ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ:
Настоящее изобретение относится к улучшенному процессу получения линезолида. Более конкретно, настоящее изобретение относится к улучшенному процессу получения (S)-N-[[3-[3-фтор-4-[4-морфолинил]фенил]-2-оксо-5-оксазолидинил]метил]фталимида и (S)-глицидилфталимида - промежуточных соединений, используемых для получения линезолида.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ:
Линезолид представляет собой синтетический антибиотик, первый из класса оксазолидинонов, который используется для лечения инфекций, вызванных мультирезистентными бактериями, включая стрептококки и метициллин-резистентный золотистый стафилококк (МРЗС). Антибактериальный эффект оксазолидинонов состоит в том, что они действуют как ингибиторы синтеза белка, нарушая его на раннем этапе, когда N-формилметионил-т-РНК связывается с рибосомой.
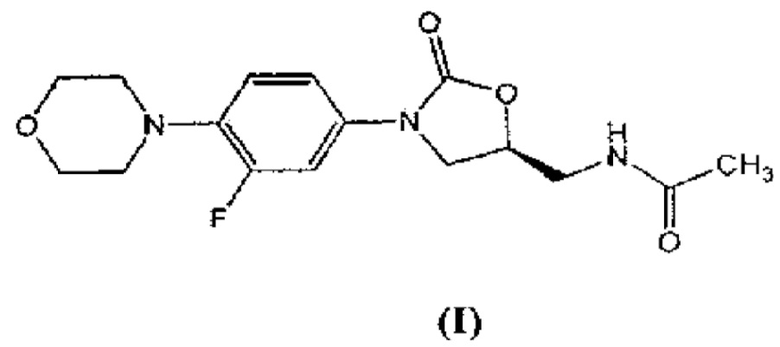
Линезолид выпускается компанией «Пфайзер» (Pfizer) под коммерческим наименованием «Зивокс» (Zyvox) и с химической точки зрения представляет собой (S)-N-[[3-(3-фтор-4-морфолинилфенил)-2-оксо-5-оксазолидинил]метил]ацетамид, имеющий формулу (I).
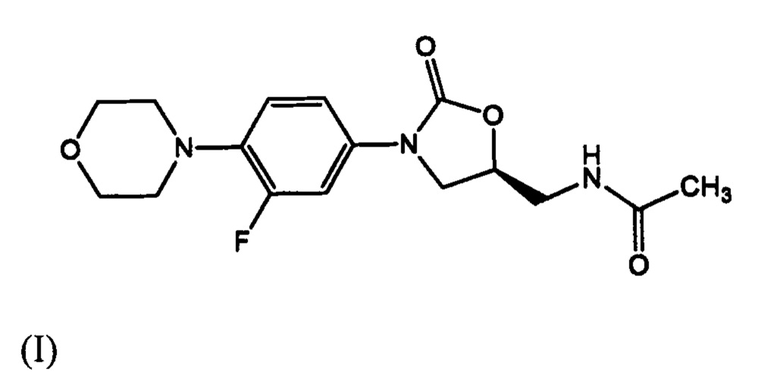
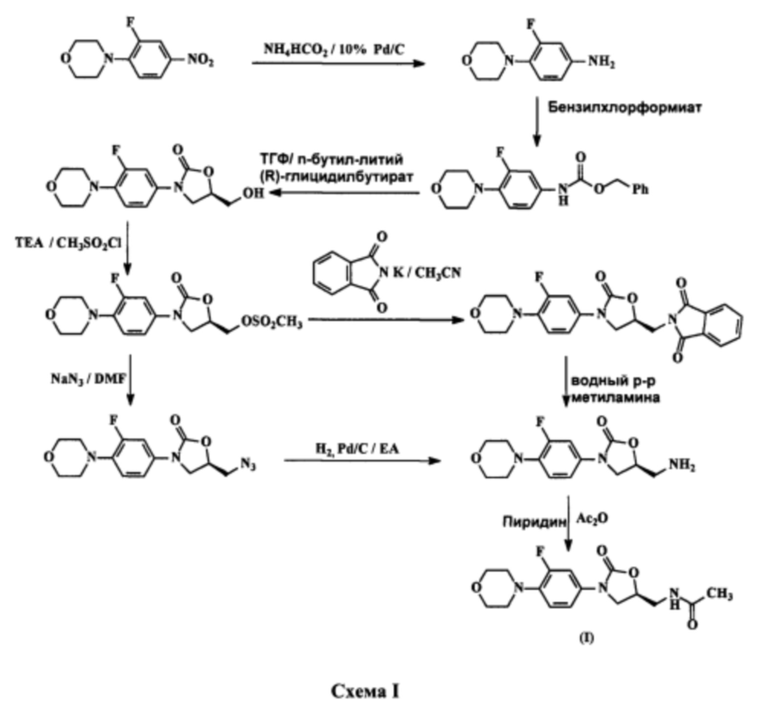
Впервые линезолид был раскрыт в патенте США №5688792, и его получение описывается как процесс с использованием R-глицилидбутирата, который приводит к образованию (R)-N-[[3-[3-фтор-4-морфолинил]фенил]-2-оксо-5-оксазолидинил]метанола, на последующих этапах синтеза претерпевающего превращения в различные промежуточные соединения, а те в конце концов приводят к получению линезолида. Указанный процесс также включает промежуточное соединение из класса азидов, получение которого в промышленных масштабах затруднено, что отражено на приведенной ниже схеме I:
Патент WO 1999/24393 А1 раскрывает процесс получения производных оксазолидинона, отраженный на приведенной ниже схеме:
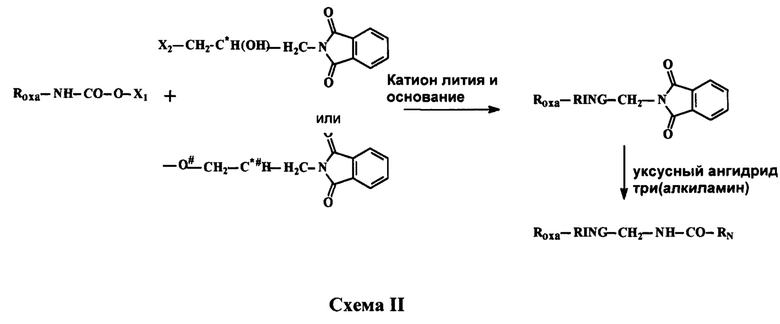
Где Roxa представляет собой фенил, замещенный одним атомом фтора и одной замещенной аминогруппой, при этом к числу замещенных аминогрупп относятся 4-(бензилоксикарбонил)-1-пиперазинил, 4-морфолинил и 4-гидроксиацетилпиперазинил.
X1 - это алкил С1-С20;
Х2 - это Cl, Br
RN - это алкил С1-С5
# означает, что атомы, помеченные символом (#), связываются друг с другом, образуя тем самым кольцо, a RING - это
Однако в документе WO' 393 не раскрываются какие бы то ни было конкретные примеры и не раскрываются условия, подходящие для получения линезолида.
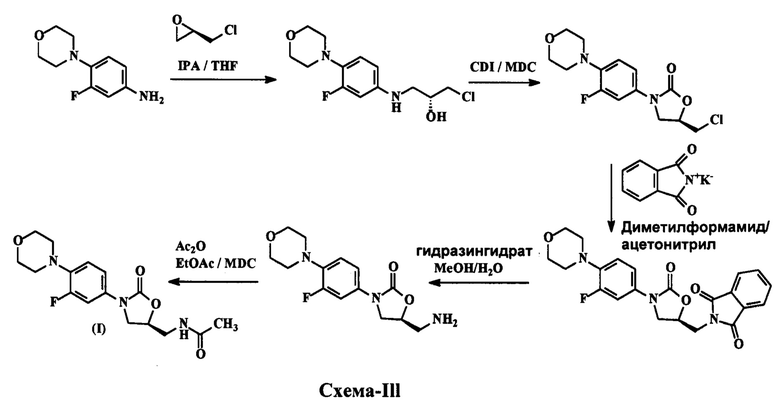
В патенте WO 2005/099353 А2 раскрывается процесс получения линезолида, показанный на схеме III ниже:
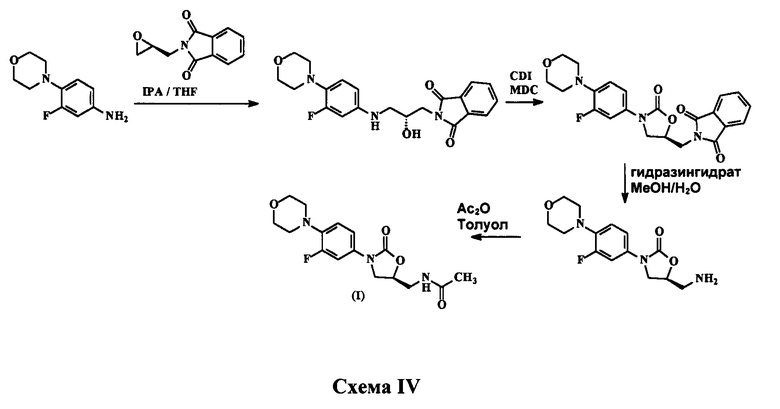
В патенте WO 2006/008754 А1 раскрывается процесс получения линезолида, показанный на схеме IV ниже:
В патенте W02007116284 раскрывается процесс получения линезолида путем реакции соединения, имеющего структуру (1), с соединением, имеющим структуру (2), в диапазоне температур от комнатной до примерно 65°С с получением соединения, имеющего структуру (3), которое подвергается гидролизу, а затем ацилированию, приводя к линезолиду, как показано на схеме V ниже:
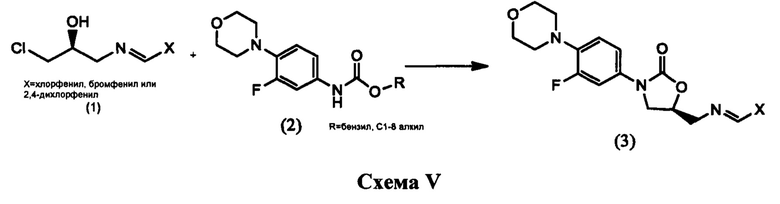
Недостатком данного процесса является необходимость проведения отдельного синтеза для получения соединения, имеющего структуру (1), а участвующие в ней основания Шиффа чувствительны к воде, из-за чего чистый продукт со структурой (1) не всегда возможно получить, что увеличивает количество примесей и способствует образованию побочных продуктов реакции.
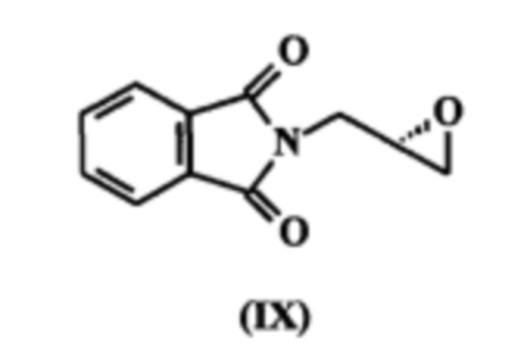
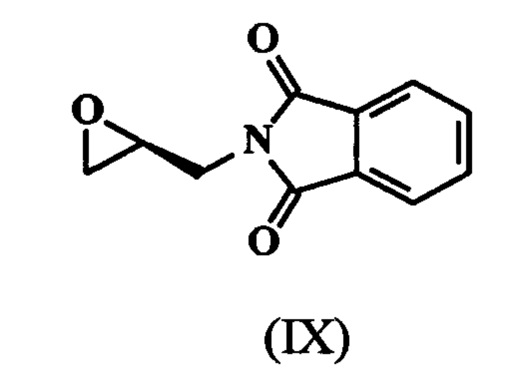
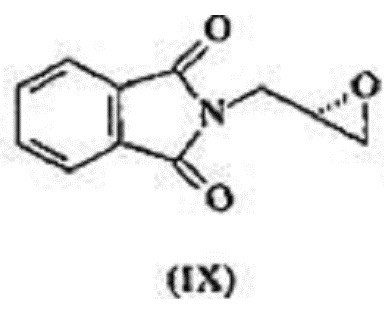
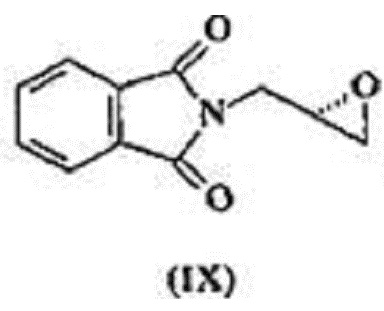
(S)-глицидилфталимид, известный также как 2-[(2S)-оксиран-2-илметил]-1Н-изоиндол-1,3(2H)-дион с формулой (DC) - это ключевое промежуточное соединение во всех путях синтеза линезолида.
2-[(2)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-дион (I) впервые был описан в публикации Compt. rend. 1930, 190, 495-6, где также описывается процесс получения 2-[(2)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-диона, в ходе которого протекает реакция между фталимидом калия и эпибромгидрином в присутствии газообразного хлороводорода или водного раствора соляной кислоты. Вышеупомянутая реакция схематически изображена ниже:

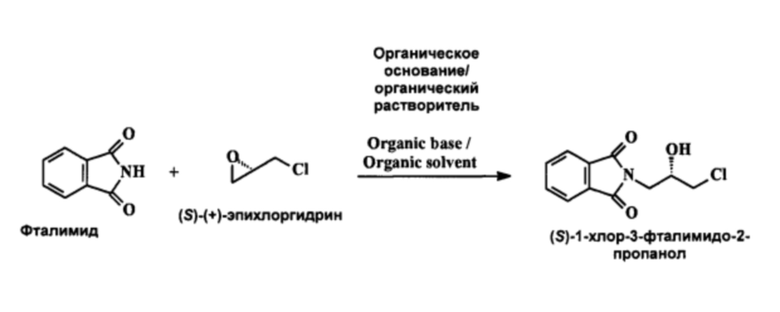
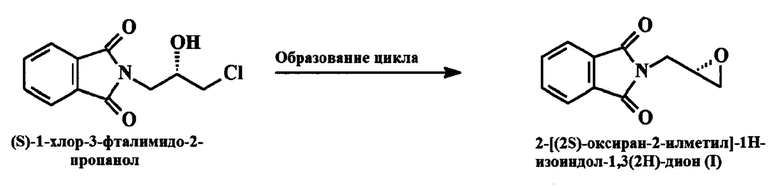
В патенте США 5608110 раскрывается процесс получения 2-[(2S)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-диона (I) в котором реакция между фталимидом и (S)-(+)-эпихлоргидрином протекает в азотной атмосфере в растворе этилацетата/гексана с возвратом флегмы через обратный холодильник, при этом получается (S)-1-хлор-3-фталимидо-2-пропанол, который претерпевает циклическое превращение в присутствии NaH/ТГФ.
Вышеупомянутая реакция схематически изображена ниже:
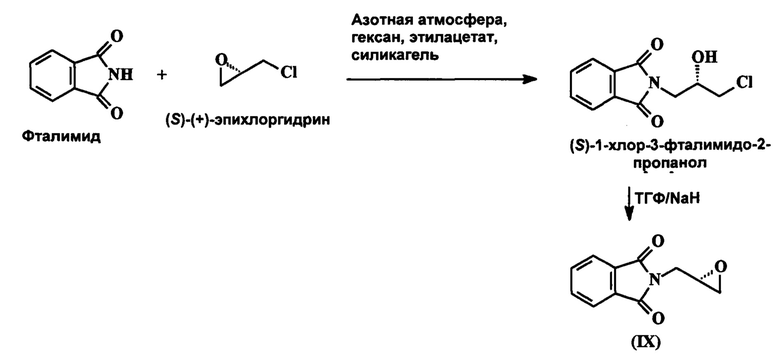
В патенте США 6875875 раскрывается процесс получения 2-[(2S)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-диона (I), в котором реакция между фталимидом и (S)-эпихлоргидрином протекает в присутствии карбонатов щелочных металлов, гидрокарбонатов щелочных металлов или четвертичной аммониевой соли, при этом получается (S)-1-хлор-3-фталимидо-2-пропанол, который претерпевает циклическое превращение в присутствии алкоксидов металлов. Вышеупомянутая реакция схематически изображена ниже:
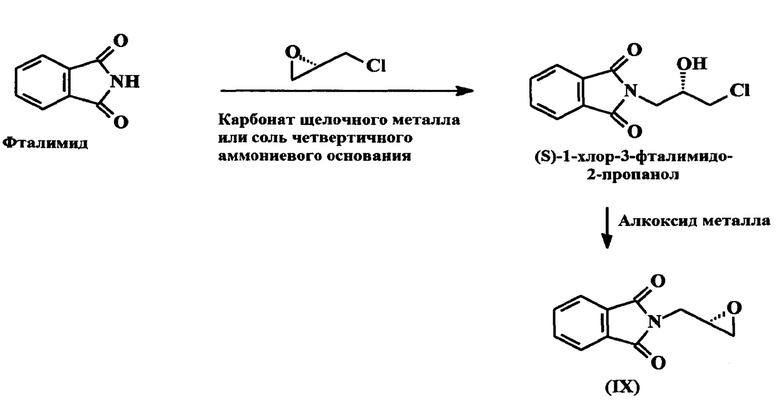
Согласно вышеописанным ранее известным процессам, так как оптическая чистота снижена и так как имеет место постоянный возврат оптически активного эпихлоргидрина в растворитель, возникает проблема того, что оптическая чистота продуктов реакции снижается. Для известных ранее процессов также характерно большое количество отходов; получение чистого продукта требует сложного процесса очистки, такого как хроматографическое разделение на колонке с силикагелем, и этот этап становится крайне важен, при том что его сложно организовать в промышленном масштабе.
Таким образом, пути с использованием газообразного хлороводорода или его водного раствора, а также с использованием карбонатов и гидрокарбонатов щелочных металлов и солей ЧАО могут оказаться экономически неприемлемыми для промышленного производства (S)-глицидилфталимида.
В поисках экологически безопасной и технологически осуществимой реакции получения (S)-глицидилфталимида наши исследователи неожиданно обнаружили реакцию конденсации со вторичными аминами.
Ни в одном из ранее известных процессов нет ни предложения, ни даже предположения относительно возможности использования первичных или вторичных органических оснований.
Следовательно, имеется потребность в таком альтернативном методе получения линезолида и промежуточных соединений, который не включал бы указанные выше недостатки.
Следовательно, на текущем уровне технологии имеется потребность в простом и легко осуществимом процессе получения линезолида, и наши изобретатели разработали экономически эффективный и промышленно осуществимый процесс его получения.
ЦЕЛЬ ИЗОБРЕТЕНИЯ
Основной целью настоящего изобретения является экономически эффективный и коммерчески осуществимый процесс получения линезолида.
Еще одной целью настоящего изобретения является процесс получения промежуточных соединений: (S)-N-[[3-[3-фтор-4-[4-морфолинил]фенил]-2-оксо-5-оксазолидинил]метил]фталимида и (S)-глицидилфталимида, в котором бы использовались менее дорогие, более доступные и экологически безопасные реагенты.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
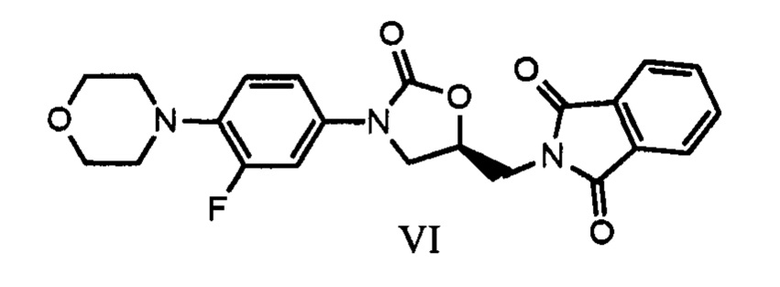
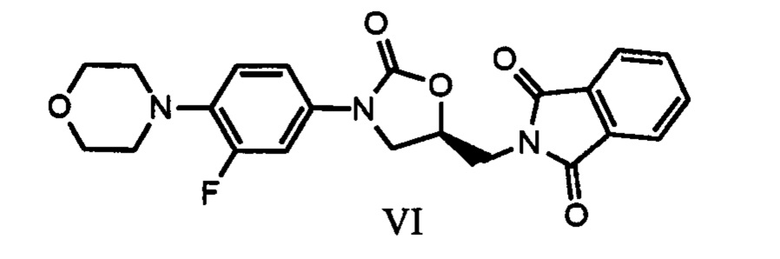
В одном из аспектов настоящего изобретения предлагается улучшенный процесс получения (S)-N-[[3-[3-фтор-4-[4-морфолинил]фенил]-2-оксо-5-оксазолидинил]метил]фталимида, имеющего формулу (IV),
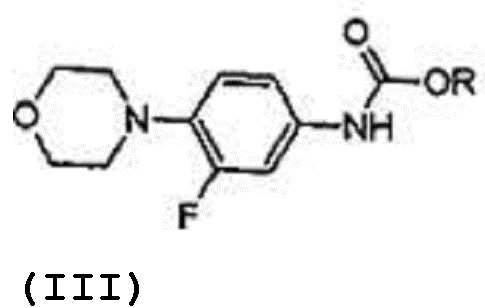
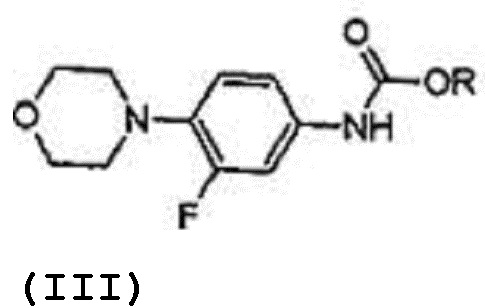
который включает реакцию карбаматного соединения с формулой (III)
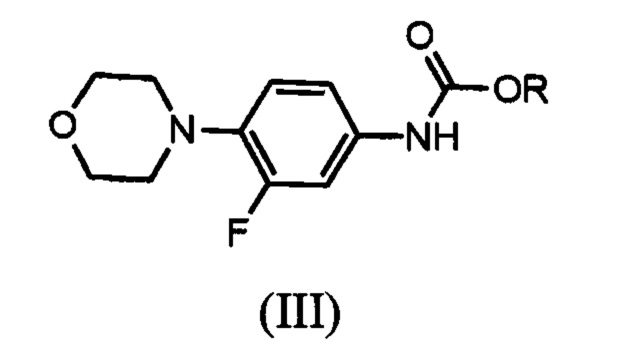
R представляет собой водород, алкил С1-С5, арил, арилалкил;
с (S)-глицидилфталимидом, обладающим формулой (IX),
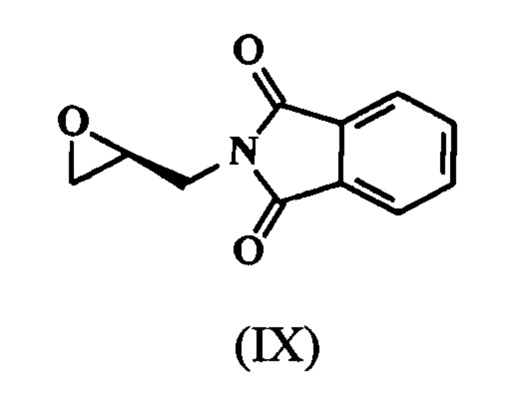
в присутствии иодидов щелочных металлов (или) металлогидридов
(или)
в присутствии третичного бутоксида лития в количестве 0,2-0,4 моль-эквивалента по отношению к карбамату, имеющему формулу (III).
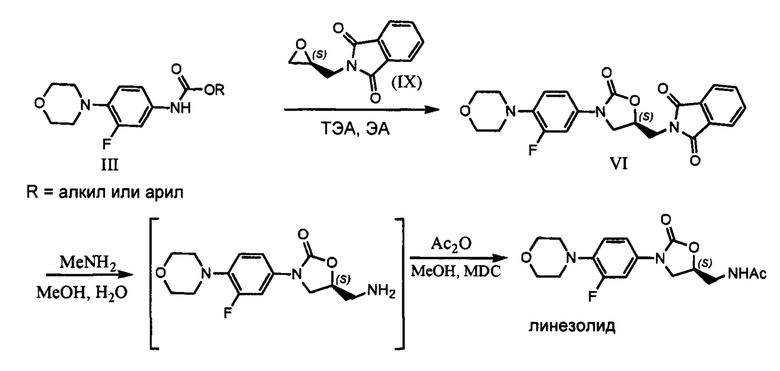
Другим аспектом настоящего изобретения является предоставление улучшенного процесса получения соединения, имеющего формулу (I), в который входят следующие шаги:
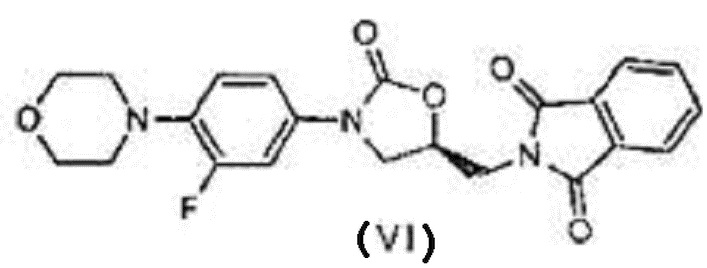
a) реакция соединения с формулой (III) с (S)-глицидилфталимидом или соединением с формулой (IX) в присутствии иодидов щелочных металлов (или) металлогидридов с получением соединения, отвечающего формуле (VI),
b) воздействие на соединение с формулой (VI) водного раствора метиламина или
гидразингидрата,
c) ацилирование продукта реакции стадии b), и
d) выделение продукта, соответствующего формуле (I).
Вышеуказанный процесс синтеза иллюстрируется приводимой далее схемой
В еще одном аспекте настоящего изобретения рассматривается улучшенный процесс получения 2-[(2S)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-диона, имеющего формулу (IX), который состоит из следующих шагов:
a) 1H-изоиндол-1,3(2H)-дион или фталимид реагирует с (S)-эпихлоргидрином в присутствии органического основания в органическом растворителе с получением (S)-1-хлор-3-фталимидо-2-пропанола.
В этом случае органическое основание представляет собой первичные или вторичные алкиламины с длиной углеродной цепи С1-С5.
b) (S)-1-хлор-3-фталимидо-2-пропанол образует циклическое соединение в присутствии алкоксидов щелочных металлов, при этом получается 2-[(2S)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-дион
Полное описание изобретения
В варианте осуществления настоящего изобретения предлагается улучшенный процесс получения (S)-N-[[3-[3-фтор-4-[4-морфолинил]фенил]-2-оксо-5-оксазолидинил]метил]фталимида, имеющего формулу (IV),
который включает реакцию карбаматного соединения с формулой (III)
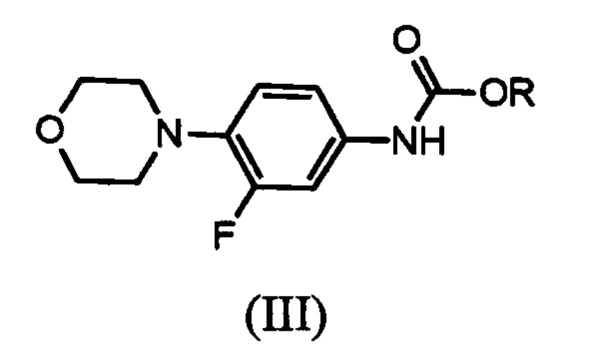
R представляет собой водород, алкил С1-С5, арил, арилалкил;
с (S)-глицидилфталимидом, обладающим формулой (IX),
в присутствии иодидов щелочных металлов (или) металлогидридов (или)
в присутствии третичного бутоксида лития, используемого в количестве 0,2-0,4 моль-эквивалента по отношению к карбамату, имеющему формулу (III).
В другом варианте осуществления реакция проводится с использованием иодидов щелочных металлов в присутствии или отсутствии растворителя в диапазоне температур от 60 до 120°С. Реакция протекает в течение 10-14 часов.
В другом варианте осуществления изобретения реакция протекает с использованием третичного бутоксида лития, который применяется в диапазоне 0,2-0,4 моль-эквивалента в присутствии соответствующего растворителя и при температуре от 40 до 100°С. Реакция протекает в течение 4-12 часов.
Согласно этому варианту осуществления настоящего изобретения, в этом случае подходящими иодидами щелочных металлов могут быть иодид лития, иодид натрия, иодид калия и тому подобные вещества; подходящим растворителем может быть спирт, такой как метанол, этанол, изопропанол и т.п., или их смесь; кетоны, такие как метилизобутилкетон, метилэтилкетон, н-бутанон и т.п.; галогенированные растворители, такие как дихлорметан, дихлорэтилен, хлороформ и т.п.; сложные эфиры, такие как этилацетат, н-пропилацетат, изопропилацетат и т.п.; углеводородные растворители, такие как толуол, ксилол, циклогексан и тому подобные; простые эфиры, такие как 1,4-диоксан, тетрагидрофуран и т.п.; и амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и тому подобные, или диметилсульфоксид или смесь указанных растворителей.
В другом варианте осуществления настоящего изобретения рассматривается предоставление улучшенного процесса получения соединения, имеющего формулу (I), в который входят следующие шаги:
a) реакция соединения с формулой (III) с (S)-глицидилфталимидом или соединением с формулой (IX) в присутствии иодидов щелочных металлов (или) металлогидридов с получением соединения, отвечающего формуле (VI),
b) воздействие на соединение с формулой (VI) водного раствора метиламина или гидразингидрата,
c) ацилирование продукта реакции стадии b), и
d) выделение продукта, соответствующего формуле (I).
Согласно варианту осуществления данного изобретения, реакция между веществом с формулой (III) и (S)-глицидилфталимидом, отвечающим формуле (IX), проводится в присутствии подходящих иодидов щелочных металлов (или металлогидридов) и растворителей при соответствующей температуре с получением соединения, отвечающего формуле (VI); в дальнейшем оно подвергается депротонированию под действием гидразингидрата или водного раствора метиламина с получением (S)-5-аминометил-3-(3-фтор-4-морфолин-4-илфенил)-оксазолидин-2-она, который затем ацилируется с помощью уксусного ангидрида или ацетилхлорида до (S)-N-[[3-(3-фтор-4-морфолинилфенил)-2-оксо-5-оксазолидинил]метил]ацетамида (линезолида), отвечающего формуле I.
Согласно этому варианту осуществления, реакция между веществом с формулой (III) и (S)-глицидилфталимидом, имеющим формулу (IX), проводится в присутствии соответствующих металлогидридов и растворителя при соответствующей температуре, в результате чего получается соединение с формулой (VI); эта реакция длится меньше и приводит к хорошему количественному выходу продукта высокой чистоты.
Согласно этому варианту осуществления, в данном случае подходящими иодидами щелочных металлов могут быть иодид лития, иодид натрия, иодид калия и тому подобные вещества; в качестве металлогидридов могут использоваться гидрид натрия, гидрид лития или гидрид магния.
Согласно этому варианту осуществления настоящего изобретения, в этом случае подходящим растворителем может быть спирт, такой как метанол, этанол, изопропанол и т.п., или их смесь; кетоны, такие как метилизобутилкетон, метилэтилкетон, н-бутанон и т.п.; галогенированные растворители, такие как дихлорметан, дихлорэтилен, хлороформ и т.п.; сложные эфиры, такие как этилацетат, н-пропилацетат, изопропилацетат и т.п.; углеводородные растворители, такие как толуол, ксилол, циклогексан и тому подобные; простые эфиры, такие как 1,4-диоксан, тетрагидрофуран и т.п.; и амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и тому подобные, или диметилсульфоксид или смесь указанных растворителей.
В варианте осуществления настоящее изобретение, кроме того, предусматривает преобразование соединения с формулой (VI) в линезолид, имеющий формулу (I), которое подразумевает превращение фталимидного соединения с формулой (VI) в амин с последующим его ацилированием, приводящим к линезолиду, что достигается обычными, уже известными методами.
Согласно варианту осуществления изобретения, ацилирование выполняется в присутствии уксусного ангидрида или ацетилхлорида.
Реакция протекает при температуре кипения используемого растворителя или ниже, более предпочтительно при температуре от 10°С до температуры кипения используемого растворителя, и еще более предпочтительно при температуре кипения растворителя. Время, необходимое для завершения реакции, зависит от факторов, таких как используемый растворитель и температура проведения реакции.
Согласно варианту осуществления, настоящее изобретение относится к улучшенному процессу получения 2-[(2S)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-диона, имеющего формулу (IX), который состоит из следующих шагов:
а) Фталимид реагирует с (S)-(+)-эпихлоргидрином в присутствии органического основания в органическом растворителе при температуре 60°С с получением (S)-1-хлор-3-фталимидо-2-пропанола.
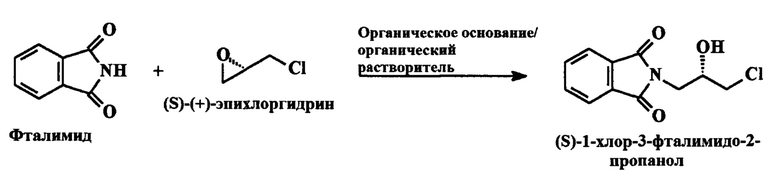
В этом случае органическое основание представляет собой первичные или вторичные алкиламины с длиной углеродной цепи С1-С5.
Органическое основание выбирается из первичных или вторичных алкиламинов с длиной цепи С1-С5, таких как метиламин, этиламин, этилметиламин, диэтиламин, дипропиламин, дибутиламин, предпочтительно - диэтиламин.
Органический растворитель выбирается из группы, включающей спирты, простые эфиры, сложные эфиры, нитрилы с длиной углеродной цепи С1-С4. Спирты предпочтительны.
Спирты выбираются из метанола, этанола, пропанола, изопропанола, бутанола; простые эфиры - из диэтилового эфира, тетрагидрофурана и т.п.; сложные эфиры выбираются из этилацетата, метилацетата и т.п., нитрилы - из ацетонитрила, пропионитрила, бутиронитрила и т.п.
b) (S)-1-хлор-3-фталимидо-2-пропанол образует циклическое соединение в присутствии алкоксидов щелочных металлов, при этом получается 2-[(2S)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-дион
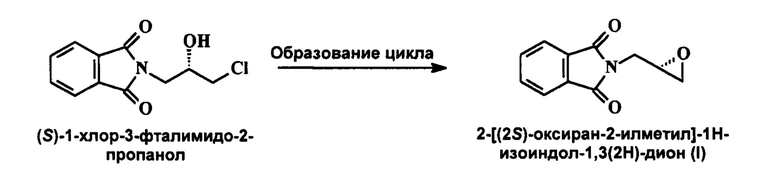
Реакция образования кольца с участием (S)-1-хлор-3-фталимид-2-пропанола приводит к 2-[(2S)-оксиран-2-илметил]-1H-изоиндол-1,3-(2H)-диону.
Реакция образования кольца проводится в присутствии алкоксидов щелочных металлов, как описано в патенте США 6875875.
Линезолид, полученный согласно настоящему изобретению, может быть аморфным или иметь форму кристаллов (форма I и форма II).
Согласно настоящему изобретению, соединение формулы (I), или линезолид, имеет ВЭЖХ-чистоту не менее 99%.
Преимущества настоящего изобретения:
1. Настоящее изобретение представляет собой простой, понятный оператору и осуществимый в промышленном масштабе процесс.
2. Процесс коммерчески перспективен и приводит к получению продукта с высоким выходом, что делает его экономичным.
3. На осуществление реакций в соответствии с настоящим изобретением нужно меньше времени.
4. Настоящее изобретение обеспечивает получение соединений высокой чистоты по формулам (I), (VI) и (IX) с очень незначительным профилем примесей.
Подробное описание процесса настоящего изобретения приводится ниже в примерах, которые носят иллюстративных характер и, следовательно, не должны читаться ограничивающими объем изобретения.
Экспериментальная процедура:
Пример 1:
Получение 2-[(2S)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-диона:
К суспензии 1H-изоиндол-1,3(2H)-диона (100 г, 0,68 моля) и метиламина (10 г) в изопропаноле (200 мл) добавили S-эпихлоргидрин (100 г, 1,08 моля) и нагрели смесь до 60°С, после чего выдержали при данной температуре в течение 5 часов. 30%-ный раствор метоксида натрия в метаноле (160 г) добавляли порционно при температуре 10-15°С к реакционной смеси, после чего температуру подняли до 25-30°С и перемешивали реакционную массу при той же температуре в течение 2-3 часов. По завершении реакции добавили воду (600 мл), перемешивали смесь в течение 10 минут, отфильтровали твердый осадок и промыли его водой, получив заявленное вещество.
Выход: 107 г (77,5%)
Пример 2:
Получение 2-[(2S)-оксиран-2-илметил]-1H-изоиндол-1,3(2H)-диона:
К суспензии 1H-изоиндол-1,3(2H)-диона (100 г, 0,68 моля) и диэтиламина (10 г) в этаноле (200 мл) добавили 3-эпихлоргидрин (100 г, 1,08 моля) и нагрели смесь до 60°С, после чего выдержали при данной температуре в течение 5 часов. 30%-ный раствор метоксида натрия в метаноле (160 г) добавляли порционно при температуре 10-15°С к реакционной смеси, после чего температуру подняли до 25-30°С и перемешивали реакционную массу при той же температуре в течение 2-3 часов. По завершении реакции добавили воду (600 мл), перемешивали смесь в течение 10 минут, отфильтровали твердый осадок и промыли его водой, получив заявленное вещество.
Выход: 107 г (77,5%)
Пример 3:
Получение (S) 2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-5-илметил]-изоиндол-1,3-диона
Смесь метил(3-фтор-4-морфолинофенил)карбамата (50 г, 0,196 моля) в этилацетате (100 мл) перемешивали в течение 10 минут при температуре 25-30°С. 2-[(2S)-оксиран-2-илметил]-1Н-изоиндол-1,3(2Н)-дион (50 г, 0,246 моля) и третичный бутоксид лития (5,0 г, 0,0624 моля) были добавлены одновременно при температуре 25-30°С, а затем температуру медленно повысили до 70-75°С и выдерживали смесь при этой температуре в течение примерно 6-8 часов. Реакционную смесь охладили до комнатной температуры, добавили этилацетат (50 мл) и полученную взвесь перемешивали в течение 30 минут при 25-30°С, после чего отфильтровали твердые вещества. К полученному осадку на фильтре добавили этилацетат (250 мл) при температуре 25-30°С и нагрели до 70-75°С, перемешивали в течение 15-20 минут, охладили смесь до 25-30°С и перемешивали в течение 30 минут. Полученное вещество отфильтровали и промыли осадок этилацетатом (50 мл), чтобы получить чистое соединение (5S)2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-изоиндол-1,3-дион.
Выход: 70 г (85% выхода от теоретического)
Пример 4:
Получение (S) 2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-5-илметил]-изоиндол-1,3-диона
Смесь метил (3-фтор-4-морфолинофенил) карбамата (50 г, 0,196 моля) в диметилформамиде (75 мл), 2-[(2S)-оксиран-2-илметил]-1Н-изоиндол-1,3(2Н)-диона (50 г, 0,246 моля) и третичного бутоксида лития (5,0 г, 0,0624 моля) нагрели до 70-75°С и выдерживали при этой температуре 4-6 часов. Реакционной смеси дали остыть до 26°С, добавили метанол (250 мл) и перемешивали полученную взвесь в течение 30 минут при 25-30°С. Полученный осадок был отфильтрован и промыт метанолом (50 мл). Полученный твердый осадок подвергли перекристаллизации с этилацетатом для получения чистого (5S) 2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-изоиндол-1,3-диона с выходом: 65 г (77,7%).
Пример 5:
Получение (S) 2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-5-илметил]-изоиндол-1,3-диона
Метил (3-фтор-4-морфолинофенил) карбамата (100 г, 0,392 моля) в диметилформамиде (200 мл), 2-[(2S)-оксиран-2-илметил]-1Н-изоиндол-1,3(2Н)-диона (100 г, 0,492 моля) и третичного бутоксида лития (5,0 г, 0,0625 моля) смешали одновременно при температуре 25-30°С, затем нагрели до 80-85°С и выдерживали при этой температуре 4-6 часов. Реакционную смесь охладили до комнатной температуры, затем добавили очищенную воду (500 мл) и перемешивали в течение 30 минут при 25-30°С. Полученное твердое вещество отфильтровали и промыли очищенной водой (100 мл). Полученный твердый осадок подвергли перекристаллизации с этилацетатом для получения чистого (5S)2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-изоиндол-1,3-диона.
Выход: 140 г (85%)
Пример 6:
Получение (S) 2-[3-(3-фтор-4-морфол ин-4-ил-фенил)-2-оксо-оксазолидин-5-илметил]-изоиндол-1,3-диона
Метил (3-фтор-4-морфолинофенил) карбамат (20 г, 0,0784 моля), 2-[(2S)-оксиран-2-илметил]-1Н-изоиндол-1,3(2Н)-дион (20 г, 0,0984 моля), диметилформамид (80 мл) и иодид натрия (4,0 г) смешали одновременно при температуре от 25 до 30°С, после чего температуру медленно повысили до 110-115°С. Реакционную смесь выдерживали при температуре 110-115°С в течение 6-8 часов. Реакционную смесь охладили до комнатной температуры (200 мл), перемешивали в течение 30 минут при 25-30°С, а затем отфильтровали твердые вещества. Полученный осадок подвергли перекристаллизации в метаноле (20 мл) с получением чистого (5S)2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-изоиндол-1,3-диона.
Выход: 25 г (75%)
Пример 7:
Получение (S) 2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-5-илметил]-изоиндол-1,3-диона
Смесь метил (3-фтор-4-морфолинфенил) карбамата (25 г, 0,098 моля) в тетрагидрофуране (50 мл), 2-[(2S)-оксиран-2-илметил]-1Н-изоиндол-1,3(2Н)-диона (25 г, 0,123 моля) перемешивали после добавления в течение 10 минут при температуре 25-30°С; после чего однократно добавили гидрид натрия (1,2 г, 0,05 моля) при 25-30°С. Реакционную смесь медленно нагрели до 50-60°С и выдержали при этой температуре в течение примерно 2-3 часов. Реакционную массу охладили до температуры ниже 20°С, залили 25 мл метанола для разложения избытка гидрида натрия, отогнали растворитель и добавили метанол (125 мл). Полученную взвесь перемешивали в течение 30 минут при 25-30°С и затем профильтровали. К полученному осадку на фильтре добавили этилацетат (125 мл), нагрели до 70-75°С, перемешивали в течение 15-20 минут, затем охладили до 25-30°С, профильтровали и промыли осадок этилацетатом (25 мл), чтобы получить чистое соединение (5S)2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-изоиндол-1,3-дион.
Выход: 34 г (77%)
Пример 8:
Получение (S) 2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-5-илметил]-изоиндол-1,3-диона
Смесь метил (3-фтор-4-морфолинфенил) карбамата (25 г, 0,098 моля) в N,N-диметилформамиде (50 мл) и 2-[(2S)-оксиран-2-илметил]-1Н-изоиндол-1,3(2Н)-диона (25 г, 0,123 моля) перемешивали после добавления в течение 10 минут при температуре 25-30°С; после чего однократно добавили гидрид натрия (1,2 г, 0,05 моля) при 25-30°С. Реакционную смесь медленно нагрели до 50-60°С и выдержали при этой температуре в течение примерно 2-3 часов. Реакционную массу охладили до температуры ниже 20°С, залили 25 г метанола для разложения избытка гидрида натрия, отогнали растворитель и добавили метанол (125 мл), а полученную взвесь перемешивали в течение 30 мин при 25-30°С, после чего профильтровали. К полученному осадку на фильтре добавили этилацетат (125 мл), нагрели до 70-75°С, перемешивали в течение 15-20 минут, затем охладили до 25-30°С, профильтровали и промыли осадок этилацетатом (25 мл), чтобы получить чистое соединение (5S)2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-изоиндол-1,3-дион.
Выход: 32 г (75%)
Пример 9:
Получение N-({(5S)-3-[3-фтор-4-(морфолин-4-ил)фенил]-2-оксо-1,3-оксазолидин-5-ил}метил)ацетамида(линезолида)
К смеси метанола (100 мл), деминерализованной воды (400 мл) и (S)2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-5-илметил]-изоиндол-1,3-диона (100 г, 0,212 моля) добавили раствор метиламина (47 г) при температуре 25-30°С, перемешали и стали медленно нагревать до 80-85°С, после чего перемешивали в течение 2-3 часов при 80-85°С. Реакционную смесь охладили до 25-30°С и добавили дихлорметан (500 мл), после чего реакционную смесь перемешивали в течение 15 минут и дали ей разделиться на два слоя. Дихлорметан отогнали при атмосферном давлении полностью и получили в остатке (5S)-5-(аминометил)-3-[3-фтор-4-(морфолин-4-ил)фенил]-1,3-оксазолидин-2-он. К остатку добавили дихлорметан (400 мл) и стали медленно приливать уксусный ангидрид (25 г) при 25-30°С в течение 60 минут. По завершении процесса 5%-ный водный раствор бикарбоната натрия медленно добавили к смеси, перемешивали в течение 15 минут, после чего смесь разделилась на два слоя. Слой с дихлорметаном промыли деминерализованной водой (200 мл). Слой с дихлорметаном пропустили через диатомовый фильтр и полностью отогнали дихлорметан под вакуумом при температуре 40°С. Циклогексан (500 мл) был добавлен к остатку, после чего смесь нагрели до 45-50°С. Полученную взвесь охладили до 20-25°С, перемешивали в течение 60 минут, отфильтровали твердое вещество, промыли осадок циклогексаном (200 мл) и высушили его при температуре 45-55°С, получив чистый N-({(5S)-3-[3-фтор-4-(морфолин-4-ил)фенил]-2-оксо-1,3-оксазолидин-5-ил}метил)ацетамид (линезолид) (53 г, 75%).
Пример 10:
Получение N-({(5S)-3-[3-фтор-4-(морфолин-4-ил)фенил]-2-оксо-1,3-оксазолидин-5-ил}метил)ацетамида (линезолида)
К смеси метанола (100 мл), деминерализованной воды (400 мл) и (S)2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-5-илметил]-изоиндол-1,3-диона (100 г, 0,212 моля) добавили гидразингидрат (50 г) при температуре 25-30°С, перемешали и стали медленно нагревать до 70-75°С, после чего перемешивали в течение 2-3 часов при 70-75°С. Реакционную смесь охладили до 25-30°С и добавили дихлорметан (250 мл), после чего реакционную смесь перемешивали в течение 15 минут и дали ей разделиться на два слоя. Дихлорметан отогнали при атмосферном давлении полностью и получили в остатке (5S)-5-(аминометил)-3-[3-фтор-4-(морфолин-4-ил)фенил]-1,3-оксазолидин-2-он. К остатку добавили дихлорметан (200 мл) и стали медленно приливать уксусный ангидрид (13 г) при 25-30°С в течение 60 минут. Реакционную смесь перемешивали в течение 60 минут при 25-30°С. По завершении процесса реакции 5%-ный водный раствор бикарбоната натрия медленно добавляли к смеси в течение 15 минут, после чего смесь разделилась на два слоя. Слой с дихлорметаном промыли водой (100 мл). Слой с дихлорметаном пропустили через диатомовый фильтр и полностью отогнали дихлорметан под вакуумом при температуре ниже 40°С. Циклогексан (250 мл) добавили к остатку и нагрели его до 45-50°С. Полученную взвесь охладили до 20-25°С, перемешивали в течение 60 минут, отфильтровали твердое вещество, промыли осадок циклогексаном (100 мл) и высушили его при температуре 45-55°С, получив чистый кристаллический N-({(5S)-3-[3-фтор-4-(морфолин-4-ил)фенил]-2-оксо-1,3-оксазолидин-5-ил}метил)ацетамид (линезолид) в количестве 55 г.
Пример 11:
Получение N-({(5S)-3-[3-фтор-4-(морфолин-4-ил)фенил]-2-оксо-1,3-оксазолидин-5-ил}метил)ацетамида (линезолида)
Раствор метиламина (50 г) смешали с метанолом (100 мл), деминерализованной водой (400 мл) и (5S)2-[3-(3-фтор-4-морфолин-4-ил-фенил)-2-оксо-оксазолидин-5-илметил]-изоиндол-1,3-дионом (100 г, 0,235 моля) при 25-30°С. Реакционную смесь перемешали, медленно нагрели до 80-85°С и перемешивали в течение 2-3 часов при 80-85°С. Реакционную смесь охладили до 25-30°С, после чего добавили дихлорметан (500 мл) и перемешивали в течение 15 минут; затем смеси дали разделиться на два слоя. К слою дихлорметана добавили очищенную воду (500 мл), и смесь подкислили до рН 2,0-3,0 разбавленной соляной кислотой, после чего перемешивали в течение 10-15 минут, затем дали слоям разделиться и подщелочили их до рН 10,0-11,0 раствором аммиака, чтобы отделить слой дихлорметана, который затем был отогнан при атмосферном давлении, а в остатке был получен (5S)-5-(аминометил)-3-[3-фтор-4-(морфолинил-4-ил)фенил]-1,3-оксазолидинил-2-он. К остатку добавили дихлорметан (400 мл) и стали медленно приливать уксусный ангидрид (25 г) при 25-30°С в течение 60 минут. Реакционную смесь перемешивали в течение 60 минут при 25-30°С. По завершении реакции 5%-ный водный раствор бикарбоната натрия медленно добавили к смеси, перемешивали в течение 15 минут, после чего смесь разделилась на два слоя. Слой с дихлорметаном промыли деминерализованной водой (200 мл). Дихлорметановый слой пропустили через диатомовый фильтр и полностью отогнали дихлорметан под вакуумом при температуре ниже 40°С. Циклогексан (500 мл) был добавлен к остатку, после чего смесь нагрели до 45-50°С. Полученную взвесь охладили до 20-25°С, перемешивали в течение 60 минут, отфильтровали полученное твердое вещество, промыли осадок циклогексаном (200 мл) и высушили его при температуре 45-55°С, получив чистый кристаллический N-({(5S)-3-[3-фтор-4-(морфолин-4-ил)фенил]-2-оксо-1,3-оксазолидин-5-ил}метил)ацетамид (линезолид) (53 г, 75%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 5-ХЛОР-N-({(5S)-2-ОКСО-3-[4-(3-ОКСО-4-МОРФОЛИНИЛ)-ФЕНИЛ]-1, 3-ОКСАЗОЛИДИН-5-ИЛ}-МЕТИЛ)-2-ТИОФЕНКАРБОКСАМИДА | 2004 |
|
RU2383540C2 |
| ОКСАЗОЛИДИНОНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2006 |
|
RU2417223C2 |
| Замещенные оксазолидиноны, способы их получения, лекарственное средство на их основе и применение замещенных оксазолидинонов | 2000 |
|
RU2836548C9 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ПОМОЩЬЮ ЗАМЕЩЕННЫХ ОКСАЗОЛИДИНОНОВ | 2002 |
|
RU2321407C9 |
| ЗАМЕЩЕННЫЕ ОКСАЗОЛИДИНОНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ ЗАМЕЩЕННЫХ ОКСАЗОЛИДИНОНОВ | 2000 |
|
RU2297415C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| ОКСАЗОЛИДИНОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ПРОТИВОБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2016 |
|
RU2794494C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| ОКСАЗОЛИДИНОН-ХИНОЛОНГИБРИДНЫЕ АНТИБИОТИКИ | 2004 |
|
RU2371443C2 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА | 2001 |
|
RU2292345C9 |
Изобретение относится к улучшенному процессу получения соединения, имеющего формулу (I)
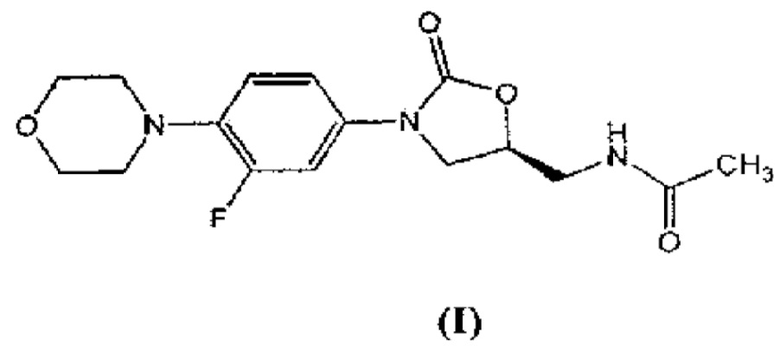 ,
,
в который входят следующие стадии:
а) реакция соединения с формулой (III) с (S)-глицидилфталимидом формулы (IX) в растворителе в присутствии металлогидрида при температуре 50-60ºC, где температура 50-60ºC выдерживается в течение 2-3 часов с получением соединения, отвечающего формуле (VI),
 ,
,  ,
,  ,
,
в вышеуказанной формуле R представляет собой метил,
b) обработка соединения формулы (VI) водным раствором метиламина или гидразингидрата,
c) ацилирование продукта реакции стадии b) ацилирующим веществом и
d) выделение продукта, соответствующего формуле (I), при этом способ по п.1 осуществляется при мольном соотношении на стадии а) соединения с формулой (III) к (S)-глицидилфталимиду формулы (IX) и металлогидриду, соответствующем 0,098:0,123:0,05. Технический результат: разработан улучшенный способ получения линезолида, который позволяет сократить время процесса и приводит к получению продукта с высоким выходом высокой чистоты. 2 з.п. ф-лы, 11 пр.
1. Улучшенный процесс получения соединения, имеющего формулу (I)
 ,
,
в который входят следующие стадии:
а) реакция соединения с формулой (III) с (S)-глицидилфталимидом формулы (IX) в растворителе в присутствии металлогидрида при температуре 50-60ºC, где температура 50-60ºC выдерживается в течение 2-3 часов с получением соединения, отвечающего формуле (VI),
 ,
,  ,
, 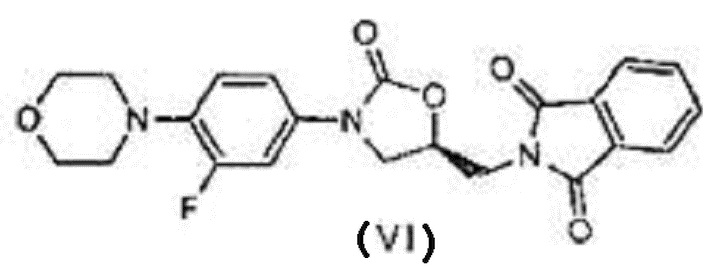 ,
,
в вышеуказанной формуле R представляет собой метил,
b) обработка соединения формулы (VI) водным раствором метиламина или гидразингидрата,
c) ацилирование продукта реакции стадии b) ацилирующим веществом и
d) выделение продукта, соответствующего формуле (I), при этом способ по п.1 осуществляется при мольном соотношении на стадии а) соединения с формулой (III) к (S)-глицидилфталимиду формулы (IX) и металлогидриду, соответствующем 0,098:0,123:0,05.
2. Улучшенный процесс по п. 1, где гидрид металла представляет собой гидрид натрия; ацилирующее вещество представляет собой уксусный ангидрид.
3. Улучшенный процесс по п. 1, где стадия а) проводится в присутствии растворителя, выбираемого из группы, которую составляют N,N-диметилформамид, тетрагидрофуран и этилацетат.
| Регистрирующий измеритель скорости движения | 1926 |
|
SU5865A1 |
| WO 2014141067 A2, 18.09.2014 | |||
| CN 103420933 A, 04.12.2013 | |||
| RU 2012151303 A, 10.06.2014. | |||

