Уровень техники
Хемокины регулируют миграцию различных типов мононуклеарных клеток. Они классифицируются на четыре подсемейства, то есть CC, CXC, CX3C и C, на основании положений консервативных цистеиновых остатков на их N-концах. Стромально клеточный фактор-1 (SDF-1 или CXCL12), хемокин CXC, состоящий из 67 аминокислотных остатков, в основном экспрессируется в костном мозге, центральной нервной системе и периферии. CXCL12 служит специфическим лигандом для рецептора хемокинов CXC типа 4 (CXCR4), рецептора, связанного с G-белком, с семью трансмембранными доменами на поверхности многих типов стволовых клеток. Ось CXCR4/CXCL12 играет ключевую роль в регуляции метастазирования рака, поиска, миграции и мобилизации стволовых клеток. Взаимодействие между CXCR4 и CXCL12 способствует возникновению множества патологических состояний, таких как ВИЧ (Schols et al., J. Exp. Med. 186:1383-1388 (1997); Wu et al., J. Med. Chem. 58:1452-1465 (2015)), ревматоидный артрит (Lenoir et al., J. Immunol. 172:7136-7143 (2004)), астма (Gonzalo et al., J. Immunol. 165:499-508 (2000)), и метастазы опухоли (Müller et al., Nature. 410:50-56 (2001); Liang et al., Cancer Res. 65:967-971 (2005)).
Антагонисты CXCR4, которые нарушают взаимодействие между CXCR4 и CXCL12, способны мобилизовать различные типы мононуклеарных клеток, включая гемопоэтические стволовые клетки, эндотелиальные клетки-предшественники и мезенхимальные стволовые клетки, из костного мозга в периферическую кровь. Например, антагонист CXCR4 AMD3100 (плериксафор) в клинической практике используется при трансплантации стволовых клеток периферической крови (PBSC), чтобы помочь пациентам с гематологическими злокачественными новообразованиями, такими как неходжкинская лимфома или множественная миелома. Трансплантация PBSC является типичной медицинской процедурой, при которой здоровые стволовые клетки, в частности, CXCR4+/CD34+ гематопоэтические стволовые клетки, мобилизуются из костного мозга в периферическую кровь для сбора при лечении антагонистами CXCR4 с последующей аутологичной трансплантацией больным раком после лучевой терапии или химиотерапии для быстрого восстановления их иммунной системы.
Соединения, которые нарушают взаимодействие между CXCR4 и CXCL12, можно использовать для лечения различных заболеваний, включая повреждение тканей (Lin et al., J. Invest. Dermatol. 134:2458-2468 (2014)), воспалительного заболевания (Lukacs et al., Am. J. Pathol. 160:1353-1360 (2002)), ишемической болезни (Huang et al., Stroke. 44:190-197 (2013); Wu et al., J. Med. Chem. 58:2315-2325 (2015); Wu et al., Cell Transplantation. in press (2017)), рака (Chen et al., Hepatology. 59:1435-1447 (2014)), и аутоиммунного заболевания (Matthys et al., J. Immunol. 167:4686-4692 (2001)).
Существует необходимость в разработке новых соединений, которые могут эффективно нарушать взаимодействие между CXCR4 и CXCL12.
Сущность изобретения
Настоящее изобретение основано на неожиданном открытии того, что некоторые гетероциклические соединения могут эффективно связываться с CXCR4, тем самым нарушая взаимодействие между CXCR4 и CXCL12.
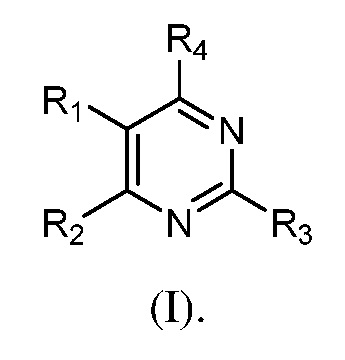
В одном аспекте настоящее изобретение относится к гетероциклическим соединениям формулы (I):
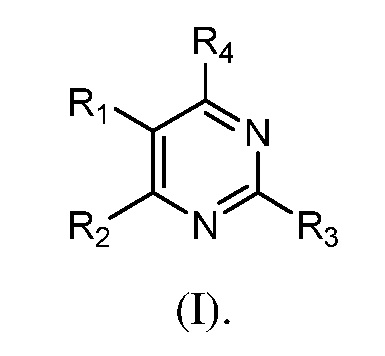
В этой формуле каждый из R1 и R2 независимо представляет собой Н, галоген, NO2, CN, NH2, C1-6 алкил, C1-6 алкоксил, C3-10 циклоалкил, C1-10 гетероциклоалкил, арил или гетероарил; или R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой C5-10 циклоалкил, C3-10 гетероциклоалкил, арил или гетероарил, где каждый из C1-6 алкила, C1-6 алкоксила, C3-10 циклоалкила, C1-10 гетероциклоалкила, C5-10 циклоалкила, C3-10 гетероциклоалкила, арила и гетероарила необязательно замещены галогеном, NO2, CN, NH2, C1-6 алкилом, C1-6 алкоксилом, арилом, гетероарилом или C(O)ORa, где Ra представляет собой H, C1-10 алкил, C3-10 циклоалкил, C3-10 гетероциклоалкил, арил или гетероарил; и каждый из R3 и R4, независимо, представляет собой 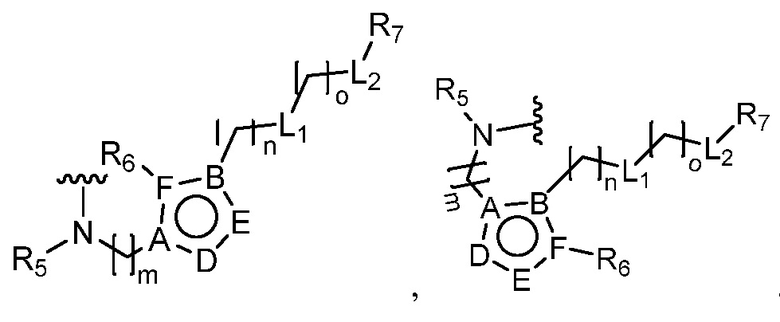 или
или 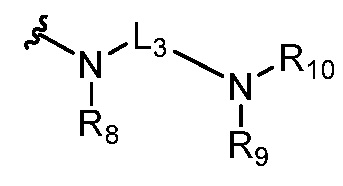 ,
,
где R5 представляет собой H, C1-6 алкил, C3-10 циклоалкил, C1-10 гетероциклоалкил, арилалкил, гетероарилалкил, арил или гетероарил, каждый из C1-6 алкила, C3-10 циклоалкила, C1-10 гетероциклоалкила, арилалкила, гетероарилалкила, арила и гетероарила необязательно замещен галогеном, нитро, циано, амино, C1-6 алкилом, C1-6 алкоксилом, C3-10 циклоалкилом, C1-10 гетероциклоалкилом, арилом или гетероарилом; R6 представляет собой H, C1-6 алкил, C1-6 алкоксил, C3-10 циклоалкил, C1-10 гетероциклоалкил, арил или гетероарил, или отсутствует, где каждый из C1-6 алкила, C1-6 алкоксила, C3-10 циклоалкила, C1-10 гетероциклоалкила, арила и гетероарила необязательно замещен гидрокси, гидрокси C1-6 алкилом, галогеном, нитро, циано или амино; R7 представляет собой H, C1-6 алкил, C1-6 алкоксил, C3-10 циклоалкил, C1-10 гетероциклоалкил, арил или гетероарил, каждый из C1-6 алкила, C1-6 алкоксила, C3-10 циклоалкила, C1-10 гетероциклоалкила, арила и гетероарила необязательно замещен гидрокси, гидрокси C1-6 алкилом, галогеном, нитро, циано, амино, амино C1-6 алкилом, амино C3-10 циклоалкилом, амино C1-10 гетероциклоалкилом, C3-10 циклоалкилом, C1-10 гетероциклоалкилом, арилом или гетероарилом; каждый из A и B, независимо, представляет собой C или N; каждый из D, E и F, независимо, представляет собой C, N, O, или S; каждый из L1 и L2, независимо, представляет собой гетероарил, C1-10 гетероциклоалкил или NRd, в котором Rd представляет собой H или C(O)(CH2)2CHNH2CO2Re, Re представляет собой H, C1-6 алкил, C3-10 циклоалкил, C3-10 гетероциклоалкил, арил или гетероарил; каждый из m, n и o, независимо, равен 1, 2, 3, 4, 5 или 6; каждый из R8 и R9, независимо, представляет собой H, C1-6 алкил, C3-10 циклоалкил, C1-10 гетероциклоалкил, арил или гетероарил, каждый из C1-6 алкила, C3-10 циклоалкила, C1-10 гетероциклоалкила, арила и гетероарила необязательно замещен C(O)ORf, где Rf представляет собой H, C1-10 алкил, C3-20 циклоалкил, C3-20 гетероциклоалкил, арил или гетероарил; или R8 и R9, вместе с атомами азота, с которыми они связаны, представляют собой C3-10 гетероциклоалкил; L3 представляет собой C1-6 алкил; или L3, вместе с R8 или R9 и атомом азота, с которым они связаны, представляет собой C4-10 гетероциклоалкил или гетероарил; и R10 представляет собой H, C1-6 алкил, C1-6 алкоксил, C3-10 циклоалкил, C1-10 гетероциклоалкил, арил, гетероарил, или 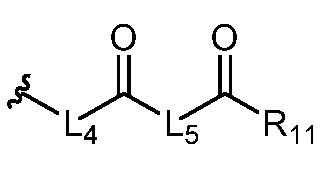 , где L4 представляет собой C1-6 алкиламино или отсутствует; L5 представляет собой C1-6 алкил, C1-6 алкиламино, или ди-C1-6 алкиламино; и R11 представляет собой гидроксил или C1-6 алкиламино; каждый из C1-6 алкила, C1-6 алкоксила, C3-10 циклоалкила, C1-10 гетероциклоалкила, C1-6 алкиламино; ди-C1-6 алкиламино, арила и гетероарила необязательно замещен гидроксилом, амино, C(O)OR12 или P(O)(OR13)2, где каждый из R12 и R13, независимо, представляет собой H или C1-6 алкил.
, где L4 представляет собой C1-6 алкиламино или отсутствует; L5 представляет собой C1-6 алкил, C1-6 алкиламино, или ди-C1-6 алкиламино; и R11 представляет собой гидроксил или C1-6 алкиламино; каждый из C1-6 алкила, C1-6 алкоксила, C3-10 циклоалкила, C1-10 гетероциклоалкила, C1-6 алкиламино; ди-C1-6 алкиламино, арила и гетероарила необязательно замещен гидроксилом, амино, C(O)OR12 или P(O)(OR13)2, где каждый из R12 и R13, независимо, представляет собой H или C1-6 алкил.
Термин «алкил» в настоящем описании относится к насыщенному, линейному или разветвленному углеводородному радикалу, такому как -CH3 или разветвленный -C3H7. Термин «циклоалкил» относится к неароматическому, моноциклическому, бициклическому, трициклическому или тетрациклическому углеводородному радикалу, такому как циклогексил, циклогексен-3-ил или адамантил. Термин «алкоксил» относится к -O-алкильному радикалу. Примеры алкокси включают, но не ограничиваются ими, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси. Термин «гетероциклоалкил» относится к неароматическому, моноциклическому, бициклическому, трициклическому или тетрациклическому радикалу, имеющему один или несколько кольцевых гетероатомов (например, N, O или S), такому как 4-тетрагидропиранил или 4-пиранил. Термин «арил» относится к углеводородному радикалу, имеющему одно или несколько ароматических колец. Примеры арила включают, но не ограничиваются ими, фенил (Ph), фенилен, нафтил, нафтилен, пиренил, антрил и фенантрил. Термин «гетероарил» относится к группе, имеющей одно или несколько ароматических колец, которые содержат по меньшей мере один гетероатом (например, N, О или S). Примеры гетероарила включают, но не ограничиваются ими, фурил, фурилен, флуоренил, пирролил, тиенил, оксазолил, имидазолил, тиазолил, пиридил, пиримидинил, хиназолинил, хинолил, изохинолил и индолил. Термин «арилалкил» относится к алкилу, который замещен по меньшей мере одной арильной группой. Примеры арилалкила включают бензил (Bn) и 1-нафтилметил. Термин «гетероарилалкил» относится к алкилу, который замещен по меньшей мере одной гетероарильной группой. Примеры гетероарилалкила включают 2-фуранилметил и 2-тиенилметил. Термин «аминоалкил» или «алкиламино» относится к алкилу, который замещен по меньшей мере одной аминогруппой. Примеры аминоалкила или алкиламино включают аминометил и 2-аминоэтил. Термин «диалкиламино» относится к аминогруппе, которая замещена двумя алкильными группами. Примеры диалкиламино включают 1,1-диметиламино и 1-метил-1-этиламино. Термин «аминоциклоалкил» относится к циклоалкилу, который замещен по меньшей мере одной аминогруппой. Примеры аминоциклоалкила включают аминоциклопропил и аминоциклопентил. Термин «аминогетероциклоалкил» относится к гетероциклоалкилу, который замещен по меньшей мере одной аминогруппой. Примеры аминогетероциклоалкила включают аминопирролидинил и аминопиперидинил. Термин «гидроксилалкил» относится к алкилу, который замещен по меньшей мере одной гидроксильной группой. Примеры гидроксилалкила включают гидроксилметил и гидроксилэтил.
Алкил, циклоалкил, гетероциклоалкил, арил, гетероарил, арилалкил и гетероарилалкил, указанные в настоящей заявке, включают как замещенные, так и незамещенные группы, если не указано иное. Возможные заместители в циклоалкиле, гетероциклоалкиле, ариле и гетероариле включают C1-10 алкил, C2-10 алкенил, C2-10 алкинил, C3-20 циклоалкил, C3-20 циклоалкенил, C1-20 гетероциклоалкил, C1-20 гетероциклоалкенил, C1-10 алкокси, арил, арилокси, гетероарил, гетероарилокси, амино, C1-10 алкиламино, C1-20 диалкиламино, ариламино, диариламино, гидроксил, галоген, тио, C1-10 алкилтио, арилтио, C1-10 алкилсульфонил, арилсульфонил, ациламино, аминоацил, аминотиоацил, амидино, гуанидин, уреидо, циано, нитро, ацил, тиоацил, ацилокси, карбоксил и эфир карбоновой кислоты. С другой стороны, возможные заместители в алкиле включают все из вышеупомянутых заместителей за исключением C1-10 алкила, C2-10 алкенила и C2-10 алкинила. Циклоалкил, гетероциклоалкил, арил и гетероарил могут также быть конденсированными друг с другом.
Гетероциклические соединения, описанные выше, включают сами соединения, а также их соли, пролекарства и сольваты, в случае необходимости. Например, соль может быть образована между анионом и положительно заряженной группой (например, амино) в гетероциклических соединениях. Подходящие анионы включают хлорид, бромид, йодид, сульфат, нитрат, фосфат, цитрат, метансульфонат, трифторацетат, ацетат, малат, тозилат, тартрат, фумурат, глутамат, глюкуронат, лактат, глутарат и малеат. Подобным образом, соль также может быть образована между катионом и отрицательно заряженной группой (например, карбоксилатом) в гетероциклическом соединении. Подходящие катионы включают ион натрия, ион калия, ион магния, ион кальция и аммониевый катион, такой как ион тетраметиламмония. Гетероциклические соединения также включают соли, содержащие четвертичный атом азота. Примеры пролекарств включают сложные эфиры и другие фармацевтически приемлемые производные, которые при их введении субъекту способны обеспечивать активные гетероциклические соединения. Сольват относится к комплексу, образованному между активным гетероциклическим соединением и фармацевтически приемлемым растворителем. Примеры фармацевтически приемлемого растворителя включают воду, этанол, изопропанол, этилацетат, уксусную кислоту и этаноламин.
Гетероциклические соединения могут содержать неароматические двойные связи, которые могут встречаться в цис- или транс-изомерных формах. Такие изомерные формы предусмотрены.
Другой аспект настоящего изобретения относится к способу мобилизации гемопоэтических стволовых клеток (HSC) и эндотелиальных клеток-предшественников (EPC) в периферическое кровообращение. Способ включает контактирование HSC и EPC с эффективным количеством одного или нескольких гетероциклических соединений формулы (I), описанных выше.
Дополнительный аспект натоящего изобретения относится к способу лечения повреждений тканей, рака, воспалительного заболевания и аутоиммунного заболевания. Способ включает введение субъекту, нуждающемуся в этом, эффективного количества одного или нескольких гетероциклических соединений формулы (I), описанных выше. Примеры повреждения тканей включают нейродегенеративное заболевание, дисфункцию пигментного эпителия сетчатки, инфаркт сердца и миокарда, ишемическую болезнь (например, ишемический инсульт и ишемия конечностей), рану, перелом кости, повреждение поджелудочной железы, почечное повреждение, повреждение кишечника и повреждение легких. Примеры рака включают острый миелоидный лейкоз, немелкоклеточный рак легкого, множественную миелому и рак поджелудочной железы. Примеры воспалительного заболевания включают воспалительное заболевание кишечника, аллергическую астму и увеит глаза. Типичным аутоиммунным заболеванием является ревматоидный артрит.
В конкретном примере способ осуществляют для лечения почечного повреждения (например, острое почечное повреждение). Способ включает введение субъекту, страдающему почечным повреждением, эффективного количества одного или нескольких гетероциклических соединений, описанных выше.
Также в объем настоящего изобретения входит фармацевтическая композиция, содержащая одно или несколько описанных выше гетероциклических соединений формулы (I). Фармацевтическая композиция может быть использована для лечения повреждения ткани (например, острое почечное повреждение), рака, воспалительного заболевания и аутоиммунного заболевания.
Настоящее изобретение также относится к применению одного или нескольких вышеописанных гетероциклических соединений формулы (I) для производства лекарственного средства для лечения повреждения ткани (например, острое почечное повреждение), рака, воспалительного заболевания и аутоиммунного заболевания.
Термин «лечить» или «лечение» относится к введению одного или нескольких гетероциклических соединений субъекту, у которого имеется вышеописанное заболевание, симптом такого заболевания или предрасположенность к такому заболеванию, с целью получения терапевтического эффекта, например, для лечения, уменьшения, изменения, воздействия, ослабления или предотвращения вышеописанного заболевания, симптома такого заболевания или предрасположенности к такому заболеванию. «Эффективное количество» относится к количеству активного соединения, которое необходимо для получения терапевтического эффекта. Эффективные дозы будут варьироваться, как это известно специалистам в данной области, в зависимости от типов подвергаемого лечению заболевания, пути введения, использования эксципиентов и возможности совместного использования с другим терапевтическим лечением.
Для осуществления способа по настоящему изобретению композицию, содержащую одно или несколько вышеописанных гетероциклических соединений, можно вводить парентерально, перорально, назально, ректально, местно или буккально. Термин "парентеральный", как используется в настоящей заявке, относится к подкожному, внутрикожному, внутривенному, интраперитонеальному, внутримышечному, внутрисуставному, интраартериальному, интрасиновиальному, интрастернальному, интратекальному, внутриочаговому или интракраниальному введению путем инъекции, а также при помощи любой подходящей инфузии.
Стерильная композиция для инъекций может представлять собой раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как раствор в 1,3-бутандиоле. Из приемлемых носителей и растворителей, которые можно использовать, можно указать маннит, воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, удобно использовать нелетучие масла в качестве растворителя или среды для суспендирования (например, синтетические моно- или диглицериды). Жирная кислота, такая как олеиновая кислота и ее глицеридные производные, являются полезными для получения препаратов для инъекций, так же как и природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Эти масляные растворы или суспензии также могут содержать длинноцепочечный спиртовой разбавитель или диспергирующее вещество, карбоксиметилцеллюлозу или подобные диспергирующие вещества. Другие традиционно используемые поверхностно-активные вещества, такие как Tweens или Spans, или другие подобные эмульгаторы или вещества, повышающие биодоступность, которые обычно используют для получения фармацевтически приемлемых твердых, жидких или других лекарственных форм, также можно использовать для формулирования композиций.
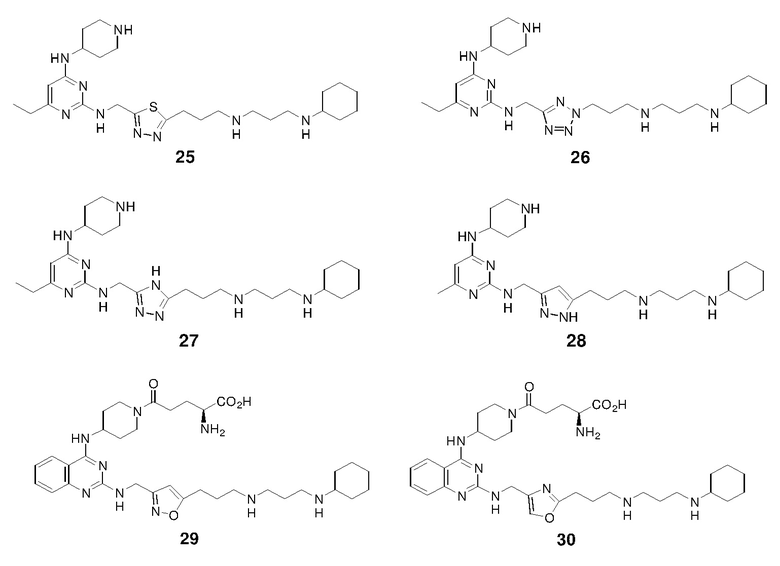
Композиция для перорального введения может представлять собой любую перорально приемлемую лекарственную форму, включая капсулы, таблетки, эмульсии и водные суспензии, дисперсии и растворы. В случае таблеток традиционно используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в форме капсул полезные разбавители включают лактозу и сухой кукурузный крахмал. Когда водные суспензии или эмульсии вводят перорально, активный ингредиент может быть суспендирован или растворен в масляной фазе вместе с эмульгаторами или суспендирующими веществами. Если это желательно, могут быть добавлены некоторые подсластители, отдушки или красители.
Назальный аэрозоль или композицию для ингаляций можно получить в соответствии со способами, хорошо известными в области формулирования фармацевтических композиций. Например, такую композицию можно получить в виде раствора в физиологическом растворе с использованием любого подходящего консерванта или промотора абсорбции (например, бензилового спирта) или любого солюбилизирующего или диспергирующего агента (например, фторуглерода).
Композицию, содержащую одно или несколько из вышеописанных гетероциклических соединений, также можно вводить в форме суппозиториев для ректального введения.
Носитель в фармацевтической композиции должен быть "приемлемым" в том смысле, что он должет быть совместимым с активным ингредиентом композиции (и предпочтительно способным стабилизировать активный ингредиент) и не оказывать вредное действие на субъекта, подлежащего лечению. Одно или несколько солюбилизирующих веществ можно использовать в качестве фармацевтических эксципиентов для доставки активного соединения 1,5-дифенилпента-1,4-диен-3-она. Примеры других носителей включают коллоидный оксид кремния, стеарат магния, целлюлозу, лаурилсульфат натрия и D&C желтый # 10.
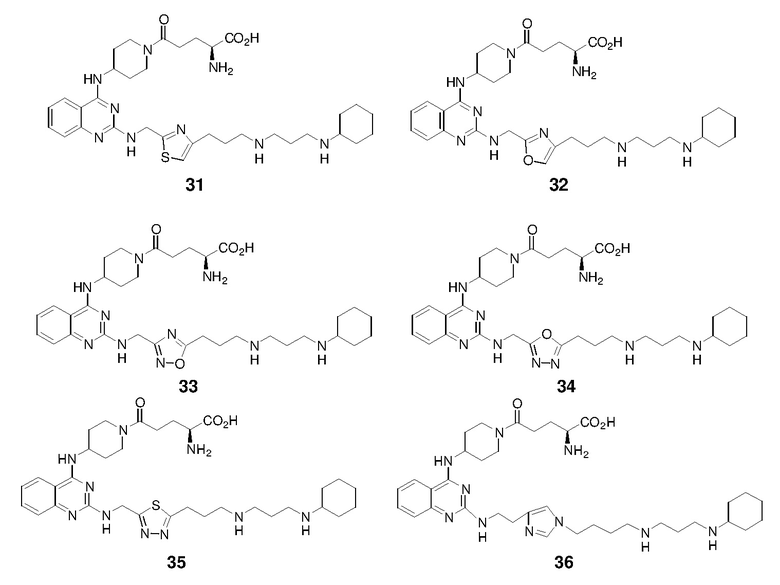
Детали одного или нескольких вариантов осуществления изобретения изложены в приведенном ниже описании. Другие признаки, объекты и преимущества изобретения будут очевидны из описания и формулы изобретения.
ПОДРОБНОЕ ОПИСАНИЕ
Ниже подробно раскрыты гетероциклические соединения формулы (I):
R1 - R4 определены в разделе ʺСущность изобретенияʺ выше.
Одна подгруппа гетероциклических соединений формулы (I) включает соединения, в которых каждый из R1 и R2, независимо, представляет собой H, NH2, C1-6 алкил или C1-10 гетероциклоалкил (например, морфолин, пиперидин или пиперазин), необязательно замещенный C1-6 алкилом или C(O)ORa, где Ra представляет собой H или C1-10 алкил. Типичные соединения в этой подгруппе включают соединения, в которых каждый из R1 и R2, независимо, представляет собой H или C1-6 алкил; и те, в которых каждый из R1 и R2, независимо, представляет собой H, NH2 или C1-10 гетероциклоалкил, необязательно замещенный C1-6 алкилом или C(O)ORa.
Другая подгруппа гетероциклических соединений формулы (I) настоящего изобретения включает те, в которых R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой арил или гетероарил. Типичные соединения в этой подгруппе включают те, в которых R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой 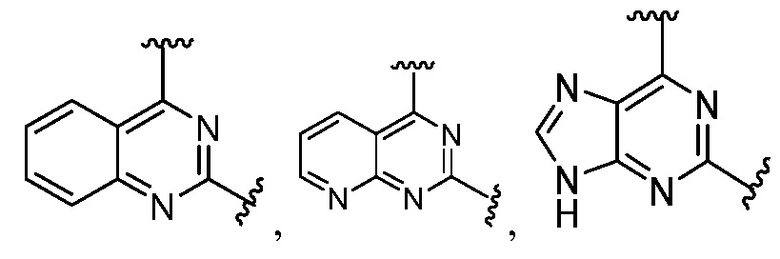 или
или 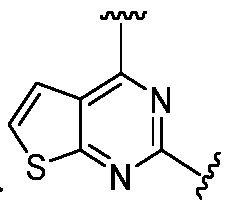 .
.
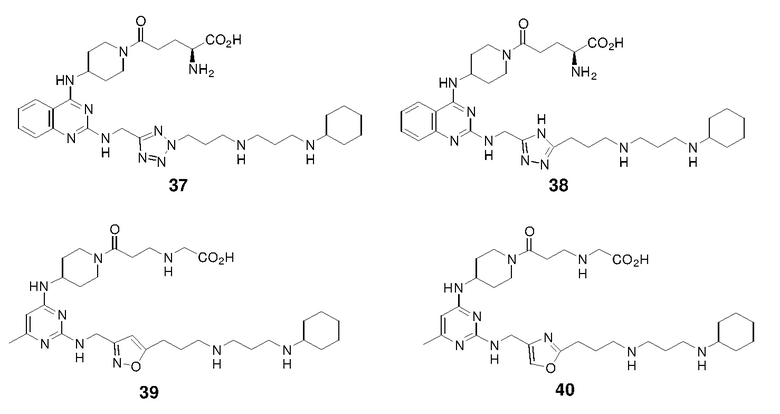
Еще одна подгруппа гетероциклических соединений формулы (I) настоящего изобретения включает те, в которых каждый из R3 и R4, независимо, представляет собой 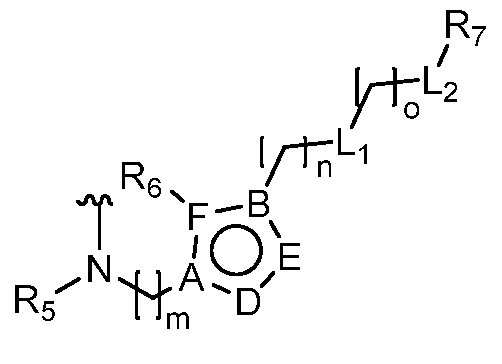 или
или 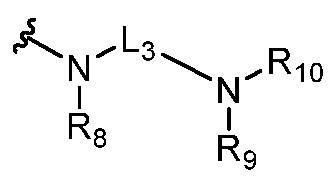 , где R5 представляет собой H; R6 отсутствует; каждый из m, n, и o, независимо, равен 1, 2, 3 или 4; и каждый из L1 и L2 представляет собой NRd. В этой подгруппе соединения могут иметь C в качестве каждого из своих A и B и иметь C, N, или S в качестве каждого из своих D, E, и F. Они также могут иметь каждый из R1 и R2, независимо, являющийся H или C1-6 алкилом (например, R1 представляет собой и R2 представляет собой C1-6 алкил); или R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой арил или гетероарил. Например, эта подгруппа включает соединения, имеющие R1 и R2, вместе с двумя атомами углерода, с которыми они связаны, являющиеся
, где R5 представляет собой H; R6 отсутствует; каждый из m, n, и o, независимо, равен 1, 2, 3 или 4; и каждый из L1 и L2 представляет собой NRd. В этой подгруппе соединения могут иметь C в качестве каждого из своих A и B и иметь C, N, или S в качестве каждого из своих D, E, и F. Они также могут иметь каждый из R1 и R2, независимо, являющийся H или C1-6 алкилом (например, R1 представляет собой и R2 представляет собой C1-6 алкил); или R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой арил или гетероарил. Например, эта подгруппа включает соединения, имеющие R1 и R2, вместе с двумя атомами углерода, с которыми они связаны, являющиеся 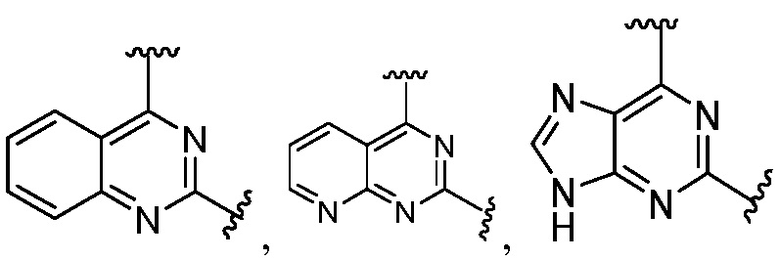 или
или 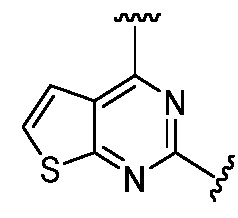 .
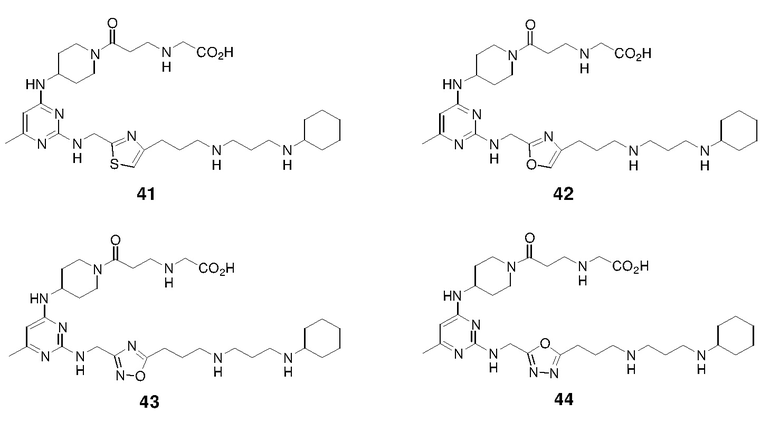
.
Еще одна подгруппа гетероциклических соединений формулы (I) включает соединения, в которых R3 представляет собой 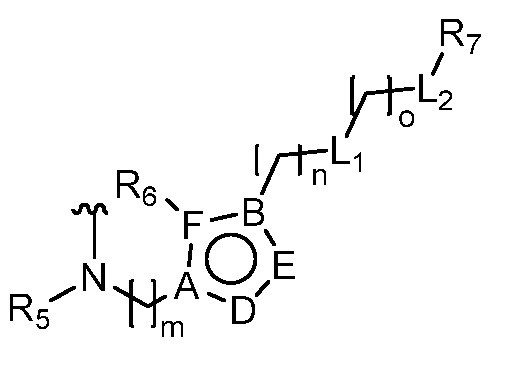 , R4 представляет собой
, R4 представляет собой 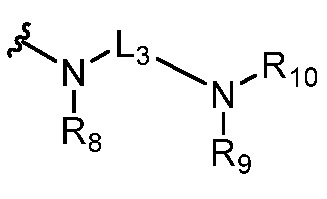 , R5 представляет собой H; R6 отсутствует; каждый из m, n, и o, независимо, равен 1, 2, 3, или 4; и каждый из L1 и L2 представляет собой NRd. В этом подмножестве соединения могут иметь каждый из R1 и R2 независимо друг от друга в виде Н или C1-6 алкила (например, R1 представляет собой Н и R2 представляет собой C1-6 алкил); или R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой арил или гетероарил. Например, эта подгруппа включает соединения, имеющие R1 и R2, вместе с двумя атомами углерода, с которыми они связаны, являющиеся
, R5 представляет собой H; R6 отсутствует; каждый из m, n, и o, независимо, равен 1, 2, 3, или 4; и каждый из L1 и L2 представляет собой NRd. В этом подмножестве соединения могут иметь каждый из R1 и R2 независимо друг от друга в виде Н или C1-6 алкила (например, R1 представляет собой Н и R2 представляет собой C1-6 алкил); или R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой арил или гетероарил. Например, эта подгруппа включает соединения, имеющие R1 и R2, вместе с двумя атомами углерода, с которыми они связаны, являющиеся 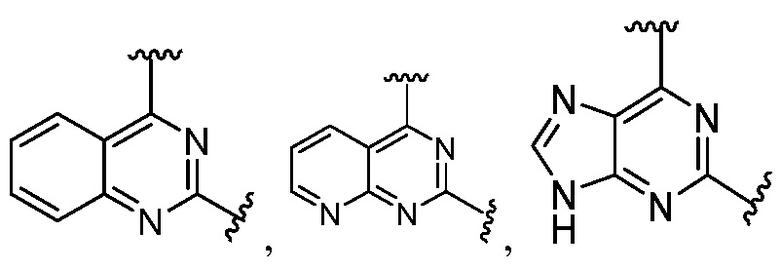 или
или  . В частности, соединения могут иметь свои R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, как
. В частности, соединения могут иметь свои R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, как  . Они также могут иметь C в качестве каждого из своих A и B и иметь C, N или S в качестве каждого из своих D, E и F. Также в этой подгруппе соединения могут иметь L3, вместе с R8 или R9 и атомом азота, с которым они связаны, являющийся C4-10 гетероциклоалкилом; и R10 является H или
. Они также могут иметь C в качестве каждого из своих A и B и иметь C, N или S в качестве каждого из своих D, E и F. Также в этой подгруппе соединения могут иметь L3, вместе с R8 или R9 и атомом азота, с которым они связаны, являющийся C4-10 гетероциклоалкилом; и R10 является H или 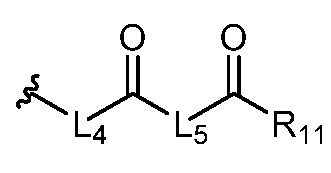 . Соединения в этой подгруппе могут иметь R8, являющийся Н и L3, вместе с R9 и атомом азота, с которым они связаны, представляющий собой C4-10 гетероциклоалкил.
. Соединения в этой подгруппе могут иметь R8, являющийся Н и L3, вместе с R9 и атомом азота, с которым они связаны, представляющий собой C4-10 гетероциклоалкил.
Примерное соединение в этой подгруппе имеет R1, являющийся Н, и R2, являющийся C1-6 алкилом, или R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, являются 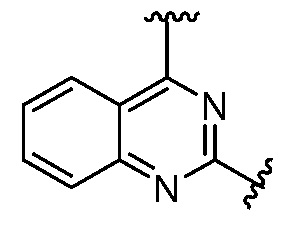 ; R10 является
; R10 является 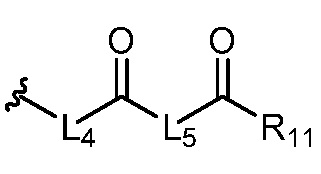 ; каждый из А и В представляет собой С; и каждый из D, E и F, независимо, представляет собой C, N или S.
; каждый из А и В представляет собой С; и каждый из D, E и F, независимо, представляет собой C, N или S.
Также в рамках настоящего изобретения находится фармацевтическая композиция, содержащая одно или несколько гетероциклических соединений формулы (I), описанных выше, для лечения повреждения ткани (например, острое почечное повреждение), рака, воспалительного заболевания и аутоиммунного заболевания.
Кроме того, настоящее изобретение охватывает способ лечения повреждения тканей (например, острое почечное повреждение), рака, воспалительного заболевания и аутоиммунного заболевания, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I).
Гетероциклические соединения формулы (I), описанные выше, могут быть получены в соответствии со способами, хорошо известными в данной области. См., например, R. Larock, Comprehensive Organic Transformations (2nd Ed., VCH Publishers 1999); P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis (4th Ed., John Wiley and Sons 2007); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis (John Wiley and Sons 1994); L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis (2nd ed., John Wiley and Sons 2009); and G. J. Yu et al., J. Med. Chem. 2008, 51, 6044-6054.
Соединения, указанные в настоящей заявке, могут содержать неароматическую двойную связь и один или несколько асимметрических центров. Таким образом, они могут существовать в виде рацематов и рацемических смесей, отдельных энантиомеров, индивидуальных диастереомеров, диастереомерных смесей и цис- или транс-изомерных форм. Все такие изомерные формы предусматриваются настоящим изобретением.
Полученные таким образом соединения формулы (I) могут быть первоначально подвергнуты скринингу с использованием анализов in vitro, например, анализа радиолигандного связывания, описанного в приведенном ниже примере 2, в отношении их эффективности в ингибировании связывания CXCL12 с CXCR4. Впоследствии они могут быть оценены с использованием анализов in vivo, например, колониеобразующего анализа, на их эффективность в усилении мобилизации гемопоэтических стволовых клеток у млекопитающего. Выбранные соединения могут быть дополнительно протестированы для проверки их эффективности при лечении повреждений тканей (например, острое почечное повреждение), рака, воспалительного заболевания и аутоиммунного заболевания. Например, соединение может быть введено животному (например, мыши), имеющему ишемическое острое почечное повреждение, и затем оцениваются его терапевтические эффекты. На основании результатов можно определить подходящий диапазон дозировки и путь введения.
Без каких-либо дополнительных уточнений полагают, что специалист в данной области может на основании приведенного выше описания использовать настоящее изобретение в самом полном его объеме. Следующие конкретные примеры, следовательно, следует рассматривать просто как иллюстративные, и никоим образом не ограничивающие остальную часть раскрытия. Все публикации, указанные в настоящей заявке, включены в настоящую заявку посредством ссылки.
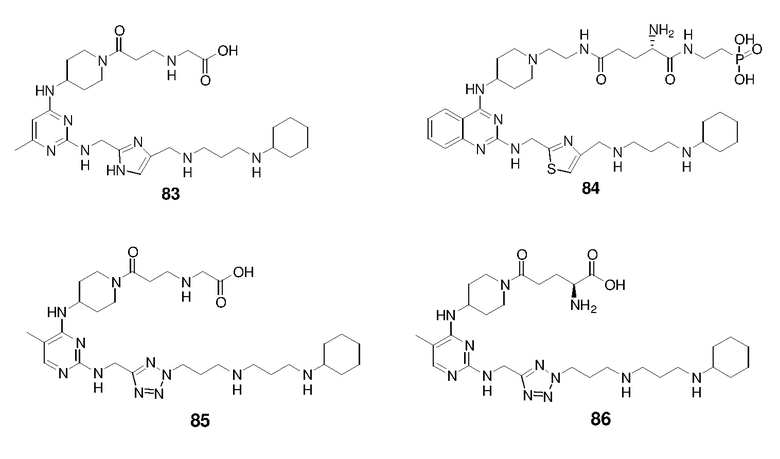
Непосредственно ниже показаны структуры 86 типичных соединений формулы (I). Способы получения этих соединений, а также аналитические данные для полученных таким образом соединений приведены в примере 1 ниже. Способы тестирования этих соединений описаны в примерах 2-5 также ниже.
Ниже описаны способы получения тринадцати боковых цепей, т.е. боковых цепей S-I - S-XIII, используемых для синтеза вышеуказанных 86 примерных соединений. Следует отметить, что все боковые цепи получали разными способами. Структуры соединений с боковой цепью S-I - S-XIII показаны ниже:
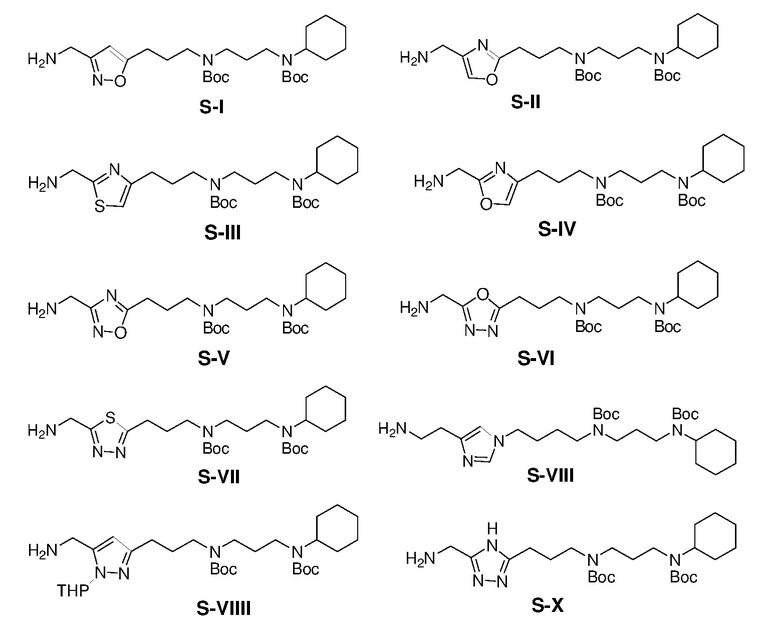
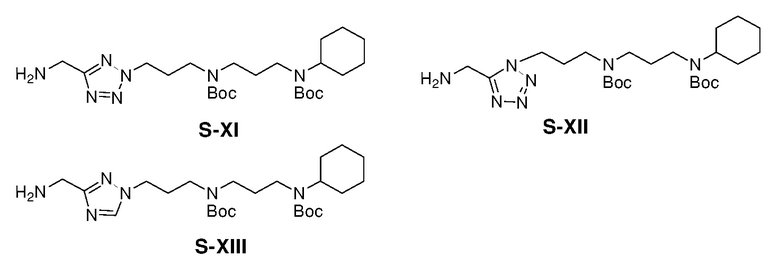
Получение S-I
Боковую цепь S-I получали по схемам, приведенным ниже:
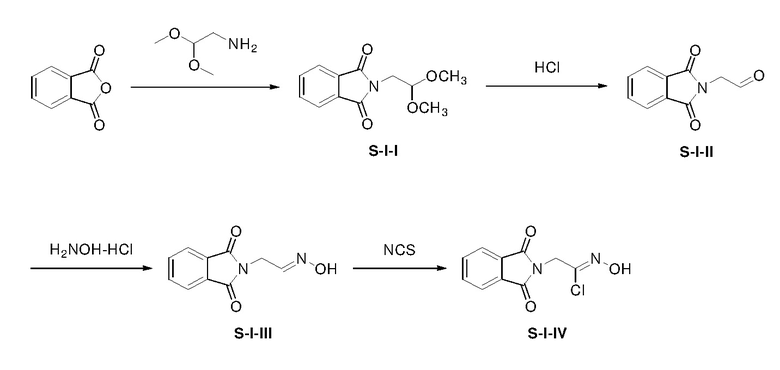
Раствор фталевого ангидрида (10,00 г), аминоацетальдегида (7,81 г) и N,N-диизопропилэтиламина (13,09 г) в толуоле в атмосфере азота нагревали при 120°C в течение 16 ч и затем гасили NH4Cl(водн) (100 мл, 2 M). Водную фазу экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением неочищенного остатка S-I-I (15,49 г, выход: 98%).
К раствору S-I-I (15,49 г) в EtOH/H2O (20 мл/40 мл) добавляли HCl(водн) (120 мл, 6 н) в атмосфере азота. Смесь нагревали при 80°C в течение 16 ч и затем концентрировали. Остаток экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали NaHCO3(водн) и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением неочищенного остатка S-I-II (6,26 г, выход: 50%).
К раствору S-I-II (6,26 г) и TEA (10,04 г) в дихлорметане (100 мл) при 5-10°C добавляли гидроксиламин гидрохлорид (2,53 г). Смесь перемешивали при комнатной температуре в течение 15 ч и затем гасили NH4Cl(водн) (50 мл, 2M). Водную фазу экстрагировали CH2Cl2 (2×50 мл). Объединенные органические экстракты промывали NaHCO3(водн) и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-I-III (4,01 г, выход: 59%).
Раствор S-I-III (4.01 г) и N-хлорсукцинимида (2,75 г) в DMF (100 мл) нагревали при 50°C в течение 5 ч и затем выливали в воду. Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-I-IV (3,64 г, выход: 78%).
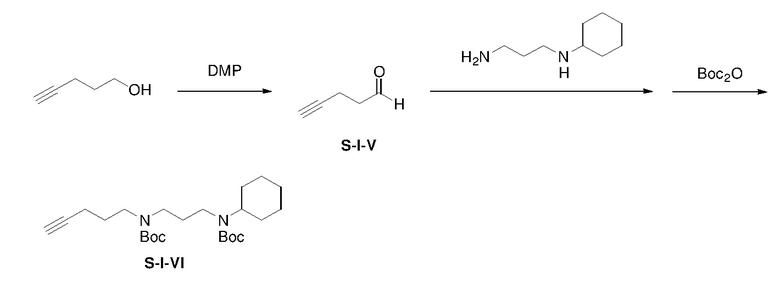
К раствору 4-пентин-1-ола (0,30 г) в дихлорметане (20 мл) при 0°C добавляли периодинан Десса-Мартина (1,66 г) в атмосфере азота. Смесь перемешивали при 0°C в течение 2 ч и затем гасили NaHCO3(водн) (50 мл, 2 M) и натрий тиосульфат Na2S2O3(водн) (50 мл, 2 M). Водную фазу экстрагировали дихлорметаном (3×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением неочищенного S-I-V (0,26 г, выход: 87%).
К перемешиваемому магнитной мешалкой раствору S-I-V (0,26 г) в MeOH (30 мл) добавляли N-циклогексил-1,3-пропандиамин (0,54 г). Затем смесь перемешивали при 25°C в течение 1 ч, к смеси медленно добавляли NaBH4 (0,24 г). Полученную смесь перемешивали в течение еще 15 ч и затем гасили NH4Cl(водн) (50 мл, 2M). Смесь концентрировали. Остаток экстрагировали CH2Cl2 (2×150 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. К перемешиваемому магнитной мешалкой фильтрату добавляли Boc2O ангидрид (0,84 г) в одной порции. Смесь перемешивали при комнатной температуре в течение 15 ч и затем концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (2:1) с получением продукта S-I-VI (0,48 г, выход: 36% за 2 стадии).
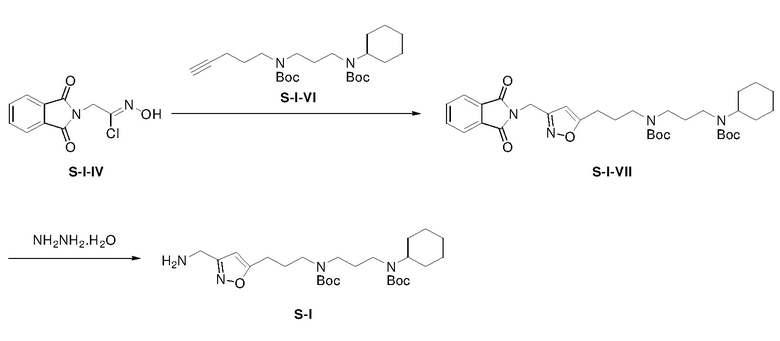
Раствор S-I-IV (0,27 г), S-I-VI (0,48 г), и триэтиламина (0,34 г) в хлороформе (30 мл) в атмосфере азота перемешивали при 25°C в течение 15 ч и затем гасили NH4Cl(водн) (50 мл, 2 M). Водную фазу экстрагировали CH2Cl2 (3×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (4:1) с получением соединения S-I-VII (0,09 г, выход: 13%).
Раствор S-I-VII (0,09 г) и гидразин моногидрата (0,02 г) в MeOH/CH2Cl2 (20 мл/20 мл) перемешивали при 25°C в течение 15 ч и затем фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-I (0,07 г, выход: 98%).
Получение S-II
Боковую цепь S-II получали по схеме, показанной ниже:
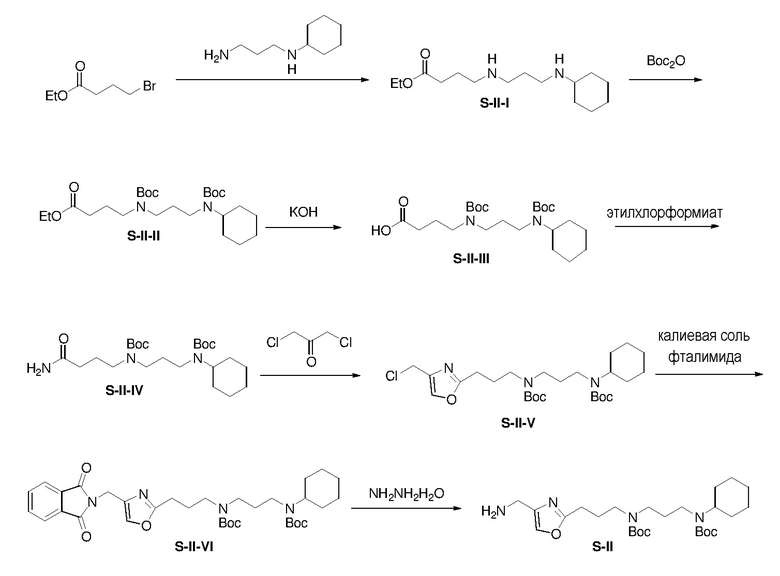
К раствору N-циклогексил-1,3-пропандиамина (4,22 г) и K2CO3 (7,09 г) в ацетонитриле (100 мл) при 0°С добавляли этил-4 бромбутират (5,00 г). Смесь перемешивали при 25°C в течение 15 ч и затем выливали в воду. Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. К перемешиваемому магнитной мешалкой фильтрату S-II-I добавляли Boc2O ангидрид (11,11 г) в одной порции. Смесь перемешивали при комнатной температуре в течение 15 ч и затем концентрировали. Полученный таким образом остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (4:1) с получением продукта S-II-II (3,60 г, выход: 30% за 2 стадии).
К раствору S-II-II (3,60 г) в THF (30 мл) в атмосфере азота добавляли раствор KOH (2,14 г) в H2O (10 мл). Смесь перемешивали при 25°C в течение 15 ч и затем подкисляли HCl (водн.) (38 мл, 1 н). Водную фазу экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением неочищенного остатка S-II-III (3,36 г, выход: 99%).
К раствору S-II-III (3,36 г) и TEA (1,16 г) в THF (30 мл) добавляли этилхлорформиат (1,00 г) при 0°C. Затем смесь перемешивали при 0°C в течение 5 ч, NH4OH(водн) (50 мл, 2M) медленно добавляли к смеси при 0°C и затем перемешивали при 25°C в течение еще 15 ч. Полученную смесь экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного S-II-IV (2,94 г, выход: 88%).
Раствор S-II-IV (2,94 г) и 1,3-дихлорацетона (1,10 г) в изопропиловом спирте (25 мл) нагревали при 100°C в течение 15 ч и затем концентрировали. Полученный таким образом остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (4:1) с получением соединения S-II-V (0,70 г, выход: 20%).
Раствор S-II-V (0,70 г) и калиевую соль фталимида (1,27 г) в DMF (20 мл) перемешивали при 25°C в течение 15 ч и затем выливали в воду. Полученную смесь экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (4:1) с получением S-II-VI (0,28 г, выход: 33%).
Раствор S-II-VI (0,28 г) и гидразин моногидрат (0,04 г) в MeOH/CH2Cl2 (20 мл/20 мл) нагревали при 25°C в течение 15 ч и затем выливали в воду. Полученную смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Фильтрат концентрировали с получением неочищенного продукта S-II (0,19 г, выход: 86%).
Получение S-III
Боковую цепь S-III получали по схеме, показанной ниже:
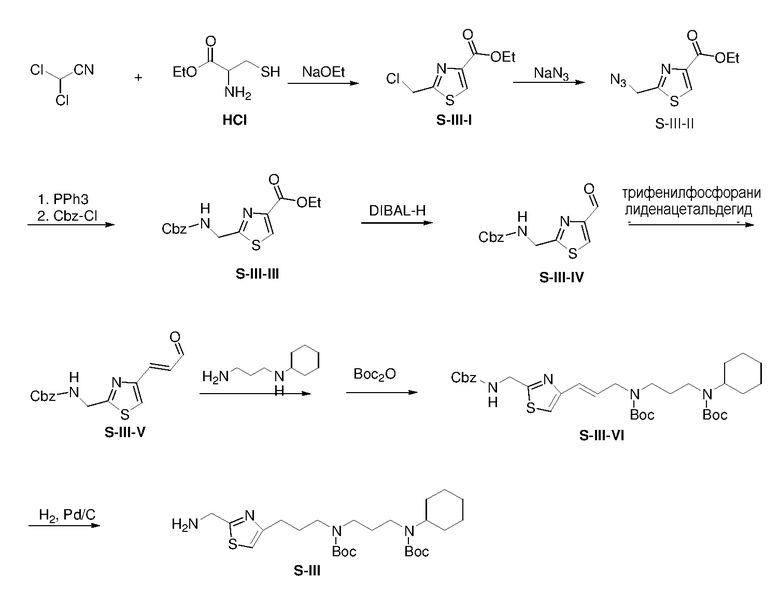
К перемешиваемому магнитной мешалкой раствору этоксида натрия (1,0 мл, 4,4 M в EtOH) в DCM (300 мл) и EtOH (35 мл) при 0°C добавляли дихлорацетонитрил (50,1 г) в течение 45 мин. Затем смесь перемешивали при 0°C в течение 1 ч, к полученной смеси добавляли L-цистеин этиловый эфир гидрохлорид (84,51 г). Реакционную смесь перемешивали при 25°C в течение 15 ч и затем гасили водой (50 мл). Полученную смесь концентрировали и затем остаток экстрагировали дихлорметаном (3×50 мл). Экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Раствор остатка и DIPEA (119 мл) в DCM (500 мл) перемешивали при 50°C в течение 15 ч и затем гасили NH4Cl(водн) (500 мл, 2M). Отделенную водную фазу экстрагировали DCM (2×100 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали с получением неочищенного S-III-I (93,62 г, выход: 100%).
Раствор S-III-I (93,62 г) и азид натрия (148,12 г) в DMF (500 мл) перемешивали при 25°C в течение 15 ч и затем гасили NH4Cl(водн) (50 мл, 2M). Полученный раствор экстрагировали Et2O (3×50 мл). Объединенные экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали с получением неочищенного S-III-II (77,11 г, выход: 80%).
Смесь S-III-II (77,11 г), трифенилфосфина (96,02 г), и воды (20 мл) в THF (1820 мл) перемешивали при 25°C в течение 15 ч. Полученную смесь экстрагировали этилацетатом (3×500 мл). Экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с MeOH/NH4OH(водн) (9:1) с получением амино продукта. К смеси амино продукта в дихлорметане (1000 мл) и NaHCO3(водн) (400 мл, 2N) при 5-10°C добавляли бензилхлорформиат (49,13 г). Смесь перемешивали при комнатной температуре в течение 15 ч и затем гасили водным NH4Cl(водн) (400 мл, 2 M). Водную фазу экстрагировали дихлорметаном (3×400 мл). Экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (3:1) с получением продукта S-III-III (74,35 г, выход: 64% за 2 стадии).
К раствору S-III-III (7,02 г) в сухом CH2Cl2 (100 мл) добавляли DIBAL-H (28,5 мл, 1,0 M в толуоле) при -78°C. Смесь перемешивали при -78°C в течение 2 ч и затем гасили метанолом (15 мл) при -78°C. К смеси добавляли HCl(водн) (80 мл, 1н) и смесь перемешивали при 0°C в течение 1 ч. Отделенный водный слой экстрагировали дихлорметаном (2×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали с получением неочищенного S-III-IV. Суспензию S-III-IV и трифенилфосфоранилиденацетальдегида (4,38 г) в толуоле (100 мл) нагревали при 80°C в течение 5 ч и затем выливали в воду (100 мл). Водную фазу экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали с получением неочищенного S-III-V (5,28 г, выход: 80% за две стадии).
Смесь S-III-V (6,02 г), N-циклогексил-1,3-пропандиамина (3,12 г), и MgSO4 (4,82 г) в CH2Cl2 (50 мл) перемешивали при 25°C в течение 2 ч и затем фильтровали и концентрировали. К раствору остатка в MeOH (40 мл) при 5-10°C добавляли NaBH4 (1,11 г). Смесь энергично перемешивали при 25°C в течение 1 ч и затем выливали в H2O. Полученную смесь концентрировали и затем остаток экстрагировали CH2Cl2 (2×150 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. К фильтрату добавляли Boc2O ангидрид (8,72 г) и TEA (5 мл) в одной порции. Смесь перемешивали при комнатной температуре в течение 2 ч и затем концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (3:1) с получением продукта S-III-VI (7,72 г, выход: 60% за 2 стадии).
Раствор S-III-VI (7,72 г) и Pd/C (0,77 г) в этаноле (200 мл) перемешивали в атмосфере H2(g) при 25°C в течение 5 ч. Полученную смесь фильтровали и затем концентрировали с получением продукта S-III (5,51 г, выход: 90%)
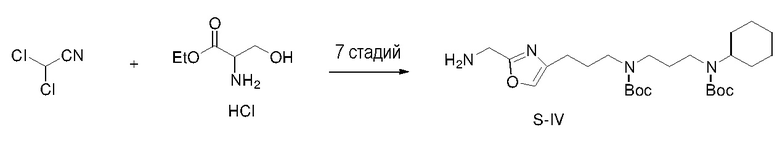
Получение S-IV
Боковую цепь S-IV получали по схеме, показанной ниже, способом, аналогичным тому, который использовали для получения S-III.
Получение S-V
Боковую цепь S-V получали по схемам, показанным ниже:
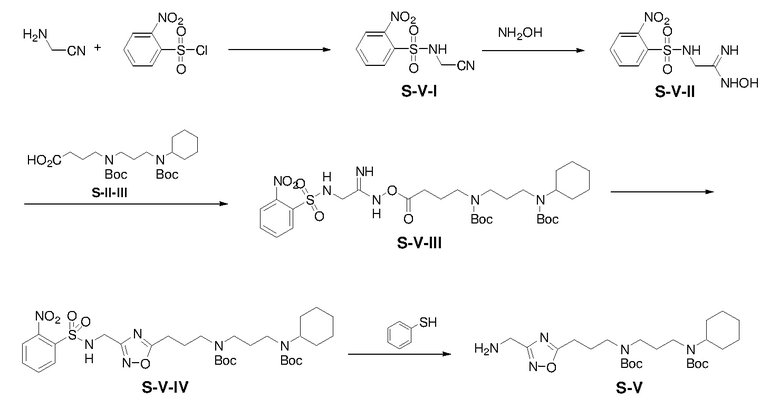
К раствору аминоацетонитрил гидрохлоридной соли (5,02 г) и TEA (16,38 г) в EtOH (100 мл) при 5-10°C добавляли раствор 2-нитробенен сульфонилхлорида (11,43 г) в сухом THF (20 мл) по каплям в течение 5 мин. Смесь перемешивали при 25°C в течение 15 ч и затем концентрировали. Остаток выливали в воду и смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением неочищенного S-V-I (9,43 г, выход: 72%).
Раствор S-V-I (4,49 г) и NH2OH (5,02 г, 50% в H2O масс/масс) в MeOH (50 мл) нагревали при 40°C в течение 1 ч и затем концентрировали. Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного S-V-II (4,14 г, выход: 81%)
Раствор S-V-II (10,02 г), S-II-III (24,32 г), EDCI (10,50 г), и DMAP (6,71 г) в сухом THF (120 мл) перемешивали при 25°C в течение 6 ч и затем выливали в воду. Полученную смесь экстрагировали этилацетатом (3×120 мл). Объединенные органические экстракты промывали NaHCO3(водн) и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (9:1) с получением продукта S-V-III (12,02 г, выход: 47%).
Раствор S-V-III (5,00 г) в толуоле (30 мл) нагревали при 120°C в течение 8 ч и затем выливали в воду. Полученную смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (3:1) с получением соединения S-V-IV (2,03 г, выход: 42%).
Раствор S-V-IV (5,56 г), тиофенола (0,9 мл), и Cs2CO3 (7,95 г) в сухом THF (40 мл) перемешивали при 25°C в течение 15 ч и затем выливали в воду. Полученную смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с MeOH/NH4OH (9:1) с получением соединения S-V (2,80 г, выход: 69%).
Получение S-VI
Боковую цепь S-VI получали по схемам, показанным ниже:
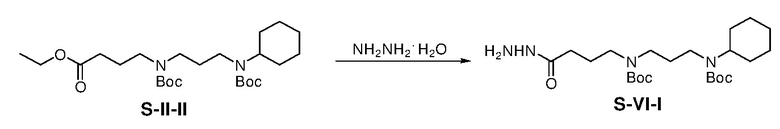
Раствор S-II-II (42,05 г) и гидразин моногидрата (31,31 г) в этаноле (420 мл) в атмосфере азота нагревали при 70°C в течение 15 ч и затем концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с MeOH/DCM (1/19) с получением продукта S-VI-I (25,30 г, выход: 62%).
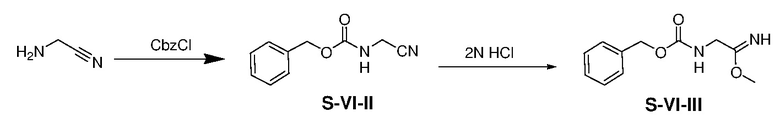
К раствору аминоацетонитрил гидрохлорида (25,27 г) и K2CO3 (109,80 г) в THF/H2O (200 мл/400 мл) при 5-10°C добавляли бензилхлорформиат (45,22 г) в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 15 ч и затем гасили NH4Cl(водн) (100 мл, 2 M). Полученную смесь экстрагировали этилацетатом (3×200 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-VI-II (46,88 г, выход: 90%).
К раствору S-VI-II (7,01 г) в метаноле (3 мл) по каплям добавляли HCl (50 мл, 2 н. в эфире). Смесь перемешивали при 25°C в течение 2 ч и затем фильтровали. Отфильтрованный осадок сушили с получением S-VI-III (8,02 г, выход: 98%).
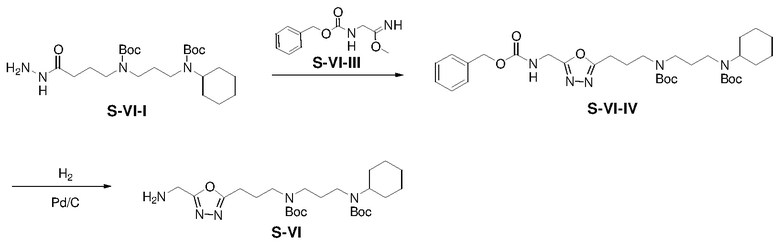
Раствор S-VI-I (3,71 г) и S-VI-III (8,02 г) в ACN (80 мл) перемешивали при 60°C в течение 48 ч и затем концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (1:1) с получением S-VI-IV (3,20 г, выход: 63%).
Раствор S-VI-IV (3,20 г) и Pd/C (0,32 г) в EtOH (20 мл) перемешивали в атмосфере H2(g) при 25°C в течение 16 ч. Полученную смесь фильтровали и концентрировали с получением S-VI (2,15 г, выход: 85%).
Получение S-VII
Боковую цепь S-VII получали по схеме, показанной ниже:
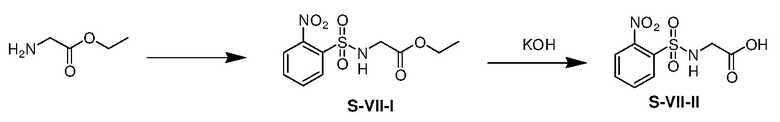
К раствору гидрохлорид этилового эфира глицина (29,81 г) и триэтиламина (64,74 г) в этаноле (600 мл) при 5-10°C в атмосфере азота добавляли раствор 2-нитробенен сульфонилхлорида (47,22 г) в тетрагидрофуране (600 мл). Смесь перемешивали при комнатной температуре в течение 15 ч и затем концентрировали. Остаток выливали в воду и полученную смесь экстрагировали этилацетатом (3×500 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-VII-I (54,22 г, выход: 88%).
К перемешиваемому магнитной мешалкой раствору соединения S-VII-I (54,22 г) в MeOH/THF (300 мл/300 мл) в атмосфере азота добавляли раствор KOH (31,63 г) в H2O (100 мл). Реакционную смесь перемешивали при 25 °C в течение 15 ч и затем подкисляли водным 4н HCl (140 мл). Полученную смесь концентрировали и остаток экстрагировали этилацетатом (3×300 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-VII-II (39,10 г, выход: 80%).
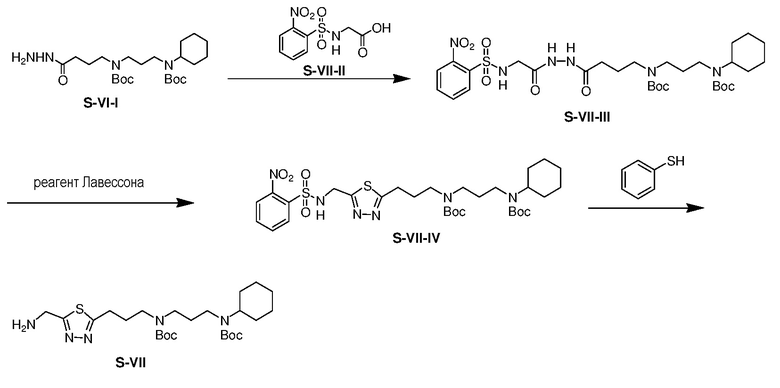
К перемешиваемому магнитной мешалкой раствору S-VII-II (6,10 г) в дихлорметане (120 мл) в атмосфере азота добавляли EDCI (4,93 г) при 25°C. Затем смесь перемешивали при 25°C в течение 1 ч, к смеси добавляли раствор соединения S-VI-I (8,23 г) в дихлорметане (20 мл) в одной порции. Реакционную смесь перемешивали в течение еще 6 ч и затем выливали в воду. Полученную смесь экстрагировали дихлорметаном (2×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1/19) с получением продукта S-VII-III (8,52 г, выход: 68%).
К перемешиваемому магнитной мешалкой раствору соединения S-VII-III (8,52 г) в дихлорметане (200 мл) добавляли реагент Лавессона (6,90 г). Смесь перемешивали при комнатной температуре в течение 15 ч и затем концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (1:1) с получением продукта S-VII-IV (4,85 г, выход: 57%).
Раствор S-VII-IV (6,40 г), карбоната цезия (5,97 г) и тиофенола (2,02 г) в ацетонитриле (120 мл) в атмосфере азота перемешивали при 25°C в течение 15 ч и затем концентрировали. Остаток выливали в воду и затем водный слой экстрагировали дихлорметаном (3×120 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/NH4OH (9:1) с получением продукта S-VII (4,55 г, выход: 97%).
Получение S-VIII
Боковую цепь S-VIII получали по схемам, показанным ниже:
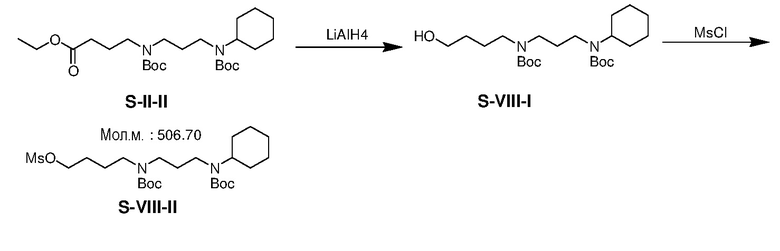
К раствору LAH (1,14 г) в THF (94 мл) при 5-10°C добавляли S-II-II (4,72 г) в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 6 ч и затем гасили хлоридом аммония NH4Cl(водн) (5,7 мл, 2 M). После добавления безводного сульфата натрия (5,71 г), полученную смесь перемешивали при 25°C в течение еще 1 ч и затем фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-VIII-I (3,85 г выход: 90%).
К раствору S-VIII-I (3,85 г) и TEA (2,02 г) в дихлорметане (180 мл) при 5-10°C добавляли MsCl (1,14 г) по каплям. Смесь перемешивали при комнатной температуре в течение 15 ч и затем гасили NH4Cl(водн). Водную фазу экстрагировали CH2Cl2 (2×50 мл). Объединенные органические экстракты промывали NaHCO3(водн) и насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-VIII-II (3,64 г, выход: 80%).
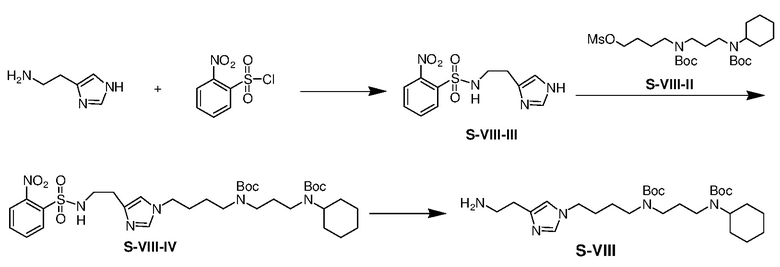
К раствору гистамина (1,02 г) и триэтиламина (2,01 г) в сухом THF (200 мл) при 5-10°C добавляли раствор 2-нитробенен сульфонилхлорида (2,21 г) в сухом THF (5 мл) по каплям в течение 5 мин. Смесь перемешивали при 25°C в течение 15 ч и затем концентрировали. Остаток выливали в воду и полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-VIII-III (1,61 г, выход: 60%).
Раствор S-VIII-III (1,61 г), K2CO3 (3,73 г) и S-VIII-II (4,01 г) в DMF (30 мл) нагревали при 80°C в течение 15 ч и затем выливали в воду. Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1/19) с получением продукта S-VIII-IV (0,76 г, выход: 20%).
Раствор S-VIII-IV (0,76 г), карбоната цезия (0,41 г), и тиофенола (0,18 г) в ацетонитриле (15 мл) в атмосфере азота перемешивали при 25°C в течение 15 ч и затем концентрировали. Остаток выливали в воду и полученную смесь экстрагировали дихлорметаном (3×50 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/NH4OH (9:1) с получением продукта S-VIII (0,51 г, выход: 91%).
Получение S-VIIII
Боковую цепь S-VIIII получали по схеме, показанной ниже:
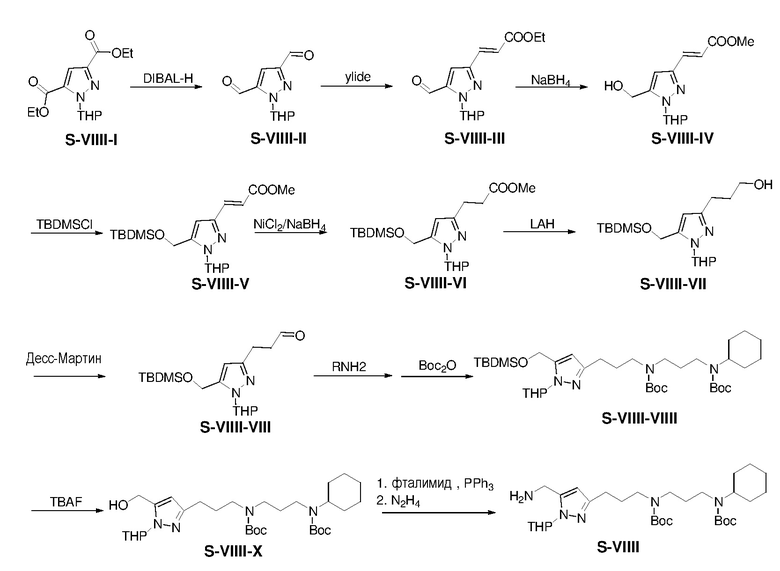
К раствору S-VIIII-I (10,02 г) в сухом CH2Cl2 (160 мл) добавляли DIBAL-H (70 мл, 1,0 M в толуоле) при -78°C. Смесь перемешивали при -78°C в течение 1 ч и затем гасили метанолом (100 мл) при -78°C. Полученную смесь фильтровали и фильтрат затем концентрировали с получением неочищенного S-VIIII-II. Суспензию (этоксикарбонилметилиден)трифенилфосфорана (6,91 г) и S-VIIII-II в толуоле (160 мл) нагревали при 80°C в течение 2 ч и затем выливали в воду (100 мл). Водную фазу экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали с получением неочищенного S-VIIII-III. Раствор соединения S-VIIII-III и NaBH4 (3,22 г) в MeOH (210 мл) перемешивали при 25°C в течение 15 ч и затем гасили NH4Cl(водн) (100 мл, 2M). Смесь концентрировали и остаток экстрагировали дихлорметаном (3×100 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на силикагеле с н-гексан/этилацетатом (1:1) с получением S-VIIII-IV (3,51 г, выход: 39% за три стадии)
Раствор S-VIIII-IV (3,5 г), имидазола (1,81 г), и TBDMSCl (2,38 г) в DCM (160 мл) перемешивали при 25°C в течение 15 ч и затем выливали в воду. Водную фазу экстрагировали дихлорметаном (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали с получением неочищенного S-VIIII-V. К раствору S-VIIII-V в MeOH (70 мл) при 0°C добавляли NiCl2 (18 мг) и NaBH4 (1,06 г). Смесь перемешивали при 0°C в течение 1 ч и затем гасили NH4Cl(водн) (1 мл, 2M). Полученную смесь фильтровали и фильтрат концентрировали с получением неочищенного S-VIIII-VI. К раствору S-VIIII-VI в THF (70 мл) при 0°C добавляли LAH (1,06 г). Смесь перемешивали при 0°C в течение 1 ч и затем гасили NaOH(водн) (4 мл, 10% масс./масс.). Полученную смесь фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (1:1) с получением S-VIIII-VII (2,03 г, выход: 43% за три стадии).
К раствору S-VIIII-VII (2,03 г) в дихлорметане (28 мл) при 0°C добавляли периодинан Десса-Мартина (2,51 г) в атмосфере азота. Смесь перемешивали при 0°C в течение 1 ч и затем гасили NaHCO3(водн) (30 мл, 2 M) и Na2S2O3(водн) (30 мл, 2 M). Водную фазу экстрагировали дихлорметаном (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного S-VIIII-VIII. Раствор S-VIIII-VIII, N-циклогексил-1,3-пропандиамина (1,07 г) и триацетоксиборгидрид натрия (2,43 г) в дихлорметане (28 мл) перемешивали при 25°C в течение 15 ч и затем выливали в NaHCO3(водн) (30 мл, 2M). Водный слой экстрагировали CH2Cl2 (2×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. К перемешиваемому магнитной мешалкой фильтрату и TEA (1,41 г) добавляли Boc2O ангидрид (3,26 г) в одной порции. Смесь перемешивали при комнатной температуре в течение 15 ч и затем концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (3:1) с получением продукта S-VIIII-VIIII. (2,27 г, выход: 57% за 2 стадии).
Раствор соединения S-VIIII-VIIII (2,27 г) и TBAF (4,9 мл, 1M в THF) в THF (16 мл) перемешивали при 25°C в течение 1 ч и затем выливали в NaHCO3(водн) (30 мл, 2M). Полученную смесь экстрагировали этилацетатом (2×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного S-VIIII-X. К раствору S-VIIII-X, фталимида (0,51 г), и PPh3 (0,91 г) в сухом THF (15 мл) при 0°C добавляли раствор DEAD (0,72 г) в сухом THF (1,5 мл) по каплям. Реакционную смесь перемешивали в атмосфере азота при 25°C в течение 15 ч и затем концентрировали. Раствор остатка и гидразин моногидрата (0,8 мл) в MeOH (20 мл) перемешивали при 25°C в течение 15 ч и затем фильтровали. Фильтрат концентрировали и полученный остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с MeOH/NH4OH (9:1) с получением S-VIIII (1,71 г, выход: 90% за две стадии)
Получение S-X
Боковую цепь S-X получали по схеме, показанной ниже:
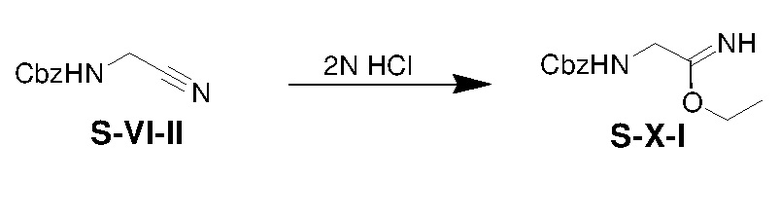
К раствору S-VI-II (10,02 г) в этаноле (3 мл) по каплям добавляли HCl (50 мл, 2 н. в эфире). Полученную смесь перемешивали при 25°C в течение 2 ч и затем фильтровали. Отфильтрованный осадок сушили при пониженном давлении с получением S-X-I (8,02 г, выход: 64%).
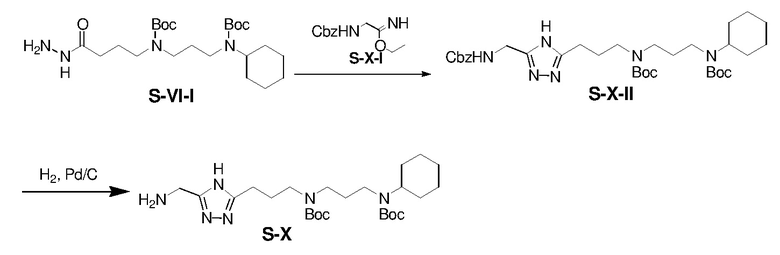
Раствор S-VI-I (4,22 г), CH3CO2K (4,13 г), и S-X-I (4,73 г) в н-BuOH (80 мл) перемешивали при 80°C в течение 1 ч, затем 125°C в течение 16 ч и затем концентрировали. Остаток очищали флэш-хроматографией на силикагеле с н-гексан/этилацетатом (1/1) с получением S-X-II (2,76 г, выход: 30%).
Раствор S-X-II (1,82 г) и 10% Pd/C (0,18 г) в EtOH (20 мл) перемешивали в атмосфере H2(g) при 25°C в течение 16 ч. Полученную смесь фильтровали и концентрировали с получением S-X (1,20 г, выход: 84%).
Получение S-XI и S-XII
Боковые цепи S-XI и S-XII получали по схеме, показанной ниже:
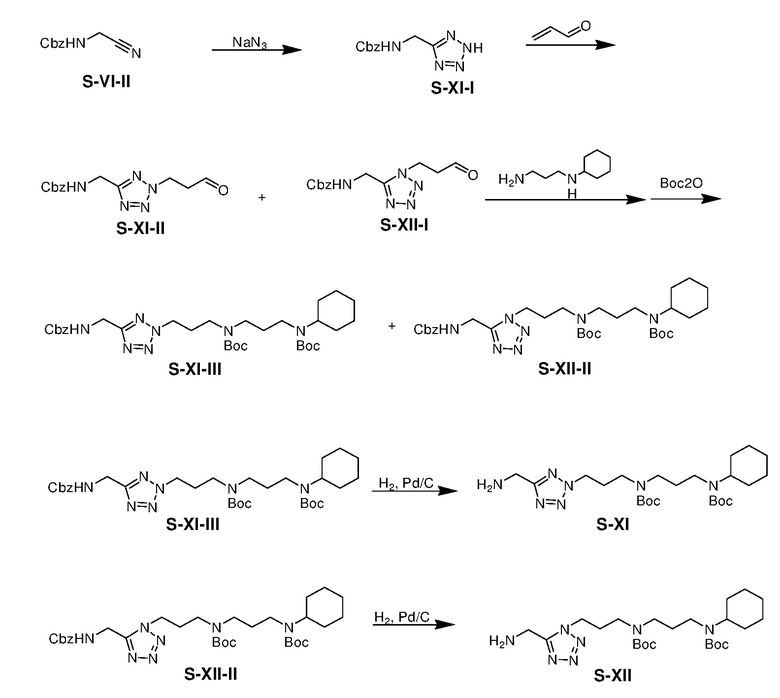
Раствор S-VI-II (37,10 г), азида натрия (31,73 г), и бромида цинка (30,75 г) в IPA/H2O (300 мл/600 мл) в атмосфере азота перемешивали при 75°C в течение 15 ч. К смеси при комнатной температуре медленно добавляли HCl(водн) (4 M) до полного растворения всего твердого вещества. Полученную смесь экстрагировали этилацетатом (3×200 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного продукта S-XI-I (43,22 г, выход: 95%).
К раствору S-XI-I (17,10 г) и TEA (29,65 г) в растворителе CH2Cl2/MeOH (320 мл/32 мл) при 5-10°C добавляли акролеин (16,43 г) по каплям. Полученную смесь перемешивали при комнатной температуре в течение 4 ч и затем гасили NH4Cl(водн) (50 мл). Полученную смесь концентрировали и затем остаток экстрагировали CH2Cl2 (3×200 мл). Объединенные органические экстракты промывали NaHCO3(водн) и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с MeOH/DCM (1/32) с получением смеси продукта S-XI-II и S-XII-I (16,90 г, выход: 80%).
К смеси S-XI-II и S-XII-I (25,10 г) в MeOH (250 мл) добавляли N-(3-аминопропил)циклогексиламин (16,26 г) в атмосфере азота. Смесь перемешивали при 0°C в течение 2 ч и к смеси медленно добавляли NaBH4 (2,78 г). Полученную смесь перемешивали в течение еще 1 ч и затем гасили NH4Cl(водн). Смесь концентрировали и остаток экстрагировали дихлорметаном (3×150 мл). Объединенные органические экстракты промывали NaHCO3(водн) и насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. К фильтрату добавляли Boc2O ангидрид (45,44 г) в одной порции. Смесь перемешивали при комнатной температуре в течение 15 ч и затем концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (1:1) с получением продукта S-XI-III (12,82 г, выход: 24% за 2 стадии) и S-XII-II (11,20 г, выход: 21% за 2 стадии).
Раствор S-XI-III (15,80 г) и 10% Pd/C (1,58 г) в 2-пропаноле (158 мл) перемешивали в атмосфере H2(g) при 60°C в течение 15 ч. Полученную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта S-XI (12,10 г, выход: 97%)
Раствор S-XII-II (11,20 г) и 10% Pd/C (1,12 г) в 2-пропаноле (112 мл) перемешивали в атмосфере H2(g) при 60°C в течение 15 ч. Полученную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта S-XII (8,37 г, выход: 95%).
Получение S-XIII
Боковую цепь S-XIII получали по схеме, показанной ниже:
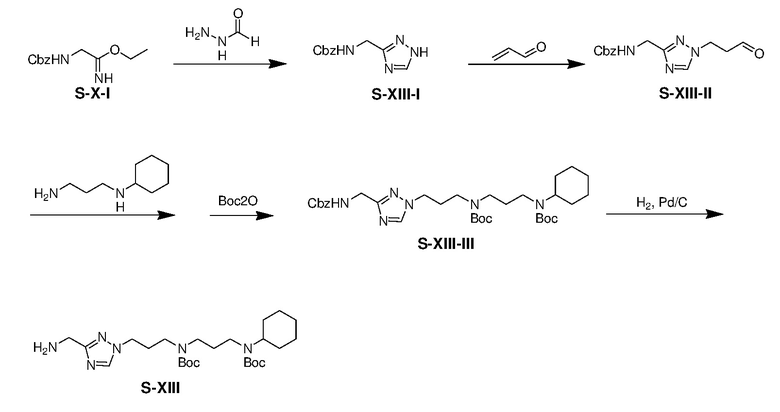
К раствору S-X-I (10,02 г) в CH2Cl2 (100 мл) при 0°С добавляли KOH(водн) (100 мл, 2,4% масс./масс.). Смесь перемешивали при 0°С в течение 10 мин и затем экстрагировали дихлорметаном (3×50 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Раствор остатка, формогидразида (3,31 г) и CH3CO2K (3,33 г) в н-BuOH (100 мл) перемешивали при 80°C в течение 1 ч, затем 125°C в течение 16 ч и затем концентрировали. Остаток кристаллизовали с н-гексан/этилацетатом (1/1) с получением S-XIII-I (7,21 г, выход: 73%).
К раствору S-XIII-I (4,05 г) и TEA (0,8 мл) в растворителе MeOH (20 мл) при -10°C добавляли акролеин (2 мл) по каплям. Полученную смесь перемешивали при -10°C в течение 3 ч и затем гасили NH4Cl(водн) (50 мл). Полученную смесь концентрировали и затем остаток экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали NaHCO3(водн) и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с MeOH/этилацетатом (1:10) с получением S-XIII-II (2,08 г, выход: 42%).
К смеси S-XIII-II (2,08 г) в MeOH (20 мл) при 0°C добавляли N-(3-аминопропил)циклогексиламин (1,6 мл) в атмосфере азота. Смесь перемешивали при 0°C в течение 2 ч и к смеси медленно добавляли NaBH4 (0,45 г). Полученную смесь перемешивали в течение еще 1 ч и затем гасили NH4Cl(водн). Смесь концентрировали и остаток экстрагировали дихлорметаном (3×150 мл). Объединенные органические экстракты промывали NaHCO3(водн) и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. К фильтрату добавляли Boc2O ангидрид (1,58 г) в одной порции. Смесь перемешивали при комнатной температуре в течение 15 ч и затем концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (1:2) с получением продукта S-XIII-III (2,42 г, выход: 54% за 2 стадии).
Раствор S-XIII-III (5,41 г) и 10% Pd/C (0,54 г) в EtOH (20 мл) перемешивали в атмосфере H2(g) при 25°C в течение 15 ч. Полученную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта S-XIII (3,69 г, выход: 87%)
Ниже приведены исходные вещества, то есть 2,4-дихлоргетероциклические производные, для получения соединений 1-86.
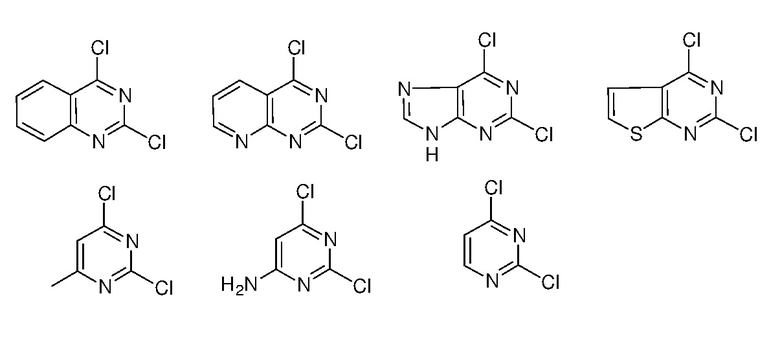
Ниже показан путь синтеза, по которому синтезировали некоторые соединения формулы (I), как показано в примере 1 ниже. Соединение А, содержащее две галогеновые группы, подвергали взаимодействию с аминосоединением R4-H, с получением соединения B, которое подвергали взаимодействию с другим аминосоединением R3-H (которое может быть таким же, как R4-H), с получением соединения C, то есть соединения формулы (I).
Соединения, синтезированные таким способом, очищали с использованием такого способа, как колоночная хроматография, высокоэффективная жидкостная хроматография или перекристаллизация.
Промежуточные соединения, используемые в синтезе, описанном выше, являются либо коммерчески доступными, либо они могут быть получены с использованием способов, известных в данной области. Способы могут также включать дополнительные стадии, либо до, либо после стадий, конкретно описанных в настоящем документе, для добавления или удаления подходящих защитных групп, если необходимо, для облегчения синтеза соединений. Кроме того, различные стадии синтеза могут быть выполнены в альтернативной последовательности или порядке для получения желаемых соединений.
Все химические вещества и растворители были приобретены у коммерческих поставщиков и использованы в том виде, в котором они были получены. Все реакции осуществляли в атмосфере сухого азота. Реакции контролировали с помощью TLC, используя пластины с силикагелем Merck 60 F254 со стеклянной подложкой (5×10 см); и зоны детектировали визуально под ультрафиолетовым излучением (254 нм) или путем опрыскивания реагентом фосфомолибденовой кислоты (Aldrich) с последующим нагреванием при 80°C. Всю колоночную флэш-хроматографию проводили с использованием Merck Kieselgel 60, No. 9385, 230-400 меш, силикагель ASTM в качестве неподвижной фазы. Спектры протонного ядерного магнитного резонанса (1H) измеряли на спектрометре Varian Mercury-300 или Varian Mercury-400. Химические сдвиги регистрировали в миллионных долях (м.д.) по шкале дельта (δ) относительно резонанса пика растворителя. Для описания характера расщепления использовали следующие сокращения: с=синглет; д=дублет; т=триплет; кв=квартет; квин= квинтет; шир.=широкий; и м=мультиплет. Данные LCMS измеряли на системе Agilent MSD-1100 ESI-MS/MS, Agilent 1200 серии LC/MSD VL и системе Waters Acquity UPLC-ESI-MS/MS.
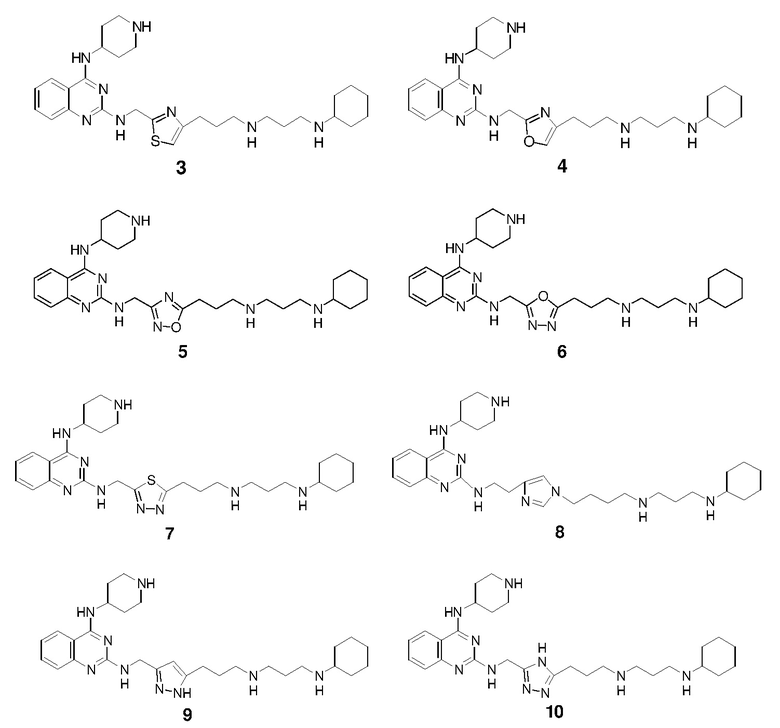
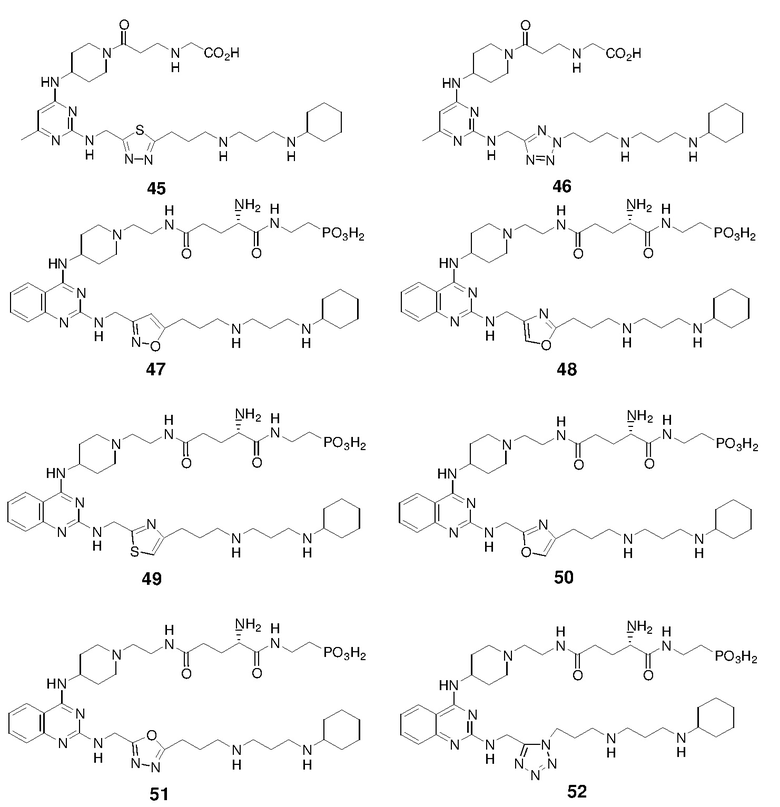
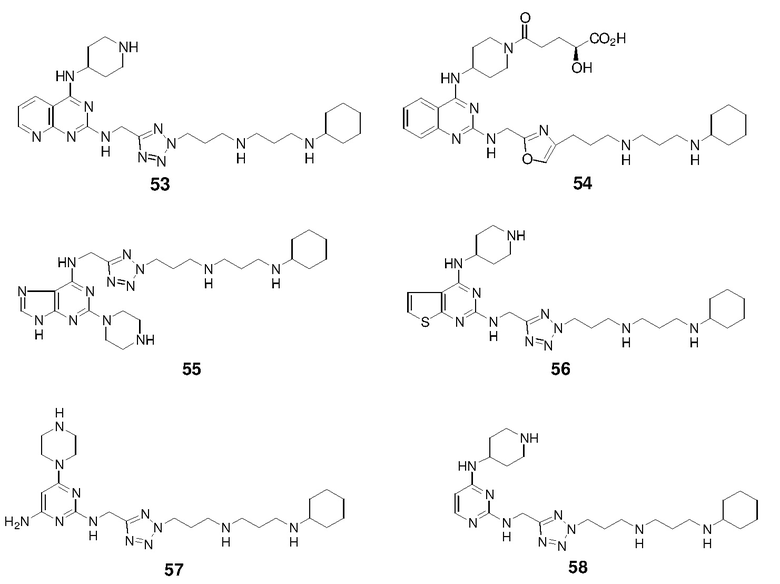
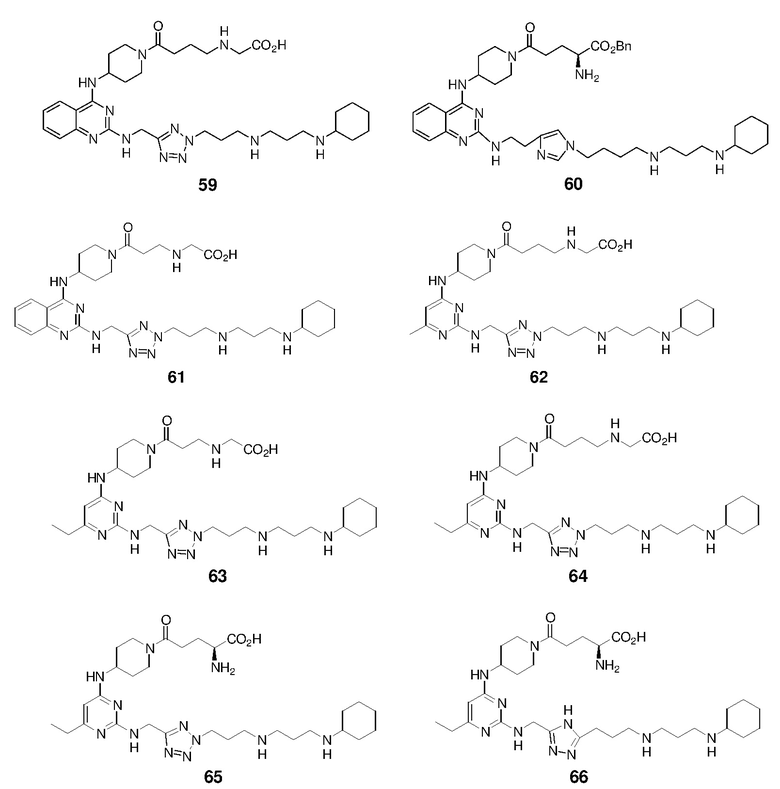
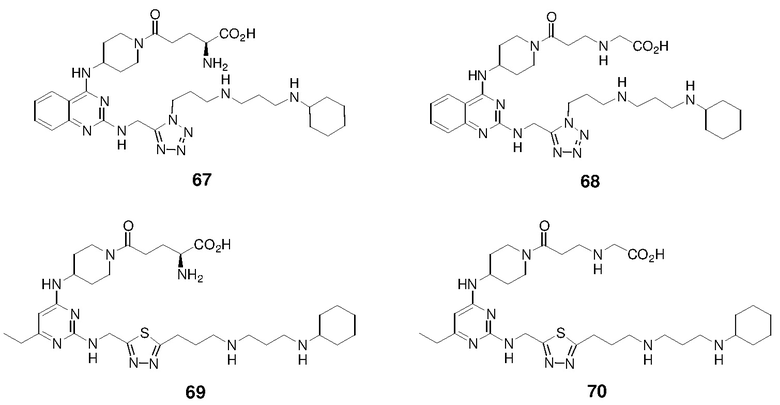
Пример 1: Синтез соединений 1-86
Соединения 1-86 синтезировали путем объединения исходных веществ и соединений с боковой цепью, указанных ниже:
Получение соединения 1
Ниже показана схема синтеза соединения 1 через промежуточные соединения 1-I и 1-II.
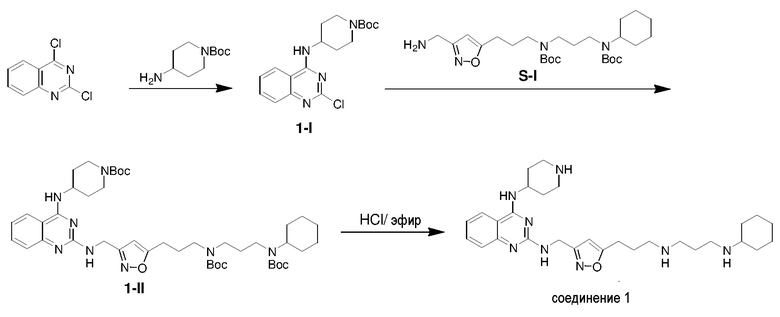
Раствор 2,4-дихлорхиназолина (1,01 г), 4-аминопиперидин-1-карбоновой кислоты трет-бутилового эфира (1,05 г) и триэтиламина (1,01 г) в THF (30 мл) в атмосфере азота перемешивали при 25°C в течение 15 ч и затем гасили водной NH4Cl (50 мл, 2 M). Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали флэш-хроматографией на силикагеле с н-гексаном/этилацетатом (1:1) с получением соединения 1-I (1,31 г, выход: 71%).
Раствор соединения 1-I (120,1 мг) и S-I (160,2 мг) в 1-пентаноле (1,4 мл) нагревали при 120°C в течение 15 мин с использованием микроволнового излучения и затем концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1/32) с получением соединения 1-II (150,1 мг, выход: 55%).
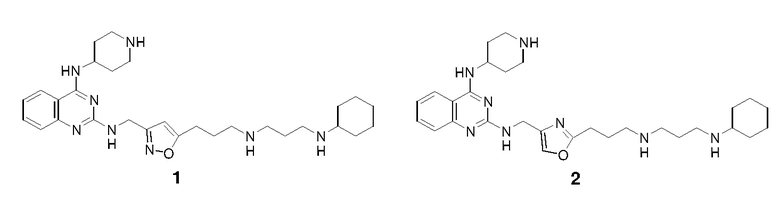
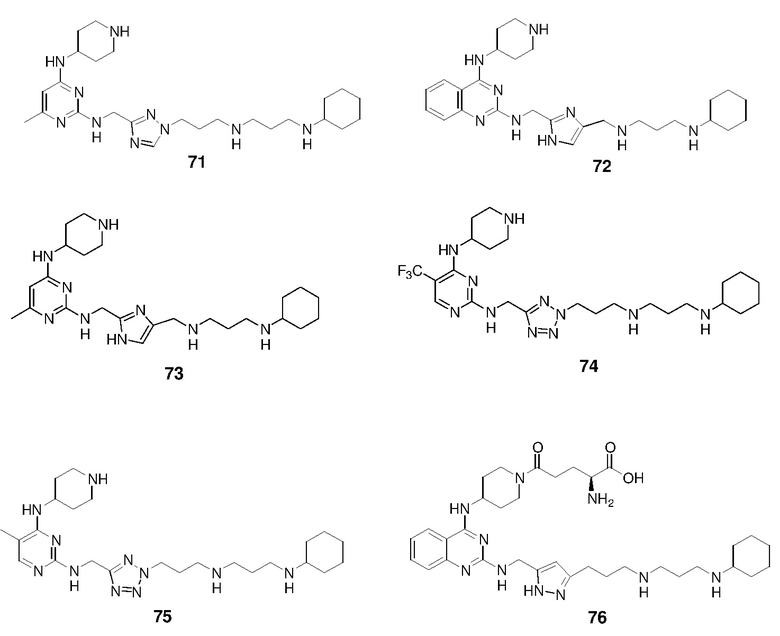
Раствор 1н HCl/диэтиловый эфир (3 мл) добавляли к раствору соединения 1-II (150,1 мг) в дихлорметане (6 мл). Реакционную смесь перемешивали при 25°C в течение 15 ч и затем концентрировали с получением гидрохлоридной соли соединения 1 (98,6 мг, выход: 86%). 1H ЯМР (400 МГц, D2O) δ 8,04 (д, 1H), 7,83 (дд, 1H), 7,49-7,43 (м, 2H), 6,38 (с, 1H), 4,77 (с, 2H), 4,46 (м, 1H), 3,58 (м, 2H), 3,25-3,13 (м, 8H), 2,93 (т, 2H), 2,21-2,03 (м, 8H), 1,99-1,81 (м, 4H), 1,69 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 521,5 (M+1).
Получение соединения 2
Соединение 2 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,07 (д, 1H), 7,87-7,80 (м, 2H), 7,51-7,43 (м, 2H), 4,69 (с, 2H), 4,58 (м, 1H), 3,56 (м, 2H), 3,20-3,02 (м, 8H), 2,96 (т, 2H), 2,33 (м, 2H), 2,21-2,03 (м, 6H), 2,01-1,81 (м, 4H), 1,70 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 521,5 (M+1).
Получение соединения 3
Соединение 3 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,01 (д, 1H), 7,79 (дд, 1H), 7,73 (с, 1H), 7,46-7,39 (м, 2H), 4,81 (с, 2H), 4,38 (м, 1H), 3,56 (м, 2H), 3,20-3,02 (м, 8H), 2,61 (т, 2H), 2,21-2,02 (м, 6H), 2,00-1,80 (м, 6H), 1,67 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 537,5 (M+1).
Получение соединения 4
Соединение 4 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,06 (д, 1H), 7,84 (дд, 1H), 7,52-7,43 (м, 2H), 7,24 (с, 1H), 5,03 (с, 2H), 4,42 (м, 1H), 3,56 (м, 2H), 3,20-3,01 (м, 8H), 2,87 (т, 2H), 2,18-2,02 (м, 8H), 1,96-1,79 (м, 4H), 1,69 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 521,5 (M+1).
Получение соединения 5
Соединение 5 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,05 (д, 1H), 7,83 (дд, 1H), 7,48-7,42 (м, 2H), 4,89 (с, 2H), 4,48 (м, 1H), 3,60 (м, 2H), 3,28-3,08 (м, 10H), 2,30-2,02 (м, 8H), 2,00-1,80 (м, 4H), 1,69 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 522,5 (M+1).
Получение соединения 6
Соединение 6 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,08 (д, 1H), 7,86 (м, 1H), 7,53-7,45 (м, 2H), 4,58 (м, 1H), 4,38 (с, 2H), 3,60 (м, 2H), 3,24-3,12 (м, 8H), 2,49 (т, 2H), 2,39 (м, 2H), 2,14-1,80 (м, 10H), 1,69 (м, 1H), 1,40-1,16 (м, 6H); EI-MS: 522,5 (M+1).
Получение соединения 7
Соединение 7 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,07 (д, 1H), 7,86 (дд, 1H), 7,53-7,45 (м, 2H), 5,13 (с, 2H), 4,40 (м, 1H), 3,58 (м, 2H), 3,30-3,11 (м, 10H), 2,24-2,02 (м, 8H), 2,00-1,82 (м, 4H), 1,69 (м, 1H), 1,40-1,16 (м, 6H); EI-MS: 538,5 (M+1).
Получение соединения 8
Соединение 8 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,80 (с, 1H), 8,06 (д, 1H), 7,82 (дд, 1H), 7,50-7,40 (м, 3H), 4,58 (м, 1H), 4,20 (т, 2H), 3,90 (т, 2H), 3,64 (м, 2H), 3,32-3,10 (м, 8H), 2,95 (м, 2H), 2,38 (м, 2H), 2,19-2,00 (м, 6H), 1,97-1,62 (м, 7H), 1,42-1,17 (м, 6H); EI-MS: 548,5 (M+1).
Получение соединения 9
Соединение 9 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,05 (д, 1H), 7,83 (дд, 1H), 7,48-7,44 (м, 2H), 6,33 (с, 1H), 4,80 (с, 2H), 4,45 (м, 1H), 3,54 (м, 2H), 3,20-3,06 (м, 8H), 2,80 (т, 2H), 2,20-2,02 (м, 8H), 2,00-1,80 (м, 4H), 1,69 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 520,5 (M+1).
Получение соединения 10
Соединение 10 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,02 (д, 1H), 7,82 (дд, 1H), 7,47-7,44 (м, 2H), 4,90 (с, 2H), 4,36 (м, 1H), 3,57 (м, 2H), 3,22-3,08 (м, 8H), 2,97 (т, 2H), 2,20-2,02 (м, 8H), 2,00-1,80 (м, 4H), 1,69 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 521,5 (M+1).
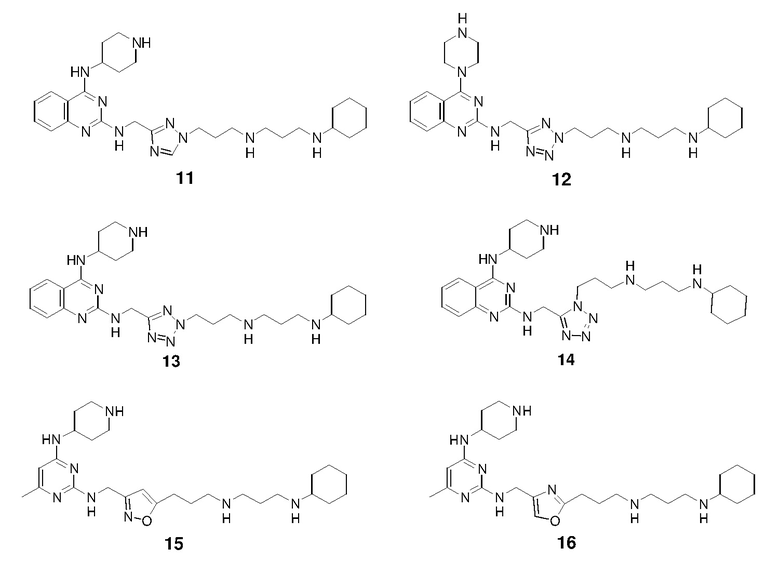
Получение соединения 11
Соединение 11 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,57 (с, 1H), 8,06 (д, 1H), 7,85 (м, 1H), 7,53-7,44 (м, 2H), 4,86 (с, 2H), 4,43 (м, 1H), 4,37 (т, 2H), 3,57 (м, 2H), 3,21-3,04 (м, 8H), 2,28 (м, 2H), 2,20-2,01 (м, 6H), 1,98-1,80 (м, 4H), 1,69 (м, 1H), 1,40-1,16 (м, 6H); EI-MS: 521,5 (M+1).
Получение соединения 12
Соединение 12 получали способом, аналогичным тому, который использовали для получения соединения 1. EI-MS: 508,5 (M+1).
Получение соединения 13
Соединение 13 получали способом, аналогичным тому, который использовали для получения соединения 1. 1H ЯМР (400 МГц, D2O) δ 8,08 (д, 1H), 7,87 (дд, 1H), 7,54-7,46 (м, 2H), 5,08 (с, 2H), 4,47 (м, 1H), 3,59 (м, 2H), 3,26-3,15 (м, 10H), 2,45 (м, 2H), 2,21-2,01 (м, 6H), 1,99-1,81 (м, 4H), 1,71 (м, 1H), 1,39-1,17 (м, 6H); EI-MS: 522,5 (M+1).
Получение соединения 14
Соединение 14 получали способом, аналогичным тому, который использовали для получения соединения 1. EI-MS: 522,5 (M+1).
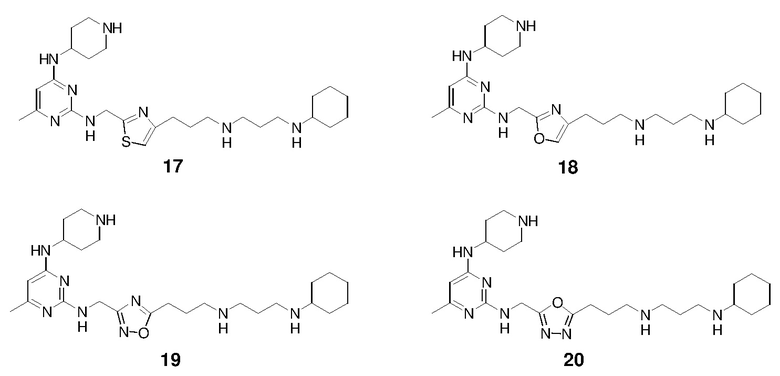
Получение соединения 15
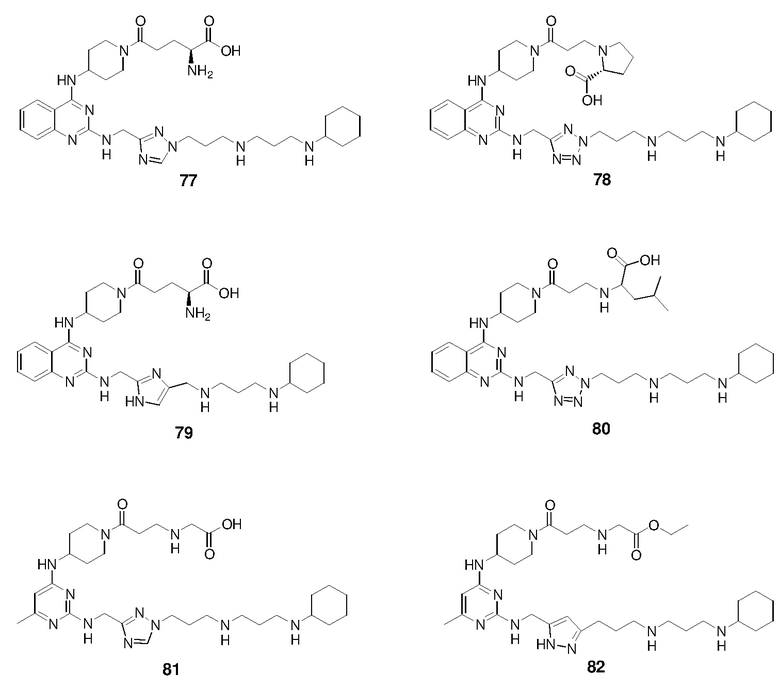
Ниже показана схема синтеза соединения 15 через промежуточные соединения 15-I и 15-II.
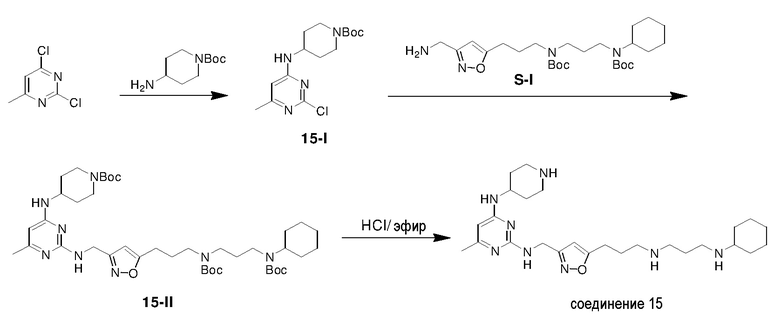
Раствор 2,4-дихлор-6-метилпиримидина (5,00 г), 4-аминопиперидин-1-карбоновой кислоты трет-бутилового эфира (8,36 г) и TEA (4,64 г) в THF (100 мл) в атмосфере азота перемешивали при 25°C в течение 15 ч и затем гасили водным NH4Cl (50 мл, 2 M). Полученную смесь экстрагировали этилацетатом (3×100 мл) и объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (3:1) с получением соединения 15-I (4,75 г, выход: 47%).
Раствор 15-I (70,2 мг) и S-I (110,3 мг) в 1-пентаноле (1,4 мл) нагревали при 140°C в течение 4 ч и затем концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1/32) с получением соединения 15-II (100,1 мг, выход: 59%).
Раствор 1н HCl/диэтиловый эфир (2 мл) добавляли к раствору соединения 15-II (100,1 мг) в дихлорметане (4 мл). Смесь перемешивали при 25°C в течение 15 ч и затем концентрировали с получением гидрохлоридной соли соединения 15 (67,8 мг, выход: 89%). 1H ЯМР (400 МГц, D2O) δ 6,33 (с, 1H), 5,95 (с, 1H), 4,69 (с, 2H), 4,16 (м, 1H), 3,49 (м, 2H), 3,27-3,07 (м, 8H), 2,93 (т, 2H), 2,28 (с, 3H), 2,19-1,99 (м, 8H), 1,87 (м, 2H), 1,79-1,64 (м, 3H), 1,42-1,17 (м, 6H); EI-MS: 485,5 (M+1).
Получение соединения 16
Соединение 16 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 7,78 (с, 1H), 5,95 (с, 1H), 4,56 (с, 2H), 4,27 (м, 1H), 3,49 (м, 2H), 3,22-3,14 (м, 8H), 2,95 (т, 2H), 2,26 (с, 3H), 2,20-2,04 (м, 8H), 1,90-1,77 (м, 4H), 1,64 (м, 1H), 1,40-1,16 (м, 6H); EI-MS: 485,5 (M+1).
Получение соединения 17
Соединение 17 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 7,31 (с, 1H), 5,97 (с, 1H), 4,95 (с, 2H), 4,11 (м, 1H), 3,43 (м, 2H), 3,21-3,00 (м, 8H), 2,88 (т, 2H), 2,35 (с, 3H), 2,18-1,99 (м, 8H), 1,85 (м, 2H), 1,76-1,62 (м, 3H), 1,41-1,17 (м, 6H); EI-MS: 501,5 (M+1).
Получение соединения 18
Соединение 18 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 7,71 (с, 1H), 5,96 (с, 1H), 4,72 (с, 2H), 4,15 (м, 1H), 3,46 (м, 2H), 3,23-3,03 (м, 8H), 2,61 (т, 2H), 2,28 (с, 3H), 2,18-1,96 (м, 8H), 1,86 (м, 2H), 1,79-1,64 (м, 3H), 1,41-1,18 (м, 6H); EI-MS: 485,5 (M+1).
Получение соединения 19
Соединение 19 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 5,96 (с, 1H), 4,77 (с, 2H), 4,15 (м, 1H), 3,50 (м, 2H), 3,24-3,08 (м, 10H), 2,28 (с, 3H), 2,26-2,03 (м, 8H), 1,87 (м, 2H), 1,80-1,63 (м, 3H), 1,40-1,17 (м, 6H); EI-MS: 486,4 (M+1).
Получение соединения 20
Соединение 20 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 6,00 (с, 1H), 4,25 (с, 2H), 4,17 (м, 1H), 3,50 (м, 2H), 3,25-3,06 (м, 10H), 2,29 (с, 3H), 2,26-2,02 (м, 8H), 1,90-1,73 (м, 4H), 1,68 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 486,4 (M+1).
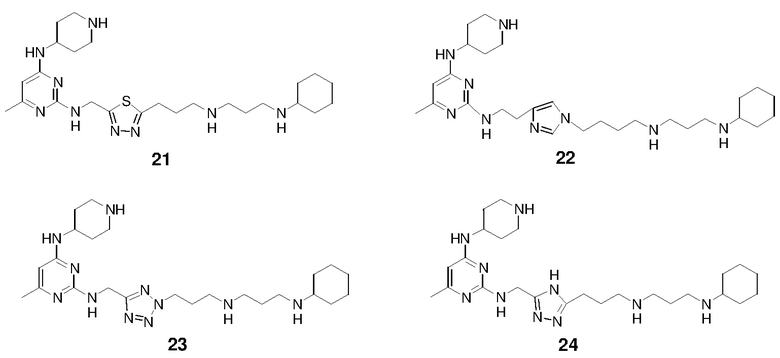
Получение соединения 21
Соединение 21 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 5,98 (с, 1H), 5,01 (с, 2H), 4,16 (м, 1H), 3,47 (м, 2H), 3,28-3,06 (м, 10H), 2,29 (с, 3H), 2,25-1,97 (м, 8H), 1,87 (м, 2H), 1,78-1,62 (м, 3H), 1,40-1,16 (м, 6H); EI-MS: 502,5 (M+1).
Получение соединения 22
Соединение 22 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 8,74 (с, 1H), 7,40 (с, 1H), 5,94 (с, 1H), 4,39-4,25 (м, 3H), 3,80 (м, 2H), 3,54 (м, 2H), 3,27-3,05 (м, 10H), 2,29 (м, 2H), 2,26 (с, 3H), 2,19-2,02 (м, 4H), 2,00-1,79 (м, 8H), 1,70 (м, 1H), 1,42-1,17 (м, 6H); EI-MS: 512,5 (M+1).
Получение соединения 23
Соединение 23 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 5,96 (с, 1H), 4,95 (с, 2H), 4,13 (м, 1H), 3,49 (м, 2H), 3,28-3,09 (м, 10H), 2,45 (м, 2H), 2,28 (с, 3H), 2,19-2,00 (м, 6H), 1,87 (м, 2H), 1,79-1,64 (м, 3H), 1,42-1,16 (м, 6H); EI-MS: 486,4 (M+1).
Получение соединения 24
Соединение 24 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 5,95 (с, 1H), 4,75 (с, 2H), 4,03 (м, 1H), 3,46 (м, 2H), 3,22-3,01 (м, 8H), 2,91 (м, 2H), 2,28 (с, 3H), 2,20-2,02 (м, 6H), 1,93 (м, 2H), 1,86 (м, 2H), 1,77-1,62 (м, 3H), 1,41-1,17 (м, 6H); EI-MS: 485,4 (M+1).
Получение соединения 25
Соединение 25 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 6,01 (с, 1H), 5,03 (с, 2H), 4,09 (м, 1H), 3,48 (м, 2H), 3,26-3,04 (м, 10H), 2,60 (кв, 2H), 2,24-1,96 (м, 9H), 1,84 (м, 2H), 1,70 (м, 2H), 1,41-1,13 (м, 9H); EI-MS: 516,5 (M+1).
Получение соединения 26
Соединение 26 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 5,98 (с, 1H), 4,97 (с, 2H), 4,14 (м, 1H), 3,46 (м, 2H), 3,22-3,12 (м, 10H), 2,58 (кв, 2H), 2,46 (м, 2H), 2,20-2,02 (м, 6H), 1,88 (м, 2H), 1,80-1,66 (м, 3H), 1,41-1,13 (м, 9H); EI-MS: 500,5 (M+1).
Получение соединения 27
Соединение 27 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (400 МГц, D2O) δ 5,98 (с, 1H), 4,77 (с, 2H), 4,06 (м, 1H), 3,47 (м, 2H), 3,28-3,02 (м, 8H), 2,93 (м, 2H), 2,58 (кв, 2H), 2,20-1,96 (м, 8H), 1,87 (м, 2H), 1,69 (м, 3H), 1,41-1,13 (м, 9H); EI-MS: 499,5 (M+1).
Получение соединения 28
Соединение 28 получали способом, аналогичным тому, который использовали для получения соединения 15.
1H ЯМР (400 МГц, D2O) δ 6,38 (шир.с, 1H), 5,96 (с, 1H), 4,72 (с, 2H), 4,16 (м, 1H), 3,48 (м, 2H), 3,21-3,04 (м, 8H), 2,82 (м, 2H), 2,29 (с, 3H), 2,17-2,00 (м, 8H), 1,87 (м, 2H), 1,77-1,64 (м, 3H), 1,42-1,17 (м, 6H); EI-MS: 484,5 (M+1).
Получение соединения 29
Ниже показана схема синтеза соединения 29 через промежуточные соединения 29-I - 29-IV.
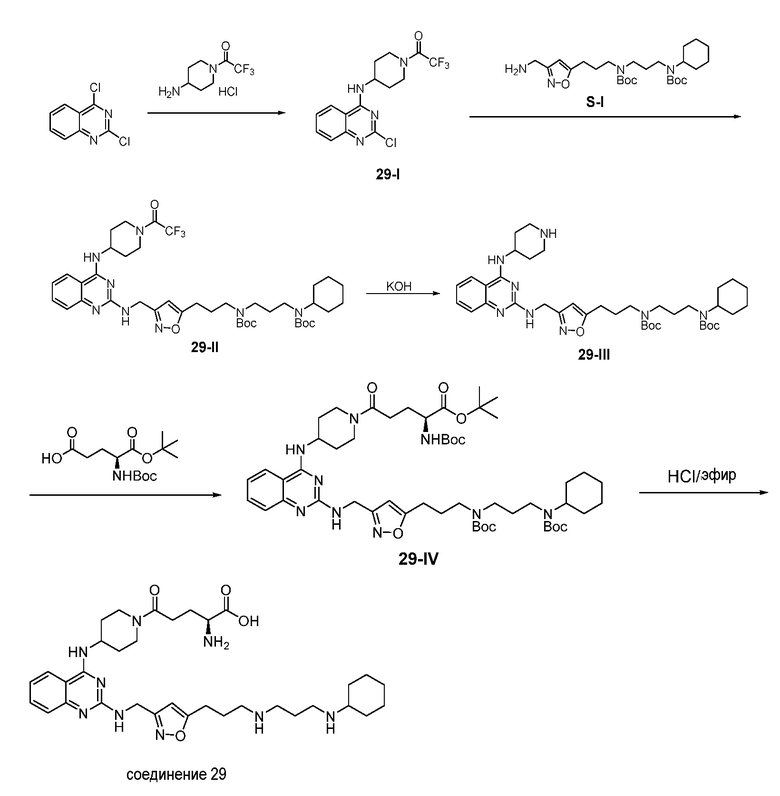
Раствор 2,4-дихлорхиназолина (1,02 г), гидрохлоридной соли 1-(4-аминопиперидин-1-ил)-2,2,2-трифторэтанона (1,21 г), и TEA (1,02 г) в THF (30 мл) в атмосфере азота перемешивали при 25°C в течение 15 ч и затем гасили водным NH4Cl (50 мл, 2 M). Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (1:1) с получением соединения 29-I (1,37 г, выход: 75%).
Раствор соединения 29-I (0,26 г) и S-I (0,36 г) в 1-пентаноле (2 мл) нагревали при 120°C в течение 15 мин с использованием микроволнового излучения и затем концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1:32) с получением соединения 29-II (0,29 г, выход: 49%).
К перемешиваемому магнитной мешалкой раствору соединения 29-II (0,29 г) в MeOH/THF (2,6 мл/2,6 мл) в атмосфере азота добавляли раствор KOH (0,05 г) в H2O (0,52 мл). Смесь перемешивали при 25°C в течение 15 ч и затем концентрировали. Полученный остаток экстрагировали дихлорметаном (3×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного соединения 29-III (0,24 г, выход: 94%).
К перемешиваемому магнитной мешалкой раствору 1-трет-бутилового эфира 2-трет-бутоксикарбониламино-пентандиовой кислоты (300,2 мг) в дихлорметане (20 мл) в атмосфере азота добавляли EDCI (120,3 мг) и HOBt (96,2 мг) при 25°C. Затем смесь перемешивали при 25°C в течение 1 ч, раствор 29-III (240,2 мг) в дихлорметане (10 мл) добавляли к смеси в одной порции. Реакционную смесь перемешивали в течение еще 6 ч и затем выливали в воду. Полученную смесь экстрагировали дихлорметаном (2×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1:19) с получением 29-IV (170,1 мг, выход: 51%).
Раствор 4н HCl/диоксан (0,85 мл) добавляли к раствору 29-IV (170,1 мг) в дихлорметане/1,4-диоксане (3,4 мл/3,4 мл). Смесь перемешивали при 25°C в течение 15 ч и концентрировали с получением гидрохлоридной соли соединения 29 (115,7 мг, выход: 90%). 1H ЯМР (400 МГц, D2O) δ 7,98 (д, 1H), 7,79 (т, 1H), 7,47-7,38 (м, 2H), 6,36 (с, 1H), 4,77 (с, 2H), 4,45 (м, 1H), 4,38 (м, 1H), 4,06-3,96 (м, 2H), 3,30 (м, 1H), 3,26-3,12 (м, 6H), 2,93 (м, 2H), 2,80 (м, 1H), 2,72 (м, 2H), 2,22 (м, 2H), 2,16-1,79 (м, 10H), 1,66 (м, 2H), 1,51 (м, 1H), 1,39-1,15 (м, 6H); EI-MS: 650,5 (M+1).
Получение соединения 30
Соединение 30 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 7,94 (м, 1H), 7,82-7,71 (м, 2H), 7,42-7,42 (м, 2H), 4,64 (с, 2H), 4,48-4,40 (м, 2H), 4,08-3,99 (м, 2H), 3,30-3,06 (м, 7H), 2,93 (м, 2H), 2,84 (м, 1H), 2,72 (м, 2H), 2,23-1,98 (м, 9H), 1,90-1,79 (м, 3H), 1,67 (м, 2H), 1,56 (м, 1H), 1,41-1,15 (м, 6H); EI-MS: 650,5 (M+1).
Получение соединения 31
Соединение 31 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 7,97 (д, 1H), 7,78 (т, 1H), 7,48 (с, 1H), 7,44-7,38 (м, 2H), 5,15 (д, 1H), 5,11 (д, 1H), 4,42 (м, 1H), 4,31 (м, 1H), 4,14 (м, 1H), 4,01 (м, 1H), 3,21-3,04 (м, 7H), 2,94 (м, 2H), 2,75-2,66 (м, 3H), 2,25 (м, 2H), 2,19-1,78 (м, 10H), 1,67 (м, 2H), 1,49 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 666,5 (M+1).
Получение соединения 32
Соединение 32 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 8,00 (д, 1H), 7,81 (т, 1H), 7,73 (с, 1H), 7,46-7,41 (м, 2H), 4,81 (с, 2H), 4,46 (м, 1H), 4,31 (м, 1H), 4,08-3,99 (м, 2H), 3,23 (м, 1H), 3,18-3,04 (м, 6H), 2,78-2,73 (м, 3H), 2,62 (м, 2H), 2,24 (м, 2H), 2,11-1,78 (м, 10H), 1,64 (м, 2H), 1,53 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 650,5 (M+1).
Получение соединения 33
Соединение 33 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 8,01 (д, 1H), 7,82 (т, 1H), 7,47-7,42 (м, 2H), 4,81 (с, 2H), 4,50 (м, 1H), 4,39 (м, 1H), 4,04-3,96 (м, 2H), 3,26-3,12 (м, 9H), 2,83 (м, 1H), 2,71 (м, 2H), 2,24-2,18 (м, 4H), 2,17-1,81 (м, 8H), 1,68 (м, 2H), 1,53 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 651,5 (M+1).
Получение соединения 34
Соединение 34 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 8,00 (д, 1H), 7,80 (т, 1H), 7,46-7,38 (м, 2H), 4,56-4,42 (м, 2H), 4,33 (с, 2H), 4,06-4,00 (м, 2H), 3,27 (м, 1H), 3,20-3,10 (м, 6H), 2,86 (м, 1H), 2,72 (м, 2H), 2,47 (м, 2H), 2,24-1,98 (м, 9H), 1,86-1,58 (м, 6H), 1,40-1,14 (м, 6H); EI-MS: 651,5 (M+1).
Получение соединения 35
Соединение 35 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 8,01 (д, 1H), 7,83 (т, 1H), 7,49-7,43 (м, 2H), 5,11 (д, 1H), 5,07 (д, 1H), 4,46 (м, 1H), 4,32 (м, 1H), 4,06-3,98 (м, 2H), 3,36-3,12 (м, 9H), 2,72-2,66 (м, 3H), 2,24-2,04 (м, 8H), 1,98-1,76 (м, 4H), 1,66 (м, 2H), 1,53 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 667,5 (M+1).
Получение соединения 36
Соединение 36 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 8,74 (с, 1H), 8,03 (д, 1H), 7,83 (т, 1H), 7,51-7,39 (м, 3H), 4,57-4,41 (м, 2H), 4,17 (м, 2H), 4,15-4,02 (м, 2H), 3,91 (м, 2H), 3,35-3,12 (м, 7H), 3,01-2,81 (м, 3H), 2,77 (м, 2H), 2,31-2,03 (м, 8H), 1,99-1,60 (м, 9H), 1,40-1,17 (м, 6H); EI-MS: 677,6 (M+1).
Получение соединения 37
Соединение 37 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 8,02 (д, 1H), 7,83 (т, 1H), 7,51-7,41 (м, 2H), 5,07 (д, 1H), 5,03 (д, 1H), 4,47 (м, 1H), 4,37 (м, 1H), 4,07-3,96 (м, 2H), 3,35-3,12 (м, 9H), 2,87 (м, 1H), 2,73 (м, 2H), 2,46 (м, 2H), 2,23 (м, 2H), 2,17-2,01 (м, 4H), 2,00-1,80 (м, 4H), 1,68 (м, 2H), 1,57 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 651,5 (M+1).
Получение соединения 38
Соединение 38 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 7,95 (д, 1H), 7,42 (т, 1H), 7,42-7,38 (м, 2H), 4,92 (д, 1H), 4,87 (д, 1H), 4,41 (м, 1H), 4,22 (м, 1H), 4,11 (м, 1H), 4,02 (м, 1H), 3,22-3,08 (м, 7H), 2,99 (т, 2H), 2,75 (м, 2H), 2,62 (м, 1H), 2,30-2,00 (м, 8H), 1,99-1,72 (м, 4H), 1,67 (м, 2H), 1,49 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 650,6 (M+1).
Получение соединения 39
Ниже показана схема синтеза соединения 39 через промежуточные соединения 39-I - 39-V.
Раствор 2,4-дихлор-6-метилпиримидина (0,82 г), гидрохлоридной соли 1-(4-аминопиперидин-1-ил)-2,2,2-трифторэтанона (1,21 г), и TEA (1,02 г) в THF (30 мл) в атмосфере азота перемешивали при 25°C в течение 15 ч и затем гасили водным NH4Cl (50 мл, 2 M). Полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с н-гексаном/этилацетатом (1:1) с получением соединения 39-I (1,07 г, выход: 66%).
Раствор соединения 39-I (0,24 г) и S-I (0,36 г) в 1-пентаноле (2 мл) нагревали при 120°C в течение 15 мин с использованием микроволнового излучения и затем концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1:32) с получением соединения 39-II (0,33 г, выход: 57%).
К перемешиваемому магнитной мешалкой раствору соединения 39-II (0,33 г) в MeOH/THF (2,6 мл/2,6 мл) в атмосфере азота добавляли раствор KOH (0,05 г) в H2O (0,52 мл). Смесь перемешивали при 25°C в течение 15 ч и затем концентрировали. Полученный остаток экстрагировали дихлорметаном (3×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного соединения 39-III (0,23 г, выход: 79%).
К перемешиваемому магнитной мешалкой раствору 3-(трет-бутоксикарбонил-этоксикарбонилметиламино)пропионовой кислоты (280,1 мг) в дихлорметане (20 мл) в атмосфере азота добавляли EDCI (116,4 мг) и HOBt (92,5 мг) при 25°C. Затем смесь перемешивали при 25°C в течение 1 ч, раствор 39-III (232,8 мг) в дихлорметане (20 мл) добавляли к смеси в одной порции. Реакционную смесь перемешивали в течение еще 6 ч и затем выливали в воду. Полученную смесь экстрагировали дихлорметаном (2×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1:19) с получением 39-IV (231,4 мг, выход: 72%).
К раствору 39-IV (231,4 мг) в THF (30 мл) в атмосфере азота добавляли раствор LiOH(водн) (1 мл, 1н). Смесь перемешивали при 25°C в течение 15 ч и затем гасили NH4Cl(водн) (20 мл, 2M). Водную фазу экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением неочищенного остатка 39-V (200,4 мг, выход: 89%).
Раствор 4н HCl/диоксан (1 мл) добавляли к раствору 39-V (200,4 мг) в дихлорметане/1,4-диоксане (4 мл/4 мл). Смесь перемешивали при 25°C в течение 15 ч и концентрировали с получением гидрохлоридной соли соединения 39 (154,7 мг, выход: 98%). 1H ЯМР (400 МГц, D2O) δ 6,34 (с, 1H), 5,92 (с, 1H), 4,73 (д, 1H), 4,65 (д, 1H), 4,30 (м, 1H), 4,10 (м, 1H), 3,90 (с, 2H), 3,88 (м, 1H), 3,41 (т, 2H), 3,31-3,12 (м, 7H), 3,00-2,82 (м, 5H), 2,30 (с, 3H), 2,18-2,00 (м, 7H), 1,98-1,77 (м, 4H), 1,70 (м, 1H), 1,53 (м, 1H), 1,46-1,16 (м, 6H); EI-MS: 614,5 (M+1).
Получение соединения 40
Соединение 40 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 7,78 (с, 1H), 5,91 (с, 1H), 4,58 (д, 1H), 4,52 (д, 1H), 4,32-4,20 (м, 2H), 3,97 (с, 2H), 3,92 (м, 1H), 3,44 (м, 2H), 3,36-3,12 (м, 7H), 3,04-2,90 (м, 5H), 2,26 (с, 3H), 2,21-1,80 (м, 10H), 1,69 (м, 1H), 1,57 (м, 1H), 1,46 (м, 1H),1,40-1,16 (м, 6H); EI-MS: 614,5 (M+1).
Получение соединения 41
Соединение 41 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 7,39 (с, 1H), 5,92 (с, 1H), 5,00 (д, 1H), 4,92 (д, 1H), 4,22 (м, 1H), 4,04 (м, 1H), 3,93 (с, 2H), 3,84 (м, 1H), 3,41 (т, 2H), 3,22-3,10 (м, 7H), 2,96 (т, 2H), 2,91 (т, 2H), 2,83 (м, 1H), 2,27 (с, 3H), 2,18-2,03 (м, 7H), 1,93-1,80 (м, 3H), 1,70 (м, 2H), 1,50 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 632,5 (M+1).
Получение соединения 42
Соединение 42 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 7,71 (с, 1H), 5,92 (с, 1H), 4,74 (д, 1H), 4,67 (д, 1H), 4,27 (м, 1H), 4,02 (м, 1H), 3,93 (м, 1H), 3,91 (с, 2H), 3,42 (м, 2H), 3,26-3,04 (м, 7H), 2,97 (м, 2H), 2,90 (м, 1H), 2,63 (т, 2H), 2,27 (с, 3H), 2,19-1,78 (м, 11H), 1,69 (м, 1H), 1,53 (м, 1H), 1,42-1,17 (м, 6H); EI-MS: 614,5 (M+1).
Получение соединения 43
Соединение 43 получали способом, аналогичным тому, который использовали для получения соединения 39.
1H ЯМР (400 МГц, D2O) δ 5,93 (с, 1H), 4,80 (д, 1H), 4,73 (д, 1H), 4,29 (м, 1H), 4,08 (м, 1H), 3,89 (м, 1H), 3,87 (с, 2H), 3,41 (т, 2H), 3,30-3,10 (м, 9H), 2,98 (т, 2H), 2,91 (м, 1H), 2,27 (с, 3H), 2,25-2,04 (м, 7H), 1,98-1,78 (м, 4H), 1,68 (м, 1H), 1,54 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 615,5 (M+1).
Получение соединения 44
Соединение 44 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 5,98 (с, 1H), 4,38-4,20 (м, 4H), 3,92 (м, 1H), 3,84 (с, 2H), 3,42 (т, 2H), 3,38-3,16 (м, 7H), 3,06 (м, 1H), 2,99 (т, 2H), 2,51 (т, 2H), 2,29 (с, 3H), 2,22-2,01 (м, 7H), 1,97-1,81 (м, 3H), 1,70 (м, 1H), 1,62-1,43 (м, 2H), 1,41-1,17 (м, 6H); EI-MS: 615,5 (M+1).
Получение соединения 45
Соединение 45 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 5,94 (с, 1H), 5,04 (д, 1H), 4,95 (д, 1H), 4,28 (м, 1H), 4,02 (м, 1H), 3,96-3,84 (м, 3H), 3,41 (т, 2H), 3,28-3,16 (м, 9H), 2,98 (т, 2H), 2,83 (м, 1H), 2,27 (с, 3H), 2,24-2,04 (м, 7H), 1,92-1,80 (м, 3H), 1,70 (м, 2H), 1,51 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 631,5 (M+1).
Получение соединения 46
Соединение 46 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 5,94 (с, 1H), 4,99 (д, 1H), 4,92 (д, 1H), 4,28 (м, 1H), 4,05 (м, 1H), 3,93 (с, 2H), 3,90 (м, 1H), 3,43 (т, 2H), 3,31-3,15 (м, 9H), 2,99 (т, 2H), 2,92 (м, 1H), 2,47 (м, 2H), 2,28 (с, 3H), 2,19-2,03 (м, 5H), 1,92-1,80 (м, 3H), 1,71 (м, 2H), 1,53 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 615,5 (M+1).
Получение соединения 47
Ниже показана схема синтеза соединения 47 через промежуточные соединения 47-I - 47-III.
К перемешиваемому магнитной мешалкой раствору соединения 29-III (241 мг) и K2CO3 (241 мг) в ацетонитриле (50 мл) в атмосфере азота добавляли 2-(2-бром-этил)изоиндол-1,3-дион (135 мг). Реакционную смесь перемешивали при 60°C в течение 15 ч и затем гасили NH4Cl(водн) (50 мл, 2 M). Полученную смесь экстрагировали дихлорметаном (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1:19) с получением 47-I (215 мг, выход: 72%).
К перемешиваемому раствору соединения 47-I (215 мг) в метаноле (5 мл) при 5°C добавляли 85% NH2NH2·H2O (40 мг) по каплям. Полученную смесь перемешивали при 25°C в течение 15 ч и затем концентрировали. Остаток выливали в K2CO3 (водн) (50 мл, 10% масс./масс.) и смесь экстрагировали CH2Cl2 (3×100 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/NH4OH (9:1) с получением 47-II (182,3 мг, выход: 99%).
К перемешиваемому магнитной мешалкой раствору 4-трет-бутоксикарбониламино-4-[2-(диэтоксифосфорил)этилкарбамоил]масляной кислоты (300,6 мг) в дихлорметане (50 мл) в атмосфере азота добавляли EDCI (91,2 мг) и HOBt (72,9 мг) при 25°C. Затем смесь перемешивали при 25°C в течение 1 ч, к смеси добавляли раствор соединения 47-II (182,3 мг) в дихлорметане (10 мл) в одной порции. Реакционную смесь перемешивали в течение еще 6 ч и затем выливали в воду. Полученную смесь экстрагировали дихлорметаном (2×50 мл). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1:19) с получением 47-III (241,2 мг, 87% выход).
TMSBr (0,8 мл) добавляли к раствору соединения 47-III (241,2 мг) в дихлорметане (15 мл). Реакционную смесь перемешивали при 25°C в течение 15 ч и концентрировали с получением гидробромидной соли соединения 47 (175,3 мг, выход: 81%). 1H ЯМР (400 МГц, D2O) δ 7,97 (д, 1H), 7,79 (дд, 1H), 7,42-7,36 (м, 2H), 6,40 (с, 1H), 4,78 (с, 2H), 4,42 (м, 1H), 4,07 (м, 1H), 3,81 (м, 2H), 3,69 (м, 2H), 3,52 (м, 2H), 3,40 (м, 2H), 3,30-3,10 (м, 8H), 2,94 (т, 2H), 2,50 (м, 2H), 2,24-1,93 (м, 14H), 1,85 (м, 2H), 1,68 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 800,5 (M+1).
Получение соединения 48
Соединение 48 получали способом, аналогичным тому, который использовали для получения соединения 47. 1H ЯМР (400 МГц, D2O) δ 8,02 (д, 1H), 7,84 (с, 1H), 7,80 (дд, 1H), 7,48-7,40 (м, 2H), 4,68 (с, 2H), 4,59 (м, 1H), 4,05 (м, 1H), 3,82 (м, 2H), 3,67 (м, 2H), 3,52 (м, 2H), 3,38 (м, 2H), 3,31-3,14 (м, 8H), 2,97 (т, 2H), 2,50 (м, 2H), 2,39 (м, 2H), 2,23-2,01 (м, 12H), 1,85 (м, 2H), 1,68 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 800,5 (M+1).
Получение соединения 49
Соединение 49 получали способом, аналогичным тому, который использовали для получения соединения 47. 1H ЯМР (400 МГц, D2O) δ 8,02 (д, 1H), 7,81 (дд, 1H), 7,48-7,40 (м, 2H), 7,24 (с, 1H), 5,01 (с, 2H), 4,42 (м, 1H), 3,99 (м, 1H), 3,69 (м, 2H), 3,65 (м, 2H), 3,54 (м, 1H), 3,45 (м, 1H), 3,34 (м, 2H), 3,20-3,04 (м, 8H), 2,87 (т, 2H), 2,47 (м, 2H), 2,24-1,97 (м, 12H), 1,92-1,80 (м, 4H), 1,68 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 816,5 (M+1).
Получение соединения 50
Соединение 50 получали способом, аналогичным тому, который использовали для получения соединения 47. 1H ЯМР (400 МГц, D2O) δ 8,00 (д, 1H), 7,80 (дд, 1H), 7,75 (с, 1H), 7,46-7,40 (м, 2H), 4,82 (с, 2H), 4,40 (м, 1H), 4,01 (м, 1H), 3,73 (м, 2H), 3,67 (м, 2H), 3,54 (м, 1H), 3,43 (м, 1H), 3,38 (м, 2H), 3,22-3,08 (м, 8H), 2,62 (т, 2H), 2,48 (м, 2H), 2,25-1,96 (м, 14H), 1,84 (м, 2H), 1,68 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 800,6 (M+1).
Получение соединения 51
Соединение 51 получали способом, аналогичным тому, который использовали для получения соединения 47. 1H ЯМР (400 МГц, D2O) δ 8,07 (д, 1H), 7,87 (дд, 1H), 7,54-7,46 (м, 2H), 5,01 (с, 2H), 4,39 (м, 1H), 4,05 (м, 1H), 3,84 (м, 2H), 3,69 (м, 2H), 3,51 (м, 2H), 3,41 (м, 2H), 3,36-3,02 (м, 10H), 2,51 (м, 2H), 2,32-1,96 (м, 14H), 1,87 (м, 2H), 1,69 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 801,6 (M+1).
Получение соединения 52
Соединение 52 получали способом, аналогичным тому, который использовали для получения соединения 47. 1H ЯМР (400 МГц, D2O) δ 8,07 (д, 1H), 7,86 (дд, 1H), 7,55-7,43 (м, 2H), 5,17 (с, 2H), 4,69 (т, 2H), 4,46 (м, 1H), 4,06 (м, 1H), 3,82 (м, 2H), 3,69 (м, 2H), 3,56-3,12 (м, 12H), 2,53-2,38 (м, 4H), 2,32-1,80 (м, 14H), 1,69 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 801,6 (M+1).
Получение соединения 53
Соединение 53 получали способом, аналогичным тому, который использовали для получения соединения 1. EI-MS: 523,5 (M+1).
Получение соединения 54
Соединение 54 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 8,02 (д, 1H), 7,83 (т, 1H), 7,75 (с, 1H), 7,50-7,42 (м, 2H), 4,82 (с, 2H), 4,47 (м, 1H), 4,40-4,24 (м, 2H), 4,06 (м, 1H), 3,25 (м, 1H), 3,20-3,04 (м, 6H), 2,80 (м, 1H), 2,70-2,60 (м, 4H), 2,20-1,78 (м, 9H), 1,92-1,80 (м, 3H), 1,71 (м, 2H), 1,53 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 651,5 (M+1).
Получение соединения 55
Ниже показана схема синтеза соединения 55 через промежуточные соединения 55-I - 55-III.
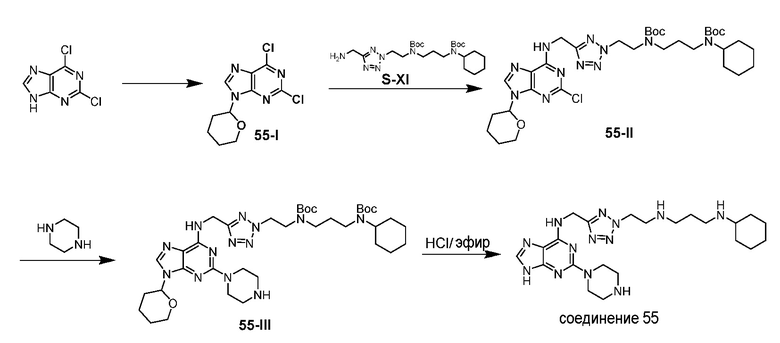
К перемешиваемому магнитной мешалкой раствору 2,6-дихлорпурина (10 г) в этилацетате (100 мл) добавляли моногидрат п-толуолсульфоновой кислоты (0,08 г). Полученную смесь нагревали до 50°C в атмосфере азота и 3,4-дигидро-2Н-пиран (7,5 мл) добавляли в течение 2 ч. Смесь перемешивали при 25°C в течение 15 ч и фильтровали с получением неочищенного твердого вещества. Твердое вещество промывали н-гексаном/этилацетатом (1:1) с получением соединения 55-I (14,4 г, выход: 100%)
К перемешиваемому магнитной мешалкой раствору соединения 55-I (0,65 г) в этилацетате (35 мл) в атмосфере азота добавляли соединение S-XI (1,15 г) и TEA (0,75 г). Смесь нагревали до 50°C в течение 4 ч, охлаждали до 25°C, и затем гасили водным NH4Cl (50 мл, 2 M). Полученный раствор экстрагировали этилацетатом (3×100 мл). Объединенные экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1:9) с получением соединения 55-II (1,16 г, выход: 68%) в виде светло-желтого твердого вещества.
Раствор соединения 55-II (1,05 г) и пиперазина (1,00 г) в 1-пентаноле (6 мл) нагревали при 100°C в течение 15 ч и затем концентрировали. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле с MeOH/DCM (1:1) с получением соединения 55-III (0,67 г, выход: 60%).
Раствор 1н HCl/диэтиловый эфир (5,3 мл) добавляли к раствору соединения 55-III (264 мг) в дихлорметане (10,6 мл). Реакционную смесь перемешивали в течение 15 ч и концентрировали с получением гидрохлоридной соли соединения 55 (204 мг, 94% выход). EI-MS: 498,5 (M+1).
Получение соединения 56
Соединение 56 получали способом, аналогичным тому, который использовали для получения соединения 1. EI-MS: 528,5 (M+1).
Получение соединения 57
Соединение 57 получали способом, аналогичным тому, который использовали для получения соединения 15. EI-MS: 473,5 (M+1).
Получение соединения 58
Соединение 58 получали способом, аналогичным тому, который использовали для получения соединения 15. EI-MS: 472,5 (M+1).
Получение соединения 59
Соединение 59 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 8,02 (д, 1H), 7,83 (т, 1H), 7,46-7,41 (м, 2H), 5,10 (д, 1H), 5,02 (м, 1H), 4,47 (м, 1H), 4,36 (м, 1H), 4,05 (м, 1H), 3,95 (с, 2H), 3,34-3,12 (м, 9H), 2,84 (м, 1H), 2,69 (т, 2H), 2,58 (т, 2H), 2,47 (м, 2H), 2,17-2,00 (м, 7H), 1,94-1,80 (м, 3H), 1,71 (м, 2H), 1,51 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 665,6 (M+1).
Получение соединения 60
Соединение 60 получали способом, аналогичным тому, который использовали для получения соединения 29. EI-MS: 767,6 (M+1).
Получение соединения 61
Соединение 61 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 8,03 (д, 1H), 7,84 (т, 1H), 7,51-7,45 (м, 2H), 5,11 (д, 1H), 5,02 (д, 1H), 4,48 (м, 1H), 4,28 (м, 1H), 4,01-3,83 (м, 3H), 3,45 (т, 2H), 3,31-3,11 (м, 7H), 3,02 (т, 2H), 2,95-2,81 (м, 3H), 2,48 (м, 2H), 2,15-1,97 (м, 5H), 1,92-1,81 (м, 3H), 1,71 (м, 2H), 1,57 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 651,5 (M+1).
Получение соединения 62
Соединение 62 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 5,93 (с, 1H), 4,99 (д, 1H), 4,92 (д, 1H), 4,28 (м, 1H), 4,05 (м, 1H), 3,96 (м, 1H), 3,87 (с, 2H), 3,34-3,14 (м, 11H), 2,90 (м, 1H), 2,66 (т, 2H), 2,47 (м, 2H), 2,28 (с, 3H), 2,19-2,00 (м, 7H), 1,94-1,81 (м, 3H), 1,71 (м, 2H), 1,51 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 629,5 (M+1).
Получение соединения 63
Соединение 63 получали способом, аналогичным тому, который использовали для получения соединения 39.
1H ЯМР (400 МГц, D2O) δ 5,95 (с, 1H), 4,98 (д, 1H), 4,91 (д, 1H), 4,29 (м, 1H), 4,04 (м, 1H), 3,98-3,86 (м, 3H), 3,43 (т, 2H), 3,38-3,18 (м, 9H), 2,99 (т, 2H), 2,92 (м, 1H), 2,59 (кв, 2H), 2,46 (м, 2H), 2,32-1,64 (м, 10H), 1,52 (м, 1H), 1,41-1,17 (м, 9H); EI-MS: 629,5 (M+1).
Получение соединения 64
Соединение 64 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 5,94 (с, 1H), 4,99 (д, 1H), 4,92 (д, 1H), 4,28 (м, 1H), 4,05 (м, 1H), 3,99-3,84 (м, 3H), 3,34-3,14 (м, 11H), 2,89 (м, 1H), 2,65 (т, 2H), 2,58 (кв, 2H), 2,46 (м, 2H), 2,19-2,00 (м, 7H), 1,94-1,81 (м, 3H), 1,70 (м, 2H), 1,51 (м, 1H), 1,41-1,17 (м, 9H); EI-MS: 643,6 (M+1).
Получение соединения 65
Соединение 65 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 5,94 (с, 1H), 4,98 (д, 1H), 4,93 (д, 1H), 4,29 (м, 1H), 4,12-4,01 (м, 2H), 3,93 (м, 1H), 3,30-3,14 (м, 9H), 2,90 (м, 1H), 2,72 (м, 2H), 2,58 (кв, 2H), 2,46 (м, 2H), 2,26-2,05 (м, 7H), 1,92-1,82 (м, 3H), 1,69 (м, 2H), 1,51 (м, 1H), 1,40-1,17 (м, 9H); EI-MS: 629,5 (M+1).
Получение соединения 66
Соединение 66 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 5,94 (с, 1H), 4,82 (с, 2H), 4,28 (м, 1H), 4,10-3,83 (м, 3H), 3,18-3,12 (м, 7H), 2,95 (м, 2H), 2,83 (м, 1H), 2,71 (м, 2H), 2,58 (кв, 2H), 2,26-2,04 (м, 9H), 1,94-1,78 (м, 3H), 1,69 (м, 2H), 1,50 (м, 1H), 1,40-1,17 (м, 9H); EI-MS: 628,5 (M+1).
Получение соединения 67
Соединение 67 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 8,04 (д, 1H), 7,86 (т, 1H), 7,51-7,43 (м, 2H), 5,12 (с, 2H), 4,70 (т, 2H), 4,47 (м, 1H), 4,27 (м, 1H), 4,12-4,01 (м, 2H), 3,31-3,13 (м, 7H), 2,86 (м, 1H), 2,75 (м, 2H), 2,43 (м, 2H), 2,28 (м, 2H), 2,17-2,04 (м, 4H), 1,95-1,80 (м, 4H), 1,71 (м, 2H), 1,57 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 651,5 (M+1).
Получение соединения 68
Соединение 68 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 8,04 (д, 1H), 7,84 (т, 1H), 7,51-7,42 (м, 2H), 5,15 (с, 2H), 4,69 (т, 2H), 4,49 (м, 1H), 4,30 (м, 1H), 4,01-3,90 (м, 3H), 3,44 (т, 2H), 3,36-3,13 (м, 7H), 3,02 (т, 2H), 2,90 (м, 1H), 2,43 (м, 2H), 2,18-2,05 (м, 4H), 1,94-1,81 (м, 4H), 1,71 (м, 2H), 1,58 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 651,5 (M+1).
Получение соединения 69
Соединение 69 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (400 МГц, D2O) δ 5,95 (с, 1H), 5,02 (д, 1H), 5,00 (д, 1H), 4,31 (м, 1H), 4,09-3,86 (м, 3H), 3,30-3,12 (м, 9H), 2,85 (м, 1H), 2,70 (м, 2H), 2,58 (кв, 2H), 2,28-2,05 (м, 9H), 1,92-1,80 (м, 3H), 1,70 (м, 2H), 1,50 (м, 1H), 1,40-1,17 (м, 9H); EI-MS: 645,5 (M+1).
Получение соединения 70
Соединение 70 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (400 МГц, D2O) δ 5,96 (с, 1H), 5,05 (д, 1H), 4,97 (д, 1H), 4,29 (м, 1H), 4,03 (м, 1H), 3,98-3,84 (м, 3H), 3,43 (т, 2H), 3,32-3,13 (м, 9H), 2,99 (т, 2H), 2,86 (м, 1H), 2,58 (кв, 2H), 2,28-2,04 (м, 7H), 1,92-1,80 (м, 3H), 1,71 (м, 2H), 1,52 (м, 1H), 1,41-1,17 (м, 9H); EI-MS: 645,5 (M+1).
Получение соединения 71
Соединение 71 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (300 МГц, D2O) δ 8,67 (с, 1H), 5,94 (с, 1H), 4,76 (с, 2H), 4,36 (т, 2H), 4,10 (м, 1H), 3,44 (м, 2H), 3,21-3,02 (м, 8H), 2,32 (м, 2H), 2,26 (с, 3H), 2,19-1,98 (м, 6H), 1,84 (м, 2H), 1,79-1,60 (м, 3H), 1,42-1,16 (м, 6H); EI-MS: 485,6 (M+1).
Получение соединения 72
Соединение 72 получали способом, аналогичным тому, который использовали для получения соединения 1. EI-MS: 492,6 (M+1).
Получение соединения 73
Соединение 73 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (300 МГц, D2O) δ 7,66 (с, 1H), 6,01 (с, 1H), 4,97 (с, 2H), 4,41 (с, 2H), 4,05 (м, 1H), 3,45 (м, 2H), 3,28- 3,03 (м, 6H), 2,29 (с, 3H), 2,18-1,96 (м, 6H), 1,86 (м, 2H), 1,79-1,60 (м, 3H), 1,41-1,18 (м, 6H); EI-MS: 456,6 (M+1).
Получение соединения 74
Compound 74 получали способом, аналогичным тому, который использовали для получения соединения 15. 1H ЯМР (300 МГц, D2O) δ 8,21 (с, 1H), 5,05 (с, 2H), 4,08 (м, 1H), 3,49 (м, 2H), 3,21-3,06 (м, 10H), 2,43 (м, 2H), 2,19-2,00 (м, 7H), 1,90-1,62 (м, 7H), 1,42-1,16 (м, 6H); EI-MS: 540,7 (M+1).
Получение соединения 75
Соединение 75 получали способом, аналогичным тому, который использовали для получения соединения 15. EI-MS: 486,6 (M+1).
Получение соединения 76
Соединение 76 получали способом, аналогичным тому, который использовали для получения соединения 29.
1H ЯМР (300 МГц, D2O) δ 7,94 (д, 1H), 7,76 (т, 1H), 7,42-7,36 (м, 2H), 6,52 (с, 1H), 4,81 (с, 2H), 4,41 (м, 1H), 4,27 (м, 1H), 4,08 (м, 1H), 3,99 (м, 1H), 3,22-3,06 (м, 7H), 2,86 (т, 2H), 2,80-2,66 (м, 3H), 2,22 (м, 2H), 2,16-2,00 (м, 6H), 1,99-1,59 (м, 4H),1,66 (м, 2H), 1,49 (м, 1H), 1,39-1,15 (м, 6H); EI-MS: 649,6 (M+1).
Получение соединения 77
Соединение 77 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (300 МГц, D2O) δ 8,65 (с, 1H), 7,93 (д, 1H), 7,76 (т, 1H), 7,41-7,34 (м, 2H), 4,80 (м, 2H), 4,44 (м, 1H), 4,37 (т, 2H), 4,25 (м, 1H), 4,11 (м, 1H), 3,99 (м, 1H), 3,25-3,02 (м, 7H), 2,77 (м, 1H), 2,73 (м, 2H), 2,31-2,20 (м, 4H), 2,17-2,01 (м, 4H), 2,00-1,80 (м, 4H), 1,68 (м, 2H), 1,57 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 650,6 (M+1).
Получение соединения 78
Соединение 78 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (300 МГц, D2O) δ 7,93 (д, 1H), 7,77 (т, 1H), 7,41-7,35 (м, 2H), 5,05 (д, 1H), 4,97 (д, 1H), 4,43 (м, 1H), 4,29 (м, 1H), 4,19 (м, 1H), 3,90 (м, 1H), 3,58 (м, 2H), 3,31-3,06 (м, 9H), 2,98 (м, 2H), 2,81 (м, 1H), 2,55-2,38 (м, 4H), 2,22-1,77 (м, 12H), 1,71 (м, 2H), 1,57 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 691,6 (M+1).
Получение соединения 79
Соединение 79 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (300 МГц, D2O) δ 8,02 (д, 1H), 7,83 (т, 1H), 7,76 (с, 1H), 7,50-7,42 (м, 2H), 5,09 (м, 2H), 4,44 (с, 2H), 4,40 (м, 1H), 4,23 (м, 1H), 4,08-3,99 (м, 2H), 3,23-3,04 (м, 5H), 2,78-2,73 (м, 3H), 2,22 (м, 2H), 2,18-2,00 (м, 4H), 1,94-1,70 (м, 4H), 1,64 (м, 2H), 1,53 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 621,7 (M+1).
Получение соединения 80
Соединение 80 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (300 МГц, D2O) δ 7,92 (д, 1H), 7,74 (т, 1H), 7,41-7,35 (м, 2H), 5,00 (м, 2H), 4,43 (м, 1H), 4,29 (м, 1H), 4,02-3,95 (м, 2H), 3,42 (м, 2H), 3,31-3,06 (м, 9H), 2,99 (м, 2H), 2,81 (м, 1H), 2,42 (м, 2H), 2,14-2,00 (м, 5H), 1,98-1,60 (м, 8H), 1,53 (м, 1H), 1,41-1,17 (м, 6H), 0,99 (д, 6H); EI-MS: 707,7 (M+1).
Получение соединения 81
Соединение 81 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (300 МГц, D2O) δ 8,76 (с, 1H), 5,91 (с, 1H), 4,77 (м, 2H), 4,39 (т, 2H), 4,22 (м, 1H), 4,05 (м, 1H), 3,99 (с, 2H), 3,84 (м, 1H), 3,43 (т, 2H), 3,24-3,08 (м, 7H), 2,97 (т, 2H), 2,87 (м, 1H), 2,31 (м, 2H), 2,25 (с, 3H), 2,19-2,03 (м, 5H), 1,92-1,61 (м, 5H), 1,53 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 614,6 (M+1).
Получение соединения 82
Соединение 82 получали способом, аналогичным тому, который использовали для получения соединения 39. EI-MS: 641,7 (M+1).
Получение соединения 83
Соединение 83 получали способом, аналогичным тому, который использовали для получения соединения 39. EI-MS: 585,6 (M+1).
Получение соединения 84
Соединение 84 получали способом, аналогичным тому, который использовали для получения соединения 47. EI-MS:788,6 (M+1).
Получение соединения 85
Соединение 85 получали способом, аналогичным тому, который использовали для получения соединения 39. 1H ЯМР (300 МГц, D2O) δ 7,45 (с, 1H), 4,92 (м, 2H), 4,40 (м, 1H), 4,16 (м, 1H), 3,95 (с, 2H), 3,91 (м, 1H), 3,42 (т, 2H), 3,25-3,10 (м, 9H), 2,99 (т, 2H), 2,76 (м, 1H), 2,44 (м, 2H), 2,19-2,03 (м, 4H), 1,96 (с, 3H), 1,92-1,61 (м, 6H), 1,53 (м, 1H), 1,41-1,17 (м, 6H); EI-MS: 615,6 (M+1).
Получение соединения 86
Соединение 86 получали способом, аналогичным тому, который использовали для получения соединения 29. 1H ЯМР (300 МГц, D2O) δ 7,44 (с, 1H), 4,92 (м, 2H), 4,40 (м, 1H), 4,21 (м, 1H), 4,06 (м, 1H), 3,96 (м, 1H), 3,26-3,10 (м, 9H), 2,80-2,64 (м, 3H), 2,41 (м, 2H), 2,22 (м, 2H), 2,16-2,02 (м, 4H), 1,95 (с, 3H), 1,90-1,60 (м, 6H), 1,51 (м, 1H), 1,40-1,17 (м, 6H); EI-MS: 615,6 (M+1).
Пример 2: Ингибирование связывания радиолиганда в человеческих CXCR4-трансфицированных клетках HEK293
Конкурентное связывание между соединениями формулы (I) и CXCL12 человека оценивали с использованием анализа связывания радиолиганда, как описано ниже.
Мембраны (2-4 мкг), полученные из человеческих CXCR4-трансфицированных клеток HEK293, в 40 мкл буфера для анализа (50 мМ HEPES-NaOH, pH 7,4, 100 мМ NaCl, 5 мМ MgCl2, 1 мМ CaCl2, 0,5% бычий сывороточный альбумин) инкубировали с 20 мкл радиоактивно меченного 125I-CXCL12 (0,16 нМ) и 20 мкл тестируемого соединения в планшете для анализа (Costar Corning, Cambridge, MA). Через 60 минут при 30°C инкубацию прекращали путем переноса полученной реакционной смеси на 96-луночный фильтровальный планшет GF/B (Millipore Corp., Billerica, MA) и фильтровали через коллектор. Планшет четыре раза промывали 100 мкл ледяного промывочного буфера (50 мМ HEPES-NaOH, pH 7,4, 100 мМ NaCl). Радиоактивность, связанная с фильтром, была измерена с помощью Topcount (PerkinElmer Inc., Waltham, MA).
Неожиданно было обнаружено, что концентрация, необходимая для ингибирования связывания 125I-CXCL12 с CXCR4 на 50% (IC50) из 25 испытываемых соединений, была ниже 50 нМ, 33 испытанных соединения имели значения IC50 50-100 нМ, а 28 испытанных соединений имели IC50 значения 100-1000 нМ. Более конкретно, список соединений, показывающих значения IC50 ниже, чем 50 нМ, включает соединения 1-7, 9, 12, 13, 15-19, 21, 23, 25, 28-30, 40, 42, 59 и 75; список соединений, показывающих значения IC50 50-100 нМ, включает соединения 8, 10, 11, 14, 20, 22, 24, 26, 27, 31-35, 37-39, 43, 45-50, 58, 61, 62, 66, 72, 73, 76, 78 и 82; и список соединений, показывающих значения IC50 100-1000 нМ, включает соединения 36, 41, 44, 51-57, 60, 63-65, 67-71, 74, 77, 79-81 и 83-86.
Эти результаты показывают, что соединения формулы (I) имеют высокую аффинность связывания с CXCR4.
Пример 3: Ингибирование хемотаксиса в клетках лимфобластного лейкоза
Ответ раковых клеток на соединения формулы (I) оценивали с использованием хемотаксического анализа, как изложено ниже.
Клетки острого Т-клеточного лимфобластного лейкоза (CCRF-CEM) в среде Roswell Park Memorial Institute (RPMI) 1640, дополненной 10% бычьим сывороточным альбумином, инкубировали с 250 мкл тестируемого соединения. Анализ осуществляли, используя вставки для культивирования клеток Millicell Hanging (размер пор 5 мкм; 24-луночный планшет; Millipore, Bedford, MA, USA). Через 10 минут при 37°C, 250 мкл клеток, предварительно инкубированных с тестируемым соединением, высевали на лунку в верхние камеры вставок с плотностью 2,5×105 клеток/лунку. 300 мкл/лунку среды, содержащей CXCL12 (10 нМ) и тестируемое соединение, высевали в нижнюю камеру вставки. Через 2,5 ч при 37°С клетки в обеих камерах вставок измеряли проточной цитометрией (Guava Technologies, Hayward, CA, USA).
Было обнаружено, что 39 испытываемых соединений неожиданно показали концентрации, необходимые для ингибирования хемотаксиса на 50% (ЕС50) со значениями ниже 50 нМ, а 4 испытываемые соединения показали значения ЕС50 50-150 нМ. Более конкретно, список соединений, показывающих значения EC50 ниже 50 нМ, включает соединения 1-8, 10, 13-18, 20-24, 26, 29-32, 35, 37-42, 45, 47-49, 59, 61 и 62; и список соединений, показывающих значения ЕС50 50-150 нМ, включает соединения 33, 34, 46 и 50.
Эти результаты показывают, что соединения формулы (I) обладают высокой эффективностью в ингибировании хемотаксиса определенных раковых клеток.
Пример 4: Влияние на мобилизацию стволовых клеток у мышей
38 соединений формулы (I) были протестированы для оценки их эффективности в усилении мобилизации стволовых клеток/клеток-предшественников следующим образом. Список этих 38 соединений включает соединения 1-3, 13, 15, 17, 24, 26, 29-31, 33, 35, 36, 38-43, 45, 46, 49, 50, 54, 59-68, 76, 78 и 83.
Каждое из 38 соединений растворяли в физиологическом растворе с образованием раствора. Раствор вводили подкожно мышам-самцам C57BL/6 (National Laboratory Animal Center, Taipei, Taiwan). Мышей, которые получали физиологический раствор, использовали в качестве контроля. Цельную кровь собирали через 2 ч после подкожной инъекции и метили следующими антителами: (i) APC-конъюгированное анти-CXCR4 (клон 2B11; eBioscience), (ii) FITC-конъюгированное анти-CD34 (клон RAM34; eBioscience), (iii) PE-конъюгированное анти-CD133 (клон 13A4; eBioscience), (iv) анти-c-kit (клон 2B8; eBioscience), (v) анти-Sca-1 (клон D7; eBioscience), (vi) анти-линейное (Mouse Hematopoietic Lineage Biotin Panel, eBioscience) и (vii) стрептавидин PE-Cy7 (eBioscience). Гемопоэтические стволовые клетки (CD34+) и эндотелиальные клетки-предшественники (CD133+) определяли количественно с помощью окрашивания поверхности антителами и проточной цитометрии (Guava Technologies, Hayward, CA, USA).
Неожиданно, эти 38 соединений значительно усилили мобилизацию гемопоэтических стволовых клеток CD34+ (до 3,7 раз) и эндотелиальных клеток-предшественников CD133+ (до 4,5 раз) в периферическую кровь по сравнению с контролем физиологическим раствором. Кроме того, было обнаружено, что 4 тестируемых соединения, то есть соединения 40, 45, 49 и 50, в сочетании с G-CSF неожиданно синергически мобилизуют гемопоэтические стволовые клетки, о чем свидетельствует значительное увеличение количества CFU-GM.
Эти результаты показывают, что соединения формулы (I) обладают высокой эффективностью в усилении мобилизации стволовых клеток/клеток-предшественников.
Пример 5: Лечение ишемически-реперфузионного повреждения почки у крыс
Эффективность пяти соединений формулы (I) при лечении ишемически-реперфузионного повреждения оценивали с использованием модели острого почечного повреждения, модели ишемического инсульта и модели ишемии конечности. Эти пять соединений представляют собой соединения 13, 35, 40, 45 и 46.
В модели острого почечного повреждения (AKI) каждое из пяти соединений растворяли в физиологическом растворе с образованием раствора. Раствор вводили подкожно самцам крыс Sprague-Dawley (National Laboratory Animal Center, Taipei, Taiwan) в дозе 6 мг/кг. Через 40 минут после подкожной инъекции у крыс индуцировали AKI путем зажима их двусторонней почечной вены и артерии в течение одного часа с последующим высвобождением зажимов сосуда для обеспечения 24-часовой реперфузии. Цельную кровь собирали через 24 часа после индукции AKI. Азот мочевины в крови (BUN) и сывороточный креатинин (Scr), два маркера, которые увеличиваются при повреждении почек, измеряли с помощью анализатора FUJI DRI-CHEM 3500s (Fujifilm, Tokyo, Japan). В качестве контроля использовали крыс без AKI и крыс с AKI, получавших физиологический раствор.
Было обнаружено, что у крыс с AKI, которым вводили тестируемые соединения, неожиданно были уровни BUN и Scr, соответственно, 11-25% и 10-56% от этих уровней, которые индуцировались у крыс с AKI, получавших физиологический раствор. Более конкретно, у крыс с AKI, которым вводили соединения 13, 35, 40, 45 и 46, соответствующие уровни BUN составляли 25%, 15%, 20%, 11% и 22% от этих уровней, индуцированных у крыс с AKI, получавших физиологический раствор; и соответствующие уровни Scr составляли 56%, 22%, 36%, 10% и 22% от этих уровней, индуцированных у крыс с AKI, получавших физиологический раствор.
Эти результаты показывают, что соединения формулы (I) обладают высокой эффективностью при лечении повреждения почек.
Пример 6: Лечение гепатоцеллюлярной карциномы (HCC) у мышей
Эффективность соединения формулы (I), то есть соединения 42, при лечении HCC оценивали с использованием модели сингенной мыши следующим образом.
Использовали клеточную линию HCA-1 HCC, полученную от C3H мыши. Клетки HCA-1 имплантировали ортотопически мышам C3H в течение 10 дней. Затем мышей лечили сорафенибом (низкомолекулярным лекарственным средством для лечения гепатоцеллюлярной карциномы; 40 мг/кг) ежедневно в течение двух недель или обрабатывали одним носителем (PBS) в качестве контроля. Тестируемые соединения, например, AMD3100 (10 мг/кг/день) и соединение 42 (10 мг/кг/день), вводили непрерывно каждой из мышей, которые получали лечение сорафенибом, используя осмотический насос Alzet (DURECT Corporation, Cupertino, CA) в течение двух недель.
Было обнаружено, что у мышей, получавших лечение соединением 42 и сорафенибом, неожиданно уменьшился размер опухоли с примерно 400 мм3 (контроль) до примерно 50 мм3 по сравнению с AMD3100 в сочетании с сорафенибом, который уменьшил размер опухоли с примерно 400 мм3 (контроль) до примерно 250 мм3. Важно отметить, что значительная потеря массы тела не наблюдалась у животных, получавших лечение соединением 42.
Эти результаты показывают, что соединение 42 обладает неожиданно более высокой эффективностью при лечении HCC по сравнению с AMD3100.
Пример 7: Лечение легкого травматического повреждения головного мозга у мышей
Травматическое повреждение головного мозга (TBI), также известное как внутричерепная травма, происходит, когда внешняя сила повреждает мозг. Это может быть классифицировано на основе серьезности, механизма или других признаков (например, происходящих в определенном месте или в широко распространенной области).
TBI приводит к физическим, когнитивным, социальным, эмоциональным и поведенческим симптомам.
Эффективность соединения формулы (I), то есть соединения 42, в лечении легкого травматического повреждения головного мозга (mTBI) оценивали с использованием мышиной модели mTBI следующим образом.
Модель легкого травматического повреждения головного мозга (mTBI)
Взрослых мышей CD1 содержали в 12-часовом темном (с 19 часов до 7 часов) и 12-часовом световом (с 7 до 19 часов) цикле. Мышей анестезировали изофлураном. mTBI получали, опуская 30 г металлический снаряд на височную часть черепа, перед правым ухом. Анестезированных мышей клали на бок. Металлическую трубку (внутренний диаметр 13 мм) помещали вертикально над головой, и металлический снаряд сбрасывали с высоты 80 см вниз по трубке, чтобы ударить в височную область черепа впереди правого уха. Стержнеобразный снаряд был изготовлен из металла со слегка закругленным концом, чтобы обеспечить плавный контакт с черепом без каких-либо внешних повреждений в месте падения груза. Использовали губчатую подушку для иммобилизации (L: 4-5 дюймов; W: 2,7 дюйма; H: 1,8 дюйма), допускающая движение головы во время травмы. Примерно через 5 минут после mTBI мышам вводили соединение 42 или носитель (физиологический раствор). Контрольные (без mTBI) животные получали изофлуран, но нет mTBI.
Измерение локомоторного поведения
Через 15 минут и на 5-й день после травмы и восстановления после анестезии мышей индивидуально помещали в камеры тестирования двигательной активности (Accuscan, Columbus, OH) на срок до 24 часов (12 часов света и 12 часов темноты/день). Еду и воду постоянно подавали в камеры, которые содержали 16 горизонтальных и 8 вертикальных инфракрасных датчиков, расположенных на расстоянии 2,5 см друг от друга. Каждую мышь помещали в открытый ящие из плексигласа 42×42×31 см. См., например, Airavaara et al., J Comp Neurol, 2009, 515:116-124. Моторные активности измеряли по количеству и порядку лучей, разбитых животными. Были записаны четыре локомоторных параметра, то есть горизонтальная активность, общее пройденное расстояние, вертикальная активность и вертикальное время.
Количественная обратная транскрипция-ПЦР (qRT-PCR)
Кору головного мозга от каждой мыши собирали на 5 день после mTBI для анализа qRTPCR. См., например, Luoetal.,AnnNeurol,2009,65:520-530;Luoetal.,AnnNeurol,2013,65:520-530;andShenetal.,JNeurosciRes,2009,87:545-555. Тотальные РНК выделяли с использованием реагентов TRIZOL (Life Technologies, #15596-026), и кДНК синтезировали из 1 мкг тотальной РНК с использованием набора для синтеза кДНК RevertAid First Strand cDNA (Thermo Scientific, #K1622). Анализы экспрессии генов с помощью TaqMan (набор праймеров и зондов) для специфического выявления IBA1 (#Rn00574125_g1) приобретали у Thermo Scientific. Праймеры и зонды, используемые в количественной ОТ-ПЦР для референсных генов, являются следующими: бета-актин прямой праймер (5'-CATTGCTGACAGGATGCAGAAGG); обратный праймер (5'-TGCTGGAAGGTGGACAGTGAGG); прямой праймер GAPDH (5'-CATCACTGCCACCCAGAAGACTG); обратный праймер (5'-ATGCCAGTGAGCTTCCCGTTCAG). Количественную ПЦР в реальном времени (qRT-PCR) проводили с использованием TaqMan Fast Advanced Master Mix (Life Technologies, #4444557) и системы ПЦР в реальном времени Applied Biosystems 7500 Fast. Экспрессию и нормализацию целевого гена IBA1 рассчитывали относительно эндогенного референсного гена (бета-актин+GAPDH) с помощью модифицированного алгоритма дельта-дельта-Ct, который учитывает специфическую эффективность амплификации конкретного гена для точного расчета. Все эксперименты были продублированы.
Результаты
Взрослых мышей CD1 анестезировали изофлураном, затем получали mTBI, как описано в Shen et al., Clinical Proteomics, 2014, 11:11. Соединение 42 или носитель вводили системно после mTBI. Раннее последующее лечение соединением 42 значительно улучшило двигательную активность после mTBI. Экспрессию маркера воспаления адаптерной молекулы-1, связывающей ионизированный кальций (IBA1), в пораженной коре исследовали с помощью qRTPCR. Было замечено, что лечение соединением 42 значительно снижало экспрессию IBA1 в мозге mTBI.
Более конкретно, 22 мыши были разделены на две группы: группа мышей mTBI, получавших носитель (n=14), и группа мышей mTBI, получавших соединение 42 (3 мг/кг, n=8). Поведение анализировали каждые 3 часа в течение 24 часов, начиная с 15 минут после травмы. Значительное снижение всей двигательной активности наблюдалось у мышей mTBI по сравнению с мышами без mTBI (p <0,001, двухфакторный дисперсионный анализ или ANOVA). Лечение соединением 42 в дозе 3 мг/кг значительно улучшило вертикальную активность у мышей mTBI (вертикальная активность (p=0,009, F1,140=6,969); и время вертикального движения (p=0,007, F1,140=8,662).
Кроме того, 15 мышей использовали для оценки влияния соединения 42 на нейровоспаление, в котором 7 мышей получали носитель и 8 мышей обрабатывали соединением 42 в дозе 3 мг/кг. Кору мозга собирали на 5 день после mTBI. Экспрессию нейровоспалительного маркера IBA1 и референсных генов (GAPDH-актин и бета-актин) измеряли для анализа qRTPCR. Было отмечено, что экспрессия как IBA1 (актин GAPDH), так и IBA1 (бета-актин) в пораженной боковой коре головного мозга была значительно подавлена у мышей mTBI, получавших соединение 42 (p=0,030, t-критерий).
Эти результаты, демонстрирующие соединением 42 нейропротективный эффект на мышиной модели mTBI, показывают, что оно эффективно при лечении легкого травматического повреждения головного мозга.
Пример 8: Эффект на инфаркт миокарда у крыс
Эффективность соединения формулы (I), то есть соединения 42, в защите от инфаркта миокарда оценивали на модели ишемического инфаркта миокарда у крыс следующим образом.
Самцам крыс SD (по 400-500 грамм) вводили одну подкожную инъекцию соединения 42 в дозе 5 мг/кг или равный объем физиологического раствора (n=18-20 на группу) за 30 минут до проведения операции. Левая передняя нисходящая артерия (LAD) была временно лигирована с использованием нейлонового шва 6-0 в течение 30-минутного ишемического периода в этой операции. Через 24 часа каждую крысу анестезировали, и LAD снова лигировали. 2 мл 5% эванс синий затем вводили в хвостовую вену и давали перфузировать в течение 2 минут. Сердце сразу же иссекали, промывали физиологическим раствором, замораживали при -80°С и разрезали в полузамороженном состоянии на срезы толщиной 2 мм. Затем срезы инкубировали в 1% растворе трифенилтетразолия хлорида в течение 10 минут при 37°С и фиксировали в 10% формалине. Размер инфаркта регистрировали после окрашивания. Было омечено, что лечение соединением 42 до ишемии/реперфузии, вызванной хирургическим вмешательством, в значительной степени защищало сердце от ишемического повреждения.
Результаты показывают, что соединение 42 эффективно защищает от инфаркта миокарда у крыс.
Другие варианты осуществления
Все признаки, раскрытые в этой спецификации, могут быть объединены в любой комбинации. Каждый признак, раскрытый в настоящем описании, можно заменить альтернативным признаком, служащим для той же самой, эквивалентной или подобной цели. Таким образом, если явно не указано иное, каждый раскрытый признак является только примером общей серии эквивалентных или сходных признаков.
Исходя из приведенного выше описания, специалист в данной области сможет легко оценить существенные характеристики настоящего изобретения и, не отступая от сути и объема изобретения, сможет осуществить различные изменения и модификации настоящего изобретения для адаптирования его к различным применениям и условиям. Таким образом, другие варианты осуществления также входят в объем представленной далее формулы изобретения.
Ссылки:
1. Schols D, Struyf S, Van Damme J, Esté JA, Henson G, De Clercq E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J. Exp. Med. 186:1383-1388 (1997).
2. Wu CH, Wang CJ, Chang CP, Cheng YC, Song JS, Jan JJ, Chou MC, Ke YY, Ma J, Wong YC, Hsieh TC, Tien YC, Gullen EA, Lo CF, Cheng CY, Liu YW, Sadani AA, Tsai CH, Hsieh HP, Tsou LK, Shia KS. Function-oriented development of CXCR4 antagonists as selective human immunodeficiency virus (HIV)-1 entry inhibitors. J. Med. Chem. 58:1452-1465 (2015).
3. Lenoir M, Djerdjouri B, Périanin A. Stroma cell-derived factor 1alpha mediates desensitization of human neutrophil respiratory burst in synovial fluid from rheumatoid arthritic patients. J. Immunol. 172:7136-7143 (2004).
4. Gonzalo JA, Lloyd CM, Peled A, Delaney T, Coyle AJ, Gutierrez-Ramos JC. Critical involvement of the chemotactic axis CXCR4/stromal cell-derived factor-1 alpha in the inflammatory component of allergic airway disease. J. Immunol. 165:499-508 (2000).
5. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 410:50-56 (2001).
6. Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 65:967-971 (2005).
7. Lin Q, Wesson RN, Maeda H, Wang Y, Cui Z, Liu JO, Cameron AM, Gao B, Montgomery RA, Williams GM, Sun Z. Pharmacological mobilization of endogenous stem cells significantly promotes skin regeneration after full-thickness excision: the synergistic activity of AMD3100 and tacrolimus. J. Invest. Dermatol. 134:2458-2468 (2014).
8. Lukacs NW, Berlin A, Schols D, Skerlj RT, Bridger GJ. AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am. J. Pathol. 160:1353-1360 (2002).
9. Huang J, Li Y, Tang Y, Tang G, Yang GY, Wang Y. CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke. 44:190-197 (2013).
10. Wu CH, Song JS, Chang KH, Jan JJ, Chen CT, Chou MC, Yeh KC, Wong YC, Tseng CT, Wu SH, Yeh CF, Huang CY, Wang MH, Sadani AA, Chang CP, Cheng CY, Tsou LK, Shia KS. Stem cell mobilizers targeting chemokine receptor CXCR4: renoprotective application in acute kidney injury. J. Med. Chem. 58:2315-2325 (2015).
11. Wu KJ, Yu SJ, Shia KS, Wu CH, Song JS, Kuan HH, Yeh KC, Chen CT, Bae E, Wang Y. A novel CXCR4 antagonist CX549 induces neuroprotection in stroke brain. Cell transplantation. in press (2017).
12. Chen Y, Huang Y, Reiberger T, Duyverman AM, Huang P, Samuel R, Hiddingh L, Roberge S, Koppel C, Lauwers GY, Zhu AX, Jain RK, Duda DG. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axisand myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 59:1435-1447 (2014).
13. Matthys P, Hatse S, Vermeire K, Wuyts A, Bridger G, Henson GW, De Clercq E, Billiau A, Schols D. AMD3100, a potent and specific antagonist of the stromal cell-derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-gamma receptor-deficient mice. J. Immunol. 167:4686-4692 (2001).
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2671571C1 |
| ПРОИЗВОДНЫЕ 1,3-ДИОКСОИНДЕНА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ АНТИВИРУСНАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2012 |
|
RU2566761C2 |
| ИНГИБИТОР LSD1, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2763898C2 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2647592C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2016 |
|
RU2744766C2 |
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2014 |
|
RU2638549C2 |
| МОДУЛЯТОРЫ АКТИВНОСТИ НЕС1 И СПОСОБЫ ДЛЯ НИХ | 2011 |
|
RU2576036C2 |
| БИЦИКЛИЧЕСКОЕ ГЕТЕРОАРОМАТИЧЕСКОЕ КОЛЬЦЕВОЕ ПРОИЗВОДНОЕ | 2019 |
|
RU2798838C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2673458C1 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
Изобретение относится к гетероциклическим соединениям формулы (I), где R1 и R2 независимо представляют собой Н, NH2 или C1-6 алкил или R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой фенил или гетероарил, выбранный из тиенила, имидазолила и пиридила; каждый из R3 и R4 независимо представляет собой 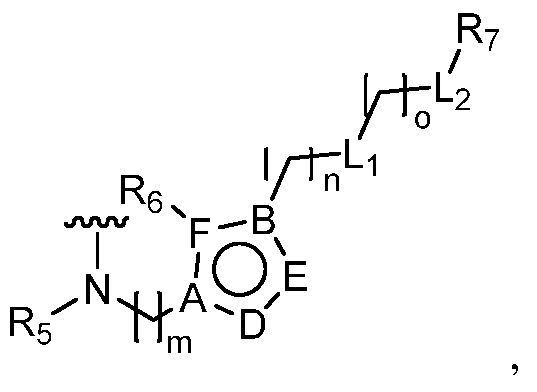
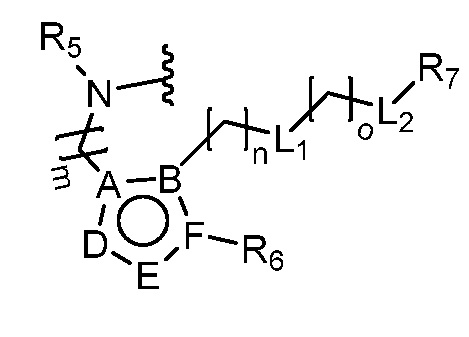 или
или 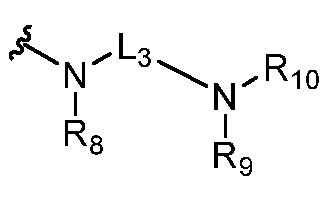 , которые являются такими, как раскрыто в формуле изобретения; R5 представляет собой H; R6 отсутствует или представляет собой H; R7 представляет собой C3-10 циклоалкил. Также изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), к способу мобилизации гемопоэтических стволовых клеток (HSC) и эндотелиальных клеток-предшественников (EPC) в периферическое кровообращение и к способу лечения гепатоцеллюлярной карциномы, почечного повреждения, инфаркта миокарда или легкого травматического повреждения головного мозга. Технический результат – соединения формулы (I), используемые для нарушения взаимодействия между CXCR4 и CXCL12. 6 н. и 25 з.п. ф-лы, 8 пр.
, которые являются такими, как раскрыто в формуле изобретения; R5 представляет собой H; R6 отсутствует или представляет собой H; R7 представляет собой C3-10 циклоалкил. Также изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), к способу мобилизации гемопоэтических стволовых клеток (HSC) и эндотелиальных клеток-предшественников (EPC) в периферическое кровообращение и к способу лечения гепатоцеллюлярной карциномы, почечного повреждения, инфаркта миокарда или легкого травматического повреждения головного мозга. Технический результат – соединения формулы (I), используемые для нарушения взаимодействия между CXCR4 и CXCL12. 6 н. и 25 з.п. ф-лы, 8 пр.
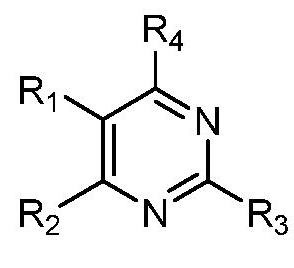
(I)
1. Соединение формулы (I):
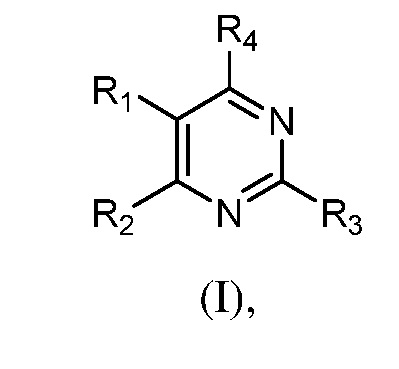
где
каждый из R1 и R2 независимо представляет собой Н, NH2 или C1-6 алкил или R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой фенил или гетероарил, выбранный из тиенила, имидазолила и пиридила; и
каждый из R3 и R4, независимо, представляет собой 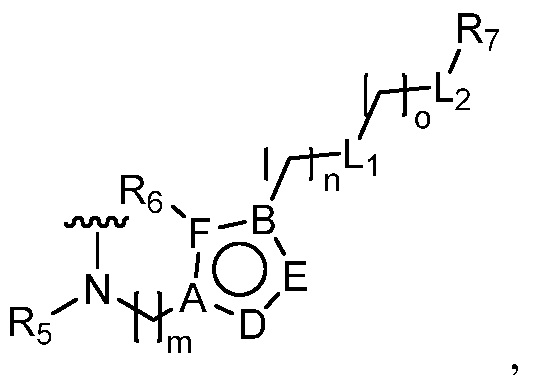
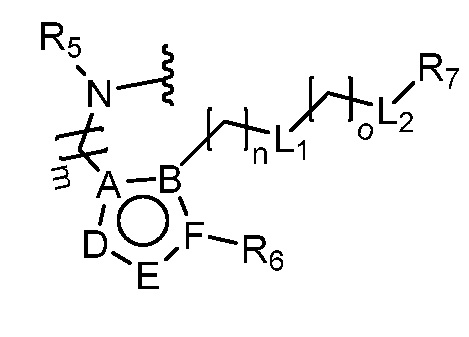 или
или 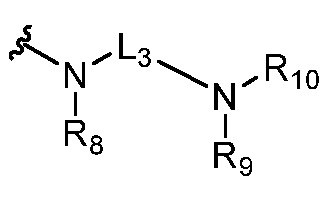 ,
,
где по меньшей мере один из R3 и R4 представляет собой 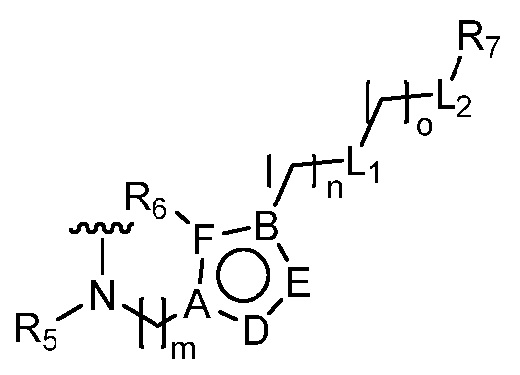 или ,
или ,
R5 представляет собой H;
R6 отсутствует или представляет собой H;
R7 представляет собой C3-10 циклоалкил;
А в представляет собой C; и
каждый из B, D, E и F в представляет собой N; и
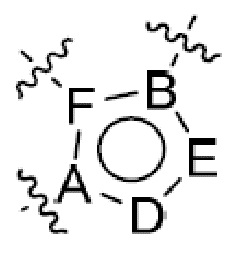 в представляет собой
в представляет собой  ,
, 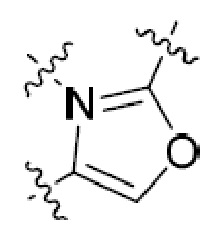 ,
, 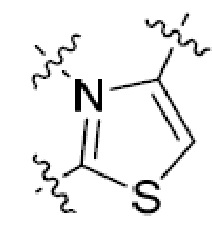 ,
, 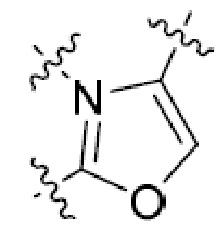 ,
, 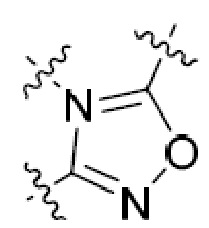 ,
, 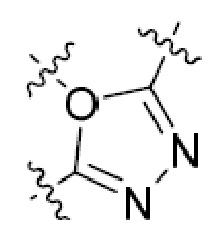 ,
, 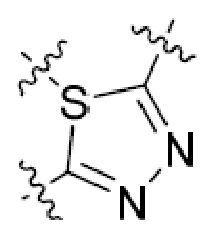 ,
, 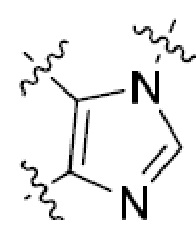 ,
, 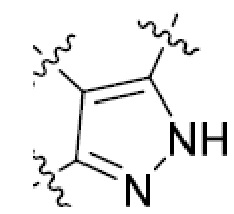 ,
, 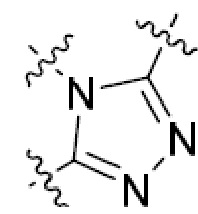 ,
, 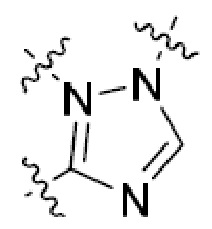 ,
, 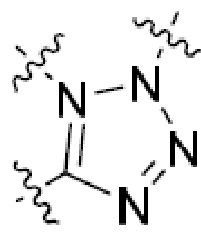 ,
, 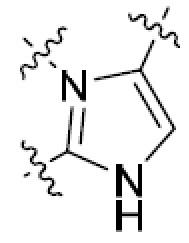 или
или 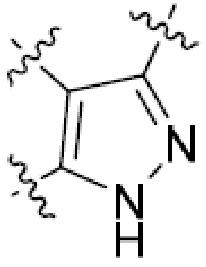 ;
;
каждый из L1 и L2 представляет собой NRd, где Rd представляет собой H;
каждый из m, n и o, независимо, равен 1, 2, 3, 4, 5 или 6;
каждый из R8 и R9 представляет собой H или R8 и R9 вместе с атомами азота, с которыми они связаны, представляют собой C6 гетероциклоалкил, содержащий 1-2 атома N;
L3 представляет собой C1-6 алкил или L3 вместе с R8 или R9 и атомом азота, с которым они связаны, представляет собой C6 гетероциклоалкил, содержащий 1-2 атома N; и
R10 представляет собой H или 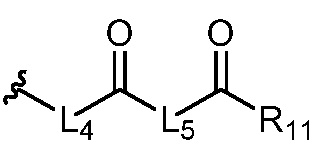 , где L4 отсутствует или представляет собой C1-6 алкиламино; L5 представляет собой C1-6 алкил, C1-6 алкиламино или ди-C1-6 алкиламино; и R11 представляет собой гидроксил или C1-6 алкиламино; каждый из C1-6 алкила и C1-6 алкиламино необязательно замещен гидроксилом, амино или P(O)(OR13)2, где R13 представляет собой H.
, где L4 отсутствует или представляет собой C1-6 алкиламино; L5 представляет собой C1-6 алкил, C1-6 алкиламино или ди-C1-6 алкиламино; и R11 представляет собой гидроксил или C1-6 алкиламино; каждый из C1-6 алкила и C1-6 алкиламино необязательно замещен гидроксилом, амино или P(O)(OR13)2, где R13 представляет собой H.
2. Соединение по п. 1, где каждый из R1 и R2, независимо, представляет собой H или C1-6 алкил.
3. Соединение по п. 1, где каждый из R1 и R2, независимо, представляет собой H или NH2.
4. Соединение по п. 1, где R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой фенил или гетероарил, выбранный из тиенила, имидазолила и пиридила.
5. Соединение по п. 4, где R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой 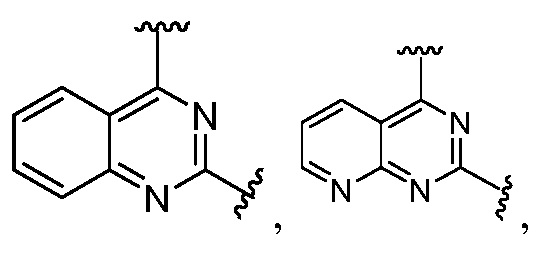
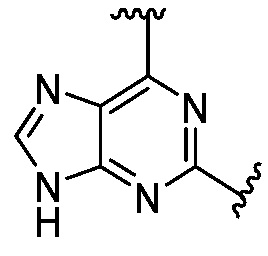 или
или 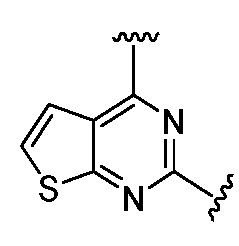 .
.
6. Соединение по п. 1, где каждый из R3 и R4, независимо, представляет собой
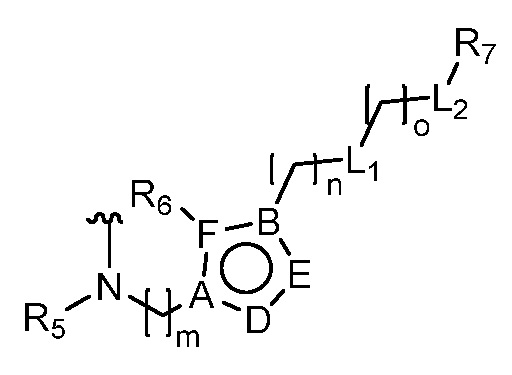 или
или 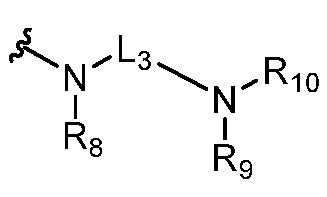 , где R6 отсутствует; и каждый из m, n и o, независимо, равен 1, 2, 3 или 4.
, где R6 отсутствует; и каждый из m, n и o, независимо, равен 1, 2, 3 или 4.
7. Соединение по п. 6, где  в представляет собой
в представляет собой 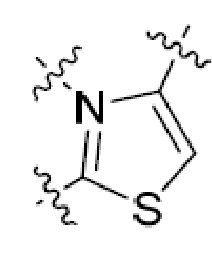 ,
, 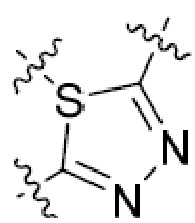 ,
, 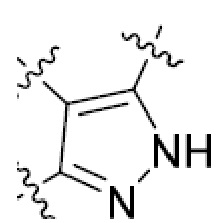 ,
, 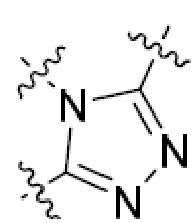 ,
, 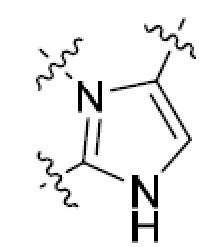 или
или 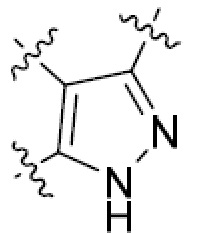 .
.
8. Соединение по п. 6, где каждый из R1 и R2, независимо, представляет собой H или C1-6 алкил.
9. Соединение по п. 8, где R1 представляет собой H и R2 представляет собой C1-6 алкил.
10. Соединение по п. 6, где R1 и R2, вместе с двумя атомами углерода, с которыми они связаны, представляют собой фенил или гетероарил, выбранный из тиенила, имидазолила и пиридила.
11. Соединение по п. 10, где R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой 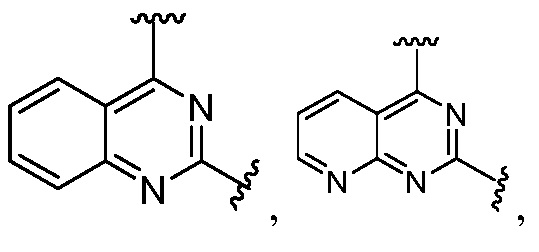
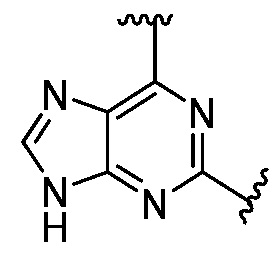 или
или 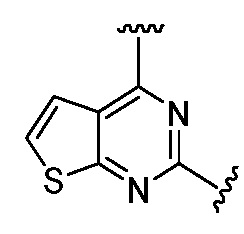 .
.
12. Соединение по п. 1, где R3 представляет собой
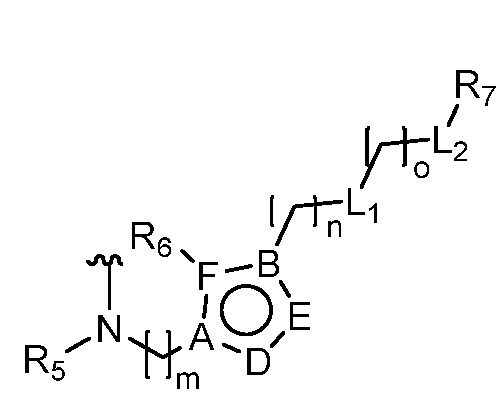 и R4 представляет собой
и R4 представляет собой 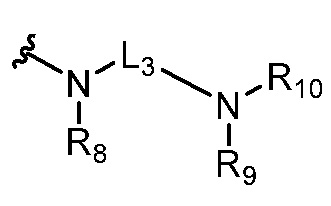 , где R6 отсутствует и каждый из m, n и o, независимо, равен 1, 2, 3, или 4.
, где R6 отсутствует и каждый из m, n и o, независимо, равен 1, 2, 3, или 4.
13. Соединение по п. 12, где каждый из R1 и R2, независимо, представляет собой H или C1-6 алкил.
14. Соединение по п. 13, где R1 представляет собой H и R2 представляет собой C1-6 алкил.
15. Соединение по п. 12, где R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой фенил или гетероарил, выбранный из тиенила, имидазолила и пиридила.
16. Соединение по п. 15, где R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой 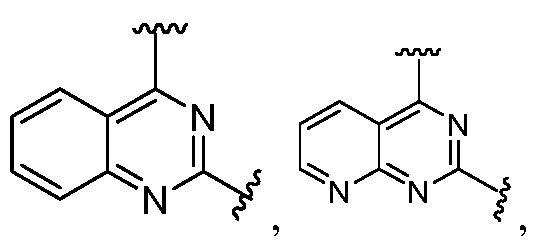
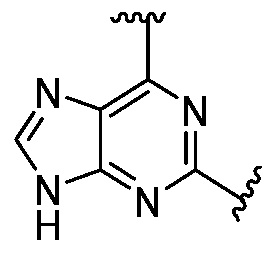 или
или 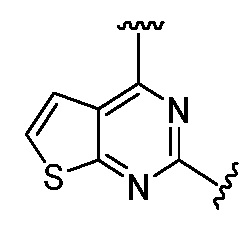 .
.
17. Соединение по п. 16, где R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой 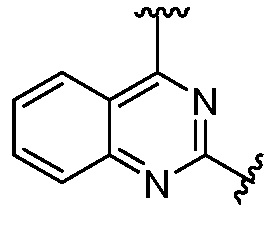 .
.
18. Соединение по п. 16, где 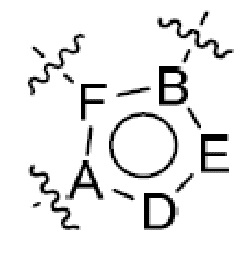 в представляет собой
в представляет собой 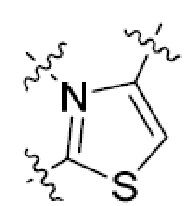 ,
, 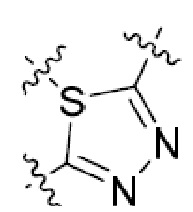 ,
, 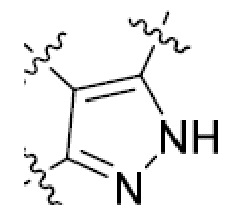 ,
, 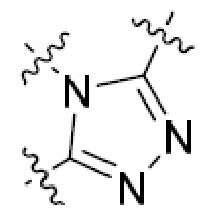 ,
, 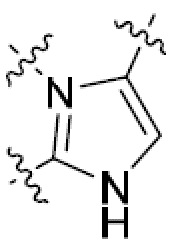 или
или 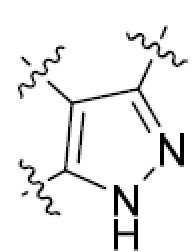 .
.
19. Соединение по п. 12, где L3 вместе с R8 или R9 и атомом азота, с которым они связаны, представляет собой C6 гетероциклоалкил, содержащий 1-2 атома N.
20. Соединение по п. 19, где R8 представляет собой H и L3 вместе с R9 и атомом азота, с которым они связаны, представляет собой C6 гетероциклоалкил, содержащий 1-2 атома N.
21. Соединение по п. 20, где R1 представляет собой H и R2 представляет собой C1-6 алкил.
22. Соединение по п. 20, где R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой 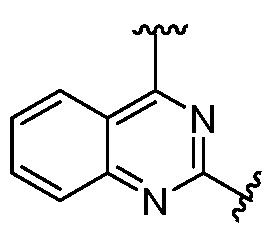 .
.
23. Соединение по п. 22, где 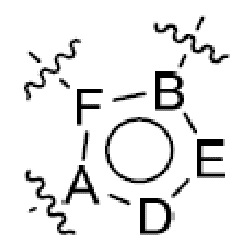 в представляет собой
в представляет собой 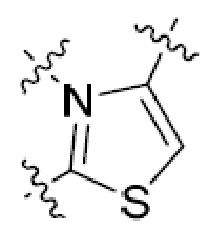 ,
, 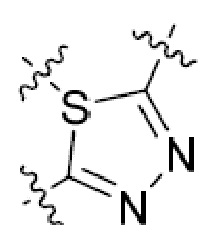 ,
, 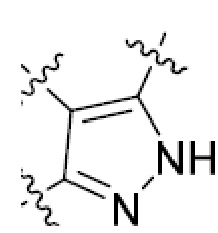 ,
, 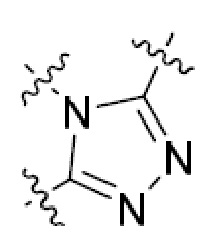 ,
, 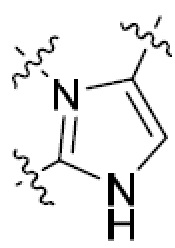 или
или 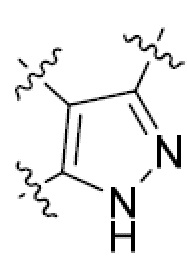 .
.
24. Соединение по п. 20, где R10 представляет собой 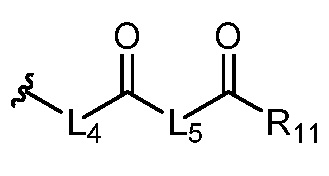 .
.
25. Соединение по п. 24, где R1 представляет собой H и R2 представляет собой C1-6 алкил или R1 и R2 вместе с двумя атомами углерода, с которыми они связаны, представляют собой  ; и
; и 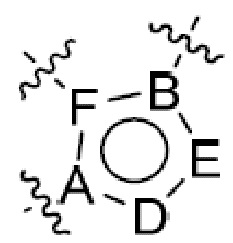 в представляет собой
в представляет собой 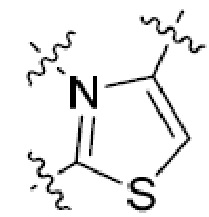 ,
, 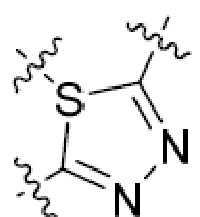 ,
, 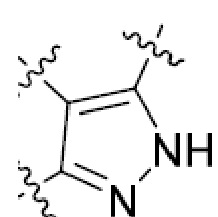 ,
, 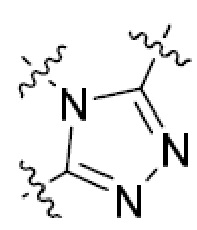 ,
, 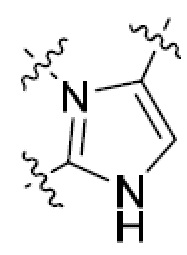 или
или 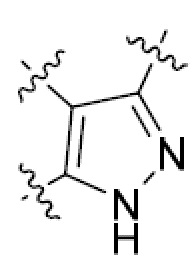 .
.
26. Соединение, имеющее одну из следующих структур:
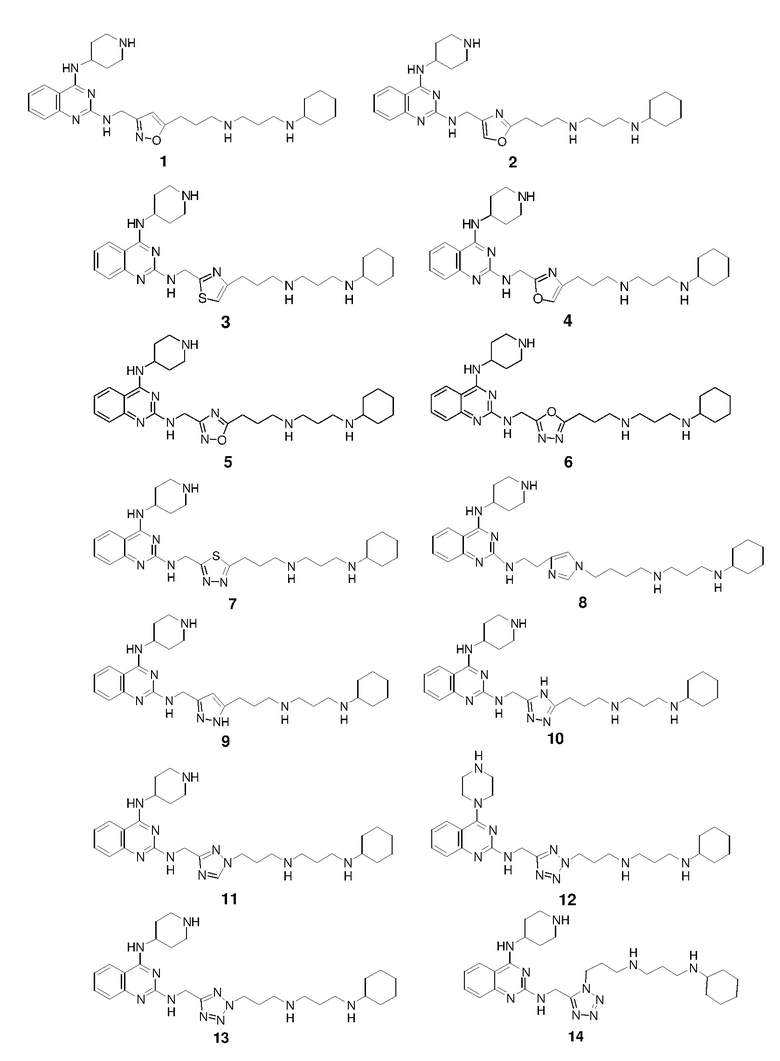
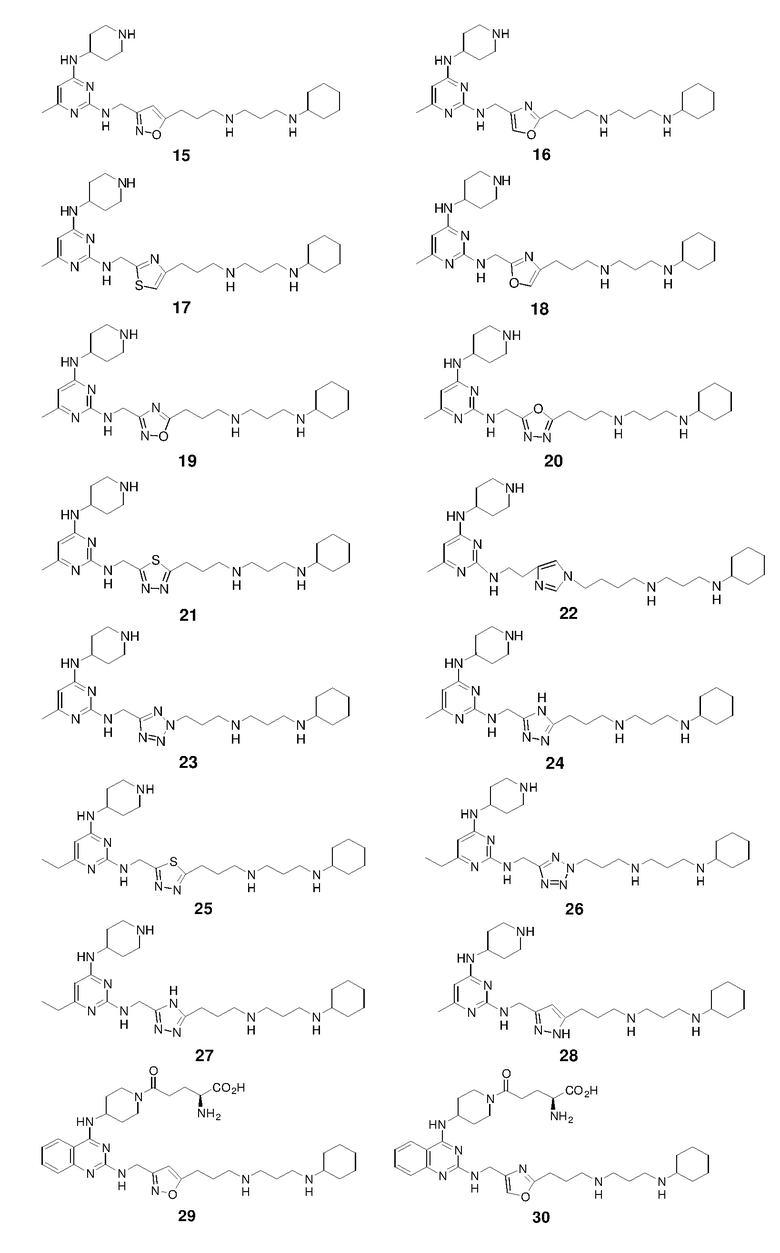
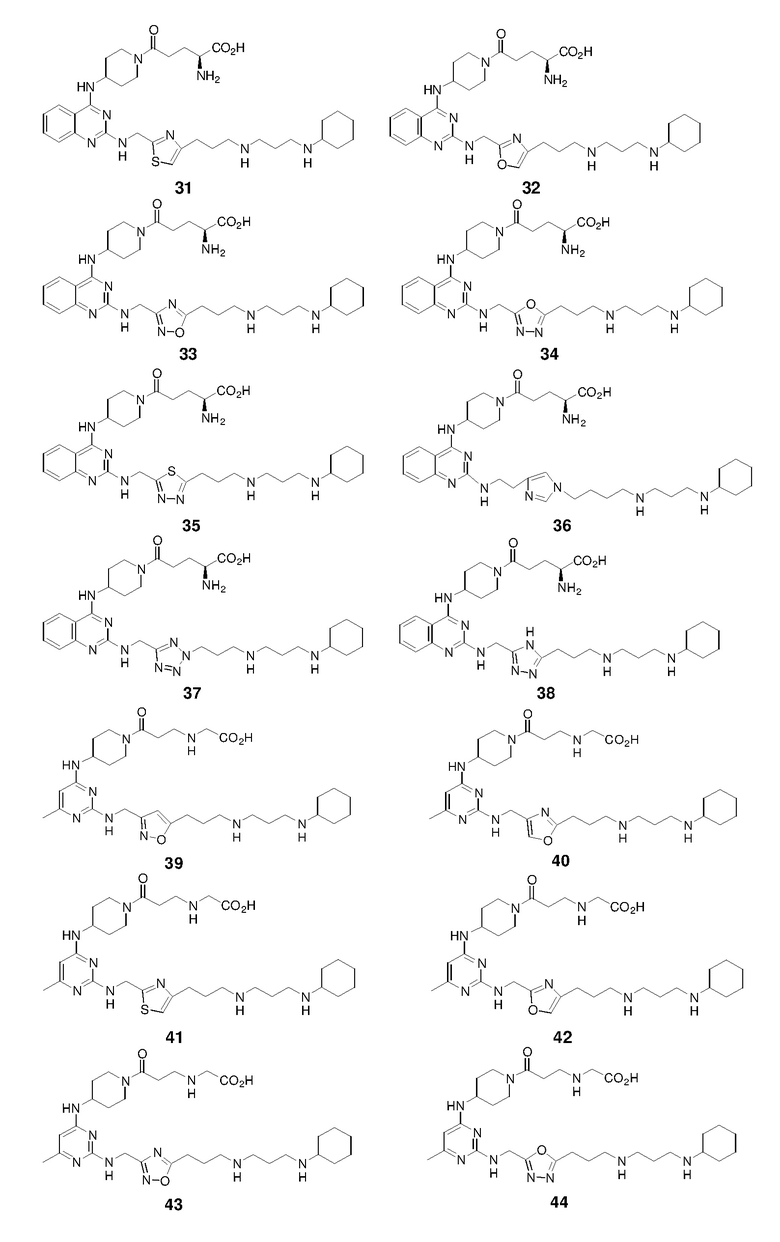
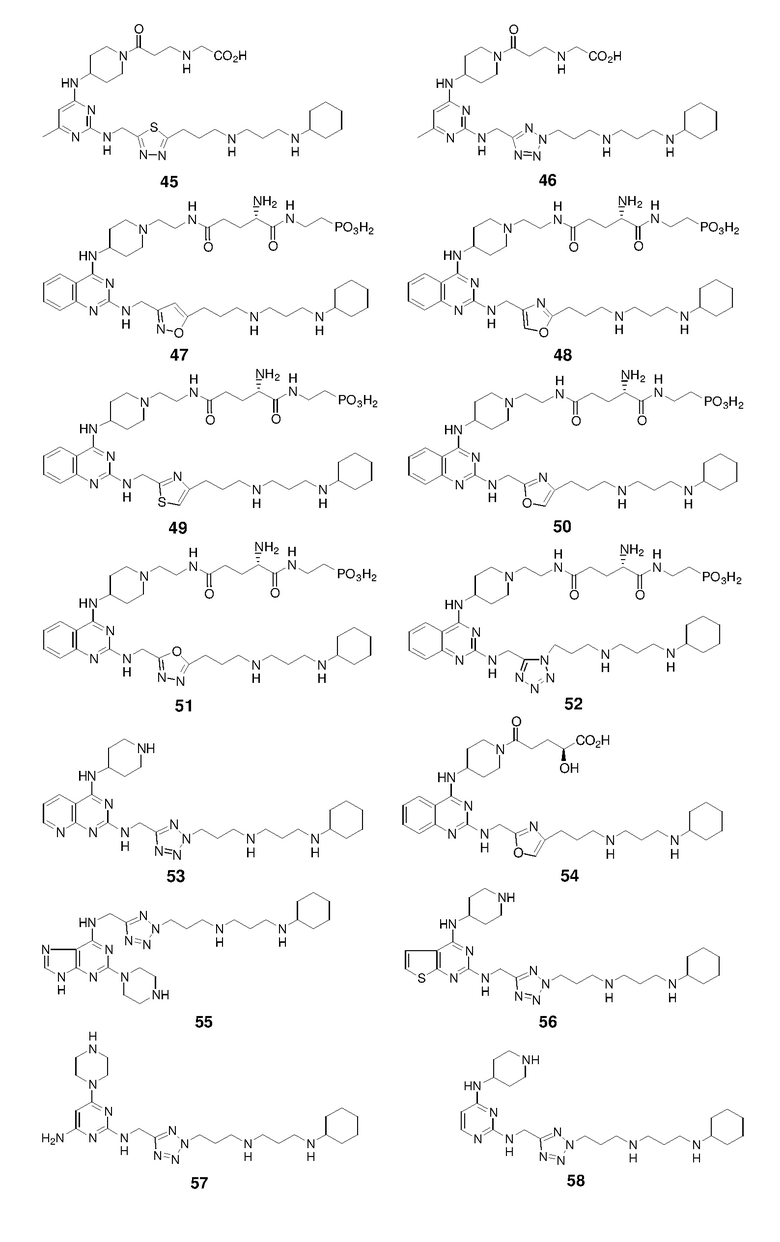
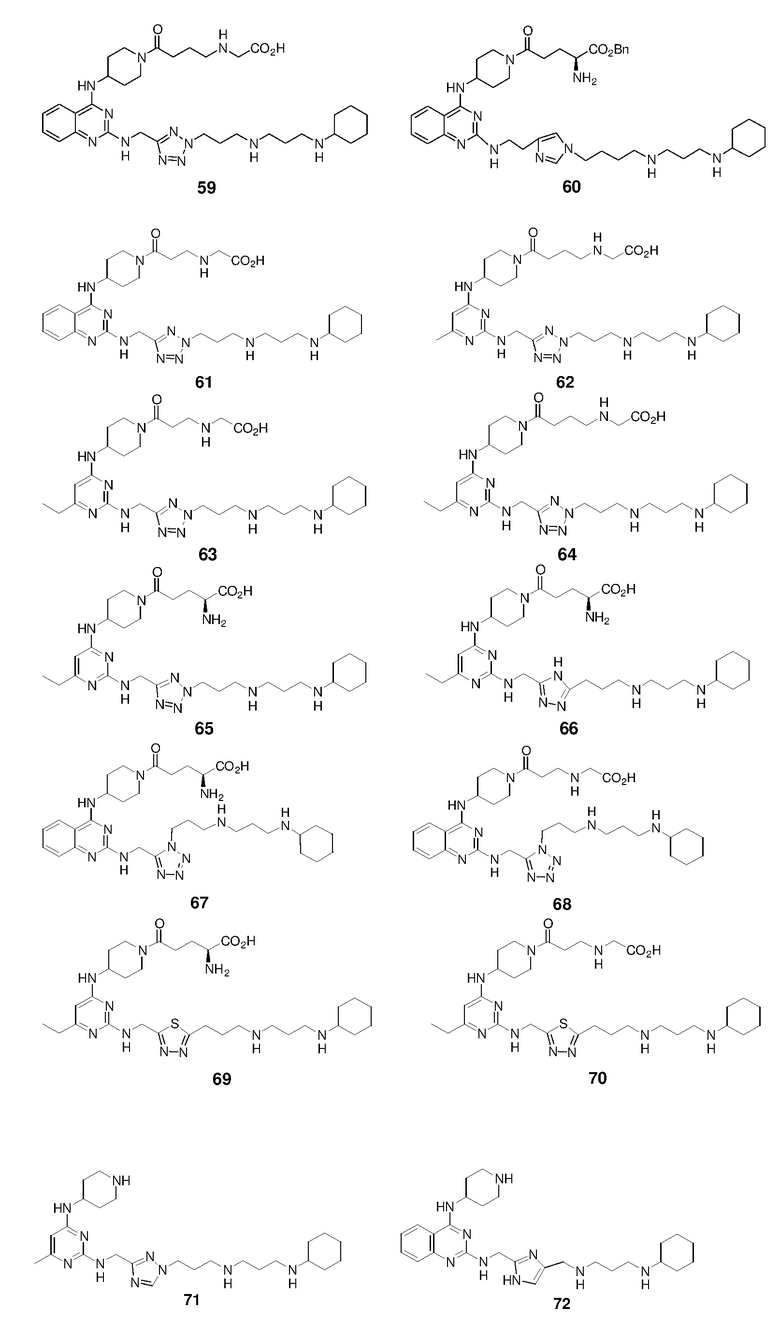
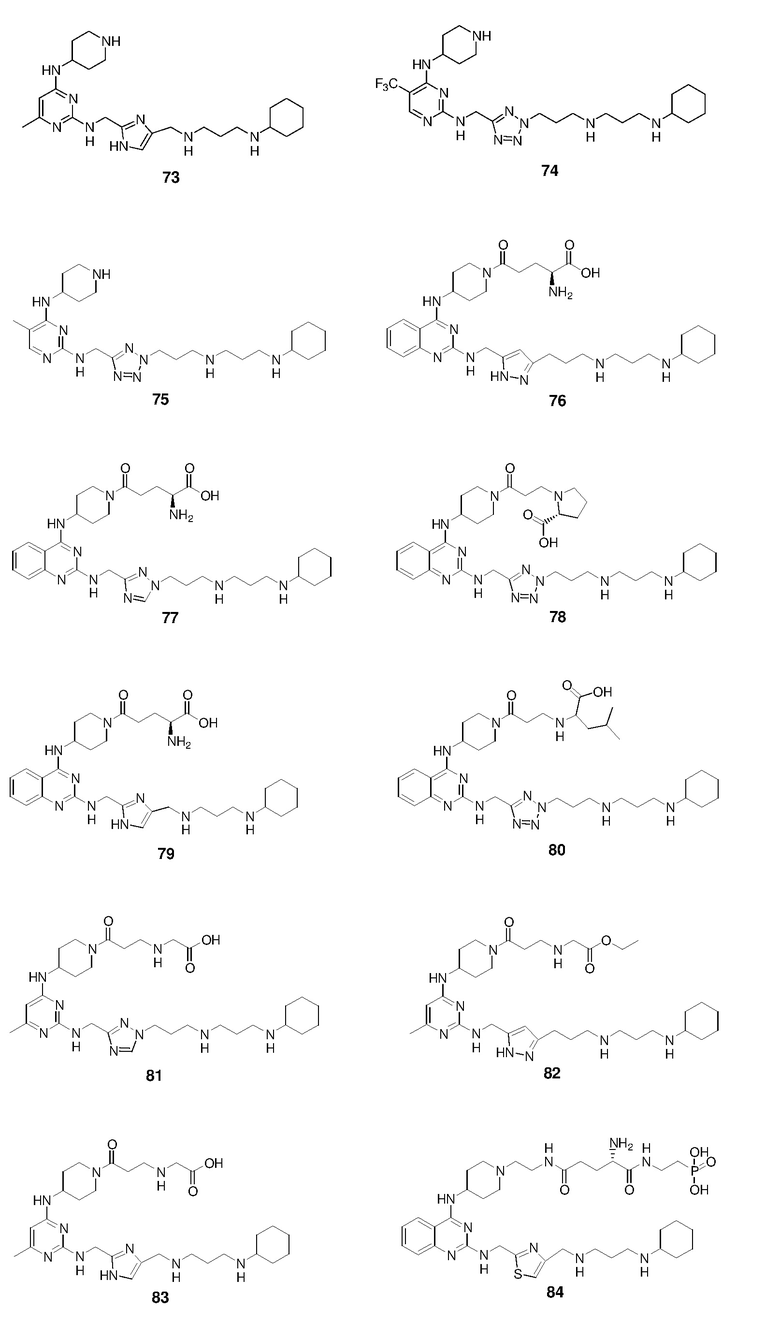
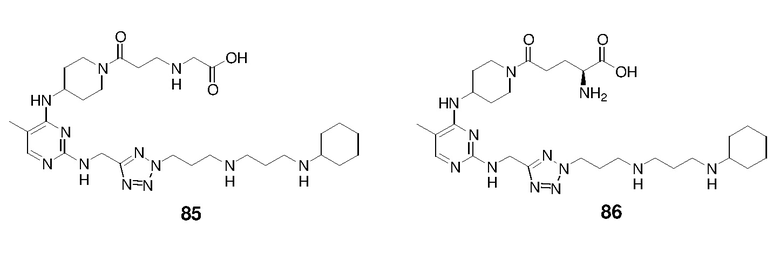
27. Соединение по п. 1, где соединение представляет собой одно из следующих соединений:
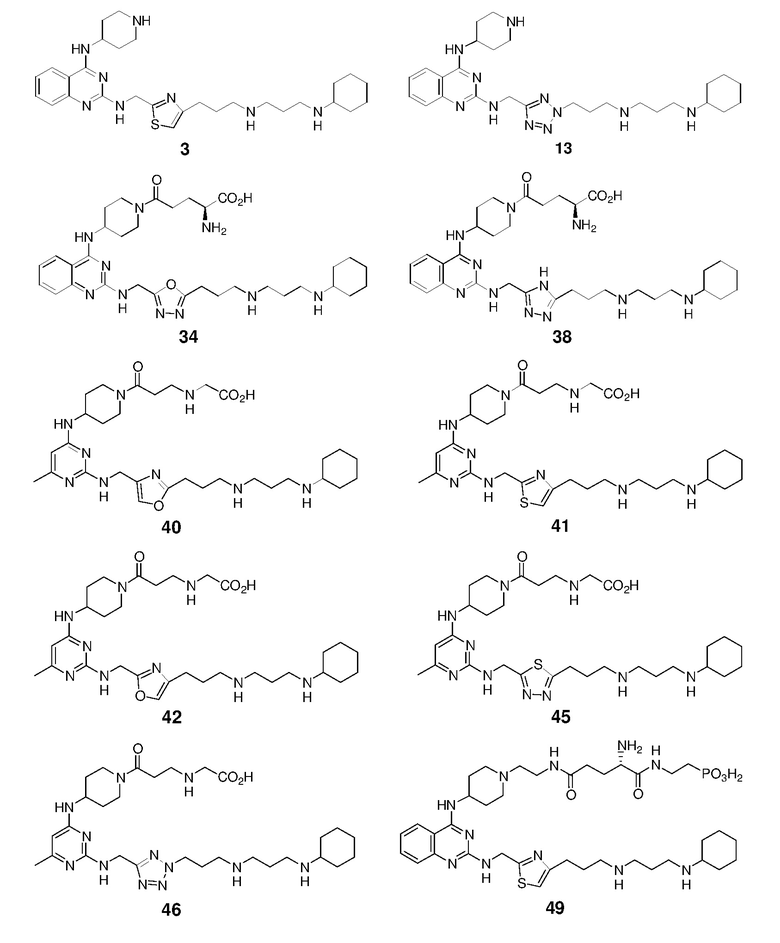
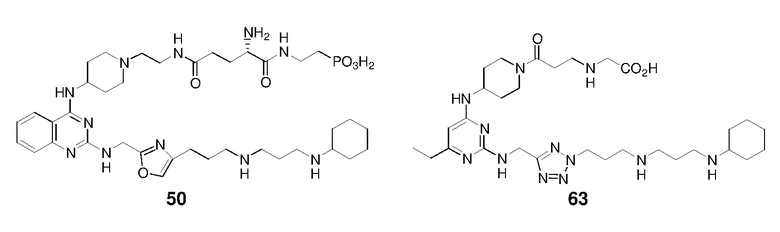
28. Фармацевтическая композиция для нарушения взаимодействия между CXCR4 и CXCL12, содержащая эффективное количество соединения по п.1 и его фармацевтически приемлемый носитель.
29. Способ мобилизации гемопоэтических стволовых клеток (HSC) и эндотелиальных клеток-предшественников (EPC) в периферическое кровообращение, включающий контактирование HSC и EPC с эффективным количеством соединения по п.1.
30. Способ лечения гепатоцеллюлярной карциномы, почечного повреждения, инфаркта миокарда или легкого травматического повреждения головного мозга, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения по п.1.
31. Способ лечения гепатоцеллюлярной карциномы, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения по п.1.
| Токарный резец | 1924 |
|
SU2016A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| ПРОИЗВОДНЫЕ 4-ФЕНИЛПИРИМИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2243221C2 |

