ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способу получения табачной ароматической жидкости, используемой в курительных изделиях, к табачной ароматической жидкости, к способу получения сложноэфирного соединения и к курительному изделию.
УРОВЕНЬ ТЕХНИКИ
Например, Японская Национальная Публикация PCT № 2017-502681, как сообщение из Японской патентной литературы, раскрывает способ получения ароматизаторов и родственных материалов. Этот документ описывает, что за последние годы были предложены различные способы обработки и добавки для модифицирования общего характера или природы табачных материалов, применяемых в табачных изделиях.
СПИСОК ЦИТИРОВАННОЙ ЛИТЕРАТУРЫ
ПАТЕНТНЫЙ ДОКУМЕНТ 1: Японская Национальная Публикация PCT № 2017-502681
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
В областях производства курительных изделий и тому подобных постоянно существовала потребность в новом способе получения применимого в качестве ароматизатора материала из табака, то есть, растения из рода Nicotiana.
Цель настоящего изобретения состоит в создании способа получения табачной ароматической жидкости, которая пригодна для применения в курительных изделиях, табачной ароматической жидкости, способа получения сложноэфирного соединения, и курительного изделия, которое содержит табачную ароматическую жидкость.
РЕШЕНИЕ ЗАДАЧИ
Способ получения табачной ароматической жидкости согласно одному варианту осуществления настоящего изобретения включает: экстракцию компонента из исходного табачного материала с использованием органического растворителя для получения тем самым содержащей табачный компонент жидкости; и смешение содержащей табачный компонент жидкости с водным раствором, содержащим оснóвное вещество и спирт, в кислородсодержащей атмосфере для реакции содержащей табачный компонент жидкости с водным раствором, с получением тем самым табачной ароматической жидкости.
Табачную ароматическую жидкость согласно одному варианту осуществления настоящего изобретения получают вышеописанным способом получения табачной ароматической жидкости.
Способ получения сложноэфирного соединения согласно одному варианту осуществления настоящего изобретения включает: экстракцию компонента из исходного табачного материала с использованием органического растворителя для получения тем самым содержащей табачный компонент жидкости; введение в реакцию содержащей табачный компонент жидкости с водным раствором, содержащим оснóвное вещество и спирт, в кислородсодержащей атмосфере, для получения тем самым табачной ароматической жидкости; и выделение сложноэфирного соединения из табачной ароматической жидкости.
Курительное изделие согласно одному варианту осуществления настоящего изобретения включает вышеуказанную табачную ароматическую жидкость.
ПРЕИМУЩЕСТВЕННЫЕ РЕЗУЛЬТАТЫ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению, возможно создание способа получения табачной ароматической жидкости, которая применима для курительных изделий, табачной ароматической жидкости, способа получения сложноэфирного соединения, и курительного изделия, которое содержит табачную ароматическую жидкость.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
ФИГ. 1 представляет технологическую блок-схему, показывающую предпочтительный вариант исполнения способа согласно настоящему изобретению.
ФИГ. 2 представляет вид в разрезе, показывающий один пример ароматического ингалятора нагревательного типа, содержащего табачный наполнитель.
ФИГ. 3 представляет вид в разрезе, показывающий один пример ароматического ингалятора нагревательного типа, содержащего табачную ароматическую жидкость
ФИГ. 4 представляет график, показывающий результаты анализа методом газовой хроматографии-масс-спектрометрии (GC-MS) эффективности обработки гидроксидом натрия.
ФИГ. 5 представляет график, показывающий результаты GC-MS-анализа в Примере 1, в котором после стадии эстерификации были получены различные этиловые сложные эфиры.
ФИГ. 6 представляет график, показывающий результаты GC-MS-анализа в Примерах 1 и 2А, в которых количества валериановых кислот и капроновых кислот, первоначально содержащихся в исходном табачном материале, сокращались после стадии эстерификации.
ФИГ. 7 представляет график, показывающий результаты GC-MS-анализа в Примере 2В, в котором количества валериановых кислот и капроновых кислот, первоначально содержащихся в исходном табачном материале, сокращались после стадии эстерификации.
ФИГ. 8 представляет график, показывающий результаты GC-MS-анализа в Примере 2С, в котором количества валериановых кислот и капроновых кислот, первоначально содержащихся в исходном табачном материале, немного сокращались после стадии эстерификации.
ФИГ. 9 представляет график, показывающий результаты GC-MS-анализа в Примере 2D, в котором количества валериановых кислот и капроновых кислот, первоначально содержащихся в исходном табачном материале, немного сокращались после стадии эстерификации.
ФИГ. 10 представляет график, показывающий результаты GC-MS-анализа в Примерах 1 и 2А, в которых после стадии эстерификации образовались различные этиловые сложные эфиры.
ФИГ. 11 представляет график, показывающий результаты GC-MS-анализа в Примере 2В, в котором после стадии эстерификации образовались различные этиловые сложные эфиры.
ФИГ. 12 представляет график, показывающий результаты GC-MS-анализа в Примере 2С, в котором после стадии эстерификации в небольших количествах образовались различные этиловые сложные эфиры.
ФИГ. 13 представляет график, показывающий результаты GC-MS-анализа в Примере 2D, в котором после стадии эстерификации различные этиловые сложные эфиры образовались в небольших количествах.
ФИГ. 14 представляет график, показывающий результаты GC-MS-анализа в Примере 1 и в Примерах 3А-3С, в которых после стадии эстерификации образовались различные этиловые сложные эфиры, и в Сравнительных Примерах 1-4, в которых после стадии эстерификации различные этиловые сложные эфиры не образовались.
ФИГ. 15 представляет график, показывающий результаты GC-MS-анализа в Примере 1 и в Примерах 4А-4D, в которых валериановые кислоты и капроновые кислоты, первоначально содержащиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные этиловые сложные эфиры.
ФИГ. 16 представляет график, показывающий численные значения высот пиков 3-MVA-этилового сложного эфира, полученные при GC-MS-анализе в Примерах 4Е-4К, когда высоту пика 3-MVA-этилового сложного эфира, полученного при GC-MS-анализе в Примере 4Е, принимают за 100.
ФИГ. 17 представляет график, показывающий результаты GC-MS-анализа в Примерах 5А и 5D, в которых валериановые кислоты и капроновые кислоты, первоначально содержащиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные этиловые сложные эфиры.
ФИГ. 18 представляет график, показывающий результаты GC-MS-анализа в Примерах 6А-6С, в которых валериановые кислоты и капроновые кислоты, первоначально содержащиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные пропиловые сложные эфиры и различные бутиловые сложные эфиры.
ФИГ. 19 представляет график, показывающий результаты GC-MS-анализа в Примерах 7А-7С, в которых валериановые кислоты и капроновые кислоты, первоначально содержащиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные этиловые сложные эфиры.
ФИГ. 20 представляет график, показывающий результаты GC-MS-анализа в Примерах 8А-8G, в которых валериановые кислоты и капроновые кислоты, первоначально содержащиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные этиловые сложные эфиры.
ФИГ. 21 представляет линейный график, показывающий результаты, приведенные в ФИГ. 20.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение будет подробно описано; однако приведенное ниже описание предназначено для охарактеризования настоящего изобретения, и не предполагается быть ограничивающим настоящее изобретение.
<1. Способ получения табачной ароматической жидкости>
Согласно одному варианту исполнения, способ получения табачной ароматической жидкости включает:
(S1) экстракцию компонента из исходного табачного материала с использованием органического растворителя для получения тем самым содержащей табачный компонент жидкости; и
(S2) смешение содержащей табачный компонент жидкости с водным раствором, содержащим оснóвное вещество и спирт, в кислородсодержащей атмосфере для реакции содержащей табачный компонент жидкости с водным раствором, с получением тем самым табачной ароматической жидкости.
Вышеуказанный вариант исполнения может дополнительно включать:
(S3) выделение сложноэфирного соединения из табачной ароматической жидкости.
Описанный выше предпочтительный вариант исполнения показан в ФИГ. 1. Далее предпочтительный вариант исполнения будет описан в порядке стадий от (S1) до (S3) со ссылкой на ФИГ. 1.
[Стадия (S1) экстракции]
Компонент, содержащийся в исходном табачном материале, экстрагируют из исходного табачного материала с использованием органического растворителя для получения содержащей табачный компонент жидкости и табачного остатка.
Исходный табачный материал может представлять собой собранный лист (табачный лист), только что собранный на ферме, табачный лист, полученный из собранных листьев, высушенных на ферме, или резаный табак, готовый для смешения с образованием табачного продукта, после подвергания обработке сушкой на ферме, с последующим длительным процессом старения в течение от одного года до нескольких лет на складе сырьевого материала, и последующей технологической обработкой на производственном предприятии. В качестве исходного табачного материала может быть надлежащим образом использован полученный на ферме свежесобранный лист. В качестве исходного табачного материала также может быть применен собранный лист по истечении предварительно определенного времени после сбора. В этом описании высушенный исходный табачный материал называется «листовым табаком», и исходный табачный материал, выращенный и высушенный, называется «табачным листом».
В качестве исходного табачного материала (собранного листа) может быть использован табачный лист, содержащий большие количества органических кислот, например, табачный лист, содержащий от 0,5 мг/г до 1,5 мг/г 3-метилвалериановой кислоты и от 0,05 мг/г до 0,3 мг/г изовалериановой кислоты. Могут быть применены любые многообразные исходные табачные материалы (собранные листья). В качестве исходного табачного материала, например, могут быть применены сортотип Ориентал, сортотип Вирджиния (желтый), сортотип Берли, местный вид, дикие сорта табака, и тому подобные. Среди них сортотип Ориентал и местный вид имеют относительно высокое содержание валериановой кислоты на единицу веса, и могут быть надлежащим образом применены в способе получения табачной ароматической жидкости согласно настоящему варианту исполнения. Из них сортотип Ориентал имеет наибольшее содержание валериановой кислоты на единицу веса, и может быть лучше всего применен в способе получения табачной ароматической жидкости согласно настоящему варианту исполнения. В качестве исходного табачного материала могут быть использованы многообразные исходные табачные материалы по отдельности, или же смеси различных многообразных исходных табачных материалов.
Стадия экстракции может быть выполнена в любой атмосфере. Стадия экстракции может быть проведена в кислородсодержащей атмосфере, такой как воздушная атмосфера. Стадия экстракции может быть выполнена в иной атмосфере, нежели кислородсодержащая атмосфера, например, в атмосфере азота. Стадия экстракции может быть проведена в воздушной атмосфере при повышенном давлении и при пониженном давлении.
Температура в стадии экстракции может быть, например, температурой от 0°С до 90°С, более предпочтительно температурой от 10°С до 70°С, например, и наиболее предпочтительно температурой от 15°С до 60°с. Температура в стадии экстракции предпочтительно является более низкой, чем температура кипения органического растворителя, применяемого для экстракции.
Стадия экстракции может быть выполнена погружением исходного табачного материала в органический растворитель в течение периода времени, например, от 30 секунд до нескольких дней, более предпочтительно от 1 минуты до 12 часов, и наиболее предпочтительно от 5 минут до 3 часов. В стадии экстракции надлежащим образом может проводиться взбалтывание или перемешивание.
Органический растворитель, используемый в стадии экстракции, предпочтительно имеет относительно низкую полярность и температуру кипения. В качестве такого органического растворителя предпочтительно применяют гексан. Используемый в стадии экстракции органический растворитель не ограничивается гексаном, и может представлять собой иной органический растворитель, нежели гексан. Например, может быть применен этилацетат.
В стадии экстракции органический растворитель предпочтительно применяют в количестве в диапазоне от 0,5 л до 3,0 л в расчете на 1 кг исходного табачного материала, и более предпочтительно применяют в количестве в диапазоне от 1,0 л до 2,0 л в расчете на 1 кг исходного табачного материала. Объем растворителя или раствора имеет отношение к объему при 25°С. Когда количество органического растворителя является слишком малым, затруднительно в достаточной мере экстрагировать компоненты, которые будут описаны позже, из исходного табачного материала. Когда количество органического растворителя является слишком большим, концентрация компонентов в первой содержащей табачный компонент жидкости, описываемой позже, становится низкой. Когда первую содержащую табачный компонент жидкость, содержащую вышеуказанные компоненты с низкой концентрацией, используют в стадии (S2) эстерификации, то затруднительно выполнить эстерификацию с высокой эффективностью. Кроме того, когда первая содержащая табачный компонент жидкость содержит вышеуказанные компоненты с низкой концентрацией, требуется удаление большого количества органического растворителя, чтобы получить сконцентрированный сухой твердый продукт из первой содержащей табачный компонент жидкости.
Компоненты, содержащиеся в исходном табачном материале, включают, например, органические кислоты, более конкретно, валериановую кислоту, капроновую кислоту, и тому подобные.
По завершении стадии экстракции содержащую табачный компонент жидкость (первую содержащую табачный компонент жидкость) и табачный остаток отделяют друг от друга. Табачный остаток используют, например, в последующей стадии как материал для повторного применения в получении табачной ароматической жидкости. Табачный остаток может быть высушен и затем использован как носитель для нанесения вкусо-ароматической добавки. В альтернативном варианте, табачный остаток может быть сформован в прессовку для использования в качестве носителя для нанесения вкусо-ароматической добавки. Прессовка табачного остатка может быть сформована в виде листа, разрезана на кусочки, полученные резкой листообразной прессовки до формы резаного табака, или преобразована в порошок измельчением листообразной прессовки. Листообразная прессовка может быть получена, например, формованием табачного остатка до формы листа с использованием технологии изготовления бумаги.
Содержащая табачный компонент жидкость (первая содержащая табачный компонент жидкость) может быть подвергнута обработке в стадии фильтрации, в которой содержащую табачный компонент жидкость профильтровывают через фильтровальную бумагу или тому подобную. Стадия фильтрации является необязательной. В альтернативном варианте, фильтрат, полученный фильтрацией содержащей табачный компонент жидкости, может быть сконцентрирован досуха для получения сконцентрированного сухого твердого продукта. Концентрирование до сухого и твердого состояния может быть выполнено высушиванием при пониженном давлении с использованием испарителя или центрифужного испарителя. В альтернативном варианте, концентрирование до сухого и твердого состояния может быть выполнено лиофильной сушкой вымораживанием или сушкой при нагревании. Сконцентрированный сухой твердый продукт может быть растворен опять в органическом растворителе (например, в гексане), использованном для экстракции, или растворен в ином органическом растворителе, для применения в качестве содержащей табачный компонент жидкости (второй содержащей табачный компонент жидкости). Количество органического растворителя, используемого для получения второй содержащей табачный компонент жидкости, предпочтительно составляет величину в диапазоне от 500 мл до 10000 мл, и более предпочтительно в диапазоне от 1000 мл до 6000 мл, в расчете на 1 г сконцентрированного сухого твердого продукта.
[Стадия (S2) эстерификации]
В стадии (S2) эстерификации, в кислородсодержащей атмосфере, к предварительно определенному количеству содержащей табачный компонент жидкости (первой содержащей табачный компонент жидкости или второй содержащей табачный компонент жидкости), полученной в стадии (S1) экстракции, добавляют водный раствор, и проводят реакцию с содержащей табачный компонент жидкостью, с образованием тем самым сложноэфирного соединения в смешанном растворе из водного раствора и содержащей табачный компонент жидкости. В результате этого может быть получена табачная ароматическая жидкость с повышенным количеством сложноэфирного соединения. Содержащую табачный компонент жидкость и водный раствор предпочтительно смешивают в объемном отношении от 4:1 до 4:5, более предпочтительно смешивают в объемном отношении от 4:1 до 4:3, и наиболее предпочтительно смешивают в объемном отношении от 5:2 до 4:3. Смешанный раствор при стоянии в неподвижном состоянии разделяется на две фазы, например, на органическую фазу, образованную содержащей табачный компонент жидкостью, и водную фазу, образованную водным раствором. В этом случае, хотя это может варьировать в зависимости от типа органического растворителя и тому подобного, согласно одному примеру фаза органического растворителя представляет собой верхний слой, и водная фаза представляет собой нижний слой.
Содержащая табачный компонент жидкость, используемая в стадии (S2) эстерификации, содержит вышеуказанные компоненты (то есть, органические кислоты, такие как валериановая кислота и капроновая кислота), экстрагированные из исходного табачного материала, предпочтительно в концентрации от 0,1 мг/л до 5000 мг/л, и более предпочтительно в концентрации от 0,5 мг/л до 500 мг/л.
В качестве органического растворителя, входящего в состав используемой в стадии (S2) эстерификации содержащей табачный компонент жидкости, может быть выбран один из группы, состоящей из гексана, пентана, диэтилового простого эфира, TBME (трет-бутилметилового простого эфира), и этилацетата. Органический растворитель, входящий в состав используемой в стадии (S2) эстерификации содержащей табачный компонент жидкости, предпочтительно имеет такое свойство, что не смешивается с водой (водным раствором), то есть, проявляет несмешиваемость с водой (водным раствором). Органический растворитель, входящий в состав используемой в стадии (S2) эстерификации содержащей табачный компонент жидкости, может быть либо таким же, либо отличающимся от органического растворителя, применяемого в качестве экстракционного растворителя в стадии (S2) экстракции.
Полярность органических растворителей, включенных в вышеуказанную группу, изменяется в ряду гексан/пентан<диэтиловый простой эфир<TBME<этилацетат. Таким образом, среди этих органических растворителей гексан и пентан имеют наименьшую полярность и имеют такое свойство не растворяться в воде (водном растворе) на наивысшем уровне, то есть, наивысшую несмешиваемость с водой (водным раствором). Среди этих органических растворителей диэтиловый простой эфир также имеет относительно низкую полярность, и имеет свойство не растворяться в воде (водном растворе), то есть, несмешиваемость с водой (водным раствором). Среди этих органических растворителей TBME имеет относительно высокую полярность, и имеет свойство хорошо смешиваться с водой (водным раствором), то есть, смешиваемость с водой (водным раствором). Среди этих органических растворителей этилацетат имеет наивысшую полярность, и имеет свойство хорошо смешиваться с водой (водным раствором) на наивысшем уровне, то есть, наивысшую смешиваемость с водой (водным раствором).
Органический растворитель, содержащийся в используемой в стадии (S2) эстерификации содержащей табачный компонент жидкости, может представлять собой смесь двух или многих, выбранных из вышеуказанной группы, и несмешивающуюся с водой (водным раствором). Каждое соединение, содержащееся в смеси, предпочтительно не смешивается с водой (водным раствором). Однако, пока смесь является несмешивающейся с водой (водным раствором), некоторые из присутствующих в смеси соединений могут растворяться в воде (водном растворе).
Водный раствор может быть сформирован из спирта и водного раствора, содержащего оснóвное вещество. Водный раствор оснóвного вещества и спирт в водном растворе предпочтительно однородно смешаны. Водный раствор оснóвного вещества и спирт могут быть смешаны, например, в объемном отношении 1:1, или смешаны в других соотношениях. Водный раствор оснóвного вещества и спирт предпочтительно смешаны в объемном отношении от 10:1 до 1:4, и более предпочтительно смешаны в объемном отношении от 5:3 до 1:2.
Спирт может быть любым спиртом. В качестве спирта предпочтительно используют первичный спирт. В качестве спирта предпочтительно применяют одноатомный спирт. Например, низший спирт, имеющий малое число атомов углерода, такое как число атомов углерода 4 или менее, или 5 или менее, может быть надлежащим образом применен как спирт. В стадии эстерификации согласно настоящему варианту исполнения надлежащим образом используют низший спирт благодаря его высокой растворимости в воде. В качестве спирта может быть применен один, выбранный из группы, состоящей из этанола, метанола, 1-пропанола и 1-бутанола. В качестве спирта может быть использована смесь двух или многих, выбранных из вышеуказанной группы.
В стадии (S2) эстерификации доля объема спирта в сумме объема содержащей табачный компонент жидкости (первой содержащей табачный компонент жидкости или второй содержащей табачный компонент жидкости) и объема водного раствора предпочтительно составляет от 2,0% до 28,6%. В стадии (S2) эстерификации доля объема спирта в сумме объема содержащей табачный компонент жидкости (первой содержащей табачный компонент жидкости или второй содержащей табачный компонент жидкости) и объема водного раствора более предпочтительно составляет от 10,7% до 28,6%.
Оснóвное вещество предпочтительно является высокооснóвным, со степенью ионизации, близкой к 1. Оснóвное вещество предпочтительно образовано из гидроксида щелочного металла или щелочноземельного металла. Например, в качестве гидроксида щелочного металла может быть использован гидроксид натрия или гидроксид калия. Например, в качестве гидроксида щелочноземельного металла может быть применен гидроксид кальция. Оснóвное вещество предпочтительно присутствует в водном растворе оснóвного вещества при концентрации от 0,01 н. до 5 н., и более предпочтительно присутствует в водном растворе оснóвного вещества при концентрации от 0,05 н. до 0,5 н.
Значение рН водного раствора оснóвного вещества предпочтительно составляет от 11,0 до 14,0, и более предпочтительно от 12,0 до 13,0.
Реакцию в стадии эстерификации проводят в кислородсодержащей атмосфере, как описано выше. Кислородсодержащая атмосфера представляет собой, например, воздушную атмосферу. Кислородсодержащая атмосфера предпочтительно содержит кислород с концентрацией 20% по объему или более.
Температура в стадии эстерификации предпочтительно составляет температуру от 0°С до 90°С, более предпочтительно температуру от 10°С до 70°С, и наиболее предпочтительно температуру от 15°С до 60°С.
Реакцию в стадии эстерификации предпочтительно проводят в течение периода времени 1 минуты или более, и более предпочтительно в течение периода времени от 1 до 60 минут. Реакцию еще более предпочтительно проводят в течение периода времени от 5 до 60 минут. Еще более предпочтительно реакцию проводят в течение периода времени от 10 до 60 минут. Реакцию предпочтительно проводят при взбалтывании, и более предпочтительно проводят при взбалтывании со скоростью от 10 об/мин до 300 об/мин. Еще более предпочтительно реакцию проводят при взбалтывании со скоростью от 100 об/мин до 200 об/мин.
Когда реакцию проводят в течение определенного периода времени в таком режиме, стимулируется реакция эстерификации с использованием органической кислоты, такой как валериановая кислота или капроновая кислота, в качестве базового вещества, и увеличивается количество сложноэфирного соединения, соответствующего типу добавленного спирта. То есть, когда в качестве спирта применяют этанол, количество органической кислоты сокращается, и вместо нее образуются различные этиловые сложные эфиры. Когда в качестве спирта применяют метанол, количество органической кислоты сокращается, и вместо нее образуются различные метиловые сложные эфиры. Когда в качестве спирта применяют 1-пропанол, количество органической кислоты сокращается, и вместо нее образуются различные пропиловые сложные эфиры. Когда в качестве спирта применяют 1-бутанол, количество органической кислоты сокращается, и вместо нее образуются различные бутиловые сложные эфиры.
Полученная таким образом табачная ароматическая жидкость содержит большое количество сложноэфирных соединений. Известно, что, как правило, сложноэфирные соединения часто имеют аромат, хотя он варьирует в зависимости от типа органической кислоты, служащей в качестве базового вещества в реакции.
Табачная ароматическая жидкость может быть получена, например, из смешанного раствора, согласно следующей методике. Сначала, по завершении взбалтывания, смешанный раствор оставляют стоять в течение предварительно определенного времени, например, от 5 до 10 минут, и из разделившегося на две фазы смешанного раствора удаляют только фазу органического растворителя как верхний слой. Фазу органического растворителя концентрируют до сухого и твердого состояния таким же образом, как описано в стадии экстракции, и сконцентрированный сухой твердый продукт растворяют в еще одном растворителе, таком как неводный растворитель, например, в органическом растворителе, в результате чего может быть получена табачная ароматическая жидкость, содержащая большое количество сложноэфирных соединений. Фаза органического растворителя может быть использована в качестве табачной ароматической жидкости. Такая табачная ароматическая жидкость либо может быть применена как источник вкусо-ароматической добавки в ароматическом ингаляторе нагревательного типа (описываемом позже), или набрызгана на табачный материал (вышеописанный табачный остаток или тому подобный) сигареты для усиления аромата.
[Стадия (S3) выделения сложноэфирного соединения]
Для выделения сложноэфирного соединения из табачной ароматической жидкости может быть использован известный способ выделения. Например, могут быть применены хроматографические методы различных типов и тому подобные как способ выделения, и более предпочтительно может быть надлежащим образом применена колоночная хроматография. Тем самым сложноэфирное соединение может быть выделено и очищено.
<2. Табачная ароматическая жидкость>
Представлена табачная ароматическая жидкость, полученная описанным выше в разделе <1. Способ получения табачной ароматической жидкости> способом. Табачная ароматическая жидкость может быть использована таким образом, что ее набрызгивают на табачный наполнитель, применяемый в описываемом позже «ароматическом ингаляторе нагревательного типа, включающем табачный наполнитель». Табачный наполнитель представляет собой остаток или тому подобный, полученный в описанной выше стадии (S2) экстракции; однако табачную ароматическую жидкость не обязательно наносят на табачный наполнитель.
В альтернативном варианте, табачная ароматическая жидкость может быть дополнительно подвергнута обработке в различных стадиях обработки, или смешана с содержащей табачный компонент жидкостью, полученной в стадии выделения, и затем использована как табачная ароматическая жидкость в описываемом позже «ароматическом ингаляторе нагревательного типа, включающем табачную ароматическую жидкость».
<3. Ароматический ингалятор нагревательного типа, включающий табачный наполнитель>
В качестве примера курительного изделия, содержащего табачную ароматическую жидкость, показан ароматический ингалятор нагревательного типа. Ароматический ингалятор 11 нагревательного типа, показанный в ФИГ. 2, не содержит табачную ароматическую жидкость в жидкостном состоянии, но включает табачный наполнитель 12, опрысканный табачной ароматической жидкостью для усиления табачного аромата.
Ароматический ингалятор 11 нагревательного типа включает корпус 13, мундштук 14 и вентиляционное отверстие (не показано). Ароматический ингалятор 11 нагревательного типа включает, в корпусе 13, табачный наполнитель 12, резервуар 15 для хранения источника аэрозоля, батарею 16, схему 17 управления, нагреватель (не показан), и чувствительный к давлению датчик (не показан). Схема 17 управления и нагреватель получают питание от батареи 16. Чувствительный к давлению датчик соединен со схемой 17 управления и снабжается электроэнергией от схемы 17 управления.
Табачный наполнитель 12 сформирован заполнением внутренности картриджа 18 или тому подобного продуктом, полученным набрызгиванием на вышеописанный табачный остаток табачной ароматической жидкости. Табачный наполнитель 12 размещен вблизи мундштука 14. Резервуар 15 для хранения источника аэрозоля включает капсулу 15А и источник аэрозоля, заполняющий капсулу 15А. Источник аэрозоля образован из жидкости, такой как пропиленгликоль, глицерин, или их смесь.
Когда пользователь начинает затяжку, удерживая мундштук 14 вблизи его или ее рта, чувствительный к давлению датчик детектирует отрицательное давление. В результате этого срабатывает схема 17 управления, включая переключатель (не показан) для подачи энергии на нагреватель. В результате этого нагреватель приводится в действие для нагревания источника аэрозоля в резервуаре 15 для хранения источника аэрозоля, и тем самым генерирует аэрозоль. Когда пользователь продолжает вдох, аэрозоль протягивается через табачный наполнитель 12 вместе с воздухом, поступающим из вентиляционного отверстия. Когда аэрозоль проходит через табачный наполнитель 12 на стороне мундштука 14, табачный аромат в табачном наполнителе 12 увлекается аэрозолем так, что пользователь может получать удовольствие от табачного аромата.
В ароматическом ингаляторе 11 нагревательного типа согласно настоящему варианту исполнения источник аэрозоля хранится в резервуаре 15 для хранения источника аэрозоля; однако размещение источника аэрозоля этим не ограничивается. Источник аэрозоля может быть нанесен и добавлен непосредственно на табачный наполнитель 12. В этом случае нагреватель размещен вблизи табачного наполнителя 12, и может непосредственно нагревать табачный наполнитель 12 до такой степени, что табачный наполнитель 12 не загорается. В результате этого аэрозоль образуется из источника аэрозоля, нанесенного и добавленного непосредственно на табачный наполнитель 12, и табачный аромат из табачного наполнителя 12 увлекается аэрозолем. Пользователь может получать удовольствие от табачного аромата при вдыхании аэрозоля вместе с воздухом.
<4. Ароматический ингалятор нагревательного типа, содержащий табачную ароматическую жидкость>
В качестве примера курительного изделия, содержащего табачную ароматическую жидкость, показан ароматический ингалятор 11’ нагревательного типа. В отличие от ароматического ингалятора нагревательного типа, описанного выше в разделе <3. Ароматический ингалятор нагревательного типа, включающий табачный наполнитель>, ароматический ингалятор 11’ нагревательного типа, показанный в ФИГ. 3, не содержит табачный наполнитель 12. Табачный аромат может быть получен непосредственно из табачной ароматической жидкости распылением табачной ароматической жидкости.
Ароматический ингалятор 11’ нагревательного типа включает корпус 13, включающий первую часть 13А и вторую часть 13В, мундштук 14 и вентиляционное отверстие 21. Ароматический ингалятор 11’ нагревательного типа включает, в корпусе 13, источник 22 табачного аромата, батарею 16, схему 17 управления, фитиль 23, нагреватель 24, намотанный вокруг фитиля 23, направляющий проток 25 для направления содержащего табачный аромат воздуха, линию 26 электропередачи для подачи электроэнергии от батареи 16 на нагреватель 24, и чувствительный к давлению датчик (не показан). Схема 17 управления и нагреватель 24 получают питание от батареи 16. Чувствительный к давлению датчик соединен со схемой 17 управления и снабжается электроэнергией от схемы 17 управления. Источник 22 табачного аромата сформирован заполнением внутренности капсулы с жидкостью. Жидкость образована смешением табачной ароматической жидкости и источника аэрозоля. Источник аэрозоля образован из жидкости, такой как пропиленгликоль, глицерин, или их смесь.
Жидкость, которая хранится внутри источника 22 табачного аромата, засасывается фитилем 23. Когда пользователь начинает затяжку, удерживая мундштук 14 вблизи его или ее рта, чувствительный к давлению датчик детектирует отрицательное давление. В результате этого срабатывает схема 17 управления, включая переключатель (не показан) для подачи энергии на нагреватель 24, чтобы нагревать фитиль 23, образуя тем самым содержащий табачный аромат аэрозоль из фитиля 23. Когда пользователь продолжает вдох, аэрозоль проходит вокруг фитиля вместе с воздухом, поступающим из вентиляционного отверстия 21. Содержащий аэрозоль воздух поступает в ротовую полость пользователя через направляющий проток 25 так, что пользователь может получать удовольствие от табачного аромата.
Табачная ароматическая жидкость, используемая в данном ароматическом ингаляторе 11’ нагревательного типа, может представлять собой табачную ароматическую жидкость, полученную способом, описанным выше в разделе <1. Способ получения табачной ароматической жидкости>, или табачную ароматическую жидкость, вновь образованную смешением отдельно полученной табачной ароматической жидкости и табачной ароматической жидкости, полученной способом, описанным выше в разделе <1. Способ получения табачной ароматической жидкости>.
В настоящем варианте исполнения ароматический ингалятор 11 нагревательного типа, содержащий опрысканный табачной ароматической жидкостью табачный наполнитель 12, и ароматический ингалятор 11’ нагревательного типа, содержащий табачную ароматическую жидкость в жидкостном состоянии, были описаны как курительные изделия, содержащие табачную ароматическую жидкость. Само собой разумеется, что курительное изделие может представлять собой так называемую «обычную сигарету (обернутую бумагой сигарету)». Например, в обычной сигарете продукт, полученный набрызгиванием вышеописанной табачной ароматической жидкости на табачный остаток (подложку), полученный описанным выше в разделе <1. Способ получения табачной ароматической жидкости> способом, может быть использован как табачный наполнитель. В этом случае объект, опрысканный табачной ароматической жидкостью, может быть несколько иным, нежели табачный остаток, полученный описанным выше в разделе <1. Способ получения табачной ароматической жидкости> способом.
<5. Преимущественные результаты изобретения>
Соответственно способу получения табачной ароматической жидкости согласно настоящему изобретению, в стадии (S1) экстракции компонент экстрагируют из исходного табачного материала с использованием органического растворителя, для получения тем самым содержащей табачный компонент жидкости; и в последующей стадии (S2) эстерификации содержащую табачный компонент жидкость смешивают и вводят в реакцию с водным раствором, содержащим оснóвное вещество и спирт, в кислородсодержащей атмосфере, с получением тем самым табачной ароматической жидкости. Поэтому табачная ароматическая жидкость с повышенным количеством сложных эфиров может быть получена исключительно простым способом (смотри описанный позже Пример 1). Применением этой табачной ароматической жидкости может быть создано курительное изделие с усиленным табачным ароматом.
В способе получения табачной ароматической жидкости согласно настоящему изобретению вышеуказанную реакцию проводят, например, при температуре от 0°С до 90°С. Водный раствор представляет собой смешанный раствор водного раствора оснóвного вещества и спирта, и значение рН водного раствора оснóвного вещества составляет от 11,0 до 14,0. Согласно этим конфигурациям, стадия эстерификации может быть проведена при низкой температуре в присутствии оснóвного вещества, имеющего значение рН от 11,0 до 14,0. В частности, традиционно требуется проведение реакции в присутствии сильной кислоты, такой как серная кислота, с базовым веществом при высокой температуре, как описано в абзаце [0062] Японской Национальной Публикации PCT № 2017-502681, что создает проблему в отношении безопасности. В дополнение, важные ароматические компоненты и прочие компоненты в содержащей табачный компонент жидкости могут быть денатурированы под действием нагревания и сильной кислоты. Также необходимо создание производственного оборудования, способного выдерживать долговременное применение нагревания и сильной кислоты, что повышает производственные издержки. Соответственно способу получения табачной ароматической жидкости согласно настоящему изобретению, температура реакции может быть сделана относительно низкой, в каковом случае табачная ароматическая жидкость может быть получена предельно простым и безопасным путем. Кроме того, возможно создание табачной ароматической жидкости, которая сохраняет первоначальный аромат исходного табачного материала и имеет усиленный сложными эфирами аромат. Также нет никаких накладных расходов на производственное оборудование.
В способе получения табачной ароматической жидкости согласно настоящему изобретению реакцию проводят на протяжении периода времени, например, от 1 до 60 минут. Согласно этой конфигурации, реакция может быть завершена за короткий период времени, табачная ароматическая жидкость может быть получена за короткий период времени, и может быть снижена нагрузка на производственное оборудование, когда получают табачную ароматическую жидкость.
Вышеуказанную реакцию проводят, например, в то же время со взбалтыванием. Согласно этой конфигурации, реакция может быть стимулирована перемешиванием реакционного раствора под действием прилагаемой снаружи силы.
В соответствии со способом получения сложноэфирного соединения согласно настоящему изобретению, в стадии (S1) экстракции компонент экстрагируют из исходного табачного материала с использованием органического растворителя, для получения тем самым содержащей табачный компонент жидкости; в последующей стадии (S2) эстерификации содержащую табачный компонент жидкость вводят в реакцию с водным раствором, содержащим оснóвное вещество и спирт, в кислородсодержащей атмосфере, для получения тем самым табачной ароматической жидкости; и в последующей стадии (S3) выделения сложноэфирное соединение выделяют из табачной ароматической жидкости. Поэтому сложноэфирное соединение может быть получено исключительно простым, безопасным и эффективным путем.
<6. Предпочтительные варианты осуществления изобретения>
Далее совместно показаны предпочтительные варианты осуществления настоящего изобретения.
[1] Способ получения табачной ароматической жидкости, причем способ включает:
экстракцию компонента из исходного табачного материала с использованием органического растворителя для получения тем самым содержащей табачный компонент жидкости; и
смешение содержащей табачный компонент жидкости с водным раствором, содержащим оснóвное вещество и спирт, в кислородсодержащей атмосфере для реакции содержащей табачный компонент жидкости с водным раствором, с получением тем самым табачной ароматической жидкости.
[2] Способ согласно пункту [1], в котором реакцию проводят при температуре от 0°С до 90°С, предпочтительно от 10°С до 70°С, и более предпочтительно от 15°С до 60°С.
[3] Способ согласно любому из пунктов [1] и [2], в котором
водный раствор представляет собой смешанный раствор водного раствора оснóвного вещества и спирта, и
значение рН водного раствора оснóвного вещества составляет от 11,0 до 14,0, предпочтительно от 11,3 до 13,0, более предпочтительно от 11,8 до 13,0, и еще более предпочтительно от 12,0 до 13,0.
[4] Способ согласно любому из пунктов [1]-[3], в котором оснóвное вещество представляет собой гидроксид щелочного металла или щелочноземельного металла.
[5] Способ согласно любому из пунктов [1]-[4], в котором оснóвное вещество является высокооснóвным.
[6] Способ согласно любому из пунктов [1]-[5], в котором
водный раствор представляет собой смешанный раствор водного раствора оснóвного вещества и спирта, и
оснóвное вещество присутствует в водном растворе оснóвного вещества при концентрации от 0,01 н. до 5 н., и предпочтительно от 0,05 н. до 0,5 н.
[7] Способ согласно любому из пунктов [1]-[6], в котором органический растворитель, входящий в состав содержащей табачный компонент жидкости, является несмешивающимся с водным раствором.
[8] Способ согласно любому из пунктов [1]-[7], в котором доля объема спирта в сумме объема содержащей табачный компонент жидкости и объема водного раствора составляет от 2,0% до 28,6%, предпочтительно от 10,7% до 28,6%.
[9] Способ согласно любому из пунктов [1]-[8], в котором реакцию проводят на протяжении периода времени от 1 до 60 минут, предпочтительно от 5 до 60 минут.
[10] Способ согласно любому из пунктов [1]-[9], в котором реакцию проводят при взбалтывании.
[11] Табачная ароматическая жидкость, полученная способом получения табачной ароматической жидкости согласно любому из пунктов [1]-[10].
[12] Способ получения сложноэфирного соединения, причем способ включает:
экстракцию компонента из исходного табачного материала с использованием органического растворителя для получения тем самым содержащей табачный компонент жидкости;
введение содержащей табачный компонент жидкости в реакцию с водным раствором, содержащим оснóвное вещество и спирт, в кислородсодержащей атмосфере, для получения тем самым табачной ароматической жидкости; и
выделение сложноэфирного соединения из табачной ароматической жидкости.
[13] Курительное изделие, включающее табачную ароматическую жидкость согласно пункту [11].
[14] Способ согласно любому из пунктов [1]-[10],
в котором исходный табачный материал представляет собой один, или смесь двух или многих, выбранных из группы, состоящей из сортотипа Ориентал, сортотипа Вирджиния (желтого), сортотипа Берли, местного вида, и диких сортов табака, и предпочтительно одного или многих, выбранных из группы, состоящей из сортотипа Ориентал и местного вида.
[15] Способ согласно любому из пунктов [1]-[10] и [14], в котором исходный табачный материал представляет собой табачный лист, содержащий от 0,5 мг/г до 1,5 мг/г 3-метилвалериановой кислоты и от 0,05 мг/г до 0,3 мг/г изовалериановой кислоты.
[16] Способ согласно любому из пунктов [1]-[12], [14] и [15], в котором исходный табачный материал представляет собой либо полученный на ферме свежесобранный лист, либо собранный лист по истечении предварительно определенного времени после сбора.
[17] Способ согласно любому из пунктов [1]-[12] и [14]-[16], в котором органический растворитель, включенный в содержащую табачный компонент жидкость, представляет собой один, или смесь двух или многих, выбранных из группы, состоящей из гексана, пентана, диэтилового простого эфира, TBME (трет-бутилметилового простого эфира), и этилацетата.
[18] Способ согласно любому из пунктов [1]-[12] и [14]-[17], в котором водный раствор представляет собой смешанный раствор водного раствора оснóвного вещества и спирта, и водный раствор оснóвного вещества и спирт предпочтительно смешаны в объемном отношении от 10:1 до 1:4, и более предпочтительно смешаны в объемном отношении от 5:3 до 1:2.
[19] Способ согласно любому из пунктов [1]-[12] и [14]-[18], в котором спирт представляет собой низший спирт, имеющий число атомов углерода 5 или менее.
[20] Способ согласно любому из пунктов [1]-[12] и [14]-[19], в котором экстракцию проводят погружением исходного табачного материала в органический растворитель в течение периода времени, например, от 30 секунд до нескольких дней, более предпочтительно от 1 минуты до 12 часов, и наиболее предпочтительно от 5 минут до 3 часов.
ПРИМЕРЫ
<Оценка эффективности обработки гидроксидом натрия>
[Пример 1]
1,5 кг собранных листьев табака сортотипа Ориентал в качестве исходного табачного материала погрузили в 3,0 л гексана при 25°С в течение 0,5 часа в воздушной атмосфере (стадия (S1) экстракции). Полученную тем самым содержащую табачный компонент жидкость (первую содержащую табачный компонент жидкость) профильтровали, и фильтрат выпарили досуха с использованием роторного испарителя. 0,02 г сконцентрированного сухого твердого продукта поместили в центрифужную пробирку, изготовленную из полипропилена, и добавили в нее 20 мл гексана, для получения содержащей табачный компонент жидкости (второй содержащей табачный компонент жидкости), в которой был растворен сконцентрированный сухой твердый продукт. Затем в ту же центрифужную пробирку добавили 5 мл 0,5н.-ного водного раствора гидроксида натрия и 5 мл этанола, с образованием смешанного раствора из гексана (органического растворителя) и водного раствора (водного раствора гидроксида натрия и этанола). Центрифужную пробирку, содержащую смешанный раствор, взбалтывали при 180 об/мин при 25°С в течение 60 минут в воздушной атмосфере (стадия (S2) эстерификации).
После взбалтывания центрифужную пробирку оставили стоять в течение 5 минут. В результате этого смешанный раствор в центрифужной пробирке разделился на две фазы из фазы органического растворителя и фазы водного раствора. Когда концентрация оснóвного вещества является низкой, то оказывается затруднительным самопроизвольное разрушение пузырьков в граничном слое. Тем самым в таком случае проводили центрифугирование в течение 1 минуты при 3000 об/мин с использованием центробежного сепаратора, чтобы разрушить пузырьки в граничном слое. Жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом газовой хроматографии-масс-спектрометрии (GC-MS). Перед стадией (S2) эстерификации измерили значение рН водного раствора оснóвного вещества до того, как смешали его со спиртом.
[Сравнительный Пример 1]
В качестве Сравнительного Примера 1 приготовили образец в таких же условиях, как условия Примера 1, за исключением того, что добавили 5 мл воды вместо 5 мл водного раствора гидроксида натрия. В Сравнительном Примере 1 также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS, таким же образом, как описано в Примере 1.
[Результаты]
Результаты показаны в ФИГУРАХ 4, 6 и 10. В диаграммах GC-MS горизонтальная ось представляет истекшее время, и вертикальная ось представляет интенсивность сигнала. В ФИГ. 4 сигналы Примера 1 и Сравнительного Примера 1 сдвинуты вверх.
В Примере 1 различные валериановые кислоты и капроновые кислоты, первоначально содержащиеся в исходном табачном материала, исчезли после обработки, как показано в ФИГ. 6. Как используемые здесь, IVA представляет изовалериановую кислоту, 3MVA представляет 3-метилвалериановую кислоту, 4MHA представляет 4-метилкапроновую кислоту, и 4MVA представляет 4-метилвалериановую кислоту.
В Примере 1 вместо различных валериановых кислот и капроновых кислот образовались различные этиловые сложные эфиры, как показано в ФИГУРАХ 4 и 10. Различные этиловые сложные эфиры включают IVA-этиловый сложный эфир, 3MVA-этиловый сложный эфир, 4MVA-этиловый сложный эфир, 4MHA-этиловый сложный эфир, и тому подобные. Согласно Примеру 1, были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. В Сравнительном Примере 1 не происходило ни образование различных этиловых сложных эфиров, ни сокращение различных валериановых кислот и капроновых кислот, как показано в ФИГУРАХ 4 и 10. В Примере 1 значение рН водного раствора оснóвного вещества перед смешением со спиртом составляло 13,0.
Еще один результат анализа (GC-MS) Примера 1 показан в ФИГ. 5. В Примере 1, в дополнение к вышеуказанным IVA-этиловому сложному эфиру, 3MVA-этиловому сложному эфиру, 4MVA-этиловому сложному эфиру и 4MHA-этиловому сложному эфиру, образовались различные этиловые сложные эфиры органических кислот, имеющих число атомов углерода от 4 до 10, как показано в ФИГ. 5. В Примере 1 образовались этиловый сложный эфир 2-метилпропионовой кислоты (2-Me-C3), этиловый сложный эфир масляной кислоты (C4), этиловый сложный эфир 3-метилмасляной кислоты (3-Me-C4), этиловый сложный эфир валериановой кислоты (C5), этиловый сложный эфир 3-метилвалериановой кислоты (3-Me-C5), этиловый сложный эфир 4-метилвалериановой кислоты (4-Me-C5), этиловый сложный эфир капроновой кислоты (C6), этиловый сложный эфир энантовой кислоты (C7), этиловый сложный эфир 4-метилкапроновой кислоты (4-Me-C6), этиловый сложный эфир каприловой кислоты (C8), этиловый сложный эфир пеларгоновой кислоты (C9), и этиловый сложный эфир каприновой кислоты (C10). В Примере 1 не был зарегистрирован сложный эфир органической кислоты, имеющей число атомов углерода 11 или более.
[Обсуждение]
Было найдено, что в стадии (S2) эстерификации было необходимым присутствие оснóвного вещества, такого как гидроксид натрия, в смешанном растворе органического растворителя и водного раствора.
<Оценка эффективности добавления иного органического растворителя, нежели гексан>
[Пример 2А]
В Примере 2А, перед стадией (S2) эстерификации, добавили 20 мл пентана как органического растворителя, вместо 20 мл гексана, для растворения 0,02 г сконцентрированного сухого твердого продукта. Прочие условия были такими же, как условия в Примере 1. В Примере 2А проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (пентановой фазы), анализировали методом GC-MS, как описано в Примере 1.
[Пример 2В]
В Примере 2В, перед стадией (S2) эстерификации, добавили 20 мл диэтилового простого эфира как органического растворителя, вместо 20 мл гексана, для растворения 0,02 г сконцентрированного сухого твердого продукта. Прочие условия были такими же, как условия в Примере 1. В Примере 2В также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (фазы диэтилового простого эфира), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Пример 2С]
В Примере 2С 20 мл TBME (трет-бутилметилового простого эфира) добавили в качестве органического растворителя вместо 20 мл гексана для растворения 0,02 г сконцентрированного сухого твердого продукта. Прочие условия были такими же, как условия в Примере 1. В Примере 2С также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (TBME-фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Пример 2D]
В Примере 2D 20 мл этилацетата добавили в качестве органического растворителя вместо 20 мл гексана для растворения 0,02 г сконцентрированного сухого твердого продукта. Прочие условия были такими же, как условия в Примере 1. В Сравнительном Примере 2D также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (фазы этилацетата), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Результаты]
Результаты показаны в ФИГУРАХ 6-13.
В Примере 2А (с растворением в пентане) валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации (смотри ФИГ. 6), и вместо них образовались различные этиловые сложные эфиры (смотри ФИГ. 10). Таким образом, результаты Примера 2А были приблизительно такими же, как результаты Примера 1. В Примере 2А были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. ФИГУРЫ 6 и 10 показывают результаты применения гексана как органического растворителя для растворения сконцентрированного сухого твердого продукта. В ФИГУРАХ 6 и 10 представлены только результаты Примера 1 как показательные для приблизительно таких же результатов, которые были получены, когда использоваоли пентан как в Примере 2А.
В Примере 2В (с растворением в диэтиловом простом эфире) количества валериановых кислот и капроновых кислот, первоначально содержавшиеся в исходном табачном материале, несколько снизились после стадии эстерификации (смотри ФИГ. 7), и вместо них образовались различные этиловые сложные эфиры (смотри ФИГ. 11). То есть, согласно Примеру 2В, были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. Однако масса различных этиловых сложных эфиров, образованных в Примере 2В, составляла примерно 28% массы различных этиловых сложных эфиров, образованных в Примере 1.
В Примере 2С (с растворением в TBME) количества валериановых кислот и капроновых кислот, первоначально содержавшиеся в исходном табачном материале, слегка сократились после стадии эстерификации (смотри ФИГ. 8), и вместо них образовались различные этиловые сложные эфиры (смотри ФИГ. 12). То есть, согласно Примеру 2С, были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. Однако масса различных этиловых сложных эфиров, образованных в Примере 2С, составляла около 10% массы различных этиловых сложных эфиров, образованных в Примере 1.
В Примере 2D (с растворением в этилацетате) количества валериановых кислот и капроновых кислот, первоначально содержавшиеся в исходном табачном материале, слегка сократились после стадии эстерификации (смотри ФИГ. 9), и вместо них образовались различные этиловые сложные эфиры (смотри ФИГ. 13). То есть, согласно Примеру 2D, были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. Однако масса различных этиловых сложных эфиров, образованных в Примере 2DF, была малой, и составляла около 7% массы различных этиловых сложных эфиров, образованных в Примере 1.
[Обсуждение]
Из результатов Примера 1, Примеров 2А-2D, и Сравнительного Примера 1 было найдено, что органический растворитель, добавленный для растворения сконцентрированного сухого твердого продукта перед стадией (S2) эстерификации, предпочтительно был несмешивающимся с водным раствором, в частности, как показано в Примере 1 и Примерах 2А-2С. С другой стороны, было найдено, что, когда 5 мл воды добавляли вместо 5 мл водного раствора гидроксида натрия, как в Сравнительном Примере 1, количества валериановых кислот и тому подобных не сокращались, и этиловые сложные эфиры не образовывались.
<Оценка температуры в стадии эстерификации>
[Пример 3А]
В Примере 3А температура реакционной смеси в стадии (S2) эстерификации составляла 15°С. Прочие условия были такими же, как в Примере 1. В Примере 3А также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Пример 3В]
В Примере 3В температура реакционной смеси в стадии (S2) эстерификации составляла 40°С. Прочие условия были такими же, как в Примере 1. В Примере 3В также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Пример 3С]
В Примере 3С температура реакционной смеси в стадии (S2) эстерификации составляла 60°С. Прочие условия были такими же, как в Примере 1. В Примере 3С также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Сравнительный Пример 2]
В Сравнительном Примере 2, перед стадией (S2) эстерификации, в центрифужную пробирку добавляли 5 мл воды вместо 5 мл водного раствора гидроксида натрия. Температура реакционной смеси в стадии (S2) эстерификации составляла 15°С. Прочие условия были такими же, как в Примере 1. В Сравнительном Примере 2 также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Сравнительный Пример 3]
В Сравнительном Примере 3, перед стадией (S2) эстерификации, в центрифужную пробирку добавляли 5 мл воды вместо 5 мл водного раствора гидроксида натрия. Температура реакционной смеси в стадии (S2) эстерификации составляла 40°С. Прочие условия были такими же, как в Примере 1. В Сравнительном Примере 3 также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Сравнительный Пример 4]
В Сравнительном Примере 4, перед стадией (S2) эстерификации, в центрифужную пробирку добавляли 5 мл воды вместо 5 мл водного раствора гидроксида натрия. Температура реакционной смеси в стадии (S2) эстерификации составляла 60°С. Прочие условия были такими же, как в Примере 1. В Сравнительном Примере 4 также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Результаты]
Результаты показаны в ФИГ. 14.
В Примерах 3А и 3В (температуры реакционной смеси в стадии эстерификации: 15°С и 40°С, соответственно) валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные этиловые сложные эфиры (смотри ФИГ. 14). Таким образом, результаты Примеров 3А и 3В были приблизительно такими же, как результаты Примера 1. В Примерах 3А и 3В были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров.
В Примере 3С (температура реакционной смеси в стадии эстерификации: 60°С) валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные этиловые сложные эфиры (смотри ФИГ. 14). Таким образом, в Примере 3С были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. Однако масса различных этиловых сложных эфиров, образованных в Примере 3С, составляла около 50% массы различных этиловых сложных эфиров, образованных в Примере 1.
В Сравнительном Примере 2 (без добавления водного раствора гидроксида натрия, температура реакционной смеси в стадии эстерификации: 15°С), количества валериановых кислот и капроновых кислот, первоначально содержавшихся в исходном табачном материале, не сократились после стадии эстерификации. Кроме того, различные этиловые сложные эфиры не образовались после стадии эстерификации (смотри ФИГ. 14).
В Сравнительном Примере 3 (без добавления водного раствора гидроксида натрия, температура реакционной смеси в стадии эстерификации: 40°С), количества валериановых кислот и капроновых кислот, первоначально содержавшихся в исходном табачном материале, не сократились после стадии эстерификации. Здесь также различные этиловые сложные эфиры не образовались после стадии эстерификации (смотри ФИГ. 14).
В Сравнительном Примере 4 (без добавления водного раствора гидроксида натрия, температура реакционной смеси в стадии эстерификации: 60°С), количества валериановых кислот и капроновых кислот, первоначально содержавшихся в исходном табачном материале, не сократились после стадии эстерификации. Здесь также различные этиловые сложные эфиры не образовались после стадии эстерификации (смотри ФИГ. 14).
[Обсуждение]
Из результатов Примеров 1 и 3А-3С было найдено, что повышение температуры реакционной смеси приводило к снижению эффективности реакции эстерификации, как показано в Примере 3С. Было найдено, что температура реакционной смеси в стадии (S2) эстерификации тем самым предпочтительно составляла от 15°С до 60°С, и более предпочтительно от 15°С до 40°С. Из результатов Сравнительных Примеров 1 и 2-4 было найдено, что присутствие оснóвного вещества было существенным в стадии (S2) эстерификации, и что его отсутствие препятствовало протеканию реакции эстерификации, даже когда варьировали температуру. Вышеуказанное наводит на мысль, что в реакции эстерификации, протекающей в стадии (S2) эстерификации, в реакционном растворе мог бы присутствовать каталитический компонент некоторого рода, производный из исходного табачного материала. Каталитический компонент не был индентифицирован. Этот каталитический компонент может стимулировать реакцию эстерификации. Как предполагается, когда температура реакционной смеси в стадии (S2) эстерификации превышает 60°С, температура отклоняется от оптимальной температуры, при которой действует каталитический компонент.
Как предполагается, каталитический компонент, как один из многих кандидатов, представляет собой фермент некоторого вида. При допущении, что каталитический компонент представляет собой фермент, его активность может значительно снижаться, когда температура реакционной смеси в стадии (S2) эстерификации превышает 60°С.
<Оценка концентрации оснóвного вещества>
[Пример 4А]
В Примере 4А, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл 0,25н.-ого водного раствора гидроксида натрия вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4А также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Перед стадией (S2) эстерификации, до смешения со спиртом, измерили значение рН водного раствора оснóвного вещества (водного раствора гидроксида натрия).
[Пример 4В]
В Примере 4В, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл 0,1н.-ого водного раствора гидроксида натрия вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4В также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Перед стадией (S2) эстерификации, до смешения со спиртом, измерили значение рН водного раствора оснóвного вещества (водного раствора гидроксида натрия).
[Пример 4С]
В Примере 4С, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл 0,05н.-ого водного раствора гидроксида натрия вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4С также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Перед стадией (S2) эстерификации, до смешения со спиртом, измерили значение рН водного раствора оснóвного вещества (водного раствора гидроксида натрия).
[Пример 4D]
В Примере 4D, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл 0,01н.-ого водного раствора гидроксида натрия вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4D также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
Результаты показаны в ФИГ. 15.
Во всех Примерах 4А-4С валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них были образованы различные этиловые сложные эфиры. Таким образом, в Примерах 4А-4D были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. Во всех Примерах 4А-4D количества различных этиловых сложных эфиров, образованных в стадии эстерификации, были эквивалентны количествам различных этиловых сложных эфиров, образованных в Примере 1. Таким образом, результаты Примеров 4А-4D были приблизительно такими же, как в Примере 1.
[Обсуждение]
Из результатов Примеров 1 и 4А-4D было найдено, что является вполне достаточным, пока концентрация водного раствора высокоосновного вещества (гидроксида натрия), используемого в стадии (S2) эстерификации, составляла 0,01 н. или более.
<Оценка величины рН водного раствора оснóвного вещества>
Взаимосвязь между значением рН водного раствора оснóвного вещества и эффективностью образования различных сложных эфиров исследовали более подробно с использованием следующих Примеров 4Е-4К.
[Пример 4Е]
В Примере 4Е, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл водного раствора гидроксида натрия, имеющего рН 12,6, вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4Е также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Пример 4F]
В Примере 4F, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл водного раствора гидроксида натрия, имеющего рН 13,0, вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4F также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Кроме того, рассчитали численное значение высоты пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4F, когда высоту пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4Е, приняли за 100.
[Пример 4G]
В Примере 4G, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл водного раствора гидроксида натрия, имеющего рН 12,2, вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4G также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Кроме того, рассчитали численное значение высоты пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4G, когда высоту пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4Е, приняли за 100.
[Пример 4H]
В Примере 4H, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл водного раствора гидроксида натрия, имеющего рН 11,8, вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4H также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Кроме того, рассчитали численное значение высоты пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4H, когда высоту пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4Е, приняли за 100 (степень эстерификации).
[Пример 4I]
В Примере 4I, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл водного раствора гидроксида натрия, имеющего рН 11,3, вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4I также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Кроме того, рассчитали численное значение высоты пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4I, когда высоту пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4Е, приняли за 100 (степень эстерификации).
[Пример 4J]
В Примере 4J, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл водного раствора гидроксида натрия, имеющего рН 11,1, вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4J также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Кроме того, рассчитали численное значение высоты пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4J, когда высоту пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4Е, приняли за 100 (степень эстерификации).
[Пример 4К]
В Примере 4К, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл водного раствора гидроксида натрия, имеющего рН 10,7, вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 4К также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Кроме того, рассчитали численное значение высоты пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4К, когда высоту пика 3-MVA-этилового сложного эфира, полученного GC-MS-анализом в Примере 4Е, приняли за 100 (степень эстерификации).
[Результаты]
Результаты показаны ниже в таблице и в ФИГ. 16.
[Таблица 1]
Во всех Примерах 4Е-4К валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них были образованы различные этиловые сложные эфиры. Таким образом, в Примерах 4Е-4К были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. Степень эстерификации в Примере 4F составляла 87,56, степень эстерификации в Примере 4G составляла 97,01, степень эстерификации в Примере 4H составляла 59,70, степень эстерификации в Примере 4I составляла 7,46, степень эстерификации в Примере 4J составляла 4,97, и степень эстерификации в Примере 4К составляла 1,99.
[Обсуждение]
Из результатов Примеров 4Е-4К было найдено, что количество образованного 3-MVA-этилового сложного эфира быстро возрастает, когда водный раствор оснóвного вещества (гидроксида натрия), использованный в стадии (S2) эстерификации, имел значение рН 11,3 или более. Таким образом, было найдено, что достаточное количество 3-MVA-этилового сложного эфира получалось, когда водный раствор оснóвного вещества (гидроксида натрия), использованный в стадии (S2) эстерификации, имел значение рН от 11,3 до 13,0. Также было найдено, что степень эстерификации составляла около 60 или более, и достаточное количество 3-MVA-этилового сложного эфира получалось, когда водный раствор оснóвного вещества (гидроксида натрия), использованный в стадии (S2) эстерификации, имел значение рН от 11,8 до 13.0.
<Оценка иных оснóвных веществ, нежели гидроксид натрия>
[Пример 5А]
В Примере 5А, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл 0,5н.-ого водного раствора гидроксида калия вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 5А также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Перед стадией (S2) эстерификации, до смешения со спиртом, измерили значение рН водного раствора оснóвного вещества (водного раствора гидроксида калия).
[Пример 5В]
В Примере 5В, перед стадией (S2) эстерификации, добавили в центрифужную пробирку 5 мл 0,5н.-ого водного раствора гидроксида кальция вместо 5 мл 0,5н.-ого водного раствора гидроксида натрия, с образованием смешанного раствора. Прочие условия были такими же, как в Примере 1. В Примере 5В также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1. Перед стадией (S2) эстерификации, до смешения со спиртом, измерили значение рН водного раствора оснóвного вещества (водного раствора гидроксида кальция).
[Результаты]
Результаты показаны в ФИГ. 17.
В Примере 5А (с добавлением 0,5н.-ого водного раствора гидроксида калия) и в Примере 5В (с добавлением 0,5н.-ого водного раствора гидроксида кальция), валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них были образованы различные этиловые сложные эфиры (смотри ФИГ. 17). Результаты Примеров 5А и 5В были приблизительно такими же, как в Примере 1. Таким образом, в Примерах 5А и 5В были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров.
Значение рН водного раствора в Примере 5А составляло 13,3. Значение рН водного раствора в Примере 5В составляло 12,3.
[Обсуждение]
Из результатов Примеров 1, 5А, 5В и 5С было найдено, что в стадии (S2) эстерификации высокоосновные вещества, имеющие близкую к 1 степень ионизации, такие как гидроксид натрия, гидроксид калия и гидроксид кальция, были эффективными в водном растворе.
Кроме того, из результатов Примеров 1, 5А и 5В было найдено, что в стадии (S2) эстерификации значение рН водных растворов составляло от 12,3 до 13,3, и что значение рН типа оснóвного вещества было эффективным. Поэтому, с учетом результатов Примеров 1, 5А и 5В, и результатов Примеров 1 и 4А-4К, определено, что в стадии (S2) эстерификации значение рН водного раствора оснóвного вещества предпочтительно составляет от 11,3 до 13,3, и что значение рН типа оснóвного вещества является эффективным.
<Оценка эффективности добавления иного спирта, нежели этанол>
[Пример 6А]
В Примере 6А, перед стадией (S2) эстерификации, добавили 5 мл метанола в качестве спирта вместо 5 мл этанола, и прочие условия были такими же, как в Примере 1. В Примере 6А также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Пример 6В]
В Примере 6В, перед стадией (S2) эстерификации, добавили 5 мл 1-пропанола в качестве спирта вместо 5 мл этанола, и прочие условия были такими же, как в Примере 1. В Примере 6В также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Пример 6С]
В Примере 6С, перед стадией (S2) эстерификации, добавили 5 мл 1-бутанола в качестве спирта вместо 5 мл этанола, и прочие условия были такими же, как в Примере 1. В Примере 6С также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Результаты]
Результаты показаны в ФИГ. 18. В ФИГ. 18 линии Примеров 1 и 6А-8С сдвинуты по вертикальному направлению, чтобы они не перекрывались друг с другом.
В Примере 6А (с добавлением метанола) валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные метиловые сложные эфиры (метиловый сложный эфир 3-метилвалериановой кислоты и метиловый сложный эфир изовалериановой кислоты). В Примере 6А были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных метиловых сложных эфиров.
В Примере 6В (с добавлением 1-пропанола) валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные пропиловые сложные эфиры (пропиловый сложный эфир 3-метилвалериановой кислоты и пропиловый сложный эфир изовалериановой кислоты). В Примере 6В были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных пропиловых сложных эфиров.
В Примере 6С (с добавлением 1-бутанола) валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные бутиловые сложные эфиры (бутиловый сложный эфир 3-метилвалериановой кислоты и бутиловый сложный эфир изовалериановой кислоты). В Примере 6С были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных бутиловых сложных эфиров.
[Обсуждение]
Из результатов Примеров 1 и 6А-6С было найдено, что спирты, добавленные к водному раствору перед стадией (S2) эстерификации, предпочтительно представляли собой низший спирт, одноатомный спирт, первичный спирт, и тому подобные.
<Оценка добавленного количества этанола>
[Пример 7А]
В Примере 7А, перед стадией (S2) эстерификации, добавили 10 мл этанола в качестве спирта вместо 5 мл этанола, и прочие условия были такими же, как в Примере 1. В Примере 7А также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Пример 7В]
В Примере 7В, перед стадией (S2) эстерификации, добавили 3 мл этанола в качестве спирта вместо 5 мл этанола, и прочие условия были такими же, как в Примере 1. В Примере 7В также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Пример 7С]
В Примере 7С, перед стадией (S2) эстерификации, добавили 0,5 мл этанола в качестве спирта вместо 5 мл этанола, и прочие условия были такими же, как в Примере 1. В Примере 7С также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Сравнительный Пример 5]
В Сравнительном Примере 5 перед стадией (S2) эстерификации в центрифужную пробирку не добавляли 5 мл этанола, и прочие условия были такими же, как в Примере 1. В Сравнительном Примере 5 также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS таким же образом, как описано в Примере 1.
[Результаты]
Результаты показаны в ФИГ. 19.
В обоих Примерах 7А и 7В (добавляли 10 мл этанола и 3 мл этанола, соответственно) валериановые кислоты и капроновые кислоты, первоначально содержавшиеся в исходном табачном материале, исчезли после стадии эстерификации, и вместо них образовались различные этиловые сложные эфиры, как показано в ФИГ. 19. Таким образом, в Примерах 7А и 7В были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. В обоих Примерах 7А и 7В количества различных этиловых сложных эфиров, образованных в стадии эстерификации, были эквивалентны количествам различных этиловых сложных эфиров, образованных в Примере 1. Таким образом, результаты Примеров 7А и 7В были приблизительно такими же, как результаты в Примере 1.
В Примере 7С (с добавлением 0,5 мл этанола) количества валериановых кислот и капроновых кислот, первоначально содержавшихся в исходном табачном материале, сократились после стадии эстерификации, и вместо них образовались различные этиловые сложные эфиры. Таким образом, в Примере 7С были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров. Однако в Примере 7С количества различных этиловых сложных эфиров, образованных в стадии эстерификации, составляли около 55% количеств различных этиловых сложных эфиров, образованных в Примере 1.
В Сравнительном Примере 5 (без добавления этанола) различные этиловые сложные эфиры не образовались, но количества различных валериановых кислот и капроновых кислот сократились, как показано в ФИГ. 19.
[Обсуждение]
Из результатов Примеров 1 и 7А-7С было найдено, что количество спирта (этанола), содержащегося в водном растворе, предпочтительно составляло от 0,5 мл до 10,0 мл в стадии (S2) эстерификации. Тем самым было найдено, что доля объема спирта в сумме объема содержащей табачный компонент жидкости и объема водного раствора предпочтительно составляла от 2,0% до 28,6%.
В альтернативном варианте, было найдено, что количество спирта (этанола), добавленного в водный раствор, более предпочтительно составляло от 3,0 мл до 10,0 мл в стадии (S2) эстерификации. Тем самым было найдено, что доля объема спирта в сумме объема содержащей табачный компонент жидкости и объема водного раствора предпочтительно составляла от 10,7% до 28,6%.
В Примере 7С, хотя образовались различные этиловые сложные эфиры, полученные количества их были меньшими, чем количества в Примере 1; поэтому сделан вывод, что количество жидкого спирта (этанола) в смешанном растворе в стадии (S2) эстерификации было малым, и реакция протекала медленно.
<Оценка продолжительности реакции в стадии эстерификации>
[Пример 8А]
В Примере 8А продолжительность взбалтывания в центрифужной пробирке, содержащей смешанный раствор, регулировали на 40 минут, вместо 60 минут в стадии (S2) эстерификации. Прочие условия были такими же, как условия в Примере 1. В Примере 8А также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS, как описано в Примере 1.
[Пример 8В]
В Примере 8В продолжительность взбалтывания в центрифужной пробирке, содержащей смешанный раствор, регулировали на 30 минут, вместо 60 минут в стадии (S2) эстерификации. Прочие условия были такими же, как условия в Примере 1. В Примере 8В также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS, как описано в Примере 1.
[Пример 8С]
В Примере 8С продолжительность взбалтывания в центрифужной пробирке, содержащей смешанный раствор, регулировали на 20 минут, вместо 60 минут в стадии (S2) эстерификации. Прочие условия были такими же, как условия в Примере 1. В Примере 8С также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS, как описано в Примере 1.
[Пример 8D]
В Примере 8D продолжительность взбалтывания в центрифужной пробирке, содержащей смешанный раствор, регулировали на 10 минут, вместо 60 минут в стадии (S2) эстерификации. Прочие условия были такими же, как условия в Примере 1. В Примере 8D также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS, как описано в Примере 1.
[Пример 8Е]
В Примере 8Е продолжительность взбалтывания в центрифужной пробирке, содержащей смешанный раствор, регулировали на 5 минут, вместо 60 минут в стадии (S2) эстерификации. Прочие условия были такими же, как условия в Примере 1. В Примере 8Е также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS, как описано в Примере 1.
[Пример 8F]
В Примере 8F продолжительность взбалтывания в центрифужной пробирке, содержащей смешанный раствор, регулировали на 1 минуту, вместо 60 минут в стадии (S2) эстерификации. Прочие условия были такими же, как условия в Примере 1. В Примере 8F также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS, как описано в Примере 1.
[Пример 8G]
В Примере 8G центрифужную пробирку, содержащую смешанный раствор, не взбалтывали в стадии (S2) эстерификации, но вместо этого оставили стоять для протекания реакции в течение нескольких секунд. Прочие условия были такими же, как условия в Примере 1. В Примере 8G также проводили стадию (S1) экстракции и стадию (S2) эстерификации, и жидкость, удаленную из верхней фазы органического растворителя (гексановой фазы), анализировали методом GC-MS, как описано в Примере 1.
[Результаты]
Результаты показаны в ФИГУРАХ 20 и 21. В ФИГ. 20 линии Примеров 1 и 8А-8G сдвинуты по вертикальному направлению, чтобы они не перекрывались друг с другом.
В Примерах 8А-8Е (взбалтывание проводили в течение времени от 40 до 5 минут) различные этиловые сложные эфиры образовались в количествах, эквивалентных количествам в Примере 1. В Примерах 8А-8Е были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров.
В Примере 8F (взбалтывание проводили в течение 1 минуты) образовались различные этиловые сложные эфиры, имеющую массу, соответствующую около 70% массы различных этиловых сложных эфиров, образованных в Примере 1. В Примере 8F были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров.
В Примере 8G (взбалтывание не выполняли) образовались различные этиловые сложные эфиры, имеющую массу, соответствующую около 10% массы различных этиловых сложных эфиров, образованных в Примере 1. В Примере 8G были получены табачная ароматическая жидкость или сложноэфирные соединения с увеличенными количествами различных этиловых сложных эфиров.
[Обсуждение]
Из результатов Примеров 8А-8F было найдено, что реакцию (взбалтывание) в стадии (S2) эстерификации предпочтительно проводили в течение периода времени 1 минуты или более, более предпочтительно проводили в течение периода времени от 1 до 60 минут, и еще более предпочтительно проводили в течение периода времени от 5 до 60 минут.
СПИСОК ССЫЛОЧНЫХ ПОЗИЦИЙ
11. Ароматический ингалятор нагревательного типа
12. Табачный наполнитель
13. Корпус
14. Всасывающий канал (мундштук)
15. Резервуар для хранения источника аэрозоля
16. Батарея
17. Схема управления
18. Картридж
21. Вентиляционное отверстие
22. Источник табачного аромата
23. Фитиль
24. Нагреватель
25. Направляющий проток
26. Линия электропередачи
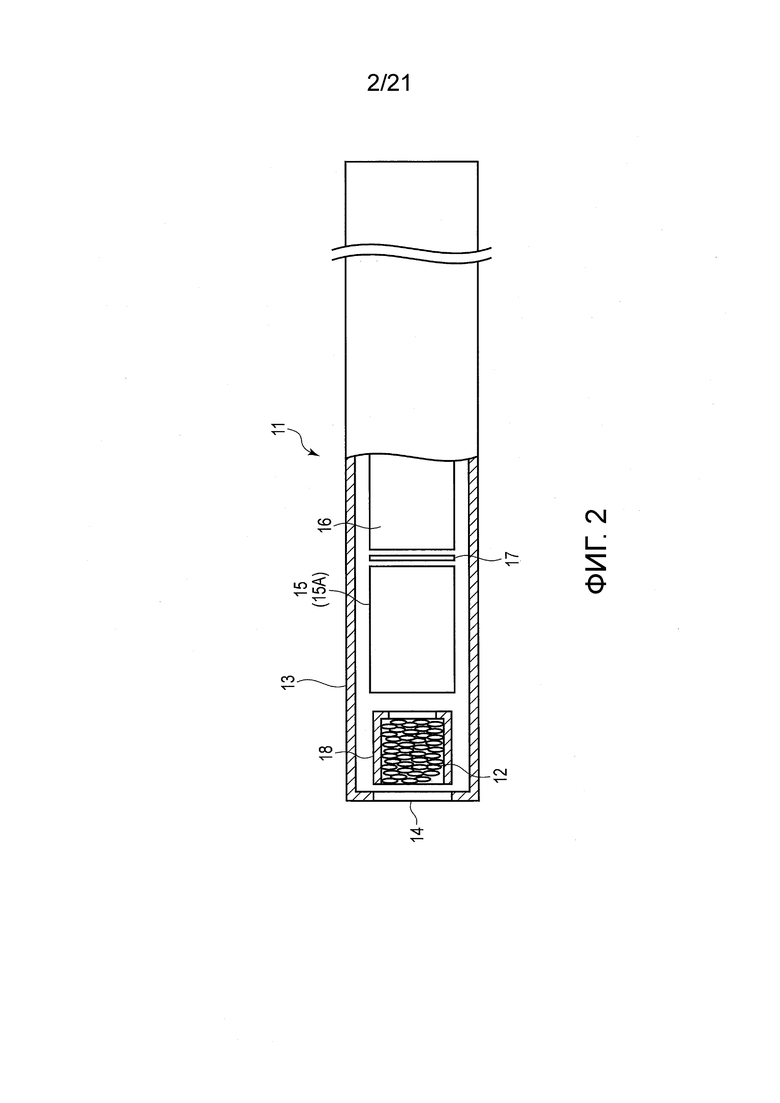
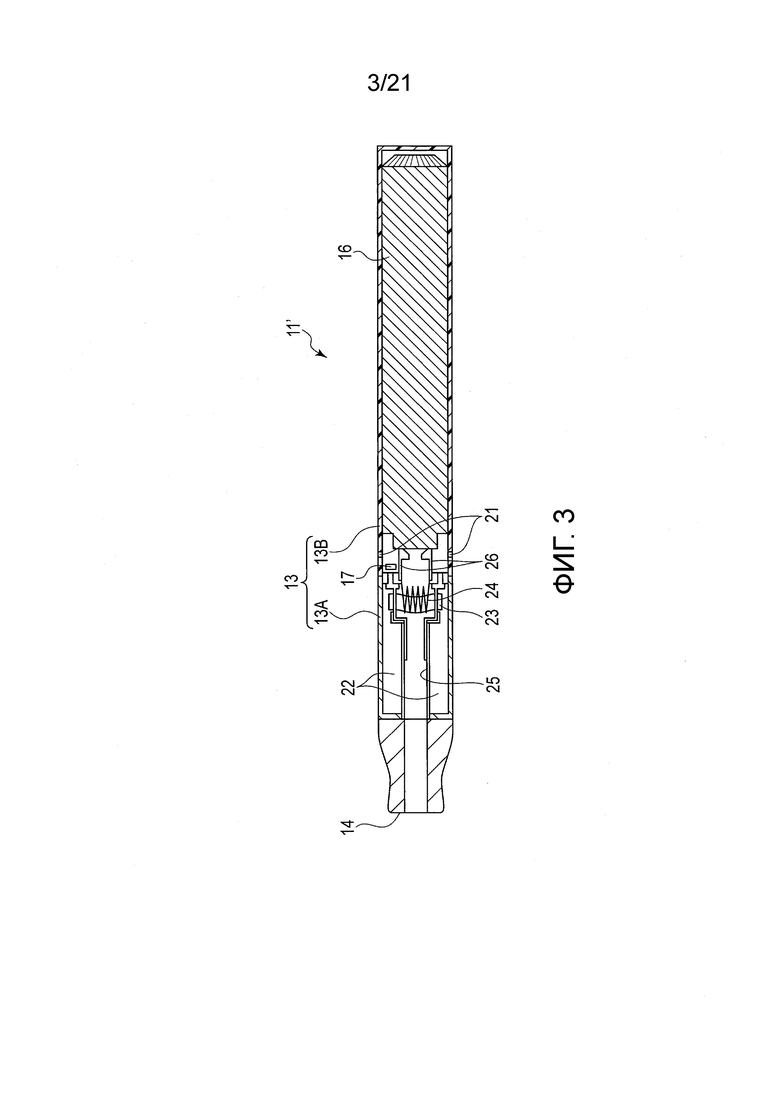
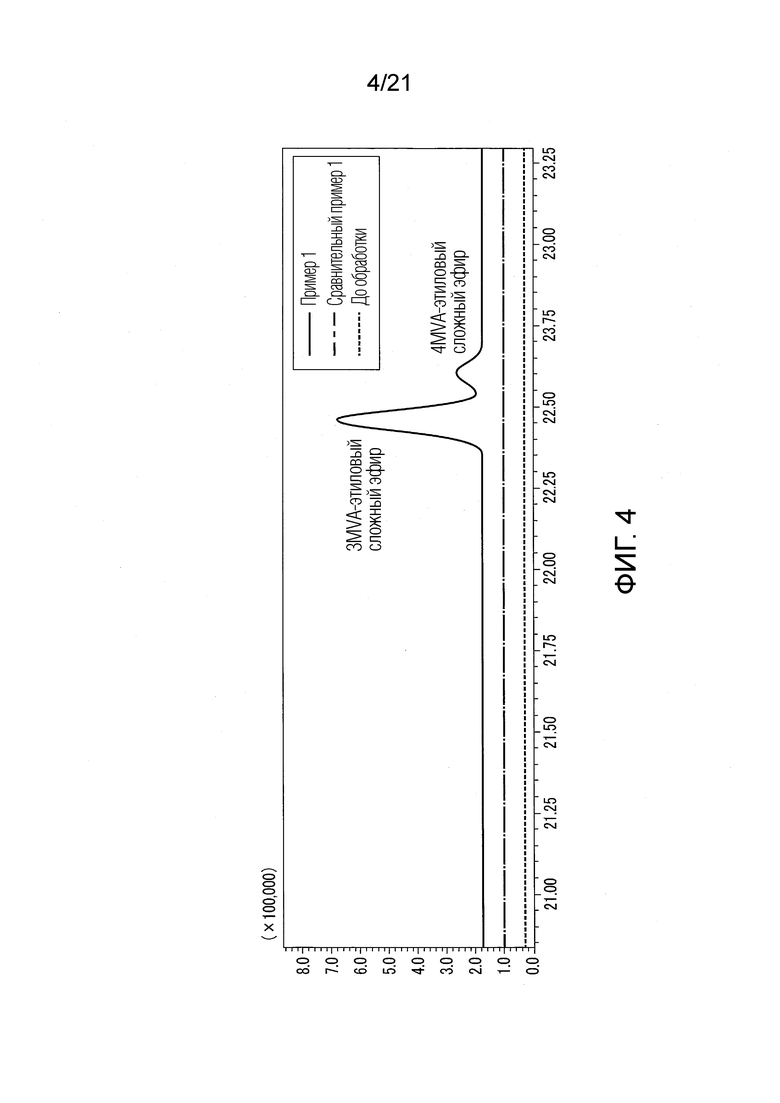
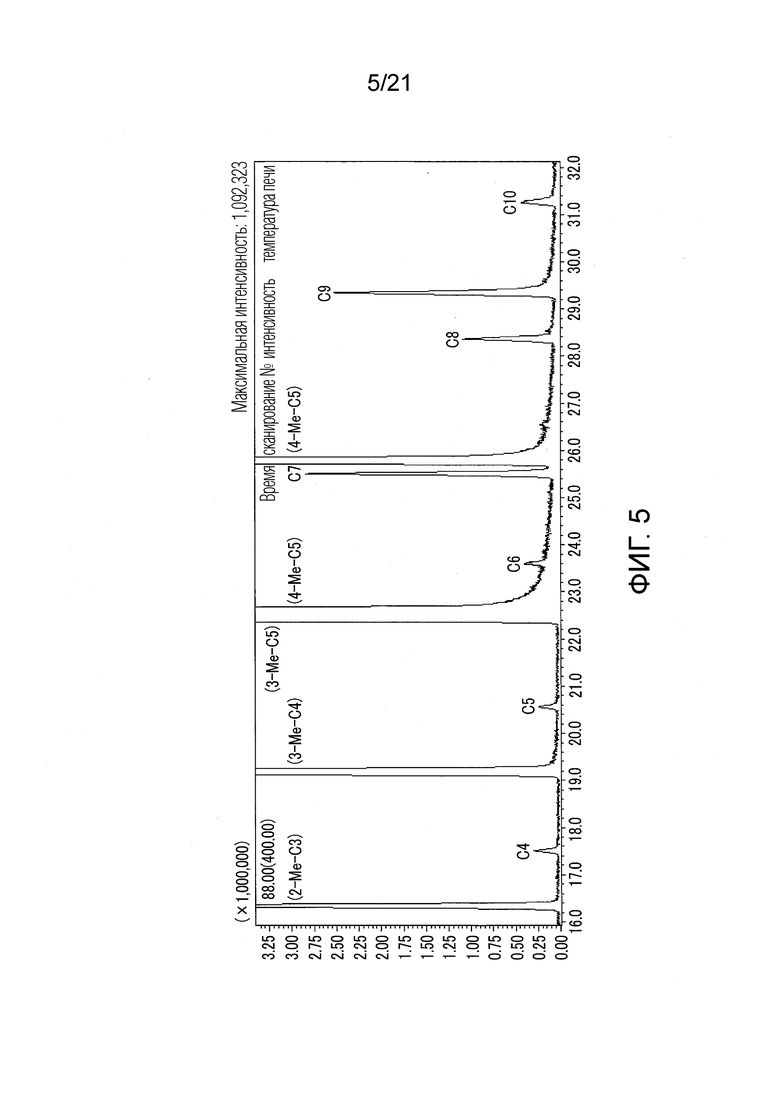

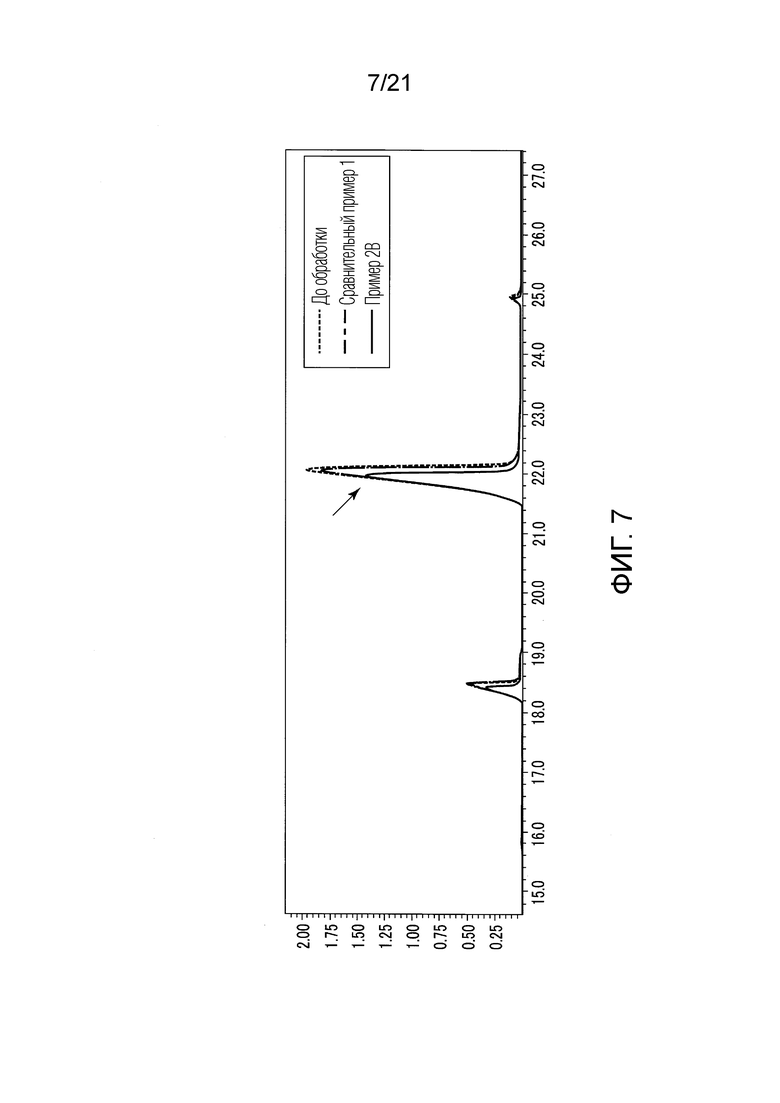
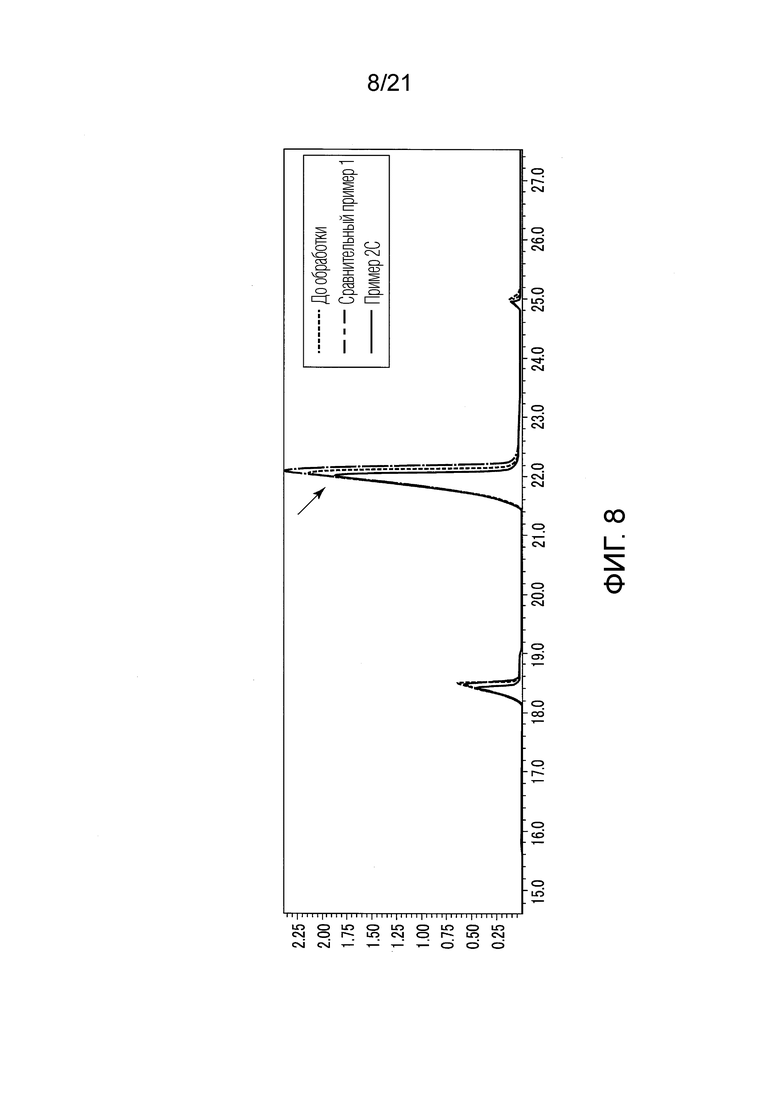
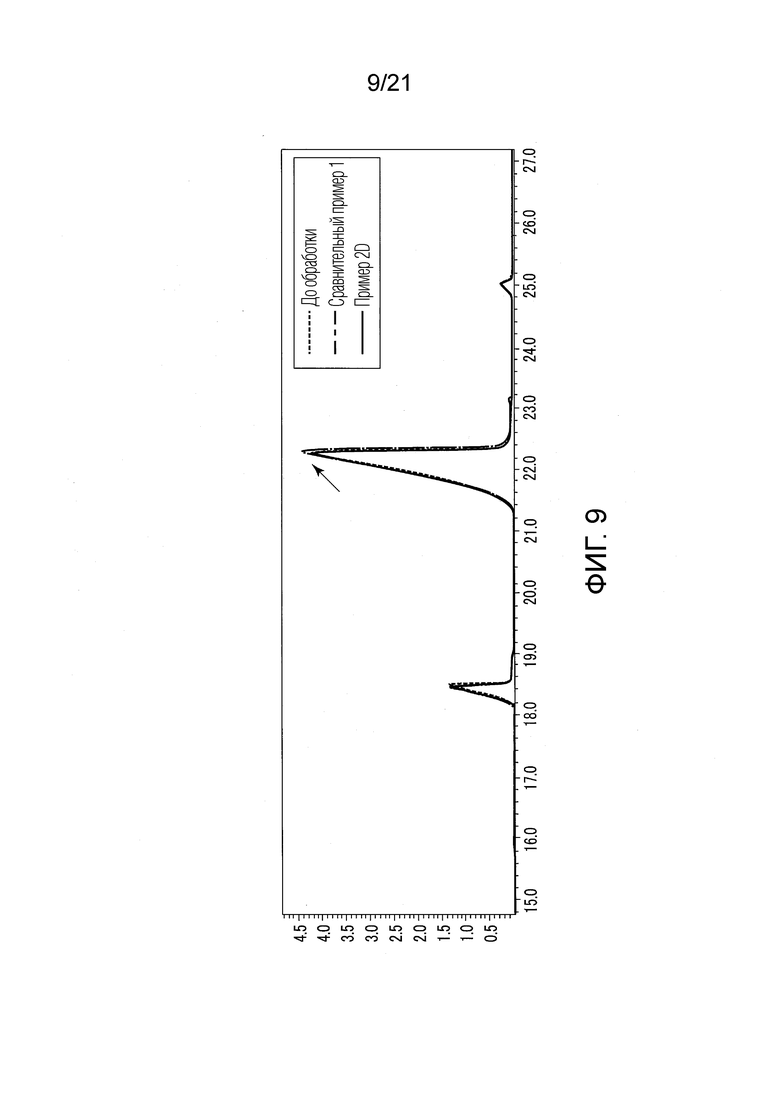
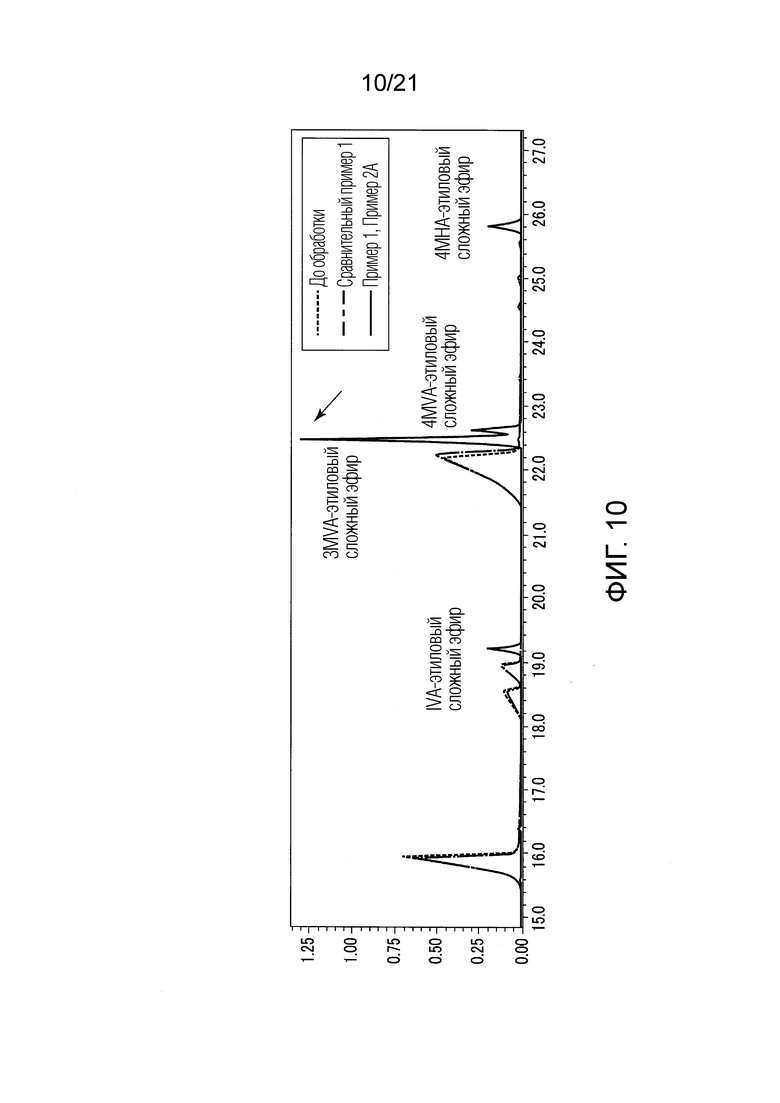
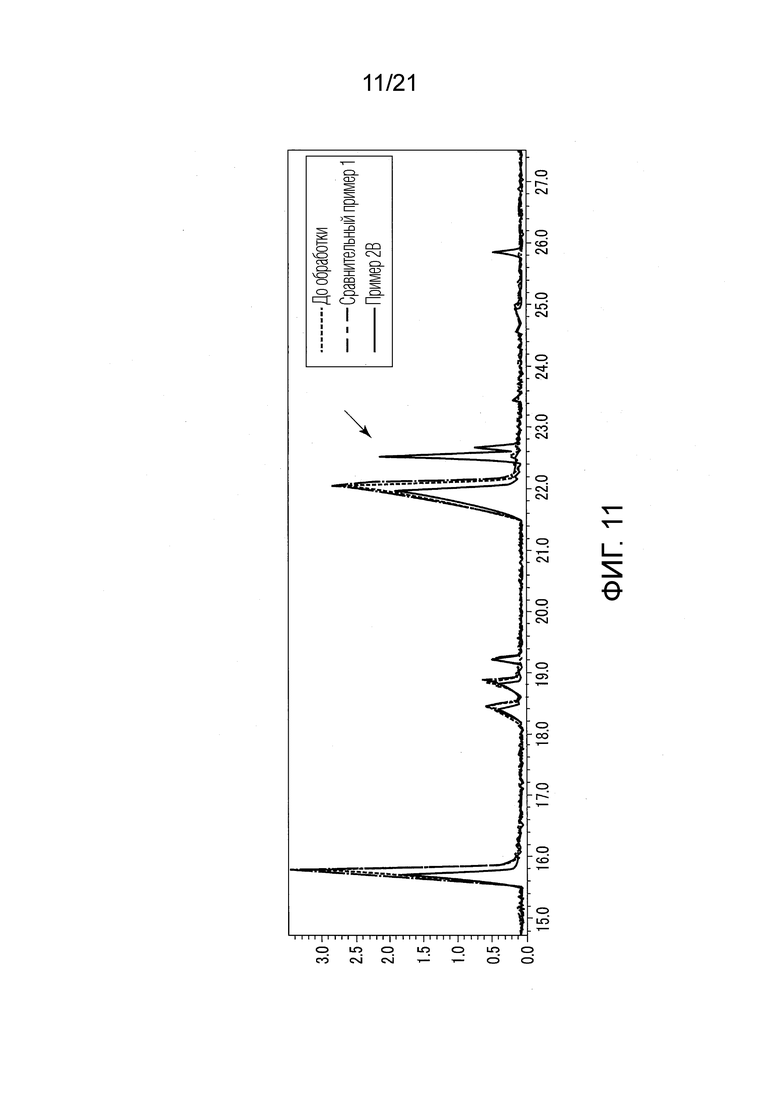
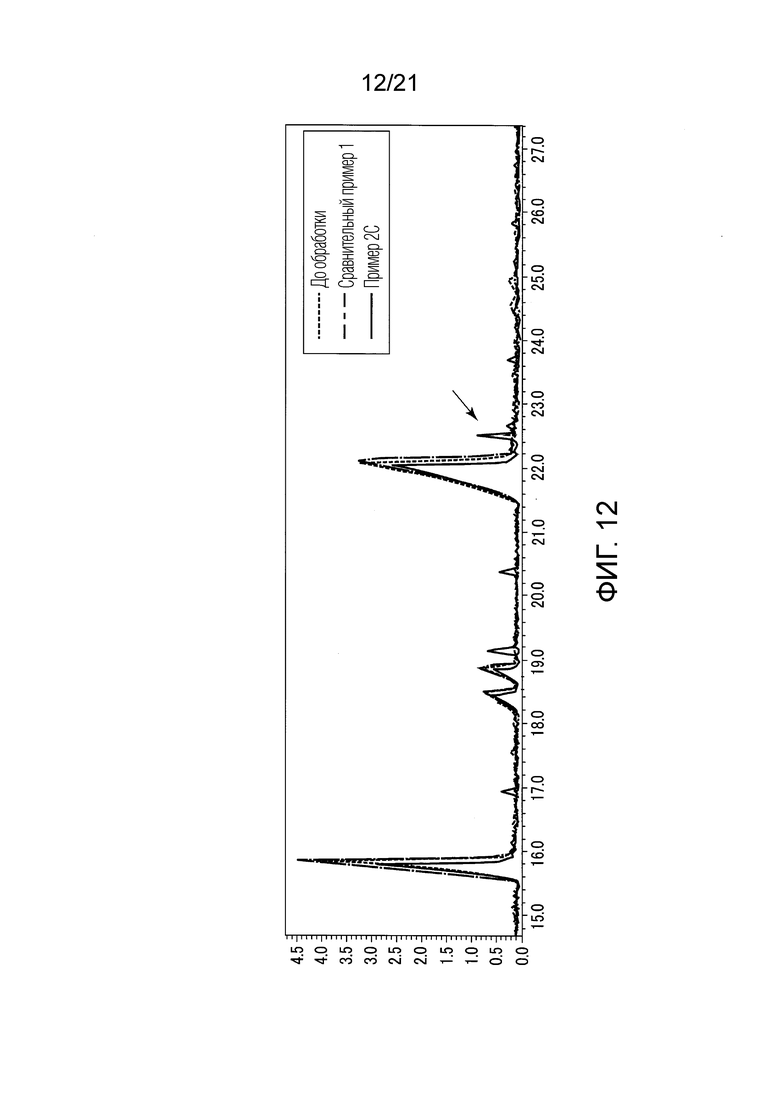
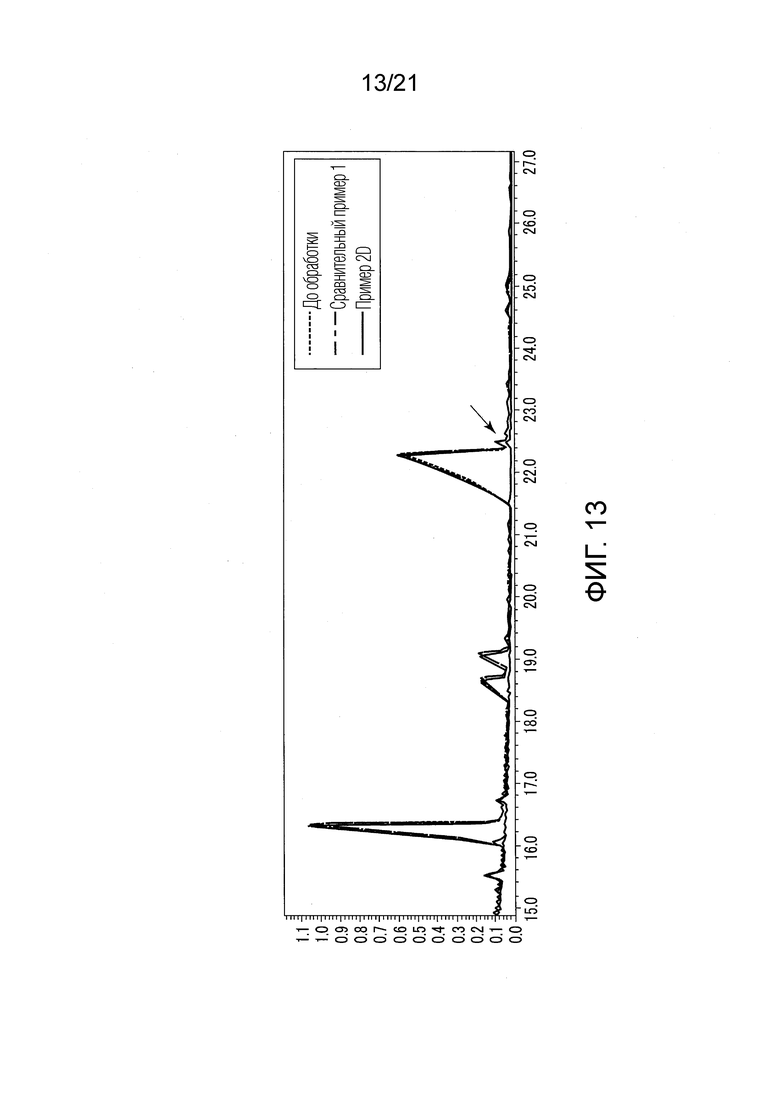
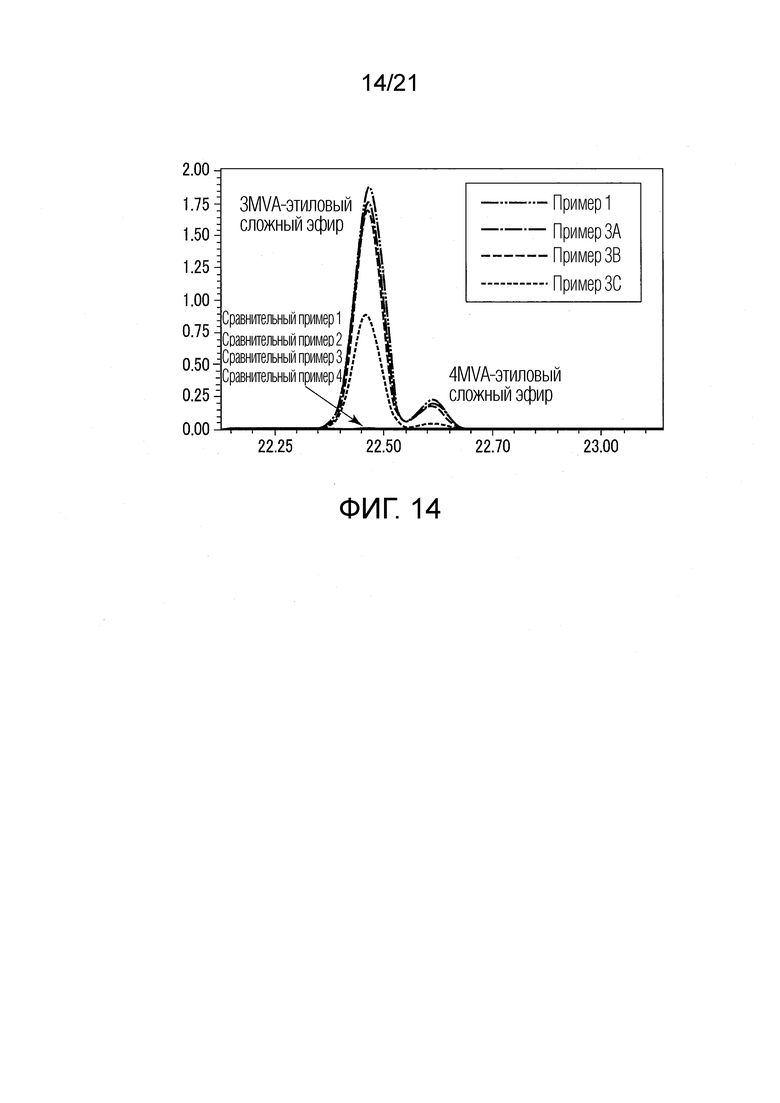
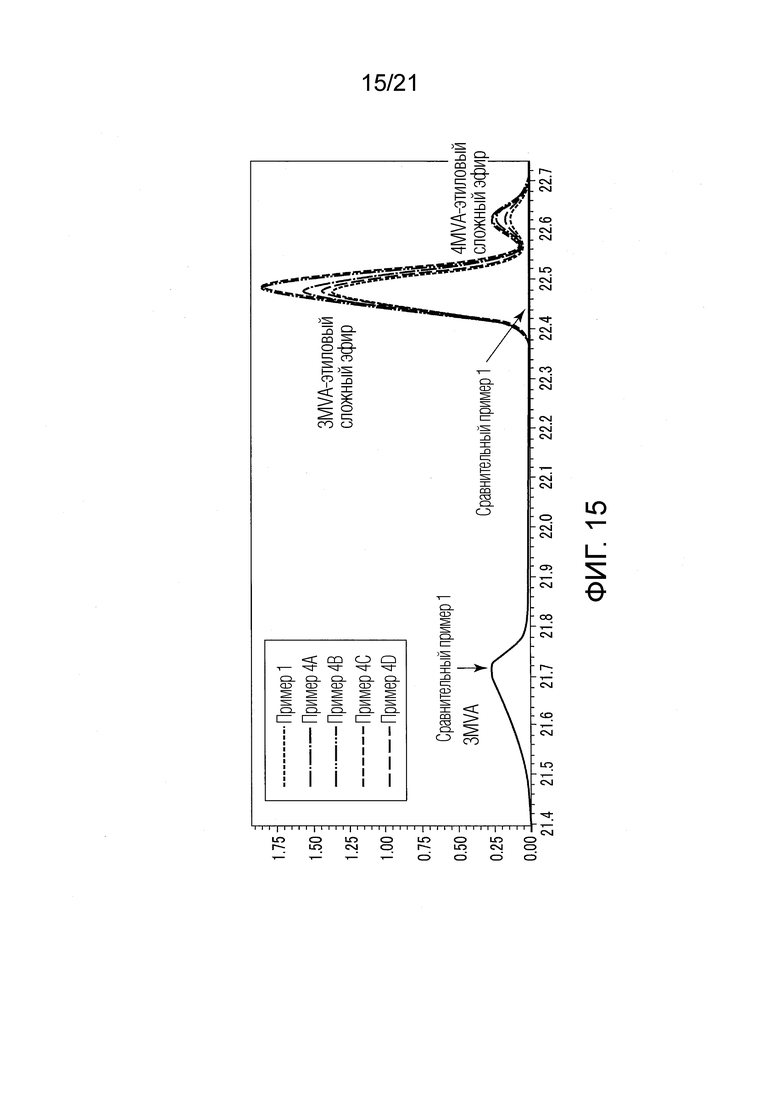
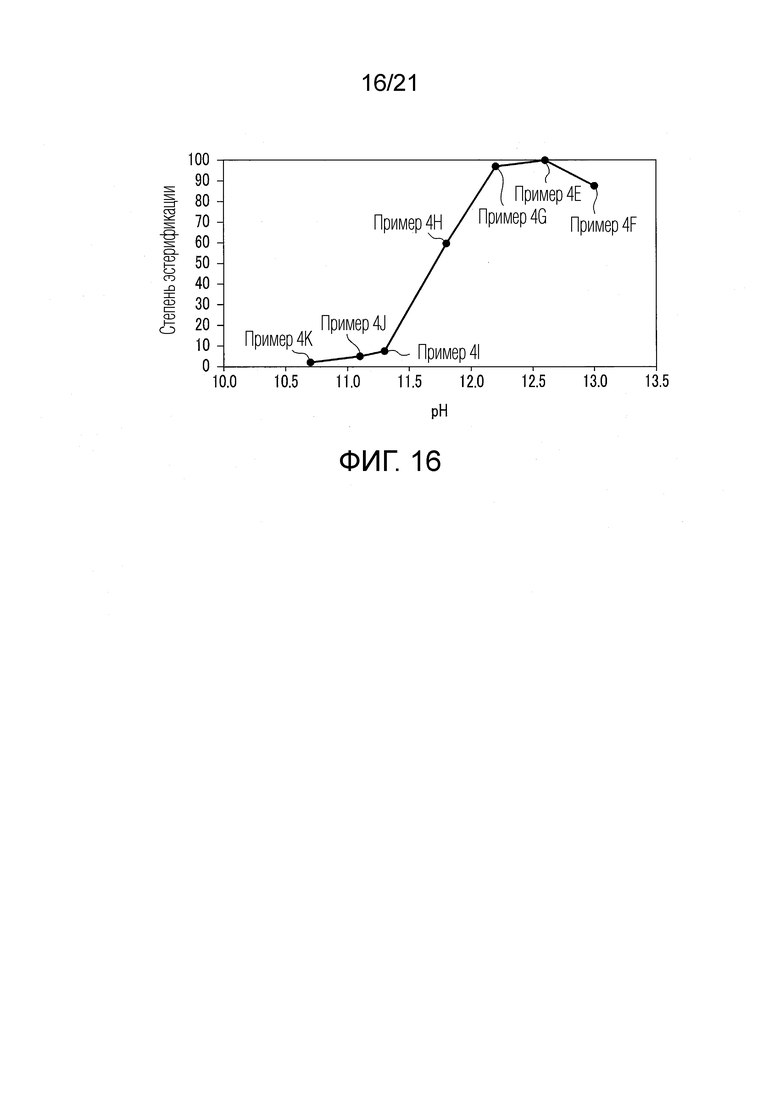

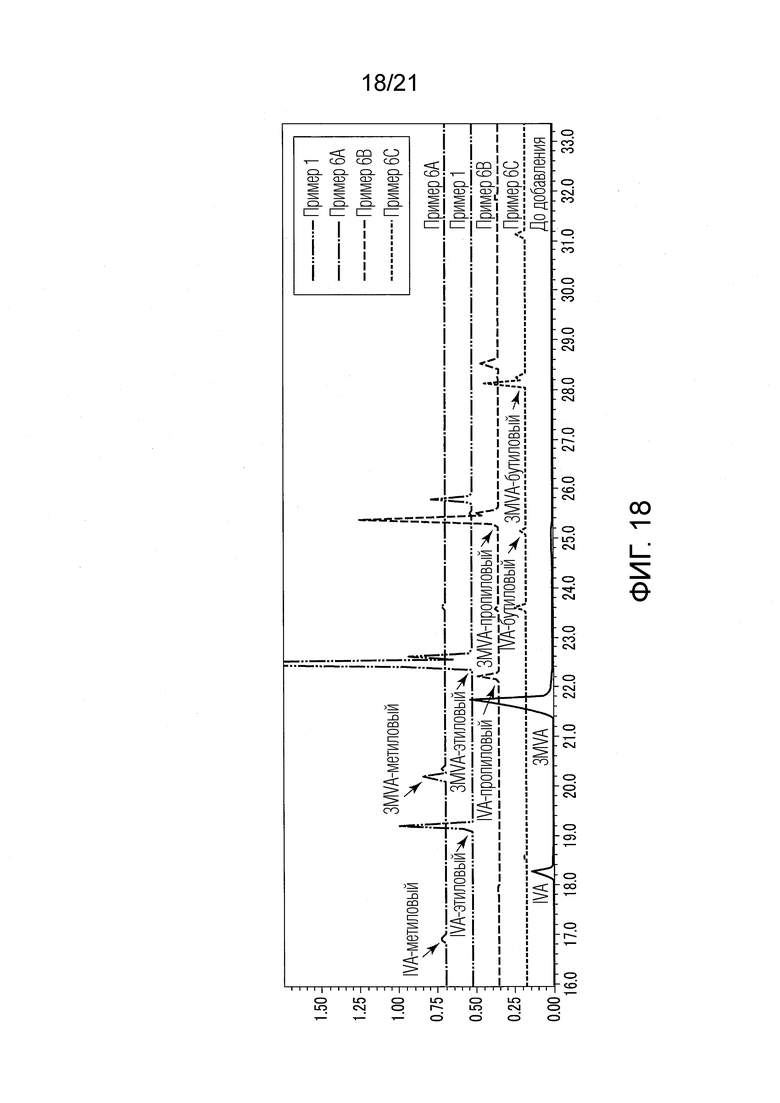
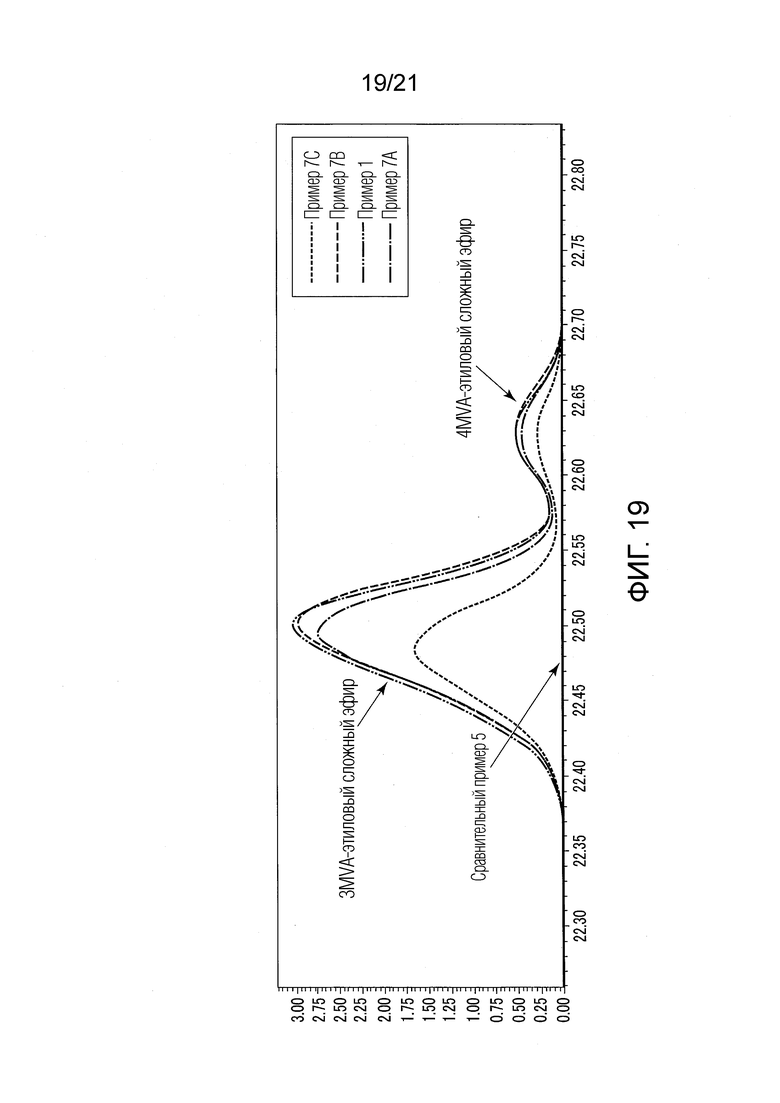
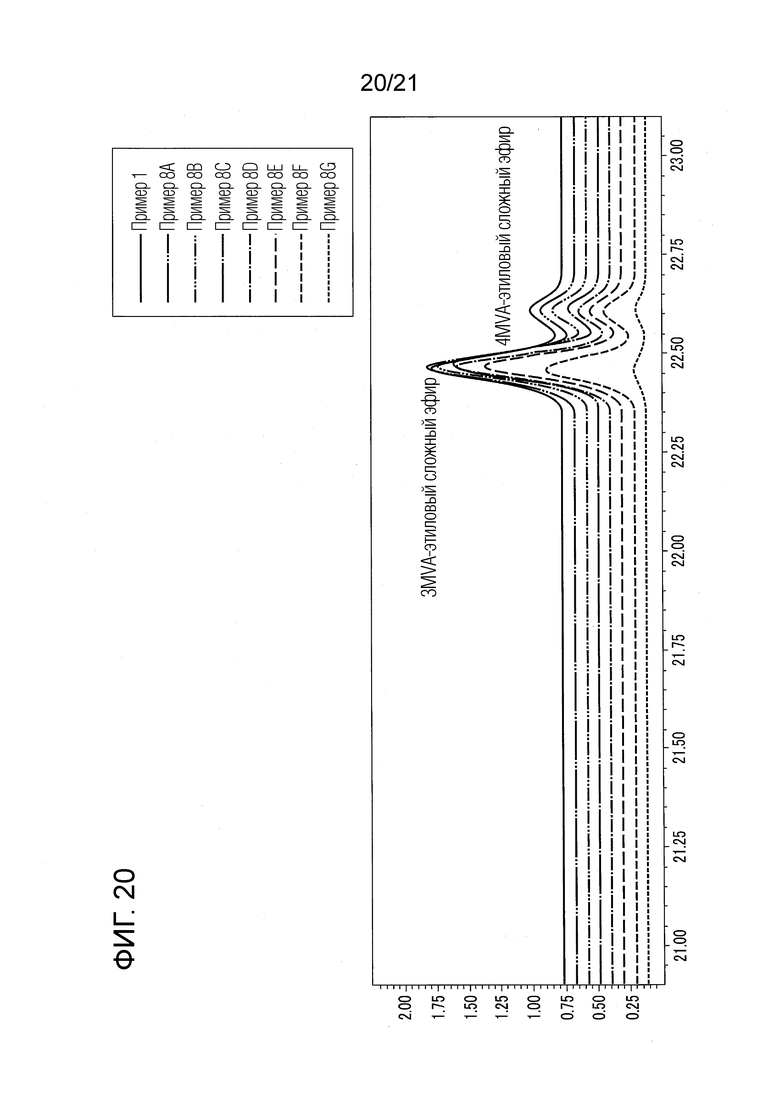
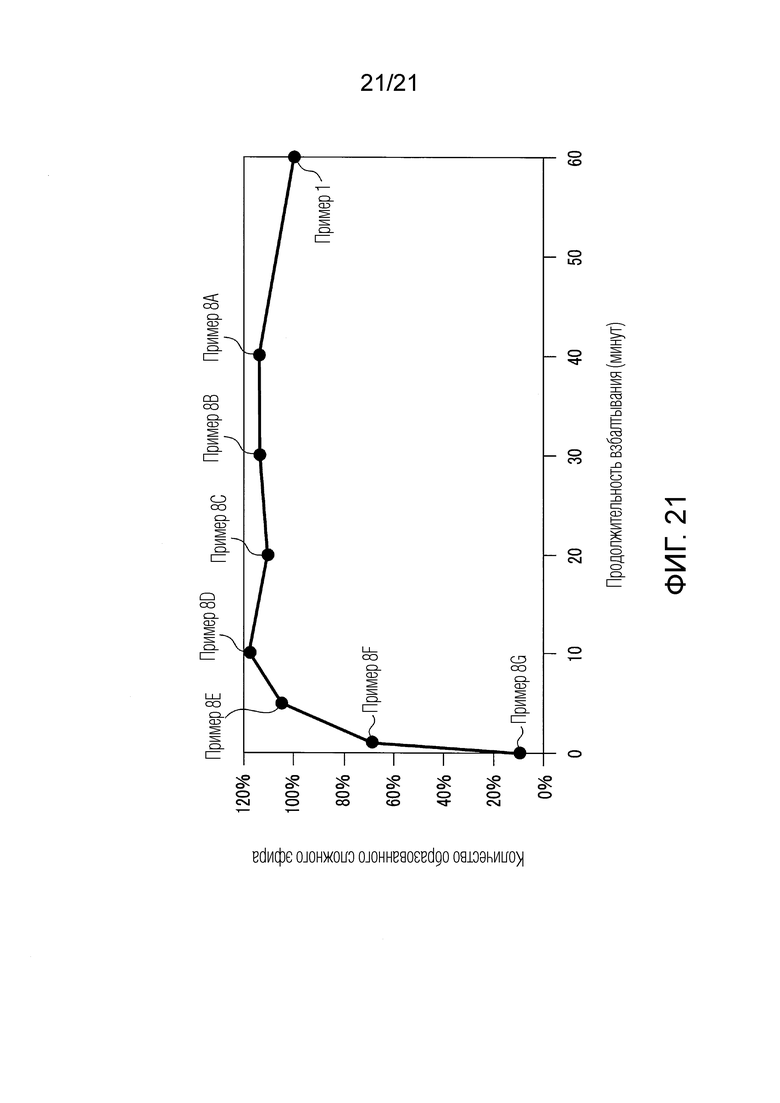
Группа изобретений относится к способу получения табачной ароматической жидкости, табачной ароматической жидкости, способу получения сложноэфирного соединения и курительному изделию, которое содержит табачную ароматическую жидкость. Способ получения табачной ароматической жидкости включает экстракцию компонента из исходного табачного материала с использованием органического растворителя для получения таким образом содержащей табачный компонент жидкости и смешение содержащей табачный компонент жидкости с водным раствором, включающим основное вещество и спирт, в кислородсодержащей атмосфере для реакции содержащей табачный компонент жидкости с водным раствором с получением таким образом табачной ароматической жидкости. Водный раствор представляет собой смешанный раствор водного раствора основного вещества и спирта, при этом основное вещество присутствует в водном растворе основного вещества при концентрации от 0,01 до 5 н. Доля объема спирта в сумме объема содержащей табачный компонент жидкости и объема водного раствора составляет от 2,0 до 28,6%. Реакцию проводят на протяжении периода времени от 1 до 60 мин при взбалтывании. Обеспечивается получение табачной ароматической жидкости с увеличенным количеством различных этиловых сложных эфиров, т.е. создание табачной ароматической жидкости, которая сохраняет первоначальный аромат исходного табачного материала и имеет усиленный сложными эфирами аромат. 4 н. и 5 з.п. ф-лы, 21 ил.
1. Способ получения табачной ароматической жидкости, включающий:
экстракцию компонента из исходного табачного материала с использованием органического растворителя для получения таким образом содержащей табачный компонент жидкости; и
смешение содержащей табачный компонент жидкости с водным раствором, включающим основное вещество и спирт, в кислородсодержащей атмосфере для реакции содержащей табачный компонент жидкости с водным раствором с получением таким образом табачной ароматической жидкости, при этом водный раствор представляет собой смешанный раствор водного раствора основного вещества и спирта, при этом основное вещество присутствует в водном растворе основного вещества при концентрации от 0,01 до 5 н, при этом доля объема спирта в сумме объема содержащей табачный компонент жидкости и объема водного раствора составляет от 2,0 до 28,6%, причем реакцию проводят на протяжении периода времени от 1 до 60 мин при взбалтывании.
2. Способ по п. 1, в котором реакцию проводят при температуре от 0 до 90°С.
3. Способ по п. 1, в котором водный раствор представляет собой смешанный раствор водного раствора основного вещества и спирта, при этом
водный раствор основного вещества имеет значение рН от 11,0 до 14,0.
4. Способ по п. 1, в котором основное вещество представляет собой гидроксид щелочного металла или щелочноземельного металла.
5. Способ по п. 1, в котором основное вещество является высокоосновным.
6. Способ по п. 1, в котором органический растворитель, входящий в состав содержащей табачный компонент жидкости, является несмешивающимся с водным раствором.
7. Табачная ароматическая жидкость, полученная способом получения табачной ароматической жидкости по любому из пп. 1-6.
8. Способ получения сложноэфирного соединения, включающий:
экстракцию компонента из исходного табачного материала с использованием органического растворителя для получения таким образом содержащей табачный компонент жидкости;
введение содержащей табачный компонент жидкости в реакцию с водным раствором, включающим основное вещество и спирт, в кислородсодержащей атмосфере для получения таким образом табачной ароматической жидкости; и
выделение сложноэфирного соединения из табачной ароматической жидкости, при этом водный раствор представляет собой смешанный раствор водного раствора основного вещества и спирта, при этом основное вещество присутствует в водном растворе основного вещества при концентрации от 0,01 до 5 н, при этом доля объема спирта в сумме объема содержащей табачный компонент жидкости и объема водного раствора составляет от 2,0 до 28,6%, причем реакцию проводят на протяжении периода времени от 1 до 60 мин при взбалтывании.
9. Курительное изделие, включающее табачную ароматическую жидкость по п. 7.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| JPS 5312899 A, 04.02.1978 | |||
| EP 2944697 A1, 18.11.2015 | |||
| CN 107660817 A, 06.02.2018 | |||
| УГОЛЬНЫЙ ИСТОЧНИК ТЕПЛА И ИНГАЛЯТОР АРОМАТА | 2013 |
|
RU2577727C1 |
| Привязь для лошадей и других животных | 1929 |
|
SU16476A1 |

