Область изобретения
В настоящем изобретении раскрыты новые 5-замещенные производные 7-амино-[1,3]тиазоло[4,5-d]пиримидина, а также способы их получения, фармацевтические композиции, содержащие их, и их применение в терапии.
Предшествующий уровень техники
Хемокины играют важную роль в иммунном и воспалительном ответах при различных заболеваниях и расстройствах, включая астму, атеросклероз и аллергические заболевания, а также при аутоиммунных патологиях, таких как ревматоидный артрит и рассеянный склероз. Эти небольшие, секретируемые молекулы являются увеличивающимся надсемейством 8-14 кДа белков, отличающихся консервативным цистеиновым мотивом. В настоящее время надсемейство хемокинов включает четыре группы, имеющие характеристические структурные мотивы, семейства С-Х-С, С-С и С-Х3-С и ХС. С-Х-С и С-С семейства обладают подобием последовательностей и отличаются друг от друга одной аминокислотной вставкой между NН-проксимальной парой цистеиновых остатков. С-Х3-С семейство отличается от других двух семейств тем, что имеет вставку из трех аминокислот между NH-проксимальной парой цистеиновых остатков. Наоборот, у членов ХС семейства отсутствует один из первых двух цистеиновых остатков.
С-Х-С хемокины включают несколько эффективных хемоаттрактантов и активаторов нейтрофилов, таких как интерлейкин-8 (IL-8) и нейтрофил-активирующий пептид 2 (NAP-2).
С-С хемокины включают эффективные хемоаттрактанты моноцитов, лимфоцитов и нейтрофилов. Примеры включают хемотаксические белки 1-3 (МСР-1, МСР-2 и МСР-3) моноцитов человека, RANTES (регулируемые при активации, экспрессируемые и секретируемые нормальными Т-клетками), эотаксин и макрофагальные воспалительные белки 1α и 1β(М1Р-1а и М1Р-1β).
С-Х3-С хемокин (также известный как фракталкин) представляет собой эффективный хемоаттрактант и активатор микроглии в центральной нервной системе (CNS), а также моноцитов, Т-клеток, NK- клеток и тучных клеток.
Исследования продемонстрировали, что действия хемокинов опосредованы подсемействами рецепторов, сопряженных с G-белком, в число которых входят рецепторы, обозначенные как CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для С-С семейства); CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для С-Х-С семейства) и СХ3СР1 для С-Х3-С семейства. Эти рецепторы являются хорошими мишенями для разработки лекарственных средств, так как агенты, которые модулируют эти рецепторы, будут полезны в лечении расстройств и заболеваний, таких как упомянутые выше.
В WO 01/58907 раскрыты некоторые 2-замещенные производные 4-аминотиазолопиримидина, которые полезны в качестве антагонистов рецепторов, связанных с семействами С-Х-С и С-С хемокинов, в частности как антагонисты CXCR2 рецептора.
Настоящее изобретение относится к группе соединений, которые близки к соединениям, раскрытым в WO 01/58907, но имеют тип структуры, конкретно не полученный там. По сравнению с примерами, раскрытыми в WO 01/58907, соединения по настоящему изобретению демонстрируют неожиданно полезные свойства как антагонисты СХ3СР1 рецептора.
Описание изобретение
В настоящем изобретении предложены соединения формулы (I)

где R1 представляет собой СН3 или СF3;
R2 представляет собой галогено, CN или C1-C6алкил;
R3 представляет собой Н или СН3;
R4 представляет собой Н или СН3;
n равен 0,1 или 2;
в виде свободного основания или его фармацевтически приемлемых соли, сольвата или сольвата его соли.
В одном воплощении изобретения предложены соединения формулы (I), где n равен 1.
В другом воплощении изобретения предложены соединения формулы (I), где R1 представляет собой СН3.
В еще одном воплощении изобретения предложены соединения формулы (I), где R2 представляет собой галогено или CN.
В еще одном воплощении изобретения предложены соединения формулы (I), где R2 представляет собой F или Cl.
В еще одном воплощении изобретения предложены соединения формулы (I), где R2 представляет собой CN.
В еще одном воплощении изобретения предложены соединения формулы (I), где n равен 1; R1 представляет собой СН3 и R2 представляет собой F, Cl или CN.
В еще одном воплощении изобретения предложены соединения формулы (I), в которых пиридин присоединен по своему положению 5 и несет Cl в положении 2.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 2 и несет CN в положении 4.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 2 и несет F в положении 5.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 2 и несет Cl по положению 5.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 2 и несет F в положении 3.
В еще одном воплощении изобретения предложены соединения формулы (I), где пиридин присоединен по своему положению 4 и несет С1 в положении 3.
В еще одном воплощении изобретения предложены соединения формулы (I), где R3 представляет собой Н.
В еще одном воплощении изобретения предложены соединения формулы (I), где R4 представляет собой СН3.
В еще одном воплощении изобретения предложены соединения формулы (I), выбранные из
(2R)-2-[(2-амино-5-{[1-(5-фторпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-амино-5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-амино-5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-амино-5-{[1-(3-хлорпиридин-4-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-амино-5-{[(1S)-1-(3-хлорпиридин-4-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-амино-5-{[(1S)-1-(3-хлорпиридин-4-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-амино-5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]пиpимидин-7-ил)aминo]-4-мeтилпeнтaн-1-oлa;
(2R)-2-[(2-амино-5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио}[1,3]тиазоло[4,5-d]пиpимидин-7-ил)(мeтил)aминo]пeнтaн-1-oлa;
(2R)-2-[(2-амино-5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ола и
2-{(1S)-1-[(2-амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}изоникотинонитрила
в виде свободного основания или его фармацевтически приемлемых соли, сольвата или сольвата его соли.
Соединения формулы (I) могут существовать в стереоизомерной и/или таутомерной формах. Следует понимать, что все энантиомеры, диастереомеры, рацематы, таутомеры и их смеси включены в объем данного изобретения.
По сравнению с соединениями, раскрытыми в WO 01/58907, соединения по настоящему изобретению отличаются присутствием разветвленного тиоалкилпиридила в положении 5 тиазолопиримидиновой кольцевой системы. То есть соединения по настоящему изобретению содержат группу R1, которая не является водородом.
В соответствии с изобретением авторы изобретения также предлагают способ получения соединения формулы (I) или его фармацевтически приемлемой соли, который включает
а) взаимодействие соединения формулы (II)

где R3 и R4 такие, как они определены в формуле (I)
с соединением формулы (III)

(111) где R1, R2, и n такие, как они определены в формуле (I), и L1 представляет собой уходящую группу, или
б) взаимодействие соединения формулы (IV)
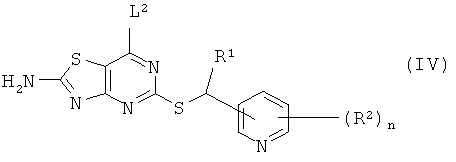
где R1, R2 и n такие, как они определены в формуле (I), и L2 представляет собой уходящую группу,
с соединением формулы (V)

где R3 и R4 такие, как они определены в формуле (I),
и, где это необходимо, превращение полученного соединения формулы (I), или другой его соли, в его фармацевтически приемлемую соль или превращение полученного соединения формулы (I) в другое соединение формулы (I) и, где это желательно, превращение полученного соединения формулы (I) в его оптический изомер.
В способе (а), реагенты (II) и (III) объединяют вместе в подходящем органическом растворителе, таком как диметилсульфоксид (DMSO), ацетонитрил или 1-метил-2-пирролидинон (NMP). Взаимодействие возможно осуществляют в присутствии добавленного органического или неорганического основания, такого как триэтиламин, N,N-диизопропилэтиламин (DIPEA) или натрия гидрид. Взаимодействие возможно осуществляют в присутствии мягкого восстановителя, такого как боргидрид натрия. Взаимодействие проводят при подходящей температуре, обычно между комнатной температурой и точкой кипения растворителя. Взаимодействие в общем случае продолжалось в течение промежутка времени от одного часа до одной недели или пока анализ не указывал, что образование целевого продукта закончено.
В способе (б) реагенты (IV) и (V) объединяют вместе в подходящем органическом растворителе, таком как тетрагидрофуран, ацетонитрил, диметилсульфоксид или 1-метил-2-пирролидинон. Взаимодействие возможно осуществляют в присутствии добавленного основания. Данное основание может быть органическим основанием, таким как триэтиламин или N,N-диизопропилэтиламин, или неорганическим основанием, таким как калия карбонат. Взаимодействие проводят при подходящей температуре, обычно между комнатной температурой и точкой кипения растворителя, но возможно при более высоких температурах, если используют герметичный реакционный сосуд. Взаимодействие в общем случае продолжается в течение периода времени от примерно одного часа до одной недели или пока анализ не указывал, что образование целевого продукта закончено.
Подходящими уходящими группами L1 и L2 являются галогеновые, в частности хлор или бром. В одном воплощении каждый из L1 и L2 представляет собой хлор.
Специалисту в данной области техники очевидно, что в указанных выше способах может быть желательно или необходимо защищать амин, гидроксил или другую потенциально реакционноспособную группу. Подходящие защитные группы и подробности способов присоединения и удаления таких групп в общем случае хорошо известны в данной области техники. Смотри, например, "Protective Groups in Organic Synthesis", 3 изд. (1999), Greene and Wuts.
Настоящее изобретение включает соединения формулы (I) в форме солей. Подходящие соли включают соли, образованные органическими или неорганическими кислотами, или органическими или неорганическими основаниями. Такие соли обычно бывают фармацевтически приемлемыми, хотя соли фармацевтически неприемлемых кислот или оснований можно использовать в получении и очистке интересующего соединения.
Соли соединения формулы (I) могут быть образованы посредством взаимодействия свободного соединения или его соли, энантиомера или рацемата, с одним или более эквивалентами соответствующей кислоты или основания. Взаимодействие можно проводить в растворителе или среде, в которой соль нерастворима, или в растворителе, в котором соль растворима, например в воде, диоксане, этаноле, тетрагидрофуране или диэтиловом эфире, или в смеси растворителей, которые могут быть удалены в вакууме или посредством лиофильной сушки. Взаимодействие также может представлять собой обменный процесс или может быть проведено на ионообменной смоле.
Соединения формулы (II) либо известны из, например, WO 01/58907, WO 01/25242 или WO 02/76990, или могут быть получены с использованием известных способов, которые очевидны специалисту в данной области техники.
Соединения формулы (IV) могут быть получены с использованием способов, аналогичных описанным, например, в WO 00/09511, или с использованием других известных способов, которые очевидны специалисту в данной области техники.
Соединения формул (III) и (V) либо имеются в продаже, либо известны в литературе, или могут быть получены с использованием известных способов, которые очевидны специалисту в данной области техники.
Подходящие конкретные способы получения соединений формул (II), (III), (IV) и (V) подробно изложены в разделе Примеры настоящей заявки, и такие способы являются конкретными воплощениями способов по изобретению.
Например, соединения формулы (II) и затем соединения формулы (I), могут быть получены, как показано на схеме 1:
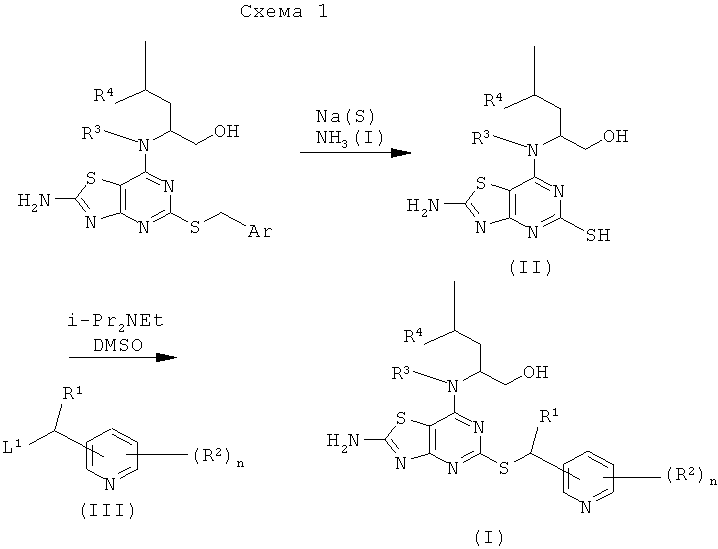
Промежуточные соединения можно использовать как таковые или в защищенной форме. Подходящие защитные группы и подробности способов присоединения и удаления таких групп в общем случае хорошо известны в данной области техники. Смотри, например, "Protective Groups in Organic Synthesis", 3 изд. (1999), Greene and Wuts.
Соединения по изобретению и промежуточные соединения могут быть выделены из реакционных смесей и, если необходимо, дополнительно очищены с использованием стандартных методик.
Соединения формулы (I) могут существовать в стереоизомерных формах. Следовательно, все энантиомеры, диастереомеры, рацематы и их смеси включены в объем изобретения. Различные оптические изомеры могут быть выделены путем разделения стереоизомерной смеси соединений с использованием традиционных методик, например фракционной кристаллизации или HPLC. Альтернативно различные оптические изомеры могут быть получены непосредственно с использованием оптически активных исходных веществ.
Соединения формулы (I) содержат два стереогенных центра и могут, таким образом, существовать в четырех отдельных стереоизомерных формах, как показано в формулах (Ia)-(Id)


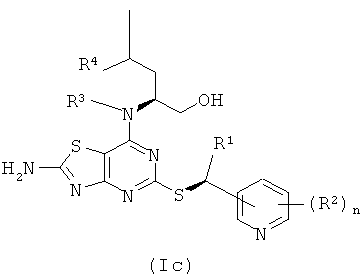
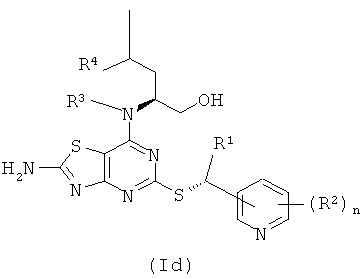
Все такие четыре стереоизомера и любые их смеси включены в объем изобретения. В одном воплощении соединения формулы (I) имеют стереохимию, показанную в формуле (Ia). В другом воплощении соединения формулы (I) имеют стереохимию, показанную в формуле (Ib).
Промежуточные соединения также могут существовать в стереоизомерных формах и их можно использовать в виде очищенных знантиомеров, диастереомеров, рацематов или их смесей.
В данной заявке термин "С1-C6-алкил" включает алкильные группы как с прямой, так и с разветвленной цепью, а также циклические алкильные группы. С1-C6-алкил содержит от 1 до 6 атомов углерода и может представлять собой, но не ограничен ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, трет-пентил, неопентил, н-гексил, изогексил или циклогексил.
В данной заявке термин "галогено" или "галоген" относится к фторо, хлоро, бромо и йодо.
Соединения формулы (I) и их фармацевтически приемлемые соли полезны, потому что они обладают фармакологической активностью как антагонисты СХ3СR1 рецепторов. В частности, по сравнению с соединениями, приведенными в виде конкретных примеров в WO 01/58907, соединения формулы (I) по настоящему изобретению обладают значительно повышенными эффективностями в отношении ингибирования СХ3СR1 рецептора и/или пониженными эффективностями в отношении ингибирования CXCR2 рецептора. Предпочтительные соединения по настоящему изобретению проявляют как улучшенную эффективность в отношении ингибирования СХ3СR1, так и уменьшенную эффективность в отношении ингибирования CXCR2.
В одном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в качестве лекарственного средства.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики заболеваний или состояний, при которых полезен антагонизм СХ3СR1 рецептора.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики нейродегенеративных расстройств, демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, болезни периферических артерий, ревматоидного артрита, легочных заболеваний, таких как COPD (хроническая обструктивная болезнь легких), астмы или боли.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики рассеянного склероза (MS).
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики атеросклероза путем предупреждения и/или уменьшения образования новых атеросклеротических поражений или бляшек и/или путем предупреждения или замедления развития существующих поражений и бляшек.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики атеросклероза путем изменения состава бляшек с целью снижения риска разрыва бляшки и атеротромботических явлений.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики инсульта или транзиторного повреждения мозга (ТВI).
В соответствии с изобретением также предложен способ лечения или уменьшения риска заболеваний или состояний, при которых полезен антагонизм CX3CR1 рецептора, включающий введение субъекту, страдающему указанным заболеванием или состоянием или имеющему риск указанного заболевания или состояния терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ лечения или уменьшения риска нейродегенеративных расстройств, демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий, ревматоидного артрита, легочных заболеваний, таких как COPD, астмы или боли, у субъекта, страдающего указанным заболеванием или состоянием или имеющего риск указанного заболевания или состояния, включающий введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ лечения или уменьшения риска рассеянного склероза (MS) у субъекта, страдающего указанным заболеванием или состоянием или имеющего риск указанного заболевания или состояния, включающий введение этому субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ лечения или уменьшения риска атеросклероза путем предупреждения и/или уменьшения образования новых атеросклеротических поражений или бляшек и/или путем предупреждения или замедления развития существующих поражений и бляшек у субъекта, страдающего указанным заболеванием или состоянием или имеющего риск указанного заболевания или состояния, включающий введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ лечения или уменьшения риска атеросклероза путем изменение состава бляшек так, чтобы снизить риск разрыва бляшки и атеротромботических явлений у субъекта, страдающего указанным заболеванием или состоянием или имеющего риск указанного заболевания или состояния, включающий введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ лечения или уменьшения риска инсульта или транзиторного повреждения мозга (ТВI) у субъекта, страдающего указанным заболеванием или состоянием или имеющего риск указанного заболевания или состояния, включающий введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Соединения можно использовать в качестве монотерапии или в комбинациях, либо как профилактическое или терапевтическое лечение воспалительных состояний и заболеваний центральной нервной системы, таких как инсульт и транзиторное повреждение мозга (TBI) (Soriano et al. J.Neuroimmunology 2002, 125, 59-65).
В другом аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем для применения в лечении или профилактике заболеваний или состояний, при котором полезен антагонизм СХ3СR1 рецептора.
В другом аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем, для применения в лечении или профилактике нейродегенеративных расстройств, демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий, ревматоидного артрита, COPD, астмы или боли.
В другом аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем, для применения в лечении или профилактике рассеянного склероза.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем для применения в лечении или профилактике атеросклероза путем предупреждения и уменьшения образования новых атеросклеротических поражений и/или бляшек и/или путем предупреждения или замедления развития существующих поражений и бляшек.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем, для применения в лечении или профилактике атеросклероза путем изменения состава бляшек, так чтобы снизить риск разрыва бляшки и атеротромботических явлений.
Соединения формулы (I) и их фармацевтически приемлемые соли показаны для применения в лечении или профилактике заболеваний или состояний, при которых желательна модуляция активности СХ3СR1 рецептора. В частности, соединения показаны для применения в лечении нейродегенеративных расстройств или демиелинизирующего заболевания у млекопитающего, включая человека. Более конкретно, соединения показаны для применения в лечении рассеянного склероза. Соединения также показаны как полезные в лечении боли, ревматоидного артрита, остеоартрита, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий и легочной артериальной гипертензии.
Состояния, который могут быть конкретно упомянуты, представляют собой нейродегенеративные заболевания и деменции, например болезнь Альцгеймера, амиотрофический боковой склероз и другие заболевания двигательных нейронов, болезнь Крейтцфельдта-Якобса и другие прионные заболевания, ВИЧ-энцефалопатию, болезнь Хантингтона, фронтотемпоральную деменцию, деменцию с тельцами Леви и сосудистую деменцию; полинейропатии, например синдром Гийена-Барре, хроническую воспалительную демиелинизирующую полирадикулопатию, множественную моторную нейропатию и плексопатии; демиелинизацию ЦНС (центральной нервной системы), например острый рассеянный/геморрагический энцефаломиелит и подострый склерозирующий панэнцефалит; нервно-мышечные расстройства, например тяжелую миастению и синдром Ламберта-Итона; спинномозговые расстройства, например тропический спастический парапарез и синдром ригидности (stiff-man); паранеопластические синдромы, например дегенерация мозжечка и энцефаломиелит; травматическое повреждение мозга; мигрень; рак; отторжение аллотрансплантата; системный склероз; вирусные инфекции; заболевания, передаваемые паразитами, например малярию; периодонтальное заболевание; инфаркт миокарда; инсульт; коронарную болезнь сердца; ишемическую болезнь сердца; рестеноз; ревматоидный артрит; легочные заболевания, такие как COPD; астму или боль.
Соединения по изобретению также показаны для применения в лечении атеросклероза путем предупреждения и/или уменьшения образования новых атеросклеротических поражений или бляшек, и/или путем предупреждения или замедления развития существующих поражений и бляшек.
Соединения по изобретению также показаны для применения в лечении атеросклероза путем изменения состава бляшек так, чтобы снизить риск разрыва бляшки и атеротромботических явлений.
Соединения по изобретению также показаны для применения в лечении воспалительной болезни кишечника (IВГ), например болезни Крона и язвенного колита, путем индуцирования ремиссии и/или поддержания ремиссии IBD.
Профилактика, как ожидается, является особенно релевантной в отношении лечения субъектов, которые страдали ранее эпизодом, или иным образом оцениваются как имеющие повышенный риск, рассматриваемого заболевания или состояния. Субъекты с риском развития конкретного заболевания или состояния в общем случае включают субъектов, имеющих в семейном анамнезе это заболевание или состояние, или субъектов, которые были идентифицированы путем генетического тестирования или скрининга как особенно подверженные развитию этого заболевания или состояния.
Для вышеупомянутых терапевтических показаний вводимые дозировки будут, конечно, варьироваться в зависимости от используемого соединения, способа введения и требуемого лечения.
Однако в общем случае удовлетворительные результаты получают, когда соединения вводят с дозировкой твердой формы от 1 мг до 2000 мг в сутки.
Соединения формулы (I) и их фармацевтически приемлемые производные можно использовать сами по себе или в виде подходящей фармацевтической композиции, в которой соединение или производное представлено в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем. Введение может происходить, без ограничения ими, энтеральным (включая пероральный, сублингвальный или ректальный), интраназальным, внутривенным, местным или другими парентеральными путями. Традиционные методики выбора и получения подходящих фармацевтических композиций описаны, например, в "Pharmaceuticals - The Science of Dosage Form Designs", M.E.Aulton, Churchill Livingston, 1988. Фармацевтическая композиция предпочтительно содержит менее 80% и более предпочтительно менее 50% соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ получения такой фармацевтической композиции, включающий смешивание ингредиентов.
Изобретение дополнительно относится к комбинационному лечению, при котором соединение формулы (I) или его фармацевтически приемлемую соль, или фармацевтическую композицию, или композицию, содержащую соединение формулы (I), вводят одновременно или последовательно с терапией и/или агентом для лечения какого-либо кардио- и цереброваскулярного атеросклеротического расстройства и заболевания периферических артерий.
В частности, соединение формулы (I) или его фармацевтически приемлемая соль могут быть введены вместе с соединениями из одной или более из следующих групп:
1) противовоспалительные агенты, например,
а) NSAID (нестероидные противовоспалительные средства) (например, ацетилсалициловая кислота, ибупрофен, напроксен, флурбипрофен, диклофенак, индометацин);
б) ингибиторы синтеза лейкотриенов (5-LO ингибиторы, например AZD4407, зилейтон, ликофелон, CJ13610, CJ13454; FLAP ингибиторы, например BAY-Y-1015, DG-031, MK591, МК886, А81834; ингибиторы LTA4-гидролазы, например SC56938, SC57461A);
в) антагонисты лейкотриеновых рецепторов (например, СР195543, амелубан, LY293111, акколат, МК571);
2) антигипертензивные агенты, например,
а) бета-блокаторы (например, метопролол, атенолол, солатолол);
б) ингибиторы ангиотензин-превращающего фермента (например, каптоприл, рамиприл, хинаприл, эналаприл);
в) блокаторы кальциевых каналов (например верапамил, дилтиазем, фелодипин, амлодипин);
г) антагонисты ангиотензиновых рецепторов II типа (например, ирберсартан, кандесартан, телемисартан, лосартан);
3) антикоагулянты, например,
а) ингибиторы тромбина (например, ксимелагатран), гепарины, ингибиторы фактора Ха;
б) ингибиторы агрегации тромбоцитов (например, клопидогрел, тиклоридин, празугель, AZ4160);
4) модуляторы липидного метаболизма, например,
а) сенсибилизаторы к инсулину, такие как агонисты PPAR (рецепторов, активируемых пролифератором пероксисом) (например, пиоглитазон, розиглитазон, Галида, мураглитазаар, гефемрозил, фенофибрат);
б) ингибиторы HMG-CoA-редуктазы, статины (например симвастатин, правастатин, аторвастатин, розувастатин, флувастатин, питавастатин);
в) ингибиторы абсорбции холестерина (например эзетимиб);
г) ингибиторы IBAT (транспорта желчных кислот в подвздошной кишке) (например, AZD-7806);
г) агонисты LXR (печеночных Х-рецепторов) (например, GW-683965A, Т-0901317);
д) модуляторы FXR (фарнезиодных Х-рецепторов);
е) ингибиторы фосфолипазы;
5) антиангинальные средства, например нитраты и нитриты;
6) модуляторы окислительного стресса, например антиокислители (пробукол), ингибиторы миелопероксидазы.
Изобретение проиллюстрировано, но не в качестве ограничения, следующими примерами.
Общие способы
Все используемые растворители имели аналитическую чистоту и для реакций обычно использовали имеющиеся в продаже безводные растворители. Взаимодействия обычно проводили в инертной атмосфере азота или аргона.
1H и 13С ЯМР-спектры регистрировали при 400 МГц для протона и 100 МГц для углерода-13 либо на Varian Unity+400 ЯМР-спектрометре, оборудованном 5 мм ВВО зондом с Z-градиентами, или BrukerAvance 400 ЯМР-спектрометре, оборудованном 60 мкл двойным зондом для инверсного потока с Z-градиентами, или Bruker DPX400 ЯМР-спектрометре, оборудованном 4-ядерным зондом, оборудованным Z-градиентами. 600 МГц 1H ЯМР-спектры регистрировали на Bruker av600 ЯМР-спектрометре, оборудованном 5 мм BBI зондовой головкой с Z-градиентами. 300 МГц 1Н ЯМР-спектры регистрировали на Varian Gemini 300 NMR, оборудованном 5 мм ВВI зондовой головкой. Если в примерах конкретно не указано, спектр регистрировали при 400 МГц для протона и 100 МГц для углерода-13. Использовали следующие эталонные сигналы: средняя линия DMSO-d6 δ 2.50 (1Н), δ 39.51 (13С); средняя линия СD3OD δ 3.31 (1Н) или δ 49.15 (13С); ацетон-d6 2.04 (1Н), 206.5 (13С), и CDCl3 δ (7.26 (1Н), средняя линия CDCl3 77.16 (13С) (если не указано иное).
Энантиомерный избыток (ее) определяли посредством ГХ (газовой хроматографии) на колонке Cyclodex В (изотермическое элюирование 100°С) или на колонке Cyclosil В (температурный градиент 110-130°С). Диастереомерный избыток (de) определяли посредством HPLC (высокоэффективная жидкостная хроматография).
Масс-спектры регистрировали на Waters LCMS, состоящем из Alliance 2795 (LC) и ZQ одноквадрупольного масс-спектрометра. Масс-спектрометр был оборудован электрораспылительным ионным источником (ESI), оперирующим в режиме положительных или отрицательных ионов. Капиллярное напряжение составляло 3 кВ, и масс-спектрометр сканировал m/z 100-700 со временем сканирования 0,3 или 0,8 с. Разделения осуществляли либо на Waters X-Terra MS, С8-колонках (3,5 мкм, 50 или 100 мм (2,1 мм вн. д. (внутренний диаметр)), или на ScatecLab's АСЕ 3 AQ колонке (100 мм (2.1 мм вн.д.). Температуру колонки устанавливали на 40°С. Применяли линейный градиент, используя нейтральную или кислотную систему подвижной фазы, пропуская от 0% до 100% органической фазы за 4-5 минут, скорость потока 0,3 мл/мин. Нейтральная система подвижной фазы: ацетонитрил/[10 мМ NH4OAc (водн.)/МеСN (95:5)], или [10 мМ NH4OAc (водн.)/МеСN (1/9)]/ [10 мМ NH4OAc (водн.)/МеСМ (9/1)]. Кислая система подвижной фазы: [133 мМ НСООН (водн.) / MeCN (5/95)]/[8 мМ НСООН (водн.) / MeCN (98/2)].
Альтернативно масс-спектры регистрировали на GC-MS (GC 6890, 5973N MSD, Agilent Technology), используя колонку VF-5 MS (вн. д. 0,25 мм ×30 м, 0,25 мкм (Varian Inc.)). Применяли линейный градиент температуры (40°С-300°С), 25°С/минута. MS был оборудован CI (химическая ионизация) источником ионов и реагентным газом являлся метан. MS сканировали между m/z 50-500 и устанавливали скорость сканирования 3,25 сканов/с. HPLC-анализы осуществляли на Agilent НР1000 системе, состоящей из G1379A Micro Vacuum Degaser, G1312A бинарного насоса, G1367A Wellplate автоматического пробоотборника, С1316А термостатируемого колоночного отделения и G1315B диодно-матричного детектора. Колонка: X-Terra MS, Waters, 4,6×50 мм, 3,5 мкм. Устанавливали температуру колонки 40°С и скорость потока 1,5 мл/мин. Диодно-матричный детектор сканировал при 210-300 нм, шаг и ширину пика устанавливали 2 нм и 0,05 мин соответственно. Применяли линейный градиент, пропуская от 0% до 100% ацетонитрила за 4 мин. Подвижная фаза: ацетонитрил/10 мМ аммония ацетат в 5%-ном ацетонитриле в MilliQ воде.
Типичный метод обработки после взаимодействия состоял в экстракции продукта растворителем, таким как этилацетат, промывки водой с последующей сушкой органической фазы над MgSO4 или Na2SO4 и концентрировании раствора в вакууме.
Тонкослойную хроматографию (TLC) проводили на Merck TCX пластинах (силикагель 60 F254) и для визуализации пятен использовали УФ (ультрафиолет). Флэш-хроматографию проводили на Combi Flash® Companion™, используя ReadyStep™ флэш-колонки с нормальной фазой или на Merck Silica Gel 60 (0,040-0,063 мм). Типичными растворителями, используемыми для флэш-хроматографии, были смеси хлороформ/метанол, толуол/этилацетат и этилацетат/гексаны.
Препаративную хроматографию проводили на Gilson аутопрепаративной HPLC с диодно-матричным детектором, используя колонку ХТеrrа MS (С8, 19×300 мм, 7 мкм), и градиент ацетонитрил/0,1М аммония ацетат в 5% ацетонитриле в MilliQ воде, пропуская от 20% до 60% ацетонитрила за 13 мин, и скорость потока 20 мл/мин, если в примерах не указано иное. Альтернативно очистку проводили на полупрепаративной Shimadzu LC-8A HPLC с Shimadzu SPD-10A UV-vis.-детектором, оборудованном Waters Symmetry® колонкой (С18, 5 мкм, 100 мм (19 мм). Градиент ацетонитрил/0,1% трифторуксусная кислота в MilliQ воде, прогон 35%-60% ацетонитрила за 20 мин. Скорость потока 10 мл/мин.
Перекристаллизацию обычно осуществляли в растворителях или смесях растворителей, таких как эфир, этилацетат/гептаны и метанол/вода.
Использовали следующие сокращения: DCM=дихлорметан; de=диастереомерный избыток; DIPCI=β-хлордиизопинокамфенилборан (DIP-Chlorid™); DIPEA=N,N-диизопропилэтиламин; DMF=N,N-диметилформамид; DMSO=диметилсульфоксид; ее=энантиомерный избыток; NCS=N-хлорсукцинимид; NMP=1-метил-2-пирролидинон; THF=тетрагидрофуран; водн.=водная; конц.=концентрированная.
Исходные используемые вещества либо имелись в продаже, либо были получены в соответствии с известными из литературы методами и имели экспериментальные данные, соответствующие сообщаемым. Ниже приведены примеры исходного вещества, которые были получены:
(2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол: WO 02/076990 (Примеры 1-5 и 8);
5-(бензилтио)-7-хлор[1,3]тиазоло[4,5-d]пиримидин-2-амин: WO 00/09511 (Пример 8);
5-фтор-2-формилпиридин: WO 2005/066155 (Пример 1);
5-Фторпиридин-2-карбонитрил WO 2005/066155 (Пример 3);
1-(3-Хлорпиридин-4-ил)этанон: Marsais, F. et al. J.Organometal. Chem. 1981, 216, 139-147 (Пример 4);
2-ацетил-изоникотинонитрил: Citterio et al. J. Chem. Res. Synopses 1982, 10, 272-273 (Пример 8) и
1-(6-хлорпиридин-3-ил)этанон: Lee, С. et al. J. Med. Chem. 2001, 44, 2133 (Примеры 6 и 7).
В общих способах, которые следуют ниже, Ру представляет собой возможно замещенный пиридил.
Общий Способ А
Натрия боргидрид (0,1 экв.), DIPEA (1.5 экв.) и соединение общей формулы (III) (1,2 экв.) добавляли к соединению общей формулы (II) (1,0 экв.) в DMSO в атмосфере азота. Полученную реакционную смесь перемешивали при 40°С, пока реакция не заканчивалась (контролировали посредством LC-MS, HPLC или TLC). Смесь выливали в ледяную воду и продукт экстрагировали DCM или ЕtOАс. Объединенные органические фазы сушили и концентрировали в вакууме. Неочищенный продукт, если необходимо, очищали, используя препаративную HPLC или посредством колоночной флэш-хроматографии.
Общий Способ В1
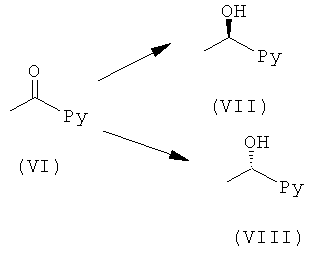
Соединение (VI) (1,0 экв.) в THF добавляли при 0°С к (+)-DIPCI (с получением (VII)) или (-)-DIPCI (с получением (VIII)) (1,5 экв.) в THF в атмосфере аргона. Реакционную смесь оставляли медленно достигать комнатной температуры в течение ночи. Растворитель выпаривали, затем добавляли EtαO и диэтаноламин (2,2 экв.). Смесь перемешивали, пока реакция не заканчивалась (контролировали посредством LC-MS, HPLC или TLC). Образовавшийся осадок отфильтровывали, промывали Et2O и фильтрат концентрировали в вакууме. Неочищенный продукт, если необходимо, очищали, используя препаративную HPLC или посредством колоночной флэш-хроматографии.
Общий способ В2
(R)-(+)-2-Метил-СВS-оксазоборолидин (1 М в толуоле, 0,1-1 экв.) растворяли в THF и охлаждали до 0°С. По каплям добавляли комплекс боран-метилсульфид (2 М в THF, 1 экв.) и реакционную смесь перемешивали в течение 1 ч. Реакционную смесь охлаждали до -10°С и добавляли по каплям (VI) (1 экв.), растворенный в THF, в течение 0,5 ч. Полученную смесь перемешивали в течение 1 ч или пока не заканчивалась реакция, и температуру медленно повышали до 10°С. Добавляли 1 М HCl водн., чтобы погасить реакцию. Добавляли насыщенный NаНСО3 водн., пока рН не составил приблизительно 8. Продукт экстрагировали DCM. Объединенные органические экстракты сушили над Na2SO4 и концентрировали в вакууме с получением (VIII). Продукт возможно очищали колоночной хроматографией.
Общий Способ С1

Трифенилфосфин (1,3 экв.) в THF добавляли при 0°С к NCS (1,3 экв.) в THF в атмосфере аргона. Полученную смесь перемешивали при температуре окружающей среды в течение 30 мин. (VII) или (VIII) (1 экв.) добавляли при 0°С и реакционную смесь перемешивали при температуре окружающей среды, пока взаимодействие не было полным (контролировали посредством LC-MS, HPLC или TLC). Растворитель выпаривали, затем добавляли гексан и удаляли осадок фильтрацией. Фильтрат концентрировали в вакууме с получением (IX) или (X). Неочищенный продукт, если необходимо, очищали, используя препаративную HPLC или посредством колоночной флэш-хроматографии.
Общий способ С2
Хлорангидрид циануровой кислоты (0,6 экв.) растворяли в этилацетате. Добавляли DMF (1,5 экв.) и смесь перемешивали при комнатной температуре в течение 10 мин. Реакционную смесь охлаждали до 0°С. (VII) или (VIII) (1 экв.) растворяли в этилацетате и добавлен по каплям в течение 10 мин. Полученную смесь перемешивали при комнатной температуре в течение ночи. Добавляли изопропанол (приблизительно 0,25 мл/ммоль (VII) или (VIII)). Осадок отфильтровывали и промывали ЕtOАс. Фильтрат концентрировали с получением указанного в заголовке соединения (IX) или (X).
Пример 1
(2R)-2-[2-Амино-5-{[1-(5-фторпиридин-2-ил)этил]тио}[1.3] тиазоло[4,5d]пиримидин-7-ил)амино]-4-метилпентан-1-ол

а) 1-(5-Фторпиридин-2-ил)этанол

К раствору 5-фтор-2-формилпиридин (0,66 г, 5,3 ммоль) в THF (20 мл) с температурой -78°С по каплям добавляли метиллитий (1,6 М в диэтиловом эфире, 4,0 мл). После перемешивания в течение 1,5 ч при -78°С добавляли насыщенный раствор аммония хлорида (25 мл), затем воду (25 мл). Смесь экстрагировали хлороформом, органическую фазу сушили над магния сульфатом и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (элюент: хлорформ : метанол 98:2) с получением указанного в заголовке соединения (0,25 г, выход 33%).
1H ЯМР (400 МГц, CDCl3): δ м.д. 8.38 (d, 1 Н), 7.42 (dt, 1 H), 7.33 (dd, 1 H), 4.90 (q, 1 H), 3.90 (s, 1 H), 1.50 (d, 3 H); MS (ESI) m/z 142 [М+1]+.
б) 2-(1-Хлорэтил)-5-фторпиридин
Смесь трифенилфосфина (0,92 г, 3.5 ммоль) и ССl4 (4 мл) перемешивали в течение 10 мин. Затем добавляли раствор 1-(5-фторпиридин-2-ил)этанола (0,25 г, 1,7 ммоль) в DCM (2 мл). Через 18 ч добавляли пентан (20 мл), твердое вещество удаляли фильтрацией и фильтрат концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (элюент: гептан : этилацетат 3:1) с получением указанного в заголовке соединения (8 мг, выход 3%).
1H ЯМР (400 МГц, CDCl3): δ м.д. 8.35 (d, 1 H), 7.44 (dd, 1 H), 7.36 (dt, 1 H). 5.09 (q,1 H),1.81 (d,3H).
в) (2R)-2-[(2-Амино-5-{[1-(5-фторпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
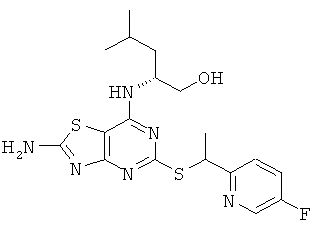
Указанное в заголовке соединение получали в соответствии с Общим способом А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (21 мг, 0,072 ммоль) и 2-(1-хлорэтил)-5-фторпиридина (8 мг, 0,048 ммоль). В результате очистки посредством препаративной HPLC получали 4 мг (21% выход) указанного в заголовке соединения в виде смеси диастереомеров.
1H ЯМР (400 МГц, DMSO-d6): δ м.д. 8.44 (t, 1H), 7.91 (s, 2H), 7.55-7.64 (m, 1H), 7.47-7.55 (m, 1H), 6.83 (d, 1H), 4.99-5.11 (m, 1H), 4.59 (q, 1H), 3.41-3.28 (m, 2H), 1.60 (dd. 3H), 1.48-1.57 (m, 1H), 1.25-1.42 (m, 2H), 0,72-0,86 (m, 6H); MS (ESI) m/z 423 [M+1]+.
Пример 2
(2R)-2-[(2-Амино-5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}[1.3]тиазоло[4,5-d]пиpимидин-7-ил)aминo]-4-мeтилпeнтaн-1-oл

а) 1-(5-Хлорпиридин-2-ил)этанон

5-Хлорпиридин-2-карбонитрил (10,7 г, 77 ммоль) растворяли в диэтиловом эфире (65 мл) и THF (35 мл) в атмосфере азота. Смесь охлаждали, пока внутренняя температура не достигала -63°С. Добавляли метилмагнийбромид (3 М в THF, 35 мл, 105 ммоль) в течение 30 мин. Реакционную смесь затем оставляли перемешиваться при -60°С в течение 45 мин и затем нагревали до комнатной температуры. Добавляли THF (50 мл) для растворения любого осажденного вещества и реакционную смесь перемешивали в течение 1 ч. Добавляли 2 М HCI водн. (100 мл) и реакционную смесь перемешивали в течение 4 ч. рН затем доводили до 7 бикарбонатом натрия. Фазы разделяли и продукт экстрагировали DCM из водной фазы. Объединенные органические экстракты сушили над натрия сульфатом и концентрировали в вакууме. Продукт очищали колоночной флэш-хроматографией (элюент: гептан : ЕtOАс, градиент) с получением 7,9 г (64% выход) указанного в заголовке соединения.
1H ЯМР (300 МГц, СDСl3) δ м.д. 8.62 (m, 1Н); 8.00 (m, 1Н); 7.80 (m, 1H); 2.70 (s. 3H).
б) (1S)-1-(5-Хлорпиридин-2-ил)этанол

Указанное в заголовке соединение получали Общим способом В2, начиная с 1-(5-хлорпиридин-2-ил)этанона (780 мг, 5 ммоль). После очистки колоночной флэш-хроматографией получали 695 мг (88% выход) указанного в заголовке соединения с ее 92%.
1H ЯМР (300 МГц, CDCl3): 8.47 (s, 1Н); 7.65 (d, 1Н); 7.26 (d, 1Н); 4.87 (q, 1Н); 3.87 (br s, 1Н); 1.47 (d, 3H); MS (ESI) m/z 140 и 142 [M+1]+.
в) 5-Хлор-2-[(1R)-1-хлорэтил]пиридин
Указанное в заголовке соединение получали Общим способом С2, начиная с (1S)-1-(5-хлорпиридин-2-ил)этанола (695 мг, 4.41 ммоль). Неочищенный продукт использовали на следующей стадии без очистки.
1H ЯМР (400 МГц, CDCl3): δ м.д. 8.46 (d, 1H), 7.64 (dd, 1H), 7.41 (d, 1H), 5.08 (q, 1H), 1.80 (d. 3 H); MS (ESI) m/z 176 и 178 [М+1]+.
г) (2R)-2-[(2-Амино-5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]]пиримидин-7-ил)амино]-4-метилпентан-1-ол

Указанное в заголовке соединение получали Общим способом А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (572 мг, 1,91 ммоль) и 5-хлор-2-[(1R)-1-хлорэтил]пиридина (376 мг, 2,1 ммоль). После очистки препаративной HPLC получали 99 мг (12% выход) указанного в заголовке соединения.
1H ЯМР (400 МГц, СD3ОD): (м.д. 8.49 (d, 1H), 7.79 (dd, 1H), 7.66 (d, 1H), 5.22 (q, 1H), 4.46 (br s, 1H), 3.57-3.40 (m, 2H), 1.78-1.66 (m, 4H), 1.61-1.40 (m, 2H), 1.03-0.93 (m, 6H); MS (ESI) m/z 439 и 441 [M+1]+.
Пример 3
(2R)-2-[(2-Амино-5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}[1,3]тиазоло[4.5-d]пиримидин-7-ил)aмино]-4-метилпентан-1-oл

а) 1-(5-Фторпиридин-2-ил)этанон
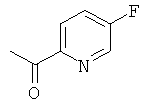
5-Фторпиридин-2-карбонитрил (29 г, 240 ммоль) растворяли в THF (150 мл) в атмосфере азота. Реакционную смесь охлаждали до температуры внутри смеси -64°С. Метилмагнийбромид (3 М в THF, 105 мл, 315 ммоль) добавляли в течение 40 мин. Реакционную смесь перемешивали при -65°С в течение 1,5 ч, затем нагревали до комнатной температуры. Добавляли THF (50 мл) и смесь перемешивали еще 3 ч. Добавляли 2 М соляную кислоту (водн., 100 мл), пока смесь не становилась слегка кислой, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли натрия бикарбонат для нейтрализации реакционной смеси. Фазы разделяли и водную фазу экстрагировали DCM. Объединенные органические экстракты промывали рассолом, сушили над натрия сульфатом и концентрировали в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией с получением 18 г (55% выход) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDCl3): 8.50 (m, 1Н); 8.10 (m. 1H); 7.52 (m, 1H); 2.70 (s, 3Н).
б) (1S)-1-(5-Фторпиридин-2-ил)этанол
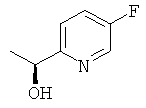
Указанное в заголовке соединение получали Общим способом В2, начиная с 1-(5-фторпиридин-2-ил)этанона (2,70 г, 19,4 ммоль). После очистки колоночной флэш-хроматографией получали 0.85 г (31% выход) указанное в заголовке соединение с 94% ее.
1H ЯМР (300 МГц, CDCl3): 8.38 (m, 1Н); 7.5-7.2 (m. 2H); 4.89 (q, 1H); 3.9 (br s,1H);1.49(d,3H).
в) 2-[(1R)-1-Хлорэтил]-5-фторпиридин
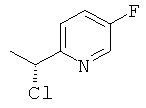
Указанное в заголовке соединение с ее 87% получали Общим способом С2, начиная с (1S)-1-(5-фторпиридин-2-ил)этанола (407 мг, 2,9 ммоль). Неочищенный продукт использовали на следующей стадии без очистки.
1H ЯМР (300 МГц, CDCl3): 8.44-8.40 (m, 1H); 7.6-7.4 (m, 2H); 5.16 (q, 1H), 1.86(d,3H).
г) (2R)-2-[(2-Амино-5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол

Указанное в заголовке соединение получали Общим способом А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (550 мг, 1,8 ммоль) и 2-[(1R)-1-хлорэтил]-5-фторпиридина (0,46 г, 2,9 ммоль). Продукт очищали колоночной флэш-хроматографией, затем препаративной HPLC (колонка: Reprosil, элюент: изопропанол : гептан 20:80, поток 16 мл/мин), собирая первый элюируемый изомер, с получением 135 мг (выход 18%) указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-d6) δ м.д. 8.51 (d, 1H), 7.98 (s, 2H), 7.65 (dt, 1H); 7.58 (dd, 1H); 6.88 (d, 1H); 5.12 (q, 1H); 4.66 (t, 1H); 4.27 (br s, 1H); 3.41-3.27 (m, 2H), 1.66 (d, 3H), 1.65-1.55 (m, 1H); 1.48-1.35 (m, 2H), 0,88 (d, 3H), 0,85 (d, 3H); MS(ESI) m/z 423 [M+1]+.
Пример 4
(2R)-2-[(2-Амино-5-{[-(3-хлорпиридин-4-ил)этил]тио}[1.3]тиазоло[4.5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол

а) (1S)-1-(3-Хлорпиридин-4-ил)этанол

Указанное в заголовке соединение с ее 90% получали, используя Общий способ В1, начиная с 1-(3-хлорпиридин-4-ил)этанона (0,90 г, 5,78 ммоль).
1H ЯМР (300 МГц, CDCl3) 8.47 (s, 1Н); 7.65 (d, 1H); 7.26 (d, 1H); 4.87 (q, 1H); 3.87 (br s, 1H); 1.47 (d, 3H); MS (ESI) m/z 158 и 160 [М+1]+.
б) 3-Хлор-4-[(1R)-1-хлорэтил]пиридин
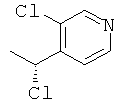
Указанное в заголовке соединение получали, используя Общий способ С2, начиная с (1S)-1-(3-хлорпиридин-4-ил)этанола (570 мг, 3,62 ммоль). Неочищенный продукт использовали на следующей стадии без очистки.
MS (ESI) m/z 176 и 178 [М+1]+.
в) (2R)-2-[(2-Амино-5-{[1-(3-хлорпиридин-4-ил)этил]тио}[1,3]тиазоло[4,5-d)]пиримидин-7-ил)амино]-4-метилпентан-1-ол (изомер 1 и изомер 2)
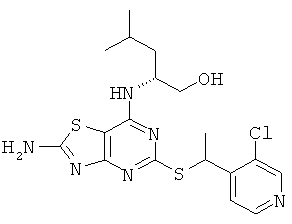
Указанное в заголовке соединение получали, используя Общий способ А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (860 мг, 2,87 ммоль) и 3-хлор-4-[(1R)-1-хлорэтил]пиридина с предыдущей стадии. После очистки флэш-хроматографией получали указанное в заголовке соединение (105 мг, выход 8%) в виде диастереомерной смеси.
MS (ESI) m/z 439 и 441 [М+1]+.
Пример 5
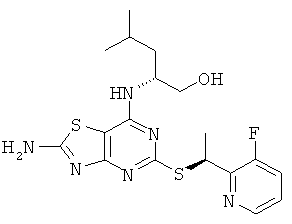
(2R)-2-[(2-Амино-5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}[1.3] тиазоло[4.5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
а) 1-(6-Бром-3-фторпиридин-2-ил)этанон

2-Бром-5-фторпиридин (11 г, 62.5 ммоль) растворяли в диэтиловом эфире при комнатной температуре в атмосфере азота. Реакционную смесь охлаждали, пока внутренняя температура не достигала -66°С. Бутиллитий (2,5 М в гексанах, 26 мл, 65 ммоль) добавляли по каплям в течение 0,5 ч. Полученную реакционную смесь оставляли при -65°С на 1 ч. N,N-Диметилацетамид (6.5 мл, 70 ммоль) добавляли в течение 10 мин.
Реакционную смесь перемешивали при -65°С в течение 2 ч. Добавляли 1 М соляную кислоту водн. (50 мл) и смесь нагревали до комнатной температуры. рН доводили до 7 дополнительным количеством соляной кислоты. Водную фазу экстрагировали диэтиловым эфиром три раза. Объединенные органические фазы промывали рассолом, сушили над натрия сульфатом и концентрировали в вакууме. После очистки колоночной флэш-хроматографией (элюент: гептан : диэтиловый эфир, градиент) получали 4,6 г (выход 34%) указанного в заголовке соединения.
1H ЯМР (300 МГц, DMSO-d6): 8.0-7.8 (m, 2H); 2.57 (s, 3H); MS (ESI) m/z 218 и 220 [М+1]+.
б) (1S)-1-(6-Бром-3-фторпиридин-2-ил)этанол
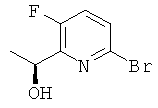
Указанное в заголовке соединение получали, используя Общий способ В2, начиная с 1-(6-бром-3-фторпиридин-2-ил)этанона (1,76 г, 8,19 ммоль). Продукт очищали колоночной флэш-хроматографией (элюент: гептан : этилацетат, градиент) с получением 1,31 г (выход 73%) указанного в заголовке соединения с ее 80%.
1H ЯМР (300 МГц, CDCl3): 7.38 (m, 1Н); 7.26 (m, 1H); 5.06 (q, 1H); 3.38 (br s, 1H); 1.47 (d, 3H); MS (ESI) m/z 220 и 222 [М+1]+, m/z 202 [M-H2-O]+.
в) (1S)-1-(3-Фторпиридин-2-ил)этанол

(1S)-1-(6-Бром-3-фторпиридин-2-ил)этанол (1,3 г, 5,9 ммоль), триэтиламин (1.6 мл, 11.5 ммоль) и палладий на углероде (0,64 г, 0,34 ммоль) смешивали в DCM (25 мл). Колбу вакуумировали/заполняли газообразным водородом за 4 цикла и затем оставляли при давлении газообразного водорода 2,5 атм (0,25 МПа) при комнатной температуре в течение 24 ч. Смесь фильтровали и твердое вещество промывали DCM. Фильтрат промывали водой и рассолом, сушили над натрия сульфатом и концентрировали в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией (элюент DСМ : метанол, градиент) с получением 0,54 г (выход 65%) указанного в заголовке соединения.
1H ЯМР (300 МГц, CDCl3): 8.38 (m, 1Н); 7.39 (m, 1H); 7.26 (m, 1H); 5.11 (q, 1H);4.16 (br s,1H);1.49(d,3H).
г) 2-((R)- 1-Хлорэтил)-3-фторпиридин
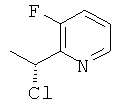
Указанное в заголовке соединение (0,24 г) получали, используя Общий способ С2, начиная с (1S)-1-(3-фторпиридин-2-ил)этанола (254 мг, 1,8 ммоль).
1H ЯМР (300 МГц, CDCl3): 8.46 (m, 1H); 7.47 (m, 1H); 7.34 (m, 1H); 5.48 (q, 1H), 1.94 (d, 3H); MS (ESI) m/z 160 и 162 [М+1]+.
д) (2R)-2-[(2-Амино-5-{[(1S)-1-(3-фторпиридин-2-ил)этил]тио}-[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол

Указанное в заголовке соединение получали, используя Общий способ А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (348 мг, 1,16 ммоль) и 2-((R)-1-хлорэтил)-3-фторпиридина (240 мг, 1,5 ммоль). После очистки колоночной флэш-хроматографией (элюент: DCM : метанол, градиент) получали 190 мг (выход 47%) указанного в заголовке соединения с диастереомерным избытком 60%. 55 мг данного вещества дополнительно очищали посредством препаративной HPLC. Последний элюируемый изомер собирали с получением 21 мг указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-d6) δ м.д. 8.40 (dt, 1H), 7.98 (s, 2H), 7.70 (m, 1H), 7.40 (m, 1H); 6.92 (d, 1H); 5.45 (q, 1H); 4.65 (t, 1H); 4 27 (br s, 1H); 345-3.30 (m, 2H), 1.69 (d, 3H), 1.66-1.58 (m, 1H), 1.50-1.35 (m, 2H), 0,88 (d, 3H), 0,85 (d, 3H); MS (ESI) m/z 423 [М+1]+.
Пример 6
(2R)-2-[(2-Амино-5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио{[1.3] тиазоло[4.5-d]пиримидин-7-ил)(метил)амино]пентан-1-ол

а) (1S)-1-(6-Хлорпиридин-3-ил)этанол

Указанное в заголовке соединение получали в соответствии с Общим способом В1, используя (-)DIPCI и 1-(6-хлорпиридин-3-ил)этанон (0,80 г, 5,14 ммоль), с получением 0,71 г (выход 88%) указанного в заголовке соединения.
1H ЯМР (CDCl3) δ м.д. 8.40-8.28 (m, 1H), 7.75-7.63 (m, 1H), 7.35-7.24 (m, 1H), 5.04-4.79 (m, 1H), 1.63-1.45 (m, 3H); MS (ESI) m/z 158 и 160 [M+1]+.
б) 2-Хлор-5-[(1R)-1-хлорэтил]пиридин

Указанное в заголовке соединение получали в соответствии с общим способом С1, используя (1S)-1-(6-хлорпиридин-3-ил)этанол (0,20 г, 1,27 ммоль), с получением 0,16 г (выход 72%) указанного в заголовке соединения.
1H ЯМР (CDCl3) δ м.д. 8.45-8.35 (m, 1H), 7.79-7.70 (m, 1H), 7.39-7.29 (m, 1H), 5.07 (q, 1H), 1.85-1.78 (m, 3H); MS (ESI) m/z 176 и 178 [М+1]+.
в) N-(Этоксикарбонил)-D-норвалин
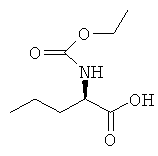
D-Норвалин (10,0 г, 85.3 ммоль) растворяли в водном растворе гидроксида натрия (4 М, 25 мл). Этилхлорформиат (10,6 мл, 111 ммоль) и водный натрия гидроксид (4М, 25 мл) добавляли в течение 15 мин при 0°С. Реакционную смесь нагревали до комнатной температуры и перемешивали при данной температуре в течение 4 ч. Реакционную смесь промывали диэтиловым эфиром три раза и затем подкисляли водной соляной кислотой (2М). Продукт экстрагировали диэтиловым эфиром три раза. Объединенные органические фазы сушили над магния сульфатом и концентрировали в вакууме с получением указанного в заголовке соединения с количественным выходом.
1H ЯМР (CDCl3) δ м.д. 6.43 (br s, 1Н). 5.22 (d, 1Н), 4.37 (q, 1H), 4.13 (q, 2H), 1.84 (m, 1Н), 1.68 (sextet, 1Н), 1.42 (sextet, 1Н), 1.25 (t, 3Н), 0,95 (t, 3H); MS (CI) 144 (100%),190 [M+1]+.
г) (2R)-2-(Метиламино)пентан-1-ол

Алюмогидрид лития (6,5 г, 171 ммоль) суспендировали в THF при 0°С в атмосфере азота. N-(Этоксикарбонил)-D-норвалин растворяли в THF и добавляли по каплям при 0°С. Реакционную смесь кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры добавляли насыщенный водный сульфат натрия с образованием взвеси. Полученную смесь фильтровали через целит.Твердое вещество промывали DCM, пока весь продукт не экстрагировался. Объединенный фильтрат сушили над натрия сульфатом и концентрировали в вакууме. В результате перегонки "из колбы в колбу" (bulb-to-bulb distiltion) при 0,1 мбар (10 Па), собирая фракция при 75-85°С, получали 7,1 г (выход 71%) указанного в заголовке соединения.
1H ЯМР (CDCl3) 3.63 (dd, 1Н); 3.30 (dd, 1Н); 2.51 (m, 1Н); 2.41 (s, 3H); 2.09 (br s, 2H); 1.50-1.28 (m, 4H); 0,93 (t, 3H); MS (CI) 86 (100%), 118 [M+1]+.
д) (2R)-2-{[2-Амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил](метил)амино}пентан-1-ол

5-(Бензилтио)-7-хлор[1,3]тиазоло[4,5-d]пиримидин-2-амин (6,0 г, 19,4 ммоль) растворяли в NMP (25 мл). Добавляли DIPEA (6,8 мл, 38,8 ммоль) и (2R)-2-(метиламино)пентан-1-ол (3,4 г, 29,1 ммоль) и смесь нагревали до 120°С в течение 3 суток. Дополнительно добавляли (2R)-2-(метиламино)пентан-1-ол (350 мг, 2,99 ммоль) и DIPEA (1 мл, 5,74 ммоль) и реакционную смесь нагревали в течение 6 ч при 120°С. После охлаждения до комнатной температуры смесь вливали в лед. Осажденный продукт собирали фильтрацией и очищали колоночной флэш-хроматографией (элюент: DCM : этилацетат, градиент) с получением указанного в заголовке соединения (5,74 г, выход 76%).
1H ЯМР (DMSO-d6) 7.98 (br s, 2H), 7.41 (m, 2H), 7.29 (m, 2H), 7.22 (m, 1H), 4.73 (t, 1H), 4.54 (br s, 1H), 4.33 (m, 2H), 3.55-3.40 (m, 2H), 3.01 (s, 3H), 1.52-1.44 (m, 2H), 1.25-1.10 (m. 2H), 0,84 (t, 3H); MS (ESI) m/z 390 [М+1]+.
e) (2R)-2[(2-Амино-5-меркапто[1,3]тиазоло[4,5-d]]пиримидин-7-ил)(метил)амино]пентан- 1-ол

Круглодонную колбу оборудовали холодильником со смесью сухой лед-этанол и погружали в охлаждающую баню сухой лед-этанол. Аммиак (200 мл) конденсировали в колбу, затем добавляли (2R)-2-{[2-амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил](метил)амино}пентан-1-ол (5,43 г, 13,9 ммоль). Полученную смесь оставляли нагреваться до -33°С и добавляли небольшими кусочками металлический натрий, пока не появлялось и не сохранялось в течение 30 секунд синее окрашивание. Реакцию затем гасили, добавляя ложку твердого аммония хлорида. Аммиак выпаривали и к остатку добавляли воду (250 мл). Полученную смесь нейтрализовали 1 М соляной кислотой (водн.). Осажденный продукт собирали фильтрацией, промывали водой и ацетонитрилом и сушили в вакууме с получением 3,38 г (выход 81%) указанного в заголовке соединения.
1H ЯМР (DMSO-d6) 12.81 (br s, 1H); 8.45 (br s. 2H), 4.84 (br s. 1H), 3.55-3.40 (m, 2H), 3.02 (s. 3H), 1.48 (m, 2H), 1.21 (m, 2H), 0,87 (t, 3H); MS (ESI) m/z 300 [M+1]+.
ж) (2R)-2-[(2-Амино-5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)(метил)амино]пентан-1-ол
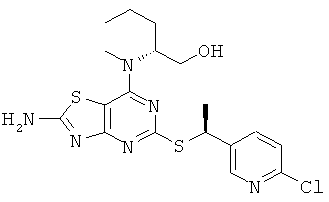
Указанное в заголовке соединение получали, используя Общий способ А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)(метил)амино]пентан-1-ола (96 мг, 0,32 ммоль) и 2-хлор-5-[(1R)-1-хлорэтил]пиридина (85 мг, 0,48 ммоль). После очистки колоночной флэш-хроматографией (элюент: ОСМ : этилацетат) получали 22 мг (16% выход) указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-d6) δ м.д. 8.51 (d, 1H), 7.99 (br s, 2H), 7.96 (dd, 1H), 7.45 (d, 1H); 4.98 (q, 1H); 4.73 (t, 1H); 4.46 (br s, 1H); 3.53-3.38 (m, 2H), 2.97 (s, 3H), 1.66 (d, 3H), 1.48 (m, 2H), 1.17 (m, 2H), 0,84 (t, 3H); MS (ESI) m/z 439 и 441 [М+1]+.
Пример 7
(2R)-2-[(2-Амино-5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол
а) (2R)-2-{[2-Амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}пентан-1-ол

5-(Бензилтио)-7-хлор[1,3]тиазоло[4,5-d]пиримидин-2-амин (6,0 г, 19,4 ммоль) растворяли в NMP (30 мл). Добавляли DIPEA (8,4 мл, 48,5 ммоль) и 2-амино-(2R)-1-пентанол (3,5 г, 33,9 ммоль), смесь нагревали до 110°С в течение 4 суток. После охлаждения до комнатный температуры смесь вливали в воду (200 мл). Осажденный продукт собирали фильтрацией, промывали водой и использовали на следующий стадии без дополнительной очистки (7,0 г, выход 97%).
MS (ESl) m/z 376 [М+1]+.
б) (2R)-2-[(2-Амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол

Круглодонную колбу оборудовали холодильником со смесью сухой лед-этанол и погружали в охлаждающую баню сухой лед-этанол. Аммиак (250 мл) конденсировали в колбу, затем добавляли (2R)-2-{[2-амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}пентан-1-ол (6,8 г, 18,1 ммоль). Полученную смесь оставляли нагреваться до -33°С и добавляли небольшими кусочками металлический натрий, пока не появлялось и не сохранялось в течение 30 секунд синее окрашивание. Реакцию затем гасили добавлением ложки твердого аммония хлорида. Аммиак выпаривали и к остатку добавляли воду (250 мл). Полученную смесь нейтрализовали 1 М соляной кислотой (водн.). Осажденный продукт собирали фильтрацией, промывали водой и сушили в вакууме с получением 4,15 г (выход 80%) указанного в заголовке соединения.
MS(ESI) m/z 286 [M+1]+.
в) (2R)-2-[(2-Амино-5-{[(1S)-1-(6-хлорпиридин-3-ил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол

Указанное в заголовке соединение получали, используя Общий способ А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]иримидин-7-ил)амино]пентан-1-ола (70 мг, 0,245 ммоль) и 2-хлор-5-[(1R)-1-хлорэтил]пиридина (Пример 6б, 52 мг, 0,29 ммоль). После очистки препаративной HPLC получали 23 мг (22% выход) указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-d6) 8.50 (d, 1Н); 7.99 (br s, 2H); 7.95 (dd, 1H); 7.44 (d, 1H); 6.91 (d, 1H); 4.95 (q, 1H); 4.11 (m, 1H); 3.41 (dd, 1H); 3.31 (dd, 1H); 1.64 (d, 3Н); 1.56 (m, 1H); 1.41 (m, 1H); 1.4-1.2 (m, 2H); 0,85 (t, 3H); MS (ESI) m/z 425 и 427 [M+1]+.
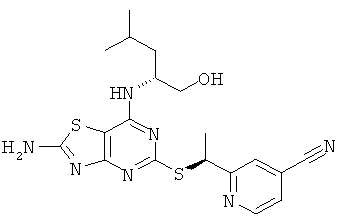
Пример 8
2-{(1S)-1-[(2-Амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}изоникотинонитрил
а) 2-((S)-1-Гидроксиэтил)изоникотинонитрил
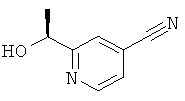
Указанное в заголовке соединение получали в соответствии с Общим способом В, начиная с 2-ацетил-изоникотинонитрила (2,00 г, 13,7 ммоль) и (-)-DIPCI (6,58 г, 20,53 ммоль) с получением указанного в заголовке соединения (625 мг, 4,22 ммоль) с ее 90%.
1H ЯМР (CDCI3) δ 8.72 (d, 1Н), 7.62 (s, 1Н), 7.44 (dd, 1H), 4.96 (q, 1H), 1.54 (d, 3H).
б) 2-((R)-1-Хлорэтил)изоникотинонитрил

Указанное в заголовке соединение получали из 2-((S)-1-гидроксиэтил)изоникотинонитрила (620 мг, 4,18 ммоль) в соответствии с Общим способом С, за исключением того, что использовали 1,0 экв. NCS и 1,0 экв. PPh3. После 24 ч взаимодействия добавляли еще по 0,2 экв. каждого из NCS и PPh3 и температуру повышали до 30°С в течение 12 ч. После обработки и очистки колоночной флэш-хроматографией получали указанное в заголовке соединение (46 мг, 7% выход) с ее 82%.
1H ЯМР (CDCl3) δ 8.74 (d, 1H), 7.76 (s, 1H), 7.46 (dd, 1H). 5.16 (q, 1H), 1.88 (d. 3H).
в) 2-{(1S)-1-[(2-Амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}изоникотинонитрил
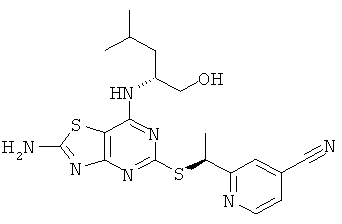
Указанное в заголовке соединение получали в соответствии с Общим Способом А, начиная с 2-((R)-1-хлорэтил)изоникотинонитрила (46 мг, 0,28 ммоль) и (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (69 мг, 0,23 ммоль). После очистки хиральной HPLC (колонка: Chiralpak AD 50×150 мм, элюент: гептан : изопропанол 85:15), скорость потока 60 мл/мин) получали указанное в заголовке соединение (20 мг, выход 20%).
1H ЯМР (400 МГц, DMSO-d6) δ м.д. 8.78 (d, 1H), 8.00 (s, 1H), 7.95 (s, 1H), 7.72 (m, 1H), 6.90 (m, 1H), 5.14 (q, 1H), 4.64 (t, 1H), 4.23 (br s, 1 H), 3.23-3.41 (m, 2H), 1.67 (d, 3H), 1.64 - 1.52 (m, 1H), 1.32-1.48 (m, 2H), 0,87 (dd, 6H); MS (ESI) m/z430 [M+1]+.
Пример 9
Пример 9а
(2R)-2-[(2-Амино-5-{[(1R)-1-(3-хлорпиридин-4-ил)этил]тио}[1,3] тиазоло[4.5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол и
Пример 9б
(2R)-2-[(2-Амино-5-{[(1S)-1-(3-хлорпиридин-4-ил)этил]тио}[1.3] тиазоло[4.5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол

Диастереомерную смесь (2R)-2-[(2-амино-5-{[1-(3-хлорпиридин-4-ил)этил]тио}[1,3]тиазоло[4,5d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (105 мг) из Примера 4 разделяли посредством препаративной HPLC с получением 14 мг первого элюируемого изомера (Пример 9а).
1H ЯМР (400 МГц, DMSO-d6) δ м.д. 8.59 (s, 1H), 8.48 (d, 1H), 8.00 (s, 2H), 7.68 (d, 1H); 6.93 (d, 1H); 5.27 (q, 1H); 4.63 (t, 1H); 4.16 (br s, 1H); 3.42-3.30 (m, 2H), 1.64 (d, 3Н), 1.58-1.30 (m, 3Н), 0,84 (d, 3H), 0,74 (d, 3H); MS (ESI) m/z 439 и 441 [М+1]+.
Также получили еще 15 мг последнего элюируемого изомера (Пример 9б).
1H ЯМР (400 МГц, DMSO-d6) δ м д. 8.60 (s, 1H), 8.48 (d, 1H), 8.00 (s, 2H), 7.67 (d, 1H); 6.92 (d, 1H); 5.28 (q, 1H); 4.56 (t, 1H); 4.22 (br s, 1H); 3.33-3.12 (m, 2H), 1.62 (d, 3Н), 1.60-1.30 (m, 3Н), 0,88 (d, 3Н), 0,85 (d, 3H); MS (ESI) m/z 439 и 441 [М+1]+.
Фармакологические скрининги
Вещества
Рекомбинантный фракталкин человека (hCX3CL1) и рекомбинантный интерлейкин-8 человека (IL-8 или hCXCL8) приобретали у PeproTech Inc., UK. Рекомбинантный [125|]-фракталкин (человека) и [125|] hlL-8 с удельной активностью 2200 Ки/ммоль приобретали у NEN® Life Science Products, Inc., UK. Fluo4-AM приобретали у Molecular Probes, US. Все другие химические реагенты имели аналитическую чистоту.
Клетки
Полные кДНК CX3CR1 человека (GenBank номер U20350) экстрагировали от мРНК человеческого мозга (Superscript, Life Technologies) и лигировали в pCR-Blunt II ТОРО вектор (InVitrogen). Вставку соответствующего hСХ3СР1 выделяли и дополнительно субклонировали в pcDNA3.1zeo. Плазмидную ДНК получали, используя Plasmid Mini Kit (Qiagen). Используя Superfect Transfection Reagent (Qiagen) в соответствии с протоколом изготовителя, экспрессионную плазмиду для hCX3CR1 затем вводили в суспензию клеточной линии почек эмбрионов человека (HEKS) 293, содержащей вектор для стабильной экспрессии химерного G-белка Gαqi5. Стабильный клон получали, используя селекцию с зеоцином (500 мкг/мл) и гигромицином (100 мкг/мл). Для дальнейшего применения клетки поддерживали в среда Игла, модифицированной Дульбекко/питательной смеси Хэма F12 (DMEM/F12), содержащей пиридоксин с добавлением 10% (об./об.) фетальной бычьей сыворотки, 2 мМ L-глутамина, 100 Ед/мл пенициллина и 100 мг/мл стрептомицина, 250 мкг/мл зеоцина и 100 мкг/мл гидромицина.
Клетки, экспрессирующие CXCR2 человека, полученные от AstraZeneca Charnwood, культивируют в ЕМЕМ, содержащей Глутамакс с добавлением 10% FBS (от РАА, Austria), 1% неэссенциальных аминокислот (NEAA), 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина (PEST) и 500 мкг/мл генетицина/G418.
Мембранный препарат
Клетки выращивают при 37°С и 5% СO2 и собирают при 60-80% слиянии в буфере, содержащем 10 мМ Трис-HCl, рН 7,4, 5 мМ EDTA (этилендиаминтерауксусной кислоты), 0,1 мг/мл бацитрацина. Клетки центрифугируют при 300х в течение 10 мин, осадок ресуспендируют в буфере для сбора (10 мМ Трис-HCl, рН 7,4, 5 мМ этилендиаминтетрауксусная кислота (EDTA) и 0,1 мг/мл бацитрацина), объединяют и гомогенизируют, используя Dounce гомогенизатор. Гомогенат центрифугируют при 48000хg в течение 10 мин и ресуспендируют в буфере для сбора, используя Ultra-Turrax Т8. Аликвоты мембран хранят при -80°С. Концентрацию белка определяли в микротитровальных планшетах, как описано в Harrington (1990, Anal. Biochem. 186,285-287).
In vitro анализ связывания рецепторов
Исследования конкурентного связывания [125|]фракталкина проводили в 2 мл 96-луночных планшетах с глубокими лунками (Beckman, Германия) в суммарном объеме 1000 мкл/лунку. Каждая лунка содержала 10 пМ [125I]-фракталкина и эквивалентное количество мембраны до концентрации рецептора 1 пМ в аналитическом буфере (50 мМ HEPES-KOH, рН 7,4, 10 мМ MgCl2, 1 мМ EDTA, 0,1% (мас./об.) желатина). Десять концентраций (2 точек/Iog единицу) тестируемого соединения предварительно растворяли в DMSO и добавляли до достижения конечной концентрации 1% (об./об.) DMSO. Анализ начинали с добавления мембран и инкубировали при 25°С в течение 24 ч. Реакции останавливали путем быстрой фильтрации при помощи Whatman GF/B стекловолоконных фильтров, предварительно обработанных 0,3% полиэтилимином, и последующей промывки смесью лед-холодный буфер (10 мМ HEPES-KOH, рН 7,4, 500 мМ NaCl), используя Brandel харвестер для рецепторного связывания. Добавляли сцинтилляционный коктейль и радиоактивность определяли в Packard 2500TR жидкостном сцинтилляционном счетчике (Perkin Elmer, USA).
Исследования [125I]-hIL-8 конкурентного связывания осуществляли в одном экземпляре в белых 96-луночных изопланшетах с прозрачным дном с конечным объемом 200 мкл, каждая лунка содержала 150 пМ [125I]-hIL-8 (удельная активность 2200 Ки/ммоль), препарат мембрана-SPA, эквивалентный 20 пМ рецепторов, и 1,5 мг SPA-гранул в аналитическом буфере [50 мМ HEPES-KOH, рН 7,4, 10 мМ MgCl2, 1 мМ EDTA, 0,5% (мас/об.) желатина]. Тестируемые соединения обрабатывали, как указано выше. Неспецифическое связывание определяли в присутствии 500 нм немеченого hIL-8. Агонист hIL-8 (кривая концентрация - ответ от 3 пМ до 30 нМ) используют в качестве эталонного соединения в каждом тесте. Пептидная кривая не содержит DMSO. Реакцию связывания начинают путем добавления 140 мкл препарата мембрана-SPA и образцы инкубируют в темноте при комнатной температуре в течение 4 ч. Аналитические планшеты обсчитывают в жидкостном сцинтилляционном счетчике (Wallac MicroBeta® TriLux 1450 jn PerkinElmer, USA).
[35S]GTPγS связывание
Исследования [35S]GTPγS связывания проводили в микротитровальных планшетах с прозрачным дном в двух параллелях с 10 концентрациями ингибитора (2 конц./log единицу), разбавленными DMSO (конечная концентрация 1%) и при комнатной температуре. Мембраны, экспрессирующие hCX3CR1 рецептор (конечная концентрация 20 мкг белка/лунка), добавляли вместе со SPA-гранулами (конечная концентрация 1 мг/лунка), все суспендировали в GTPγS связывающем буфере (50 мМ Трис-HCl, 100 мМ NaCl, 0,1% желатина, 15 мкг сапонина/мл и 3 мкМ GDP (гуанозин-5'-дифосфат), рН 7,4 при комнатной температуре). Мембраны, SPA гранулы и лекарственные средства предварительно инкубировали 30 мин, затем добавляли 310 пМ фракталкин для максимальной стимуляции. Исходную активность определяли как активность, обнаруживаемую без стимуляции фракталкином (GTPγS связывающий буфер). Через еще 30 мин реакцию начинали добавлением [35S]GTPγS до конечной концентрации 0,1 нм и конечного аналитического объема 0,2 мл. Эксперимент заканчивали через 30 минут путем центрифугирования при 2000 об/мин в течение 2×5 минут (разные направления) и радиоактивность определяли в жидкостном сцинтилляционном счетчике (Wallac MicroBeta® TriLux 1450).
Результаты
Типичные СХ3СR1 Ki значения для соединений по настоящему изобретению находятся в диапазоне от примерно 0,1 до примерно 1000 нм. Другие значения СХ3СR1 Ki находятся в диапазоне от примерно 0,1 нм до примерно 500 нм. Другие значения СХ3СК1 Ki находятся в диапазоне от примерно 0,1 нм до примерно 25 нм. Результаты in vitro анализа hСХ3СR1 связывания для конечных соединений показаны в таблице.
Соединения по настоящему изобретению, где R1 представляет собой Me (содержащие разветвленную тиоалкилпиридильную группу в положении 5), являются как более эффективными антагонистами СХ3СР1 рецептора, так и/или менее эффективными антагонистами CXCR2 рецептора, чем соответствующие эталонные соединения, где R1 представляет собой Н. Такая улучшенная селективность в отношении антагонизма CX3CR1 рецептора, как ожидается, будет приносить значительную терапевтическую пользу.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ 5,7-ДИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ [1,3]ТИАЗОЛО[4,5-d]ПИРИМИДИН-2(3Н)-ОНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2007 |
|
RU2441012C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 5,7-ДИЗАМЕЩЕННОГО [1,3] ТИАЗОЛО[4,5-D]ПИРИМИДИН-2(3H)-ОНА | 2006 |
|
RU2411245C9 |
| НОВЫЕ ПРОИЗВОДНЫЕ 5-ЗАМЕЩЕННОГО 7-АМИНО-[1,3]ТИАЗОЛО[4,5-d] ПИРИМИДИНА | 2006 |
|
RU2419623C2 |
| ФОСФАТНЫЕ И ФОСФОНАТНЫЕ ПРОИЗВОДНЫЕ 7-АМИНО-5-ТИОТИАЗОЛО[4,5-d]ПИРИМИДИНОВ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ, СВЯЗАННЫХ С ПОВЫШЕННЫМИ УРОВНЯМИ CX3CR1 И/ИЛИ CX3CL1 | 2019 |
|
RU2801664C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| СПОСОБ СТЕРЕОСЕЛЕКТИВНОГО ПОЛУЧЕНИЯ ХИРАЛЬНЫХ 2-[(ГЕТЕРО)АРИЛАЛКИЛСУЛЬФАНИЛ]ПИРИМИДИНОВ И ПРОДУКТОВ, ПОЛУЧЕННЫХ ИЗ НИХ | 2019 |
|
RU2813355C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| ЦИКЛОАЛКАНОВОЕ ПРОИЗВОДНОЕ | 2013 |
|
RU2635354C2 |
| ПРОЛЕКАРСТВО ПРОИЗВОДНОГО АМИНОКИСЛОТЫ | 2017 |
|
RU2739318C2 |
Изобретение относится к соединениям общей формулы (I), где R1 представляет собой СН3; R2 представляет собой галогено или CN; R3 представляет собой Н или СН3; R4 представляет собой Н или СН3; n представляет собой 1, и к их фармацевтически приемлемым солям. Также изобретение относится к фармацевтической композиции и к применению соединений формулы (I) в изготовлении лекарственного средства, обладающих антагонистической активностью в отношении CX3CR1 рецептора. Технический результат - соединения формулы (I) в качестве антагонистов CX3CR1 рецептора. 7 н. и 6 з.п. ф-лы, 1 табл.
1. Соединение формулы (I)
где R1 представляет собой СН3;
R2 представляет собой галогено или CN;
R3 представляет собой Н или СН3;
R4 представляет собой Н или СН3;
n представляет собой 1;
в виде свободного основания или его фармацевтически приемлемой соли.
2. Соединение по п.1, где R2 представляет собой F или Cl.
3. Соединение по п.1, где R2 представляет собой CN.
4. Соединение по п.1, где R2 представляет собой F, Сl или CN.
5. Соединение по п.1, где R3 представляет собой Н.
6. Соединение по п.1, где R4 представляет собой СН3.
7. Соединение, выбранное из:
(2R)-2-[(2-амино-5-{[1-(5-фторпиридин-2-ил)этил]тио}[1,3]тиазоло[4,5-d]-пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-амино-5-{[(1S)-1-(5-хлорпиридин-2-ил)этил]тио}[1,3]тиазоло-[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-амино-5-{[(1S)-1-(5-фторпиридин-2-ил)этил]тио}[1,3]тиазоло-[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1 -ола;
(2R)-2-[(2-амино-5-{[(1S)-(3-хлорпиридин-4-ил)этил]тио}[1,3]тиазоло[4,5-d]-пиримидин-7-ил)амино]-4-метилпентан-1 -ола;
(2R)-2-[(2-амино-5-{[(1S)-1-(3-хлорпиридин-4-ил)этил]тио}[1,3]тиазоло-[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-амино-5-{[(1R)-1-(3-хлорпиридин-4-ил)этил]тио}[1,3]тиазоло-[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-aминo-5-{[(1S)-1-(3-фтopпиpидин-2-ил)этил]тиo}[1,3]тиaзoлo-[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола;
(2R)-2-[(2-aминo-5-{[(1S)-1-(6-xлopпиpидин-3-ил)этил]тиo}[l,3]тиaзoлo-[4,5-d]пиримидин-7-ил)(метил)амино]пентан-1-ола;
(2R)-2-[(2-aминo-5-{[(1S)-1-(6-xлopпиpидин-3-ил)этил]тиo}[1,3]тиaзoлo-[4,5-d]пиримидин-7-ил)амино]пентан-1-ола; и
2-{(1S)-1-[(2-амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}изоникотинонитрила;
в виде свободного основания или его фармацевтически приемлемой соли.
8. Соединение по любому из пп.1-7 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства для лечения или профилактики заболеваний или состояний, при которых благоприятным является антагонизм в отношении CX3CR1 рецептора.
9. Фармацевтическая композиция, обладающая антагонистической активностью в отношении CX3CR1 рецептора, содержащая терапевтически эффективное количество соединения по любому из пп.1-7 или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым разбавителем или носителем.
10. Применение соединения формулы (I), как оно определено в любом из пп.1-7, или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, болезни периферических артерий, ревматоидного артрита или астмы.
11. Применение соединения формулы (I), как оно определено в любом из пп.1-7, или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики рассеянного склероза.
12. Применение соединения формулы (I), как оно определено в любом из пп.1-7, или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики атеросклероза путем изменения состава бляшек с целью уменьшения риска разрыва бляшки и атеротромботических явлений.
13. Применение соединения формулы (I), как оно определено в любом из пп.1-7, или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики заболеваний или состояний, при которых благоприятным является антагонизм в отношении CX3CR1 рецептора.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 00/09511 А1, 24.02.2000 | |||
| RU 2002125451 А, 10.01.2004 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

