Настоящее изобретение относится к новым соединениям, представляющим собой замещенные тиазолы и оксазолы формулы (I), обладающим антагонистическими свойствами в отношении Р2Х7 рецепторов (Р2Х7), фармацевтическим композициям, содержащим эти соединения, химическим способам получения этих соединений и их применению в лечении или профилактике заболеваний, ассоциированных с активностью Р2Х7 рецепторов, у животных, в частности у людей.
Р2Х7 принадлежит к семейству ионотропных Р2Х рецепторов. Р2Х7 активируется внеклеточными нуклеотидами, в особенности аденозинтрифосфатом (АТФ). Р2Х7 отличается от других членов Р2Х семейства специфической локализацией (в частности, в центральной нервной системе (ЦНС) и иммунокомпетентных клетках), высокой концентрацией АТФ (в миллимолярном диапазоне), необходимой для его активации, и его способностью формировать большую пору при длительной или повторной стимуляции. Р2Х7 представляет собой лиганд-управляемый ионный канал и присутствует на клетках различных типов, главным образом на клетках, вовлеченных в воспалительный и/или иммунный процесс, в особенности, на макрофагах, тучных клетках и лимфоцитах (Т и В). Активация Р2Х7 рецептора внеклеточными нуклеотидами, например АТФ, приводит к высвобождению интерлейкина-1β (IL-1β) и образованию гигантских клеток (макрофагов/микроглиальных клеток), дегранулированию (тучных клеток) и шеддингу молекул L-селектина (у лимфоцитов). Кроме того, Р2Х7 рецепторы локализованы на антигенпредставляющих клетках (АРС), кератиноцитах, ацинарных клетках слюнных желез (клетках околоушной железы), гепатоцитах, эритроцитах, эритролейкозных клетках, моноцитах, фибробластах, клетках костного мозга, нейронах и мезангиальных клетках почки. Также известно, что Р2Х7 рецептор является болевым сенсором в нервной системе. В экспериментах с использованием Р2Х7-дефицитных мышей была продемонстрирована роль Р2Х7 в развитии боли, поскольку у таких мышей не отмечалось развития ни индуцированной адъювантами воспалительной боли, ни индуцированной частичным лигированием нерва невропатической боли. Также отмечается растущее число фактов того, что Р2Х7 или его нижележащие эффекторы, такие как интерлейкин-1β (IL-1β), вовлечены в патофизиологию некоторых неврологических расстройств, таких как болезнь Альцгеймера (J.I. Diaz-Hernandez et al., Neurobiol. Aging, 2012, 1816-1828: "In vivo P2X7 inhibition reduces Aβ plaques in AD through GSK3β"). Полагают, что P2X7 осуществляет важную функцию в нейротрансмиссии в ЦНС, оказывая активирующее воздействие на постсинаптические и/или пресинаптические нейроны и нейроглию. С использованием данных гибридизации in situ было выяснено, что мРНК Р2Х7 рецептора широко распределена по всему головному мозгу крысы. В особенности, области с высокой экспрессией мРНК Р2Х7 были обнаружены в переднем обонятельном ядре, коре головного мозга, грушевидной коре (Pir), латеральном ядре перегородки мозга (LS), пирамидальных клетках слоев СА1, СА3, СА4 гиппокампа, мостовых ядрах, наружном клиновидном ядре и медиальном вестибулярном ядре. Сигналы гибридизации Р2Х7 также наблюдали в двигательных нейронах двигательного ядра тройничного нерва, ядра лицевого нерва, ядра подъязычного нерва и переднего рога спинного мозга.
Таким образом, существует терапевтическое основание для применения Р2Х7 антагонистов в лечении ряда болезненных состояний. Эти состояния включают, но не ограничиваются этим, заболевания, ассоциированные с ЦНС, такие как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, боковой амиотрофический склероз, травма спинного мозга, церебральная ишемия, травма головы, менингит, расстройства сна, расстройства настроения и тревожные расстройства, эпилепсия, ВИЧ-индуцированное нейровоспаление и поражение ЦНС, а также хроническая невропатическая и воспалительная боль. Кроме того, воспалительные расстройства периферической нервной системы и аутоиммунные заболевания, включая, но не ограничиваются этим, ревматоидный артрит, остеоартрит, псориаз, аллергический дерматит, астму, хроническую обструктивную болезнь легких, гиперчувствительность дыхательных путей, септический шок, бронхит, гломерулонефрит, синдром раздраженного кишечника, жировую болезнь печени, фиброз печени, повреждение кожных покровов, эмфизему легких, мышечную дистрофию, фиброз, атеросклероз, ожоговое повреждение, болезнь Крона, неспецифический язвенный колит, возрастную дегенерацию желтого пятна, рост и метастазирование раковых клеток, синдром Шегрена, миобластический лейкоз, диабет, остеопороз, ишемическое заболевание сердца, все являются примерами заболеваний, при которых подразумевается вовлечение Р2Х7 рецепторов. Ввиду клинической значимости Р2Х7, идентификация соединений, которые модулируют функцию Р2Х7 рецепторов, представляет собой привлекательное направление в области разработки новых терапевтических агентов.
Ингибиторы Р2Х7 описаны в различных патентных заявках, таких как:
WO 2004/099146, в которой раскрыты бензамидные ингибиторы Р2Х7 рецептора и их применение в лечении воспалительных заболеваний;
WO 2009/108551, в которой раскрыты аналоги гетероариламидов и их применение для опосредуемых Р2Х7 рецепторами состояний;
WO 2009/132000, в которой раскрыты замещенные хинолины и изохинолины в качестве антагонистов Р2Х7 рецепторов и их применение для опосредуемых Р2Х7 рецепторами состояний.
Однако все еще имеется неудовлетворенная потребность в соединениях, которые способны оказывать эффективное антагонизирующее действие в отношении Р2Х7 и которые могут быть доставлены в разные целевые органы, являющиеся очагами развития Р2Х7-опосредуемой патологии, включая головной мозг. Такие соединения предложены в данной заявке.
Далее представлены различные воплощения данного изобретения.
Настоящее изобретение относится к соединениям, представляющим собой тиазолы или оксазолы следующей формулы (I), или их фармацевтически приемлемой соли:
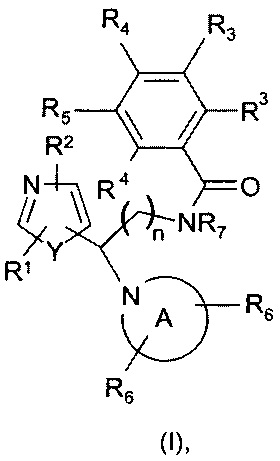
включая их любую стереохимически изомерную форму, где
n равно 1 или 2;
Y представляет собой кислород или серу;
каждый из R1 и R2 независимо выбран из группы, состоящей из водорода, дейтерия, галогена, С1-С4алкила (возможно замещенного гидрокси или галогеном, такого как гидроксиметил, фторметил, дифторметил, трифторметил), С3-С6циклоалкила (возможно замещенного гидрокси или галогеном) или С1-С4алкилокси, каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, галогена, С1-С4алкила, дифторметила, трифторметила, С1-С4алкилокси, NR9R10, где R9 и R10 независимо представляют собой водород или С1-С4алкил, или 2-тиазолидин-1,1-диона; либо две группы R3 или группы R3 и R4 вместе образуют шестичленное гетероциклическое кольцо, содержащее атом азота;
R5 выбран из водорода, галогена или представляет собой гетероциклическое кольцо, выбранное из пиримидин-2-ила, пиридин-2-ила или пиразин-2-ила, возможно замещенное галогеном, С1-С4алкилом, фторметилом, дифторметилом, трифторметилом или С1-С4алкилокси;
R7 представляет собой водород или С1-С4алкил, предпочтительно метил и этил;
радикал
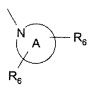
представляет собой возможно замещенное азетидиновое, пирролидиновое, пиперидиновое, морфолиновое, оксазепановое или 1,2,3,4-тетрагидроизохинолиновое кольцо, где каждый из R6 независимо выбран из группы, состоящей из водорода, галогена, С1-С4алкила, С3-С6циклоалкила, С3-С6спироциклоалкила, дифторметила, трифторметила, С1-С4алкилокси, арила, гетероарила, С1-С4арилокси или С1-С4арилалкокси, где арильная или гетероарильная группа возможно замещена галогеном, С1-С4алкилом, фторметилом, дифторметилом, трифторметилом или С1-С4алкилокси.
Две группы R6 могут быть связаны с одним и тем же атомом углерода.
Как использовано в приведенных выше определениях.
Термины «галогено», «галоген» и «галогенид», которые могут быть применены взаимозаменяемо, относятся к замещающему атому фтора, хлора, брома или йода.
Термин «стереохимически изомерные формы», использованный ранее, определяет все возможные изомерные формы, которые могут быть у соединений формулы (I). Если не упомянуто или не указано иное, химическое наименование соединений означает смесь всех возможных стереохимически изомерных форм, причем указанные смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. Более конкретно, стереогенные центры могут иметь R- или S-конфигурацию; заместители в бивалентных циклических (частично) насыщенных радикалах могут находиться либо в цис-, либо в транс-конфигурации.
Очевидно, что стереохимически изомерные формы соединений формулы (I) включены в объем настоящего изобретения.
Абсолютная стереохимическая конфигурация соединений формулы (I) и промежуточных соединений, использованных для их получения, может быть легко определена специалистами в данной области техники с применением хорошо известных методов, таких как, например, дифракция рентгеновских лучей.
Кроме того, некоторые соединения формулы (I) и некоторые из промежуточных соединений, использованных для их получения, могут проявлять полиморфизм. Следует понимать, что настоящее изобретение охватывает любые полиморфные формы, обладающие свойствами, полезными при лечении указанных выше состояний.
Подразумевается, что упомянутые выше фармацевтически приемлемые соли представляют собой терапевтически активные нетоксичные формы солей присоединения кислоты, которые способны образовываться на основе соединений формулы (I). Эти фармацевтически приемлемые соли присоединения кислоты могут быть легко получены путем обработки формы в виде основания определенной подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галоген водородные кислоты, например соляная или бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные кислоты; или органические кислоты, такие как, например, уксусная кислота, пропановая кислота, гидроксиуксусная кислота, молочная кислота, пировиноградная кислота, щавелевая (т.е. этандиовая) кислота, малоновая кислота, янтарная кислота (т.е. бутандиовая кислота), малеиновая кислота, фумаровая кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, трифторметансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, цикламовая кислота, салициловая кислота, п-аминосалициловая кислота, памовая кислота и подобные кислоты.
В свою очередь, указанные солевые формы могут быть превращены в форму свободного основания путем обработки соответствующим основанием.
Соединения формулы (I) могут существовать как в несольватированной, так и в сольватированной формах. Термин «сольват» используется в данной заявке для описания ассоциации молекул, содержащей молекулу соединения по изобретению и одну или более чем одну молекулу фармацевтически приемлемого растворителя, например воды или этанола. Термин «гидрат» используют, когда указанным растворителем является вода.
Предпочтительное воплощение изобретения относится к соединениям формулы (I), определенным выше, где Y и R1-R6 являются такими, как определено выше, R7 представляет собой водород, и n равно 1.
Другое воплощение изобретения относится к соединениям формулы (I), определенным выше, где n, Y и R3-R7 являются такими, как определено выше, и оба R1 и R2 представляют собой водород или один из них представляет собой водород, а другой представляет собой метил, этил, пропил, трет-бутил, возможно замещенные гидрокси или фтором, С3-С6циклоалкил, возможно замещенный гидрокси или фтором.
Другое воплощение изобретения относится к соединениям формулы (I), определенным выше, где n, Y и R1, R2, R6 являются такими, как определено выше, R5 представляет собой водород, R7 представляет собой водород, и каждый из R3 и R4 независимо представляет собой водород, галоген, предпочтительно Cl или F, С1-С4алкил, предпочтительно метил, С1-С4алкилокси, предпочтительно метокси, NR9R10, где R9 и R10 независимо представляют собой водород или С1-С4алкил, или 2-тиазолидин-1,1-дион, либо две группы R3 вместе образуют шестичленное гетероциклическое кольцо, содержащее атом азота.
Другое воплощение изобретения относится к соединениям формулы (I), определенным выше, где n, Y и R1, R2, R6 являются такими, как определено выше, R7 представляет собой водород, R4 представляет собой водород, R3 в мета-положении представляет собой водород, и R3 в орто-положении выбран из группы, состоящей из галогена, предпочтительно Cl или F, или С1-С4алкила, предпочтительно метила, и R5 представляет собой гетероциклическое кольцо, выбранное из пиримидин-2-ила, пиридин-2-ила или пиразин-2-ила, возможно замещенное галогеном, предпочтительно пиримидин-2-ила, возможно замещенного фтором.
Другое воплощение изобретения относится к соединениям формулы (I), определенным выше, где n, Y и R1-R5, являются такими, как определено выше, R7 представляет собой водород, и кольцо А выбрано из группы, состоящей из
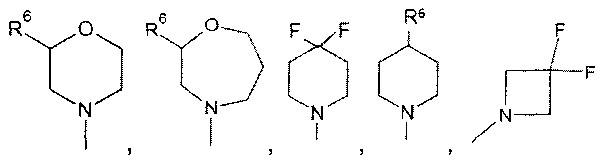 или
или 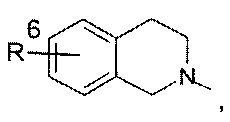
где R6 представляет собой водород, галоген, бензилокси или фенокси, фенил, пиразол, С3-С6циклоалкил, возможно замещенный галогеном, предпочтительно замещенный фтором.
Наиболее предпочтительно соединение формулы 1 по настоящему изобретению выбрано из группы, состоящей из:




Как правило, соединения формулы (I) могут быть получены в результате взаимодействия соединения формулы (II):
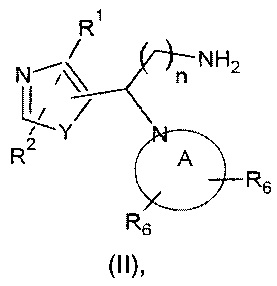
где значения n, Y, А и R1, R2 и R6 являются такими, как определено выше, с соединением формулы (III):
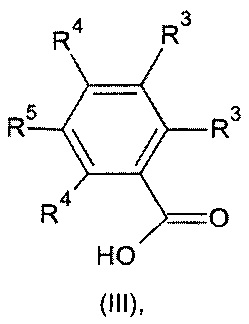
где значения R3, R4 и R5 являются такими, как определено выше; или
с соединением формулы (IIIa):
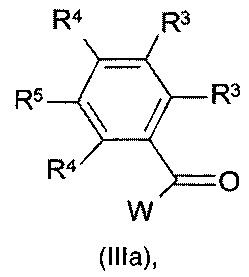
где значения R3, R4 и R5 являются такими, как определено выше, и W представляет собой подходящую уходящую группу;
и возможно превращения полученного соединения формулы (I) в соль присоединения, и/или получения его стереохимически изомерных форм.
Взаимодействие соединения формулы (II) с соединением формулы (III) может быть осуществлено по меньшей мере в одном реакционно-инертном растворителе и возможно в присутствии по меньшей мере одного подходящего реагента сочетания и/или его подходящего основания. Для активации карбоновой кислоты формулы (III) может быть удобным добавление эффективного количества активатора реакции. Неограничивающие примеры таких активаторов реакции включают карбонилдиимидазол, N,N'-дициклогексил-карбодиимид или 1-(3-диметиламинопропил)-3-этилкарбодиимид, гидроксибензотриазол, гексафторфосфат бензотриазолил-окситрис-(диметиламино)-фосфония, гексафторфосфат тетрапирролидинофосфония, гексафторфосфат бромтрипирролидинофосфония или их функциональное производное, как например, описано в D. Hudson (J. Org. Chem. (1988), 53, 617).
W в соединении формулы (IIIa) представляет собой подходящую уходящую группу, такую как, например, галоген, например фтор, хлор, бром, йод, или в некоторых случаях W также может представлять собой сульфонилоксигруппу, например метансульфонилокси, трифторметансульфонилокси, бензолсульфонилокси и подобные реакционно-способные уходящие группы. Взаимодействие соединения формулы (II) с соединением формулы (III) может быть осуществлено в реакционно-инертном растворителе, таком как, например, ацетонитрил, диметилацетамид, N-метил-пирролидон или DMF, и возможно в присутствии подходящего основания, такого как, например, карбонат натрия, карбонат калия или триэтиламин. Скорость реакции можно увеличить посредством перемешивания. Реакцию можно подходящим образом осуществлять при температуре в диапазоне от комнатной температуры до температуры дефлегмации реакционной смеси.
Соединения формулы (III) и (IIIa) известны в данной области техники, или их можно получить, следуя способам, изложенным в примерах.
Соединения формулы (II) могут быть получены в соответствии с приведенной ниже схемой:
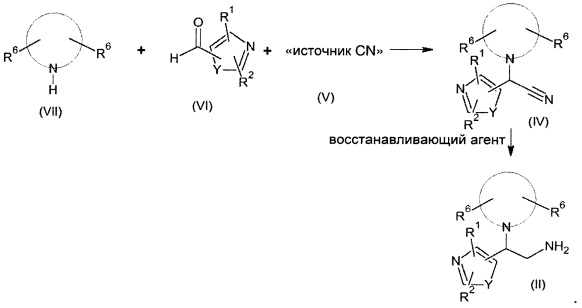
Первичные амины (II) могут быть получены путем восстановления соответствующих нитрильных производных (IV) в реакции образования связи азот-водород. Неограничивающие примеры такой реакции включают восстановление с использованием:
- водорода или источника водорода в присутствии металла, такого как никель, платина, палладий и кобальт, или его производного, такого как Ni Ренея, оксид платины, оксид палладия или кобальт Ренея, в качестве катализатора;
- гидрида, такого как алюмогидрид лития, диизобутилалюминийгидрид (DIBAL), боргидрид или их функциональное производное.
Реакция может быть проведена в подходящем растворителе, таком как метанол, тетрагидрофуран, уксусная кислота, диэтиловый эфир, толуол или метанольный раствор аммиака, предпочтительно при температурах от -78°С до комнатной температуры (КТ).
Соединения формулы (IV), где R1, R2 и R6 являются такими, как определено в формуле (I), могут быть получены из альдегидов (VI) в реакции конденсации Штреккера при взаимодействии с соответствующим гетероциклильным промежуточным соединением (VII) в присутствии источника цианида (V), например триметилсилилцианида (TMSCN) или его функционального производного, в растворителе, таком как АсОН или MeCN, предпочтительно при температурах от 0°С до КТ.
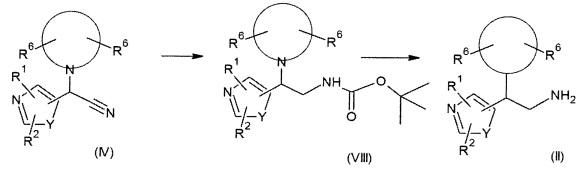
Альтернативно, соединения формулы (II) также могут быть получены с применением двухстадийной методики, как сообщалось выше. В результате взаимодействия соединений формулы (IV) с восстанавливающим реагентом, предпочтительно боргидридом натрия, в присутствии гексагидрата хлорида никеля(II) или гексагидрата хлорида кобальта(II) и Boc2O в растворителе, таком как МеОН, предпочтительно при температурах от 0°С до КТ, получают Boc-защищенный первичный амин формулы (VIII). После удаления защиты с использованием подходящей кислоты, предпочтительно трифторуксусной кислоты (TFA), получают соединения (II).
Примеры соединений формулы (VI) представлены на следующей схеме:
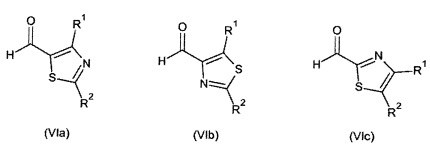
Скорость реакции конденсации Штреккера можно увеличить посредством перемешивания. Исходные вещества и некоторые из промежуточных соединений являются известными соединениями и имеются в продаже или могут быть получены в соответствии с традиционными реакционными методами, общеизвестными в данной области техники.
Указанный способ возможно дополнительно включает асимметричную реакцию синтеза с применением хиральных вспомогательных веществ (с использованием углевода, хирального амина или циклического кетимина) и/или каталитический асимметричный синтез Штреккера (с использованием гуанидина, хирального шиффова основания или катализатора на основе 1,1'-би-2-нафтола (BINOL)).
Соединения формулы (I), полученные описанными выше способами, могут быть синтезированы в форме рацемических смесей энантиомеров, которые можно отделить друг от друга, следуя известным в данной области техники методам разделения. Те соединения формулы (I), которые получают в рацемической форме, могут быть превращены в соответствующие диастереомерные солевые формы путем взаимодействия с подходящей хиральной кислотой. Указанные диастереомерные солевые формы впоследствии разделяют, например, посредством селективной или фракционной кристаллизации, и энантиомеры высвобождают из них с использованием щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (1) включает жидкостную хроматографию с использованием хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы также могут быть получены из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ, при условии, что реакция протекает стереоспецифично. Предпочтительно, чтобы в случае, если желателен конкретный стереоизомер, указанное соединение было бы синтезировано с использованием стереоспецифических способов получения. В этих способах предпочтительно использовать энантиомерно чистые исходные вещества.
Соединения формулы (I), их фармацевтически приемлемые соли и стереоизомерные формы обладают антагонистическими свойствами в отношении Р2Х7 рецептора, что продемонстрировано в фармакологических примерах. Другими примерами известных в данной области техники реакций трансформации групп с целью превращения соединений формулы (I) в другие соединения формулы (I) являются: гидролиз сложных эфиров карбоновых кислот до соответствующей карбоновой кислоты или соответствующего спирта; гидролиз амидов до соответствующих карбоновых кислот или аминов; спирты могут быть превращены в сложные эфиры и простые эфиры; первичные амины могут быть превращены во вторичные или третичные амины; можно провести гидрирование по двойным связям до соответствующей одинарной связи. Исходные вещества и некоторые из промежуточных соединений являются известными соединениями и имеются в продаже или могут быть получены в соответствии с традиционными реакционными методиками, общеизвестными в данной области техники. Соединения формулы (I), полученные описанными выше способами, могут быть синтезированы в форме рацемических смесей энантиомеров, которые можно отделить друг от друга, следуя известным в данной области техники методикам разделения. Те соединения формулы (I), которые получают в рацемической форме, могут быть превращены в соответствующие диастереомерные солевые формы путем взаимодействия с подходящей хиральной кислотой. Указанные диастереомерные солевые формы впоследствии разделяют, например, посредством селективной или фракционной кристаллизации, и энантиомеры высвобождают из них с использованием щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает жидкостную хроматографию с применением хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы также могут быть получены из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ, при условии, что реакция протекает стереоспецифично. Предпочтительно, чтобы в случае, если желателен конкретный стереоизомер, указанное соединение было бы синтезировано с использованием стереоспецифических способов получения. В этих способах предпочтительно использовать энантиомерно чистые исходные вещества. При получении соединений формулы (I) и исходных веществ и/или промежуточных соединений, описанных в данной заявке, может быть полезным защитить определенные группы, которые являются чувствительными к реакционным условиям. Оценка полезности возможной защиты, а также выбор подходящего для защиты агента в соответствии с реакцией, проводимой при получении соединений по изобретению, и функциональной группы, подлежащей защите, находятся в пределах общеизвестных знаний специалиста. Удаление возможных защитных групп проводят в соответствии с традиционными методами. В качестве основной ссылки на применение защитных групп в органической химии см. Theodora W. Greene and Peter G.M. Wuts, "Protective groups in organic synthesis", John Wiley & Sons, Inc., II Ed., 1991.
Получение солей соединений формулы (I) осуществляют в соответствии с известными методами. Следовательно, соединения по настоящему изобретению формулы (I) полезны в качестве лекарственного средства, в особенности при лечении состояния или заболевания, опосредуемого Р2Х7 рецептором, в частности, лекарственного средства с антагонистической активностью в отношении Р2Х7 рецептора. Следовательно, соединения по настоящему изобретению могут быть использованы для изготовления лекарственного средства для лечения состояния или заболевания, опосредуемого активностью Р2Х7 рецептора, в частности, лекарственного средства с антагонистической активностью в отношении Р2Х7 рецептора.
Согласно настоящему изобретению также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения состояний или заболеваний, выбранных из опосредуемых Р2Х7 рецептором состояний или заболеваний. В одном из воплощений согласно настоящему изобретению предложено соединение формулы (I) для применения в качестве лекарственного средства или для применения при лечении состояний или заболеваний, выбранных из опосредуемых Р2Х7 рецептором состояний или заболеваний. Кроме того, согласно настоящему изобретению также предложен способ лечения состояния, опосредуемого активностью Р2Х7 рецептора, у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. С учетом вышеописанных механизмов действия соединения по изобретению полезны для лечения нейродегенеративных расстройств различного происхождения, таких как болезнь Альцгеймера и другие связанные с деменцией состояния, как например, деменция с тельцами Леви, лобно-височная деменция и таупатии; амиотрофический боковой склероз, рассеянный склероз, болезнь Паркинсона и другие синдромы паркинсонизма; ВИЧ-индуцированное нейровоспаление; эссенциальный тремор; другие спиноцеребеллярные дегенерации и нейропатия Шарко-Мари-Тута. Соединения по изобретению также полезны для лечения неврологических состояний, таких как эпилепсия, включая простой парциальный припадок, сложный парциальный припадок, вторично-генерализованный припадок, дополнительно включающий малый эпилептический припадок, миоклонические судороги, клонические судороги, тонические судороги, тонико-клонические судороги и атонические судороги, и также для предупреждения и лечения эпилептического статуса (SE).
Соединения по изобретению также полезны при лечении когнитивных расстройств и психических расстройств. Психические расстройства включают, но не ограничиваются этим, глубокую депрессию, дистимию, манию, биполярное расстройство (такое как биполярное расстройство I типа, биполярное расстройство II типа), циклотимическое расстройство, «быструю» цикличность, чрезвычайно быструю смену маниакальной и депрессивной фаз, манию, гипоманию, шизофрению, шизофреноморфные расстройства, шизоаффективные расстройства, расстройства личности, расстройства внимания с гиперактивным поведением или без него, бредовые расстройства, кратковременные психотические расстройства, индуцированные психотические расстройства, психотическое расстройство, обусловленное общим состоянием здоровья, вызванные веществами психотические расстройства или не идентифицированное психотическое расстройство, тревожные расстройства, такие как генерализованное тревожное расстройство, панические расстройства, посттравматическое стрессовое расстройство, расстройства контроля над побуждениями, фобические расстройства, диссоциативные состояния и, кроме того, зависимость от курения, лекарственную зависимость и алкоголизм. В частности, биполярные расстройства, психоз, тревога и зависимость.
Соединения по настоящему изобретению полезны в предупреждении или лечении нейровоспаления и поражения ЦНС, индуцированных ВИЧ-инфекцией, и ВИЧ-ассоциированных нейрокогнитивных нарушений. Соединения по настоящему изобретению полезны в предупреждении или лечении невропатической боли. Синдромы невропатической боли включают, но не ограничиваются этим: диабетическую нейропатию; ишиалгию; неспецифическую боль в поясничном отделе позвоночника; боль при рассеянном склерозе; фибромиалгию; ВИЧ-обусловленную нейропатию; невралгию, такую как постгерпетическая невралгия и невралгия тройничного нерва, невралгия Мортона, каузалгия; и боль, обусловленную физической травмой, ампутацией, фантомом конечности, раковым заболеванием, токсинами или хроническими воспалительными состояниями; центральную боль, такую как боль, наблюдаемую при таламических синдромах; смешанные центральные и периферические формы боли, такие как комплексные региональные болевые синдромы (CRPS), также называемые симпатическими рефлекторными дистрофиями.
Соединения по изобретению также полезны в лечении хронической боли. Хроническая боль включает, но не ограничивается этим, хроническую боль, вызванную воспалением или связанным с воспалением состоянием, остеоартритом, ревматоидным артритом, острым повреждением или травмой, боль в верхнем отделе позвоночника и боль в поясничном отделе позвоночника (являющуюся результатом системного, регионарного или первичного заболевания позвоночника, как например, при радикулопатии), боль в костях (обусловленную остеоартритом, остеопорозом, костным метастазированием или неизвестными причинами), тазовую боль, боль, ассоциированную с травмой спинного мозга, кардиальную боль в грудной клетке, некардиальную боль в грудной клетке, центральную постинсультную боль, миофасциальный болевой синдром, боль при серповидноклеточной анемии, боль при раковом заболевании, боль при болезни Фабри, боль при синдроме приобретенного иммунодефицита (СПИД), боль в старческом возрасте или боль, причиной которой являются головная боль, синдром височно-нижнечелюстного сустава, подагра, фиброз или компрессионные синдромы верхней апертуры грудной клетки, в частности, ревматоидный артрит и остеоартрит.
Соединения по изобретению также полезны при лечении острой боли, вызванной острым повреждением, болезнью, спортивно-медицинскими повреждениями, синдромом запястного канала, ожогами, растяжениями и напряжениями в скелетно-мышечной системе, мышечно-сухожильным растяжением, шейно-плечевыми болевыми синдромами, диспепсией, язвой желудка, язвой двенадцатиперстной кишки, дисменореей, эндометриозом или хирургическим вмешательством (таком как, при операции на открытом сердце или коронарном шунтировании), в лечении послеоперационной боли, боли при камнях в почках, боли в желчном пузыре, боли при камнях в желчном пузыре, родовой боли или зубной боли.
Соединения по изобретению также полезны в лечении головных болей, таких как мигрень, головная боль напряжения, трансформированная мигрень или развивающаяся головная боль, кластерная головная боль, а также расстройств, связанных с вторичной головной болью, таких как расстройства, вызванные инфекциями, метаболическими расстройствами или другими системными болезнями, и других острых головных болей, пароксизмальной гемикрании и тому подобных, являющихся результатом ухудшения вышеупомянутых первичной и вторичной головных болей.
Соединения по изобретению также полезны в лечении таких заболеваний, как головокружение, шум в ушах, мышечный спазм и другие расстройства, включая, но не ограничиваясь ими, сердечно-сосудистые заболевания (такие как сердечная аритмия, инфаркт миокарда или стенокардия, гипертензия, сердечная ишемия, церебральная ишемия), эндокринные нарушения (такие как акромегалия или несахарный диабет), заболевания, при которых патофизиология расстройства включает избыточную секрецию или гиперсекрецию или другую неподходящую клеточную секрецию эндогенного вещества (такого как катехоламин, гормон или фактор роста).
Соединения по изобретению также полезны в селективном лечении заболевания печени, такого как воспалительные заболевания печени, например, хронический вирусный гепатит В, хронический вирусный гепатит С, алкогольное поражение печени, первичный билиарный цирроз, аутоиммунный гепатит, фиброз печени, неалкогольный стеатогепатит и отторжение трансплантата печени.
Соединения по изобретению ингибируют воспалительные процессы, затрагивающие все системы организма. Поэтому они полезны в лечении воспалительных процессов скелетно-мышечной системы, для которых ниже приведен список примеров, не являющийся исчерпывающим списком всех целевых расстройств: артритоподобные состояния, такие как, анкилозирующий спондилит, артрит шейного отдела позвоночника, фибромиалгия, подагра, ювенильный ревматоидный артрит, пояснично-крестцовый артрит, остеоартрит, остеопороз, псориатический артрит, ревматическая болезнь; расстройства, затрагивающих кожу и родственные ткани: экзема, псориаз, дерматит и воспалительные состояния, такие как солнечный ожог; расстройства дыхательной системы: астма, аллергический ринит и респираторный дистресс-синдром, легочные расстройства, в которые вовлечено такое восплание, как астма и бронхит; хроническая обструктивная болезнь легких; расстройства иммунной и эндокринной систем: узелковый периартрит, тиреоидит, апластическая анемия, склеродермия, тяжелая миастения, рассеянный склероз и другие демиелинизирующие заболевания, энцефаломиелит, саркоидоз, почечный синдром, синдром Бехчета, полимиозит, гингивит.
Соединения по изобретению также полезны в лечении расстройств желудочно-кишечного тракта (ЖКТ), таких как воспалительные заболевания кишечника (IBD), включая, но не ограничиваясь этим, неспецифический язвенный колит, болезнь Крона, илеит, проктит, глютеновую болезнь, энтеропатии, микроскопичесий или коллагенозный колит, эозинофильный гастроэнтерит или паучит, возникающий после проктоколэктомии и постиленатального анастомоза, и синдром раздраженного кишечника, включающий любые расстройства, ассоциированные с абдоминальной болью и/или абдоминальным дискомфортом, такие как пилороспазм, нервная диспепсия, синдром раздраженной толстой кишки, спастический колит, спастический кишечник, невроз кишечника, функциональный колит, слизистый колит, колит, вызванный приемом слабительных средств, и функциональная диспепсия; а также для лечения атрофического гастрита, гастрита различной этиологии (gastritis varialoforme), неспецифического язвенного колита, пептической язвы, изжоги и другого поражения ЖКТ, например, в результате инфекции Helicobacter pylori, гастроэзофагеальной рефлюксной болезни, гастропареза, такого как диабетический гастропарез; и других функциональных расстройств кишечника, таких как неязвенная диспепсия (NUD); рвота, диарея и висцеральное воспаление внутренних органов.
Соединения по изобретению также полезны в лечении расстройств мочеполовых путей, таких как гиперактивность мочевого пузыря, простатит (хронический бактериальный и хронический небактериальный простатит), простатодиния, интерстициальный цистит, недержание мочи и доброкачественная гиперплазия предстательной железы, аднексит, пельвиоперитонит, бартолинит и вагинит. В частности, гиперактивность мочевого пузыря и недержание мочи.
Соединения по изобретению также полезны в лечении офтальмологических заболеваний, таких как ретинит, ретинопатия, увеит и острое повреждение ткани глаза, возрастная макулярная дегенерация или глаукома, конъюнктивит.
Соединения по изобретению также полезны в лечении расстройств приема пищи, таких как нервная анорексия, включая подтипы ограничительного типа и типа с компульсивным перееданием/очищением кишечника (при помощи слабительных средств); нервная булимия, включая подтипы булимии с очищением кишечника и булимии без очищения кишечника; ожирение; компульсивные расстройства приема пищи; компульсивное переедание; и не классифицированное расстройство приема пищи.
Соединения по изобретению также полезны в лечении аллергического дерматита, гипервосприимчивости дыхательных путей, хронической обструктивной болезни легких (COPD), бронхита, септического шока, синдрома Шегрена, гломерулонефрита, атеросклероза, роста и метастазирования клеток злокачественного образования, миобластического лейкоза, диабета, менингита, остеопороза, ожогового повреждения, ишемической болезни сердца, инсульта, заболевания периферических кровеносных сосудов, варикозного расширения вен, глаукомы.
Термин «подвергание лечению» и «лечение», использованный в данном описании, относится к терапевтическому, паллиативному и профилактическому лечению, включая реверсирование, облегчение, подавление развития или предупреждение заболевания, расстройства или состояния, к которому применяется такой термин, или одного или более симптомов такого заболевания, расстройства или состояния.
Кроме того, согласно настоящему изобретению предложены фармацевтические композиции, содержащие по меньшей мере один фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы (I).
Чтобы приготовить фармацевтические композиции по настоящему изобретению, эффективное количество конкретного соединения, в форме основания или соли присоединения кислоты, в качестве активного ингредиента объединяют в однородной смеси по меньшей мере с одним фармацевтически приемлемым носителем, который может иметь самые разнообразные формы в зависимости от формы препарата, необходимого для введения. Эти фармацевтические композиции желательно находятся в стандартной лекарственной форме, предпочтительно, для перорального введения, ректального введения, чрескожного введения или парентеральной инъекции.
Например, при изготовлении композиций в пероральной лекарственной форме можно использовать любой из обычных жидких фармацевтических носителей, таких как, например, вода, гликоли, масла, спирты и тому подобное, в случае пероральных препаратов в жидкой форме, таких как суспензии, сиропы, эликсиры и растворы; или можно использовать любой из твердых фармацевтических носителей, таких как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, разрыхлители и тому подобное, в случае порошков, пилюль, капсул и таблеток. Ввиду легкости их введения таблетки и капсулы представляют собой наиболее удобную пероральную стандартную лекарственную форму, и в этом случае, несомненно, используют твердые фармацевтические носители. В случае композиций для парентеральной инъекции фармацевтический носитель будет преимущественно содержать стерильную воду, хотя для улучшения растворимости активного ингредиента могут быть включены другие компоненты.
Растворы для инъекций могут быть приготовлены, например, с использованием фармацевтически приемлемого носителя, содержащего физиологический раствор, раствор глюкозы или смесь их обоих. Суспензии для инъекций также могут быть приготовлены с использованием подходящих жидких носителей, суспендирующих агентов и тому подобного. Фармацевтический носитель в композициях, подходящих для чрескожного введения, возможно может содержать усиливающий проникновение через кожу агент и/или пригодный увлажняющий агент, возможно в комбинации с небольшими количествами пригодных добавок, которые не оказывают существенного вредного воздействия на кожу. Указанные добавки могут быть выбраны с целью облегчения введения активного ингредиента в кожу и/или быть полезными при изготовлении требуемых композиций. Эти композиции для местного применения можно вводить различными способами, например, в виде трансдермального пластыря, препарата для капельного нанесения («spot-on») или мази. Соли присоединения соединений формулы (1), благодаря своей повышенной растворимости в воде по сравнению с соответствующей формой в виде основания, несомненно, являются более подходящими для приготовления водных композиций.
Особенно предпочтительным является приготовление фармацевтических композиций по изобретению в стандартной лекарственной форме для удобства введения и однородности дозирования.
Термин «стандартная лекарственная форма», используемый в данном описании, относится к физически дискретным единицам, подходящим в качестве одинарных дозировок, причем каждая единица содержит предварительно заданное количество активного ингредиента, рассчитанное для оказания желаемого терапевтического эффекта, совместно с необходимым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (в том числе делимые таблетки или таблетки, покрытые оболочкой), капсулы, пилюли, пакетики с порошком, облатки, растворы или суспензии для инъекций, мерные дозы, соответствующие чайной ложке, мерные дозы, соответствующие столовой ложке и тому подобное и их отделенные кратные количества.
Для целей перорального введения фармацевтические композиции по настоящему изобретению могут иметь вид твердых лекарственных форм, например, таблеток (как в проглатываемых, так и в жевательных формах), капсул или желатиновых капсул, приготовленных традиционными способами, вместе с фармацевтически приемлемыми эксципиентами и носителями, такими как связующие вещества (например, прежелатинизированный кукурузный крахмал, поливинилпирролидон, гидроксипропилметилцеллюлоза и тому подобное), наполнители (например, лактоза, микрокристаллическая целлюлоза, фосфат кальция и тому подобное), смазывающие вещества (например, стеарат магния, тальк, диоксид кремния и тому подобное), разрыхлители (например, картофельный крахмал, натрия крахмала гликолят и тому подобное), увлажняющие агенты (например, лаурилсульфат натрия) и тому подобное. Такие таблетки также могут быть покрыты оболочкой способами, хорошо известными в данной области техники.
Препараты для перорального введения в жидкой форме могут иметь вид, например, растворов, сиропов или суспензий, или они могут быть приготовлены в виде сухого продукта для смешивания с водой и/или другим подходящим жидким носителем перед использованием. Такие препараты в жидкой форме могут быть приготовлены традиционными методами, возможно вместе с другими фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, метилцеллюлоза, гидроксипропилметилцеллюлоза или гидрированные пищевые жиры), эмульгирующие агенты (например, лецитин или аравийская камедь), неводные носители (например, миндальное масло, сложные эфиры жирных кислот или этиловый спирт), подсластители, корригенты, маскирующие вкус и запах агенты и консерванты (например, метил- или пропил-п-гидроксибензоаты или сорбиновая кислота).
Фармацевтически приемлемые подсластители, пригодные для фармацевтических композициях по изобретению, предпочтительно включают по меньшей мере один подсластитель с интенсивным вкусом, такой как аспартам, ацесульфам калия, цикламат натрия, элитам, дигидрохалконовый подсластитель, монеллин, стевиозид-сукралоза (4,1',6'-трихлор-4,1',6'-тридезоксигалактосахароза) или, предпочтительно, сахарин, натриевая или кальциевая соль сахарина, и возможно по меньшей мере один объемный подсластитель, такой как сорбит, маннит, фруктоза, сахароза, мальтоза, изомальт, глюкоза, гидрогенизированный глюкозный сироп, ксилит, карамель или мед. Подсластители с интенсивным вкусом обычно используют в низких концентрациях. Например, в случае натриевой соли сахарина указанная концентрация может меняться в диапазоне от примерно 0,04% до 0,1% (масса/объем) от конечной композиции. Объемный подсластитель можно эффективно использовать в более высоких концентрациях, изменяющихся в диапазоне от примерно 10% до примерно 35%, предпочтительно от примерно 10% до примерно 15% (масса/объем). Фармацевтически приемлемые корригенты, которые могут маскировать ингредиенты с горьким вкусом в композициях с низкими дозами, предпочтительно включают фруктовые корригенты, такие как вишневые, малиновые, черносмородиновые или клубничные корригенты. Сочетание двух корригентов может обеспечить очень хорошие результаты. В композициях с высокими дозами могут потребоваться более сильные фармацевтически приемлемые корригенты, такие как карамель-шоколад (Caramel Chocolate), освежающая мята (Mint Cool), «фантазия» (Fantasy) и тому подобное.
Каждый корригент может присутствовать в конечной композиции в концентрации, изменяющейся в диапазоне от примерно 0,05% до 1% (масса/объем). Предпочтительно используются сочетания указанных сильных корригентов. Предпочтительно используют корригент, который не подвергается какому-либо изменению или не теряет свой вкус и/или цвет в условиях данной композиции.
На основе соединений формулы (I) могут быть изготовлены композиции для парентерального введения путем инъекции, обычно внутривенной, внутримышечной или подкожной инъекции, например, болюсной инъекции или непрерывной внутривенной инфузии. Композиции для инъекции могут быть представлены в стандартной лекарственной форме, например, в ампулах или многодозовых контейнерах, включающей добавленный в нее консервант. Они могут находиться в таких формах, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать такие включаемые при изготовлении композиций агенты, как средства, способствующие поддержанию изотоничности, суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может присутствовать в порошковой форме для смешивания с подходящим носителем, например стерильной апирогенной водой, перед использованием.
На основе соединений формулы (I) также могут быть изготовлены ректальные композиции, такие как суппозитории или удерживающие клизмы, например, содержащие традиционные основы для суппозиториев, такие как масло какао и/или другие глицериды.
Специалисты в области лечения заболеваний, связанных с регулированием лиганд-управляемых ионных каналов, легко определят терапевтически эффективное количество соединения формулы (I) на основании представленных далее результатов тестирования. Обычно подразумевается, что терапевтически эффективная доза будет составлять от примерно 0,001 мг/кг до примерно 50 мг/кг массы тела, более предпочтительно от примерно 0,01 мг/кг до примерно 10 мг/кг массы тела подлежащего лечению пациента. Может оказаться приемлемым введение терапевтически эффективной дозы в форме двух или более субдоз с подходящими интервалами в течение суток. Указанные субдозы могут быть приготовлены в виде стандартных лекарственных форм, при этом, например, каждая из них содержит от примерно 0,1 мг до примерно 1000 мг, более конкретно от примерно 1 до примерно 500 мг активного ингредиента на одну стандартную лекарственную форму.
Как использовано в настоящем описании, «терапевтически эффективное количество» соединения представляет собой количество соединения, в результате введения которого индивидууму или животному уровень такого соединения у индивидуума или животного оказывается достаточно высоким, чтобы вызвать заметный антагонистический ответ в отношении Р2Х7 рецепторов.
Точная дозировка и частота введения зависит от конкретного используемого соединения формулы (I), конкретного подвергаемого лечению состояния, степени тяжести подвергаемого лечению состояния, возраста, массы и общего физического состояния конкретного пациента, а также другого препарата, который этот пациент может принимать, что хорошо известно специалистам в данной области техники. Кроме того, указанное «терапевтически эффективное количество» можно уменьшать или увеличивать в зависимости от реакции подвергаемого лечению пациента и/или в зависимости от оценки врача, назначающего соединения по настоящему изобретению. Поэтому упомянутые выше диапазоны эффективных количеств, принимаемых в течение одних суток, представляют собой всего лишь рекомендации.
Номенклатура и структуры
В общем случае использованная в этой заявке номенклатура основывается на ChemSketch™ (ACDLabs), и при этом генерируются химические названия, соответствующие систематической номенклатуре IUPAC. Химические структуры, показанные в данном описании, получали с использованием ISIS®, версия 2.2. Любая свободная валентность, показанная на атоме углерода, кислорода, серы или азота в приведенных в данном описании структурах, указывает на присутствие атома водорода, если не указано иное. Если азот-содержащее гетероарильное кольцо показано со свободной валентностью на атоме азота, и на этом гетероарильном кольце показаны переменные, такие как R1, R2, R3 и т.д., то такие переменные могут быть связаны с атомом азота, имеющим свободную валентность, или присоединены к нему. Если в структуре имеется хиральный центр, но для этого хирального центра не показана никакая конкретная стереохимия, то данной структурой охватываются оба энантиомера, ассоциированные с данным хиральным центром. Если структура, показанная в данном описании, может существовать в нескольких таутомерных формах, то все такие таутомеры охватываются данной структурой. Подразумевается, что атомы, представленные в структуре, приведенной в данном описании, охватывают все существующие в природе изотопы таких атомов. Так, например, подразумевается, что атомы водорода, представленные в данном описании, включают дейтерий и тритий, и подразумевается, что атомы углерода включают изотопы С13 и С14.
Сокращения
В описании схем и примеров использованы следующие сокращения:


Экспериментальная часть
Следующие далее примеры иллюстрируют настоящее изобретение. Если конкретно не указано иное, все показатели (в особенности процентные содержания и количества) относятся к массе.
А. Синтез промежуточных соединений
Пример А.1
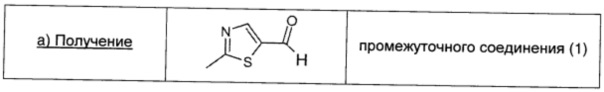
2-Броммалональдегид (1,5 г; 9,94 ммоль; 1 экв.) и тиоацетамид (0,83 г; 11,05 ммоль; 1,11 экв.) суспендировали в DCM (10 мл) и охлаждали до 0°С; затем по каплям добавляли DIPEA (1,75 мл; 10,05 ммоль; 1,01 экв.). Полученный коричневый раствор оставляли перемешиваться при комнатной температуре в течение 3 суток. Затем реакционную смесь разбавляли водой (50 мл) и собирали органическую фазу. Водную фазу трижды экстрагировали DCM (10 мл). Органическую фазу сушили над безв. Na2SO4, фильтровали и упаривали. Коричневый остаток растворяли в Et2O (20 мл), дважды промывали насыщенным раствором NaHCO3 (20 мл) и рассолом (20 мл), сушили над безв. Na2SO4, фильтровали и окончательно упаривали, получая чистый указанный в заголовке продукт в виде коричневого масла (0,40 г; выход 31%).
Пример А.2
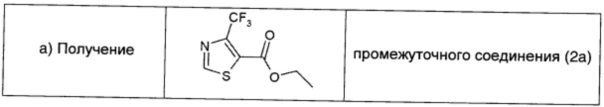
Этил-2-амино-4-(трифторметил)-5-тиазол-карбоксилат (1,15 г; 4,79 ммоль; 1 экв.) растворяли в 1,3-диоксане (27 мл) и по каплям при комнатной температуре добавляли изоамилнитрит (1,51 г; 12,93 ммоль; 2,7 экв.). Реакционную смесь нагревали при 80°С и через 1 ч с использованием TLC (80/20 петролейный эфир/EtOAc) наблюдали полное превращение в продукт исходного вещества. Растворитель удаляли в вакууме и неочищенный продукт очищали прямофазовой флэш-хроматографией (95/5→90/10 петролейный эфир/EtOAc), получая чистое соединение 2а (0,94 г; выход 87%) в виде желтого масла.
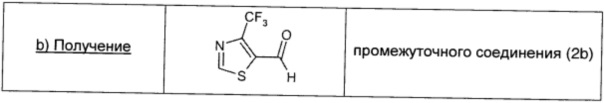
Промежуточное соединение 2а (0,82 г; 3,66 ммоль; 1 экв.) растворяли в безводном DCM (18 мл) в атмосфере аргона и охлаждали до -70°С. Затем по каплям в течение 10 минут добавляли 1 М DIBAL в DCM (4,1 мл; 4,10 ммоль; 1,12 экв.) и смесь перемешивали при этой же температуре в течение 1,5 ч. Температуру реакционной смеси подводили до 0°С, последовательно добавляли воду (0,186 мл), 15% раствор NaOH (0,186 мл) и вторую порцию воды (0,186 мл), и смесь перемешивали до полного выпадения в осадок соли алюминия (5 мин). Смесь сушили над безводным Na2SO4 и фильтровали. После выпаривания растворителя неочищенный продукт очищали прямофазовой флэш-хроматографией (20/80→50/50 DCM/петролейный эфир), получая чистое промежуточное соединение (2b) в виде желтого масла (0,3 г; выход 45%).
Пример А.3
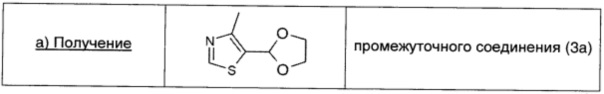
Моногидрат п-толуолсульфоновой кислоты (0,03 г; 0,16 ммоль; 0,08 экв.) добавляли к смеси 4-метил-1,3-тиазол-5-карбальдегида (0,25 г; 1,97 ммоль; 1 экв.) и 1,2-этандиола (0,38 мл; 6,88 ммоль; 3,5 экв.) в безводном толуоле (5,5 мл). Колбу соединяли с ловушкой Дина-Старка, и смесь нагревали до температуры дефлегмации в течение 6 ч. После охлаждения до температуры окружающей среды реакцию гасили 10% раствором Na2CO3 (15 мл). Водный слой экстрагировали этилацетатом (10 мл × 3). Объединенные экстракты сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали на колонке с силикагелем (100% DCM → 30/70 EtOAc/DCM), получая промежуточное соединение (3а) в виде желтого масла (0,29 г; выход 86%).
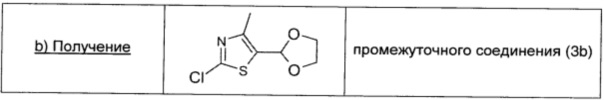
Раствор 1,6 М н-бутиллития в гексане (1,28 мл; 2,05 ммоль; 1,5 экв.) по каплям при -70°С в атмосфере аргона добавляли к раствору промежуточного соединения (3а) (0,23 г; 1,37 ммоль; 1 экв.) в безводном THF (4,5 мл). Полученный темный раствор перемешивали при -70°С в течение 30 мин, затем по каплям при этой же температуре добавляли 1,05 М четыреххлористый углерод в безводном THF (2 мл; 2,10 ммоль; 1,5 экв.). Через 1 ч реакцию гасили насыщенным водным раствором NH4Cl (1 мл) и температуру реакционной смеси доводили до комнатной. Смесь распределяли между водой (10 мл) и AcOEt (10 мл), и водный слой экстрагировали AcOEt (10 мл ×3). Объединенные экстракты сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле (100% DCM → 5/95 EtOAc/DCM 0%), получая промежуточное соединение (3b) в виде темного масла (0,208 г; выход 74%).
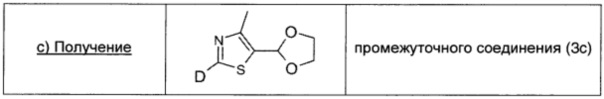
Раствор 1,6 М н-бутиллития в гексане (0,632 мл; 1,01 ммоль; 2 экв.) по каплям при -70°С в атмосфере аргона добавляли к раствору промежуточного соединения (3b) (0,10 г; 0,51 ммоль; 1 экв.) в безводном THF (2,4 мл). Полученный темный раствор перемешивали при -70°С в течение 30 мин. Реакцию гасили оксидом дейтерия (2 мл) и температуру реакционной смеси подводили до комнатной. Смесь распределяли между рассолом (10 мл) и AcOEt (10 мл), и водный слой экстрагировали AcOEt (10 мл × 2). Объединенные экстракты сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали на колонке с силикагелем с прямой фазой (20/80 AcOEt/DCM), получая промежуточное соединение (3с) в виде желтого масла (0,79 г; выход 90%).
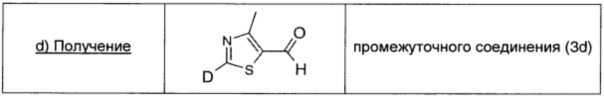
Водный 5,0 М HCl (0,19 мл; 0,96 ммоль; 2,5 экв.) добавляли к раствору промежуточного соединения (3с) (0,07 г; 0,38 ммоль; 1,0 экв.) в THF (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 1,5 ч. Смесь распределяли между рассолом (10 мл) и AcOEt (10 мл) и водный слой экстрагировали AcOEt (10 мл × 2). Объединенные органические экстракты промывали насыщенным раствором бикарбоната натрия (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали, получая промежуточное соединение (3d) в виде желтого твердого вещества (0,37 г; 74,7%).
Пример А.4
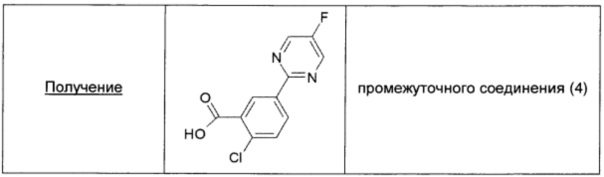
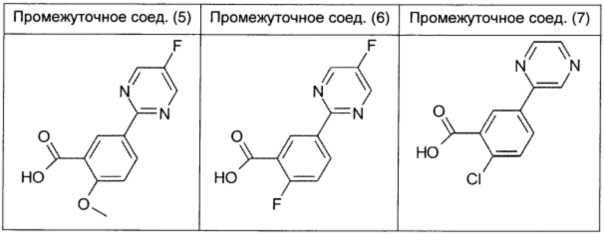
Во флаконе для микроволнового реактора (объем 20 мл) суспендировали 3-карбокси-4-хлорфенилбороновую кислоту (0,210 г; 1 ммоль; 1,0 экв.), 2-хлор-5-фтор-1,3-пиримидин (0,175 г; 1,25 ммоль; 1,25 экв.), тетракис(трифенилфосфин)палладий(0) (0,023 г; 0,02 ммоль; 0,02 экв.) и карбонат цезия (0,5 г; 1,5 ммоль; 1,5 экв.) в дегазированном растворе 5/1 DMF/H2O (2,5 мл). Флакон герметично закрывали, продували азотом и содержимое механически перемешивали в течение 5 мин. Затем смесь нагревали в течение 4 ч при 80°С в микроволновом реакторе. Полученную желтую суспензию упаривали в вакууме, добавляли воду (20 мл), затем смесь 1/1 DCM/AcOEt (20 мл) и 37% раствор HCl (10 мл). Полученный раствор выливали в делительную воронку и дважды экстрагировали смесью 1/1 DCM/EtOAc. Объединенные органические экстракты сушили над безводным Na2SO4, фильтровали и упаривали, получая промежуточное соединение (4) в виде белого порошка (0,214 г; выход 85%).
Используя аналогичную методику, получали промежуточное соединение (5), исходя из 3-карбокси-4-метоксифенилбороновой кислоты (0,248 г; выход 99%), и промежуточное соединение (6) получали, исходя из 3-карбокси-4-фторфенилбороновой кислоты (0,235 г; выход 99%).
Используя аналогичную методику, но заменяя 2-хлор-5-фтор-1,3-пиримидин на 2-хлорпиразин, получали промежуточное соединение (7), исходя из 3-карбокси-4-хлорфенилбороновой кислоты (0,1 г; выход 85%).
Пример А.5
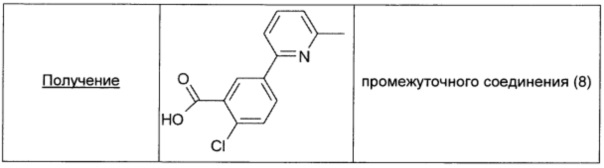
Во флаконе для микроволнового реактора (объем 20 мл) суспендировали 3-карбокси-4-хлорфенилбороновую кислоту (0,218 г; 0,92 ммоль; 2,0 экв.), 2-хлор-6-метилпиридин (0,05 мл; 0,46 ммоль; 1 экв.), ацетат палладия (0,017 г; 0,08 ммоль; 0,17 экв.), XPhos (0,09 г; 0,157 ммоль; 0,34 экв.) и карбонат натрия (0,147 г; 3 ммоль; 3 экв.) в дегазированном растворе 10/1 диоксан/H2O (2,5 мл). Флакон герметично закрывали, продували азотом и содержимое механически перемешивали в течение 5 мин. Затем смесь нагревали в течение 2 ч при 80°С в микроволновом реакторе. Полученную темную суспензию упаривали в вакууме, добавляли воду (20 мл), затем смесь 1/1 DCM/AcOEt (20 мл) и 37% раствор HCl (10 мл). Полученный раствор выливали в делительную воронку, водный слой отделяли и упаривали. Полученный белый порошок обрабатывали МеОН (3 мл), фильтровали и окончательно упаривали, получая промежуточное соединение (8) в виде белого порошка (0,11 г; выход 95%).
Пример А.6
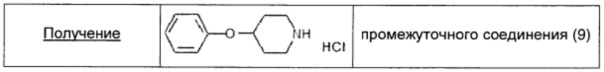
Трифенилфосфин (1,69 г; 6,46 ммоль; 1,3 экв.) добавляли к смеси 4-гидрокси-N-Boc-пиперидина (1,00 г; 4,97 ммоль; 1 экв.) и фенола (0,51 г; 5,47 ммоль; 1,1 экв.) в безводном THF (8,5 мл), затем медленно в течение 10 минут добавляли DIAD (1,27 мл; 6,46 ммоль; 1,3 экв.). Смесь перемешивали при комнатной температуре в течение ночи, затем растворитель выпаривали и остаток очищали флэш-хроматографией на силикагеле (элюент: петролейный эфир/EtOAc от 95/5 до 90/10). 4-Фенокси-N-Boc-пиперидин получали в виде бледно-розового масла (0,72 г; 2,61 ммоль; выход 52%).
4-Фенокси-N-Boc-пиперидин (0,72 г; 2,61 ммоль; 1 экв.) растворяли в 4 М растворе HCl в диоксане (5 мл) и раствор перемешивали при комнатной температуре в течение 2 часов. Раствор упаривали и остаток сушили под высоким вакуумом. Затем остаток растирали с MeCN (5 мл), фильтровали и промывали MeCN (1-2 мл), получая промежуточное соединение (9) в виде белого твердого вещества (0,45 г; 2,09 ммоль; выход 81%).
Пример А.7
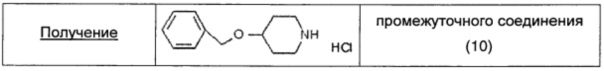
4-Гидрокси-N-Boc-пиперидин (1,18 г; 4,97 ммоль; 1 экв.) растворяли в безводном THF (10 мл) и раствор охлаждали до 0°С, затем порциями добавляли NaH (0,248 г; 60% дисперсия в минеральном масле; 5,96 ммоль; 1,2 экв.). Суспензию энергично перемешивали при комнатной температуре в течение 30 мин. Затем по каплям добавляли бензилбромид (1,1 г; 6,46 ммоль; 1,3 экв.) и смесь нагревали до температуры дефлегмации. Через 1 час добавляли последовательно NaH (0,103 г; 60% дисперсия в минеральном масле; 2,48 ммоль; 0,5 экв.) и бензилбромид (0,423 г; 2,48 ммоль; 0,5 экв.), и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли еще одну порцию NaH (0,207 г; 60% дисперсия в минеральном масле; 5 ммоль; 1 экв.), и реакционную смесь кипятили с обратным холодильником в течение 1 часа и перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в водный насыщенный раствор NH4Cl (50 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические экстракты сушили над безв. Na2SO4, фильтровали и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией на силикагеле (элюент: 90/10 петролейный эфир/AcOEt), получая чистый 4-бензилокси-N-boc-пиперидин (1,4 г; 4,83 ммоль; выход 97%) в виде бесцветного масла. 4-Бензилокси-N-boc-пиперидин (1,4 г; 4,83 ммоль; 1 экв.) растворяли в диоксане (10 мл). По каплям добавляли 4 М раствор HCl в диоксане (5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Затем добавляли 4 М раствор HCl в диоксане (3 мл) и реакционную смесь далее перемешивали при комнатной температуре в течение ночи. Наконец, растворитель удаляли в вакууме, получая чистое промежуточное соединение (10) в виде беловатого твердого вещества (1 г; 4,38 ммоль; выход 99%).
Пример А.8
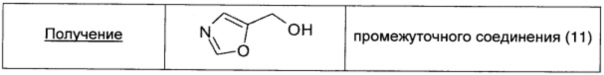
Этил-оксазол-5-карбоксилат (1 г; 7,09 ммоль; 1 экв.) растворяли в EtOH (14 мл) и смесь охлаждали до 0°С. Порциями с перемешиванием добавляли боргидрид натрия (0,54 г; 14,17 ммоль; 2 экв.) и затем смесь оставляли нагреваться до кт. После перемешивания в течение ночи при кт происходило полное превращение в продукт по данным TLC (95/5 DCM/MeOH). Смесь охлаждали до 0°С и по каплям добавляли 2 н раствор HCl до тех пор, пока не прекращалось выделение газа (pH 5-6). Полученную суспензию концентрировали при пониженном давлении и остаток очищали флэш-хроматографией (SiO2), используя в качестве элюента смесь 95/5 DCM/MeOH. Получали чистое промежуточное соединение (11) в виде бесцветного масла (0,49 г; выход 70%).
Пример А.9
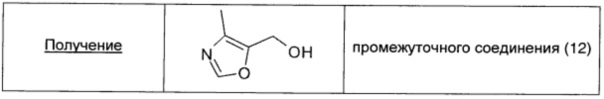
Подходящее производное, этил-оксазол-5-карбоксилат (1,1 г; 7,09 ммоль; 1 экв.), растворяли в EtOH (14 мл) и смесь охлаждали до 0°С. Порциями с перемешиванием добавляли NaBH4 (14,17 ммоль; 2 экв.) и затем смесь оставляли нагреваться до кт. Через 2 ч выдерживания при температуре дефлегмации происходило полное превращение в продукт по данным TLC (95/5 DCM/MeOH). Смесь охлаждали до 0°С и по каплям добавляли 2 н раствор HCl до тех пор, пока не прекращалось выделение газа (pH 5-6). Полученную суспензию концентрировали при пониженном давлении, и остаток очищали флэш-хроматографией (SiO2), используя в качестве элюента смесь DCM/MeOH = 95/5. Получали чистое промежуточное соединение (12) в виде бесцветного масла (0,46 г; выход 58%).
Пример А.10
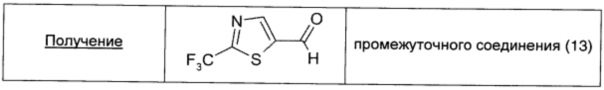
Этил-2-(трифторметил)тиазол-5-карбоксилат (0,50 г; 2,22 ммоль; 1 экв.) растворяли в безводном DCM (11 мл) в атмосфере аргона и охлаждали до -70°С. Затем по каплям в течение 10 минут добавляли 1 М DIBAL в DCM (2,5 мл; 2,49 ммоль; 1,12 экв.) и смесь перемешивали при этой же температуре в течение 1,5 ч. Температуру реакционной смеси подводили до 0°С, добавляли последовательно воду (0,10 мл), 15% раствор NaOH (0,10 мл) и вторую порцию воды (0,25 мл) и смесь перемешивали до полного выпадения в осадок соли алюминия (5 минут). Смесь сушили над безводным Na2SO4 и фильтровали. После выпаривания растворителей неочищенный продукт очищали прямофазовой флэш-хроматографией (30/70 DCM/петролейный эфир → 100% DCM), получая чистое промежуточное соединение (13) в виде желтого масла (0,3 г; выход 75%).
Пример А.11
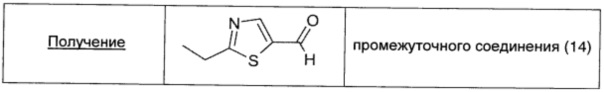
2-Броммалональдегид (0,71 г; 4,70 ммоль; 1 экв.) и пропантиоамид (0,42 г; 4,71 ммоль; 1 экв.) суспендировали в DCM (10 мл) и охлаждали до 0°С; затем добавляли DIPEA (0,82 мл; 4,71 ммоль; 1 экв.) двумя порциями. Полученный коричневый раствор оставляли перемешиваться при комнатной температуре в течение 2 суток. Растворитель удаляли выпариванием, коричневый остаток растворяли в Et2O (20 мл), дважды промывали насыщенным раствором NaHCO3 (20 мл) и рассолом (20 мл), сушили над безв. Na2SO4, фильтровали и окончательно упаривали. Неочищенный продукт очищали прямофазовой флэш-хроматографией (20/80 EtOAc/петролейный эфир), получая чистое промежуточное соединение (14) в виде коричневого масла (0,13 г; выход 20%).
Пример А.12
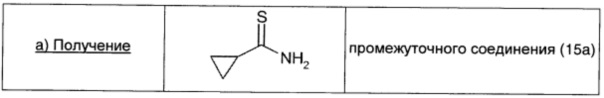
Циклопропанкарбоксамид (0,5 г; 5,87 ммоль; 1 экв.), карбонат натрия (0,62 г; 5,87 ммоль; 1 экв.) и реагент Лавессона (2,37 г; 5,87 ммоль; 1 экв.) в THF (25 мл) кипятили с обратным холодильником в течение 2,5 ч. Растворитель удаляли в вакууме и неочищенный продукт распределяли между водой (20 мл) и диэтиловым эфиром (20 мл). Органический слой сушили над безв. Na2SO4, фильтровали и окончательно упаривали, получая промежуточное соединение (15а) в виде белого твердого вещества (0,44 г; выход 74%).
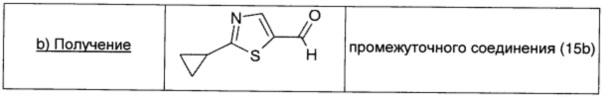
2-Броммалональдегид (0,66 г; 4,35 ммоль; 1 экв.), растворенный в безводном THF (2 мл), добавляли к раствору промежуточного соединения 15а (0,44 г; 4,35 ммоль; 1 экв.) в безводном DCM (10 мл). Смесь охлаждали до -15°С; затем порциями добавляли DIPEA (0,76 мл; 4,35 ммоль; 1 экв.). Полученный желтый раствор оставляли перемешиваться при комнатной температуре в течение 4 суток. Растворитель удаляли выпариванием, коричневый остаток растворяли в Et2O (20 мл), дважды промывали насыщенным раствором NaHCO3 (20 мл) и рассолом (20 мл), сушили над безв. Na2SO4, фильтровали и окончательно упаривали. Неочищенный продукт очищали прямофазовой флэш-хроматографией (10/90 EtOAc/петролейный эфир), получая чистое промежуточное соединение (15b) в виде коричневого масла (0,25 г; выход 38%).
Пример А.13
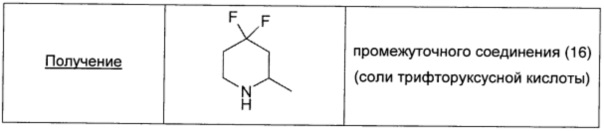
Раствор 1-Boc-2-метилпиперидин-4-она (0,55 г; 2,6 ммоль; 1 экв.) в безводном DCM (7,5 мл) охлаждали при 0°С и по каплям добавляли DAST (0,68 мл; 5,2 ммоль; 2 экв.). Реакционную смесь перемешивали в течение ночи при 10°С, затем разбавляли DCM (10 мл), промывали насыщенным раствором NaHCO3 (10 мл), 5% раствором лимонной кислоты в воде (10 мл) и окончательно рассолом (10 мл). Органический слой сушили над безв. Na2SO4, фильтровали и упаривали. Остаток очищали флэш-хроматографией на силикагеле (элюент: 10/90 EtOAc/петролейный эфир), получая 0,53 г чистого 1-N-Boc-4,4-дифторметилпиперидина в виде белого твердого вещества.
TFA (2 мл; 26 ммоль; 10 экв.) добавляли с перемешиванием к раствору 1-N-Boc-4,4-дифторметилпиперидина (0,53 г; 2,25 ммоль) в DCM (8 мл), охлаждали с использованием ледяной бани. Реакционную смесь оставляли нагреваться при комнатной температуре и перемешивали в течение еще 30 минут. Растворитель удаляли при пониженном давлении, получая 0,73 г (выход 70% за две стадии) промежуточного соединения 16 в виде соли TFA.
Пример А.14
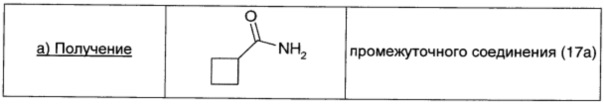
Раствор циклобутанкарбоновой кислоты (1,91 мл; 16,6 ммоль; 1 экв.) в безводном THF обрабатывали тионилхлоридом (4 мл; 50 ммоль; 3 экв.) и кипятили с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли DCM (5 мл) и упаривали при пониженном давлении. Остаток растворяли в ацетонитриле (31 мл), по каплям добавляли к перемешиваемому раствору гидроксида аммония (59 мл) при 0°С и перемешивали при этой температуре в течение 1 ч. Затем реакционную смесь выливали в делительную воронку и экстрагировали EtOAc (15 мл × 2). Объединенные органические экстракты промывали 0,1 М водным раствором HCl (20 мл), водой (20 мл) и рассолом (20 мл), сушили над безв. Na2SO4, фильтровали и окончательно упаривали, получая промежуточное соединение 17а (0,31 г; выход 16%) в виде белого твердого вещества.
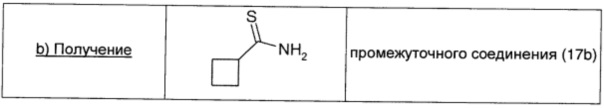
Промежуточное соединение 17а (0,31 г; 3,17 ммоль; 1 экв.), карбонат натрия (0,34 г; 3,17 ммоль; 1 экв.) и реагент Лавессона (1,38 г; 3,17 ммоль; 1 экв.) в THF (16 мл) кипятили с обратным холодильником в течение 3 ч. Растворитель удаляли в вакууме и неочищенный продукт распределяли между водой (20 мл) и диэтиловым эфиром (20 мл). Органический слой сушили над безв. Na2SO4, фильтровали и окончательно упаривали, получая промежуточное соединение (17b) в виде желтой жидкости (0,36 г; выход 99%).
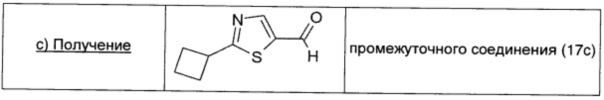
2-Броммалональдегид (0,51 г; 3,15 ммоль; 1 экв.), растворенный в безводном THF (5 мл), добавляли к раствору промежуточного соединения 19 (0,36 г; 3,15 ммоль; 1 экв.) в безводном DCM (8 мл). Смесь охлаждали до -15°С, затем порциями в атмосфере Ar добавляли DIPEA (0,55 мл; 3,15 ммоль; 1 экв.). Полученный коричневый раствор оставляли перемешиваться при комнатной температуре в течение 2 суток. Растворитель удаляли выпариванием, коричневый остаток растворяли в Et2O (20 мл), дважды промывали насыщенным раствором NaHCO3 (20 мл) и рассолом (20 мл), сушили над безв. Na2SO4, фильтровали и окончательно упаривали. Неочищенный продукт очищали прямофазовой флэш-хроматографией (20/80 EtOAc/петролейный эфир), получая чистое промежуточное соединение (17с) в виде желтой жидкости (0,13 г; выход 26%).
Пример А.15
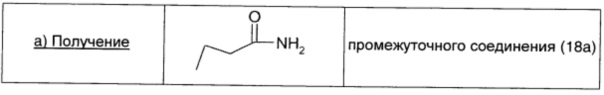
Раствор гидроксида аммония в воде (28%-ный; 31 мл) добавляли к перемешиваемому раствору бутирилхлорида (0,97 мл; 9,3 ммоль; 1 экв.) в ацетонитриле (15,5 мл) при 0°С. Через 15 мин реакционную смесь выливали в делительную воронку и экстрагировали EtOAc (30 мл × 3). Объединенный органический экстракт промывали 0,1 М водным раствором HCl (20 мл), водой (20 мл) и рассолом (20 мл), сушили над безв. Na2SO4, фильтровали и окончательно упаривали, получая промежуточное соединение 18а (0,32 г; выход 40%) в виде белого твердого вещества.
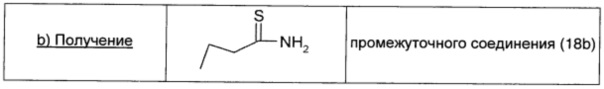
Промежуточное соединение 18а (1,24 г; 14 ммоль; 1 экв.), карбонат натрия (1,48 г; 14 ммоль; 1 экв.) и реагент Лавессона (5,66 г; 14 ммоль; 1 экв.) в THF (17 мл) кипятили с обратным холодильником в течение 3 ч. Растворитель удаляли в вакууме и неочищенный продукт распределяли между водой (20 мл) и диэтиловым эфиром (20 мл). Органический слой сушили над безв. Na2SO4, фильтровали и окончательно упаривали, получая промежуточное соединение 18b в виде желтой жидкости (1,23 г; выход 89%).
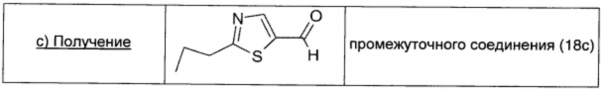
2-Броммалональдегид (1,88 г; 12 ммоль; 1 экв.), растворенный в безводном THF (15 мл), добавляли к раствору промежуточного соединения 18b (1,23 г; 12 ммоль; 1 экв.) в безводном DCM (30 мл). Смесь охлаждали до -15°С; затем порциями в атмосфере Ar добавляли DIPEA (2,16 мл; 12 ммоль; 1 экв.). Полученный коричневый раствор оставляли перемешиваться при комнатной температуре в течение 3 суток. Растворитель удаляли выпариванием, коричневый остаток растворяли в Et2O (20 мл), дважды промывали насыщенным раствором NaHCO3 (20 мл) и рассолом (20 мл), сушили над безводным Na2SO4, фильтровали и окончательно упаривали. Неочищенный продукт очищали прямофазовой флэш-хроматографией (20/80 EtOAc/петролейный эфир), получая чистое промежуточное соединение 18с в виде желтой жидкости (0,46 г; выход 25%).
Пример А.16
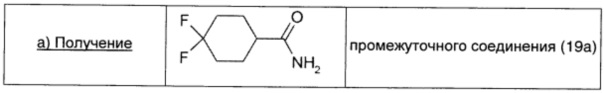
4,4-Дифторциклопропанкарбоновую кислоту (1 г; 6,09 ммоль; 1 экв.) растворяли в безводном THF (37 мл), охлаждали до -70°С и обрабатывали 4-метилморфолином (0,67 мл; 6,09 моль; 1 экв.) в атмосфере Ar. Затем по каплям при -70°С добавляли бутилхлорформиат (0,79 мл; 6,09 ммоль; 1 экв.). Через 15 мин добавляли 28% раствор гидроксида аммония в воде (7,4 мл) и реакционную смесь нагревали до комнатной температуры. Растворитель удаляли при пониженном давлении, остаток растворяли в EtOAc, промывали водой (20 мл), сушили над безв. Na2SO4, фильтровали и окончательно упаривали, получая промежуточное соединение 19а (0,86 г; выход 86%) в виде белого твердого вещества.
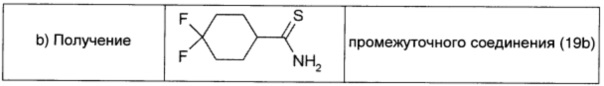
Промежуточное соединение 19а (0,86 г; 5,29 ммоль; 1 экв.), карбонат натрия (0,56 г; 5,29 ммоль; 1 экв.) и реагент Лавессона (2,14 г; 5,29 ммоль; 1 экв.) в THF (26 мл) кипятили с обратным холодильником в течение 3 ч. Растворитель удаляли в вакууме и неочищенный продукт распределяли между водой (20 мл) и диэтиловым эфиром (20 мл). Органический слой сушили над безв. Na2SO4, фильтровали и окончательно упаривали, получая промежуточное соединение 19b в виде беловатого твердого вещества (1,09 г; выход 99%).
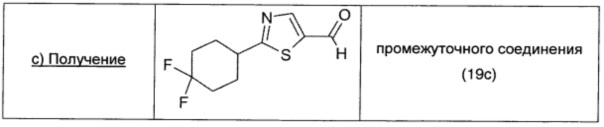
2-Броммалональдегид (0,97 г; 6,11 ммоль; 1 экв.), растворенный в безводном THF (10 мл), добавляли к раствору промежуточного соединения 19b (1,09 г; 6,11 ммоль; 1 экв.) в безводном DCM (15 мл). Смесь охлаждали до -15°С, затем порциями в атмосфере Ar добавляли DIPEA (1,06 мл; 6,11 ммоль; 1 экв.). Полученный коричневый раствор оставляли перемешиваться при комнатной температуре в течение 1 суток. Растворитель удаляли выпариванием, коричневый остаток растворяли в Et2O (20 мл), дважды промывали насыщенным раствором NaHCO3 (20 мл) и рассолом (20 мл), сушили над безводным Na2SO4, фильтровали и окончательно упаривали. Неочищенный продукт очищали прямофазовой флэш-хроматографией (30/70 EtOAc/петролейный эфир), получая чистое промежуточное соединение 19с в виде бесцветной жидкости (0,34 г; выход 24%).
Пример А.17
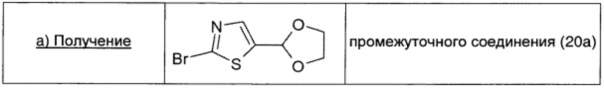
Смесь 2-бром-5-формил-1,3-тиазола (0,384 г; 2 ммоль; 1 экв.), п-толуолсульфоновой кислоты (0,031 г; 0,16 ммоль; 0,08 экв.) и этиленгликоля (0,334 мл; 6 ммоль; 3 экв.) в безводном толуоле (12 мл) кипятили с обратным холодильником с использованием аппарата Дина-Старка в течение 3 ч. Затем растворитель удаляли и остаток очищали флэш-хроматографией на силикагеле (100% петролейный эфир → 20/80 EtOAc/петролейный эфир), получая промежуточное соединение 20а (0,33 г; выход 70%) в виде бесцветного масла.
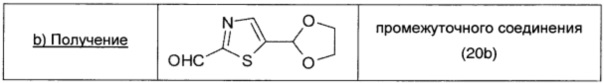
Промежуточное соединение 20а (0,33 г; 1,4 ммоль; 1 экв.) растворяли в безводном THF (2 мл) и охлаждали до -70°С. Затем по каплям в атмосфере Ar добавляли раствор 1,6 М н-BuLi в гексане (0,96 мл; 1,54 ммоль; 1,1 экв.). Через 50 минут по каплям при -70°С добавляли DMF (0,08 мл; 3 ммоль; 1,6 экв.) и реакционную смесь перемешивали при этой температуре в течение 50 мин. Затем добавляли NH4Cl (насыщенный водный раствор, 10 мл) и реакционную смесь нагревали до комнатной температуры. Далее реакционную смесь экстрагировали DCM (20 мл × 2). Объединенные органические экстракты сушили над безв. Na2SO4, фильтровали и упаривали, получая промежуточное соединение 20b (0,225 г; выход 87%) в виде оранжевого масла.
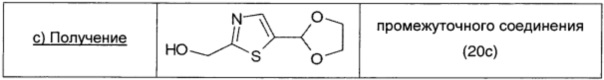
Боргидрид натрия (0,046 г; 1,215 ммоль; 1 экв.) при 0°С и в атмосфере азота добавляли порциями к перемешиваемому раствору промежуточного соединения 20b (0,225 г; 1,25 ммоль; 1 экв.) в метаноле (2 мл). Реакционную смесь перемешивали в течение 30 мин при 0°С, затем растворитель выпаривали при пониженном давлении. Остаток распределяли между смесью 2/1 EtOAc/DCM (10 мл) и водой (10 мл), органический слой сушили над безв. Na2SO4, фильтровали и упаривали, получая промежуточное соединение 20с (0,19 г; выход 84%) в виде оранжевого масла.
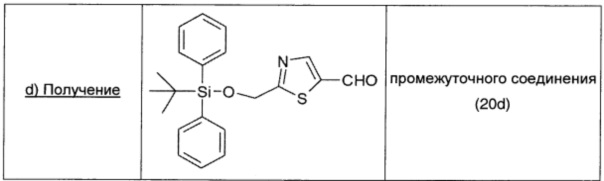
трет-Бутилдифенилсилилхлорид (0,30 г; 1,01 ммоль; 1,1 экв.) добавляли при 0°С в атмосфере азота к перемешиваемому на магнитной мешалке раствору промежуточного соединения 20 с (0,19 г; 1 ммоль; 1 экв.) и имидазола (0,072 г; 1,05 ммоль; 1,05 экв.) в безводном DCM (1,5 мл). Через 2 ч реакционную смесь нагревали до комнатной температуры и выливали в насыщенным раствор NaHCO3 (5 мл). Органический слой сушили над безв. Na2SO4, фильтровали и упаривали. Остаток (0,47 г) растворяли в THF (10 мл) и обрабатывали 5 н раствором HCl (3 мл) при комнатной температуре. Через 2 ч реакционную смесь подщелачивали насыщенным раствором NaHCO3 (3 мл) и экстрагировали DCM (10 мл × 2). Объединенные органические экстракты сушили над безводным Na2SO4, фильтровали и упаривали, получая промежуточное соединение 20d (0,194 г; выход 51%) в виде бесцветного масла.
Пример А.18
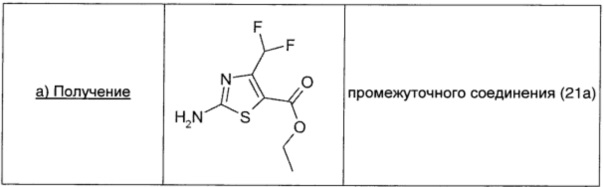
Сульфурилхлорид (1,23 мл; 15,2 ммоль; 1,01 экв.) по каплям в атмосфере азота добавляли при 0°С к этил-4,4-дифторацетоацетату (2,5 г; 15,0 ммоль; 1 экв.) и перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли EtOAc (20 мл) и выливали в смесь лед/вода (20 мл). Органический слой сушили над безв. Na2SO4, фильтровали и упаривали, получая 3,2 г неочищенного продукта в 2-хлор-4,4-дифторацетоацетате в виде желтого масла. Неочищенный продукт растворяли в этаноле (10 мл), обрабатывали тиомочевиной (3,2 г; 30 ммоль; 2 экв.) и нагревали в микроволновом реакторе в течение 1 ч при 101°С. Затем растворитель удаляли в вакууме и остаток распределяли между насыщенным раствором NaHCO3 (10 мл) и EtOAc (10 мл). Органический слой промывали рассолом (20 мл), сушили над безв. Na2SO4, фильтровали и упаривали. Неочищенный продукт обрабатывали диэтиловым эфиром, фильтровали и сушили в вакууме, получая 1,37 г (выход 41%) промежуточного соединения 21а в виде желтого твердого вещества.
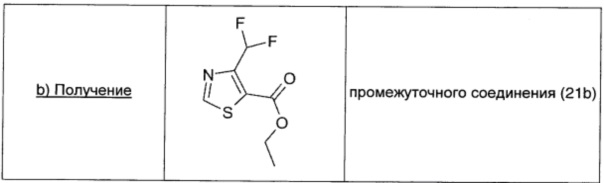
Промежуточное соединение 21а (1,37 г; 6,16 ммоль; 1 экв.) растворяли в диоксане (35 мл), добавляли изоамилнитрит (2,24 мл; 16,64 ммоль; 2,7 экв.) и реакционную смесь нагревали в течение 1 часа при 80°С. Растворитель удаляли выпариванием при пониженном давлении, и остаток очищали флэш-хроматографией на силикагеле (EtOAc/петролейный эфир, 10/90), получая промежуточное соединение 21b (1,02 г; выход 80%) в виде желтого твердого вещества.
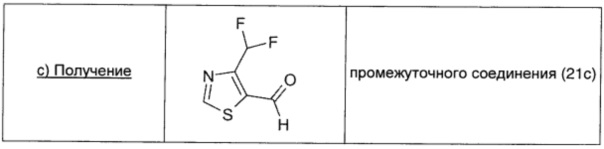
Промежуточное соединение 21b (0,758 г; 3,66 ммоль; 1 экв.) растворяли в безводном DCM (18,5 мл) в атмосфере аргона и охлаждали до -75°С. По каплям добавляли 1 М гидрид диизобутилалюминия в DCM (4,1 мл; 4,1 ммоль; 1,12 экв.) и реакционную смесь перемешивали при -70°С. Через 1,5 ч по каплям добавляли 1 М гидрид диизобутилалюминия в DCM (2,5 мл; 2,5 ммоль; 0,68 экв.) и реакционную смесь перемешивали еще в течение 1 ч при -70°. Реакционную смесь нагревали до 0°С и обрабатывали водой (0,264 мл), 15% раствором NaOH (0,264 мл) и водой (0,66 мл) в указанном порядке. Затем ее перемешивали в течение 5 минут при 0°С, далее в течение 30 минут при комнатной температуре. Добавляли последовательно воду (0,24 мл), затем 15% раствор NaOH (0,130 мл) и реакционную смесь перемешивали при комнатной температуре до образования осадка. Смесь фильтровали и затем растворитель упаривали. Остаток очищали флэш-хроматографией на силикагеле (DCM/петролейный эфир, 80/20→100% DCM), получая желтое масло (0,34 мг; выход 40%), содержащее промежуточное соединение 21с (чистота приблизительно 70%), которое использовали в таком виде.
Пример А.19
Общая методика получения тиазол-содержащих промежуточных соединений
СТАДИЯ а). Получение α-аминонитрилов
Способ а1)
Альдегид (2,21 ммоль; 1 экв.) растворяли в ледяной АсОН (6,8 мл). Последовательно добавляли AcONa (3,315 ммоль; 1,5 экв.) и амин (2,652 ммоль; 1,2 экв.), перемешивая при комнатной температуре в атмосфере N2. Желтый раствор перемешивали в течение 1 ч и затем охлаждали до 0°С. По каплям добавляли TMSCN (4,42 ммоль; 2 экв.) и смесь оставляли нагреваться до комнатной температуры. В последующие часы, при необходимости, добавляли в виде двух порций 1 эквивалент TMSCN (1,1 ммоль × 2). Когда по данным UPLC-MS происходило полное превращение в продукт, добавляли воду (5 мл) и раствор упаривали. К остатку добавляли насыщенный раствор NaHCO3 (20 мл) и смесь экстрагировали DCM (15 мл × 3). Объединенные органические фазы сушили (безв. Na2SO4) и упаривали. Неочищенный продукт очищали флэш-хроматографией (SiO2) с использованием смеси петролейный эфир/AcOEt, получая чистый α-аминонитрил (выход в среднем 65%).
Используя способ а1, получали А0013_15_01 (выход 59%), А0013_24_01 (выход 60%), А0011_48_01 (выход 71%), исходя из 2-тиазолкарбоксальдегида и 1,2,3,4-тетрагидроизохинолина, 4,4-дифторпиперидина гидрохлорида и морфолина, соответственно; промежуточные соединения А0013_23_01 (выход 60%), А0015_24_01 (выход 61%) получали, исходя из 4-тиазол-карбальдегида и 4,4-дифторпиперидина, гомоморфолина гидрохлорида, соответственно; промежуточные соединения А0015_04_01 (выход 65%), А0013_41_01 (выход 83%), А0013_41_02 (выход 50%), А0013_83_01 (выход 64%), А0015_85_01 (выход 79%), А0016_13_01 (выход 74,5%) получали, исходя из 5-тиазол-карбальдегида и морфолина, 4,4-дифторпиперидина гидрохлорида, 1,2,3,4-тетрагидроизохинолина, промежуточного соединения (10), промежуточного соединения (9) и пиперидина, соответственно; промежуточные соединения А0015_48_01 (выход 50%), А0015_47_01 (выход 85%), А0015_46_01 (выход 22%), А0018_42_01 (выход 83%), А0018_41_01 (выход 90%), А0017_69_01 (выход 72%) и А0017_70_01 (выход 81%) получали, исходя из тиазол-4-метил-5-илкарбальдегида и 1,2,3,4-тетрагидроизохинолина, 4,4-дифторпиперидина гидрохлорида, морфолина, промежуточного соединения (10), промежуточного соединения (9), 3,3-дифторпиперидина гидрохлорида и 3,3-дифторазетидина гидрохлорида, соответственно; промежуточные соединения А0012_57_01 (выход 79%), А0012_58_01 (выход 69%) и А0018_14_01 (выход 73%) получали, исходя из промежуточного соединения (1) и 4,4-дифторпиперидина гидрохлорида, 1,2,3,4-тетрагидроизохинолина и промежуточного соединения (10); промежуточное соединение А0018_57_01 получали, исходя из промежуточного соединения (2b) и 4,4-дифторпиперидина гидрохлорида; промежуточное соединение А0018_72_01 получали, исходя из промежуточного соединения (3d) и 4,4-дифторпиперидина гидрохлорида; промежуточное соединение А0018_91_01 (выход 71%) получали, исходя из промежуточного соединения (13) и 4,4-дифторпиперидина гидрохлорида; промежуточное соединение А0020_17_01 (выход 84%) получали, исходя из промежуточного соединения (14) и 4,4-дифторпиперидина гидрохлорида; промежуточное соединение А0020_25_01 (выход 67%) получали, исходя из промежуточного соединения (15b) и 4,4-дифторпиперидина гидрохлорида; промежуточное соединение А0020_10_02 (выход 45%) получали, исходя из промежуточного соединения (1) и гомоморфолина гидрохлорида; промежуточное соединение А0021_05_01 (выход 81%) получали, исходя из 4-метил-5-тиазолкарбоксальдегида и 2-[2-(трифторметил)фенил]морфолина; промежуточное соединение А0021_06_02 (выход 87%) получали, исходя из 4-метил-5-тиазолкарбоксальдегида и 2-(2,4-дифторфенил)морфолина; промежуточное соединение А0021_06_04 (выход 95%) получали, исходя из 4-метил-5-тиазолкарбоксальдегида и 2-(1-метил-1H-пиразол-4-ил)морфолина; промежуточное соединение А0021_06_03 (выход 77%) получали, исходя из метил-5-тиазолкарбоксальдегида и 5-окса-8-азаспиро[3.5]нонана; промежуточное соединение А0020_33_01 (выход 61%) получали, исходя из метил-5-тиазолкарбоксальдегида и промежуточного соединения 16; промежуточное соединение А0016_39_01 (выход 72%) получали, исходя из промежуточного соединения 17с и 4,4-дифторпиперидина гидрохлорида; промежуточное соединение А0016_40_01 (выход 64%) получали, исходя из промежуточного соединения 18с и 4,4-дифторпиперидина гидрохлорида; промежуточное соединение А0017_98_01 (выход 19%) получали, исходя из промежуточного соединения 1 и промежуточного соединения 16; А0016_46_01 (выход 51%) получали, исходя из промежуточного соединения 19с и морфолина; А0016_45_01 (выход 45%) получали, исходя из промежуточного соединения 19с и 4,4-дифторпиперидина гидрохлорида; промежуточное соединение А0020_60_01 (выход 42%) получали, исходя из промежуточного соединения 20d и 4,4-дифторпиперидина гидрохлорида; А0018_98_01 (выход 39%) получали, исходя из промежуточного соединения 21с и 4,4-дифторпиперидина гидрохлорида; А0016_55_05 (выход 67%) получали, исходя из 2-трет-бутил-1,3-тиазол-5-карбальдегида и 4,4-дифторпиперидина гидрохлорида; А0021_41_01 (выход 79%) получали, исходя из 4-циклопропил-1,3-тиазол-5-карбальдегида и 4,4-дифторпиперидина гидрохлорида; А0016_96_01 (выход 58%) получали, исходя из промежуточного соединения 21с и морфолина.
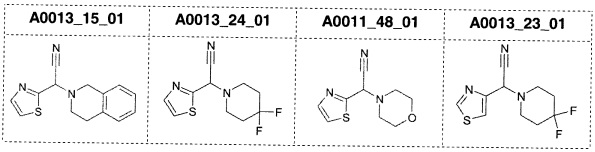
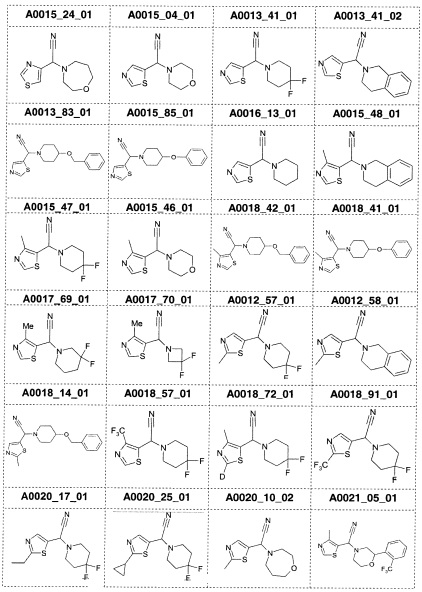
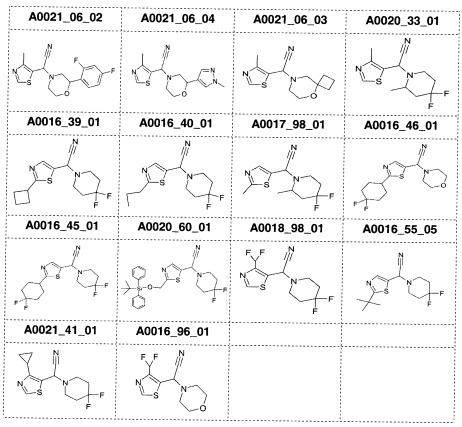
Способ а2)
Альдегид (1,33 ммоль; 1 экв.) растворяли в безводном MeCN (3 мл) и при комнатной температуре в атмосфере азота добавляли ледяную АсОН (приблизительно 10 капель). Через 10 минут по каплям добавляли амин (1,33 ммоль; 1 экв.) и полученный оранжевый раствор перемешивали при комнатной температуре в течение 30 минут. Затем смесь охлаждали до 0°С с использованием ледяной бани и по каплям добавляли TMSCN (2 ммоль; 1,5 экв.). Перемешивание продолжали при комнатной температуре до полного превращения альдегида в продукт (обычно приблизительно 1,5 ч). Реакцию гасили насыщенным водным раствором NaHCO3 и растворитель удаляли в вакууме. Полученную водную смесь экстрагировали DCM (20 мл × 3) и объединенные органические фазы сушили над безв. Na2SO4, фильтровали и упаривали. Наконец, остаток очищали флэш-хроматографией на силикагеле (элюент: 50/50 петролейный эфир/EtOAc), получая чистые α-аминонитрилы.
Используя способ а2, промежуточное соединение А0015_02_01 (выход 67%) получали, исходя из 4-тиазол-карбальдегида и морфолина. Аналогичным образом, А0015_01_01 (выход 53%) получали, исходя из 4-тиазол-карбальдегида и 1,2,3,4-тетрагидроизохинолина.
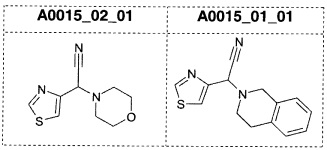
Стадия b). Получение диаминов
Способ b1)
α-Аминонитрил (0,478 ммоль; 1 экв.) растворяли в 3 М NH3 в МеОН (16 мл; 100 экв.) и раствор гидрировали в проточном реакторе H-Cube™ с применением картриджа с Ni Ренея (CatCart®, длиной 55 мм), используя скорость потока 0,7 мл/мин. Давление водорода варьировали от 30 до 60 бар (от 3 до 6 МПа), а температуру от 30°С до 40°С в зависимости от субстрата. После протекания реакции в течение соответствующего времени (обычно через 2 ч) раствор упаривали, получая первичный амин, который использовали в таком виде на следующей стадии без какой-либо очистки (выход в среднем 65%).
Используя способ b1, промежуточное соединение А0013_31_01 (выход 91%) получали, исходя из А0015_02_01; промежуточное соединение А0015_11_01 (выход 34%) получали, исходя из А0015_01_01; промежуточное соединение А0015_25_01 (выход 27%) получали, исходя из А0015_24_01; промежуточное соединение А0013_30_01 (выход 64%) получали, исходя из А0013_23_01; промежуточное соединение А0013_33_01 (выход 55%) получали, исходя из А0013_41_02; промежуточное соединение А0013_40_01 (выход 41%) получали, исходя из А0015_04_01; промежуточное соединение А0013_54_03 (выход 65%) получали, исходя из А0013_41_01; промежуточное соединение А0017_01_01 (выход 45%) получали, исходя из А0013_83_01; промежуточное соединение А0016_09_01 (выход 39%) получали, исходя из А0015_85_01; промежуточное соединение А0016_17_01 (выход 9%) получали, исходя из А0016_13_01; промежуточное соединение А0015_52_01 (выход 50%) получали, исходя из А0015_48_01; промежуточное соединение А0015_56_01 (выход 76%) получали, исходя из А0015_47_01; промежуточное соединение А0015_54_01 (выход 75%) получали, исходя из А0015_46_01; промежуточное соединение А0017_59_01 (выход 84%) получали, исходя из А0018_42_01; промежуточное соединение А0017_58_01 (выход 76%) получали, исходя из А0018_41_01; промежуточное соединение А0017_73_01 (выход 88%) получали, исходя из А0017_69_01; промежуточное соединение А0017_72_01 (выход 86%) получали, исходя из А0017_70_01; промежуточное соединение А0018_58_01 (выход 81%) получали, исходя из А0018_57_01; промежуточное соединение А0018_75_01 (выход 77%) получали, исходя из А0018_72_1; промежуточное соединение А0021_07_01 (выход 91%) получали, исходя из А0021_05_01; промежуточное соединение А0021_07_02 (выход 74%) получали, исходя из А0021_06_02; промежуточное соединение А0021_07_04 (выход 52%) получали, исходя из А0021_06_04; промежуточное соединение А0021_07_03 (выход 33%) получали, исходя из А0021_06_03; А0020_66_01 (выход 81%) получали, исходя из А0016_39_01; А0020_69_01 (выход 70%) получали, исходя из А0016_40_01; А0016_48_01 (выход: 81%) получали, исходя из А0016_46_01; А0016_47_01 (выход 66%) получали, исходя из А0016_45_01; А0020_63_01 (выход 86%) получали, исходя из А0020_60_01; А0016_59_01 (выход 88%) получали, исходя из А0016_55_05.
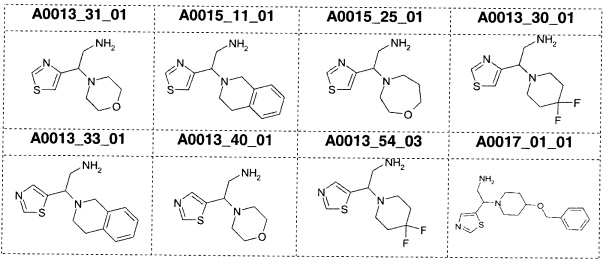
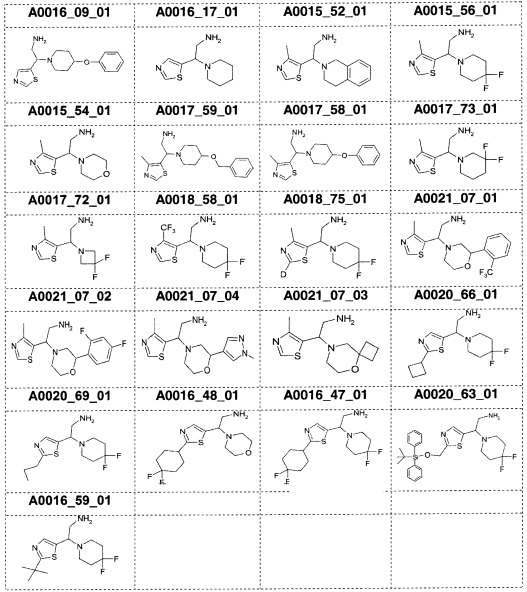
Способ b2)
Способ b2 состоит из двух стадий.
Стадия b2-1
α-Аминонитрил (1 экв.), Boc2O (2 экв.) и гексагидрат хлорида никеля(II) (0,05 экв.) переносили в безводный метанол (0,75 мл) и охлаждали до 0°С. Затем порциями в течение 45 минут при перемешивании добавляли боргидрид натрия (7 экв.). Реакционную смесь перемешивали в течение 3 ч, после чего добавляли этилендиамин (3 экв.) и реакционную смесь оставляли нагреваться до комнатной температуры. После перемешивания в течение дополнительных 0,5 ч растворитель удаляли в вакууме, и полученное твердое вещество переносили в насыщенный водный раствор бикарбоната натрия (10 мл) и EtOAc (10 мл). Органический слой промывали рассолом и сушили (Na2SO4). Полученный N-Boc-α-аминонитрил очищали флэш-хроматографией на силикагеле.
Используя методику, описанную для стадии b2-1, промежуточное соединение А0013_16_01 (выход 35%) получали, исходя из А0013_15_01; промежуточное соединение А0013_26_01 (выход 28%) получали, исходя из А0013_24_01; промежуточное соединение А0011_52_01 (выход 51%) получали, исходя из А0011_48_01.
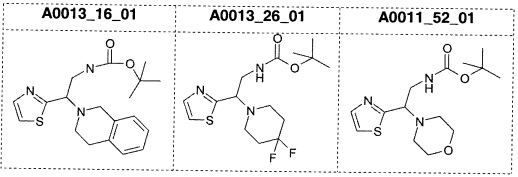
Стадия b2-2
N-Boc-α-аминонитрил растворяли в смеси 1/1 DCM/TFA (1-2 мл) и перемешивали при комнатной температуре вплоть до завершения реакции. Затем растворитель удаляли в вакууме, получая чистый α-аминонитрил в виде его соли TFA, которую использовали на следующих стадиях синтеза без какой-либо очистки.
Используя методику, описанную для стадии b2-2, промежуточное соединение А0013_28_01 получали, исходя из А0013_16_01; промежуточное соединение А0013_28_03 получали, исходя из А0013_26_01; промежуточное соединение А0011_54_01 получали, исходя из А0011_52_01.
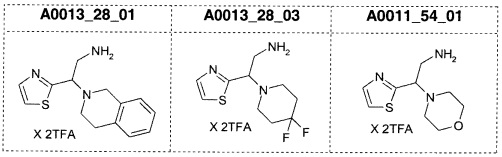
Способ b3)
α-Аминонитрил (1 экв.) растворяли в безводном THF в атмосфере азота, и раствор охлаждали до 0°С, используя ледяную баню. По каплям добавляли 1 М суспензию LiAlH4 в безводном THF (1 экв.) и реакционную смесь перемешивали в течение 30 мин при 0°С. Эту процедуру повторяли (обычно 3 раза) до полного расходования нитрила, наблюдаемого посредством TLC. Реакцию гасили путем медленного добавления МеОН при 0°С до полного прекращения выделения газа. Смесь упаривали и неочищенный продукт очищали флэш-хроматографией на силикагеле (элюент: DCM/MeOH/NH4OH), получая чистый α-аминонитрил в виде масла.
Используя способ b3, промежуточное соединение А0012_61_02 (выход 45%) получали, исходя из А0012_57_01; промежуточное соединение А0012_63_01 (выход 45%) получали, исходя из А0012_58_01; промежуточное соединение А0018_16_01 (выход 42%) получали, исходя из А0018_14_01; промежуточное соединение А0018_93_01 (выход 14%) получали, исходя из А0018_91_01; промежуточное соединение А0020_19_01 (выход 41%) получали, исходя из А0020_17_01; промежуточное соединение А0020_26_01 (выход 42%) получали, исходя из А0020_25_01; промежуточное соединение А0020_37_01 (выход 49%) получали, исходя из А0020_10_02; А0020_38_01 (выход 38%) получали, исходя из А0020_33_01; А0020_31_01 (выход 50%) получали, исходя из А0017_98_01; А0016_57_01 (выход 40%) получали, исходя из А0021_41_01.
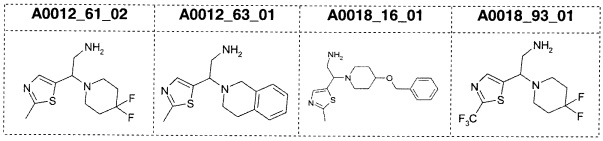
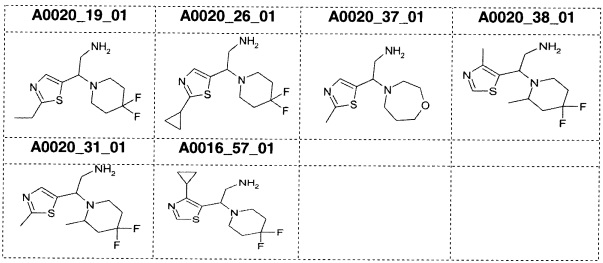
Способ b4)
А0016_96_01 (0,10 г; 0,405 ммоль) растворяли в безводном DCM (4 мл) в атмосфере азота и раствор охлаждали до 0°С. По каплям при этой же температуре добавляли 1 М DIBAL-H в DCM (1,6 мл; 1,62 ммоль; 4 экв.) и реакционную смесь перемешивали в течение 30 мин. Реакцию останавливали путем добавления 1 М раствора HCl в МеОН (3 мл), реакционную смесь фильтровали и растворители удаляли в вакууме. Остаток растворяли в насыщенном растворе NaHCO3 (10 мл) и экстрагировали DCM (10 мл × 3) и EtOAc (10 мл × 3). Объединенные органические экстракты сушили над безв. Na2SO4, фильтровали и упаривали, получая А0022_01_01 (0,069 г; выход 65%) в виде коричневого твердого вещества.
Пример А.20
Промежуточное соединение А0016_13_01 (0,235 г; 1,134 ммоль; 1 экв.) растворяли в МеОН (11 мл) и бесцветный раствор охлаждали до 0°С, используя ледяную баню. Добавляли CoCl2⋅6H2O (405 г; 1,7 ммоль; 1,5 экв.) и получали фиолетовый раствор. Затем порциями добавляли NaBH4 (0,214 г; 5,67 ммоль; 5 экв.) (ОСТОРОЖНО: интенсивное выделение пузырьков газа!). Сразу же получали темную смесь. Через 1 ч выдерживания при этой же температуре наблюдали почти полное превращение исходного вещества в продукт, используя TLC (95/5/0,5 CM/MeOH/NH4OH). Добавляли NaBH4 (0,107 г; 2,5 ммоль; 2,2 экв.) и через 1 ч реакцию гасили добавлением NH4OH.
Смесь фильтровали через набивку целита, промывая МеОН. Раствор упаривали и остаток суспендировали в DCM (15 мл). Смесь фильтровали через набивку целита, промывая DCM. Раствор упаривали и неочищенное вещество (темное масло, сложная смесь по данным TLC) очищали флэш-хроматографией (SNAP®, 10 г; SiO2, Biotage) с использованием смеси 95/5/0,5 DCM/MeOH/NH4OH, получая А0016_17_01 (0,022 г; выход 9%) в виде темного масла.
Пример А.21
Общая методика получения 5-оксазолил-содержащих производных
СТАДИЯ а). Получение α-аминонитрилов
Способ а1)
Соответствующее оксазол-спиртовое промежуточное соединение (11) или промежуточное соединение (12) (0,858 ммоль; 1 экв.) растворяли в DCM (1,5 мл) и раствор охлаждали до 0°С. Добавляли периодинан Десса-Мартина (0,943 ммоль; 1,1 экв.) и смесь оставляли перемешиваться при этой же температуре. Через несколько минут образовывалась белая суспензия, и через 30 мин происходило полное превращение в продукт, по данным TLC (95/5 DCM/MeOH). Промежуточный альдегид не выделяли. К суспензии добавляли последовательно ледяную АсОН (5 мл), AcONa (1,93 ммоль; 2,25 экв.) и соответствующий амин (1,54 ммоль; 1,8 экв.) и смесь оставляли нагреваться до кт. Через 1,5 ч добавляли TMSCN (2,57 ммоль; 3 экв.) и смесь оставляли перемешиваться при этой же температуре в течение ночи. Затем летучие вещества упаривали и к остатку добавляли насыщенный раствор NaHCO3 (40 мл). Смесь экстрагировали EtOAc (20 мл × 3), объединенные органические фазы сушили (безв. Na2SO4) и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (SiO2), используя смесь петролейного эфира и AcOEt с изменением соотношения от 9/1 до 4/6. Чистый α-аминонитрил получали в виде бесцветного масла (выход в среднем 53%).
Используя аналогичную методику, промежуточные соединения А0015_60_01 (выход 45%), А0015_64_01 (выход 51%), А0015_65_01 (выход 40%) и А0017_43_01 (выход 37%) получали, исходя из промежуточного соединения (11), с использованием морфолина, 4,4-дифторпиперидина гидрохлорида, 1,2,3,4-тетрагидроизохинолина или промежуточного соединения (10), соответственно; промежуточное соединение А0016_31__01 (выход 45%), А0016_29_01, (выход 67%) и А0016_30_01 (выход 71%) получали, исходя из промежуточного соединения (12), с использованием морфолина, 4,4-дифторпиперидина гидрохлорида или 1,2,3,4-тетрагидроизохинолина, соответственно.
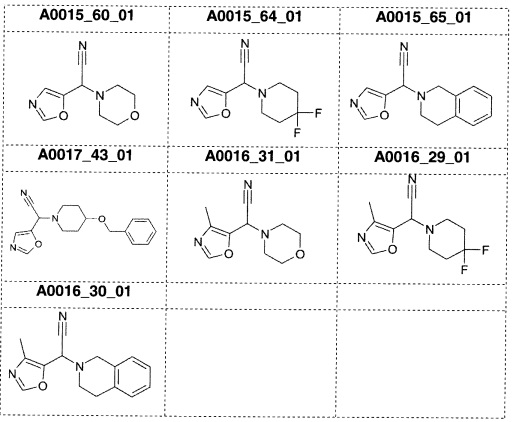
Способ а2)
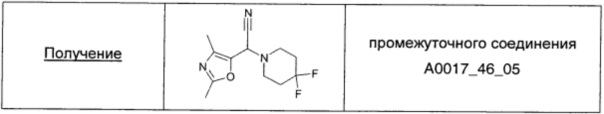
2,4-Диметилоксазол-5-карбальдегид (0,1 г; 0,8 ммоль; 1 экв.) растворяли в ледяной АсОН (2 мл). Добавляли последовательно AcONa (1,91 ммоль; 2,4 экв.) и 4,4-дифторпиперидина гидрохлорид (0,151 г; 0,96 ммоль; 1,2 экв.), перемешивая при комнатной температуре в атмосфере N2. Желтый раствор перемешивали в течение 2 ч и затем охлаждали до 0°С. По каплям добавляли TMSCN (0,3 мл; 2,4 ммоль; 3 экв.), смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Затем реакцию останавливали путем добавления МеОН и растворитель удаляли выпариванием.
Неочищенный продукт растворяли в DCM, добавляли насыщенный раствор NaHCO3 (20 мл) и смесь экстрагировали DCM (15 мл × 3). Объединенные органические фазы сушили (безв. Na2SO4) и упаривали. Неочищенный продукт очищали флэш-хроматографией (SiO2) с элюированием градиентом 100% петролейный эфир → 1/1 петролейный эфир/этилацетат, получая чистый указанный в заголовке α-аминонитрил А0017_46_05 (0,237 г; выход 79%).
Стадия b). Получение диаминов
Способ b1)
α-Аминонитрил (1 экв.) растворяли в 3 М NH3 в МеОН (100 экв.) и раствор гидрировали в проточном реакторе H-Cube™ с применением картриджа с Ni Ренея (30 мм, CatCart®, ThalesNano), используя скорость потока 1 мл/мин. Давление водорода варьировали от 30 до 60 бар (от 3 до 6 МПа), а температуру от 30°С до 40°С в зависимости от субстрата. По окончании соответствующего времени (обычно через 2 ч) раствор упаривали, получая чистый первичный амин, который использовали на следующей стадии без какой-либо очистки (выход в среднем 75%).
Используя аналогичную методику, промежуточные соединения А0015_61_01 (выход 83%), А0015_66_01 (выход 83%), А0015_67_02 (выход 79%) и А0017_67_01 (выход 80%) получали, исходя из А0015_60_01, А0015_64_01, А0015_65_01 и А0017_43_01, соответственно. Промежуточные соединения А0017_27_01 (выход 65%), А0017_28_01 (выход 77%) и А0017_34_01 (выход 73%) получали, исходя из А0016_31_01, А0016_29_01 и А0016_30_01, соответственно.
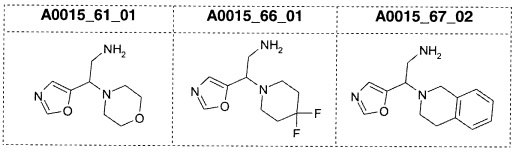
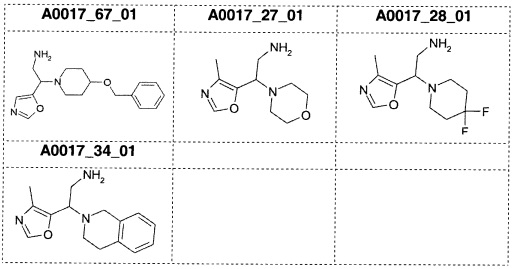
Способ b2)
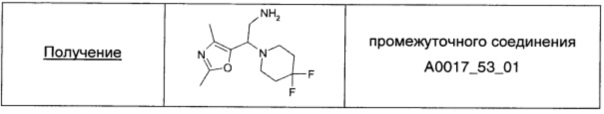
α-Аминонитрил А0017_46_05 (0,22 г; 0,86 ммоль; 1 экв.) растворяли в 3 М NH3 в МеОН (100 экв.) и раствор гидрировали в проточном реакторе H-Cube™ с применением картриджа с Ni Ренея (30 мм, CatCart®, ThalesNano), используя скорость потока 1 мл/мин. Давление водорода составляло 50 бар (5 МПа), и температура от 30°С до 40°С. Через 3 ч гидрирование останавливали и раствор упаривали, получая неочищенный продукт, содержащий первичный амин А0017_53_01, который использовали на следующей стадии без какой-либо очистки (0,175 г; выход 78%).
В. Получение конечных соединений
Пример В.1
Способ а)
Получение конечных продуктов А0015_08_01, А0015_10_02 и А0015_12_01
Смесь 2-хлор-6-фторбензойной кислоты (0,281 ммоль; 1 экв.) и HBTU (0,281 ммоль; 1 экв.) растворяли в безводном DMF (1,4 мл). Бледно-желтый раствор охлаждали до 0°С в атмосфере N2 в герметично закрытой пробирке и по каплям добавляли DIPEA (1,125 ммоль; 4 экв.). Через 16 ч выдерживания при этой же температуре добавляли по каплям раствор первичного амина (А0013_40_01, или А0013_54_03, или А0013_33_01; 0,281 ммоль; 1 экв.) в безводном DMF (1,4 мл). Затем смесь оставляли нагреваться до комнатной температуры, и после прохождения соответствующего периода времени (30 мин - 1 ч) реакция завершалась. Растворитель выпаривали и остаток распределяли между насыщенным раствором NaHCO3 (20 мл) и DCM (15 мл). Водную фазу дополнительно экстрагировали DCM (15 мл × 2) и объединенные органические фазы сушили (Na2SO4) и упаривали. Неочищенный продукт очищали флэш-хроматографией (SiO2) с элюированием смесью 85/15 DCM/EtOAc или 50/50 петролейный эфир/этилацетат. Остаток очищали препаративной LC-MS (см. Аналитическую часть). Объединенные собранные фракции упаривали до небольшого объема (1-2 мл). Удаление противоиона TFA проводили, используя многоцелевой картридж для твердофазной экстракции (MP SPE cartridge; multi-purpose solid phase extraction cartridge) PL-НСО3 (Agilent Technologies; 0,1 г; объем 6 мл). Наконец, сублимационную сушку проводили с использованием системы от Martin Christ, получая указанные в заголовке соединения А0015_08_01, А0015_10_02 или А0015_12_01 (для стадии сочетания выход в среднем 60%).
Используя аналогичную методику, соединение:
А0013_29_01 получали, исходя из А0013_28_01;
А0013_29_03 получали, исходя из А0013_28_03;
А0015_09_02 получали, исходя из А0011_54_01;
А0013_29_02 получали, исходя из А0013_30_01;
А0013_32_01 получали, исходя из А0013_31_01;
А0015_13_01 получали, исходя из А0015_11_01;
А0015_28_03 получали, исходя из А0015_25_01 и щавелевой кислоты;
А0017_05_01 получали, исходя из А0017_01_01;
А0016_10_01 получали, исходя из А0016_09_01;
А0016_20_03 получали, исходя из А0016_17_01;
А0015_55_01 получали, исходя из А0015_52_01;
А0015_58_02 получали, исходя из А0015_56_01;
А0015_57_02 получали, исходя из А0015_54_01;
А0012_60_01 получали, исходя из А0012_61_02;
А0012_64_01 получали, исходя из А0012_63_01;
А0015_62_03 получали, исходя из А0015_61_01;
А0015_68_02 получали, исходя из А0015_66_01;
А0015_69_02 получали, исходя из А0015_67_02.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на 2,6-диметилбензойную кислоту, соединения:
А0013_42_05 получали, исходя из А0013_40_01;
А0013_55_05 получали, исходя из А0013_54_03;
А0013_58_03 получали, исходя из А0013_33_01;
А0017_05_03 получали, исходя из А0017_01_01;
А0016_11_02 получали, исходя из А0016_09_01;
А0015_73_01 получали, исходя из А0015_52_01;
А0015_72_01 получали, исходя из А0015_56_01;
А0015_71_02 получали, исходя из А0015_54_01;
А0012_62_02 получали, исходя из А0012_61_02;
А0012_65_01 получали, исходя из А0012_63_01;
А0016_24_02 получали, исходя из А0015_66_01;
А0016_25_02 получали, исходя из А0015_67_02.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на 5-амино-2-хлорбензойную кислоту, соединения:
А0013_42_04 получали, исходя из А0013_40_01;
А0013_55_04 получали, исходя из А0013_54_03;
А0013_58_02 получали, исходя из А0013_33_01.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на 2-хлор-6-метилбензойную кислоту, соединения:
А0013_42_02 получали, исходя из А0013_40_01;
А0013_55_02 получали, исходя из А0013_54_03;
А0013_58_01 получали, исходя из А0013_33_01;
А0017_05_02 получали, исходя из А0017_01_01;
А0012_62_01 получали, исходя из А0012_61_02;
А0012_66_01 получали, исходя из А0012_63_01;
А0021_26_04 получали в виде смеси двух диастереоизомеров, исходя из А0021_07_02;
А0021_26_03 получали в виде единственного диастереоизомера, исходя из А0021_26_04, с использованием препаративной LC-MS (см. Аналитическую часть).
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на промежуточное соединение (4), соединения:
А0013_82_01 получали, исходя из А0013_54_03;
А0016_23_02 получали, исходя из А0015_56_01;
А0017_13_01 получали, исходя из А0015_54_01;
А0016_26_02 получали, исходя из А0015_66_01;
А0017_37_04 получали, исходя из А0017_28_01;
А0017_37_05 получали, исходя из А0017_27_01;
А0017_37_06 получали, исходя из А0017_34_01;
А0017_50_01 получали, исходя из А0012_61_02;
А0017_55_01 получали, исходя из А0017_53_01;
А0017_75_02 получали, исходя из А0017_73_01;
А0017_75_01 получали, исходя из А0017_72_01;
А0018_60_01 получали, исходя из А0018_58_01;
А0018_76_01 получали, исходя из А0018_75_01;
А0017_83_01 получали, исходя из А0017_58_01;
А0018_94_01 получали, исходя из А0018_93_01;
А0020_21_01 получали, исходя из А0020_19_01;
А0021_17_01 получали, исходя из А0020_26_01;
А0021_24_01 получали, исходя из А0020_37_01;
А0021_09_01 получали, исходя из А0021_07_03;
А0021_10_01 получали, исходя из А0021_07_04;
А0021_24_02 получали, исходя из А0020_38_01, без удаления противоиона TFA;
А0021_39_01 получали, исходя из А0020_69_01;
А0020_67_01 получали, исходя из А0020_66_01;
А0020_32_01 получали, исходя из А0020_31_01;
А0016_67_01 получали, исходя из А0016_57_01;
А0016_64_01 получали, исходя из А0016_59_01;
А0016_53_01 получали, исходя из А0016_48_01;
А0016_50_01 получали, исходя из А0016_47_01.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на промежуточное соединение (5), соединение А0017_81_03 получали, исходя из А0015_56_01.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на промежуточное соединение (6), соединение А0017_81_02 получали, исходя из А0015_56_01; соединение А0018_88_01 получали, исходя из А0012_61_02; соединение А0020_21_02 получали, исходя из А0020_19_01; соединение А0020_28_01 получали, исходя из А0020_26_01; соединение А0021_09_02 получали, исходя из А0021_07_03; соединение А0021_10_02 получали, исходя из А0021_07_04; соединение А0021_38_02 получали, исходя из А0020_66_01.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на промежуточное соединение (7), соединение А0017_85_01 получали, исходя из А0015_56_01; соединение А0018_89_01 получали, исходя из А0012_61_02.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на промежуточное соединение (8) соединение А0018_69_01 получали, исходя из А0015_56_01; соединение А0018_89_02 получали, исходя из А0012_61_02.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на 5-хинолинкарбоновую кислоту, соединение А0016_21_02 получали, исходя из А0015_56_01; соединение А0021_26_02 получали, исходя из А0021_07_02; А0021_38_01 получали, исходя из А0020_66_01; А0021_39_02 получали, исходя из А0020_69_01; соединение А0016_54_01 получали, исходя из А0016_48_01; А0016_68_01 получали, исходя из А0016_57_01; А0016_65_01 получали, исходя из А0016_59_01.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на 2,3-диметоксибензойную кислоту, соединения:
А0017_09_03 получали, исходя из А0013_54_03;
А0017_37_01 получали, исходя из А0017_28_01;
А0017_37_02 получали, исходя из А0017_27_01;
А0017_37_03 получали, исходя из А0017_34_01.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на 2-хлор-4-(1,1-диоксидо-2-изотиазолидинил)-бензойную кислоту, соединение А0016_60_01 получали, исходя из А0015_56_01.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензойную кислоту на 7-фтор-2-оксо-1,2,3,4-тетрагидрохинолин-6-карбоновую кислоту, соединение А0016_61_01 получали, исходя из А0015_56_01.
Способ b)
2-Хлор-6-фторбензоилхлорид (0,013 мл; 0,09 ммоль; 1,05 экв.) добавляли к перемешиваемому раствору первичного амина (0,08 ммоль) в безводном DCM (1 мл) и TEA (0,063 мл; 0,45 ммоль; 5 экв.). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем распределяли между 2% раствором KOH и DCM. Органические слои сушили над Na2SO4 (сухим), фильтровали и окончательно упаривали, получая неочищенный конечный продукт, который очищали препаративной LC-MS (см. Аналитическую часть). Объединенные собранные фракции упаривали до небольшого объема (1-2 мл). Удаление противоиона TFA проводили, используя MP SPE картридж PL-НСО3 (Agilent Technologies; 0,1 г; объем 6 мл). Наконец, сублимационную сушку проводили с использованием системы от Martin Christ, получая конечный продукт в виде свободного основания.
А0017_33_01 получали, исходя из А0017_28_01;
А0017_33_02 получали, исходя из А0017_27_01;
А0017_60_02 получали, исходя из А0017_59_01;
А0017_60_01 получали, исходя из А0017_58_01;
А0018_17_01 получали, исходя из А0018_16_01;
А0017_68_01 получали, исходя из А0017_67_01;
А0017_74_02 получали, исходя из А0017_73_01;
А0018_59_01 получали, исходя из А0018_58_01;
А0018_95_01 получали, исходя из А0018_93_01;
А0020_20_01 получали, исходя из А0020_19_01;
А0020_27_01 получали, исходя из А0020_26_01;
А0021_07_11 получали, исходя из А0021_07_01;
А0021_07_22 получали, исходя из А0021_07_02;
А0021_07_44 получали, исходя из А0021_07_04;
А0021_07_33 получали, исходя из А0021_07_03;
А0021_25_02 получали, исходя из А0020_38_01, без удаления противоиона TFA;
А0021_40_01 получали, исходя из А0020_69_01;
А0020_68_01 получали, исходя из А0020_66_01;
А0016_52_01 получали, исходя из А0016_48_01;
А0016_49_01 получали, исходя из А0016_47_01;
А0016_63_01 получали, исходя из А0016_59_01;
А0016_66_01 получали, исходя из А0016_57_01;
А0022_02_01 получали, исходя из А0022_01_01.
Используя аналогичную методику, но заменяя 2-хлор-6-фторбензоилхлорид на 2,6-дифторбензоилхлорид, А0017_09_02 получали, исходя из А0013_54_03.
Пример В.2
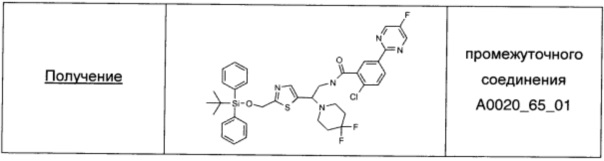
Смесь промежуточного соединения 4 (0,056 г; 0,22 ммоль; 1,2 экв.) и HBTU (0,08 г; 0,28 ммоль; 1,15 экв.) растворяли в безводном DMF (3 мл). Бледно-желтый раствор охлаждали до 0°С в атмосфере N2 в герметично закрытой пробирке и по каплям добавляли DIPEA (0,093 ммоль; 3 экв.). Через 1 ч выдерживания при этой же температуре добавляли по каплям раствор А0020_63_01 (0,095 г; 0,18 ммоль; 1 экв.) в безводном DMF (1 мл). Затем смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Растворитель выпаривали и остаток распределяли между насыщенным раствором NaHCO3 (20 мл) и DCM (15 мл). Водную фазу дополнительно экстрагировали DCM (15 мл × 2) и объединенные органические фазы сушили (Na2SO4) и упаривали. Неочищенный продукт (0,17 г) очищали флэш-хроматографией (SiO2) с элюированием смесью 50/50 петролейный эфир/этилацетат, получая промежуточное соединение А0020_65_01 (0,09 г; выход 66%).
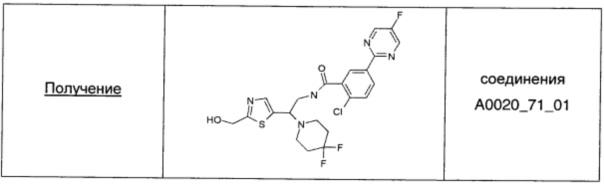
А0020_65_01 (0,09 г; 0,120 ммоль; 1 экв.) растворяли в безводном THF (2 мл) и обрабатывали 1 М фторидом тетрабутиламмония в THF (1,32 мл; 1,32 ммоль; 1,2 экв.) при комнатной температуре. Через 1 ч добавляли насыщенным раствор NH4Cl (1 мл), растворитель удаляли при пониженном давлении и остаток распределяли между насыщенным раствором NaHCO3 (4 мл) и DCM (4 мл). Объединенный органический экстракт сушили над безв. Na2SO4, фильтровали и упаривали, получая неочищенный продукт (0,08 г), который очищали препаративной LC-MS (см. Аналитическую часть), получая А0020_71_01 (0,01 г; выход 16%) в виде белого твердого вещества.
Пример В.3
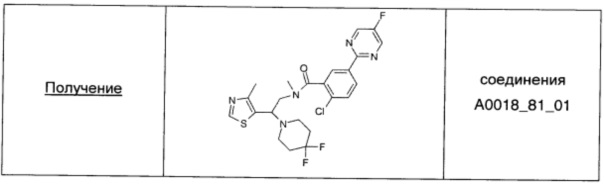
А0016_23_02 (0,33 г; 0,067 ммоль; 1 экв.) растворяли в безводном DMF (0,7 мл) и охлаждали до 0-5°С в атмосфере аргона. Добавляли NaH (60% суспензия в минеральном масле, 0,003 г; 0,074 ммоль; 1,1 экв.) одной порцией и полученный желтый раствор перемешивали при 0°С в течение 30 мин. Далее добавляли раствор метилиодида (0,0086 г; 0,061 ммоль; 0,9 экв.) в безводном DMF (0,1 мл), и реакционную смесь перемешивали при 0°С в течение 2 минут, затем при комнатной температуре в течение 30 мин. Реакцию останавливали путем добавления воды (0,1 мл) и МеОН (0,1 мл). Растворитель удаляли выпариванием при пониженном давлении. Остаток растворяли в DCM (3 мл) и промывали 5% раствором лимонной кислоты в воде (3 мл), насыщенным раствором бикарбоната натрия (3 мл) и рассолом (3 мл). Органический слой сушили над безв. Na2SO4, фильтровали и упаривали, получая 0,043 г неочищенного продукта, который очищали препаративной LC-MS (см. Аналитическую часть), получая А0018_81_01 (19 мг; выход 28%) в виде белого твердого вещества.
Пример В.4
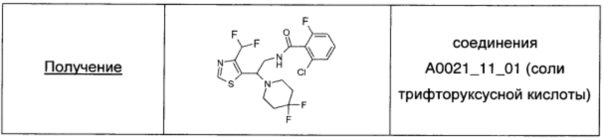
А0018_98_01 (0,040 г; 0,136 ммоль; 1 экв.) растворяли в МеОН (4,5 мл) и раствор гидрировали в проточном реакторе H-Cube™ с применением картриджа с Ni Ренея (CatCart®, длиной 55 мм), используя скорость потока 0,7 мл/мин. Давление водорода составляло 50 бар (5 МПа), и температура составляла 35°С. Через 1 ч раствор обрабатывали щавелевой кислотой (0,037 г; 0,408 ммоль; 3 экв.) и перемешивали в течение 30 мин при комнатной температуре. Растворитель удаляли при пониженном давлении, и неочищенный продукт кристаллизовали, добавляя диэтиловый эфир (1 мл). Полученное твердое вещество промывали 3 раза диэтиловым эфиром, получая светло-желтый порошок, который использовали в таком виде.
Неочищенную желтую оксалатную соль суспендировали в THF (2 мл) и добавляли 2-хлор-5-фтор-бензоилхлорид (0,077 г; 0,4 ммоль; 2,94 экв.), затем водн. насыщенный раствор NaHCO3 (1 мл). Полученную смесь перемешивали в течение 1 ч при комнатной температуре. Растворители удаляли при пониженном давлении и водный слой экстрагировали DCM (10 мл × 2). Объединенные органические экстракты сушили над безв. Na2SO4, фильтровали и упаривали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (EtOAc/петролейный эфир, 50/50), получая неочищенное масло (30 мг), которое очищали препаративной LC-MS (см. Аналитическую часть). Собранные фракции упаривали до небольшого объема (1-2 мл). Удаление противоиона TFA проводили, используя MP SPE-картридж PL-НСО3 (Agilent Technologies; 0,1 г; объем 6 мл). Сублимационную сушку проводили с использованием системы от Martin Christ, получая белое твердое вещество, которое растворяли в диэтиловом эфире (1 мл) и обрабатывали щавелевой кислотой (1,5 экв.), полученное твердое вещество отфильтровывали и промывали диэтиловым эфиром (5 мл ×3), получая А0021_11_01 (0,019 г; выход 23%) в виде соли трифторуксусной кислоты.
Пример В.5
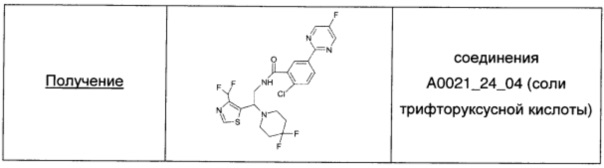
А0018_98_01 (0,07 г; 0,238 ммоль; 1 экв.) растворяли в МеОН (3 мл) и раствор гидрировали в проточном реакторе H-Cube™ с применением картриджа с Ni Ренея (CatCart®, длиной 55 мм), используя скорость потока 1 мл/мин. Давление водорода составляло 40 бар (4 МПа), а температура 35°С. Через 1,5 часа раствор обрабатывали щавелевой кислотой (0,15 г; 1,66 ммоль; 6,9 экв.) и растворитель удаляли при пониженном давлении, получая белый порошок, содержащий в основном первичный амин в виде оксалатной соли, который использовали в таком виде.
Смесь промежуточного соединения 4 (0,033 г; 0,13 ммоль; 1,1 экв.) и HBTU (0,049 г; 0,13 ммоль; 1,1 экв.) растворяли в безводном DMF (0,5 мл). Бледно-желтый раствор охлаждали до 0°С в атмосфере N2 в герметично закрытой пробирке и по каплям добавляли безводный TEA (0,105 мл; 0,75 ммоль; 3 экв.). Через 2,5 ч выдерживания при этой же температуре добавляли раствор первичного амина в виде оксалатной соли (0,033 г; 0,13 ммоль; 1 экв.) в безводном DMF (1 мл) и реакционную смесь перемешивали в течение 15 минут при 0°С, затем при комнатной температуре в течение ночи. Реакцию гасили, добавляя воду (0,1 мл), и реакционную смесь упаривали. Неочищенный продукт растворяли в DCM (3 мл), промывали насыщенным водным раствором карбоната натрия (23 мл × 2) и рассолом (20 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (EtOAc/петролейный эфир, 50/50), получая 0,011 г твердого вещества, содержащего А0021_24_04.
Данную процедуру повторяли, исходя из А0018_98_01 (0,07 г; 0,238 ммоль), с получением 0,012 г твердого вещества, содержащего А0021_24_04. Эти две партии, содержащие А0021_24_04, объединяли и очищали препаративной LC-MS (см. Аналитическую часть). Объединенные фракции упаривали до небольшого объема (1-2 мл). Наконец, сублимационную сушку проводили с использованием системы от Martin Christ, получая А0021_24_04 (0,015 г; выход 10%) в виде соли трифторуксусной кислоты.
В Таблице F-1 приведены конечные соединения, которые получали и тестировали в соответствии с экспериментальной методикой, описанной в примерах В.1, В.2, В.3, В.4 и В.5.
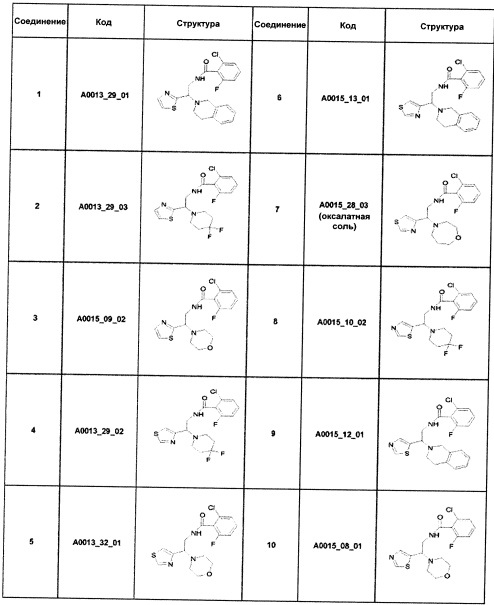
(продолжение следует)
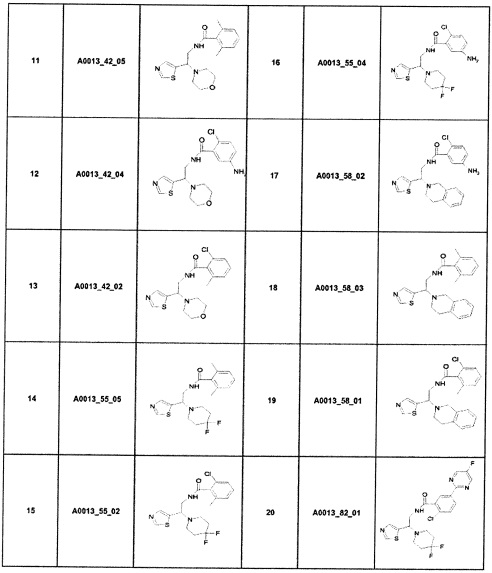
(продолжение следует)
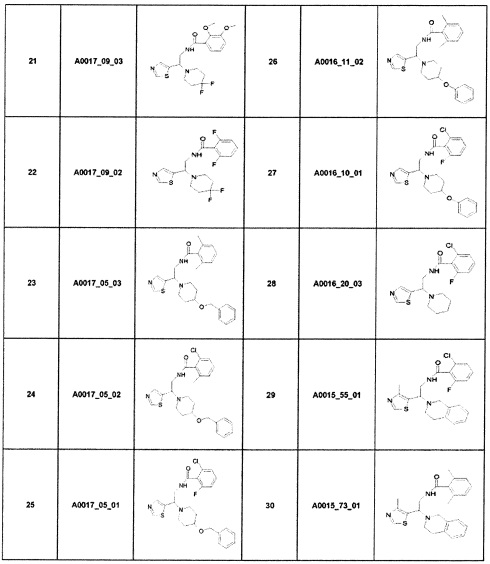
(продолжение следует)

(продолжение следует)
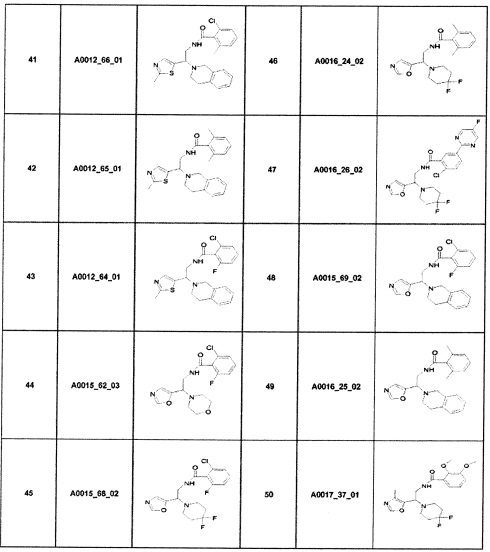
(продолжение следует)
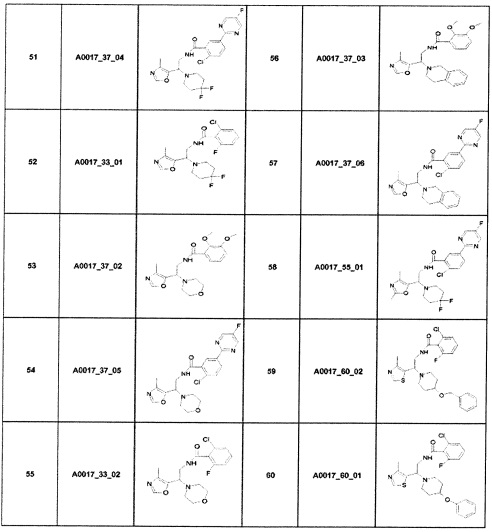
(продолжение следует)
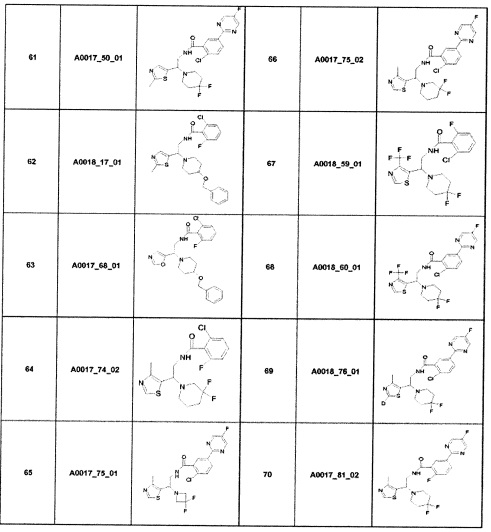
(продолжение следует)
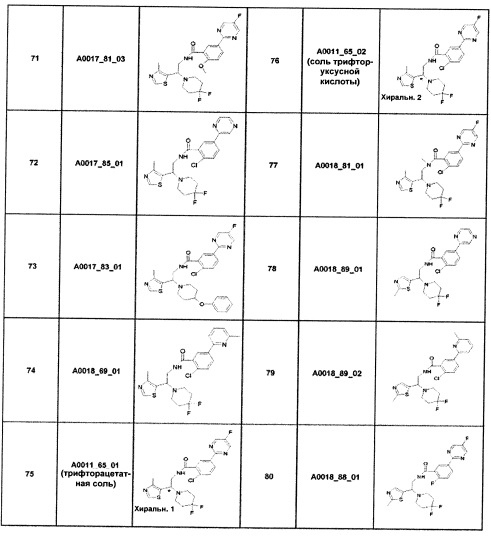
(продолжение следует)
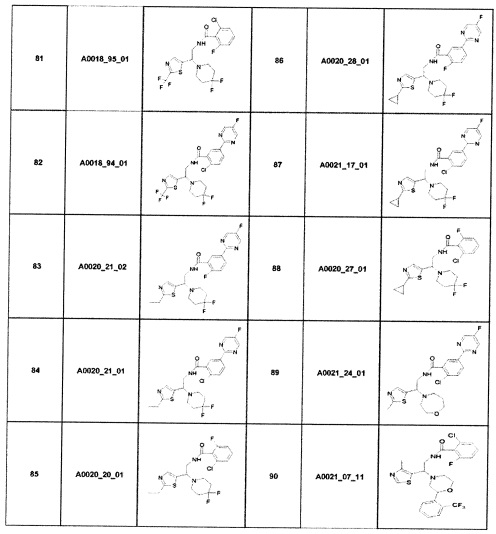
(продолжение следует)
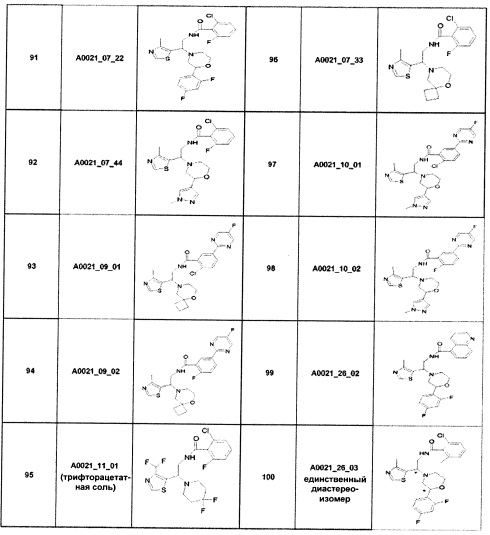
(продолжение следует)
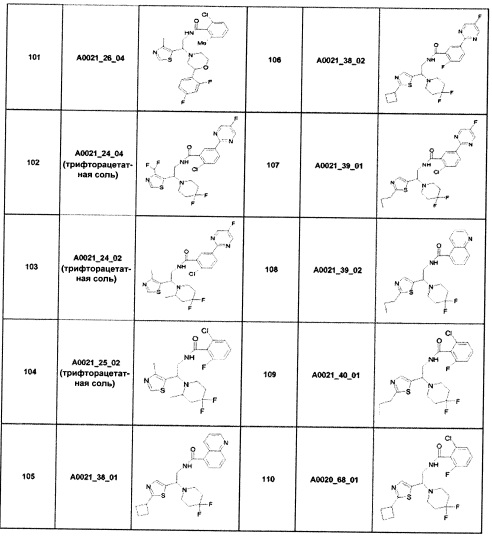
(продолжение следует)
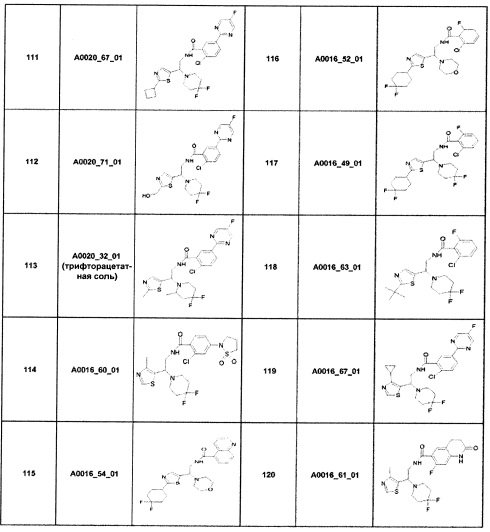
(продолжение следует)
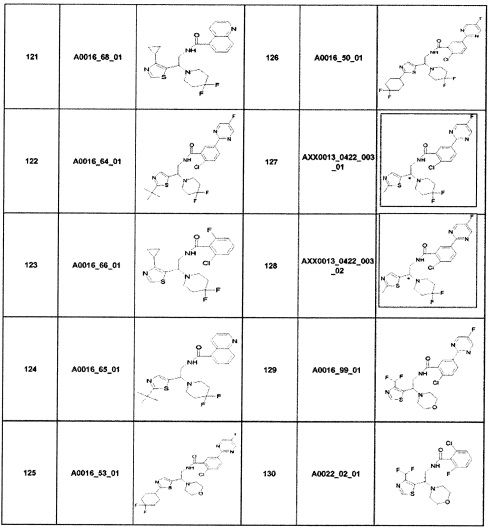
Аналитическая часть
Методология очистки
Препаративная HPLC-MS
Система для высокоэффективной жидкостной хроматографии (HPLC) от Waters с насосом 2525 от Waters, устройством ввода пробы 2767 от Waters, держателем колонки для жидкостной хроматографии с насосом 515 для LC, фотодиодным матричным детектором (PDA) 2996 от Waters и масс-спектрометром ZQ Micromass с источником ESI и одиночным квадрупольным детектором. Использовали две подвижные фазы, подвижную фазу А: воду (MilliQ) с 0,1% TFA; подвижную фазу В: ацетонитрил (Chromasolv, Sigma-Aldrich) с 0,1% TFA, и для каждого конкретного соединения определяли условия градиентного элюирования. Использовали две препаративные колонки: X-Bridge С18, 100×19 мм, 5 мкм, от Waters для липофильных соединений и Atlantis С18, 100×19 мм, 5 мкм, от Waters для очень полярных соединений. Использовали объем вводимой пробы от 20 до 900 мкл, и скорость потока составляла 20 мл/минута. Удаление противоиона проводили, используя MP картридж PL-HCO3, устройство на основе четвертичного амина SAX (от англ. strong anion exchanger - сильный анионообменник) (форма HCO3-) для удаления TFA из элюатов после HPLC и переведения солей TFA в форму свободного основания. Сублимационную сушку проводили с использованием системы от Martin Christ.
Разделение рацематов
Способ 1. Рацемическую смесь соединения 34 обрабатывали, используя модуль Agilent 1100, содержащий насос для четырехкомпонентных смесей с дегазатором, автосэмплер, термостат для колонок (установленный на 40°С), диодный матричный детектор (DAD) (использовали длину волны 220 нм), чтобы собрать оба энантиомера с достаточной степенью чистоты. Полупрепаративную обращенно-фазовую HPLC проводили на хиральной С18-колонке Cyclobond I 2000 HP-RSI Supelco (5 мкм; 4,6×150 мм) со скоростью потока 1,2 мл/мин. Использовали две подвижные фазы, подвижную фазу А: воду (MilliQ) с 0,1% TFA; подвижную фазу В: ацетонитрил (Chromasolv, Sigma-Aldrich) с 0,1% TFA, и их применяли для осуществления хроматографирования в изократическом режиме с элюированием 10% В в течение 20 минут. Для введения использовали объем 20 мкл раствора в концентрации 2,2 мг/мл.
Способ 2. Рацемическую смесь соединения 34 при необходимости обрабатывали, используя четырехкомпонентный градиентный модуль 2535 от WATERS, оборудованный детектором 2489 от WATERS, работающим в ультрафиолетовой (УФ)/видимой области спектра (использовали две длины волны 240 и 360 нм), чтобы собрать оба энантиомера с достаточной степенью чистоты. Аналитическую нормально-фазовую HPLC проводили на колонке Chiral Kromasil 5-Amycoat (5 мкм; 4,6×250 мм) со скоростью потока 1,0 мл/мин. Использовали две подвижные фазы, подвижную фазу А: гексан (Chromasolv для HPLC Sigma-Aldrich); подвижную фазу В: изопропанол (Chromasolv для HPLC Sigma-Aldrich), и их применяли для осуществления хроматографирования в изократическом режиме с элюированием 40% В в течение 20 минут. Для введения использовали объем 100 мкл раствора в концентрации 10,0 мг/мл.
Способ 3. Рацемическую смесь соединения 61 обрабатывали, используя четырехкомпонентный градиентный модуль 2535 от WATERS, оборудованный детектором 2489 от WATERS, работающим в УФ/видимой части спектра (использовали две длины волны 240 и 360 нм), чтобы собрать оба энантиомера с достаточной степенью чистоты. Аналитическую нормально-фазовую HPLC проводили на колонке Chiral Kromasil 5-Amycoat (5 мкм; 4,6×250 мм) со скоростью потока 1,0 мл/мин. Использовали две подвижные фазы, подвижную фазу А: гексан (Chromasolv для HPLC Sigma-Aldrich); подвижную фазу В: этанол (Chromasolv для HPLC Sigma-Aldrich), и их применяли для осуществления хроматографирования в изократическом режиме с элюированием 15% В в течение 90 минут. Для введения использовали объем 100 мкл раствора в концентрации 10,0 мг/мл.
Соединения 75 и 76 получали в виде единственного энантиомера из рацемата 34.
Соединения 127 и 128 получали в виде единственного энантиомера из рацемата 61.
Жидкостная хроматография в сочетании с масс-спектрометрией (LCMS)
Общая методика LCMS
HPLC-анализ проводили, используя модуль Agilent 1100, содержащий насос для четырехкомпонентных смесей с дегазатором, автосэмплер, термостат для колонок (установленный на 40°С), диодный матричный детектор (DAD) (использовали длину волны 215 нм) и колонку, специально уточненную для соответствующего метода ниже. Часть потока с колонки отводили в MS спектрометр. MS детектор (анализатор с ионной ловушкой Esquire 3000 plus от Bruker) был оснащен источником электрораспылительной ионизации. Масс-спектры получали путем сканирования в диапазоне от 50 до 1500 в течение 0,2 секунды. Напряжение на капиллярной игле составляло 4 кВ в режиме положительной ионизации и температуру в источнике поддерживали при 365°С. В качестве распыляющего газа использовали азот, скорость потока составляла 10 л/мин. Обработку данных осуществляли, используя программу для анализа данных от Bruker.
LCMS - методика 1
В дополнение к общей методике: обращенно-фазовую HPLC проводили на С18-колонке Discovery, Supelco (5 мкм; 4,6×150 мм) со скоростью потока 1,0 мл/мин. Использовали две подвижные фазы, подвижную фазу А: воду (MilliQ) с 0,05% TFA; подвижную фазу В: ацетонитрил (Chromasolv, Sigma-Aldrich) с 0,05% TFA, и их применяли для хроматографирования в следующих условиях градиента: от 20% В до 90% в течение 15 минут, 100% В в течение 0,9 минуты, 20% В в течение 0,1 минуты и выдерживание в этих условиях в течение 4 минут для повторного уравновешивания колонки. Для введения использовали объем 5 мкл.
LCMS - методика 2
В дополнение к общей методике: обращенно-фазовую HPLC проводили на С18-колонке Discovery, Supelco (5 мкм; 4,6×150 мм) со скоростью потока 1,0 мл/мин. Использовали две подвижные фазы, подвижную фазу А: воду (MilliQ) с 0,05% TFA; подвижную фазу В: ацетонитрил (Chromasolv, Sigma-Aldrich) с 0,05% TFA, и их применяли для хроматографирования в следующих условиях градиента: от 5% В до 50% в течение 15 минут, 100% В в течение 0,9 минуты, 5% В в течение 0,1 минуты и выдерживание в этих условиях в течение 4 минут для повторного уравновешивания колонки. Для введения использовали объем 5 мкл.
LCMS - методика 3
В дополнение к общей методике: обращенно-фазовую HPLC проводили на С18-колонке Atlantis от Waters (3 мкм; 4,6×100 мм) со скоростью потока 1,0 мл/мин. Использовали две подвижные фазы, подвижную фазу А: воду (MilliQ) с 0,05% TFA; подвижную фазу В: ацетонитрил (Chromasolv, Sigma-Aldrich) с 0,05% TFA, и их применяли для хроматографирования в следующих условиях градиента: от 0% В до 30% в течение 12 минут, 50% В в течение 0,9 минуты, 0% В в течение 0,1 минуты и выдерживание в этих условиях в течение 3 минут для повторного уравновешивания колонки. Для введения использовали объем 5 мкл.
LCMS - методика 4
В дополнение к общей методике: обращенно-фазовую HPLC проводили на хиральной С18-колонке Cyclobond I 2000 HP-RSI Supelco (5 мкм; 4,6×150 мм) со скоростью потока 1,0 мл/мин. Использовали две подвижные фазы, подвижную фазу А: воду (MilliQ) с 0,1% TFA; подвижную фазу В: ацетонитрил (Chromasolv, Sigma-Aldrich) с 0,1% TFA, и их применяли для хроматографирования в следующих условиях градиента: от 10% В до 20% в течение 15 минут. Для введения использовали объем 5 мкл.
LCMS - методика 5
В дополнение к общей методике: обращенно-фазовую HPLC проводили на экспресс-колонке Ascentis (3 мкм; 4,6×150 мм) со скоростью потока 1,0 мл/мин. Использовали две подвижные фазы, подвижную фазу А: воду (MilliQ) с 0,1% TFA; подвижную фазу В: ацетонитрил (Chromasolv, Sigma-Aldrich) с 0,1% TFA, и их применяли для хроматографирования в следующих условиях градиента: от 10% В до 20% в течение 15 минут. Для введения использовали объем 5 мкл.
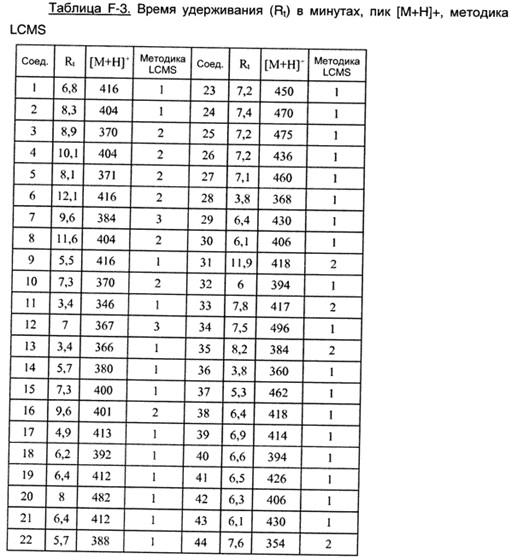
(продолжение следует)
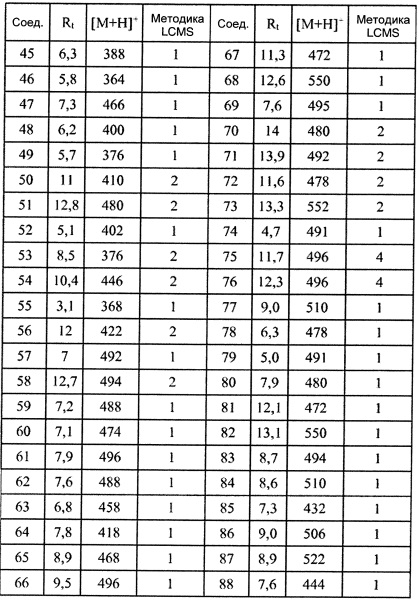
(продолжение следует)
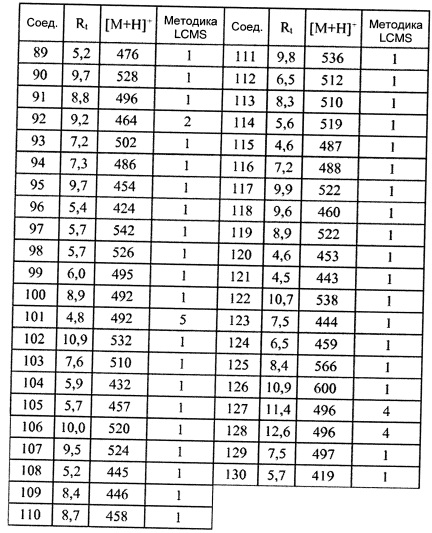
Определение характеристик ЯМР
Для ряда соединений регистрировали 1Н ЯМР-спектры на спектрометре Avance (400 МГц) от Bruker, используя в качестве растворителей DMSO-d6 или DMSO-d6 с каплей трифторуксусной кислоты. Химические сдвиги (δ) приведены в виде числа частей на миллион (млн-1) относительно тетраметилсилана (TMS), который использовали в качестве внутреннего стандарта.
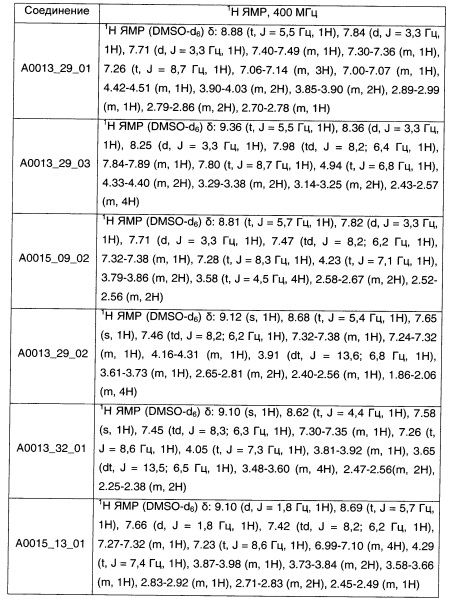
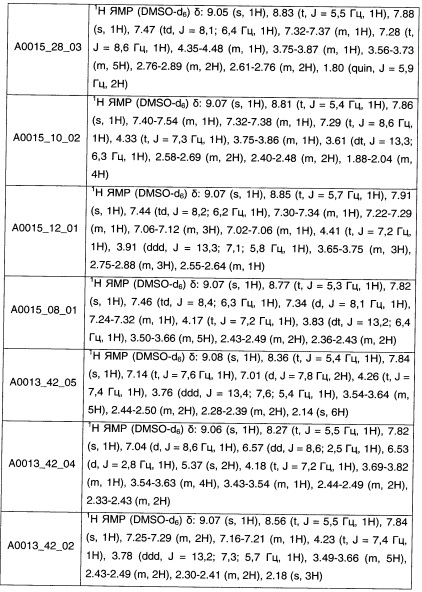
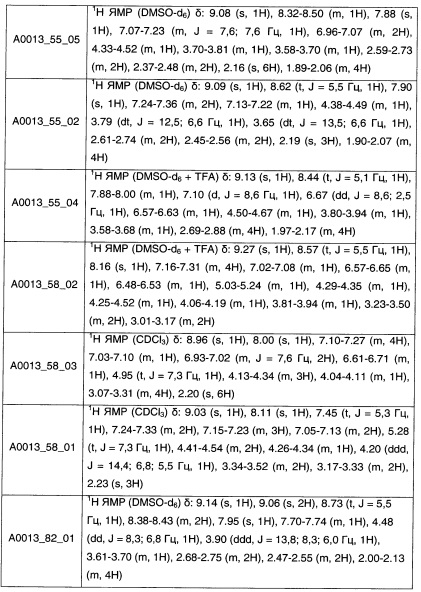
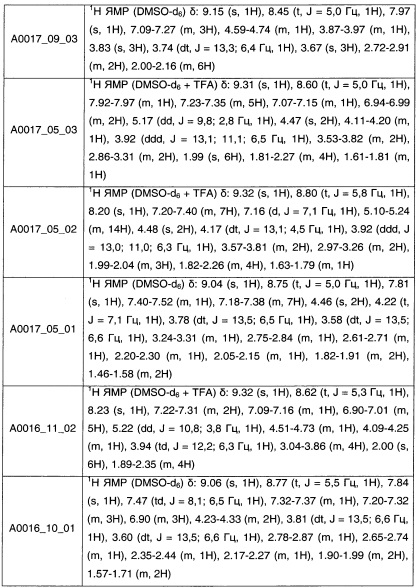
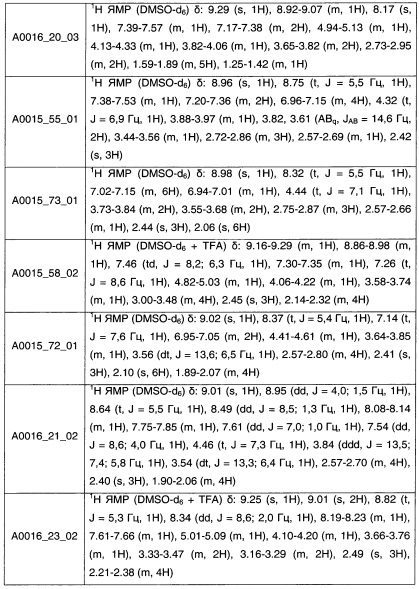
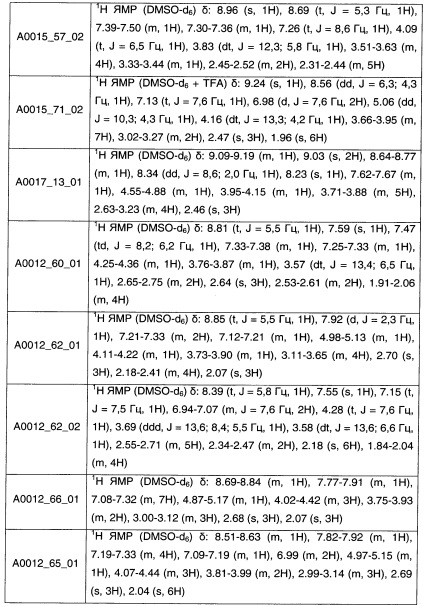
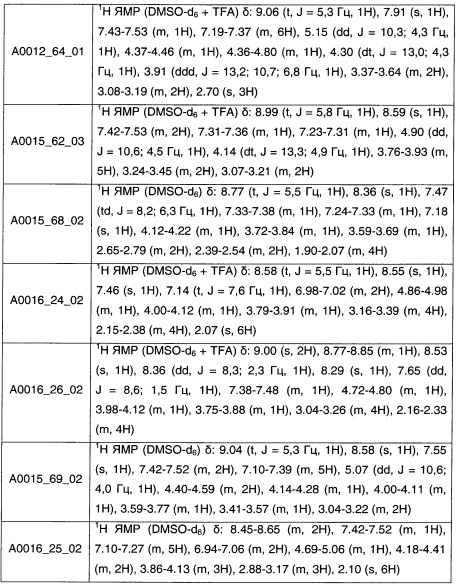
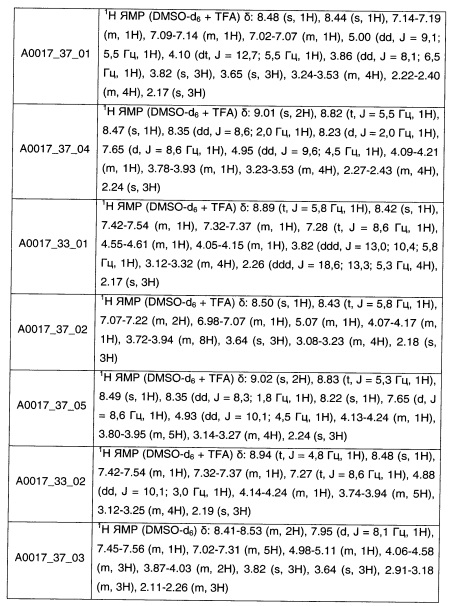
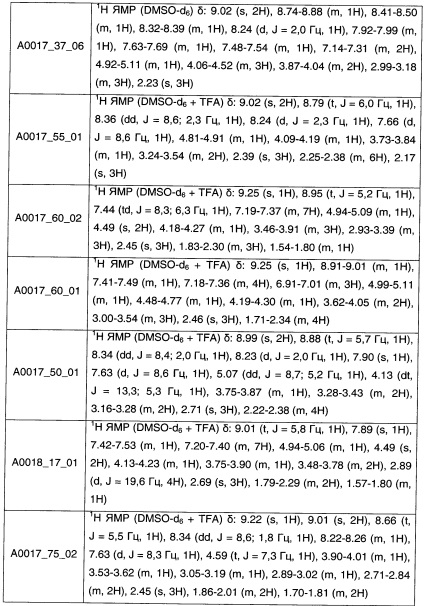
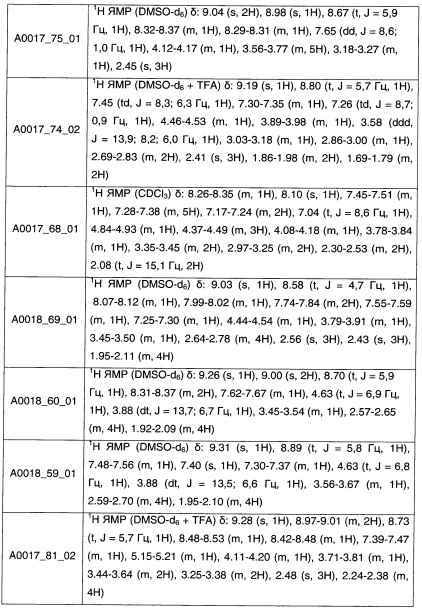
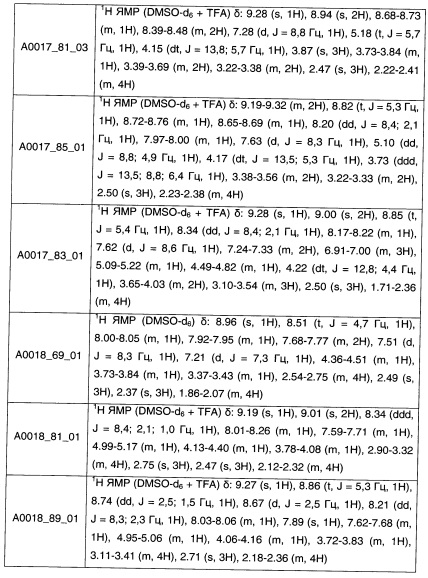
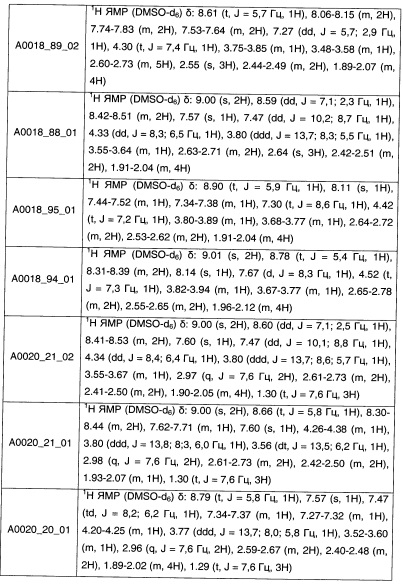
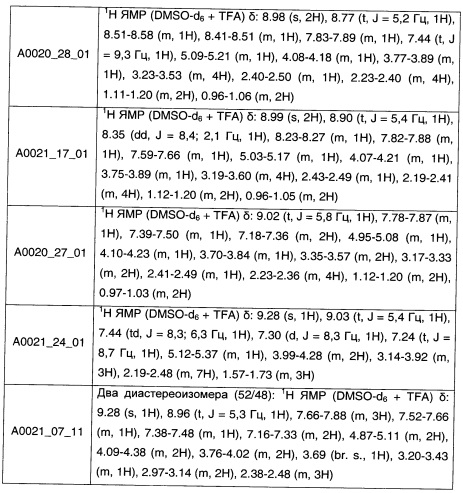
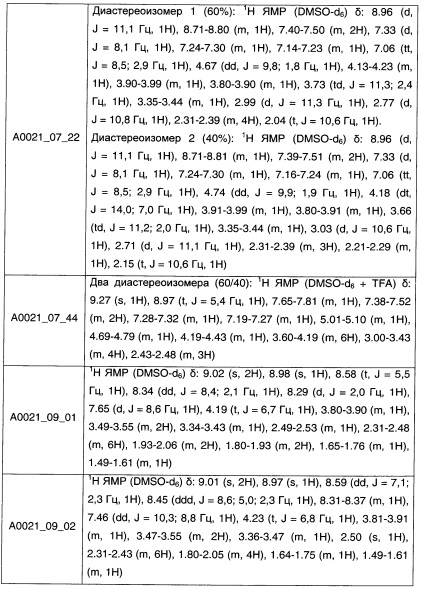
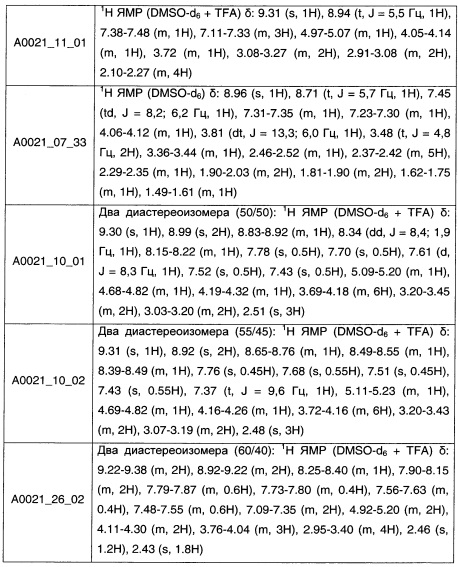
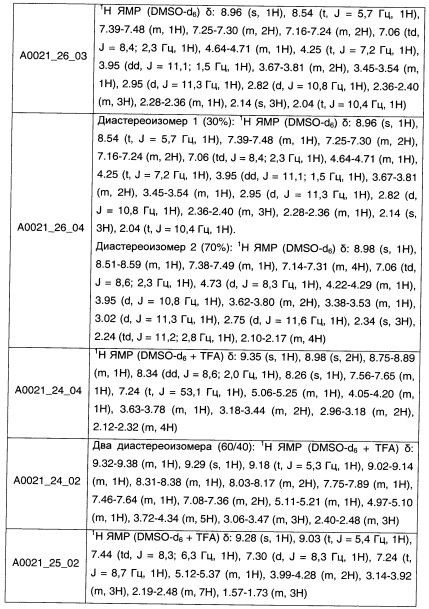
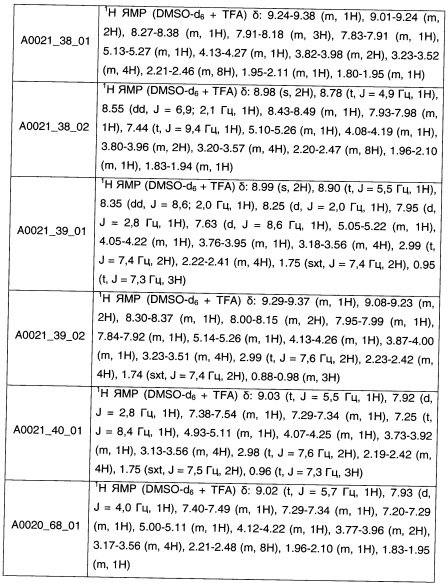
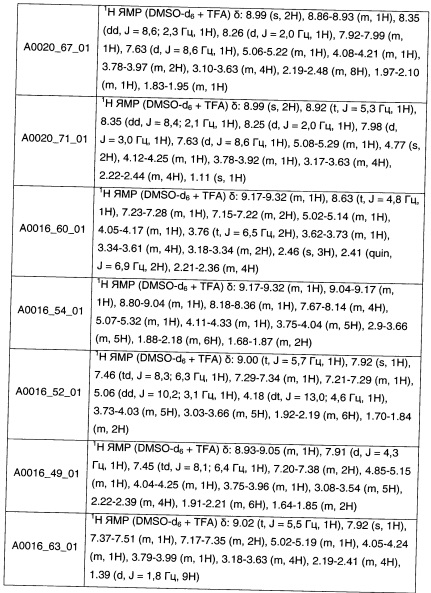
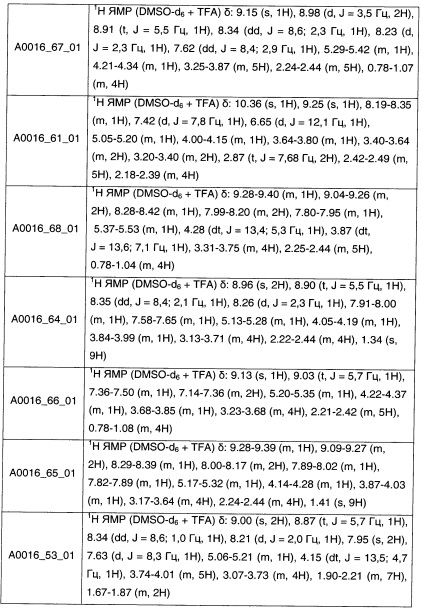

Фармакологические примеры
С использованием набора для определения содержания кальция с применением Fluo-8 без стадии отмывки Screen Quest™ было обнаружено, что описанные в примерах соединения по изобретению являются ингибиторами Р2Х7.
Связывание находящейся вне клетки триэтиламмониевой соли 2'(3')-O-(4-бензоилбензоил)аденозин 5'-трифосфата (Bz-ATP) с Р2Х7 рецептором открывает канал и дает возможность для поступления Ca2+ в клетки. Такое поступление Ca2+ измеряли в клетках HEK-293, стабильно трансфицированных геном, кодирующим Р2Х7 рецептор, используя набор для определения содержания кальция с применением Fluo-8 без стадии отмывки Screen Quest™ (AAt Bioquest®, № по кат. 36316). После попадания внутрь клетки липофильные блокирующие группы Fluo-8 отщепляются под действием неспецифических клеточных эстераз с образованием в результате этого отрицательно заряженного флуоресцентного красителя, который остается внутри клеток. Его флуоресценция усиливается при связывании с ионами кальция. Когда клетки HEK-293/Р2Х7 стимулируют Bz-ATP, Са2+ поступает в клетки и сигнал флуоресценции от Fluo-8 NW (без стадии отмывки) возрастает. Этот краситель имеет спектр поглощения, сочетаемый с возбуждением при 488 нм под действием в качестве источников возбуждения аргоновых лазеров, и длина волны его излучения находится в диапазоне 515-575 нм.
Клетки HEK-293, стабильно трансфицированные геном, кодирующим Р2Х7 рецептор, рассевали в течение ночи в ростовой среде в количестве 10000-20000 клеток/лунка в 384-луночном планшете. Через 24 часа среду удаляли, и клетки предварительно нагружали Fluo-8 NW в количестве 20 мкл/лунка в течение 1 часа при КТ. Затем с использованием системы флуоресцентного лазерного визуализирующего планшетного ридера FLIPRTETRA вводили тестируемые соединения в количестве 10 мкл/лунка и референсный антагонист А438079 в 3-кратной концентрации и в течение пяти минут проводили мониторинг кинетики ответа. С применением FLIPRTETRA осуществляли второе введение, используя референсный активатор в количестве 15 мкл/лунка в 3-кратной концентрации относительно ЕС80 (концентрация, которая приводит к 80% максимального ответа) для Bz-ATP, и регистрировали сигнал испускаемой флуоресценции в течение еще трех минут. Весь эксперимент проводили в буфере для анализа с низким содержанием двухвалентных катионов (0,3 мМ Са2+ и 0 мМ Mg2+). Эффект тестируемых соединений определяли в виде процента ингибирования относительно референсного антагониста и соответственно рассчитывали значения IC50 (концентрация полумаксимального ингибирования).
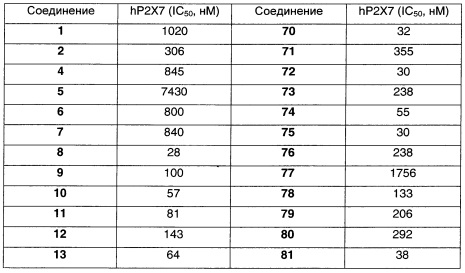
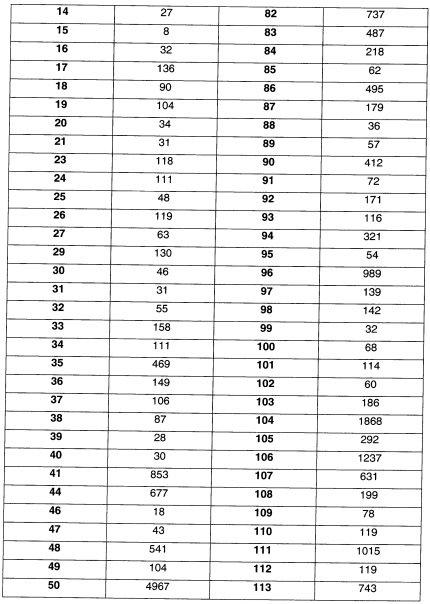
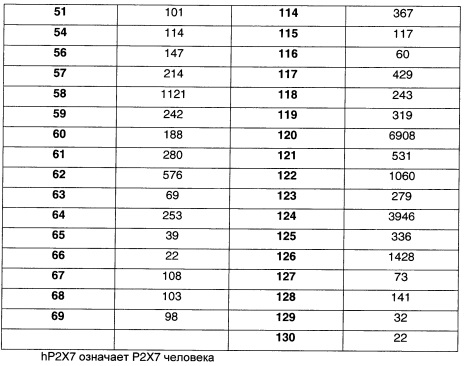
С использованием анализа поглощения YO-PRO®-1 было обнаружено, что описанные в примерах соединения по изобретению являются ингибиторами P2X7.c
YO-PRO®-1 представляет собой флуоресцентный ДНК-связывающий краситель с молекулярной массой (MW) 374 Да (Molecular Probes®, №по кат. Y3603). В основе этого метода лежит предполагаемая способность YO-PRO®-1 проникать через расширенную пору или «форму большой поры (large pore form)», образуемую Р2Х7 рецептором, и связываться с внутриклеточной ДНК, вследствие чего многократно усиливается интенсивность его флуоресценции. Этот краситель имеет спектр поглощения, сочетаемый с возбуждением при 488 нм под действием в качестве источников возбуждения аргоновых лазеров, и длина волны его излучения находится в диапазоне 515-575 нм. Цель этого анализа заключалась в подтверждении взаимодействия антагонистов с Р2Х7 рецепторами посредством использования альтернативного считывания показаний от Са2+-чувствительных флуоресцентных красителей.
Клетки HEK-293, стабильно трансфицированные геном, кодирующим Р2Х7 рецептор, рассевали в течение ночи в ростовой среде в количестве 20000 клеток/лунка в 384-луночном планшете. Через 24 часа среду удаляли, клетки промывали буфером для анализа с низким содержанием двухвалентных катионов (0,3 мМ Са2+ и 0 мМ Mg2+) и затем предварительно нагружали 5 мкМ красителем YO-PRO®-1 в количестве 20 мкл/лунка. Сразу же начинали измерение флуоресценции на системе FLIPRTETRA. Затем с использованием FLIPRTETRA вводили тестируемые соединения в количестве 10 мкл/лунка и референсный антагонист А438079 в 3-кратной концентрации и в течение пяти минут проводили мониторинг кинетики ответа. С применением FLIPRTETRA осуществляли второе введение, используя Bz-ATP в количестве 10 мкл/лунка в концентрации 3×ЕС80 (30 мкМ), и регистрировали сигнал испускаемой флуоресценции в течение еще 60 минут. Весь эксперимент проводили в буфере для анализа с низким содержанием двухвалентных катионов (0,3 мМ Са2+ и 0 мМ Mg2+).
Эффект тестируемых соединений определяли в виде процента ингибирования относительно референсного антагониста и соответственно рассчитывали значения IC50.
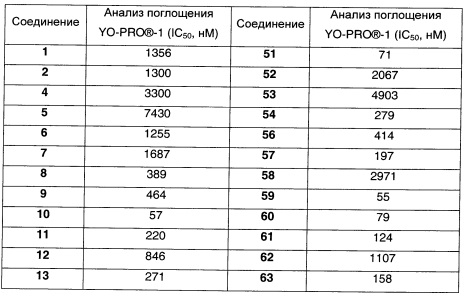
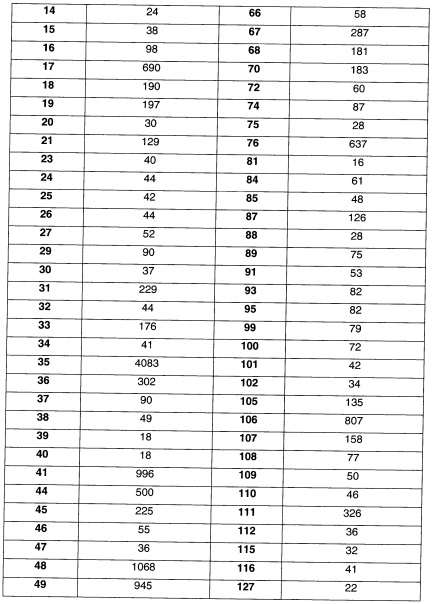

В анализе с применением автоматизированного метода локальной фиксации потенциал (пэтч-кламп) было обнаружено, что описанные в примерах соединения по изобретению активны в отношении канала Р2Х7 человека.
Чтобы провести прямой мониторинг блокирования канала Р2Х7, был разработан электрофизиологический анализ, который был осуществлен на автоматизированном приборе для электрофизиологических исследований QPatch16X.
Клетки HEK-293, экспрессирующие каналы Р2Х7, культивировали в модифицированной минимальной поддерживающей среде Игла (ЕМЕМ).
За 72 часа до начала эксперимента во флаконы Т225 рассевали 5 миллионов клеток. Непосредственно перед экспериментом клетки дважды промывали, открепляли от стенок флаконов, используя смесь трипсина и EDTA (этилендиаминтетрауксусная кислота), ресуспендировали в растворе для суспендирования и помещали в QPatch16X.
В день эксперимента из растворов соединений (20 мМ в 100%-ном DMSO), которые хранили при -20°С, готовили разведенные растворы (первое разведение 1:20 в 100% DMSO для получения 1 мМ концентрированного раствора, затем следует получение 1 мкМ раствора во внеклеточном растворе + серийное разведение 1:10).
Стандартные эксперименты с фиксацией потенциала в конфигурации «целая клетка» проводили при комнатной температуре. Для этих экспериментов использовали многоканальную технологию и данные регистрировали при 2 кГц.
Внутриклеточный раствор содержал (в мМ): 135 CsF, 10 NaCl, 1 EGTA (этиленгликольтетрауксусная кислота), 10 HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), (подводили до pH 7,2 с использованием CsOH), в то время как внеклеточный раствор содержал (в мМ): 145 NaCl, 4 KCl, 0,5 MgCl2, 1 CaCl2, 10 HEPES, 10 Glc (глюкоза) (подводили до pH 7,2 с использованием NaOH).
После установления герметичного контакта и перехода к конфигурации «целая клетка» потенциал клеток фиксировали при -80 мВ. Ток через канал P2XR7 вызывали, апплицируя только 100 мкМ BzATP (4 раза), а затем в присутствии возрастающих концентраций исследуемых соединений (1, 10, 100 и 1000 нМ).
Периоды 5-8 преинкубации соответствуют возрастающим концентрациям представляющего интерес соединения (1, 10, 100 и 1000 нМ), как показано на фигуре (протокол аппликации).
Измеряли максимальный входящий ток, вызванный BzATP в отсутствие или в присутствии возрастающих концентраций исследуемых соединений, и выполняли нормирование. Возможный агонистический эффект измеряли в виде % от контроля и в виде величины IC50, определенной посредством аппроксимации данных для кривых зависимости доза-ответ, используя следующее уравнение:
Y = 100 / (1 + 10 ^ ((LogIC50 - Х) * коэффициент Хилла)),
где:
X: логарифм концентрации;
Y: нормированный ответ, уменьшается от 100% до 0% по мере увеличения X;
LogIC50: в тех же логарифмических единицах, что и X;
коэффициент Хилла (HS): или коэффициент наклона, безразмерная величина.
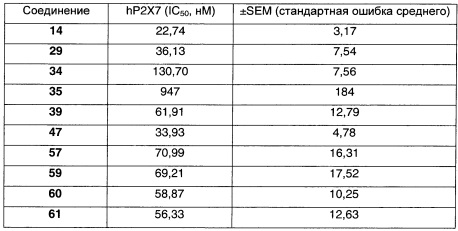
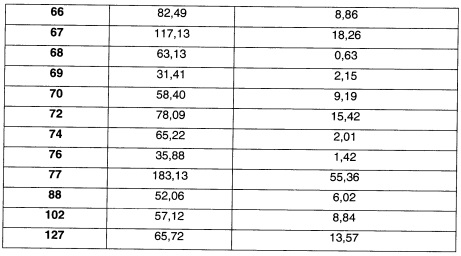
| название | год | авторы | номер документа |
|---|---|---|---|
| Замещенные производные N-[2-(4-феноксипиперидин-1-ил)-2-(1,3-тиазол-5-ил)этил]бензамида и N-[2-(4-бензилоксипиперидин-1-ил)-2-(1,3-тиазол-5-ил)этил]бензамида в качестве антагонистов P2X7 рецептора | 2017 |
|
RU2755705C2 |
| ИНДОЛЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-HT | 2007 |
|
RU2449990C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И БЕНЗОКСАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2512283C2 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| АНТАГОНИСТЫ CRF-РЕЦЕПТОРОВ И СПОСОБЫ, ОТНОСЯЩИЕСЯ К НИМ | 2004 |
|
RU2394035C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И БЕНЗОМОРФОЛИНА В КАЧЕСТВЕ МОДУЛЯТОРА МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2517181C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ ПАРАЗИТАРНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2793122C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ГИСТОНДЕЗАЦЕТИЛАЗЫ 6 | 2018 |
|
RU2797613C2 |
| ТИАЗОЛИЛФЕНИЛБЕНЗОЛСУЛЬФОНАМИДОПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2012 |
|
RU2606497C2 |
| НАПАДИЗИЛАТНАЯ СОЛЬ АНТАГОНИСТА МУСКАРИНОВОГО М3-РЕЦЕПТОРА | 2008 |
|
RU2459810C2 |

Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, включая их любую стереохимически изомерную форму, где n равно 1 или 2; Y представляет собой кислород или серу; каждый из R1 и R2 независимо выбран из группы, состоящей из водорода, дейтерия, С1-С4алкила, возможно замещенного гидрокси или галогеном, такого как гидроксиметил, фторметил, дифторметил, трифторметил, С3-С6циклоалкила, возможно замещенного гидрокси или галогеном, или С1-С4алкилокси; каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, галогена, С1-С4алкила, С1-С4алкилокси, NR9R10, где R9 и R10 независимо представляют собой водород или С1-С4алкил, или 1,2-тиазолидин-1,1-диона; либо две группы R3 или группы R3 и R4 вместе образуют шестичленное гетероциклическое кольцо, содержащее атом азота; R5 выбран из водорода, галогена или представляет собой гетероциклическое кольцо, выбранное из пиримидин-2-ила, пиридин-2-ила или пиразин-2-ила, возможно замещенное галогеном, С1-С4алкилом; R7 представляет собой водород или С1-С4алкил; радикал формулы (а) представляет собой возможно замещенное азетидиновое, пиперидиновое, морфолиновое, оксазепановое или 1,2,3,4-тетрагидроизохинолиновое кольцо, где каждый из R6 независимо выбран из группы, состоящей из водорода, галогена, С3-С6спироциклоалкила, фенила, пиразолила, фенокси или бензилокси, где фенильная или пиразолильная группа возможно замещена галогеном, С1-С4алкилом, фторметилом, дифторметилом, трифторметилом. Соединения формулы (I) обладают антагонистическими свойствами в отношении Р2Х7 рецептора. 4 н. и 11 з.п. ф-лы, 1 ил., 2 табл., 26 пр.
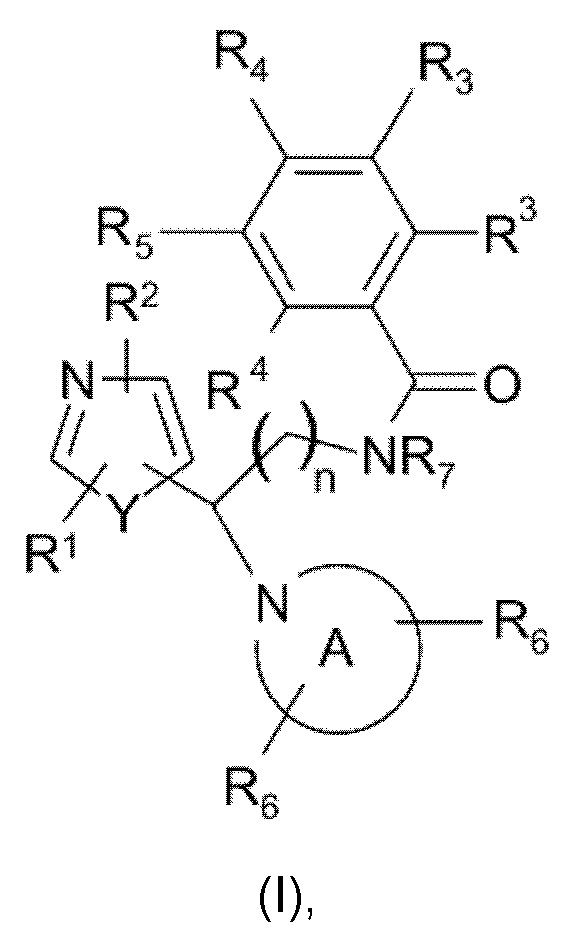 ,
,  (а)
(а)
1. Соединение следующей формулы (I) или его фармацевтически приемлемая соль:
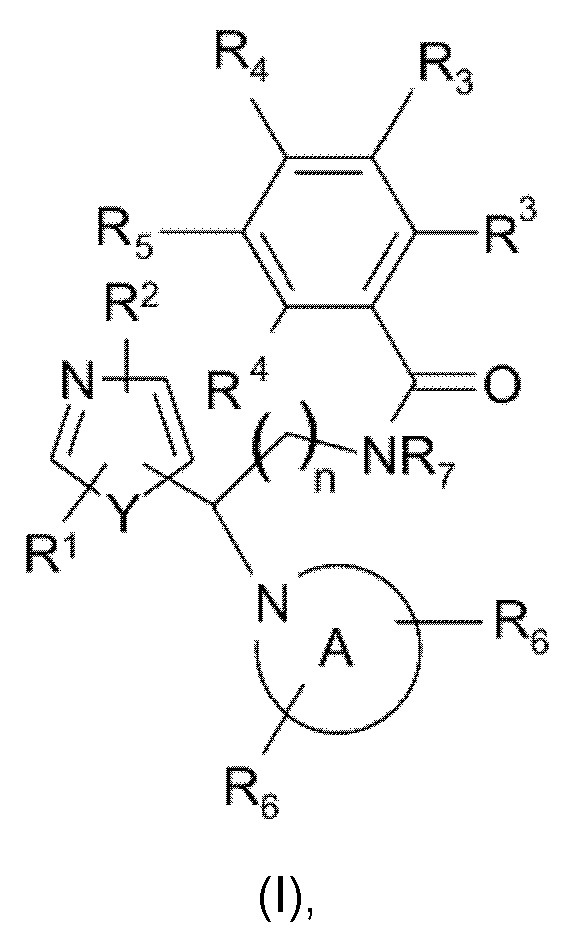
включая их любую стереохимически изомерную форму, где
n равно 1 или 2;
Y представляет собой кислород или серу;
каждый из R1 и R2 независимо выбран из группы, состоящей из водорода, дейтерия, С1-С4алкила, возможно замещенного гидрокси или галогеном, такого как гидроксиметил, фторметил, дифторметил, трифторметил, С3-С6циклоалкила, возможно замещенного гидрокси или галогеном, или С1-С4алкилокси;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, галогена, С1-С4алкила, С1-С4алкилокси, NR9R10, где R9 и R10 независимо представляют собой водород или С1-С4алкил, или 1,2-тиазолидин-1,1-диона; либо две группы R3 или группы R3 и R4 вместе образуют шестичленное гетероциклическое кольцо, содержащее атом азота;
R5 выбран из водорода, галогена или представляет собой гетероциклическое кольцо, выбранное из пиримидин-2-ила, пиридин-2-ила или пиразин-2-ила, возможно замещенное галогеном, С1-С4алкилом;
R7 представляет собой водород или С1-С4алкил;
радикал
представляет собой возможно замещенное азетидиновое, пиперидиновое, морфолиновое, оксазепановое или 1,2,3,4-тетрагидроизохинолиновое кольцо, где каждый из R6 независимо выбран из группы, состоящей из водорода, галогена, С3-С6спироциклоалкила, фенила, пиразолила, фенокси или бензилокси, где фенильная или пиразолильная группа возможно замещена галогеном, С1-С4алкилом, фторметилом, дифторметилом, трифторметилом.
2. Соединение формулы (I) по п. 1, где R7 представляет собой водород и n равно 1.
3. Соединение формулы (I) по п. 1, где оба R1 и R2 представляют собой водород или один из них представляет собой водород, а другой представляет собой метил, этил, пропил, трет-бутил, возможно замещенные гидрокси или фтором, С3-С6циклоалкил, возможно замещенный гидрокси или фтором.
4. Соединение формулы (I) по п. 1, где R5 представляет собой водород и каждый из R3 и R4 независимо представляет собой водород, галоген, С1-С4алкил, С1-С4алкилокси, NR9R10, где R9 и R10 независимо представляют собой водород или С1-С4алкил, или 1,2-тиазолидин-1,1-дион; либо две группы R3 или группы R3 и R4 вместе образуют шестичленное гетероциклическое кольцо, содержащее атом азота.
5. Соединение формулы (I) по п. 1, где R4 представляет собой водород, R3 в мета-положении представляет собой водород и R3 в орто-положении выбран из группы, состоящей из галогена или С1-С4алкила, и R5 представляет собой гетероциклическое кольцо, выбранное из пиримидин-2-ила, пиридин-2-ила или пиразин-2-ила, возможно замещенное галогеном.
6. Соединение формулы (I) по любому из пп. 1-5, где кольцо А выбрано из группы, состоящей из
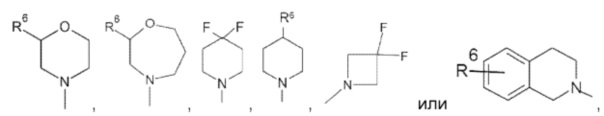 где R6 представляет собой водород, галоген, бензилокси или фенокси, фенил, пиразолил, где фенильная или пиразолильная группа возможно замещена галогеном, предпочтительно фтором.
где R6 представляет собой водород, галоген, бензилокси или фенокси, фенил, пиразолил, где фенильная или пиразолильная группа возможно замещена галогеном, предпочтительно фтором.
7. Соединение, выбранное из группы, состоящей из:

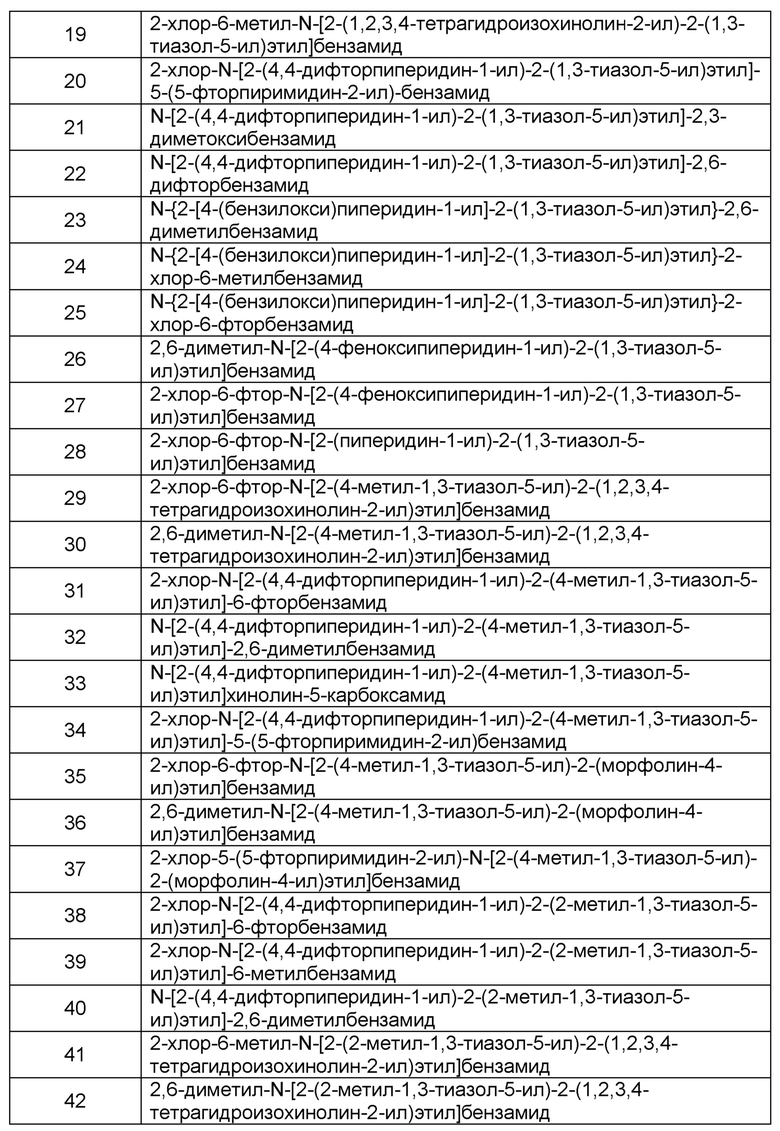

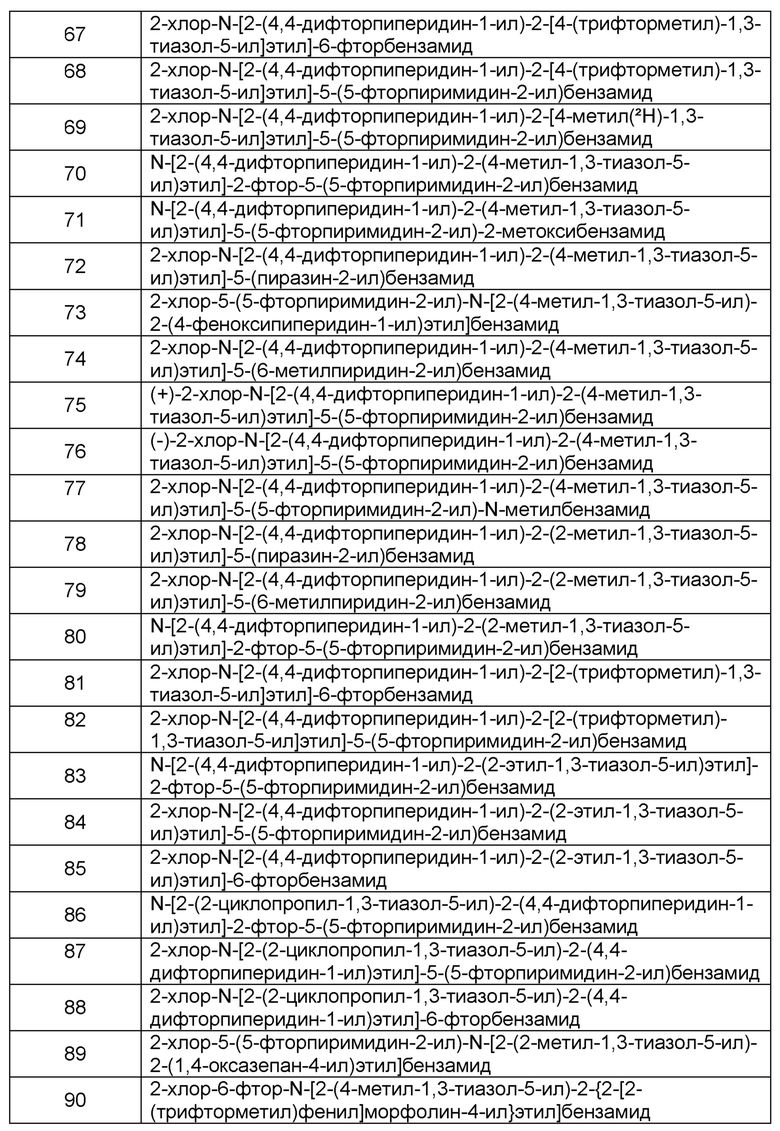

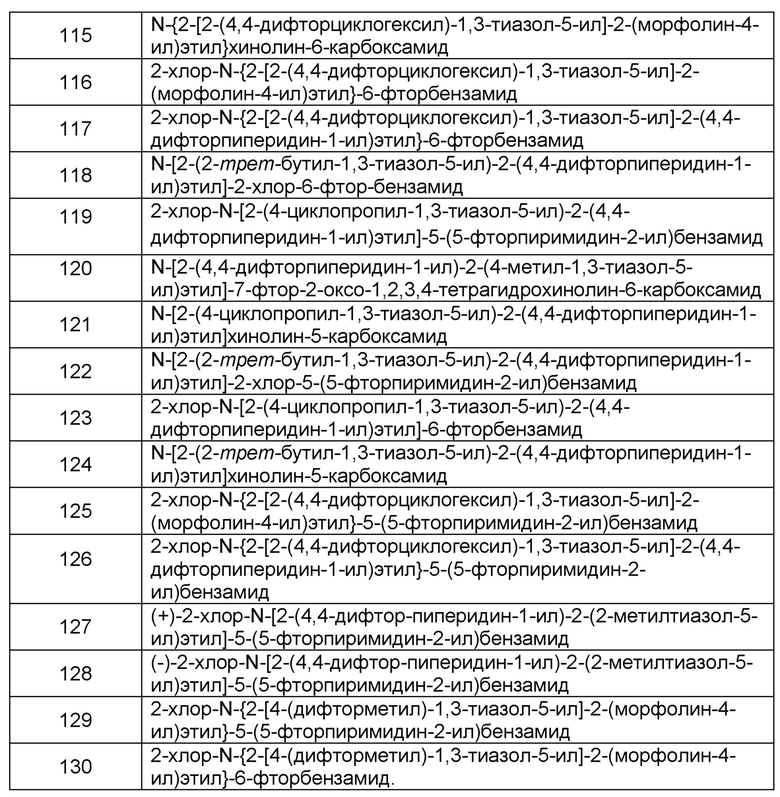
8. Способ получения соединения формулы (I) по п. 1, включающий стадию взаимодействия соединения формулы (II):
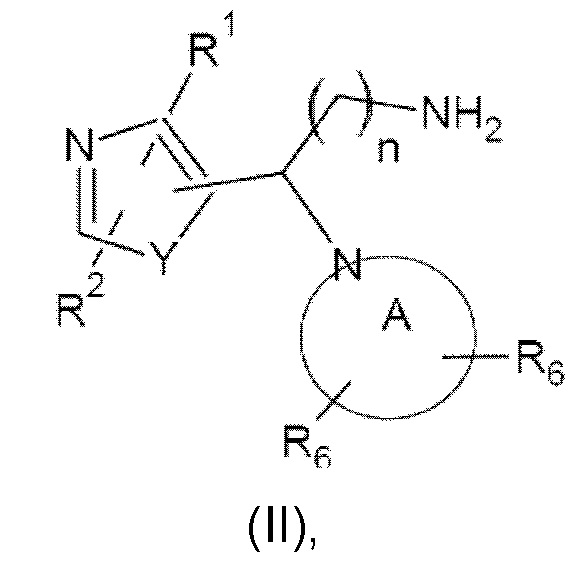
где значения n, Y, А и R1, R2 и R6 являются такими, как определено в п. 1, с соединением формулы (III)
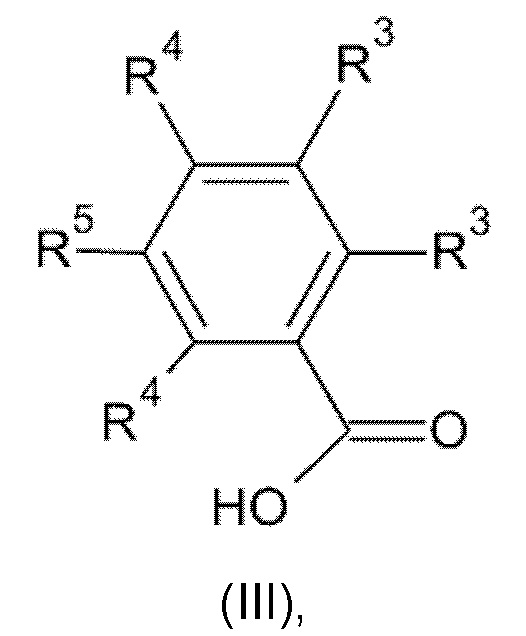
где значения R3, R4 и R5 являются такими, как определено в п. 1; или с соединением формулы (IIIа):
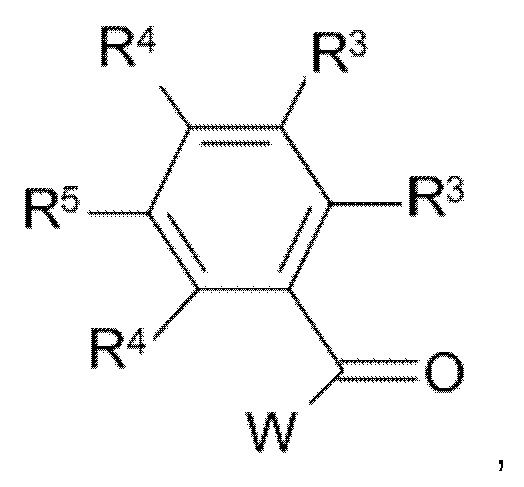
где значения R3, R4 и R5 являются такими, как определено в п. 1, и W представляет собой подходящую уходящую группу, и возможно превращение полученного соединения формулы (I) в соль присоединения и/или получение его стереохимически изомерных форм.
9. Фармацевтическая композиция, обладающая антагонистической активностью в отношении рецептора Р2Х7, содержащая эффективное количество соединения формулы (I) по любому из пп. 1-7 и фармацевтически приемлемый разбавитель и/или носитель.
10. Соединение формулы (I) по пп. 1-7 для использования в качестве лекарственного средства, обладающего антагонистической активностью в отношении рецептора Р2Х7.
11. Соединение формулы (I) по пп. 1-7 для использования в лечении состояний или заболеваний, выбранных из опосредуемых Р2Х7 рецепторами состояний или заболеваний.
12. Соединение формулы (I) по п. 11 для использования в предупреждении и/или лечении болезни Альцгеймера, деменции с тельцами Леви, лобно-височной деменции, таупатий, амиотрофического бокового склероза, рассеянного склероза, болезни Паркинсона и синдромов паркинсонизма, болезни Хантингтона, нейропатии Шарко-Мари-Тута; травмы головы, церебральной ишемии, менингита, когнитивных и психических расстройств; расстройств сна; невропатической и воспалительной боли, хронической боли, мигрени, боли при рассеянном склерозе, ВИЧ-обусловленной нейропатии, ВИЧ-индуцированного нейровоспаления и поражения ЦНС, эпилепсии и эпилептического статуса, воспалительных процессов скелетно-мышечной системы, фиброза печени, расстройств желудочно-кишечного тракта, расстройства мочеполовых путей, офтальмологических заболеваний, хронической обструктивной болезни легких (COPD), аутоиммунных заболеваний, почечного синдрома; мышечной дистрофии.
13. Соединение формулы (I) по п. 12, где психическое расстройство выбрано из депрессии, биполярных расстройств, тревоги и шизофрении.
14. Соединение формулы (I) по п. 12, где расстройство желудочно-кишечного тракта выбрано из неспецифического язвенного колита, синдрома раздраженного кишечника и болезни Крона.
15. Соединение формулы (I) по п. 12, где офтальмологическое заболевание выбрано из возрастной макулярной дегенерации и ретинопатий.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| ПРОИЗВОДНЫЕ ИНДОЛА ИЛИ БЕНЗИМИДАЗОЛА ДЛЯ МОДУЛЯЦИИ IkB-КИНАЗЫ | 2003 |
|
RU2318820C2 |

