Область техники
Группа изобретений относится к полимерным композиционным материалам (ПКМ), а именно к получению покрытий на полимерных композиционных материалах с фталонитрильными матрицами, защищающими их от окисления в атмосфере воздуха при повышенных температурах, для применения в авиационно-космической промышленности при изготовлении деталей двигателей, для высокотемпературной защиты электроники, а также для применения в кораблестроении при изготовлении негорючих деталей интерьера подводных лодок.
Уровень техники
Полимерные композиционные материалы с фталонитрильными матрицами известны устойчивостью к высоким температурам, а именно температурами стеклования выше 400°С и температурами разложения выше 500°С. Однако при длительном экспонировании ПКМ с фталонитрильными матрицами при температурах выше 300°С механические свойства теряются полностью достаточно быстро, ограничивая возможный ресурс применения таких материалов. Для увеличения ресурса деталей из ПКМ, эксплуатируемых при высоких температурах, необходимо создавать барьерные покрытия, перекрывающие доступ кислорода к композиту.
Для защиты полимерных композиционных материалов (ПКМ) с термостойкими матрицами (полииимдные, бис-малеимидные) от окисления при длительной эксплуатации при высоких температурах известно несколько неорганических защитных покрытий и способов их нанесения, представленных далее.
Известен способ защиты изделий из ПКМ от окисления при высоких температурах, заключающийся в нанесении защитного покрытия на изделия из ПКМ методом атомно-слоевого осаждения (EP3540002). Данные покрытия защищают ПКМ при температурах до 316°С (600 F). В качестве ПКМ использованы композиты, состоящие из углеродного, стеклянного, графитового, арамидного, полимерного, керамического волокон или их смесей, в качестве матрицы использованы как термореактивные, так и термопластичные материалы, например: бис-малеимиды, циановые эфиры, эпоксидные смолы, фенолформальдегидные смолы, полиэфирэфиркетон, полиимиды, а в качестве защитного покрытия использованы Al2O3, CeO2, MgO, NiO, TiO2, V2O5, ZrO2 и их смеси, и/или нитриды: AlN, HfN, InN, MgN, NbN, SiN, TaN, TiN, ZrN и их смеси, и/или оксинитриды, такие как оксинитрид кремния, SiOxNy, оксинитрид алюминия, (AlN)x ⋅ (Al2O3)1-x, а также их смеси. Недостатками представленных покрытий являются малая адгезия к поверхности ПКМ и большая разница в коэффициентах термического расширения, что приводит к отслаиванию и растрескиванию покрытия.
Известно защитное покрытие из оксида алюминия, жаропрочного сплава никеля, хрома, алюминия и иттрия (NiCrAlY) и нитрида кремния, нанесенное на углепластик с полиимидной матрицей методами осаждения из газовой фазы (D. R. Harding et al.: Oxidation protective barrier coatings for high-temperature polymer matrix composites. J. Mater. Res., Vol. 9, No. 6, Jun 1994, PP 1584-1595). Покрытия на основе нитрида кремния при термическом старении в течение 200 ч при 371°С показали на 33-50% меньшую потерю массы, чем непокрытые образцы. Покрытия на основе оксида алюминия отклеивались от поверхности ПКМ сразу же, после нанесения, покрытия на основе NiCrAlY отслаивались от ПКМ при старении при 371°С. Таким образом известные покрытия характеризуются слабой адгезией к ПКМ, что не обеспечивает защитную функцию покрытия. Также из-за невозможности нанести покрытие на торцы образцов ПКМ увеличение времени работы композита оказалось невозможным.
Известно защитное покрытие, состоящее из серебра, алюминия, титана и оксида алюминия, нанесенное на углепластик с бис-малеимидной матрицей газотермическим и электронно-лучевым напылением (N. An et al.: Thermo-oxidative performance of metal-coated polymers and composites, Surface & Coatings Technology 232 (2013) 166-172). Были получены покрытия толщиной 55-2800 нм, требовавшие также нанесения промежуточных слоёв хрома для снижения разницы между коэффициентами термического расширения материалов защитного покрытия и углепластика с бис-малеимидной матрицей. Наилучшие результаты по сохранению свойств получены для серебряных покрытий толщиной 580-700 нм. Старение проводили при 177°С, при этом площадь окисленной поверхности композита оказалась на 20% ниже, чем у непокрытого образца сравнения.
Однако, все известные защитные покрытия неорганической природы для нанесения на ПКМ требуют нанесения дополнительных слоёв для нивелирования разницы в коэффициентах термического расширения композита (ПКМ) и покрытия. Также проблемой нанесения таких слоев является сложность их нанесения, требующая специального оборудования. При этом покрытие крупных деталей сложной формы затруднено.
Наиболее близким к заявляемому решению является многослойное защитное покрытие для ПКМ с полиимидной матрицей, состоящее из защитного слоя карбида ванадия-кобальта, среднего слоя из композита карбида ванадия-кобальта и полиимида, и внутреннего слоя, состоящего из полиимида, необходимых для снижения разницы между коэффициентами теплового расширения материалов. Нижний и средний слои покрытия наносят на поверхность ПКМ методом высокоскоростного газопламенного напыления, а внешний слой - методом термического напыления порошка в пламени, что позволяет избежать уноса первых двух слоев потоком газа (M. Ivosevic et al.: Adhesive/Cohesive Properties of Thermally Sprayed Functionally Graded Coatings for Polymer Matrix Composites, Journal of Thermal Spray Technology Volume 14(1) March 2005-45). Толщина покрытий составляет 600 мкм. Сведения о защитных свойствах покрытия в публикации отсутствуют. Однако следует отметить, что защитный слой карбида ванадия-кобальта при нанесении на ПКМ с фталонитрильными матрицами не будет иметь высокую адгезию к материалу композита, при этом разница между коэффициентами термического расширения материалов защитного покрытия и углепластика будет значительной, что повлечет за собой растрескивание покрытия. Кроме того, использование данного покрытия ограничено сложностью нанесения, т.к. способы высокоскоростного газопламенного напыления и термического напыления порошка в пламени реализуются только с помощью специальных роботизированных установок.
Техническая проблема, решаемая посредством заявляемого изобретения, заключается в необходимости преодоления недостатков, присущих аналогам и прототипу, а именно защиты изделий из ПКМ от окисления при высоких температурах при упрощении способа нанесения защитного покрытия на ПКМ за счет исключения из покрытия дополнительных слоев, которые наносили в известных решениях для снижения разницы между коэффициентами теплового расширения материалов.
Раскрытие изобретения
Техническим результатом заявляемой группы изобретений является получение нового соединения - фторсодержащего олигомера, которое при использовании в композиции для получения термореактивной смолы, предназначенной для нанесения на поверхность ПКМ с фталонитрильной матрицей, обеспечивает высокую адгезию защитного слоя к матрице и минимальную разницу в коэффициентах термического расширения материалов покрытия и композита, что позволяет получить защитное покрытие, которое при длительном температурном воздействии позволяет сохранять эксплуатационные характеристики, такие как прочность при межслоевом сдвиге, прочность при растяжении, сжатии. За счет устойчивости такого покрытия к окислению при повышенных температурах, эксплуатационные характеристики ПКМ сохраняются на ~90% даже после нагрева в течение 200 ч при 350°C по сравнению с материалами, покрытыми композицией, включающей не фторсодержащий олигомер (фталонитрильной смолой), которые в тех же условиях позволяют сохранить лишь ~70% от начальных эксплуатационных характеристик композита.
Также заявляемые композиции характеризуются простотой нанесения покрытия на поверхность детали, поскольку не требуют нанесения дополнительных слоев для снижения разницы между коэффициентами термического расширения, и использования сложных установок.
Технический результат достигается олигомером фталонитрила в качестве компонента полимеризуемой смолы для защитных покрытий полимерной композиционной матрицы, представляющий собой соединение формул (I)
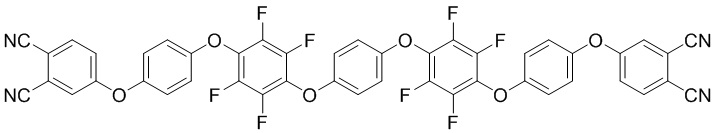 (I)
(I)
или формулы (II):
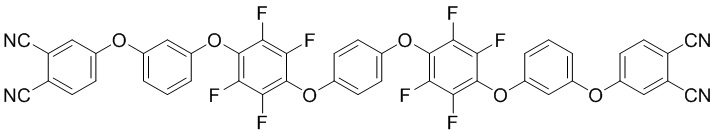 (II)
(II)
или формулы (III):
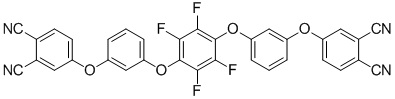 (III)
(III)
Технический результат также достигается способом получения олигомера фталонитрила по п. 1, характеризующийся тем, что проводят последовательное нуклеофильное замещение атомов фтора в гексафторбензоле и нитро-группы в 4-нитрофталонитриле гидроксилсодержащими ароматическими соединениями.
Для получения соединения формулы (I) для нуклеофильного замещения атомов фтора растворяют гидрохинон в высококипящем полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, к раствору добавляют неорганическую соль взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут, а затем добавляют гексафторбензол, взятый из расчета 2 мольных эквивалента гексафторбензола на 3 мольных эквивалента гидрохинона, полученную реакционную смесь нагревают до 60±5°C и оставляют при перемешивании на 24±2 часов с последующим нагревом до 110±5°C и выдержкой при перемешивании в течение 36±2 часов, затем реакционную смесь добавляют в большой избыток воды комнатной температуры, перемешивают и оставляют для выпадения осадка, выпавший осадок отфильтровывают и промывают от остатков растворителя, основания и непрореагировавшего гидрохинона, затем высушивают. Для замещения нитро-группы в 4-нитрофталонитриле, полученный после нуклеофильного замещения атомов фтора, бисфенол растворяют в высококипящем полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, затем добавляют неорганическую соль, взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут, после этого добавляют 4-нитрофталонитрил, взятый из расчета 1 моль 4-нитрофталонитрила на 1 моль участвующей в реакции гидроксильной группы, нагревают реакционную смесь до 80±5°C и оставляют при перемешивании на 24±2 часа, затем реакционную смесь добавляют в большой избыток воды комнатной температуры, перемешивают и оставляют при комнатной температуре для выпадения осадка, выпавший осадок отфильтровывают и промывают от непрореагировавших остатков и высушивают. При этом в качестве высококипящего апротонного растворителя используют N-метил-2-пирролидон, N,N-диметилацетамид или N,N-диметилформамид, а в качестве неорганической соли используют фторид калия, карбонат калия, фосфат калия. Для выпадения осадка реакционную смесь после добавления в воду комнатной температуры, взятую в объеме в 2,5-3,0 превышающую объем органического растворителя, выдерживают 2±0.5 часа, а для высушивания осадка его помещают на 12±1 часов в сушильный шкаф, разогретый до 100°C.
Для получения соединения формулы (II) для нуклеофильного замещения атомов фтора растворяют 4-(3-гидроксифенокси)фталонитрил или гидрохинон в полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, к раствору добавляют неорганическую соль взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут, а затем добавляют гексафторбензол, взятый из расчета 1 мольный эквивалент гексафторбензола на 1 мольный эквивалент 4-(3-гидроксифенокси)фталонитрила или гидрохинона, полученную реакционную смесь нагревают до 30±5°C и оставляют при перемешивании на 96±2 часов, затем реакционную смесь фильтруют через тонкий слой силикагеля, осадок промывают, фильтрат упаривают и проводят флэш-хроматографию с использованием смеси метиленхлорила/петролейный эфир, взятых в объемном соотношении 1/1. Для замещения нитро-группы в 4-нитрофталонитриле, полученное после нуклеофильного замещения атомов фтора соединение, взятое из расчета 1 моль на 1 моль участвующей в реакции гидроксильной группы, добавляют к раствору гидрохинона или 4-(3-гидроксифенокси)фталонитрил в полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, с добавлением неорганической соли, взятой из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, полученную реакционную смесь нагревают до 100±5°C и оставляют при перемешивании на 120±2 часов, затем реакционную смесь добавляют в большой избыток воды комнатной температуры, перемешивают и оставляют (выдерживают) до выпадения осадка, выпавший осадок отфильтровывают и промывают от непрореагировавших остатков 3 раза по 10 мл ацетона и высушивают. При этом в качестве полярного апротонного растворителя используют ацетон, N-метил-2-пирролидон, N,N-диметилацетамид или N,N-диметилформамид, а в качестве неорганической соли используют фторид калия, карбонат калия, фосфат калия. Для получения осадка реакционную смесь добавляют в воду комнатной температуры, взятую в объеме в 2,5-3,0 превышающую объем органического растворителя и выдерживают 2±0.5 часа, для высушивания осадка его помещают на 12±1 часов в сушильный шкаф, разогретый до 100°C.
Для получения соединения формулы (III) растворяют 4-(3-гидроксифенокси)фталонитрил в полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, затем добавляют неорганическую соль, взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут, добавляют гексафторбензол, взятый из расчета 1 мольный эквивалент гексафторбензола на 2 мольных эквивалента 4-(3-гидроксифенокси)фталонитрила, полученную реакционную смесь нагревают до 60±5°C и оставляют при перемешивании на 96±2 часов, затем реакционную смесь фильтруют через тонкий слой силикагеля, полученный осадок промывают 3 раза по 10 мл ацетона, фильтрат упаривают, проводят флэш-хроматографию с использованием смеси метиленхлорила/петролейный эфир, взятых в объемном соотношении 1/1. При этом в качестве полярного апротонного растворителя используют ацетон, N,N-диметилацетамид, N,N-диметилформамид или N-метил-2-пирролидон, в качестве неорганической соли используют фторид калия, карбонат калия, фосфат калия. Для получения осадка реакционную смесь добавляют в воду комнатной температуры, взятую в объеме в 2,5-3,0 превышающую объем органического растворителя и выдерживают 2±0.5 часа. Для высушивания осадка его помещают на 12±1 часов в сушильный шкаф, разогретый до 100°C.
Технический результат также достигается композицией для приготовления термореактивной смолы, включающей, по меньшей мере, одно соединение формулы I-III и отвердитель, выбранный из группы, включающей 4-(4-аминофенокси)фталонитрил, 4-(3-аминофенокси)фталонитрил, 4,4'-((перфтор-1,4-фенилен)бис(окси))дианилин, 3,3'-((перфтор-1,4-фенилен)бис(окси))дианилин взятых в концентрации от 4 до 20 масс. %.
Технический результат также достигается способом приготовления термореактивной смолы, заключающемся в том, что соединение формулы I-III расплавляют при температуре 120-180°С и при перемешивании добавляют отвердитель, полученную смесь перемешивают в течение 15-60 минут, после чего смесь охлаждают до отвердевания.
Также технический результат достигается способом приготовления термореактивного лака из термореактивной смолы, полученной заявляемым способом заключающемся в том, что смолу растворяют в растворителе, выбранном из группы ацетон, метилэтилкетон, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, взятом в количестве, обеспечивающем получение раствора для нанесения на поверхность ПКМ.
Осуществление изобретения
Заявляемые фторсодержащие олигомеры формулы (I - III) получают путем последовательного нуклеофильного замещения атомов фтора в гексафторбензоле (или производных гексафторбензола) и нитро-группы в 4-нитрофталонитриле различными гидроксилсодержащими ароматическими соединениями.
Для получения соединения формулы (I) растворяют гидрохинон в высококипящем полярном апротонном растворителе, выбранном из группы, включающей N-метил-2-пирролидон, N,N-диметилацетамид или N,N-диметилформамид, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов. Затем добавляют неорганическую соль, которая выступает в роли основания, но при этом является слабым нуклеофилом, выбранным из группы, включающей K2CO3 или K3PO4, или фторид калия (KF) - является сильным нуклеофилом, но не образует побочных продуктов, взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут. После этого добавляют гексафторбензол, взятый из расчета 2 мольных эквивалента гексафторбензола на 3 эквивалента гидрохинона, полученную реакционную смесь нагревают до 60±5°C и оставляют при перемешивании на 24±2 часов с последующим нагревом до 110±5°C и выдержкой при перемешивании в течение 36±2 часов. Затем реакционную смесь добавляют в большой избыток воды комнатной температуры (в 2,5-3,0 превышающий объем используемого растворителя), перемешивают и оставляют (выдерживают) на 2±0.5 часа при комнатной температуре. Выпавший осадок отфильтровывают и несколько раз промывают горячей водой (85-95°C), чтобы смыть остатки растворителя, основания и непрореагировавшего гидрохинона, затем помещают на 12±1 часов в сушильный шкаф, разогретый до 100°C. Полученный бисфенол растворяют в высококипящем полярном апротонном растворителе, выбранном из группы, включающей N-метил-2-пирролидон, N,N-диметилацетамид или N,N-диметилформамид, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов. Затем добавляют неорганическую соль, которая выступает в роли основания, но при этом является слабым нуклеофилом, выбранную из группы, включающей K2CO3 или K3PO4, или фторид калия (KF) - является сильным нуклеофилом, но не образует побочных продуктов, взятого из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут. После этого добавляют 4-нитрофталонитрил, взятый из расчета 1 моль 4-нитрофталонитрила на 1 моль участвующей в реакции гидроксильной группы, нагревают реакционную смесь до 80±5°C и оставляют при перемешивании на 24±2 часа. Затем реакционную смесь добавляют в большой избыток воды комнатной температуры (в 2,5-3,0 превышающий объем органического растворителя), перемешивают и выдерживают (оставляют) на 2±0.5 часа при комнатной температуре. Выпавший осадок отфильтровывают и несколько раз промывают горячей водой (85-95°C), чтобы смыть остатки растворителя или основания, затем помещают на 12±1 часов в сушильный шкаф, разогретый до 100°C.
Для получения соединения формулы (II) растворяют 4-(3-гидроксифенокси)фталонитрил в полярном апротонном растворителе, выбранном из группы, включающей ацетон, N,N-диметилацетамид, N,N-диметилформамид или N-метил-2-пирролидон, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов. Затем добавляют неорганическую соль, выступающую в роли основания и являющуюся слабым нуклеофилом, например, K3PO4, карбонат калия, или KF, который является сильным нуклеофилом, но не образует побочных продуктов, взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут. После этого добавляют гексафторбензол, взятый из расчета 1 мольный эквивалент гексафторбензола на 1 эквивалент 4-(3-гидроксифенокси)фталонитрила, полученную реакционную смесь нагревают до 30±5°C и оставляют при перемешивании на 96±2 часов. Затем реакционную смесь фильтруют через тонкий слой силикагеля (чтобы неорганические соединения и непрореагировавший 4-(3-гидроксифенокси)фталонитрил остались на силикагеле), осадок промывают 3 раза по 10 мл ацетона. Фильтрат упаривают, в результате чего образуется смесь продуктов моно- и ди-замещения. Для получения чистого целевого соединения (4-(3-(перфторфенокси)фенокси)фталонитрил) проводят флэш-хроматографию (элюент - смесь метиленхлорила/петролейный эфир 1/1). На второй стадии процесса получения (II) растворяют гидрохинон в полярном апротонном растворителе, выбранном из группы, включающей ацетон, N,N-диметилацетамид, N,N-диметилформамид или N-метил-2-пирролидон, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов. Затем добавляют неорганическую соль, выступающую в роли основания и являющуюся слабым нуклеофилом, например, K3PO4, карбонат калия или KF (сильный нуклеофил, но не образует побочных продуктов), взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут. После этого добавляют 4-(3-(перфтофенокси)фенокси)фталонитрил, полученный ранее, взятый из расчета 1 моль на 1 моль участвующей в реакции гидроксильной группы, нагревают реакционную смесь до 100±5°C и оставляют при перемешивании на 120±2 часов. Затем реакционную смесь добавляют в большой избыток воды комнатной температуры (в 2,5-3,0 превышающий объем органического растворителя), перемешивают и выдерживают (оставляют на) 2±0.5 часа при комнатной температуре. Выпавший осадок отфильтровывают и несколько раз промывают горячей водой (85-95°C), чтобы смыть остатки растворителя, основания или непрореагировавшего гидрохинона, затем помещают на 12±1 часов в сушильный шкаф, разогретый до 100°C.
Альтернативный метод получения соединения (II) также подразумевает использование реакций нуклеофильного замещения, но на первом этапе вместо фенолят-иона 4-(3-гидроксифенокси)фталонитрила используется фенолят-ион гидрохинона в качестве нуклеофила. И наоборот, на втором этапе используется фенолят-ион 4-(3-гидроксифенокси)фталонитрила вместо фенолят-иона гидрохинона.
Для получения соединения формулы (III) растворяют 4-(3-гидроксифенокси)фталонитрил в полярном апротонном растворителе, выбранном из группы, включающей, ацетон, N,N-диметилацетамид, N,N-диметилформамид, N-метил-2-пирролидон, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов. Затем добавляют неорганическую соль, которая выступает в роли основания, но при этом является слабым нуклеофилом, например, карбонат калия, фосфат калия, KF (сильный нуклеофил, но не образует побочных продуктов), взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут. После этого добавляют гексафторбензол, взятый из расчета 1 мольный эквивалент гексафторбензола на 2 эквивалента 4-(3-гидроксифенокси)фталонитрила, полученную реакционную смесь нагревают до 60±5°C и оставляют при перемешивании на 96±2 часов. Затем реакционную смесь фильтруют через тонкий слой силикагеля (чтобы неорганические соединения и непрореагировавший 4-(3-гидроксифенокси)фталонитрил остались на силикагеле), осадок промывают 3 раза по 10 мл ацетона. Фильтрат упаривают, в результате чего образуется смесь продуктов моно- и ди-замещения. Для получения чистого целевого соединения (III) проводят флэш-хроматографию (элюент - смесь метиленхлорила/петролейный эфир 1/1).
Для приготовления термореактивной смолы или лака фторсодержащие олигомеры формулы (I-III) смешивают с отвердителями. Для этого олигомер фталонитрила расплавляют при температуре 120-180℃ и при перемешивании добавляют отвердитель, полученную смесь перемешивают в течение 15-60 минут. После этого смесь выливают в ровную металлическую или стеклянную емкость, обработанную антиадгезивом, и оставляют при комнатной температуре для остывания.
В качестве отвердителя используют соединения, выбранные из группы, включающей 4-(4-аминофенокси)фталонитрил, 4-(3-аминофенокси)фталонитрил, 4,4'-((перфтор-1,4-фенилен)бис(окси))дианилин, 3,3'-((перфтор-1,4-фенилен)бис(окси))дианилин взятые в концентрации от 4 до 20% (масс).
Нанесение покрытия возможно в виде расплава, предварительно разогретого до температуры 100-160°С, кистью или распылением сжатым воздухом.
В другом варианте осуществления изобретения термореактивные смолы можно растворять в растворителях, выбранных из группы ацетон, метилэтилкетон, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, для получения лака. Лак наносят на поверхность ПКМ при помощи молярной кисти или распыления равномерно покрывая всю поверхность ПКМ, затем нагревают при температуре, обеспечивающей удаление растворителя.
Нанесение подобного рода покрытий возможно на ПКМ, удовлетворяющим следующим условиям: химическое родство материала матрицы композита и покрытия, которое обеспечивает высокую адгезию и минимальное различие в коэффициентах термического расширения (предотвращает растрескивание покрытия); температура эксплуатации ПКМ сопоставима с температурой эксплуатации покрытия (но не выше). Этим условиям соответствуют ПКМ на основе фталонитрилов.
ПКМ получали двумя способами: методом вакуумной инфузии (А) и методом вакуумного прессования (Б). При этом применение покрытий не ограничено ПКМ, полученными данными способами, также могут быть использованы ПКМ, сформованные другими способами, но использующие матрицу на основе фталонитрилов.
Метод А. Вакуумный пакет собирали на оснастке из нержавеющей стали с нанесенным на поверхность силиконовым антиадгезивным покрытием. Углеродную ткань саржевого плетения 2×2 плотностью 200 г/м2 (волокно HTA-40, 3k https://www.teijincarbon.com/ru/produkci%D1%8F/uglerodn%D1%8Be-volokna-tenax%C2%AE/zzgut%D1%8B-tenax%C2%AE/) резали на куски размером 30×30 см (10 слоев) и выкладывали по формуле [0]12. Далее собрали вакуумный пакет и помещали его в термошкаф, нагревали до 120°C. Связующее (600 г) ФНИ350 (http://itecma.ru/products/svyazuyushchie/do-450/46/) дегазировали в стеклянном реакторе при температуре 120°С и пониженном давлении, после выливали в металлический стакан, переносили в термошкаф и помещали в него входную трубку из пакета и начинали пропитку. Через 10-15 минут связующее выходило из пакета, что означало окончание пропитки. Пакет герметизировали, повышали температуру в шкафу до 180°C со скоростью не более 2°C/мин и выдерживали в течение 8 часов. После этого пакет разбирали, предотвержденный образец извлекали и постотверждали в свободном виде по следующей программе: нагрев до 180°C со скоростью не выше 2°C/мин, 30 минут выдержки и нагрев до температуры постотверждения (330-375°C) со скоростью 10°C/ч, 8 часов выдержки.
Метод Б. Пакет собирали на оснастке из нержавеющей стали с нанесенным на поверхность силиконовым антиадгезивным покрытием. Листы препрега РНТ450 (на основе углеродной ткани саржевого плетения 2×2 плотностью 240 г/м2, волокно HTA-40, 3k https://www.teijincarbon.com/ru/produkci%D1%8F/uglerodn%D1%8Be-volokna-tenax%C2%AE/zzgut%D1%8B-tenax%C2%AE/, ТУ 23.99.14-044-73047899-2018) резали на куски размером 30×30 см (9 слоев) и выкладывали на оснастке внутри силиконовой рамки 30×30 см так, чтобы слой нанесенного связующего на каждом из слоев находился сверху. Сверху листы препрега накрывали сухим слоем ткани и стальной пластиной (цулагой) размером 30×30 см. Затем поверх образованного слоя выкладывали дренажный материал марки Ниалон® ИП 340 А с размерами 45×55 см. Вокруг оснастки собирали вакуумный пакет, затем помещали в термопресс Langzauner LZT-L 250 и вакуумировали. Температуру пресса повышали до 140°C со скоростью 2°C/мин, прикладывали давление к образцу в 8.75 бар, после чего следовал нагрев до 180°C со скоростью 2°C/мин, повышение давления до 21.8 бар и выдержка в течение 8 часов. После этого пакет разбирали, предотвержденный образец извлекали и постотверждали в свободном виде по следующей программе: нагрев до 180°C со скоростью не выше 2°C/мин, 30 минут выдержки и нагрев до температуры постотверждения (330-375°C) со скоростью 10°C/ч, 8 часов выдержки.
Покрытие может наноситься на ПКМ путём расплавления и нанесения расплава распылением или кистью.
Кроме того, из смолы может быть приготовлен лак путём её растворения в растворителях, выбранных из группы: диметилацетамид, диметилформамид, N-метил-2-пирролидон, ацетон, метилэтилкетон и нанесен на поверхность ПКМ кистью или распылением.
После нанесения на композит лак отверждается по следующей программе: нагрев до 180°C со скоростью не более 2°C/мин и выдержкой в течение 3 часов, после чего нагрев до температуры постотверждения (330-375°C) со скоростью 10°C/ч и выдержкой в течение 8 часов для получения высокотермостойкого защитного покрытия толщиной 10-100 мкм.
Ввиду того, что фторированные полимеры обладают более высокой термоокислительной стабильностью, покрытие окисляется медленнее, чем матрица углепластика.
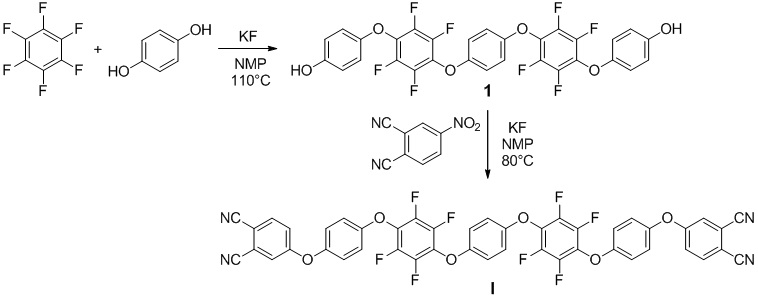
Пример 1. Получение 4,4'-(((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))дифенол (1). В одногорлой колбе (100 мл), оснащенной обратным холодильником, в атмосфере аргона растворили 4.44 г (0.0400 моль) гидрохинона в 50 мл сухого N-метил-2-пирролидона. Затем был добавлен фторид калия (3.90 г, 0.0670 моль), после чего реакционную смесь перемешивали в течение 30 минут. После этого добавили гексафторбензол (5.00 г, 0.0270 моль), нагрели реакционную смесь до 60°C и оставили при перемешивании на 24 часов с последующим нагревом до 110°C и выдержкой при перемешивании в течение 36 часов. Реакционную смесь добавили в большой избыток воды (около 250 мл), перемешали и оставили на 2 часа. Выпавший осадок отфильтровали и несколько раз промыли горячей водой, затем поместили в сушильный шкаф. Для получения чистого целевого соединения провели флэш-хроматографию. В качестве элюента использовали смесь метиленхлорид: метанол в соотношении 60:1. Выход - 7.06 г (84.0 %).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 6.83 (д, J=7.41 Гц, 4 H), 7.09 (с, 4 H), 7.31 (д, J=7.89 Гц 4 H), 9.43 (с, 2 H).
13C ЯМР (151 МГц, ДМСО-d6) δ м.д. 115.73, 115.95, 116.38, 116.52, 116.79, 129.91, 141.06, 142.70, 149.70, 152.57, 152.90, 153.64.
Элементный анализ для C30H14F8O6: Расчетные значения: C (57.89), H (2.27), F (24.42). Экспериментальные значения: C (57.83), H (2.31), F (24.37).
Пример 2. Получение 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(4,1-фенилен))бис(окси))дифталонитрил (I). В одногорлой колбе (100 мл), оснащенной обратным холодильником, в атмосфере аргона растворили 4,4'-(((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))дифенол (1) (4.00 г, 0.0064 моль) в 50 мл сухого N-метил-2-пирролидона. Затем был добавлен фторид калия (0.75 г, 0.0129 моль), после чего реакционную смесь перемешивали в течение 30 минут. После этого добавили 4-нитрофталонитрил (2.23 г, 0.0129 моль), нагрели реакционную смесь до 80°C и оставили при перемешивании на 24 часа. Реакционную смесь добавили в большой избыток воды (около 250 мл), перемешали и оставили на 2,5 часа. Выпавший осадок, который представлял целевой продукт, отфильтровали и несколько раз промыли горячей водой, затем поместили в сушильный шкаф. Выход - 5.42 г (96.4%).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 7.04 (д, J=6.53 Гц, 4 H), 7.17 (с, 4 H), 7.30 (д, J=7.55 Гц, 4 H), 7.41 (с, 2 H), 7.76 (д, J=8.87 Гц, 2 H), 8.30 (д, J=8.53 Гц, 2 H).
13C ЯМР (151 МГц, ДМСО-d6) δ м.д. 108.08, 110.65, 115.70, 115.93, 116.41, 116.61, 116.85, 122.20, 122.37, 124.70, 124.76, 129.92, 136.28, 136.60, 141.11, 142.76, 149.48, 152.64, 152.91, 161.30.
Элементный анализ для C46H18F8N4O6: Расчетные значения: C (63.17), H (2.07), F (17.38), N (6.41). Экспериментальные значения: C (63.11), H (2.05), F (17.32), N (6.43).
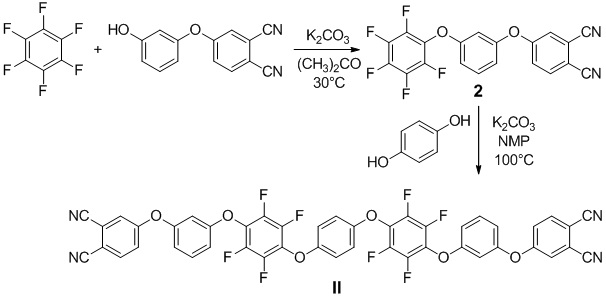
Пример 3. Получение 4-(3-(перфторфенокси)фенокси)фталонитрил (2). В одногорлой колбе (100 мл), оснащенной обратным холодильником, в атмосфере аргона растворили 4-(3-гидроксифенокси)фталонитрил (синтез соединения описан в работе A. Lyubimtsev et al.: Synthesis of novel covalently linked dimeric phthalocyanines, Eur. J. Org. Chem. (12) (2007) 2000-2005) (2.54 г, 0.0108 моль) в 50 мл сухого ацетона. Затем был добавлен карбонат калия (1.78 г, 0.0129 моль), после чего реакционную смесь перемешивали в течение 30 минут. После этого добавили гексафторбензол (2.00 г, 0.0108 моль), нагрели реакционную смесь до 30°C и оставили при перемешивании на 96 часов. Реакционную смесь отфильтровали через тонкий слой силикагеля, осадок несколько раз промыли ацетоном. Упарив фильтрат, получили смесь целевого и побочного соединений. Для получения чистого целевого соединения провели флэш-хроматографию. В качестве элюента использовали смесь метиленхлорид:петролейный эфир в соотношении 1:1. Выход - 3.51 г (82.5 %).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 6.98 (дд, J=8.11, 1.25 Гц, 1 H), 7.06 - 7.11 (м, 2 H), 7.42 (дд, J=8.70, 2.68 Гц, 1 H), 7.50 (т, J=8.53 Гц, 1 H), 7.83 (д, J=2.51 Гц, 1 H), 8.11 (д, J=8.70 Гц, 1 H).
13C ЯМР (151 МГц, ДМСО-d6) δ м.д. 107.70, 108.64, 112.40, 115.29, 115.51, 115.80, 116.73, 122.31, 123.00, 128.28, 131.83, 136.34, 137.19, 137.87, 138.84, 139.51, 140.77, 142.42, 155.15, 158.03, 160.46.
Элементный анализ для C20H7F5N2O2: Расчетные значения: C (59.71), H (1.75), F (23.61), N (6.96). Экспериментальные значения: C (59.67), H (1.74), F (23.63), N (6.94).
Пример 4. Получали 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(3,1-фенилен))бис(окси))дифталонтрил (II). В одногорлой колбе (50 мл), оснащенной обратным холодильником, в атмосфере аргона растворили гидрохинон (0.05 г, 0.0005 моль) в 25 мл сухого N-метил-2-пирролидона. Затем был добавлен карбонат калия (0.14 г, 0.001 моль), после чего реакционную смесь перемешивали в течение 35 минут. После этого добавили 4-(3-(перфторфенокси)фенокси)фталонитрил (2) (0.37 г, 0.0009 моль), нагрели реакционную смесь до 100°C и оставили при перемешивании на 120 часов. Реакционную смесь добавили в большой избыток воды (около 125 мл), перемешали и оставили на 1,5 часа. Выпавший осадок, который представлял целевой продукт, отфильтровали и несколько раз промыли горячей водой, затем поместили в сушильный шкаф. Выход - 0.37 г (93.2 %).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 7.05 (дд, J=8.13, 1.27 Гц, 2 H), 7.12 - 7.17 (м, 4 H), 7.22 (с, 4 H), 7.49 (дд, J=8.69, 2.72 Гц, 2 H), 7.57 (т, J=8.44 Гц, 2 H), 7.90 (д, J=2.53 Гц, 2 H), 8.18 (д, J=8.70 Гц, 2 H).
13C ЯМР (151 МГц, ДМСО-d6) δ м.д. 107.76 (с), 108.70 (с), 112.46 (с), 115.35 (с), 115.57 (с), 115.86 (с), 116.79 (с), 116.82 (с), 122.37 (с), 123.06 (с), 128.34 (с), 129.07 (с), 131.89 (с), 136.40 (с), 137.25 (с), 138.90 (с), 140.87 (с), 142.44 (с), 152.92 (с), 155.21 (с), 158.09 (с), 160.52 (с).
Элементный анализ для C46H18F8N4O6: Расчетные значения: C (63.17), H (2.07), F (17.38), N (6.41). Экспериментальные значения: C (63.12), H (2.04), F (17.33), N (6.40).
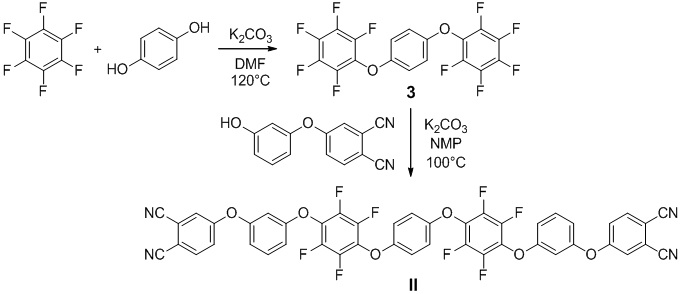
Пример 5. Получение 1,4-бис(перфторфенокси)бензол (3). Данное соединение получали способом, описанным в работе I.M. Tkachenko, et al. Synthesis of fluorinated poly(arylene ether)s with dibenzodioxin and spirobisindane units from new bis(pentafluorophenyl)- and bis(nonafluorobiphenyl)-containing monomers // J. Fluor. Chem. 2017. Vol. 195. P. 1-12. В одногорлой колбе (250 мл), оснащенной обратным холодильником, в атмосфере аргона растворили 2.00 г (0.018 моль) гидрохинона в 55 мл сухого N,N-диметилформамида. Затем был добавлен карбонат калия (5.00 г, 0.036 моль), после чего реакционную смесь перемешивали в течение 25 минут. После этого добавили гексафторбензол (27.00 г, 0.145 моль), нагрели реакционную смесь до 120°C и оставили при перемешивании на 8 часов. После охлаждения до комнатной температуры смесь отфильтровали. Из фильтрата отогнали избыток гексафторбензола при атмосферном давлении, после чего оставшийся раствор добавили к избытку холодной воды (250 мл). Выпавший осадок отфильтровали, промыли несколько раз водой. Для получения чистого целевого соединения провели флэш-хроматографию. В качестве элюента использовали смесь метиленхлорид:петролейный эфир в соотношении 1:1. Выход - 6.83 г (85%).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 7.15 (с, 4 H).
13C ЯМР (151 МГц, ДМСО-d6) δ м.д. 116.76 (с), 129.01 (с), 137.15 (с), 137.61 (с), 138.79 (с), 139.26 (с), 140.83 (с), 142.49 (с), 152.86 (с).
Элементный анализ для C18H4F10O2: Расчетные значения: C (48.89), H (0.91), F (42.96). Экспериментальные значения: C (48.83), H (0.89), F (42.89).
Пример 6. Получали 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(3,1-фенилен))бис(окси))дифталонтрил (II). В одногорлой колбе (100 мл), оснащенной обратным холодильником, в атмосфере аргона растворили 4-(3-гидроксифенокси)фталонитрил (1.068 г, 0.0045 моль) в 50 мл сухого N-метил-2-пирролидона. Затем был добавлен карбонат калия (0.688 г, 0.0049 моль), после чего реакционную смесь перемешивали в течение 30 минут. После этого добавили 1,4-бис(перфторфенокси)бензол (3) (1.00 г, 0.0023 моль), нагрели реакционную смесь до 105°C и оставили при перемешивании на 122 часа. Реакционную смесь добавили в большой избыток воды (около 250 мл), перемешали и оставили на 1,5 часа. Выпавший осадок, который представлял целевой продукт, отфильтровали и несколько раз промыли горячей водой, затем поместили в сушильный шкаф. Выход - 1.84 г (93 %).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 7.05 (дд, J=8.13, 1.27 Гц, 2 H), 7.12 - 7.17 (м, 4 H), 7.22 (с, 4 H), 7.49 (дд, J=8.69, 2.72 Гц, 2 H), 7.57 (т, J=8.44 Гц, 2 H), 7.90 (д, J=2.53 Гц, 2 H), 8.18 (д, J=8.70 Гц, 2 H).
13C ЯМР (151 МГц, ДМСО-d6) δ м.д. 107.76 (с), 108.70 (с), 112.46 (с), 115.35 (с), 115.57 (с), 115.86 (с), 116.79 (с), 116.82 (с), 122.37 (с), 123.06 (с), 128.34 (с), 129.07 (с), 131.89 (с), 136.40 (с), 137.25 (с), 138.90 (с), 140.87 (с), 142.44 (с), 152.92 (с), 155.21 (с), 158.09 (с), 160.52 (с).
Элементный анализ для C46H18F8N4O6: Расчетные значения: C (63.17), H (2.07), F (17.38), N (6.41). Экспериментальные значения: C (63.15), H (2.09), F (17.33), N (6.37).
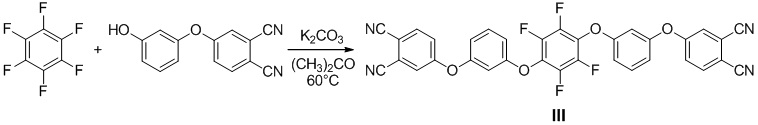
Пример 7. Получали 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрил (III). В одногорлой колбе (100 мл), оснащенной обратным холодильником, в атмосфере аргона растворили 4-(3-гидроксифенокси)фталонитрил (5.08 г, 0.0216 моль) в 50 мл сухого ацетона. Затем был добавлен карбонат калия (3.56 г, 0.0258 моль), после чего реакционную смесь перемешивали в течение 30 минут. После этого добавили гексафторбензол (2.00 г, 0.0108 моль), нагрели реакционную смесь до 60°C и оставили при перемешивании на 96 часов. Реакционную смесь отфильтровали через тонкий слой силикагеля, осадок несколько раз промыли ацетоном. Упарив фильтрат, получили смесь целевого и побочного соединений. Для получения чистого целевого соединения провели флэш-хроматографию. В качестве элюента использовали смесь метиленхлорид:петролейный эфир в соотношении 1:1. Выход - 5.46 г (82%).
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 7.06 (дд, J=8.11, 1.42 Гц, 2 H), 7.22 - 7.28 (м, 4 H), 7.52 (дд, J=8.78, 2.43 Гц, 2 H), 7.59 (т, J=8.11 Гц, 2 H), 7.93 (д, J=2.34 Гц, 2 H), 8.20 (д, J=8.70 Гц, 2 H).
13C ЯМР (151 МГц, ДМСО-d6) δ м.д. 107.84 (с), 108.68 (с), 112.49 (с), 115.32 (с), 115.36 (с), 115.82 (с), 116.74 (с), 122.42 (с), 123.09 (с), 129.44 (с), 131.77 (с), 136.35 (с), 141.10 (с), 142.79 (с), 155.21 (с), 158.07 (с), 160.43 (с).
Элементный анализ для C34H14F4N4O4: Расчетные значения: C (66.03), H (2.28), F (12.29), N (9.06). Экспериментальные значения: C (59.97), H (2.32), F (12.25), N (9.00).
Получение смол:
Пример 8. В колбу объемом 500 мл, оснащенную магнитной мешалкой, поместили 192 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(4,1-фенилен))бис(окси))дифталонитрила. Колбу нагрели до 150°C. После того, как вещество расплавилось, включили перемешивание и добавили 8 г 4-(4-аминофенокси)фталонитрила и перемешивали в течение 30 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 9. В колбу объемом 500 мл, оснащенную магнитной мешалкой, поместили 180 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(4,1-фенилен))бис(окси))дифталонитрила. Колбу нагрели до 120°C. После того, как вещество расплавилось, включили перемешивание и добавили 20 г 3,3'-((перфтор-1,4-фенилен)бис(окси))дианилина и перемешивали в течение 60 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 10. В колбу объемом 500 мл, оснащенную магнитной мешалкой, поместили 170 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(3,1-фенилен))бис(окси))дифталонтрила. Колбу нагрели до 180°C. После того, как вещество расплавилось, включили перемешивание и добавили 30 г 4-(4-аминофенокси)фталонитрила и перемешивали в течение 15 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 11. В колбу объемом 500 мл, оснащенную магнитной мешалкой, поместили 160 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(3,1-фенилен))бис(окси))дифталонтрила. Колбу нагрели до 150 °C. После того, как вещество расплавилось, включили перемешивание и добавили 40 г 4-(3-аминофенокси)фталонитрила и перемешивали в течение 30 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 12. В колбу объемом 500 мл, оснащенную механической мешалкой, поместили 192 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(4,1-фенилен))бис(окси))дифталонитрила. Колбу нагрели до 180°C. После того, как вещество расплавилось, включили перемешивание и добавили 8 г 4-(3-аминофенокси)фталонитрила и перемешивали в течение 15 минут, затем добавили 2 г пиролитического оксида кремния (AEROSIL 200) и перемешивали при максимальной скорости вращения мешалки в течение 15 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 13. В колбу объемом 500 мл, оснащенную механической мешалкой, поместили 192 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(4,1-фенилен))бис(окси))дифталонитрила. Колбу нагрели до 140°C. После того, как вещество расплавилось, включили перемешивание и добавили 8 г 4-(3-аминофенокси)фталонитрила и перемешивали в течение 40 минут, затем добавили 20 г пиролитического оксида кремния (AEROSIL R202) и перемешивали при максимальной скорости вращения мешалки в течение 15 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 14. В колбу объемом 500 мл, оснащенную магнитной мешалкой, поместили 160 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(3,1-фенилен))бис(окси))дифталонтрила. Колбу нагрели до 150°C. После того, как вещество расплавилось, включили перемешивание и добавили 40 г 4-(4-аминофенокси)фталонитрила и перемешивали в течение 30 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 15. В колбу объемом 500 мл, оснащенную механической мешалкой, поместили 180 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила. Колбу нагрели до 150°C. После того, как вещество расплавилось, включили перемешивание и добавили 20 г 4,4'-((перфтор-1,4-фенилен)бис(окси))дианилина и перемешивали в течение 30 минут, затем добавили 2 г пиролитического оксида кремния (AEROSIL 200) и перемешивали при максимальной скорости вращения мешалки в течение 15 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 16. В колбу объемом 500 мл, оснащенную магнитной мешалкой, поместили 180 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(4,1-фенилен))бис(окси))дифталонитрила. Колбу нагрели до 150°C. После того, как вещество расплавилось, включили перемешивание и добавили 20 г 4,4'-((перфтор-1,4-фенилен)бис(окси))дианилина и перемешивали в течение 30 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 17. В колбу объемом 500 мл, оснащенную механической мешалкой, поместили 170 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила. Колбу нагрели до 150°C. После того, как вещество расплавилось, включили перемешивание и добавили 30 г 3,3'-((перфтор-1,4-фенилен)бис(окси))дианилина и перемешивали в течение 30 минут, затем добавили 10 г пиролитического оксида кремния (AEROSIL R202) и перемешивали при максимальной скорости вращения мешалки в течение 15 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 18. В колбу объемом 500 мл, оснащенную магнитной мешалкой, поместили 96 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(4,1-фенилен))бис(окси))дифталонитрила и 96 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила. Колбу нагрели до 150°C. После того, как вещество расплавилось, включили перемешивание и добавили 8 г 4-(4-аминофенокси)фталонитрила и перемешивали в течение 30 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Пример 19. В колбу объемом 500 мл, оснащенную магнитной мешалкой, поместили 126 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила и 54 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила. Колбу нагрели до 170°C. После того, как вещество расплавилось, включили перемешивание и добавили 20 г 3,3'-((перфтор-1,4-фенилен)бис(окси))дианилина и перемешивали в течение 60 минут. После этого смесь вылили на металлический поднос, обработанный антиадгезивом, и дали остыть. Получили аморфную стеклообразную смолу.
Нанесение безрастворных полимерных покрытий:
Пример 20. Смолу, полученную в примере 8, расплавили при 120°C в открытой алюминиевой емкости объемом 500 мл. Расплав тонким слоем с помощью малярной кисти нанесли на ПКМ (метод Б), постоотвержденный при 330°C.
Пример 21. Смолу, полученную в примере 12, расплавили при 150°C в открытой алюминиевой емкости объемом 500 мл. Расплав тонким слоем с помощью малярной кисти нанесли на ПКМ (метод А), предотвержденный при 180°C.
Пример 22. Смолу, полученную в примере 13, расплавили при 130°C в открытой алюминиевой емкости объемом 500 мл. Расплав тонким слоем с помощью малярной кисти нанесли на ПКМ (метод А), постоотвержденный при 375°C.
Пример 23. Смолу, полученную в примере 14, расплавили при 150°C в открытой алюминиевой емкости объемом 500 мл. Расплав тонким слоем с помощью малярной кисти нанесли на ПКМ (метод Б), предотвержденный при 180°C.
Пример 24. Смолу, полученную в примере 17, расплавили при 150°C в открытой алюминиевой емкости объемом 500 мл. Расплав тонким слоем с помощью малярной кисти нанесли на ПКМ (метод А), постоотвержденный при 350°C.
Пример 25. Смолу, полученную в примере 9, расплавили при 150°C в открытой алюминиевой емкости объемом 500 мл. Расплав тонким слоем с помощью распыления сжатым воздухом нанесли на ПКМ (метод Б), постоотвержденный при 350°C.
Пример 26. Смолу, полученную в примере 10, расплавили при 150°C в открытой алюминиевой емкости объемом 500 мл. Расплав тонким слоем с помощью распыления сжатым воздухом нанесли на ПКМ (метод А), постоотвержденный при 330°C.
Пример 27. Смолу, полученную в примере 9, расплавили при 160°C в открытой алюминиевой емкости объемом 500 мл. Расплав тонким слоем с помощью распыления сжатым воздухом нанесли на ПКМ (метод Б), постоотвержденный при 375°C.
Нанесение покрытий в виде лака:
Пример 28. Смолу, полученную в примере 8, в количестве 50 г растворили в 36 мл ДМАА. Полученный лак тонким слоем с помощью малярной кисти нанесли на ПКМ (метод А), предотвержденный при 180°C. Покрытие высушили в конвекционном программируемом шкафу при 170°C в течение 1 часа.
Пример 29. Смолу, полученную в примере 9, в количестве 50 г растворили в 42 мл ацетона. Полученный лак тонким слоем с помощью малярной кисти нанесли на ПКМ (метод Б), постотвержденный при 330°C. Покрытие высушили в конвекционном программируемом шкафу при 60°C в течение 1 часа.
Пример 30. Смолу, полученную в примере 10, в количестве 50 г растворили в 41 мл метилэтилкетона. Полученный лак тонким слоем с помощью малярной кисти нанесли на ПКМ (метод Б), постотвержденный при 375°C. Покрытие высушили в конвекционном программируемом шкафу при 80°C в течение 1 часа.
Пример 31. Смолу, полученную в примере 11, в количестве 50 г растворили в 32 мл N-метил-2-пирролидоне. Полученный лак тонким слоем с помощью малярной кисти нанесли на ПКМ (метод А), постотвержденный при 350°C. Покрытие высушили в конвекционном программируемом шкафу при 205°C в течение 1 часа.
Пример 32. Смолу, полученную в примере 14, в количестве 50 г растворили в 35 мл диметилформамиде. Полученный лак тонким слоем с помощью малярной кисти нанесли на ПКМ (метод Б), предотвержденный при 180°C. Покрытие высушили в конвекционном программируемом шкафу при 155°C в течение 1 часа.
Пример 33. Смолу, полученную в примере 16, в количестве 50 г растворили в 42 мл ацетона. Полученный лак тонким слоем с помощью малярной кисти нанесли на ПКМ (метод Б), постотвержденный при 330°C. Покрытие высушили в конвекционном программируемом шкафу при 60°C в течение 1 часа.
Пример 34. Смолу, полученную в примере 8, в количестве 50 г растворили в 36 мл ДМАА. Полученный лак тонким слоем с помощью распыления ручным пульверизатором нанесли на ПКМ (метод А), предотвержденный при 180°C. Покрытие высушили в конвекционном программируемом шкафу при 170°C в течение 1 часа.
Пример 35. Смолу, полученную в примере 9, в количестве 50 г растворили в 42 мл ацетона. Полученный лак тонким слоем с помощью распыления ручным пульверизатором нанесли на ПКМ (метод Б), постотвержденный при 330°C. Покрытие высушили в конвекционном программируемом шкафу при 60°C в течение 1 часа.
Пример 36. Смолу, полученную в примере 11, в количестве 50 г растворили в 32 мл N-метил-2-пирролидоне. Полученный лак тонким слоем с помощью распыления ручным пульверизатором нанесли на ПКМ (метод А), постотвержденный при 350°C. Покрытие высушили в конвекционном программируемом шкафу при 205°C в течение 1 часа.
Пример 37. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 96 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила, 4 г 4-(4-аминофенокси)фталонитрила и 71 мл диметилацетамида. Смесь перемешивали до полного растворения компонентов, чтобы получить лак. После этого лак нанесли тонким слоем с помощью малярной кисти на ПКМ (метод А), постотвержденный при 330°C. Затем покрытие высушили в программируемом конвекционном шкафу при 170°C в течение 1 часа.
Пример 38. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 90 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила, 10 г 4,4'-((перфтор-1,4-фенилен)бис(окси))дианилина и 65 мл N-метил-2-пирролидона. Смесь перемешивали до полного растворения компонентов, чтобы получить лак. После этого лак нанесли тонким слоем с помощью малярной кисти на ПКМ (метод А), постотвержденный при 350°C. Затем покрытие высушили в программируемом конвекционном шкафу при 205°C в течение 1 часа.
Пример 39. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 96 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила, 4 г 3,3'-((перфтор-1,4-фенилен)бис(окси))дианилина и 71 мл диметилформамида. Смесь перемешивали до полного растворения компонентов, чтобы получить лак. После этого лак нанесли тонким слоем с помощью малярной кисти на ПКМ (метод А), постотвержденный при 375°C. Затем покрытие высушили в программируемом конвекционном шкафу при 155°C в течение 1 часа.
Пример 40. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 80 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила, 20 г 4-(3-аминофенокси)фталонитрила и 84 мл ацетона. Смесь перемешивали до полного растворения компонентов, чтобы получить лак. После этого лак нанесли тонким слоем с помощью малярной кисти на ПКМ (метод А), предотвержденный при 180°C. Затем покрытие высушили в программируемом конвекционном шкафу при 60°C в течение 1 часа.
Пример 41. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 85 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила, 15 г 4-(4-аминофенокси)фталонитрила и 83 мл метилэтилкетона. Смесь перемешивали до полного растворения компонентов, чтобы получить лак. После этого лак нанесли тонким слоем с помощью малярной кисти на ПКМ (метод Б), постотвержденный при 330°C. Затем покрытие высушили в программируемом конвекционном шкафу при 80°C в течение 1 часа.
Пример 42. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 90 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила, 10 г 4-(3-аминофенокси)фталонитрила и 83 мл метилэтилкетона. Смесь перемешивали до полного растворения компонентов, чтобы получить лак. После этого лак нанесли тонким слоем с помощью малярной кисти на ПКМ (метод Б), предотвержденный при 180°C. Затем покрытие высушили в программируемом конвекционном шкафу при 80°C в течение 1 часа.
Пример 43. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 90 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила, 10 г 4-(4-аминофенокси)фталонитрила и 65 мл N-метил-2-пирролидона. Смесь перемешивали до полного растворения компонентов, чтобы получить лак. После этого лак нанесли тонким слоем с помощью распыления ручным пульверизатором на ПКМ (метод Б), постотвержденный при 375°C. Затем покрытие высушили в программируемом конвекционном шкафу при 205°C в течение 1 часа.
Пример 44. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 90 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила, 10 г 3,3'-((перфтор-1,4-фенилен)бис(окси))дианилин и 83 мл метилэтилкетона. Смесь перемешивали до полного растворения компонентов, чтобы получить лак. После этого лак нанесли тонким слоем с помощью распыления ручным пульверизатором на ПКМ (метод Б), постотвержденный при 350°C. Затем покрытие высушили в программируемом конвекционном шкафу при 80°C в течение 1 часа.
Пример 45. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 16 г 4,4'-((((перфтор-1,4-фенилен)бис(окси))бис(3,1-фенилен))бис(окси))дифталонитрила, 64 г 4,4'-(((((1,4-фениленбис(окси))бис(2,3,5,6-тетрафтор-4,1-фенилен))бис(окси))бис(4,1-фенилен))бис(окси))дифталонитрила, 20 г 4-(3-аминофенокси)фталонитрила и 84 мл ацетона. Смесь перемешивали до полного растворения компонентов, чтобы получить лак. После этого лак нанесли тонким слоем с помощью малярной кисти на ПКМ (метод А), постотвержденный при 330°C. Затем покрытие высушили в программируемом конвекционном шкафу при 60°C в течение 1 часа.
Отверждение покрытий:
Образцы ПКМ с нанесенным покрытием помещали в термошкаф. Температуру повышали до 180°C со скоростью не более 2°C/мин и выдерживали в течение 3 часов. Затем нагревали термошкаф до температуры постотверждения (330-375°C) со скоростью 10 °C/ч и выдерживали в течение 8 часов. Температура постотверждения зависит от выбранного в каждом конкретном случае ПКМ. Если использовали ПКМ с определенной температурой постотверждения, то конечная температура обработки покрытия была такой же. Если же использовали предотвержденный при 180°C ПКМ, то конечную температуру обработки покрытия варьировали в интервале 330-375°C.
Пример 46. ПКМ с нанесенным покрытием из примера 20 поместили в термошкаф. Образец выдерживали по следующей программе: нагрев до 180°C со скоростью 2°C/мин, выдержка 3 часа и нагрев до 330°C со скоростью 10°C/ч, выдержка 8 часов.
Пример 47. ПКМ с нанесенным покрытием из примера 22 поместили в термошкаф. Образец выдерживали по следующей программе: нагрев до 180°C со скоростью 2°C/мин, выдержка 3 часа и нагрев до 375 °C со скоростью 10°C/ч, выдержка 8 часов.
Пример 48. ПКМ с нанесенным покрытием из примера 29 поместили в термошкаф. Образец выдерживали по следующей программе: нагрев до 180°C со скоростью 2°C/мин, выдержка 3 часа и нагрев до 330°C со скоростью 10°C/ч, выдержка 8 часов.
Пример 49. ПКМ с нанесенным покрытием из примера 32 поместили в термошкаф. Образец выдерживали по следующей программе: нагрев до 180°C со скоростью 2°C/мин, выдержка 3 часа и нагрев до 350 °C со скоростью 10 °C/ч, выдержка 8 часов.
Пример 50. ПКМ с нанесенным покрытием из примера 38 поместили в термошкаф. Образец выдерживали по следующей программе: нагрев до 180°C со скоростью 2°C/мин, выдержка 3 часа и нагрев до 350 °C со скоростью 10 °C/ч, выдержка 8 часов.
Пример 51. ПКМ с нанесенным покрытием из примера 42 поместили в термошкаф. Образец выдерживали по следующей программе: нагрев до 180°C со скоростью 2°C/мин, выдержка 3 часа и нагрев до 375°C со скоростью 10°C/ч, выдержка 8 часов.
Для сравнения эффективности представленных покрытий на ПКМ также наносили защитные покрытия на основе нефторированного фталонитрила 4,4'-(1,3-фениленбис(окси))дифталонитрила в смеси с отвердителем 4,4'-(1,3-фениленбис(окси))дианилином (является типичным отвердителем фталонитрильных смол) и на неорганической основе.
Сравнительный пример 1. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 96 г 4,4'-(1,3-фениленбис(окси))дифталонитрила, 4 г 4,4'-(1,3-фениленбис(окси))дианилина и 71 мл диметилацетамида. Смесь перемешивали до полного растворения компонентов. После этого раствор нанесли тонким слоем с помощью малярной кисти на ПКМ (метод А), постотвержденный при 330°C. Затем покрытие высушили в программируемом конвекционном шкафу при 170°C в течение 1 часа.
Сравнительный пример 2. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 96 г 4,4'-(1,3-фениленбис(окси))дифталонитрил, 4 г 4,4'-(1,3-фениленбис(окси))дианилин и 71 мл диметилацетамида. Смесь перемешивали до полного растворения компонентов. После этого раствор нанесли тонким слоем с помощью распыления ручным пульверизатором на ПКМ (метод Б), постотвержденный при 375 °C. Затем покрытие высушили в программируемом конвекционном шкафу при 170°C в течение 1 часа.
Сравнительный пример 3. В колбу объемом 250 мл, оснащенную магнитной мешалкой, поместили 198 г ортофосфорной кислоты и 33.5 г гидроксида алюминия. Смесь перемешивали 15 минут. После этого смесь нанесли тонким слоем с помощью малярной кисти на ПКМ (метод А), постотвержденный при 350°C. Затем покрытие высушили в программируемом конвекционном шкафу при 60°C в течение 4 часов, после чего повысили температуру до 350°C и выдерживали еще 6 часа.
Сравнительный пример 4. ПКМ с нанесенным покрытием из сравнительного примера 1 поместили в термошкаф. Образец выдерживали по следующей программе: нагрев до 180°C со скоростью 2°C/мин, выдержка 3 часа и нагрев до 330°C со скоростью 10°C/ч, выдержка 8 часов.
Сравнительный пример 5. ПКМ с нанесенным покрытием из сравнительного примера 2 поместили в термошкаф. Образец выдерживали по следующей программе: нагрев до 180°C со скоростью 2°C/мин, выдержка 3 часа и нагрев до 375°C со скоростью 10°C/ч, выдержка 8 часов.
Старение при 350℃:
Образцы ПКМ помещали в заранее разогретый до 350°C программируемый конвекционный шкаф. По истечении времени выдержки (200 или 1000 часов) образцы извлекали. Полученные результаты представлены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕАКТИВНЫЙ РАЗБАВИТЕЛЬ ФТАЛОНИТРИЛЬНЫХ СМОЛ И ТЕРМООТВЕРЖДАЕМАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2018 |
|
RU2712547C1 |
| 4,4'-(((1,4-ФЕНИЛЕНБИС(ОКСИ))БИС(4,1-ФЕНИЛЕН))БИС(ОКСИ))ДИФТАЛОНИТРИЛ | 2018 |
|
RU2708399C1 |
| ТРИФУНКЦИОНАЛЬНЫЙ ФТАЛОНИТРИЛЬНЫЙ МОНОМЕР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И КОМПОЗИЦИЯ СВЯЗУЮЩЕГО НА ЕГО ОСНОВЕ | 2019 |
|
RU2744165C1 |
| МОДИФИЦИРОВАННЫЙ КРЕМНИЙОРГАНИЧЕСКИМИ ФРАГМЕНТАМИ ФТАЛОНИТРИЛЬНЫЙ МОНОМЕР, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СВЯЗУЮЩЕЕ НА ЕГО ОСНОВЕ И ПРЕПРЕГ | 2014 |
|
RU2580927C1 |
| МОДИФИЦИРОВАННЫЙ ФОСФОРОРГАНИЧЕСКИМИ ФРАГМЕНТАМИ МОНОМЕР ФТАЛОНИТРИЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СВЯЗУЮЩЕЕ НА ЕГО ОСНОВЕ И ПРЕПРЕГ | 2016 |
|
RU2638307C1 |
| КОМПОЗИЦИЯ ФТАЛОНИТРИЛЬНОГО СВЯЗУЮЩЕГО ДЛЯ ПОЛИМЕРНЫХ КОМПОЗИЦИОННЫХ МАТЕРИАЛОВ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРНОГО КОМПОЗИЦИОННОГО МАТЕРИАЛА И МАТЕРИАЛ, ПОЛУЧЕННЫЙ ЭТИМ СПОСОБОМ | 2018 |
|
RU2695606C1 |
| Теплостойкое низковязкое связующее для изготовления изделий методами вакуумной инфузии и пропитки под давлением и способ его получения | 2021 |
|
RU2762559C1 |
| ФТАЛОНИТРИЛЬНОЕ СВЯЗУЮЩЕЕ И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ПРЕДНАЗНАЧЕННАЯ ДЛЯ ИЗГОТОВЛЕНИЯ ПОЛИМЕРНОГО КОМПОЗИЦИОННОГО МАТЕРИАЛА | 2021 |
|
RU2789601C1 |
| СПОСОБ ПОЛУЧЕНИЯ МОНО- И ДИФТАЛОНИТРИЛОВ | 1995 |
|
RU2079488C1 |
| БЕЗРАСТВОРНЫЙ СПОСОБ ПОЛУЧЕНИЯ ФТАЛОНИТРИЛЬНОГО ПРЕПРЕГА И ПОЛИМЕРНЫЙ КОМПОЗИЦИОННЫЙ МАТЕРИАЛ НА ЕГО ОСНОВЕ | 2019 |
|
RU2740286C1 |
Группа изобретений относится к полимерным композиционным материалам, полученным из олигомеров фталонитрила в качестве компонента полимеризуемой смолы для защитных покрытий полимерной композиционной матрицы. Материалы с фталонитрильными матрицами могут применяться в деталях двигателей в авиационно-космической промышленности, для высокотемпературной защиты электроники, а также для применения в кораблестроении при изготовлении негорючих деталей интерьера подводных лодок. Олигомер фталонитрила в качестве компонента полимеризуемой смолы для защитных покрытий полимерной композиционной матрицы представляет собой соединение формул (I)
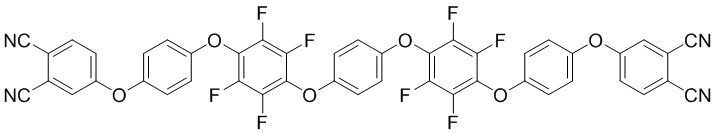
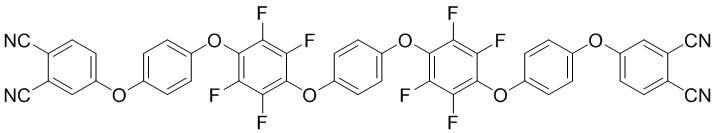 (I),
(I),
или формулы (II)
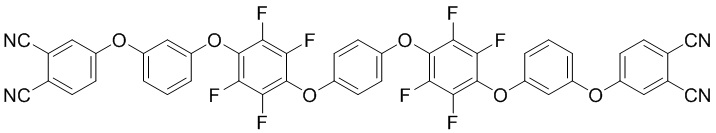
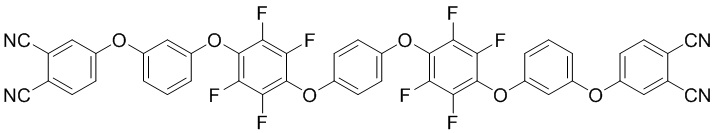 (II),
(II),
или формулы (III)
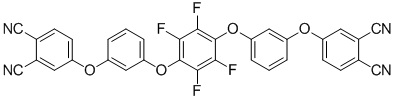 (III)
(III)
Описан способ получения олигомера фталонитрила, характеризующийся последовательным нуклеофильным замещением атомов фтора в гексафторбензоле и/или нитрогруппы в 4-нитрофталонитриле гидроксилсодержащими ароматическими соединениями. Описаны также композиция для приготовления термореактивной смолы, включающая по меньшей мере одно соединение (I) или (II) или их смесь с соединением формулы (III) и отвердитель, способ приготовления термореактивной смолы и способ приготовления термореактивного лака. Термореактивная смола может наноситься на поверхность композита в виде расплава или лака (раствора смолы в органическом растворителе) для создания защитных покрытий. Техническим результатом заявляемой группы изобретений является получение термореактивной смолы, полученной на основе фторсодержащих олигомеров, в отверждённом виде обладающей повышенной термоокислительной стабильностью, которая в качестве защитного покрытия позволяет сохранить (~90%) эксплуатационные характеристики ПКМ даже после 200 ч при 350°C; в отличие от известных покрытий на основе фталонитрильных смол, которые в тех же условиях позволяют сохранить лишь ~70% от начальных эксплуатационных характеристик композита, и характеризующейся простотой нанесения. 5 н. и 24 з.п. ф-лы, 1 табл., 56 пр.
1. Олигомер фталонитрила в качестве компонента полимеризуемой смолы для защитных покрытий полимерной композиционной матрицы, представляющий собой соединение формул (I)
 (I),
(I),
или формулы (II):
 (II),
(II),
или формулы (III):
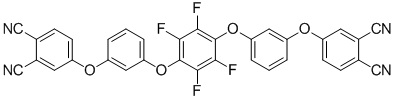 (III)
(III)
2. Способ получения олигомера фталонитрила по п.1, характеризующийся тем, что проводят последовательное нуклеофильное замещение атомов фтора в гексафторбензоле и/или нитрогруппы в 4-нитрофталонитриле гидроксилсодержащими ароматическими соединениями, при этом для получения соединения формулы (I) проводят последовательное нуклеофильное замещение атомов фтора в гексафторбензоле гидрохиноном и нитрогруппы в 4-нитрофталонитриле; для получения соединения формулы (II) проводят последовательное нуклеофильное замещение атомов фтора в гексафторбензоле 4-(3-гидроксифенокси)фталонитрилом или гидрохиноном и атомов фтора в полученном промежуточном соединении гидрохиноном или 4-(3-гидроксифенокси)фталонитрилом; для получения соединения формулы (III) проводят последовательное нуклеофильное замещение атомов фтора в гексафторбензоле 4-(3-гидроксифенокси)фталонитрилом.
3. Способ по п.2, характеризующийся тем, что для получения соединения формулы (I) для нуклеофильного замещения атомов фтора растворяют гидрохинон в высококипящем полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, к раствору добавляют неорганическую соль, взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут, а затем добавляют гексафторбензол, взятый из расчета 2 мольных эквивалента гексафторбензола на 3 мольных эквивалента гидрохинона, полученную реакционную смесь нагревают до 60±5°C и оставляют при перемешивании на 24±2 часа с последующим нагревом до 110±5°C и выдержкой при перемешивании в течение 36±2 часа, затем реакционную смесь добавляют в избыток воды комнатной температуры, взятой в объеме, в 2,5-3,0 превышающем объем используемого растворителя, перемешивают и выдерживают для выпадения осадка, выпавший осадок отфильтровывают и промывают от остатков растворителя, основания и непрореагировавшего гидрохинона, затем высушивают.
4. Способ по п.2, характеризующийся тем, что для получения соединения формулы (I) для замещения нитрогруппы в 4-нитрофталонитриле полученный после нуклеофильного замещения атомов фтора бисфенол растворяют в высококипящем полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, затем добавляют неорганическую соль, взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут, после этого добавляют 4-нитрофталонитрил, взятый из расчета 1 моль 4-нитрофталонитрила на 1 моль участвующей в реакции гидроксильной группы, нагревают реакционную смесь до 80±5°C и оставляют при перемешивании на 24±2 часа, затем реакционную смесь добавляют в избыток воды комнатной температуры, взятой в объеме, в 2,5-3,0 превышающем объем используемого растворителя, перемешивают и выдерживают при комнатной температуре для выпадения осадка, выпавший осадок отфильтровывают и промывают от непрореагировавших остатков и высушивают.
5. Способ по п.3, характеризующийся тем, что для выпадения осадка реакционную смесь после добавления в воду выдерживают 2±0.5 часа.
6. Способ по п.3 или 4, характеризующийся тем, что в качестве высококипящего апротонного растворителя используют N-метил-2-пирролидон, N,N-диметилацетамид или N,N-диметилформамид.
7. Способ по п.3 или 4, характеризующийся тем, что в качестве неорганической соли используют фторид калия, карбонат калия, фосфат калия.
8. Способ по п.3 или 4, характеризующийся тем, что для получения осадка реакционную смесь добавляют в воду комнатной температуры, взятую в объеме, в 2,5-3,0 превышающем объем органического растворителя.
9. Способ по п.3 или 4, характеризующийся тем, что для высушивания осадка его помещают на 12±1 час в сушильный шкаф, разогретый до 100°C.
10. Способ по п.2, характеризующийся тем, что для получения соединения формулы (II) для нуклеофильного замещения атомов фтора растворяют 4-(3-гидроксифенокси)фталонитрил или гидрохинон в полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, к раствору добавляют неорганическую соль, взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут, а затем добавляют гексафторбензол, взятый из расчета 1 мольный эквивалент гексафторбензола на 1 мольный эквивалент 4-(3-гидроксифенокси)фталонитрила или гидрохинона, полученную реакционную смесь нагревают до 30±5°C и оставляют при перемешивании на 96±2 часа, затем реакционную смесь фильтруют через тонкий слой силикагеля, осадок промывают, фильтрат упаривают и проводят флэш-хроматографию.
11. Способ по п.2, характеризующийся тем, что для получения соединения формулы (II) для замещения нитрогруппы в 4-нитрофталонитриле полученное после нуклеофильного замещения атомов фтора соединение, взятое из расчета 1 моль на 1 моль участвующей в реакции гидроксильной группы, добавляют к раствору гидрохинона или 4-(3-гидроксифенокси)фталонитрил в полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, с добавлением неорганической соли, взятой из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, полученную реакционную смесь нагревают до 100±5°C и оставляют при перемешивании на 120±2 часа, затем реакционную смесь добавляют в большой избыток воды комнатной температуры, перемешивают и выдерживают до выпадения осадка, выпавший осадок отфильтровывают и промывают от непрореагировавших остатков и высушивают.
12. Способ по п.10 или 11, характеризующийся тем, что для выпадения осадка реакционную смесь после добавления в воду выдерживают 2±0.5 часа.
13. Способ по п.10 или 11, характеризующийся тем, что в качестве полярного апротонного растворителя используют ацетон, N-метил-2-пирролидон, N,N-диметилацетамид или N,N-диметилформамид.
14. Способ по п.10 или 11, характеризующийся тем, что в качестве неорганической соли используют фторид калия, карбонат калия, фосфат калия.
15. Способ по п.10 или 11, характеризующийся тем, что для получения осадка реакционную смесь добавляют в воду комнатной температуры, взятую в объеме, в 2,5-3,0 превышающем объем органического растворителя.
16. Способ по п.10 или 11, характеризующийся тем, что для высушивания осадка его помещают на 12±1 час в сушильный шкаф, разогретый до 100°C.
17. Способ по п.10 или 11, характеризующийся тем, что отфильтрованный осадок промывают 3 раза по 10 мл ацетона.
18. Способ по п.10, характеризующийся тем, что флэш-хроматографию проводят с использованием смеси метиленхлорила/петролейный эфир, взятых в объемном соотношении 1/1.
19. Способ по п.2, характеризующийся тем, что для получения соединения формулы (III) растворяют 4-(3-гидроксифенокси)фталонитрил в полярном апротонном растворителе, взятом из расчета ~5-10 мл на 1 г смеси растворимых компонентов, затем добавляют неорганическую соль, взятую из расчета 1-1.25 моль на 1 моль участвующей в реакции гидроксильной группы, после чего реакционную смесь перемешивают в течение 30±5 минут, добавляют гексафторбензол, взятый из расчета 1 мольный эквивалент гексафторбензола на 2 мольных эквивалента 4-(3-гидроксифенокси)фталонитрила, полученную реакционную смесь нагревают до 60±5°C и оставляют при перемешивании на 96±2 часа, затем реакционную смесь фильтруют через тонкий слой силикагеля, полученный осадок промывают, фильтрат упаривают, проводят флэш-хроматографию.
20. Способ по п.19, характеризующийся тем, что в качестве полярного апротонного растворителя используют ацетон, N,N-диметилацетамид, N,N-диметилформамид или N-метил-2-пирролидон.
21. Способ по п.19, характеризующийся тем, что в качестве неорганической соли используют фторид калия, карбонат калия, фосфат калия.
22. Способ по п.19, характеризующийся тем, что для выпадения осадка реакционную смесь после добавления в воду выдерживают 2±0.5 часа.
23. Способ по п.19, характеризующийся тем, что для получения осадка реакционную смесь добавляют в воду комнатной температуры, взятую в объеме, в 2,5-3,0 превышающем объем органического растворителя.
24. Способ по п.19, характеризующийся тем, что для высушивания осадка его помещают на 12±1 час в сушильный шкаф, разогретый до 100°C.
25. Способ по п.19, характеризующийся тем, что отфильтрованный осадок промывают 3 раза по 10 мл ацетона.
26. Способ по п.19, характеризующийся тем, что флэш-хроматографию проводят с использованием смеси метиленхлорила/петролейный эфир, взятых в объемном соотношении 1/1.
27. Композиция для приготовления термореактивной смолы, характеризующаяся тем, что включает по меньшей мере одно соединение формулы (I), или (II), или их смесь с соединением формулы (III) по п.1 и отвердитель, выбранный из группы, включающей 4-(4-аминофенокси)фталонитрил, 4-(3-аминофенокси)фталонитрил, 4,4'-((перфтор-1,4-фенилен)бис(окси))дианилин, 3,3'-((перфтор-1,4-фенилен)бис(окси))дианилин, взятые в концентрации от 4 до 20 масс.%.
28. Способ приготовления термореактивной смолы, характеризующийся тем, что по меньшей мере одно соединение формулы (I), или (II), или их смесь с соединением формулы (III) по п.1 расплавляют при температуре 120-180° и при перемешивании добавляют отвердитель, полученную смесь перемешивают в течение 15-60 минут, после чего смесь охлаждают до отвердевания, при этом отвердитель выбирают из группы, включающей 4-(4-аминофенокси)фталонитрил, 4-(3-аминофенокси)фталонитрил, 4,4'-((перфтор-1,4-фенилен)бис(окси))дианилин, 3,3'-((перфтор-1,4-фенилен)бис(окси))дианилин, взятые в концентрации от 4 до 20 масс.%.
29. Способ приготовления термореактивного лака из термореактивной смолы, полученной способом по п.28, характеризующийся тем, что смолу растворяют в растворителе, выбранном из группы ацетон, метилэтилкетон, диметилформамид, диметилацетамид, н-метилпирролидон, взятом в количестве, обеспечивающем получение раствора для нанесения на поверхность ПКМ.
| MINJIE WU ET AL | |||
| HIGH-PERFORMANCE FUNCTIONAL PHTHALONITRILE RESIN WITH A LOW MELTING POINT AND LOW DIELECTRIC CONSTANT, SOFT MATTER, 11.01.2020 | |||
| МОДИФИЦИРОВАННЫЙ КРЕМНИЙОРГАНИЧЕСКИМИ ФРАГМЕНТАМИ ФТАЛОНИТРИЛЬНЫЙ МОНОМЕР, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СВЯЗУЮЩЕЕ НА ЕГО ОСНОВЕ И ПРЕПРЕГ | 2014 |
|
RU2580927C1 |
| КОМПОЗИЦИЯ ФТАЛОНИТРИЛЬНОГО СВЯЗУЮЩЕГО ДЛЯ ПОЛИМЕРНЫХ КОМПОЗИЦИОННЫХ МАТЕРИАЛОВ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРНОГО КОМПОЗИЦИОННОГО МАТЕРИАЛА И МАТЕРИАЛ, ПОЛУЧЕННЫЙ ЭТИМ СПОСОБОМ | 2018 |
|
RU2695606C1 |
| МОДИФИЦИРОВАННЫЙ ФОСФОРОРГАНИЧЕСКИМИ ФРАГМЕНТАМИ МОНОМЕР ФТАЛОНИТРИЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СВЯЗУЮЩЕЕ НА ЕГО ОСНОВЕ И ПРЕПРЕГ | 2016 |
|
RU2638307C1 |
| РЕАКТИВНЫЙ РАЗБАВИТЕЛЬ ФТАЛОНИТРИЛЬНЫХ СМОЛ И ТЕРМООТВЕРЖДАЕМАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2018 |
|
RU2712547C1 |
| WO 2017172515 A1, 05.10.2017 | |||
| JP 5221952 A, 31.08.1993 | |||
| JP 2006199599 A, 03.08.2006 | |||
| JP 8333322 | |||

