ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящая заявка относится к замещенным океаном имидазопиразиновым и имидазотриазиновым ингибиторам тирозинкиназы Брутона (ВТК), включая мутантную ВТК, полезным в лечении заболеваний или расстройств, связанных с киназой ВТК. Эти соединения имеют потенциальное применение при лечении иммунных расстройств, рака, сердечно-сосудистых заболеваний, вирусных инфекций, воспалений, нарушений метаболизма/функций эндокринных желез и неврологических расстройств.
В частности, заявка относится к соединениям и их композициям, которые ингибируют ВТК, способам лечения заболеваний или нарушений, связанных с ВТК, и способам синтеза этих соединений.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Тирозинкиназа Брутона (ВТК), также известная как тирозин-протеинкиназа ВТК, является членом семейства Тес тирозинкиназ и играет важную роль в регуляции раннего развития В-клеток, активации и выживания зрелых В-клеток. (Hunter, Cell, 87, 50, 823-829). Фермент ВТК кодируется геном ВТК, и, как было показано, инициирует ряд клеточных процессов, включая клеточную пролиферацию, выживание, дифференцировку, подвижность, ангиогенез, продукцию цитокинов и презентацию антигена.
На моделях мышей с дефицитом ВТК было показано, что ВТК играет роль в аллергических заболеваниях, и/или аутоиммунных заболеваниях, и/или воспалительных заболеваниях; а ингибирование ВТК имеет потенциальную пользу при лечении таких заболеваний, как системная красная волчанка (СКВ), крапивница/синдром Шегрена, ревматоидный артрит, васкулит, идиопатическая тромбоцитопеническая пурпура (ИТП), тяжелая миастения, аллергический ринит и астма.
Роль ВТК в апоптозе также демонстрирует пользу ингибирования активности ВТК для лечения рака, например, В-клеточной лимфомы, лейкоза и других гематологических злокачественных новообразований. Кроме того, ВТК играет роль в функции остеокластов, поэтому ингибирование активности ВТК обладает потенциальной пользой при лечении заболеваний костей, таких как остеопороз.
Одобренные соединения, которые ингибируют ВТК, включают ибрутиниб (В-клеточные неоплазии, например мантийноклеточная лимфома, хронический лимфоцитарный лейкоз (ХЛЛ), макроглобулинемия Вальденстрема); акалабрутиниб (мантийноклеточная лимфома и ХЛЛ); и занубрутиниб (мантийноклеточная лимфома). Кроме того, клинические испытания проходят несколько ингибиторов ВТК, включая эвобрутиниб (рассеянный склероз); ABBV-105 (системная красная волчанка (СКВ)); ONO-4059/GS-4059 (неходжкинская лимфома и ХЛЛ); спебрутиниб (spebrutinib) (рецидивирующая или рефрактерная В-клеточная неходжкинская лимфома, ХЛЛ и макроглобулинемия Вальденстрема); и НМ71224 (аутоиммунные заболевания).
Несмотря на значительные терапевтические достижения в лечении В-клеточных неоплазий с использованием ингибиторов ВТК, возникали случаи первичной и вторичной резистентности с неблагоприятными исходами и ограниченными возможностями лечения.
Ковалентные (необратимые) ингибиторы ВТК, такие как ибрутиниб и акалабрутиниб, связываются с сайтом С481 ВТК, делая его неактивным в отношении киназы. Это связывание является постоянным до тех пор, пока белок ВТК не распадется. Преимущество этих необратимых ингибиторов состоит в том, что они являются сильнодействующими и обычно эффективны только в течение короткого периода времени. Однако их клиническая польза ограничена нецелевой токсичностью, что приводит к высокой частоте прекращения лечения и приобретенной резистентности из-за мутаций ВТК С481, которые нарушают ковалентное связывание с ВТК, снижая сродство соединений к связыванию и уменьшая их способность ингибировать ферментативную активность ВТК (Leukaemia 2015, Apr; 29(4):895-900). Большинство (более 50%) пациентов с ХЛЛ, у которых отмечается прогресс при лечении ковалентными ингибиторами ВТК, становятся резистентными к лечению из-за развития мутации C481S (N. Engl. J. Med. 370; 24, 2014; JAMA Oncol. 2015; 1(1):80-87 и J. Clin. Oncol. 35:1437-1443, 2017).
Первичная лимфома центральной нервной системы (ПЛЦНС) представляет собой заболевание, при котором злокачественные (раковые) клетки образуются в лимфатической ткани головного и/или спинного мозга, и на ее долю приходится приблизительно 1% всех лимфом и от 2% до 5% всех первичных опухолей головного мозга. Подавляющее большинство (приблизительно 95%) ПЛЦНС представляют собой диффузные крупноклеточные В-клеточные лимфомы (ДККЛ). Мутации в генах CD79B и MYD88 часто (приблизительно на 30-80%) совпадают с ПЛЦНС (Neuropathol. Appl. Neurobiol. 2016 Apr; 42(3):279-90). Хотя на сегодняшний день ингибиторы ВТК не были одобрены для лечения ДККЛ, данные свидетельствуют о том, что ДККЛ с мутациями CD79B и MYD88 более чувствительны к ингибированию ВТК (Nat. Med. 2015 Aug; 21(8):922-6).
Вторичная лимфома ЦНС (ВЛЦНС) относится к распространению в центральную нервную систему лимфомы, возникшей в другом месте (в отличие от первичной лимфомы ЦНС). Как правило, она представляет собой неходжкинскую лимфому и может представлять собой изолированный рецидив или может быть частью системного заболевания во время проявления. В отличие от первичной лимфомы ЦНС, она чаще поражает лептоменинкс.
PRN2246 (SAR442168), проникающий через гематоэнцефалический барьер (ГЭБ) ковалентный ингибитор ВТК, хорошо переносился в I фазе испытаний рассеянного склероза (PC). Кроме того, в некоторых испытаниях (Grommes С, et al., Cancer Discov. 2017 Sep;7(9): 1018-1029; Grommes C, et al., Blood. 2019; 133(5):436-445; Lionakis et al. al., 2017, Cancer Cell 31, 833-843 и Soussain С et al., Eur J Cancer. 2019 Aug; 117:121-130) предполагалось, что высокая доза ибрутиниба (840 мг) будет эффективна при лимфомах ЦНС (как ПЛЦНС, так и вторичной лимфомы нервной системы (ВЛЦНС)), но на сегодняшний день не одобрены к применению никакие ингибиторы ВТК, нацеленные на мутацию ВТК-С481 или обладающие активностью при ПЛЦНС.Оба они остаются неудовлетворенной медицинской потребностью.
В WO 2009/143051 раскрыты некоторые замещенные имидазопиразины и имидазотриазины, включая некоторые замещенные гексаном имидазопиразины и имидазотриазины, в качестве ингибиторов активированной p21cdc42Hs-ассоциированной киназы (АСК1). Однако соединения по WO 2009/143051 демонстрируют высокий клиренс гепатоцитов человека, что означает, что эти соединения потенциально не могут обеспечить достаточное устойчивое действие лекарственного средства даже при максимальной абсорбируемой дозе (что делает соединения неэффективными); и/или необходимы очень высокие дозы для ингибирования мишени, что приводит к высокой максимальной концентрации лекарственного средства (потенциально приводя к вторичным фармакологическим (то есть неблагоприятным) эффектам и проблемам с токсичностью).
В WO 2017111787А1 раскрыт тетрагидропираниламино-пирролопиримидинон, который модулирует активность ВТК; в WO 2018039310А1 раскрыты амино-пирролопиримидиноновые соединения и способы их применения; в WO 2017103611А1 раскрыты соединения, полезные в качестве ингибиторов ВТК; и в WO 2011152351 раскрыты производные пуринона, обладающие селективной ингибирующей активностью в отношении ВТК. Однако ни одно из этих соединений не обладает сочетанием желаемых свойств, которыми обладают соединения по настоящему изобретению.
Здесь раскрыты некоторые новые замещенные океанами имидазопиразины и имидазотриазины, которые являются сильнодействующими селективными ингибиторами ВТК, как ВТК дикого типа, так и ВТК с мутацией С481 (например мутацией C481S, C481Y, C481R или C481F). Эти соединения являются нековалентными обратимыми ингибиторами, демонстрируют низкий клиренс гепатоцитов человека и проникают через гематоэнцефалический барьер (ГЭБ).
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Здесь раскрыты соединения формулы (I):
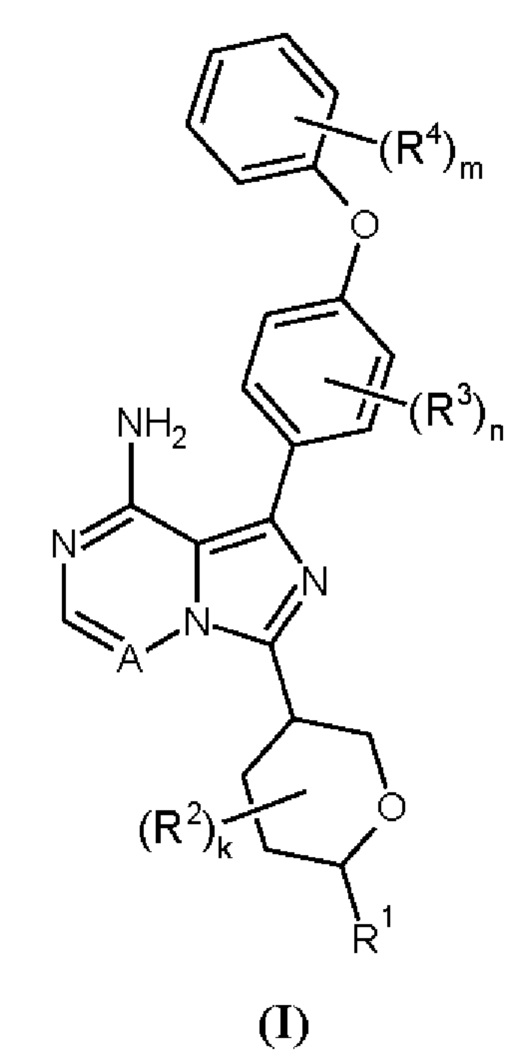
и их фармацевтически приемлемые соли и их применение в качестве ингибиторов ВТК, в частности в терапии.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Многие воплощения изобретения подробно описаны в описании и будут очевидны для специалиста в данной области техники. Изобретение не следует интерпретировать как ограниченное каким-либо из приведенных воплощений, а пункты формулы изобретения являются воплощениями. Следует понимать, что некоторые признаки настоящего изобретения, которые для ясности описаны в контексте отдельных воплощений, также могут быть представлены в комбинации в одном воплощении. И наоборот, различные признаки настоящего изобретения, которые для краткости описаны в контексте одного воплощения, также могут быть представлены по отдельности или в любой подходящей подкомбинации.
Здесь раскрыто соединение формулы (I):
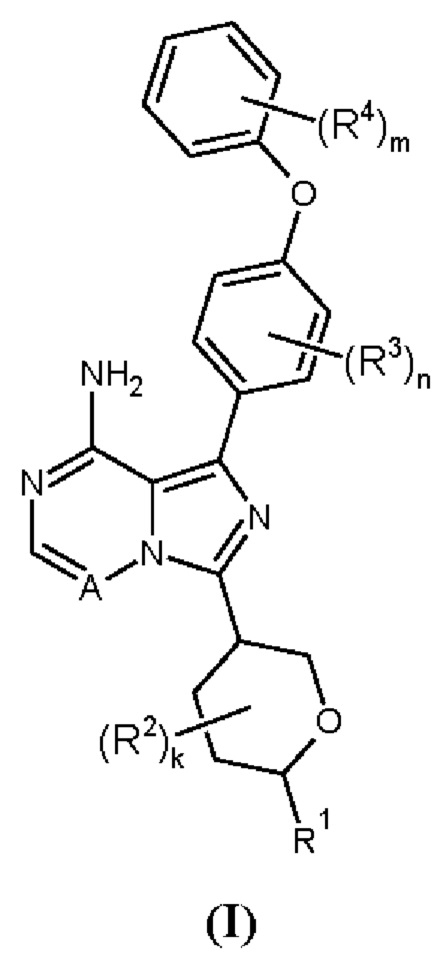
где:
R1 выбран из водорода, С1-6алкила, C1-6алкокси, N-С1-6алкиламино, N,N-(C1-6алкил)2амино, карбоциклила и гетероциклила; где R1 может быть возможно замещен одним или более чем одним R5;
R2 выбран из галогена, С1-3алкила, C1-3алкокси, карбоциклила и гетероциклила; или два R2, либо при одном атоме, либо при соседних атомах, могут вместе с атомами, к которым они присоединены, образовывать 3-7-членное кольцо;
k равен 0-4;
R3 выбран из галогена, С1-3алкила и С1-3алкокси;
n равен 0-4;
R4 выбран из галогена, С1-3алкила и С1-3алкокси;
m равен 0-5;
А представляет собой =N- или =C(R6)-;
R5 выбран из галогена, гидрокси, С1-6алкокси, амино, N-С1-6алкиламино, N,N-(C1-6алкил)2амино, карбоциклила и гетероциклила; где R5 может быть независимо возможно замещен одним или более чем одним R7;
R6 выбран из водорода и галогена;
R7 выбран из галогена, гидрокси, амино, С1-3алкила и С1-3алкокси;
или его фармацевтически приемлемая соль.
В одном воплощении R1 выбран из водорода и С1-6алкила; где R1 может быть возможно замещен одним R5; где R5 выбран из гидрокси, С1-6алкокси, N,N-(С1-6алкил)2амино и гетероциклила.
В одном воплощении R1 выбран из водорода и С1-3алкила; где R1 может быть возможно замещен одним R5; где R5 выбран из гидрокси, С1-3алкокси, N,N-(C1-2алкил)2амино и азетидинила.
В одном воплощении R1 выбран из водорода, метила, гидроксиметила, метоксиметила, N,N-диметиламинометила и азетидин-1-илметила.
В одном воплощении R1 представляет собой гидроксиметил.
В одном воплощении R2 выбран из галогена или С1-3алкокси.
В одном воплощении R2 выбран из фтора или метокси.
В одном воплощении или два R2, либо при одном атоме, либо при соседних атомах, могут вместе с атомами, к которым они присоединены, образовывать 3-7-членное кольцо.
В одном воплощении или два R2 при одном атоме могут вместе с атомом, к которому они присоединены, образовывать 3-7-членное кольцо.
В одном воплощении или два R2 при соседних атомах, могут вместе с атомами, к которым они присоединены, образовывать 3-7-членное кольцо.
В одном воплощении k равен 0.
В одном воплощении k равен 1.
В одном воплощении k равен 2.
В одном воплощении k равен 3.
В одном воплощении k равен 4.
В одном воплощении R3 представляет собой галоген.
В одном воплощении R3 представляет собой фтор.
В одном воплощении n равен 0-2.
В одном воплощении n равен 0.
В одном воплощении n равен 1.
В одном воплощении n равен 2.
В одном воплощении n равен 3.
В одном воплощении n равен 4.
В одном воплощении R4 представляет собой галоген.
В одном воплощении R4 представляет собой фтор.
В одном воплощении m равен 0-2.
В одном воплощении m равен 0.
В одном воплощении m равен 1.
В одном воплощении m равен 2.
В одном воплощении m равен 3.
В одном воплощении m равен 4.
В одном воплощении m равен 5.
В одном воплощении А представляет собой =N- или=С(Н)-.
В одном воплощении А представляет собой =N-.
В одном воплощении А представляет собой =C(R6)-.
В одном воплощении А представляет собой =С(Н)-.
Соединение формулы (I) (где R1 не является водородом) содержит два хиральных центра (отмечены “*”):
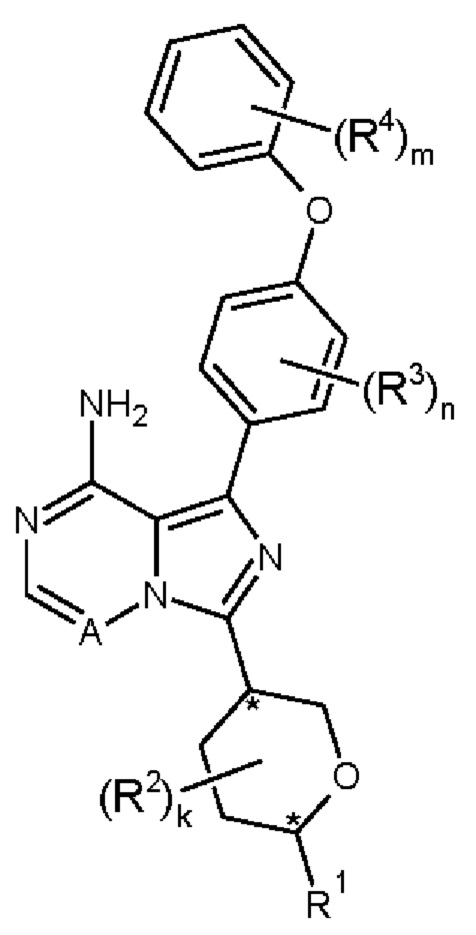
Эти хиральные центры могут существовать в «транс»-конфигурации (это означает, что два заместителя в оксановом кольце расположены на противоположных сторонах оксанового кольца); и «цис»-конфигурация (что означает, что два заместителя в оксановом кольце расположены по одну и ту же сторону оксанового кольца). Структуры (IA) и (IB) ниже представляют цис-изомеры соединений формулы (I), структуры (IC) и (ID) ниже представляют транс-изомеры соединений формулы (I).
В одном аспекте изобретения соединение формулы (I) представляет собой транс-соединение формулы (I).
В одном аспекте изобретения соединение формулы (I) представляет собой цис-соединение формулы (I).
В одном аспекте изобретения соединение формулы (I) представляет собой соединение формулы (IA):
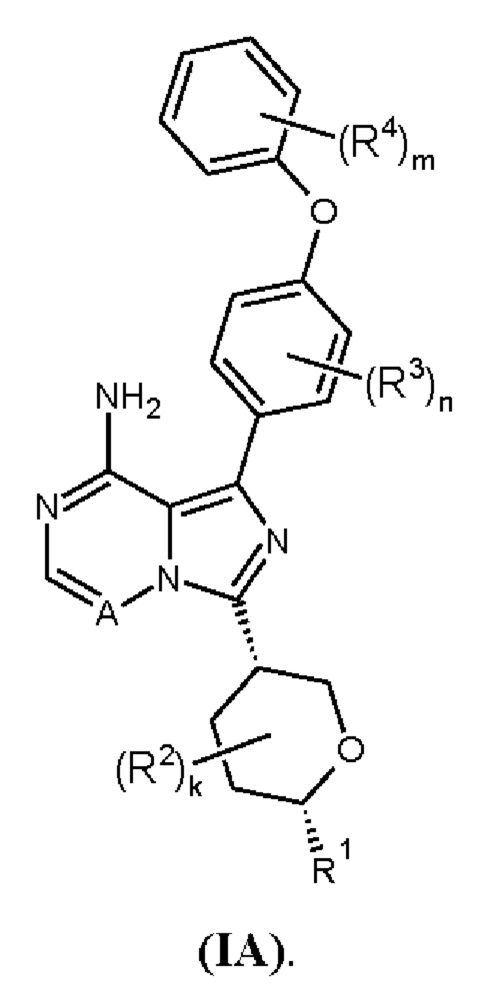
В одном аспекте изобретения соединение формулы (I) представляет собой соединение формулы (IB):
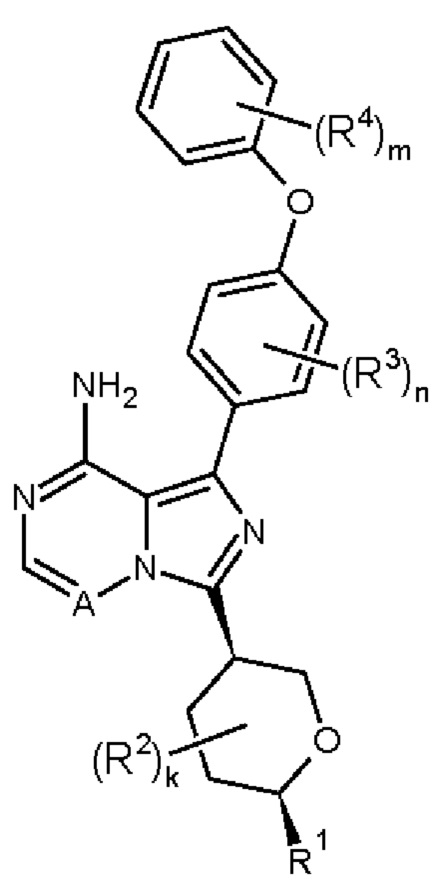
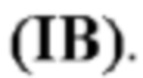
В одном аспекте изобретения соединение формулы (I) представляет собой соединение формулы (IC):
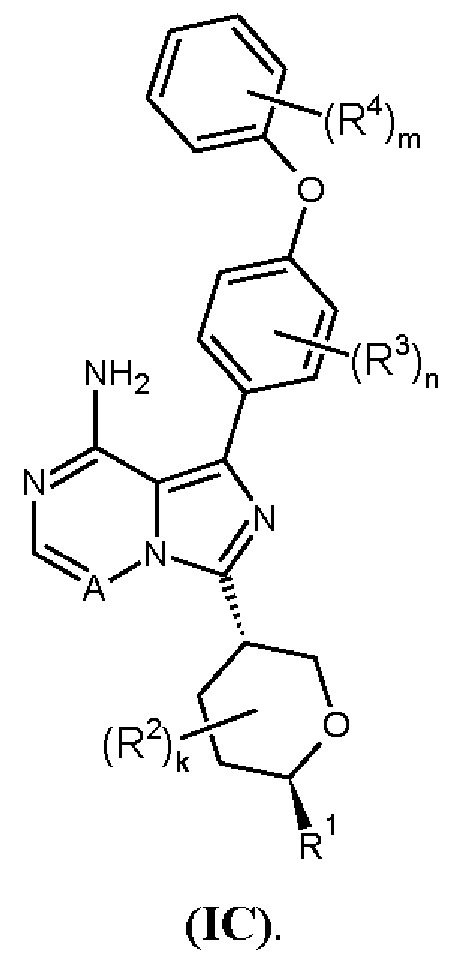
В одном аспекте изобретения соединение формулы (I) представляет собой соединение формулы (ID):
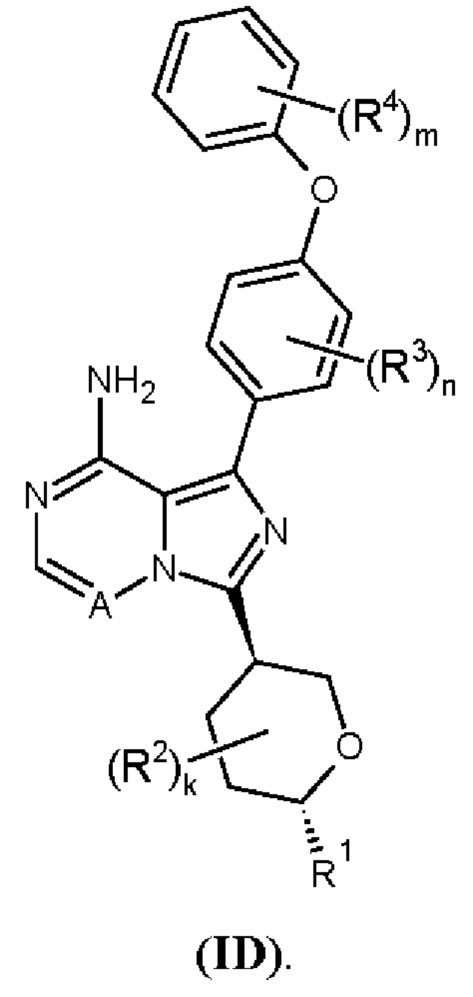
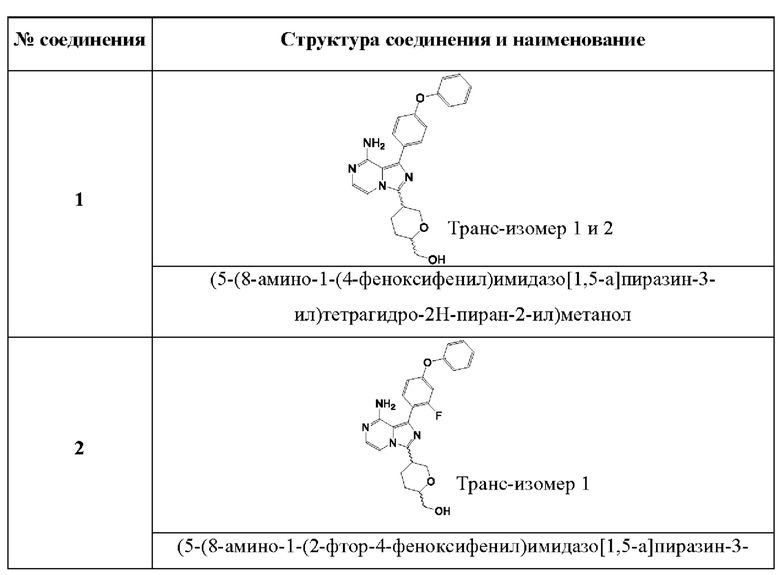
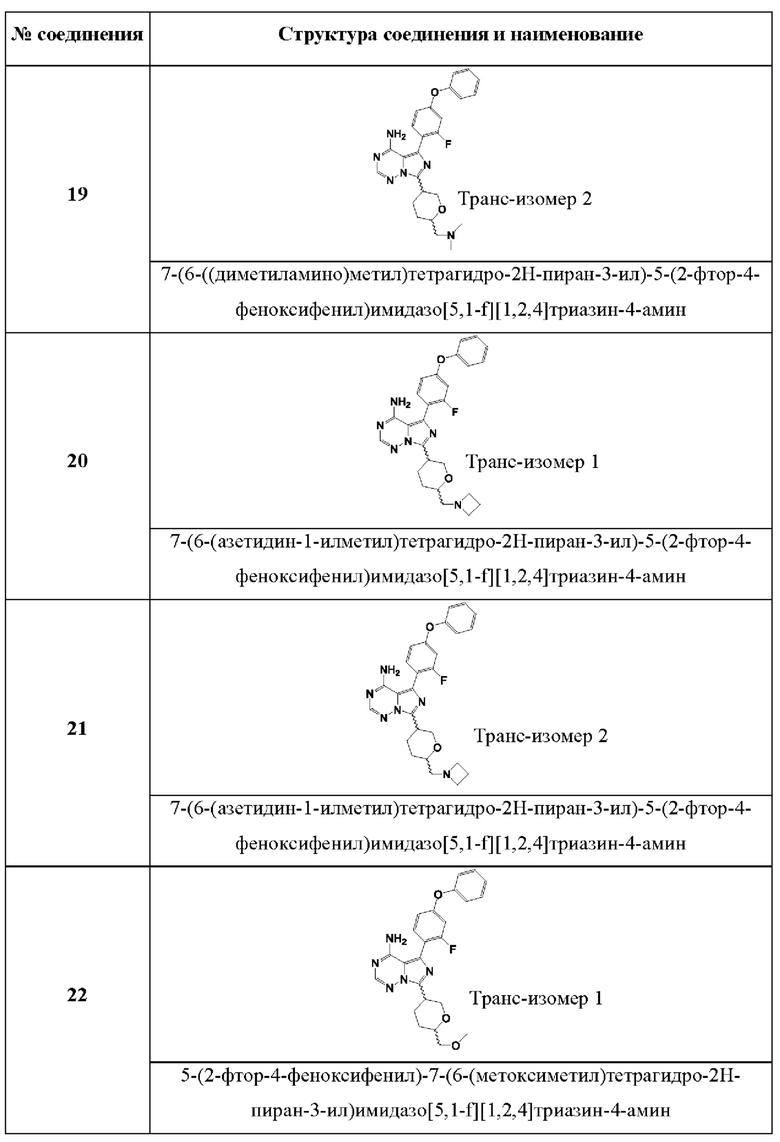
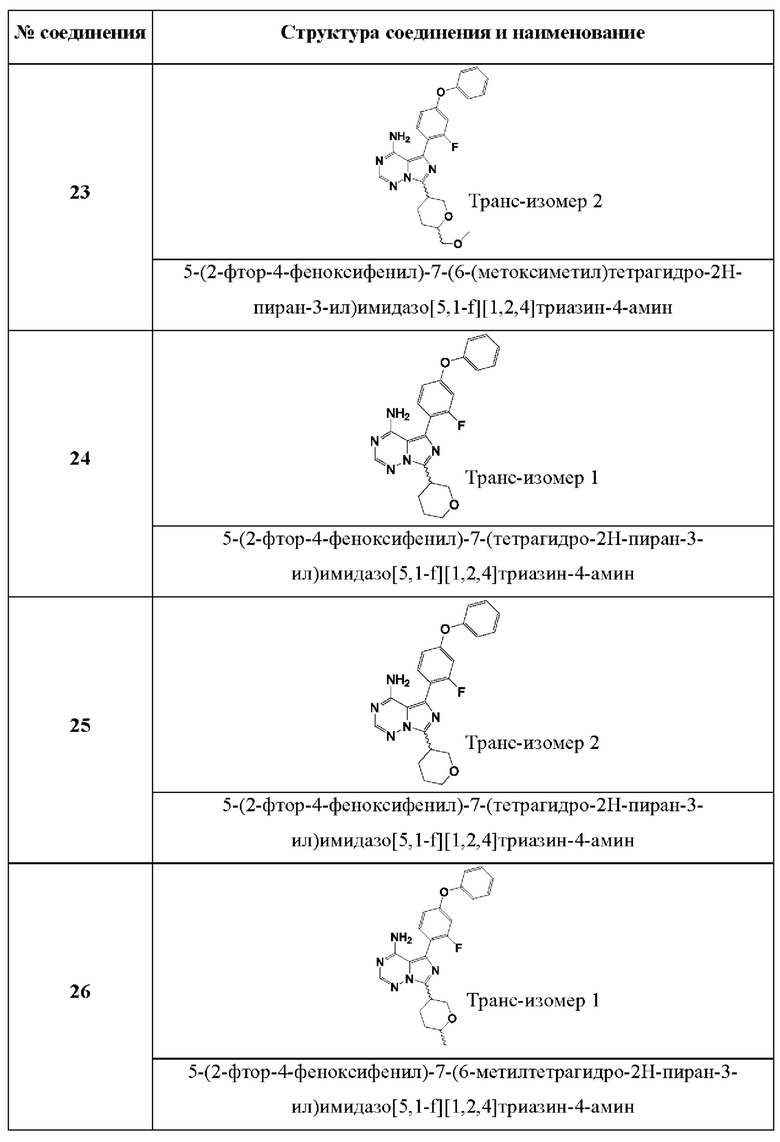
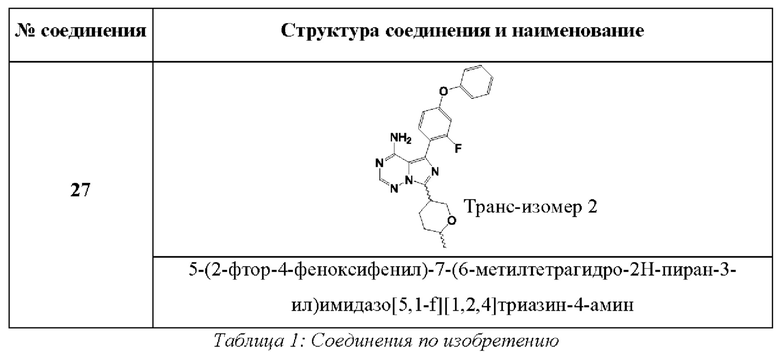
В одном аспекте изобретения соединение формулы (I) выбрано из
(5-(8-амино-1-(4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(8-амино-1-(4-(2,3-дифторфенокси)фенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
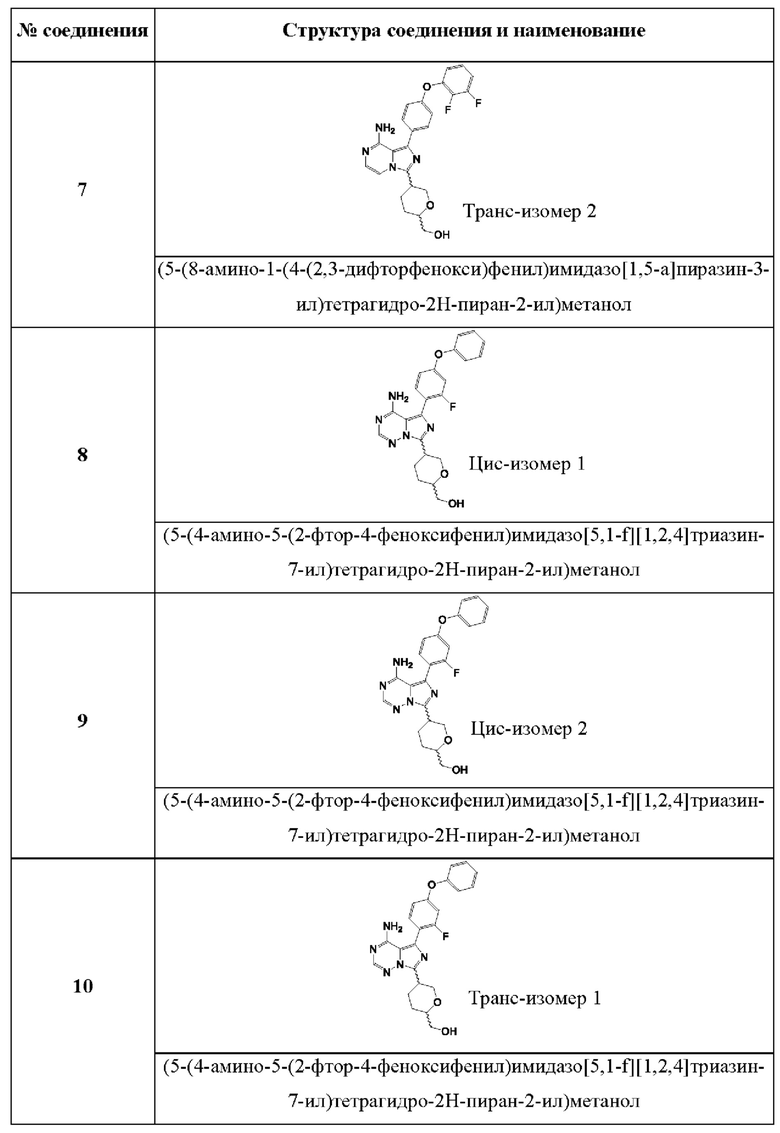
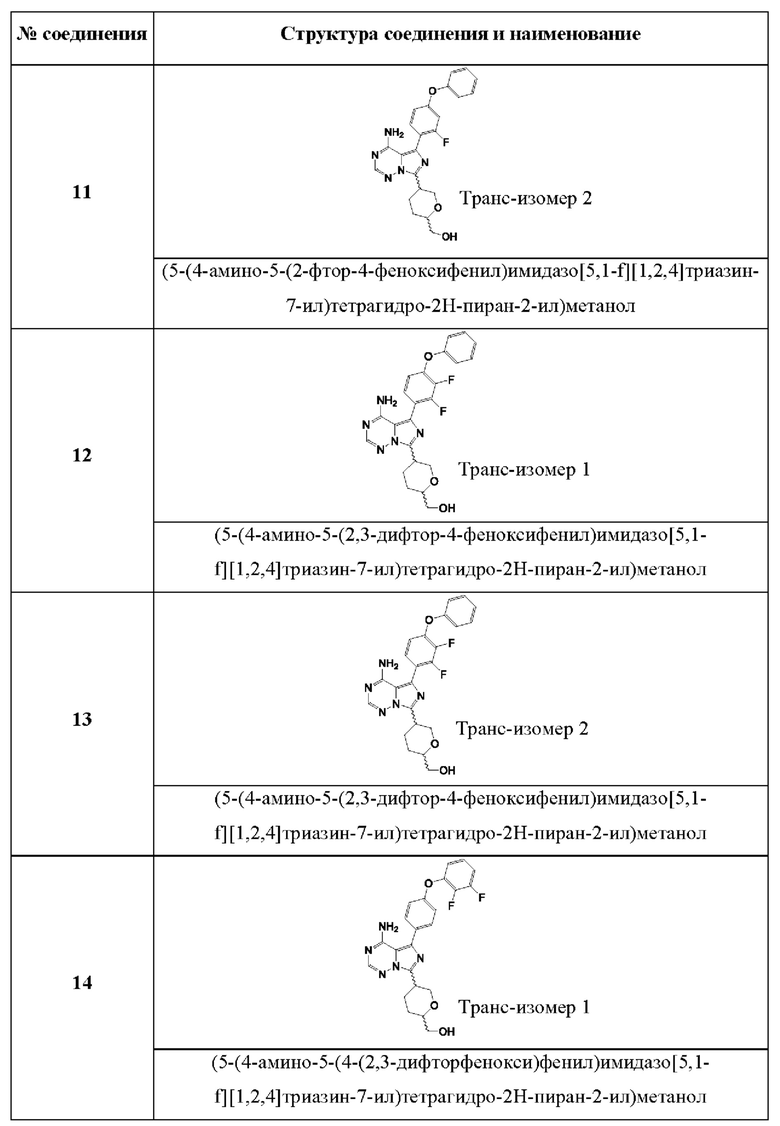
(5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(4-амино-5-(4-(2,3-Дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
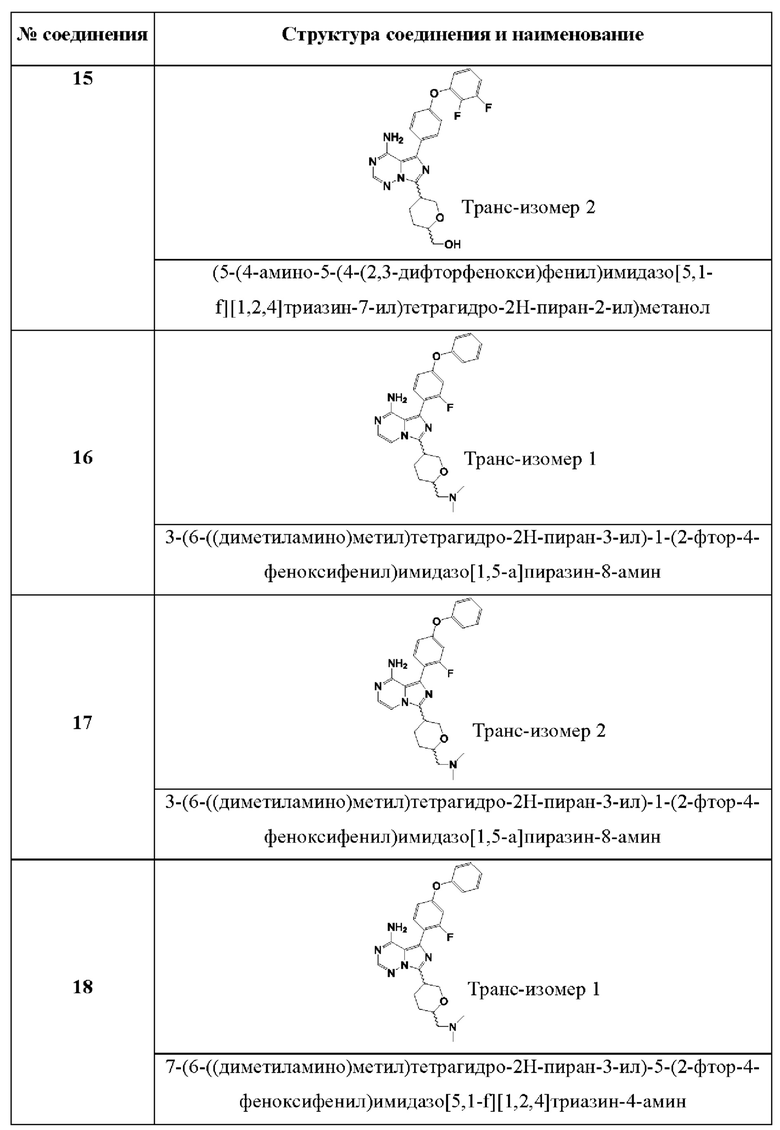
3-(6-((диметиламино)метил)тетрагидро-2Н-пиран-3-ил)-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-8-амина;
7-(6-((диметиламино)метил)тетрагидро-2Н-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-4-амина;
7-(6-(азетидин-1-илметил)тетрагидро-2Н-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[51,-f][1,2,4]триазин-4-амина;
5-(2-фтор-4-феноксифенил)-7-(6-(метоксиметил)тетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина;
5-(2-фтор-4-феноксифенил)-7-(тетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4- амина; и
5-(2-фтор-4-феноксифенил)-7-(6-метилтетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина.
В одном аспекте изобретения соединение формулы (I) выбрано из
(5-(8-амино-1-(4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(8-амино-1-(4-(2,3-дифторфенокси)фенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
(5-(4-амино-5-(4-(2,3-дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
3-(6-((диметиламино)метил)тетрагидро-2Н-пиран-3-ил)-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-8-амина;
7-(6-((диметиламино)метил)тетрагидро-2Н-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-4-амина;
7-(6-(азетидин-1-илметил)тетрагидро-2Н-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-4-амина;
5-(2-фтор-4-феноксифенил)-7-(6-(метоксиметил)тетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина;
5-(2-фтор-4-феноксифенил)-7-(тетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина; и
5-(2-фтор-4-феноксифенил)-7-(6-метилтетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина;
или его фармацевтически приемлемой соли.
В одном аспекте изобретения соединение формулы (I) выбрано из
((2R,5R)-5-(8-амино-1-(4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5R)-5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5R)-5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5R)-5-(8-амино-1-(4-(2,3-дифторфенокси)фенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5R)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5R)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5R)-5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5R)-5-(4-амино-5-(4-(2,3-Дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
3-((3R,6R)-6-((диметиламино)метил)тетрагидро-2Н-пиран-3-ил)-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-8-амина;
7-((3R,6R)-6-((диметиламино)метил)тетрагидро-2H-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-4-амина;
7-((3R,6R)-6-(азетидин-l-илметил)тетрагидро-2H-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f],[1,2,4]триазин-4-амина;
5-(2-фтор-4-феноксифенил)-7-((3R,6R)-6-(метоксиметил)тетрагидро-2H-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина;
(R)-5-(2-фтор-4-феноксифенил)-7-(тетрагидро-2H-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина; и
5-(2-фтор-4-феноксифенил)-7-((3R,6R)-6-метилтетрагидро-2H-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина;
или его фармацевтически приемлемой соли.
В одном аспекте изобретения соединение формулы (I) выбрано из
((2S,5S)-5-(8-амино-1-(4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5S)-5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5S)-5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5S)-5-(8-амино-1-(4-(2,3-Дифторфенокси)фенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5S)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5S)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5S)-5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5S)-5-(4-амино-5-(4-(2,3-дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
3-((3S,6S)-6-((диметиламино)метил)тетрагидро-2Н-пиран-3-ил)-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-8-амина;
7-((3S,6S)-6-((диметиламино)метил)тетрагидро-2Н-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-4-амина;
7-((3S,6S)-6-(азетидин-1-илметил)тетрагидро-2Н-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-4-амина;
5-(2-фтор-4-феноксифенил)-7-((3S,6S)-6-(метоксиметил)тетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина;
(S)-5-(2-фтор-4-феноксифенил)-7-(тетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина; и
5-(2-фтор-4-феноксифенил)-7-((3S,6S)-6-метилтетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина;
или его фармацевтически приемлемой соли.
В одном аспекте изобретения соединение формулы (I) выбрано из ((2S,5R)-5-(8-амино-1-(4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5R)-5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5R)-5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5R)-5-(8-амино-1-(4-(2,3-дифторфенокси)фенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5R)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5R)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5R)-5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2S,5R)-5-(4-амино-5-(4-(2,3-Дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
3-((3S,6R)-6-((диметиламино)метил)тетрагидро-2Н-пиран-3-ил)-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-8-амина;
7-((3S,6R)-6-((диметиламино)метил)тетрагидро-2Н-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-4-амина;
7-((3S,6R)-6-(азетидин-1-илметил)тетрагидро-2Н-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f],[1,2,4]триазин-4-амина;
5-(2-фтор-4-феноксифенил)-7-((3S,6R)-6-(метоксиметил)тетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина; и
5-(2-фтор-4-феноксифенил)-7-((3S,6R)-6-метилтетрагидро-2Н-пиран-3-ил)имидазо[5,1-f] [1,2,4]триазин-4-амина;
или его фармацевтически приемлемой соли.
В одном аспекте изобретения соединение формулы (I) выбрано из ((2R,5S)-5-(8-амино-1-(4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5S)-5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5S)-5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5S)-5-(8-амино-1-(4-(2,3-Дифторфенокси)фенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5S)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5S)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5S)-5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
((2R,5S)-5-(4-амино-5-(4-(2,3-дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанола;
3-((3R,6S)-6-((диметиламино)метил)тетрагидро-2H-пиран-3-ил)-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-8-амина;
7-((3R,6S)-6-((диметиламино)метил)тетрагидро-2H-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-4-амина;
7-((3R,6S)-6-(азетидин-1-илметил)тетрагидро-2Н-пиран-3-ил)-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-4-амина;
5-(2-фтор-4-феноксифенил)-7-((3R,6S)-6-(метоксиметил)тетрагидро-2H-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина; и
5-(2-фтор-4-феноксифенил)-7-((3R,6S)-6-метилтетрагидро-2Н-пиран-3-ил)имидазо[5,1-f][1,2,4]триазин-4-амина;
или его фармацевтически приемлемой соли.
В одном аспекте изобретения предложено любое соединение формулы (I), раскрытое здесь.
В одном аспекте изобретения предложено любое соединение формулы (I), раскрытое здесь, или его фармацевтически приемлемая соль.
В одном аспекте изобретения предложено синтетическое промежуточное соединение, используемое для получения соединения формулы (I), как раскрыто здесь.
В одном аспекте изобретения предложено синтетическое промежуточное соединение, используемое для получения соединения формулы (I), как раскрыто здесь, или его фармацевтически приемлемая соль.
В различных частях настоящего изобретения описаны связывающие заместители. В случаях, когда структура явно требует наличия связывающей группы, тогда подразумевают, что переменные Маркуша, перечисленные для этой группы, являются связывающими группами. Например, если для структуры требуется связывающая группа и в определении группы Маркуша для этой переменной перечислен «алкил», то подразумевают, что «алкил» представляет собой связывающую алкиленовую группу.
Использованный здесь термин «замещенный», когда он относится к химической группе, означает, что химическая группа имеет один или более чем один атом водорода, который удален и заменен заместителями. Использованный здесь термин «заместитель» имеет обычное значение, известное в данной области техники, и относится к химической группировке, которая ковалентно присоединена или, если целесообразно, конденсирована с исходной группой. Использованный здесь термин «возможно замещенный» или «возможно… замещенный» означает, что химическая группа может не иметь заместителей (то есть быть незамещенной) или может иметь один или более чем один заместитель (то есть быть замещенной). Следует понимать, что замещение по данному атому ограничено валентностью.
Использованный здесь термин «Ci-j» обозначает диапазон количества атомов углерода, где i и j являются целыми числами, а диапазон количества атомов углерода включает конечные числа (то есть i и j) и каждое целое число между ними, и где j больше i. Например С1-6 обозначает диапазон от одного до шести атомов углерода, включая один атом углерода, два атома углерода, три атома углерода, четыре атома углерода, пять атомов углерода и шесть атомов углерода. В некоторых воплощениях термин «C1-6» обозначает от 1 до 6, в частности от 1 до 5, в частности от 1 до 4, в частности от 1 до 3 или в частности от 1 до 2 атомов углерода.
Использованный здесь термин «алкил», независимо от того, используется ли он как часть другого термина или сам по себе, относится к насыщенной углеводородной цепи. Упомянутая выше углеводородная цепь может быть линейной или разветвленной. Термин «Ci-jалкил» относится к алкилу, имеющему от i до j атомов углерода. Примеры С1-6алкила включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил; высшие гомологи, такие как 2-метил-1-бутил, н-пентил, 3-пентил, н-гексил, 1,2,2-триметилпропил и тому подобные, но не ограничиваются ими. Примеры группы «С1-3алкил» включают метил, этил, пропил и изопропил.
Использованный здесь термин «галоген» относится к атому, выбранному из фтора, хлора, брома и иода.
Использованный здесь термин «алкокси», независимо от того, используется ли он как часть другого термина или сам по себе, относится к группе формулы -О-алкил. Термин «Ci-jалкокси» означает, что алкильная группировка алкоксигруппы имеет от i до j атомов углерода. Примеры алкоксигрупп включают метокси, этокси, пропокси (например н-пропокси и изопропокси), трет-бутокси и тому подобные, но не ограничиваются ими. Примеры группы «О-валкоксил» представляют собой метокси, этокси и пропокси. Примеры группы «С1-3алкоксил» представляют собой метокси, этокси и пропокси.
Примеры группы "N-(С1-6алкил)амино" представляют собой метиламино и этиламино. Примеры группы "N,N-(С1-6алкил)2амино" представляют собой N,N-диметиламино, N,N-диэтиламино и N-этил-N-метиламино.
Использованный здесь термин «карбоциклил», независимо от того, используется ли он как часть другого термина или сам по себе, относится к насыщенному моноциклическому кольцу, в котором все атомы кольца представляют собой углерод и которое содержит по меньшей мере три атома углерода, образующие кольцо. В некоторых воплощениях карбоциклил может содержать от 3 до 7 атомов углерода, образующих кольцо, или от 3 до 6 атомов углерода, образующих кольцо. В некоторых воплощениях -CH2- группа кольца может быть заменена -С(О)- группой кольца. Примеры карбоциклильных групп включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, но не ограничиваются ими.
Использованный здесь термин «гетероциклил» относится к моноциклической, насыщенной карбоциклильной группе, где один или более чем один (например 1, 2 или 3) кольцевой атом заменен гетероатомами, которые включают кислород, серу, азот, фосфор и тому подобные, но не ограничиваются ими. В некоторых воплощениях -CH2-группа кольца может быть заменена -С(О)- группой кольца. В некоторых воплощениях кольцевой атом серы может быть возможно окислен с образованием S-оксидов. В некоторых воплощениях гетероциклил присоединен через углерод. В некоторых воплощениях гетероциклил присоединен через азот. Примеры гетероциклильных групп включают азетидинил, пиперидил, пирролидинил, тетрагидрофурил, пиперидинил, пиперазинил, морфолинил и тому подобные, но не ограничиваются ими.
В одном воплощении два R2 при одном атоме вместе с атомом, к которому они присоединены, образуют 3-7-членное кольцо. Образующиеся в результате «спирокольца» имеют два кольца (одно из которых представляет собой океан формулы (I)), соединенных через один единственный общий атом. Неоксановое кольцо может представлять собой 3-7-членное карбоциклильное кольцо или 3-7-членное гетероциклическое кольцо. Примеры двух R2 при одном атоме, вместе с атомом, к которому они присоединены, образующие 3-7-членное кольцо (показаны с океаном формулы (I)), включают:
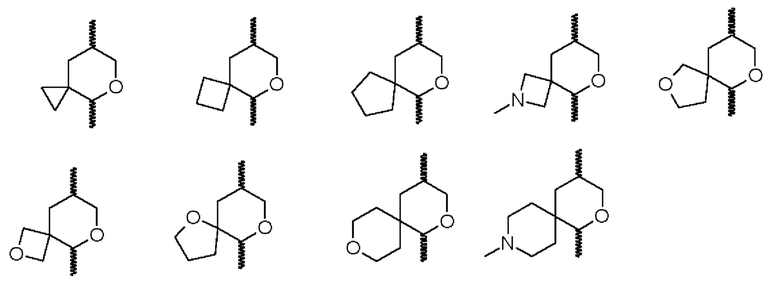
(где  показывает присоединение к остальной части молекулы).
показывает присоединение к остальной части молекулы).
В одном воплощении два R2 при соседних атомах вместе с атомами, к которым они присоединены, образуют 3-7-членное кольцо. Образующиеся в результате «конденсированные кольца» имеют два кольца (одно из которых представляет собой океан формулы (I)), делящие два соседних атома. Неоксановое кольцо может представлять собой 3-7-членное карбоциклильное кольцо или 3-7-членное гетероциклическое кольцо. Примеры двух R2 при соседних атомах, вместе с атомами, к которым они присоединены, образующие 3-7-членное кольцо (показаны с океаном формулы (I)), включают:
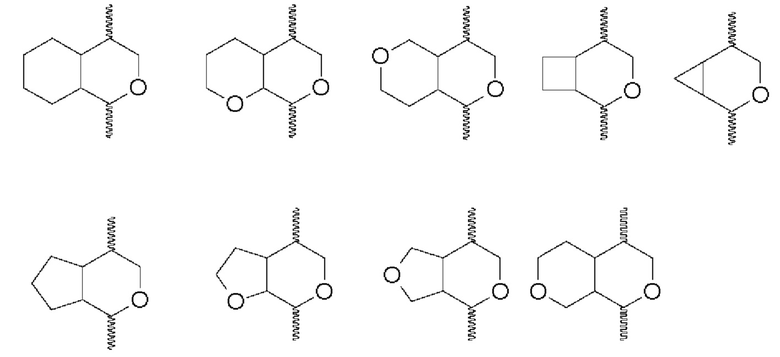
(где  показывает присоединение к остальной части молекулы).
показывает присоединение к остальной части молекулы).
Подразумевается, что «соединение» по настоящему изобретению включает все стереоизомеры, геометрические изомеры и таутомеры предстаавленных структур, если не оговорено особо.
Термин «стереоизомер» относится к любой из различных стереоизомерных конфигураций (например к энантиомерам, диастереомерам и рацематам) асимметричного соединения (например к тем, которые имеют один или более чем один асимметрично замещенный атом углерода или «асимметрический центр»). Соединения по настоящему изобретению, которые содержат асимметрические центры, могут быть выделены в оптически активных (энантиомеры или диастереомеры) или оптически неактивных (рацемических) формах. Термин «энантиомер» включает пары стереоизомеров, которые являются не налагающимися зеркальными изображениями друг друга. Смесь пары энантиомеров в соотношении 1:1 представляет собой «рацемическую смесь». Термины «диастереомеры» или «диастереоизомеры» включают стереоизомеры, которые имеют по меньшей мере два асимметрических атома, но не являются зеркальными отражениями друг друга. Некоторые соединения, содержащие один или более чем один асимметрический центр, могут давать энантиомеры, диастереомеры или другие стереоизомерные формы, которые могут быть определены на основании абсолютной конфигурации как (R)- или (S)- при каждом асимметрическом центре согласно R-S системе Кана-Ингольда-Прелога. Разделенные соединения, абсолютная конфигурация которых неизвестна, могут быть обозначены с использованием термина «или» по асимметрическому центру. Способы получения оптически активных форм из рацемических смесей, такие как разделение посредством ВЭЖХ (высокоэффективная жидкостная хроматография) или стереоселективный синтез, известны в данной области техники.
Термины «геометрические изомеры» или «цис- и транс-изомеры» относятся к соединениям с одинаковой формулой, но их функциональные группы повернуты в другую ориентацию в трехмерном пространстве.
Термин «таутомеры» включает прототропные таутомеры, которые представляют собой изомерные состояния протонирования соединений, имеющих ту же формулу и общий заряд. Примеры прототропных таутомеров включают пары кетон-енол, пары амид-имидовая кислота, пары лактам-лактим, пары енамин-имин и кольцевые формы, в которых протон может занимать два или более положений гетероциклической системы, например 1H- и 3Н-имидазол, 1Н-, 2Н- и 4Н- 1,2,4-триазол, 1Н- и 2Н-изоиндол и 1Н- и 2Н-пиразол, но не ограничиваются ими. Таутомеры могут находиться в равновесии или быть стерически заблокированы в одну форму посредством соответствующего замещения. Подразумевается, что соединения по настоящему изобретению, идентифицированные по названию или структуре как одна конкретная таутомерная форма, включают другие таутомерные формы, если не оговорено особо.
Подразумевается также, что «соединение» по настоящему изобретению также включает все изотопы атомов в соединениях. Изотопы атома включают атомы, имеющие одинаковый атомный номер, но разные массовые числа. Например, если не оговорено особо, предусмотрено, что водород, углерод, азот, кислород, фосфор, сера, фтор, хлор, бром или йод в «соединении» по настоящему изобретению также включают изотопы, такие как: 1Н, 2Н, 3Н, 11С, 12С, 13С, 14С, 14N, 15N, 16О, 17O, l8O, 31Р, 32Р, 32S, 33S, 34S, 36S, 17F, 19F, 35Cl, 37Cl, 79Br, 81Br, 127I и 131I, но не ограничиваются ими. В некоторых воплощениях водород включает протий, дейтерий и тритий. В некоторых воплощениях термин «замещенный дейтерием» или «дейтерийзамещенный» означает замену другой изоформы водорода (например протия) в химической группе дейтерием. В некоторых воплощениях углерод включает 12С и 13С. В некоторых воплощениях «соединение» по настоящему изобретению включает только изотопы водорода в соединении. В некоторых воплощениях «соединение» по настоящему изобретению включает только распространенные в природе изотопы атомов.
Также следует понимать, что «соединение» по настоящему изобретению может существовать в сольватированных, а также в несольватированных формах, таких как, например гидратированные формы, твердые формы, и настоящее изобретение, как подразумевается, включает все такие сольватированные и несольватированные формы.
Кроме того, следует понимать, что «соединение» по настоящему изобретению может существовать в виде фармацевтически приемлемых солей.
Использованный здесь термин «фармацевтически приемлемый» относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые в рамках здравого медицинского суждения являются подходящими для применения в контакте с тканями человека и животного без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений, соизмеримы с разумным соотношением польза/риск. В некоторых воплощениях соединения, вещества, композиции и/или лекарственные формы, которые являются фармацевтически приемлемыми, относятся к тем, которые одобрены регулирующим органом (таким как Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов Китая или Европейское агентство лекарственных средств) или перечислены в общепризнанной фармакопее (такой как Фармакопея США, Китайская фармакопея или Европейская фармакопея) для применения у животных и, более конкретно, у людей.
Использованный здесь термин «фармацевтически приемлемые соли» относится к производным соединений по настоящему изобретению, в которых исходное соединение модифицировано посредством превращения существующей кислотной группировки (например карбоксила и тому подобного) или основной группировки (например амина, щелочи и тому подобного) в его солевую форму. Во многих случаях соединения по настоящему изобретению способны образовывать кислые и/или основные соли благодаря присутствию амино- и/или карбоксильных групп или подобных им групп. И фармацевтически приемлемые соли представляют собой соли кислот и/или оснований, которые сохраняют биологическую эффективность и свойства исходного соединения, которые обычно не являются нежелательными с биологической или иной точки зрения. Подходящие фармацевтически приемлемые соли соединения по настоящему изобретению включают, например, соль присоединения кислоты, которая может быть образована, например, из неорганической кислоты (например соляной, бромистоводородной, серной, азотной, фосфорной кислоты и тому подобных) или органической кислоты (например муравьиной, уксусной, пропионовой, гликолевой, щавелевой, малеиновой, малоновой, янтарной, фумаровой, винной, тримезиновой, лимонной, молочной, фенилуксусной, бензойной, миндальной, метансульфоновой, нафталиндисульфоновой, этансульфоновой, толуолсульфоновой, трифторуксусной, салициловой, сульфосалициловой кислот и тому подобных).
Подходящие фармацевтически приемлемые соли соединения по настоящему изобретению также включают, например, соль присоединения основания, которая может быть образована, например, из неорганических оснований (например солей натрия, калия, аммония и гидроксидов, карбонатов, бикарбонатов металлов с I по XII группу периодической таблицы, таких как кальций, магний, железо, серебро, цинк, медь и тому подобных) или органических оснований (например первичных, вторичных и третичных аминов, замещенных аминов, включая встречающиеся в природе замещенные амины, циклических аминов, основных ионообменных смол и тому подобных). Некоторые органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин, но не ограничиваются ими. Специалисту будет понятно, что также возможно добавление кислот или оснований для образования солей присоединения кислот/оснований, отличных от показанных в примерах. Перечни дополнительных подходящих солей можно найти, например в "Remington's Pharmaceutical Sciences", 20th ed., Mack Publishing Company, Easton, Pa., (1985); и в "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Мы обнаружили, что соединения по настоящему изобретению или их фармацевтически приемлемые соли являются эффективными ингибиторами ВТК и могут быть использованы для получения ингибирующего эффекта в отношении ВТК у теплокровного животного, нуждающегося в таком лечении. Соответственно, ожидается, что соединения по настоящему изобретению будут полезны при лечении заболеваний или медицинских состояний, опосредованных полностью или частично ВТК.
Соответственно, соединения по настоящему изобретению, как ожидают, будут полезны при лечении иммунных расстройств, рака, сердечно-сосудистых заболеваний, вирусных инфекций, нарушений метаболизма/функций эндокринных желез и неврологических расстройств, аллергических заболеваний, аутоиммунных заболеваний и воспалительных заболеваний, включая крапивницу/синдром Шегрена, ревматоидный артрит, остеопороз, васкулит, идиопатическую тромбоцитопеническую пурпуру (ИТП), тяжелую миастению, аллергический ринит, астму, рассеянный склероз и системную красную волчанку.
Ожидается, что исходя из того, что они обладают свойствами ингибитора ВТК, соединения по изобретению будут обладать широким спектром противораковых свойств в отношении ВТК опосредованного роста, наблюдаемого при раке человека, включая В-клеточные неоплазии, но не ограничиваясь этим. В частности, ожидается, что такие соединения по изобретению могут быть полезны для лечения лимфом и лейкозов. Более конкретно, ожидается, что такие соединения по изобретению или их фармацевтически приемлемые соли будут полезны при лечении мелкоклеточной лимфоцитарной лимфомы (МЛЛ), фолликулярной лимфомы, трансформации Рихтера, мантийноклеточной лимфомы, хронического лимфоцитарного лейкоза (ХЛЛ), макроглобулинемии Вальденстрема, неходжкинской лимфомы, первичной лимфомы центральной нервной системы, вторичной лимфомы центральной нервной системы или диффузной крупноклеточной В-клеточной лимфомы. В частности, соединения по настоящему изобретению полезны при лечении диффузной крупноклеточной В-клеточной лимфомы с метастазами в головной мозг, первичной лимфомы центральной нервной системы или вторичной лимфомы центральной нервной системы. В частности, соединения по настоящему изобретению полезны при лечении хронического лимфоцитарного лейкоза. В частности, соединения по настоящему изобретению полезны при терапии второй линии хронического лимфоцитарного лейкоза. В частности, соединения по настоящему изобретению полезны при терапии первой линии хронического лимфоцитарного лейкоза. В частности, соединения по настоящему изобретению полезны при лечении диффузной крупноклеточной В-клеточной лимфомы. В частности, соединения по настоящему изобретению полезны при лечении первичной лимфомы центральной нервной системы.
В некоторых воплощениях соединения или их фармацевтически приемлемые соли по настоящему изобретению обладают противораковой активностью на ранней стадии активно прогрессирующего, метастатического и/или фармакорезистентного рака. В некоторых воплощениях рак относится к местно-распространенному раку. В некоторых воплощениях рак относится к местно-распространенному и/или метастатическому раку. В некоторых воплощениях рак относится к метастатическому раку. В некоторых воплощениях рак относится к инвазивному раку. В некоторых воплощениях рак относится к резистентному к ибрутинибу раку.
В одном воплощении изобретения, где упоминается ингибирование ВТК, это относится как к ВТК дикого типа, так и к ВТК с мутацией С481 (например мутацией C481S, C481Y, C481R или C481F).
В одном воплощении изобретения, где упоминается ингибирование ВТК, это относится к ВТК дикого типа.
В одном воплощении изобретения, где упоминается ингибирование ВТК, это относится к ВТК с мутацией С481.
В одном воплощении изобретения, где упоминается ингибирование ВТК, это относится к ВТК с мутацией C481S.
В одном воплощении изобретения, где упоминается ингибирование ВТК, это относится к ВТК с мутацией C481Y.
В одном воплощении изобретения, где упоминается ингибирование ВТК, это относится к ВТК с мутацией C481R.
В одном воплощении изобретения, где упоминается ингибирование ВТК, это относится к ВТК с мутацией C481F.
Фармацевтическая композиция, дозировка и введение
Согласно настоящему изобретению предложены фармацевтические композиции, содержащие по меньшей мере одно соединение по настоящему изобретению или его фармацевтически приемлемую соль. В некоторых воплощениях фармацевтическая композиция содержит более одного соединения по настоящему изобретению или его фармацевтически приемлемой соли. В некоторых воплощениях фармацевтическая композиция содержит одно или более чем одно соединение по настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Как правило, фармацевтически приемлемые носители представляют собой обычные в данной области техники носители для лекарственных средств, которые могут быть получены посредством способов, хорошо известных в области фармацевтики. В некоторых воплощениях соединения по настоящему изобретению или их фармацевтически приемлемые соли могут быть смешаны с фармацевтически приемлемым носителем для приготовления фармацевтической композиции.
Форма фармацевтических композиций зависит от ряда критериев, включая способ введения, степень заболевания или дозу, которую необходимо ввести, но не ограничиваясь ими. Фармацевтические композиции могут быть приготовлены в виде препаратов для перорального, назального, ректального, чрескожного, внутривенного или внутримышечного введения. В соответствии с требуемым путем введения фармацевтические композиции могут быть приготовлены в виде таблеток, капсул, пилюль, драже, порошка, гранул, саше, облаток, лепешек, суспензий, эмульсий, растворов, сиропов, аэрозолей (в виде твердого вещества или в жидкой среде), спрея, мази, пасты, крема, лосьона, геля, пластыря, ингалятора или суппозиториев.
В некоторых воплощениях фармацевтические композиции содержат от приблизительно 1 мг до приблизительно 500 мг соединений по настоящему изобретению или их фармацевтически приемлемых солей, в частности, от 1 мг до приблизительно 200 мг. Фармацевтическая композиция также может быть введена один, два, три или даже четыре раза в сутки. Однако суточная доза обязательно будет варьироваться в зависимости от получающего лечение хозяина, конкретного пути введения и тяжести заболевания, которое лечат. Соответственно, оптимальная дозировка может быть определена практикующим врачом, который лечит любого конкретного пациента.
Терапевтически эффективное количество предложенного здесь соединения или его фармацевтически приемлемых солей будет зависеть от различных факторов, известных в данной области техники, таких, например, как масса тела, возраст, история болезни, применяемые лекарственные препараты, состояние здоровья субъекта и возможность перекрестного взаимодействия, аллергические реакции, чувствительность и побочные эффекты, а также путь введения и степень развития заболевания. Дозировки могут быть пропорционально уменьшены или увеличены специалистом в данной области техники (например врачом или ветеринаром) в соответствии с этими и другими обстоятельствами или требованиями.
В дополнительном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в сочетании с фармацевтически приемлемым разбавителем или носителем.
В дополнительном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в сочетании с фармацевтически приемлемым разбавителем или носителем для применения при обеспечении ингибирующего эффекта в отношении ВТК у теплокровного животного, такого как человек.
В дополнительном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в сочетании с фармацевтически приемлемым разбавителем или носителем для применения при обеспечении противоракового эффекта у теплокровного животного, такого как человек.
В дополнительном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в сочетании с фармацевтически приемлемым разбавителем или носителем для применения в лечении мелкоклеточной лимфоцитарной лимфомы (МЛЛ), фолликулярной лимфомы, трансформации Рихтера, мантийноклеточной лимфомы, хронического лимфоцитарного лейкоза (ХЛЛ), макроглобулинемии Вальденстрема, неходжкинской лимфомы, первичной лимфомы центральной нервной системы, вторичной лимфомы центральной нервной системы или диффузной крупноклеточной В-клеточной лимфомы у теплокровного животного, такого как человек.
В дополнительном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в сочетании с фармацевтически приемлемым разбавителем или носителем для применения в лечении диффузной крупноклеточной В-клеточной лимфомы с метастазами в головной мозг, первичной лимфомы центральной нервной системы или вторичной лимфомы центральной нервной системы.
Комбинации
В некоторых воплощениях фармацевтические композиции содержат одно или более чем одно соединение по настоящему изобретению или его фармацевтически приемлемую соль в качестве первого активного ингредиента и дополнительно содержат второй активный ингредиент. Второй активный ингредиент может представлять собой любой противоопухолевый агент, известный в данной области техники, например, ингибиторы PI3K (фосфатидилинозитол-3-киназы), анти-CD20-антитела, анти-PD-1/L1-антитела и другие одобренные лекарственные средства или комбинации лекарственных средств для лечения неходжкинской лимфомы. Типичные примеры противоопухолевых агентов в качестве второго активного ингредиента включают иделалисиб, дювелисиб, обинутузумаб, офатумумаб, ритуксимаб, алемтузумаб, блеомицин, брентуксимаб, ведотин, кармустин, циклофосфамид, хлорамбуцил, дакарбазин, дексаметазон, доксорубицин, ломустин, мехлорэтамин, прокарбазин, преднизон, бендамустин, венетоклакс, преднизон, CVP (комбинированное лечение С - циклофосфамидом, химиотерапевтическим препаратом, V - винкристином, химиотерапевтическим препаратом и Р - преднизолоном, стероидом), мидостаурин и винбластин, но не ограничиваются ими,
Следует понимать, что использованный здесь термин «комбинация» относится к одновременному, раздельному или последовательному введению. В одном аспекте настоящего изобретения «комбинация» относится к одновременному введению. В другом аспекте настоящего изобретения «комбинация» относится к раздельному введению. В дополнительном аспекте настоящего изобретения «комбинация» относится к последовательному введению. Если введение является последовательным или раздельным, отсрочка введения второго компонента не должна быть такой, чтобы положительный эффект комбинации был потерян.
Поэтому, в дополнительном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь, в комбинации с противоопухолевым агентом, выбранным из одного из перечисленных здесь выше.
Поэтому, в дополнительном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь, в комбинации с противоопухолевым агентом, выбранным из одного из перечисленных здесь выше, для применения при получении противоракового эффекта.
Поэтому, в дополнительном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь, в комбинации с противоопухолевым агентом, выбранным из одного из перечисленных здесь выше, для применения в лечении мелкоклеточной лимфоцитарной лимфомы (МЛЛ), фолликулярной лимфомы, трансформации Рихтера, мантийноклеточной лимфомы, хронического лимфоцитарного лейкоза (ХЛЛ), макроглобулинемии Вальденстрема, неходжкинской лимфомы, первичной лимфомы центральной нервной системы, вторичной лимфомы центральной нервной системы или диффузной крупноклеточной В-клеточной лимфомы.
Поэтому, в дополнительном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь, в комбинации с противоопухолевым агентом, выбранным из одного из перечисленных здесь выше, для применения в лечении диффузной крупноклеточной В-клеточной лимфомы с метастазами в головной мозг, первичной лимфомы центральной нервной системы или вторичной лимфомы центральной нервной системы.
Согласно этому аспекту настоящего изобретения предложена комбинация, подходящая для применения при лечении рака, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, и любой из противоопухолевых агентов, перечисленных выше.
Согласно дополнительному аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в комбинации с противоопухолевым агентом, выбранным из одного из перечисленных здесь выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
Согласно дополнительному аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в комбинации с противоопухолевым агентом, выбранным из одного из перечисленных здесь выше, в сочетании с фармацевтически приемлемым разбавителем или носителем для применения при получении противоракового эффекта.
Согласно дополнительному аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в комбинации с противоопухолевым агентом, выбранным из одного из перечисленных здесь выше, в сочетании с фармацевтически приемлемым разбавителем или носителем для применения в лечении мелкоклеточной лимфоцитарной лимфомы (МЛЛ), фолликулярной лимфомы, трансформации Рихтера, мантийноклеточной лимфомы, хронического лимфоцитарного лейкоза (ХЛЛ), макроглобулинемии Вальденстрема, неходжкинской лимфомы, первичной лимфомы центральной нервной системы, вторичной лимфомы центральной нервной системы или диффузной крупноклеточной В-клеточной лимфомы.
Согласно дополнительному аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в комбинации с противоопухолевым агентом, выбранным из одного из перечисленных здесь выше, в сочетании с фармацевтически приемлемым разбавителем или носителем для применения в лечении диффузной крупноклеточной В-клеточной лимфомы с метастазами в головной мозг, первичной лимфомы центральной нервной системы или вторичной лимфомы центральной нервной системы.
Согласно дополнительному аспекту настоящего изобретения предложен набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в комбинации с противоопухолевым агентом, выбранным из одного из перечисленных здесь выше.
Согласно дополнительному аспекту настоящего изобретения предложен набор, содержащий:
a) соединение формулы (I) или его фармацевтически приемлемую соль, как определено здесь, в первой стандартной лекарственной форме;
b) противоопухолевый агент, выбранный из одного из перечисленных здесь выше, во второй стандартной лекарственной форме; и
c) контейнер, предназначенный для содержания указанных первой и второй лекарственных форм.
Фармакологические инструменты
Помимо их применения в лечебной медицине соединения формулы (I) или их фармацевтически приемлемые соли также полезны в качестве фармакологических инструментов при разработке и стандартизации in vitro и in vivo тест-систем для оценки эффектов ингибирования ВТК на лабораторных животных, таких как кошки, собаки, кролики, обезьяны, крысы и мыши, при осуществлении поиска новых терапевтических агентов.
Способ лечения
Согласно дополнительному аспекту настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь, для применения в способе лечения человека или животного посредством терапии.
Согласно дополнительному признаку этого аспекта изобретения предложен способ обеспечения ингибирующего эффекта в отношении ВТК у теплокровного животного, такого как человек, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь.
Согласно дополнительному признаку этого аспекта изобретения предложен способ лечения рака у теплокровного животного, такого как человек, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь.
Согласно дополнительному признаку этого аспекта изобретения предложен способ лечения мелкоклеточной лимфоцитарной лимфомы (МЛЛ), фолликулярной лимфомы, трансформации Рихтера, мантийноклеточной лимфомы, хронического лимфоцитарного лейкоза (ХЛЛ), макроглобулинемии Вальденстрема, неходжкинской лимфомы, первичной лимфомы центральной нервной системы, вторичной лимфомы центральной нервной системы или диффузной крупноклеточной В-клеточной лимфомы у теплокровного животного, такого как человек, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь.
Согласно дополнительному признаку этого аспекта изобретения предложен способ лечения диффузной крупноклеточной В-клеточной лимфомы с метастазами в головной мозг, первичной лимфомы центральной нервной системы или вторичной лимфомы центральной нервной системы у теплокровного животного, такого как человек, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь.
Использованные здесь термины «лечение» и «лечить» относятся к реверсированию, облегчению, задержке начала или ингибированию развития заболевания или расстройства или одного или более чем одного его симптома, как описано здесь. В некоторых воплощениях лечение можно проводить после развития одного или более чем одного симптома. В других воплощениях лечение можно проводить в отсутствие симптомов. Например, лечение может быть проведено восприимчивому индивидууму до появления симптомов (например с учетом характера развития симптомов и/или с учетом генетических или других факторов восприимчивости). Лечение также может быть продолжено после устранения симптомов, например, чтобы предотвратить или отсрочить их повторное появление.
Согласно настоящему изобретению также предложен способ скрининга пациента, подходящего для лечения соединением формулы (I) или его фармацевтически приемлемой солью, как определено в настоящем документе. Способ включает секвенирование образцов опухолей пациентов и обнаружение накопления ВТК или наличия мутаций ВТК.
Согласно дополнительному признаку этого аспекта настоящего изобретения предложен способ лечения рака у теплокровного животного, такого как человек, включающий (1) определение того, имеется ли у теплокровного животного рак, восприимчивый к ингибированию ВТК или нет и (2) если имеется, то введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь.
Применение соединений
В некоторых воплощениях согласно настоящему изобретению предложено применение соединений, их фармацевтически приемлемых солей или фармацевтической композиции по настоящему изобретению в изготовлении лекарственных средств для лечения заболеваний или состояний, опосредованных или зависимых от ВТК.
Таким образом, согласно этому аспекту изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь, для применения в качестве лекарственного средства.
Таким образом, согласно этому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь, в качестве лекарственного средства.
Таким образом, согласно этому аспекту изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь, для применения в терапии.
Согласно дополнительному аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь, в изготовлении лекарственного средства для обеспечения ингибирующего эффекта в отношении ВТК у теплокровного животного, такого как человек.
Согласно этому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь, в изготовлении лекарственного средства для обеспечения противоракового эффекта у теплокровного животного, такого как человек.
Согласно дополнительному признаку изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь, в изготовлении лекарственного средства для лечения мелкоклеточной лимфоцитарной лимфомы (МЛЛ), фолликулярной лимфомы, трансформации Рихтера, мантийноклеточной лимфомы, хронического лимфоцитарного лейкоза (ХЛЛ), макроглобулинемии Вальденстрема, неходжкинской лимфомы, первичной лимфомы центральной нервной системы, вторичной лимфомы центральной нервной системы или диффузной крупноклеточной В-клеточной лимфомы
Согласно дополнительному признаку изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь, в изготовлении лекарственного средства для лечения диффузной крупноклеточной В-клеточной лимфомы с метастазами в головной мозг, первичной лимфомы центральной нервной системы или вторичной лимфомы центральной нервной системы.
Согласно дополнительному аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь, для применения при обеспечении ингибирующего эффекта в отношении ВТК у теплокровного животного, такого как человек.
Согласно этому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено здесь, для применения при обеспечении противоракового эффекта у теплокровного животного, такого как человек.
Согласно дополнительному признаку изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь для применения в лечении мелкоклеточной лимфоцитарной лимфомы (МЛЛ), фолликулярной лимфомы, трансформации Рихтера, мантийноклеточной лимфомы, хронического лимфоцитарного лейкоза (ХЛЛ), макроглобулинемии Вальденстрема, неходжкинской лимфомы, первичной лимфомы центральной нервной системы, вторичной лимфомы центральной нервной системы или диффузной крупноклеточной В-клеточной лимфомы
Согласно дополнительному признаку изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено здесь, для применения в лечении диффузной крупноклеточной В-клеточной лимфомы с метастазами в головной мозг, первичной лимфомы центральной нервной системы или вторичной лимфомы центральной нервной системы.
В вышеуказанных признаках фармацевтических композиций, способов, применений и получения лекарственных средств также применяются альтернативные и предпочтительные воплощения соединений по настоящему изобретению, описанные здесь.
ПРИМЕРЫ
Общие эксперименты
Аббревиатуры
Синтез предложенных здесь соединений, включая их фармацевтически приемлемые соли, проиллюстрирован на схемах синтеза в примерах. Предложенные здесь соединения могут быть получены с использованием любых известных методов органического синтеза и могут быть синтезированы в соответствии с любым из многочисленных возможных путей синтеза, и, таким образом, эти схемы являются только иллюстративными и не предназначены для ограничения других возможных способов, которые могут быть использованы для получения соединений, предложенных здесь. Кроме того, стадии в способах предназначены для лучшей иллюстрации и могут быть изменены, как целесообразно. Воплощения соединений в примерах были синтезированы для целей исследования и возможно для представления в регулирующие органы.
Реакции получения соединений по настоящему изобретению могут быть проведены в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящие растворители могут быть по существу инертными в отношении исходных веществ (реагентов), промежуточных соединений или продуктов при температурах, при которых проводят взаимодействия, например при температурах, которые могут находиться в диапазоне от температуры замерзания растворителя до температуры кипения растворителя. Данное взаимодействие может быть проведено в одном растворителе или в смеси более чем одного растворителя. В зависимости от конкретной стадии взаимодействия квалифицированный специалист может выбрать подходящие растворители для конкретной стадии взаимодействия.
Получение соединений по настоящему изобретению может включать защиту и снятие защиты с различных химических групп.Необходимость защиты и снятия защиты и выбор соответствующих защитных групп могут быть легко определены специалистом в данной области техники. Химию защитных групп можно найти, например в T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999), которая включена здесь посредством ссылки во всей своей полноте.
Ход взаимодействий можно контролировать посредством любого подходящего способа, известного в данной области техники. Например, образование продукта можно контролировать с помощью методов спектроскопии, таких как спектроскопия ядерного магнитного резонанса (например 1Н или 13С), инфракрасная спектроскопия, спектрофотометрия (например в УФ (ультрафиолетовой) и видимой областях спектра), масс-спектрометрия, или с помощью методов хроматографии, таких как высокоэффективная жидкостная хроматография (ВЭЖХ), жидкостная хроматография-масс-спектроскопия (ЖХ-МС) или тонкослойная хроматография (ТСХ). Соединения могут быть очищены специалистами в данной области техники посредством различных способов, включая высокоэффективную жидкостную хроматографию (ВЭЖХ) («Preparative LC-MS Purification: Improved Compound Specific Method Optimization)) Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883, которая включена здесь посредством ссылки во всей своей полноте), сверхкритическую флюидную хроматографию (СФХ) и нормально-фазовую хроматографию на диоксиде кремния.
Структуры соединений в примерах охарактеризованы с помощью ядерного магнитного резонанса (ЯМР) или/и жидкостной хроматографии-масс-спектрометрии (ЖХ-МС). Химический сдвиг ЯМР (δ) дан в единицах 10-6 (м.д.). Спектры 1H-ЯМР регистрируют в растворителе диметилсульфоксид-d6 (DMSO-d6), или CDCl3, или CD3OD, или D2O, или ацетон-d6, или CD3CN (от Aldrich, или Cambridge Isotope Lab., Inc.) на спектрометрах Bruker AVANCE NMR (400 МГц) при использовании ICON-NMR (под контролем программы TopSpin) или спектрометрах Varian 400MR NMR или Varian VNMR400 NMR (400 МГц) (под контролем программы VnmrJ) с тетраметилспланом в качестве внутреннего стандарта.
Измерение МС проводят при использовании масс-спектрометра Shimadzu 2010, или Agilent 6110А MSD, или времяпролетного масс-спектрометра 1969А при использовании методов электрораспыления, химической ионизации и ионизации электронным ударом с помощью ряда приборов. Подробные способы, используемые в данном изобретении, включают:
ЖХ-МС способ A: 10-80AB_7min_220&254_Shimadzu.lcm
Подвижная фаза: 1,5 мл/4 л TFA в воде (растворитель А) и 0,75 мл/4 л TFA в ацетонитриле (растворитель В), с использованием градиента элюирования 10%-80% (растворитель В) за 6 минут и удерживания при 80% в течение 0,5 минуты при скорости потока 0,8 мл/мин;
Колонка: Xtimate С18 2,1×30 мм, 3 мкм;
Длина волны: УФ 220 нм, 254 нм;
Температура колонки: 50°С;
МС ионизация: ЭРИ (ионизация электрораспылением).
ЖХ-МС способ В: 10-80AB_4min_220&254_Shimadzu.lcm
Подвижная фаза: 1,5 мл/4 л TFA в воде (растворитель А) и 0,75 мл/4 л TFA в ацетонитриле (растворитель В), с использованием градиента элюирования 10%-80% (растворитель В) за 3 минуты и удерживания при 80% в течение 0,5 минуты при скорости потока 0,8 мл/мин;
Колонка: Xtimate С18 2,1×30 мм, 3 мкм;
Длина волны: УФ 220 нм, 254 нм;
Температура колонки: 50°С;
МС ионизация: ЭРИ.
ЖХ-МС способ С: 10-80CD_7min_220&254_Agilent.lcm
Подвижная фаза: 0,2 мл/1 л NH3⋅H2O в воде (растворитель А) и ацетонитрил (растворитель В), с использованием градиента элюирования 10%-80% (растворитель В) за 6 минут и удерживания при 80% в течение 0,5 минуты при скорости потока 0,8 мл/мин;
Колонка: Xbrige Shield RP-18, 5 мкм, 2,1×50 мм;
Длина волны: УФ 220 нм и 254 нм;
Температура колонки: 30°С;
МС ионизация: ЭРИ.
Измерение посредством метода высокоэффективной жидкостной хроматографии (ВЭЖХ) проводят на системах Shimadzu LC-20A или Shimadzu серии LC-2010HT, или Agilent 1200 LC, или Agilent серии 1100 при использовании колонки Ultimate ХВ-С18 (3,0×50 мм, 3 мкм или 3,0×150 мм, 3 мкм), или колонки Xbridge shieldRP18 (5 мкм, 50 мм×2,1 мм), или колонки Xtimate С18 (3 мкм, 2,1×30 мм), или MERCK RP18 2,5-2 мм и так далее. Подробные способы, используемые в данном изобретении, включают:
ВЭЖХ способ А: 10-80АВ_8min.met
Подвижная фаза: 2,75 мл/4 л TFA в воде (растворитель А) и 2,5 мл/4 л TFA в ацетонитриле (растворитель В), при использовании градиента элюирования 10%-80% (растворитель В) за 6 минут и удерживания при 80% в течение 2 минут при скорости потока 1,2 мл/мин;
Колонка: Ultimate С18 3,0×50 мм, 3 мкм;
Длина волны: УФ 220 нм, 215 нм, 254 нм;
Температура колонки: 40°С.
ВЭЖХ способ В: 10-80CD_8min.met
Подвижная фаза: 2,0 мл/4 л NH3⋅H2O в воде (растворитель А) и ацетонитрил (растворитель В), при использовании градиента элюирования 10%-80% (растворитель В) за 4 минуты и удерживания при 80% в течение 2 минут при скорости потока 1,2 мл/мин;
Колонка: Xbrige Shield RP-18, 2,1×50 мм, 5 мкм;
Длина волны: УФ 220 нм, 215 нм, 254 нм;
Температура колонки: 40°С.
Измерение посредством метода сверхкритической флюидной хроматографии (СФХ) проводят на приборах Agilent серии 1260, или Waters серии UPCC, или Shimadzu серии LC-20AB при использовании колонки ChiralPak AD-3 (3 мкм, 150×4,6 мм), или колонки Chiralcel OJ-3 (3 мкм, 150×4,6 мм), или колонки Chiralpak IG-3 (3 мкм, 50 ммх4,6 мм) и так далее. Подробные способы, используемые в данном изобретении, включают:
СФХ способ А: подвижная фаза: А: СО2, В: этанол (0,05% DEA); градиент: от 5% до 40% В за 5,5 мин и удерживание при 40% в течение 3 мин, затем 5% В в течение 1,5 мин; скорость потока: 2,5 мл/мин;
Колонка: ChiralPak AD-3 150×4,6 мм ID. (внутренний диаметр), 3 мкм;
Температура колонки: 40°С;
Обратное давление: 100 бар (10 МПа).
СФХ способ В: подвижная фаза: А: СО2, В: метанол (0,05% DEA); градиент: от 5% до 40% В за 5 мин и от 40% до 5% В за 0,5 мин, удерживание при 5% В в течение 1,5 мин; скорость потока: 2,5 мл/мин
Колонка: Chiralcel OJ-3 150×4,6 мм I.D., 3 мкм;
Температура колонки: 35°С;
ABPR (регулируемое обратное давление): 1500 ф/кв дюйм (10,34 МПа).
СФХ способ С: подвижная фаза: А: СО2, В: метанол (0,05% DEA), изократическое элюирование: 40% В, скорость потока: 4 мл/мин;
Колонка: Chiralpak IG-3 50 мм×4,6 мм I.D., 3 мкм;
Температура колонки: 35°С;
ABPR: 1500 ф/кв дюйм (10,34 МПа).
Тонкослойную хроматографию проводят при использовании силикагеля Yantai Huanghai HSGF254 или пластин Anhui Liang Chen Gui Yuan. Пластины с силикагелем, используемые для тонкослойной хроматографии (ТСХ), имеют толщину слоя от 0,15 мм до приблизительно 0,2 мм. Пластины с силикагелем, используемые для разделения и очистки продуктов посредством ТСХ, имеют толщину слоя от 0,4 мм до приблизительно 0,5 мм.
В хроматографической колонке для очистки используется в качестве носителя силикагель (100~200, 200~300 или 300~400 меш, производства Yantai Huanghai со. или Anhui Liang Chen Gui Yuan со. и так далее), или флэш-колонка (флэш-колонка silica-CS 40-60 мкм или колонка С18 с обращенной фазой 20-35 мкм, производства Agela Technologies и так далее), или флэш-колонка silica-CS (40-60 мкм), или колонка С18 (20-40 мкм) от Agela Technologies в комбинированной флэш-системе Teledyne ISCO или флэш-системе Biotage. Размер колонок регулируется в зависимости от количества соединений.
Известные исходные вещества по настоящему изобретению могут быть синтезированы при использовании или в соответствии с известными в данной области техники способами, либо их можно приобрести у Alfa Aesar, TCI, Sigma-Aldrich, Bepharm и Scochem (или PharmaBlock, Bide, Amatek, Stru Chem, Firster Pharmaceutical, Titan (Adamas) и так далее).
Если не оговорено особо, все взаимодействия проводят в атмосфере аргона или азота. Атмосфера аргона или азота означает, что реакционная колба соединена с баллоном с аргоном или азотом объемом около 1 л. Гидрирование обычно проводят под давлением. Если не оговорено особо, температура взаимодействий в примерах соответствует температуре окружающей среды, которая составляет 10°С~30°С.
За ходом взаимодействий следят с помощью ТСХ или/и ЖХ-МС.Системы элюентов, используемые для взаимодействий, включают систему дихлорметан-метанол и систему петролейный эфир-этилацетат.Объемные соотношения растворителей регулируют в соответствии с различной полярностью соединений.
Система элюирования колоночной хроматографии, используемая для очистки соединений, и система элюентов ТСХ включают систему дихлорметан-метанол и систему петролейный эфир-этилацетат.Объемные соотношения растворителей регулируют в соответствии с различной полярностью соединений. Для корректировки может быть добавлено небольшое количество щелочных или кислотных агентов (0,1%~1%), таких как муравьиная кислота, или уксусная кислота, или TFA, или аммиак.
Соединения по изобретению

Способы синтеза
Соединения по изобретению могут быть получены согласно следующим двум способам синтеза.
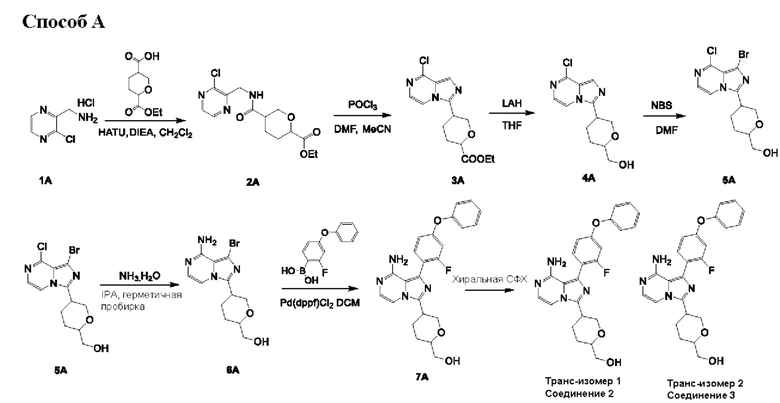
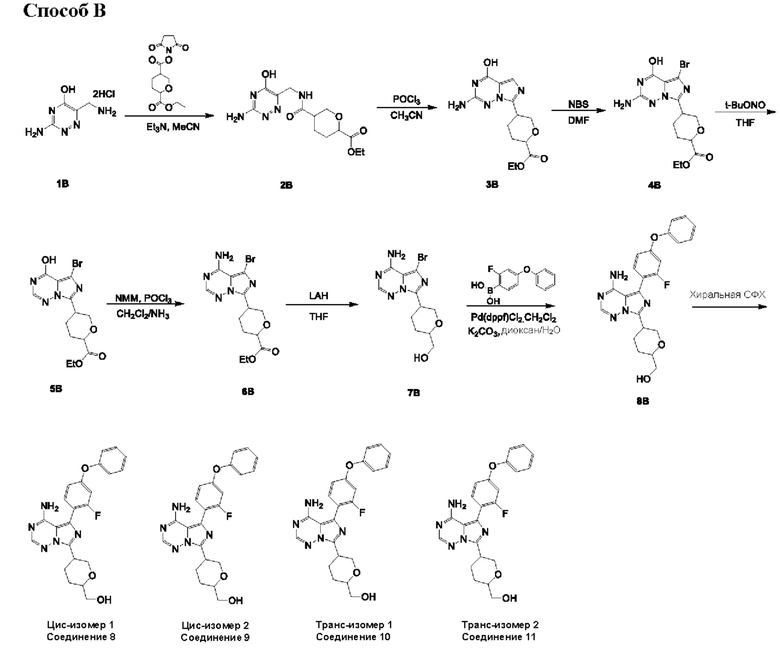
Способ А
Соединение 2А: этил-5-(((3-хлорпиразин-2-ил)метил)карбамоил)тетрагидро-2Н-пиран-2-карбоксилат
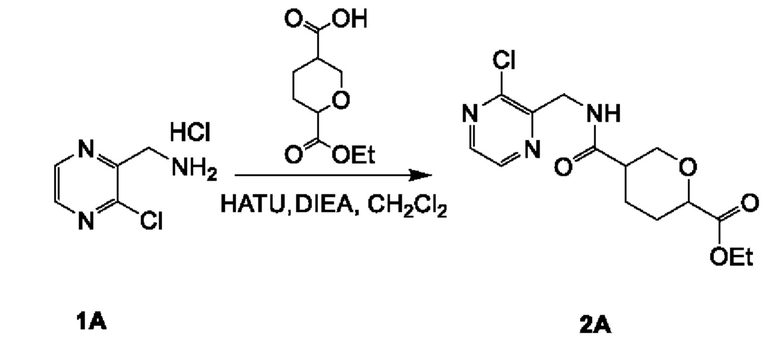
К смеси 6-(этоксикарбонил)тетрагидро-2Н-пиран-3-карбоновой кислоты (смотри WO 2019001420A1) (5,90 г; 29,17 ммоль) и соединения 1А (5,25 г; 29,17 ммоль) в дихлорметане (150 мл) добавляли HATU (16,64 г; 43,76 ммоль) и DIEA (11,31 г; 87,51 ммоль). Смесь перемешивали при комнатной температуре (18-22°С) в течение 12 часов. Реакционную смесь концентрировали и разбавляли водой (100 мл), экстрагировали этилацетатом (200 мл×3). Объединенные органические слои промывали рассолом (200 млх4), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (2,5-3,5% метанол в дихлорметане) с получением соединения 2А (9,0 г; выход 94,0%) в виде желтого масла.
ЖХ-МС: tR (время удерживания) составляет 0,689 мин в хроматографическом способе 5-95АВ_1.5min_220&254_Shimadzu.lcm (Agilent Pursuit 5 C18 20×2,0 мм), МС (ЭРИ) m/z составляет 328,2 [М+Н]+.
Соединение 3А: этил-5-(8-хлоримидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-карбоксилат
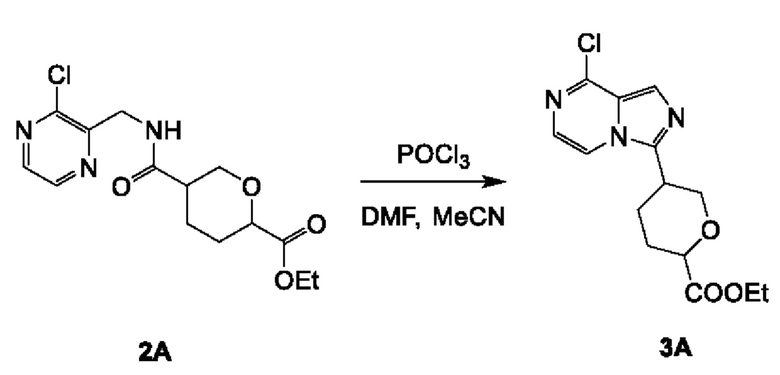
К смеси соединения 2А (9,0 г; 27,46 ммоль) и DMF (600 мкл) в MeCN (150 мл) добавляли POCl3 (21,05 г; 137,30 ммоль). Смесь перемешивали при 70°С в течение 1 часа. Реакционную смесь концентрировали и промывали водой (50 мл) и насыщенным NaHCO3 (50 мл), экстрагировали этилацетатом (100 мл×3). Объединенные органические слои промывали рассолом (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (52~62% этилацетата в петролейном эфире) с получением соединения 3А (3,5 г; выход 41,2%) в виде желтого масла.
ЖХ-МС: tR составляет 0,742 мин в хроматографическом способе 5-95AB_1.5min_220&254_Shimadzu.lcm (Agilent Pursuit 5 С18 20×2,0 мм), МС (ЭРИ) m/z составляет 310,2 [М+Н]+.
1H ЯМР (400 МГц, CDCl3): δ 7.80 (d, J=0,4 Гц, 1H), 7.70-7.64 (m, 1Н), 7.37 (d, J=4,8 Гц, 0.3Н), 7.35 (d, J=4,8 Гц, 0.7Н), 4.44 (t, J=4,4 Гц, 0.7Н), 4.30-4.25 (m, 2Н), 4.23-4.14 (m, 1H), 4.00 (dd, J=3,2, 12,0 Гц, 1H), 3.79 (t, J=11,2 Гц, 0.4Н), 3.42-3.19 (m, 1H), 2.47-2.37 (m, 0.8Н), 2.29-2.06 (m, ЗН), 1.91-1.77 (m, 0.4Н), 1.35-1.31 (m, 3Н).
Соединение 4А: (5-(8-хлоримидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанол
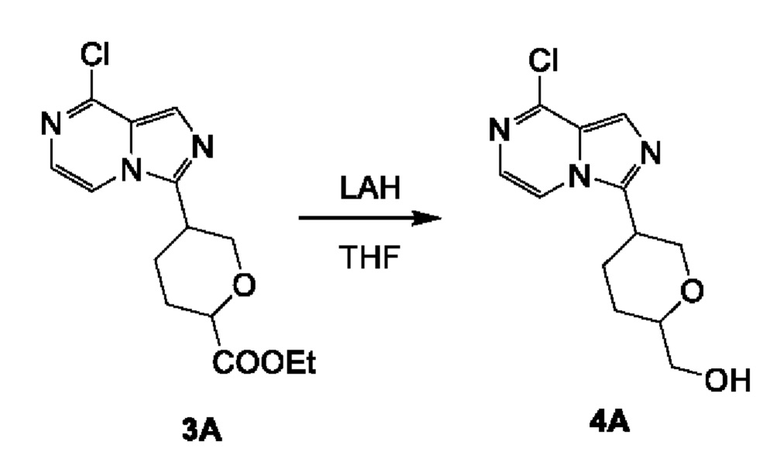
К смеси LiAlH4 (860 мг; 22,60 ммоль) в THF (20 мл) добавляли соединение 3А (3,5 г; 11,30 ммоль) в THF (20 мл) при 0°С. Смесь перемешивали при 0°С в течение 1 часа. Реакционную смесь гасили водой (860 мкл), 15% NaOH (860 мкл) и водой (2580 мкл). Смесь сушили над безводным сульфатом натрия и перемешивали при комнатной температуре в течение 0,5 часа, фильтровали и концентрировали с получением соединения 4А (2,7 г; выход 89,4%) в виде желтого твердого вещества.
ЖХ-МС: tR составляет 0,635 мин в хроматографическом способе 5-95AB_1.5min_220&254_Shimadzu.lcm (Agilent Pursuit 5 С18 20×2,0 мм), МС (ЭРИ) m/z составляет 267,8 [М+Н]+.
Соединение 5А: (5-(1-бром-8-хлоримидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанол
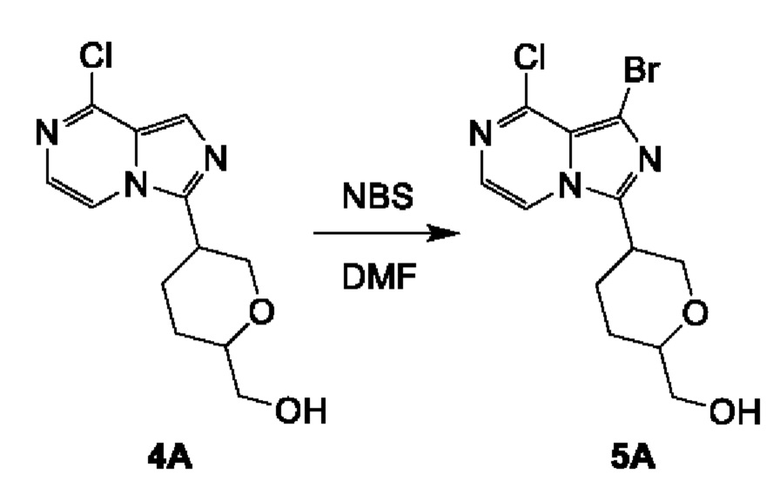
К смеси соединения 4А (2,7 г; 10,11 ммоль) в MeCN (100 мл) добавляли NBS (2,34 г; 13,14 ммоль). Смесь перемешивали при комнатной температуре (16-20°С) в течение 1 часа. Реакционную смесь концентрировали с получением неочищенного продукта, который очищали посредством колоночной хроматографии на силикагеле (2,2% метанол в дихлорметане) с получением соединения 5А (2,5 г; выход 71,63%) в виде желтого твердого вещества.
ЖХ-МС: tR составляет 0,717 мин в хроматографическом способе 5-95AB_1.5min_220&254_Shimadzu.lcm (Agilent Pursuit 5 С18 20×2,0 мм), МС (ЭРИ) m/z составляет 347,8 [М+Н]+.
Соединение 6А: (5-(8-амино-1-бромимидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанол
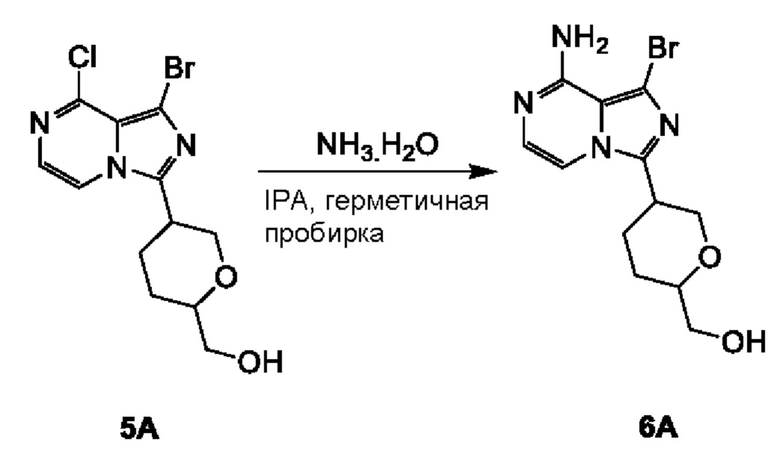
К смеси соединения 5А (2,5 г; 7,21 ммоль) в IPA (15 мл) добавляли NH3⋅H2O (15 мл). Смесь перемешивали при 100°С в течение 12 часов в герметичной пробирке на 100 мл. Реакционную смесь концентрировали с получением соединения 6А (2,4 г; чистота 98,0%) в виде желтого масла.
ЖХ-МС: tR составляет 0,436 мин в хроматографическом способе 10-80AB_3min_220&254_Shimadzu.lcm (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z составляет 327,1 [М+Н]+.
Соединение 7А: (5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил)метанол
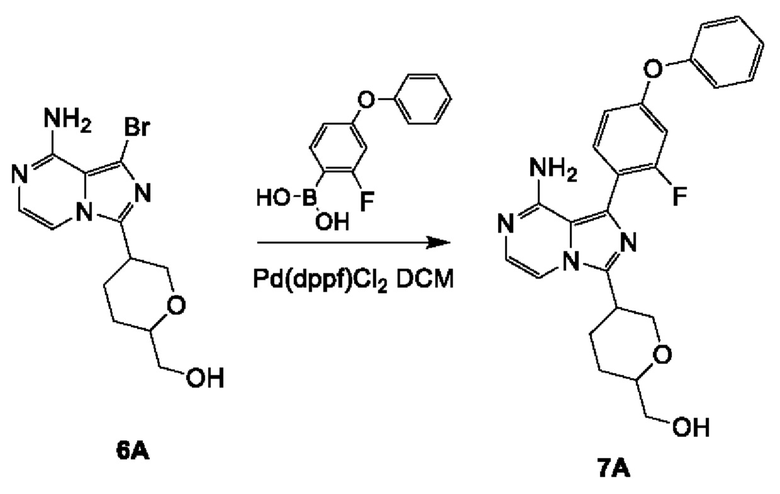
К смеси соединения 6А (500 мг; 1,31 ммоль; чистота 97,99%) и (2-фтор-4-феноксифенил)бороновой кислоты (450 мг; 1,95 ммоль) в смеси 1,4-диоксан (15 мл)/H2O (5 мл) добавляли Pd(dppf)Cl2⋅CH2Cl2 (32 мг; 0,04 ммоль) и К2СО3 (375 мг; 2,62 ммоль) в атмосфере азота. Смесь перемешивали при 100°С в течение 12 часов в атмосфере азота. Реакционную смесь разбавляли водой (20 мл), экстрагировали этилацетатом (50 мл×3). Объединенные органические слои промывали рассолом (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (2,2% метанол в дихлорметане) с получением соединения 7А (смесь цис/транс рацемата) (150 мг; выход 26,4%) в виде желтого твердого вещества.
ЖХ-МС: tR составляет 0,723 мин в хроматографическом способе 5-95AB_1.5min_220&254_Shimadzu.lcm (Agilent Pursuit 5 С18 20×2,0 мм), МС (ЭРИ) m/z составляет 435,1 [М+Н]+.
(2-фтор-4-феноксифенил)6ороновая кислота:
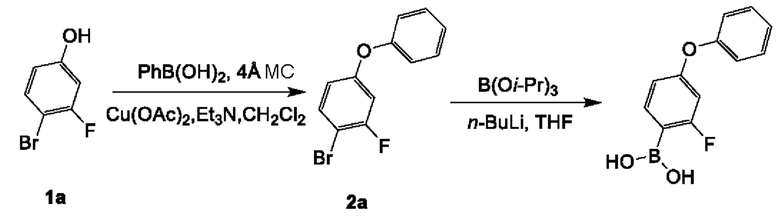
К смеси соединения 1а (10 г; 52,35 ммоль) в CH2Cl2 (480 мл) добавляли PhB(OH)2 (12,8 г; 104,71 ммоль), Cu(ОАс)2 (9,5 г; 52,35 ммоль), NEt3 (21 мл; 151,05 ммоль) и 4Å МС (молекулярное сито) (5 г) при комнатной температуре (25-30°С). Смесь перемешивали при комнатной температуре (25-30°С) в течение 16 часов в воздушной атмосфере. Смесь фильтровали через набивку из целита. Фильтрат концентрировали под вакуумом с получением неочищенного вещества, которое очищали посредством колоночной хроматографии на силикагеле (петролейный эфир) с получением соединения 2а (11,3 г; выход 80,8%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 7.47 (t, J=8,4 Гц, 1H), 7.42-7.36 (m, 2Н), 7.19 (t, J=8,4 Гц, 1H), 7.05 (d, J=8,0 Гц, 2Н), 6.78 (dd, J=10,0, 2,8 Гц, 1H), 6.71 (dd, J=8,8, 2,0 Гц, 1H).
К раствору соединения 2а (11,3 г; 42,30 ммоль) в THF (150 мл) при -65°С добавляли n-BuLi (19 мл; 46,53 ммоль; 2,5 н в н-гексане) при -65°С. Смесь перемешивали при -65°С в течение 0,5 часа. Затем добавляли В(Oi-Pr)3 (9,5 г; 50,76 ммоль) при -65°С. Смесь перемешивали при -65°С в течение 2 часов. Смесь гасили насыщенным раствором хлорида аммония (50 мл), экстрагировали этилацетатом (50 мл×3), промывали рассолом (100 мл×2). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного вещества, которое растирали с петролейным эфиром (100 мл), фильтровали, и осадок на фильтре концентрировали с получением (2-фтор-4-феноксифенил)бороновой кислоты (4,3 г) в виде желтого масла. Фильтрат концентрировали и очищали посредством колоночной хроматографии на силикагеле (0~20% этилацетат в петролейном эфире) с получением 2-фтор-4-феноксифенил)бороновой кислоты (3 г) в виде желтого масла.
1Н ЯМР (400 МГц, DMSO-d6): δ 8.10 (s, 2Н), 7.58 (t, J=8,0 Гц, 1H), 7.47-7.41 (m, 2Н), 7.22 (t, J=7,6 Гц, 1H), 7.09 (d, J=7,6 Гц, 2Н), 6.78-6.68 (m, 2Н).
Транс-изомер 1, соединение 2: транс-(5-(8-амино-1-(2-фтор-4-феноксифенил)-имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил) метанол
Транс-изомер 2, соединение 3: транс-(5-(8-амино-1-(2-фтор-4-феноксифенил)-имидазо[1,5-а]пиразин-3-ил)тетрагидро-2Н-пиран-2-ил) метанол
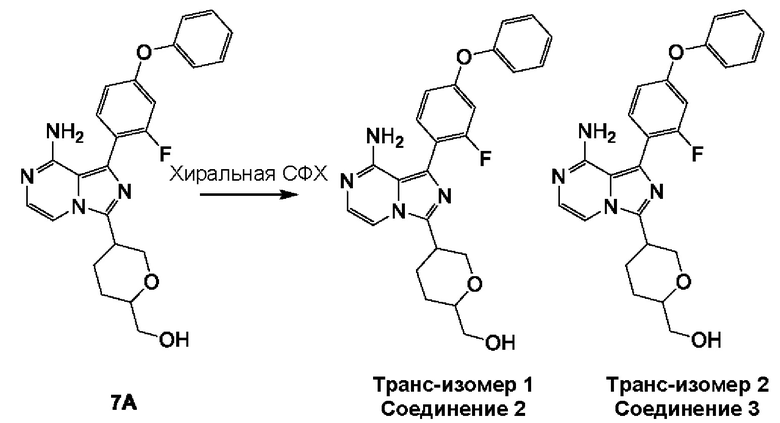
Смесь соединения 7А (150 мг; 0,35 ммоль) очищали посредством хиральной СФХ (колонка: DAICEL CHIRALPAK AD-H) (250 мм×30 мм, 10 мкм); условия: 0,1% NH3⋅H2O в EtOH, 40%, скорость потока 80 мл/мин). Фракции, содержащие требуемые соединения, концентрировали, разбавляли H2O (10 мл) и CH3CN (10 мл), лиофилизировали с получением соединения 2 (34,8 мг; выход 23,2%) в виде белого твердого вещества и соединения 3 (29,4 мг; 19,6%) в виде белого твердого вещества. Два цис-энантиомера не собирали.
Спектры соединения 2:
ЖХ-МС: tR составляет 2,037 мин в хроматографическом способе 10-80АВ_7min_220&254_Shimadzu.lcm (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z составляет 435,3 [М+Н]+.
ВЭЖХ: tR составляет 3,03 мин в 10-80АВ_8min.met (ВЭЖХ-BJ Ultimate 3,0×50 мм 3мкм).
СФХ: tR составляет 6,119 мин, оптическая чистота: 96,3%. Подробности способа: колонка: ChiralPak AD-3 150×4,6 мм I.D., 3 мкм; подвижная фаза: А: СО2, В: этанол (0,05% DEA); градиент: от 5% до 40% В за 5,5 мин и удерживание при 40% в течение 3 мин, затем 5% В в течение 1,5 мин; скорость потока: 2,5 мл/мин; температура колонки: 40°С; обратное давление: 100 бар (10 МПа).
1Н ЯМР (400 МГц, CD3OD) δ 7.63 (d, J=5,2 Гц, 1H), 7.54-7.36 (m, 3Н), 7.27-7.18 (m, 1H), 7.14 (dd, J=8,8, 0,8 Гц, 2Н), 7.02 (d, J=4,8 Гц, 1H), 6.98-6.82 (m, 2Н), 4.23-4.07 (m, 1Н), 3.75 (t, J=11,2 Гц, 1H), 3.62-3.51 (m, 3H), 3.46-3.36 (m, 1H), 2.25-2.15 (m, 1H), 2.13-1.96 (m, 1H), 1.79 (d, J=13,6 Гц, 1H), 1.66-1.49 (m, 1H).
19F HMP (CD3OD) δ -112,240.
Спектры соединения 3:
ЖХ-МС: tR составляет 2,030 мин в хроматографическом способе 10-80AB_7min_220&254_ Shimadzu.lcm (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z составляет 435,3 [М+Н]+.
ВЭЖХ: tR составляет 3,02 мин в 10-80АВ_8min.met (ВЭЖХ-BJ Ultimate 3,0×50 мм 3 мкм).
СФХ: tR составляет 7,334, оптическая чистота: 92,7%. Подробности способа: колонка: ChiralPak AD-3 150×4,6 мм I.D., 3 мкм; подвижная фаза: А: СО2, В: этанол (0,05% DEA); градиент: от 5% до 40% В за 5,5 мин и удерживание при 40% в течение 3 мин, затем 5% В в течение 1,5 мин; скорость потока: 2,5 мл/мин; температура колонки: 40°С; обратное давление: 100 бар (10 МПа).
1H ЯМР (400 МГц, CD3OD) δ 7.65 (d, J=5,2 Гц, 1Н), 7.52-7.37 (m, ЗН), 7.29-7.21 (m, 1Н), 7.20-7.11 (m, 2Н), 7.04 (d,.7=4,8 Гц, 1H), 6.99-6.84 (m, 2Н), 4.24-4.13 (m, 1H), 3.77 (t, J=11,2 Гц, 1H), 3.62-3.51 (m, 3Н), 3.48-3.77 (m, 1H), 2.31-2.17 (m, 1H), 2.15-2.01 (m, 1H), 1.91-1.74 (m, 1H), 1.69-1.51 (m, 1H).
19F HMP (400 МГц, CD3OD) δ -112,258.
Следующие соединения синтезировали согласно способу А:
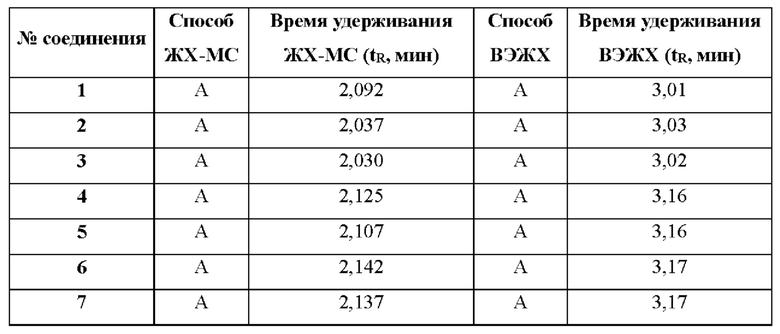
СПОСОБ В
Соединение 2В: этил-5-(((3-амино-5-гидрокси-1,2,4-триазин-6-ил)метил)карбамоил)-тетрагидро-2Н-пиран-2-карбоксилат
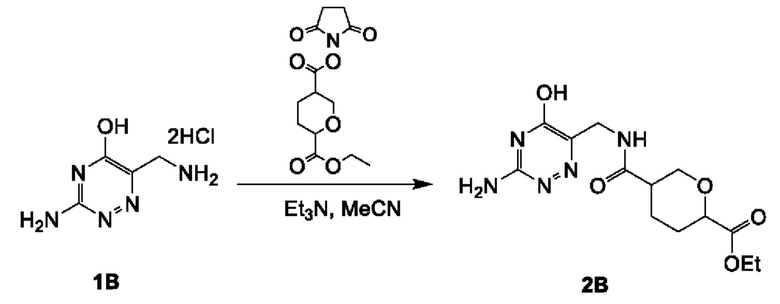
К смеси 5-(2,5-диоксопирролидин-1-ил)-2-этил-тетрагидро-2Н-пиран-2,5-дикарбоксилата (20 г, неочищенный; 60,88 ммоль) в ацетонитриле (30 мл) добавляли соединение 1 В (13 г; 60,88 ммоль) и триэтиламин (25 мл; 182,6 ммоль). Смесь перемешивали при 50°С в течение 16 часов. Смесь концентрировали с получением соединения 2 В (35 г, неочищенный) в виде коричневого твердого вещества.
ЖХ-МС: tR составляет 0,582 мин в хроматографическом способе 5-95АВ_1.5min_220&254_Shimadzu.lcm (Merck RP18 25-3 мм), МС (ЭРИ) m/z составляет 325,9 [М+Н]+.
5-(2,5-диоксопирролидин-1-ил)-2-этил тетрагидро-2Н-пирап-2,5-дикарбоксилат:
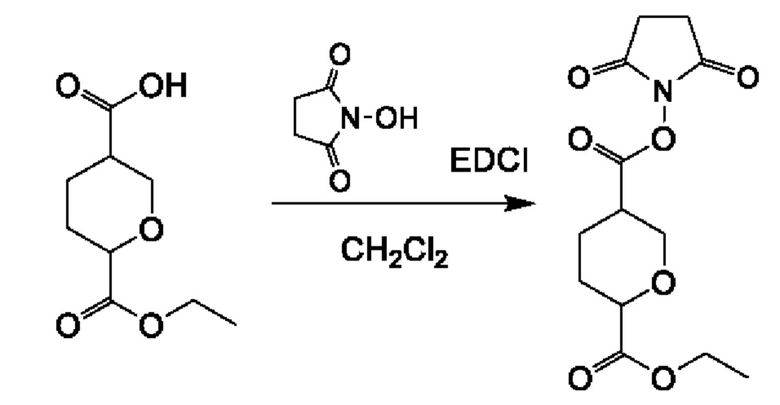
К раствору 6-(этоксикарбонил)тетрагидро-2Н-пиран-3-карбоновой кислоты (1,4 г; 6,92 ммоль) в CH2Cl2 (30 мл) добавляли 1-гидроксипирролидин-2,5-дион (877 мг; 7,61 ммоль) и EDCI (1,6 г; 8,30 ммоль). Смесь перемешивали при 22-26°С в течение 1,5 часа. Смесь разбавляли дихлорметаном (30 мл), промывали рассолом (50 мл×3). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением 5-(2,5-диоксопирролидин-1-ил)-2-этил-тетрагидро-2Н-пиран-2,5-дикарбоксилата в виде бесцветного масла, которое непосредственно использовали.
Соединение 3В: этил-5-(2-амино-4-гидроксиимидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-карбоксилат
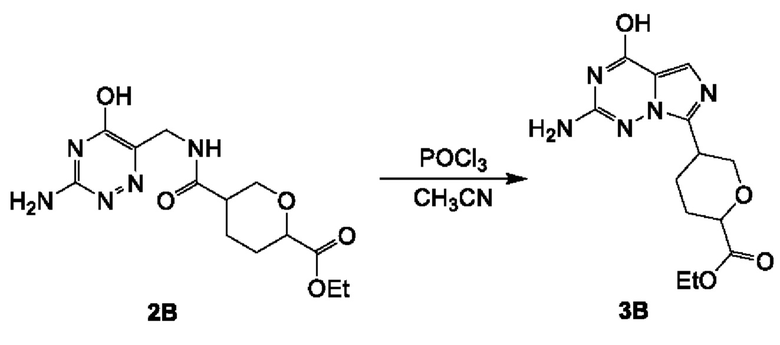
К раствору соединения 2 В (35 г, неочищенный; 60,88 ммоль) в ацетонитриле (300 мл) добавляли хлорангидрид фосфорной кислоты (23 мл; 243,52 ммоль). Затем смесь перемешивали при 70°С в течение 16 часов. Смесь концентрировали, остаток разбавляли дихлорметаном (150 мл), выливали в охлажденный насыщенный раствор бикарбоната натрия (300 мл) до значения рН 8, экстрагировали смесью дихлорметан/метанол (10:1; 200 мл×4) с последующей экстракцией смесью IPA/CHCl3 (150 млх4). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения 3В (45 г, неочищенное) в виде черного масла.
ЖХ-МС: tR составляет 0,13 мин в хроматографическом способе 5-95AB_1.5min_220&254_Shimadzu.lcm (Merck RP18 25-3 мм), МС (ЭРИ) m/z составляет 308,0 [М+Н]+.
Соединение 4В: этил-5-(2-амино-5-бром-4-гидроксиимидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-карбоксилат
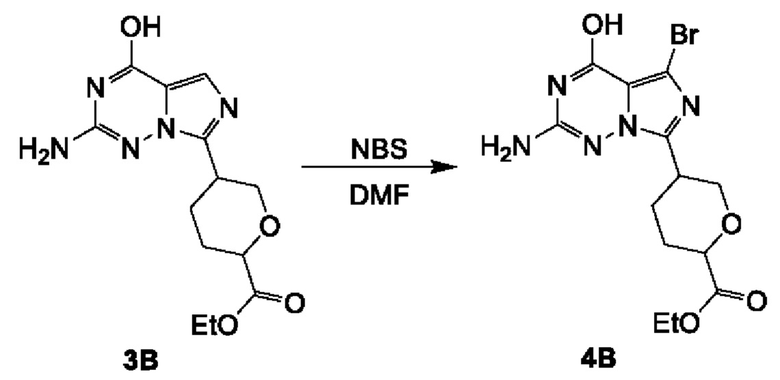
К раствору соединения 3В (45 г, неочищенное; 60,88 ммоль) в N,N-диметилформамиде (200 мл) добавляли NBS (11,8 г; 66,96 ммоль). Смесь перемешивали при комнатной температуре (16-21°С) в течение 0,5 часа. Смесь выливали в воду (300 мл), экстрагировали этилацетатом (300 мл×4), промывали рассолом (500 мл×4). Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения 4 В (19 г, неочищенное) в виде черного масла.
ЖХ-МС: tR составляет 0,602 мин в хроматографическом способе 5-95АВ_lmin_220&254_Agilent chromatography (Agilent Poroshell 120 EC-C18 2,7 мкм 3,0×30 мм), МС (ЭРИ) m/z составляет 388,0 [М+Н+2]+ (Изотоп брома).
Соединение 5В: этил-5-(5-6ром-4-гидроксиимидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-карбоксилат
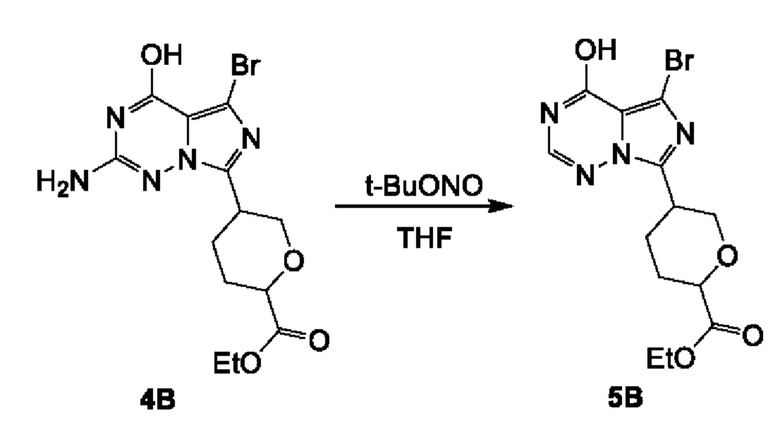
К раствору соединения 4В (19 г, неочищенное; 49,19 ммоль) в тетрагидрофуране (300 мл) добавляли трет-бутилнитрит (12 мл; 98,39 ммоль) при 0-5°С, и смесь перемешивали при комнатной температуре (16-21°С) в течение 16 часов. Смесь объединяли с другой порцией (7,2 г неочищенного соединения 4 В) и концентрировали с получением неочищенного вещества, которое очищали посредством колоночной хроматографии на силикагеле (0-60% этилацетат в петролейном эфире) с получением соединения 5 В (14,3 г; выход 44% за 5 стадий) в виде желтого твердого вещества.
ЖХ-МС: tR составляет 0,753 мин в хроматографическом способе 5-95AB_1.5min_220&254_Shimadzu.lcm (Agilent Pursuit 5 С18 20x×2,0 мм), МС (ЭРИ) m/z составляет 370,9 [М+Н]+.
1Н ЯМР (400 МГц, CD3OD): δ 7.74 (s, 0.3Н), 7.72 (s, 0.5Н), 4.37 (t, J=4,8 Гц, 0.7Н), 4.29-4.16 (m, 3Н), 4.12 (dd, J=11,6, 2,0 Гц, 0.3Н), 3.92 (dd, J=11,6, 4,0 Гц, 0.7Н), 3.70 (t, J=11,2 Гц, 0.4Н), 3.57-3.45 (m, 1H), 2.35-1.92 (m, 4Н), 1.33-1.27 (m, 3Н).
Соединение 6В: этил-5-(4-амино-5-бромимидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-карбоксилат
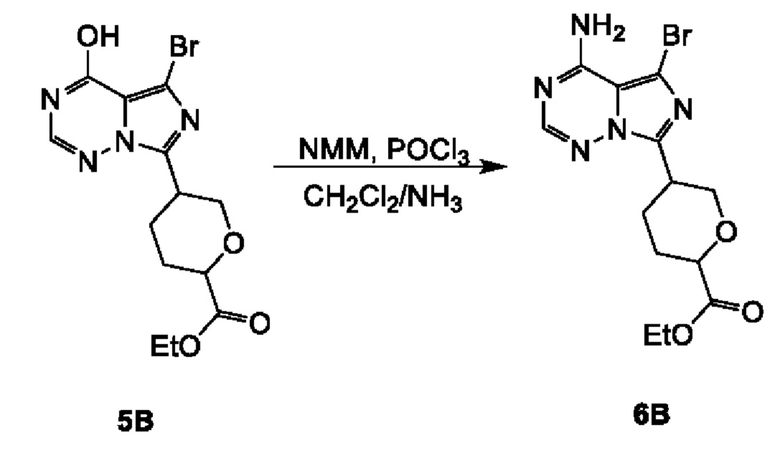
К раствору 1,2,4-триазола (28 г; 404,1 ммоль) в ацетонитриле (200 мл) добавляли POCl3 (12,5 мл; 134,7 моль) при температуре ниже 10°С с последующим добавлением триметиламина (56 мл; 404,1 ммоль). Смесь перемешивали в течение 20 минут при 10°С, добавляли соединение 5 В (5 г; 13,47 ммоль), и реакционную смесь перемешивали в течение 1,5 часа при 90°С. Смесь охлаждали до 10°С, добавляли аммиак (30 мл; 28%), поддерживая температуру ниже 20°С и перемешивали в течение 0,5 часа при 10°С. Получали еще одну порцию в таком же масштабе. Смеси объединяли и разбавляли водой (200 мл) и экстрагировали этилацетатом (500 мл×3), промывали рассолом (500 мл×3), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом, затем очищали посредством колоночной хроматографии на силикагеле (2-4% метанол в дихлорметане) с получением соединения 6 В (9 г; 90% выход) в виде желтого твердого вещества.
ЖХ-МС: tR составляет 0,614 мин в хроматографическом способе \5-95AB_lmin_220&254_Agilent (Agilent Poroshell 120 EC-C18 2,7 мкм 3,0×30 мм), МС (ЭРИ) m/z составляет 372,0 [М+Н+2]+(Изотоп брома).
1H ЯМР (400 МГц, CD3OD): δ 7.81 (s, 0.3H), 7.79 (s, 0.6Н), 4.36 (t, J=5,2 Гц, 0.6H), 4.29-4.19 (m, 3Н), 4.12 (dd, J=11,6, 2,0 Гц, 0.3H), 3.93 (dd, J=11,6, 4,0 Гц, 0.7H), 3.71 (t, J=11,2 Гц, 0.3H), 3.63-3.48 (m, 1H), 2.35-2.20 (m, 1H), 2.17-1.94 (m, 2.7Н), 1.79-1.68 (m, 0.3H), 1.34-1.26 (m, 3Н).
Соединение 7B: (5-(4-амино-5-бромимидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил) метанол
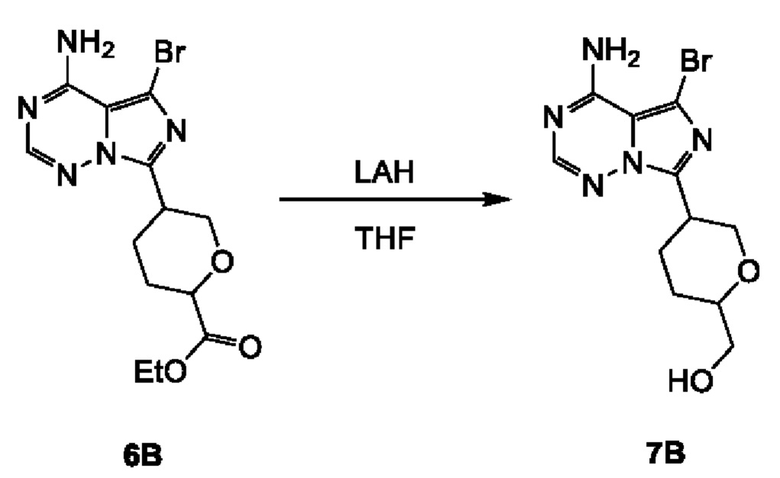
К раствору соединения 6В (6,1 г; 16,48 ммоль) в тетрагидрофуране (120 мл), охлажденному при 0-5°С, добавляли LiAlH4 (1,20 г; 32,95 ммоль), поддерживая температуру ниже 10°С. Смесь перемешивали при 0-10°С в течение 1,5 часа. К смеси добавляли 25 г Na2SO4⋅10H2O при 0-5°С, и перемешивали в течение 2 часов, и фильтровали. Осадок на фильтре дважды суспендировали в смеси дихлорметан/метанол (100 мл смеси 10:1) и фильтровали. Объединенный фильтрат концентрировали под вакуумом, остаток очищали посредством колоночной хроматографии на диоксиде кремния (0~10% метанол в дихлорметане) с получением соединения 7 В (3,9 г; выход 72%) в виде белого твердого вещества и 0,6 г побочного продукта дебромирования в виде желтого твердого вещества.
ЖХ-МС: tR составляет 1,958 и 2,016 мин в хроматографическом способе \0-60AB_7min_220&254_Shimadzu.lcm (Xtimate С18 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z составляет 328,1 [М+Н]+.
1H ЯМР (400 МГц, метанол-d4): δ 7.80 (s, 0.3Н), 7.78 (s, 0.6Н), 4.47 (dt, J=10,0, 2,0 Гц, 0.6Н), 4.20-4.13 (m, 0.3Н), 3.84 (dd, J=11,6, 3,2 Гц, 0.6Н), 3.64 (t, J=10,8 Гц, 0.4H), 3.60-3.38 (m, 4H), 2.45-2.36 (m, 0.6H), 2.22-2.13 (m, 0.3Н), 2.09-1.96 (m, 1H), 1.93-1.73 (m, 1H), 1.62-1.45 (m, 1H).
Соединение 8B: (5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанол
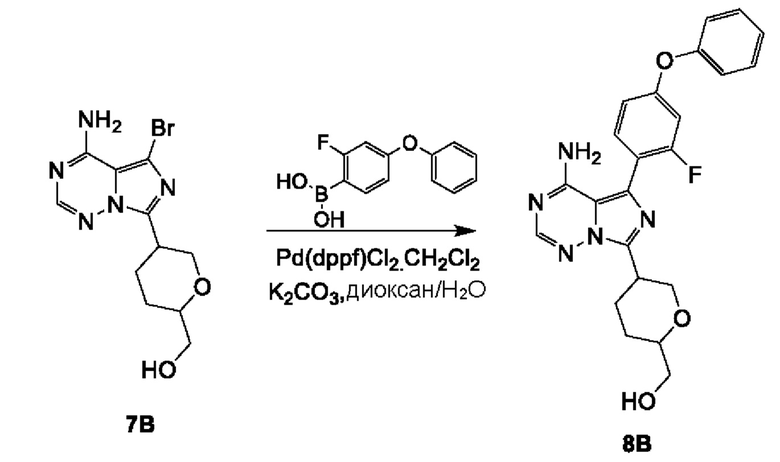
Соединение 7 В (106 мг; 0,457 ммоль), Pd(dppf)Cl2⋅CH2Cl2 (25 мг; 0,0304 ммоль) и карбонат калия (84 мг; 0,608 ммоль) помещали в реакционную пробирку, и продували азотом 3 раза, и добавляли (2-фтор-4-феноксифенил)бороновую кислоту (100 мг; 0,304 ммоль) в 1,4-диоксане (3 мл) и воде (1 мл). Полученную смесь перемешивали при 100°С в атмосфере азота в течение 2 часов. Смесь концентрировали с получением неочищенного вещества, которое очищали посредством колоночной хроматографии на силикагеле (0-100% этилацетат в петролейном эфире) с получением соединения 8 В (80 мг, неочищенное; смесь транс/цис-рацематов) в виде желтого масла.
ЖХ-МС: tR составляет 2,274 мин и 2,340 мин в хроматографическом способе 10-80АВ_7min_220&254_Shimadzu.lcm (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z составляет 436,2 [М+Н]+.
СФХ: tR составляет 4,171 мин, 4,286 мин, 4,454 мин и 5,581 мин. Способ: колонка: Chiralcel OJ-3 150×4,6 мм I.D., 3 мкм; подвижная фаза: А: СО2, В: метанол; градиент: от 5% до 40% В за 5 мин и от 40% до 5% В за 0,5 мин, удерживание при 5% В в течение 1,5 мин; скорость потока: 2,5 мл/мин, температура колонки: 35°С; ABPR: 1500 ф/кв дюйм (10,34 МПа).
Цис-изомер 1, соединение 8: цис-(5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанол
Цис-изомер 2, соединение 9: цис-(5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанол
Транс-изомер 1, соединение 10: транс-(5-(4-амино-5-(2-фтор-4-феноксифенил)-имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанол
Транс-изомер 2, соединение 11: транс-(5-(4-амино-5-(2-фтор-4-феноксифенил)-имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2Н-пиран-2-ил)метанол
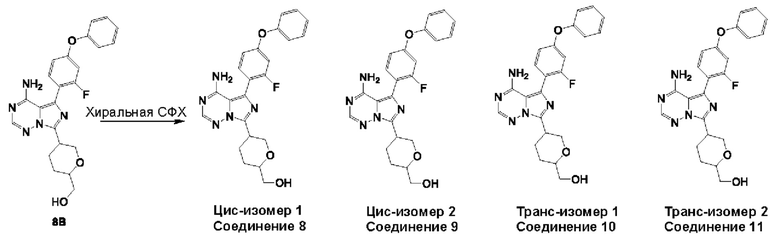
Соединение 8 В (80 мг) далее разделяли посредством препаративной СФХ (колонка: DAICEL CHIRALCEL OJ-H (250 мм×30 мм, 5 мкм); условия: 30% МеОН (0,1% NH3⋅H2O) в СО2; скорость потока: 60 мл/мин) с получением 6,8 мг неочищенного соединения 11, которое дополнительно очищали посредством препаративной ВЭЖХ (колонка: Welch Xtimate С18 100×40 мм, 3 мкм, условия: 25-55% (А: вода(0,225%ЕА), В: CH3CN), скорость потока: 25 мл/мин) с получением соединения 11 (1,7 мг; выход 1% за 2 стадии) в виде белого твердого вещества. После проведения СФХ смесь других трех пиков (40 мг) дополнительно очищали посредством хиральной СФХ (колонка: DAICEL CHIRALPAK IG (250 мм×30 мм, 10 мкм); условия: 55% МеОН (0,1% NH3⋅H2O) в СО2; скорость потока: 80 мл/мин) с получением соединения 8 (8,6 мг; выход 6,4% за 2 стадии) в виде белого твердого вещества, соединения 9 (6,7 мг; выход 5% за 2 стадии) в виде белого твердого вещества и соединения 10 (5,4 мг; выход 4% за 2 стадии) в виде белого твердого вещества.
Спектры соединения 8:
ЖХ-МС: tR составляет 2,356 мин в хроматографическом способе 10-80АВ_7min_220&254_Shimadzu.lcm (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z составляет 436,2 [М+Н]+.
ВЭЖХ: tR составляет 3,00 мин в хроматографическом способе 10-80CD_8min.met. (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР (400 МГц, CD3OD): δ 7.84 (s, m), 7.54 (t, J=8,4 Гц, 1H), 7.47-7.41 (m, 2Н), 7.26-7.20 (m, 1H), 7.17-7.12 (m, 2Н), 6.94 (dd, J=8,0, 2,0 Гц, 1Н), 6.88 (dd, J=10,8, 2,4 Гц, 1H), 4.56-4.51 (m, 1H), 3.89 (dd, J=11,6, 3,6 Гц, 1H), 3.65-3.48 (m, 4Н), 2.51-2.42 (m, 1H), 2.12-2.02 (m, 1H), 1.99-1.85 (m, 1H), 1.64-1.55 (m, 1H).
СФХ: tR составляет 4,059 мин, оптическая чистота 99,94%.
Способ: колонка: Chiralcel OJ-3 150×4,6 мм I.D., 3 мкм; подвижная фаза: А: СО2, В: метанол (0,05% DEA); градиент: от 5% до 40% В за 5 мин и от 40% до 5% В за 0,5 мин, удерживание при 5% В в течение 1,5 мин; скорость потока: 2,5 мл/мин; температура колонки: 35°С; ABPR: 1500 ф/кв дюйм (10,34 МПа).
Спектры соединения 9:
ЖХ-МС: tR составляет 2,340 мин в хроматографическом способе 10-80AB_7min_220&254_Shimadzu.lcm (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z составляет 436,3 [М+Н]+.
ВЭЖХ: tR составляет 2,99 мин в хроматографическом способе 10-80CD_8min.met. (XBridge Shield RP 18 2,1×50 мм; 5 мкм).
1Н ЯМР (400 МГц, CD3OD): δ 7.84 (s, m), 7.53 (t, J=8,4 Гц, 1H), 7.47-7.41 (m, 2Н), 7.25-7.19 (m, 1H), 7.17-7.12 (m, 2Н), 6.94 (dd, J=8,4, 2,0 Гц, 1Н), 6.87 (dd, J=11,2, 2,4 Гц, 1H), 4.56-4.50 (m, 1Н), 3.88 (dd, J=12,0, 3,6 Гц, 1H), 3.65-3.48 (m, 4Н), 2.50-2.41 (m, 1H), 2.12-2.02 (m, 1Н), 1.97-1.86 (m, 1H), 1.64-1.55 (m, 1Н).
СФХ: tR составляет 4,161 мин, оптическая чистота 100%.
Способ: колонка: Chiralcel OJ-3 150×4,6 мм I.D., 3 мкм; подвижная фаза: А: СО2, В: метанол (0,05% DEA); градиент: от 5% до 40% В за 5 мин и от 40% до 5% В за 0,5 мин, удерживание при 5% В в течение 1,5 мин; скорость потока: 2,5 мл/мин; температура колонки: 35°С; ABPR: 1500 ф/кв дюйм (10,34 МПа).
Спектры соединения 10:
ЖХ-МС: tR составляет 2,410 мин в хроматографическом способе 10-80AB_7min_220&254_Shimadzu.lcm (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z составляет 436,3 [М+Н]+.
ВЭЖХ: tR составляет 2,98 мин в хроматографическом способе 10-80CD_8min.met. (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР (400 МГц, CD3OD): δ 7.86 (s, 1Н), 7.50 (t, J=8,4 Гц, 1H), 7.47-7.41 (m, 2H), 7.26-7.20 (m, 1H), 7.17-7.12 (m, 2H), 6.94 (dd, J=8,4, 2,4 Гц, 1H), 6.88 (dd, J=10,8, 2,4 Гц, 1H), 4.24-4.18 (m, 1H), 3.75 (t, J=11,2 Гц, 1H), 3.68-3.48 (m, 4H), 2.27-2.19 (m, 1H), 2.16-2.04 (m, 1H), 1.84-1.76 (m, 1H), 1.59-1.47 (m, 1H).
СФХ: tR составляет 4,405 мин, оптическая чистота 99,83%.
Способ: колонка: Chiralcel OJ-3 150×4,6 мм I.D., 3 мкм; подвижная фаза: А: СО2, В: метанол (0,05% DEA); градиент: от 5% до 40% В за 5 мин и от 40% до 5% В за 0,5 мин, удерживание при 5% В в течение 1,5 мин; скорость потока: 2,5 мл/мин; температура колонки: 35°С; ABPR: 1500 ф/кв дюйм (10,34 МПа).
Спектры соединения 11:
ЖХ-МС: tR составлчяет 2,365 мин в хроматографическом способе 10-80АВ_7min_220&254_Shimadzu.lcm (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z составляет 436,2 [М+Н]+.
ВЭЖХ: tR составляет 3,05 мин в хроматографическом способе 10-80CD_8min.met. (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР (400 МГц, CD3OD): δ 7.86 (s, 1Н), 7.50 (t, J=8,4 Гц, 1H), 7.47-7.41 (m, 2H), 7.26-7.21 (m, 1H), 7.17-7.12 (m, 2H), 6.94 (dd, J=8,4, 2,4 Гц, 1H), 6.88 (dd, J=10,8, 2,4 Гц, 1H), 4.24-4.18 (m, 1H), 3.75 (t, J=10,8 Гц, 1H), 3.68-3.47 (m, 4H), 2.26-2.18 (m, 1H), 2.16-2.04 (m, 1H), 1.84-1.76 (m, 1H), 1.59-1.47 (m, 1H).
СФХ: tR составляет 5,802 мин, оптическая чистота 100%.
Способ: колонка: Chiralcel OJ-3 150×4,6 мм I.D., 3 мкм; подвижная фаза: А: СО2, В: метанол (0,05% DEA); градиент: от 5% до 40% В за 5 мин и от 40% до 5% В за 0,5 мин, удерживание при 5% В в течение 1,5 мин; скорость потока: 2,5 мл/мин; температура колонки: 35°С; ABPR: 1500 ф/кв дюйм (10,34 МПа). Следующие соединения синтезировали согласно способу В:

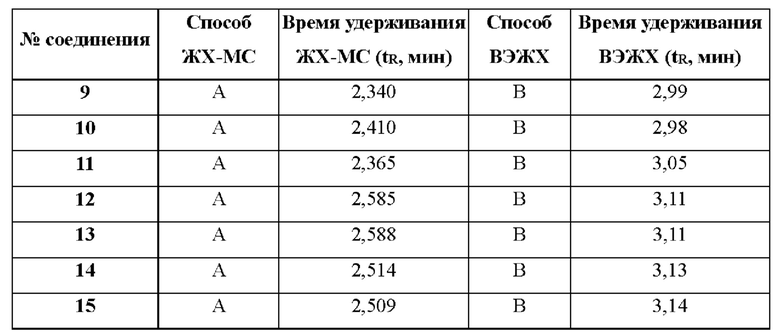
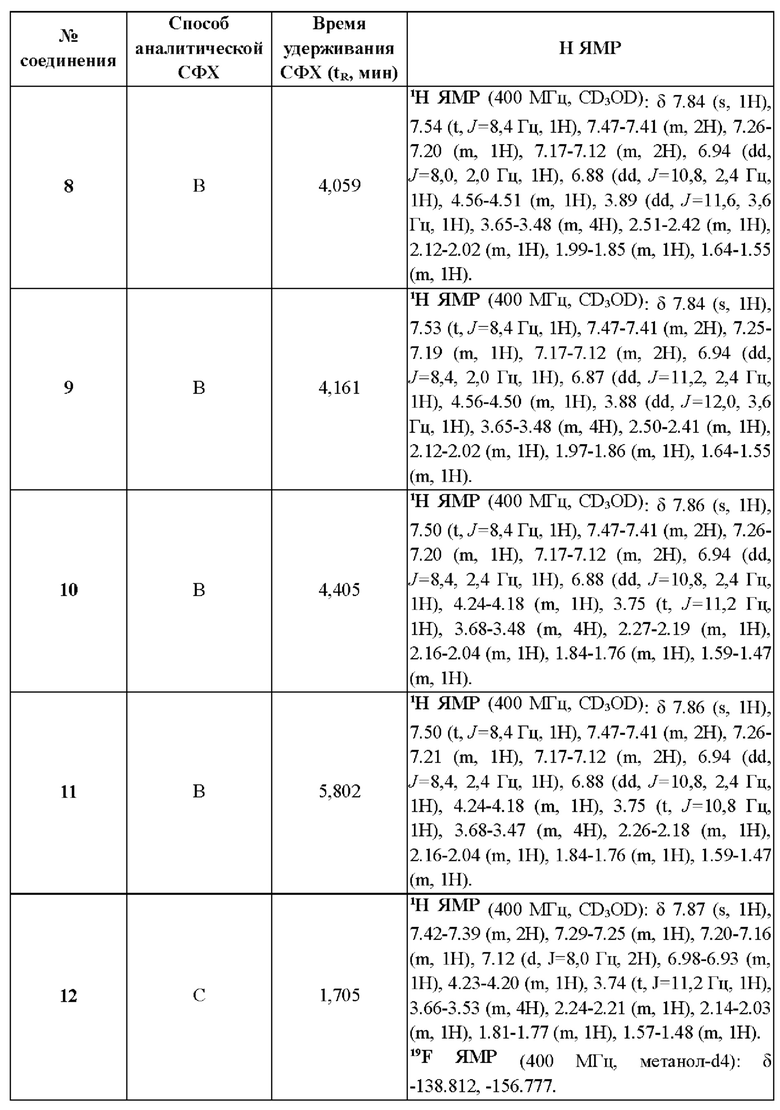
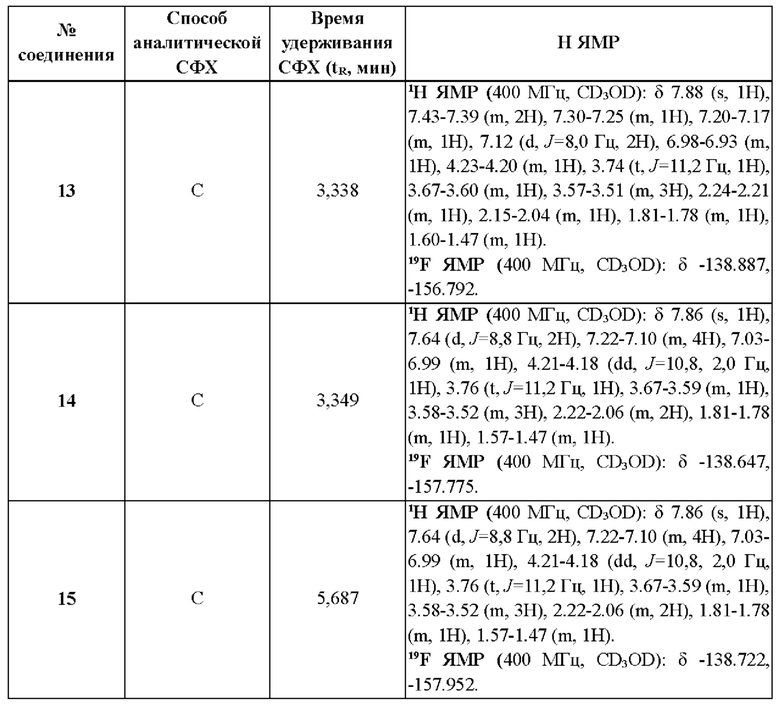
Биологические данные
Анализ киназы ВТК WT и ВТК C481S HTRF
Рекомбинантная ВТК дикого типа (ВТК WT) была приобретена у Thermo fisher. Рекомбинантная BTK(C481S) была приобретена у SignalChem. Ингибирующую активность соединений в отношении ВТК и BTK(C481S) оценивали с использованием метода гомогенной флуоресценции с временным разрешением.
Вкратце, рекомбинантные киназы предварительно инкубировали в присутствии или в отсутствие соединения при комнатной температуре в течение 30 минут. Реакцию инициировали посредством добавления АТФ и пептидного субстрата, который в ходе реакции мог быть фосфорилирован киназами. После инкубации в течение 120 минут реакцию останавливали посредством добавления смеси реагентов для обнаружения, содержащей EDTA. Флуоресценцию измеряли при 615 нм и 665 нм соответственно с длиной волны возбуждения 320 нм. Рассчитанное соотношение сигналов 665 нм/615 нм пропорционально активности киназы. Концентрацию соединения, обеспечивающую 50% ингибирование соответствующей киназы (IC50), рассчитывали с использованием четырехпараметрической логистической модели подгонки с XL-подгонкой.
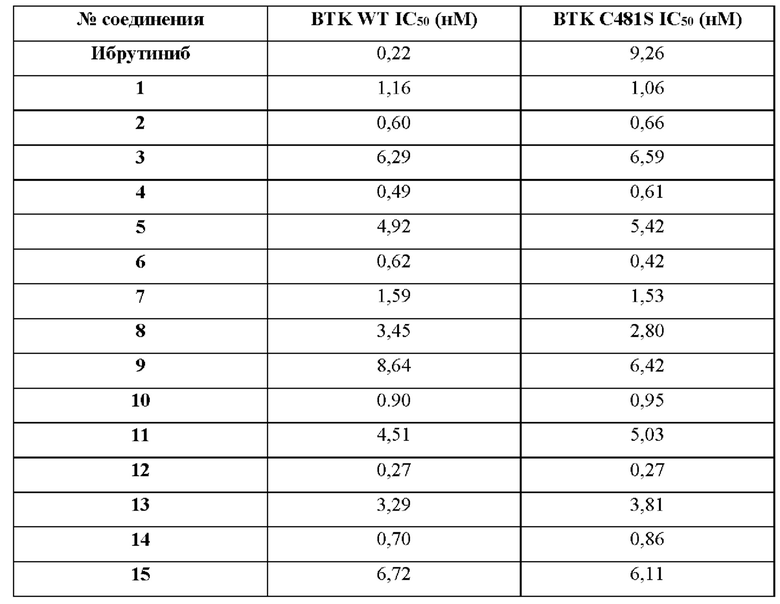
Анализ ИФА (иммуноферментный анализ) р-ВТК клеточной линии TMD8
Клетки TMD-8 высевали в 96-луночные планшеты с плотностью 30000 клеток/лунка со средой RPMI1640 с 1,5% фетальной бычьей сыворотки. К клеткам добавляли исследуемые соединения и клетки инкубировали в течение 0,5 часа при 37°С, 5% СО2. Затем к клеткам добавляли раствор перванадата до создания конечной концентрации 100 мкМ, и клетки инкубировали в течение еще 1 часа при 37°С, 5% СО2. После обработки соединением клетки лизировали, и сигнал р-ВТК детектировали в соответствии с методикой, точно соответствующей набору для анализа сендвич-ИФА (#23843) PathScan® Phospho-Btk (Туг223). Планшет считывали на ридере Multiscan Spectrum, настроенном на длину волны 450 нм. Эти данные были обработаны в программе GraphPad Prism.
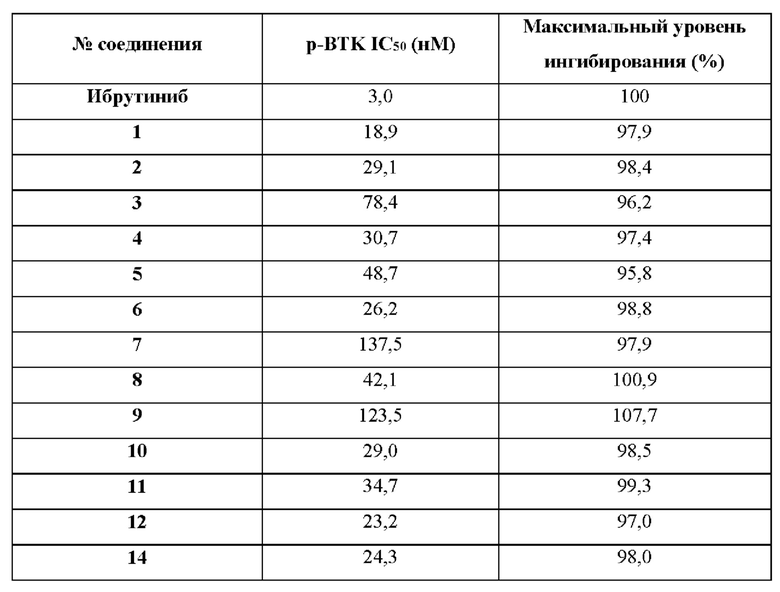
HEK293-BTK(WT) и HEK293-BTK(C481S)
Полноразмерные кДНК (комплементарные дезоксирибонуклеиновые кислоты) ВТК, содержащие мутацию C481S, были созданы посредством мутагенеза с использованием набора для сайт-направленного мутагенеза QuikChange II XL. Мутантные кДНК ВТК были подтверждены секвенированием. Затем кДНК BTK(WT), BTK-C481S клонировали в лентивирусный вектор PLVX-Puro. Лентивирусы упаковывали в клетки 293Т путем трансфекции лентивирусными векторами и смесью упаковок. Лентивирусы BTK(WT), BTK-C481S трансфицировали в клетки НЕК293. Трансфицированные клетки отбирали в 2 мкг/мл пуромицина. Стабильные поликлональные клеточные линии были подтверждены WB и использованы для дальнейшего исследования. Клетки HEK293-BTK(WT), HEK293-BTK(C481S) культивировали в среде DMEM (Gibco; 12430), с 10% FBS (фетальная бычья сыворотка) (Gibco; 10099), с 1 мкг/мл пуромицина. Все клетки содержали в инкубаторе с контролем влажности при 37°С с 5% СО2.
Клетки HEK293-BTK(WT) и HEK293-BTK(C481S) высевали в 96-луночные планшеты с плотностью 5000 клеток/лунка со средой RPMI1640 с 1,5% фетальной бычьей сыворотки. К клеткам добавляли исследуемые соединения, и клетки инкубировали в течение 1,5 часа при 37°С, 5% СО2. После обработки соединением клетки лизировали, и сигнал р-ВТК детектировали в соответствии с методикой, точно соответствующей набору для анализа сендвич-ИФА (#23843) PathScan® Phospho-Btk (Tyr223). Планшет считывали на ридере Multiscan Spectrum, настроенном на длину волны 450 нм. Данные были обработаны в программе GraphPad Prism.
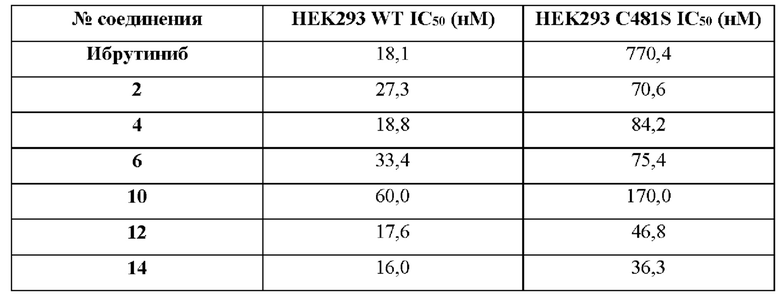
Анализ анти-пролиферации TMD-8
Клетки TMD-8 готовили в среде RPMI1640, содержащей 10% фетальной бычьей сыворотки, и высевали в 384-луночные планшеты с плотностью 1500 клеток на лунку. Клетки инкубировали в течение ночи при 37°С, 5% СО2. После инкубации в планшеты для анализа добавляли соединения с различной концентрацией и инкубировали клетки в течение еще 72 часов при 37°С, 5% СО2. После инкубации в течение 72 часов в каждую лунку добавляли 15 мкл реагента CellTiter 96® AQueous One Solution Reagent, и планшеты инкубировали при комнатной температуре в течение 30 минут.Жизнеспособность клеток определяли при использовании набора CellTiter-Glo (Promega, США). Анализ CellTiter-Glo проводили в соответствии с инструкциями производителя, а люминесценцию определяли в многомодовом планшет-ридере (Envision, PerkinElmer, США). Данные обрабатывали в программе XLfit.

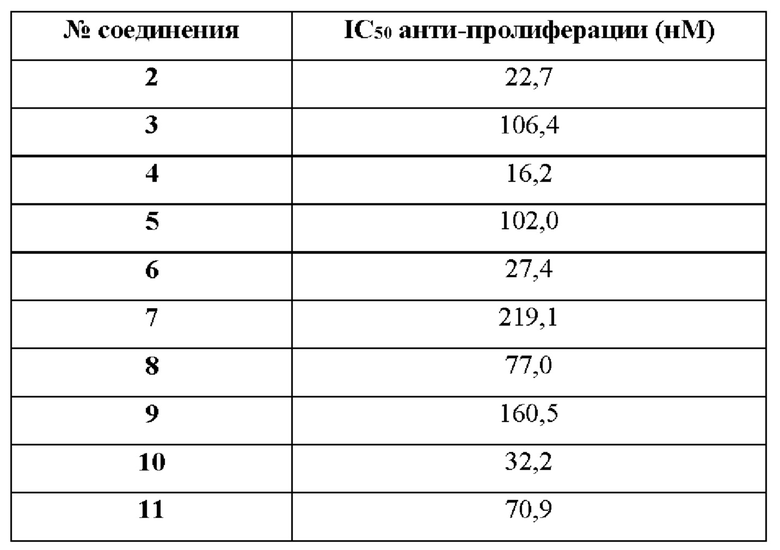
In vitro анализ клиренса гепатоцитов крысы/человека
Гепатоциты самцов крысы и гепатоциты человека обоих полов были получены от коммерческих поставщиков (например, BioreclamationlVT) и хранились при -150°С до использования. Готовили 10 мМ исходные растворы исследуемых соединений в DMSO. Среду для оттаивания и инкубационную среду с добавками (бессывороточную) перед использованием помещали на водяную баню при 37°С по меньшей мере на 15 минут. Исходные растворы разбавляли до 100 мкМ посредством объединения 198 мкл ацетонитрила и 2 мкл 10 мМ исходного раствора.
Флаконы с криоконсервированными гепатоцитами изымали из хранилища, следя за тем, чтобы флаконы оставались при криогенных температурах. Флаконы оттаивали на водяной бане при 37°С при осторожном встряхивании. Флаконы выдерживали на водяной бане до тех пор, пока все кристаллы льда не растворялись и не исчезали. Флаконы опрыскивали 70% этанолом перед переносом в бокс биологической безопасности. А затем содержимое выливали в коническую пробирку со средой для оттаивания объемом 50 мл. Флаконы центрифугировали при 100 g в течение 10 минут при комнатной температуре. Среду для оттаивания отсасывали, а гепатоциты ресуспендировали в бессывороточной инкубационной среде с получением приблизительно 1,5×106 клеток/мл.
Жизнеспособность и плотность клеток подсчитывали, используя исключение трипанового синего, а затем клетки разводили бессывороточной инкубационной средой до рабочей плотности клеток 1×106 жизнеспособных клеток/мл. Часть гепатоцитов с концентрацией 1×106 жизнеспособных клеток/мл кипятили в течение 10 мин перед добавлением в планшет в качестве отрицательного контроля для устранения ферментативной активности, чтобы наблюдалось лишь незначительное превращение субстрата или его отсутствие. Инактивированные гепатоциты использовали для приготовления отрицательных образцов, которые использовали для исключения вводящего в заблуждение фактора, связанного с нестабильностью самого химического вещества.
Аликвоты по 247,5 мкл гепатоцитов распределяли в каждую лунку 96-луночного непокрытого планшета. Планшет помещали в инкубатор на орбитальном шейкере примерно на 10 минут. Аликвоты по 2,5 мкл 100 мкМ исследуемых соединений добавляли в соответствующие лунки непокрытого 96-луночного планшета для начала реакции. Этот анализ проводили в двух повторностях. Планшет инкубировали в инкубаторе на орбитальном шейкере в течение расчетных моментов времени. 20 мкл содержимого переносили и смешивали с 6 объемами (120 мкл) холодного ацетонитрила с внутренним стандартом для прекращения реакции в моменты времени 5, 15, 30, 45, 60, 80 и 100 минут. Образцы центрифугировали в течение 20 минут при 4000 g, и аликвоты по 100 мкл супернатантов использовали для анализа ЖХ-МС/МС для измерения исследуемых соединений.
Клиренс гепатоцитов in vitro оценивали на основании определения периода полувыведения (Т1/2) соединений от их исходных концентраций. Рассчитывали отношения площадей пиков каждого соединения (тестируемого или контрольного) к IS (внутреннему стандарту). Строили кривую зависимости Ln (% контроля) от времени инкубации (мин) и рассчитывали наклон линейной аппроксимирующей прямой. Константу скорости выведения лекарственного средства k (мин-1), Т1/2 (мин) и внутренний клиренс CLint (мкл/мин/106 клеток) in vitro рассчитывали согласно следующим уравнениям:
k= -угловой коэффициент
Т1/2=0,693/k
CLint=k/Chep
где Chep (клетки × мкл-1) представляет собой концентрацию клеток в инкубационной системе.
Методика определения Log D
10 мкл рабочего раствора из каждой кассеты помещают по порядку в соответствующую позицию 96-луночного штатива (планшет Log D). Добавляют 500 мкл насыщенного октанола в каждую лунку вышеуказанного планшета Log D без крышки с последующим добавлением 500 мкл насыщенного фосфатного буфера. Закрывают герметично самоуплотняющейся крышкой PTFE/SIL для 96-луночного планшета.
Планшет Log D переносят в шейкер для планшетов Eppendorf Thermomixer Comfort и встряхивают при 25°С, 2000 об/мин в течение 2 часов.
Образцы центрифугируют при 4000 об/мин при 25°С в течение 30 минут для разделения фаз. Пипетку и шприц используют для пипетирования приблизительно 100 мкл из октаноловой и буферной фаз в новый 96-луночный планшет соответственно.
5 мкл октанольного образца переносят в новый 96-луночный планшет с последующим добавлением 495 мкл смеси H2O и ацетонитрила, содержащей внутренний стандарт (1:1), в виде разведенных в 100 раз октанольных образцов. Перемешивали с помощью вихревой мешалки в течение 5 минут при 1000 об/мин.
50 мкл разведенных в 100 раз образцов переносят в новый 96-луночный планшет с последующим добавлением 450 мкл смеси H2O и ацетонитрила, содержащей внутренний стандарт (1:1), в виде разведенных в 1000 раз октанольных образцов. Перемешивали с помощью вихревой мешалки в течение 5 минут при 1000 об/мин.
Разведенные в 1000 раз октанольные образцы последовательно разбавляют в 10000, 100000 и 1000000 раз смесью H2O и ацетонитрила, содержащей внутренний стандарт (1:1).
50 мкл образцов буфера переносят в новый 96-луночный планшет с последующим добавлением 450 мкл смеси H2O и ацетонитрила, содержащей внутренний стандарт (1:1), в виде разведенных в 10 раз образцов буфера. Перемешивали с помощью вихревой мешалки в течение 5 минут при 1000 об/мин.
Разведенные в 10 раз образцы буфера последовательно разбавляют в 100, 1000 и 10000 раз смесью H2O и ацетонитрила, содержащей внутренний стандарт (1:1). Образцы оценивают с помощью анализа ЖХ/МС/МС. Все соединения тестируют в одной повторности.
Все расчеты проводят с помощью Microsoft Excel. Концентрации исследуемого соединения в растворе октанол/буфер оценивают посредством анализа ЖХ/МС/МС. Значение Log D исследуемого соединения рассчитывают следующим образом:
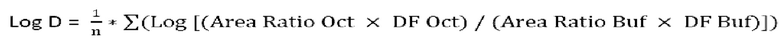
DF означает коэффициент разведения.
Area Ratio Oct означает отношение площадей октанольных образцов.
Area Ratio Buf означает отношение площадей образцов буфера.
Методика измерения связывания белков в плазме человека с использованием равновесного диализа
Добавляют 597 мкл контрольной плазмы в каждую лунку нового пластикового планшета или отдельную пластиковую пробирку, добавив 3 мкл рабочего раствора из каждой кассеты, перемешивали с помощью вихревой мешалки при 1000 об/мин в течение 5 минут. Конечный процент объема органического растворителя составляет 0,5%, а конечная концентрация исследуемого соединения составляет 5 мкМ. Немедленно переносят 50 мкл суспензии плазмы с добавками в 96-луночный планшет, чтобы действовать в качестве контрольного образца Т=0. Образцы обрабатывают так же, как и образцы после инкубации. Помещают всю оставшуюся плазму с добавками в инкубатор на время исследования.
Помещают вставки открытым концом вверх в лунки основного планшета. Добавляют 500 мкл фосфатного буфера (рН 7,4) в буферную камеру, обозначенную белым кольцом. Добавляют 300 мкл образца плазмы с добавками в камеру для образцов, которая обозначена красным кольцом. Накрывают прибор газопроницаемой крышкой и инкубируют в течение 18 часов при 37°С при 300 об/мин с 5% СО2 на орбитальном шейкере в CO2-инкубаторе. В конце инкубации удаляют крышку и пипетируют 50 мкл образцов после диализа как из буферной, так и плазменной камер в отдельный 96-луночный планшет для анализа соответственно.
В то же время оставшийся образец плазмы с добавками в пластиковом планшете или отдельной пластиковой пробирке инкубируют в течение 18 часов при 37°С с 5% СО2 в CO2-инкубаторе. В момент времени Т, равный 18 часов, переносят 50 мкл исходной суспензии плазмы с добавками в 96-луночный планшет для анализа.
Добавляют 50 мкл плазмы человека в образцы буфера и равный объем PBS (фосфатно-солевой буфер) в собранные образцы плазмы. Планшет перемешивают с помощью вихревой мешалки при 1000 об/мин в течение 2 минут и добавляют 400 мкл ацетонитрила, содержащего соответствующий внутренний стандарт (IS) для осаждения белка и высвобождения соединения. Перемешивают с помощью вихревой мешалки при 1000 об/мин в течение 10 минут. Центрифугируют в течение 30 минут при 4000 об/мин. Переносят 250 мкл супернатанта в новые 96-луночные планшеты и снова центрифугируют (4000 об/мин, 30 минут). Затем переносят 100 мкл супернатанта в новые 96-луночные планшеты для анализа. Добавляют в каждый образец по 100 мкл дистиллированной воды и перемешивают с помощью вихревой мешалки в течение 5 минут при 1000 об/мин для анализа посредством ЖХ-МС/МС. Все соединения тестируют в одной повторности при концентрации 5 мкМ в плазме человека.
Все расчеты производят с помощью Microsoft Excel
Рассчитывают процент несвязанного, процент связанного и восстановление исследуемого соединения следующим образом:

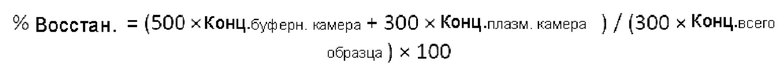
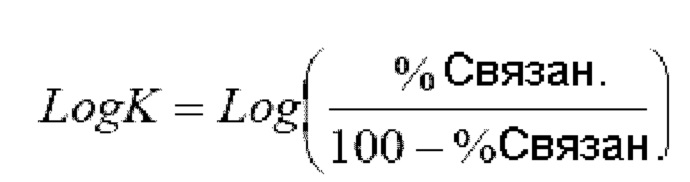
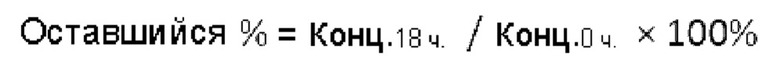
Результаты
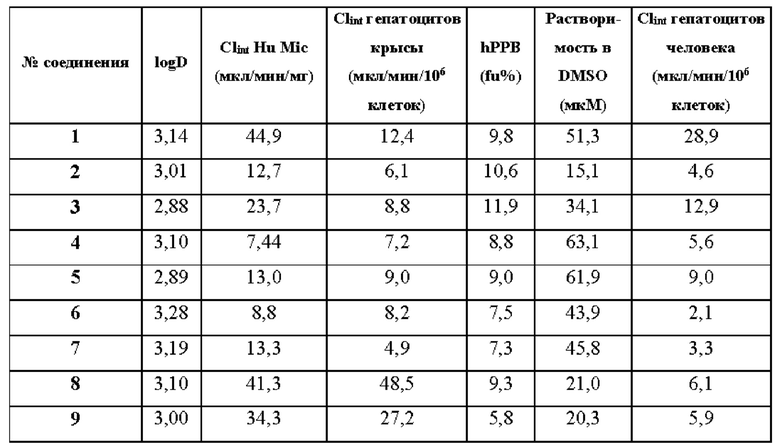
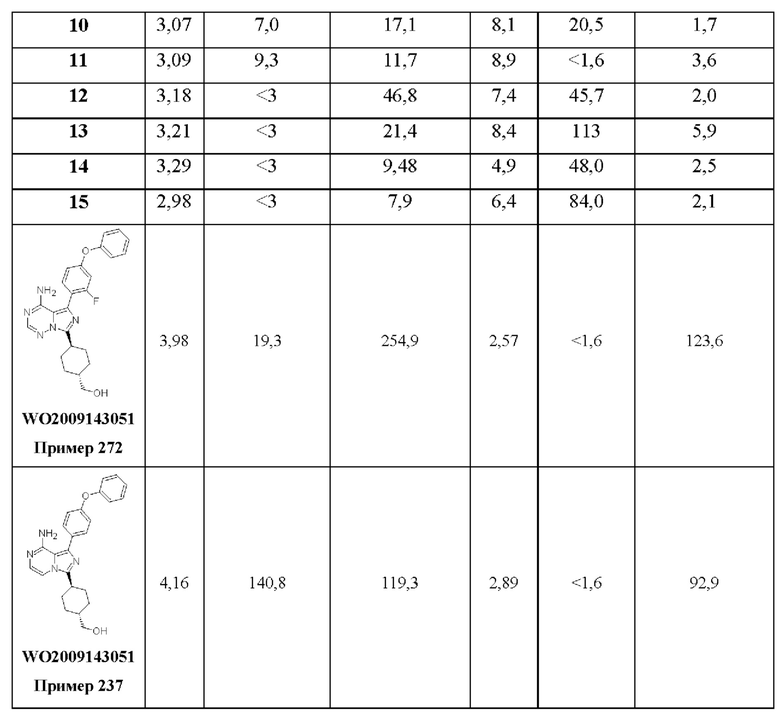
Анализ быстрой пероральной абсорбции (SOA) у крыс
Модель быстрой пероральной абсорбции (SOA) представляет собой модель скрининга in vivo для выявления проникновения соединения в мозг. Чтобы оценить способность соединения проникать через гематоэнцефалический барьер у крыс, измеряли общее соотношение мозг/плазма (Кр, мозг) с помощью АUСмозг/АUСплазма. Соотношение СМЖ (спинномозговая жидкость)/плазма (Кр,СМЖ) определяли либо по АUССМЖ/AUCплазма, либо по среднему соотношению СМЖплазма в доступные моменты времени после перорального введения соответственно. Свободные фракции в биологической матрице определяли с помощью in vitro анализа связывания в плазме и мозге в отдельном исследовании. (1) Кр,uu,мозг и Кр,uu,СМЖ рассчитывали по следующим уравнениям: 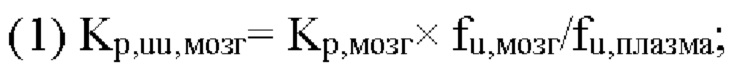
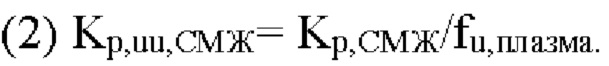
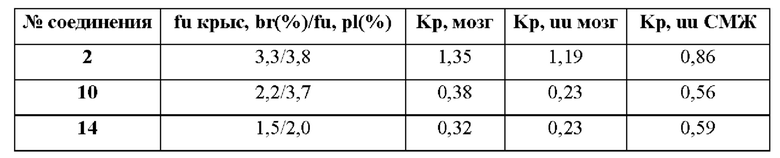
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ИМИДАЗОТРИАЗИНОНА | 2011 |
|
RU2603140C2 |
| ИНГИБИТОРЫ mTOR КИНАЗЫ ДЛЯ ОНКОЛОГИЧЕСКИХ ПОКАЗАНИЙ И ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С mTOR/PI3K/AKT ПУТЕМ МЕТАБОЛИЗМА | 2009 |
|
RU2546658C2 |
| Новые производные имидазо[4,5-с]хинолина в качестве ингибиторов LRRK2 | 2018 |
|
RU2773516C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ПИПЕРАЗИНА | 2012 |
|
RU2628616C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ПИРРОЛОТРИАЗИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2726632C1 |
| ИНГИБИТОРЫ mTOR КИНАЗЫ ДЛЯ ОНКОЛОГИЧЕСКИХ ПОКАЗАНИЙ И ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С mTOR/PI3K/AKT ПУТЕМ МЕТАБОЛИЗМА | 2009 |
|
RU2692796C2 |
| Новые производные имидазо[4,5-c] хинолинов и имидазо[4,5-c][1,5] нафтиридинов в качестве ингибиторов LRRK2 | 2016 |
|
RU2722149C1 |
| Соединение, ингибирующее активности киназ ВТК и/или JAK3 | 2014 |
|
RU2650512C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ И ОЧИСТКИ ГЕТЕРОАРИЛЬНЫХ СОЕДИНЕНИЙ | 2010 |
|
RU2557242C2 |
Изобретение относится к соединениям формулы (I)
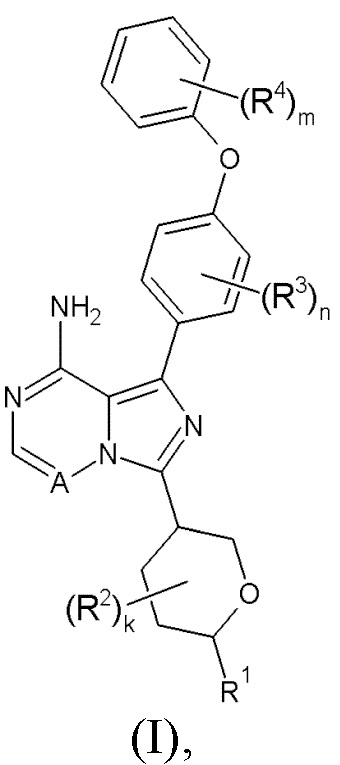
где R1 представляет собой C1-6алкил, где R1 может быть замещен одним R5; k равен 0; R3 представляет собой галоген; n равен 0-2; R4 представляет собой галоген; m равен 0-2; A представляет собой =N- или =C(R6)-; R5 представляет собой гидрокси; R6 представляет собой водород; или его фармацевтически приемлемой соли, а также фармацевтической композиции и способу лечения заболеваний или расстройств, опосредованных полностью или частично BTK, и способу ингибирования BTK. Технический результат: получены новые соединения, которые обладают ингибирующей активностью в отношении BTK, где BTK представляет собой BTK дикого типа или BTK с мутацией C481. 4 н. и 13 з.п. ф-лы, 15 пр.
1. Соединение формулы (I)
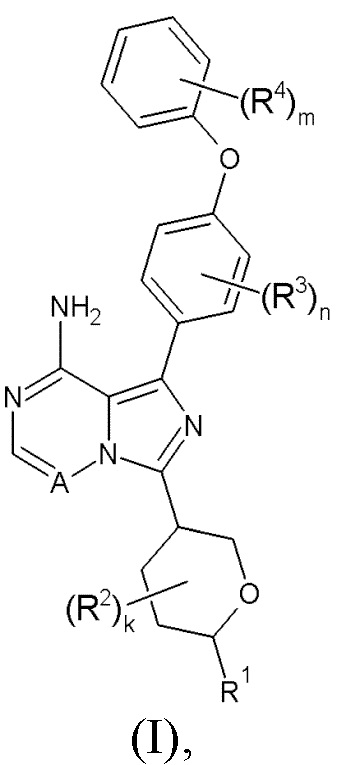
где R1 представляет собой C1-6алкил, где R1 может быть замещен одним R5;
k равен 0;
R3 представляет собой галоген;
n равен 0-2;
R4 представляет собой галоген;
m равен 0-2;
A представляет собой =N- или =C(R6)-;
R5 представляет собой гидрокси;
R6 представляет собой водород;
или его фармацевтически приемлемая соль.
2. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой метил или гидроксиметил.
3. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1 или 2, где R3 представляет собой фтор.
4. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-3, где R4 представляет собой фтор.
5. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-4, где A представляет собой =N-.
6. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-5, выбранное из:
(5-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
(5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
(5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
(5-(8-амино-1-(4-(2,3-дифторфенокси)фенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
(5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
(5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола; и
(5-(4-амино-5-(4-(2,3-дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола.
7. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-5, выбранное из:
((2R,5R)-5-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5R)-5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5R)-5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5R)-5-(8-амино-1-(4-(2,3-дифторфенокси)фенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5R)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5R)-5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5R)-5-(4-амино-5-(4-(2,3-дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5S)-5-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5S)-5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5S)-5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5S)-5-(8-амино-1-(4-(2,3-дифторфенокси)фенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5S)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5S)-5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5S)-5-(4-амино-5-(4-(2,3-дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5R)-5-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5R)-5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5R)-5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5R)-5-(8-амино-1-(4-(2,3-дифторфенокси)фенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5R)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5R)-5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2S,5R)-5-(4-амино-5-(4-(2,3-дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5S)-5-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5S)-5-(8-амино-1-(2-фтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5S)-5-(8-амино-1-(2,3-дифтор-4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5S)-5-(8-амино-1-(4-(2,3-дифторфенокси)фенил)имидазо[1,5-a]пиразин-3-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5S)-5-(4-амино-5-(2-фтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола;
((2R,5S)-5-(4-амино-5-(2,3-дифтор-4-феноксифенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола; и
((2R,5S)-5-(4-амино-5-(4-(2,3-дифторфенокси)фенил)имидазо[5,1-f][1,2,4]триазин-7-ил)тетрагидро-2H-пиран-2-ил)метанола.
8. Фармацевтическая композиция для ингибирования тирозинкиназы Брутона (BTK), содержащая эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-7 в сочетании с фармацевтически приемлемым разбавителем или носителем.
9. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-7 для применения в качестве лекарственного средства для ингибирования BTK.
10. Способ лечения заболеваний или расстройств, опосредованных полностью или частично BTK, у теплокровного животного, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-7, где указанное заболевание или расстройство выбрано из иммунных расстройств, рака, сердечно-сосудистых заболеваний, вирусных инфекций, нарушений метаболизма/функций эндокринных желез, неврологических расстройств, аллергических заболеваний, аутоиммунных заболеваний и воспалительных заболеваний.
11. Способ по п. 10, где рак выбран из мелкоклеточной лимфоцитарной лимфомы (МЛЛ), фолликулярной лимфомы, трансформации Рихтера, мантийноклеточной лимфомы, хронического лимфоцитарного лейкоза (ХЛЛ), макроглобулинемии Вальденстрёма, неходжкинской лимфомы, первичной лимфомы центральной нервной системы, вторичной лимфомы центральной нервной системы или диффузной крупноклеточной В-клеточной лимфомы.
12. Способ по п. 10, где заболевание или расстройство выбрано из крапивницы/синдрома Шегрена, ревматоидного артрита, остеопороза, васкулита, идиопатической тромбоцитопенической пурпуры (ИТП), тяжелой миастении, аллергического ринита, астмы, рассеянного склероза и системной красной волчанки.
13. Способ по п. 10, где теплокровное животное представляет собой человека.
14. Способ по п. 12, где заболевание или расстройство представляет собой рассеянный склероз.
15. Способ ингибирования BTK, включающий введение животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-7.
16. Способ по п. 15, где BTK представляет собой BTK дикого типа или BTK с мутацией C481.
17. Способ по п. 16, где BTK c мутацией C481 выбрана из группы, состоящей из BTK с мутацией C481S, BTK с мутацией C481Y, BTK с мутацией C481R и BTK с мутацией C481F.
| WO 2015074138 A1, 28.05.2015 | |||
| WO 2018001331 A1, 04.01.2018 | |||
| WO 2012158795 A1, 22.11.2012 | |||
| ИМИДАЗОПИРАЗИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗ | 2004 |
|
RU2405784C2 |

