Настоящее изобретение относится к соли и кристаллическим формам 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты, которые подходят для получения в промышленном масштабе, а также к фармацевтическим композициям, содержащим эти вещества, способам их получения и их применению в терапии.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
В течение последних нескольких десятилетий частота устойчивости к противомикробным препаратам и ее ассоциация с тяжелыми инфекционными заболеваниями увеличивались с пугающими скоростями. Особенно огорчает возрастающая распространенность устойчивости у нозокомиальных патогенных микроорганизмов. Из более чем 2 миллионов случаев нозокомиальной (полученной в медицинском учреждении) инфекции, происходящих каждый год в Соединенных Штатах Америки, от 50 до 60% вызваны устойчивыми к противомикробным препаратам штаммами бактерий. Высокая степень устойчивости к широко используемым противобактериальным средствам увеличивает заболеваемость, смертность и затраты, связанные с нозокомиальными инфекциями. Полагают, что в Соединенных Штатах Америки нозокомиальные инфекции способствуют или являются причиной более 77000 смертельных исходов в год, а ежегодные затраты составляют приблизительно от 5 до 10 миллиардов долларов.
Значимые причины устойчивости грамотрицательных бактерий включают β-лактамазы расширенного спектра действия (ESBL), сериновые карбапенемазы (KPC) и металло-β-лактамазы (например, NDM-1) у Klebsiella pneumoniae, Escherichia coli и Proteus mirabilis, высокоуровневую обусловленную β-лактамазой (AmpC) устойчивость к цефалоспоринам третьего поколения у видов Enterobacter и Citrobacter freundii и гены множественной лекарственной устойчивости, выявленные у Pseudomonas, Acinetobacter и Stenotrophomonas. Проблема противобактериальной устойчивости осложняется существованием бактериальных штаммов, устойчивых к нескольким противобактериальным препаратам. Например, Klebsiella pneumonia, несущая ген металло-β-лактамазы NDM-1, на той же плазмиде, которая несет NDM-1, часто несет дополнительные сериновые β-лактамазы.
Таким образом, существует необходимость в новых противобактериальных соединениях, в частности, в противобактериальных соединениях, которые эффективны против существующих устойчивых к лекарственным средствам микроорганизмов или менее предрасположены к развитию новой бактериальной устойчивости. По настоящему изобретению предоставлены твердые формы таких соединений, которые особенно хорошо подходят для промышленного производства вследствие их пригодности к обработке в подходящих для производства рабочих условиях.
В неопубликованной патентной заявке номер PCT/US2015/022011 описаны определенные монобактамные антибиотики. В этой заявке одним из соединений, которое демонстрирует высокую активность против грамотрицательных бактерий, включая штаммы, которые демонстрируют устойчивость к другим монобактамам, является 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновая кислота, которое в настоящем документе обозначают как соединение X:
 (Соединение X).
(Соединение X).
Для производства фармацевтических соединений и их составов важно, чтобы активное соединение находилось в форме, с которой можно подходящим образом обращаться и которую можно подходящим образом обрабатывать с получением рентабельного, надежного и воспроизводимого производственного процесса. Соединение X и многие из его солей являются твердыми при комнатной температуре, и их можно получать в различных твердых формах в зависимости от условий, используемых для получения, очистки или кристаллизации вещества. Существование нескольких твердых форм, часто обозначаемых как полиморфные формы, хорошо известно для твердых фармацевтических соединений, и химическая и физическая стабильность таких соединений, а также их пригодность к обработке часто зависит от того, какую твердую форму используют. Таким образом, выбор конкретной твердой формы активного лекарственного вещества (например, формы соли, гидратированной или сольватированной формы или полиморфной формы) при разработке надежного и воспроизводимого способа получения и при хранении, обработке и распространении безопасной и эффективной формы лекарственного вещества часто является очень важным.
В основном выявлено, что существуют преимущества при производстве конкретной твердой формы фармацевтического ингредиента, и они описаны в "Handbook of Pharmaceutical Salts; Properties, Selection and Use", P. Heinrich Stahl, Camille G. Wermuth (Eds.) (Verlag Helvetica Chimica Acta, Zurich). Способы производства твердых формы также описаны в "Practical Process Research and Development", Neal G. Anderson (Academic Press, San Diego) и "Polymorphism: In the Pharmaceutical Industry", Rolf Hilfiker (Ed) (Wiley VCH).
Авторы настоящего изобретения выявили определенные соли и полиморфные формы соединения X, которые особенно пригодны для использования при производстве, хранении или введение соединения X, как описано в настоящем документе.
Для лекарственного продукта важно, чтобы он был достаточно стабилен, чтобы избежать значительного разрушения при транспортировке и хранении в коммерчески приемлемых условиях. Авторы изобретения выявили, что соединение X в растворе для максимальной стабильности во влажных условиях предпочтительно использовать и хранить при pH от 4 до 6, предпочтительно от 4,0 до 5,5. Оптимальная стабильность раствора достигается при pH приблизительно от 4,0 до 5,5 Таким образом, изобретение относится к фармацевтическим композициям, содержащим соединение X при pH от 4 до 6, предпочтительно от 4,0 до 5,5 и более предпочтительно при pH приблизительно 5±0,5 или при pH 5±0,2. Подходящие композиции содержат соединение X в водном растворе, таком как раствор декстрозы или солевой раствор, который может быть изотоническим и может содержать другие вещества, такие как стабилизаторы, антиоксиданты, буферы или модификаторы pH, питательные вещества и т.п. В некоторых из этих вариантов осуществления требуемого pH достигают посредством комбинирования соединение X и модификатора pH, подходящего для достижения требуемого pH в водном растворе. Подходящие модификаторы pH в качестве неограничивающих примеров включают гидроксид натрия, карбонат натрия, бикарбонат натрия, карбонат калия, гидроксид калия, амины, такие как TRIS (трис(гидроксиметил)аминометан), и аминокислоты, такие как аргинин, лизин, гистидин, и т.п. Известные монобактамы, включая азтреонам, сформулированы с аргинином.
Подходящего pH можно достигать, добавляя модификатор pH в водный раствор соединения X или добавляя соединение X в водный раствор, содержащий модификатор pH. Специалист может легко определить подходящие количества модификатора pH и соединения X. Подходящие модификаторы pH включают гидроксид натрия и аргинин. Таким образом, раствор или суспензию соединения X можно обрабатывать гидроксидом натрия или аргинином с получением раствора, содержащего натриевую соль или аргининовую соль соединения. Кроме того, этот раствор можно лиофилизировать с удалением воды и любых присутствующих сорастворителей, оставляя лиофилизированное твердое вещество, которое содержит соединение X и модификатор pH или соль, сформированную соединением X и модификатором pH, например, натриевую соль или аргининовую соль соединения X.
Кроме того, по настоящему изобретению предоставлен ряд твердых форм соединения X, которые обеспечивают пригодность к обработке, подходящую для производства в промышленном масштабе, вместе со способами получения этих полиморфных форм.
Репрезентативными являются перечисленные ниже варианты осуществления изобретения:
1. Аргининовая соль 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты. В определенных вариантах осуществления соль представляет собой соль (L)-аргинина.
2. Натриевая соль 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты.
3. Гидратированная твердая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты.
4. Гидратированная твердая форма по варианту осуществления 3, которая в основном состоит из тригидрата 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты.
5. Способ получения гидратированной твердой формы по варианту осуществления 4, который включает приведение 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты в контакт с атмосферой с относительной влажностью от 25% до 50% при температуре от 20°C до 30°C.
6. Фармацевтическая композиция, содержащая соединение по любому из вариантов осуществления 1-4 и по меньшей мере один фармацевтически приемлемый носитель или эксципиент.
7. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 1), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 6,6, 13/4 и 18,8. В определенных вариантах осуществления у формы 1 присутствуют дополнительные пики XRPD, как описано ниже.
8. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 2), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 7,5, 19,3 и 20,0. В определенных вариантах осуществления у формы 2 присутствуют дополнительные пики XRPD, как описано ниже.
9. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 3), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 7,3, 18,9 и 21,2. В определенных вариантах осуществления у формы 3 присутствуют дополнительные пики XRPD, как описано ниже.
10. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 4), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 7,0, 8,6, 19,3 и 20,9. В определенных вариантах осуществления у формы 4 присутствуют дополнительные пики XRPD, как описано ниже.
11. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 5), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 7,3, 9,3 и 27,8. В определенных вариантах осуществления у формы 5 присутствуют дополнительные пики XRPD, как описано ниже.
12. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 6), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 8,1, 9,2 и 12,8; и необязательно дополнительные пики при 21,2 и 24,7. В определенных вариантах осуществления у формы 6 присутствуют дополнительные пики XRPD, как описано ниже.
13. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 7), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 6,7, 7,3 и 20,3. В определенных вариантах осуществления у формы 7 присутствуют дополнительные пики XRPD, как описано ниже.
14. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 8), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 6,2, 21,8 и 25,9. В определенных вариантах осуществления у формы 8 присутствуют дополнительные пики XRPD, как описано ниже.
15. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 9), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 6,3, 12,6 и 22,3 и необязательно один или несколько дополнительных пиков, выбранных из 22,1, 23,1, 27,0 и 27,5. В определенных вариантах осуществления у формы 9 присутствуют дополнительные пики XRPD, как описано ниже.
16. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 10), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 6,6, 11,0 и 16,5 и необязательно один или несколько дополнительных пиков, выбранных из 22,2 и 23,4. В определенных вариантах осуществления у формы 10 присутствуют дополнительные пики XRPD, как описано ниже.
17. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 11), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 7,4, 9,7 и 29,3 и необязательно один или несколько дополнительных пиков, выбранных из пиков при 17,0,19,5, 22,2, 26,3 и 28,1. В определенных вариантах осуществления у формы 11 присутствуют дополнительные пики XRPD, как описано ниже.
18. Кристаллическая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты (форма 12), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 19,0, 20,4 и 24,0 и необязательно один или несколько дополнительных пиков, выбранных из пиков при 7,3, 24,7 и 27,2. В определенных вариантах осуществления у формы 12 присутствуют дополнительные пики XRPD, как описано ниже.
19. Фармацевтическая композиция, содержащая 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновую кислоту и аргинин.
20. Фармацевтическая композиция по п. 19, которая дополнительно содержит фармацевтически приемлемый носитель.
21. Фармацевтическая композиция по п. 20, где носитель является водным.
22. Фармацевтическая композиция по п. 21, которая находится при pH приблизительно 5,0.
23. Фармацевтическая композиция по п. 21 или 22, которая дополнительно содержит по меньшей мере один эксципиент, выбранный из сахарозы, фруктозы, трегалозы, маннита и лактозы.
24. Соединение по любому из пп. 1-4 или его кристаллическая форма по любому из пп. 7-18, для применения в терапии.
25. Соединение по п. 24, где терапия представляет собой лечение инфекции грамотрицательных бактерий.
26. Соединение по п. 24, где бактерия, вызывающая инфекцию грамотрицательных бактерий выбрана из бактерий Citrobacter, Enterobacter, Escherichia, Haemophilus, Klebsiella, Morganella, Moraxella, Pseudomonas, Proteus, Salmonella, Serratia, Shigella и Neisseria.
27. Применение соединения по любому из пп. 1-4 или его кристаллической формы по любому из пп. 7-18 в производстве лекарственного средства для лечения инфекции грамотрицательных бактерий.
28. Применение по п. 27, где бактерия, вызывающая инфекцию грамотрицательных бактерий представляет собой вид, выбранный из бактерий Citrobacter, Enterobacter, Escherichia, Haemophilus, Klebsiella, Morganella, Moraxella, Pseudomonas, Proteus, Salmonella, Serratia, Shigella и Neisseria.
29. Способ лечения инфекции грамотрицательных бактерий, включающий введение нуждающемуся в этом индивидууму терапевтически эффективного количества соединения по любому из пп. 1-4, или кристаллической формы по любому из пп. 7-18, или фармацевтической композиции по любому из пп. 19-23.
30. Способ по п. 29, где бактерия, вызывающая инфекцию грамотрицательных бактерий представляет собой вид, выбранный из бактерий Citrobacter, Enterobacter, Escherichia, Haemophilus, Klebsiella, Morganella, Moraxella, Pseudomonas, Proteus, Salmonella, Serratia, Shigella или Neisseria.
Таким образом, в одном из аспектов изобретение относится к кристаллической форме соединения X (форма 1), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 6,6, 13,4 и 18,8. В одном из вариантов осуществления форма 1 демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции: 6,6, 13,4, 16,6, 17,9, 18,8, 20,3, 25,1 и 28,9. В другом варианте осуществления форма 1 демонстрирует по меньшей мере характеристические пики рентгеновской порошковой дифракции, приведенные в списке 1, или любое подмножество по меньшей мере из пяти пиков, выбранных из списка 1. В еще одном варианте осуществления форма 1 демонстрирует по существу такой же профиль рентгеновской порошковой дифракции, как приведен на фигуре 1.
В другом аспекте изобретение относится к кристаллической форме соединения X (форма 2), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 7,5, 19,3 и 20,0. В одном из вариантов осуществления форма 2 демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции: 6,7, 7,5, 13,3, 13,7, 15,3, 17,9, 18,7, 19,3 и 20,0. В другом варианте осуществления форма 2 демонстрирует по меньшей мере характеристические пики рентгеновской порошковой дифракции, приведенные в списке 2, или любое подмножество по меньшей мере из пяти пиков, выбранных из списка 2. В еще одном варианте осуществления форма 2 демонстрирует по существу такой же профиль рентгеновской порошковой дифракции, как приведен на фигуре 2.
В другом аспекте изобретение относится к кристаллической форме соединения X (форма 3), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 7,3, 18,9 и 21,2; и необязательно дополнительно включая пик при 8,3. В одном из вариантов осуществления форма 3 демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции: 7,3, 13,9, 16,7, 18,9, 20,3, 21,2 и 24,6. В другом варианте осуществления форма 3 демонстрирует по меньшей мере характеристические пики рентгеновской порошковой дифракции, приведенные в списке 3, или любое подмножество по меньшей мере из пяти пиков, выбранных из списка 3. В еще одном варианте осуществления форма C демонстрирует по существу такой же профиль рентгеновской порошковой дифракции, как приведен на фигуре 4.
В другом аспекте изобретение относится к кристаллической форме соединения X (форма 4), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 7,0, 8,6, 19,3 и 20,9. В одном из вариантов осуществления форма 4 демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции: 7,0, 8,6, 15,2, 17,0, 18,0, 19,3, 20,9, 24,5 и 26,9. В другом варианте осуществления форма 4 демонстрирует по меньшей мере характеристические пики рентгеновской порошковой дифракции, приведенные в списке 4, или любое подмножество по меньшей мере из пяти пиков, выбранных из списка 4. В еще одном варианте осуществления форма 4 демонстрирует по существу такой же профиль рентгеновской порошковой дифракции, как приведен на фигуре 5.
В другом аспекте изобретение относится к кристаллической форме соединения X (форма 5), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 7,3, 9,3 и 27,8; и необязательно дополнительно включая пик при 19,9. В одном из вариантов осуществления форма 5 демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции: 7,3, 9,3, 16,3, 18,6, 19,2, 19,9, 21,7, 24,1, 24,9, 27,3, 27,8 и 29,8. В другом варианте осуществления форма 5 демонстрирует по меньшей мере характеристические пики рентгеновской порошковой дифракции, приведенные в списке 5, или любое подмножество по меньшей мере из пяти пиков, выбранных из списка 5. В еще одном варианте осуществления форма 5 демонстрирует по существу такой же профиль рентгеновской порошковой дифракции, как приведен на фигуре 6.
В другом аспекте изобретение относится к кристаллической форме соединения X (форма 6), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 8,1, 9,2, 12,8, 21,2 и 24,7. В одном из вариантов осуществления форма 6 демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции: 8,1, 9,2, 12,8, 13,9, 14,4, 16,7, 20,1, 21,2, 24,7 и 26,6. В другом варианте осуществления форма 6 демонстрирует по меньшей мере характеристические пики рентгеновской порошковой дифракции, приведенные в списке 6, или любое подмножество по меньшей мере из пяти пиков, выбранных из списка 6. В еще одном варианте осуществления форма 6 демонстрирует по существу такой же профиль рентгеновской порошковой дифракции, как приведен на фигуре 7.
В другом аспекте изобретение относится к кристаллической форме соединения X (форма 7), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 6,7, 7,3 и 20,3. В одном из вариантов осуществления форма 7 демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции: 6,7, 7,3, 17,6, 18,0, 20,3 и 24,9. В другом варианте осуществления форма 7 демонстрирует по меньшей мере характеристические пики рентгеновской порошковой дифракции, приведенные в списке 7, или любое подмножество по меньшей мере из пяти пиков, выбранных из списка 7. В еще одном варианте осуществления форма 7 демонстрирует по существу такой же профиль рентгеновской порошковой дифракции, как приведен на фигуре 8.
В другом аспекте изобретение относится к кристаллической форме соединения X (форма 8), которая демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции (выраженные в градусах 2θ): 6,2, 21,8 и 25,9. В одном из вариантов осуществления форма 8 демонстрирует по меньшей мере следующие характеристические пики рентгеновской порошковой дифракции: 6,2, 17,8, 20,7, 21,8 и 25,9. В другом варианте осуществления форма 8 демонстрирует по меньшей мере характеристические пики рентгеновской порошковой дифракции, приведенные в списке 8, или любое подмножество по меньшей мере из пяти пиков, выбранных из списка 8. В еще одном варианте осуществления форма 8 демонстрирует по существу такой же профиль рентгеновской порошковой дифракции, как приведен на фигуре 9.
В одном из аспектов изобретения полиморфные формы по изобретению обладают кристаллическими свойствами и предпочтительно являются по меньшей мере на 50% кристаллическими, более предпочтительно по меньшей мере на 60% кристаллическими, еще более предпочтительно по меньшей мере на 70% кристаллическими и наиболее предпочтительно по меньшей мере на 80% кристаллическими. Степень кристаллизации можно определять общепринятыми способами дифрактометрии рентгеновских лучей или способами инфракрасной спектроскопии.
В определенных вариантах осуществления твердая форма соединения X содержит одну или несколько из форм, описываемых в настоящем документе. Твердая форма соединения X может включать две или более из этих форм, т.е., она может представлять собой смесь из двух или более форм. В определенных вариантах осуществления образец твердой формы в основном состоит из одной формы, выбранной из форм 1-8, что означает, что 50% или более вещества представляют собой одну твердую форму. Относительные количества различных форм в смеси можно определять по данным XRPD. Как описано в настоящем документе, определенные формы могут формироваться или взаимопреобразовываться в подходящих условиях, например, формы 4 и 5, которые могут находиться в виде смеси и могут взаимопреобразовываться в зависимости от относительной влажности и температуры при которых хранят вещество.
В одном из аспектов изобретения полиморфные формы по изобретению являются от 50%, 60%, 70%, 80% или 90% до 95%, 96%, 97%, 98%, 99% или 100% кристаллическими.
По настоящему описанию пики рентгеновской порошковой дифракции (выраженные в градусах 2θ) определяют с использованием рентгеновских лучей из меди с длиной волны 1,5406 Å (альфа1) и 1,5444 Å (альфа2). Положения пиков регистрируют в градусах 2θ, и понятно, что они подвержены небольшим числовым вариациям и, таким образом, следует понимать, что углы подвержены вариации на величину ±0,2°.
Кристаллические формы по настоящему изобретению могут существовать в несольватированной или сольватированной формах. Термин "сольват" в настоящем документе используют для описания молекулярного комплекса, содержащего соединение по изобретению и определенное количество одного или нескольких фармацевтически приемлемых растворителей. Примеры фармацевтически приемлемых растворителей включают этанол и воду. Термин "гидрат" используют, когда растворитель представляет собой воду. Несколько полиморфных форм соединения X, как описано в настоящем документе, представляют собой гидраты.
В одном из аспектов изобретение относится к соли или кристаллической форме, определенных в настоящем документе, для применения в терапии. В другом аспекте изобретение относится к способу лечения посредством терапии, включающей введение нуждающемуся в этом индивидууму фармацевтически приемлемого количества соли или кристаллической формы по изобретению.
В одном из аспектов изобретение относится к применению соли или кристаллической формы, определенных в настоящем документе, в производстве лекарственного средства для применения в терапии.
В одном из вариантов осуществления терапия представляет собой лечение инфекции, вызываемой грамотрицательными бактериями.
Изобретение также относится к способам получения кристаллических форм, описываемых в настоящем документе. Таким образом, в одном из аспектов изобретение относится к способу получения любой из форм 1, 2, 3, 4, 5, 6, 7 и 8, как описано в настоящем документе, включающему кристаллизацию требуемой формы из раствора соединения X с использованием систем растворителя и условий, описанных в примерах, предоставляемых в настоящем документе.
В контексте настоящего изобретения, указания в настоящем документе на "лечения", включают указания на приводящее к выздоровлению, паллиативное и профилактическое лечение, если конкретно не указано противоположное. Термины "терапия, "терапевтический" и "терапевтически" следует трактовать подобным образом.
Как правило, соединение X вводят посредством инъекции или инфузии, и его можно получать для введения посредством растворения соединения X в подходящем количестве воды или водного раствора, такого как раствор декстрозы или солевой раствор, например, изотонический раствор декстрозы или соли. Необязательно сформулированная композиция также может включать модификатор pH, такой как гидроксид натрия, бикарбонат натрия, карбонат натрия, гидроксид калия, карбонат калия, гидроксид кальция, гидроксид магния, меглумин, TRIS или аминокислота (например, лизин, гистидин или аргинин) в количестве, достаточном для обеспечения требуемого pH, такого как pH от 4 до 6. Хотя аминокислоты, используемые в примерах, представляли собой (L)-аминокислоты, вместо них можно использовать (D)-аминокислоты или рацемические смеси. Фармацевтические композиции, содержащие соединение X в растворе, как правило, доводят до pH от 4,5 до 5,5, часто или приблизительно при pH 5, например, от pH 4,8 до 5,2.
В определенных вариантах осуществления соединение X формулируют с модификатором pH в водном растворе, и затем лиофилизируют в твердую форму для хранения и распространения. Для введения можно восстанавливать лиофилизированный лекарственный продукт, добавляя водный носитель, как правило, стерильный водный носитель, такой как вода или изотонический раствор декстрозы или соли или другой в/в раствор, такой как раствор Рингера, лактат Рингера или раствор Хартмана. Таким образом, изобретение также относится к лиофилизату (твердому веществу, полученному посредством лиофилизации), содержащему соединение X и модификатор pH, такой как модификаторы, указанные выше, например, (L)-аргинин, (L)-лизин, меглумин, TRIS или гидроксид натрия. Необязательно можно включать другие эксципиенты, такие как сахароза. В одном из вариантов осуществления получают раствор соединения X и аргинина (два эквивалента), и pH доводят до pH от 4,8 до 5,2 с использованием, например, гидроксида натрия или соляной кислоты по мере необходимости. Затем раствор лиофилизируют до белого или желтоватого твердого вещества, которое стабильно при хранении и которое можно легко восстанавливать подходящим стерильным водным раствором для внутривенного введения.
В другом варианте осуществления получают раствор соединения X, сахарозы и аргинина (два эквивалента) в воде, подходящей для инъекции, и pH доводят до pH от 4,8 до 5,2 с использованием, например, гидроксида натрия или соляной кислоты по мере необходимости. Затем раствор лиофилизируют до белого или желтоватого твердого вещества, которое стабильно при хранении и которое можно легко восстанавливать подходящим стерильным водным раствором для внутривенного введения.
Соли и кристаллические формы по настоящему изобретению можно вводить отдельно или в комбинации с одним или несколькими другими лекарственными средствами. Как правило, их вводят в виде состава в ассоциации с одним или несколькими фармацевтически приемлемыми эксципиентами. Термин "эксципиент" используют в настоящем документе для описания любого ингредиента, отличного от соединения(ий) по изобретению, который может придавать составам функциональные (т.е., контроль скорости высвобождения лекарственного средства) и/или нефункциональные (т.е., технологическая добавка или разбавитель) характеристики. Выбор эксципиента в большой степени зависит от таких факторов, как конкретный способ введения, действие эксципиента на растворимость и стабильность и характер лекарственной формы. Фармацевтические композиции также могут содержать носитель, который по существу представляет собой инертный материал, часто жидкость, используемый для растворения активного ингредиента(ов). Подходящие носители известны в данной области и включают, например, стерильную воду и стерильные растворы солей или декстрозы.
Фармацевтические композиции подходящие для доставки твердых форм соединения X и способы их получения очевидны специалистам в данной области. Такие композиции и способы их получения можно найти, например, в Remington's Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
Как правило, для введения пациентам-людям общая суточная доза соли или кристаллической формы находится в диапазоне от 1000 мг до 10,000 мг, или от 2000 мг до 8000 мг, или от 3000 мг до 8000 мг, или от 4000 мг до 6000 мг в зависимости от состояния индивидуума и таких параметров, как масса тела, возраст и пол индивидуума. Общую суточную дозу можно вводить в однократной дозе или ее можно делить на две или более доз, и она по решению врача может находится вне типичного диапазона, приводимого в настоящем документе. Как правило, суточную дозу доставляют посредством внутривенной инъекции или инфузии, и ее можно вводить в одной, двух, трех или четыре дозах, которые суммарно обеспечивают требую общую суточную дозу. Инфузия может быть быстрой, или ее можно проводить в течение периода продолжительностью приблизительно от 15 минут до 4 часов, как правило, в течение периода продолжительностью 1-3 часа. Типичная схема дозирования предусматривает три или четыре инфузии ежедневно, где каждая длится 0,25-2 часа или 0,25-3 часа, с доставкой от 1 до 2 или от 1 до 2,5 грамм соединения X на дозу, а типичная общая суточная доза составляет 3-8 грамм. Например, по схеме дозирования можно доставлять 2 грамма соединения X на инфузию с тремя часовыми инфузиями в сутки. Альтернативно можно использовать одну инфузию 2-6 грамм или 3-5 грамм в течение 1, или 1,5, или 2, или 2,5, или 3 часов. Приведенные выше дозы основаны на среднем индивидууме-человеке с массой приблизительно от 60 кг до 70 кг, и их можно соответствующим образом масштабировать для других индивидуумов. Лечащий врач может легко определить дозы для индивидуумов, масса которых находится вне этого диапазона, например, грудного ребенка и пожилого человека.
Краткое описание чертежей
Далее изобретение проиллюстрировано приводимыми ниже неограничивающими примерами. В примерах представлены следующие фигуры:
Фигура 1: профиль рентгеновской порошковой дифракции формы 1.
Фигура 2: профиль рентгеновской порошковой дифракции формы 2.
Фигура 3: профиль рентгеновской порошковой дифракции формы 1 после перемешивания с ацетоном, с наложением XRPD для формы 1 для сравнения.
Фигура 4: профиль рентгеновской порошковой дифракции формы 3.
Фигура 5: профиль рентгеновской порошковой дифракции формы 4.
Фигура 6A: профиль рентгеновской порошковой дифракции формы 5 с наложением XPRD для фигуры 4.
Фигура 6B: профиль рентгеновской порошковой дифракции формы 5 для получения в большем масштабе.
Фигура 7: профиль рентгеновской порошковой дифракции формы 6.
Фигура 8: профиль рентгеновской порошковой дифракции формы 7.
Фигура 9: профиль рентгеновской порошковой дифракции формы 8.
Фигура 10: профиль рентгеновской порошковой дифракции формы 9.
Фигура 11: профиль рентгеновской порошковой дифракции формы 10.
Фигура 12: профиль рентгеновской порошковой дифракции формы 11.
Фигура 13: профиль рентгеновской порошковой дифракции формы 12.
Общие экспериментальные характеристики
Каждый образец (несколько миллиграммов) помещают между тремя полимерными пленками (Kapton® и/или полипропилен). Следует отметить, что Kapton® демонстрирует широкий пик со слабой интенсивностью приблизительно при 2θ=5,5°.
Затем образец помещают в дифрактометр PANALYTICAL X'PERT PRO MPD, сконфигурированный в трансмиссионном режиме, и анализируют с использованием указанных ниже условий.
Анализы проводят от 2° до 50° (если не указано иначе).
Излучение: CuKα
Настройки генератора: 40 кВ и 40 мА
Величина шага: 0,026°
Шаги: 1828
Тип измерения: повторное сканирование (3/5/20 раз)
Все исходные вещества, составляющие блоки, реагенты, кислоты, основания, дегидратирующие средства, растворители и катализаторы, используемые для синтеза соединения по настоящему изобретению, являются коммерчески доступными, или их можно получать способами органического синтеза, известными специалисту в данной области (Houben-Weyl 4th Ed. 1952, Methods of Organic Synthesis, Thieme, Volume 21).
Общие условия
Масс-спектры получали на системах LC-MS, SFC-MS или GC-MS с использованием способов ионизации электрораспылением, химического способа и способа электронного удара на ряде устройств со следующими конфигурациями: система СЭЖХ Waters ACQUITY и оборудованная системой ZQ 2000 или SQD MS, где (M+1) относится к протонированному молекулярному иону химической молекулы, (M+) относится к непротонированному четвертичному аммонийному катиону, (M+Na) относится к иону с присоединенным натрием и (M-1) относится к депротонированному молекулярному иону химической молекулы.
Спектры ЯМР получали на спектрометрах ЯМР Bruker AVANCE 500MHz или Varian 400MHz с использованием ICON-ЯМР, под контролем программы TopSpin. Спектры определяли при 298K, если не указано иначе, и соотносили с резонансом растворителя.
Оборудование
Способы MS: с использованием систем ВЭЖХ Agilent 1100 с масс-спектрометром Agilent 6110
Способ 2m_acidic:
Колонка Kinetex C18 50×2,1 мм, 2,6 мкм
Температура колонки 50°C
Элюенты A: H2O, B: ацетонитрил, оба содержат 0,1% TFA
Скорость потока 1,2 мл/мин
Градиент от 2% до 88% B в течение 1,30 мин, 0,15 мин 95% B
Способ 2m_acidic_polar:
Колонка Kinetex C18 50×2,1 мм, 2,6 мкм
Температура колонки 50°C
Элюенты A: H2O, B: ацетонитрил, оба содержат 0,1% TFA
Скорость потока 1,2 мл/мин
Градиент от 1% до 30% B в течение 1,30 мин, 0,15 мин 98% B
Получение соединения X
Промежуточное соединение A: ((2S,3S)-3-(((бензилокси)карбонил)амино)-1-(2,4-диметоксибензил)-4-оксоазетидин-2-ил)метилметансульфонат.
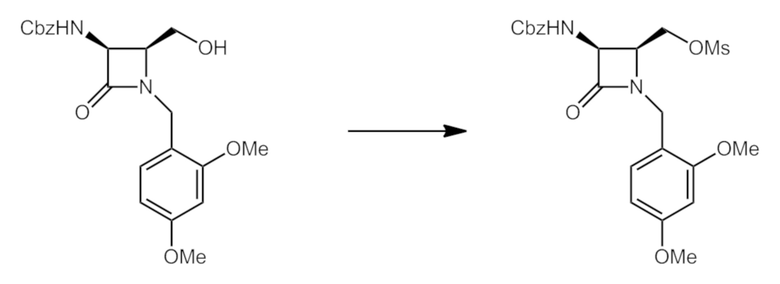
К раствору бензил-((2S,3S)-1-(2,4-диметоксибензил)-2-(гидроксиметил)-4-оксоазетидин-3-ил)карбамата (5,37 г, 13,41 ммоль) и TEA (3,72 мл, 26,8 ммоль) в DCM при 0°C добавляли метансульфонилхлорид (MsCl) (1,15 мл, 14,75 ммоль). После перемешивания при 0°C в течение 1 часа смесь разбавляли водой/DCM и разделяли слои. Водный слой экстрагировали DCM (2×) и объединенные органические слои отмывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Затем неочищенный остаток растворяли в толуоле и концентрировали (2×), получая указанное в заголовке соединение в виде почти белого твердого вещества. В таком виде его использовали в последующих реакциях. LCMS: Rt=0,86 мин, масса/заряд=479,2 (M+1) способ 2m_acidic.
Исходное вещество для этого этапа можно получать с использованием приводимых ниже подходов или определенной комбинации этапов на основе этих подходов. В первом подходе защищенный хиральный альдегид конденсируют с 2,4-диметоксибензиламином с получением хирального имина.
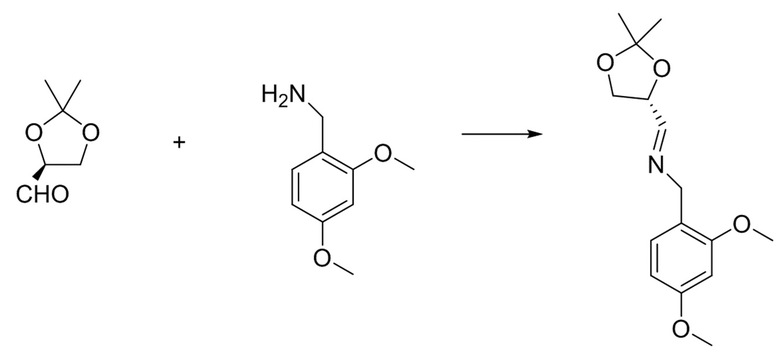
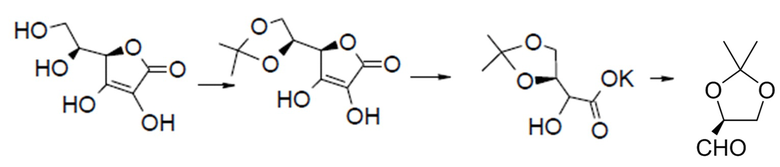
Хиральный альдегид известен, и его можно получать из лимонной кислоты:
Хиральный имин можно подвергать реакции с защищенными формами глицина, такими как эти:
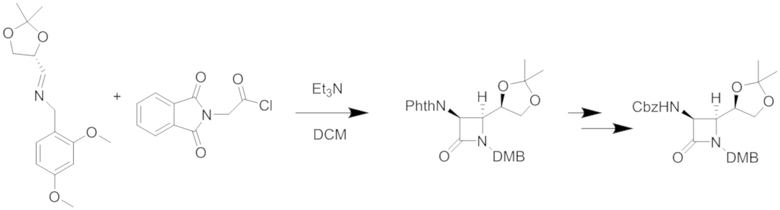
и
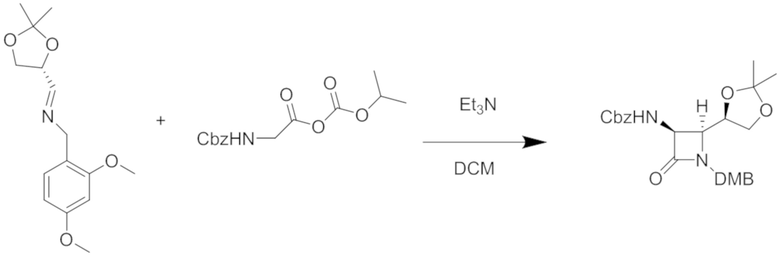
Требуемый смешанный ангидрид для второго варианта можно получать из защищенного CBZ глицина и изопропилхлорформиата в дихлорметане. (DMB относится к 2,4-диметоксибензильной группе). Затем диоксолан этого защищенного промежуточного соединения гидролизуют в умеренных кислых условиях и окисляют до альдегида, который затем можно легко восстанавливать, например, борогидридом натрия с получением двузащещенного первичного спирта, как представлено.

Первичный спирт этого промежуточного соединения можно преобразовывать в уходящую группу, например, обрабатывая йодом и трифенилфосфином, с получением первичного йодида, или обрабатывая основанием и сульфонилхлоридом, с получением, например, мезилата или тозилата. Это активированное промежуточное соединение легко реагирует с гидроксиэтиламином, как приведено на этапе 1 ниже.
Промежуточное соединение B: 3-(((2R,3S)-3-амино-4-оксоазетидин-2-ил)метил)оксазолидин-2-он.
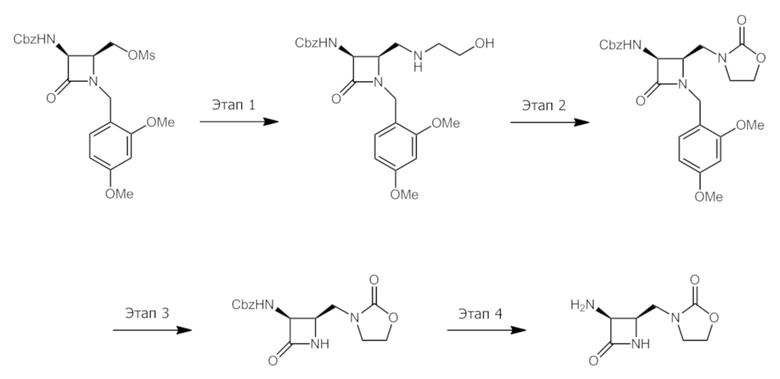
Этап 1: Бензил-((2R,3S)-1-(2,4-диметоксибензил)-2-(((2-гидроксиэтил)амино)метил)-4-оксоазетидин-3-ил)карбамат. К раствору ((2S,3S)-3-(((бензилокси)карбонил)амино)-1-(2,4-диметоксибензил)-4-оксоазетидин-2-ил)метилметансульфоната (6,43 г, 13,4 ммоль) в ацетонитриле (44,8 мл) добавляли этаноламин (8,13 мл, 134 ммоль) с последующим DIEA (7,0 мл, 40 ммоль). Раствор нагревали до 80°C в течение 20 часов, после чего его охлаждали до комнатной температуры, разбавляли EtOAc, промывали водой, сушили над Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке соединения (4,47 г, 75%) в виде белого твердого вещества. LCMS: Rt=0,60 мин, масса/заряд=444,2 (M+1).
Этап 2: Бензил-((3S,4R)-1-(2,4-диметоксибензил)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)азетидин-3-ил)карбамат. К раствору бензил-((2R,3S)-1-(2,4-диметоксибензил)-2-(((2-гидроксиэтил)амино)метил)-4-оксоазетидин-3-ил)карбамата (4,47 г, 10,08 ммоль) в хлороформе (50 мл) добавляли карбонилдиимидазол (CDI) (4,90 г, 30,2 ммоль). После перемешивания при комнатной температуре в течение 30 минут реакционную смесь концентрировали при пониженном давлении. Неочищенный остаток очищали посредством хроматографии на силикагеле (MeOH-DCM, 0-5%) с получением указанного в заголовке соединения (3,84 г, 81%) в виде белой пены. LCMS: Rt=0,76 мин, масса/заряд=470,1 (M+1).
Этап 3: Бензил-((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)азетидин-3-ил)карбамат. Получали аналогично получению в Mastalerz et al. J. Med. Chem. 1988, 31, 1190 с использованием бензил-((3S,4R)-1-(2,4-диметоксибензил)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)азетидин-3-ил)карбамата (3,84 г, 8,18 ммоль), K2S2O8 (3,10 г, 11,5 ммоль) и K2HPO4 (1,852 г, 10,6 ммоль) в ACN:вода (2:1, 136 мл) и нагревая в течение 40 мин при 90°C. Добавляли еще K2S2O8 (663 г, 2,45 ммоль) и K2HPO4 (370 мг, 2,13 ммоль) и смесь нагревали в течение дополнительных 3 часов. Добавляли еще K2S2O8 (332 мг, 1,23 ммоль) и K2HPO4 (185 мг, 1,06 ммоль) и ее нагревали в течение дополнительных 2 часов, после чего ее концентрировали при пониженном давлении, удаляя большинство ACN. Смесь разбавляли насыщенным солевым раствором/EtOAc и слои разделяли. Водный слой экстрагировали EtOAc (3x) и объединенные органические слои сушили над Na2SO4. Неочищенный остаток очищали посредством хроматографии на силикагеле (EtOAc-Гептан, 0-100% затем MeOH-DCM, 10%) с получением указанного в заголовке соединения (1,61 г, 62%) в виде бежевой пены. LCMS: Rt=0,51 мин, масса/заряд=320,0 (M+1) Способ 2m_acidic.
Этап 4: 3-(((2R,3S)-3-амино-4-оксоазетидин-2-ил)метил)оксазолидин-2-он. Получали по Malmström et al. Bioorg. Med. Chem. Lett. 2012, 22, 5293 с использованием бензил-((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)азетидин-3-ил)карбамата (96 мг, 0,30 ммоль) и Pd/C 10% Degussa типа 101 (10%, 64 мг) и водорода в EtOH:MeOH (4:1, 1,5 мл) в течение 1 часа. Неочищенный остаток в таком виде использовали на следующем этапе. LCMS: Rt=0,11 мин, масса/заряд=186,0 (M+1) Способ 2m_acidic.
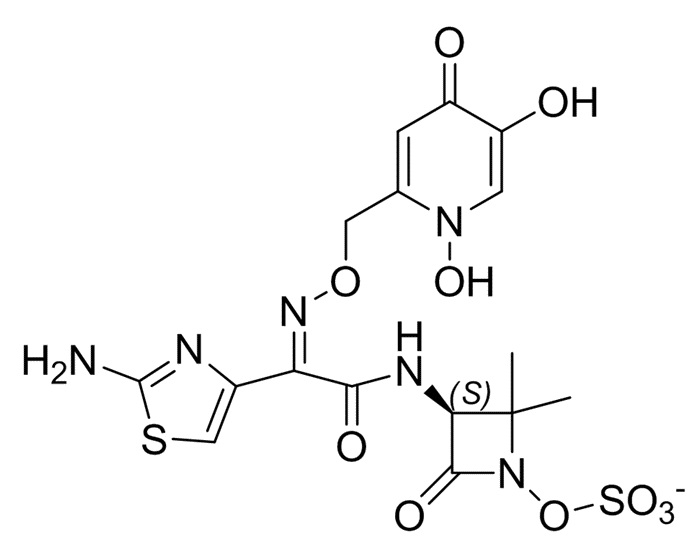
Соединение X: 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновая кислота.
Этап 1: Бензгидрил-1-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)азетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоксилат. К раствору (Z)-2-((1-((бензгидрилокси)карбонил)циклопропокси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты (854 мг, 1,59 ммоль), полученному по опубликованной патентной заявке US2011/0190254, промежуточному соединению B (324 мг, 1,75 ммоль) и HATU (785 мг, 2,07 ммоль) в DMF (7,9 мл) добавляли DIPEA (832 мкл, 4,77 ммоль). Через 1 час перемешивания смесь выливали в воду и экстрагировали EtOAc. В водный слой добавляли насыщенный солевой раствор и его дополнительно экстрагировали этилацетатом (EtOAc) (3×). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный остаток очищали посредством хроматографии на силикагеле (0-10% MeOH-DCM) с получением указанного в заголовке соединения (1,09 г, 97%) в виде бежевой пены. LCMS: Rt=0,97 мин, масса/заряд=705,3 (M+1) Способ 2m_acidic.
Вместо HATU можно использовать ряд других связывающих реагентов, таких как любой из типичных карбодиимидов или CDMT (2-хлор-4,6-диметокси-1,3,5-триазин) и N-метилморфолин с формированием амидной связи, получаемой на этапе 1.
Этап 2: (3S,4R)-3-((Z)-2-((1-((бензгидрилокси)карбонил)циклопропокси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)азетидин-1-сульфоновая кислота. Бензгидрил-1-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)азетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоксилат (1,00 г, 1,42 ммоль) в DMF (7,0 мл) при 0°C обрабатывали SO3•DMF (448 мг, 2,84 ммоль). Через 2 часа перемешивания при комнатной температуре раствор выливали в ледяной насыщенный солевой раствор и экстрагировали EtOAc (3×). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке соединения (условно количественно) в виде белого твердого вещества. LCMS: Rt=0,90 мин, масса/заряд=785,2 (M+1) Способ 2m_acidic.
Этап 3: 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновая кислота.

К раствору (3S,4R)-3-((Z)-2-((1-((бензгидрилокси)карбонил)циклопропокси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)азетидин-1-сульфоновой кислоты (1,10 г, 1,40 ммоль) в DCM (1,5 мл) при 0°C добавляли TFA (5,39 мл, 70,0 ммоль) и после 10 минут ледяную баню удаляли. Через 1 час при комнатной температуре добавляли дополнительный TFA (3,24 мл, 42,0 ммоль) и раствор разбавляли DCM и после дополнительных 30 минут концентрировали при пониженном давлении. Необязательно для реакции с TFA можно добавлять анизол для помощи в уменьшении образования побочного продукта, что может увеличить выход требуемого продукта на этом этапе. Неочищенный остаток очищали посредством препаративной ВЭЖХ с обращенной фазой (XSelect CSH, 30×100 мм, 5 мкм, колонка C18; ACN-вода с модификатором 0,1% муравьиной кислотой, 60 мл/мин) с получением указанного в заголовке соединения (178 мг, 23%) в виде белого порошка. LCMS: Rt=0,30 мин, масса/заряд=518,9 (M+1) Способ 2m_acidic; 1H ЯМР (400 МГц, DMSO-d6) δ 9,27 (д, J=9,0 Гц, 1H) 6,92 (с, 1H) 5,23 (дд, J=9,1, 5,7 Гц, 1H) 4,12-4,23 (м, 3H) 3,72-3,62 (м, предположительно 2H; замаскированный водой) 3,61-3,52 (м, предположительно 1H; замаскированный водой) 3,26 (дд, J=14,5, 5,9 Гц, 1H) 1,36 (с, 4H). 1H ЯМР (400 МГц, D2O) δ 7,23 (с, 1H), 5,48 (д, J=5,8 Гц, 1H), 4,71-4,65 (м, 1H), 4,44 (т, J=8,2 Гц, 2H), 3,89-3,73 (м, 3H), 3,54 (дд, J=14,9, 4,9 Гц, 1H), 1,65-1,56 (м, 2H), 1,56-1,46 (м, 2H). Продукт этого процесса является аморфным. Соединение X, в дополнение к растворителям, описанным ниже, можно кристаллизовать из ацетона, этанола, цитратного буфера при pH 3 (50 мМ) или ацетатного буфера при pH 4,5 (50 мМ).
Оборудование
DSC: Pyris Diamond DSC, газообразн азот (20 мл/мин)
TGA: Pyris 1 TGA, газообразный азот (20 мл/мин), сканирование с 10°C/мин
XRPD: X'Pert Pro MPD Panalytical, анод из Cu, 40кВ при токе 40 мА
Анод трубки: (Cu)
Напряжение генератора: 40 кВ
Ток трубки: 40 мА
Стартовый угол [2θ]: 3
Конечный угол [2θ]: 40
Время сканирования 2 минуты
Форма 1 и форма 2
Соединение X получали, как описано выше, и кристаллизовали из растворителя с получением веществ с профилем рентгеновской порошковой дифракции (XRPD) на фигуре 1 или фигура 2. Они представляют два отдельные партии кристаллического вещества, и их в настоящем документе обозначают как форма 1 и форма 2. Формы 1 и 2 демонстрируют некоторые сходные пики XRPD, но XRPD формы 2 демонстрирует уширенные линия, так что продукт, идентифицируемый как форма 2, может содержать определенное вещество формы 1, или оба образца могут представлять собой смеси кристаллических форм. Эти две формы по существу являлись безводными, содержа менее чем приблизительно 1% воды по массе в соответствии с анализом Karl Fischer.
Соединение X формы 1 (1,2 г) суспендировали в 12 мл ацетона и перемешивали в течение 3 суток при 20°C. По-видимому, образец не изменяется: хотя происходило определенное уширение линий XRPD продукта (фигура 3) все еще в основном соответствовала форме 1.
Образец с XRPD на фигуре 1 демонстрировал сильный экзотермический эффект при DSC приблизительно при 205°C и постепенную потерю массы при TGA, составляя приблизительно до 2% потери при 180°C.
Образец, дающий XRPD на фигуре 2 (форма 2), демонстрировал сильный экзотермический эффект при DSC приблизительно при 203°C, и немного большую потерю массы (3,7%) при 160°C по сравнению с формой 1.
Список 1: список пиков XRPD для формы 1 (2тета: наиболее интенсивные пики подчеркнуты)
1 6,6
2 11,1
3 13,4
4 14,4
5 15,2
6 16,6
7 17,9
8 18,8
9 20,3
10 22,5
11 23,3
12 25,1
13 27,7
14 28,9
15 30,2
Форму 1 также можно охарактеризовать подмножеством этих пиков, например, пиками при 6,6, 13,4 и 18,8.
Список 2: список пиков XRPD для формы 2 (2тета: наиболее интенсивные пики подчеркнуты)
1 6,7
2 7,5
3 11,3
4 13,3
5 13,7
6 15,3
7 17,9
8 18,7
9 19,3
10 20,0
11 22,8
12 24,8
13 27,9
Форму 2 также можно охарактеризовать подмножеством этих пиков, например, пиками при 7,5, 19,3 и 20,0.
Форма 3
Соединение X формы 1 суспендировали в метаноле и в течение нескольких минут оно преобразовывалось в липкий твердый продукт. Продукт демонстрировал профиль XRPD, приведенный на фигуре 4, и его в настоящем документе обозначают как форма 3. Следует отметить, что после более длительного перемешивания в метаноле в большем масштабе получали другую форму (форма 8), как описано ниже; таким образом, форма 3 может представлять собой переходную форму или смесь, формируемую по мере преобразования формы 1 в этих условиях.
Список 3: список пиков XRPD для формы 3 (2тета: наиболее интенсивные пики подчеркнуты)
1 6,6
2 7,3
3 8,3
4 12,7
5 13,3
6 13,9
7 16,7
8 18,9
9 20,3
10 21,2
11 22,2
12 23,9
13 24,6
14 25,1
15 27,2
16 28,0
Форму 3 также можно охарактеризовать подмножеством этих пиков, например, пиками при 7,3, 18,9 и 21,2.
Форма 4
Соединение X формы 1 (1,2 г) суспендировали в воде (12 мл) и перемешивали при 20°C в течение 3 суток. Продукт демонстрировал профиль XRPD, приведенный на фигуре 5, и его в настоящем документе обозначают как форма 4. XRPD получали у образца, высушенного фильтровальной бумагой. Анализ TGA для формы 4 демонстрирует постепенную потерю массы, начиная приблизительно с 40°C и становясь быстрой приблизительно при 100°C до плато приблизительно при 60% от исходной массы при 120-170°C. Выше этой температуры наблюдают постепенную потерю массы. Ранняя потеря массы соответствует потери воды из формы 4, и с DSC, которая демонстрирует сильный эндотермический эффект в том же диапазоне температур и плато приблизительно при 110-180°C. Подобным образом, анализ динамической сорбции паров (DVS) для формы 4 демонстрирует быструю потерю приблизительно 20% массы образца, когда относительная влажность падала приблизительно до 50%, после чего циклически изменение до более низкой, затем более высокой, затем более низкой, а затем более высокой относительной влажности приводило к соответствующему уменьшению, увеличению, уменьшению и увеличению массы образца с минимальной массой приблизительно 65% от исходной и максимумом приблизительно при 80% от исходной массы. Таким образом, форма 4 является гидратированной формой и изменяется в зависимости от относительной влажности. Степень гидратации образца варьирует в зависимости от относительной влажности от безводной формы при очень низкой влажности до тригидрата при относительной влажности приблизительно 20-50%, до гексагидрата при относительной влажности выше 60%. Образец формы 4, высушенный при 0% относительной влажности, демонстрирует XRPD в соответствии с формой 1.
Список 4: список пиков XRPD для формы 4 (2тета: наиболее интенсивные пики подчеркнуты)
1 7,0
2 8,6
3 13,7
4 15,2
5 16,1
6 17,0
7 18,0
8 19,3
9 20,9
10 24,5
11 26,9
Форму 4 также можно охарактеризовать подмножеством этих пиков, например, пиками при 7,0, 8,6, 19,3 и 20,9.
Форма 5
Образец формы 4 сушили в потоке сухого воздуха в течение суток, получая порошок, который демонстрировал XRPD, приведенную на фигуре 6A. Это вещество в настоящем документе обозначают как форма 5. DSC для формы 5 демонстрирует крутую экзотерму, приблизительно при 204°C, и приблизительно при этой температура образец начинает чернеть, что указывает на разрушение. TGA демонстрирует приблизительно 7% потерю массы от 45°C до 160°C. Анализ Karl Fisher демонстрирует содержание воды для формы 5 9,6%, что соответствует тригидрату, но образец демонстрирует потерю массы при относительной влажности приблизительно ниже 30%; а при относительной влажности приблизительно 80% он преобразуется в форма 4 в пределах одних суток.
Таким образом, формы 1, 4 и 5 соединения X, по видимому, взаимопреобразуются по мере изменения относительной влажности, и может существовать смесь этих гидратированных форм. Форма 5 представляет собой предпочтительную форму для обработки при температуре окружающей среды приблизительно 25°C при производстве, так как она кристаллизуется в виде хорошо ведущего себя твердого вещества, которое является более стабильным при хранении и переработке, чем другие формы, при условии поддержания подходящей относительной влажности приблизительно 20-50%, предпочтительно 30-40%. В пределах этих диапазонов относительной влажности вещество преимущественно представляет собой тригидрат и соответственно хорошо себя ведет и стабилен при обработке и хранении без особых мер предосторожности.
Получение формы 5 в более крупном масштабе
Воду (20 мл) и THF (40 мл) смешивали в колбе и добавляли соединение X (10 г, чистоты приблизительно 91% по данным ВЭЖХ) с перемешиванием при 25°C с получением прозрачного желтого раствора. 20 мг образца формы 5 (см. выше) добавляли в качестве затравки и смесь перемешивали в течение 50 минут. Затем медленно добавляли THF (140 мл) в течение 1 часа и смесь перемешивали в течение дополнительных 2 часов. Суспензию фильтровали и сырой осадок отмывали холодной (<5°C) водой, затем сушили при 20-25°C в течение 11 часов при давлении 10 кПа с получением 6,6 г тригидрата формы 5, чистота которого составляла 97,8% по данным ВЭЖХ. Образец демонстрировал профиль XRPD, приведенный на фигуре 6B. Он демонстрирует стабильность при хранении при относительной влажности 25-50%, предпочтительно при относительной влажности 30-40%; при меньшей или большей относительной влажности он может преобразовываться в другие гидратированные состояния, как описано в настоящем документе. Пересушивание приводит к форме, которая разрушается при комнатной температуре в пределах нескольких часов, дополнительно демонстрируя преимущество гидратированных форм в стабильности.
Список 5: список пиков XRPD для формы 5 (2тета: наиболее интенсивные пики подчеркнуты)
1 7,3
2 9,3
3 12,0
4 13,6
5 15,8
6 16,3
7 18,0
8 18,6
9 19,2
10 19,9
11 20,2
12 21,7
13 23,0
14 23,5
15 24,1
16 24,9
17 27,3
18 27,8
19 29,8
Форму 5 также можно охарактеризовать подмножеством этих пиков, например, пиками при 7,3, 9,3 и 27,8; и необязательно пиками при 7,3, 9,3, 19,9 и 27,8.
Форму 5 (тригидрат) альтернативно можно охарактеризовать монокристалической рентгеновской структурой, которая является моноклинической, с пространственной группой P2(1), с постоянными решетки a=13,121(4) Å; b=7,400(3) Å; c=25,017(8) Å (α=90°; β=96,037°; γ=90°); и объемом элементарной ячейки 2415,6(14) Å3. Данные для данной структуры получали при длине волны 1,54178 Å, 100°K; диапазоне тета от 3,99° до 68,44°; получали 47822 отображений. Монокристаллическая структура подтверждает присутствие трех молекул воды на молекулу соединения X в элементарной ячейке, с молекулами воды, расположенными в каналах.
Форма 6
Соединение X формы 1 (1,2 г) суспендировали в DMSO:воде (отношение 25:75 об./об., 12 мл) и перемешивали при 20°C в течение 3 суток. Образец твердого вещества собирали и сушили фильтровальной бумагой. Продукт демонстрировал профиль XRPD, приведенный на фигуре 7, и его в настоящем документе обозначают как форма 6. DSC и TGA соответствуют потере растворителя приблизительно до 130°C и снова от 150°C до 200°C с последующим разрушением выше 200°C.
Список 6: список пиков XRPD для формы 6 (2тета: наиболее интенсивные пики подчеркнуты)
1 6,6
2 8,1
3 9,2
4 12,8
5 13,9
6 14,4
7 16,7
8 17,8
9 19,4
10 20,1
11 21,2
12 24,7
13 26,6
14 27,3
Форму 6 также можно охарактеризовать подмножеством этих пиков, например, пиками при 8,1, 9,2 и 12,8; и необязательно дополнительными пиками при 21,2 и 24,7.
Форма 7
Получали соединение X формы 6, как описано выше, сушили в течение суток в потоке сухого воздуха. Полученный порошок демонстрировал профиль XRPD, приведенный на фигуре 8, и его в настоящем документе обозначают как форма 7. DSC демонстрирует большой экзотермический эффект, начиная приблизительно при 134°C и продолжая приблизительно до 170°C. 1H ЯМР демонстрирует присутствие приблизительно 2 эквивалентов DMSO и определенного к количества воды (3,6%). Титрование Karl Fisher подтверждает присутствие 3,6% воды, соответствующих 1 эквиваленту. Таким образом, эта форма представляет собой сольват.
Список 7: список пиков XRPD для формы 7 (2тета: наиболее интенсивные пики подчеркнуты)
1 6,7
2 7,3
3 9,2
4 10,6
5 12,8
6 14,4
7 16,0
8 17,6
9 18,0
10 19,5
11 20,0
12 20,3
13 24,9
14 27,0
15 27,6
Форму 7 также можно охарактеризовать подмножеством этих пиков, например, пиками при 6,7, 7,3 и 20,3.
Форма 8
Соединение X формы 1 (1,2 г) суспендировали в метаноле (12 мл) и перемешивали при 20°C в течение 3 суток. Образец твердого вещества собирали и получали профиль XRPD, приведенный на фигуре 9 (без сушки), который в настоящем документе обозначают как форма 8. Когда образец формы 8 сушили в потоке сухого воздуха пики значительно уширялись, но, как правило, оставались приблизительно в тех же положениях. TGA для высушенного образца демонстрирует постепенную потерю массы (приблизительно 4%) до 140°C и более резкую потерю массы, начиная приблизительно со 170°C. DSC демонстрирует сильный экзотермический эффект приблизительно при 172°C that, что может быть ассоциировано с разрушением образца.
Список 8: список пиков XRPD для формы 8 (2тета: наиболее интенсивные пики подчеркнуты)
1 6,2
2 12,5
3 16,8
4 17,8
5 19,5
6 20,7
7 21,8
8 25,9
Форму 8 также можно охарактеризовать подмножеством этих пиков, например, пиками при 6,2, 21,8 и 25,9.
Форма 9
Соединение X формы 3 суспендировали в ацетоне и воде при температурах от 25°C до 40°C. Отношение воды и ацетона варьировали от 2:98 до 10:90. После уравновешивания в течение 24 часов в каждом случае получали форму низкокристаллического гетеросольвата. Хотя XRPD варьировал при изменении отношения воды и ацетона, все образцы демонстрировали спектры XRPD с широкими максимумами, а не резкими пиками. На фигуре 10 представлена XRPD для образца уравновешенного в воде/ацетоне 10:90 при 40°C в течение 24 часов, и в приводимой ниже таблице обобщены данные XRPD для этого образца. Эту твердую форму в настоящем документе обозначают как форма 9.
Форма 9 можно охарактеризовать подмножеством этих пиков, например, пиками определяемыми в таблице выше как "сильная" относительная интенсивность, например, пиками при 6,3 и 12,6 и необязательно одним или несколькими пиками в таблице со средней относительной интенсивностью, такими как пики при 22,1, 22,3, 23,1, 27,0 и 27,5
Форма 10
Соединение X формы 1 (ангидрат) подвергали воздействию 43% относительной влажности в течение одних суток или более. Кристаллический продукт, на основе определения содержания воды по-видимому, представляет собой сесквигидрат (соединение X-1,5 H2O). Следует отметить, что форма 5, тригидрат, в подобных условиях является стабильной, начиная с ангидрита, по-видимому, она в этих условиях уравновешивается в виде сесквигидрата и остается в этой форме при хранении при той же относительной влажности в течение по меньшей мере 14 суток. Эта кристаллическая форма демонстрирует спектр XRPD, приведенный на фигуре 11, и ее в настоящем документе обозначают как форма 10. В таблице ниже обобщены основные пики в XRPD этого образца.
Форму 10 можно охарактеризовать пиками XRPD в приводимой выше таблице с относительными интенсивностями от средней до сильной, например, пиками при 6,6, 11,0, 13,3, 15,6, 16,5, 18,6, 22,2, 23,4 и 27,4. Также ее можно охарактеризовать подмножеством этих пиков, например, пиками с относительной интенсивностью 40 или более, например, пиками при 6,6, 11,0, 16,5, 22,2 и 23,4, или подмножеством по меньшей мере из 3 или 4 этих пиков.
Форма 11
Соединение X формы 5, тригидрат, подвергали относительной влажности 22% в течение приблизительно 3 суток с получением кристаллического твердого вещества, характеризуемого на основе анализа содержания воды как дигидрат (соединение X-2H2O). Это вещество легко преобразуется в тригидрат (форма 5), если его подвергать воздействию относительной влажности приблизительно выше 40%. Дигидрат, обозначаемый в настоящем документе как форма 11, демонстрировал спектр XRPD, приведенный на фигуре 12: основные пики этого спектра XRPD перечислены в приводимой ниже таблице.
Форма 11 характеризуется пиками XRPD в этой таблице с относительными интенсивностями 20 или более, или альтернативно этими пиками с относительной интенсивностью по меньшей мере 25, например, пиками при 7,4, 9,7, 17,0, 19,5, 22,2, 26,3, 28,1 и 29,3, или подмножеством из 3, 4 или 5 этих пиков с относительной интенсивностью в таблице по меньшей мере 26 или по меньшей мере 28.
Форма 12
Соединение X формы 5 подвергали воздействию воздуха с относительной влажностью 65% в течение приблизительно 3 суток, получая кристаллическое вещество, характеризуемое на основе анализа содержания воды как тетрагидрат. Это вещество легко преобразуется в тригидрат (форма 5), если относительную влажность уменьшить приблизительно до 40%. Тетрагидрат, обозначаемый в настоящем документе как форма 12, демонстрировал спектр XRPD, приведенный на фигуре 13: основные пики этого спектра XRPD перечислены в приводимой ниже таблице.
Форму 12 можно охарактеризовать пиками XRPD с относительной интенсивностью в таблице выше по меньшей мере 40, т.е. пиками XRPD при 7,3, 17,8, 19,0, 19,8, 20,4, 24,0, 24,7, 24,9, 27,2, 27,8 и 32,1. Альтернативно, ее можно охарактеризовать подмножеством по меньшей мере из 3, или по меньшей мере из 4, или по меньшей мере из 5 этих пиков. Форму 12 также можно охарактеризовать пиками XRPD, описываемыми в таблице как пики с сильной относительной интенсивностью, т.е., пиками при 7,3, 19,0, 20,4, 24,0, 24,7 и 27,2.
Показано, что образцы гидратов соединения X, включая формы 5, 11 и 12 легко взаимопреобразуются по мере изменения относительной влажности приблизительно от 22% до приблизительно 92%. Следует отметить, что при 92% относительной влажности соединение X, по-видимому, представляет собой смесь кристаллической формы, характеризуемой как гексагидрат, смешанной с тетрагидратом формы 12. Для сравнения ангидрид (форма 1) предпочтительно преобразуется в сесквигидрат при возрастании относительной влажности до 43%, и преобразуется в тригидрат и тетрагидрат при более высокой относительной влажности.
Фармацевтические композиции соединения X
Соединение X (500 мг) и L-аргинин (332,5 мг) комбинируют во флаконе. Добавляют сахарозу (кристаллическую, не содержащую пирогенов сахарозу: 1000 мг) вместе с водой, подходящей для инъекции (8,00 мл). Если необходимо, pH раствора доводят с использованием 1,0Н HCl или 1,0Н NaOH с получением pH 5,0±0,5, предпочтительно pH 5,0±0,2. Это приводит к получению раствора, содержащего приблизительно 62,5 мг/мл соединения X в виде аргининовой соли. Если необходимо, этот раствор можно фильтровать и можно лиофилизировать с получением белого или почти белого твердого вещества (лиофилизата). Лиофилизированное твердое вещество можно восстанавливать стерильной водой или фармацевтически приемлемым водным носителем, таким как изотонический солевой раствор или декстроза с получением раствора, подходящего для внутривенного введения. Лиофилизат следует хранить в контейнере, который исключает воздействие света, для защиты лиофилизата от фотодеструкции. Этот процесс можно проводить в увеличенном или уменьшенном масштабе с получением единиц дозирования для хранения и распространения, или большого количества вещества, которое можно дополнительно обрабатывать, как требуется. Для проведения в увеличенном масштабе важен контроль температуры: соединение X в растворе перед добавлением аргинина или другого основания следует поддерживать при температуре ниже 10°C, предпочтительно от 0°C до 8°C, и более предпочтительно от 2°C до 8°C, так как соединение подвержено гидролитическому разрушению в воде в отсутствие основания или вне диапазона pH 4-6.
В одном из вариантов осуществления смесь по приводимому выше примеру получают, как описано, с использованием тригидрата (формы 5) соединения X в количестве, которое составляет приблизительно 500 мг безводного соединения X и лиофилизируют во флакон, которые затем герметизируют для хранения и распространения, предпочтительно с использованием пробки из бутилрезины (например, пробки D777), где лиофилизат в каждом флаконе содержит приблизительно 500 мг соединения X. Флаконы с лиофилизатом до использования хранят при комнатной температуре или ниже.
Альтернативный состав, подходящий для в/в инъекции, содержит соединение X (100 мг) и 0,5Н бикарбонат натрия (0,75 мл), и, если необходимо, регулятор pH (1 Н NaOH или 1 Н HCl по мере необходимости) с доведением pH приблизительно до 5,5 (от pH 5 до pH 6)с определенным количеством воды для инъекций, достаточным для получения конечной концентрации 100 мг/мл.
Стабильность соединения X
Соединение X является стабильным в твердой форме, но соли соединения X в растворе являются более стабильными, чем форма свободной кислоты; таким образом, важно определить подходящие фармацевтически приемлемые соли для использования для введения. Соли соединения X получали, добавляя основание (1,0 или 2,0 эквив.) к соединению X в воде и лиофилизируя раствор. Полученные, таким образом, твердые вещества по данным XRPD являются аморфными. Таким образом, формирование солей соединения X пытались проводить с использованием гидроксида натрия, (L)-лизина, (L)-аргинина, гидроксида кальция, меглумина и гидроксида магния. Выявлено, что натриевые соли и аргининовые соли являются особенно стабильными и, таким образом, желательными в качестве форм для введения в водных средах, как правило, используемых для внутривенных инъекций и инфузий.
Образцы динатриевой соли и двойной соли (L)-аргинина в виде твердых веществ подвергали тестам стабильности при 25°C и 40°C. Исходно образец натриевой соли по данным ВЭЖХ являлся на 97,2% чистым, и через 6 недель при 25°C, он все еще был на 96,2% чистым. То же вещество, хранимое при 40°C через 3 недели было на 94,8% чистым, и через 6 недель на 93,6% чистым. Значимые примеси, которые появляются или увеличиваются в количестве при исследовании выявляют с относительным временем удержания (RRT) 0,34 и 1,11 (с соединением X, определенным как RRT=1).
Исходно аргининовая соль по данным ВЭЖХ была на 97,3% чистой, и через 6 недель при 25°C была чистой на 96,3%. При 40°C ее чистота падала до 95,1% через 3 недели и до 94,2% через 6 недель. Значимые примеси, которые появляются или увеличиваются в количестве в течение этого исследования, выявляют с относительным временем удержания (RRT) 1,09, 1,11 и 1,13 (с соединением X, определенным как RRT=1). Профили ВЭЖХ этих образцов в этих исследованиях стабильности через 6 недель при 25°C и 40°C, соответственно, приведены на фигурах 10 и 11. Нижний профиль на каждой фигуре представляет собой образец, используемой для определения предела количественного определения (LOQ); следующий профиль выше, который представляет собой образец соединения X, используемый для формирования соли; следующий профиль выше, который представляет собой профиль для аргининовой соли через 6 недели; и следующий (верхний) профиль представляет собой натриевую соль через 6 недель.
Условия ВЭЖХ для исследований стабильности (фигуры 10 и 11):
Система Agilent 1290 с УФ-детектором при 260 нм
Колонка Acquity HSS T3, с ID 100 мм × 2,1 мм; размер частиц 1,8 мкм (поставляемая Waters)
Температура колонки: 40°C
Подвижные фазы
A: 0,05% TFA в воде
B: 0,05% TFA в метаноле
Скорость потока: 0,45 мл/мин
Градиент (отношение A/B): 97:3 в течение 8 минут; 75:25 в течение 3 минут; 0:100 в течение 1 минуты
Также выявлено, что соединение X подвержено фотохимическому разрушению; таким образом, соединение X для наилучшего срока хранения следует хранить в темных или непрозрачных контейнерах. В одном из вариантов осуществления изобретения соединение X упаковывают в контейнере, который в значительной степени уменьшает воздействие света, предпочтительно в атмосфере при относительной влажности 25-50% и более предпочтительно при относительной влажности 30-40% или 30-45%.
Биологическая активность
Скрининг и культуры бактерий
Бактериальные изоляты культивировали из замороженных при -70°C хранимых культур посредством двух последовательных пересевов ночных культур при 35°C в воздухе окружающей среды на 5% кровяном агаре (Remel, Lenexa, Kans.). Контроль качества и P. aeruginosa ATCC 27853) получали в American Type Culture Collection (ATCC; Rockville, Md.), а PAO1 получали у Dr. K. Poole.
Конструирование изогенных штаммов Escherichia coli штаммов NB27273-CDY0026 (исходный), NB27273-CDY0033 (KPC2) и NB27273-CDY0030 (SHV12)
Штамм NB27273 (BW25113 pspB::Kmr) получали из коллекции вставок траспозонов Keio. Штамм содержал ген pspB, замененный маркером устойчивости к канамицину (BW25113 pspB::Kmr). Этот штамм избавляли от транспозона pspB посредством рекомбиназы FLP по опубликованной методологии. Полученный штамм, BW25113 pspB, использовали в качестве хозяина для мультикопийных векторов, экспрессирующих ключевые β-лактамазы. Мультикопийные плазмиды, обуславливающие конститутивную экспрессию β-лактамаз получали следующим образом: синтетические, гены с оптимизированными кодонами, кодирующие β-лактамазы KPC2 и SHV12 were E. coli получали посредством DNA2.0 (Palo Alto, CA). Каждый из синтетических фрагментов конструировали так, чтобы он содержал участки рестрикции NotI и NcoI на своих концах, обеспечивая лигирование в расщепленное NotI/NcoI производное pET28a(+) для экспрессии белка. Вставки в эти векторы служили матричной ДНК для амплификации посредством ПЦР гена, кодирующего KPC2 и SHV12, с использованием пар праймеров E225 (tcgcCTCGAGgcgactgcgctgacgaatttgg) (SEQ ID NO:1) и E202 (aatcGAATTCttactgaccattaacgcccaagc) (SEQ ID NO:2) и E227 (tcgcCTCGAGgcgagcccgcaaccgctgga) (SEQ ID NO:3) и E204 (aatcGAATTCttaacgctgccagtgctcaatc) (SEQ ID NO:4), соответственно. Нуклеотидные последовательности с оптимизированными кодонами и соответствующая информация для распознавания праймеров, приведены ниже:
SHV12
atgggccatcatcatcatcatcacagcagcggcctggaagttctgttccaggggcccgcgagcccgcaaccgctggagcagatcaagcagtctgagagccagctgagcggccgtgtgggtatgatcgagatggatctggcttccggccgtacgctgacggcatggcgtgccgacgaacgtttcccgatgatgtcgacctttaaagttgttctgtgtggtgcggtcttggcacgtgtagacgcgggtgacgaacaactggagcgcaagatccattaccgccaacaggacttggtcgactacagcccggttagcgaaaagcacctggcggatggcatgaccgtgggtgaattgtgcgccgctgcgattaccatgagcgacaatagcgcggctaatctgctgttggcgaccgttggtggcccagcgggcttgaccgcatttctgcgtcaaatcggcgataatgttacgcgtctggatcgctgggaaacggagctgaacgaggcactgccgggtgatgcccgtgataccacgactcctgctagcatggcagcgaccctgcgtaaactgctgaccagccagcgtctgagcgcacgtagccaacgccagctgctgcaatggatggtggatgaccgcgtggcgggtccgctgatccgctccgtcctgccagcaggctggttcattgcggacaaaactggtgcctctaagcgtggtgcgcgtggtatcgtcgcgctgctgggtccgaacaacaaagccgaacgtattgtggttatctatctgcgcgacaccccggcaagcatggccgagcgcaaccagcaaattgcgggcattggtgcggcactgattgagcactggcagcgttaaCGCCGGCG (SEQ ID NO:5)
E227 tcgcCTCGAGgcgagcccgcaaccgctgga (SEQ ID NO:6)
E204 aatcGAATTCttaacgctgccagtgctcaatc (SEQ ID NO:7)
Обратная комплементарная последовательность E204 gattgagcactggcagcgttaaGAATTCgatt (SEQ ID NO:8)
KPC2
ATGGGCCATCATCATCATCATCACAGCAGCGGCCTGGAAGTTCTGTTCCAGGGGCCCGCGACTGCGCTGA
CGAATTTGGTGGCCGAGCCGTTCGCGAAATTGGAGCAAGATTTTGGTGGTTCGATCGGTGTCTACGCGAT
GGACACCGGTAGCGGTGCCACCGTGAGCTACCGTGCCGAAGAGCGTTTTCCGCTGTGTAGCTCTTTCAAG
GGTTTTCTGGCCGCAGCCGTGCTGGCACGCAGCCAACAGCAAGCGGGCCTGCTGGACACCCCGATCCGTT
ACGGCAAAAATGCGCTGGTTCCGTGGAGCCCGATTAGCGAAAAGTACCTGACCACCGGCATGACGGTGGC
GGAGTTGAGCGCTGCGGCGGTTCAGTATTCCGATAACGCTGCGGCAAATCTGCTGCTGAAAGAACTGGGC
GGTCCAGCGGGTCTGACGGCTTTCATGCGTTCTATTGGCGACACCACCTTTCGCTTGGACCGCTGGGAGC
TGGAGCTGAACAGCGCGATTCCGGGCGACGCACGTGATACGAGCAGCCCGCGTGCAGTGACCGAGAGCCT
GCAGAAGCTGACCCTGGGCAGCGCACTGGCCGCACCGCAGCGCCAACAGTTCGTCGATTGGCTGAAGGGT
AACACCACCGGTAACCATCGTATTCGCGCAGCGGTCCCGGCTGATTGGGCAGTTGGTGACAAGACTGGTA
CGTGCGGCGTTTATGGTACGGCGAATGACTACGCGGTTGTTTGGCCTACGGGTCGTGCGCCGATCGTCCT
GGCGGTGTATACCCGTGCTCCGAACAAAGACGATAAACACTCCGAAGCGGTCATCGCCGCAGCAGCGCGT
CTGGCCCTGGAAGGCTTGGGCGTTAATGGTCAGTAACGCCGGCG (SEQ ID NO:9)
E225 tcgcCTCGAGgcgactgcgctgacgaatttgg (SEQ ID NO:10)
E202 aatcGAATTCttactgaccattaacgcccaagc (SEQ ID NO:11)
Обратная комплементарная последовательность E202 gcttgggcgttaatggtcagtaaGAATTCgatt (SEQ ID NO:12)
ПОДЧЕРКНУТО=ДНК, кодирующая BL
Затем продукты ПЦР расщепляли XhoI и EcoRI и лигировали в расщепленную подобным образом плазмиду pAH63-pstS(BlaP). Плазмида pAH63-pstS(BlaP) представляет собой производное плазмиды pAH63 (J. Bacteriol:183(21): 6384-6393), полученное клонированием кодирующей промотор и сигнальный пептид TEM-1 (bla) области плазмиды pBAD (J Bacteriol. 1995 Jul. 177(14):4121-30) в плазмиду pAH63. Этот фрагмент клонировали посредством ПЦР с pBAD с использованием пары праймеров E192 (ttcaCTGCAGtgaacgttgcgaagcaacggC) (SEQ ID NO:13) и E194 (TCGAggatcctcgagagcaaaaacaggaaggcaaaatgccg) (SEQ ID NO:14), расщепляли PstI и BamHI и вставляли в расщепленную подобным образом плазмиду pAH63. Таким образом, экспрессия β-лактамаз с основанных на pAH63-pstS(BlaP) конструкций является конститутивной, и добавлена сигнальная последовательность для направления этих белков в периплазму. Основанные на плазмиде pAH63 векторы используют для вставки в геном одной копии, однако для обеспечения более высоких уровней экспрессии для обеспечения более чувствительной детекции восприимчивости соединений к β-лактамазам, экспрессионные вставки, содержащиеся в этих векторах, перемешали в репликационный мультикопийный вектор pBAD-Kan (J Bacteriol. 1995 Jul. 177(14):4121-30). Для осуществления этого вставки, содержащие гены β-лактамаз с ассоциированным промотором и сигнальными последовательностями TEM амплифицировали посредством ПЦР с их соответствующих векторов с использованием праймеров E269 (ccgTCTAGAcggatggcctttttgcgtttc) (SEQ ID NO:15) и E202 (aatcGAATTCttactgaccattaacgcccaagc) (SEQ ID NO:16) для конструкции KPC2 и E204 (aatcGAATTCttaacgctgccagtgctcaatc) (SEQ ID NO:17) для конструкции SHV12. Затем эти фрагменты расщепляли XbaI и EcoRI и каждый вставляли в pBAD18-kan, которую расщепляли теми же ферментами с получением пары мультикопийных векторов, экспрессирующих KPC2 и SHV12, соответственно. Эти векторы трансформировали в BW25113 pspB с получением штаммов NB27273-CDY0033 (экспрессирующего KPC2) и NB27273-CDY0030 (экспрессирующего SHV12). Вектор pBAD18-kan также содержит промоторную область и сигнальную последовательность TEM, (но в нем отсутствуют любые интактные гены β-лактамаз), и его трансформировали в BW25113 pspB с использованием стандартных протоколов с получением контрольного штамма NB27273-CDY0026. Экспрессию β-лактамаз подтверждали проверкой сниженной чувствительности к тестовым антибиотикам примера, которые являются известными субстратами KPC2 или SHV12.
Тестирование чувствительности
Минимальные ингибирующие концентрации (MIC) определяли способом микроразведений в бульоне по руководствам Clinical and Laboratories Institute (CLSI). В кратком изложении, свежие ночные бактериальные культуры суспендировали в стерильном солевом растворе и доводили до стандарта мутности по McFarland 0,5. Затем бактериальные суспензии разбавляли в бульоне Мюллера-Хинтона со стандартизированным содержанием катионов (MHB II; BBL) с получением конечного инокулята приблизительно 5×105 колониеобразующих единиц (КОЕ)/мл. Получали основную панель антибиотиков с концентрацией, эквивалентной стократной наибольшей требуемой конечной концентрации в 100% диметилсульфоксиде (DMSO). Затем основную панель антибиотиков разбавляли посредством серийных двукратных разведений многоканальной пипеткой. Полученные серийные разведения соединений разбавляли 1:10 стерильной водой, что приводило к 10% конечной концентрации DMSO. Серийные разведения лекарственных средств в объеме 10 мкл переносили в 96-луночные планшеты для анализа. В планшеты для анализа инокулировали 90 мкл бактериальных суспензий и инкубировали при 35-37°C в течение 20 часов. Планшеты для анализа сканировали с использованием сканера для планшетом для микротитрования (Molecular Devices) при 600 нм, а также посредством визуального осмотра с использованием считывающего зеркала. Наименьшую концентрация соединения, которая предотвращала видимый рост, регистрировали как MIC. Производительность анализа контролировали посредством тестирования азтреонама по отношению к лабораторным штаммам контроля качества по руководствам CLSI.
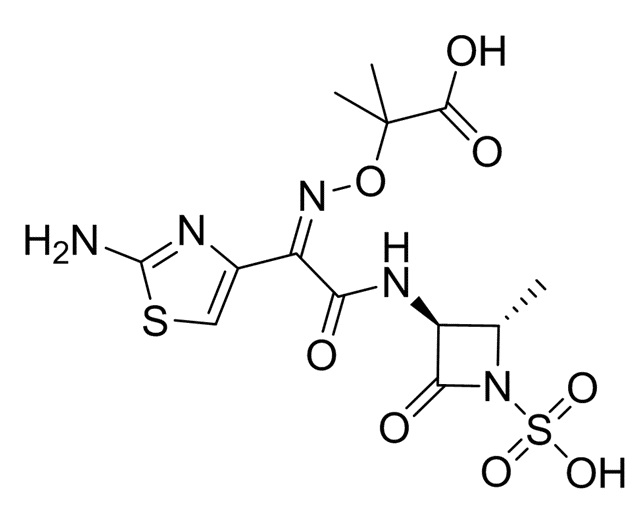
Контрольные соединения: для сравнения по настоящему документу используют следующие монобактамные соединения:
Контрольное соединение 1: Азтреонам
Контрольное соединение 2: Карумонам
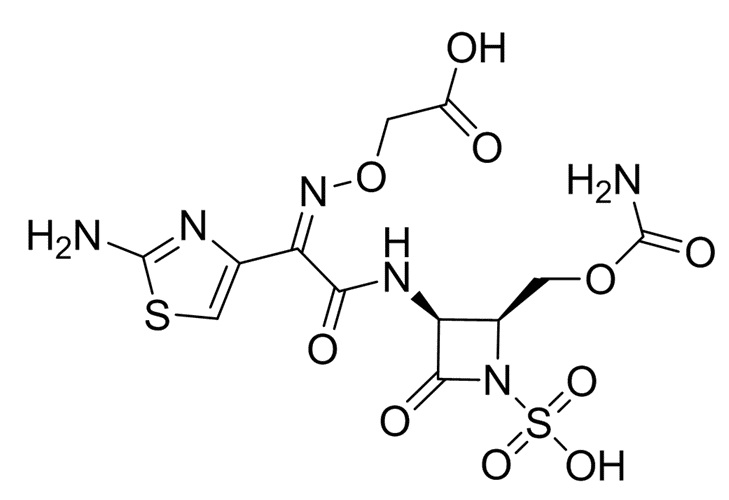
Контрольное соединение 3: BAL30072
Контрольное соединение 4: Айкурис WO2013110643

Таблица A: Минимальные ингибирующие концентрации (MIC) в отношении к изогенным штаммам E. coli, несущим различные детерминанты устойчивости.
Штамм 1: NB27273-CDY0026 E.coli (исходный)
Штамм 2: NB27273-CDY0033 E.coli (KPC2)
Штамм 3: NB27273-CDY0030 E.coli (SHV12)
Данные в таблице A демонстрируют, что соединение X обладает хорошей противобактериальной активностью в отношении E. coli, включая штаммы, которые демонстрируют высокую устойчивость к различным известным монобактамным и сульфактамным антибиотикам.
В таблице ниже приведены дополнительные данные по активности соединения X. Соединение X тестировали на штамме E. coli 25922 и E. coli, содержащей KPC-2 (штамм 2 выше, который представляет собой известную карбапенемазу из Klebsiella pneumoniae), и приведены эти ингибирующие концентрации (MIC) в мг/мл.

| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА LTA4H | 2019 |
|
RU2808992C2 |
| НОВЫЕ ПОЛИМОРФЫ И СОЛИ | 2011 |
|
RU2573384C2 |
| АМИННЫЙ СОЛЬВАТ ИНГИБИТОРА НАТРИЙ-ГЛЮКОЗНОГО КОТРАНСПОРТЕРА И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2710230C1 |
| СРЕДСТВА, СТИМУЛИРУЮЩИЕ РОСТ ВОЛОС | 2005 |
|
RU2342384C2 |
| СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ С ТРЕМЯ КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ СОЛЬ ИЛИ КРИСТАЛЛИЧЕСКАЯ ФОРМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2020 |
|
RU2835105C1 |
| СОЛЬ АГОНИСТА ОПИОИДНОГО РЕЦЕПТОРА (MOR), КРИСТАЛЛИЧЕСКАЯ ФОРМА I ЕГО ФУМАРАТНОЙ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2779119C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА TLR7/TLR8 | 2019 |
|
RU2792005C2 |
| Твёрдые формы цефтолозана | 2014 |
|
RU2703457C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА ФОСФОДИЭСТЕРАЗЫ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2814498C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ГЛИКОПИРРОНИЯ ХЛОРИДА | 2011 |
|
RU2569852C2 |

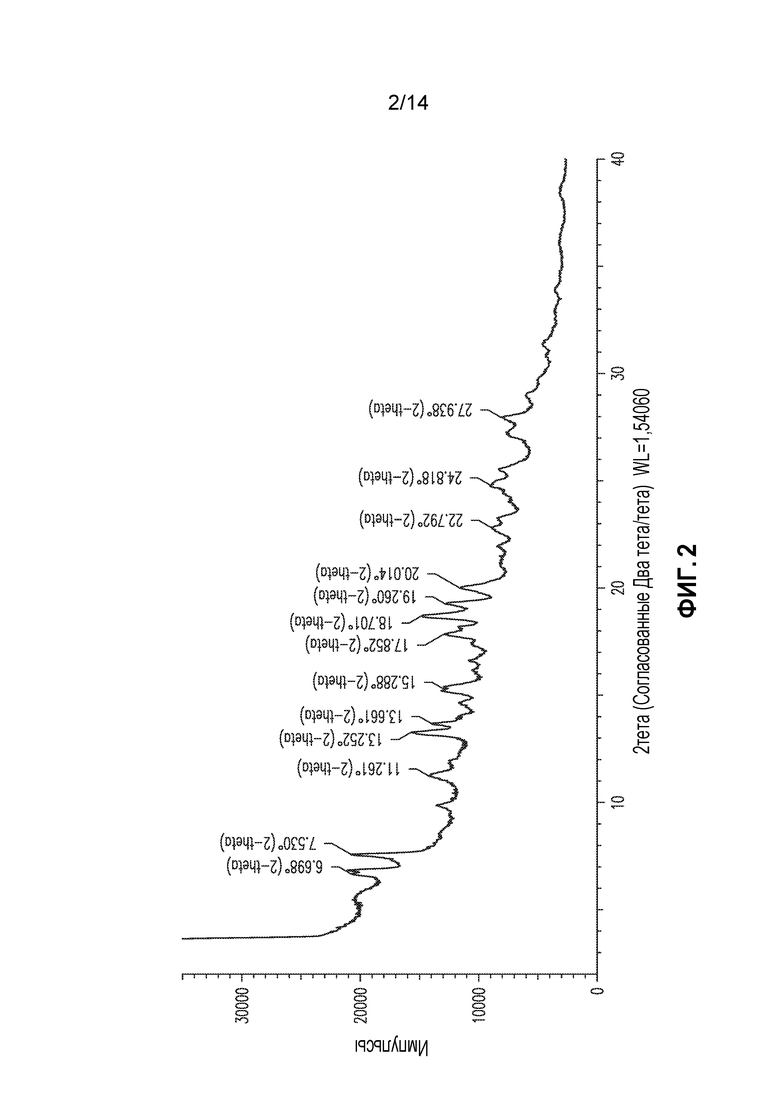


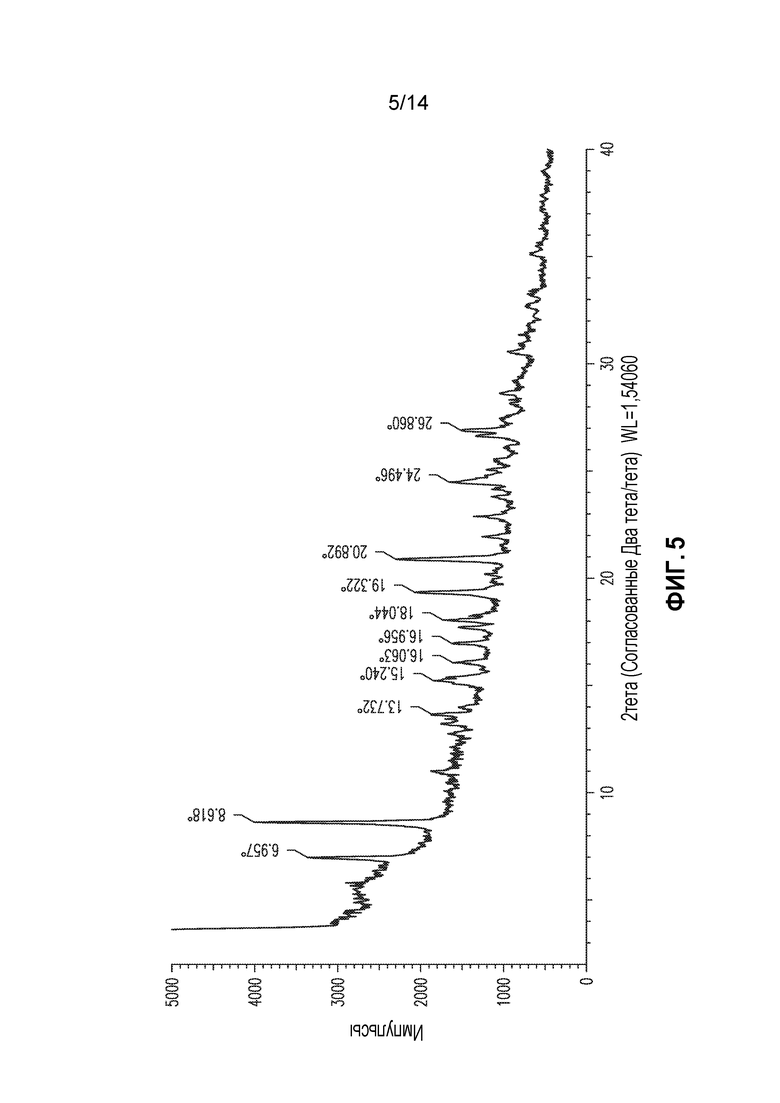

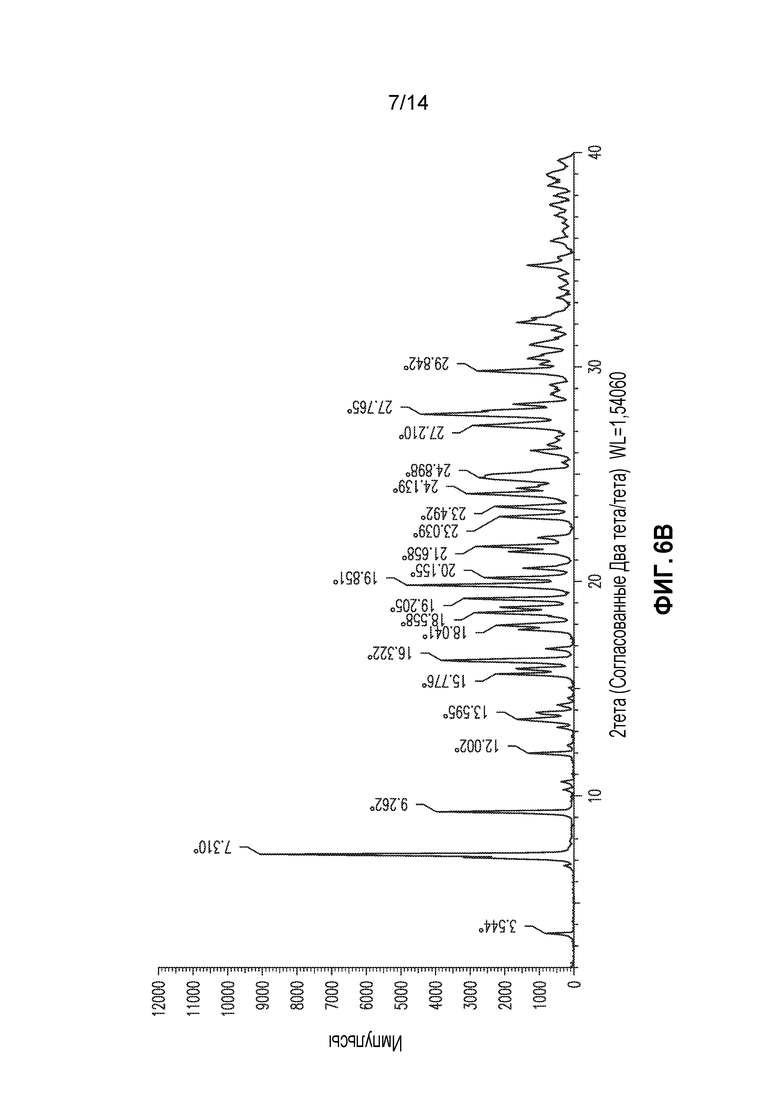

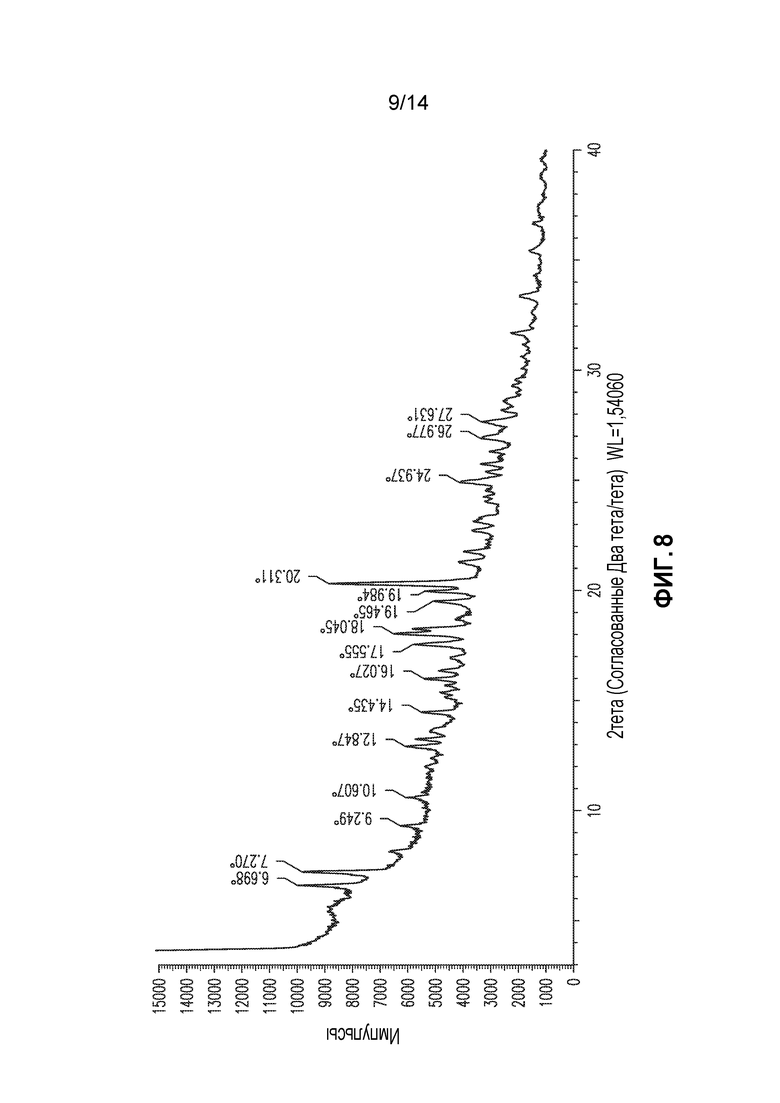
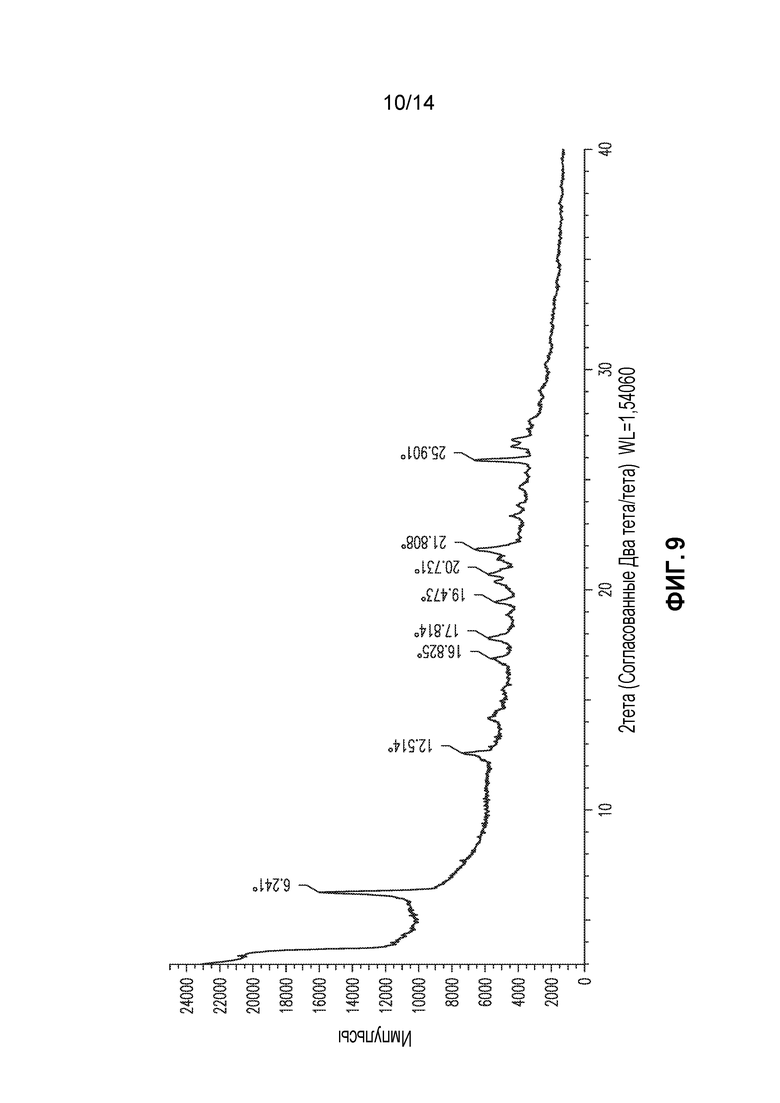
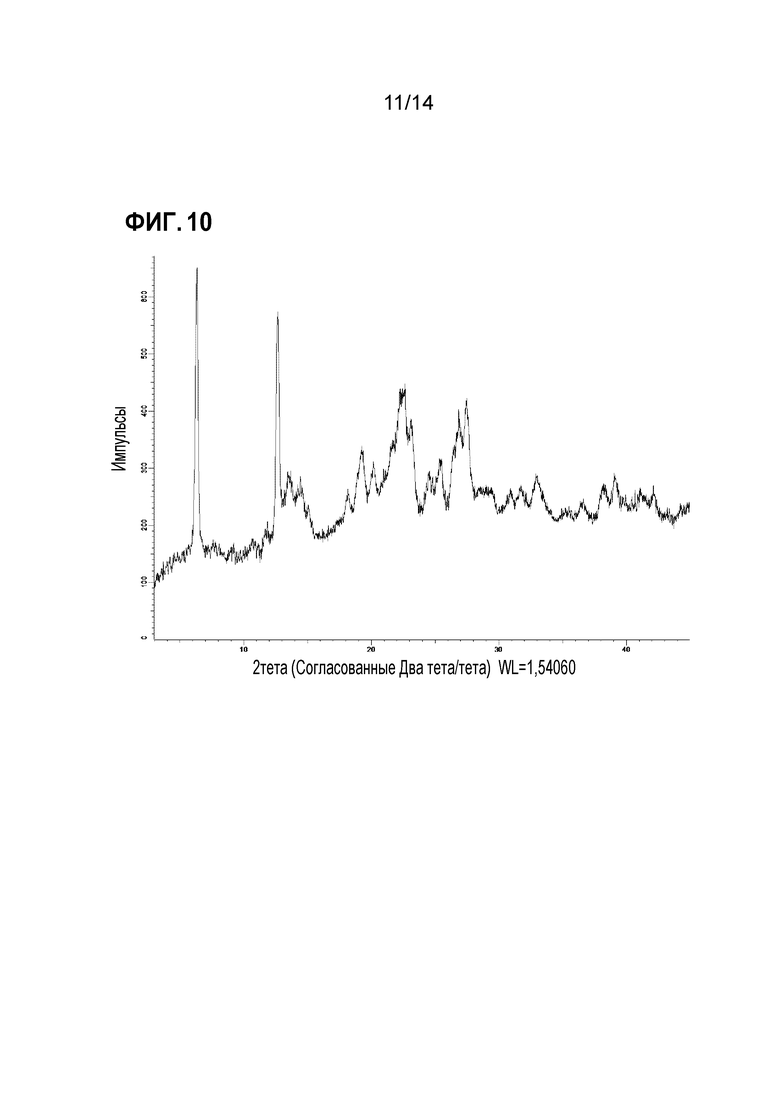


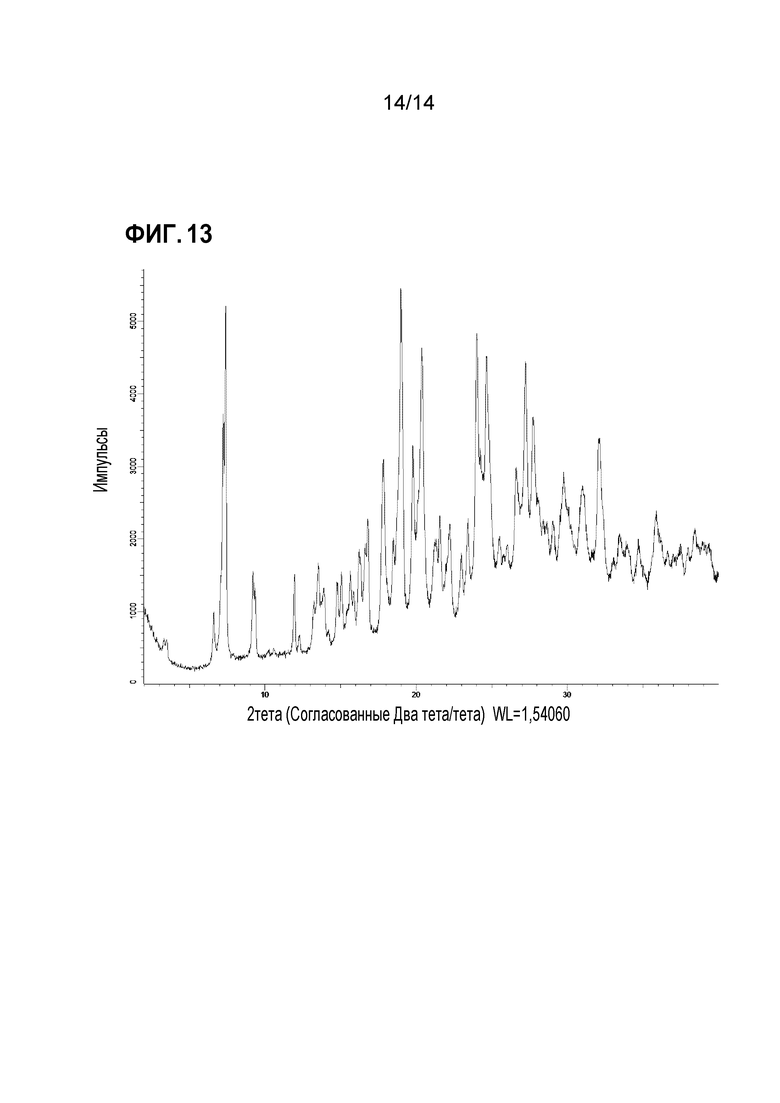
Изобретение относится к соли 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты, выбранной из соли (L)-аргинина и натриевой соли. Также изобретение относится к гидратированной твердой форме 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты. Соединения по изобретению предназначены для применения в производстве лекарственного средства для лечения инфекции грамотрицательных бактерий. 7 н. и 1 з.п. ф-лы, 5 табл., 14 ил.
1. Соль (L)-аргинина 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты.
2. Натриевая соль 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты.
3. Гидратированная твердая форма 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты.
4. Гидратированная твердая форма по п. 3, которая состоит на 50% или более из тригидрата 1-(((Z)-(1-(2-аминотиазол-4-ил)-2-оксо-2-(((3S,4R)-2-оксо-4-((2-оксооксазолидин-3-ил)метил)-1-сульфоазетидин-3-ил)амино)этилиден)амино)окси)циклопропанкарбоновой кислоты.
5. Фармацевтическая композиция, предназначенная для лечения инфекции, вызванной грамотрицательными бактериями, содержащая терапевтически эффективное количество соединения по любому из пп. 1-4 и по меньшей мере один фармацевтически приемлемый носитель или эксципиент.
6. Применение соединения по любому из пп. 1-4 для лечения инфекции, вызванной грамотрицательными бактериями.
7. Применение соединения по любому из пп. 1-4 в производстве лекарственного средства для лечения инфекции, вызванной грамотрицательными бактериями.
8. Способ лечения инфекции, вызванной грамотрицательными бактериями, включающий введение нуждающемуся в этом индивидууму терапевтически эффективного количества соединения по любому из пп. 1-4.
| Способ ведения температурного режима печи | 1930 |
|
SU30850A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Способ получения заменителей кожи | 1950 |
|
SU93376A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| STEPHEN M.BERGE et al | |||
| Pharmaceuticals Salts, JOURNAL of PHARMACEUTICAL SCIENCES, 1977, v.66(1), p.1-19 | |||
| Sherry L.Morissette et al | |||
| High-throughput crystallization: polymorphs, salts, co-crystals and | |||

