирать фильтрацией или ее можно получать путем выпаривания растворителя.
Приемлемые кислотно-аддитивные соли получают из кислот, которые образуют нетоксичные соли, их примерами являются такие соли, как гидрохлорид, гидробромид, гидройодид, сульфат, бисульфат, нитрат, фосфат, вторичный кислый фосфат, ацетат, малеат, фумарат, лактат, тартрат, цитрат, глюконат, сукцинат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, пара-толуолсульфонат и памоат.
Приемлемые соли присоединения оснований получают из оснований, которые образуют нетоксичные соли, их примерами являются такие соли, как натриевая, калиевая, алюминиевая, кальциевая, магниевая и цинковая соли диэтаноламина.
Болезненные состояния
Соединение или композицию по настоящему изобретению можно применять для лечения нарушений, указанных в WO-A-98/05635. Для простоты ссылки ниже перечислены некоторые из них: рак, воспаление или воспалительное заболевание, дерматологические нарушения, лихорадка, сердечно-сосудистые нарушения, кровотечение, свертывание и острая фаза реакции, кахексия, анорексия, острое инфекционное заболевание, ВИЧ-инфекция, состояния шока, реакции «трансплантат против хозяина», аутоиммунное заболевание, повреждение, связанное с реперфузией, менингит, мигрень и зависящий от аспирина тромбоз; рост, инвазия и распространение опухоли, ангиогенез, метастаз, злокачественный асцит и злокачественный плевральный выпот; церебральная ишемия, ишемическая болезнь сердца, остеоартрит, ревматоидный артрит, остеопороз, астма, рассеянный склероз, нейродегенеративные заболевания, болезнь Альцгеймера, атеросклеоз, «удар», васкулит, болезнь Крона и неспецифический язвенный колит; периодонтит, гингивит, псориаз; атопический дерматит, хронические язвы, врожденный буллезный эпидермолиз; изъязвление роговицы, ретинопатия и заживление ран, связанных с хирургическим вмешательством; ринит, аллергический конъюнктивит, экзема, анафилактическая реакция; рестеноз, застойная сердечная недостаточность, эндометриоз, атеросклероз или эндосклероз.
Помимо перечисленных или в дополнение к ним соединение или композицию по настоящему изобретению можно применять для лечения нарушений, указанных в WO-A-98/07859. Для простоты ссылки ниже перечислены некоторые виды активности соединения или композиции:
активность цитокинов и активность в отношении клеточной пролиферации/дифференциации; иммуносуперессорная или иммуностимулирующая активность (например, для лечения иммунного дефицита, включая инфекцию, связанную с вирусом иммунодефицита человека;
регуляция роста лимфоцитов; лечение рака и многих аутоиммунных заболеваний и для предупреждения отторжения трансплантата или индуцированного опухолью иммунитета); регуляция гематопоэза, например, при лечении миелоидных или лимфоидных заболеваний; усиление роста костной ткани, хряща, сухожилия, связки и нервной ткани, например, для заживления ран, лечения ожогов, язв и болезней периодонта и нейродегенеративных болезней;
ингибирование или активация фолликулостимулирующего гормона (модуляция фертильности); хемотактическая/хемокинетическая активность (например, с целью мобилизации специфических типов клеток к местам повреждения или инфекции); гемостатическая и тромболитическая активность (например, для лечения гемофилии и «удара»); противовоспалительная активность (например, для лечения септического шока или болезни Крона); активность в качестве противомикробного агента; модуляторная активность в отношении, например, метаболизма или поведения; в качестве аналгетиков; активность в отношении специфических, связанных с дефицитом нарушений; активность в отношении, например, псориаза, в терапии человека или в ветеринарии.
Помимо перечисленных или в дополнение к ним композицию, предлагаемую в настоящем изобретении, можно применять для лечения нарушений, указанных в WO-A-98/09985. Для простоты ссылки ниже перечислены некоторые виды активности композиции: ингибирующая активность в отношении макрофагов и/или ингибирующая активность в отношении Т-клеток и связанная с ними противовоспалительная активность; антииммунная активность, т.е. ингибирующие воздействия на клеточный и/или гуморальный иммунный ответ, включая ответ, не связанный с воспалением; ингибирование способности макрофагов и Т-клеток прикрепляться к компонентам внеклеточного матрикса и фибронектину, а также повышающая регуляция экспрессии рецепторов в Т-клетках; ингибирование нежелательного иммунного ответа и воспаления, включая артрит, в том числе ревматоидный артрит, воспаления, связанного с гиперчувствительностью, аллергических реакций, астмы, системной красной волчанки, связанных с коллагеном заболеваний и других аутоиммуннных заболеваний, воспаления, связанного с атеросклерозом, артериосклерозом, атеросклеротическим заболеванием сердца, повреждением, вызванным реперфузией, остановкой сердца, инфарктом миокарда, сосудистыми воспалительными заболеваниями, респираторным дистресс-синдромом или другими сердечно-легочными заболеваниями, воспаления, связанного с пептической язвой, неспецифическим язвенным колитом и другими заболеваниями желудочно-кишечного тракта, гепатическим фиброзом, циррозом печени или другими болезнями печени, тиреоидитом или другими заболеваниями желез, гломерулонефритом или другими почечными и урологическими заболеваниями, отитом или другими оториноларингологическими заболеваниями, дерматитами или другими кожными болезнями, заболеваниями периодонта или другими зубными болезнями, орхитами или эпидидимоорхитами, бесплодием, травмой яичка или другими, близкими к иммунным заболеваниями, относящими к яичку, дисфункцией плаценты, плацентарной недостаточностью, привычным выкидышем, эклампсией, преэклампсией и другими иммунными и/или связанными с воспалением гинекологическими заболеваниями, задним увеитом, промежуточными увеитом, передним увеитом, конъюнктивитом, хориоретинитом, увеоретинитом, оптическим невритом, внутриглазным воспалением, например ринитом или кистозным отеком желтого пятна, симпатетической офталмией, склеритом, пигментным ретинитом, иммунными или воспалительными компонентами дегенеративного заболевания, воспалительными компонентами, связанными с травмой глаза, воспалением глаза, вызванным инфекцией, пролиферативными витреоретинопатиями, острой ишемической глазной невропатией, чрезмерными шрамами, например, после операции по фильтрации глаукомы, иммунной и/или воспалительной реакцией против глазного имплантата и другими иммунными и связанными с воспалением офталмическими заболеваниями, воспаления, связанного с аутоиммунными заболеваниями, или состояниями, или нарушениями, при которых в центральной нервной системе (ЦНС) или и в любых других органах показаны подавление иммунного ответа и/или воспаления, с болезнью Паркинсона, осложнением и/или побочными действиями, связанным с лечением болезни Паркинсона, связанной со СПИДом деменцией, комлексной связанной с ВИЧ энцефалопатией, болезнью Девика, хореей Сиденгама, болезнью Альцгеймера и другими дегенеративными заболеваниями, состояниями или нарушениями ЦНС, воспалительными компонентами «удара», постполимиелитным синдромом, иммунными и воспалительными компонентами психиатрических заболеваний, миелитами, энцефалитами, подострыми склерозирующими панэнцефалитами, энцефаломиелитами, острой невропатией, подострой невропатией, хронической невропатией, синдромом Гийена-Барре-Штроля, хореей Сиденгама, тяжелой псевдопаралитической миастенией, псевдоопухолевым энцефалитом, синдромом Дауна, болезнью Гентингтона, боковыми амиотрофическим склерозом, воспалительными компонентами сдавления ЦНС или травмы ЦНС или инфекции ЦНС, воспалительными компонентами мышечной атрофии и дистрофии, и заболеваниями, связанными с иммунной системой или воспалениями, состояниями или нарушениями центральной и периферической нервных систем, посттравматическим воспалением, септическим шоком, инфекционными заболеваниями, воспалительными осложнениями или побочными воздействиями хирургического вмешательства, трансплантацией костного мозга или другими связанными с трансплантацией осложнениями и/или побочными воздействиями, воспалительными и/или иммунными осложнениями или побочными воздействиями, связанными с генной терапией, например, с инфекцией, связанной с вирусным носителем, или воспалением, связанным со СПИДом, для подавления или ингибирования гуморального и/или клеточного иммунного ответа, для лечения или облегчения связанных с моноцитами или лейкоцитами пролиферативных заболеваний, например лейкоза, путем снижения количества моноцитов или лимфоцитов, для предупреждения и/или лечения отторжения трансплантата при трансплантации природных или искусственных клеток, тканей или органов, таких как роговица, костный мозг, органы, хрусталики, пейсмекеры, природная или искусственная кожная ткань.
Лечение
Лечение включает любую терапевтическую обработку, которая может быть полезной для человека или животного, кроме человека. Особенно предпочтительным является лечение млекопитающих. Под объем настоящего изобретения подпадают как лечение человека, так и лечение животных.
Лечение может касаться уже существующего состояния или может быть профилактическим. Оно может затрагивать взрослую особь, ювенильную стадию, детскую стадию, плод или часть каждого из указанных объектов (например, орган, ткань, клетку или молекулу нуклеиновой кислоты).
При лечении действующее вещество можно вводить любым приемлемым путем и в любой приемлемой дозе. Дозы могут варьироваться в широких пределах в зависимости от природы лечения, возраста и состояния индивидуума, подлежащего лечению, и т.д., и лечащий врач может определять оптимальную для применения дозу. Однако, не ограничиваясь какими-либо конкретными дозами, можно рекомендовать в качестве приемлемой суточную дозу соединения по настоящему изобретению от 1 мкг до 1 мг/кг веса тела. Дозу можно повторять так часто, как это требуется. При проявлении побочных воздействий количество и/или частоту приема доз можно снижать согласно эффективной клинической практике.
Полиморфная(ые) форма(ы)/асимметричный(ые) атом(ы) углерода
Агент, предлагаемый в настоящем изобретении, может находиться в полиморфной форме.
Агент, предлагаемый в настоящем изобретении, может содержать один или несколько асимметричных атомов углерода и поэтому может находиться в двух или в большем количестве стереоизомерных форм. Когда агент содержит алкенил или алкенилен, то может иметь место также цис- (Е)- и транс- (Z)-изомерия. Настоящее изобретение включает индивидуальные стереоизомеры агента и, если они существуют, индивидуальные таутомерные формы, а также их смеси.
Разделение диастереоизомеров или цис- и транс-изомеров можно осуществлять с помощью общепринятых методов, например фракционированной кристаллизации, хроматографии, ЖХВР смеси стереоизомеров агента или приемлемой соли или его производного.
Индивидуальный энантиомер соединения, представляющего собой агент, предлагаемого в изобретении, можно получать также из соответствующего оптически чистого промежуточного продукта или путем разделения, например, с помощью ЖХВР, соответствующего рацемата с использованием пригодного хирального носителя или при необходимости путем фракционированной кристаллизации диастереоизомерных солей, полученных с помощью взаимодействия соответствующего рацемата с приемлемыми оптически активными кислотой или основанием.
Изотопные варианты
Настоящее изобретение включает также все приемлемые изотопные варианты агента или его фармацевтически приемлемой соли. Под изотопным вариантом агента по настоящему изобретению или его фармацевтически приемлемой соли понимают вариант, в котором по меньшей мере один атом заменен атомом, который имеет тот же самый атомный номер, но атомная масса которого отличается от атомной массы, обычно встречающейся в естественных условиях. Примерами изотопов, которые можно включать в агент или его фармацевтически приемлемую соль, являются изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2Н, 3Н, 13С, 14С, 15N, 17O, 18O, 31P, 32P,35S, 18F и 36Cl соответственно. Определенные изотопные варианты агента и его фармацевтически приемлемой соли, например, варианты, в которые включен такой радиоактивный изотоп, как 3Н или 14Cl, можно применять в опытах по распределению лекарственного средства и/или субстрата в ткани. Изотоп трития, т.е. 3Н, и углерода-14, т.е. 14С, являются особенно предпочтительными из-за простоты их получения и обнаружения. Кроме того, замена на изотопы, такие как дейтерий, т.е. 2Н, может обеспечивать определенные терапевтические преимущества, приводящие к большей метаболической стабильности, например увеличению времени полужизни in vivo или снижению требований к дозе, что может быть предпочтительным при определенных обстоятельствах. Изотопные варианты агента, предлагаемого в настоящем изобретении, и его фармацевтически приемлемых солей по изобретению можно получать общепринятыми методами с использованием изотопных вариантов соответствующих реагентов.
Пролекарство
Специалисту в данной области должно быть очевидно, что агент, предлагаемый в настоящем изобретении, можно получать из пролекарства. Примеры пролекарств включают соединения, которые имеют определенную(ые) защитную(еы) группу(ы) и которые могут не обладать фармакологической активностью сами по себе, но после их введения в определенных условиях (например, перорально или парентерально) в результате метаболизма в организме они образуют агент, предлагаемый в настоящем изобретении, который обладает фармакологической активностью.
Профрагмент
Также должно быть очевидно, что некоторые фрагменты, известные как «профрагменты», например, описанные в "Design ofProdmgs" у Н.Bundgaad, Elsevier, 1985 (публикация включена в настоящее описание в качестве ссылки), можно помещать на соответствующие функциональные группы агентов. Такие пролекарства также подпадают под объем изобретения.
Производное
Понятие «производное» или «дериватизация» в контексте настоящего описания включает химическую модификацию агента. Примером таких химических модификаций может быть замена водорода галогеном, алкильной группой, ацильной группой или аминогруппой.
Химическая модификация
Согласно одному из вариантов осуществления настоящего изобретения агент может представлять собой химически модифицированный агент.
Химическая модификация агента, предлагаемого в настоящем изобретении, может либо повышать, либо понижать взаимодействие водородных связей, взаимодействие зарядов, гидрофобное взаимодействие, силы Ван-дер-Ваальса или дипольное взаимодействие между агентом и мишенью.
Согласно одному из объектов выявленный агент может являться моделью (например, матрицей) для создания других соединений.
Ниже настоящее изобретение проиллюстрировано на примерах, не ограничивающих его объем, со ссылкой на чертежи, на которых показано:
на фиг.1 - схематическая диаграмма;
на фиг.2 - схема;
на фиг.3 - структурное и графическое изображение;
на фиг.4 - схема;
на фиг.5 - схема;
на фиг.6 - схема;
на фиг.7 - схема;
на фиг.8 - схема;
на фиг.9 - схема;
на фиг.10 - график;
на фиг.11 - график;
на фиг.12 - график;
на фиг.13 - график;
на фиг.14 - график;
на фиг.15 - график;
на фиг.16 - график;
на фиг.17 - график;
на фиг.18 - график;
на фиг.19 - график;
на фиг.20 - график;
на фиг.21 - график;
на фиг.22 - график;
на фиг.23 - график;
на фиг.24 - график.
Примеры
Пример 1
ДОФЭ, ДСФЭ (дистеароилфосфатидилэтаноламин) и ДСФХ (дистеароилфосфатидилхолин) покупали непосредственно у фирмы Sigma-Aldrich, Пул, графство Дорсет, Великобритания; ДО(14-ин)ФЭ и ДО(14-ин)ФХ синтезировали согласно методу, приведенному на схеме 4, фиг.7 и фиг.2. Оценивали способность к трансфекции этих соединений в катионных липосомах, несущих хелперный липид: ДК-Хол в молярном соотношении 1:1. Данные этих исследований представлены графически на фиг.9.
Синтез моноацетиленовых жирных кислот
Синтезировали 5 моноацетиленовых жирных кислот (см. ниже): 3 жирные кислоты с 18 атомами углерода (С 18) (соединения 5, 6 и 7), которые отличаются положением тройной связи (4, 9 и 14 соответственно), и 2 жирные кислоты с 17 атомов углерода (С 17) (соединения 8 и 9, тройная связь в положениях 9 и 14 соответственно).
С использованием 5 жирных кислот затем получали моноацетиленовые аналоги каждого из приведенного ниже липидов:
Это позволило получить серию, состоящую в целом из 20 липидов
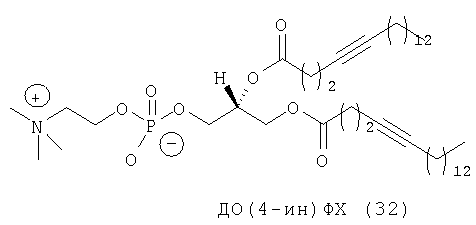
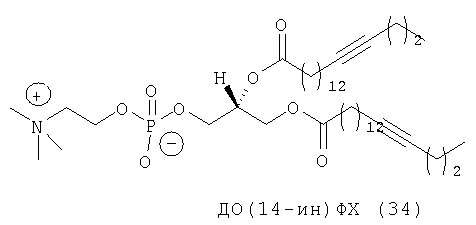
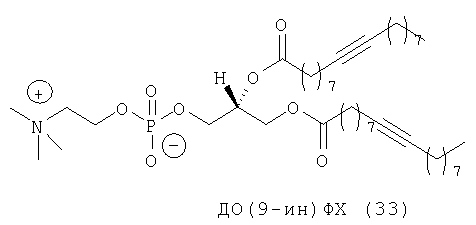
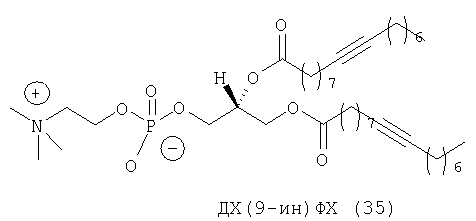
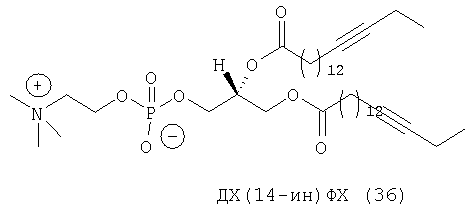
Пять моноацетиленовых аналогов ДОФХ
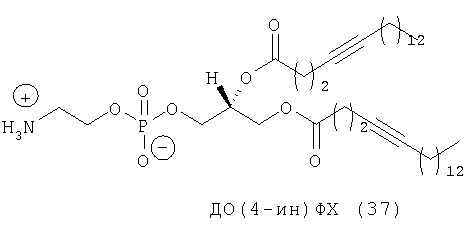
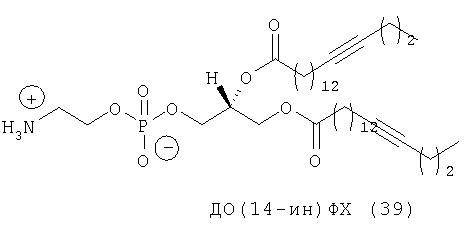
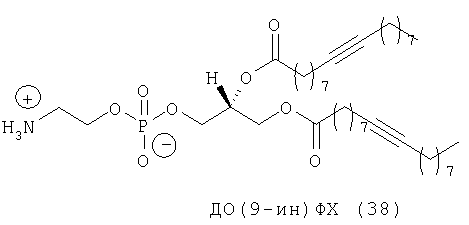
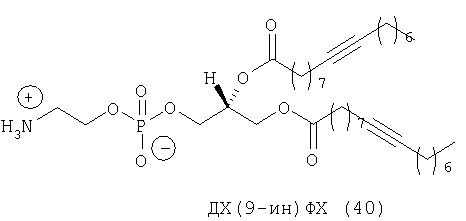
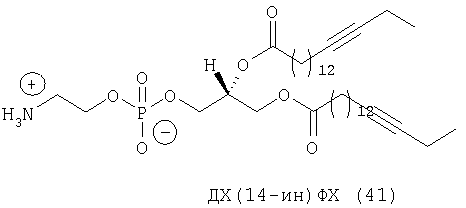
Пять моноацетиленовых аналогов ДОФЭ
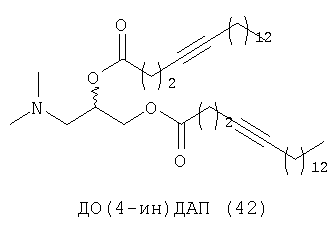
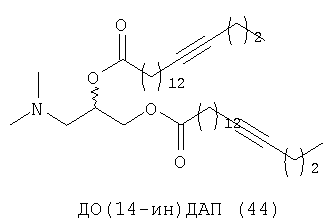

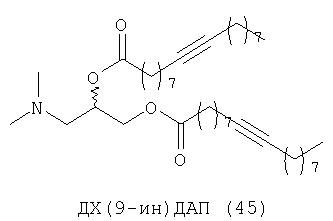
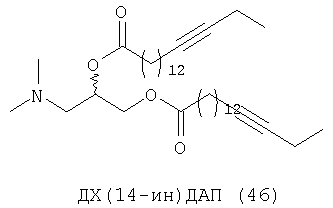
Пять моноацетиленовых аналогов ДОДАП
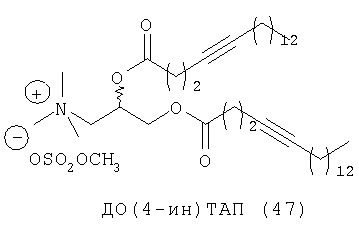
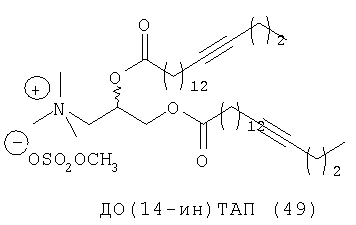
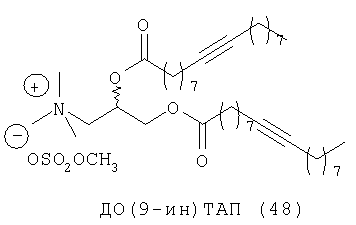
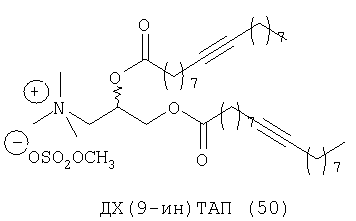
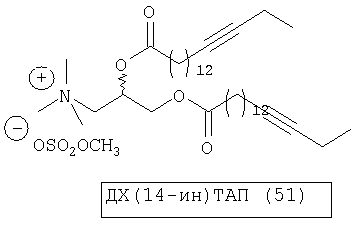
Пять моноацетиленовых аналогов ДОТАП
Синтез октадека-4-иноевой кислоты (5), октадека-9-иноевой кислоты (6) и гептадека-9-иноевой кислоты (8)
Октадека-4-иноевую кислоту (5) получали следующим образом (схема 1, фиг.4). Сначала получали ТНР-защищенное производное пен-4-ин-1-ола (10) путем реакции с DHP в условиях мягкого кислотного катализа с PPTS, выход которой составлял 91%. Тем временем 1-бромтридекан превращали в его более реакционноспособный йодированный аналог (11, выход 80%) путем хорошо известной реакции замещения галогена Финкельштейна.
Депротонирование терминального алкина 10 с помощью BuLi в присутствии НМРА (ГМПК) позволяло получать анион алкина, необходимый для SN2 - реакции на основе 1-йодтридекана. Эта стадия давала невысокий выход (49%). С помощью катализируемого кислотой гидролиза соединения 12 незащищенную первичную спиртовую группу, которую затем окисляли с помощью реагента Джонса (CrO3, конц. H2SO4) в ацетоне, получали октадека-4-иноевую кислоту (5) с выходом 69%.
Октадека-9-иноевую кислоту синтезировали аналогичным методом (схема 2, фиг.5). Сначала гидроксигруппу 8-бромоктан-1-ола защищали в виде ТНР-эфира с помощью DHP и PPTS, получая практически количественный выход (91%). Реакция сочетания 1-децина с более реакционноспособным соединением 15 (ср.14) позволяла получать продукт в виде внутреннего алкина (16) с хорошим выходом (72%). ТНР-гидролиз с помощью TsOH в МеОН приводил к получению алкинола 17, выход которого составлял 92%, после окисления которого с помощью реагента Джонса получали кислоту 6 в виде порошка белого цвета (76%).
Синтез гептадека-4-иноевой кислоты (8, схема 3, фиг.6) осуществляли практически аналогично получению октадека-9-иноевой кислоты за исключением того, что в качестве реагента для введения внутренней тройной связи С-С применяли 1-нонин (вместо 1-децина).
Синтез октадека-14-иноевой кислоты (7) и гептадека-14-иноевой кислоты (9)
Работая аналогично описанному в предыдущем разделе методу, в качестве исходного продукта для получения октадека-14-иноевой кислоты (7) применяли 12-бромдодекан-1-ол (схема 4, фиг.7). ТНР-защиту первичной спиртовой группы осуществляли количественно, получая соединение 22. И в этом случае для получения более реакционноспособного йодированного аналога (23) применяли реакцию Финкельштайна. Пент-1-ин подвергали взаимодействию с соединением 23 с использованием усовершенствованной авторами изобретения реакции депротонирования алкина - алкилирования, получая соединение 24 с хорошим выходом (59%). Функционально активную группу ТНР-эфира превращали непосредственно в бромид (25) с помощью PPh3Br3/PPh3 (выход 76%); полученный бромалкин (25) затем подвергали SN2 - реакции с CN-, получая гомологичную цепь жирного ацила. Сильный щелочной гидролиз нитрильной группы (26) позволял осуществлять введение карбоксильной группы с получением октадека-14-иноевой кислоты (7) с очень высоким выходом (94%).
Гептадека-14-иноевую кислоту (9) получали практически аналогично (схема 5, фиг.8). Единственным различием была необходимость введения функционально активной группы внутреннего алкина с помощью двух стадий (в противоположность одной стадии), что было связано с доступностью исходных продуктов, и превращения функционально активной группы ТНР-эфира в бромид с помощью двух стадий (кататализирумый кислотой гидролиз высвобождал первичную спиртовую группу, которую подвергали SN2 - реакции с CBr4 и PPh3 с получением требуемого бромида).
Синтез аналогов ДОФХ, ДОДАП и ДОТАП
Синтез аналогов ДОФХ (например, соединения 33) включал активацию жирных кислот (например, 6) в виде ацилимидазолидов, последующее взаимодействие sn-глицеро-3-фосфохолина (ГФХ) в присутствии ДБУ. Выход реакции составлял в среднем 60%. Синтез аналогов ДОФЭ (например, соединения 38) осуществляли путем двухфазного (хлороформ/вода) ферментативного трансфосфатидилирования аналогов ДОФХ с этаноламином с выходом примерно 90% (схема 6, фиг.9).
Аналогично этому синтезировали аналоги ДОДАП (например, соединение 43) с количественными выходами. Обработка аналогов ДОДАП диметилсульфатом позволяла получать аналоги ДОТАП (например, соединение 48) в виде метилсульфатов, выход которых составлял 70-80% (схема 7, фиг.10).
Данные о трансфекции
Введение
Моноацетиленовые аналоги ДОФХ, ДОФЭ, ДОДАП и ДОТАП синтезировали согласно описанному выше методу. Стандартные липиды, предназначенные для сравнительного анализа (ДОФХ, ДОФЭ и ДОТАП (хлорид)), получали от фирмы Sigma-Aldrich (Пул, графство Дорсет, Великобритания). Применяли катионный, включающий в качестве основы холестерин липид N1-холестериноксикарбонил-3,7-диазанононан-1,9-диамин (ХДАН) по изобретению, чистый заряд которого =+1,6 при рН 7,4. ХДАН (ниже) группой авторов настоящего изобретения был синтезирован ранее22, и в настоящее время в продажу поступает композиция на основе ХДАН и ДОФЭ в молярном соотношении 1:1 под названием Trojene™ (Avanti Polar Lipids Inc., Алабастер, шт.Алабама, США).
Чистоту липида оценивали с помощью ТСХ или ЖХВР. Все липиды хранили в виде маточных растворов в безводном CH2Cl2 в концентрациях либо 5, либо 10 мг/мл при -80°С в атмосфере Ar. Для получения катионных липосом липиды вносили с помощью шприца в круглодонную колбу в атмосфере Ar и при необходимости добавляли новую порцию дистиллированного CH2Cl2, получая концентрацию примерно 2,5 мг/мл. Затем добавляли 4 мМ HEPES, рН 7,0 (1 мл) и двухфазную систему перемешивали вращением. Суспензию липосом получали путем удаления органического растворителя при пониженном давлении при 25°С с последующей обработкой ультразвуком в течение 2-5 мин в источнике ультразвука в водяной бане. При необходимости добавляли дважды дистиллированную воду, получая прежний общий объем 1 мл. Все содержащие липосомы растворы получали с конечной концентрацией 5 мг/мл. Значение рН суспензии липосом определяли с помощью рН-метра Boy (фирма Camlab Ltd., Овер, Кэмбриджшир, Великобритания) и доводили до рН 7,0±0,1 с помощью концентрированных водных растворов HCl и NaOH.
Липосомы экструдировали (экструдер фирмы Northern Lipids Inc., Ванкувер, провинция Британская Колумбия, Канада), пропуская 10 раз через два поликарбонатных фильтра с размером пор 100 нм (IsoporeТМ Membrane Filteres, Millipore (UK) Ltd., Ватфорд, графство Хертфордшир, Великобритания). Для каждого препарата оценивали распределение размеров липосом с помощью фотонно-корреляционной спектроскопии (ФКС) (устройство Coulter® N4 Plus Submicron Particle Sizer, фирма Beckman Coulter, Хай-Уиком, графство Букингемшир, Великобритания). Для содержащих фосфолипиды липосом концентрацию фосфолипидов оценивали с помощью метода Стьюарта23. Во всех случаях конечная концентрация липида составляла 4,7±0,1 мг/мл. Для несодержащих фосфолипиды липосом потеря липидов при экструзии также была неизбежной. Для всех суспензий липосом, подвергающихся быстрым экструзиям, установлено, что концентрации общих липидов составляли 4,7 мг/мл. При более медленных экструзиях установлено, что концентрации общих липидов составляли 4,3 мг/мл. При очень медленных экструзиях липосомы экструдировали только три раза и концентрации общих липидов составляли 4,7 мг/мл.
Получение LMD (липосома: Mu:ДНК)
Плазмидную ДНК, содержащую ген β-галактозидазы (pNGVLl-nt-beta-gal; размером 7,53 т.п.н.) хранили в виде замороженных аликвот при -80°С в концентрации 1,2 мг/мл; синтезировали 1 микроединицу (стандартная единица Mu) пептида (коровый пептид аденовируса [H2N-Met-Arg-Arg-Ala-His-His-Arg-Arg-Arg-Arg-Ala-Ser-His-Arg-Arg-Met-Arg-Gly-Gly-CO2H] согласно описанному ранее методу и выдерживали при 4°С в виде аликвот 1 мг/мл. Комплексы Mu-ДНК получали, добавляя плазмидную ДНК к Mu в 4 мМ HEPES при соотношении 1:0,6 (мас/мас) при быстром перемешивании. LMD получали, создавая комплексы частиц Mu-ДНК с катионными липосомами (как описано выше) в соотношении 1:12 (мас/мас), так, чтобы все LMD, применяемые во всех композициях, были практически катионными. При каждом процессе получения распределение размеров LMD оценивали с помощью ФКС.
Трансфекции клеток
Клетки линии Рапс-1 (линия клеток рака поджелудочной железы человека) поддерживали в среде RPMI, дополненной 10% ФТС и 1% пенициллина-стрептомицина (Gibco™, Invitrogen Corporation, Пейсли, Шотландия, Великобритания) в увлажненной атмосфере, содержащей 5% СО2, при 37°С. За 24 ч до трансфекции вносили по 30000 клеток на лунку 48-луночных титрационных микропланшетов (тип Corning Costar, фирма Merk Ltd., Люттерворф, графство Лестершир, Великобритания) в 500 мкл среды. В качестве контроля в лунку добавляли плазмидную ДНК (0,5 мкг в 200 мкл среды). В качестве положительного контроля в лунки вносили 1,5 мкл Transfast™ (Promega Corporation, Мэдисон, шт.Висконсин, США; полученный согласно протоколу поставщика) в 200 мкл среды в виде комплекса с плазмидной ДНК (0,5 мкг) вносили в лунку. В каждую лунку добавляли по 5 мкл (0,5 мкг ДНК) содержащей LMD композиции. Все эксперименты проводили в 4 повторностях. Планшет вращали 30 с и инкубировали в течение 1 ч при 37°С. Затем среду удаляли и добавляли 500 мкл свежей среды. Клетки инкубировали при 37°С в течение еще 24 ч.
Клетки линии COS-7 поддерживали в среде DMEM, дополненной 10% ФТС и 1% пенициллина-стрептомицина (Gibco™) в увлажненной атмосфере, содержащей 5% СО2, при 37°С. За 24 ч до трансфекции вносили по 10000 клеток на лунку 48-луночных титрационных микропланшетов (тип Corning Costar) в 500 мкл среды. Применяемые в качестве контролей плазмидную ДНК и Transfast™ приготавливали согласно описанному ранее методу. В каждую лунку добавляли по 5 мкл (0,5 мкг ДНК) содержащей LMD композиции. Все эксперименты проводили в 4 повторностях. Планшет вращали 30 с и инкубировали в течение 1 ч при 37°С. Затем среду удаляли и добавляли 500 мкл свежей среды. Клетки инкубировали при 37°С в течение еще 24 ч.
Оценка β-галактозидазы и определение общего протеина
Среду отбирали из лунок с помощью аспирации и клеточный слой промывали забуференным фосфатом физиологическим раствором (ЗФР) (Gibco™). Клетки из каждой лунки лизировали в 150 мкл реагента для лизиса (приготовленного согласно протоколу поставщика; набор для анализа репортерного гена β-галактозидазы (β-Gal Reporter Gene Assay) фирмы Roche Diagnostics GmbH, D-68305, Маннгейм, Германия) при комнатной температуре в течение 30 мин. После лизиса одинаковые количества 50 и 20 мкл каждой клеточной суспензии использовали для определения активности β-галактозидазы и для определения общего клеточного протеина (для стандартизации результатов) соответственно.
При анализе β-галактозидазы 100 мкл субстрата добавляли к 50 мкл клеточной суспензии в 96-луночном микропланшете (тип Corning Costar). Планшет инкубировали 30 мин при комнатой температуре. Автоматическую инициацию (повышение ферментативной активности, которая образуется после добавления субстрата) осуществляли с помощью люминометра для микропланшетов (Anthos Lucy 1, фирма Labtech International Ltd., Рингмер, графство Восточный Суссекс, Великобритания), при этом инъецировали 50 мкл реагента для инициации и ферментативную активность оценивали в течение последующих 30 с.
Количество клеточного протеина оценивали с помощью ВСА-анализа (фирма Pierce, Рокфорд, штат Иллинойс, США) с использованием 20 мкл клеточного лизиса или бычьего сывороточного альбумина в качестве внутреннего стандарта для калибровки, добавляя 200 мкл реагента ВСА (согласно протоколу поставщика). После инкубации в течение 30 мин при комнатной темперауре осуществляли колориметрический анализ при 570 нм с помощью планшет-ридера (Anthos Lucy 1). Активность β-галактозидазы выражали в виде RLU/мкг протеина. На приведенных ниже чертежах на оси абсцисс представлены данные, полученные для контрольных и хелперных липидов или катионных липидов (указанных в заголовках чертежей).
Аналоги ДОФХ
Использование в композициях катионных липосом на основе ДОФХ, как правило, устраняет или в значительной степени ослабляет трансфекцию по сравнению с использованием ДОФЭ24. Результаты трансфекции (клетки линии Panc-1) с использованием LMD, в которых липосомы включали ХДАН: хелперный липид, 1:1 (моль/моль), представлены ниже (фиг.12). Во всех случаях уровни трансфекции оказались очень низкими. Замена ДОФХ на ДО(4-ин)ФХ привела к почти полной потере способности к трансфекции. Однако при использовании таких аналогов, как ДО(9-ин)ФХ и ДО(14-ин)ФХ, уровень трансфекции оказался таким же и даже более высоким, чем при использовании ДОФХ.
Результаты трансфекции клеток линии COS-7 с использованием таких же LMD представлены на фиг.13. ДОФХ и контроль Transfast одинаково хорошо осуществляли трансфекцию. И в этом случае замена ДОФХ на ДО(4-ин)ФХ приводила к почти полной потере способности к трансфекции. При использовании ДО(14-ин)ФХ уровень трансфекции оказался примерно в 2 раза более высоким, чем при использовании ДОФХ. Для обеих линий клеток (Panc-1 и COS-7) установлено, что при применении ДО(9-ин)ФХ уровень трансфекции оказался таким же и даже более высоким, чем при использовании ДОФХ.
Аналоги ДОФЭ
На фиг.14 представлены данные о трансфекции (клетки линии COS-7) при использовании LMD, в которых липосомы представляли собой ХДАН: хелперный липид, 1:1. Когда хелперный липид представлял собой либо ДОФЭ, либо ДО(9-ин)ФЭ, уровни трансфекции были высокими и примерно одинаковыми; замена либо на ДО(4-ин)ФЭ, либо на ДО(14-ин)ФЭ приводила к снижению уровней трансфекции примерно на 67%.
Ниже представлены результаты, полученные на клетках линии Panc-1 (фиг.15), с использованием LMD, в которых липосомы представляли собой ХДАН: хелперный липид, взятые в соотношении 3:2. Уровень трансфекции с использованием ДОФЭ оказался примерно в 2 раза выше, чем при использовании ближайшего ацетиленового аналога (ДО(9-ин)ФЭ), хотя абсолютные уровни трансфекции оказались весьма низкими. Трансфекция при применении как ДО(4-ин)ФЭ, так и ДО(14-ин)ФЭ оказалась очень низкой. Аналогичные данные, полученные при применении ацетиленовых аналогов, представлены на фиг.14.
Результаты, полученные на клетках линии Panc-1, с использованием LMD, в которых липосомы представляли собой ХДАН:хелперный липид, взятые в соотношении 1:1, представлены на фиг.16. С учетом «усов» (отрезков прямой, характеризующих значение погрешности результатов) при использовании ДОФЭ, ДО(4-ин)ФЭ и ДО(9-ин)ФЭ уровень трансфекции был высоким и примерно одинаковым. Уровень трансфекции при применении ДО(14-ин)ФЭ составлял примерно половину от уровня при применении ДОФЭ.
Ниже (фиг.17) представлены результаты, полученные с учетом дальнейшего развития тенденции снижения соотношения ХДАН:хелперный липид, что приводило к снижению зарядов липосом и LMD, с использованием LMD, в липосомах которых соотношение ХДАН: хелперный липид составляло 2:3 (клетки линии Panc-1). В этом случае уровень трансфекции при использовании ДОФЭ оказался ниже, чем у контроля Transfast. Интересно отметить, что при использовании всех моноацетиленовых аналогов уровень трансфекции оказался выше, чем при использовании ДОФЭ. Уровень трансфекции при применении ДО(4-ин)ФЭ и ДО(9-ин)ФЭ превышал до 5 раз уровень трансфекции при использовании ДОФЭ. Из изученных моноацетиленовых аналогов наименее высокий уровень трансфекции обнаружен при применении ДО(14-ин)ФЭ.
На фиг.18 представлены результаты повторения предыдущего эксперимента (вновь проведенного на клетках линии Panc-1). Для этого опыта применяли те же липосомы, но LMD готовили вновь. При этом была получена аналогичная картина за исключением того, что уровень трансфекции при использовании ДО(4-ин)ФЭ понизился примерно до уровня, полученного при использовании Transfast. Кроме того, относительное повышение трансфекции при замене ДОФЭ на ДО(9-ин)ФЭ достигло примерно 6-кратного уровня. Абсолютные значения величин, получаемых от эксперимента к эксперименту, трудно сравнивать из-за таких факторов, как стадия клеточного цикла. Для этой цели в эксперименты был включен «контроль», представляющий собой LMD, в которых липосомы представляли собой ХДАН:ДОФЭ, 1:1. При применении этих LMD уровень трансфекции был таким же, что и при использовании LMD, состоящих из липосом, которые представляли собой ХДАН:ДО(9-ин)ФЭ, 2:3.
Аналоги ДОТАП
На фиг.19 представлены результаты трансфекции клеток линии Panc-1 с использованием LMD, в которых липосомы полностью состояли из ДОТАП или аналогов ДОТАП. При использовании всех аналогов (включая исходный ДОТАП) уровень трансфекции оказался ниже, чем при применении положительного контроля Transfast, за исключением ДО(14-ин)ТАП. Все аналоги осуществляли трансфекцию лучше чем ДОТАП; при использовании ДО(14-ин)ТАП уровень трансфекции в шесть раз превышал уровень трансфекции, получаемый с помощью стандартного ДОТАП.
Данные о трансфекции клеток линии COS-7 с помощью LMD, в которых липосомы состояли из ДОТАП, представлены ниже (фиг.20). С учетом «усов» уровень трансфекции Transfast и ДОТАП оказался одинаковым. Замена ДОТАП на ДО(14-ин)ТАП приводила к снижению уровня трансфекции приблизительно на 40%; замена на ДО(14-ин)ТАП приводила к практически полному снижению способности к трансфекции. При применении ДО(9-ин)ТАР уровень трансфекции оказался примерно в 1,5 раз выше, чем при использовании стандартного ДОТАП. Видно, что изменение активности моноацетиленовых аналогов при переходе от ДО(4-ин)ТАП к ДО(9-ин)ТАП и к ДО(14-ин)ТАП напоминает схему, обнаруженную для аналогов ДОФЭ.
Ниже (на фиг.21) представлены данные о трансфекции с использованием LMD, в которых липосомы представляли собой ДОФЭ: катионный липид, 1:1 (клетки линии Panc-1). Все уровни трансфекции оказались ниже, чем при использовании Transfast, но выше, чем при использовании LMD только на основе ДОТАП. Все уровни трансфекции были очень низкими. Замена ДОТАП на ДО(4-ин)ТАП или ДО(9-ин)ТАП приводила к небольшому повышению трансфекции, однако при замене ДОТАП на ДО(14-ин)ТАП в липосомах, состоящих из ДОФЭ:ДОТАП, уровень трансфекции повышался в 7 раз.
Замена ДОФЭ на холестерин привела к получению представленных ниже результатов (фиг.22). И в этом случае были обнаружены низкие уровни трансфекции (клетки линии Panc-1). Замена ДОТАП на ДО(4-ин)ТАП приводила практическому к полному прекращению трансфекции, а при замене на ДО(14-ин)ТАП способность к трансфекции снижалась примерно на 30%. Однако при использовании ДО(9-ин)ТАП уровень трансфекции соответствовал уровню трансфекции при использовании ДОТАП.
Заключительные эксперименты по оценке трансфекции с использованием LMD, липосомы которых состояли из комплексов либо ДОФЭ:катионный липид, 1:1 (фиг.23), либо холестерин:катионный липид, 1:1 (фиг.24), проведены на клетках линии COS-7. При использовании липосом, состоящих только из чистого ДОТАП (100% ДОТАП), уровень трансфекции оказался выше, чем при использовании ДОТАП: Хол, которые оказались более эффективными в отношении трансфекции, чем ДОТАП:ДОФЭ. При использовании всех аналогов ДОТАП уровень трансфекции оказался более низким или примерно таким же, что и при применении композиций на основе ДОТАП:ДОФЭ, 1:1.
Способность к трансфекции липосом, состоящих из комплексов ДО(4-ин)ТАП: холестерин, 1:1 и ДО(14-ин)ТАП: холестернин, 1:1, оказалась такой же, что и у липосом, состоящих из чистого ДОТАП. Способность к трансфекции клеток линии COS-7 липосомами, состоящими из смеси ДОТАП: холестерин, 1:1 и ДО(9-ин)ТАП: холестерин, 1:1, составляла лишь половину от активности липосом, включающих ДО(4-ин)ТАП: холестерин, 1:1.
Заключение и выводы
Способность ДО(9-ен)ФХ и ДО(14-ен)ФХ осуществлять трансфекцию клеток линии Panc-1 такая же, как у ДОФХ при включении в липосомы в виде комплекса ХДАН: хелперный липид, 1,1 (молярное соотношение). Способность ДО(9-ен)ФХ трансфектировать клетки линии COS-7 в 2 раза превышает способность ДОФХ трансфектировать клетки.
ДОФЭ и ДО(9-ен)ФЭ очень эффективно трансфектируют клетки линии COS-7, и при их применении уровень трансфекции аналогичен уровню, полученному при использовании липосом, включающих ХДАН: хелперный липид, 1:1. Уровень трансфекции при использовании ДО(4-ен)ФЭ и ДО(14-ен)ФЭ составляет примерно 1/3 относительно ДОФЭ.
При включении в липосомы, состоящие из комплекса ХДАН: хелперный липид, взятые в молярном соотношении 3:2, при использовании ДОФЭ уровень трансфекции более высокий, чем при применении всех моноацетиленовых аналогов (клетки линии Panc-1). У наиболее эффективного аналога, т.е. ДО(9-ен)ФЭ, способность к трансфекции составляла половину от способности к трансфекции ДОФЭ. Однако при этом все уровни трансфекции являются низкими.
С учетом «усов» способность к трансфекции клеток линии Panc-1 у ДОФЭ, ДО(4-ин)ФЭ и ДО(9-ин)ФЭ является одинаково высокой при включении в липосомы ХДАН: хелперный липид, 1:1.
При оценке дальнейшего снижения положительных зарядов LMD установлено, что липосомы, состоящие из комплекса ХДАН: хелперный липид, 2:3, позволяли осуществлять трансфекцию также эффективно, как и соответствующие липосомы, в которых соотношение ингредиентов составляо 1,1, кроме варианта, когда хелперный липид представлял собой ДОФЭ. ДО(4-ин)ФЭ и ДО(9-ин)ФЭ осуществляли трансфекцию до 5 раз более эффективно, чем ДОФЭ.
Липосомы, включающие ХДАН:ДО(9-ин)ФЭ, осуществляли трансфекцию также эффективно, как липосомы, включающие ХДАН:ДОФЭ, 1:1.
Липосомы на основе ДОТАП и ДОТАП:ДОФЭ, 1:1, осуществляли трансфекцию хуже, чем липосомы ХДАН:ДОФЭ, 1:1. При использовании ДОТАП уровни трансфекции, как правило, были низкими.
Липосомы на основе чистого аналога ДОТАП осуществляли трансфекцию лучше, чем липосомы на основе чистого ДОТАП. Липосомы на основе ДО(14-ин)ТАП осуществляли трансфекцию в 6 раз лучше, чем на основе ДОТАП (клетки линии Panc-1).
Трансфекцию клеток линии COS-7 только липосомы на основе чистого ДО(9-ин)ТАП осуществляли также эффективно, как и липосомы на основе ДОТАП. У ДО(14-ин)ТАП практически отсутствовала способность осуществлять трансфекцию.
Липосомы на основе чистых ДОТАП или аналога ДОТАП и соответствующие липосомы на основе ДОТАП:ДОФЭ, 1:1 или ДОТАП: аналог ДОФЭ, 1:1, обладали одинаковой способностью к трансфекции. Включение в композицию ДОФЭ, вероятно, снижает трансфекцию. Способность к трансфекции липосом на основе ДО(14-ин)ТАП:ДОФЭ, 1:1 примерно в 7 раз выше, чем у липосом ДОТАП:ДОФЭ, 1:1 (клетки линии Panc-1).
Липосомы на основе ДОТАП: холестерин, 1:1, ДО(9-ин)ТАП осуществляют трансфекцию так же, как и ДОТАП (клетки линии Panc-1).
Способность к трансфекции липосом ДОТАП:ДОФЭ или ДОТАП: аналог ДОФЭ, 1:1 примерно в половину ниже, чем у липосом на основе чистого ДОТАП при изучении на клетках линии COS-7. У всех липосом ДОТАП:ДОФЭ, 1:1 способность к трансфекции одинаковая.
У липосом ДОТАП:холестерин и ДО(9-ин)ТАП:холестерин, 1:1 способность осуществлять трансфекцию составляет половину по сравнению с липосомами на основе чистого ДОТАП (клетки линии COS-7). У липосом ДО(4-ин)ТАП:холестерин и ДО(14-ин)ТАП:холестерин, 1:1 способность осуществлять трансфекцию такая же, как у липосом на основе чистого ДОТАП.
При создании изобретения синтезировали и оценивали способность осуществлять трансфекцию in vitro серий моноацетиленовых аналогов известных олеоильных липидов. При биологической оценке способности этих липидов осуществлять трансфекцию был обнаружен по меньшей мере один моноацетиленовый аналог жирного ацила в каждой серии липидов (кроме серии, включающей аналоги ДОДАП), который обладал такой же, если не более высокой способностью осуществлять трансфекцию, что и соответствующий стандартный олеоильный липид. Исходя из предпосылки, что тройная связь является более стабильной к окислению, чем двойная связь, эти результаты позволяют предположить, что те аналоги, у которых способность к трансфекции такая же, как у стандартов, могут являться их приемлемыми заменителями in vitro. Кроме того, тройная связь придает большую жесткость липосомам, удлиняя тем самым время полужизни этих липосом в кровотоке in vivo. Ясно, что оба эти аспекта являются важными.
ДО(9-ин)ФХ (33) и ДО(9-ин)ФЭ (38) представляют собой наиболее эффективные фосфолипидные аналоги, что позволяет высказать предположение о том, что непосредственная замена двойной связи на тройную связь не ухудшает способность к трансфекции in vitro. Кроме того, эти липиды не только обладают выраженной способностью к трансфекции, но было установлено также, что получение липосом, включающих аналог ДОФЭ:ХДАН, проще, чем соответствующих липосом ДОФЭ:ХДАН. Это может быть результатом противоположных воздействий имеющей существенное значение петли в цепях кислого ацила ДОФЭ (поддерживание текучести) и сдвига, характерного для ХДАН на основе холестерина (поддерживание жесткости); при замене двойной связи ДОФЭ на тройную связь эта степень закрученности существенно снижается. Это может определять меньшую устойчивость к включению ХДАН, что облегчает приготовление липосом. Кроме того, это может облегчать включение более высоких количеств аналогов ДОФЭ относительно ХДАН.
Более важным является выявление того, что возрастающий уровень нейтрального аналога ДОФЭ относительно ХДАН (что снижает положительные заряды катионных липосом в LMD) в отличие от стандартного ДОФЭ не снижает способность осуществлять трансфекцию. Такие LMD, несущие меньшее количество положительных зарядов, обладают третьей степенью стабильности. Анионные элементы крови могут в результате действия электростатических сил привлекаться к LMD, потенциально способствуя их разрушению или в более простом случае вытесняя комплекс Mu-ДНК. LMD с более низким зарядом должны иметь более длительное время полужизни в кровотоке, что дает частицам шанс достать их ткани/клетки-мишени.
Данные о трансфекции ДОТАП: ДО(14-ин)ТАП обладает более высокой способностью трансфектировать клетки линии Panc-1, чем ДОТАП; ДО(9-ин)ТАП обладает более высокой способностью трансфектировать клетки линии COS-7. Для других хелперных липидов были получены другие результаты.
Все упомянутые в описании публикации включены в него в качестве ссылки. Как должно быть очевидно специалистам в данной области, могут быть сделаны различные модификации и вариации описанных методов и системы, предлагаемых в изобретении, без отклонения от сущности и объема изобретения. Хотя изобретение описано со ссылкой на конкретные предпочтительные варианты осуществления изобретения, очевидно, что заявленный объем изобретения в целом не ограничен этими конкретными вариантами его осуществления. Так, под объем приведенной ниже формулы изобретения подпадают различные модификации осуществления изобретения, которые очевидны специалистам в области химии или родственных областях.
Список литературы
1a. W.F.Anderson, Science, 256, 1992, с.808.
2а. F.D.Ledley, Current Opinion in Biotechnology, 5, 1994, с.626; K.F. Kozarsky и др., ibid, 3, 1993, с.499; Gordon и др., ibid, 5, 1994, с.611.
3а. С.Р.Hodgon, Bio Tech, 13, 1995, с.222.
4а. Р.L.Felgner и др, Proc Nati Acad Sci USA, 84, 1987, с.7413; Felgner и др., Nature, 337, 1989, с.387; H-J. Burger и др, Proc Nati Acad Sci USA, 89, 1992, с.2145.
5a. Malone и др., Proc Nati Acad Sci USA, 86, 1989, с.6077.
6а. M-Y.Chiang и др., J Biol Chem, 226, 1991, с.18162.
7a. R.J.Debs и др., J Biol Chem, 265, 1990, с.10189; С.Walker и др., Proc Nati Acad Sci USA, 89, 1992, с.7915.
8a. A.D.Bangham, Hospital Practice, 27, 1992, с.51.
9a. J-P.Behr и др., Proc Nati Acad Sci USA, 86, 1989, с.6982; R.Leventis и др., Biochim Biophys Acta, 1023, 1990, с.124.
10а. X.Gao и др., Gene Therapy, 2, 1995, с.710.
11a. R.Stribling и др., Proc Nati Acad Sci USA, 89, 1992, с.11277.
12a. E.W. F.W.Alton и др. Nature Genetics, 5, 1993, с.135.
1. M.Ogris, S.Brunner, S.Schuller, R.Kircheis, E.Wagner, Gene Therapy 6, 1999, с.595.
2. A.L.Bailey, S.M.Sullivan, Biochimica Et Biophysica Acta-Biomembranes 1468, 2000, с.239.
3. J.L.Coll, P.Chollet, E.Brambilla, D.Desplanques, J.P.Behr, M.Favrot, Hum Gene Ther 10,1999, с.1659.
4. P.Erbacher, T.Bettinger, P.Belguise-Valladier, S.Zou, J.L. Coll, J.P.Behr, J.S.Remy, J Gene Medl, 1999, с.210.
5. S.M.Zou, P.Erbacher, J.S.Remy, J.P.Behr, J Gene Med 2, 2000, с.128.
6. A.Bragonzi, G.Dina, A.Villa, G.Calori, A.Biffi, C.Bordignon, B.M.Assael, M.Conese, Gene Therapy 7, 2000, с.1753.
7. S.Li, M.A.Rizzo, S.Bhattacharya, L.Huang, Gene Therapy 5,1998, с.930.
8. M.I.Papisov, Advanced Drug Delivery Reviews 32,1998, с.119.
9. D.V.Devine, A.J.Bradley, Advanced Drug Delivery Reviews 32, 1998, с.19.
10. С.Kitson, B.Angel, D.Judd, S.Rothery, N.J.Severs, A.Dewar, L.Huang, S.C.Wadsworth, S.H.Cheng, D.M.Geddes, E.Alton, Gene Therapy 6, 1999, с.534.
11. S.Li, W.C.Tseng, D.B.Stolz, S.P.Wu, S.C.Watkins, L.Huang, Gene Therapy 6, 1999, с.585.
12. N.Duzgunes, S.Nir, Advanced Drug Delivery Reviews 40, 1999, с.3.
13. D.Needham, D.H.Kirn, Colloids and Surfaces B-Biointerfaces 18, 2000, с.183.
14. I.M.Hafez, P.R.Cullis, Adv Drug Deliv Rev 47, 2000, с.139.
15. D.Kirpotin, К.L.Hong, N. Mullah, D. Papahadjopoulos, S.Zaiipsky, Febs Letters 388, 1996, с.115.
16. M.N.Jones, Advanced Drug Delivery Reviews 13, 1994, с.215.
17. S.Medda, S.Mukherjee, N.Das, К.Naskar, S.B.Mahato, M.K.Basu, Biotechnology and Applied Biochemistry 17, 1993, с.37.
(1б) Mountain A. Trends Biotechnol. 18, 2000, сс.119-128.
(2б) Hafez I.M. и Cullis P.R., Adv. Drug Deliv. Rev.47, 2001, cc.139-148.
(2в) Bailey A.L. и Cullis P.R., Biochemistry 33,1994, сс.12573-12580.
(3б) Barve J.A., Gunstone F.D. Chem. Phys. Lipids, 7, 1971, сс.311-323.
(4б) Rürup J., Mannova M., Brezesinski G. Schmid R.D., Chem. Phys. Lipids 1994, 70, 187-198.
(56) Pisch S., Bornscheuer U.T., Meyer H.H., Schmid R.D. Tetrahedron 53, 1997, с.14627-14634.
(6б) Sharma A., Sharma U.S., Int. J. Pharm. 154,1997, сс.123-140.
(7б) Murray K.D., Etheridge C.J., Shah S.I., Matthews D.A., Russell W., Gurling H.M.D., Miller A. D. Gene Ther. 8, 2001, сс.453-460.
(8б) Berry D.E., Chan J.A., MacKenzie L., Hecht S.M., Chem. Res. Toxicol. 4, 1991, сс.195-198.
(9б) Schilstra M.J., Nieuwenhuizen W.F., Veldink G.A., Vliegenthart J.F.G., Biochemistry 35,1996, с.3396-3401.
(10б) Aitken A.E., Roman L.J., Loughran P.A., de la Garza M., Masters B.S.S., Arch. Biochem. Biophys. 393, 2001, сс.329-338.
(11б) Fatope M.O., Adoum O.A., Takeda Y., Journ. Agricul. Food Chem. 48, 2000, сс.1872-1874.
(12б) Lenart J., Pikula S., Acta Biochim. Polon. 46, 1999, сс.203-210.
(13б) Kraus C.M., Neszmelyi A., Holly S., Wiedemann В., Nenninger A., Torssell K.
B.G., Bohlin L., Wagner H., Journ. Nat. Prod. 61, 1998, сс.422-427.
(14б) Datta K., Kulkarni A.P., Toxicol. Lett. 73,1994, сс.157-165.
(156) Roy В., Arruda A., Mallik S., Campiglia A., 222nd Amer. Chem. Soc. Nat. Meeting 26th-30th August 2001.
(16б) Hennies P.Т., Santana M.H.C., Correia C.R.D, Journ. Brazilian Chem. Soc.12, 2001,сс.64-72.
(17б) Okada J., Cohen S., Langer R.S., PCT Int. Appl. (1995), WO 9503035.
(18б) Hayward J.A., Johnston D.S., Chapman D., Annals New YorkAcad. Sciences 446, 1985, сс.267-281.
(19б) Patel M., Vivien E., Hauchecorne M., Oudrhiri N., Ramasawmy R., Vigneron J.-P., Lehn P., Lehn J.-M., Biochem. and Biophys. Res. Commun. 281, 2001, сс.536-543.
(20б) Fieser L.F., Fieser M., Advanced Organic Chemistry 1961, cc.227.
(21б) Fieser L.F., Fieser M., Advanced Organic Chemistry 1961, cc.236-237.
(21в) Clayden J., Greeves N., Warren S. и Wothers P. Organic Chemistry 2001, cc.326-331.
(21г) Lewis R.N.A.H., Mannock D.A., McElhaney R.N., Biochemistry 28, 1989, cc. 541-548.
(22б) Cooper R.G., Etheridge C.J., Stewart L., Marshall J., Rudginsky S., Cheng S.H., Miller A.D., Chem. Eur. J. 4, 1998, cc.137-151.
(23б) Stewart J.C.M., Anal. Biochem. 104, 1959, cc.10.
(24б) Koltover I., Salditt T., Radler J.O., Safinya C.R., Science 281, 1998, cc.78-81.
| название | год | авторы | номер документа |
|---|---|---|---|
| НЕВИРУСНЫЙ ВЕКТОР ДЛЯ ВВЕДЕНИЯ НУКЛЕИНОВОЙ КИСЛОТЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, КОНДЕНСИРОВАННЫЙ КОМПЛЕКС ПОЛИПЕПТИД/НУКЛЕИНОВАЯ КИСЛОТА И СПОСОБ ВСТРАИВАНИЯ ПРЕДСТАВЛЯЮЩЕЙ ИНТЕРЕС НУКЛЕИНОВОЙ КИСЛОТЫ В ЭУКАРИОТИЧЕСКУЮ КЛЕТКУ | 2000 |
|
RU2267536C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, КОМПОЗИЦИЯ ДЛЯ ПРИГОТОВЛЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2001 |
|
RU2290409C2 |
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ, НЕЙТРАЛЬНЫЙ ЛИПОПОЛИМЕР И СПОСОБ УВЕЛИЧЕНИЯ ВРЕМЕНИ ЦИРКУЛЯЦИИ ЛИПОСОМЫ | 2000 |
|
RU2250911C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РИНИТА И РОДСТВЕННЫХ ЗАБОЛЕВАНИЙ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2006 |
|
RU2444351C2 |
| ФОТОХИМИЧЕСКАЯ ИНТЕРНАЛИЗАЦИЯ ДЛЯ ДОСТАВКИ МОЛЕКУЛ В ЦИТОЗОЛЬ | 2001 |
|
RU2333246C2 |
| СТАБИЛИЗИРОВАННЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2732567C2 |
| СТАБИЛИЗИРУЮЩИЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2833053C2 |
| ПЕГИЛИРОВАННЫЕ ЛИПОСОМЫ ДЛЯ ДОСТАВКИ КОДИРУЮЩЕЙ ИММУНОГЕН РНК | 2012 |
|
RU2628705C2 |
| ГЕТЕРОВЕЗИКУЛЯРНЫЕ ЛИПОСОМЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2120795C1 |
| ВЫСОКОЭФФЕКТИВНАЯ ИНКАПСУЛЯЦИЯ ГИДРОФИЛЬНЫХ СОЕДИНЕНИЙ В УНИЛАМЕЛЛЯРНЫЕ ЛИПОСОМЫ | 2018 |
|
RU2788456C2 |
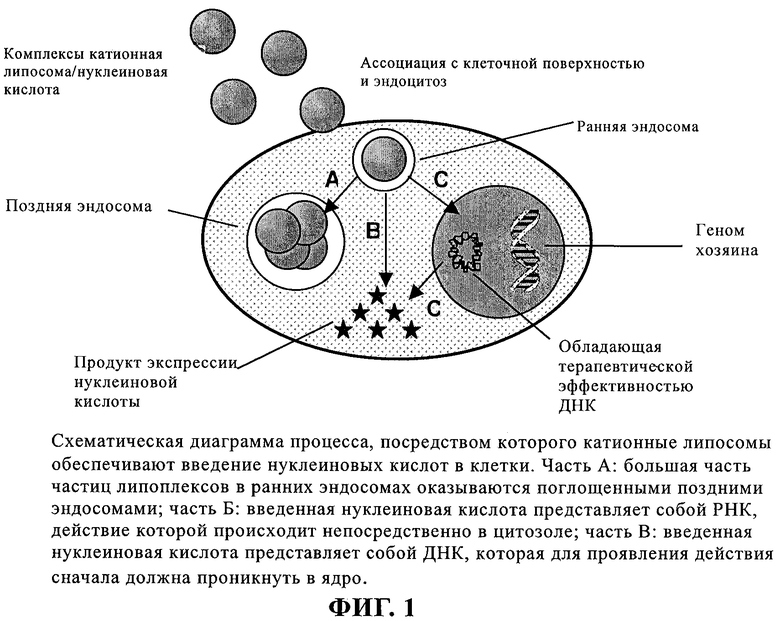
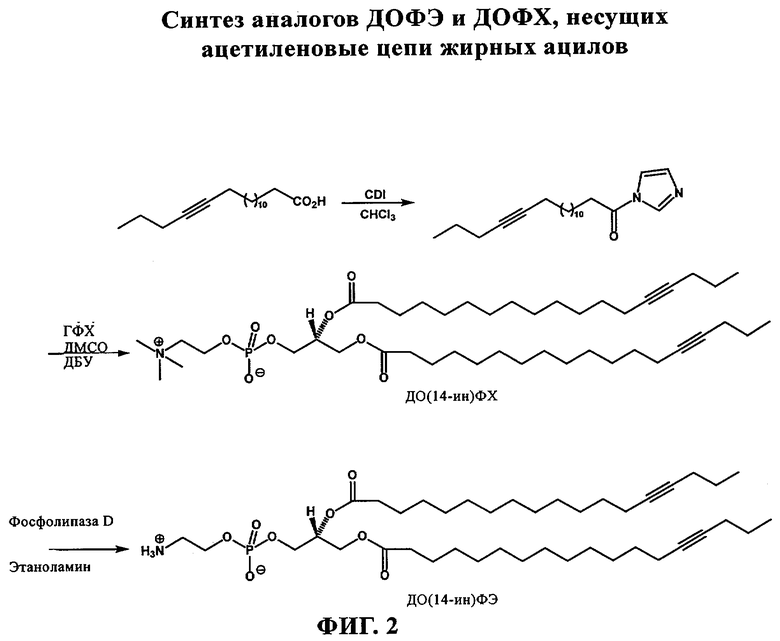
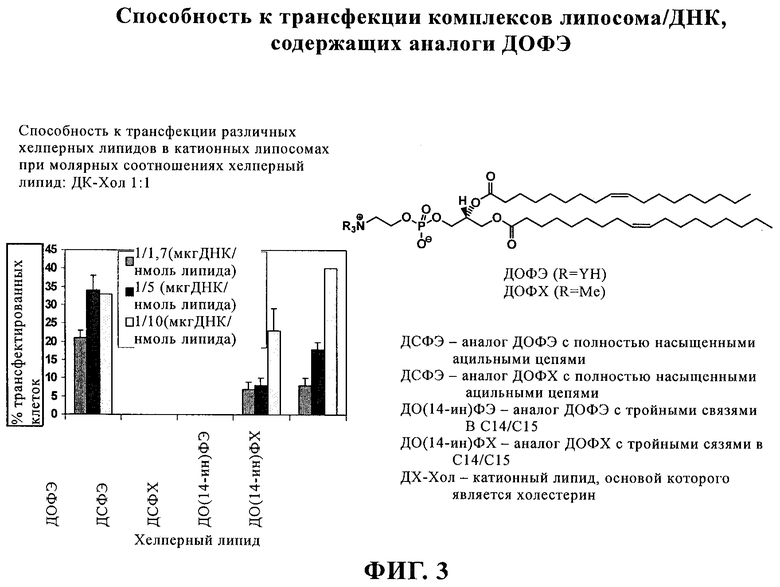
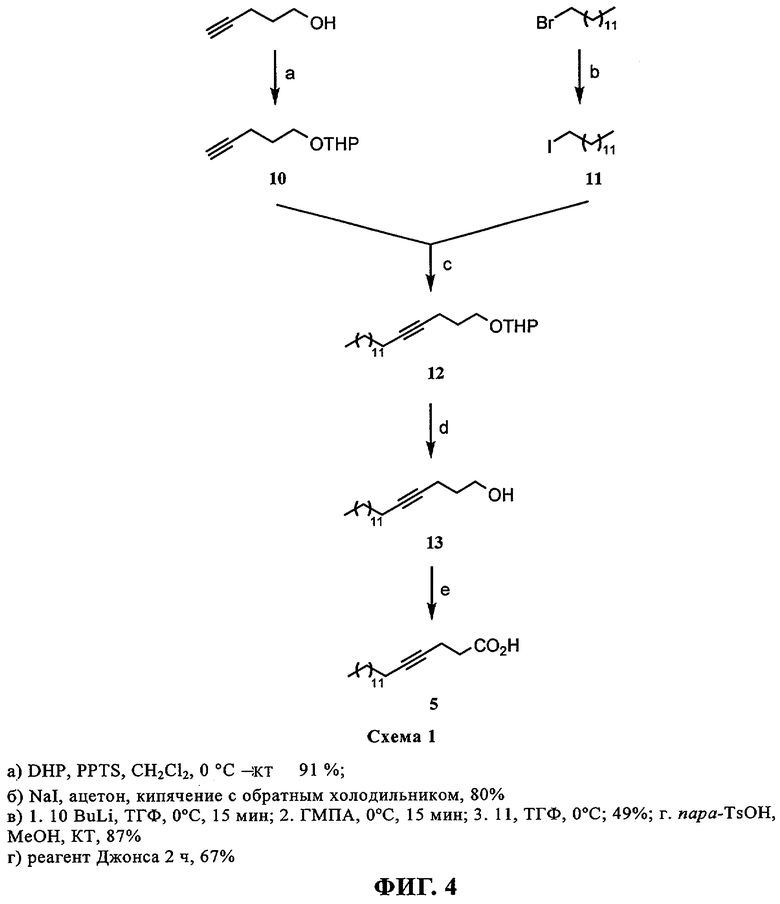
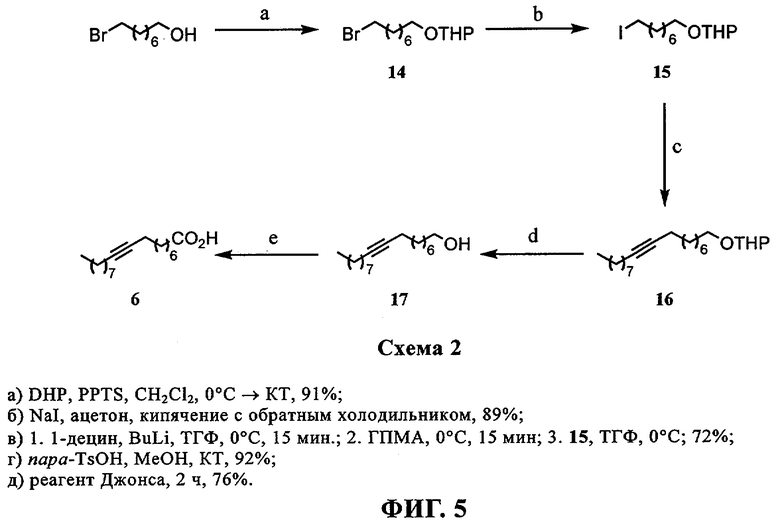
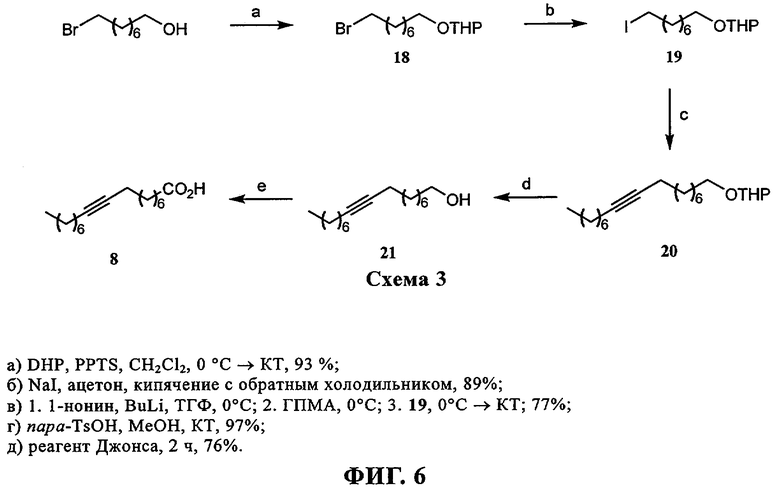
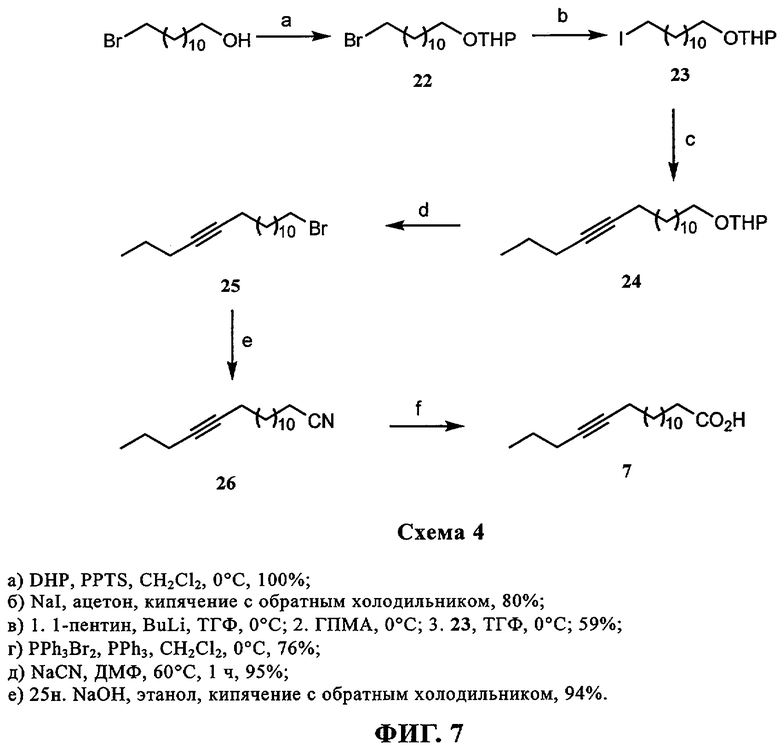
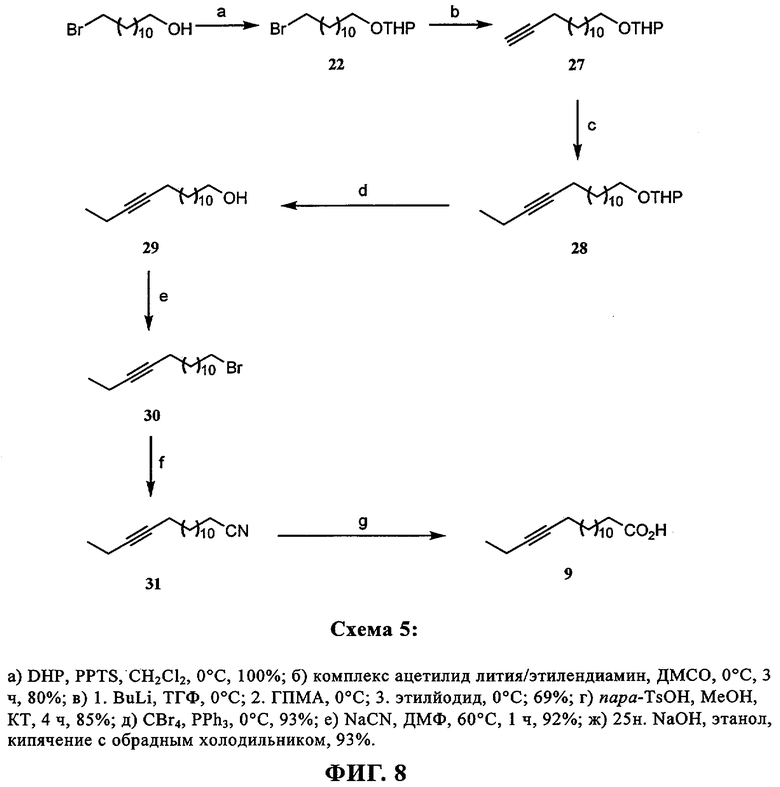
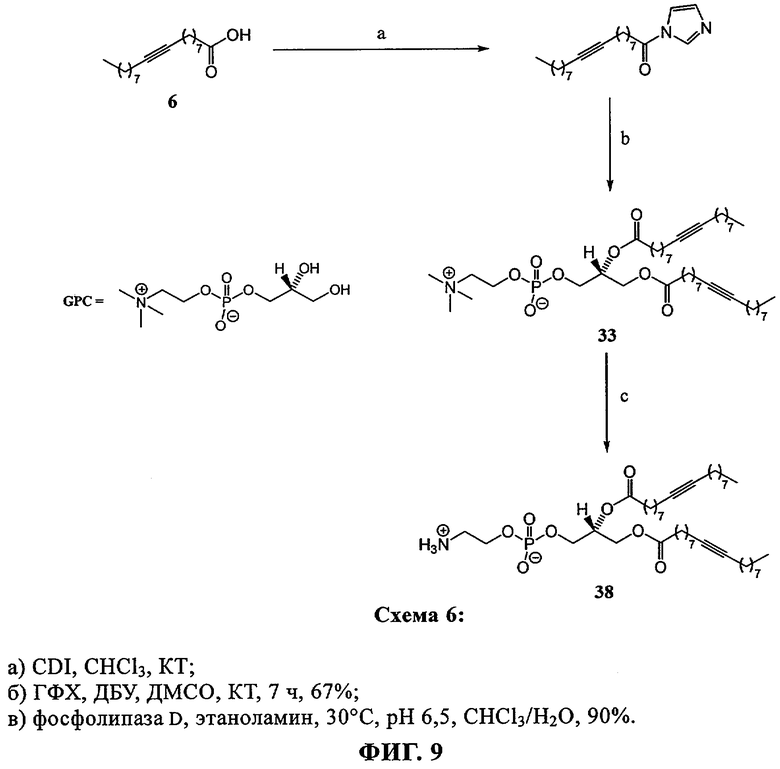
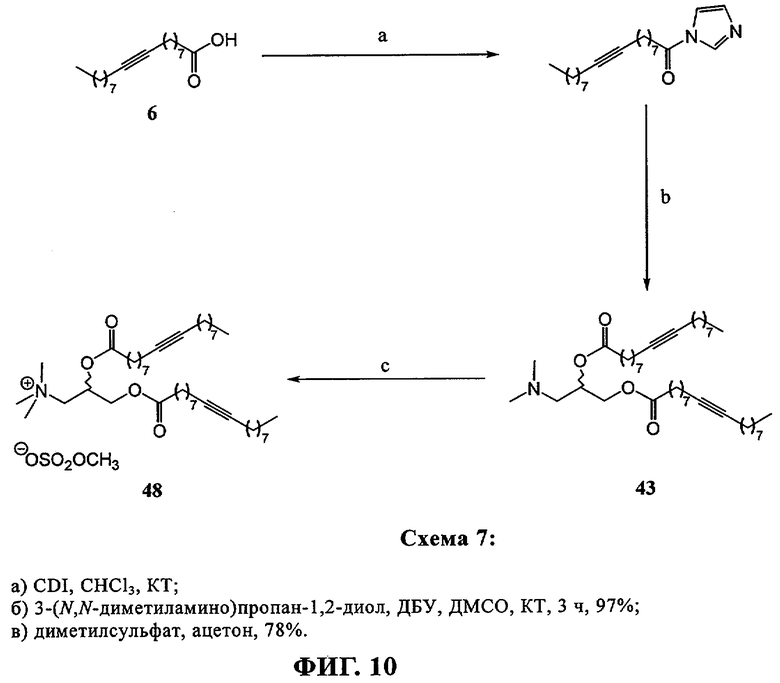
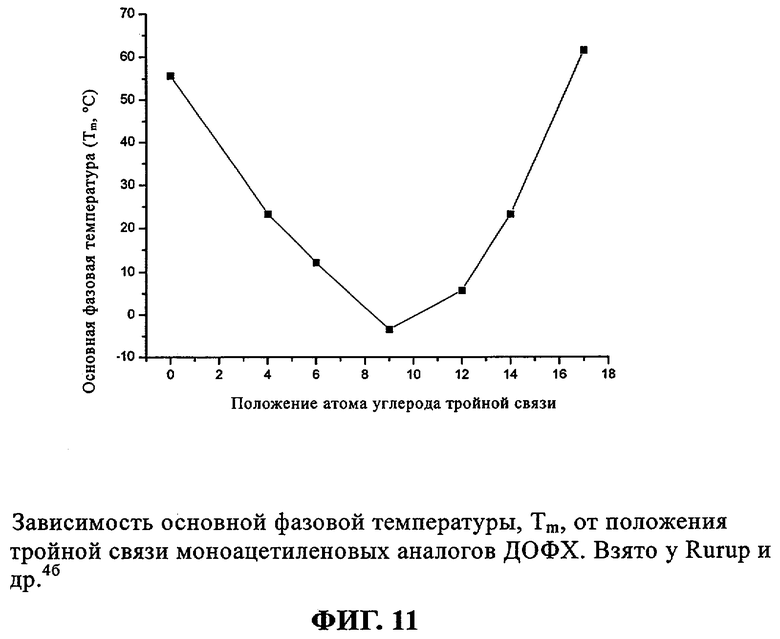
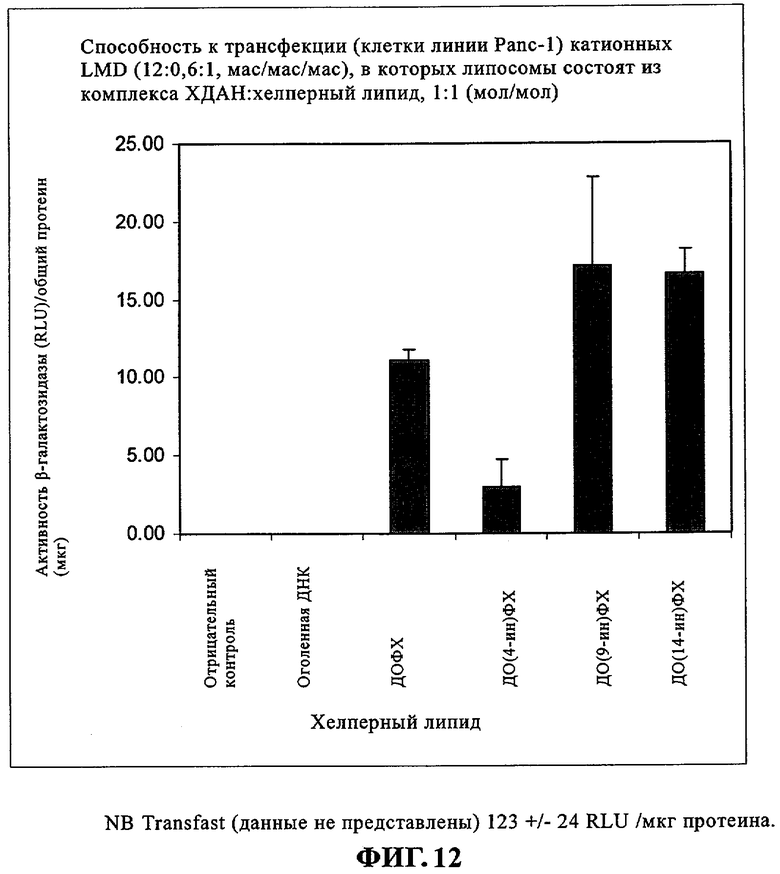
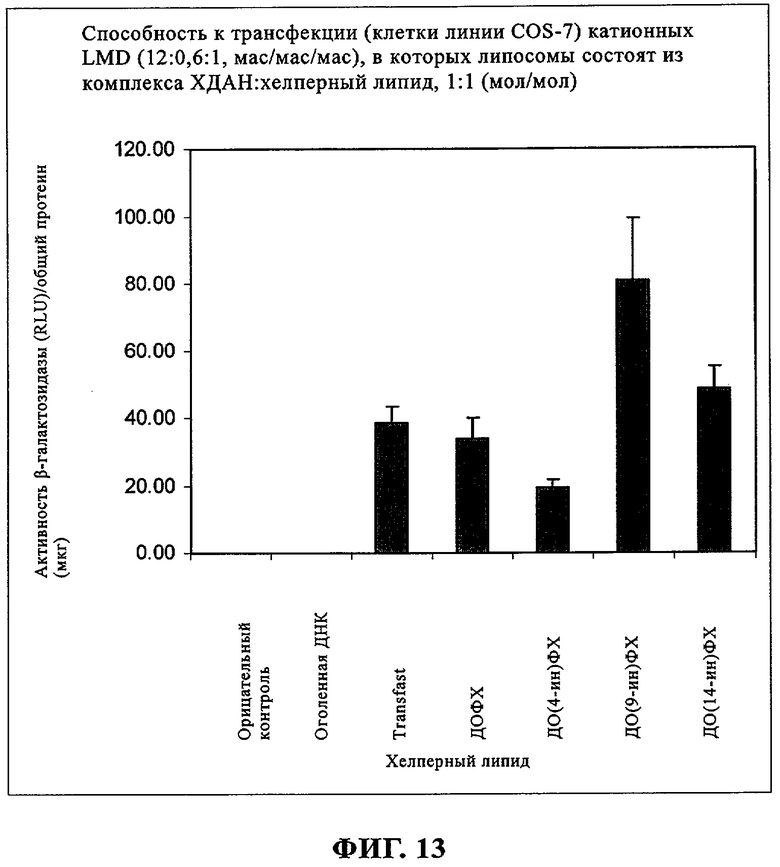
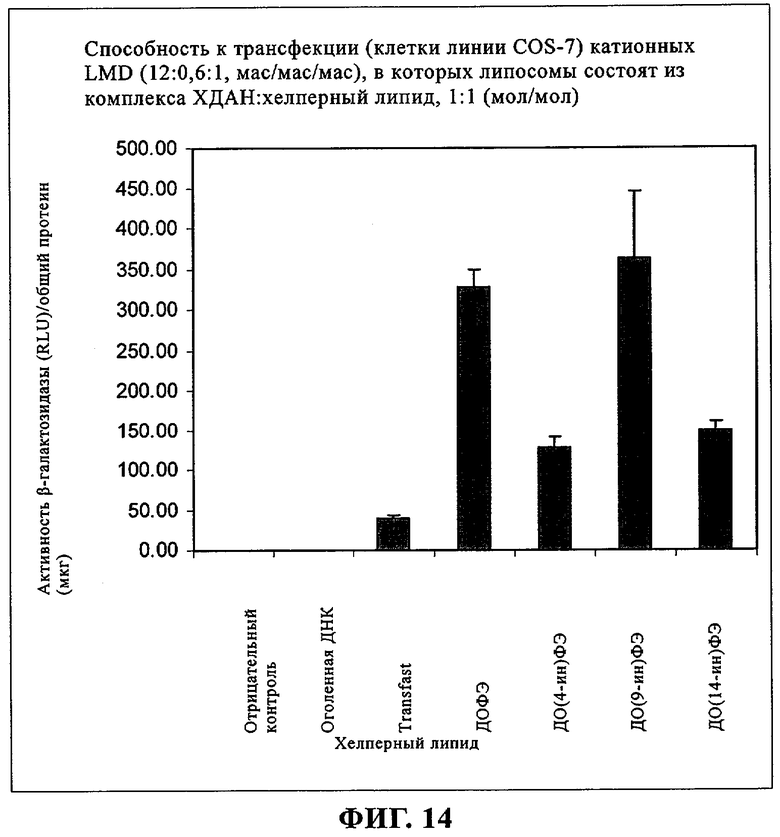
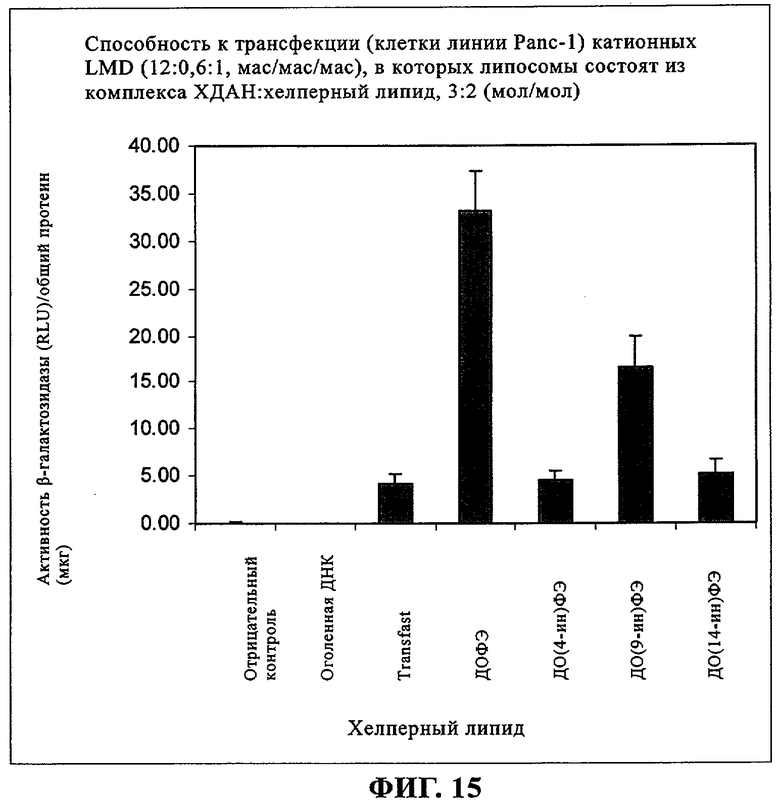
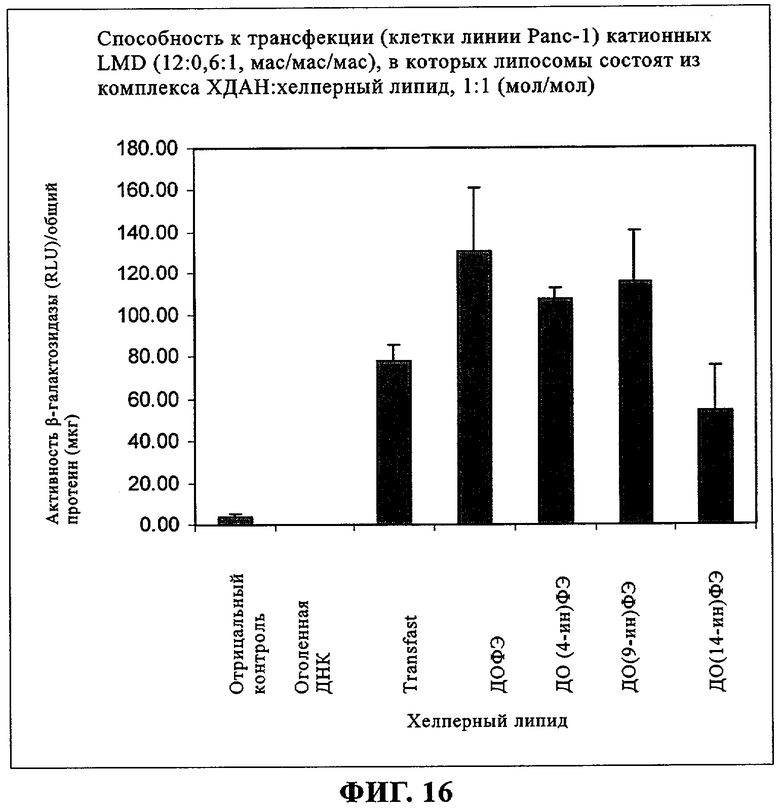
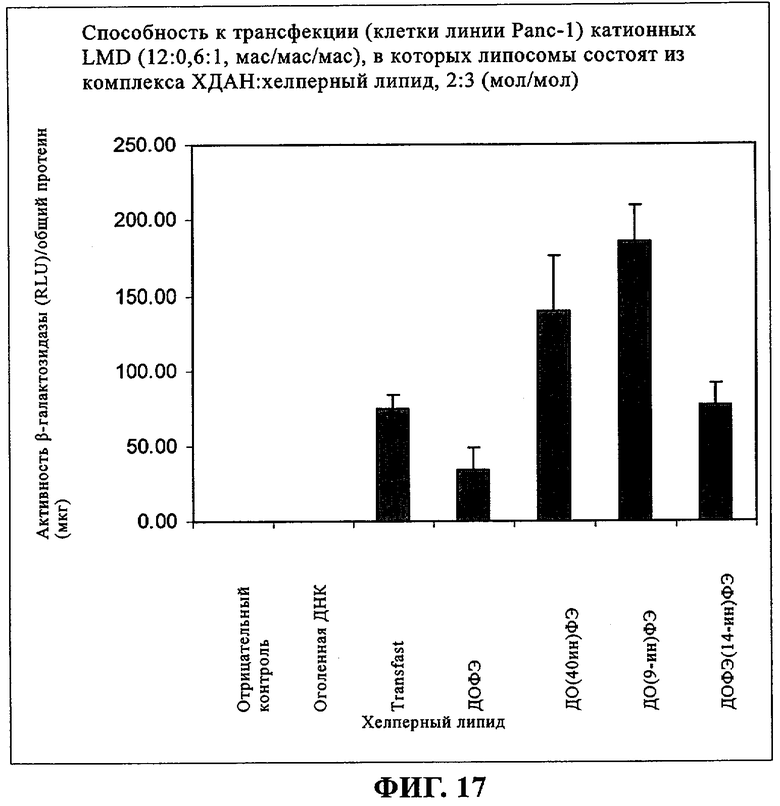
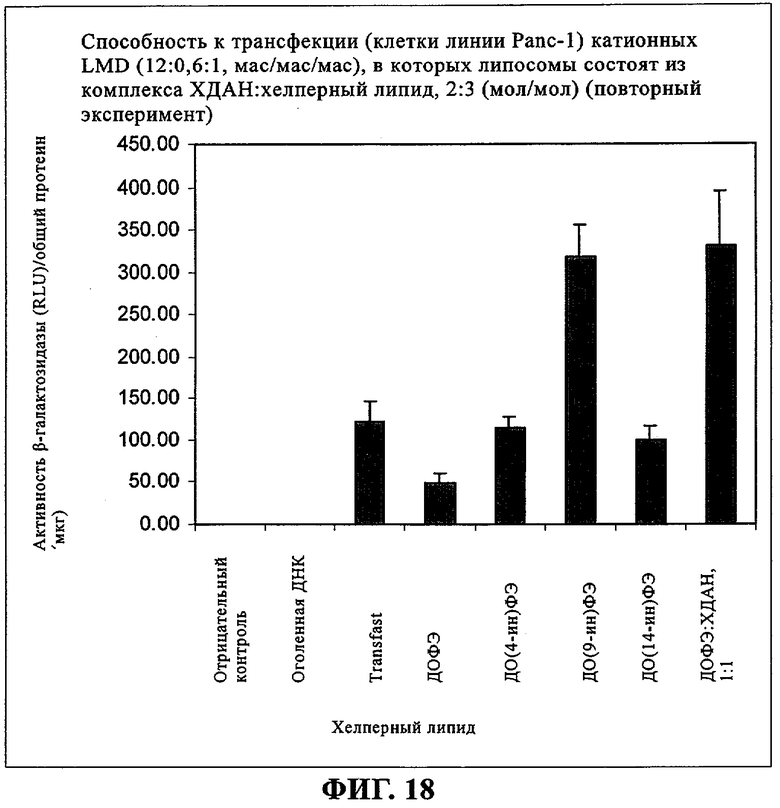
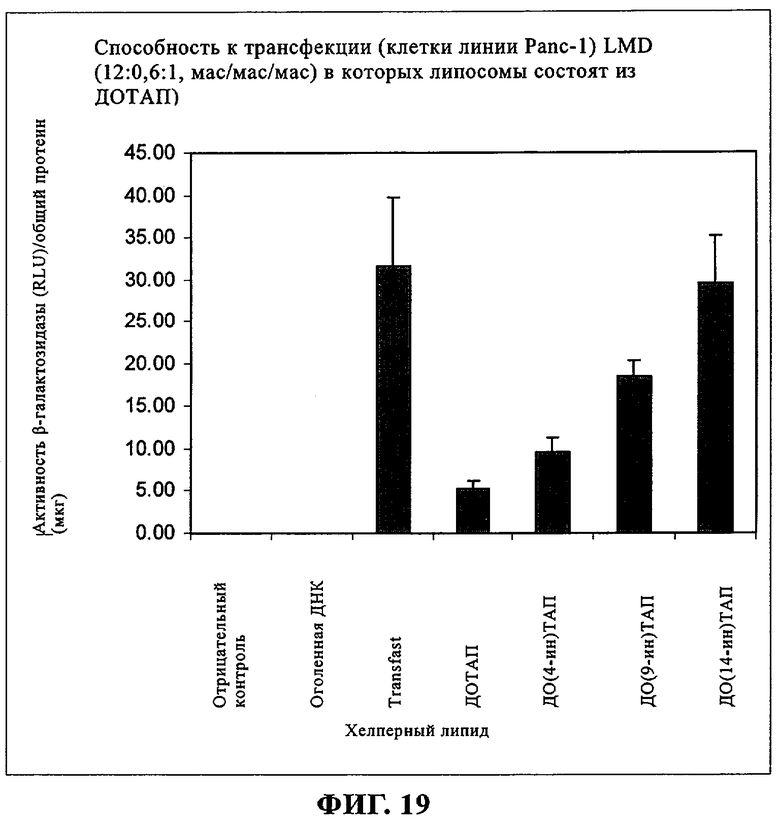
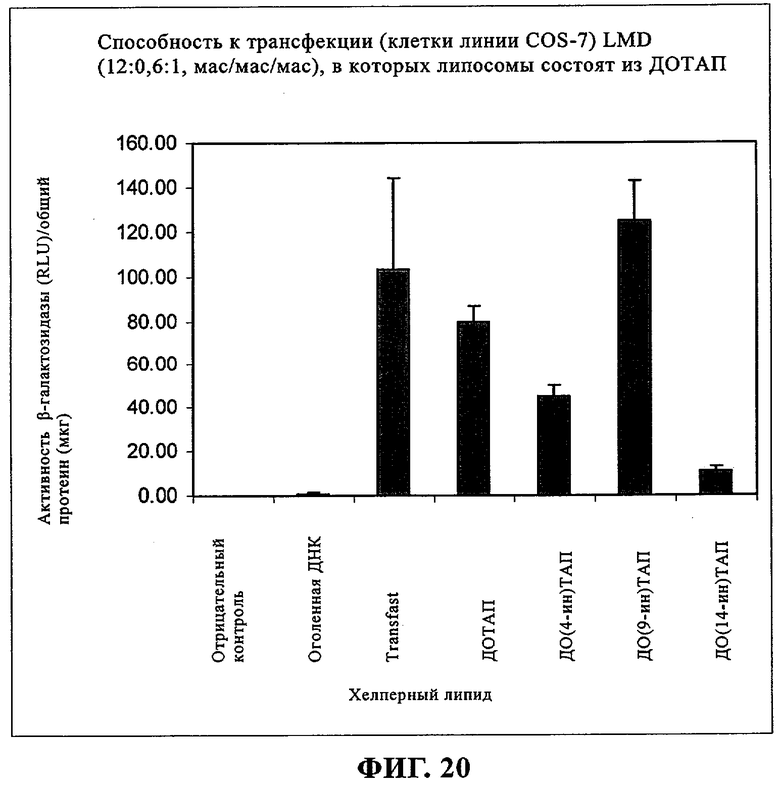
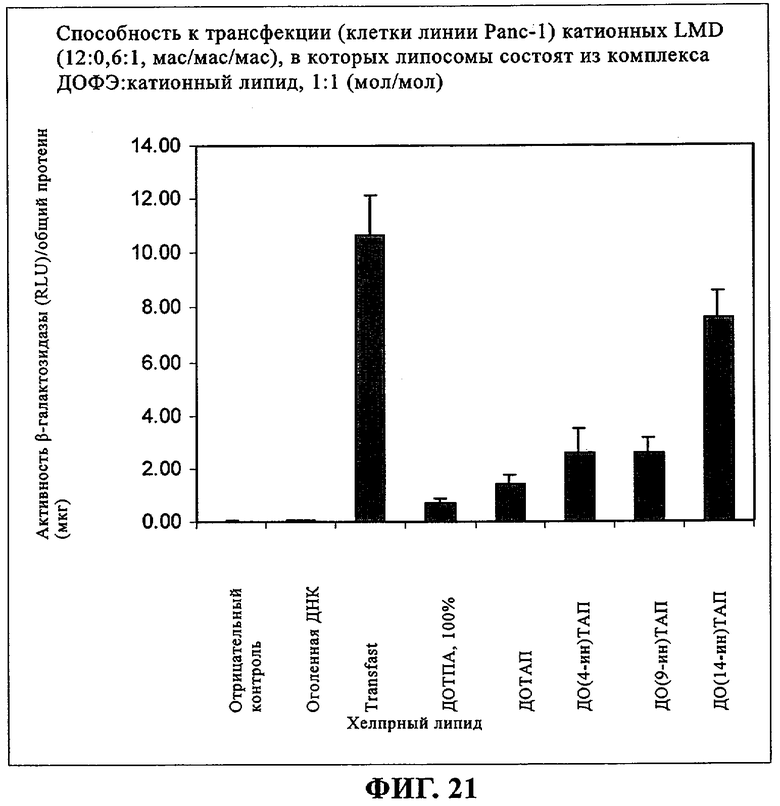
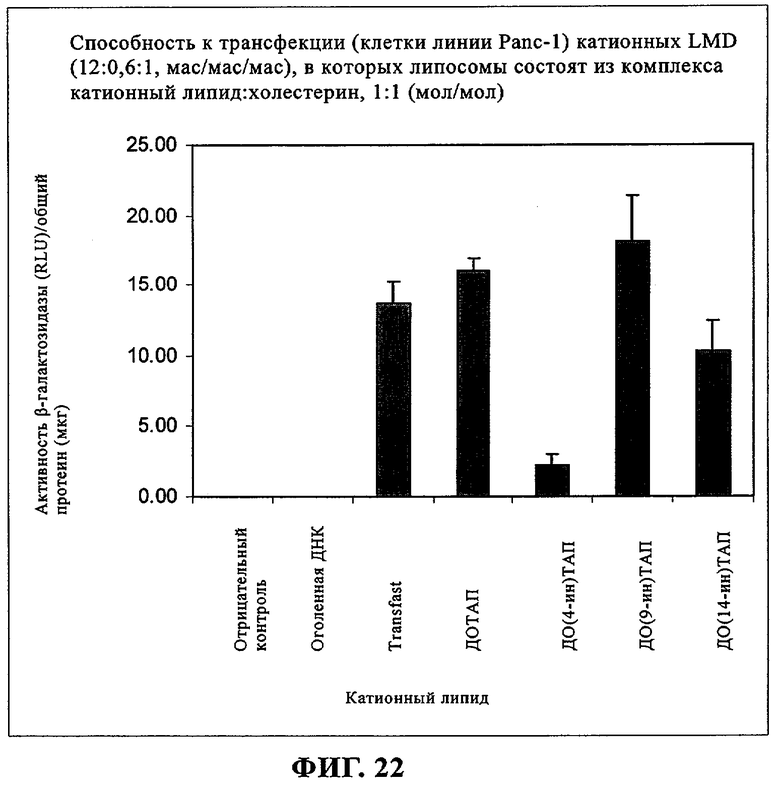
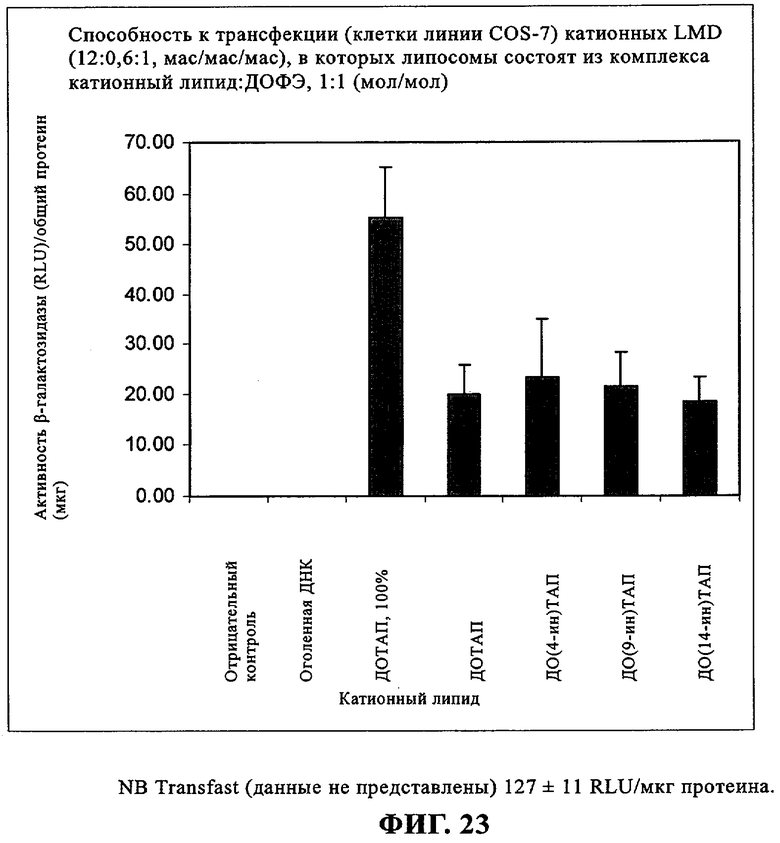
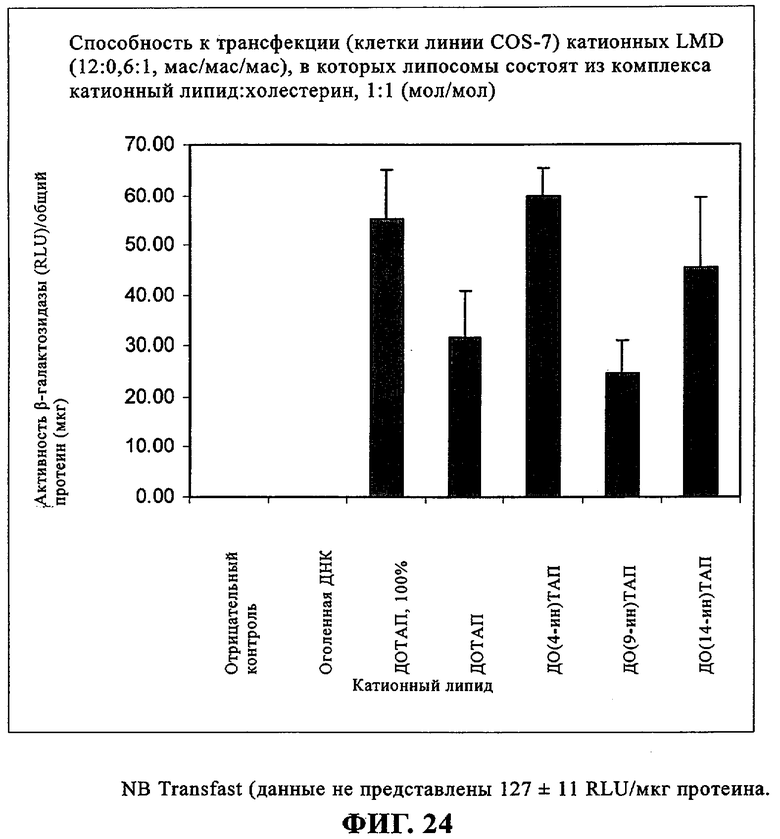
Изобретение относится к области фармакологи и касается композиции, содержащей липид и терапевтический агент, ее применения для изготовления лекарственного средства, нового липида, его применения для изготовления лекарственного средства и фармацевтической композиции. Изобретение расширяет арсенал средств переноса терапевтических агентов в клетки. 5 н. и 33 з.п. ф-лы, 24 ил.
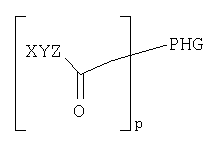
где р обозначает целое число от 1 до 10 и где каждый из X, Y и Z выбран независимо друг от друга,
причем Х обозначает ацетиленовую группу, содержащую одну связь С≡С и от 3 до 30 атомов углерода, и связь С≡С ацетиленовой группы расположена на расстоянии 2-15 атомов углерода от концевого атома ацетиленовой группы;
Y обозначает О или СН2; и
Z обозначает необязательную группу C1-С10алкил,
где PHG обозначает полярную головную группу, полученную из одного из фосфолипидов, церамидов, триацилглицеринов, лизофосфолипидов, фосфатидилсеринов, глицеринов, спиртов, соединений, содержащих алкоксигруппу, моноацилглицеринов, ганглиозидов, сфингомиелинов, цереброзидов, фосфатидилхолинов, фосфатидилэтаноламинов, фосфатидилинозитов (ФИ), диацилглицеринов, фосфатидиновых кислот, глицероуглеводов, полиспиртов и фосфатидилглицеринов, и
(II) терапевтический агент.
где р обозначает целое число от 1 до 5 и где каждый из X, Y и Z выбран независимо друг от друга.
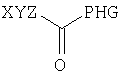
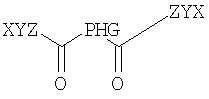
где каждый X, Y и Z выбирают независимо друг от друга.
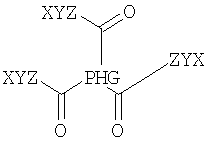
где каждый X, Y и Z выбирают независимо друг от друга.
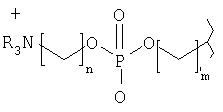
или формулу
где R независимо друг от друга выбирают из Н и гидрокарбила, m обозначает 1-10 и n обозначает 1-10.
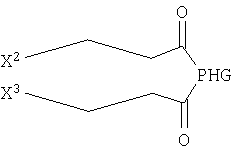
где X2 и X3 независимо друг от друга выбирают из незамещенного С10-С18алкинила.
где Х2 и Х3 независимо друг от друга выбирают из незамещенного С14алкинила и незамещенного С15алкинила.
где Х2 и Х3 независимо друг от друга выбирают из СН3(СН2)12С≡С-, СН3СН2СН2С≡С(СН2)10-, СН3(СН2)7С≡С(СН2)5-, СН3(СН2)6С≡С(СН2)5- и СН3СН2С≡С(СН2)10-.
где р обозначает целое число от 1 до 10 и где каждый из X, Y и Z выбран независимо друг от друга,
причем Х обозначает ацетиленовую группу, содержащую одну связь С≡С и от 3 до 30 атомов углерода, и связь С≡С ацетиленовой группы расположена на расстоянии 2-15 атомов углерода от концевого атома ацетиленовой группы;
Y обозначает О или СН3; и
Z обозначает необязательную группу C1-С10алкил,
где PHG обозначает полярную головную группу, полученную из одного из фосфолипидов, церамидов, триацилглицеринов, лизофосфолипидов, фосфатидилсеринов, глицеринов, спиртов, соединений, содержащих алкоксигруппу, моноацилглицеринов, ганглиозидов, сфингомиелинов, цереброзидов, фосфатидилхолинов, фосфатидилэтаноламинов, фосфатидилинозитов (ФИ), диацилглицеринов, фосфатидиновых кислот, глицероуглеводов, полиспиртов и фосфатидилглицеринов, и причем соединение отлично от ДО(4-ин)ФХ, ДО(9-ин)ФХ, ДО(14-ин)ФХ, ДО(4-ин)ФЭ и ДО(14-ин)ФЭ.
где р обозначает целое число от 1 до 10 и где каждый из X, Y и Z выбран независимо друг от друга,
причем Х обозначает ацетиленовую группу, содержащую одну связь С≡С и от 3 до 30 атомов углерода, и связь С≡С ацетиленовой группы расположена на расстоянии 2-15 атомов углерода от концевого атома ацетиленовой группы;
Y обозначает О или СН; и
Z обозначает необязательную группу C1-С10алкил,
где PHG обозначает полярную головную группу, полученную из одного из фосфолипидов, церамидов, триацилглицеринов, лизофосфолипидов, фосфатидилсеринов, глицеринов, спиртов, соединений, содержащих алкоксигруппу, моноацилглицеринов, ганглиозидов, сфингомиелинов, цереброзидов, фосфатидилхолинов, фосфатидилэтаноламинов, фосфатидилинозитов (ФИ), диацилглицеринов, фосфатидиновых кислот, глицероуглеводов, полиспиртов и фосфатидилглицеринов.
| IVAN STANISH et all, High stable vesicles composed of a new chain-terminus acetylenic photopolymeric phospholipids | |||
| Chemistry and Physics of lipids, 2001, v.112, p.99-109 | |||
| Трансформатор с регулированием на постоянную силу тока | 1932 |
|
SU32622A1 |
| ЛИПОСОМЫ, ОСУЩЕСТВЛЯЮЩИЕ ТИМУС-ЗАВИСИМУЮ ПОМОЩЬ "СЛАБЫМ" АНТИГЕНАМ, ИСПОЛЬЗУЕМЫМ ДЛЯ ПРИГОТОВЛЕНИЯ ВАКЦИНЫ | 1993 |
|
RU2107493C1 |
| ФОСФОЛИПИДНАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПРИГОТОВЛЕНИЯ | 1994 |
|
RU2119791C1 |

