Область техники, к которой относится изобретение
Настоящее изобретение относится к области медицины, и в частности относится к кристаллической форме ингибитора фосфодиэстеразы, способу ее получения и ее применению.
Предпосылки создания изобретения
Фосфодиэстераза 9 является важным членом семейства PDE и обладает очень высокой селективностью в отношении cGMP. Ингибиторы фосфодиэстеразы 9 можно применять для лечения заболеваний, связанных с когнитивными нарушениями, которые вызваны нарушениями центральной нервной системы, такими как болезнь Альцгеймера и шизофрения, а также нейродегенеративное заболевание головного мозга.
В ходе разработки лекарственных препаратов изучение кристаллических форм является очень важным, и кристаллические формы соединения сильно отличаются от других его форм с точки зрения стабильности, растворимости и т.д. Авторы настоящего изобретения провели исследования в отношении соединений, представляющих собой ингибиторы PDE9, с целью получения фармацевтически приемлемых кристаллических форм.
Краткое описание изобретения
Целью настоящего изобретения является обеспечение кристаллической формы соединения формулы (I) и способа ее получения.
Настоящее изобретение предусматривает кристаллическую форму I соединения, представленного формулой (I):
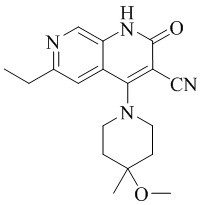
кристаллическая форма I соединения, представленного формулой (I), 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики при значениях 2θ 7,3 ± 0,2°, 13,6 ± 0,2°, 14,5 ± 0,2°, 18,0 ± 0,2°, 19,1 ± 0,2°, 22,0 ± 0,2° и 23,4 ± 0,2°, как определено с применением излучения Cu-Kα,
 … (I).
… (I).
В одном варианте осуществления настоящего изобретения в дополнение к вышеуказанным характеристическим пикам рентгеновская порошковая дифрактограмма кристаллической формы I соединения, представленного формулой (I), содержит характеристические пики при значениях 2θ 14,2 ± 0,2°, 16,1 ± 0,2°, 19,4 ± 0,2° и 25,6 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения в дополнение к вышеуказанным характеристическим пикам рентгеновская порошковая дифрактограмма кристаллической формы I соединения, представленного формулой (I), содержит характеристические пики при значениях 2θ 15,1 ± 0,2° и 17,6 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения кристаллическая форма I соединения, представленного формулой (I), характеризуется рентгеновской порошковой дифрактограммой, которая по сути изображена на фиг. 1, при применении излучения Cu-Kα.
Настоящее изобретение дополнительно предусматривает способ получения кристаллической формы I соединения, представленного формулой (I):
способ включает растворение соединения формулы (I) в одном или смешанном растворителе, повышение температуры до температуры образования флегмы до полного растворения и медленное снижение температуры до осаждения кристаллической формы I.
В одном варианте осуществления настоящего изобретения один или смешанный растворитель выбран из одного из метанола, этанола, изопропанола, толуола, ацетона, тетрагидрофурана, дихлорметана, дихлорэтана, этилацетата, ацетонитрила, метил-трет-бутилового эфира, 2-метилтетрагидрофурана, диметилсульфоксида и воды или их смеси.
В одном варианте осуществления настоящего изобретения один или смешанный растворитель выбран из метанола, этанола, изопропанола, толуола, ацетона, тетрагидрофурана, смеси вода/этанол, вода/изопропанол, дихлорметана, этилацетата и ацетонитрила.
В одном варианте осуществления настоящего изобретения выражение "снижение температуры" относится к снижению температуры до менее 30°C, предпочтительно до комнатной температуры. Выражение "комнатная температура" относится к температуре 15°C-25°C или относится к 10°C-30°C в соответствии с фармакопеей Китайской Народной Республики.
В одном варианте осуществления настоящего изобретения один или смешанный растворитель применяют в 20-30-кратном объеме относительно соединения формулы (I).
Настоящее изобретение дополнительно предусматривает другой способ получения кристаллической формы I соединения, представленного формулой (I):
способ включает полное растворение соединения формулы (I) в одном или смешанном растворителе, и выпаривание одного или смешанного растворителя до насыщения системы и осаждения кристаллической формы I.
В одном варианте осуществления настоящего изобретения один или смешанный растворитель выбран из одного из метанола, этанола, изопропанола, толуола, ацетона, тетрагидрофурана, дихлорметана, дихлорэтана, этилацетата, ацетонитрила, метил-трет-бутилового эфира, 2-метилтетрагидрофурана, диметилсульфоксида и воды или их смеси.
В одном варианте осуществления настоящего изобретения один или смешанный растворитель выбран из метанола, этанола, изопропанола, толуола, ацетона, тетрагидрофурана, смеси вода/этанол, вода/изопропанол, дихлорметана, этилацетата, ацетонитрила, смеси дихлорметан/ацетон, дихлорметан/ацетонитрил, дихлорметан/этилацетат, дихлорметан/метил-трет-бутиловый эфир, дихлорметан/тетрагидрофуран, дихлорметан/этанол, дихлорметан/изопропанол, дихлорметан/толуол, дихлорметан/вода/этанол и дихлорметан/вода/изопропанол.
В одном варианте осуществления настоящего изобретения один растворитель выбран из метанола, этанола, изопропанола, толуола, ацетона, тетрагидрофурана, дихлорметана, этилацетата и ацетонитрила.
В одном варианте осуществления настоящего изобретения смешанный растворитель выбран из смеси дихлорметана и ацетона, ацетонитрила, этилацетата, метил-трет-бутилового эфира, тетрагидрофурана, этанола, изопропанола, толуола, смеси вода/этанол и вода/изопропанол в объемном соотношении 1: 0,5-3, предпочтительно 1: 1,5-3.
В одном варианте осуществления настоящего изобретения один или смешанный растворитель применяют в 30-кратном объеме относительно соединения формулы (I).
В одном варианте осуществления настоящего изобретения выражение "выпаривание" может относиться к выпариванию в естественных условиях или выпариванию при нагревании до определенной температуры.
Настоящее изобретение предусматривает кристаллическую форму II гидрохлорида соединения, представленного формулой (I):
кристаллическая форма II гидрохлорида соединения, представленного формулой (I), 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики при значениях 2θ 4,0 ± 0,2°, 6,7 ± 0,2°, 7,9 ± 0,2°, 13,5 ± 0,2°, 14,2 ± 0,2°, 15,4 ± 0,2°, 20,2 ± 0,2° и 22,0 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения в дополнение к вышеуказанным характеристическим пикам рентгеновская порошковая дифрактограмма кристаллической формы II соединения, представленного формулой (I), содержит характеристические пики при значениях 2θ 10,2 ± 0,2°, 13,8 ± 0,2°, 14,6 ± 0,2° и 26,40 ± 0,2° и 26,80 ± 0,2° при применении излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения кристаллическая форма II соединения, представленного формулой (I), характеризуется рентгеновской порошковой дифрактограммой, которая по сути изображена на фиг. 6, при применении излучения Cu-Kα.
Настоящее изобретение дополнительно предусматривает способ получения кристаллической формы II соединения, представленного формулой (I):
способ включает добавление одного или смешанного растворителя к гидрохлориду соединения формулы (I), нагревание смеси до полного растворения и медленное охлаждение нагретой смеси до осаждения кристаллической формы II.
В одном варианте осуществления настоящего изобретения один или смешанный растворитель выбран из одного из метанола, этанола, изопропанола и воды или их смеси; предпочтительно один или смешанный растворитель выбран из метанола, этанола, изопропанола, смеси вода/метанол, вода/этанол и вода/изопропанол.
В одном варианте осуществления настоящего изобретения выражение "охлаждение" относится к охлаждению до комнатной температуры. Выражение "комнатная температура" относится к температуре 15°C-25°C или относится к 10°C-30°C в соответствии с фармакопеей Китайской Народной Республики.
Настоящее изобретение предусматривает кристаллическую форму III гидрохлорида соединения, представленного формулой (I):
кристаллическая форма III гидрохлорида соединения, представленного формулой (I), 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики при значениях 2θ 5,2 ± 0,2°, 6,4 ± 0,2°, 15,3 ± 0,2°, 18,6 ± 0,2°, 22,0 ± 0,2° и 26,4 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения в дополнение к вышеуказанным характеристическим пикам рентгеновская порошковая дифрактограмма кристаллической формы III соединения, представленного формулой (I), содержит характеристические пики при значениях 2θ 8,0 ± 0,2°, 10,3 ± 0,2°, 13,5 ± 0,2° и 25,0 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения кристаллическая форма III соединения, представленного формулой (I), характеризуется рентгеновской порошковой дифрактограммой, которая по сути изображена на фиг. 9, при применении излучения Cu-Kα.
Настоящее изобретение дополнительно предусматривает способ получения кристаллической формы III соединения, представленного формулой (I):
способ включает добавление одного или смешанного растворителя к гидрохлориду соединения формулы (I), нагревание смеси до полного растворения, проведение горячего фильтрования и концентрирование фильтрата до осаждения кристаллической формы III.
В одном варианте осуществления настоящего изобретения один или смешанный растворитель выбран из одного из ацетонитрила, ацетона, тетрагидрофурана и этилацетата или их смеси.
Настоящее изобретение предусматривает кристаллическую форму IV п-толуолсульфоната соединения, представленного формулой (I):
кристаллическая форма IV п-толуолсульфоната соединения, представленного формулой (I), 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики при значениях 2θ 5,8 ± 0,2°, 7,8 ± 0,2°, 9,3 ± 0,2°, 11,3 ± 0,2°, 13,7 ± 0,2°, 14,8 ± 0,2° и 15,7 ± 0,2°, как определено с применением излучения Cu-Kα,
 … (I).
… (I).
В одном варианте осуществления настоящего изобретения в дополнение к вышеуказанным характеристическим пикам рентгеновская порошковая дифрактограмма кристаллической формы IV соединения, представленного формулой (I), содержит характеристические пики при значениях 2θ 17,1 ± 0,2°, 18,7 ± 0,2°, 20,0 ± 0,2° и 22,4 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения кристаллическая форма IV соединения, представленного формулой (I), характеризуется рентгеновской порошковой дифрактограммой, которая по сути изображена на фиг. 12, при применении излучения Cu-Kα.
Настоящее изобретение дополнительно предусматривает способ получения кристаллической формы IV соединения, представленного формулой (I):
способ включает добавление соединения формулы (I) к п-толуолсульфоновой кислоте, нагревание смеси до полного растворения и медленное снижение температуры до осаждения кристаллической формы IV.
В одном варианте осуществления настоящего изобретения выражение "снижение температуры" относится к снижению температуры до менее 30°C, предпочтительно до комнатной температуры. Выражение "комнатная температура" предпочтительно относится к температуре 15°C-25°C или 10°C-30°C в соответствии с фармакопеей Китайской Народной Республики.
В одном варианте осуществления настоящего изобретения п-толуолсульфоновую кислоту применяют в 5-30-кратном объеме относительно соединения формулы (I).
В одном варианте осуществления настоящего изобретения п-толуолсульфоновая кислота может представлять собой п-толуолсульфоновую кислоту различной концентрации или водный раствор п-толуолсульфоновой кислоты.
Настоящее изобретение предусматривает кристаллическую форму V метансульфоната соединения, представленного формулой (I):
кристаллическая форма V метансульфоната соединения, представленного формулой (I), 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики при значениях 2θ 7,0 ± 0,2°, 9,7 ± 0,2°, 13,2 ± 0,2°, 18,1 ± 0,2°, 19,5 ± 0,2°, 20,7 ± 0,2° и 21,7 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения в дополнение к вышеуказанным характеристическим пикам рентгеновская порошковая дифрактограмма кристаллической формы V соединения, представленного формулой (I), содержит характеристические пики при значениях 2θ 16,9 ± 0,2°, 18,6 ± 0,2°, 19,1 ± 0,2° и 20,2 ± 0,2° и 28,0 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения кристаллическая форма V соединения, представленного формулой (I), характеризуется рентгеновской порошковой дифрактограммой, которая по сути изображена на фиг. 13, при применении излучения Cu-Kα.
Настоящее изобретение дополнительно предусматривает способ получения кристаллической формы V соединения, представленного формулой (I):
способ включает добавление соединения формулы (I) к метансульфоновой кислоте, нагревание смеси до полного растворения, а затем добавление растворителя, подвергание смеси фильтрованию с отсасыванием и высушивание подвергнутой фильтрованию с отсасыванием смеси с получением V, где растворитель представляет собой метанол, этанол или изопропанол.
В одном варианте осуществления настоящего изобретения метансульфоновую кислоту применяют в 5-30-кратном объеме относительно соединения формулы (I).
В одном варианте осуществления настоящего изобретения метансульфоновая кислота может представлять собой метансульфоновую кислоту различной концентрации или водный раствор метансульфоновой кислоты.
Настоящее изобретение предусматривает кристаллическую форму VI сульфата соединения, представленного формулой (I):
кристаллическая форма VI сульфата соединения, представленного формулой (I), 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики при значениях 2θ 4,4 ± 0,2°, 7,1 ± 0,2°, 8,8 ± 0,2°, 14,3 ± 0,2°, 17,8 ± 0,2°, 19,6 ± 0,2° и 21,6 ± 0,2°, как определено с применением излучения Cu-Kα,
… (I).
В одном варианте осуществления настоящего изобретения в дополнение к вышеуказанным характеристическим пикам рентгеновская порошковая дифрактограмма кристаллической формы VI соединения, представленного формулой (I), содержит характеристические пики при значениях 2θ 25,5 ± 0,2° и 27,7 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения кристаллическая форма VI соединения, представленного формулой (I), характеризуется рентгеновской порошковой дифрактограммой, которая по сути изображена на фиг. 14, при применении излучения Cu-Kα.
Настоящее изобретение дополнительно предусматривает способ получения кристаллической формы VI соединения, представленного формулой (I):
способ включает добавление соединения формулы (I) к водному раствору серной кислоты, нагревание смеси до полного растворения и медленное снижение температуры до осаждения кристаллической формы VI сульфата соединения, представленного формулой (I); или
добавление сульфата соединения формулы (I) к одному или смешанному растворителю, нагревание смеси до полного растворения и медленное снижение температуры до осаждения кристаллической формы VI.
В одном варианте осуществления настоящего изобретения выражение "снижение температуры" относится к снижению температуры до менее 30°C, предпочтительно до комнатной температуры. Выражение "комнатная температура" относится к температуре 10°C-30°C, предпочтительно к температуре 15°C-25°C.
В одном варианте осуществления настоящего изобретения водный раствор серной кислоты применяют в 2-30-кратном, предпочтительно в 2-10-кратном, более предпочтительно в 4-кратном, 5-кратном и 6-кратном объеме относительно соединения формулы (I).
В одном варианте осуществления настоящего изобретения один или смешанный растворитель применяют в 5-60-кратном, предпочтительно в 5-50-кратном, более предпочтительно в 10-кратном, 15-кратном и 50-кратном объеме относительно сульфата соединения формулы (I).
В одном варианте осуществления настоящего изобретения один или смешанный растворитель выбран из одного из ацетона, этанола, метанола и тетрагидрофурана или их смеси; предпочтительно растворитель представляет собой один растворитель; более предпочтительно один растворитель выбран из ацетона, этанола, метанола и тетрагидрофурана.
Настоящее изобретение предусматривает кристаллическую форму VII сульфата соединения, представленного формулой (I):
кристаллическая форма VII сульфата соединения, представленного формулой (I), 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики при значениях 2θ 7,1 ± 0,2°, 13,6 ± 0,2°, 14,5 ± 0,2°, 18,0 ± 0,2°, 19,0 ± 0,2°, 22,0 ± 0,2° и 23,4 ± 0,2°, как определено с применением излучения Cu-Kα,
… (I).
В одном варианте осуществления настоящего изобретения в дополнение к вышеуказанным характеристическим пикам рентгеновская порошковая дифрактограмма кристаллической формы VII соединения, представленного формулой (I), содержит характеристические пики при значениях 2θ 15,0 ± 0,2°, 16,1 ± 0,2°, 17,4 ± 0,2° и 19,4 ± 0,2°, как определено с применением излучения Cu-Kα.
В одном варианте осуществления настоящего изобретения кристаллическая форма VII соединения, представленного формулой (I), характеризуется рентгеновской порошковой дифрактограммой, которая по сути изображена на фиг. 18, при применении излучения Cu-Kα.
Настоящее изобретение дополнительно предусматривает способ получения кристаллической формы VII соединения, представленного формулой (I):
способ включает добавление сульфата соединения формулы (I) к изопропанолу, нагревание смеси до полного растворения и медленное снижение температуры до осаждения кристаллической формы VII.
В одном варианте осуществления настоящего изобретения выражение "снижение температуры" относится к снижению температуры до менее 30°C, предпочтительно до комнатной температуры. Выражение "комнатная температура" относится к температуре 10°C-30°C, предпочтительно к температуре 15°C-25°C.
В одном варианте осуществления настоящего изобретения изопропанол применяют в 5-60-кратном, предпочтительно в 10-50-кратном, более предпочтительно в 30-50-кратном объеме относительно кристаллической формы VI.
Настоящее изобретение дополнительно предусматривает фармацевтическую композицию, содержащую кристаллическую форму I, II, III, IV, V, VI или VII соединения, представленного формулой (I), и одно или несколько дополнительных терапевтически активных средств.
Настоящее изобретение дополнительно предусматривает фармацевтический препарат, содержащий кристаллическую форму I, II, III, IV, V, VI или VII соединения, представленного формулой (I).
В одном варианте осуществления настоящего изобретения фармацевтический препарат может содержать один или несколько фармацевтических носителей.
Фармацевтический носитель по настоящему изобретению может предусматривать один или несколько твердых или жидких наполнителей, подходящих для медицинского применения. Фармацевтический носитель предпочтительно имеет достаточную чистоту и достаточно низкую токсичность и совместим с соединением, предусмотренным в настоящем изобретении, без значительного снижения эффективности соединения. Например, фармацевтический носитель может представлять собой наполнитель, связующее вещество, разрыхлитель, смазывающее вещество, водный растворитель, неводный растворитель и т.д.
Фармацевтический препарат по настоящему изобретению может быть получен в любой фармацевтически приемлемой лекарственной форме, и "терапевтически эффективное количество" кристаллической формы I, II, III, IV, V, VI или VII соединения формулы (I), как описано выше, вводят пациенту или субъекту, нуждающемуся в таком лечении, любым подходящим способом введения, как например посредством перорального, парентерального, ректального или легочного введения. При применении для перорального введения фармацевтический препарат может быть получен в виде таблетки, капсулы, пилюли, гранулы и т.д. При применении для парентерального введения фармацевтический препарат может быть получен в виде инъекции, стерильного порошка для инъекции и т.д.
Настоящее изобретение дополнительно предусматривает применение кристаллической формы I, II, III, IV, V, VI или VII соединения, представленного формулой (I), фармацевтического препарата или фармацевтической композиции, содержащих кристаллическую форму I, II, III, IV, V, VI или VII, в изготовлении лекарственного препарата для лечения или предупреждения заболеваний, опосредованных PDE9. В частности, заболевания, опосредованные PDE9, включают заболевания CNS и, более конкретно, включают нарушения, связанные с восприятием, вниманием, памятью и обучением, старческую деменцию, шизофрению, возрастную потерю памяти, сосудистую деменцию, черепно-мозговую травму, инсульт, постинсультную деменцию, посттравматическую деменцию, общий дефицит внимания, дефицит внимания с проблемами обучения и памяти у детей, болезнь Альцгеймера, деменцию с тельцами Леви, деменцию с дегенерацией лобно-височной доли, деменцию с кортикобазальной ганглиозной дегенерацией, боковой амиотрофический склероз, болезнь Хантингтона, рассеянный склероз, таламическую дегенерацию, деменцию при болезни Крейтцфельдта-Якоба, деменцию при HIV, шизофрению, эпилепсию, психоз Корсакова, депрессию, биполярное аффективное расстройство и т.д. В частности, заболевания, опосредованные PDE9, включают заболевания, связанные с CNS, а точнее, нарушение сна, метаболический синдром, ожирение, диабет, гипергликемию, дислипидемию, нарушение толерантности к глюкозе и т.д. В частности, заболевания, опосредованные PDE9, включают сердечно-сосудистые заболевания и заболевания крови, а более конкретно, сердечную недостаточность, анемию, серповидноклеточную анемию и т.д.
Настоящее изобретение дополнительно предусматривает применение кристаллической формы I, II, III, IV, V, VI или VII соединения, представленного формулой (I), фармацевтического препарата или фармацевтической композиции, содержащих кристаллическую форму I, II, IV, V, VI или VII, для лечения или предупреждения заболеваний, опосредованных PDE9.
Настоящее изобретение дополнительно предусматривает способ лечения или предупреждения заболеваний, опосредованных PDE9, при этом способ включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллической формы I, II, III, IV, V, VI или VII формулы (I), как описано выше, фармацевтического препарата или фармацевтической композиции, содержащих кристаллическую форму I, II, IV, V, VI или VII.
Подробное описание изобретения
Выражение "комнатная температура" в настоящем изобретении относится к температуре внутри помещения, которая, как правило, составляет 15°C-25°C, или относится к температуре 10°C-30°C в соответствии с фармакопеей Китайской Народной Республики.
Выражение "кратный объем" в настоящем изобретении относится к объему растворителя (мл), необходимому для растворения 1 г вещества; например, если объем растворителя, необходимый для растворения 1 г соединения формулы (I), составляет 20 мл, его называют 20-кратным объемом.
Выражение "терапевтически эффективное количество" в настоящем изобретении относится к количеству вышеуказанного соединения или его фармацевтически приемлемой соли и стереоизомера, композиции или фармацевтического препарата, которое при введении пациенту может по меньшей мере облегчить симптомы состояния пациента. Фактическое количество, включающее "терапевтически эффективное количество", будет варьировать в зависимости от различных состояний, включающих без ограничения конкретные состояния, подлежащие лечению, тяжесть состояния, физическое состояние и состояние здоровья пациента и путь введения. Подходящее количество может быть легко определено квалифицированным практикующим врачом с применением способов, известных в области медицины.
Превосходный эффект настоящего изобретения
В ходе исследования настоящего изобретения было обнаружено, что кристаллические формы по настоящему изобретению обладают превосходными физико-химическими свойствами и фармацевтической стабильностью, а также обладают лучшими фармакодинамическими и фармакокинетическими свойствами, что в большей степени способствует поиску биологической мишени, с которой может связываться лекарственный препарат.
Краткое описание графических материалов
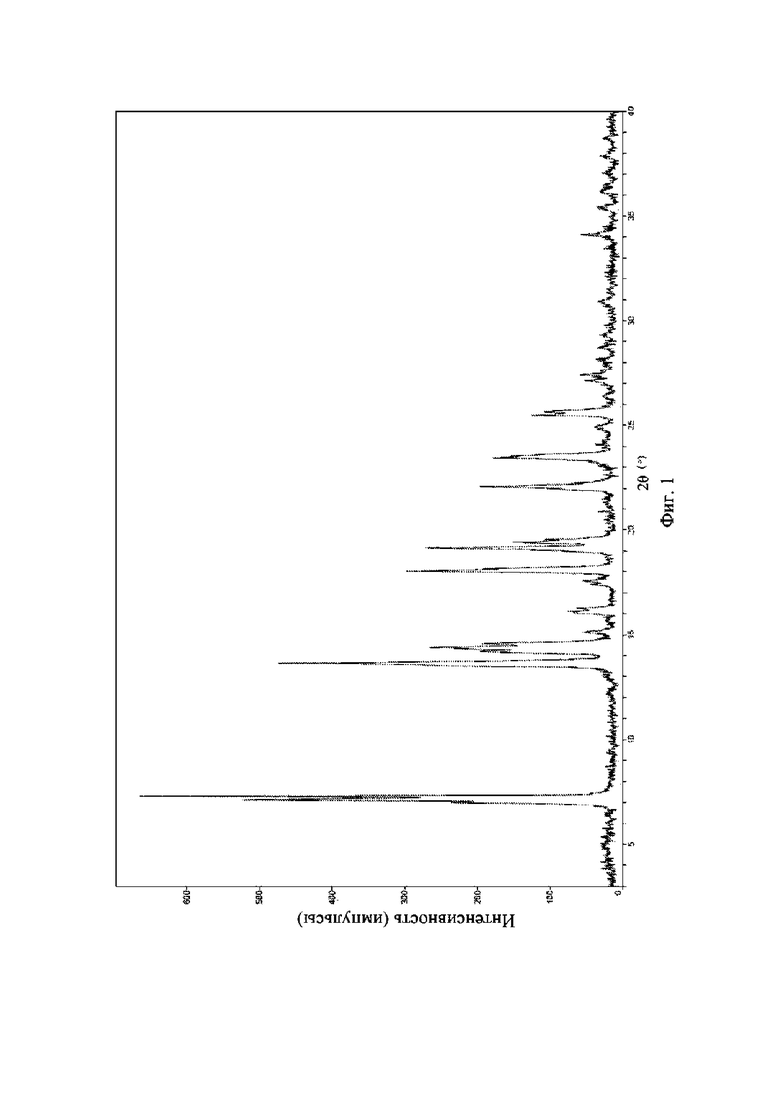
На фиг. 1 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы I соединения формулы (I).
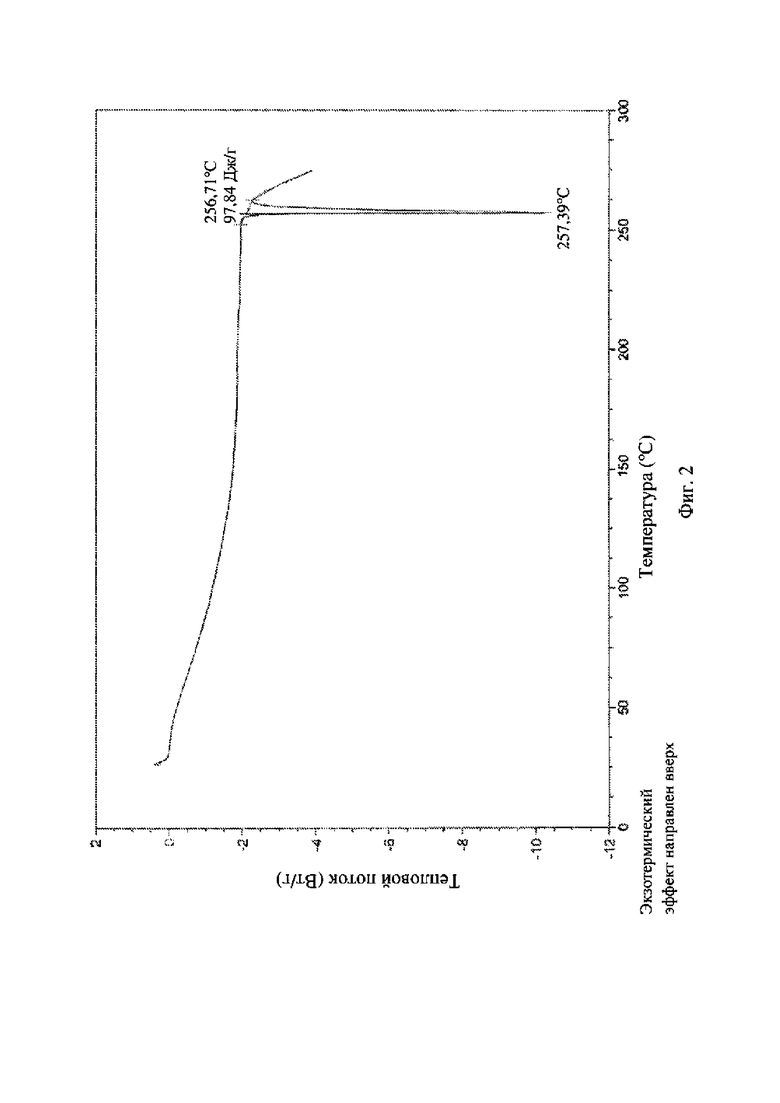
На фиг. 2 представлена термограмма дифференциальной сканирующей калориметрии (DSC) кристаллической формы I соединения формулы (I).
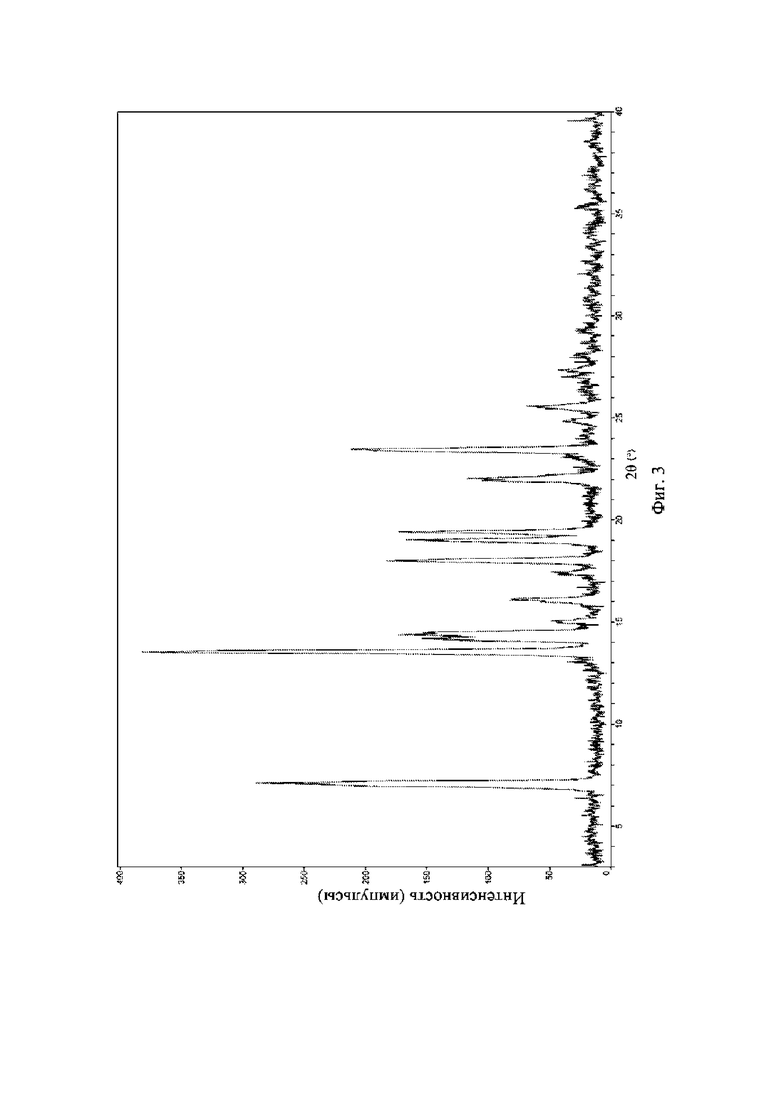
На фиг. 3 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы I соединения формулы (I).
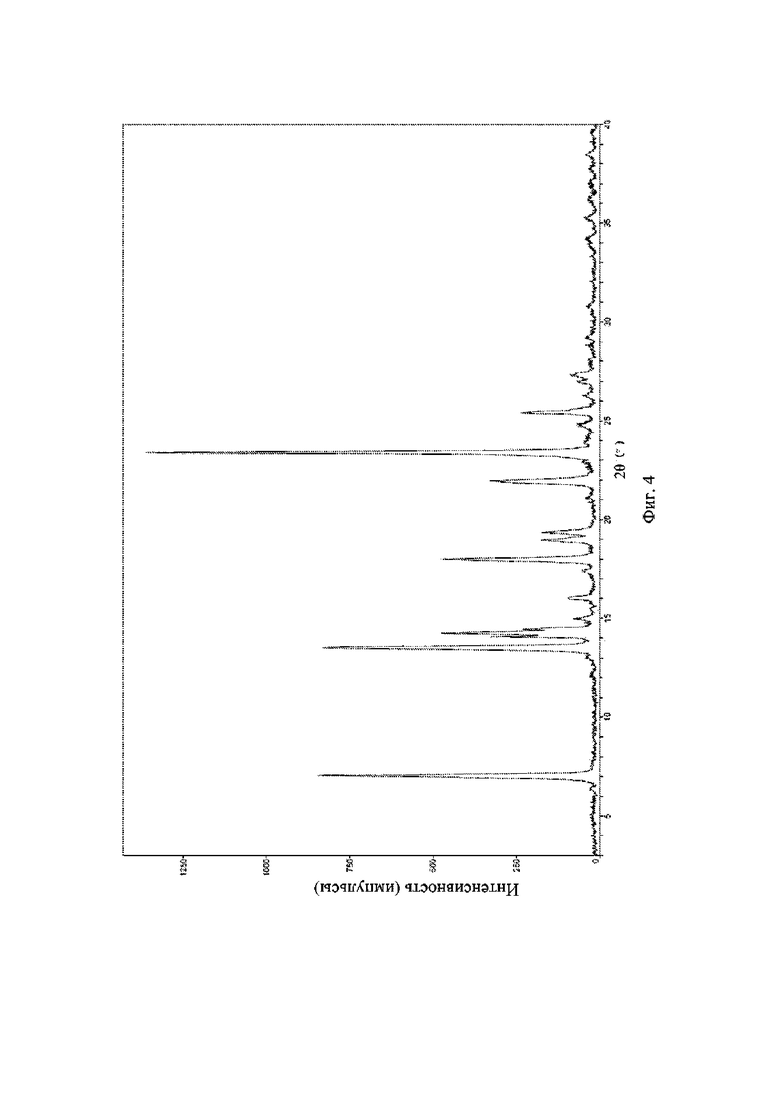
На фиг. 4 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы I соединения формулы (I).
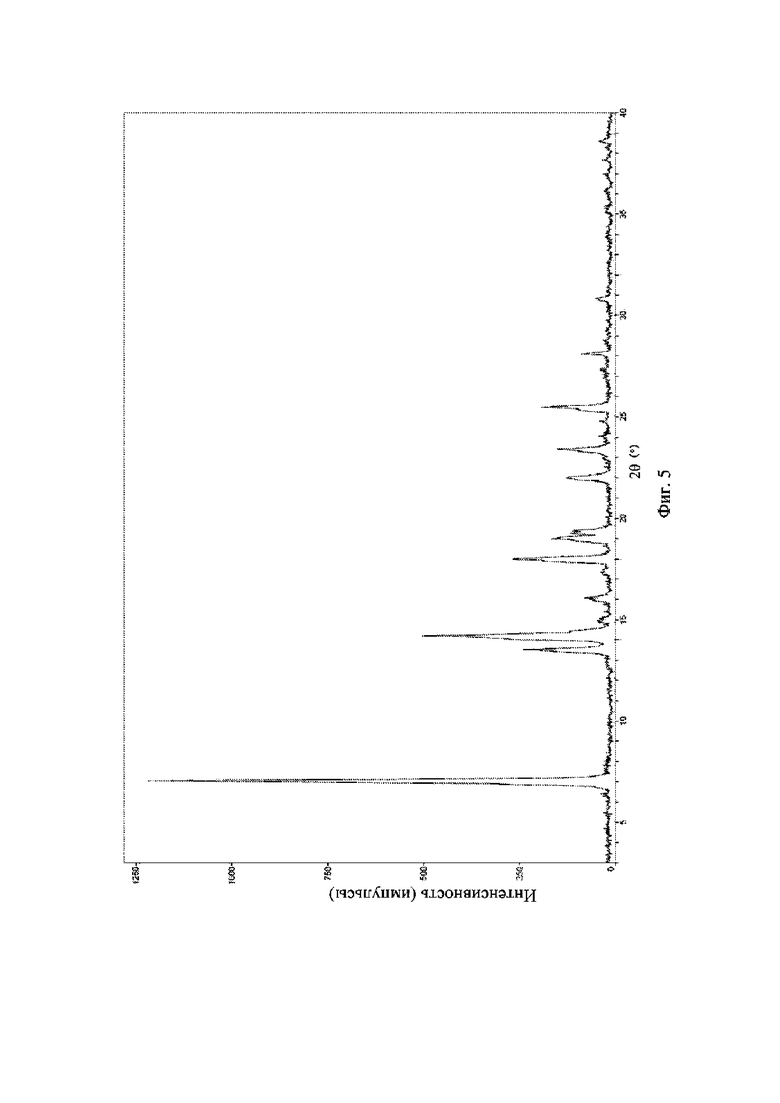
На фиг. 5 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы I соединения формулы (I).
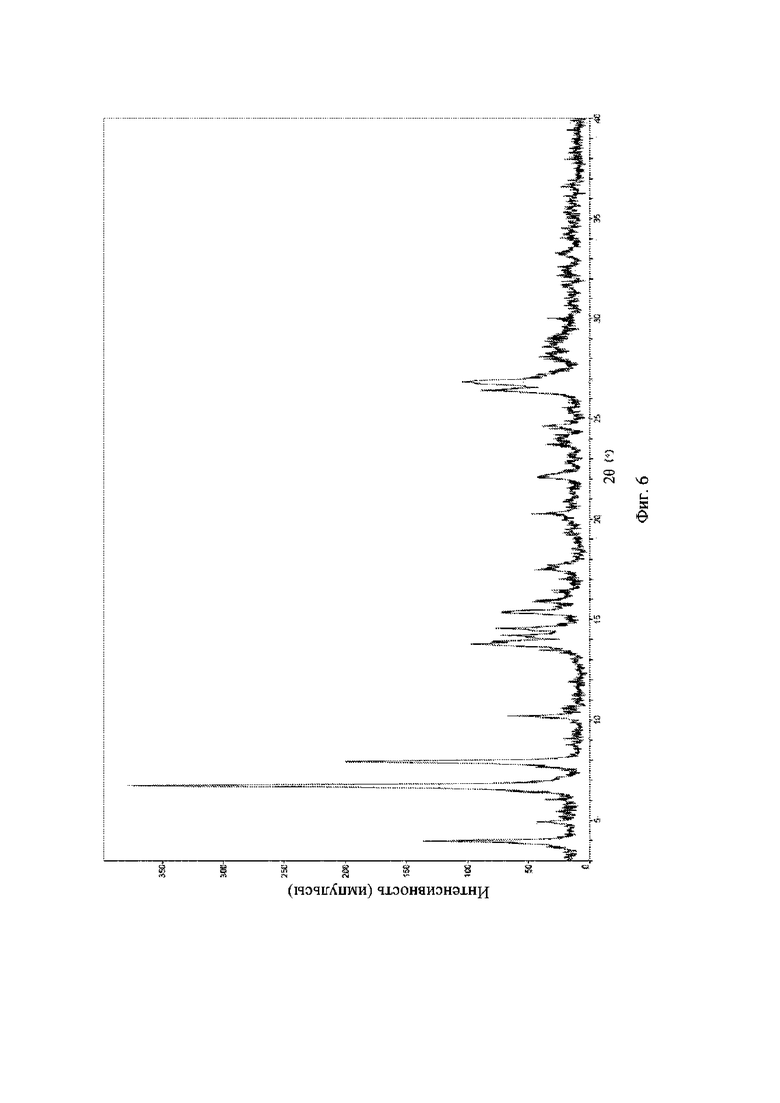
На фиг. 6 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы II соединения формулы (I).
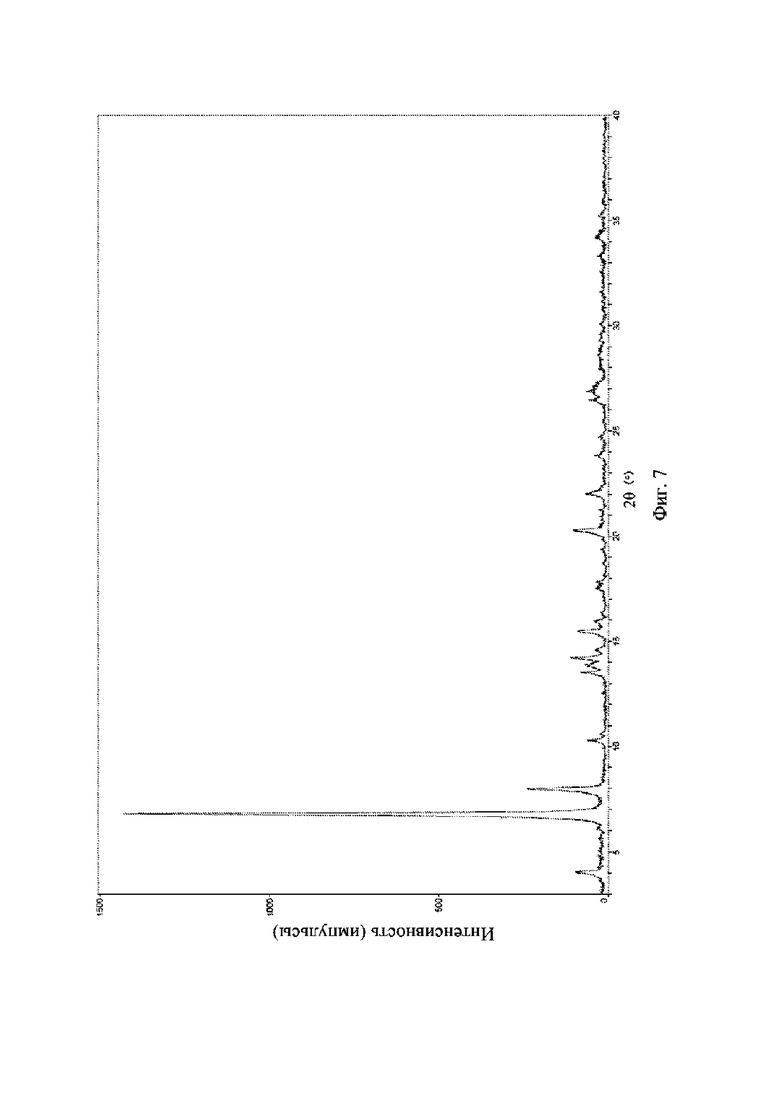
На фиг. 7 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы II соединения формулы (I).
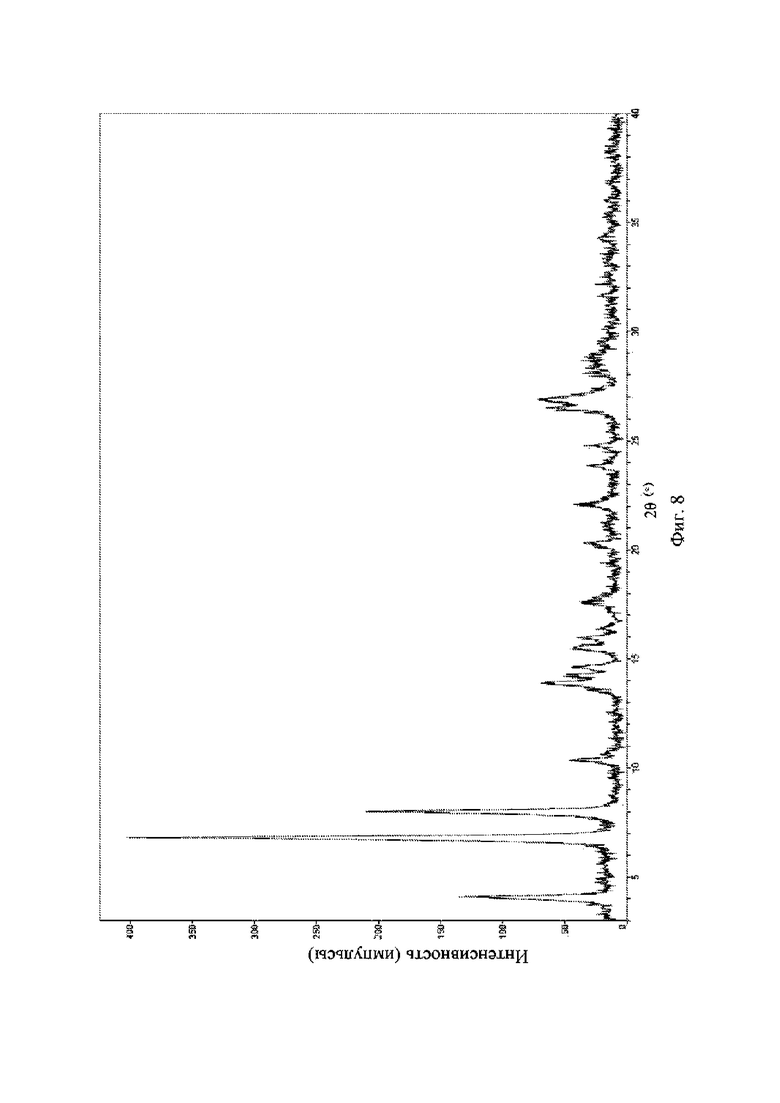
На фиг. 8 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы II соединения формулы (I).
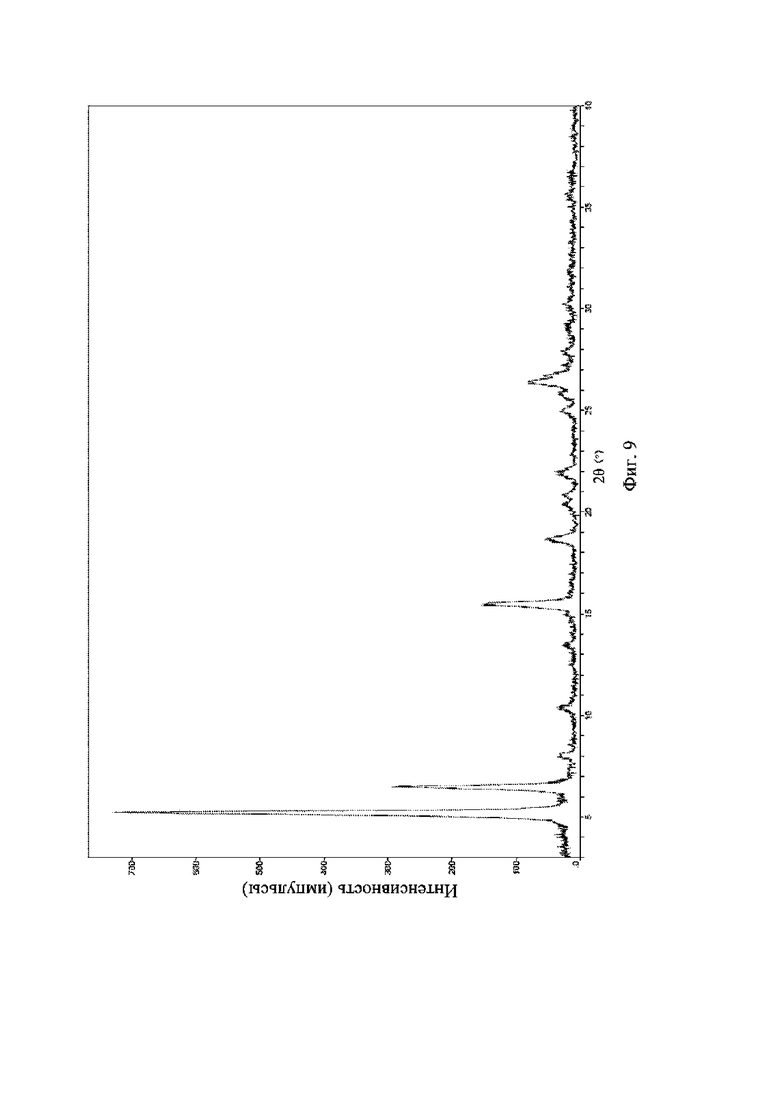
На фиг. 9 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы III соединения формулы (I).
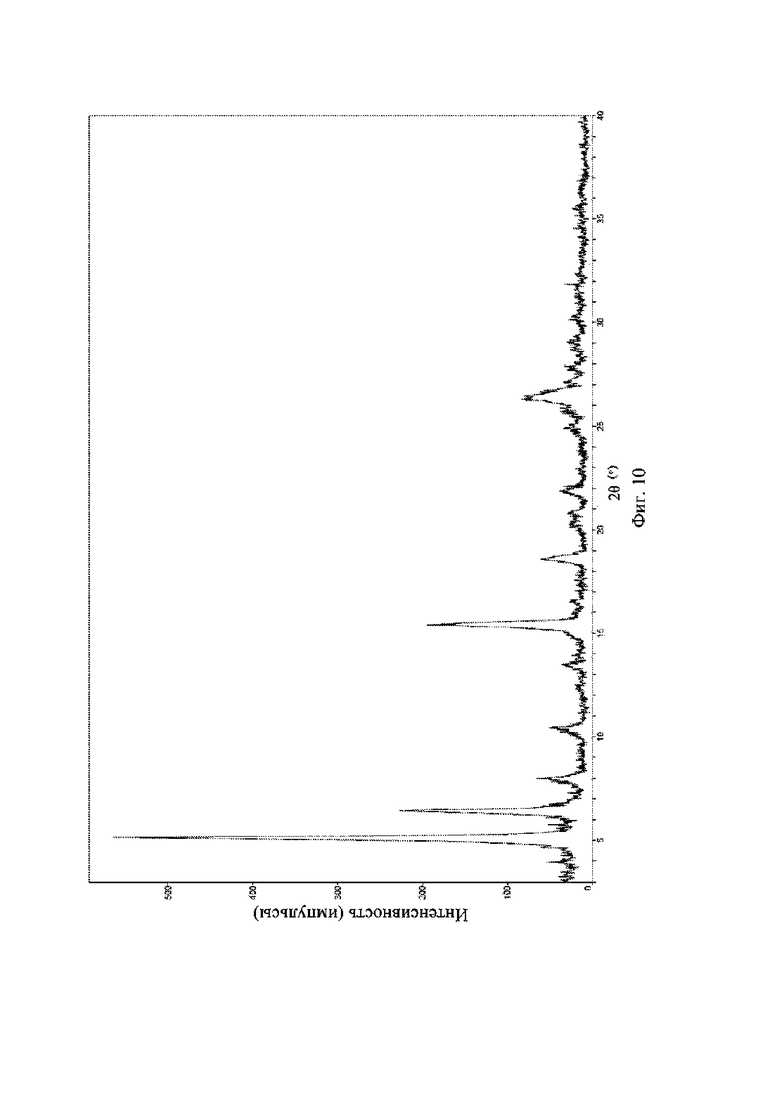
На фиг. 10 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы III соединения формулы (I).
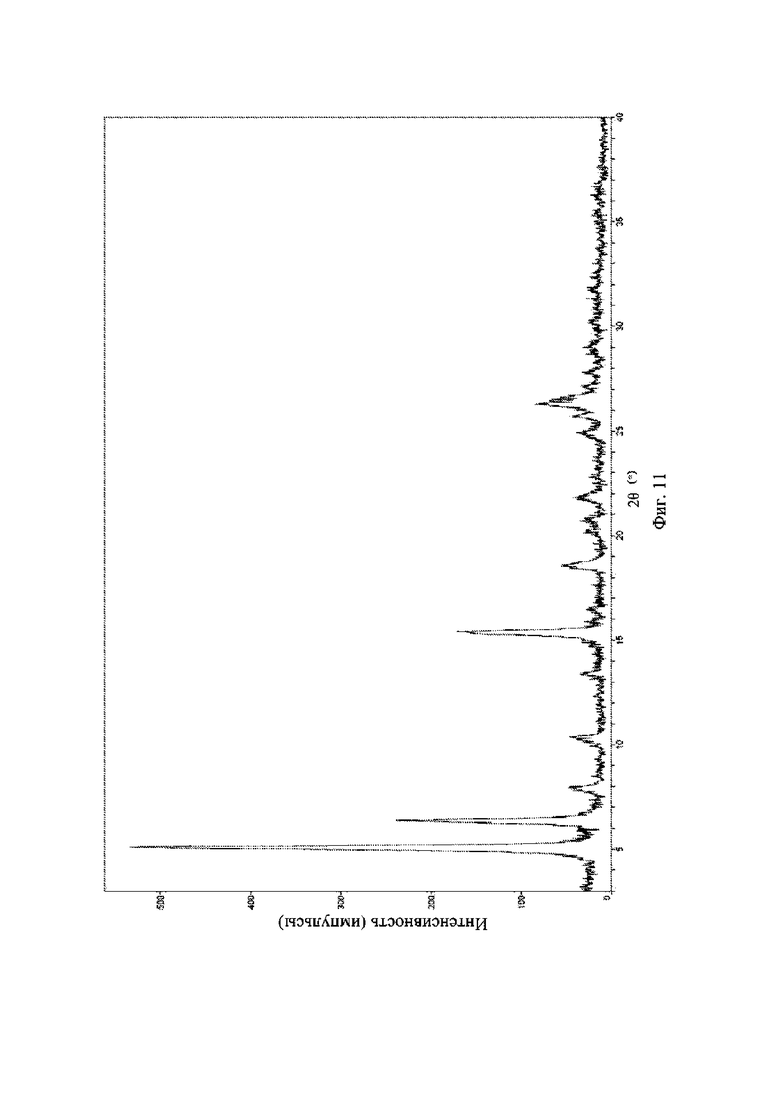
На фиг. 11 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы III соединения формулы (I).
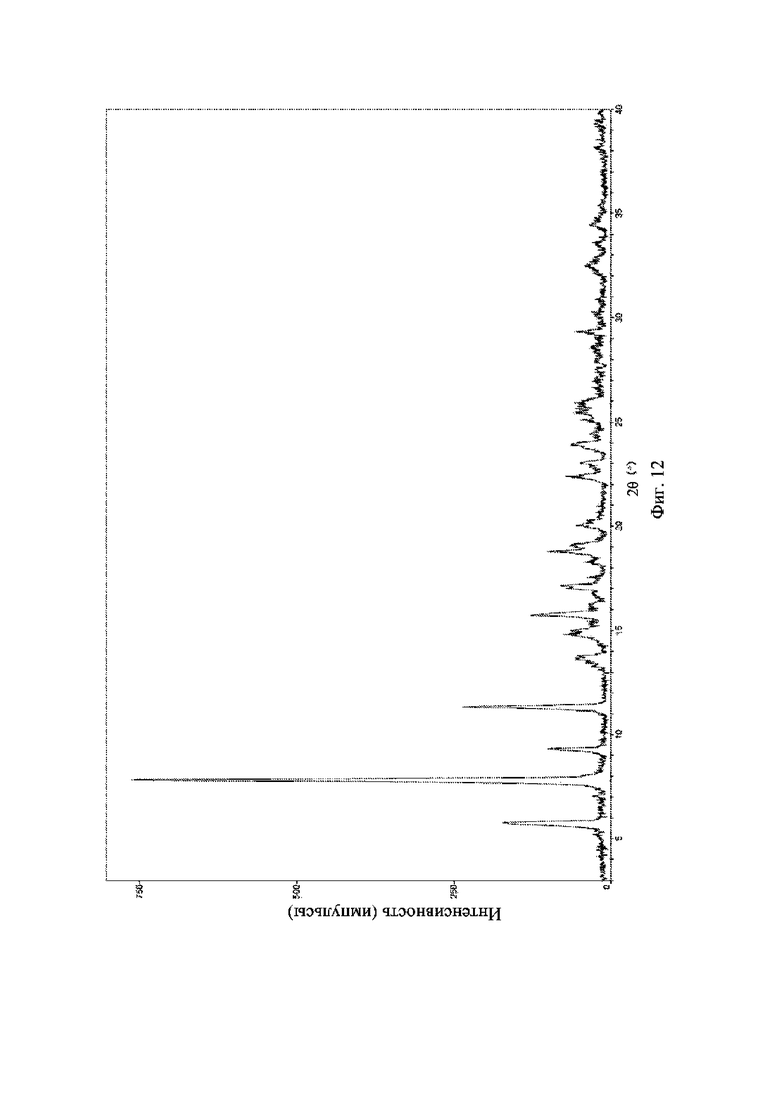
На фиг. 12 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы IV соединения формулы (I).
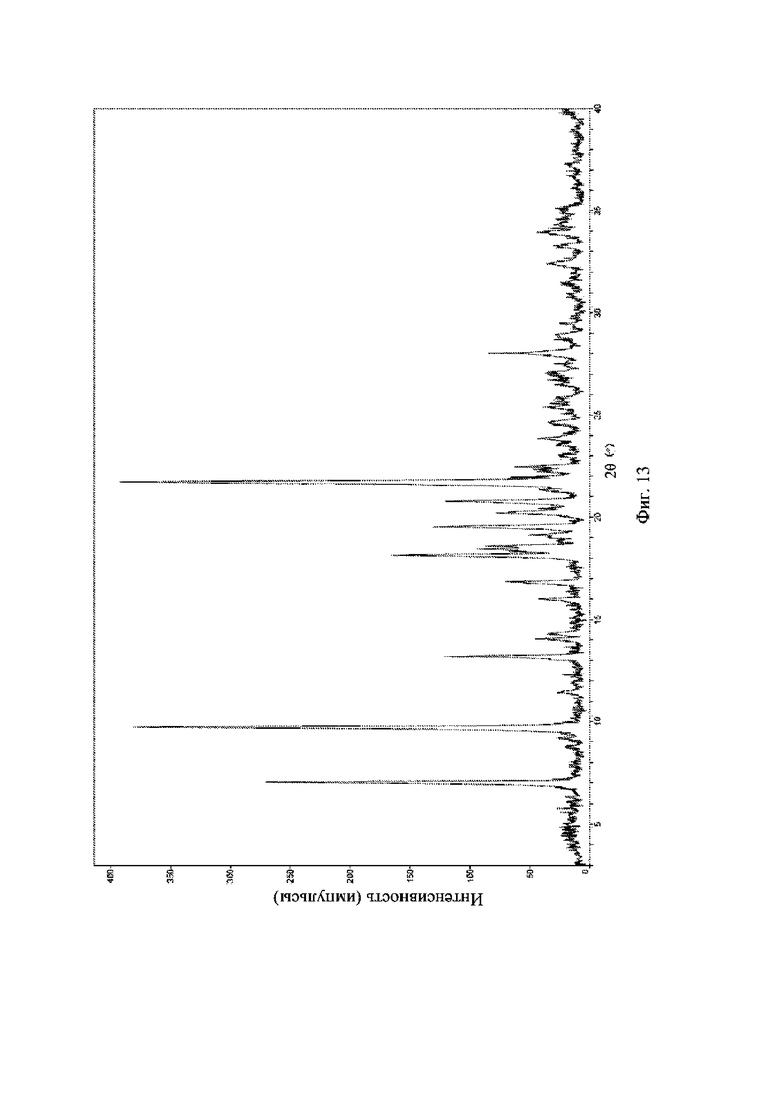
На фиг. 13 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы V соединения формулы (I).
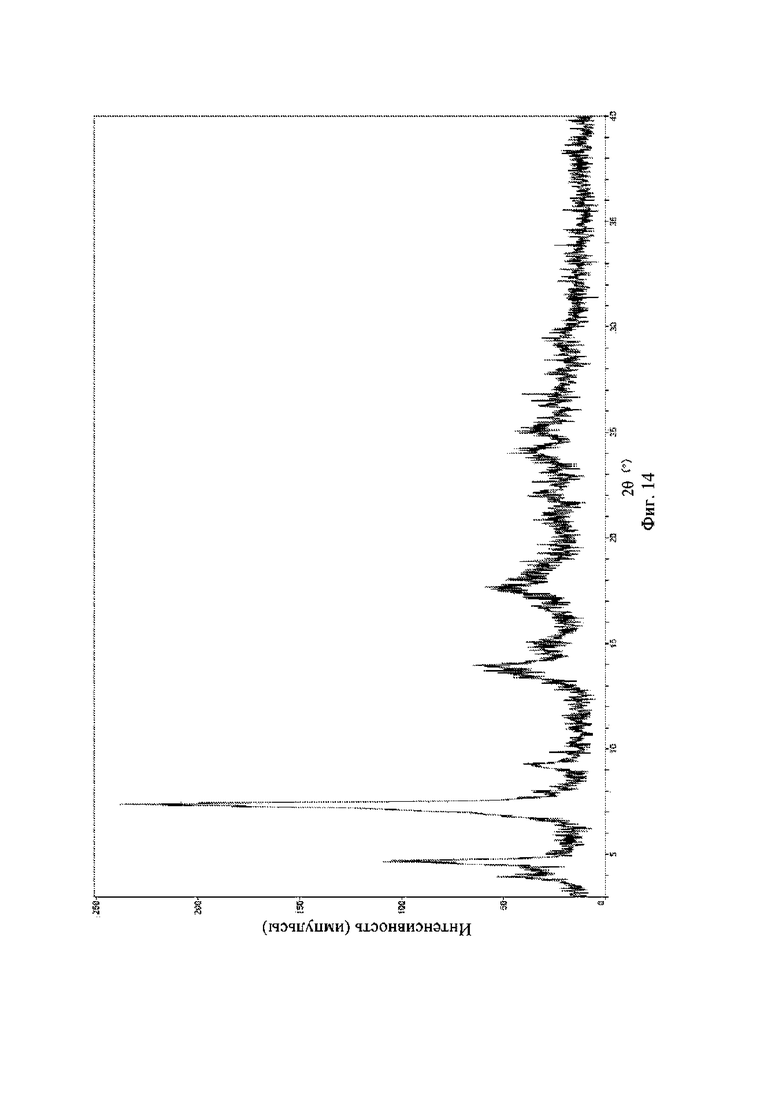
На фиг. 14 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы VI соединения формулы (I).
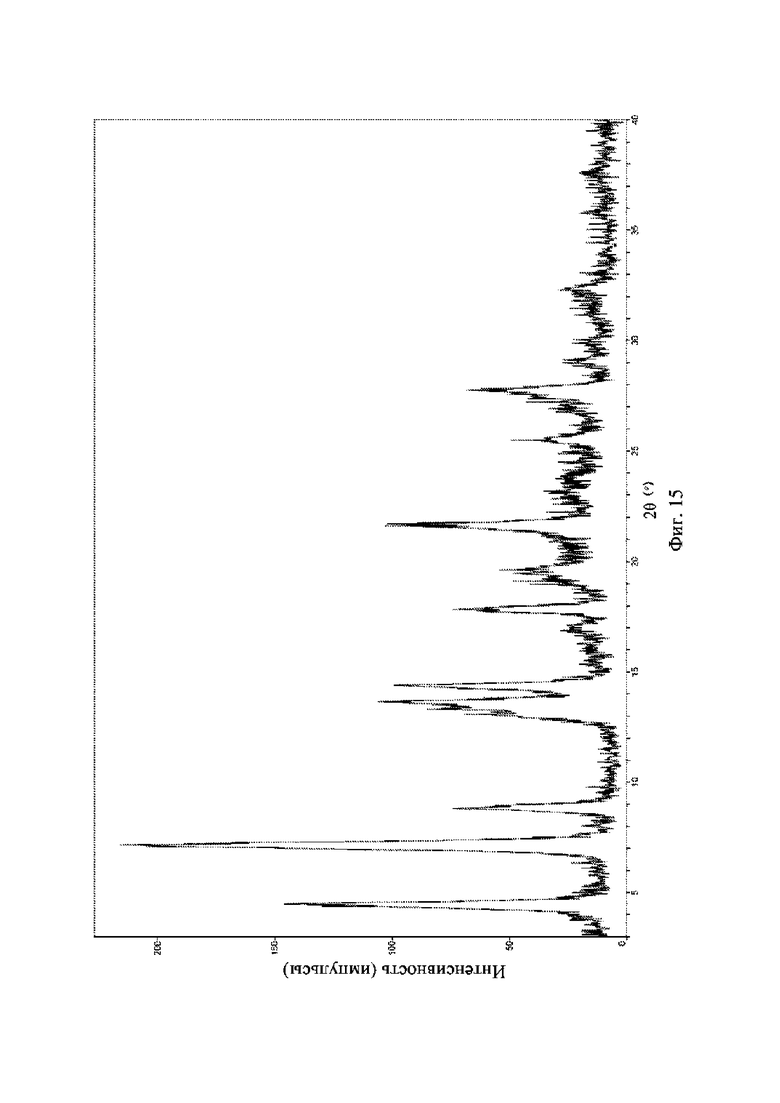
На фиг. 15 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы VI соединения формулы (I).
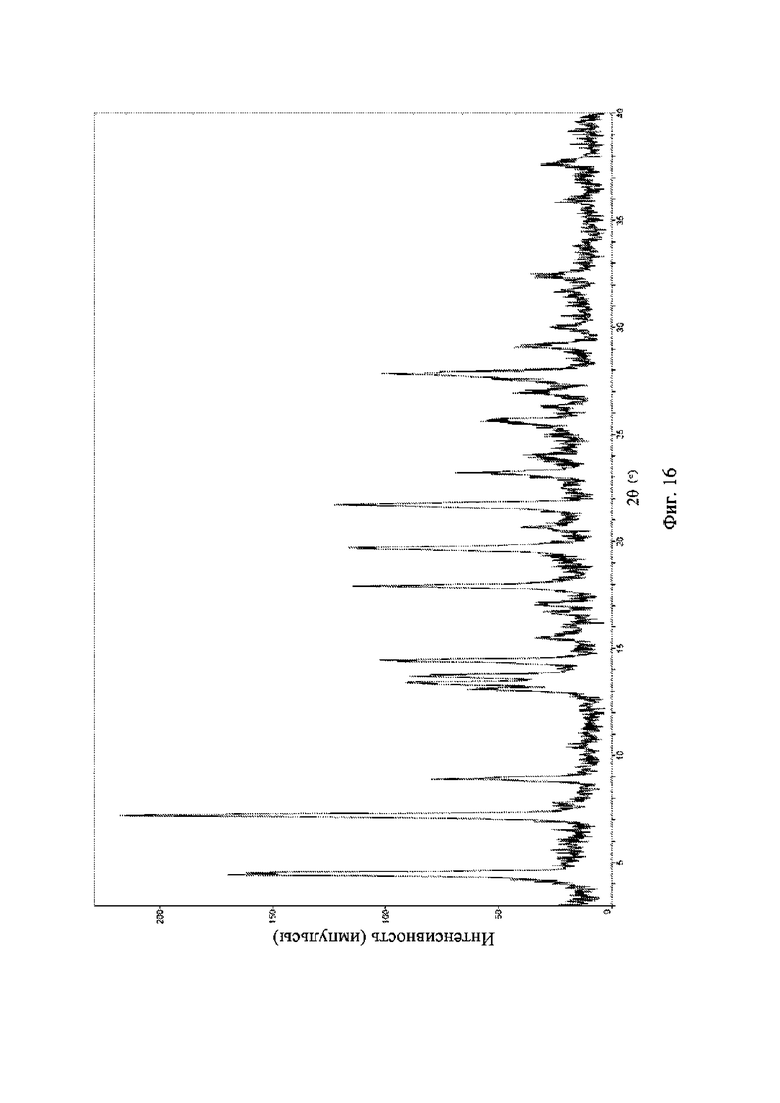
На фиг. 16 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы VI соединения формулы (I).
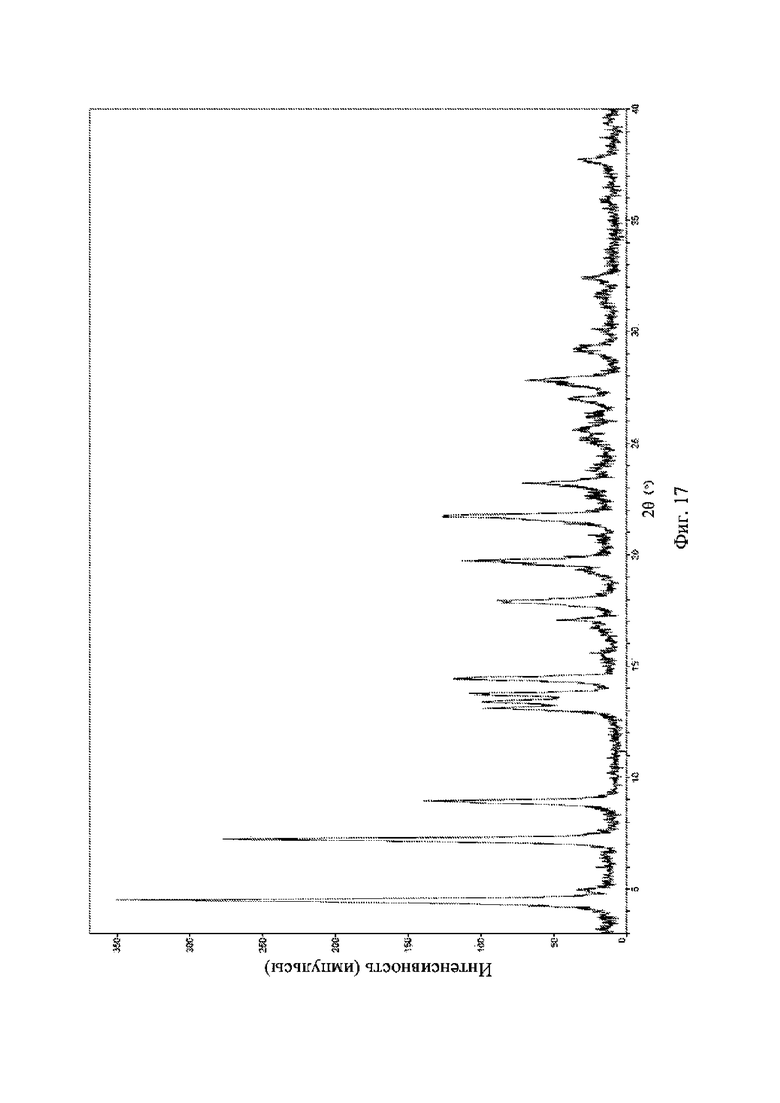
На фиг. 17 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы VI соединения формулы (I).
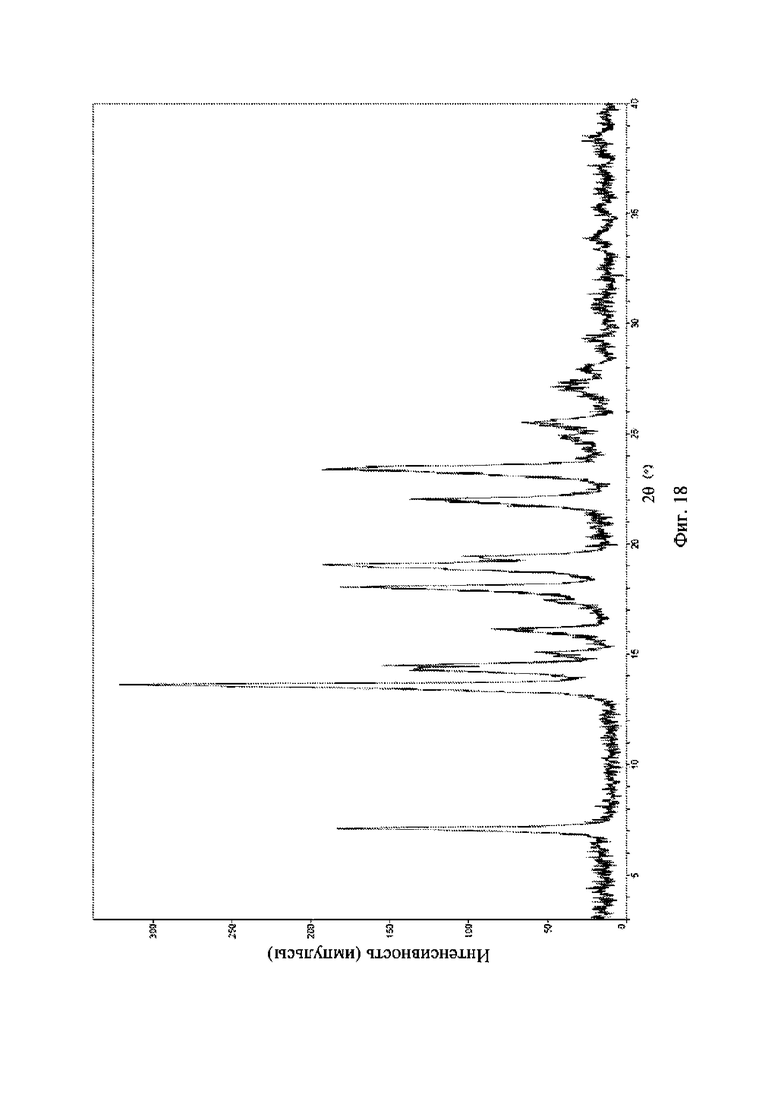
На фиг. 18 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы VII соединения формулы (I).
Подробное описание вариантов осуществления
Вышеуказанное содержание настоящего изобретения будет далее подробно описано с помощью конкретных примеров, но это не следует трактовать как ограничение объема вышеуказанного объекта настоящего изобретения следующими примерами. Все методики, реализованные согласно вышеуказанному содержанию настоящего изобретения, находятся в пределах объема настоящего изобретения.
Сокращения, применяемые в данном документе, являются следующими:
"DIPEA" означает N,N-диизопропилэтиламин;
"RH" означает относительную влажность;
"DMF" означает N,N-диметилформамид;
"DCM" означает дихлорметан;
"DMSO" означает диметилсульфоксид.
Препаративный пример 1. Синтез промежуточного соединения 4,6-дихлор-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила
Стадия 1. Синтез 6-хлор-2H-пиридо[3,4-d][1,3]оксазин-2,4(1H)-диона
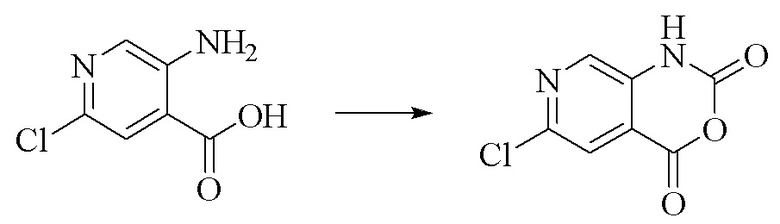
5-Амино-2-хлоризоникотиновую кислоту (30 г, 0,1738 моль, 1,0 экв.) растворяли в N,N-диметилформамиде (300 мл), порциями добавляли N,N'-карбонилдиимидазол (48 г, 0,2955 моль, 1,7 экв.) при 0°C и смесь медленно нагревали до комнатной температуры в течение ночи. LC-MS показывала завершение реакции, полученное охлаждали до комнатной температуры и непосредственно применяли на следующей стадии без обработки.
Стадия 2.
Синтез 6-хлор-4-гидроксил-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила
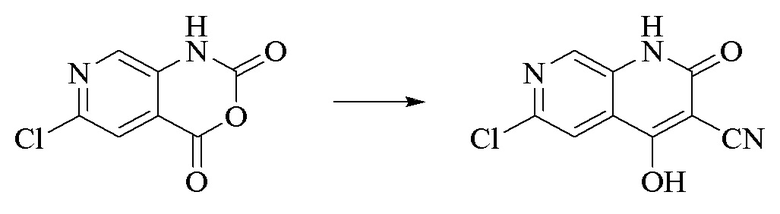
К вышеуказанной реакционной смеси добавляли триэтиламин (35,182 г, 0,3478 моль, 2 экв.) и этилцианоацетат (19,665 г, 0,1738 моль) и обеспечивали осуществление реакции при 150°C в течение 3 ч.; контроль посредством LC-MS продемонстрировал завершение реакции, а затем реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении; добавляли воду (200 мл) и смесь регулировали до pH 1 хлористоводородной кислотой (1 моль/л), перемешивали в течение 15 минут, и фильтровали путем отсасывания, и осадок на фильтре дважды промывали с помощью EA и высушивали при 40°C с получением продукта в виде светло-кирпичного твердого вещества (25,655 г, выход: 66%).
Стадия 3.
Синтез 4,6-дихлор-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила
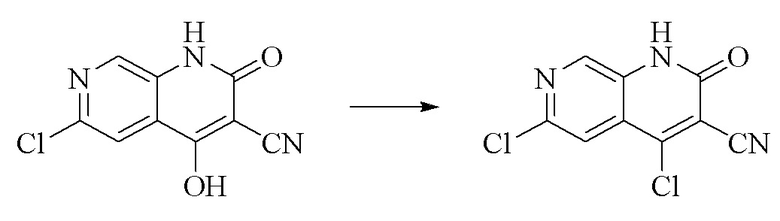
В реакционную колбу добавляли 6-хлор-4-гидроксил-2- оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрил (5,0 г, 0,0226 моль, 1 экв.) и оксихлорид фосфора (15 мл); реакционную колбу помещали на масляную баню, уже нагретую до 100°C, для осуществления реакции в течение приблизительно 6 мин. и твердое вещество начало медленно растворяться и цвет от светло-желтого постепенно становился более насыщенным. Обнаружение посредством TLC продемонстрировало завершение реакции и полученное охлаждали до комнатной температуры; в колбу добавляли подходящее количество DCM и смесь выливали в ледяную воду (100 мл), перемешивали в течение 10 мин., и фильтровали путем отсасывания, и осадок на фильтре промывали метил-трет-бутиловым эфиром, сливали жидкость и высушивали в вакууме при 40°C с получением продукта в виде светло-желтого твердого вещества. Материалы подавали пятью порциями и в общей сложности подавали 25,655 г (0,1157 моль) 6-хлор-4-гидроксил-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила с получением 19,486 г продукта (выход: 70,1%).
Пример 1. Получение соединения формулы (I)
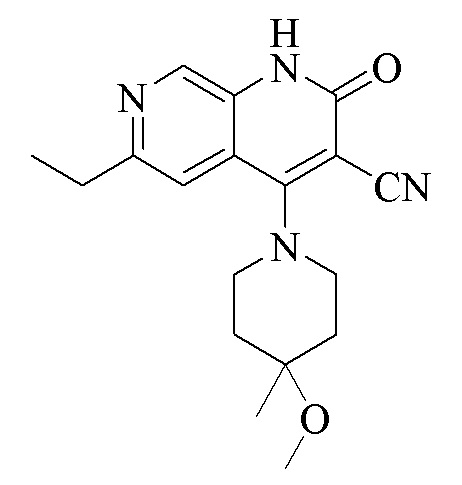
Стадия 1.
Синтез 6-хлор-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила
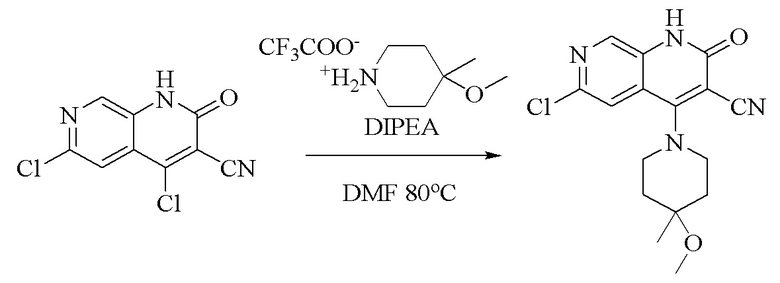
Промежуточное соединение 4,6-дихлор-2-оксо-1,2-дигидро-1,7-диазанафталин- 3-карбонитрил (2,0 г, 8,33 ммоль, 1,0 экв.) растворяли в DMF (10 мл); добавляли DIPEA (6,45 г, 50 ммоль, 6,0 экв.) и трифторацетат 4-метокси-4-метилпиперидина (2,2 г, 9,16 ммоль, 1,1 экв.) и смесь подвергали реакции при 80°C в течение 2 часов. Обнаружение посредством LC-MS продемонстрировало завершение реакции; добавляли воду (10 мл) и смесь экстрагировали дихлорметаном (10 мл × 3); органическую фазу промывали водой (10 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением продукта в виде желтого твердого вещества (2,7 г, неочищенный).
1H ЯМР (400 МГц, DMSO-d6) δ(ppm): 12,11 (s, 1H), 8,45 (s, 1H), 7,61 (s, 1H), 3,61-3,59 (m, 4H), 3,18 (s, 3H),1,91-1,88 (m, 2H), 1,81-1,76 (m, 2H), 1,21 (s, 3H).
Молекулярная формула: C16H17N4O2Cl, молекулярная масса: 332,79 LC-MS (положит., масса/заряд) = 333,7[M+H]+
Стадия 2.
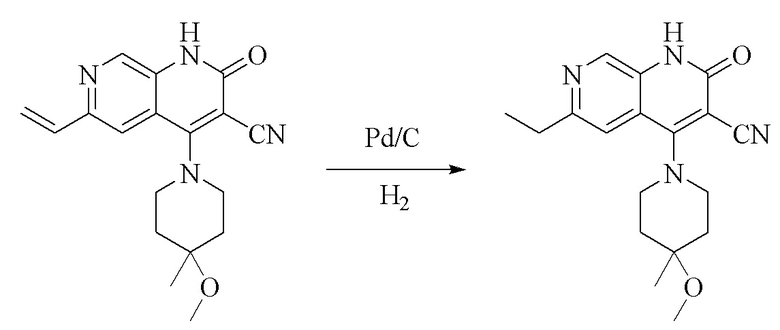
Синтез 4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-6-винил-1,2-дигидро-1,7-диазанафталин-3-карбонитрила
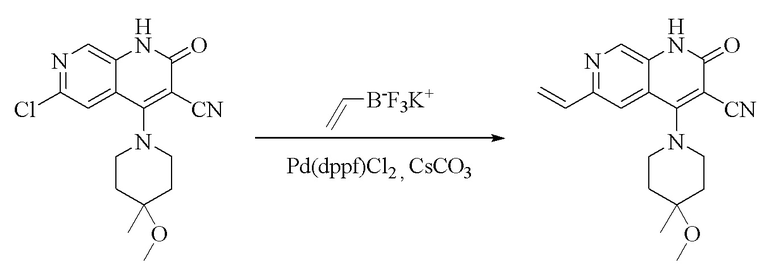
Промежуточное соединение 6-хлор-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-
1,2-дигидро-1,7-диазанафталин-3-карбонитрил (2,7 г, неочищенный, 8,11 ммоль, 1,0 экв.) растворяли в 1,4-диоксане (20 мл) и H2O (5 мл); добавляли винилтрифторборат калия (1,63 г, 12,17 ммоль, 1,5 экв.), карбонат цезия (3,965 г, 12,17 ммоль, 1,5 экв.) и дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (297 мг, 0,41 ммоль, 0,05 экв.) и смесь подвергали реакции в течение 8 часов в защитной атмосфере азота при 100°C. Обнаружение посредством LC-MS продемонстрировало завершение реакции; добавляли воду (20 мл) и смесь экстрагировали дихлорметаном (30 мл × 3); органическую фазу высушивали над безводным сульфатом натрия, фильтровали, и концентрировали при пониженном давлении, и неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM: MeOH = 70: 1) с получением продукта в виде желтого твердого вещества (1,15 г, выход: 43%).
Стадия 3.
Синтез 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила
 … Формула (I)
… Формула (I)
Промежуточное соединение 4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо- 6-винил-1,2-дигидро-1,7-диазанафталин-3-карбонитрил (150 мг, 0,46 ммоль, 1,0 экв.) растворяли в метаноле (5 мл) и добавляли Pd/C (100 мг); смесь три раза подвергали замене атмосферы на водород, и обеспечивали осуществление реакции в течение 1 часа в атмосфере водорода, и обнаружение посредством LC-MS продемонстрировало завершение реакции. Смесь фильтровали путем отсасывания, и фильтрат концентрировали при пониженном давлении с получением продукта (120 мг, выход: 80%).
1HЯМР (400 МГц, DMSO-d6) δ(ppm): 11,89 (s, 1H), 8,59 (s, 1H), 7,41 (s, 1H), 3,60-3,62 (m, 4H), 3,19 (s, 3H), 2,79-2,84 (m, 2H), 1,89-1,93(m, 2H), 1,75-1,82 (m, 2H), 1,22-1,27 (m, 6H).
Молекулярная формула: C18H22N4O2, молекулярная масса: 326,40 LC-MS (положит., масса/заряд) = 327,26[M+H]+.
Пример 2. Получение кристаллической формы I соединения формулы (I)
В реакционный сосуд объемом 50 л добавляли этанол (21 л, 30 V) и добавляли 985 г соединения формулы (I) в реакционную систему; реакционную систему нагревали с обратным холодильником и перемешивали в течение 1 часа до полного растворения системы; в систему добавляли активированный уголь (70 г, 10%), перемешивали в течение получаса и проводили горячее фильтрование для удаления активированного угля и примесей; повышали температуру фильтрата до полного растворения системы; этанол отгоняли при пониженном давлении и полученный продукт нагревали с обратным холодильником, перемешивали и охлаждали естественным путем до комнатной температуры для кристаллизации.
Рентгеновская порошковая дифрактограмма кристаллической формы I содержит характеристические пики при значениях2θ (°) 7,3 ± 0,2°, 13,6 ± 0,2°, 14,5 ± 0,2°, 18,0 ± 0,2°, 19,1 ± 0,2°, 22,0 ± 0,2° и 23,4 ± 0,2°, содержит характеристические пики при значениях 2θ (°) 14,2 ± 0,2°, 16,1 ± 0,2°, 19,4 ± 0,2° и 25,6 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 15,1 ± 0,2° и 17,6 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 1.
Температура плавления кристаллической формы I, измеренная с помощью дифференциального сканирующего калориметра, составляет приблизительно 256°C-258°C, как показано на фиг. 2.
Пример 3. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл метанола; смесь нагревали с обратным холодильником до полного растворения системы и полученный раствор выпаривали до сухого состояния при 50°C с получением кристаллической формы I.
Рентгеновская порошковая дифрактограмма кристаллической формы I содержит характеристические пики при значениях2θ (°) 7,3 ± 0,2°, 13,6 ± 0,2°, 14,5 ± 0,2°, 18,0 ± 0,2°, 19,1 ± 0,2°, 22,0 ± 0,2° и 23,4 ± 0,2°, содержит характеристические пики при значениях 2θ (°) 14,2 ± 0,2°, 16,1 ± 0,2°, 19,4 ± 0,2° и 25,6 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 15,1 ± 0,2° и 17,6 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 3.
Пример 4. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 30 мл ацетона и смесь выпаривали при 50°C с удалением 30 мл растворителя, добавляли дополнительные 30 мл ацетона и выпаривали до сухого состояния при 50°C с получением кристаллической формы I.
Пример 5. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 30 мл ацетонитрила и смесь выпаривали при 50°C с удалением 30 мл растворителя, добавляли еще 30 мл ацетонитрила и выпаривали до сухого состояния при 50°C с получением кристаллической формы I.
Пример 6. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 20 мл этилацетата; смесь выпаривали в естественных условиях при 15°C-20°C в течение 5 дней до осаждения твердого вещества и твердое вещество фильтровали путем отсасывания с получением кристаллической формы I.
Пример 7. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 20 мл метил-трет-бутилового эфира; смесь выпаривали в естественных условиях при 15°C-20°C в течение 5 дней до осаждения твердого вещества и твердое вещество фильтровали путем отсасывания с получением кристаллической формы I.
Пример 8. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 20 мл дихлорметана и смесь выпаривали в естественных условиях при 15°C-20°C в течение 5 дней с непосредственным получением кристаллической формы I.
Рентгеновская порошковая дифрактограмма кристаллической формы I содержит характеристические пики при значениях 2θ (°) 7,3 ± 0,2°, 13,6 ± 0,2°, 14,5 ± 0,2°, 18,0 ± 0,2°, 19,1 ± 0,2°, 22,0 ± 0,2° и 23,4 ± 0,2°, содержит характеристические пики при значениях 2θ (°) 14,2 ± 0,2°, 16,1 ± 0,2°, 19,4 ± 0,2° и 25,6 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 15,1 ± 0,2° и 17,6 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 4.
Пример 9. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 20 мл тетрагидрофурана; смесь выпаривали в естественных условиях при 15°C-20°C в течение 5 дней до осаждения твердого вещества и твердое вещество фильтровали путем отсасывания с получением кристаллической формы I.
Пример 10. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 20 мл этанола; смесь выпаривали в естественных условиях при 15°C-20°C в течение 5 дней до осаждения твердого вещества и твердое вещество фильтровали путем отсасывания с получением кристаллической формы I.
Пример 11. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 20 мл изопропанола; смесь выпаривали в естественных условиях при 15°C-20°C в течение 5 дней до осаждения твердого вещества и твердое вещество фильтровали путем отсасывания с получением кристаллической формы I.
Пример 12. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 20 мл толуола; смесь выпаривали в естественных условиях при 15°C-20°C в течение 5 дней до осаждения твердого вещества и твердое вещество фильтровали путем отсасывания с получением кристаллической формы I.
Пример 13. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 20 мл (вода : этанол = 1: 4); смесь выпаривали в естественных условиях при 15°C-20°C в течение 5 дней до осаждения твердого вещества и твердое вещество фильтровали путем отсасывания с получением кристаллической формы I.
Пример 14. Получение кристаллической формы I соединения формулы (I)
Брали 1 г соединения формулы (I) и растворяли в 20 мл дихлорметана, а затем добавляли 20 мл (вода : изопропанол = 3: 2); смесь выпаривали в естественных условиях при 15°C-20°C в течение 5 дней до осаждения твердого вещества и твердое вещество фильтровали путем отсасывания с получением кристаллической формы I.
Рентгеновская порошковая дифрактограмма кристаллической формы I содержит характеристические пики при значениях 2θ (°) 7,3 ± 0,2°, 13,6 ± 0,2°, 14,5 ± 0,2°, 18,0 ± 0,2°, 19,1 ± 0,2°, 22,0 ± 0,2° и 23,4 ± 0,2°, содержит характеристические пики при значениях 2θ (°) 14,2 ± 0,2°, 16,1 ± 0,2°, 19,4 ± 0,2° и 25,6 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 15,1 ± 0,2° и 17,6 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 5.
Пример 15. Получение гидрохлорида соединения формулы (I)
2 г соединения формулы (I) добавляли к 1 моль/л раствору хлористоводородной кислоты (50 мл) при 45°C; смесь перемешивали в течение 15 мин. до полного растворения твердого вещества с получением прозрачного желтого раствора и через 2 мин. осаждали желтое твердое вещество. Раствор медленно охлаждали до комнатной температуры до осаждения большого количества желтых твердых веществ, и фильтровали путем отсасывания, и осадок на фильтре высушивали при пониженном давлении при 40°C с получением 1,3 г гидрохлорида соединения формулы (I).
Пример 16. Получение кристаллической формы II соединения формулы (I)
К 200 мг гидрохлорида соединения формулы (I) добавляли воду; смесь нагревали до 60°C до полного растворения, медленно охлаждали при перемешивании для кристаллизации, и фильтровали путем отсасывания, и осадок на фильтре высушивали при пониженном давлении при 40°C с получением кристаллической формы II.
Рентгеновская порошковая дифрактограмма кристаллической формы II содержит характеристические пики при значениях 2θ (°) 4,0 ± 0,2°, 6,7 ± 0,2°, 7,9 ± 0,2°, 13,5 ± 0,2°, 14,2 ± 0,2°, 15,4 ± 0,2°, 20,2 ± 0,2° и 22,0 ± 0,2°, и дополнительно содержит характеристические пики при значениях 2θ (°) 10,2 ± 0,2°, 13,8 ± 0,2°, 14,6 ± 0,2°, 26,40 ± 0,2° и 26,80 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 6.
Пример 17. Получение кристаллической формы II соединения формулы (I)
К 200 мг гидрохлорида соединения формулы (I) добавляли 80% изопропанол (вода : изопропанол = 1: 4); смесь нагревали до 60°C до полного растворения, медленно охлаждали при перемешивании для кристаллизации, и фильтровали путем отсасывания, и осадок на фильтре высушивали при пониженном давлении при 40°C с получением кристаллической формы II.
Рентгеновская порошковая дифрактограмма кристаллической формы II содержит характеристические пики при значениях 2θ (°) 4,0 ± 0,2°, 6,7 ± 0,2°, 7,9 ± 0,2°, 13,5 ± 0,2°, 14,2 ± 0,2°, 15,4 ± 0,2°, 20,2 ± 0,2° и 22,0 ± 0,2°, и дополнительно содержит характеристические пики при значениях 2θ (°) 10,2 ± 0,2°, 13,8 ± 0,2°, 14,6 ± 0,2°, 26,40 ± 0,2° и 26,80 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 7.
Пример 18. Получение кристаллической формы II соединения формулы (I)
К 200 мг гидрохлорида соединения формулы (I) добавляли метанол; смесь нагревали до 60°C до полного растворения, медленно охлаждали при перемешивании для кристаллизации, и фильтровали путем отсасывания, и осадок на фильтре высушивали при пониженном давлении при 40°C с получением кристаллической формы II.
Рентгеновская порошковая дифрактограмма кристаллической формы II содержит характеристические пики при значениях 2θ (°) 4,0 ± 0,2°, 6,7 ± 0,2°, 7,9 ± 0,2°, 13,5 ± 0,2°, 14,2 ± 0,2°, 15,4 ± 0,2°, 20,2 ± 0,2° и 22,0 ± 0,2°, и дополнительно содержит характеристические пики при значениях 2θ (°) 10,2 ± 0,2°, 13,8 ± 0,2°, 14,6 ± 0,2°, 26,40 ± 0,2° и 26,80 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 8.
Пример 19. Получение кристаллической формы II соединения формулы (I)
К 200 мг гидрохлорида соединения формулы (I) добавляли этанол; смесь нагревали до 60°C до полного растворения, медленно охлаждали при перемешивании для кристаллизации, и фильтровали путем отсасывания, и осадок на фильтре высушивали при пониженном давлении при 40°C с получением кристаллической формы II.
Пример 20. Получение кристаллической формы III соединения формулы (I)
К 200 мг гидрохлорида соединения формулы (I) добавляли ацетонитрил; смесь нагревали с обратным холодильником до растворения большей части твердых веществ и проводили горячее фильтрование путем отсасывания; фильтрат концентрировали при пониженном давлении до осаждения большого количества твердых веществ, и фильтровали путем отсасывания, и осадок на фильтре высушивали при пониженном давлении при 40°C с получением кристаллической формы III.
Рентгеновская порошковая дифрактограмма кристаллической формы III содержит характеристические пики при значениях 2θ (°) 5,2 ± 0,2°, 6,4 ± 0,2°, 15,3 ± 0,2°, 18,6 ± 0,2°, 22,0 ± 0,2° и 26,4 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 8,0 ± 0,2°, 10,3 ± 0,2°, 13,5 ± 0,2° и 25,0 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 9.
Пример 21. Получение кристаллической формы III соединения формулы (I)
К 200 мг гидрохлорида соединения формулы (I) добавляли ацетон; смесь нагревали до 60°C до растворения большей части твердых веществ и проводили горячее фильтрование путем отсасывания; фильтрат концентрировали при пониженном давлении до осаждения большого количества твердых веществ, и фильтровали путем отсасывания, и осадок на фильтре высушивали при пониженном давлении при 40°C с получением кристаллической формы III.
Пример 22. Получение кристаллической формы III соединения формулы (I)
К 200 мг гидрохлорида соединения формулы (I) добавляли этилацетат; смесь нагревали до 60°C до растворения большей части твердых веществ и проводили горячее фильтрование путем отсасывания; фильтрат концентрировали при пониженном давлении до осаждения большого количества твердых веществ, и фильтровали путем отсасывания, и осадок на фильтре высушивали при пониженном давлении при 40°C с получением кристаллической формы III.
Рентгеновская порошковая дифрактограмма кристаллической формы III содержит характеристические пики при значениях 2θ (°) 5,2 ± 0,2°, 6,4 ± 0,2°, 15,3 ± 0,2°, 18,6 ± 0,2°, 22,0 ± 0,2° и 26,4 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 8,0 ± 0,2°, 10,3 ± 0,2°, 13,5 ± 0,2° и 25,0 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 10.
Пример 23. Получение кристаллической формы III соединения формулы (I)
К 200 мг гидрохлорида соединения формулы (I) добавляли тетрагидрофуран; смесь нагревали до 60°C до растворения большей части твердых веществ и проводили горячее фильтрование путем отсасывания; фильтрат концентрировали при пониженном давлении до осаждения большого количества твердых веществ, и фильтровали путем отсасывания, и осадок на фильтре высушивали при пониженном давлении при 40°C с получением кристаллической формы III.
Рентгеновская порошковая дифрактограмма кристаллической формы III содержит характеристические пики при значениях 2θ (°) 5,2 ± 0,2°, 6,4 ± 0,2°, 15,3 ± 0,2°, 18,6 ± 0,2°, 22,0 ± 0,2° и 26,4 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 8,0 ± 0,2°, 10,3 ± 0,2°, 13,5 ± 0,2° и 25,0 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 11.
Пример 24. Получение кристаллической формы IV соединения формулы (I)
1,3 г соединения формулы (I) добавляли к 10 мл 1 моль/л водного раствора п-толуолсульфоновой кислоты, и смесь нагревали до 50°C до полного растворения, и медленно охлаждали до комнатной температуры при перемешивании до осаждения желтого твердого вещества. Осадок фильтровали путем отсасывания и осадок на фильтре промывали небольшим количеством воды, фильтровали путем отсасывания в течение 1 часа и высушивали при пониженном давлении при 40°C с получением кристаллической формы IV.
Рентгеновская порошковая дифрактограмма кристаллической формы IV содержит характеристические пики при значениях 2θ (°)5,8 ± 0,2°, 7,8 ± 0,2°, 9,3 ± 0,2°, 11,3 ± 0,2°, 13,7 ± 0,2°, 14,8 ± 0,2° и 15,7 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 17,1 ± 0,2°, 18,7 ± 0,2°, 20,0 ± 0,2° и 22,4 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 12.
Пример 25. Получение кристаллической формы V соединения формулы (I)
1,3 г соединения формулы (I) добавляли к 10 мл 1 моль/л водного раствора метансульфоновой кислоты; смесь нагревали до 50°C до полного растворения, медленно охлаждали до комнатной температуры при перемешивании, добавляли метанол, и фильтровали путем отсасывания, и осадок на фильтре промывали небольшим количеством метанола, фильтровали путем отсасывания в течение 1 часа и высушивали при пониженном давлении при 40°C с получением кристаллической формы V.
Рентгеновская порошковая дифрактограмма кристаллической формы V содержит характеристические пики при значениях 2θ (°) 7,0 ± 0,2°, 9,7 ± 0,2°, 13,2 ± 0,2°, 18,1 ± 0,2°, 19,5 ± 0,2°, 20,7 ± 0,2° и 21,7 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°)16,9 ± 0,2°, 18,6 ± 0,2°, 19,1 ± 0,2°, 20,2 ± 0,2° и 28,0 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 13.
Экспериментальный пример 26. Получение кристаллической формы VI соединения формулы (I)
Исходный материал 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)- 2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрил (10 г, 30,63 ммоль, 1,0 экв.) порциями добавляли к серной кислоте (1 н.) (40 мл), и растворяли, и смесь подвергали реакции в течение 1 часа; прекращали нагревание после растворения исходного материала с получением прозрачного раствора; раствор охлаждали до комнатной температуры до осаждения большого количества твердых веществ, и фильтровали путем отсасывания, и осадок на фильтре высушивали при 50°C с получением 6,9 г сульфата соединения формулы (I), т.е. кристаллической формы VI.
Рентгеновская порошковая дифрактограмма кристаллической формы VI содержит характеристические пики при значениях 2θ (°) 4,4 ± 0,2°, 7,1 ± 0,2°, 8,8 ± 0,2°, 14,3 ± 0,2°, 17,8 ± 0,2°, 19,6 ± 0,2° и 21,6 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 25,5 ± 0,2° и 27,7 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 14.
Пример 27. Получение кристаллической формы VI соединения формулы (I)
Сульфат соединения формулы (I) (1 г, 2,36 ммоль, 1,0 экв.) растворяли в ацетоне (50 мл), нагревали с обратным холодильником, смесь перемешивали в течение 3 ч., но обеспечить получения прозрачного раствора не удалось, прекращали нагревание, раствор охлаждали до комнатной температуры, и фильтровали путем отсасывания, и осадок на фильтре высушивали при 50°C с получением кристаллической формы VI.
Рентгеновская порошковая дифрактограмма кристаллической формы VI содержит характеристические пики при значениях 2θ (°) 4,4 ± 0,2°, 7,1 ± 0,2°, 8,8 ± 0,2°, 14,3 ± 0,2°, 17,8 ± 0,2°, 19,6 ± 0,2° и 21,6 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 25,5 ± 0,2° и 27,7 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 15.
Пример 28. Получение кристаллической формы VI соединения формулы (I)
Сульфат соединения формулы (I) (1 г, 2,36 ммоль, 1,0 экв.) растворяли в этаноле (15 мл), нагревали с обратным холодильником и смесь перемешивали в течение 3 ч.; после растворения сульфата с получением прозрачного раствора прекращали нагревание; раствор охлаждали до комнатной температуры до осаждения большого количества твердых веществ, и фильтровали путем отсасывания, и осадок на фильтре высушивали при 50°C с получением кристаллической формы VI.
Рентгеновская порошковая дифрактограмма кристаллической формы VI содержит характеристические пики при значениях 2θ (°) 4,4 ± 0,2°, 7,1 ± 0,2°, 8,8 ± 0,2°, 14,3 ± 0,2°, 17,8 ± 0,2°, 19,6 ± 0,2° и 21,6 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 25,5 ± 0,2° и 27,7 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 16.
Пример 29. Получение кристаллической формы VI соединения формулы (I)
Сульфат соединения формулы (I) (1 г, 2,36 ммоль, 1,0 экв.) растворяли в метаноле (10 мл), нагревали с обратным холодильником и смесь перемешивали в течение 3 ч.; после растворения сульфата с получением прозрачного раствора прекращали нагревание; раствор охлаждали до комнатной температуры до осаждения большого количества твердых веществ, и фильтровали путем отсасывания, и осадок на фильтре высушивали при 50°C с получением кристаллической формы VI.
Пример 30. Получение кристаллической формы VI соединения формулы (I)
Сульфат соединения формулы (I) (1 г, 2,36 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (50 мл), нагревали с обратным холодильником и смесь перемешивали в течение 3 ч.; прекращали нагревание, если не удавалось обеспечить получение прозрачной реакционной смеси после растворения; раствор охлаждали до комнатной температуры, и фильтровали путем отсасывания, и осадок на фильтре высушивали при 50°C с получением кристаллической формы VI.
Рентгеновская порошковая дифрактограмма кристаллической формы VI содержит характеристические пики при значениях 2θ (°) 4,4 ± 0,2°, 7,1 ± 0,2°, 8,8 ± 0,2°, 14,3 ± 0,2°, 17,8 ± 0,2°, 19,6 ± 0,2° и 21,6 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 25,5 ± 0,2° и 27,7 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 17.
Пример 31. Получение кристаллической формы VII соединения формулы (I)
Сульфат соединения формулы (I) (1 г, 2,36 ммоль, 1,0 экв.) растворяли в изопропаноле (50 мл), нагревали с обратным холодильником и смесь перемешивали в течение 3 ч.; после растворения сульфата с получением прозрачного раствора прекращали нагревание; раствор охлаждали до комнатной температуры до осаждения большого количества твердых веществ, и фильтровали путем отсасывания, и осадок на фильтре высушивали при 50°C с получением кристаллической формы VII.
Рентгеновская порошковая дифрактограмма кристаллической формы VII содержит характеристические пики при значениях 2θ (°) 7,1 ± 0,2°, 13,6 ± 0,2°, 14,5 ± 0,2°, 18,0 ± 0,2°, 19,0 ± 0,2°, 22,0 ± 0,2° и 23,4 ± 0,2° и дополнительно содержит характеристические пики при значениях 2θ (°) 15,0 ± 0,2°, 16,1 ± 0,2°, 17,4 ± 0,2° и 19,4 ± 0,2°, как определено с применением излучения Cu-Kα. Анализ посредством XRPD представлен на фиг. 18.
В соответствии со следующими экспериментальными примерами настоящее изобретение можно понять лучше. Однако специалист в данной области техники сможет легко понять, что содержание, описанное в экспериментальных примерах, применяется только для иллюстрации настоящего изобретения и не должно и не будет ограничивать настоящее изобретение, подробно описанное в формуле изобретения.
Экспериментальный пример 1. Оценка PDE9 с помощью ферментативного способа
Тестируемые вещества: соединение формулы (I) получали в соответствии с примером 1 настоящего изобретения, кристаллическую форму I получали в соответствии с примером 2 настоящего изобретения, и кристаллическую форму II получали в соответствии с примером 16 настоящего изобретения.
1. Экспериментальные материалы и приборы
Фермент PDE9A2 (BPS, № по кат. 60090)
384-луночный планшет (Perkin Elmer, № по кат. 6007279)
Набор для оценки PDE IMAP FP (набор для обнаружения ферментативной активности) (Molecular Devices, № по кат. R8175)
2. Стадии теста
Приготовление растворов тестируемых веществ. Тестируемые вещества получали в виде 10 мМ исходного раствора для длительного хранения с применением DMSO; исходный раствор разбавляли в 100 раз с помощью DMSO с получением 100 мкМ рабочего раствора тестируемых веществ; и рабочий раствор соединений разбавляли в 3 раза с помощью DMSO с получением в целом градиентов с 8-10 концентрациями разбавленного раствора тестируемых веществ (100×).
Инкубация с обработкой. Разбавленный раствор тестируемых веществ отмеряли пипеткой в 384-луночный планшет с применением Echo, системы для отмеривания пипеткой очень небольшого количества жидкости; 200 нл разбавленного раствора тестируемых веществ и 10 мкл раствора фермента PDE9A2 добавляли в лунку с каждым тестируемым веществом, и планшет центрифугировали при 1000 об./мин. в течение 1 мин., и затем инкубировали при комнатной температуре в течение 15 мин. В соответствии с инструкциями к набору для оценки PDE IMAP FP (набору для обнаружения ферментативной активности) добавляли 10 мкл смеси субстратов, и смесь центрифугировали при 1000 об./мин. в течение 1 мин., и инкубировали со встряхиванием при комнатной температуре в течение 30 мин. Наконец, добавляли стоп-раствор для завершения реакции в системе и смесь инкубировали со встряхиванием при комнатной температуре в течение 60 мин. В лунке с максимальным считанным значением (Max) соединение заменяли на растворитель DMSO; и в лунке с минимальным считанным значением (Min) растворы тестируемых веществ и фермента заменяли на растворитель DMSO.
Обнаружение. Применяли микропланшет-ридер для определения считанного значения флуоресценции (F) при 480 нм/535 нм.
Расчет. Степень ингибирования рассчитывали в соответствии со следующей формулой и применяли GraphPad Prism 5.0 для подбора IC50:
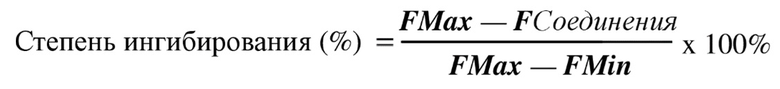
3. Результаты экспериментов представлены в таблице 1 ниже.
Таблица 1. Ингибирующая активность в отношении ферментативной активности PDE9
Как можно увидеть из таблицы 1, все из соединения формулы (I), кристаллической формы I и кристаллической формы II по настоящему изобретению обладают превосходной ингибирующей активностью в отношении ферментативной активности PDE9 и имеют потенциальное значение для клинического применения при лечении заболеваний, опосредованных PDE9.
Экспериментальный пример 2. Эксперимент по оценке фармакокинетики соединения и кристаллической формы I по настоящему изобретению у крыс
Животные самцы крыс SD
Тестируемые вещества: соединение формулы (I) получали в соответствии с примером 1 настоящего изобретения, и кристаллическую форму I получали в соответствии с примером 2 настоящего изобретения.
Введение животным и сбор образцов
Соединение формулы (I) для эксперимента растворяли в физиологическом растворе 2% DMSO + 10% PEG400 + 88% (28% HP-β-CD) с получением раствора; раствор тестируемого вещества вводили крысам SD через желудочный зонд в дозе 5,0 мг/кг; и временные точки сбора образцов крови составляли 15 мин., 30 мин., 1 ч., 2 ч., 4 ч., 6 ч., 8 ч. и 24 ч. после введения.
Соединение формулы (I) для эксперимента растворяли в физиологическом растворе 2% DMSO + 10% PEG400 + 88% (28% HP-β-CD) с получением раствора; раствор тестируемого вещества вводили крысам SD внутривенно болюсно в дозе 1 мг/кг; и временные точки сбора образцов крови составляли 5 мин., 15 мин., 30 мин., 1 ч., 2 ч., 4 ч., 6 ч., 8 ч. и 24 ч. после введения.
Кристаллическую форму I для эксперимента растворяли в физиологическом растворе 5% DMSO + 10% PEG400 + 85% (20% HP-β-CD) с получением раствора; раствор тестируемого вещества вводили крысам SD через желудочный зонд в дозе 60,0 мг/кг; и временные точки сбора образцов крови составляли 15 мин., 30 мин., 1 ч., 2 ч., 4 ч., 8 ч., 24 ч., 30 ч. и 48 ч. после введения.
Кристаллическую форму I для эксперимента растворяли в физиологическом растворе 2% DMSO + 10% PEG400 + 88% (28% HP-β-CD) с получением раствора; раствор тестируемого вещества вводили крысам SD внутривенно болюсно в дозе 5 мг/кг; и временные точки сбора образцов крови составляли 5 мин., 15 мин., 30 мин., 1 ч., 4 ч., 8 ч., 12 ч. и 24 ч. после введения.
Соединение формулы (I). Применяли и фиксировали обычных крыс SD; хвосты крыс нагревали на водяной бане за 10 минут до каждой временной точки; и отбирали приблизительно 100 мкл крови из хвостовой вены, а затем помещали в пробирку, содержащую антикоагулянт EDTA-K2. Образец крови центрифугировали при 8000 об./мин. при 4°C в течение 6 мин. с получением образца плазмы крови, который получали в течение 30 минут после забора крови. Перед тестом плазму крови хранили в холодильнике при -80°C.
Кристаллическая форма I. Крысам проводили катетеризацию яремной вены (JVC); в каждой точке отбирали приблизительно 0,35 мл крови у крыс через катетер яремной вены в центрифужную пробирку, содержащую 5 мкл 15% EDTA-K2; смесь осторожно перемешивали с получением однородной смеси, а затем центрифужную пробирку немедленно помещали на ледяную баню. В течение 1 часа после сбора цельной крови смесь центрифугировали при 4°C и 2400× g в течение 5 минут, и сразу же отбирали надосадочную жидкость с получением образца плазмы крови. Перед тестом плазму крови хранили в холодильнике при -80°C.
Способ анализа образца
Соединение формулы (I)
Подлежащий тестированию образец плазмы крови извлекали из холодильника при -80°C, подвергали естественному плавлению при комнатной температуре, а затем перемешивали вихревым способом в течение 5 мин.; 20 мкл образца плазмы крови точно отмеряли пипеткой в центрифужную пробирку объемом 1,5 мл; добавляли 200 мкл рабочего раствора внутреннего стандарта (толбутамида в метаноле) с концентрацией 100 нг/мл и смесь перемешивали до однородного состояния; однородную смесь перемешивали вихревым способом в течение 5 мин., а затем центрифугировали при 12000 об./мин. в течение 5 мин.; 50 мкл надосадочной жидкости точно отмеряли пипеткой в 96-луночный планшет, предварительно заполненный 150 мкл воды/лунку; и содержимое планшета перемешивали вихревым способом в течение 5 мин. для получения однородной смеси и подвергали анализу LC-MS/MS с объемом вводимого образца 5 мкл.
Кристаллическая форма I
Подлежащий тестированию образец плазмы крови извлекали из холодильника при -80°C, подвергали естественному плавлению при комнатной температуре, а затем перемешивали вихревым способом в течение приблизительно 30 секунд для получения однородной смеси; 50 мкл образца (50 мкл холостой плазмы крови крысы, отобранной в качестве холостого образца и холостого образца внутреннего стандарта) пипеткой отмеряли в 96-луночный планшет (планшет-1); добавляли 20 мкл раствора IS-W [для холостого образца добавляли 20 мкл метанола] и смесь перемешивали до однородного состояния; добавляли 400 мкл ацетонитрила и смесь перемешивали вихревым способом в течение приблизительно 3 мин., при этом 96-луночный планшет накрывали крышкой, и центрифугировали при 3200× g и 4°C в течение 5 мин. Отбирали 100 мкл надосадочной жидкости и помещали в 96-луночный планшет (планшет-2), и высушивали продувкой потоком азота при 40°C; добавляли 200 мкл восстановленного раствора [0,4% водный раствор муравьиной кислоты (pH 3,2):ацетонитрил (65: 35)] и смесь перемешивали вихревым способом в течение приблизительно 3 мин., при этом 96-луночный планшет накрывали крышкой; и проводили анализ посредством LC-MS/MS.
Способ обработки данных
В качестве концентраций тестируемых веществ применяли выходные результаты Analyst 1.6.3 (компания AB). Такие параметры, как среднее значение, стандартное отклонение и коэффициент вариации, рассчитывали с помощью Microsoft Excel (для прямого вывода результатов из Analyst 1.6.3 расчет не требовался); и фармакокинетические параметры рассчитывали с применением программного обеспечения Pharsight Phoenix 6.1 NCA (Tmax выражали в виде медианы).
Результаты экспериментов
Таблица 2. Результаты экспериментов по оценке фармакокинетики соединения формулы (I) и кристаллической формы I
(ч.)
(л/кг)
(ч.*нг/мл)
/[AUClast PO/доза] (ч.*нг/мл)
Примечания. tz1/2: конечный период полувыведения; Cl_наблюд.: клиренс; Vz_наблюд.: кажущийся объем распределения; Tmax: время достижения максимальной концентрации вещества в плазме крови; AUClast: площадь под кривой зависимости концентрации в плазме крови от времени от нуля до бесконечности.
Как можно увидеть из вышеприведенной таблицы, соединение формулы (I) и кристаллическая форма I по настоящему изобретению обладают превосходными фармакокинетическими характеристиками.
Экспериментальный пример 3. Испытание стабильности кристаллической формы I по настоящему изобретению
Тестируемые вещества: кристаллическую форму I получали в соответствии с примером 2 настоящего изобретения.
Способ: брали подходящее количество образцов кристаллической формы I и помещали на открытый воздух при 60°C и RH 92,5% при освещении; и отбирали образцы на 5 день, 10 день и 30 день для изучения изменений свойств, содержания и содержания влаги образцов.
Результаты
Таблица 3. Результаты испытания влияющих факторов
Заключение: кристаллическая форма I по настоящему изобретению демонстрирует отсутствие очевидных изменений в свойствах, родственных примесях, содержании и содержании влаги после помещения на 30 дней в условия различных влияющих факторов, что указывает на хорошую стабильность.
Экспериментальный пример 4. Определение растворимости соединения по настоящему изобретению в тестовых растворах с различными значениями pH и воде
Тестируемые вещества: соединение формулы (I) получали в соответствии с примером 1 настоящего изобретения, кристаллическую форму I получали в соответствии с примером 2 настоящего изобретения, кристаллическую форму II получали в соответствии с примером 16 настоящего изобретения, кристаллическую форму III получали в соответствии с примером 20 настоящего изобретения, кристаллическую форму IV получали в соответствии с примером 24 настоящего изобретения, кристаллическую форму V получали в соответствии с примером 25 настоящего изобретения, кристаллическую форму VI получали в соответствии с примером 27 настоящего изобретения, и кристаллическую форму VII получали в соответствии с примером 31 настоящего изобретения.
Получение буферных солевых растворов с различными значениями pH
Раствор хлористоводородной кислоты с pH 1,0: пипеткой отмеряли 9 мл хлористоводородной кислоты и добавляли воду для разбавления до 1000 мл; смесь перемешивали до однородного состояния с получением раствора хлористоводородной кислоты с pH 1,03. Фосфатный буферный раствор с рН 4,5: отвешивали 1,56 г дигидрата дигидрофосфата натрия и добавляли 200 мл воды с получением фосфатного буферного раствора с pH 4,50. Фосфатный буферный раствор с рН 6,8: отвешивали 1,56 г дигидрата дигидрофосфата натрия и добавляли 200 мл воды; и смесь доводили до pH 6,80 с помощью 1 моль/л раствора гидроксида натрия.
Порядок проведения эксперимента
Получение буферных растворов с различными значениями pH (образцы для определения растворимости)
Отвешивали 50 мг каждого тестируемого вещества, и добавляли 5 мл раствора хлористоводородной кислоты (pH 1,0), и смесь перемешивали до однородного состояния со встряхиванием. Отвешивали приблизительно 2,5 мг каждого тестируемого вещества и добавляли 5 мл фосфатного буферного раствора (pH 4,5), 5 мл фосфатного буферного раствора (pH 6,8) и 5 мл воды высшей степени очистки, соответственно; смесь перемешивали до однородного состояния со встряхиванием и затем помещали на водяную баню со встряхиванием при 37°C для встряхивания; отбирали образцы через 24 часа и подвергали центрифугированию; и отбирали надосадочную жидкость и непосредственно вводили (если растворимость была высокой, надосадочную жидкость разбавляли 0,1 моль/л раствором хлористоводородной кислоты в подходящее количество раз, и затем вводили).
Растворимость образцов определяли количественно методом внешнего стандарта с калибровкой по одной точке. Результаты экспериментов представлены ниже.
Таблица 4. Растворимость соединения формулы (I) и различных кристаллических форм в тестовых растворах с различными значениями pH/воде
Заключение: растворимость соединения формулы (I) и различных кристаллических форм увеличивается с увеличением кислотности раствора, и их растворимость лучше в тестовом растворе с pH 1,0.
Экспериментальный пример 5. Испытание стабильности различных кристаллических форм соединения по настоящему изобретению
Тестируемые вещества: кристаллическую форму I получали в соответствии с примером 2 настоящего изобретения, кристаллическую форму II получали в соответствии с примером 16 настоящего изобретения, кристаллическую форму III получали в соответствии с примером 20 настоящего изобретения, кристаллическую форму IV получали в соответствии с примером 24 настоящего изобретения, кристаллическую форму V получали в соответствии с примером 25 настоящего изобретения, кристаллическую форму VI получали в соответствии с примером 27 настоящего изобретения, и кристаллическую форму VII получали в соответствии с примером 31 настоящего изобретения.
Способ: брали подходящее количество различных тестируемых веществ и помещали в условия при 105°C на 3 дня и при 60°C и RH 92,5% при освещении на 5 дней и 10 дней, соответственно; и отбирали образцы на 0 день, 3 день, 5 день и 10 день для изучения изменений свойств и содержания образцов.
Результаты
В таблице 5 и таблице 6 представлены результаты в отношении свойств и содержания после помещения образцов в условия при 105°C на 3 дня. В таблице 7 и таблице 8 представлены результаты в отношении свойств и содержания после помещения образцов в условия при 60°C и RH 92,5% при освещении на 5 дней. В таблице 9 и таблице 10 представлены результаты в отношении свойств и содержания после помещения образцов в условия при RH 92,5% при освещении на 10 дней.
Таблица 5. Результаты в отношении свойств различных тестируемых веществ после помещения в вышеуказанные условия на 3 дня
Таблица 6. Результаты в отношении содержания различных тестируемых веществ после помещения в вышеуказанные условия на 3 дня
на 3 дня
Заключение: различные кристаллические формы по настоящему изобретению демонстрируют отсутствие очевидных изменений свойств и содержания после помещения в условия при 105°C на 3 дня, что указывает на хорошую стабильность.
Таблица 7. Результаты в отношении свойств различных тестируемых веществ после помещения в условия различных влияющих факторов на 5 дней
Таблица 8. Результаты в отношении содержания различных тестируемых веществ после помещения в условия различных влияющих факторов на 5 дней
на 5 дней
на 5 дней
Заключение: различные кристаллические формы по настоящему изобретению демонстрируют отсутствие очевидных изменений свойств и содержания после помещения в условия различных влияющих факторов на 5 дней, что указывает на хорошую стабильность.
Таблица 9. Результаты в отношении свойств различных тестируемых веществ после помещения в условия различных влияющих факторов на 10 дней
Таблица 10. Результаты в отношении содержания различных тестируемых веществ после помещения в условия различных влияющих факторов на 10 дней
на 10 дней
Заключение: различные кристаллические формы по настоящему изобретению демонстрируют отсутствие очевидных изменений свойств и содержания после помещения в условия различных влияющих факторов на 10 дней, что указывает на хорошую стабильность.
Приведенные описания являются всего лишь предпочтительными вариантами осуществления настоящего изобретения и не предназначены для ограничения настоящего изобретения, и при этом любые модификации, эквивалентные замены, улучшения и т.д., сделанные в рамках сущности и принципа настоящего изобретения, должны быть включены в объем защиты настоящего изобретения.
Изобретение относится к кристаллической форме I соединения, представленного формулой (I), 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, где кристаллическая форма характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики при значениях 2θ 7,3 ± 0,2°, 13,6 ± 0,2°, 14,5 ± 0,2°, 18,0 ± 0,2°, 19,1 ± 0,2°, 22,0 ± 0,2° и 23,4 ± 0,2°, как определено с применением излучения Cu-Kα. Кристаллическую форму I по изобретению получают путем растворения соединения формулы (I) в метаноле или этаноле, повышения температуры до температуры образования флегмы до полного растворения и охлаждения естественным путем до комнатной температуры для кристаллизации; или полным растворением соединения формулы (I) в одном растворителе – дихлорметане или в смешанном растворителе – дихлорметан/вода/изопропанол, и выпариванием одного растворителя или смешанного растворителя до насыщения системы и осаждения кристаллической формы I. Также изобретение относится к фармацевтическому препарату для лечения или предупреждения заболеваний, опосредованных PDE9, содержащему кристаллическую форму I по изобретению и один или несколько фармацевтических носителей. Технический результат - кристаллическая форма 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, обладающая превосходными физико-химическими свойствами и фармацевтической стабильностью, а также фармакодинамическими и фармакокинетическими свойствами. 4 н. и 2 з.п. ф-лы, 18 ил., 10 табл., 36 пр.
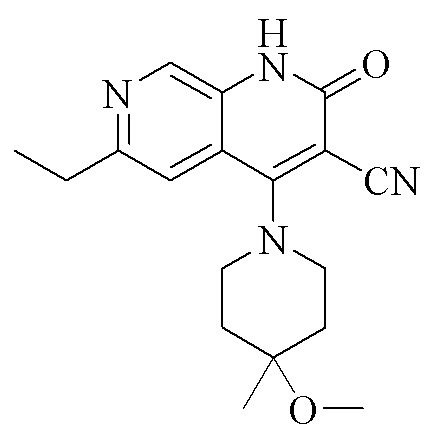 (I)
(I)
1. Кристаллическая форма I соединения, представленного формулой (I), 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-диазанафталин-3-карбонитрила, где кристаллическая форма характеризуется рентгеновской порошковой дифрактограммой, содержащей характеристические пики при значениях 2θ 7,3 ± 0,2°, 13,6 ± 0,2°, 14,5 ± 0,2°, 18,0 ± 0,2°, 19,1 ± 0,2°, 22,0 ± 0,2° и 23,4 ± 0,2°, как определено с применением излучения Cu-Kα,
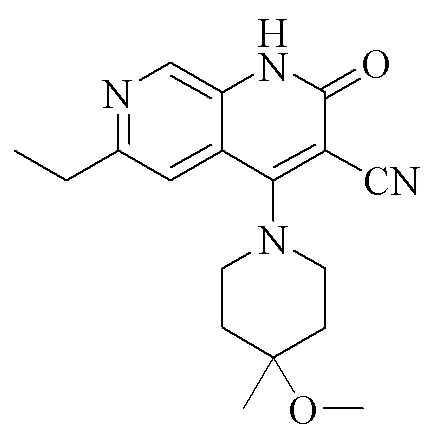 (I).
(I).
2. Кристаллическая форма I по п. 1, где рентгеновская порошковая дифрактограмма дополнительно содержит характеристические пики при значениях 2θ 14,2 ± 0,2°, 16,1 ± 0,2°, 19,4 ± 0,2° и 25,6 ± 0,2°, как определено с применением излучения Cu-Kα.
3. Кристаллическая форма I по п. 2, где кристаллическая форма характеризуется рентгеновской порошковой дифрактограммой, которая по сути изображена на фиг. 1, при применении излучения Cu-Kα.
4. Способ получения кристаллической формы I по п. 1,
где способ включает растворение соединения формулы (I) в метаноле или этаноле, повышение температуры до температуры образования флегмы до полного растворения и охлаждение естественным путем до комнатной температуры для кристаллизации;
или полное растворение соединения формулы (I) в одном растворителе – дихлорметане или в смешанном растворителе – дихлорметан/вода/изопропанол, и выпаривание одного растворителя или смешанного растворителя до насыщения системы и осаждения кристаллической формы I.
5. Фармацевтический препарат для лечения или предупреждения заболеваний, опосредованных PDE9, содержащий кристаллическую форму I по любому из пп. 1-3 и один или несколько фармацевтических носителей.
6. Применение кристаллической формы I по любому из пп. 1-3 для изготовления лекарственного препарата для лечения или предупреждения заболеваний, опосредованных PDE9.
| Способ получения 4-замещенных производных амино-3-карбэтокси-или 3-циано-1,2-дигидро-2-оксо-1,8-нафтиридина или их солей | 1980 |
|
SU1131469A3 |
| MINO R.CAIRA, Crystalline Polymorphism of Organic Compounds, 1998, p.163-208 | |||
| Sherry L.Morissette | |||
| et al | |||
| "High-through put crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids", ADVANCED DRUG DELIVERY REVIEWS, 2004, v.56, p.275-300 | |||
| Richard J.Bastin | |||
| et al | |||
| "Salt selection and | |||

