ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящая заявка относится к области медицины и химии, и, в частности, она относится к кристаллической или аморфной форме производного стероидов в качестве агониста FXR, кристаллической композиции, содержащей кристаллическую или аморфную форму, фармацевтической композиции, а также к их путям применения в медицине.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Фарнезоидный X-рецептор (FXR) представляет собой орфанный ядерный рецептор, изначально идентифицированный из библиотеки cDNA печени крысы (BM. Forman, et al., Cell 81: 687-693 (1995)), который наиболее тесно связан с рецептором экдизона насекомого. FXR является членом семейства ядерных рецепторов с лиганд-активированными факторами транскрипции, которое включает рецепторы для стероидных, ретиноидных и тиреоидных гормонов (DJ. Mangelsdorf, et al., Cell 83: 841-850 (1995)). Нозерн-блоттинг и анализ in situ показали, что FXR наиболее широко экспрессируется в печени, кишечном тракте, почке и надпочечной железе (BM. Forman, et al., Cell 81: 687-693 (1995) и W. Seol, et al., Mol. Endocrinnol, 9: 72-85 (1995)). FXR связывается с ДНК в виде гетеродимера с рецептором 9-цис-ретиноевой кислоты (RXR). Гетеродимер FXR/RXR предпочтительно связывается с элементами, которые состоят из двух полусайтов ядерного рецептора с консенсусной последовательностью AG(G/T)TCA, организованной в виде инвертированного повтора и разделенной одним нуклеотидом (мотив IR-1) (BM. Forman, et al., Cell 81: 687-693 (1995)). Однако такие соединений не способны активировать FXR мыши и человека, вследствие чего природа эндогенного лиганда FXR вызывает сомнения. Несколько встречающихся в природе желчных кислот связывают и активируют FXR при физиологических концентрациях (PCT WO 00/37077, опубликовано 29 июня 2000 года). Как описано в этом документе, желчные кислоты, которые выступают в качестве лигандов FXR, включают хенодезоксихолевую кислоту (CDCA), дезоксихолевую кислоту (DCA), литохолевую кислоту (LCA) и таурининовые и глициновые конъюгаты таких желчных кислот
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте в настоящей заявке предусмотрена кристаллическая форма A соединения, представленного посредством формулы I,
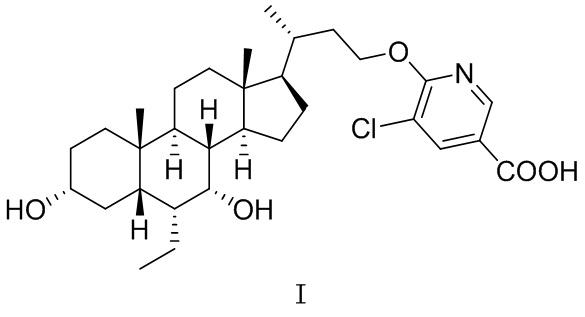 ,
,
где ее порошковая рентгеновская дифрактограмма (XRPD) содержит дифракционные пики при 2θ 5,95°, 10,10°, 15,14°, 18,83°, 20,23°; преимущественно дифракционные пики при 2θ 5,95°, 7,95°, 10,10°, 13,32°, 15,14°, 15,85°, 18,83°, 20,23°; более преимущественно дифракционные пики при 2θ 5,95°, 7,95°, 10,10°, 13,32°, 14,17°, 15,14°, 15,85°, 18,83°, 19,18°, 20,23°, 24,69°; еще более преимущественно дифракционные пики при 2θ 5,95°, 7,95°, 10,10°, 13,32°, 14,17°, 14,58°, 15,14°, 15,85°, 18,25°, 18,83°, 19,18°, 20,23°, 24,69°, 25,81°, где диапазон погрешности 2θ составляет ±0,2°.
В некоторых вариантах осуществления в соответствии с настоящей заявкой порошковая рентгеновская дифрактограмма (XRPD) кристаллической формы A содержит дифракционные пики при 2θ 5,9°, 10,1°, 15,1°, 18,8°, 20,2°; преимущественно дифракционные пики при 2θ 5,9°, 7,9°, 10,1°, 13,3°, 15,1°, 15,8°, 18,8°, 20,2°; более преимущественно дифракционные пики при 2θ 5,9°, 7,9°, 10,1°, 13,3°, 14,1°, 15,1°, 15,8°, 18,8°, 19,1°, 20,2°, 24,6°; еще более преимущественно дифракционные пики при 2θ 5,9°, 7,9°, 10,1°, 13,3°, 14,1°, 14,5°, 15,1°, 15,8°, 18,2°, 18,8°, 19,1°, 20,2°, 24,6°, 25,8°, где диапазон погрешности 2θ составляет ±0,3°, предпочтительно ±0,2°.
В некоторых вариантах осуществления в соответствии с настоящей заявкой пики порошковой рентгеновской дифрактограммы кристаллической формы A в соответствии с настоящей заявкой обладают следующими характеристиками:
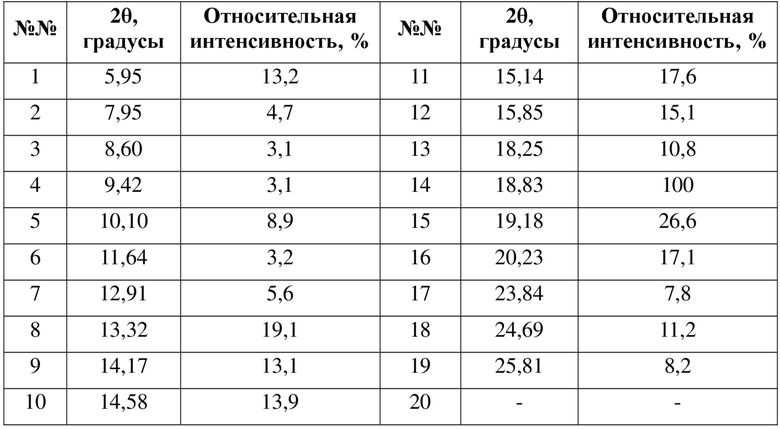
где диапазон погрешности 2θ составляет ±0,3°, предпочтительно ±0,2°.
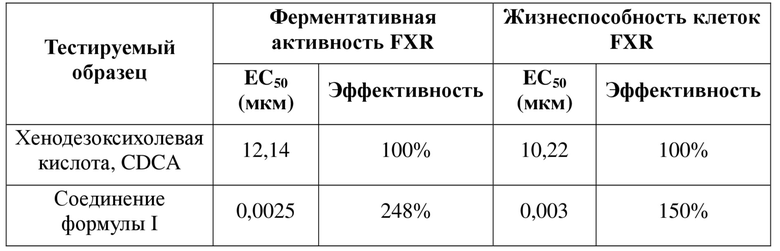
В некоторых вариантах осуществления в соответствии с настоящей заявкой порошковая рентгеновская дифрактограмма кристаллической формы A является такой, как показано на фиг. 1.
В некоторых вариантах осуществления в соответствии с настоящей заявкой DSC-термограмма кристаллической формы A является такой, как показано на фиг. 2.
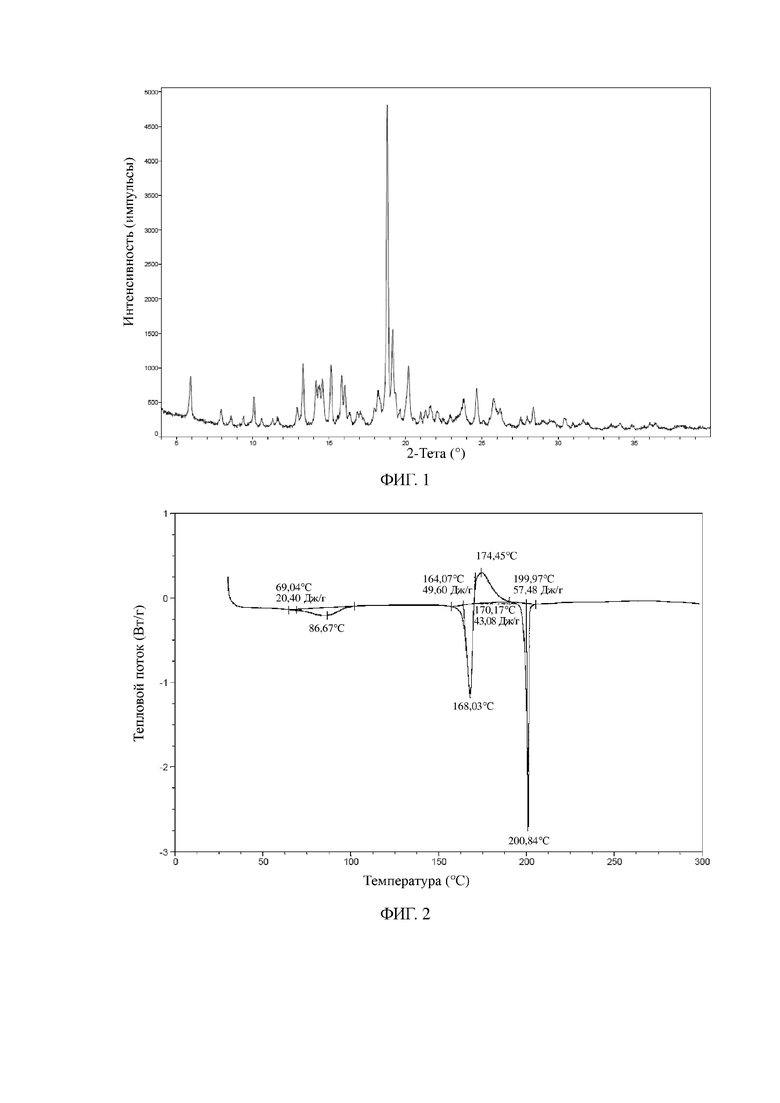
В некоторых вариантах осуществления в соответствии с настоящей заявкой TGA-термограмма кристаллической формы A является такой, как показано на фиг. 3.
В некоторых вариантах осуществления в соответствии с настоящей заявкой другие пики порошковой рентгеновской дифрактограммы кристаллической формы A в соответствии с настоящей заявкой обладают следующими характеристиками:
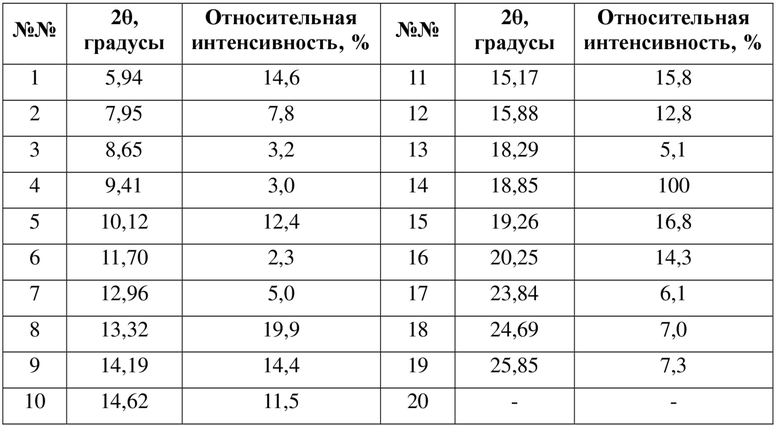
где диапазон погрешности 2θ составляет ±0,3°, предпочтительно ±0,2°.
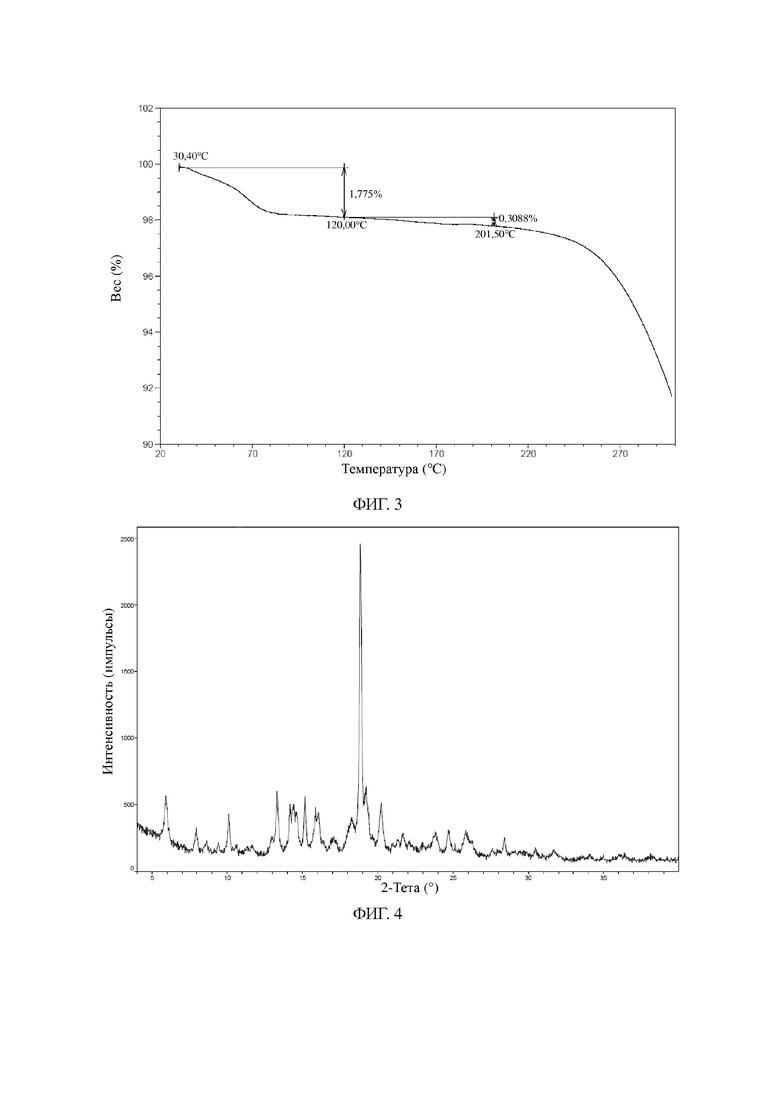
В некоторых вариантах осуществления в соответствии с настоящей заявкой другая порошковая рентгеновская дифрактограмма кристаллической формы A является такой, как показано на фиг. 4.
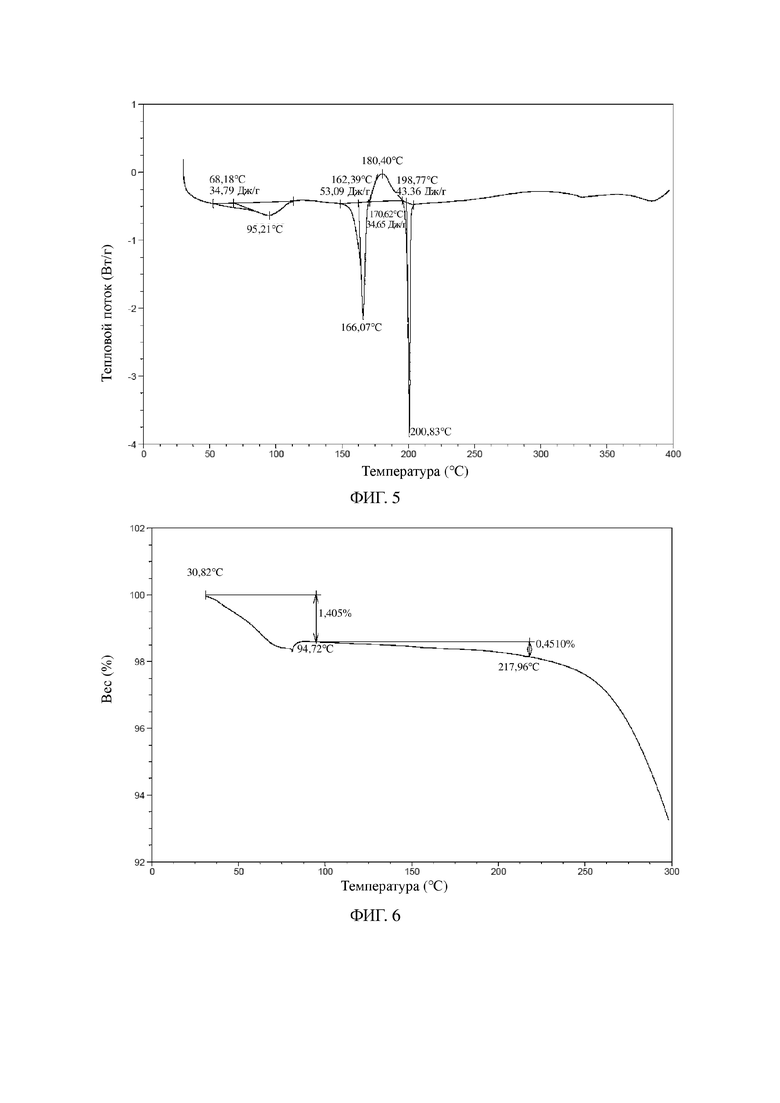
В некоторых вариантах осуществления в соответствии с настоящей заявкой другая DSC-термограмма кристаллической формы A является такой, как показано на фиг. 5.
В некоторых вариантах осуществления в соответствии с настоящей заявкой другая TGA-термограмма кристаллической формы A является такой, как показано на фиг. 6.
В некоторых вариантах осуществления в соответствии с настоящей заявкой в кристаллической форме A присутствует(присутствуют) молекула(молекулы) H2O, и соотношение эквивалентов молекулы H2O и соединения формулы I (в молях) выбрано из диапазона от 0,1 до 2,0 экв.; в некоторых вариантах осуществления соотношение эквивалентов предпочтительно выбрано из диапазона от 0,1 до 1,0 экв., от 0,2 до 0,8 экв., от 0,3 до 0,7 экв. или от 0,4 до 0,6 экв.; в некоторых вариантах осуществления соотношение эквивалентов предпочтительно выбрано из 0,1 экв., 0,2 экв., 0,3 экв., 0,4 экв., 0,5 экв., 0,6 экв., 0,7 экв., 0,8 экв., 0,9 экв., 1,0 экв., 1,1 экв., 1,2 экв., 1,3 экв., 1,4 экв., 1,5 экв., 1,6 экв., 1,7 экв., 1,8 экв., 1,9 экв. или 2,0 экв. или диапазонов между любыми двумя вышеуказанными значениями, например, от 0,2 до 0,8 экв., от 0,3 до 0,7 экв., от 0,4 до 0,6 экв., от 0,6 до 1,0 экв., от 0,7 до 0,8 экв., от 0,8 до 1,2 экв., от 0,9 до 1,1 экв., от 1,3 до 1,7 экв., от 1,4 до 1,6 экв., от 0,4 экв. до 1,8 экв., от 0,6 до 1,4 экв. или от 0,8 до 1,2 экв.
В настоящей заявке дополнительно представлен способ получения кристаллической формы A, который включает следующие стадии:
1) суспендирование или растворение соединения формулы I в растворителе для кристаллизации при перемешивании, при этом растворитель для кристаллизации выбран из воды или смешанного растворителя, содержащего воду; и
2) фильтрование и необязательно промывание и/или высушивание.
В некоторых вариантах осуществления в соответствии с настоящей заявкой в способе получения кристаллической формы A неводный растворитель в смешанном растворителе, содержащем воду, выбран из смешиваемого с водой органического растворителя; предпочтительно смешиваемый с водой органический растворитель выбран из C1-4спиртов, тетрагидрофурана, ацетона, ацетонитрила или DMF; предпочтительно из метанола, этанола, н-пропанола, изопропанола, н-бутанола, изобутанола или трет-бутанола и более предпочтительно представляет собой этанол.
В некоторых вариантах осуществления в соответствии с настоящей заявкой в способе получения кристаллической формы A смешиваемый с водой органический растворитель и вода в смешанном растворителе, содержащем воду, могут присутствовать в любом пригодном соотношении. В некоторых вариантах осуществления соотношение смешиваемого с водой органического растворителя и воды (по объему) выбрано из диапазона от 0,1:1 до 10:1, предпочтительно от 0,2:1 до 8:1, от 0,4:1 до 6:1, от 0,5:1 до 5:1, от 0,6:1 до 3:1 или от 0,8:1 до 2:1.
В некоторых вариантах осуществления в соответствии с настоящей заявкой в способе получения кристаллической формы A количество применяемого растворителя для кристаллизации можно выбрать из более широкого диапазона. В некоторых вариантах осуществления количество растворителя для кристаллизации (в единицах мл) на грамм соединения формулы I выбрано из диапазона от 0,1 мл до 100 мл, предпочтительно от 1 мл до 50 мл и более предпочтительно от 2 мл до 20 мл.
В некоторых вариантах осуществления в соответствии с настоящей заявкой в способе получения кристаллической формы A температура реакции во время кристаллизации может быть выбрана из более широкого диапазона. В некоторых вариантах осуществления стадию 1) проводят при температуре, выбранной из диапазона от 10°C до 80°C, предпочтительно от 20°C до 60°C и более предпочтительно от 25°C до 50°C.
В настоящей заявке дополнительно представлена кристаллическая композиция, содержащая кристаллическую форму A соединения формулы I. В некоторых вариантах осуществления в соответствии с настоящей заявкой кристаллическая форма A соединения формулы I составляет 50% или больше, предпочтительно 80% или больше, более предпочтительно 90% или больше и наиболее предпочтительно 95% или больше от веса кристаллической композиции.
В настоящей заявке дополнительно представлена фармацевтическая композиция, содержащая кристаллическую форму A соединения формулы I, которая содержит эффективное количество кристаллической формы A соединения формулы I или кристаллической композиции, содержащей кристаллическую форму A соединения формулы I. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носитель, вспомогательное вещество и/или среду или может не содержать их.
В настоящей заявке дополнительно представлено применение кристаллической формы A соединения формулы I, или кристаллической композиции, описанной выше, или фармацевтической композиции, описанной выше, в изготовлении лекарственного препарата для лечения или предупреждения заболевания, связанного с фарнезоидным X-рецептором.
В настоящей заявке дополнительно предусмотрены способ или применение для лечения или предупреждения заболевания, связанного с фарнезоидным X-рецептором, предусматривающие введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества кристаллической формы A соединения формулы I или кристаллической композиции, или фармацевтической композиции, описанных выше.
В настоящей заявке дополнительно представлена кристаллическая форма A соединения формулы I, или кристаллическая композиция, или фармацевтическая композиция, описанные выше, для применения в лечении или предупреждении заболевания, связанного с фарнезоидным X-рецептором.
В другом аспекте в настоящей заявке предусмотрена кристаллическая форма B соединения, представленного посредством формулы I,
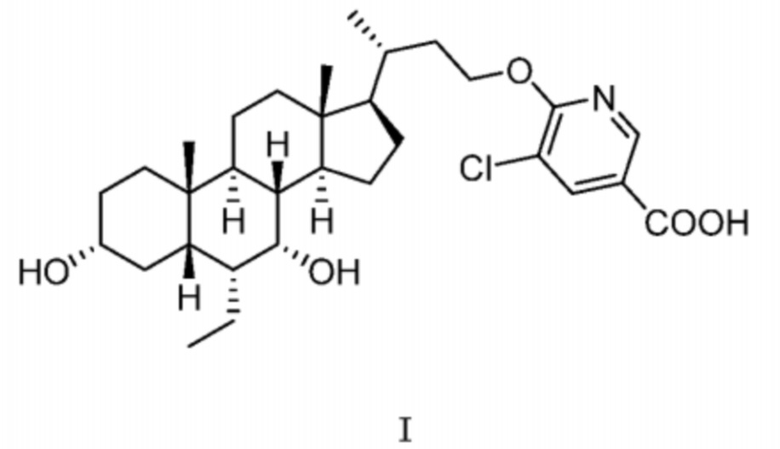 ,
,
где ее порошковая рентгеновская дифрактограмма (XRPD) содержит дифракционные пики при 2θ 6,21°, 9,77°, 10,71°, 12,33°, 13,04°; преимущественно дифракционные пики при 2θ 6,21°, 9,49°, 9,77°, 10,71°, 12,33°, 13,04°, 14,29°, 15,13°; более преимущественно дифракционные пики при 2θ 6,21°, 9,00°, 9,77°, 10,71°, 12,33°, 13,04°, 14,29°, 14,72°, 15,13°, 15,59°; еще более преимущественно дифракционные пики при 2θ 6,21°, 9,00°, 9,77°, 10,71°, 12,33°, 13,04°, 14,29°, 14,72°, 15,13°, 15,59°, 18,14°, 20,09°, 21,41°, где диапазон погрешности 2θ составляет ±0,2°.
В некоторых вариантах осуществления в соответствии с настоящей заявкой порошковая рентгеновская дифрактограмма (XRPD) кристаллической формы B содержит дифракционные пики при 2θ 6,2°, 9,7°, 10,7°, 12,3°, 13,0°; преимущественно дифракционные пики при 2θ 6,2°, 9,4°, 9,7°, 10,7°, 12,3°, 13,0°, 14,2°, 15,1°; более преимущественно дифракционные пики при 2θ 6,2°, 9,0°, 9,4°, 9,7°, 10,7°, 12,3°, 13,0°, 14,2°, 14,7°, 15,1°, 15,5°; еще более преимущественно дифракционные пики при 2θ 6,2°, 9,0°, 9,4°, 9,7°, 10,7°, 12,3°, 13,0°, 14,2°, 14,7°, 15,1°, 15,5°, 18,1°, 20,0°, 21,4°, где диапазон погрешности 2θ составляет ±0,3°, предпочтительно ±0,2°.
В некоторых вариантах осуществления в соответствии с настоящей заявкой пики порошковой рентгеновской дифрактограммы кристаллической формы B в соответствии с настоящей заявкой обладают следующими характеристиками:
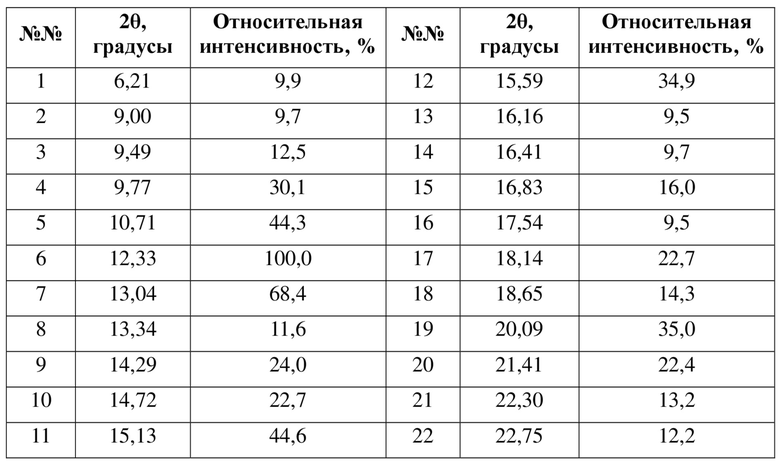
где диапазон погрешности 2θ составляет ±0,3°, предпочтительно ±0,2°.
В некоторых вариантах осуществления в соответствии с настоящей заявкой порошковая рентгеновская дифрактограмма кристаллической формы B является такой, как показано на фиг. 7.
В некоторых вариантах осуществления в соответствии с настоящей заявкой DSC-термограмма кристаллической формы B является такой, как показано на фиг. 8.
В некоторых вариантах осуществления в соответствии с настоящей заявкой TGA-термограмма кристаллической формы B является такой, как показано на фиг. 9.
В некоторых вариантах осуществления в соответствии с настоящей заявкой в кристаллической форме B присутствует(присутствуют) молекула(молекулы) этилацетата, и соотношение эквивалентов (в молях) молекулы этилацетата и соединения формулы I выбрано из диапазона от 0,1 до 0,5 экв., предпочтительно от 0,2 до 0,4 экв., более предпочтительно от 0,25 до 0,35 экв. и еще более предпочтительно выбрано из 0,25 экв., 0,26 экв., 0,27 экв., 0,28 экв., 0,29 экв., 0,30 экв., 0,31 экв., 0,32 экв., 0,33 экв., 0,34 экв. или 0,35 экв.
В настоящей заявке дополнительно представлен способ получения кристаллической формы B, включающий следующие стадии:
1) суспендирование или растворение соединения формулы I в этилацетате и
2) обеспечение кристаллизации и необязательно фильтрование, промывание и/или высушивание.
В некоторых вариантах осуществления в соответствии с настоящей заявкой в способе получения кристаллической формы B количество применяемого растворителя для кристаллизации можно выбрать из более широкого диапазона. В некоторых вариантах осуществления количество растворителя для кристаллизации (в единицах мл) на грамм соединения формулы I выбрано из диапазона от 0,1 мл до 100 мл, предпочтительно от 1 мл до 50 мл и более предпочтительно от 2 мл до 20 мл.
В настоящей заявке дополнительно представлена кристаллическая композиция, содержащая кристаллическую форму B соединения формулы I. В некоторых вариантах осуществления в соответствии с настоящей заявкой кристаллическая форма B соединения формулы I составляет 50% или больше, более предпочтительно 80% или больше, еще более предпочтительно 90% или больше и наиболее предпочтительно 95% или больше от веса кристаллической композиции.
В настоящей заявке дополнительно представлена фармацевтическая композиция, содержащая кристаллическую форму B соединения формулы I, которая содержит эффективное количество кристаллической формы B соединения формулы I или кристаллической композиции, содержащей кристаллическую форму B соединения формулы I. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носитель, вспомогательное вещество и/или среду или может не содержать их.
В настоящей заявке дополнительно представлено применение кристаллической формы B соединения формулы I, или кристаллической композиции, описанной выше, или фармацевтической композиции, описанной выше, в изготовлении лекарственного препарата для лечения или предупреждения заболевания, связанного с фарнезоидным X-рецептором.
В настоящей заявка дополнительно представлены способ или применение для лечения или предупреждения заболевания, связанного с фарнезоидным X-рецептором, предусматривающие введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества кристаллической формы B соединения формулы I, описанной выше, или кристаллической композиции, описанной выше, или фармацевтической композиции, описанной выше.
В настоящей заявке дополнительно представлена кристаллическая форма B соединения формулы I, или кристаллическая композиция, описанная выше, или фармацевтическая композиция, описанная выше, для применения в лечении или предупреждении заболевания, связанного с фарнезоидным X-рецептором.
В другом аспекте в соответствии с настоящей заявкой предусмотрена твердая аморфная форма соединения, представленного посредством формулы I,
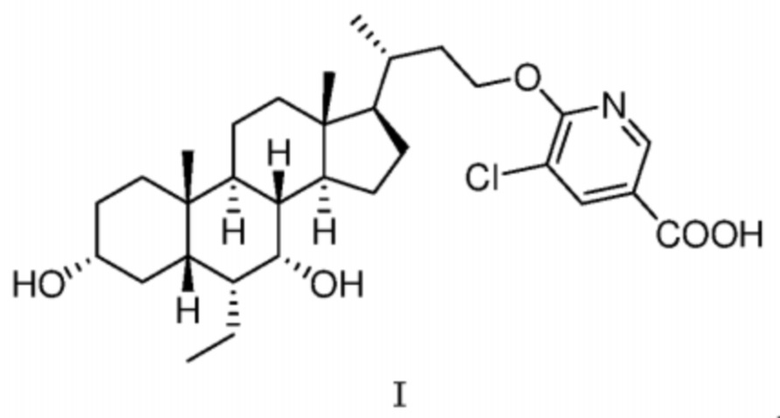
В некоторых вариантах осуществления в соответствии с настоящей заявкой твердая аморфная форма соединения формулы I не характеризуется типичными дифракционными пиками на XRPD-дифрактограмме, показанной на фиг. 10.
В некоторых вариантах осуществления в соответствии с настоящей заявкой MDSC-термограмма твердой аморфной формы соединения формулы I является такой, как показано на фиг. 11.
В некоторых вариантах осуществления в соответствии с настоящей заявкой TGA-термограмма твердой аморфной формы соединения формулы I является такой, как показано на фиг. 12.
В некоторых вариантах осуществления в соответствии с настоящей заявкой твердую аморфную форму соединения формулы I получают с помощью следующего безводного растворителя: метанола, этанола, н-пропанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, тетрагидрофурана, ацетона, DMF или смешанного растворителя на их основе. В некоторых вариантах осуществления растворитель предпочтительно выбран из этанола или изопропанола.
В настоящей заявке дополнительно представлен способ получения твердой аморфной формы соединения формулы I, включающий:
1) растворение соединения формулы I в безводном растворителе, выбранном из метанола, этанола, н-пропанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, тетрагидрофурана, ацетона, DMF или смешанного растворителя на их основе, и
2) охлаждение с осаждением твердого вещества или выпаривание растворителя до сухого состояния и необязательно фильтрование, промывание и/или высушивание.
В некоторых вариантах осуществления в соответствии с настоящей заявкой на стадии 1) способа получения твердой аморфной формы соединения формулы I растворитель предпочтительно выбран из этанола или изопропанола.
В некоторых вариантах осуществления в соответствии с настоящей заявкой в способе получения твердой аморфной формы соединения формулы I растворитель может применяться в количестве, выбранном из более широкого диапазона. В некоторых вариантах осуществления количество растворителя (в единицах мл) на грамм соединения формулы I выбрано из диапазона от 0,1 мл до 100 мл, предпочтительно от 1 мл до 50 мл и более предпочтительно от 2 мл до 20 мл.
В настоящей заявке дополнительно представлена фармацевтическая композиция, содержащая твердую аморфную форму соединения формулы I, которая содержит эффективное количество твердой аморфной формы соединения формулы I. Кроме того, фармацевтическая композиция может содержать фармацевтически приемлемые носитель, вспомогательное вещество и/или среду или может не содержать их.
В настоящей заявке дополнительно представлено применение твердой аморфной формы соединения формулы I или фармацевтической композиции, описанной выше, в изготовлении лекарственного препарата для лечения или предупреждения заболевания, связанного с фарнезоидным X-рецептором.
В настоящей заявке дополнительно представлены способ или применение для лечения или предупреждения заболевания, связанного с фарнезоидным X-рецептором, предусматривающие введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества твердой аморфной формы соединения формулы I, описанной выше, или фармацевтической композиции, описанной выше.
В настоящей заявке заболевание, связанное с фарнезоидным X-рецептором, включает неалкогольную жировую болезнь печени (NAFLD), неалкогольный стеатогепатит (NASH), первичный билиарный цирроз (PBC), холестатическую гепатопатию, хроническое заболевание печени, инфекцию, вызванную вирусом гепатита С, алкогольную болезнь печени, фиброз печени, первичный склерозирующий холангит (PSC), камень в желчном пузыре, билиарную атрезию, симптом нижних мочевыводящих путей и доброкачественную гиперплазию предстательной железы (BPH), камни в мочеточнике, ожирение, диабет 2 типа, атеросклероз, артериосклероз, гиперхолестеринемию, гиперлипидемию или нарушение функции печени, обусловленное гиперхолестеринемией и гиперлипидемией.
В соответствии с настоящей заявкой порошковые рентгеновские дифрактограммы измеряют с помощью следующего способа: измерительный прибор: рентгеновский дифрактометр Bruker D8 ADVANCE; способ: мишень: Cu: K-альфа; длина волны: λ=1,54179 Ǻ; напряжение на рентгеновской трубке: 40 кВ; щель расходимости: 0,60 мм; щель детектора: 10,50 мм; противорассеивающая щель: 7,10 мм; сила тока на рентгеновской трубке: 40 мА; диапазон сканирования: от 4° до 40°; скорость сканирования: 0,12 сек./шаг, 0,02°/шаг; скорость вращения лотка для образцов: 15 об./мин.
Следует отметить, что в отношении спектра рентгеновской дифракции, дифрактограмма, полученная для кристаллического соединения, обычно является характеристической для конкретной кристаллической формы, при этом значения относительной интенсивности полос (особенно при низких значениях угла) могут изменяться в зависимости от эффектов предпочтительной ориентации, возникающих из-за различий в условиях кристаллизации, размерах частиц и других условиях измерения. Следовательно, значения относительной интенсивности дифракционных пиков не являются характеристическими для конкретной кристаллической формы. При оценке того, является ли кристаллическая форма идентичной уже известной кристаллической форме, следует обращать больше внимания на взаимное расположение пиков, а не на их значения относительной интенсивности. Кроме того, в случае любой конкретной кристаллической формы может иметь место небольшая погрешность в расположении пиков, что также хорошо известно в области кристаллографии. Например, положение пика может сдвигаться вследствие изменения температуры, изменения положения образца или калибровки измерительного прибора и так далее во время анализа образца, при этом погрешность измерения значения 2θ в некоторых случаях составляет приблизительно ±0,3° или ±0,2°. Соответственно, при идентификации кристаллической структуры следует учитывать такую погрешность. Обычно положение пика выражается в виде угла 2θ или межплоскостного расстояния d на XRPD-дифрактограмме, и между ними существует простое соотношение преобразования d=λ/2sin θ, где d представляет собой межплоскостное расстояние, λ представляет собой длину волны падающих рентгеновских лучей, и θ представляет собой угол отклонения при дифракции. В случае одинаковой кристаллической формы одного и того же соединения расположение пиков на их XRPD-спектре в целом имеет сходство, а погрешность значений относительной интенсивности может быть больше. Кроме того, необходимо отметить, что при идентификации смеси вследствие некоторых факторов, таких как низкие содержания, части дифракционных линий могут отсутствовать. В данное время даже одна полоса может быть характеристической для данной кристаллической формы вне зависимости от всех полос образца высокой чистоты. Из уровня техники известно, что относительную интенсивность пика (I%) можно рассчитать на основе высоты пика или также можно рассчитать на основе площади пика. В соответствии с настоящей заявкой преимущественно применяют способ, основанный на высоте пика.
В соответствии с настоящей заявкой применяют следующий способ дифференциальной сканирующей калориметрии (DSC): измерительный прибор: дифференциальный сканирующий калориметр TA Q2000; способ: образцы (~1 мг) тестируют в алюминиевом тигле для DSC при температуре от 30°C (комнатная температура) до 300°C (или 350°C) при скорости нагревания 10°C/мин. (или 5°C/мин.) в атмосфере N2, 50 мл/мин.
В соответствии с настоящей заявкой применяют следующий способ дифференциальной сканирующей калориметрии с модуляцией по температуре (MDSC): измерительный прибор: дифференциальный сканирующий калориметр TA Q2000; способ: образцы (~2 мг) тестируют в алюминиевом тигле для DSC при температуре от 0°C до 200°C и при скорости нагревания 2°C/мин., в атмосфере N2, 50 мл/мин., амплитуда 2°C и период 60 с.
Следует отметить, что DSC можно применять для измерения температуры теплового перехода кристалла при поглощении или высвобождении тепла вследствие изменения его кристаллической структуры или плавления кристалла. В непрерывном анализе одинаковой кристаллической формы одного и того же соединения погрешность температуры теплового перехода и точки плавления преимущественно находится в пределах диапазона приблизительно 5°C. Если сказано, что соединение характеризуется указанным DSC-пиком или точкой плавления, то это означает DSC-пик или точку плавления ±5°C. DSC представляет собой вспомогательный способ различения разных кристаллических форм. Различные кристаллические формы могут быть идентифицированы по их характеристически различным температурам фазового перехода.
В соответствии с настоящей заявкой применяют следующий способ термогравиметрического анализа (TGA): измерительный прибор: термогравиметрический анализатор TA Q5000; образцы (2-5 мг) тестируют в платиновом тигле для TGA, в котором образцы нагревают от комнатной температуры до 300°C или до потери 20% веса при скорости нагревания 10°C/мин. в атмосфере N2, 25 мл/мин.
Следует отметить, что в ходе получения кристаллической формы лекарственного средства, если молекулы лекарственного средства и молекулы растворителя находятся в контакте друг с другом, трудно избежать того, что молекулы растворителя будут образовывать эвтектические смеси с молекулами соединения и оставаться в твердом состоянии вследствие внешних условий и внутренних факторов, образуя тем самым сольват, включая, в частности, стехиометрический и нестехиометрический сольват. Такие сольваты охватываются объемом настоящего изобретения.
В соответствии с настоящей заявкой, если молекулы растворителя включены в кристалл, то он представлен с помощью соотношения эквивалентов (в молях) молекулы растворителя и соединения формулы I. Например, если соотношение эквивалентов молекулы H2O и соединения формулы I (в молях) выбрано из диапазона от 0,1 до 2,0 экв., это означает, что мольное соотношение соединения формулы I и молекулы H2O в кристалле составляет от 1:0,1 до 2,0.
В настоящей заявке термин «фармацевтическая композиция» относится к составу из одного или больше соединений по настоящей заявке и носителя, вспомогательного вещества и/или среды, в целом приемлемых в данной области для доставки биоактивного соединения в организм, такой как организм человека. Задача фармацевтической композиции заключается в способствовании введению соединения в соответствии с настоящей заявкой в организм.
Термин «носитель» определен как соединение, которое способствует введению соединения в клетку или ткань. Например, обычно в качестве носителя применяют диметилсульфоксид (DMSO) , поскольку его легко применять для введения некоторых органических соединений в клетки или ткани организма.
Термин «фармацевтически приемлемый носитель» включает без ограничения любые адъювант, вспомогательное вещество, вещество, способствующее скольжению, подсластитель, разбавитель, консервант, краситель/красящее вещество, ароматизатор, поверхностно-активное вещество, смачивающее средство, диспергирующее средство, суспендирующее средство, стабилизатор, изотоническое средство, растворитель или эмульгатор, одобренные национальным управлением по надзору за лекарственными средствами как приемлемые для применения в отношении человека или домашнего скота.
Термин «терапевтически эффективное количество» относится к количеству соединения в соответствии с настоящей заявкой, и при его введению млекопитающему, предпочтительно человеку, его достаточно для обеспечения лечения вирусной инфекции у млекопитающего, предпочтительно человека, как определено далее в данном документе. Количество соединения по настоящей заявке, которое образует «терапевтически эффективное количество» изменяется в зависимости от соединения, болезненного состояния и его тяжести, пути введения и возраста млекопитающего, подлежащего лечению, но его обычно могут определить специалисты средней квалификации в данной области, исходя из их собственных знаний и раскрытия настоящей заявки.
Все растворители, применяемые в настоящей заявке, являются коммерчески доступными и их можно применять без дополнительной очистки. Реакции, как правило, проводят в атмосфере инертного азота в безводном растворителе.
Кристаллические формы A и B соединения формулы I, представленного в настоящей заявке, обладают следующими преимуществами: высокой чистотой, высокой кристалличностью, хорошей стабильностью, низкой гигроскопичностью и т. д.; а твердая аморфная форма соединения формулы I, представленного в настоящей заявке, обладает таким преимуществом, как низкая гигроскопичность. Кроме того, способ получения кристаллических форм A и B, а также твердой аморфной формы соединения формулы I, представленного в настоящей заявке, является простым и в нем применяется дешевый и легко получаемый растворитель с мягкими условиями кристаллизации, при этом он является подходящим для промышленного производства.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1 показана XRPD-дифрактограмма для кристаллической формы A соединения формулы I (способ 1 в примере 2).
На фиг. 2 показана DSC-термограмма для кристаллической формы A соединения формулы I (способ 1 в примере 2).
На фиг. 3 показана TGA-термограмма для кристаллической формы A соединения формулы I (способ 1 в примере 2).
На фиг. 4 показана XRPD-дифрактограмма для кристаллической формы A соединения формулы I (способ 2 в примере 2).
На фиг. 5 показана DSC-термограмма для кристаллической формы A соединения формулы I (способ 2 в примере 2).
На фиг. 6 показана TGA-термограмма для кристаллической формы A соединения формулы I (способ 2 в примере 2).
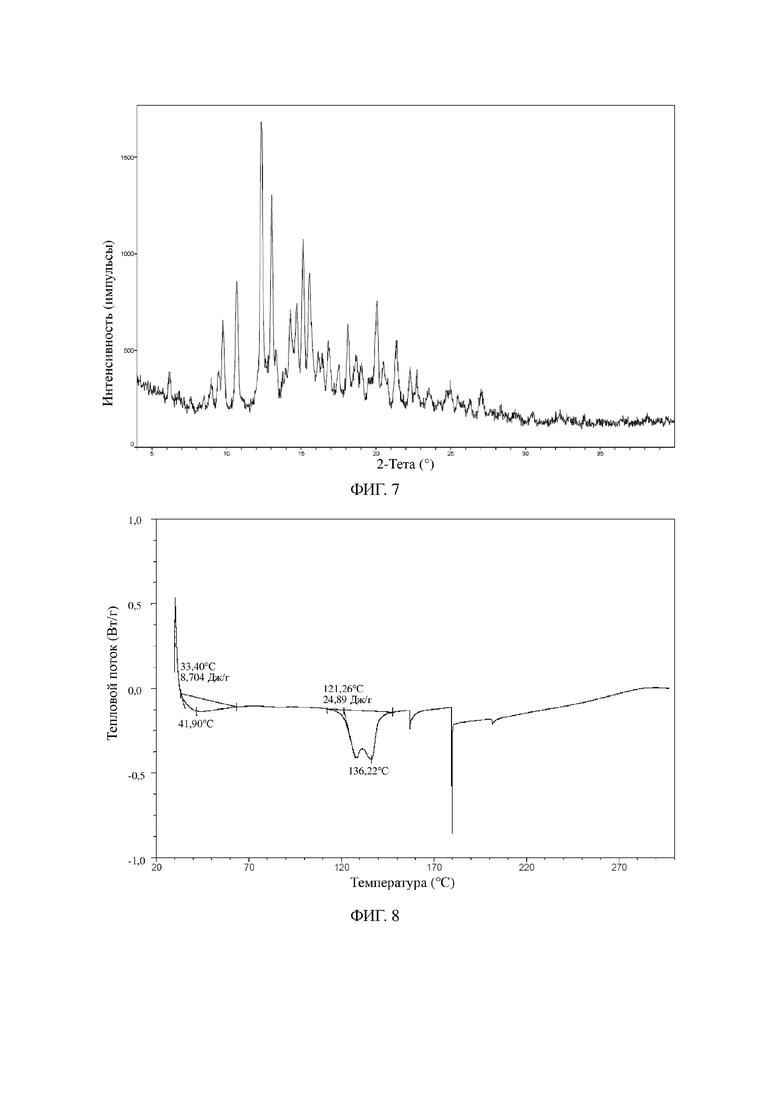
На фиг. 7 показана XRPD-дифрактограмма для кристаллической формы B соединения формулы I.
На фиг. 8 показана DSC-термограмма для кристаллической формы B соединения формулы I.
На фиг. 9 показана TGA-термограмма для кристаллической формы B соединения формулы I.
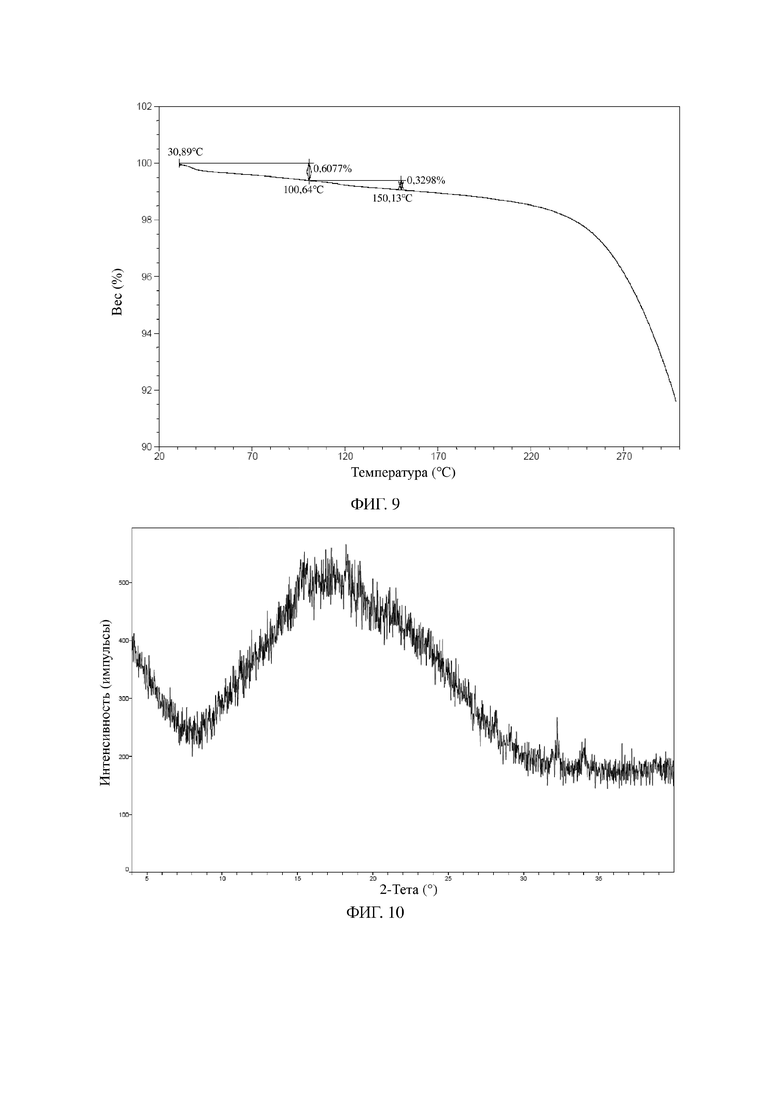
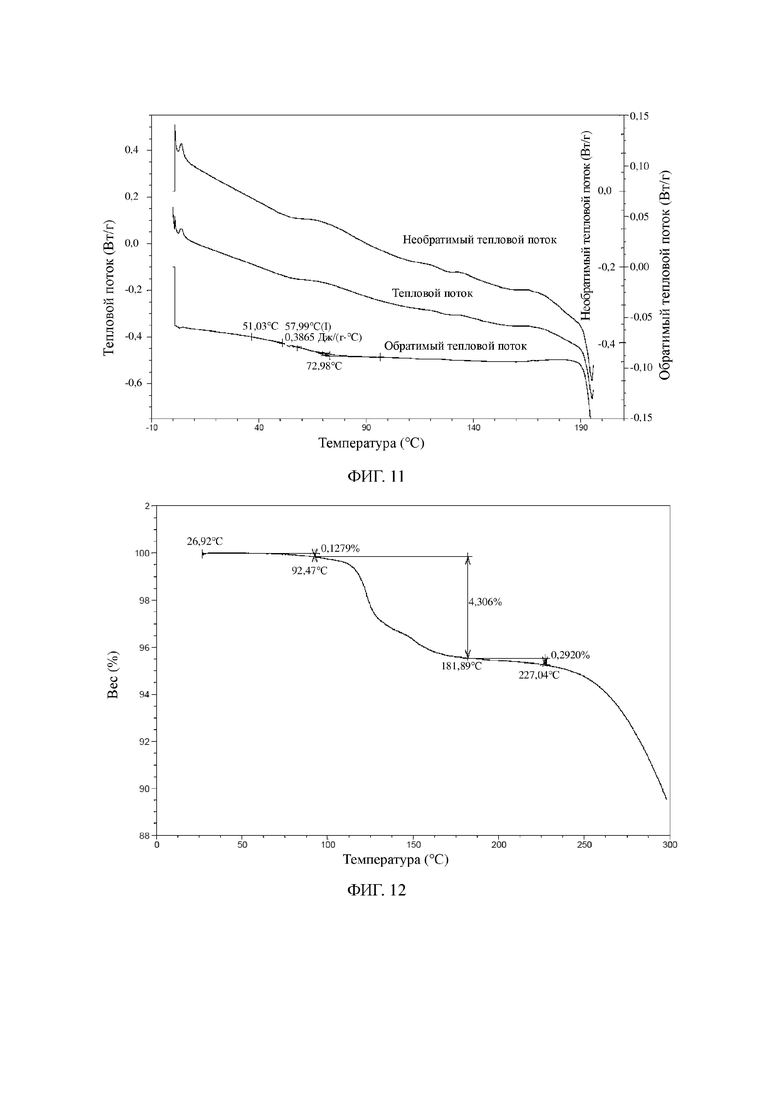
На фиг. 10 показана XRPD-дифрактограмма для твердой аморфной формы соединения формулы I.
На фиг. 11 показана MDSC-термограмма для твердой аморфной формы соединения формулы I.
На фиг. 12 показана TGA-термограмма для твердой аморфной формы соединения формулы I.
ПОДРОБНОЕ ОПИСАНИЕ КОНКРЕТНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Варианты осуществления в соответствии с настоящей заявкой будут подробно описаны с помощью следующих примеров без ограничения. Они не должны рассматриваться как ограничение объема настоящей заявки, а лишь как иллюстративные описания и типичные варианты реализации настоящей заявки. Все растворители, реагенты и исходные материалы, применяемые в соответствии с настоящей заявкой, представляли собой коммерчески доступные химически чистые или чистые для анализа продукты.
Пример 1. Получение соединения формулы I
Стадия 1-1. Получение соединения 3
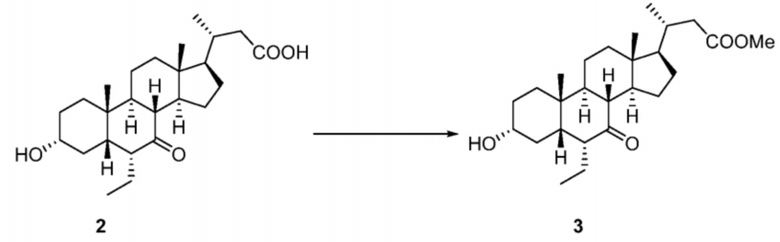
Метанол (33 л) добавляли в реактор объемом 50 л при 25°C и добавляли в реактор субстрат 2 (3,330 кг, 8,23 моль) с последующим добавлением моногидрата п-толуолсульфоновой кислоты (156,6 г, 0,823 моль). Реакционный раствор нагревали до 60°C при перемешивании в течение 12 часов. Реакцию контролировали с помощью TLC, и TLC показала исчезновение исходных материалов. HPLC показала, что образовался приблизительно 100%-ый продукт. Реакционный раствор охлаждали до комнатной температуры, затем регулировали до pH приблизительно 9 с помощью насыщенного раствора бикарбоната натрия и подвергали ротационному выпариванию до сухого состояния с получением неочищенного продукта. Неочищенный продукт растворяли в этилацетате (30 л), последовательно промывали насыщенным раствором бикарбоната натрия (9 л), водой (9 л) и насыщенным солевым раствором (9 л). Органическую фазу подвергали ротационному выпариванию до сухого состояния с получением продукта в виде коричневой маслянистой жидкости.
1H ЯМР (400 МГц, CDCl3) δ 3,66 (s, 3H), 3,61-3,49 (m, 1H), 2,74-2,66 (m, 1H), 2,48-2,33 (m, 2H), 2,24-2,15 (m, 1H), 2,07-1,61 (m, 13H), 1,54-1,40 (m, 3H), 1,31-1,07 (m, 6H), 1,02-0,77 (m, 9H), 0,69 (s, 3H).
Стадия 1-2. Получение соединения 4
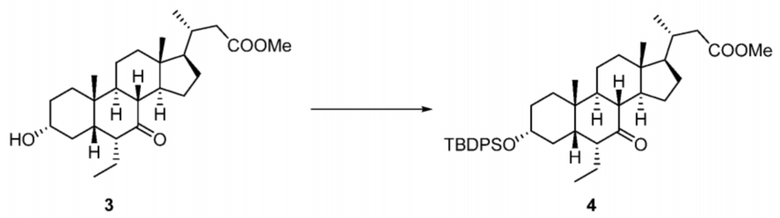
Соединение 3 (3100 г) растворяли в дихлорметане (30 л) и затем последовательно добавляли имидазол (529,4 г) и триэтиламин (786,8 г). Температуру в реакторе снижали (внутренняя температура составляла 5°C), медленно по каплям добавляли TBDPSCl (2140 г) при данной температуре, и температура во время добавления по каплям не превышала 10°C. После завершения добавления по каплям реакционную смесь перемешивали при комнатной температуре в течение 16 часов. TLC показывала, что исходные материалы полностью прореагировали, и к реакционному раствору медленно по каплям добавляли 15 л воды для гашения реакции. Обеспечивали отстаивание раствора и его разделяли. Нижнюю фазу на основе дихлорметана отделяли и промывали насыщенным солевым раствором (10 л). Полученную органическую фазу концентрировали с получением продукта в виде коричневой маслянистой жидкости.
1H ЯМР (400 МГц, CDCl3) δ 7,69-7,63 (m, 4H), 7,45-7,34 (m, 6H), 3,77 (br t, J = 6,1 Гц, 1H), 3,69 (s, 3H), 3,54-3,44 (m, 1H), 2,57 (q, J = 6,1 Гц, 1H), 2,46 (br dd, J = 3,0, 14,6 Гц, 1H), 2,36-2,21 (m, 2H), 2,08-1,67 (m, 9H), 1,62-1,17 (m, 12H), 1,12-0,87 (m, 14H), 0,70-0,62 (m, 6H).
Стадия 1-3. Получение соединения 5
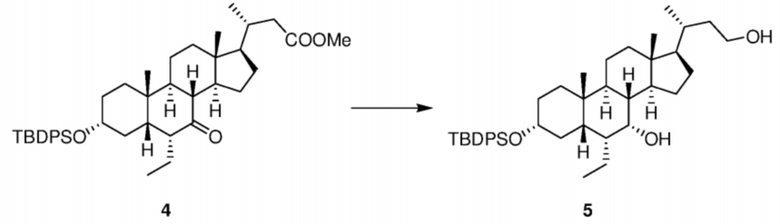
Тетрагидрофуран (10 л) добавляли в реактор объемом 50 л при 15°C, в реактор добавляли LiAlH4 (235 г, 6,2 моль) в защитной атмосфере N2 и реакционную смесь охлаждали до внутренней температуры 5°C. После растворения соединения 4 (2,04 кг) с помощью тетрагидрофурана его медленно по каплям добавляли к раствору LiAlH4 в тетрагидрофуране в течение приблизительно 2,5 часа. Реакционную смесь перемешивали при 15°C в течение 2 часов и протекание реакции контролировали с помощью TLC, и TLC показала, что исходные материалы исчезли. К реакционному раствору медленно по каплям добавляли H2O (235 мл) для гашения реакции, затем к реакционному раствору добавляли раствор на основе тетрагидрофурана (20 л) и к реакционному раствору медленно по каплям добавляли 15% раствор NaOH (235 мл) при перемешивании в течение 12 часов. Реакционную смесь фильтровали и осадок на фильтре промывали дихлорметаном (3 л). Фильтрат подвергали ротационному выпариванию до сухого состояния с получением маслянистого вещества. После растворения маслянистого вещества в DCM (15 л) органическую фазу промывали один раз водой (5 л) и насыщенным солевым раствором (5 л) в указанном порядке и фильтрат подвергали ротационному выпариванию до сухого состояния с получением белого твердого вещества (1,8 кг). Реакционную смесь охлаждали до комнатной температуры (приблизительно 16°C), затем регулировали до pH приблизительно 9 с помощью насыщенного раствора бикарбоната натрия и подвергали ротационному выпариванию (осталось небольшое количество) с получением неочищенного продукта. Неочищенный продукт растворяли в этилацетате (30 л) и последовательно промывали насыщенным раствором бикарбоната натрия (9 л), водой (9 л) и насыщенным солевым раствором (9 л). Органическую фазу подвергали ротационному выпариванию до сухого состояния с получением продукта в виде коричневой маслянистой жидкости.
1H ЯМР (400 МГц, CDCl3) δ 7,64-7,58 (m, 4H), 7,37-7,25 (m, 6H), 3,68-3,52 (m, 3H), 3,38-3,28 (m, 1H), 1,91-1,03 (m, 25H), 1,02-0,93 (m, 11H), 0,88 (d, J = 6,5 Гц, 3H), 0,72-0,64 (m, 6H), 0,57 (s, 3H).
Стадия 1-4. Получение соединения 6
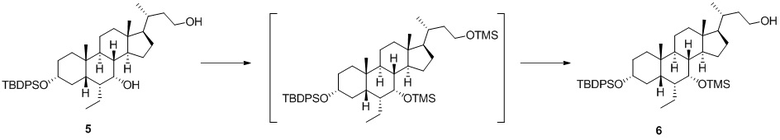
Имидазол (1,14 кг, 16,73 моль) добавляли к раствору соединения 5 (3,52 кг, 5,58 моль) в безводном дихлорметане (35 л). При 5°C в реакционную систему добавляли по каплям триметилхлорсилан (1770 мл, 13,95 моль) в течение двух часов. Реакционную систему перемешивали при 15°C в течение 3 часов. Выявление с помощью TLC показало, что реакция была практически полностью завершена. К реакционной системе добавляли 10 л воды при 15°C, перемешивали и разделяли. Органическую фазу один раз последовательно промывали с помощью 10 л воды и 10 л насыщенного солевого раствора.
Органическую фазу концентрировали до приблизительно 5 л и добавляли 30 л этанола. К раствору добавляли карбонат калия (1,93 кг, 13,95 моль) при 15°C. Реакционную систему перемешивали при 15°C в течение 14 часов. Выявление с помощью TLC показало, что реакция была практически полностью завершена. Реакционный раствор фильтровали. Осадок на фильтре ополаскивали с помощью 3 л дихлорметана. Фильтрат концентрировали с получением маслянистого вещества. Маслянистое вещество растворяли в 20 л дихлорметана и один раз последовательно промывали с помощью 10 л воды и 10 л насыщенного солевого раствора. Органическую фазу высушивали над 3 кг безводного сульфата натрия и фильтровали. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (100-200 меш, 230 мм × 800 мм) с помощью смеси н-гептан:этилацетат = от 30:1 до 20:1 для элюирования с получением указанного в заголовке соединения 6 (3,20 кг, чистота 87%).
1H ЯМР (400 МГц, CDCl3): δ 7,77-7,64 (m, 4H), 7,45-7,32 (m, 6H), 3,78-3,56 (m, 3H), 3,43-3,31 (m, 1H), 1,98-1,13 (m, 24H), 1,07 (s, 9H), 0,97 (d, J = 6,5 Гц, 3H), 0,83-0,74 (m, 4H), 0,68-0,55 (m, 6H), 0,17-0,05 (m, 9H).
Стадия 1-5. Получение соединения 8
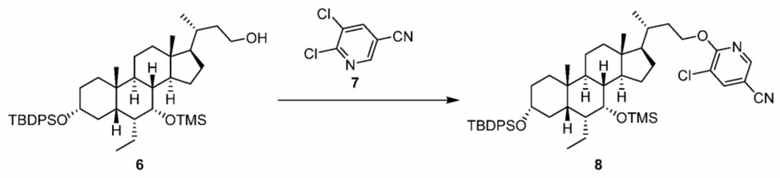
Соединение 6 (2498,0 г, 3,10 моль) добавляли в реактор, растворяли в THF (12,5 л) и при контролируемой внутренней температуре от 5°C до 10°C медленно добавляли t-BuONa (614,2 г, 6,20 моль) в течение приблизительно 40 минут. После перемешивания в течение 10 минут реакционную смесь нагревали до температуры от 20°C до 23°C при перемешивании в течение 1,5 часа и затем охлаждали до температуры от 5°C до 10°C. К реакционной смеси добавляли по каплям раствор соединения 7 (12,5 л, 6,20 моль, 1073,1 г) в THF при вышеуказанной внутренней температуре и затем смесь нагревали до 60°C. После перемешивания в течение 1,5 часа выявление с помощью TLC и HPLC показало, что реакция была полностью завершена. Затем реакционную смесь охлаждали до 20°C, гасили путем добавления 25 л воды и экстрагировали этилацетатом (25 л × 2). Органические фазы объединяли и промывали три раза насыщенным солевым раствором (25 л × 3). Полученный раствор подвергали ротационному выпариванию до сухого состояния с получением неочищенного маслянистого продукта. Неочищенный продукт растворяли в 2,5 л ацетона и раствор неочищенного продукта медленно добавляли по каплям соответственно в три трехгорлые колбы объемом 10 л с 6,6 × 3 л метанола при внутренней температуре от -10°C до -15°C, при перемешивании, и осаждалось большое количество твердого вещества. После фильтрации осадок на фильтре промывали с помощью 3,0 л метанола с получением желтого твердого вещества (невысушенное), которое затем добавляли к 18,0 л метанола и суспендировали в течение ночи. После фильтрации осадок на фильтре промывали с помощью 3,0 л метанола с получением желтого твердого вещества (невысушенное), которое затем добавляли к 18,0 л метанола, суспендировали в течение ночи и фильтровали. Осадок на фильтре промывали с помощью 2,0 л метанола, высушивали под вакуумом в течение 24 часов с получением 2522,0 г желтого твердого вещества, т. е. соединения 8 (2522,0 г, выход 90%, чистота 92,9%).
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ = 8,25 (d, J = 2,0 Гц, 1H), 7,73 (d, J = 2,0 Гц, 1H), 7,63-7,51 (m, 4H), 7,33-7,21 (m, 6H), 4,48-4,27 (m, 2H), 3,50 (s, 1H), 3,31-3,18 (m, 1H), 1,98-1,03 (m, 27H), 0,95 (s, 9H), 0,73-0,64 (m, 4H), 0,58-0,46 (m, 6H), 0,00 (s, 9H).
Стадия 2-2. Получение соединения 9

В реактор (20 л) добавляли соединение 8 (2520,0 г, 2,79 моль) и затем добавляли EtOH (13,0 л) при перемешивании для растворения. При внутренней температуре, контролируемой на уровне приблизительно 10°C, порциями добавляли водный раствор (13,0 л) NaOH (2232,0 г, 55,8 моль). Реакционный раствор нагревали до температуры 105°C при перемешивании в течение 2,8 часа. Реакция была полностью завершена, как показало выявление с помощью TLC и HPLC. Реакционный раствор охлаждали до 10°C, обеспечивали его отстаивание в течение двух часов и твердое вещество осаждали на дне емкости. Удаляли 19,5 л надосадочной жидкости, затем к реакционной смеси добавляли 39,0 л воды и перемешивали в течение 36 часов при контролируемой внутренней температуре не выше 12°C. После фильтрации твердое вещество последовательно промывали с помощью 6,0 л воды и 6,0 л ацетонитрила. Твердое вещество суспендировали с помощью 10,0 л ацетонитрила в течение 2 часов и фильтровали с получением другого твердого вещества. Его суспендировали с помощью 12,0 л ацетона в течение 16 часов и фильтровали с получением еще одного твердого вещества. Еще одно твердое вещество снова суспендировали с помощью 12,0 л ацетона в течение 16 часов, затем фильтровали и высушивали с получением 2332,3 г соединения 9 в виде белого твердого вещества (2332,3 г, выход 94,7%, чистота 99,7%).
1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ = 8,48 (d, J = 2,0 Гц, 1H), 8,07 (d, J = 2,0 Гц, 1H), 7,55 (br dd, J = 6,5, 12,5 Гц, 4H), 7,41-7,11 (m, 6H), 4,52-4,15 (m, 2H), 3,54 (br s, 1H), 3,34-3,22 (m, 1H), 2,04-1,14 (m, 28H), 0,93 (s, 9H), 0,69 (s, 4H), 0,60-0,43 (m, 6H), 0,00 (s, 9H).
Стадия 2-3. Получение соединения формулы I

В реактор (50 л) добавляли соединение 9 (2330,3 г, 2,65 ммоль) и для его растворения затем добавляли THF (24,0 л). При внутренней температуре, контролируемой на уровне 10°C, медленно по каплям добавляли концентрированную HCl (10,0 л, 120,00 моль) в течение 2 ч. и смесь нагревали до 13°C (комнатная температура) при перемешивании в течение 90 часов. При проведении выявления с помощью TLC медленно добавляли 75 л раствора на основе гидроксида натрия (6000 г) при температуре от 8°C до 10°C для регулирования pH до значения 10, перемешивали в течение получаса и экстрагировали метил-трет-бутиловым эфиром (30 л × 4). Полученный раствор регулировали до pH 5 с помощью концентрированной HCl (3000 мл) и экстрагировали этилацетатом (30 л × 2). Органическую фазу промывали водой (30 л × 4) и концентрировали с получением 1350 г продукта. Полученный продукт суспендировали с помощью смешанного растворителя, состоящего из 2,0 л этилацетата и 5,0 л н-гептана, в течение ночи и фильтровали с получением 1280 г другого продукта. После полного растворения с помощью 9,0 л этилацетата (80°C) полученный раствор медленно охлаждали до комнатной температуры (10°C) с получением 1222 г соединения формулы I.
1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ = 8,69 (d, J = 2,0 Гц, 1H), 8,23 (d, J = 2,0 Гц, 1H), 4,67-4,30 (m, 2H), 3,67 (br s, 1H), 3,34-3,22 (m, 1H), 2,10-1,11 (m, 25H), 1,09-0,97 (m, 3H), 0,96-0,86 (m, 6H), 0,73 (s, 3H).
Продукт, полученный на данной стадии, представлял собой кристаллическую форму B соединения формулы I, и его иллюстративные XRPD-дифрактограмма, DSC- и TGA-термограмма показаны на фиг. 7, 8 и 9.
Пример 2. Кристаллическая форма A соединения формулы I
Способ 1
58 г соединения формулы I суспендировали в смешанном растворителе, состоящем из этанола (225 мл) и воды (175 мл), и перемешивали при 45°C в течение 18 часов с получением большого количества белого твердого вещества. После фильтрации осадок на фильтре высушивали с получением 48 г продукта.
Способ 2
200 мг соединения формулы I добавляли к воде (2 мл) с образованием суспензии, которую перемешивали при 40°C в течение 1,5 дня, затем нагревали до температуры 50°C и перемешивали в течение еще одного дня. Полученное вещество центрифугировали и затем помещали в вакуумный сушильный шкаф при 30°C для высушивания с получением продукта.
Иллюстративные XRPD-дифрактограмма, DSC- и TGA-термограмма кристаллической формы A соединения формулы I (полученной с помощью способа 1 в примере 2) показаны на фиг. 1, 2 и 3.
Другие иллюстративные XRPD-дифрактограмма, DSC-термограмма, TGA-термограмма кристаллической формы A соединения формулы I (полученной с помощью способа 2 в примере 2) показаны на фиг. 4, 5 и 6.
Пример 3. Твердая аморфная форма соединения формулы I
122 г соединения формулы I растворяли в безводном этаноле (500 мл) и перемешивали при 15°C в течение 30 минут для растворения с образованием прозрачного раствора. Раствор высушивали, подвергали ротационному выпариванию и дополнительно высушивали с применением масляного насоса до постоянного веса с получением 119 г белого твердого вещества.
Иллюстративная XRPD-дифрактограмма твердой аморфной формы соединения, представленного посредством формулы I, показана на фиг. 8.
Экспериментальный пример 1. Тест в отношении стабильности в твердом состоянии кристаллической формы A
Стабильность в твердом состоянии кристаллической формы A исследовали при следующих условиях: 1) 40°C (открытый), 2) 60°C (открытый), 3) комнатная температура/92,5% RH (открытый), 4) комнатная температура/75% RH (открытый), 5) 40°C/75% RH (открытый) и 6) 60°C/75% RH (открытый), где комнатная температура выбрана из диапазона от 20°C до 30°C.
Отбирали несколько образцов кристаллической формы A в подходящем количестве, помещали на дно стеклянных емкостей для образцов и распределяли в тонкий слой. Емкости с образцами, подлежащими помещению в вышеуказанные условия, накрывали алюминиевой фольгой, в которой было сделано несколько небольших отверстий, обеспечивающих полный контакт образцов с воздухом окружающей среды. Отбор образцов для проведения выявления с помощью XRPD осуществляли в день 5 и день 10 и результаты выявления сравнивали с исходными результатами в день 0, которые указывали на то, что кристаллическая форма образцов оставалась без изменений.
Экспериментальный пример 2. Тест в отношении стабильности в твердом состоянии кристаллической формы B
Стабильность в твердом состоянии кристаллической формы B исследовали при следующих условиях: 1) комнатная температура/92,5% RH (открытый), 2) комнатная температура/75% RH (открытый), 3) 40°C/75% RH (открытый) и 4) 60°C/75% RH (открытый), при этом комнатная температура выбрана из диапазона от 20°C до 30°C.
Отбирали несколько образцов кристаллической формы B в подходящем количестве, помещали на дно стеклянных емкостей для образцов и распределяли в тонкий слой. Емкости с образцами, подлежащими помещению в вышеуказанные условия, накрывали алюминиевой фольгой, в которой было сделано несколько небольших отверстий, обеспечивающих полный контакт образцов с воздухом окружающей среды. Отбор образцов для проведения выявления с помощью XRPD осуществляли в день 5 и день 10 и результаты выявления сравнивали с исходными результатами в день 0, которые указывали на то, что кристаллическая форма образцов оставалась без изменений.
Экспериментальный пример 3. Тест в отношении стабильности в твердом состоянии твердой аморфной формы соединений формулы I
Стабильность в твердом состоянии твердой аморфной формы соединения формулы I исследовали при следующих условиях: 1) комнатная температура/75% RH (открытый), 2) 40°C/75% RH (открытый), 3) 60°C/75% RH (открытый), где комнатная температура выбрана из диапазона от 20°C до 30°C.
Отбирали несколько образцов твердой аморфной формы в подходящем количестве, помещали соответственно на дно стеклянных емкостей для образцов и распределяли в тонкий слой. Емкости с образцами, подлежащими помещению в вышеуказанные условия, накрывали алюминиевой фольгой, в которой было сделано несколько небольших отверстий, обеспечивающих полный контакт образцов с воздухом окружающей среды. Отбор образцов для проведения выявления с помощью XRPD осуществляли в день 10 и через один месяц и результаты выявления сравнивали с исходными результатами в день 0, которые, исходя из выявления с помощью XRPD, указывали на то, что образцы оставались без изменений.
Экспериментальный пример 4. Тест в отношении гигроскопичности
Проводили анализ посредством динамической сорбции паров (DVS) на примере кристаллической формы A и твердой аморфной формы соединения формулы I с помощью следующих способа и условий: образцы (10-15 мг) помещали в тигель для образца; модель измерительного прибора: анализатор динамической сорбции паров SMS DVS Advantage; температура: 25°C; критерий равновесия: dm/dt = 0,01%/мин. (Min: 10 мин., Max: 180 мин.); высушивание: в течение 120 мин. при 0% RH; градиент RH (%) для тестирования: 10%; диапазон RH (%) для теста с градиентом: 0% - 90% - 0%. Гигроскопичность оценивали с применением следующей шкалы:
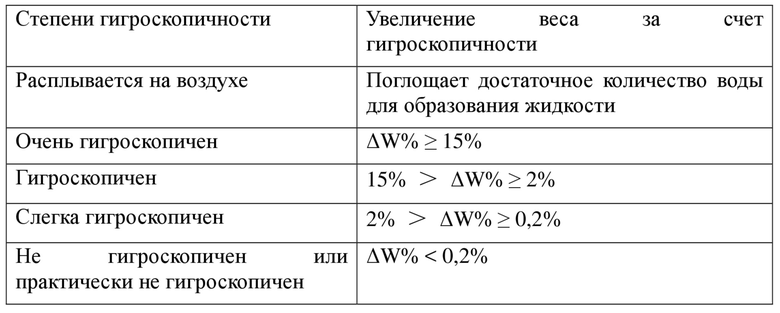
Результаты показали, что 1) увеличение веса за счет гигроскопичности кристаллической формы A при 25±1°C и при 80±2% RH составляло 0,835%, указывая на то, что образец слегка гигроскопичен; и 2) увеличение веса за счет гигроскопичности твердой аморфной формы соединения формулы I при 25±1°C и при 80±2% RH составляло 1,775%, указывая на то, что образец слегка гигроскопичен.
Экспериментальный пример 5. Оценивание in vitro
Биохимический эксперимент в отношении FXR
Цель эксперимента
Активационный эффект соединения в отношении реакции связывания с FXR определяли с помощью AlphaScreen.
Экспериментальные материалы
1. Белок: белок FXR человека, меченный глутатион-S-трансферазой (Invitrogen)
2. Коактиватор: коактиватор стероидных рецепторов, меченный биотином (Anaspec)
3. Реагент для выявления: набор для выявления AlphaScreen (PerkinElmer)
Экспериментальный способ
1. Разбавление соединений. Соединение, подлежащее тестированию, получали в виде 40 мкM раствора в DMSO и затем 3-кратно разбавляли до 10 точек концентрации. Эталонное соединение получали в виде 400 мкM раствора в DMSO и затем 1,5-кратно разбавляли до 10 точек концентрации. Разбавленный раствор в DMSO добавляли в лунки 384-луночного планшета в объеме 150 нл на лунку.
2. Белок FXR человека, меченный глутатион-S-трансферазой, и коактиватор стероидных рецепторов, меченный биотином, составляли в виде смешанного раствора с концентрациями 0,4 нM и 30 нM, соответственно, добавляли в лунки 384-луночного планшета в объеме 15 мкл на лунку и инкубировали в течение 1 часа при комнатной температуре.
4. Смешанный раствор акцепторных гранул в наборе для выявления AlphaScreen разбавляли 125-кратно и добавляли в лунки 384-луночного планшета в объеме 7,5 мкл на лунку. Процесс в ходе экспериментального способа защищали от света. Инкубирование проводили в течение 1 часа при комнатной температуре.
5. Смешанный раствор донорных гранул в наборе для выявления AlphaScreen разбавляли 125-кратно и добавляли в лунки 384-луночного планшета в объеме 7,5 мкл на лунку. Процесс в ходе экспериментального способа защищали от света. Инкубирование проводили в течение 1 часа при комнатной температуре.
6. Тест в отношении EC50. Envision применяли при длине волны возбуждения 680 нм со считыванием сигналов поглощения при 520-620 нм.
7. Аналитические данные. Данные анализировали посредством применения Prism 5.0 и рассчитывали значения EC50, демонстрирующие активационные эффекты соединения. Затем применяли соотношение наибольшего значения сигнала соединения и значения эталонного соединения для получения процентного значения эффективности активации соединением.
Клеточный эксперимент в отношении FXR
Цель эксперимента
Влияние соединения на клеточную функциональную активность определяли посредством методики с применением репортерного гена β-лактамазы.
Экспериментальные материалы
1. Клеточная линия: FXR HEK 293T DA
2. Среда для культивирования клеток: среда DMEM, дополненная 10% сывороткой и смесью пенициллин/стрептомицин (1x)
3. Реагент для выявления: набор для выявления репортерных генов GeneBLAzer®(Invitrogen)
Экспериментальный способ
1. Разбавление соединений. Соединение, подлежащее тестированию, получали в виде 100 мкM раствора в DMSO и затем соединение 3-кратно разбавляли до 10 точек концентрации. Эталонное соединение получали в виде 100 мкM раствора в DMSO и затем 1,3-кратно разбавляли до 10 точек концентрации. Разбавленный раствор в DMSO добавляли в лунки 384-луночного планшета в объеме 200 нл на лунку.
2. Инокуляция клеток. Клетки FXR HEK 293T DA восстанавливали, ресуспендировали в среде для культивирования, разбавляли до плотности 5 x 105 клеток/мл и добавляли в лунки 384-луночного планшета в объеме 40 мкл на лунку.
3. 384-луночный планшет инкубировали при 37℃, 5% CO2 в течение 16 часов.
4. 6 мкл 1 мM субстрата LiveBLAzer™-FRET B/G (CCF4-AM) смешивали с 60 мкл раствора B и 934 мкл раствора C и добавляли в лунки 384-луночного планшета в объеме 8 мкл на лунку.
5. 384-луночный планшет инкубировали в темноте в течение 2 часов при комнатной температуре.
6. Тест в отношении EC50. Envision применяли при длине волны возбуждения 409 нм со считыванием сигналов поглощения при 460 и 530 нм.
7. Аналитические данные. Данные анализировали посредством применения Prism 5.0 и рассчитывали значения EC50, демонстрирующие активационные эффекты соединения. Затем применяли соотношение наибольшего значения сигнала соединения и значения эталонного соединения (хенодезоксихолевая кислота, CDCA) для получения процентного значения эффективности активации соединением.
Таблица 1. Результаты тестирования в отношении EC50 для биохимического эксперимента и клеточного эксперимента
Вывод: агонистический эффект соединения по настоящему изобретению в отношении рецептора FXR является значимым и агонистический эффект в отношении рецептора FXR на клеточном уровне также является значимым.
Экспериментальный пример 6. Исследование in vivo
Фармакокинетика у мышей, которым вводили одно соединение
12 самцов мышей (C57BL/6J) произвольным образом разделяли на две группы, т. е. по 6 мышей в каждой группе. Первая группа представляла собой группу внутривенного введения, предусматривающего введение дозы 2 мг/кг, 2 мл/кг путем инъекции через хвостовую вену (среда-носитель представляла собой 10% водный раствор HPbCD, и если растворимость лекарственного средства не была удовлетворительной, добавляли coрастворитель); вторая группа представляла собой группу перорального введения, предусматривающего внутрижелудочное введение дозы 10 мг/кг, 10 мл/кг (среда-носитель представляла собой 0,5% водный раствор HPMC). В группе внутривенного введения образцы плазмы крови (с применением K2-EDTA в качестве антикоагулянта) отбирали через 0,083, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа после введения; и в группе перорального введения образцы плазмы крови отбирали через 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа после введения. В случае 6 животных в каждой группе забор образцов крови осуществляли у 3 животных в один момент времени. Забор образцов у 3 животных первой партии производили поочередно с 3 животными второй партии. Анализ образцов плазмы крови выполняли с применением LC-MS/MS. Полученные в результате значения концентрации в плазме крови представляли в виде графика зависимости от времени и параметры PK рассчитывали с применением Phoenix WinNonlin 6.3.
Таблица 2

Вывод: как показано в таблице 2, после перорального введения одной и той же дозы пиковая концентрация и лекарственное воздействие соединения формулы I были выше, чем таковые обетихолевой кислоты, представляющей собой контрольное соединение.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО ТИОФЕНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2022 |
|
RU2830948C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА TLR7/TLR8 | 2019 |
|
RU2792005C2 |
| АМОРФНОЕ ПРОИЗВОДНОЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТА PPAR И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2749056C1 |
| СОЕДИНЕНИЕ ИНГИБИТОРА BRD4 В ТВЕРДОЙ ФОРМЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2793346C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА ФОСФОДИЭСТЕРАЗЫ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2814498C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА МИМЕТИКА SMAC, ПРИМЕНЯЕМОГО В КАЧЕСТВЕ ИНГИБИТОРА IAP, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2020 |
|
RU2819398C2 |
| СОЛИ И ПОЛИМОРФЫ ЗАМЕЩЕННОГО ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНА | 2015 |
|
RU2732125C2 |
| ТВЕРДАЯ ФОРМА, КРИСТАЛЛИЧЕСКАЯ ФОРМА И КРИСТАЛЛИЧЕСКАЯ ФОРМА А АГОНИСТА FXR, И СПОСОБ ИХ ПОЛУЧЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2804320C2 |
| АНАЛОГ ПИРИДО[1,2-A]ПИРИМИДОНА, ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2753696C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СПОСОБ ЕЕ ОЧИСТКИ | 2011 |
|
RU2604734C2 |
Изобретение относится к кристаллической форме A соединения формулы I, где ее порошковая рентгеновская дифрактограмма содержит дифракционные пики при 2θ 5,95°, 10,10°, 15,14°, 18,83°, 20,23°, где диапазон погрешности 2θ составляет ±0,2°, а также к способу ее получения и ее применению для лечения или предупреждения заболевания, связанного с фарнезоидным Х-рецептором. Технический результат – получена кристаллическая форма нового соединения, которая является стабильной, чистой и негигроскопичной, и может найти применение в медицине для лечения заболеваний, связанных с фарнезоидным Х-рецептором (FXR), например, таких как неалкогольная жировая болезнь печени (NAFLD), неалкогольный стеатогепатит (NASH), первичный билиарный цирроз (PBC), холестатическая гепатопатия, хроническое заболевание печени, ожирение, диабет 2 типа и др. 3 н. и 4 з.п. ф-лы, 12 ил., 5 табл., 3 пр.
1. Кристаллическая форма A соединения формулы I
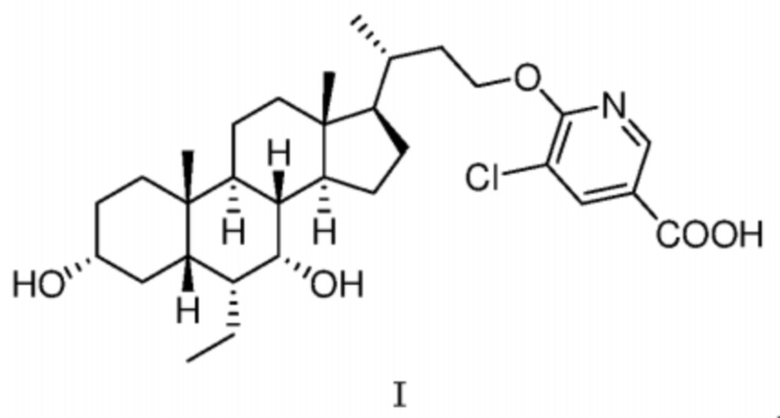 ,
,
где ее порошковая рентгеновская дифрактограмма содержит дифракционные пики при 2θ 5,95°, 10,10°, 15,14°, 18,83°, 20,23°, где диапазон погрешности 2θ составляет ±0,2°.
2. Кристаллическая форма A по п. 1, где порошковая рентгеновская дифрактограмма содержит дифракционные пики при 2θ 5,95°, 7,95°, 10,10°, 13,32°, 15,14°, 15,85°, 18,83°, 20,23°, где диапазон погрешности 2θ составляет ±0,2°.
3. Кристаллическая форма A по п. 1, где порошковая рентгеновская дифрактограмма содержит дифракционные пики при 2θ 5,95°, 7,95°, 10,10°, 13,32°, 14,17°, 15,14°, 15,85°, 18,83°, 19,18°, 20,23°, 24,69°, где диапазон погрешности 2θ составляет ±0,2°.
4. Кристаллическая форма A по п. 1, где порошковая рентгеновская дифрактограмма содержит дифракционные пики при 2θ 5,95°, 7,95°, 10,10°, 13,32°, 14,17°, 14,58°, 15,14°, 15,85°, 18,25°, 18,83°, 19,18°, 20,23°, 24,69°, 25,81°, где диапазон погрешности 2θ составляет ±0,2°.
5. Способ получения кристаллической формы A соединения формулы I по любому из пп. 1-4, включающий следующие стадии:
1) суспендирование/растворение соединения формулы I в растворителе для кристаллизации, выбранном из воды или смешанного растворителя, содержащего воду, при перемешивании; и
2) фильтрование и необязательно промывание и/или высушивание,
где неводный растворитель в смешанном растворителе, содержащем воду, представляет собой этанол.
6. Применение кристаллической формы A соединения формулы I по любому из пп. 1-4 в изготовлении лекарственного препарата для лечения или предупреждения заболевания, связанного с фарнезоидным X-рецептором.
7. Применение по п. 6, где заболевание, связанное с фарнезоидным X-рецептором, включает неалкогольную жировую болезнь печени, неалкогольный стеатогепатит, первичный билиарный цирроз, холестатическую гепатопатию, хроническое заболевание печени, инфекцию, вызванную вирусом гепатита С, алкогольную болезнь печени, фиброз печени, первичный склерозирующий холангит, камень в желчном пузыре, билиарную атрезию, симптом нижних мочевыводящих путей и доброкачественную гиперплазию предстательной железы (BPH), камни в мочеточнике, ожирение, диабет 2 типа, атеросклероз, артериосклероз, гиперхолестеринемию, гиперлипидемию или нарушение функции печени, обусловленное гиперхолестеринемией и гиперлипидемией.
| WO 2017129125 A1, 03.08.2017 | |||
| WO 2016107575 A1, 07.07.2016 | |||
| WO 2016173397 A1, 03.11.2016 | |||
| WO 2016086169 A1, 02.06.2016 | |||
| US 7932244 B2, 26.04.2011 | |||
| Электромагнитный прижим для обрабатываемых на станках деревянных частей | 1929 |
|
SU17714A1 |

