Область, к которой относится изобретение
Настоящее изобретение относится к производным нафталинкарбоксамида (нафтамида), которые обладают активностью ингибирования протеинкиназ и ингибирования гистондеацетилаз, к способам их получения и их клинического применения при лечении заболеваний, связанных с нарушением активности протеинкиназы и нарушением активности гистондеацетилазы.
Предшествующий уровень техники
Протеинкиназы представляют собой семейство ферментов, которые катализируют фосфорилирование белков, в частности, гидроксигруппы определенных остатков тирозина, серина и треонина в белках. Протеинкиназы играют ключевую роль в регуляции широкого разнообразия клеточных процессов, включая метаболизм, клеточную пролиферацию, клеточную дифференциацию, выживание клеток, реакцию организма хозяина на факторы окружающей среды, иммунный ответ и ангиогенез. Многие заболевания связаны с патологическими клеточными реакциями, запускаемыми регуляцией протеинкиназы. Эти заболевания включают воспалительные заболевания, аутоиммунные заболевания, онкологические заболевания, заболевания нервной системы и нейродегенеративные заболевания, сердечно-сосудистые заболевания, метаболические заболевания, аллергии, астму и заболевания, связанные с гормональными нарушениями (Tan, S-L., 2006, J. Immunol., 176: 2872-2879; Healy, A. et al., 2006, J. Immunol., 177: 1886-1893; Salek-Ardakani, S. et al., 2005, J. Immunol., 175: 7635-7641; Kim, J. et al., 2004, J. Clin. Invest., 114: 823-827). Поэтому предпринимались значительные усилия для идентификации ингибиторов протеинкиназы, которые эффективны в качестве терапевтических средств против указанных заболеваний.
Протеинкиназы могут обычно делиться на два класса: протеинтирозинкиназы (PTK) и серин-треонинкиназы (STK).
Протеинтирозинкиназы (PTK) могут делиться на два класса: нетрансмембранные тирозинкиназы и имеющие тирозинкиназную активность трансмембранные рецепторы факторов роста (RTK). В настоящее время были идентифицированы, по меньшей мере, 19 отчетливых подсемейств RTK, такие как рецептор эпидермального фактора роста (EGFR), рецептор сосудистого эндотелиального фактора роста (VEGFR), рецептор тромбоцитарного фактора роста (PDGFR) и рецептор фактора роста фибробластов (FGFR).
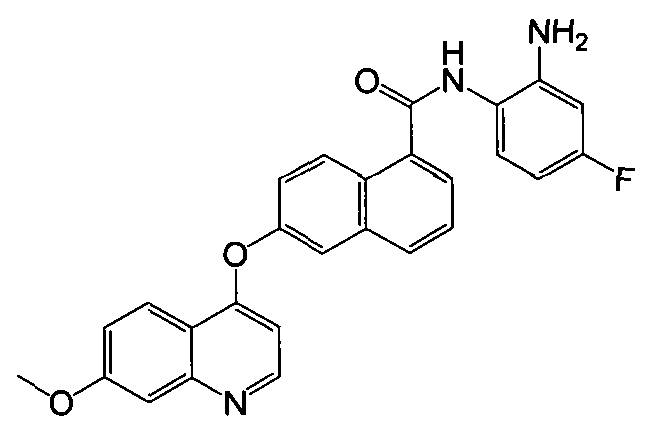
Семейство рецептора эпидермального фактора роста (EGFR) включает четыре имеющих тирозинкиназную активность трансмембранных рецепторов факторов роста: HER1, HER2, HER3 и HER4. Связывание специфического набора лигандов рецептора содействует димеризации EGFR и приводит к аутофосфорилированию рецепторов на остатках тирозина (Arteaga, C-L., 2001, Curr. Opin. Oncol., 6: 491-498). После аутофосфорилирования рецептора, становятся активированными несколько находящихся ниже по ходу транскрипции путей передачи сигналов EGFR. Пути передачи сигналов EGFR связывали с неопластическими процессами, включая развитие клеточного цикла, ингибирование апоптоза, подвижность, инвазию и метастазирование опухолевых клеток. Активация EGFR также стимулирует сосудистый эндотелиальный фактор роста (VEGF), который представляет собой первичный индуктор ангиогенеза (Petit, A-M. et al., 1997, Am. J. Pathol., 151: 1523-1530). В экспериментальных моделях, разрегуляция опосредованных EGFR путей передачи сигналов связана с онкогенезом (Wikstrand, C-J. et al., 1998, J Natl Cancer Inst., 90: 799-800). Мутации, ведущие к непрерывной активации амплификации и избыточной экспрессии протеинов EGFR, наблюдаются при многих опухолях человека, включая опухоли молочных желез, легких, яичников и почек. Данные мутации являются определяющими агрессивность опухолей (Wikstrand, C-J. et al.,1998, J Natl Cancer Inst., 90: 799-800). Избыточная экспрессия EGFR часто наблюдается при немелкоклеточном раке легких (NSCLC). Активность EGFR может ингибироваться или блокированием внеклеточного домена связывания лиганда с использованием анти-EGFR антител, или путем использования мелких молекул, которые ингибируют тирозинкиназу EGFR, таким образом приводя к ингибированию находящихся ниже по ходу транскрипции компонентов пути EGFR (Mendelsohn, J., 1997, Clin. Can. Res., 3: 2707-2707).
Сосудистый эндотелиальный фактор роста (VEGF) секретируется почти всеми сулидными опухолями и связанной с опухолью стромой в ответ на гипоксию. Он высоко специфичен для сосудистого эндотелия и регулирует и пролиферацию, и проницаемость сосудов. Избыточная экспрессия уровней VEGF коррелируется с увеличенной микрососудистой плотностью, рецидивом онкологических заболеваний и уменьшенным выживанием (Parikh, A-A., 2004;, Hematol. Oncol. Clin. N. Am., 18:951-971). Существуют 6 различных лигандов рецептора VEGF, с VEGF-A по -E и плацентарный фактор роста. Лиганды связываются со специфическими рецепторами на эндотелиальных клетках, главным образом, VEGFR-2. Связывание VEGF-A с VEGFR-1 вызывает миграцию эндотелиальных клеток. Связывание с VEGFR-2 вызывает пролиферацию, проницаемость и выживание эндотелиальных клеток. Считается, что VEGFR-3 опосредует лимфангиогенез. Связывание VEGF с рецепторами VEGFR-2 приводит к активации и аутофосфорилированию внутриклеточных доменов тирозинкиназы, которые далее запускают другие внутриклеточные каскады процессов передачи сигналов (Parikh, A-A., 2004, Hematol. Oncol. Clin. N. Am., 18:951-971).
Серин-треонинкиназы (STK) являются преимущественно внутриклеточными, хотя существует несколько рецепторных киназ типа STK. STK представляют собой самые распространенные формы цитозольных киназ, которые выполняют свою функцию в части цитоплазмы, отличной от цитоплазматических органелл и цитоскелета.
Гликоген синтаза-киназа-3 (GSK-3) представляет собой серин-треонинпротеинкиназу, состоящую из α- и β-изоформ, каждая из которых кодируется определенными генами. Было обнаружено, что GSK-3 фосфорилирует и модулирует активность ряда регуляторных белков. GSK-3 связывали с развитием различных заболеваний, включая сахарный диабет, болезнь Альцгеймера, расстройства ЦНС, такие как маниакально-депрессивное расстройство и нейродегенеративные заболевания, и гипертрофия кардиомиоцитов (Haq, et al., 2000, J. Cell Biol., 151: 117).
Aurora-2 представляет собой серин-треонинпротеинкиназу, которую связывали с онкологическими заболеваниями у людей, такими как рак ободочной кишки, рак молочной железы и другие сулидные опухоли. Считают, что данная киназа участвует в фосфорилировании белков, которые регулируют клеточный цикл. В частности, Aurora-2 может играть роль в регуляции точной сегрегации хромосом во время митоза.
Нарушение регуляции клеточного цикла может привести к клеточной пролиферации и другим патологическим процессам. Было обнаружено, что в ткани рака ободочной кишки человека, белок Aurora-2 избыточно экспрессирован (Schumacher, et al., 1998, J. Cell Biol., 143: 1635-1646; Kimura et al., 1997, J. Biol. Chem., 272: 13766-13771).
Циклин-зависимые киназы (CDK) представляют собой серин-треонинпротеинкиназу, которая регулирует деление клеток млекопитающих. К настоящему времени были идентифицированы 9 субъединиц киназ (CDK 1-9). Каждая киназа ассоциируется с определенным регуляторным партнером и вместе составляет активную каталитическую часть. Неконтролируемая пролиферация представляет собой отличительный признак злокачественных клеток, и нарушение регуляции функции CDK происходит с высокой частотой при многих важных сулидных опухолях. CDK2 и CDK4 представляют особый интерес, потому что их активность часто нарушается при широком разнообразии онкологических заболеваний людей.
Raf-киназа, находящийся ниже по ходу транскрипции эффектор онкопротеина ras, представляет собой ключевой медиатор путей трансдукции сигналов от клеточной поверхности к ядру клетки. Ингибирование raf-киназы коррелировалось in vitro и in vivo с ингибированием роста разнообразных типов опухолей человека (Monia et al., 1996, Nat. Med., 2: 668-675).
Другие серин-треонинкиназы включают протеинкиназы A, B и C. Указанные киназы, известные как PKA, PKB и PKC, играют ключевые роли в путях трансдукции сигналов.
Предпринималось множество попыток идентификации небольших молекул, которые действуют в качестве ингибиторов протеинкиназ, которые можно применять при лечении заболеваний, связанных с аномальной активностью протеинкиназ. Например, циклические соединения (патент США № 7151096), бициклические соединения (патент США № 7189721), трициклические соединения (патент США № 7132533), (2-оксиндол-3-метилиден), производные уксусной кислоты (патент США № 7214700), производные 3-(4-амидопиррол-2-илметилиден)-2-индолинона (патент США № 7179910), конденсированные производные пиразола (патент США № 7166597), соединения аминофуразана (патент США № 7157476), пиррол-замещенные 2-индолиноновые соединения (патент США № 7125905), триазольные соединения (патент США № 7115739), пиразолиламин-замещенные хиназолиновые соединения (патент США № 7098330) и индазольные соединения (патент США № 7041687) - все были описаны в качестве ингибиторов протеинкиназы. Несколько ингибиторов протеинкиназы, таких как гливек, сутен и сорафениб, были успешно утверждены FDA (Администрацией пищевых продуктов и лекарственных средств США) для противораковой терапии. Их клиническое применение продемонстрировало явные преимущества перед существующими видами химиотерапевтического лечения, стимулируя сохраняющиеся интересы к инновации основанных на механизмах способов лечения и усовершенствованию химических каркасов для выявления новых соединений с превосходной пероральной биодоступностью, лучшей противоопухолевой активностью и более низкой токсичностью.
Краткое изложение сущности изобретения
Одной целью настоящего изобретения является получение определенных производных нафтамида, которые способны селективно ингибировать протеинкиназы и гистондеацетилазы. Другой целью настоящего изобретения является разработка способов получения указанных соединений.
Другой целью настоящего изобретения является обеспечение клинического применения указанных соединений при лечении заболеваний, связанных с аномальной активностью протеинкиназ и аномальной активностью гистондеацетилаз.
Краткое описание чертежей
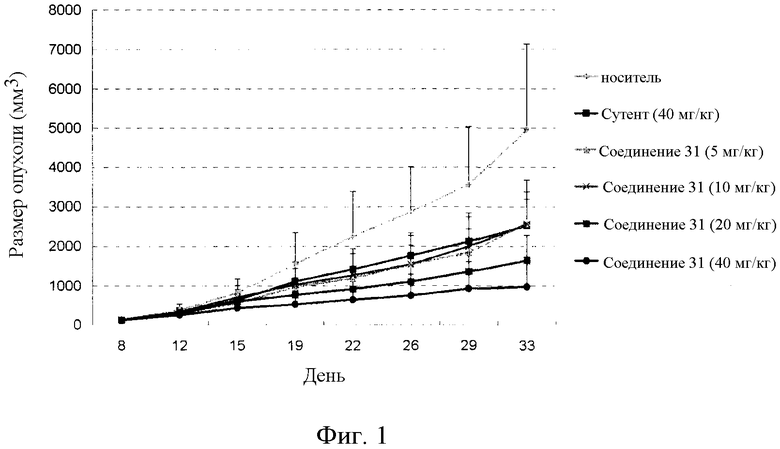
Фиг.1 графически иллюстрирует противоопухолевую активность соединения 31 у голой мыши с трансплантированной опухолью рака легких человека A549, где Vehicle представляет носитель, Sutent представляет существующее лекарственное средство сунитиниб и comp 31 представляет соединение 31.
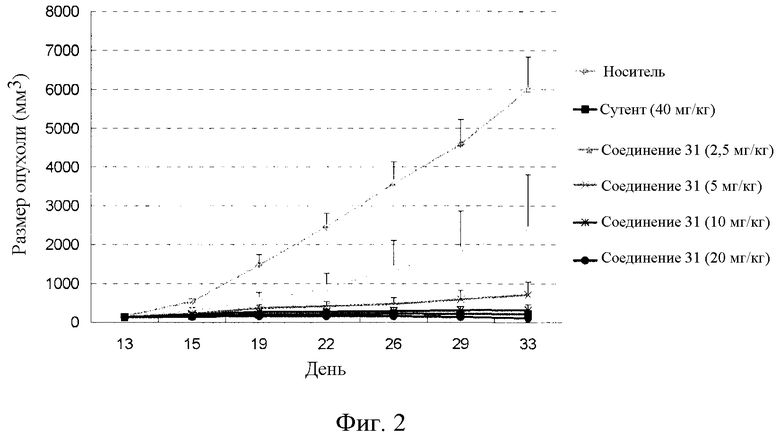
Фиг.2 графически иллюстрирует противоопухолевую активность соединения 31 у голой мыши с трансплантированной опухолью рака ободочной кишки человека HCT-8, где Vehicle представляет носитель, Sutent представляет существующее лекарственное средство сунитиниб и comp 31 представляет соединение 31.
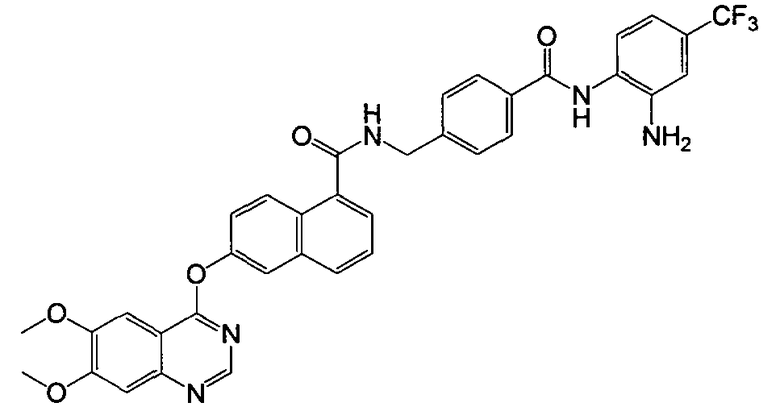
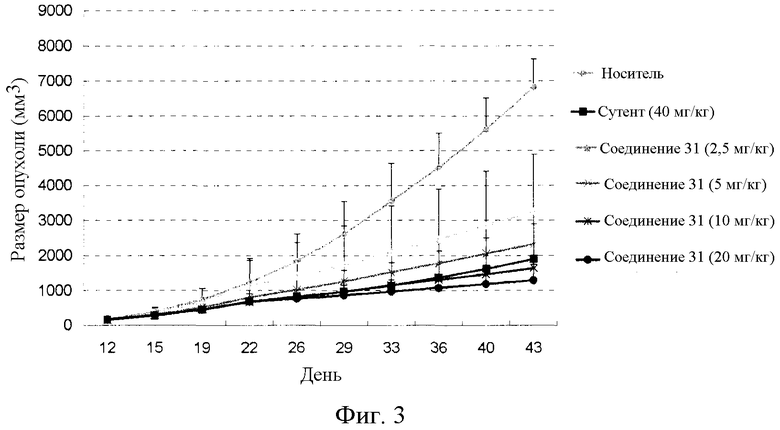
Фиг.3 графически иллюстрирует противоопухолевую активность соединения 31 у голой мыши с трансплантированной опухолью рака печени человека SSMC7721, где Vehicle представляет носитель, Sutent представляет существующее лекарственное средство сунитиниб и comp 31 представляет соединение 31.
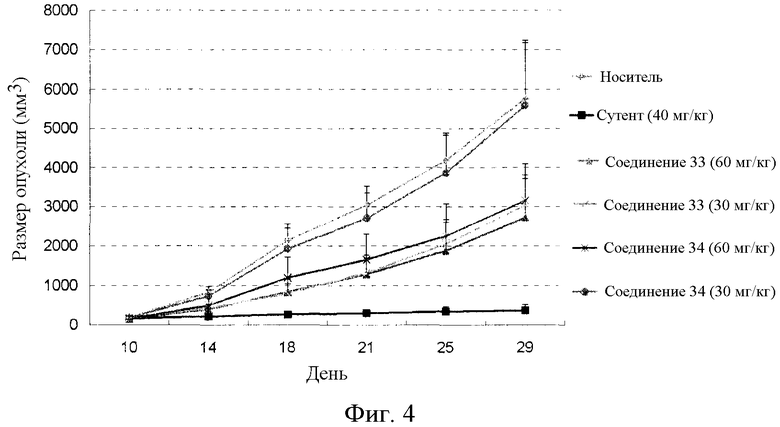
Фиг.4 графически иллюстрирует противоопухолевую активность соединения 33 и соединения 34 у голой мыши с трансплантированной опухолью рака ободочной кишки человека HCT-8, где Vehicle представляет носитель, Sutent представляет существующее лекарственное средство сунитиниб, comp 33 представляет соединение 33, и com 34 представляет соединение 34.
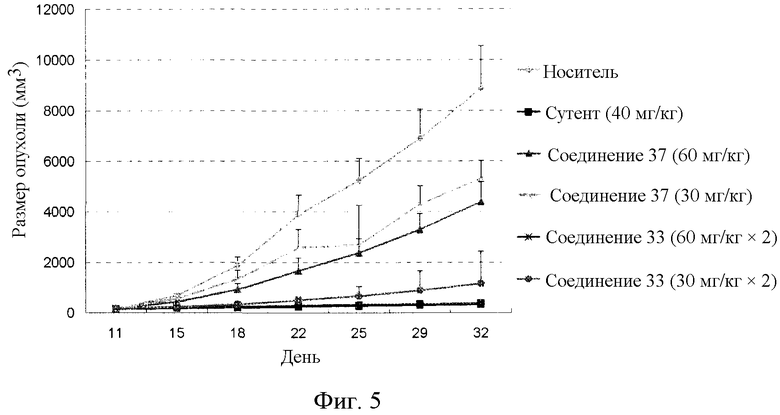
Фиг.5 графически иллюстрирует противоопухолевую активность соединения 33 и соединения 37 у голой мыши с трансплантированной опухолью рака ободочной кишки человека HCT-8, где Vehicle представляет носитель, Sutent представляет существующее лекарственное средство сунитиниб, comp 33 представляет соединение 33, и com 37 представляет соединение 37.
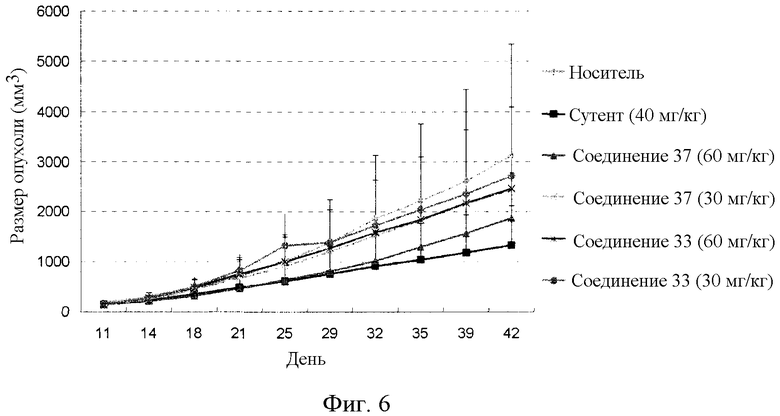
Фиг.6 графически иллюстрирует противоопухолевую активность соединения 33 и соединения 37 у голой мыши с трансплантированной опухолью рака печени человека SSMC7721, где Vehicle представляет носитель, Sutent представляет существующее лекарственное средство сунитиниб, comp 33 представляет соединение 33, и com 37 представляет соединение 37.
Детальное описание изобретения
Белки гистондеацетилазы (HDAC) играют ключевую роль в регуляции генной экспрессии in vivo путем изменения доступности транскрипционных факторов для геномной ДНК. В частности, белки HDAC удаляют ацетильную группу остатков ацетиллизина на гистонах, что может привести к нуклеосомальному ремоделированию (Grunstein, M., 1997, Nature, 389: 349-352). Белки HDAC, ввиду их руководящей роли в генной экспрессии, связаны с разнообразными клеточными явлениями, включая регуляцию клеточного цикла, клеточную пролиферацию, дифференциацию, перепрограммирование генной экспрессии и развитие онкологических заболеваний (Ruijter, A-J-M., 2003, Biochem. J., 370: 737-749; Grignani, F., 1998, Nature, 391: 815-818; Lin, R-J., 1998, 391: 811-814; Marks, P-A., 2001, Nature Reviews Cancer, 1: 194). Патологическое деацетилирование, вызванное нарушением регуляции деацетилирования гистонов, связывали с различными заболеваниями, такими как синдром Рубинштейна-Тэйби, синдром хрупкой X-хромосомы, нейродегенеративные заболевания, сердечно-сосудистые и метаболические заболевания, ревматоидное заболевание, лейкоз и другие виды онкологических заболеваний (Langley B et al., 2005, Current Drug Targets-CNS & Neurological Disorders, 4: 41-50). В экспериментах было продемонстрировано, что у людей и животных ингибиторы HDAC уменьшают рост опухолей, включая рак легких, рак желудка, рак молочной железы, рак предстательной железы, лимфому и тому подобные (Dokmanovic, M.,2005, J. Cell Biochenm., 96: 293-304).
HDAC млекопитающих можно разделить на три класса в соответствии с гомологией последовательности. Класс I состоит из белков, подобных дрожжевой Rpd3 (HDAC 1, 2, 3, 8 и 11). Класс II состоит из белков, подобных дрожжевой HDA1 (HDAC 4, 5, 6, 7, 9 и 10). Класс III состоит из белков, подобных дрожжевой SIR2 (SIRT 1, 2, 3, 4, 5, 6 и 7).
Активность HDAC1 связана с клеточной пролиферацией, характерным признаком онкологического заболевания. В частности, клетки млекопитающих с нокдауном экспрессии HDAC1 с использованием siРНК (малой интерферирующей РНК) обладали антипролиферативным действием (Glaser, K-B., 2003, Biochem. Biophys. Res. Comm., 310: 529-536). Хотя у мыши с нокдауном HDAC1 была эмбриональная летальная мутация, полученные стволовые клетки проявляли измененный клеточный рост (Lagger, G., 2002, EMBO J., 21: 2672-2681). Мышиные клетки, избыточно экспрессирующие HDAC1, продемонстрировали удлинение фаз G2 и M и сниженную скорость роста (Bartl. S., 1997, Mol. Cell Biol., 17: 5033-5043). Поэтому опубликованные данные подразумевают участие HDAC1 в регуляции клеточного цикла и пролиферации клеток.
HDAC2 регулирует экспрессию многих плодных миокардиальных белковых изоформ. Недостаточность HDAC2 или химическое ингибирование гистондеацетилазы могут предотвратить повторную экспрессию фетальных генов и ослабляют сердечную гипертрофию. Устойчивость к гипертрофии связана с увеличенной экспрессией гена, кодирующего инозитол полифосфат-5-фосфатазу f (Inpp5f), приводя к активации гликоген синтазы киназы 3β (Gsk3β) посредством инактивации прото-онкогена тимомы (Akt) и 3-фосфоинозитид-зависимой протеинкиназы-1 (Pdk1). Напротив, у HDAC2 трансгенных мышей имелась усиленная гипертрофия, связанная с инактивированной Gsk3β. Химическое ингибирование активированной Gsk3β позволило взрослым с недостаточностью HDAC2 стать чувствительной к гипертрофической стимуляции. Данные результаты свидетельствуют о том, что HDAC2 представляет собой важную молекулярную мишень ингибиторов HDAC в сердце и что и HDAC2, и Gsk3β представляют собой компоненты регуляторного пути, обеспечивая привлекательную терапевтическую мишень для лечения сердечной гипертрофии и сердечной недостаточности (Trivedi, C-M., 2007, Nat. Med,. 13: 324-331).
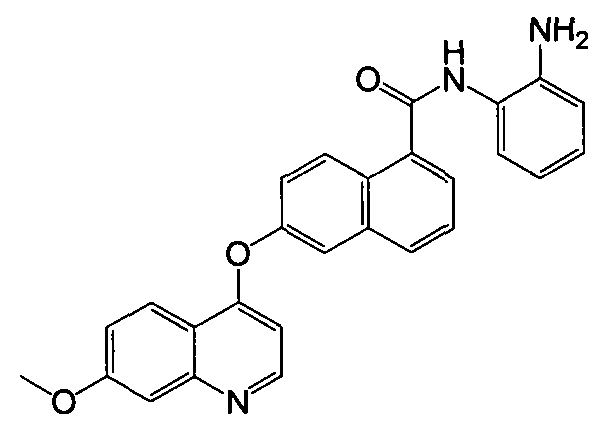
HDAC3 максимально экспрессированы в пролиферирующих клетках крипт в здоровом кишечнике. Сайленсинг экспрессии HDAC3 в линиях клеток рака ободочной кишки приводил к ингибированию роста клеток, уменьшению выживания клеток и увеличенному апоптозу. Подобные результаты наблюдались для HDAC2 и, в меньшей степени, для HDAC1. Сайленсинг гена HDAC3 также селективно вызывал экспрессию щелочной фосфатазы, маркера созревания клеток ободочной кишки. Сверхэкспрессия HDAC3 ингибировала базальную и вызванную бутиратом транскрипцию p21, тогда как сайленсинг HDAC3 стимулировал промотерную активность и экспрессию p21. Указанные данные идентифицируют HDAC3 в качестве гена, регуляция которого нарушается при раке ободочной кишки человека и в качестве нового регулятора созревания клеток ободочной кишки и экспрессии p21 (Wilson, A-J., 2006, J. Biol. Chem., 281: 13548-13558).
HDAC6 представляет собой подтип семейства HDAC, который деацетилирует альфа-тубулин и увеличивает подвижность клеток. С использованием количественной полимеразной цепной реакции с обратной транскрипцией и вестерн-блоттинга на девяти линиях клеток, полученных из плоскоклеточной карциномы ротовой полости (OSCC) и нормальных кератиноцитов ротовой полости (NOK), мРНК HDAC6 и экспрессия белков обычно стимулировались во всех клеточных линиях, по сравнению с NOK. Анализ иммунофлюоресценции выявил белок HDAC6 в цитоплазме линий клеток OSCC. Подобно линиям клеток OSCC, стимуляция HDAC6 была очевидна по уровню и мРНК (74%), и белка (51%) первичных опухолей OSCC человека. Среди анализируемых клинических переменных величин было обнаружено, что клиническая стадия опухоли связана с состояниями экспрессии HDAC6. Анализ указал на значительное различие уровня экспрессии HDAC6 между опухолями на ранней стадии (стадии I и II) и на поздних стадиях (стадии III и IV) (P=0,014). Данные результаты свидетельствуют о том, что экспрессия HDAC6 может коррелировать с агрессивностью опухоли, и предоставляют ключи для планирования новых видов лечения (Sakuma, T., 2006, Int. J. Oncol., 29: 117-124).
Эпигенетический сайленсинг функциональных хромосом HDAC представляет собой один из основных механизмов, происходящих при многих патологических процессах, при которых связанные с функцией гены репрессируются или перепрограммируются активностью HDAC, что ведет к утрате фенотипов при регуляции конечной дифференциации, созревания и роста и к утрате функциональной активности тканей. Например, гены-суппрессоры опухолей часто подвергаются сайленсингу во время развития онкологических заболеваний, и ингибитор HDAC может подавлять экспрессию этих генов-суппрессоров опухолей, приводя к ингибированию роста и дифференциации клеток (Glaros S et al., 2007, Oncogene June 4, в печати; Mai, A, et al., 2007, Int J. Biochem Cell Bio., April 4, в печати; Vincent A. et al., 2007, Oncogene, April 30, в печати; неопубликованные данные заявителей). Репрессия структурных генов, таких как FXN при атаксии Фридриха и SMN при спинномозговой мышечной атрофии, может устраняться ингибиторами HDAC, что приводит к реэкспрессии генов FXN и SMN и возобновлению их функций в тканях (Herman D et al., 2006, Nature Chemical Biology, 2(10): 551-8; Avila AM et al., 2007, J Clinic Investigation, 117(3): 659-71; de Bore J, 2006, Tissue Eng. 12(10): 2927-37). Индукция экспрессии всего семейства генов MHC II посредством перепрограммирования «горячего пятна» HDAC в хромосоме 6p21-22 ингибитором HDAC дополнительно расширяет эпигенетическую модуляцию иммунного распознавания и иммунного ответа (Gialitakis M et al., 2007, Nucleic Acids Res., 34(1);765-72).
Были идентифицированы несколько классов ингибиторов HDAC, включая (1) короткоцепочечные жирные кислоты, например, бутират и фенилбутират; (2) органические гидроксамовые кислоты, например, субероиланилидгидроксамовую кислоту (SAHA) и трихостатин A (TSA); (3) циклические тетрапептиды, содержащие часть в виде 2-амино-8-оксо-9,10-экспоксидеканоила (AOE), например, трапоксин и HC-токсин; (4) циклические тетрапептиды без части в виде AOE, например, апицидин и FK228; и (5) бензамиды, например, MS-275 (EP0847992A1, US2002/0103192A1, WO02/26696A1, WO01/70675A2, WO01/18171A2). Хотя HDAC унаследовал очень перспективные биологические роли в качестве лекарственной мишени, успех препарата SAHA, выпускаемого компанией Merck, в настоящее время ограничивается лишь лечением кожной T-клеточной лимфомы, в то время как не было сообщений о высокой эффективности данного лечения при основных сулидных опухолях. Поэтому еще имеется потребность в обнаружении новых соединений с более высокой ингибирующей активностью в отношении HDAC и активностью против онкологических заболеваний, с более селективным ингибированием различных подтипов HDAC и более низкой токсичностью.
Любимой метафорой разработчиков лекарственных средств против онкологических заболеваний в течение длительного времени была прицельная терапия. Была надежда на создание лекарственного средства, которое может поражать опухолевые клетки как одну специфическую мишень, уничтожать опухолевые клетки, в то же время нормальные клетки оставляя неповрежденными. Однако злокачественные клетки могут использовать множественные биологические пусковые механизмы и пути для роста и распространения по всему организму. Поражение их в виде одной мишени также вызовет их перегруппировку и перераспределение по новым путям роста. Осознание этого привело к разработке способов комбинированной прицельной терапии, которые становятся новой парадигмой для лечения онкологических заболеваний. В настоящее время разрабатываются несколько ингибиторов киназы, имеющих множество мишеней, и два из них - сорафениб и сутен, уже утверждены в США. Например, сорафениб, разработанный компанией Bayer Pharmaceuticals, представляет собой первый лекарственный препарат, нацеленный и на путь RAF/MEK/ERK (участвующий в клеточной пролиферации) и каскаде передачи сигналов VEGFR2/PDGFRβ (участвующем в ангиогенезе). Этот лекарственный препарат был впервые утвержден в декабре 2005 г. по поводу запущенного рака почек. Однако данные прицельные терапии, хотя и являются эффективными против некоторых сулидных опухолей, но далеки от удовлетворения с точки зрения достижения лучшей эффективности при сохранении приемлемых побочных эффектов, связанных со способами лечения против других сулидных опухолей.
Настоящее изобретение относится к новым химическим соединениям, которые комбинируют антиангиогенную и антипролифератиную активность ингибиторов RTK вместе с индуцирующей дифференциацию, иммуномодулирующей, останавливающей клеточный цикл и индуцирующей апоптоз активностью ингибиторов HDAC, для достижения лучшей эффективности против сулидных опухолей, одновременно преодолевая побочные эффекты, такие как гипертензия, удлинение интервала QT на ЭКГ, регрессия щитовидной железы, сыпь и изменение цвета кожи и боли, связанные с имеющимися в настоящее время на рынке ингибиторами RTK.
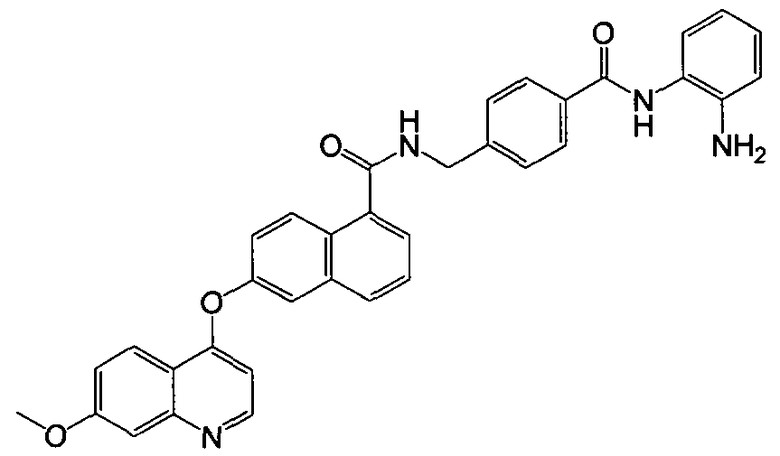
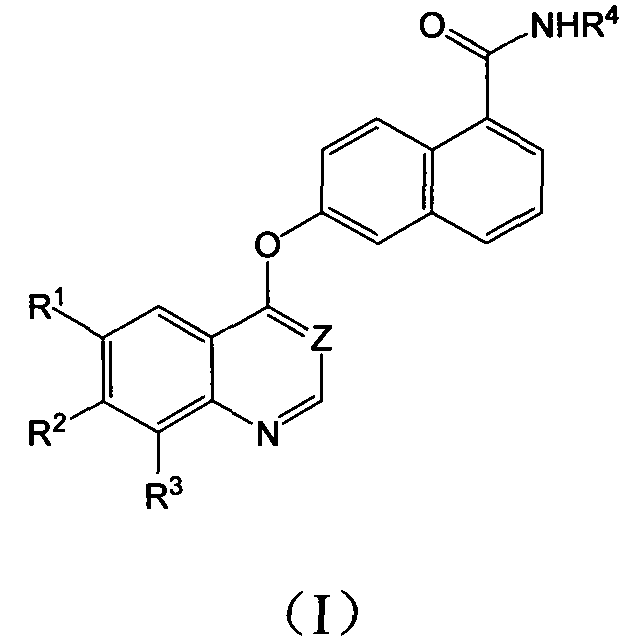
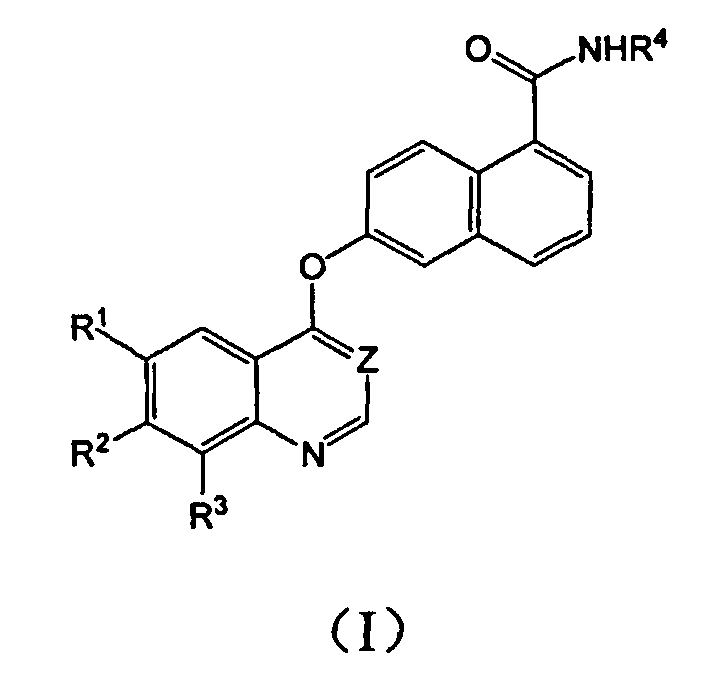
В частности, настоящее изобретение относится к соединению, имеющему структуру, представленную формулой (I):
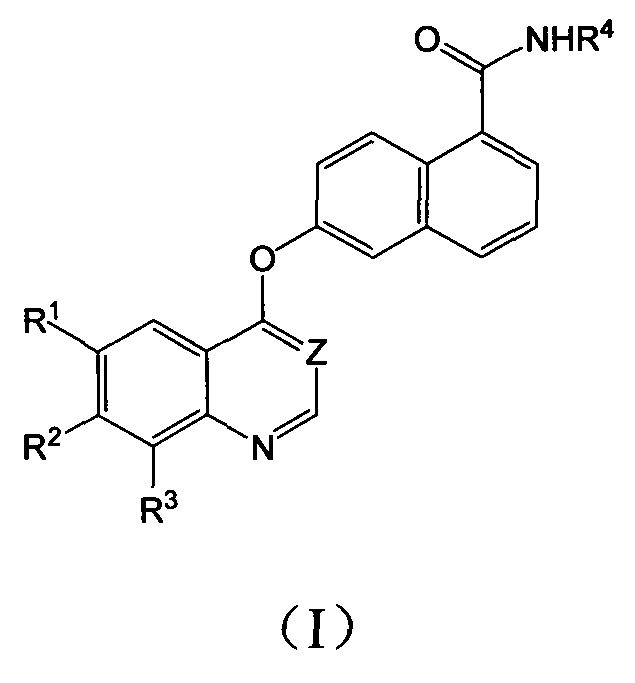
включая его свободную форму, солевую форму, энантиомер, диастереомер или гидрат,
где
Z представляет CH или N;
каждая из групп R1, R2 и R3 представляет водород, галоген, алкил, алкокси или трифторметил;
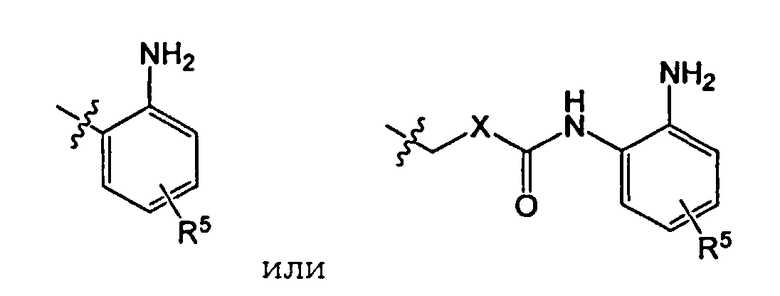
R4 представляет
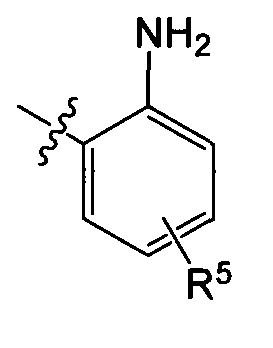 или
или 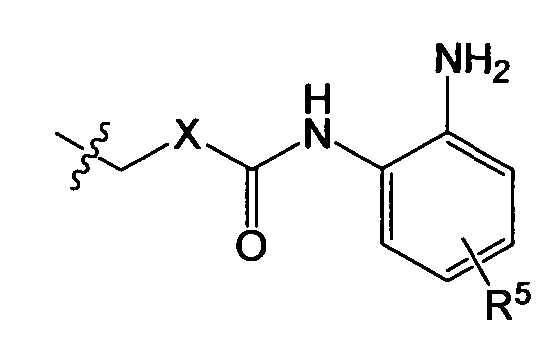
X представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила.
В предпочтительном варианте осуществления, соединения по настоящему изобретению представляют собой соединения формулы (I), где
Z представляет CH;
каждая из групп R1, R2 и R3 представляет водород, галоген, алкил, алкокси или трифторметил;
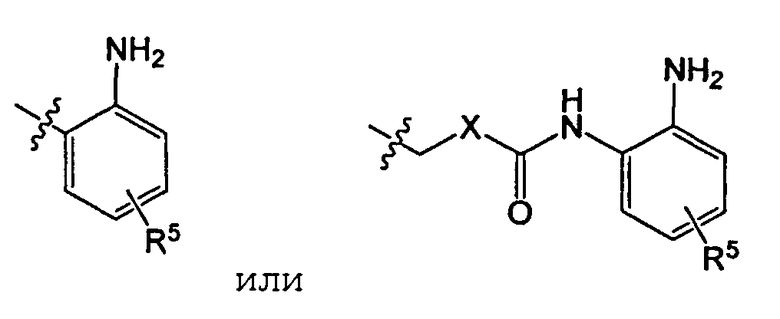
R4 представляет
или
X представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила.
В другом предпочтительном варианте осуществления, соединения по изобретению представляют собой соединения формулы (I), где
Z представляет CH;
каждая из групп R1, R2 и R3 представляет водород или алкокси;
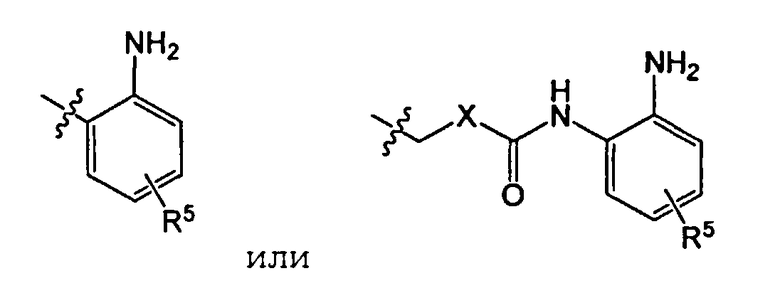
R4 представляет
или
X представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила.
В другом предпочтительном варианте осуществления, соединения по изобретению представляют собой соединения формулы (I), где
Z представляет CH;
каждая из групп R1 и R2 представляет водород или метокси;
R3 представляет H;
R4 представляет
или
X представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила.
В другом предпочтительном варианте осуществления, соединения по изобретению представляют собой соединения формулы (I), где
Z представляет CH;
каждая из групп R1 и R2 представляет водород или метокси;
R3 представляет H;
R4 представляет
или
X представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет H или F.
Используемый в настоящем описании термин «галоген» означает фтор, хлор, бром или йод.
Используемый в настоящем описании термин «алкил» включает линейные, разветвленные или циклические алкилы, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, изопентил, н-гексил, изогексил, циклопропил, циклобутил, циклопентил, циклогексил и тому подобные.
Используемый в настоящем описании термин «алкокси» означает группу, образованную присоединением алкильного радикала к атому кислорода, где атом кислорода имеет способность свободного связывания. Его примеры включают метокси, этокси, пропокси, бутокси, пентокси, изопропокси, трет-бутокси, циклопропокси, циклогексилокси и тому подобные.
Соединения по настоящему изобретению могут быть получены следующим образом:
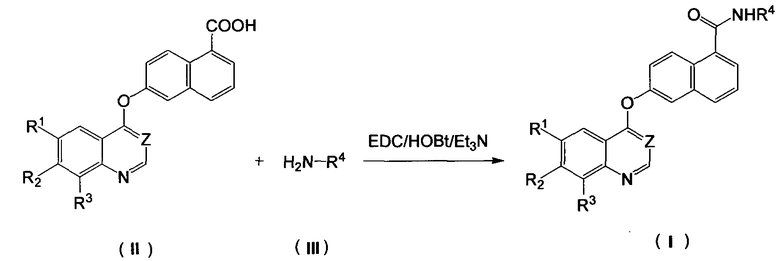
Соединение формулы (II) конденсируется соединением формулы (III) для получения соединения (I). Реакция конденсации проводится путем использования пептидного конденсирующего агента в качестве катализатора, такого как 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), N,N'-дициклогексилкарбодиимид (DCC), N,N'-карбонилдиимидазол (CDI) и т.д. Реакция может проводиться при температуре от 0 до 80°C в течение от 4 до 72 часов. Растворители, которые могут использоваться, представляют собой нормальные растворители, такие как бензол, толуол, тетрагидрофуран, диоксан, дихлорметан, хлороформ, N,N-диметилформамид и т.д. При необходимости, основание, такое как гидроксид натрия, триэтиламин или пиридин, может добавляться к реакционной системе.
Соединения формулы (II) могут быть получены следующим образом:
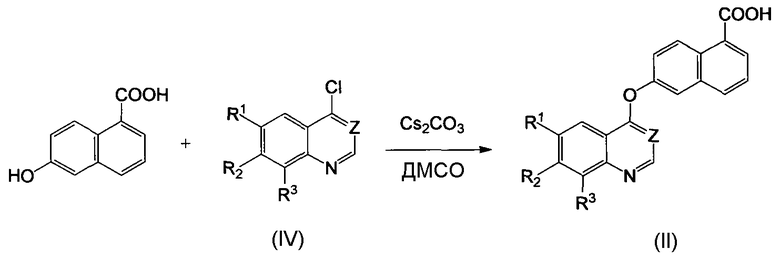
Коммерчески доступная 6-гидроксинафтоевая кислота нагревается в присутствии карбоната цезия и соответствующим образом замещенного 4-хлорхинолина (IV) в ДМСО (диметелсульфоксида) для получения нафтоевых кислот (II). Реакция может проводиться при температуре от 130 до 140°C в течение 3-24 часов.
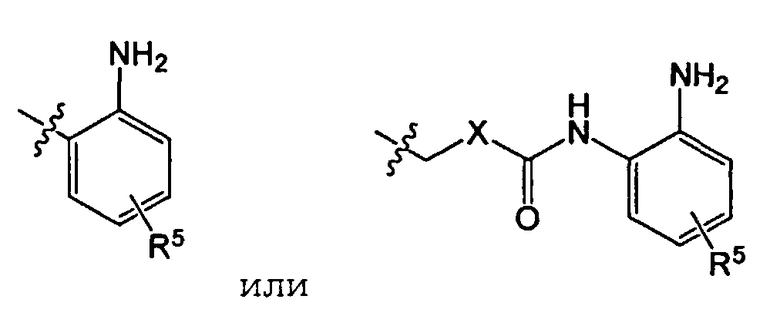
Соединения формулы (III) коммерчески доступны или получаются следующим образом:
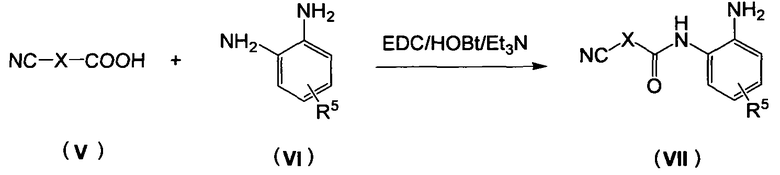
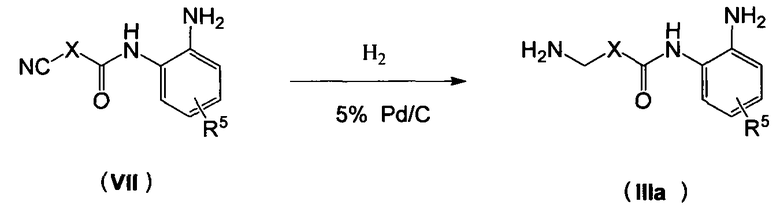
Коммерчески доступное соединение (V) конденсируется с коммерчески доступным соединением (VI) для получения соединения (VII). Реакция конденсации проводится с использованием пептидного конденсирующего агента в качестве катализатора, такого как 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), N,N'-дициклогексилкарбодиимид (DCC), N,N'-карбонилдиимидазол (CDI) и т.д. Реакция может проводиться при температуре от 0 до 60°C в течение 2-72 часов. Растворители, которые могут использоваться, представляют собой нормальные растворители, такие как бензол, толуол, тетрагидрофуран, диоксан, дихлорметан, хлороформ, N,N-диметилформамид и т.д. При необходимости, основание, такое как гидроксид натрия, триэтиламин или пиридин, могут добавляться к реакционной системе.
Соединение (VII) растворяется в метаноле и гидрируется с использованием 5% палладия на активированном угле в качестве катализатора для выхода соединения (IIIa). Реакция может проводиться при комнатной температуре. При необходимости, к реакционной системе может добавляться кислота, такая как серная кислота.
Соединения, представленные формулой (I), могут очищаться обычными способами выделения, такими как экстракция, перекристаллизация, колоночная хроматография и тому подобные.
Соединения, представленные формулой (I), способны одновременно ингибировать протеинкиназы и гистондеацетилазы, и поэтому могут применяться при лечении заболеваний, связанных с аномальными видами активности протеинкиназы и аномальными видами активности гистондеацетилазы. В частности, они высокоэффективны против гематологических злокачественных заболеваний и сулидных опухолей.
Соединения, представленные формулой (I), могут составляться в обычные фармацевтические препараты, такие как таблетки, капсулы, порошки, сиропы, растворы, суспензии, инъекционные препараты, мази и тому подобные. Препараты могут содержать соединение формулы (I) в качестве активного ингредиента вместе с фармацевтически приемлемыми носителями, эксципиентами и разбавителями. Такой препарат обычно содержит от 0,5 до 70%, предпочтительно от 1 до 20% масс. активного ингредиента.
В настоящем изобретении фармацевтически приемлемые носители, эксципиенты и разбавители включают без ограничения те, которые перечислены в руководстве «Hubook of Pharmaceutical Excipients» (American Pharmaceutical Association, October, 1986).
Соединения, представленные в настоящем описании формулой (I), могут клинически вводиться млекопитающим, включая людей, перорально или путем инъекций. Предпочтительно введение пероральным путем. Вводимая дозировка находится в диапазоне от 0,0001 до 200 мг/кг массы тела в день, предпочтительно в диапазоне от 0,01 до 100 мг/кг массы тела в день и, наиболее предпочтительно, в диапазоне от 0,1 до 50 мг/кг массы тела в день. Однако оптимальная дозировка варьируется у различных получающих лечение индивидов, в целом, первоначально вводится меньшая доза, и затем производится ее ступенчатое увеличение.
Репрезентативные соединения по настоящему изобретению показаны ниже в таблице 1. Номера соединений соответствуют «номерам примеров» в разделе «Примеры». То есть синтез соединения 1, как показано в таблице 1, описан в «Примере 1», а синтез соединения 44, как показано в таблице 1, описан в «Примере 44».

Далее настоящее изобретение будет проиллюстрировано в комбинации с приведенными ниже примерами, однако объем защиты настоящего изобретения не ограничивается приведенными примерами. Далее, пока нет иных определений, приведенные в настоящем описании процентные доли даны на массу. Любой диапазон приведенных в описании чисел, таких как единицы измерения, условия реакций, физические состояния соединения или процентные доли, предназначены для предоставления буквальной и определенной ссылкой. Специалисты в данной области при осуществлении настоящего изобретения, используя температуры, концентрации, количества, числа атомов углерода и тому подобные, которые выпадают из диапазона или отличаются от одной величины, достигнут желаемого результата.
Пример 1
Получение 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты
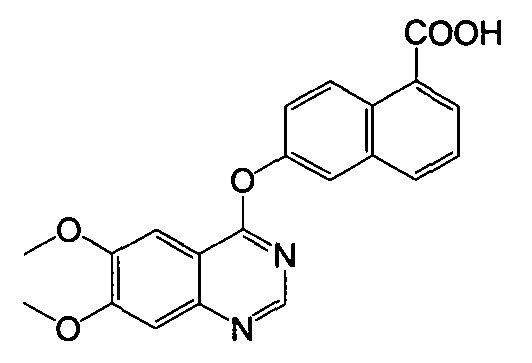
6-Гидрокси-1-нафтоевую кислоту (1,43 г, 7,6 ммоль) растворяли в 38 мл ДМСО, затем добавляли карбонат цезия (7,5 г, 22,9 ммоль) и 4-хлор-6,7-диметоксихиназолина (2,05 г, 9,14 ммоль). Смесь нагревали при 140°C в течение 3 часов. Когда реакция завершалась, смесь охлаждали до комнатной температуры и разбавляли 40 мл H2O. Смесь нейтрализовали 2 н. HCl до pH=6,5. Осажденные твердые вещества фильтровали, промывали H2O, сушили и перекристаллизовывали из метанола для получения указанного в заголовке соединения (1,68 г, выход 59%) в виде твердого вещества коричневого цвета. LC-MS (жидкостная хроматография-масс-спектроскопия) (m/z) 377 (M+1).
Пример 2
Получение 6-(7-метоксихинолин-4-илокси)-1-нафтоевой кислоты
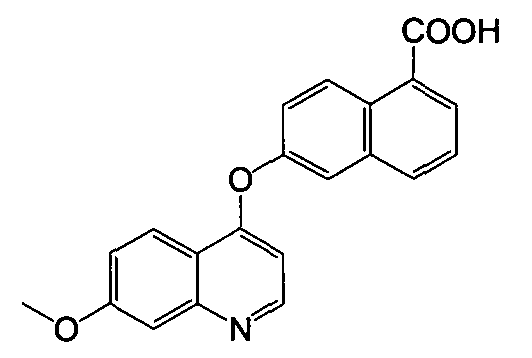
Указанное в заголовке соединение (1,73 г, выход 66%) получали в виде твердого вещества коричневого цвета из 6-гидрокси-1-нафтоевой кислоты (1,43 г, 7,6 ммоль) и 4-хлор-7-метоксихинолина (1,77 г, 9,14 ммоль) процедурой, аналогичной процедуре, описанной в примере 1. LC-MS (m/z) 346 (M+1).
Пример 3
Получение 6-(6,7-диметоксихинолин-4-илокси)-1-нафтоевой кислоты
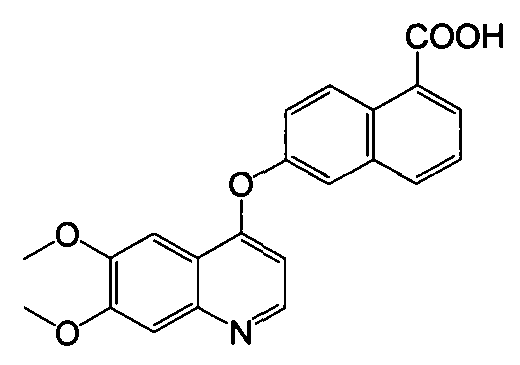
Указанное в заголовке соединение (1,95 г, выход 68%) получали в виде твердого вещества коричневого цвета из 6-гидрокси-1-нафтоевой кислоты (1,43 г, 7,6 ммоль) и 4-хлор-6,7-диметоксихинолина (2,04 г, 9,14 ммоль) процедурой, аналогичной процедуре, описанной в примере 1. LC-MS (m/z) 376 (M+1).
Пример 4
Получение 6-(хинолин-4-илокси)-1-нафтоевой кислоты
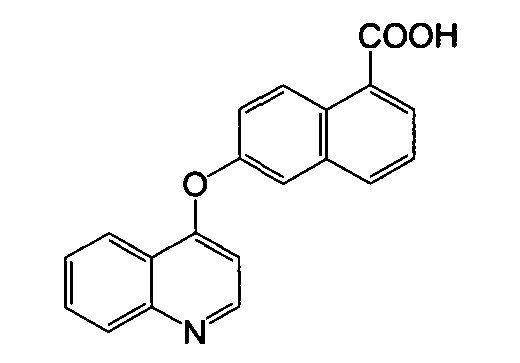
Указанное в заголовке соединение (1,24 г, выход 52%) получали в виде твердого вещества коричневого цвета из 6-гидрокси-1-нафтоевой кислоты (1,43 г, 7,6 ммоль) и 4-хлорхинолина (1,49 г, 9,14 ммоль) процедурой, аналогичной процедуре, описанной в примере 1. LC-MS (m/z) 316 (M+1).
Пример 5
Получение 6-(8-метилхинолин-4-илокси)-1-нафтоевой кислоты
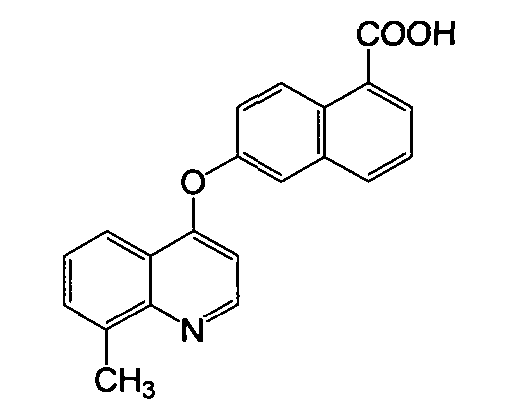
Указанное в заголовке соединение (1,25 г, выход 55%) получали в виде твердого вещества коричневого цвета из 6-гидрокси-1-нафтоевой кислоты (1,43 г, 7,6 ммоль) и 4-хлор-8-метилхинолина (1,62 г, 9,14 ммоль) процедурой, аналогичной процедуре, описанной в примере 1. LC-MS (m/z) 330 (M+1).
Пример 6
Получение 6-(7-хлорхинолин-4-илокси)-1-нафтоевой кислоты
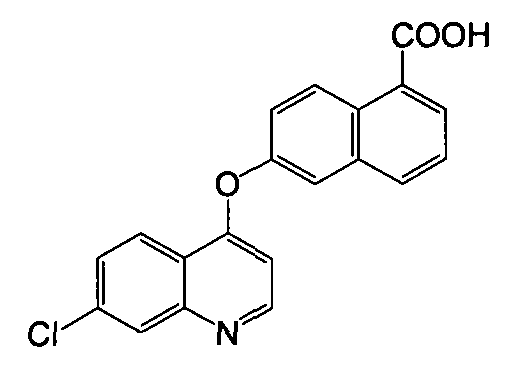
Указанное в заголовке соединение (1,57 г, выход 59%) получали в виде твердого вещества коричневого цвета из 6-гидрокси-1-нафтоевой кислоты (1,43 г, 7,6 ммоль) и 4,7-дихлорхинолина (1,81 г, 9,14 ммоль) процедурой, аналогичной процедуре, описанной в примере 1. LC-MS (m/z) 350 (M+1).
Пример 7
Получение 6-(8-(трифторметил)хинолин-4-илокси)-1-нафтоевой кислоты
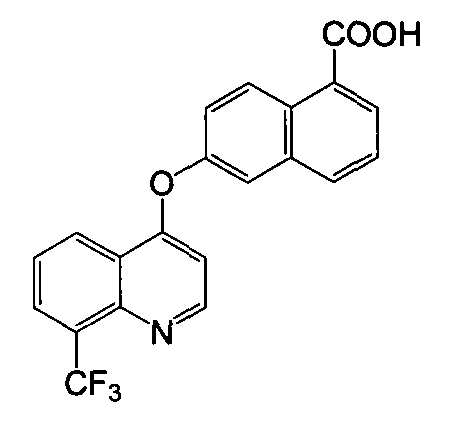
Указанное в заголовке соединение (1,43 г, выход 49%) получали в виде твердого вещества коричневого цвета из 6-гидрокси-1-нафтоевой кислоты (1,43 г, 7,6 ммоль) и 4-хлор-8-(трифторметил)хинолина (2,12 г, 9,14 ммоль) процедурой, аналогичной процедуре, описанной в примере 1. LC-MS (m/z) 384 (M+1).
Пример 8
Получение 4-(аминометил)-N-(2-аминофенил)бензамида
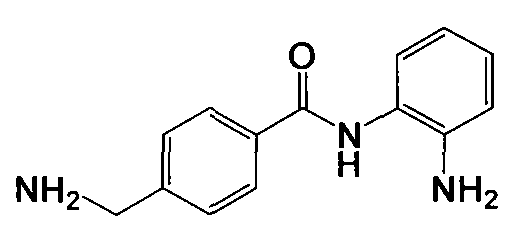
4-Цианобензойную кислоту (294 мг, 2 ммоль) растворяли в 8 мл ДМФА, затем добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (768 мг, 4 ммоль), 1-гидроксибензотриазол (324 мг, 2,4 ммоль), триэтиламин (808 мг, 8 ммоль) и о-фенилендиамин (432 мг, 4 ммоль). Смесь перемешивали в течение 20 часов при комнатной температуре. Смесь разбавляли 400 мл концентрированного раствора соли. Твердые вещества собирали вакуумной фильтрацией, промывали водой и сушили в вакууме для получения N-(2-аминофенил)-4-цианобензамида (364 мг, 77%) в виде твердого вещества серого цвета. LC-MS (m/z) 238 (M+1).
N-(2-аминофенил)-4-цианобензамид (237 мг, 1 ммоль) растворяли в метаноле (40 мл), затем добавляли серную кислоту (196 мг, 1 ммоль) и 5% палладий на активированном угле (0,20 г). Смесь перемешивали в атмосфере водорода до завершения реакции. Смесь фильтровали через целит и фильтрат нейтрализовали 1 н. раствором NaOH (2 мл). Полученную смесь фильтровали, и фильтрат концентрировали в вакууме для получения указанного в заголовке соединения (232 мг, выход 96%) в виде твердого вещества серого цвета. LC-MS (m/z) 242 (M+1).
Пример 9
Получение 4-(аминометил)-N-(2-амино-4-фторфенил)бензамида
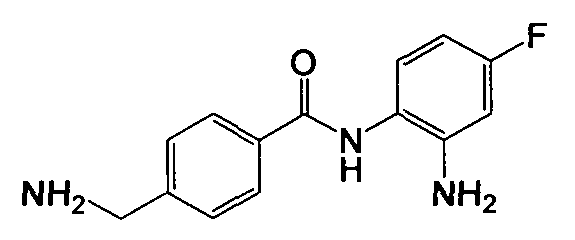
Указанное в заголовке соединение (186 мг, выход 72%) получали в виде твердого вещества коричневого цвета из 4-цианобензойной кислоты (294 мг, 2 ммоль) и 4-фтор-о-фенилендиамина (302 мг, 2,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 8. LC-MS (m/z) 260 (M+1).
Пример 10
Получение 4-(аминометил)-N-(2-амино-4-метилфенил)бензамида
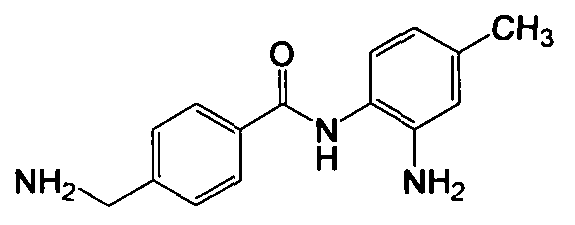
Указанное в заголовке соединение (173 мг, выход 68%) получали в виде твердого вещества серого цвета из 4-цианобензойной кислоты (294 мг, 2 ммоль) и 4-метил-о-фенилендиамина (293 мг, 2,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 8. LC-MS (m/z) 256 (M+1).
Пример 11
Получение 4-(аминометил)-N-(2-амино-4-метоксифенил)бензамида
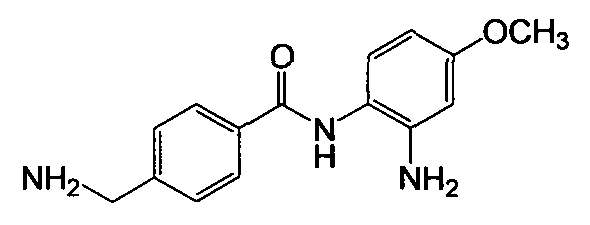
Указанное в заголовке соединение (192 мг, выход 71%) получали в виде твердого вещества серого цвета из 4-цианобензойной кислоты (294 мг, 2 ммоль) и 4-метокси-о-фенилендиамина (331 мг, 2,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 8. LC-MS (m/z) 272 (M+1).
Пример 12
Получение 4-(аминометил)-N-(2-амино-4-трифторметилфенил)бензамида
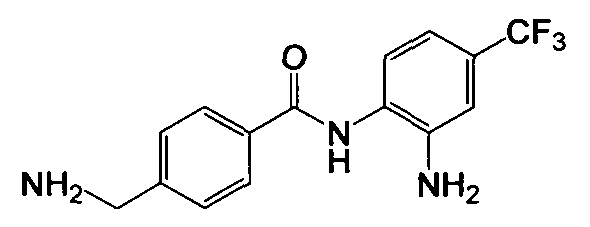
Указанное в заголовке соединение (195 мг, выход 63%) получали в виде твердого вещества серого цвета из 4-цианобензойной кислоты (294 мг, 2 ммоль) и 4-трифторметил-о-фенилендиамина (422 мг, 2,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 8. LC-MS (m/z) 310 (M+1).
Пример 13
Получение 3-(аминометил)-N-(2-аминофенил)бензамида
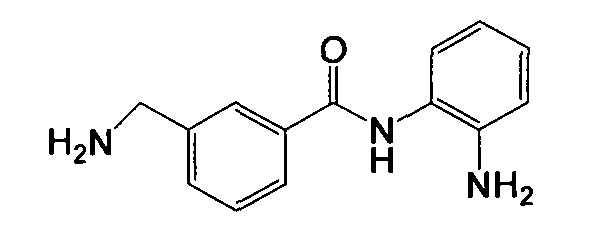
Указанное в заголовке соединение (140 мг, выход 58%) получали в виде твердого вещества серого цвета из 3-цианобензойной кислоты (294 мг, 2 ммоль) и о-фенилендиамина (432 мг, 4 ммоль) процедурой, аналогичной процедуре, описанной в примере 8. LC-MS (m/z) 242 (M+1).
Пример 14
Получение 6-(аминометил)-N-(2-аминофенил)никотинамида
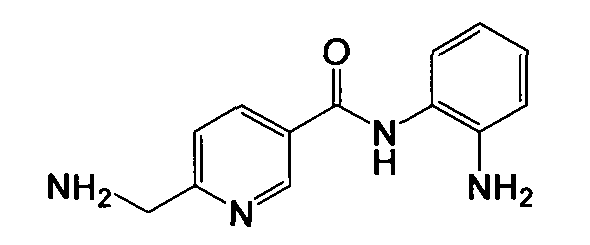
Указанное в заголовке соединение (157 мг, выход 65%) получали в виде твердого вещества серого цвета из 6-цианоникотиновой кислоты (296 мг, 2 ммоль) и о-фенилендиамина (864 мг, 8 ммоль) процедурой, аналогичной процедуре, описанной в примере 8. LC-MS (m/z) 243 (M+1).
Пример 15
Получение 6-(аминометил)-N-(2-амино-4-фторфенил)никотинамида
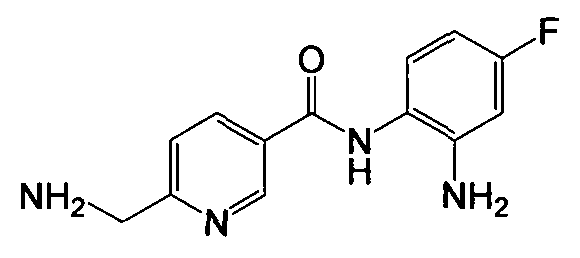
Указанное в заголовке соединение (135 мг, выход 52%) получали в виде твердого вещества серого цвета из 6-цианоникотиновой кислоты (296 мг, 2 ммоль) и 4-фтор-о-фенилендиамина (302 мг, 2,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 8. LC-MS (m/z) 261 (M+1).
Пример 16
Получение N-(2-аминофенил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
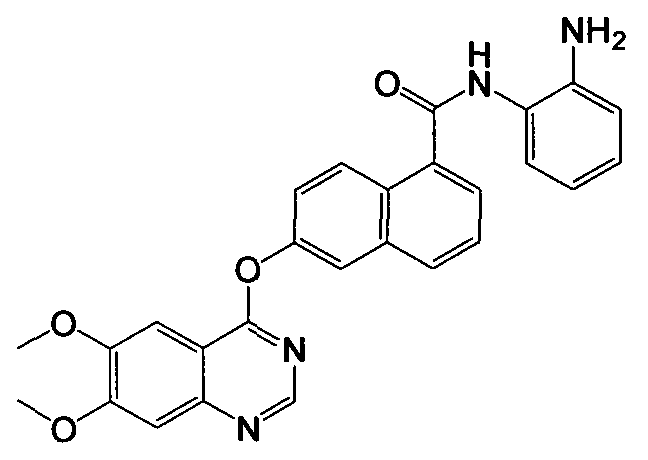
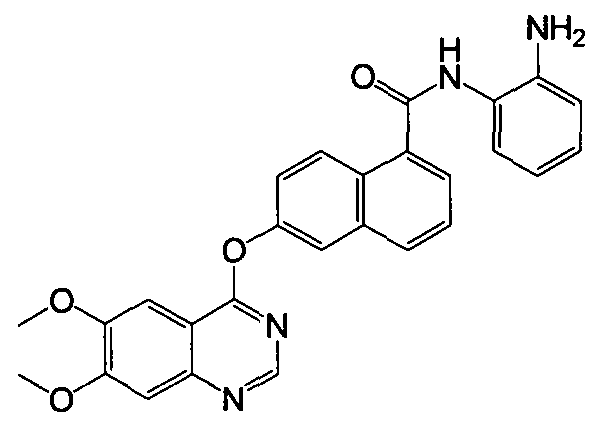
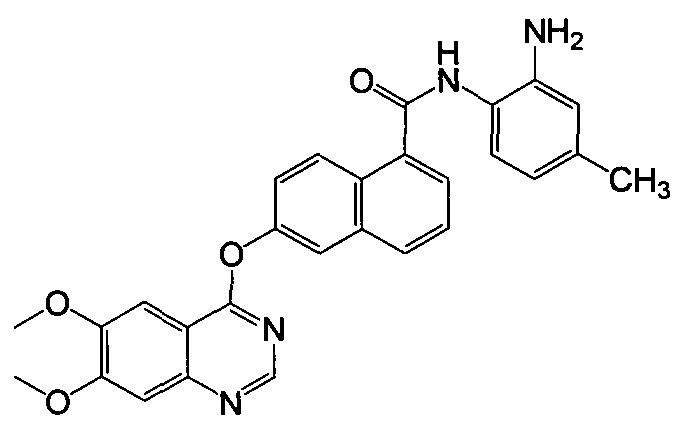
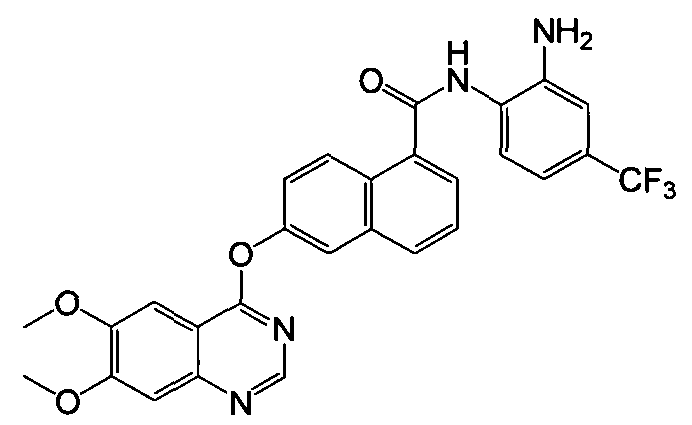
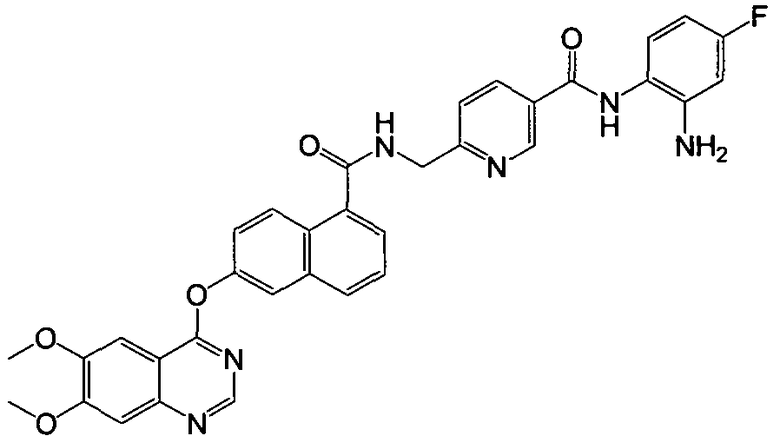
6-(6,7-Диметоксихиназолин-4-илокси)-1-нафтоевую кислоту (37,6 мг, 0,1 ммоль) растворяли в 4 мл ДМФА, затем добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (38,4 мг, 0,2 ммоль), 1-гидроксибензотриазол (16,2 мг, 0,12 ммоль), триэтиламин (40,4 мг, 0,4 ммоль) и о-фенилендиамин (43,2 мг, 0,4 ммоль). Смесь перемешивали в течение 20 часов при комнатной температуре. Смесь разбавляли 200 мл концентрированного раствора соли. Твердые вещества собирали вакуумной фильтрацией, промывали водой и сушили в вакууме для получения указанного в заголовке соединения (39,1 мг, 84%) в виде твердого вещества коричневого цвета. 1H-ЯМР (ДМСО-d6) δ 4,01 (с, 6H, 2×OCH3), 4,97 (с, 2H, бензол-NH2), 6,65 (т, J=7,2 Гц, 1H, Ar-H), 6,82 (д, J=7,0 Гц, 1H, Ar-H), 7,00 (т, J=7,1 Гц, 1H, Ar-H), 7,38 (д, J=7,1 Гц, 1H, Ar-H), 7,42 (с, 1H, Ar-H), 7,60 (дд, J=2,4 и 9,2 Гц, 1H, Ar-H), 7,64-7,68 (м, 2H, Ar-H), 7,87 (д, J=6,7 Гц, 1H, Ar-H), 7,97 (д, J=2,3 Гц, 1H, Ar-H), 8,09 (д, J=8,2 Гц, 1H, Ar-H), 8,38 (д, J=9,2 Гц, 1H, Ar-H), 8,54 (с, 1H, Ar-H), 9,85 (с, 1H, бензол-NH). LC-MS (m/z) 467 (M+1).
Пример 17
Получение N-(2-амино-4-фторфенил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
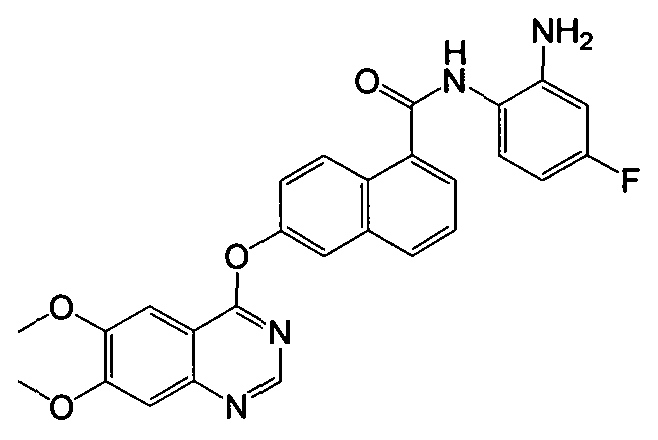
Указанное в заголовке соединение (43,1 мг, выход 89%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-фтор-о-фенилендиамина (15,1 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 4,01 (с, 6H, 2×ОСН3), 5,28 (с, 2Н, бензол-NH2), 6,41 (тд, J=2,6 и 8,5 Гц, 1H, Ar-H), 6,59 (дд, J=2,6 и 11,2 Гц, 1H, Ar-H), 7,35 (тд, J=1,8 и 7,5 Гц, 1H, Ar-H), 7,41 (с, 1H, Ar-H), 7,59 (дд, J=2,2 и 8,4 Гц, 1H, Ar-H), 7,63-7,67 (м, 2H, Ar-H), 7,89 (д, J=6,9 Гц, 1H, Ar-H), 7,96 (д, J=1,9 Гц, 1H, Ar-H), 8,08 (д, J=8,2 Гц, 1H, Ar-H), 8,38 (д, J=9,2 Гц, 1H, Ar-H), 8,54 (с, 1H, Ar-H), 9,77 (с, 1H, бензол-NH). LC-MS (m/z) 485 (M+1).
Пример 18
Получение N-(2-амино-4-метилфенил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
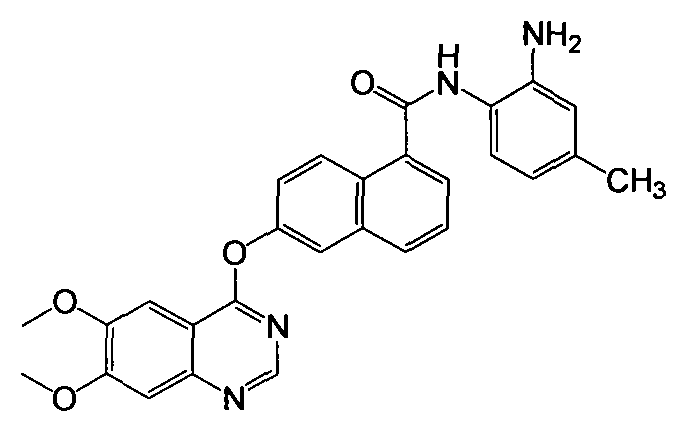
Указанное в заголовке соединение (39,4 мг, выход 82%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-метил-о-фенилендиамина (14,6 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) (отношение изомеров 0,77/0,23) δ 2,21 (с, 1H, Ar-CH3), 4,01 (с, 6H, 2×ОСН3), 4,77 (с, 0,23×2H, бензол-NH2), 4,89 (с, 0,77×2H, бензол-NH2), 6,46 (д, J=7,6 Гц, 0,77×1H, Ar-H), 6,64 (с, 0,77×1H, Ar-H), 6,73 (д, J=7,9 Гц, 0,23×1H, Ar-H), 6,81 (с, 0,23×1H, Ar-H), 7,24 (д, J=8,1 Гц, 1H, Ar-H), 7,41 (с, 1H, Ar-H), 7,58-7,66 (м, 3H, Ar-H), 7,85 (д, J=6,7 Гц, 1H, Ar-H), 7,97 (с, 1H, Ar-H), 8,08 (д, J=7,9 Гц, 1H, Ar-H), 8,38 (д, J=9,0 Гц, 1H, Ar-H), 8,54 (с, 1H, Ar-H), 9,77 (с, 1H, бензол-NH). LC-MS (m/z) 481 (M+1).
Пример 19
Получение N-(2-амино-4-метоксифенил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
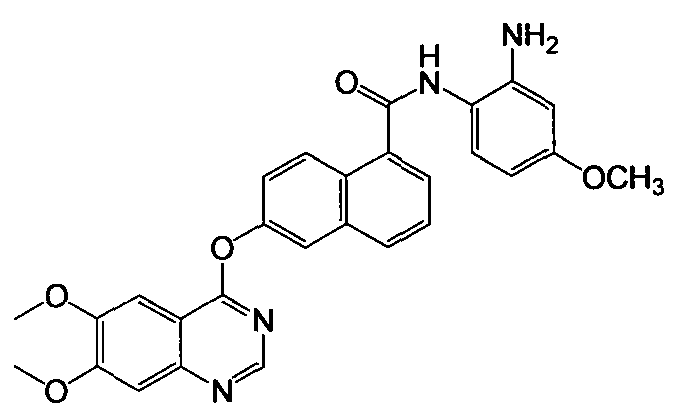
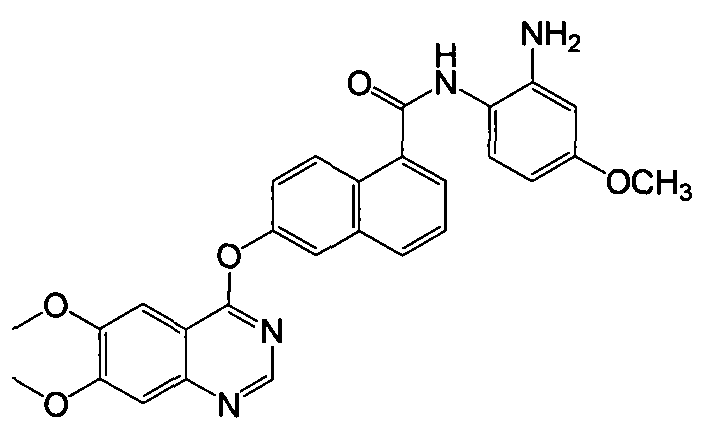
Указанное в заголовке соединение (43,2 мг, выход 87%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-метокси-о-фенилендиамина (16,5 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 3,70 (с, 3H, -ОСН3), 4,01 (с, 6H, 2×ОСН3), 5,00 (с, 2H, бензол-NH2), 6,23 (дд, J=2,6 и 8,6 Гц, 1H, Ar-H), 6,40 (д, J=2,6 Гц, 1H, Ar-H), 7,22 (д, J=8,6 Гц, 1H, Ar-H), 7,41 (с, 1H, Ar-H), 7,59 (дд, J=2,2 и 9,1 Гц, 1H, Ar-H), 7,62-7,66 (м, 2H, Ar-H), 7,86 (д, J=6,9 Гц, 1H, Ar-H), 7,96 (д, J=2,0 Гц, 1H, Ar-H), 8,07 (д, J=8,2 Гц, 1H, Ar-H), 8,38 (д, J=9,2 Гц, 1H, Ar-H), 8,54 (с, 1H, Ar-H), 9,70 (с, 1H, бензол-NH). LC-MS (m/z) 497 (M+1).
Пример 20
Получение N-(2-амино-4-хлорфенил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
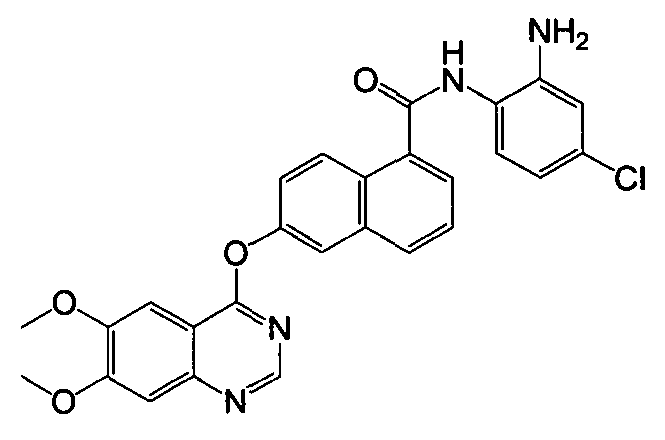
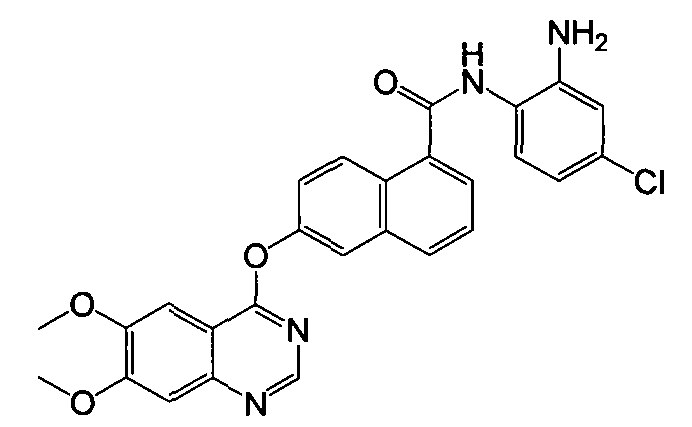
Указанное в заголовке соединение (42,9 мг, выход 83%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-хлор-о-фенилендиамина (17,1 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 4,01 (с, 6H, 2×ОСН3), 5,31 (с, 2H, бензол-NH2), 6,65 (д, J=8,3 Гц, 1H, Ar-H), 6,86 (д, J=1,9 Гц, 1H, Ar-H), 7,41 (с, 1H, Ar-H), 7,58-7,67 (м, 4H, Ar-H), 7,89 (д, J=6,8 Гц, 1H, Ar-H), 8,01 (с, 1H, Ar-H), 8,09 (д, J=8,1 Гц, 1H, Ar-H), 8,37 (д, J=9,2 Гц, 1H, Ar-H), 8,55 (с, 1H, Ar-H), 9,84 (с, 1H, бензол-NH). LC-MS (m/z) 501 (M+1).
Пример 21
Получение N-(2-амино-4-бромфенил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
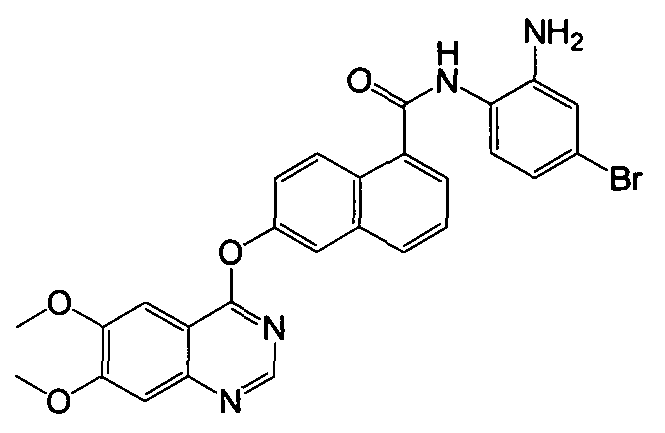
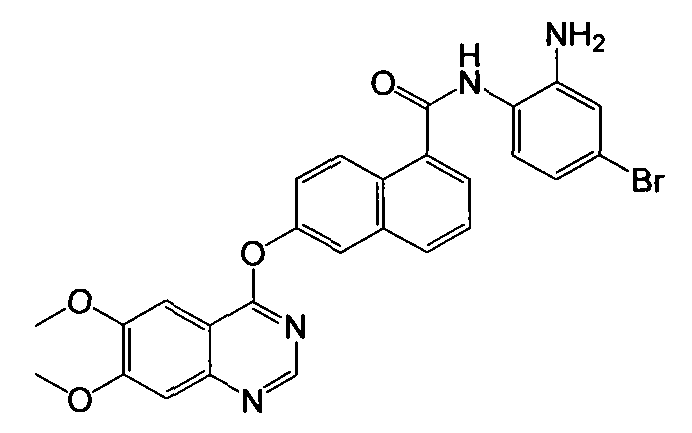
Указанное в заголовке соединение (42,0 мг, выход 77%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-бром-о-фенилендиамина (22,4 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 4,01 (с, 6H, 2×ОСН3), 5,31 (с, 2H, бензол-NH2), 6,77 (д, J=8,3 Гц, 1H, Ar-H), 7,01 (с, 1H, Ar-H), 7,41 (с, 1H, Ar-H), 7,58-7,65 (м, 5H, Ar-H), 7,89 (д, J=7,0 Гц, 1H, Ar-H), 8,00 (с, 1H, Ar-H), 8,14 (д, J=10,2 Гц, 1H, Ar-H), 8,37 (д, J=9,1 Гц, 1H, Ar-H), 8,54 (с, 1H, Ar-H), 9,84 (с, 1H, бензол-NH). LC-MS (m/z) 545 (M+1).
Пример 22
Получение N-(2-амино-4-трифторметилфенил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
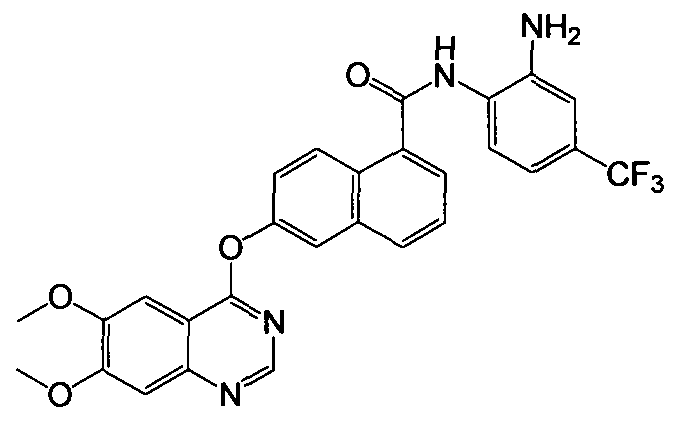
Указанное в заголовке соединение (42,3 мг, выход 79%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-трифторметил-о-фенилендиамина (21,1 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 4,01 (с, 6H, 2×ОСН3), 5,72 (с, 2Н, бензол-NH2), 6,92 (д, J=8,5 Гц, 1H, Ar-H), 7,42 (с, 1H, Ar-H), 7,59-7,65 (м, 3H, Ar-H), 7,90-7,96 (м, 2H, Ar-H), 7,98 (с, 1H, Ar-H), 8,10 (д, J=8,3 Гц, 1H, Ar-H), 8,17 (д, J=7,3 Гц, 1H, Ar-H), 8,39 (д, J=9,2 Гц, 1H, Ar-H), 8,54 (с, 1H, Ar-H), 9,90 (с, 1H, бензол-NH). LC-MS (m/z) 535 (M+1).
Пример 23
Получение N-(4-((2-аминофенил)карбамоил)бензил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
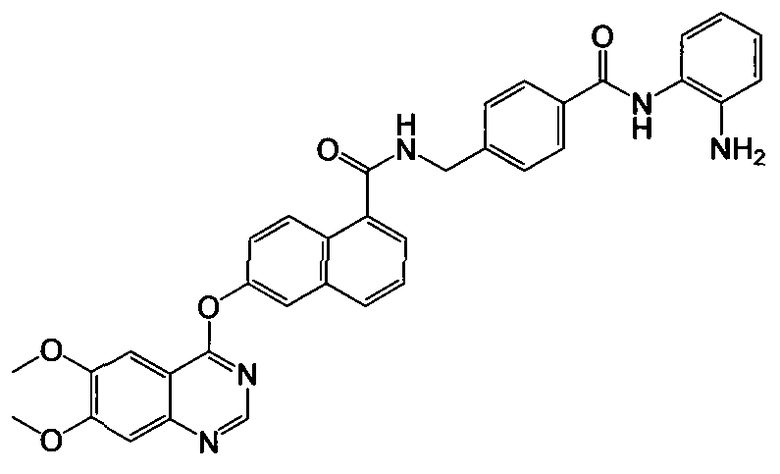
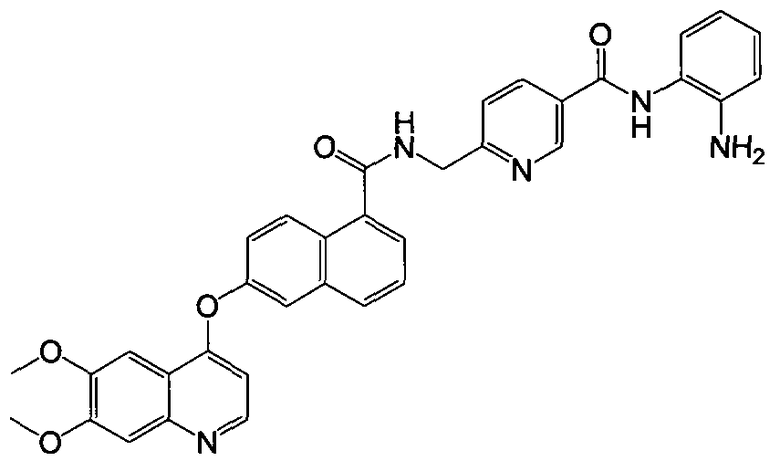
Указанное в заголовке соединение (43,1 мг, выход 72%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-(аминометил)-N-(2-аминофенил)бензамида (28,9 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. LC-MS (m/z) 600 (M+1).
Пример 24
Получение N-(4-((2-амино-4-фторфенил)карбамоил)бензил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
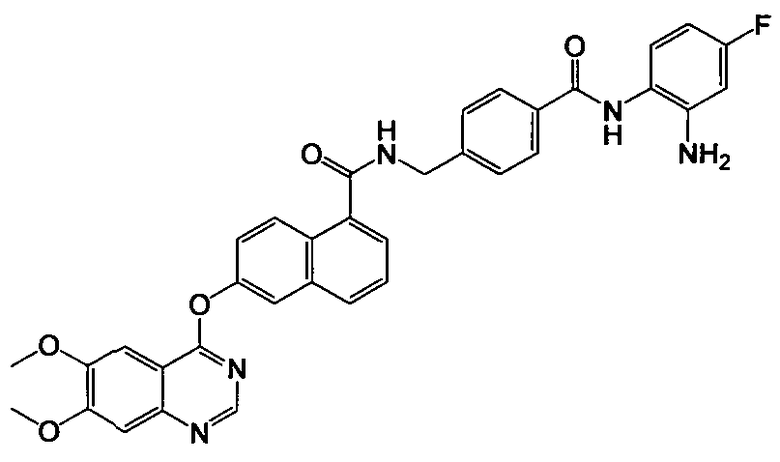
Указанное в заголовке соединение (46,3 мг, выход 75%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-(аминометил)-N-(2-амино-4-фторфенил)бензамида (31,1 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. LC-MS (m/z) 618 (M+1).
Пример 25
Получение N-(2-аминофенил)-6-((2-(6,7-диметоксихиназолин-4-илокси)-1-нафтамидо)метил)никотинамида
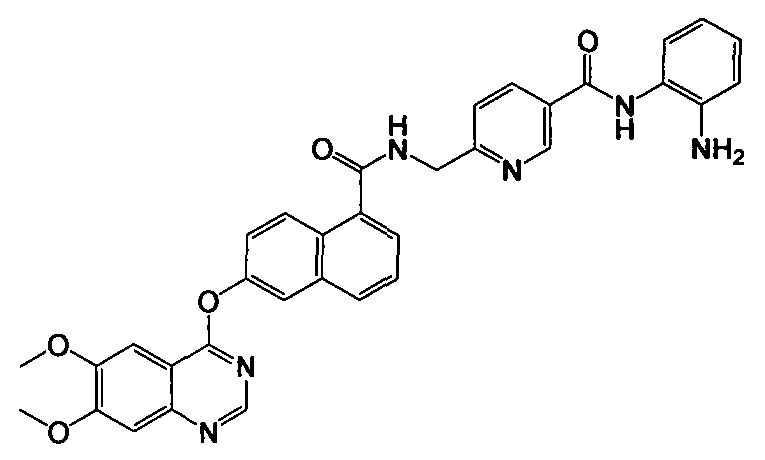
Указанное в заголовке соединение (41,4 мг, выход 69%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 6-(аминометил)-N-(2-аминофенил)никотинамида (29,0 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. LC-MS (m/z) 601 (M+1).
Пример 26
Получение N-(2-амино-4-фторфенил)-6-((2-(6,7-диметоксихиназолин-4-илокси)-1-нафтамидо)метил)никотинамида
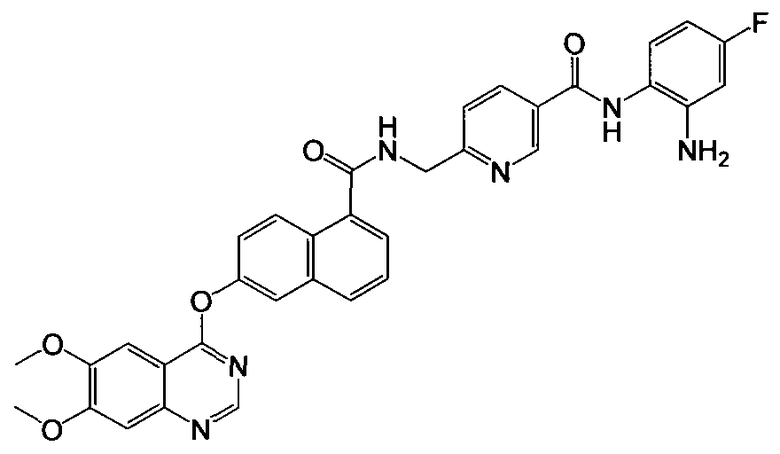
Указанное в заголовке соединение (43,3 мг, выход 77%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 6-(аминометил)-N-(2-амино-4-фторфенил)никотинамида (31,2 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. LC-MS (m/z) 619 (M+1).
Пример 27
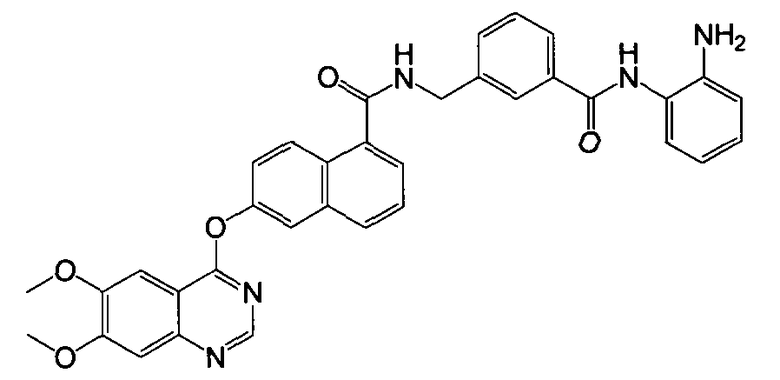
Получение N-(3-((2-аминофенил)карбамоил)бензил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
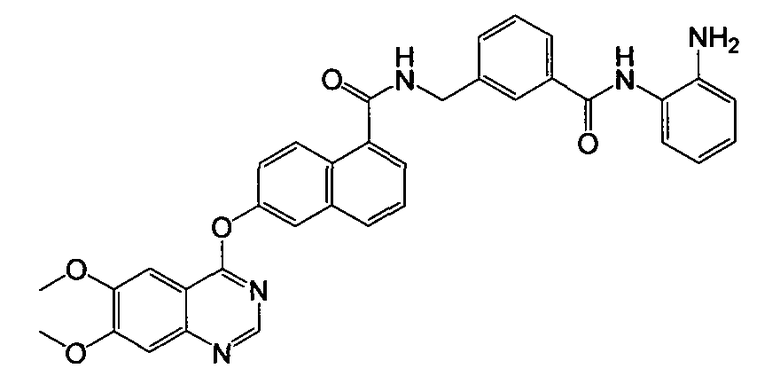
Указанное в заголовке соединение (48,5 мг, выход 81%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 3-(аминометил)-N-(2-аминофенил)бензамида (28,9 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. LC-MS (m/z) 600 (M+1).
Пример 28
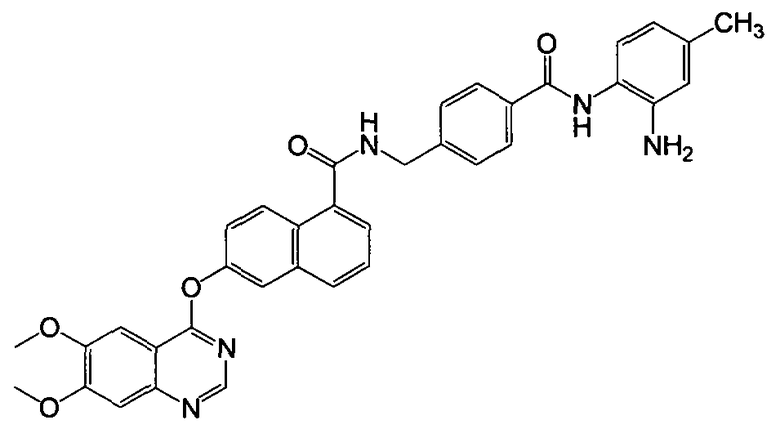
Получение N-(4-((2-амино-4-метилфенил)карбамоил)бензил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
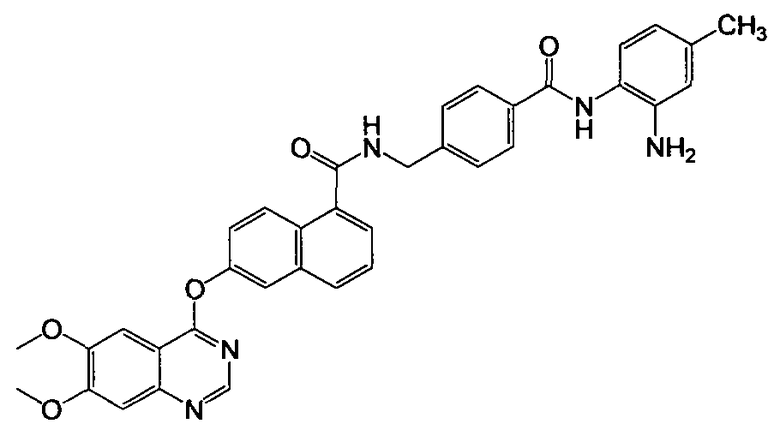
Указанное в заголовке соединение (52,7 мг, выход 86%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-(аминометил)-N-(2-амино-4-метилфенил)бензамида (30,6 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. LC-MS (m/z) 614 (M+1).
Пример 29
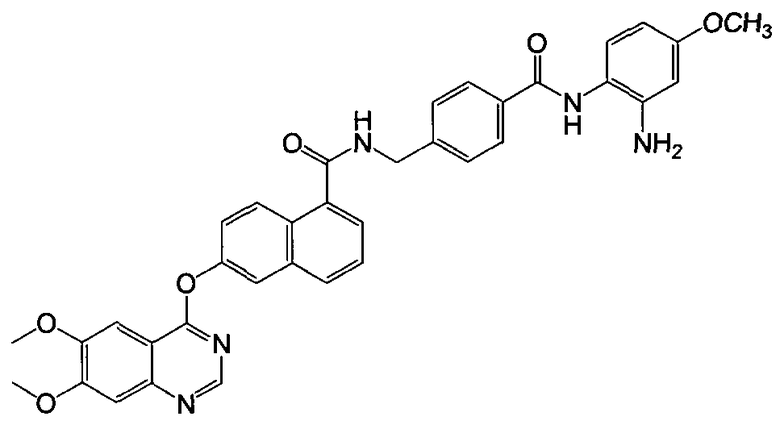
Получение N-(4-((2-амино-4-метоксифенил)карбамоил)бензил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
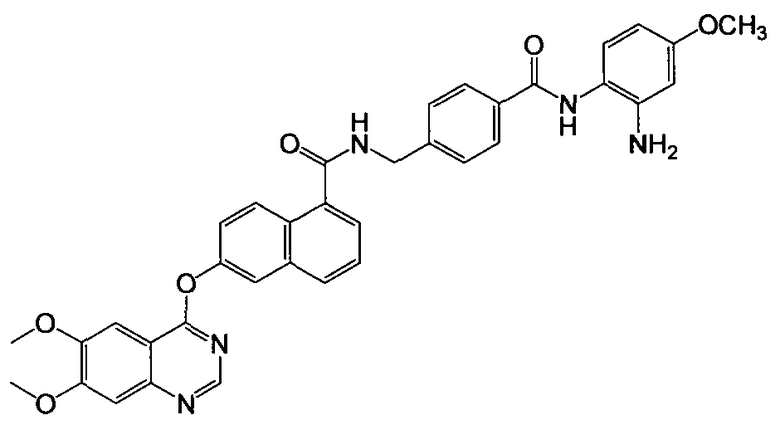
Указанное в заголовке соединение (51,6 мг, выход 82%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-(аминометил)-N-(2-амино-4-метоксифенил)бензамида (32,5 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. LC-MS (m/z) 630 (M+1).
Пример 30
Получение N-(4-((2-амино-4-трифторметилфенил)карбамоил)бензил)-6-(6,7-диметоксихиназолин-4-илокси)-1-нафтамида
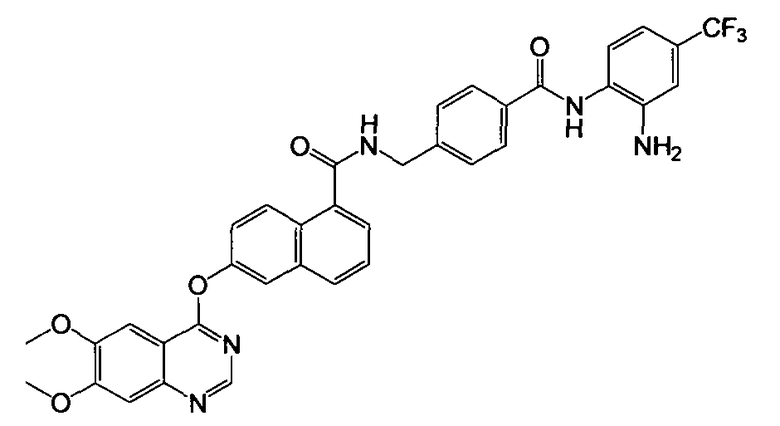
Указанное в заголовке соединение (46,7 мг, выход 70%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихиназолин-4-илокси)-1-нафтоевой кислоты (37,6 мг, 0,1 ммоль) и 4-(аминометил)-N-(2-амино-4-трифторметилфенил)бензамида (37,1 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. LC-MS (m/z) 668 (M+1).
Пример 31
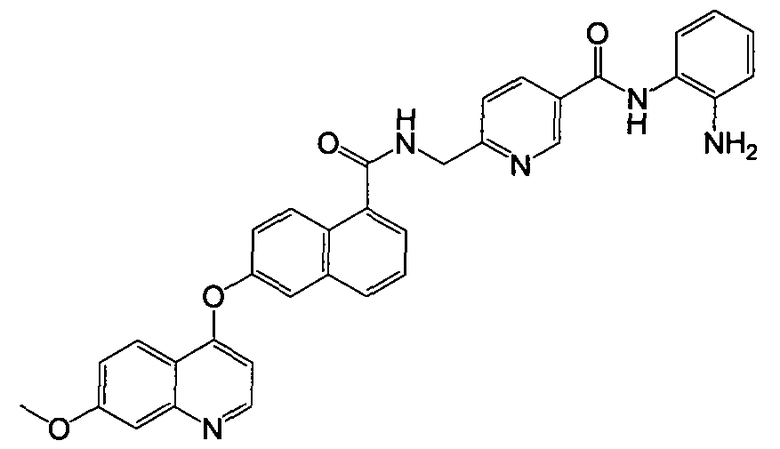
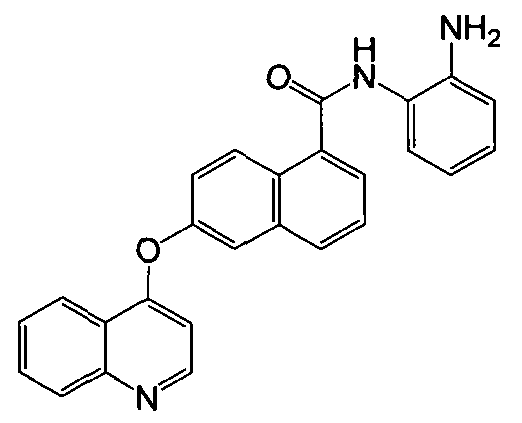
Получение N-(2-аминофенил)-6-(7-метоксихинолин-4-илокси)-1-нафтамида
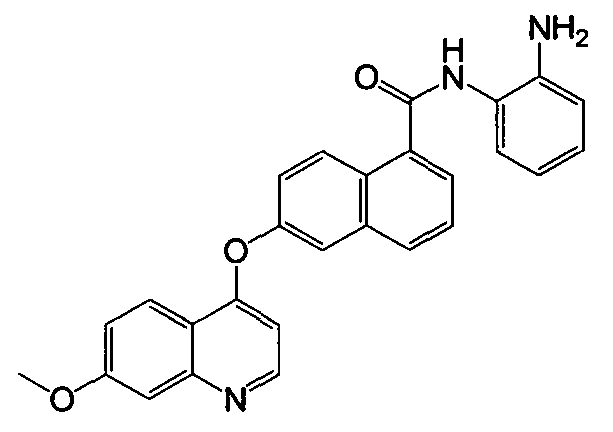
Указанное в заголовке соединение (39,6 мг, выход 91%) получали в виде твердого вещества коричневого цвета из 6-(7-метоксихинолин-4-илокси)-1-нафтоевой кислоты (34,5 мг, 0,1 ммоль) и о-фенилендиамина (43,2 мг, 0,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 3,95 (с, 3H, -OCH3), 4,97 (с, 2H, бензол-NH2), 6,60 (д, J=5,2 Гц, 1H, Ar-H), 6,64 (т, J=7,6 Гц, 1H, Ar-H), 6,82 (д, J=7,8 Гц, 1H, Ar-H), 6,99 (т, J=7,4 Гц, 1H, Ar-H), 7,31 (дд, J=2,5 и 9,1 Гц, 1H, Ar-H), 7,38 (д, J=7,6 Гц, 1H, Ar-H), 7,45 (д, J=2,4 Гц, 1H, Ar-H), 7,57 (дд, J=2,4 и 9,2 Гц, 1H, Ar-H), 7,65 (т, J=7,8 Гц, 1H, Ar-H), 7,87-7,88 (м, 2H, Ar-H), 8,07 (д, J=8,2 Гц, 1H, Ar-H), 8,25 (д, J=9,2 Гц, 1H, Ar-H), 8,43 (д, J=9,2 Гц, 1H, Ar-H), 8,65 (д, J=5,2 Гц, 1H, Ar-H), 9,84 (с, 1H, бензол-NH). LC-MS (m/z) 436 (М+1).
Пример 32
Получение N-(2-амино-4-фторфенил)-6-(7-метоксихинолин-4-илокси)-1-нафтамида
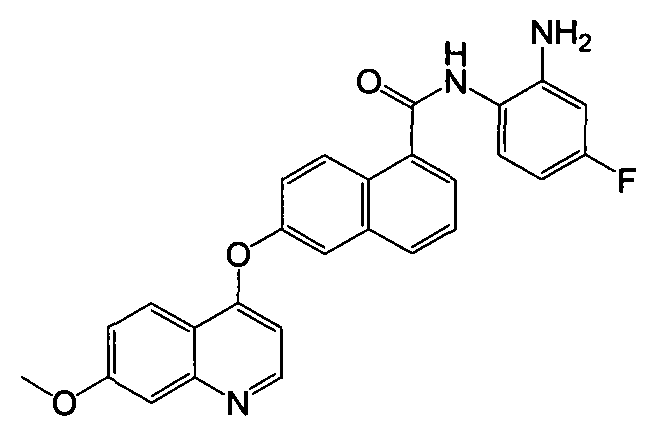
Указанное в заголовке соединение (33,1 мг, выход 73%) получали в виде твердого вещества коричневого цвета из 6-(7-метоксихинолин-4-илокси)-1-нафтоевой кислоты (34,5 мг, 0,1 ммоль) и 4-фтор-о-фенилендиамина (15,1 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 3,95 (с, 3Н, -ОСН3), 5,27 (с, 2Н, бензол-NH2), 6,41 (тд, J=2,5 и 8,4 Гц, 1H, Ar-H), 6,57-6,61 (м, 2H, Ar-H), 7,30-7,36 (м, 2H, Ar-H), 7,45 (д, J=2,2 Гц, 1H, Ar-H), 7,56 (дд, J=2,2 и 9,2 Гц, 1H, Ar-H), 7,65 (т, J=7,6 Гц, 1H, Ar-H), 7,87-7,91 (м, 2H, Ar-H), 8,07 (д, J=8,3 Гц, 1H, Ar-H), 8,24 (д, J=9,1 Гц, 1H, Ar-H), 8,43 (д, J=9,2 Гц, 1H, Ar-H), 8,65 (д, J=5,1 Гц, 1H, Ar-H), 9,75 (с, 1H, бензол-NH). LC-MS (m/z) 454 (М+1).
Пример 33
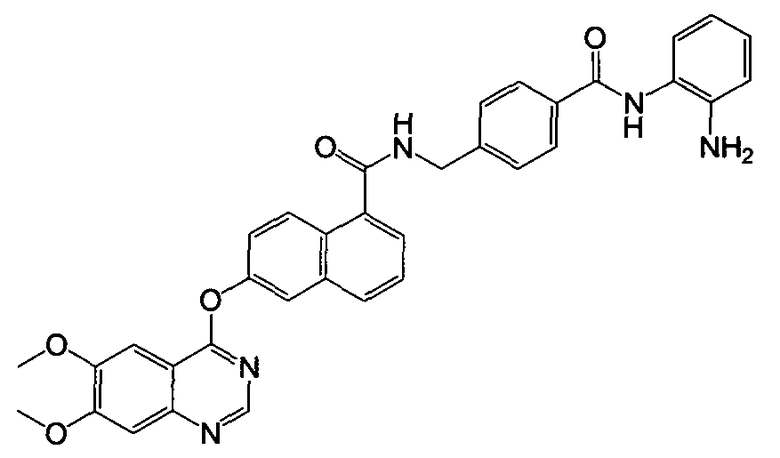
Получение N-(4-((2-аминофенил)карбамоил)бензил)-6-(7-метоксихинолин-4-илокси)-1-нафтамида
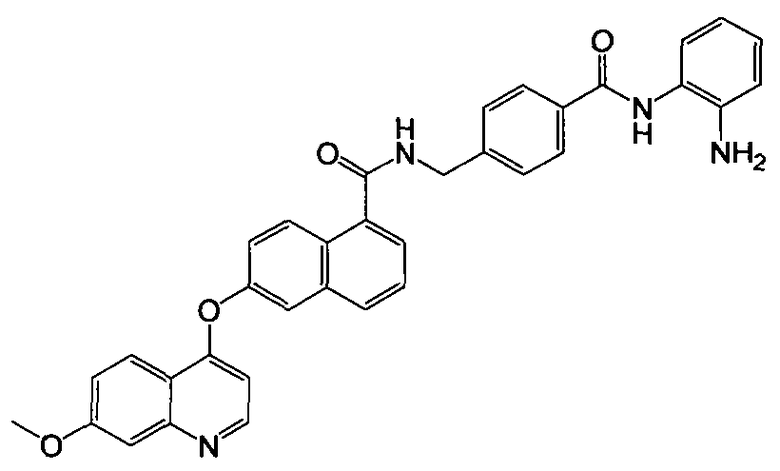
Указанное в заголовке соединение (48,3 мг, выход 85%) получали в виде твердого вещества коричневого цвета из 6-(7-метоксихинолин-4-илокси)-1-нафтоевой кислоты (34,5 мг, 0,1 ммоль) и 4-(аминометил)-N-(2-аминофенил)бензамида (28,9 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 3,95 (с, 3Н, -ОСН3), 4,64 (д, J=5,6 Гц, 2Н, -СН2), 4,87 (с, 2H, бензол-NH2), 6,58-6,62 (т, 2Н, Ar-Н), 6,78 (дд, J=1,2 и 7,8 Гц, 1H, Ar-H), 6,97 (тд, J=1,4 и 8,1 Гц, 1H, Ar-H), 7,18 (д, J=7,0 Гц, 1H, Ar-H), 7,31 (дд, J=2,5 и 9,2 Гц, 1H, Ar-H), 7,44 (д, J=2,4 Гц, 1H, Ar-H), 7,53-7,56 (м, 3H, Ar-H), 7,62 (т, J=8,0 Гц, 1H, Ar-H), 7,72 (д, J=6,1 Гц, 1H, Ar-H), 7,86 (д, J=2,5 Гц, 1H, Ar-H), 7,98-8,06 (м, 3H, Ar-H), 8,24 (д, J=9,1 Гц, 1H, Ar-H), 8,39 (д, J=9,2 Гц, 1H, Ar-H), 8,64 (д, J=5,2 Гц, 1H, Ar-H), 9,21 (т, J=6,0 Гц, 1H, -CONH), 9,61 (с, 1H, бензол-NH). LC-MS (m/z) 569 (М+1).
Пример 34
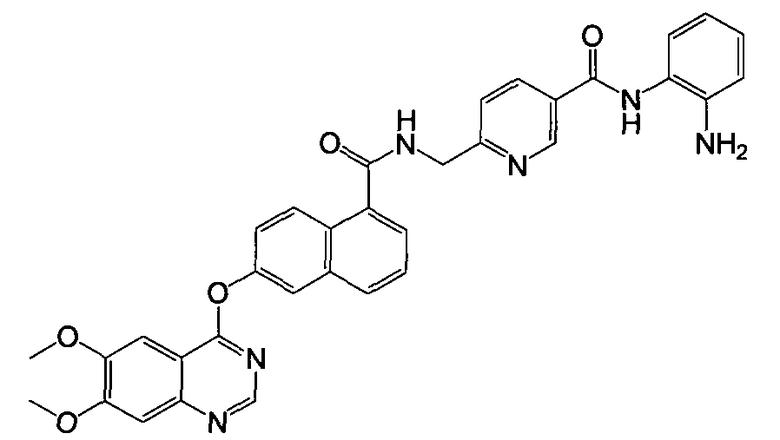
Получение N-(2-аминофенил)-6-((2-(7-метоксихинолин-4-илокси)-1-нафтамидо)метил)никотинамида
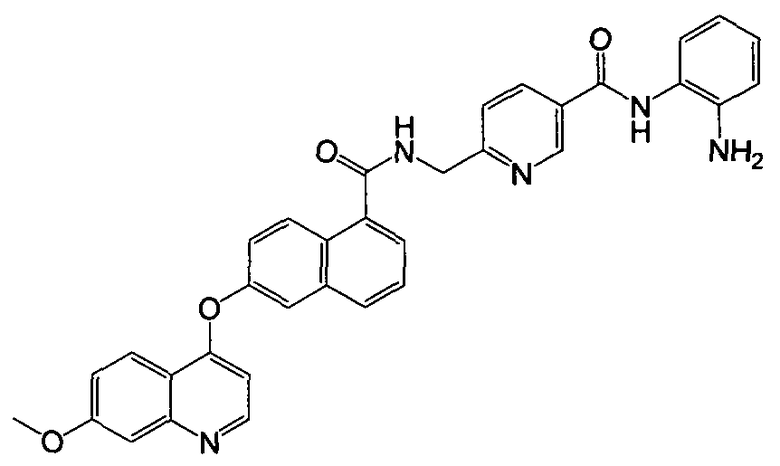
Указанное в заголовке соединение (46,6 мг, выход 82%) получали в виде твердого вещества коричневого цвета из 6-(7-метоксихинолин-4-илокси)-1-нафтоевой кислоты (34,5 мг, 0,1 ммоль) и 6-(аминометил)-N-(2-аминофенил)никотинамида (29,0 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 3,95 (с, 3H, -OCH3), 4,74 (с, 2H, -CH2), 4,95 (с, 2H, бензол-NH2), 6,60 (м, 2H, Ar-H), 6,79 (с, 1H, Ar-H), 6,98 (с, 1H, Ar-H), 7,17 (с, 1H, Ar-H), 7,31 (д, J=8,6 Гц, 1H, Ar-H), 7,44 (с, 1H, Ar-H), 7,58-7,63 (м, 3H, Ar-H), 7,77 (с, 1H, Ar-H), 7,87 (с, 1H, Ar-H), 8,05 (д, J=5,6 Гц, 1H, Ar-H), 8,24 (д, J=8,3 Гц, 1H, Ar-H), 8,33 (с, 1H, Ar-H), 8,47 (д, J=7,5 Гц, 1H, Ar-H), 9,13 (с, 1H, Ar-H), 9,25 (с, 1H, -CONH), 9,77 (с, 1H, бензол-NH). LC-MS (m/z) 570 (M+1).
Пример 35
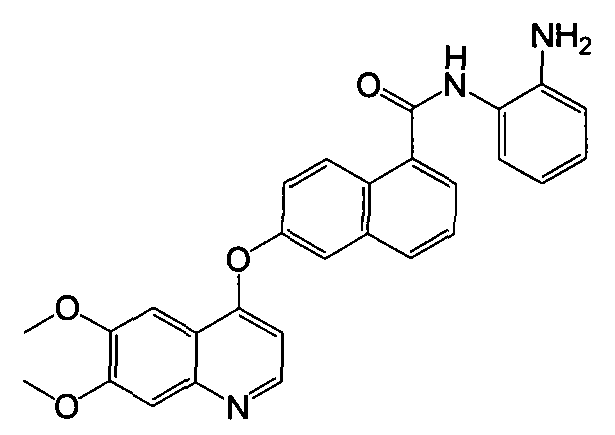
Получение N-(2-аминофенил)-6-(6,7-диметоксихинолин-4-илокси)-1-нафтамида
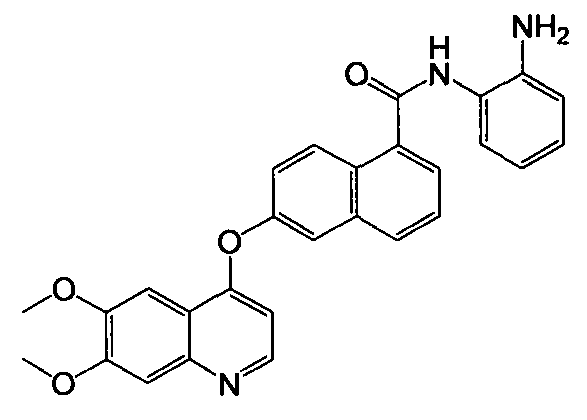
Указанное в заголовке соединение (40,0 мг, выход 86%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихинолин-4-илокси)-1-нафтоевой кислоты (37,5 мг, 0,1 ммоль) и о-фенилендиамина (43,2 мг, 0,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 3,93 (с, 3Н, -ОСН3), 3,95 (с, 3Н, -ОСН3), 4,99 (с, 2H, бензол-NH2), 6,56 (д, J=5,2 Гц, 1H, Ar-Н), 6,63 (т, J=7,6 Гц, 1H, Ar-H), 6,81 (д, J=7,6 Гц, 1H, Ar-H), 6,98 (т, J=7,2 Гц, 1H, Ar-H), 7,36 (д, J=7,6 Гц, 1H, Ar-H), 7,43 (с, 1H, Ar-H), 7,56-7,58 (м, 2H, Ar-H), 7,65 (т, J=7,6 Гц, 1H, Ar-H), 7,87-7,90 (м, 2H, Ar-H), 8,08 (д, J=8,0 Гц, 1H, Ar-H), 8,43 (д, J=9,2 Гц, 1H, Ar-H), 8,49 (д, J=5,2 Гц, 1H, Ar-H), 9,87 (с, 1H, бензол-NH). LC-MS (m/z) 466 (M+1).
Пример 36
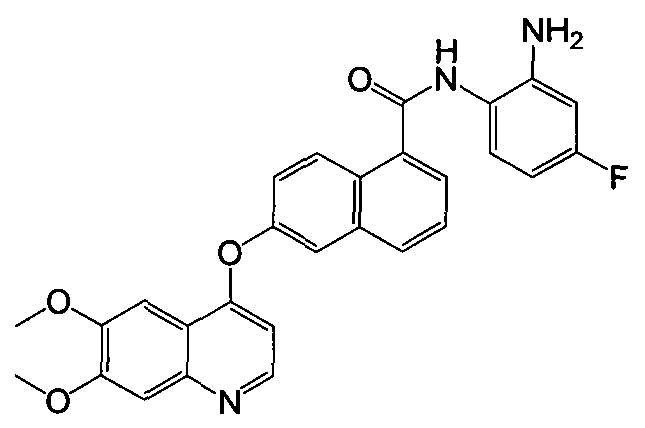
Получение N-(2-амино-4-фторфенил)-6-(6,7-диметоксихинолин-4-илокси)-1-нафтамида
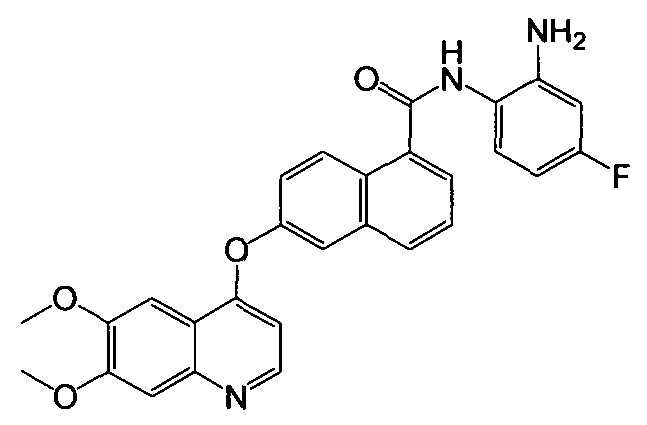
Указанное в заголовке соединение (39,1 мг, выход 81%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихинолин-4-илокси)-1-нафтоевой кислоты (37,5 мг, 0,1 ммоль) и 4-фтор-о-фенилендиамина (15,1 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 3,93 (с, 3Н, -ОСН3), 3,95 (с, 3Н, -ОСН3), 5,31 (с, 2H, бензол-NH2), 6,40 (с, 1H, Ar-H), 6,55-6,59 (т, 2Н, Ar-Н), 7,30 (д, J=7,6 Гц, 1Н, Ar-H), 7,42 (с, 1H, Ar-H), 7,54-7,57 (м, 2H, Ar-H), 7,64 (т, J=8,0 Гц, 1H, Ar-H), 7,89-7,91 (м, 2H, Ar-H), 8,07 (д, J=8,0 Гц, 1H, Ar-H), 8,42 (д, J=9,2 Гц, 1H, Ar-H), 8,49 (д, J=5,2 Гц, 1H, Ar-H), 9,79 (с, 1H, бензол-NH). LC-MS (m/z) 484 (M+1).
Пример 37
Получение N-(4-((2-аминофенил)карбамоил)бензил)-6-(6,7-диметоксихинолин-4-илокси)-1-нафтамида
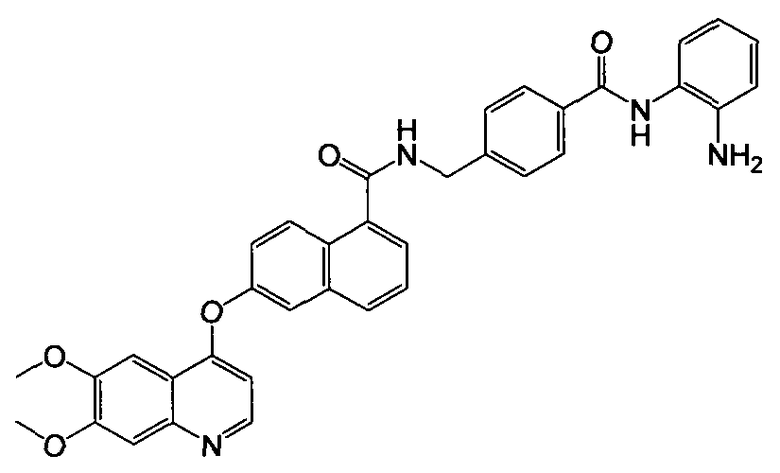
Указанное в заголовке соединение (49,0 мг, выход 82%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихинолин-4-илокси)-1-нафтоевой кислоты (37,5 мг, 0,1 ммоль) и 4-(аминометил)-N-(2-аминофенил)бензамида (28,9 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 3,93 (с, 3H, -OCH3), 3,95 (с, 3H, -OCH3), 4,63 (д, J=5,6 Гц, 2H, -CH2), 4,90 (с, 2H, бензол-NH2), 6,56-6,59 (м, 2H, Ar-H), 6,78 (д, J=7,6 Гц, 1H, Ar-H), 6,96 (т, J=8,1 Гц, 1H, Ar-H), 7,17 (д, J=7,6 Гц, 1H, Ar-H), 7,42 (с, 1H, Ar-H), 7,53-7,55 (м, 4H, Ar-H), 7,62 (т, J=8,0 Гц, 1H, Ar-H), 7,71 (д, J=6,8 Гц, 1H, Ar-H), 7,87 (с, 1H, Ar-H), 7,98-8,06 (м, 3H, Ar-H), 8,39 (д, J=9,2 Гц, 1H, Ar-H), 8,49 (д, J=5,2 Гц, 1H, Ar-H), 9,26 (т, J=6,0 Гц, 1H, -CONH), 9,66 (с, 1H, бензол-NH). LC-MS (m/z) 599 (M+1).
Пример 38
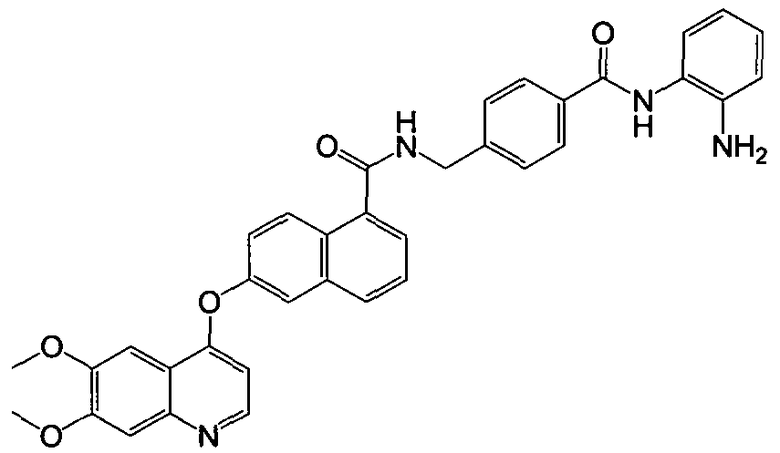
Получение N-(2-аминофенил)-6-((2-(6,7-диметоксихинолин-4-илокси)-1-нафтамидо)метил)никотинамида
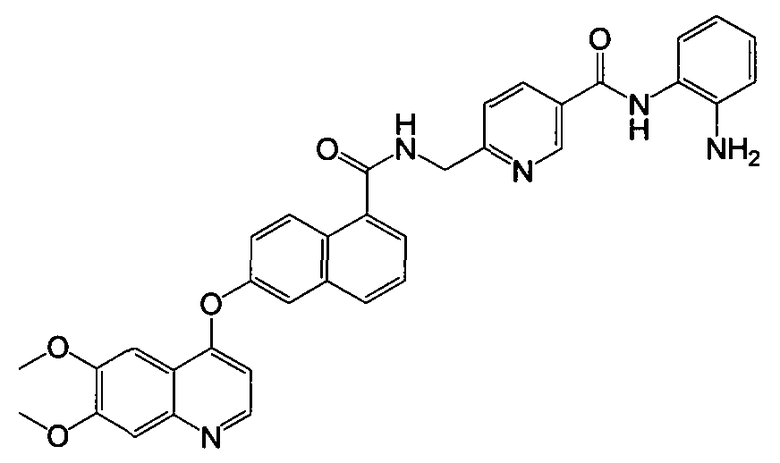
Указанное в заголовке соединение (47,9 мг, выход 80%) получали в виде твердого вещества коричневого цвета из 6-(6,7-диметоксихинолин-4-илокси)-1-нафтоевой кислоты (37,5 мг, 0,1 ммоль) и 6-(аминометил)-N-(2-аминофенил)никотинамида (29,0 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 3,93 (с, 3H, -OCH3), 3,95 (с, 3H, -OCH3), 4,73 (д, J=5,6 Гц, 2H, -CH2), 4,97 (с, 2H, бензол-NH2), 6,57 (м, 2H, Ar-H), 6,77 (д, J=6,4 Гц, 1H, Ar-H), 6,98 (т, J=8,1 Гц, 1H, Ar-H), 7,16 (д, J=5,6 Гц, 1H, Ar-H), 7,42 (с, 1H, Ar-H), 7,55-7,63 (м, 4H, Ar-H), 7,62 (т, J=8,0 Гц, 1H, Ar-H), 7,76 (д, J=6,8 Гц, 1H, Ar-H), 7,88 (с, 1H, Ar-H), 8,06 (с, 1H, Ar-H), 8,33 (с, 1H, Ar-H), 8,45-8,48 (м, 2H, Ar-H), 9,12 (с, 1H, Ar-H), 9,30 (т, J=6,0 Гц, 1H, -CONH), 9,80 (с, 1H, бензол-NH). LC-MS (m/z) 600 (M+1).
Пример 39
Получение N-(2-аминофенил)-6-(хинолин-4-илокси)-1-нафтамида
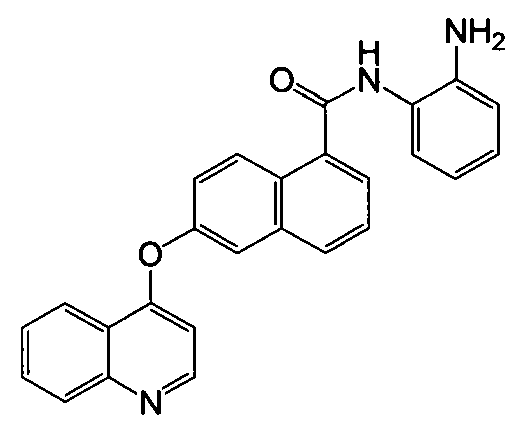
Указанное в заголовке соединение (35,6 мг, выход 88%) получали в виде твердого вещества коричневого цвета из 6-(хинолин-4-илокси)-1-нафтоевой кислоты (31,5 мг, 0,1 ммоль) и о-фенилендиамина (43,2 мг, 0,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 4,97 (с, 2Н, бензол-NH2), 6,65 (т, J=7,3 Гц, 1H, Ar-H), 6,75 (д, J=5,1 Гц, 1H, Ar-H), 6,82 (д, J=7,8 Гц, 1H, Ar-H), 7,00 (т, J=7,1 Гц, 1H, Ar-H), 7,38 (д, J=7,5 Гц, 1H, Ar-H), 7,59 (дд, J=2,3 и 9,2 Гц, 1H, Ar-H), 7,64-7,71 (м, 2H, Ar-H), 7,83-7,92 (м, 3H, Ar-H), 8,08 (д, J=8,4 Гц, 2H, Ar-H), 8,37 (д, J=7,9 Гц, 1H, Ar-H), 8,45 (д, J=9,2 Гц, 1H, Ar-H), 8,73 (д, J=5,1 Гц, 1H, Ar-H), 9,85 (с, 1H, бензол-NH). LC-MS (m/z) 406 (M+1).
Пример 40
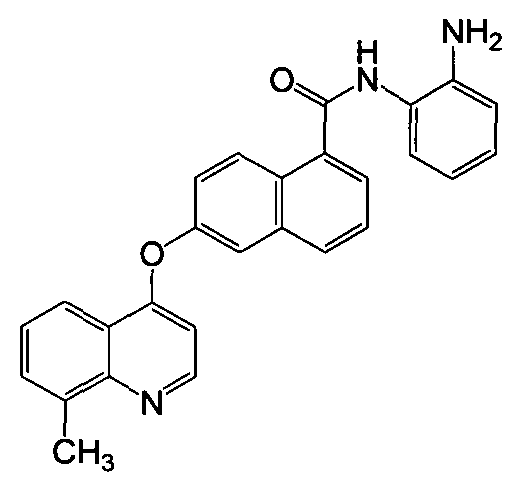
Получение N-(2-аминофенил)-6-(8-метилхинолин-4-илокси)-1-нафтамида
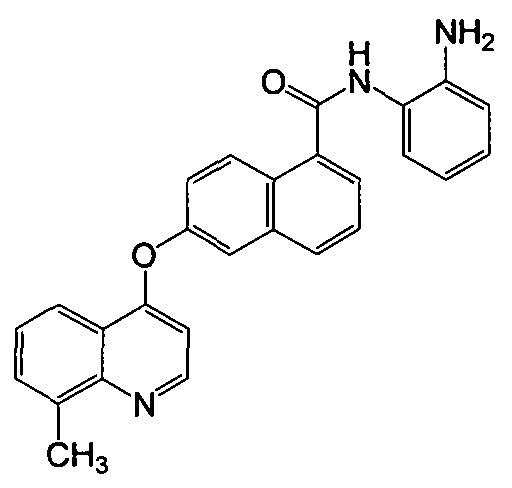
Указанное в заголовке соединение (37,7 мг, выход 90%) получали в виде твердого вещества коричневого цвета из 6-(8-метилхинолин-4-илокси)-1-нафтоевой кислоты (32,9 мг, 0,1 ммоль) и о-фенилендиамина (43,2 мг, 0,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 2,76 (с, 3Н, Ar-СН3), 4,97 (с, 2H, бензол-NH2), 6,64 (т, J=7,1 Гц, 1H, Ar-H), 6,78 (д, J=5,0 Гц, 1H, Ar-H), 6,82 (д, J=7,8 Гц, 1H, Ar-H), 6,99 (т, J=7,3 Гц, 1H, Ar-H), 7,38 (д, J=7,5 Гц, 1H, Ar-H), 7,55-7,58 (м, 2H, Ar-H), 7,65 (т, J=7,6 Гц, 1H, Ar-H), 7,71 (д, J=7,0 Гц, 1H, Ar-H), 7,87-7,89 (м, 2H, Ar-H), 8,07 (д, J=8,2 Гц, 1H, Ar-H), 8,20 (д, J=7,9 Гц, 1H, Ar-H), 8,44 (д, J=9,2 Гц, 1H, Ar-H), 8,76 (д, J=5,0 Гц, 1H, Ar-H), 9,84 (с, 1H, бензол-NH). LC-MS (m/z) 420 (M+1).
Пример 41
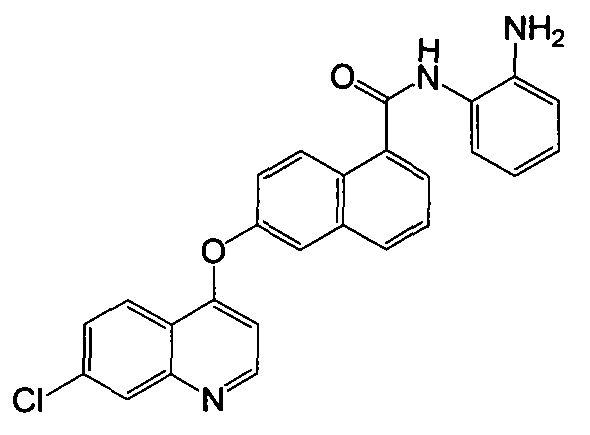
Получение N-(2-аминофенил)-6-(7-хлорхинолин-4-илокси)-1-нафтамида
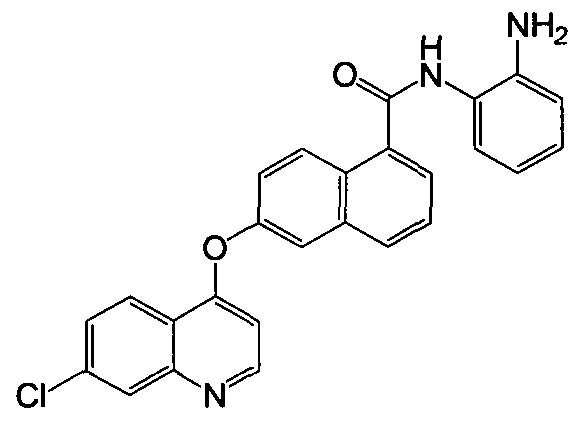
Указанное в заголовке соединение (33,2 мг, выход 83%) получали в виде твердого вещества коричневого цвета из 6-(7-хлорхинолин-4-илокси)-1-нафтоевой кислоты (35,0 мг, 0,1 ммоль) и о-фенилендиамина (43,2 мг, 0,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 4,97 (с, 2H, бензол-NH2), 6,65 (т, J=7,4 Гц, Ar-H), 6,77 (д, J=5,5 Гц, 1H, Ar-H), 6,82 (д, J=7,2 Гц, 1H, Ar-H), 7,00 (т, J=7,0 Гц, 1H, Ar-H), 7,38 (д, J=7,2 Гц, 1H, Ar-H), 7,60 (дд, J=2,6 и 9,2 Гц, 1H, Ar-H), 7,67-7,74 (м, 2H, Ar-H), 7,89 (д, J=7,4 Гц, 1H, Ar-H), 7,94 (д, J=2,4 Гц, 1H, Ar-H), 8,09 (д, J=8,2 Гц, 1H, Ar-H), 8,13 (д, J=2,1 Гц, 1H, Ar-H), 8,41 (д, J=9,0 Гц, 1H, Ar-H), 8,46 (д, J=9,6 Гц, 1H, Ar-H), 8,76 (д, J=5,2 Гц, 1H, Ar-H), 9,85 (с, 1H, бензол-NH). LC-MS (m/z) 440 (M+1).
Пример 42
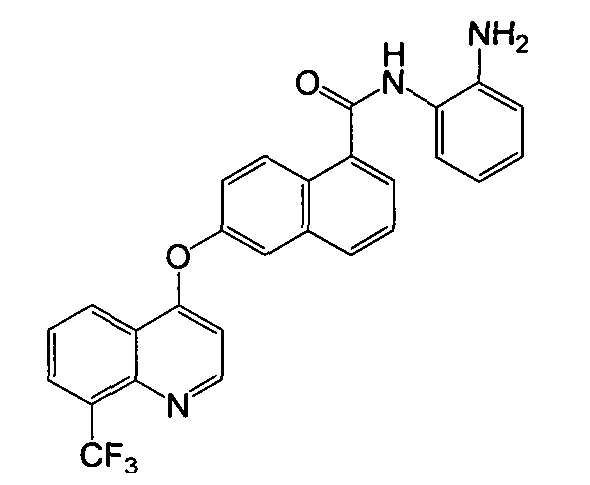
Получение N-(2-аминофенил)-6-(8-трифторметилхинолин-4-илокси)-1-нафтамида
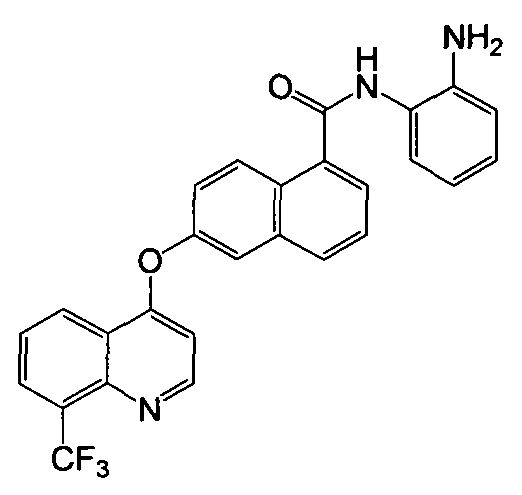
Указанное в заголовке соединение (38,3 мг, выход 81%) получали в виде твердого вещества коричневого цвета из 6-(8-трифторметилхинолин-4-илокси)-1-нафтоевой кислоты (39,8 мг, 0,1 ммоль) и о-фенилендиамина (43,2 мг, 0,4 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 4,98 (с, 2H, бензол-NH2), 6,65 (т, J=7,3 Гц, 1H, Ar-H), 6,83 (д, J=7,6 Гц, 1H, Ar-H), 6,89 (д, J=5,2 Гц, 1H, Ar-H), 7,00 (т, J=7,2 Гц, 1H, Ar-H), 7,38 (д, J=7,5 Гц, 1H, Ar-H), 7,62 (дд, J=2,4 и 9,2 Гц, 1H, Ar-H), 7,68 (т, J=7,7 Гц, 1H, Ar-H), 7,83 (т, J=7,9 Гц, 1H, Ar-H), 7,90 (д, J=7,0 Гц, 1H, Ar-H), 7,97 (д, J=2,3 Гц, 1H, Ar-H), 8,10 (д, J=8,3 Гц, 1H, Ar-H), 8,29 (д, J=7,1 Гц, 1H, Ar-H), 8,47 (д, J=9,2 Гц, 1H, Ar-H), 8,70 (д, J=7,8 Гц, 1H, Ar-H), 8,87 (д, J=5,2 Гц, 1H, Ar-H), 9,86 (с, 1H, бензол-NH). LC-MS (m/z) 474 (M+1).
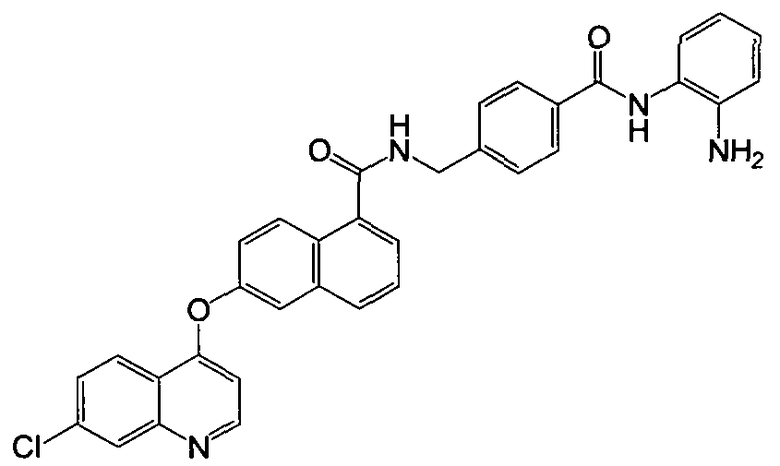
Пример 43
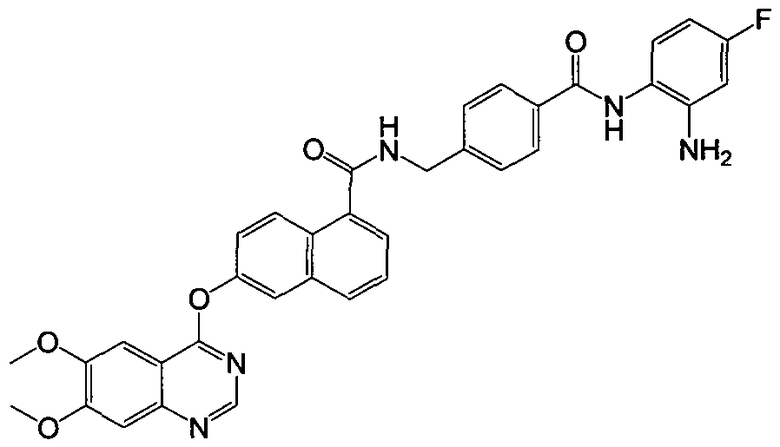
Получение N-(4-((2-аминофенил)карбамоил)бензил)-6-(7-хлорхинолин-4-илокси)-1-нафтамида
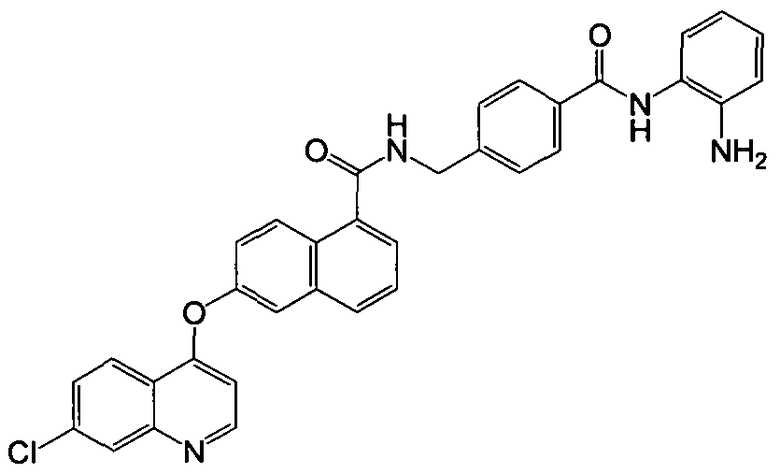
Указанное в заголовке соединение (42,4 мг, выход 74%) получали в виде твердого вещества коричневого цвета из 6-(7-хлорхинолин-4-илокси)-1-нафтоевой кислоты (35,0 мг, 0,1 ммоль) и 4-(аминометил)-N-(2-аминофенил)бензамида (28,9 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 4,64 (д, J=5,8 Гц, 2H, -CH2), 4,87 (с, 2H, бензол-NH2), 6,60 (т, J=7,0 Гц, 1H, Ar-H), 6,75-6,79 (м, 2H, Ar-H), 6,97 (т, J=7,5 Гц, 1H, Ar-H), 7,18 (д, J=7,7 Гц, 1H, Ar-H), 7,53-7,59 (м, 3H, Ar-H), 7,66 (т, J=8,0 Гц, 1H, Ar-H), 7,70-7,74 (м, 2H, Ar-H), 7,92 (д, J=2,0 Гц, 1H, Ar-H), 7,99 (д, J=7,9 Гц, 2H, Ar-H), 8,06 (д, J=8,2 Гц, 1H, Ar-H), 8,13 (с, 1H, Ar-H), 8,39-8,42 (м, 2H, Ar-H), 8,75 (д, J=5,1 Гц, 1H, Ar-H), 9,22 (т, J=5,6 Гц, 1H, -CONH), 9,62 (с, 1H, бензол-NH). LC-MS (m/z) 573 (M+1).
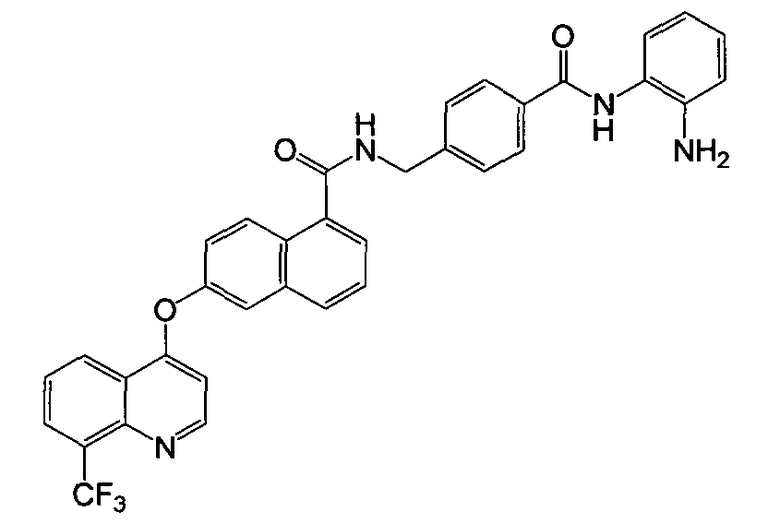
Пример 44
Получение N-(4-(((2-аминофенил)карбамоил)бензил)-6-(8-трифторметилхинолин-4-илокси)-1-нафтамида
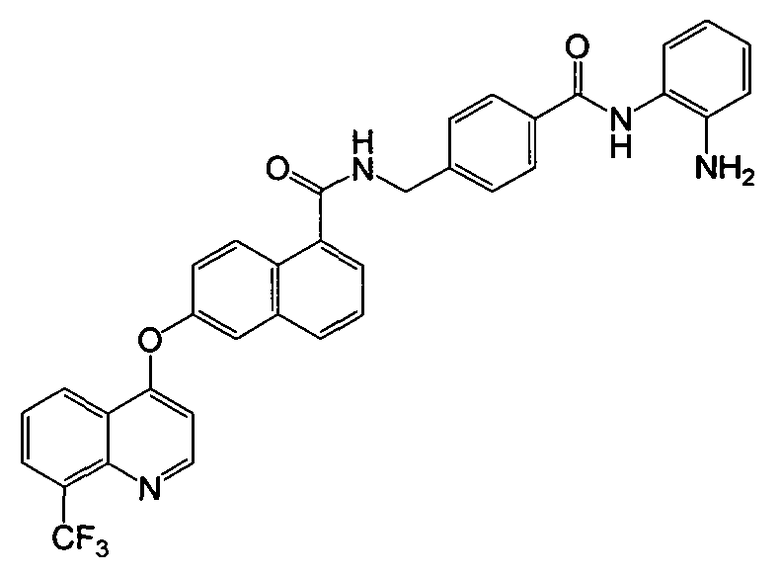
Указанное в заголовке соединение (47,3 мг, выход 78%) получали в виде твердого вещества коричневого цвета из 6-(8-трифторметилхинолин-4-илокси)-1-нафтоевой кислоты (38,3 мг, 0,1 ммоль) и 6-(аминометил)-N-(2-аминофенил)никотинамида (29,0 мг, 0,12 ммоль) процедурой, аналогичной процедуре, описанной в примере 16. 1H-ЯМР (ДМСО-d6) δ 4,64 (д, J=5,6 Гц, 2H, -CH2), 4,87 (с, 2H, бензол-NH2), 6,60 (т, J=7,2 Гц, 1H, Ar-H), 6,78 (д, J=7,8 Гц, 1H, Ar-H), 6,89 (д, J=5,1 Гц, 1H, Ar-H), 6,97 (т, J=7,2 Гц, 1H, Ar-H), 7,18 (д, J=7,9 Гц, 1H, Ar-H), 7,53-7,66 (м, 4H, Ar-H), 7,74 (д, J=6,9 Гц, 1H, Ar-H), 7,83 (т, J=7,9 Гц, 1H, Ar-H), 7,95-8,08 (м, 4H, Ar-H), 8,29 (д, J=7,0 Гц, 1H, Ar-H), 8,42 (д, J=9,1 Гц, 1H, Ar-H), 8,69 (д, J=8,3 Гц, 1H, Ar-H), 8,86 (д, J=5,0 Гц, 1H, Ar-H), 9,22 (т, J=5,5 Гц, 1H, -CONH), 9,61 (с, 1H, бензол-NH). LC-MS (m/z) 607 (M+1).
Пример 45
Получение таблеток
Состав (1000 таблеток):
Соединение 31 просеивали через сито 100 меш. Микрокристаллическую целлюлозу, карбоксиметилкрахмал натрия и порошок талька соответственно просеивали через сито 80 меш. Микрокристаллическую целлюлозу и карбоксиметилкрахмал натрия отвешивали во включаемом в состав количестве и однородно смешивали с соединением 31 способом ступенчатого прибавления. Для получения влажных гранул добавляли подходящее количество 4% раствора поливидона (K30) в абсолютном этаноле. Гранулы сушили и добавляли порошок талька во включаемом в состав количестве. Затем проводили прессование для получения таблеток.
Пример 46
Получение капсул
Состав (1000 капсул):
Соединение 31 просеивали через сито 100 меш. Микрокристаллическую целлюлозу, лактозу, карбоксиметилкрахмал натрия и стеарат магния соответственно просеивали через сито 80 меш. Микрокристаллическую целлюлозу, лактозу и карбоксиметилкрахмал натрия отвешивали во включаемом в состав количестве и однородно смешивали с соединением 31 способом ступенчатого прибавления. Затем стеарат магния добавляли во включаемом в состав количестве и однородно смешивали. Затем проводили заполнение капсул для получения готовых к употреблению капсул.
Пример 47
Получение инъекционного препарата
Состав:
Соединение 31 растворяли в ДМСО и затем добавляли этанол для получения инъекционного препарата.
Пример 48
Аанализ зависимой от лигандов PDGF и VEGF клеточной пролиферации под действием соединений формулы (I)
Измерение ингибирования in vivo зависимой от рецепторных лигандов клеточной пролиферации:
1. PDGF-зависимая клеточная пролиферация:
Методами инженерии конструировали линию клеток мышиных фибробластов NIH-3T3 для стабильной экспрессии человеческого PDGFRβ и использовали для оценки PDGF-зависимой клеточной пролиферации. Экспрессирующие человеческие PDGFRβ клетки NIH-3T3 высевали в 96-луночные планшеты в количестве 5000 на лунку и инкубировали в течение ночи в бессывороточной среде через 24 часа. Добавляли подлежащие тестированию соединения и PDGF BB (50 нг/мл) и клетки инкубировали в течение 72 часов в бессывороточной среде. Воздействия на пролиферацию определяли способом MTS (Promega), в соответствии с инструкцией. Инкубацию выполняли в течение 2 часов при 37°C в инкубаторе в атмосфере CO2, и спектральную поглощательную способность при 490 нм измеряли с использованием считывающего устройства планшет ELISA (иммуноферментного анализа).
2. VEGF-зависимая клеточная пролиферация:
Клетки HUVEC высевали в 96-луночные планшеты в количестве 6000 на лунку и через 24 часа инкубировали в бессывороточной среде в течение 2 часов. Добавляли подлежащие тестированию соединения и VEGF 165 (50 нг/мл) и инкубировали в течение 72 часов в бессывороточной среде. Воздействия на пролиферацию определяли способом MTS (Promega), в соответствии с инструкцией. Инкубацию выполняли в течение 2 часов при 37°C в инкубаторе в атмосфере CO2, и спектральную поглощательную способность при 490 нм измеряли с использованием считывающего устройства планшет ELISA.
Экспериментальные результаты показаны в таблице 2.
(зависимая от лиганда PDGF клеточная пролиферация)
(зависимая от лиганда VEGF клеточная пролиферация)
Пример 49
Ингибирование in vitro общей ферментативной активности HDAC и ингибирование in vivo ферментной активности подтипа HDAC соединениями формулы (I)
Анализ общей ферментативной активности HDAC in vitro:
Общую ферментативную активность HDAC in vitro определяли с использованием набора для флуорометрического анализа HDAC/обнаружения лекарственных средств (BIOMOL), в соответствии с инструкцией производителя.
Принцип эксперимента следующий: под действием гистондеацетилазы (в эксперименте использовался ядерный экстракт клеток HeLa, который был богат разнообразными подтипами HDAC), ацетильная группа удаляется из специального субстрата Fluor de Lys, таким образом, открывается свободная аминогруппа. После добавления проявителя, субстрат продуцирует флуоресценцию. Для флуоресценции длина волн возбуждения составляет 360 нм, и длина волн эмиссии 460 нм. Чем полнее деацетилирование субстрата, тем выше вызываемая флуоресценция. В отсутствие ингибиторов, величина флуоресценции принимается в качестве контроля; когда ингибитор возбуждается, величина вызванной флуоресценции будет снижена, тогда как в случае отсутствия возбуждения фермента (соответствующего полному ингибированию активности фермента), величина флуоресценции не определяется. В целом, после подавления, величины флуоресценции будут составлять от контрольной величины до отсутствия флуоресценции. При проведении анализа субстрат с отсутствием флуоресценции используется как 0, и контроль используется как 1. Меньшая величина означает более высокую ингибиторную активность.
1. Добавить аналитический буфер, разбавленный трихостатин A и тестируемый ингибитор в соответствующие лунки титрационного микропланшета. В следующей таблице представлены количества, используемые для каждого реагента различных типов анализов.
2. Добавить разбавленный экстракт нуклеопротеина HeLa во все лунки, кроме тех, которые маркированы как «пустые».
3. Дать возможность субстрату Fluor de Lys™ и образцам в титрационном микропланшете уравновеситься до 25°C.
4. Инициировать реакции HDAC добавлением разбавленного субстрата (25 мкл) в каждую лунку и тщательно смешать.
5. Дать возможность реакциям продолжаться в течение 30 минут и затем остановить их добавлением проявителя Fluor de Lys™ (50 мкл). Инкубировать планшет при комнатной температуре (25°C) в течение 10-15 мин.
6. Считывать величины флуоресценции образцов на считывающем флуориметре титрационных микропланшетов при длине волн возбуждения 369 нм и длине волн эмиссии 451 нм.
Анализ селективности ингибиторов в отношении подтипов HDAC с использованием гена-репортера:
Различные подтипы HDAC могут связываться с различными транскрипционными факторами и участвовать в регуляции экспрессии различных генов. Подходящие регуляционные элементы для транскрипционных факторов выбираются для конструкции генов-репортеров, которые могут использоваться для оценки селективного ингибирования подтипов HDAC ингибиторами. Вкратце, клетки HeLa высевали в 96-луночные планшеты за день до трансфекции для обеспечения слияния 50-80% при трансфекции. Клетки трансфецировали плазмидой, содержащей промотерную последовательность p21 или реактивный элемент выше по ходу транскрипции конструкта гена люциферазы с использованием реагента трансфекции FuGene6, в соответствии с инструкцией производителя (Roche). Для нормализации эффективности трансфекции, котрансфецировали экспрессионную плазмиду GFP. Через 24 часа добавляли соединения или контроль носителя (ДМСО). Через 24 часа, клетки собирали и лизировали и количество люциферазы оценивали с использованием наборов анализа люциферазы (Promega), в соответствии с инструкцией производителя.
Экспериментальные результаты показаны в таблице 3.
(анализ репортера p21)
Кратность индукции при 10 мкМ
Пример 50
Ингибирование пролиферации опухолевых клеток соединениями формулы (I)
Опухолевые клетки трипсинизировали и высевали в 96-луночные планшеты в количестве 3000 на лунку и инкубировали в полной среде с 10% FBS (фетальной телячьей сывороткой) в течение 24 часов. Добавляли подлежащие тестированию соединения и носитель, и конечная концентрация соединений находилась в диапазоне от 100 нмоль/л до 100 мкмоль/л. Соединения инкубировали в течение 72 часов в полной среде. Добавляли реагент MTS (Promega) в соответствии с инструкцией, инкубировали в течение 2 часов при 37єC в инкубаторе в атмосфере CO2. Затем показания спектральной поглощательной способности при 490 нм считывали с использованием считывающего устройства планшет ELISA.
Экспериментальные результаты представлены в таблице 4.
в A-498
в A549
в Bel-7402
в HCT-8
в MCF-7
CS055: хидамид, ингибитор HDAC, разработанный компанией Chipscreen Biosciences, обладает превосходной противоопухолевой активностью и в настоящее время проходит II стадию клинических испытаний.
Пример 51
Ингибирование соединением 31 клеток раковой опухоли легких человека A549, трансплантированных голым мышам
Самок мышей nu/nu с массой тела примерно 14-16 г кормили нормальным рационом в течение 3 дней. Затем культивированные клетки раковой опухоли легких человека A549 имплантировали в подмышечную впадину 50 мышам. Когда опухоли достигали диаметра более 6 мм, мышей делили на 6 групп случайным методом. Каждая группа включала 8 мышей. Одну группу лечили носителем. Одну группу лечили сутентом, лекарственным средством положительного контроля. Другие четыре группы лечили соединением 31 в дозах 5, 10, 20 и 40 мг/кг массы тела. Каждая группа получала вводимое средство перорально один раз в день в течение 24 дней. Объем опухолей наряду с массой тела регистрировали дважды в неделю. На следующий день после введения 24 доз мышей умерщвляли, и опухоли взвешивали. Ингибирование роста опухолей в каждой группе рассчитывали, используя следующую формулу:
{[(средняя масса опухолей контрольной группы)-(средняя масса опухолей тестируемой группы)]/(среднюю массу опухолей контрольной группы)}×100%.
Экспериментальные результаты представлены в таблице 5 и на фиг.1.
Пример 52
Ингибирование соединением 31 клеток раковой опухоли ободочной кишки человека HCT-8, трансплантированных голым мышам
Самок мышей nu/nu с массой тела примерно 18-20 г кормили нормальным рационом в течение 3 дней. Затем культивированные клетки раковой опухоли ободочной кишки человека HCT-8 имплантировали в подмышечную впадину 50 мышам. Когда опухоли достигали объема не менее 100 мм3, мышей делили на 6 групп случайным методом. Каждая группа включала 8 мышей. Одну группу лечили носителем. Одну группу лечили сутентом, лекарственным средством положительного контроля. Другие четыре группы лечили соединением 31 в дозах 2,5, 5, 10 и 20 мг/кг массы тела. Каждая группа получала вводимое средство перорально один раз в день в течение 24 дней. Объем опухолей наряду с массой тела регистрировали дважды в неделю. На следующий день после введения 20 доз мышей умерщвляли, и опухоли взвешивали. Ингибирование роста опухолей в каждой группе рассчитывали, используя следующую формулу:
{[(средняя масса опухолей контрольной группы)-(средняя масса опухолей тестируемой группы)]/(среднюю массу опухолей контрольной группы)}×100%.
Экспериментальные результаты представлены в таблице 6 и на фиг.2.
Пример 53
Ингибирование соединением 31 клеток раковой опухоли ободочной кишки человека SSMC7721, трансплантированных голым мышам
Самок мышей nu/nu с массой тела примерно 18-20 г кормили нормальным рационом в течение 3 дней. Затем культивированные клетки раковой опухоли ободочной кишки человека SSMC7721 имплантировали в подмышечную впадину 50 мышам. Когда опухоли достигали объема не менее 100 мм3, мышей делили на 6 групп случайным методом. Каждая группа включала 8 мышей. Одну группу лечили носителем. Одну группу лечили сутентом, лекарственным средством положительного контроля. Другие четыре группы лечили соединением 31 в дозах 2,5, 5, 10 и 20 мг/кг массы тела. Каждая группа получала вводимое средство перорально один раз в день в течение 24 дней. Объем опухолей наряду с массой тела регистрировали дважды в неделю. На следующий день после введения 24 доз мышей умерщвляли, и опухоли взвешивали. Ингибирование роста опухолей в каждой группе рассчитывали, используя следующую формулу:
{[(средняя масса опухолей контрольной группы)-(средняя масса опухолей тестируемой группы)]/(среднюю массу опухолей контрольной группы)} ×100%.
Экспериментальные результаты представлены в таблице 7 и фиг.3.
Пример 54
Ингибирование соединением 33 и соединением 34 клеток раковой опухоли ободочной кишки человека HCT-8, трансплантированных голым мышам
Самок мышей nu/nu с массой тела примерно 18-20 г кормили нормальным рационом в течение 3 дней. Затем культивированные клетки раковой опухоли ободочной кишки человека HCT-8 имплантировали в подмышечную впадину 50 мышам. Когда опухоли достигали объема не менее 100 мм3, мышей делили на 6 групп случайным методом. Каждая группа включала 8 мышей. Одну группу лечили носителем. Одну группу лечили сутентом, лекарственным средством положительного контроля. Две группы лечили соединением 33 в различных концентрациях. Две другие группы лечили соединением 34 в различных концентрациях. Каждая группа получала вводимое средство перорально один раз в день в течение 20 дней. Объем опухолей наряду с массой тела регистрировали дважды в неделю. На следующий день после введения 20 доз мышей умерщвляли, и опухоли взвешивали. Ингибирование роста опухолей в каждой группе рассчитывали, используя следующую формулу:
{[(средняя масса опухолей контрольной группы)-(средняя масса опухолей тестируемой группы)]/(среднюю массу опухолей контрольной группы)}×100%.
Экспериментальные результаты представлены в таблице 8 и на фиг.4.
Пример 55
Ингибирование соединением 33 и соединением 37 клеток раковой опухоли ободочной кишки человека HCT-8, трансплантированных голым мышам
Самок мышей nu/nu с массой тела примерно 18-20 г кормили нормальным рационом в течение 3 дней. Затем культивированные клетки раковой опухоли ободочной кишки человека HCT-8 имплантировали в подмышечную впадину 50 мышам. Когда опухоли достигали объема не менее 100 мм3, мышей делили на 6 групп случайным методом. Каждая группа включала 8 мышей. Одну группу лечили носителем. Одну группу лечили сутентом, лекарственным средством положительного контроля. Две группы лечили соединением 33 в различных концентрациях. Две другие группы лечили соединением 37 в различных концентрациях. Соединение 33 вводили дважды в день с интервалом 6 часов. Для других групп введение выполняли один раз в день. Каждая группа получала вводимое средство перорально один раз в день в течение 20 дней. Объем опухолей наряду с массой тела регистрировали дважды в неделю. На следующий день после введения 20 доз мышей умерщвляли, и опухоли взвешивали. Ингибирование роста опухолей в каждой группе рассчитывали, используя следующую формулу:
{[(средняя масса опухолей контрольной группы)-(средняя масса опухолей тестируемой группы)]/(среднюю массу опухолей контрольной группы)}×100%.
Экспериментальные результаты представлены в таблице 9 и на фиг.5.
Пример 56
Ингибирование соединением 33 и соединением 37 клеток раковой опухоли печени человека SSMC7721, трансплантированных голым мышам
Самок мышей nu/nu с массой тела примерно 18-20 г кормили нормальным рационом в течение 3 дней. Затем культивированные клетки раковой опухоли печени человека SSMC7721 имплантировали в подмышечную впадину 50 мышам. Когда опухоли достигали объема не менее 100 мм3, мышей делили на 6 групп случайным методом. Каждая группа включала 8 мышей. Одну группу лечили носителем. Одну группу лечили сутентом, лекарственным средством положительного контроля. Две группы лечили соединением 33 в различных концентрациях. Две другие группы лечили соединением 37 в различных концентрациях. Каждая группа получала вводимое средство перорально один раз в день в течение 30 дней. Объем опухолей наряду с массой тела регистрировали дважды в неделю. На следующий день после введения 30 доз мышей умерщвляли, и опухоли взвешивали. Ингибирование роста опухолей в каждой группе рассчитывали, используя следующую формулу:
{[(средняя масса опухолей контрольной группы)-(средняя масса опухолей тестируемой группы)]/(среднюю массу опухолей контрольной группы)}×100%.
Экспериментальные результаты представлены в таблице 10 и на фиг.6.
| название | год | авторы | номер документа |
|---|---|---|---|
| Производные 3-гидроксихиназолин-4(3Н)-она в качестве ингибиторов гистондеацетилазы и способ их получения | 2020 |
|
RU2740503C1 |
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2014 |
|
RU2673819C2 |
| ХИНОЛИНИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2803116C2 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2671571C1 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2647592C2 |
| КОМБИНАЦИЯ, СОДЕРЖАЩАЯ ПАЛБОЦИКЛИБ И 6-(2,4-ДИХЛОРФЕНИЛ)-5-[4-[(3S)-1-(3-ФТОРПРОПИЛ)ПИРРРОЛИДИН-3-ИЛ]-8,9-ДИГИДРО-7H-БЕНЗО[7]АННУЛЕН-2-КАРБОНОВУЮ КИСЛОТУ, И ЕЕ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2764116C2 |
| 6,7-ДИГИДРО-5H-ПИРАЗОЛО[1,2-a]ПИРАЗОЛ-1-ОНЫ (ВАРИАНТЫ), ИНГИБИРУЮЩИЕ ВЫСВОБОЖДЕНИЕ ВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ | 2002 |
|
RU2299885C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| Способ получения N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)-бензамидов | 2019 |
|
RU2722694C1 |
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
Настоящее изобретение относится к производным нафталинкарбоксамида общей формулы I, которые обладают свойствами ингибиторов протеинкиназы или гистондеацетилазы. Соединения могут найти применение для получения лекарственного средства для лечения воспалительных заболеваний, аутоиммунных заболеваний, онкологических заболеваний, заболеваний нервной системы и нейродегенеративных заболеваний, аллергий, астмы, сердечно-сосудистых заболеваний и метаболических заболевания или заболеваний, связанных с гормональными расстройствами. В общей формуле I
Z представляет CH или N; каждая из групп R1, R2 и R3 представляет водород, галоген, алкил, алкокси или трифторметил; R4 представляет
или
X представляет бензольное кольцо или пиридиновое кольцо; R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила. Изобретение также относится к способу получения указанных соединений, фармацевтическому препарату и их применению. 5 н. и 8 з.п. ф-лы, 10 табл., 6 ил.
1. Соединение формулы I
где
Z представляет СН или N;
каждая из групп R1, R2 и R3 представляет водород, галоген, алкил, алкокси или трифторметил;
R4 представляет
Х представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила.
2. Соединение по п.1, где
Z представляет СН;
каждая из групп R1, R2 и R3 представляет водород, галоген, алкил, алкокси или трифторметил;
R4 представляет
Х представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила.
3. Соединение по п.1, где
Z представляет СН;
каждая из групп R1, R2 и R3 представляет водород или алкокси;
R4 представляет
Х представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила.
4. Соединение по п.1, где
Z представляет СН;
каждая из, групп R1 и R2 представляет водород или метокси;
R3 представляет Н;
R4 представляет
Х представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила.
5. Соединение по п.1, где
Z представляет СН;
каждая из групп R1 и R2 представляет водород или метокси;
R3 представляет Н;
R4 представляет
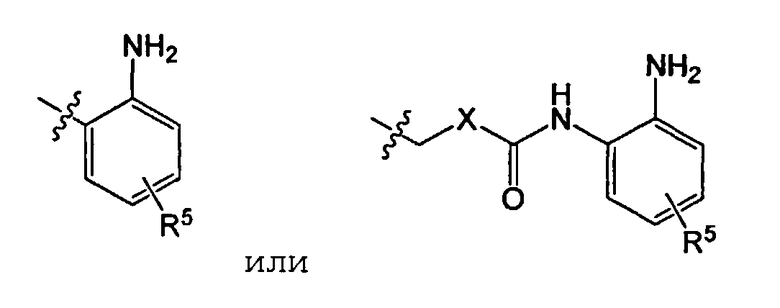
Х представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет Н или F.
6. Способ получения соединения формулы I
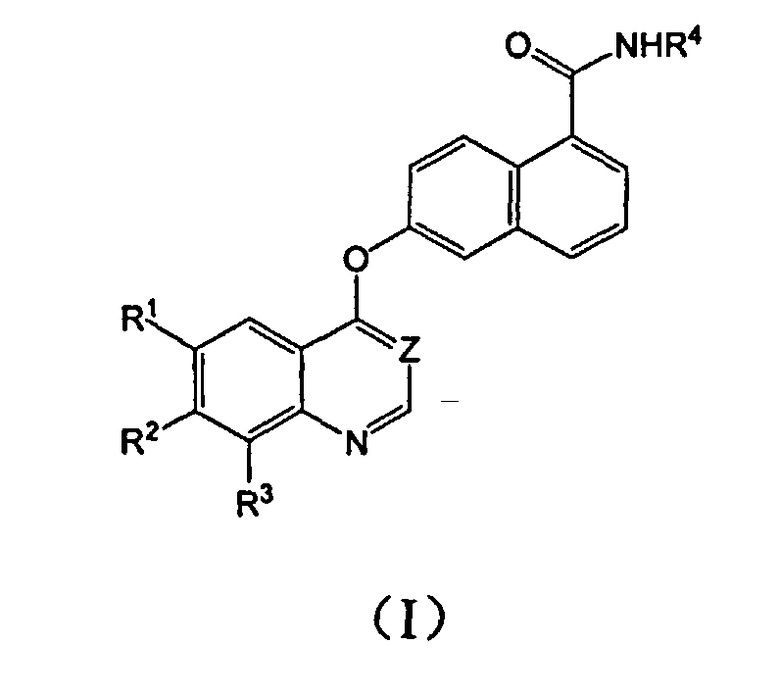
где
Z представляет СН или N;
каждая из групп R1, R2 и R3 представляет водород, галоген, алкил, алкокси или трифторметил;
R4 представляет
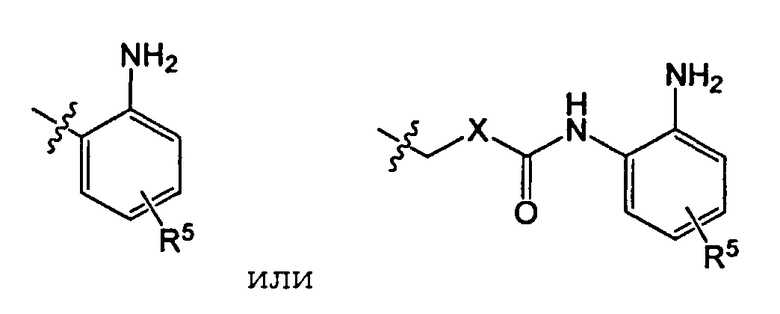
Х представляет бензольное кольцо или пиридиновое кольцо;
R5 представляет один или более заместителей, выбранных из группы, состоящей из водорода, галогена, алкила, алкокси и трифторметила;
включающий взаимодействие
соединения формулы (II)
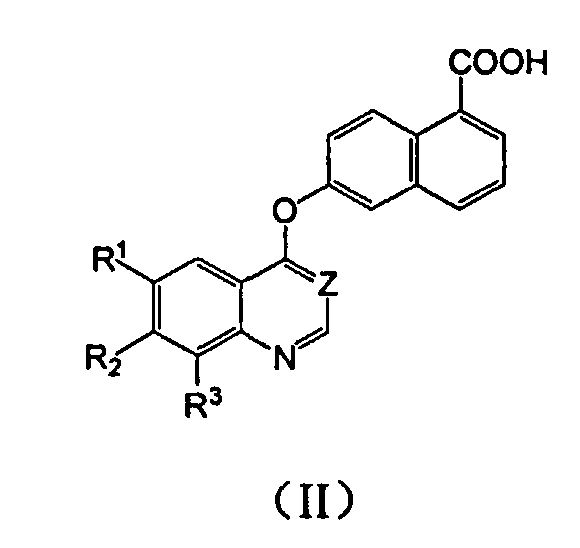
с соединением формулы (III)
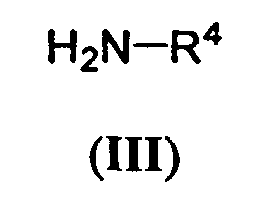
в присутствии органического растворителя и пептидного конденсирующего агента, для образования соединения формулы (I).
7. Способ по п.6, где указанный пептидный конденсирующий агент выбран из группы, состоящей из 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC), N,N'-дициклогексилкарбодиимида (DCC) и N,N'-карбонилдиимидазола (CDI).
8. Способ по п.6, где указанный органический растворитель выбран из группы, состоящей из бензола, толуола, тетрагидрофурана, 1,4-диоксана, дихлорметана, хлороформа и N,N-диметилформамида.
9. Фармацевтический препарат для лечения заболеваний, связанных с аномальной активностью протеинкиназы или аномальной активностью гистондеацетилазы, включающий пригодное количество соединения формулы (I) по п.1 и фармацевтически приемлемый носитель, эксципиент или разбавитель.
10. Фармацевтический препарат по п.9 в форме таблетки, капсулы, порошка, сиропа, раствора, суспензии, инъекции или мази.
11. Применение соединения по п.1 при получении лекарственного средства для лечения воспалительных заболеваний, аутоиммунных заболеваний, онкологических заболеваний, заболеваний нервной системы и нейродегенеративных заболеваний, аллергий, астмы, сердечно-сосудистых заболеваний и метаболических заболевания или заболеваний, связанных с гормональными расстройствами.
12. Применение фармацевтического препарата по п.9 при получении лекарственного средства для лечения воспалительных заболеваний, аутоиммунных заболеваний, онкологических заболеваний, заболеваний нервной системы и нейродегенеративных заболеваний, аллергий, астмы, сердечно-сосудистых заболеваний и метаболических заболевания или заболеваний, связанных с гормональными расстройствами.
13. Фармацевтический препарат по п.9, содержащий указанное соединение формулы (I) в количестве в диапазоне от 0,001 до 200 мг.
| Устройство для сигнализирования о начале и конце уроков в школе | 1928 |
|
SU11402A1 |
| WO 2005021553 A1, 10.03.2005 | |||
| JEAN-CHRISTOPHE HARMANGE et al., Naphthalamides as Novel and Potent Vascular Endotelial Growth Factor ReceptorTyrosine Kinase Inhibitors:Desigh,Synthesis and Evalution, J | |||
| Med.Chem., 2008, 51, 1649-1667 | |||
| JUAN DU et al., Molecular modeling studies of vascular endothelial growth factor receptor | |||

