Настоящее изобретение относится к ингибирующему гликозилтрансферазу иминосахару в кристаллической форме, который можно применять в лечении болезни Гоше, способу его получения и содержащей его фармацевтической композиции.
УРОВЕНЬ ТЕХНИКИ
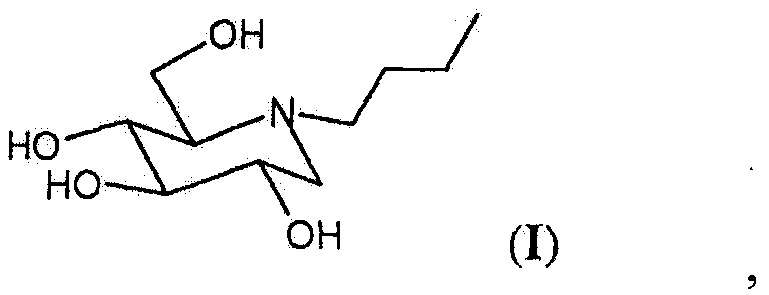
N-бутил-1,5-дидезокси-1,5-имино-D-глюцит формулы (I), также известный как N-бутил-1-дезоксинойиримицин или миглустат, является мощным ингибитором гликозилтрансферазы и применяется главным образом в лечении болезни Гоше.
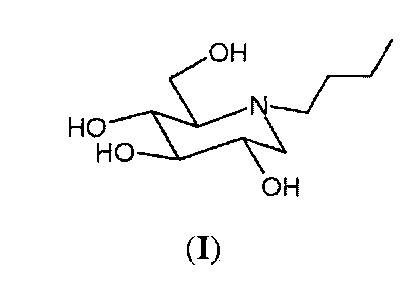
Миглустат относится к классу азасахаров или иминосахаров, то есть соединений с множеством видов биологической активности, характеризующихся присутствием атома азота вместо атома кислорода в фуранозном или пиранозном кольце сахара. Синтез азасахаров в качестве аналогов углеводов начался более 50 лет назад. Первый азасахар был синтезирован более сорока лет назад и представлял собой 1-дезоксинойиримицин формулы (II), который был выделен из природных источников только спустя несколько лет и проявлял значительную биологическую активность.
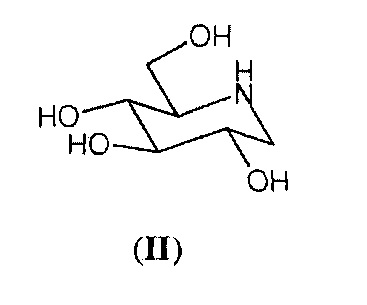
В 1980-х ряд исследований, проводимых в отношении биологической активности N-алкилированных производных 1-дезоксинойиримицина формулы (II), показал, что указанные соединения обладают большей активностью, чем 1-дезоксинойиримицин, и N-бутильное производное формулы (I) оказалось одним из лучших. Поскольку оно являлось синтетическим производным 1-дезоксинойиримицина, первые синтезы миглустата, разумеется, осуществляли путем введения бутильной цепи в 1-дезоксинойиримицин формулы (II) или его производные, защищенные с помощью функциональных групп, посредством восстановительного аминирования с использованием бутиральдегида (см., например, US 4639436 и ЕР 367748).
Указанный синтез, очевидно, сместил проблему синтеза при получении N-алкилированного производного на эффективный синтез 1-дезоксинойиримицина, который, хотя и присутствует в природе во многих растениях и микроорганизмах, не может быть выделен в количествах, достаточных для того, чтобы обеспечить его промышленное использование, поэтому должен быть получен посредством химического синтеза. На протяжении ряда лет сообщалось о различных способах получения 1-дезоксинойиримицина, некоторые из которых являлись полностью химическими или биохимическими, при помощи более или менее сложных микроорганизмов, как правило, начиная с сахаров, таких как глюкоза и рибоза. Вызывающий интерес синтез N-алкилированных производных 1-дезоксинойиримицина, включая миглустат, был опубликован Baxter и Reitz в J. Org. Chem. 1994, 59, 3175-3185. В данном синтезе используют один из классических способов получения пиперидина и пирролидина, а именно двойное восстановительное аминирование 1,5-дикарбонильных производных с помощью первичных аминов.
Существует три основные проблемы, связанные с разработкой в промышленном масштабе способа Baxter и Reitz, к которым относится 1) получение 5-кетоглюкозы, которое включает несколько этапов синтеза, применение соединений на основе олова и малый выход продукта; 2) стереохимия восстановительного аминирования, которое является селективным для получения изомера со глюко-стереохимией только для некоторых типов заместителей на гидроксилах исходного дикарбонила; и последнее, но не менее важное, 3) критические этапы, относящиеся к обработке и очистке конечного продукта, который очищают с помощью флэш-хроматографии.
Первые две проблемы были частично преодолены со временем с помощью синтеза, описанного Matos C.R.R. et al. (Synthesis 1999, 571-573), в котором используется защищенное промежуточное соединение миглустата формулы (III),
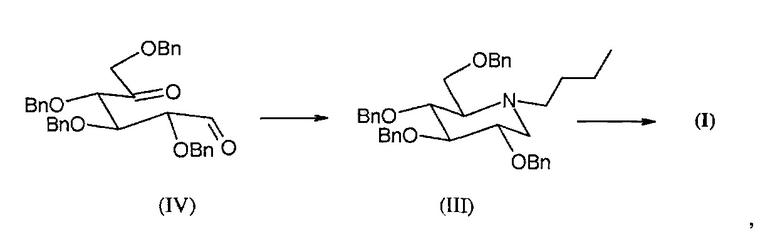
полученное из защищенного дикарбонила формулы (IV), который может быть получен без применения производных олова и с получением большого выхода продукта, начиная с коммерчески доступного 2,3,4,6-тетра-O-бензил-D-глюцита, или путем восстановления 2,3,4,6-тетра-O-бензил-D-глюкозы, также коммерчески доступной, которая в свою очередь может быть получена из D-глюкозы с помощью известных способов. Из промежуточного соединения формулы (III) в результате реакции дебензилирования получают миглустат формулы (I).
Реакцию восстановительного аминирования соединения формулы (IV), описанного выше, повторяли в лабораториях компании и полученный по окончании реакции неочищенный продукт анализировали с помощью ВЭЖХ.
Таким образом, было продемонстрировано, что в действительности восстановительное аминирование не является полностью селективным, и образование диастереоизомера с идо-конфигурацией формулы (V),

происходит наряду с образованием желаемого производного с глюко-конфигурацией формулы (III).
Как сообщалось Matos C.R.R. et al., промежуточное соединение формулы (III) очищают флэш-хроматографией на силикагеле и после выпаривания фракций, содержащих продукт, получают твердое вещество с температурой плавления 64-65°С.
Когда процесс повторили и проанализировали полученное в результате твердое вещество формулы (III), его недостаточная чистота была подтверждена с помощью анализа ВЭЖХ, и твердое вещество было аморфным с температурой плавления приблизительно 64°С.
Следовательно, существует потребность в более эффективном альтернативном способе получения миглустата и, в частности, его защищенного промежуточного соединения формулы (III). Указанный новый способ должен быть, в частности, более применимым в промышленном масштабе и, следовательно, должен включать эффективный способ очистки промежуточного соединения (III) без применения очистки с помощью хроматографии с получением миглустата со степенью чистоты, достаточной для того, чтобы позволить его применение в области фармации и в то же время обеспечить желаемые соединения с большим выходом продукта.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ И СПОСОБОВ АНАЛИЗА
Данные спектров рентгеновской дифракции (XRPD) для N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюцита формулы (III) в кристаллической форме, называемого в данном документе форма А, и N-бутил-1,5-дидезокси-1,5-имино-D-глюцита формулы I, называемого в данном документе кристаллическая форма I, собирали с помощью автоматического порошкового дифрактометра APD-2000, изготовленного Ital-Structures, со следующими рабочими параметрами: геометрия Брегга-Брентано, CuKa-излучение (λ=1,54 Å), сканирование с диапазоном значений угла 2θ 3-40°, с размером шага 0,03° в 1 с. Использованный детектор представлял собой сцинтиллятор.
DSC-термограммы для N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюцита формулы (III) в кристаллической форме, называемого в данном документе форма А, получали с помощью дифференциального сканирующего калориметра DSC 822е от Mettler-Toledo со следующими рабочими параметрами: открытый алюминиевый тигель, диапазон 30-400°С при скорости 10°С/мин с азотом в качестве продувочного газа (80 мл/мин).
DSC-термограммы для N-бутил 1,5-дидезокси-1,5-имино-D-глюцита формулы (I), называемого в данном документе кристаллическая форма I, были получены с помощью дифференциального сканирующего калориметра DSC 822е от Mettler-Toledo со следующими рабочими параметрами: алюминиевый тигель с перфорированной крышкой, диапазон 30-300°С при скорости 10°С/мин с азотом в качестве продувочного газа (80 мл/мин.).
Содержание воды в N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюците в кристаллической форме, называемом в данном документе форма А, определяли путем титрования по методу Карла Фишера.
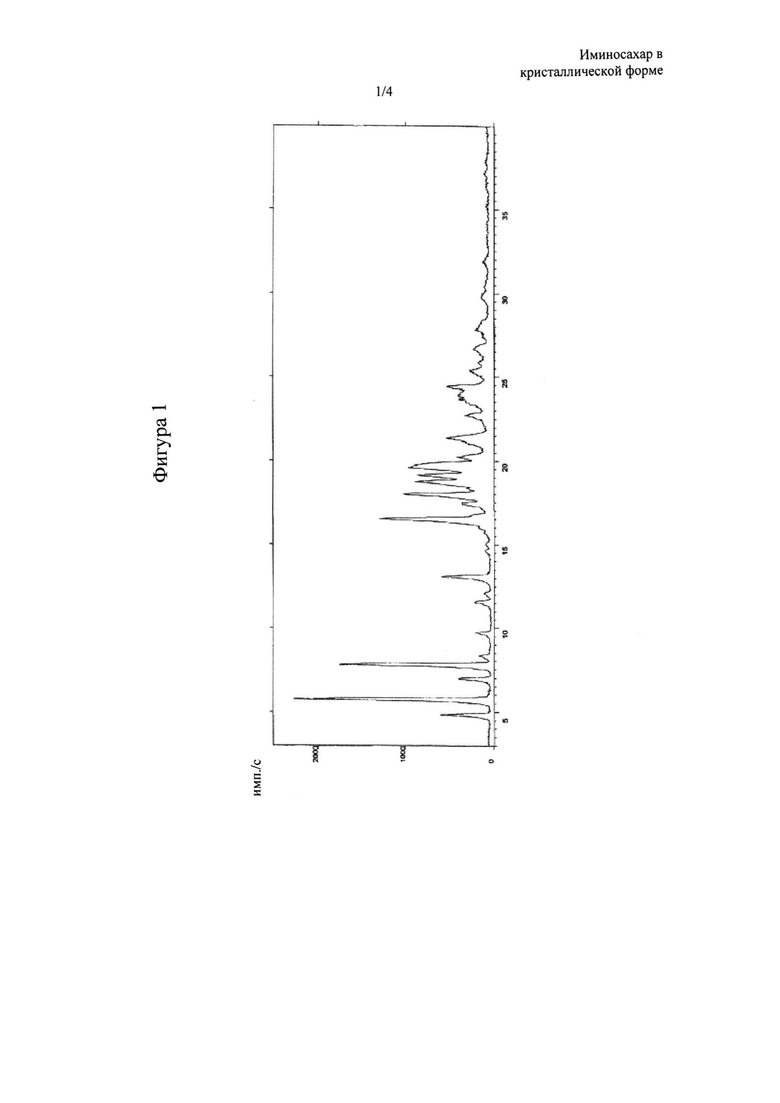
Фигура 1: XRPD-спектр N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюцита в кристаллической форме, называемого в данном документе форма А, где основные пики (выраженные в 2θ°) обнаружены при 4,83, 5,76, 6,96, 7,80, 13,08, 16,50, 17,97, 18,75, 19,14, 19,62+0,2°.
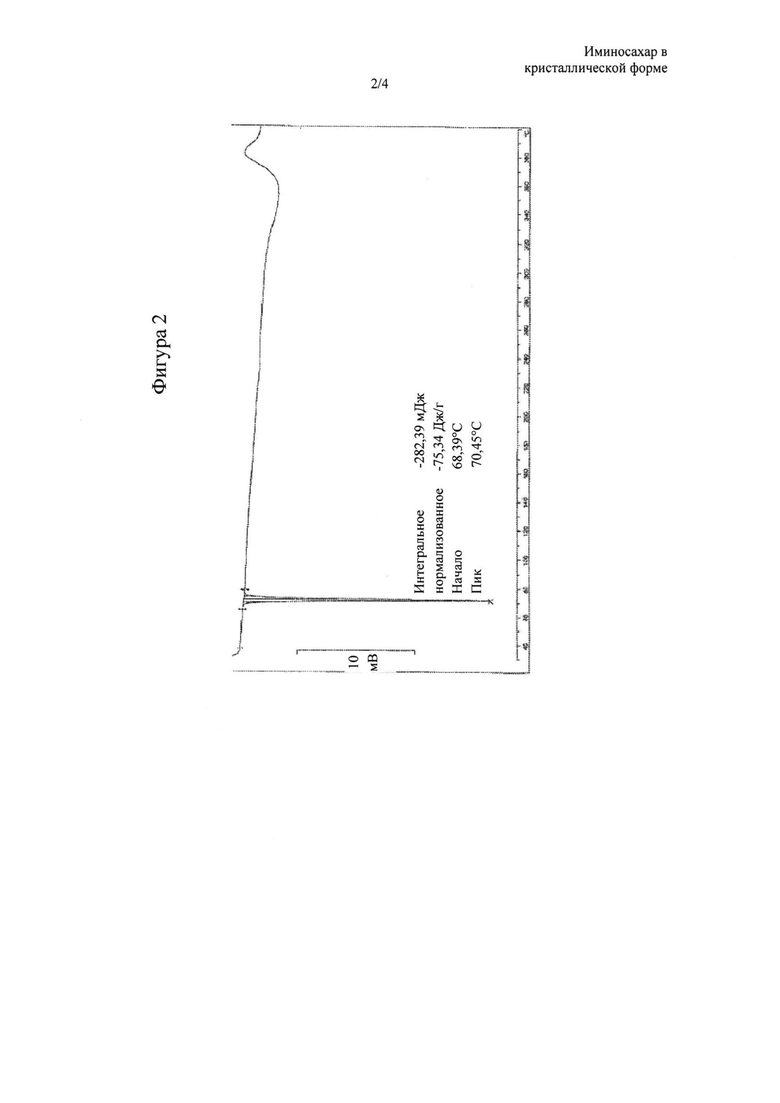
Фигура 2: DSC-термограмма N-бутил-2,3,4,6-тетра-О-бензил-1,5-дидезокси-1,5-имино-D-глюцита в кристаллической форме, называемого в данном документе форма А. Эндотермический пик при приблизительно 70°С указывает на процесс конденсации.
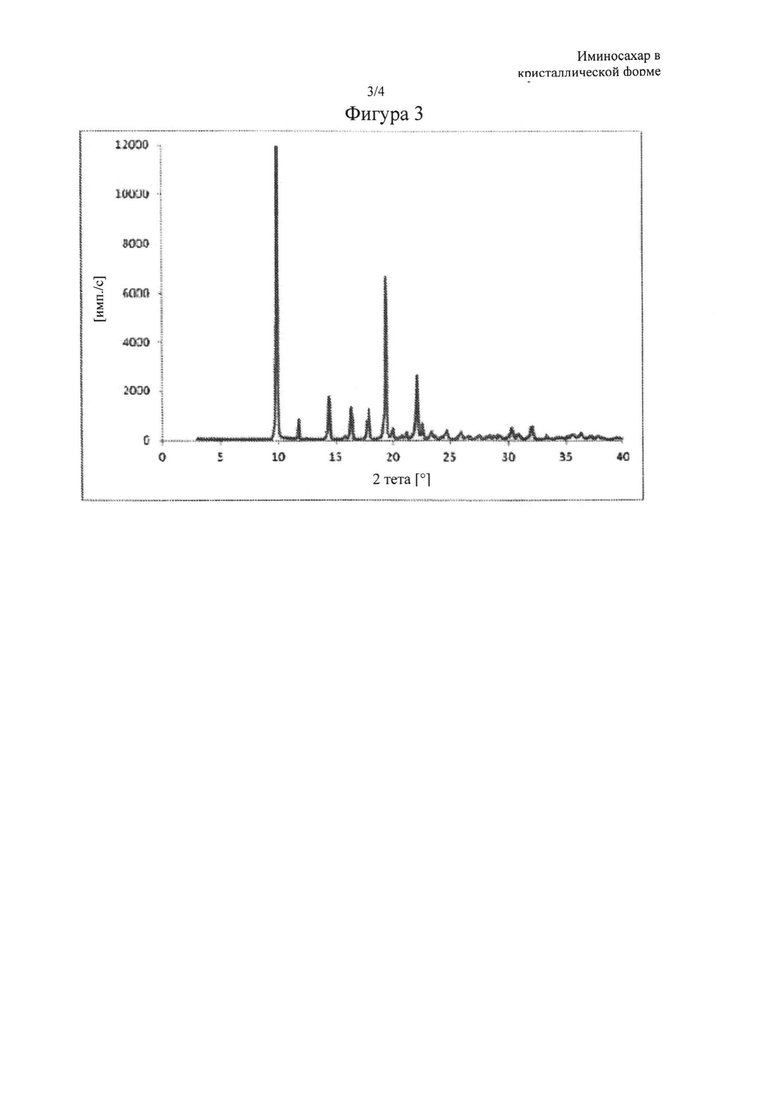
Фигура 3: XRPD-спектр N-бутил-1,5-дидезокси-1,5-имино-D-глюцита в кристаллической форме, называемого в данном документе форма I, где основные пики (выраженные в 2θ°) обнаружены при 9,93, 11,82, 14,46, 15,84, 16,41, 17,76, 17,94, 19,41, 20,01, 20,79, 21,21, 22,14, 22,62, 23,40, 24,75, 26,04 и 30,27±0,2° в 2θ.
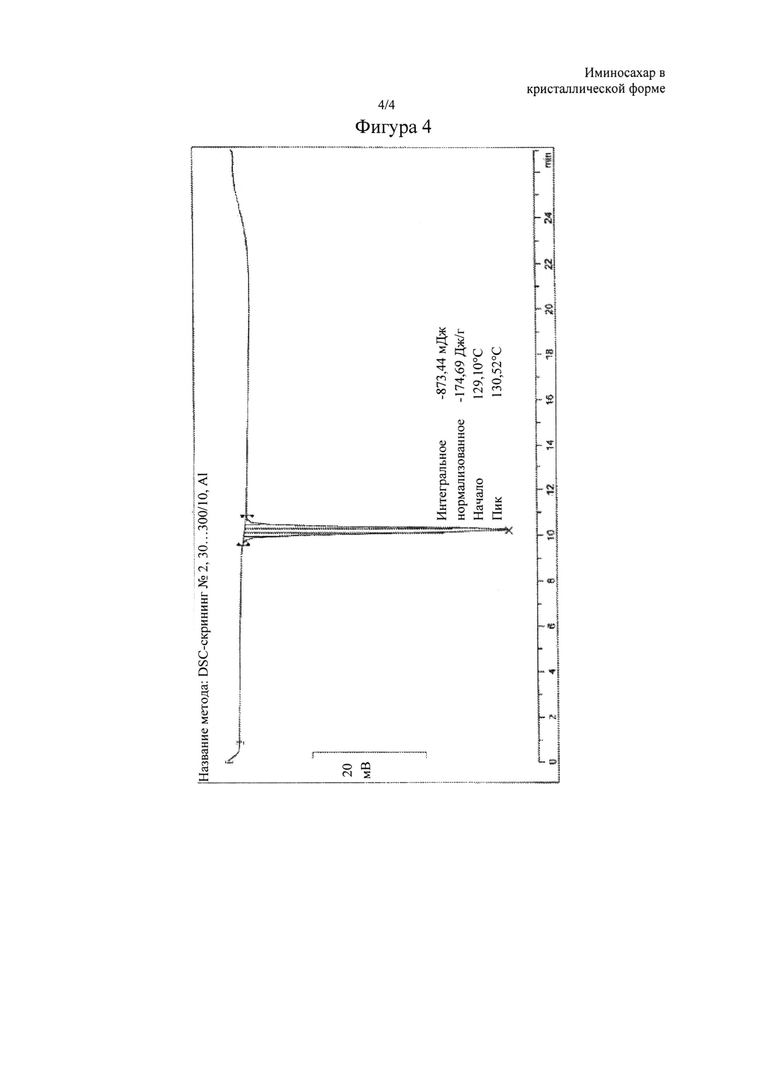
Фигура 4: DSC-термограмма N-бутил-1,5-дидезокси-1,5-имино-D-глюцита в кристаллической форме, называемого в данном документе форма I. Эндотермический пик при 129-130°С указывает на процесс конденсации.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение предусматривает N-бутил-1,5-дидезокси-1,5-имино-d-глюцит в кристаллической форме, называемый в данном документе форма I, способ его получения и содержащую его фармацевтическую композицию.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Первым объектом настоящего изобретения является способ очистки соединения формулы (III),
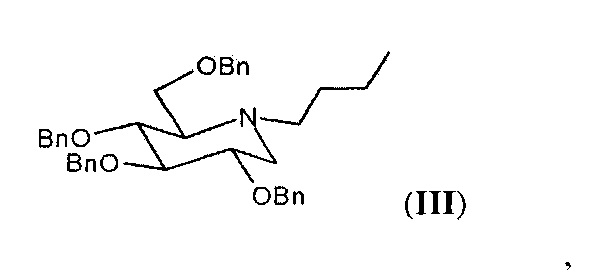
а именно N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюцита, в форме кристаллического твердого вещества, включающий его кристаллизацию из растворяющей среды, содержащей протонный растворитель. Согласно предпочтительному аспекту указанный способ очистки включает:
- растворение соединения формулы (III) в растворяющей среде, содержащей протонный растворитель,
- образование осадка и
- извлечение кристаллического твердого вещества.
Очищенный продукт, получаемый с помощью способа очистки согласно настоящему изобретению, представляет собой твердое вещество в кристаллической форме, N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюцит, в частности, в форме, обозначенной в данном документе как форма А.
Протонный растворитель в растворяющей среде может представлять собой С1-С5алканол с прямой или разветвленной цепью, такой как метанол, этанол или изопропанол, как правило, изопропанол, карбоновую кислоту, такую как уксусная кислота, воду или смесь двух или более, как правило, двух или трех из указанных растворителей.
Концентрация соединения формулы (III) в исходной дисперсии может варьировать в диапазоне от приблизительно 2 до 90% вес/вес, предпочтительно от приблизительно 30 до 70%.
При необходимости для того, чтобы ускорить растворение соединения формулы (III), дисперсию, содержащую указанное соединение, можно нагревать до полного растворения.
Образования осадка можно достичь путем поддержания перемешивания раствора, например, в течение периода времени в диапазоне от приблизительно 5 до 20 часов. При необходимости для того, чтобы ускорить образование осадка, раствор можно охладить, например, до температуры в диапазоне от приблизительно -5° до 5°С. Кроме того, для того, чтобы ускорить образование осадка, также можно вводить затравку в виде ранее полученных кристаллов кристаллической формы А.
Кристаллическое твердое вещество может быть извлечено с помощью известных методик, таких как фильтрация или центрифугирование. В частности, при необходимости извлечение может быть ускорено посредством необязательного добавления растворителя, подходящего для разжижения дисперсии, такого как C1-С5алканол, идентичного или отличающегося от такового, присутствующего в растворяющей среде.
Твердое вещество может быть затем высушено с помощью известных способов, например, высушено в сушильной камере, при температуре в диапазоне от приблизительно 30°С до 55°С в вакууме.
Неочищенный исходный материал, который подлежит очистке с помощью способа согласно настоящему изобретению, может представлять собой неочищенное соединение формулы (III), полученное с помощью любых известных способов, описанных в литературе, например, описанного Matos C.R.R. et al. в Synthesis 1999, 571-573.
Неочищенное соединение формулы (III), используемое в качестве исходного материала, таким образом, характеризуется содержанием вещества по результатам анализа, как правило, в диапазоне от приблизительно 10 до 90% вес/вес, предпочтительно от приблизительно 30% до 70% вес/вес.
Твердое вещество в кристаллической форме, соединение (III), обозначенное в данном документе как форма А, получаемое с помощью способа очистки согласно настоящему изобретению, характеризуется XRPD, изображенной на фигуре 1, где наиболее интенсивные пики (выраженные в 2θ°) обнаруживают при 4,83, 5,76, 6,96, 7,80, 13,08, 16,50, 17,97, 18,75, 19,14, 19,62±0,2°. Оно также характеризуется DSC-термограммой, изображенной на фигуре 2, где эндотермический пик при приблизительно 70°С указывает на процесс конденсации. Поскольку указанная кристаллическая форма А характеризуется содержанием воды менее 0,2%, предпочтительно менее 0,1%, ее можно охарактеризовать как фактически безводную.
Еще одним объектом настоящего изобретения, таким образом, является соединение формулы (III), а именно N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюцит, в кристаллической форме, в частности, в кристаллической форме А, как определено выше.
Величина кристаллов соединения (III) в кристаллической форме А, получаемых с помощью способа, описанного выше, характеризуется значением D50 в диапазоне от приблизительно 25 до 250 мкм. При необходимости указанное значение может быть уменьшено посредством микронизации или тонкого измельчения.
Полученный по окончании реакции неочищенный продукт получения соединения (III), как правило, характеризующийся содержанием вещества по результатам анализа в диапазоне от приблизительно 10 до 90% вес/вес, предпочтительно от приблизительно 30% до 70% вес/вес, может затем быть подвергнут очистке с помощью способа согласно настоящему изобретению с получением его кристаллической формы, в частности, кристаллической формы А, со степенью химической чистоты, оцененной с помощью ВЭЖХ, более или равной 95%, предпочтительно более или равной 98%.
В частности, соединение формулы (III) в кристаллической форме, в частности, в форме А, полученное с помощью способа согласно настоящему изобретению, содержит соединение в идо-конфигурации формулы (V),
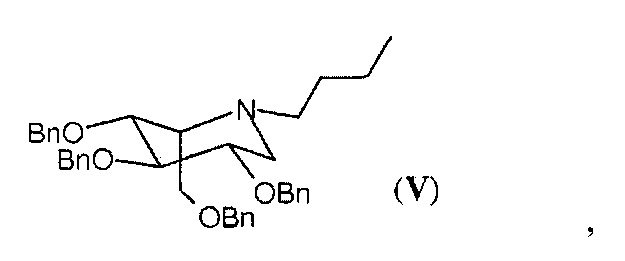
в количествах менее 0,2%, предпочтительно менее 0,1%, рассчитанных с помощью ВЭЖХ.
Полученное таким образом соединение формулы (III) в кристаллической форме, в частности, в кристаллической форме А, может быть подвергнуто реакции дебензилирования с получением миглустата с большим выходом продукта и высокой степенью чистоты, который может затем быть очищен с получением его в кристаллической форме, называемой в данном документе форма I.
Реакцию дебензилирования можно осуществлять в соответствии с известными способами путем удаления бензильной защитной группы от гидроксильных функциональных групп, предпочтительно посредством каталитического гидрирования.
Согласно предпочтительному аспекту настоящего изобретения миглустат может быть получен в кристаллической форме I с помощью способа очистки, включающего:
- образование раствора миглустата в С1-С5алканоле с прямой или разветвленной цепью, предпочтительно метаноле или этаноле;
- добавление С3-С7 с прямой или разветвленной цепью, предпочтительно ацетона или метилэтилкетона;
- охлаждение смеси
- и извлечение твердого вещества.
Раствор миглустата в С1-С5алканоле может быть образован при температуре, как правило, в диапазоне от приблизительно 40 до 60°С.
Смесь можно охлаждать со скоростью, как правило, в диапазоне от 0,1 до 5°С/мин, предпочтительно от 0,1 до 0,3°С/мин, до тех пор, пока ее температура не будет находиться в диапазоне от температуры окружающей среды до приблизительно -5°С. Поддерживают перемешивание смеси на протяжении указанного охлаждения и в общей сложности от приблизительно 2 до 20 часов после того.
Кристаллическое твердое вещество может быть извлечено с помощью известных методик, таких как фильтрация или центрифугирование, и необязательно сушки, такой как сушка в сушильной камере, при низком давлении.
Продукт миглустат, а именно N-бутил-1,5-дидезокси-1,5-имино-D-глюцит, полученный с помощью способа очистки согласно настоящему изобретению, находится в кристаллической форме, называемой в данном документе форма I, характеризуется XRPD-спектром, изображенным на фигуре 3, где основные пики (выраженные в 2θ°) обнаружены при 9,93, 11,82, 14,46, 15,84, 16,41, 17,76, 17,94, 19,41, 20,01, 20,79, 21,21, 22,14, 22,62, 23,40, 24,75, 26,04 и 30,27±0,2° в 2θ; a DSC-термограмма, изображенная на фигуре 4, имеет эндотермический пик при 129-130°С, что указывает на процесс конденсации.
Полученный таким образом миглустат, в частности, в кристаллической форме I, характеризуется степенью химической чистоты более или равной 98%, предпочтительно более или равной 99%, рассчитанной с помощью ВЭЖХ.
Еще одним объектом настоящего изобретения, таким образом, является способ получения миглустата формулы (I), в частности, в кристаллической форме I,
включающий применение в качестве исходного материала соединения формулы (III) в кристаллической форме, в частности, в кристаллической форме А, как определено в данном документе.
Продукт миглустат, в качестве API (активного фармацевтического ингредиента), полученный с помощью способа согласно настоящему изобретению, в частности, в кристаллической форме I, содержит соединение в идо-конфигурации формулы (V),
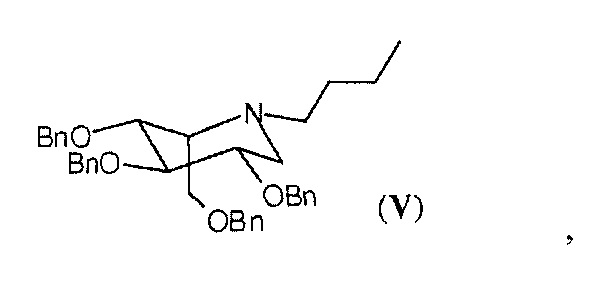
в количествах менее 0,1%, предпочтительно менее 0,05%, рассчитанных с помощью ВЭЖХ.
Еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая миглустат в качестве активного ингредиента, в частности, в кристаллической форме I, соединение в идо-конфигурации формулы (V),
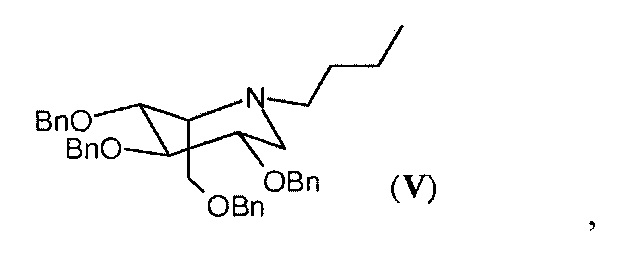
в количествах менее 0,1%, предпочтительно менее 0,05%, рассчитанных с помощью ВЭЖХ, и фармацевтически приемлемый носитель и/или разбавитель.
Еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая миглустат в качестве активного ингредиента, в частности, в кристаллической форме I, и фармацевтически приемлемый носитель и/или разбавитель.
Количество активного вещества, в частности, в виде кристаллической формы I, которое предполагается вводить млекопитающему, как правило, человеку, может, как правило, находиться в диапазоне от приблизительно 70 до 150 мг, предпочтительно 100 мг.
Предпочтительным путем введения является пероральный в форме капсул, таблеток, сиропов, хотя также может применяться парентеральное введение.
Фармацевтическая композиция может быть получена в соответствии с известными способами, например, как раскрыто в патентном документе США 5472969.
Размер кристаллов миглустата, получаемых с помощью способа, описанного выше, в частности, в кристаллической форме I, характеризуется значением D50 от приблизительно 25 до 250 мкм. При необходимости указанное значение может быть уменьшено посредством микронизации или тонкого измельчения.
Следующие примеры иллюстрируют настоящее изобретение.
Пример 1. Синтез N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюцита (III)
Раствор оксалилхлорида (99,8 г, 0,79 моль) в дихлорметане (300 мл) охлаждали до -75°С, последовательно обрабатывали в инертной атмосфере раствором DMSO (77,1 г, 0,99 моль) в дихлорметане (100 мл), добавленным медленно по каплям, а затем спустя приблизительно 1 ч. раствором, полученным путем растворения 2,3,4,6-тетра-O-бензил-D-глюцита, полученного, как описано в Synthesis 1999, 571-573 (анализ ВЭЖХ, 94,5%, 105,9 г, 0,18 моль) в дихлорметане (100 мл), добавленным медленно по каплям. Поддерживали перемешивание реакционной смеси при температуре, не превышавшей 65°С, и обрабатывали спустя приблизительно 2 часа триэтиламином (187 г, 1,85 моль), добавленным медленно по каплям, поддерживали перемешивание реакционной смеси при температуре, не превышавшей 50°С, в течение по меньшей мере 4 часов. Полученную по окончании реакции смесь затем добавляли к смеси, в которой поддерживали перемешивание в инертной атмосфере при температуре 0°С, полученной путем смешивания н-бутиламина (135 г, 1,84 моль), уксусной кислоты (111 г, 1,85 моль), сульфата натрия (32,5 г, 0,51 моль) и цианоборогидрида натрия (31,7 г, 0,48 моль) в метаноле (400 мл). Значение рН полученной таким образом реакционной смеси корректировали путем добавления дополнительной уксусной кислоты до того момента, пока не получали значение рН 6, и поддерживали перемешивание смеси при приблизительно 20°С в течение 15 часов. Полученную по окончании реакции смесь затем последовательно обрабатывали 20% водным раствором NaOH, 3М НСl до рН 6, 11%) раствором NaClO, 10% раствором Na2SO3, насыщенным раствором NaHCO3 и, наконец, нейтральной водой. Полученную таким образом органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали при низком давлении с получением маслянистого остатка с весом приблизительно 120 г. В полученном таким образом неочищенном продукте, анализированном с помощью ВЭЖХ, соотношение между продуктом формулы (III) и продуктом формулы (V) составляло приблизительно 4:1.
Пример 2. Кристаллизация N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюцита (III)
Неочищенное соединение формулы (III), полученное, как в примере 1, растворяли в изопропаноле (120 мл) и полученный в результате раствор охлаждали в ледяной бане и обрабатывали водой (18 мл). Поддерживали перемешивание полученной суспензии при приблизительно 20°С в течение 15 часов, а затем фильтровали через воронку Бюхнера и панель промывали изопропанолом. Влажное твердое вещество сушили в сушильной камере при температуре 50°С в вакууме до постоянного веса с получением 50 г соединения формулы (III) с высокой степенью химический чистоты в кристаллической форме А, где основные пики (выраженные в 2θ°) обнаружены при 4,83, 5,76, 6,96, 7,80, 13,08, 16,50, 17,97, 18,75, 19,14, 19,62. Указанный кристаллический продукт характеризуется DSC-термограммой, изображенной на фигуре 2, и содержанием воды менее 0,1%.
Соединение формулы (III) может быть рекристаллизовано из изопропанола само с получением соединения формулы (III) со степенью чистоты, рассчитанной с помощью ВЭЖХ, превышающей 99%.
Пример 3. Синтез миглустата формулы (I)
Раствор, полученный путем смешивания N-бутил-2,3,4,6-тетра-O-бензил-1,5-дидезокси-1,5-имино-D-глюцита формулы (III), полученного, как в примере 2 (105,1 г, 0,17 моль), в метаноле (500 мл) в присутствии 32% НСl (43,5 г), обрабатывали 16% Pd/C (10,2 г). Поддерживали интенсивное перемешивание смеси в атмосфере водорода при 4 бар в течение приблизительно 4 часов, а затем фильтровали через перлитовую панель и концентрировали полученный раствор при низком давлении. Полученный таким образом остаток твердого вещества растворяли в воде (100 мл) и полученный кислый раствор пропускали через колонку на ионообменной смоле, активируемой основной формой (Amberlite IRA 900Сl). Фракции, в которых нингидриновая проба была положительной, объединяли и концентрировали при низком давлении с получением 50 г миглустата в виде маслянистого остатка, имеющего степень химической чистоты, превышающую 98%, рассчитанную с помощью ВЭЖХ.
Пример 4. Кристаллизация миглустата формулы (I)
Разбавляли 200 г миглустата формулы (I), полученного, как в примере 3, в метаноле и обрабатывали ацетоном при медленном добавлении по каплям при перемешивании при приблизительно 20°С. Поддерживали перемешивание образовавшейся суспензии при одинаковой температуре в течение 5 часов, а затем фильтровали через воронку Бюхнера и промывали полученное твердое вещество ацетоном и сушили при 50°С в вакууме до постоянного веса. Получали 131 г миглустата со степенью чистоты, превышающей 99,5%, рассчитанной с помощью ВЭЖХ.
Полученный таким образом продукт находился в кристаллической форме, называемой в данном документе форма I, характеризовался XRPD-спектром, изображенным на фигуре 3, где основные пики (выраженные в 2θ°) обнаружены при 9,93, 11,82, 14,46, 15,84, 16,41, 17,76, 17,94, 19,41, 20,01, 20,79, 21,21, 22,14, 22,62, 23,40, 24,75, 26,04 и 30,27+0,2° в 2θ, как показано на фигуре 3; a DSC-термограмма имеет эндотермический пик при 129-130°С, что указывает на процесс конденсации, как показано на фигуре 4.
Пример 5. Кристаллизация миглустата формулы (I)
Разбавляли 200 г миглустата в форме масла, полученного, как в примере 3, с 100 мл метанола и оставляли перемешиваться при 50°С. Поддерживая температуру, добавляли по каплям 1000 мл ацетона в течение приблизительно 1 ч. Кристаллизовалось белое твердое вещество. По окончании добавления смесь охлаждали до температуры окружающей среды за приблизительно 2 ч и оставляли перемешиваться при этой температуре в течение приблизительно 15 ч. Твердое вещество извлекали путем фильтрации через воронку Бюхнера и промывали ацетоном. Твердое вещество сушили при низком давление при приблизительно 50°С до получения постоянной массы. Извлекали 131 г кристаллического миглустата.
Полученный таким образом продукт находился в кристаллической форме, называемой в данном документе форма I, характеризующейся XRPD-спектром, изображенным на фигуре 3, где основные пики (выраженные в 2θ°) обнаружены при 9,93, 11,82, 14,46, 15,84, 16,41, 17,76, 17,94, 19,41, 20,01, 20,79, 21,21, 22,14, 22,62, 23,40, 24,75, 26,04 и 30,27±0,2° в 2θ, как показано на фигуре 3; a DSC-термограмма имеет эндотермический пик при 129-130°С, что указывает на процесс конденсации, как показано на фигуре 4.
| название | год | авторы | номер документа |
|---|---|---|---|
| Миглустат N-бутил-1,5-дидезокси-1,5-имино-D-глюцит | 2019 |
|
RU2743694C1 |
| Способ получения (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола | 2020 |
|
RU2762605C1 |
| Способ получения производных 3,4,5-триоксипиперидина | 1981 |
|
SU1050563A3 |
| Способ получения производных 3,4,5-триоксипиперидина | 1980 |
|
SU1017168A3 |
| СОКРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО С-ГЛИКОЗИДА C L-ПРОЛИНОМ | 2007 |
|
RU2408595C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНЫХ ДИГИДРОПИРИМИДИНА | 2013 |
|
RU2646599C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА TLR7/TLR8 | 2019 |
|
RU2792005C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА BTK | 2020 |
|
RU2828460C2 |
| КРИСТАЛЛИЧЕСКАЯ ИЛИ АМОРФНАЯ ФОРМА АГОНИСТОВ FXR, ПРЕДСТАВЛЯЮЩИХ СОБОЙ ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2800751C2 |
| С-ГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ И ИХ СОЛИ | 2004 |
|
RU2317288C2 |
Изобретение относится к применимому в медицине миглустату, а именно N-бутил-1,5-дидезокси-1,5-имино-D-глюциту в кристаллической форме I, характеризующейся XRPD-спектром, изображенным на фигуре 3, в котором наиболее интенсивные пики (выраженные в 2θ°) должны обнаруживаться при 9,93, 11,82, 14,46, 15,84, 16,41, 17,76, 17,94, 19,41, 20,01, 20,79, 21,21, 22,14, 22,62, 23,40, 24,75, 26,04 и 30,27±0,2° в 2θ; и DSC-термограммой, изображенной на фигуре 4, которая имеет эндотермический пик при 129-130°С, а также способу его получения путем дебензилирования 2,3,4,6-тетра-O-бензил-D-глюцита с последующей кристаллизацией в С1-С5 алканоле при добавлении С3-С7 кетона с охлаждением смеси и извлечением твердого вещества. Предложена новая кристаллическая форма ценного соединения и фармацевтические композиции на ее основе, эффективные для лечения болезни Гоше. 5 н. и 2 з.п. ф-лы, 5 пр., 4 ил.
1. Миглустат, а именно N-бутил-1,5-дидезокси-1,5-имино-D-глюцит в кристаллической форме, называемый в данном документе форма I, характеризующийся XRPD-спектром, изображенным на фигуре 3, в котором наиболее интенсивные пики (выраженные в 2θ°) должны обнаруживаться при 9,93, 11,82, 14,46, 15,84, 16,41, 17,76, 17,94, 19,41, 20,01, 20,79, 21,21, 22,14, 22,62, 23,40, 24,75, 26,04 и 30,27±0,2° в 2θ; и DSC-термограммой, изображенной на фигуре 4, которая имеет эндотермический пик при 129-130°С.
2. Миглустат в кристаллической форме I по п. 1, где кристаллы имеют значение D50 в диапазоне от 25 до 250 мкм.
3. Способ получения миглустата формулы (I) в кристаллической форме I по п. 1,
включающий проведение реакции дебензилирования соединения формулы (III) в кристаллической форме, в частности в кристаллической форме А,
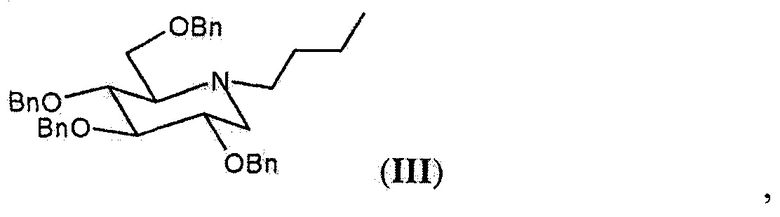
характеризующегося XRPD, изображенной на фигуре 1, где наиболее интенсивные пики (выраженные в 2θ°) должны обнаруживаться при 4,83, 5,76, 6,96, 7,80, 13,08, 16,50, 17,97, 18,75, 19,14, 19,62±0,2°, путем удаления бензильной защитной группы от гидроксильных функциональных групп посредством каталитического гидрирования в метаноле;
и процесс очистки для получения миглустата в кристаллической форме I по п. 1, включающий
получение раствора миглустата в C1-C5 алканоле с прямой или разветвленной цепью, предпочтительно в метаноле или этаноле; добавление С3-С7 кетона с прямой или разветвленной цепью, предпочтительно ацетона или метилэтилкетона; охлаждение смеси; и извлечение твердого вещества.
4. Способ получения миглустата в кристаллической форме I по п. 1 с помощью способа очистки, включающего:
- получение раствора миглустата в С1-С5 алканоле с прямой или разветвленной цепью, предпочтительно метаноле или этаноле;
- добавление С3-С7 кетона с прямой или разветвленной цепью, предпочтительно ацетона или метилэтилкетона;
- охлаждение смеси и
- извлечение твердого вещества.
5. Фармацевтическая композиция для лечения болезни Гоше, содержащая по меньшей мере 99% по весу миглустата в кристаллической форме I по п. 1.
6. Фармацевтическая композиция для лечения болезни Гоше, содержащая миглустат в кристаллической форме I по п. 1 в качестве активного ингредиента и фармацевтически приемлемый носитель и/или разбавитель.
7. Фармацевтическая композиция по п. 6, где кристаллы миглустата имеют значение D50 в диапазоне от 25 до 250 мкм.
| EP 367748 B1, 25.01.1995 | |||
| EP 367748 B1, 25.01.1995 | |||
| Carlos R.r | |||
| et al, Synthesis, 1999, N 4, 571-573 | |||
| US 5472969 A1, 05.12.1995 | |||
| Способ получения производных 3,4,5-триоксипиридина или их солей | 1978 |
|
SU917697A3 |

