ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым производным дигидроптеридинона, к способу их получения, к фармацевтическим композициям, содержащим указанные производные, и к их терапевтическим применениям, в частности, к их фармацевтическому применению в качестве ингибитора Plk киназы.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Семейство циклинзависимых киназ (Cdks) долгое время считали основным регулятором клеточного цикла, но появляется все больше и больше разнообразных протеинкиназ, которые играют важную роль в прохождении клеточного цикла. Одной из них является семейство Polo-подобных киназ (Plk).
Киназы Plk являются серин-треонин киназами, которые играют важную роль в регуляции клеточного цикла. На уровне техники раскрыто четыре Plk, то есть Plk1, Plk2, Plk3 и Plk4. Plk играют важную роль в регуляции цикла эукариотической клетки (например, регуляция митоза в клетках млекопитающих). В частности, Plk1 играет центральную роль в регуляции митоза (Glover et al. 1998, Genes Dev. 12: 3777-87; Qian et al. 2001, Mol Biol Cell. 12: 1791-9). По-видимому, гиперэкспрессия Plk1 сильно ассоциирована с опухолевыми клетками, включающими раки (WO 2004014899). Доказано, что гиперэкспрессия Plk1 ассоциирована с различными типами опухолей, такими как немелкоклеточный рак легкого, чешуйчато-клеточные раки, карциномы молочной железы, яичника или папиллярная карцинома, а также с колоректальными раками (Wolf et al. 1997, Oncogene 14: 543-549; Knecht et al. 1999, Cancer Res. 59: 2794-2797; Wolf etal. 2000, Pathol Res Pract. 196: 753-759; Weichert et al. 2004, Br. J. Cancer 90: 815-821; Ito et al. 2004, Br. J. Cancer 90: 414-418; Takahashi et al. 2003, Cancer Sci. 94: 148-152).
Сообщают, что Plk1 является консервативной от дрожжей до человека и вовлечена в различные митотические процессы, включающие активацию Cdc25C и Cdk1/циклина В при транзиции G2-M, созревание центросомы, образование и сборка веретена. На последних стадиях митоза Plk1 также вовлечена в разделение сестринских хроматид, активацию компонентов комплекса, стимулирующего анафазу, и регуляцию септина во время цитокинеза.
В предшествующем уровне техники было раскрыто много производных птеридинона в качестве ингибиторов Plk с антипролиферативной активностью. Например, в WO 2003020722 и WO 2004076454 описаны производные птеридинона, способ их получения и фармацевтические композиции, которые применяли для лечения заболеваний, связанных с активностью циклинзависимой киназы и характеризующихся гиперэкспрессией или аномальной клеточной пролиферацией. В WO 01/019825 описано применение производных птеридинона для лечения опухолевых и вирусных заболеваний. В связи с лекарственной устойчивостью различных типов опухолей для лечения опухолей крайне необходимы новые лекарства. В других заявках на патенты, таких как WO 2004076454, WO 2006018220, US 20040176380, WO 2007135374, WO 2006018185, WO 2006058876, WO 2006018222 и WO 2006018182, также раскрыты соединения в качестве ингибиторов Plk.
Однако, хотя несколько ингибиторов Plk Киназы было раскрыто, необходимы также безопасные ингибиторы Plk с улучшенной фармакокинетикой.
Целью настоящего изобретения является разработка ингибитора Plk Киназы новой структуры с более эффективными активностями, большей безопасностью и меньшей токсичностью, которые применяют для лечения нарушения клеточной пролиферации, такого как рак, инфекции, воспалительное и аутоиммунное заболевание.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
С целью преодоления недостаточности предшествующего уровня техники настоящее изобретение направлено на производные дигидроптеридинона формулы (I) и их таутомеры, энантиомеры, диастереомеры, рацематы и их смеси, а также фармацевтически приемлемые соли, гидраты или сольваты, а также их метаболиты или пролекарства:
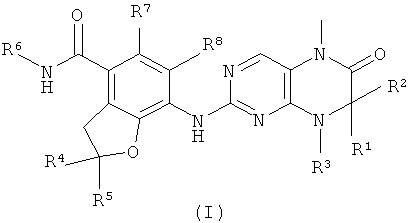
где:
R1 и R2 каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкенила, алкинила, циклоалкила, гетероциклического алкила, арила, гетероарила, карбокси или эфира карбоновой кислоты, где алкил, алкенил, алкинил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, арила, сульфурила, карбокси или эфира карбоновой кислоты;
или R1 и R2 вместе с атомом, к которому они присоединены, образуют 3-6 членное кольцо, где 3-6-членное кольцо необязательно содержит от 1 до 2 гетероатомов N, О или S(O)n;
R3 выбран из группы, состоящей из атома водорода, алкила, алкенила, алкинила, циклоалкила, гетероциклического алкила, арила или гетероарила, где алкил, алкенил, алкинил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, алкоксила, атома галогена, гидроксила, арила, сульфурила, карбокси или эфира карбоновой кислоты;
или R1 и R3 или R2 и R3 вместе с атомом, к которому они присоединены, образуют 3-6 членное кольцо, где 3-6-членное кольцо содержит от 1 до 2 гетероатомов N, О или S(O)n, и каждое 3-6-членное кольцо необязательно замещено одной или более группами, выбранными из группы, состоящей из алкила, алкоксила, атома галогена, карбонила, арила, бензила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты;
R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкоксила, циано, гидроксила, атома галогена, алкенила, алкинила, циклоалкила, гетероциклического алкила, арила, гетероарила, карбокси или эфира карбоновой кислоты, где алкил, алкоксил, алкенил, алкинил, циклоалкил, гетероциклический алкил, арил и гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, алкоксила, атома галогена, гидроксила, арила, сульфурила, -NR9R10, карбокси или эфира карбоновой кислоты;
R6 выбран из группы, состоящей из алкила, циклоалкила, гетероциклического алкила, арила или гетероарила, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, алкоксила, гидроксила, сульфурила, карбонила, циклоалкила, циклоалкилалкила, гетероциклического алкила, арила, бензила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты;
R7 и R8 каждый независимо выбран из группы, состоящей из атома водорода, алкила или атома галогена;
R9 и R10 каждый независимо выбран из группы, состоящей из атома водорода, алкила, циклоалкила, гетероциклического алкила, арила, гетероарила, карбокси или эфира карбоновой кислоты, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, циано, алкоксила, арилоксила, циклоалкила, циклоалкилалкила, гетероциклического алкила, арила, гетероарила, сульфурила, карбокси или эфира карбоновой кислоты;
или R9 и R10 вместе с атомом азота, к которому они присоединены, образуют 4-8 членный гетероцикл, где 4-8-членный гетероцикл содержит один или более гетероатомов N, О или S(O)n, и каждый 4-8-членный гетероцикл необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, галогеноалкила, циано, алкоксила, арилоксила, гидроксилалкила, циклоалкила, циклоалкилалкила, гетероциклического алкила, арила, гетероарила, сульфурила, карбокси или эфира карбоновой кислоты; и
n равно 0, 1 или 2.
В одной предпочтительной форме осуществления изобретения находятся производные дигидроптеридинона формулы (I), где
R1 и R2 каждый независимо выбран из группы, состоящей из атома водорода или алкила;
R3 выбран из группы, состоящей из атома водорода, алкила или циклоалкила.
Кроме того, в одной предпочтительной форме осуществления изобретения находятся производные дигидроптеридинона формулы (I), где
R1 и R2 каждый независимо выбран из группы, состоящей из атома водорода или алкила;
R3 выбран из группы, состоящей из алкила или циклоалкила;
R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода или алкила, где алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкоксила или -NR9R10;
R6 выбран из группы, состоящей из алкила, циклоалкила или гетероциклического алкила, где алкил, циклоалкил или гетероциклический алкил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, гидроксила, циклоалкилалкила, гетероциклического алкила, -NR9R10, карбокси или эфира карбоновой кислоты;
R7 и R8 каждый независимо представляет собой атом водорода;
R9 и R10 каждый независимо выбран из группы, состоящей из атома водорода, алкила, циклоалкила, гетероциклического алкила, арила, гетероарила, карбокси или эфира карбоновой кислоты, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, циано, алкоксила, арилоксила, гетероциклического алкила, арила, гетероарила, сульфурила, карбокси или эфира карбоновой кислоты;
или R9 и R10 вместе с атомом азота, к которому они присоединены, образуют 4-8 членный гетероцикл, где 4-8-членный гетероцикл содержит один или более гетероатомов N, О или S(O)n, и каждый 4-8-членный гетероцикл необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, галогеноалкила, циано, алкоксила, арилоксила, гидроксилалкила, циклоалкила, циклоалкилалкила, гетероциклического алкила, арила, гетероарила, сульфурила, карбокси или эфира карбоновой кислоты; и
n равно 0, 1 или 2.
Соединения по изобретению включают, но не ограничены ими, приведенные ниже:
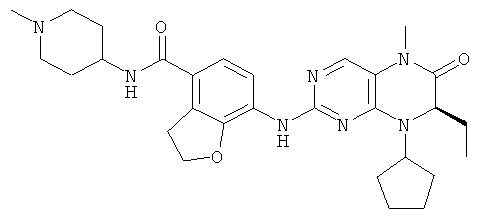
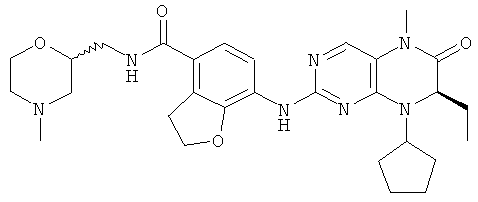
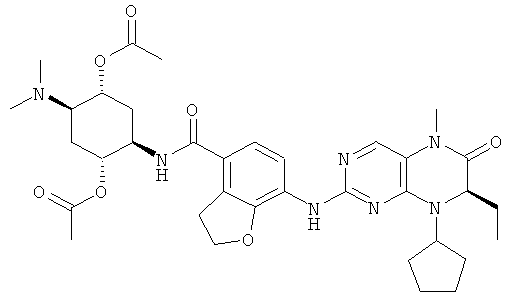
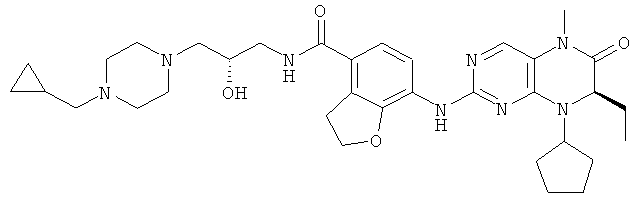
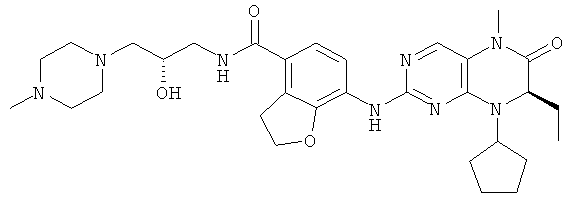

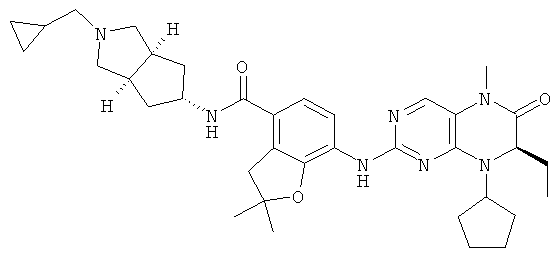
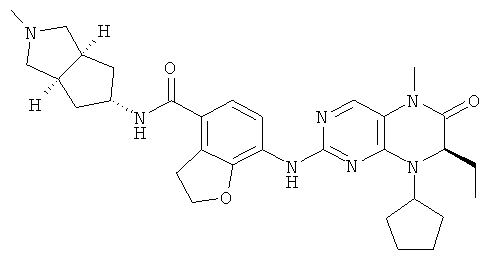
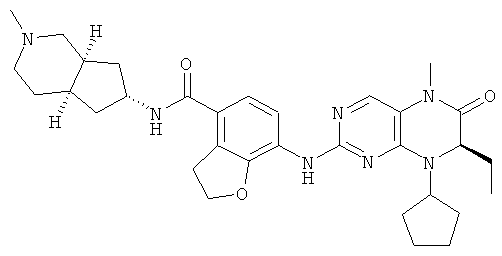


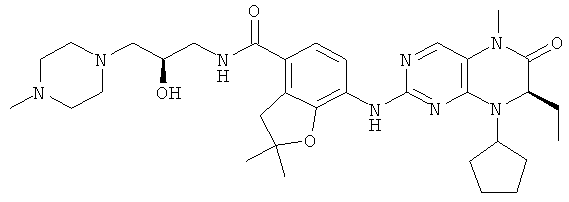
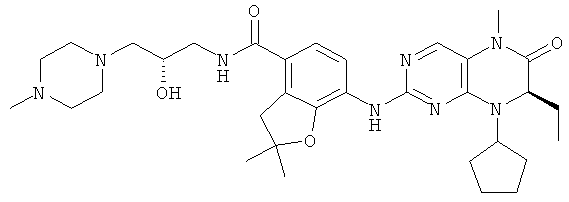


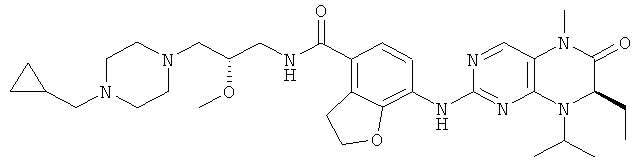
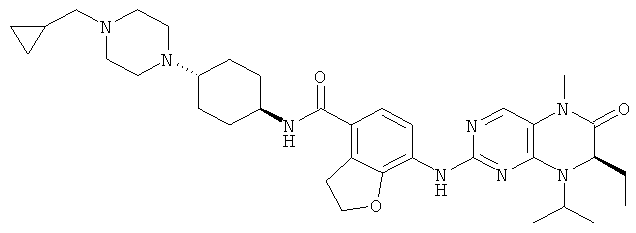

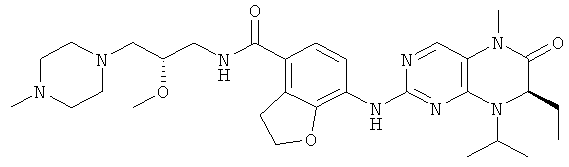
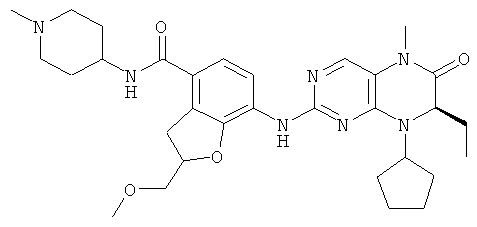
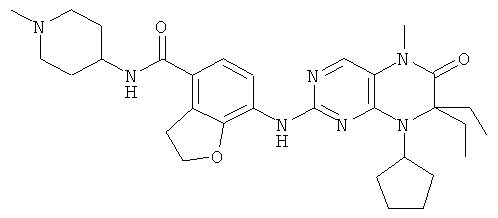
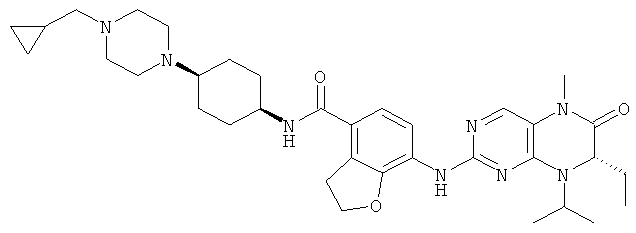
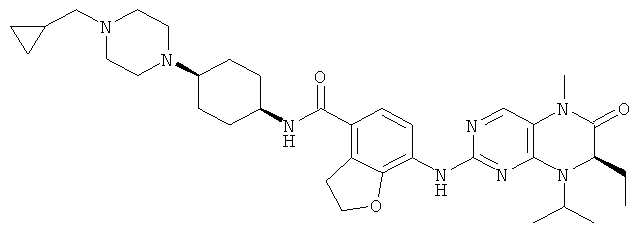
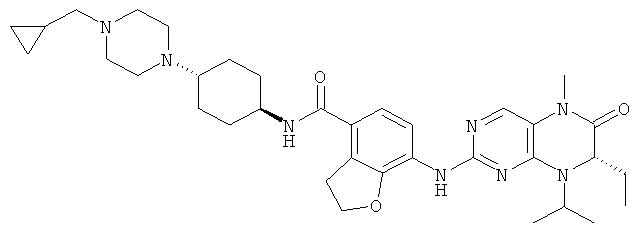
или их таутомеры, рацематы, энантиомеры, диастереоизомеры и их смеси, а также их фармацевтически приемлемые соли.
В другом аспекте данное изобретение относится к соединениям приведенной ниже формулы (IA) в качестве промежуточных соединений для синтеза соединений формулы (I):
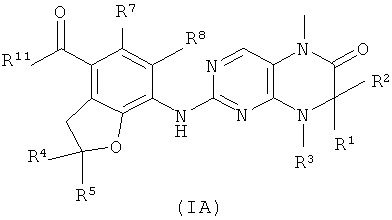
где:
R1 и R2 каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкенила, алкинила, циклоалкила, гетероциклического алкила, арила, гетероарила, карбокси или эфира карбоновой кислоты, где алкил, алкенил, алкинил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, арила, сульфурила, карбокси или эфира карбоновой кислоты;
или R1 и R2 вместе с атомом, к которому они присоединены, образуют 3-6 членное кольцо, где 3-6-членное кольцо необязательно содержит от 1 до 2 гетероатомов N, О или S(O)n;
R3 выбран из группы, состоящей из атома водорода, алкила, алкенила, алкинила, циклоалкила, гетероциклического алкила, арила или гетероарила, где алкил, алкенил, алкинил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, алкоксила, атома галогена, гидроксила, арила, сульфурила, карбокси или эфира карбоновой кислоты;
или R1 и R3 или R2 и R3 вместе с атомом, к которому они присоединены, образуют 3-6 членное кольцо, где 3-6-членное кольцо содержит от 1 до 2 гетероатомов N, О или S(O)n, и каждое 3-6-членное кольцо необязательно замещено одной или более группами, выбранными из группы, состоящей из алкила, алкоксила, атома галогена, карбонила, арила, бензила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты;
R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкоксила, циано, гидроксила, атома галогена, алкенила, алкинила, циклоалкила, гетероциклического алкила, арила, гетероарила, карбокси или эфира карбоновой кислоты, где алкил, алкоксил, алкенил, алкинил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, алкоксила, атома галогена, гидроксила, арила, сульфурила, -NR9R10, карбокси или эфира карбоновой кислоты;
R7 и R8 каждый независимо выбран из группы, состоящей из атома водорода, алкила или атома галогена;
R9 и R10 каждый независимо выбран из группы, состоящей из атома водорода, алкила, циклоалкила, гетероциклического алкила, арила, гетероарила, карбокси или эфира карбоновой кислоты, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, циано, алкоксила, арилоксила, циклоалкила, циклоалкилалкила, гетероциклического алкила, арила, гетероарила, сульфурила, карбокси или эфира карбоновой кислоты;
или R9 и R10 вместе с атомом азота, к которому они присоединены, образуют 4-8 членный гетероцикл, где 4-8-членный гетероцикл содержит один или более гетероатомов N, О или S(O)n, и каждый 4-8-членный гетероцикл необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, галогеноалкила, циано, алкоксила, арилоксила, гидроксилалкила, циклоалкила, циклоалкилалкила, гетероциклического алкила, арила, гетероарила, сульфурила, карбокси или эфира карбоновой кислоты;
R11 выбран из группы, состоящей из гидроксила или алкоксила; и
n равно 0, 1 или 2.
Предпочтительны соединения формулы (IA), где R1 и R2 каждый независимо выбран из группы, состоящей из атома водорода или алкила; R3 выбран из группы, состоящей из атома водорода, алкила или циклоалкила.
Предпочтительны соединения формулы (IA), где R1 и R2 каждый независимо выбран из группы, состоящей из атома водорода или алкила;
R3 выбран из группы, состоящей из алкила или циклоалкила;
R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода или алкила, где алкил необязательно замещен одной или более группами, выбранными из алкоксила или -NR9R10;
R6 выбран из группы, состоящей из алкила, циклоалкила или гетероциклического алкила, где алкил, циклоалкил или гетероциклический алкил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, гидроксила, циклоалкилалкила, гетероциклического алкила, -NR9R10, карбокси или эфира карбоновой кислоты;
R7 и R8 независимо представляют собой атом водорода;
R9 и R10 каждый независимо выбран из группы, состоящей из атома водорода, алкила, циклоалкила, гетероциклического алкила, арила, гетероарила, карбокси или эфира карбоновой кислоты, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, циано, алкоксила, арилоксила, гетероциклического алкила, арила, гетероарила, сульфурила, карбокси или эфира карбоновой кислоты;
или R9 и R10 вместе с атомом азота, к которому они присоединены, образуют 4-8 членный гетероцикл, где 4-8-членный гетероцикл содержит один или более гетероатомов N, О или S(O)n, и каждый 4-8-членный гетероцикл необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидроксила, галогеноалкила, циано, алкоксила, арилоксила, гидроксилалкила, циклоалкила, циклоалкилалкила, гетероциклического алкила, арила, гетероарила, сульфурила, карбокси или эфира карбоновой кислоты;
R11 выбран из группы, состоящей из гидроксила или алкоксила; и
n равно 0, 1 или 2.
В другом аспекте данное изобретение относится к способу получения соединения формулы (I), включающему приведенные ниже стадии:
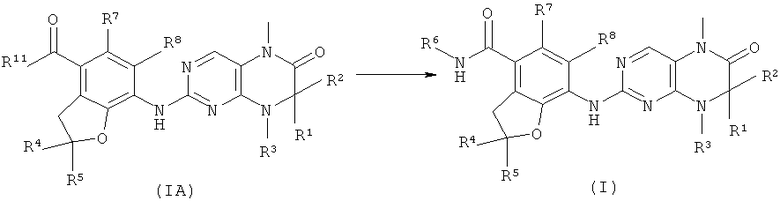
Взаимодействие соединений формулы (IA) с R6NH2 с получением соединений формулы (I);
где R1-R8 такие, как в формуле (I), и R11 такой, как в формуле (IA).
В другом аспекте данное изобретение относится к применению соединений формулы (I) или их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей, а также их фармацевтически приемлемых солей для получения лекарственного средства для лечения нарушения клеточной пролиферации, где нарушение клеточной пролиферации выбрано из группы, состоящей из рака, инфекции, воспаления или аутоиммунного заболевания; где рак выбран из группы, состоящей из немелкоклеточного рака легкого, чешуйчато-клеточного рака, рака молочной железы, рака яичника, рака шейки матки, папиллярной карциномы или колоректальной карциномы; предпочтительно рака шейки матки или колоректальной карциномы.
Кроме того, данное изобретение также относится к применению соединений формулы (I) или их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей, а также их фармацевтически приемлемых солей для получения лекарственного средства в качестве ингибитора Plk.
Еще в одном другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединений формулы (I) в соответствии с настоящим изобретением или их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей, а также их фармацевтически приемлемых солей вместе с фармацевтически приемлемыми носителями или эксципиентами. Также настоящее изобретение относится к применению данной фармацевтической композиции для получения лекарственного средства для лечения рака, инфекции, воспаления и аутоиммунного заболевания; где рак выбран из группы, состоящей из немелкоклеточного рака легкого, чешуйчато-клеточного рака, рака молочной железы, рака яичника, рака шейки матки, папиллярной карциномы или колоректальной карциномы; предпочтительно рака шейки матки или колоректальной карциномы. Также настоящее изобретение относится к применению данной фармацевтической композиции для получения лекарственного средства в качестве ингибитора Plk. Также настоящее изобретение относится к данной фармацевтической композиции для применения в качестве лекарственного средства для лечения рака. Также настоящее изобретение относится к к способу получения данной фармацевтической композиции, включающему стадию объединения соединений формулы (I) с фармацевтически приемлемым носителем или эксципиентом.
Настоящее изобретение относится к способу лечения нарушения клеточной пролиферации, где этот способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединений формулы (I), их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей, а также их фармацевтически приемлемых солей или содержащей их фармацевтической композиции; где нарушение клеточной пролиферации выбрано из группы, состоящей из рака, инфекции, воспаления или аутоиммунного заболевания, где рак выбран из группы, состоящей из немелкоклеточного рака легкого, чешуйчато-клеточного рака, рака молочной железы, рака яичника, рака шейки матки, папиллярной карциномы или колоректальной карциномы; предпочтительно рака шейки матки или колоректальной карциномы.
Настоящее изобретение относится к способу модулирования активности Plk киназы, где этот способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединений формулы (I), их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей, а также их фармацевтически приемлемых солей или содержащей их фармацевтической композиции.
Настоящее изобретение относится к соединению формулы (I) или его таутомерам, рацематам, энантиомерам, диастереоизомерам и их смесям, а также к его фармацевтически приемлемым солям или к содержащей их фармацевтической композиции для применения в качестве лекарственного средства для лечения нарушения клеточной пролиферации, где нарушение клеточной пролиферации выбрано из группы, состоящей из рака, инфекции, воспаления или аутоиммунного заболевания; где рак выбран из группы, состоящей из немелкоклеточного рака легкого, чешуйчато-клеточного рака, рака молочной железы, рака яичника, рака шейки матки, папиллярной карциномы или колоректальной карциномы; предпочтительно рака шейки матки или колоректальной карциномы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, приведенные ниже термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
"Алкил" относится к насыщенной алифатической углеводородной группе, включающей C1-C20 прямоцепочечные и разветвленные группы. Предпочтительно алкильная группа представляет собой алкил, имеющий от 1 до 12 атомов углерода. Репрезентативные примеры включают, но не ограничены ими метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их изомеры разветвленной цепи. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода. Репрезентативные примеры включают, но не ограничены ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 2-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.д. Алкильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты.
"Алкенил" относится к алкилу, определенному, как выше, который имеет по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную двойную связь, например, винил, 1-пропенил, 2-пропенил, 1-, 2- или 3-бутенил и т.д. Алкенильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты.
"Алкинил" относится к алкилу, определенному, как выше, который имеет по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную двойную связь, например, этинил, 1-пропинил, 2-пропинил, 1-, 2- или 3-бутинил и т.д. Алкинильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты.
"Циклоалкил" относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе и имеет от 3 до 20 атомов углерода. Предпочтительно циклоалкильная группа представляет собой циклоалкил, имеющий от 3 до 12 атомов углерода. Более предпочтительно циклоалкильная группа представляет собой циклоалкил, имеющий от 3 до 10 атомов углерода. Репрезентативные примеры моноциклического циклоалкила включают, но не ограничены ими, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.д. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо и кольцо с внутренним мостиком.
"Спиро-циклоалкил" относится к 5-20-членной полициклической углеводородной группе с кольцами, соединенными через один общий атом углерода (называемый спиро-атомом), где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы. Предпочтительно спиро-циклоалкил является 6-14-членным, более предпочтительно является 7-10-членным. В соответствии с числом общих спиро-атомов спиро-циклоалкил делят на моноциклическое спиро-кольцо, бициклическое спиро-кольцо или полициклическое спиро-кольцо, предпочтительно оно относится к моноциклическому спиро-кольцу или бициклическому спиро-кольцу. Более предпочтительно спиро-циклоалкил представляет собой 4-членное/4-членное, 4-членное/5-членное, 4-членное/6-членное, 5-членное/5-членное или 5-членное/6-членное моноциклическое спиро-кольцо. Репрезентативные примеры спиро-циклоалкила включают, но не ограничены ими, приведенные ниже группы:
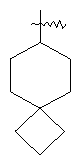
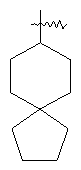
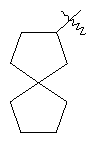
 и
и  .
.
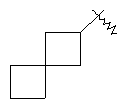
"Конденсированный циклоалкил" относится к 5-20-членной полициклической углеводородной группе, где каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы. Предпочтительно конденсированная циклоалкильная группа является 6-14-членной, более предпочтительно 7-10-членной. В соответствии с числом колец-членов конденсированный циклоалкил делят на конденсированное бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или пол и циклическое кольцо, Предпочтительно он относится к конденсированному бициклическому или трициклическому кольцу. Более предпочтительно конденсированный циклоалкил представляет собой 5-членное/5-членное или 5-членное/6-членное конденсированное бициклическое кольцо. Репрезентативные примеры конденсированного циклоалкила включают, но не ограничены ими, приведенные ниже группы:
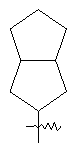

 и
и 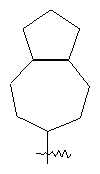
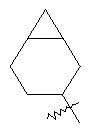
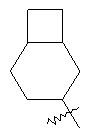
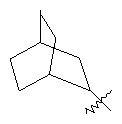
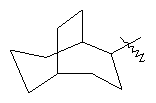
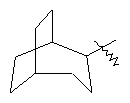
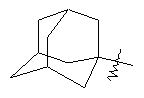
"Циклоалкил с внутренним мостиком" относится к 5-20-членной полициклической углеводородной группе, где каждые два кольца в системе имеют два общих разъединенных атома углерода. Эти кольца могут иметь одну или более чем одну двойную связь, но не имеют полностью конъюгированной пи-электронной системы. Предпочтительно циклоалкил с внутренним мостиком является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов циклоалкил с внутренним мостиком делят на бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или полициклическое кольцо с внутренним мостиком, предпочтительно он относится к бициклическому, трициклическому или тетрациклическому циклоалкилу с внутренним мостиком, более предпочтительно относится к бициклическому или трициклическому циклоалкилу с внутренним мостиком. Репрезентативные примеры циклоалкила с внутренним мостиком включают, но не ограничены ими, приведенные ниже группы:


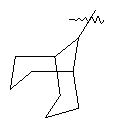

 и
и 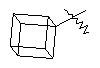 .
.
Циклоалкил может быть конденсирован с арилом, гетероарилом или гетероциклическим алкилом, где кольцом, соединенным с исходной структурой, является циклоалкил. Репрезентативные примеры циклоалкила с внутренним мостиком включают, но не ограничены ими, инданилуксусную кислоту, тетрагидронафталин, бензоциклогептил и т.д. Циклоалкил может быть замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляют собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты.
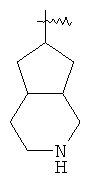
"Гетероциклический алкил" относится к 3-20-членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более гетероатомов, выбранный из группы, состоящей из N, О или S(O)n (где n равно 0, 1 или 2), в качестве кольцевых атомов, но исключая -O-O-, -O-S- или -S-S- в кольце, где остальными кольцевыми атомами являются атомы С. Предпочтительно гетероциклический алкил является 3-12-членным, имеющим от 1 до 4 указанных гетероатомов, более предпочтительно 3-10-членным. Репрезентативные примеры моноциклического гетероциклического алкила включают, но не ограничены ими, пирролидил, пиперидил, пиперазинил, морфолинил, сульфоморфолинил, гомопиперазинил и т.д. Полициклический гетероциклический алкил включает гетероциклический алкил, имеющий спиро-кольцо, конденсированное кольцо и кольцо с внутренним мостиком.
"Спиро-гетероциклоалкил" относится к 5-20-членной полициклической гетероциклической алкильной группе с кольцами, соединенными через один общий атом углерода (называемый спиро-атомом), где кольца имеют один или более гетероатомов, выбранный из группы, состоящей из N, О или S(O)p (где р равно 0,1 или 2) в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы. Предпочтительно спиро-гетероциклоалкил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом общих атомов спиро-гетероциклоалкил делят на моноциклический спиро-гетероциклоалкил, бициклический спиро-гетероциклоалкил или полициклический спиро-гетероциклоалкил, предпочтительно он относится к моноциклическому спиро-гетероциклоалкилу или бициклическому спиро-гетероциклоалкилу. Более предпочтительно спиро-гетероциклоалкил представляет собой 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноциклический спиро-гетероциклоалкил. Репрезентативные примеры спиро-гетероциклоалкила включают, но не ограничены ими, приведенные ниже группы:
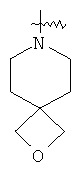
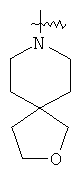
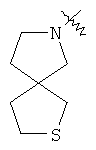
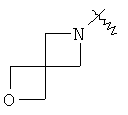 и
и 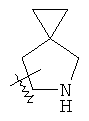 .
.
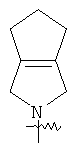
"Конденсированный гетероциклический алкил" относится к 5-20-членной полициклической гетероциклической алкильной группе, где каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, и где кольца имеют один или более гетероатомов, выбранный из группы, состоящей из N, О или S(O)p (где р равно 0, 1 или 2) в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С. Предпочтительно конденсированный гетероциклический алкил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов конденсированный гетероциклический алкил делят на конденсированное бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или полициклическое кольцо, предпочтительно он относится к конденсированному бициклическому или трициклическому кольцу. Более предпочтительно конденсированный гетероциклический алкил представляет собой 5-членное/5-членное или 5-членное/6-членное конденсированное бициклическое кольцо. Репрезентативные примеры конденсированного гетероциклического алкила включают, но не ограничены ими, приведенные ниже группы:
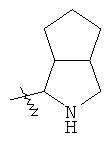
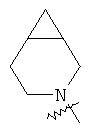

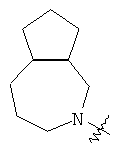
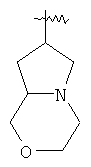
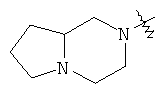 и
и 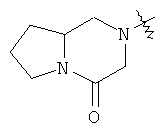 .
.
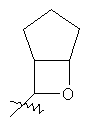
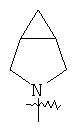
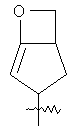
"Гетероциклический алкил с внутренним мостиком" относится к 5-14-членной полициклической гетероциклической алкильной группе, где каждые два кольца в системе имеют два общих разъединенных атома углерода, кольца могут иметь одну или более чем одну двойную связь, но не имеют полностью конъюгированной пи-электронной системы, кольца имеют один или более гетероатомов, выбранный из группы, состоящей из N, О или S(O)p (где р равно 0, 1 или 2) в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С. Предпочтительно гетероциклический алкил с внутренним мостиком является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов гетероциклический алкил с внутренним мостиком делят на бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или полициклическое кольцо с внутренним мостиком, предпочтительно он относится к бициклическому, трициклическому или тетрациклическому гетероциклическому алкилу с внутренним мостиком, более предпочтительно относится к бициклическому или трициклическому гетероциклическому алкилу с внутренним мостиком. Репрезентативные примеры гетероциклического алкила с внутренним мостиком включают, но не ограничены ими, приведенные ниже группы:
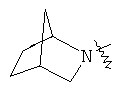
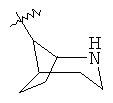
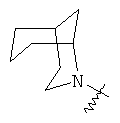
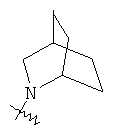
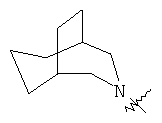
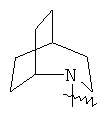
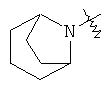 и
и  .
.
Гетероциклический алкил может быть конденсирован с арилом, гетероциклическим алкилом или циклоалкилом, где кольцом, соединенным с исходной структурой, является гетероциклический алкил. Репрезентативные примеры гетероциклического алкила включают, но не ограничены ими, приведенные ниже группы:
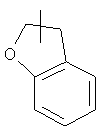
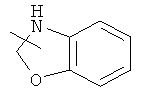
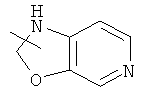 и
и 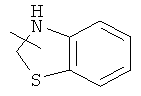 .
.
Гетероциклический алкил может быть замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляют собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты.
"Арил" относится к 6-14-членной полностью углеродной моноциклической кольцевой или к полициклической конденсированной кольцевой ("конденсированная" кольцевая система означает, что каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом в системе) группе и имеет полностью конъюгированную пи-электронную систему. Предпочтительно арил является 6-10-членным, таким как фенил и нафтил. Арил может быть конденсирован с гетероарилом, гетероциклическим алкилом или циклоалкилом, где кольцом, соединенным с исходной структурой, является арил. Репрезентативные примеры арила включают, но не ограничены ими, приведенные ниже группы:
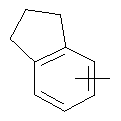
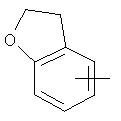
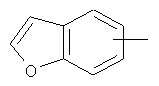
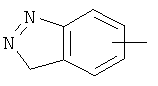
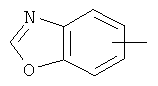

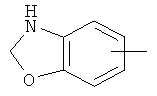
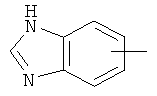
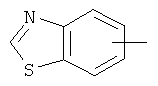
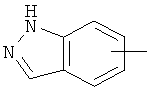
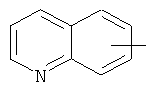
 и
и 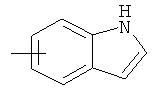 .
.
Арильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляют собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты.
"Гетероарил" относится к гетероарилу, имеющему от 1 до 4 гетероатомов, выбранных из группы, состоящей из N, О или S в качестве кольцевых атомов и имеющему от 5 до 14 кольцевых атомов, предпочтительно 5-10-членное кольцо, более предпочтительно 5- или 6-членное кольцо. Примеры гетероарильных групп включают фурил, тиенил, пиридил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил и тому подобное. Гетероарил может быть конденсирован с кольцом арила, гетероциклической группы или циклоалкила, где кольцом, соединенным с исходной структурой, является гетероарил. Репрезентативные примеры включают, но не ограничены ими, приведенные ниже группы:
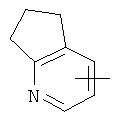


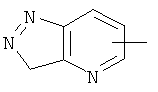
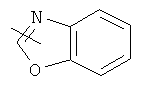
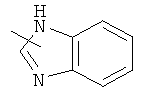
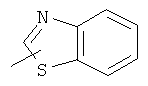
 и
и  .
.
Гетероарильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляют собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты.
"Алкоксил" относится как к группе -О-(алкил), так и к группе -O-(незамещенный циклоалкил), где алкил определен выше. Репрезентативные примеры включают, но не ограничены ими, метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и тому подобное. Алкоксил может быть замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляют собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, -C(O)R9, -C(O)NR9R10, -NR9R10, карбокси или эфира карбоновой кислоты.
"Арилоксил" относится как к группе -О-(арил), так и к группе -О-(гетероарил), где арил и гетероарил определены выше. Репрезентативные примеры включают фенокси, пиридинилокси, фурилокси, тиенилокси, пиримидинилокси, пиразинилокси и их производные.
"Гидрокси" относится к -ОН группе.
"Атом галогена" относится к фторо, хлоро, бромо или йодо.
"Амино" относится к группе -NH2.
"Циано" относится к группе -CN.
"Нитро" относится к группе -NO2.
"Сульфурил" относится к группе (группа)-S(=O)2-(группа).
"Карбонил" относится к группе (группа)-С(=O)-(группа).
"Гидроксилалкил" относится к группе -алкил-ОН, где алкил определен выше.
"Бензил" относится к группе -СН2-(фенил), где фенил определен выше.
"Карбокси" относится к группе (алкил)С(=O)ОН, где алкил определен выше.
"Эфир карбоновой кислоты" относится к группе (алкил)С(=O)O(алкил), где алкил определен выше.
"Фармацевтическая композиция" относится к смеси одного или более чем одного из соединений, описанных в данной заявке, или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Целью фармацевтической композиции является облегчение введения соединения в организм.
СПОСОБ СИНТЕЗА СОЕДИНЕНИЯ ПО ИЗОБРЕТЕНИЮ
Чтобы осуществить цель изобретения, в изобретении применено приведенное ниже техническое решение:
Способ получения соединения формулы (I) по изобретению, включающий приведенные ниже стадии:
Соединения формулы (I) необязательно подвергают гидролизу, а затем конденсируют с R6NH2 в присутствии O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората с получением соединений формулы (I),
где R1-R8 такие, как в формуле (I), и R11 такой, как в формуле (IA).
ПРЕДПОЧТИТЕЛЬНЫЕ ФОРМЫ ОСУЩЕСТВЛЕНИЯ
Приведенные ниже примеры служат для иллюстрации изобретения, но примеры не следует рассматривать как ограничивающие объем изобретения.
Примеры
Структуру соединения идентифицировали с помощью ЯМР и/или МС. Химические сдвиги ЯМР (δ) приведены в млн-1. ЯМР определяли с помощью прибора Bruker AVANCE-400. Растворителями являлись дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD) с тетраметилсиланом (ТМС) в качестве внутреннего стандарта.
МС определяли с помощью масс-спектрометра FINNIGAN LCQAd (ИЭР) (изготовитель: Thermo, тип: Finnigan LCQ advantage MAX);
ВЭЖХ определяли на спектрометре жидкостной хроматографии высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и на спектрометре жидкостной хроматографии высокого давления Waters 2695-2996 (хроматографическая колонка Gimini С18 150×4,6 мм).
IC50 определяли на NovoStar ELISA (BMG Co. German).
В качестве тонкослойного силикагеля использовали пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер пластин, используемых в ТСХ, составлял от 0,15 мм до 0,2 мм, а размер пластин, используемых для очистки продукта, составлял от 0,4 мм до 0,5 мм.
При колоночной хроматографии в качестве носителя, как правило, использовали силикагель Yantai Huanghai с размером ячеек от 200 до 300 меш.
Известный исходный материал по изобретению может быть получен общепринятым способом синтеза предшествующего уровня техники или приобретен от фирмы ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc или Dari chemical Company и т.д.
Если не указано иное, нижеописанные реакционные смеси помещали в атмосферу азота или в атмосферу аргона.
Термин "атмосфера азота" или "атмосфера аргона" относится к тому, что реакционная колба оборудована 1 л баллоном азота или аргона.
Термин "атмосфера водорода" относится к тому, что реакционная колба оборудована 1 л баллоном водорода.
Реакции гидрогенизации под давлением проводили с помощью спектрометра гидрогенизации Parr 3916ЕКХ и генератора водорода QL-500.
При реакциях гидрогенизации реакционную систему обычно помещали в вакуум и заполняли водородом, повторяя вышеуказанную операцию три раза.
Препаративная хроматография ВЭЖХ: Gilson GX-281.
Микроволновые реакции проводили в микроволновом реакторе СЕМ Discover-S 908860.
Если не указано иное, раствор, используемый в описанных ниже реакциях, относится к водному раствору.
Если не указано иное, температурой реакций в описанных ниже реакциях является комнатная температура.
Комнатная температура представляет собой предельно допустимую температуру окружающей среды, которая составляет 20°С-30°С.
За ходом реакций примеров следили с помощью тонкослойной хроматографии (ТСХ), хроматографическая система растворителей включала: дихлорметан и метанол, гексан и этилацетат, петролейный эфир и этилацетат, ацетон. Соотношение объемов растворителей регулировали в соответствии с полярностью соединений.
Система элюирования очистки соединений колоночной хроматографией и тонкослойной хроматографией включала: А: систему дихлорметана и метанола, В: систему гексана и этилацетата, С: систему этилацетата и метанола, D: гексан, Е: этилацетат. Соотношение объемов растворителей регулировали в соответствии с полярностью соединений, и иногда также добавляли небольшое количество щелочного агента, такого как триэтиламин, или кислотного агента, такого как уксусная кислота.
Примеры получения:
Пример 1
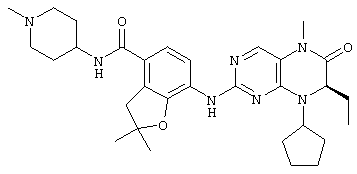
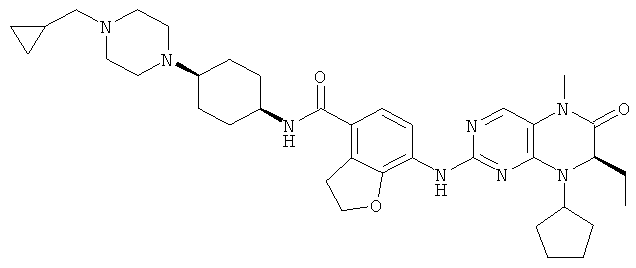
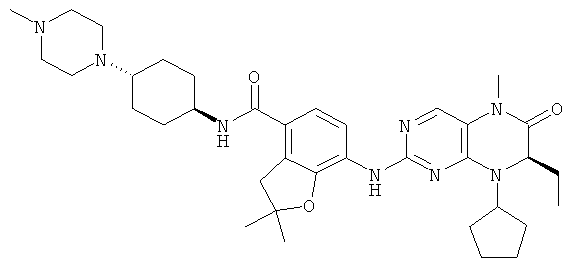
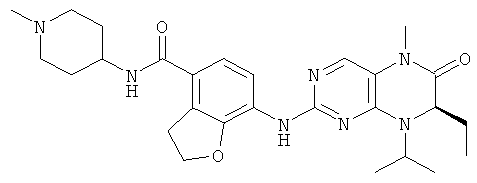
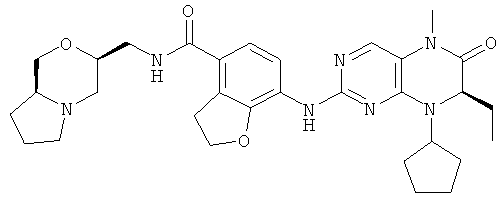
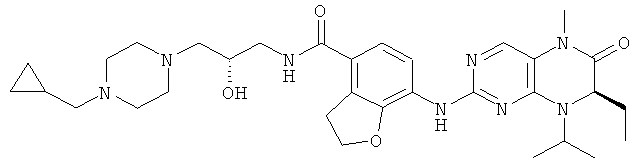
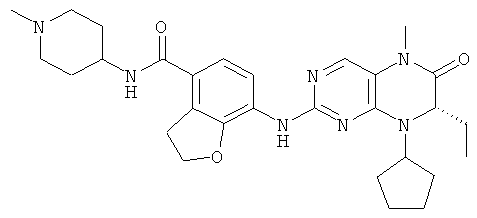
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(1-метилпиперидил)-2,3-дигидробензофуран-4-карбоксамид
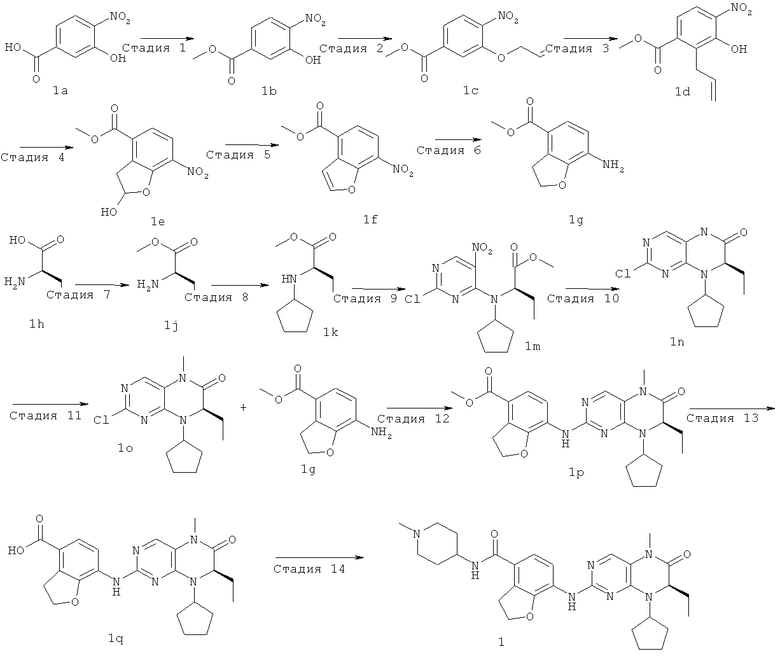
Стадия 1
Метил-3-гидрокси-4-нитробензоат
3-Гидрокси-4-нитробензойную кислоту 1а (3,17 г, 17,32 ммоль) растворяли в 40 мл безводного метанола. Реакционный раствор охлаждали до 0°С и добавляли по каплям тионилхлорид (3,09 г, 25,98 ммоль) при перемешивании. После завершения добавления полученный в результате раствор нагревали до образования флегмы в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, экстрагировали этилацетатом (200 мл). Объединенную органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия (100 мл×3), насыщенным раствором хлорида натрия (100 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, метил 3-гидрокси-4-нитробензоата 1b (3,30 г в виде желтого твердого вещества), выход: 96,7%.
МС m/z(ИЭР): 195,8 [М-1]
Стадия 2
Метил-3-аллилокси-4-нитробензоат
Метил-3-гидрокси-4-нитробензоат 1b (7 г, 35,50 ммоль) растворяли в 100 мл безводного ацетонитрила с последующим добавлением карбоната калия (14,70 г, 106,50 ммоль) и 3-бромпропена (6,2 мл, 71 ммоль) последовательно, реакционную смесь нагревали до образования флегмы в течение 3 часов при перемешивании. Полученную в результате смесь фильтровали и фильтрат концентрировали при пониженном давлении. К сырому остатку добавляли 150 мл этилацетата, последовательно промывали водой (100 мл×3) и насыщенным раствором хлорида натрия (100 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-3-аллилокси-4-нитробензоата 1 с (7,45 г, выход: 89,0%) в виде желтого твердого вещества.
МС m/z(ИЭР): 235,9 [М-1]
Стадия 3
Метил-2-аллил-3-гидрокси-4-нитробензоат
Метил-3-аллилокси-4-нитробензоат 1 с (6 г, 25,30 ммоль) добавляли в трехгорлую колбу, нагревали до 190°С в течение 3 часов и охлаждали до комнатной температуры. К полученной в результате смеси добавляли 150 мл этилацетата, концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-2-аллил-3-гидрокси-4-нитробензоата 1d (4,06 г, выход: 68,0%) в виде желтой жидкости.
МС m/z(ИЭР): 235,9 [М-1]
Стадия 4
Метил 2-гидрокси-7-нитро-2,3-дигидробензофуран-4-карбоксилат Метил 2-аллил-3-гидрокси-4-нитробензоат 1d (5,39 г, 22,70 ммоль) растворяли в 110 мл смеси растворителей дихлорметана и метанола (об./об. = 10:1), реакционный раствор охлаждали до -78°С и заполняли оксоном. После перемешивания в течение 50 минут оксон удаляли из заполнения. Реакционную смесь перемешивали еще в течение 20 минут, затем заполняли аргоном и перемешивали в течение 10 минут. Добавляли трифенилфосфин (17,80 г, 68,10 ммоль). После удаления бани с сухим льдом полученной в результате смеси давали нагреться до комнатной температуры и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли к нему 200 мл этилацетата, последовательно промывали водой (100 мл×3) и насыщенным раствором хлорида натрия (100 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-2-гидрокси-7-нитро-2,3-дигидробензофуран-4-карбоксилата 1е (5,20 г, выход: 96,0%) в виде коричневого твердого вещества.
МС m/z(ИЭР): 237,9 [М-1]
Стадия 5
Метил-7-нитробензофуран-4-карбоксилат
Метил-2-гидрокси-7-нитро-2,3-дигидробензофуран-4-карбоксилат 1е (2 г, 8,40 ммоль) суспендировали в 250 мл 85% фосфорной кислоты, перемешивали в течение 10 минут, помещали в масляную баню на 100°С и перемешивали еще в течение 20 минут. К полученной в результате реакционной смеси добавляли 50 мл воды, экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу последовательно промывали насыщенным раствором карбоната натрия (50 мл×3) и насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-7-нитробензофуран-4-карбоксилата 1f (0,63 г, выход: 34,0%) в виде светло-желтого твердого вещества.
МС m/z(ИЭР): 220,7 [М-1]
Стадия 6
Метил-7-амино-2,3-дигидробензофуран-4-карбоксилат Метил-7-нитробензофуран-4-карбоксилат 1f (820 мг, 3,70 ммоль) растворяли в 150 мл метанола в бане лед-вода, добавляли (164 мг, 10%) палладий/углерод, метанол (0,3 мл). Полученный в результате раствор подвергали гидрогенизации в течение 16 часов при 3 атмосферах при комнатной температуре. Полученную в результате смесь фильтровали и промывали 50 мл метанола, концентрировали при пониженном давлении. Сырой остаток перекристаллизовали 25 мл смеси растворителей этилацетата и н-гексана (об./об. = 1:4) с получением соединения, указанного в заголовке, метил-7-амино-2,3-дигидробензофуран-4-карбоксилата 1g (446 мг, выход: 62,0%) в виде серого твердого вещества.
МС m/z(ИЭР): 194,1 [М+1]
Стадия 7 Метил(2.R)-2-аминобутаноат
(R)-2-Аминобутановую кислоту 1h (10 г, 0,10 моль) растворяли в 50 мл метанола. Раствор охлаждали до -10°С в бане лед-соль, добавляли по каплям тионилхлорид (13 мл, 0,17 моль). После завершения добавления полученную в результате смесь нагревали до образования флегмы в течение 1 часа при перемешивании и охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, метил(2.Рч)-2-аминобутаноата 1j в виде бесцветной маслянистой жидкости, которое использовали в следующей стадии без дальнейшей очистки.
МС m/z(ИЭР): 118,0 [М+1]
Стадия 8
Метил-(2R)-2-(циклопентиламино)бутаноат
Сырое соединение метил-(2R)-2-аминобутаноат 1j (11,24 г, 0,10 моль) и циклопентанон (8,24 г, 0,10 моль) растворяли в 150 мл дихлорметана, перемешивали в течение 1,5 часов с последующим добавлением ацетата натрия (8,04 г, 0,10 моль) и триацетоксиборгидрида натрия (30,52 г, 0,14 моль) и перемешивали в течение 3 часов. Реакционный раствор наливали в 150 мл 10% раствора бикарбоната натрия, экстрагировали дихлорметаном (100 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (100 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил(2.R)-2-(циклопентиламино)бутаноата 1к (6,04 г, выход: 34,0%) в виде светлой жидкости.
МС m/z(ИЭР): 186,1 [М+1]
Стадия 9
Метил-(2R)-2-[(2-хлор-5-нитропиримидин-4-ил)-(1-циклопентил)амино]бутаноат
Метил-(2R)-2-(циклопентиламино)бутаноат 1k (2,50 г, 13,50 ммоль) и бикарбонат натрия (4,54 г, 54 ммоль) растворяли в 100 мл циклогексана, перемешивали в течение 30 минут с последующим добавлением 2,4-дихлор-5-нитропиримидина (2,88 г, 14,84 ммоль), нагревали до 60°С и перемешивали еще в течение 12 часов. Реакционную смесь фильтровали, промывали дихлорметаном (50 мл), и фильтрат концентрировали при пониженном давлении, полученный в результате остаток перекристаллизовали 150 мл смеси растворителей этилацетата и н-гексана (об./об. = 1:4) с получением соединения, указанного в заголовке, метил-(2R)-2-[(2-хлор-5-нитропиримидин-4-ил)-(1-циклопентил)амино]бутаноата 1m (3,36 г, выход: 72,6%) в виде светло-желтого твердого вещества.
МС m/z(ИЭР): 343,1 [М+1]
Стадия 10
(7R)-2-Хлор-8-циклопентил-7-этил-7,8-дигидро-5H-птеридин-6-он
Метил-(2R)-2-[(2-хлор-5-нитропиримидин-4-ил)-циклопентиламино]бутаноат 1m (1 г, 3 ммоль) растворяли в 10 мл уксусной кислоты с последующим добавлением никеля Ренея (0,50 г), фильтровали с водородом три раза, затем нагревали до 75-80°С и перемешивали в течение 12 часов. Реакционную смесь фильтровали, промывали дихлорметаном (50 мл). Фильтрат концентрировали при пониженном давлении, добавляли 100 мл этилацетата, последовательно промывали водой (50 мл×3) и насыщенным раствором хлорида натрия (50 мл×3). Объединенную органическую фазу высушивали над безводным сульфатом магния, фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (7R)-2-хлор-8-циклопентил-7-этил-7,8-дигидро-5H-птеридин-6-она 1n (0,56 г, выход: 66,7%) в виде белого твердого вещества.
МС m/z (ИЭР): 281,2 [М+1]
Стадия 11
(7R)-2-хлор-8-циклопентил-7-этил-5-метил-5H-птеридин-6-он
(7R)-2-Хлор-8-циклопентил-7-этил-7,8-дигидро-5Н-птеридин-6-он 1n (3,50 г, 12,50 ммоль) растворяли в 80 мл ацетона с последующим добавлением метил-пара-толуолсульфоната (3,40 г, 18,70 ммоль) и карбоната калия (3,45 г, 25 ммоль). Полученную в результате смесь нагревали до образования флегмы в течение 2 часов при перемешивании, а затем охлаждали до комнатной температуры. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (7R)-2-хлор-8-циклопентил-7-этил-5-метил-5Н-птеридин-6-она 1о (3,40 г, выход: 93,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 295,4 [М+1]
Стадия 12
Метил-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксилат
(7R)-2-Хлор-8-циклопентил-7-этил-5-метил-5Н-птеридин-6-он 1о (670 мг, 2,30 ммоль), метил-7-амино-2,3-дигидробензофуран-4-карбоксилат 1g (440 мг, 2,30 ммоль) и пара-толуолсульфоновую кислоту (700 мг, 3,68 ммоль) растворяли в 25 мл 3-метил-2-пентанола. Полученный в результате раствор нагревали до образования флегмы в течение 6 часов при перемешивании. К реакционному раствору добавляли 25 мл насыщенного раствора бикарбоната натрия, экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу последовательно промывали водой (50 мл×3) и насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксилата 1р (0,77 г, выход: 74,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 452,3 [М+1]
Стадия 13
7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин- 2-ил]амино]-2,3-дигидробензофуран-4-карбоновая кислота
Метил-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксилат 1р (5 г, 11 ммоль) растворяли в 180 мл смешанного раствора 1 моль/л раствора гидроксида лития и метанола (об./об. = 1:1). Полученную в результате смесь нагревали до образования флегмы в течение 3 часов при перемешивании. К реакционному раствору добавляли 50 мл воды, концентрировали при пониженном давлении и экстрагировали этилацетатом (50 мл×3). 1 М соляную кислоту добавляли по каплям до доведения рН водной фазы до 2 с образованием в результате осадка. Этот осадок фильтровали и высушивали с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновой кислоты 1q (4,70 г, выход: 97,0%) в виде белого твердого вещества.
МС m/z(ИЭР): 436,3 [М-1]
Стадия 14
7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(1-метилпиперидил)-2,3-дигидробензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (300 мг, 0,68 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (148 мг, 0,46 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением диизопропилэтиламина (131 мг, 1 ммоль). Реакционный раствор перемешивали в течение 10 минут с последующим добавлением 1-метилпиперидин-4-иламина (52 мг, 0,46 ммоль), и перемешивали еще в течение 3 часов. К реакционной смеси последовательно добавляли 30 мл насыщенного раствора хлорида натрия и 30 мл дихлорметана, затем разделяли. Органическую фазу последовательно промывали насыщенным раствором карбоната натрия (50 мл×2), водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(1-метилпиперидил)-2,3-дигидробензофуран-4-карбоксамида 1 (194 мг, выход: 79%) в виде белого твердого вещества.
МС m/z (ИЭР): 534,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.28 (d, 1Н), 7.67 (s, 1Н), 7.10-7.00 (m, 2Н), 5.89 (d, 1Н), 4.68 (t, 2Н), 4.47 (t, 1Н), 4.21 (dd, 1Н), 4.07-3.91 (m, 1Н), 3.58 (t, 2Н), 3.32 (s, 3Н), 2.91 (d, 2Н), 2.36 (s, 3Н), 2.24 (t, 2Н), 2.16-1.83 (m, 4Н), 1.86-1.78 (m, 4Н), 1,76-1.62 (m, 6Н), 0.88 (t, 3Н).
Пример 2
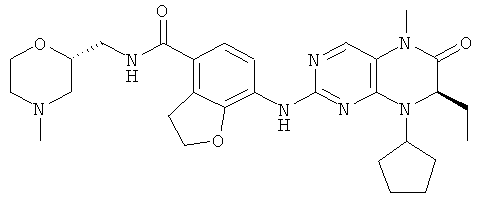
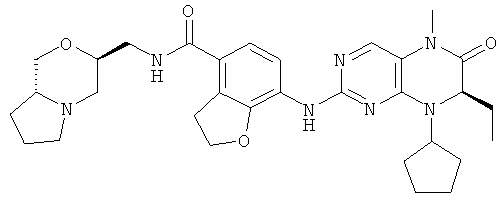
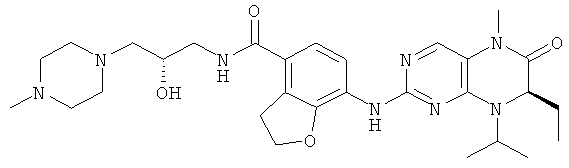
7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(4-метилморфолин-2-ил)метил]-2,3-дигидробензофуран-4-карбоксамид

Стадия 1
3-(Бензил(2-гидроксиэтил)амино)-2-хлорпропаннитрил
2-(Бензиламино)этанол 2a (5 г, 0,03 моль) и 2-хлорпропеннитрил (3 г, 0,03 моль) растворяли в 50 мл безводного этилового эфира в бане лед-вода, нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, 3-(бензил(2-гидроксиэтил)амино)-2-хлорпропаннитрила 2b (7 г) в виде красной маслянистой жидкости, который использовали в следующей стадии без дальнейшей очистки.
Стадия 2 4-Бензилморфолин-2-карбонитрил Сырое соединение 3-(бензил(2-гидроксиэтил)амино)-2-хлорпропаннитрил 2b (8 г, 0,03 моль) растворяли в 70 мл тетрагидрофурана с последующим добавлением трет-бутанолята калия (5,50 г, 0,05 моль) в бане лед-вода. Полученный в результате раствор перемешивали в течение 2 часов, нагревали до образования флегмы в течение 1 часа, добавляли 50 мл насыщенного раствора карбоната натрия и экстрагировали этилацетатом (40 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 4-бензилморфолин-2-карбонитрила 2 с (2,71 г, выход: 41,0%) в виде желтой маслянистой жидкости.
МС m/z(ИЭР): 203,2 [М+1]
Стадия 3
(4-Бензилморфолин-2-ил)метанамин
4-Бензилморфолин-2-карбонитрил 2с (0,80 г, 4 ммоль) растворяли в 30 мл метанола с последующим добавлением никеля Ренея (0,50 г), заполняли водородом два раза, и полученный в результате раствор перемешивали в течение 12 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (4-бензилморфолин-2-ил)метанамина 2d (0,60 г) в виде бесцветной маслянистой жидкости, который использовали на следующей стадии без дальнейшей очистки.
Стадия 4
N-[(4-Бензилморфолин-2-ил)метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
Сырое соединение (4-бензилморфолин-2-ил)метанамин 2d (141 мг, 0,69 ммоль), 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (300 мг, 0,69 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (221 мг, 0,69 ммоль) и диизопропилэтиламин (260 мл, 1,52 ммоль) растворяли в 40 мл дихлорметана, перемешивали в течение 12 часов. К полученной в результате смеси добавляли 50 мл насыщенного раствора бикарбоната натрия, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[(4-бензилморфолин-2-ил)метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 2е (190 мг, выход: 44,0%) в виде белого твердого вещества.
МС m/z(ИЭР): 626,5 [М+1]
Стадия 5
7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(морфолин-2-илметил)-2,3-дигидробензофуран-4-карбоксамид N-[(4-бензилморфолин-2-ил)метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид 2е (0,19 г, 0,30 ммоль) растворяли в 30 мл метанола с последующим добавлением палладия/углерода (40 мг, 10%), заполняли водородом два раза. Реакционный раствор перемешивали в течение 2 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(морфолин-2-илметил)-2,3-дигидробензофуран-4-карбоксамида 2f (163 мг) в виде белого твердого вещества, который использовали в следующей стадии без дальнейшей очистки.
Стадия 6
7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(4-метилморфолин-2-ил)метил]-2,3-дигидробензофуран-4-карбоксамид
Сырое соединение 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(морфолин-2-илметил)-2,3-дигидробензофуран-4-карбоксамид 2f (163 мг, 0,30 ммоль) растворяли в 60 мл смеси растворителей ацетонитрила и воды (об./об. = 1:1) в бане лед-вода, реакционный раствор охлаждали до 0°С с последующим добавлением формальдегида (18 мг, 0,60 ммоль) и триацетоксиборгидрида натрия (191 мг, 0,90 ммоль), медленно подогревали до комнатной температуры и перемешивали в течение 2 часов. К полученной в результате смеси добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 9-10, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(4-метилморфолин-2-ил)метил]-2,3-дигидробензофуран-4-карбоксамида 2 (62 мг, выход: 38,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 550,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.67 (s, 1Н), 7.13 (d, 1Н), 7.04 (s, 1Н), 6.46-6.35 (m, 1Н), 4.68 (t, 2Н), 4.47 (t, 1Н), 4.21 (dd, 1Н), 3.96-3.86 (m, 1Н), 3.78-3.66 (m, 3Н), 3.59 (t, 2H), 3.37 (td, 1H), 3.32 (s, 3Н), 2.80 (d, 1H), 2.69 (d, 1H), 2.32 (s, 3Н), 2.21-2.07 (m, 2H), 2.04-1.90 (m, 2H), 1.89-1.76 (m, 4H), 1.76-1.62 (m, 4н), 0.87 (t, 3H).
Пример 3
[(1R,2R,3R,4R,5R)-4-Ацетокси-5-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]-2-диметиламиноциклогексил]ацетат

Стадия 1
7-Оксабицикло[4.1.0]гепт-3-ен
1,4-Циклогександиен 3а (8 г, 0,10 моль), 75% мета-хлорпербензойную кислоту (16,40 г, 0,10 моль) и бикарбонат натрия (8,80 г, 0,11 моль) растворяли в 400 мл дихлорметана, перемешивали в течение 12 часов. К полученному в результате раствору добавляли 100 мл дихлорметана и 20 мл воды, экстрагировали и разделяли. Органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при нормальном давлении, сырое масло перегоняли, и фракцию 119-122°С собирали с получением соединения, указанного в заголовке, 7-оксабицикло[4.1.0]гепт-3-ена 3b (4 г, выход: 41,7%) в виде бесцветной маслянистой жидкости.
Стадия 2
(1R,6R)-6-Азидоциклогекс-3-ен-1-ол
7-Оксабицикло[4.1.0]гепт-3-ен 3b (3,50 г, 36,40 ммоль), хлорид аммония (4,90 г, 91 ммоль) и азид натрия (5,90 г, 91 ммоль) растворяли в 200 мл смеси растворителей метанола и воды (об./об. = 9:1), реакционный раствор нагревали до образования флегмы в течение 2 часов. К полученному в результате раствору последовательно добавляли 500 мл этилацетата, 200 мл этилового эфира и 50 мл воды, разделяли. Органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (1R,6R)-6-азидоциклогекс-3-ен-1-ола 3с (3 г, выход: 60,0%) в виде желтого масла.
Стадия 3
(1S,3R,4R)-4-Азидо-7-оксабицикло[4.1.0]гептан-3-ол
(1R,6R)-6-Азидоциклогекс-3-ен-1-ол 3с (500 мг, 3,60 ммоль) растворяли в 150 мл дихлорметана с последующим добавлением бикарбоната натрия (600 мг, 7.20 ммоль) и мета-хлорпербензойную кислоту (2,40 г, 9,60 ммоль), реакционный раствор перемешивали в течение 12 часов. Полученный в результате раствор последовательно промывали насыщенным раствором бикарбоната натрия (50 мл), насыщенным раствором тиосульфата натрия (50 мл) и насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении, остаток очищали ВЭЖХ с получением соединения, указанного в заголовке, (1S,3R,4R)-4-азидо-7-оксабицикло[4.1.0]гептан-3-ола 3d (360 мг, выход: 64,6%) в виде белого твердого вещества.
Стадия 4
(1R,2R,4R,5R)-2-Амино-5-азидоциклогексан-1,4-диол
(1S,3R,4R)-4-Азидо-7-оксабицикло[4.1.0]гептан-3-ол 3d (600 мг, 3,87 ммоль) растворяли в 20 мл этанола с последующим добавлением 10 мл водного аммиака. Полученный в результате раствор нагревали до образования флегмы в течение 4 часов, затем охлаждали до комнатной температуры и перемешивали еще в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (1R,2R,4R,5R)-2-амино-5-азидоциклогексан-1,4-диола 3е (665 мг, в виде желтого масла), который использовали в следующей стадии без дальнейшей очистки.
МС m/z(ИЭР): 173,1 [М+1]
Стадия 5
трет-Бутил-N-[(1R,2R,4R,5R)-4-азидо-2,5-дигидроксициклогексил]карбамат
Сырое соединение (1R,2R,4R,5R)-2-амино-5-азидоциклогексан-1,4-диол 3е (665 мг, 3,87 ммоль), ди-трет-бутилдикарбонат (1,10 г, 5 ммоль) и триэтиламин (1,6 мл, 11,60 ммоль) растворяли в 40 мл дихлорметана, затем реакционный раствор перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли 50 мл этилацетата и 20 мл воды, затем добавляли по каплям 1 М соляную кислоту до доведения рН до 3-4, разделяли. Органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении, остаток очищали ВЭЖХ с получением соединения, указанного в заголовке, трет-бутил-N-[(1R,2R,4R,5R)-4-азидо-2,5-дигидроксициклогексил]карбамата 3f (350 мг, выход: 35,0%) в виде белого твердого вещества.
Стадия 6
[(1R,2R,4R,5R)-4-Ацетокси-2-азидо-5-(трет-бутоксикарбониламино)циклогексил]ацетат
трет-Бутил-N-[(1R,2R,4R,5R)-4-азидо-2,5-дигидроксициклогексил]карбамат 3f (350 мг, 1,28 ммоль) растворяли в 10 мл уксусного ангидрида с последующим добавлением 1 мл пиридина и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли 50 мл этилацетата и 10 мл воды, затем добавляли по каплям 1 М соляную кислоту до доведения рН до 3-4, разделяли, органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, [(1R,2R,4R,5R)-4-ацетокси-2-азидо-5-(трет-бутоксикарбониламино)циклогексил]ацетата 3g (450 мг, выход: 100,0%) в виде желтого масла.
МС m/z (ИЭР): 379,2 [М+23]
Стадия 7
[(1R,2R,4R,5R)-4-Ацетокси-2-амино-5-(трет-бутоксикарбониламино)циклогексил]ацетат
[(1R,2R,4R,5R)-4-Ацетокси-2-азидо-5-(трет-бутоксикарбониламино)циклогексил]ацетат 3g (450 мг, 1,26 ммоль) растворяли в 30 мл метанола с последующим добавлением палладия/углерода (50 мг, 10%), заполняли водородом три раза. Реакционную смесь перемешивали в течение 1,5 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, [(1R,2R,4R,5R)-4-ацетокси-2-амино-5-(трет-бутоксикарбониламино)циклогексил]ацетата 3h (350 мг, выход: 84,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 331,0 [М+1]
Стадия 8
[(1R,2R,4R,5R)-4-Ацетокси-5-(трет-бутоксикарбониламино)-2-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]циклогексил]ацетат
[(1R,2R,4R,5R)-4-Ацетокси-2-амино-5-(трет-бутоксикарбониламино)циклогексил]ацетат 3h (100 мг, 0,30 ммоль), 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (132 мг, 0,30 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (96 мг, 0,30 ммоль) и диизопропилэтиламин (0,1 мл, 0,66 ммоль) растворяли в 30 мл дихлорметана. Полученный в результате раствор перемешивали в течение 2 часов, добавляли 10 мл воды, экстрагировали дихлорметаном (30 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (20 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, [(1R,2R,4R,5R)-4-ацетокси-5-(трет-бутоксикарбониламино)-2-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]циклогексил]ацетат 3j (130 мг, выход: 58,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 750,6 [М+1]
Стадия 9
[(1R,2R,4R,5R)-4-Ацетокси-2-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]-5-диметиламиноциклогексил]ацетат
[(1R,2R,4R,5R)-4-Ацетокси-5-(трет-бутоксикарбониламино)-2-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]циклогексил]ацетат 3j (130 мг, 0,17 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением 20 мл 4 М раствора 1,4-диоксана в растворе соляной кислоты, перемешивали в течение 0,5 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в 60 мл смеси растворителей ацетонитрила и 30 мл воды (об./об. = 1:1), добавляли 30% формальдегид (68 мг, 0,68 ммоль), перемешивали 0,5 часа с последующим добавлением триацетоксиборгидрида натрия (216 мг, 1,02 ммоль), перемешивали в течение 12 часов. К полученному в результате раствору добавляли 10 мл водного аммиака, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, [(1R,2R,4R,5R)-4-ацетокси-2-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]-5-диметиламиноциклогексил]ацетата 3 (40 мг, выход: 34,8%) в виде белого твердого вещества.
МС m/z (ИЭР): 678,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.27 (d, 1Н), 7.67 (s, 1Н), 7.10-7.01 (m, 2Н), 5.32-5.27 (m, 1Н), 4.67 (t, 2Н), 4.46 (d, 2Н), 4.21 (dd, 1Н), 3.59 (dd, 2Н), 3.32 (s, 3Н), 2.45-2.30 (m, 6Н), 2.27-2.17 (m, 2Н), 2.18-2.08 (m, 6Н), 2.05-1.91 (m, 7Н), 1.86 (d, 4Н), 1.71 (dd, 4Н), 0.88 (t, 3Н).
Пример 4
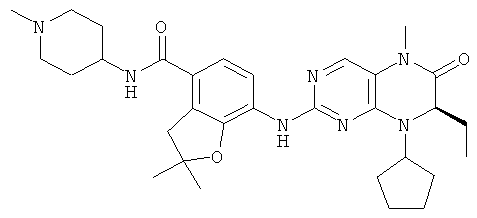
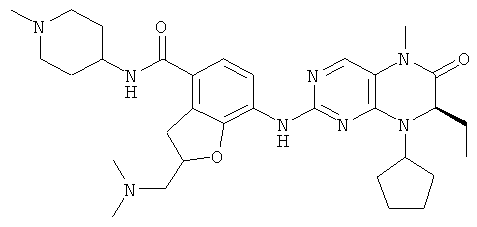
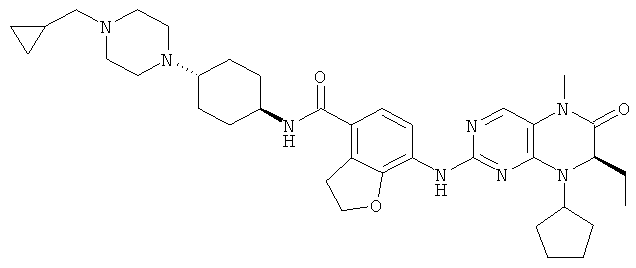
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-N-(1-метил-4-пиперидил)-3Н-бензофуран-4-карбоксамид
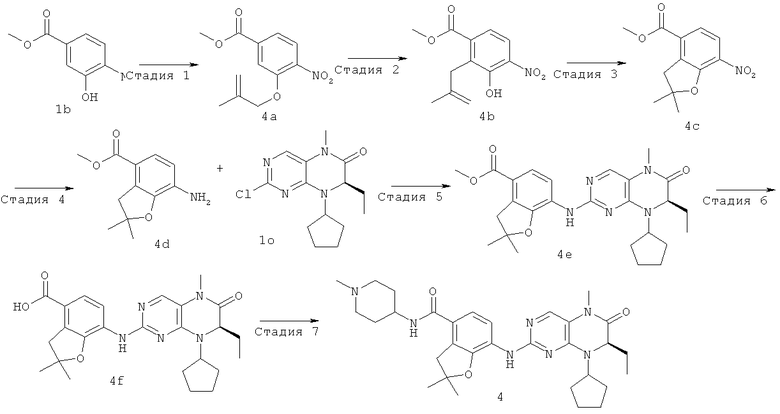
Стадия 1
Метил-3-(2-метилаллилокси)-4-нитробензоат
Трифенилфосфин (7,87 г, 30 ммоль) растворяли в 80 мл тетрагидрофурана с последующим добавлением диэтилазодикарбоксилата (5,23 г, 30 ммоль) в бане с сухим льдом. Реакционную смесь перемешивали 30 минут, добавляли по каплям раствор метил-3-гидрокси-4-нитробензоата 1b (2,96 г, 15 ммоль) в тетрагидрофуране и перемешивали 10 минут с последующим добавлением 2-метилаллилового спирта (1,41 г, 19,50 ммоль), затем перемешивали еще в течение 1 часа. Полученную в результате смесь нагревали до комнатной температуры и перемешивали в течение 12 часов. К полученной в результате смеси добавляли 10 мл воды, добавляли по каплям 1 М соляную кислоту до доведения рН до 2-3 и экстрагировали этилацетатом (50 мл×3). К объединенной органической фазе добавляли по каплям 1 М раствор гидроксида натрия до доведения рН до 9-10, экстрагировали этилацетатом (50 мл×2). Объединенную органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-3-(2-метилаллилокси)-4-нитробензоата 4а (3 г, выход: 79,6%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 252,1 [М+1]
Стадия 2
Метил-3-гидрокси-2-(2-метилаллил)-4-нитробензоат
Метил-3-(2-метилаллилокси)-4-нитробензоат 4а (4,52 г, 18 ммоль) нагревали до 190°С и перемешивали в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры с последующим добавлением 50 мл дихлорметана. Реакционный раствор концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-3-гидрокси-2-(2-метилаллил)-4-нитробензоата 4b (2,60 г, выход: 57,7%) в виде желтого масла. МС m/z (ИЭР): 252,1 [М+1]
Стадия 3
Метил-2,2-диметил-7-нитро-3Н-бензофуран-4-карбоксилат
Метил-3-гидрокси-2-(2-метилаллил)-4-нитробензоат 4b (2,58 г, 10,30 ммоль) растворяли в 50 мл 1,2-дихлорпентана с последующим добавлением трифторметансульфоновой кислоты (77 мг, 0,50 ммоль). Реакционный раствор перемешивали в течение 2 часов и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-2,2-диметил-7-нитро-3Н-бензофуран-4-карбоксилата 4 с (0,83 г, выход: 32,1%) в виде желтого твердого вещества.
МС m/z (ИЭР): 252,1 [М+1]
Стадия 4
Метил-7-амино-2,2-диметил-3Н-бензофуран-4-карбоксилат
2,2-Диметил-7-нитро-3Н-бензофуран-4-карбоксилат 4 с (1,20 г, 4,77 ммоль) и порошок железа (0,80 г, 14,32 ммоль) растворяли в 25 мл уксусной кислоты. Реакционную смесь перемешивали в течение 12 часов. К полученной в результате смеси добавляли карбонат натрия и твердый бикарбонат натрия до доведения рН до 7-8, экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу последовательно промывали водой (30 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-7-амино-2,2-диметил-3Н-бензофуран-4-карбоксилата 4d (789 мг, выход: 74,6%) в виде белого твердого вещества.
МС m/z (ИЭР): 222,2 [М+1]
Стадия 5
Метил-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н -птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксилат
Метил-7-амино-2,2-диметил-3Н-бензофуран-4-карбоксилат 4d (770 мг, 3,48 ммоль) растворяли в 20 мл 1,3-диметилбутанола с последующим добавлением (7R)-2-хлор-8-циклопентил-7-этил-5-метил-5Н-птеридин-6-она 1о (1,23 г, 4,18 ммоль) и пара-толуолсульфоновой кислоты (1,06 г, 5,57 ммоль) последовательно. Реакционный раствор нагревали до образования флегмы в течение 2 часов при перемешивании. К полученному в результате раствору добавляли 50 мл насыщенного раствора бикарбоната натрия, экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу последовательно промывали водой (30 мл), насыщенным раствором хлорида натрия (20 мл) и высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Я-птеридин-2-ил]амино]-2,2-диметил-3H-бензофуран-4-карбоксилата 4е (1,78 г, выход: 100%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 480,4 [М+1]
Стадия 6
7-[[(7Р)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин- 2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновая кислота
Метил-7-[[(7Р)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксилат 4е (1,67 г, 3,48 ммоль) растворяли в 50 мл метанола с последующим добавлением 1 М раствора гидроксида лития (17,4 мл, 17,40 ммоль). Реакционный раствор нагревали до 50°С и перемешивали в течение 12 часов. К полученному в результате раствору добавляли 10 мл воды и добавляли по каплям 1 М соляную кислоту до доведения рН до 2-3. Реакционный раствор концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновой кислоты 4f (1,89 г, выход: 100%) в виде белого твердого вещества.
МС m/z (ИЭР): 464,3 [М-1]
Стадия 7
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-N-(1-метил-4-пиперидил)-3Н-бензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (931 мг, 2 ммоль) растворяли в 50 мл дихлорметана с последующим добавлением 1-метилпиперидил-4-иламина (228 мг, 2 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (642 мг, 2 ммоль) и диизопропилэтиламина (775 мг, 6 ммоль). Реакционный раствор перемешивали в течение 1,5 часов. К полученной в результате смеси добавляли 50 мл насыщенного раствора бикарбоната натрия, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали водой (15 мл), насыщенным раствором хлорида натрия (15 мл), высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-N-(1-метил-4-пиперидил)-3Н-бензофуран-4-карбоксамида 4 (793 мг, выход: 70,8%) в виде белого твердого вещества. МС m/z (ИЭР): 562,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.27 (d, 1Н), 7.67 (s, 1Н), 7.11-7.00 (m, 2Н), 5.85 (d, 1Н), 4.56 (t, 1Н), 4.21 (dd, 1Н), 3.95 (dd, 1Н), 3.38 (s, 2Н), 3.33 (s, 3Н), 2.84 (d, 2Н), 2.32 (s, 3Н), 2.25-2.10 (m, 3Н), 2.08-1.95 (m, 3Н), 1.89-1.75 (m, 4Н), 1.75-1.64 (m, 4н), 1.64-1.54 (m, 2Н), 1.51 (s, 6Н), 0.88 (t, 3Н).
Пример 5
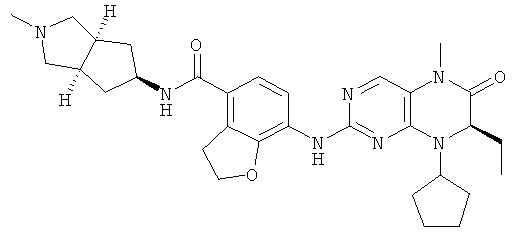
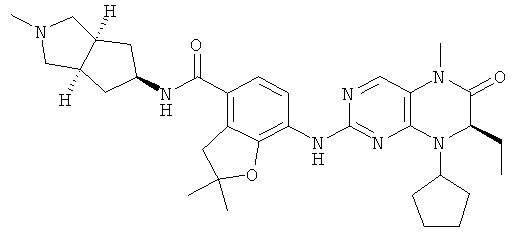
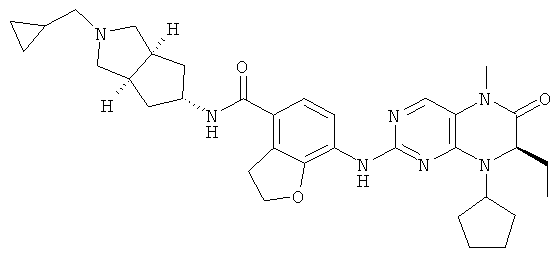
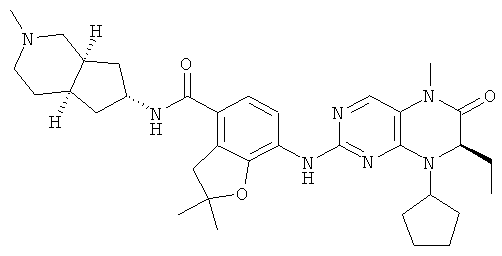
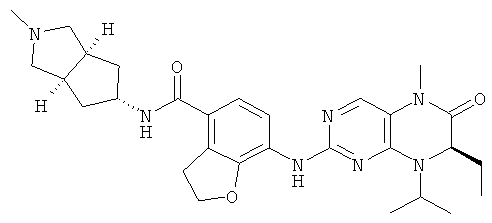
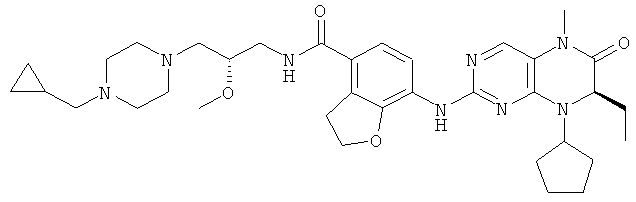
(цис-экзо)-N-2-Метил-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
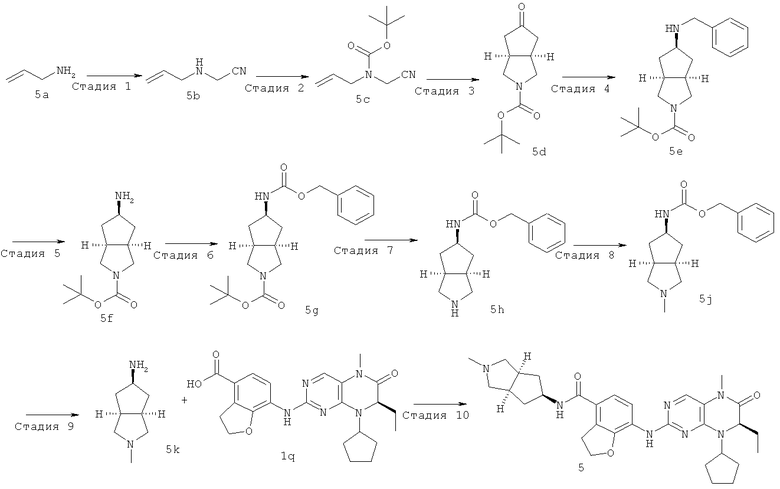
Стадия 1
N-аллил-2-пропиниламин
Пропениламин 5а (225 мл, 3 моль) растворяли в 100 мл раствора 2 моль/л гидроксида натрия, медленно добавляли по каплям пропаргилбромид (89,1 мл, 1 моль) при перемешивании в течение 1 часа, затем нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, N-аллил-2-пропинил-1-амина 5b, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 96,2 [М+1]
Стадия 2
трет-Бутил-N-аллил-N-проп-2-инкарбамат
N-Аллил-2-пропинил-1-амин 5b (90,05 г, 0,95 моль), карбонат калия (130,75 г, 0,95 моль) и ди-трет-бутилдикарбонат (120 г, 0,55 моль) растворяли в 200 мл дихлорметана. Полученный в результате раствор перемешивали в течение 12 часов с последующим добавлением 10 мл воды, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-N-аллил-N-проп-2-инкарбамата 5с (76,97 г, выход: 41,7%).
Стадия 3
трет-Бутил-(цис)-5-оксо-1,3,3a,4,6,6a-гексагидроциклопента[c]пиррол-2-карбоксилат
трет-Бутил-N-аллил-N-проп-2-инкарбамат 5с (16,70 г, 0,86 моль) растворяли в 100 мл дихлорметана с последующим добавлением дикобальта октакарбонила (29,40 г, 0,86 моль) и 31 мл воды. Полученный в результате раствор нагревали до образования флегмы и перемешивали в течение 3 часов, добавляли 10 мл воды. Реакционный раствор концентрировали при пониженном давлении, добавляли 100 мл этилацетата, 100 мл воды и 50 мл 1 М соляной кислоты, экстрагировали этилацетатом (100 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил(цис)-5-оксо-1,3,3а,4,6,6а-гексагидроциклопента[с]пиррол-2-карбоксилата 5d (7,69 г, выход: 40,0%).
Стадия 4
трет-Бутил(цис-экзо)-5-(бензиламино)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-2-карбоксилат
трет-Бутил(цис)-5-оксо-1,3,3а,4,6,6а-гексагидроциклопента[с]пиррол-2-карбоксилат 5d (3,37 г, 15 ммоль), бензиламин (1,60 г, 15 ммоль) и уксусную кислоту (0,90 г, 15 ммоль) растворяли в 60 мл дихлорметана в бане лед-вода, перемешивали в течение 0,5 часа с последующим добавлением триацетоксиборгидрида натрия (6,40 г, 30 ммоль), перемешивали в течение 12 часов. К полученному в результате раствору добавляли 50 мл насыщенного раствора бикарбоната натрия и 100 мл дихлорметана, разделяли. Органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-(цис-экзо)-5-(бензиламино)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-2-карбоксилата 5е (4,70 г, выход: 100%) в виде белого твердого вещества.
МС m/z (ИЭР): 317,3 [М+1]
Стадия 5
трет-Бутил-(цис-экзо)-5-(амино)-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-карбоксилат
трет-Бутил-(цис-экзо)-5-(бензиламино)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-2-карбоксилат 5е (4,70 г, 14,80 ммоль) и уксусную кислоту (1 г, 14,80 ммоль) растворяли в 100 мл метанола с последующим добавлением палладия/углерода (500 мг, 10%), заполняли водородом три раза. Реакционную смесь перемешивали в течение 12 часов и фильтровали. Реакционный раствор концентрировали при пониженном давлении, добавляли 50 мл насыщенного раствора бикарбоната натрия и 100 мл дихлорметана, разделяли. Органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, трет-бутил-(цис-экзо)-5-(амино)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-2-карбоксилата 5f (1,70 г, выход: 51,5%) в виде белого твердого вещества.
Стадия 6
трет-Бутил-(цис-экзо)-5-бензилоксикарбониламино-3,3а,4,5,6,6а- гексагидро-1Н-циклопента[с]пиррол-2-карбоксилат
трет-Бутил-(цис-экзо)-5-(амино)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-2-карбоксилат 5f (1,70 г, 7,50 ммоль) растворяли в 60 мл дихлорметана с последующим добавлением бензилоксихлорида (1,41 г, 8,26 ммоль) и триэтиламина (1,52 г, 15 ммоль), перемешивали в течение 3 часов. К полученному в результате раствору добавляли 50 мл насыщенного раствора бикарбоната натрия и 100 мл дихлорметана. Органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-(цис-экзо)-5-бензилоксикарбониламино-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-2-карбоксилата 5g (1,98 г, выход: 73,3%) в виде бесцветной вязкой жидкости.
Стадия 7
Бензил-(цис-экзо)-N-[1,2,3,3а,4,5,6,6а-октагидроциклопента[с]пиррол-5-ил]карбамат
трет-Бутил-(цис-экзо)-5-бензилоксикарбониламино-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-карбоксилат 5g (1,96 г, 5,44 ммоль) растворяли в 10 мл 1,4-диоксана с последующим добавлением 10 мл 2 М соляной кислоты, нагревали до 50°С, перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, к нему добавляли по каплям 2 М раствор гидроксида натрия до доведения рН до 2, экстрагировали дихлорметаном (50 мл). Органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, бензил-(цис-экзо)-N-[1,2,3,3а,4,5,6,6а-октагидроциклопента[с]пиррол-5-ил]карбамата 5h (635 мг, выход: 45,0%) в виде светло-желтой маслянистой жидкости.
МС m/z (ИЭР): 261,2 [М+1]
Стадия 8
Бензил-(цис-экзо)-N-[2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]карбамат
Бензил-(цис-экзо)-N-[1,2,3,3а,4,5,6,6а-октагидроциклопента[с]пиррол-5-ил]карбамат 5h (635 мг, 2,40 ммоль) растворяли в 20 мл смеси растворителей ацетонитрила и воды (об./об. = 9:1) в бане лед-вода с последующим добавлением 37% водного раствора формальдегида (110 мг, 3,66 ммоль), перемешивали 10 минут, добавляли триацетоксиборгидрид натрия (1,52 г, 7,20 ммоль), перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли 50 мл насыщенного раствора бикарбоната натрия и 100 мл дихлорметана, разделяли. Органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-(цис-экзо)-N-[2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]карбамата 5j (559 мг, выход: 85%) в виде желтой маслянистой жидкости.
Стадия 9
(цис-экзо)-2-Метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амин
Бензил-(цис-экзо)-N-[2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]карбамат 5j (550 мг, 2 ммоль) растворяли в 20 мл метанола с последующим добавлением палладия/углерода (55 мг, 10%), заполняли водородом три раза. Реакционную смесь перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (цис-экзо)-2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амина 5k (246 мг, выход: 87,9%) в виде бесцветной маслянистой жидкости.
МС m/z (ИЭР): 141,2 [М+1]
Стадия 10
(цис-экзо)-N-2-Метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (187 мг, 0,43 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (138 мг, 0,43 ммоль) растворяли в 40 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,2 мл, 0,95 ммоль), перемешивали до тех пор, пока раствор не становился прозрачным, с последующим добавлением 5 мл раствора (цис-экзо)-2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амина 5k (60 мг, 0,43 ммоль) в дихлорметане и перемешивали еще в течение 2 часов. К полученному в результате раствору добавляли 30 мл дихлорметана, последовательно промывали разбавленным водным аммиаком (20 мл), водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-экзо)-N-2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 5 (170 мг, выход: 71,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 560,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.25 (d, 1Н), 7.67 (s, 1Н), 7.12-6.99 (m, 2Н), 4.66 (t, 2Н), 4.59 (dd, 1Н), 4.58-4.35 (m, 1Н), 4.20 (dd, 1Н), 3.66 (t, 2Н), 3.32 (s, 3H), 3.10-2.80 (m, 2Н), 2.80-2.70 (m, 2Н), 2.47 (s, 3H), 2.40-2.25 (m, 2Н), 2.24-2.06 (m, 3H), 2.05-1.92 (m, 2Н), 1.91-1.74 (m, 4Н), 1.73-1.59 (m, 6Н), 0.88 (t, 3H).
Пример 6
(цис-экзо)-N-[2-Метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксамид
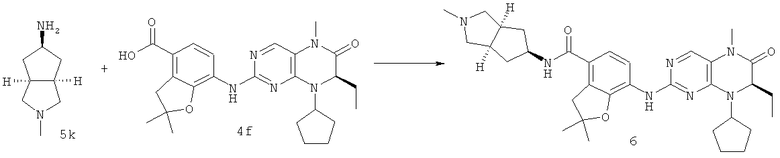
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (163 мг, 0,35 ммоль) растворяли в 15 мл дихлорметана с последующим добавлением (цис-экзо)-2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амина 5к (59 мг, 0,42 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (112 мг, 0,35 ммоль) и диизопропилэтиламина (135 мг, 1,05 ммоль). Реакционный раствор перемешивали в течение 1,5 часов. К полученному в результате раствору добавляли 10 мл насыщенного раствора бикарбоната натрия, экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу последовательно промывали водой (10 мл), насыщенным раствором хлорида натрия (10 мл) и высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-экзо)-N-[2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксамида 6 (40 мг, выход: 19,5%) в виде белого твердого вещества.
МС m/z (ИЭР): 588,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.21 (d, 1Н), 7.65 (s, 1Н), 7.26-7.16 (m, 2Н), 6.99 (s, 1Н), 4.59-4.41 (m, 2Н), 4.22 (dd, 1Н), 3.57 (s, 2Н), 3.43 (s, 2Н), 3.32 (s, 3H), 2.96 (s, 3H), 2.87 (s, 3H), 2.47-2.35 (m, 2Н), 2.14 (s, 3H), 2.06-1.94 (m, 2Н), 1.89-1.63 (m, 8Н),1.51 (s, 6Н), 0.88 (t, 3H).
Пример 7
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-гидроксипропил]-2,3-дигидробензофуран-4-карбоксамид
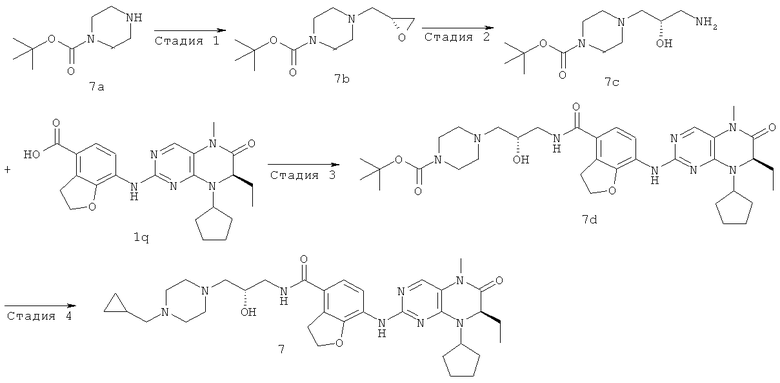
Стадия 1
трет-Бутил-4-[[(2R)-оксиран-2-ил]метил]пиперазин-1-карбоксилат
трет-Бутилпиперазин-1-карбоксилат 7a (3,72 г, 20 ммоль) растворяли в 40 мл этанола с последующим добавлением (R)-2-хлорметилоксирана (1,85 г, 20 ммоль). Реакционный раствор перемешивали в течение 5 часов и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-4-[[(2R)-оксиран-2-ил]метил]пиперазин-1-карбоксилата 7b (3,55 г, выход: 74,0%) в виде бесцветного масла.
МС m/z (ИЭР): 243,2 [М+1]
Стадия 2
трет-Бутил-4-[(2S)-3-амино-2-гидроксипропил]пиперазин-1-карбоксилат трет-Бутил-4-[[(2R)-оксиран-2-ил]метил]пиперазин-1-карбоксилат 7b (3,55 г, 14,70 ммоль) растворяли в 40 мл этанола с последующим добавлением 40 мл водного аммиака, перемешивали в течение 12 часов. Полученный в результате раствор нагревали до 50°С и перемешивали еще в течение 1 часа, затем концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, трет-бутил-4-[(2S)-3-амино-2-гидроксипропил]пиперазин-1-карбоксилата 7 с (3,40 г) в виде светло-желтого твердого вещества, который использовали в следующей стадии без дальнейшей очистки.
Стадия 3
трет-Бутил-4-[(2S)-3-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]-2-гидроксипропил]пиперазин-1-карбоксилат
трет-Бутил-4-[(2S)-3-амино-2-гидроксипропил]пиперазин-1-карбоксилат 7с (355 мг, 1,37 ммоль), 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (600 мг, 1,37 ммоль), O-(бензотриазол-1ил)-N,N,N',N'-тетраметилурония тетрафторборат (440 мг, 1,37 ммоль) и диизопропилэтиламин (390 мг, 3,01 ммоль) растворяли в 40 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору последовательно добавляли 50 мл воды и 10 мл водного аммиака, перемешивали в течение 0,5 часа, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-4-[(2S)-3-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]-2-гидроксипропил]пиперазин-1-карбоксилата 7d (300 мг, выход: 32,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 679,6 [М+1]
Стадия 4
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-гидроксипропил]-2,3-дигидробензофуран-4-карбоксамид
трет-Бутил-4-[(2S)-3-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]-2-гидроксипропил]пиперазин-1-карбоксилат 7d (300 мг, 0,44 ммоль) растворяли в 40 мл дихлорметана. Реакционный раствор заполняли газом хлоридом водорода и перемешивали в течение 0,5 часа. Реакционный раствор концентрировали при пониженном давлении. Полученный в результате остаток растворяли в 41 мл ацетонитрила с последующим добавлением 183 мл триэтиламина, бикарбоната натрия (148 мг, 1,76 ммоль) и бромметилциклопропана (130 мг, 0,97 ммоль). Полученный в результате раствор перемешивали в течение 12 часов и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-Н-[(2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-гидроксипропил]-2,3-дигидробензофуран-4-карбоксамида 7 (200 мг, выход: 72,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 633,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.26 (d, 1Н), 7.65 (s, 1Н), 7.25 (d, 1Н), 7.13 (s, 1Н), 7.04 (s, 1Н), 4.67 (t, 2Н), 4.44 (t, 1Н), 4.21 (dd, 1Н), 4.15-4.08 (m, 1Н), 3.74-3.66 (m, 1Н), 3.59 (t, 2Н), 3.52-3.41 (m, 2Н), 3.31 (s, 3H), 3.41-3.01 (m, 6Н), 2.85-2.52 (m, 4Н), 2.17-2.07 (m, 1Н), 2.01-1.92 (m, 1Н), 1.88-1.76 (m, 4H), 1.74-1.64 (m, 4н), 1.38 (t, 1H), 1.26 (t, 1H), 1.18 (s, 1н), 0.87 (t, 3H), 0.71 (d, 2H), 0.40-0.31 (m, 2н)
Пример 8
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамид
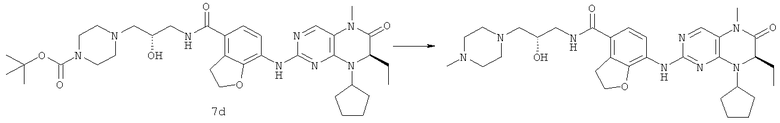
трет-Бутил-4-[(2S)-3-[[7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбонил]амино]-2-гидроксипропил]пиперазин-1-карбоксилат 7d (580 мг, 0,85 ммоль) растворяли в 50 мл дихлорметана с последующим добавлением 20 мл 4 М раствора хлорида водорода в диоксане. Реакционный раствор перемешивали в течение 0,5 часа и концентрировали при пониженном давлении. Полученный в результате остаток растворяли в 40 мл ацетонитрила с последующим добавлением бикарбоната натрия (285 мг, 3,40 ммоль) и метил-пара-толуолсульфоната (316 мг, 1,70 ммоль) и перемешивали в течение 12 часов. К полученному в результате раствору добавляли 50 мл воды и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-Н-[(2S)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамида 8 (150 мг, выход: 30,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 593,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.67 (s, 1Н), 7.15 (d, 1Н), 7.04 (s, 1Н), 6.66-6.54 (m, 1Н), 4.68 (t, 2Н), 4.53-4.41 (m, 1Н), 4.21 (dd, 1Н), 4.02-3.90 (m, 1Н), 3.75-3.65 (m, 1Н), 3.61-3.55 (m, 2Н), 3.45-3.36 (m, 1Н), 3.32 (s, 3H), 2.90-2.76 (m, 2Н), 2.70-2.45 (m, 7Н), 2.38 (s, 3H), 2.20-2.08 (m, 2Н), 2.04-1.92 (m, 2Н), 1.90-1.79 (m, 4Н), 1.77-1.63 (m, 4Н), 0.88 (t, 3H).
Пример 9
(цис-экзо)-N-[2-Метил-3,3a,4,5,6,6a-гексагидро-1H-циклопента[c]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксамид
Стадия 1
трет-Бутил-(цис-экзо)-5-гидрокси-3,3а,4,5,6,6а-гексагидро-1H-циклопента[c]пиррол-2-карбоксилат
трет-Бутил-(цис)-5-оксо-1,3,3а,4,6,6а-гексагидроциклопента[с]пиррол-2-карбоксилат 5d (1,80 г, 8 ммоль) растворяли в 30 мл тетрагидрофурана с последующим добавлением боргидрида натрия (0,60 г, 16 ммоль), перемешивали в течение 12 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора бикарбоната натрия и экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-(цис-экзо)-5-гидрокси-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-карбоксилата 9а (1,64 г, выход: 90,0%) в виде желтой вязкой жидкости.
МС m/z (ИЭР): 228,1 [М+1]
Стадия 2
трет-Бутил-(цис-экзо)-5-метилсульфонилокси-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-карбоксилат
трет-Бутил-(цис-экзо)-5-гидрокси-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-карбоксилат 9а (1,64 г, 7,20 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением метансульфонилхлорида (0,9 мл, 11 ммоль) и триэтиламина (2 мл, 14,40 ммоль) в бане лед-вода, перемешивали в течение 2 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-(цис-экзо)-5-метилсульфонилокси-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-2-карбоксилата 9b (1,76 г, выход: 80,0%) в виде желтой жидкости.
МС m/z (ИЭР): 305,9 [М+1]
Стадия 3
трет-Бутил-(цис-эндо)-5-азидо-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-карбоксилат
трет-Бутил-(цис-экзо)-5-метилсульфонилокси-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-карбоксилат 9b (1,76 г, 5,76 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением азида натрия (0,94 г, 14,40 ммоль), нагревали до 70-80°С и перемешивали в течение 4 часов. К полученному в результате раствору добавляли 5 мл воды и экстрагировали этилацетатом (30 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (30 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-(цис-эндо)-5-азидо-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-2-карбоксилата 9 с (1,15 г, выход: 79,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 253,0 [М+1]
Стадия 4
(цис-эндо)-5-Азидо-1,2,3,3а,4,5,6,6а-октагидроциклопента[с]пиррола гидрохлорид
трет-Бутил-(цис-эндо)-5-азидо-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-2-карбоксилат 9 с (0,62 г, 2,44 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением 10 мл 4 М раствора [1,4]диоксана в растворе соляной кислоты и перемешивали в течение 0,5 часа. Реакционный раствор концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (цис-эндо)-5-азидо-1,2,3,3а,4,5,6,6а-октагидроциклопента[с]пиррола гидрохлорида 9d (0,47 г, выход: 100%) в виде белого твердого вещества.
МС m/z (ИЭР): 153,1 [М+1]
Стадия 5
(цис-эндо)-5-Азидо-2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол
(цис-эндо)-5-Азидо-1,2,3,3а,4,5,6,6а-октагидроциклопента[с]пиррола гидрохлорид 9d (0,45 г, 2,38 ммоль) растворяли в 10 мл смеси растворителей ацетонитрила и воды (об:об = 9:1). Реакционный раствор охлаждали до 0°С с последующим добавлением формальдегида (0,4 мл, 4,76 ммоль) и триацетоксиборгидрида натрия (1,51 г, 7,14 ммоль) и перемешивали в течение 2 часов. К полученному в результате раствору добавляли по каплям насыщенный раствор карбоната натрия до доведения рН до 10, перемешивали в течение 10 минут и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (20 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-эндо)-5-азидо-2-метил-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррола 9е (0,28 г, выход: 71,0%) в виде светло-желтой жидкости.
МС m/z (ИЭР): 167,1 [М+1]
Стадия 6
(цис-эндо)-2-Метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амин
(цис-эндо)-5-Азидо-2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол 9е (150 мг, 0,90 ммоль) растворяли в 20 мл метанола с последующим добавлением палладия/углерода (30 мг, 10%), заполняли водородом два раза. Реакционную смесь перемешивали в течение 2 часов и фильтровали. Фильтрационный кек промывали 10 мл метанола. Объединенный фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (цис-эндо)-2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амина 9f (0,08 г, выход: 63,0%) в виде светло-желтой жидкости.
МС m/z (ИЭР): 141,4 [М+1]
Стадия 7
(цис-экзо)-N-[2-Метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (163 мг, 0,35 ммоль) растворяли в 15 мл дихлорметана с последующим добавлением (цис-эндо)-2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амина 9f (59 мг, 0,35 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (112 мг, 0,35 ммоль) и диизопропилэтиламина (135 мг, 1,05 ммоль), перемешивали в течение 2 часов. К полученному в результате раствору добавляли 10 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали водой (50 мл), насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-экзо)-N-[2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксамида 9 (141 мг, выход: 68,8%) в виде белого твердого вещества.
МС m/z (ИЭР): 588,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.26 (d, 1Н), 7.67 (s, 1Н), 7.09-6.95 (m, 2Н), 5.86 (d, 1Н), 4.62-4.45 (m, 2Н), 4.21 (dd, 1Н), 3.39 (s, 2Н), 3.32 (s, 3H), 2.86-2.68 (m, 4Н), 2.32 (s, 3H), 2.26-2.08 (m, 4Н), 2.01 (d, 1Н), 1.93 (dd, 2Н), 1.88-1.60 (m, 9Н), 1.51 (s, 6Н), 0.88 (t, 3H).
Пример 10
N-[(цис-эндо)-2-(Циклопропилметил)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3H-бензофуран-4-карбоксамид
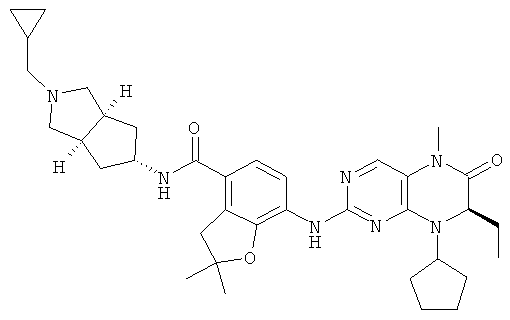
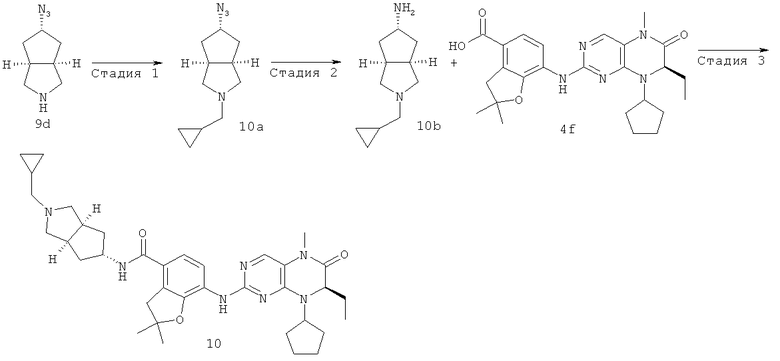
Стадия 1
(цис-эндо)-5-Азидо-2-(циклопропилметил)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол
(цис-эндо)-5-Азидо-1,2,3,3а,4,5,6,6а-октагидроциклопента[с]пиррола гидрохлорид 9d (693 мг, 3,67 ммоль) растворяли в 25 мл ацетонитрила с последующим добавлением карбоната калия (1,50 г, 11 ммоль) и бромметилциклопропана (644 мг, 4,77 ммоль). Реакционный раствор нагревали до образования флегмы в течение 2 часов. Полученный в результате раствор концентрировали при пониженном давлении, добавляли 20 мл воды и экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу последовательно промывали водой (50 мл), насыщенным раствором хлорида натрия (50 мл) и высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-эндо)-5-азидо-2-(циклопропилметил)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррола 10а (0,54 г, выход: 70,9%) в виде светло-желтого твердого вещества.
Стадия 2
(цис-эндо)-2-(Циклопропилметил)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амин
(цис-эндо)-5-Азидо-2-(циклопропилметил)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол 10а (517 мг, 2,51 ммоль) растворяли в 20 мл метанола с последующим добавлением палладия/углерода (60 мг, 10%), заполняли водородом три раза. Реакционную смесь перемешивали в течение 1,5 часов и фильтровали. Фильтрационный кек промывали метанолом. Объединенный фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (цис-эндо)-2-(циклопропилметил)-3,3а,4,5,6,6а-гексагидро-1Н- циклопента[с]пиррол-5-амина 10b (0,43 г, выход: 95,6%) в виде желтой жидкости.
МС m/z (ИЭР): 181,2 [М+1]
Стадия 3
N-[(цис-эндо)-2-(Циклопропилметил)-31За,415,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (163 мг, 0,35 ммоль) растворяли в 15 мл дихлорметана с последующим добавлением (цис-эндо)-2-(циклопропилметил)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амина 10b (63 мг, 0,35 ммоль), О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (112 мг, 0,35 ммоль) и диизопропилэтиламина (135 мг, 1,05 ммоль). Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 10 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали водой (50 мл), насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[(цис-эндо)-2-(циклопропилметил)-3,3a,4,5,6,6a-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксамида 10 (153 мг, выход: 69,5%) в виде белого твердого вещества.
МС m/z (ИЭР): 628,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.27 (d, 1Н), 7.67 (s, 1Н), 7.08-6.95 (m, 2Н), 5.96 (d, 1Н), 5.30 (s, 1Н), 4.66-4.43 (m, 2Н), 4.21 (dd, 1Н), 3.55-3.36 (m, 4Н), 3.32 (s, 3H), 3.01 (m, 2Н), 2.56 (m, 2Н), 2.33 (m, 2Н), 2.15 (m, 1Н), 2.01 (dd, 3H), 1.90-1.60 (m, 9Н), 1.51 (s, 6Н), 1.15-1.05 (m, 1Н), 0.88 (t, 3H), 0.63 (d, 2Н), 0.26 (d, 2Н).
Пример 11
(цис-эндо)-N-[2-Метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
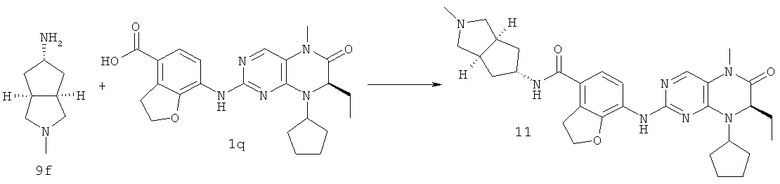
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (187 мг, 0,43 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (138 мг, 0,43 ммоль) растворяли в 20 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,2 мл, 0,95 ммоль), перемешивали до тех пор, пока раствор не становился прозрачным. Затем к реакционному раствору добавляли (цис-эндо)-2-метил-3,3а,4,5,6,6а-гексагидро-1H-циклопента[с]пиррол-5-амин 9f (60 мг, 0,43 ммоль) и перемешивали еще в течение 3 часов. К полученной в результате смеси добавляли 10 мл насыщенного раствора карбоната натрия и 10 мл дихлорметана. Органическую фазу последовательно промывали насыщенным раствором карбоната натрия (50 мл×2), водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-эндо)-N-[2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 11 (160 мг, выход: 67,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 560,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.27 (d, 1Н), 7.67 (s, 1Н), 7.10-6.94 (m, 2Н), 5.88 (d, 1Н), 4.68 (t, 2Н), 4.56 (d, 1Н), 4.47 (t, 1Н), 4.21 (dd, 1Н), 3.59 (t, 2Н), 3.32 (s, 3H), 2.90-2.67 (m, 4Н), 2.33 (s, 3H), 2.25-2.19 (m, 2H), 2.18-2.05 (m, 2H), 2.05-1.89 (m, 3H), 1.89-1.60 (m, 9H), 0.88 (t, 3H).
Пример 12
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(цис)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,3-дигидробензофуран-4-карбоксамид
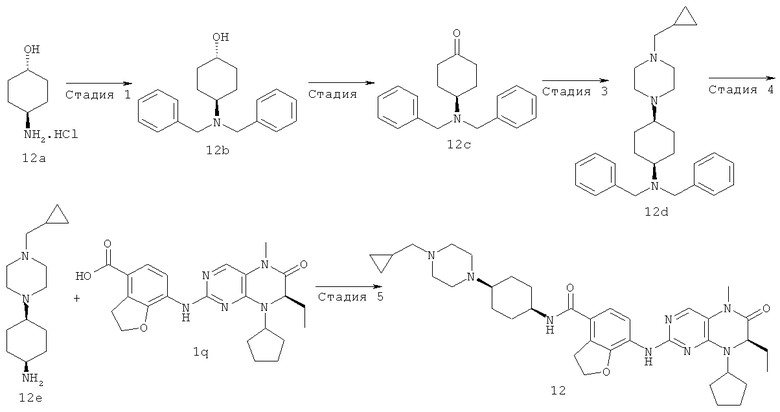
Стадия 1
(транс)-4-4-(Дибензиламино)циклогексанол
(транс)-4-Аминоциклогексанола гидрохлорид 12а (3,08 г, 20,31 ммоль) растворяли в 100 мл ацетонитрила при комнатной температуре с последующим добавлением карбоната калия (14,04 г, 101,57 ммоль) и бензилбромида (7,02 г, 41,03 ммоль). Реакционный раствор перемешивали в течение 12 часов и фильтровали. Фильтрационный кек промывали дихлорметаном (20 мл×3). Фильтрат концентрировали при пониженном давлении, и остаток растворяли вышеуказанным дихлорметаном, последовательно промывали насыщенным раствором хлорида аммония (50 мл) и насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (транс)-4-4-(дибензиламино)циклогексанола 12b (5,93 г, выход: 98,8%) в виде белого твердого вещества.
МС m/z (ИЭР): 296,3 [М+1]
Стадия 2
(R)-4-(Дибензиламино)циклогексанон
(транс)-4-(Дибензиламино)циклогексанол 12b (5,93 г, 20 ммоль) растворяли в 120 мл ацетона в бане лед-соль с последующим добавлением раствора 2,5 моль/л триоксида хрома в растворе серной кислоты (24 мл, 60 ммоль), перемешивали в течение 5 минут. Реакционный раствор нагревали до комнатной температуры и перемешивали еще в течение 30 минут. К полученной в результате смеси добавляли 400 мл дихлорметана, добавляли по каплям насыщенный раствор карбоната калия до доведения рН до 8, фильтровали и разделяли. Органическую фазу концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (R)-4-(дибензиламино)циклогексанона 12 с (3,09 г, выход: 70,0%) в виде белого масла.
МС m/z (ИЭР): 294,3 [М+1]
Стадия 3
(цис)-N,N-Дибензил-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамин
Циклопропилметилпиперазина дигидрохлорид (5,16 г, 24,20 ммоль) растворяли в 280 мл ацетонитрила, добавляли твердый ацетат натрия до доведения рН до 6-7 с последующим добавлением (R)-4-(дибензиламино)циклогексанона 12 с (6,48 г, 22 ммоль) и триацетоксиборгидрида натрия (11,66 г, 55 ммоль) последовательно, перемешивали в течение 12 часов. К полученному в результате раствору добавляли 100 мл воды, затем добавляли твердый карбонат натрия до доведения рН до 8 и разделяли. Водную фазу экстрагировали дихлорметаном (250 мл×2). Объединенную органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия (100 мл) и насыщенным раствором хлорида натрия (100 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис)-N,N-дибензил-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамин 12d (3,13 г, выход: 34,1%) в виде белого твердого вещества.
МС m/z (ИЭР): 418,4 [М+1]
Стадия 4
(цис)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексанамин (цис)-N,N-Дибензил-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамин 12d (2,44 г, 5,83 ммоль) растворяли в 30 мл смеси растворителей дихлорметана и метанола (об./об. = 1:2) с последующим добавлением палладия/углерода (1,22 г, 10%) и 0,1 мл уксусной кислоты, заполняли водородом три раза. Реакционную смесь подвергали взаимодействию при 3 атмосферах в течение 12 часов. К полученному в результате раствору добавляли 4 г щелочного оксида алюминия (200-300 меш) и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (цис)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамин 12е (2,38 г, выход: 98,0%) в виде бесцветного твердого вещества.
Стадия 5
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(цис)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,3-дигидробензофуран-4-карбоксамид
(цис)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексанамин 12е (139 мг, 0,40 ммоль), 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,2-дигидробензофуран-4-карбоновую кислоту 1q (175 мг, 0,40 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (128 мг, 0,40 ммоль) и диизопропилэтиламин (258 мг, 2,08 ммоль) растворяли в 50 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов с последующим добавлением 50 мл воды и 10 мл водного аммиака последовательно и перемешивали еще в течение 0,5 часа. Полученный в результате раствор экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(цис)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,3-дигидробензофуран-4-карбоксамида 12 (160 мг, выход: 61,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 657,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.28 (d, 1Н), 7.67 (s, 1Н), 7.13 (d, 1Н), 7.03 (s, 1Н), 6.18-6.08 (m, 1Н), 4.68 (t, 2Н), 4.52-4.48 (m, 1Н), 4.28-4.19 (m, 2Н), 3.60 (t, 2Н), 3.32 (s, 3H), 3.15-2.64 (m, 8Н), 2.63-2.45 (m, 4Н), 2.16-2.08 (m, 1Н), 2.08-1.92 (m, 3H), 1.92-1.75 (m, 6Н), 1.75-1.60 (m, 8Н), 1.07-0.92 (m, 1Н), 0.88 (t, 3H), 0.61-0.50 (m, 2Н), 0.23-0.15 (m, 2Н).
Пример 13
(цис-эндо)-N-[2-(Циклопропилметил)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
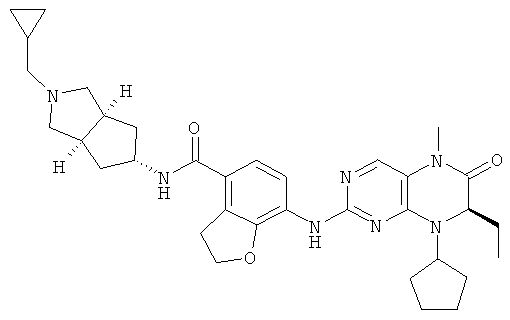
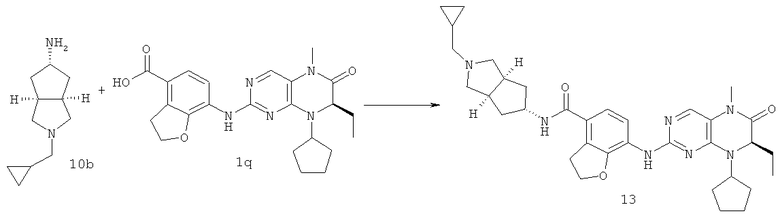
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (187 мг, 0,43 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (138 мг, 0,43 ммоль) растворяли в 20 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,2 мл, 0,95 ммоль), перемешивали до тех пор, пока раствор не становился прозрачным, добавляли (цис-эндо)-2-(циклопропилметил)-3, За, 4,5,6,6а-гексагидро-1Н-
циклопента[с]пиррол-5-амин 10b (77 мг, 0,43 ммоль), перемешивали в течение 3 часов. К полученному в результате раствору добавляли 10 мл насыщенного раствора карбоната натрия и 20 мл дихлорметана и разделяли. Органическую фазу последовательно промывали насыщенным раствором карбоната натрия (20 мл×2), водой (30 мл) и насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-эндо)-N-[2-(циклопропилметил)-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 13 (210 мг, выход: 81,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 600,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.27 (d, 1Н), 7.67 (s, 1Н), 7.10-6.92 (m, 2Н), 5.90 (d, 1Н), 4.68 (t, 2Н), 4.47 (s, 2Н), 4.21 (dd, 1Н), 3.59 (t, 2Н), 3.32 (s, 3H), 3.06 (s, 2Н), 2.80 (s, 2Н), 2.31 (d, 2Н), 2.23-2.06 (m, 3H), 2.06-1.91 (m, 3H), 1.91-1.77 (m, 4Н), 1.77-1.52 (m, 6н), 1.39-1.15 (m, 1Н), 0.88 (t, 3H), 0.58-0.36 (m, 2Н), 0.20-0.08 (m, 2Н).
Пример 14
(цис-эндо)-N-[2-Метил-1,3,3а,4,5,6,7,7а-октагидроциклопента[с]пиридин-6-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
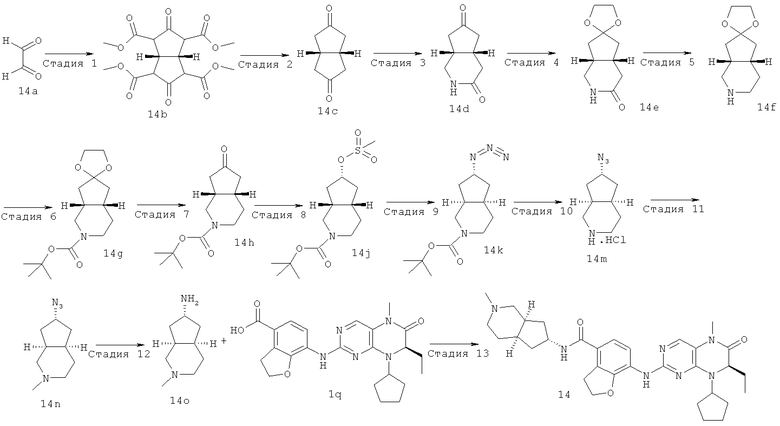
Стадия 1
(цис)-Тетраметилбицикло[3.3.0]октан-3,7-диоксо-2,4,6,8-тетракарбоксилат
Гидроксид натрия (6,40 г, 0,16 моль) растворяли в 115 мл метанола в бане лед-вода, добавляли по каплям диметил-1,3-ацетондикарбоксилат (22,6 мл, 0,16 моль) при перемешивании. Реакционный раствор нагревали до образования флегмы до тех пор, пока соль не растворялась полностью, быстро добавляли по каплям глиоксаль 14а (12,85 г, 0,09 моль) при 65°С, затем охлаждали до комнатной температуры, перемешивали в течение 12 часов и фильтровали. Фильтрационный кек промывали 50 мл метанола и растворяли в 180 мл смеси растворителей дихлорметана и воды (об./об. = 5:4). Полученный в результате раствор охлаждали до 0°С, добавляли по каплям 1 М соляную кислоту до доведения рН до 6 и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (цис)-тетраметилбицикло[3.3.0]октан-3,7-диоксо-2,4,6,8-тетракарбоксилата 14b (20 г, выход: 61,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 371,3 [М+1]
Стадия 2
(цис)-Бицикло[3.3.0]октан-3,7-дион
(цис)-Тетраметилбицикло[3.3.0]октан-3,7-диоксо-2,4,6,8-тетракарбоксилат 14b (6,75 г, 0,02 моль) растворяли в 3,3 мл уксусной кислоты с последующим добавлением 30 мл 1 М соляной кислоты. Реакционный раствор нагревали до образования флегмы в течение 3,5 часов, а затем охлаждали до комнатной температуры. Реакционный раствор экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу концентрировали при пониженном давлении, добавляли 100 мл дихлорметана, добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 7 и разделяли. Органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (цис)-бицикло[3.3.0]октан-3,7-диона 14 с (2 г, выход: 80,0%) в виде белого твердого вещества.
Стадия 3
(цис)-2,4,4а,5,7,7а-гексагидро-1Н-циклопента[с]пиридин-3,6-дион
(цис)-Бицикло[3.3.0]октан-3,7-дион 14 с (1,80 г, 13 ммоль) растворяли в 25 мл концентрированной соляной кислоты в бане лед-вода, добавляли азид натрия (1,10 г, 16,90 ммоль) порциями, затем нагревали до комнатной температуры и перемешивали в течение 12 часов. К реакционному раствору добавляли по каплям 20% раствор гидроксида натрия до доведения рН до 10-11, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (цис)-2,4,4а,5,7,7а-гексагидро-1Н-циклопента[с]пиридин-3,6-диона 14d (3 г, выход: 100,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 154,1 [М+1]
Стадия 4
(цис)-спиро[1,3-Диоксолан-2,6'-2,4,4а,5,7,7а-гексагидро-1Н-циклопента[с]пиридин]-3'-он
(цис)-2,4,4а,5,7,7а-Гексагидро-1Н-циклопента[с]пиридин-3,6-дион 14d (1,20 г, 7,80 ммоль), этандиол (1,34 г, 21,50 ммоль) и пара-толуолсульфоновую кислоту (24 мг, 0,13 ммоль) растворяли в 60 мл толуола, нагревали до образования флегмы в течение 8 часов при перемешивании, затем добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН примерно до 7 и экстрагировали дихлорметаном (50 мл). Водную фазу экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис)-спиро[1,3-диоксолан-2,6'-2,4,4а,5,7,7а-гексагидро-1Н-циклопента[с]пиридин]-3'-она 14е (1,10 г, выход: 70,0%) в виде белого твердого вещества
МС m/z (ИЭР): 198,1 [М+1]
Стадия 5
(цис)-спиро[1,2,3,4,4а,5,7,7а-октагидроциклопента[с]пиридин-6,2'-1,3-диоксолан]
В бане лед-вода алюмогидрид лития (47 мг, 1,27 ммоль) растворяли в 10 мл тетрагидрофурана с последующим добавлением 10 мл раствора (цис)-спиро[1,3-диоксолан-2,6'-2,4,4а,5,7,7а-гексагидро-1Н-циклопента[с]пиридин]-3'-она 14е (120 мг, 0,60 ммоль) в тетрагидрофуране. Полученный в результате раствор перемешивали в течение 1 часа, добавляли порциями 1 мкл воды, 1 мкл 15% раствора гидроксида натрия, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (цис)-спиро[1,2,3,4,4а,5,7,7а-октагидроциклопента[с]пиридин-6,2'-1,3-диоксолан] 14f (150 мг) в виде желтой маслянистой жидкости, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 184,2 [М+1]
Стадия 6
(цис)-трет-Бутил-спиро[1,3-диоксолан-2,6'-3,4,4а,5,7,7а-гексагидро-1Н-циклопента[с]пиридин]-2'-карбоксилат
(цис)-спиро[1,2,3,4,4а,5,7,7а-Октагидроциклопента[c]пиридин-6,2'-1,3-диоксолан] 14f (100 мг, 0,54 ммоль) растворяли в 15 мл дихлорметана в бане лед-вода. К полученному в результате раствору последовательно добавляли триэтиламин (109 мг, 1,08 ммоль) и ди-трет-бутилдикарбонат (130 мг, 0,60 ммоль), перемешивали в течение 2 часов, затем добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 8-9, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис)-трет-бутил-спиро[1.3-диоксолан-2,6'-3,4,4а,5,7,7а-гексагидро-1Н-циклопента[с]пиридин]-2'-карбоксилата 14g (100 мг, выход: 66,0%) в виде бесцветной маслянистой жидкости.
Стадия 7
(цис)-трет-Бутил-6-оксо-3,4,4а,5,7,7а-гексагидро-1Н-циклопента[с]пиридин-2-карбоксилат
(цис)-трет-Бутил-спиро[1,3-диоксолан-2,6'-3,4,4а,5,7,7а-гексагидро-1Н-циклопента[с] пиридин]-2'-карбоксилат 14g (620 мг, 2,20 ммоль) растворяли в 50 мл ацетона с последующим добавлением пара-толуолсульфоновой кислоты (200 мг, 1,10 ммоль). Полученный в результате раствор нагревали до образования флегмы в течение 30 минут, затем охлаждали до комнатной температуры, добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 8-9, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис)-трет-бутил-6-оксо-3,4,4а,5,7,7а-гексагидро-1Н-циклопента[с]пиридин-2-карбоксилата 14h (450 мг, выход: 85,0%) в виде бесцветной маслянистой жидкости.
Стадия 8
(цис-экзо)-трет-Бутил-6-метилсульфонилокси-1,3,4,4а, 5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат (цис)-трет-Бутил-6-оксо-3,4,4а,5,7,7а-гексагидро-1H-циклопента[с]пиридин-2-карбоксилат 14h (22 мг, 0,10 ммоль) растворяли в 10 мл тетрагидрофурана с последующим добавлением боргидрида натрия (10 мг, 0,14 ммоль), перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с последующим добавлением 20 мл дихлорметана, затем добавляли триэтиламин (40 мкл, 0,29 ммоль) и метансульфонилхлорид (20 мкл, 0,14 ммоль), перемешивали в течение 2 часов. К полученному в результате раствору добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 8-9, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-экзо)-трет-бутил-6-метилсульфонилокси-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилата 14j (28 мг, выход: 98,0%) в виде бесцветной маслянистой жидкости.
Стадия 9
(цис-эндо)-трет-бутил-6-азидо-1,3)4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат
(цис-экзо)-трет-Бутил-6-метилсульфонилокси-1,3,4,4а, 5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат 14j (32 мг, 0,10 ммоль) растворяли в 10 мл N,N-диметилформамида, медленно добавляли азид натрия (20 мг, 0,25 ммоль), затем нагревали до 70-80°С и перемешивали в течение 3,5 часов. К полученному в результате раствору добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 8-9, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-эндо)-трет-бутил-6-азидо-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилата 14k (40 мг, выход: 100,0%) в виде бесцветной маслянистой жидкости.
Стадия 10
(цис-эндо)-6-Азидо-2,3,4,4а,5,6,7,7а-октагидро-1H-циклопента[с]пиридина гидрохлорид
(цис-эндо)-трет-Бутил-6-азидо-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат 14k (1,15 г, 4,32 ммоль) растворяли в 5 мл дихлорметана с последующим добавлением 10 мл 4 М раствора хлорида водорода в диоксане, перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (цис-эндо)-6-азидо-2,3,4,4а,5,6,7,7а-октагидро-1Н-циклопента[с]пиридина гидрохлорида 14m (880 мг) в виде бесцветной маслянистой жидкости, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 167,2 [М+1]
Стадия 11
(цис-эндо)-6-Азидо-2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин
(цис-эндо)-6-Азидо-2,3,4,4а,5,6,7,7а-октагидро-7Н-циклопента[с]пиридина гидрохлорид 14m (880 мг, 4,32 ммоль) растворяли в 20 мл смеси растворителей ацетонитрила и воды (об./об. = 1:1) в бане лед-вода, последовательно добавляли формальдегид (0,7 мл, 8,64 ммоль) и триацетоксиборгидрид натрия (2,70 г, 12,96 ммоль), затем перемешивали в течение 2 часов с последующим добавлением 10 мл 1 М соляной кислоты, и добавляли по каплям 15 мл 5% раствора гидроксида натрия до доведения рН до 9. Полученный в результате раствор перемешивали 10 минут, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-эндо)-6-азидо-2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридина 14n (650 мг, выход: 84,0%) в виде бесцветного масла.
МС m/z (ИЭР): 181,1 [М+1]
Стадия 12
(цис-эндо)-2-Метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-амин (цис-эндо)-6-Азидо-2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин 14т (650 мг, 3,60 ммоль) растворяли в 20 мл метанола с последующим добавлением палладия/углерода (70 мг, 10%), заполняли водородом три раза. Реакционную смесь перемешивали в течение 2 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (цис-эндо)-2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-амина 14о (505 мг, выход: 91,0%) в виде светло-желтой маслянистой жидкости.
МС m/z (ИЭР): 155,2 [М+1]
Стадия 13
N-[(цис-эндо)-2-Метил-1,3,3а,4,5,6,7,7а-октагидроциклопента[с]пиридин-6-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (187 мг, 0,43 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (138 мг, 0,43 ммоль) растворяли в 30 мл безводного дихлорметана, последовательно добавляли диизопропилэтиламин (0,2 мл, 0,95 ммоль) и (цис-эндо)-2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-амин 14о (66 мг, 0,43 ммоль), перемешивали в течение 3 часов. К полученному в результате раствору добавляли 10 мл насыщенного раствора карбоната натрия и 30 мл дихлорметана, разделяли. Органическую фазу последовательно промывали раствором карбоната натрия (50 мл×2), водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[(цис-эндо)-2-метил-1,3,3а,4,5,6,7,7а-октагидроциклопента[с]пиридин-6-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 14 (120 мг, выход: 48,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 574,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.27 (d, 1Н), 7.67 (s, 1Н), 7.10-6.94 (m, 2Н), 6.04 (d, 1Н), 4.67 (t, 2Н), 4.58 (d, 1Н), 4.47 (t, 1Н), 4.21 (dd, 1Н), 3.58 (t, 2Н), 3.32 (s, 3H), 2.70-2.45 (m, 4Н), 2.41 (s, 3H), 2.36-2.07 (m, 5Н), 2.06-1.92 (m, 2Н), 1.90-1.60 (m, 11Н), 0.88 (t, 3H)
Пример 15
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[[(2R)-4-метилморфолин-2-ил]метил]-2,3-дигидробензофуран-4-карбоксамид
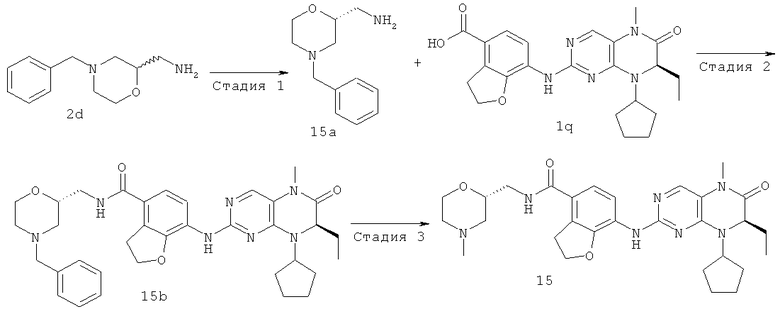
Стадия 1
[(2R)-4-Бензилморфолин-2-ил]метанамин
(4-Бензилморфолин-2-ил)метанамин 2d (1,35 г, 6,50 ммоль) растворяли в 30 мл этанола с последующим добавлением L-(-)-дибензоилвинной кислоты (2,34 г, 6,50 ммоль). Реакционный раствор нагревали до образования флегмы в течение 10 минут, затем охлаждали до комнатной температуры, помещали на 16 часов, что привело в результате к образованию кристаллов, и фильтровали. Фильтрационный кек растворяли в 10 мл воды, добавляли 1 М водный раствор гидроксида натрия до доведения рН до 9 и экстрагировали дихлорметаном (30 мл×3). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, [(2R)-4-бензилморфолин-2-ил]метанамина 15а (0,28 г, выход: 28,0%) в виде светло-желтой маслянистой жидкости.
МС m/z (ИЭР): 207,2 [М+1]
Стадия 2
N-[[(2R)-4-Бензилморфолин-2-ил]метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид 7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (276 мг, 0,63 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (202 мг, 0,63 ммоль) растворяли в 20 мл безводного дихлорметана, последовательно добавляли диизопропилэтиламин (0,2 мл, 1,4 ммоль), [(2R)-4-бензилморфолин-2-ил]метанамин 15а (130 мг, 0,63 ммоль) при перемешивании. Реакционный раствор перемешивали в течение 3 часов. К полученному в результате раствору последовательно добавляли 10 мл насыщенного раствора карбоната натрия и 10 мл дихлорметана и разделяли. Органическую фазу последовательно промывали раствором карбоната натрия (50 мл×2), водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[[(2R)-4-бензилморфолин-2-ил]метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 15b (370 мг, выход: 94,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 626,5 [М+1]
Стадия 3
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-N-[[(2R)-4-метилморфолин-2-ил]метил]-2,3-дигидробензофуран-4-карбоксамид
N-[[(2R)-4-Бензилморфолин-2-ил]метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид 15b (370 мг, 0,59 ммоль) растворяли в 30 мл метанола. Реакционный раствор подвергали гидрогенизации H-Cube (колонка катализатора: 10% палладий/углерод; давление водорода: 10 атмосфер; температура: 40°С) в течение 8 часов. Реакционный раствор концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5- метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[[(2R)-4-метилморфолин-2-ил]метил]-2,3-дигидробензофуран-4-карбоксамида 15 (60 мг, выход: 18,5%) в виде белого твердого вещества.
МС m/z (ИЭР): 550,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.25 (d, 1Н), 7.67 (s, 1Н), 7.11-7.02 (m, 2Н), 4.66 (t, 2Н), 4.62-4.53 (m, 1Н), 4.50 (t, 1Н), 4.20 (dd, 1Н), 3.66 (t, 2Н), 3.32 (s, 3H), 2.91 (s, 2н), 2.78 (s, 2Н), 2.47 (s, 3H), 2.34 (s, 2Н), 2.24-2.06 (m, 3H), 2.06-1.94 (m, 1Н), 1.89-1.75 (m, 4Н), 1.72-1.62 (m, 5Н), 0.88 (t, 3H).
Пример 16
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,2-диметил-3Н-бензофуран-4-карбоксамид
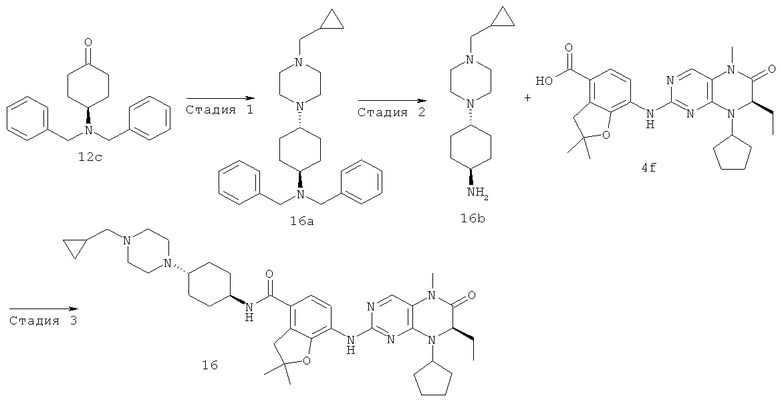
Стадия 1
(транс)-N,N-Дибензил-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамин
Циклопропилметилпиперазина дигидрохлорид (5,16 г, 24,20 ммоль) растворяли в 280 мл ацетонитрила с последующим добавлением 5 г твердого ацетата натрия до доведения рН до 6-7, затем последовательно добавляли (R)-4-(дибензиламино)циклогексанон 12с (6,48 г, 22 ммоль) и триацетоксиборгидрид натрия (11,66 г, 55 ммоль). Реакционный раствор перемешивали в течение 12 часов. К полученному в результате раствору последовательно добавляли 100 мл воды и твердый карбонат натрия до доведения рН до 8, разделяли. Водную фазу экстрагировали дихлорметаном (250 мл×2). Объединенную органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия (50 мл×3) и насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (транс)-N,N-дибензил-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамина 16а (1,14 г, выход: 15,4%) в виде белого твердого вещества.
МС m/z (ИЭР): 418,4 [М+1]
Стадия 2
(транс)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексанамин (транс)-N,N-Дибензил-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамин 16а (1,41 г, 3,38 ммоль) растворяли в 15 мл метанола с последующим добавлением палладия/углерода (706 мг, 10%) и 0,1 мл уксусной кислоты, заполняли водородом три раза. Реакционную смесь подвергали взаимодействию в течение 12 часов при 3 атмосферах водорода, добавляли 3 г щелочного оксида алюминия (200-300 меш) и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамина 16b (900 мг, выход: 100%) в виде бесцветного твердого вещества.
МС m/z (ИЭР): 238,2 [М+1]
Стадия 3
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,2-диметил-3H-бензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (150 мг, 0,32 ммоль), (транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамин 16b (76 мг, 0,32 ммоль) и О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (103 мг, 0,32 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением диизопропилэтиламина (103 мг, 0,80 ммоль). Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 20 мл насыщенного раствора хлорида аммония, разделяли, органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью препаративной пластины с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,2-диметил-3H-бензофуран-4-карбоксамида 16 (130 мг, выход: 59,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 685,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.28 (d, 1Н), 7.70 (s, 1Н), 7.23-7.00 (m, 2Н), 5.92 (d, 1Н), 4.58 (t, 1Н), 4.25 (dd, 1Н), 4.00-3.81 (m, 1Н), 3.41 (s, 2Н), 3.35 (s, 3H), 3.10 (s, 5н), 2.80-2.55 (m, 3H), 2.30-2.11 (m, 5Н), 2.07-1.97 (m, 2Н), 1.93-1.66 (m, 8н), 1.54 (s, 6Н), 1.48 (s, 1Н), 1.44-1.21 (m, 5Н), 1.21-1.05 (m, 1Н), 0.91 (t, 3H), 0.71 (d, 2Н), 0.34 (d, 2Н).
Пример 17
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-N-[(транс)-4-(4-метилпиперазин-1-ил)циклогексил]-3Н-бензофуран-4-карбоксамид
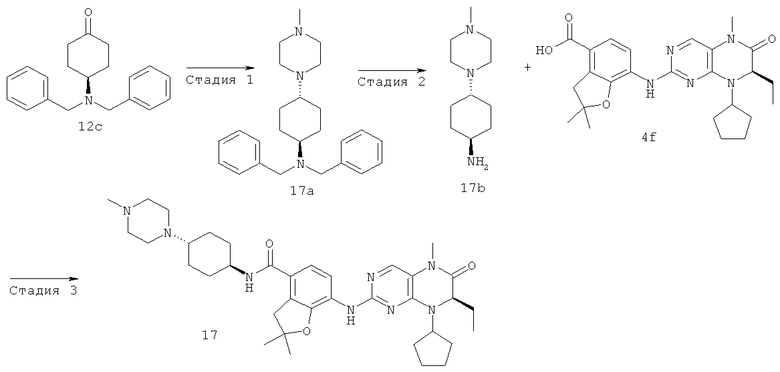
Стадия 1
(транс)-N,N-Дибензил-4-(4-метилпиперазин-1-ил)циклогексанамин
(R)-4-(Дибензиламино)циклогексанон 12с (4,61 г, 15,71 ммоль) растворяли в 150 мл дихлорметана с последующим добавлением 1-метилпиперазина (1,73 г, 17,28 ммоль), последовательно добавляли по каплям 0,1 мл уксусной кислоты, затем добавляли триацетоксиборгидрид натрия (6,66 г, 31,42 ммоль). Реакционный раствор перемешивали в течение 12 часов. К полученному в результате раствору добавляли 15 мл 1 М разбавленную соляную кислоту и по каплям насыщенный раствор карбоната натрия до доведения рН до 8 и разделяли. Органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (транс)-N,N-дибензил-4-(4-метилпиперазин-1-ил)циклогексанамина 17а (3,80 г, выход: 64,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 378,4 [М+1]
Стадия 2
(транс)-4-(4-Метилпиперазин-1-ил)циклогексанамин
(транс)-N,N-Дибензил-4-(4-метилпиперазин-1-ил)циклогексанамин 17а (3,35 г, 8,90 ммоль) растворяли в 20 мл метанола с последующим добавлением палладия/углерода (335 мг, 10%), заполняли водородом три раза. Реакционный раствор подвергали взаимодействию при 3 атмосферах в течение 12 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с последующим добавлением 10 мл 1 М соляной кислоты и экстрагировали дихлорметаном (30 мл×3). К водной фазе добавляли по каплям насыщенный водный раствор карбоната натрия до доведения рН до 11, экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (транс)-4-(4-метилпиперазин-1-ил)циклогексанамина 17b (0,82 г, выход: 46,7%) в виде желтой маслянистой жидкости, который использовали в следующей стадии без дальнейшей очистки.
Стадия 3
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-N-[(транс)-4-(4-метилпиперазин-1-ил)циклогексил]-3H-бензофуран-4-карбоксамид
Сырое соединение (транс)-4-(4-метилпиперазин-1-ил)циклогексанамин 17b (60 мг, 0,30 ммоль), 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (153 мг, 0,33 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (96 мг, 0,30 ммоль) и диизопропилэтиламин (96 мг, 0,75 ммоль) растворяли в 25 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 8-9, экстрагировали дихлорметаном (30 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-N-[(транс)-4-(4-метилпиперазин-1-ил)циклогексил]-3Н-бензофуран-4-карбоксамида 17 (70 мг, выход: 33,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 645,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.26 (d, 1Н), 7.68 (s, 1Н), 7.13-7.91 (m, 2Н), 6.08 (d, 1Н), 4.58 (t, 1Н), 4.22 (dd, 2Н), 3.39 (s, 2Н), 3.33 (s, 3H), 2.75-2.35 (m, 8Н), 2.31 (s, 3H), 2.30-2.10 (m, 2Н), 2.09-1.95 (m, 2Н), 1.93-1.63 (m, 12Н), 1.50 (s, 6Н), 0.88 (t, 6Н).
Пример 18
(цис-экзо)-N-[2-Метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксамид
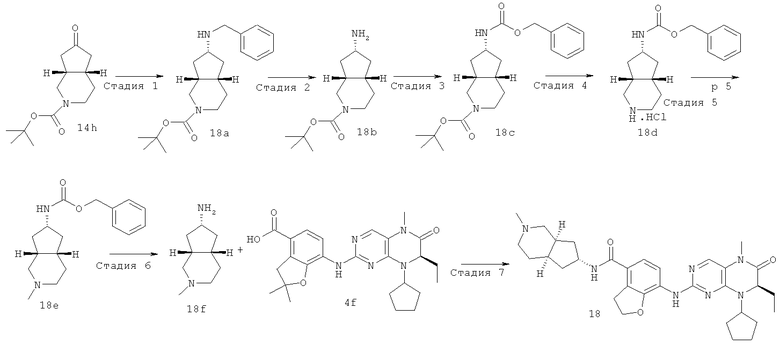
Стадия 1
трет-Бутил-(цис-экзо)-6-(бензиламино)-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат
трет-Бутил(цис)-6-оксо-3,4,4а,5,7,7а-гексагидро-4Н-циклопента[c]пиридин-2-карбоксилат 14h (1,10 г, 4,60 ммоль) и бензиламин (492 мг, 4.60 ммоль) растворяли в 50 мл дихлорметана в бане лед-вода с последующим добавлением уксусной кислоты (276 мг, 4,60 ммоль), перемешивали в течение 30 минут. К реакционному раствору добавляли триацетоксиборгидрид натрия (1,95 г, 9,20 ммоль), нагревали до комнатной температуры и перемешивали еще в течение 12 часов. К полученному в результате раствору добавляли по каплям насыщенный раствор карбоната натрия до доведения рН до 9 и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-(цис-экзо)-6-(бензиламино)-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилата 18а (1,25 г, выход: 83,0%) в виде светло-желтого масла.
МС m/z (ИЭР): 331,3 [М+1]
Стадия 2
трет-Бутил-(цис-экзо)-6-амино-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат
трет-Бутил-(цис-экзо)-6-(бензиламино)-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат 18а (1,25 г, 3,78 ммоль) растворяли в 20 мл метанола с последующим добавлением палладия/углерода (125 мг, 10%), заполняли водородом три раза. Реакционную смесь перемешивали в течение 12 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, трет-бутил-(цис-экзо)-6-амино-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилата 18b (910 мг, выход: 100,0%) в виде светло-желтого масла.
МС m/z (ИЭР): 241,2 [М+1]
Стадия 3
трет-Бутил-(цис-экзо)-6-бензилоксикарбониламино-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат
трет-Бутил-(цис-экзо)-6-амино-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат 18b (909 мг, 3,78 ммоль) растворяли в 25 мл дихлорметана с последующим добавлением триэтиламина (763 мг, 7,56 ммоль) и бензилхлорформиат (707 мг, 4,16 ммоль) в бане лед-вода, затем нагревали до комнатной температуры и перемешивали в течение 2 часов. К полученному в результате раствору добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 8-9 и экстрагировали дихлорметаном (30 мл×3). Объединенную органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-(цис-экзо)-6-бензилоксикарбониламино-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилата 18 с (520 мг, выход: 37,0%) в виде бесцветного масла.
Стадия 4
Бензил(цис-экзо)-N-[2,3,4,4а,5,6,7,7а-октагидро-7Н-циклопента[с]пиридин-6-ил]карбамата гидрохлорид
трет-Бутил-(цис-экзо)-6-бензилоксикарбониламино-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-2-карбоксилат 18 с (513 мг, 1,37 ммоль) растворяли в 6 мл дихлорметана с последующим добавлением 6 мл 4 М раствора 1,4-диоксана в хлориде водорода. Реакционный раствор перемешивали в течение 30 минут. Полученный в результате раствор концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, бензил-(цис-экзо)-N-[2,3,4,4а,5,6,7,7а-октагидро-7Н-циклопента[с]пиридин-6-ил]карбамата гидрохлорида 18d (430 мг) в виде белой вязкой жидкости, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 275,2 [М+1]
Стадия 5
Бензил-(цис-экзо)-N-[2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-ил]карбамат
Сырое соединение бензил-(цис-экзо)-N-[2,3,4,4а,5,6,7,7а-октагидро-1H-циклопента[с]пиридин-6-ил]карбамата гидрохлорид 18d (425 мг, 1,37 ммоль) растворяли в 20 мл смеси растворителей ацетонитрила и воды (об./об. = 1: 1) в бане лед-вода с последующим добавлением триацетоксиборгидрида натрия (871 мг, 4,11 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 12 часов. К полученному в результате раствору добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН примерно до 9, и экстрагировали дихлорметаном (20 мл×3), последовательно промывали водой (20 мл), насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, бензил-(цис-экзо)-N-[2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-ил]карбамата 18е (374 мг) в виде светло-желтого масла, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 289,2 [М+1]
Стадия 6
(цис-экзо)-2-Метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-амин
Сырое соединение бензил-(цис-экзо)-N-[2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-ил]карбамат 18е (374 мг, 1,29 ммоль) растворяли в 25 мл метанола с последующим добавлением палладия/углерода (40 мг, 10%), заполняли водородом три раза. Реакционную смесь перемешивали в течение 2 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (цис-экзо)-2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-амин 18f (198 мг) в виде белого твердого вещества, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 155,2 [М+1]
Стадия 7
(цис-экзо)-N-[2-Метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,2-диметил-3H-бензофуран-4-карбоксамид
Сырое соединение (цис-экзо)-2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-амин 18f (60 мг, 0,39 ммоль), 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7R-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (199 мг, 0,43 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (125 мг, 0,39 ммоль) и диизопропилэтиламин (125 мг, 0.98 ммоль) растворяли в 25 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 25 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (30 мл×3), последовательно промывали водой (30 мл), насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-экзо)-N-[2-метил-1,3,4,4а,5,6,7,7а-октагидроциклопента[с]пиридин-6-ил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоксамида 18 (120 мг, выход: 50,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 602,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.71 (s, 1Н), 7.11-7.03 (m, 2Н), 6.12 (d, 1Н), 4.60 (t, 1Н), 4.55-4.46 (m, 1Н), 4.25 (dd, 1Н), 3.43 (s, 2Н), 3.36 (s, 3H), 2.57-2.45 (m, 2Н), 2.43-2.33 (m, 2Н), 2.32-2.28 (m, 4Н), 2.25-2.13 (m, 4н), 2.11-1.96 (m, 3H), 1.91-1.62 (m, 8Н), 1.53 (s, 6Н), 1.51-1.45 (m, 2Н), 0.92 (t, 3H).
Пример 19
6-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(транс)-4-(4-метилпиперазин-1-ил)циклогексил]-2,3-дигидробензофуран-4-карбоксамид
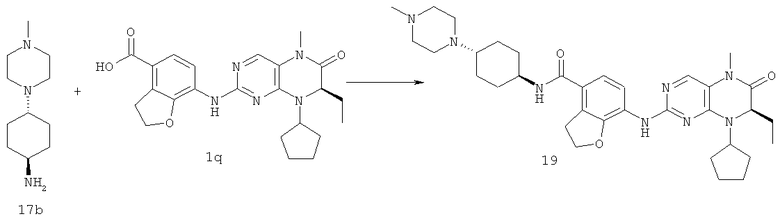
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (200 мг, 0,46 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (147 мг, 0,46 ммоль) растворяли в 25 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,2 мл, 1 ммоль) и (транс)-4-(4-метилпиперазин-1-ил)циклогексанамина 17b (90 мг, 0,46 ммоль) последовательно. Реакционный раствор перемешивали в течение 3 часов. К полученному в результате раствору добавляли 20 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (30 мл×3). Объединенную органическую фазу последовательно промывали насыщенным раствором карбоната натрия (30 мл×2), вода (30 мл) и насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(транс)-4-(4-метилпиперазин-1-ил)циклогексил]-2,3-дигидробензофуран-4-карбоксамида 19 (190 мг, выход: 67,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 617,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.28 (d, 1Н), 7.67 (s, 1Н), 7.11 (d, 1Н), 7.03 (s, 1Н), 6.14 (s, 1Н), 4.68 (t, 2Н), 4.49 (t. 1H), 4.21 (dd, 2H), 3.60 (t, 2H), 3.32 (s, 3H), 2.90-2.55 (m, 8H), 2.37 (s, 3H), 2.38-2.30 (m, 1H), 2.12 (d, 1H), 2.08-1.94 (m, 2H), 0.94-1.78 (m, 7H), 1.78-1.55 (m, 8H), 0.88 (t, 3H).
Пример 20
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2R)-3-(4-метилпиперазин-1-ил)-2-гидроксипропил]-2,2-диметил-3H-бензофуран-4-карбоксамид
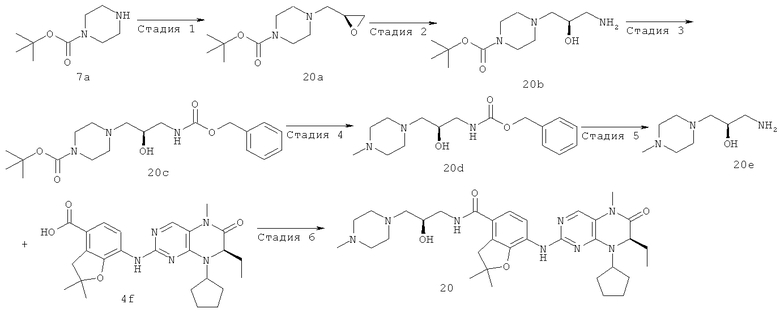
Стадия 1
трет-Бутил-4-[[(2S)-оксиран-2-ил]метил]пиперазин-1-карбоксилат
трет-Бутилпиперазин-1-карбоксилат 7a (7,44 г, 40 ммоль) и (S)-2-хлорметилоксиран (3,70 г, 40 ммоль) растворяли в 60 мл этанола. Реакционный раствор перемешивали в течение 12 часов. Полученный в результате раствор концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-4-[[(2S)-оксиран-2-ил]метил]пиперазин-1-карбоксилата 20а (9,20 г, выход: 95,0%) в виде белого твердого вещества.
Стадия 2
трет-Бутил 4-[(2R)-3-амино-2-гидрокси-пропил]пиперазине-1-карбоксилат
трет-Бутил-4-[[(2S)-оксиран-2-ил]метил]пиперазин-1-карбоксилат 20а (9,20 г, 38 ммоль) растворяли в 60 мл этанола с последующим добавлением 50 мл водного аммиака. Реакционный раствор перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, трет-бутил-4-[(2R)-3-амино-2-гидроксипропил]пиперазин-1-карбоксилата 20b (9 г, в виде белого твердого вещества), который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 260,2 [М+1]
Стадия 3
трет-Бутил-4-[(2R)-3-бензилоксикарбониламино-2-гидроксипропил]пиперазин-1-карбоксилат
Сырое соединение трет-бутил-4-[(2R)-3-амино-2-гидроксипропил]пиперазин-1-карбоксилат 20b (4,50 г, 17,40 ммоль) растворяли в 80 мл дихлорметана в бане лед-вода с последующим добавлением диизопропилэтиламина (2,50 г, 19,10 ммоль) и добавлением по каплям 20 мл раствора карбобензоксихлорида (2 г, 19,10 ммоль) в дихлорметане последовательно, перемешивали в течение 12 часов. К полученной в результате смеси добавляли 20 мл воды и перемешивали в течение 10 минут. Реакционный раствор концентрировали при пониженном давлении, добавляли 100 мл воды и экстрагировали этилацетатом (100 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-4-[(2R)-3-бензилоксикарбониламино-2-гидроксипропил]пиперазин-1-карбоксилата 20с (1,80 г, выход: 26,0%) в виде светло-желтого масла.
МС m/z (ИЭР): 394,3 [М+1]
Стадия 4
Бензил-N-[(2R)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]карбамат
трет-Бутил-4-[(2R)-3-бензилоксикарбониламино-2-гидроксипропил]пиперазин-1-карбоксилат 20с (1,80 г, 4,80 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением 20 мл раствора хлорида водорода в диоксане. Реакционный раствор перемешивали в течение 0,5 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли 30 мл воды и 30 мл ацетонитрила, формальдегид (288 мг, 9,6 ммоль) и добавляли по каплям 0,1 мл уксусной кислоты. Полученный в результате раствор перемешивали в течение 0,5 часов с последующим добавлением триацетоксиборгидрида натрия (3,05 г, 14,40 ммоль). Реакционный раствор перемешивали в течение 12 часов, добавляли по каплям насыщенный раствор карбоната натрия до доведения рН до 10 и экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-N-[(2R)-2-гидрокси-3-(4-метил пиперазин-1-ил)пропил]карбамата 20d (830 мг, выход: 56,0%) в виде желтого масла.
МС m/z (ИЭР): 308,2 [М+1]
Стадия 5
(2R)-1 -Амино-3-(4-метил пиперазин-1-ил)пропан-2-ол
Бензил-N-[(2R)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]карбамат 20d (830 мг, 2,70 ммоль) растворяли в 40 мл метанола с последующим добавлением палладия/углерода (85 мг, 10%), заполняли водородом три раза. Реакционный раствор перемешивали в течение 12 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (2R)-1-амино-3-(4-метилпиперазин-1-ил)пропан-2-ола 20е (500 мг) в виде светло-желтого масла, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 174,2 [М+1]
Стадия 6
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин- 2-ил]амино]-N-[(2R)-3-(4-метилпиперазин-1-ил)-2-гидроксипропил]-2,2-диметил-3Н-бензофуран-4-карбоксамид 7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (116 мг, 0,25 ммоль) растворяли в 15 мл дихлорметана с последующим добавлением (2R)-1-амино-3-(4-метил пиперазин-1-ил)пропан-2-ола 20е (48 мг, 0,28 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (80 мг, 0,25 ммоль) и диизопропилэтиламина (97 мг, 0,75 ммоль). Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 20 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали водой (50 мл), насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2R)-3-(4-метилпиперазин-1-ил)-2-гидроксипропил]-2,2-диметил-3Н-бензофуран-4-карбоксамида 20 (120 мг, выход: 77,4%) в виде белого твердого вещества.
МС m/z (ИЭР): 621,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.27 (d, 1Н), 7.67 (s, 1Н), 7.13 (d, 1Н), 7.05 (s, 1Н), 6.56 (s, 1Н), 4.55 (t, 1Н), 4.22 (dd, 1Н), 3.91 (d, 1Н), 3.76-3.61 (m, 1Н), 3.45-3.28 (m, 6Н), 2.80-2.65 (m, 2Н), 2.62-2.34 (m, 7Н), 2.30 (s, 3H), 2.20-2.10 (m, 2Н), 2.1 (dd, 2Н), 1.92-1.62 (m, 8Н), 1.51 (s, 6Н), 0.88 (t, 3H).
Пример 21
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-3-(4-метилпиперазин-1-ил)-2-гидроксипропил]-2,2-диметил-3Н-бензофуран-4-карбоксамид
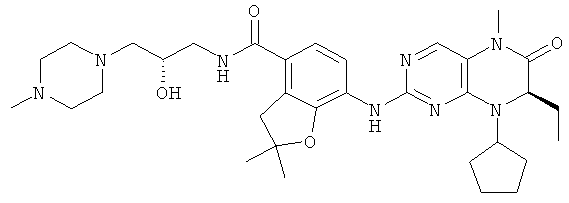
Стадия 1
трет-Бутил-4-[(2S)-3-бензилоксикарбониламино-2-гидроксипропил]пиперазин-1-карбоксилат
трет-Бутил-4-[(2S)-3-амино-2-гидроксипропил]пиперазин-1-карбоксилат 7 с (1,24 г, 4,80 ммоль) растворяли в 60 мл дихлорметана с последующим добавлением триэтиламина (970 мг, 9,60 ммоль) и карбобензоксихлорида (0,90 г, 5,28 ммоль). Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 50 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-4-[(2S)-3-бензилоксикарбониламино-2-гидроксипропил]пиперазин-1-карбоксилата 21а (1,45 г, выход: 76,0%) в виде бесцветного масла.
МС m/z (ИЭР): 394,3 [М+1]
Стадия 2
Бензил-N-[(2S)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]карбамат
трет-Бутил-4-[(2S)-3-бензилоксикарбониламино-2-гидроксипропил]пиперазин-1-карбоксилат 21а (1,45 г, 3,70 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением 20 мл 4 М раствора хлорида водорода в диоксане. Реакционный раствор перемешивали в течение 0,5 часов и концентрировали при пониженном давлении. К остатку последовательно добавляли 40 мл дихлорметана, формальдегид (222 мг, 7,40 ммоль) и 0,1 мл уксусной кислоты. Полученный в результате раствор перемешивали в течение 0,5 часа с последующим добавлением триацетоксиборгидрида натрия (2,35 г, 11,10 ммоль), перемешивали еще в течение 12 часов. К полученному в результате раствору добавляли 50 мл воды и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-N-[(2S)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]карбамата 21 b (840 мг, выход: 74,0%) в виде желтого масла.
МС m/z (ИЭР): 308,3 [М+1]
Стадия 3
(2S)-1 -Амино-3-(4-метил пиперазин-1-ил)пропан-2-ол
Бензил-N-[(2S)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]карбамат 21b (840 мг, 2,70 ммоль) растворяли в 50 мл метанола с получением раствора 0,01 моль/л. Реакционный раствор подвергали гидрогенизации с помощью H-Cube (колонка катализатора: 10% палладий/углерод; температура: 30°С; скорость тока: 1 мл/мин; давление: 1 атмосфера) в течение 50 минут. Реакционный раствор концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (2S)-1-амино-3-(4-метилпиперазин-1-ил)пропан-2-ола 21 с (500 мг) в виде желтого масла, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 174,2 [М+1]
Стадия 4
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]-2,2-диметил-3Н-бензофуран-4-карбоксамид
Сырое соединение 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (160 мг, 0,35 ммоль), (2S)-1-амино-3-(4-метилпиперазин-1-ил)пропан-2-ол 21 с (60 мг, 0,35 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (111 мг, 0,35 ммоль) и диизопропилэтиламин (89 мг, 0,69 ммоль) растворяли в 25 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 8-9 и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали водой (50 мл), насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-N-[(2S)-3-(4-метилпиперазин-1-ил)-2-гидрокси-пропил]-1,2-диметил-3Н-бензофуран-4-карбоксамида 21 (90 мг, выход: 45,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 621,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.26 (d, 1Н), 7.67 (s, 1Н), 7.19-6.97 (m, 2Н), 6.61 (s, 1Н), 4.55 (t, 1Н), 4.22 (dd, 1Н), 3.99-3.83 (m, 1Н), 3.77-3.62 (m, 1Н), 3.45-3.27 (m, 6Н), 2.80-2.65 (m, 2Н), 2.63-2.34 (m, 8Н), 2.30 (s, 3H), 2.20-2.10 (m, 2Н), 2.08-1.92 (m, 1Н), 1.90-1.61 (m, 8Н), 1.51 (s, 6Н), 0.88 (t, 3H).
Пример 22
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
Стадия 1
Метил-(2R)-2-(изопропиламино)бутаноат
Метил-(2R)-2-амино-бутаноат 1j (28,78 г, 0,19 моль) растворяли в 200 мл дихлорметана с последующим добавлением ацетона (11,96 г, 0,21 моль) и ацетата натрия (30,76 г, 0,38 моль), перемешивали 3 часа. Добавляли триацетоксиборгидрид натрия (59,61 г, 0,28 моль). Реакционный раствор перемешивали в течение 12 часов с последующим добавлением 100 мл 1 М соляной кислоты и добавлением по каплям насыщенного раствора карбоната натрия до доведения рН до 8-9, экстрагировали дихлорметаном (100 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-(2R)-2-(изопропиламино)бутаноата 22а (16,40 г, выход: 55,0%) в виде светло-желтой маслянистой жидкости.
Стадия 2
Метил-(2R)-2-[(2-хлор-5-нитропиримидин-4-ил)-изопропиламино] бутаноат
2,4-Дихлор-5-нитропиримидин (19,90 г, 103 ммоль) растворяли в 300 мл циклогексана с последующим добавлением метил-(2R)-2-(изопропиламино)бутаноата 22а (16,40 г, 103 ммоль) и бикарбоната натрия (36,61 г, 412 ммоль). Реакционный раствор нагревали до 80°С, перемешивали в течение 4 часов и фильтровали. Фильтрат концентрировали при пониженном давлении, добавляли 100 мл воды и экстрагировали дихлорметаном (100 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (100 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-(2R)-2-[(2-хлор-5-нитропиримидин-4-ил)-изопропиламино]бутаноата 22b (20,50 г, выход: 62,8%) в виде желтого твердого вещества.
Стадия 3
(7R)-2-Хлор-7-этил-8-изопропил-5,7-дигидроптеридин-6-он
Метил-(2R)-2-[(2-хлор-5-нитропиримидин-4-ил)-изопропиламино]бутаноат 22b (7,30 г, 23 ммоль) растворяли в 100 мл уксусной кислоты с последующим добавлением 5 г никеля Ренея, повторяли заполнение водородом три раза. Реакционную смесь нагревали до 75°С, перемешивали в течение 2 часов и фильтровали. Фильтрат концентрировали при пониженном давлении, добавляли 30 мл ледяной воды, что привело в результате к образованию осадка. Осадок фильтровали, фильтрационный кек высушивали нагреванием с получением сырого соединения, указанного в заголовке, (7R)-2-хлор-7-этил-8-изопропил-5,7-дигидроптеридин-6-она 22с (12,10 г, выход: 71,7%) в виде серого твердого вещества, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 255,1 [М+1]
Стадия 4
(7R)-2-Хлор-7-этил-8-изопропил-5-метил-7Н-птеридин-6-он
Сырое соединение (7R)-2-хлор-7-этил-8-изопропил-5,7-дигидроптеридин-6-он 22с (12,10 г, 47,50 ммоль) растворяли в 180 мл ацетона с последующим добавлением метил-пара-толуолсульфоната (13,28 г, 71,30 ммоль) и карбоната калия (13,11 г, 95 ммоль). Реакционный раствор нагревали до образования флегмы в течение 3 часов при перемешивании и фильтровали. Фильтрат концентрировали при пониженном давлении, добавляли 100 мл воды и экстрагировали дихлорметаном (100 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (100 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (7R)-2-хлор-7-этил-8-изопропил-5-метил-7Н-птеридин-6-она 22d (10,80 г, выход: 84,7%) в виде белого твердого вещества.
Стадия 5
7-Амино-2,3-дигидробензофуран-4-карбоновая кислота
Метил-7-амино-2,3-дигидробензофуран-4-карбоксилат 1д (2,30 г, 11,90 ммоль) растворяли в 80 мл смеси растворителей метанола и тетрагидрофурана (об./об. = 1:1) с последующим добавлением 1 М раствора гидроксида лития (47,6 мл, 47,60 ммоль) в тетрагидрофуране. Реакционный раствор нагревали до образования флегмы в течение 5 часов, затем охлаждали до комнатной температуры, перемешивали в течение 12 часов. К реакционному раствору добавляли 20 мл воды и концентрировали при пониженном давлении. К полученному в результате остатку добавляли 100 мл воды, экстрагировали этилацетатом (50 мл×2), к объединенной водной фазе добавляли по каплям 1 М соляную кислоту до доведения рН до 3, что привело в результате к образованию осадка. Осадок фильтровали с получением соединения, указанного в заголовке, 7-амино-2,3-дигидробензофуран-4-карбоновой кислоты 22е (1,70 г, выход: 80,0%) в виде серого твердого вещества.
МС m/z (ИЭР): 178,1 [М-1]
Стадия 6
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновая кислота
(7R)-2-Хлор-7-этил-8-изопропил-5-метил-7Н-птеридин-6-он 22d (82 мг, 3,07 ммоль) и 7-амино-2,3-дигидробензофуран-4-карбоновую кислоту 22е (500 мг, 2,80 ммоль) растворяли в 34 мл смеси растворителей этанола и воды (об./об. = 1:2,4) с последующим добавлением 0,4 мл концентрированной соляной кислоты. Реакционный раствор нагревали до образования флегмы в течение 15 часов и охлаждали, что привело в результате к образованию осадка. Осадок фильтровали с получением соединения, указанного в заголовке, 7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновой кислоты 22f (0,75 г, выход: 65,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 410,2 [М-1]
Стадия 7
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 22f (120 мг, 0,29 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (93 мг, 0,29 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением диизопропилэтиламина (83 мг, 0,64 ммоль) и 1-метилпиперидил-4-иламина (33 мг, 0,29 ммоль) последовательно. Реакционный раствор перемешивали в течение 3 часов. Полученный в результате раствор последовательно промывали насыщенным раствором карбоната натрия (30 мл) и насыщенным раствором хлорида натрия (30 мл). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамида 22 (117 мг, выход: 80,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 508,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.31 (d, 1Н), 7.67 (s, 1Н), 7.13-7.01 (m, 2Н), 5.96 (d, 1Н), 4.82-4.61 (m, 3H), 4.29 (dd, 1Н), 4.06 (d, 1Н), 3.59 (t, 2Н), 3.32 (s, 3H), 3.06 (d, 2н), 2.54-2.30 (т,5Н), 2.10 (d,2H), 1.98-1.80 (m, 2Н), 1.73 (td, 2Н), 1.43 (d, 3H), 1.36 (d, 3H), 0.88 (t, 3H).
Пример 23
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(цис)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,2-диметил-3Н-бензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,2-диметил-3Н-бензофуран-4-карбоновую кислоту 4f (116 мг, 0,25 ммоль) растворяли в 15 мл дихлорметана с последующим добавлением (цис)-4-[4-(цикпопропилметил)пиперазин-1-ил]циклогексанамина 12е (65 мг, 0,28 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (80 мг, 0,25 ммоль) и диизопропилэтиламина (97 мг, 0,75 ммоль). Реакционный раствор перемешивали в течение 3 часов. К полученному в результате раствору добавляли 20 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали водой (50 мл), насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(цис)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,2-диметил-3H-бензофуран-4-карбоксамида 23 (135 мг, выход: 78,9%) в виде белого твердого вещества.
МС m/z (ИЭР): 685,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.27 (d, 1Н), 7.68 (s, 1Н), 7.08-6.98 (m, 2Н), 6.09 (d, 1Н), 4.58 (t, 1Н), 4.22 (dd, 2Н), 3.40 (s, 2Н), 3.33 (s, 3H), 2.95-2.40 (m, 7Н), 2.33-2.24 (m, 3H), 2.19-2.13 (m, 1Н), 2.13-1.95 (m, 1Н), 1.94-1.64 (m, 16Н), 1.62-1.55 (m, 2н), 1.51 (s, 6Н), 0.87 (t, 3H), 0.57-0.48 (m, 2Н), 0.19-0.10 (m, 2Н).
Пример 24
(цис-эндо)-N-[2-Метил-3,3а,4,5,6,6а-гексагидро-7H-циклопента[с]пиррол-5-ил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
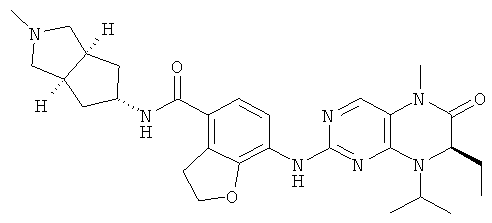

7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 22f (120 мг, 0,29 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (93 мг, 0,29 ммоль) растворяли в 30 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,64 ммоль) и (цис-эндо)-2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-амина 9f (41 мг, 0,29 ммоль). Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора карбоната натрия, перемешивали 10 минут и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали насыщенным раствором карбоната натрия (50 мл), водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (цис-эндо)-N-[2-метил-3,3а,4,5,6,6а-гексагидро-1Н-циклопента[с]пиррол-5-ил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 24 (80 мг, выход: 52,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 534,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.66 (s, 1Н), 7.11-7.00 (m, 2Н), 5.98 (d, 1Н), 4.73-4.67 (m, 3H), 4.62-4.54 (m, 1Н), 4.28 (dd, 1Н), 3.59 (t, 2Н), 3.32 (s, 3H), 3.22-3.12 (m, 2Н), 3.05-2.90 (m, 2Н), 2.52 (s, 3H), 2.39 (dd, 2Н), 1.98 (dd, 2Н), 1.91 (ddd, 1Н), 1.79-1.66 (m, 3H), 1.42 (d, 3H), 1.25 (d, 3H), 0.88 (t, 3H).
Пример 25
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2R)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамид
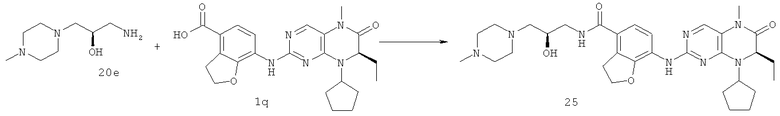
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (150 мг, 0,34 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (110 мг, 0,34 ммоль) растворяли в 30 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,75 ммоль) и (2R)-1-амино-3-(4-метилпиперазин-1-ил)пропан-2-ола 20е (59 мг, 0,34 ммоль). Реакционный раствор перемешивали в течение 3 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали насыщенным раствором карбоната натрия (50 мл×3), водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2R)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамида 25 (120 мг, выход: 60,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 593,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.67 (s, 1Н), 7.15 (d, 1Н), 7.05 (s, 1Н), 6.62 (t, 1Н), 4.68 (t, 2Н), 4.47 (t, 1Н), 4.21 (dd, 1Н), 3.98-3.92 (m, 1Н), 3.72-3.65 (m, 1Н), 3.60 (t, 2Н), 3.42-3.35 (m, 1Н), 3.32 (s, 3H), 2.85-2.70 (m, 3H), 2.62-2.43 (m, 8Н), 2.34 (s, 3H), 2.15-2.08 (m, 1Н), 2.03-1.94 (m, 1Н), 1.88-1.78 (m, 4Н), 1.70 (m, 4Н), 0.88 (t, 3H).
Пример 26
N-[[(38,8а5)-3,4,6,7,8,8а-Гексагидро-7H-пирроло[2,1-с][1,4]оксазин-3-ил]метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
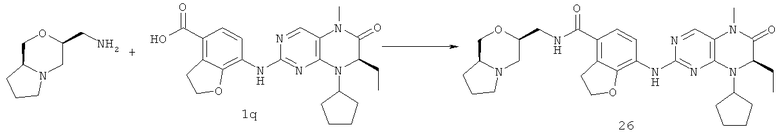
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (160 мг, 0,36 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (117 мг, 0,36 ммоль) растворяли в 30 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,80 ммоль) и (3S,8aS)-гексагидропирроло[2,1-с][1,4]оксазин-3-ил]метанамина (57 мг, 0,36 ммоль, полученного хорошо известным способом, раскрытым в заявке на патент CN 101392001). Реакционный раствор перемешивали в течение 2 часов. Полученный в результате раствор последовательно промывали насыщенным раствором карбоната натрия (30 мл), насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом натрия, фильтровали и, фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[[(3S,8aS)-3,4,6,7,8,8а-гексагидро-1Н-пирроло[2,1-с][1,4]оксазин-3-ил]метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 26 (140 мг, выход: 67,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 576,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.67 (s, 1Н), 7.13 (d, 1Н), 7.04 (s, 1Н), 6.45-6.37 (m, 1Н), 4.68 (t, 2Н), 4.47 (t, 1Н), 4.21 (dd, 1Н), 4.05 (dd, 1Н), 3.78-3.69 (m, 2Н), 3.59 (t, 2Н), 3.50-3.38 (m, 2Н), 3.32 (s, 3H), 3.14 (s, 2Н), 2.31-2.08 (m, 3H), 2.05-1.95 (m, 2Н), 1.89-1.76 (m, 6Н), 1.69 (m, 6Н), 0.88 (m, 3H).
Пример 27
N-[[(3S,8aR)-3,4,6,7,818а-Гексагидро-7Н-пирроло[2,1-с][1,4]оксазин-3-ил]метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
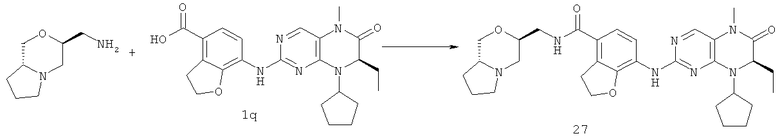
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (160 мг, 0,36 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (117 мг, 0,36 ммоль) растворяли в 30 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,80 ммоль) и (38,8аЯ?)-гексагидропирроло[2,1-с][1,4]оксазин-3-ил]метанамина (57 мг, 0,36 ммоль, получен хорошо известным способом, раскрытым в заявке на патент CN 101392001). Реакционный раствор перемешивали в течение 12 часов. Полученный в результате раствор последовательно промывали насыщенным раствором карбоната натрия (30 мл), насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[[(3S,8aR)-3,4,6,7,8,8а-гексагидро-1Н-пирроло[2,1-с][1,4]оксазин-3-ил]метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7R-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 27 (120 мг, выход: 57,4%) в виде белого твердого вещества.
МС m/z (ИЭР): 576,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.28 (d, 1Н), 7.67 (s, 1Н), 7.11 (d, 1Н), 7.04 (s, 1Н), 6.86-6.74 (m, 1Н), 4.68 (t, 2Н), 4.48 (t, 1Н), 4.21 (dd, 1Н), 3.97-3.85 (m, 2Н), 3.75-3.68 (m, 3H), 3.61 (t, 2Н), 3.32 (s, 3H), 3.03-2.95 (m, 1Н), 2.80 (dd, 1Н), 2.55 (dd, 1Н), 2.39-2.25 (m, 2Н), 2.12 (m, 1Н), 1.99 (m, 1Н), 1.91-1.78 (т,5Н), 1.76-1.64 (m, 7Н), 0.88 (t, 3H).
Пример 28
N-[[(3R,8aR)-3,4,6,7,8,8а-Гексагидро-7Н-пирроло[2,1-с][1,4]оксазин-3-ил]метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
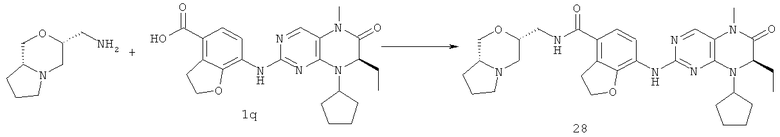
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (160 мг, 0,36 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (117 мг, 0,36 ммоль) растворяли в 30 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,80 ммоль) и (3£?,8aR)-гексагидропирроло[2,1-с][1,4]оксазин-3-ил]метанамина (57 мг, 0,36 ммоль, получен хорошо известным способом, раскрытым в заявке на патент CN 101392001) последовательно. Реакционный раствор перемешивали в течение 12 часов. Полученный в результате раствор последовательно промывали насыщенным раствором карбоната натрия (30 мл), насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[[(3R,8aR)-3,4,6,7,8,8а-гексагидро-1Н-пирроло[2,1-с][1,4]оксазин-3-ил]метил]-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 28 (128 мг, выход: 61,2%) в виде белого твердого вещества.
МС m/z (ИЭР): 576,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.67 (s, 1Н), 7.13 (d, 1Н), 7.04 (s, 1Н), 6.42 (s, 1Н), 4.68 (t, 2Н), 4.46 (d, 1Н), 4.21 (dd, 1Н), 4.04 (dd, 1Н), 3.79-3.67 (m, 2Н), 3.59 (t, 2Н), 3.41 (dd, 2Н), 3.32 (s, 3H), 3.10 (d, 2Н), 2.27-2.06 (m, 3H), 2.05-1.95 (m, 2Н), 1.91-1.75 (m, 6Н), 1.75-1.57 (m, 6Н), 0.88 (t, 3H).
Пример 29
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(диметиламинометил)-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
Стадия 1
Бензил-3-гидрокси-4-нитробензоат
3-Гидрокси-4-нитробензойную кислоту 1а (10 г, 54,60 ммоль) растворяли в 125 мл толуола с последующим добавлением бензилового спирта (9 г, 83,20 ммоль) и пара-толуолсульфоновой кислоты (1 г, 5,30 ммоль). Реакционный раствор нагревали до образования флегмы в течение 16 часов в водоотделителе. К полученному в результате раствору добавляли 50 мл этилацетата, последовательно промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-3-гидрокси-4-нитробензоата 29а (8 г, выход: 53,7%) в виде желтого твердого вещества.
МС m/z (ИЭР): 272,1 [М-1]
Стадия 2
Бензил-3-аллилокси-4-нитробензоат
Бензил-3-гидрокси-4-нитробензоат 29а (4,50 г, 16,47 ммоль) растворяли в 75 мл безводного ацетонитрила. К полученному в результате раствору последовательно добавляли карбонат калия (6,83 г, 49,41 ммоль) и 3-бромпропен (2,9 мл, 32,94 ммоль), нагревали до образования флегмы в течение 3 часов, затем фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-3-аллилокси-4-нитробензоата 29b (4,86 г, выход: 94,2%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ (млн-1) 7.85-7.81 (d, 1Н), 7.80 (s, 1Н), 7.77-7.74 (m, 1Н), 7.50-7.39 (m, 5Н), 6.12-6.02 (m, 1Н), 5.56-5.51 (d, 2Н), 5.50-5.38 (m, 2н), 4.79-4.77 (m, 2Н).
Стадия 3
Бензил-2-аллил-3-гидрокси-4-нитробензоат
Бензил-3-аллилокси-4-нитробензоат 29b (2,19 г, 7 ммоль) помещали в трехгорлую колбу, нагревали до 190°С и перемешивали в течение 3,5 часов. Полученный в результате раствор охлаждали до комнатной температуры, добавляли 50 мл дихлорметана и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-2-аллил-3-гидрокси-4-нитробензоата 29 с (1,36 г, выход: 62,1%) в виде желтой жидкости.
МС m/z (ИЭР): 312,1 [М-1]
Стадия 4
Бензил-2-(гидроксиметил)-7-нитро-2,3-дигидробензофуран-4-карбоксилат
Бензил-2-аллил-3-гидрокси-4-нитробензоат 29 с (1,36 г, 4,30 ммоль) растворяли в 100 мл 1,2-дихлорэтана с последующим добавлением мета-хлорпербензойной кислоты (1,18 г, 4,77 ммоль). Реакционный раствор нагревали до 70°С и перемешивали в течение 3 часов. Полученный в результате раствор охлаждали до комнатной температуры, добавляли 20 мл насыщенного раствора тиосульфата натрия, перемешивали в течение 30 минут и экстрагировали этилацетатом (20 мл×3). Объединенную органическую фазу последовательно промывали водой (20 мл), насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-2-(гидроксиметил)-7-нитро-2,3-дигидробензофуран-4-карбоксилата 29d (430 мг, выход: 30,1%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 330,2 [М+1]
Стадия 5
Бензил-7-нитро-2-(толуил-4-сульфонилоксиметил)-2,3-дигидробензофуран-4-карбоксилат
Бензил-2-(гидроксиметил)-7-нитро-2,3-дигидробензофуран-4-карбоксилат 29d (420 мг, 1,27 ммоль) растворяли в 25 мл дихлорметана с последующим добавлением пара-толуолсульфонилхлорида (729 мг, 3,83 ммоль) и триэтиламина (514 мг, 5,08 ммоль) в бане лед-вода. Реакционный раствор подогревали до комнатной температуры и перемешивали в течение 2 часов. К полученному в результате раствору добавляли 5 мл воды и экстрагировали этилацетатом (20 мл×3). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-7-нитро-2-(толуил-4-сульфонилоксиметил)-2,3-дигидробензофуран-4-карбоксилата 29е (524 мг, выход: 85,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 501,3 [М+18]
Стадия 6
Бензил-2-(диметиламинометил)-7-нитро-2,3-дигидробензофуран-4-карбоксилат
Диметиламина гидрохлорид (424 мг, 5,20 ммоль) растворяли в 20 мл ацетонитрила с последующим добавлением триэтиламина (737 мг, 7,28 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 20 минут с последующим добавлением бензил-7-нитро-2-(толуил-4-сульфонилоксиметил)-2,3-дигидробензофуран-4-карбоксилата 29е (510 мг, 1,04 ммоль) и 10 мг йодида калия. Реакционный раствор нагревали до образования флегмы в течение 3 часов. К полученному в результате раствору добавляли 5 мл воды и экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу последовательно промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-2-(диметиламинометил)-7-нитро-2,3-дигидробензофуран-4-карбоксилата 29f (249 мг, выход: 67,1%) в виде желтой маслянистой жидкости.
МС m/z (ИЭР): 357,2 [М+1]
Стадия 7
Бензил-7-амино-2-(диметиламинометил)-2,3-дигидробензофуран-4-карбоксилат
Бензил-2-(диметиламинометил)-7-нитро-2,3-дигидробензофуран-4-карбоксилат 29f (240 мг, 0,67 ммоль) растворяли в 30 мл уксусной кислоты с последующим добавлением порошка железа (189 мг, 3,37 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный в результате раствор концентрировали при пониженном давлении, добавляли 20 мл воды и 20 мл дихлорметана, затем добавляли по каплям 2 М раствор карбоната натрия до доведения рН до примерно 10 и экстрагировали дихлорметаном (30 мл×3). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, бензил-7-амино-2-(диметиламинометил)-2,3-дигидробензофуран-4-карбоксилата 29g (212 мг, выход: 97,2%) в виде желтой маслянистой жидкости.
МС m/z (ИЭР): 327,2 [М+1]
Стадия 8
Бензил-7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(диметиламинометил)-2,3-дигидробензофуран-4-карбоксилат
Бензил-7-амино-2-(диметиламинометил)-2,3-дигидробензофуран-4-карбоксилат 29g (212 мг, 0,65 ммоль) растворяли в 20 мл 4-метил-2-пентанола с последующим добавлением (7R)-2-хлор-8-циклопентил-7-этил-5-метил-7Н-птеридин-6-она 1о (230 мг, 0,78 ммоль) и пара-толуолсульфоновой кислоты моногидрата (198 мг, 1,04 ммоль). Реакционный раствор нагревали до образования флегмы в течение 3 часов при перемешивании и концентрировали при пониженном давлении. К полученному в результате остатку добавляли 30 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали водой (30 мл) и насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, бензил-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(диметиламинометил)-2,3-дигидробензофуран-4-карбоксилата 29п (0,13 г, выход: 34,2%) в виде белого твердого вещества.
МС m/z (ИЭР): 585,5 [М+1]
Стадия 9
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(диметиламинометил)-2,3-дигидробензофуран-4-карбоновая кислота
Бензил-7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(диметиламинометил)-2,3-дигидробензофуран-4-карбоксилат 29h (120 мг, 0,20 ммоль) растворяли в 20 мл метанола с последующим добавлением палладия/углерода (12 мг, 10%). Реакционный раствор перемешивали в течение 12 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(диметиламинометил)-2,3-дигидробензофуран-4-карбоновой кислоты 29j (30 мг, выход: 25,8%) в виде белого твердого вещества.
МС m/z (ИЭР): 493,3 [М-1]
Стадия 10
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2-(диметиламинометил)-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид 7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(диметиламинометил)-2,3-дигидробензофуран-4-карбоновую кислоту 29j (30 мг, 0,06 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением 1-метилпиперидил-4-иламина (7 мг, 0,06 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (20 мг, 0,06 ммоль) и диизопропилэтиламина (24 мг, 0,18 ммоль). Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 10 мл насыщенного раствора хлорида натрия и экстрагировали дихлорметаном (10 мл×3). Объединенную органическую фазу последовательно промывали водой (10 мл) и насыщенным раствором хлорида натрия (10 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(диметиламинометил)-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамида 29 (27 мг, выход: 77,1%) в виде белого твердого вещества.
МС m/z (ИЭР): 591,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.22 (d, 1Н), 7.79 (s, 1Н), 7.33 (d, 1Н), 5.34 (d, 1Н), 4.56 (d, 1Н), 4.27 (dd, 1Н), 4.11 (t, 1Н), 3.78 (dd, 1Н), 3.51 (d, 2Н), 3.44-3.36 (m, 1Н), 3.40-3.20 (m, 7Н), 3.19-3.10 (m, 2Н), 2.93 (s, 6Н), 2.84 (s, 3Н), 2.23-2.10 (m, 3Н), 2.05-1.94 (m, 3Н), 1.91-1.81 (m, 4Н), 1.81-1.66 (m, 4Н), 0.88 (t, 3Н).
Пример 30
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-метил-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
Стадия 1
Метил-2-метил-7-нитро-2,3-дигидробензофуран-4-карбоксилат
Метил-2-аллил-3-гидрокси-4-нитробензоат 1d (800 мг, 3,30 ммоль) растворяли в 10 мл 1,2-дихлорэтана с последующим добавлением 1 мл пара-толуолсульфоновой кислоты. Реакционный раствор перемешивали в течение 48 часов. К полученному в результате раствору добавляли 10 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-2-метил-7-нитро-2,3-дигидробензофуран-4-карбоксилата 30а (100 мг, выход: 12,5%) в виде желтого твердого вещества.
Стадия 2
Метил-7-амино-2-метил-2,3-дигидробензофуран-4-карбоксилат
Метил-2-метил-7-нитро-2,3-дигидробензофуран-4-карбоксилат 30а (100 мг, 0,42 ммоль) растворяли в 20 мл метанола с последующим добавлением палладия/углерода (10 мг, 10%), заполняли водородом три раза. Реакционный раствор перемешивали в течение 12 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, метил-7-амино-2-метил-2,3-дигидробензофуран-4-карбоксилата 30B (80 мг, выход: 91,0%) в виде бесцветной маслянистой жидкости.
Стадия 3
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2-метил-2,3-дигидробензофуран-4-карбоксилат
Метил-7-амино-2-метил-2,3-дигидробензофуран-4-карбоксилат 30B (80 мг, 0,38 ммоль), (7R)-2-хлор-8-циклопентил-7-этил-5-метил-7Н-птеридин-6-он 1о (113 мг, 0,38 ммоль) и пара-толуолсульфоновой кислоты моногидрат (115 мг, 0,60 ммоль) растворяли в 20 мл 4-метил-2-пентанола. Реакционный раствор нагревали до образования флегмы в течение 2,5 часов при перемешивании. Полученный в результате раствор концентрировали при пониженном давлении с последующим добавлением 30 мл насыщенного раствора бикарбоната натрия и экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу последовательно промывали водой (30 мл) и насыщенным раствором хлорида натрия (30 мл) затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали на препаративной пластине с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-метил-2,3-дигидробензофуран-4-карбоксилата 30 с (0,10 г, выход: 56,0%) в виде белого твердого вещества.
Стадия 4
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-метил-2,3-дигидробензофуран-4-карбоновая кислота
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-метил-2,3-дигидробензофуран-4-карбоксилат 30 с (100 мг, 0,21 ммоль) и 1 М раствор гидроксида лития растворяли в 50 мл смеси растворителей метанола и тетрагидрофурана (об./об. = 1:1). Реакционный раствор перемешивали в течение 12 часов с последующим добавлением 1 М соляной кислоты до доведения рН до 6-7. Полученный в результате раствор концентрировали при пониженном давлении. К остатку добавляли по каплям 1 М раствор гидроксида лития до доведения рН до 3-4 и экстрагировали этилацетатом (30 мл×3). Объединенную органическую фазу концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-метил-2,3-дигидробензофуран-4-карбоновой кислоты 30d (90 мг, выход: 95,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 450,2 [М-1]
Стадия 5
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-метил-А/-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид 7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-метил-2,3-дигидробензофуран-4-карбоновую кислоту 30d (70 мг, 0,20 ммоль), 1-метилпиперидил-4-иламин (23 мг, 0,20 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (64 мг, 0,20 ммоль) и диизопропилэтиламин (65 мг, 0,50 ммоль) растворяли в 30 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 20 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали водой (30 мл) и насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-метил-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамида 30 (40 мг, выход: 36,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 548,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.32-8.23 (m, 1Н), 7.67 (s, 1Н), 7.11-7.01 (m, 2Н), 5.94-5.84 (m, 1Н), 5.10-4.97 (m, 1Н), 4.59-4.45 (m, 1Н), 4.28-4.16 (m, 1Н), 3.99 (s, 1Н), 3.78-3.61 (m, 2Н), 3.33 (m, 3Н), 3.23-3.12 (m, 1Н), 2.97-2.81 (m, 2Н), 2.34 (s, 3Н), 2.29-2.15 (m, 2Н), 2.13-1.89 (m, 6Н), 1.88-1.78 (m, 3Н), 1.77-1.57 (m, 4Н), 1.50 (d, 3Н), 0.88 (t, 3Н).
Пример 31
N-[(2S)-3-[4-(Циклопропилметил)пиперазин-1-ил]-2-гидроксипропил]-7-[[(7Н)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
Стадия 1
трет-Бутил-4-(циклопропилметил)пиперазин-1-карбоксилат
трет-Бутил пиперазин-1-карбоксилат 7а (6,14 г, 33 ммоль), бромметилциклопропан (4,05 г, 30 ммоль) и триэтиламин (6,06 г, 60 ммоль) растворяли в 70 мл дихлорметана. Реакционный раствор перемешивали в течение 12 часов. К полученному в результате раствору добавляли 50 мл водного насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-4-(циклопропилметил)пиперазин-1-карбоксилата 31а (4,50 г, выход: 62,5%) в виде желтого масла.
МС m/z (ИЭР): 241,2 [М+1]
Стадия 2
1-(Циклопропилметил)-4-[[(2R)-оксиран-2-ил]метил]пиперазин
трет-Бутил-4-(циклопропилметил)пиперазин-1-карбоксилат 31а (2 г, 8,30 ммоль) растворяли в 40 мл дихлорметана с последующим добавлением 20 мл 4 М раствора хлорида водорода в диоксане. Реакционный раствор перемешивали в течение 0,5 часа. Реакционный раствор концентрировали при пониженном давлении с последующим добавлением 40 мл дихлорметана и добавлением по каплям 20 мл триэтиламина до доведения рН до 10-11. Полученный в результате раствор концентрировали при пониженном давлении. К остатку добавляли 50 мл этилацетата и фильтровали. Фильтрат концентрировали при пониженном давлении, добавляли 50 мл этилацетата и 20 мл воды, затем добавляли (£?)-2-хлорметилоксиран (0,92 г, 9,96 ммоль) и гидроксид натрия (0,66 г, 16,60 ммоль). Реакционный раствор перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли 50 мл воды и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 1-(циклопропилметил)-4-[[(2R)-оксиран-2-ил]метил]пиперазина 31 b (0,91 г, выход: 57,0%) в виде желтого масла.
МС m/z (ИЭР): 197,3 [М+1]
Стадия 3
(2S)-1-Амино-3-[4-(циклопропилметил)пиперазин-1-ил]-изопропанол
1-(Циклопропилметил)-4-[[(2R)-оксиран-2-ил]метил]пиперазин 31 b (908 мг, 4,62 ммоль) растворяли в 40 мл этанола с последующим добавлением 10 мл водного аммиака. Реакционный раствор перемешивали в течение 12 часов. Полученный в результате раствор концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (2S)-1-амино-3-[4-(циклопропилметил)пиперазин-1-ил]-изопропанола 31 с (0,74 г) в виде светло-желтого масла, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 214,2 [М+1]
Стадия 4
N-[(2S)-3-[4-(Циклопропилметил)пиперазин-1-ил]-2-гидроксипропил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 22f (150 мг, 0,36 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (117 мг, 0,36 ммоль) растворяли в 30 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,80 ммоль), перемешивали до тех пор, пока раствор не становился прозрачным, затем добавляли сырое соединение (2S)-1-амино-3-[4-(циклопропилметил)пиперазин-1-ил]-изопропанол 31 с (78 мг, 0,36 ммоль), перемешивали в течение 2 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[(2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-гидроксипропил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 31 (50 мг, выход: 23,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 607,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.30 (d, 1Н), 7.67 (s, 1Н), 7.16 (d, 1Н), 7.08 (s, 1Н), 6.54-6.61 (m, 1Н), 4.74-4.62 (m, 3Н), 4.29 (dd, 1Н), 3.97-3.88 (m, 1Н), 3.76-3.65 (m, 1Н), 3.61 (t, 2Н), 3.37 (td, 1Н), 3.33 (s, 3Н), 2.78-2.63 (m, 4Н), 2.59-2.38 (m, 5Н), 2.30 (d, 2Н), 1.91 (m, 2Н), 1.74 (qd, 2Н), 1.43 (d, 3Н), 1.35 (d, 3Н), 1.32-1.23 (m, 1Н), 0.88 (t, 3Н), 0.58-0.49 (m, 2Н), 0.13 (q, 2Н).
Пример 32
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-N-[(2S)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамид
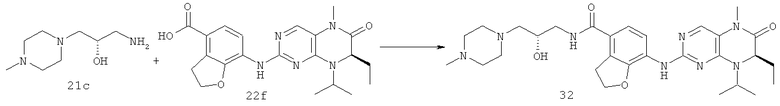
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 22f (150 мг, 0,36 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (117 мг, 0,36 ммоль) растворяли в 30 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,8 ммоль) и (2S)-1-амино-3-(4-метилпиперазин-1-ил)пропан-2-ола 21 с (63 мг, 0,36 ммоль) последовательно. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 20 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамида 32 (40 мг, выход: 20,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 567,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.31 (d, 1Н), 7.67 (s, 1Н), 7.16 (d, 1Н), 7.08 (s, 1Н), 6.61-6.55 (m, 1Н), 4.74-4.63 (m, 3Н), 4.29 (dd, 1Н), 3.94 (d, 1Н), 3.74-3.65 (m, 1Н), 3.61 (t, 2Н), 3.42-3.29 (m, 4Н), 2.77-2.53 (m, 2Н), 2.66-2.39 (m, 7Н), 2.34 (s, 3Н), 1.91 (dtd, 2Н), 1.74 (dt, 2Н), 1.35 (d, 3Н), 1.28 (d, 3Н), 0.88 (t, 3Н).
Пример 33
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2R)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамид
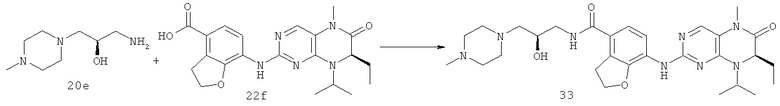
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 22f (150 мг, 0,36 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (117 мг, 0,36 ммоль) растворяли в 30 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,80 ммоль) и (2R)-1-амино-3-(4-метилпиперазин-1-ил)пропан-2-ола 20е (63 мг, 0,36 ммоль) последовательно. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2R)-2-гидрокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамида 33 (78 мг, выход: 40,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 567,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.66 (s, 1Н), 7.15 (d, 1Н), 7.06 (s, 1Н), 6.59 (t, 1Н), 4.72-4.63 (m, 3Н), 4.28 (dd, 1Н), 3.94 (d, 1Н), 3.75-3.65 (m, 1Н), 3.59 (t, 2Н), 3.37 (d, 1Н), 3.31 (s, 3Н), 2.79-2.56 (m, 6Н), 2.53-2.38 (m, 3Н), 2.34 (s, 3Н), 1.90 (dt, 2н), 1.73 (dt, 2Н), 1.41 (d, 3Н), 1.35 (d,3H), 0.88 (t, 3Н).
Пример 34
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,3-дигидробензофуран-4-карбоксамид
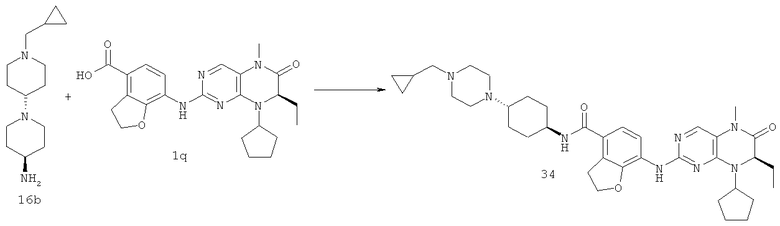
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (140 мг, 0,346 ммоль), (транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамин 16b (76 мг, 0,35 ммоль), О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (102 мг, 0,32 ммоль) и диизопропилэтиламин (103 мг, 0,80 ммоль) растворяли в 25 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли по каплям насыщенный раствор бикарбоната натрия до доведения рН до 8-9 и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-N-[(транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-2,3-дигидробензофуран-4-карбоксамида 34 (50 мг, выход: 40,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 329,4 [М/2+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.26 (d, 1Н), 7.67 (s, 1Н), 7.07-6.99 (m, 2Н), 5.79 (d, 1Н), 4.74-4.63 (m, 3Н), 4.47 (s, 1Н), 4.21 (dd, 1Н), 3.59 (t, 2Н), 3.32 (s, 3Н), 2.75-2.40 (m, 7Н), 2.30-2.19 (m, 2Н), 2.18-2.16 (m, 2Н), 2.01-1.85 (m, 2Н), 1.92-1.76 (m, 3Н), 1.76-1.50 (m, ЮН), 1.44 (d, 2Н), 1.36-1.16 (m, 2Н), 0.87 (m, 4Н), 0.56-0.47 (m, 2Н), 0.17-0.07 (m, 2Н).
Пример 35
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-метоксипропил]-2,3-дигидробензофуран-4-карбоксамид
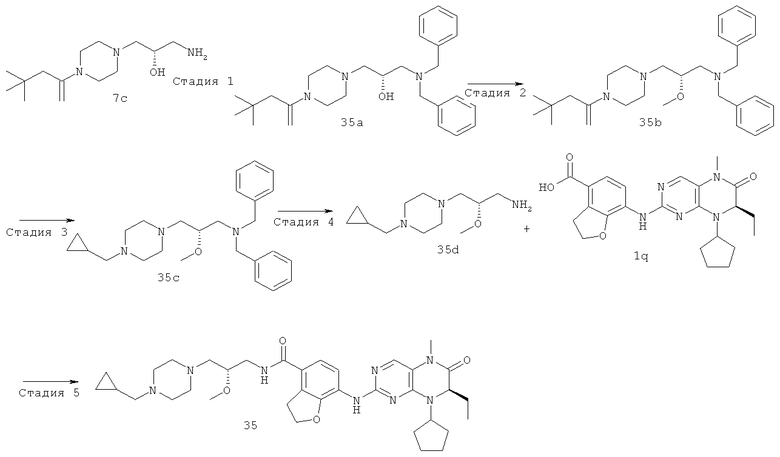
Стадия 1
трет-Бутил-4-[(2S)-3-(дибензиламино)-2-гидроксипропил]пиперазин-1-карбоксилат
трет-Бутил-4-[(2S)-3-амино-2-гидрокси-пропил]пиперазин-1-карбоксилат 7 с (5 г, 19,30 ммоль), карбонат калия (10,70 г, 77,20 ммоль) и бензилбромид (7,90 г, 46,30 ммоль) растворяли в 120 мл смеси растворителей этанола и воды (об./об. = 5:1). Реакционный раствор нагревали до 60°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли 100 мл воды и экстрагировали этилацетатом (100 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трет-бутил-4-[(2S)-3-(дибензиламино)-2-гидроксипропил]пиперазин-1-карбоксилата 35а (3,65 г, выход: 43,0%) в виде светло-желтого масла.
МС m/z (ИЭР): 440,4 [М+1]
Стадия 2
трет-Бутил-4-[(2S)-3-(дибензиламино)-2-метоксипропил]пиперазин-1-карбоксилат
Гидрид натрия (333 мг, 8,30 ммоль) растворяли в 50 мл тетрагидрофурана, добавляли по каплям раствор трет-бутил-4-[(2S)-3-(дибензиламино)-2-гидроксипропил]пиперазин-1-карбоксилата 35а (3,65 г, 8,30 ммоль) в тетрагидрофуране в бане лед-вода. Реакционный раствор перемешивали в течение 10 минут и подогревали до комнатной температуры. Реакционный раствор перемешивали в течение 1 часа и охлаждали до 0°С с последующим добавлением метилиодида (1,18 г, 8,30 ммоль). Реакционный раствор перемешивали в течение 12 часов. К полученному в результате раствору добавляли 10 мл насыщенного раствора бикарбоната натрия и экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, трея?-бутил-4-[(2S)-3-(дибензиламино)-2-метоксипропил]пиперазин-1-карбоксилата 35b (2,10 г, выход: 56,0%) в виде желтого масла.
МС m/z (ИЭР): 454,5 [М+1]
Стадия 3
(2S)-N,N-Дибензил-3-[4-(циклопропилметил)пиперазин-1-ил]-2-метоксипропан-1-амин
трет-Бутил-4-[(2S)-3-(дибензиламино)-2-метоксипропил]пиперазин-1-карбоксилат 35b (2,10 г, 4,60 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением 20 мл 4 М раствора хлорида водорода в диоксане. Реакционный раствор перемешивали в течение 0,5 часа. Полученный в результате раствор концентрировали при пониженном давлении. К остатку добавляли 40 мл ацетонитрила. В бане лед-вода к полученному в результате раствору добавляли триэтиламин (2,09 г, 20,70 ммоль), перемешивали в течение 0,5 часа с последующим добавлением бромметилциклопропана (0,88 г, 6,47 ммоль). Реакционный раствор перемешивали в течение 12 часов. Полученный в результате раствор концентрировали при пониженном давлении, добавляли 20 мл насыщенного раствора бикарбоната натрия и экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (50 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (2S)-N,N-дибензил-3-[4-(циклопропилметил)пиперазин-1 -ил]-метоксипропан-1 -амина 35 с (1,52 г, выход: 81,3%) в виде коричневого масла.
МС m/z (ИЭР): 408,4 [М+1]
Стадия 4
(2S)-3-[4-(Циклопропилметил)пиперазин-1-ил]-2-метоксипропан-1-амин
(2S)-N,N-Дибензил-3-[4-(циклопропилметил)пиперазин-1-ил]-2-метоксипропан-1-амин 35 с (500 мг, 1,23 ммоль) растворяли в 30 мл метанола с последующим добавлением палладия/углерода (100 мг, 10%), заполняли водородом три раза. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-метоксипропан-1-амина 35d (299 мг) в виде желтой маслянистой жидкости, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 226,2 [М-1]
Стадия 5
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-метоксипропил]-2,3-дигидробензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (115 мг, 0,26 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (85 мг, 0,26 ммоль) растворяли в 30 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,58 ммоль) и сырого соединения (2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-метоксипропан-1-амина 35d (60 мг, 0,26 ммоль) последовательно. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-N-[(2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-метоксипропил]-2,3-дигидробензофуран-4-карбоксамида 35 (80 мг, выход: 47,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 647,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.66 (s, 1Н), 7.21-7.09 (m, 2Н), 7.02 (s, 1Н), 4.68 (t, 2Н), 4.46 (t, 1Н), 4.21 (dd, 1Н), 3.66-3.57 (m, 3Н), 3.56-3.48 (m, 1Н), 3.44 (s, 3Н), 3.32 (s, 3Н), 2.75-2.30 (m, 9Н), 2.19-2.04 (m, 5Н), 2.04-1.92 (m, 1Н), 1.87-1.57 (m, 8Н), 0.93-0.88 (m, 4Н), 0.53 (d, 2Н), 0.17-0.05 (m, 2Н).
Пример 36
N-[(2S)-3-[4-(Циклопропилметил)пиперазин-1-ил]-2-метокси-пропил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
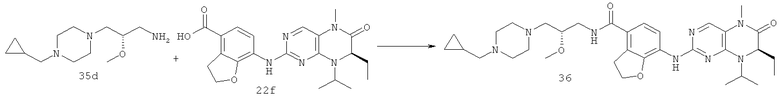
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 22f (70 мг, 0,17 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (55 мг, 0,17 ммоль) растворяли в 15 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,58 ммоль) и (2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-метоксипропан-1-амина 35d (39 мг, 0,17 ммоль) последовательно. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (30 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[(2S)-3-[4-(циклопропилметил)пиперазин-1-ил]-2-метоксипропил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 36 (40 мг, выход: 38,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 621,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.31 (d, 1Н), 7.67 (s, 1Н), 7.15 (d, 1Н), 7.06 (s, 1Н), 4.74-4.62 (m, 3Н), 4.29 (dd, 1Н), 3.72-3.57 (m, 4Н), 3.45 (s, 3Н), 3.33 (s, 3Н), 2.79-2.49 (m, 8Н), 2.30-2.25 (m, 2Н), 2.00-1.85 (m, 1Н), 1.83-1.67 (m, 2Н), 1.65-1.55 (m, 3Н), 1.43 (d, 3Н), 1.36 (d, 3Н), 0.87 (m, 4Н), 0.55 (d, 2Н), 0.17-0.07 (m, 2Н).
Пример 37
N-[(транс)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
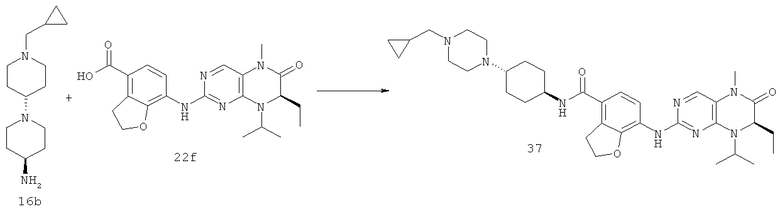
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 22f (70 мг, 0,17 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (55 мг, 0,17 ммоль) растворяли в 15 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,37 ммоль) и (транс)4-[4-(циклопропилметил)пиперазин-1-ил]циклогексанамина 16b (40 мг, 0,17 ммоль) последовательно. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, N-[(транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 37 (25 мг, выход: 23,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 631,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.28 (d, 1Н), 7.66 (s, 1Н), 7.10-7.02 (m, 2Н), 4.73-4.63 (m, 3Н), 4.28 (d, 1Н), 3.59 (t, 2Н), 3.32 (s, 3Н), 2.75-2.55 (m, 6Н), 2.40-2.24 (m, 3Н), 1.78-1.66 (m, 3Н), 1.65-1.55 (m, 6Н), 1.47-1.37 (m, 4Н), 1.35 (d, 3Н), 1.31-1.20 (m, 4Н), 0.86 (m, 4Н), 0.54 (d, 2Н), 0.14 (d,2H).
Пример 38
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-2-метокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамид
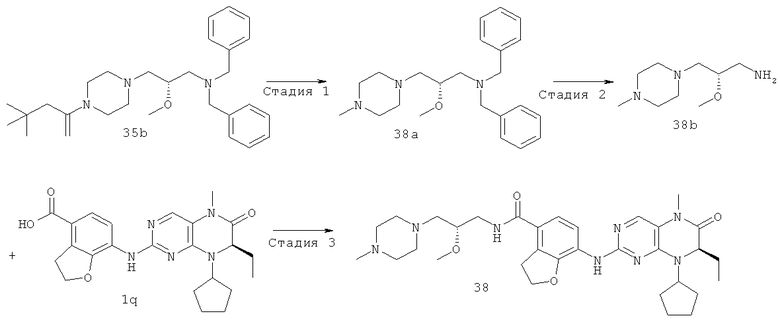
Стадия 1
(2S)-N,N-Дибензил-2-метокси-3-(4-метилпиперазин-1-ил)пропан-1-амин
трет-Бутил-4-[(2S)-3-(дибензиламино)-2-метоксипропил]пиперазин-1-карбоксилат 35b (4,36 г, 9,60 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением 20 мл 4 М раствора хлорида водорода в диоксане. Реакционный раствор перемешивали в течение 0,5 часа. Полученный в результате раствор концентрировали при пониженном давлении. К остатку последовательно добавляли 30 мл ацетонитрила, 30 мл воды и формальдегид (0,58 г, 19,20 ммоль). Реакционный раствор перемешивали в течение 0,5 часа, добавляли триацетоксиборгидрид натрия (6,10 г, 28,80 ммоль). Реакционный раствор перемешивали в течение 1 часа. К полученному в результате раствору добавляли по каплям водный аммиак до доведения рН до 9-10 и экстрагировали этилацетатом (50 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (2S)-N,N-дибензил-2-метокси-3-(4-метилпиперазин-1-ил)пропан-1-амина 38а (2,90 г, выход: 83,0%) в виде коричневого масла.
МС m/z (ИЭР): 368,3 [М+1]
Стадия 2
(2S)-2-Метокси-3-(4-метил пиперазин-1 -ил)пропан-1 -амин
(2S)-N,N-Дибензил-2-метокси-3-(4-метилпиперазин-1-ил)пропан-1-амин 38а (500 мг, 1,36 ммоль) растворяли в 20 мл метанола с последующим добавлением палладия/углерода (100 мг, 10%), заполняли водородом три раза. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого соединения, указанного в заголовке, (2S)-2-метокси-3-(4-метилпиперазин-1-ил)пропан-1-амина 38b (250 мг, в виде желтой маслянистой жидкости), который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 188,2 [М-1]
Стадия 3
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-2-метокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 1q (100 мг, 0,23 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (74 мг, 0,23 ммоль) растворяли в 20 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,50 ммоль) и сырого соединения (2S)-2-метокси-3-(4-метилпиперазин-1-ил)пропан-1-амина 38b (43 мг, 0,23 ммоль) последовательно. Реакционный раствор перемешивали в течение 2 часов, добавляли 30 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-2-метокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамида 38 (40 мг, выход: 30,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 607,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d, 1Н), 7.71-7.63 (m, 1Н), 7.13 (d, 1Н), 7.03 (s, 1Н), 4.74-4.63 (m, 2Н), 4.47 (s, 1Н), 4.21 (dd, 1Н), 3.67-3.57 (m, 3Н), 3.55-3.50 (m, 1Н), 3.45 (s, 3Н), 3.32 (s, 3Н), 2.71-2.41 (m, 8Н), 2.33 (s, 3Н), 2.15-1.95 (m, 2Н), 1.91-1.77 (m, 4Н), 1.77-1.57 (m, 8Н), 0.88 (t, 3Н).
Пример 39
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-2-метокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамид
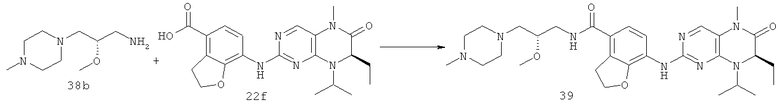
7-[[(7R)-7-Этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 22f (94 мг, 0,23 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (74 мг, 0,23 ммоль) растворяли в 20 мл безводного дихлорметана с последующим добавлением диизопропилэтиламина (0,1 мл, 0,50 ммоль) и (2S)-2-метокси-3-(4-метилпиперазин-1-ил)пропан-1 -амина 38b (43 мг, 0,23 ммоль) последовательно. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 30 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-[(2S)-2-метокси-3-(4-метилпиперазин-1-ил)пропил]-2,3-дигидробензофуран-4-карбоксамида 39 (35 мг, выход: 26,5%) в виде белого твердого вещества.
МС m/z (ИЭР): 581,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.31 (d, 1Н), 7.66 (s, 1Н), 7.14 (d, 1Н), 7.07 (s, 1Н), 4.77-4.61 (m, 3Н), 4.28 (dd, 1H), 3.75-3.49 (m, 4H), 3.45 (s, 3Н), 3.32 (s, 3Н), 2.95-2.60 (m, 5H), 2.55-2.40 (m, 3Н), 1.95-1.80 (m, 1H), 1.68-1.45 (m, 8H), 1.42 (d, 3Н), 1.35 (d, 3Н), 0.88 (t, 3Н).
Пример 40
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(метоксиметил)-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
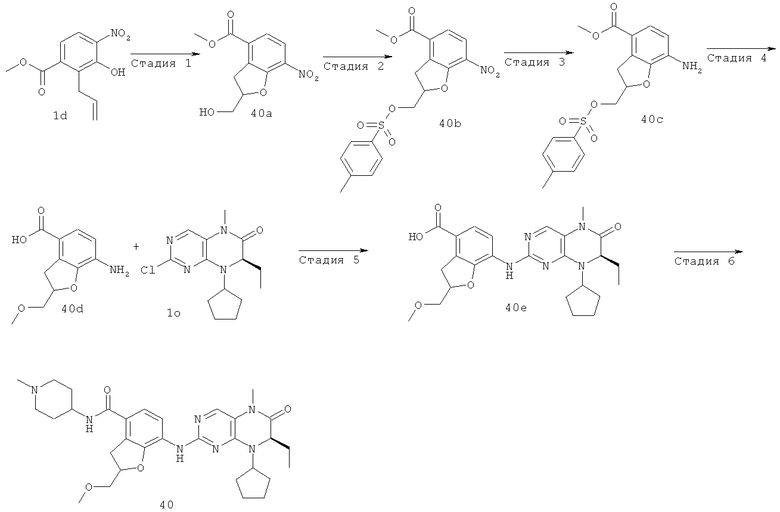
Стадия 1
Метил 2-(гидроксиметил)-7-нитро-2,3-дигидробензофуран-4-карбоксилат
Метил 2-аллил-3-гидрокси-4-нитробензоат 1d (0,47 г, 2 ммоль) растворяли в 80 мл дихлорметана с последующим добавлением мета-хлорпербензойной кислоты (0,99 г, 4 ммоль). Реакционный раствор перемешивали в течение 24 часов. К полученному в результате раствору последовательно добавляли 20 мл насыщенного раствора тиосульфата натрия и 20 мл насыщенного раствора бикарбоната натрия, перемешивали в течение 5 минут и экстрагировали этилацетатом (20 мл×3). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-2-(гидроксиметил)-7-нитро-2,3-дигидробензофуран-4-карбоксилата 40а (440 мг, выход: 87,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 252,1 [М-1]
Стадия 2
Метил-7-нитро-2-(толуил-4-сульфонилоксиметил)-2,3-дигидробензофуран-4-карбоксилат
Метил-2-(гидроксиметил)-7-нитро-2,3-дигидробензофуран-4-карбоксилат 40а (253 мг, 1 ммоль) растворяли в 40 мл дихлорметана с последующим добавлением диизопропилэтиламина (0,3 мл, 2 ммоль), 4-диметиламинопиридина (24 мг, 0,20 ммоль) и пара-толуолсульфонилхлорида (286 мг, 1,50 ммоль). Реакционный раствор перемешивали в течение 12 часов. К полученному в результате раствору добавляли 5 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (20 мл×3). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, метил-7-нитро-2-(толуил-4-сульфонилоксиметил)-2,3-дигидробензофуран-4-карбоксилата 40b (160 мг, выход: 39,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 408,2 [М+1]
Стадия 3
Метил-7-амино-2-(толуил-4-сульфонилоксиметил)-2,3-дигидробензофуран-4-карбоксилат
Метил-7-нитро-2-(толуил-4-сульфонилоксиметил)-2,3-дигидробензофуран-4-карбоксилат (360 мг, 0,88 ммоль) растворяли в 20 мл смеси растворителей метанола и дихлорметана (об./об. = 1:1). Реакционный раствор подвергали гидрогенизации с помощью H-Cube (колонка катализатора: 10% палладий/углерод; температура: 30°С; скорость тока: 1 мл/мин; давление: 1 атмосфера) в течение 20 минут. Полученный в результате раствор концентрировали при пониженном давлении с получением соединения, указанного в заголовке, метил-7-амино-2-(толуил-4-сульфонилоксиметил)-2,3-дигидробензофуран-4-карбоксилата 40 с (280 мг, выход: 84,0%) в виде светло-красного твердого вещества.
МС m/z (ИЭР): 378,2 [М+1]
Стадия 4
7-Амино-2-(метоксиметил)-2,3-дигидробензофуран-4-карбоновая кислота
Метил-7-амино-2-(толуил-4-сульфонилоксиметил)-2,3-дигидробензофуран-4-карбоксилат (320 мг, 0,85 ммоль) растворяли в 16 мл метанола с последующим добавлением 4 мл 50% раствора метанолата натрия в метаноле. Реакционный раствор нагревали до 50°С и перемешивали в течение 5 часов. К полученному в результате раствору добавляли 100 мл воды, концентрировали при пониженном давлении и экстрагировали дихлорметаном (50 мл×2). К водной фазе добавляли по каплям разбавленную соляную кислоту до доведения рН до 4 и экстрагировали дихлорметаном (50 мл×2). Органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 7-амино-2-(метоксиметил)-2,3-дигидробензофуран-4-карбоновой кислоты 40d (160 мг, выход: 84,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 222,1 [М-1]
Стадия 5
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(метоксиметил)-2,3-дигидробензофуран-4-карбоновая кислота
7-Амино-2-(метоксиметил)-2,3-дигидробензофуран-4-карбоновую кислоту 40d (160 мг, 0,70 ммоль) и (7R)-2-хлор-8-циклопентил-7-этил-5-метил-7,8-дигидро-5Н-птеридин-6-он 1о (232 мг, 0,80 ммоль) растворяли в 34 мл смеси растворителей этанола и воды (об./об. = 1:2,4) с последующим добавлением 0,4 мл концентрированной соляной кислоты. Реакционный раствор нагревали до образования флегмы в течение 16 часов при перемешивании, затем охлаждали, что приводило в результате к образованию осадка. Осадок фильтровали с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(метоксиметил)-2,3-дигидробензофуран-4-карбоновой кислоты 40е (0,15 г, выход: 43,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 480,3 [М-1]
Стадия 6
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2-(метоксиметил)-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
7-[[(7R)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2-(метоксиметил)-2,3-дигидробензофуран-4-карбоновую кислоту 40е (50 мг, 0,10 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (33 мг, 0,10 ммоль) растворяли в 40 мл дихлорметана с последующим добавлением диизопропилэтиламина (38 мкл, 0,23 ммоль) и 1-метилпиперидил-4-иламина (12 мг, 0,10 ммоль) последовательно. Реакционный раствор перемешивали в течение 1 часа, добавляли 20 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (30 мл×3). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 7-[[(7R)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2-(метоксиметил)-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамида 40 (40 мг, выход: 69,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 578,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.32 (d, 1Н), 7.67 (s, 1Н), 7.14 (s, 1Н), 7.07 (d, 1Н), 6.05 (d, 1Н), 5.13-5.00 (m, 1Н), 4.63-4.47 (m, 1Н), 4.21 (dd, 1Н), 4.14-3.95 (m, 1Н), 3.71-3.57 (m, 3Н), 3.46 (s, 3Н), 3.38-3.25 (m, 4Н), 3.08 (d, 3Н), 2.52-2.37 (m, 5Н), 2.11 (d, 3Н), 2.00 (d, 1Н), 1.92-1.63 (m, 9Н), 0.88 (t, 3Н).
Пример 41
7-[[(7S)-8-Циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
Стадия 1
Метил-(2S)-2-аминобутаноат
(2S)-2-Аминобутановую кислоту 41а (21,56 г, 0,21 моль) растворяли в 120 мл метанола. Реакционный раствор охлаждали до 0°С в бане лед-вода, добавляли по каплям тионилхлорид (30,5 мл, 0,42 моль) при перемешивании. После завершения добавления реакционную смесь нагревали до образования флегмы в течение 2 часов, затем охлаждали до комнатной температуры. Реакционный раствор концентрировали при пониженном давлении с получением соединения, указанного в заголовке, метил-(2S)-2-аминобутаноата 41b (31,87 г, выход: 99,3%) в виде белого твердого вещества.
Стадия 2
Метил-(2S)-2-(циклопентиламино)бутаноат
Метил-(2S)-2-аминобутаноат 41b (18,74 г, 0,12 моль) растворяли в 280 мл дихлорметана. Реакционный раствор охлаждали до 0°С в бане лед-вода с последующим добавлением ацетата натрия (5 г, 0,061 моль), циклопентанона (10,78 г, 0,13 моль) и триацетоксиборгидрида натрия (31 г, 0,16 моль) последовательно. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 4 часов. Реакционный раствор наливали в 150 мл воды, добавляли по каплям насыщенный раствор карбоната натрия до доведения рН до 11-12 и разделяли. Водную фазу экстрагировали дихлорметаном (150 мл×2). Объединенную органическую фазу промывали насыщенным рассолом (100 мл×2), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, метил-(2S)-2-(циклопентиламино)бутаноата 41 с (17,42 г, выход: 77,1%) в виде светло-желтого масла.
МС m/z (ИЭР): 186,1 [М+1]
Стадия 3
Метил-(2S)-2-[(2-хлор-5-нитропиримидин-4-ил)-циклопентиламино]бутаноат
Метил-(2S)-2-(циклопентиламино)бутаноат 41 с (17,36 г, 93,69 ммоль) растворяли в 180 мл циклогексана с последующим добавлением бикарбоната натрия (31,48 г, 0,37 моль) и 2,4-дихлор-5-нитропиримидина (21,81 г, 0,11 моль) последовательно. Реакционный раствор нагревали до 85°С и перемешивали в течение 2 часов. Полученный в результате раствор охлаждали до комнатной температуры, добавляли 300 мл дихлорметана, затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования В с получением соединения, указанного в заголовке, метил-(2S)-2-[(2-хлор-5-нитропиримидин-4-ил)-циклопентиламино]бутаноата 41 d (14,24 г, выход: 44,3%) в виде желтого твердого вещества.
МС m/z (ИЭР): 343,1 [М+1]
Стадия 4
(7S)-2-Хлор-8-циклопентил-7-этил-5,7-дигидроптеридин-6-он Метил-(2S)-2-[(2-хлор-5-нитропиримидин-4-ил)-циклопентиламино]бутаноат 41d (14,17 г, 41,33 ммоль) растворяли в 300 мл уксусной кислоты с последующим добавлением порошка железа (9 г, 0,17 моль). После завершения экзотермической реакции реакционный раствор нагревали до 65°С и перемешивали в течение 1,5 часов. Реакционную смесь фильтровали, и фильтрационный кек промывали дихлорметаном (350 мл). Фильтрат концентрировали при пониженном давлении, добавляли 150 мл воды и фильтровали. Фильтрационный кек последовательно промывали водой (60 мл×3) и смесью растворителей гексана и ацетона (об./об. = 6:1) (140 мл). Объединенную органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (7S)-2-хлор-8-циклопентил-7-этил-5,7-дигидроптеридин-6-она 41 е (10,67 г, выход: 92,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 281,1 [М+1]
Стадия 5
(7S)-2-Хлор-8-циклопентил-7-этил-5-метил-7Н-птеридин-6-он
(7S)-2-Хлор-8-циклопентил-7-этил-5,7-дигидроптеридин-6-он 41 е (2,80 г, 10 ммоль) растворяли в 50 мл ацетона с последующим добавлением метил-пара-толуолсульфоната (2,80 г, 15 ммоль) и карбоната калия (2,76 г, 20 ммоль) последовательно, перемешивали в течение 12 часов. К полученному в результате раствору добавляли 50 мл воды и экстрагировали этилацетатом (100 мл×3). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении, Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования В с получением соединения, указанного в заголовке, (7S)-2-хлор-8-циклопентил-7-этил-5-метил-7Н-птеридин-6-она 41f (2,50 г, выход: 85,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 295,1 [М+1]
Стадия 6
7-[[(7S)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновая кислота
(7S)-2-Хлор-8-циклопентил-7-этил-5-метил-7Н-птеридин-6-опе 41 f (300 мг, 1.70 ммоль) и метил-7-амино-2,3-дигидробензофуран-4-карбоксилат 1д (543 мг, 1.80 ммоль) растворяли в 34 мл смеси растворителей этанола и воды (об./об. = 1:2,4) с последующим добавлением 0,4 мл концентрированной соляной кислоты и нагревали до образования флегмы в течение 16 часов. Реакционный раствор охлаждали до комнатной температуры, помещали в холодильник на 1 час и фильтровали. Фильтрационный кек промывали водой (50 мл) и растворяли в 20 мл смеси растворителей воды и 1 М гидроксида лития (об./об. = 1:1), экстрагировали этилацетатом (50 мл×2), к водной фазе добавляли по каплям 1 М соляную кислоту до доведения рН до 2, что приводило в результате к образованию осадка. Осадок фильтровали с получением соединения, указанного в заголовке, 7-[[(7S)-8-циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновой кислоты 41 g (0,45 г, выход: 62,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 438,2 [М+1]
Стадия 7
7-[[(7S)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
7-[[(7S)-8-Циклопентил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 41 g (150 мг, 0,34 ммоль) и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (111 мг, 0,34 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением диизопропилэтиламина (0,13 мл, 0,76 ммоль), перемешивали в течение 10 минут, затем добавляли 1-метилпиперидил-4-иламин
(40 мг, 0,34 ммоль), перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли 20 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (100 мл×2). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, 7-[[(7S)-8-циклопентил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамида 41 (120 мг, выход: 65,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 267,7 [М/2+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.28 (d, 1Н), 7.67 (s, 1Н), 7.10-7.00 (m, 2Н), 5.91 (d, 1Н), 4.68 (t, 2Н), 4.47 (t, 1Н), 4.21 (d, 1Н), 4.02-3.93 (m, 1Н), 3.58 (t, 2Н), 3.32 (s, 3Н), 2.86 (d, 2Н), 2.33 (s, 3Н), 2.23-1.96 (m, 6Н), 1.89-1.76 (m, 4Н), 1.74-1.58 (m, 6Н), 0.90-0.83 (t, 3Н).
Пример 42
7-[(8-Циклопентил-7,7-диэтил-5-метил-6-оксоптеридин-2-ил)амино]-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
Стадия 1
2-Хлор-8-циклопентил-7,7-диэтил-5-метил-птеридин-6-он
(7S)-2-Хлор-8-циклопентил-7-этил-5-метил-7Н-птеридин-6-он 41f (148 мг, 0,50 ммоль) растворяли в 20 мл тетрагидрофурана. Реакционный раствор охлаждали до -78°С с последующим добавлением диизопропиламида лития (0,5 мл, 1 ммоль), перемешивали в течение 30 минут, затем добавляли этилиодид (156 мг, 1 ммоль) и перемешивали еще в течение 4 часов. К реакционной смеси добавляли 50 мл насыщенного раствора хлорида аммония, концентрировали при пониженном давлении и экстрагировали дихлорметаном (100 мл×2). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования В с получением соединения, указанного в заголовке, 2-хлор-8-циклопентил-7,7-диэтил-5-метилптеридин-6-она 42а (100 мг, выход: 62,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 323,2 [М+1]
Стадия 2
7-[(8-Циклопентил-7,7-диэтил-5-метил-6-оксо-птеридин-2-ил)амино]-2,3-дигидробензофуран-4-карбоновая кислота
2-Хлор-8-циклопентил-7,7-диэтил-5-метил-птеридин-6-он 42а (100 мг, 0,31 ммоль) и метил-7-амино-2,3-дигидробензофуран-4-карбоновую кислоту 1д (50 мг, 0,28 ммоль) растворяли в 17 мл смеси растворителей этанола и воды (об./об. = 5:12) с последующим добавлением 0,2 мл концентрированной соляной кислоты. Реакционную смесь нагревали до образования флегмы в течение 16 часов при перемешивании. Полученный в результате раствор концентрировали при пониженном давлении, добавляли по каплям 1 М раствор гидроксида лития до доведения рН до 12 и экстрагировали этилацетатом (50 мл×2), к объединенной водной фазе добавляли по каплям 1 М соляную кислоту до доведения рН до 1 и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением 7-[(8-циклопентил-7,7-диэтил-5-метил-6-оксо-птеридин-2-ил)амино]-2,3-дигидробензофуран-4-карбоновой кислоты 42b (50 мг, выход: 38,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 466,3 [М+1]
Стадия 3
7-[(8-Циклопентил-7,7-диэтил-5-метил-6-оксоптеридин-2-ил)амино]-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамид
7-[(8-Циклопентил-7,7-диэтил-5-метил-6-оксо-птеридин-2-ил)амино]-2,3-дигидробензофуран-4-карбоновую кислоту 42b (50 мг, 0,11 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (35 мг, 0,11 ммоль), диизопропилэтиламина (31 мг, 0,24 ммоль) и 1-метилпиперидил-4-иламина (13 мг, 0,11 ммоль) последовательно. Реакционный раствор перемешивали при комнатной температуре в течение 1 часа. К полученному в результате раствору добавляли 50 мл насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (100 мл×2). Объединенную органическую фазу последовательно промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, 7-[(8-циклопентил-7,7-диэтил-5-метил-6-оксоптеридин-2-ил)амино]-N-(1-метил-4-пиперидил)-2,3-дигидробензофуран-4-карбоксамида 42 (10 мг, выход: 16,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 281,7 [М/2+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.25 (d, 1Н), 7.56 (s, 1Н), 7.06 (d, 1Н), 6.78 (s, 1Н), 5.89 (d, 1Н), 4.68 (t, 2Н), 4.06-3.92 (m, 1Н), 3.83-3.71 (m, 1Н), 3.58 (t, 2Н), 3.32 (s, 3Н), 2.89 (d, 2н), 2.58 (d, 2Н), 2.35 (s, 3Н), 2.28-2.14 (m, 4Н), 2.10-2.00 (m, 4Н), 1.80 (d, 2Н), 1.74-1.56 (m, 6Н), 0.84 (t, 6Н)
Пример 43
N-[(цис)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексил]-7-[[(7S)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
Стадия 1
Метил-(2S)-2-(изопропиламино)бутаноат
(2S)-2-Аминобутаноат 41b (13,14 г, 85,52 ммоль) растворяли в 200 мл дихлорметана. Реакционный раствор охлаждали до 0°С в бане лед-вода с последующим добавлением ацетата натрия (3,51 г, 42,76 ммоль), ацетона (5,96 г, 0,10 моль) и триацетоксиборгидрида натрия (21,75 г, 0,10 моль) последовательно. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 4 часов. К полученному в результате раствору добавляли 100 мл воды, добавляли по каплям насыщенный раствор карбоната натрия до доведения рН до 11-12 и разделяли. Водную фазу экстрагировали дихлорметаном (150 мл×2). Объединенную органическую фазу промывали насыщенным рассолом (100 мл×2), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением метил-(2S)-2-(изопропиламино)бутаноата 43а (10,85 г, выход: 79,7%) в виде светло-желтого масла.
МС m/z (ИЭР): 160,2 [М+1]
Стадия 2
Метил-(2S)-2-[(2-хлор-5-нитропиримидин-4-ил)-изопропиламино]бутаноат
Метил-(2S)-2-(изопропиламино)бутаноат 43а (10,75 г, 67,50 ммоль) растворяли в 180 мл циклогексана с последующим добавлением бикарбоната натрия (22,68 г, 0,27 моль) и 2,4-дихлор-5-нитропиримидина (15,71 г, 0,081 моль) последовательно, реакционную смесь нагревали до 85°С и перемешивали в течение 2,5 часов. Полученную в результате смесь охлаждали до комнатной температуры, добавляли 200 мл дихлорметана, затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования В с получением метил-(2S)-2-[(2-хлор-5-нитропиримидин-4-ил)-изопропиламино]бутаноата 43b (8,78 г, выход: 41,1%) в виде желтого твердого вещества.
МС m/z (ИЭР): 317,1 [М+1]
Стадия 3
(7S)-2-Хлор-8-изопропил-7-этил-5,7-дигидроптеридин-6-он
Метил-(2S)-2-[(2-хлор-5-нитропиримидин-4-ил)-изопропиламино]бутаноат 43b (8,74 г, 27,7 ммоль) растворяли в 100 мл уксусной кислоты с последующим добавлением порошка железа (6,20 г, 0,11 моль). Реакционную смесь нагревали до 65°С, перемешивали в течение 4 часов и фильтровали. Фильтрационный кек промывали дихлорметаном (100 мл). Фильтрат концентрировали при пониженном давлении, затем добавляли 100 мл воды и фильтровали. Фильтрационный кек высушивали нагреванием с получением сырого соединения (7S)-2-хлор-8-изопропил-7-этил-5,7-дигидроптеридин-6-она 43 с (7,17 г) в виде серого твердого вещества, который использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 255,1 [М+1]
Стадия 4
(7S)-2-Хлор-8-изопропил-7-этил-5-метил-7H-птеридин-6-он
Сырое соединение (7S)-2-хлор-8-изопропил-7-этил-5,7-дигидроптеридин-6-он 43с (7,17 г, 28,21 ммоль), метил-пара-толуолсульфонат (5,77 г, 31,03 ммоль) и карбонат калия (11,70 г, 84,60 ммоль) растворяли в 40 мл ацетона. Реакционный раствор перемешивали в течение 12 часов и осадок фильтровали. Фильтрационный кек промывали дихлорметаном (100 мл). Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования В с получением (7S)-2-хлор-8-изопропил-7-этил-5-метил-7Н-птеридин-6-она 43d (6,14 г, выход: 81,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 269,1 [М+1]
Стадия 5
7-[[(7S)-8-Изопропил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-дигидробензофуран-4-карбоновая кислота
(7S)-2-Хлор-8-изопропил-7-этил-5-метил-7Н-птеридин-6-он 43d (2,50 г, 9,24 ммоль) и метил-7-амино-2,3-дигидробензофуран-4-карбоновую кислоту 1д (1,50 г, 8,40 ммоль) растворяли в 120 мл смеси растворителей этанола и воды (об./об. = 1:2) с последующим добавлением 1,5 мл концентрированной соляной кислоты. Реакционную смесь нагревали до образования флегмы в течение 24 часов при перемешивании и осадок фильтровали. Фильтрационный кек промывали дихлорметаном (50 мл×3) и высушивали нагреванием с получением сырого соединения 7-[[(7S)-8-изопропил-7-этил-5-метил-6-оксо-7H-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновой кислоты 43е (2 г) в виде серого твердого вещества, которую использовали в следующей стадии без дальнейшей очистки.
МС m/z (ИЭР): 412,2 [М+1]
Стадия 6
N-[(цис)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексил]-7-[[(7S)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
(цис)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексанамин 12е (577 мг, 2,43 ммоль), сырое соединение 7-[[(7S)-8-изопропил-7-этил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 43е (1 г, 2,43 ммоль), О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (780 мг, 2,43 ммоль) и диизопропилэтиламин (690 мг, 5,35 ммоль) растворяли в 60 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов. К полученной в результате смеси добавляли 50 мл водного аммиака и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным рассолом (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, N-[(цис)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-7-[[(7S)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 43 (1,12 г, выход: 74,6%) в виде белого твердого вещества.
МС m/z (ИЭР): 631,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.29 (d. 1Н), 7.67 (s, 1Н), 7.12-7.04 (m, 2Н), 6.13 (d, 1Н), 4.77-4.64 (m, 3Н), 4.29 (d, 1Н), 4.27-4.19 (m, 1Н), 3.60 (t, 2Н), 3.32 (s, 3Н), 2.64 (s, 7Н), 2.31-2.19 (m, 3Н), 1.98-1.79 (m, 5Н), 1.78-1.63 (m, 3Н), 1.56 (q, 2Н), 1.44 (d, 3Н), 1.37 (d, 3Н), 0.87 (t, 4Н), 0.55-0.49 (m, 2Н), 0.14-0.07 (m, 2Н).
Пример 44
N-[(цис)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
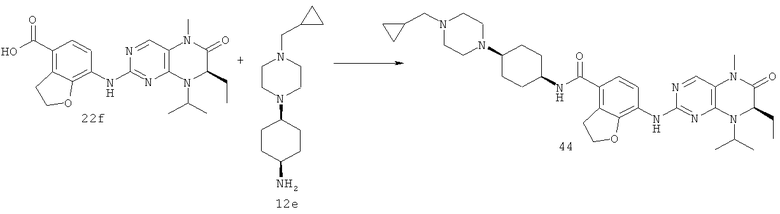
(цис)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексанамин 12e (577 мг, 2,43 ммоль), 7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 22f (1 г, 2,43 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (780 мг, 2,43 ммоль) и диизопропилэтиламин (690 мг, 5,35 ммоль) растворяли в 40 мл дихлорметана. Реакционную смесь перемешивали в течение 2 часов. К полученному в результате раствору добавляли 50 мл водного аммиака и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным рассолом (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, N-[(цис)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-7-[[(7R)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 44 (576 мг, выход: 38,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 631,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.28 (d, 1Н), 7.66 (s, 1Н), 7.15-7.08 (m, 2Н), 6.17 (d, 1Н), 4.75-4.64 (m, 3Н), 4.31-4.20 (m, 2H), 3.60 (t, 2H), 3.32 (s, 3Н), 2.69 (s, 7H), 2.36-2.22 (m, 3Н), 2.01 (s, 1H), 1.95-1.80 (m, 5H), 1.77-1.56 (m, 5H), 1.44 (d, 3Н), 1.37 (d, 3Н), 0.92-0.84 (m, 4H), 0.58-0.50 (m, 2H), 0.13 (q,2H).
Пример 45
N-[(транс)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексил]-7-[[(7S)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамид
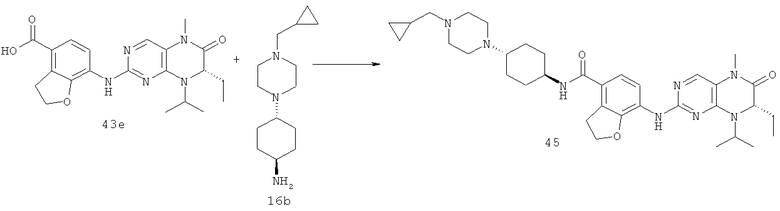
(транс)-4-[4-(Циклопропилметил)пиперазин-1-ил]циклогексанамин 16b (350 мг, 1,48 ммоль), 7-[[(7S)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоновую кислоту 43е (607 мг, 1,48 ммоль), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (475 мг, 1,48 ммоль) и диизопропилэтиламин (420 мг, 3,30 ммоль) растворяли в 40 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов. К полученному в результате раствору добавляли 50 мл водного аммиака и экстрагировали дихлорметаном (50 мл×3). Объединенную органическую фазу промывали насыщенным рассолом (50 мл×3), затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, N-[(транс)-4-[4-(циклопропилметил)пиперазин-1-ил]циклогексил]-7-[[(7S)-7-этил-8-изопропил-5-метил-6-оксо-7Н-птеридин-2-ил]амино]-2,3-дигидробензофуран-4-карбоксамида 45 (360 мг, выход: 38,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 316,2 [М/2+1]
1Н ЯМР (400 МГц, CDCl3) δ 8.27 (d, 1Н), 7.66 (s, 1Н), 7.09-7.02 (m, 2Н), 5.82 (d, 1Н), 4.73-4.63 (m, 3Н), 4.28 (d, 1Н), 3.96-3.82 (m, 1Н), 3.59 (t, 2Н), 3.32 (s, 3Н), 2.66 (s, 8Н), 2.35-2.25 (m, 3Н), 2.17 (d, 2H), 2.04-1.86 (m, 5H), 1.76-1.68 (m, 1H), 1.50-1.39 (m, 5H), 1.35 (d, 3Н), 1.26 (d, 2H), 0.86 (t, 4H), 0.55-0.48 (m, 2H), 0.11 (q, 2H).
ТЕСТОВЫЕ ПРИМЕРЫ
БИОЛОГИЧЕСКИЕ АНАЛИЗЫ
Тестовый Пример 1: Анализ ингибирования пролиферации клеток Plk
Приведенный ниже анализ in vitro предназначен для определения активности соединений по настоящему изобретению по ингибированию пролиферации клеток рака шейки матки человека Hela, которые обладают высокой экспрессией Plk.
Клеточный анализ in vitro, описанный в данной заявке, предназначен для определения активности тестируемых соединений по ингибированию пролиферации раковых клеток, которые обладают высокой экспрессией Plk. Активность представлена как IC50. Общие методы анализа были такими, как приведено ниже: Клетки Hela с высокой экспрессией Plk (Институт биохимии и клеточной биологии) высевали в 96-луночный культуральный планшет при подходящей концентрации (экспоненциальная фаза 3000 клеток/мл среды). Затем клетки культивировали в инкубаторе с диоксидом углерода (CO2) и выращивали в течение ночи. Затем культуральную среду клеток заменяли свежей средой с тестируемыми соединениями, добавленными в нее в серийных концентрациях (как правило, от 7 до 9 концентраций). Затем клетки помещали обратно в инкубатор и культивировали непрерывно в течение 72 часов. Спустя 72 часа активность тестируемых соединений по ингибированию клеточной пролиферации определяли путем использования метода ССК8 (набора-8 для подсчета клеток, Dojindo, NO:CK04). Значение IC50 на тестируемых клетках вычисляли на основании данных коэффициентов ингибирования для серийных концентраций тестируемых соединений.
Активность соединений по изобретению:
Биологическую активность соединений по изобретению тестировали путем использования вышеописанного анализа. Значения IC50 измерены и представлены в приведенной ниже таблице:
Вывод: соединения по настоящему изобретению обладают очевидной активностью по ингибированию пролиферации клеток Hela.
ФАРМАКОКИНЕТИЧЕСКИЙ АНАЛИЗ
Тестовый Пример 2: фармакокинетический анализ соединения Примера 37 настоящего изобретения
1. Реферат
Соединение 37 настоящего изобретения вводили путем внутривенной инъекции или внутрижелудочно крысам, чтобы определить концентрацию лекарства в плазме в различные моменты времени методом ЖХ-МС/МС. Фармакокинетические свойства соединений по настоящему изобретению исследовали и оценивали на крысах.
2. Протокол
2.1. Образцы Соединение Примера 37.
2.2. Подопытные животные
8 здоровых взрослых крыс SD, половина самцов и половина самок, были приобретены в фирме SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО, номер лицензии: SCXK (Шанхай) 2003-0016.
2.3. Приготовление тестируемых соединений
Соответствующее количество тестируемого соединения растворяли в 0,5 мл ДМСО, добавляли 1,5 мл 0,1 М HCl, затем разводили нормальным физиологическим раствором до 2,5 мг/мл суспензии перед применением.
2.4. Введение
8 здоровых взрослых крыс SD, половина самцов и половина самок, были разделены на две группы. После ночного голодания крысам вводили внутрижелудочно или вводили путем инъекции в хвостовую вену в дозе 25,0 мг/кг.
3. Проведение эксперимента
25 мкл плазмы крыс, взятой в различные моменты времени после введения, смешивали с 20 мкл раствора внутреннего стандарта и 150 мкл метанола в течение 3 минут путем использования вортекса, и смесь центрифугировали в течение 10 минут при 13500 об/мин. 5 мкл супернатанта анализировали с помощью ЖХ-МС/МС.
Концентрацию в плазме наносили на ось абсцисс, отношение площади хроматографического пика между образцом и внутренним стандартом наносили на ось ординат, линейную регрессию осуществляли взвешенным методом наименьших квадратов (w=1/x2). Основные фармакокинетические параметры вычисляли с помощью программного обеспечения DAS 2.0.
4. Результаты фармакокинетических параметров
Фармакокинетические параметры соединений по настоящему изобретению представлены ниже:
Вывод: соединение примера 37 обладает хорошим всасыванием в фармакокинетике и высокой пероральной биодоступностью.
Терапевтические эффекты против ксенотрансплантатов рака ободочной кишки человека НТ-116 у бестимусных мышей
1. Реферат
Оценивали терапевтический эффект Примера 1 против ксенотрансплантатов рака ободочной кишки человека НТ-116 у бестимусных мышей. В качестве ингибитора Plk-1 соединение Примера 1 при меньшей токсичности заметно ингибировало рост рака ободочной кишки человека НТ-116 у бестимусных мышей.
2. Цель
Оценивали и сравнивали терапевтический эффект Примера 1 против ксенотрансплантатов рака ободочной кишки человека НТ-116 у бестимусных мышей.
3. Тестируемое лекарство
Название лекарства: соединение Примера 1
Способ приготовления: соединение Примера 1 готовили до соответствующей концентрации путем использования нормального физиологического раствора.
4. Подопытные животные
Бестимусные мыши BALB/cA в возрасте от 6 до 7 недель были приобретены у фирмы Slaccas Experimei Animal LTD., CO.
№сертификата: SCXK 2007to0005. Окружающая среда: уровень SPF (свободный от специфических патогенов).
5. Экспериментальный протокол
Бестимусным мышам инокулировали подкожно клетки рака ободочной кишки человека НТ-116. Опухоли вырастали до 60-150 мм, и мышей случайным образом делили на группы (d0).
Объем опухоли и массу мышей измеряли и регистрировали 2-3 раза в неделю. Формула вычисления объема опухоли (V):V=1/2×a×b2, а: длина опухоли, b: ширина опухоли.
6. Результат
Соединение Примера 1 заметно ингибировало рост рака ободочной кишки человека НТ-116 и обладало меньшей токсичностью.
Ингибирование соединением 37 пролиферации различных раковых клеток, культивируемых in vitro
I. Цель
Приведенный ниже анализ in vitro предназначен для определения активности соединения 37 по настоящему изобретению по ингибированию пролиферации различных раковых клеток, культивируемых in vitro.
II. Материалы
i. Тестируемое лекарство: Соединение 37 по настоящему изобретению (см. Пример 37)
ii. Клеточная линия
Происхождение и условия культивирования клеток показаны в Таблице 1.
АТСС обозначает American Type Culture Collection (Американская коллекция типовых культур)
iii. Реагенты и оборудование
DMEM, RPMI-1640, McCoy′s5a, F-12 были приобретены у Gibco BRL;
FBS был приобретен у JRH Ltd.;
Считывающее устройство для микропланшетов SPECTRA МАХ 190 было приобретено у Molecular Devices LLC;
SRB и MTT были приобретены у Sigma;
Инкубатор с диоксидом углерода был приобретен у Thermo;
Инвертированный микроскоп был приобретен у Chongqing Photoelectric Instrument Co., Ltd.
III. Способ
i. Способ SRB
Определенное число клеток в фазе логарифмического роста высевали в 96-луночный культуральный планшет. Клеточную линию выращивали с адгезией в течение 24 часов, добавляли лекарство в серийных концентрациях и культивировали в инкубаторе в течение 72 часов. После обработки лекарством клетки фиксировали трихлоруксусной кислотой, а затем окрашивали раствором SRB. В конце добавляли трис-раствор для растворения SRB. Значение OD измеряли при длине волны 510 нм на считывающем устройстве для микропланшетов (SPECTRA МАХ 190) и рассчитывали скорость ингибирования клеточного роста по следующей формуле:
Скорость ингибирования = (ODконтрольной лунки - ODлунки с лекарством)/ODконтрольной лунки × 100%
Значение IC50 вычисляли по данным о скоростях ингибирования серийными концентрациями тестируемого соединения.
ii. Способ МТТ
Определенное число клеток в фазе логарифмического роста высевали в 96-луночный культуральный планшет. Добавляли лекарство в серийных концентрациях и культивировали в инкубаторе в течение 72 часов. После обработки лекарством в каждую лунку добавляли рабочий раствор МТТ. Спустя 4 часа добавляли смесь HCI, SDS и изопропанола и выдерживали в течение ночи при 37°C. На следующий день измеряли значение OD при длине волны 570 нм на считывающем устройстве для микропланшетов (SPECTRA МАХ 190) и рассчитывали скорость ингибирования клеточного роста по следующей формуле:
Скорость ингибирования = (ODконтрольной лунки - ODлунки с лекарством)/ODконтрольной лунки × 100%
Значение IC50 вычисляли по данным о скоростях ингибирования серийными концентрациями тестируемого соединения.
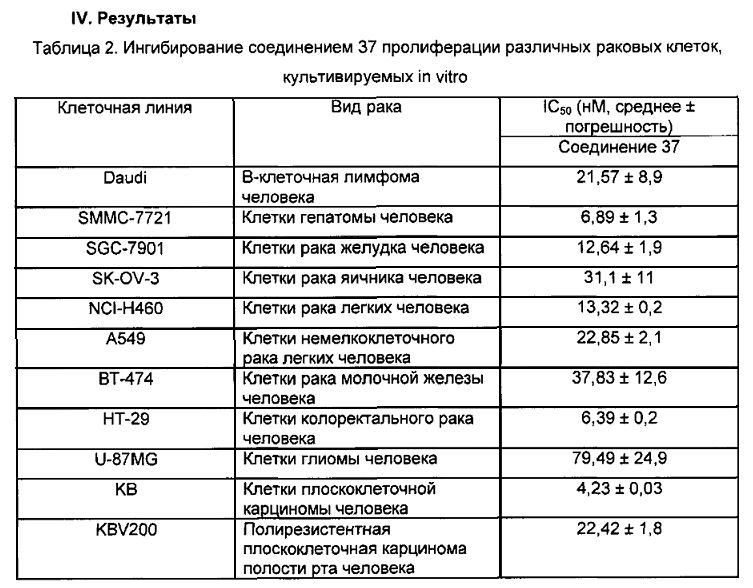
Как показано в Таблице 2, соединение 37 обладало высокой активностью по ингибированию пролиферации 11 раковых клеток и IC50 от 4 нМ до 200 нМ. Это соединение слабо действовало на клетки глиомы, но оказывало сильное действие на другие опухоли, такие как В-клеточная лимфома, рак печени, рак желудка, рак легких, рак молочной железы, колоректальный рак и т.п. В целом, выраженная селективность по клеточным типам не отмечалась.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2012 |
|
RU2621039C1 |
| БИСУЛЬФАТ ИНГИБИТОРА ЯНУС-КИНАЗЫ (JAK) И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2014 |
|
RU2665680C2 |
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ | 2019 |
|
RU2783414C2 |
| ПРОИЗВОДНОЕ БЕНЗОФУРАНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2016 |
|
RU2727198C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2730508C2 |
| ПРОИЗВОДНЫЕ ПИРИДИЛКЕТОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2014 |
|
RU2667892C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ГРИППА, СПОСОБЫ ИХ ПРИМЕНЕНИЯ И ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2737190C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, ИХ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2007 |
|
RU2474582C2 |
Изобретение относится к новым производным дигидроптеридинона формулы (I), способу их получения (вариантам) и их фармацевтическому применению в качестве ингибиторов Plk киназы. Соединения могут быть использованы для получения лекарства для лечения нарушения клеточной пролиферации, в частности при нарушении, которое является раком, который выбран из группы, состоящей из немелкоклеточного рака легкого, чешуйчато-клеточного рака, рака молочной железы, рака яичника, рака шейки матки, папиллярной карциномы или колоректальной карциномы, предпочтительно рака шейки матки или колоректальной карциномы. В формуле (I)
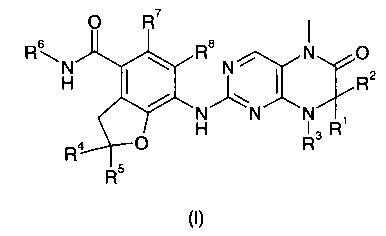
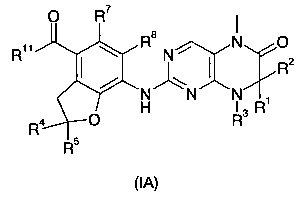
R1 и R2 каждый независимо выбран из группы, состоящей из атома водорода и C1-6алкила; R3 выбран из группы, состоящей из C1-6алкила и С5-6циклоалкила; R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода, C1-6алкила, где алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из C1-6алкоксила и -NR9R10; R6 выбран из группы, состоящей из С1-3алкила, С5-6циклоалкила, гетероциклического алкила, выбранного из 6-членного гетероциклического алкила с 1 атомом азота, гексагидроциклопента(с)пиррола или циклопента(с)пиперидина, где C1-3алкил, С5-6циклоалкил или гетероциклический алкил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из С1-6алкила, С1-6алкоксила, гидроксила, С3-6циклоалкила, С3-6циклоалкилС1-6алкила, гетероциклического алкила, N-метилморфолина, гексагидропирроло[2,1-с][1,4]оксазина, -C(O)R9, -NR9R10 и C1-6алкилкарбонилоксигруппы; где R9 и R10 вместе с атомом азота, к которому они присоединены, образуют 4-8-членный гетероцикл, где 4-8-членный гетероцикл содержит один или более гетероатомов N или О и 4-8 членный гетероцикл необязательно замещен C1-6алкилом, необязательно замещенным С3-6циклоалкилом; R7 и R8 каждый независимо выбран из атома водорода или С1-6алкила; R9 и R10 каждый независимо выбран из атома водорода или C1-6алкила. Изобретение также относится к новому промежуточному соединению формулы (IA) где R1, R2 R3 R4, R5, R7, R8 R9 и R10 имеют вышеуказанные значения; R11 выбран из группы, состоящей из гидроксила или С1-6алкоксила. Способ получения соединения формулы I заключается во взаимодействии соединения формулы (IA) с соединением R6NH2, либо во взаимодействии соединения формулы (IB) с соединением формулы (IC). Формулы (IB) и (IС) указаны в формуле изобретения. 8 н. и 3 з.п. ф-лы, 2 табл., 47 пр.
1. Соединения формулы (I) или их таутомеры, рацематы, энантиомеры, диастереоизомеры и их смеси, а также их фармацевтически приемлемые соли
где:
R1 и R2 каждый независимо выбран из группы, состоящей из атома водорода и C1-6алкила;
R3 выбран из группы, состоящей из C1-6алкила и С5-6циклоалкила;
R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода, C1-6алкила, где алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из C1-6алкоксила и -NR9R10;
R6 выбран из группы, состоящей из С1-3алкила, С5-6циклоалкила, гетероциклического алкила, выбранного из 6-членного гетероциклического алкила с 1 атомом азота, гексагидроциклопента(с)пиррола или циклопента(с)пиперидина, где
C1-3алкил, С5-6циклоалкил или гетероциклический алкил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из С1-6алкила, С1-6алкоксила, гидроксила, С3-6циклоалкила, С3-6циклоалкилС1-6алкила, гетероциклического алкила, N-метилморфолина, гексагидропирроло[2,1-с][1,4]оксазина, -C(O)R9, -NR9R10 и C1-6алкилкарбонилоксигруппы; где R9 и R10 вместе с атомом азота, к которому они присоединены, образуют 4-8-членный гетероцикл, где 4-8-членный гетероцикл содержит один или более гетероатомов N или О, и 4-8 членный гетероцикл необязательно замещен C1-6алкилом, необязательно замещенным
С3-6циклоалкилом;
R7 и R8 каждый независимо выбран из атома водорода или С1-6алкила;
R9 и R10 каждый независимо выбран из атома водорода или C1-6алкила.
2. Соединения по п. 1 или их таутомеры, рацематы, энантиомеры, диастереоизомеры и их смеси, а также их фармацевтически приемлемые соли, где соединения выбраны из группы, состоящей из
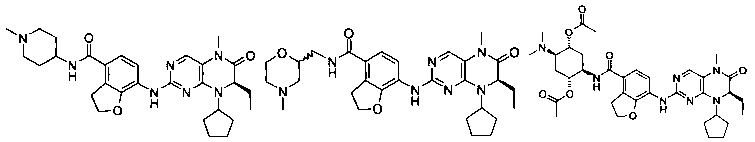
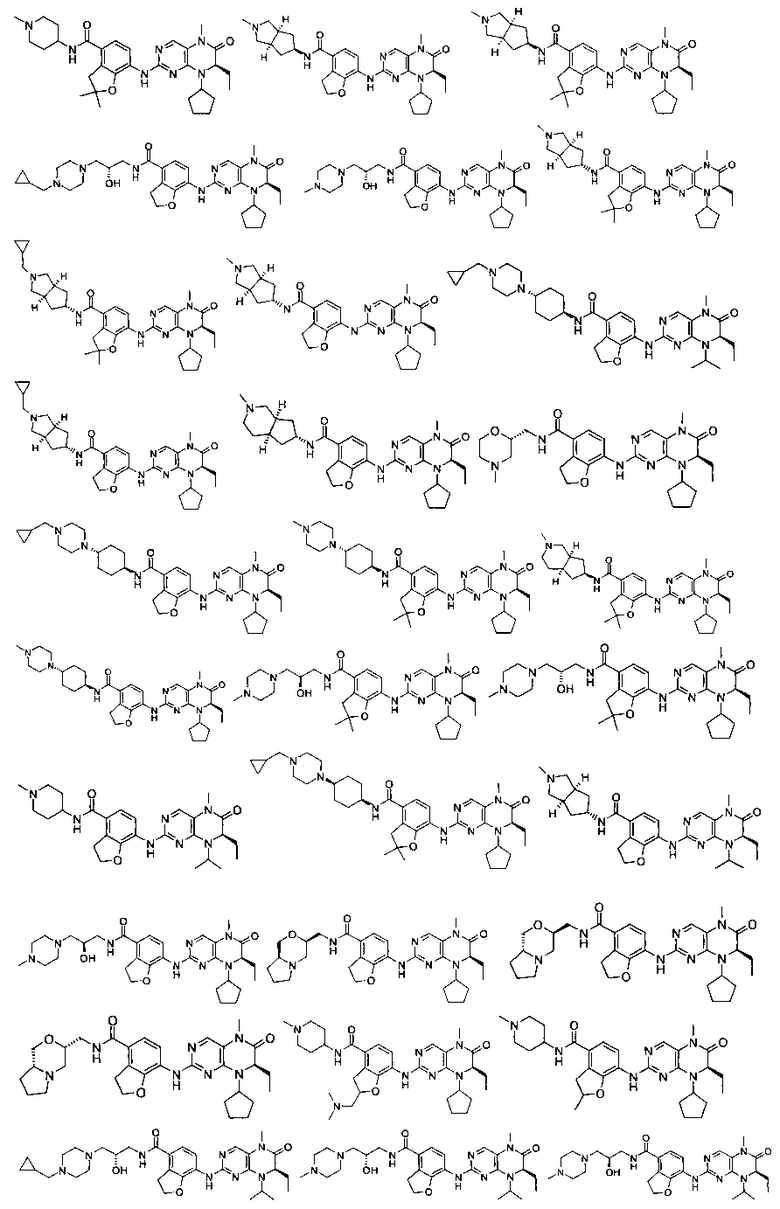
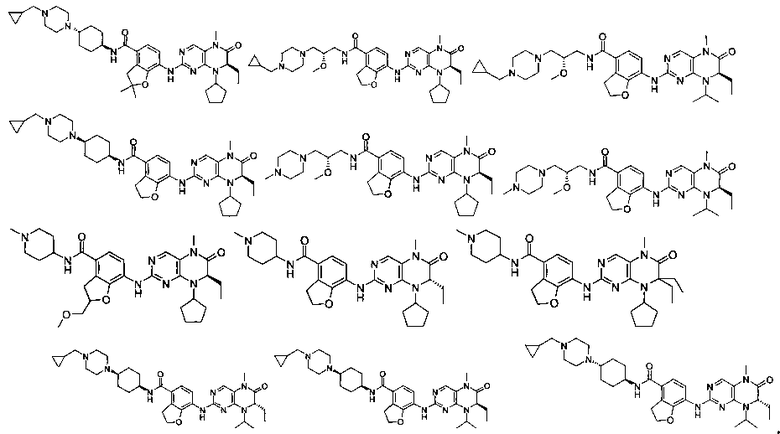
3. Соединения формулы (IA)
где:
R1 и R2 каждый независимо выбран из группы, состоящей из атома водорода и C1-6алкила;
R3 выбран из группы, состоящей из С1-6алкила и С5-6циклоалкила;
R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода, С1-6алкила, где алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из С1-6алкоксила и -NR9R10;
R7 и R8 каждый независимо выбран из атома водорода или С1-6алкила;
R9 и R10 каждый независимо выбран из атома водорода или С1-6алкила;
R11 выбран из группы, состоящей из гидроксила или С1-6алкоксила.
4. Способ получения соединений формулы (I) по любому из пп. 1-2 согласно приведенной ниже стадии
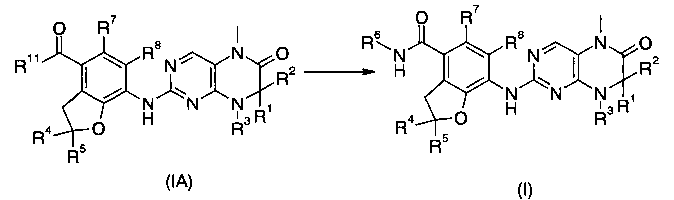
взаимодействия соединения формулы (IA) с R6NH2 с получением соединения формулы (I),
где R1-R8 являются такими, как определено в п. 1, и R11 является таким, как определено в п. 3.
5. Способ получения соединений формулы (IA) по п. 3 согласно приведенной ниже стадии
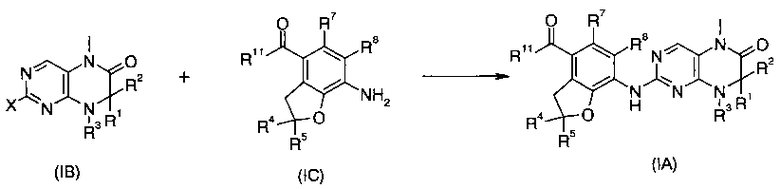
взаимодействия соединения формулы (IB) с соединением формулы (IC) с получением соединения формулы (IA),
где R1-R8 являются такими, как определено в п. 1, и R11 является таким, как определено в п. 3.
6. Фармацевтическая композиция для лечения нарушения клеточной пролиферации, содержащая терапевтически эффективное количество соединений по любому из пп. 1-2 или их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей, а также их фармацевтически приемлемых солей вместе с фармацевтически приемлемыми носителями или эксципиентами.
7. Способ получения фармацевтической композиции по п. 6, включающий стадию объединения соединений формулы (I) по п. 1 с фармацевтически приемлемыми носителями или эксципиентами.
8. Применение соединений по любому из пп. 1-2 или их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей, а также их фармацевтически приемлемых солей для получения лекарства для лечения нарушения клеточной пролиферации.
9. Применение по п. 8, где нарушение клеточной пролиферации представляет собой рак.
10. Применение по п. 9, где рак выбран из группы, состоящей из немелкоклеточного рака легкого, чешуйчато-клеточного рака, рака молочной железы, рака яичника, рака шейки матки, папиллярной карциномы или колоректальной карциномы, предпочтительно рака шейки матки или колоректальной карциномы.
11. Применение соединений по любому из пп. 1-2 или их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей, а также их фармацевтически приемлемых солей для получения лекарства, обладающего свойствами ингибитора Plk для лечения опухолевых заболеваний.
| Стыковое соединение концов резинотросовой ленты и способ его изготовления | 1988 |
|
SU1551881A1 |
| EP 1953163 A1, 06.08.2008 | |||
| Установка для перемещения свежеотформованных керамических дренажных труб | 1958 |
|
SU119825A1 |
| WO 2004076454 A1,10.09.2004 | |||
| WO 2006018220 A2, 23.02.2006 | |||
| US 6861422 B2, 01.03.2005 | |||
| WO 2006018185 A2, 23.02.2006 | |||

