Изобретение относится к способам количественного анализа органических соединений в реакциях, прежде всего – реакциях безводородного гидрирования ненасыщенных углеводородов (ароматических и алифатических) с помощью низших спиртов. Способ заключается отборе проб реакционной смеси без остановки реакционного процесса с применением инфракрасной спектроскопии (ИК-спектроскопии) для мониторинга количественных изменений в химическом составе жидких реакционных смесей и основан на детектировании интенсивности характеристических полос поглощения избранных функциональных групп компонентов реакции – спиртов и кетонов.
Аналогом данного изобретения является американский патент на использование метода ИК–Фурье (инфракрасной-Фурье) спектроскопии ближней ИК-области (FT–NIR или БИК-спектроскопия) для количественного определения ненасыщенных цис-/транс-жирных кислот (т.е. кислот, молекулы которых содержат кратные С=С связи в цис- или транс-конфигурации) в нативных и частично гидрированных растительных маслах [H. Azizian, FT–NIR fatty acid determination method, US 7329547 B2, 2008]. Для получения количественных оценок было предложено измерять и сравнивать между собой интенсивности отдельных полос поглощения цис-/транс-изомеров ненасыщенных жирных кислот. При этом полагалось, что в регистрируемых инфракрасных спектрах (ИК-спектрах) разница в положении полос поглощения цис-/транс-изомеров является значимой для их надёжной дифференциации и идентификации, что на самом деле далеко не так. Поэтому для верификации количественных оценок, полученных методом БИК-спектроскопии, был осуществлён количественный хроматографический анализ растительных масел или модельных смесей с известной концентрацией ненасыщенных жирных кислот. Для такого детального анализа в патенте был применён целый комплекс хроматографических методов, включающий комбинацию стандартной газовой хроматографии (ГХ), ГХ с использованием серебросодержащей неподвижной фазы (ГХ–Ag+), ГХ–ВЭЖХ, ГХ, сопряжённой с тонкослойной хроматографией (ГХ–ТСХ), а также газовую хроматомасс-спектрометрию (ГХ–МС). Предложенный количественный хроматографический анализ был необходим для построения калибровочной матрицы, элементы которой однозначно связывали изменения в интенсивности избранных полос поглощения в спектрах FT–NIR и количественным содержанием цис-/транс-изомерных жирных кислот в анализируемом образце.
Для достижения обозначенной цели изобретения-аналога было предложено осуществлять сканирование в рабочем диапазоне частот 5400–9000 см–1 [H. Azizian, FT–NIR fatty acid determination method, US 7329547 B2, 2008], что, на наш взгляд, следует считать слишком широким для удобства наблюдения и быстрого детектирования на практике и последующего анализа спектра. Один из главных недостатков изобретения-аналога связан с известным фактом, что и БИК-, и ИК-спектроскопия среднего диапазона характеризуются низкой чувствительностью по отношению к С=С связям олефинов. Именно поэтому метод БИК-спектроскопии традиционно используют не для детектирования олефинов (каковыми являются ненасыщенные жирные кислоты), а для определения количеств водородно-связанных химических веществ, например остаточных количеств воды в анализируемых образцах [R.L. Long, R.C. Yeh, B.C. Devoy, Appatatus dewatering an elastomeric polymer, US 6517335B1, 2003]. Кроме того, предложенный хроматографический анализ жирных кислот является трудновыполнимым в подавляющем большинстве аналитических химических лабораторий, оборудованных стандартным приборным парком. Проведение такой количественной оценки с обязательным применением мультихроматографического анализа нельзя считать рутинным, простым или быстрым.
В другом патенте-аналоге метод ИК–Фурье спектроскопии средней области ИК-спектра был применён для количественного определения цис-, транс- и терминальных С=С связей в сополимерах на основе бутадиена [J. Kurazumi, E. Suzuki, Y. Ozawa, Modified conjugated diene copolymer, rubber compositions and tires, US 7968652 B2, 2011]. Здесь детектирование методом ИК–Фурье спектроскопии проводилось в довольно узком диапазоне при четырёх длинах волн – 1130, 967, 911 и 736 см–1. В этом патенте-аналоге для верификации количественных оценок, полученных методом ИК-спектроскопии, была применена 1H и 13C ЯМР-спектроскопия. В патенте указывалось, как минимум, на два недостатка-ограничения для применения предложенного подхода. Во-первых, отмечалось, что ИК-спектроскопия средней области ИК-спектра позволяет количественно определить с высокой точностью терминальные С=С связи (т.е. концевые), но не интернальные (в цис- или транс-конфигурации). Во-вторых, рекомендуемое количество С=С связей в цис-конфигурации, определяемое в сополимере, должно быть не ниже 94% от общего количества С=С связей. Такое требование подразумевает применения альтернативных методов исследования или источников информации для уточнения количества С=С связей в цис-конфигурации в полимере. По нашему мнению, имеется ещё одно ограничение предложенного подхода: запатентованный способ не даёт возможности проведения количественных измерений в режиме in situ, что является важным при проведении реальных органических реакций или при другой необходимости быстрого анализа.
В изобретениях-аналогах [H. Azizian, FT–NIR fatty acid determination method, US 7329547 B2, 2008; J. Kurazumi, E. Suzuki, Y. Ozawa, Modified conjugated diene copolymer, rubber compositions and tires, US 7968652 B2, 2011] метод ИК-спектроскопии использовался для количественного определения ненасыщенных органических соединений, содержащих в составе своих молекул С=С связи, т.е. детектировались вещества из той же группы органических веществ, что и в нашем патенте. Параллельно использовалась хроматография или ЯМР-спектроскопия в качестве альтернативного количественного метода, и этот момент являлся принципиальным: альтернативный количественный метод позволил провести строгую корреляцию между экспериментальными количественными данными, полученными двумя разными аналитическими методами. При этом оба аналога обладают недостатками и ограничениями, уже перечисленными нами выше или отмеченными самими авторами в тексте патентов, что серьёзно сужает область применения предложенных подходов на практике.
Прототипом данного изобретения является патент РФ, RU 2736503 C1, дата публикации 17.11.2020 [И.В. Кожевников, А.М. Чибиряев, О.Н. Мартьянов, Способ безводородного гидрирования сульфатного скипидара в проточном режиме]. В этом изобретении впервые были решены две важные технические задачи: 1) проведение безводородного гидрирования ненасыщенных углеводородов сульфатного скипидара в проточном режиме; 2) осуществление онлайн-контроля конверсии скипидара методом ИК-спектроскопии. Для решения второй задачи применялось ИК-детектирование для определения количеств изопропанола и ацетона в реакционной смеси, через которые оценивалась конверсия исходных ненасыщенных углеводородов скипидара.
Заявляемое изобретение решает задачу онлайн-контроля количественного определения конверсии ненасыщенных ароматических и алициклических углеводородов и выхода продуктов их гидрирования непосредственно в процессе химической реакции.
Принципиальным отличием предлагаемого технического решения от ранее применённых в изобретениях-аналогах является отказ от обязательного ИК-детектирования С=С связей исходных ненасыщенных органических соединений. Причина – в пониженной чувствительности ИК-спектроскопии ближней и средней области ИК-диапазона к С=С связям олефинов. Для решения проблемы количественной оценки конверсии ненасыщенных органических соединений и выхода продуктов их гидрирования предложено техническое решение по ИК-детектированию сопутствующих химических компонентов реакционной смеси, а именно – изопропанола и ацетона, образующегося из изопропанола в ходе безводородного гидрирования. Основная идея заключается в том, что при реализации безводородного гидрирования ненасыщенных углеводородов точное определение концентраций изопропанола и ацетона в реакционной смеси, полученное методом ИК - спектроскопии, позволяет точно рассчитать количество водорода, выделившегося при дегидрировании изопропанола и поглощённого затем продуктами гидрирования. Это даёт возможность оценить как конверсию исходного углеводорода, так и выход продукта (или продуктов) гидрирования.
Важным отличием заявляемого способа является проведение его в онлайн-режиме - отбор проб реакционной смеси в ИК-кювете (кювете для инфракрасной спектроскопии) без остановки реакционного процесса для определения не только конверсии исходных углеводородов, но и выхода продуктов их гидрирования. При этом отбор проб возможно производить как в проточной ИК-кювете так и в жидкостных ИК-кюветах (ГОСТ Р 57939—2017).
Техническим результатом заявляемого изобретения является возможность количественного определения одновременно конверсии ненасыщенных углеводородов (ароматических и/или алифатических) и выхода продуктов их гидрирования в реакции безводородного гидрирования с использованием изопропанола без остановки реакционного процесса методом ИК-Фурье спектроскопии. Таким образом достигнуто снижение технических требований к применяемому оборудованию и упрощение всей процедуры количественного анализа реакционной смеси.
Предложенный метод количественного определения с помощью ИК - спектроскопии даёт точные значения концентрации не только для превращений алициклических олефинов, но и ароматических углеводородов.
Кроме того, предложенным способом осуществляется экспресс-определение концентрации ацетона в растворах изопропанола, что может быть использовано для решения других подобных задач, например, при каталитическом дегидрировании изопропанола целевым образом в ацетон [R. Yamaguchi, K.-i. Fujita, Dehydrogenation catalyst, and carbonyl compound and hydrogen production method using said catalyst, PCT WO 2013125712 A1, 2013].
Способ количественного определения конверсии ненасыщенных ароматических и алициклических углеводородов и продуктов их гидрирования в реакции безводородного гидрирования с использованием изопропанола, характеризуется тем, что определение конверсии и конечного выхода продукта проводят методом ИК-Фурье спектроскопии без остановки реакционного процесса в кювете для инфракрасной спектроскопии с проведением трёхкратного инфракрасного сканирования реакционной смеси и определением в ней количественного содержания изопропанола и образующегося в процессе реакции ацетона в интервале частот спектра 1640–1810 см–1 для ацетона и 798–833 см–1 для изопропанола.
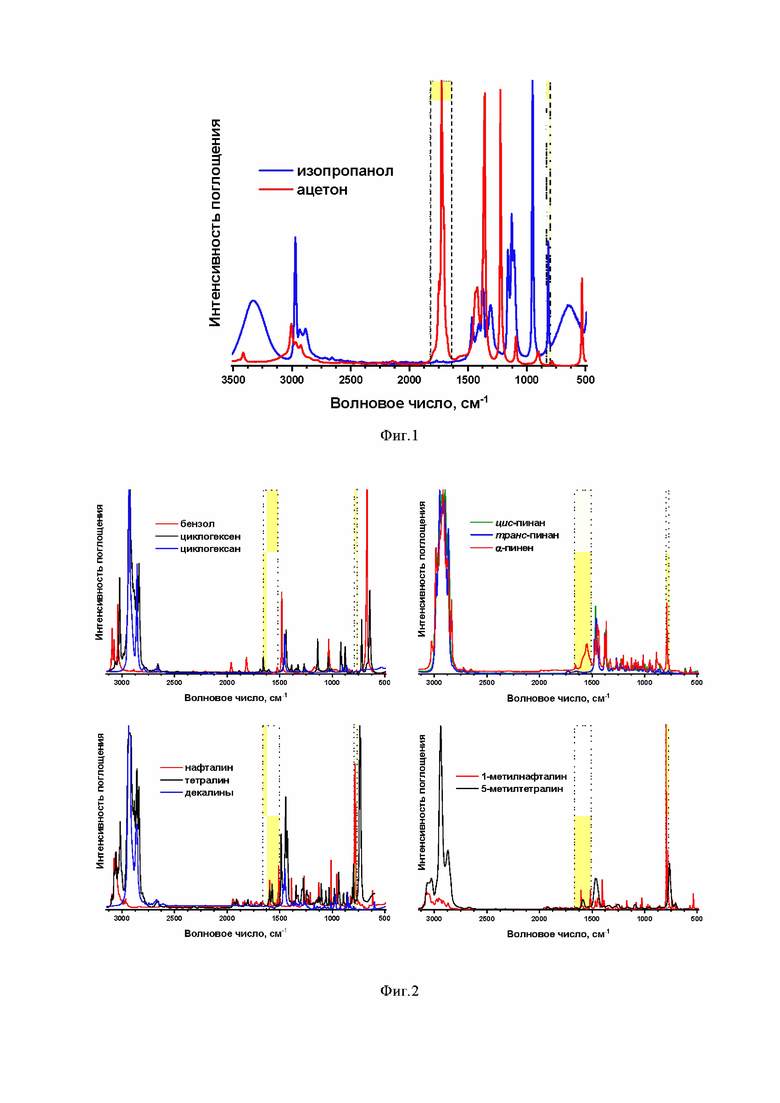
Фиг.1 ИК-спектры изопропанола (синий) и ацетона (красный) в чистом виде. Жёлтым выделены выбранные диапазоны для количественных оценок;
Фиг.2 ИК-спектры исходных ненасыщенных углеводородов и продуктов их гидрирования. Жёлтым выделены выбранные диапазоны ИК-спектров изопропанола и ацетона;
Фиг.3 Калибровочная кривая для растворов ацетона в изопропаноле.
Одним из технических решений, обеспечивающим достижение технического результата данного изобретения, является выбор детектируемой характеристической полосы поглощения компонентов реакционной смеси – изопропанола и ацетона: диапазоны частот 798–833 см–1 для изопропанола и 1640–1810 см–1 для ацетона (Фиг. 1). Только эти диапазоны являются практически незатронутыми характеристическими полосами поглощения субстратов и продуктов, т.е. в этих диапазонах ИК-спектра не наблюдается пересечений (наложений) полос (Фиг. 2), что значительно повышает точность ИК-измерений. Кроме того, сканирование только этих двух узких участков среднего ИК-диапазона значительно сокращает время регистрации и анализа ИК-спектра смеси изопропанола и ацетона, ускоряя весь процесс количественных измерений.
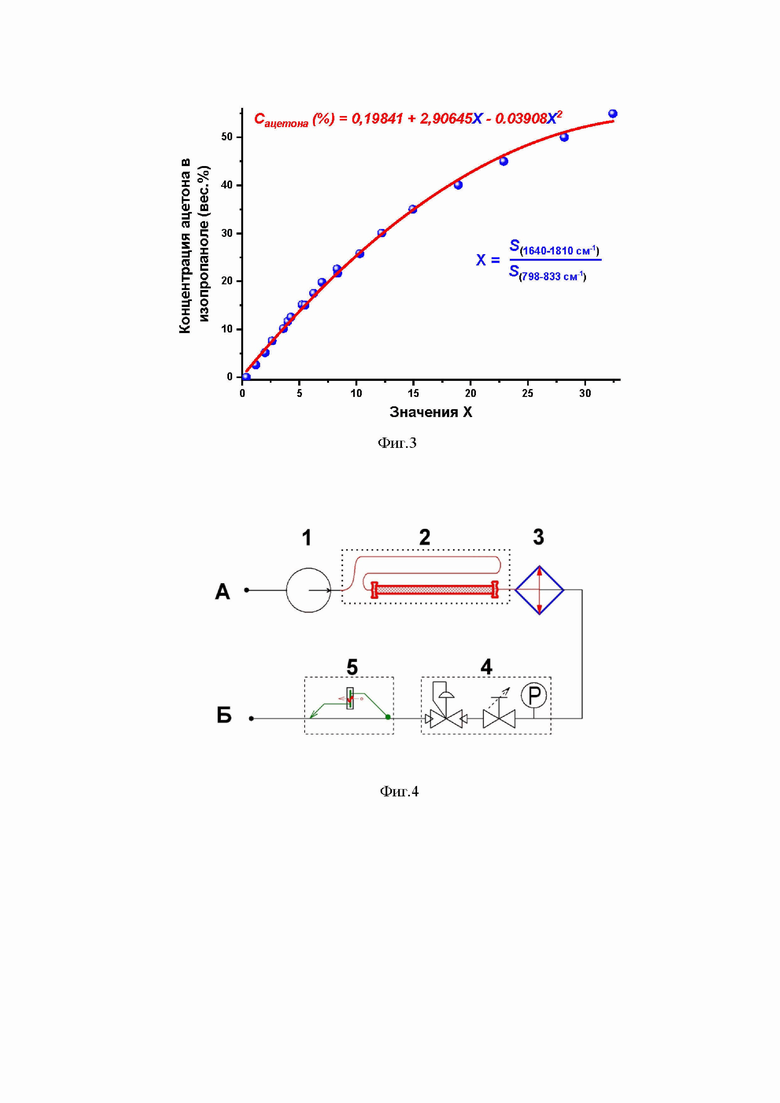
Для количественного определения концентрации ацетона в изопропаноле методом ИК-Фурье-спектроскопии построена калибровочная кривая, которая демонстрирует строгую корреляцию между количественным содержанием ацетона в растворе и количественным соотношением площадей выбранных полос поглощения ацетона и изопропанола в ИК-спектре. Для этих целей гравиметрическим способом была приготовлена серия калибровочных растворов с диапазоном концентраций от 1 до 55 вес.% ацетона в изопропаноле ( Фиг.3). Для каждого из этих растворов трёхкратно был записан ИК-спектр, из которых рассчитаны значения Х – усреднённые численные отношения площадей
S1640–1810/S798–833. Полученные экспериментальные данные были аппроксимированы функцией квадратичного полинома: Сацетона(вес.%) = 0.19841 + 2.90645Х – 0.03908Х2 (Фиг.3). Далее эта калибровочная кривая и функция были использованы для определения концентрации ацетона и изопропанола в реальных реакционных растворах.
Для демонстрации универсальности предложенного подхода расширен круг исходных ненасыщенных углеводородов для реакции безводородного гидрирования, и на примерах превращений алициклических олефинов (α-пинена и циклогексена) и ароматических углеводородов (нафталина и 1-метилнафталина) показано успешное применение предложенного способа.
Для достижения указанного технического результата важным условием является или обеспечение непрерывности процесса ИК-детектирования реакционной смеси, или возможность быстрого отбора жидкой реакционной пробы и последующей регистрации её ИК-спектра. Оба этих условия связаны с высокой скоростью выбранных реакций безводородного гидрирования, что требует короткого времени контакта реакционной смеси со слоем катализатора. Время контакта для выбранного типа реактора, как известно, зависит от объёмной скорости подачи реакционной смеси в реактор (варьировалась от 1,0 мл/мин для реакции 1-метилнафталина до 2,5 мл/мин для реакции нафталина) и объёма каталитической зоны (~6 мл). Поэтому условные времена контакта у нас изменялись от ~2,5 минут для реакции нафталина до ~6 минут для реакции 1-метилнафталина. Выполнение упомянутого условия ИК-детектирования в онлайн-режиме достигнуто за счёт применения реактора проточного типа и проточной ИК-кюветы, что продемонстрировано в Примерах 1, 3, 5 и 7. Достаточность для достижения заявленного технического результата быстрого отбора пробы и быстрой же регистрации ИК-спектра продемонстрирована в Примерах 2, 4, 6 и 8, поскольку использованный проточный реактор и жидкостная ИК-кювета позволяли осуществлять необходимый отбор проб реакционной смеси без остановки химического процесса. В результате этого временной интервал от начала отбора пробы до окончания регистрации её ИК-спектра не превышал 4 минуты, что оказалось вполне достаточным для успешной реализации предложенного способа.
Для демонстрации основного технического результата изобретения проведены реакции безводородного гидрирования двух ароматических (нафталин и 1-метилнафталин) и двух алициклических (α-пинен и циклогексен) ненасыщенных углеводородов при использовании изопропанола в качестве донора водорода и растворителя одновременно. В соответствие со способом, изложенным в патенте-прототипе (RU 2736503C1), раствор ненасыщенного углеводорода в изопропаноле, предварительно нагретый до 120°С (α-пинен, нафталин, 1-метилнафталин) или 80°С (циклогексен), поступает в нагретый до такой же температуры трубчатый реактор с каталитическим картриджем при реакционном давлении от 27 до 29 атм. Химическая суть процесса заключается в следующем: изопропанол на катализаторе дегидрировался до ацетона, а выделяющийся водород оставался адсорбированным на металлическом катализаторе и участвовал в восстановлении (гидрировании) С=С связи молекулы ненасыщенного углеводорода. Далее реакционная смесь, после предварительного охлаждения до 35°С и декомпрессии, поступала в ИК-кювету, где регистрировался её ИК-спектр. Одновременно с регистрацией ИК-спектра производился отбор проб реакционной смеси для проведения параллельного количественного анализа методом ГХ–МС (газовая хроматомасс-спектрометрия). ГХ–МС анализ применялся в качестве альтернативного метода для подтверждения правильности количественных оценок, полученных предложенным методом ИК-Фурье спектроскопии, т.е. с помощью ГХ–МС производилась верификация результатов, полученных при ИК-детектировании. Для этого в ходе анализа оценивалась интегральная площадь хроматографических пиков суммарного ионного тока относительно пиков внутреннего стандарта додекана с использованием в расчётах коррелирующих коэффициентов чувствительности, полученных для калибровочных растворов разной концентрации. ГХ–МС анализ дал количественные характеристики наблюдаемой химической реакции – конверсии и выхода продуктов гидрирования, после чего количественные оценки двух методов сравнивались между собой с последующим заключением о совпадении результатов анализа. Таким образом, на примере безводородного гидрирования нафталина (Примеры 1–2, Таблица 1), 1-метилнафталина (Примеры 3–4, Таблица 2), α-пинена (Примеры 5–6, Таблица 3) и циклогексена (Примеры 7–8, Таблица 4) успешно реализован предложенный нами метод количественных оценок конверсии и выхода продуктов гидрирования с помощью ИК-Фурье-спектроскопии.
На Фиг.4 представлена схема реакционной установки. А – ёмкость с раствором исходного ненасыщенного углеводорода в изопропаноле; (1) насос непрерывного действия, снабжённый датчиком давления; (2) металлический трубчатый реактор, состоящий из петли предварительного нагрева и реакционного каталитического картриджа, помещённые в нагревательную печь или баню; (3) теплообменник для быстрого охлаждения реакционного потока; (4) блок регулировки давления проточной системы, состоящий из последовательно соединённых игольчатого крана, поршневого крана обратного давления и манометра; (5) ИК-кювета ИК–Фурье спектрометра; Б – ёмкость-сборник реакционной смеси.
Пример 1.
Раствор нафталина в изопропаноле (5 вес.%) подавался насосом (1, Фиг. 4) в термостатированный при 120 ± 2°С трубчатый реактор (2). Подача осуществлялась с объёмной скоростью 2,5 мл/мин при давлении 27–29 атм. Количество использованного свежеприготовленного никеля Ренея для катализа безводородного гидрирования нафталина – 2,70 г.
Для количественного определения концентраций ацетона и изопропанола методом ИК-Фурье спектроскопии реакционная смесь подавалась в проточную ИК-кювету, в которой спектр записывался трёхкратно для каждого образца с разрешением 4 см–1, площади полос поглощения ИК-спектров интегрировались в диапазонах 1640–1810 см–1 и 798–833 см–1, а результат численного отношения площадей S1640–1810/S798–833 усреднялся по всем трём измерениям для каждого образца. Количественное содержание ацетона и изопропанола в реакционных смесях вычислялось согласно экспериментальной формуле Сацетона(вес.%) = 0.19841 + 2.90645Х – 0.03908Х2.
Альтернативным методом ГХ–МС анализа было показано, что в ходе безводородного гидрирования нафталина с помощью изопропанола при 100%-ной конверсии образовались тетралин и цис-/транс-декалины, как и в случае традиционного гидрирования нафталина газообразным Н2 (Таблица 1), т.е. водород, выделившийся из изопропанола, не расходовался на образование других продуктов.
Из сравнительных данных Таблицы 1 видно, что в реакции безводородного гидрирования нафталина наблюдалась высокая корреляция между количественными данными, полученными двумя аналитическими методами – ИК-Фурье спектроскопией и газовой хроматомасс-спектрометрией. Отношение количества продуктов гидрирования нафталина, установленного методом ГХ–МС, к количеству образовавшегося ацетона, определённому методом ИК-Фурье спектроскопии, варьировалось в интервале величин 0.930–0.985. Таким образом, отклонение в экспериментальных данных не превышало 7,0 отн.% при определении массовой доли продуктов этими двумя аналитическими методами. Это означает, что концентрация образовавшегося ацетона в реакционной смеси коррелировала с высокой точностью не только с конверсией исходного нафталина, но и с количеством образованных продуктов гидрирования, т.е. количественное определение концентрации ацетона методом ИК-Фурье спектроскопии является достаточным для определения конверсии нафталина и выхода продуктов его гидрирования в реакции безводородного гидрирования с использованием изопропанола.
Таким образом, Пример 1 демонстрирует успешное достижение основного технического результата – точное количественное определение конверсии нафталина и выхода продуктов его гидрирования (тетралина и декалинов) методом ИК-спектроскопии в ИК-кювете проточного типа без общей остановки процесса реакции.
Таблица 1. Безводородное гидрирование нафталина изопропанолом. Условия: 120°С, 27–29 атм, объёмная скорость подачи реагентов – 2,5 мл/мин, исходная концентрация нафталина в изопропаноле – 5 вес.%.
ГХ–МС
mтетралин×100/(mостаток нафталина + ∑mостальные продукты)
mдекалины×100/(mостаток нафталина + ∑mостальные продукты)
ИК-Фурье спектроскопии
Пример 2.
Данный Пример также относится к безводородному гидрированию нафталина с помощью изопропанола, но демонстрирует возможность ИК-детектирования реакционной смеси без использования проточной ИК-кюветы, а применяя жидкостную ИК-кювету. Технически процесс проводился аналогично Примеру 1.
Для количественного определения текущих концентраций ацетона и изопропанола методом ИК-Фурье спектроскопии, по ходу реакции производился периодический отбор проб реакционной смеси объёмом 0,2–0,4 мл. Отбор осуществлялся после прохождения реакционным потоком блока регулировки давления (4,Фиг.4). Интервал отбора проб совпадал со временем, указанным в Таблице 1. Каждая отобранная проба сразу же помещалась в жидкостную ИК-кювету, в которой спектр каждого образца в ИК-диапазонах 1640–1810 см–1 и 798–833 см–1 записывался трёхкратно с разрешением 4 см–1. Время, затраченное на отбор пробы и регистрацию ИК-спектра в периодическом режиме, составляло около 4 минут, что близко соответствовало аналогичному времени регистрации ИК-спектра в ИК-кювете проточного типа (Пример 1). Площади полос поглощения ИК-спектров в диапазонах 1640–1810 см–1 и 798–833 см–1 интегрировались, а результат отношения площадей S1640–1810/S798–833 усреднялся по всем трём повторностям для каждого образца. Количественное содержание ацетона и изопропанола в реакционных смесях вычислялось согласно экспериментальной функции Сацетона(вес.%) = 0.19841 + 2.90645Х – 0.03908Х2. Параллельно для каждой пробы был проведён ГХ–МС анализ, результаты которого практически совпали с приведёнными в Таблице 1 по каждому из времён отбора.
В Примере 2 на реакции безводородного гидрирования нафталина показано, что отклонение в соотношении количества продуктов гидрирования, определённого методом ГХ–МС, к количеству образовавшегося ацетона, определённому методом ИК-Фурье спектроскопии, не превышало 7,0 отн.% (Таблица 1). Это означает, что количественные данные ИК-детектирования, полученные в проточной и жидкостной ИК-кюветах, были практически идентичными между собой. Таким образом, Пример 2 демонстрирует успешное достижение основного технического результата – точное количественное определение конверсии нафталина и выхода продуктов его гидрирования (тетралина и декалинов) методом ИК-спектроскопии в жидкостной ИК-кювете без общей остановки процесса реакции.
Пример 3.
В данном Примере достижение основного технического результата было продемонстрировано в реакции безводородного гидрирования 1-метилнафталина с помощью изопропанола. Для этого раствор 1-метилнафталина в изопропаноле (20 вес.%) подавался насосом (1, Фиг. 4) в термостатированный при 120 ± 2°С трубчатый реактор (2). Подача осуществлялась с объёмной скоростью 1,0 мл/мин при давлении 27–29 атм. Количество использованного свежеприготовленного никеля Ренея для катализа безводородного гидрирования – 2,70 г.
Процедура количественных оценок конверсии 1-метилнафталина, выхода продуктов его гидрирования, а также ацетона, образованного из изопропанола, технически была выполнена аналогично Примеру 1, включая применение ГХ–МС метода в качестве сравнительного количественного анализа. ИК-детектирование проводилось в проточной ИК-кювете без остановки процесса реакции путём трёхкратной регистрации спектра в ИК-диапазонах 1640–1810 см–1 и 798–833 см–1 с разрешением 4 см–1. Изменения в процедуре касались только сокращения общего числа отобранных и проанализированных проб, что было связано с ускоренной дезактивацией катализатора в этой реакции (если сравнивать с реакцией нафталина) и, как следствие, с недостижением 100%-ной конверсии исходного 1-метилнафталина ни в одной из проб.
Альтернативным методом ГХ–МС анализа было определено в реакционной смеси количество 1-метил- и 5-метилтетралинов – ожидаемых [Ch. Liu, Z. Rong, Zh. Sun, Y. Wang, W. Du, Y. Wang, L. Lu, Quenched skeletal Ni as the effective catalyst for selective partial hydrogenation of polycyclic aromatic hydrocarbons. RSC Adv., 2013, 3 (46), 23984–23988. DOI:10.1039/c3ra44871a] изомерных продуктов безводородного гидрирования 1-метилнафталина с помощью изопропанола (Таблица 2).
Таблица 2. Безводородное гидрирование 1-метилнафталина изопропанолом. Условия: 120°С, 27–29 атм, объёмная скорость подачи реагентов – 1,0 мл/мин, исходная концентрация 1-метилнафталина в изопропаноле – 20 вес.%.
ГХ–МС
m1-метилтетралин×100/(mостаток 1-метилнафталина+ ∑mостальные продукты)
M5-метилтетралин×100/(mостаток 1-метилнафталина+ ∑mостальные продукты)
ИК-Фурье спектроскопии
ацетона к изопропанолу, вес.%
Из данных Таблицы 2 видно, что и в условиях неполной конверсии 1-метилнафталина наблюдалась высокая корреляция между сравнительными количественными данными, полученными двумя разными методами – ИК-Фурье спектроскопии и ГХ–МС: отношение количества продуктов гидрирования 1-метилнафталина, полученного из данных ГХ–МС анализа, к количеству образовавшегося ацетона, определённому методом ИК-Фурье спектроскопии, варьировалось в интервале величин 0.952–1.049. Таким образом, отклонение в этих экспериментальных данных не превышало 4,9 отн.% (Таблица 2) при определении массовой доли продуктов двумя аналитическими методами. Это означает, что концентрация образовавшегося ацетона в реакционной смеси коррелировала с высокой точностью и с конверсией исходного 1-метилнафталина, и с количеством образованных продуктов гидрирования, т.е. количественное определение концентрации ацетона методом ИК-Фурье спектроскопии является достаточным для определения конверсии 1-метилнафталина и выхода продуктов его гидрирования в реакции безводородного гидрирования с использованием изопропанола.
Пример 4.
Данный Пример относится к безводородному гидрированию 1-метилнафталина с помощью изопропанола и демонстрирует возможность ИК-детектирования реакционной смеси без использования проточной ИК-кюветы, а применяя жидкостную ИК-кювету. Технически процесс проводился аналогично Примеру 3.
Для количественного определения текущих концентраций ацетона и изопропанола методом ИК-Фурье спектроскопии по ходу реакции производился периодический отбор проб реакционной смеси объёмом 0,2–0,4 мл. Отбор осуществлялся после прохождения реакционным потоком блока регулировки давления (4, Фиг.4). Интервал отбора проб совпадал со временем, указанным в Таблице 2. Каждая отобранная проба сразу же помещалась в жидкостную ИК-кювету, в которой спектр каждого образца в ИК-диапазонах 1640–1810 см–1 и 798–833 см–1 записывался трёхкратно с разрешением 4 см–1. Время, затраченное на отбор пробы и регистрацию ИК-спектра в периодическом режиме, составляло около 4 минут, что близко соответствовало аналогичному времени регистрации ИК-спектра в ИК-кювете проточного типа (Пример 3). Площади полос поглощения ИК-спектров в диапазонах 1640–1810 см–1 и 798–833 см–1 интегрировались, а результат отношения площадей S1640–1810/S798–833 усреднялся по всем трём повторностям для каждого образца. Количественное содержание ацетона и изопропанола в реакционных смесях вычислялось согласно экспериментальной функции Сацетона(вес.%) = 0.19841 + 2.90645Х – 0.03908Х2. Параллельно для каждой пробы был проведён ГХ–МС анализ, результаты которого практически совпали с приведёнными в Таблице 2 по каждому из времён отбора.
В Примере 4 на реакции безводородного гидрирования 1-метилнафталина показано, что отклонение в соотношении количества продуктов гидрирования, определённого методом ГХ–МС, к количеству образовавшегося ацетона, определённому методом ИК-Фурье спектроскопии, не превышало 4,8 отн.% (Таблица 2). Это означает, что количественные данные ИК-детектирования, полученные в проточной и жидкостной ИК-кюветах, были практически идентичными. Таким образом, Пример 4 демонстрирует успешное достижение основного технического результата – точное количественное определение конверсии 1-метилнафталина и выхода продуктов его гидрирования (1-метил- и 5-метилтетралинов) методом ИК-спектроскопии в жидкостной ИК-кювете без общей остановки процесса реакции.
Пример 5
В данном Примере достижение основного технического результата продемонстрировано в реакции безводородного гидрирования α-пинена с помощью изопропанола. Для этого раствор α-пинена в изопропаноле (20 вес.%) подавался насосом (1, Фиг. 4) в термостатированный при 120 ± 2°С трубчатый реактор (2). Подача осуществлялась с объёмной скоростью 2,0 мл/мин при давлении 27–29 атм. Количество использованного свежеприготовленного никеля Ренея для катализа безводородного гидрирования – 2,70 г.
Процедура количественных оценок конверсии α-пинена, выхода продуктов его гидрирования, а также ацетона, образованного из изопропанола, технически была выполнена аналогично Примеру 1, включая применение ГХ–МС метода в качестве сравнительного количественного анализа. ИК-детектирование проводилось в проточной ИК-кювете без остановки процесса реакции путём трёхкратной регистрации спектра в ИК-диапазонах 1640–1810 см–1 и 798–833 см–1 с разрешением 4 см–1. Изменения в процедуре касались только сокращения общего числа отобранных и проанализированных проб.
Альтернативным методом ГХ–МС анализа определено в реакционной смеси количество цис-/транс-пинанов – продуктов безводородного гидрирования α-пинена с помощью изопропанола (Таблица 3).
Из данных Таблицы 3 видно, что для реакции безводородного гидрирования α-пинена с использованием изопропанола наблюдалась самая высокая (из всех изученных нами реакций) корреляция между сравнительными аналитическими данными, полученными методами ГХ–МС и ИК-Фурье спектроскопии. Отношение количества гидрированных продуктов, полученного из данных ГХ–МС, к количеству образовавшегося ацетона, определённому методом ИК-Фурье спектроскопии, варьировалось в интервале величин 0.987–1.006. Таким образом, отклонение между экспериментальными данными не превышало 1,1 отн.% (Таблица 3) при определении массовой доли продуктов этими двумя аналитическими методами. Это означает, что концентрация образовавшегося ацетона в реакционной смеси коррелировала с высокой точностью и с конверсией исходного α-пинена, и с количеством образованных продуктов гидрирования (цис-/транс-пинанов), т.е. количественное определение концентрации ацетона методом ИК-Фурье спектроскопии является достаточным для определения конверсии α-пинена и выхода продуктов его гидрирования в реакции безводородного гидрирования с использованием изопропанола.
Таблица 3. Безводородное гидрирование α-пинена изопропанолом. Условия: 120°С, 27–29 атм, объёмная скорость подачи реагентов – 2,0 мл/мин, исходная концентрация α-пинена в изопропаноле – 20 вес.%.
ИК-Фурье спектроскопии
ГХ–МС, к количеству образовавшегося ацетона, определённому методом ИК-Фурье спектроскопии
Пример 6.
Данный Пример относится к безводородному гидрированию α-пинена с помощью изопропанола и демонстрирует возможность ИК-детектирования реакционной смеси без использования проточной ИК-кюветы, а применяя только жидкостную ИК-кювету. Технически процесс проводился аналогично Примеру 5.
Для количественного определения текущих концентраций ацетона и изопропанола методом ИК-Фурье спектроскопии по ходу реакции производился периодический отбор проб реакционной смеси объёмом 0,2–0,4 мл. Отбор осуществлялся после прохождения реакционным потоком блока регулировки давления (4, Фиг.4). Интервал отбора проб совпадал со временем, указанным в Таблице 3. Каждая отобранная проба сразу же помещалась в жидкостную ИК-кювету, в которой спектр каждого образца в ИК-диапазонах 1640–1810 см–1 и 798–833 см–1 записывался трёхкратно с разрешением 4 см–1. Время, затраченное на отбор пробы и регистрацию ИК-спектра в периодическом режиме, составляло около 4 минут, что близко соответствовало аналогичному времени регистрации ИК-спектра в ИК-кювете проточного типа (Пример 5). Площади полос поглощения ИК-спектров в диапазонах 1640–1810 см–1 и 798–833 см–1 интегрировались, а результат отношения площадей S1640–1810/S798–833 усреднялся по всем трём повторностям для каждого образца. Количественное содержание ацетона и изопропанола в реакционных смесях вычислялось согласно экспериментальной функции Сацетона(вес.%) = 0.19841 + 2.90645Х – 0.03908Х2. Параллельно для каждой пробы был проведён ГХ–МС анализ, результаты которого практически совпали с приведёнными в Таблице 3 по каждому из времён отбора.
В Примере 6 на реакции безводородного гидрирования α-пинена показано, что отклонение в соотношении количества продуктов гидрирования, определённого методом ГХ–МС, к количеству образовавшегося ацетона, определённому методом ИК-Фурье спектроскопии, не превышало 1,1 отн.% (Таблица 3). Это означает, что количественные данные ИК-детектирования, полученные в проточной и жидкостной ИК-кюветах, были практически идентичными. Таким образом, Пример 6 демонстрирует успешное достижение основного технического результата – точное количественное определение конверсии α-пинена и выхода продуктов его гидрирования (цис-/транс-пинанов) методом ИК-спектроскопии в жидкостной ИК-кювете без общей остановки процесса реакции.
Пример 7.
В данном Примере достижение основного технического результата продемонстрировано в реакции безводородного гидрирования циклогексена с помощью изопропанола. Для этого раствор циклогексена в изопропаноле (25 вес.%) подавался насосом (1, Фиг. 4) в термостатированный при 80 ± 2°С трубчатый реактор (2). Подача осуществлялась с объёмной скоростью 2,0 мл/мин при давлении 27–29 атм. Количество использованного свежеприготовленного никеля Ренея для катализа безводородного гидрирования – 2,70 г.
Для демонстрации основного технического результата на примере реакции безводородного гидрирования циклогексена авторам потребовалось решение серьёзной технической задачи – исключить влияние побочной реакции на конечный результат. Как уже отмечалось нами ранее, количество ацетона в реакционной смеси равно количеству водорода, образованного из изопропанола и поглощённого затем продуктами безводородного гидрирования. Поэтому количество ацетона в реакционной смеси точно коррелировало с конверсией и выходом продуктов гидрирования в Примерах 1–6. В реакции безводородного гидрирования циклогексена основным продуктом являлся циклогексан, а побочным процессом являлось каталитическое диспропорционирование циклогексена на металлическом никеле, в результате которого из трёх молекул циклогексена образуются две молекулы циклогексана и одна молекула бензола [Y. Shu, L.E. Murillo, J.P. Bosco, W. Huang, A.I. Frenkel, J.G. Chen, The effect of impregnation sequence on the hydrogenation activity and selectivity of supported Pt/Ni bimetallic catalysts. Appl. Catal. A: Gen., 2008, 339 (2), 169–179. DOI: 10.1016/j.apcata.2008.01.024]. Это означало, что и в основной реакции циклогексена, и в побочном процессе образовывался один и тот же главный продукт – циклогексан. Предложенный авторами способ определения концентрации ацетона методом ИК-Фурье спектроскопии даёт оценку количества лишь того циклогексана, который образовался в результате безводородного гидрирования циклогексена, но не в результате диспропорционирования.
Чтобы уменьшить влияние побочной реакции в образование циклогексана, реакционную температуру безводородного гидрирования циклогексена понизили до 80°С по сравнению с безводородным гидрированием нафталина, 1-метилнафталина и α-пинена [сравни температуры процесса: D.M. Rebhan, V. Haensel, A kinetic and mechanistic study of cyclohexene disproportionation: an example of irreversible hydrogen transfer. J. Catal., 1988, 111 (2), 397–408. DOI: 10.1016/0021-9517(88)90098-X]. Однако такой приём не позволил полностью подавить диспропорционирование циклогексена, поэтому главной задачей, решение которой обеспечивало авторам достижение основного технического результата, стало точное определение количества циклогексана, образованного в результате диспропорционирования. Эта задача была успешно решена с помощью метода ГХ–МС через определение количества бензола в реакционной смеси (Таблица 4). После этого стало возможным продемонстрировать достижение основного технического результата.
Далее процедура количественных оценок конверсии циклогексена в результате безводородного гидрирования, выхода продукта его гидрирования, а также ацетона, образованного из изопропанола, технически была выполнена аналогично Примеру 1. ИК-детектирование проводилось в проточной ИК-кювете без остановки процесса реакции путём трёхкратной регистрации спектра в ИК-диапазонах 1640–1810 см–1 и 798–833 см–1 с разрешением 4 см–1. Как уже отмечалось, изменения в процедуре касались не только сокращения общего числа отобранных и проанализированных проб, но и температуры реакции.
Из данных Таблицы 4 видно, что для реакции безводородного гидрирования циклогексена с использованием изопропанола наблюдалась высокая корреляция между сравнительными аналитическими данными, полученными методами ГХ–МС и ИК-Фурье спектроскопии. Отклонение между количеством циклогексана, образованного в результате безводородного гидрирования циклогексена, и количеством образовавшегося ацетона, определённого методом ИК-Фурье спектроскопии, не превышало 4,8 отн.% (Таблица 4), что свидетельствует об успешном решении авторами сопутствующей задачи разделения всего циклогексана, образовавшегося в реакционной смеси, на «гидрированный» и «диспропорционированный» продукт. Так, предложенный способ позволил надёжно установить, что количество ацетона в реакционной смеси за 3 часа процесса в проточном режиме снижается с 14,5 вес.% до 11,6 вес.%, что соответствует снижению количества циклогексана, образованного в результате безводородного гидрирования, с 59,6 вес.% до 48,3 вес.% (Таблица 4). Комбинированием этого результата с количественными данными ГХ–МС анализа было показано, что вклад процесса диспропорционирования в общую конверсию циклогексена возрастает с 32% для 15 минут до 42% для 3 часов реакции (Таблица 4). Поэтому авторы констатируют, что, в рамках демонстрации основного технического результата, предложенный способ с применением ИК-Фурье спектроскопии позволяет успешно решать другие сложные научные задачи, такие как разделение вкладов основной и побочной реакции.
Таблица 4. Безводородное гидрирование циклогексена изопропанолом. Условия: 80°С, 27–29 атм, объёмная скорость подачи реагентов – 2,0 мл/мин, исходная концентрация циклогексена в изопропаноле – 25 вес.%.
ГХ–МС
(рассчитано из количества бензола)
(рассчитано с учётом образованного бензола)
к изопропанолу, вес.%
(по ацетону, ИК-Фурье) и поглощённого водорода
(по гидрированному продукту, ГХ–МС), отн.%
Пример 8.
Данный Пример относится к безводородному гидрированию циклогексена с помощью изопропанола и демонстрирует возможность ИК-детектирования реакционной смеси без использования ИК-кюветы проточного типа, а применяя только жидкостную ИК-кювету. Технически процесс проводился аналогично Примеру 7.
Для количественного определения текущих концентраций ацетона и изопропанола методом ИК-Фурье спектроскопии по ходу реакции производился периодический отбор проб реакционной смеси объёмом 0,2–0,4 мл. Отбор осуществлялся после прохождения реакционным потоком блока регулировки давления (4, Фиг. 4). Интервал отбора проб совпадал со временем, указанным в Таблице 4.
Каждая отобранная проба сразу же помещалась в жидкостную ИК-кювету, в которой спектр каждого образца в ИК-диапазонах 1640–1810 см–1 и 798–833 см–1 регистрировался трёхкратно с разрешением 4 см–1. Время, затраченное на отбор пробы и регистрацию ИК-спектра в периодическом режиме, составляло около 4 минут, что близко соответствовало аналогичному времени регистрации ИК-спектра в проточной ИК-кювете (см. Пример 7). Площади полос поглощения ИК-спектров в диапазонах 1640–1810 см–1 и 798–833 см–1 интегрировались, а результат отношения площадей S1640–1810/S798–833 усреднялся по всем трём повторностям для каждого образца. Количественное содержание ацетона и изопропанола в реакционных смесях вычислялось согласно экспериментальной функции Сацетона(вес.%) = 0.19841 + 2.90645Х – 0.03908Х2. Параллельно для каждой пробы был проведён ГХ–МС анализ, результаты которого практически совпали с приведёнными в Таблице 4 по каждому из времён отбора.
В Примере 8 на реакции безводородного гидрирования циклогексена показано, что отклонение в соотношении количества циклогексана, определённого методом ГХ–МС, к количеству образовавшегося ацетона, определённому методом ИК-Фурье спектроскопии, не превышало 4,7 отн.% (Таблица 4). Это означает, что количественные данные ИК-детектирования, полученные в проточной и жидкостной ИК-кюветах, были практически идентичными. Таким образом, Пример 8 демонстрирует успешное достижение основного технического результата – точное количественное определение конверсии циклогексена через реакцию безводородного гидрирования (в отличие от диспропорционирования) и выхода циклогексана методом ИК-спектроскопии в жидкостной ИК-кювете без общей остановки процесса реакции.
Таким образом, на Примерах 1–6 в выбранных реакционных условиях безводородного гидрирования ненасыщенных углеводородов нафталина, 1-метилнафталина и α-пинена с помощью изопропанола продемонстрировано, что ИК-детектирование в проточной и жидкостной ИК-кюветах в диапазоне частот 798–833 см–1 и 1640–1810 см–1 позволяет точно определять конверсию исходных ненасыщенных углеводородов и количество образовавшихся гидрированных продуктов без остановки реакционного процесса и без привлечения дополнительных аналитических методов. Применение ИК-Фурье спектроскопии в указанном диапазоне ИК-частот нацелено на определение количественного соотношения изопропанол/ацетон в соответствии с экспериментальной зависимостью, описываемой следующей функцией: Сацетона(вес.%) = 0.19841 + 2.90645Х – 0.03908Х2, где Х – это усреднённые численные отношения площадей S1640–1810/S798–833. Представленная функция зависимости концентрации ацетона (Сацетона) в изопропаноле от Х точно описывает количественные характеристики растворов ацетон–изопропанол, как минимум, в интервале концентраций ацетона от 1 до 55 весовых процента.
В рамках демонстрации основного технического результата на Примерах 7–8 для реакции безводородного гидрирования циклогексена с помощью изопропанола показано, что ИК-детектирование в проточной и жидкостной ИК-кюветах, обеспечивая точное определение концентрации ацетона и изопропанола, позволяет точно установить количество циклогексана, образованного именно по основному маршруту безводородного гидрирования циклогексена. С помощью предложенного способа и с привлечением количественных данных ГХ–МС анализа реакционных смесей можно успешно решать сложные научные задачи, например, разделение вкладов основной и побочной реакции.
Кроме того, на Примерах 1–8 показано, что предложенный способ может быть успешно применён для экспресс-определения концентрации ацетона в растворах изопропанола, что может быть совсем не связано с реакцией безводородного гидрирования.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ безводородного гидрирования сульфатного скипидара в проточном режиме | 2020 |
|
RU2736503C1 |
| Способ получения анилина из нитробензола | 2024 |
|
RU2834863C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛОГЕКСАНОНА ИЗ БЕНЗОЛА | 2002 |
|
RU2205819C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТИЛ-1,4-НАФТОХИНОНА | 1990 |
|
RU2022958C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ СОСТАВА ГЛИФОСАТСОДЕРЖАЩИХ СМЕСЕЙ | 2022 |
|
RU2787117C1 |
| КАТАЛИЗАТОР И СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА КОНВЕРСИЕЙ УГЛЕВОДОРОДОВ | 2001 |
|
RU2194572C2 |
| Способ получения тетраалкилортосиликатов из кремнезёма | 2019 |
|
RU2698701C1 |
| Способ количественного определения N-(фосфонометил)-глицина и N-(фосфонометил)-иминодиуксусной кислоты | 2021 |
|
RU2775230C1 |
| Способ получения изопропилового спирта | 2023 |
|
RU2813540C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛОГЕКСАНОНА | 2002 |
|
RU2205175C1 |
Изобретение относится к способу количественного определения конверсии ненасыщенных ароматических или алициклических углеводородов и выхода продуктов их гидрирования в реакции безводородного гидрирования с использованием изопропанола. Определение конверсии исходного углеводорода и выхода конечного продукта проводят методом ИК-Фурье спектроскопии без остановки реакционного процесса в кювете для инфракрасной спектроскопии с проведением трёхкратного инфракрасного сканирования реакционной смеси и определением в ней количественного содержания изопропанола и образующегося в процессе реакции ацетона в интервале частот спектра 798–833 см–1 для изопропанола и 1640–1810 см–1 для ацетона по формуле, применимой для интервала концентраций ацетона в изопропаноле от 1 до 55 вес.%: Сацетона(вес.%) = 0.19841 + 2.90645Х – 0.03908Х2, где Х – численное отношение площадей S1640–1810/S798–833, усреднённое по сумме трёх результатов измерений. Технический результат - возможность количественного определения одновременно конверсии ненасыщенных углеводородов (ароматических и/или алифатических) и выхода продуктов их гидрирования в реакции безводородного гидрирования с использованием изопропанола без остановки реакционного процесса методом ИК-Фурье спектроскопии в ИК-кювете, снижение технических требований к применяемому оборудованию и упрощение всей процедуры количественного анализа реакционной смеси. 2 з.п. ф-лы, 4 ил., 4 табл., 8 пр.
1. Способ количественного определения конверсии ненасыщенных ароматических или алициклических углеводородов и выхода продуктов их гидрирования в реакции безводородного гидрирования с использованием изопропанола, характеризующийся тем, что определение конверсии исходного углеводорода и выхода конечного продукта проводят методом ИК-Фурье спектроскопии без остановки реакционного процесса в кювете для инфракрасной спектроскопии с проведением трёхкратного инфракрасного сканирования реакционной смеси и определением в ней количественного содержания изопропанола и образующегося в процессе реакции ацетона в интервале частот спектра 798–833 см–1 для изопропанола и 1640–1810 см–1 для ацетона по формуле, применимой для интервала концентраций ацетона в изопропаноле от 1 до 55 вес.%:
Сацетона(вес.%) = 0.19841 + 2.90645Х – 0.03908Х2, где Х – численное отношение площадей S1640–1810/S798–833, усреднённое по сумме трёх результатов измерений.
2. Способ по п. 1, отличающийся тем, что в качестве алициклических углеводородов выбраны циклогексен и α-пинен.
3. Способ по п. 1, отличающийся тем, что в качестве ароматических углеводородов выбраны нафталин и 1-метилнафталин.
| Способ безводородного гидрирования сульфатного скипидара в проточном режиме | 2020 |
|
RU2736503C1 |
| US 7968652 B2, 28.06.2011 | |||
| US 7329547 B2, 12.02.2008. | |||

