[1] Настоящая заявка испрашивает право на приоритет относительно следующей заявки:
[2] CN 201711331447.7, дата подачи: 13 декабря 2017.
ОБЛАСТЬ ТЕХНИКИ
[3] Настоящее изобретение относится к кристаллической форме и солевой форме ингибитора TGF-βRI и способу их получения и дополнительно включает применение кристаллической формы и солевой формы при получении лекарственного средства для лечения рака.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
[4] Трансформирующий фактор роста-β (TGF-β) представляет собой многофункциональное суперсемейство факторов роста с широким спектром биологических активностей, включая раннее эмбриональное развитие, образование хрящей и костей, синтез внеклеточного матрикса, воспаление, интерстициальный фиброз, регуляцию иммунитета и эндокринные функции, а также образование и развитие опухоли.
[5] Суперсемейство TGF-β состоит из класса структурно и функционально близких полипептидных факторов роста, включая TGF-β (т.е. TGF-β в узком смысле), активины, ингибиторы и костные морфогенетические белки (BMP) (т.е. муллериан), где TGF-β является одним из важных членов этого семейства. У млекопитающих TGF-β в основном существует в трех формах: TGF-β1, TGF-β2 и TGF-β3, которые расположены на разных хромосомах, среди которых TGF-β1 составляет наибольшую долю (>90%) в соматических клетках и обладает самой сильной активностью, большинством функций и наиболее широким распространением. Вновь синтезированный TGF-β возникает как неактивный предшественник, который состоит из трех частей: сигнального пептида, латентно-ассоциированного полипептида (LAP) и зрелого TGF-β. После ферментативного гидролиза образуется активный TGF-β, который затем связывается со своим рецептором для оказания биологического эффекта.
[6] Сигнальные молекулы TGF-β осуществляют передачу сигнала через трансмембранный рецепторный комплекс. TGF-β рецепторы представляют собой трансмембранные белки, которые находятся на поверхности клеток. Они подразделяются на рецепторы типа I (TGF-βRI), рецепторы типа II (TGF-βRII) и рецепторы типа III (TGF-βRIII), где TGF-βRI также называют киназой, подобной рецептору активина, 5 (ALK5). TGF-βRIII не обладает внутренней активностью, что в основном связано с накоплением TGF-β. TGF-βRI и TGF-βRII принадлежат к семейству серин/треонин киназ. Рецепторы типа II могут связываться с TGF-β-лигандами с более высокой аффинностью и образовывать гетерологичные рецепторные комплексы с рецепторами типа I. Область, богатая глициновыми и сериновыми остатками рецепторов типа I (домен GS) вблизи мембраны, фосфорилируется для инициирования внутриклеточных сигнальных каскадных реакций.
[7] Smads является важной молекулой для передачи и регуляции сигнала TGF-β в клетке, которая может напрямую передавать сигнал TGF-β от клеточной мембраны в ядро клетки. Сигнальный путь TGF-β/Smads играет важную роль в возникновении и развитии опухолей. При передаче сигнала TGF-β/Smads активированный TGF-β сначала связывается с TGF-βRII на поверхности клеточной мембраны с образованием гетеродимерного комплекса, а TGF-βRI распознает и связывается с бинарным комплексом.
[8] TGF-βRII фосфорилирует серин/треонин в GS-домене цитоплазматической области TGF-βRI, тем самым активируя TGF-βRI; активированный TGF-βRI впоследствии фосфорилирует белок R-Smads (Smad2/Smad3), который затем связывается с Co-Smad (Smad4), образуя гетеротримерный комплекс, где комплекс входит в ядро и взаимодействует с другими коактиваторами и коингибиторами для того, чтобы регулировать транскрипцию генов-мишеней. Изменения в любой части пути передачи сигналов TGF-β/Smads приведут к отклонениям в пути передачи сигналов.
[9] Современные исследования показывают, что в опухолевых клетках TGF-β может непосредственно влиять на рост опухоли (внешние эффекты передачи сигналов TGF-β) или может косвенно влиять на рост опухоли (внутренние эффекты TGF-β), индуцируя эпителиально-мезенхимальную трансдифференциацию, блокирование противоопухолевых иммунных реакций, увеличение фиброза, связанного с опухолью, и усиление регенерации сосудов. Кроме того, TGF-β обладает сильным эффектом индукции фиброза и является активатором фибробластов, связанных с опухолями. Эти фибробласты являются основным источником коллагена типа I и других фиброзных факторов. Индуцированные продукты фибробластов и других фиброзных факторов могут продолжать создавать микроокружение, в котором снижается иммунный ответ, повышается резистентность к лекарствам и усиливается ангиогенез опухоли. Кроме того, TGF-β влияет на ангиогенез во время индивидуального развития и роста опухоли. Например, TGF-βRI-дефицитные мышиные эмбрионы показали серьезные дефекты развития сосудов, доказывая то, что сигнальный путь TGF-β является ключевым регулятором в развитии эндотелия сосудов и клеток гладких мышц.
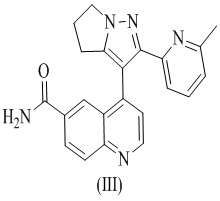
[10] В 2013 году FDA одобрило низкомолекулярный ингибитор TGF-βRI LY2157299 (WO 2002/094833) от Eli Lilly для лечения глиомы и рака печени. LY2157299 представляет собой находящийся в разработке препарат для лечения редких заболеваний под названием Галунисертиб. Галунисертиб может не только ингибировать инвазию и метастазирование опухолевых клеток, но также ингибировать инфильтрацию опухолевых клеток в кровеносные сосуды. В фазе 2 клинических испытаний для лечения пациентов с раком печени после лечения Галунисертибом примерно у 23% пациентов было отмечено снижение уровней альфа-фетопротеина (АФП) в сыворотке по меньшей мере на 20%. По сравнению с пациентами, которые не отвечали на АФП, у этих пациентов наблюдалось более медленное прогрессирование опухоли и более длительный период выживаемости, а также повышенная экспрессия кадгерина в эпителиальных клетках, что указывает на то, что Галунисертиб может регулировать ENT путем ингибирования пути передачи сигналов TGF-β, подавляя тем самым прогрессирование рака печени.
[11] Структура Галунисертиба (LY2157299) является такой, как показано в формуле (III):
[12] В дополнение к терапевтической эффективности, разработчики лекарств предпринимают попытки обеспечить подходящие формы активных молекул с фармацевтическими свойствами, причем свойства включают обработку, производство, стабильность при хранении и т.д. Таким образом, было обнаружено, что для разработки лекарств необходимы формы с желаемыми свойствами.
Содержание настоящего изобретения
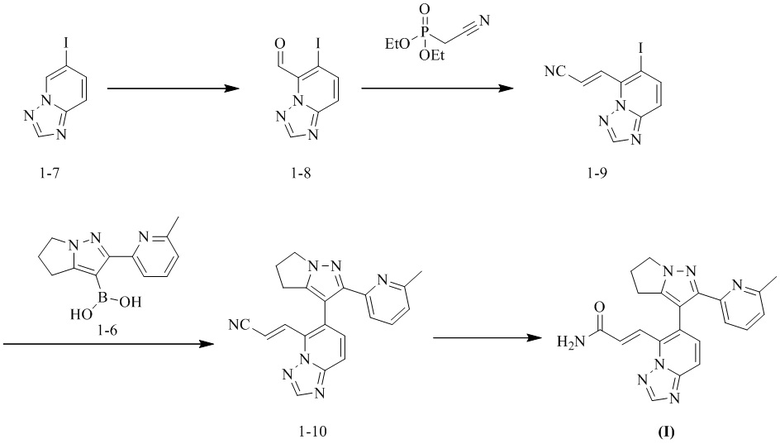
[13] Настоящее изобретение предусматривает кристаллическую форму А соединения формулы (I), характеризующуюся тем, что ее порошковая дифракционная рентгенограмма имеет характерные дифракционные пики при следующих углах 2θ: 11,894°±0,2°, 17,502°±0,2°, 19,785°±0,2°, 24,072°±0,2° и 24,664°±0,2°.
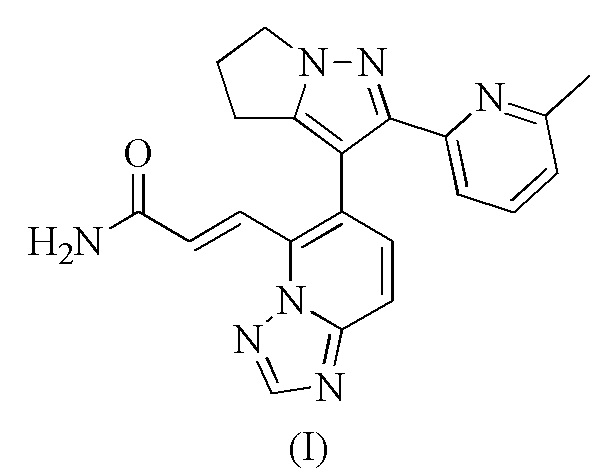
[14] В некоторых вариантах воплощения настоящего изобретения порошковая дифракционная рентгенограмма (ПДР) вышеупомянутой кристаллической формы A имеет характерные дифракционные пики при следующих углах 2θ: 9,553°±0,2°, 11,894°±0,2°, 15,370°±0,2°, 17,502°±0,2°, 19,785°±0,2°, 20,283°±0,2°, 24,072°±0,2° и 24,664°±0,2°.
[15] В некоторых вариантах воплощения настоящего изобретения порошковая дифракционная рентгенограмма вышеупомянутой кристаллической формы A является такой, как показано на фиг. 1.
[16] В некоторых вариантах воплощения настоящего изобретения данные анализа ПДР вышеупомянутой кристаллической формы A являются такими, как показано в таблице 1.
Таблица 1
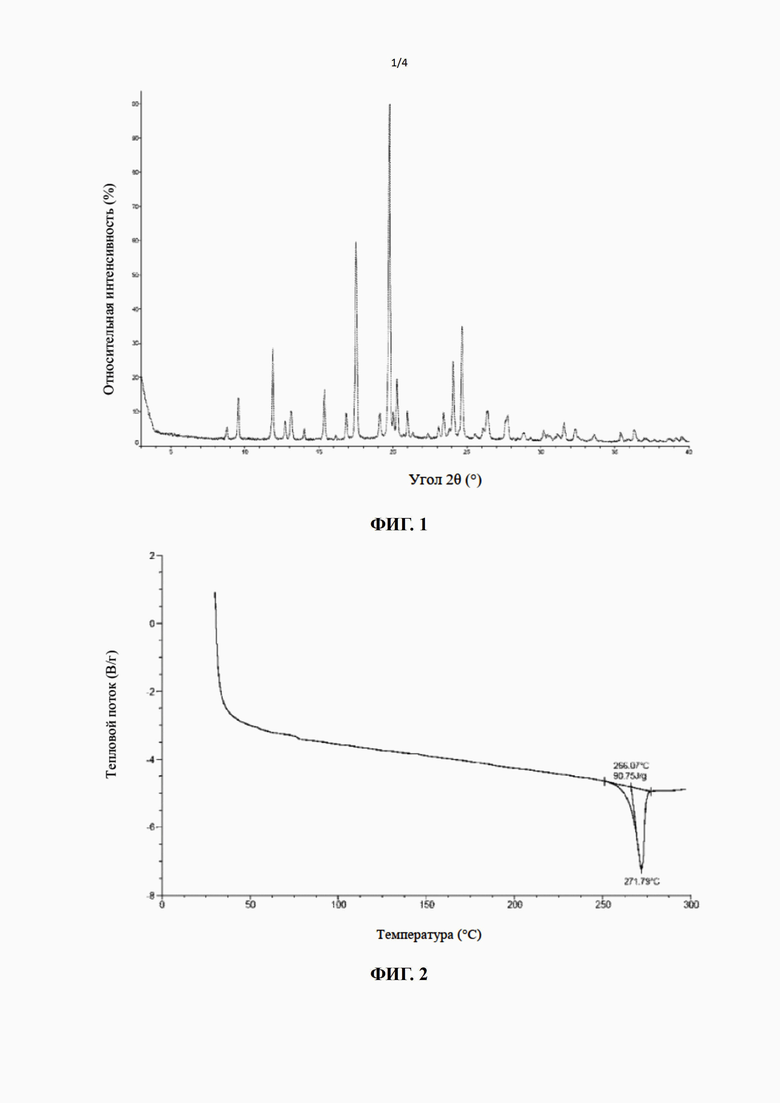
[17] В некоторых вариантах воплощения настоящего изобретения вышеупомянутая кристаллическая форма A также может характеризоваться с помощью ДСК с начальной температурой 266,07°С и пиковой температурой 271,79°С.
[18] В некоторых вариантах воплощения настоящего изобретения профиль дифференциальной сканирующей калориметрии вышеупомянутой кристаллической формы A имеет эндотермический пик при 271,79±3°С.
[19] В некоторых вариантах воплощения настоящего изобретения диаграмма профиля дифференциальной сканирующей калориметрии вышеупомянутой кристаллической формы A является такой, как показано на фиг. 2.
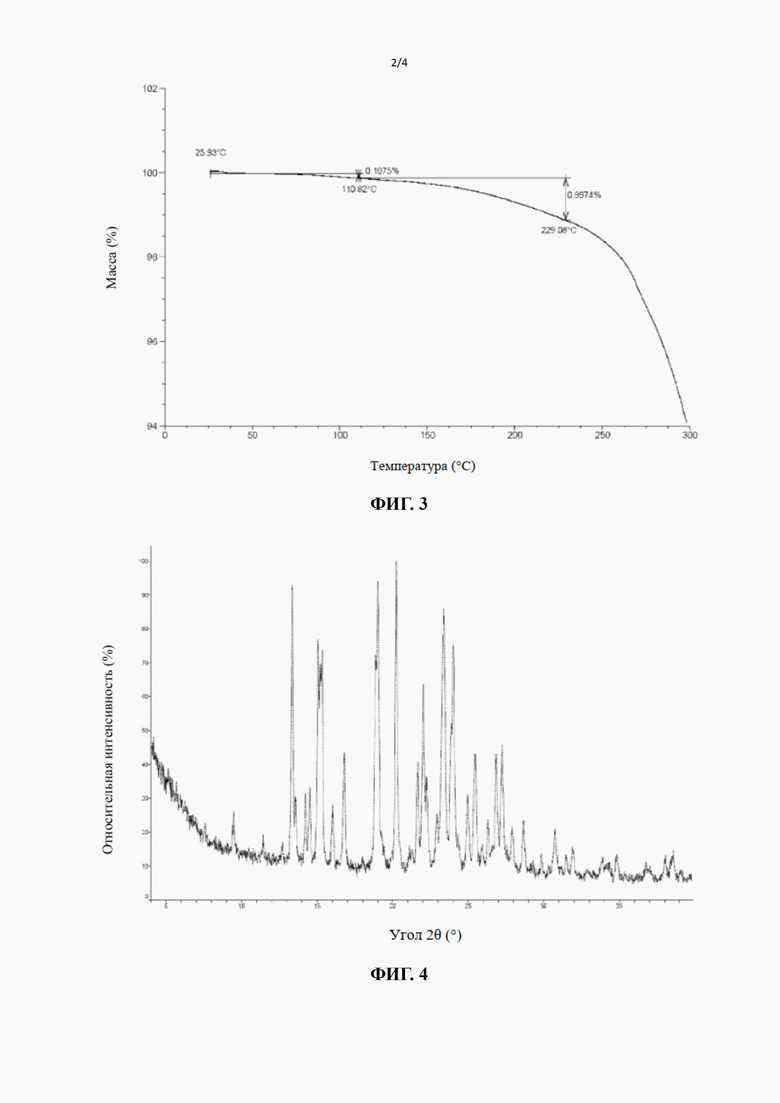
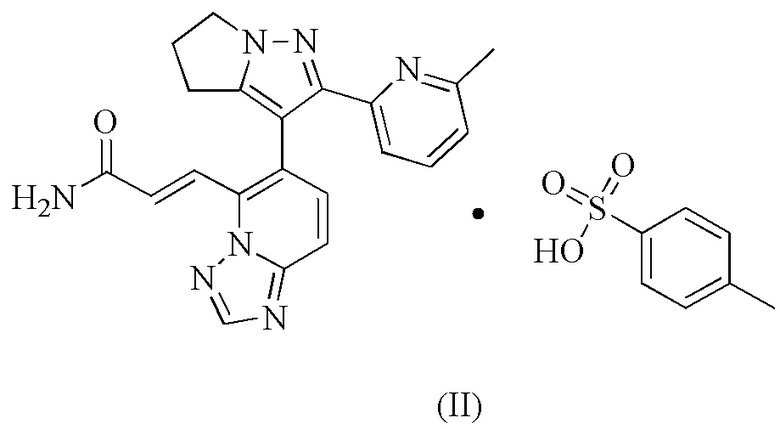
[20] В некоторых вариантах воплощения настоящего изобретения вышеупомянутая кристаллическая форма A также может характеризоваться с помощью ТГА, где диаграмма ТГА показывает, что когда кристаллическая форма нагревается до 110,82°С, масса уменьшается на 0,1075%; при нагревании до 229,08°С масса дополнительно уменьшается на 0,9974%; происходит большая потеря массы после 229,08°С.
[21] В некоторых вариантах воплощения настоящего изобретения профиль термогравиметрического анализа вышеупомянутой кристаллической формы A показывает 0,1075% потери массы при 110,82±3°С и 1,105% потери массы при 229,08±3°С.
[22] В некоторых вариантах воплощения настоящего изобретения диаграмма профиля термогравиметрического анализа вышеупомянутой кристаллической формы A является такой, как показано на фиг. 3.
[23] Настоящее изобретение дополнительно предусматривает соединение формулы (II).
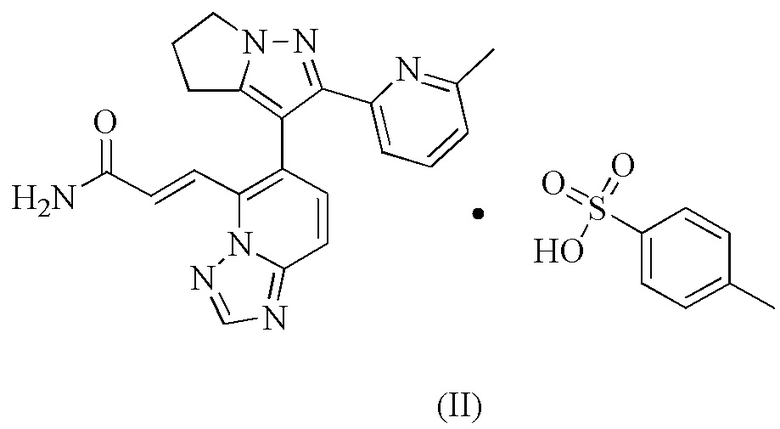
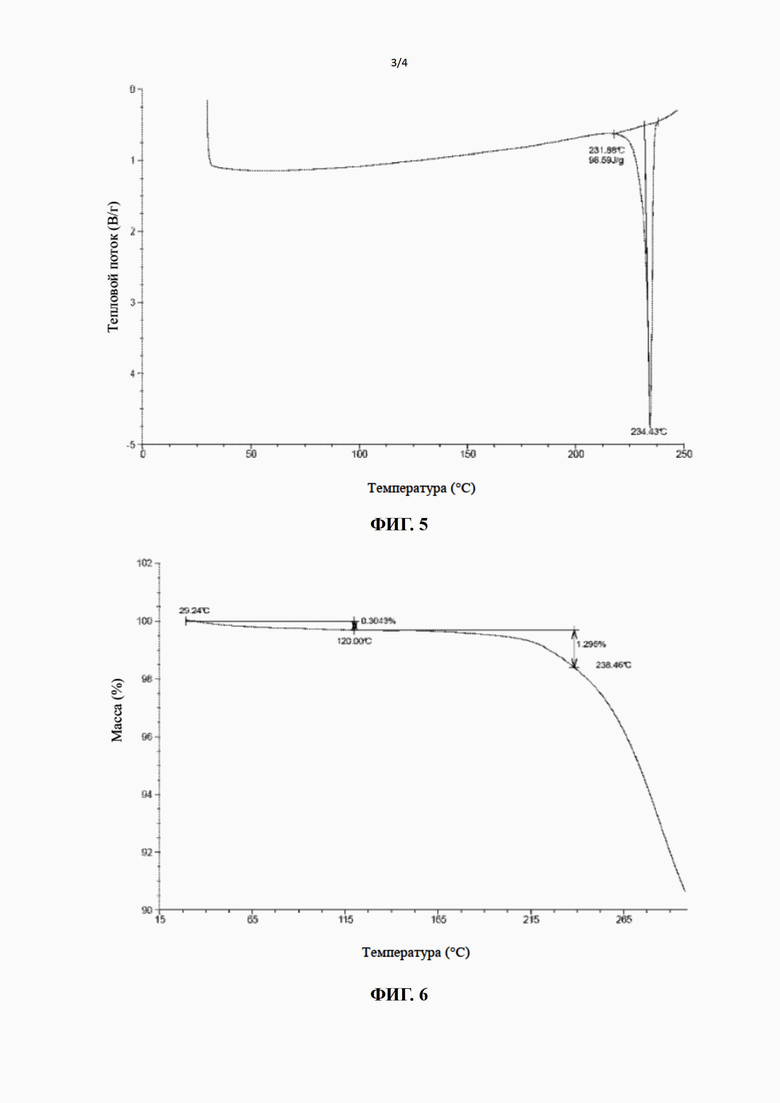
[24] Настоящее изобретение дополнительно предусматривает кристаллическую форму B соединения формулы (II), характеризующуюся тем, что ее порошковая дифракционная рентгенограмма имеет характерные дифракционные пики при следующих углах 2θ: 13,349±0,2°, 19,012±0,2°, 20,235±0,2° и 23,370±0,2°.
[25] В некоторых вариантах воплощения настоящего изобретения порошковая дифракционная рентгенограмма вышеупомянутой кристаллической формы B имеет характерные дифракционные пики при следующих углах 2θ: 13,349±0,2°, 15,066±0,2°, 16,782±0,2°, 19,012±0,2°, 20,235±0,2°, 22,027±0,2°, 23,370±0,2° и 27,253±0,2°.
[26] В некоторых вариантах воплощения настоящего изобретения порошковая дифракционная рентгенограмма вышеупомянутой кристаллической формы B является такой, как показано на фиг. 4.
[27] В некоторых вариантах воплощения настоящего изобретения данные анализа ПДР вышеупомянутой кристаллической формы B являются такими, как показано в таблице 2.
Таблица 2
[28] В некоторых вариантах воплощения настоящего изобретения вышеупомянутая кристаллическая форма B также может характеризоваться с помощью ДСК, которая имеет эндотермический пик при 234,43±3°С.
[29] В некоторых вариантах воплощения настоящего изобретения профиль дифференциальной сканирующей калориметрии вышеупомянутой кристаллической формы B имеет эндотермический пик при 234,43±3°С.
[30] В некоторых вариантах воплощения настоящего изобретения диаграмма профиля дифференциальной сканирующей калориметрии вышеупомянутой кристаллической формы B является такой, как показано на фиг. 5.
[31] В некоторых вариантах воплощения настоящего изобретения вышеупомянутая кристаллическая B также может характеризоваться с помощью ТГА, где диаграмма ТГА показывает, что когда кристаллическая форма нагревается до 120°С, масса уменьшается на 0,3043%; при нагревании до 238,46°С масса дополнительно уменьшается на 1,295%.
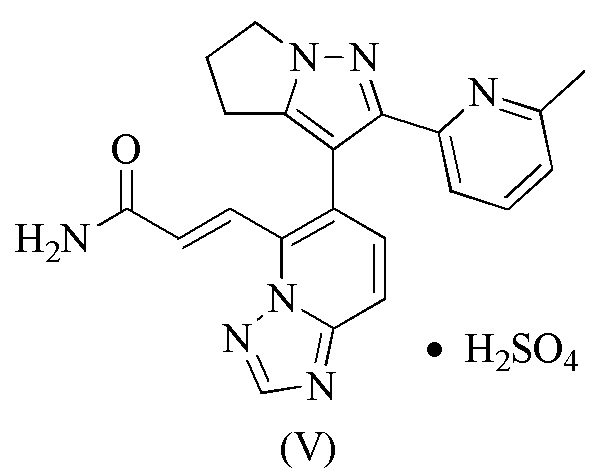
[32] В некоторых вариантах воплощения настоящего изобретения профиль термогравиметрического анализа вышеупомянутой кристаллической формы B показывает 0,3043% потери массы при 120±3°С и 1,599% потери массы при 238,46±3°С.
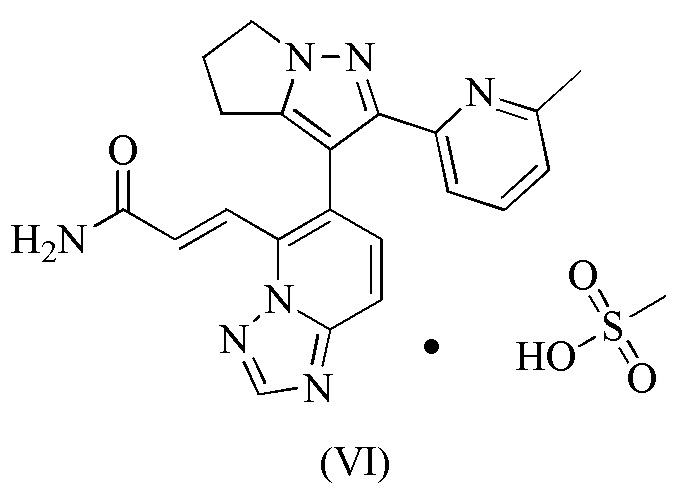
[33] В некоторых вариантах воплощения настоящего изобретения диаграмма профиля термогравиметрического анализа вышеупомянутой кристаллической формы B является такой, как показано на фиг. 6.
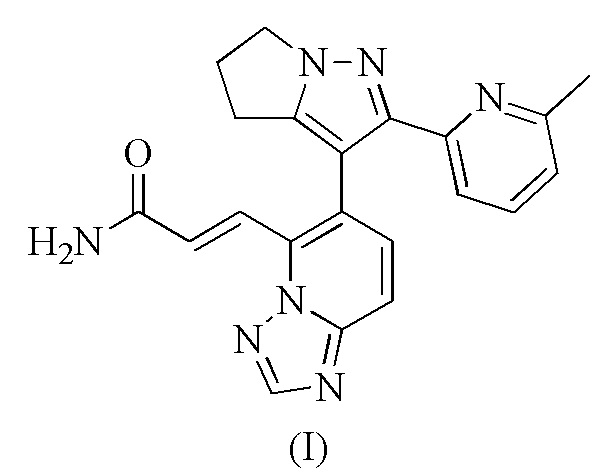
[34] Настоящее изобретение дополнительно предусматривает гидрохлорид, сульфат и метансульфонат соединения формулы (I).
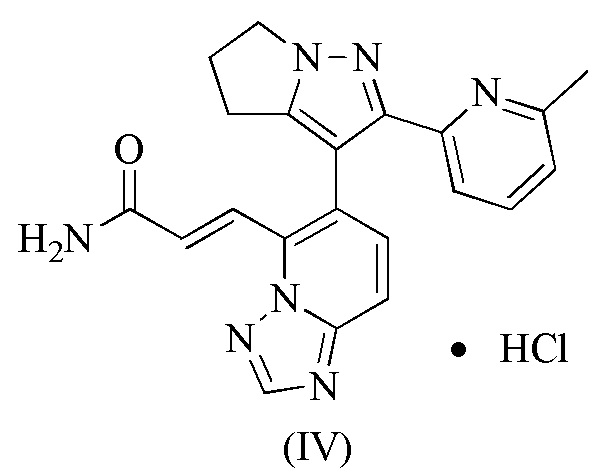
[35] В некоторых вариантах воплощения настоящего изобретения вышеупомянутый гидрохлорид представляет собой
 .
.
[36] В некоторых вариантах воплощения настоящего изобретения вышеупомянутый сульфат представляет собой
 .
.
[37] В некоторых вариантах воплощения настоящего изобретения вышеупомянутый метансульфонат представляет собой
 .
.
[38] Настоящее изобретение дополнительно предусматривает применение вышеупомянутых соединений или кристаллических форм при получении лекарственного средства для лечения рака.
[39] Технологические эффекты
[40] Способ получения солевой формы и кристаллической формы, предлагаемый в настоящем изобретении, является простым; кроме того, кристаллическая форма стабильна в условиях высокой температуры и высокой влажности и является слегка гигроскопичной, а солевая форма имеет хорошую растворимость в чистой воде и биологическом носителе и имеет хорошие перспективы в изготовлении.
[41] Определения и описание
[42] Если не указано иное, следующие термины и фразы, используемые в данном документе, предполагают следующие значения. Конкретную фразу или термин не следует считать неопределенными или неясными, если они специально не оговорены, их следует понимать в обычном смысле. Когда торговое наименование упоминается в данном документе, оно предназначено для обозначения соответствующего товара или его активного ингредиента.
[43] Промежуточные соединения по настоящему изобретению могут быть получены различными синтетическими способами, хорошо известными специалисту в данной области техники, включая конкретные варианты воплощения, перечисленные ниже, варианты воплощения, образованные комбинацией с другими способами химического синтеза, и эквивалентные альтернативные варианты воплощения, хорошо известные специалисту в данной области техники, причем предпочтительные варианты воплощения включают, но не ограничиваются ими, примеры настоящего изобретения.
[44] Химические реакции, описанные в конкретных вариантах воплощения настоящего изобретения, проводятся в подходящем растворителе, причем этот растворитель должен быть подходящим для химических изменений по настоящему изобретению и для требуемых для этого реагентов и материалов. Чтобы получить соединения по настоящему изобретению, специалисту в данной области техники иногда необходимо модифицировать или выбирать стадии синтеза или схемы реакций на основе существующих вариантов воплощения.
[45] Настоящее изобретение будет конкретно описано ниже посредством примеров, которые никоим образом не предназначены для ограничения настоящего изобретения.
[46] Все растворители, используемые в настоящем изобретении, являются коммерчески доступными и могут использоваться без дальнейшей очистки.
[47] В настоящем изобретении используются следующие сокращения: к.т. обозначает комнатную температуру; в.р. обозначает водный раствор; экв. обозначает эквивалент; ДХМ обозначает дихлорметан; ТГФ обозначает тетрагидрофуран; ДМСО обозначает диметилсульфоксид; ДМФА обозначает N,N-диметилформамид; EtOAc обозначает этилацетат; EtOH обозначает этанол; MeOH обозначает метанол; диоксан обозначает диоксан; HOAc обозначает уксусную кислоту; DIPEA обозначает диизопропилэтиламин; TEA или Et3N обозначает триэтиламин; Na2CO3 обозначает карбонат натрия; K2CO3 обозначает карбонат калия; NaHCO3 обозначает бикарбонат натрия; Na2SO4 обозначает сульфат натрия; NaOH обозначает гидроксид натрия; LiHMDS обозначает литий-бис(триметилсилил)амин; Pd(dppf)Cl2 обозначает [1,1'-бис(дифенилфосфино)ферроцен]палладий дихлорид; Xphos обозначает 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил; Xphos-PD-G2 обозначает хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил) [2-(2'-амино-1,1'-бифенил)палладий (II); NBS обозначает N-бромсукцинимид; HCl обозначает соляную кислоту; H2SO4 обозначает серную кислоту; °С обозначает градус Цельсия.
[48] Соединения названы заявителем или программным обеспечением ChemDraw®, а коммерчески доступные соединения названы именами в каталоге поставщика.
[49] Приборы и способы анализа
[50] В настоящем изобретении порошковый рентгеновский дифрактометр (ПДР) определяется с использованием следующего способа:
[51] модель прибора: рентгеновский дифрактометр Bruker D8 advance;
[52] способ тестирования: приблизительно 10 - 20 мг образца используется для ПДР детекции.
[53] Подробные параметры ПДР следующие:
[54] Рентгеновская трубка: Cu, kα, (λ=1,54056 );
);
[55] вольтаж трубки: 40 кВ, ток в трубке: 40 мА;
[56] щель расходимости: 0,60 мм;
[57] щель детектора: 10,50 мм;
[58] антирассеивающая щель: 7,10 мм;
[59] диапазон сканирования: 3-40 град или 4-40 град;
[60] размер шага: 0,02 град;
[61] длина шага: 0,12 с;
[62] скорость вращения диска образцов: 15 об/мин.
[63] В настоящем изобретении дифференциальная сканирующая калориметрия (ДСК) определяется с использованием следующего способа:
[64] модель прибора: дифференциальный сканирующий калориметр TA Q2000.
[65] Способ тестирования: поместить около 1 мг образца в алюминиевый поддон для тестирования ДСК; в условиях 50 мл/мин азота, нагреть образец от 30°С (комнатная температура) до 300°С (или 350°С) со скоростью нагрева 10 °С/мин.
[66] В настоящем изобретении термогравиметрический анализатор (ТГА) определяют с использованием следующего способа:
[67] модель прибора: термогравиметрический анализатор TA Q5000.
[68] Способ тестирования: поместить 2-5 мг образца в платиновую чашку для тестирования ТГА; при условии 25 мл/мин N2, нагревать образец от комнатной температуры до 300°С или до 20% потери массы при скорости нагрева 10°С/мин.
[69] В настоящем изобретении динамическая сорбция паров (DVS) определяется с использованием следующего способа:
[70] модель прибора: прибор динамической сорбции паров SMS DVS Advantage
[71] Условия тестирования: поместить 10-15 мг образца в диск с образцами для тестирования DVS.
[72] Подробные параметры DVS следующие:
[73] температура: 25°С
[74] баланс: dm/dt=0,01%/мин (минимум: 10 мин; максимум: 180 мин)
[75] сушка: сушить в течение 120 мин при 0% RH
[76] RH (%) этапа тестирования: 10%
[77] диапазон RH (%) этапа тестирования: 0%-90%-0%
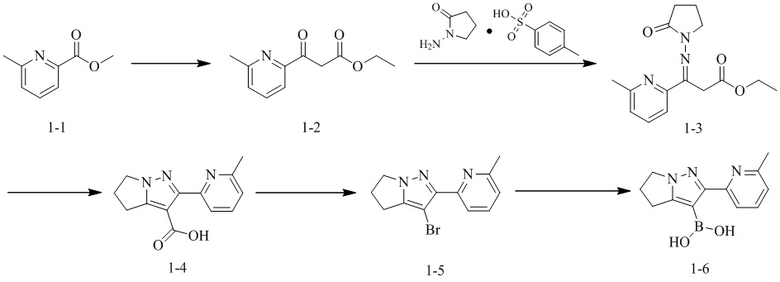
[78] Классификация оценки гигроскопичности выглядит следующим образом:
[79] * Гигроскопическое увеличение веса (δW%) при 25±1°С и 80%±2% RH
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[80] Фиг. 1 представляет собой ПДР кристаллической формы А соединения формулы (I).
[81] Фиг. 2 представляет собой диаграмму ДСК кристаллической формы А соединения формулы (I).
[82] Фиг. 3 представляет собой диаграмму ТГА кристаллической формы А соединения формулы (I).
[83] Фиг. 4 представляет собой ПДР кристаллической формы B соединения формулы (II).
[84] Фиг. 5 представляет собой диаграмму ДСК кристаллической формы B соединения формулы (II).
[85] Фиг. 6 представляет собой диаграмму ТГА кристаллической формы B соединения формулы (II).
[86] Фиг. 7 представляет собой диаграмму DVS кристаллической формы B соединения формулы (II).
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ВОПЛОЩЕНИЯ
[87] Чтобы лучше понять содержание настоящего изобретения, следующие конкретные примеры используются для дальнейшего описания, но конкретные варианты воплощения не ограничивают содержание настоящего изобретения.
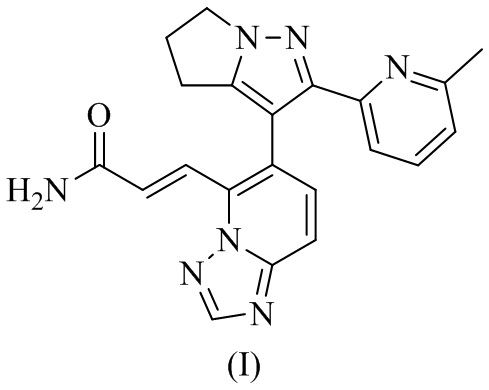
[88] Пример 1. Получение соединения формулы (I).
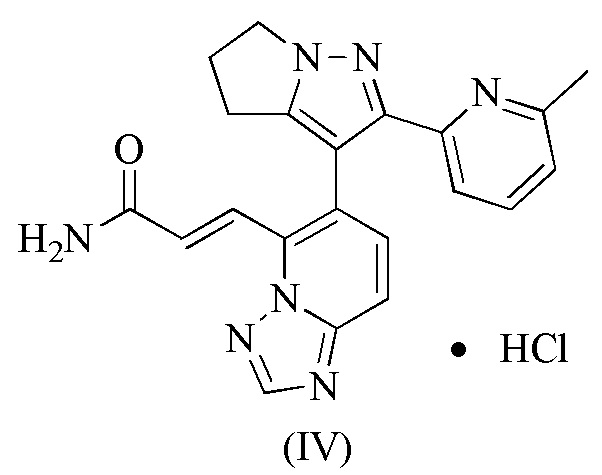
[89] Приготовление промежуточных продуктов 1-6:
[90] Этап A: Этилацетат (291,41 мл, 2,98 моль) растворяли в толуоле (750,00 мл) и затем порциями при комнатной температуре добавляли этоксид натрия (135,06 г, 1,98 моль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. 1-1 (150,00 г, 992,33 ммоль) добавляли к вышеупомянутому реакционному раствору при 25°C, а затем нагревали до 95°C и перемешивали в течение 15 часов. Реакционную смесь охлаждали до около 30°С, доводили до рН 7 уксусной кислотой, разбавляли водой (500 мл) и затем экстрагировали этилацетатом (500 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем (элюент : петролейный эфир/этилацетат об/об = 50/1) с получением 1-2.
[91] Этап B: 1-2 (120,00 г, 579,07 ммоль) растворяли в пиридине (300 мл) и затем добавляли p-толуолсульфонатную соль (172,01 г, 631,66 ммоль) 1-аминопирролидин-2-она. Реакционную смесь перемешивали при 25°С в течение 16 часов и затем концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (300 мл) и затем экстрагировали этилацетатом (300 мл × 2). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, с получением 1-3.
[92] Этап C: 1-3 (155,00 г, 535,72 ммоль) растворяли в толуоле и затем к нему добавляли этоксид натрия (72,91 г, 1,07 моль). Реакционную смесь нагревали до 100°С и перемешивали в течение 16 часов, а затем охлаждали до комнатной температуры. Реакционную смесь медленно разбавляли водой (1,5 л), доводили до рН 4 концентрированной соляной кислотой и экстрагировали смесью дихлорметан/изопропанол (об./об. = 10/1, 1 л × 7). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток суспендировали с петролейным эфиром/этилацетатом (об./об. = 10/1, 200 мл) и фильтровали, а твердое вещество собирали. Твердое вещество сушили при пониженном давлении, с получением 1-4.
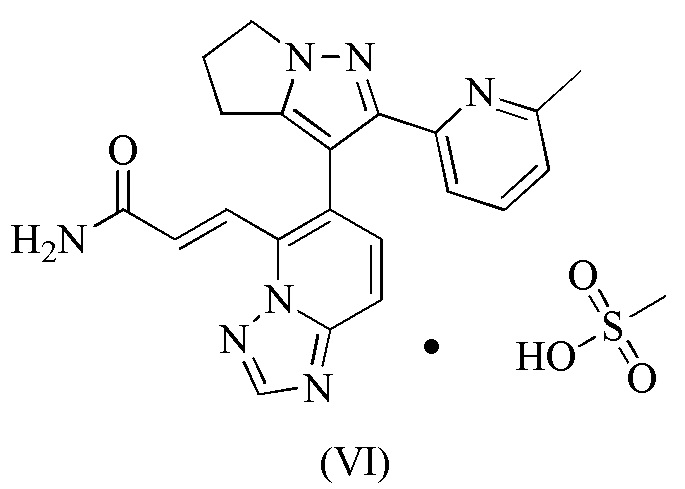
[93] Этап D: 1-4 (45,00 г, 184,99 ммоль) растворяли в N,N-диметилформамиде (650,00 мл), а затем к нему добавляли NBS (49,09 г, 258,99 ммоль). Реакционную смесь перемешивали при 30°С-40°С в течение 60 часов, а затем разбавляли водой (600 мл) и экстрагировали дихлорметаном/изопропанолом (об./об. = 10/1, 500 мл × 3). Объединенную органическую фазу промывали один раз гидроксидом натрия (0,5 моль/л, 800 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученное твердое вещество суспендировали с петролейным эфиром/этилацетатом (об./об. = 10/1, 200 мл) и фильтровали, а твердое вещество собирали. Твердое вещество сушили при пониженном давлении, с получением 1-5.
[94] Этап E: 1-5 (1,00 г, 3,60 ммоль) и триизопропилборат (1,79 г, 9,54 ммоль) растворяли в тетрагидрофуране (20,00 мл). Реакционную смесь охлаждали до минус 70°С и затем по каплям добавляли n-бутиллитий (2,5 М, 3,74 мл). После завершения добавления по каплям реакционную смесь перемешивали при 25°С в течение 1 часа и затем доводили до рН 7 водной соляной кислотой (0,5 моль/л). Затем реакционную смесь концентрировали при пониженном давлении для удаления тетрагидрофурана и затем охлаждали до 15°С. Смесь фильтровали и осадок на фильтре суспендировали в петролейном эфире/этилацетате (об./об. = 10/1, 5,5 мл) и фильтровали, а твердое вещество собирали, которое сушили при пониженном давлении, получая 1-6.
[95] Получение соединения формулы (I):
[96] Этап A: 1-7 (16,00 г, 65,30 ммоль) растворяли в тетрагидрофуране (800,00 мл) и после охлаждения до минус 60°С - минус 70°С к нему по каплям добавляли гексаметилдисилазид лития (1 моль/л, 130,60 мл, 65,30 ммоль). Реакционную смесь перемешивали при минус 60°С - минус 70°С в течение 15 минут и к ней добавляли N,N-диметилформамид (14,32 г, 195,90 ммоль, 15,07 мл). Затем реакционную смесь непрерывно перемешивали при минус 60°С - минус 70°С в течение 15 минут и затем гасили насыщенным водным раствором хлорида аммония (500 мл). Реакционную смесь нагревали до комнатной температуры и затем экстрагировали этилацетатом (500 мл × 2). Объединенную органическую фазу промывали солевым раствором (500 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем (элюент: дихлорметан/этилацетат об./об. = 10/1) с получением 1-8. 1H ЯМР (400 МГц, DMSO-d6) δ 10,46 (s, 1H), 8,62 (s, 1H), 8,16 (d, J=9,3 Гц, 1H), 7,88 (d, J=9,3 Гц, 1H).
[97] Этап B: В трехгорлую колбу на 500 мл, снабженную термометром и баллоном с азотом, добавляли 2-диэтоксифосфорилацетонитрил (3,83 г, 21,61 ммоль, 3,48 мл) и тетрагидрофуран (80 мл). Смесь охлаждали до 0°C и затем порциями добавляли трет-бутоксид калия (2,42 г, 21,61 ммоль). Реакционную смесь перемешивали при 0°С в течение 15 минут и затем добавляли по каплям к другой суспензии (1-8, диспергировали в тетрагидрофуране (120 мл) и охлаждали до 0°С) через капельную воронку. Реакционную смесь перемешивали при 0°С в течение 15 минут, а затем вливали в воду (300 мл), гасили и экстрагировали этилацетатом (200 мл) и дихлорметаном (200 мл × 2). Объединенную органическую фазу промывали солевым раствором (300 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем (элюент: дихлорметан/этилацетат об./об. = 200/1 до 10/1) с получением 1-9. 1H ЯМР (400 МГц, CDCl3) δ 8,42 (s, 1H), 8,03 (d, J=9,3 Гц, 1H), 7,98-7,91 (m, 1H), 7,85-7,78 (m, 1H), 7,60 (d, J=9,2 Гц, 1H).
[98] Этап C: 1-9 (4,50 г, 15,20 ммоль), 1-6 (4,43 г, 18,24 ммоль), карбонат натрия (4,83 г, 45,60 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]палладий дихлорид (556,07 мг, 759,96 мкмоль), 2-дициклогексилфосфино-2',6'-диметоксибифенил (311,98 мг, 759,96 мкмоль) и [2-(2-аминофенил)фенил]-хлор-палладий-циклогексил-[2-(2,6-диметоксифенил)фенил]фосфин (547,64 мг, 759,96 мкмоль) добавляли к смешанному растворителю из диоксана (100 мл) и воды (20 мл). Смесь трижды вентилировали азотом, затем нагревали до 90°С - 100°С и перемешивали в течение 2 часов. Реакционную смесь вливали в воду (200 мл), гасили и экстрагировали дихлорметаном (200 мл × 2). Объединенную органическую фазу промывали солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем (элюент: дихлорметан/метанол, об./об. = 30/1) для получения сырого продукта, который перемешивали в течение 12 часов в смешанном растворителе петролейного эфира/этилацетата (об./об. = 5/1) и отфильтровывали, а твердое вещество собирали, концентрировали и сушили при пониженном давлении, чтобы получить 1-10. 1H ЯМР (400 МГц, CDCl3) δ 8,49 (s, 1H), 7,82-7,74 (m, 2H), 7,59-7,46 (m, 4H), 6,99 (dd, J=2,6, 6,1 Гц, 1H), 4,39 (d, J=6,3 Гц, 2H), 2,90-2,70 (m, 4H), 2,20 (s, 3H).
[99] Этап D: 1-10 (5,37 г, 14,62 ммоль) растворяли в смешанном растворителе из дихлорметана (20 мл), диметилсульфоксида (70 мл) и воды (20 мл), а затем перекиси водорода (8,29 г, 73,10 ммоль, 7,02 мл, 30%) и гидроксид натрия (2 моль/л, 14,62 мл) соответственно. Смесь перемешивали при 15°С -20°С в течение 12 часов. Смесь вливали в воду (200 мл), гасили и экстрагировали смешанным растворителем дихлорметана/изопропанола (3/1) (200 мл × 1). Органическую фазу промывали насыщенным водным раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной высокоэффективной жидкостной хроматографией (колонка: Phenomenex Gemini C18 250×50 мм×10 мкм; мобильная фаза: [вода (0,05% аммиака по объему)-ацетонитрила]; градиент: 5% -32%, 33; 80% минут), чтобы получить соединение формулы (I). 1H ЯМР (400 МГц, CDCl3) δ 8,45 (s, 1H), 8,09 (d, J=15,6 Гц, 1H), 7,85 (d, J=15,6 Гц, 1H), 7,69 (d, J=9,2 Гц, 1H), 7,55-7,45 (m, 2H), 7,37 (d, J=7,8 Гц, 1H), 6,99 (d, J=7,7 Гц, 1H), 5,93-5,65 (m, 2H), 4,35 (br. s., 2H), 2,99-2,64 (m, 4H), 2,33 (s, 3H).
[100] Пример 2. Получение соединения формулы (II).
[101] 115 мг соединения формулы (I) добавляли в стеклянную бутылку объемом 8 мл и добавляли к нему 4 мл тетрагидрофурана, которые образовывали суспензию путем растворения с помощью ультразвука; и затем медленно добавляли 1,05 эквивалента моногидрата p-толуолсульфоновой кислоты. Вышеупомянутый суспендированный образец помещали на магнитную мешалку (40°С) и перемешивали в течение 16 часов. Раствор образца центрифугировали, твердое вещество отбирали и помещали в вакуумную печь при 35°С для сушки на 16 часов, получая соединение формулы (II). 1H ЯМР (400 МГц, CD3OD) δ 8,61 (s, 1H), 8,14 (t, J=8,0 Гц, 1H), 8,05 (d, J=15,6 Гц, 1H), 7,90 (d, J=8,8 Гц, 1H), 7,70 (dd, J=8,4, 15,6 Гц, 4H), 7,54 (d, J=15,6 Гц, 1H), 7,39 (d, J=8,0 Гц, 1H), 7,20 (d, J=7,6 Гц, 2H), 4,42 (m, 2H), 3,05-2,87 (m, 2H), 2,82 (s, 3H), 2,81-2,74 (m, 2H), 2,35 (s, 3H).
[102] Пример 3. Получение соединения формулы (IV).
[103] 115 мг соединения формулы (I) добавляли в стеклянную бутылку объемом 8 мл и добавляли к нему 4 мл тетрагидрофурана, которые образовывали суспензию путем растворения с помощью ультразвука; и затем медленно добавляли 1,05 эквивалента соляной кислоты. Вышеупомянутый суспендированный образец помещали на магнитную мешалку (40°С) и перемешивали в течение 16 часов. Раствор образца центрифугировали, твердое вещество отбирали и помещали в вакуумную печь при 35°С для сушки на 16 часов. Полученное твердое вещество добавляли к соответствующему количеству ацетона для приготовления суспензии, перемешивали при 40°C и затем центрифугировали для удаления супернатанта, а твердый образец сушили с помощью масляного насоса при комнатной температуре для получения соединения формулы (IV).
[104] Пример 4. Получение соединения формулы (V).
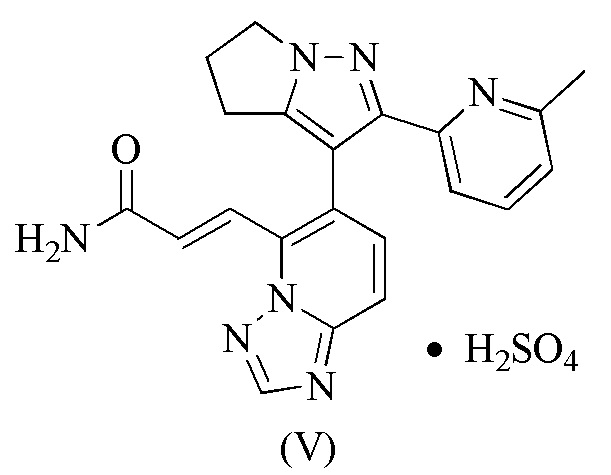
[105] 115 мг соединения формулы (I) добавляли в стеклянную бутылку объемом 8 мл и добавляли туда 4 мл тетрагидрофурана, которые образовывали суспензию путем растворения с помощью ультразвука; и затем медленно добавляли 1,05 эквивалента серной кислоты. Вышеупомянутый суспендированный образец помещали на магнитную мешалку (40°С) и перемешивали в течение 16 часов. Раствор образца центрифугировали, твердое вещество отбирали и помещали в вакуумную печь при 35°С для сушки на 16 часов, получая соединение формулы (V).
[106] Пример 5. Получение соединения формулы (VI).
[107] 115 мг соединения формулы (I) добавляли в стеклянную бутылку объемом 8 мл и добавляли туда 4 мл тетрагидрофурана, которые образовывали суспензию путем растворения с помощью ультразвука; и затем медленно добавляли 1,05 эквивалента метансульфоновой кислоты. Вышеупомянутый суспендированный образец помещали на магнитную мешалку (40°С) и перемешивали в течение 16 часов. Раствор образца центрифугировали, твердое вещество отбирали и помещали в вакуумную печь при 35°С для сушки на 16 часов, получая соединение формулы (VI).
[108] Пример 6. Получение кристаллической формы А соединения формулы (I).
[109] 10 г соединения формулы (I) отбирали и помещали в смешанный растворитель этанола (80 мл) и воды (40 мл), нагревали до 70°С-75°С и перемешивали до прозрачности, а затем фильтровали в горячем состоянии; фильтрат перегоняли при пониженном давлении до тех пор, пока объем оставшегося раствора не составлял около 50 мл, а затем охлаждали и оставляли стоять для кристаллизации и фильтровали; полученный осадок на фильтре сушили при пониженном давлении, и полученное из него твердое вещество представляло собой кристаллическую форму А соединения формулы (I).
[110] Пример 7. Получение кристаллической формы B соединения формулы (II).
[111] 192 мг соединения формулы (I) взвешивали и добавляли в стеклянную бутылку. Добавляли 10 мл смешанного растворителя тетрагидрофурана: уксусная кислота (об./об. = 9/1), а после 30 минут солюбилизации с помощью ультразвука образец растворяли в прозрачном растворе. Раствор помещали на магнитную мешалку (40°С) и перемешивали. После медленного добавления 1,05 эквивалента моногидрата p-толуолсульфоновой кислоты образец перемешивали в течение ночи. После естественного охлаждения до комнатной температуры супернатант удаляли центрифугированием; 10 мл тетрагидрофурана добавляли и перемешивали в течение получаса, а затем супернатант снова удаляли центрифугированием; этот процесс был повторен дважды. Полученное твердое вещество помещали в вакуумную печь при 40°C для сушки в течение 1 часа и дополнительно сушили в вакуумной печи при 30°C в течение 16 часов после дробления, получая кристаллическую форму B соединения формулы (II).
[112] Пример 8. Протокол скрининга активности связывания рецептора TGFβ-RI in vitro.
[113] 1. Экспериментальный способ:
[114] 1) Соединение для тестирования: IC50 определяли способом из 10 точек градиента с каждым трехкратным разведением, и начальная концентрация составляла 5 мкМ.
[115] 2) Реакционная система содержала 10 мкМ АТФ.
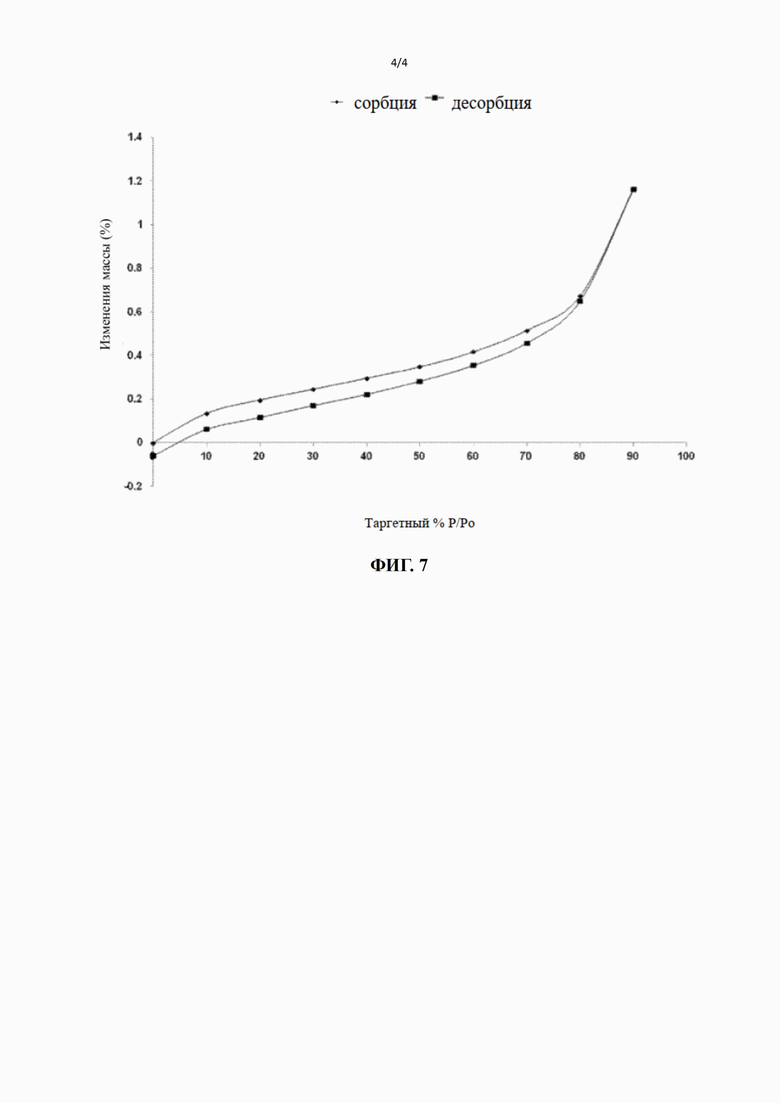
[116] 3) Когда процентная доля активности фермента в образце при самой высокой концентрации (по сравнению с группой растворителей) составляла менее 65%, для расчета значения IC50 проводили аппроксимацию кривой.
[117] 2. Результаты экспериментов показаны в таблице ниже:
[118] Вывод: Активность ингибирования TGF-βRI соединения формулы (I) лучше, чем активность LY2157299 в тех же экспериментальных условиях, что описаны выше.
[119] Пример 9. Исследование растворимости различных типов солей соединения формулы (I) в биологическом носителе
[120] 1 мл раствора биологического носителя (FaSSIF, FeSSIF и SGF) пипетировали соответственно в стеклянную бутылку объемом 1,5 мл и затем добавляли к вышеупомянутому раствору с градиентом 2 мг до 10 мг или смесь была насыщенной. Смесь готовили в 2-х частях параллельно, а затем встряхивали при 37°С. Образцы отбирали через 4 и 24 часа соответственно. Отобранные образцы быстро центрифугировали, а надосадочную жидкость измеряли на оценки рН и разбавляли разбавителем до подходящего кратного значения и затем определяли концентрацию с помощью ВЭЖХ. Результаты тестирований приведены в Таблице 3 ниже.
Таблица 3 Растворимость различных типов солей соединения формулы (I) в биологическом носителе
(свободное основание)
(п-толуолсульфонат)
[121] [Примечание]: SGF обозначает смоделированную желудочную жидкость; FaSSIF обозначает смоделированную кишечную жидкость в состоянии натощак; FeSSIF обозначает смоделированную кишечную жидкость в состоянии сытости.
[122] Вывод: Из результатов в приведенной выше таблице видно, что растворимость соединения формулы (II) в биологическом носителе значительно улучшена по сравнению с соединением формулы (I).
[123] Пример 10. Исследование растворимости соединений формулы (I) и формулы (II) в воде.
[124] 2 мг образца каждого соединения взвешивали и добавляли в стеклянный флакон объемом 1,5 мл, пипеткой добавляли определенный объем чистой воды и соответствующим образом проводили ультразвуковую солюбилизацию. Исследование проводилось при комнатной температуре и проверено на солюбилизацию. Приблизительные результаты растворимости следующие:
[125] Вывод: Приблизительная растворимость соединения формулы (II) в чистой воде значительно улучшена по сравнению с соединением формулы (I).
[126] Пример 11. Исследование гигроскопичности кристаллической формы B соединения формулы (II).
[127] 1. Экспериментальный материал: прибор динамической сорбции паров SMS DVS Advantage
[128] 2. Экспериментальный способ: соответствующее количество кристаллической формы B соединения формулы (II) помещали в диск с образцами DVS для анализа DVS.
[129] 3. Результат эксперимента: Диаграмма DVS кристаллической формы B соединения формулы (II) показана на фиг. 7 с ΔW=0,673%.
[130] Вывод: Гигроскопический прирост массы кристаллической формы B соединения формулы (II) при 25°C/80% RH составил 0,673%, что является слегка гигроскопичным.
[131] Пример 12. Тестирование на стабильность кристаллической формы A соединения формулы (I) при высокой температуре
[132] В соответствии с «Руководством по тестированию стабильности активных фармацевтических ингредиентов и фармацевтических препаратов» (Chinese Pharmacopoeia 2010 Appendix XIXC) была исследована стабильность кристаллической формы А соединения формулы (I) в условиях ускоренного теста при высокой температуре (60°С).
[133] Кристаллическая форма А соединения формулы (I) была помещена в открытый чистый контейнер при 60°С, и образцы были отобраны на 30, 60 и 90 сутки соответственно для тестирования. По сравнению с первоначальными результатами теста в день 0 в Таблице 4 ниже показаны результаты теста.
Таблица 4 Тестирование стабильности кристаллизации соединения формулы (I) при высокой температуре
[134] Вывод: Тестирование на стабильность при высокой температуре показывает, что кристаллическая форма А соединения формулы (I) имеет хорошую стабильность в условиях высоких температур.
[135] Пример 13. Тестирование на стабильность кристаллической формы A соединения формулы (I) при высокой влажности
[136] В соответствии с «Руководством по тестированию стабильности активных фармацевтических ингредиентов и фармацевтических препаратов» (Chinese Pharmacopoeia 2010 Appendix XIXC) была исследована стабильность кристаллической формы А соединения формулы (I) в условиях ускоренного теста при высокой влажности (40°С/75% влажности (в открытых условиях)).
[137] Кристаллическая форма А соединения формулы (I) была помещена в открытый контейнер с постоянной температурой и влажностью для ускоренного тестирования в условиях 40°С/75% влажности (открытые условия), а образцы для тестирования были взяты на 30-й, 60-й и 90-й дни. По сравнению с первоначальными результатами теста в день 0 в Таблице 5 ниже показаны результаты тестирования:
Таблица 5. Тестирование стабильности кристаллизации соединения формулы (I) при высокой влажности
(открытые условия)
[138] Вывод: Тестирование на стабильность при высокой влажности показывает, что кристаллическая форма А соединения формулы (I) имеет хорошую стабильность в условиях высокой влажности.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединение ингибитора FGFR в твердой форме и способ его получения | 2020 |
|
RU2810067C2 |
| ТВЕРДАЯ ФОРМА, КРИСТАЛЛИЧЕСКАЯ ФОРМА И КРИСТАЛЛИЧЕСКАЯ ФОРМА А АГОНИСТА FXR, И СПОСОБ ИХ ПОЛУЧЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2804320C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО ТИОФЕНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2022 |
|
RU2830948C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА ATR И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2832707C2 |
| КРИСТАЛЛИЧЕСКАЯ ИЛИ АМОРФНАЯ ФОРМА АГОНИСТОВ FXR, ПРЕДСТАВЛЯЮЩИХ СОБОЙ ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2800751C2 |
| АНАЛОГ ПИРИДО[1,2-A]ПИРИМИДОНА, ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2753696C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ДИАЗАБЕНЗОФЛУОРАНТРЕНОВЫХ СОЕДИНЕНИЙ | 2017 |
|
RU2762189C2 |
| СОЛЬ И ПОЛИМОРФ ФЕНИЛ-ПИРИМИДОНОВОГО СОЕДИНЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2761213C2 |
| АЛЬФА- И БЕТА-НЕНАСЫЩЕННОЕ АМИДНОЕ СОЕДИНЕНИЕ - ПРОИЗВОДНОЕ БЕНЗОТРИАЗОЛА, ПРИМЕНЯЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА TGF-βRI | 2017 |
|
RU2737737C2 |
| СОЛЬ ИНГИБИТОРА LSD1 И ЕЁ ПОЛИМОРФНАЯ ФОРМА | 2019 |
|
RU2794977C2 |
Изобретение относится к новым кристаллическим формам и солевым формам ингибитора TGF-βR1, а также их применению при получении лекарственных средств для лечения рака. Технический результат: получены новые кристаллические формы соединения формулы (I), а также соединения формул (II), (IV), (V), (VI), которые обладают свойствами ингибитора TGF-βR1 и могут быть применимы для лечения рака, опосредованного ингибированием TGF-βR1. 7 н. и 12 з.п. ф-лы, 7 ил., 5 табл., 13 пр.
1. Кристаллическая форма А соединения формулы (I), отличающаяся тем, что ее порошковая дифракционная рентгенограмма имеет характерные дифракционные пики при следующих углах 2θ: 11,894±0,2°, 17,502±0,2°, 19,785±0,2°, 24,072±0,2° и 24,664±0,2°
 .
.
2. Кристаллическая форма А по п. 1, отличающаяся тем, что ее порошковая дифракционная рентгенограмма имеет характерные дифракционные пики при следующих углах 2θ: 9,553±0,2°, 11,894±0,2°, 15,370±0,2°, 17,502±0,2°, 19,785±0,2°, 20,283±0,2°, 24,072±0,2° и 24,664±0,2°.
3 Кристаллическая форма А по п. 2, отличающаяся тем, что ее порошковая дифракционная рентгенограмма является такой, как показано на фиг. 1.
4. Кристаллическая форма А по любому из пп. 1-3, отличающаяся тем, что ее диаграмма дифференциальной сканирующей калориметрии имеет эндотермический пик при 271,79°С±3°С.
5. Кристаллическая форма А по п. 4, отличающаяся тем, что ее диаграмма дифференциальной сканирующей калориметрии является такой, как показано на фиг. 2.
6. Кристаллическая форма А по любому из пп. 1-3, отличающаяся тем, что ее диаграмма термогравиметрического анализа показывает 0,1075% потери массы при 110,82°С±3°С и 1,105% потери массы при 229,08°С±3°С.
7. Кристаллическая форма А по п. 6, отличающаяся тем, что ее диаграмма термогравиметрического анализа является такой, как показано на фиг. 3.
8. Соединение формулы (II)
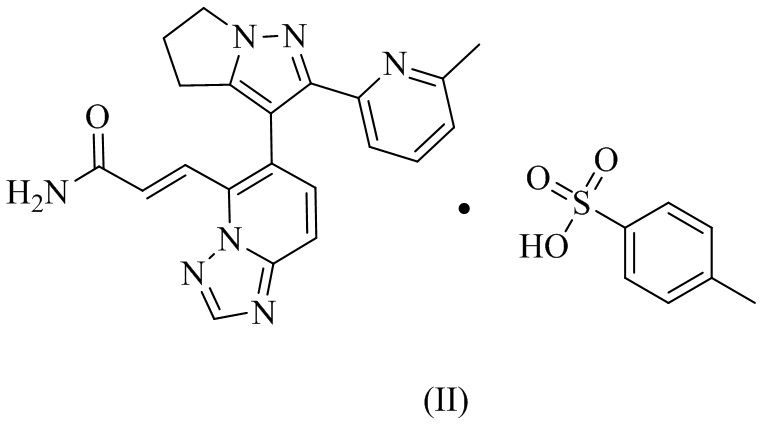 .
.
9. Кристаллическая форма B соединения формулы (II) ,
отличающаяся тем, что ее порошковая дифракционная рентгенограмма имеет характерные дифракционные пики при следующих углах 2θ: 13,349±0,2°, 19,012±0,2°, 20,235±0,2° и 23,370±0,2°.
10. Кристаллическая форма B по п. 9, отличающаяся тем, что ее порошковая дифракционная рентгенограмма имеет характерные дифракционные пики при следующих углах 2θ: 13,349±0,2°, 15,066±0,2°, 16,782±0,2°, 19,012±0,2°, 20,235±0,2°, 22,027±0,2°, 23,370±0,2° и 27,253±0,2°.
11. Кристаллическая форма B по п. 10, отличающаяся тем, что ее порошковая дифракционная рентгенограмма является такой, как показано на фиг. 4.
12. Кристаллическая форма B по любому из пп. 9-11, отличающаяся тем, что ее диаграмма дифференциальной сканирующей калориметрии имеет эндотермический пик при 234,43°С±3°С.
13. Кристаллическая форма B по п. 12, отличающаяся тем, что ее диаграмма дифференциальной сканирующей калориметрии является такой, как показано на фиг. 5.
14. Кристаллическая форма B по любому из пп. 9-11, отличающаяся тем, что ее диаграмма термогравиметрического анализа показывает 0,3043% потери массы при 120°С±3°С и 1,599% потери массы при 238,46°С±3°С.
15. Кристаллическая форма B по п. 14, отличающаяся тем, что ее диаграмма термогравиметрического анализа является такой, как показано на фиг. 6.
16. Соединение формулы (IV)
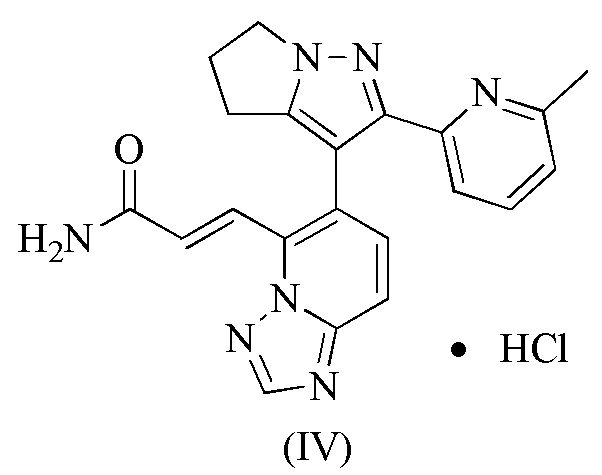 .
.
17. Соединение формулы (V)
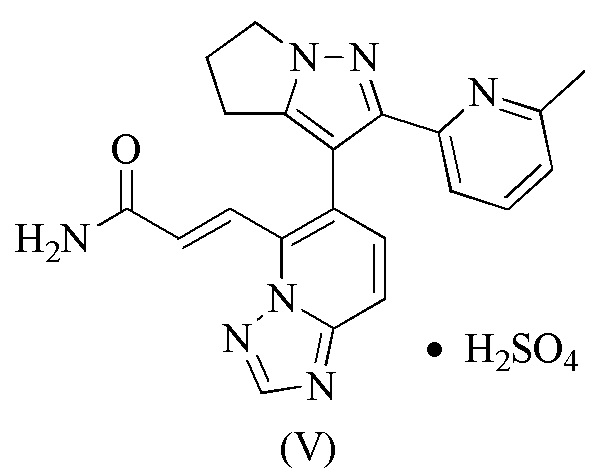 .
.
18. Соединение формулы (VI)
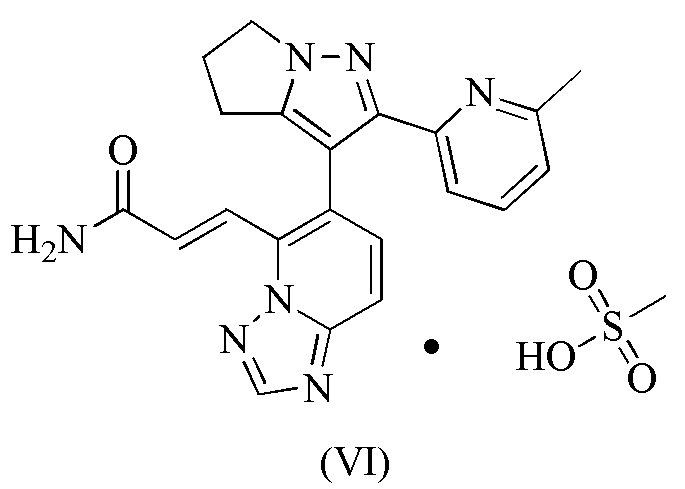 .
.
19. Применение соединения по любому из пп. 8 и 16-18 или кристаллической формы по любому из пп. 1-7 и 9-15 при получении лекарственного средства для лечения рака, опосредованного ингибированием TGF-βRI.
| УКАЗАТЕЛЬНОЕ ПРИСПОСОБЛЕНИЕ ДЛЯ ИЗМЕРИТЕЛЬНЫХ ПРИБОРОВ | 1927 |
|
SU7782A1 |
| EA 200500377 A1, 25.08.2005 | |||
| WO 2004050659 A1, 17.06.2004 | |||
| ИНГИБИТОРЫ КИНАЗ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2442777C2 |

