Предмет изобретения
Настоящее изобретение относится к процессу нитрозирования фенола с получением 4-нитрозофенола. Более конкретно, изобретение относится к непрерывному способу получения 4-нитрозофенола взаимодействием фенола с нитритом натрия и серной кислотой в водной среде, позволяющему получать 4-нитрозофенол высокой степени чистоты, а также получать глауберову соль в качестве ценного попутного продукта.
Предшествующий уровень техники
4-Нитрозофенол (пара-нитрозофенол), является промежуточным продуктом во многочисленных реакциях нитро- и аминосоединений и широко используется в производстве продуктов органического синтеза, красителей, пластмасс, фармацевтических препаратов и других продуктов.
В промышленности для получения 4-нитрозофенола обычно используется процесс нитрозирования фенола реакцией с нитритом натрия и серной кислоты в водном растворе. Общее (балансовое) уравнение такой реакции нитрозирования фенола выглядит следующим образом:
С6Н5ОН+0,5H2SO4+NaNO2=NOC6H4OH+0,5Na2SO4+H2O.
Согласно приведенному уравнению, на реакцию с 1 молем фенола C6H5OH уходит 1 моль нитрита натрия NaNO2 и 0,5 молей серной кислоты H2SO4. При этом на 1 моль прореагировавшего фенола наряду с 1 молем 4-нитрозофенола образуется 0,5 молей сульфата натрия Na2SO4 и 1 моль воды Н2О.
В соответствии с известными литературными данными, полагают, что в основе механизма реакции нитрозирования фенола лежит первоначальная электрофильная атака ароматического кольца (лимитирующая стадия) нитрозирующим агентом. Все нитрозирующие электрофильные агенты обычно образуются из азотистой кислоты. Последняя неустойчива, и ее обычно получают в момент реакции из нитрита натрия в присутствии сильных минеральных кислот при низкой температуре (0-5°С).
Наименее активным нитрозирующим агентом является неионизированная молекула азотистой кислоты HO-N=O, которая под действием минеральной кислоты превращается в активную электрофильную форму (Э+). При этом активность нитрозирующих агентов в реакции нитрозирования фенола уменьшается в следующем ряду:
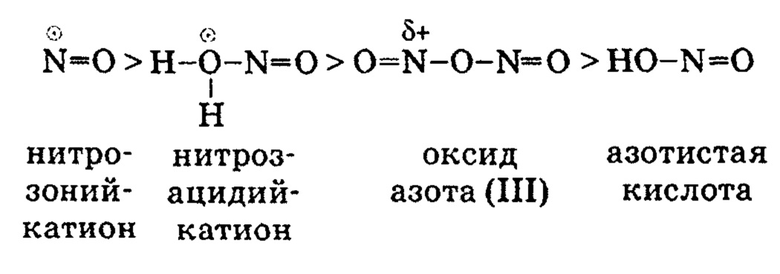
Концентрация активных электрофильных частиц в растворе зависит от условий реакции, таких как рН, температура, избыток азотистой кислоты. Так, самая активная нитрозирующая частица - нитрозоний-катион - образуется в ощутимых количествах лишь в концентрированной серной кислоте (рН<0). Высокая активность нитрозоний-катиона обусловлена наличием незаполненной электронной оболочки у атома азота.
В разбавленной серной кислоте (рН=1÷3) активными агентами являются нитрозацидий-катион и оксид азота (III), который образуется при взаимодействии нитрозацидий-катиона с нитрит-анионом: 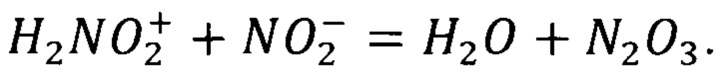
В растворе с высоким значением рН (более 5) концентрация нитрозирующих частиц резко снижается и тем самым подавляются процессы с их участием.
Электрофильный нитрозирующий агент атакует катион водорода, расположенный в пара- и орто-положениях ароматического кольца фенола. В результате в процессе нитрозирования наряду с пара-нитрозофенолом образуется также менее устойчивый орто-нитрозофенол в равновесном соотношении изомеров орто : пара = 1:15. В водном растворе данное соотношение сохраняется неизменным, поэтому родственная примесь орто-нитрозофенола не мешает целевому синтезу пара-нитрозофенола (здесь также называемого «4-нитрозофенол»).
Процесс обычно осуществляют при поддержании температуры в интервале 0-5°С, чтобы не допустить разложения термически неустойчивого 4-нитрозофенола. Кроме того, при повышенной температуре также повышается скорость побочных реакций, инициируемых активными нитрозирующими агентами, в частности реакций с участием 4-нитрозофенола и непрореагировавшего фенола с образованием индофенола, хиноидных соединений и других хромофорных соединений. При пониженных температурах 0-5°С скорость побочных реакций с образованием хромофоров обычно незначительна.
При этом необходимо учитывать, что нитрозирование фенола - экзотермическая реакция, сопровождающаяся выделением большого количества теплоты (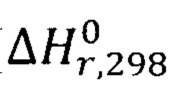 =-239,2 кДж/моль), поэтому для поддержания температуры реакции в интервале 0-5°С процесс следует проводить при охлаждении.
=-239,2 кДж/моль), поэтому для поддержания температуры реакции в интервале 0-5°С процесс следует проводить при охлаждении.
В уровне техники известен непрерывный способ получения п-нитрозофенола взаимодействием водных растворов нитрита натрия, фенола и серной кислоты при 0-15°С, рН<5 и интенсивном перемешивании в реакционной среде, содержащей сульфат натрия (документ US 3320324). В указанном способе нитрит натрия, фенол и серную кислоту берут в химически эквивалентном соотношении (1,2-1,4):1,0:(1,2-1,4), используя 10-85%-ный водный раствор H2SO4, 10-50%-ный водный раствор NaNO2 и 85-90%-ный фенол. В реактор загружают воду и при 5°С и интенсивном перемешивании непрерывно вводят серную кислоту, нитрит натрия и фенол. Примерно через 1 ч, при содержании кристаллического п-нитрозофенола в реакционной смеси в количестве ≈ 10% кристаллы п-нитрозофенола отделяют, и процесс продолжают, непрерывно возвращая маточный раствор в производственный цикл. Выход п-нитрозофенола составляет 73-85 мол., не включая продукт, растворенный в маточной жидкости. [1]
Недостатками данного способа являются относительно невысокий выход целевого продукта, неполная конверсия фенола в целевой продукт, в результате чего его потери с маточной жидкостью составляют 5-10 мол. Кроме того, в результате выведения избытка маточного раствора из процесса для сохранения водно-солевого баланса имеют место потери с маточной жидкостью и других веществ, в том числе, в расчете на 1 моль фенола: Na2SO4 - 0.5 моль; H2SO4 - 0,8 моль; и NaNO2 -0,3 моль. Кроме того, продукты побочных реакций фенола с п-нитрозофенолом (индофенол, хиноидные соединения - хромофоры), растворенные в маточном растворе, частично возвращаемом в цикл, возвращаются в цикл вместе с ним и загрязняют выводимый из процесса продукт - п-нитрозофенол, так что может быть необходима дополнительная стадия очистки п-нитрозофенола от побочных продуктов синтеза. Наконец, в ходе взаимодействия реагентов при указанных молярных соотношениях фенол: нитрит натрия: серная кислота (например, при соотношении = 1,0:1,3:1,3) в реакционной среде, молярное отношение свободного нитрита натрия и серной кислоты составляет 0,3:0,8. Этому соотношению соответствует значение рН среды 2,57, поэтому из реакционного раствора при незначительном избыточном давлении инертного газа могут выделяться окислы азота, ухудшая экологические параметры процесса в целом.
Известен также способ непрерывного получения п-нитрозофенола реакцией фенола с нитритом натрия в водном растворе серной кислоты, который характеризуется тем, что процесс проводят в две стадии (патент Румынии №60020). На первой стадии в реактор непрерывно при перемешивании подают водные растворы фенола, нитрита натрия и серной кислоты при их эквивалентном соотношении (1,0-1,1):1,0:(3,2-3,6) и при массовом соотношении фенол вода 1,0:(17-19) при температуре -1+4°С в течение 110-130 мин. На второй стадии реакционную смесь непрерывно подают во второй реактор с последующим добавлением водного раствора нитрита натрия при соотношении нитрит натрия исходный фенол (0,2-0,3): 1,0, при -2°С 0°С в течение 90-150 мин. В реактор загружают водный раствор серной кислоты, а затем подают водные растворы нитрита натрия и фенола в течение, например, 2 ч, после чего дополнительно добавляют водный раствор нитрита натрия. Выход п-нитрозофенола составляет 85% в расчете на фенол.
Недостатками данного процесса являются относительно невысокие выход и чистота получаемого п-нитрозофенола. Кроме того, данный процесс характеризуется высоким расходным коэффициентом по серной кислоте, что приводит к улучшению растворимости п-нитрозофенола в реакционной смеси. С учетом того, что маточный раствор после отделения кристаллов п-нитрозофенола повторно не используется, а полностью выводится из процесса, это ведет к увеличенным потерям п-нитрозофенола с маточной жидкостью. Также в результате такого выведения происходит потеря с выводимой маточной жидкостью значительных количеств других веществ, в том числе, в расчете на 1 моль вводимого в реакцию фенола потери могут составлять: Na2SO4 - 0,5 моль; H2SO4 - 2,9 моль; NaNO2 - 0,2 моль. Помимо этого, в условиях данного процесса, при взаимодействии непрореагировавшего фенола, который присутствует в постоянном избытке по отношению к нитриту натрия, с образовавшимся п-нитрозофенолом достаточно легко образуются побочные продукты синтеза, загрязняющие целевой продукт. Наконец, на второй стадии процесса достигается конечное молярное отношение фенол: нитрит натрия: серная кислота примерно 1,0:1,2:3,4, которому соответствует значение рН среды около 1,82, так что из реакционного раствора при незначительном избыточном давлении инертного газа могут выделяться окислы азота, ухудшая экологические параметры процесса в целом.
Известен также способ получения п-нитрозофенола (документ RU 2076096) взаимодействием фенола, нитрита натрия и серной кислоты в водной среде, при молярном соотношении фенол: нитрит натрия: серная кислота: вода 1,0:(1,05-1,4):(2,00-2,30):(80-100), проводимым в атмосфере инертного газа. При этом сначала проводят взаимодействие расчетного количества водного раствора нитрита натрия с водными растворами фенола и серной кислоты, взятыми в количестве 45-55% от расчетного, при 0-10°С в течение 30-60 мин, затем в полученную реакционную смесь вводят остальное количество водных растворов фенола и серной кислоты при 0-10° с в течение 30-60 мин. и выдерживают реакционную смесь при 5-15°С в течение 30-60 мин. В данном способе выход п-нитрозофенола достигает 90-95% в расчете на фенол, а чистота целевого продукта составляет 98-99%.
Однако, использование трехстадийного этапа нитрозирования в таком способе приводит к его усложнению. При этом, как и в предыдущем случае, маточный раствор после отделения кристаллов п-нитрозофенола повторно не используется, а полностью выводится из процесса, в результате чего происходит значительная потеря веществ с маточной жидкостью в том числе, в расчете на 1 моль вводимого в реакцию фенола потери могут составлять: Na2SO4 - 0,5 моль; H2SO4 - 1,7 моль; NaNO2 - 0,3 моль. Кроме того, промывной раствор после отмывки п-нитрозофенола захоложенной водой при 5°С также повторно не используется. Наконец, в ходе взаимодействия реагентов на третьей стадии процесса достигается конечное молярное отношение фенол: нитрит натрия: серная кислота примерно 1,0:1,3:2,2, которому соответствует значение рН среды около 2,24, поэтому и в данном случае возможно выделение окислов азота из реакционного раствора при незначительном избыточном давлении инертного газа, что ухудшает экологические параметры процесса в целом.
Следует заметить, что теряемый в вышеуказанных процессах известного уровня нитрит натрия NaNO2 теоретически мог бы повторно использоваться для нитрозирования фенола, а теряемый сульфат натрия Na2SO4 представляет собой полезный продукт, применяемый в стекольном и содовом производстве, и особенно в медицине.
Сущность изобретения
Таким образом, задача, решаемая настоящим изобретением, состоит в том, чтобы предложить непрерывный и достаточно простой способ нитрозирования фенола, который обеспечивает достижение таких технических результатов, как получение п-нитрозофенола с высоким выходом и чистотой, снижение расходных коэффициентов по сырью, уменьшение газообразных выбросов токсичных окислов азота и сокращение отходов производства, в частности сернокислотных отходов, а также получение побочного продукта реакции - 10-ти водного сульфата натрия Na2SO4⋅10H2O (далее по тексту, мирабилит, или глауберова соль) с достаточно высокой чистотой, позволяющей его дальнейшее использование в качестве полезного попутного продукта, пригодного, в частности, для медицинского применения.
Поставленная техническая задача решается и желаемые технические результаты достигаются с помощью предлагаемого способа непрерывного синтеза 4-нитрозофенола, в котором осуществляют взаимодействие нитрита натрия, фенола и серной кислоты в водной среде, причем способ включает нижеследующие стадии, на которых:
а) - готовят реакционную смесь путем смешения в турбулентном смесителе без доступа воздуха следующих компонентов:
- жидкая смесь воды и фенола при температуре от 20°С и выше, содержащая 92,2 мас. % фенола С6Н5ОН и 7,8 мас. % воды Н2О (далее по тексту, «жидкий фенол»),
- 75÷85 мас. % раствор серной кислоты H2SO4,
оборотный раствор, содержащий нитрит натрия NaNO2 и подаваемый со стадии (к),
причем общее количество NaNO2 в указанном подаваемом оборотном растворе составляет 3,0÷4,0 моль NaNO2 в расчете на 1 моль подаваемого С6Н5ОН, и
количество H2SO4 составляет 0,7÷0,9 моль в расчете на 1 моль подаваемого С6Н5ОН;
б) реакционную смесь, полученную на стадии (а), подают в реактор,
в) проводят нитрозирование фенола в реакторе в атмосфере инертного газа при избыточном давлении инертного газа 20÷50 кПа, температуре 0÷5°С и рН=3,9÷4,4;
г) образовавшуюся в ходе нитрозирования фенола на стадии (в) суспензию, содержащую кристаллы 4-нитрозофенола в маточном растворе, непрерывно подают в восходящий поток очищенной, обескислороженной и охлажденной до 0÷5°С воды, в котором кристаллы 4-нитрозофенола отмываются от окклюдированных примесей; отмытые кристаллы 4-нитрозофенола отделяют от первой жидкой фазы, представляющей собой смесь указанного маточного раствора и промывного раствора стадии (г);
д) первую жидкую фазу со стадии (г) пропускают через слой сорбента - мезопористого материала (согласно классификации IUPAC), содержащего полости или каналы с диаметром в интервале 2÷50 нм, для очистки раствора от побочных продуктов реакции с получением очищенного раствора;
е) очищенный раствор со стадии (д) с рН=3,9÷4,4 нейтрализуют едким натром до рН=6÷9 с получением нейтрализованного раствора;
ж) нейтрализованный раствор со стадии (е) подают в кристаллизатор;
з) в кристаллизаторе растворяют в указанном нейтрализованном растворе твердый нитрит натрия, подаваемый в стехиометрическом количестве из расчета 1 моль NaNO2 на 1 моль С6Н5ОН, подаваемого на стадии (а), с высаливанием при этом кристаллов мирабилита Na2SO4⋅10H2O;
причем растворение осуществляют в атмосфере инертного газа при атмосферном давлении, температуре 0÷5°С и рН=6÷9,
и) образовавшуюся в ходе растворения нитрита натрия на стадии (з) суспензию, содержащую кристаллы мирабилита в маточном растворе, непрерывно подают в восходящий поток очищенной и охлажденной до 0÷5°С воды, в котором кристаллы Na2SO4⋅10H2O отмываются от окклюдированных примесей; затем отмытые кристаллы глауберовой соли отделяют от второй жидкой фазы, представляющей собой смесь указанного маточного раствора и промывного раствора стадии (и);
к) вторую жидкую фазу со стадии (и) с растворенным в ней нитритом натрия непрерывно подают в качестве оборотного раствора на стадию (а).
Предлагаемый способ позволяет осуществлять нитрозирование фенола в условиях замкнутой системы без образования сточных вод и солевых отходов. При этом способ предусматривает возможность получения попутного продукта, а именно 10-ти водного сульфата натрия (глауберовой соли) фармакопейного качества, обеспечивает уменьшение расходных коэффициентов по сырью, а также снижает выбросы вредных веществ, таких как пары фенола, NOx, в окружающую среду.
Описание фигур
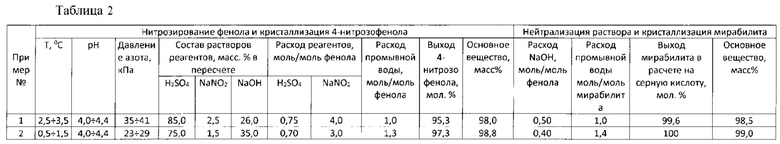
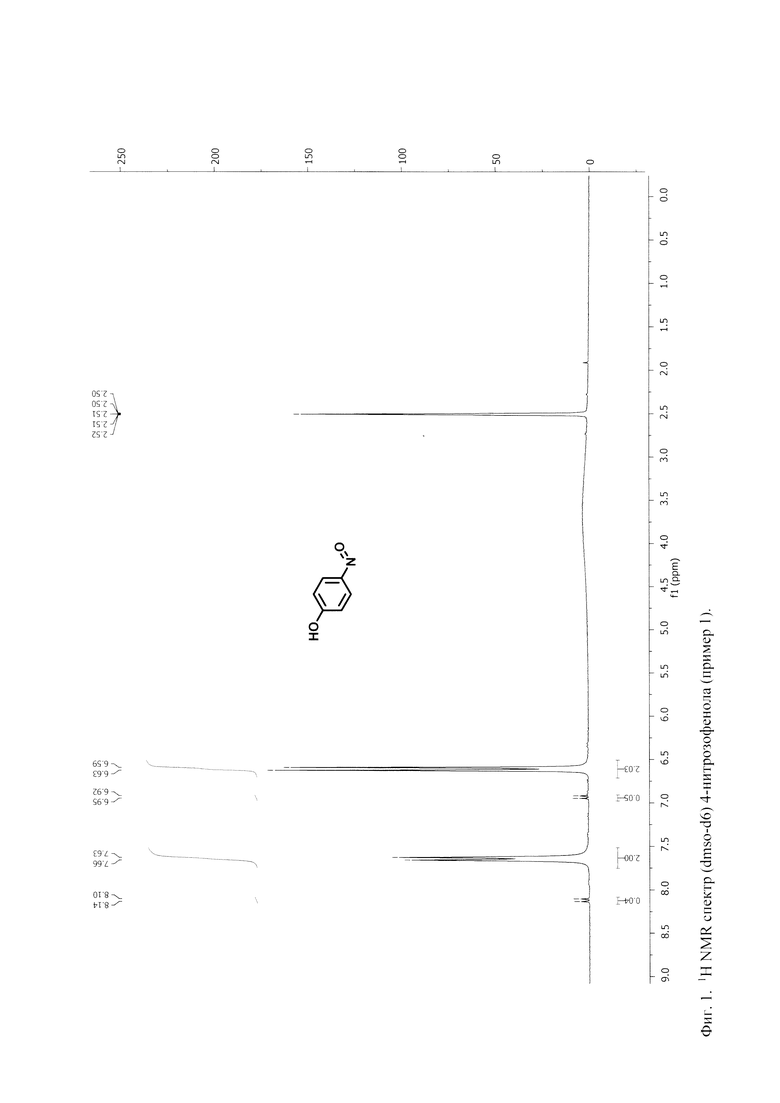
На Фигурах 1 и 2 представлены 1Н-ЯМР спектры (dmso-d6) 4-нитрозофенола, синтезированного в примерах 1 и 2 по настоящему изобретению.
Подробное описание изобретения
Настоящее изобретение обеспечивает способ непрерывного нитрозирования фенола, позволяющий преодолеть указанные выше недостатки известных способов.
Основное отличие заявляемого способа состоит в том, что нитрозирование фенола осуществляют при рН 3,9÷4,4, в избытке нитрита натрия, который подается в реакционную систему в 3÷4-х кратном количестве по отношению к стехиометрическому количеству, при избыточном давлении инертного газа 20÷50 кПа, что обеспечивает благоприятные эффекты настоящего изобретения, так как в условиях существенного избытка нитрозирующего агента фенол достаточно быстро превращается в 4-нитрозофенол, тем самым снижается вероятность образования побочных продуктов конденсации свободного фенола с 4-нитрозофенолом.
Непрореагировавший избыток нитрита натрия отделяют от продуктов реакции и в виде оборотного раствора возвращают на этап нитрозирования фенола, причем перед подачей оборотного раствора на этап нитрозирования в нем восполняют количество нитрита натрия, ранее израсходованное в реакции нитрозирования.
Далее более подробно рассматриваются стадии способа согласно настоящему изобретению.
i) На первой стадии (стадия (а)) заявляемого способа готовят смесь реакционных компонентов - фенола C6H5OH, серной кислоты H2SO4 и нитрита натрия NaNO2.
Используемый в способе фенол представляет собой стандартный фенол, используемый в процессе нитрозирования. В частности, может применяться стандартный фенол технический (марка А по ГОСТ 23519-93), представляющий собой белое кристаллическое вещество с температурой кристаллизации не ниже 40,7°С и содержанием органических примесей не более 0,01 масс. %.
Для подачи в реакционную смесь предварительно готовят жидкую смесь воды и фенола, содержащую примерно 92,2 мас. % фенола и примерно 7,8 мас. % воды, которая представляет собой эвтектическую смесь (расплав) фенола и воды.
Такая смесь может быть приготовлена, например, следующим образом: в расплавленный при температуре 50°С фенол в указанном соотношении добавляют очищенную теплую воду (под слой фенола) и перемешивают смесь до получения однородной жидкости.
Жидкий фенол, подаваемый на смешение, имеет температуру от 20°С и выше.
Используемая в способе по изобретению для получения реакционной смеси серная кислота подается на смешение в виде 75÷85 мас. % водного раствора серной кислоты. Такой раствор может быть получен из концентрированной серной кислоты. В типичном варианте реализации способа используется серная кислота техническая контактная (улучшенная по ГОСТ 2184-2013), содержащая 92,5÷94,0 мас. % моногидрата H2SO4. Концентрированную серную кислоту разбавляют очищенной водой в емкости с мешалкой. Очищенную воду подают во всасывающую линию насоса и смешивают с серной кислотой в массовом соотношении, необходимом для достижения требуемой концентрации серной кислоты 75÷85 мас. %.
Количество подаваемого раствора серной кислоты обычно устанавливается таким образом, чтобы обеспечить подачу 0,7÷0,9 моль H2SO4 в расчете на 1 моль подаваемого C6H5OH.
Используемый в способе нитрит натрия представляет собой обычно используемый в процессах нитрозирования нитрит натрия. Как правило, используется нитрит натрия высшего сорта по ГОСТ 19906-74, представляющий собой белые кристаллы с желтоватым или сероватым оттенком и содержащий не менее 99,0 мас. % NaNO2. При приготовлении исходной реакционной смеси нитрит натрия подают в виде оборотного раствора со стадии (к).
Такой оборотный раствор представляет собой водный раствор, содержащий нитрит натрия, обычно с концентрацией 1,5÷2,5%. При этом общее количество NaNO2 в подаваемом оборотном растворе составляет 3,0÷4,0 моль NaNO2 в расчете на 1 моль подаваемого С6Н5ОН. Данное общее количество NaNO2 в указанном подаваемом оборотном растворе соответствует стехиометрическому количеству 1 моль NaNO2 в расчете на 1 моль подаваемого С6Н5ОН плюс постоянный избыток из расчета 2,0÷3,0 моль NaNO2 в расчете на 1 моль подаваемого С6Н5ОН.
Вышеуказанные жидкий фенол, водный раствор серной кислоты и оборотный раствор, содержащий нитрит натрия, подают на смешение в указанных количествах.
Смешение осуществляют в турбулентном смесителе. В качестве такого смесителя может использоваться любой подходящий турбулентный смеситель, например, винтовой насос, центробежный насос, миксер. Предпочтительно используют винтовой насос.
Смешение компонентов в турбулентном смесителе производят без доступа воздуха.
В результате указанного смешения получают реакционную смесь, содержащую исходные компоненты реакции нитрозирования.
ii) Полученную на стадии (а) реакционную смесь подают в реактор нитрозирования (стадия (б)).
В указанном реакторе осуществляют нитрозирование фенола в атмосфере инертного газа (стадия (в)). В качестве инертного газа могут использоваться, в частности, такие газы, как азот, углекислый газ, аргон. Возможно, нитрозирование осуществляют в атмосфере азота.
Реактор нитрозирования может представлять собой любой подходящий для данного процесса реактор, известный специалистам и обеспечивающий возможность поддержания требуемых давления и температуры реакции, в частности из группы изотермических реакторов смешения непрерывного или полупериодического действия.
Нитрозирование фенола в реакторе проводят при избыточном давлении инертного газа 20÷50 кПа, предпочтительно 24÷30 кПа, и при температуре 0÷5°С, предпочтительно 0,5÷1,5°С. рН реакционной смеси в процессе нитрозирования составляет 3,9÷4,4.
Не намереваясь ограничиваться какими-либо теоретическими воззрениями, заявитель предполагает, что в ходе реакции ароматическое кольцо фенола непрерывно нитрозируется электрофильными частицами нитрозацидий-катиона (Н2О*NO+) и оксида азота (III) (N2O3), образующихся в реакции нитрита натрия с серной кислотой при 0-5°С и рН=3,9÷4,4. Осуществление нитрозирования фенола при рН=3,9÷4,4 под давлением инертного газа 20÷50 кПа препятствует разложению электрофильных частиц до окислов азота NO и NO2, практически исключает выделение последних из раствора в газовую фазу, что ускоряет основную реакцию образования 4-нитрозофенола, уменьшает вероятность побочных реакций конденсации фенола с 4-нитрозофенолом. Тем самым, существенно снижается образование загрязняющих примесей и повышается чистота основного и попутного продуктов реакции.
Время пребывания реакционной смеси в реакторе, достаточное для полного завершения основной реакции, обычно составляет:
- не более 2,5 ч при температуре 0°С и рН=4,4;
- не более 1 ч при температуре 5°С и рН=3,9.
В результате протекания реакции (1) нитрозирования фенола на стадии (в) в качестве основного продукта образуется 4-нитрозофенол, который, ввиду относительно слабой растворимости в воде, выделяется из реакционной смеси в виде кристаллов, в результате чего образуется суспензия кристаллов 4-нитрозофенола в маточном растворе. Маточный раствор содержит также образовавшийся сульфат натрия Na2SO4, растворенную часть 4-нитрозофенола и непрореагировавшие серную кислоту и нитрит натрия. Что касается фенола, то весь фенол, вводимый в реакционную смесь, практически полностью нитрозируется, и лишь незначительное количество фенола (1÷1,5 мас. %) вступает в побочные реакции конденсации, так что маточный раствор, по существу, не содержит свободного фенола.
iii) Из образовавшейся в ходе нитрозирования фенола на стадии (в) суспензии кристаллов 4-нитрозофенола в маточном растворе выделяют очищенные кристаллы 4-нитрозофенола.
Для этого указанную суспензию непрерывно подают в восходящий поток очищенной, обескислороженной и охлажденной до 0÷5°С воды. В указанном потоке кристаллы 4-нитрозофенола отмываются от окклюдированных примесей.
Данную операцию можно осуществить в любом аппарате колонного типа, обеспечивающем отсутствие продольного (вдоль оси) перемешивания. В частности, отмывание кристаллов нитрозофенола может быть проведено в пульсационной колонне. Подходящие пульсационные колонны в целом известны специалистам и описаны, например, в работе Карпачева С.М., Рябчиков Б.Е., Пульсационная аппаратура в химической технологии, М., 1983.
При осуществлении отмывки от окклюдированных примесей расход указанной промывной воды на стадии (г) обычно составляет 1,0÷1,5 моль в расчете на 1 моль С6Н5ОН, подаваемого на стадии (а).
Отмытые кристаллы 4-нитрозофенола отделяют в подходящем аппарате от первой жидкой фазы. Подходящие для данной операции аппараты в целом известны специалистам и могут представлять собой, например, центробежные декантеры, ультрацентрифуги и т.п. Так, подходящие центробежные декантеры описаны, например, в следующих работах: A. Records and K. Sutherland, "Decanter Centrifuge Handbook", Elsevier Science, 2001; Wallace Woon-Fong Leung, "Industrial Centrifugation Technology", McGraw-Hill Professional, 1998; Dr.-Ing. A. Karolis, "Die Technologie der Vollmantel-Schneckenzentrifugen", Zentrifugen in der grosstechnischen Anwendung, 07-08/12/1999 Haus der Technik, Essen; W.J. Sokolow, "Moderne Industriezentrifugen", VEB-Verlag Technik, Berlin, 1971.
Отделенная как указано выше от кристаллов 4-нитрозофенола первая жидкая фаза представляет собой смесь вышеуказанного маточного раствора, из которого происходит кристаллизация 4-нитрозофенола, и промывного раствора, использованного для промывки кристаллов 4-нитрозофенола
iv) Указанная отделенная от кристаллов 4-нитрозофенола жидкая фаза со стадии (г) может содержать побочные продукты реакции синтеза 4-нитрозофенола, в частности продукты конденсации фенола с 4-нитрозофенолом, в том числе органические хромофоры, в частности такие, как индофенол и хиноидные соединения, загрязняющие продукты реакции и придающие им нежелательную окраску.
При вышеуказанных условиях реакции нитрозирования фенола (рН, температура, давление) скорость побочных реакций с участием 4-нитрозофенола и непрореагировавшего фенола с образованием индофенола, хиноидных соединений и других хромофоров незначительна. Тем не менее, в оборотном растворе может происходить их накопление, при этом цвет раствора меняется от розового до темно-вишневого.
Поэтому после отделения от кристаллов 4-нитрозофенола жидкую фазу со стадии (г) пропускают через слой сорбента.
В качестве сорбента может использоваться любой сорбент, содержащий наряду с микропорами диаметром менее 2 нм (не участвуют в процессе адсорбции индофенола) достаточное количество мезопор диаметром 2÷50 нм (классификация IUPAC). Чем больше объемная доля мезопор в сорбенте, тем выше его адсорбционная емкость в отношении органических красителей, в том числе индофенола (OC6H4NC6H4OH). Подходящие сорбенты, как правило, должны содержать не менее 60% по объему пор диаметром 2÷50 нм с удельной поверхностью 85÷200 м2/г, предпочтительно содержат не менее 80% по объему пор диаметром 2÷50 нм. В частности, в качестве подходящего сорбента могут использоваться активные угли, например активированный уголь марки АГ-3, ОУ-Г или МИУ-С.
Операция может осуществляться в любых подходящих устройствах, известных специалистам, например в картридж-блоках.
В результате данной стадии (д) получают очищенный раствор.
v) Очищенный раствор со стадии (д) все еще содержит избыток серной кислоты и нитрита натрия, в результате чего в нем может продолжаться образование активных электрофильных нитрозирующих агентов, которые могут генерировать побочные реакции с образованием нежелательных примесей. Поэтому указанный очищенный раствор подвергают нейтрализации до рН=6÷9 на стадии (е).
Нейтрализацию осуществляют едким натром, предпочтительно 25÷35% водным раствором едкого натра, путем смешения очищенного раствора и едкого натра без доступа воздуха. Такое смешение может осуществляться в различных подходящих устройствах для смешения, например, в трубчатом смесителе, эжекторе типа Вентури. При этом расход NaOH предпочтительно составляет 0,3÷0,7 моль в расчете на 1 моль С6Н5ОН, подаваемого на стадии (а).
В результате данной операции получают нейтрализованный раствор.
vi) Далее нейтрализованный раствор со стадии (е) подают в кристаллизатор.
В кристаллизаторе производят растворение в указанном нейтрализованном растворе твердого нитрита натрия NaNO2. За счет этого осуществляется восполнение количества нитрита натрия, израсходованного в реакции нитрозирования фенола на стадии (в).
Кроме того, при растворении в нейтрализованном растворе твердого нитрита натрия снижается растворимость сульфата натрия, образовавшегося в реакции нитрозирования фенола на стадии (в) и в реакции нейтрализации едким натром на стадии (е), и происходит высаливание из образующегося при этом маточного раствора кристаллов мирабилита в качестве дополнительного продукта способа по настоящему изобретению.
Данную стадию (стадия (з)) можно осуществлять в любом подходящем кристаллизаторе, снабженном устройствами для подачи нейтрализованного раствора и твердого нитрита натрия. В частности, для подачи в кристаллизатор твердого нитрита натрия может использоваться шнековый дозатор.
Количество подаваемого в кристаллизатор на стадии (з) нитрита натрия задается как стехиометрическое количество, т.е. из расчета 1 моль NaNO2 на 1 моль С6Н5ОН, подаваемого на стадии (а). Растворение нитрита натрия в нейтрализованном маточном растворе осуществляют в атмосфере инертного газа при атмосферном давлении, температуре 0÷5°С и рН=6÷9.
В качестве инертного газа могут использоваться различные инертные газы, известные специалистам, возможно использование азота.
В результате проведения стадии (з) образуется суспензия, содержащая кристаллы мирабилита в маточном растворе, в котором также растворен нитрит натрия.
vii) Из образовавшейся на стадии (з) суспензии кристаллов мирабилита в маточном растворе выделяют очищенные кристаллы глауберовой соли.
Для этого указанную суспензию непрерывно подают в восходящий поток очищенной и охлажденной до 0÷5°С воды. В указанном потоке кристаллы Na2SO4÷10H2O отмываются от окклюдированных примесей.
Данную операцию можно осуществить в любом аппарате колонного типа, обеспечивающем отсутствие продольного (вдоль оси) перемешивания. В частности, отмывание глауберовой соли может быть проведено в пульсационной колонне. Подходящие пульсационные колонны в целом известны специалистам и описаны, например, в работе Карпачева С.М., Рябчиков Б.Е., Пульсационная аппаратура в химической технологии, М, 1983.
При осуществлении отмывки от окклюдированных примесей расход указанной промывной воды на стадии (и) обычно составляет 1,0÷1,5 моль в расчете на 1 моль твердого Na2SO4÷10Н2О.
Отмытые кристаллы глауберовой соли отделяют в подходящем аппарате от второй жидкой фазы. Подходящие для данной операции аппараты в целом известны специалистам и могут представлять собой, например, центробежные декантеры, ультрацентрифуги и т.п. Так, например, подходящие декантеры описаны в работах, указанных выше на стр. 11 настоящего описания.
В результате осуществления данной стадии (и) получают отмытые кристаллы глауберовой соли. При этом выход твердого Na2SO4⋅10H2O обычно составляет 0,7÷0,9 моль в расчете на 1 моль подаваемого С6Н5ОН.
Кроме того, в результате осуществления стадии (и) получают вторую жидкую фазу, которая представляет собой смесь вышеуказанного маточного раствора, из которого происходит кристаллизация мирабилита, и промывного раствора, использованного для промывки кристаллов мирабилита. Данная вторая жидкая фаза содержит растворенные в ней нитрит натрия и сульфат натрия Na2SO4.
viii) Вторую жидкую фазу со стадии (и) с растворенным в ней нитритом натрия рециркулируют и непрерывно подают на стадию (а) в качестве оборотного раствора.
Указанный оборотный раствор обеспечивает поступление на стадию (а) нужного количества нитрита натрия, необходимого для образования реакционной смеси исходных компонентов реакции нитрозирования, как описано здесь выше.
В результате реализации способа, согласно настоящему изобретению, получают в качестве основного продукта 4-нитрозофенол высокой степени чистоты, составляющей 98*99% основного вещества в пересчете на сухой продукт, с весьма высоким выходом, который составляет 0,95-0,98 моль на моль фенола, подаваемого на стадию (а), т.е. 95-98%.
При реализации предлагаемого способа нитрозирования фенола при рН=3,9-4,4 в газовую фазу практически не поступают вредные вещества (пары фенола и окислы азота), реакционная и промывная вода выводится из процесса в виде кристаллизационной воды вместе с сульфатом натрия, а побочные продукты синтеза (индофенол, хиноидные соединения - хромофоры) выводятся из процесса сорбентами. Противоточная промывка целевого и попутного продуктов - 4-нитрозофенола и 10-водного кристаллогидрата сульфата натрия - водой в пульсационной колонне обеспечивает их высокую чистоту, составляющую 98÷99% основного вещества в пересчете на сухой продукт. В способе по изобретению также достигается высокий выход продуктов, составляющий в расчете на фенол, подаваемый на стадию (а): для целевого продукта (4-нитрозофенола) - 95÷98 мол. %, а для попутного продукта (глауберовой соли) - 70÷87 мол. %.
Реализация описываемого здесь технического решения позволяет достичь высокой (на уровне 98%) степени нитрозирования фенола в 4-нитрозофенол по реакции 1, т.е. практически полного отсутствия потерь фенола в процессе. Также обеспечивается повышение выхода целевого продукта, так как не происходит потерь 4-нитрозофенола с маточным раствором и промывной водой. Заявляемый способ также позволяет осуществить практически 100%-ное извлечение попутного продукта - глауберовой соли, а также обеспечивает снижение содержания хромофорных примесей в целевом и попутном продукте, так как они выводятся из маточного раствора сорбентом. Наконец, снижение выделения окислов азота позволяет улучшить экологические параметры процесса в целом и снизить его вредное воздействие на окружающую среду.
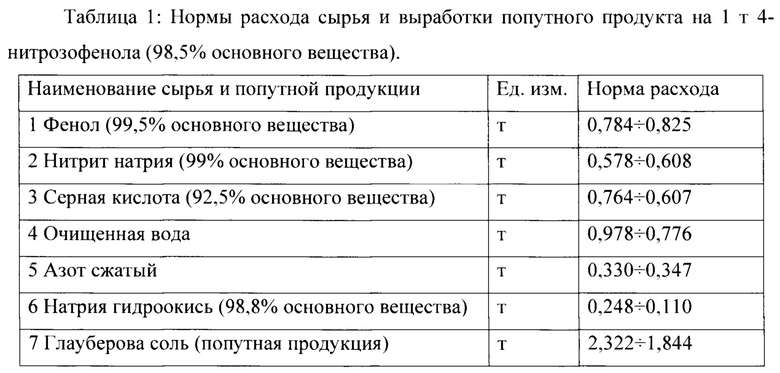
Реализация предлагаемого способа, согласно настоящему изобретению, позволяет осуществлять получение 4-нитрозофенола, в частности, при следующих ориентировочных нормах расхода сырья и выработки попутного продукта, приведенных в таблице 1:
Заявляемое изобретение далее демонстрируется нижеследующими примерами, которые представлены только для иллюстрации и не предназначены для какого-либо ограничения объема настоящего изобретения.
Примеры реализации способа.
Пример 1.
В турбулентный смеситель (винтовой насос) непрерывно подают оборотный раствор в количестве 52,0 кг/ч, содержащий: NaNO2 - 2,5 мас. %, Na2SO4 - 1,8 мас. % и 4-нитрозофенол - 0,4 мас. %; жидкий фенол в количестве 0,478 кг/ч; и 85% серную кислоту в количестве 0,404 кг/ч. Полученную реакционную смесь подают винтовым насосом в реактор-кристаллизатор под слой жидкости со скоростью подачи 52,9 кг/ч.
Нитрозирование фенола проводят в реакторе-кристаллизаторе с рабочим объемом 20 дм3, снабженном змеевиковым теплообменником, датчиками температуры и рН. Суспензия 4-нитрозофенола, находящаяся в реакторе, перемешивается за счет циркуляции, создаваемой винтовым насосом. Для создания азотной подушки в реактор непрерывно подают азот со скоростью 60 дм3/ч. При этом в реакторе устанавливается избыточное давление 38±3 кПа.
В указанном реакторе-кристаллизаторе реакционную смесь охлаждают до температуры 3±0,5°С; рН реакционной среды автоматически регулируют на уровне рН=4,2±0,2 посредством регулирования расхода серной кислоты. Образуются кристаллы 4-нитрозофенола с эффективным диаметром 80±15 мкм в количестве 0,550 кг/ч (95,3 мол. % от расхода фенола), которые поступают в противоточную пульсационную колонну. В пульсационной колонне кристаллы 4-нитрозофенола промываются восходящим потоком очищенной и обескислороженной воды. Вода, которую подают в нижнюю часть колонны со скоростью 0,080 кг/ч, вымывает с поверхности кристаллов 4-нитрозофенола окклюдированный раствор, вместе с которым поступает в реактор-кристаллизатор.
Отмытые кристаллы 4-нитрозофенола декантируют от жидкой фазы в центрифуге и высушивают под разрежением при температуре 45÷50°С. Выход целевого продукта с содержанием основного вещества 98,0% составляет 0,56 кг/ч.
Маточный раствор, содержащий около 0,05 мас. % хромофорных примесей (в пересчете на индофенол), подают в картридж с активированным углем марки АГ-3 со скоростью 52,4 кг/ч. Органические примеси - хромофоры задерживаются в порах сорбента, а очищенный раствор поступает в трубчатый смеситель, в котором нейтрализуется 26% раствором NaOH до рН=7±1. Раствор NaOH подают в смеситель со скоростью 0,357 кг/ч.
Нейтрализованный раствор со скоростью 52,8 кг/ч поступает в кристаллизатор объемом 15 дм3, снабженный змеевиковым теплообменником, лопастной мешалкой, датчиками температуры и рН и азотной подушкой. В кристаллизатор через шнековый дозатор подают порошкообразный нитрит натрия со скоростью 0,326 кг/ч. При растворении нитрита натрия в нейтрализованном растворе при температуре 3±0,5°С и рН=7,5±1 из раствора высаливаются кристаллы 10-ти водного сульфата натрия (глауберова соль), которые поступают в противоточную пульсационную колонну со скоростью 1,134 кг/ч (74,7 мол. % от расхода фенола, т.е. 99,6 мол. % в расчете на подаваемую серную кислоту). В пульсационной колонне кристаллы глауберовой соли промываются восходящим потоком очищенной воды. Вода, которую подают в нижнюю часть колонны со скоростью 0,064 кг/ч, вымывает с поверхности кристаллов окклюдированный раствор и поступает в кристаллизатор.
Отмытые кристаллы Na2SO4⋅10Н2О отжимают от жидкой фазы в центрифуге до влажности 4,5%. Выход глауберовой соли с содержанием основного вещества 98,5 мас. % в расчете на сухое вещество составляет 1,15 кг/ч.
Маточный раствор, содержащий NaNO2 - 2,5 мас. %, Na2SO4 - 1,8 мас. %, 4-нитрозофенол - 0,4 мас. %, подают перистальтическим насосом в винтовой насос на смешение с жидким фенолом и 85 мас. % серной кислотой, т.е. на начальную стадию способа.
На фиг 1 представлен 1Н ЯМР спектр (dmso-d6) синтезированного 4-нитрозофенола.
Как показывает оценка на основании данного спектра, примеси (пики с координатой 8,10÷8,40 и 6,92÷6,95) в полученном 4-нитрозофеноле составляют не более 2% масс%.
Пример 2.
В турбулентный смеситель (винтовой насос) непрерывно подают оборотный раствор в количестве 64,5 кг/ч, содержащий: NaNO2 - 1,5 мас. %, Na2SO4 - 1,8 мас. % и 4-нитрозофенол - 0,4 мас. %; жидкий фенол в количестве 0,478 кг/ч; и 75,0% серную кислоту в количестве 0,429 кг/ч. Полученную реакционную смесь подают винтовым насосом в реактор-кристаллизатор под слой жидкости со скоростью подачи 65,4 кг/ч.
Нитрозирование фенола проводят в реакторе-кристаллизаторе с рабочим объемом 20 дм3, снабженном змеевиковым теплообменником, датчиками температуры и рН. Суспензия 4-нитрозофенола, находящаяся в реакторе, перемешивается за счет циркуляции, создаваемой винтовым насосом. Для создания азотной подушки в реактор непрерывно подают азот со скоростью 60 дм3/ч. При этом в реакторе устанавливается избыточное давление 26±3 кПа.
В указанном реакторе-кристаллизаторе реакционную смесь охлаждают до температуры 1±0,5°С; рН реакционной среды автоматически регулируют на уровне рН=4,2±0,2 посредством регулирования расхода серной кислоты. Образуются кристаллы 4-нитрозофенола с эффективным диаметром 80±15 мкм в количестве 0,561 кг/ч (97,3 мол. % от расхода фенола), которые поступают в противоточную пульсационную колонну. В пульсационной колонне кристаллы 4-нитрозофенола промываются восходящим потоком очищенной и обескислороженной воды. Вода, которую подают в нижнюю часть колонны со скоростью 0,107 кг/ч, вымывает с поверхности кристаллов 4-нитрозофенола окклюдированный раствор, вместе с которым поступает в реактор-кристаллизатор.
Отмытые кристаллы 4-нитрозофенола декантируют от жидкой фазы в центрифуге и высушивают под разрежением при температуре 45÷50°С. Выход целевого продукта с содержанием основного вещества 98,8 мас. % составляет 0,567 кг/ч.
Маточный раствор, содержащий около 0,02 мас. % хромофорных примесей (в пересчете на индофенол), подают в картридж с активированным углем марки АГ-3 со скоростью 65,0 кг/ч. Органические примеси - хромофоры задерживаются в порах сорбента, а очищенный раствор поступает в трубчатый смеситель, в котором нейтрализуется 35% раствором NaOH до рН=7±1. Раствор NaOH подают в смеситель со скоростью 0,214 кг/ч.
Нейтрализованный раствор со скоростью 65,2 кг/ч поступает в кристаллизатор объемом 15 дм3, снабженный змеевиковым теплообменником, лопастной мешалкой, датчиками температуры и рН и азотной подушкой. В кристаллизатор через шнековый дозатор подают порошкообразный нитрит натрия со скоростью 0,326 кг/ч. При растворении нитрита натрия в нейтрализованном растворе при температуре 1±0,5°С и рН=7,5±1 из раствора высаливаются кристаллы 10-ти водного сульфата натрия (глауберова соль), которые поступают в противоточную пульсационную колонну со скоростью 1,065 кг/ч (70,0 мол. % от расхода фенола, т.е. практически 100 мол. % в расчете на подаваемую серную кислоту). В пульсационной колонне кристаллы глауберовой соли промываются восходящим потоком очищенной воды. Вода, которую подают в нижнюю часть колонны со скоростью 0,083 кг/ч, вымывает с поверхности кристаллов окклюдированный раствор и поступает в кристаллизатор.
Отмытые кристаллы Na2SO4⋅10H2O отжимают от жидкой фазы в центрифуге до влажности 4,5%. Выход глауберовой соли с содержанием основного вещества 99 мас. % в расчете на сухое вещество составляет 1,076 кг/ч.
Маточный раствор, содержащий NaNO2 - 1,5 мас. %, Na2SO4 - 1,8 мас. %, 4-нитрозофенол - 0,4 мас. %, подают перистальтическим насосом в винтовой насос на смешение с жидким фенолом и 75,0 мас. % серной кислотой, т.е. на начальную стадию способа.
На фиг 2 представлен 1Н ЯМР спектр (dmso-d6) синтезированного 4-нитрозофенола.
Как показывает оценка на основании данного спектра, примеси в полученном 4-нитрозофеноле не обнаружены.
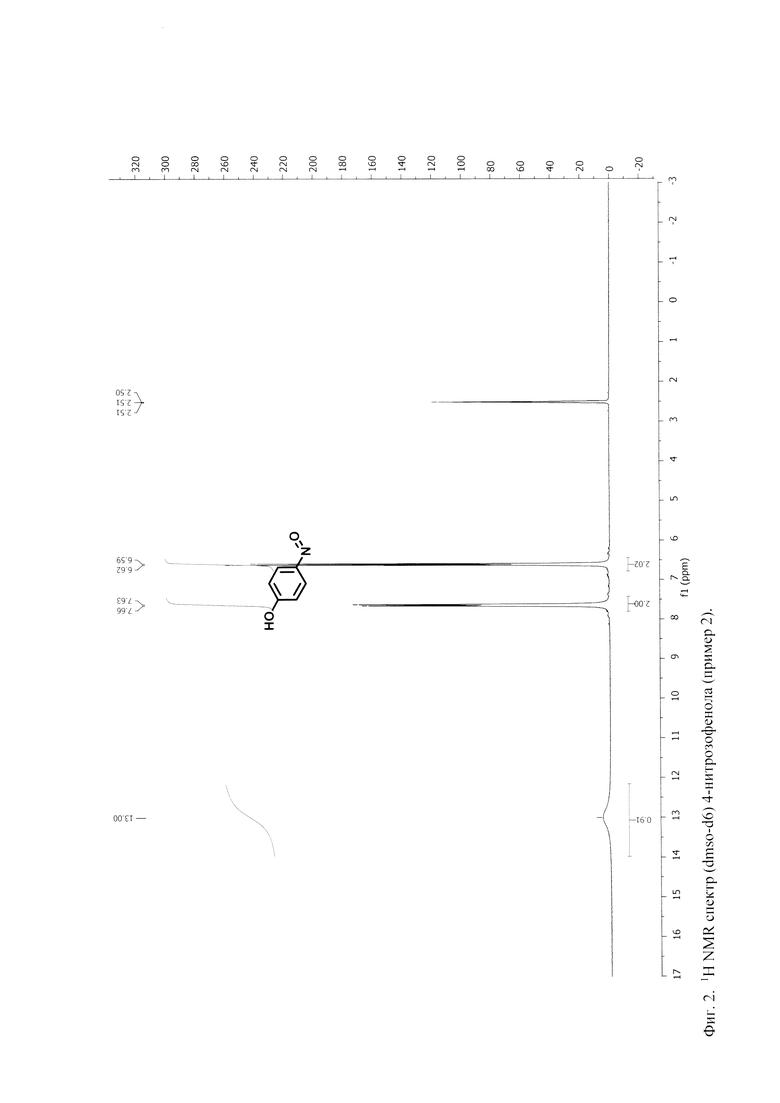
Условия и технологические параметры процессов, а также характеристики продуктов в Примерах 1 и 2 приведены в таблице 2 ниже.
Таким образом, в приведенных выше примерах в результате осуществления способа по настоящему изобретению получают нитрозофенол высокой степени чистоты (не менее 98 мас.%) с высоким выходом, составляющим более 95 мас.%. Одновременно получают полезный побочный продукт - глауберову соль, с чистотой не менее 98,5 мас.% и выходом свыше 99 мас.% (в расчете на подаваемую серную кислоту). При этом практически отсутствуют потери фенола, нитрита натрия и серной кислоты.
Специалисту будет понятно, что описанное здесь изобретение не ограничивается только приведенными выше вариантами реализации и может быть осуществлено с использованием различных комбинаций признаков, характеризующих настоящее изобретение, или их эквивалентов. Действительный объем изобретения, таким образом, определяется только приводимой ниже формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СИНТЕЗА п-НИТРОЗОФЕНОЛА И УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ п-НИТРОЗОФЕНОЛА ЭТИМ СПОСОБОМ | 2023 |
|
RU2813692C1 |
| СПОСОБ ПОЛУЧЕНИЯ п-АМИНОФЕНОЛА И УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ п-АМИНОФЕНОЛА ЭТИМ СПОСОБОМ | 2023 |
|
RU2822065C1 |
| СПОСОБ ПОЛУЧЕНИЯ П-НИТРОЗОФЕНОЛА | 1995 |
|
RU2076096C1 |
| СПОСОБ И УСТАНОВКА ДЛЯ ПОЛУЧЕНИЯ ПАРА-АМИНОФЕНОЛА ИЗ ФЕНОЛА ПУТЁМ ПОСЛЕДОВАТЕЛЬНОГО НИТРОЗИРОВАНИЯ И ВОССТАНОВЛЕНИЯ СУЛЬФИДОМ АММОНИЯ | 2023 |
|
RU2801692C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРОФЕНОЛОВ | 1994 |
|
RU2105754C1 |
| СПОСОБ И УСТАНОВКА ПОЛУЧЕНИЯ ПАРАЦЕТАМОЛА ИЗ ФЕНОЛА | 2023 |
|
RU2814270C1 |
| УСТАНОВКА ДЛЯ НИТРОЗИРОВАНИЯ ФЕНОЛА | 2024 |
|
RU2834723C1 |
| СПОСОБ ПОЛУЧЕНИЯ П-НИТРОЗОФЕНОЛА | 1997 |
|
RU2129117C1 |
| Способ получения нитрозофенола | 1936 |
|
SU48325A1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ СУЛЬФАТА НАТРИЯ И НИТРАТОВ МЕТАЛЛОВ | 2015 |
|
RU2610076C1 |
Изобретение относится к способу непрерывного синтеза 4-нитрозофенола взаимодействием нитрита натрия, фенола и серной кислоты в водной среде. Способ включает следующие стадии а)-к). На стадии а) смешением в турбулентном смесителе без доступа воздуха готовят реакционную смесь, содержащую следующие компоненты: жидкий фенол, представляющий собой жидкую смесь воды и фенола, содержащую 92,2 мас.% фенола С6Н5ОН и 7,8 мас.% воды Н2О; 75-85 мас.% раствор серной кислоты H2SO4, причем количество подаваемой H2SO4 составляет 0,7-0,9 моль в расчете на 1 моль подаваемого C6H5OH; оборотный раствор, содержащий нитрит натрия NaNO2, подаваемый со стадии к), причем общее количество NaNO2 в указанном подаваемом оборотном растворе составляет 3,0-4,0 моль NaNO2 в расчете на 1 моль подаваемого С6Н5ОН. На стадии б) реакционную смесь, полученную на стадии а), подают в реактор. На стадии в) проводят нитрозирование фенола в реакторе в атмосфере инертного газа при избыточном давлении инертного газа 20-50 кПа, температуре 0-5°С и рН = 3,9-4,4. На стадии г) образовавшуюся в ходе нитрозирования на стадии в) реакционную смесь, содержащую кристаллы 4-нитрозофенола, непрерывно подают в восходящий поток очищенной, обескислороженной и охлажденной до 0-5°С воды, в котором кристаллы 4-нитрозофенола отмываются от окклюдированных примесей, и отмытые кристаллы 4-нитрозофенола отделяют от первой жидкой фазы, представляющей собой смесь указанного маточного раствора и промывного раствора. На стадии д) первую жидкую фазу со стадии г) пропускают через слой сорбента, представляющего собой мезопористый материал, содержащий полости или каналы с диаметром в интервале от 2 до 50 нм, для очистки раствора от побочных продуктов реакции с получением очищенного раствора. На стадии е) очищенный раствор со стадии д) с рН = 3,9-4,4 нейтрализуют едким натром до рН = 6-9 с получением нейтрализованного раствора. На стадии ж) нейтрализованный раствор со стадии е) подают в кристаллизатор. На стадии з) в указанном кристаллизаторе растворяют в указанном нейтрализованном растворе твердый нитрит натрия, подаваемый в стехиометрическом количестве из расчета 1 моль NaNO2 на 1 моль фенола C6H5OH, подаваемого на стадии а), причем растворение осуществляют в атмосфере инертного газа при 0-5°С и рН = 6-9 с высаливанием при этом кристаллов мирабилита Na2SO4⋅10H2O. На стадии и) образовавшуюся в ходе растворения нитрита натрия на стадии з) суспензию, содержащую кристаллы мирабилита Na2SO4⋅10H2O в маточном растворе, непрерывно подают в восходящий поток очищенной и охлажденной до 0-5°С воды, в котором кристаллы Na2SO4⋅10H2O отмываются от окклюдированных примесей; затем отмытые кристаллы глауберовой соли отделяют от второй жидкой фазы, представляющей собой смесь указанного маточного раствора и промывного раствора. На стадии к) вторую жидкую фазу со стадии и) с растворенным в ней нитритом натрия непрерывно подают в качестве оборотного раствора на стадию а). Предлагаемый способ позволяет синтезировать 4-нитрозофенол с высокой степенью чистоты и высоким выходом, снизить расходные коэффициенты по сырью, сократить количество отходов, а также получать ценный попутный продукт реакции - глауберову соль - с высокой чистотой. 13 з.п. ф-лы, 2 табл., 2 ил., 2 пр.
1. Способ непрерывного синтеза 4-нитрозофенола взаимодействием нитрита натрия, фенола и серной кислоты в водной среде, включающий стадии, на которых:
а) смешением в турбулентном смесителе без доступа воздуха готовят реакционную смесь, содержащую следующие компоненты:
- жидкий фенол, представляющий собой жидкую смесь воды и фенола, содержащую 92,2 мас.% фенола С6Н5ОН и 7,8 мас.% воды Н2О,
- 75-85 мас.% раствор серной кислоты H2SO4, причем количество подаваемой H2SO4 составляет 0,7-0,9 моль в расчете на 1 моль подаваемого C6H5OH; и
- оборотный раствор, содержащий нитрит натрия NaNO2 и подаваемый со стадии к),
причем общее количество NaNO2 в указанном подаваемом оборотном растворе составляет 3,0-4,0 моль NaNO2 в расчете на 1 моль подаваемого С6Н5ОН;
б) реакционную смесь, полученную на стадии а), подают в реактор;
в) проводят нитрозирование фенола в реакторе в атмосфере инертного газа при избыточном давлении инертного газа 20-50 кПа, температуре 0-5°С и рН = 3,9-4,4;
г) образовавшуюся в ходе нитрозирования на стадии в) реакционную смесь, содержащую кристаллы 4-нитрозофенола, непрерывно подают в восходящий поток очищенной, обескислороженной и охлажденной до 0-5°С воды, в котором кристаллы 4-нитрозофенола отмываются от окклюдированных примесей, и отмытые кристаллы 4-нитрозофенола отделяют от первой жидкой фазы, представляющей собой смесь указанного маточного раствора и промывного раствора;
д) первую жидкую фазу со стадии г) пропускают через слой сорбента, представляющего собой мезопористый материал, содержащий полости или каналы с диаметром в интервале от 2 до 50 нм, для очистки раствора от побочных продуктов реакции с получением очищенного раствора;
е) очищенный раствор со стадии д) с рН = 3,9-4,4 нейтрализуют едким натром до рН = 6-9 с получением нейтрализованного раствора;
ж) нейтрализованный раствор со стадии е) подают в кристаллизатор;
з) в указанном кристаллизаторе растворяют в указанном нейтрализованном растворе твердый нитрит натрия, подаваемый в стехиометрическом количестве из расчета 1 моль NaNO2 на 1 моль фенола C6H5OH, подаваемого на стадии а), причем растворение осуществляют в атмосфере инертного газа при 0-5°С и рН = 6-9 с высаливанием при этом кристаллов мирабилита Na2SO4⋅10H2O;
и) образовавшуюся в ходе растворения нитрита натрия на стадии з) суспензию, содержащую кристаллы мирабилита Na2SO4⋅10H2O в маточном растворе, непрерывно подают в восходящий поток очищенной и охлажденной до 0-5°С воды, в котором кристаллы Na2SO4⋅10H2O отмываются от окклюдированных примесей; затем отмытые кристаллы глауберовой соли отделяют от второй жидкой фазы, представляющей собой смесь указанного маточного раствора и промывного раствора;
к) вторую жидкую фазу со стадии и) с растворенным в ней нитритом натрия непрерывно подают в качестве оборотного раствора на стадию а).
2. Способ по п. 1, где турбулентный смеситель представляет собой винтовой насос.
3. Способ по п. 1 или 2, где в качестве инертного газа на стадии в) используется азот.
4. Способ по любому из пп. 1-3, в котором отмывание кристаллов 4-нитрозофенола от окклюдированных примесей осуществляют в пульсационной колонне.
5. Способ по любому из пп. 1-4, в котором расход указанной промывной воды на стадии г) составляет 1,0-1,5 моль в расчете на 1 моль подаваемого С6Н5ОН.
6. Способ по любому из пп. 1-5, в котором побочные продукты реакции, отделяемые на стадии д), представляют собой продукты конденсации фенола с 4-нитрозофенолом, в частности хромофорные соединения, например такие, как индофенол.
7. Способ по любому из пп. 1-6, в котором в сорбент содержит не менее 60 об.% мезопор, предпочтительно содержит не менее 80 об.% мезопор.
8. Способ по любому из пп. 1-7, в котором в качестве сорбента используют активированный уголь, предпочтительно активированный уголь марки МИУ-С или марки АГ-3.
9. Способ по любому из пп. 1-8, где нейтрализацию едким натром осуществляют в трубчатом смесителе.
10. Способ по любому из пп. 1-9, где нейтрализацию очищенного промывного раствора на стадии е) осуществляют 25-35%-ным раствором едкого натра.
11. Способ по любому из пп. 1-10, где расход NaOH составляет 0,3-0,7 моль в расчете на 1 моль С6Н5ОН, подаваемого на стадии а).
12. Способ по любому из пп. 1-11, где в качестве инертного газа на стадии з) используется азот.
13. Способ по любому из пп. 1-12, в котором отмывание кристаллов глауберовой соли от окклюдированных примесей на стадии и) осуществляют в пульсационной колонне.
14. Способ по любому из пп. 1-13, в котором расход указанной промывной воды на стадии и) составляет 1,0-1,5 моль в расчете на 1 моль подаваемого С6Н5ОН.
| US 3320324 A, 16.05.1967 | |||
| СПОСОБ ПОЛУЧЕНИЯ П-НИТРОЗОФЕНОЛА | 1995 |
|
RU2076096C1 |
| СПОСОБ ПОЛУЧЕНИЯ П-НИТРОЗОФЕНОЛА | 1997 |
|
RU2129117C1 |

