Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к новым оксазолидиноновым соединениям, применимым для лечения бактериальных инфекций, в частности, микобактериальных инфекций. Настоящее изобретение также относится к способам применения оксазолидиноновых соединений для лечения микобактериальных инфекций, таких как вызванные Mycobacteria tuberculosis.
Предшествующий уровень техники настоящего изобретения
Mycobacterium является родом бактерий, которые не являются ни истинно грамположительными, ни истинно грамотрицательными и включают в себя патогены, отвечающие за туберкулез (M. tuberculosis) и лепру (M. leprae). Туберкулез (TB), в частности, несмотря на доступность лекартственных средств против TB, таких как изониазид и рифампин, считается одним из самых смертоносных заболеваний в мире. По данным Всемирной организации охраны здоровья в 2012 году насчитывалось 8,6 миллиона новых случаев TB и 1,3 миллиона смертей от TB. См. мировой отчет по туберкулезу за 2013 год, опубликованный Всемирной организацией охраны здоровья. Усугубление эпидемии TB заключается в нарастающем потоке устойчивых к нескольким лекарственным средствам штаммов и в смертельной ассоциации с HIV. У людей, которые являются HIV-позитивными и инфицированными TB, в 30 раз вероятнее развитие активного TB, чем у HIV-отрицательных, и TB является причиной смерти одного из трех человек с HIV/AIDS во всем мире. См., например, Kaufmann et al., Trends Microbiol. 1: 2-5 (1993), и Bloom et al., N. Engl. J. Med. 338: 677-678 (1998).
Микобактерии, отличные от M. tuberculosis, все чаще встречаются при оппортунистических инфекциях, которые поражают больного AIDS. Организмы комплекса M. avium-intracellulare (MAC), особенно серотипы четыре и восемь, составляют 68% микобактериальных изолятов от больных AIDS. Обнаружено огромное количество MAC (до 1010 кислотоустойчивых бацилл на грамм ткани), и, следовательно, прогноз для инфицированного больного AIDS является плохим.
Оксазолидиноны составляют класс соединений, содержащих 2-оксазолидон, 5-членное кольцо, содержащее азот и кислород, которые используют в качестве противомикробных средств. См., например, WO2009157423. В целом, оксазолидиноны, как известно, являются ингибиторами моноаминоксидазы и обладают активностью против грамположительных микроорганизмов. WO2006022794, Suzuki et al., Med.Chem. Lett. 4:1074-1078 (2013), Yang et al., J. Med. Chem. 58:6389-6409 (2015), Shaw et al., Ann. N.Y. Acad. Sci. 1241:48-70 (2011). Некоторые оксазолидиноновые антибиотики были одобрены или проходят клинические испытания для лечения грамположительных бактериальных инфекций, таких как вызванные устойчивым к метициллину Staphylococcus aureus. Примеры оксазолидиноновых антибиотиков включают в себя линезолид (Zyvox™, Pfizer Inc., New York, NY) и тедизолид (Sivextro™, Merck Sharp & Dohme Corp., Kenilworth, NJ). Тедизолид используют для лечения острых бактериальных инфекций кожи и кожных структур, вызванных особенно восприимчивыми грамположительными бактериями. Линезолид назначают для лечения некоторых инфекций, вызванных восприимчивыми штаммами грамположительных микроорганизмов, в том числе нозокомиальной пневмонии, осложненных инфекций кожи и кожных структур, а также внебольничной пневмонии. Кроме того, в настоящее время его тестируют в условиях клинических испытаний для лечения устойчивых к нескольким лекарственным средствам (MDR) и обладающих широкой лекарственной устойчивостью (XDR) Mycobacterium tuberculosis (Mtb). Lee et al., N. Engl. J. Med 367: 1508-18 (2012). Несмотря на клиническую эффективность в лечении этих заболеваний длительное применение линезолида ассоциировалось с неблагоприятными явлениями, в том числе с миелосупрессией (включающей в себя анемию и лейкопению) (Hickey et al., Therapy 3(4):521-526 (2006), нейропатией и серотониновым синдромом. Предполагают, что такие неблагоприятные явления ассоциируются с ингибированием синтеза митохондриального белка. Flanagan et al., Antimicrobial Agents and Chemotherapy 59(1):178-185 (2015).
Разработка оксазолидининоновых антибиотиков, которые более безопасны, чем одобренные оксазолидиноны, но по меньшей мере столь же эффективны, принесет большую пользу для больных Mtb.
Краткое описание настоящего изобретения
Настоящее изобретение относится к определенным новым оксазолидиноновым соединениям, обладающим антибактериальной активностью. Соединения и их фармацевтически приемлемые соли могут быть применимы, например, для лечения бактериальных инфекций, например, микобактериальных инфекций. Более конкретно, настоящее изобретение включает в себя соединения формулы I или их фармацевтически приемлемые соли:
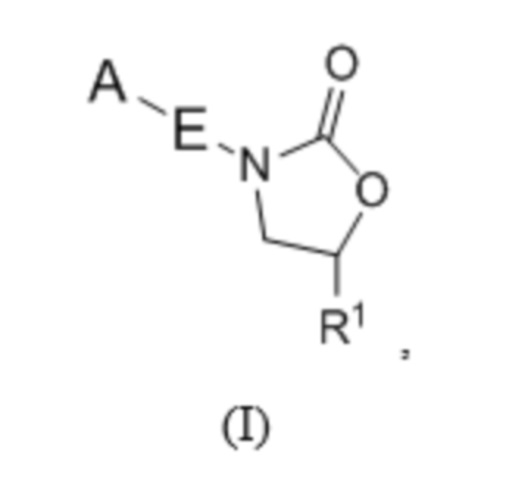
где:
R1 представляет собой -CH2N(R2)2, -CH2NR2COR3, -CH2NR2COOR3, -CH2NR2CON(R2)2, -CH2NR2CONR2N(R2)2, -CH2NR2SO2R3, - CON(R2)2, -C=NOR3, -CH2OR4, -CH2NR2R4 или -CH2R6;
в каждом случае R2 независимо выбран из H, C1-C6 алкила, C2-C6 алкенила и C3-C6 циклоалкила, причем указанный C1-C6 алкил, указанный C2-C6 алкенил и указанный C3-C6 циклоалкил может быть необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, NH2, NHCH3 и N(CH3)2;
R3 представляет собой H, C1-C6 алкил, C2-C6 алкенил и C3-C6 циклоалкил, причем указанный C1-C6 алкил, указанный C2-C6 алкенил и указанный C3-C6 циклоалкил может быть необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, NH2, NHCH3 и N(CH3)2;
R4 представляет собой 5- или 6-членный гетероцикл, который необязательно замещен R5;
R5 выбран из H, галогена, C1-C6 алкила, C3-C6 алкенила и C3-C6 циклоалкила, причем указанный C1-C6 алкил, указанный C3-C6 алкенил и указанный C3-C6 циклоалкил может быть необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, NH2, NHCH3 и N(CH3)2;
R6 представляет собой H, C1-C6 алкил или 5-членный гетероцикл, причем указанный 5-членный гетероцикл необязательно замещен R7 в количестве до двух;
R7 представляет собой H, галоген, оксо, C1-C6 алкил, C2-C6 алкенил или C3-C6 циклоалкил, причем указанный C1-C6 алкил и указанный C3-C6 циклоалкил может быть необязательно замещен заместителями в количестве от одного до четырех, которые независимо выбраны из галогена OCH3, OH, NH2, NHCH3 и N(CH3)2;
E представляет собой 6-членный арил или 5- или 6-членный гетероарил, содержащий от одного до трех гетероатомов, независимо выбранных из S, O и N, причем указанный арил и указанный гетероарил необязательно замещены заместителями в количестве до четырех, которые независимо выбраны из галогена -CN, -CF3, -CHF2, -CH2NH2, -CH2NHCOCH3, -OCF3, -OCHF2, -OH, -O-(C1-C6)алкила, C1-C6 алкила и C3-C6 циклоалкила;
A представляет собой гетероцикл, необязательно замещенный R8 в количестве до четырех, или арил, замещенный R8 в количестве до четырех;
в каждом случае R8 независимо выбран из галогена, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 гетероциклоалкила, бензила, -OCF3, -OCHF2, -OR3, =O, -CN, -NO2, -SR3, -SF5, -SCF3, -SOR3, -SO2R3, -S(=O)(=NH)R2, -N(R2)2, -NR2COR3, -SO2N(R2)2, -NR2SO2R3, -COOH, -COR9, -COOR3, -CON(R2)2 и -C(R9)2N(R2)2, причем указанный C1-C6 алкил, C3-C6 циклоалкил, C3-C6 гетероциклоалкил и бензил необязательно замещены F, -OCH3, -OH, =O, NH2, NHCH3 и N(CH3)2 в количестве до четырех; и
в каждом случае R9 независимо выбран из H, C1-C6 алкила и C3-C6 циклоалкила.
Настоящее изобретение также относится к фармацевтической композиции для лечения бактериальной инфекции у субъекта, в частности инфекции M. tuberculosis, содержащей оксазолидиноновое соединение по настоящему изобретению и фармацевтически приемлемый носитель, разбавитель или наполнитель.
Соединения формулы (I) (также в настоящем описании они упоминаются как «оксазолидиноновые соединения») и их фармацевтически приемлемые соли могут быть применимы, например, для ингибирования роста Mycobacterium tuberculosis и/или для лечения или предупреждения туберкулеза у пациента. Не связываясь любой конкретной теорией полагали, что применение оксазолидиноновых соединений по настоящему изобретению для лечения туберкулеза по всей вероятности вызывает меньше миелосупрессии, чем известные оксазолидиноновые соединения, такие как линезолид, поскольку они не связаны с высокой степенью ингибирования митохондриального белкового синтеза (см. пример 65). Кроме того, оксазолидиноновые соединения по настоящему изобретению являются более селективными, чем известные оксазолидиноновые соединения с более слабой грам-положительной антибактериальной активностью и высокой активностью по отношению к M. tuberculosis.
Настоящее изобретение также относится к 1) способам лечения туберкулеза у субъекта, нуждающегося в таком лечении, предусматривающим введение субъекту эффективного количества оксазолидинонового соединения; и 2) применениям оксазолидинонового соединения для лечения туберкулеза.
Варианты осуществления, подварианты осуществления и признаки настоящего изобретения или описаны далее, или будут выявлены в дальнейшем описании, примерах и приложенной формуле изобретения.
Подробное раскрытие настоящего изобретения
Оксазолидиноны изначально разрабатывались для применения в лечении инфекций грамположительных бактерий, в частности, инфекций устойчивого к метициллину S. aureus. Как показано в примерах, in vitro тестирование оксазолидиноновых соединений формулы I продемонстрировало, что такие соединения обладают высокой эффективностью в ингибировании роста Mycobacteria tuberculosis, но не показывают сильную активность против грамположительных бактерий. Кроме того, оксазолидиноновые соединения в соответствии с настоящим изобретением не ассоциируются с высокой степенью ингибирования синтеза митохондриального белка. Таким образом, соединения формулы I и их фармацевтически приемлемые соли, как предполагают, применимы для лечения микобактериального туберкулеза (Mtb), но не вызвают побочных эффектов, таких как миелосупрессия, которые ассоциируются с оксазолидиноновым линезолидом, одобренным для лечения грамположительных инфекций. Поэтому, такие соединения будут обладать значительными преимуществами над линезолидом и аналогами в качестве терапевтических средств против Mtb.
Соединения формулы (I)
В одном аспекте настоящее изобретение включает в себя соединения формулы I:
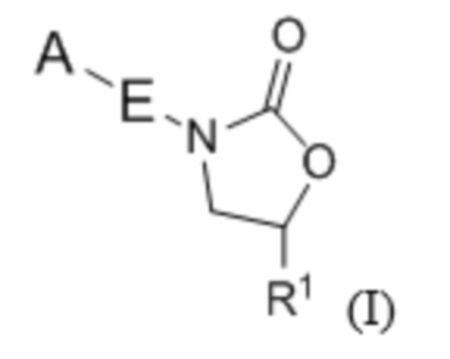
и их фармацевтически приемлемые соли, где A, E и R1 определены выше для соединений формулы (I); причем соединения могут подходить для применения для лечения бактериальных инфекций, в частности, микобактериальный инфекций.
Первый вариант осуществления настоящего изобретения (вариант осуществления E1) относится к соединению формулы I или его фармацевтически приемлемой соли, где A представляет собой арил, необязательно замещенный R8 в количестве до четырех; и где все другие переменные определены в начале (т. е. как определено в формуле I в кратком описании настоящего изобретения).
Согласно подварианту осуществления варианта осуществления E1 A представляет собой 5-членный арил. Согласно дополнительному подварианту осуществления A представляет собой 6-членный арил. Согласно еще одним дополнительным подвариантам осуществления A представляет собой 7-, 8-, 9-, 10-, 11-, 12-, 13- или 14-членный арил.
Второй вариант осуществления (вариант осуществления E2) относится к соединению формулы I или его фармацевтически приемлемой соли, где A представляет собой гетероарил, где от 1 до 4 кольцевых атомов независимо представляют собой O, N или S, а оставшиеся кольцевые атомы представляют собой атомы углерода, и все другие переменные определены в кратком описании настоящего изобретения.
Согласно подварианту осуществления варианта осуществления E2 A представляет собой 5-членный гетероарил, содержащий один гетероатом. Согласно дополнительному подварианту осуществления A представляет собой 5-членный гетероарил, содержащий два гетероатома. Согласно другому подварианту осуществления A представляет собой 5-членный гетероарил, содержащий три гетероатома. Согласно еще одному подварианту осуществления A представляет собой 5-членный гетероарил, содержащий четыре гетероатома. Согласно другому подварианту осуществления A представляет собой 6-членный гетероарил, содержащий один гетероатом. Согласно дополнительному подварианту осуществления A представляет собой 6-членный гетероарил, содержащий два гетероатома. Согласно другому подварианту осуществления A представляет собой 6-членный гетероарил, содержащий три гетероатома. Согласно еще одному подварианту осуществления A представляет собой 6-членный гетероарил, содержащий четыре гетероатома. Согласно еще одним дополнительным подвариантам осуществления A представляет собой 7-, 8-, 9-, 10-, 11-, 12-, 13- или 14-членный гетероарил, содержащий от одного до четырех гетероатомов, выбранных из N, O и S.
Третий вариант осуществления (вариант осуществления E3) относится к соединению формулы I или его фармацевтически приемлемой соли, где A представляет собой моноциклическое насыщенное или частично ненасыщенное кольцо, необязательно замещенное R8 в количестве до четырех, где от 1 до 4 кольцевых атомов независимо представляют собой O, N или S, а оставшиеся кольцевые атомы представляют собой атомы углерода, и все другие переменные определены в кратком описании настоящего изобретения.
Согласно подварианту осуществления варианта осуществления E3 A представляет собой 5-членное моноциклическое насыщенное или частично ненасыщенное кольцо, содержащее один гетероатом. Согласно дополнительному подварианту осуществления A представляет собой 5-членное моноциклическое насыщенное или частично ненасыщенное кольцо, содержащее два гетероатома. Согласно другому подварианту осуществления A представляет собой 5-членное моноциклическое насыщенное или частично ненасыщенное кольцо, содержащее три гетероатома. Согласно еще одному подварианту осуществления A представляет собой 5-членное моноциклическое насыщенное или частично ненасыщенное кольцо, содержащее четыре гетероатома. Согласно другому подварианту осуществления A представляет собой 6-членное моноциклическое насыщенное или частично ненасыщенное кольцо, содержащее один гетероатом. Согласно дополнительному подварианту осуществления A представляет собой 6-членное моноциклическое насыщенное или частично ненасыщенное кольцо, содержащее два гетероатома. Согласно другому подварианту осуществления A представляет собой 6-членное моноциклическое насыщенное или частично ненасыщенное кольцо, содержащее три гетероатома. Согласно еще одному подварианту осуществления A представляет собой 6-членное моноциклическое насыщенное или частично ненасыщенное кольцо, содержащее четыре гетероатома.
Четвертый вариант осуществления (вариант осуществления E4) относится к соединению формулы I или его фармацевтически приемлемой соли, где A представляет собой бициклическую насыщенную или частично ненасыщенную кольцевую систему, необязательно замещенную R8 в количестве до четырех, где от 1 до 4 кольцевых атомов независимо представляют собой O, N или S, а оставшиеся кольцевые атомы представляют собой атомы углерода, и все другие переменные определены в кратком описании настоящего изобретения.
Согласно подварианту осуществления варианта осуществления E4 A представляет собой бициклическую насыщенную или частично ненасыщенную кольцевую систему, содержащую один гетероатом. Согласно дополнительному подварианту осуществления A представляет собой бициклическую насыщенную или частично ненасыщенную кольцевую систему, содержащую два гетероатома. Согласно другому подварианту осуществления A представляет собой бициклическую насыщенную или частично ненасыщенную кольцевую систему, содержащую три гетероатома. Согласно еще одному подварианту осуществления A представляет собой бициклическую насыщенную или частично ненасыщенную кольцевую систему, содержащую четыре гетероатома.
Согласно вариантам осуществления E1-E4 и подвариантам осуществления вариантов осуществления E1-E4 A необязательно замещен заместителями R8 в количестве до четырех. Согласно подвариантам осуществления вариантов осуществления E1-E4 и их предыдущим подвариантам осуществления A замещен четырьмя заместителями R8, которые определены в начале. Согласно другому подварианту осуществления A замещен тремя заместителями R8. Согласно дополнительному подварианту осуществления A замещен двумя заместителями R8. Согласно еще одному дополнительному подварианту осуществления A замещен одним заместителем R8. Согласно альтернативным вариантам осуществления вариантов осуществления E1-E4 A является незамещенным.
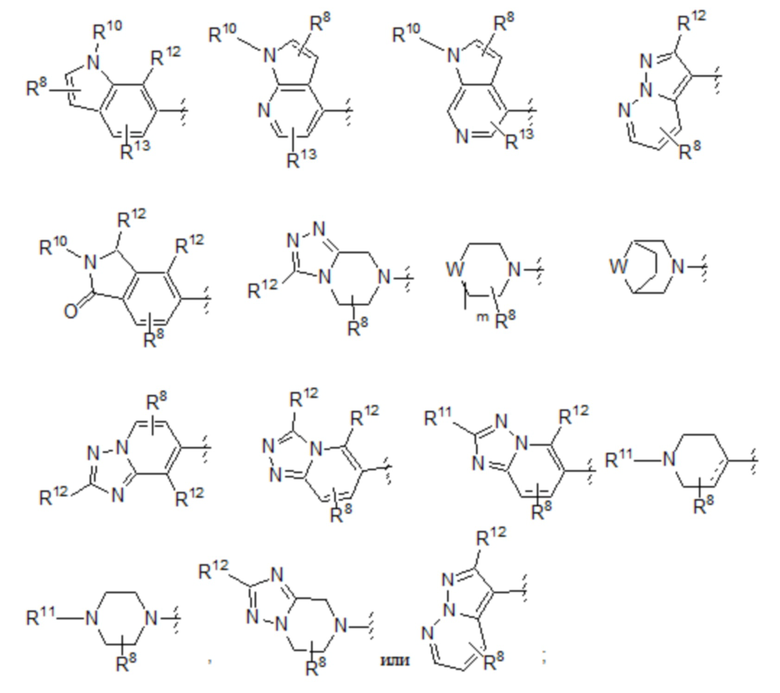
Пятый вариант осуществления (вариант осуществления E5) относится к соединению формулы I или его фармацевтически приемлемой соли, где A выбран из группы, состоящей из: 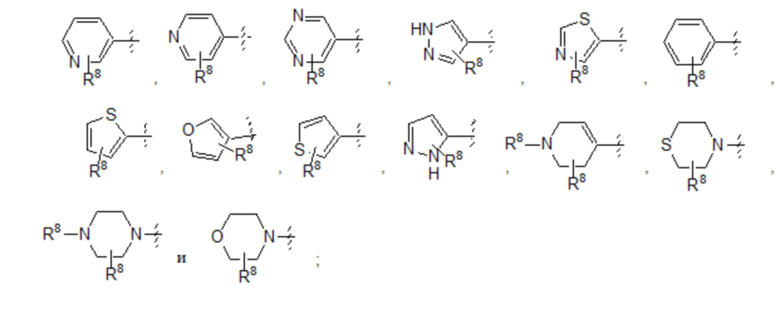
где R8 представляет собой необязательные заместители в количестве до четырех, которые могут быть одинаковыми или разными; и где R8 и все другие переменные определены в кратком описании настоящего изобретения.
Согласно подвариантам осуществления вариантов осуществления E5 A замещен четырьмя заместителями R8. Согласно другому подварианту осуществления A замещен тремя заместителями R8. Согласно дополнительному подварианту осуществления A замещен двумя заместителями R8. Согласно еще одному дополнительному подварианту осуществления A замещен одним заместителем R8. Согласно альтернативному подварианту осуществления варианта осуществления E5 A является незамещенным.
Шестой вариант осуществления (вариант осуществления E6) относится к соединению формулы I или его фармацевтически приемлемой соли, где A выбран из группы, состоящей из:
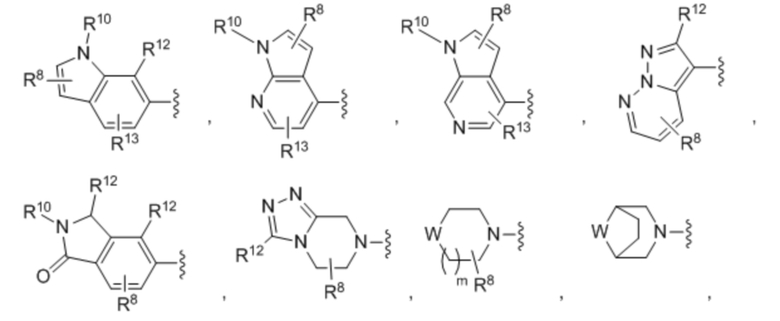
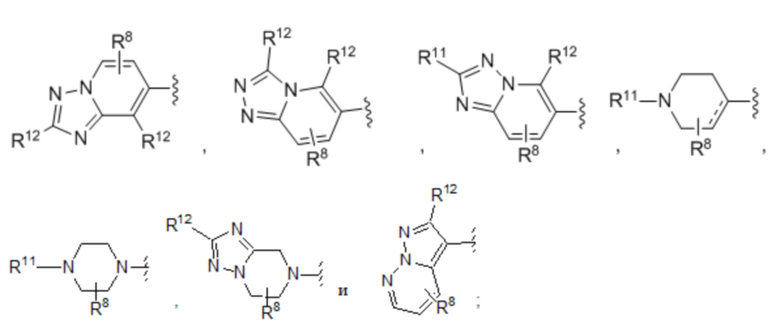
где:
m=равно 0, 1, 2 или 3;
в каждом случае R8 независимо выбран из H, галогена, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 гетероциклоалкила, бензила, -OCF3, -OCHF2, -OR3, =O, -CN, -NO2, -SR3, -SF5, -SCF3, -SOR3, -SO2R3, -S(=O)(=NH)R2, -N(R2)2, -NR2COR3, -SO2N(R2)2, -NR2SO2R3, -COOH, -COR9, -COOR3, -CON(R2)2 и -C(R9)2N(R2)2, причем указанный C1-C6 алкил, C3-C6 циклоалкил, C3-C6 гетероциклоалкил и бензил необязательно замещены F, -OCH3, -OH, =O, NH2, NHCH3 и N(CH3)2 в количестве до четырех;
R10 выбран из H, C1-C6 алкила и C3-C6 циклоалкила, причем указанный C1-C6 алкил и указанный C3-C6 циклоалкил необязательно замещены заместителями в количестве от одного до четырех, которые независимо выбраны из F, -OCH3, -OH, NH2, NHCH3 и N(CH3)2;
R11 выбран из H, C1-C6 алкила и C3-C6 циклоалкила, -COR9, -COOR9, -CON(R9)2 и –SO2R9;
в каждом случае R12 независимо выбран из H, галогена, C1-C6 алкила, C3-C6 циклоалкила, бензила, -OCF3, -OCHF2, -OR3, -CN, -NO2, -SR3, -SF5, -SCF3, -SOR3, -SO2R3, -S(=O)(=NH)R2, -N(R2)2, -NR2COR3, -SO2N(R2)2, -NR2SO2R3, -COOH, -COR9, -COOR3, -CON(R2)2 и -C(R9)2N(R2)2, причем указанный C1-C6 алкил, C3-C6 циклоалкил и бензил необязательно замещены F, -OCH3, -OH, NH2, NHCH3 и N(CH3)2 в количестве до четырех;
R13 выбран из H, галогена, C1-C6 алкила, C3-C6 циклоалкила, бензила, -OCF3, -OCHF2, -OR3, -CN, -NO2, -SR3, -SF5, -SCF3, -SOR3, -SO2R3, -S(=O)(=NH)R2, -N(R2)2, -NR2COR3, -SO2N(R2)2, -NR2SO2R3, -COOH, -COR9, -COOR3, -CON(R2)2 и -C(R9)2N(R2)2, причем указанный C1-C6 алкил, C3-C6 циклоалкил и бензил необязательно замещены F, -OCH3, -OH, NH2, NHCH3 и N(CH3)2 в количестве до четырех; и
W выбран из O, S, SO, SO2 и S(=O)(=NH); и
где --- представляет собой двойную или простую связь,
где все другие переменные определены в кратком описании настоящего изобретения.
Согласно вариантам осуществления E1-E6 и подвариантам осуществления вариантов осуществления E1-E6 в каждом случае R8 независимо выбран из галогена, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 гетероциклоалкила, бензила, -OCF3, -OCHF2, -OR3, =O, -CN, -NO2, -SR3, -SF5, -SCF3, -SOR3, -SO2R3, -S(=CH2)C1-C6 алкила, -S(=O)(=NH)R2, -N(R2)2, -NR2COR3, -SO2N(R2)2, -NR2SO2R3, -COOH, -COR9, -COOR3, -CON(R2)2 и -C(R9)2N(R2)2, причем указанный C1-C6 алкил, C3-C6 циклоалкил, C3-C6 гетероциклоалкил и бензил необязательно замещены F, -OCH3, -OH, =O, NH2, NHCH3 и N(CH3)2 в количестве до четырех.
Согласно конкретному подварианту осуществления вариантов осуществления E1-E6 один или несколько заместителей R8 выбрано из: -H, -OH, -CN, -CH3,-CH2NH2, -CONH2, C(CH3)2OH, -COOC(CH3)2CH3, -SCH3, -OCH3, -Cl, -F, -CHF2, =O, -циклопропила, NO2, -NHSO2CH3, -SO2N(CH3)2, -S(=O)(=NH)CH3, -SO2CH3, -SOCH3 и -SO2NH2.
Согласно другому конкретному подварианту осуществления вариантов осуществления E1-E6 один или несколько заместителей R8 выбрано из:
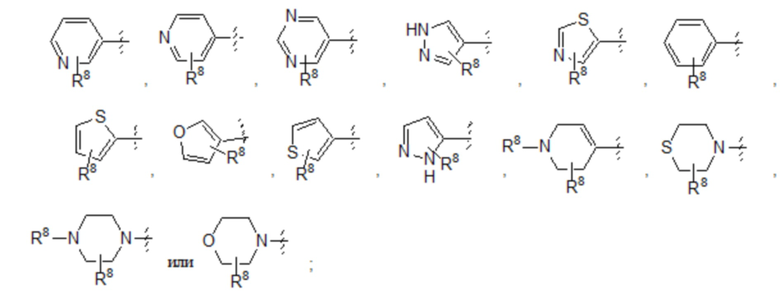
Седьмой вариант осуществления (вариант осуществления E7) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E представляет собой 6-членный арил, причем указанный арил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -CN, -CF3, -CHF2, -CH2NH2, -CH2NHCOCH3, -OCF3, -OCHF2, -OH, -O-(C1-C6)алкила, C1-C6 алкила и C3-C6 циклоалкила, и все другие переменные определены в кратком описании настоящего изобретения.
Восьмой вариант осуществления (вариант осуществления E8) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E представляет собой 5-членный гетероарил, содержащий от одного до трех гетероатомов, независимо выбранных из S, O и N, причем указанный гетероарил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -CN, -CF3, -CHF2, -CH2NH2, -CH2NHCOCH3, -OCF3, -OCHF2, -OH, -O-(C1-C6)алкила, C1-C6 алкила и C3-C6 циклоалкила, и все другие переменные определены в кратком описании настоящего изобретения.
Девятый вариант осуществления (вариант осуществления E9) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E представляет собой 6-членный гетероарил, содержащий от одного до трех гетероатомов, независимо выбранных из S, O и N, причем указанный арил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -CN, -CF3, -CHF2, -CH2NH2, -CH2NHCOCH3, -OCF3, -OCHF2, -OH, -O-(C1-C6)алкила, C1-C6 алкила и C3-C6 циклоалкила, и все другие переменные определены в кратком описании настоящего изобретения.
Десятый вариант осуществления (вариант осуществления E10) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E представляет собой:
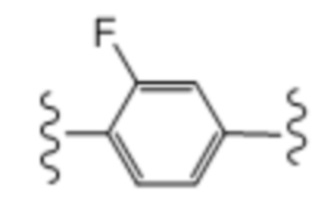 ; и все другие переменные определены в кратком описании настоящего изобретения.
; и все другие переменные определены в кратком описании настоящего изобретения.
Одиннадцатый вариант осуществления (вариант осуществления E11) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E представляет собой:
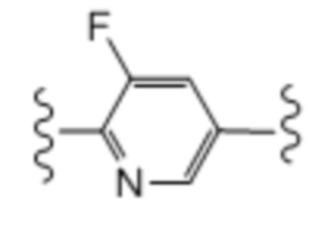 ; и все другие переменные определены в кратком описании настоящего изобретения.
; и все другие переменные определены в кратком описании настоящего изобретения.
Двенадцатый вариант осуществления (вариант осуществления E12) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E представляет собой:
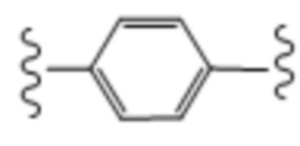 ; и все переменные определены в начале.
; и все переменные определены в начале.
Тринадцатый вариант осуществления (вариант осуществления E13) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 выбран из-CH2N(R2)2, -CH2NR2COR3, -CH2NR2COOR3, -CH2NR2CON(R2)2, -CH2NR2CONR2N(R2)2, -CH2NSO2R3, -CON(R2)2, -C=NOR3, -CH2OR4, -CH2NR2R4 и -CH2R6.
Четырнадцатый вариант осуществления (вариант осуществления E14) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2N(R2)2.
Согласно подварианту осуществления варианта осуществления E14 в одном случае R2 представляет собой H и в одном случае R2 представляет собой C1-C6 алкил, причем указанный C1-C6 алкил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, -NH2, -NHCH3 и –N(CH3)2.
Согласно дополнительному подварианту осуществления варианта осуществления E14 в одном случае R2 представляет собой H и в одном случае R2 представляет собой C2-C6 алкенил, причем указанный C2-C6 алкенил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, -NH2, -NHCH3 и –N(CH3)2.
Согласно еще одному подварианту осуществления варианта осуществления E14 в одном случае R2 представляет собой H и в одном случае R2 представляет собой C3-C6 циклоалкил, причем указанный C3-C6 циклоалкил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, -NH2, -NHCH3 и –N(CH3)2.
Пятнадцатый вариант осуществления (вариант осуществления E15) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NR2COR3.
Шестнадцатый вариант осуществления (вариант осуществления E16) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NR2COOR3.
Семнадцатый вариант осуществления (вариант осуществления E17) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NR2CON(R2)2.
Согласно подварианту осуществления варианта осуществления E17 R1 представляет собой -CH2NHCON(R2)2.
Согласно дополнительному подварианту осуществления R1 представляет собой -CH2NR2CONH(R2).
Согласно другому подварианту осуществления R1 представляет собой -CH2NR2CONH2.
Восемнадцатый вариант осуществления (вариант осуществления E18) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NR2CONR2N(R2)2.
Согласно подварианту осуществления варианта осуществления E18 R1 представляет собой -CH2NHCONR2N(R2)2.
Согласно подварианту осуществления варианта осуществления E18 R1 представляет собой -CH2NR2CONHN(R2)2.
Согласно другому подварианту осуществления варианта осуществления E18 R1 представляет собой -CH2NR2CONR2NH(R2).
Согласно дополнительному подварианту осуществления варианта осуществления E18 R1 представляет собой -CH2NHCONHN(R2)2.
Согласно подварианту осуществления варианта осуществления E18 R1 представляет собой -CH2NR2CONR2NH2.
Согласно подварианту осуществления варианта осуществления E18 R1 представляет собой -CH2NR2CONHNH2.
Согласно подварианту осуществления варианта осуществления E18 R1 представляет собой -CH2NHCONR2NH2.
Девятнадцатый вариант осуществления (вариант осуществления E19) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NR2SO2R3.
Двадцатый вариант осуществления (вариант осуществления E20) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CON(R2)2.
Согласно подварианту осуществления варианта осуществления E20 в одном случае R2 представляет собой H и в одном случае R2 представляет собой C1-C6 алкил, причем указанный C1-C6 алкил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, -NH2, -NHCH3 и –N(CH3)2.
Согласно дополнительному подварианту осуществления варианта осуществления E20 в одном случае R2 представляет собой H и в одном случае R2 представляет собой C2-C6 алкенил, причем указанный C2-C6 алкенил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, -NH2, -NHCH3 и –N(CH3)2.
Согласно еще одному подварианту осуществления варианта осуществления E20 в одном случае R2 представляет собой H и в одном случае R2 представляет собой C3-C6 циклоалкил, причем указанный C3-C6 циклоалкил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, -NH2, -NHCH3 и –N(CH3)2.
Двадцать первый вариант осуществления (вариант осуществления E21) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -C=NOR3.
Согласно подвариантам осуществления вариантов осуществления E15, E16, E19 и E21 R3 представляет собой H.
Согласно подвариантам осуществления вариантов осуществления E15, E16, E19 и E21 R3 представляет собой C1-C6 алкил, причем указанный C1-C6 алкил может быть необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, -NH2, -NHCH3 и –N(CH3)2.
Согласно подвариантам осуществления вариантов осуществления E15, E16, E19 и E21 R3 представляет собой C2-C6 алкенил, причем указанный C2-C6 алкенил может быть необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, -NH2, -NHCH3 и –N(CH3)2.
Согласно подвариантам осуществления вариантов осуществления E15, E16, E19 и E21 R3 представляет собой C3-C6 циклоалкил, причем указанный C3-C6 циклоалкил может быть необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, -NH2, -NHCH3 и –N(CH3)2.
Двадцать второй вариант осуществления (вариант осуществления E22) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2OR4.
Двадцать третий вариант осуществления (вариант осуществления E23) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NR2R4.
Согласно подвариантам осуществления вариантов осуществления E22 и E23 R4 представляет собой 5-членный гетероцикл, необязательно замещенный R5.
Согласно подвариантам осуществления вариантов осуществления E22 и E23 R4 представляет собой 6-членный гетероцикл, необязательно замещенный R5.
Согласно подвариантам осуществления вариантов осуществления E15, E16, E19 и E23 R2 представляет собой H.
Согласно дополнительным подвариантам осуществления вариантов осуществления E15, E16, E19 и E23 R2 представляет собой C1-C6 алкил, причем указанный C1-C6 алкил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, NH2, NHCH3 и N(CH3)2.
Согласно другим подвариантам осуществления вариантов осуществления E15, E16, E19 и E23 R2 представляет собой C2-C6 алкенил, причем указанный C2-C6 алкенил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, NH2, NHCH3 и N(CH3)2.
Согласно альтернативным подвариантам осуществления вариантов осуществления E15, E16, E19 и E23 R2 представляет собой C3-C6 циклоалкил, причем указанный C3-C6 циклоалкил необязательно замещен заместителями в количестве до четырех, которые независимо выбраны из галогена -OCH3, -OH, NH2, NHCH3 и N(CH3)2.
Двадцать четвертый вариант осуществления (вариант осуществления E24) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2R6.
Согласно подварианту осуществления варианта осуществления E24 R6 представляет собой H.
Согласно подварианту осуществления варианта осуществления E24 R6 представляет собой C1-C6 алкил.
Согласно подварианту осуществления варианта осуществления E24 R6 представляет собой 5-членный гетероцикл, причем указанный 5-членный гетероцикл необязательно замещен одним или двумя R7.
Двадцать пятый вариант осуществления (вариант осуществления E25) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NH2.
Двадцать шестой вариант осуществления (вариант осуществления E26) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NHC(O)CH3.
Двадцать седьмой вариант осуществления (вариант осуществления E27) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NHC(O)-циклопропил.
Двадцать восьмой вариант осуществления (вариант осуществления E28) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NHCOOCH3.
Двадцать девятый вариант осуществления (вариант осуществления E29) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой -CH2NHCOCH2NH2.
Тридцатый вариант осуществления (вариант осуществления E30) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой CH2NHCOCHCH2CH3.
Тридцать первый вариант осуществления (вариант осуществления E31) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой CH2NHC(O)NHN(CH3)2.
Тридцать второй вариант осуществления (вариант осуществления E32) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12 и R1 представляет собой:
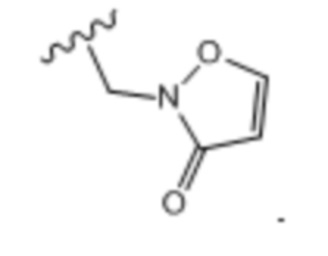
Тридцать третий вариант осуществления (вариант осуществления E33) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E13 и R1 представляет собой:
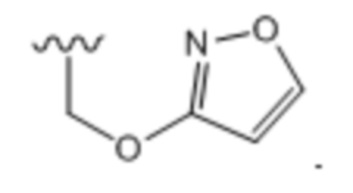
Тридцать четвертый вариант осуществления (вариант осуществления E34) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E13 и R1 представляет собой:
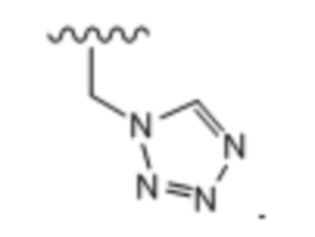
Тридцать пятый вариант осуществления (вариант осуществления E35) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E13 и R1 представляет собой:
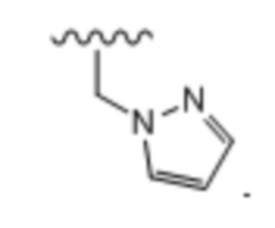
Тридцать шестой вариант осуществления (вариант осуществления E36) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E13 и R1 представляет собой:
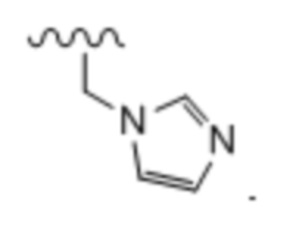
Тридцать седьмой вариант осуществления (вариант осуществления E37) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в кратком описании настоящего изобретения или в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E13 и R1 представляет собой:
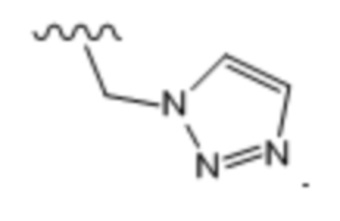
Тридцать восьмой вариант осуществления (вариант осуществления E38) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12, R1 представляет собой -CH2OR4 или -CH2NR2R4; и
R4 выбран из:
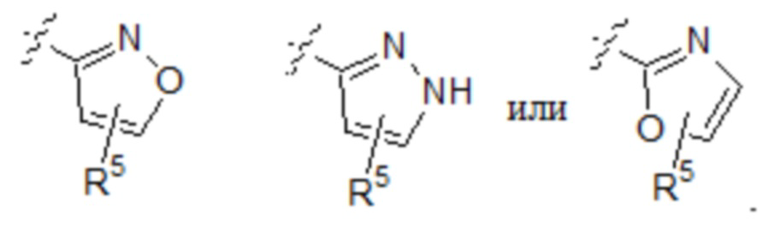
Тридцать девятый вариант осуществления (вариант осуществления E39) относится к соединению формулы I или его фармацевтически приемлемой соли, где A определен в любом из вариантов осуществления E1-E6, E определен в любом из вариантов осуществления E7-E12, R1 представляет собой CH2R6 и R6 выбран из:
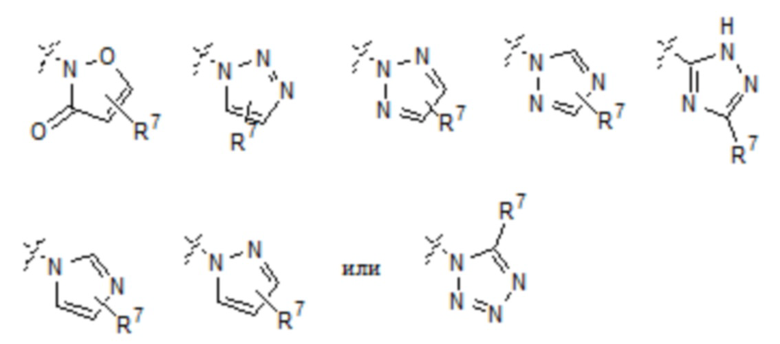
Сороковой вариант осуществления (вариант осуществления E40) относится к соединению формулы I или его фармацевтически приемлемой соли, где A представляет собой:
где R8 представляет собой заместители углеродного кольца в количестве до четырех, которые могут быть одинаковыми или разными, E определен в любом из вариантов осуществления E7-E12 и R1 определен в любом из вариантов осуществления E13-E37.
Сорок первый вариант осуществления (вариант осуществления E41) относится к соединению формулы I или его фармацевтически приемлемой соли, где E определен в любом из вариантов осуществления E7-E12, R1 определен в любом из вариантов осуществления E13-E37, A представляет собой:
где:
m=равно 0, 1, 2 или 3;
в каждом случае R8 независимо выбран из H, галогена, C1-C6 алкила, C3-C6 циклоалкила, C3-C6 гетероциклоалкила, бензила, -OCF3, -OCHF2, -OR3, =O, -CN, -NO2, -SR3, -SF5, -SCF3, -SOR3, -SO2R3, -S(=O)(=NH)R2, -N(R2)2, -NR2COR3, -SO2N(R2)2, -NR2SO2R3, -COOH, -COR9, -COOR3, -CON(R2)2 и -C(R9)2N(R2)2, причем указанный C1-C6 алкил, C3-C6 циклоалкил, C3-C6 гетероциклоалкил и бензил необязательно замещены F, -OCH3, -OH, =O, NH2, NHCH3 и N(CH3)2 в количестве до четырех;
R10 выбран из H, C1-C6 алкила и C3-C6 циклоалкила, причем указанный C1-C6 алкил и указанный C3-C6 циклоалкил необязательно замещены заместителями в количестве от одного до четырех, которые независимо выбраны из F, -OCH3, -OH, NH2, NHCH3 и N(CH3)2;
R11 выбран из H, C1-C6 алкила и C3-C6 циклоалкила, -COR9, -COOR9, -CON(R9)2 и –SO2R9;
в каждом случае R12 независимо выбран из H, галогена, C1-C6 алкила, C3-C6 циклоалкила, бензила, -OCF3, -OCHF2, -OR3, -CN, -NO2, -SR3, -SF5, -SCF3, -SOR3, -SO2R3, -S(=O)(=NH)R2, -N(R2)2, -NR2COR3, -SO2N(R2)2, -NR2SO2R3, -COOH, -COR9, -COOR3, -CON(R2)2 и -C(R9)2N(R2)2, причем указанный C1-C6 алкил, C3-C6 циклоалкил и бензил необязательно замещены F, -OCH3, -OH, NH2, NHCH3 и N(CH3)2 в количестве до четырех;
R13 выбран из H, галогена, C1-C6 алкила, C3-C6 циклоалкила, бензила, -OCF3, -OCHF2, -OR3, -CN, -NO2, -SR3, -SF5, -SCF3, -SOR3, -SO2R3, -S(=O)(=NH)R2, -N(R2)2, -NR2COR3, -SO2N(R2)2, -NR2SO2R3, -COOH, -COR9, -COOR3, -CON(R2)2 и -C(R9)2N(R2)2, причем указанный C1-C6 алкил, C3-C6 циклоалкил и бензил необязательно замещены F, -OCH3, -OH, NH2, NHCH3 и N(CH3)2 в количестве до четырех; и
W выбран из O, S, SO, SO2 и S(=O)(=NH); и
где --- представляет собой двойную или простую связь и где все другие переменные определены в кратком описании настоящего изобретения.
Сорок второй вариант осуществления (вариант осуществления E42) относится к соединению формулы IA или его фармацевтически приемлемой соли формулы:
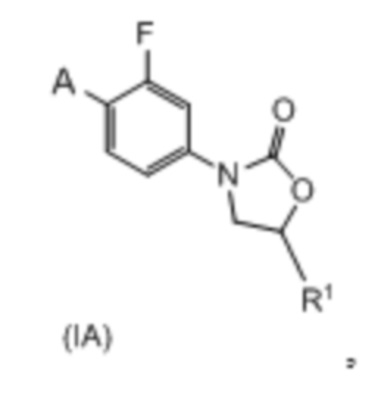
где:
R1 представляет собой -CH2NH2, -CH2NHC(O)CH3, -CH2NHC(O)-C3-C6циклоалкил или CH2R6; A определен в любом из вариантов осуществления E1-E6; и R6 определен в кратком описании настоящего изобретения.
Сорок третий вариант осуществления (вариант осуществления E43) относится к соединению формулы IB или его фармацевтически приемлемой соли формулы:
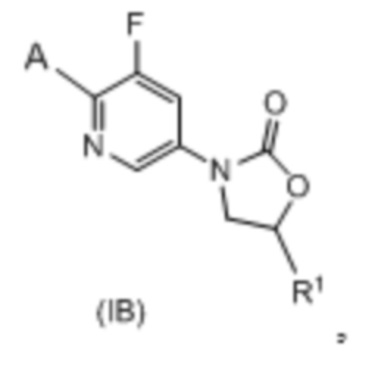
где:
R1 представляет собой -CH2NH2, -CH2NHC(O)CH3, -CH2NHC(O)-C3-C6циклоалкил или CH2R6; A определен в любом из вариантов осуществления E1-E6; и R6 определен в кратком описании настоящего изобретения.
Согласно подвариантам осуществления вариантов осуществления E42 и E43 R1 представляет собой -CH2NH2.
Согласно дополнительным подвариантам осуществления вариантов осуществления E42 и E43 A представляет собой: 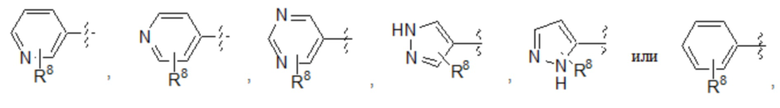 где R8 представляет собой заместители углеродного кольца в количестве до четырех, которые могут быть одинаковыми или разными.
где R8 представляет собой заместители углеродного кольца в количестве до четырех, которые могут быть одинаковыми или разными.
Ссылка на различные варианты осуществления в отношении соединений формулы I в частности включает в себя различные варианты осуществления формулы I, например, формулы IA и формулы IB, подварианты осуществления формулы IA и формулы IB, другие варианты осуществления, представленные в настоящем описании, и отдельные соединения, описанные в настоящем изобретении.
Другие варианты осуществления настоящего изобретения включают в себя следующее:
(a) фармацевтическую композицию, содержащую эффективное количество соединения формулы I, IA или IB, как определено в настоящем описании, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
(b) фармацевтическую композицию (a), дополнительно содержащую второе соединение, причем второе соединение является антибиотиком.
(c) фармацевтическую композицию (b), причем второе соединение выбрано из группы, состоящей из: этамбутола, пиразинамида, изониазида, левофлоксацина, моксифлоксацина, гатифлоксацина, офлоксацина, канамицина, амикацина, капреомицина, стрептомицина, этионамида, протионамида, циклосерина, теридидона, парааминосалициловой кислоты, клофазимина, кларитромицина, амоксициллина-клавуланата, тиоацетазона, меропенема-клавуланата и тиоридазина.
(d) фармацевтическую композицию, содержащую (i) соединение формулы I, IA или IB или его фармацевтически приемлемую соль, и (ii) второе соединение, причем второе соединение является антибиотиком, где каждое соединение формулы I, IA или IB и каждое второе соединение использовали в количестве, которое делает комбинацию эффективной для лечения или предупреждения бактериальной инфекции.
(e) комбинацию (d), где второе соединение выбрано из группы, состоящей из: этамбутола, пиразинамида, изониазида, левофлоксацина, моксифлоксацина, гатифлоксацина, офлоксацина, канамицина, амикацина, капреомицина, стрептомицина, этионамида, протионамида, циклосерина, теридидона, парааминосалициловой кислоты, клофазимина, кларитромицина, амоксициллина-клавуланата, тиоацетазона, меропенема-клавуланата и тиоридазина.
(f) способ лечения бактериальной инфекции у субъекта, который включает в себя введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения формулы I, Ia или Ib или его фармацевтически приемлемой соли.
(g) способ предупреждения и/или лечения бактериальной инфекции, который включает в себя введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения формулы I, Ia или Ib или его фармацевтически приемлемой соли.
(h) способ лечения бактериальной инфекции, который включает в себя введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества композиции (a), (b), (c), (d) или (e).
(i) способ лечения бактериальной инфекции, как изложено в (f), (g) или (h), при этом бактериальная инфекция возникает в результате Mycobacterium tuberculosis.
(j) способ предупреждения и/или лечения микобактериальной инфекции, который включает в себя введение субъекту, нуждающемуся в таком лечении, эффективного количества композиции, содержащей оксазолидиноновое соединение или его фармацевтически приемлемую соль.
(k) способ лечения микобактериальной инфекции, как изложено в (j), где микобактериальная инфекция вызвана M. tuberculosis.
(l) способ лечения микобактериальной инфекции, как изложено в (j), где композиция является композицией (a), (b), (c), (d) или (e).
Настоящее изобретение также включает в себя соединение формулы I, Ia или Ib или его фармацевтически приемлемую соль (i) для применения в (ii) для использования в качестве лекарственного препарата для или (iii) для использования при получении (или изготовлении) лекарственного препарата для терапии или лечения бактериальной инфекции, в частности микобактериальной инфекции. При таких применениях соединения по настоящему изобретению необязательно могут быть использованы в комбинации с одним или несколькими вторыми терапевтическими средствами, включая этамбутол, пиразинамид, изониазид, левофлоксацин, моксифлоксацин, гатифлоксацин, офлоксацин, канамицин, амикацин, капреомицин, стрептомицин, этионамид, протионамид, циклосерин, теридидон, парааминосалициловую кислоту, клофазимин, кларитромицин, амоксициллин-клавуланат, тиоацетазон, меропенем-клавуланат и тиоридазин.
Дополнительные варианты осуществления настоящего изобретения включают в себя фармацевтические композиции, комбинации и способы, изложенные в (a)-(l) выше, и применения, изложенные в предыдущем параграфе, где используемое соединение по настоящему изобретению является соединением одного из описанных вариантов осуществления, подвариантов осуществления, классов или подклассов. Соединение необязательно может быть использовано в форме фармацевтически приемлемой соли в таких вариантах осуществления.
Согласно вариантам осуществления представленных в настоящем описании соединений и солей являлось понятным, что каждый вариант осуществления может быть объединен с одним или несколькими другими вариантами осуществления до такой степени, что такая комбинация обеспечивает стабильное соединение или соль и соответствует описанию вариантов осуществления. Кроме того, является понятным, что варианты осуществления композиций и способов, представленных выше как (a)-(l), включают в себя все варианты осуществления соединений и/или солей, включая такие варианты осуществления, которые являются результатом комбинацией вариантов осуществления.
Дополнительные варианты осуществления настоящего изобретения включают в себя каждую из фармацевтических композиций, комбинаций, способов и применений, изложенных в предыдущих параграфах, причем используемое соединение по настоящему изобретению или его соль являются в основном чистыми. В отношении фармацевтической композиции, содержащей соединение формулы I, IA или IB или его соль и фармацевтически приемлемый носитель и необязательно один или несколько наполнителей, подразумевается, что термин «в основном чистый» относится к соединению формулы I IA, или IB или его соли непосредственно; т. e., к чистоте активного вещества в композиции.
Определения и аббревиатуры
Используемые в настоящем описании термины обладают своим обычным значением, и значение таких терминов в каждом случае является независимым. При этом и за исключением, если не отмечено иное, следующие определения используются по всему описанию и формуле изобретения. Химические названия, общие названия и химические структуры могут быть использованы взаимозаменяемо для описания той же структуры. Если химическое соединение направлено к применению как химической структуры, так и химического названия и между структурой и названием существует двусмысленность, структура является преобладающей. Эти определения используются независимо от того, будет ли термин использоваться сам по себе или в комбинации с другими терминами, если не отмечено иное. Следовательно, определение «алкила» относится к «алкилу», а также к частям «алкила» в «гидроксиалкиле», «галогеналкиле»,
«-O-алкиле» и т. п.
Используемые в настоящем описании и про всему раскрытию следующие термины, если не отмечено иное, следует понимать как обладающие следующими значениями:
«Алкил» означает насыщенные углеродные цепи, которые могут быть неразветвленными или разветвленными или их комбинациями, если углеродная цепь не определена иным образом. Другие группы с приставкой «алк», такие как алкокси и алканоил, также могут быть неразветвленными или разветвленными или их комбинациями, если углеродная цепь не определена иным образом. Примеры алкильных групп включают в себя метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил и т. п.
«Алкенил» означает углеродные цепи, которые содержат по меньшей мере одну двойную связь углерод-углерод и которые могут быть неразветвленными или разветвленными или их комбинациями, если не отмечено иное. Примеры алкенила включают в себя винил, аллил, изопропенил, пентенил, гексенил, гептенил, 1-пропенил, 2-бутенил, 2-метил-2-бутенил и т. п.
«Антибиотик» относится к соединению или композиции, которая понижает жизнеспособность микроорганизма или которая ингибирует рост или пролиферацию микроорганизма. Выражение «ингибирует рост или пролиферацию» означает увеличение времени генерации (т. e., времени, необходимое для разделения бактериальной клетки или для удваивания популяции) по меньшей мере в 2 раза. Предпочтительными антибиотиками являются такие антибиотики, которые могут увеличивать время генерации по меньшей мере в 10 раз или больше (например, по меньшей мере в 100 раз или даже безгранично, как при полной гибели клетки). Предусмотрено, что используемый в настоящем раскрытии антибиотик дополнительно включает в себя антибактериальное, бактериостатическое или бактерицидное средство.
«Приблизительно» при модификации количества (например, кг, л или эквиваленты) вещества или композиции или значения физического свойства или значения параметра, характеризующего стадию процесса (например, температуры, при которой проводили стадию процесса) или т. п. относится к вариации в численной величине, которая может возникать, например, при обычном измерении, процедурах обработки и отбора, которые включены в получение, характеристику и/или применение вещества или композиции; при неизбежной ошибке при таких процедурах; при различиях в изготовлении, источнике или чистоте ингредиентов, используемых для получения или применения композиций или проведении процедур; и т. п. Согласно определенным вариантам осуществления «приблизительно» может означать вариацию ± 0,1, 0,2, 0,3, 0,4, 0,5, 1,0, 2,0, 3,0, 4,0 или 5,0 соответствующей единицы. Согласно определенным вариантам осуществления «приблизительно» может означать вариацию ± 1%, 2%, 3%, 4%, 5%, 10% или 20%.
«Ароматическая кольцевая система» означает моноциклическое, бициклическое или трициклическое ароматическое кольцо или кольцевую систему, содержащую 5-14 кольцевых атомов, где по меньшей мере одно из колец является ароматическим. Используемые в настоящем описании ароматические кольцевые системы охватывают арилы и гетероарилы. Термин может быть использован для описания карбоциклического кольца, конденсированного с арильной группой. Например, 5-7-членный циклоалкил может быть конденсирован через два смежных атома кольца с 5-6-членным гетероарилом, содержащим 1, 2 или 3 гетероатома в кольцевых атомах, выбранных из N, O и S. В другом примере гетеромоноциклическое кольцо конденсировано через два кольцевых атома с фенилом или 5-6-членным гетероарилом, содержащим 1, 2 или 3 гетероатома, выбранные из N, O и S.
«Арил» означает моноциклическое, бициклическое или трициклическое карбоциклическое ароматическое кольцо или кольцевую систему, содержащую 5-14 атомов углерода, где по меньшей мере одно из колец является ароматическим. Примеры арила включают в себя фенил и нафтил.
«Циклоалкил» означает насыщенное моноциклическое, бициклическое или с мостиковыми связями карбоциклическое кольцо, содержащее конкретное число атомов углерода. Примеры циклоалкила включают в себя циклопропил, циклобутил, циклопентил, циклогексил и т. п.
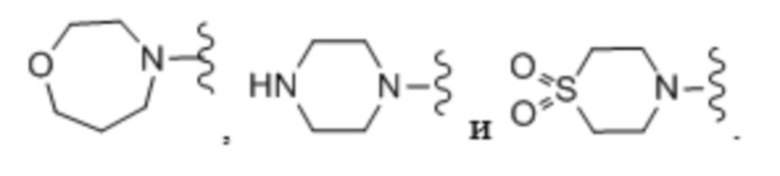
Используемый в настоящем описании термин «гетероциклоалкил» относится к неароматической насыщенной моноциклической или полициклической кольцевой системе, содержащей от 3 до 11 кольцевых атомов, где от 1 до 4 кольцевых атомов независимо представляют собой O, S или N, а остаток кольцевых атомов является атомами углерода. Гетероциклоалкильная группа может быть присоединена при помощи кольцевого атома углерода или кольцевого атома азота (если присутствует). Согласно одному варианту осуществления гетероциклоалкильная группа является моноциклической и содержит от приблизительно 3 до приблизительно 7 кольцевых атомов. Согласно другому варианту осуществления гетероциклоалкильная группа является моноциклической и содержит от приблизительно 4 до приблизительно 7 кольцевых атомов. Согласно другому варианту осуществления гетероциклоалкильная группа является бициклической и содержит от приблизительно 7 до приблизительно 11 кольцевых атомов. Согласно еще одному варианту осуществления гетероциклоалкильная группа является моноциклической и содержит 5 или 6 кольцевых атомов. Согласно одному варианту осуществления гетероциклоалкильная группа является моноциклической. Согласно другому варианту осуществления гетероциклоалкильная группа является бициклической. В кольцевой системе отсутствуют смежные атомы кислорода и/или серы. Любая –NH группа в гетероциклоалкильном кольце может существовать как защищенная группа, такая как, например, -N(BOC), -N(Cbz), -N(Tos) группа и т. п.; такие защищенные гетероциклоалкильные группы рассматриваются как часть настоящего изобретения. Термин «гетероциклоалкил» также охватывает гетероциклоалкильную группу, как определено выше, которая конденсирована с арильным (например, бензольным) или гетероарильным кольцом. Гетероциклоалкильная группа необязательно может быть замещена одним или несколькими «заместителями кольцевой системы», которые могут быть одинаковыми или разными и они определены ниже в настоящем описании. Атом азота или серы гетероциклоалкила (если присутствуют) необязательно могут быть окислены до соответствующего N-оксида, S-оксида или S,S-диоксида. Неограничивающие примеры моноциклических гетероциклоалкильных колец включают в себя оксетанил, пиперидил, пирролидинил, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,4-диоксанил, тетрагидрофуранил, тетрагидротиофенил, дельта-лактам, дельта-лактон, силaциклопентан, силапирролидин и т. п., и все их изомеры.
Используемый в настоящем описании, например, в определении R1,
«-CH2NR2COR3» означает «-CH2NR2C=OR3».
«Устойчивость к воздействию лекарств» по отношению к микобактерии означает микобактерию, которая больше не чувствительна по меньшей мере к одному ранее эффективному лекарственному средству; у которой развита способность противостоять воздействию антибиотика по меньшей мере у одного ранее эффективного лекарственного средства. Штамм, устойчивый к лекарственным средствам, может передать своему потомству такую способность противостоять. Указанная устойчивость может возникать вследствие случайных генетических мутаций в бактериальной клетке, которые изменяют ее чувствительность к одному лекарственному средству или к разным лекарственным средствам.
«Галоген» включает в себя фтор, хлор, бром и йод.
Используемый в настоящем описании «гетероарил» относится к моноциклической или полициклической кольцевой системе, содержащей от приблизительно 5 до приблизительно 14 кольцевых атомов, где от 1 до 4 кольцевых атомов независимо представляют собой O, N или S, а оставшиеся кольцевые атомы представляют собой атомы углерода, где по меньшей мере одно из содержащих гетероатом колец является ароматическим. Согласно одному варианту осуществления гетероарильная группа содержит от 5 до 10 кольцевых атомов. Согласно другому варианту осуществления гетероарильная группа является моноциклической и содержит 5 или 6 кольцевых атомов. Согласно другому варианту осуществления гетероарильная группа является бициклической. Гетероарильная группа может быть необязательно замещена одним или несколькими «заместителями кольцевой системы», которые могут быть одинаковыми или разными, и определены ниже в настоящем описании. Гетероарильная группа присоединена при помощи кольцевого атома углерода и любой атом азота гетероарила необязательно может быть окислен до соответствующего N-оксида. Термин «гетероарил» также охватывает гетероарильную группу, как определено выше, которая конденсирована с бензольным кольцом. Неограничивающие примеры гетероарилов включают в себя пиридил, пиразинил, фуранил, тиенил, пиримидинил, пиридон (включая N-замещенные пиридоны), изоксазолил, изотиазолил, оксазолил, оксадиазолил, тиазолил, пиразолил, фуразанил, пирролил, триазолил, 1,2,4-тиадиазолил, пиразинил, пиридазинил, хиноксалинил, фталазинил, оксиндолил, имидазо[1,2-a]пиридинил, имидазо[2,1-b]тиазолил, бензофуразанил, индолил, азaиндолил, бензимидазолил, бензотиенил, хинолинил, имидазолил, бензимидазолил, тиенопиридил, хиназолинил, тиенопиримидил, пирролопиридил, имидазопиридил, изохинолинил, бензоазаиндолил, 1,2,4-триазинил, бензотиазолил и т. п. и все их изомерные формы. Согласно другому варианту осуществления настоящего изобретения гетероарил представляет собой пиридин. Примеры бициклических колец включают в себя:
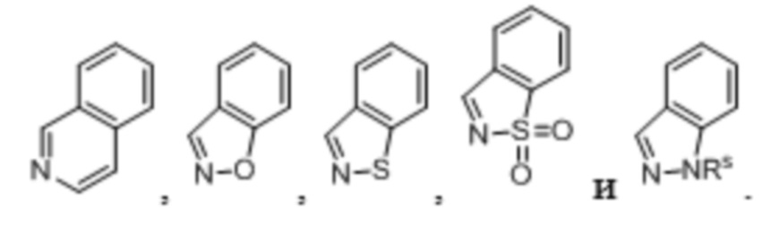
«Гетероцикл» означает моноциклическую или бициклическую насыщенную, частично ненасыщенную или ненасыщенную кольцевую систему, содержащую 5-10 атомов и содержащую по меньшей мере один кольцевой гетероатом, выбранный из N, S и O. Согласно выбранным вариантам осуществления кольцевая система содержит 1-4 гетероатома, выбранные из N, S и O. Если гетероцикл содержит два кольца, кольца могут быть конденсированными, с мостиковыми связями или спироциклическими. Примеры моноциклических гетероциклических колец включают в себя пиперазин, пиперидин и морфолин. Примеры бициклических гетероциклических колец включают в себя 1,4-диазабицикло[2,2,2]октан и 2,6-диазаспирогептан.
«Оксо» означает атом кислорода, соединенный с другим атомом при помощи двойной связи и он может быть представлен как «=O».
«Туберкулез» включает в себя болезненное состояние, обычно связанное с инфекциями, вызванными видами микобактерии, включающими в себя комплекс M. tuberculosis. Термин «туберкулез» также связан с микобактериальными инфекциями, вызванными микобактерией, отличной от M. tuberculosis (MOTT). Другие виды микобактерий включают в себя M. avium-intracellulare, M. kansarii, M. fortuitum, M. chelonae, M. leprae, M. africanum и M. microti, M. avium paratuberculosis, M. intracellulare, M. scrofulaceum, M. xenopi, M. marinum и M. ulcerans.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I или его фармацевтически приемлемой соли, как определено в начале или как определено в любом из вышеописанных вариантов осуществления, подвариантов осуществления, аспектов, классов или подклассов, где соединение или его соль находятся в основной чистой форме. Используемый в настоящем описании «в основном чистый» означает, что соответственно по меньшей мере приблизительно 60 масс.%, типично по меньшей мере приблизительно 70 масс.%, предпочтительно по меньшей мере приблизительно 80 масс.%, более предпочтительно по меньшей мере приблизительно 90 масс.% (например, от приблизительно 90 масс.% до приблизительно 99 масс.%), даже более предпочтительно по меньшей мере приблизительно 95 масс.% (например, от приблизительно 95 масс.% до приблизительно 99 масс.% или от приблизительно 98 масс.% до 100 масс.%) и наиболее предпочтительно по меньшей мере приблизительно 99 масс.% (например, 100 масс.%) продукта, содержащего соединение формулы I, IA или IB или соль формулы I, IA или IB (например, продукта, выделенного из реакционной смеси с получением соединения или соли), состоит из соединения или соли. Уровень чистоты соединений и солей может быть определен с применением стандартного способа анализа, такого как тонкослойная хроматография, гель-электрофорез, высокоэффективная жидкостная хроматография и/или масс-спектрометрия. Если использовали более одного способа анализа и способы обеспечивали экспериментально существенные различия на уровне определенной чистоты, тогда преимущественным является способ, который обеспечивает самый высокий уровень чистоты. Соединение или соль 100% чистоты являются такими, которые не содержат обнаруживаемых примесей, как было определено стандартным способом анализа. В отношении соединения по настоящему изобретению, которое обладает одним или несколькими центрами асимметрии и может встречаться в виде смесей стереоизомеров, в основном чистое соединение может быть или в основном чистой смесью стереоизомеров, или в основном чистым отдельным диастереомером или энантиомером.
Настоящее изобретение охватывает все стереоизомерные формы соединений формулы I, формулы IA и формулы IB. Если не отмечена конкретная стереохимия, подразумевается, что настоящее изобретение охватывает все такие изомерные формы таких соединений. Центры асимметрии, которые присутствуют в соединениях формулы I, формулы IA и формулы IB, все могут независимо друг от друга обладать (R) конфигурацией или (S) конфигурацией. Если связи с хиральным атомом углерода в структурных формулах по настоящему изобретению изображены в виде прямых линий, является понятным, что обе конфигурации (R) и (S) хирального атома углерода и, таким образом, оба энантиомера и их смеси охватываются в пределах формулы. Подобным образом, если название соединения указано без хирального обозначения хирального атома углерода, является понятным, что обе конфигурации (R) и (S) хирального атома углерода и, таким образом, отдельные энантиомеры и их смеси охватываются названием. Образование конкретных стереоизомеров или их смесей может быть определено в примерах, в которых такие стереоизомеры или смеси были получены, но это никак не ограничивает включение всех стереоизомеров или их смесей от того, что они находятся в пределах объема настоящего изобретения.
Настоящее изобретение включает в себя все возможные энантиомеры и диастереомеры и смеси двух или нескольких стереоизомеров, например, смеси энантиомеров и/или диастереомеров во всех соотношениях. Таким образом, энантиомеры являются объектом настоящего изобретения в энантиомерно чистой форме, оба как левовращающие и как правовращающие антиподы, в форме рацематов и в форме смесей двух энантиомеров во всех соотношениях. В случае цис/транс изомерии настоящее изобретение включает в себя как цис форму, так и транс форму, а также смеси этих форм во всех соотношениях. Получение отдельных стереоизомеров может быть проведено, при необходимости, разделением смеси обычными способами, например, методом хроматографии или кристаллизации с применением стереохимически однородных исходных веществ для синтеза или путем стереоселективного синтеза. Перед разделением стереоизомеров необязательно может быть проведена дериватизация. Разделение смеси стереоизомеров может быть проведено на промежуточной стадии в течение синтеза соединения формулы I, формулы IA или формулы IB или может быть проведено с конечным рацемическим продуктом. Абсолютная стереохимия может быть определена методом рентгеноструктурной кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые дериватизированы, если необходимо, реагентом, содержащим стереогенный центр известной конфигурации. Если соединения по настоящему изобретению способны к таутомеризации, все отдельные таутомеры, а также их смеси, включены в объем настоящего изобретения. Если не отмечен конкретный изомер, соль, сольват (включая гидраты) или сольватированная соль такого рацемата, энантиомера, диастереомера или таутомера, настоящее изобретение включает в себя все такие изомеры, а также соли, сольваты (включая гидраты) и сольватированные соли таких рацематов, энантиомеров, диастереомеров и таутомеров и их смеси.
Если любая переменная (например, R1, Ra и т. п.) встречается более одного раза в любой составляющей или в формуле I, ее определение в каждом случае не зависит от ее определения в каждом другом случае. Также допустимы комбинации заместителей и/или переменных, только если такие комбинации приводят к стабильным соединениям. Волнистая линия поперек связи в переменной заместителя представляет собой точку присоединения.
При стандартной номенклатуре, используемой по всему раскрытию, концевая часть указанной боковой цепи описана последней, ей предшествует смежная функциональная группа около точки присоединения.
При выборе соединений по настоящему изобретению специалист настоящей области техники будет принимать во внимание, что различные заместители, т. e. R1, R2 и т. п., выбраны в соответствии с хорошо известными принципами способности присоединения и стабильности химической структуры.
Термин «замещенный» означает, что один или несколько атомов водорода на одном обозначенном атоме заменены выбранным вариантом из указанной группы, при условии, что нормальная валентность обозначенного атома при существующих условиях не превышена и что замещение приводит к стабильному соединению. Считается, что термин замещенный включает в себя несколько степеней замещения указанным заместителем. Если несколько фрагментов заместителя раскрыты или заявлены, замещенное соединение независимо может быть замещено одним или несколькими из раскрытых или заявленных фрагментов заместителя, отдельно или во множественном виде. Под независимо замещенным подразумевается, что (два или несколько) заместители могут быть одинаковыми или разными.
Подразумевается, что «стабильное соединение» и «стабильная структура» обозначают соединение, которое является достаточно устойчивым, чтобы сохраняться после выделения до приемлемой степени чистоты из реакционной смеси, и структура и свойства которого остаются или могут служить поводом, чтобы оставаться без значительных изменений в течение времени, которого достаточно, чтобы позволить применение соединения в целях, описанных в настоящем изобретении (например, терапевтическое введение субъекту). Соединения по настоящему изобретению ограничены стабильными соединениями, которые включены в формулу I.
Если группа, например, C1-C8 алкил, обозначена как замещенная, такие замещения также могут возникать, если такая группа является частью большего заместителя, например, –C1-C6алкил-C3-C7циклоалкил и –C1-C8алкиларил.
В соединениях формулы I, IA и IB атомы могут проявлять свой натуральный изотопный состав или один или несколько атомов могут быть искусственно обогащены конкретным изотопом с таким же атомным числом, но с атомной массой или массовым числом, отличным от атомной массы или массового числа, преимущественно встречающихся в природе. Подразумевается, что настоящее изобретение включает в себя все приемлемые изотопные варианты соединений формулы I, IA и IB. Например, различные изотопные формы водорода (H) включают в себя протий (1H) и дейтерий (2H или D). Протий представляет собой преобладающий изотоп водорода, встречающийся в природе. Обогащение дейтерием может давать определенные терапевтические преимущества, такие как увеличение in vivo периода полураспада или снижение дозировки, или может обеспечивать соединение, применимое в качестве стандарта для характеристики биологических образцов. Изотопно-обогащенные соединения формулы I, IA и IB могут быть получены без несоответствующего проведения экспериментов традиционными методиками, хорошо известными специалистам настоящей области техники, или способами, аналогичными описанным в примерах настоящего описания с применением соответствующих изотопно-обогащенных реагентов и/или промежуточных соединений.
Если в конкретном контексте прямо не указано обратное, любое из различных циклических колец или кольцевых систем, описанных в настоящем изобретении, может быть присоединено к остатку соединения при любом кольцевом атоме (т.e., любом атоме углерода или любом гетероатоме), при условии, что это приводит к стабильному соединению.
Если прямо не указано обратное, все упомянутые в настоящем описании диапазоны являются включительными. Например, гетероароматическое кольцо, описанное как содержащее от «1 до 4 гетероатомов», означает, что кольцо может содержать 1, 2, 3 или 4 гетероатома. Также является понятным, что любой упомянутый в настоящем описании диапазон включает в свой объем все поддиапазоны в пределах этого диапазона. Таким образом, например, подразумевается, что гетероциклическое кольцо, описанное как содержащее от «1 до 4 гетероатомов» включает в себя в качестве его аспектов гетероциклические кольца, содержащие от 2 до 4 гетероатомов, 3 или 4 гетероатома, от 1 до 3 гетероатомов, 2 или 3 гетероатома, 1 или 2 гетероатома, 1 гетероатом, 2 гетероатома, 3 гетероатома и 4 гетероатома. Подобным образом, C1-6 при использовании с цепью, например, алкильной цепью, означает, что цепь может содержать 1, 2, 3, 4, 5 или 6 атомов углерода. Также он включает в себя все содержащиеся в нем диапазоны, включая C1-5, C1-4, C1-3, C1-2, C2-6, C3-6, C4-6, C5-6 и все другие возможные комбинации.
Также следует отметить, что предполагается, что любой атом углерода, а также гетероатом с ненасыщенными валентностями в представленном тексте, схемах, примерах и таблицах содержит достаточное число атома(ов) водорода для насыщения валентностей.
Если любой заместитель или переменная (например, алкил, R8, R11 и т. п.) встречаются более одного раза в любой составляющей или в формуле (I), их определение в каждом случае не зависит от его определения в каждом другом случае, если не отмечено иное.
Термин «соединение» относится к свободному соединению и, до степени, до которой они стабильны, к любому его гидрату или сольвату. Гидрат представляет собой соединение, образующее комплекс с водой, а сольват представляет собой соединение, образующее комплекс с органическим растворителем.
Как указано выше, соединения по настоящему изобретению могут быть использованы в форме фармацевтически приемлемых солей. Будет понятно, что, при использовании в настоящем описании, соединения по настоящему изобретению также могут включать в себя фармацевтически приемлемые соли, а также соли, которые не являются фармацевтически приемлемыми при использовании в качестве исходных продуктов по отношению к свободным соединениям или их фармацевтически приемлемым солям или при других манипуляциях при синтезе.
Термин «фармацевтически приемлемая соль» относится к соли, которая обладает эффективностью исходного соединения и которая не является биологически или иным образом нежелательной (например, не является ни токсичной, ни вредной иным образом для пациента). Термин «фармацевтически приемлемая соль» относится к солям, полученным из фармацевтически приемлемых не токсичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли основных соединений, которые охватываются термином «фармацевтически приемлемая соль», относится к не токсичным солям соединений по настоящему изобретению, которые обычно получали путем осуществления взаимодействия свободного основания с подходящей органической или неорганической кислотой. Типичные соли основных соединений по настоящему изобретению включают в себя без ограничения следующее: ацетат, аскорбат, адипат, альгинат, аспират, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, бутират, камфорат, камфорсульфонат, камзилат, карбонат, хлорид, клавуланат, цитрат, циклопентанпропионат, соль диэтилуксусной кислоты, диглюконат, дигидрохлорид, додецилсульфанат, эдетат, эдизилат, эстолат, эзилат, этансульфонат, соль муравьиной кислоты, фумарат, глюцептат, глюкогептаноат, глюконат, глутамат, глицерофосфат, гликолиларсанилат, гемисульфат, гептаноат, гексаноат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, 2-гидроксиэтансульфонат, гидроксинафтоат, йодид, соль изоникотиновой кислоты, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, метансульфонат, мукат, 2-нафталинсульфонат, напсилат, никотинат, нитрат, N-метилглюкамин аммониевую соль, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, пектинат, персульфат, фосфат/дифосфат, соль пимелиновой кислоты, соль фенилпропионовой кислоты, полигалактуронат, пропионат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тиоцианат, тозилат, триэтйодид, трифторацетат, ундеконат, валерат и т. п. Более того, если соединения по настоящему изобретению несут кислотный фрагмент, их фармацевтически приемлемые соли включают в себя без ограничения соли, полученные из неорганических оснований, включая алюминий, аммоний, кальций, медь, трехвалентное железо, двухвалентное железо, литий, магний, трехвалентный марганец, двухвалентный марганец, калий, натрий, цинк и т. п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических не токсичных оснований, включают в себя соли первичных, вторичных и третичных аминов, циклических аминов, дициклогексиламинов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этиламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминновые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т. п. Также включены основные азотсодержащие группы, которые могут быть кватернизированы такими средствами, как низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, -бромиды и -йодиды; диалкилсульфаты, такие как
диметил-, диэтил-, дибутил- и диамилсульфаты, длинноцепочечные галогениды, такие как децил-, лаурил-, миристил- и стераилхлориды, бромиды и -йодиды, аралкилгалогениды, такие как бензил- и фенэтилбромиды, и другие.
Такие соли могут быть получены известными способами, например, смешиванием соединения по настоящему изобретению с эквивалентным количеством и раствором, содержащим требуемую кислоту, основание или т. п., а затем собиранием требуемой соли методом фильтрации соли или отгонки растворителя. Соединения по настоящему изобретению и их соли могут образовывать сольваты с растворителем, таким как вода, этанол или глицерин. Соединения по настоящему изобретению могут образовывать кислотно-аддитивную соль и соль с основанием в это же время согласно типу заместителя боковой цепи.
Соединения по настоящему изобретению также могут быть использованы в форме пролекарства. Например, атом водорода в –COOH может быть замене любой из следующих групп: C1-6 алкил, C3-6 циклоалкил, –C1-6алкил-C3-6циклоалкил, C3-7 циклогетероалкил, –C1-6алкил-C3-7циклогетероалкил, арил, –C1-10алкиларил, гетероарил и –C1-10 алкил-гетероарил. Любой C1-6 алкил, C3-6 циклоалкил или C3-7 циклогетероалкил также может быть замещен. Любой арил или гетероарил также может быть замещен, как отмечено.
Как изложено выше, настоящее изобретение включает в себя фармацевтические композиции, содержащие соединение формулы I, IA или IB по настоящему изобретению, необязательно один или несколько других активных компонентов и фармацевтически приемлемый носитель. Характерные особенности носителя будут зависеть от пути введения. Под «фармацевтически приемлемым» подразумевается, что ингредиенты фармацевтической композиции должны соответствовать друг другу, не препятствовать эффективности активного(ых) ингредиента(ов) и не быть вредными (например, токсичными) для пациента. Таким образом, композиции согласно настоящему изобретению могут, в дополнение к ингибитору, содержать разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы и другие вещества, хорошо известные из области техники.
Также как изложено выше, настоящее изобретение включает в себя способ лечения бактериальной инфекции, причем способ предусматривает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы I, IA или IB или его фармацевтически приемлемой соли. Используемый в настоящем описании термин «субъект» (или, альтернативно, «пациент») относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который был объектом лечения, наблюдения или эксперимента. Термин «введение» и его вариации (например, «введение» соединения) в отношении соединения формулы I, IA или IB означает обеспечение соединения или его фармацевтически приемлемой соли для пациента при необходимости лечения. Если соединение или его соль обеспечены в комбинации с одним или несколькими другими активными средствами, под «введением» и его вариациями подразумевается включение предоставления соединения или его соли и других средств в одно время или в разное время. Если средства комбинации вводили в одно время, они могут быть введены вместе в одной композиции или они могут быть введены отдельно. Является понятным, что «комбинация» активных средств может быть одной композицией, содержащей все активные средства, или несколькими композициями, каждая содержит одно или несколько активных средств. В случае двух активных средств комбинация может быть или в виде одной композицией, содержащей оба средства, или в виде двух отдельных композиций, каждая содержит одно из средств; в случае трех активных средств комбинация может быть или в виде одной композиции, содержащей все три агента, трех отдельных композиций, каждая содержит одно из средств, или двух композиций, одна из которых содержит два средства, а другие содержат третье средство; и так далее.
Композиции и комбинации по настоящему изобретению соответственно вводили в эффективных количествах. Термин «эффективное количество», используемый в настоящем описании по отношению к оксазолидиноновому соединению, означает количество активного соединения, достаточного, чтобы вызывать бактерицидный или бактериостатический эффект. Согласно одному варианту осуществления эффективное количество представляет собой «терапевтически эффективное количество», что означает количество активного соединения, которое может преодолеть бактериальную устойчивость к лекарственному средству и которое является достаточным для ингибирования бактериальной редупликации и/или приводит к уничтожению бактерий. Если активное соединение (т. e., активный ингредиент) вводили в виде соли, ссылки на количество активного ингредиента относятся к форме свободной кислоты или свободного основания соединения.
Подходящим введением для композиции в соответствии с настоящим изобретением образом является парентеральное, пероральное, подъязычное, чрескожное, местное, интраназальное, интратрахеальное, интраокулярное или внутриректальное, при этом композицию надлежащим образом составляют для введения выбранным путем с использованием способов составления, хорошо известных в уровне техники, в том числе, например, способов получения и введения составов, описанных в главах 39, 41, 42, 44 и 45 Remington – The Science and Practice of Pharmacy, 21st edition, 2006. Согласно одному варианту осуществления соединения в соответствии с настоящим изобретением вводят внутривенно в больничных условиях. Согласно другому варианту осуществления введение является пероральным в форме таблетки или капсулы, или т.п. Дозировка соединений в соответствии с настоящим изобретением и их фармацевтически приемлемых солей может варьировать в широких переделах и, естественно, должна подбираться в каждом конкретном случае в соответствии с отдельными условиями и патогенными агентами, подлежащими контролю. В целом, для применеия в лечении бактериальных инфекций суточная доза может составлять от 0,005 мг/кг до 100 мг/кг, от 0,01 мг/кг до 10 мг/кг, от 0,05 мг/кг до 5 мг/кг, от 0,05 мг/кг до 1 мг/кг.
Согласно некоторым вариантам осуществления соединение в соответствии с настоящим изобретением обеспечивают в фармацевтическом составе для перорального, внутривенного, внутримышечного, назального или местного введения. Таким образом, согласно некоторым вариантам осуществления состав может быть получен в дозированной форме, такой как без ограничения таблетка, капсула, жидкость (раствор или суспензия), суппозиторий, мазь, крем или аэрозоль. Согласно некоторым вариантам осуществления раскрываемый в настоящем документе объект обеспечивает такие соединения и/или составы, которые были лиофилизированы и которые могут быть восстановлены с образованием фармацевтически приемлемых составов для введения, например, внутривенной или внутримышечной инъекцией.
Внутривенное введение соединения в соответствии с настоящим изобретением может быть выполнено путем восстановления порошковой формы соединения в приемлемом растворителе. Подходящие растворители включают в себя, например, солевые растворы (например, 0,9% физиологический раствор для инъекции) и стерильную воду (например, стерильную воду для инъекции, бактериостатическую воду для инъекции с метилпарабеном и пропилпарабеном или бактериостатическую воду для инъекции с 0,9% бензиловым спиртом). Порошковая форма соединения может быть получена с помощью обработки соединения гамма-излучением или с помощью лиофилизации раствора соединения, после чего порошок может храниться (например, в закупоренном флаконе) при комнатной или более низкой температуре до восстановления. Концентрация соединения в восстановленном IV растворе может находиться, например, в диапзоне от приблизительно 0,1 мг/мл до приблизительно 20 мг/мл.
Способы раскрываемого в настоящем документе объекта применимым для лечения таких состояний, при которых они подавляют проявление, развитие или распространение состояния, вызывают регрессию состояния, излечивают состояние или иным образом улучшают общее самочувствие субъекта, пораженного состоянием или подвергающегося риску заразиться состоянием. Таким образом, в соответствии с раскрываемым в настоящем документе объектом термины «лечить», «лечение» и их грамматические вариации, а также фраза «способ лечения» охватывают любое желаемое терапевтическое вмешательство, в том числе без ограничения способ лечения имеющейся инфекции у субъекта и способ профилактики (т.e. предупреждения) инфекции, например, у субъекта, который подвергался воздействию раскрываемого в настоящем документе микроба или в отношении которого предполагается воздействие раскрываемого в настоящем документе микроба.
Инфекции, которые можно лечить соединениями в соответствии с настоящим изобретением, могут быть вызваны рядом микробов, в том числе грибками, водорослями, простейшими, бактериями и вирусами. Согласно некоторым вариантам осуществления инфекцией является бактериальная инфекция. Типичные микробные инфекции, которые можно лечить способами в соответствии с настоящим изобретением, включают в себя без ограничения инфекции, вызванные одним или несколькими из Staphylococcus aureaus, Enterococcus faecalis, Bacillus anthracis, видов Streptococcus (например, Streptococcus pyogenes и Streptococcus pneumoniae), Escherichia coli, Pseudomonas aeruginosa, Burkholderia cepacia, видов Proteus (например, Proteus mirabilis и Proteus vulgaris), Klebsiella pneumoniae, Acinetobacter baumannii, Strenotrophomonas maltophillia, Mycobacterium tuberculosis, Mycobacterium bovis, другие микобактерии туберкулезного комплекса, а также нетуберкулезные микобактерии, в том числе Mycobacterium ulcerans.
Согласно некоторым вариантам осуществления инфекцией является инфекция грамположительной бактерии. Согласно некоторым вариантам осуществления инфекция выбрана из микобактериальной инфекции, инфекции Bacillus anthracis, инфекции Enterococcus faecalis и инфекции Streptococcus pneumoniae.
Согласно некоторым вариантам осуществления соединение формулы I, IA или IB вводят профилактически для предупреждения или снижения заболеваемости одним из (a) инфекции Mycobacterium tuberculosis у субъекта с риском инфекции; (b) рецидива инфекции Mycobacterium tuberculosis и (c) их комбинаций. Согласно некоторым вариантам осуществления соединение формулы I, IA или IB вводят для лечения имеющейся инфекции Mycobacterium tuberculosis. Согласно некоторым вариантам осуществления соединение формулы I, IA или IB вводят для лечения инфекции устойчивого к нескольким лекарственным средствам штамма Mycobacterium tuberculosis (т.e. штамма, который является устойчивым к двум или более ранее известным противотуберкулезным лекарственным средствам, таким как изониазид, этамбутол, рифампицин, канамицин, капреомицин, линезолид и стрептомицин). Согласно некоторым вариантам осуществления соединение формулы (I) имеет минимальную ингибиторную концентрацию (MIC) в отношении Mycobacterium tuberculosis 25 мкг/мл или меньше. Согласно некоторым вариантам осуществления соединение формулы I, IA или IB вводят для лечения инфекции устойчивого к нескольким лекарственным средствам штамма Mycobacterium tuberculosis.
Таким образом, способы раскрываемого в настоящем документе объекта могут быть применимыми для лечения туберкулеза за счет того, что они подавляют проявление, развитие или распространение инфекции TB, вызывают регрессию инфекции TB, излечивают инфекцию TB или иным образом улучшают общее самочуствие субъекта, пораженного туберкулезом или подвергающегося риску заразиться таковым.
Субъекты, страдающие инфекцией M. tuberculosis или другой связанной с туберкулезом инфекцией, могут быть выявлены с помощью ряда методик, таких как мазок мокроты, рентгенография грудной клетки, туберкулиновая кожная проба (т.e. реакция Манту или PPD проба), и/или по наличию других клинических симптомов (например, боль в груди, откашливание крови, лихорадка, ночная потливость, потеря аппетита, утомление и т.д.). При необходимости бактериальные РНК, ДНК или белки можно выделить из субъекта, предположительно страдающего TB, проанализировать способами, известными в уровне техники, и сравнить с известными последовательностями нуклеиновых кислот или аминокислот бактериальных РНК, ДНК или белка.
Согласно некоторым вариантам осуществления соединение формулы I, IA или IB обладает минимальной ингибиторной концентрацией (MIC) в отношении Mycobacterium tuberculosis 25 мкг/мл или меньше. MIC могут быть определены способами, известными в уровне техники, например, описанными в Hurdle et al., 2008, J. Antimicrob. Chemother. 62:1037-1045.
Согласно некоторым вариантам осуществления способы в соответствии с настоящим изобретением дополнительно предусматривают введение субъекту дополнительного терапевтического соединения. Согласно некоторым вариантам осуществления соединение в соответствии с настоящим изобретением вводят субъекту до, после или одновременно с одним или несколькими дополнительными терапевтическими соединениями. Согласно некоторым вариантам осуществления дополнительным терапевтическим соединением является антибиотик. Согласно некоторым вариантам осуществления дополнительным терапевтическим соединением является противотуберкулезное терапевтическое средство. Согласно некоторым вариантам осуществления дополнительное терапевтическое соединение выбрано из группы, содержащей изониазид, этамбутол, рифампицин, канамицин, капреомицин, линезолид и стрептомицин.
Настоящее изобретение, таким образом, согласно следующему аспекту относится к комбинации, содержащей соединение формулы I, IA или IB или его фармацевтически приемлемую соль вместе с одним или несколькими дополнительными терапевтическими средствами. Примерами таких одного или нескольких дополнительных терапевтических средств являются противотуберкулезные средства, в том числе без ограничения амикацин, аминосалициловая кислота, капреомицин, циклосерин, этамбутол, этионамид, изониазид, канамицин, пиразинамид, рифамицины (такие как рифампин, рифапентин и рифабутин), стрептомицин, кларитромицин, азитромицин, оксазолидиноны и фторхинолоны (такие как офлоксацин, ципрофлоксацин, моксифлоксацин и гатифлоксацин). Такая химиотерапия определяется на усмотрение лечащего врача с использованием предпочтительных комбинаций лекарственный средств. Химиотерапевтические средства «первой линии», используемые для лечений инфекции Mycobacterium tuberculosis, которая не является устойчивой к лекарственным средствам, включают в себя изониазид, рифампин, этамбутол, стрептомицин и пиразинамид. Химиотерапевтические средства «второй линии», используемые для лечений инфекции Mycobacterium tuberculosis, которая демонстрировала устойчивость к одному или нескольким лекарственным средствам «первой линии», включают в себя офлоксацин, ципрофлоксацин, этионамид, аминосалициловую кислоту, циклосерин, амикацин, канамицин и капреомицин. Помимо вышеупомянутых существует ряд новых противотуберкулезных терапевтических средств, появляющихся после клинических исследований, которые также могут быть использованы в качестве одного или нескольких дополнительных терапевтических средств в комбинации с соединением формулы I, IA или IB, в том числе без ограничения TMC-207, OPC-67683, PA-824, LL-3858 и SQ-109.
Таким образом, другим антибиотиком, который может быть скомбинирован с соединениями формулы I, IA или IB, является, например, рифампицин (=рифампин); изониазид; пиразинамид; амикацин; этионамид; моксифлоксацин; этамбутол; стрептомицин; пара-аминосалициловая кислота; циклосерин; капреомицин; канамицин; тиоацетазон; хинолоны/фторхинолоны, такие как, например, офлоксацин, ципрофлоксацин, спарфлоксацин; макролиды, такие как, например, кларитромицин, клофазимин, амоксициллин с клавулановой кислотой; рифамицины; рифабутин; рифапентин.
Согласно следующему аспекту одним или несколькими дополнительными терапевтическими средствами является, например, средство, применимое для лечения туберкулеза у млекопитающего, терапевтические вакцины, противобактериальные средства, противовирусные средства; антибиотики и/или средства для лечения HIV/AIDS. Примеры таких терапевтических средств включают в себя изониазид (INH), этамбутол, рифампин, пиразинамид, стрептомицин, капреомицин, ципрофлоксацин и клофазимин.
Согласно одному аспекту одним или несколькими дополнительными терапевтическими средствами является терапевтическая вакцина. Соединение формулы I, IA или IB или его фармацевтически приемлемая соль, таким образом, могут быть введены в сочетании с вакцинацией против микобактериальной инфекции, в частности, вакцинацией против инфекции Mycobacterium tuberculosis. Существующие вакцины против микобактериальной инфекции включают в себя бациллу Кальметта-Герена (BCG). Вакцины, которые в настоящее время разрабатываются для лечения, профилактики или облегчения микобактериальной инфекции, включают в себя модифицированные штаммы BCG, которые рекомбинантно экспрессируют дополнительные антигены, цитокины и другие средства, предназначенные для улучшения эффективности или безопасности; аттенуированные микобактерии, которые экспрессируют ряд антигенов, более подобных Mycobacterium tuberculosis, чем BCG; и субъединичные вакцины. Субъединичные вакцины могут быть введены в форме одного или нескольких отдельных белковых антигенов или в форме слияния или слияний нескольких белковых антигенов, которые либо могут быть необязательно добавлены в качестве адъюванта, либо могут иметь форму полинуклеотида, кодирующего один или несколько отдельных белковых антигенов или кодирующего слияние или слияния нескольких белковых антигенов, например, если полинуклеотид вводят в векторе экспрессии. Примеры субъединичных вакцин включают в себя без ограничения M72, белок слияния, полученный из антигенов Mtb32a и Mtb39; HyVac-1, белок слияния, полученный из антигена 85b и ESAT-6; HyVac-4, белок слияния, полученный из антигена 85b и Tb10.4; MVA85a, модифицированный вирус вакцины Анкара, экспрессирующий антиген 85a; и Aeras-402, аденовирус 35, экспрессирующий белок слияния, полученный из антигена 85a, антигена 85b и Tb10.4.
Используемые в настоящем описании аббревиатуры включают в себя следующее:
ACN=ацетонитрил; CBZ-Cl=бензилхлорформиат; CDCl3=дейтерированный хлороформ; DCM=дихлорметан; DIAD=диизопропилдиазодикарбоксилат, DIEA=N,N-диизопропилэтиламин; DMF=N,N-диметилформамид; DMSO=диметилсульфоксид; Et=этил; EtOAc=этилацетат; EtOH=этанол; GFP=зеленый флуоресцентный белок; HATU=(1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксидгексафторфосфат), HET=гетероцикл; H2=газообразный водород, HPLC=высокоэффективная жидкостная хроматография; LC-MS=жидкостная хроматография/масс-спектрометрия; Me=метил; MeOH=метанол; MIC=минимальная ингибирующая концентрация; MW=молекулярная масса; MS=масс-спектрометрия; Mtb=Mycobacterium tuberculosis; Pd-C=палладиевый катализатор на углеродном носителе; к. т.=комнатная температура; TB=туберкулез; TEA=триэтиламин; TFA=трифторуксусная кислота; THF=тетрагидрофуран и TBDMS=трет-бутилдиметилсилил.
Способы получения соединений формулы (I):
Соединения формулы (I) могут быть получены согласно следующим схемам реакций и примерам или их модификациям с применением легко доступных исходных веществ, реагентов и традиционными методиками синтеза. При таких реакциях также возможно использование вариаций, которые сами по себе известны специалистам настоящей области техники, но не упомянуты более подробно. Альтернативные пути синтеза и аналогичные структуры будут очевидны специалистам в области органического синтеза с учетом следующих реакционных схем и примеров.
На схеме A показан способ, применимый для получения оксазолидиноновых соединений формулы A7, которые соответствуют соединениям формулы (I), где R1 представляет собой CH2NH2 и A определен в кратком описании настоящего изобретения. На схеме ниже показан синтез оксазолидиноновых аналогов, где A введен при помощи реакции сочетания. Схема синтеза обсуждалась подробно в литературе (Org. Proc. Res. Dev. 2003, 533). Оксизолидинон A3 может быть может быть составлен одной стадией из оксирана S1 и карбамата A2. Дальнейшей модификацией гидроксигруппы при помощи азидного промежуточного соединения A5 может быть получен амин A6. На заключительной стадии при помощи реакции сочетания Сузуки получали требуемое соединение A7, которое может быть использовано для синтеза соединений, где R1 представляет собой производное CH2NH2.
Схема A
На схеме B показан способ, применимый для получения оксазолидиноновых соединений формулы B4, которые соответствуют соединениям формулы (I), где R1 представляет собой гетероцикл, такой как имидазол, триазол или тетразол, и A определен в кратком описании настоящего изобретения. На схеме B соединение B1 превращали в соединение B2 при помощи реакции сочетания Сузуки с последующим введением гетероцикла путем замены мезилатного промежуточного соединения B3 с получением соединения B4.
Схема B
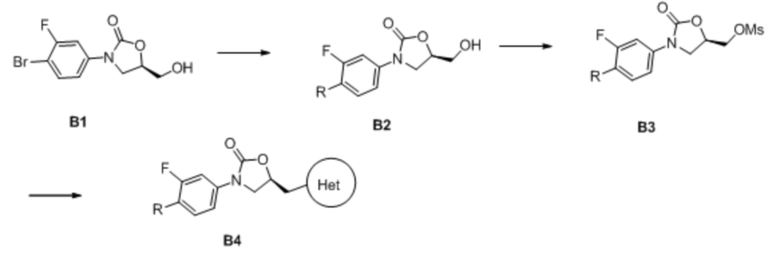
Соединения формулы A7 и B4 могут быть дополнительно обработаны с применением способов, которые будут хорошо известны специалистам в области органического синтеза или, например, способов, описанных в примерах ниже, с получением полного объема соединений формулы (I).
Пример 1
Синтез (S)-5-(аминометил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-она.
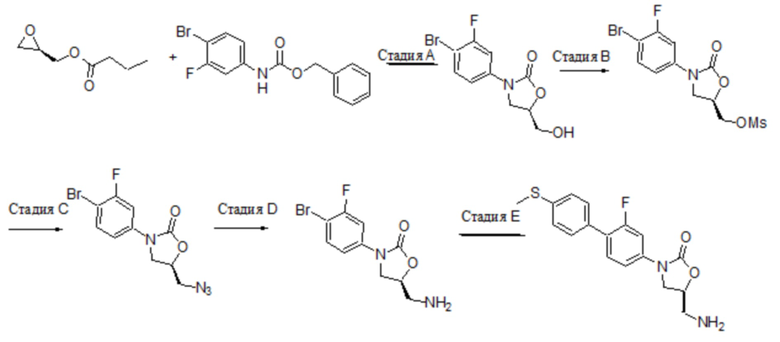
Стадия A: Синтез (R)-3-(4-бром-3-фторфенил)-5-(гидроксиметил)оксазолидин-2-она.
К раствору бензил(4-бром-3-фторфенил)карбамата (50,0 г, 154 ммоль) в THF (600 мл) добавляли трет-бутилат лития (37,0 г, 463 ммоль) при 0°C. Смесь перемешивали при 20°C в течение 2 часов, а затем добавляли (R)-оксиран-2-илметилбутират (44,5 г, 309 ммоль) при 20°C. Полученную смесь перемешивали при 20°C в течение 16 часов, а затем добавляли концентрированную HCl для доведения значения pH до 7. Органический растворитель удаляли при пониженном давлении, а остаток разбавляли EtOAc (400 мл), промывали водой (400 мл x 2) и солевым раствором (500 мл), сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали методом колоночной хроматографии (SiO2, PE:EtOAc=1:1) с получением продукта (R)-3-(4-бром-3-фторфенил)-5-(гидроксиметил)оксазолидин-2-она в виде твердого вещества.
Стадия B: Синтез (R)-(3-(4-бром-3-фторфенил)-2-оксооксазолидин-5-ил)метилметансульфоната.
К раствору (R)-3-(4-бром-3-фторфенил)-5-(гидроксиметил)оксазолидин-2-она (1,0 г, 3,45 ммоль) в DCM (10 мл) добавляли N-этил-N-изопропилпропан-2-амин (1,2 мл, 6,89 ммоль) и метансульфохлорид (5,17 ммоль) при 0°C. Реакционную смесь перемешивали при 28°C в течение 2 часов, а затем разбавляли водой (20 мл), экстрагировали DCM (20 мл x 3). Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением (R)-(3-(4-бром-3-фторфенил)-2-оксооксазолидин-5-ил)метилметансульфоната в виде масла. MS (ESI) m/z: 368,2, 370,2 [M+H+].
Стадия C: (R)-5-(азидометил)-3-(4-бром-3-фторфенил)оксазолидин-2-он
К раствору (R)-(3-(4-бром-3-фторфенил)-2-оксооксазолидин-5-ил)метилметансульфоната (13,0 г, 31,8 ммоль) в DMF (150 мл) добавляли азид натрия (2,5 г, 38,1 ммоль) и полученную смесь перемешивали при 70°C в течение 4 часов. Затем смесь разбавляли EtOAc (200 мл), промывали водой (200 мл x 2) и солевым раствором (200 мл), сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного (R)-5-(азидометил)-3-(4-бром-3-фторфенил)оксазолидин-2-она, который переносили на следующую стадию без очистки.
Стадия D: (S)-5-(аминометил)-3-(4-бром-3-фторфенил)оксазолидин-2-он
К раствору (R)-5-(азидометил)-3-(4-бром-3-фторфенил)оксазолидин-2-она (13,0 г, 31,7 ммоль) в THF (50 мл) добавляли воду (5,72 мл, 317 ммоль) и трифенилфосфин (20,81 г, 79 ммоль), затем смесь перемешивали при 70°C в течение 12 часов. Реакционную смесь выпаривали с удалением органического растворителя, затем добавляли воду (20 мл) и значение pH доводили до 4 с применением 1 M HCl. Полученную смесь экстрагировали EtOAc (30 мл x3), затем значение pH водного слоя доводили до 10 при помощи Na2CO3 (3M). Водный слой экстрагировали EtOAc (40 мл x4), объединенные органические растворители сушили над Na2SO4, фильтровали и концентрировали досуха с получением неочищенного (S)-5-(аминометил)-3-(4-бром-3-фторфенил)оксазолидин-2-она в виде твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS (ESI) m/z: 288,9, 290,9 [M+H+].
Стадия E: (S)-5-(аминометил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-он
К раствору (S)-5-(аминометил)-3-(4-бром-3-фторфенил)оксазолидин-2-она (3,0 г, 9,34 ммоль), (4-(метилтио)фенил)бороновой кислоты (1,9 г, 11,21 ммоль) и трехосновного фосфата калия (9,3 мл, 18,68 ммоль) в THF (40 мл) добавляли предварительный каталитический нейтрализатор xphos 2го поколения (0,7 г, 0,93 ммоль) в атмосфере N2. Смесь дегазировали и повторно заполняли N2 (3x). Смесь перемешивали при 60°C в течение 16 часов. Затем смесь концентрировали с получением неочищенного продукта, который очищали методом колоночной хроматографии (SiO2, PE:EtOAc=1:1 к DCM: MeOH=10:1) с получением требуемого продукта (S)-5-(аминометил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-она 1 в виде твердого вещества. MS (ESI) m/z: 333,1 [M+H+] (рассчит. 333,1).
Следующие соединения получали с использованием способов, описанных в примере 1, с замещением соответствующих реактивов и/или реагентов.
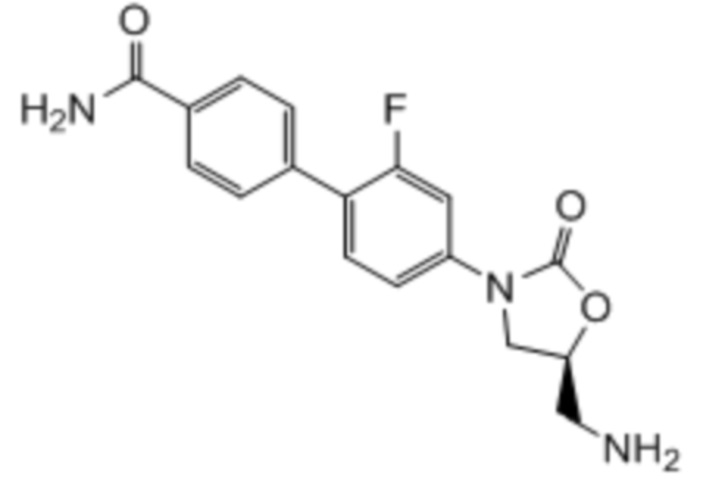
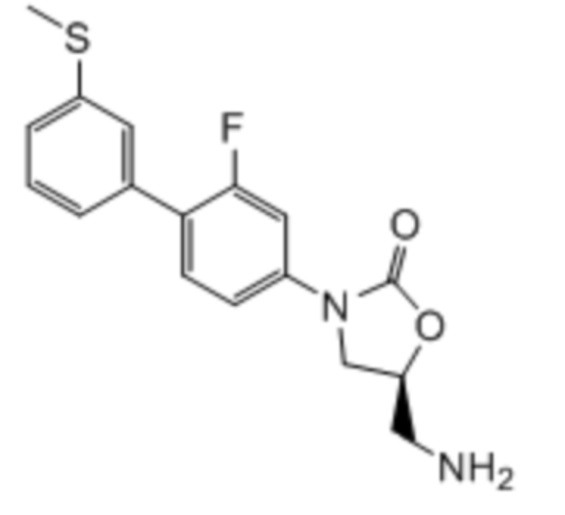
бифенил-4-ил]-1,3-оксазолидин-2-он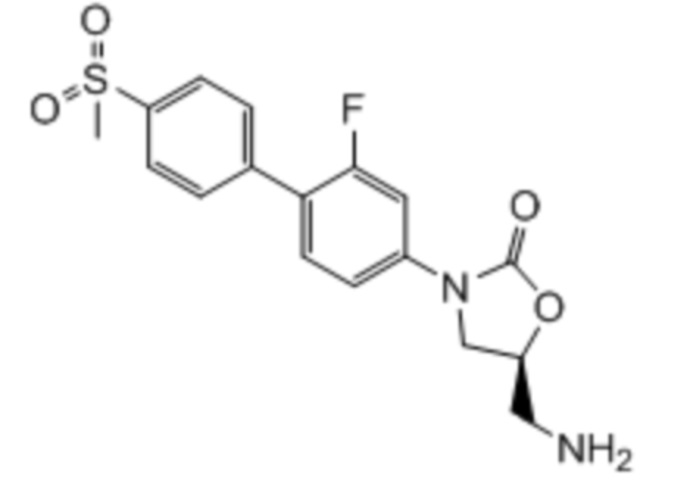
бифенил-4-ил]-1,3-оксазолидин-2-он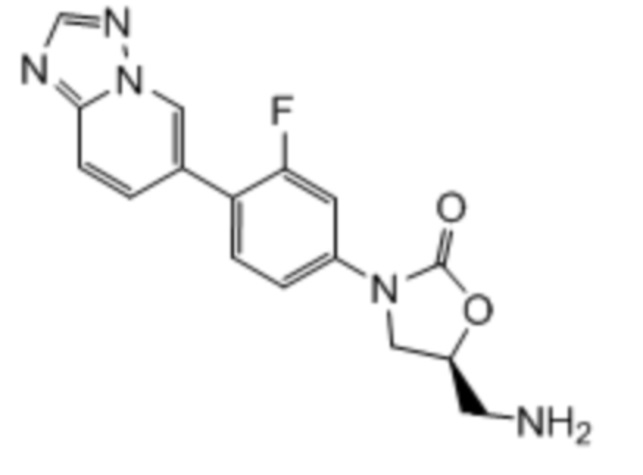
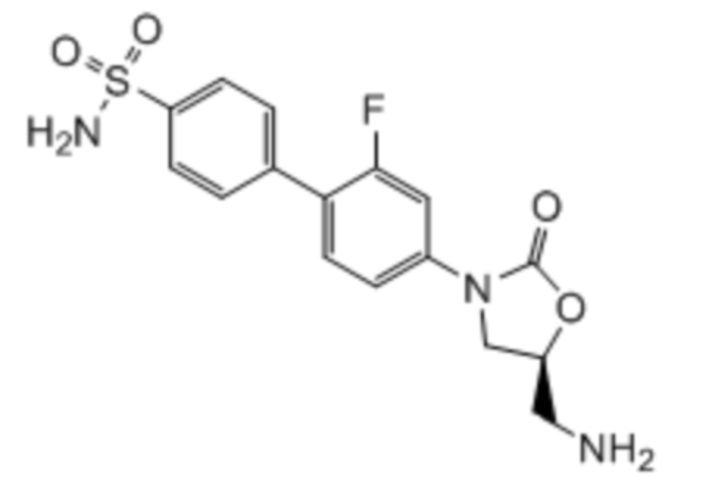
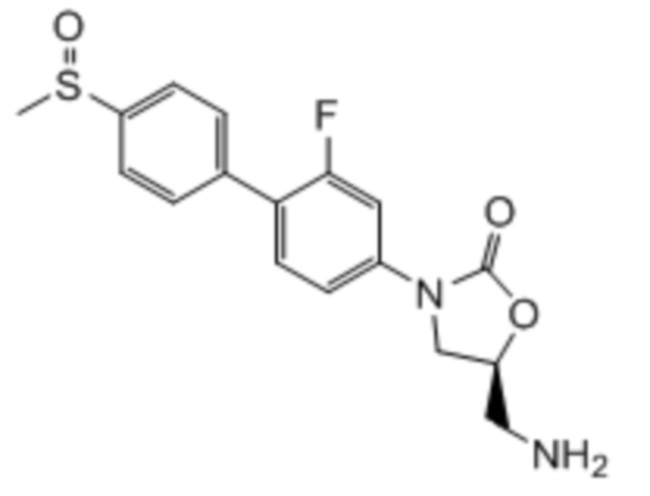
бифенил-4-ил]-1,3-оксазолидин-2-он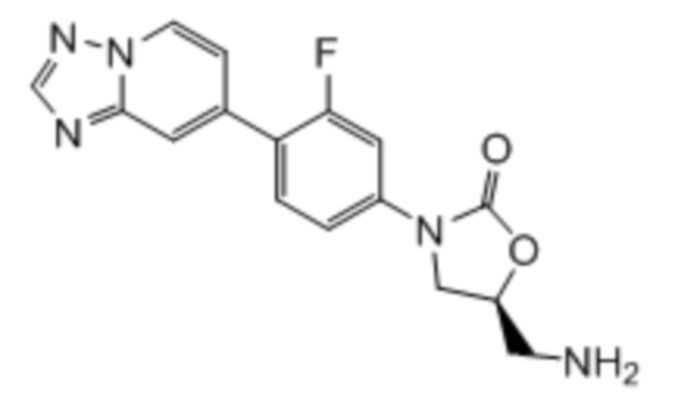
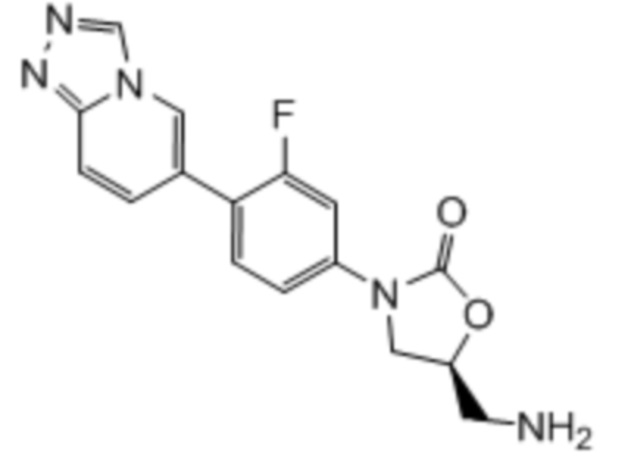
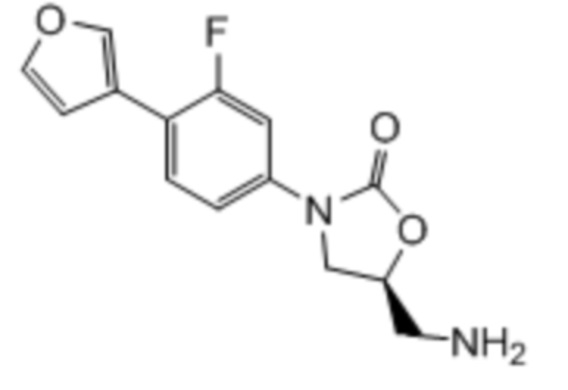
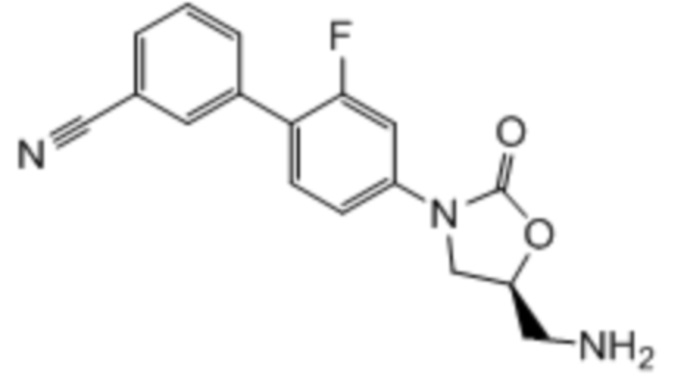
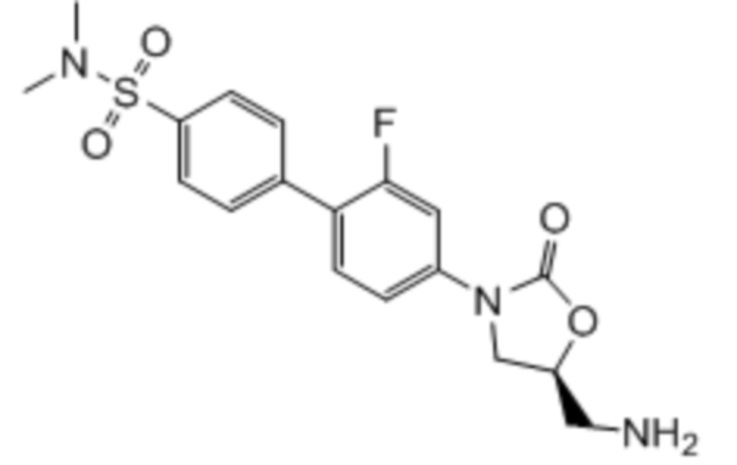
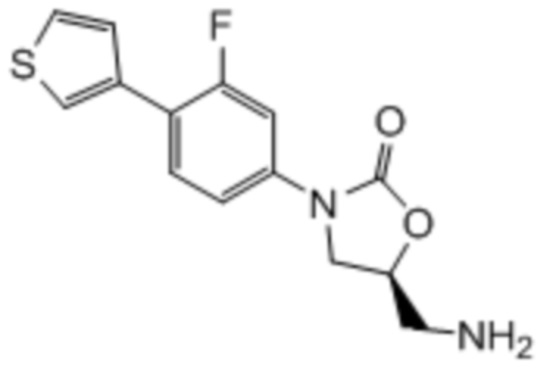
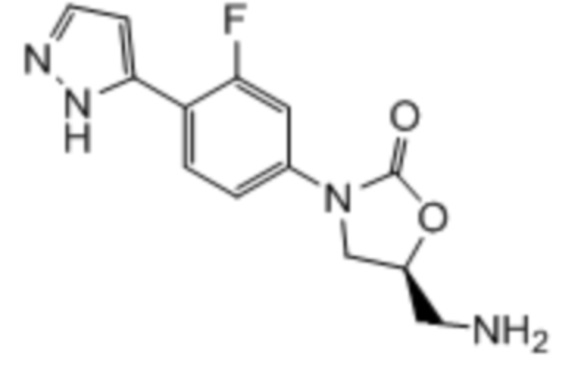
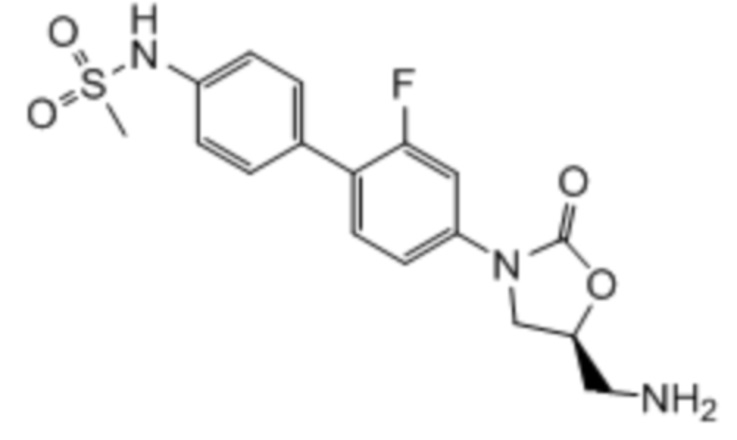
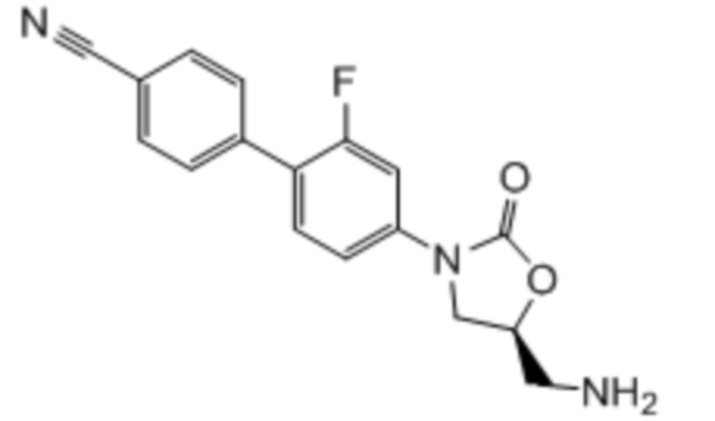
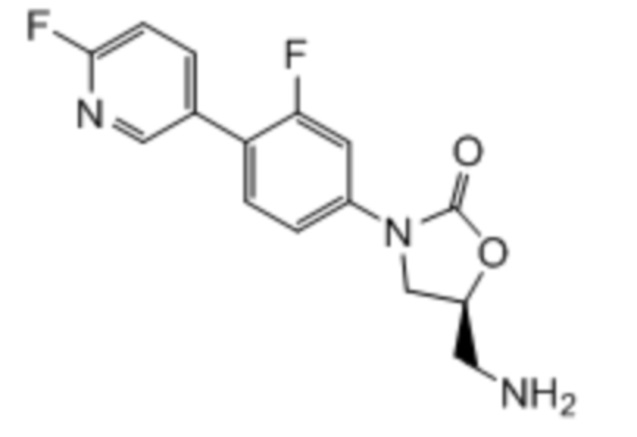
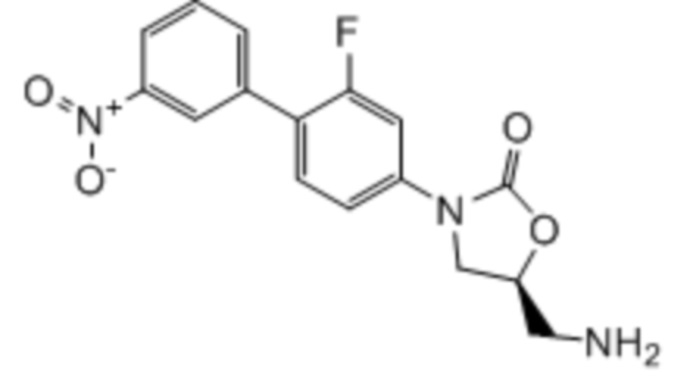
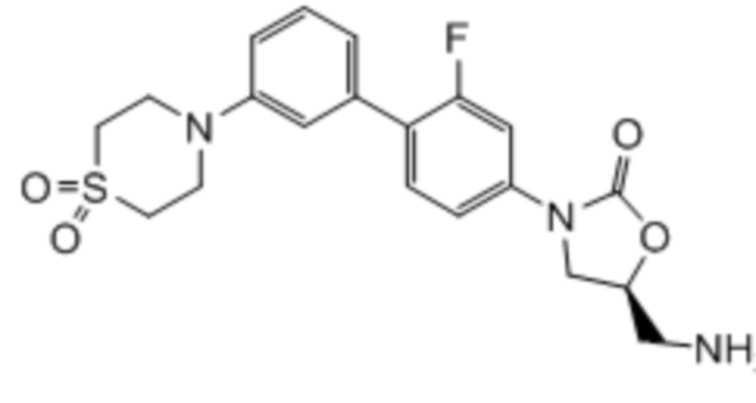
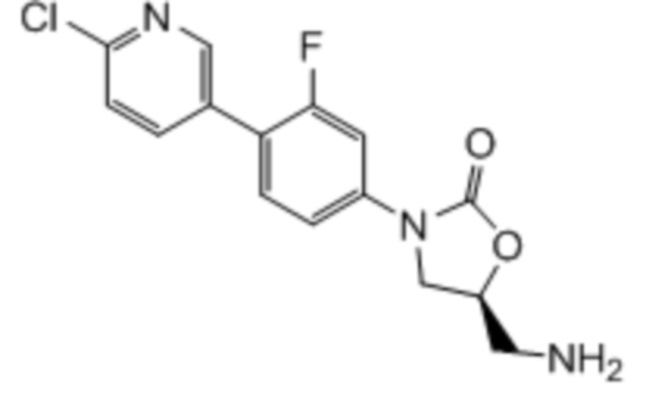
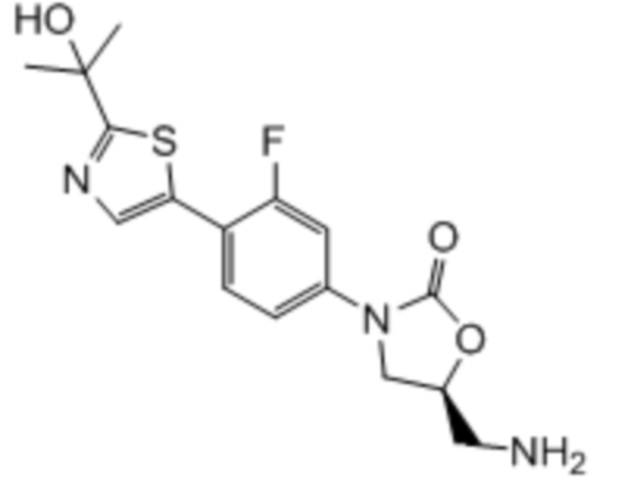

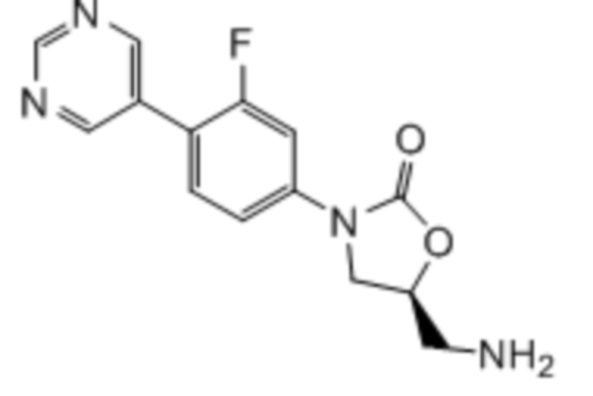
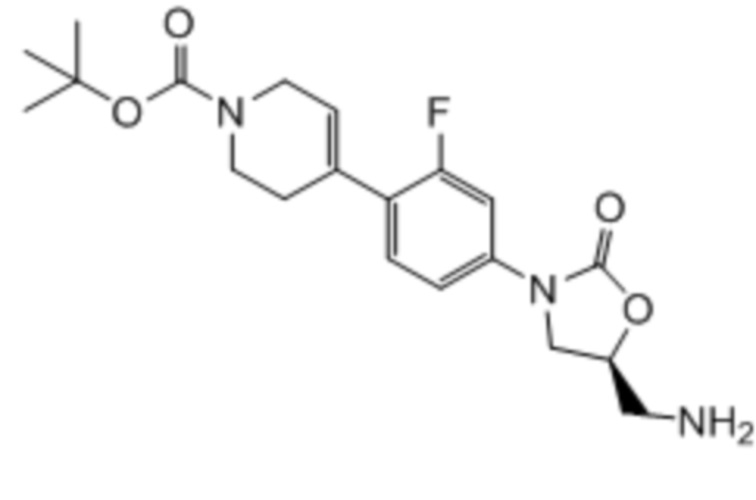
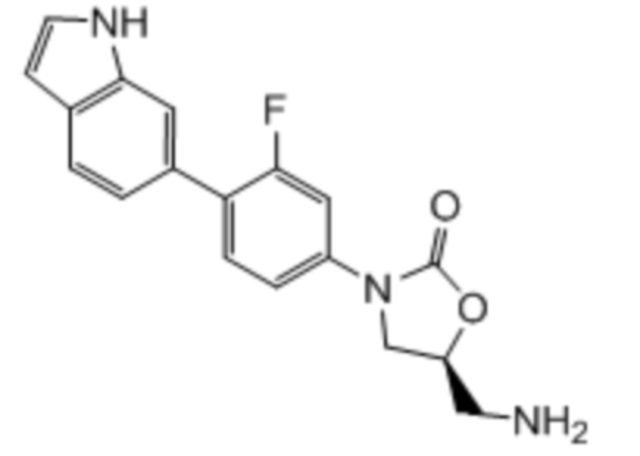
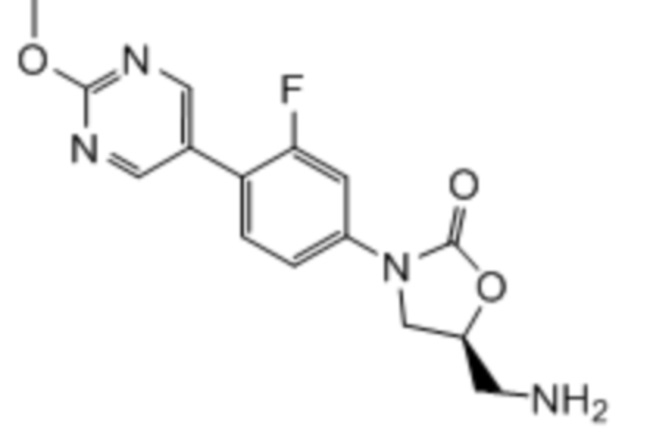
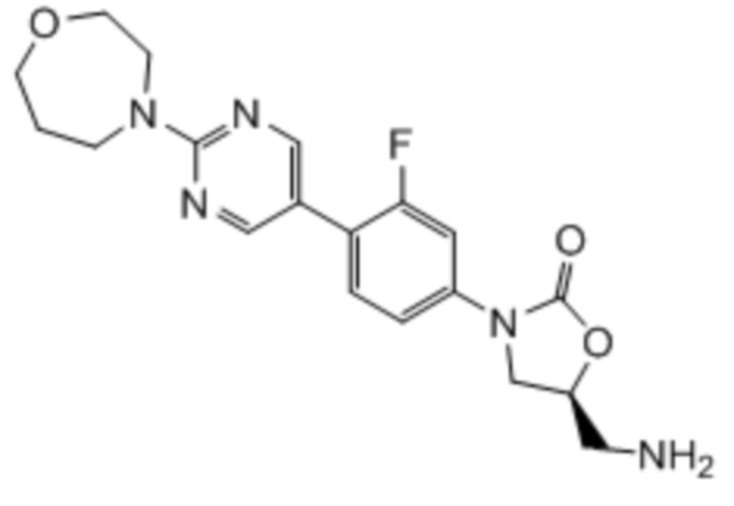
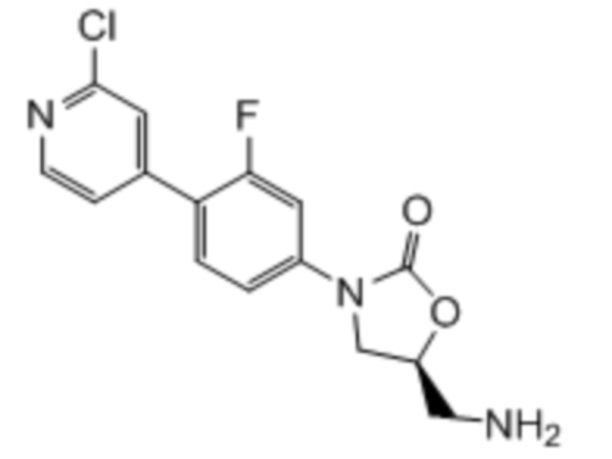
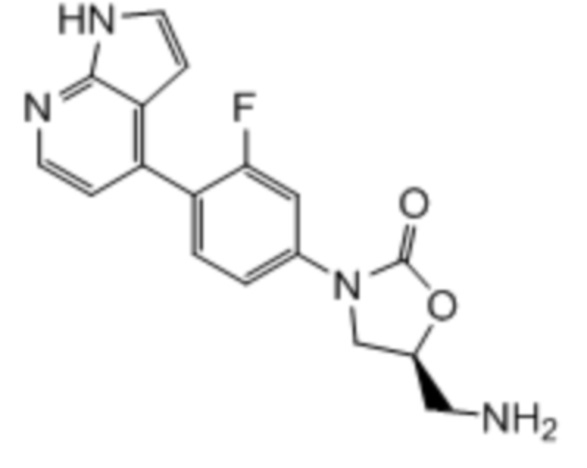
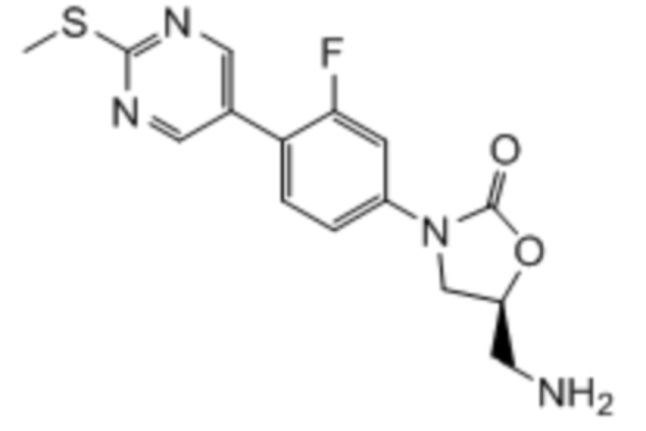
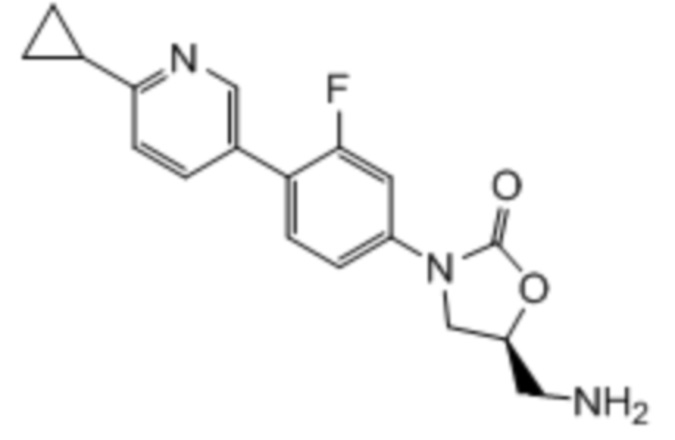
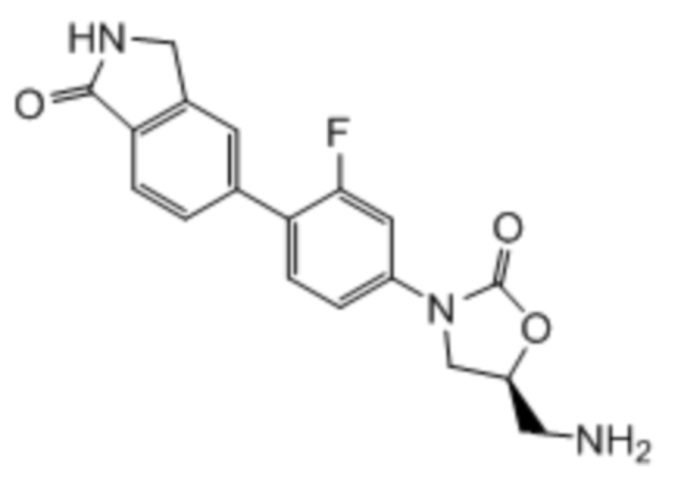
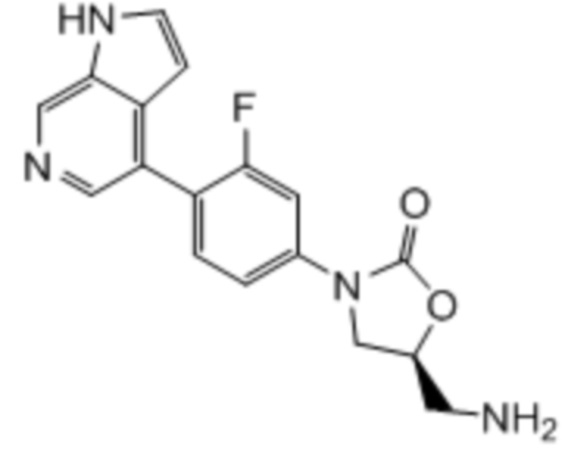
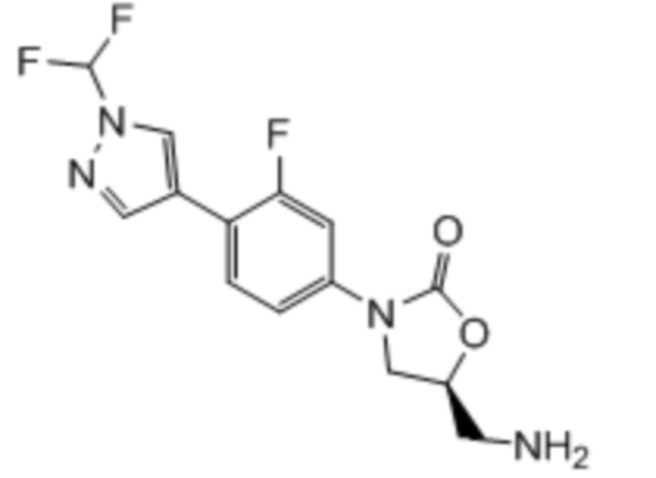
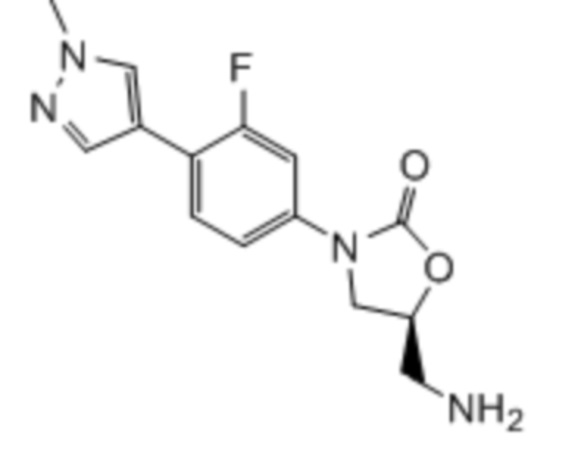
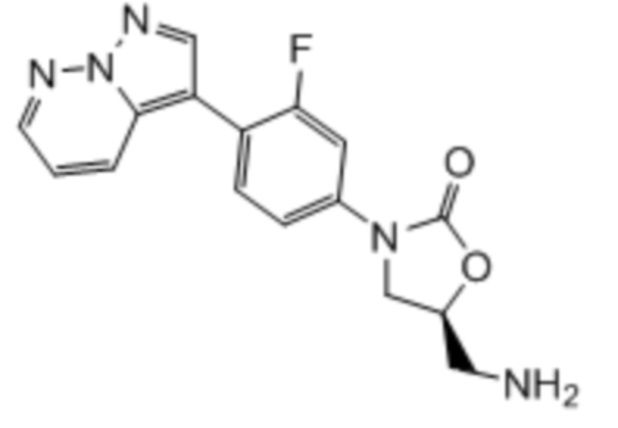
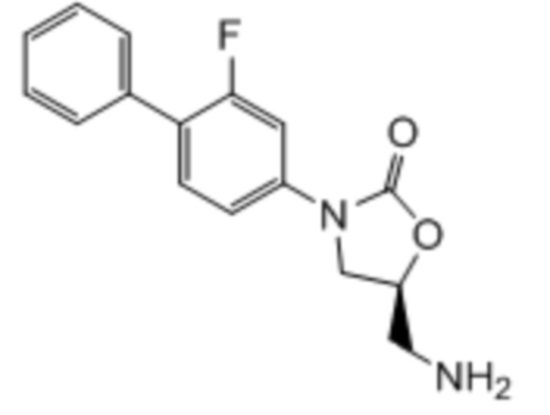
Пример 40
Синтез (S)-N-((3-(5-фтор-6-(4-(метилтио)фенил)пиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида.
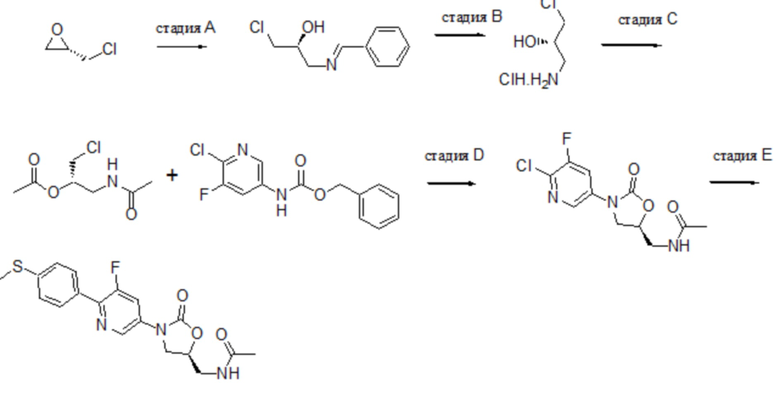
Стадия A: Синтез (S,E)-1-(бензилиденамино)-3-хлорпропан-2-ола.
К раствору (S)-2-(хлорметил)оксирана (5,0 г, 54,3 ммоль) в EtOH (15 мл) добавляли гидрат аммиака (4,9 г, 140 ммоль) и бензальдегид (5,98 г, 56,4 ммоль) и полученную смесь перемешивали при 40°C в течение 16 часов. Реакционную смесь концентрировали с получением (S,E)-1-(бензилиденамино)-3-хлорпропан-2-ола в виде твердого вещества, которое использовали на следующей стадии без очистки.
Стадия B: Синтез (S)-1-амино-3-хлорпропан-2-ола гидрохлорида
К раствору (S,E)-1-(бензилиденамино)-3-хлорпропан-2-ола (6,2 г, 31,4 ммоль) в EtOH (13 мл) и толуоле (12 мл) добавляли HCl (6,8 мл, 83 ммоль) и полученную смесь перемешивали при 35°C в течение 4 часов. Реакционную смесь концентрировали до меньшего объема, промывали водой (20 мл) и водный экстракт лиофилизировали с получением (S)-1-амино-3-хлорпропан-2-ола гидрохлорида в виде твердого вещества. Неочищенный продукт переносили на следующую стадию без дополнительной очистки.
Стадия C: Синтез (S)-1-ацетамидо-3-хлорпропан-2-ила ацетата
К раствору (S)-1-амино-3-хлорпропан-2-ола гидрохлорида (28,0 г, 153 ммоль) в DCM (100 мл) добавляли уксусный ангидрид (36,0 г, 353 ммоль), пиридин (15,3 г, 193 ммоль) и полученную смесь перемешивали при 35°C в течение 16 часов. Затем порционно добавляли K2CO3 (42,0 г, 304 ммоль), поддерживая температуру 5-7°C, затем добавляли 0,5 н HCl (250 мл), полученную смесь разбавляли H2O (250 мл) и экстрагировали DCM (200 мл x 3). Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением (S)-1-ацетамидо-3-хлорпропан-2-ила ацетата в виде твердого вещества. Неочищенный продукт использовали сразу на следующей стадии без дополнительной очистки. MS (ESI) m/z: 217,3 [M+Na+].
Стадия D: Синтез (S)-N-((3-(6-хлор-5-фторпиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида
К раствору бензил(6-хлор-5-фторпиридин-3-ил)карбамата (6 г, 21,38 ммоль) в DMF (20 мл) добавляли MeOH (1,730 мл, 42,8 ммоль), а затем по каплям добавляли раствор трет-бутилата лития (5,1 г, 64,1 ммоль) в THF (30 мл) при 0°C. Полученную смесь медленно нагревали до 16°C и перемешивали в течение 2 часов. Затем к этой реакционной смеси добавляли (S)-1-ацетамидо-3-хлорпропан-2-ила ацетат (8,3 г, 42,8 ммоль) и перемешивали при 16°C в течение 12 часов. Смесь концентрировали с удалением THF и разбавляли EtOAc (20 мл), органический слой промывали H2O (25 мл x 3), сушили над Na2SO4, фильтровали и концентрировали досуха с получением неочищенного продукта, который очищали методом колоночной хроматографии на силикагеле (0-90% DCM/MeOH) с получением (S)-N-((3-(6-хлор-5-фторпиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида в виде твердого вещества. MS (ESI) m/z:287,8[M+H+].
Стадия E: Синтез (S)-N-((3-(5-фтор-6-(4-(метилтио)фенил)пиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида 40
К раствору (S)-N-((3-(6-хлор-5-фторпиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида (80,0 мг, 0,278 ммоль), (4-(метилтио)фенил)бороновой кислоты (46,7 мг, 0,278 ммоль) и трехосновного фосфата калия (118,0 мг, 0,556 ммоль) в THF (2 мл) добавляли предварительный каталитический нейтрализатор xphos 2-го поколения (21,9 мг, 0,028 ммоль) в атмосфере N2. Смесь дегазировали и повторно заполняли N2 (3x). Смесь перемешивали при 60°C в течение 16 часов. Реакционную смесь концентрировали с получением неочищенного продукта, который очищали методом преп-HPLC с обращенной фазой с получением (S)-N-((3-(5-фтор-6-(4-(метилтио)фенил)пиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида 40 в виде твердого вещества. LC/MS=376,1 [M+H+].
Пример 41
Синтез (S)-N-((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)циклопропанкарбоксамида.

К раствору циклопропанкарбоновой кислоты (14,2 мг, 0,165 ммоль) в DMF (2 мл) добавляли DIEA (53 мл, 0,301 ммоль) и HATU (62,9 мг, 0,165 ммоль). Смесь перемешивали при 20°C в течение 15 минут а затем добавляли (S)-5-(аминометил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-он (50,0 мг, 0,150 ммоль). Смесь перемешивали при 20°C в течение 16 часов. Реакционную смесь фильтровали и концентрировали с получением остатка, который очищали методом преп-HPLC с обращенной фазой с получением (S)-N-((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)циклопропанкарбоксамида в виде твердого вещества. LC/MS=401,1 [M+H+].
Пример 42
Синтез (S)-N-((3-(4-(5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамида.
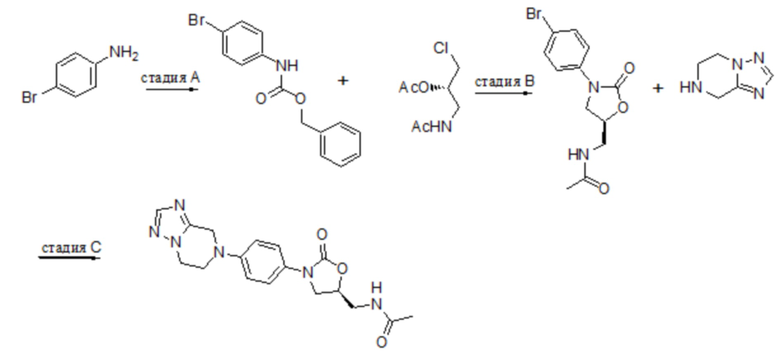
Стадия A: Синтез бензил(4-бромфенил)карбамата:
К раствору 4-броманилина (50,0 г, 291 ммоль) в ацетоне (500 мл) при 0°C добавляли бензилхлорформиат (54,5 г, 320 ммоль) с образованием белой взвеси. Затем медленно добавляли раствор бикарбоната натрия (581 мл, 581 ммоль). После добавления смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 16 часов. Органический растворитель удаляли при пониженном давлении, а осажденное твердое вещество собирали фильтрацией и промывали водой (200 мл Ч 2). Твердое вещество затем сушили с получением бензил(4-бромфенил)карбамата в виде твердого вещества.
Стадия B: Синтез (S)-N-((3-(4-бромфенил)-2-оксооксазолидин-5-ил)метил)ацетамида:
К раствору бензил(4-бромфенил)карбамата (10,0 г, 32,7 ммоль) в смеси DMF (40 мл) и MeOH (2,6 мл, 65,3 ммоль) добавляли раствор 2-метилпропан-2-олята лития (7,8 г, 98,0 ммоль) в THF (60 мл) при 0°C, а затем добавляли (S)-1-ацетамидо-3-хлорпропан-2-ила ацетат (12,6 г, 65,3 ммоль). Смесь нагревали до 20°C и перемешивали в течение 16 часов. Смесь концентрировали с получением остатка, который выливали в ледяную воду (100 мл), и твердое вещество осаждали. Смесь фильтровали и твердое вещество промывали 100 мл PE и сушили в вакууме с получением (S)-N-((3-(4-бромфенил)-2-оксооксазолидин-5-ил)метил)ацетамида в виде твердого вещества. MS (ESI) m/z:313,0 [M+H+]. 1H ЯМР (400MГц, DMSO-d6) д 8.30 - 8.22 (m, 1H), 7.60 - 7.54 (m, 2H), 7.54 - 7.48 (m, 2H), 4.72 (dd, J=6.0, 8.2 Гц, 1H), 4.10 (t, J=8.9 Гц, 1H), 3.73 (dd, J=6.6, 8.8 Гц, 1H), 3.41 (t, J=5.4 Гц, 2H), 1.83 (s, 3H). MS (ESI) m/z: 313,0, 315,0 [M+H+].
Стадия C: Синтез (S)-N-((3-(4-(5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамида
К раствору (S)-N-((3-(4-бромфенил)-2-оксооксазолидин-5-ил)метил)ацетамида (100,0 мг, 0,319 ммоль), 5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-a]пиразина (150,0 мг, 1,208 ммоль) и Cs2CO3 (312,0 мг, 0,958 ммоль) в 1,4-диоксане (2 мл) добавляли предварительный каталитический нейтрализатор ruphos 2-го поколения (12,4 мг, 0,016 ммоль) в атмосфере N2. Смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь концентрировали с получением остатка, который очищали методом преп-HPLC с обращенной фазой с получением (S)-N-((3-(4-(5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамида в виде твердого вещества. MS(ESI) m/z: 357,1 [M+H+].
Пример 43
Синтез (R)-5-((1H-1,2,4-триазол-1-ил)метил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-она.
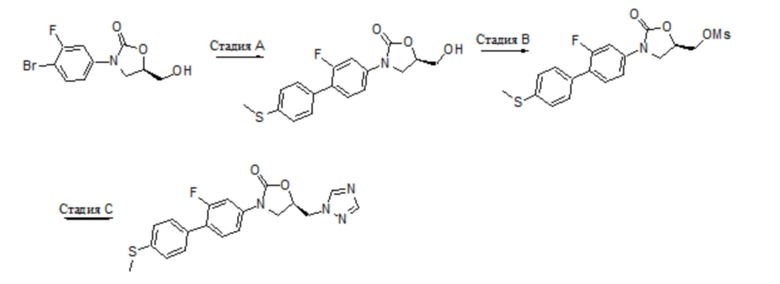
Стадия A: Синтез (R)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-5-(гидроксиметил)оксазолидин-2-она.
К раствору (R)-3-(4-бром-3-фторфенил)-5-(гидроксиметил)оксазолидин-2-она (5,0 г, 17,24 ммоль) и (4-(метилтио)фенил)бороновой кислоты (3,5 г, 20,68 ммоль) в THF (60 мл) добавляли водный фосфат калия (17,2 мл, 34,5 ммоль), а затем предварительный каталитический нейтрализатор xphos 2-го поколения (0,7 г, 0,862 ммоль) в атмосфере N2. Полученную смесь перемешивали при 60°C в течение 16 часов. Затем смесь фильтровали и концентрировали с получением остатка, который очищали методом хроматографии на силикагеле (20%-100% EtOAc в PE) с получением (R)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-5-(гидроксиметил)оксазолидин-2-она в виде твердого вещества. 1H ЯМР (400 MГц, DMSO-d6) д 7.66-7.32 (m, 7H), 5.25-5.22 (m, 1H), 4.74-4.70 (m, 1H), 4.13-4.08 (m, 1H), 3.87-3.83 (m, 1H), 3.70-3.65 (m, 1H), 3.58-3.54 (m, 1H), 2.47 (s, 3H).
Стадия B: (R)-(3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метила метансульфонат
К раствору (R)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-5-(гидроксиметил)оксазолидин-2-она (500,0 мг, 1,50 ммоль) в DCM (10 мл) добавляли DIEA (0,5 мл, 3,00 ммоль) и MsCl (0,2 мл, 2,25 ммоль) при 0°C. Полученную смесь перемешивали при 20°C в течение 2 часов. Реакционную смесь гасили водой (5 мл), а затем разбавляли DCM (20 мл), промывали водой (20 мл x 2), а затем солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта (R)-(3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метила метансульфоната в виде твердого вещества. Неочищенное вещество использовали на следующей стадии синтеза без дополнительной очистки.
Стадия C: Синтез (R)-5-((1H-1,2,4-триазол-1-ил)метил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-она.
К раствору 4H-1,2,4-триазола (12,6 мг, 0,18 ммоль) в DMF (4 мл) добавляли K2CO3 (50,4 мг, 0,36 ммоль) и (R)-(3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метила метансульфонат (50,0 мг, 0,12 ммоль). Полученную смесь перемешивали при 20°C в течение 24 часов. Реакционную смесь гасили насыщенным NH4Cl (5 мл), а затем разбавляли EtOAc (20 мл), промывали водой (20 мл x 2) и солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали с получением остатка, который очищали методом преп-HPLC с обращенной фазой с получением (R)-5-((1H-1,2,4-триазол-1-ил)метил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-она в виде твердого вещества. LC/MS, m/z: 385,0 [M+H+].
Следующие соединения получали с использованием способов, описанных в примере 43, с замещением соответствующих реактивов и/или реагентов.
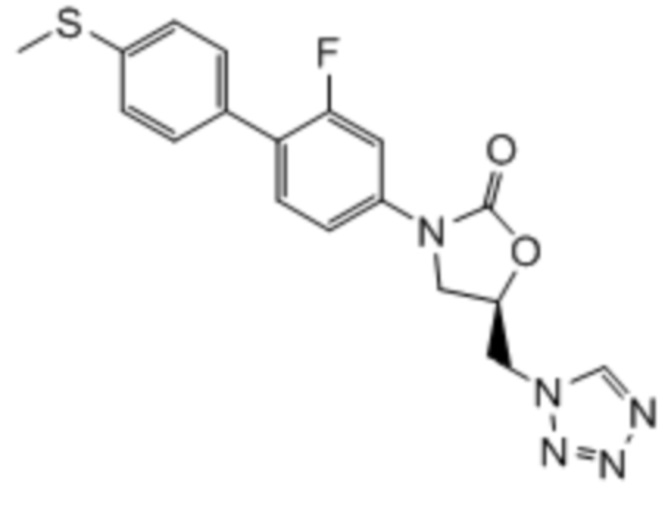
бифенил-4-ил]-5-(1H-тетразол-1-илметил)-1,3-оксазолидин-2-он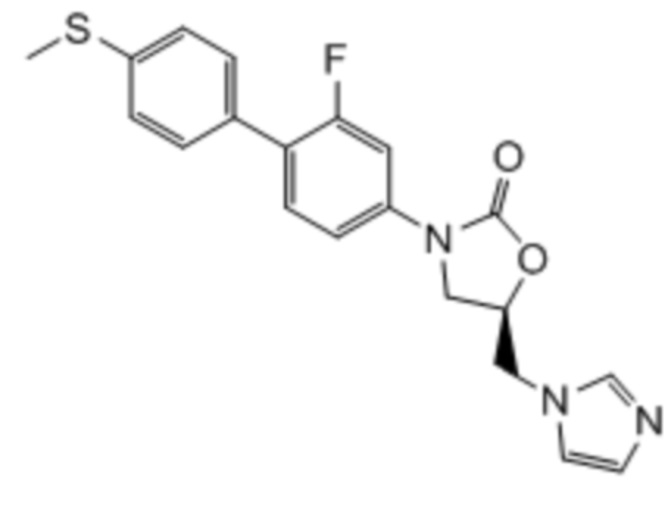
бифенил-4-ил]-5-(1H-имидазол-1-илметил)-1,3-оксазолидин-2-он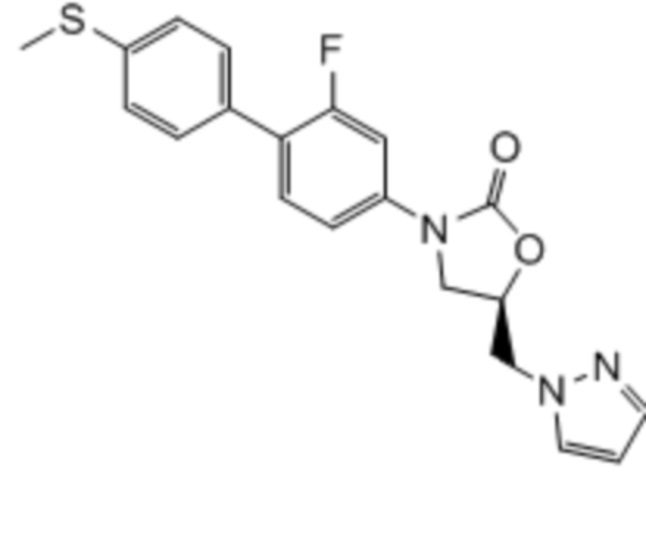
бифенил-4-ил]-5-(1H-пиразол-1-илметил)-1,3-оксазолидин-2-он
Пример 47
Синтез (S)-N-((3-(2,6-дифтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)ацетамида.
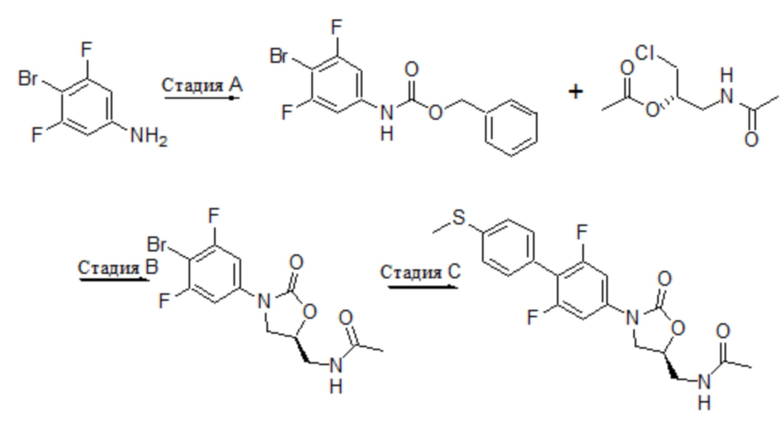
Стадия A: Синтез бензил(4-бром-3,5-дифторфенил)карбамата.
К перемешиваемому раствору 4-бром-3,5-дифторанилина (20 г, 96 ммоль) в ацетоне (200 мл) при 0°C добавляли бензилхлорформиат (18,04 г, 106 ммоль) с образованием белой взвеси. Затем частями добавляли бикарбонат натрия (192 мл, 192 ммоль). После добавления смесь оставляли нагреваться до 25°C и перемешивали в течение 16 часов. Органический растворитель удаляли при пониженном давлении, осажденное твердое вещество собирали фильтрацией и промывали водой (50 мл Ч 2). Твердое вещество затем сушили с получением бензил(4-бром-3,5-дифторфенил)карбамата в виде твердого вещества. 1HЯМР (CDCl3, 400 MГц) д 7.39-7.37 (m, 5H), 7.11-7.09 (dd, J=2.0, 6 Гц, 2H), 5.20 (s, 2H).
Стадия B: (S)-N-((3-(4-бром-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамид:
К раствору бензил(4-бром-3,5-дифторфенил)карбамата (8 г, 23,38 ммоль)) в смеси DMF (24 мл) и метанола (1,513 г, 47,2 ммоль) добавляли раствор 2-метилпропан-2-олята лития (5,62 г, 70,1 ммоль) в THF (48 мл). Полученную смесь охлаждали до 5°C, затем добавляли (S)-1-ацетамидо-3-хлорпропан-2-ила ацетат (9,06 г, 46,8 ммоль). Реакционную смесь перемешивали при 20°C в течение 16 часов, а затем гасили добавлением насыщенного NH4Cl (50,0 мл). Осажденное твердое вещество собирали, промывали водой (50 мл) и сушили с получением (S)-N-((3-(4-бром-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида в виде твердого вещества. MS (ESI) m/z: 348,8, 350,8 [M+H+].
Стадия C: (S)-N-((3-(2,6-дифтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)ацетамид 47 :
К раствору (S)-N-((3-(4-бром-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида (40,0 мг, 0,12 ммоль), (4-(метилтио)фенил)бороновой кислоты (28,9 мг, 0,17 ммоль), водного фосфата калия (0,23 мл, 0,23 ммоль) в THF (1 мл) добавляли предварительный каталитический нейтрализатор xphos 2-го поколения (4,5 мг, 5,73 мкмоль) в атмосфере N2. Смесь перемешивали при 60°C в течение 16 часов. Реакционную смесь фильтровали и концентрировали с получением остатка, который очищали методом преп-HPLC с обращенной фазой с получением (S)-N-((3-(2,6-дифтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)ацетамида в виде твердого вещества. LC/MS, m/z: 393,1 [M+H+].
Пример 48
Синтез (S)-5-(аминометил)-3-(6-(1,1-диоксидотиоморфолинo)-5-фторпиридин-3-ил)оксазолидин-2-она.
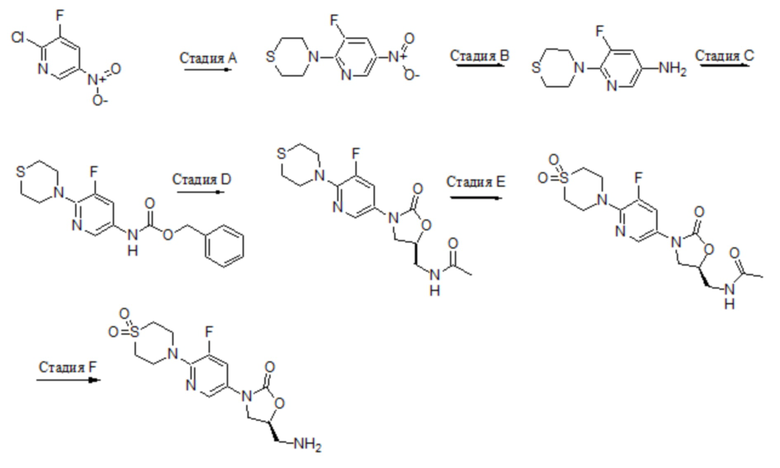
Стадия A: Синтез 4-(3-фтор-5-нитропиридин-2-ил)тиоморфолина.
К раствору 2-хлор-3-фтор-5-нитропиридина (3,0 г, 15,3 ммоль) в DMF (30 мл) добавляли тиоморфолин (1,6 г, 15,3 ммоль) и DIEA (5,3 мл, 30,6 ммоль). Смесь перемешивали при 60°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, а затем разбавляли водой (50 мл). Осажденное твердое вещество собирали фильтрацией и сушили с получением 4-(3-фтор-5-нитропиридин-2-ил)тиоморфолина в виде твердого вещества. 1HЯМР д 8.87 (s, 1H), 7.98 (dd, J=2.0, 13.6Гц, 1H), 4.15-4.12 (m, 4H), 2.76-2.74 (m, 4H).
Стадия B: Синтез 5-фтор-6-тиоморфолинoпиридин-3-амина.
К раствору 4-(3-фтор-5-нитропиридин-2-ил)тиоморфолина (4,0 г, 14,8 ммоль) в воде (20 мл) и HOAc (20 мл) добавляли железо (4,1 г, 74,0 ммоль). Полученную смесь перемешивали при 60°C в течение 4 часов. Реакционную смесь концентрировали, а затем разбавляли EtOAc (100 мл), промывали насыщенным NaHCO3 (80 мл x 2), а затем солевым раствором (80 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением 5-фтор-6-тиоморфолинoпиридин-3-амина в виде твердого вещества. Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Стадия C: Синтез бензил(5-фтор-6-тиоморфолинoпиридин-3-ил)карбамата.
К перемешиваемому раствору 5-фтор-6-тиоморфолинoпиридин-3-амина (3,1 г, 13,1 ммоль) в ацетоне (30 мл) при 0°C добавляли CBZ-Cl (2,2 мл, 15,7 ммоль) с образованием взвеси. Затем медленно добавляли раствор бикарбоната натрия (26,2 мл, 26,2 ммоль). После добавления смесь оставляли нагреваться до 20°C и перемешивали в течение 16 часов. Органический растворитель удаляли при пониженном давлении, а осажденное твердое вещество собирали фильтрацией, промывали водой (40 мл Ч 2) и сушили с получением бензил(5-фтор-6-тиоморфолинoпиридин-3-ил)карбамата в виде твердого вещества. 1HЯМР (CDCl3, 400 MГц) д 7.83 (s, 1H), 7.75-7.72 (m, 1H), 7.39-7.35 (m, 5H), 6.61 (s, 1H), 5.20 (s, 2H), 3.71-3.68 (m, 4H), 2.75-2.67 (m, 4H).
Стадия D: Синтез (S)-N-((3-(5-фтор-6-тиоморфолинoпиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида.
К раствору бензил(5-фтор-6-тиоморфолинoпиридин-3-ил)карбамата (4,1 г, 10,6 ммоль) в смеси DMF (15 мл) и MeOH (0,9 мл, 21,2 ммоль) добавляли раствор трет-бутилата лития (2,6 г, 31,9 ммоль) в THF (30 мл) при 0°C. Полученную смесь перемешивали при 20°C в течение 0,5 часа, а затем добавляли (S)-1-ацетамидо-3-хлорпропан-2-ила ацетат (4,1 г, 21,2 ммоль) при 20°C. Реакционную смесь перемешивали при 20°C в течение 16 часов. Смесь гасили концентрированной HCl (10 мл), а затем концентрировали с получением неочищенного продукта, который очищали методом колоночной хроматографии (SiO2, PE:EtOAc=1:1-DCM:MeOH=10:1) с получением (S)-N-((3-(5-фтор-6-тиоморфолинoпиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида в виде твердого вещества. 1HЯМР (CDCl3, 400 MГц) д 8.06 (d, J=2Гц, 1H), 7.90 (dd, J=2.4, 14.8Гц, 1H), 4.84-4.78 (m, 1H), 4.14-4.12 (m, 1H), 3.81-3.70 (m, 5H), 3.57-3.56 (m, 2H), 2.74-2.71 (m, 4H), 1.97 (s, 3H).
Стадия E: Синтез (S)-N-((3-(6-(1,1-диоксидотиоморфолинo)-5-фторпиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида.
К раствору (S)-N-((3-(5-фтор-6-тиоморфолинoпиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида (300 мг, 0,76 ммоль) в MeOH (3 мл) и воде (1,5 мл) добавляли оксон (703 мг, 1,14 ммоль) при 0°C. Смесь перемешивали при 20°C в течение 2 часов. Реакционную смесь концентрировали с получением неочищенного продукта, который очищали методом преп-HPLC с получением (S)-N-((3-(6-(1,1-диоксидотиоморфолинo)-5-фторпиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида в виде твердого вещества. 1HЯМР (DMSO-d6, 400 MГц) д 8.25-8.22 (m, 1H), 8.13 (s, 1H), 7.93 (dd, J=1.6, 14.8Гц, 1H), 4.77-4.72 (m, 1H), 4.11-4.07 (m, 1H), 3.84 (s, 4H), 3.73-3.69 (m, 1H), 3.41-3.39 (m, 2H), 3.20 (s, 4H), 1.82 (s, 3H). MS (ESI) m/z: 387.0 [M+H+].
Стадия F: (S)-5-(аминометил)-3-(6-(1,1-диоксидотиоморфолинo)-5-фторпиридин-3-ил)оксазолидин-2-он 48
К раствору (S)-N-((3-(6-(1,1-диоксидотиоморфолинo)-5-фторпиридин-3-ил)-2-оксооксазолидин-5-ил)метил)ацетамида (150 мг, 0,39 ммоль) в MeOH (2 мл) и воде (0,5 мл) добавляли HCl (0,5 мл, 6,09 ммоль). Смесь перемешивали при 70°C в течение 16 часов. Смесь концентрировали с получением неочищенного продукта, который очищали методом преп-HPLC с обращенной фазой с получением (S)-5-(аминометил)-3-(6-(1,1-диоксидотиоморфолинo)-5-фторпиридин-3-ил)оксазолидин-2-она 48 в виде твердого вещества. 1HЯМР (CDCl3, 400 MГц) д 8.21 (s, 3H), 8.11 (s, 1H), 7.92 (dd, J=2.0, 14.8Гц, 1H), 4.95-4.88 (m, 1H), 4.18-4.14 (m, 1H), 3.84-3.77 (m, 5H), 3.23-3.19 (m, 6H). MS (ESI) m/z: 345.0 [M+H+].
Примеры 49 и 50
Синтез (R)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-5-((изоксазол-3-илокси)метил)оксазолидин-2-она.
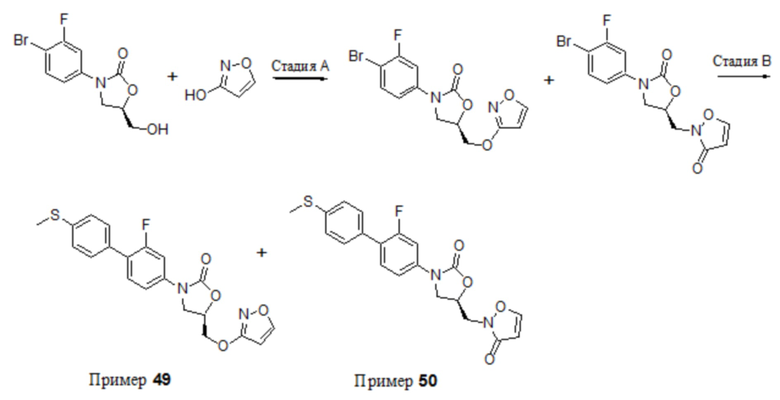
Стадия A: (R)-3-(4-бром-3-фторфенил)-5-((изоксазол-3-илокси)метил)оксазолидин-2-он и (R)-2-((3-(4-бром-3-фторфенил)-2-оксооксазолидин-5-ил)метил)изоксазол-3(2H)-он
К раствору трифенилфосфина (2,2 г, 8,3 ммоль) в сухом THF (50 мл) добавляли DIAD (1,6 мл, 8,3 ммоль) и полученную смесь перемешивали в течение 0,5 часа. К этой реакционной смеси добавляли изоксазол-3-ол (645 мг, 7,6 ммоль) и смесь перемешивали в течение 0,5 часа, а затем добавляли (R)-3-(4-бром-3-фторфенил)-5-(гидроксиметил)оксазолидин-2-он (2,0 г, 6,9 ммоль). Полученную смесь перемешивали при 20°C в течение 16 часов. Реакционную смесь разбавляли EtOAc (200 мл), промывали соляной кислотой (1M, 50 мл), затем водой (50 мл) и солевым раствором (50 мл), сушили над Na2SO4 и фильтровали. Фильтрат выпаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии (SiO2, PE-EtOAc) с получением (R)-3-(4-бром-3-фторфенил)-5-((изоксазол-3-илокси)метил)оксазолидин-2-она в виде твердого вещества (основной продукт) и (R)-2-((3-(4-бром-3-фторфенил)-2-оксооксазолидин-5-ил)метил)изоксазол-3(2H)-она в виде твердого вещества (побочный продукт).
(R)-3-(4-бром-3-фторфенил)-5-((изоксазол-3-илокси)метил)оксазолидин-2-он 1H ЯМР (CDCl3, 400 MГц) д 8.16 (d, J=1.6 Гц, 1H), 7.66 (dd, J=7.2, 11.6 Гц, 1H), 7.39-7.58 (m, 4H), 7.17 (dd, J=1.6, 8.8 Гц, 1H), 6.00 (d, J=1.6 Гц, 1H), 4.89–5.11 (m, 2H), 4.43-4.63 (m, 2H), 4.15 (t, J=8.8 Гц, 1H), 3.96 (dd, J=6.4, 8.8 Гц, 1H).
(R)-2-((3-(4-бром-3-фторфенил)-2-оксооксазолидин-5-ил)метил)изоксазол-3(2H)-он 1H ЯМР (CDCl3, 400 MГц) д 7.88 (d, J=2.4 Гц, 1H), 7.40-7.59 (m, 3H), 7.04-7.17 (m, 1H), 5.82 (d, J=2.4 Гц, 1H), 4.91–5.02 (m, 1H), 4.17-4.38 (m, 2H), 4.11 (t, J=8.8 Гц, 1H), 3.96 (dd, J=5.6, 9.2 Гц, 1H).
Стадия B: (R)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-5-((изоксазол-3-илокси)метил)оксазолидин-2-он:
Предварительный каталитический нейтрализатор xphos 2-го поколения (66,1 мг, 0,08 ммоль) добавляли к смеси (R)-3-(4-бром-3-фторфенил)-5-((изоксазол-3-илокси)метил)оксазолидин-2-она (300,0 мг, 0,84 ммоль), (4-(метилтио)фенил)бороновой кислоты (155 мг, 0,924 ммоль), трехосновного фосфата калия (357,0 мг, 1,68 ммоль) в THF (5 мл) и воде (1,5 мл) в атмосфере N2. Смесь перемешивали при 70°C в течение 16 часов. При помощи LCMS наблюдали израсходование большей части исходного вещества и образовывалось 50% требуемого продукта. Смесь охлаждали, добавляли воду (10 мл) и смесь экстрагировали этилацетатом (20 мл Ч 2). Объединенные органические фракции промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали методом преп-HPLC с обращенной фазой с получением (R)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-5-((изоксазол-3-илокси)метил)оксазолидин-2-она, соединения примера 49, в виде твердого вещества. 1H ЯМР (CDCl3, 400 MГц) д 8.71 (d, J=1.6 Гц, 1H), 7.64-7.58 (m, 2H), 7.51-7.47 (m, 3H), 7.36 (d, J=8.4 Гц, 2H), 6.40 (d, J=1.6 Гц, 1H), 5.12-5.11 (m, 1H), 4.53-4.48 (m, 2H), 4.25-4.23 (m, 1H), 4.00-3.96 (m, 1H), 2.50 (s, 3H). MS (ESI) m/z: 401,0 [M+H+].
(R)-2-((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)изоксазол-3(2H)-он, соединение примера 50, получали из (R)-2-((3-(4-бром-3-фторфенил)-2-оксооксазолидин-5-ил)метил)изоксазол-3(2H)-она на основе протокола, описанного на стадии B в способе выше, в виде твердого вещества. (CDCl3, 400 MГц) д 8.56 (d, J=2.4 Гц, 1H), 7.58-7.48 (m, 4H), 7.36-7.34 (m, 3H), 5.98 (d, J=2.4 Гц, 1H), 4.91–5.02 (m, 1H), 4.27-4.20 (m, 3H), 3.90-3.88 (m, 1H), 2.50 (s, 3H). MS (ESI) m/z: 401,1 [M+H+].
Пример 54
Синтез (R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(3-фтор-4-(6-(пиперазин-1-ил)пиридин-3-ил)фенил)оксазолидин-2-она.
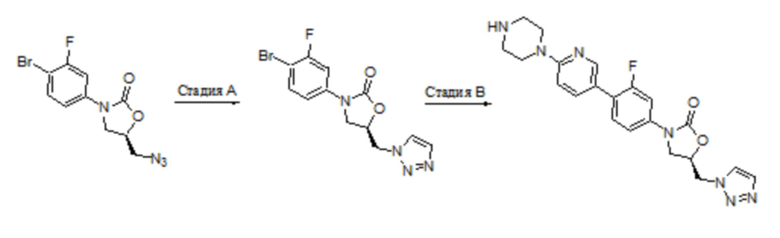
Стадия A: Синтез (R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-бром-3-фторфенил)оксазолидин-2-она.
2,5-Норборнадиен (13,2 г, 143 ммоль) добавляли к раствору (R)-5-(азидометил)-3-(4-бром-3-фторфенил)оксазолидин-2-она (9,0 г, 28,6 ммоль) в диоксане (200 мл), затем смесь перемешивали при 110°C в течение 17 часов. Затем реакционную смесь концентрировали, а неочищенный продукт очищали методом колоночной хроматографии с применением силикагеля (SiO2, DCM-DCM:MeOH=95:5) с получением (R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-бром-3-фторфенил)оксазолидин-2-она в виде твердого вещества, а также 2,0 г неочищенного продукта (<80% чистоты). MS (ESI) m/z: 382,1, 384,1[M+ACN+H+].
Стадия B: (R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(3-фтор-4-(6-(пиперазин-1-ил)пиридин-3-ил)фенил)оксазолидин-2-он
К раствору (R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-бром-3-фторфенил)оксазолидин-2-она (50,0 мг, 0,15 ммоль), (6-(пиперазин-1-ил)пиридин-3-ил)бороновой кислоты (36,4 мг, 0,18 ммоль) и водного трехосновного фосфата калия (1M, 0,23 мл, 0,29 ммоль) в THF (1 мл) добавляли предварительный каталитический нейтрализатор xphos 2-го поколения (5,8 мг, 7,33 мкмоль) в атмосфере N2. Смесь перемешивали при 60°C в течение 16 часов. Смесь фильтровали и концентрировали с получением остатка, который очищали методом преп-HPLC с обращенной фазой с получением (R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(3-фтор-4-(6-(пиперазин-1-ил)пиридин-3-ил)фенил)оксазолидин-2-она, соединения примера 54, в виде твердого вещества. MS (ESI) m/z: 424,2 [M+H+].
Следующее соединение получали с использованием способов, описанных в примере 54, с замещением соответствующих реактивов и/или реагентов.
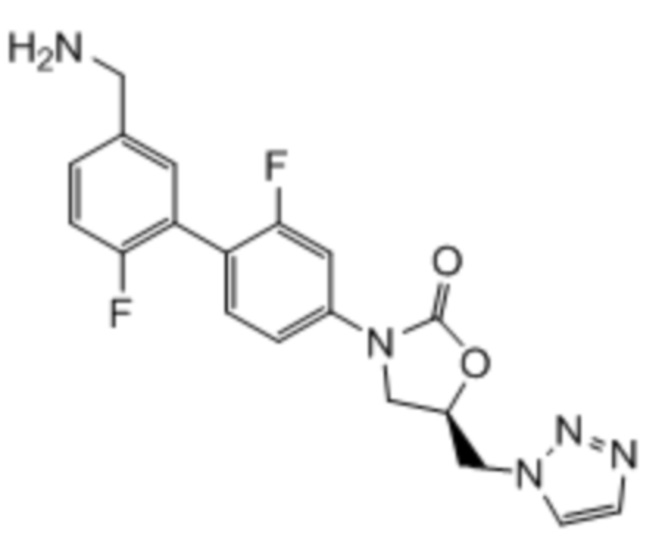
Пример 56
Синтез (S)-5-(аминометил)-3-(3-фтор-4-(тиофен-2-ил)фенил)оксазолидин-2-она
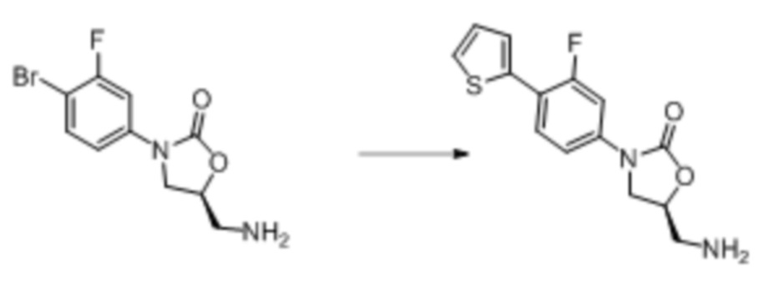
К раствору (S)-5-(аминометил)-3-(4-бром-3-фторфенил)оксазолидин-2-она (70,0 мг, 0,24 ммоль), трибутил(тиофен-2-ил)станнана (108,0 мг, 0,29 ммоль) и трехосновного фосфата калия (0,48 мл, 0,48 ммоль) в THF (2 мл) добавляли предварительный каталитический нейтрализатор xphos 2-го поколения (19,0 мг, 0,02 ммоль) в атмосфере N2. Смесь дегазировали и повторно трижды заполняли N2. Реакционную смесь перемешивали при 60°C в течение 16 часов. Реакционную смесь концентрировали с получением неочищенного продукта, который очищали методом преп-HPLC с обращенной фазой с получением (S)-5-(аминометил)-3-(3-фтор-4-(тиофен-2-ил)фенил)оксазолидин-2-она, соединения примера 54, в виде твердого вещества. MS (ESI) m/z: 293,0 [M+H+].
Пример 57
Синтез (S)-4'-(5-((2-аминоацетамидо)метил)-2-оксооксазолидин-3-ил)-2'-фтор-[1,1'-бифенил]-4-карбоксамида.
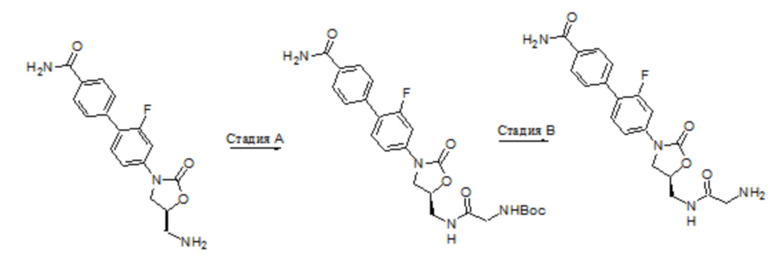
Стадия A: Синтез (S)-трет-бутил(2-(((3-(4'-карбамоил-2-фтор-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)амино)-2-оксоэтил)карбамата.
К раствору 2-((трет-бутоксикарбонил)амино)уксусной кислоты (35,9 мг, 0,20 ммоль) в DMF (1,5 мл) добавляли DIEA (0,05 мл, 0,27 ммоль) и HATU (78,0 мг, 0,20 ммоль). Смесь перемешивали при 20°C в течение 10 минут, затем добавляли (S)-4'-(5-(аминометил)-2-оксооксазолидин-3-ил)-2'-фтор-[1,1'-бифенил]-4-карбоксамид, соединение примера 2 (50,0 мг, 0,14 ммоль). Полученную смесь перемешивали при 20°C в течение 16 часов. Реакционную смесь фильтровали и концентрировали с получением остатка, который очищали методом преп-HPLC с обращенной фазой с получением (S)-трет-бутил(2-(((3-(4'-карбамоил-2-фтор-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)амино)-2-оксоэтил)карбамата в виде твердого вещества.
Стадия B: (S)-4'-(5-((2-аминоацетамидо)метил)-2-оксооксазолидин-3-ил)-2'-фтор-[1,1'-бифенил]-4-карбоксамид
К раствору (S)-трет-бутил(2-(((3-(4'-карбамоил-2-фтор-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)амино)-2-оксоэтил)карбамата (70,0 мг, 0,13 ммоль) в DCM (5 мл) добавляли TFA (0,1 мл, 1,30 ммоль). Полученную смесь перемешивали при 20°C в течение 16 часов. Реакционную смесь фильтровали и концентрировали с получением остатка, который очищали методом преп-HPLC с обращенной фазой с получением (S)-4'-(5-((2-аминоацетамидо)метил)-2-оксооксазолидин-3-ил)-2'-фтор-[1,1'-бифенил]-4-карбоксамида в виде твердого вещества. MS (ESI) m/z: 386,9 [M+H+].
Пример 58
Синтез (S)-трет-бутил(2-(((3-(4'-карбамоил-2-фтор-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)амино)-2-оксоэтил)карбамата.
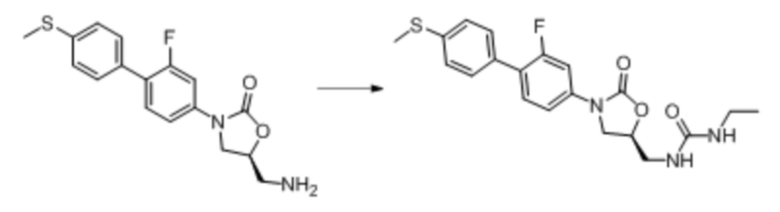
К раствору (S)-5-(аминометил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-она (50,0 мг, 0,15 ммоль) в DMF (2 мл) добавляли DIEA (0,05 мл, 0,30 ммоль) и изоцианатоэтан (12,8 мг, 0,18 ммоль). Полученную смесь перемешивали при 20°C в течение 16 часов. Реакционную смесь концентрировали с получением остатка, который очищали методом преп-HPLC с обращенной фазой с получением (S)-1-этил-3-((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)мочевины, соединения примера 56, в виде твердого вещества. MS (ESI) m/z: 403,9 [M+H+].
Пример 59
Синтез (S)-5-(аминометил)-3-(3,5-дифтор-4-морфолинофенил)оксазолидин-2-она (соединение примера 59).
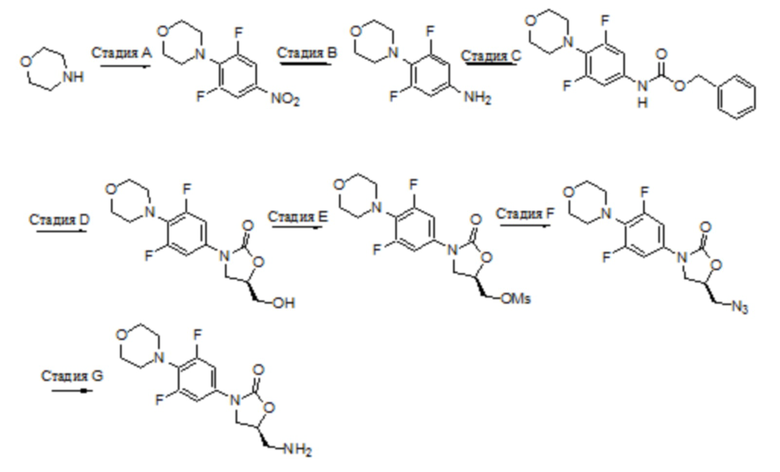
Стадия A: Синтез 4-(2,6-дифтор-4-нитрофенил)морфолина.
К раствору 1,2,3-трифтор-5-нитробензола (1,6 г, 8,75 ммоль) в DMF (10 мл) добавляли карбонат калия (1,3 г, 9,63 ммоль) и морфолин (0,8 г, 8,75 ммоль) при 20°C. Реакционную смесь перемешивали при 50°C в течение 10 часов. Реакционную смесь выливали в EtOAc (50 мл) и промывали солевым раствором (50 мл × 3). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением 4-(2,6-дифтор-4-нитрофенил)морфолина в виде твердого вещества. 1H ЯМР (CDCl3, 400 MГц) д 7.22 (m, 4H), 4.98 (br s, 2H), 3.97 (m, 2H), 3.00 (m, 2H), 1.59 (m, 4H). MS (ESI) m/z: 244,8 [M+H+].
Стадия B: Синтез 3,5-дифтор-4-морфолиноанилина.
К раствору 4-(2,6-дифтор-4-нитрофенил)морфолина (2,3 г, 9,42 ммоль) в MeOH (25 мл) добавляли Pd-C (10% масс., 0,3 г, 2,82 ммоль) при 20°C. Реакционную смесь перемешивали в атмосфере H2 при 20°C в течение 10 часов. Реакционную смесь фильтровали и фильтрат концентрировали с получением 3,5-дифтор-4-морфолиноанилина в виде твердого вещества. 1H ЯМР (CDCl3, 400 MГц) д 6.17 (m, 2H), 3.79 (m, 4H), 3.07 (m, 4H). MS (ESI) m/z: 214,9 [M+H+].
Стадия C: Синтез бензил(3,5-дифтор-4-морфолинофенил)карбамата
К раствору 3,5-дифтор-4-морфолиноанилина (1,8 г, 8,40 ммоль) в ацетоне (20 мл) добавляли бензилхлорформиат (2,2 г, 12,60 ммоль) при 20°C, а затем раствор гидрокарбоната натрия (1,4 г, 16,81 ммоль) в воде (20 мл). Реакционную смесь перемешивали при 20°C в течение 10 часов, а затем выпаривали и растворяли в EtOAc (50 мл). Органическую фазу промывали водой (50 мл × 3), сушили над Na2SO4 и концентрировали с получением бензил(3,5-дифтор-4-морфолинофенил)карбамата в виде твердого вещества. 1H ЯМР (CDCl3, 400 MГц) д 7.37 (m, 7H), 6.96 (d, J=10.4 Гц, 2H), 5.19 (s, 2H), 4.70 (s, 1H), 3.79 (m, 4H), 3.13 (br s, 4H). MS (ESI) m/z: 349,3[M+H+].
Стадия D: Синтез (R)-3-(3,5-дифтор-4-морфолинофенил)-5-(гидроксиметил)оксазолидин-2-она
К раствору бензил(3,5-дифтор-4-морфолинофенил)карбамата (2,1 г, 6,03 ммоль) в THF (30 мл) добавляли 2-метилпропан-2-олят лития (1,4 г, 18,09 ммоль). Реакционную смесь перемешивали при 20°C в течение 0,5 часа, а затем добавляли (R)-оксиран-2-илметила бутират (1,7 г, 12,06 ммоль). Полученную смесь перемешивали при 20°C в течение 10 часов. Растворитель удаляли, а неочищенный продукт очищали методом колоночной хроматографии (SiO2, EtOAc:PE=2:1) с получением (R)-3-(3,5-дифтор-4-морфолинофенил)-5-(гидроксиметил)оксазолидин-2-она в виде твердого вещества. 1H ЯМР (CDCl3, 400 MГц) д 7.08 (m, 2H), 4.73 (m, 1H), 3.97 (m, 3H), 3.80 (m, 4H), 3.72 (dd, J=3.5, 12.9 Гц, 1H), 3.22 (br s, 2H), 3.13 (br s, 4H). MS (ESI) m/z: 314,9 [M+H+].
Стадия E: Синтез (R)-(3-(3,5-дифтор-4-морфолинофенил)-2-оксооксазолидин-5-ил)метилметансульфоната.
К раствору (R)-3-(3,5-дифтор-4-морфолинофенил)-5-(гидроксиметил)оксазолидин-2-она (350 мг, 1,11 ммоль) в DCM (3 мл) добавляли метансульфохлорид (191 мг, 1,67 ммоль) и триэтиламин (225 мг, 2,227 ммоль) при 0°C. Реакционную смесь перемешивали при 23°C в течение 10 часов. Смесь промывали водой (20 мл × 3). Затем органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением (R)-(3-(3,5-дифтор-4-морфолинофенил)-2-оксооксазолидин-5-ил)метилметансульфоната в виде твердого вещества. MS (ESI) m/z: 392,9 [M+H+].
Стадия F: Синтез (R)-5-(азидометил)-3-(3,5-дифтор-4-морфолинофенил)оксазолидин-2-она.
К раствору (R)-(3-(3,5-дифтор-4-морфолинофенил)-2-оксооксазолидин-5-ил)метилметансульфоната (100,0 мг, 0,26 ммоль) в DMF (1 мл) добавляли азид натрия (21,5 мг, 0,33 ммоль). Реакционную смесь перемешивали при 70°C в течение 5 часов. Смесь разбавляли EtOAc (20 мл), промывали солевым раствором (15 мл × 3). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением (R)-5-(азидометил)-3-(3,5-дифтор-4-морфолинофенил)оксазолидин-2-она в виде масла. MS (ESI) m/z: 339,9 [M+H+].
Стадия G: Синтез (S)-5-(аминометил)-3-(3,5-дифтор-4-морфолинофенил)оксазолидин-2-она.
К раствору (R)-5-(азидометил)-3-(3,5-дифтор-4-морфолинофенил)оксазолидин-2-она (86 мг, 0,23 ммоль) в смеси THF (2 мл) и воды (0,1 мл) добавляли трифенилфосфин (120 мг, 0,46 ммоль). Реакционную смесь перемешивали при 70°C в течение 10 часов. Затем растворитель удаляли под вакуумом и к остатку добавляли EtOAc (20 мл). Органическую фазу промывали солевым раствором (15 мл × 3), сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали методом преп-HPLC с обращенной фазой с получением (S)-5-(аминометил)-3-(3,5-дифтор-4-морфолинофенил)оксазолидин-2-она в виде твердого вещества. 1H ЯМР (CDCl3, 400 MГц) д 7.22 (m, 2H), 4.2 (t, J=9.2 Гц, 1H), 3.77 (m, 4H), 3.35 (m, 3H), 3.11 (br s, 3H). MS (ESI) m/z: 313,9 [M+H+].
Пример 60
Синтез (S)-N-((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)-2,2-диметилгидразинкарбоксамида (соединение примера 60)
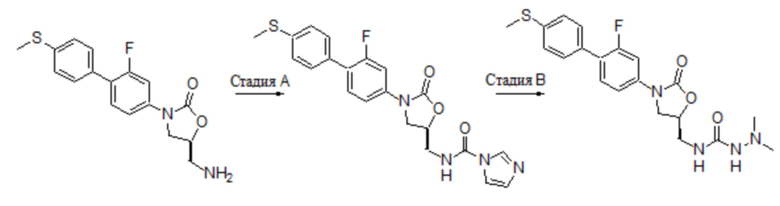
Стадия A: Синтез (S)-N-((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)-1H-имидазол-1-карбоксамида.
К раствору (S)-5-(аминометил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-она (350 мг, 1,05 ммоль) в DMF (5 мл) добавляли DIEA (817 мг, 6,32 ммоль) и CDI и полученную смесь перемешивали при 20°C в течение 16 часов. Неочищенное вещество использовали сразу на следующей стадии. MS (ESI) m/z: 426,9[M+H+].
Стадия B: Синтез (S)-N-((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)-2,2-диметилгидразинкарбоксамида.
К раствору (S)-N-((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)-1H-имидазол-1-карбоксамида (100 мг, 0,23 ммоль) в DMF (2 мл) добавляли 1,1-диметилгидразин (85 мг, 1,41 ммоль) и полученную смесь перемешивали при 40°C в течение 12 часов. Реакционную смесь выпаривали досуха и неочищенный продукт очищали методом преп-HPLC с обращенной фазой с получением (S)-N-((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)-2,2-диметилгидразинкарбоксамида в виде твердого вещества. MS (ESI) m/z:441,0 [M+Na+].
Пример 62
Синтез (S)-метил((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)карбамата (соединение примера 62).
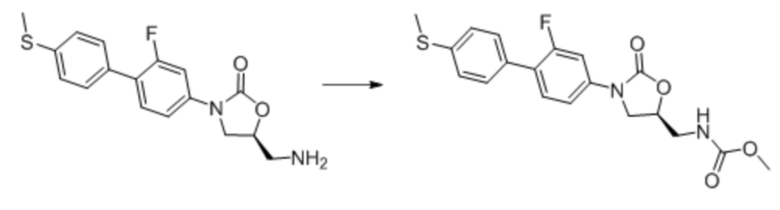
К раствору (S)-5-(аминометил)-3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)оксазолидин-2-она в DMF (2 мл) добавляли DIEA (0,08 мл, 0,451 ммоль) и метилхлорформиат в атмосфере N2. Полученную смесь дегазировали и повторно трижды заполняли N2. Реакционную смесь перемешивали при 15°C в течение 16 часов, затем разбавляли водой (5 мл) и экстрагировали EtOAc (10 мл x3). Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали методом преп-HPLC с обращенной фазой с получением (S)-метил((3-(2-фтор-4'-(метилтио)-[1,1'-бифенил]-4-ил)-2-оксооксазолидин-5-ил)метил)карбамата в виде твердого вещества. LC/MS:MS (ESI) m/z: 391,1 [M+H+]
Следующее соединение получали с применением способов, описанных в примере 62, с замещением соответствующих реактивов и/или реагентов.
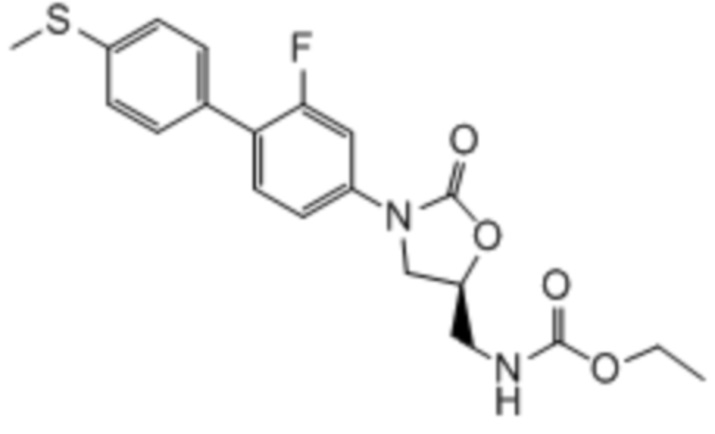
бифенил-4-ил]-2-оксо-1,3-оксазолидин-5-ил}метил)карбамат
Пример 64
Синтез (5R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(2-фтор-4'-(S-метилсульфонимидоил)-[1,1'-бифенил]-4-ил)оксазолидин-2-она (соединение 64).
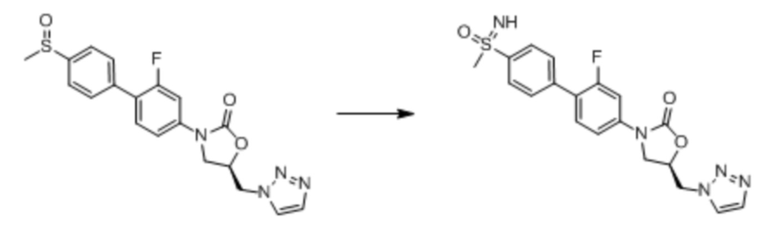
К раствору (5R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(2-фтор-4'-(метилсульфинил)-[1,1'-бифенил]-4-ил)оксазолидин-2-она (40,0 мг, 0,10 ммоль) (обычно полученного с применением способа примера 54) и азида натрия (9,7 мг, 0,15 ммоль) в CHCl3 (2 мл) добавляли H2SO4 (1 мкл, 0,02 ммоль). Смесь перемешивали при 45°C в течение 5 часов. Смесь гасили насыщенным Na2CO3(1 мл). Смесь концентрировали и очищали методом преп-HPLC с обращенной фазой с получением (5R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(2-фтор-4'-(S-метилсульфонимидоил)-[1,1'-бифенил]-4-ил)оксазолидин-2-она в виде твердого вещества. 1H ЯМР (400MГц, CD3OD) д 8.40 (s, 1H), 8.25 (d, J=8.4 Гц, 2H), 8.14 (s, 1H), 8.00 (d, J=7.9 Гц, 2H), 7.69-7.59 (m, 2H), 7.42 (d, J=7.9 Гц, 1H), 5.25 (br s., 1H), 5.10-5.00 (m, 2H), 4.38 (t, J=8.9 Гц, 1H), 4.08 (dd, J=5.3, 8.6 Гц, 1H), 3.93 (s, 3H). MS (ESI) m/z: 416.2 [M+H+].
Пример 65
Биологические анализы
Анализ роста Mycobacterium tuberculosis (Mtb)
Ингибирование роста Mycobacterium tuberculosis (Mtb) оценивали на двух in vivo-соответствующих источниках углерода - глюкозе и холестерине, при pH 6,8. Для глюкозы в качестве источника углерода среда состояла из бульона Middlebrook 7H9, дополненного 4 г/л глюкозы, 0,08 г/л NaCl, 5 г/л BSA фракции V и 0,05% тилоксапола. Для холестерина в качестве источника углерода среда состояла из бульона Middlebrook 7H9, дополненного 97 мг/л холестерина, 0,08 г/л NaCl, 5 г/л BSA фракции V и 0,05% тилоксапола. Mtb, экспрессирующую зеленый флуоресцентный белок (Mtb-GFP; H37Rv pMSP12::GFP), предварительно адаптировали для роста на соответствующем источнике углерода в основе бульона Middlebrook 7H9, дополненной альбумином бычьей сыворотки и тилоксаполом, перед скринингом. Бактерии распределяли в 384-луночные микротитровальные планшеты при приблизительно 2 Ч 104 активно растущих клеток в 24-мкл объемах на лунку. В микротитровальные планшеты предварительно распределяли 0,2 мкл соединения, диметилсульфоксида (отрицательный контроль) или рифампицина (25 мкM; положительный контроль). На клетки воздействовали соединениями с 2-кратными серийными разведениями от 50 мкM до 0,049 мкM. В некоторых экспериментах соединения тестировали при более низких концентрациях. Ингибирование роста оценивали после 7-суточного периода роста путем измерения флуоресценции с использованием спектрофотометра. В лунках отрицательного контроля клетки по-прежнему активно росли на момент считывания. Самую низкую концентрацию тестируемого соединения, необходимую для ингибирования 95% роста бактерий, определяли как MITC95. Все исследования выполняли в лаборатории BSL3.
Анализ роста Staphylococcus aureus
Ингибирование роста Staphylococcus aureus (устойчивого к метициллину, MB5393, COL) оценивали в бульоне Мюллера-Хинтона II со стандартизированным содержанием катионов (CAMHB) в 384-луночных микротитровальных планшетах. Бактерии распределяли в микротитровальные планшеты при приблизительно 5 Ч 105 активно растущих клеток в 49-мкл объемах на лунку. На клетки воздействовали соединениями с 2-кратными серийными разведениями от 200 мкM до 0,195 мкM, а рост сравнивали с контролем без лекарственного средства. Разведения соединений готовили в 100% диметилсульфоксиде и добавляли в микротитровальные планшеты в объемах 1 мкл на лунку. Контрольные лунки с полным ростом содержали диметилсульфоксид и бактерии. Контрольные лунки с отсутствием роста содержали диметилсульфоксид и CAMHB, но не бактерии. Ингибирование роста оценивали после 22-часового периода роста путем измерения оптической плотности с использованием спектрофотометра. Самую низкую концентрацию тестируемого соединения, необходимую для ингибирования 95% роста бактерий, определяли как MITC95. Все исследования выполняли в лаборатории BSL2.
Анализ синтеза митохондриального белка
Ингибирование синтеза митохондриального белка оценивали в клетках HepG2 путем сравнения содержаний двух субъединиц комплексов ферментов окислительного фосфорилирования, субъединицы I комплекса IV (COX-I) и 70-кДа субъединицы комплекса II (SDH-A). COX-I кодируется митохондриальной ДНК, а SDH-A кодируется ядерной ДНК. Клетки HepG2 высевали в 96-луночные покрытые коллагеном планшеты при 8000 клеток на лунку и подвергали соединениям с 2-кратными серийными разведениями от 100 мкM до 6,25 мкM. Микротитровальные планшеты инкубировали на протяжении приблизительно 5 репликативных циклов (4 суток) перед оцениванием содержаний белков с использованием набора, как предписывалось изготовителем (ab110217 MitoBiogenesis In Cell ELISA Kit, Abcam, Cambridge, MA). Ингибирование синтеза митохондриального белка выражали как отношение содержаний COX-1 к SDH-A.
Анализ роста Mycobacterium tuberculosis (Mtb)
Ингибирование роста Mycobacterium tuberculosis (Mtb) оценивали на двух in vivo-соответствующих источниках углерода - глюкозе и холестерине, при pH 6,8. Для глюкозы в качестве источника углерода среда состояла из бульона Middlebrook 7H9, дополненного 4 г/л глюкозы, 0,08 г/л NaCl, 5 г/л BSA фракции V и 0,05% тилоксапола. Для холестерина в качестве источника углерода среда состояла из бульона Middlebrook 7H9, дополненного 97 мг/л холестерина, 0,08 г/л NaCl, 5 г/л BSA фракции V и 0,05% тилоксапола. Mtb, экспрессирующую зеленый флуоресцентный белок (Mtb-GFP; H37Rv pMSP12::GFP), предварительно адаптировали для роста на соответствующем источнике углерода в основе бульона Middlebrook 7H9, дополненной альбумином бычьей сыворотки и тилоксаполом, перед скринингом. Бактерии распределяли в 384-луночные микротитровальные планшеты при приблизительно 2 Ч 104 активно растущих клеток в 24-мкл объемах на лунку. В микротитровальные планшеты предварительно распределяли 0,2 мкл соединения, диметилсульфоксида (отрицательный контроль) или рифампицина (25 мкM; положительный контроль). На клетки воздействовали соединениями с 2-кратными серийными разведениями от 50 мкM до 0,049 мкM. В некоторых экспериментах соединения тестировали при более низких концентрациях. Ингибирование роста оценивали после 7-суточного периода роста путем измерения флуоресценции с использованием спектрофотометра. В лунках отрицательного контроля клетки по-прежнему активно росли на момент считывания. Самую низкую концентрацию тестируемого соединения, необходимую для ингибирования 95% роста бактерий, определяли как MITC95. Все исследования выполняли в лаборатории BSL3.
Анализ роста Staphylococcus aureus
Ингибирование роста Staphylococcus aureus (устойчивого к метициллину, MB5393, COL) оценивали в бульоне Мюллера-Хинтона II со стандартизированным содержанием катионов (CAMHB) в 384-луночных микротитровальных планшетах. Бактерии распределяли в микротитровальные планшеты при приблизительно 5 Ч 105 активно растущих клеток в 49-мкл объемах на лунку. На клетки воздействовали соединениями с 2-кратными серийными разведениями от 200 мкM до 0,195 мкM, а рост сравнивали с контролем без лекарственного средства. Разведения соединений готовили в 100% диметилсульфоксиде и добавляли в микротитровальные планшеты в объемах 1 мкл на лунку. Контрольные лунки с полным ростом содержали диметилсульфоксид и бактерии. Контрольные лунки с отсутствием роста содержали диметилсульфоксид и CAMHB, но не бактерии. Ингибирование роста оценивали после 22-часового периода роста путем измерения оптической плотности с использованием спектрофотометра. Самую низкую концентрацию тестируемого соединения, необходимую для ингибирования 95% роста бактерий, определяли как MITC95. Все исследования выполняли в лаборатории BSL2.
Анализ синтеза митохондриального белка
Ингибирование синтеза митохондриального белка оценивали в клетках HepG2 путем сравнения содержаний двух субъединиц комплексов ферментов окислительного фосфорилирования, субъединицы I комплекса IV (COX-I) и 70-кДа субъединицы комплекса II (SDH-A). COX-I кодируется митохондриальной ДНК, а SDH-A кодируется ядерной ДНК. Клетки HepG2 высевали в 96-луночные покрытые коллагеном планшеты при 8000 клеток на лунку и подвергали соединениям с 2-кратными серийными разведениями от 100 мкM до 6,25 мкM. Микротитровальные планшеты инкубировали на протяжении приблизительно 5 репликативных циклов (4 суток) перед оцениванием содержаний белков с использованием набора, как предписывалось изготовителем (ab110217 MitoBiogenesis In Cell ELISA Kit, Abcam, Cambridge, MA). Ингибирование синтеза митохондриального белка выражали как отношение содержаний COX-1 к SDH-A.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МЛЕКОПИТАЮЩИХ | 2019 |
|
RU2798336C2 |
| ПРОИЗВОДНЫЕ 5-АМИНОЦИКЛИЛМЕТИЛОКСАЗОЛИДИН-2-ОНА | 2008 |
|
RU2492169C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
| ТРИЦИКЛИЧЕСКИЕ ОКСАЗОЛИДИНОНОВЫЕ АНТИБИОТИЧЕСКИЕ СОЕДИНЕНИЯ | 2009 |
|
RU2530884C2 |
| ОКСАЗОЛИДИНИЛОВЫЕ АНТИБИОТИКИ | 2009 |
|
RU2516701C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2563634C2 |
| БИЦИКЛИЧЕСКИЕ АРИЛЬНЫЕ МОНОБАКТАМОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2017 |
|
RU2733402C2 |
| НОВЫЕ ПРОТИВОМИКРОБНЫЕ СРЕДСТВА | 2009 |
|
RU2522582C2 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2801647C2 |
Изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R1 представляет собой -CH2N(R2)2, или -CH2NR2COOR3; в каждом случае R2 независимо выбран из Н или C1-С6 алкила; R3 представляет собой Н или C1-C6 алкил; Е представляет собой пиридин, где указанный пиридин замещен одним или двумя атомами фтора; А представляет собой фрагмент (а). Также изобретение относится к фармацевтической композиции для лечения микобактериальной инфекции, которая содержит терапевтически эффективное количество соединения по изобретению или его фармацевтически приемлемую соль, и фармацевтически приемлемому носителю. Соединение по изобретению применяют для лечения микобактериальной инфекции. Технический результат - оксазолидиноновые соединения, применимые для лечения микобактериальных инфекций. 3 н. и 6 з.п. ф-лы, 1 табл., 65 пр.
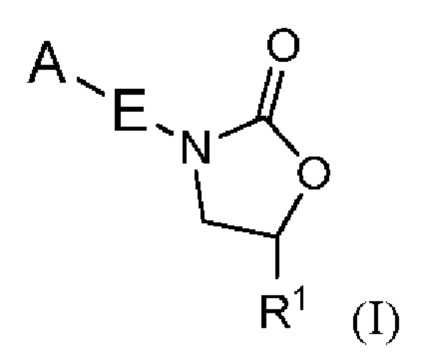 ,
, 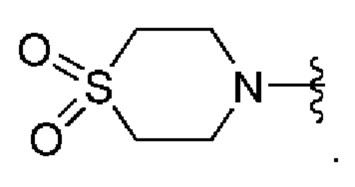 (а)
(а)
1. Соединение формулы I
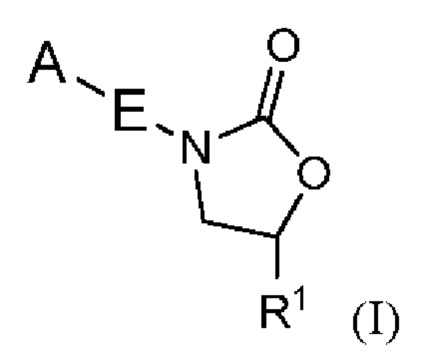
или его фармацевтически приемлемая соль, где:
R1 представляет собой -CH2N(R2)2, или -CH2NR2COOR3;
в каждом случае R2 независимо выбран из Н или C1-С6 алкила;
R3 представляет собой Н или C1-C6 алкил;
Е представляет собой пиридин, где указанный пиридин замещен одним или двумя атомами фтора;
А представляет собой 
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где: R1 представляет собой -CH2NR2COOR3 и в каждом случае R2 и R3 независимо выбираются из водорода и C1-С6алкила.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 представляет собой -CH2NHCOOCH3.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, имеющее структуру:
5. Фармацевтическая композиция для лечения микобактериальной инфекции, которая содержит терапевтически эффективное количество соединения по п. 1 или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель.
6. Применение терапевтически эффективного количества соединения по п. 1 или его фармацевтически приемлемой соли для лечения микобактериальной инфекции.
7. Применение по п. 6, при котором микобактериальная инфекция вызвана Mycobacterium tuberculosis.
8. Применение по п. 7, при котором М. tuberculosis представляет собой устойчивый к лекарственным средствам микобактериальный штамм.
9. Применение по пп. 6-8, при котором соединение или его фармацевтически приемлемую соль вводят перорально, парентерально или местно.

