Область изобретения
Настоящее изобретение принадлежит к области медицины и относится к производному бензофурана, способу его получения и его применению в медицинских исследованиях. В настоящем изобретении раскрыто применение производного в качестве ингибитора EZH2 в предупреждении и/или лечении заболевания, такого как опухоль и рак и т.д., в частности, в предупреждении и/или лечении неходжкинской лимфомы, диффузной крупноклеточной В-клеточной лимфомы, фолликулярной лимфомы и синовиальной саркомы и т.д.
Предшествующий уровень техники
Возникновение опухоли представляет собой процесс эволюции многочисленных факторов и многочисленных этапов, в который вовлечены мутационные и эпигенетические изменения множественных генов, таких как онкоген, антионкоген и ген репарации повреждений ДНК. Строго говоря, эпигенетику определяют как комбинацию генетических механизмов, способных к изменению функции генома в дополнение к прямому изменению последовательности ДНК. В настоящем документе «эпигенетика» в широком смысле относится к элементам структуры хроматина, которые контролируют функцию генома независимо от того, является ли этот контроль наследуемым. Эпигенетика или регуляция на уровне хроматина играет важную роль в экспрессии генов в нормальной физиологической функции или в развитии рака.
Белок группы polycomb (PcG, от англ. - Polycomb group protein) является важным белковым фактором, вовлеченным в отрицательную эпигенетическую регуляцию хроматиновых генов. У млекопитающих PcG главным образом делится на два типа с различными структурами и функциями: репрессивный комплекс polycomb 2 (PRC2, от англ. - polycomb repressive complex 2) и репрессивный комплекс polycomb 1 (PRC1). Метилтрансфераза гистонов, кодируемая геном EZH2, является каталитичнеским компонентом репрессивного комплекса polycomb 2 (PRC2) и проявляет метилтрансферазную активность на остатке лизина 27 (H3K27) гистона Н3 с образованием H3K27me3 посредством домена SET субъединицы EZH2. Это в результате приводит к транскрипционной репрессии посредством различных механизмов, включая экстренный рекрутмент метилтрансфераз ДНК (DNMT, от англ. - DNA methyltransferases) и PRC1, которые осуществляют убиквитинирование H2AK119. Кодонная мутация в кодоне 641 EZH2, наиболее частой горячей точке мутаций, представляет собой мутацию, при которой белковый продукт экспрессии мутантного гена приобретает новые и патологические функции, приводящую к усиленному метилированию H3K27, и играет важную роль в туморогенезе диффузной крупноклеточной В-клеточной лимфомы GCB-типа (DLBCL, от англ. - diffuse large B-cell lymphoma) и фолликулярной лимфомы (FL). Возвратные соматические мутации в домене SET гена Ezh2 характерны у пациентов с диффузными крупноклеточными В-клеточными лимфомами (DLBCL). Кроме того, сверхэкспрессия EZH2 характерна для ряда типов опухоли с неблагоприятным прогнозом, включая злокачественные (раковые) опухоли, лимфомы и саркомы мягких тканей и т.д. Экспрессия EZH2 в синовиальной саркоме связана с высокой степенью триметилирования H3K27. Концентрация белка EZH2 обычно повышена в раковых тканях по сравнению с нормальными тканями, и наиболее высокая экспрессия EZH2 отмечена в распространенных злокачественных опухолях или опухолях с неблагоприятным прогнозом. В некоторых типах злокачественных опухолей сверхэкспрессия EZH2 происходит одновременно с амплификацией гена EZH2. В многочисленных исследованиях малых интерферирующих (ми)/коротких шпилечных (кш) РНК показано, что снижение экспрессии Ezh2 в линиях опухолевых клеток может приводить к ингибированию пролиферации, миграции и инвазии опухолевых клеток или ангиогенеза и к апоптозу.
Заявки на патенты, в которых раскрыты селективные ингибиторы EZH2, включают WO 2012005805, WO 2012050532, WO 2012118812, WO 2012142513, WO 2012142504, WO 2013049770, WO 2013039988, WO 2013067300, WO 2015141616, WO 2011140325 и т.д.
Ингибиторы EZH2 в качестве лекарственных средств имеют многообещающие перспективы применения в фармацевтической промышленности. Однако все же существует потребность в разработке новых ингибиторов EZH2 для достижения лучшего терапевтического эффекта при опухоли или раке и лучшего соответствия требованиям рынка.
Краткое изложение сущности изобретения
Настоящее изобретение относится к соединению формулы (I) или к его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси или к его фармацевтически приемлемой соли, где структура соединения формулы (I) является такой, как показано ниже:
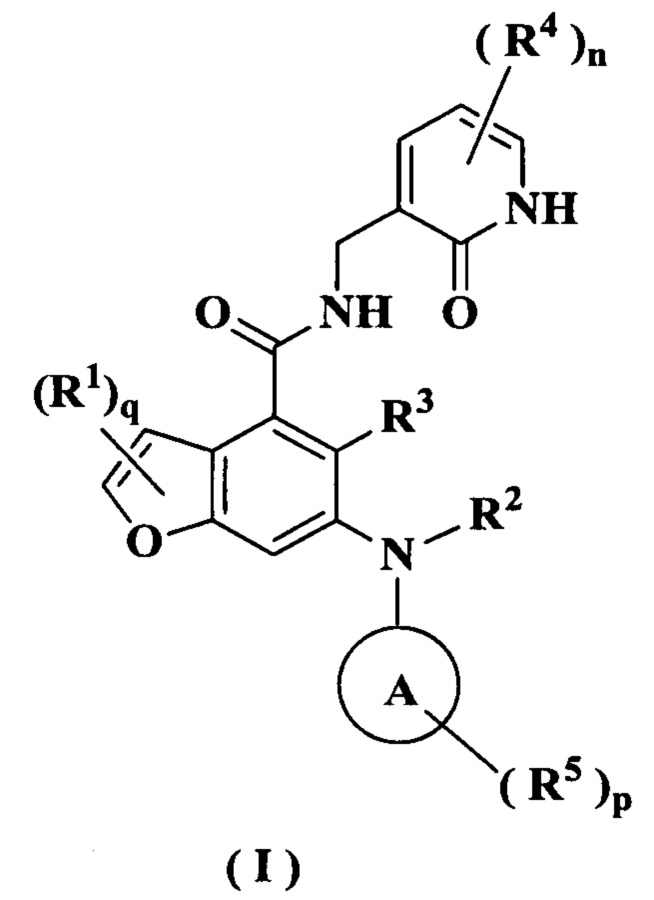
где:
кольцо А выбрано из группы, состоящей из гетероциклила и циклоалкила;
каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, атома галогена, алкила, галогеналкила, алкокси, галогеналкокси, амино, нитро, гидрокси, циано, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)mR6, -S(O)mNR7R8 и -(CH2)xRa, где алкил, галогеналкил, гетероциклил, арил и гетероарил каждый независимо и необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, галогеналкила, атома галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
Ra выбран из группы, состоящей из атома галогена, циклоалкила, гетероциклила и -NR7R8, где циклоалкил и гетероциклил каждый независимо и необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, галогеналкила, атома галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R2 представляет собой атом водорода или алкил, где алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из атома галогена, гидрокси, циано, циклоалкила и гетероциклила;
R3 выбран из группы, состоящей из атома водорода, алкила, атома галогена, циано, алкокси и галогеналкила;
каждый R4 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидрокси, амино, алкокси, галогеналкокси, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)mR6, -S(O)mNR7R8 и -NR7R8;
каждый R5 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, оксо, атома галогена, галогеналкила, гидрокси, амино, алкокси, галогеналкокси, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)mR6, -S(O)mNR7R8 и -NR7R8;
R6 выбран из группы, состоящей из атома водорода, алкила, галогеналкила, алкокси, гидроксиалкила, гидрокси, амино, циклоалкила, гетероциклила, арила и гетероарила;
R7 и R8 идентичны или отличаются друг от друга, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкокси, гидроксиалкила, гидрокси, амино, алкоксикарбонила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, амино, циклоалкил, гетероциклил, арил и гетероарил каждый независимо и необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидрокси, амино, алкоксикарбонила, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
m равно 0, 1 или 2;
n равно 0, 1, 2 или 3;
р равно 0, 1, 2, 3, 4 или 5;
q равно 0, 1 или 2; и
х равно 0, 1, 2 или 3.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо его фармацевтически приемлемой соли n равно 2
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо его фармацевтически приемлемой соли q равно 0 или 1.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо его фармацевтически приемлемой соли р равно 0 или 1.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо его фармацевтически приемлемой соли
каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, атома галогена, алкила, галогеналкила, циано, циклоалкила, гетероциклила и -(CH2)xRa, где алкил и гетероциклил каждый независимо и необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, гидроксиалкила и атома галогена; и Ra выбран из группы, состоящей из атома галогена, циклоалкила, гетероциклила и -NR7R8, где циклоалкил и гетероциклил каждый независимо и необязательно замещен одной или более группами, выбранными из группы, состоящей из гидроксиалкила, алкила и атома галогена; и R7, R8 и х являются такими, как определено в формуле (I).
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо его фармацевтически приемлемой соли, и каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, галогеналкила, циано и -(CH2)xRa, где х равно 0, a Ra представляет собой атом галогена или циклоалкил.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо его фармацевтически приемлемой соли R2 представляет собой алкил, необязательно замещенный циклоалкилом.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо его фармацевтически приемлемой соли R3 выбран из группы, состоящей из алкила, атома галогена и галогеналкила.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо его фармацевтически приемлемой соли каждый R4 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила и алкокси.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо его фармацевтически приемлемой соли каждый R5 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, атома галогена, оксо, галогеналкила, -C(O)R6, -S(O)mR6 и -NR7R8, где радикалы с R6 по R8 и m являются такими, как определено в формуле (I), предпочтительно представляют собой атом водорода.
В предпочтительном воплощении настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемая соль представляет собой соединение формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемую соль:
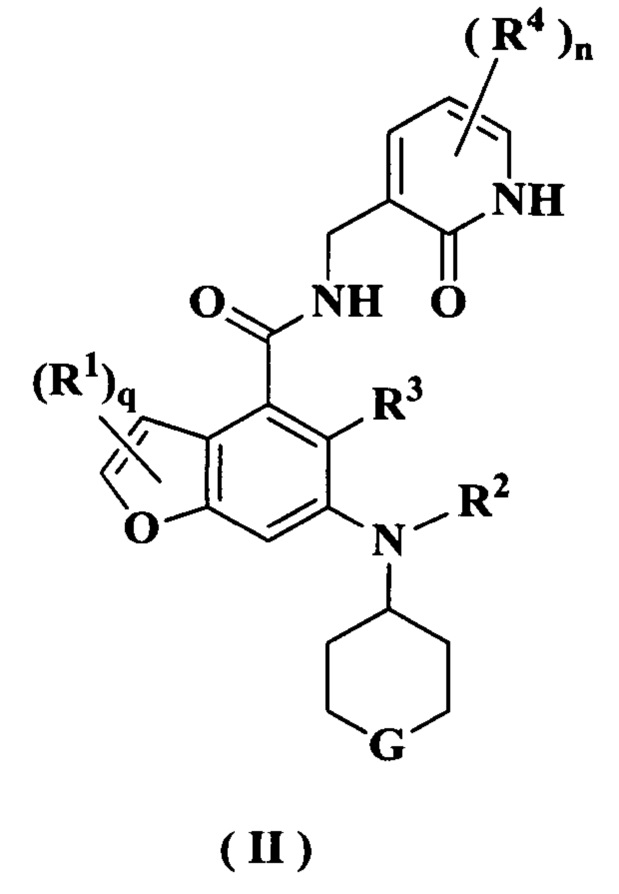
где:
G выбран из группы, состоящей из CRbRc, C=O, S(O)m, NRd и атома кислорода;
Rb и Rc каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкокси, атома галогена, амино, нитро, гидрокси, циано, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)mR6 и -NR7R8;
Rd выбран из группы, состоящей из атома водорода, алкила, циклоалкила, галогеналкила, гидроксиалкила, гетероциклила, арила, гетероарила, -C(O)R6, -C(O)OR6 и S(O)mR6; и
радикалы с R1 по R4, с R6 по R8, n, m и q являются такими, как определено в формуле (I).
В предпочтительном воплощении настоящего изобретения соединение формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемая соль представляет собой соединение формулы (III) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемую соль:
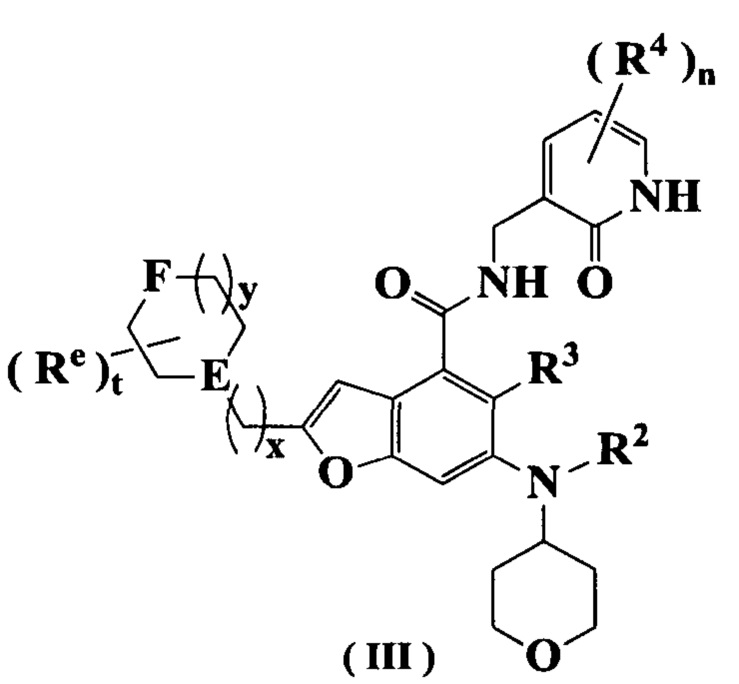
где:
Е представляет собой СН или атом N;
F выбран из группы, состоящей из CRbRb, C=O, NRd атома кислорода;
Rb и Rc каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкокси, атома галогена, амино, нитро, гидрокси, циано, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)R6 и NR7R8;
Rd выбран из группы, состоящей из атома водорода, алкила, циклоалкила, галогеналкила, гидроксиалкила, гетероциклила, арила, гетероарила, -C(O)R6, -C(O)OR6 и -S(O)mR6;
каждый Re идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, галогеналкила, атома галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
t равно 0, 1, 2, 3, 4 или 5;
х равно 0, 1, 2 или 3;
y равно 0, 1, 2 или 3; и
R1-R4, R6-R8, m и n являются такими, как определено в формуле (I).
В предпочтительном воплощении настоящего изобретения соединение формулы (III) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемая соль представляет собой соединение формулы (IV) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемую соль:
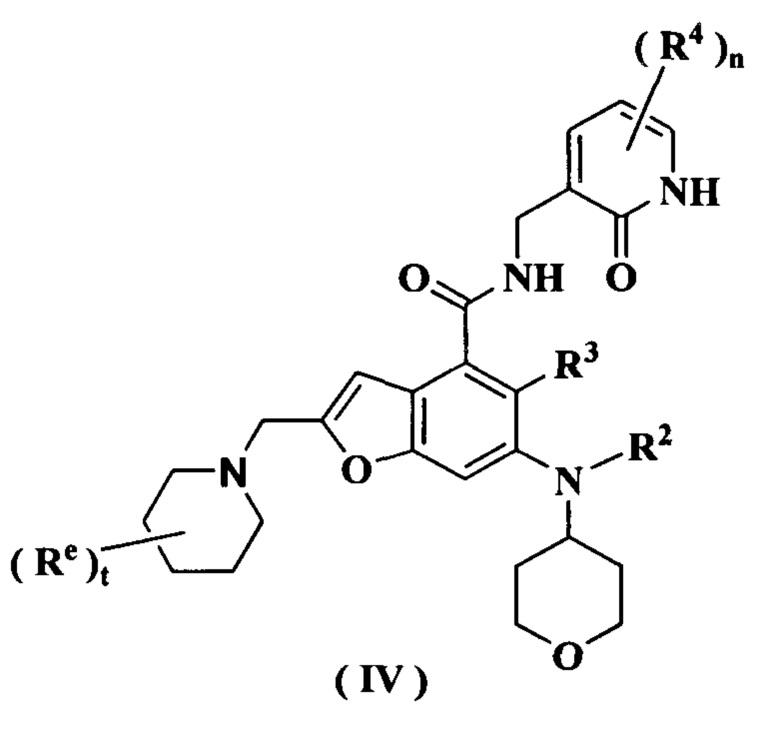
где:
каждый Re идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила и атома галогена;
t равно 0, 1, 2, 3, 4 или 5; и
радикалы с R2 по R4 и n являются такими, как определено в формуле (I).
В предпочтительном воплощении настоящего изобретения соединение формулы (III) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемая соль представляет собой соединение формулы (V) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемую соль:
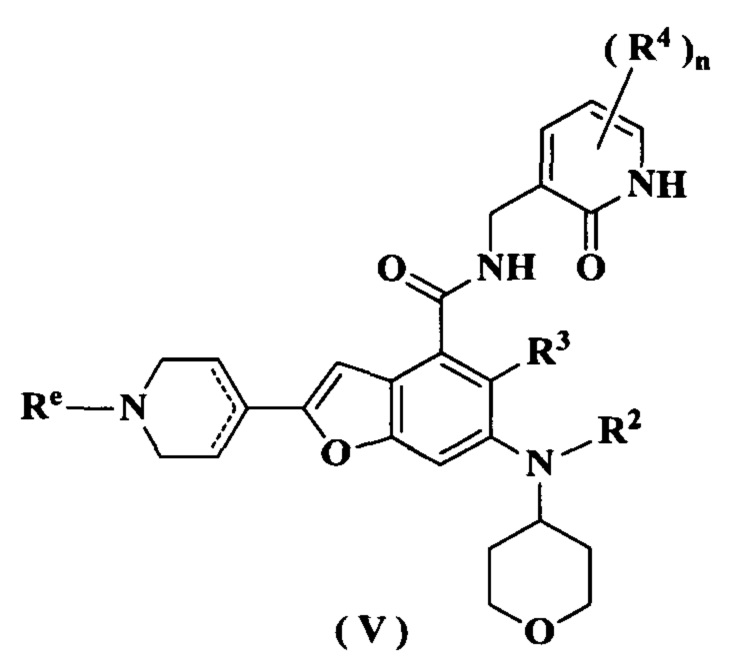
где:
Re выбран из группы, состоящей из атома водорода, алкила, галогеналкила, атома галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила; и
радикалы с R2 по R4 и n являются такими, как определено в формуле (I).
Характерные соединения формулы (I) или их таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемая соль включают без ограничений:
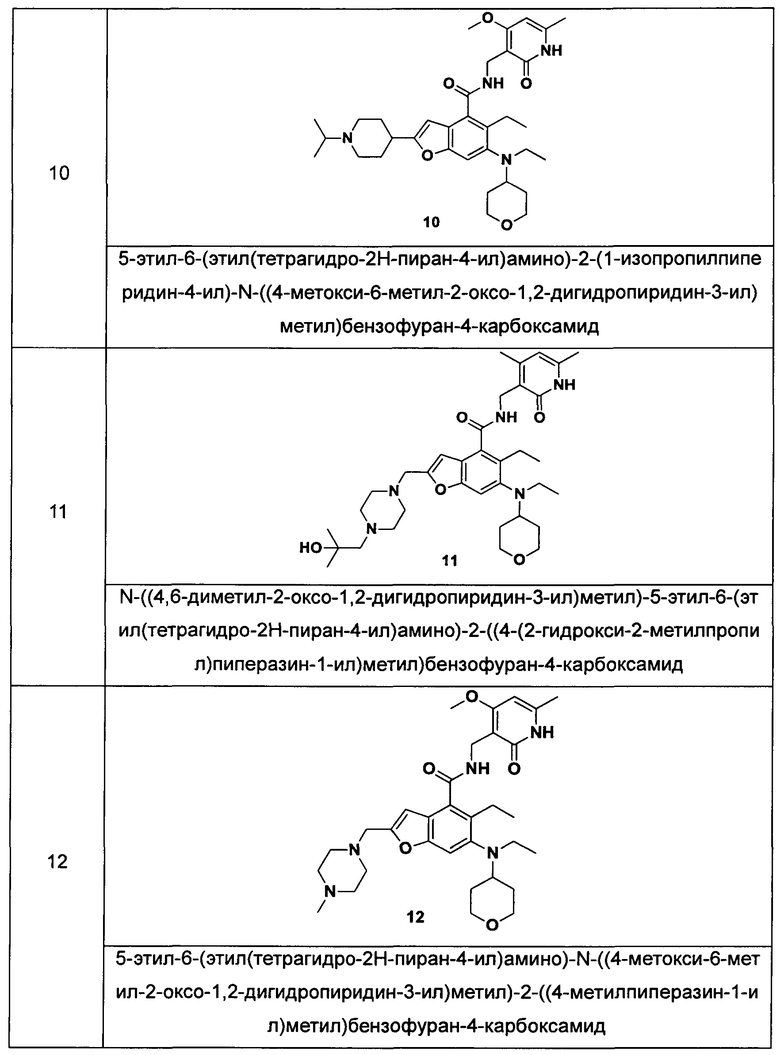
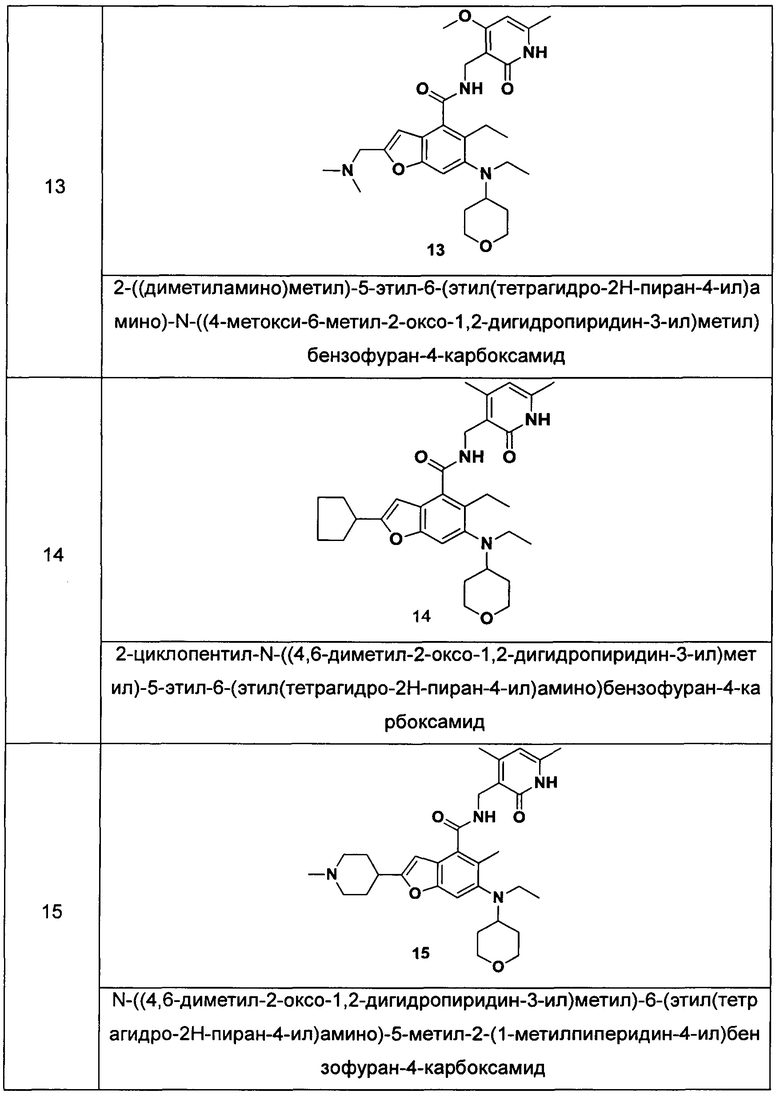
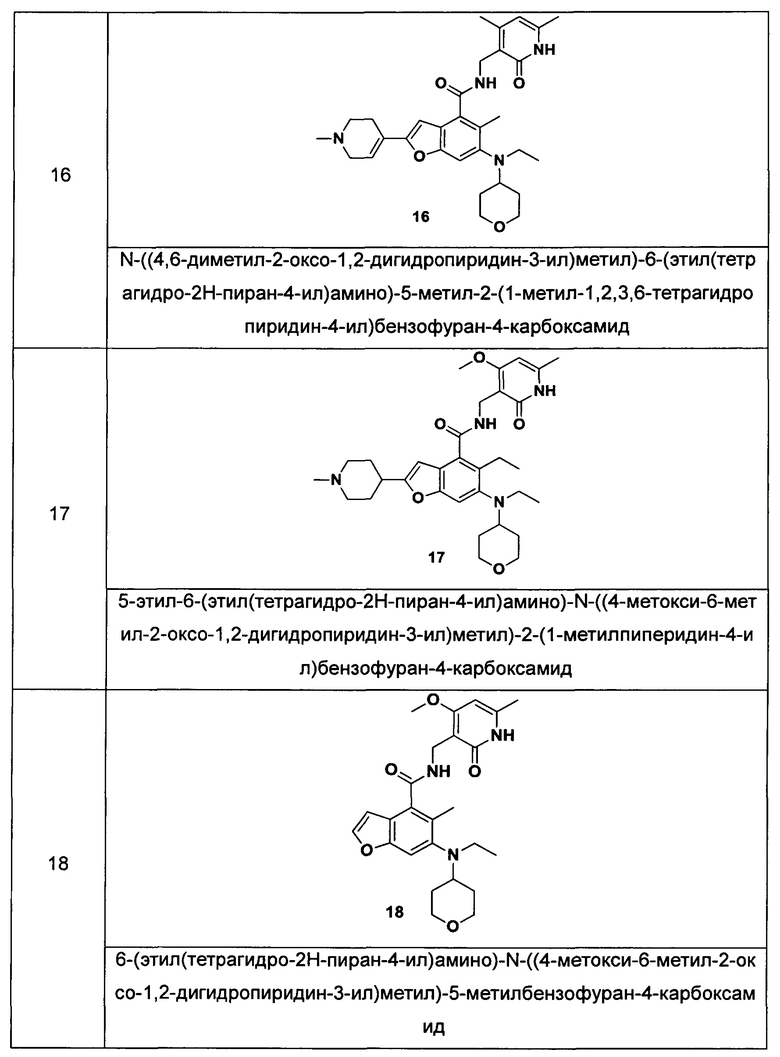
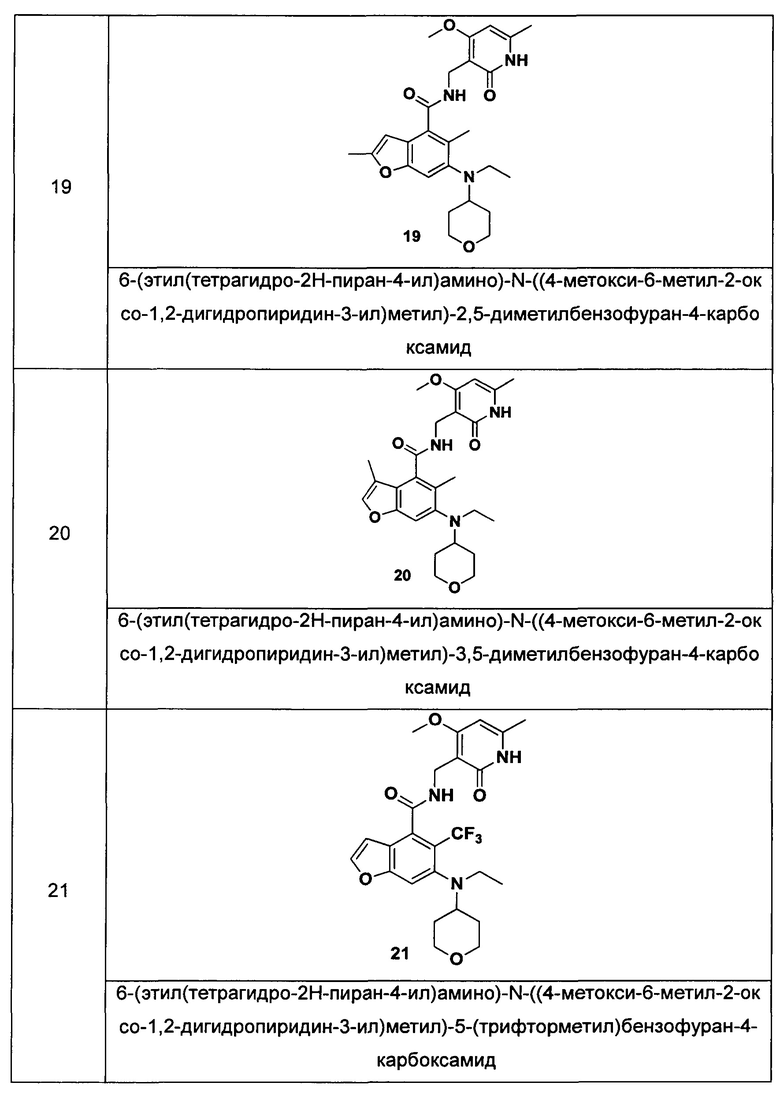
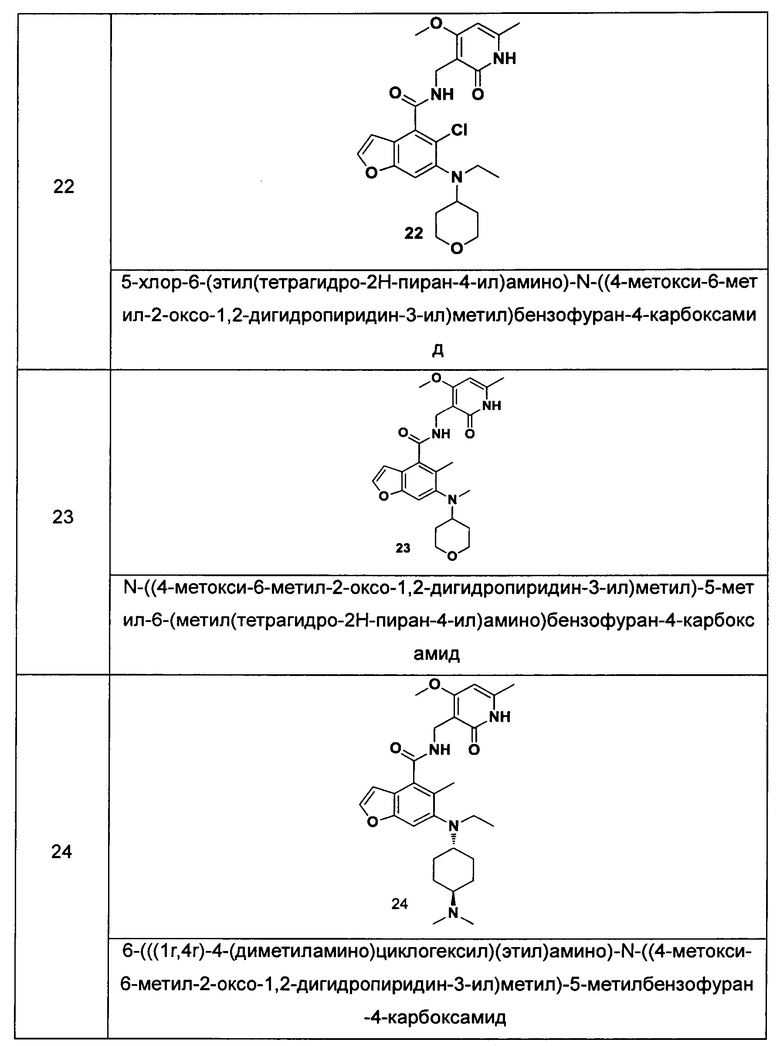
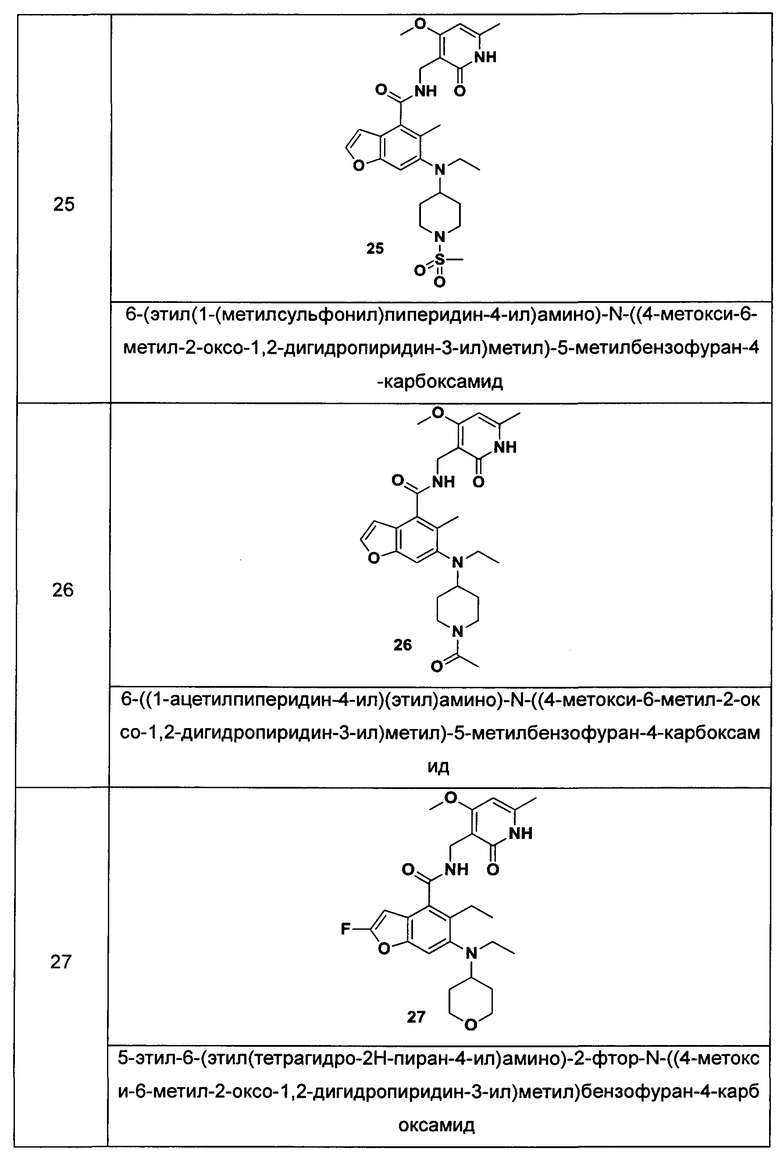
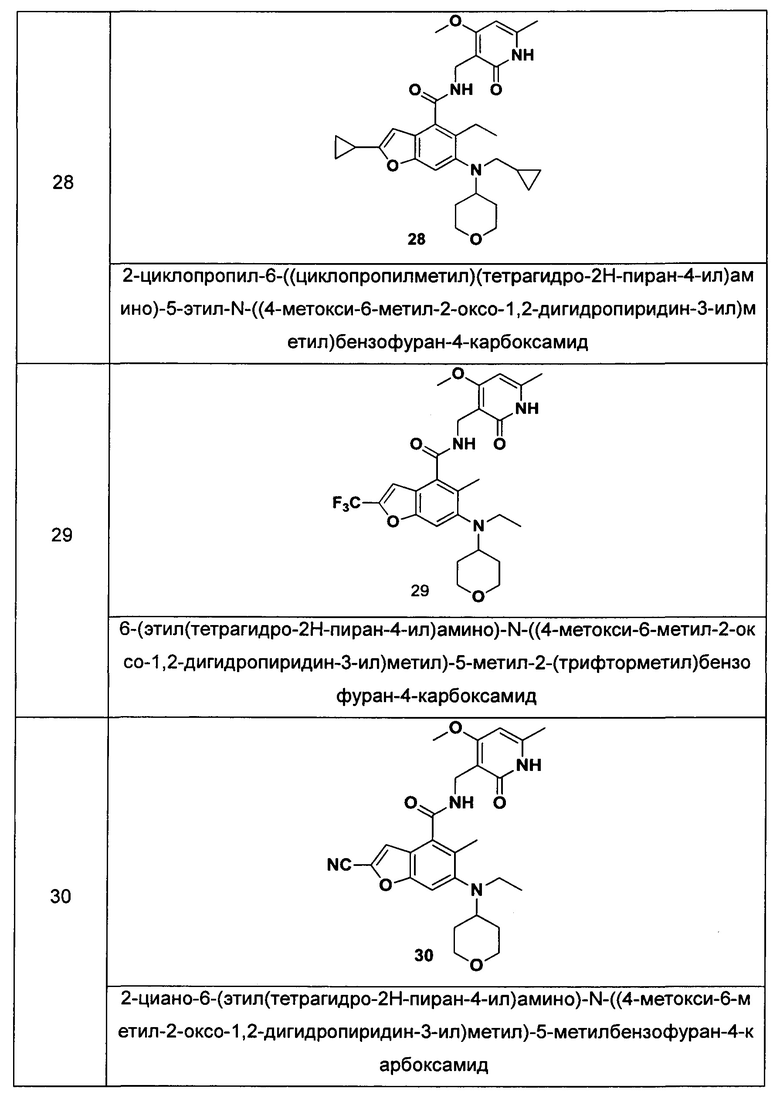
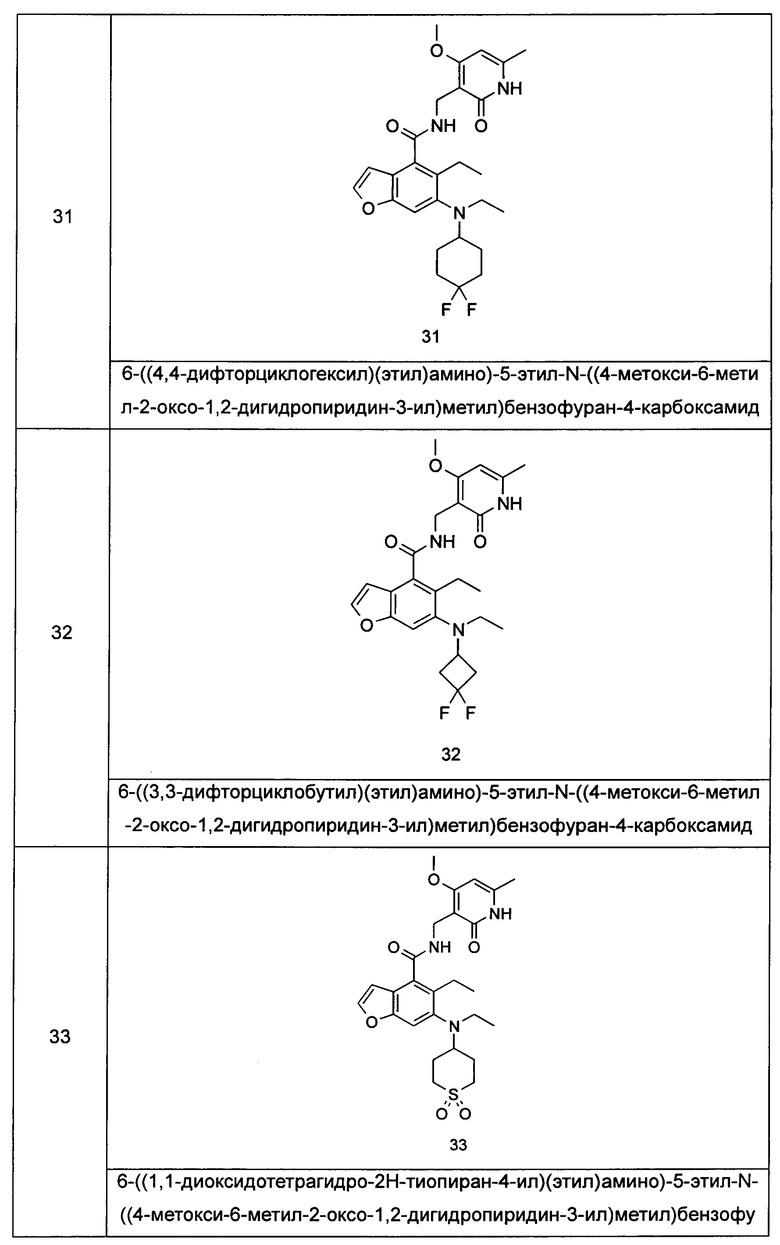
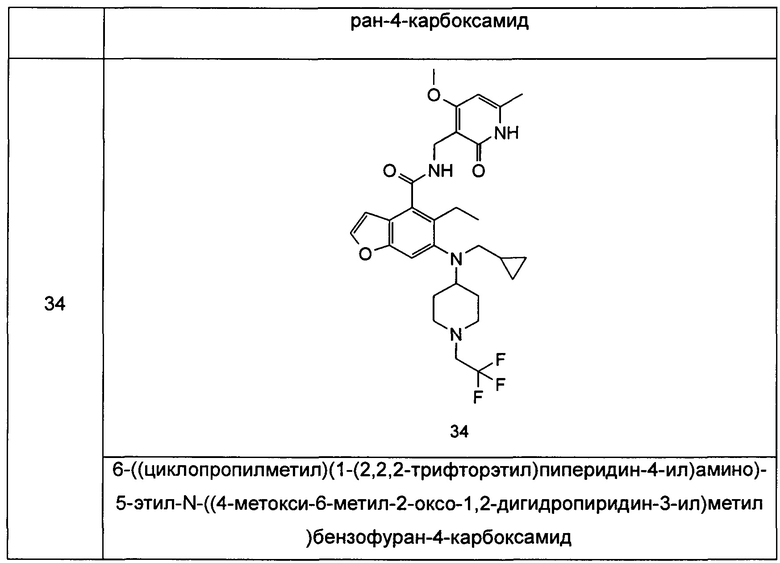
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или его фармацевтически приемлемая соль.
В настоящем изобретении дополнительно предложено промежуточное соединение для получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо его фармацевтически приемлемой соли, т.е. соединение формулы (VI):
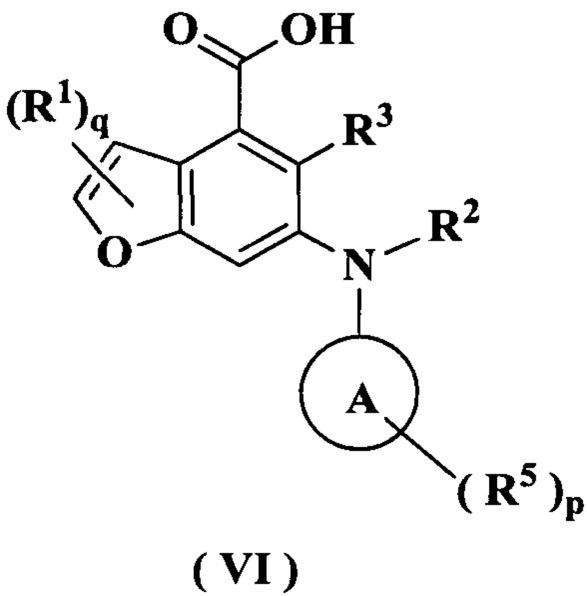
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемая соль,
где:
кольцо A, R1-R3, R5, р и q являются такими, как определено в формуле (I).
В другом аспекте настоящее изобретение также относится к способу получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо его фармацевтически приемлемой соли, включающему стадию:
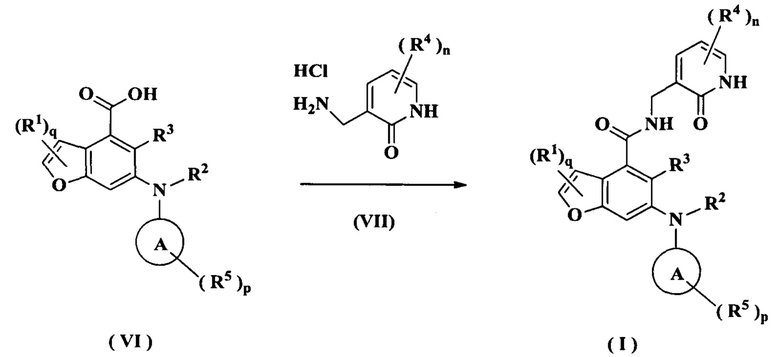
конденсации соединения формулы (VI) с соединением формулы (VII) при комнатной температуре с получением соединения формулы (I);
где:
R1-R5, кольца А, р, q и n являются такими, как определено в формуле (I).
В другом аспекте настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I), (II), (III), (IV) или (V) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо его фармацевтически приемлемую соль и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Настоящее изобретение также относится к способу получения указанной выше композиции, включающий стадию смешивания соединения формулы (I), (II), (III), (IV) или (V) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо его фармацевтически приемлемой соли с одним или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо его фармацевтически приемлемой соли или содержащей их фармацевтической композиции в получении лекарственного средства для профилактики и/или лечения опухоли и рака, где рак выбран из группы, состоящей из лимфомы, лейкоза, рака молочной железы, рака легкого, рака предстательной железы, рака яичника, рака печени, меланомы, рабдомиосаркомы, синовиальной саркомы, мезотелиомы, рака шейки матки, рака толстой кишки, рака прямой кишки, рака желудка, рака поджелудочной железы, рака головного мозга, рака кожи, рака полости рта, рака костей, рака почек, рака мочевого пузыря, опухолей фаллопиевой трубы, опухоли яичника, опухоли брюшной полости, глиомы, глиобластомы, рака головы и шеи и миеломы; предпочтительно лимфомы, лейкоза, рака молочной железы, рака легкого, рака предстательной железы, рака яичника, рака печени, меланомы, рабдомиосаркомы, синовиальной саркомы и мезотелиомы; где рак легкого выбран из группы, состоящей из мелкоклеточного рака легкого и немелкоклеточного рака легкого; где лейкоз выбран из группы, состоящей из хронического миелоидного лейкоза, острого миелоидного лейкоза и лейкоза смешанного происхождения; где лимфома выбрана из неходжкинской лимфомы, диффузной крупноклеточной В-клеточной лимфомы и фолликулярной лимфомы.
Настоящее изобретение дополнительно относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, либо его фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции для применения в качестве лекарственного средства для профилактики и/или лечения опухоли и рака, где опухоль и рак являются такими, как определено выше.
Настоящее изобретение также относится к способу профилактики и/или лечения опухоли и рака, включающему стадию введения, нуждающемуся в этом, пациенту терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо его фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции, где опухоль и рак являются такими, как определено выше.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо его фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции в качестве ингибитора EZH2 в получении лекарственного средства для профилактики и/или лечения опухоли и рака, где опухоль и рак являются такими, как определено выше.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или его фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции в получении лекарственного средства для ингибирования EZH2.
Настоящее изобретение дополнительно относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, либо его фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции для применения в качестве лекарственного средства для ингибирования EZH2.
Настоящее изобретение также относится к способу ингибирования EZH2, включающему стадию введения, нуждающемуся в этом, пациенту терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо его фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции.
Содержащие активный ингредиент фармацевтические композиции могут принимать форму, приемлемую для перорального применения, например, таблетки, пастилки, лепешки, водной или масляной суспензии, диспергируемого порошка или гранулы, эмульсии, твердой или мягкой капсулы, либо сиропа или эликсира. Композиции для перорального применения можно готовить в соответствии с любым известным в данной области техники способом приготовления фармацевтических композиций. Такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из подсластителей, корригентов, красящих веществ и консервантов, с целью получения фармацевтического препарата привлекательного внешнего вида и вкуса. Таблетка содержит активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, пригодными для производства таблетки.
Водная суспензия содержит активный ингредиент в смеси с эксципиентами, приемлемыми для производства водной суспензии. Водная суспензия также может содержать один или более консервантов, таких как этилпарабен или н-пропилпарабен, одно или более красящих веществ, один или более корригентов и один или более подсластителей, таких как сахароза, сахарин или аспартам.
Масляную суспензию можно готовить в виде лекарственной формы путем суспендирования активного ингредиента в растительном масле. Масляная суспензия может содержать загуститель. Для обеспечения приемлемого вкуса препарата можно добавлять указанные выше подсластители и корригенты.
Активный ингредиент в смеси с диспергирующими или смачивающими агентами, суспендирующим агентом или одним или более консервантом можно готовить в виде диспергируемого порошка или гранулы, приемлемых для приготовления водной суспензии путем добавления воды. Примеры приемлемых диспергирующих или смачивающих агентов и суспендирующих агентов уже упомянуты выше. Могут быть также добавлены дополнительные эксципиенты, такие как подсластители, корригенты и красящие вещества. Эти композиции можно сохранять путем добавления антиоксиданта, такого как аскорбиновая кислота.
Настоящая фармацевтическая композиция может также принимать форму эмульсии масло-в-воде.
Фармацевтическая композиция может иметь форму стерильного инъекционного водного раствора. Приемлемыми носителями и растворителями, которые можно применять, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Стерильный инъекционный препарат может также представлять собой стерильную инъекционную микроэмульсию масло-в-воде, в которой активный ингредиент растворен в масляной фазе. Например, активный ингредиент можно сначала растворять в смеси соевого масла и лецитина, впоследствии вводить масляный раствор в смеси воды и глицерина и впоследствии обрабатывать с образованием микроэмульсии. Инъекционный раствор или микроэмульсию можно вводить в кровоток индивида путем локальной болюсной инъекции. Альтернативно преимуществом может обладать введение раствора микроэмульсии таким путем, чтобы поддерживать постоянную концентрацию настоящего соединения в кровотоке. Для поддержания такой постоянной концентрации можно использовать устройство для непрерывной внутривенной доставки. Примером такого устройства является насос для внутривенных инъекций Deltec CADD-PLUS. ТМ. 5400.
Фармацевтическая композиция может принимать форму стерильной инъекционной водной или масляной суспензии для внутримышечного и подкожного введения. Такую суспензию можно готовить в лекарственной форме с приемлемыми диспергирующими или смачивающими агентами и суспендирующими агентами, как описано выше, в соответствии с известными методами. Стерильный инъекционный препарат может также представлять собой стерильный инъекционный раствор или суспензию, приготовленный в нетоксичном разбавителе или растворителе, приемлемом для парентерального применения. Кроме того, в качестве растворителя или суспензионной среды можно легко использовать стерильные нелетучие масла.
Настоящее соединение можно вводить в форме суппозитория для ректального введения. Эти фармацевтические композиции можно готовить путем смешивания лекарственного средства с приемлемым нераздражающим эксципиентом, который является твердым при обычных температурах, но жидким в прямой кишке, таким образом, он плавится в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао, глицерин, желатин, гидрогенизированные растительные масла, смеси полиэтиленгликолей различных молекулярных масс и сложные эфиры жирных кислот и полиэтиленгликолей.
Специалистам в данной области техники хорошо известно, что дозировка лекарственного средства зависит от ряда факторов, включающих без ограничений следующие факторы: активность конкретного соединения, возраст пациента, массу тела пациента, общее состояние здоровья пациента, поведение пациента, рацион питания пациента, время введения, путь введения, скорость выведения, комбинацию лекарственных средств и т.п. Кроме того, лучший вариант лечения, такой как метод лечения, суточная доза соединения формулы (I) или тип его фармацевтически приемлемой соли, может быть подтвержден на основании традиционных схем терапии.
Подробное описание изобретения
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют описанные ниже значения.
«Алкил» относится к насыщенной алифатической углеводородной группе, включающей С1-С20 прямоцепочечные и разветвленные группы, предпочтительно алкил, имеющий от 1 до 12 атомов углерода, и более предпочтительно алкил, имеющий от 1 до 6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексили их разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, п-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкильная группа может быть замещенной или незамещенной. В случае ее замещения группа (-ы) заместителей может (-гут) быть замещена (-ы) в любой доступной точке соединения. Группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкилнила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Алкилен» относится к алкилу, атом водорода которого дополнительно замещен, например, «метилен» относится к -CH2-, «этилен» относится к -(CH2)2-, «пропилен» относится к -(CH2)3-, «бутилен» относится к -(CH2)4- и т.п.
«Алкенил» относится к алкилу, как определено выше, имеющему по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную двойную связь, например, этенил, 1-пропенил, 2-пропенил, 1-, 2- или 3-бутенил и т.п. Алкенильная группа может быть замещенной или незамещенной. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкилнила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио и гетероциклического алкилтио.
«Циклоалкил» относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода и более предпочтительно от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.п. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
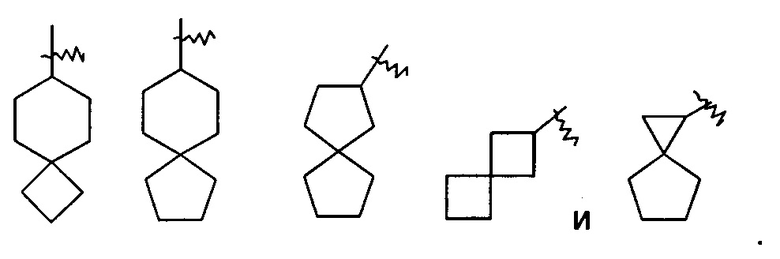
«Спиро-циклоалкил» относится к 5-20-членной полициклической группе, в которой кольца соединены посредством одного общего атома углерода (называемого спиро-атомом), где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно к 6-14-членному спиро-циклоалкилу и более предпочтительно к 7-10-членному спиро-циклоалкилу. В зависимости от числа спиро-атомов, общих для колец, спиро-цикпоалкил можно разделить на моно-спиро-циклоалкил, ди-спиро-циклоалкил или поли-спиро-циклоалкил, предпочтительно моно-спиро-циклоалкил или ди-спиро-циклоалкил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-циклоалкил. Неограничивающие примеры спиро-циклоалкилов включают:
«Конденсированный циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, в которой каждое кольцо в системе имеет общую пару примыкающих атомов углерода с другим кольцом, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно к 6-14-членному конденсированному циклоалкилу, более предпочтительно к 7-10-членному конденсированному циклоалкилу. В зависимости от числа колец, содержащих члены, конденсированный циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, предпочтительно бициклический или трициклический конденсированный циклоалкил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:
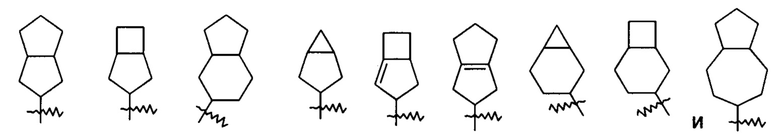
«Мостиковый циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, в которой каждые два кольца в системе имеют общую пару не соединенных атомов углерода, где кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно к 6-14-членному мостиковому циклоалкилу и более предпочтительно к 7-10-членному мостиковому циклоалкилу. В соответствии с числом колец, содержащих кольцевые атомы, мостиковый циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, предпочтительно бициклический, трициклический или тетрациклический мостиковый циклоалкил и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостиковых циклоалкилов включают:

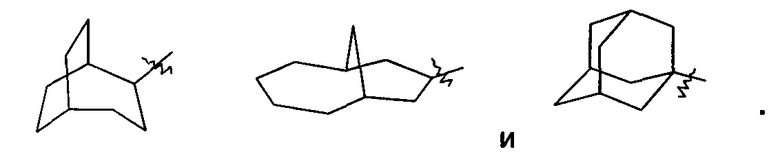
Циклоалкильное кольцо может быть конденсировано с кольцом арила, гетероарила или гетероциклила, где кольцо, связанное с исходной структурой, представляет собой циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и т.п. Циклоалкил может быть необязательно замещенным или незамещенным. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкилнила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Гетероциклил» относится к 3-20-членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве кольцевых атомов, но за исключением -O-O-, -O-S- или -S-S- в кольце, где остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно гетероциклил имеет от 3 до 12 атомов, где от 1 до 4 атомов представляют собой гетероатомы, более предпочтительно от 3 до 8 атомов, где от 1 до 3 атомов представляют собой гетероатомы, и наиболее предпочтительно от 3 до 6 атомов, где 1-2 атома представляют собой гетероатомы. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагидротиенил, дигидроимидазолил, дигидрофурил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил и т.п., предпочтительно пиперидинил, пирролидинил, пиранил, морфолинил или 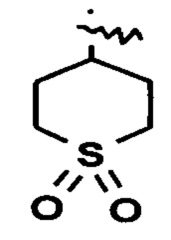 . Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
«Спиро-гетероциклил» относится к 5-20-членному полициклическому гетероциклилу, кольца которого соединены через один общий атом (называемый спиро-атомом), где кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, где остальные кольцевые атомы представляют собой атомы углерода, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно к 6-14-членному спиро-гетероциклилу и более предпочтительно к 7-10-членному спиро-гетероциклилу. В соответствии с числом общих для колец спиро-атомов спиро-гетероциклил можно разделить на моно-спиро-гетероциклил, ди-спиро-гетероциклил или поли-спиро-гетероциклил и предпочтительно моно-спиро-гетероциклил или ди-спиро-гетероциклил, более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-гетероциклил. Неограничивающие примеры спиро-гетероциклилов включают:
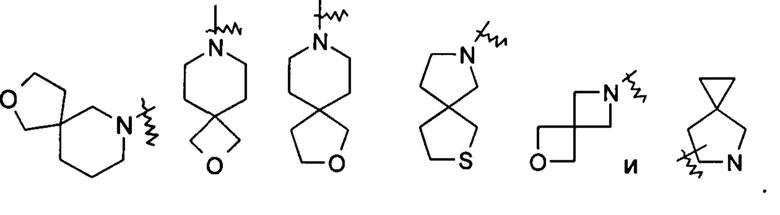
«Конденсированный гетероциклил» относится к 5-20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую пару смежных атомов с другим кольцом, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, и где кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, где остальные кольцевые атомы представляют собой атомы углерода, предпочтительно к 6-14-членному конденсированному гетероциклилу и более предпочтительно к 7-10-членному конденсированному гетероциклилу. В зависимости от числа колец, содержащих члены, конденсированный гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, предпочтительно бициклический или трициклический конденсированный гетероциклил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:
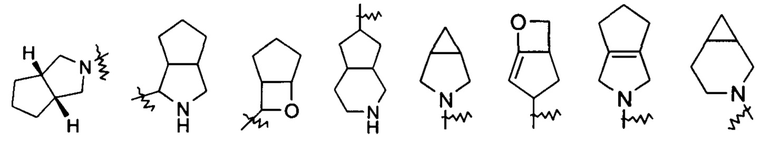
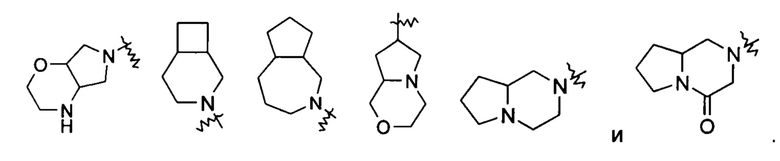
«Мостиковый гетероциклил» относится к 5-14-членной полициклической гетероциклильной группе, где каждые два кольца в системе имеет общую пару не соединенных друг с другом атомов с другим кольцом, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, и кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, где остальные кольцевые атомы представляют собой атомы углерода, предпочтительно к 6-14-членному мостиковому гетероциклилу и более предпочтительно к 7-10-членному мостиковому гетероциклилу. В соответствии с числом колец, содержащих кольцевые атомы, мостиковый гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, и предпочтительно бициклический, трициклический или тетрациклический мостиковый гетероциклил и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостиковых гетероциклилов включают:
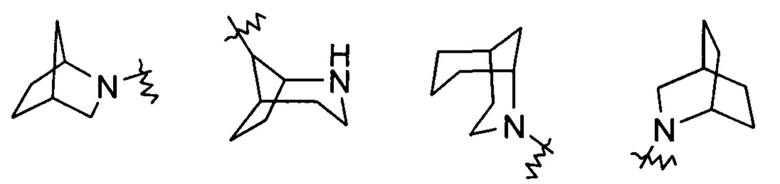
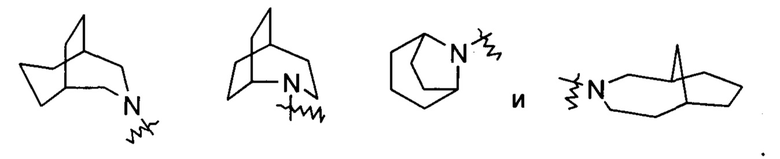
Гетероциклильное кольцо может быть конденсировано с кольцом арила, гетероарила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероциклил. Неограничивающие примеры включают:
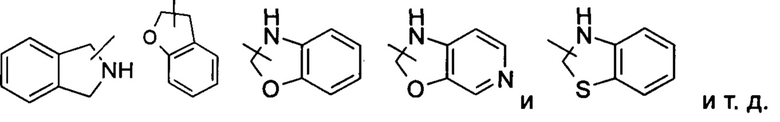
Гетероциклил может быть необязательно замещенным или незамещенным. В случае замещения групп(-ы) заместител(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкилнила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, цикпоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Арил» относится к 6-14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо в системе имеет общую пару смежных атомов углерода с другим кольцом в системе), имеющему полностью конъюгированную пи-электронную систему, предпочтительно к 6-10-членному арилу, например фенилу и нафтилу, и более предпочтительно к фенилу. Арильное кольцо может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой арильное кольцо. Неограничивающие примеры включают:
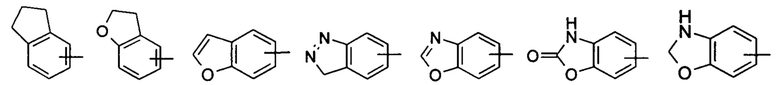
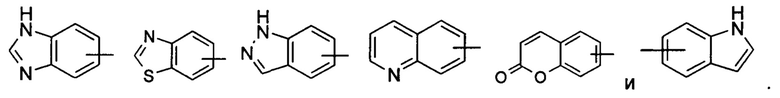
Арил может быть необязательно замещенным или незамещенным. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкилнила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси и алкоксикарбонила.
«Гетероарил» относится к 5-14-членной гетероароматической системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N в качестве кольцевых атомов, предпочтительно к 5-10-членному гетероарилу с 1-3 гетероатомами, более предпочтительно к 5- или 6-членному гетероарилу с 1-2 гетероатомами, например, к имидазолилу, фурилу, тиенилу, тиазолилу, пиразолилу, оксазолилу, пирролилу, тетразолилу, пиридинилу, пиримидинилу, тиадиазолу, пиразинилу и т.п., предпочтительно к имидазолилу, тетразолилу, тиенилу, пиразолилу, пиримидинилу или тиазолилу и более предпочтительно к пиразолилу или тиазолилу. Гетероарильное кольцо может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо. Неограничивающие примеры включают:
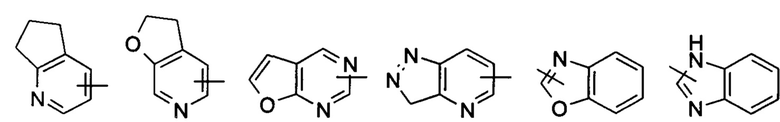
Гетероарил может быть необязательно замещенным или незамещенным. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкилнила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси и алкоксикарбонила.
«Алкокси» относится к группе -О-(алкил) или -O-(незамещенный циклоалкил), где алкил является таким, как определено выше. Неограничивающие примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и т.п. Алкоксигруппа может быть необязательно замещенной или незамещенной. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкилнила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси и алкоксикарбонила.
«Галогеналкил» относится к алкильной группе, замещенной одним или более атомов галогена, где алкил является таким, как определено выше.
«Галогеналкокси» относится к алкоксигруппе, замещенной одним или более атомов галогена, где алкил является таким, как определено выше.
«Гидроксиалкил» относится к алкильной группе, замещенной гидроксигруппой, где алкил является таким, как определено выше.
«Гидрокси» относится к группе -ОН.
«Атом галогена» относится к атому фтора, хлора, брома или йода.
«Амино» относится к группе -NH2.
«Циано» относится к группе -CN.
«Нитро» относится к группе -NO2.
«Оксо» относится к группе =O.
«Карбонил» относится к группе С=O.
«Карбокси» относится к группе -С(O)ОН.
«Изоцианато» относится к группе -NCO.
«Гидроксиимино» относится к группе =N-ОН.
«Алкоксикарбонил» относится к группе -С(O)O(алкил) или -С(O)O(циклоалкил), где алкил и циклоалкил являются такими, как определено выше.
«Ацилгалогенид» относится к соединению, содержащему группу -С(O)-атом галогена.
«Необязательный» или «необязательно» означает, что описанное впоследствии событие или обстоятельство может произойти, но необязательно произойдет, и такое описание включает ситуацию, где это событие или обстоятельство происходит или не происходит. Например, «гетероциклильная группа, необязательно замещенная алкилом» означает, что алкильная группа может присутствовать, но необязательно присутствует, и такое описание включает ситуацию замещения гетероциклильной группы алкилом и гетероциклильной группы, не замещенной алкилом.
«Замещенный» относится к одному или более атомов водорода в группе, предпочтительно вплоть до 5, более предпочтительно 1-3 атома водорода, независимо замещены соответствующим количеством заместителей. Безусловно, заместители могут существовать в их возможном химическом положении. Специалист в данной области техники способен определить, является ли замещение возможным или невозможным, с помощью экспериментов или теории, не прилагая слишком больших усилий. Например, комбинация амино- или гидроксигруппы, имеющих свободные атомы водорода, и атомов углерода, имеющих ненасыщенные связи (такие как олефиновые связи), может быть нестабильной.
«Фармацевтическая композиция» относится к смеси одного или более соединений в соответствии с настоящим изобретением или его физиологически/фармацевтически приемлемых солей или пролекарств и других химических компонентов, а также Других компонентов, таких как физиологически/фармацевтически приемлемые носители и эксципиенты. Цель фармацевтической композиции состоит в том, чтобы способствовать введению соединения в организм, дающему возможность абсорбции и, следовательно, проявления биологической активности активного ингредиента.
«Фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которая является безопасной и эффективной у млекопитающего и обладает желаемой биологической активностью.
Способ синтеза соединения по настоящему изобретению Для достижения цели настоящего изобретения в настоящем изобретении применяют следующие технические решения синтеза.
Способ получения соединения формулы (I) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо его фармацевтически приемлемой соли включает следующие стадии:
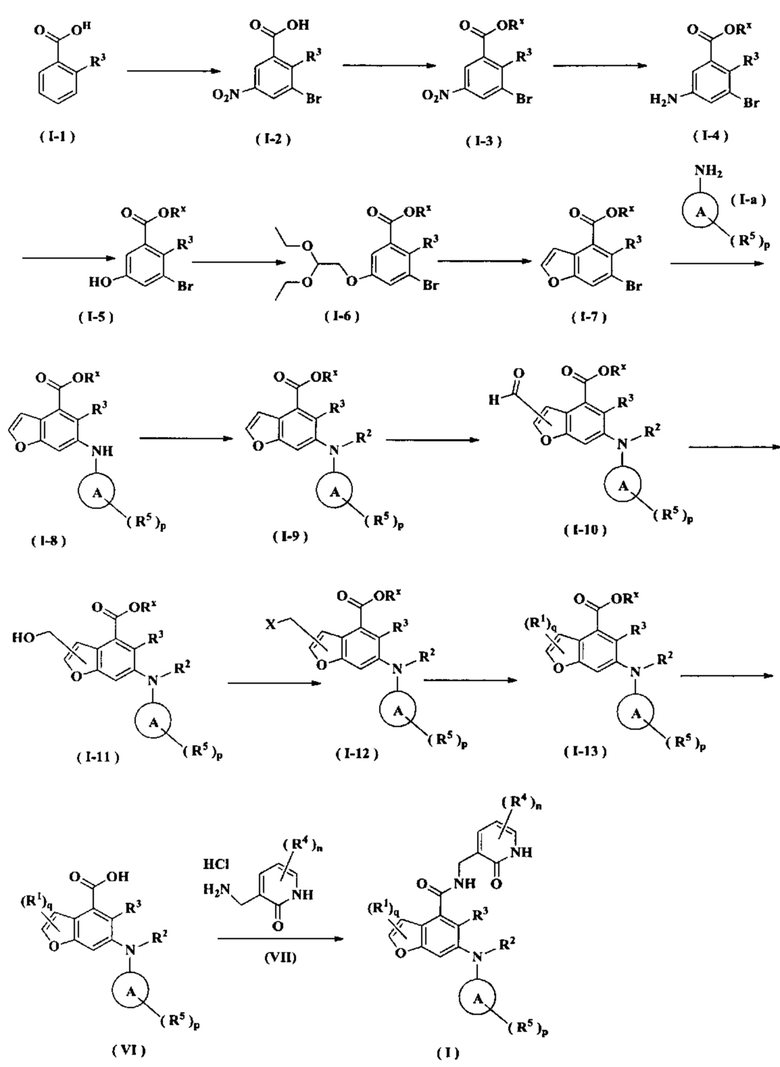
Соединение формулы (I-1) добавляют к серной кислоте в ледяной бане, добавляют порциями нитрат натрия и N-бромсукцинимид с получением соединения формулы (I-2) при нагревании. Соединение формулы (I-2) подвергают реакции эстерификации с хлоридом в щелочных условиях с получением соединения формулы (I-3), где щелочной реагент, обеспечивающий щелочные условия, предпочтительно представляет собой карбонат калия. Соединение формулы (I-3) восстанавливают с получением соединения формулы (I-4). Соединение (I-5) получают в присутствии серной кислоты и нитрита натрия из соединения формулы (I-4). Соединение формулы (I-5) подвергают взаимодействию с 2-бром-1,1-диэтоксиэтаном в щелочных условиях с получением соединения формулы (I-6). Соединение формулы (I-6) подвергают реакции замыкания цикла с 3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоатом при нагревании и в кислых условиях с получением соединения формулы (I-7). Соединение формулы (I-7) подвергают взаимодействию с соединением формулы (Ia) в присутствии катализатора при нагревании и в щелочных условиях с получением соединения формулы (I-8), где щелочной реагент, обеспечивающий щелочные условия для этой реакции, предпочтительно представляет собой карбонат калия, а катализатор предпочтительно представляет собой трис(дибензилиденацетон)дипалладий или (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин. Соединение формулы (I-8) подвергают взаимодействию с алкилгалогенидом в щелочных условиях с получением соединения формулы (I-9). Соединение формулы (I-9) подвергают взаимодействию с N,N-диметилформамидом в щелочных условиях с получением соединения формулы (I-10), где щелочной реагент, обеспечивающий щелочные условия для данной реакции, предпочтительно представляет собой диизопропиламид лития. Соединение формулы (I-10) восстанавливают до соединения формулы (I-11) в присутствии восстанавливающего агента, где восстанавливающий агент в этих условиях предпочтительно представляет собой боргидрид натрия. Соединение формулы (I-11) подвергают взаимодействию с трибромидом фосфора с получением соединения формулы (I-12). Соединение формулы (I-12) подвергают взаимодействию с R1H с получением соединения формулы (I-13). Соединение формулы (I-13) подвергают гидролизу в щелочных условиях с получением соединения формулы (VI), где щелочной реагент, обеспечивающий щелочные условия для данной реакции, предпочтительно представляет собой гидроксид натрия. Соединение формулы (VI) подвергают реакции ацилирования с соединением формулы (VII) с получением соединения формулы (I).
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания, где органические основания включают без ограничений триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия, и где неорганические основания включают без ограничений гидрид натрия, фосфат калия, карбонат натрия, карбонат калия и карбонат цезия.
Катализатор включает без ограничений 2-дициклогексилфосфино-2',4',6'-триизопропилдифенил, (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин, трис(дибензилиденацетон)дипалладий, ацетат палладия, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, трифенилфосфин, тетракистрифенилфосфин палладий,
где:
Rx выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, циклоалкил, гетероциклил, арил и гетероарил каждый независимо и необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидрокси, амино, алкоксикарбонила, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
X представляет собой атом галогена;
радикалы с R1 по R5, кольца А, р, q и n являются такими, как определено в формуле (I).
Предпочтительные воплощения изобретения
Далее настоящее изобретение описано со ссылкой на следующие примеры, но эти примеры не следует истолковывать как ограничивающие объем изобретения.
Примеры
Структуры соединений идентифицированы по данным ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). ЯМР определяют с помощью прибора Bruker AVANCE-400. Растворители для определения представляют собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), внутренний стандарт представляет собой триметилсилан (ТМС), и химические сдвиги ЯМР (δ) приведены в 10-6 (частей на миллион (ч. н. м.)).
МС определяют с помощью спектрометра FINNIGAN LCQAd (ионизация электрораспылением (ИЭР)) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Данные высокоэффективной жидкостной хроматографии (ВЭЖХ) определяют на спектрометре для жидкостной хроматографии высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и на спектрометре для жидкостной хроматографии высокого давления Waters 2695-2996 (хроматографическая колонка Gimini С18 150×4,6 мм).
Значения IC50 средней скорости ингибирования киназы определяют с помощью набора реагентов для твердофазного иммуносорбентного ферментного анализа (ИФА) NovoStar (BMG Co., Германия).
Для тонкослойной хроматографии на силикагеле (ТСХ) используют пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размеры пластины силикагеля, используемой для ТСХ, составляют от 0,15 мм до 0,2 мм, а размеры пластины силикагеля, используемой для очистки продукта, составляют от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии используют силикагель Yantai Huanghai от 200 до 300 меш.
Известные исходные вещества по настоящему изобретению могут быть получены традиционными способами синтеза в данной области техники или приобретены у компаний ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc. или Dari Chemical Company и т.д.
Если не указано иное, реакции проводят в атмосфере аргона или в атмосфере азота.
Термин «атмосфера аргона» или «атмосфера азота» означает, что реакционная колба оборудована баллоном аргона или азота емкостью 1 л.
Термин «атмосфера водорода» означает, что реакционная колба оборудована баллоном водорода емкостью 1 л.
Реакции гидрогенизации под давлением проводят с помощью аппарата Парра для гидрогенизации Parr 3916EKX и генератора водорода QL-500 или аппарата для гидрогенизации HC2-SS.
В реакциях гидрогенизации в реакционной системе обычно создают вакуум и заполняют ее водородом, и описанную выше операцию повторяют три раза.
В микроволновой реакции используют микроволновой реактор типа СЕМ Discover-S 908860.
Если не указано иное, раствор, используемый в реакциях, относится к водному раствору.
Если не указано иное, температура реакции в реакциях относится к комнатной температуре.
Комнатная температура является наиболее подходящей температурой реакции и находится в диапазоне от 20°С до 30°С.
Мониторинг хода реакции проводят с помощью тонкослойной хроматографии (ТСХ), и система растворителей для проявления включает: А: система дихлорметана и метанола, В: система н-гексана и этилацетата, С: система петролейного эфира и этилацетата, D: ацетон. Отношение объема растворителей можно регулировать в соответствии с полярностью соединений.
Система элюции для очистки соединений колоночной хроматографией и тонкослойной хроматографией включает: А: система дихлорметана и метанола, В: система н-гексана и этилацетата, С: система н-гексана, этилацетата и дихлорметана, D: система петролейного эфира и этилацетата, Е: этилацетат. Объемное отношение растворителей можно регулировать в соответствии с полярностью соединений, и иногда можно добавлять слабощелочной реагент, такой как триэтиламин, или кислый реагент.
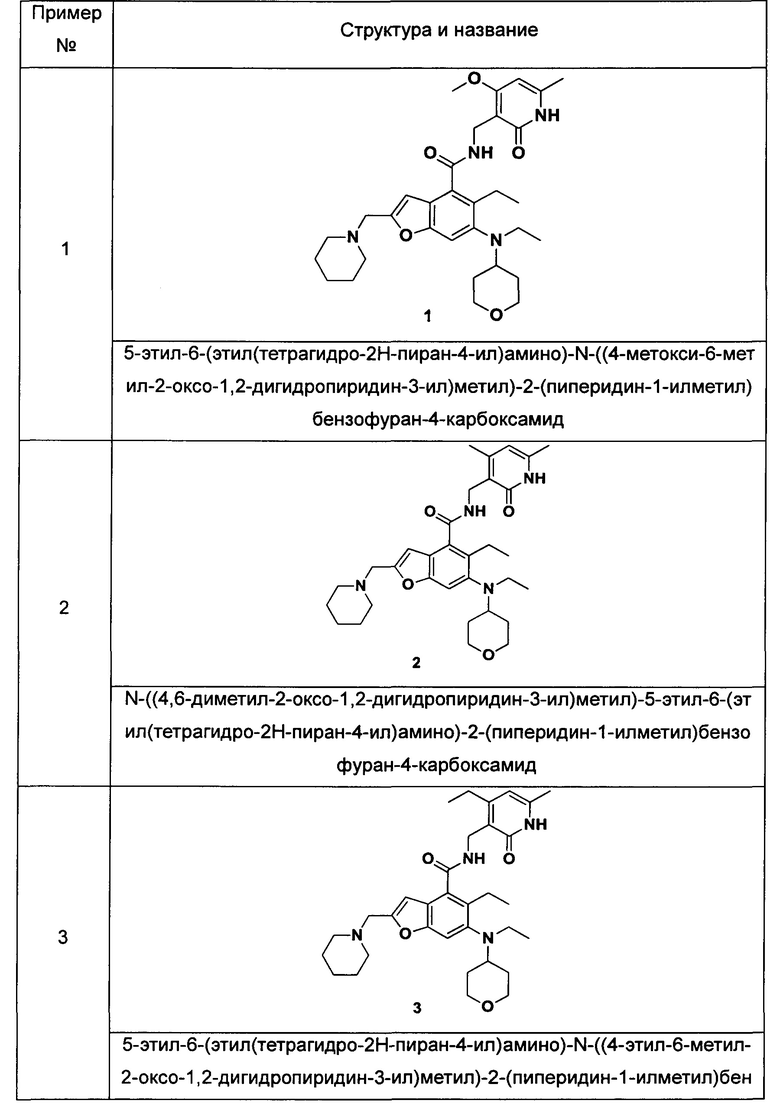
Пример 1
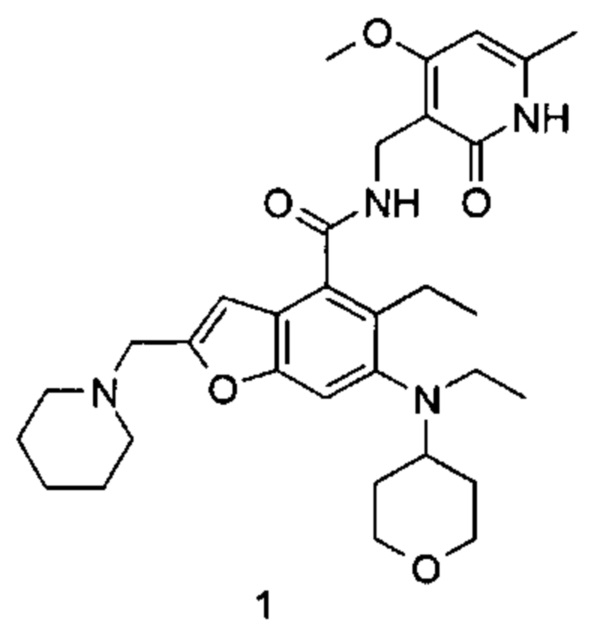
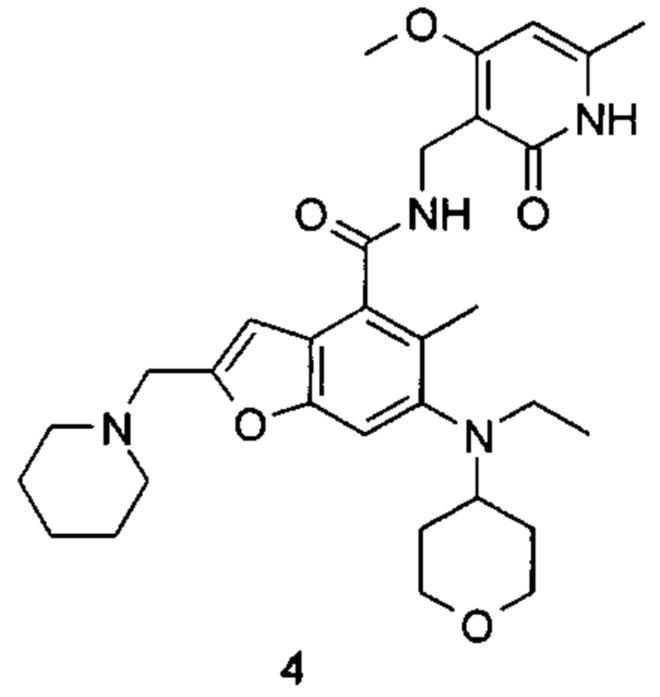
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(пиперидин-1-илметил)бензофуран-4-карбоксамид
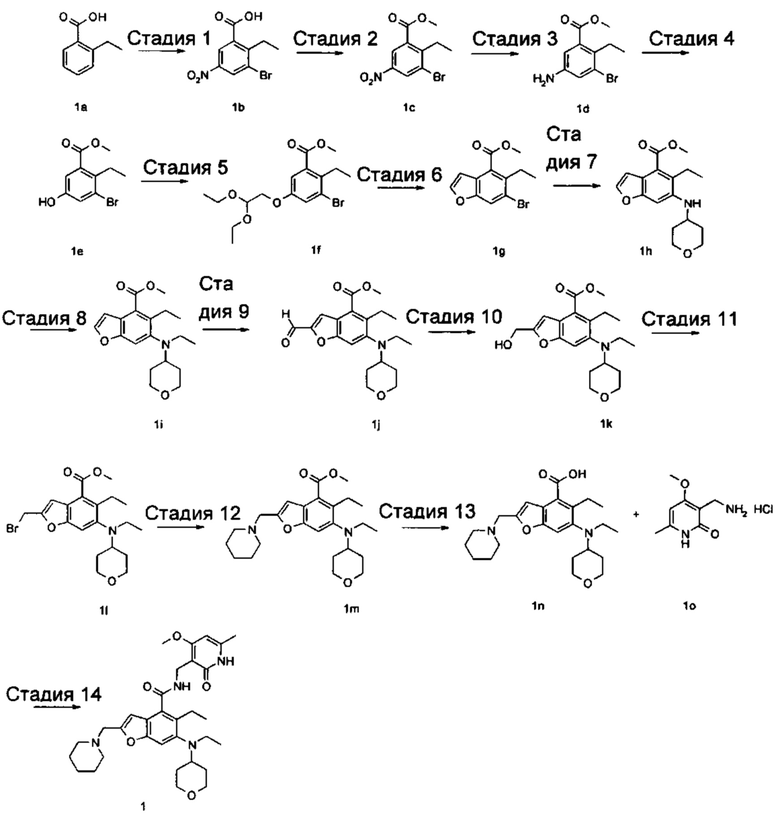
Стадия 1
3-Бром-2-этил-5-нитробензойная кислота
2-Этилбензойную кислоту 1а (20,0 г, 133 ммоль, получена способом, описанным в Journal of the American Chemical Society, 1991, 113 (13), 4931-6) добавляли к 150 мл серной кислоты, впоследствии добавляли порциями нитрат натрия (11,3 г, 133 ммоль) в ледяной бане. Смесь перемешивали в течение 3 часов, впоследствии добавляли порциями N-бромсукцинимид (2,6 г, 14,5 ммоль). Реакционную систему перемешивали в течение 1 часа при 60°С. После завершения реакции реакционный раствор наливали в ледяную воду, тщательно перемешивали и фильтровали. Фильтрат промывали водой и концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 3-бром-2-этил-5-нитробензойной кислоты 1b (35 г), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
Метил 3-бром-2-этил-5-нитробензоат
Неочищенную 3-бром-2-этил-5-нитробензойную кислоту 1b (35 г, 128 ммоль) растворяли в 200 мл N,N-диметилформамида, впоследствии добавляли йодметан (21,8 г, 153 ммоль) и карбонат калия (35,3 г, 255 ммоль). Реакционную систему перемешивали в течение 2 часов при комнатной температуре. После завершения реакции реакционный раствор концентрировали при пониженном давлении. Добавляли избыток воды, и смесь экстрагировали этилацетатом. Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения метил-3-бром-2-этил-5-нитробензоата 1с (36 г), в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
Метил-5-амино-3-бром-2-этилбензоат
Неочищенный метил-3-бром-2-этил-5-нитробензоат 1с (35,0 г, 121 ммоль) добавляли к 250 мл этанола и 150 мл воды. Смесь нагревали до 70°С, добавляли хлорид аммония (52,8 г, 969 ммоль), впоследствии добавляли порциями порошок железа (34 г, 606 ммоль). Реакционную систему перемешивали в течение 2 часов при 70°С. После завершения реакции смесь фильтровали через целит в горячем виде. Фильтрационный кек промывали горячим этанолом, впоследствии фильтрат объединяли и концентрировали под пониженным давлением. Добавляли этилацетат и насыщенный раствор бикарбоната натрия. Две фазы разделяли, и водную фазу экстрагировали этилацетатом (30 мл).. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения метил-5-амино-3-бром-2-этилбензоата 1d (171 мг, выход 70%), в виде желтого твердого вещества.
Стадия 4
Метил-3-бром-2-этил-5-гидроксибензоат
Метил-5-амино-3-бром-2-этилбензоат 1d (15,0 г, 58 ммоль) растворяли в 10 мл ацетонитрила, впоследствии добавляли 200 мл 10% серной кислоты. Смесь тщательно перемешивали и охлаждали до 3°С в бане с ледяной солью, впоследствии добавляли 10 мл предварительно приготовленного раствора нитрита натрия (4,4 г, 64 ммоль). Смесь перемешивали в течение 4 часов при указанной выше температуре, добавляли по каплям 200 мл 50% серной кислоты, впоследствии перемешивали в течение 1 часа при 90°С. После завершения реакции реакционный раствор экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения метил-3-бром-2-этил-5-гидроксибензоата 1е (5,5 г, выход 37%), в виде коричневого твердого вещества.
Стадия 5
Метил-3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоат
Метил-3-бром-2-этил-5-гидроксибензоат 1е (35 г, 135 ммоль) растворяли в 200 мл N,N-диметилформамида, впоследствии добавляли 2-бром-1,1-диэтоксиэтан (40 г, 202 ммоль) и карбонат калия (37 г, 269 ммоль). Реакционную систему перемешивали при 120°С в течение 12 часов. После завершения реакции реакционный раствор концентрировали под пониженным давлением для удаления N,N-диметилформамида. Добавляли воду, и смесь экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения метил-3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоата 1f (40 г, выход 80%), в виде светло-желтого масла.
Стадия 6
Метил-6-бром-5-этилбензофуран-4-карбоксилат
Полифосфорную кислоту (30 г) добавляли к 400 мл толуола. Смесь нагревали до 100°С, добавляли 50 мл предварительно приготовленного раствора метил-3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоат 1f (40 г, 107 ммоль) в толуоле при перемешивании. Смесь перемешивали в течение 16 часов при 100°С. После завершения реакции супернатант сливали. К остатку добавляли воду и этилацетат. Две фазы разделяли, и водную фазу экстрагировали этилацетатом (30 мл).. Органические фазы объединяли, промывали насыщенным раствором карбоната натрия и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения метил-6-бром-5-этилбензофуран-4-карбоксилата 1g (171 мг, выход 39%), в виде желтого твердого вещества.
Стадия 7
Метил-5-этил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-6-бром-5-этилбензофуран-4-карбоксилат 1g (11,0 г, 39 ммоль), тетрагидро-2Н-пиран-4-амин (5,89 г, 58 ммоль), трис(дибензилиденацетон)дипалладий (3,6 г, 3,9 ммоль), (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин (4,86 г, 7,8 ммоль) и карбонат цезия (38 г, 117 ммоль) растворяли в 100 мл толуола. Смесь перемешивали в течение 12 часов при 100°С. После завершения реакции смесь фильтровали через слой целита, и фильтрационный кек промывали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 5-этил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилата 1h (10,0 г, выход 85%), в виде желтого твердого вещества.
Стадия 8
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-5-этил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 1h (10,0 г, 0,033 ммоль) растворяли в 150 мл 1,2-дихлорэтана, впоследствии добавляли ацетальдегид (7,2 г, 0,165 ммоль) и уксусную кислоту (9,9 г, 0,165 ммоль). Смесь перемешивали в течение 1 часа, впоследствии добавляли триацетоксиборгидрид натрия (20,8 г, 0,1 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения
5-этил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилата 1i (7,8 г, выход 71%), в виде белого твердого вещества.
МС m/z (ЖХ-МС): 332,4 [М+1]
Стадия 9
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 1i (1,6 г, 4,8 ммоль) растворяли в 25 мл тетрагидрофурана. Смесь охлаждали до -70°С, впоследствии добавляли по каплям 2,0 М раствор диизопропиламида лития (3,6 мл, 7,3 ммоль) в атмосфере аргона. Смесь перемешивали в течение 90 минут, впоследствии добавляли N.N-диметилформамид (536 мг, 7,3 ммоль). Смесь перемешивали в течение 2 часов, впоследствии медленно подогревали до комнатной температуры. Добавляли избыток хлорида аммония. Смесь тщательно перемешивали и экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилата 1j (1,3 г, выход 75%) в виде желтого масла.
МС m/z (ИЭР): 360,2 [М+1]
Стадия 10
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(гидроксиметил)бензофуран-4-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилат 1j (1,4 г, 3,9 ммоль) растворяли в смеси 5 мл тетрагидрофурана и 10 мл метанола, впоследствии добавляли боргидрид натрия (222 мг, 5,8 ммоль). Смесь перемешивали в течение 30 минут при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением, добавляли воду и насыщенный раствор бикарбоната натрия, и смесь экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(гидроксиметил)бензофуран-4-карбоксилата 1k (1,4 г, выход 99%) в виде желтого масла.
Стадия 11
Метил-2-(бромметил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(гидроксиметил) бензофуран-4-карбоксилат 1k (1,0 г, 2,8 ммоль) растворяли в 30 мл тетрагидрофурана, впоследствии добавляли по каплям трибромид фосфора (1,12 г, 4,2 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции смесь нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения метил-2-(бромметил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилата 1l, в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 12
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксилат
Неочищенный метил-2-(бромметил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 1l (1,15 г, 2,7 ммоль) растворяли в 15 мл ацетонитрила, впоследствии добавляли по каплям 10 мл предварительно приготовленного раствора пиперидина (362 мг, 4,3 ммоль) в ацетонитриле. Смесь перемешивали в течение 30 минут при комнатной температуре. После завершения реакции реакционный раствор концентрировали при пониженном давлении. Добавляли этилацетат и насыщенный раствор бикарбоната натрия. Две фазы разделяли, и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксилата 1m (1,2 г, выход 99%) в виде желтого масла.
МС m/z (ЖХ-МС): 429,2 [М+1]
Стадия 13
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоновая кислота
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-ил метил)бензофуран-4-карбоксилат 1m (1,2 г, 2,7 ммоль) растворяли в 5 мл тетрагидрофурана и добавляли 20 мл метанола, впоследствии добавляли 5 мл 4М раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. После завершения реакции добавляли концентрированную хлористоводородную кислоту для доведения рН реакционного раствора до 4. Смесь концентрировали под пониженным давлением, и остаток растворяли в смеси дихлорметана и метанола (об. : об. составляет 5:1) и фильтровали. Фильтрационный кек промывали смесью дихлорметана и метанола (об. : об. составляет 5:1). Фильтрат объединяли и концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоновой кислоты 1n (1,1 г) в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ЖХ-МС): 415,2 [М+1]
Стадия 14
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(пиперидин-1-илметил)бензофуран-4-карбоксамид
Неочищенную 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоновую кислоту 1n (150 мг, 0,36 ммоль) растворяли в 5 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (104 мг, 0,54 ммоль), 1-гидроксибензотриазол (73 мг, 0,54 ммоль) и N,N-диизопропилэтиламин (232 мг, 1,8 ммоль). Смесь перемешивали 1 час, впоследствии добавляли 3-(аминометил)-4-метокси-6-метилпиридин-2(1Н)-она гидрохлорид 1о (96 мг, 0,47 ммоль, получен способом, раскрытым в заявке на патент WO 2014177982). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции, добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(пиперидин-1-илметил)бензофуран-4-карбоксамида 1 (155 мг, выход 76%) в виде белого твердого вещества.
МС m/z(H3P): 565,3 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 11.44 (s, 1Н), 8.00 (s, 1Н), 7.40 (s, 1Н), 6.53 (brs, 1Н), 6.10 (s, 1Н), 4.29 (d, 2Н), 3.85 (brs, 2Н), 3.83 (s, 3Н), 3.56 (brs, 2Н), 3.22 (t, 2Н), 3.03-3.08 (m, 2Н), 2.93-2.98 (m, 1Н), 2.78-2.84 (m, 2Н), 2.42 (brs, 4Н), 2.18 (s, 3Н), 1.64-1.67 (brd, 2Н), 1.47-1.56 (m, 6Н), 1.38 (brs, 2Н), 1.06 (t, 3Н), 0.83 (t, 3Н).
Пример 2
N-((4,6-Диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксамид
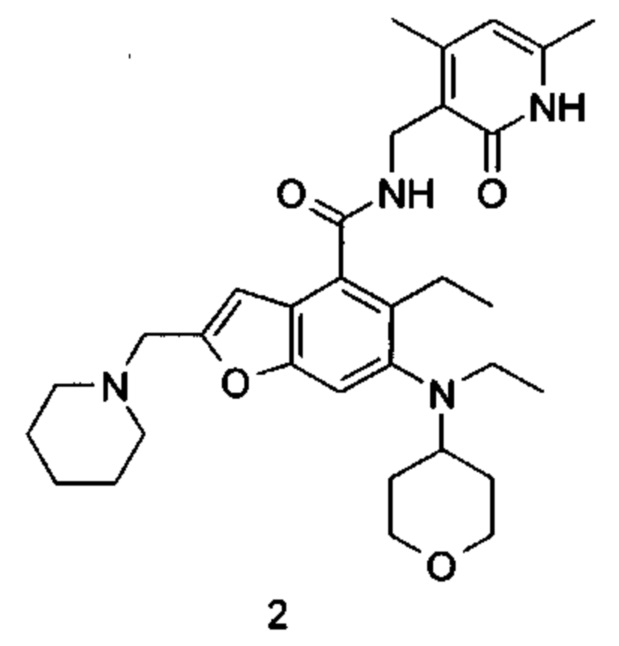
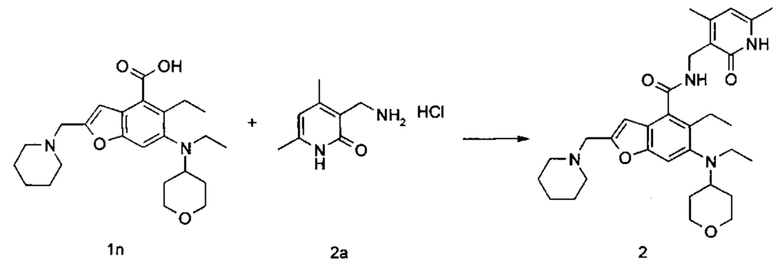
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоновую кислоту 1n (1,0 г, 2,4 ммоль) растворяли в 30 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (696 мг, 3,6 ммоль), 1-гидроксибензотриазол (490 мг, 3,6 ммоль) и N,N-диизопропилэтиламин (1,56 г, 12,1 ммоль). Смесь перемешивали в течение 1 часа, впоследствии добавляли 3-(аминометил)-4,6-диметилпиперидин-2(1Н)-она гидрохлорид 2а (593 мг, 3,0 ммоль, получен способом, раскрытым в заявке на патент WO 2014097041). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции, добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксамида 2 (750 мг, выход 57%) в виде белого твердого вещества.
МС m/z(H3P): 549,7 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 11.48 (s, 1Н), 8.15 (t, 1Н), 7.39 (s, 1Н), 6.46 (s, 1Н), 5.86 (s, 1Н), 4.32 (d, 2Н), 3.83 (d, 2Н), 3.54 (s, 2Н), 3.21 (t, 2Н), 3.01-3.07 (m, 2Н), 2.92-2.97 (m, 1Н), 2.77-2.82 (m, 2Н), 2.39 (brs, 4Н), 2.23 (s, 3Н), 2.11 (s, 3Н), 1.64-1.67 (brd, 2Н), 1.47-1.55 (m, 6Н), 1.36-1.37 (brd, 2Н), 1.02 (t, 3Н), 0.82 (t, 3Н).
Пример 3
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-этил-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(пиперидин-1-илметил)бензофуран-4-карбоксамид
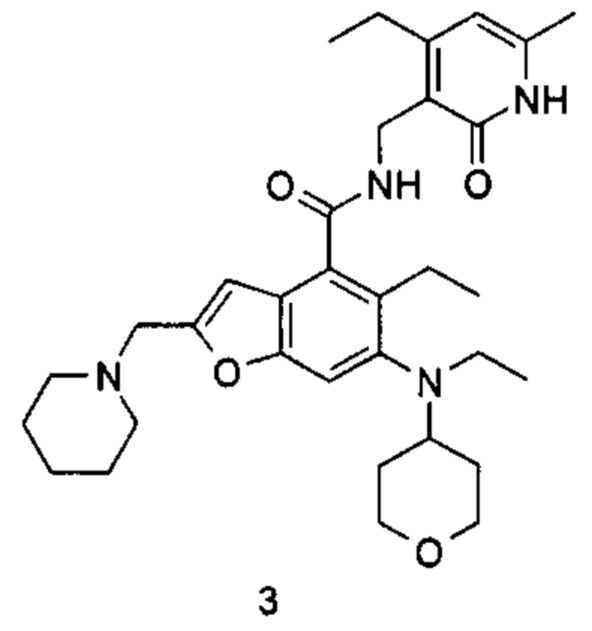
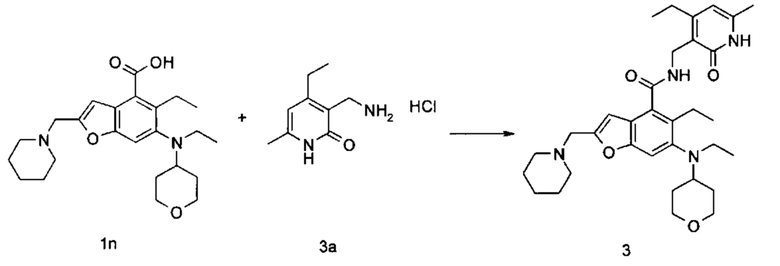
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил) бензофуран-4-карбоновую кислоту 1n (50 мг, 0,12 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (35 мг, 0,18 ммоль), 1-гидроксибензотриазол (24 мг, 0,18 ммоль) и N,N-диизопропилэтиламин (78 мг, 0,60 ммоль). Смесь перемешивали в течение 1 часа, впоследствии добавляли 3-(аминометил)-4-этил-6-метилпиридин-2(1Н)-она гидрохлорид 3а (36 мг, 0,18 ммоль, получен способом, раскрытым в заявке на патент WO 2013173441). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции, добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-этил-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(пиперидин-1-илметил)бензофуран-4-карбоксамид а 3 (58 мг, выход 85%) в виде белого твердого вещества.
МС m/z (ИЭР): 563,7 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 11.51 (s, 1Н), 8.12 (t, 1Н), 7.39 (s, 1Н), 6.47 (s, 1Н), 5.91 (s, 1Н), 4.33 (d, 2Н), 3.81-3.83 (brd, 2Н), 3.54 (brs, 2Н), 3.20 (t, 2Н), 3.01-3.06 (m, 2Н), 2.91-2.97 (m, 1Н), 2.76-2.82 (m, 2Н), 2.59 (q, 2Н), 2.39 (brs, 4Н), 2.13 (s, 3Н), 1.63-1.66 (brd, 2Н), 1.48-1.50 (m, 6Н), 1.36 (brs, 2Н), 1.37 (t, 3Н), 1.01 (t, 3Н), 0.81 (t, 3Н).
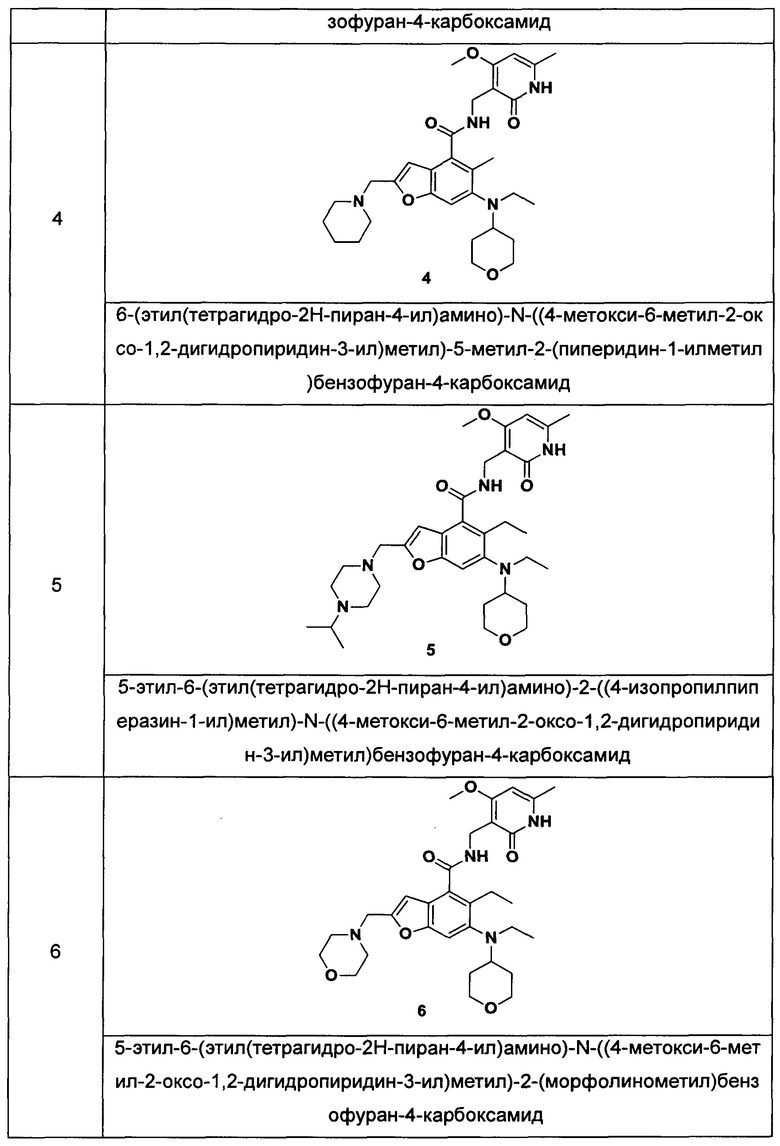
Пример 4
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метил-2-(пиперидин-1-илметил)бензофуран-4-карбоксамид
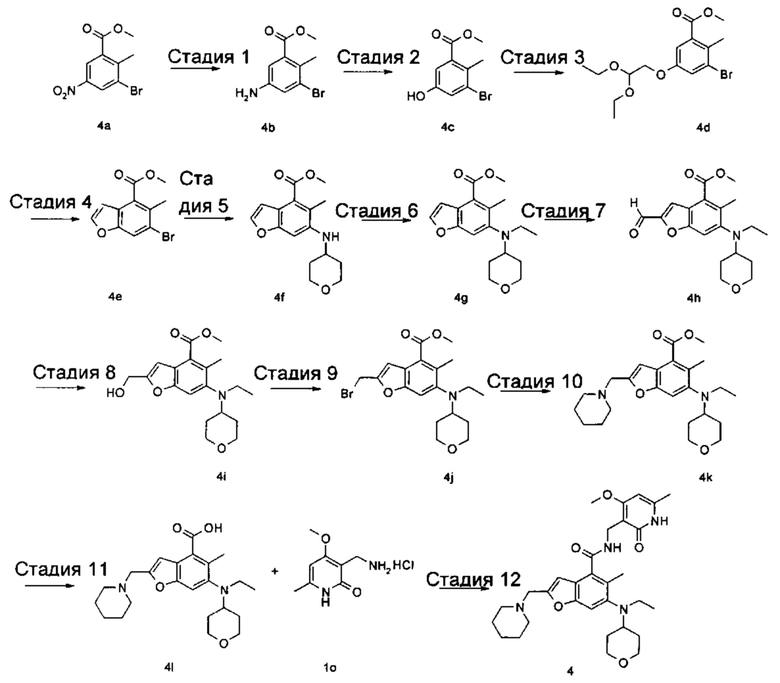
Стадия 1
Метил-5-амино-3-бром-2-метилбензоат
Метил-3-бром-2-метил-5-нитробензоат 4а (13 г, 47,3 ммоль, получен способом, раскрытым в заявке на патент WO 2012061602) добавляли к смеси 200 мл этанола и 50 мл воды. Смесь нагревали до 70°С, впоследствии добавляли хлорид аммония (20,6 г, 378 ммоль) и добавляли порциями порошок железа (13,3 г, 236 ммоль). Реакционную систему перемешивали в течение 2 часов при 70°С. После завершения реакции смесь фильтровали через слой целита в горячем виде. Фильтрационный кек промывали горячим этанолом, впоследствии фильтрат объединяли и концентрировали под пониженным давлением. Остаток нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали три раза этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения метил 5-амино-3-бром-2-метилбензоата 4b (11,0 г, выход 95%), в виде желтого твердого вещества.
Стадия 2
Метил-3-бром-5-гидрокси-2-метилбензоат
Метил-5-амино-3-бром-2-метилбензоат 4b (3,0 г, 0,012 ммоль) суспендировали в 20 мл 10% серной кислоты. Смесь охлаждали до 0°С, затем добавляли 5 мл предварительно приготовленного раствора нитрита натрия (1,0 г, 14,7 ммоль). Смесь перемешивали в течение 3 часов при указанной выше температуре, реакционный раствор наливали в 30 мл предварительно приготовленного 10% раствора серной кислоты при 80°С, впоследствии перемешивали в течение 1 часа при 80°С. После завершения реакции реакционный раствор экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения метил-3-бром-5-гидрокси-2-метилбензоата 4с (1,1 г, выход 32%), в виде желтого твердого вещества.
Стадия 3
Метил-3-бром-5-(2,2-диэтоксиэтокси)-2-метилбензоат
Метил-3-бром-5-гидрокси-2-метилбензоат 4с (800 мг, 3,3 ммоль) растворяли в 15 мл N,N-диметилформамида, впоследствии добавляли 2-бром-1,1-диэтоксиэтан (965 мг, 4,9 ммоль) и карбонат калия (900 мг, 6,5 ммоль). Реакционную систему перемешивали в течение 12 часов при 120°С. После завершения реакции добавляли избыток воды, и смесь экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения метил-3-бром-5-(2,2-диэтоксиэтокси)-2-метилбензоата 4d (820 мг, выход 69%), в виде светло-желтого масла.
Стадия 4
Метил-6-бром-5-метилбензофуран-4-карбоксилат
Метил-3-бром-5-(2,2-диэтоксиэтокси)-2-метилбензоат 4d (650 мг, 1,8 ммоль) растворяли в 10 мл толуола, впоследствии добавляли 10 мл предварительно приготовленного раствора полифосфорной кислоты (10 г) в толуоле. Смесь перемешивали в течение 5 часов при 100°С. После завершения реакции верхнюю органическую фазу сливали. К остатку добавляли воду и экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором карбоната натрия и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения метил-6-бром-5-метилбензофуран-4-карбоксилата 4е (220 мг, выход 45%), в виде желтого твердого вещества.
Стадия 5
Метил-5-метил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-6-бром-5-метилбензофуран-4-карбоксилат 4е (250 мг, 0,93 ммоль), тетрагидро-2Н-пиран-4-амин (141 мг, 1,4 ммоль), трис(дибензилиденацетон)дипалладий (85 мг, 0,09 ммоль), (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин (116 мг, 0,19 ммоль) и карбонат цезия (909 мг, 2,79 ммоль) растворяли в 10 мл толуола. Смесь перемешивали в течение 12 часов при 100°С. После завершения реакции смесь фильтровали через слой целита, и фильтрационный кек промывали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения метил-5-метил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилата 4f (250 мг, выход 90%), в виде желтого твердого вещества.
Стадия 6
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метилбензофуран-4-карбоксилат
Метил-5-метил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 4f (250 мг, 0,87 ммоль) растворяли в 10 мл 1,2-дихлорэтана, впоследствии добавляли ацетальдегид (190 мг, 4,3 ммоль) и уксусную кислоту (260 г, 4,3 ммоль). Смесь перемешивали в течение 1 часа, впоследствии добавляли триацетоксиборгидрид натрия (545 мг, 2,6 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением. Добавляли этилацетат и насыщенный раствор бикарбоната натрия. Две фазы разделяли, и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метилбензофуран-4-карбоксилата 4g (180 мг, выход 56%) в виде желтого масла.
МС m/z (ЖХ-МС): 318,2 [М+1]
Стадия 7
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формил-5-метилбензофуран-4-карбоксилат
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метилбензофуран-4-карбоксилат 4g (180 мг, 0,57 ммоль) растворяли в 5 мл тетрагидрофурана. Смесь охлаждали до -70°С, впоследствии добавляли по каплям 2,0 М раствор диизопропиламида лития (0,57 мл, 1,14 ммоль) в атмосфере аргона. Смесь перемешивали в течение 1 часа, впоследствии добавляли 4-формилморфолин (98 мг, 0,85 ммоль). Смесь перемешивали в течение 1 часа, впоследствии медленно подогревали до комнатной температуры и добавляли раствор хлорида аммония. Смесь перемешивали в течение 20 минут и экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формил-5-метилбензофуран-4-карбоксилата 4h (130 мг, выход 66%), в виде желтого масла.
Стадия 8
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(гидроксиметил)-5-метилбензофуран-4-карбоксилат
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формил-5-метилбензофуран-4-карбоксилат 4h (130 мг, 0,38 ммоль) растворяли в смеси 1 мл тетрагидрофурана и 5 мл метанола, впоследствии добавляли порциями боргидрид натрия (22 мг, 0,57 ммоль). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением. Добавляли насыщенный раствор бикарбоната натрия, и смесь экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(гидроксиметил)-5-метилбензофуран-4-карбоксилата 4i (120 мг), в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ЖХ-МС): 348,0 [М+1]
Стадия 9
Метил-2-(бромметил)-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метилбензофуран-4-карбоксилат
Неочищенный метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(гидроксиметил)-5-метилбензофуран-4-карбоксилат 4i (130 мг, 0,36 ммоль) растворяли в 5 мл тетрагидрофурана, впоследствии добавляли по каплям трибромид фосфора (146 мг, 0,54 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции добавляли воду, и смесь экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения метил-2-(бромметил)-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метилбензофуран-4-карбоксилата 4j (152 мг), в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 10
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(пиперидин-1-илметил)бензофуран-4-карбоксилат
Неочищенный метил-2-(бромметил)-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метилбензофуран-4-карбоксилат 4j (140 мг, 0,33 ммоль) растворяли в 5 мл ацетонитрила, впоследствии добавляли по каплям 5 мл предварительно приготовленного раствора пиперидина (56 мг, 0,66 ммоль) в ацетонитриле. Смесь перемешивали в течение 1 часа при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением, впоследствии добавляли этилацетат и насыщенный раствор бикарбоната натрия. Две фазы разделяли, и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(пиперидин-1-илметил)бензофуран-4-карбоксилата 4k (120 мг, выход 86%), в виде бесцветного масла.
Стадия 11
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(пиперидин-1-илметил)бензофуран-4-карбоновая кислота
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(пиперидин-1-илметил)бензофуран-4-карбоксилат 4k (120 мг, 0,29 ммоль) растворяли в 10 мл метанола, впоследствии добавляли 3 мл 4М раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. После завершения реакции добавляли концентрированную хлористоводородную кислоту для доведения рН реакционного раствора до 4. Смесь концентрировали под пониженным давлением, и остаток растворяли в смеси дихлорметана и метанола (об.: об. составляет 5:1) и фильтровали. Фильтрационный кек промывали смесью дихлорметана и метанола (об. : об. составляет 5:1). Фильтрат объединяли и концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(пиперидин-1-илметил)бензофуран-4-карбоновой кислоты 4i (120 мг), в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ЖХ-МС): 399,0 [М+1]
Стадия 12
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метил-2-(пиперидин-1-илметил)бензофуран-4-карбоксамид
Неочищенную 6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(пиперидин-1-илметил)бензофуран-4-карбоновую кислоту 4i (40 мг, 0,1 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (29 мг, 0,15 ммоль), 1-гидроксибензотриазол (20 мг, 0,15 ммоль) и N,N-диизопропилэтиламин (63 мг, 0,48 ммоль). Смесь перемешивали в течение 16 часов, впоследствии добавляли 3-(аминометил)-4-метокси-6-метилпиридин-2(1Н)-она гидрохлорид 1о (26 мг, 0,13 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метил-2-(пиперидин-1-илметил)бензофуран-4-карбоксамида 4 (35 мг, выход 64%), в виде белого твердого вещества.
МС m/z(ИЭP): 551,7 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.45 (s, 1Н), 8.01 (s, 1Н), 7.40 (s, 1Н), 6.91 (brs, 1Н), 6.12 (s, 1Н), 4.30 (d, 2Н), 3.84 (brs, 5Н), 3.23 (t, 2Н), 3.04-3.09 (m, 2Н), 2.95-3.01 (m, 1Н), 2.62 (s, 3Н), 2.19 (s, 3Н), 1.64-1.81 (brd, 6Н), 1.50-1.55 (m, 4Н), 1.38 (brs, 2Н), 0.81 (t, 3Н).
Пример 5
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-изопропилпиперазин-1-ил)метил)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид
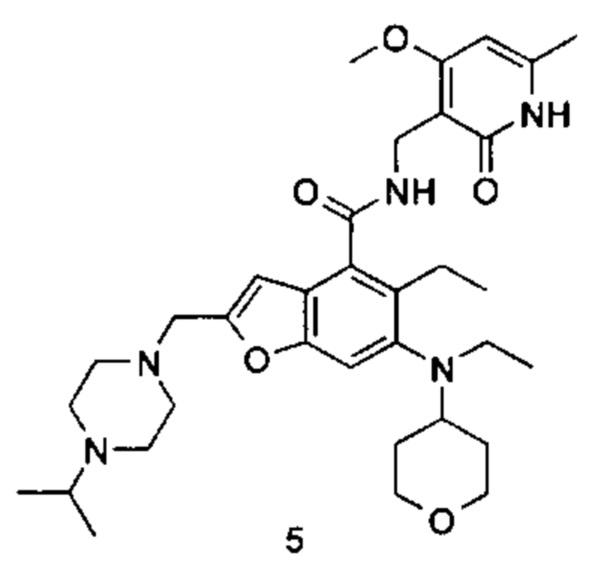
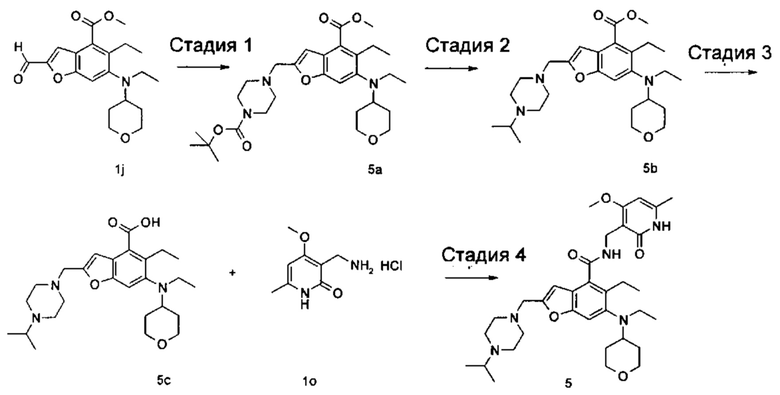
Стадия 1
трет-Бутил-4-((5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)бензофуран-2-ил)метил)пиперазин-1-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилат 1j (30 мг, 0,083 ммоль), 1-(трет-бутоксикарбонил)пиперазин (24 мг, 0,13 ммоль) и уксусную кислоту (25 мг, 0,42 ммоль) добавляли к 5 мл метанола. Смесь перемешивали в течение 1 часа, впоследствии добавляли триацетоксиборгидрид натрия (53 мг, 0,25 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции реакционный раствор концентрировали под пониженным давлением, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения трет-бутил-4-((5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)бензофуран-2-ил)метил)пиперазин-1-карбоксилат 5а (40 мг, выход 90%), в виде желтого масла.
МС m/z (ЖХ-МС): 530,3 [М+1]
Стадия 2
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-изопропилпиперазин-1-ил)метил)бензофуран-4-карбоксилат
трет-Бутил-4-((5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)бензофуран-2-ил)метил)пиперазин-1-карбоксилат 5а (40 мг, 0,075 ммоль) добавляли к 10 мл трифторуксусной кислоты. Смесь перемешивали в течение 1 часа при комнатной температуре, впоследствии концентрировали под пониженным давлением. К остатку добавляли 5 мл N,N-диметилформамида и карбонат калия (21 мг, 0,15 ммоль), и смесь перемешивали в течение 30 минут. Затем добавляли 1-йодпропан (20 мг, 0,11 ммоль), и реакционную систему перемешивали в течение 1 часа при 70°С. После завершения реакции реакционный раствор наливали в избыток воды и экстрагировали три раза смесью дихлорметана и метанола (об.: об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-изопропилпиперазин-1-ил)метил)бензофуран-4-карбоксилата 5b (25 мг, выход 70%), в виде бесцветного масла.
МС m/z (ЖХ-МС): 472,3 [М+1]
Стадия 3
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-изопропилпиперазин-1-ил)метил)бензофуран-4-карбоновая кислота
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-изопропилпиперазин-1-ил)метил)бензофуран-4-карбоксилат 5b (10 мг, 0,021 ммоль) растворяли в смеси 3 мл метанола и 1 мл тетрагидрофурана, впоследствии добавляли 13 мл 2М раствора гидроксида натрия. Реакционную систему перемешивали в течение 12 часов при 60°С. После завершения реакции реакционный раствор нейтрализовали концентрированной серной кислотой, впоследствии концентрировали под пониженным давлением. Смесь растворяли в смеси дихлорметана и метанола (об. : об. составляет 5:1) и фильтровали. Фильтрационный кек промывали смесью дихлорметана и метанола (об. : об. составляет 5:1). Фильтрат объединяли и концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-изопропилпиперазин-1-ил)метил)бензофуран-4-карбоновой кислоты 5с (9 мг), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ЖХ-МС): 458,4 [М+1]
Стадия 4
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-изопропилпиперазин-1-ил)метил)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид
Неочищенную 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-изопропилпиперазин-1-ил)метил)бензофуран-4-карбоновую кислоту 5с (10 мг, 0,022 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (7 мг, 0,033 ммоль), 1-гидроксибензотриазол (5 мг, 0,033 ммоль) и N,N-диизопропилэтиламин (6 мг, 0,044 ммоль). Смесь перемешивали в течение 30 минут, впоследствии добавляли 3-(аминометил)-4-метокси-6-метилпиридин-2(1Н)-она гидрохлорид 1о (5,8 мг, 0,028 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-изопропилпиперазин-1-ил)метил)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамида 5 (9 мг, выход 69%), в виде белого твердого вещества.
МС m/z (ИЭР): 608,6 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.45 (s, 1Н), 8.01 (s, 1Н), 7.39 (s, 1Н), 6.56 (brs, 1Н), 6.11 (s, 1Н), 4.29 (d, 2Н), 3.85 (brs, 2Н), 3.83 (s, 3Н), 3.63 (brs, 3Н), 3.22 (t, 2Н), 3.03-3.07 (m, 2Н), 2.93-2.99 (m, 2Н), 2.79-2.82 (m, 2Н), 2.34-2.68 (brs, 8Н), 2.19 (s, 3Н), 1.64-1.67 (brd, 2Н), 1.50-1.52 (m, 2Н), 1.24-1.26 (brd, 2Н), 1.06 (t, 3Н), 0.98 (brs, 6Н), 0.83 (t, 3Н).
Пример 6
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(морфолинометил)бензофуран-4-карбоксамид
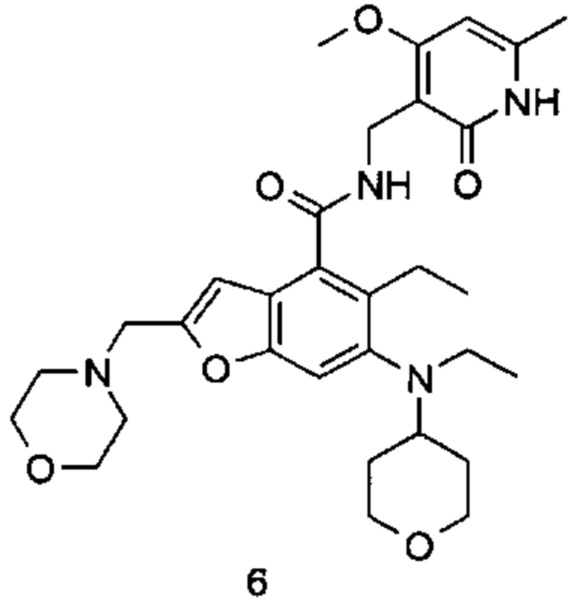
Стадия 1
Метил-6-бром-5-этил-2-формилбензофуран-4-карбоксилат
Метил-6-бром-5-этилбензофуран-4-карбоксилат 1g (260 мг, 0,92 ммоль) растворяли в 8 мл тетрагидрофурана. Смесь охлаждали до -70°С, впоследствии добавляли по каплям 2,0 М раствор диизопропиламида лития (0,92 мл, 1,84 ммоль) в атмосфере аргона. Смесь перемешивали в течение 1 часа, впоследствии добавляли 4-формилморфолин (158 мг, 1,38 ммоль). Реакционную смесь медленно подогревали до комнатной температуры и добавляли избыток хлорида аммония. Смесь тщательно перемешивали и экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения метил-6-бром-5-этил-2-формилбензофуран-4-карбоксилата 6а (80 мг, выход 48%), в виде желтого и белого твердого вещества.
Стадия 2
Метил-6-бром-5-этил-2-(гидроксиметил)бензофуран-4-карбоксилат
Метил-6-бром-5-этил-2-формилбензофуран-4-карбоксилат 6а (80 мг, 0,26 ммоль)) растворяли в смеси 4 мл метанола и 0,5 мл тетрагидрофурана, впоследствии добавляли порциями боргидрид натрия (20 мг, 0,51 ммоль) при комнатной температуре. Смесь перемешивали в течение 3 минут. После завершения реакции добавляли этилацетат и насыщенный раствор бикарбоната натрия. Фазы разделяли, и смесь экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения метил-6-бром-5-этил-2-(гидроксиметил)бензофуран-4-карбоксилата 6b (80 мг), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
Метил-6-бром-5-этил-2-(морфолинометил)бензофуран-4-карбоксилат
Неочищенный метил-6-бром-5-этил-2-(гидроксиметил)бензофуран-4-карбоксилат 6b (80 мг, 0,26 ммоль) растворяли в 5 мл дихлорметана, впоследствии добавляли метансульфонилхлорид (45 мг, 0,38 ммоль) и трифторуксусную кислоту (130 мг, 1,29 ммоль). Смесь перемешивали в течение 12 часов. Смесь концентрировали под пониженным давлением, впоследствии добавляли 5 мл N,N-диметилформамид, карбонат калия (71 мг, 0,51 ммоль) и морфолин (40 мг, 0,51 ммоль). Реакционную систему перемешивали в течение 1 часов при 80°С. После завершения реакции добавляли воду, и смесь экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения метил-6-бром-5-этил-2-(морфолинометил)бензофуран-4-карбоксилата 6с (65 мг, выход 64%), в виде желтого твердого вещества.
МС m/z (ИЭР): 382,0 [М+1]
Стадия 4
Метил-5-этил-2-(морфолинометил)-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-6-бром-5-этил-2-(морфолинометил)бензофуран-4-карбоксилат 6с (90 мг, 0,24 ммоль), тетрагидро-2Н-пиран-4-амин (36 мг, 0,35 ммоль), трис(дибензилиденацетон)дипалладий (22 мг, 0,02 ммоль), (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин (30 мг, 0,05 ммоль) и карбонат цезия (230 мг, 0,71 ммоль) растворяли в 5 мл толуола. Смесь перемешивали в течение 12 часов при 100°С. После завершения реакции смесь фильтровали через слой целита. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения метил-5-этил-2-(морфолинометил)-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилата 6d (85 мг, выход 89%), в виде белого твердого вещества.
МС m/z (ЖХ-МС): 401,2 [М+1]
Стадия 5
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(морфолинометил)бензофуран-4-карбоксилат
Метил-5-этил-2-(морфолинометил)-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 6d (85 мг, 0,21 ммоль) растворяли в 5 мл 1,2-дихлорэтана, впоследствии добавляли ацетальдегид (93 мг, 2,1 ммоль) и уксусную кислоту (63 мг, 1,06 ммоль). Смесь перемешивали в течение 2 часов, впоследствии добавляли триацетоксиборгидрид натрия (133 мг, 0,63 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(морфолинометил)бензофуран-4-карбоксилата 6е (75 мг, выход 82%), в виде бесцветного масла.
МС m/z (ЖХ-МС): 344,1 [М-86]
Стадия 6
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(морфолинометил)бензофуран-4-карбоновая кислота
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(морфолинометил)бензофуран-4-карбоксилат 6е (75 мг, 0,17 ммоль) растворяли в смеси 1 мл тетрагидрофурана и 3 мл метанола, впоследствии добавляли 3 мл 4М раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. После завершения реакции добавляли концентрированную хлористоводородную кислоту для доведения рН реакционного раствора до 4. Смесь концентрировали при пониженном давлении. Остаток промывали метанолом и фильтровали для удаления белого твердого вещества. Фильтрат объединяли и концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(морфолинометил)бензофуран-4-карбоновой кислоты 6f (71 мг), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 7
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(морфолинометил)бензофуран-4-карбоксамид
Неочищенную 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(морфолинометил)бензофуран-4-карбоновую кислоту 6f (45 мг, 0,11 ммоль) растворяли в 5 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (31 мг, 0,16 ммоль), 1-гидроксибензотриазол (22 мг, 0,16 ммоль) и N,N-диизопропилэтиламин (70 мг, 0,54 ммоль). Смесь перемешивали в течение 1 часа, впоследствии добавляли 3-(аминометил)-4-метокси-6-метилпиридин-2(1Н)-она гидрохлорид 1о (33 мг, 0,16 ммоль). Смесь перемешивали в течение 36 часов при комнатной температуре. После завершения реакции добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(морфолинометил)бензофуран-4-карбоксамида 6 (35 мг, выход 57%) в виде белого твердого вещества.
МС m/z (ИЭР): 567,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.43 (s, 1Н), 7.99 (t, 1Н), 7.39 (s, 1Н), 6.54 (s, 1Н), 6.10 (s, 1Н), 4.28 (d, 2Н), 3.84 (brs, 2Н), 3.82 (s, 3Н), 3.59 (s, 2Н), 3.55-3.57 (m, 4Н), 3.21 (t, 2Н), 3.02-3.07 (m, 2Н), 2.92-2.97 (m, 1Н), 2.78-2.83 (m, 2Н), 2.43 (brs, 4Н), 2.18 (s, 3Н), 1.64-1.67 (brd, 2Н), 1.45-1.55 (m, 2Н), 1.05 (t, 3Н), 0.82 (t, 3Н).
Пример 7
2-((4,4-Дифторпиперидин-1-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид
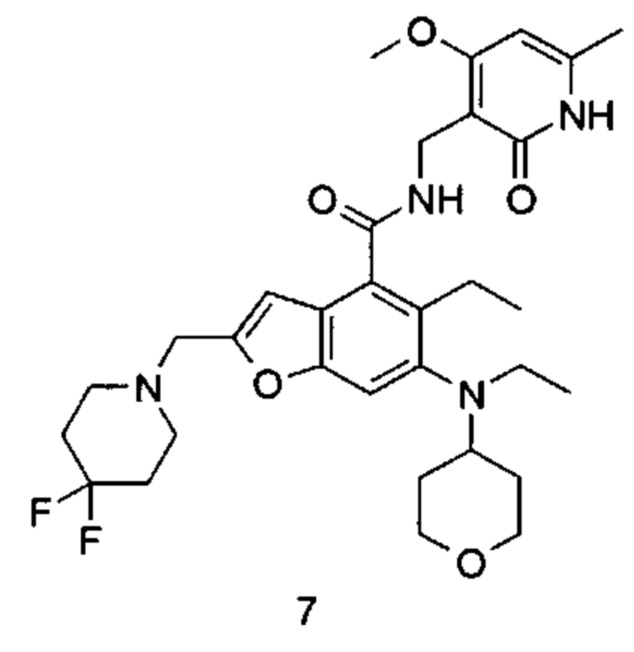
Стадия 1
Метил-2-((4,4-дифторпиперидин-1-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилат 1j (50 мг, 0,14 ммоль), 4,4-дифторпиперидина гидрохлорид (33 мг, 0,21 ммоль) и уксусную кислоту (42 мг, 0,49 ммоль) добавляли к 5 мл метанола. Смесь перемешивали 1 час, впоследствии добавляли молекулярные сита и триацетоксиборгидрид натрия (88 мг, 0,42 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции смесь нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали смесью дихлорметана и метанола (об. : об. составляет 5:1). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения метил-2-((4,4-дифторпиперидин-1-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилата 7а (25 мг, выход 45%), в виде бесцветного масла.
МС m/z (ИЭР): 465,1 [М+1]
Стадия 2
2-((4,4-Дифторпиперидин-1-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоновая кислота
Метил-2-((4,4-дифторпиперидин-1-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 7а (20 мг, 0,043 ммоль) растворяли в смеси 1 мл тетрагидрофурана и 3 мл метанола, впоследствии добавляли 3 мл 4М раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. После завершения реакции реакционный раствор нейтрализовали концентрированной хлористоводородной кислотой, впоследствии концентрировали под пониженным давлением. Смесь растворяли в смеси дихлорметана и метанола (об.: об. составляет 5:1) и фильтровали. Фильтрационный кек промывали смесью дихлорметана и метанола (об. : об. составляет 5:1). Фильтрат объединяли и концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 2-((4,4-дифторпиперидин-1-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоновой кислоты 7b (20 мг), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
2-((4,4-Дифторпиперидин-1-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид
Неочищенную 2-((4,4-дифторпиперидин-1-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоновую кислоту 7b (20 мг, 0,044 ммоль) растворяли в 5 мл N,N-диметилформамиде, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (13 мг, 0,066 ммоль), 1-гидроксибензотриазол (9 мг, 0,066 ммоль) и N,N-диизопропилэтиламин (29 мг, 0,22 ммоль). Смесь перемешивали 1 час, затем добавляли 3-(аминометил)-4-метокси-6-метилпиридин-2(1Н)-она гидрохлорид 1о (12 мг, 0,058 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции, добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 2-((4,4-дифторпиперидин-1-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамида 7 (21 мг, выход 78%), в виде белого твердого вещества.
МС m/z (ИЭР): 601,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.43 (s, 1Н), 8.00 (t, 1Н), 7.39 (s, 1Н), 6.55 (s, 1Н), 6.10 (s, 1Н), 4.28 (d, 2Н), 3.84 (brs, 2Н), 3.82 (s, 3Н), 3.69 (s, 2Н), 3.21 (t, 2Н), 3.02-3.07 (m, 2Н), 2.92-2.98 (m, 1Н), 2.78-2.83 (m, 2Н), 2.57 (brs, 4Н), 2.17 (s, 3Н), 1.95-1.98 (m, 4Н), 1.64-1.66 (brd, 2Н), 1.46-1.55 (m, 2Н), 1.05 (t, 3Н), 0.82 (t, 3Н).
Пример 8
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(пирролидин-1-илметил)бензофуран-4-карбоксамид
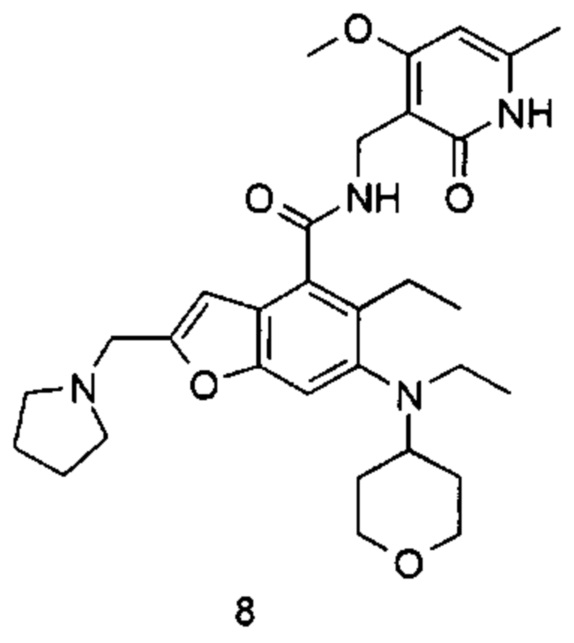
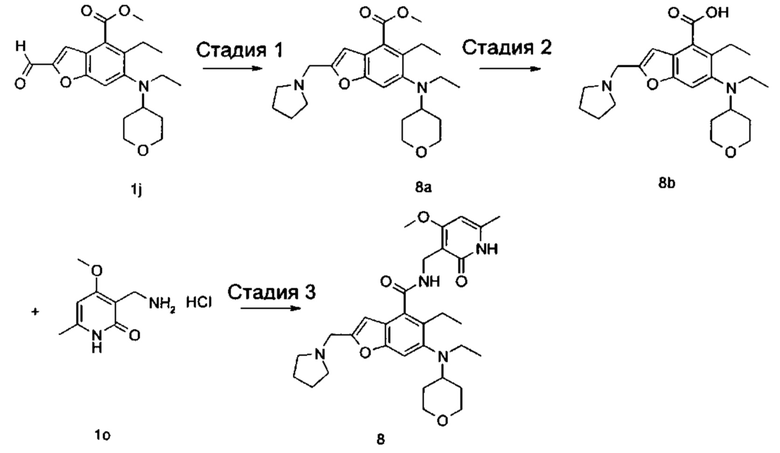
Стадия 1
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пирролидин-1-илметил)бензофуран-4-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формилбензофуран-4- карбоксилат 1j (40 мг, 0,11 ммоль), пирролидин (15 мг, 0,22 ммоль) и уксусную кислоту (30 мг, 0,55 ммоль) растворяли в 5 мл метанола. Смесь перемешивали в течение 1 часа, впоследствии добавляли триацетоксиборгидрид натрия (70 мг, 0,33 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции смесь нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пирролидин-1-илметил)бензофуран-4-карбоксилата 8а (44 мг, выход 95%), в виде бесцветного масла.
Стадия 2
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пирролидин-1-илметил)бензофуран-4- карбоновая кислота
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пирролидин-1-илметил) бензофуран-4-карбоксилат 8а (56 мг, 0,13 ммоль) растворяли в смеси 1 мл тетрагидрофурана и 5 мл метанола, впоследствии добавляли 2 мл 4 М раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. После завершения реакции добавляли концентрированную хлористоводородную кислоту для доведения рН до 4, затем смесь концентрировали под пониженным давлением. Смесь растворяли в смеси дихлорметана и метанола (об.: об. составляет 5:1) и фильтровали. Фильтрационный кек промывали смесью дихлорметана и метанола (об.: об. составляет 5:1). Фильтрат объединяли и концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пирролидин-1-илметил)бензофуран-4-карбоновой кислоты 8b (50 мг), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 399,0 [М-1]
Стадия 3
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(пирролидин-1-илметил)бензофуран-4-карбоксамид
Неочищенную
5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пирролидин-1-илметил)бензофуран-4-карбоновую кислоту 8b (25 мг, 0,60 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (15 мг, 0,09 ммоль), 1-гидроксибензотриазол (12 мг, 0,09 ммоль) и N,N-диизопропилэтиламин (387 мг, 3,0 ммоль). Смесь перемешивали 1 час, затем добавляли 3-(аминометил)-4-метокси-6-метилпиридин-2(1Н)-она гидрохлорид 1о (16 мг, 0,078 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об.: об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(пирролидин-1-илметил)бензофуран-4-карбоксамида 8 (12 мг, выход 36%) в виде белого твердого вещества.
МС m/z(ИЭР): 551,6 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.43 (s, 1Н), 7.99 (t, 1Н), 7.37 (s, 1Н), 6.50 (s, 1Н), 6.10 (s, 1Н), 4.28 (d, 2Н), 3.84 (brs, 2Н), 3.82 (s, 3Н), 3.71 (brs, 2Н), 3.18-3.26 (m, 4Н), 3.03-3.07 (m, 2Н), 2.92-2.97 (m, 1Н), 2.78-2.81 (m, 2Н), 2.17 (s, 3Н), 1.70 (brs, 4Н), 1.64 (brs, 2Н), 1.46-1.54 (m, 2Н), 1.23 (brs, 2Н), 1.05 (t, 3Н), 0.82 (t, 3Н).
Пример 9
N-((4,6-Диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)бензофуран-4-карбоксамид
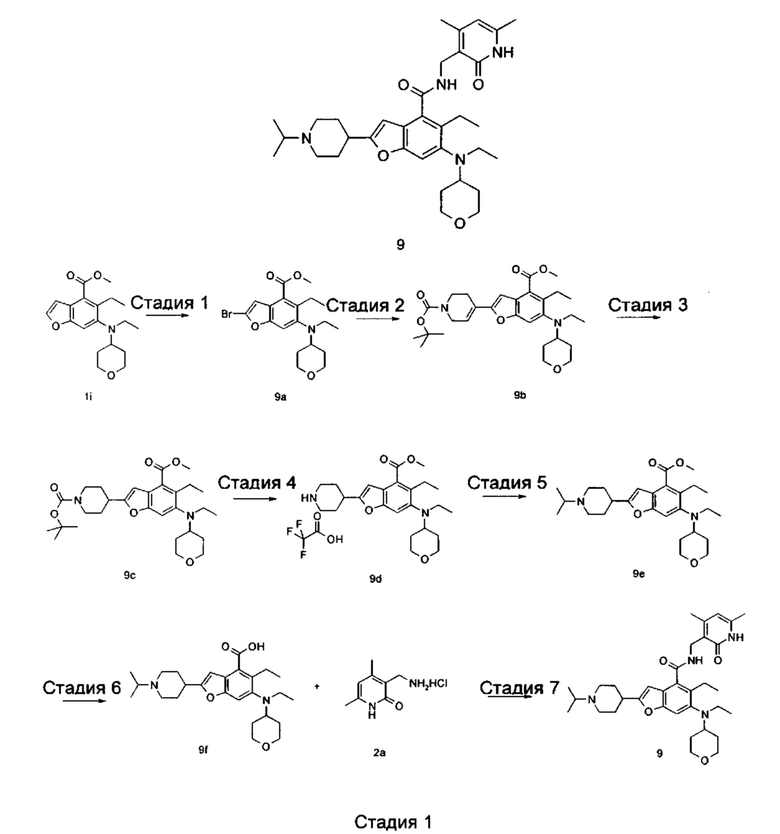
Метил-2-бром-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 1i (1 г, 3,0 ммоль) растворяли в 15 мл тетрагидрофурана, впоследствии добавляли по каплям 1,8 мл 2М диизопропиламида лития при -70°С. смесь перемешивали 90 минут, впоследствии добавляли 1,2-дибромтетрахлорэтан (978 мг, 3 ммоль). Реакционный раствор перемешивали в течение 1 часа и подогревали до комнатной температуры. Добавляли раствор хлорида аммония, и смесь экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения метил-2-бром-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карб оксилата 9а (320 мг, выход 40%), в виде желтого масла.
Стадия 2
трет-Бутил-4-(5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)бензофуран-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат
Метил-2-бром-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 9а (1,4 г, 3,4 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридине-1(2Н)-карбоксилат (1.6 г, 5.1 ммоль), трис(дибензилиденацетон)дипалладий (312 мг, 0.34 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилдифенил (325 мг, 0,68 ммоль) и карбонат цезия (3,4 г, 10,2 ммоль) смешивали в 50 мл смеси 1,4-диоксана и воды (об.: об. составляет 4:1). Смесь подогревали до 80°С и перемешивали в течение 16 часов. Реакционный раствор фильтровали через слой целита и промывали этилацетатом. К фильтрату добавляли воду, две фазы разделяли, и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения трет-бутил-4-(5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)бензофуран-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата 9b (1,67 г, выход 95%), в виде масла пурпурного цвета.
Стадия 3
трет-Бутил-4-(5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)бензофуран-2-ил)пиперидин-1-карбоксилат
трет-Бутил-4-(5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)бензофуран-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат 9b (1,7 г, 3,3 ммоль) растворяли в 23 мл смеси метанола и тетрагидрофурана (об.: об. составляет 20:3), впоследствии добавляли Pd/C (200 мг, 10%). Реакционную систему продували три раза водородом и перемешивали в течение 1 часа. Реакционный раствор фильтровали через слой целита. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения трет-бутил-4-(5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)бензофуран-2-ил)пиперидин-1-карбоксилата 9с (1,6 г, выход 94%), в виде светло-желтого масла.
МС m/z (ИЭР): 515,0 [М+1]
Стадия 4
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-4-ил)бензофуран-4-карбоксилата 2,2,2-трифторацетат
трет-Бутил-4-(5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)бензофуран-2-ил)пиперидин-1-карбоксилат 9 с (1,6 г, 3,3 ммоль) растворяли в 20 мл дихлорметана, впоследствии добавляли 4 мл трифторуксусной кислоты. Смесь перемешивали в течение 2 часов, впоследствии концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-4-ил)бензофуран-4-карбоксилата 2,2,2-трифторацетата 9d (1,3 г), в виде светло-желтого масла, которое непосредственно использовали в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 415,3 [М+1]
Стадия 5
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)бензофуран-4-карбоксилат
Неочищенный метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-4-ил)бензофуран-4-карбоксилата 2,2,2-трифторацетат 9d (1,3 г, 3,1 ммоль) растворяли в 30 мл N,N-диметилформамида, впоследствии добавляли карбонат калия (1,3 г, 8,4 ммоль). Смесь перемешивали 10 минут, затем добавляли 2-бромпропан (575 мг, 4,7 ммоль) и йодид калия (261 мг, 1,5 ммоль). Смесь перемешивали в течение 2 часов при 70°С, впоследствии добавляли воду, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об.: об. составляет 5:1). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)бензофуран-4-карбоксилата 9е (1,4 г, выход 90%), в виде бесцветного масла.
МС m/z (ИЭР): 457,0 [М+1]
Стадия 6
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)бензофуран-4-карбоновая кислота
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)бензофуран-4-карбоксилат 9е (1,1 г, 2,4 ммоль) растворяли в 30 мл метанола, затем добавляли 5 мл 4 М раствора гидроксида натрия. Смесь перемешивали в течение 16 часов при 60°С, впоследствии добавляли концентрированную хлористоводородную кислоту для доведения рН реакционного раствора до 4. Смесь концентрировали под пониженным давлением, и остаток растворяли в смеси дихлорметана и метанола (об.: об. составляет 5:1) и фильтровали. Фильтрационный кек промывали смесью дихлорметана и метанола (об.: об. составляет 5:1). Фильтрат объединяли и концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)бензофуран-4-карбоновой кислоты 9f (1,4 г), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 443,3 [М+1]
Стадия 7
N-((4,6-Диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)бензофуран-4-карбоксамид
Неочищенную
5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил) бензофуран-4-карбоновую кислоту 9f (1,1 г, 2,5 ммоль) растворяли в 20 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (715 мг, 3,7 ммоль), 1-гидроксибензотриазол (510 мг, 3,7 ммоль) и N,N-диизопропилэтиламин (1,6 г, 12,5 ммоль). Смесь перемешивали 1 час, затем добавляли 3-(аминометил)-4,6-метокси-6-диметилпиперидин-2(1Н)-она гидрохлорид 2а (470 мг, 2,5 ммоль). Смесь перемешивали в течение 16 часов. После завершения реакции, добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об.: об. составляет 8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)бензофуран-4-карбоксамида 9 (850 мг, выход 59%), в виде белого твердого вещества.
МС m/z (ИЭР): 577,7 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.50 (s, 1Н), 8.16 (t, 1Н), 7.38 (s, 1Н), 6.37 (brs, 1Н), 5.87 (s, 1Н), 4.31 (d, 2Н), 3.82 (d, 2Н), 3.21 (t, 2Н), 3.01-3.07 (m, 4Н), 2.91-2.96 (m, 1Н), 2.79-2.81 (m, 2Н), 2.24 (s, 3Н), 2.17 (brs, 2Н), 2.12 (s, 3Н), 2.02 (brs, 2Н), 1.63-1.66 (brd, 2Н), 1.45-1.54 (m, 2Н), 1.25 (brs, 6Н), 1.01 (t, 3Н), 0.81 (t, 3Н).
Пример 10
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид
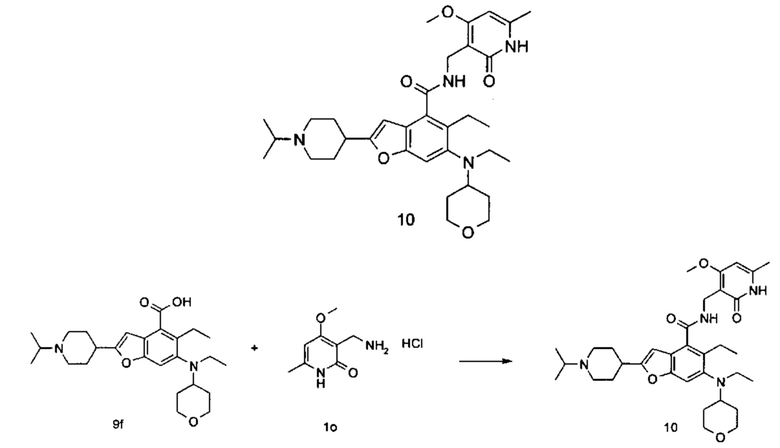
Неочищенную
5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил) бензофуран-4-карбоновую кислоту 9f (30 мг, 0,07 ммоль) растворяли в 5 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (20 мг, 0,1 ммоль), 1-гидроксибензотриазол (15 мг, 0,1 ммоль) и N,N-диизопропилэтиламин (45 мг, 0,34 ммоль). Смесь перемешивали 1 час, впоследствии добавляли 3-(аминометил)-4-метокси-6-метилпиридин-2(1Н)-она гидрохлорид 1о (21 мг, 0,1 ммоль). Смесь перемешивали в течение 16 часов. После завершения реакции, добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об.: об. составляет 8: 1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(1-изопропилпиперидин-4-ил)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамида 10 (31 мг, выход 77%), в виде белого твердого вещества.
МС m/z (ИЭР): 593,3 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.44 (s, 1Н), 7.95 (brs, 1Н), 7.36 (s, 1Н), 6.37 (brs, 1Н), 6.11 (s, 1Н), 4.28 (d, 2Н), 3.84 (brs, 2Н), 3.82 (s, 3Н), 3.21 (t, 3Н), 3.01-3.06 (m, 3Н), 2.90-2.96 (m, 2Н), 2.77-2.83 (m, 3Н), 2.19 (s, 3Н), 2.01 (brs, 4Н), 1.63-1.67 (brd, 2Н), 1.45-1.53 (m, 2Н),1.22 (brs, 6Н), 1.04 (t, 3Н), 0.81 (t, 3Н).
Пример 11
N-((4,6-Диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-(2-гидрокси-2-метилпропил)пиперазин-1-ил)метил)бензофуран-4-карбоксамид 11
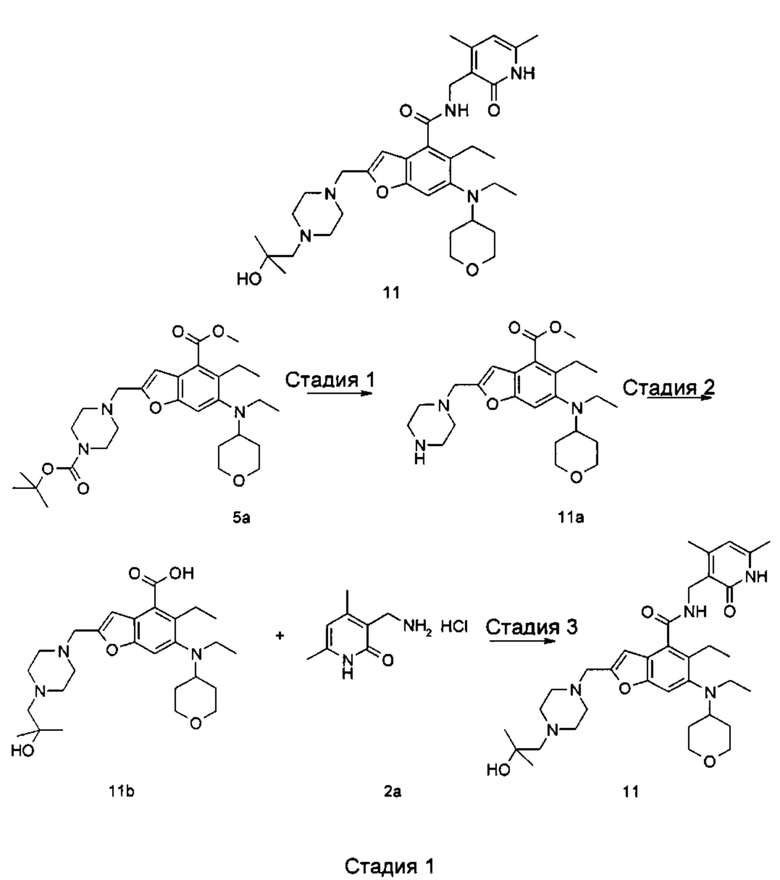
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперазин-1-ил метил)бензофуран-4-карбоксилат 11а
Соединение 5а (60 мг, 0,12 ммоль) растворяли в 5 мл дихлорметана, впоследствии добавляли 0,5 мл трифторуксусной кислоты. После перемешивания в течение 2 часов смесь концентрировали под пониженным давлением, нейтрализовали насыщенным раствором карбоната натрия и экстрагировали смесью дихлорметана и метанола. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 11а (45 мг), в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-(2-гидрокси-2-метилпропил) пиперазин-1-ил)метил)бензофуран-4-карбоновая кислота 11b
Неочищенное соединение 11а (25 мг, 0,06 ммоль) растворяли в 5 мл этанола, впоследствии добавляли 2,2-диметилоксиран (6,5 мг, 0,09 ммоль) и карбонат калия (17 мг, 0,12 ммоль). Смесь перемешивали при 60°С в течение 12 часов. После завершения реакции смесь фильтровали и добавляли 12 М раствор хлористоводородной кислоты для доведения рН до 4. Смесь концентрировали под пониженным давлением, и полученный в результате остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке соединения 11b (15 мг, выход 52%), в виде белого твердого вещества.
Стадия 3
N-((4,6-Диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-(2-гидрокси-2-метилпропил)пиперазин-1-ил)метил)бензофуран-4-карбоксамид 11
Соединение 11b (15 мг, 0,031 ммоль) растворяли в 4 мл N,N-диметилформамида, впоследствии в указанный выше реакционный раствор добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (9 мг, 0,046 ммоль), 1-гидроксибензотриазол (7 мг, 0,046 ммоль) и N,N-диизопропилэтиламин (20 мг, 0,155 ммоль). Смесь перемешивали 1 час, впоследствии в указанный выше реакционный раствор добавляли соединение 2а (9 мг, 0,046 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об.: об. составляет 10:1) (10 мл). Органические фазы объединяли, промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 11 (13 мг, выход 70%), в виде белого твердого вещества.
МС m/z (ИЭР): 622,7 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.48 (brs, 1Н), 8.17 (t, 1Н), 7.39 (s, 1Н), 6.48 (s, 1Н), 5.86 (s, 1Н), 4.32 (d, 2Н), 4.04 (brs, 1Н), 3.83 (brd, 2Н), 3.58 (s, 2Н), 3.21 (t, 2Н), 3.04 (q, 2Н), 2.91-2.97 (m, 1Н), 2.79 (q, 2Н), 2.56 (brs, 4Н), 2.45 (brs, 4Н), 2.23 (s, 3Н), 2.16 (brs, 2Н), 2.11 (s, 3Н), 1.63-1.66 (m, 2Н), 1.48-1.52 (m, 2Н), 1.00-1.06 (m, 9Н), 0.82 (t, 3Н).
Пример 12
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-((4-метилпиперазин-1-ил)метил)бензофуран-4-карбоксамид 12
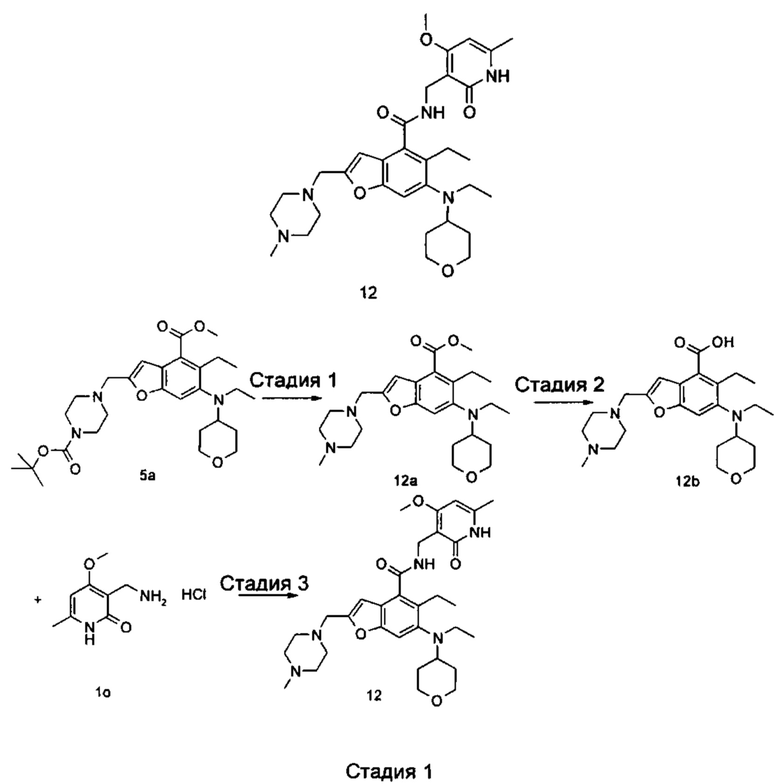
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-метилпиперазин-1-ил)метил)бензофуран-4-карбоксилат 12а
Соединение 5а (74 мг, 0,14 ммоль) растворяли в 10 мл 1,2-дихлорэтана, впоследствии добавляли 1,0 мл трифторуксусной кислоты. После перемешивания в течение 1 часа смесь концентрировали при пониженном давлении. Впоследствии к остатку последовательно добавляли 5 мл метанола, формальдегид (61 мг, 1,4 ммоль) и триацетоксиборгидрид натрия (88 мг, 0,42 ммоль). Смесь перемешивали в течение 3 часов. После завершения реакции смесь концентрировали под пониженным давлением, нейтрализовали 20 мл насыщенного раствора бикарбоната натрия и экстрагировали смесью дихлорметана и метанола (об.: об. составляет 8:1) (10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 12а (60 мг, выход 97%), в виде белого твердого вещества.
Стадия 2
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-((4-метилпиперазин-1-ил)метил)бензофуран-4-карбоновая кислота 12b
Соединение 12а (60 мг, 0,13 ммоль) растворяли в 4 мл метанола, впоследствии добавляли 4 мл 20% раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. После завершения реакции добавляли 12 М хлористоводородную кислоту для доведения рН до 4. Смесь концентрировали под пониженным давлением, впоследствии остаток растворяли в 10 мл метанола и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 12b (55 мг), в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-((4-метилпиперазин-1-ил)метил)бензофуран-4-карбоксамид 12
Неочищенное соединение 12b (25 мг, 0,058 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (17 мг, 0,087 ммоль), 1-гидроксибензотриазол (12 мг, 0,087 ммоль) и N,N-диизопропилэтиламин (38 мг, 0,29 ммоль). Смесь перемешивали 2 час, впоследствии к реакционному раствору добавляли соединение 1о (18 мг, 0,087 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции добавляли 20 мл воды, реакционный раствор экстрагировали смесью дихлорметана и метанола (об.: об. составляет 8: 1) (10 мл×3). Органические фазы объединяли, промывали водой (10 мл) и насыщенным раствором хлорида натрия (10 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 12 (22 мг, выход 65%), в виде белого твердого вещества.
МС m/z (ИЭР): 580,3 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.43 (brs, 1Н), 7.99 (s, 1Н), 7.38 (s, 1Н), 6.56 (s, 1Н), 6.10 (s, 1Н), 4.28 (d, 2Н), 3.85 (brs, 2Н), 3.82 (s, 3Н), 3.63 (s, 2Н), 3.21 (t, 2Н), 3.02-3.07 (m, 2Н), 2.92-2.97 (m, 1Н), 2.79-2.81 (m, 2Н), 2.58 (brs, 4Н), 2.40 (brs, 4Н), 2.18 (s, 3Н), 1.64-1.66 (m, 2Н), 1.46-1.55 (m, 2Н), 1.05 (t, 3Н), 0.82 (t, 3Н).
Пример 13
2-((Диметиламино)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид 13
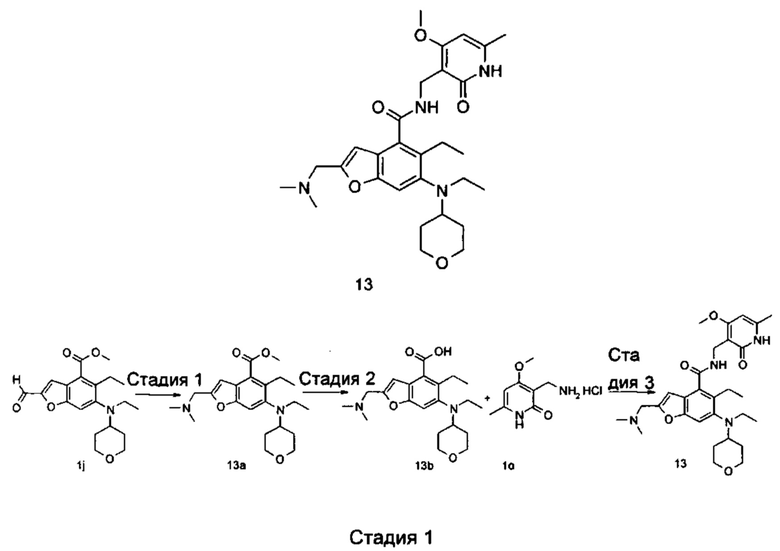
Метил-2-((диметиламино)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 13а
Соединение 1j (30 мг, 0,084 ммоль) растворяли в 5 мл метанола, впоследствии добавляли диметиламина гидрохлорид (14 мг, 0,17 ммоль). Смесь перемешивали 1 час, впоследствии добавляли триацетоксиборгидрид натрия (53 мг, 0,25 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции смесь концентрировали под пониженным давлением, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали смесью дихлорметана и метанола. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 13а (30 мг, выход 90%), в виде желтого масла.
Стадия 2
2-((Диметиламино)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоновая кислота 13b
Соединение 13а (30 мг, 0,077 ммоль) растворяли в смеси 3 мл этанола и 1 мл тетрагидрофурана, впоследствии добавляли 5 мл 2 н. раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. После завершения реакции реакционный раствор доводили до слабокислого состояния концентрированной хлористоводородной кислотой. Смесь концентрировали под пониженным давлением, и остаток растворяли в смеси дихлорметана и метанола (об.: об. составляет 1:1) и фильтровали, чтобы удалить неорганические соли. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 13b (25 мг), в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
2-((Диметиламино)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид 13
Неочищенное соединение 13b (15 мг, 0,040 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (7,7 мг, 0,04 ммоль), 1-гидроксибензотриазол (8 мг, 0,060 ммоль) и N,N-диизопропилэтиламин (26 мг, 0,20 ммоль). Смесь перемешивали 0,5 часа, впоследствии добавляли 1о (11 мг, 0,052 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции добавляли избыток воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об.: об. составляет 8:1) (10 мл). Органические фазы объединяли, промывали водой (10 мл) и насыщенным раствором хлорида натрия (10 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 13 (7 мг, выход 30%), в виде белого твердого вещества.
МС m/z (ИЭР): 525,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.44 (brs, 1Н), 8.00 (brs, 1Н), 7.37 (s, 1Н), 6.51 (s, 1Н), 6.10 (s, 1Н), 4.28 (d, 2Н), 3.85 (brs, 2Н), 3.82 (s, 3Н), 3.52 (s, 2Н), 3.21 (t, 2Н), 3.02-3.06 (m, 2Н), 2.92-2.96 (m, 1Н), 2.77-2.83 (m, 2Н), 2.19 (s, 6Н), 2.17 (s, 3Н), 1.63-1.67 (m, 2Н), 1.46-1.53 (m, 2Н), 1.06 (t, 3Н), 0.82 (t, 3Н).
Пример 14
2-Циклопентил-N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксамид 14
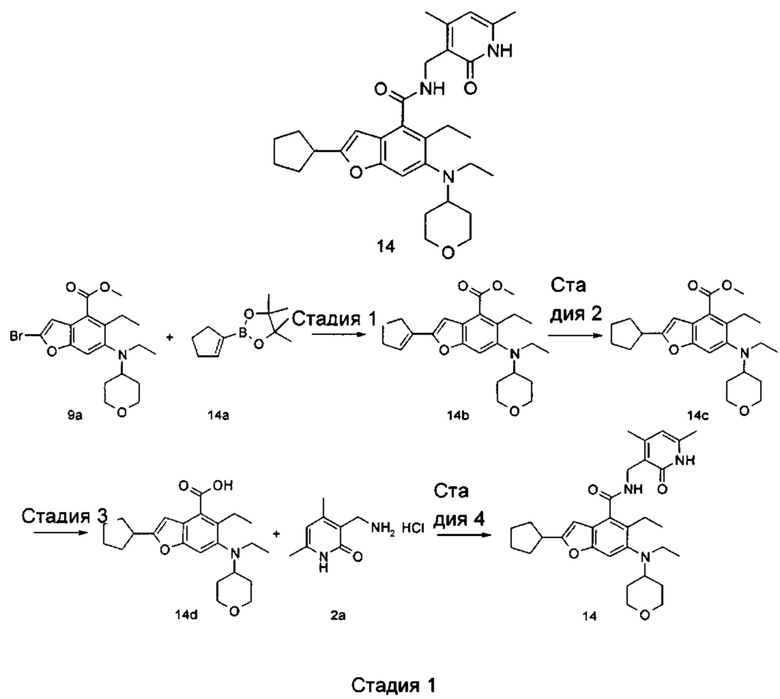
Метил-2-(циклопент-1-ен-1-ил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 14b
Соединение 9а (70 мг, 0,17 ммоль), 2-(циклопент-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан 14а (40 мг, 0,20 ммоль), трис(дибензилиденацетон)дипалладий (16 мг, 0,017 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилдифенил (16 мг, 0,034 ммоль) и карбонат натрия (52 мг, 0,51 ммоль) растворяли в 5 мл толуола. Смесь перемешивали в течение 12 часов при 100°С в атмосфере аргона. После завершения реакции смесь фильтровали через целит. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 14b (50 мг, выход 75%), в виде желтого масла.
Стадия 2
Метил-2-циклопентил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 14с
Соединение 14b (75 мг, 0,19 ммоль) растворяли в 5 мл метанола, добавляли 20 мг 10% Pd/C. Реакционную систему продували три раза водородом и перемешивали в течение 1 часа. После завершения реакции смесь фильтровали через целит. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 14с (55 мг, выход 73%), в виде бесцветного масла.
Стадия 3
2-Циклопентил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоновая кислота 14d
Соединение 14с (55 мг, 0,14 ммоль) растворяли в 5 мл смеси метанола и тетрагидрофурана (об.: об. составляет 3:2), впоследствии добавляли 2 мл 2 н. раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. После завершения реакции смесь концентрировали под пониженным давлением, чтобы удалить органические растворители, нейтрализовали 12 М раствором хлористоводородной кислоты для доведения рН до 3 и экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 14d (45 мг, выход 85%)), в виде бесцветного масла.
Стадия 4
2-Циклопентил-N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксамид 14
Соединение 14d (15 мг, 0,065 ммоль) растворяли в 4 мл N,N-диметилформамида, впоследствии в реакционный раствор добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (20 мг, 0,097 ммоль), 1-гидроксибензотриазол (15 мг, 0,097 ммоль) и N,N-диизопропилэтиламин (50 мг, 0,33 ммоль). Смесь перемешивали 1 час, впоследствии добавляли соединение 2а (18 мг, 0,097 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции добавляли 20 мл воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об.: об. составляет 8:1) (10 мл×3). Органические фазы объединяли, последовательно промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 14 (18 мг, выход 53%), в виде белого твердого вещества.
МС m/z (ИЭР): 520,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.49 (brs, 1Н), 8.09 (s, 1Н), 7.35 (s, 1Н), 6.30 (s, 1Н), 5.87 (s, 1Н), 4.31 (d, 2Н), 3.82 (brd, 2Н), 3.16-3.23 (m, 3Н), 3.03 (q, 2Н), 2.90-2.96 (m, 1Н), 2.79 (q, 2Н), 2.24 (s, 3Н), 2.11 (s, 3Н), 2.00 (brd, 2Н), 1.67 (brs, 8Н), 1.43-1.53 (m, 2Н), 1.01 (t, 3Н), 0.81 (t, 3Н).
Пример 15
N-((4,6-Диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(1-метилпиперидин-4-ил)бензофуран-4-карбоксамид
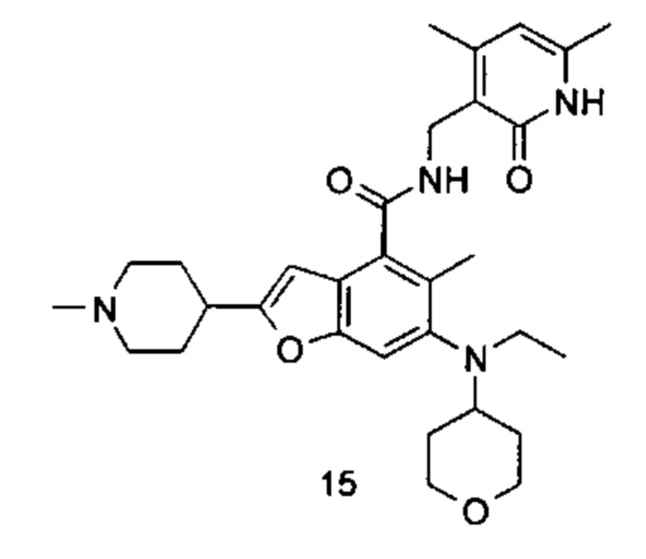
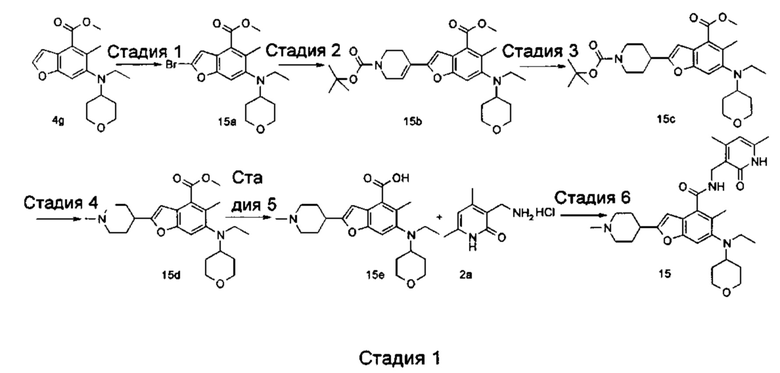
Метил-2-бром-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метилбензофуран-4-карбоксилат 15а
Соединение 4g (330 мг, 1,04 ммоль) растворяли в 6 мл тетрагидрофурана, впоследствии добавляли по каплям 0,63 мл 2М раствора диизопропиламида лития при -70°С в атмосфере аргона. Смесь перемешивали 1 час, впоследствии добавляли 20 мл предварительно приготовленного раствора 1,2-дибромтетрахлорэтана (170 мг, 0,53 ммоль) в тетрагидрофуране. Смесь медленно подогревали и перемешивали в течение 1 часа. После завершения реакции добавляли 5 мл насыщенного раствора хлорида аммония для гашения реакции, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 15а (145 мг, выход 35%), в виде желтого масла.
Стадия 2
трет-Бутил-4-(6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)-5-метилбензофуран-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат 15b
Соединение 15а (90 мг, 0,23 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат (105 мг, 0,34 ммоль), трис(дибензилиденацетон)дипалладий (21 мг, 0,023 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилдифенил (22 мг, 0,045 ммоль) и карбонат натрия (72 мг, 0,68 ммоль) растворяли в 5 мл смеси 1,4-диоксана и воды (об.: об. составляет 4:1). Смесь перемешивали в течение 12 часов при 90°С в атмосфере аргона. После завершения реакции смесь фильтровали через слой целита, и две фазы разделяли. Органическую фазу последовательно промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 15b (100 мг, выход 88%), в виде желтого твердого вещества.
Стадия 3
трет-Бутил-4-(6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-4-(метоксикарбонил)-5-метилбензофуран-2-ил)пиперидин-1-карбоксилат 15с
Соединение 15b (100 мг, 0,2 ммоль) растворяли в 6 мл этанола, добавляли 30 мг 10% Pd/C. Реакционную систему продували три раза водородом и перемешивали в течение 0,5 часа. Реакционный раствор фильтровали, фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 15с (80 мг, выход 80%), в виде желтого масла.
Стадия 4
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(1-метилпиперидин-4-ил)бензофуран-4-карбоксилат 15d
Соединение 15с (80 мг, 0,16 ммоль) растворяли в 10 мл дихлорметана, впоследствии добавляли 1 мл трифторуксусной кислоты. После перемешивания в течение 1 часа смесь концентрировали под пониженным давлением. Добавляли 5 мл метанола и 5 мл формальдегида, и смесь перемешивали в течение 1 часа, затем добавляли триацетоксиборгидрид натрия (100 мг, 0,48 ммоль). После перемешивания в течение 3 часов реакционный раствор концентрировали под пониженным давлением, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали смесью дихлорметана и метанола (об.: об. составляет 8:1). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 15d (60 мг, выход 90%) в виде желтого масла.
Стадия 5
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(1-метилпиперидин-4-ил)бензофуран-4-карбоновая кислота
Соединение 15d (30 мг, 0,072 ммоль) растворяли в смеси 5 мл метанола и 2 мл тетрагидрофурана, впоследствии добавляли 2 мл 4М раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. Добавляли 12 М концентрированную хлористоводородную кислоту для доведения рН реакционного раствора до 4, и смесь концентрировали под пониженным давлением. Остаток растворяли в 10 мл метанола и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 15е (30 мг), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 6
N-((4,6-Диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(1-метилпиперидин-4-ил)бензофуран-4-карбоксамид 15
Неочищенное соединение 15е (25 мг, 0,06 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (18 мг, 0,094 ммоль), 1-гидроксибензотриазол (15 мг, 0,094 ммоль) и N,N-диизопропилэтиламин (40 мг, 0,31 ммоль). Смесь перемешивали 1 час, впоследствии добавляли соединение 2а (18 мг, 0,094 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции добавляли 20 мл воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола. Органические фазы объединяли, последовательно промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 15 (24 мг, выход 73%), в виде белого твердого вещества.
МС m/z (ИЭР): 535,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.50 (brs, 1Н), 8.10 (s, 1Н), 7.34 (s, 1Н), 6.29 (s, 1Н), 5.87 (s, 1Н), 4.31 (d, 2Н), 3.81 (brd, 2Н), 3.22 (t, 2Н), 3.03 (q, 2Н), 2.91-2.94 (m, 1Н), 2.80 (brd, 2Н), 2.67 (brs, 1Н), 2.23 (s, 3Н), 2.22 (s, 3Н), 2.18 (s, 3Н), 2.11 (s, 3Н), 1.92-2.02 (m, 4Н), 1.61-1.64 (m, 4Н), 1.45-1.48 (m, 2Н), 0.78 (t, 3Н).
Пример 16
N-((4,6-Диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)бензофуран-4-карбоксамид 16
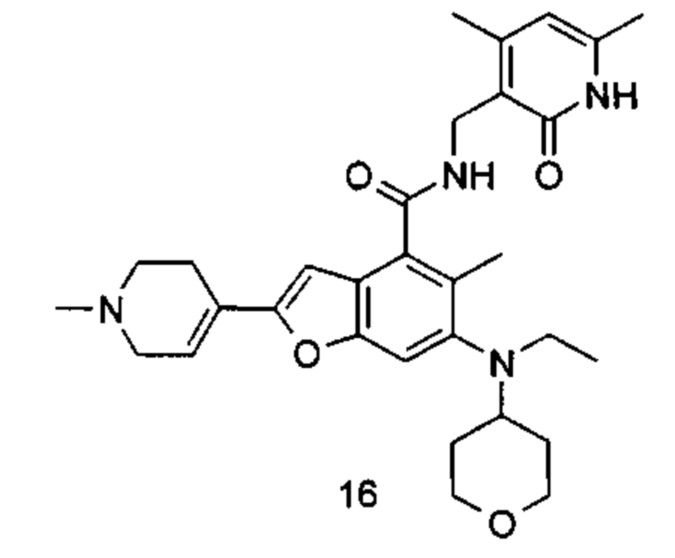
В соответствии с методом синтеза примера 12 исходное вещество 5а заменяли 15b, а 1о заменяли 2а, соответственно, получили соединение, указанное в заголовке, 16 (18 мг, выход 62%), в виде белого твердого вещества.
МС m/z (ИЭР): 533,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.51 (brs, 1Н), 8.17 (t, 1Н), 7.35 (s, 1Н), 6.58 (s, 1Н), 6.35 (s, 1Н), 5.87 (s, 1Н), 4.31 (d, 2Н), 3.81 (brd, 2Н), 3.19-3.25 (m, 3Н), 3.02-3.07 (m, 3Н), 2.90-2.97 (m, 1Н), 2.43 (brs, 2Н), 2.23 (s, 3Н), 2.22 (s, 3Н), 2.11 (s, 3Н), 1.62-1.64 (m, 2Н), 1.44-1.51 (m, 2Н), 1.17 (t, 2Н), 0.79 (t, 3Н).
Пример 17
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2-(1-метилпиперидин-4-ил)бензофуран-4-карбоксамид 17
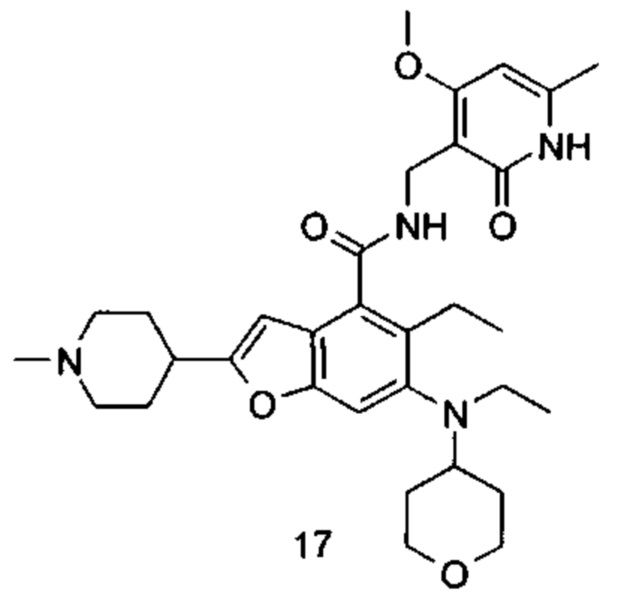
В соответствии с методом синтеза примера 12 исходное вещество 5а заменяли 9с, соответственно, получили соединение, указанное в заголовке, 17 (18 мг, выход 50%), в виде белого твердого вещества.
МС m/z (ИЭР): 565,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.43 (brs, 1Н), 7.94 (brs, 1Н), 7.35 (s, 1Н), 6.39 (brs, 1Н), 6.10 (s, 1Н), 4.27 (d, 2Н), 3.83 (brs, 2Н), 3.81 (s, 3Н), 3.20 (t, 2Н), 3.02 (q, 2Н), 2.90-2.95 (m, 2Н), 2.79 (q, 2Н), 2.44 (brs, 4Н), 2.18 (s, 3Н), 2.07 (brs, 2Н), 1.81 (brs, 2Н), 1.62-1.64 (m, 2Н), 1.43-1.52 (m, 2Н), 1.03 (t, 3Н), 0.80 (t, 3Н).
Пример 18
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид 18
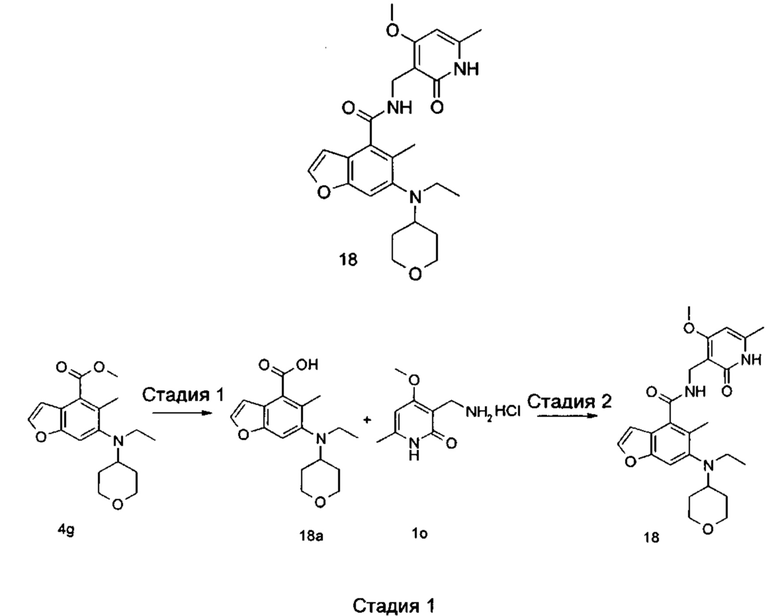
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метилбензофуран-4-карбоновая кислота 18а
Соединение 4g (45 мг, 0,142 ммоль) растворяли в 4 мл смеси метанола и воды (об.: об. составляет 1:1), впоследствии добавляли гидроксид натрия (60 мг, 1,58 ммоль). Смесь перемешивали в течение 12 часов при 70°С. После завершения реакции реакционный раствор концентрировали под пониженным давлением, чтобы удалить метанол. Добавляли по каплям 10% хлористоводородную кислоту для доведения рН до 3, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×1) и высушивали над безводным сульфатом натрия. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 18а (40 мг, выход 93%), в виде белого твердого вещества.
Стадия 2
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид 18
Соединение 18а (15 мг, 0,049 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии добавляли N,N-диизопропилэтиламин (18,9 мг, 0,147 ммоль) и 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (27,9 мг, 0,0735 ммоль). Реакционную систему перемешивали в течение 1 часа при 0°С. Добавляли соединение 1о (12 мг, 0,0558 ммоль) в атмосфере азота. Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции добавляли 30 мл воды, смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×1), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 18 (4,4 мг, выход 20%), в виде белого твердого вещества.
МС m/z (ИЭР): 454,2 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 10.90 (s, 1Н), 7.43 (s, 1Н), 7.27 (s, 1Н), 6.76 (s, 1Н), 6.61 (s, 1Н), 5.85 (s, 1Н), 4.52 (brs, 2Н), 3.90-3.93 (brd, 2Н), 3.82 (s, 3Н), 3.28 (t, 2Н), 3.02-3.07 (m, 2Н), 2.91-2.95 (m, 1Н), 2.29 (s, 3Н), 1.83 (s, 3Н), 1.56-1.68 (m, 4Н), 0.83 (t, 3Н).
Пример 19
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2,5-диметилбензофуран-4-карбоксамид 19
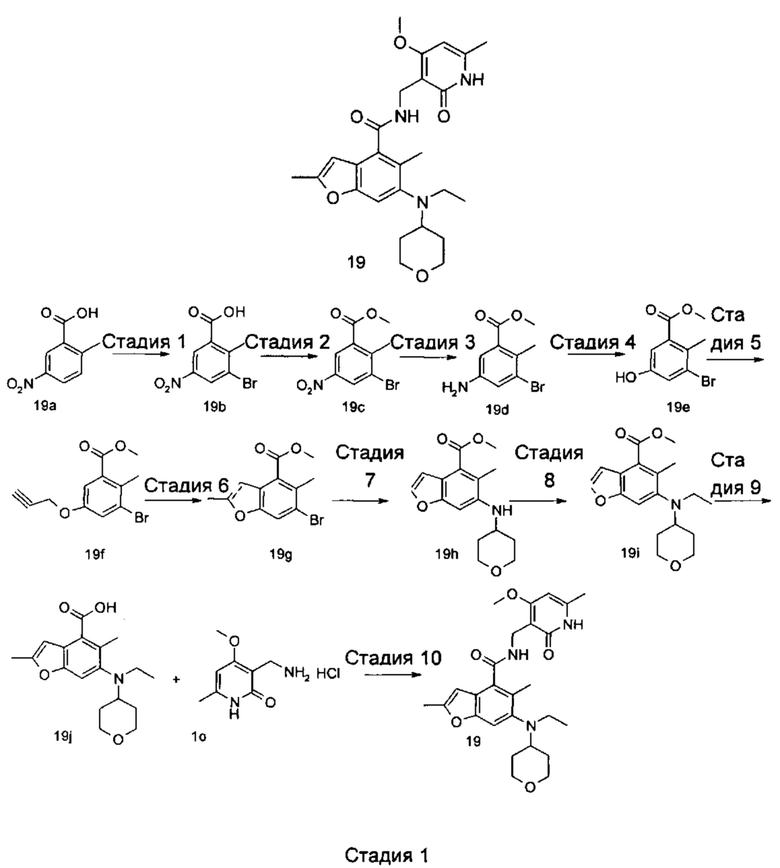
3-Бром-2-метил-5-нитробензойная кислота 19b
2-Метил-5-нитробензойную кислоту (30 г, 0,166 моль, Bide) растворяли в 70 мл концентрированной серной кислоты, впоследствии добавляли порциями N-бромсукцинимид (35,4 г, 0,199 моль). Реакционную систему перемешивали в течение 3 часов при 60°С. После завершения реакции получили реакционный раствор указанного в заголовке соединения 19b, который использовали непосредственно в следующей стадии без дополнительной обработки.
Стадия 2
Метил-3-бром-2-метил-5-нитробензоат 19с
К реакционному раствору полученного, как описано выше, соединения 19b добавляли 100 мл метанола и непрерывно перемешивали в течение 6 часов. После завершения реакции реакционный раствор наливали в 1 л воды и экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным раствором хлорида натрия (200 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 19с (53 мг), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
Метил-5-амино-3-бром-2-метилбензоат 19d
Неочищенный 19с (52 г, 0.189 моль) растворяли в 200 мл этанола, впоследствии добавляли порошок железа (31,7 г, 0,569 моль) и медленно добавляли по каплям 16 мл 4 н. раствора хлористоводородной кислоты. Смесь нагревали до 75°С и перемешивали в течение 4 часов. После завершения реакции добавляли 200 мл воды, и смесь экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (200 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 19d (27 г, выход 59%), в виде желтого твердого вещества.
Стадия 4
Метил-3-бром-5-гидрокси-2-метилбензоат 19е
Соединение 19d (5 г, 0,02 моль) растворяли в 50 мл 10% серной кислоты, впоследствии добавляли 10 мл предварительно приготовленного раствора нитрита натрия (1,55 г, 0,023 моль) при 0°С. смесь перемешивали 1 час, впоследствии добавляли 50 мл 50% серной кислоты. Смесь нагревали до 100°С и перемешивали в течение 1 часа. После завершения реакции реакционный раствор экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 19е (27 г, выход 29%), в виде желтого твердого вещества.
Стадия 5
Метил-3-бром-2-метил-5-(проп-2-ил-1-илоху)бензоат 19f
Соединение 19е (0,5 г, 2,04 ммоль) растворяли в 20 мл ацетона, впоследствии добавляли 3-бромпропин (0,73 г, 6,12 ммоль) и карбонат калия (0,85 г, 6,12 ммоль). Реакционную систему перемешивали при 80°С в течение 12 часов. После завершения реакции реакционный раствор концентрировали под пониженным давлением, впоследствии добавляли 20 мл воды, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 19f (0,5 г, выход 86,7%), в виде желтой жидкости.
Стадия 6
Метил-6-бром-2,5-диметилбензофуран-4-карбоксилат 19g
Соединение 19f (0,5 г, 1,76 ммоль) и фторид цезия (0,79 г, 5,28 ммоль) добавляли к 5 мл N,N-диэтиланилина. Смесь перемешивали в течение 2 часов при 200°С в микроволновом реакторе. После завершения реакции добавляли 20 мл воды, и смесь экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 19g (0,2 г, выход 40%), в виде желтой жидкости.
Стадия 7
Метил-2,5-диметил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 19h
Соединение 19h (0,2 г, 0,70 ммоль), тетрагидро-2Н-пиран-4-амин (14 мг, 0,14 ммоль), трис(дибензилиденацетон)дипалладий (64 мг, 0,07 ммоль), (±)-2,2-бис(дифенилфосфино)-1,1'-динафталин (87,15 мг, 0,14 ммоль) и карбонат цезия (455 мг, 1,4 ммоль) растворяли в 5 мл толуола. Смесь перемешивали в течение 12 часов при 110°С в атмосфере азота. После завершения реакции добавляли 10 мл воды, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 19h (140 мг, выход 67%), в виде желтого твердого вещества.
Стадия 8
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2,5-диметилбензофуран-4-карбоксилат 19i
Соединение 19h (90,0 г, 0,297 ммоль), ацетальдегид (39,2 мг, 0,89 ммоль) и уксусную кислоту (89,1 мг, 1,48 ммоль) растворяли в 8 мл метанола. Смесь перемешивали в течение 1 часа при 0°С и еще 4 часов при комнатной температуре в атмосфере азота, впоследствии добавляли цианоборгидрид натрия (55,9 мг, 0,891 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции реакционный раствор концентрировали под пониженным давлением, добавляли 20 мл воды, и смесь экстрагировали этил ацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 19i (60 мг, выход 61%), в виде бесцветной жидкости.
Стадия 9
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-2,5-диметилбензофуран-4-карбоновая кислота 19j
Соединение 19i (60 мг, 0,18 ммоль) и гидроксид калия (50,6 мг, 0,90 ммоль) добавляли к 4 мл смеси метанола и воды (об.: об. составляет 1:1). Смесь перемешивали в течение 16 часов при 75°С. После завершения реакции смесь охлаждали и добавляли 2 н. хлористоводородную кислоту для доведения рН реакционного раствора до 3-4. Смесь экстрагировали смесью дихлорметана и метанола (об.: об. составляет 10:1) (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 19j (30 мг), в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 10
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-2,5-диметилбензофуран-4-карбоксамид 19
Неочищенное соединение 19j (15 мг, 0,047 ммоль) растворяли в 2 мл N,N-диметилформамида, впоследствии добавляли N,N-диизопропилэтиламин (18,3 мг, 0,14 ммоль) и 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (47,7 мг, 0,125 ммоль). Смесь перемешивали 1 час при 0°С, впоследствии добавляли 1о (11,58 мг, 0,057 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции добавляли 50 мл воды, смесь экстрагировали смесью дихлорметана и метанола (об.: об. составляет 10:1) (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 19 (20 мг, выход 91%), в виде белого твердого вещества.
МС m/z (ИЭР): 468,2 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 11.57 (s, 1Н), 7.19 (s, 1Н), 6.38 (s, 1Н), 5.93 (s, 1Н), 5.36 (s, 1Н), 4.64 (d, 2Н), 3.95 (brs, 2Н), 3.91 (s, 3Н), 3.28-3.33 (m, 2Н), 3.03-3.05 (m, 2Н), 2.92-2.96 (m, 1Н), 2.37 (s, 3Н), 2.34 (s, 3Н), 2.19 (s, 3Н), 1.69 (brs, 2Н), 1.25 (brs, 2Н), 0.85 (t, 3Н).
Пример 20
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-3,5-диметилбензофуран-4-карбоксамид 20
В соответствии с методом синтеза примера 19 исходное вещество 3-бромпиридин, используемое на стадии 5, заменяли 1-бромпропан-2-оном, соответственно, получили соединение, указанное в заголовке, 20 (17 мг, выход 11%), в виде белого твердого вещества.
МС m/z (ИЭР): 468,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 11.22 (s, 1Н), 7.27 (s, 1Н), 7.21 (s, 1Н), 6.94 (s, 1Н), 5.94 (s, 1Н), 4.61-4.62 (brd, 2Н), 3.92-3.95 (brd, 2Н), 3.89 (s, 3Н), 3.29 (dt, 2Н), 3.16-3.20 (m, 3Н), 2.29 (s, 3Н), 2.22 (s, 3Н), 2.11 (s, 3Н), 1.65 (brs, 2Н), 1.31 (brs, 2Н), 0.86 (t, 3Н).
Пример 21
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-(трифторметил)бензофуран-4-карбоксамид
Метил-3-бром-5-(2,2-диэтоксиэтокси)бензоат 21b
Метил-3-бром-5-гидроксибензоат 21а (1,9 г, 8,23 ммоль, получен способом, раскрытым в Journal of Medicinal Chemistry, 2013, 56(8), 3217-3227) растворяли в 60 мл N,N-диметилформамида, впоследствии добавляли 2-бром-1,1-диэтоксиэтан (3,23 г, 16,4 ммоль) и карбонат цезия (5,35 г, 16,4 ммоль). Реакционную систему перемешивали в течение 4 часов при 95°С. После завершения реакции реакционный раствор наливали в ледяную воду, экстрагировали этилацетатом (50 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 21b (25 мг), в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
Метил-6-бромбензофуран-4-карбоксилат 21с
Полифосфорную кислоту (5,46 г, 16,4 ммоль) добавляли к 20 мл толуола. Смесь нагревали до 115°С, добавляли 40 мл предварительно приготовленного раствора соединения 21b (3,5 г, 10 ммоль) в толуоле при перемешивании. Смесь перемешивали в течение 5 часов. После завершения реакции смесь наливали в ледяную воду, нейтрализовали насыщенным раствором бикарбоната натрия, экстрагировали этилацетатом (20 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 21с (1,08 г, выход 52%), в виде желтого твердого вещества.
Стадия 3
Метил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 21d
Соединение 21с (1,08 г, 4,25 ммоль), тетрагидро-2Н-пиран-4-амин (0,644 г, 6,38 ммоль), трис(дибензилиденацетон)дипалладий (393 мг, 0,43 ммоль), (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин (267 мг, 0,43 ммоль) и карбонат цезия (2,6 г, 8,5 ммоль) растворяли в 60 мл толуола. Смесь перемешивали в течение 36 часов в атмосфере азота. После завершения реакции реакционный раствор наливали в ледяную воду, экстрагировали этилацетатом (30 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 21d (200 мг, выход 17%), в виде желтого твердого вещества.
Стадия 4
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 21е
Соединение 21d (200 мг, 0,727 ммоль), ацетальдегид (160 мг, 3,64 ммоль) и 0,2 мл уксусной кислоты растворяли в 4 мл метанола. После перемешивания в течение 24 часов смесь охлаждали до температуры ниже 0°С. Добавляли цианоборгидрид натрия (227 мг, 3,6 ммоль), и смесь естественным путем подогревали до комнатной температуры. За ходом реакции следили по ТСХ до тех пор, пока не исчезала основная часть исходного вещества. После завершения реакции реакционный раствор охлаждали до 0°С и добавляли 20 мл насыщенного раствора бикарбоната натрия для гашения реакции. Смесь экстрагировали этилацетатом (10 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 21е (140 мг, выход 64%), в виде желто-зеленого твердого вещества.
Стадия 5
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-йодобензофуран-4-карбоксилат 21f
Соединение 21е (13 мг, 0,043 ммоль) растворяли в 2 мл N,N-диметилформамида, впоследствии добавляли 5 капель трифторуксусной кислоты, и при 0°С добавляли N-йодсукцинимид (12 мг, 0,052 мл). Смесь подогревали до комнатной температуры естественным путем. За ходом реакции следили по ТСХ до тех пор, пока не исчезала основная часть исходного вещества. После завершения реакции реакционный раствор охлаждали до 0°С, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 21f (16 мг, выход 86%), в виде желтого масла.
Стадия 6
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-(трифторметил)бензофуран-4-карбоксилат 21g
Соединение 21f (16 мг, 0,0373 ммоль), йодид меди (36 мг, 0,187 ммоль) и метилфторсульфонилдифторацетат (140 мг, 0,75 ммоль) растворяли в 2 мл N,N-диметилформамида. Смесь перемешивали в течение 12 часов при 80°С в атмосфере азота. После завершения реакции смесь концентрировали под пониженным давлением. Полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 21g (10 мг, выход 72%) в виде желто-зеленого твердого вещества.
Стадия 7
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-5-(трифторметил)бензофуран-4-карбоновая кислота 21h
Соединение 21g (10 мг, 0,027 ммоль) растворяли в 4 мл смеси метанола и тетрагидрофурана (об.: об. составляет 1:1), впоследствии добавляли 0,27 мл 2 н. раствора гидроксида натрия. Смесь перемешивали в течение 4 часов при 60°С. После завершения реакции смесь концентрировали под пониженным давлением, чтобы удалить основную часть растворителей. К остатку добавляли 2 мл тетрагидрофурана при 0°С и добавляли 1 н. хлористоводородную кислоту для доведения рН до 3-4. Смесь концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 21h (9 мг), в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 8
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-(трифторметил)бензофуран-4-карбоксамид 21
Неочищенное соединение 21h (9 мг, 0,025 ммоль) растворяли в 3 мл N,N-диметилформамида, затем добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (14 мг, 0,041 ммоль) и N,N-диизопропилэтиламин (17 мг, 0,135 ммоль). Смесь перемешивали 20 минут, впоследствии к реакционному раствору добавляли соединение 1о (8,3 мг, 0,041 ммоль). Смесь перемешивали в течение 5 часов. После завершения реакции добавляли 20 мл насыщенного раствора хлорида натрия, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 21 (10 мг, выход 78%), в виде белого твердого вещества.
МС (ИЭР) m/z: 508,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.98 (brs, 1Н), 7.53 (m, 1Н), 7.32 (m, 1Н), 6.92 (s, 1Н), 5.96 (s, 1Н), 4.65 (d, 2Н), 4.09-4.05 (m, 2Н), 3.92 (s, 3Н), 3.89-3.85 (m, 1Н), 3.55-3.50 (m, 2Н), 3.37 (q, 2Н), 2.27 (s, 3Н), 1.85-1.79 (m, 4Н), 1.20 (t, 3Н).
Пример 22
5-Хлор-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид
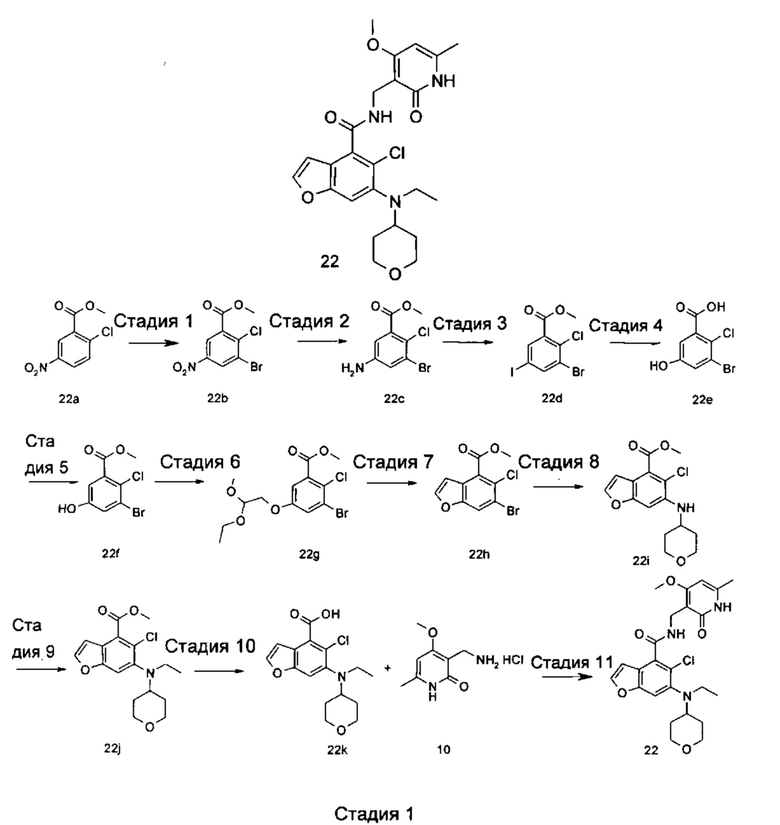
Метил-3-бром-2-хлор-5-нитробензоат 22b
Метил-2-хлор-5-нитробензоат (6,0 г, 27,8 ммоль) растворяли в 30 мл концентрированной серной кислоты, впоследствии к описанному выше реакционному раствору добавляли N-бромсукцинимид (5,2 г, 29,2 ммоль). Реакционную систему перемешивали в течение 1 часа при 60°С. После завершения реакции реакционную систему наливали в 300 мл ледяной воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором бикарбоната натрия (100 мл) и насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 22b (7,58 г), в виде желто-зеленого твердого вещества, которое использовали непосредственно в следующей реакции без очистки.
Стадия 2
Метил-5-амино-3-бром-2-хлорбензоат 22с
Неочищенное соединение 22b (7,5 г, 25 ммоль) растворяли в 55 мл смеси этанола и воды (об.: об. составляет 8:3) и добавляли хлорид аммония (11,2 г, 203 ммоль). Реакционную систему нагревали до 70°С, добавляли порошок железа (7,1 г, 127 ммоль) и перемешивали в течение 2 часов. После завершения реакции смесь фильтровали через слой целита. Фильтрат концентрировали под пониженным давлением, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 22с (5,0 г), в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
Метил-3-бром-2-хлор-5-йодбензоат 22d
Неочищенное соединение 22с (7,0 г, 26,5 ммоль) растворяли в смеси 50 мл 40% серной кислоты и 10 мл N,N-диметилформамида. Смесь охлаждали до 0°С и добавляли по каплям 5 мл предварительно приготовленного раствора нитрита натрия (2,0 г, 29,1 ммоль). Смесь перемешивали 1 час, впоследствии в описанный выше реакционный раствор добавляли 50 мл предварительно приготовленного раствора йодида калия (22 г, 132 ммоль). Смесь нагревали до 70°С и перемешивали в течение 30 минут. После завершения реакции смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали водой (100 мл) и насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 22d (8,0 г, выход 80%), в виде светло-желтого твердого вещества.
Стадия 4
3-Бром-2-хлор-5-гидроксибензойная кислота 22е
Соединение 22d (7,0 г, 18,7 ммоль) растворяли в 10 мл тетрагидрофурана, впоследствии добавляли 70 мл 4 н. раствора гидроксида натрия. Смесь перемешивали в течение 2 часов при 60°С, впоследствии концентрировали под пониженным давлением, чтобы удалить органические растворители, и добавляли оксид меди (7,7 г, 53,5 ммоль). Смесь перемешивали в течение 1 часа при 120°С в микроволновом реакторе. После завершения реакции смесь фильтровали. Фильтрат нейтрализовали 12 М раствором хлористоводородной кислоты и экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 22е (6,0 г), в виде коричневого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 5
Метил-3-бром-2-хлор-5-гидроксибензоат 22f
Неочищенное соединение 22е (6,0 г, 24 ммоль) растворяли в 30 мл метанола, впоследствии к реакционному раствору добавляли по каплям 2 мл концентрированной серной кислоты. Реакционную систему перемешивали в течение 12 часов при 60°С. После завершения реакции смесь концентрировали под пониженным давлением и растворяли в 50 мл этилацетата. Добавляли 100 мл воды, и смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 22f (3,3 г, выход 52%), в виде белого твердого вещества.
Стадия 6
Метил-3-бром-2-хлор-5-(2,2-диэтоксиэтокси)бензоат 22g
Соединение 22f (3,3 г, 12,5 ммоль) растворяли в 30 мл N,N-диметилформамида, впоследствии добавляли 2-бром-1,1-диэтоксиэтан (2,96 г, 15 ммоль) и карбонат калия (3,4 г, 25 ммоль). Смесь перемешивали в течение 3 часов при 120°С. После завершения реакции добавляли 150 мл воды, и смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 22 g (3,5 г, выход 74%), в виде бесцветного масла.
Стадия 7
Метил-6-бром-5-хлорбензофуран-4-карбоксилат 22h
Полифосфорную кислоту (1,0 г) добавляли к 15 мл толуола. Смесь нагревали до 100°С и добавляли 10 мл предварительно приготовленного раствора соединения 22g (780 мг, 2,1 ммоль) в толуоле при перемешивании. Смесь перемешивали в течение 4 часов. После завершения реакции супернатант отделяли, и к остатку добавляли воду и экстрагировали этилацетатом (20 мл×3). Этилацетатную фазу и супернатант объединяли, последовательно промывали насыщенным раствором бикарбоната натрия (30 мл) и насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 22h (150 г, выход 25%), в виде светло-желтого твердого вещества.
Стадия 8
Метил-5-хлор-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 22i
Соединение 22h (120 мг, 0,41 ммоль), тетрагидро-2Н-пиран-4-амин (60 мг, 0,6 ммоль), трис(дибензилиденацетон)дипалладий (38 г, 0,04 ммоль), (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин (26 мг, 0,04 ммоль) и карбонат цезия (405 мг, 1,24 ммоль) растворяли в 2 мл толуола. Реакционную систему перемешивали в течение 16 часов при 80°С в атмосфере аргона. После завершения реакции смесь фильтровали через слой целита. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 22i (125 мг, выход 87%), в виде желтого твердого вещества.
Стадия 9
Метил-5-хлор-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 22j
Соединение 22i (50 мг, 0,16 ммоль), ацетальдегид (36 мг, 0,81 ммоль) и уксусную кислоту (49 мг, 0,81 ммоль) добавляли к 5 мл 1,2-дихлорэтана. Смесь перемешивали 2 часа, впоследствии добавляли триацетоксиборгидрид натрия (102 мг, 0,48 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции реакционный раствор концентрировали под пониженным давлением, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, последовательно промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 22j (26 мг, выход 50%), в виде желтого твердого вещества.
Стадия 10
5-Хлор-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоновая кислота 22k
Соединение 22j (51 мг, 0,16 ммоль) растворяли в 3 мл смеси тетрагидрофурана и метанола (об. : об. составляет 1:2), впоследствии добавляли 3 мл 2 н. раствора гидроксида натрия. Смесь перемешивали в течение 1 часа при 60°С. После завершения реакции смесь концентрировали под пониженным давлением, чтобы удалить органические растворители, нейтрализовали 12 М раствором хлористоводородной кислоты для доведения рН реакционного раствора до 5, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 22k (42 мг), в виде красно-коричневого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 11
5-Хлор-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид 22
Неочищенное соединение 22k (8 мг, 0,025 ммоль) растворяли в 1 мл N.N-диметилформамида, впоследствии к описанному выше раствору добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (14 мг, 0,037 ммоль), N,N-диизопропилэтиламин (16 мг, 0,12 ммоль) и соединение 1о (7 мг, 0,032 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции добавляли 10 мл воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1) (10 мл×3). Органические фазы объединяли, промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 22 (8 мг, выход 70%), в виде белого твердого вещества.
МС (ИЭР) m/z: 474,3 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.46 (brs, 1Н), 8.17 (s, 1Н), 7.97 (s, 1Н), 7.56 (s, 1Н), 6.78 (s, 1Н), 6.10 (s, 1Н), 4.27 (d, 2Н), 3.85 (brs, 2Н), 3.82 (s, 3H), 3.23 (t, 2Н), 3.11-3.15 (m, 3H), 2.18 (s, 3H), 1.66-1.69 (m, 2Н), 1.50-1.57 (m, 2Н), 0.83 (t, 3H).
Пример 23
М-((4-Метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метил-6-(метил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксамид
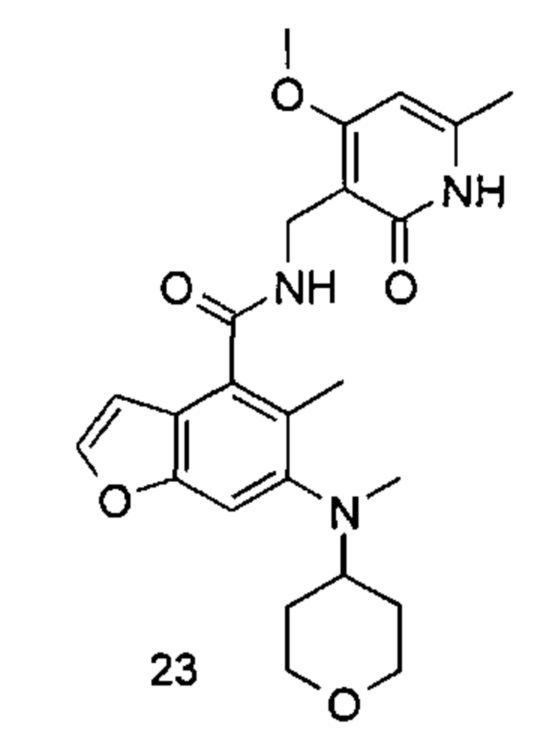
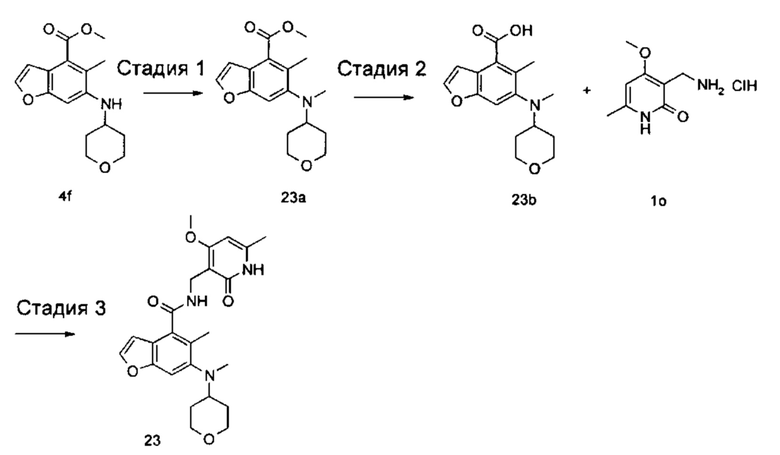
Стадия 1
Метил-5-метил-6-(метил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 23а
Соединение 4f (60 мг, 0,208 ммоль) растворяли в 5 мл метанола, впоследствии добавляли формальдегид (18,7 мг, 0,623 ммоль) и уксусную кислоту (62,4 мг, 1,04 ммоль). Смесь перемешивали в течение 1 часа при 0°С, впоследствии подогревали до комнатной температуры. Смесь перемешивали 12 часов, впоследствии к описанному выше раствору добавляли цианоборгидрид натрия (39,2 мг, 0,624 ммоль). Реакционную смесь перемешивали в течение 6 часов. После завершения реакции реакционный раствор концентрировали под пониженным давлением, впоследствии добавляли 20 мл воды и экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 23а (50 мг, выход 75%), в виде бесцветной жидкости.
Стадия 2
5-Метил-6-(метил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоновая кислота 23b
Соединение 23а (50 мг, 0,165 ммоль) растворяли в 2 мл смеси метанола и воды (об. : об. составляет 1:1), впоследствии добавляли гидроксид натрия (33 мг, 0,825 ммоль). Реакционную систему перемешивали в течение 16 часов при 75°С.
После завершения реакции смесь охлаждали до комнатной температуры. Добавляли 2 н. хлористоводородную кислоту для доведения рН реакционного раствора до 3-4, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), высушивали над безводным сульфатом натрия и фильтровали. Полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 23b (20 мг, выход 48%) в виде бесцветной жидкости.
Стадия 3
N-((4-Метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метил-6-метил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксамид 23
Соединение 23b (15 мг, 0,052 ммоль) растворяли в 2 мл N,N-диметилформамида, затем добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (47,7 мг, 0,125 ммоль) и N,N-диизопропилэтиламин (18,3 мг, 0,14 ммоль). Смесь перемешивали 1 час при 0°С, впоследствии добавляли 1о (11,58 мг, 0,057 ммоль). Смесь перемешивали в течение 12 часов. После завершения реакции добавляли 50 мл воды, и реакционный раствор экстрагировали смесью дихлорметана и метанола (об. : об. составляет 10:1) (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 23 (21 мг, выход 91%), в виде белого твердого вещества.
МС (ИЭР) m/z: 440,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 12.30 (brs, 1Н), 7.48 (d, 1Н), 7.18 (brs, 1Н), 6.74 (s, 1Н), 5.94 (s, 1Н), 4.64 (d, 2Н), 3.95-3.98 (m, 2Н), 3.91 (s, 3H), 3.30-3.36 (m, 2Н), 2.96-3.01 (m, 1Н), 2.65 (s, 3H), 2.39 (s, 3H), 2.19 (s, 3H), 1.69-1.73 (m, 2Н), 1.45 (brs, 2Н).
Пример 24
6-(((1r,4r)-4-(Диметиламино)циклогексил)(этил)амино)-N-((4-метокси-6-метил-2-оксо-1,2- дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид
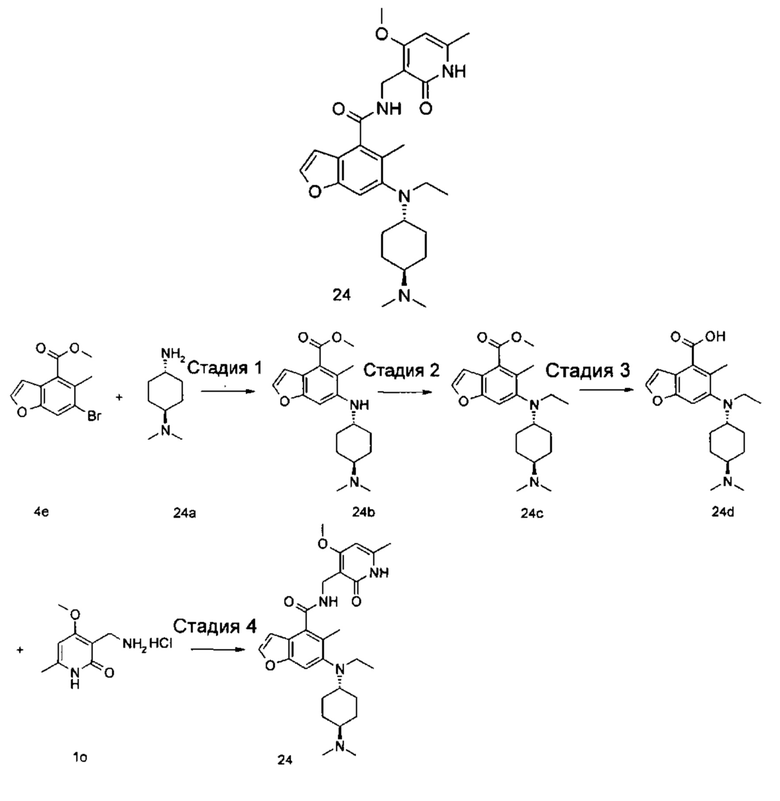
Стадия 1
Метил-6-(((1r,4r)-4-(диметиламино)циклогексил)амино)-5-метилбензофуран-4-карбоксилат 24b
Соединение 4е (200 мг, 0,75 ммоль), (1r, 4r)-N1,N1-диметилциклогексан-1,4-диамин 24а (260 мг, 1,49 ммоль, получен способом, раскрытым в Bioorganic & Medicinal Chemistry Letters, 2011, 21(15), 4622-4628), трис(дибензилиденацетон)дипалладий (137 мг, 0,15 ммоль), (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин (140 мг, 0,22 ммоль) и карбонат цезия (731,25 мг, 2,25 ммоль) растворяли в 20 мл толуола. Реакционную систему перемешивали при кипячении с обратным холодильником в течение 12 часов в атмосфере азота. После завершения реакции в реакционную систему добавляли 100 мл воды, и смесь экстрагировали смесью дихлорметана и метанола (об. : об. составляет 10:1) (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 24b (15 мг, выход 6,1%), в виде желтого твердого вещества.
Стадия 2
Метил-6-(((1r,4r)-4-(диметиламино)циклогексил)(этил)амино)-5-метилбензофуран-4-карбоксилат 24с
Соединение 24b (15 мг, 0,045 ммоль), ацетальдегид (6,3 мг, 0,136 ммоль) и уксусную кислоту (13,3 мг, 0,22 ммоль) добавляли к 3 мл этанола. Смесь перемешивали в течение 1 часа при 0°С, впоследствии медленно подогревали до комнатной температуры и перемешивали в течение 12 часов. Добавляли цианоборгидрид натрия (8,5 мг, 0,135 ммоль), и смесь перемешивали в течение 10 часов. После завершения реакции к реакционной системе добавляли 20 мл воды и экстрагировали смесью дихлорметана и метанола (об. : об. составляет 10:1) (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 24с (20 мг), в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
6-(((1r,4r)-4-(Диметиламино)циклогексил)(этил)амино)-5-метилбензофуран-4-карбоновая кислота 24d
Неочищенное соединение 24с (20 мг, 0,056 ммоль) растворяли в 4 мл смеси метанола и воды (об. : об. составляет 1:1), впоследствии добавляли гидроксид калия (18,7 мг, 0,335 ммоль). Смесь перемешивали в течение 12 часов при 80°С. После завершения реакции добавляли 2 н. хлористоводородную кислоту для доведения рН реакционного раствора до 3-4. После этого смесь концентрировали под пониженным давлением, неочищенный продукт растворяли в 8 мл смеси метиленхлорида и тетрагидрофурана (об. :об. составляет 1:1) и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 24d (25 мг), в виде коричневого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 4
6-(((1r,4r)-4-(Диметиламино)циклогексил)(этил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид 24
Неочищенное соединение 24d (25 мг, 0,073 ммоль) растворяли в 2 мл N,N-диметилформамида, впоследствии добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (41,8 мг, 0,110 ммоль) и N,N-диизопропилэтиламин (28 мг, 0,219 ммоль). Реакционную смесь перемешивали в течение 1 часа при 0°С. Добавляли соединение 1о (11,58 мг, 0,057 ммоль), и смесь перемешивали в течение 12 часов. После завершения реакции добавляли 2 мл воды для гашения реакции, и смесь фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 24 (4,9 мг, выход 14%), в виде белого твердого вещества.
МС (ИЭР) m/z: 495,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 11.62 (brs, 1Н), 7.49 (s, 1Н), 7.20 (brs, 1Н), 6.77 (s, 1Н), 5.93 (s, 1Н), 5.35 (brs, 1Н), 4.66 (d, 2Н), 3.91 (s, 3H), 3.07 (q, 2Н), 2.65-2.71 (m, 1Н), 2.39 (s, 3H), 2.21 (s, 6Н), 2.15 (s, 3H), 2.02 (brs, 1Н), 1.85-1.93 (m, 4Н), 1.32-1.43 (m, 4Н), 0.86 (t, 3H).
Пример 25
6-(Этил(1-(метилсульфонил)пиперидин-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид
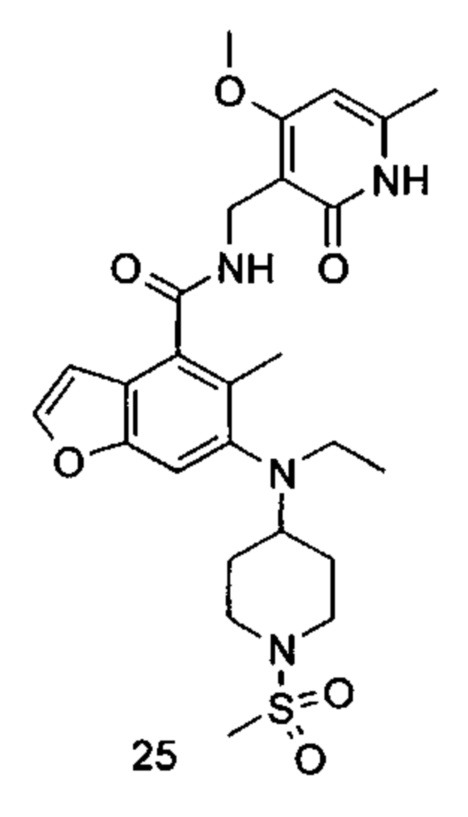
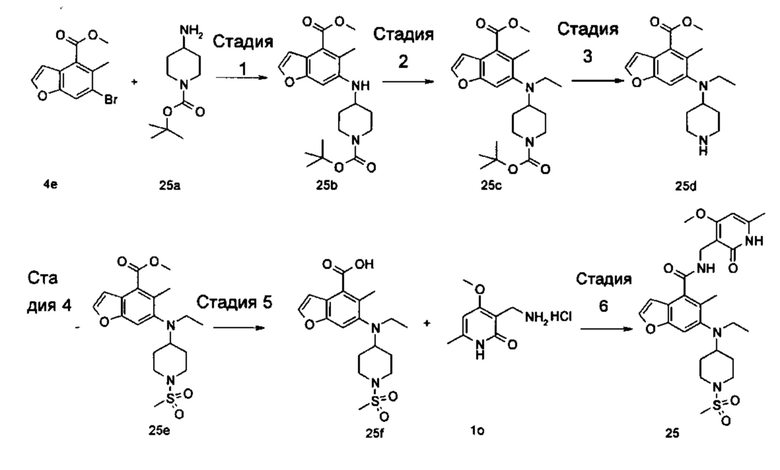
Стадия 1
трет-Бутил-4-((4-(метоксикарбонил)-5-метилбензофуран-6-ил)амино)пиперидин-1-карбоксилат 25b
Соединение 4е (810 мг, 3,02 ммоль), трет-бутил-4-аминопиперидин-1-карбоксилат 25а (1200 мг, 6,04 ммоль, получен способом, раскрытым в Bioorganic & Medicinal Chemistry Letters, 2011, 21(3), 983-988), трис(дибензилиденацетон)дипалладий (553 мг, 0,604 ммоль), (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин (564 мг, 6,04 ммоль) и карбонат цезия (1,96 г, 6,04 ммоль) растворяли в 10 мл толуола. Смесь перемешивали в течение 72 часов при 110°С. После завершения реакции смесь фильтровали через целит. Добавляли 60 мл этилацетата, и смесь промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 25b (1 г, выход 83%), в виде желтой жидкости.
Стадия 2
трет-Бутил-4-(этил(4-(метоксикарбонил)-5-метилбензофуран-6-ил)амино)пиперидине-1-карбоксилат 25с
Соединение 25b (1 г, 2,58 ммоль) растворяли в 10 мл метанола, впоследствии добавляли ацетальдегид (360 мг, 7,73 ммоль) и уксусную кислоту (774 мг, 12,9 ммоль). Смесь перемешивали при 0°С в течение 1 часа, впоследствии подогревали до комнатной температуры и перемешивали в течение 12 часов. Добавляли цианоборгидрид натрия (486 мг, 7,74 ммоль), и смесь перемешивали в течение 10 часов. После завершения реакции к реакционному раствору добавляли 50 мл воды, и смесь экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 25с (573,8 мг, выход 62,8%), в виде желтой жидкости.
Стадия 3
Метил-6-(этил(пиперидин-4-ил)амино)-5-метилбензофуран-4-карбоксилат 25d
Соединение 25с (184 мг, 0,58 ммоль) добавляли к 12 мл дихлорметана, впоследствии добавляли 3 мл трифторуксусной кислоты. Смесь перемешивали в течение 2 часов. После завершения реакции смесь нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 25d (156 мг), в виде коричневой жидкости, которую использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 4
Метил-6-(этил(1-(метилсульфонил)пиперидин-4-ил)амино)-5-метилбензофуран-4-карбоксилат 25е
Неочищенное соединение 25d (52 мг, 0,16 ммоль) добавляли к 10 мл дихлорметана, добавляли триэтиламин (64,6 мг, 0,64 ммоль), впоследствии добавляли метансульфонилхлорид (37,8 мг, 0,33 ммоль) при 0°С. Смесь медленно подогревали до комнатной температуры и перемешивали в течение 12 часов. После завершения реакции к реакционному раствору добавляли 50 мл воды, и смесь экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 25е (20 мг, выход 33%), в виде бесцветной жидкости.
Стадия 5
6-(Этил(1-(метилсульфонил)пиперидин-4-ил)амино)-5-метилбензофуран-4-карбоновая кислота 25f
Соединение 25е (20 мг, 0,05 ммоль) растворяли в смеси 1 мл метанола и 1 мл воды, впоследствии добавляли гидроксид калия (14,2 мг, 0,25 ммоль). Смесь перемешивали в течение 12 часов при 70°С. После завершения реакции добавляли 2 н. хлористоводородную кислоту для доведения рН реакционного раствора до 3-4, и смесь экстрагировали смесью дихлорметана и тетрагидрофурана (об. : об. составляет 1:1) (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 25f (16 мг), в виде бесцветной жидкости, которую использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 6
6-(Этил(1-(метилсульфонил)пиперидин-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид 25
Неочищенное соединение 25f (8 мг, 0,21 ммоль) растворяли в 2 мл N,N-диметилформамида, впоследствии к описанному выше раствору добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (12 мг, 0,0315 ммоль), N,N-диизопропилэтиламин (8 мг, 0,063 ммоль). Смесь перемешивали в течение 1 часа при 0°С. Добавляли 1о (5,2 мг, 0,025 ммоль), и реакционную смесь перемешивали в течение 48 часов при комнатной температуре. После завершения реакции добавляли 20 мл воды, и смесь экстрагировали смесью дихлорметана и метанола (об. : об. составляет 10:1) (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 25 (12,5 мг, выход 11%), в виде белого твердого вещества.
МС (ИЭР) m/z: 531,2 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 12.5 (brs, 1Н), 7.51 (d, 1Н), 7.29 (s, 1Н), 7.22 (brs, 1Н), 6.77 (s, 1H), 5.96 (s, 1H), 4.65 (d, 2H), 3.92 (s, 3H), 3.72-3.69 (m, 2H), 3.17-3.11 (m, 1H), 3.07 (q, 2H), 2.96-2.89 (m, 2H), 2.74 (s, 3H), 2.67 (t, 2H), 2.39 (s, 3H), 2.19 (s, 3H), 1.90-1.71 (m, 4H), 0.88 (t, 3H).
Пример 26
6-((1-Ацетилпиперидин-4-ил)(этил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид 26
В соответствии с методом синтеза примера 25 исходное вещество метансульфонилхлорид, используемое на стадии 4, заменяли уксусным ангидридом, соответственно, получили соединение, указанное в заголовке, 26 (10,1 мг, выход 50%), в виде белого твердого вещества.
МС (ИЭР) m/z: 495,3 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.76 (brs, 1Н), 7.45 (brs, 1Н), 6.80 (brs, 1Н), 6.30 (s, 1Н), 4.53 (s, 2Н), 3.96 (s, 3H), 3.91 (brs, 2Н), 3.36-3.30 (m, 2Н), 3.23 (q, 2Н), 3.11-3.05 (m, 3H), 2.41 (brs, 3H), 2.33 (s, 3H), 2.07 (s, 3H), 1.85 (brs, 2Н), 1.60 (brs, 2Н), 0.90 (t, 3H).
Пример 27
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-фтор-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид
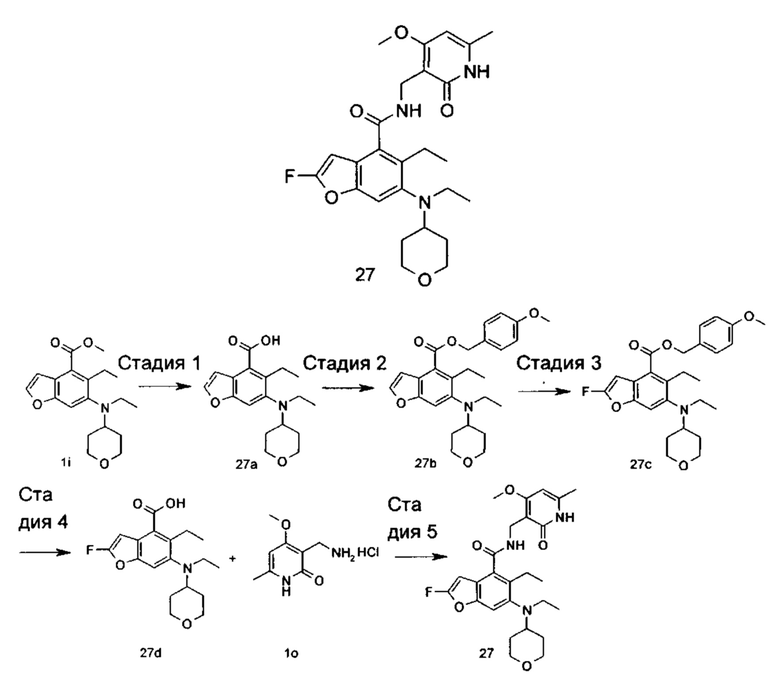
Стадия 1
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоновая кислота 27а
Соединение 1i (260 мг, 0,79 ммоль) растворяли в 15 мл смеси метанола и тетрагидрофурана (об. : об. составляет 2:1), впоследствии добавляли 5 мл 4 н. раствора гидроксида натрия. Смесь перемешивали в течение 12 часов при 60°С. После завершения реакции добавляли 12 М хлористоводородную кислоту для доведения рН реакционного раствора до 4, и смесь экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 27а (220 мг), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
4-Метоксибензил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 27b
Неочищенное соединение 27а (170 мг, 0,54 ммоль) растворяли в 15 мл ацетона, впоследствии к описанному выше раствору добавляли 1-(хлорметил)-4-метоксибензол (168 мг, 1,07 моль, Accela) и карбонат калия (148 мг, 1,07 ммоль). Смесь перемешивали в течение 48 часов. После завершения реакции смесь концентрировали при пониженном давлении, последовательно добавляли 30 мл воды и 20 мл этилацетата. Две фазы разделяли, и водную фазу экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 27b (230 мг, выход 99%), в виде бесцветного масла.
Стадия 3
4-Метоксибензил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-фторбензофуран-4-карбоксилат 27с
Соединение 27b (230 мг, 0,53 ммоль) добавляли к 8 мл тетрагидрофурана. Смесь охлаждали до -70°С, добавляли 0,79 мл 2,0 М диизопропиламида лития, и смесь перемешивали в течение 1 часа при -70°С. Добавляли N-фтордибензилиденсульфонамид (182 мг, 0,58 ммоль, Bepharm), и смесь перемешивали в течение 1 часа при -70°С. После завершения реакции смесь медленно подогревали до комнатной температуры, впоследствии добавляли 20 мл насыщенного раствора хлорида аммония для гашения реакции. Смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 27с (17 мг, выход 7%), в виде желтого масла.
Стадия 4
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-фторбензофуран-4-карбоновая кислота 27d
Соединение 27с (17 мг, 0,037 ммоль) растворяли в 3 мл дихлорметана, впоследствии добавляли 0,5 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. После завершения реакции смесь концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 27d (20 мг), в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 5
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-фтор-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид 27
Неочищенное соединение 27d (20 мг, 0,06 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии к описанному выше раствору добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (32,6 мг, 0,85 ммоль) и N,N-диизопропилэтиламин (39 мг, 0,30 ммоль). Смесь перемешивали в течение 1 часа. Впоследствии добавляли 1о (18 мг, 0,00 ммоль), и смесь перемешивали в течение 12 часов. После завершения реакции добавляли 20 мл воды, и смесь экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 27 (12 мг, выход 41%), в виде белого твердого вещества.
МС (ИЭР) m/z: 486,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.48 (brs, 1Н), 8.06 (t, 1Н), 7.43 (s, 1Н), 1.55 (d, 1Н), 6.10 (s, 1Н), 4.25 (d, 2Н), 3.83 (brd, 2Н), 3.81 (s, 3H), 3.21 (t, 2Н), 3.03 (q, 2Н), 3.90-3.96 (m, 1Н), 2.82 (q, 2Н), 2.18 (s, 3H), 1.63-1.67 (brd, 2Н), 1.45-1.53 (m, 2Н), 1.05 (t, 3H), 0.81 (t, 3H).
Пример 28
2-Циклопропил-6-((циклопропилметил)(тетрагидро-2Н-пиран-4-ил)амино)-5-этил-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид
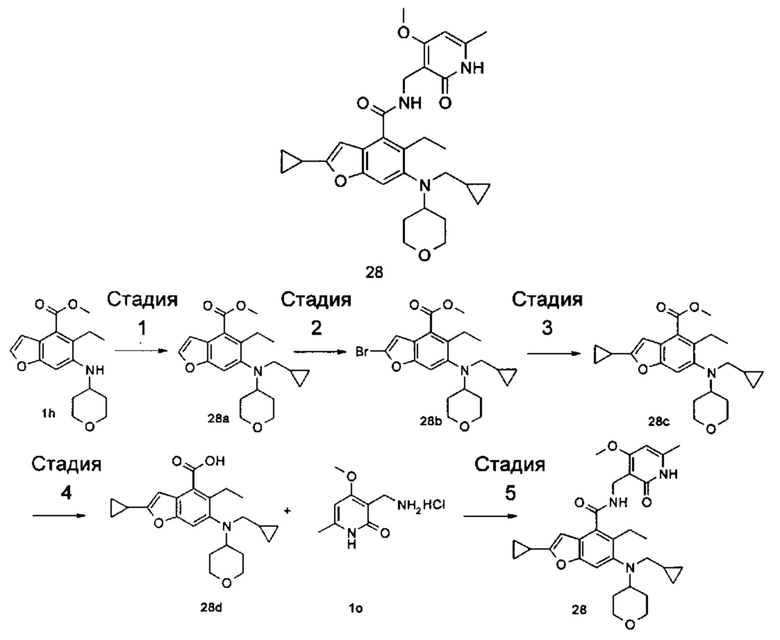
Стадия 1
Метил-6-((циклопропилметил)(тетрагидро-2Н-пиран-4-ил)амино)-5-этилбензофуран-4- карбоксилат 28а
Соединение 1h (950 мг, 3,03 ммоль) растворяли в 50 мл 1,2-дихлорэтана, впоследствии к описанному выше раствору добавляли циклопропанкарбальдегид (1,1 г, 15,7 ммоль) и уксусную кислоту (940 мг, 15,7 ммоль). Смесь перемешивали 12 часов, впоследствии добавляли триацетоксиборгидрид натрия (1,97 г, 9,4 ммоль). Смесь перемешивали в течение 3 часов. После завершения реакции реакционный раствор концентрировали под пониженным давлением, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, последовательно промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 28а (850 мг, выход 76%), в виде светло-желтого масла.
Стадия 2
Метил-2-бром-6-((циклопропилметил)(тетрагидро-2Н-пиран-4-ил)амино)-5-этилбензофуран-4-карбоксилат 28b
Соединение 28а (400 мг, 1,1 ммоль) растворяли в 15 мл тетрагидрофурана. Смесь охлаждали до -70°С, впоследствии к описанному выше раствору добавляли 1,65 мл 2,0 М раствора диизопропиламида лития. Смесь перемешивали в течение 1 часа при -70°С. К реакционному раствору добавляли по каплям 15 мл предварительно приготовленного раствора 1,2-дибром-1,1,2,2-тетрахлорэтана (429 мг, 1,3 ммоль) в тетрагидрофуране. Реакционную смесь перемешивали в течение 1 часа при -70°С. После завершения реакции добавляли 50 мл насыщенного раствора хлорида аммония для гашения реакции, и смесь экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 28b (150 г, выход 31%), в виде светло-желтого масла.
Стадия 3
Метил-2-циклопропил-6-((циклопропилметил)(тетрагидро-2Н-пиран-4-ил)амино)-5-этилбензофуран-4-карбоксилат 28с
Соединение 28b (50 мг, 0,11 ммоль) растворяли в 5 мл толуола, впоследствии к описанному выше раствору добавляли циклопропилбороновую кислоту (20 мг, 0,23 ммоль), ацетат палладия (5 мг, 0,2 ммоль), трициклогексилфосфин (10 мг, 0,03 ммоль) и тригидрат фосфата калия (91 мг, 0,34 ммоль). Реакционную смесь перемешивали в течение 12 часов при 100°С в атмосфере аргона. После завершения реакции смесь фильтровали через целит. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 28с (33 мг, выход 73%), в виде бесцветного масла.
Стадия 4
2-Циклопропил-6-((циклопропилметил)(тетрагидро-2Н-пиран-4-ил)амино)-5-этилбензофуран-4-карбоновая кислота 28d
Соединение 28с (33 мг, 0,08 ммоль) добавляли к смеси 5 мл тетрагидрофурана и 5 мл метанола, впоследствии добавляли 5 мл 4 М раствора гидроксида натрия. Реакционную систему перемешивали в течение 12 часов при 60°С. После завершения реакции смесь концентрировали для удаления органических растворителей. Добавляли небольшое количество воды, 12 М хлористоводородную кислоту для доведения рН до 3, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 28d (25 мг), в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 5
2-Циклопропил-6-((циклопропилметил)(тетрагидро-2Н-пиран-4-ил)амино)-5-этил-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид 28
Неочищенное соединение 28d (25 мг, 0,065 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии к описанному выше раствору добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (47 мг, 0,098 ммоль) и N,N-диизопропилэтиламин (42 мг, 0,33 ммоль). Смесь перемешивали в течение 1 часа. Добавляли соединение 1о (20 мг, 0.098 ммоль), и смесь перемешивали в течение 12 часов. После завершения реакции к реакционному раствору добавляли 20 мл воды, и смесь экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1). Органические фазы объединяли, последовательно промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 28 (12 мг, выход 34%), в виде светло-желтого твердого вещества.
МС (ИЭР) m/z: 534,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.48 (brs, 1Н), 7.91 (s, 1Н), 7.38 (s, 1Н), 6.36 (s, 1Н), 6.12 (s, 1Н), 4.27 (brs, 2Н), 3.83 (s, 3H), 3.79 (brs, 2Н), 3.28 (brs, 2Н), 3.17-3.23 (m, 2Н), 2.96-3.00 (m, 1Н), 2.83-2.86 (m, 2Н), 2.19 (s, 3H), 2.06 (brs, 1Н), 1.38-1.46 (m, 2Н), 1.24 (brs, 2Н), 1.06 (t, 3H), 0.98 (brs, 2Н), 0.83 (brs, 2Н), 0.64 (brs, 1Н), 0.28 (brs, 2Н), 0.03 (brs, 2Н).
Пример 29
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метил-2-(трифторметил)бензофуран-4-карбоксами
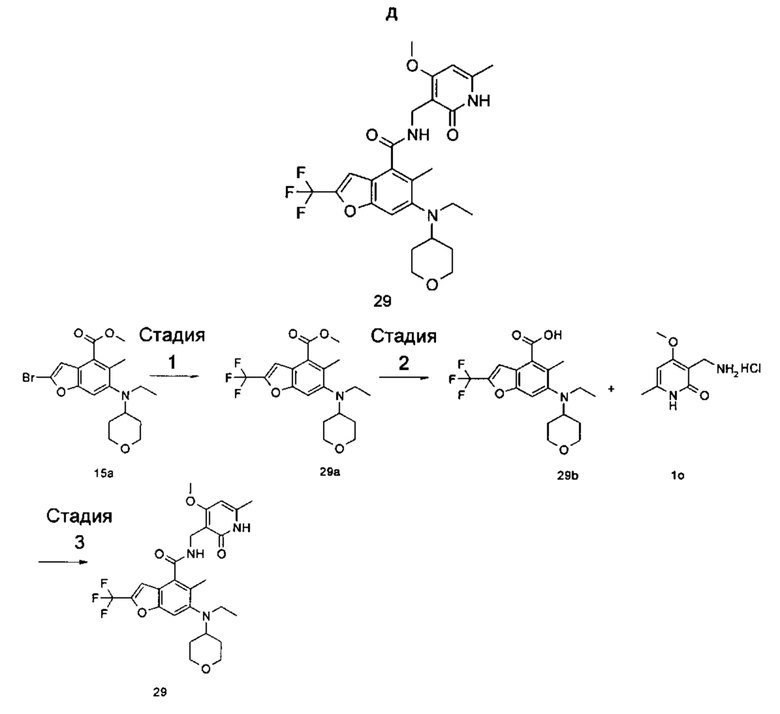
Стадия 1
Метил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(трифторметил)бензофуран-4-карбоксилат 29а
Соединение 15а (50 мг, 0,126 ммоль) растворяли в 8 мл N,N-диметилформамида, впоследствии к описанному выше раствору добавляли метилфторсульфонилдифторацетат (500 мг, 2,52 ммоль) и йодид меди (120 мг, 0,63 ммоль). Реакционную смесь перемешивали в течение 24 часов при 120°С в атмосфере азота. После завершения реакции к реакционному раствору добавляли 50 мл воды, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением указанного в заголовке соединения 29а (10 мг, выход 20,8%), в виде желтой жидкости.
Стадия 2
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метил-2-(трифторметил)бензофуран-4-карбоновая кислота 29b
Соединение 29а (10 мг, 0,026 ммоль) добавляли к 2 мл смеси воды и метанола (об. : об. составляет 1:1), впоследствии добавляли гидроксид натрия (5,16 мг, 0,129 ммоль). Реакционную систему перемешивали в течение 16 часов при 70°С. После завершения реакции реакционный раствор охлаждали до 5°С, добавляли 2 н. хлористоводородную кислоту для доведения рН до 3-4, и смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 29b (10 мг), в виде белого твердого вещества, которое непосредственно использовали в следующей стадии без дополнительной очистки.
Стадия 3
6-(Этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метил-2-(трифторметил)бензофуран-4-карбоксами д 29
Неочищенное соединение 29b (10 мг, 0,027 ммоль) растворяли в 3 мл N,N-диметилформамида, впоследствии добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (15,4 мг, 0,041 ммоль) и N,N-диизопропилэтиламин (10,5 мг, 0,081 ммоль). Реакционную систему охлаждали до 0°С и перемешивали в течение 1 часа. Добавляли соединение 1о (6,6 мг, 0,032 ммоль), и смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции добавляли 40 мл воды, и смесь экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1) (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 29 (1,6 мг, выход 11,4%), в виде белого твердого вещества.
МС (ИЭР) m/z: 522,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.52 (s, 1Н), 7.33 (s, 1Н), 7.19 (brs, 1Н), 5.98 (s, 1Н), 4.66 (s, 2Н), 4.00-3.95 (m, 2Н), 3.95 (s, 3H), 3.36-3.30 (m, 2Н), 3.12 (q, 2Н), 3.04-2.99 (m, 1H), 2.41 (s, 3H), 2.28 (s, 3H), 1.72 (brs, 4H), 0.89 (t, 3H).
Пример 30
2-Циано-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид
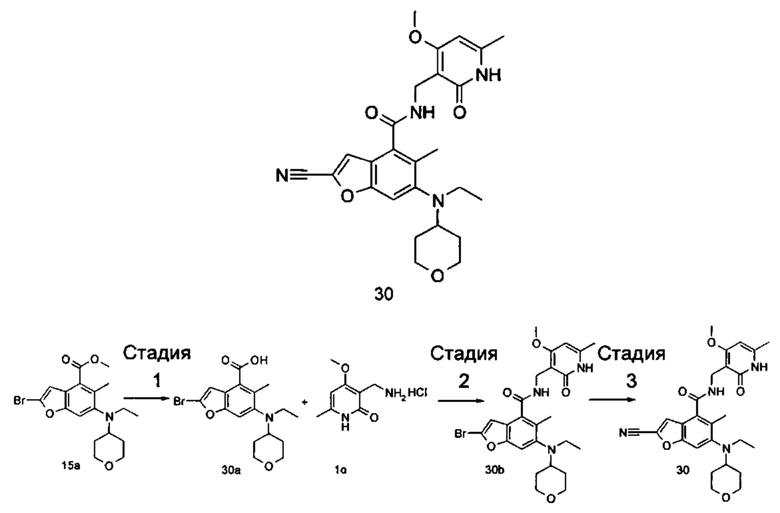
Стадия 1
2-Бром-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-5-метилбензофуран-4-карбоновая кислота
Соединение 15а (30 мг, 0,076 ммоль) добавляли к 2 мл смеси воды и метанола (об. : об. составляет 1:1), впоследствии добавляли гидроксид натрия (15,1 мг, 0,378 ммоль). Реакционную систему перемешивали в течение 12 часов при 70°С. После завершения реакции реакционный раствор охлаждали до 5°С, добавляли 2 н. хлористоводородную кислоту для доведения рН до 3-4, и смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 30а (30 мг), в виде белого твердого вещества, которое непосредственно использовали в следующей стадии без дополнительной очистки.
Стадия 2
2-Бром-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2- дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид 30b
Неочищенное соединение 30а (30 мг, 0,078 ммоль) растворяли в 5 мл N,N-диметилформамида, впоследствии к описанному выше раствору добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (44,5 мг, 0,117 ммоль) и N,N-диизопропилэтиламин (30,2 мг, 0,234 ммоль). Реакционную систему охлаждали до 0°С и перемешивали в течение 1 часа. Добавляли соединение 1о (6,6 мг, 0.032 ммоль), и смесь перемешивали в течение 12 часов. После завершения реакции добавляли 50 мл воды, и смесь экстрагировали смесью дихлорметана и метанола (об. : об. составляет 8:1) (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением неочищенного указанного в заголовке соединения 30b (50 мг), в виде желтой жидкости, которую использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
2-Циано-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-N-((4-метокси-6-метил-2-оксо-1,2- дигидропиридин-3-ил)метил)-5-метилбензофуран-4-карбоксамид 30
Неочищенное соединение 30b (40 мг, 0,075 ммоль) растворяли в 5 мл N,N-диметилацетамида, впоследствии к описанному выше раствору добавляли цианид меди (13,6 мг, 0,15 ммоль) и йодид меди (14,3 мг, 0,075 ммоль). Реакционную систему перемешивали в течение 1 часа при 170°С. После завершения реакции смесь фильтровали, и фильтрат промывали смесью дихлорметана и метанола (об. : об. составляет 10:1) (10 мл). Органические фазы объединяли, концентрировали под пониженным давлением, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением указанного в заголовке соединения 30 (4,7 мг, выход 12,5%), в виде белого твердого вещества.
МС (ИЭР) m/z: 479,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 11.5 (brs, 1Н), 7.55 (s, 1Н), 7.26 (s, 1Н), 7.16 (brs, 1Н), 5.97 (s, 1Н), 4.66 (s, 2Н), 3.98-3.96 (m, 2Н), 3.93 (s, 3H), 3.35-3.30 (m, 2Н), 3.11 (q, 2Н), 3.04-2.99 (m, 1Н), 2.41 (s, 3H), 2.27 (s, 3H), 1.75-1.66 (m, 4Н), 0.89 (t, 3H).
Пример 31
6-((4,4-Дифторциклогексил)(этил)амино)-5-этил-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид 31
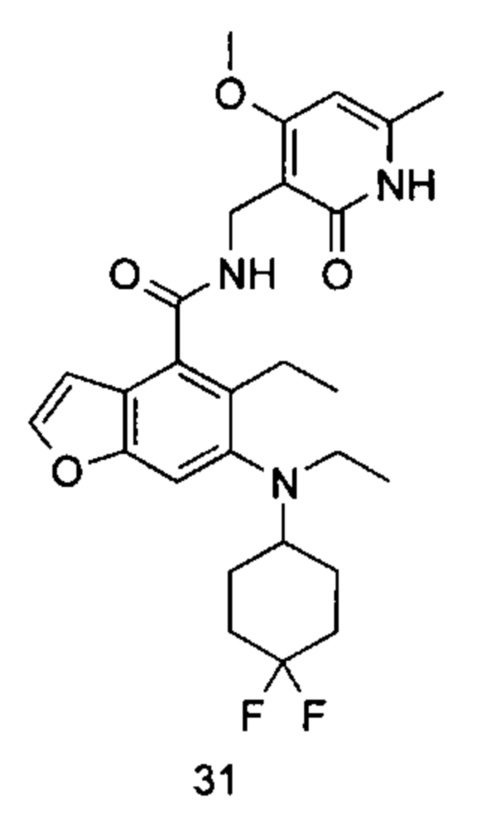
В соответствии с методом синтеза примера 24 исходное вещество 4е заменяли 1g, а 24а заменяли 4,4-дифторциклогексиламином, соответственно, получили соединение, указанное в заголовке, 31 (18 мг, выход 77%), в виде белого твердого вещества.
МС (ИЭР) m/z: 502,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.47 (brs, 1Н), 8.02 (t, 1Н), 7.88 (d, 1Н), 7.46 (s, 1Н), 6.75 (d, 1Н), 6.11 (s, 1Н), 4.29 (d, 2Н), 3.82 (s, 3H), 3.05 (brs, 2Н), 2.97 (m, 1Н), 2.82 (q, 2Н), 2.18 (s, 3H), 1.98 (brs, 2Н), 1.81 (brs, 2Н), 1.73 (m, 2Н), 1.58 (q, 2Н), 1.08 (t, 3H), 0.83 (t, 3H).
Пример 32
6-((3,3-Дифторциклобутил)(этил)амино)-5-этил-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид 32
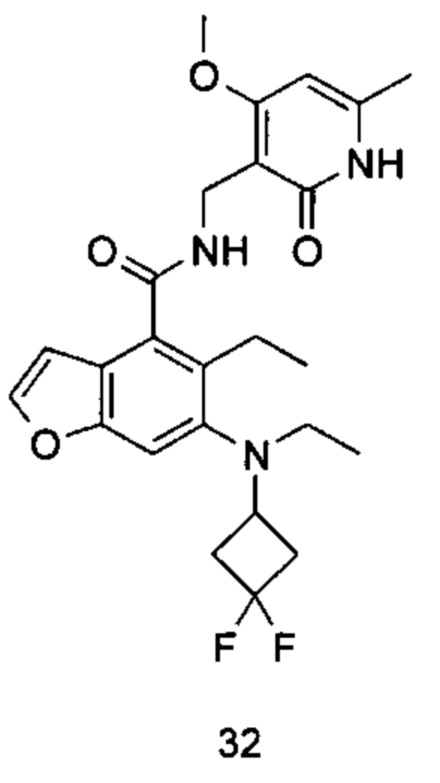
В соответствии с методом синтеза примера 24 исходное вещество 4е заменяли 1g, а 24а заменяли 3,3-дифторциклогексиламином, соответственно, получили соединение, указанное в заголовке, 32 (15 мг, выход 83%), в виде белого твердого вещества.
МС (ИЭР) m/z: 474,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.45 (brs, 1Н), 8.04 (t, 1Н), 7.88 (d, 1Н), 7.41 (s, 1Н), 6.75 (d, 1Н), 6.10 (s, 1Н), 4.28 (d, 2Н), 3.81 (s, 3H), 3.79 (brs, 1Н), 2.93 (q, 2Н), 2.81 (q, 2Н), 2.70 (brs, 2Н), 2.25 (brs, 2Н), 2.18 (s, 3H), 1.07 (t, 3H), 0.86 (t, 3H).
Пример 33
6-((1,1-Диоксидотетрагидро-2Н-тиопиран-4-ил)(этил)амино)-5-этил-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид 33
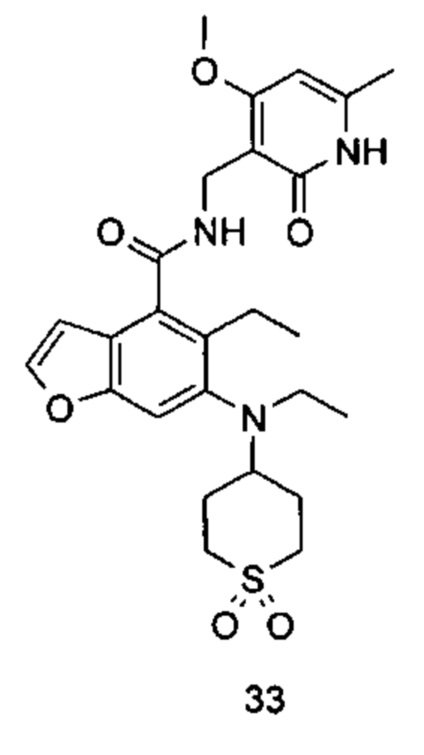
В соответствии с методом синтеза примера 24 исходное вещество 4е заменяли 1g, а 24а заменяли 4-аминотетрагидро-2Н-тиопиран-1,1-диоксидом, соответственно, получили соединение, указанное в заголовке, 33 (9 мг, выход 64%), в виде белого твердого вещества.
МС (ИЭР) m/z: 516,3 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.46 (brs, 1Н), 8.02 (t, 1Н), 7.88 (d, 1Н), 7.47 (s, 1Н), 6.75 (d, 1Н), 6.10 (s, 1Н), 4.29 (d, 2Н), 3.81 (s, 3H), 2.98-3.07 (m, 5Н), 3.01 (q, 2Н), 2.80 (q, 2Н), 2.18 (s, 3H), 2.07 (brs, 4Н), 1.09 (t, 3H), 0.83 (t, 3H).
Пример 34
6-((Циклопропилметил)(1-(2,2,2-трифторэтил)пиперидин-4-ил)амино)-5-этил-N-((4-метокси-6-метил-2-оксо-1,2-дигидропиридин-3-ил)метил)бензофуран-4-карбоксамид 34
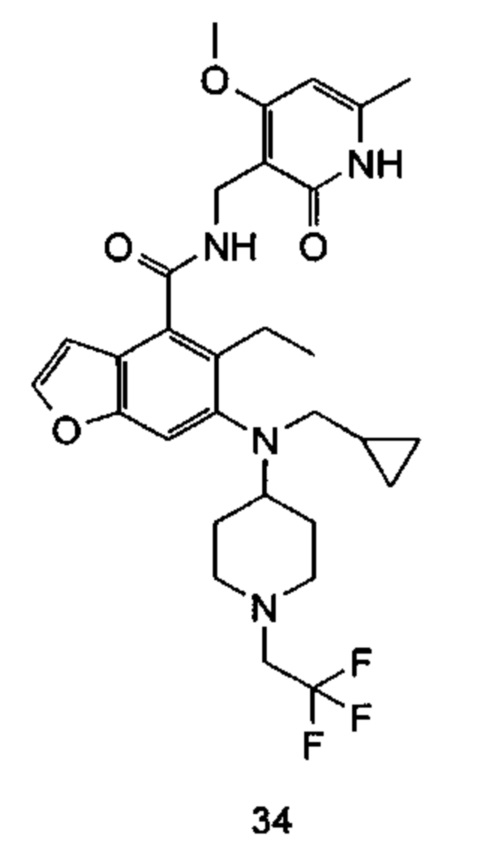
В соответствии с методом синтеза примера 25 исходное вещество 4е заменяли 1g, исходное вещество ацетальдегид, используемое на стадии 2, заменяли циклопропанкарбоксальдегидом, исходное вещество метансульфонилхлорид, используемое на стадии 4, заменяли трихлорметил(2,2,2-трифторэтил)сульфатом, соответственно, получили соединение, указанное в заголовке, 34 (14 мг, выход 65%) в виде белого твердого вещества.
МС (ИЭР) m/z: 575,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.01 (brs, 1Н), 7.87 (s, 1Н), 7.50 (s, 1Н), 6.75 (s, 1Н), 6.11 (s, 1Н), 4.29 (d, 2Н), 3.83 (s, 3H), 3.10 (q, 2Н), 2.83-2.91 (m, 4Н), 2.76 (brs, 1Н), 2.26 (t, 2Н), 2.19 (3H), 1.70 (brs, 2Н), 1.49 (q, 2Н), 1.24 (brs, 2Н), 1.10 (t, 3H), 0.65 (brs, 1Н), 0.28 (d, 2Н), 0.003 (d, 2Н).
Биологический анализ
Далее настоящее изобретение описано со ссылкой на следующие тестовые примеры, но эти примеры не следует истолковывать как ограничивающие объем изобретения.
Тестовый пример 1. Анализ для определения активности соединений примеров настоящего изобретения в отношении фермента EZH2 (мутант A677G или мутант Y641F)
Тестирование активности фермента EZH2 (с мутантом A677G или с мутантом Y641F) проводили с помощью описанного ниже способа.
Этот способ использовали для определения ингибиторного действия соединений по настоящему изобретению на активность мутанта EZH2-A677G или мутанта EZH2-Y641F.
1. Экспериментальные материалы и приборы
(1). EZH2-A677G (BPS Bioscience)
(2). EZH2-Y641F (BPS Bioscience)
(3). Гистон Н3, меченый биотином (AnaSpec)
(4). S-аденозилметионин (сокращенно SAM, Sigma)
(5). Моноклональное антитело к гистону H3K27 Ме3 (Cisbio)
(6). Стрептавидин-XL665 (Cisbio)
(7). Буфер HTRF для детектирования (Cisbio)
(8). Многофункциональное устройство для считывания микропланшетов (Tecan)
2. Методика эксперимента
Мутант EZH2-A677G (или EZH2-Y641F) разводили до концентрации 15 нг/мкл, используя буферный раствор для киназы (5×буферный раствор: 5 мг/мг мл бычьего сывороточного альбумина (БСА), 150 ммоль/л трис-Cl, 100 ммоль/л MgCl2) и добавляли в лунки 384-луночного микротитрационного планшета по 2 мкл/лунка. Меченый биотином гистон Н3 и S-аденозилметионин соответственно разводили до 50 нмоль/л и 50 мкмоль/л буферным раствором для киназы, впоследствии добавляли в лунки 384-луночного микротитрационного планшета по 4 мкл/лунка. Тестируемое соединение разводили буферным раствором для киназы (его разводили из самой высокой концентрации 30 мкмоль/л с 10-кратным градиентом концентрации до 7 точек концентрации), впоследствии добавляли в лунки 384-луночного микротитрационного планшета по 4 мкл/лунка. Планшет инкубировали при комнатной температуре в течение 2 часов. Моноклональное антитело Ме3 к гистону H3K27 и Стрептавидин-XL665 разводили до 30 нмоль/л и 500 нмоль/л буферным раствором HTRF для детектирования, впоследствии добавляли в лунки 384-луночного микротитрационного планшета по 10 мкл/лунка и инкубировали в течение 1 часа. В качестве отрицательного контроля использовали лунку бз фермента EZH2 и без соединения, а в качестве положительного контроля - лунку с ферментом EZH2, но без соединения. Показания флуоресценции снимали на многофункциональном считывающем устройстве для планшетов при длине волны испускания 620 нм и 665 нм. Строили кривую зависимости процента ингибирования от логарифма концентрации соединения относительно положительной контрольной лунки, используя программное обеспечение GraphPad Prism, впоследствии рассчитывали значения IC50.
Активность соединений по настоящему изобретению в отношении фермента EZH2-A677G тестировали с помощью описанной выше методики анализа, и значения IC50 представлены в таблице 1.
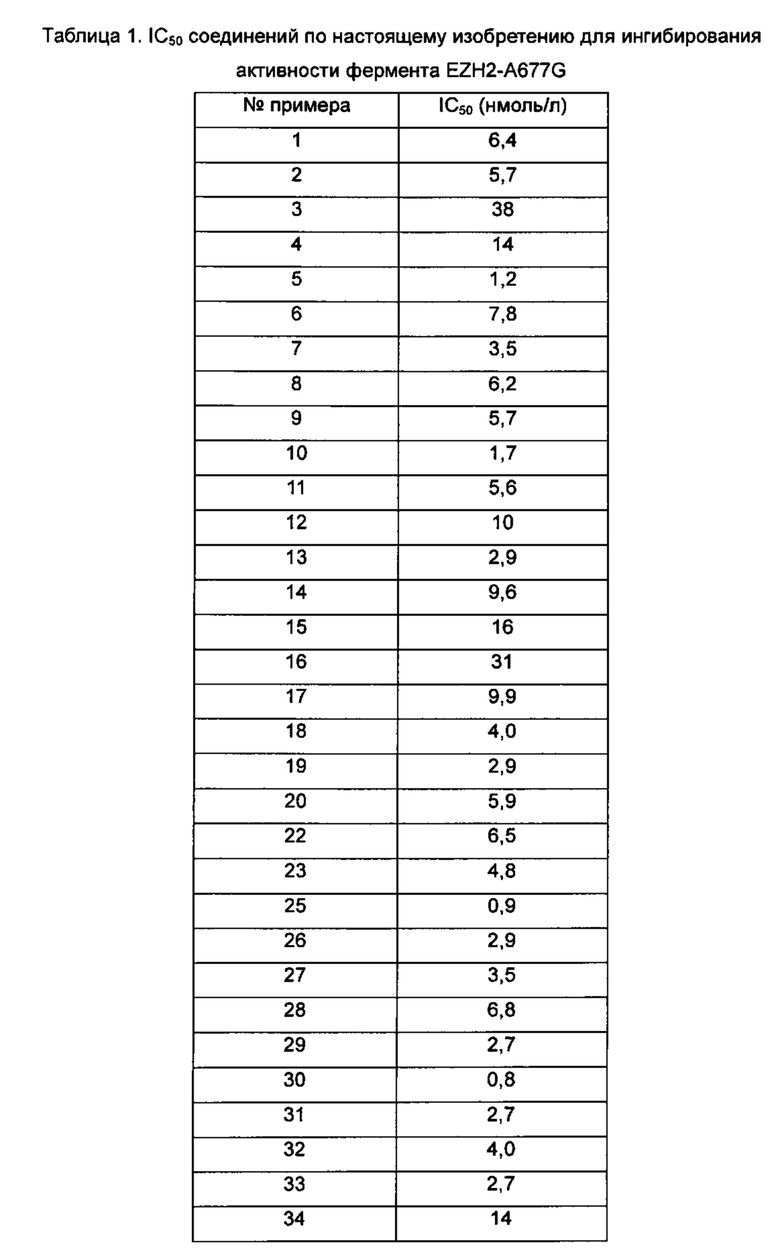
Вывод: Соединения по настоящему изобретению характеризуются статистически значимым ингибирующим действием на активность фермента EZH2-A677G.
Активность соединений по настоящему изобретению в отношении фермента EZH2-Y641F тестировали с помощью описанной выше методики анализа, и значения IC50 представлены в таблице 2.
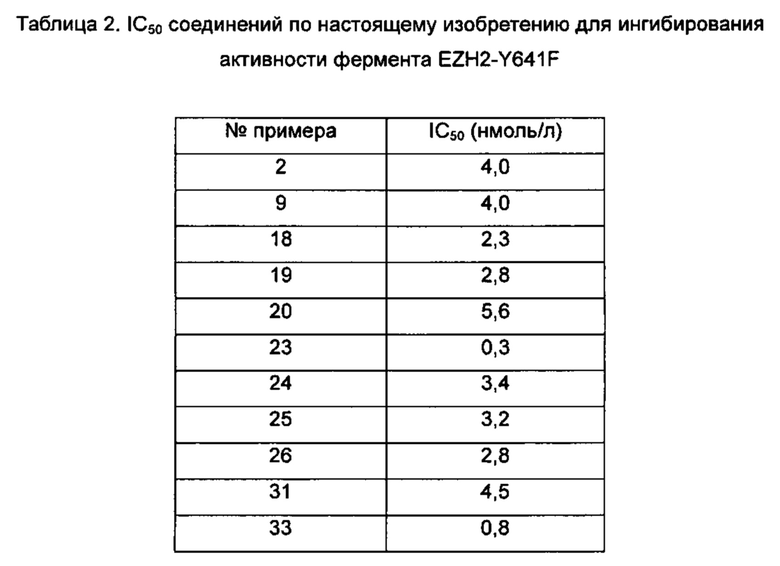
Тестовый пример 2. Анализ для определения противоопухолевой активности соединений примеров настоящего изобретения в отношении роста опухолевых клеток in vitro
Этот способ использовали для определения ингибиторного действия соединений по настоящему изобретению на активность роста опухолевых клеток (Pfeiffer или WSU-DLCL2) in vitro. Клетки Pfeiffer несут мутацию A677G, а клетки WSU-DLCL2 несут мутацию Y641F.
1. Экспериментальные материалы и приборы
(1). RPMI-1640, инактивированная фетальная бычья сыворотка, пенициллин, стрептомицин (Life technology)
(2). Линии клеток Pfeiffer и WSU-DLCL2 (АТСС)
(3). 96-луночный планшет для клеточных культур (Fisher Scientific)
(4). Инкубатор для клеточных культур (Fisher Scientific)
(5). Набор реагентов для определения жизнеспособности клеток люминесцентным методом CellTiter - Glo® (Promega)
(6). Устройство для считывания микропланшетов (Tecan)
2. Методика эксперимента
Суспензионные культуры линий клеток лимфомы (Pfeiffer или WSU-DLCL2) выращивали в питательной среде RPMI-1640, содержащей 10% инактивированной фетальной бычьей сыворотки, 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина, инкубаровали в инкубаторе в атмосфере 5% диоксида углерода при 37°С в условиях насыщенной влажности и пересевали один раз в 3-4 дня.
Суспензию клеток готовили в свежей питательной среде для культуры клеток, вносили в лунки 96-луночного планшета для культур клеток в количестве 8000 клеток/лунка (Pfeiffer) или 2000 клеток/лунка (WSU-DLCL2) и инкубировали в инкубаторе в атмосфере 5% диоксида углерода при 37°С в течение ночи. В день эксперимента в экспериментальные лунки добавляли тестируемые соединения на EZH2 (конечная концентрация: 20000, 2000, 200, 20, 2, 0,2 нмоль/л) для 3 параллельных лунок на группу. Для получения фонового показания люминесценции ставили контрольную лунку, содержащую только питательную среду без клеток. Планшет для культур клеток инкубировали в инкубаторе с 5% диоксидом углерода при 37°С в течение 5 дней.
После воздействия соединений на клетки в течение 5 дней планшет с культурами клеток помещали в условия комнатной температуры на 30 минут, впоследствии в каждую лунку добавляли объем реагента CellTiter-Glo, равный объему клеток. Содержимое лунок перемешивали на шейкере в течение 10 минут и регистрировали показания сигналов флуоресценции. Рассчитывали коэффициент ингибирования роста клеток по формуле: коэффициент ингибирования роста клеток = [(отрицательная контрольная группа -экспериментальная группа)/(отрицательная контрольная группа - фоновое показание люминесценции)] × 100%. Значения IC50 (IC50 половинного коэффициента ингибирования, т.е. концентрацию лекарственного средства, необходимую для 50% коэффициента ингибирования роста клеток) рассчитывали с помощью программного обеспечения на основании коэффициента ингибирования роста клеток и соответствующей концентрации.
Тестирование противоопухолевой активности in vitro (Pfeiffer или WSU-DLCL2) соединений по настоящему изобретению проводили с помощью описанного выше анализа, и значения IC50 приведены в таблице 3 и 4.
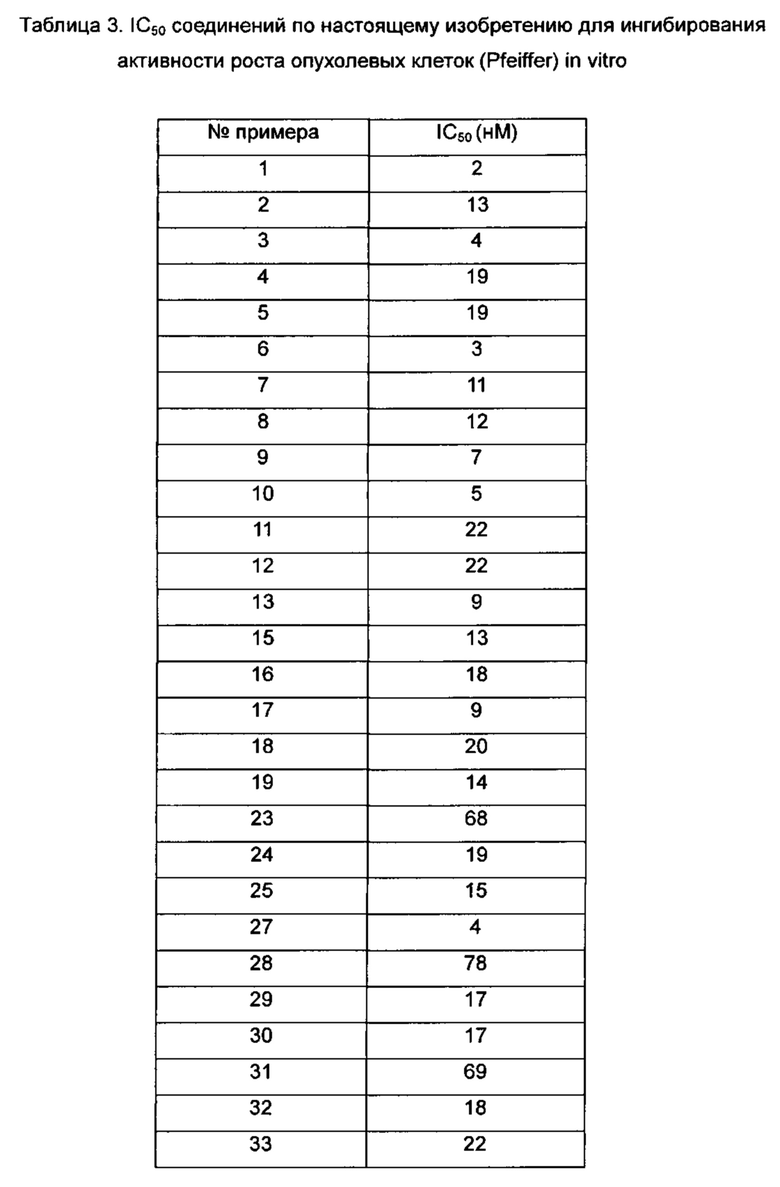
Вывод: Соединения по настоящему изобретению характеризуются статистически значимым ингибирующим действием на рост клеток лимфомы (Pfeiffer) in vitro.
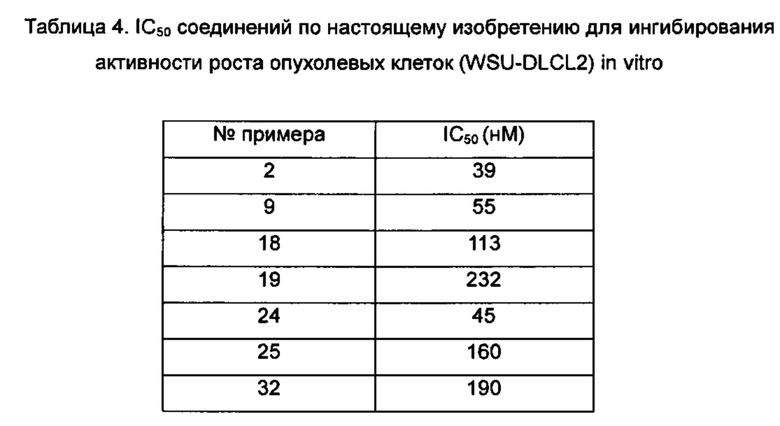
Вывод: Соединения по настоящему изобретению характеризуются статистически значимым ингибирующим действием на рост клеток лимфомы (WSU-DLCL2) in vitro.
Фармакокинетический анализ
Тестовый пример 3. Анализ фармакокинетики примеров 1, 2, 3 и 9 по настоящему изобретению.
1. Краткое описание
В качестве экспериментальных животных использовали крыс линии Спраг-Доули (SD). Концентрацию лекарственного средства в плазме крови в различные моменты времени определяли методом жидкостной хроматографии с тандемной масс-спектрометрией (ЖХ/МС/МС) после внутрижелудочного введения крысам соединений примеров 1, 2, 3 и 9. Фармакокинетические свойства соединений по настоящему изобретению изучали и оценивали у крыс.
2. Протокол
2.1 Исследуемые соединения
Соединения примеров 1, 2, 3 и 9
2.2 Экспериментальные животные
16 здоровых взрослых крыс Спраг-Доули (SD), половина самцов и половина самок, были приобретены в компании SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО, № сертификата: SCXK (Шанхай) 2008-0016.
2.3 Приготовление исследуемых соединений
Готовили соответствующую навеску каждого соединения и добавляли к ней 0,5% карбоксиметилцеллюлозу (КМЦ)-Na (содержащую 1% Tween 80) с получением суспензии 1,0 мг/мл путем измельчения. 2.4 Введение
После голодания в течение ночи 16 крыс SD делили поровну на 4 группы, половина самцов и половина самок, и вводили им исследуемые соединения внутрижелудочным путем в объеме введения 5 мл/кг.
3. Способ
Забор крови (0,2 мл) осуществляли из орбитального синуса до введения и через 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 11,0 и 24,0 часа после введения. Образцы хранили в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об/мин, чтобы отделить плазму крови. Образцы плазмы крови хранили при -20°С.
Концентрацию исследуемых соединений в плазме крови крыс после внутрижелудочного введения определяли с помощью ЖХ-МС/МС.
4. Результаты определения параметров фармакокинетики
Параметры фармакокинетики соединений примеров 1, 2, 3 и 9 по настоящему изобретению приведены ниже.
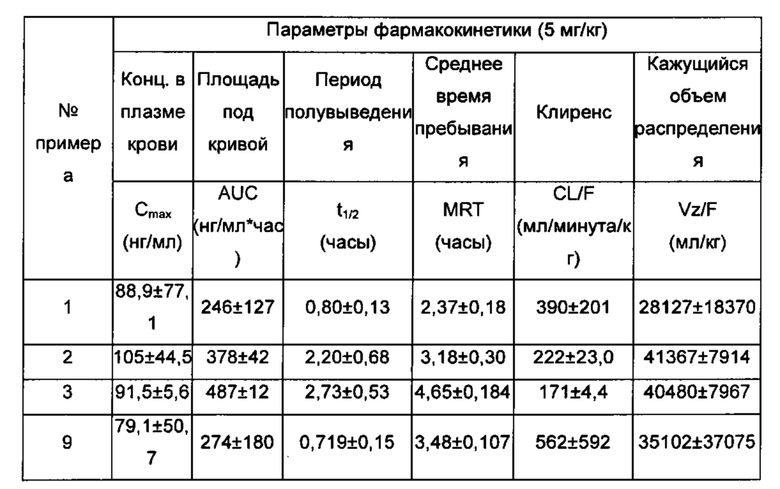
Вывод: Соединения по настоящему изобретению характеризуются высокой абсорбцией и обладают значительным фармакологическим эффектом абсорбции.
Изобретение относится к производному бензофурана, представленному общей формулой (I), или к его фармацевтически приемлемой соли, в котором кольцо А выбрано из группы, состоящей из гетероциклила, который представляет собой 5-6-членную насыщенную моноциклическую углеводородную группу, имеющую один гетероатом, выбранный из N, О и S, и 3-6-членного циклоалкила; каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, атома галогена, С1-6-алкила, галоген-С1-6-алкила, циано, 3-6-членного циклоалкила, 5-6-членной насыщенной или частично ненасыщенной моноциклической углеводородной группы, имеющей один атом N, и -(CH2)xRa, где 5-6-членная насыщенная или частично ненасыщенная моноциклическая углеводородная группа необязательно замещена одной или более группами, выбранными из группы, состоящей из С1-6-алкила; Ra выбран из группы, состоящей из гетероциклила, который представляет собой 5-6-членную насыщенную моноциклическую углеводородную группу, имеющую один или два гетероатома, выбранных из N и О, и -NR7R8, где каждый гетероциклил независимо и необязательно замещен одной или более группами, выбранными из группы, состоящей из С1-6-алкила, атома галогена и гидрокси-С1-6-алкила; R2 представляет собой С1-6-алкил, где алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из 3-6-членного циклоалкила; R3 выбран из группы, состоящей из С1-6-алкила, атома галогена и галоген-С1-6-алкила; каждый R4 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, С1-6-алкила и С1-6-алкокси; каждый R5 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, оксо, атома галогена, галоген-С1-6алкила, -C(O)R6, -S(O)mR6 и -NR7R8; R6 выбран из С1-6-алкила; R7 и R8 идентичны или отличаются друг от друга, и каждый независимо выбран из группы, состоящей из С1-6-алкила; m равно 2; n равно 2; р равно 0, 1 или 2; q равно 0 или 1; и х равно 1, 2 или 3. Изобретение также относится к способу получения соединения формулы (I) и к фармацевтической композиции, обладающей ингибиторной активностью в отношении EZH2, на его основе. Технический результат – получено новое соединение и фармацевтическая композиция на его основе, которые могут найти применение в медицине для профилактики и/или лечения болезней, таких как опухоли и рак. 6 н. и 7 з.п. ф-лы, 4 табл., 34 пр.
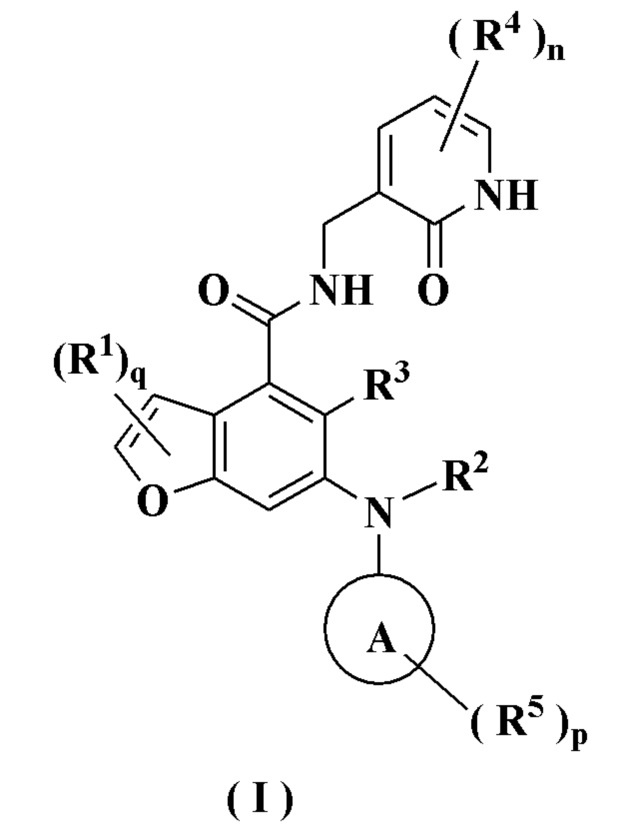
1. Соединение формулы (I):
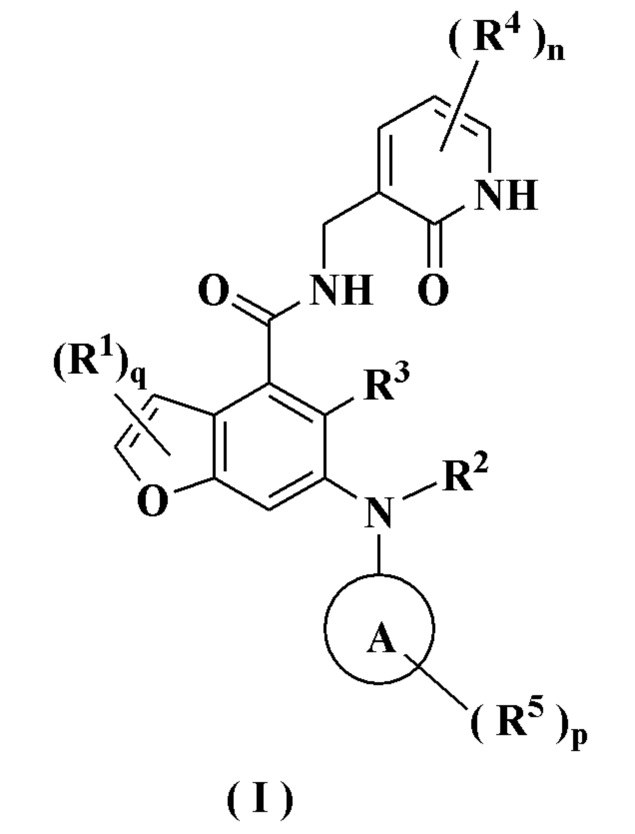
или его фармацевтически приемлемая соль,
где:
кольцо А выбрано из группы, состоящей из гетероциклила, который представляет собой 5-6-членную насыщенную моноциклическую углеводородную группу, имеющую один гетероатом, выбранный из N, О и S, и 3-6-членного циклоалкила;
каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, атома галогена, С1-6-алкила, галоген-С1-6-алкила, циано, 3-6-членного циклоалкила, 5-6-членной насыщенной или частично ненасыщенной моноциклической углеводородной группы, имеющей один атом N, и -(CH2)xRa, где 5-6-членная насыщенная или частично ненасыщенная моноциклическая углеводородная группа необязательно замещена одной или более группами, выбранными из группы, состоящей из С1-6-алкила;
Ra выбран из группы, состоящей из гетероциклила, который представляет собой 5-6-членную насыщенную моноциклическую углеводородную группу, имеющую один или два гетероатома, выбранных из N и О, и -NR7R8, где каждый гетероциклил независимо и необязательно замещен одной или более группами, выбранными из группы, состоящей из С1-6-алкила, атома галогена и гидрокси-С1-6-алкила;
R2 представляет собой С1-6-алкил, где алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из 3-6-членного циклоалкила;
R3 выбран из группы, состоящей из С1-6-алкила, атома галогена и галоген-С1-6-алкила;
каждый R4 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, С1-6-алкила и С1-6-алкокси;
каждый R5 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, оксо, атома галогена, галоген-С1-6алкила, -C(O)R6, -S(O)mR6 и -NR7R8;
R6 выбран из С1-6-алкила;
R7 и R8 идентичны или отличаются друг от друга, и каждый независимо выбран из группы, состоящей из С1-6-алкила;
m равно 2;
n равно 2;
р равно 0, 1 или 2;
q равно 0 или 1; и
х равно 1, 2 или 3.
2. Соединение формулы (I) по п. 1, где каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, С1-6-алкила, галоген-С1-6-алкила, циано, атома галогена и 3-6-членного циклоалкила.
3. Соединение формулы (I) по п. 1, представляющее собой соединение формулы (II):
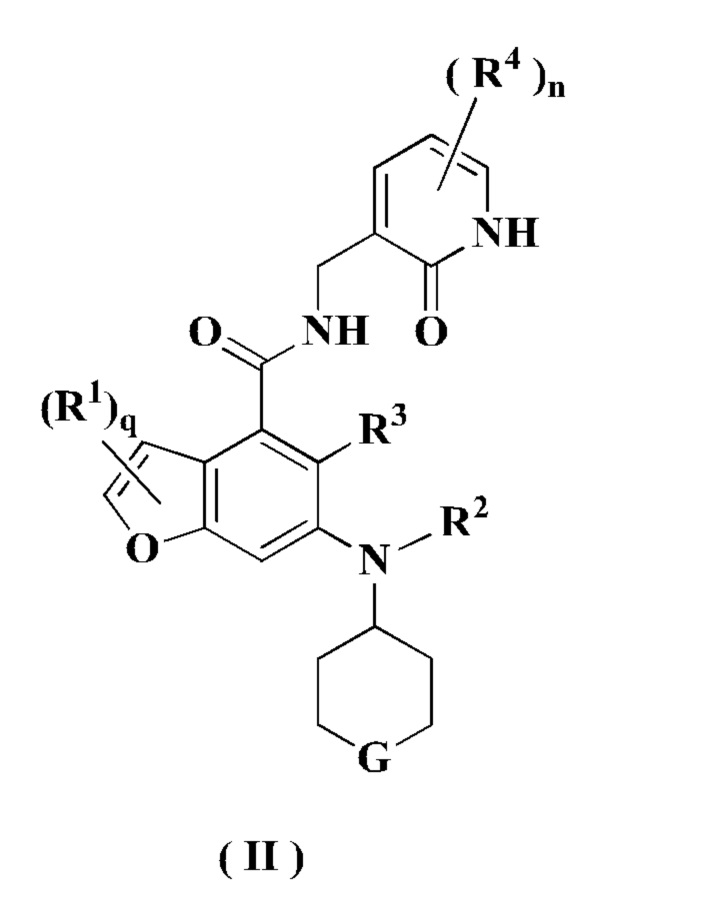
или его фармацевтически приемлемая соль,
где:
G выбран из группы, состоящей из CRbRc, NRd, S(O)m и атома кислорода; Rb и Rc каждый независимо выбран из группы, состоящей из атома водорода, атома галогена и -NR7R8;
Rd выбран из группы, состоящей из галоген-С1-6-алкила, -C(O)R6 и -S(O)mR6; и радикалы с R1 по R4, с R6 по R8, n, m и q являются такими, как определено в п. 1.
4. Соединение формулы (II) по п. 3, представляющее собой соединение формулы (III):
или его фармацевтически приемлемая соль, где:
Е представляет собой СН или атом N;
F выбран из группы, состоящей из CRbRc, NRd и атома кислорода; Rb и Rc каждый независимо выбран из группы, состоящей из атома водорода и атома галогена;
Rd выбран из группы, состоящей из С1-6-алкила и гидрокси-С1-6-алкила;
каждый Re идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из С1-6-алкила, атома галогена и гидрокси-С1-6-алкила;
t равно 0, 1 или 2;
х равно 1;
у равно 0 или 1; и
радикалы с R1 по R4, с R6 по R8, m и n являются такими, как определено в п. 1.
5. Соединение формулы (III) по п. 4, представляющее собой соединение формулы (IV):
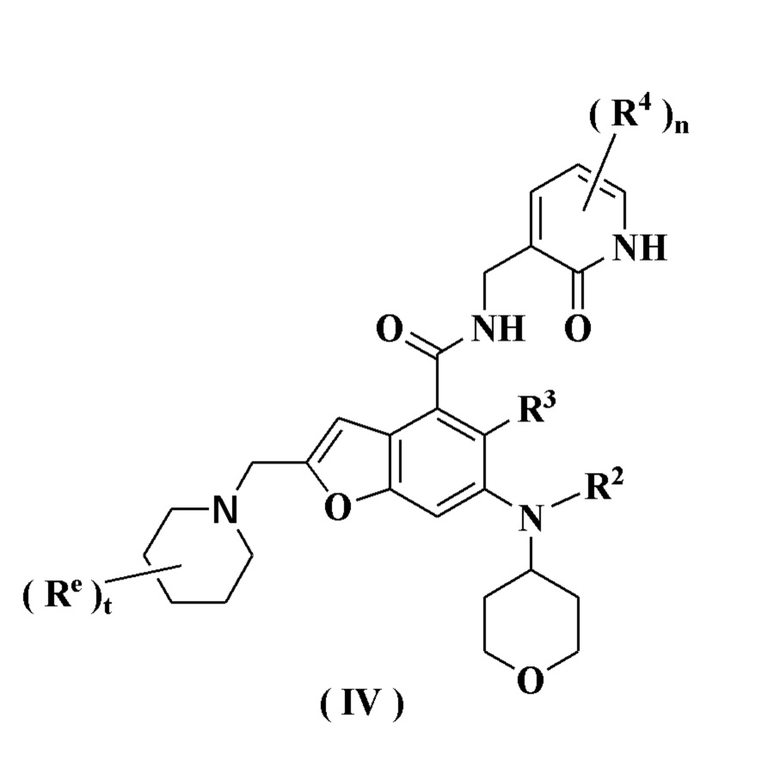
или его фармацевтически приемлемая соль, где:
каждый Re идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода и атома галогена; t равно 0, 1 или 2; и
радикалы с R2 по R4 и n являются такими, как определено в п. 1.
6. Соединение формулы (III) по п. 4, представляющее собой соединение формулы (V):
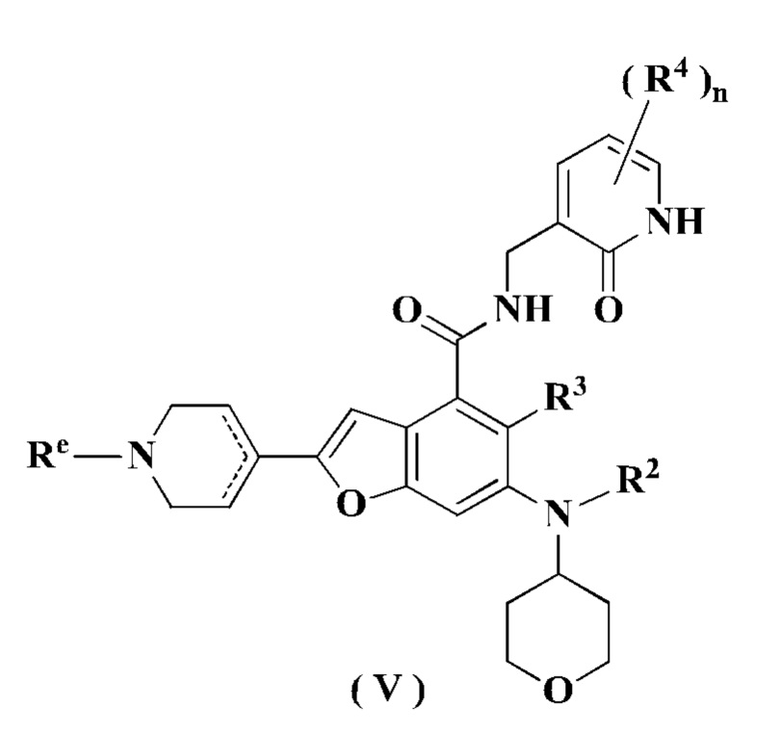
или его фармацевтически приемлемая соль, где:
Re представляет собой С1-6-алкил; и
радикалы с R2 по R4 и n являются такими, как определено в п. 1.
7. Соединение по любому из пп. 1-6, выбранное из группы, состоящей из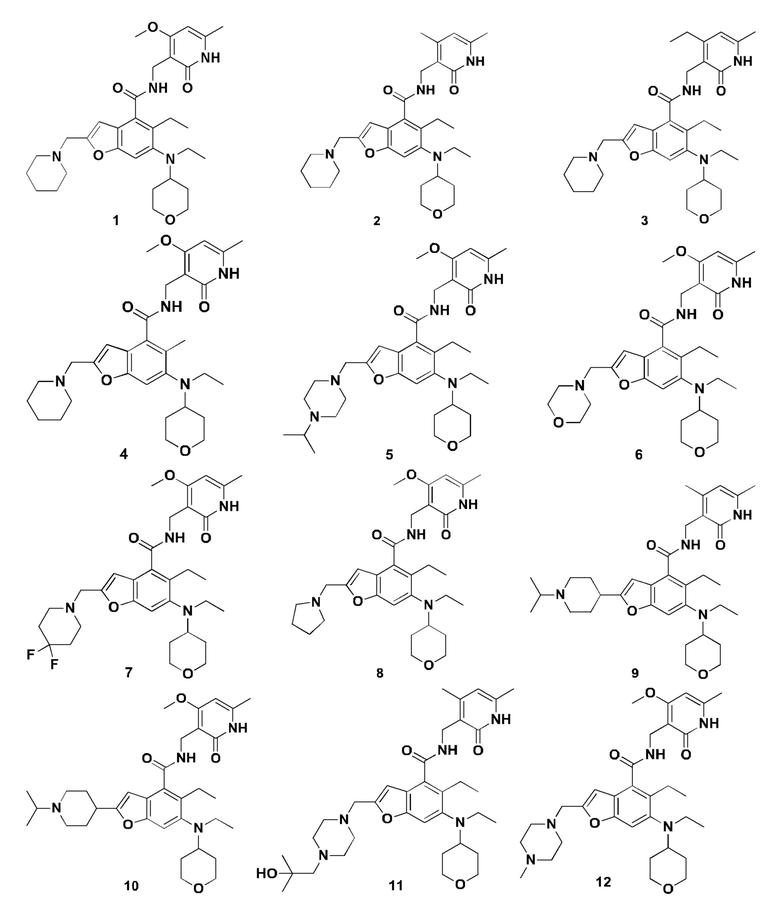
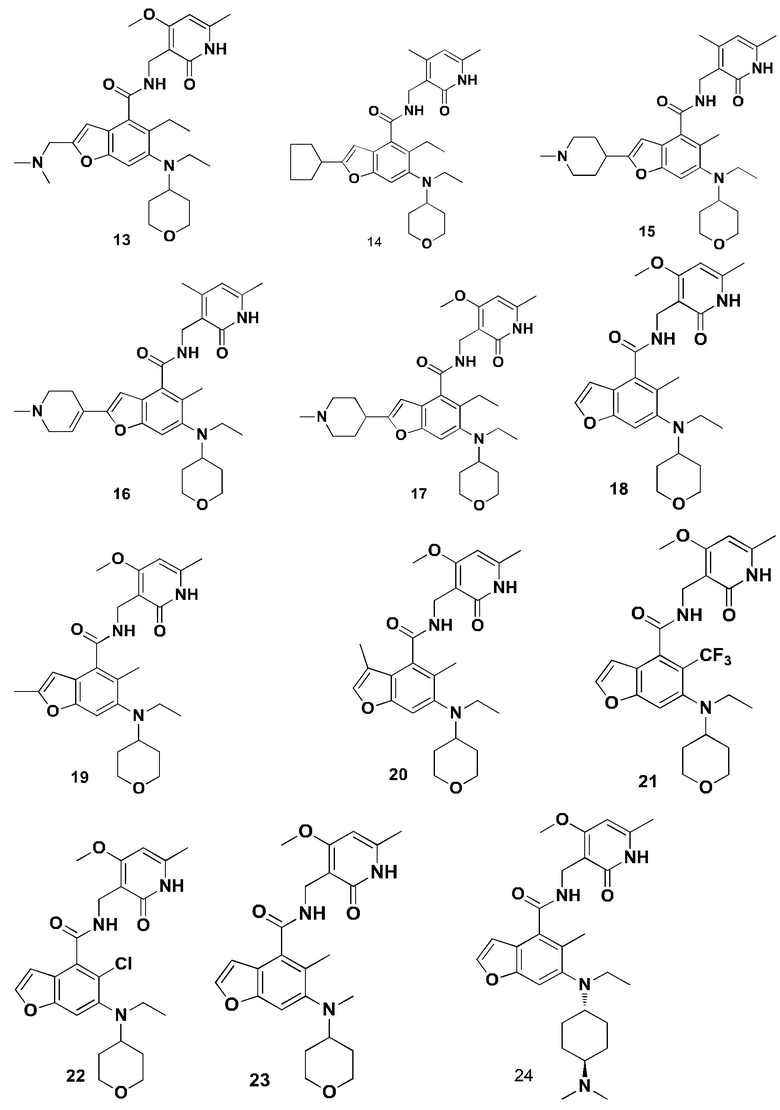
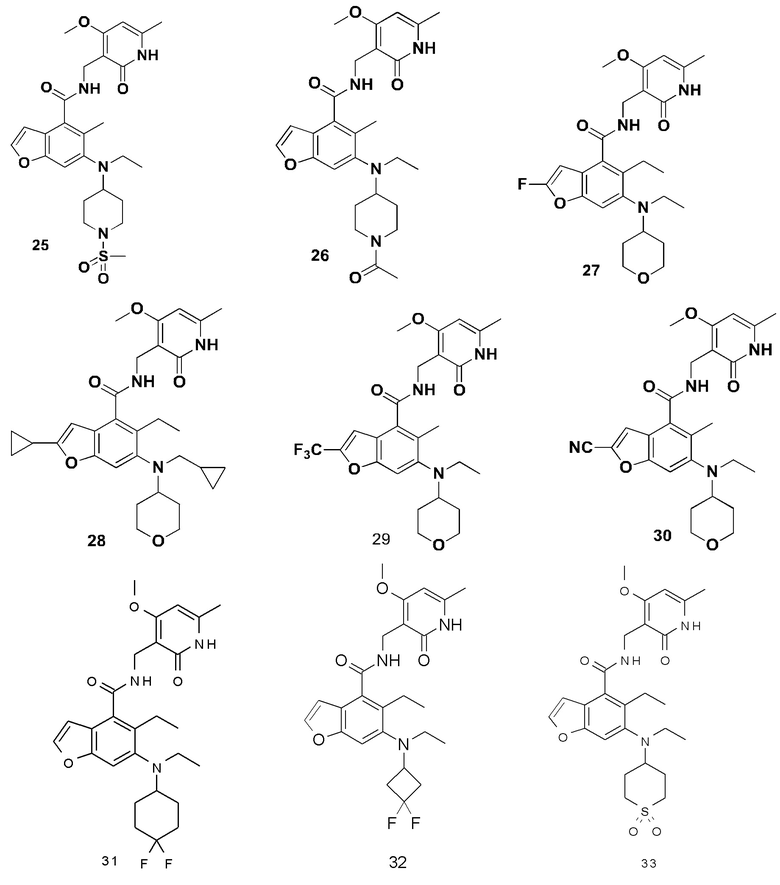
и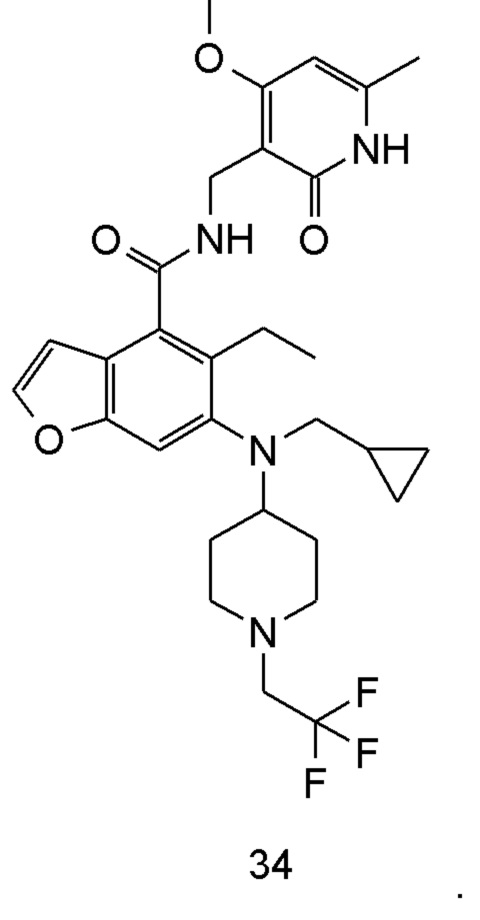
8. Соединение формулы (VI):
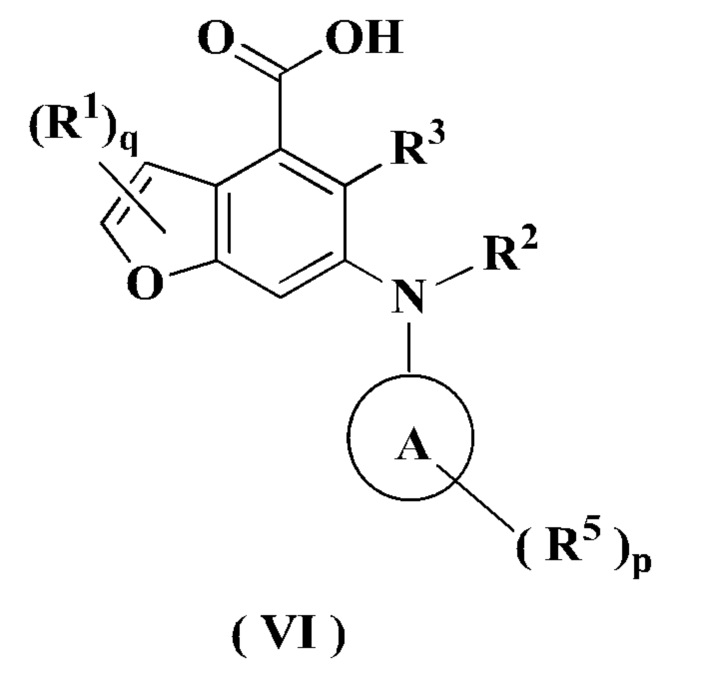
или его фармацевтически приемлемая соль, где:
кольцо А, радикалы с R1 по R3, R5, р и q являются такими, как определено в п. 1.
9. Способ получения соединения формулы (I) по п. 1, включающий стадию: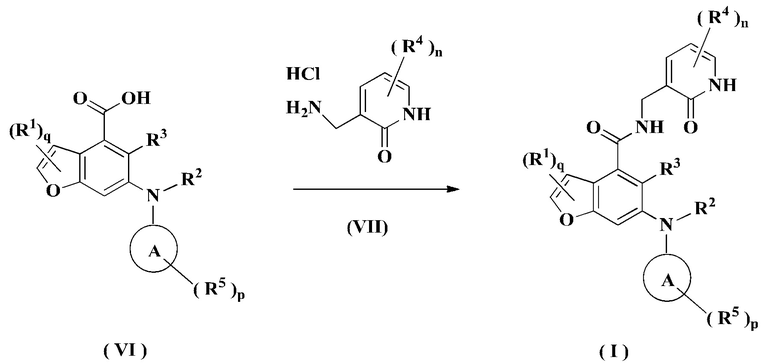
конденсации соединения формулы (VI) с соединением формулы (VII) при комнатной температуре с получением соединения формулы (I); где:
радикалы с R1 по R5, кольца А, р, q и n являются такими, как определено в п. 1.
10. Фармацевтическая композиция, обладающая ингибиторной активностью в отношении EZH2, содержащая терапевтически эффективное количество соединения формулы (I) по любому из пп. 1-7 и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
11. Применение соединения формулы (I) по любому из пп. 1-7 или фармацевтической композиции по п. 10 в качестве ингибитора EZH2 в получении лекарственного средства для профилактики и/или лечения опухоли и рака.
12. Применение по п. 11, где опухоль и рак выбраны из группы, состоящей из лимфомы, лейкоза, рака молочной железы, рака легкого, рака предстательной железы, рака яичника, рабдомиосаркомы, синовиальной саркомы, рака толстой кишки, рака поджелудочной железы, рака мочевого пузыря, опухоли яичника, глиомы, глиобластомы и миеломы; предпочтительно лимфомы, лейкоза, рака молочной железы, рака легкого, рака предстательной железы, рака яичника, рабдомиосаркомы и синовиальной саркомы;
где лейкоз предпочтительно представляет собой хронический миелоидный лейкоз, острый миелоидный лейкоз и лейкоз смешанного происхождения; где лимфома предпочтительно представляет собой неходжкинскую лимфому, диффузную крупноклеточную В-клеточную лимфому и фолликулярную лимфому.
13. Применение соединения формулы (I) по любому из пп. 1-7 или фармацевтической композиции по п. 10 в получении лекарственного средства для ингибирования EZH2.
| WO 2012118812 A2, 07.09.2012 | |||
| WO 2012142513 A1, 18.10.2012 | |||
| WO 2015141616 A1, 24.09.2015 | |||
| RU 2014117632 A, 10.11.2015. |

