Область техники
[0001]
Настоящее изобретение относится к производному циклического амина и его фармацевтическому применению.
Предпосылки изобретения
[0002]
Боль является неприятным сенсорным и эмоциональным переживанием, связанным с фактическим или возможным повреждением тканей. В соответствии с причиной боль классифицируется на ноцицептивную боль, невропатическую боль и психогенную боль. В качестве боли, вызванной неизвестной причиной, известен синдром фибромиалгии.
[0003]
Невропатическая боль является патологической болью, вызванной дисфункцией периферической или центральной нервной системы, более конкретно, болью, вызванной, например, прямым повреждением и угнетением нервной ткани, несмотря на не ноцицептивный раздражитель ноцицептора. В качестве терапевтического агента при невропатической боли используется противосудорожное средство, антидепрессант, анксиолитическое средство или противоэпилептическое средство, такие как габапентин или прегабалин.
[0004]
Синдром фибромиалгии представляет собой расстройство, в котором системная боль является ведущим симптомом, а психоневрологические и нейровегетативные симптомы являются вторичными симптомами. В качестве терапевтических средств для лечения синдрома фибромиалгии, в основном, используются прегабалин, который был одобрен в Соединенных Штатах и Японии, дулоксетин и милнаципран, которые были одобрены в Соединенных Штатах. Кроме того, используются лекарственные средства, которые не утверждены в качестве терапевтического средства для лечения синдрома фибромиалгии, т.е. нестероидное противовоспалительное средство, опиоидное соединение, антидепрессант, противосудорожное средство и противоэпилептическое лекарственное средство. Тем не менее, обычно утверждают, что нестероидные противовоспалительные средства и опиумные соединения имеют низкий терапевтический эффект (непатентная литература 1).
[0005]
Помимо этого, в патентной литературе 1 описано, что замещенные пиперидины обладают кардиотоническое активностью; в патентной литературе 2 описано, что производные имидазола оказывают ингибирующее FXa действие; и в патентной литературе 3 высказано предположение, что замещенные пиперидины имеют потенциальную лекарственную эффективность против избыточного веса или ожирения.
Список цитируемой литературы
Патентная литература
[0006]
Патентная литература 1: патент Франции 2567885
Патентная литература 2: патентная публикация JP (Kokai) № 2006-008664
Патентная литература 3: международная публикация WO 2003/031432
Непатентная литература
[0007]
Непатентная литература 1: Recla, Journal of Pain Research, vol. 3, p. 89-103, 2010.
Сущность изобретения
Техническая задача
[0008]
Однако терапия с использованием обычного терапевтического средства для лечения невропатической боли чрезвычайно часто связана с побочными реакциями центральной нервной системы, такими как головокружение, тошнота или рвота. Из-за этого является сложным принимать обычный терапевтический агент в течение длительного времени. В связи с этим была желательна разработка нового терапевтического средства для лечения невропатической боли.
[0009]
Даже прегабалин, дулоксетин и милнаципран, которые были утверждены в качестве терапевтических средств для лечения синдрома фибромиалгии, не в состоянии обеспечить клинически удовлетворительный терапевтический эффект при синдроме фибромиалгии, и эффективность препарата значительно меняется среди пациентов. В связи с этим чрезвычайно желательным является разработка нового терапевтического агента для лечения синдрома фибромиалгии, который оказывал бы достаточный терапевтический эффект.
[0010]
Следует отметить, что в патентной литературе 1 высказано предположение о том, что описанные там замещенные пиперидины обладают эффективностью при лечении мигрени; однако в литературе не описано соединение, обезболивающее действие которого было обнаружено в настоящей заявке, и не было сделано предположения о релевантности анальгезирующего действия по отношению к химической структуре. В патентной литературе 2, где описаны производные имидазола, и в патентной литературе 3, где описаны замещенные пиперидины, не описаны и не предполагается возможность анальгезирующего действия, которыми обладают эти соединения.
[0011]
В этих обстоятельствах целью настоящего изобретения является разработка соединения, имеющего сильное анальгезирующее действие, для лечения боли, в частности, невропатической боли и/или синдрома фибромиалгии.
Решение задачи
[0012]
Авторы настоящего изобретения провели интенсивные исследования с целью решения вышеупомянутых проблем. В результате они обнаружили производное циклического амина, имеющее сильный анальгезирующий эффект против боли, в частности, невропатической боли и/или синдрома фибромиалгии.
[0013]
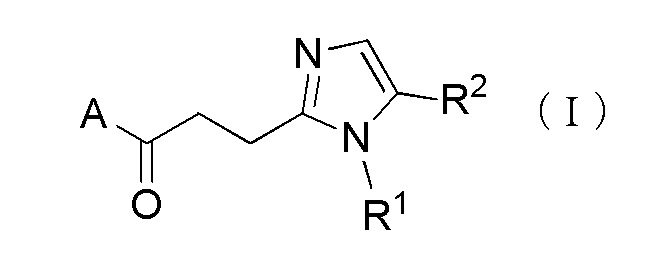
Более конкретно, настоящее изобретение относится к производному циклического амина, представленного следующей общей формулы, (I) или его фармакологически приемлемой соли.
[Формула 1]
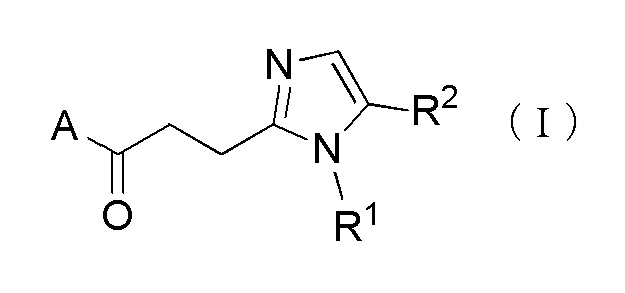
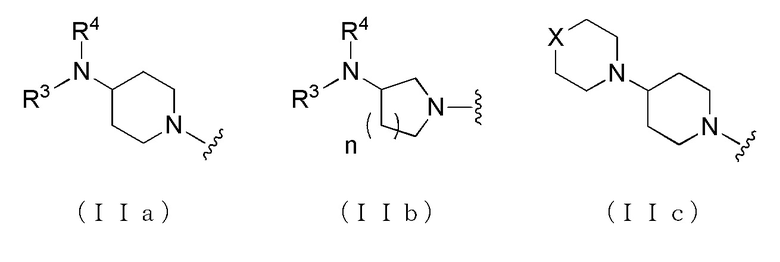
где A представляет собой группу, представленную общей формулой (IIa), (IIb) или (IIc).
[Формула 2]
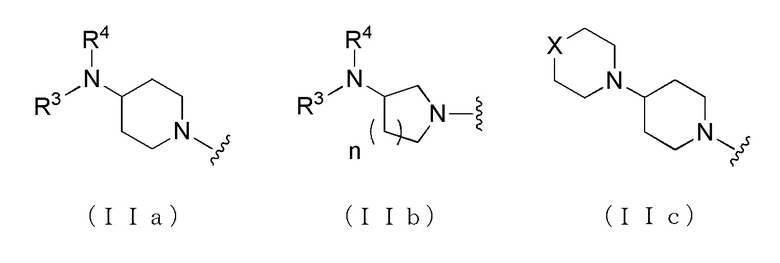
где, когда A является группой, представленной общей формулой (IIa) или (IIb), R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную атомом галогена, гидроксильной группой, амино группой или карбоксильной группой, R2 представляет собой атом водорода или атом галогена, R3 представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, R4 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 6 атомов углерода, или алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную алкилкарбониламино группой, содержащей от 2 до 6 атомов углерода, и n обозначает 1 или 2, в которой, когда R3 и R4, каждый, независимо, представляют собой алкильную группу, содержащую от 1 до 6 атомов углерода, R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную гидроксильной группой, амино группой или карбоксильной группой; и, когда A представляет собой группу, представленную общей формулой (IIc), R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную карбоксильной группой, R2 представляет собой атом водорода или атом галогена, X представляет собой CH2, O или -NR5, и R5 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода.
[0014]
В вышеуказанном производном циклического амина предпочтительным является, когда A представлен общей формулой (IIa) или (IIb), в которой, более предпочтительно, R3 представляет собой атом водорода, метильную группу или этильную группу; и далее, предпочтительно, когда R2 представляет собой атом водорода или атом хлора, R3 представляет собой атом водорода или метильную группу, и R4 представляет собой атом водорода, метилкарбонильную группу или алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную метилкарбониламино группой.
[0015]
Более конкретно, например, предпочтительным является производное циклического амина, представленное следующей общей формулы (Ia) или (Ib), или его фармакологически приемлемая соль. Особенно предпочтительным является, когда R2 представляет собой атом водорода или атом хлора, R3 представляет собой атом водорода или метильную группу, R4 представляет собой атом водорода, метилкарбонильную группу или алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную метилкарбониламино группой, в которой, когда R3 и R4, оба, представляют собой метильные группы, R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную гидроксильной группой, амино группой или карбоксильной группой.
[Формула 3]
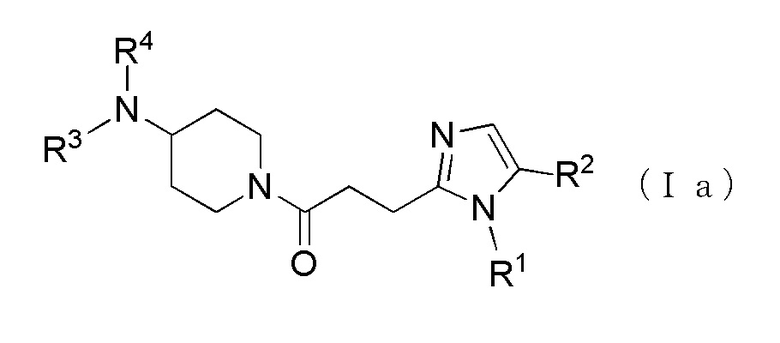
где R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную атомом галогена, гидроксильной группой, амино группой или карбоксильной группой, R2 представляет собой атом водорода или атом галогена, R3 представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, R4 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 6 атомов углерода, или алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную алкилкарбониламино группой, содержащей от 2 до 6 атомов углерода, в которой, когда R3 и R4, каждый, независимо, представляют собой алкильную группу, содержащую от 1 до 6 атомов углерода, R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную гидроксильной группой, амино группой или карбоксильной группой.
[Формула 4]
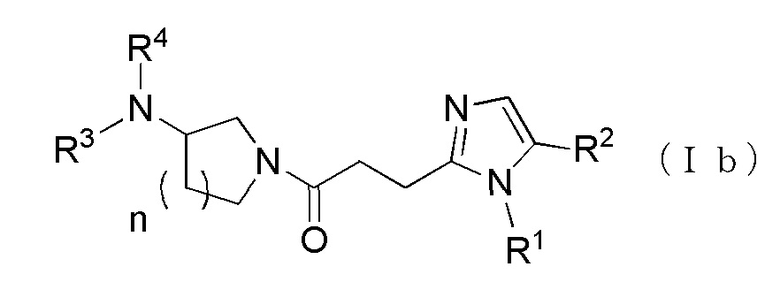
где R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную атомом галогена, гидроксильной группой, амино группой или карбоксильной группой, R2 представляет собой атом водорода или атом галогена, R3 представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, R4 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 6 атомов углерода, или алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную алкилкарбониламино группой, содержащей от 2 до 6 атомов углерода, в которой, когда R3 и R4, каждый, независимо, представляют собой алкильную группу, содержащую от 1 до 6 атомов углерода, R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную гидроксильной группой, амино группой или карбоксильной группой.
[0016]
Анальгезирующее действие может быть улучшено из-за значений определений, указанных выше.
[0017]
В вышеуказанном производном циклического амина предпочтительным является, когда A представлен общей формулой (IIc), и, более предпочтительным является, когда R2 представляет собой атом водорода или атом хлора, и R5 представляет собой метильную группу.
[0018]
Более конкретно, например, предпочтительным является производное циклического амина, представленное следующей общей формулы (Ic), или его фармакологически приемлемая соль. Более предпочтительным является, когда R2 представляет собой атом водорода или атом хлора, и R5 представляет собой метильную группу.
[Формула 5]
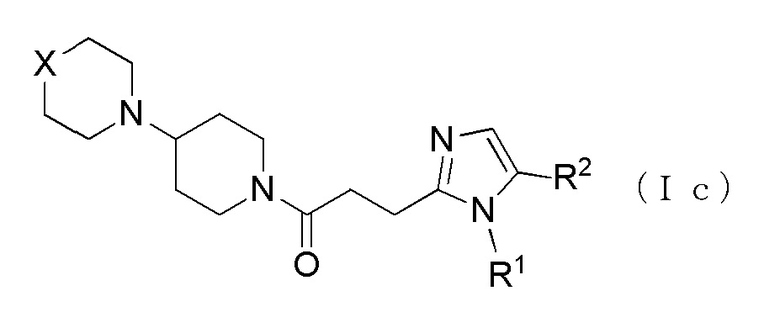
где R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную карбоксильной группой, R2 представляет собой атом водорода или атом галогена, X представляет собой CH2, O или -NR5, и R5 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода.
[0019]
Анальгезирующее действие может быть улучшено.
[0020]
Настоящее изобретение относится к пролекарству вышеуказанного производного циклического амина или его фармакологически приемлемой соли. Пролекарство, предпочтительно, представляет собой пролекарство, полученное путем этерификации карбоксильной группы вышеуказанного производного циклического амина.
[0021]
Превосходную фармакокинетику можно ожидать на крысах при пероральном введении из-за значений определений, указанных выше.
[0022]
Настоящее изобретение относится также к лекарственному средству, содержащему в качестве активного ингредиента производное циклического амина, представленное вышеуказанной общей формулой (I), пролекарство производного циклического амина или его фармакологически приемлемую соль.
[0023]
Лекарственное средство, предпочтительно, представляет собой анальгетический агент, и, особенно предпочтительно, терапевтический агент при невропатической боли или терапевтический агент при синдроме фибромиалгии.
Положительный эффект изобретения
[0024]
Производное циклического амина по настоящему изобретению или его пролекарство, или его фармакологически приемлемая соль обладает сильным обезболивающим действием против боли, в частности, невропатической боли и синдрома фибромиалгии, и может быть использовано в качестве анальгезирующего средства, в частности, терапевтического средства для лечения невропатической боли или синдрома фибромиалгии, которое может в соответствии с ожиданиями уменьшить побочные эффекты центральной нервной системы и быть подходящим для длительного применения.
Краткое описание рисунков
[0025]
[Фигура 1] На фигуре 1 представлен график, показывающий влияние соединения по примеру 8 на моделях частичного лигирования седалищного нерва на мышах (пероральное введение).
[Фигура 2] На фигуре 2 представлен график, показывающий влияние соединения по примеру 2 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 3] На фигуре 31 представлен график, показывающий влияние соединения по примеру 4 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 4] На фигуре 4 представлен график, показывающий влияние соединения по примеру 2 на моделях частичного лигирования седалищного нерва на мышах (интравентрикулярное введение).
[Фигура 5] На фигуре 5 представлен график, показывающий влияние соединения по примеру 4 на моделях частичного лигирования седалищного нерва на мышах (интравентрикулярное введение).
[Фигура 6] На фигуре 6 представлен график, показывающий влияние соединения по примеру 6 на моделях частичного лигирования седалищного нерва на мышах (интравентрикулярное введение).
[Фигура 7] На фигуре 7 представлен график, показывающий влияние соединения по примеру 10 на моделях частичного лигирования седалищного нерва на мышах (интравентрикулярное введение).
[Фигура 8] На фигуре 8 представлен график, показывающий влияние соединения по примеру 12 на моделях частичного лигирования седалищного нерва на мышах (интравентрикулярное введение).
[Фигура 9] На фигуре 9 представлен график, показывающий влияние соединения по примеру 13 на моделях частичного лигирования седалищного нерва на мышах (интравентрикулярное введение).
[Фигура 10] На фигуре 10 представлен график, показывающий влияние соединения по примеру 15 на моделях частичного лигирования седалищного нерва на мышах (интравентрикулярное введение).
[Фигура 11] На фигуре 11 представлен график, показывающий влияние соединения по примеру 38 на моделях частичного лигирования седалищного нерва на мышах (пероральное введение).
[Фигура 12] На фигуре 12 представлен график, показывающий влияние соединения по примеру 13 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 13] На фигуре 13 представлен график, показывающий влияние соединения по примеру 18 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 14] На фигуре 14 представлен график, показывающий влияние соединения по примеру 22 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 15] На фигуре 15 представлен график, показывающий влияние соединения по примеру 25 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 16] На фигуре 16 представлен график, показывающий влияние соединения по примеру 27 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 17] На фигуре 17 представлен график, показывающий влияние соединения по примеру 29 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 18] На фигуре 18 представлен график, показывающий влияние соединения по примеру 31 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 19] На фигуре 19 представлен график, показывающий влияние соединения по примеру 33 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 20] На фигуре 20 представлен график, показывающий влияние соединения по примеру 66 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 21] На фигуре 21 представлен график, показывающий влияние соединения по примеру 68 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 22] На фигуре 22 представлен график, показывающий влияние соединения по примеру 70 на моделях частичного лигирования седалищного нерва на мышах (внутривенное введение).
[Фигура 23] На фигуре 23 представлен график, показывающий влияние соединения по примеру 13 на моделях синдрома фибромиалгии на крысах (внутривенное введение).
[Фигура 24] На фигуре 24 представлен график, показывающий влияние соединения по примеру 38 на моделях синдрома фибромиалгии на крысах (пероральное введение).
Описание вариантов изобретения
[0026]
Следующие термины, используемые в описании, если не указано иное, определяются следующим образом.
[0027]
В описании производное циклического амина по настоящему изобретению представлено следующей общей формулы (I).
[Формула 6]
где A представляет собой группу, представленную общей формулой (IIa), (IIb) или (IIc),
[Формула 7]
где, когда A представляет собой группу, представленную общей формулой (IIa) или (IIb), R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную атомом галогена, гидроксильной группой, амино группой или карбоксильной группой, R2 представляет собой атом водорода или атом галогена, R3 представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, R4 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 6 атомов углерода, или алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную алкилкарбониламино группой, содержащей от 2 до 6 атомов углерода, и n обозначает 1 или 2, в которой, когда R3 и R4, каждый, независимо, представляют собой алкильную группу, содержащую от 1 до 6 атомов углерода, R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную гидроксильной группой, амино группой или карбоксильной группой; и когда A представляет собой группу, представленную общей формулой (IIc), R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную карбоксильной группой, R2 представляет собой атом водорода или атом галогена, X представляет собой CH2, O или -NR5, и R5 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода.
[0028]
В производном циклического амина предпочтительным является, когда A представлен общей формулой (IIa) или (IIb) и R3 представляет собой атом водорода, метильную группу или этильную группу; и более предпочтительным является, когда R2 представляет собой атом водорода или атом хлора, R3 представляет собой атом водорода или метильную группу, и R4 представляет собой атом водорода или метилкарбонильную группу, или алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную метилкарбониламино группой.
[0029]
В производном циклического амина предпочтительным является, когда A представлен общей формулой (IIc), R2 представляет собой атом водорода или атом хлора, и R5 представляет собой метильную группу.
[0030]
Термин «атом галогена» относится к атому фтора, атому хлора, атому брома или атому йода.
[0031]
Термин «алкильная группа, содержащая от 1 до 6 атомов углерода» относится к линейной, разветвленной или циклической насыщенной углеводородной группе, содержащей от 1 до 6 атомов углерода. Например, может быть указана метильная группа, этильная группа, н-пропильная группа, изопропильная группа, циклопропильная группа, циклопропилметильная группа, н-бутильная группа, втор-бутильная группа, трет-бутильная группа, н-пентильная группа, изопентильная группа, н-гексильная группа, изогексильная группа или циклогексильная группа.
[0032]
Термин «алкильная группа, содержащая от 1 до 6 атомов углерода и необязательно замещенная атомом галогена, гидроксильной группой, амино группой или карбоксильной группой», относится к алкильной группе, содержащей от 1 до 6 атомов углерода, в которой атомы водорода, каждый, независимо и необязательно заменены на вышеупомянутые атом галогена или гидроксильную группу, амино группу или карбоксильную группу, и представляет собой, например, метильную группу, этильную группу, н-пропильную группу, изопропильную группу, циклопропильную группу, циклопропилметильную группу, н-бутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу, изопентильную группу, н-гексильную группу, изогексильную группу или циклогексильную группу или 2-хлорэтильнуюя группу, 2,2-дифторэтильную группу, 2,2,2-трифторэтильную группу, 2-гидроксиэтильную группу, 3-гидроксипропильную группу, 2-аминоэтильную группу, 3-аминопропильную группу, 1-карбоксиметильную группу или 2-карбоксиэтильную группу.
[0033]
Термин «алкилкарбонильная группа, содержащая от 2 до 6 атомов углерода», относится к группе, полученной путем присоединения линейной, разветвленной или циклической насыщенной углеводородной группе, содержащей от 1 до 5 атомов углерода, к карбонильной группе. Например, может быть указана ацетильная группа, н-пропионильная группа, н-бутирильная группа или изобутирильная группа.
[0034]
Термин «алкилкарбониламино группа, содержащая от 2 до 6 атомов углерода», относится к группе, полученной путем присоединения вышеупомянутой алкилкарбонильной группы, содержащей от 2 до 6 атомов углерода, к амино группе. Например, могут быть указаны метилкарбониламино группа, этилкарбониламино группа, н-пропилкарбониламино группа или изопропилкарбониламино группа.
[0035]
Термин «алкильная группа, содержащая от 1 до 6 атомов углерода и необязательно замещенная алкилкарбониламино группой, содержащей от 2 до 6 атомов углерода», относится к вышеупомянутой алкильной группе, содержащей от 1 до 6 атомов углерода и необязательно замещенной вышеупомянутой алкилкарбониламино группой, содержащей от 2 до 6 атомов углерода. Например, метильная группа, этильная группа, н-пропильная группа, изопропильная группа, циклопропильная группа, циклопропилметильная группа, н-бутильная группа, втор-бутильная группа, трет-бутильная группа, н-пентильная группа, изопентильная группа, н-гексильная группа, изогексильная или циклогексильная группа или 2-(метилкарбониламино)этильная группа, 2-(этилкарбониламино)этильная группа, 2-(н-пропилкарбониламино)этильная группа, 2-(изопропилкарбониламино)этильная группа, 3-(метилкарбониламино)пропильная группа или 4-(метилкарбониламино)бутильная группа.
[0036]
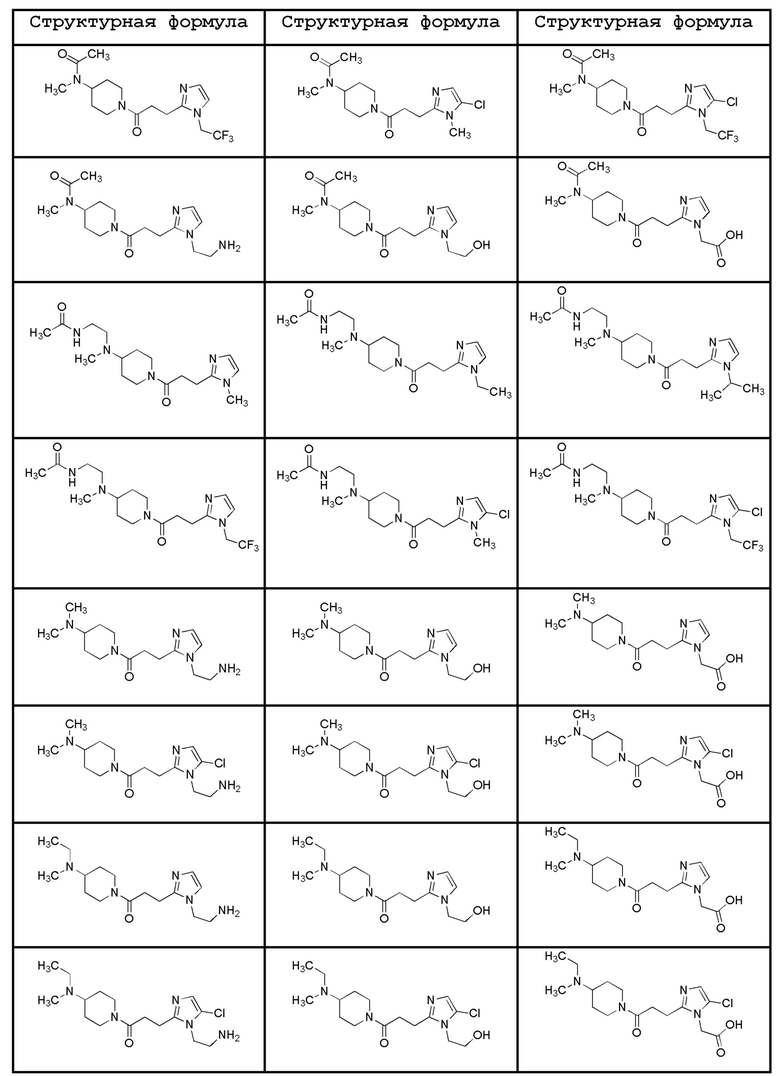
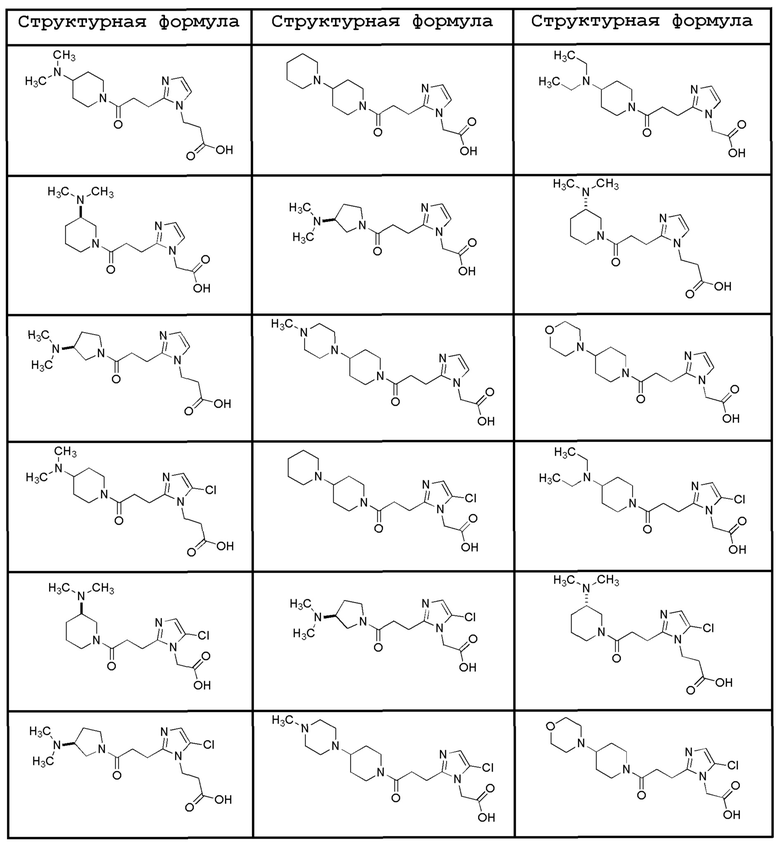
Конкретные примеры предпочтительного соединения в качестве производного циклического амина, представленного вышеуказанной общей формулой (I) (указываемого далее как производное циклического амина (I)), показаны в таблицах от 1-1 до 1-3; однако, настоящее изобретение ими не ограничивается.
[0037]
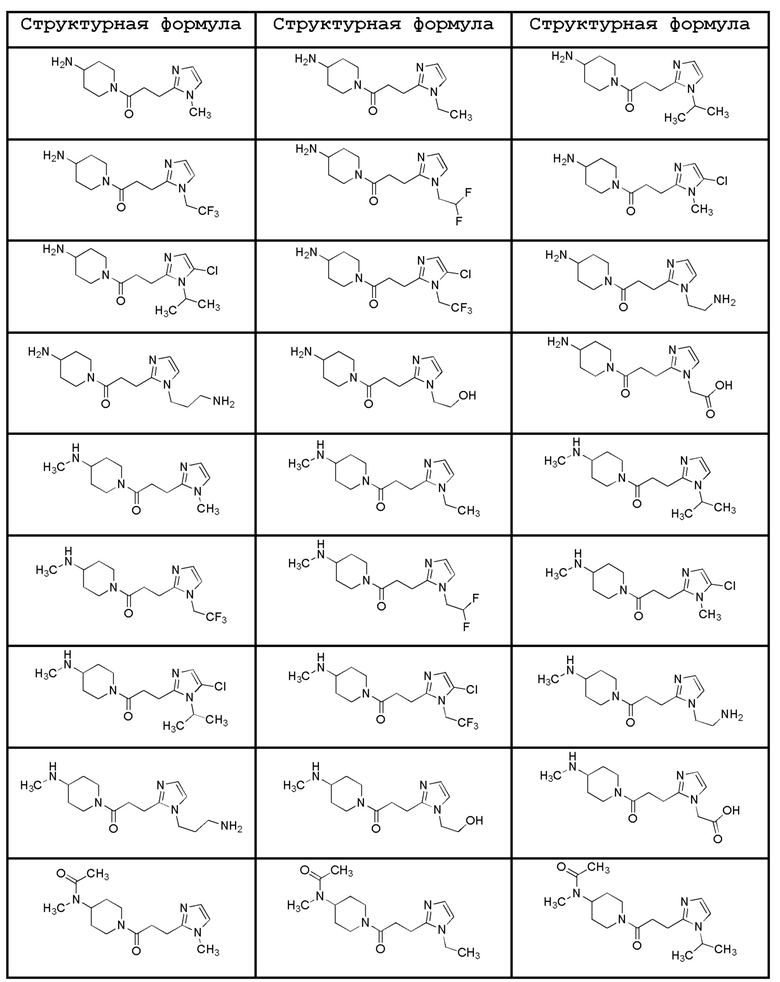
[0038]
[0039]
[0040]
Следует учесть, что, когда в производном циклического амина (I) имеется асимметрический атом углерода, изобретением охватываются все энантиомеры и их смеси. Когда имеется стереоизомер, включены все стереоизомеры и их смеси.
[0041]
Пролекарство производного циклического амина (I) или его фармакологически приемлемая соль также включены в объем настоящего изобретения. Пролекарство производного циклического амина (I) относится к соединению, которое in vivo ферментативно или химически преобразуется в производное циклического амина (I). Активной формой пролекарства производного циклического амина (I) является производное циклического амина (I); однако пролекарство производного циклического амина (I) само по себе может обладать активностью.
[0042]
В качестве пролекарства производного циклического амина (I) могут быть указаны, например, соединения, полученные путем ацилирования, алкилирования, фосфорилирования или борирования гидроксильной группы или амино группы производного циклического амина (I), или соединения, полученные путем этерификации или амидирования карбоксильной группы производного циклического амина (I); однако предпочтительным является соединение, полученное путем этерификации карбоксильной группы. Указанные соединения, каждое, могут быть синтезированы исходя из производного циклического амина (I) в соответствии с известным способом.
[0043]
В качестве соединения, полученного путем этерификации карбоксильной группы производного циклического амина (I), может быть указано метил этерифицированное, этил этерифицированное, н-пропил этерифицированное, изопропил этерифицированное, циклопропил этерифицированное, н-бутил этерифицированное, изобутил этерифицированное, втор-бутил этерифицированное, трет-бутил этерифицированное, циклопропилметил этерифицированное, н-пентил этерифицированное, изопентил этерифицированное, циклопентил этерифицированное, н-гексил этерифицированное, изогексил этерифицированное, циклогексил этерифицированное, н-гептил этерифицированное, н-октил этерифицированное, (5-метил-2-оксо-1,3-диоксолен-4-ил)метил этерифицированное, ацетилоксиметил этерифицированное, 1-ацетилоксиэтил этерифицированное, пропионилоксиметил этерифицированное, 1-пропионилоксиэтил этерифицированное, валерилоксиметил этерифицированное, 1-валерилоксиэтил этерифицированное, изобутирилоксиметил этерифицированное, 1-изобутилилоксиэтил этерифицированное, пивалоилоксиметил этерифицированное, 1-пивалоилоксиэтил этерифицированное, этоксикарбонилоксиметил этерифицированное, 1-((этоксикарбонил)окси)этил этерифицированное, 1-((этоксикарбонил)окси)изобутил этерифицированное, циклогексилоксикарбонилметил этерифицированное, 1-((циклогексил)карбонил)оксиэтил этерифицированное, 2-(диметиламино)-2-оксоэтил этерифицированное, фталидил этерифицированное или инданил этерифицированное соединение; однако предпочтительным является метил этерифицированное, этил этерифицированное, н-пропил этерифицированное, изопропил этерифицированное, циклопропил этерифицированное, н-бутил этерифицированное, изобутил этерифицированное, втор-бутил этерифицированное, трет-бутил этерифицированное, циклопропилметил этерифицированное, н-октил этерифицированное, (5-метил-2-оксо-1,3-диоксолен-4-ил)метил этерифицированное, пивалоилоксиметил этерифицированное, 2-(диметиламино)-2-оксоэтил этерифицированное, фталидил этерифицированное или инданил этерифицированное соединение.
[0044]
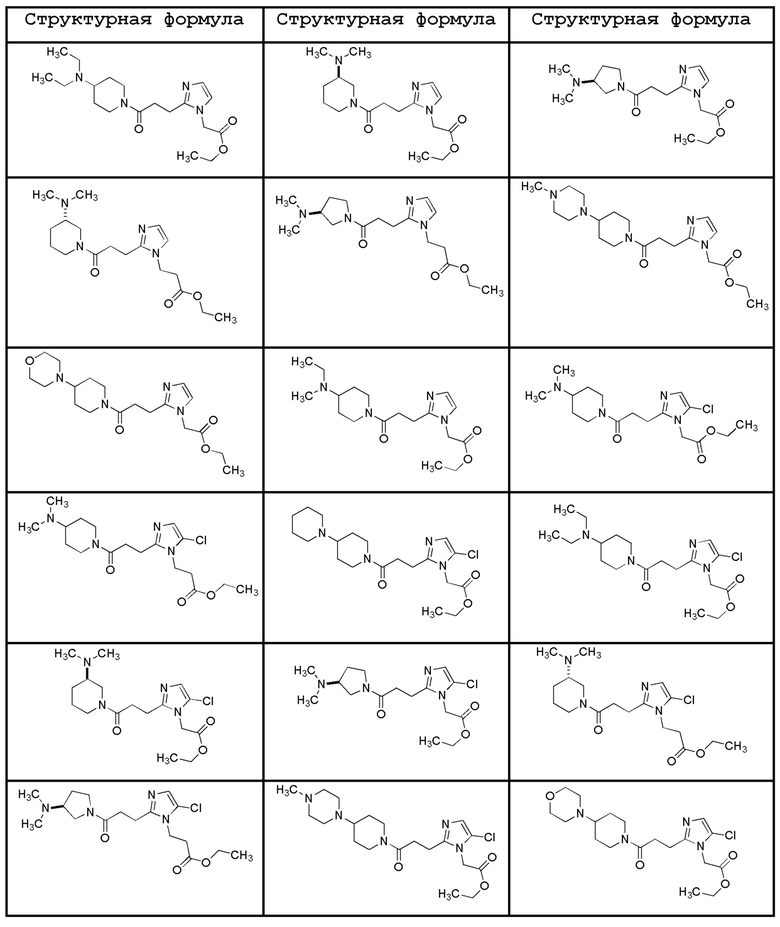
Конкретные примеры предпочтительного соединения в качестве пролекарства производного циклического амина (I) показаны в таблице 2-1 и таблице 2-2; однако настоящее изобретение ими не ограничивается.
[0045]
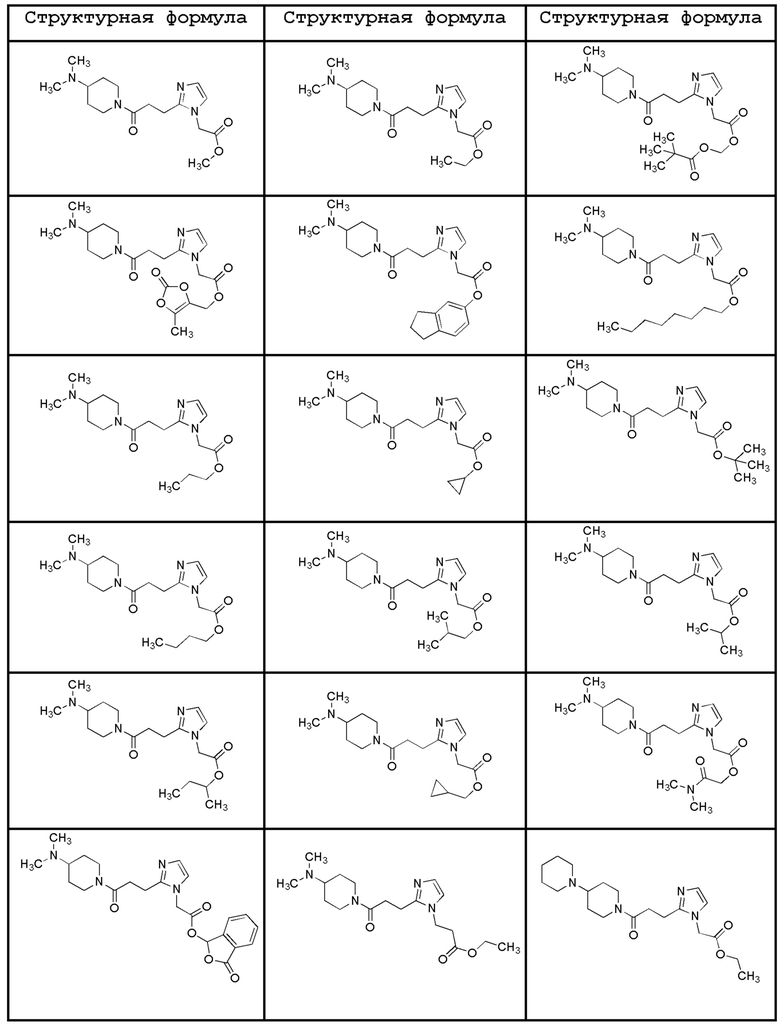
[0046]
[0047]
Пролекарство производного циклического амина (I) может быть преобразовано в производное циклического амина (I) в физиологических условиях, описанных в известных литературных источниках («Development of pharmaceutical products», Hirokawa-Shoten Ltd., vol. 7, p. 163 to 198, 1990, и Prog. Med. 5, p. 2157 to 2161, 1985).
[0048]
Производное циклического амина (I) или его пролекарство могут быть мечены радиоизотопом. Примеры радиоизотопа для использования при введении метки включают 3H, 14C и/или 125I.
[0049]
Производное циклического амина (I) или его пролекарство может быть дейтерировано.
[0050]
В качестве фармакологически приемлемой соли производного циклического амина (I) можно указать, например, неорганическую соль, такую как гидрохлорид, сульфат, фосфат или гидробромид; или органическую соль, такую как оксалат, малонат, цитрат, фумарат, лактат, малат, сукцинат, тартрат, ацетат, трифторацетат, малеат, глюконат, бензоат, салицилат, ксинафоат, памоат, аскорбат, адипат, метансульфонат, п-толуолсульфонат или циннамат. Эти соли могут существовать в форме гидрата, сольвата или кристаллического полиморфа.
[0051]
Производное циклического амина (I) или его пролекарство может быть синтезировано способами получения, которые описаны далее. Следует отметить, что, производные циклического амина (I) или их пролекарства, полученные с помощью следующих методов производства, каждый, могут быть выделены/очищены с помощью известных средств, таких как экстракция растворителем, перекристаллизация и/или с помощью хроматографии и преобразованы в желаемые соли с помощью известных способов или аналогичных способов. Когда производное циклического амина (I) или его пролекарство получают в виде соли, оно может быть преобразовано в производное циклического амина (I) или его пролекарство, или другую желаемую соль известным способом или аналогичным способом.
[0052]
В отдельных реакциях способов получения, которые описаны далее, когда исходное соединение имеет гидроксильную группу, аминогруппу или карбоксильную группу, в этих группы могут быть введены защитные группы. Желаемое соединение может быть получено удалением защитной группы, если это необходимо, после реакции.
[0053]
В отдельных реакциях способов получения, которые описаны далее, когда исходное соединение имеет гидроксильную группу, аминогруппу или карбоксильную группу, пролекарство производного циклического амина (I) можно получить без удаления защитной группы, введенной в эти группы.
[0054]
В качестве защитной группы гидроксильной группы могут быть упомянуты, например, тритильная группа, аралкильная группа, содержащая от 7 до 10 атомов углерода (например, бензильная группой), или замещенная силильная группа (например, триметилсилильная группа, триэтилсилильная группа или трет-бутилдиметилсилильная группа).
[0055]
В качестве защитной группы аминогруппы может быть указана, например, алкилкарбонильная группа, имеющая от 2 до 6 атомов углерода (например, ацетильная группа), бензоильная группа, алкоксикарбонильная группа, содержащая от 1 до 6 атомов углерода (например, трет-бутоксикарбонильная группа или бензилоксикарбонильная группа), аралкильная группа, содержащая от 7 до 10 атомов углерода (например, бензильная группа), или фталоильная группа.
[0056]
В качестве защитной группы карбоксильной группы может быть указана, например, алкильная группа, содержащая от 1 до 6 атомов углерода (например, метильная группа, этильная группа или трет-бутильная группа) или аралкильная группа, содержащая от 7 до 10 атомов углерода (например, бензильная группа).
[0057]
Удаление защитной группы, которое варьируется в зависимости от вида защитной группы, может быть осуществлено в соответствии с известным способом (например, Greene, T. W., «Greene’s Protective Groups in Organic Synthesis», Wiley-Interscience) или подобным способом.
[0058]
1. Соединение (Ia) может быть синтезировано в соответствии со способом получения, который описан далее.
[0059]
1-1. Способ получения соединения (Ia-a):
[Формула 8]
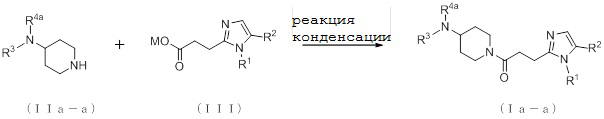
где M представляет собой атом водорода или щелочной металл, такой как литий или натрий, R4a представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную алкилкарбониламино группой, содержащей от 2 до 6 атомов углерода, и другие условные символы имеют такие же значения, которые определены выше.
Соединение (Ia-a), которое представляет собой производное циклического амина (I), где A представляет собой группу, представленную общей формулой (IIa), и R4 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную алкилкарбониламино группой, содержащей от 2 до 6 атомов углерода, может быть получено, например, путем реакции конденсации соединения (IIa-a) и соединения (III) с использованием конденсирующего агента в присутствии или в отсутствие основания.
[0060]
В реакции конденсации может быть использовано соединение (IIa-a) и его соль. В качестве соли в настоящем документе может быть указана, например, такая соль, как фармакологически приемлемая соль, которая упоминалась выше.
[0061]
В качестве соединения (IIa-a) и соединения (III) для использования в реакции конденсации могут быть непосредственно использованы коммерчески доступные соединения; однако они могут быть синтезированы, например, в соответствии со способами получения, которые описаны далее.
[0062]
В качестве основания для использования в реакции конденсации, может быть указан, например, ароматический амин, такой как пиридин или лутидин, или третичный амин, такой как триэтиламин, триизопропиламин, трибутиламин, циклогексилдиметиламин, 4-диметиламинопиридин, N,N-диметиланилин, N-метилпиперидин, N-метилпирролидин, N-метилморфолин или диизопропилэтиламин (DIEA).
[0063]
Количество основания для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIa-a) и, более предпочтительно, от 0,8 до 5,0 молей.
[0064]
В качестве конденсирующего агента для использования в реакции конденсации может быть указан, например, O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HBTU), циклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N'-этилкарбодиимид (EDC) или его гидрохлорид, 2-этокси-1-этоксикарбонил-1,2-дигидроксихинолин (EEDQ), карбонилдиимидазол (CDI), диэтилфосфорил цианид, бензотриазол-1-илокситриспирролидинoфосфония гексафторфосфат (PyBOP), дифенилфосфорилазид (DPPA), 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (DMTMM), изобутил хлорформиат, диэтилацетил хлорид или триметилацетил хлорид. Эти конденсирующие агенты используются отдельно или в комбинации с добавкой, такой как N-гидроксисукцинимид (HONSu), гидроксибензотриазол (HOBT), 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазин (HOOBT) или 4-диметиламинопиридин (DMAP).
[0065]
Количество конденсирующего агента для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIa-a) и, более предпочтительно, от 0,8 до 5,0 молей.
[0066]
Количество соединения (III) для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 3 молей относительно 1 моля соединения (IIa-a) и, более предпочтительно, от 0,8 до 1,5 молей.
[0067]
Реакцию конденсации обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; простой эфир, такой как тетрагидрофуран или 1,4-диоксан; амид, такой как N,N-диметилформамид или N-метилпирролидон; спирт, такой как метанол, этанол или 2-пропанол; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакция конденсации может быть осуществлена в отсутствие основания.
[0068]
В реакции конденсации температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0069]
В реакции конденсации время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0070]
1-2. Способ получения соединения (Ia-b), (Ia-c) и (Ia-d):
[Формула 9]
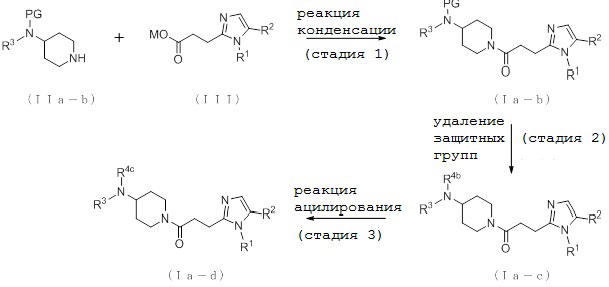
где PG представляет собой защитную группу, R4b представляет собой атом водорода, R4c представляет собой алкилкарбонильную группу, содержащую от 2 до 6 атомов углерода, и другие условные символы имеют такие же значения, которые определены выше.
(Стадия 1)
Соединение (Ia-b) может быть получено, например, путем реакции конденсации соединения (IIa-b) и соединения (III) с конденсирующим агентом в присутствии или в отсутствие основания.
[0071]
В реакции конденсации может быть использовано соединение (IIa-b) и его соль. В качестве соли в настоящем документе может быть указана, например, такая соль, как фармакологически приемлемая соль, которая упоминалась выше.
[0072]
В качестве соединения (IIa-b) и соединения (III) для использования в реакции конденсации, могут быть непосредственно использованы коммерчески доступные соединения; однако, они могут быть синтезированы, например, в соответствии со способами получения, которые описаны далее.
[0073]
В качестве основания для использования в реакции конденсации может быть указан, например, ароматический амин, такой как пиридин или лутидин, или третичный амин, такой как триэтиламин, триизопропиламин, трибутиламин, циклогексилдиметиламин, 4-диметиламинопиридин, N,N-диметиланилин, N-метилпиперидин, N-метилпирролидин, N-метилморфолин или диизопропилэтиламин (DIEA).
[0074]
Количество основания для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIa-b) и, более предпочтительно, от 0,8 до 5,0 молей.
[0075]
В качестве конденсирующего агента для использования в реакции конденсации может быть указан, например, O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HBTU), циклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N'-этил карбодиимид (EDC) или его гидрохлорид, 2-этокси-1-этоксикарбонил-1,2-дигидроксихинолин (EEDQ), карбонилдиимидазол (CDI), диэтилфосфорил цианид, бензотриазол-1-илокситриспирролидинoфосфония гексафторфосфат (PyBOP), дифенилфосфорилазид (DPPA), 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (DMTMM), изобутил хлорформиат, диэтилацетил хлорид или триметилацетил хлорид. Эти конденсирующие агенты используются отдельно или в комбинации с добавкой, такой как N-гидроксисукцинимид (HONSu), гидроксибензотриазол (HOBT), 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазин (HOOBT) или 4-диметиламинопиридин (DMAP).
[0076]
Количество конденсирующего агента для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIa-b) и, более предпочтительно, от 0,8 до 5,0 молей.
[0077]
Количество соединения (III) для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 3 молей относительно 1 моля соединения (IIa-b) и, более предпочтительно, от 0,8 до 1,5 молей.
[0078]
Реакцию конденсации обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; простой эфир, такой как тетрагидрофуран или 1,4-диоксан; амид, такой как N,N-диметилформамид или N-метилпирролидон; спирт, такой как метанол, этанол или 2-пропанол; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакция конденсации может быть осуществлена в отсутствие основания.
[0079]
В реакции конденсации температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0080]
В реакции конденсации время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0081]
(Стадия 2)
Соединение (Ia-c), которое представляет собой производное циклического амина (I), где A представляет собой группу, представленную общей формулой (IIa), и R4 представляет собой атом водорода, может быть получено путем удаления защитных групп у соединения (Ia-b).
[0082]
Удаление защитной группы, которое варьируется в зависимости от вида защитной группы, может быть осуществлено в соответствии с известным способом (например, Greene, T. W., «Greene’s Protective Groups in Organic Synthesis», Wiley-Interscience) или подобным способом.
[0083]
(Стадия 3)
Соединение (Ia-d), которое представляет собой производное циклического амина (I), где A представляет собой группу, представленную общей формулой (IIa), и R4 представляет собой алкилкарбонильную группу, содержащую от 2 до 6 атомов углерода, может быть получено путем взаимодействия соединения (Ia-c) и ацилирующего агента, такого как галогенангидрид карбоновой кислоты, содержащий от 2 до 6 атомов углерода, или ангидрид кислоты, в присутствии основания.
[0084]
В реакции ацилирования может быть использовано соединение (Ia-c) и его соль. В качестве соли в настоящем документе может быть указана, например, такая соль, как фармакологически приемлемая соль, которая упоминалась выше.
[0085]
В качестве основания, которое может быть использовано в реакции ацилирования, может быть указан, например, пиридин, триэтиламин, диизопропилэтиламин или N,N-диметиламинопиридин.
[0086]
Количество основания, которое может быть использовано в реакции ацилирования, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (Ia-c) и, более предпочтительно, от 0,8 до 5,0 молей.
[0087]
Реакцию ацилирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; простой эфир, такой как тетрагидрофуран или 1,4-диоксан; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакция ацилирования может быть осуществлена в отсутствие основания.
[0088]
1-3. Стадии образования солей соединения (Ia-a), (Ia-b), (Ia-c) и (Ia-d):
Фармакологически приемлемые соли соединения (Ia-a), (Ia-b), (Ia-c) и (Ia-d) могут быть получены посредством реакции образования солей, осуществляемой путем смешивания, осуществляемой путем смешивания, например, соединения (Ia-a), (Ia-b), (Ia-c) или (Ia-d) и кислоты.
[0089]
В качестве кислоты, которая может быть использована в реакции образования солей, может быть указана, например, неорганическая кислота, такая как хлористоводородная кислота, серная кислота, фосфорная кислота или бромистоводородная кислота; и органическая кислота, такая как щавелевая кислота, малоновая кислота, лимонная кислота, фумаровая кислота, молочная кислота, яблочная кислота, янтарная кислота, винная кислота, уксусная кислота, трифторуксусная кислота, малеиновая кислота, глюконовая кислота, бензойная кислота, салициловая кислота, 1-гидрокси-2-нафтойная кислота, памоевая кислота, аскорбиновая кислота, адипиновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота или коричная кислота.
[0090]
Реакцию образования соли обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, алифатический спирт, такой как метанол, этанол или изопропанол; простой эфир, такой как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан или диметиловый эфир этиленгликоля; амид, такой как N,N-диметилформамид или N-метилпирролидон; сульфоксид, такой как диметил сульфоксид; алифатический нитрил, такой как ацетонитрил или пропионитрил; кетон, такой как ацетон или 2-бутанон; сложный эфир, такой как этилацетат, метил ацетат или н-бутил ацетат или вода. Может быть использована смесь этих растворителей.
[0091]
2. Соединение (IIa) может быть синтезировано в соответствии со способом получения, который описан далее.
2-1. Способ получения соединения (IIa-a):
[Формула 10]
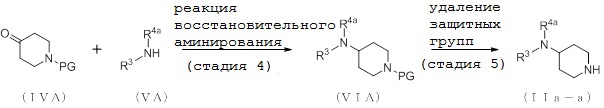
где отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 4)
Соединение (VIA) может быть получено путем реакции восстановительного аминирования между соединением (IVA) и соединением (VA).
[0092]
В качестве соединения (VA), которое может быть использовано при реакции восстановительного аминирования, может быть непосредственно использовано коммерчески доступное соединение.
[0093]
Реакция восстановительного аминирования может быть осуществлена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или подобным способом.
[0094]
(Стадия 5)
Соединение (IIa-a) может быть получено путем удаления защитных групп у соединения (VIA).
[0095]
Удаление защитной группы, которое варьируется в зависимости от вида защитной группы, может быть осуществлено в соответствии с известным способом (например, Greene, T. W., «Greene's Protective Groups in Organic Synthesis», Wiley-Interscience) или подобным способом.
[0096]
2-2. Способ получения соединения (IIa-b):
[Формула 11]
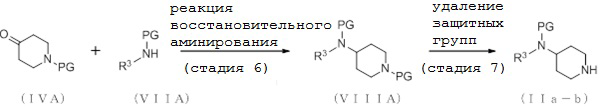
где отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 6)
Соединение (VIIIA) может быть получено путем реакции восстановительного аминирования между соединением (IVA) и соединения (VIIA).
[0097]
В качестве соединения (VIIA), которое может быть использовано в реакции восстановительного аминирования, может быть непосредственно использовано коммерчески доступное соединение.
[0098]
Реакция восстановительного аминирования может быть осуществлена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или подобным способом.
[0099]
(Стадия 7)
Соединение (IIa-b) может быть получено путем удаления защитных групп у соединения (VIIIA).
[0100]
Удаление защитной группы, которое варьируется в зависимости от вида защитной группы, может быть осуществлено в соответствии с известным способом (например, Greene, T. W., «Greene's Protective Groups in Organic Synthesis», Wiley-Interscience) или подобным способом.
[0101]
3. Способ получения соединения (IIIa):
[Формула 12]
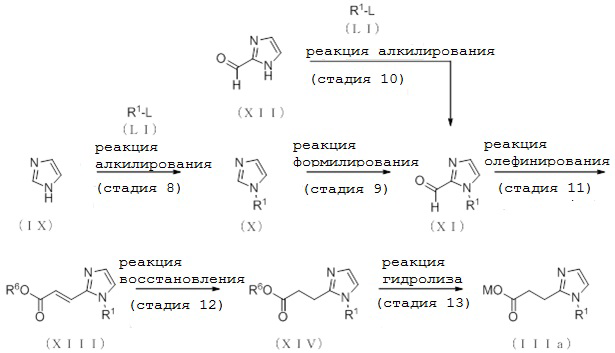
где L представляет собой удаляемую группу, такую как атом хлора, атом брома или атом йода, R6 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 10 атомов углерода, такую как метильная группа, этильная группа, пропильная группа, н-бутильная группа или бензильная группа, и другие отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 8)
Соединение (X) может быть получено депротонированием соединения (IX) с помощью основания с последующей реакцией алкилирования с помощью алкилирующего реагента (LI).
[0102]
В качестве соединения (IX), которое может быть использовано в реакции алкилирования, может быть использовано коммерчески доступное соединение.
[0103]
В качестве основания, которое может быть использовано в реакции алкилирования, могут быть указаны, например, гидрид щелочного металла, такой как натрия гидрид или гидрид калия или бутиллитий, такой как н-бутиллитий, втор-бутиллитий или трет-бутиллитий.
[0104]
Количество основания, которое может быть использовано в реакции алкилирования, предпочтительно, представляет собой от 0,5 до 3,0 молей относительно 1 моля соединения (IX) и, более предпочтительно, от 0,8 до 2,0 молей.
[0105]
В качестве алкилирующего реагента (LI), которое может быть использовано в реакции алкилирования, может быть использовано коммерчески доступное соединение.
[0106]
Количество алкилирующего реагента (LI), которое может быть использовано в реакции алкилирования, предпочтительно, представляет собой от 0,5 до 10,0 молей относительно 1 моля соединения (IX) и, более предпочтительно, от 0,8 до 5,0 молей.
[0107]
Реакцию алкилирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, алифатический углеводород, такой как гептан или гексан, или простой эфир, такой как тетрагидрофуран, диэтиловый эфир или 1,4-диоксан. Может быть использована смесь этих растворителей.
[0108]
В реакции алкилирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0109]
В реакции алкилирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0110]
(Стадия 9)
Соединение (XI) может быть получено депротонированием соединения (X) с помощью основания, с последующей реакцией формилирования с реагентом, вводящим формильную группу.
[0111]
В качестве основания, которое может быть использовано в реакции формилирования, может быть указан, например, н-бутиллитий, втор-бутиллитий или трет-бутиллитий.
[0112]
Количество основания, которое может быть использовано в реакции формилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (X) и, более предпочтительно, от 0,8 до 2,0 молей.
[0113]
В качестве реагента, вводящего формильную группу, который может быть использован в реакции формилирования, может быть указан, например, N,N-диметилформамид. В качестве N,N-диметилформамида может быть использовано коммерчески доступное соединение.
[0114]
Количество реагента, вводящего формильную группу, которое может быть использовано в реакции формилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (X) и, более предпочтительно, от 0,8 до 2,0 молей
[0115]
Реакцию формилирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, алифатический углеводород, такой как гептан или гексан, или простой эфир, такой как тетрагидрофуран, диэтиловый эфир или 1,4-диоксан. Может быть использована смесь этих растворителей.
[0116]
При депротонировании в реакции формилирования температура реакции, предпочтительно, составляет от -100 до 0°C и, более предпочтительно, от -80 до -20°C. При формилировании в реакция формилирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0117]
В реакции формилирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0118]
(Стадия 10)
Соединение (XI) может быть получено депротонированием соединения (XII) с помощью основания с последующей реакцией алкилирования посредством алкилирующего реагента (LI).
[0119]
В качестве соединения (XII), которое может быть использовано в реакции алкилирования, может быть использовано коммерчески доступное соединение.
[0120]
В качестве основания, которое может быть использовано в реакции алкилирования, может быть указан, например, карбонат металла, такой как карбонат натрия, карбонат калия или карбонат цезия, или гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия.
[0121]
Количество основания, которое может быть использовано в реакции алкилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XII) и, более предпочтительно, от 0,8 до 2,0 молей.
[0122]
Количество алкилирующего реагента (LI), которое может быть использовано в реакции алкилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XII) и, более предпочтительно, от 0,8 до 2,0 молей.
[0123]
Реакцию алкилирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0124]
В реакции алкилирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0125]
В реакции алкилирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0126]
(Стадия 11)
Соединение (XIII) может быть получено реакцией олефинирования соединения (XI).
[0127]
В качестве реагента, который может быть использован в реакции олефинирования, может быть указан, например, реагент Виттига, такой как метил 2-(трифенилфосфоранилиден)ацетат, или реагент Хорнера-Эммонса, такой как этил диэтилфосфоноацетат. В качестве реагента Виттига или реагента Хорнера-Эммонса может быть непосредственно использовано коммерчески доступное соединение.
[0128]
Количество реагента Виттига или реагента Хорнера-Эммонса, которое может быть использовано в реакции олефинирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XI) и, более предпочтительно, от 0,8 до 2,0 молей.
[0129]
Реакцию олефинирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический углеводород, такой как толуол, хлорбензол или ксилол, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0130]
В реакции олефинирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0131]
В реакции олефинирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
(Стадия 12)
[0132]
Соединение (XIV) может быть получено реакцией восстановления соединения (XIII) в присутствии катализатора на основе переходного металла в атмосфере водорода.
[0133]
В качестве катализатора на основе переходного металла, которое может быть использовано в реакции восстановления, например, может быть указан палладий-углерод.
[0134]
Количество катализатора на основе переходного металла, которое может быть использовано в реакции восстановления, предпочтительно, представляет собой 0,1 до 100 мас.% относительно соединения (XIII) и, более предпочтительно, от 1 до 50 мас.%
[0135]
Реакцию восстановления обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, алифатический углеводород, такой как гептан или гексан, или алифатический спирт, такой как метанол, этанол или пропанол. Может быть использована смесь этих растворителей.
[0136]
В реакции восстановления температура реакции, предпочтительно, составляет от 0 до 80°C и, более предпочтительно, от 10 до 40°C.
[0137]
В реакции восстановления время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0138]
(Стадия 13)
Соединение (IIIa), которое представляет собой соединение (III), где R2 представляет собой атом водорода, может быть получено реакцией гидролиза соединения (XIV).
[0139]
В качестве основания, которое может быть использовано в реакции гидролиза, может быть указан, например, гидроксид лития, гидроксид калия или гидроксид натрия.
[0140]
Количество основания, которое может быть использовано в реакции гидролиза, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XIV) и, более предпочтительно, от 0,8 до 2,0 молей.
[0141]
Реакцию гидролиза обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, алифатический спирт, такой как метанол, этанол или пропанол, или вода. Может быть использована смесь этих растворителей.
[0142]
В реакции гидролиза температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0143]
В реакции гидролиза время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0144]
4. Способ получения соединения (III):
[Формула 13]
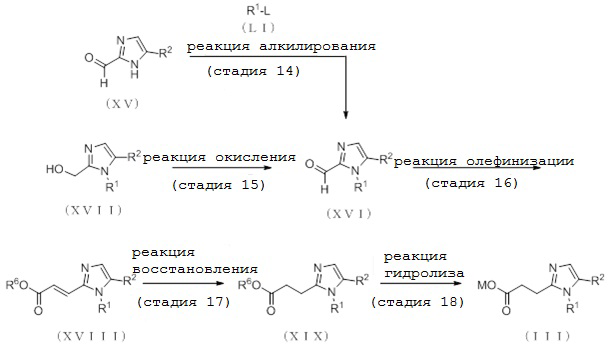
где отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 14)
Соединение (XVI) может быть получено депротонированием соединения (XV) с помощью основания с последующей реакцией алкилирования посредством алкилирующего реагента (LI).
[0145]
В качестве соединения (XV), которое может быть использовано в реакции алкилирования, может быть непосредственно использовано коммерчески доступное соединение.
[0146]
В качестве основания, которое может быть использовано в реакции алкилирования, может быть указан, например, карбонат металла, такой как карбонат натрия, карбонат калия или карбонат цезия или гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия.
[0147]
Количество основания, которое может быть использовано в реакции алкилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XV) и, более предпочтительно, от 0,8 до 2,0 молей.
[0148]
Количество алкилирующего реагента (LI), которое может быть использовано в реакции алкилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XV) и, более предпочтительно, от 0,8 до 2,0 молей.
[0149]
Реакцию алкилирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0150]
В реакции алкилирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0151]
В реакции алкилирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0152]
(Стадия 15)
Соединение (XVI) может быть получено путем реакции окисления соединения (XVII).
[0153]
В качестве соединения (XVII), которое может быть использовано в реакции окисления, может быть непосредственно использовано коммерчески доступное соединение; однако, оно может быть также синтезировано способом, известным специалистам в данной области техники.
[0154]
В качестве окислителя, который может быть использован в реакции окисления, может быть указан, например, триоксид серы-пиридин, активированный диметил сульфоксид или реагент Десс-Мартина.
[0155]
Количество окислителя, которое может быть использовано в реакции окисления, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XVII) и, более предпочтительно, от 0,8 до 2,0 молей.
[0156]
Реакцию окисления обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический амин, такой как пиридин, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0157]
В реакции окисления температура реакции, предпочтительно, составляет от -78°C до 100°C и, более предпочтительно, от -78°C до 40°C.
[0158]
В реакции окисления время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0159]
(Стадия 16)
Соединение (XVIII) может быть получено реакцией олефинирования соединения (XVI).
[0160]
В качестве реагента, который может быть использован в реакции олефинирования, может быть указан, например, реагент Виттига, такой как метил 2-(трифенилфосфоранилиден)ацетат, или реагент Хорнера-Эммонса, такой как этил диэтилфосфоноацетат. В качестве реагента Виттига или реагента Хорнера-Эммонса, может быть непосредственно использовано коммерчески доступное соединение.
[0161]
Количество реагента Виттига или реагента Хорнера-Эммонса, которое может быть использовано в реакции олефинирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XVI) и, более предпочтительно, от 0,8 до 2,0 молей.
[0162]
Реакцию олефинирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический углеводород, такой как толуол, хлорбензол или ксилол, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0163]
В реакции олефинирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0164]
В реакции олефинирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0165]
(Стадия 17)
Соединение (XIX) может быть получено реакцией восстановления соединения (XVIII) в присутствии катализатора на основе переходного металла в атмосфере водорода.
[0166]
В качестве катализатора на основе переходного металла, который может быть использован в реакции восстановления, может быть указан, например, палладий-углерод.
[0167]
Количество катализатора на основе переходного металла, которое может быть использовано в реакции восстановления, предпочтительно, представляет собой 0,1 до 100 мас.% относительно соединения (XVIII) и, более предпочтительно, от 1 до 50 мас.%.
[0168]
Реакцию восстановления обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, алифатический углеводород, такой как гептан или гексан, или алифатический спирт, такой как метанол, этанол или пропанол. Может быть использована смесь этих растворителей.
[0169]
В реакции восстановления температура реакции, предпочтительно, составляет от 0 до 80°C и, более предпочтительно, от 10 до 40°C.
[0170]
В реакции восстановления время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0171]
(Стадия 18)
Соединение (III) может быть получено путем реакции гидролиза соединения (XIX).
[0172]
В качестве основания, которое может быть использовано в реакции гидролиза, может быть указан, например, гидроксид лития, гидроксид калия или гидроксид натрия.
[0173]
Количество основания, которое может быть использовано в реакции гидролиза, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XIX) и, более предпочтительно, от 0,8 до 2,0 молей.
[0174]
Реакцию гидролиза обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, алифатический спирт, такой как метанол, этанол или пропанол, или вода. Может быть использована смесь этих растворителей.
[0175]
В реакции гидролиза температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0176]
В реакции гидролиза время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0177]
5. Способ получения соединения (XIII):
[Формула 14]
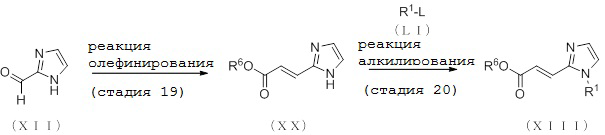
где отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 19)
Соединение (XX) может быть получено реакцией олефинирования соединения (XII).
[0178]
В качестве реагента, который может быть использован в реакции олефинирования, может быть указан, например, реагент Виттига, такой как метил 2-(трифенилфосфоранилиден)ацетат, или реагент Хорнера-Эммонса, такой как этил диэтилфосфоноацетат. В качестве реагента Виттига или реагента Хорнера-Эммонса, может быть непосредственно использовано коммерчески доступное соединение.
[0179]
Количество реагента Виттига или реагента Хорнера-Эммонса, которое может быть использовано в реакции олефинирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XII) и, более предпочтительно, от 0,8 до 2,0 молей.
[0180]
Реакцию олефинирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический углеводород, такой как толуол, хлорбензол или ксилол, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0181]
В реакции олефинирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0182]
В реакции олефинирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0183]
(Стадия 20)
Соединение (XIII) может быть получено депротонированием соединения (XX) с помощью основания с последующей реакцией алкилирования посредством алкилирующего реагента (LI).
[0184]
В качестве основания, которое может быть использовано в реакции алкилирования, может быть указан, например, карбонат металла, такой как карбонат натрия, карбонат калия или карбонат цезия или гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия.
[0185]
Количество основания, которое может быть использовано в реакции алкилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XX) и, более предпочтительно, от 0,8 до 2,0 молей.
[0186]
Количество алкилирующего реагента (LI), которое может быть использовано в реакции алкилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XX) и, более предпочтительно, от 0,8 до 2,0 молей.
[0187]
Реакцию алкилирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0188]
В реакции алкилирования температура реакции, предпочтительно, составляет от,-20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0189]
В реакции алкилирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0190]
6. Способ получения соединения (XVIII):
[Формула 15]
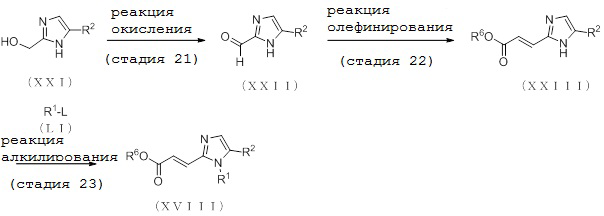
где отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 21)
Соединение (XXII) может быть получено путем реакции окисления соединения (XXI).
[0191]
В качестве соединения (XXI), которое может быть использовано в реакции окисления, может быть непосредственно использовано коммерчески доступное соединение; однако, оно также может быть синтезировано способом, известным специалистам в данной области техники.
[0192]
В качестве окислителя, которое может быть использовано в реакции окисления, может быть указан, например, триоксид серы-пиридин, активированный диметил сульфоксид или реагент Десс-Мартина.
[0193]
Количество окислителя, которое может быть использовано в реакции окисления, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XXI) и, более предпочтительно, от 0,8 до 2,0 молей.
[0194]
Реакцию окисления обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический амин, такой как пиридин, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0195]
В реакции окисления температура реакции, предпочтительно, составляет от -78°C до 100°C и, более предпочтительно, от -78°C до 40°C.
[0196]
В реакции окисления время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0197]
(Стадия 22)
Соединение (XXIII) может быть получено реакцией олефинирования соединения (XXII).
[0198]
В качестве реагента, которое может быть использован в реакции олефинирования, может быть указан, например, реагент Виттига, такой как метил 2-(трифенилфосфоранилиден)ацетат, или реагент Хорнера-Эммонса, такой как этил диэтилфосфоноацетат. В качестве реагента Виттига или реагента Хорнера-Эммонса может быть непосредственно использовано коммерчески доступное соединение.
[0199]
Количество реагента Виттига или реагента Хорнера-Эммонса, которое может быть использовано в реакции олефинирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XXII) и, более предпочтительно, от 0,8 до 2,0 молей.
[0200]
Реакцию олефинирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический углеводород, такой как толуол, хлорбензол или ксилол, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0201]
В реакции олефинирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0202]
В реакции олефинирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0203]
(Стадия 23)
Соединение (XVIII) может быть получено депротонированием соединения (XXIII) с помощью основания, с последующей реакцией алкилирования посредством алкилирующего реагента (LI).
[0204]
В качестве основания, которое может быть использовано в реакции алкилирования, может быть указан, например, карбонат металла, такой как карбонат натрия, карбонат калия или карбонат цезия, или гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия.
[0205]
Количество основания, которое может быть использовано в реакции алкилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XXIII) и, более предпочтительно, от 0,8 до 2,0 молей.
[0206]
Количество алкилирующего реагента (LI), которое может быть использовано в реакции алкилирования, предпочтительно, составляет от 0,5 до 3,0 молей относительно 1 моля соединения (XXIII) и, более предпочтительно, от 0,8 до 2,0 молей.
[0207]
Реакцию алкилирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей.
[0208]
В реакции алкилирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0209]
В реакции алкилирования время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0210]
7. Соединение (Ib) может быть синтезировано в соответствии со способом получения, который описан далее.
7-1. Способ получения соединения (Ib-a):
[Формула 16]
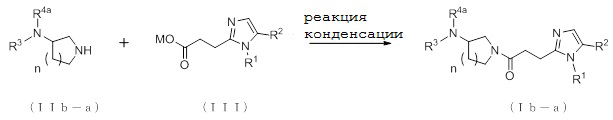
где отдельные условные символы имеют те же значения, которые определены выше.
Соединение (Ib-a), которое представляет собой производное циклического амина (I), где A представляет собой группу, представленную общей формулой (IIb), и R4 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода и необязательно замещенную алкилкарбониламино группой, содержащей от 2 до 6 атомов углерода, может быть получено, например, путем реакции конденсации между соединением (IIb-a) и соединением (III) с конденсирующим агентом, в присутствии или в отсутствие основания.
[0211]
В реакции конденсации может быть использовано соединение (IIb-a) и его соль. В качестве соли в настоящем документе может быть указана, например, такая соль, как фармакологически приемлемая соль, которая упоминалась выше.
[0212]
В качестве соединения (IIb-a) и соединения (III) для использования в реакции конденсации могут быть непосредственно использованы коммерчески доступные соединения; однако, например, соединение (IIb-a) может быть синтезировано в соответствии со способом получения, который описан далее, и соединение (III) может быть синтезировано в соответствии с описанным выше способом получения.
[0213]
В качестве основания для использования в реакции конденсации, может быть указан, например, ароматический амин, такой как пиридин или лутидин, или третичный амин, такой как триэтиламин, триизопропиламин, трибутиламин, циклогексилдиметиламин, 4-диметиламинопиридин, N,N-диметиланилин, N-метилпиперидин, N-метилпирролидин, N-метилморфолин или диизопропилэтиламин (DIEA).
[0214]
Количество основания для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIb-a) и, более предпочтительно, от 0,8 до 5,0 молей.
[0215]
В качестве конденсирующего агента для использования в реакции конденсации, может быть указан, например, O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HBTU), циклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N'-этил карбодиимид (EDC) или его гидрохлорид, 2-этокси-1-этоксикарбонил-1,2-дигидроксихинолин (EEDQ), карбонилдиимидазол (CDI), диэтилфосфорил цианид, бензотриазол-1-илокситриспирролидинoфосфония гексафторфосфат (PyBOP), дифенилфосфорил азид (DPPA), 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (DMTMM), изобутил хлорформиат, диэтилацетил хлорид или триметилацетил хлорид. Эти конденсирующие агенты могут быть использованы отдельно или в комбинации с добавкой, такой как N-гидроксисукцинимид (HONSu), гидроксибензотриазол (HOBT), 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазин (HOOBT) или 4-диметиламинопиридин (DMAP).
[0216]
Количество конденсирующего агента для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIb-a) и, более предпочтительно, от 0,8 до 5,0 молей.
[0217]
Количество соединения (III) для использования в реакции конденсации, предпочтительно, представляет собой от 0,5 до 3 молей относительно 1 моля соединения (IIb-a) и, более предпочтительно, от 0,8 до 1,5 молей.
[0218]
Реакцию конденсации обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический амин, такой как пиридин, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, спирт, такой как метанол, этанол или 2-пропанол, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакция конденсации может быть проведена в отсутствие основания.
[0219]
В реакции конденсации температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0220]
В реакции конденсации время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0221]
7-2. Способы получения соединений (Ib-b), (Ib-c) и (Ib-d):
[Формула 17]
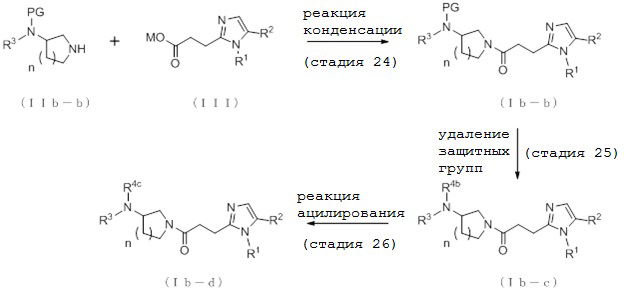
где отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 24)
Соединение (Ib-b) может быть получено, например, путем реакции конденсации между соединением (IIb-b) и соединением (III) с конденсирующим агентом в присутствии или в отсутствие основания.
[0222]
В реакции конденсации может быть использовано соединение (IIb-b) и его соль. В качестве соли в настоящем документе может быть указана, например, такая соль, как фармакологически приемлемая соль, которая упоминалась выше.
[0223]
В качестве соединения (IIb-b) и соединения (III) для использования в реакции конденсации могут быть непосредственно использованы коммерчески доступные соединения; однако, например, соединение (IIb-b) может быть синтезировано в соответствии со способом получения, который описан далее, и соединение (III) может быть синтезировано в соответствии с описанным выше способом получения.
[0224]
В качестве основания для использования в реакции конденсации, может быть указан, например, ароматический амин, такой как пиридин или лутидин, или третичный амин, такой как триэтиламин, триизопропиламин, трибутиламин, циклогексилдиметиламин, 4-диметиламинопиридин, N,N-диметиланилин, N-метилпиперидин, N-метилпирролидин, N-метилморфолин или диизопропилэтиламин (DIEA).
[0225]
Количество основания для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIb-b) и, более предпочтительно, от 0,8 до 5,0 молей.
[0226]
В качестве конденсирующего агента для использования в реакции конденсации, может быть указан, например, O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HBTU), циклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N'-этил карбодиимид (EDC) или его гидрохлорид, 2-этокси-1-этоксикарбонил-1,2-дигидроксихинолин (EEDQ), карбонилдиимидазол (CDI), диэтилфосфорил цианид, бензотриазол-1-илокситриспирролидинoфосфония гексафторфосфат (PyBOP), дифенилфосфорил азид (DPPA), 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (DMTMM), изобутил хлорформиат, диэтилацетил хлорид или триметилацетил хлорид. Эти конденсирующие агенты могут быть использованы отдельно или в комбинации с добавкой, такой как N-гидроксисукцинимид (HONSu), гидроксибензотриазол (HOBT), 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазин (HOOBT) или 4-диметиламинопиридин(DMAP).
[0227]
Количество конденсирующего агента для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIb-b) и, более предпочтительно, от 0,8 до 5,0 молей.
[0228]
Количество соединения (III) для использования в реакции конденсации, предпочтительно, представляет собой от 0,5 до 3 молей относительно 1 моля соединения (IIb-b) и, более предпочтительно, от 0,8 до 1,5 молей.
[0229]
Реакцию конденсации обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический амин, такой как пиридин, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, спирт, такой как метанол, этанол или 2-пропанол, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакция конденсации может быть проведена в отсутствие основания.
[0230]
В реакции конденсации температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0231]
В реакции конденсации время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0232]
(Стадия 25)
Соединение (Ib-c), которое представляет собой производное циклического амина (I), где A представляет собой группу, представленную общей формулой (IIb), и R4 представляет собой атом водорода, может быть получено путем удаления защитных групп у соединения (Ib-b).
[0233]
Удаление защитной группы, которое варьируется в зависимости от вида защитной группы, может быть осуществлено в соответствии с известным способом (например, Greene, T. W., «Greene’s Protective Groups in Organic Synthesis», Wiley-Interscience) или подобным способом.
[0234]
(Стадия 26)
Соединение (Ib-d), которое представляет собой производное циклического амина (I), где A представляет собой группу, представленную общей формулой (IIb), и R4 представляет собой алкилкарбонильную группу, содержащую от 2 до 6 атомов углерода, может быть получено, например, путем взаимодействия соединения (Ib-c) и ацилирующего агента, такого как галогенангидрид карбоновой кислоты, содержащей от 2 до 6 атомов углерода, или ангидрид кислоты, в присутствии основания.
[0235]
В реакции ацилирования может быть использовано соединение (Ib-c) и его соль. В качестве соли в настоящем документе может быть указана, например, такая соль, как фармакологически приемлемая соль, которая упоминалась выше.
[0236]
В качестве основания, которое может быть использовано в реакции ацилирования, может быть указан, например, пиридин, триэтиламин, диизопропилэтиламин или N,N-диметиламинопиридин.
[0237]
Количество основания, которое может быть использовано в реакции ацилирования предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (Ib-c) и, более предпочтительно, от 0,8 до 5,0 молей.
[0238]
Реакцию ацилирования обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический амин, такой как пиридин, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакция ацилирования может быть осуществлена в отсутствие основания.
[0239]
7-3. Стадии образования солей соединения (Ib-a), (Ib-b), (Ib-c) и (Ib-d):
Фармакологически приемлемые соли соединения (Ib-a), (Ib-b), (Ib-c) и (Ib-d) могут быть получены посредством реакции образования солей, осуществляемой путем смешивания, например, соединения (Ib-a), (Ib-b), (Ib-c) или (Ib-d) и кислоты.
[0240]
В качестве кислоты, которая может быть использована в реакции образования солей, может быть указана, например, неорганическая кислота, такая как хлористоводородная кислота, серная кислота, фосфорная кислота или бромистоводородная кислота; или органическая кислота, такая как щавелевая кислота, малоновая кислота, лимонная кислота, фумаровая кислота, молочная кислота, яблочная кислота, янтарная кислота, винная кислота, уксусная кислота, трифторуксусная кислота, малеиновая кислота, глюконовая кислота, бензойная кислота, салициловая кислота, ксинафоевая кислота, памоевая кислота, аскорбиновая кислота, адипиновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота или коричная кислота.
[0241]
Реакцию образования соли обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, алифатический спирт, такой как метанол, этанол или изопропанол, простой эфир, такой как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан или диметиловый эфир этиленгликоля, амид, такой как N,N-диметилформамид или N-метилпирролидон, сульфоксид, такой как диметил сульфоксид, алифатический нитрил, такой как ацетонитрил или пропионитрил, кетон, такой как ацетон или 2-бутанон, сложный эфир, такой как этилацетат, метил ацетат или н-бутил ацетат, или вода. Может быть использована смесь этих растворителей.
[0242]
8. Соединение (IIb) может быть синтезировано в соответствии со способом получения, который описан далее.
8-1. Способ получения соединения (IIb-a):
[Формула 18]
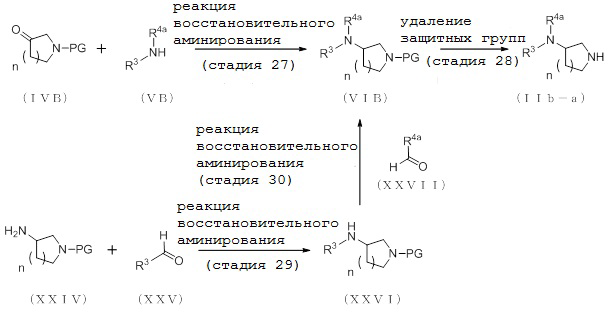
где отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 27)
Соединение (VIB) может быть получено путем реакции восстановительного аминирования между соединением (IVB) и соединение (VB).
[0243]
В качестве соединения (VB), которое может быть использовано в реакции восстановительного аминирования, может быть непосредственно использовано коммерчески доступное соединение.
[0244]
Реакция восстановительного аминирования может быть осуществлена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или подобным способом.
[0245]
(Стадия 28)
Соединение (IIb-a) может быть получено путем удаления защитных групп у соединения (VIB).
[0246]
Удаление защитной группы, которое варьируется в зависимости от вида защитной группы, может быть осуществлено в соответствии с известным способом (например, Greene, T. W., «Greene’s Protective Groups in Organic Synthesis», Wiley-Interscience) или подобным способом.
(Стадия 29)
Соединение (XXV1) может быть получено путем реакции восстановительного аминирования между соединением (XXIV) и соединением (XXV).
[0247]
В качестве соединения (XXV), которое может быть использовано в реакции восстановительного аминирования, может быть непосредственно использовано коммерчески доступное соединение.
[0248]
Реакция восстановительного аминирования может быть осуществлена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или подобным способом.
(Стадия 30)
Соединение (VIB) может быть получено путем реакции восстановительного аминирования между соединением (XXVI) и соединением (XXVII).
[0249]
В качестве соединения (XXVII), которое может быть использовано в реакции восстановительного аминирования, может быть непосредственно использовано коммерчески доступное соединение.
[0250]
Реакция восстановительного аминирования может быть осуществлена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или подобным способом.
[0251]
8-2. Способ получения соединения (IIb-b):
[Формула 19]
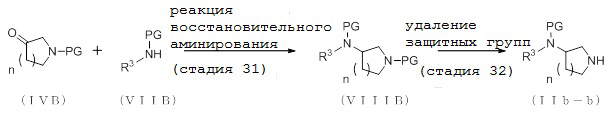
где отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 31)
Соединение (VIIIB) может быть получено путем реакции восстановительного аминирования между соединением (IVB) и соединением (VIIB).
[0252]
В качестве соединения (VIIB), которое может быть использовано в реакции восстановительного аминирования, может быть непосредственно использовано коммерчески доступное соединение.
[0253]
Реакция восстановительного аминирования может быть осуществлена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или подобным способом.
[0254]
(Стадия 32)
Соединение (IIb-b) может быть получено путем удаления защитных групп у соединения (VIIIB).
[0255]
Удаление защитной группы, которое варьируется в зависимости от вида защитной группы, может быть осуществлено в соответствии с известным способом (например, Greene, T. W., «Greene’s Protective Groups in Organic Synthesis», Wiley-Interscience) или подобным способом.
[0256]
9. Соединение (Ic) может быть синтезировано в соответствии со способом получения, который описан далее.
9-1. Способ получения соединения (Ic):
[Формула 20]
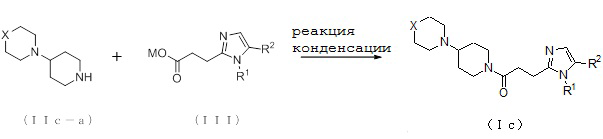
где отдельные условные символы имеют те же значения, которые определены выше.
Соединение (Ic), которое представляет собой производное циклического амина (I), где A представляет собой группу, представленную общей формулой (IIc), может быть получено, например, путем реакции конденсации между соединением (IIc-a) и соединением (III) с конденсирующим агентом в присутствии или в отсутствие основания.
[0257]
В реакции конденсации может быть использовано соединение (IIc-a) и его соль. В качестве соли в настоящем документе может быть указана, например, такая соль, как фармакологически приемлемая соль, которая упоминалась выше.
[0258]
В качестве соединения (IIc-a) и соединения (III) для использования в реакции конденсации могут быть непосредственно использованы коммерчески доступные соединения; однако, например, соединение (IIc-a) может быть синтезировано в соответствии со способом получения, который описан далее, и соединение (III) может быть синтезировано в соответствии с описанным выше способом получения.
[0259]
В качестве основания для использования в реакции конденсации, может быть указан, например, ароматический амин, такой как пиридин или лутидин, или третичный амин, такой как триэтиламин, триизопропиламин, трибутиламин, циклогексилдиметиламин, 4-диметиламинопиридин, N,N-диметиланилин, N-метилпиперидин, N-метилпирролидин, N-метилморфолин или диизопропилэтиламин (DIEA).
[0260]
Количество основания для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIc-a) и, более предпочтительно, от 0,8 до 5,0 молей.
[0261]
В качестве конденсирующего агента для использования в реакции конденсации, может быть указан, например, O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HBTU), циклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N'-этил карбодиимид (EDC) или его гидрохлорид, 2-этокси-1-этоксикарбонил-1,2-дигидроксихинолин (EEDQ), карбонилдиимидазол (CDI), диэтилфосфорил цианид, бензотриазол-1-илокситриспирролидинoфосфония гексафторфосфат (PyBOP), дифенилфосфорил азид (DPPA), 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метил морфолиния хлорид (DMTMM), изобутил хлорформиат, диэтилацетил хлорид или триметил ацетил хлорид. Эти конденсирующие агенты могут быть использованы отдельно или в комбинации с добавкой, такой как N-гидроксисукцинимид (HONSu), гидроксибензотриазол (HOBT), 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазин (HOOBT) или 4-диметиламинопиридин (DMAP).
[0262]
Количество конденсирующего агента для использования в реакции конденсации, предпочтительно, составляет от 0,5 до 10 молей относительно 1 моля соединения (IIc-a) и, более предпочтительно, от 0,8 до 5,0 молей.
[0263]
Количество соединения (III) для использования в реакции конденсации, предпочтительно, представляет собой от 0,5 до 3 молей относительно 1 моля соединения (IIc-a) и, более предпочтительно, от 0,8 до 1,5 молей.
[0264]
Реакцию конденсации обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, ароматический амин, такой как пиридин, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, амид, такой как N,N-диметилформамид или N-метилпирролидон, спирт, такой как метанол, этанол или 2-пропанол, или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь этих растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакция конденсации может быть проведена в отсутствие основания.
[0265]
В реакции конденсации температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0 до 100°C.
[0266]
В реакции конденсации время реакции, которое варьируется в зависимости от условий реакций, предпочтительно, составляет от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0267]
9-2. Стадии образования солей соединения (Ic):
Фармакологически приемлемая соль соединения (Ic) может быть получена, например, путем реакции образования соли, осуществляемой путем смешивания соединение (Ic) и кислоты.
[0268]
В качестве кислоты, которая может быть использована в реакции образования соли, можно указать, например, неорганическую кислоту, такую как хлористоводородная кислота, серная кислота, фосфорная кислота или бромистоводородная кислота, или органическую кислоту, такую как щавелевая кислота, малоновая кислота, лимонная кислота, фумаровая кислота, молочная кислота, яблочная кислота, янтарная кислота, винная кислота, уксусная кислота, трифторуксусная кислота, малеиновая кислота, глюконовая кислота, бензойная кислота, салициловая кислота, ксинафоата, памоевая кислота, аскорбиновая кислота, адипиновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота или коричная кислота.
[0269]
Реакцию образования соли обычно осуществляют в растворителе, и, соответственно, в качестве растворителя может быть указан такой растворитель, как, например, алифатический спирт, такой как метанол, этанол или изопропанол, простой эфир, такой как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан или диметиловый эфир этиленгликоля, амид, такой как N,N-диметилформамид или N-метилпирролидон, сульфоксид, такой как диметил сульфоксид, алифатический нитрил, такой как ацетонитрил или пропионитрил, кетон, такой как ацетон или 2-бутанон, сложный эфир, такой как этилацетат, метил ацетат или н-бутил ацетат, или вода. Может быть использована смесь этих растворителей.
[0270]
10. Способ получения соединения (IIc-a):
[Формула 21]
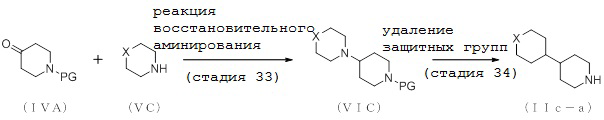
где отдельные условные символы имеют те же значения, которые определены выше.
(Стадия 33)
Соединение (VIC) может быть получено путем реакции восстановительного аминирования между соединением (IVA) и соединением (VC).
[0271]
В качестве соединения (VC), которое может быть использовано в реакции восстановительного аминирования, может быть непосредственно использовано коммерчески доступное соединение.
[0272]
Реакция восстановительного аминирования может быть осуществлена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или подобным способом.
[0273]
(Стадия 34)
Соединение (IIc-a) может быть получено путем удаления защитных групп у соединения (VIC).
[0274]
Удаление защитной группы, которое варьируется в зависимости от вида защитной группы, может быть осуществлено в соответствии с известным способом (например, Greene, T. W., «Greene’s Protective Groups in Organic Synthesis», Wiley-Interscience) или подобным способом.
[0275]
Обезболивающее действие циклического амина (I) или его пролекарства или его фармакологически приемлемой соли, в частности, терапевтическое действие на невропатическую боль и синдром фибромиалгии могут быть оценены с использованием соответствующей модели животного. В качестве подходящей животной модели при невропатической боли может быть указаны, например, модель частичного лигирования седалищного нерва на мышах или крысах (Malmberg et al., Pain, vol. 76, p. 215-222, 1998) или модель лигирования спинального нерва на мышах или крысах (Kim et al., Pain, vol. 50, p. 355-363, 1992). В качестве подходящей животной модели при синдроме фибромиалгии могут быть указаны, например, модели синдром фибромиалгии на крысах (Sluka et al., Journal of Pharmacology and Experimental Therapeutics, vol. 302, p. 1146-50, 2002; Nagakura et al., Pain, vol. 146, p. 26-33, 2009; Sluka et al., Pain, vol. 146, p. 3-4, 2009).
[0276]
Производное циклического амина (I) или его пролекарство, или его фармакологически приемлемая соль, поскольку оно обладает превосходным анальгезирующим действием, в частности, терапевтическим эффектом на невропатическую боль или синдром фибромиалгии, может быть использовано в качестве лекарственного средства, предпочтительно, использовано в качестве анальгезирующего средства, и, особенно предпочтительно, в качестве терапевтического средства для лечения невропатической боли или синдрома фибромиалгии. Следует отметить, что пролекарство производного циклического амина (I) оказывает превосходное анальгетическое действие, когда оно in vivo преобразуется в производное циклического амина (I); однако, пролекарство производного циклического амина (I) само по себе может обладать анальгезирующим действием.
[0277]
В качестве невропатической боли в данном описании могут быть указаны, например, боли при раке, боли при опоясывающем лишае, постгерпетической невралгии, невралгии, связанной со СПИДом невралгии, диабетическая невропатическая боль или невралгия тройничного нерва.
[0278]
«Синдром фибромиалгии» является симптомом, диагностированным врачом-специалистом как синдром фибромиалгии. Диагноз врача-специалиста, как правило, делается со ссылкой на стандарт классификации Американского колледжа ревматологии.
[0279]
Производное циклического амина (I) или его пролекарство или его фармакологически приемлемая соль могут быть использованы для лечения острой и хронической боли. Острая боль обычно длится в течение короткого периода времени, и, например, могут быть упомянуты послеоперационная боль, боль после экстракции или невралгия тройничного нерва. Хроническая боль определяется как боль обычно длящаяся в течение от 3-х до 6-ти месяцев и включает в себя невропатическую боль и психогенную боль организма, и могут быть упомянуты, например, хронический ревматоидный артрит, остеоартрит или постгерпетической невралгии.
[0280]
Лекарственное средство, содержащее производное циклического амина (I) или его пролекарство или фармакологически приемлемую соль в качестве активного ингредиента, оказывает превосходное анальгетическое действие, в частности, терапевтический эффект на невропатическую боль или синдром фибромиалгии при введении в организм млекопитающего (например, мыши, крысы, хомяка, кролика, кошки, собаки, коровы, овцы, обезьяны или человека), особенно, человека.
[0281]
Когда производное циклического амина (I) или его пролекарство или его фармакологически приемлемая соль используется в качестве лекарственного средства, производное циклического амина (I) или его пролекарство или его фармакологически приемлемая соль непосредственно или в комбинации с фармацевтически приемлемым носителем может быть введено перорально или парентерально. Когда в качестве лекарственного средства используется пролекарство или его фармакологически приемлемая соль производного циклического амина (I), предпочтительным является пероральное введение.
[0282]
Когда лекарственное средство, содержащее в качестве активного ингредиента производное циклического амина (I) или его пролекарство или его фармакологически приемлемую соль, вводят перорально, в качестве лекарственной формы могут быть упомянуты, например, таблетки (включая таблетки с сахарным покрытием и таблетки с пленочным покрытием), пилюли, гранулы, порошки, капсулы (включая мягкие капсулы и микрокапсулы), сиропы, эмульсии или суспензии. Когда лекарственное средство, содержащее в качестве активного ингредиента производное циклического амина (I) или его пролекарство или его фармакологически приемлемую соль, вводят парентерально, в качестве лекарственной формы могут быть упомянуты, например, инъекции, инфузии, капли, суппозитории, внутрикожные мази или пластыри. Является также эффективным получение препарата с замедленным высвобождением с использованием в комбинации подходящего основания (например, полимера бутановой кислоты, полимера гликолевой кислоты, сополимера бутановой кислоты-гликолевой кислоты, смеси полимера бутановой кислоты и полимера гликолевой кислоты или полиглицериновый эфир жирных кислот).
[0283]
Композиции, имеющие вышеуказанные лекарственные формы, могут быть получены в соответствии со способами получения, известными в области лекарственных средств. В этом случае, при необходимости, получение может быть осуществлено путем добавления вспомогательного вещества, связующего вещества, смазывающего вещества, разрыхляющего агента, подсластителя, поверхностно-активное вещество, суспендирующий агент или эмульгирующий агент, которые обычно используются в области получения лекарственных препаратов.
[0284]
Таблетки могут быть получены, например, путем добавления вспомогательного вещества, связующего, разрыхлителя или смазывающего вещества. Пилюли и гранулы могут быть получены путем добавления, например, вспомогательного вещества, связующего или разрыхлителя. Порошки и капсулы могут быть получены путем добавления, например, вспомогательного вещества. Сиропы могут быть получены путем добавления, например, подсластителя. Эмульсии или суспензии могут быть получены путем добавления, например, поверхностно-активного вещества, суспендирующего агента или эмульгатора.
[0285]
В качестве вспомогательного вещества могут быть указаны, например, лактоза, глюкоза, крахмал, сахароза, микрокристаллическая целлюлоза, порошок солодки, маннит, гидрокарбонат натрия, фосфат кальция или сульфат кальция.
[0286]
В качестве связующего вещества могут быть указаны, например, раствор крахмальной пасты, раствор гуммиарабика, раствор желатина, раствор трагаканта, раствор карбоксиметилцеллюлозы, раствор альгината натрия или глицерина.
[0287]
В качестве разрыхлителя может быть указан, например, крахмал или карбонат кальция.
[0288]
В качестве смазывающего вещества может быть указан, например, стеарат магния, стеариновая кислота, стеарат кальция или очищенный тальк.
[0289]
В качестве подсластителя могут быть указаны, например, глюкоза, фруктоза, инвертный сахар, сорбит, ксилит, глицерин или простой сироп.
[0290]
В качестве поверхностно-активного вещества могут быть указаны, например, лаурилсульфат натрия, полисорбат 80, моноэфир сорбитана и жирной кислоты или полиоксил 40 стеариновой кислоты.
[0291]
В качестве суспендирующего агента могут быть указаны, например, гуммиарабик, альгинат натрия, натрий карбоксиметилцеллюлоза, метил целлюлоза или бентонит.
[0292]
В качестве эмульгатора могут быть указаны, например, гуммиарабик, трагакант, желатин или полисорбат 80.
[0293]
Когда лекарственное средство, содержащее производное циклического амина (I) или его пролекарство или его фармакологически приемлемая соль в качестве активного ингредиента получают в виде вышеупомянутых лекарственных форм, могут быть добавлены окрашивающий агент, консервант, ароматизатор, вкусовой агент, стабилизатор или загуститель, которые обычно используются в области получения лекарственных препаратов.
[0294]
Суточная доза лекарственного средства, содержащего качестве активного ингредиента производное циклического амина (I) или его пролекарство или его фармакологически приемлемую соль, меняется в зависимости, например, от состояния или массы тела пациента или вида или пути введения соединения. Например, в случае перорального введения взрослому (масса: около 60 кг), количество производного циклического амина (I) или его пролекарства или его фармакологически приемлемой соли, представленного в качестве активного ингредиента, находится в пределах диапазона от 1 до 1000 мг, и введение, предпочтительно, осуществляют от 1 до 3 разделенными дозами. В случае парентерального введения взрослому (масса: около 60 кг) с помощью инъекционного раствора, количество производного циклического амина (I) или его пролекарства или его фармакологически приемлемой соли, представленного в качестве активного ингредиента, например, в инъекции, попадает в диапазон от 0,01 до 100 мг на массу тела (1 кг). Раствор для инъекций предпочтительно вводят внутривенно.
[0295]
Производное циклического амина (I) или его пролекарство или его фармакологически приемлемая соль могут быть использованы в комбинации с другими лекарственными средствами при соответствующем соотношении смешивания для того, чтобы дополнить или усилить терапевтический или профилактический эффект или уменьшить дозу. В этом случае, в качестве других лекарственных средств можно указать, например, антидепрессант, такой как амитриптилин, милнаципран или дулоксетин, анксиолитики, такие как алпразолам, противосудорожное средство, такое как карбамазепин, местный анестетик, такой как лидокаин, симпатический агонист, такой как адреналин, антагонист рецептора NMDA, такой как кетамин, ингибитор трансаминазы GABA, такой как вальпроат натрия, блокатор кальциевых каналов, такой как прегабалин, антагонист рецептора серотонина, такой как рисперидон, усилитель функции рецептора GABA, такой как диазепам, или противовоспалительное лекарственное средство, такое как диклофенак.
Примеры
[0296]
Настоящее изобретение описано подробно далее с отсылкой к примерам и ссылочным примерам; однако, настоящее изобретение ими не ограничивается.
[0297]
В следующем описании названия растворителей, указанным в данных ЯМР, представляют растворители, используемые при измерении. Спектр ЯМР 400 МГц получали с использованием спектрометра ядерного магнитного резонанса (ЯМР) серии JNM-AL 400 (JEOL, Ltd.). Химические сдвиги выражены в d (единица: м.д.) с использование тетраметилсилана в качестве стандарта, и соответствующие сигналы, соответственно, имеют следующие значения: с (синглет), д (дублет), т (триплет), кв (квартет), квин (квинтет), септ (септет), м (мультиплет), ушир (уширенный), дд (дублет дублетов), дт (дублет триплетов), ддд (дублет дублетов дублетов), дкв (дублет квартетов), тд (триплет дублетов) и тт (триплет триплетов). Спектр ESI-MS регистрировали с использованием Agilent Technologies 1200 Series, G6130A (от фирмы Agilent Technology). В качестве всех растворителей использовались коммерчески доступные продукты. Для флеш-хроматографии использовался YFLC W-prep2XY (от фирмы YAMAZEN).
[0298]
Исходные и промежуточные продукты для производных циклического амина (I) и их пролекарства были синтезированы способами, описанными в следующих ссылочных примерах. Следует отметить, что для соединений, используемых в синтезе соединений по ссылочным примерам были использованы коммерчески доступные продукты, методы синтеза которых не описаны далее.
[0299]
(Ссылочный пример 1) Синтез бензил 4-((2-((трет-бутоксикарбонил)амино)этил)(метил)амино)пиперидин-1-карбоксилата:
[Формула 22]
Гидрохлорид трет-бутил (2-(метиламино)этил)карбамата (0,564 г, 2,68 ммоль) и триацетоксиборгидрида натрия (0,681 г, 3,22 ммоль) добавляли к раствору бензил 4-оксопиперидин-1-карбоксилата (0,500 г, 2,14 ммоль) в дихлорметане (3,0 мл) при температуре 0°C, и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до температуры 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, гексан/этилацетат) с получением бензил 4-((2-((трет-бутоксикарбонил)амино)этил)(метил)амино)пиперидин-1-карбоксилата (0,590 г, 1,51 ммоль, 70%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,38-1,46 (11H, м), 1,67-1,76 (2H, м), 2,22 (3H, с), 2,47-2,55 (3H, м), 2,64-2,82 (2H, м), 3,12-3,21 (2H, м), 4,16-4,32 (2H, м), 4,90-5,00 (1H, м), 5,12 (2H, с), 7,30-7,37 (5H, м).
ESI-MS: m/z= 392 (M+H)+.
[0300]
(Ссылочный пример 2) Синтез сырого трет-бутил (2-(метил(пиперидин-4-ил)амино)этил)карбамата:
[Формула 23]
К раствору бензил 4-((2-((трет-бутоксикарбонил)амино)этил)(метил)амино)пиперидин-1-карбоксилата (0,300 г, 0,766 ммоль) в метаноле (4,0 мл) при комнатной температуре добавляли палладий/углерод (10%-ной влажности, 0,0815 г, 0,0766 ммоль), и полученную смесь перемешивали в атмосфере водорода в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением сырого продукта трет-бутил (2-(метил(пиперидин-4-ил)амино)этил)карбамата.
[0301]
(Ссылочный пример 3) Синтез трет-бутил 4-(N-бензил-N-метиламино)пиперидин-1-карбоксилата:
[Формула 24]
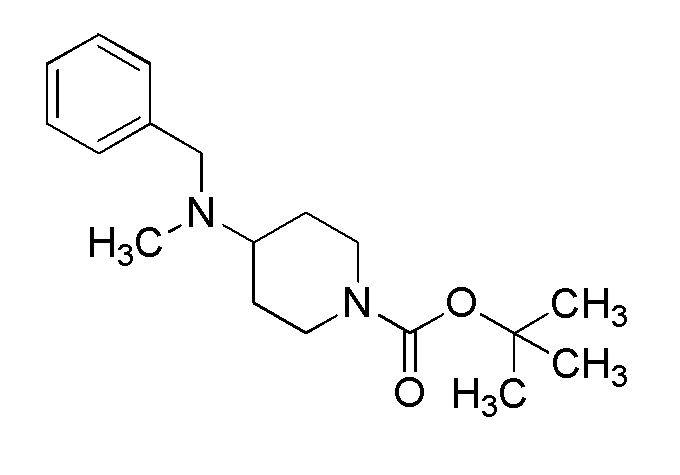
N-Бензил-N-метиламин (2,43 мл, 18,8 ммоль), уксусную кислоту (0,0860 мл, 1,51 ммоль) и триацетоксиборгидрид натрия (1,20 г, 5,66 ммоль) добавляли к раствору трет-бутил 4-оксопиперидин-1-карбоксилата (3,00 г, 15,1 ммоль) в дихлорметане (20,0 мл) при температуре 0°C. Реакционную жидкость перемешивали при той же температуре в течение 30 минут, и затем при температуре 0°C добавляли триацетоксиборгидрид натрия (1,20 г, 5,66 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 30 минут, и затем при температуре 0°C добавляли триацетоксиборгидрид натрия (2,40 г, 11,3 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до температуры 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, гексан/этилацетат) с получением трет-бутил 4-(N-бензил-N-метиламино)пиперидин-1-карбоксилата (4,49 г, 14,7 ммоль, 98%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46(9H, с), 1,48-1,58(2H, м), 1,76-1,84(2H, м), 2,19(3H, с), 2,54-2,75(3H, м), 3,57(2H, с), 4,05-4,25(2H, м), 7,21-7,32(5H, м).
ESI-MS: m/z= 305 (M+H)+.
[0302]
(Ссылочный пример 4) Синтез N-бензил-N-метилпиперидин-4-амина:
[Формула 25]
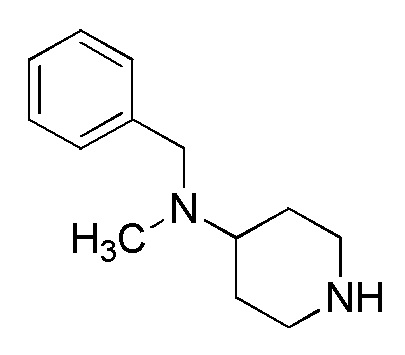
Раствор хлористого водорода в 1,4-диоксане (4,0 н, 3,28 мл, 13,1 ммоль) при комнатной температуре добавляли к перемешиваемому раствору трет-бутил 4-(N-бензил-N-метиламино)пиперидин-1-карбоксилата (1,00 г, 3,28 ммоль) в смеси 1,4-диоксан/метанол (1:1, 8,0 мл), и реакционную жидкость перемешивали при той же температуре в течение 6 часов. Реакционную жидкость концентрировали при пониженном давлении и добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, хлороформ/метанол) с получением N-бензил-N-метилпиперидин-4-амина (0,650 г, 0,318 ммоль, 97%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,44-1,56 (3H, м), 1,80-1,88 (2H, м), 2,21 (3H, с), 2,49-2,63 (3H, м), 3,12-3,19 (2H, м), 3,58 (2H, с), 7,22-7,32 (5H, м).
ESI-MS: m/z= 205 (M+H)+.
[0303]
(Ссылочный пример 5) Синтез 1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазола:
[Формула 26]
Диизопропилэтиламин (3,55 мл, 20,33 ммоль) и трет-бутилдиметилхлорсилан (2,81 г, 18,64 ммоль) добавляли к раствору 2-(1H-имидазол-1-ил)этанола (1,90 г, 16,94 ммоль) в дихлорметане (56,5 мл) при температуре 0°C, и температуру полученной смеси повышали до комнатной температуры, и полученную смесь перемешивали в течение 1 часа. В реакционную жидкость добавляли воду, и полученную смесь экстрагировали хлороформом. Органический слой промывали 10%-ным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, н-гексан/этилацетат) с получением 1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазола (3,62 г, 15,99 ммоль, 94%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: -0,03 (6H, с), 0,86 (9H, с), 3,84 (2H, т, J=5,1 Гц), 4,03 (2H, т, J=5,1 Гц), 6,95 (1H, с), 7,05 (1H, с), 7,51 (1H, с).
ESI-MS: m/z= 227 (M+H)+.
[0304]
(Ссылочный пример 6) Синтез 1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-карбальдегида:
[Формула 27]
Раствор н-бутиллития в н-гексане (1,62 M, 10,80 мл, 17,49 ммоль) добавляли по каплям к раствору 1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазола (3,60 г, 15,90 ммоль) в тетрагидрофуране (31,8 мл) при температуре -78°C, и полученную смесь перемешивали при той же температуре в течение 1 часа. При той же температуре в реакционную жидкость добавляли ДМФ (1,46 мл, 19,08 ммоль), и полученную смесь перемешивали в течение 1 часа, и затем температуру реакционной жидкости повышали до комнатной температуры. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония и этилацетат, и затем полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (силикагель, н-гексан/этилацетат) с получением 1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-карбальдегида (3,96 г, 15,67 ммоль, 98%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: -0,09 (6H, с), 0,83 (9H, с), 3,88 (2H, т, J=4,9 Гц), 4,51 (2H, т, J=4,9 Гц), 7,23 (1H, с), 7,27 (1H, с), 9,81 (1H, с).
ESI-MS: m/z= 255 (M+H)+.
[0305]
(Ссылочный пример 7) Синтез трет-бутил 2-(2-формил-1H-имидазол-1-ил)ацетата:
[Формула 28]
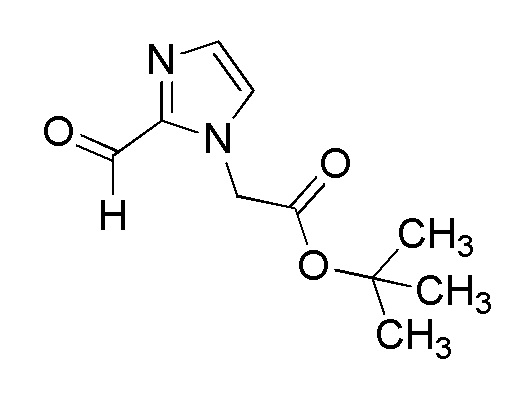
К раствору 1H-имидазол-2-карбальдегида (1,00 г, 10,41 ммоль) в N,N-диметилформамиде (52,0 мл) при комнатной температуре добавляли карбонат калия (1,73 г, 12,49 ммоль) и трет-бутил 2-бромацетат (1,83 мл, 12,49 ммоль), и полученную смесь перемешивали при той же температуре в течение 18 часов. В реакционную жидкость добавляли этилацетат и дистиллированную воду, и затем полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (силикагель, н-гексан/этилацетат) с получением трет-бутил 2-(2-формил-1H-имидазол-1-ил)ацетата (1,05 г, 0,733 ммоль, 48%) в виде масла желтого цвета
1H-ЯМР (400 МГц, CDCl3) δ: 1,48 (9H, с), 5,03 (2H, с), 7,13 (1H, с), 7,32 (1H, с), 9,80 (1H, с).
ESI-MS: m/z= 211 (M+H)+.
[0306]
(Ссылочный пример 8) Синтез (E)-этил 3-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-ил)акрилата:
[Формула 29]
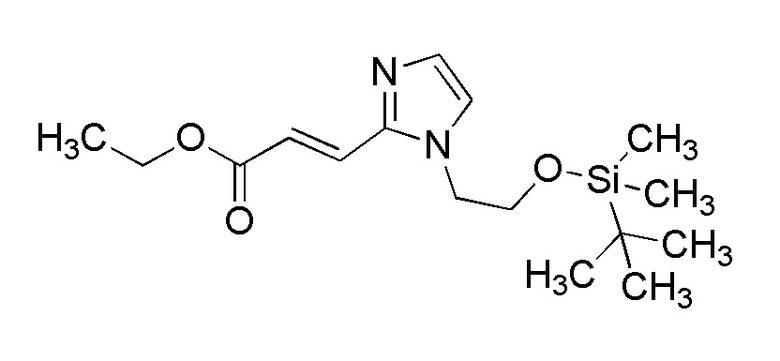
К суспензии гидрида натрия (0,702 г, 16,10 ммоль, 55%) в тетрагидрофуране (40,0 мл) при температуре 0°C добавляли этил диэтилфосфоноацетат (2,76 мл, 13,80 ммоль). Полученную смесь перемешивали при той же температуре в течение 1 часа, и затем к ней добавляли раствор 1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-карбальдегида (3,90 г, 15,33 ммоль) в тетрагидрофуране (36,7 мл), и температуру полученной смеси повышали до комнатной температуры, а затем реакционную жидкость перемешивали в течение 1 часа. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония, и полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (NH силикагель, н-гексан/этилацетат) с получением (E)-этил 3-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-ил)акрилата (3,42 г, 10,54 ммоль, 69%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: -0,08 (6H, с), 0,86 (9H, с), 1,32 (3H, т, J=7,1 Гц), 3,84 (2H, т, J=5,1 Гц), 4,15 (2H, т, J=5,1 Гц), 4,26 (3H, кв, J=7,1 Гц), 6,84 (1H, д, J=15,4 Гц), 7,04 (1H, с), 7,16 (1H, с), 7,52 (1H, д, J=15,4 Гц).
ESI-MS: m/z= 325 (M+H)+.
[0307]
(Ссылочный пример 9) Синтез (E)-бензил 3-(1-(2-(трет-бутокси)-2-оксоэтил)-1H-имидазол-2-ил)акрилата:
[Формула 30]
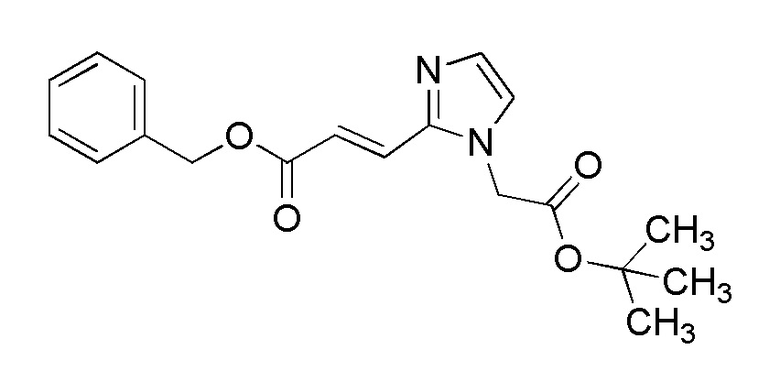
К суспензии гидрида натрия (0,125 г, 2,85 ммоль, 55%) в тетрагидрофуране (3,5 мл) при температуре 0°C добавляли раствор бензил диметилфосфоноацетата (0,700 г, 2,71 ммоль) в тетрагидрофуране (3,0 мл). Полученную смесь перемешивали при той же температуре в течение 30 минут, и затем добавляли раствор трет-бутил 2-(2-формил-1H-имидазол-1-ил)ацетата (0,600 г, 2,85 ммоль) в тетрагидрофуране (3,0 мл), температуру полученной смеси повышали до комнатной температуры, и затем реакционную жидкость перемешивали в течение 15 часов. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония, и затем полученную смесь экстрагировали хлороформом. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (NH силикагель, н-гексан/этилацетат) с получением (E)-бензил 3-(1-(2-(трет-бутокси)-2-оксоэтил)-1H-имидазол-2-ил)акрилата (0,82 г, 2,39 ммоль, 82%) в виде масла желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,45 (9H, с), 4,67 (2H, с), 5,25 (2H, с), 6,90 (1H, д, J=15,4 Гц), 7,01 (1H, с), 7,20 (1H, с), 7,31-7,44 (6H, с).
ESI-MS: m/z= 343 (M+H)+.
[0308]
(Ссылочный пример 10) Синтез сырой 3-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-ил)пропановой кислоты:
[Формула 31]
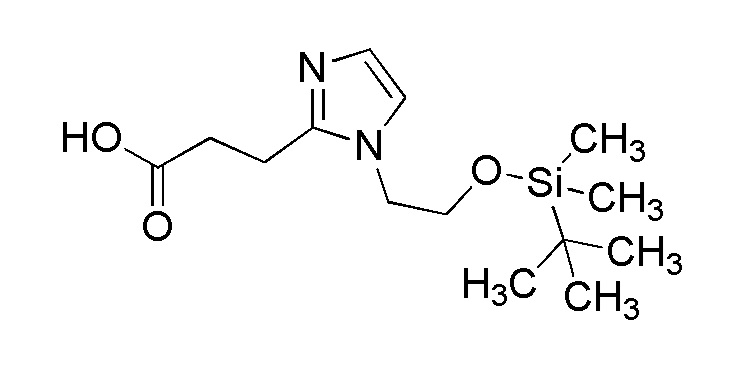
К раствору (E)-этил 3-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-ил)акрилата (3,42 г, 10,54 ммоль) в метаноле (42,2 мл) при комнатной температуре добавляли палладий-углерод (10%-ной влажности, 342 мг), и полученную смесь перемешивали в атмосфере водорода в течение 18 в течение. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. К полученному остатку при комнатной температуре добавляли метанол (21,0 мл), и полученный остаток растворялся. Реакционный раствор охлаждали до температуры 0°C, и затем в реакционную жидкость при той же температуре добавляли водный раствор гидроксида натрия (1,0 н, 11,59 мл, 11,59 ммоль), и температуру полученной смеси повышали до комнатной температуры, и реакционную жидкость перемешивали в течение 5 часов. При комнатной температуре в реакционную жидкость добавляли водный раствор гидроксида натрия (1,0 н, 5,80 мл, 5,80 ммоль), и полученную смесь перемешивали в течение 1 часа. Реакционную жидкость охлаждали до температуры 0°C, и затем в реакционную жидкость для нейтрализации добавляли соляную кислоту (1,0 н, 17,4 мл), и затем реакционную жидкость концентрировали при пониженном давлении. Остаток подвергали азеотропной отгонке с толуолом и добавляли этанол. Осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением сырого продукта 3-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-ил)пропановой кислоты (3,30 г) в виде бесцветного масла.
[0309]
(Ссылочный пример 11) Синтез сырой 3-(1-(2-(трет-бутокси)-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты:
[Формула 32]
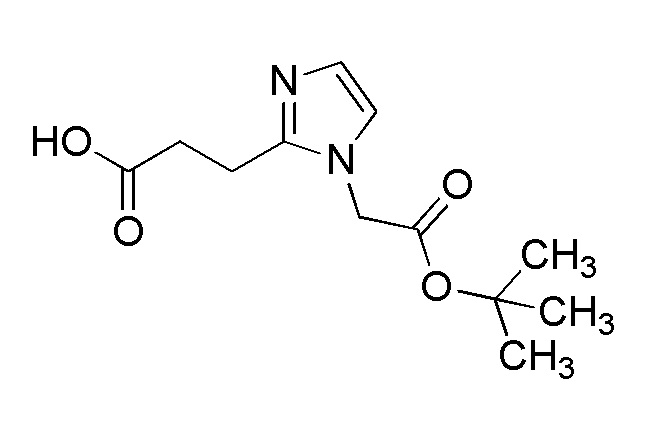
К раствору (E)-бензил 3-(1-(2-(трет-бутокси)-2-оксоэтил)-1H-имидазол-2-ил)акрилата (0,818 г, 2,39 ммоль) в метаноле (9,6 мл) при комнатной температуре добавляли палладий-углерод (10%-ной влажности, 81,8 мг), и полученную смесь перемешивали в атмосфере водорода в течение 16 часов. Реакционную жидкость фильтровали через целит, и затем фильтрат концентрировали при пониженном давлении с получением сырого продукта 3-(1-(2-(трет-бутокси)-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (0,590 г).
[0310]
(Ссылочный пример 12) Синтез 1-(4-бензил(метил)аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 33]
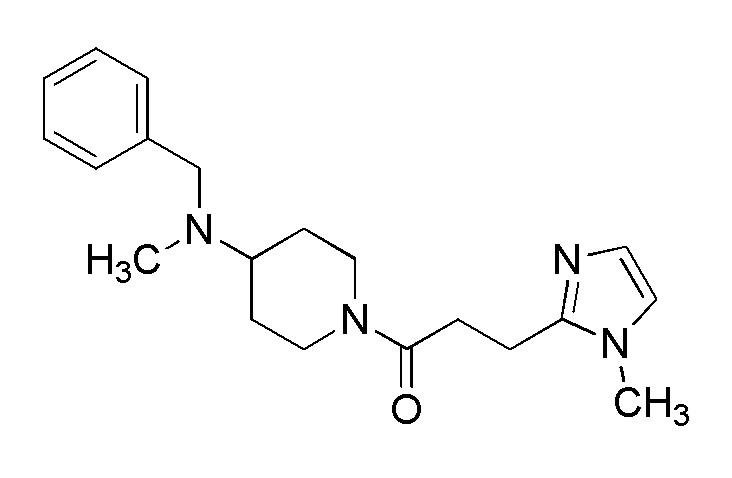
К раствору 3-(1-метил-1H-имидазол-2-ил)пропановой кислоты (0,300 г, 1,95 ммоль) в хлороформе (17,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,892 мл, 5,11 ммоль), HBTU (0,969 г, 2,55 ммоль) и N-бензил-N-метилпиперидин-4-амин (0,348 г, 1,70 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 60 часов. В реакционную жидкость добавляли метанол, и полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-бензил(метил)аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,204 г, 0,599 ммоль, 35%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,43-1,56 (2H, м), 1,80-1,88 (2H, м), 2,18 (3H, с), 2,51-2,70 (2H, м), 2,88-3,05 (5H, м), 3,56 (2H, с), 3,62 (3H, с), 4,00-4,07 (1H, м), 4,62-4,69 (1H, м), 6,79 (1H, д, J=1,2 Гц), 6,91 (1H, д, J=1,2 Гц), 7,22-7,34 (5H, м).
ESI-MS: m/z= 341 (M+H)+.
[0311]
(Ссылочный пример 13) Синтез 3-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)пропан-1-она:
[Формула 34]
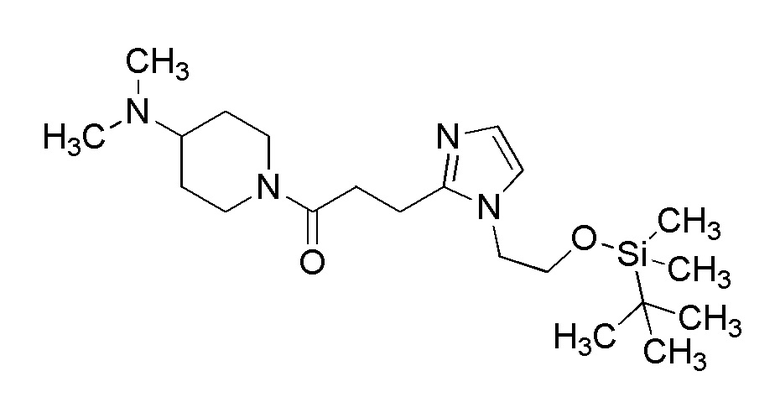
К раствору сырого продукта 3-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-ил)пропановой кислоты (3,15 г) в хлороформе (56,5 мл) при комнатной температуре добавляли диизопропилэтиламин (2,76 мл, 15,81 ммоль), HBTU (4,80 г, 12,65 ммоль) и 4-(диметиламино)пиперидин (1,12 мл, 10,01 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 12 часов. В реакционную жидкость добавляли насыщенный водный раствор карбоната калия и 10%-ный водный раствор хлорида натрия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и затем фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (NH силикагель, хлороформ/метанол) с получением 3-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)пропан-1-она (2,88 г, 7,05 ммоль, 70%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: -0,05 (6H, с), 0,84 (9H, с), 1,30-1,42 (2H, м), 1,82-1,85 (2H, м), 2,27-2,36 (7H, м), 2,55-2,63 (1H, м), 2,90-3,03 (5H, м), 3,82 (2H, т, J=5,4 Гц), 4,01-4,05 (3H, м), 4,60-4,63 (1H, м), 6,88 (1H, д, J=1,2 Гц), 6,92 (1H, д, J=1,2 Гц).
ESI-MS: m/z= 409 (M+H)+.
[0312]
(Ссылочный пример 14) Синтез трет-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата
[Формула 35]
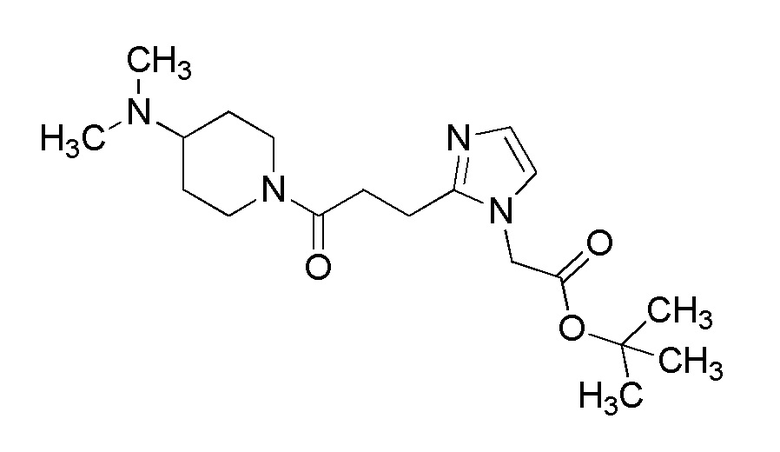
К раствору сырого продукта 3-(1-(2-(трет-бутокси)-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (0,380 г) в хлороформе (15,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,392 мл, 2,24 ммоль), HBTU (0,680 г, 1,79 ммоль) и 4-(диметиламино)пиперидин (0,167 мл, 1,42 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 12 часов. В реакционную жидкость добавляли насыщенный водный раствор карбоната калия и 10%-ный водный раствор хлорида натрия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и затем фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (NH силикагель, хлороформ/этилацетат) с получением трет-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,349 г, 0,957 ммоль, 62%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,29-1,42 (2H, м), 1,47 (9H, с), 1,81-1,83 (2H, м), 2,27-2,36 (7H, м), 2,55-2,62 (1H, м), 2,91 (4H, с), 2,96-3,03 (1H, м), 3,98-4,01 (1H, м), 4,57-4,60 (1H, м), 4,63 (2H, с), 6,81 (1H, д, J=1,2 Гц), 6,96 (1H, д, J=1,2 Гц).
[0313]
(Ссылочный пример 15) Синтез трет-бутил (2-(метил(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)амино)этил)карбамата:
[Формула 36]
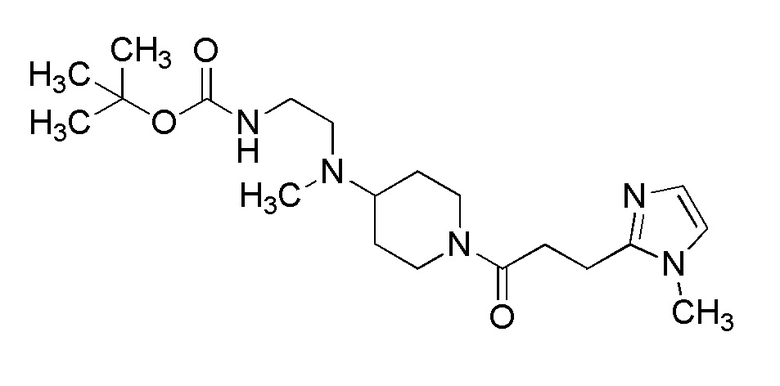
К раствору 3-(1-метил-1H-имидазол-2-ил)пропановой кислоты (0,118 г,0,765 ммоль) в хлороформе (8,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,401 мл, 2,30 ммоль), HBTU (0,348 г, 0,919 ммоль) и сырой трет-бутил (2-(метил(пиперидин-4-ил)амино)этил)карбамат (0,197 г, 0,765 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли метанол, и полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением трет-бутил (2-(метил(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)амино)этил)карбамата (0,260 г, 0,661 ммоль, 86%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,34-1,46 (11H, м), 1,71-1,80 (2H, м), 2,19-2,23 (3H, м), 2,47-2,60 (4H, м), 2,88-3,00 (5H, м), 3,12-3,20 (2H, м), 3,62 (3H, с), 4,00-4,08 (1H, м), 4,62-4,70 (1H, м), 4,91-4,98 (1H, м), 6,79 (1H, д, J=1,2 Гц), 6,91 (1H, д, J=1,2 Гц).
ESI-MS: m/z= 394 (M+H)+.
[0314]
(Ссылочный пример 16) Синтез 1-(4-((2-аминоэтил)(метил)амино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 37]
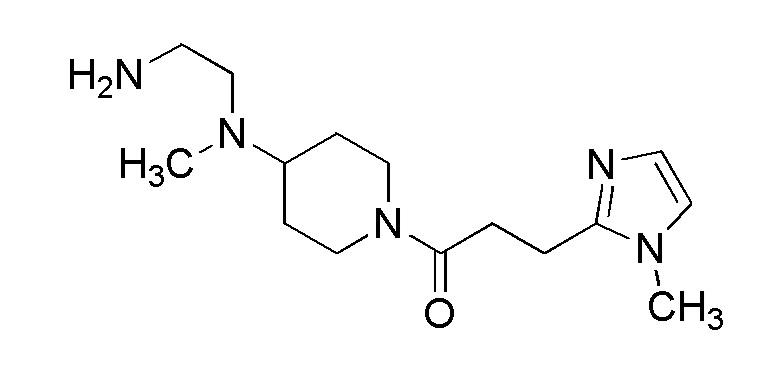
К раствору трет-бутил (2-(метил(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)амино)этил)карбамата (0,100 г, 0,254 ммоль) в 1,4-диоксане (3,0 мл) при комнатной температуре добавляли раствор хлористого водорода в 1,4-диоксане (4,0 н, 0,762 мл, 3,05 ммоль), и полученную смесь перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-((2-аминоэтил)(метил)амино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0498 г, 0,170 ммоль, 67%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,31-1,46 (2H, м), 1,64-1,85 (2H, м), 2,20 (3H, м), 2,43-2,60 (4H, м), 2,68-2,74 (2H, м), 2,86-3,00 (5H, м), 3,60 (3H, с), 3,96-4,06 (1H, м), 4,60-4,68 (1H, м), 6,77 (1H, ушир. с), 6,88 (1H, ушир. с).
ESI-MS: m/z= 294 (M+H)+.
[0315]
(Ссылочный пример 17) Синтез сырого 4-этилметиламинопиперидина:
[Формула 38]
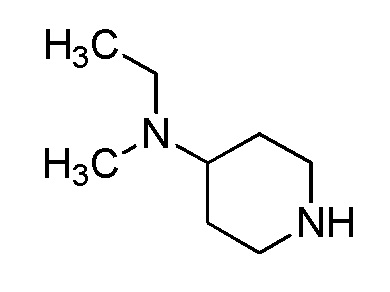
К раствору бензил 4-оксопиперидин-1-карбоксилата (0,500 г, 2,14 ммоль) в дихлорметане (12,0 мл) при температуре 0°C добавляли этилметиламин (0,230 мл, 2,68 ммоль), уксусную кислоту (0,0120 мл, 0,214 ммоль) и триацетоксиборгидрид натрия (0,681 г, 3,22 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до температуры 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, хлороформ/метанол). Полученный грубо очищенный продукт растворяли в метаноле (8,0 мл) и при комнатной температуре добавляли палладий/углерод (10%-ной влажности, 0,185 г, 0,174 ммоль), и полученную смесь перемешивали в атмосфере водорода в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением сырого продукта 4-этилметиламинопиперидина.
[0316]
(Ссылочный пример 18) Синтез сырого 4-диэтиламинопиперидина:
[Формула 39]
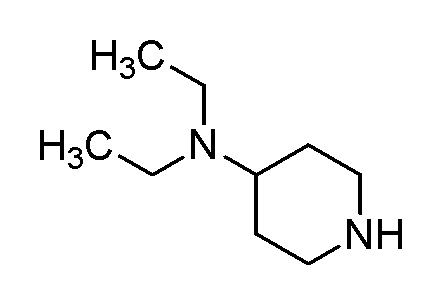
К раствору бензил 4-оксопиперидин-1-карбоксилата (0,500 г, 2,14 ммоль) в дихлорметане (12,0 мл) при температуре 0°C добавляли диэтиламин (0,276 мл, 2,68 ммоль), уксусную кислоту (0,0120 мл, 0,214 ммоль) и триацетоксиборгидрид натрия (0,681 г, 3,22 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до температуры 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, хлороформ/метанол). Полученный грубо очищенный продукт растворяли в метаноле (8,0 мл), и при комнатной температуре добавляли палладий/углерод (10%-ной влажности, 0,180 г, 0,169 ммоль), и полученную смесь перемешивали в атмосфере водорода в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением сырого продукта 4-диэтиламинопиперидина.
[0317]
(Ссылочный пример 19) Синтез 4-(пиперидин-1-ил)пиперидина:
[Формула 40]
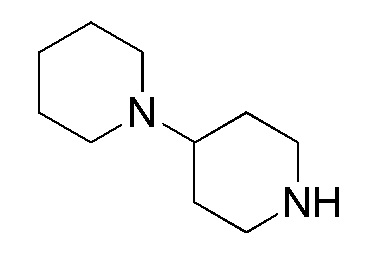
К раствору 1-трет-бутоксикарбонил-4-пиперидинона (3,02 г, 15,2 ммоль) в дихлорметане (25,0 мл) при температуре 0°C добавляли пиперидин (1,549 г, 18,19 ммоль), триацетоксиборгидрид натрия (3,85 г, 19,2 ммоль) и уксусную кислоту (0,0910 г, 1,52 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до температуры 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в соляной кислоте (1,0 н), и полученную смесь экстрагировали этилацетатом. В водный слой для подщелачивания добавляли 48%-ный водный раствор гидроксида натрия, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в метаноле (25,0 мл), и добавляли концентрированную соляную кислоту (5,0 мл), и затем полученную смесь перемешивали при температуре 40°C в течение 12 часов. Реакционную жидкость концентрировали и высушивали, и затем остаток растворяли в дистиллированной воде. Добавляли 48%-ный водный раствор гидроксида натрия для подщелачивания, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. 4-(Пиперидин-1-ил)пиперидин (2,04 г, 12,1 ммоль, 80%) получали в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,35-1,50 (4H, м), 1,53-1,67 (4H, м), 1,82 (2H, д, J=12,4 Гц), 2,34 (1H, тт, J=11,2, 4,0 Гц), 2,45-2,65(6H, м), 3,13 (2H, д, J=12,4 Гц).
ESI-MS: m/z= 169 (M+H)+.
[0318]
(Ссылочный пример 20) Синтез 4-(морфолин-4-ил)пиперидина:
[Формула 41]
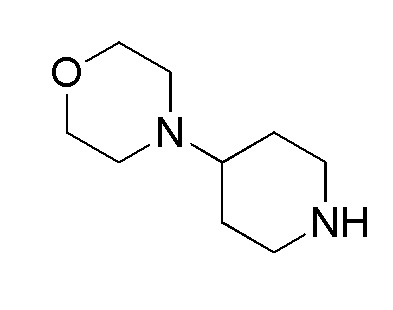
К раствору 1-трет-бутоксикарбонил-4-пиперидинона (1,51 г, 7,58 ммоль) в дихлорметане (25,0 мл) при температуре 0°C добавляли морфолин (0,792 г, 9,09 ммоль), триацетоксиборгидрид натрия (1,93 г, 9,09 ммоль) и уксусную кислоту (0,0460 г, 0,758 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до температуры 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в соляной кислоте (1,0 н), и полученную смесь экстрагировали этилацетатом. В водный слой для подщелачивания добавляли 48%-ный водный раствор гидроксида натрия, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в метаноле (25,0 мл) и добавляли концентрированную соляную кислоту (5,0 мл), и затем полученную смесь перемешивали при температуре 40°C в течение 12 часов. Реакционную жидкость концентрировали и высушивали, и затем остаток растворяли в дистиллированной воде. Добавляли 48%-ный водный раствор гидроксида натрия для подщелачивания, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. 4-(Морфолин-4-ил)пиперидин (1,52 г, 5,63 ммоль, 74%) получали в виде твердого вещества желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,34 (2H, дд, J=12,0, 4,0 Гц), 1,40 (2H, дд, J=12,0, 4,0 Гц), 1,85 (2H, д, J=12,4 Гц), 2,28 (1H, тт, J=11,2, 4,0 Гц), 3,53-3,63 (6H, м), 3,15 (2H, д, J=12,4 Гц), 3,73 (4H, т, J=4,4 Гц).
ESI-MS: m/z= 171 (M+H)+.
[0319]
(Ссылочный пример 21) Синтез 4-(1-метилпиперазин-4-ил)пиперидина:
[Формула 42]
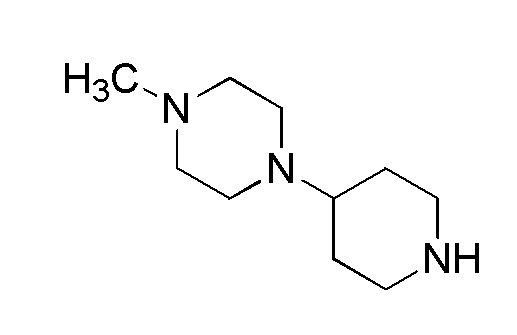
К раствору 1-трет-бутоксикарбонил-4-пиперидинона (1,50 г, 7,53 ммоль) в дихлорметане (25,0 мл) при температуре 0°C добавляли 1-метилпиперазин (0,905 г, 9,03 ммоль), триацетоксиборгидрид натрия (1,92 г, 9,03 ммоль) и уксусную кислоту (0,497 г, 8,28 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до температуры 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в соляной кислоте (1,0 н), и полученную смесь экстрагировали этилацетатом. В водный слой для подщелачивания добавляли 48%-ный водный раствор гидроксида натрия, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в метаноле (25,0 мл) и добавляли концентрированную соляную кислоту (5,0 мл), и затем полученную смесь перемешивали при температуре 40°C в течение 12 часов. Реакционную жидкость концентрировали и высушивали, и затем остаток растворяли в дистиллированной воде. Добавляли 48%-ный водный раствор гидроксида натрия для подщелачивания, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. 4-(1-Метилпиперазин-4-ил)пиперидин (0,826 г, 4,51 ммоль, 60%) получали в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,35 (2H, дд, J=12,0, 3,6 Гц), 1,41 (2H, дд, J=12,0, 3,6 Гц), 1,85 (2H, д, J=12,8 Гц), 1,96-2,06 (2H, ушир.), 2,28 (3H, с), 2,32 (1H, тт, J=11,6, 3,6 Гц), 3,37-3,70 (8H, м), 3,14 (2H, д, J=12,8 Гц).
ESI-MS: m/z= 169 (M+H)+.
[0320]
(Ссылочный пример 22) Синтез (R)-3-диметиламинопиперидина:
[Формула 43]
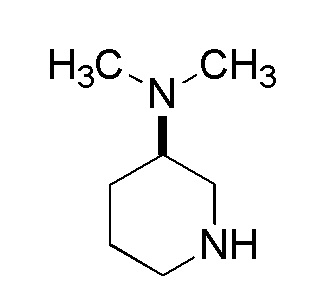
К раствору (R)-3-амино-1-трет-бутоксикарбонилпиперидина (1,00 г, 4,99 ммоль) в дихлорметане (10,0 мл) при температуре 0°C добавляли водный раствор формалина (36-38 масс %, 2,08 г, 25,0 ммоль), триацетоксиборгидрида натрия (2,12 г, 9,99 ммоль) и уксусной кислоты (0,0300 г, 0,500 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до температуры 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в соляной кислоте (1,0 н), и полученную смесь экстрагировали этилацетатом. В водный слой для подщелачивания добавляли 48%-ный водный раствор гидроксида натрия, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в метаноле (25,0 мл) и добавляли концентрированную соляную кислоту (5,0 мл), и затем полученную смесь перемешивали при температуре 40°C в течение 12 часов. Реакционную жидкость концентрировали и высушивали, и затем остаток растворяли в дистиллированной воде. Добавляли 48%-ный водный раствор гидроксида натрия для подщелачивания, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. (R)-3-Диметиламинопиперидин (0,384 г, 3,00 ммоль, 60%) получали в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,22-1,50 (2H, м), 1,73-1,78 (1H, м), 1,93-2,01 (1H, м), 2,15 (1H, тт, J=10,0, 3,6 Гц), 2,29 (6H, с), 2,45-2,53 (2H, м), 2,92-2,96 (1H, м), 3,15-3,22 (1H, м).
ESI-MS: m/z= 129 (M+H)+.
[0321]
(Ссылочный пример 23) Синтез (S)-3-диметиламинопиперидина:
[Формула 44]
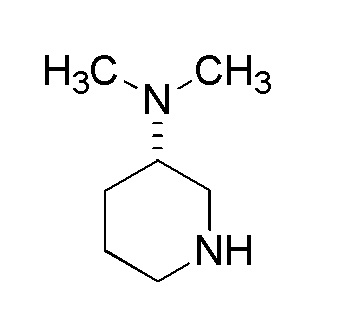
К раствору (S)-3-амино-1-трет-бутоксикарбонилпиперидина (1,00 г, 4,99 ммоль) в дихлорметане (10,0 мл) при температуре 0°C добавляли водный раствор формалина (36-38 масс %, 2,10 г, 25,2 ммоль), триацетоксиборгидрид натрия (2,13 г, 10,0 ммоль) и уксусную кислоту (0,0300 г, 0,500 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до температуры 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в соляной кислоте (1,0 н), и полученную смесь экстрагировали этилацетатом. В водный слой для подщелачивания добавляли 48%-ный водный раствор гидроксида натрия, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в метаноле (25,0 мл), и добавляли концентрированную соляную кислоту (5,0 мл), и затем полученную смесь перемешивали при температуре 40°C в течение 12 часов. Реакционную жидкость концентрировали и высушивали, и затем остаток растворяли в дистиллированной воде. Добавляли 48%-ный водный раствор гидроксида натрия для подщелачивания, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. (S)-3-Диметиламинопиперидин (0,351 г, 2,74 ммоль, 55%) получали в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,22-1,50 (2H, м), 1,73-1,78 (1H, м), 1,93-2,01 (1H, м), 2,15 (1H, тт, J=10,0, 3,6 Гц), 2,29 (6H, с), 2,45-2,53 (2H, м), 2,92-2,96 (1H, м), 3,15-3,22 (1H, м).
ESI-MS: m/z= 129 (M+H)+.
[0322]
(Ссылочный пример 24) Синтез сырого циклопропанола:
[Формула 45]
К циклопропилбороновой кислоте (0,300 г, 3,49 ммоль) при температуре 0°C добавляли водный раствор гидроксида натрия (10%, 1,29 мл, 3,49 ммоль) и водный раствор перекиси водорода (30%, 9,90 мл, 87,0 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. В реакционную жидкость добавляли насыщенный водный раствор тиосульфата натрия, и полученную смесь экстрагировали диэтиловым эфиром. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением сырого продукта циклопропанола.
[0323]
(Ссылочный пример 25) Синтез сырого 4-(гидроксиметил)-5-метил-1,3-диоксол-2-она:
[Формула 46]
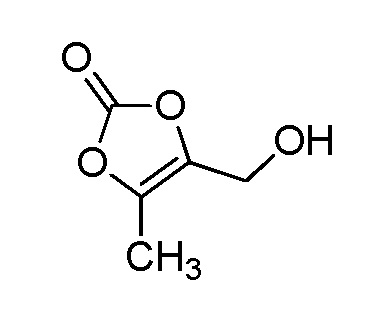
К раствору 4-(хлорметил)-5-метил-1,3-диоксол-2-она (1,00 г, 6,73 ммоль) в ацетонитриле (10,0 мл) при температуре 0°C добавляли муравьиную кислоту (0,750 мл, 19,6 ммоль) и триэтиламин (2,62 мл, 18,8 ммоль), температуру реакционной жидкости повышали до 65°C, и реакционную жидкость перемешивали в течение 3 часов. Осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении. К остатку добавляли дистиллированную воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. К полученному остатку при комнатной температуре добавляли метанол (10,0 мл), и полученный остаток растворялся. При комнатной температуре в реакционную жидкость добавляли дистиллированную воду (3,0 мл) и концентрированную соляную кислоту (0,120 мл, 3,95 ммоль), и полученную смесь перемешивали при той же температуре в течение 3 часов. В реакционную жидкость добавляли дистиллированную воду, и реакционную жидкость концентрировали при пониженном давлении. К остатку добавляли дистиллированную воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением сырого продукта 4-(гидроксиметил)-5-метил-1,3-диоксол-2-она.
[0324]
(Ссылочный пример 26) Синтез 2-гидрокси-N,N-диметилацетамида:
[Формула 47]
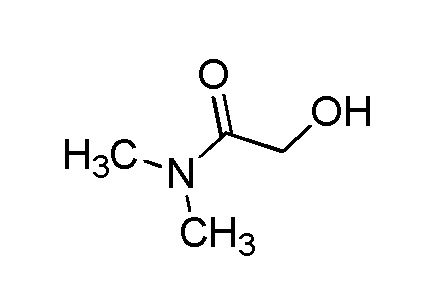
К раствору 2-(бензилокси)уксусной кислоты (1,00 г, 6,02 ммоль) в хлороформе (30,0 мл) при комнатной температуре добавляли диизопропилэтиламин (2,10 мл, 12,0 ммоль), HBTU (2,74 г, 7,22 ммоль) и раствор диметиламина в ТГФ (2,0 M, 3,61 мл, 7,22 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, хлороформ/метанол). Полученный остаток растворялся в метаноле (30,0 мл), и при комнатной температуре добавляли палладий-углерод (10%-ной влажности, 0,640 г, 0,602 ммоль), и полученную смесь перемешивали в атмосфере водорода при той же температуре в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, хлороформ/метанол) с получением 2-гидрокси-N,N-диметилацетамида (0,255 г, 2,47 ммоль, 41%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 2,88 (3H, с), 3,03 (3H, с), 4,14 (2H, ушир. с).
[0325]
(Ссылочный пример 27) Синтез этил 2-(2-формил-1H-имидазол-1-ил)ацетата:
[Формула 48]
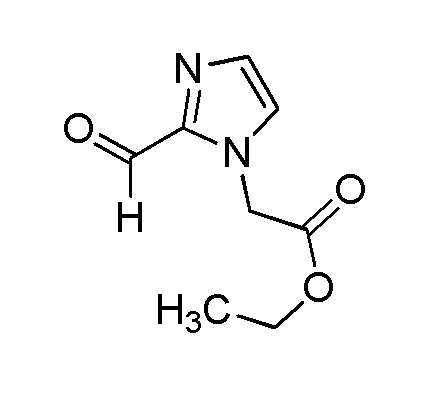
К раствору 1H-имидазол-2-карбальдегида (0,500 г, 5,20 ммоль) в N,N-диметилформамиде (10,0 мл) при комнатной температуре добавляли карбонат калия (1,44 г, 10,4 ммоль), этил хлорацетат (0,585 мл, 5,46 ммоль) и йодид калия (0,864 г, 5,20 ммоль), температуру реакционной жидкости повышали до 90°C, и реакционную жидкость перемешивали течение 4 часов. В реакционную жидкость добавляли дистиллированную воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, хлороформ/метанол) с получением этил 2-(2-формил-1H-имидазол-1-ил)ацетата (0,269 г, 1,48 ммоль, 28%) в виде масла желтого цвета
1H-ЯМР (400 МГц, CDCl3) δ: 1,29 (3H, т, J=7,2 Гц), 4,25 (2H, кв, J=7,2 Гц), 5,14 (2H, с), 7,15 (1H, ушир. с), 7,33 (1H, с), 9,79-9,91 (1H, м).
ESI-MS: m/z= 183 (M+H)+.
[0326]
(Ссылочный пример 28) Синтез (E)-бензил 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)акрилата:
[Формула 49]
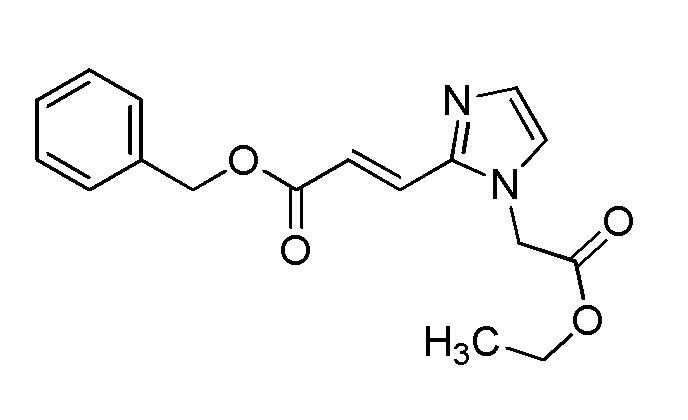
К суспензии гидрида натрия (55%, 0,958 г, 22,0 ммоль) в тетрагидрофуране (30,0 мл) при температуре 0°C добавляли бензил диметилфосфоноацетат (4,61 мл, 22,0 ммоль), и полученную смесь перемешивали при той же температуре в течение 1 часа. В реакционную жидкость добавляли этил 2-(2-формил-1H-имидазол-1-ил)ацетат (4,00 г, 22,0 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 3 часов. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония, и полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, н-гексан/этилацетат) с получением (E)-бензил 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)акрилата (4,31 г, 13,7 ммоль, 62%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,28 (3H, т, J=7,2 Гц), 4,24 (2H, кв, J=7,2 Гц), 4,77 (2H, с), 5,25 (2H, с), 6,92 (1H, д, J=15,6 Гц), 7,02 (1H, ушир. с), 7,21 (1H, ушир. с), 7,28-7,45 (6H, м).
ESI-MS: m/z= 315 (M+H)+.
[0327]
(Ссылочный пример 29) Синтез сырого 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты:
[Формула 50]
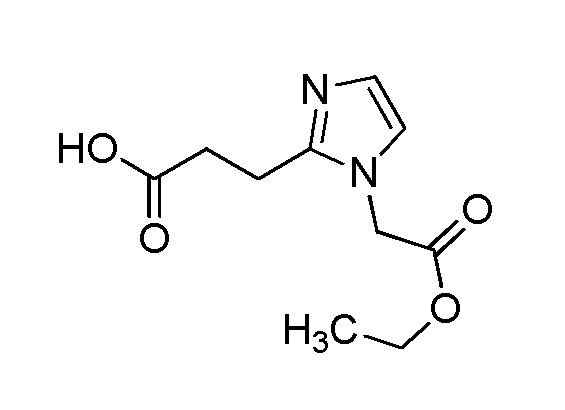
К раствору (E)-бензил 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)акрилата (4,31 г, 13,7 ммоль) в этаноле (80,0 мл) при комнатной температуре добавляли палладий-углерод (10%-ной влажности, 1,46 г, 1,37 ммоль), и реакционную жидкость перемешивали в атмосфере водорода при той же температуре в течение 24 часов. Температуру реакционной жидкости повышали до 40°C, и реакционную жидкость перемешивали в течение 1 часа. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением сырого продукта 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты.
[0328]
(Ссылочный пример 30) Синтез (E)-бензил 3-(1H-имидазол-2-ил)акрилата:
[Формула 51]
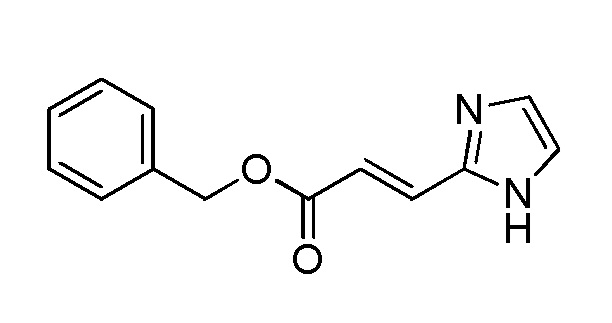
К суспензии гидрида натрия (55%, 1,12 г, 25,6 ммоль) в тетрагидрофуране (40,0 мл) при температуре 0°C добавляли бензил диметилфосфоноацетат (5,12 мл, 24,4 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. В реакционную жидкость добавляли при температуре 0°C 1H-имидазол-2-карбальдегид (2,46 г, 25,6 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 60 часов. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония, и полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, хлороформ/метанол) с получением (E)-бензил 3-(1H-имидазол-2-ил)акрилата (0,380 г, 1,66 ммоль, 7%) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 5,25 (2H, с), 6,62 (1H, д, J=15,6 Гц), 7,14-7,23(2H, м), 7,28-7,43 (5H, м), 7,57 (1H, д, J=16,0 Гц).
ESI-MS: m/z= 229 (M+H)+.
[0329]
(Ссылочный пример 31) Синтез (E)-бензил 3-(1-(3-этокси-3-оксопропил)-1H-имидазол-2-ил)акрилата:
[Формула 52]
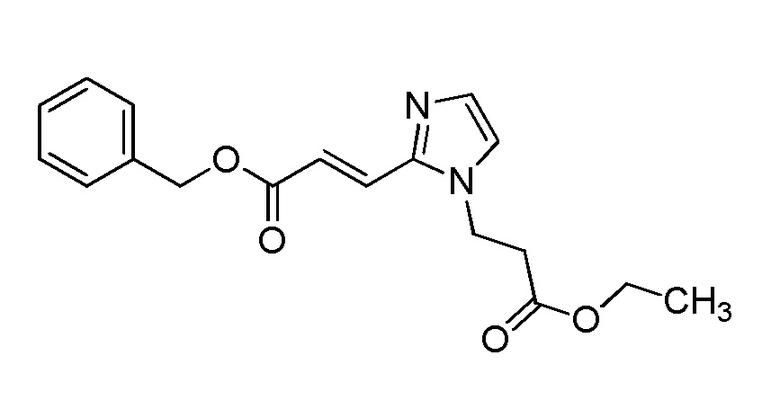
К раствору (E)-бензил 3-(1H-имидазол-2-ил)акрилата (0,500 г, 2,19 ммоль) в ДМФ (7,3 мл) при комнатной температуре добавляли карбонат калия (0,606 г, 4,38 ммоль), этил 3-бромпропаноат (0,419 мл, 3,29 ммоль) и йодид калия (0,364 г, 2,19 ммоль), температуру реакционной жидкости повышали до 90°C, и реакционную жидкость перемешивали в течение 4 часов. В реакционную жидкость добавляли дистиллированную воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали 10%-ным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (силикагель, н-гексан/этилацетат) с получением (E)-бензил 3-(1-(3-этокси-3-оксопропил)-1H-имидазол-2-ил)акрилата (0,520 г, 1,59 ммоль, 72%) в виде масла желтого цвета
1H-ЯМР (400 МГц, CDCl3) δ: 1,23 (3H, т, J=7,2 Гц), 2,76 (2H, т, J=7,2 Гц), 4,13 (2H, кв, J=7,2 Гц), 4,35 (2H, т, J=7,2 Гц), 5,26 (2H, с), 6,91 (1H, д, J=15,6 Гц), 7,06 (1H, ушир. с), 7,15 (1H, ушир. с), 7,30-7,42 (5H, м), 7,55 (1H, д, J=15,6 Гц).
ESI-MS: m/z= 329 (M+H)+.
[0330]
(Ссылочный пример 32) Синтез сырого 3-(1-(3-этокси-3-оксопропил)-1H-имидазол-2-ил)пропановой кислоты:
[Формула 53]
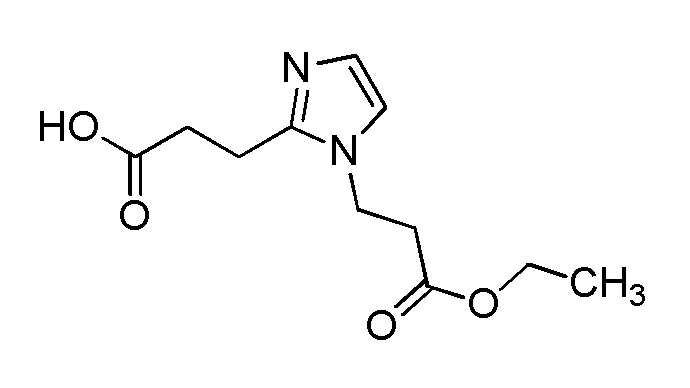
К раствору (E)-бензил 3-(1-(3-этокси-3-оксопропил)-1H-имидазол-2-ил)акрилата (0,520 г, 1,59 ммоль) в этаноле (9,0 мл) при комнатной температуре добавляли палладий-углерод (10%-ной влажности, 0,169 г, 0,159 ммоль), и реакционную жидкость перемешивали в атмосфере водорода при той же температуре в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением сырого продукта 3-(1-(3-этокси-3-оксопропил)-1H-имидазол-2-ил)пропановой кислоты.
[0331]
(Пример 1) Синтез 3-(1-метил-1H-имидазол-2-ил)-1-(4-(метиламино)пиперидин-1-ил)пропан-1-она:
[Формула 54]
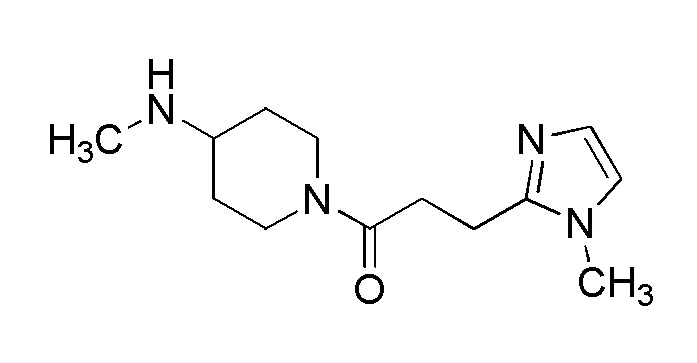
К раствору 1-(4-бензил(метил)аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,200 г, 0,587 ммоль) в этаноле (2,0 мл) при комнатной температуре добавляли гидроксид палладия на углероде (10%-ной влажности, 0,0820 г, 0,0587 ммоль), и полученную смесь перемешивали в атмосфере водорода в течение 3 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии на силикагеле (хлороформ/метанол) с получением 3-(1-метил-1H-имидазол-2-ил)-1-(4-(метиламино)пиперидин-1-ил)пропан-1-она (0,131 г, 0,523 ммоль, 89%) (здесь и далее указывается как «соединение по примеру 1») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,17-1,28 (2H, м), 1,85-1,94 (2H, м), 2,44 (3H, с), 2,54-2,62 (1H, м), 2,72-2,81 (1H, м), 2,88-3,00 (4H, м), 3,03-3,13 (1H, м), 3,62 (3H, с), 3,90-3,98 (1H, м), 4,41-4,49 (1H, м), 6,79 (1H, д, J=1,2 Гц), 6,91 (1H, д, J=1,2 Гц).
ESI-MS: m/z= 251 (M+H)+.
[0332]
(Пример 2) Синтез гидрохлорида 3-(1-метил-1H-имидазол-2-ил)-1-(4-(метиламино)пиперидин-1-ил)пропан-1-она:
[Формула 55]
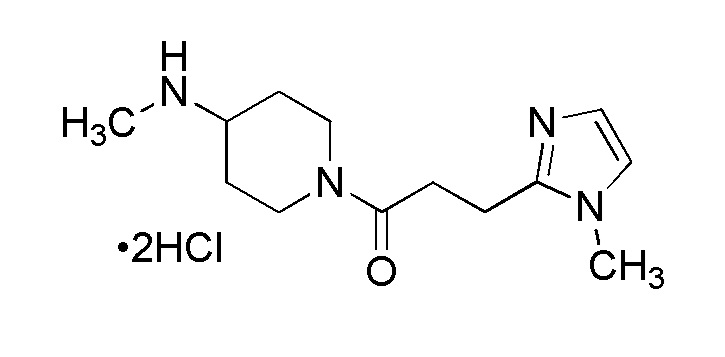
К раствору 3-(1-метил-1H-имидазол-2-ил)-1-(4-метиламинопиперидин-1-ил)пропан-1-она (0,131 г, 0,523 ммоль) в диэтиловом эфире (5,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,675 мл, 1,35 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (19,5 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида 3-(1-метил-1H-имидазол-2-ил)-1-(4-(метиламино)пиперидин-1-ил)пропан-1-она (0,0635 г, 0,196 ммоль, 38%) (здесь и далее указывается как «соединение по примеру 2») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,40-1,68 (2H, м), 2,13-2,26 (2H, м), 2,72-2,80 (4H, м), 3,01-3,08 (2H, м), 3,15-3,26 (3H, м), 3,33-3,43 (1H, м), 3,82 (3H, с), 4,01-4,13 (1H, м), 4,43-4,52 (1H, м), 7,28-7,34 (2H, м).
ESI-MS: в виде 3-(1-метил-1H-имидазол-2-ил)-1-(4-(метиламино)пиперидин-1-ил)пропан-1-она: m/z= 251 (M+H)+.
[0333]
(Пример 3) Синтез N-метил-N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида:
[Формула 56]
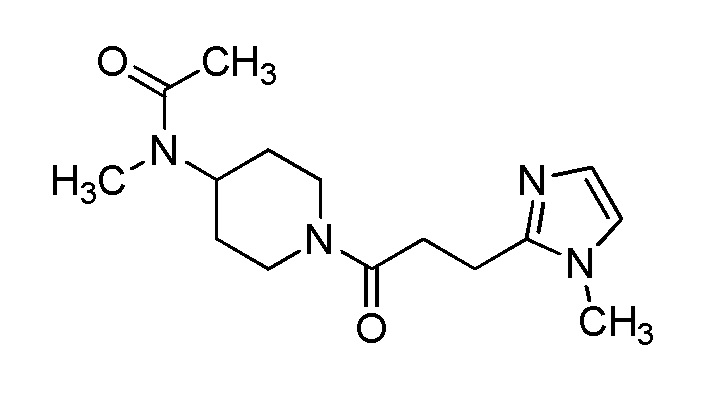
К раствору 3-(1-метил-1H-имидазол-2-ил)-1-(4-метиламинопиперидин-1-ил)пропан-1-она (0,0500 г, 0,200 ммоль) в дихлорметане (1,0 мл) при комнатной температуре добавляли триэтиламин (0,0842 мл, 0,599 ммоль) и уксусный ангидрид (0,0565 мл, 0,599 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали хлороформом. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением N-метил-N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида (0,0499 г, 0,171 ммоль, 85%) (здесь и далее указывается как «соединение по примеру 3») в виде бесцветного масла.
1H-ЯМР (400 МГц, CD3OD) δ: 1,43-1,72 (4H, м), 2,01-2,12 (3H, м), 2,52-2,93 (8H, м), 3,00-3,14 (1H, м), 3,59-3,61 (3H, м), 3,96-4,05 (1H, м), 4,47-4,60 (2H, м), 6,74-6,78 (1H, м), 6,87-6,90 (1H, м).
ESI-MS: m/z= 293 (M+H)+.
[0334]
(Пример 4) Синтез гидрохлорида N-метил-N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида:
[Формула 57]
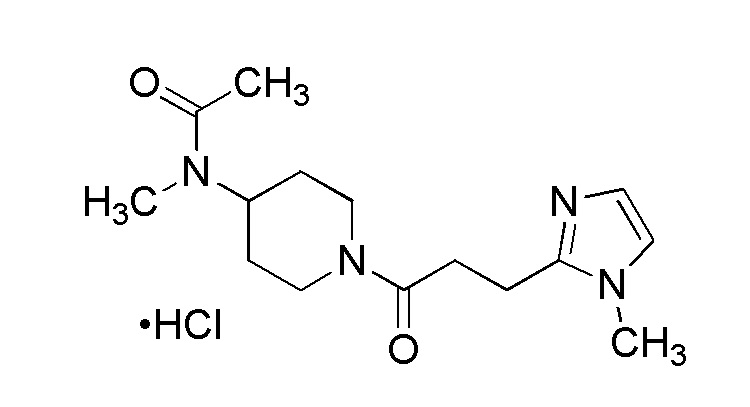
К раствору N-метил-N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида (0,0499 г, 0,171 ммоль) в диэтиловом эфире (2,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,111 мл, 0,222 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа, и затем реакционную жидкость перемешивали при комнатной температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (6,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида N-метил-N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида (0,0397 г, 0,121 ммоль, 71%) (здесь и далее указывается как «соединение по примеру 4») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,55-1,85 (4H, м), 2,08-2,19 (3H, м), 2,66-2,89 (4H, м), 2,98-3,05 (2H, м), 3,13-3,25 (3H, м), 3,80 (3H, с), 3,95-4,05 (1H, м), 4,38-4,53 (2H, м), 7,27-7,30 (2H, м).
ESI-MS: в виде N-метил-N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида: m/z= 293 (M+H)+.
[0335]
(Пример 5) Синтез N-(2-(метил(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)амино)этил)ацетамида:
[Формула 58]
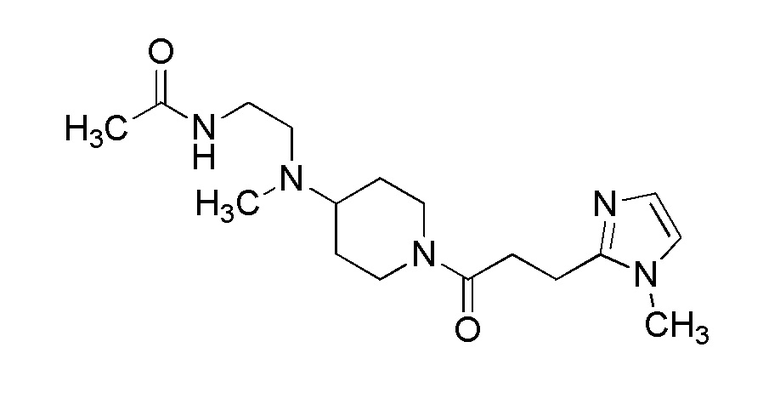
К раствору 1-(4-((2-аминоэтил)(метил)амино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0500 г, 0,170 ммоль) в дихлорметане (2,0 мл) при комнатной температуре добавляли пиридин (0,0410 мл, 0,511 ммоль) и уксусный ангидрид (0,0480 мл, 0,511 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением N-(2-(метил(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)амино)этил)ацетамида (0,0451 г, 0,134 ммоль, 79%) (здесь и далее указывается как «соединение по примеру 5») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,30-1,42(2H, м), 1,70-1,80(2H, м), 1,97 (3H, с), 2,21(3H, м), 2,46-2,62(4H, м), 2,87-3,01(5H, м), 3,25-3,32(2H, м), 3,61(3H, с), 4,00-4,08(1H, м), 4,63-4,72(1H, м), 5,97-6,05(1H, м), 6,78(1H, д, J=1,2 Гц), 6,90(1H, д, J=1,2 Гц).
ESI-MS: m/z= 336 (M+H)+.
[0336]
(Пример 6) Синтез гидрохлорида N-(2-(метил(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)амино)этил)ацетамида:
[Формула 59]
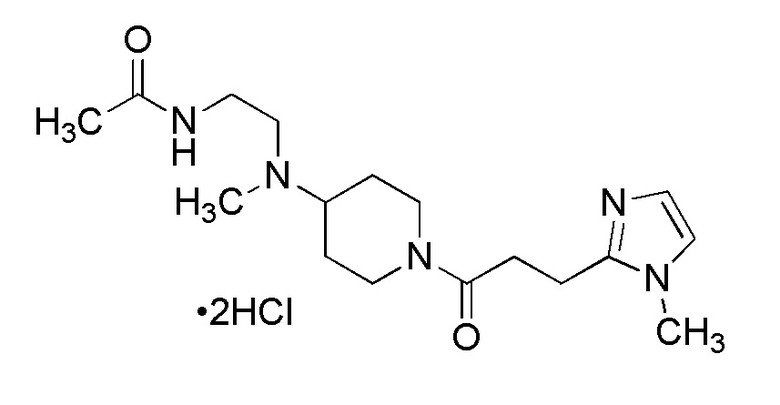
К раствору N-(2-(метил(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)амино)этил)ацетамида (0,0451 г, 0,134 ммоль) в диэтиловом эфире (2,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,174 мл, 0,349 ммоль), и полученную смесь перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (6,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида N-(2-(метил(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)амино)этил)ацетамида (0,0211 г, 0,0517 ммоль, 39%) (здесь и далее указывается как «соединение по примеру 6») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,60-1,85 (2H, м), 2,04 (3H, с), 2,07-2,20 (2H, м), 2,70-2,80 (1H, м), 2,88 (3H, с), 3,02-3,10 (2H, м), 3,18-3,30 (4H, м), 3,40-3,52 (1H, м), 3,60-3,75 (3H, м), 3,84 (3H, с), 4,10-4,18 (1H, м), 7,31-7,35 (2H, м), 7,52-7,60 (1H, м).
ESI-MS: в виде N-(2-(метил(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)амино)этил)ацетамида: m/z= 336 (M+H)+.
[0337]
(Пример 7) Синтез 1-(4-аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 60]
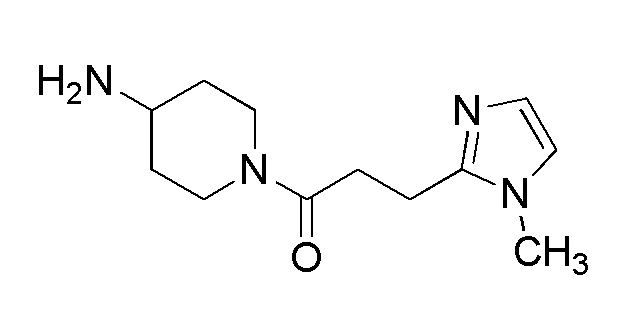
К раствору 3-(1-метил-1H-имидазол-2-ил)пропановой кислоты (0,160 г, 1,04 ммоль) в хлороформе (6,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,544 мл, 3,11 ммоль), HBTU (0,472 г, 1,25 ммоль) и трет-бутил пиперидин-4-илкарбамат (0,208 г, 1,04 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 60 часов. В реакционную жидкость добавляли метанол, и полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол). К полученному остатку при комнатной температуре добавляли 1,4-диоксан (11,0 мл), и полученный остаток растворялся. При комнатной температуре в реакционную жидкость добавляли раствор хлористого водорода в 1,4-диоксане (4,0 н, 3,11 мл, 12,5 ммоль), и полученную смесь перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли 1,0 н водный раствор гидроксида натрия, и полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,231 г, 0,978 ммоль, 94%) (здесь и далее указывается как «соединение по примеру 7») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,16-1,28 (2H, м), 1,79-1,89 (2H, м), 2,65-2,75 (1H, м), 2,85-3,10 (6H, м), 3,62 (3H, с), 3,90-3,98 (1H, м), 4,45-4,54 (1H, м), 6,79 (1H, д, J=1,2 Гц), 6,91 (1H, д, J=1,2 Гц).
ESI-MS: m/z= 237 (M+H)+.
[0338]
(Пример 8) Синтез гидрохлорида 1-(4-аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 61]
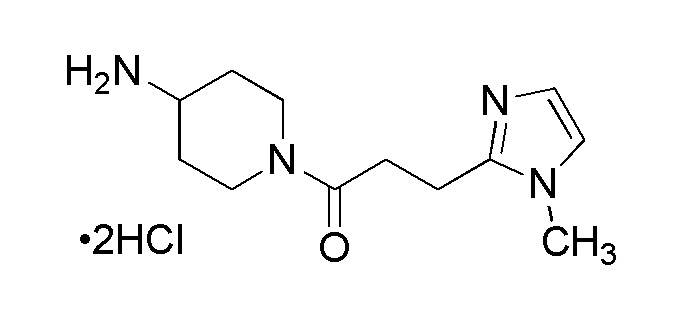
К раствору 1-(4-аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0430 г, 0,182 ммоль) в диэтиловом эфире (2,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,227 мл, 0,455 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 30 минут, и затем реакционную жидкость перемешивали при комнатной температуре в течение 1 часа. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром и сушили при комнатной температуре с получением гидрохлорида 1-(4-аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0420 г, 0,136 ммоль, 75%) (здесь и далее указывается как «соединение по примеру 8») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,46-1,69 (2H, м), 2,09-2,16 (2H, м), 2,76-2,83 (1H, м), 3,04-3,07 (2H, м), 3,20-3,25 (3H, м), 3,48-3,53 (1H, м), 3,84 (3H, с), 4,02-4,06 (1H, м), 4,43-4,46 (2H, м), 7,26 (2H, с).
ESI-MS: в виде 1-(4-аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она: m/z= 237 (M+H)+.
[0339]
(Пример 9) Синтез N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида:
[Формула 62]
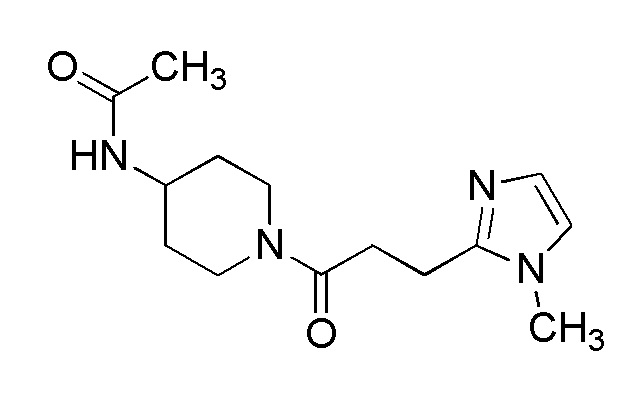
К раствору 1-(4-аминопиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0500 г, 0,212 ммоль) в дихлорметане (2,0 мл) при комнатной температуре добавляли пиридин (0,0510 мл, 0,635 ммоль) и уксусный ангидрид (0,0600 мл, 0,635 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида (0,0510 г, 0,183 ммоль, 86%) (здесь и далее указывается как «соединение по примеру 9») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,19-1,34 (2H, м), 1,88-2,02 (4H, м), 2,07-2,20 (1H, м), 2,65-2,75 (1H, м), 2,82-3,02 (4H, м), 3,05-3,15 (1H, м), 3,60 (3H, с), 3,88-4,02 (2H, м), 4,45-4,55 (1H, м), 5,68-5,82 (1H, м), 6,77 (1H, д, J=1,2 Гц), 6,87 (1H, д, J=1,2 Гц).
[0340]
(Пример 10) Синтез гидрохлорида N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида:
[Формула 63]
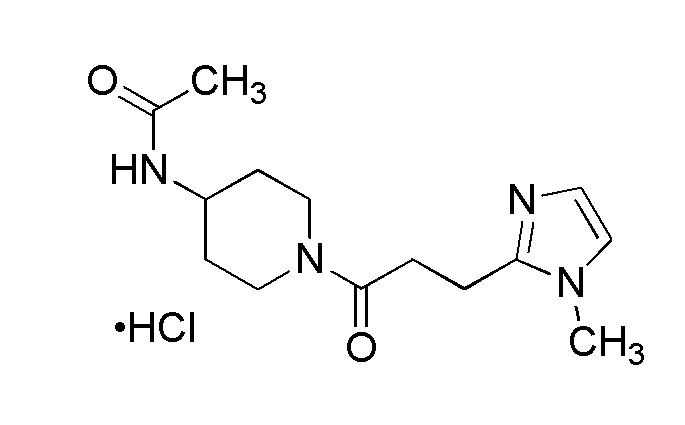
К раствору N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида (0,0510 г, 0,183 ммоль) в диэтиловом эфире (2,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,119 мл, 0,238 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (6,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида (0,0344 г, 0,109 ммоль, 60%) (здесь и далее указывается как «соединение по примеру 10») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,30-1,50 (2H, м), 1,85-1,99 (5H, м), 2,83-2,94 (1H, м), 2,97-3,06 (2H, м), 3,17-3,30 (3H, м), 3,79-3,93 (5H, м), 4,17-4,27 (1H, м), 7,27-7,33 (2H, м).
ESI-MS: в виде N-(1-(3-(1-метил-1H-имидазол-2-ил)пропаноил)пиперидин-4-ил)ацетамида: m/z= 279 (M+H)+.
[0341]
(Пример 11) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-(2-гидроксиэтил)-1H-имидазол-2-ил)пропан-1-она:
[Формула 64]
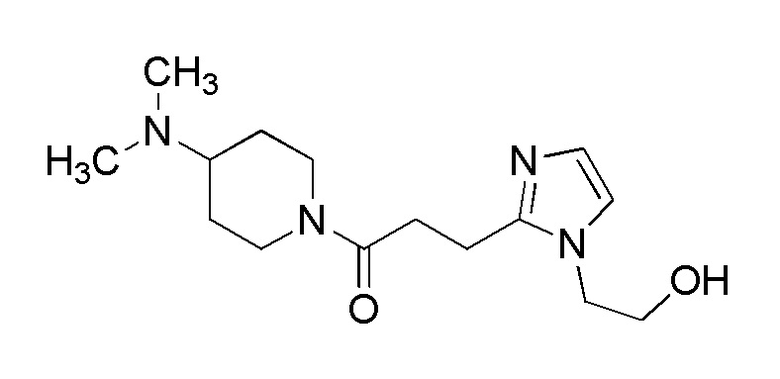
К раствору 3-(1-(2-((трет-бутилдиметилсилил)окси)этил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)пропан-1-она (1,00 г, 2,45 ммоль) в тетрагидрофуране (12,2 мл) при температуре 0°C добавляли раствор тетрабутиламмоний фторида в тетрагидрофуране (1,0 M, 3,06 мл, 3,06 ммоль), и температуру полученной смеси повышали до комнатной температуры, и полученную смесь перемешивали в течение 2 часов. Реакционную жидкость концентрировали при пониженном давлении, и затем остаток очищали с помощью колоночной хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-(2-гидроксиэтил)-1H-имидазол-2-ил)пропан-1-она (0,605 г, 2,06 ммоль, 84%) (здесь и далее указывается как «соединение по примеру 11») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,26-1,41 (2H, м), 1,78-1,86 (2H, м), 2,26-2,36 (7H, м), 2,52-2,59 (1H, м), 2,95-3,03 (5H, м), 3,88 (2H, т, J=4,9 Гц), 3,96-4,00 (1H, м), 4,09 (2H, т, J=4,9 Гц), 4,51-4,55 (1H, м), 6,85 (1H, с), 6,90 (1H, с).
ESI-MS: m/z= 295 (M+H)+.
[0342]
(Пример 12) Синтез гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-(2-гидроксиэтил)-1H-имидазол-2-ил)пропан-1-она:
[Формула 65]
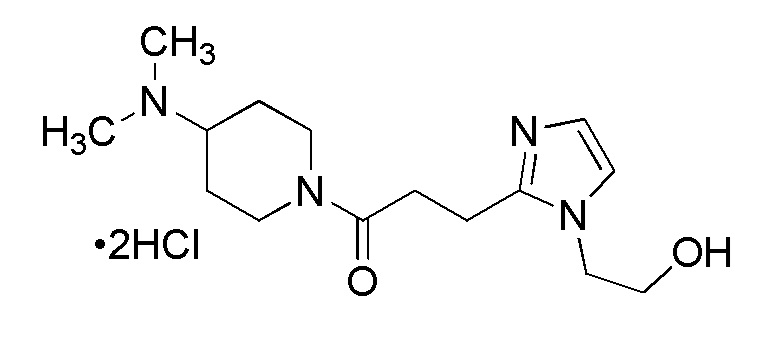
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-(2-гидроксиэтил)-1H-имидазол-2-ил)пропан-1-она (37,7 мг, 0,128 ммоль) в смеси диэтиловый эфир (2,5 мл)-дихлорметан при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,16 мл, 0,32 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа, и температуру реакционной жидкости повышали до комнатной температуры, и реакционную жидкость перемешивали в течение 1 часа. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром, и сушили при комнатной температуре с получением гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-(2-гидроксиэтил)-1H-имидазол-2-ил)пропан-1-она (43,1 мг, 0,117 ммоль, 92%) (здесь и далее указывается как «соединение по примеру 12») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,54-1,76 (2H, м), 2,13-2,20 (2H, м), 2,70-2,78 (1H, м), 2,87 (6H, с), 3,05-3,08 (2H, м), 3,16-3,30 (3H, м), 3,52 (2H, тт, J=12,0, 4,0 Гц), 3,96 (2H, т, J=5,0 Гц), 4,09-4,12 (1H, м), 4,33 (2H, т, J=5,0 Гц), 4,53-4,57 (1H, м), 7,37-7,44 (2H, м).
ESI-MS: в виде 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-(2-гидроксиэтил)-1H-имидазол-2-ил)пропан-1-она: m/z= 295 (M+H)+.
[0343]
(Пример 13) Синтез гидрохлорида 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 66]
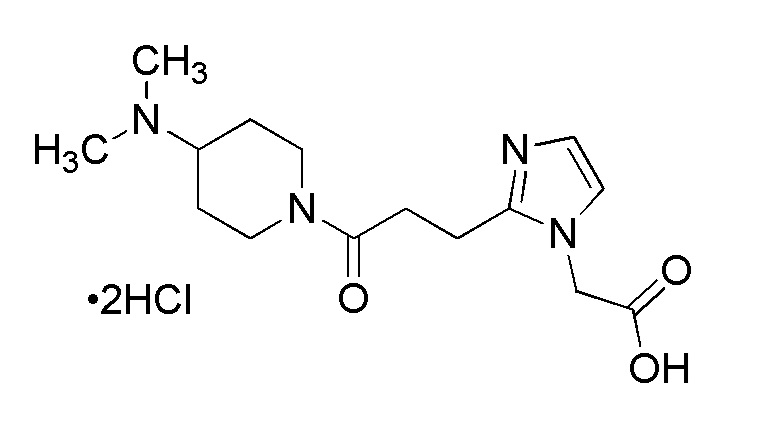
К раствору трет-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,104 г, 0,287 ммоль) в 1,4-диоксане (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в 1,4-диоксане (4,0 н, 0,861 мл, 3,45 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали с получением гидрохлорида 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (колич.) (здесь и далее указывается как «соединение по примеру 13») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,54-1,76 (2H, м), 2,13-2,19 (2H, м), 2,70-2,76 (1H, м), 2,87 (6H, с), 2,99-3,02 (2H, м), 3,15-3,24 (3H, м), 3,47-3,55 (1H, м), 4,06-4,10 (1H, м), 4,53-4,56 (1H, м), 5,02 (2H, с), 7,39 (2H, с).
ESI-MS: в виде 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты: m/z= 307 (M-H)-.
[0344]
(Пример 14) Синтез 3-(1-(2-аминоэтил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)пропан-1-она:
[Формула 67]
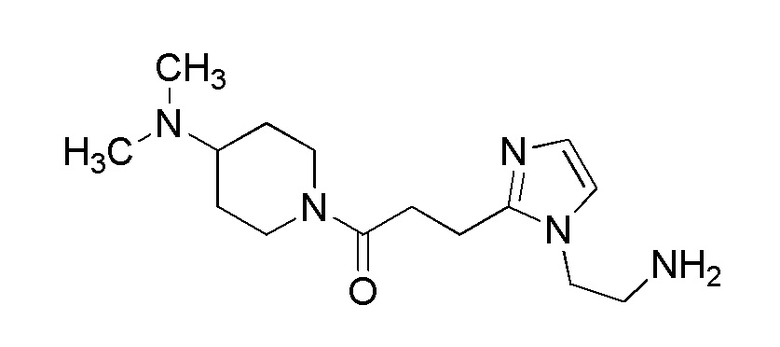
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-(2-гидроксиэтил)-1H-имидазол-2-ил)пропан-1-она (0,660 г, 2,24 ммоль) в дихлорметане (11,0 мл) при температуре 0°C добавляли триэтиламин (0,342 мл, 2,47 ммоль) и метансульфонилхлорид (0,191 мл, 2,47 ммоль), и температуру полученной смеси повышали до комнатной температуры, и полученную смесь перемешивали в течение 1 часа. В реакционную жидкость добавляли насыщенный водный раствор карбоната калия, и полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и затем фильтровали, и фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли ацетонитрил (10,8 мл), и полученный остаток растворялся. При комнатной температуре добавляли фталимид калия (0,452 г, 2,44 ммоль), и полученную смесь нагревали при кипении с обратным холодильником в течение 3 часов. Реакционную жидкость концентрировали при пониженном давлении, и затем к остатку добавляли дихлорметан и насыщенный водный раствор карбоната калия, и полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и затем фильтровали, и фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли метанол (10,8 мл), и полученный остаток растворялся. При комнатной температуре добавляли гидразин гидрат (0,158 мл, 3,25 ммоль), и полученную смесь нагревали при кипении с обратным холодильником в течение 1 часа. Температуру реакционной жидкости охлаждали до комнатной температуры, и реакционную жидкость фильтровали, и затем фильтрат концентрировали при пониженном давлении. К остатку добавляли дихлорметан и водный раствор соляной кислоты (1,0 н, 8,0 мл), и затем водный слой промывали дихлорметаном. К водному слою добавляли водный раствор гидроксида натрия (1 н, 8,0 мл), и полученную смесь концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (NH силикагель, хлороформ/метанол) с получением 3-(1-(2-аминоэтил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)пропан-1-она (0,338 г, 1,15 ммоль, 51%) (здесь и далее указывается как «соединение по примеру 14») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,30-1,43 (2H, м), 1,81-1,87 (2H, м), 2,27 (6H, с), 2,34 (1H, тт, J=11,0, 3,8 Гц) 2,56-2,63 (1H, м), 2,92-3,05 (7H, м), 3,98-4,04 (3H, м), 4,59-4,62 (1H, м), 6,88 (1H, т, J=1,2 Гц), 6,96 (1H, т, J=1,2 Гц).
ESI-MS: m/z= 294 (M+H)+.
[0345]
(Пример 15) Синтез гидрохлорида 3-(1-(2-аминоэтил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)пропан-1-она:
[Формула 68]
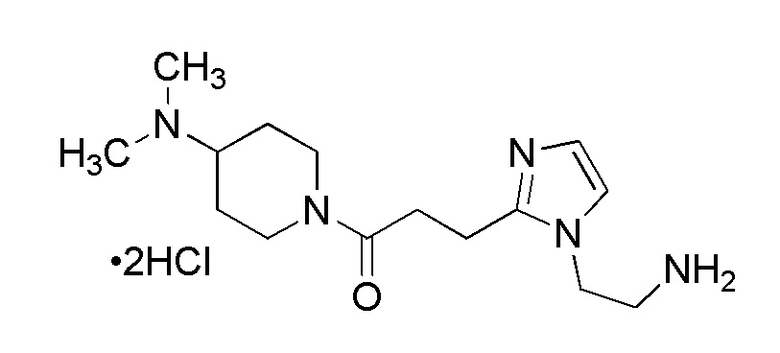
К раствору 3-(1-(2-аминоэтил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)пропан-1-она (0,0576 г, 0,196 ммоль) в диэтиловом эфире (3,9 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,245 мл, 0,491 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа, и температуру реакционной жидкости повышали до комнатной температуры, и затем реакционную жидкость перемешивали в течение 1 часа. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром, и сушили при комнатной температуре с получением гидрохлорида 3-(1-(2-аминоэтил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)пропан-1-она (0,0633 г, 0,173 ммоль, 88%) (здесь и далее указывается как «соединение по примеру 15») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,55-1,81 (2H, м), 2,14-2,22 (2H, м), 2,71-2,77 (1H, м), 2,88 (6H, с), 3,06-3,31 (5H, м), 3,50-3,60 (3H, м), 4,12-4,15 (1H, м), 4,52-4,55 (1H, м), 4,61 (2H, т, J=6,6 Гц), 7,44-7,51 (2H, м).
ESI-MS: в виде 3-(1-(2-(аминоэтил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)пропан-1-она: m/z= 294 (M+H)+.
[0346]
(Пример 16) Синтез этил 2-(2-(3-(4-(этилметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 69]
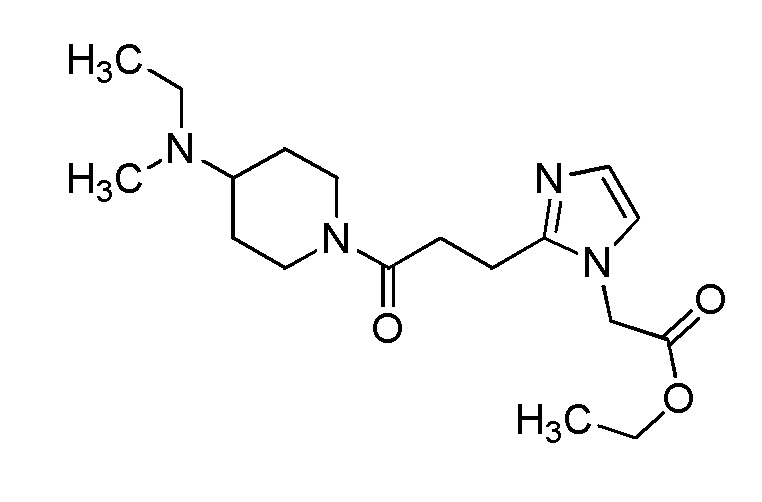
К раствору сырой 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (0,0500 г, 0,221 ммоль) в хлороформе (2,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,116 мл, 0,663 ммоль), HBTU (0,126 г, 0,332 ммоль) и сырой 4-этилметиламинопиперидин (0,0310 г, 0,221 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением этил 2-(2-(3-(4-(этилметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0250 г, 0,0713 ммоль, 32%) (здесь и далее указывается как «соединение по примеру 16») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,06 (3H, т, J=7,2 Гц), 1,25-1,45 (5H, м), 1,73-1,83 (2H, м), 2,23 (3H, с), 2,48-2,63 (4H, м), 2,88-3,03 (5H, м), 3,97-4,05 (1H, м), 4,19-4,26 (2H, м), 4,58-4,65 (1H, м), 4,75 (2H, с), 6,80-6,82 (1H, м), 6,95-6,97 (1H, м).
ESI-MS: m/z= 351 (M+H)+.
[0347]
(Пример 17) Синтез 2-(2-(3-(4-(этилметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 70]
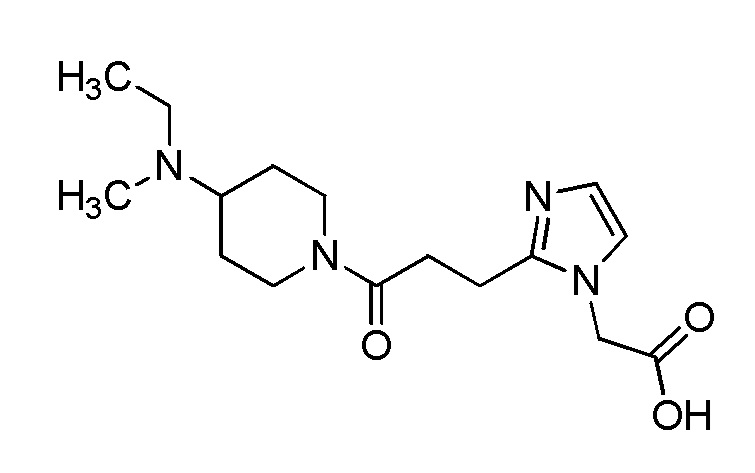
К раствору этил 2-(2-(3-(4-(этилметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,180 г, 0,514 ммоль) в этаноле (3,0 мл) при комнатной температуре добавляли водный раствор гидроксида натрия (1,0 н, 0,565 мл, 0,565 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 4 часов. Реакционную жидкость охлаждали до температуры 0°C, и затем в реакционную жидкость для нейтрализации добавляли соляную кислоту (1,0 н), и затем реакционную жидкость концентрировали при пониженном давлении. Остаток подвергали азеотропной отгонке с толуолом и добавляли этанол. Осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением 2-(2-(3-(4-(этилметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,161 г, 0,499 ммоль, 97%) (здесь и далее указывается как «соединение по примеру 17») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, DMSO-d6) δ: 1,15-1,24 (3H, м), 1,33-1,65 (2H, м), 1,86-1,97 (2H, м), 2,25-2,70 (6H, м), 2,72-2,80 (3H, м), 2,95-3,12 (3H, м), 3,95-4,05 (1H, м), 4,44-4,54 (1H, м), 4,76-4,83 (2H, м), 6,74-6,85 (1H, м), 7,00-7,09 (1H, м).
ESI-MS: m/z= 323 (M+H)+.
[0348]
(Пример 18) Синтез гидрохлорида 2-(2-(3-(4-(этилметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 71]
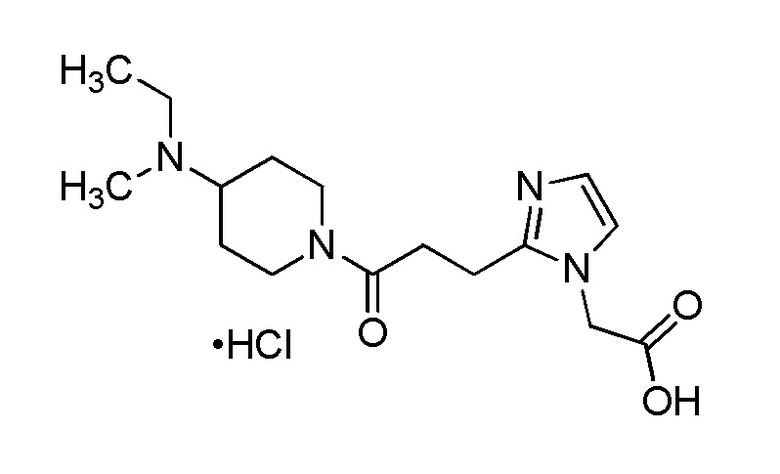
К раствору 2-(2-(3-(4-(этилметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,161 г, 0,499 ммоль) в воде (1,0 мл) при температуре 0°C добавляли соляную кислоту (1,0 н, 0,354 мл, 0,354 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 2 часов. Реакционную жидкость концентрировали при пониженном давлении, и выпавший в осадок твердый продукт фильтровали и собирали с получением гидрохлорида 2-(2-(3-(4-(этилметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,160 г, 0,446 ммоль, 89%) (здесь и далее указывается как «соединение по примеру 18») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,28-1,35 (3H, м), 1,54-1,82 (2H, м), 2,05-2,16 (2H, м), 2,68-2,81 (4H, м), 2,96-3,04 (2H, м), 3,12-3,24 (4H, м), 3,28-3,38 (1H, м), 3,54-3,64 (1H, м), 4,02-4,10 (1H, м), 4,48-4,58 (1H, м), 4,98 (2H, с), 7,35-7,38 (2H, м).
ESI-MS: в виде 2-(2-(3-(4-(этилметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты: m/z= 323 (M+H)+.
[0349]
(Пример 19) Синтез этил 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 72]
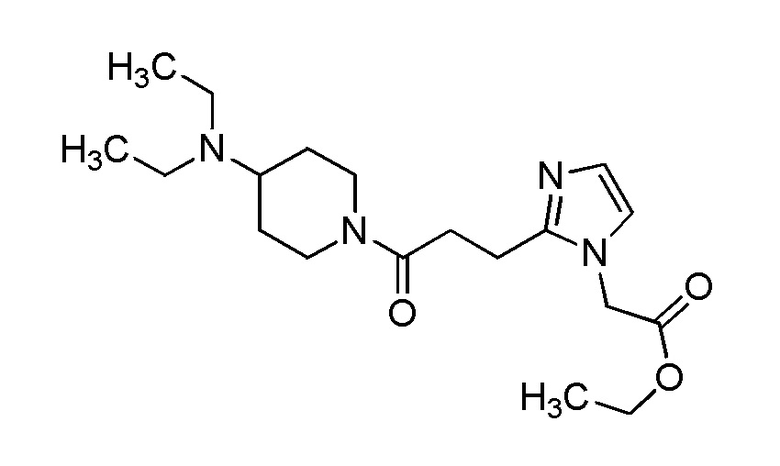
К раствору сырой 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (0,0500 г, 0,221 ммоль) в хлороформе (2,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,116 мл, 0,663 ммоль), HBTU (0,126 г, 0,332 ммоль) и сырой 4-диэтиламинопиперидин (0,0350 г, 0,221 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением этил 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0700 г, 0,192 ммоль, 87%) (здесь и далее указывается как «соединение по примеру 19») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,03 (6H, т, J=7,2 Гц), 1,25-1,43 (5H, м), 1,72-1,82 (2H, м), 2,47-2,58 (4H, м), 2,65-2,77 (1H, м), 2,88-3,00 (6H, м), 3,95-4,04 (1H, м), 4,23 (2H, кв, J=6,8 Гц), 4,58-4,65 (1H, м), 4,75 (2H, с), 6,80-6,83 (1H, м),6,95-6,97 (1H, м).
ESI-MS: m/z= 365 (M+H)+.
[0350]
(Пример 20) Синтез гидрохлорида этил 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 73]
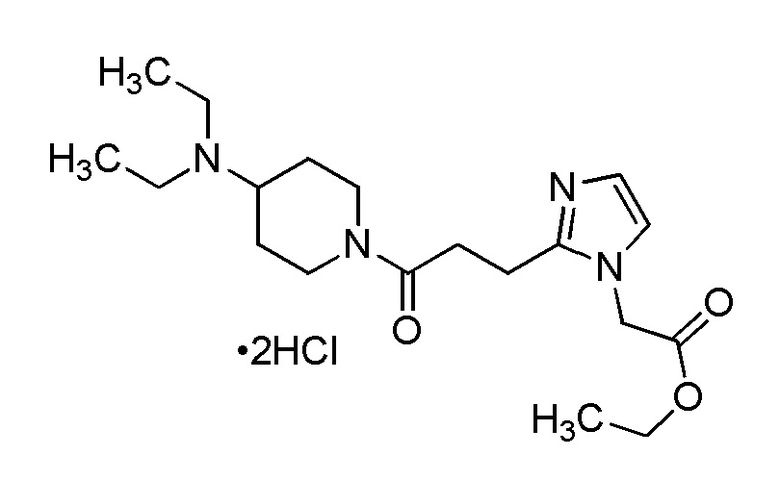
К раствору этил 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0350 г, 0,0960 ммоль) в диэтиловом эфире (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,106 мл, 0,211 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (4,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида этил 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0217 г, 0,0496 ммоль, 24%) (здесь и далее указывается как «соединение по примеру 20») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,25-1,36 (9H, м), 1,55-1,78 (2H, м), 2,08-2,18 (2H, м), 2,68-2,77 (1H, м), 2,95-3,05 (2H, м), 3,13-3,35 (7H, м),3,60-3,70 (1H, м), 4,02-4,08 (1H, м), 4,29 (2H, кв, J=7,6 Гц), 4,48-4,55 (1H, м), 5,17 (2H, м), 7,34-7,40 (2H, м).
ESI-MS: в виде этил 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z=365 (M+H)+
[0351]
(Пример 21) Синтез 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 74]
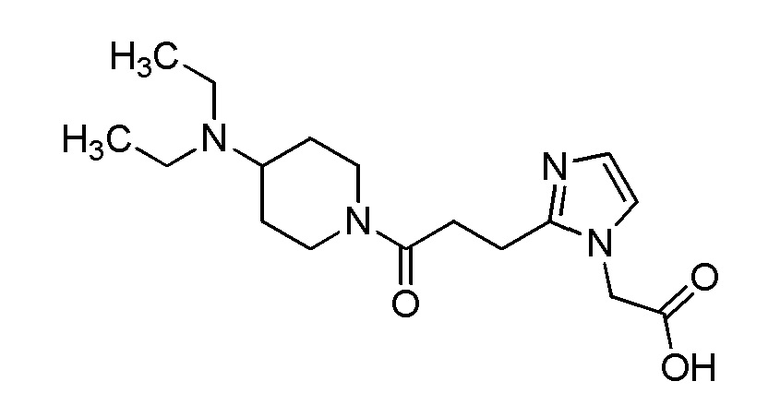
К раствору этил 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,150 г, 0,412 ммоль) в этаноле (2,0 мл) при комнатной температуре добавляли водный раствор гидроксида натрия (1,0 н, 0,452 мл, 0,452 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 4 часов. Реакционную жидкость охлаждали до температуры 0°C, в реакционную жидкость для нейтрализации добавляли соляную кислоту (1,0 н), и затем реакционную жидкость концентрировали при пониженном давлении. Остаток подвергали азеотропной отгонке с толуолом и добавляли этанол. Осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,124 г, 0,369 ммоль, 89%) (здесь и далее указывается как «соединение по примеру 21») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ: 1,36 (3H, т, J=7,6 Гц), 1,56-1,86 (2H, м), 2,03-2,13 (2H, м), 2,64-2,75 (1H, м), 2,87-3,06 (2H, м), 3,12-3,28 (10H, м), 3,58-3,66 (1H, м), 4,02-4,10 (1H, м), 4,62-4,70 (1H, м), 5,05 (2H, с), 7,42-7,47 (2H, м).
ESI-MS: m/z= 337 (M+H)+.
[0352]
(Пример 22) Синтез гидрохлорида 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 75]
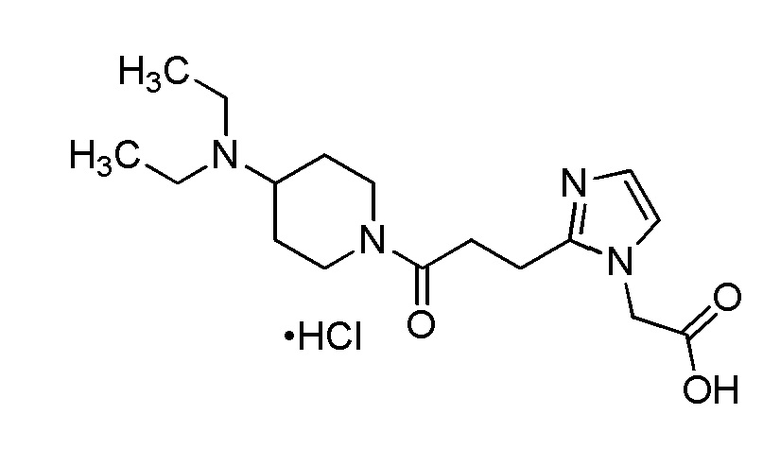
К раствору 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,124 г, 0,369 ммоль) в воде (1,0 мл) при температуре 0°C добавляли соляную кислоту (1,0 н, 0,785 мл, 0,785 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 2 часов. Реакционную жидкость концентрировали при пониженном давлении, и выпавший в осадок твердый продукт фильтровали и собирали с получением гидрохлорида 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,104 г, 0,278 ммоль, 75%) (здесь и далее указывается как «соединение по примеру 22») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,27-1,34 (6H, м), 1,55-1,80 (2H, м), 2,06-2,17 (2H, м), 2,68-2,76 (1H, м), 2,95-3,02 (2H, м), 3,13-3,35 (7H, м), 3,59-3,69 (1H, м), 4,01-4,08 (1H, м), 4,48-4,56 (1H, м), 4,95 (2H, с), 7,33-7,36 (2H, м).
ESI-MS: в виде 2-(2-(3-(4-(диэтиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты: m/z= 337 (M+H)+.
[0353]
(Пример 23) Синтез этил 2-(2-(3-(4-(пиперидин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 76]
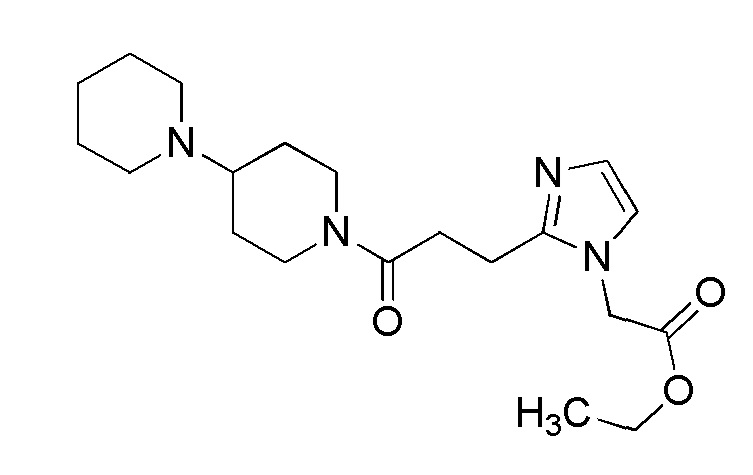
К раствору сырой 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (0,200 г, 0,884 ммоль) в дихлорметане (10,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,171 г, 1,33 ммоль), HBTU (0,402 г, 1,06 ммоль) и 4-(пиперидин-1-ил)пиперидин (0,149 г, 0,884 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 12 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, гексан/этилацетата и хлороформ/метанол) с получением этил 2-(2-(3-(4-(пиперидин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,266 г, 0,708 ммоль, 80%) (здесь и далее указывается как «соединение по примеру 23») в виде красновато-коричневого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,29 (3H, т, J=7,2 Гц), 1,36-1,48 (4H, м), 1,54-1,63 (4H, м), 1,7-1,86 (2H, м), 2,40-2,58 (6H, м), 2,85-3,00 (1H, м), 2,91 (4H, с), 3,96-4,03 (1H, м), 4,23 (2H, т, J=7,2 Гц), 4,57-4,65 (1H, м), 4,75 (2H, кв, J=7,2 Гц), 6,82 (1H, д, J=1,2 Гц), 6,97 (1H, д, J=1,2 Гц).
ESI-MS: m/z= 377 (M+H)+.
[0354]
(Пример 24) Синтез 2-(2-(3-(4-(пиперидин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 77]
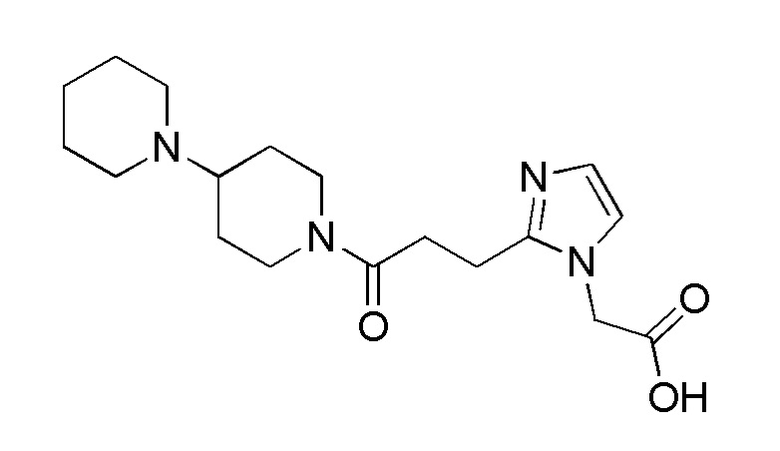
Раствор этил 2-(2-(3-(4-(пиперидин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0550 г, 0,146 ммоль) в воде (5,0 мл) нагревали до температуры 40°C, и реакционную жидкость перемешивали при той же температуре в течение 12 часов. Реакционную жидкость концентрировали и высушивали, и затем полученный остаток сушили при пониженном давлении с получением 2-(2-(3-(4-(пиперидин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,0510 г, 0,146 ммоль, 100%) (здесь и далее указывается как «соединение по примеру 24») в виде твердого вещества красновато-коричневого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,35-1,90 (8H, м), 1,93-2,03 (2H, м), 2,55 (1H, т, J=12,0 Гц), 3,30 (1H, тт, J=12,0, 3,6 Гц), 3,88-4,00 (1H, м), 4,36-4,45 (1H, м), 4,48 (1H, с), 6,84 (1H, д, J=1,2 Гц), 6,92 (1H, д, J=1,2 Гц).
ESI-MS: m/z= 349 (M+H)+.
[0355]
(Пример 25) Синтез гидрохлорида 2-(2-(3-(4-(пиперидин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 78]
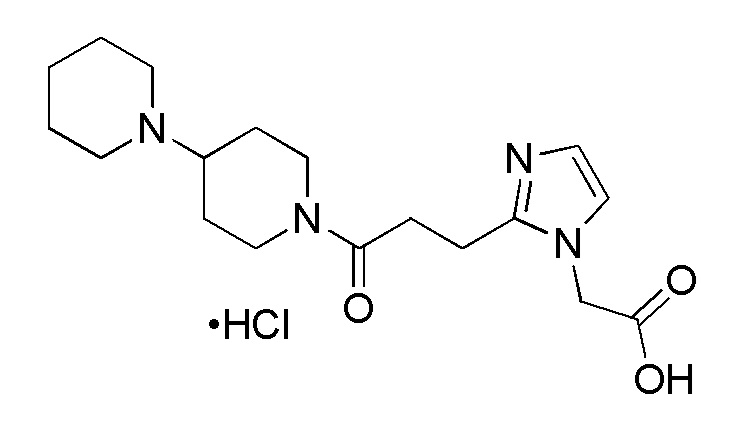
К раствору этил 2-(2-(3-(4-(пиперидин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0650 г, 0,173 ммоль) в воде (5,0 мл) при температуре 0°C добавляли водный раствор гидроксида натрия (1,0 н, 0,345 мл, 0,345 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа. Добавляли соляную кислоту (1,0 н, 0,518 мл, 0,518 ммоль) и затем полученную смесь концентрировали и высушивали. Полученный твердый продукт промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Полученный твердый продукт опять промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Полученный остаток сушили при пониженном давлении с получением гидрохлорида 2-(2-(3-(4-(пиперидин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,0510 г, 0,133 ммоль, 77%) (здесь и далее указывается как «соединение по примеру 25») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,26-1,72 (6H, м), 1,83 (2H, д, J=14,4 Гц), 2,02 (2H, т, J=12,8 Гц), 2,57 (1H, т, J=12,8 Гц), 2,83-2,95 (4H, м), 2,97-3,14 (3H, м), 3,27-3,43 (3H, м), 3,88-3,98 (1H, м), 4,33-4,43 (1H, м), 4,99 (2H, с), 7,27 (2H, с).
ESI-MS: в виде 2-(2-(3-(4-(пиперидин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты: 349 (M+H)+.
[0356]
(Пример 26) Синтез этил 2-(2-(3-(4-(морфолин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 79]
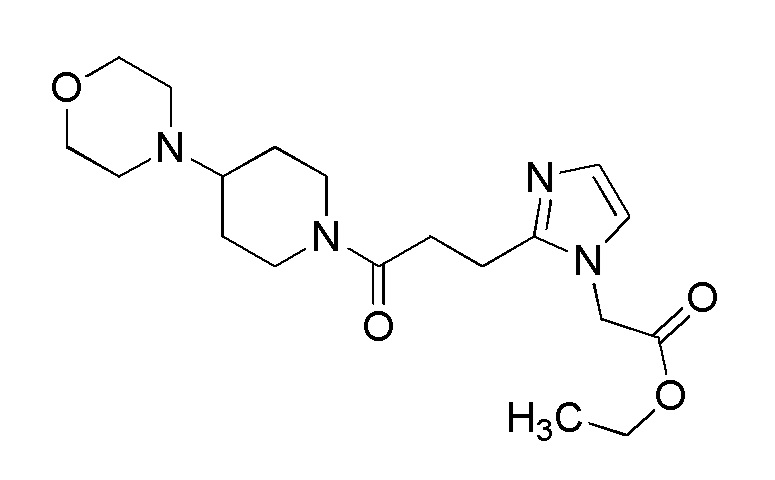
К раствору сырой 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (0,200 г, 0,884 ммоль) в дихлорметане (10,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,171 г, 1,33 ммоль), HBTU (0,402 г, 1,06 ммоль) и 4-(морфолин-4-ил)пиперидин (0,151 г, 0,884 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 12 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, гексан/этилацетата и хлороформ/метанол) с получением этил 2-(2-(3-(4-(морфолин-1-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,265 г, 0,700 ммоль, 79%) (здесь и далее указывается как «соединение по примеру 26») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,29 (3H, т, J=7,2 Гц), 1,30-1,45 (2H, м), 1,81-1,92 (2H, м), 2,39 (1H, тт, J=10,8, 3,6 Гц), 2,53 (4H, т, J=4,8 Гц), 2,59 (1H, тд, J=13,2, 2,8 Гц), 2,91 (4H, с), 3,01 (1H, тд, J=13,2, 2,8 Гц), 3,71 (4H, т, J=4,8 Гц), 3,97-4,04 (1H, м), 4,23 (2H, кв, J=7,2 Гц), 4,54-4,62 (1H, м), 4,75 (2H, с), 6,82 (1H, д, J=1,6 Гц), 6,96 (1H, д, J=1,6 Гц).
ESI-MS: m/z= 379 (M+H)+.
[0357]
(Пример 27) Синтез гидрохлорида 2-(2-(3-(4-(морфолин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 80]
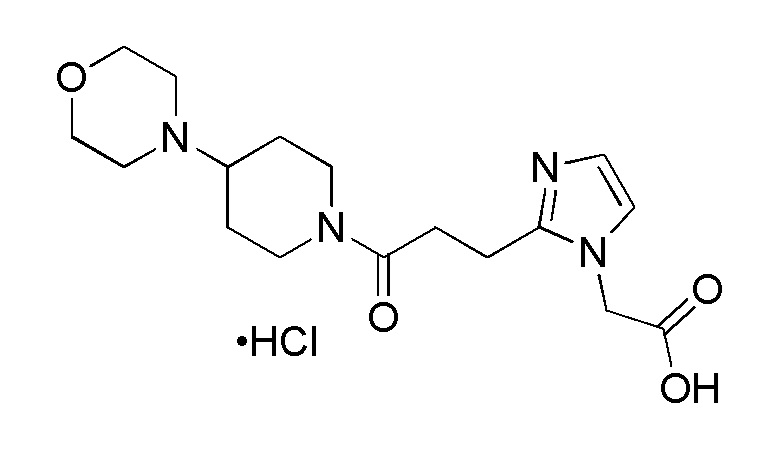
К раствору этил 2-(2-(3-(4-(морфолин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,080 г, 0,211 ммоль) в воде (5,0 мл) при температуре 0°C добавляли водный раствор гидроксида натрия (1,0 н, 0,423 мл, 0,423 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа. Добавляли соляную кислоту (1,0 н, 0,634 мл, 0,634 ммоль), и затем полученную смесь концентрировали и высушивали. Полученный твердый продукт промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Полученный твердый продукт опять промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Полученный остаток сушили при пониженном давлении с получением гидрохлорида 2-(2-(3-(4-(морфолин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,0580 г, 0,150 ммоль, 71%) (здесь и далее указывается как «соединение по примеру 27») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,44 (1H, ддд, J=25,4, 12,4, 4,4 Гц), 1,57 (1H, ддд, J=25,4, 12,4, 4,4 Гц), 2,11 (2H, т, J=12,8 Гц), 2,58 (1H, т, J=13,2 Гц), 2,98-3,17 (5H, м), 3,35-3,47 (3H, м), 3,68 (2H, т, J=12,4 Гц), 3,89-3,96 (1H, м), 3,97-4,06 (2H, м), 4,38-4,45 (1H, м), 4,68 (2H, с), 7,20 (1H, д, J=1,6 Гц), 7,21(1H, д, 1,6 Гц).
ESI-MS: в виде 2-(2-(3-(4-(морфолин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты: 351 (M+H)+.
[0358]
(Пример 28) Синтез этил 2-(2-(3-(4-(1-метилпиперазин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 81]
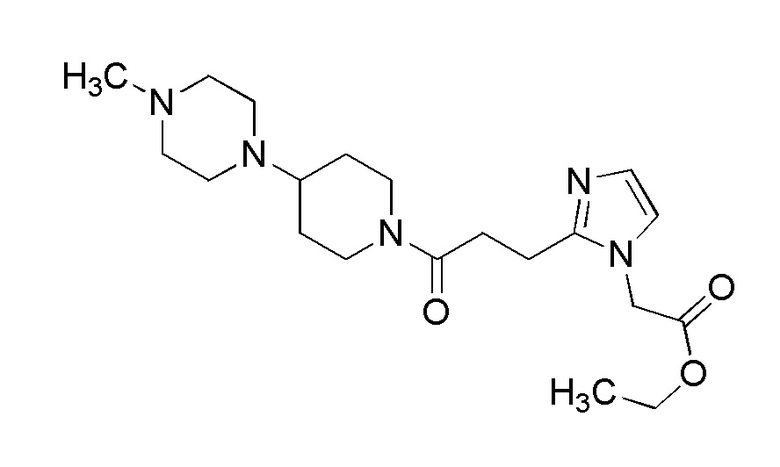
К раствору сырой 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (0,200 г, 0,884 ммоль) в дихлорметане (10,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,171 г, 1,33 ммоль), HBTU (0,402 г, 1,06 ммоль) и 4-(1-метилпиперазин-4-ил)пиперидин (0,162 г, 0,884 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 12 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, гексан/этилацетата и хлороформ/метанол) с получением этил 2-(2-(3-(4-(1-метилпиперазин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,280 г, 0,715 ммоль, 81%) (здесь и далее указывается как «соединение по примеру 28») в виде масла бледно-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,29 (3H, т, J=7,6 Гц), 1,32-1,46 (2H, м), 1,81-1,91 (2H, м), 2,28 (3H, с), 2,36-2,64 (10H, м), 2,91 (4H, с), 2,95-3,03 (1H, м), 3,97-4,04 (1H, м), 4,23 (2H, кв, J=7,6 Гц), 4,54-4,62 (1H, м), 4,75 (2H, с), 6,82 (1H, д, J=1,2 Гц), 6,96 (1H, д, J=1,2 Гц).
ESI-MS: m/z= 392 (M+H)+.
[0359]
(Пример 29) Синтез гидрохлорида 2-(2-(3-(4-(1-метилпиперазин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 82]
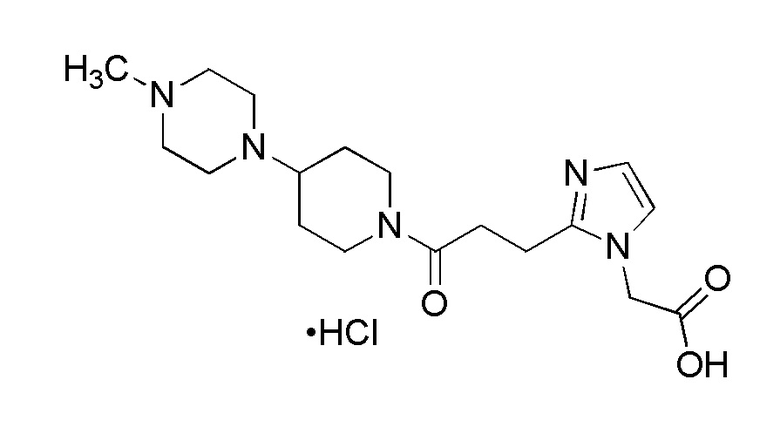
К раствору этил 2-(2-(3-(4-(1-метилпиперазин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,118 г, 0,303 ммоль) в воде (5,0 мл) при температуре 0°C добавляли водный раствор гидроксида натрия (1,0 н, 0,605 мл, 0,605 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа. Добавляли соляную кислоту (1,0 н, 0,910 мл, 0,910 ммоль), и затем полученную смесь концентрировали и высушивали. Полученный твердый продукт промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Полученный твердый продукт опять промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Полученный остаток сушили при пониженном давлении с получением гидрохлорида 2-(2-(3-(4-(1-метилпиперазин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,0850 г, 0,213 ммоль, 70%) (здесь и далее указывается как «соединение по примеру 29») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,26(1H, ддд, J=24,4, 12,4, 4,0 Гц), 1,36(1H, ддд, J=24,4, 12,4, 4,0 Гц), 1,91(2H, т, J=13,2 Гц), 2,55(1H, т, 12,4 Гц), 2,60-3,30(9H, м), 2,70(1H, с), 2,83(2H, т, J=6,8 Гц), 3,04(2H, т, J=6,8 Гц), 3,82-3,89(1H, м), 4,27-4,36 (1H, с), 4,66(2H, с), 7,19(1H, д, J=1,6 Гц), 7,21(1H, д, J=1,6 Гц).
ESI-MS: в виде 2-(2-(3-(4-(1-метилпиперазин-4-ил)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты: 364 (M+H)+.
[0360]
(Пример 30) Синтез этил 2-(2-(3-((R)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 83]
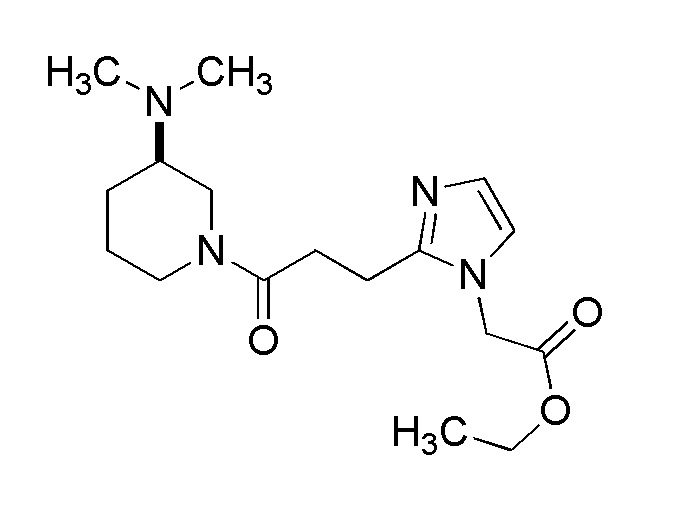
К раствору сырой 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (0,300 г, 1,33 ммоль) в дихлорметане (15,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,257 г, 1,99 ммоль), HBTU (0,603 г, 1,59 ммоль) и (R)-3-диметиламинопиперидин (0,149 г, 0,880 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 12 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, гексан/этилацетата и хлороформ/метанол) с получением этил 2-(2-(3-((R)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,325 г, 0,966 ммоль, 73%) (здесь и далее указывается как «соединение по примеру 30») в виде масла желтого цвета
1H-ЯМР (400 МГц, CDCl3) δ: 1,29 (3H, т, J=7,2 Гц), 1,36-1,48 (2H, м), 1,95-2,00 (2H, м), 2,32 (6H, с), 2,40-2,55 (1H, м), 2,78-3,00 (6H, м), 3,80-4,05 (1H, м), 4,23 (2H, кв, J=7,2), 4,45-4,67 (1H, м), 4,71-4,80 (2H, м), 6,80-6,85 (1H, м), 6,97 (1H, д, J=1,2 Гц).
ESI-MS: m/z= 337 (M+H)+.
[0361]
(Пример 31) Синтез гидрохлорида 2-(2-(3-((R)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 84]
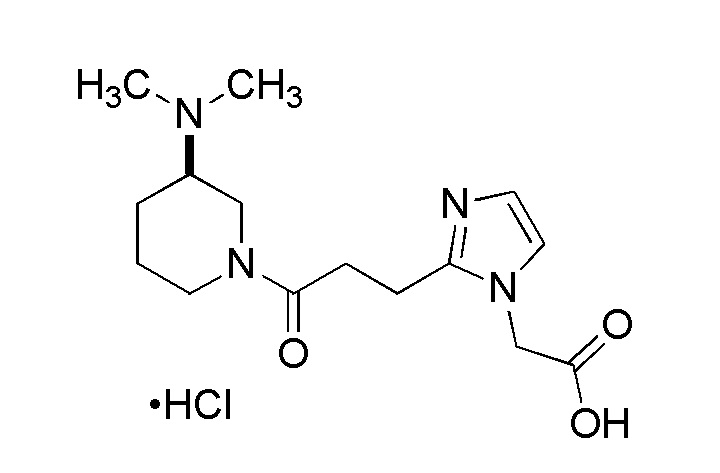
К раствору этил 2-(2-(3-((R)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0550 г, 0,165 ммоль) в воде (5,0 мл) при температуре 0°C добавляли водный раствор гидроксида натрия (1,0 н, 0,329 мл, 0,329 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа. Добавляли соляную кислоту (1,0 н, 0,494 мл, 0,494 ммоль) и затем полученную смесь концентрировали и высушивали. Полученный твердый продукт промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Полученный твердый продукт опять промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Полученный остаток сушили при пониженном давлении с получением гидрохлорида 2-(2-(3-((R)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,0400 г, 0,116 ммоль, 70%) (здесь и далее указывается как «соединение по примеру 31») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,02-1,87 (3H, м), 2,00-2,15 (1H, м), 2,55-2,95 (7H, м), 3,05-3,30 (5H, м), 3,47-3,62 (1H, м), 3,95-4,20 (1H, м), 4,99 (2H, с), 7,26 (2H, с).
ESI-MS: в виде 2-(2-(3-((R)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты: 309 (M+H)+.
[0362]
(Пример 32) Синтез этил (S)-2-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 85]
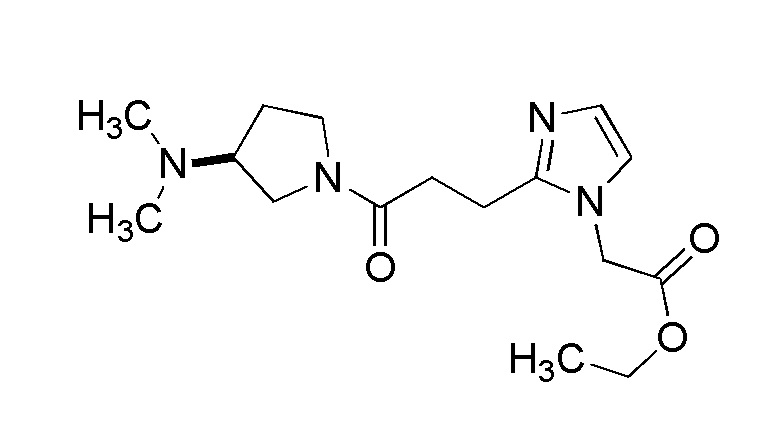
К раствору сырой 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (0,150 г, 0,663 ммоль) в дихлорметане (3,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,174 мл, 0,995 ммоль), HBTU (0,302 г, 0,796 ммоль) и (S)-3-(диметиламино)пирролидин (0,0840 мл, 0,663 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением этил (S)-2-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,200 г, 0,620 ммоль, 93%) (здесь и далее указывается как «соединение по примеру 32») в виде масла желтого цвета
1H-ЯМР (400 МГц, CDCl3) δ: 1,30 (3H, т, J =7,3 Гц), 1,71-1,84 (1H, м), 2,09-2,28 (7H, м), 2,61-2,96 (5H, м), 3,02-3,46 (2H, м), 3,64-3,82 (2H, м), 4,23 (2H, кв, J=7,3 Гц), 4,72-4,77 (2H, м), 6,81-6,82 (1H, м), 6,95-6,98 (1H, м).
ESI-MS: m/z= 323 (M+H)+.
[0363]
(Пример 33) Синтез гидрохлорида (S)-2-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 86]
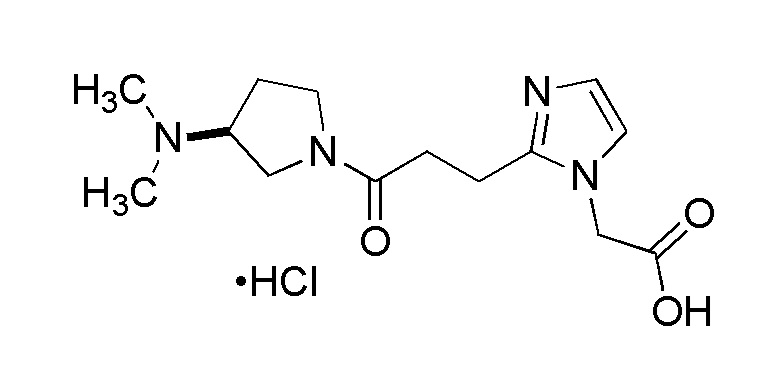
К этил (S)-2-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетату (0,160 г, 0,496 ммоль) при комнатной температуре добавляли водный раствор гидроксида натрия (1,0 н, 0,744 мл, 0,744 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 4 часов. При комнатной температуре в реакционную жидкость добавляли соляную кислоту (1,0 н, 0,744 мл, 0,744 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 5 минут. Реакционную жидкость концентрировали при пониженном давлении. Полученный остаток промывали этанолом (5,0 мл). Полученную смесь фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток опять промывали этанолом (5,0 мл). Полученную смесь фильтровали, и затем фильтрат концентрировали при пониженном давлении. К полученному остатку при комнатной температуре добавляли соляную кислоту (1,0 н, 0,595 мл, 0,595 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. Реакционную жидкость концентрировали при пониженном давлении и сушили при комнатной температуре с получением гидрохлорида (S)-2-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,120 г, 0,363 ммоль, 73%) (здесь и далее указывается как «соединение по примеру 33») в виде твердого вещества красного цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,95-2,18 (1H, м), 2,34-2,44 (1H, м), 2,67-2,90 (8H, м), 3,05-3,11 (2H, м),3,29-3,67 (3H, м), 3,78-3,93 (2H, м), 4,80 (2H, с), 7,22-7,26 (2H, м).
[0364]
(Пример 34) Синтез гидрохлорида трет-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)]ацетата:
[Формула 87]
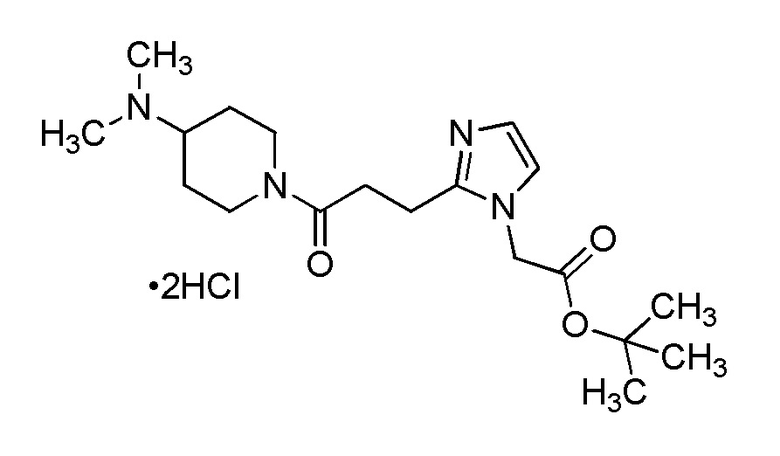
К раствору трет-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,100 г, 0,274 ммоль) в диэтиловом эфире (5,4 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,302 мл, 0,604 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (20,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида трет-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,111 г, 0,254 ммоль, 93%) (здесь и далее указывается как «соединение по примеру 34») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,48-1,72 (11H, м), 2,10-2,17 (2H, м), 2,66-2,74 (1H, м), 2,84 (6H, с), 2,90-3,25 (5H, м), 3,45-3,55 (1H, м), 4,00-4,10 (1H, м), 4,45-4,55 (1H, м), 5,08 (2H, с), 7,37-7,39 (2H, м).
ESI-MS: в виде трет-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 365 (M+H)+.
[0365]
(Пример 35) Синтез метил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 88]
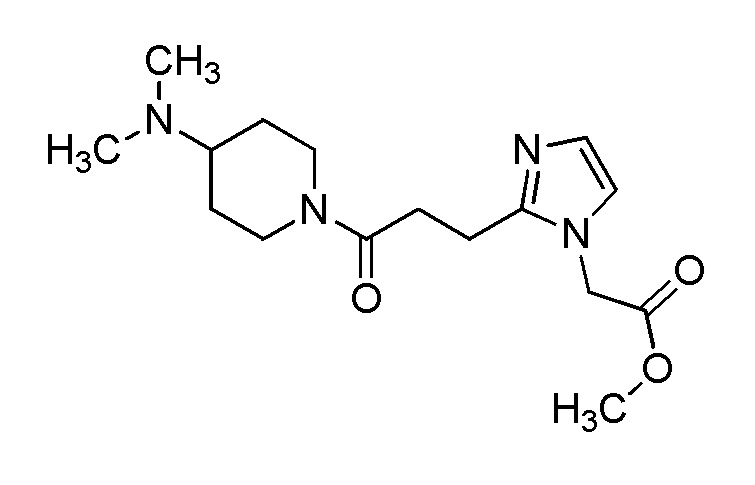
К раствору трет-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (105 мг, 0,287 ммоль) в смеси 1,4-диоксан (3,0 мл)-метанол (3,0 мл) при комнатной температуре добавляли раствор хлористого водорода в 1,4-диоксане (4,0 н, 0,718 мл, 2,87 ммоль), и полученную смесь перемешивали при той же температуре в течение 12 часов. Температуру реакционной жидкости повышали до 60°C, и реакционную жидкость перемешивали в течение 1 часа. В реакционную жидкость добавляли насыщенный водный раствор карбоната калия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и затем фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением метил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0370 г, 0,113 ммоль, 39%) (здесь и далее указывается как «соединение по примеру 35») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 1,29-1,42 (2H, м), 1,81-1,86 (2H, м), 2,27 (6H, с), 2,33 (1H, тт, J=11,1, 3,5 Гц), 2,55-2,62 (1H, м), 2,92 (4H, с), 2,96-3,03 (1H, м), 3,78 (3H, с), 3,97-4,01 (1H, м), 4,56-4,59 (1H, м), 4,78 (2H, с), 6,82 (1H, д, J=0,7 Гц), 6,97 (1H, д, J=0,7 Гц).
ESI-MS: m/z= 323 (M+H)+.
[0366]
(Пример 36) Синтез гидрохлорида метил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 89]
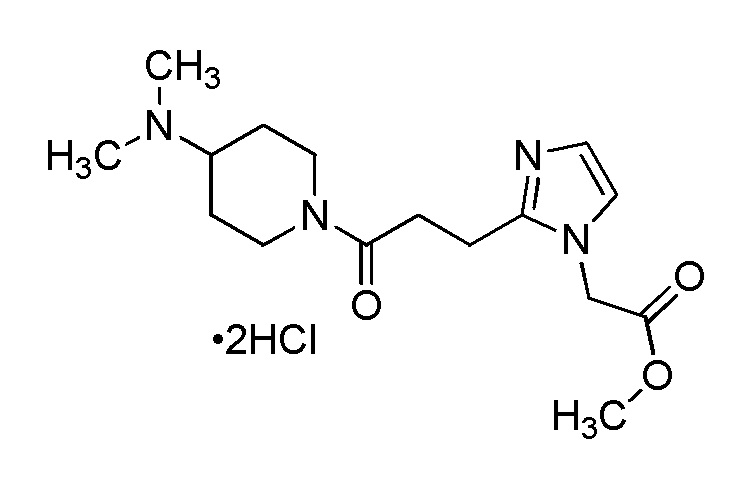
К раствору метил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (36,0 мг, 0,112 ммоль) в диэтиловом эфире (2,2 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,140 мл, 0,279 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. Температуру реакционной жидкости повышали до комнатной температуры, и реакционную жидкость перемешивали в течение 1 часа. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром, и сушили при комнатной температуре с получением гидрохлорида метил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (34,7 мг, 0,0880 ммоль, 78%) (здесь и далее указывается как «соединение по примеру 36») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,54-1,77 (2H, м), 2,13-2,20 (2H, м), 2,69-2,76 (1H, м), 2,87 (6H, с), 3,04 (2H, т, J=6,6 Гц), 3,16-3,25 (3H, м), 3,52 (1H, тт, J=12,1, 3,7 Гц), 3,85 (3H, с), 4,08-4,11 (1H, м), 4,52-4,55 (1H, м), 5,22 (2H, с), 7,41-7,42 (2H, м).
ESI-MS: в виде метил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 323 (M+H)+.
[0367]
(Пример 37) Синтез этил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 90]
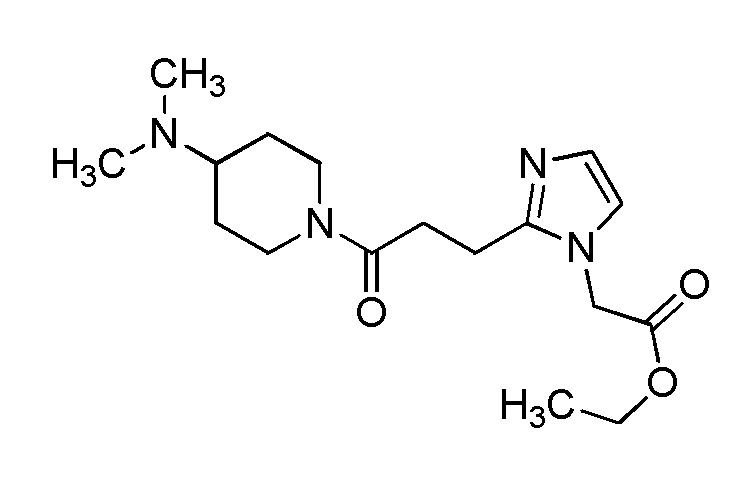
К раствору сырой 3-(1-(2-этокси-2-оксоэтил)-1H-имидазол-2-ил)пропановой кислоты (1,20 г, 5,30 ммоль) в хлороформе (24,0 мл) при комнатной температуре добавляли диизопропилэтиламин (1,39 мл, 7,96 ммоль), HBTU (2,41 г, 6,37 ммоль) и 4-диметиламинопиперидин (0,624 мл, 5,30 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением этил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,630 г, 1,87 ммоль, 35%) (здесь и далее указывается как «соединение по примеру 37») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,22-1,42 (5H, м), 1,77-1,87 (2H, м), 2,24-2,36 (7H, м), 2,54-2,64 (1H, м), 2,89-3,04 (5H, м), 3,95-4,04 (1H, м), 4,22 (2H, кв, J=7,2 Гц), 4,53-4,62 (1H, м), 4,74 (2H, с), 6,80-6,82 (1H, м), 6,96-6,97 (1H, м).
ESI-MS: m/z= 337 (M+H)+.
[0368]
(Пример 38) Синтез гидрохлорида этил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 91]
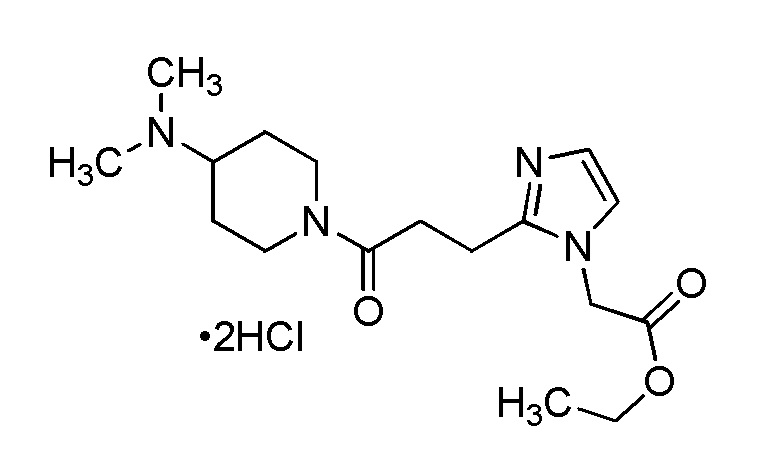
К раствору этил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,182 г, 0,541 ммоль) в диэтиловом эфире (10,8 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,595 мл, 1,19 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (40,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида этил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,182 г, 0,445 ммоль, 82%) (здесь и далее указывается как «соединение по примеру 38») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,27 (3H, т, J=7,2 Гц), 1,50-1,73 (2H, м), 2,09-2,17 (2H, м), 2,66-2,73 (1H, м), 2,84 (6H, с), 2,98-3,05 (5H, м), 3,45-3,55 (1H, м), 4,02-4,09 (1H, м), 4,28 (2H, кв, J=7,2 Гц), 4,47-4,53 (1H, м), 5,17 (2H, с), 7,37-7,39 (2H, м).
ESI-MS: в виде этил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 337 (M+H)+.
[0369]
(Пример 39) Синтез 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты:
[Формула 92]
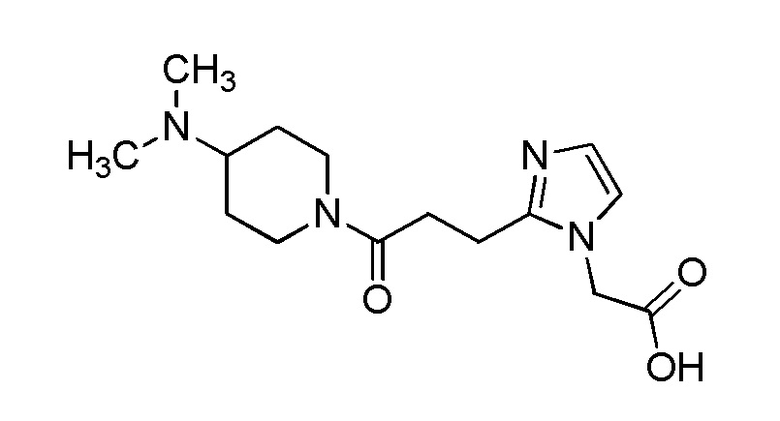
К раствору этил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,510 г, 1,52 ммоль) в этаноле (3,0 мл) при комнатной температуре добавляли водный раствор гидроксида натрия (1,0 н, 3,03 мл, 3,03 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. В реакционную жидкость для нейтрализации добавляли реакционную жидкость охлаждали до температуры 0°C и затем соляную кислоту (1,0 н), и затем реакционную жидкость концентрировали при пониженном давлении. Остаток подвергали азеотропной отгонке с толуолом и добавляли этанол. Осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,430 г, 1,39 ммоль, 92%) (здесь и далее указывается как «соединение по примеру 39») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ: 1,40-1,67 (2H, м), 1,95-2,04 (2H, м), 2,50-2,60 (1H, м), 2,73-2,88 (8H, м), 2,95-3,12 (3H, м), 3,20-3,35 (1H, м), 3,93-4,03 (1H, м), 4,54-4,64 (3H, м), 7,12-7,15 (1H, м), 7,18-7,21 (1H, м).
ESI-MS: m/z= 307 (M-H)-.
[0370]
(Пример 40) Синтез пропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 93]
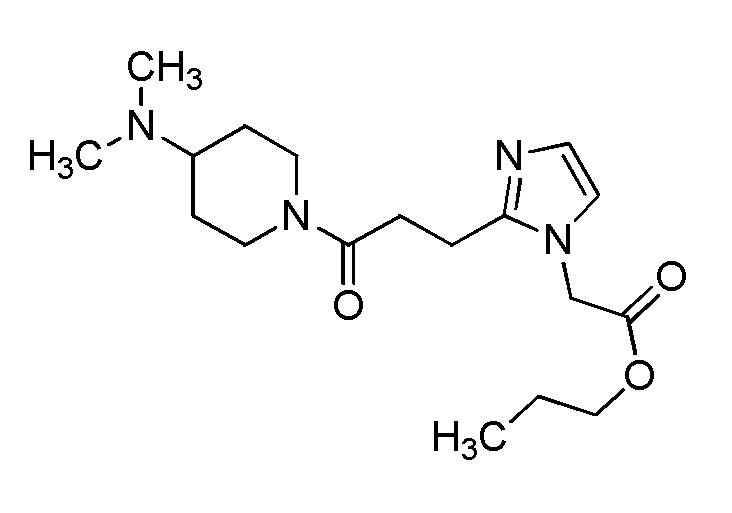
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,0500 г, 0,162 ммоль) в хлороформе (3,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,0710 мл, 0,405 ммоль), HBTU (0,0920 г, 0,243 ммоль), и пропан-1-ол (0,0300 мл, 0,405 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением пропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0499 г, 0,142 ммоль, 88%) (здесь и далее указывается как «соединение по примеру 40») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 0,92 (3H, т, J=7,6 Гц), 1,22-1,44 (2H, м), 1,60-1,70 (2H, м), 1,76-1,86 (2H, м), 2,23-2,34 (7H, м), 2,52-2,62 (1H, м), 2,88-3,02 (5H, м), 3,92-4,02 (1H, м), 4,11 (2H, т, J=7,2 Гц), 4,51-4,61 (1H, м), 6,72-6,76 (2H, м), 6,80-6,81 (1H, м), 6,94-6,95 (1H, м).
ESI-MS: m/z= 351 (M+H)+.
[0371]
(Пример 41) Синтез гидрохлорида пропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 94]
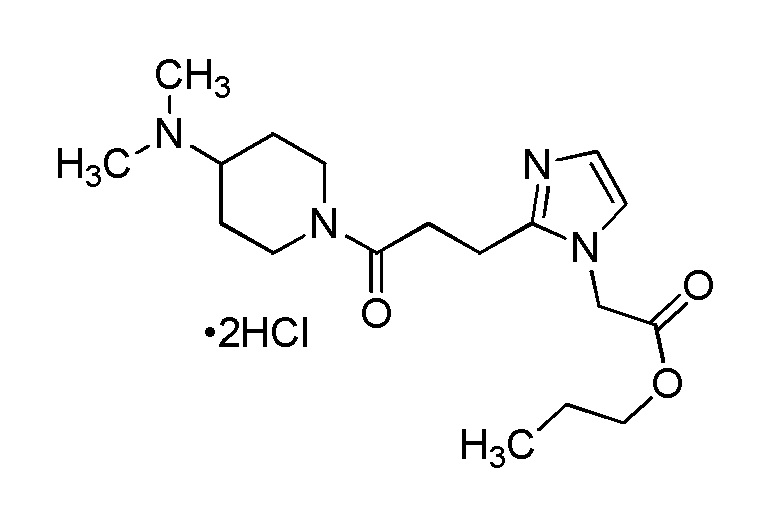
К раствору пропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0499 г, 0,142 ммоль) в диэтиловом эфире (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,157 мл, 0,314 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (4,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида пропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0552 г, 0,130 ммоль, 91%) (здесь и далее указывается как «соединение по примеру 41») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 0,91 (3H, т, J=7,2 Гц), 1,52-1,75 (4H, м), 2,08-2,24 (2H, м), 2,68-2,76 (1H, м), 2,86 (6H, с), 2,99-3,06 (2H, м), 3,13-3,26 (3H, м), 3,45-3,60 (1H, м), 4,02-4,12 (1H, м), 4,18-4,24 (2H, м), 4,48-4,58 (1H, м), 5,21 (2H, с), 7,39-7,43 (2H, м).
ESI-MS: в виде пропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 351 (M+H)+.
[0372]
(Пример 42) Синтез изопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 95]
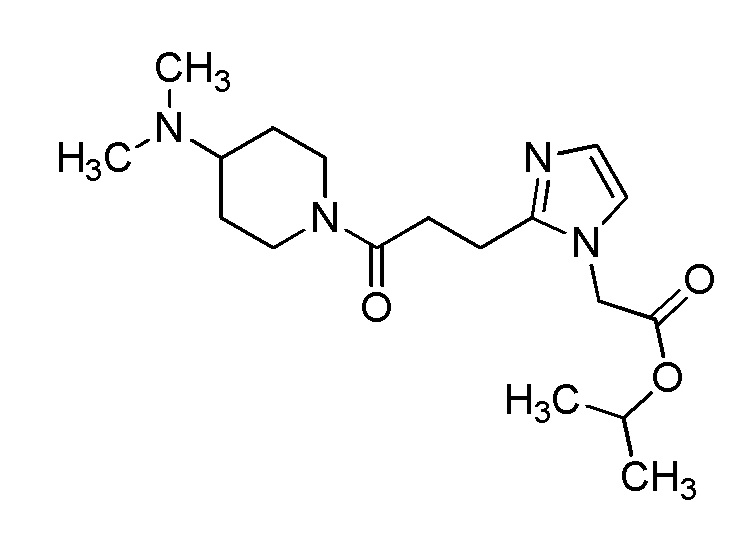
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в хлороформе (6,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,142 мл, 0,811 ммоль), HBTU (0,184 г, 0,486 ммоль) и пропан-2-ол (0,0620 мл, 0,811 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением изопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0610 г, 0,174 ммоль, 54%) (здесь и далее указывается как «соединение по примеру 42») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,22-1,38 (8H, м), 1,76-1,88 (2H, м), 2,24-2,38 (7H, м), 2,52-2,62 (1H, м), 2,88-3,02 (5H, м), 3,94-4,04 (1H, м), 4,52-4,62 (1H, м), 4,69 (2H, с), 5,00-5,10 (1H, м), 6,78-6,82 (1H, м), 6,92-6,96 (1H, м).
ESI-MS: m/z= 351 (M+H)+.
[0373]
(Пример 43) Синтез гидрохлорида изопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 96]
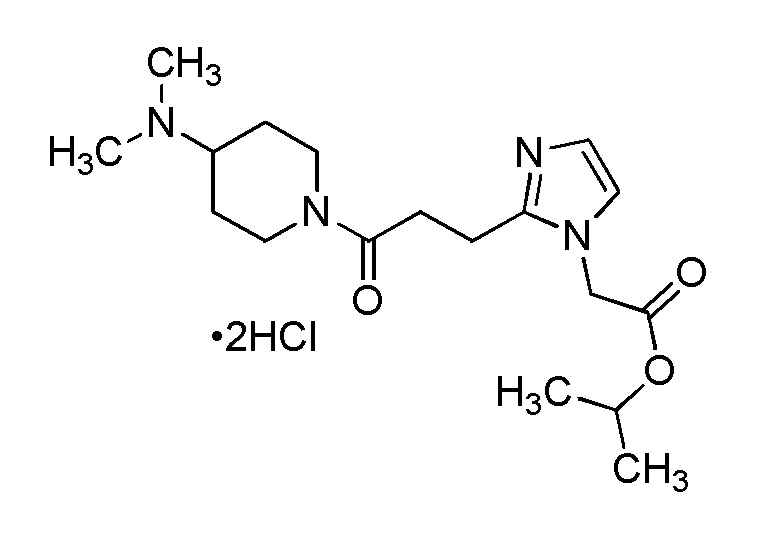
К раствору изопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0600 г, 0,171 ммоль) в диэтиловом эфире (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,188 мл, 0,376 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (4,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида изопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0359 г, 0,0928 ммоль, 54%) (здесь и далее указывается как «соединение по примеру 43») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,30 (6H, д, J=6,4 Гц), 1,52-1,76 (2H, м), 2,10-2,22 (2H, м), 2,68-2,78 (1H, м), 2,87 (6H, с), 2,98-3,05 (2H, м), 3,14-3,24 (3H, м), 3,46-3,56 (1H, м), 4,04-4,14 (1H, м), 4,50-4,57 (1H, м), 5,08-5,18 (3H, м), 7,36-7,42 (2H, м).
ESI-MS: в виде изопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 351 (M+H)+.
[0374]
(Пример 44) Синтез циклопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 97]
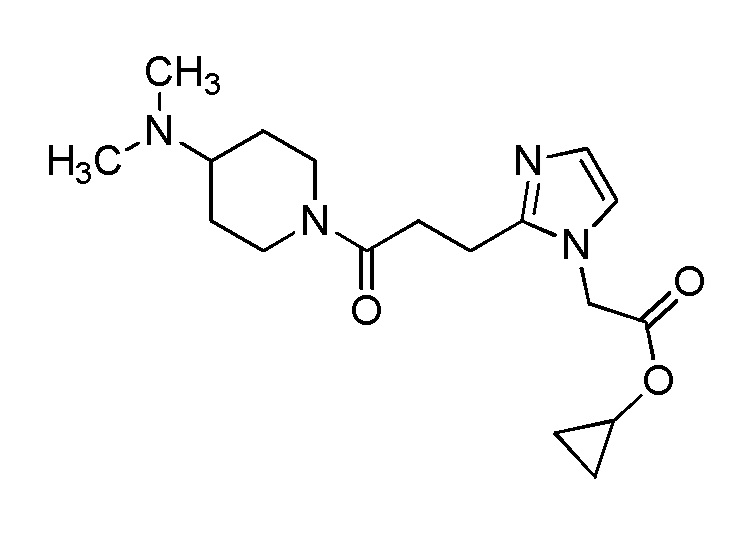
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в хлороформе (6,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,142 мл, 0,811 ммоль), HBTU (0,184 г, 0,486 ммоль) и сырой циклопропанол (0,0470 г, 0,811 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли соляную кислоту (1,0 н), и затем реакционную жидкость вновь экстрагировали. В полученный водный слой для нейтрализации добавляли насыщенный водный раствор гидрокарбоната натрия, и затем полученную смесь экстрагировали хлороформом. Органический слой промывали 10%-ным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением циклопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0610 г, 0,175 ммоль, 54%) (здесь и далее указывается как «соединение по примеру 44») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 0,70-0,78 (4H, м), 1,25-1,45 (2H, м), 1,78-1,87 (2H, м), 2,25-2,38 (7H, м), 2,52-2,62 (1H, м), 2,88-3,05 (5H, м), 3,94-4,04 (1H, м), 4,18-4,26 (1H, м), 4,54-4,62 (1H, м), 4,73 (2H, с), 6,80 (1H, ушир. с), 6,96 (1H, ушир. с).
ESI-MS: m/z= 349 (M+H)+.
[0375]
(Пример 45) Синтез гидрохлорида циклопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 98]
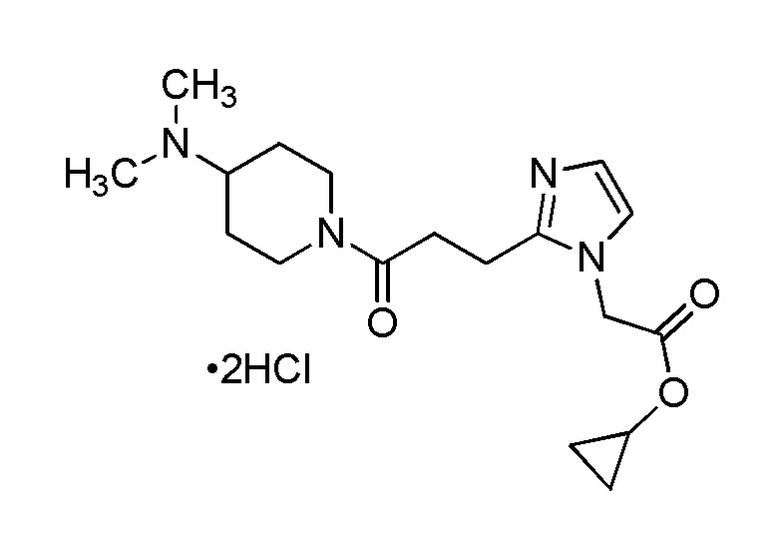
К раствору циклопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0610 г, 0,175 ммоль) в диэтиловом эфире (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,189 мл, 0,378 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (4,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида циклопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0540 г, 0,128 ммоль, 73%) (здесь и далее указывается как «соединение по примеру 45») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 0,78-0,85 (4H, м), 1,53-1,80 (2H, м), 2,10-2,24 (2H, м), 2,68-2,90 (7H, м), 2,99-3,06 (2H, м), 3,13-3,27 (3H, м), 3,45-3,60 (1H, м), 4,04-4,14 (1H, м), 4,22-4,28 (1H, м), 4,50-4,58 (1H, м), 5,19 (2H, с), 7,38-7,45 (2H, м).
ESI-MS: в виде циклопропил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 349 (M+H)+.
[0376]
(Пример 46) Синтез бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 99]
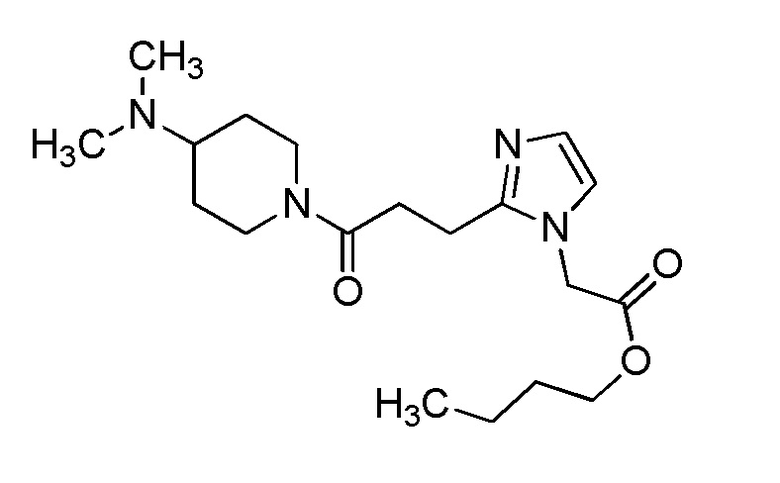
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,0500 г, 0,162 ммоль) в хлороформе (3,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,0710 мл, 0,405 ммоль), HBTU (0,0920 г, 0,243 ммоль) и бутан-1-ол (0,0370 мл, 0,405 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0314 г, 0,0861 ммоль, 53%) (здесь и далее указывается как «соединение по примеру 46») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 0,92 (3H, т, J=7,2 Гц), 1,27-1,42 (4H, м), 1,57-1,65 (2H, м), 1,76-1,86 (2H, м), 2,22-2,34 (7H, м), 2,53-2,62 (1H, м), 2,88-3,03 (5H, м), 3,93-4,02 (1H, м), 4,15 (2H, т, J=6,4 Гц), 4,52-4,60 (1H, м), 4,74 (2H, с), 6,76-6,80 (1H, м), 6,95-6,96 (1H, м).
ESI-MS: m/z= 365 (M+H)+.
[0377]
(Пример 47) Синтез гидрохлорида бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 100]
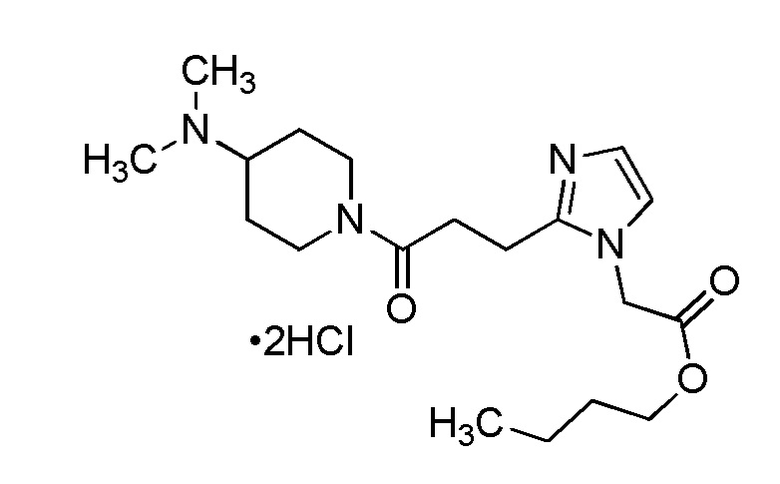
К раствору бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0600 г, 0,165 ммоль) в диэтиловом эфире (2,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,182 мл, 0,364 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (8,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0554 г, 0,127 ммоль, 77%) (здесь и далее указывается как «соединение по примеру 47») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 0,89 (3H, т, J=7,2 Гц), 1,28-1,40 (2H, м), 1,52-1,75 (4H, м), 2,10-2,20 (2H, м), 2,66-2,76 (1H, м), 2,86 (6H, с), 2,96-3,04 (2H, м), 3,11-3,22 (3H, м), 3,45-3,56 (1H, м), 4,34-4,43 (1H, м), 4,26 (2H, т, J=6,0 Гц), 4,49-4,58 (1H, м), 5,15 (2H, с), 7,26-7,38 (2H, м).
ESI-MS: в виде бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 365 (M+H)+.
[0378]
(Пример 48) Синтез изобутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 101]
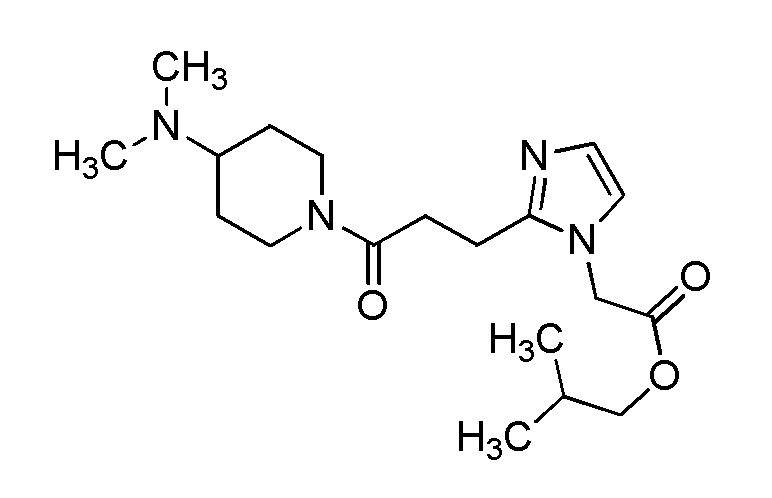
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в хлороформе (6,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,142 мл, 0,811 ммоль), HBTU (0,184 г, 0,486 ммоль) и 2-метилпропан-1-ол (0,0760 мл, 0,811 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением изобутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,101 г, 0,277 ммоль, 86%) (здесь и далее указывается как «соединение по примеру 48») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 0,85-0,89 (6H, м), 1,20-1,36 (2H, м), 1,74-1,95 (3H, м), 2,22-2,34 (7H, м), 2,48-2,58 (1H, м), 2,86-3,00 (5H, м), 3,88-3,98 (3H, м), 4,50-4,57 (1H, м), 4,73 (2H, с), 6,76-6,80 (1H, м), 6,90-6,94 (1H, м).
ESI-MS: m/z= 365 (M+H)+.
[0379]
(Пример 49) Синтез гидрохлорида изобутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 102]
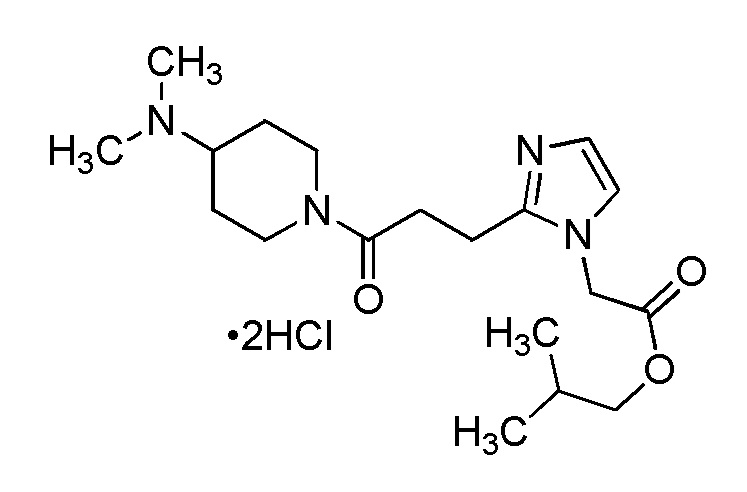
К раствору изобутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,100 г, 0,274 ммоль) в диэтиловом эфире (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,302 мл, 0,604 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (4,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида изобутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0709 г, 0,177 ммоль, 65%) (здесь и далее указывается как «соединение по примеру 49») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 0,88-0,94 (6H, м), 1,52-1,77 (2H, м), 1,92-2,02 (1H, м), 2,12-2,22 (2H, м), 2,68-2,78 (1H, м), 2,86 (6H, с), 2,98-3,05 (2H, м), 3,12-3,28 (3H, м), 3,46-3,56 (1H, м), 4,32-4,42 (3H, м), 4,48-4,58 (1H, м), 5,23 (2H, с), 7,40-7,44 (2H, м).
ESI-MS: в виде изобутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 365 (M+H)+.
[0380]
(Пример 50) Синтез циклопропилметил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 103]
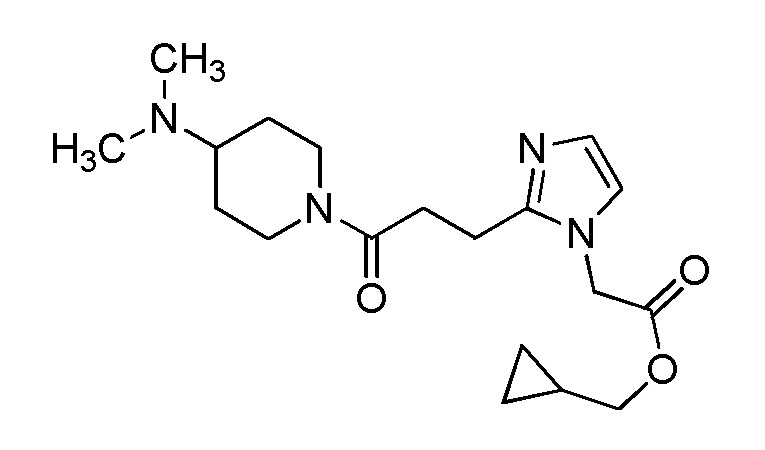
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в хлороформе (6,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,142 мл, 0,811 ммоль), HBTU (0,184 г, 0,486 ммоль) и циклопропил метанол (0,0580 г, 0,811 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением циклопропилметил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0720 г, 0,199 ммоль, 61%) (здесь и далее указывается как «соединение по примеру 50») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 0,23-0,29 (2H, м), 0,53-0,60 (2H, м), 1,05-1,16 (1H, м), 1,20-1,40 (2H, м), 1,74-1,85 (2H, м), 2,20-2,34 (7H, м), 2,50-2,60 (1H, м), 2,87-3,02 (5H, м), 3,93-4,00 (3H, м), 4,52-4,60 (1H, м), 4,75 (2H, с), 6,78-6,82 (1H, м), 6,92-6,96 (1H, м).
ESI-MS: m/z= 363 (M+H)+.
[0381]
(Пример 51) Синтез гидрохлорида циклопропилметил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 104]
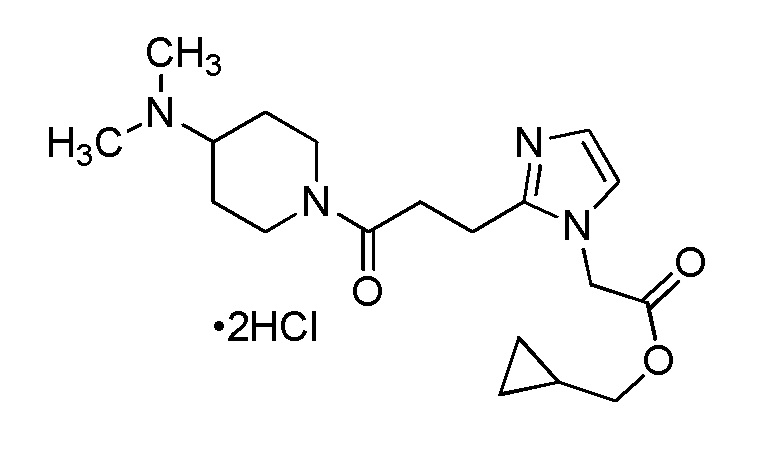
К раствору циклопропилметил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0720 г, 0,199 ммоль) в диэтиловом эфире (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,218 мл, 0,436 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (4,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида циклопропилметил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0652 г, 0,180 ммоль, 90%) (здесь и далее указывается как «соединение по примеру 51») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 0,30-0,35 (2H, м), 0,58-0,65 (2H, м), 1,15-1,25 (1H, м), 1,52-1,75 (2H, м), 2,10-2,20 (2H, м), 2,67-2,76 (1H, м), 2,86 (6H, с), 2,99-3,06 (2H, м), 3,13-3,25 (3H, м), 3,44-3,56 (1H, м), 4,05-4,12 (3H, м), 4,49-4,57 (1H, м), 5,20 (2H, с), 7,39-7,41 (2H, м).
ESI-MS: в виде циклопропилметил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 363 (M+H)+.
[0382]
(Пример 52) Синтез втор-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 105]
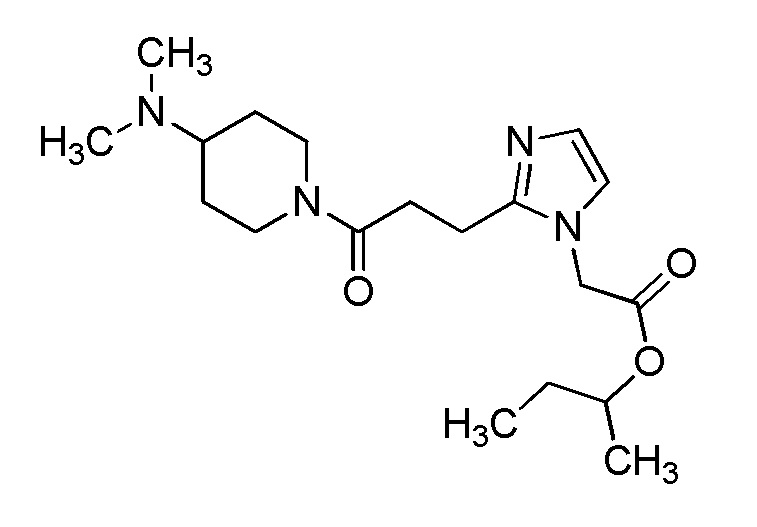
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в хлороформе (6,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,142 мл, 0,811 ммоль), HBTU (0,184 г, 0,486 ммоль) и бутан-2-ол (0,0740 мл, 0,811 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 60 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением втор-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0700 г, 0,192 ммоль, 59%) (здесь и далее указывается как «соединение по примеру 52») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 0,87 (3H, т, J=7,6 Гц), 1,20-1,40 (5H, м), 1,50-1,65 (2H, м), 1,78-1,86 (2H, м), 2,23-2,35 (7H, м), 2,54-2,62 (1H, м), 2,88-3,04 (5H, м), 3,95-4,02 (1H, м), 4,54-4,60 (1H, м), 4,72 (2H, с), 4,85-4,95 (1H, м), 6,81 (1H, ушир. с), 6,96 (1H, ушир. с).
ESI-MS: m/z= 365 (M+H)+.
[0383]
(Пример 53) Синтез гидрохлорида втор-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 106]
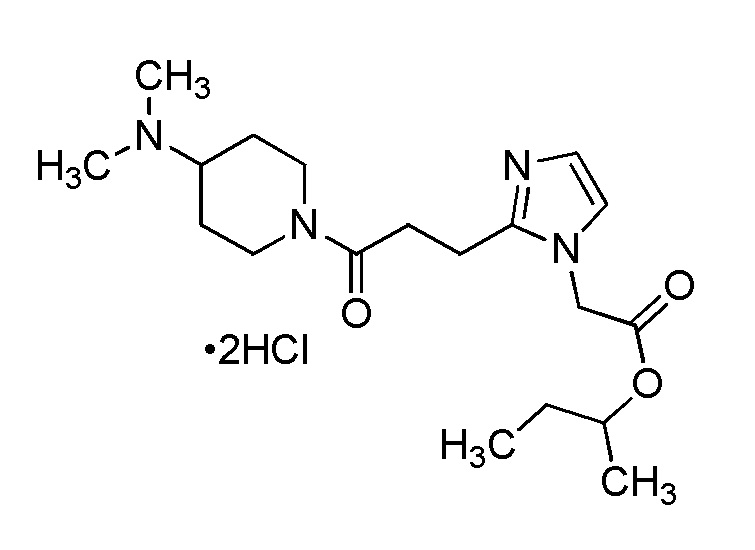
К раствору втор-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0700 г, 0,192 ммоль) в диэтиловом эфире (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,211 мл, 0,423 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (4,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида втор-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0582 г, 0,133 ммоль, 69%) (здесь и далее указывается как «соединение по примеру 53») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 0,87 (3H, т, J=7,6 Гц), 1,27 (3H, д, J=6,0 Гц). 1,50-1,76 (4H, м), 2,66-2,75 (2H, м), 2,80-2,90 (7H, м), 2,98-3,05 (2H, м), 3,13-3,25 (3H, м), 3,45-3,57 (1H, м), 4,03-4,12 (1H, м), 4,48-4,58 (1H, м), 4,93-5,00 (1H, м), 5,19 (2H, с), 7,37-7,42 (2H, м).
ESI-MS: в виде втор-бутил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 365 (M+H)+.
[0384]
(Пример 54) Синтез октил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 107]
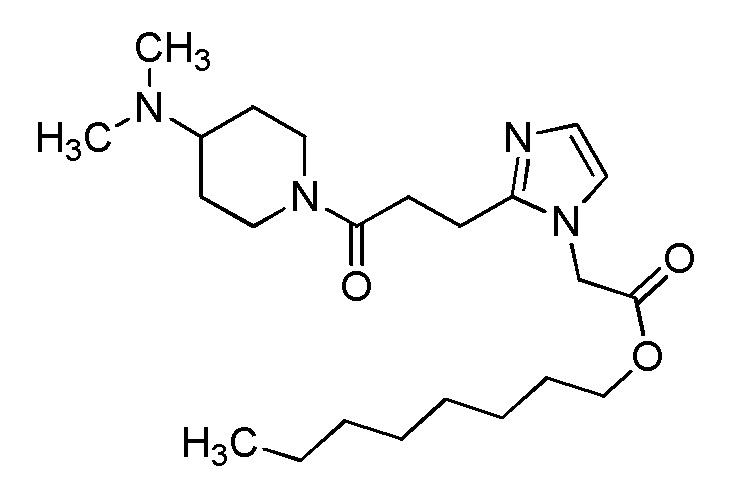
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в хлороформе (3,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,113 мл, 0,649 ммоль), HBTU (0,184 г, 0,486 ммоль) и октан-1-ол (0,103 мл, 0,649 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением октил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0602 г, 0,143 ммоль, 44%) (здесь и далее указывается как «соединение по примеру 54») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 0,85-0,93 (3H, м), 1,23-1,43 (12H, м), 1,58-1,68 (2H, м), 1,77-1,87 (2H, м), 2,25-2,40 (7H, м), 2,54-2,64 (1H, м), 2,88-3,04 (5H, м), 3,94-4,04 (1H, м), 4,12-4,17 (2H, м), 4,53-4,65 (1H, м), 4,74 (2H, с), 6,80-6,83 (1H, м), 6,94-6,98 (1H, м).
ESI-MS: m/z= 421 (M+H)+.
[0385]
(Пример 55) Синтез гидрохлорида октил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 108]
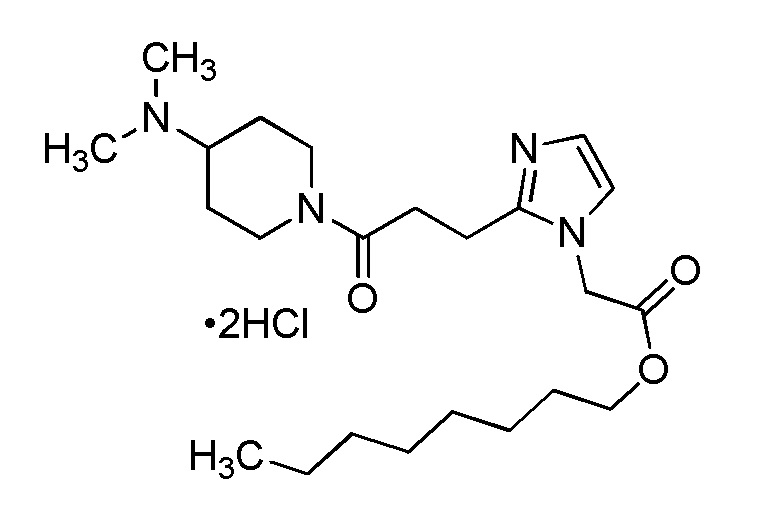
К раствору октил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0400 г, 0,0950 ммоль) в диэтиловом эфире (2,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,105 мл, 0,209 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (8,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида октил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0109 г, 0,0221 ммоль, 23%) (здесь и далее указывается как «соединение по примеру 55») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 0,84-0,89 (3H, м), 1,14-1,37 (10H, м), 1,54-1,76 (4H, м), 2,10-2,22 (2H, м), 2,65-2,77 (1H, м), 2,87 (6H, с), 3,00-3,05 (2H, м), 3,13-3,28 (3H, м), 3,46-3,58 (1H, м), 4,03-4,11 (1H, м), 4,26 (2H, т, J=6,8 Гц), 4,49-4,57 (1H, м), 5,21 (2H, с), 7,40-7,44 (2H, м).
ESI-MS: в виде октил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 421 (M+H)+.
[0386]
(Пример 56) Синтез 2,3-дигидро-1H-инден-5-ил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 109]
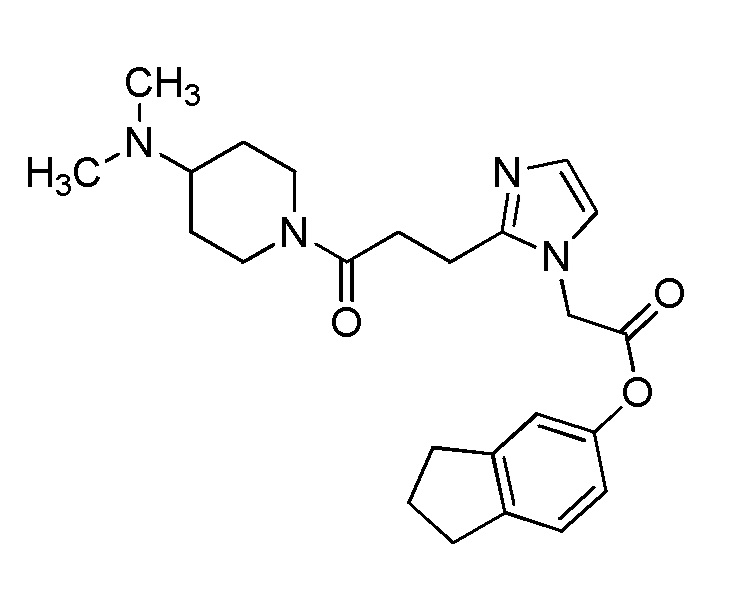
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в хлороформе (3,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,113 мл, 0,649 ммоль), HBTU (0,184 г, 0,486 ммоль) и 2,3-дигидро-1H-инден-5-ол (0,0870 г, 0,649 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли соляную кислоту (1,0 н), и затем реакционную жидкость вновь экстрагировали. В полученный водный слой для нейтрализации добавляли насыщенный водный раствор гидрокарбоната натрия, и затем полученную смесь экстрагировали хлороформом. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением 2,3-дигидро-1H-инден-5-ил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0644 г, 0,152 ммоль, 47%) (здесь и далее указывается как «соединение по примеру 56») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,19-1,42 (2H, м), 1,76-1,87 (2H, м), 2,02-2,14 (2H, м), 2,24-2,40 (7H, м), 2,50-2,64 (1H, м), 2,83-3,03 (9H, м), 3,93-4,03 (1H, м), 4,53-4,62 (1H, м), 4,98-5,03 (2H, м), 6,82-7,02 (4H, м), 7,18 (1H, д, J=8,0 Гц).
ESI-MS: m/z= 425 (M+H)+.
[0387]
(Пример 57) Синтез гидрохлорида 2,3-дигидро-1H-инден-5-ил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 110]
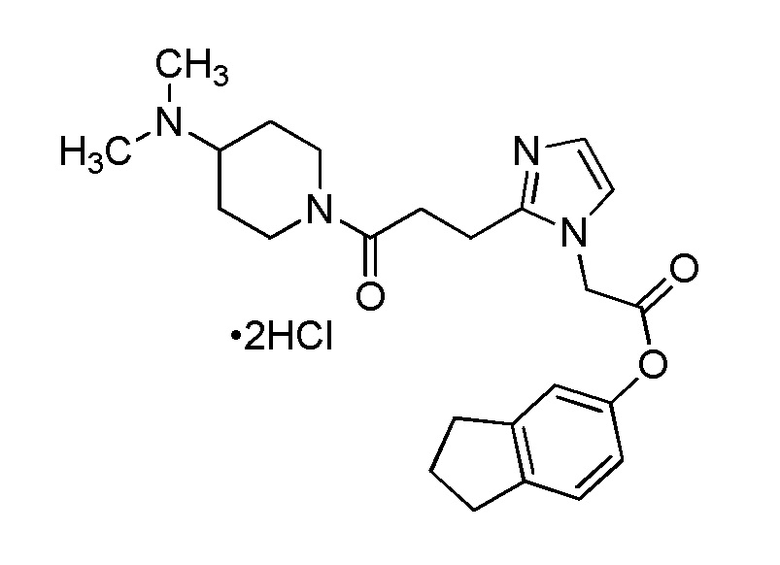
К раствору 2,3-дигидро-1H-инден-5-ил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0500 г, 0,118 ммоль) в диэтиловом эфире (2,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,130 мл, 0,259 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (8,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида 2,3-дигидро-1H-инден-5-ил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0475 г, 0,0955 ммоль, 81%) (здесь и далее указывается как «соединение по примеру 57») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,30-1,62 (2H, м), 1,94-2,16 (4H, м), 2,58-2,78 (7H, м), 2,87-3,15 (7H, м), 3,25-3,50 (3H, м), 3,97-4,05 (1H, м), 4,43-4,50 (1H, м), 5,50 (2H, с), 6,97-7,02 (1H, м), 7,11-7,14 (1H, м), 7,33-7,37 (1H, м), 7,43-7,51 (2H, м).
ESI-MS: в виде 2,3-дигидро-1H-инден-5-ил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 425 (M+H)+.
[0388]
(Пример 58) Синтез (2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетокси)метил пивалата:
[Формула 111]
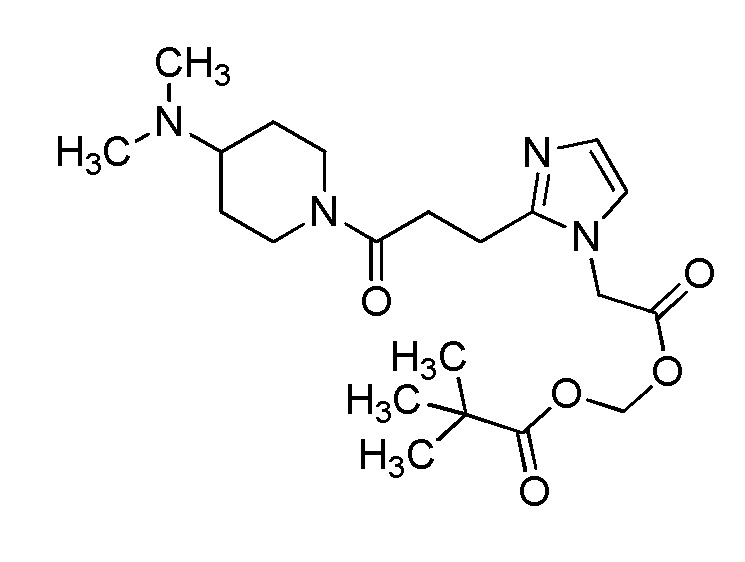
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,0980 г, 0,276 ммоль) в ДМФ (2,0 мл) при комнатной температуре добавляли карбонат калия (0,0760 г, 0,553 ммоль), хлорметил пивалат (0,0400 мл, 0,276 ммоль) и йодид натрия (0,0414 г, 0,276 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 12 часов. В реакционную жидкость добавляли этилацетат, и осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении. К остатку добавляли этилацетата, и осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении. К остатку добавляли хлороформ, и осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением (2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетокси)метил пивалата (0,0300 г, 0,0710 ммоль, 26%) (здесь и далее указывается как «соединение по примеру 58») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,19-1,45 (11H, м), 1,75-1,90 (2H, м), 2,22-2,40 (7H, м), 2,53-2,63 (1H, м), 2,88-3,02 (5H, м), 3,92-4,02 (1H, м), 4,52-4,62 (1H, м), 4,84 (2H, с), 5,81 (2H, с), 6,81 (1H, ушир. с), 6,97 (1H, ушир. с).
ESI-MS: m/z= 423 (M+H)+.
[0389]
(Пример 59) Синтез (5-метил-2-оксо-1,3-диоксол-4-ил)метил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 112]
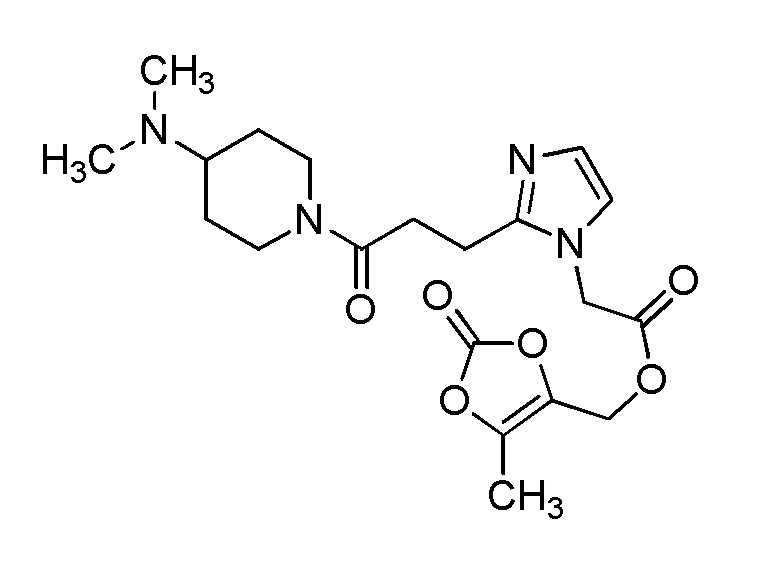
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в хлороформе (3,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,113 мл, 0,649 ммоль), HBTU (0,184 г, 0,486 ммоль) и сырой 4-(гидроксиметил)-5-метил-1,3-диоксол-2-он (0,0840 г, 0,649 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли соляную кислоту (1,0 н), и затем реакционную жидкость вновь экстрагировали. В полученный водный слой для нейтрализации добавляли насыщенный водный раствор гидрокарбоната натрия, и затем полученную смесь экстрагировали хлороформом. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением (5-метил-2-оксо-1,3-диоксол-4-ил)метил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0501 г, 0,119 ммоль, 37%) (здесь и далее указывается как «соединение по примеру 59») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,16-1,44 (2H, м), 1,77-1,90 (2H, м), 2,16 (3H, с), 2,20-2,42 (7H, м), 2,52-2,62 (1H, м), 2,86-3,04 (5H, м), 3,92-4,04 (1H, м), 4,50-4,62 (1H, м), 4,84 (2H, с), 4,92 (2H, с), 6,78-6,83 (1H, м), 6,95-6,98 (1H, м).
ESI-MS: m/z= 421 (M+H)+.
[0390]
(Пример 60) Синтез 2-(диметиламино)-2-оксоэтил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 113]
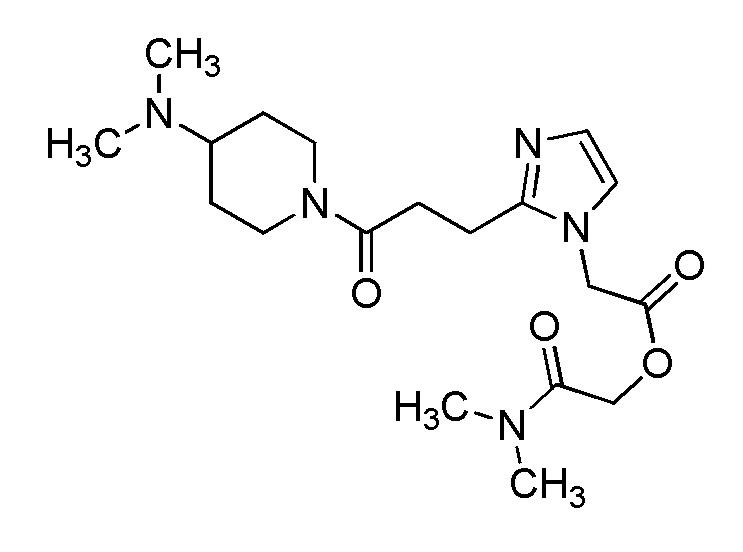
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в хлороформе (6,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,142 мл, 0,811 ммоль), HBTU (0,184 г, 0,486 ммоль) и сырой 2-гидрокси-N,N-диметилацетамид (0,0500 г, 0,486 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли соляную кислоту (1,0 н), и затем реакционную жидкость вновь экстрагировали. В полученный водный слой для нейтрализации добавляли насыщенный водный раствор гидрокарбоната натрия, и затем полученную смесь экстрагировали хлороформом. Органический слой промывали 10%-ным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением 2-(диметиламино)-2-оксоэтил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,100 г, 0,254 ммоль, 78%) (здесь и далее указывается как «соединение по примеру 60») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,24-1,44 (2H, м), 1,78-1,86 (2H, м), 2,25-2,40 (7H, м), 2,52-2,62 (1H, м), 2,78-3,05 (12H, м), 3,95-4,05 (1H, м), 4,79 (2H, м), 4,90-4,94 (2H, м), 6,86-6,88 (1H, м), 6,94-6,96 (1H, м).
ESI-MS: m/z= 394 (M+H)+.
[0391]
(Пример 61) Синтез гидрохлорида 2-(диметиламино)-2-оксоэтил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 114]
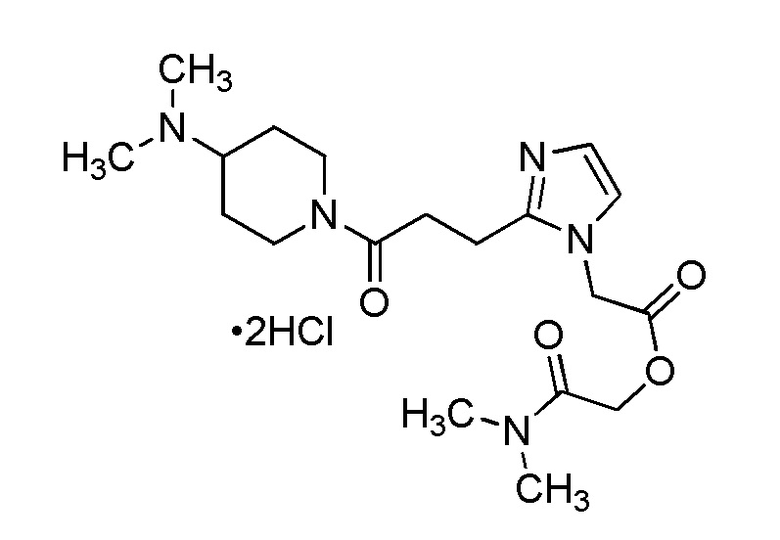
К раствору 2-(диметиламино)-2-оксоэтил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,100 г, 0,254 ммоль) в диэтиловом эфире (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,188 мл, 0,376 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (4,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида 2-(диметиламино)-2-оксоэтил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0455 г, 0,0976 ммоль, 38%) (здесь и далее указывается как «соединение по примеру 61») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,54-1,78 (2H, м), 2,10-2,23 (2H, м), 2,67-2,80 (1H, м), 2,85-3,05 (14H, м), 3,13-3,28 (3H, м), 3,47-3,57 (1H, м), 4,05-4,15 (1H, м), 4,50-4,60 (1H, м), 5,05 (2H, с), 5,36 (2H, м), 7,40-7,46 (2H, м).
ESI-MS: в виде 2-(диметиламино)-2-оксоэтил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата: m/z= 394 (M+H)+.
[0392]
(Пример 62) Синтез 3-оксо-2,3-дигидроизобензофуран-2-ил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата:
[Формула 115]
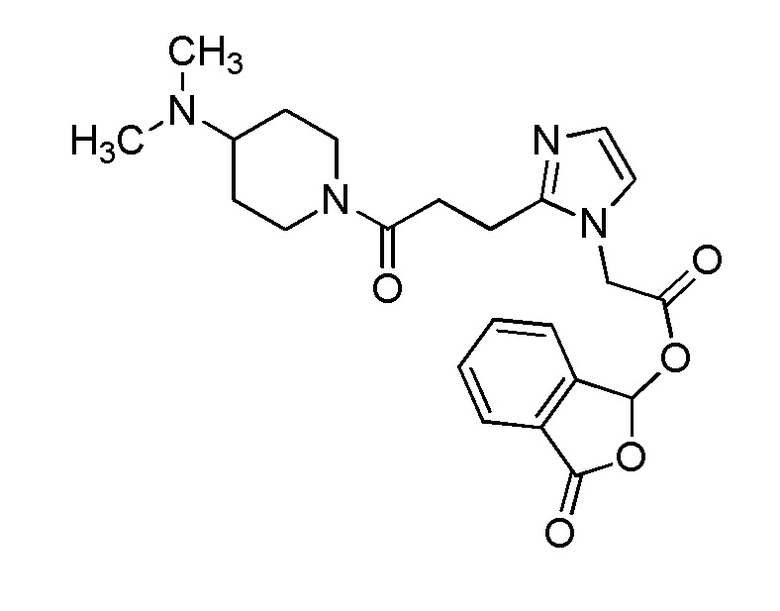
К раствору 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)уксусной кислоты (0,100 г, 0,324 ммоль) в ДМФ (3,2 мл) при комнатной температуре добавляли карбонат калия (0,0900 г, 0,649 ммоль), 3-бромфталид (0,0691 г, 0,324 ммоль) и йодид натрия (0,0486 г, 0,324 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 14 часов. В реакционную жидкость добавляли этилацетат, и осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении. К остатку добавляли этилацетат, и осадок фильтровали и собирали с получением 3-оксо-2,3-дигидроизобензофуран-2-ил 2-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)ацетата (0,0320 г, 0,0726 ммоль, 22%) (здесь и далее указывается как «соединение по примеру 62») в виде твердого вещества коричневого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ: 1,10-1,38 (2H, м), 1,65-1,80 (2H, м), 2,15 (6H, с), 2,22-2,36 (1H, м), 2,45-2,81 (5H, м), 2,90-3,03 (1H, м), 3,85-3,93 (1H, м), 4,15 (2H, с), 4,30-4,40 (1H, м), 6,62 (1H, с), 6,85 (1H, с), 7,36 (1H, т, J=7,2 Гц), 7,45-7,55 (2H, м), 7,71 (1H, д, J=7,2 Гц), 10,64 (1H, с).
[0393]
(Пример 63) Синтез этил 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата:
[Формула 116]
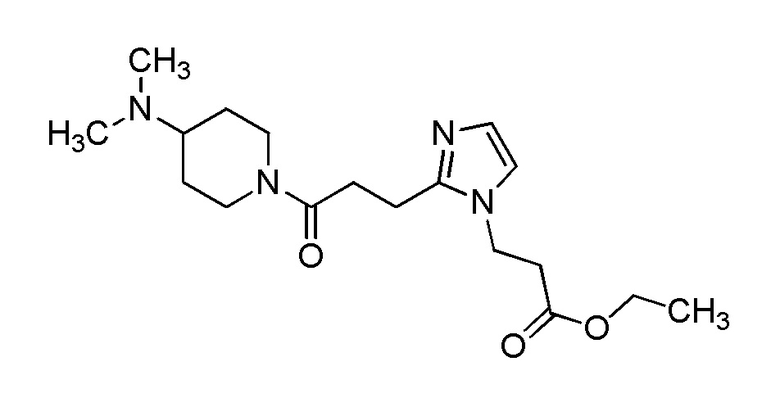
К раствору сырой 3-(1-(3-этокси-3-оксопропил)-1H-имидазол-2-ил)пропановой кислоты (0,330 г, 0,931 ммоль) в хлороформе (10,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,651 мл, 3,73 ммоль), HBTU (0,883 г, 2,33 ммоль) и 4-диметиламинопиперидин (0,208 мл, 1,77 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением этил 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата (0,172 г, 0,491 ммоль, 53%) (здесь и далее указывается как «соединение по примеру 63») в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,21-1,45 (5H, м), 1,78-1,88 (2H, м), 2,25-2,38 (7H, м), 2,55-2,64 (1H, м), 2,74 (2H, т, J=7,2 Гц), 2,90-3,05 (5H, м), 3,98-4,18 (3H, м), 4,24 (2H, т, J=7,2 Гц), 4,56-4,65 (1H, м), 6,84-6,86 (1H, м), 6,90-6,92 (1H, м).
ESI-MS: m/z= 351 (M+H)+.
[0394]
(Пример 64) Синтез гидрохлорида этил 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата:
[Формула 117]
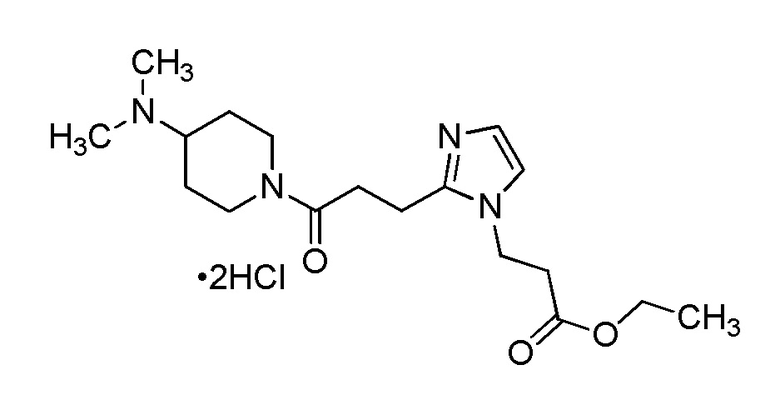
К раствору этил 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата (0,0500 г, 0,143 ммоль) в диэтиловом эфире (1,0 мл) при температуре 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 0,214 мл, 0,428 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 30 минут. Выпавший в осадок твердый продукт фильтровали и собирали, и промывали диэтиловым эфиром (4,0 мл), и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида этил 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата (0,0541 г, 0,128 ммоль, 89%) (здесь и далее указывается как «соединение по примеру 64») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,19-1,25 (3H, м), 1,53-1,80 (2H, м), 2,10-2,23 (2H, м), 2,67-2,78 (1H, м), 2,87 (6H, с), 3,00-3,10 (4H, м), 3,15-3,34 (3H, м), 3,47-3,57 (1H, м), 4,07-4,20 (3H, м), 4,45-4,58 (3H, м), 7,32-7,36 (1H, м), 7,42-7,45 (1H, м).
ESI-MS: в виде этил 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата: m/z= 351 (M+H)+.
[0395]
(Пример 65) Синтез 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты:
[Формула 118]
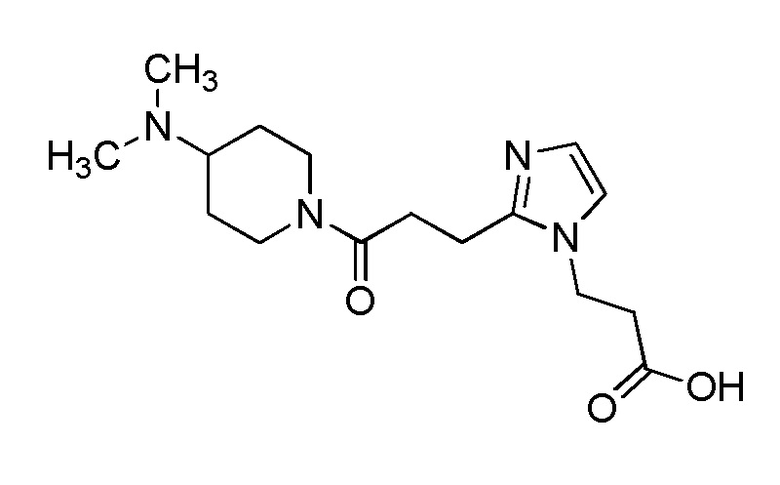
К раствору этил 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата (0,120 г, 0,342 ммоль) в этаноле (3,0 мл) при комнатной температуре добавляли водный раствор гидроксида натрия (1,0 н, 0,377 мл, 0,377 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 4 часов. Реакционную жидкость охлаждали до температуры 0°C, и в реакционную жидкость для нейтрализации добавляли соляную кислоту (1,0 н), и затем реакционную жидкость концентрировали при пониженном давлении. Остаток подвергали азеотропной отгонке с толуолом и добавляли этанол. Осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты (0,105 г, 0,326 ммоль, 95%) (здесь и далее указывается как «соединение по примеру 65») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ: 1,34-1,47 (1H, м), 1,54-1,69 (1H, м), 1,95-2,08 (2H, м), 2,60-2,68 (8H, м), 2,75-3,05 (7H, м), 3,96-4,06 (1H, м), 4,21 (2H, т, J=6,8 Гц), 4,42-4,51 (1H, м), 7,13 (1H, ушир. с), 7,33 (1H, ушир. с).
ESI-MS: m/z= 323 (M+H)+.
[0396]
(Пример 66) Синтез гидрохлорида 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты:
[Формула 119]
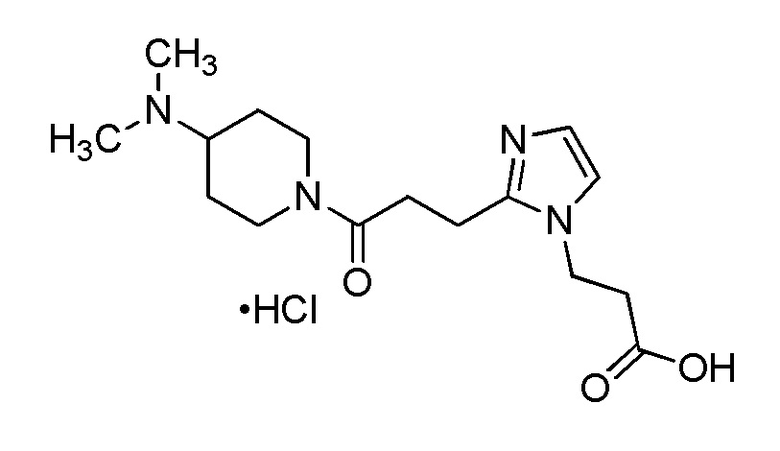
К раствору 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты (0,105 г, 0,326 ммоль) в воде (1,0 мл) при температуре 0°C добавляли соляную кислоту (1,0 н, 1,02 мл, 1,02 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 2 часов. Реакционную жидкость концентрировали при пониженном давлении, и выпавший в осадок твердый продукт фильтровали и собирали с получением гидрохлорида 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты (0,0418 г, 0,116 ммоль, 36%) (здесь и далее указывается как «соединение по примеру 66») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,53-1,80 (2H, м), 2,12-2,24 (2H, м), 2,68-2,78 (1H, м), 2,87 (6H, с), 2,98-3,34 (7H, м), 3,45-3,58 (1H, м), 4,07-4,17 (1H, м), 4,43-4,58 (3H, м), 7,34 (1H, ушир. с), 7,43 (1H, ушир. с).
ESI-MS: в виде 3-(2-(3-(4-(диметиламино)пиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты: m/z= 323 (M+H)+.
[0397]
(Пример 67) Синтез этил 3-(2-(3-((S)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата:
[Формула 120]
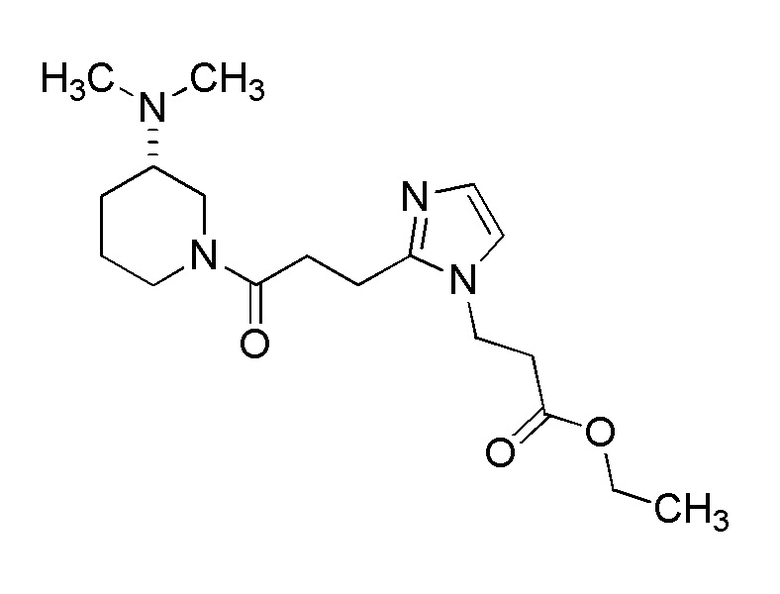
К раствору сырой 3-(1-(3-этокси-3-оксопропил)-1H-имидазол-2-ил)пропановой кислоты (0,100 г, 0,420 ммоль) в дихлорметане (10,0 мл) при комнатной температуре добавляли диизопропилэтиламин (0,0810 г, 0,620 ммоль), HBTU (0,189 г, 0,500 ммоль) и (S)-3-диметиламинопиперидин (0,0530 г, 0,420 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 12 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, гексан/этилацетата и хлороформ/метанол) с получением этил 3-(2-(3-((S)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата (0,120 г, 0,340 ммоль, 82%) (здесь и далее указывается как «соединение по примеру 67») в виде масла желтого цвета
1H-ЯМР (400 МГц, CDCl3) δ: 1,24 (3H, т, J=7,2 Гц), 1,36-1,50 (2H, м), 1,70-1,85 (1H, м), 1,95-2,05 (1H, м), 2,07-2,23 (2H, м), 2,32 (6H, с), 2,43-2,60 (1H, м), 2,73-2,78 (2H, м), 2,81-3,03 (5H, м), 3,83-4,06 (1H, м), 4,14 (2H, кв, J=7,2 Гц), 4,22-4,28 (2H, м), 4,47-4,68 (1H, м), 6,84-6,88 (1H, м), 6,91 (1H, д, J=1,2 Гц).
ESI-MS: m/z= 351 (M+H)+.
[0398]
(Пример 68) Синтез гидрохлорида 3-(2-(3-((S)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты:
[Формула 121]
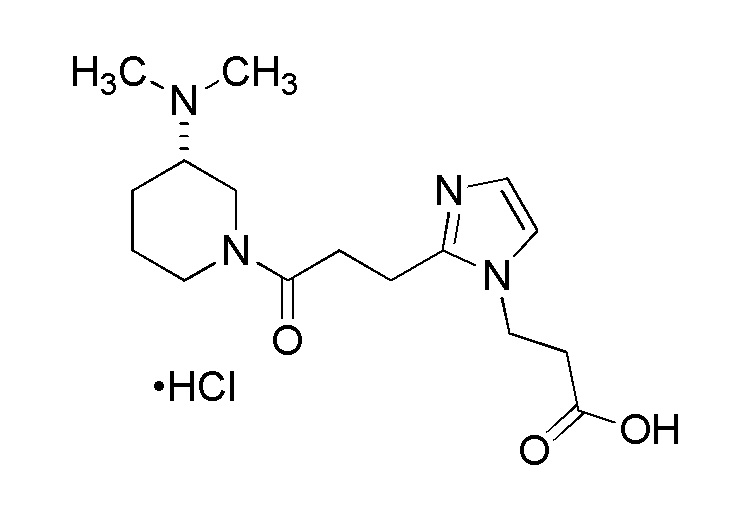
К раствору этил 3-(2-(3-((S)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата (0,062 г, 0,175 ммоль) в воде (5,0 мл) при температуре 0°C добавляли водный раствор гидроксида натрия (1,0 н, 0,351 мл, 0,351 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 2 часов. Добавляли соляную кислоту (1,0 н, 0,526 мл, 0,526 ммоль) добавляли, и затем полученную смесь концентрировали и высушивали. Полученный твердый продукт промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Полученный твердый продукт опять промывали этанолом и фильтровали, и затем фильтрат концентрировали и высушивали. Остаток сушили при пониженном давлении с получением гидрохлорида 3-(2-(3-((S)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты (0,0430 г, 0,120 ммоль, 68%) (здесь и далее указывается как «соединение по примеру 68») в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,02-1,87 (3H, м), 2,00-2,15 (1H, м), 2,55-2,95 (7H, м), 3,05-3,30 (7H, м), 3,47-3,62 (1H, м), 3,95-4,20 (1H, м), 4,99 (2H, с), 7,26 (2H, с).
ESI-MS: в виде 3-(2-(3-((S)-3-диметиламинопиперидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты: 323 (M+H)+.
[0399]
(Пример 69) Синтез этил (S)-3-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата:
[Формула 122]
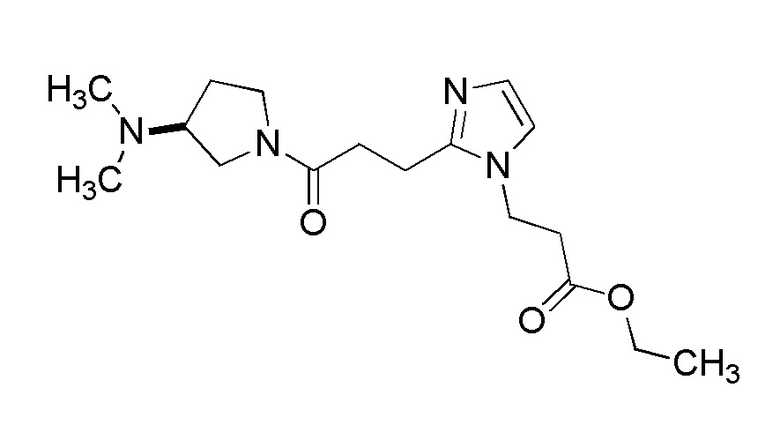
К раствору сырой 3-(1-(3-этокси-3-оксопропил)-1H-имидазол-2-ил)пропановой кислоты (0,0800 г, 0,333 ммоль) в дихлорметане (1,6 мл) при комнатной температуре добавляли диизопропилэтиламин (0,0870 мл, 0,499 ммоль), HBTU (0,152 г, 0,400 ммоль) и (S)-3-(диметиламино)пирролидин (0,0420 мл, 0,333 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 5 часов. Реакционную жидкость концентрировали при пониженном давлении. Остаток очищали посредством флеш-хроматографии (NH силикагель, хлороформ/метанол) с получением этил (S)-3-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноата (0,103 г, 0,306 ммоль, 92%) (здесь и далее указывается как «соединение по примеру 69») в виде масла красновато-коричневого цвета.
1H-ЯМР (400 МГц, CDCl3) δ:1,23-1,27 (3H, м), 1,67-1,91 (1H, м), 2,06-2,26 (7H, м), 2,58-3,36 (9H, м), 3,43-3,83 (2H, м), 4,12-4,28 (4H, м), 6,85-6,93 (2H, м).
ESI-MS: m/z= 337 (M+H)+.
[0400]
(Пример 70) Синтез гидрохлорида (S)-3-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропановой кислоты:
[Формула 123]
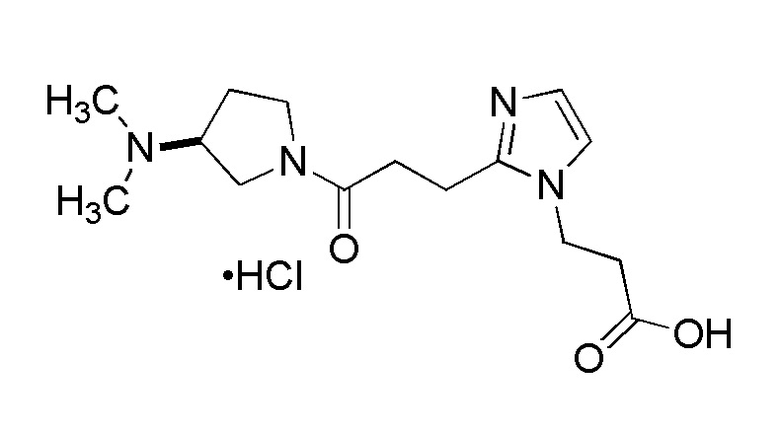
К этил (S)-3-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропаноату (0,100 г, 0,297 ммоль) при комнатной температуре добавляли водный раствор гидроксида натрия (1,0 н, 0,446 мл, 0,446 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 4 часов. При комнатной температуре в реакционную жидкость добавляли соляную кислоту (1,0 н, 0,446 мл, 0,446 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 5 минут. Реакционную жидкость концентрировали при пониженном давлении. Полученный остаток промывали этанолом (5,0 мл). Полученную смесь фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток опять промывали этанолом (5,0 мл). Полученную смесь фильтровали, и затем фильтрат концентрировали при пониженном давлении. К полученному остатку при комнатной температуре добавляли соляную кислоту (1,0 н, 0,358 мл, 0,358 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. Реакционную жидкость концентрировали при пониженном давлении и сушили при комнатной температуре с получением гидрохлорида (S)-2-(2-(3-(3-(диметиламино)пирролидин-1-ил)-3-оксопропил)-1H-имидазол-1-ил)пропана (0,0900 г, 0,259 ммоль, 87%) (здесь и далее указывается как «соединение по примеру 70») в виде твердого вещества коричневого цвета.
1H-ЯМР (400 МГц, D2O) δ: 1,95-2,21 (1H, м), 2,35-2,49 (1H, м), 2,64-2,94 (10H, м), 3,15-3,19 (2H, м), 3,29-4,08 (5H, м), 4,30-4,33 (2H, м), 7,23 (1H, с), 7,31 (1H, с).
[0401]
(Пример 71) Действие на мышиной модели частичного лигирования седалищного нерва:
Используя модель частичного лигирования седалищного нерва (модель Зельтцера) на мышах, с помощью которой может быть оценена невропатическая боль, было исследовано анальгезирующее действие производного циклического амина (I) или его пролекарства или его фармакологически приемлемой соли.
[0402]
В качестве производного циклического амина (I) или его фармакологически приемлемой для оценки были использованы соли соединения по примеру 2, 4, 6, 8, 10, 12, 13, 15, 18, 22, 25, 27, 29, 31, 33, 66, 68 или 70. В качестве пролекарства производного циклического амина (I) или его фармакологически приемлемая соли для оценки были использованы соединения по примеру 38. Соединение по примеру 38 является гидрохлоридом пролекарства, полученным путем этерификации карбоксильной группы соединения по примеру 39 этильной группой.
[0403]
1. Экспериментальный метод
Модель частичного лигирования седалищного нерва на мышах была подготовлена в соответствии со способом Seltzer et al. (Malmberg et al., Pain, vol. 76, p. 215-222, 1998).
[0404]
Slc: Мышей ICR (5-недельного возраста, самцы; от фирмы Japan SLC, Inc.) или Crl: мышей CD1 (ICR) (5-недельного возраста, самцы; от фирмы CHARLES RIVER LABORATORIES JAPAN, INC.) анестезировали фенобарбиталом натрия (70 мг/кг, внутрибрюшинное введение). Седалищный нерв в бедренной области правой задней лапы каждой мыши выводили и трехкратно лигировали шелковым швом 8-0 (от фирмы NATSUME SEISAKUSHO CO., LTD.) под стереомикроскопом таким образом, чтобы только половина толщины нерва оказалась в петле лигатуры. Группа мышей, обработанных таким образом, была определена в качестве группы частичного лигирования седалищного нерва. Группа мышей, седалищного нерва только что был выведен, и не лигирован, была определена как группа ложной хирургии.
[0405]
Оценку невропатической боли (указывается далее как тест фон Фрея) проводили следующим образом. Мышей выдерживали в течение не менее двух часов в акриловой клетке для измерения (от фирмы Natsume SEISAKUSHO CO. LTD.), помещая на проволочную сетку. После этого, используя нить (от фирмы North Coast Medical или нейробиологии), оказывающую давление 0,16 г, мышей подвергали механическому тактильному раздражению, накладывая нить на подошвенную поверхности правой задней лапы 3 раза, каждый раз в течение 3 секунд с интервалом в 3 секунды. Учитывали реакцию одергивания, наблюдаемую в течение каждого механического тактильного стимула (0, нет ответа; 1, в ответ на стимуляцию показан медленный и или слабый ответ одергивания; 2, в ответ на стимуляцию показан быстрый ответ одергивания как без передергивания (встряхивания лапами быстро и непрерывно), так и без облизывания (облизывание лап); 3, показал быстрый ответ одергивания с передергиванием и/или облизыванием), и сумму баллов, полученную при трехкратном повторении исследования (указывается далее как общий балл) использовали в качестве болевого индекса.
[0406]
(1) Пероральное введение
На 7 день после хирургии лигирования седалищного нерва соединение по примеру 8 (соединение по примеру 8: 10 мг/кг) или прегабалин (10 мг/кг; Bosche Scientific), выступающей в качестве положительного контроля, растворяли в дистиллированной воде и затем вводили перорально мышам группы частичного лигирования седалищного нерва. Группа, в которой мышам группы частичного лигирования седалищного нерва вводили соединение по примеру 8, обозначалась как группа «частичного лигирования седалищного нерва + соединение по примеру 8». Группа, в которой мышам группы частичного лигирования седалищного нерва вводили прегабалин, обозначалась как группа «частичного лигирования седалищного нерва + прегабалин». Группа, в которой мышам группы частичного лигирования седалищного нерва перорально вводили дистиллированную воду, обозначалась как группа «частичного лигирования седалищного нерва + дистиллированная вода». Группа, в которой мышам группы ложной хирургии перорально вводили дистиллированную воду, обозначалась как группа «ложной хирургии + дистиллированная вода».
[0407]
На 8 день после хирургии лигирования седалищного нерва, соединение по примеру 38 (соединение по примеру 38: 0,1 до 10 мг/кг) или прегабалин (10 мг/кг; от фирмы KEMPROTEC), выступающей в качестве положительного контроля, растворяли в дистиллированной воде и затем перорально вводили мышам группы частичного лигирования седалищного нерва. Группа, в которой мышам группы частичного лигирования седалищного нерва вводили соединение по примеру 38, обозначалась как группа «частичного лигирования седалищного нерва + соединение по примеру 38». Группа, в которой мышам группы частичного лигирования седалищного нерва вводили прегабалин, обозначалась как группа «частичного лигирования седалищного нерва + прегабалин». Группа, в которой мышам группы частичного лигирования седалищного нерва перорально вводили дистиллированную воду, обозначалась как группа «частичного лигирования седалищного нерва + дистиллированная вода». Группа, в которой мышам группы ложной хирургии перорально вводили дистиллированную воду, обозначалась как группа «ложной хирургии + дистиллированная вода».
[0408]
Тест фон Фрея проводили перед пероральным введением исследуемого соединения (предварительное значение), и спустя один час, два часа и 3 часа после перорального введения исследуемого соединения.
[0409]
(2) Внутривенное введение
На 7 день после хирургии лигирования седалищного нерва, соединение по примеру 2, 4, 13, 18, 22, 25, 27, 29, 31, 33, 66, 68 или 70 (каждое соединение по примеру 2 и 4: от 0,1 до 10 мг/кг, соединение по примеру 13: от 0,01 до 1 мг/кг, каждое соединение по примеру 18 и 22: 0,1 и 1 мг/кг, каждое соединение по примеру 25, 27, 29, 31, 33, 66, 68 и 70: 0,1 мг/кг) или прегабалин, выступающей в качестве положительного контроля (1 мг/кг; от фирмы Bosche Scientific, или 3 мг/кг; от фирмы KEMPROTEC), растворяли в физиологическом растворе и затем вводили мышам группы частичного лигирования седалищного нерва через хвостовую вену. Группа, в которой мышам группы частичного лигирования седалищного нерва вводили соединение по примеру 2, 4, 13, 18, 22, 25, 27, 29, 31, 33, 66, 68 или 70, обозначалась как группа «частичного лигирования седалищного нерва + соединение по примеру 2», группа «частичного лигирования седалищного нерва + соединение по примеру 4», группа «частичного лигирования седалищного нерва + соединение по примеру 13», группа «частичного лигирования седалищного нерва + соединение по примеру 18», группа «частичного лигирования седалищного нерва + соединение по примеру 22», группа «частичного лигирования седалищного нерва + соединение по примеру 25», группа «частичного лигирования седалищного нерва + соединение по примеру 27», группа «частичного лигирования седалищного нерва + соединение по примеру 29», группа «частичного лигирования седалищного нерва + соединение по примеру 31», группа «частичного лигирования седалищного нерва + соединение по примеру 33», группа «частичного лигирования седалищного нерва + соединение по примеру 66», группа «частичного лигирования седалищного нерва + соединение по примеру 68» или группа «частичного лигирования седалищного нерва + соединение по примеру 70», соответственно. Группа, в которой мышам группы частичного лигирования седалищного нерва вводили прегабалин, обозначалась как группа «частичного лигирования седалищного нерва + прегабалин». Группа, в которой мышам группы частичного лигирования седалищного нерва внутривенно вводили физиологический раствор, обозначалась как группа «частичного лигирования седалищного нерва + физиологический раствор». Группа, в которой мышам группы ложной хирургии внутривенно вводили физиологический раствор, обозначалась как группа «ложной хирургии + физиологический раствор».
[0410]
Тест фон Фрея для соединения по примеру 2 или 4 проводили перед внутривенным введением исследуемого соединения (предварительное значение), спустя 30 минут и 60 минут после внутривенного введения исследуемого соединения. Тест фон Фрея для соединения по примеру 13, 18, 22, 25, 27, 29, 31, 33, 66, 68 или 70, проводили перед внутривенным введением исследуемого соединения (предварительное значение), спустя 30 минут и 2 часа после внутривенного введения исследуемого соединения.
[0411]
(3) Интравентрикулярное введение
На 7 день после хирургии лигирования седалищного нерва, соединение по примеру 2, 4, 6, 10, 12, 13 или 15 (10 мкг/участок) растворяли в физиологическом растворе и затем вводили интравентрикулярно мышам группы частичного лигирования седалищного нерва. Группа, в которой мышам группы частичного лигирования седалищного нерва вводили соединение по примеру 2, 4, 6, 10, 12, 13 или 15, обозначалась как группа «частичного лигирования седалищного нерва + соединение по примеру 2», группа «частичного лигирования седалищного нерва + соединение по примеру 4» а, группа «частичного лигирования седалищного нерва + соединение по примеру 6», группа «частичного лигирования седалищного нерва + соединение по примеру 10», группа «частичного лигирования седалищного нерва + соединение по примеру 12», группа «частичного лигирования седалищного нерва + соединение по примеру 13» или группа «частичного лигирования седалищного нерва + соединение по примеру 15», соответственно. Группа, в которой мышам группы частичного лигирования седалищного нерва интравентрикулярно вводили физиологический раствор, обозначалась как группа «частичного лигирования седалищного нерва + физиологический раствор». Группа, в которой мышам группы ложной хирургии интравентрикулярно вводили физиологический раствор, обозначалась как группа «ложной хирургии + физиологический раствор».
[0412]
Тест фон Фрея проводили перед интравентрикулярным введением исследуемого соединения (предварительное значение), спустя 15 минут, 30 минут и 60 минут после интравентрикулярного введения исследуемого соединения.
[0413]
2. Результаты
(1) Пероральное введение
Результаты показаны на фигуре 1 и фигуре 11. На фигурах вертикальная ось представляет общий балл (среднее значение ± стандартная ошибка; n = 5 на фигуре 1, n = от 4 до 5 на фигуре 11) по тесту фон Фрея. Более высокое численное значение указывает на то, что боль сильнее. На горизонтальной оси указано время (час) после введения исследуемого соединения. Эффективность статистически оценивали с помощью непарного t-теста двух выборок или теста Уэлча (фигура 1) или непарного t-теста множественных выборок (уточненный Дуннеттом) (фигура 11) с использованием группы «частичного лигирования седалищного нерва + дистиллированная вода» («частичное лигирование седалищного нерва + дистиллированная вода» на фигуре) в качестве контроля для каждого времени измерения. На фигурах знак «*» указывает на то, что значение является статистически значимым (p <0,05) в сравнении с группой «частичного лигирования седалищного нерва + дистиллированная вода».
[0414]
Согласно результатам теста фон Фрея, пероральное введение соединения по примеру 8 или 38 («частичного лигирования седалищного нерва + соединение по примеру 8» или «частичного лигирования седалищного нерва + соединение по примеру 38» на фигурах) показало статистически значимое анальгезирующее действие подобное положительному контролю прегабалину («частичное лигирование седалищного нерва + прегабалин» на фигурах).
[0415]
(2) Внутривенное введение
Результаты показаны на фигуре 2, фигуре 3 и фигурах 12-22. На фигурах вертикальная ось представляет общий балл (среднее значение ± стандартная ошибка; n = от 5 до 6 на фигуре 2 и фигуре 3, n = от 4 до 7 на фигурах 12-22) по тесту фон Фрея. Более высокое численное значение указывает на то, что боль сильнее. На горизонтальной оси указано время (мин или час) после введения исследуемого соединения. Эффективность в группах, получавших прегабалин («частичного лигирования седалищного нерва + прегабалин» на фигуре) статистически оценивали с помощью непарного t-теста двух выборок или теста Уэлча с использованием группы «частичного лигирования седалищного нерва + физиологический раствор» («частичное лигирование седалищного нерва + физиологический раствор» на фигуре) в качестве контроля для каждого времени измерения. Эффективность в группах, получавших соединение по примеру 2 или 4 («частичное лигирование седалищного нерва + соединение по примеру 2» или «частичное лигирование седалищного нерва + соединение по примеру 4» на фигурах), статистически оценивали с помощью теста Уильямса или теста Ширли-Уильямса с использованием группы «частичного лигирования седалищного нерва + физиологический раствор» («частичное лигирование седалищного нерва + физиологический раствор» на фигурах) в качестве контроля для каждого времени измерения. Группа, получавшая соединение по примеру 13, 18, 22, 25, 27, 29, 31, 33, 66, 68 или 70 («частичное лигирование седалищного нерва + соединение по примеру 13, 18, 22, 25, 27, 29, 31, 33, 66, 68 или 70» на фигурах) статистически оценивали с помощью теста Ширли-Уильямса или теста Уэлча с использованием группы «частичного лигирования седалищного нерва + физиологический раствор» («частичное лигирование седалищного нерва + физиологический раствор» на фигурах) в качестве контроля для каждого времени измерения. На фигурах знак «*» указывает на то, что значение является статистически значимым (p <0,05) в сравнении с группой «частичного лигирования седалищного нерва + физиологический раствор» (двух-групповой непарный t-тест или тест Уэлча). На фигурах знак «#» указывает на то, что значение является статистически значимым в сравнении с группой «частичного лигирования седалищного нерва + физиологический раствор» группа (тест Уильямса или тест Ширли-Уильямса (p <0,025) или тест Уэлча (p <0,05)).
[0416]
Согласно тесту фон Фрея внутривенное введение соединения по примеру 2, 4, 13, 18, 22, 25, 27, 29, 31, 33, 66, 68 или 70 («частичное лигирование седалищного нерва + соединение по примеру 2, 4, 13, 18, 22, 25, 27, 29, 31, 33, 66, 68 или 70» на фигурах) показало статистически значимое анальгезирующее действие подобное положительному контролю прегабалину («частичное лигирование седалищного нерва + прегабалин» на фигуре).
[0417]
(3) Интравентрикулярное введение
Результаты показаны на фигурах 4 до 10. На фигурах вертикальная ось представляет общий балл (среднее значение ± стандартная ошибка; n = от 4 до 5 на фигурах 4 до 10) по тесту фон Фрея. Более высокое численное значение указывает на то, что боль сильнее. На горизонтальной оси указано время (мин) после введения исследуемого соединения. Эффективность статистически оценивали с помощью двух выборок непарного Т-теста или теста Уэлча с использованием группы «частичного лигирования седалищного нерва + физиологический раствор» группа («частичного лигирования седалищного нерва + физиологический раствор» на фигурах) в качестве контроля для каждого времени измерения. На фигурах знак «*» указывает на то, что значение является статистически значимым (p <0,05) в сравнении с группой «частичного лигирования седалищного нерва + физиологический раствор».
[0418]
Согласно результатам теста фон Фрея, пероральное введение соединения по примеру 2, 4, 6, 10, 12, 13 или 15 («частичного лигирования седалищного нерва + соединение по примеру 2, 4, 6, 10, 12, 13 или 15» на фигурах) показало статистически значимое анальгезирующее действие.
[0419]
Исходя из этих результатов, было четко продемонстрировано, что производное циклического амина (I) или его пролекарство или его фармакологически приемлемая соль обладает сильным обезболивающим действием против невропатической боли.
[0420]
(Пример 72) Эффект на модели синдрома фибромиалгии на крысах:
Использование модели синдрома фибромиалгии на крысах, с помощью которой может быть оценен синдром фибромиалгии, было исследовано анальгезирующее действие производного циклического амина (I) или его пролекарства или его фармакологически приемлемой соли.
[0421]
В качестве производного циклического амина (I) или его фармакологически приемлемой соли соединение по примеру 13. В качестве пролекарства производного циклического амина (I) или его фармакологически приемлемая соль для оценки было использовано соединение по примеру 38. Соединение по примеру 38 представляет собой гидрохлорид пролекарства, полученного путем этерификации карбоксильной группы соединения по примеру 39 этильной группой.
[0422]
1. Экспериментальный метод
Для подготовки модели синдрома фибромиалгии на крысах (Sluka et al., Journal of Pharmacology and Experimental Therapeutics, vol. 302, p. 1146-50, 2002; Nagakura et al., Pain, vol. 146, p. 26-33, 2009; Sluka et al., Pain, vol. 146, p. 3-4, 2009), которая обычно широко используется в области фундаментальных исследований синдрома фибромиалгии, кислотный солевой раствор (100 мкл), с pH4,0, вводили внутримышечной инъекцией в икроножную мышцу правой задней лапки Crl: CD(SD) крыс (возраста от 6 до 7 недель, самцы; от фирмы CHARLES RIVER LABORATORIES JAPAN, INC.) при непрерывном ингаляционной анестезии изофлураном, два раза (по одному разу в день 1 и день 6, где день 1 был датой, когда изначально вводили кислотный солевой раствор). Крыс, подготовленных таким образом, помещали в селекционную комнату с контролируемой температурой внутри помещения от 21 до 25°C и влажностью в помещении от 40 до 70% в условиях добровольного приема пищи и воды. Таким же образом подготавливали крыс, которым вместо кислотного раствора внутримышечно вводили физиологический раствор. Подготовленные таким образом крысы и не страдающие синдромом фибромиалгии (группа «физиологический раствор-несущая среда» на фигуре 23 и фигуре 24) также были использованы в эксперименте.
[0423]
На 7 день после начального введения кислотного солевого раствора, оценивали аллодинию у каждой крысы. Крысы, которые демонстрируют 50%-ный порог реагирования (среднее значение правой задней лапки и левой задней лапки) от 2 г или более до 6 г или менее, были выбраны в качестве крыс для модели синдрома фибромиалгии с началом синдрома фибромиалгии и их подвергали последующему эксперименту введения. Следует отметить, что измерение аллодинии проводили с использованием нити фон Фрея (произведенной North Coast Medical) в соответствии с методом, описанным в известной литературе (Chaplan et al., Journal of Neuroscience Methods, vol, 53, p. 55-63, 1994).
[0424]
Подготовленных таким образом крыс с моделью синдрома фибромиалгии разбивали на группы таким образом, чтобы 50% порог реагирования (среднее значение для правой задней лапы и левой задней лапы) в отдельных группах был равным, и исследуемо соединение вводили крысам с моделью синдрома фибромиалгии на 7 день после начального введения кислотного солевого раствора.
[0425]
Соединение по примеру 13 (3 и 10 мг/кг) растворяли в физиологическом растворе и затем вводили (внутривенное введение) крысам с моделью синдрома фибромиалгии через хвостовую вену («кислотный солевой раствор-соединение по примеру 13» на фигуре 23). Прегабалин, выступающей в качестве положительного контроля (10 мг/кг; от фирмы KEMPROTEC), растворяли в физиологическом растворе и затем вводили внутривенно («кислотный солевой раствор-прегабалин» на фигуре 23). В качестве контроля крысам с моделью синдрома фибромиалгии внутривенно вводили физиологический раствор («кислотный солевой раствор-несущая среда» на фигуре 23). Кроме того, крысам, не страдающим симптомом фибромиалгии, внутривенно вводили физиологический раствор («физиологический раствор-несущая среда» на фигуре 23). Через тридцать минут после внутривенного введения для оценки анальгезирующего действия определяли аллодинию у отдельных крыс.
[0426]
Соединение по примеру 38 (10 мг/кг) растворяли в дистиллированной воде и затем вводили перорально крысам с моделью синдрома фибромиалгии («кислотный солевой раствор-соединение по примеру 38» на фигуре 24). Прегабалин, выступающей в качестве положительного контроля (10 мг/кг; от фирмы KEMPROTEC), растворяли в дистиллированной воде и затем вводили перорально («кислотный солевой раствор-прегабалин» на фигуре 24). В качестве контроля крысам с моделью синдрома фибромиалгии перорально вводили дистиллированную воду («кислотный солевой раствор-несущая среда» на фигуре 24). Кроме того, дистиллированную воду перорально вводили крысам, не страдающим симптомом фибромиалгии («физиологический раствор-несущая среда» на фигуре 24). Через один час и через три часа после перорального введения для оценки анальгезирующего действия определяли аллодинию у отдельных крыс. При этом в качестве предварительного значения при определении аллодинии перед пероральным введением исследуемого соединения на 7 день после начального введения кислотного солевого раствора определяли значение 50% порога реагирования.
[0427]
2. Результаты
Результаты показаны на фигуре 23 и фигуре 24. На фигурах на вертикальной оси представлен 50%-ный порог реагирования (среднее значение правой задней лапки и левой задней лапки) (г) (среднее значение ± стандартная ошибка, n = от 5 до 6). Более высокое численное значение указывает на то, что на модели синдрома фибромиалгии на крысах аллодиния улучшается.
[0428]
На фигуре 23 показаны результаты через 30 минут после внутривенного введение соединения по примеру 13. На фигуре знак «$» указывает на то, что значение является статистически значимым ($: p <0,05) в качестве результата непарного t-теста с двумя выборками с использованием группы «физиологический раствор-несущая среда» ("физиологический раствор-несущая среда» на фигуре) в качестве контроля. На фигуре, знак «#» указывает на то, что значение является статистически значимым (#: p <0,025) по результатам теста Ширли-Уильямса с использованием группы «кислотный солевой раствор-несущая среда» («кислотный солевой раствор-несущая среда» на фигуре) в качестве контроля. На фигуре, знак «*» указывает на то, что значение является статистически значимым (*: p <0,05) по результатам теста Уэлча с использованием группы «кислотный солевой раствор-несущая среда» («кислотный солевой раствор-несущая среда» на фигуре) в качестве контроля.
[0429]
На фигуре 24 показаны результаты перорального введения соединения по примеру 38. На фигуре горизонтальная ось указывает время перед пероральным введением соединения по примеру 38 (предварительное значение) и время (час) после перорального введения. На фигуре знак «*» указывает на то, что значение является статистически значимым (*: p <0,05) в качестве результата мульти-группового непарного t-теста (уточненный Дуннеттом) с использованием группы «кислотный солевой раствор-несущая среда» («кислотный солевой раствор-несущая среда» на фигуре) для каждого времени измерения в качестве контроля.
[0430]
В группе, которой внутривенно вводили соединение по примеру 13 («кислотный солевой раствор-соединение по примеру 13» на фигуре 23), и в группе, которой перорально вводили соединение по примеру 38 («кислотный солевой раствор-соединение по примеру 38» на фигуре 24), наблюдаемая аллодиния на модели синдрома фибромиалгии на крысах была статистически значимо улучшенной по сравнению с группой «кислотный солевой раствор-несущая среда», подобно положительному контролю, т.е. группе, которой внутривенно или перорально вводили прегабалин («кислотный солевой раствор-прегабалин» на фигуре 23 и фигуре 24).
[0431]
Исходя из этих результатов, было четко продемонстрировано, что производное циклического амина (I) или его пролекарство или его фармакологически приемлемая соль являются эффективными при синдроме фибромиалгии.
[0432]
(Пример 73) Фармакокинетика пролекарства производного циклического амина (I) или его фармакологически приемлемой соли на крысах:
Фармакологически приемлемая соль пролекарства, полученного путем этерификации карбоксильной группы производного циклического амина (I), то есть, соединение по примеру 38, перорально вводили крысам и жидкую часть лимфы анализировали с помощью LC/MS/MS анализа. В результате было подтверждено, что соединение по примеру 38 преобразуется in-vivo у крыс в производное циклического амина (I), то есть, соединение по примеру 39.
Промышленная применимость
[0433]
Производное циклического амина по настоящему изобретению или его фармакологически приемлемая соль могут быть использованы в качестве лекарственных средств при болевых симптомах, поскольку они могут проявлять анальгезирующее действие против боли, в частности, невропатической боли или синдрома фибромиалгии.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2667062C1 |
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА В КАЧЕСТВЕ СРЕДСТВА СТИМУЛИРОВАНИЯ ДЕЙСТВИЯ АДВИЛЛИНА И НОВОЕ ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА И ЕГО ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2019 |
|
RU2792056C2 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2675375C1 |
| N-(1H-ИМИДАЗОЛ-2-ИЛ)БЕНЗАМИД И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2020 |
|
RU2806754C1 |
| ПРОИЗВОДНЫЕ ДИГИДРОИНДОЛОНА | 2009 |
|
RU2468022C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ИЛИ ПРОФИЛАКТИЧЕСКОЕ СРЕДСТВО ПРОТИВ ГНЕЗДНОЙ АЛОПЕЦИИ | 2018 |
|
RU2772507C2 |
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ, ИСПОЛЬЗУЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ДИАЦИЛГЛИЦЕРОЛ O-АЦИЛТРАНСФЕРАЗЫ 2, И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2810064C1 |
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2696862C1 |
| ПРОИЗВОДНЫЕ АРИЛ- И ГЕТЕРОАРИЛЭТИЛАЦИЛГУАНИДИНА, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2007 |
|
RU2446165C2 |
| ПРОИЗВОДНЫЕ АРИЛСУЛЬФОНИЛАМИНОГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ МАТРИЧНЫХ МЕТАЛЛОПРОТЕИНАЗ И СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2145597C1 |
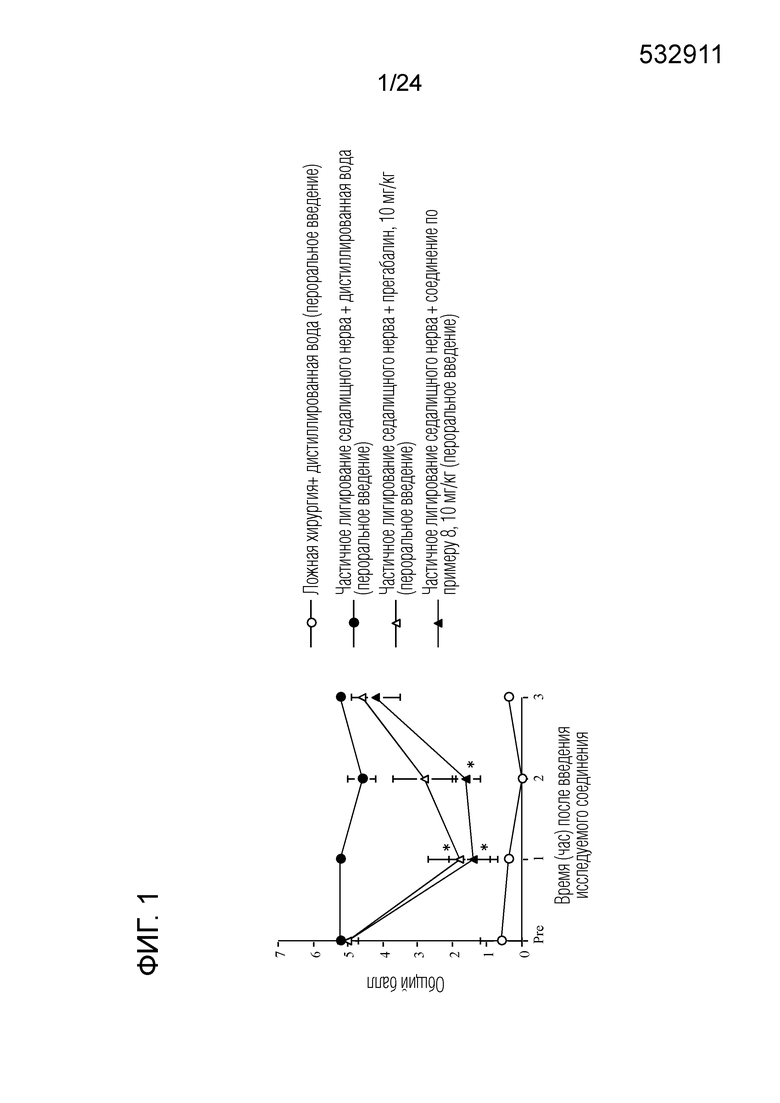
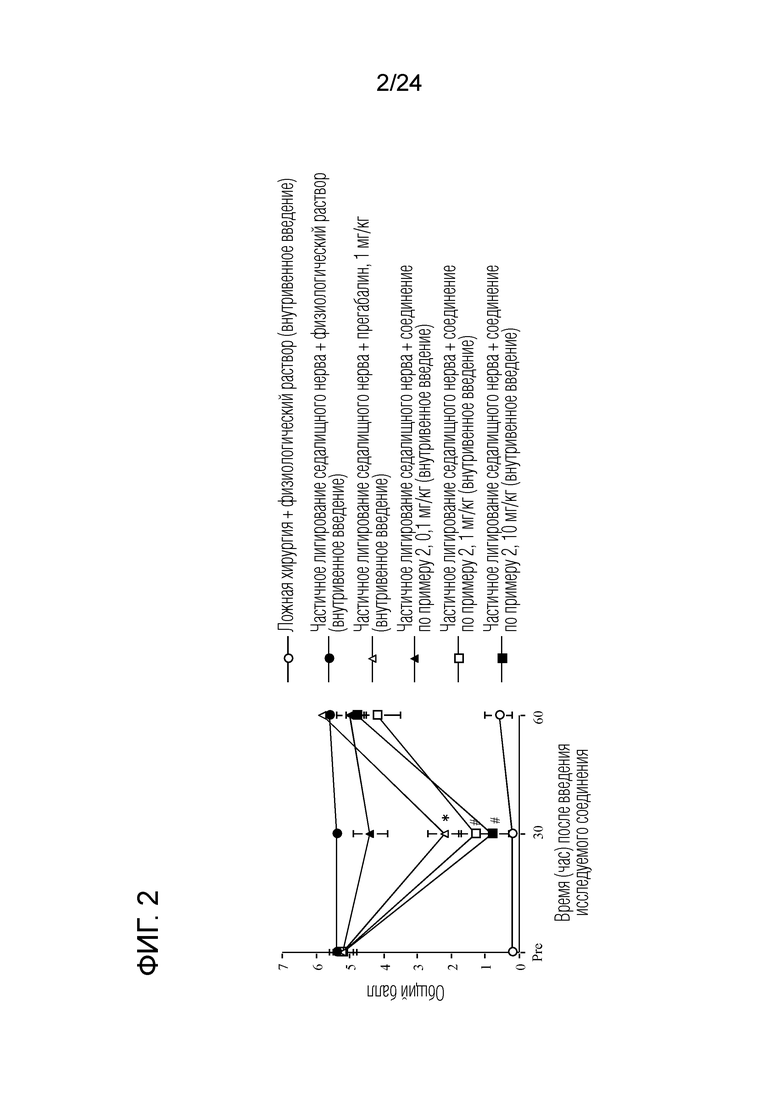
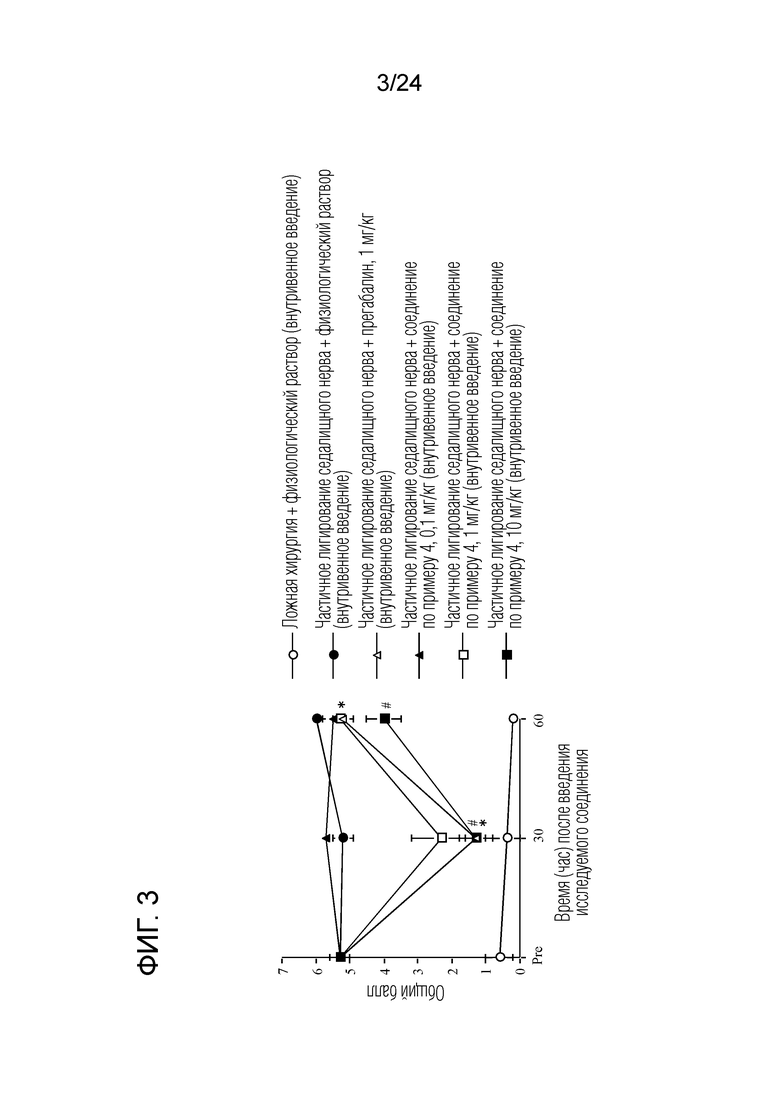
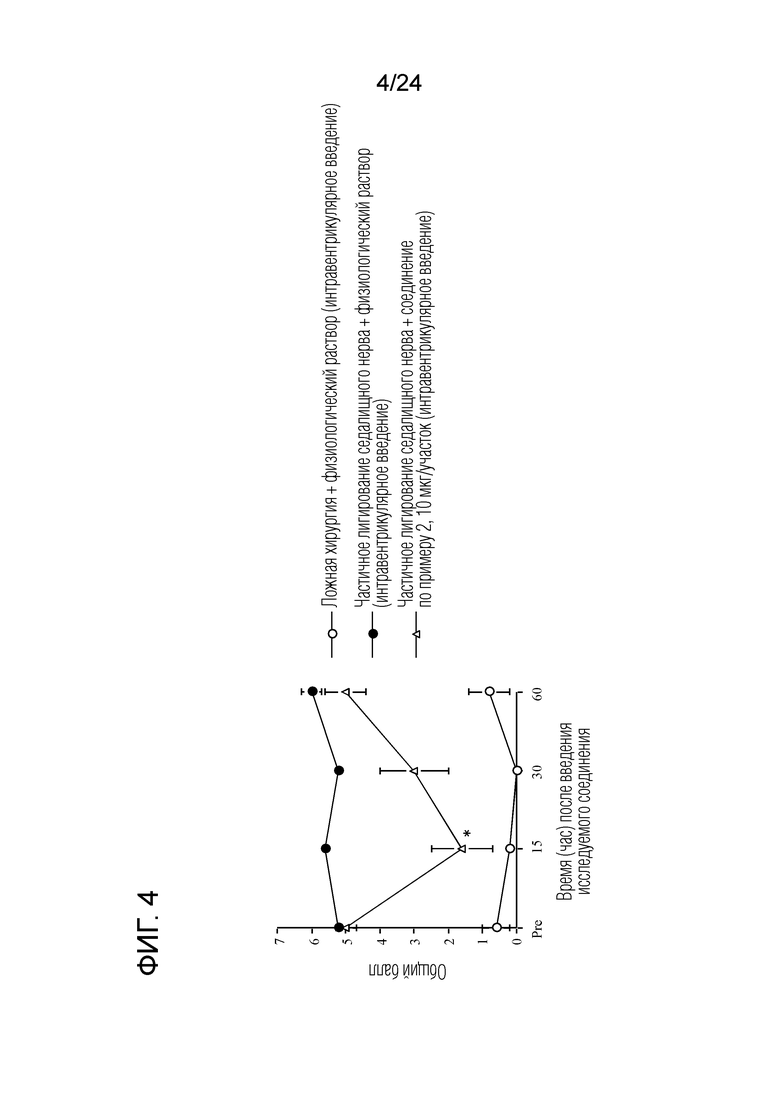
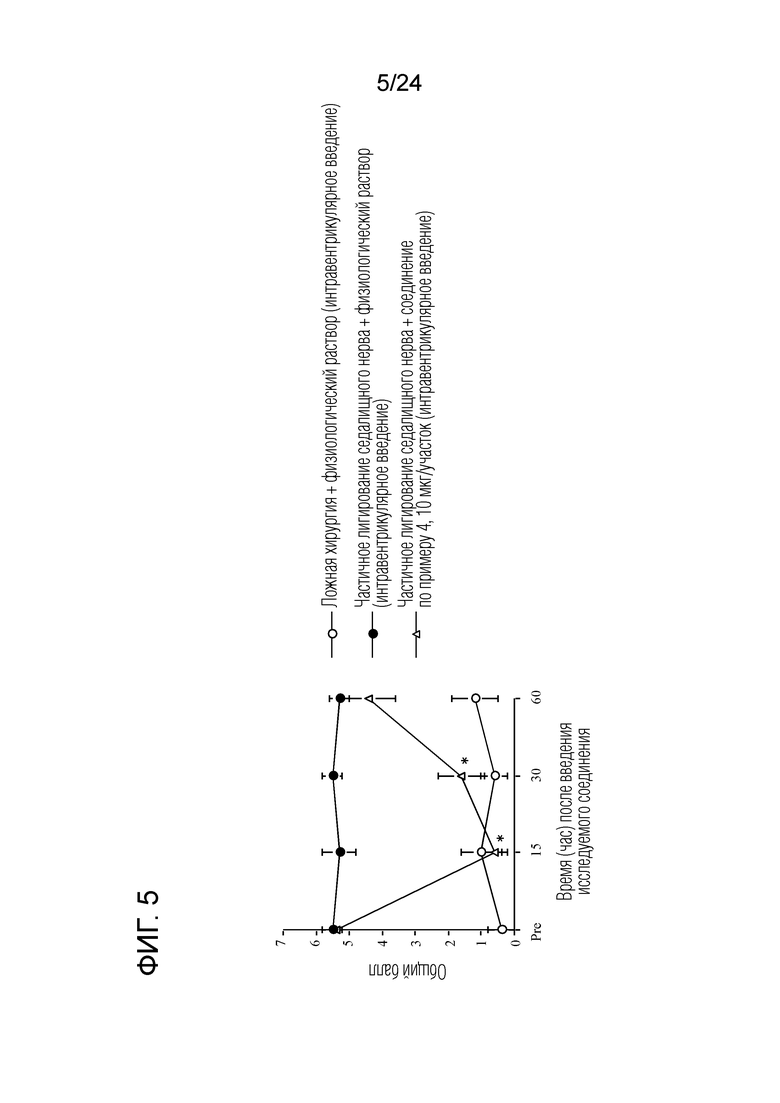
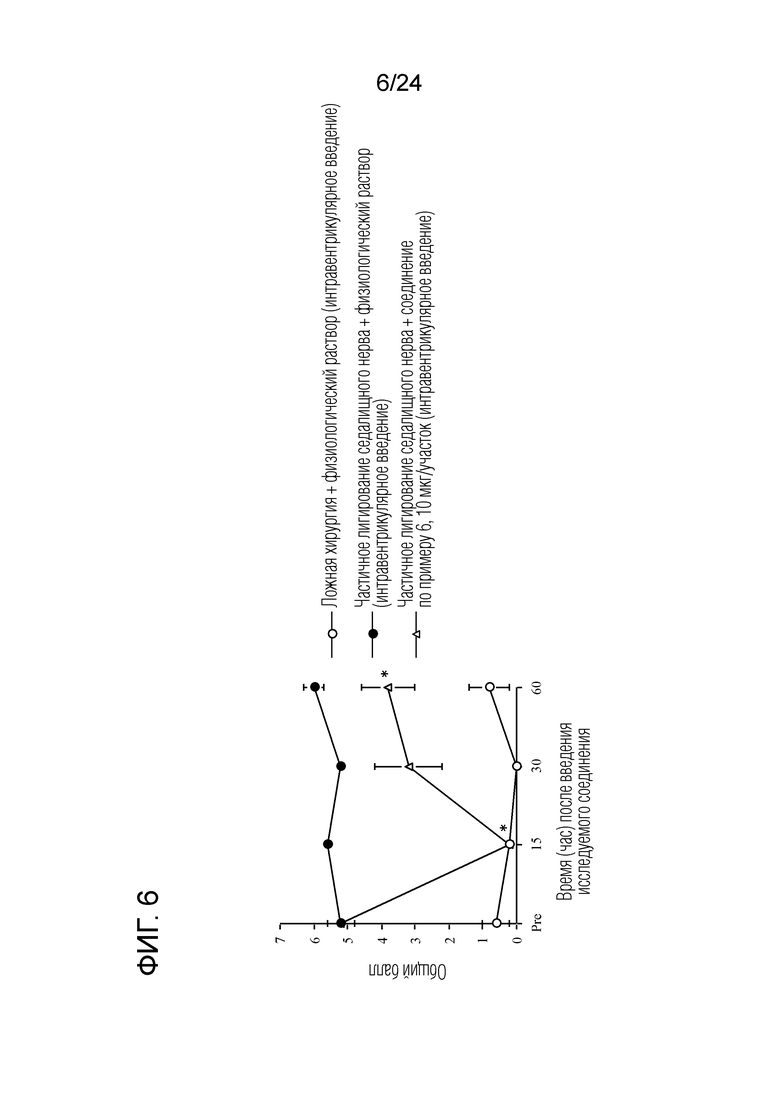
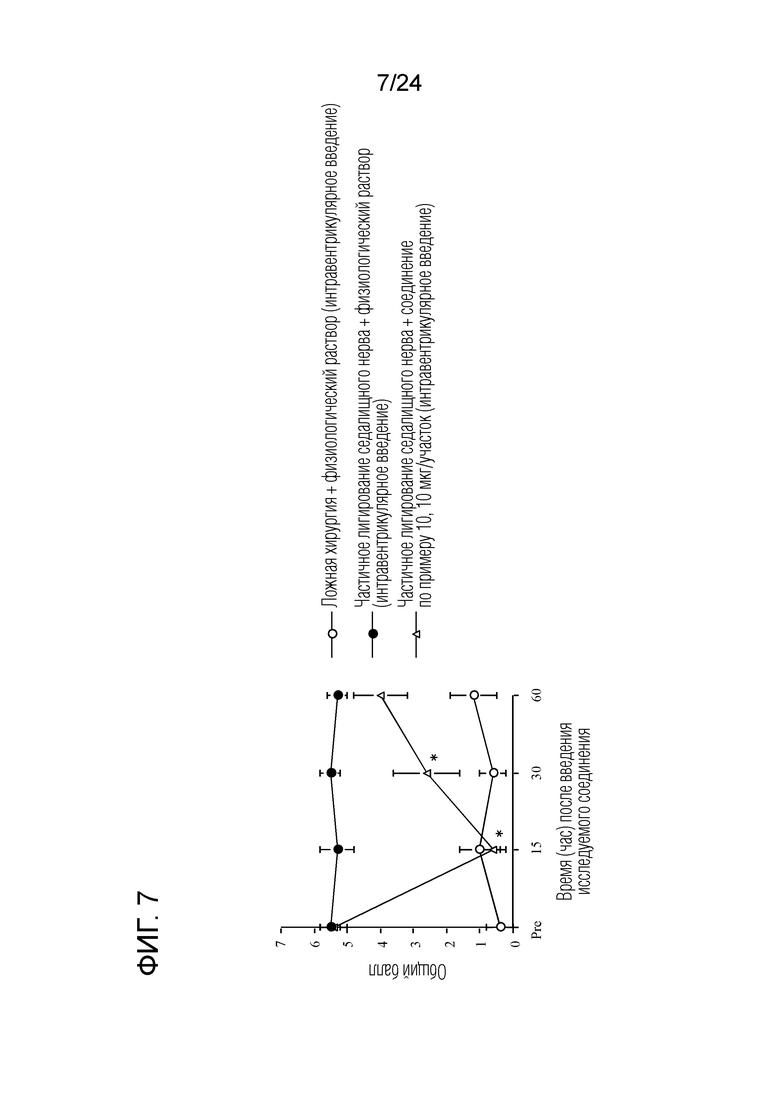
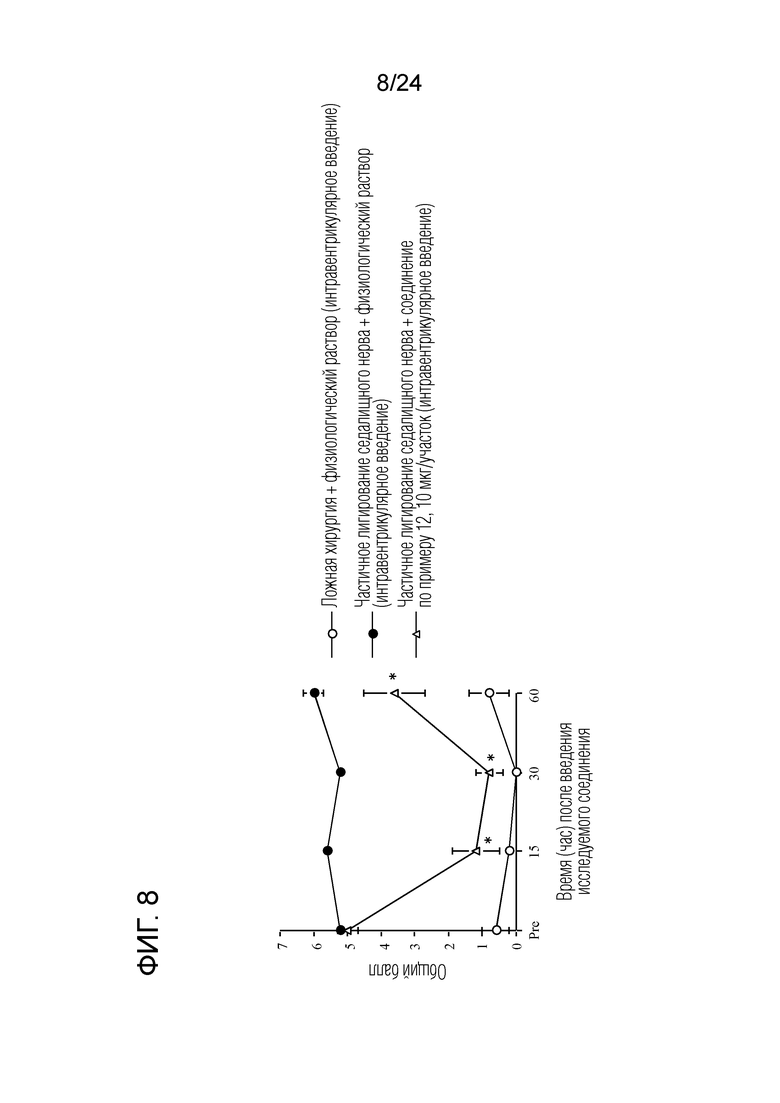
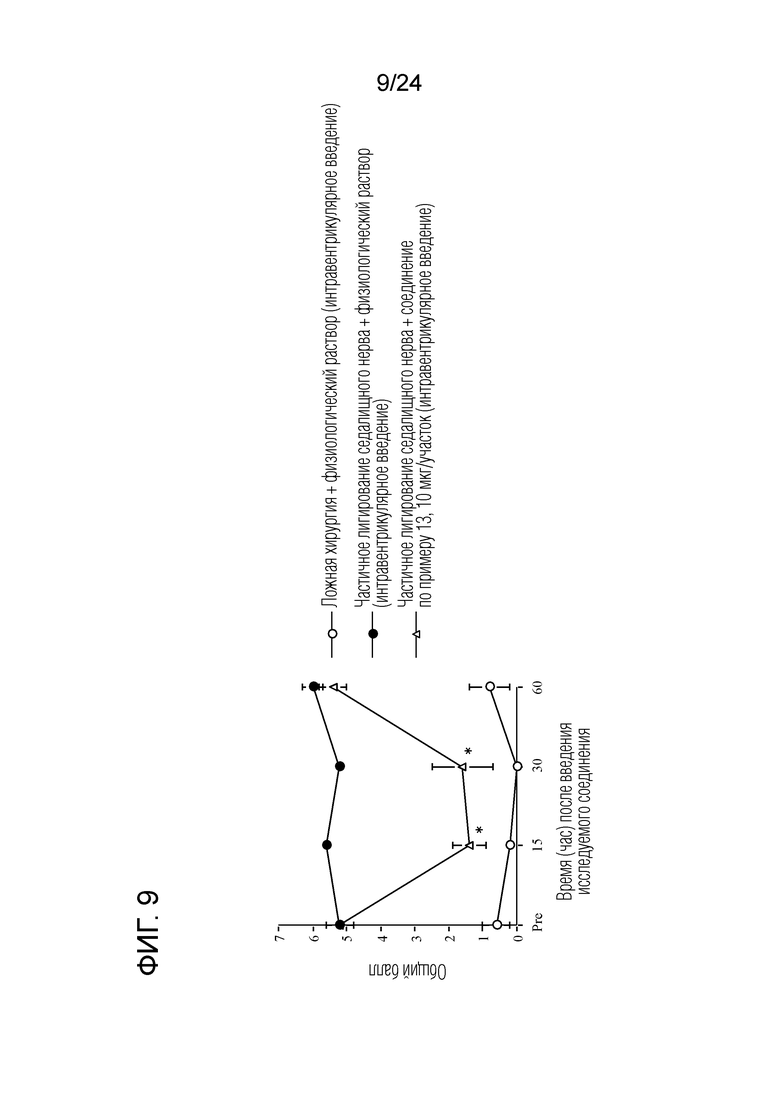
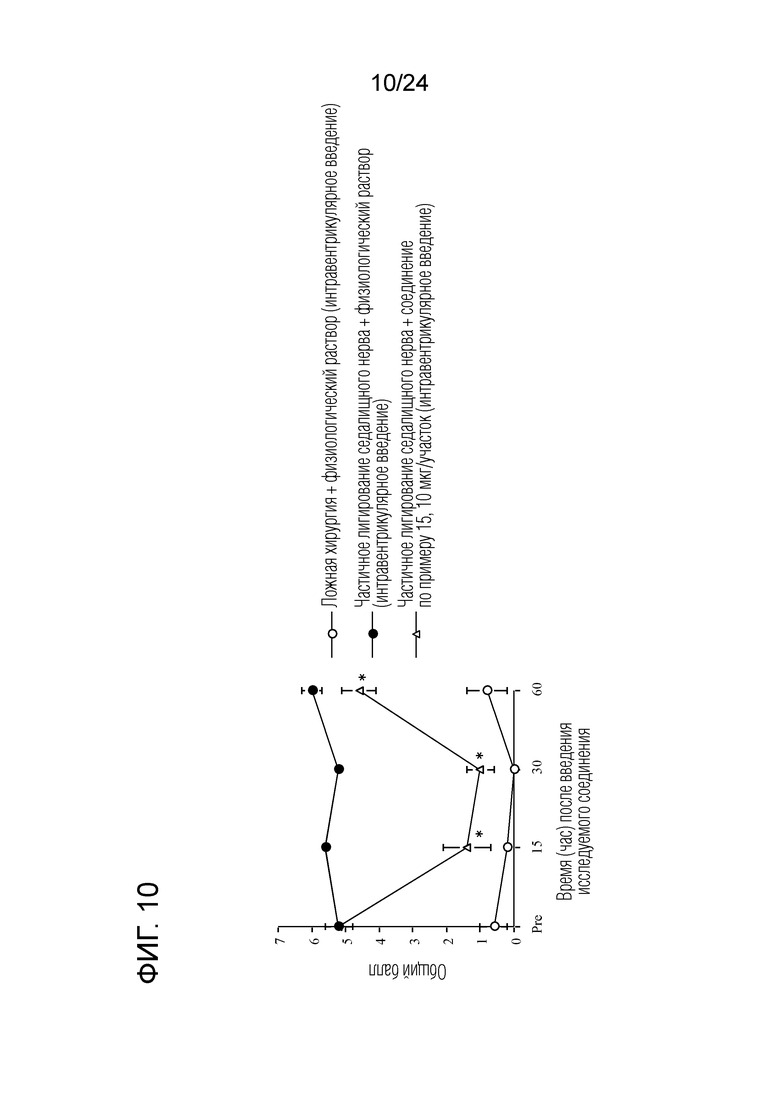
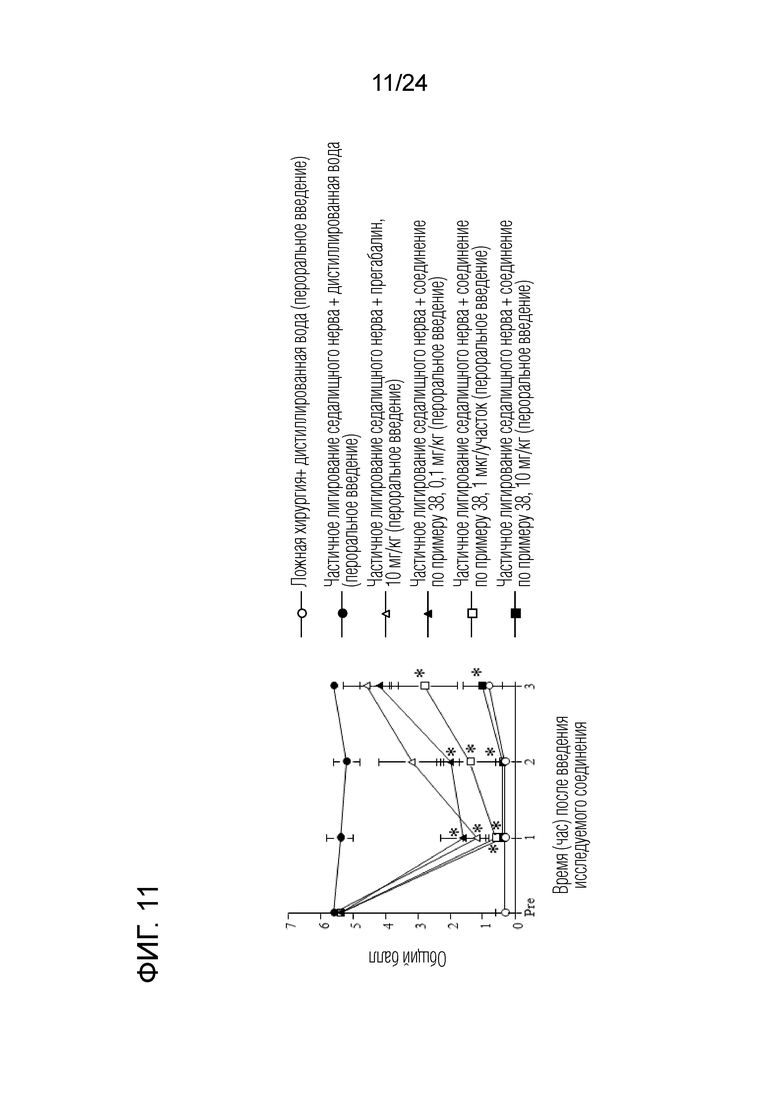

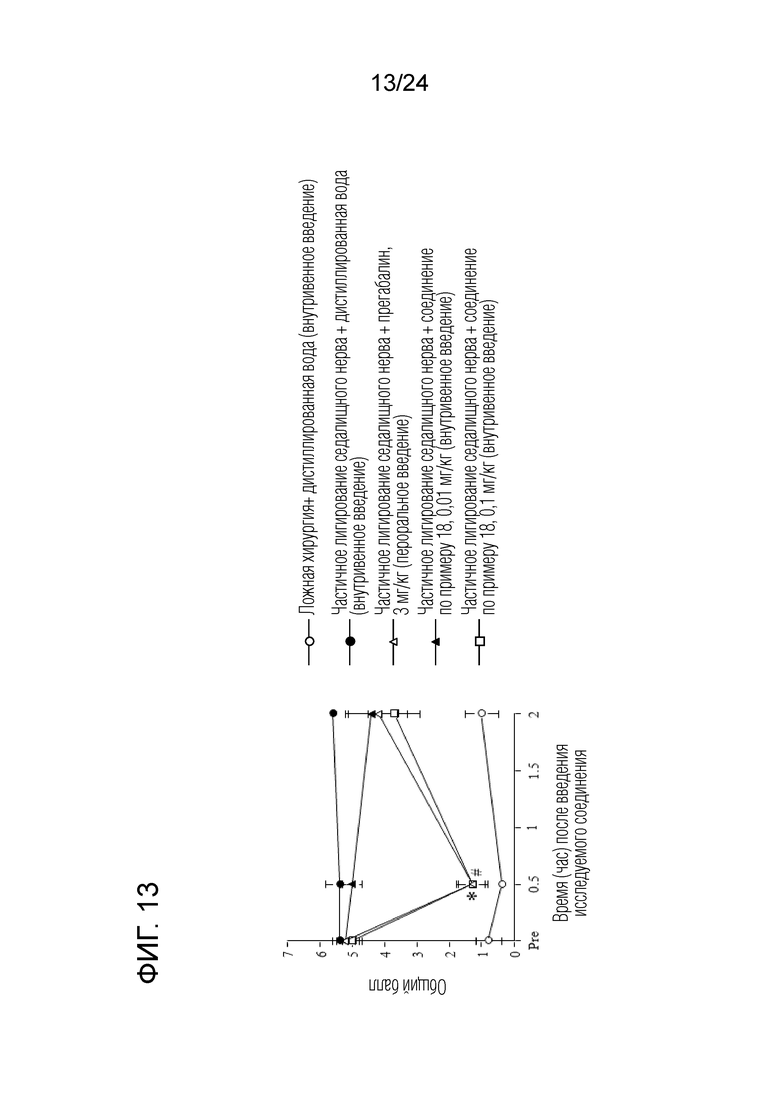
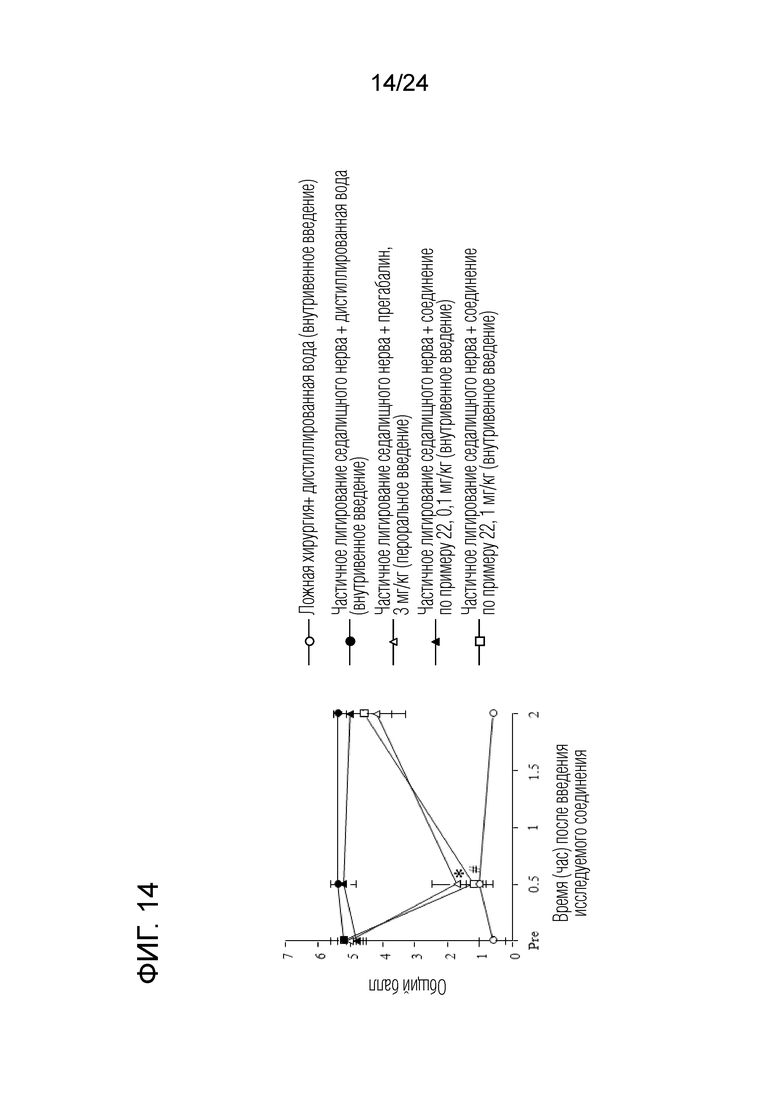
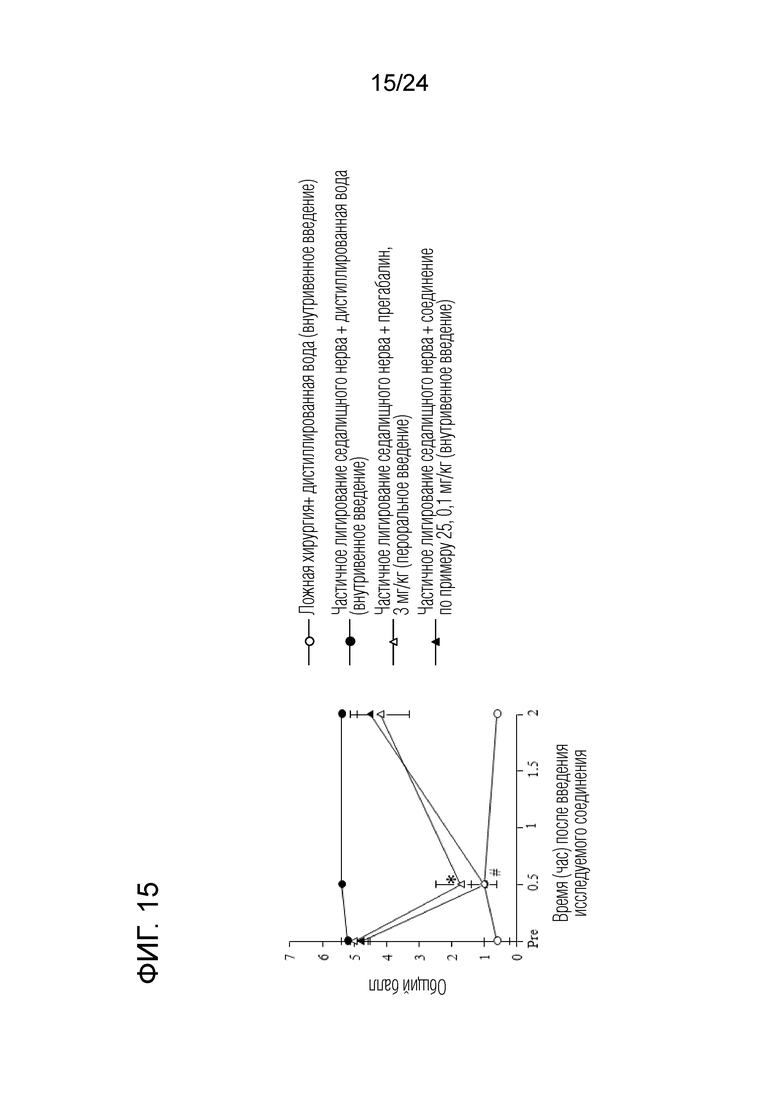
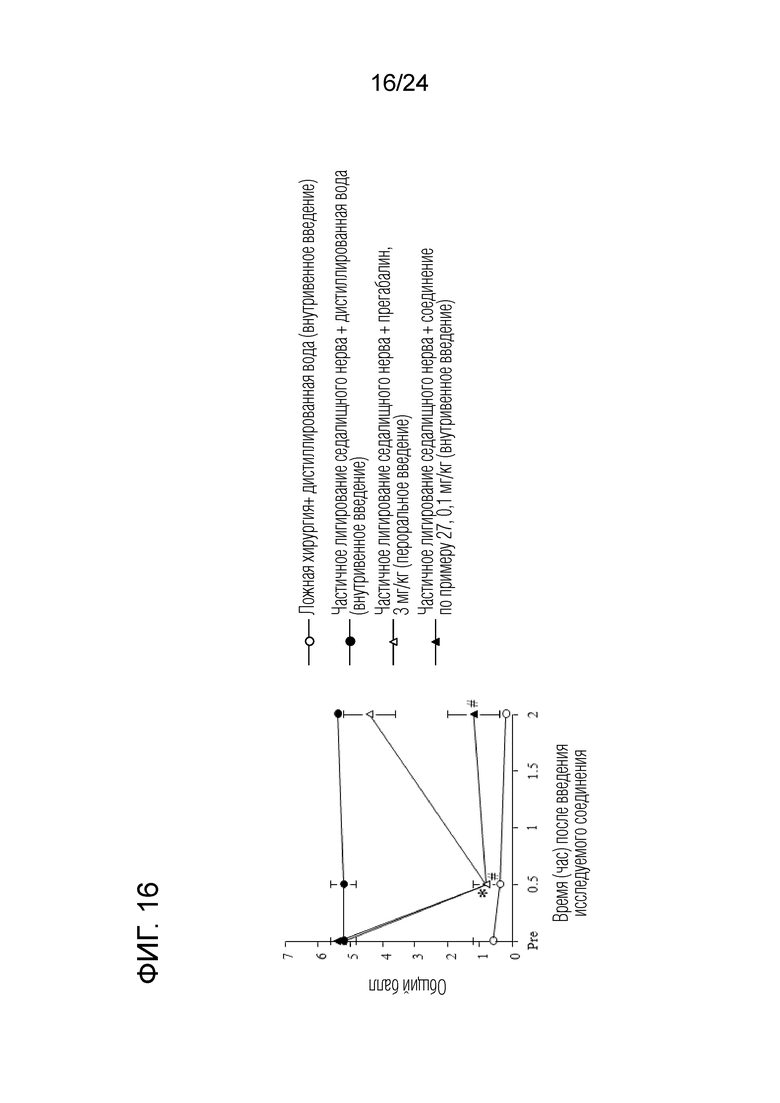
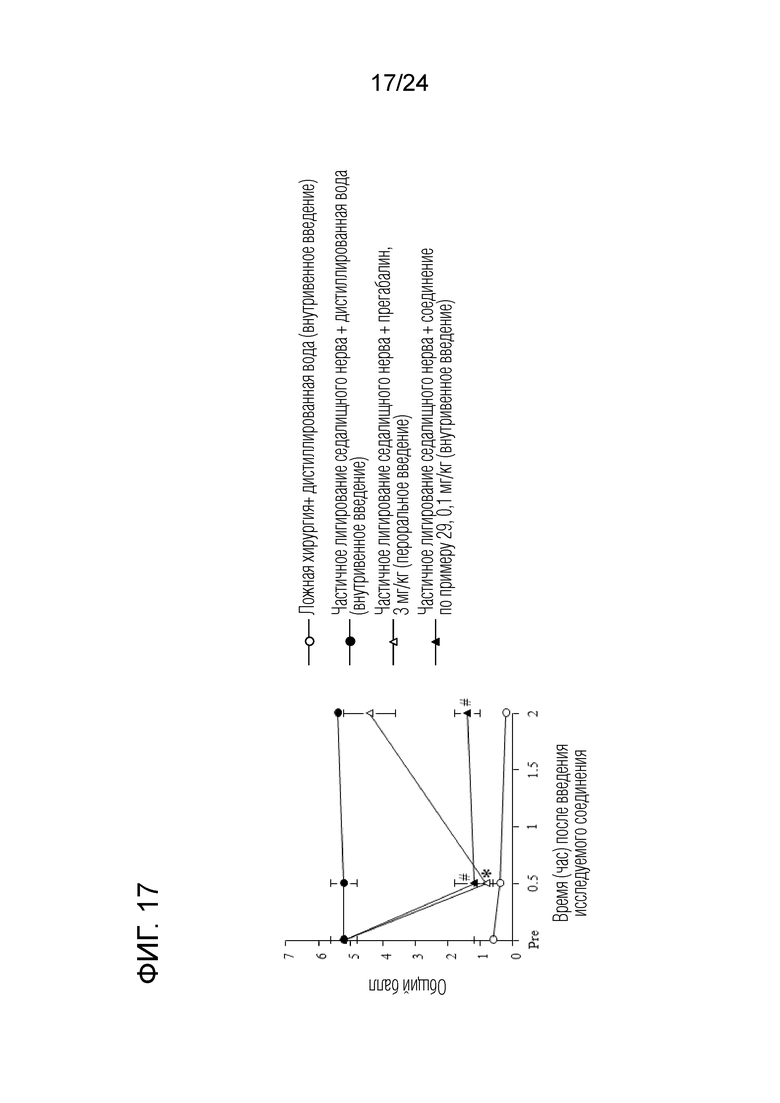
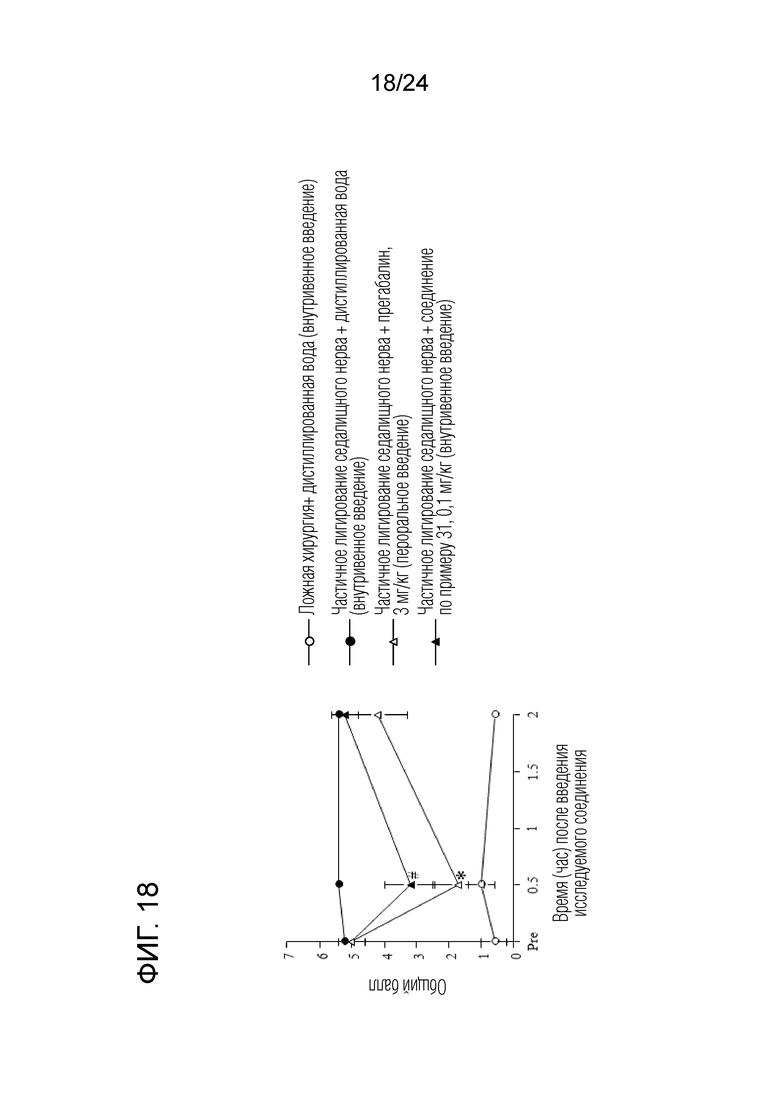
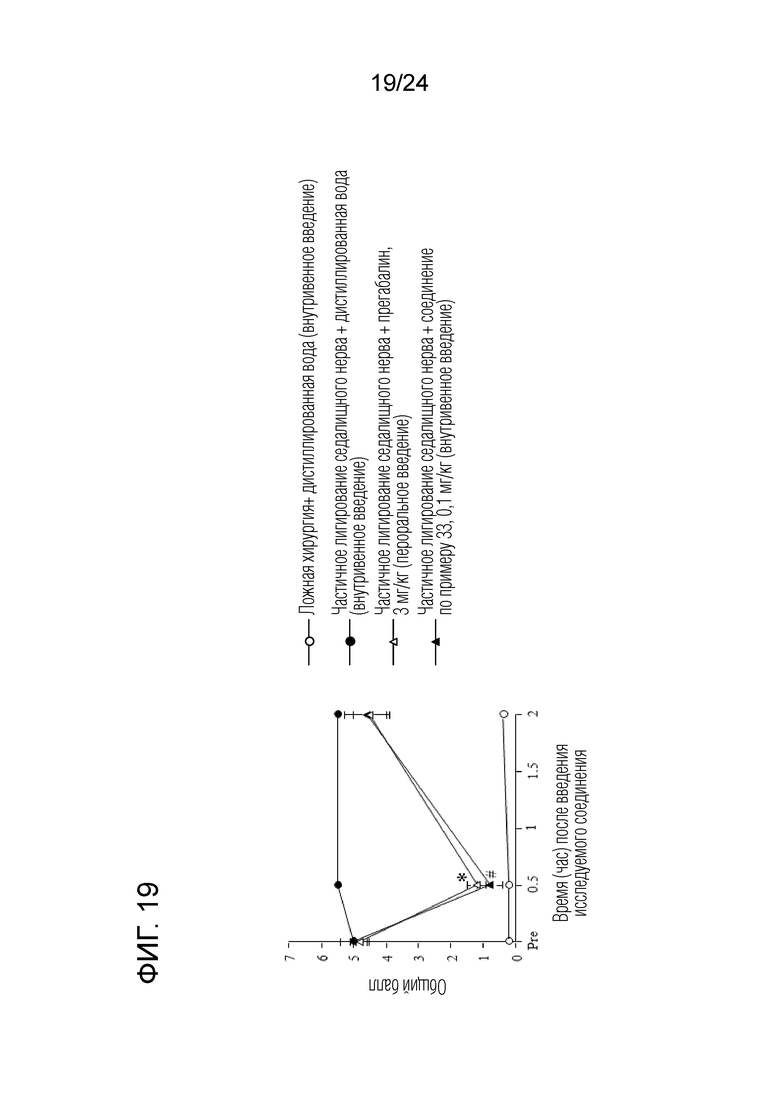
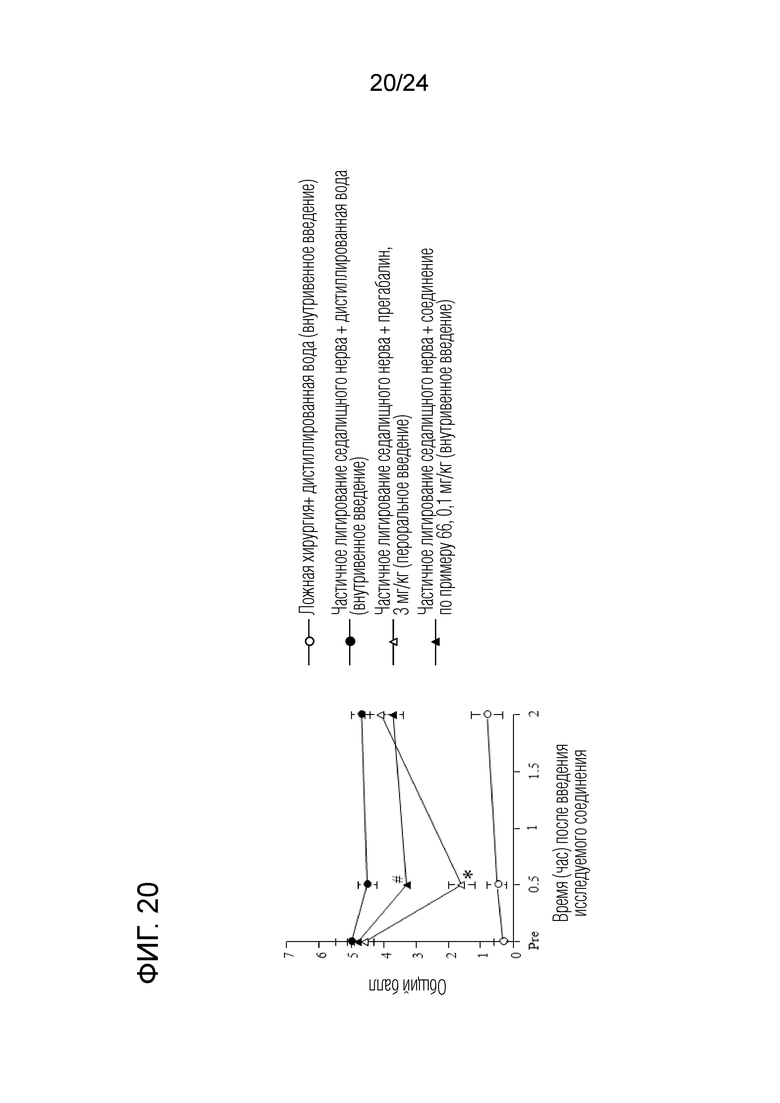
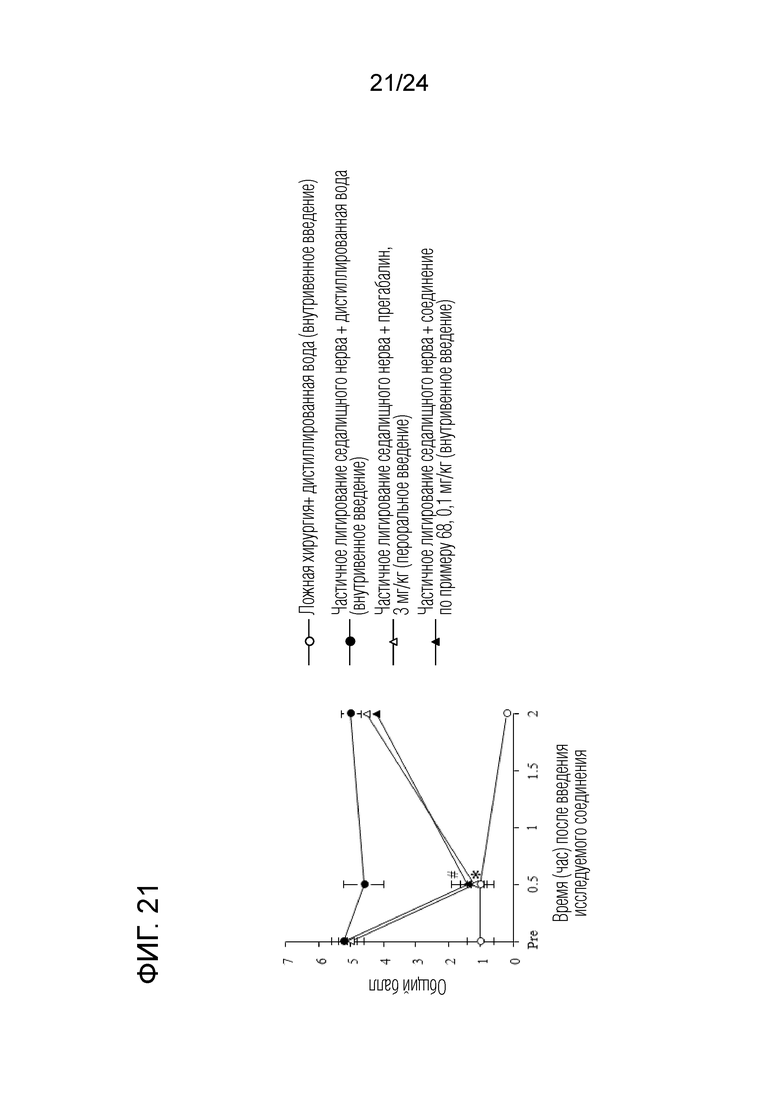
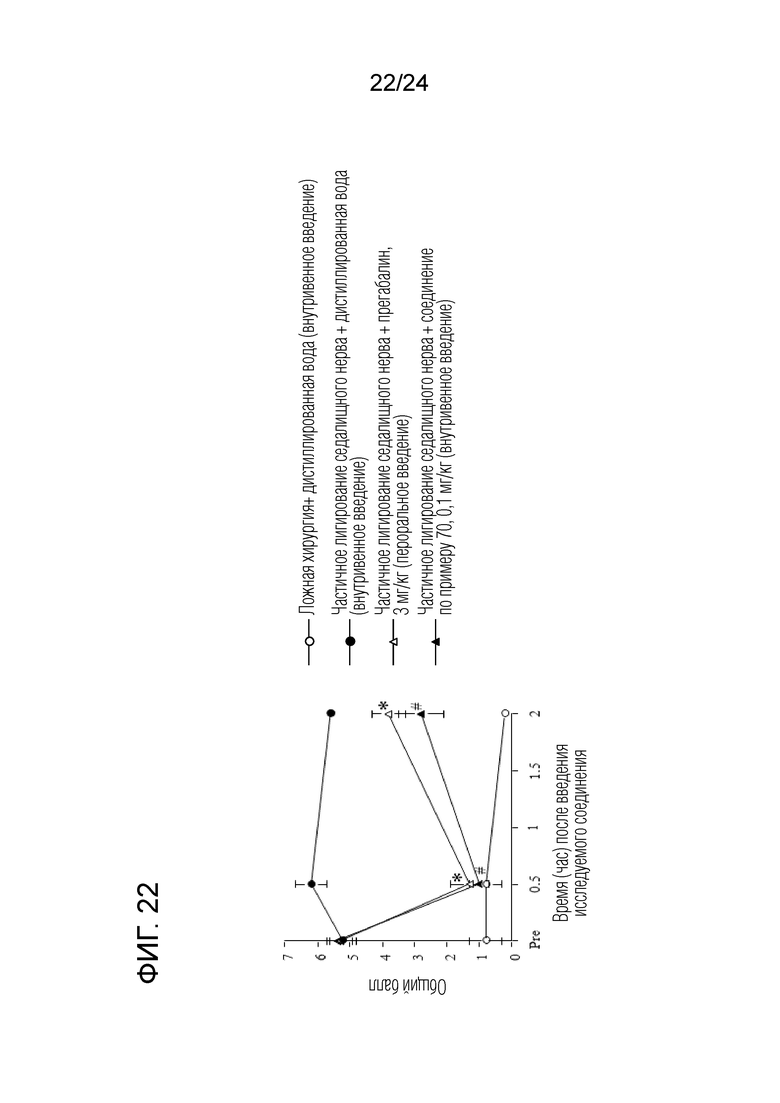
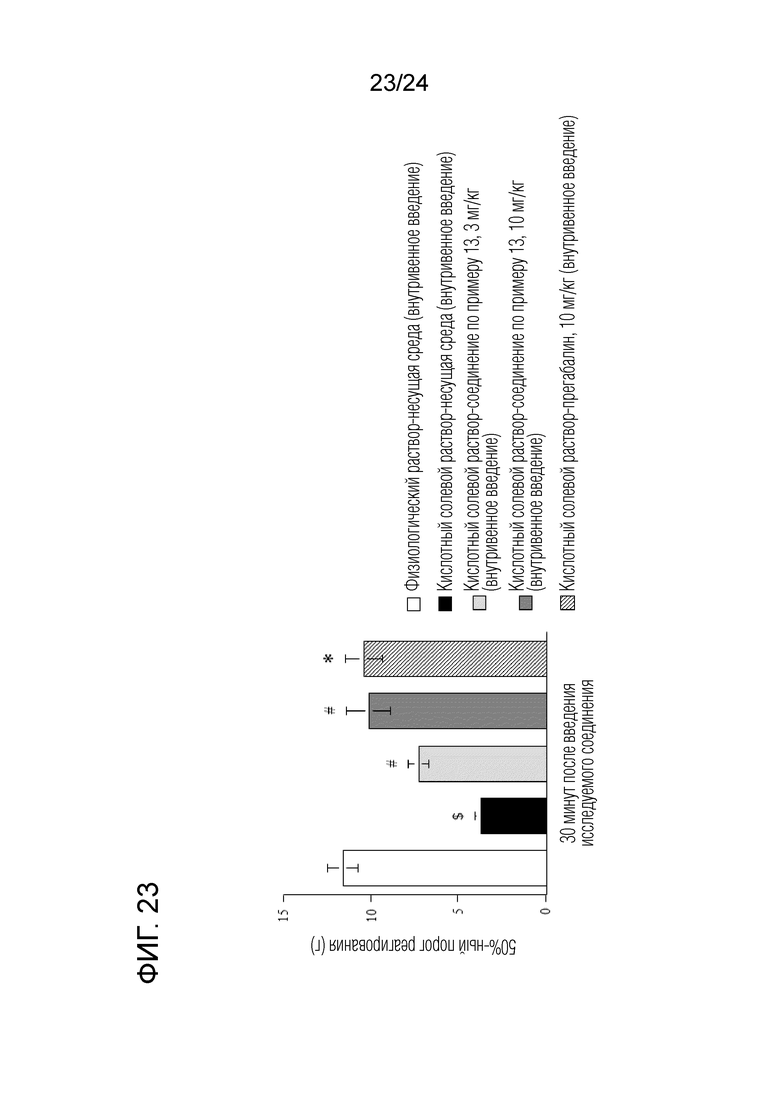
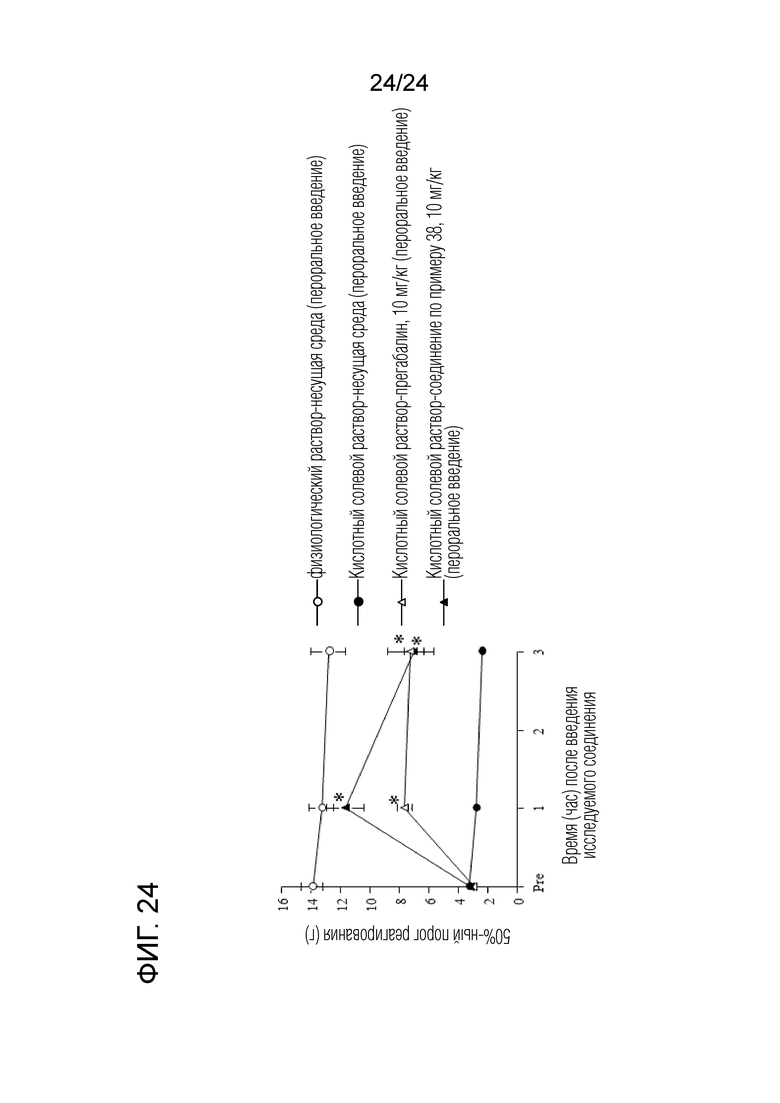
Изобретение относится к области органической химии, а именно к производным циклического амина формулы (I), где A представляет собой группу, представленную общей формулой (IIa), (IIb) или (IIc), где, когда A представляет собой группу, представленную общей формулой (IIa) или (IIb), R1 представляет собой алкильную группу, содержащую 1 или 2 атома углерода и необязательно замещенную гидроксильной группой, аминогруппой или карбоксильной группой, R2 представляет собой атом водорода, R3 представляет собой атом водорода или алкильную группу, содержащую 1 или 2 атома углерода, R4 представляет собой атом водорода или алкилкарбонильную группу, содержащую 2 атома углерода, или алкильную группу, содержащую 1 или 2 атома углерода и необязательно замещенную алкилкарбониламиногруппой, содержащей 2 атома углерода, и n обозначает 1 или 2, в которой, когда R3 и R4, каждый, независимо, представляют собой алкильную группу, содержащую 1 или 2 атома углерода, R1 представляет собой алкильную группу, содержащую 1 или 2 атома углерода и замещенную гидроксильной группой, аминогруппой или карбоксильной группой; и когда A представляет собой группу, представленную общей формулой (IIc), R1 представляет собой алкильную группу, содержащую 1 атом углерода и замещенную карбоксильной группой, R2 представляет собой атом водорода, X представляет собой CH2, O или -NR5 и R5 представляет собой алкильную группу, содержащую 1 атом углерода. Также изобретение относится к пролекарству соединения формулы (I), лекарственному средству, анальгетическому агенту и терапевтическому агенту на основе соединения формулы (I) или его пролекарства. Технический результат: получены новые производные имидазола, полезные при лечении боли. 6 н. и 5 з.п. ф-лы, 24 ил., 5 табл., 73 пр.
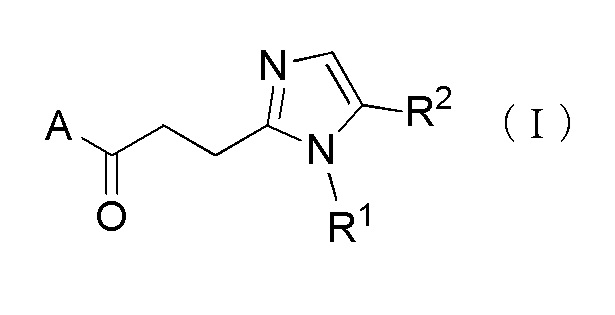
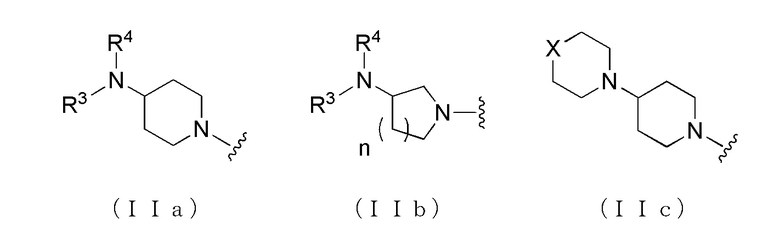
1. Производное циклического амина, представленное общей формулой (I), или его фармакологически приемлемая соль:
[Формула 1]
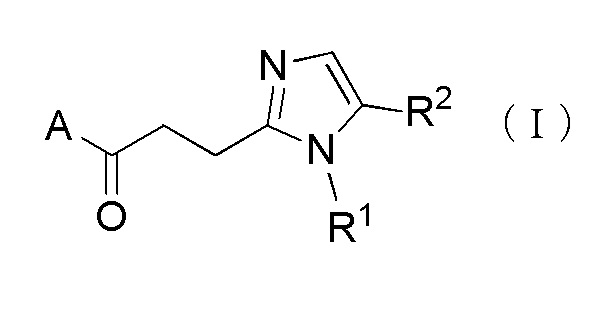
где A представляет собой группу, представленную общей формулой (IIa), (IIb) или (IIc):
[Формула 2]
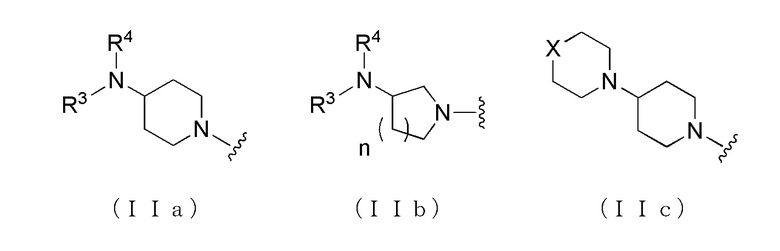
где,
когда A представляет собой группу, представленную общей формулой (IIa) или (IIb), R1 представляет собой алкильную группу, содержащую 1 или 2 атома углерода и необязательно замещенную гидроксильной группой, аминогруппой или карбоксильной группой, R2 представляет собой атом водорода, R3 представляет собой атом водорода или алкильную группу, содержащую 1 или 2 атома углерода, R4 представляет собой атом водорода или алкилкарбонильную группу, содержащую 2 атома углерода, или алкильную группу, содержащую 1 или 2 атома углерода и необязательно замещенную алкилкарбониламиногруппой, содержащей 2 атома углерода, и n обозначает 1 или 2, в которой, когда R3 и R4, каждый, независимо, представляют собой алкильную группу, содержащую 1 или 2 атома углерода, R1 представляет собой алкильную группу, содержащую 1 или 2 атома углерода и замещенную гидроксильной группой, аминогруппой или карбоксильной группой, и,
когда A представляет собой группу, представленную общей формулой (IIc), R1 представляет собой алкильную группу, содержащую 1 атом углерода и замещенную карбоксильной группой, R2 представляет собой атом водорода, X представляет собой CH2, O или -NR5 и R5 представляет собой алкильную группу, содержащую 1 атом углерода.
2. Производное циклического амина или его фармакологически приемлемая соль по п.1, где A соответствует общей формуле (IIa) или (IIb).
3. Производное циклического амина или его фармакологически приемлемая соль по п.2, где R3 представляет собой атом водорода или метильную группу.
4. Производное циклического амина или его фармакологически приемлемая соль по п.1, где A соответствует общей формуле (IIc).
5. Пролекарство производного циклического амина по любому из пп.1-4 или его фармакологически приемлемая соль, где пролекарство получают путем этерификации карбоксильной группы производного циклического амина.
6. Пролекарство или его фармакологически приемлемая соль по п.5, выбранное из группы, включающей метил этерифицированное, этил этерифицированное, н-пропил этерифицированное, изопропил этерифицированное, циклопропил этерифицированное, н-бутил этерифицированное, изобутил этерифицированное, втор-бутил этерифицированное, трет-бутил этерифицированное, циклопропилметил этерифицированное, н-пентил этерифицированное, изопентил этерифицированное, циклопентил этерифицированное, н-гексил этерифицированное, изогексил этерифицированное, циклогексил этерифицированное, н-гептил этерифицированное, н-октил этерифицированное, (5-метил-2-оксо-1,3-диоксолен-4-ил)метил этерифицированное, ацетилоксиметил этерифицированное, 1-ацетилоксиэтил этерифицированное, пропионилоксиметил этерифицированное, 1-пропионилоксиэтил этерифицированное, валерилоксиметил этерифицированное, 1-валерилоксиэтил этерифицированное, изобутирилоксиметил этерифицированное, 1-изобутилилоксиэтил этерифицированное, пивалоилоксиметил этерифицированное, 1-пивалоилоксиэтил этерифицированное, этоксикарбонилоксиметил этерифицированное, 1-((этоксикарбонил)окси)этил этерифицированное, 1-((этоксикарбонил)окси)изобутил этерифицированное, циклогексилоксикарбонилметил этерифицированное, 1-((циклогексил)карбонил)оксиэтил этерифицированное, 2-(диметиламино)-2-оксоэтил этерифицированное, фталидил этерифицированное или инданил этерифицированное соединение, и его фармацевтически приемлемая соль.
7. Пролекарство или его фармакологически приемлемая соль по п.5, выбранное из группы, включающей
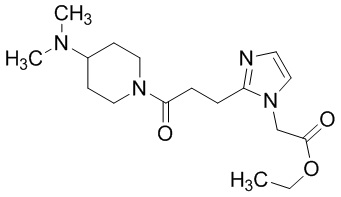
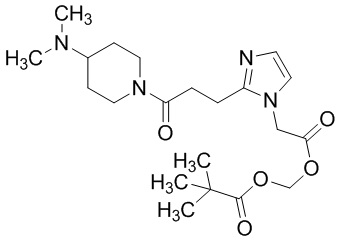
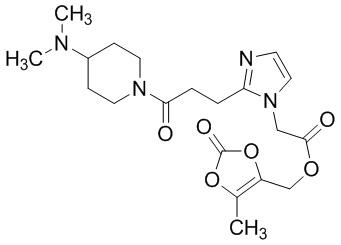
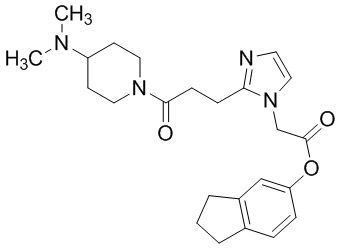
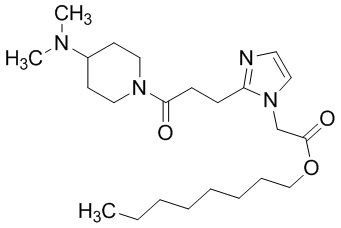
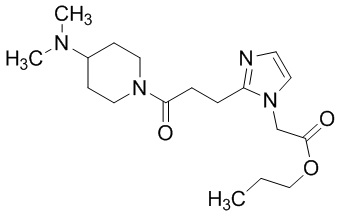
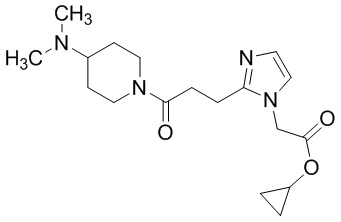
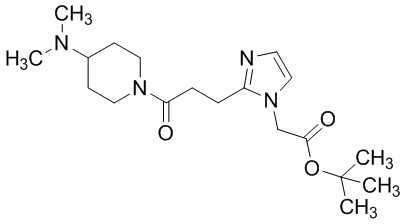
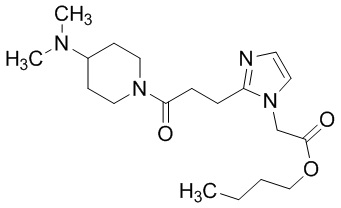
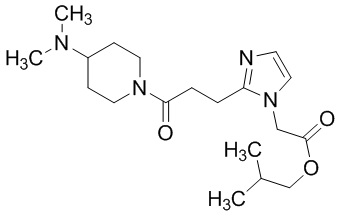
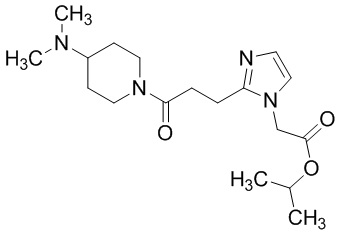
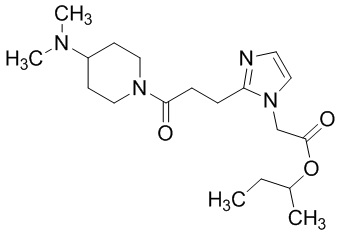
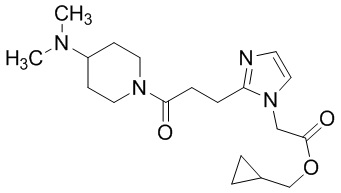
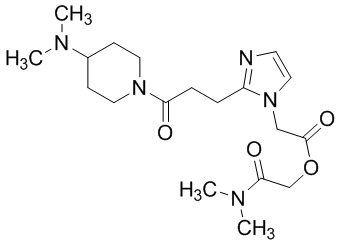
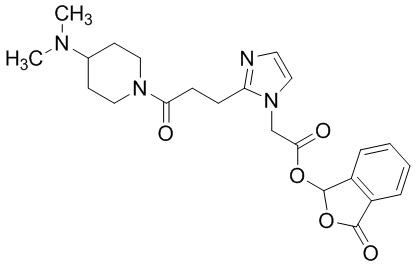
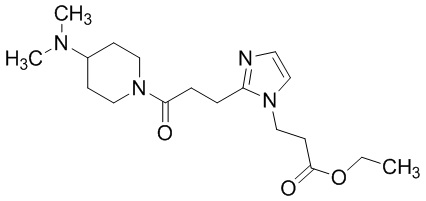
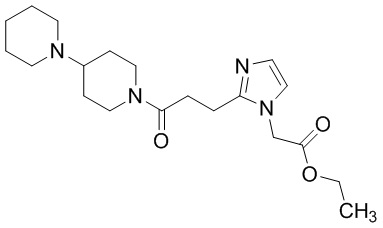
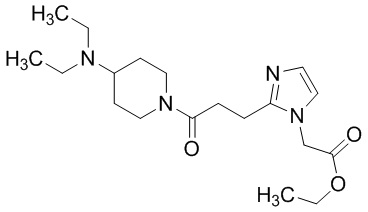
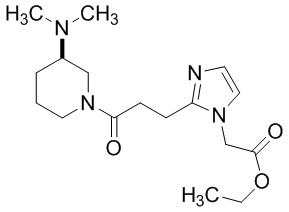
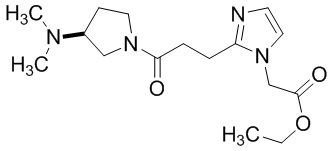
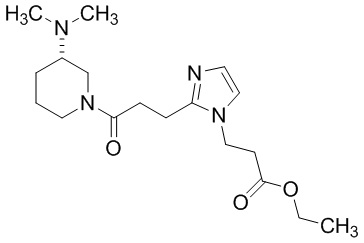
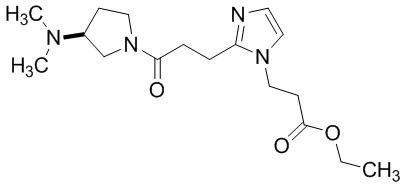
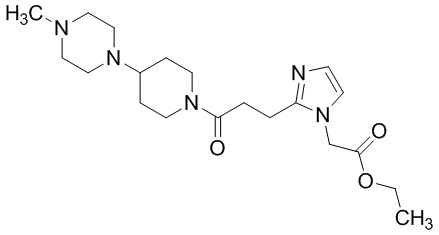
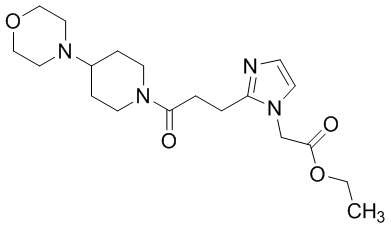
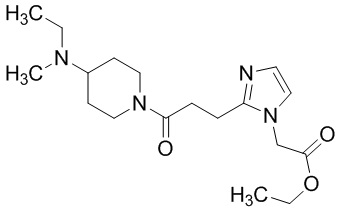
8. Лекарственное средство, обладающее анальгезирующим эффектом против боли, содержащее эффективное количество производного циклического амина, пролекарства производного циклического амина или его фармакологически приемлемой соли по любому из пп.1-7 в качестве активного ингредиента.
9. Анальгетический агент, содержащий производное циклического амина, пролекарство производного циклического амина или его фармакологически приемлемую соль по любому из пп.1-7 в качестве активного ингредиента.
10. Терапевтический агент при невропатической боли, содержащий производное циклического амина, пролекарство производного циклического амина или его фармакологически приемлемую соль по любому из пп.1-7 в качестве активного ингредиента.
11. Терапевтический агент при синдроме фибромиалгии, содержащий производное циклического амина, пролекарство производного циклического амина или его фармакологически приемлемую соль по любому из пп.1-7 в качестве активного ингредиента.
| US 2003236282 A1, 25.12.2003 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| КЛАПАН ДЛЯ ИЗМЕНЕНИЯ НАПРАВЛЕНИЯ ВОЗДУХА ИЛИ ГАЗОВ В ВЕНТИЛЯЦИОННЫХ УСТРОЙСТВАХ, СУШИЛЬНЯХ И Т.П. | 1926 |
|
SU6507A1 |
| ЦИС-ИМИДАЗОЛИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ MDM2 | 2004 |
|
RU2354649C2 |

