Настоящая заявка испрашивает приоритет по заявке на патент Китая № CN201710242119.3, поданной 14 апреля 2017 г., содержание которой включено в данный документ посредством ссылки во всей своей полноте.
Область техники, к которой относится изобретение
Настоящее изобретение относится к соли (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина, кристаллической форме I его фумаратной соли и способу их получения, а также к применению соли и кристаллической формы I фумаратной соли в фармацевтической композиции и применению указанной соли, кристаллической формы I фумаратной соли и композиции в изготовлении лекарственного препарата для лечения и/или предупреждения заболеваний, связанных с агонистом опиоидного рецептора (MOR).
Предшествующий уровень техники
Опиоидные рецепторы представляют собой важный класс сопряженных с G-белком рецепторов (GPCR), которые являются мишенями для связывания эндогенных опиоидных пептидов и опиоидного лекарственного средства, причем активированные опиоидные рецепторы оказывают регуляторное действие в отношении иммунитета нервной системы и в отношении эндокринной системы, а опиоидное лекарственное средство является сильнейшим и широко применяемым аналгетиком центрального действия. Эндогенные опиоидные пептиды представляют собой продуцируемые естественным образом у млекопитающих опиоидные активные вещества, при этом известные в настоящее время эндогенные опиоидные пептиды в целом классифицируют на энкефалины, эндорфины, динорфины и неоэндорфины (Pharmacol Rev 2007; 59: 88-123). В центральной нервной системе находятся соответствующие опиоидные рецепторы, а именно рецепторы μ (MOR), δ (DOR), κ (KOR) и т.п. MOR является мишенью для эндогенных энкефалинов и опиоидных анальгетиков, таких как морфин.
Длительное применение опиоидных лекарственных средств может привести к возникновению устойчивости и побочных эффектов, таких как угнетение дыхания и запор, причем было показано, что такие побочные эффекты тесно связаны с функционированием β-аррестина. С целью уменьшения побочных эффектов опиоидов можно разработать лекарственные средства на основе лиганда MOR, характеризующегося отрицательной смещенной активностью в отношении β-аррестина, с помощью которых можно уменьшить побочные эффекты, опосредованные β-аррестином, и повысить терапевтический эффект; в исследовании оксаспиро-производных согласно настоящему изобретению, действующих в качестве селективного в отношении MOR лекарственного средства, в компании TrevenaInc обнаружили, что арильное замещение в положении бензила приводит к снижению активности (J. Med. Chem. 2013, 56, 8019-8031), однако в WO 2017063509 (заявка на патент № PCT/CN2016/101064, дата подачи 30 сентября 2016 г.) раскрыто воздействующее на MOR соединение, демонстрирующее высокую активность, значительное повышение показателя Emax, значительное улучшение в отношении hERG и характеризующееся единственной конфигурацией после циклизации арила в положении бензила, структура которого представлена формулой (II),
 .
.
Поскольку растворимость соединения, представленного формулой (II), является низкой, то для дополнительного улучшения растворимости соединения авторы настоящего изобретения провели исследования в отношении получения солей соединения, представленного формулой (II), при этом исследуемые кислоты включают фумаровую кислоту; к настоящему времени отсутствуют сообщения касательно соли соединения, представленного формулой (II), или ее кристаллической формы, при этом хорошо известно, что структура кристалла, выступающего в качестве фармацевтически активного ингредиента, часто затрагивает химическую и физическую стабильность лекарственного средства, а различия в условиях кристаллизации и условиях хранения могут привести к изменению кристаллической структуры соединения, что также иногда сопровождается образованием других разновидностей кристаллической формы. Как правило, аморфным фармацевтическим продуктам не свойственна монокристаллическая структура, и они часто характеризуются другими недостатками, такими как недостаточная стабильность продукта, сложность фильтрации, чрезмерная способность к агломерации и плохая текучесть. Из этого следует, что необходимо улучшить различные аспекты вышеуказанных продуктов.
Содержание настоящего изобретения
Техническое задание, которое предстоит решить с помощью настоящего изобретения, заключается в обеспечении фумаратной соли (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина (представлен формулой (I)), ее кристаллической формы I и способа их получения, при этом соль характеризуется хорошей растворимостью, а кристаллическая форма характеризуется хорошей стабильностью.
Технические решения согласно настоящему изобретению являются следующими.
Настоящее изобретение предусматривает фумаратную соль (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина в виде соединения, представленного формулой (II),
.
В одном варианте осуществления стехиометрическое соотношение между (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амином и фумаровой кислотой составляет 1:1, при этом их структура представлена формулой (I),
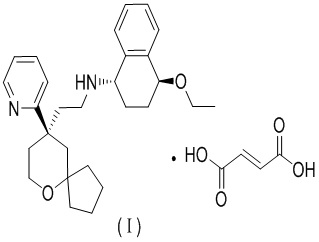 .
.
Настоящее изобретение также предусматривает способ получения соли, при этом способ включает стадию проведения реакции (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина с фумаровой кислотой.
В одном варианте осуществления солеобразование проводят в растворителе, где указанный растворитель выбран из спиртов, простых эфиров или сложных эфиров, при этом указанный растворитель на основе спирта предпочтительно представляет собой метанол, этанол или изопропанол, указанный простой эфир предпочтительно представляет собой диэтиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран или диоксан, и указанный растворитель на основе сложного эфира выбран из этилацетата, изопропилацетата и бутилацетата.
В другом варианте осуществления температура указанной реакции составляет 10-80°C.
Настоящее изобретение дополнительно предусматривает кристаллическую форму I соединения, представленного формулой (I), где порошковая рентгеновская дифрактограмма, представленная посредством угла дифракции 2θ, полученная с использованием Cu-Kα-излучения, демонстрирует характеристические пики при значениях угла дифракции 2θ, составляющих 5,76, 10,82, 11,47, 12,69, 13,86, 14,77, 15,27, 15,74, 17,26, 17,61, 18,34, 22,39, 23,06, 23,75 и 24,23, при этом диапазон погрешности для каждого из характеристических пиков 2θ составляет ±0,2.
В одном варианте осуществления характеристические пики находятся при значениях угла дифракции 2θ, составляющих 5,76, 10,82, 11,47, 12,69, 13,86, 14,77, 15,27, 15,74, 17,26, 17,61, 18,34, 19,27, 19,94, 20,37, 21,42, 21,73, 22,02, 22,39, 23,06, 23,75, 24,23 и 24,73, при этом диапазон погрешности для каждого из характеристических пиков 2θ составляет ±0,2.
В другом варианте осуществления характеристические пики находятся при значениях угла дифракции 2θ, составляющих 5,76, 7,86, 10,82, 11,47, 12,28, 12,69, 13,86, 14,77, 15,27, 15,74, 16,26, 17,26, 17,61, 18,34, 19,27, 19,94, 20,37, 21,42, 21,73, 22,02, 22,39, 23,06, 23,75, 24,23, 24,73, 25,54, 26,68, 28,59, 29,48, 31,04, 32,90 и 35,73, при этом диапазон погрешности для каждого из характеристических пиков 2θ составляет ±0,2.
Настоящее изобретение также предусматривает способ получения кристаллической формы I, где указанный способ включает растворение соединения, представленного формулой (I), в растворителе, кристаллизацию, фильтрование и высушивание с получением целевой кристаллической формы I; при этом растворитель предпочтительно представляет собой тетрагидрофуран; и кристаллизация представляет собой кристаллизацию при охлаждении, при этом охлаждение представляет собой комнатную температуру.
Настоящее изобретение дополнительно относится к фармацевтической композиции на основе соединения, представленного формулой (I), его кристаллической формы I, которая при этом содержит одно или более из фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ.
Настоящее изобретение дополнительно относится к применению соединения, представленного формулой (I), его кристаллической формы I, фармацевтической композиции на их основе в изготовлении лекарственного препарата для лечения соответствующих заболеваний, опосредованных агонистом опиоидного рецептора (MOR).
Соответствующие заболевания, опосредованные агонистом опиоидного рецептора MOR согласно настоящему изобретению, выбраны из группы, состоящей из боли, иммунной дисфункции, воспаления, гастроэзофагеального рефлюкса, неврологических и психических заболеваний, заболеваний мочевыделительной и репродуктивной систем, сердечно-сосудистых заболеваний и респираторных заболеваний, предпочтительно представляют собой боль.
Настоящее изобретение дополнительно предусматривает применение соединения, представленного формулой (I), его кристаллической формы I, фармацевтической композиции на основе соединения, представленного формулой (I), фармацевтической композиции на основе кристаллической формы I в изготовлении лекарственного препарата для предупреждения или лечения боли и заболеваний, связанных с болью.
Боль согласно настоящему изобретению выбрана из послеоперационной боли, боли, обусловленной раком, нейропатической боли, травматической боли или боли, обусловленной воспалением.
Рак согласно настоящему изобретению выбран из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака предстательной железы, рака яичника, опухоли фаллопиевой трубы, опухоли яичника, гемофилии и лейкоза.
Настоящее изобретение дополнительно предусматривает применение соединения, представленного формулой (I), его кристаллической формы I, фармацевтической композиции на основе соединения, представленного формулой (I), фармацевтической композиции на основе кристаллической формы I в изготовлении лекарственного препарата для агонистического или антагонистического воздействия на рецептор MOR.
Определение структуры и исследование формы кристалла полученной кристаллической формы I соединения, представленного формулой (I), проводили с применением порошковой рентгеновской дифрактограммы (XRPD), дифференциальной сканирующей калориметрии (DSC) и термогравиметрического анализатора (TGA).
Способ перекристаллизации, применяемый в отношении кристаллической формы I, конкретно не ограничивается, и его можно осуществлять с применением общепринятых методик проведения перекристаллизации. Например, посредством растворения соединения, представленного формулой (I), в органическом растворителе и затем кристаллизации за счет добавления в антирастворитель, а после завершения кристаллизации – фильтрования и высушивания с получением требуемого кристалла.
Способ кристаллизации согласно настоящему изобретению включает кристаллизацию при выпаривании, кристаллизацию при условиях окружающей среды, кристаллизацию при охлаждении, индуцированную затравкой кристаллизацию и т.п.
Исходным материалом, применяемым в способе получения кристаллической формы согласно настоящему изобретению, может быть любая форма соединения, представленного формулой (I), причем конкретная форма включает без ограничения аморфную форму, любую кристаллическую форму и т.п.
В описании и формуле изобретения настоящей заявки, если не указано иное, то научные и технические термины, применяемые в данном документе, имеют такие значения, которые обычно понятны специалистам в данной области. Однако для лучшего понимания настоящего изобретения ниже предоставлены определения и пояснения некоторых соответствующих терминов. Кроме того, если определения и пояснения терминов, предусмотренные настоящим изобретением, не соответствуют значениям, обычно понимаемым специалистами в данной области, то определения и пояснения терминов, предусмотренные настоящей заявкой, будут иметь преимущественную силу.
Термин «C1-6алкил» в настоящем изобретении обозначает линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода, и конкретные примеры включают без ограничения метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, 1,2-диметилпропил и т.п.
Термин «гидроксильная группа», применяемый в настоящем изобретении, обозначает группу -OH и т.п.
Термин «цианогруппа», применяемый в настоящем изобретении, обозначает группу -CN и т.п.
Термин «растворитель на основе кетона», применяемый в настоящем изобретении, обозначает соединение, в котором карбонильная группа (-C(=O)-) связана с двумя гидрокарбонильными группами, и кетон может быть классифицирован как алифатический кетон, алициклический кетон, ароматический кетон, насыщенный кетон и ненасыщенный кетон в соответствии с различными гидрокарбонильными группами в молекуле, при этом конкретные примеры включают без ограничения ацетон, ацетофенон, метилизобутилкетон или метилпирролидон.
Термин «растворитель на основе простого эфира», применяемый в настоящем изобретении, обозначает соединение с открытой цепью или циклическое соединение, содержащее эфирную связь -O- и 1-10 атомов углерода, при этом конкретные примеры включают без ограничения тетрагидрофуран, диэтиловый эфир, монометиловый эфир пропиленгликоля, метил-трет-бутиловый эфир или 1,4-диоксан.
Термин «растворитель на основе спирта», применяемый в настоящем изобретении, обозначает группу, полученную путем замещения одного или более атомов водорода в «C1-6алкиле» одной или более «гидроксильными группами», причем «гидроксильная группа» и «C1-6алкил» определены выше, а конкретные примеры включают без ограничения метанол, этанол, изопропанол, н-пропанол, изопентиловый спирт или трифторэтанол.
Термин «растворитель на основе нитрила», применяемый в настоящем изобретении, обозначает группу, полученную путем замещения одного или более атомов водорода в «C1-6алкиле» одной или более «цианогруппами», причем «цианогруппа» и «C1-6алкил» определены выше, а конкретные примеры включают без ограничения ацетонитрил или пропионитрил.
Термин «растворитель на основе сложного эфира», применяемый в настоящем изобретении, обозначает комбинацию низшей органической кислоты, содержащей 1-4 атома углерода, и низшего спирта, содержащего 1-6 атомов углерода, причем конкретные примеры включают без ограничения этилацетат, изопропилацетат или бутилацетат.
Термин «смешанный растворитель», применяемый в настоящем изобретении, обозначает растворитель, полученный путем смешивания одного или более органических растворителей различных видов в определенном соотношении, или растворитель, полученный путем смешивания органического растворителя с водой в определенном соотношении; при этом смешанный растворитель предпочтительно представляет собой смешанный растворитель на основе спирта и простого эфира; причем смешанный растворитель на основе спирта и простого эфира предпочтительно представляет собой смешанный растворитель на основе метанола и диэтилового эфира, а их соотношение предпочтительно составляет 1:10.
«Порошковая рентгеновская дифрактограмма или XRPD» в настоящем изобретении означает, что в соответствии с уравнением Брэгга 2d sin θ = nλ (где λ – длина волны рентгеновского излучения, λ=1,54056 Å, порядок дифракции n представляет собой любое положительное целое число, причем, как правило, регистрируют дифракционный пик первого порядка, n=1); если рентгеновское излучение падает при угле поворота θ (угол, представляющий собой угол падения, также называемый углом Брэгга) на атомную плоскость кристалла или часть образца кристалла с расстоянием между плоскостями матрицы d, то могут быть удовлетворены условия уравнения Брэгга и измерена порошковая рентгеновская дифрактограмма.
Применяемая в настоящем изобретении «дифференциальная сканирующая калориметрия или DSC» относится к определению с помощью измерения разницы температур и разницы теплового потока между образцом и эталоном во время повышения температуры или при постоянной температуре образца для характеристики всех физических и химических изменений, связанных с температурными эффектами, и для получения информации касательно фазового перехода образца.
Применяемый в настоящем изобретении «2θ или угол 2θ» относится к углу дифракции, при этом θ представляет собой угол Брэгга, единицей измерения является ° или градус, и диапазон погрешности 2θ составляет от ±0,1 до ±0,5, предпочтительно от ±0,1 до ±0,3, более предпочтительно ±0,2.
Применяемое в настоящем изобретении «расстояние между плоскостями или межплоскостное расстояние (показатель d)» отображает три единичных вектора a, b, c, выбранных из пространственной решетки, которые не параллельны друг другу и которые соединяют две смежные точки решетки, причем такие точки разделяют решетку на размещенные рядом единицы в форме параллелепипедов, называемые расстоянием между плоскостями. Пространственную решетку линейно разделяют в соответствии с установленной единицей в форме параллелепипеда и получают группу линейных сеток, которую называют пространственной решеткой или решеткой. Решетка отражает регулярность кристаллической структуры посредством геометрических точек и линий, а межплоскостное расстояние (т.е. расстояние между двумя смежными параллельными плоскостями) для плоскостей разных решеток является различной; при этом единицей измерения является Å или ангстрем.
Настоящее изобретение дополнительно относится к фармацевтической композиции, содержащей соединение, представленное формулой (I), его кристаллическую форму I и необязательно один или более фармацевтически приемлемых носителей и/или разбавителей. Фармацевтическая композиция может быть составлена в виде любой из фармацевтически приемлемых лекарственных форм. Например, соединение, представленное формулой (I), согласно настоящему изобретению, кристаллическая форма I или фармацевтический препарат могут быть составлены в виде таблетки, капсулы, пилюли, гранул, растворов, суспензий, сиропов, инъекционных форм (включая инъекционные растворы, стерильный порошок для инъекции и концентрированный раствор для инъекции), суппозитория, лекарственной формы для ингаляции или спрея.
Кроме того, фармацевтическую композицию согласно настоящему изобретению можно также вводить пациенту или субъекту, нуждающемуся в таком лечении, любым подходящим способом введения, таким как пероральное, парентеральное, ректальное, ингаляционное или местное введение. В случае использования фармацевтической композиции для перорального введения она может быть составлена в виде препарата для перорального применения, такого как твердый препарат для перорального применения, как например таблетка, капсула, пилюля, гранула или т.п.; или жидкий препарат для перорального применения, как например раствор для перорального применения, суспензия для перорального применения, сироп и т.п. В случае составления фармацевтического препарата в виде препарата для перорального применения он может дополнительно содержать подходящий наполнитель, связующее, разрыхлитель, смазывающее средство и т.п. В случае использования фармацевтического препарата для парентерального введения он может быть получен в виде инъекционного препарата, включая инъекционный раствор, стерильный порошок для инъекции и концентрированный раствор для инъекции. В случае составления фармацевтической композиции в виде инъекционного препарата она может быть получена путем общепринятых в современной фармацевтической области способов. В случае составления инъекционного препарата можно не осуществлять добавление какого-либо дополнительного средства к фармацевтическому препарату или же подходящее дополнительное средство можно добавлять в зависимости от природы лекарственного средства. В случае использования фармацевтического препарата для ректального введения он может быть составлен в суппозиторий или т. п. Для ингаляционного введения фармацевтический препарат может быть составлен в виде средства для ингаляции или спрея. В некоторых предпочтительных вариантах осуществления соединение, представленное формулой (I), согласно настоящему изобретению или его кристаллическая форма I присутствует в фармацевтической композиции или лекарственном препарате в терапевтически и/или профилактически эффективном количестве. В некоторых предпочтительных вариантах осуществления соединение, представленное формулой (I), согласно настоящему изобретению или его кристаллическая форма I присутствует в фармацевтической композиции или лекарственном препарате в стандартной лекарственной форме.
Соединение, представленное формулой (I), согласно настоящему изобретению и его кристаллическую форму I можно использовать для изготовления лекарственного препарата для лечения заболевания, связанного с агонистом опиоидного рецептора (MOR). Соответственно, настоящая заявка также относится к применению соединения, представленного формулой (I), согласно настоящему изобретению и его кристаллической формы I для изготовления лекарственного препарата, причем указанный лекарственный препарат применяют для лечения заболеваний, связанных с агонистом опиоидного рецептора (MOR). Кроме того, настоящая заявка также относится к способу подавления заболевания, связанного с агонистом опиоидного рецептора (MOR), который включает введение субъекту, нуждающемуся в этом, терапевтически и/или профилактически эффективного количества соединения, представленного формулой (I), согласно настоящему изобретению, его кристаллической формы I или фармацевтической композиции согласно настоящему изобретению.
В некоторых предпочтительных вариантах осуществления заболевание представляет собой заболевание, связанное с агонистом опиоидного рецептора (MOR), которое выбрано из разновидностей боли.
Полезный эффект настоящего изобретения
По сравнению с предшествующим уровнем техники техническое решение согласно настоящему изобретению обладает следующими преимуществами.
Исследования показали, что соединение, представленное формулой (I), полученное согласно настоящему изобретению, характеризуется превосходной растворимостью.
Кристаллическая форма I демонстрирует более высокую точку плавления, лучшую растворимость, более высокую чистоту и отсутствие какого-либо изменения формы кристалла (определено посредством XRPD) в условиях высокой температуры и высокой влажности; кристаллическая форма I соединения, представленного формулой (I), полученная с помощью технического решения согласно настоящему изобретению может соответствовать медицинским требованиям в отношении производства, транспортировки и хранения, а способ получения является стабильным, воспроизводимым и контролируемым, при этом его можно адаптировать для промышленного производства.
Краткое описание графических материалов
На фиг. 1 изображена XRPD-спектрограмма кристаллической формы I соединения, представленного формулой (I).
На фиг. 2 изображена DSC-спектрограмма кристаллической формы I соединения, представленного формулой (I).
На фиг. 3 изображена TGA-спектрограмма кристаллической формы I соединения, представленного формулой (I).
Подробное описание предпочтительного варианта осуществления
Ниже по тексту настоящее изобретение дополнительно и более подробно поясняется с помощью вариантов осуществления, которые предназначены лишь для иллюстрации технического решения согласно настоящему изобретению, а не для ограничения сущности и объема настоящего изобретения.
Условия проведения испытаний для оборудования, применяемого в эксперименте, являются следующими.
1. Дифференциальный сканирующий калориметр (DSC)
Модель прибора: система Mettler Toledo DSC3+ STARe
Продувочный газ: азот (50 мл/мин.)
Скорость нагревания: 10,0°C/мин.
Диапазон температур: 20-250°C
2. Порошковая рентгеновская дифракция (XRPD)
Модель прибора: Порошковый рентгеновский дифрактометр Rigaku UltimaIV
Излучение: монохромное Cu-Kα-излучение (λ=1,5418 Å)
Способ сканирования: θ/2θ, диапазон сканирования: 3-45°
Напряжение: 40 кВ, сила тока: 40 мA
3. Термогравиметрический анализ (TGA)
Модель прибора: система Mettler Toledo TGA2 STARe
Продувочный газ: азот
Скорость нагревания: 10,0°C/мин.
Диапазон температур: 20-250°C.
Сравнительный эксперимент 1. Получение (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина (соединение 19 или соединение, представленное формулой (II))

Стадия 1
(S)-1,2,3,4-тетрагидронафтален-1-трет-бутилкарбамат 11a
(S)-1,2,3,4-тетрагидро-1-нафтиламин 10a (3 г, 20,41 ммоль, полученный с помощью способа, раскрытого в «Angewandte Chemie-International Edition, 45(28), 4641-4644, 2006») растворяли в 100 мл дихлорметана, затем в раствор добавляли триэтиламин (5,7 мл, 40,82 ммоль) и Ошибка! Недопустимый объект гиперссылки.(4,9 г, 22,45 ммоль) и перемешивали с обеспечением протекания реакции в течение 12 часов. Реакционную смесь последовательно промывали водой (100 мл) и насыщенным раствором бикарбоната (100 мл), органическую фазу высушивали над безводным сульфатом натрия, затем фильтровали, фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, представляющего собой указанное в заголовке соединение 11a (5,6 г, бледно-желтое масло), который применяли непосредственно на следующей стадии без дополнительной очистки.
MS, масса/заряд (ESI): 248,3 [M+1].
Стадия 2
(S)-трет-бутил(4-оксо-1,2,3,4-тетрагидронафтален-1-ил)карбамат 11b
Неочищенный продукт, представляющий собой (S)-1,2,3,4-тетрагидронафтален-1-трет-бутилкарбамат 11a (5,6 г, 20,41 ммоль), растворяли в 90 мл смеси ацетона и воды (объем/объем = 2:1), добавляли сульфат магния (5,5 г, 45,66 ммоль), затем медленно добавляли перманганат калия (7,22 г, 45,66 ммоль) при перемешивании, смесь перемешивали с обеспечением протекания реакции в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле с применением н-гексана и EtOAc в качестве элюента с получением указанного в заголовке продукта 11b (3,1 г, грязно-белое твердое вещество) с выходом 52%.
MS, масса/заряд (ESI): 262,3 [M+1].
Стадия 3
(1S,4S)-4-гидрокси-1,2,3,4-тетрагидронафтален-1-трет-бутилкарбамат 14a
(S)-трет-бутил(4-оксо-1,2,3,4-тетрагидронафтален-1-ил)карбамат 11b (100 мг, 0,883 ммоль) растворяли в 5 мл толуола, температуру понижали до 0°C, затем добавляли (R)-2-метил-CBS-оксазол-боран (0,1 мл, 0,076 ммоль) и перемешивали в течение 5 мин., затем добавляли комплекс боран-метилсульфид (0,88 мл, 0,76 ммоль) и перемешивали с обеспечением протекания реакции в течение 2 часов. Для гашения реакции добавляли 50 мл насыщенного раствора хлорида натрия, затем экстрагировали с помощью EtOAc (30 мл × 3), органические фазы объединяли и промывали с помощью насыщенного раствора хлорида натрия (30 мл × 3), высушивали над безводным сульфатом натрия, фильтровали, фильтрат концентрировали при пониженном давлении, остаток очищали с помощью тонкослойной хроматографии с применением дихлорметана и метанола в качестве элюента с получением указанного в заголовке продукта 14a (60 мг, белое твердое вещество) с выходом 60%.
MS, масса/заряд (ESI): 208,3 [M-55].
Стадия 4
(1S,4S)-4-этокси-1,2,3,4-тетрагидронафтален-1-трет-бутилкарбамат 19a
Неочищенный продукт, представляющий собой (1S)-4-гидрокси-1,2,3,4-тетрагидронафтален-1-трет-бутилкарбамат 14a (850 мг, 3,23 ммоль), оксид серебра (76 мг, 0,33 ммоль) и этилиодид (1,3 мл, 16,15 ммоль) растворяли в дихлорметане (30 мл), реакционную смесь перемешивали в течение 48 часов, затем фильтровали, фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, представляющего собой указанное в заголовке соединение 19a (800 мг, желтое масло), который применяли непосредственно на следующей стадии без дополнительной очистки.
MS, масса/заряд (ESI): 236,1 [M-55].
Стадия 5
(1S,4S)-4-этокси-1,2,3,4-тетрагидронафтален-1-амин 19b
Неочищенный продукт, представляющий собой соединение 19a (698 мг, 2,4 ммоль), растворяли в 4 мл дихлорметана, добавляли 8 мл 4 М хлороводорода в 1,4-диоксане, реакционную смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, растирали в EtOAc (30 мл), фильтровали, осадок на фильтре растворяли в смешанном растворе дихлорметана и метанола (20 мл, объем:объем=5:1), pH реакционной смеси регулировали с помощью насыщенного раствора бикарбоната до 7-8, реакционную смесь концентрировали при пониженном давлении, промывали с помощью смешанного раствора (30 мл × 2) дихлорметана и метанола (объем:объем=5:1), фильтровали, фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, представляющего собой указанное в заголовке соединение 19b (310 мг, желтая жидкость), который применяли непосредственно на следующей стадии без дополнительной очистки.
MS, масса/заряд (ESI): 191,1 [M+1].
Стадия 6
(1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амин 19
(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этаналь 5a (500 мг, 1,85 ммоль, получен с помощью способа, раскрытого в заявка на патент № WO 2012129495), неочищенный продукт, представляющий собой соединение 19b (310 мг, 1,85 ммоль), растворяли в дихлорэтане (30 мл), перемешивали в течение 40 мин, добавляли триацетоксиборгидрид натрия (980 мг, 4,63 ммоль) и реакционную смесь перемешивали в течение 2 часов. Смесь последовательно промывали насыщенным раствором бикарбоната (30 мл × 3) и насыщенным раствором хлорида натрия (30 мл × 3), органическую фазу высушивали над безводным сульфатом натрия, фильтровали, фильтрат концентрировали при пониженном давлении, остаток очищали с помощью тонкослойной хроматографии с применением дихлорметана и метанола в качестве элюента с получением указанного в заголовке продукта 19 (280 мг, желтое вязкое твердое вещество) с выходом 35%.
Сравнительный эксперимент 2. Очистка (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина (соединение, представленное формулой (II))
Продукт сравнительного эксперимента 1 (100 мг) помещали в реакционную колбу, затем добавляли этанол (0,1 мл), смесь нагревали и кипятили с обратным холодильником до растворения, затем охлаждали до к. т. для осаждения, фильтровали, высушивали; выход составлял 55%.
Вариант осуществления 1. Получение кристаллической формы I
Получение кристаллической формы I фумаратной соли (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина
Фумаратную соль (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина (50 мг, 0,09 ммоль) растворяли в тетрагидрофуране (2,5 мл), затем фумаровую кислоту (23,2 мг, 0,2 ммоль) растворяли в тетрагидрофуране (0,25 мл) и по каплям добавляли в вышеуказанный раствор; после перемешивания раствор становился прозрачным, его нагревали до слабого кипения и перемешивали до растворения. Затем раствор естественным образом охлаждали до к. т., перемешивали в течение 16 часов. Реакционную смесь затем фильтровали, осадок на фильтре капельно промывали с помощью EtOAc (1 мл × 3) и собирали, высушивали в вакууме с получением твердого вещества (25 мг, выход 50%); XRPD-спектрограмма кристаллического образца показана на фиг. 1, а его DSC-спектрограмма показана на фиг. 2, при наличии 2 эндотермических пиков и 1 экзотермического пика; подробные данные представлены в таблице ниже.
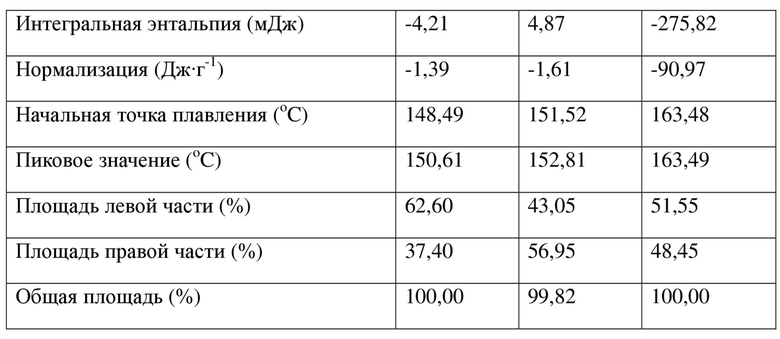
Можно увидеть, что значение начальной точки максимального эндотермического пика составляет приблизительно 162,48oC, при этом пиковое значение составляет приблизительно 163,49°C; характеристические пики при 2θ представлены в таблице ниже.
Таблица 1. Характеристические пики кристаллической формы I

TGA-спектрограмма, представленная на фиг. 3, указывает на то, что кристаллическая форма I представляла собой ангидрид.
MS, масса/заряд (ESI): 435,5 [M+1].
1H-ЯМР (400 MГц, DMSO-d6) δ 8,43-8,66 (m, 1H), 7,69-7,80 (m, 1H), 7,42-7,52 (m, 1H), 7,28-7,36 (m, 1H), 7,23 (s, 4H), 6,51 (s, 2H), 4,26-4,35 (m, 1H), 3,85-3,97 (m, 1H), 3,60 (m, 3H), 3,39-3,51 (m, 1H), 2,52-2,61 (m, 1H), 2,30-2,45 (m, 2H), 2,07-2,20 (m, 1H), 1,85-2,07 (m, 3H), 1,20-1,84 (m, 12H), 1,11 (t, 3H), 0,93-1,03 (m, 1H), 0,57-0,72 (m, 1H).
Вариант осуществления 2. Сравнение растворимости соли согласно настоящему изобретению и свободного основания в воде
Тестируемый образец: соединение, представленное формулой (II) (свободное основание), полученное в сравнительном эксперименте 2, и продукт, полученный в варианте осуществления 1 (соединение, представленное формулой (I)).
Растворитель: чистая вода.
Способ проведения эксперимента
Тестируемые образцы взвешивали и добавляли в чистую воду, затем перемешивали посредством магнитной мешалки в течение ночи, затем фильтровали и разбавляли до определенного объема для осуществления ввода образцов.
Хроматографические условия HPLC: в качестве подвижной фазы применяли смесь ацетонитрил – 0,1% водный раствор трифторуксусной кислоты (50:50), длина волны обнаружения – 264 нм, объем вводимой пробы – 10 мкл, скорость потока – 1,0 мл/мин.
Результаты эксперимента
Таблица 2. Сравнение растворимости фумаратной соли согласно настоящему изобретению и свободного основания в воде

Заключение эксперимента
В соответствии с таблицей 2 фумаратная соль согласно настоящему изобретению обладает лучшей растворимостью в воде, чем свободное основание.
Вариант осуществления 3. Исследование стабильности кристаллической формы I
Образец кристаллической формы I, полученный посредством варианта осуществления 2, помещали на плоскую поверхность в защищенных от света герметичных условиях и проводили исследование на стабильность образца при длительном хранении (25°C, 60% RH), при этом период между отбором проб составлял 5 дней, и XRPD применяли для определения происходило ли изменение кристаллической формы.
Результаты эксперимента
Дифрактограмма XRPD-пиков образца кристаллической формы I оставалась без изменений при условии длительного хранения образца (25°C, 60% RH).
Несмотря на то, что выше по тексту описаны конкретные варианты осуществления настоящего изобретения, специалистам в данной области будет понятно, что они являются лишь примерами реализации, при этом в отношении вариантов осуществления можно выполнять различные модификации и изменения без отступления от принципов и сути настоящего изобретения. Таким образом, объем настоящего изобретения определяется прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОКСАСПИРОПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЯ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 2016 |
|
RU2733373C2 |
| Замещенные производные бисфенилового эфира масляной кислоты в качестве ингибиторов NEP | 2019 |
|
RU2784522C2 |
| НОВЫЕ ФУМАРАТНЫЕ СОЛИ АНТАГОНИСТА ГИСТАМИНОВОГО РЕЦЕПТОРА Н3 | 2010 |
|
RU2537847C2 |
| АМИНОВАЯ СОЛЬ И ЕЕ КРИСТАЛЛЫ | 2013 |
|
RU2658823C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2014 |
|
RU2654855C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСАСПИРОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2018 |
|
RU2777983C2 |
| ОПТИЧЕСКИ ЧИСТОЕ ОКСАСПИРО-ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ПИРРОЛОПИРАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2800295C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЕДИНЕНИЯ 3-КАРБОКСИПРОПИЛ-АМИНОТЕТРАЛИНА | 2009 |
|
RU2512390C2 |
| СОЛИ ПРОИЗВОДНОГО ИНДОЛА ПРОТИВ МИГРЕНИ | 1995 |
|
RU2159241C2 |
| НЕЙРОГЕНЕЗ, ОПОСРЕДОВАННЫЙ ПРОИЗВОДНЫМ 4-АЦИЛАМИНОРИРИДИНА | 2007 |
|
RU2451512C2 |



Изобретение относится к фумаратной соли (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина, его кристаллической форме I. Кристаллическая форма I фумаратной соли (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина, охарактеризована порошковой рентгеновской дифрактограммой, представленной посредством угла дифракции 2θ, полученной с использованием Cu-Kα-излучения, демонстрирует характеристические пики при значениях угла дифракции 2θ, составляющих 5,76, 10,82, 11,47, 12,69, 13,86, 14,77, 15,27, 15,74, 17,26, 17,61, 18,34, 22,39, 23,06, 23,75 и 24,23, при этом диапазон погрешности для каждого из характеристических пиков 2θ составляет ±0,2. Способ получения фумаратной соли по изобретению включает стадию проведения реакции (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина с фумаровой кислотой. Способ получения кристаллической формы I соединения по изобретению, включает растворение соединения, представленного формулой (I), в растворителе, кристаллизацию, фильтрование и высушивание с получением целевой кристаллической формы I; при этом растворитель представляет собой тетрагидрофуран; и кристаллизация предствляет собой кристаллизацию при охлаждении, при этом температура охлаждения представляет собой комнатную температуру. Технический результат - фумаратная соль (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина, применимая в изготовлении лекарственного препарата для лечения соответствующих заболеваний, опосредованных агонистом опиоидного рецептора (MOR), где указанные соответствующие заболевания, опосредованные агонистом рецептора (MOR), представляют собой боль. 10 н. и 6 з.п. ф-лы, 2 табл., 3 ил.

1. Фумаратная соль соединения, представленного формулой (II),
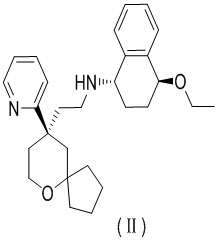 .
.
2. Соль по п. 1, где стехиометрическое соотношение между соединением, представленным формулой (II), и фумаровой кислотой составляет 1:1, при этом ее структура представлена формулой (I),
 .
.
3. Способ получения соли по п. 1, где способ включает стадию проведения реакции (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина с фумаровой кислотой.
4. Способ получения соли по п. 2, где способ включает стадию проведения реакции (1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дека-9-ил)этил)-1,2,3,4-тетрагидронафтален-1-амина с фумаровой кислотой.
5. Способ по п. 3, где солеобразование проводят в растворителе, при этом растворитель выбран из простых эфиров.
6. Способ по п. 5, где указанный простой эфир представляет собой диэтиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран или диоксан.
7. Способ по п. 5, где температура указанной реакции составляет 10-80°C.
8. Кристаллическая форма I соединения, представленного формулой (I), по п. 2, где порошковая рентгеновская дифрактограмма, представленная посредством угла дифракции 2θ, полученная с использованием Cu-Kα-излучения, демонстрирует характеристические пики при значениях угла дифракции 2θ, составляющих 5,76, 10,82, 11,47, 12,69, 13,86, 14,77, 15,27, 15,74, 17,26, 17,61, 18,34, 22,39, 23,06, 23,75 и 24,23, при этом диапазон погрешности для каждого из характеристических пиков 2θ составляет ±0,2.
9. Кристаллическая форма I по п. 8, где ее характеристические пики находятся при значениях угла дифракции 2θ, составляющих 5,76, 10,82, 11,47, 12,69, 13,86, 14,77, 15,27, 15,74, 17,26, 17,61, 18,34, 19,27, 19,94, 20,37, 21,42, 21,73, 22,02, 22,39, 23,06, 23,75, 24,23 и 24,73, при этом диапазон погрешности для каждого из характеристических пиков 2θ составляет ±0,2.
10. Кристаллическая форма I по п. 9, где ее характеристические пики находятся при значениях угла дифракции 2θ, составляющих 5,76, 7,86, 10,82, 11,47, 12,28, 12,69, 13,86, 14,77, 15,27, 15,74, 16,26, 17,26, 17,61, 18,34, 19,27, 19,94, 20,37, 21,42, 21,73, 22,02, 22,39, 23,06, 23,75, 24,23, 24,73, 25,54, 26,68, 28,59, 29,48, 31,04, 32,90 и 35,73, при этом диапазон погрешности для каждого из характеристических пиков 2θ составляет ±0,2.
11. Способ получения кристаллической формы I по п. 8, где указанный способ включает растворение соединения, представленного формулой (I), в растворителе, кристаллизацию, фильтрование и высушивание с получением целевой кристаллической формы I; при этом растворитель представляет собой тетрагидрофуран; и кристаллизация представляет собой кристаллизацию при охлаждении, при этом температура охлаждения представляет собой комнатную температуру.
12. Фармацевтическая композиция, характеризующаяся свойствами агониста опиоидного рецептора (MOR), содержащая соль по п. 1, в качестве активного ингредиента, где композиция содержит один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ.
13. Фармацевтическая композиция, характеризующаяся свойствами агониста опиоидного рецептора (MOR), содержащая кристаллическую форму I по п. 8, в качестве активного ингредиента, где композиция содержит один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ.
14. Применение соли по п. 1 в изготовлении лекарственного препарата для лечения соответствующих заболеваний, опосредованных агонистом опиоидного рецептора (MOR), где указанные соответствующие заболевания, опосредованные агонистом рецептора (MOR), представляют собой боль.
15. Применение кристаллической формы I по п. 8 в изготовлении лекарственного препарата для лечения соответствующих заболеваний, опосредованных агонистом опиоидного рецептора (MOR), где указанные соответствующие заболевания, опосредованные агонистом рецептора MOR, представляют собой боль.
16. Применение фармацевтической композиции по п. 12 или 13 в изготовлении лекарственного препарата для лечения соответствующих заболеваний, опосредованных агонистом опиоидного рецептора (MOR), где указанные соответствующие заболевания, опосредованные агонистом рецептора MOR, представляют собой боль.
| Xiao-Tao Chen, Philip Pitis, et al.: "Structure-Activity Relationships and Discovery of a G Protein Biased μ Opioid Receptor Ligand, [(3-Methoxythiophen-2-yl)methyl]({ 2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl} )amine (TRV130), for the Treatment of Acute Severe Pain", Journal of Medicinal Chemistry, 2013, v.56(20), |

