Изобретение относится к органическому синтезу, а именно к усовершенствованному способу получения 2-метил-1,4-нафтохинона (менадиона) формулы (I), обладающего свойствами витамина К, получаемого окислением 4-R-2-метил-1-нафтолов, где R = а) Н, б) СН3.
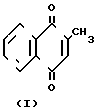

Менадион формулы I широко применяют в медицинской практике, животноводстве и птицеводстве с целью профилактики и лечения ряда заболеваний, он также является промежуточным продуктом в получении водорастворимых кровоостанавливающих препаратов.
Известны три группы способов получения менадиона. Способ 1.1 первой группы основан на конденсации толухиона с дивинилом. Последовательная цепочка окислительно-восстановительных превращений приводит к соединению (I) с 72%-ным выходом
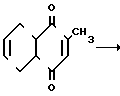
 (I)
(I)
Список принятых обозначений I 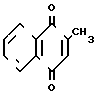 - 2-метил-1,4-нафтохинон IIа
- 2-метил-1,4-нафтохинон IIа  - 2-метил-1-нафтол IIб
- 2-метил-1-нафтол IIб  - 2,4-диметил-1-нафтол III
- 2,4-диметил-1-нафтол III  - 2-метил-1,4-бензохинон(толухинон) IV
- 2-метил-1,4-бензохинон(толухинон) IV  - метиловый эфир 2-метил-1-нафтола V
- метиловый эфир 2-метил-1-нафтола V 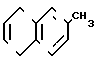 - 2-метилнафталин VI
- 2-метилнафталин VI  - 6-метил-1,4-нафтохинон VII
- 6-метил-1,4-нафтохинон VII 
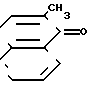 - динафтохинон VIII
- динафтохинон VIII  - 4,4-ди(2-метил-1-нафтол), динафтол IX
- 4,4-ди(2-метил-1-нафтол), динафтол IX  - 2,4-диметил-4-гидроксинафтал-2ен-1-он, гидроксинафталенон Х
- 2,4-диметил-4-гидроксинафтал-2ен-1-он, гидроксинафталенон Х 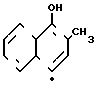 - радикал из частицы (IIа) ГПК - ванадомолибдофосфорная гетерополикислота или ее соль.
- радикал из частицы (IIа) ГПК - ванадомолибдофосфорная гетерополикислота или ее соль.
Состав кислоты описывается общей формулой Н(3+n)PMo(12-n) VnO40, где n варьируется от n = 1 до n = 6 (ГПК-n), например ГПК-3 = H6PMo9V3O40.
Состав солей ГПК описывается формулой
MeaH(3+n-a)PMo(12-n)VnO40 (МеаГПК-n), где Ме = Na+, K+, 1/2 Mg2+, 1/2Ca2+, 1/2Zn2+, например
Na1,8ГПК -3 = Na1,8H4,2PMo9V3O40,
Mg1,2ГПК- 4 = Mg1,2H4,6PMo8V4O40.
HmГПК - восстановленная на m электронов молекула ГПК или ее соль.
Состав HmГПК описывается общей формулой H(3+n+m)PMo(12-n) V(n-m)VVmIVO40. Состав восстановленной на m электронов соли описывается общей формулой MeaH(3+n+m-a) PMo(12-n)-V(n-m)V VmIVO40. Например Н4ГПК - восстановленная на 4 электрона форма ГПК или ее соли.
Способ 1.1 используют в современной промышленности тонкого органического синтеза (ТОС). Однако он не удовлетворяет современным экологическим требованиям, так как приводит к появлению большего количества отходов. Например, в способе 1.1 количество сточных вод в 15 и более раз превышает массу продукта, а количество токсичных отходов - восстановленной формы окислителя (Cr2O3) превышает массу продукта в 25 раз.
Вторая группа способов основана на использовании метилового эфира 2-метил-1-нафтола (IV) (см. список принятых обозначений). В способе 2.1 менадион получают окислением эфира (IV) перекисью водорода в присутствии K3[Fe(CN)6] в водно-уксусном растворе.
В способе 2.2 тот же эфир окисляют в две стадии. На первой стадии его бромируют в растворе дихлорэтана, а на второй стадии - бромпроизводное окисляют в целевой продукт действием азотной кислоты. Основными недостатками способов второй группы являются: невысокий выход целевого продукта в первом случае (61%) и наличие кислых отходов производства - во втором. Отходы вызывают необходимость применения нейтрализующих средств и влекут образование промышленных стоков.
Третья группа способов основана на окислении доступного сырья - 2-метил-нафталина (V). В разновидностях третьего способа в качестве окислителя используют перекись водорода в уксусной кислоте, в том числе с применением катализирующей палладиевой добавки, соединения хрома в кислой среде, в том числе с катализирующей добавкой соли церия, различные соединения церия в присутствии серной кислоты. В этой группе известны также способы электрохимического окисления, отличающиеся низкими выходами целевого продукта (10-35%).
Известен также способ газофазного окисления соединения (V) в присутствии гетерогенного катализатора на основе V2O5, K2S2O7 и K2SO4. По этому способу реакцию ведут при высокой температуре (400оС), что приводит к низкому выходу продукта (10%). Основным общим недостатком всех разновидностей третьей группы способов является низкая избирательность в целевой реакции. Наряду с 2-метил-1,4-нафтохиноном при окислении соединения (V) всегда образуется 6-метил-1,4-нафтохинон (VI) в разных соотношениях к целевому продукту от 3:1 до 6:1 и даже 20:1, в зависимости от катализаторов и окислителей. Присутствие соединения (VI) существенно снижает выход главного продукта при его очистке.
Наиболее близким решением поставленной технической задачи является способ окисления 2-метилнафталина (V) кислородом в присутствии ванадомолибдофосфорных гетерополикислот общей формулы H(3+n)PMo(12-n)VnO40 (ГПК-n), где n = 2, 4, 6. Способ реализуют при 120-140оС под давлением кислорода 3-8 ати в гомогенных водно-уксуснокислых растворах, содержащих 50-90 об.% уксусной кислоты, 0,2-0,8 моль/л серной кислоты, 0,02-0,2 моль/л ГПК-n, а также субстрат (V) в молярном отношении (V) К (ГПК-n) 2-10. Способ обеспечивает частичную конверсию исходного соединения (V) (83% превращения) при селективности до 64%.
Прототип имеет ряд существенных недостатков:
использование довольно дорогого субстрата (V);
неполная конверсия и невысокая селективность, усложняющие выделение целевого продукта;
дорогая технология, обусловленная высокой коррозионной активностью катализатора, повышенным давлением кислорода и повышенными температурами реакции, обусловившими жесткие требования в аппаратуре и соблюдению мер техники безопасности;
проведение реакции в гомогенных условиях (в одной фазе) создавало сложность в отделении продуктов реакции от катализатора и не позволяло полностью исключить появление вредных отходов производства.
Целью изобретения является увеличение выхода целевого продукта и упрощение технологии процесса.
Поставленная цель достигается способом получения 2-метил-1,4-нафтохинона путем окисления кислородом производного нафталина при повышенной температуре в присутствии в качестве катализатора ванадомолибдофосфорной гетерополикислоты общей формулы
H(3+n)PMo(12-n)VnO40,
где n = 1-6, а отличительной особенностью является то, что в качестве производных нафталина используют 2-метил-1-нафтол или его смесь с 2,4-диметил-1-нафтолом, а процесс проводят в двухфазной системе, в которой катализатор входит в состав водной фазы в количестве, соответствующем его концентрации 0,1-0,5 моль/л, а производное нафталина - в состав органической фазы, несмешивающейся с водной фазой, при интенсивном перемешивании фаз при температуре 20-70оС в атмосфере инертного газа в течение 0,5-3 ч с последующим выделением целевого продукта из органической фазы и окислением водной фазы, содержащей восстановленную форму катализатора, кислородом или кислородсодержащим газом. Предпочтительно в качестве катализатора используют кислые соли ванадомолибдофосфорной кислоты общей формулы
MeaH(3+n-a)PMo(12-n)VnO40,
где Me = Na+, K+, 1/2 Mg2+, 1/2Ca2+, 1/2Zn2+;
n = 1-6.
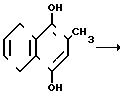
В основе изобретения лежит разработка нового высокоизбирательного способа получения 2-метил-1,4-нафтохинона окислением соединений формулы II, где R = а) Н, б) СН3, либо окислением смеси этих соединений. Последнее важно, так как смеси веществ IIа и IIб оказываются существенно дешевле и доступнее, нежели каждый из этих компонентов в чистом виде. Предполагается, что данный способ может быть положен в основу безотходной технологии синтеза продукта формулы I.
Это достигается применением в стадиях синтеза менадиона ряда рассмотренных ниже новых катализаторов и новых способов их использования.
В данном способе менадион (I) получают из нафтолов (IIа) и (IIб) по суммарным реакциям (2) и (2а), состоящим из этапов (3), (4) и (3а), (4) соответственно: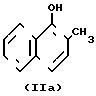 +O2_____→
+O2_____→  +H2O (2)
+H2O (2) +3/2 O2____→
+3/2 O2____→  +CH2O+H2O (2a)
+CH2O+H2O (2a)
(IIa)+ГПК+H2O ____→ (I)+H4ГПК (3)
(IIб)+3/2 ГПК+2H2O ____→ (I)+CH2O+3/2 H4ГПК (3a)
Из стехиометрии суммарных реакций (2) и (2а) видно, что ванадомолибдофосфорная гетерополикислота или ее соль (ГПК), являются окислительными катализаторами превращения. Расходуемым окислителем в суммарном процессе оказывается кислород или кислородсодержащий газ. Последний факт исключает необходимость использования других неорганических окислителей, оказывающихся всегда главным источником вредных отходов производства. Способ проведения каталитических реакций (2) и (2а) методом межфазного катализа предложен впервые. Он позволяет упростить задачу отделения продуктов реакции от катализатора и ликвидировать основные источники возможных экологических загрязнений при реализации процесса. Однако, проведение реакций (2) и (2а) существенно осложняются побочным превращением (5), продуктом которого был хинон (VII)

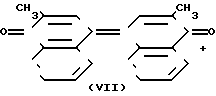 H2O (5)
H2O (5)
Хинон (VII) и соединения, образующиеся при его дальнейшем окислении, серьезно затрудняют выделение и очистку целевого продукта (I).
Превращение (5) протекает через стадии (6), (7) и (4)
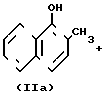


 H2ГПК (6)
H2ГПК (6)
(VIII)+H2ГПК ___→ (VII)+H4ГПК (7)
Соотношение скоростей стадий (3) и (6), определяющее избирательность катализатора в отношении продуктов (I) и (VIII), зависит от состава молекулы ГПК, от метода и условий осуществления реакции (4). Подбором перечисленных параметров удается добиться выгодного отношения скоростей стадий (3) и (6) и этим достичь высокой избирательности катализатора в отношении целевого продукта (I) с перспективами реализации непрерывного процесса. Большим достоинством метода является возможность получения чистого менадиона из смеси нафтолов (IIа) и (IIб). При этом нафтол (IIб) окисляется в две стадии по схеме (8) с промежуточным образованием гидроксинафталенона (IX):
 (8)
(8)
Поскольку формальдегид СН2О в этих условиях может окисляться дальше в муравьиную кислоту по реакции (9), то окисление субстрата (IIб) по реакции (3а) требует вдвое большего количества катализатора, нежели окисление субстрата (IIа) по реакции (3).
Благодаря тому, что удалось подобрать оптимальные условия для полного окисления нафтола (IIб) в целевой продукт (I), предложено использовать для синтеза менадиона либо отдельные нафтолы (IIа) или (IIб), либо их смеси.
Таким образом, в данном способе использованы следующие элементы новизны:
для синтеза менадиона предлагается использовать нафтолы (Iа) или (IIб), или их смеси;
применены новые катализаторы синтеза менадиона;
синтез осуществлен методом межфазного катализа;
найдены оптимальные условия синтеза, обеспечивающие найбольшую избирательность катализатора в отношении реакции (2) и наименьшую - в отношении побочной реакции (5).
Обоснование необходимости использования перечисленных элементов новизны и детализация их сущности;
использование смесей нафтолов (IIа) и (IIб) практически не снижает качества целевого продукта (I). Поэтому оно значительно удешевляет и расширяет сырьевую базу для нового метода синтеза (I), обеспечивает его высокую конкурентноспособность по сравнению со всеми другими известными промышленными методами получения (I) из других видов сырья;
в качестве катализаторов реакции (2) и (3а) используют водные растворы гетерополикислот общей формулы H(3+n)PMo(12-n) VnO40 (ПК-n), где n может меняться от n = 1 до n=6, или водные растворы их кислых солей с катионами Ме, имеющие общую формулу
MeaH(3+n-a)PMo(12-n)VnO40(ГПК-n),
где Me = Na+, K+, 1/2Mg2+, 1/2Ca2+, 1/2Zn2+ и других ГПК-n готовились известными методами, а МеаГПК-n - также известными методами.
Все перечисленные катализаторы впервые применены для окисления соединений (IIа) и (IIб) в (I), а соли этих ГПК-n были вообще впервые применены для окисления ароматических соединений.
Отсутствие добавок уксусной и серной кислот в данном способе, в отличие от прототипа, а также использование солей МеаГПК-n, вместо самих кислот ГПК-n, существенно снижает требования к коррозионной устойчивости применяемой аппаратуры. Невысокие температуры реакции (не выше 50оС) и отсутствие необходимости использования чистого кислорода или кислорода под давлением существенно упрощали и удешевляли технологию по сравнению с прототипом.
Синтез методом межфазного катализатора состоит в проведении реакций (2) и (2а) на границе раздела фаз двух несмешивающихся жидкостей. Одной из таких жидкостей является водный раствор ГПК-n или МеаГПК-n, являющийся катализатором. Второй жидкостью является раствор субстрата в органическом растворителе, не смешивающимся с водой (бензоле, ксилоле, ином растворителе, инертном в отношении ГПК).
Реакции (2) и (2а) начинаются путем включения реакторной мешалки, они протекают на поверхности капелек эмульсии, в которой водная фаза распределена внутри органической фазы. Со стороны органической фазы в стадиях (3) и (3а) участвуют молекулы субстратов (IIа) и (IIб), а со стороны водной фазы - молекулы ГПК и воды. Прекращение перемешивания приводит к быстрому разрушению эмульсии с ее разделением на два слоя. При этом автоматически останавливаются все стадии реакций (2) и (2а). Регенерацию окисленной формы катализатора осуществляют по реакции (4) как отдельную стадию. Ее протекание возможно благодаря свойству обратимой окисляемости ванадомолибдофосфорных ГПК (и их солей). Возможность раздельного проведения стадий (3)-(3а) и (4) является важным свойством предложенной многофазной системы. Осуществление стадии (3)-(3а) в отсутствии кислорода или воздуха позволяет в 3 раза повысить избирательность катализатора в направлении синтеза целевого продукта. Этот факт, по-видимому, обусловлен тем, что комплекс молекулярного кислорода с восстановленной формой ГПК (HmГПК) реагирует с субстратом (IIа) (или IIб) через промежуточное образование свободных радикалов вида (X)
(X)
Их рекомбинация дает соединение (VIII), превращающееся в (VII), а затем в полимер.
В рассматриваемых примерах межфазного катализа задача отделения катализатора от продуктов реакции легко и полно решается простым разделением водной и органической фаз. Катализатор окисления (ГПК), отделенный в виде основной фазы, используется повторно неограниченное число раз. Целевой продукт выделяют из органической фазы путем отгонки растворителя. Последний используют повторно, а продукт подвергают тонкой очистке, например путем перекристаллизации, сублимации или перегонки с водяным паром.
Данный способ позволяет заложить основы безотходной технологии синтеза менадиона.
Оптимальные условия синтеза включают выбор следующих параметров проведения реакции:
температуры 0-100оС;
природы органического растворителя - жидкого парафина или иного растворителя, плохо растворяющего вещества (VII) и (VIII), в качестве растворителя может быть использован также сам расплавленный субстрат (IIа) или (IIб);
состав газовой среды при проведении стадий (3)-(3а) и (4). При проведении стадий (3)-(3а) газовая фаза должна состоять из СО2 или инертных газов, а при проведении стадии (4) она должна состоять из O2или кислородсодержащего газа;
концентрации катализатора (0,2-0,5 М) и субстрата (II) (менее 0,1 М) в процессе проведения стадий (3)-(3а).
Проведение реакции в оптимальных условиях решает не только проблемы высокой избирательности и производительности катализатора, но и задачи его отделения от продуктов реакции. Например, продукт (VII) побочной реакции (5) мешает разделению фаз и очистке целевого продукта. Поэтому без высокой избирательности катализатора не удается решить всех технологических проблем нового способа. В результате реализация данного способа потребовала сочетания всех перечисленных элементов новизны.
П р и м е р 1 выявляет полный возможный ассортимент продуктов реакции и позволяет выбрать методы их идентификации.
П р и м е р 2 показывает, что смола, как побочный продукт окисления нафтолов (II) образуется через динафтол (VIII). Уменьшение выхода последнего, а вместе с тем и смолы, можно добиться снижением концентрации субстратов (II) во время реакции. Это было учтено в методике последующих примеров, где субстраты (II) в растворе петролейного эфира постепенно прикапывают к катализатору. В результате до минимума снижались концентрации (II) в растворе во время реакции.
Сопоставление примеров 2 и 3 показывает, что стадийное проведение целевой реакции, когда стадия (3) протекает в отсутствие кислорода, резко повышает избирательность катализатора в направлении образования продукта I за счет снижения выхода смолы. Изменения природы внешнесферного катиона в соли ГПК-n (в эквивалентных соотношениях) практически не влияет на избирательность в отношении I. Это видно из примеров 3-3в. Из примеров 3б-3в следует, что изменение природы инертного газа (CO2, N2O, N2, Ar) на стадии реакции (3) не влияет на избирательность катализатора.
Сопоставление примеров 3 и 4 показывает, что повышение избирательности катализатора в отношении целевого продукта (I) достигается увеличением молярной доли катализатора в реакционной смеси (т.е. уменьшением молярного соотношения субстрата и катализатора).
Из примеров 3, 5-7 (см. далее) видно, что для проведения целевой реакции (3) пригодны ГПК-n с разным числом n атомов ванадия в молекуле от n = 1 до n = 6, но по ряду технологических причин оптимальным является n = 3.
Сопоставление результатов примеров 3-4 позволяет оптимизировать способ проведения реакции (2). В описываемом ниже виде он используется в балансовых опытах и подтвержден в опытах на пилотной установке, где была выпущена опытная партия продукта I (4 кг).
Сопоставление примеров 4 и 8 показывает, что уменьшение длительности введения раствора субстратов (II) в катализатор снижает выход целевого продукта I. Этот факт еще раз подтверждает, что выход побочных продуктов растет с повышением концентрации непрореагировавших нафтолов (IIа) и (IIб).
Процесс осуществляют следующим образом. В реактор с мешалкой, термостатированный при выбранной температуре (оптимальная Т = 50оС) заливают водный раствор катализатора (например Мg9, ГПК-3). Реактор продувают инертным газом, например CO2, и в слабом его токе при хорошем перемешивании (300-500 об/мин) в реактор равномерно вливают (прикапывают) в течение 2 ч раствор субстрата II (или IIа) в органическом растворителе, например петролейном эфире. На каждые 0,8-1 г субстрата берут около 50 мл растворителя. Молярное соотношение (II) и ГПК-n (или Меа ГПК-n) поддерживают в пределах 1:8 < < 11: ГПК-n ≅ 1: 3. После вливания всего раствора субстрата его выдерживают 10-20 мин, после чего мешалку останавливают и раствор выливают из реактора. Водный и органический слои разделяют. Водный слой кипятят, упаривают и окисляют в реакторе при перемешивании с поддувом воздуха в течение 3-4 ч при 90-100оС. После доведения объема катализатора до исходного и достижения исходного ярко-красного цвета, катализатор используют повторно.
Органический слой промывают водой (10% от объема катализатора), промывную воду после отделения от органического слоя добавляют к катализатору, а затем отпаривают от него, как описано выше. Органический растворитель отгоняют от продуктов реакции и используют повторно. Маслянистый кубовый остаток перегоняют с водяным паром. При этом целевой продукт желтых хлопьев собирают вместе с водой в конденсаторе. Для полного выделения 1 г продукта из содержащей смолу реакционной массы требуется 50-100 г водяного пара. Побочный продукт реакции (VIII) и образующаяся из него смола остаются в кубе перегонного аппарата. Они в виде массы, похожей на сургуч, в дальнейшем уничтожаются полным сжиганием (до CO2 и H2O). Однако смола, как показывает качественная оценка, способна вулканизовать полимер, содержащий двойные связи и может найти полезное применение.
Из собранной в конденсаторе водной пульпы продукт I экстрагируют небольшой порцией хлороформа (5-10 мл на 1 г продукта). Воду сливают, а хлороформный экстракт выпаривают. Отогнанный хлороформ используют повторно, а оставшийся твердый остаток является чистым целевым продуктом I.
Из лабораторных опытов и результатов проверочных опытных работ на пилотной установке, проведенных по этой же методике, следует, что данный способ может быть положен в основу экологически чистой технологии почти без сточных вод и без вредных газовых выбросов.
Большинство приводимых примеров послужили основой подбора катализатора и метода проведения реакции, поэтому условия проведения реакции в них отличаются от оптимальных и требуют комментариев приведенных в текстовой части. Количественные данные всех опытов из примеров приведены в табл.1. Опыты примеров 1 и 2 являются методическими. П р и м е р 1. В трехгорловую колбу объемом 200 мл загружают 1,12 г смеси нафтола в 70 мл бензола (состав смеси по ГЖХ : 85,5% (IIа), 9% 2,4- и 2,8-диметил-1-нафтолов (3:1), 1,2% 2,4,8-триметил-1-нафтола, 4,3% смеси 1-нафтола, метиловых эфиров нафтолов (IIа) (IIб) и 1-нафтола). К полученному раствору при размешивании добавляют 30 мл водного раствора ГПК-3 (С = 0,2 моль/л). Реакционную смесь интенсивно перемешивая, выдерживают 3 ч при 60оС. Катализатор (ГПК-3) в виде водного слоя отделяют, окисляют при 100оС воздухом 3 ч по реакции (4) и используют повторно (см. пример 2). Органический слой отделяют, промывают водой, сушат CaCl2, бензол выпаривают. После дополнительной осушки в вакууме полученное масло темно-красного цвета разделяют методом колоночной хроматографии на силикагеле (элюент хлороформ - диэтиловый эфир 120:1). Получают: фракция I - 0,9 г нафтохинона (I), т.пл. 105-106оС (ИК-, УФ-, ИМР-спектры соответствуют спектрам эталонного образца); фракция 2 - 0,895 г смесь нафтохинона (I) и динафтола (VIII) с соотношением 1:1; фракция 3 - 0,2 г динафтола (VIII); фракция 4 - 0,14 г хинона (IX), 4,4-ди-(2-метил-1-нафтол) (VIII) - т.пл. 243-244оС (из спирта). УФ-спектр λмакс, нм (lg ε): 223 (4,912), 308 (4,140), 329 (4,026). ИК-спектр (CCl4, см-1): 3620 (ОН). Спектр ПМР (CDCl3, δ, м.д., 60 МГц): 2,46 с (6Н, СН3), 5,20 с (2Н, OH), 7,12-7,66 м (6Н, СН), 8,10-8,44 (4Н, СН). Найдено: M 314.1316. C22H18O2. Вычислено: М 314.1307.
2,4-диметил-4-гидроксинафтален-2-он-1 (IX)-масло. УФ-спектр, λмакс, нм (lg ε): 211 (4,330), 231 (4,314), 278 (3,880). ИК-спектр (CCl4, см-1): 1600 (С= С), 1650, 1670 (С=С, С=О), 3600 (OH). Спектр ПМР (CDCl3, δ, м.д., 60 МГц): 1,47 с (3Н, СН3), 1,76 с (3Н, СН3), 3,19 с (1Н, OH), 6,62 м (1Н, СН), 7,04-7,97 м (4Н, СН). Найдено: М 188,0830. С12Н12O2. Вычислено: М 188,0837.
П р и м е р 2. 0,1 г димера (VIII) растворяют в 7 мл бензола, добавляют 3 мл водного раствора [Mg0,9ГПК-3], с концентрацией 0,5 моль/л. Реакционную смесь интенсивно перемешивают 1,5 ч при 80оС. Катализатор отделяют, окисляют как в опыте 1 и используют в примере 3. Бензольный слой промывают водой, сушат CaCl2, бензол упаривают. Получают 0,093 г (94%) динафтохинона (VII) в виде осадка темно-красного цвета, т.пл. 190оС (с разлож.). ИК-спектр (KBr, см-1): 1585, 1620 С=С, C=O). ПМР-спектр (CDCl3, δ, м.д., 200 МГц): 2,18 с (6Н, СН3), 7,64-7,66 м (4Н, СН), 7,93 с (2Н, СН), 8,33 с (2Н, СН).
П р и м е р 3. В примерах 3-7 используют фракцию нафтолов, содержащую по данным ГЖХ 91% нафтола (IIа), 9% смеси 2,4- и 2,8-диметил-1-нафтолов и 2,4,8-триметил-1-нафтола.
В трехгорловую колбу объемом 300 мл, снабженную мешалкой, холодильником, термометром и капельной воронкой загружают 80 мл водного раствора Mg0,9ГПК-3 (С= = 0,5 моль/л, часть этого раствора используют в примерах 2). Затем при интенсивном перемешивании в токе CO2 в течение 2 ч при 50оС прикапывают, раствор 0,8 г фракции нафтолов в 60 мл петроелйного эфира (точка кипения 40-70оС). Реакционную смесь выдерживают еще 10 мин. Отделяют эфирный слой. Оставшийся катализатор экстрагируют хлороформом для полного удаления смолы из водного слоя. Водный раствор катализатора окисляют как в опыте 1 используют в примере 4. Органические фракции объединяют, выпаривают. Полученное масло перегоняют с паром. Получают 0,67 г нафтохинона (I) (содержание основного продукта 98% здесь и далее по данным ГЖХ) и 0,18 г смолы, состоящей из (VII), (VIII) и их полимеров. Выход продукта I 84,5% (в расчете на 2-метил-1-нафтол). Соотношение субстрата и катализатора 1:8.
П р и м е р 3а. В опыте этого примера, проведенного по методике примера 3, использована в качестве катализатора кислая кальциевая соль Са0,9ГПК-3. Выход целевого продукта I составил 80%= смолы - около 20%
П р и м е р 3б. В опыте этого примера, проведенном по методике предыдущего примера, в качестве катализатора использован раствор Na1,8ГПК-3, а в качестве инертного газа, продуваемого через реактор во время опыта, вместо CO2 использована закись азота. Результаты этого опыта практически не отличались от предыдущего. Выход продукта I 77%, смолы 23%.
П р и м е р 3в. Практически такие же результаты, как в примере 3б получены при использовании в качестве инертной атмосферы N2 или Ar:
79 I, 21% смолы - c N2,
80% I, 20% смолы - Ar.
П р и м е р 4. К 80 мл раствора Mg0,9ГПК-3 (С = 0,5 моль/л) при перемешивании в токе СO2 при 52,5оС в течение 3,5 ч прикапывают раствор 1,6 г фракции нафтолов в 120 мл гексана. Реакционную смесь и катализатор обрабатывают здесь и далее аналогично примеру 3. Получают 1,15 г нафтохинона (I) (содержание 97%) и 0,53 г смолы. Выход 69%. Соотношение субстрата и катализатора 1:4.
П р и м е р 5. К 50 мл раствора ГПК-6 (С= = 0,2 моль/л) при перемешивании прикапывают в токе CO2 при 50оС в течение 80 мин 0,3 г фракции нафтолов в 30 мл гексана. После реакции водный слой окисляют как в примере 3. После обработки реакционной смеси получают 0,23 г нафтохинона (I) (содержание 97% ) и 0,1 г смолы. Выход 69%.
П р и м е р 6. К 80 мл раствора ГПК-1 (С= = 0,2 моль/л) при перемешивании прикапывают в токе CO2 при 20оС за 80 мин 0,3 г нафтолов в 30 мл гексана. После обработки реакционной смеси получают 0,17 г нафтохинона (I) (содержание 97,5% ) и 0,14 г смолы. Выход 52%. Восстановленная форма ГПК-1 не окисляется воздухом, не может быть окислена действием других кислородсодержащих окислителей.
П р и м е р 7. В примерах 7-10 используют фракцию, содержащую по данным ГЖХ: 88,4% нафтола (IIа); 6% 2,4- и 2,8-диметил-нафтолов; 2% 1-нафтола; 3,6% эфиров нафтолов.
К 125 мл раствора Mg1,2ГПК-4 (С = 0,5 моль/л) при перемешивании прикапывают в токе CO2 при 20оС в течение 30 мин 3 г фракции нафтолов в 95 мл гексана. После обработки реакционной смеси получают 1,57 г нафтохинона (I) (содержание 85% ) и 1,4 г смолы. Выход 46%. Катализатор после опыта регенерирован как в примере 3.
П р и м е р 8. К 100 мл раствора Mg0,9ГПК-3 (С = 0,4 моль/л) при перемешивании прикапывают в токе CO2 при 20оС в течение 30 мин 1,6 г фракции нафтолов в 80 мл гексана. После обработки реакционной смеси получают 0,65 г нафтохинона (I) (содержание 885% и 0,95 г смолы. Выход 37%. Соотношение субстрата и катализатора 1:4.
П р и м е р 9. К 160 мл раствора Mg0,9ГПК-3 (С = 0,5 моль/л) при перемешивании прикапывают в токе CO2 при 20оС в течение 80 мин 2,5 г фракции нафтолов в 100 мл гексана После обработки реакционной смеси получают 1,19 г нафтохинона (I) (содержание 86%) и 1,3 г смолы. Выход 42%. Соотношение субстрата и катализатора 1:5. Этот пример подтверждает вывод из предыдущего примера.
П р и м е р 10. К 80 мл раствора Mg0,9ГПК3 (С = 0,5 моль/л) при перемешивании прикапывают в токе CO2 при 20оС в течение 0,8 г фракции нафтолов в 50 мл гексана. После обработки реакционной смеси получают 0,52 г нафтохинона (I) (содержание 92%) и 0,28 г смолы. Выход 61%. Соотношение субстрата и катализатора 1:8. Этот пример подтверждает тот факт, что величина последнего соотношения очень важна для избирательности. Сопоставление выходов этого примера и примера 3 указывает на выгодность повышения температуры реакции до 50оС.
П р и м е р 11. В примерах 11-12 используют смесь, содержащую по данным ГЖХ : :49,6% соединения (IIа), 39,8% соединения (IIб); 3,4% эфира нафтолов и 1-нафтола; 4,5% 2,4,8-три-1-нафтола.
К 80 мл раствора Mg0,9ГПК-3 (С = 0,5 моль/л) при перемешивании прикапывают в токе CO2 при 52оС в течение 3 ч 1,6 г смеси нафтолов в 120 мл гексана. После обработки реакционной смеси получают 1,12 г нафтохинона (I) и 0,49 г смолы. Выход 62%. Соотношение субстрата и катализатора 1:4.
П р и м е р 12. К 80 мл раствора Mg0,9ГПК-3 (С = 0,5 моль/л) при перемешивании прикапывают в токе CO2 при 52о в течение 2 ч 0,8 г смеси нафтолов в 60 мл гексана. После обработки реакционной смеси получают 0,65 г нафтохинона (I) (содержание 88%) и 0,2 г смолы. Выход 73%. Соотношение субстрата и катализатора 1:8.
П р и м е р 12а и 12б. Этот пример повторяет пример 12 лишь с тем различием, что в качестве катализатора используют кислые соли ГПК-3:К1,8ГПК-3, Zn0,9ГПК-3. Соответственно получают следующие выходы продукта (I) и смолы: К 68% и 0,3 г, Zn 66% и 0,21 г.
П р и м е р 13. В примерах 13-18 используют смесь, содержащую по данным ГЖХ: 88,4% нафтола (IIа); 6% 2,4- и 2,8-диметил-1-нафтолов; 2% 1-нафтола; 3,6% эфиров нафтолов.
К 15 мл раствора [Mg2ГПК-4] (С = 0,2 моль/л) при перемешивании прикапывают 1,28 г фракции нафтолов в 31,5 мл о-ксилола. При температуре 0оС реакционную смесь выдерживают 3 ч. После обработки смесь содержит по данным ГЖХ: 72% исходных метилированных 1-нафтолов; 14% нафтохинона (I); 14% динафтола (VIII). Этот пример показывает, что снижение концентрации раствора ГПК и снижение температуры реакции приводит к резкому уменьшению скорости превращения метилированных 1-нафтолов в нафтохинон (I).
П р и м е р 14. К 150 мл водного раствора [Mg3ГПК-4] (С = 0,4 моль/л) прикапывают 3,16 г фракции нафтолов в 20 мл толуола при температуре 101оС в течение 50 мин с одновременной отгонкой с водяным паром образующегося нафтохинона (I). Получено 0,237 г нафтохинона (содержание 90%) и 3,17 г смолы. Выход 6%. Этот пример показывает, что увеличение числа атомов Mg в молекуле ГПК-4 и повышение температуры реакции резко снижает выход целевого продукта.
П р и м е р ы 15-18. Окисление 2-метил-1-нафтола проводят с ГПК-4 или ее магниевой солью, как описано в примере 3.
Обобщенные результаты проведения процесса по примерам 1-18 приведены в таблице.
П р и м е р 19. В примерах 19-21 используют фракцию нафтолов того же состава, что и в примерах 3-7.
К 100 мл 0,1 М раствора [Mg0,9ГПК-3] (10-2 моль) при перемешивании прикапывают в токе CO2 при 50оС за 80 мин 0,320 (2 x x 10-3 моль) фракции нафтолов в 20 мл петролейного эфира. По окончании реакции из органического слоя выделяют 0,120 г I (35%) и 0,220 г (64%) смолы. Соотношение субстрата и катализатора 1:5. Из сопоставления выходов этого примера и примера 11 (в которых условия близки) видно, что снижение концентрации катализатора в водном слое снижает избирательность процесса.
П р и м е р 20. К 50 мл 0,2 М раствора Mg0,3ГПК-1 (10-2 моль) при перемешивании в токе CO2 при 50оС прикапывают за 90мин 10 мл петролейного эфира с 0,160 г (10-3 моль) фракции нафтолов. Соотношение субстрата и катализатора 1:10. После реакции из органического слоя выделяют 0,086 г (50%) целевого продукта I и 0,091 г смолы (53%).
Сопоставление этих результатов с данными примера 12, где условия близки, показывает, что снижение содержания ванадия в молекуле ГПК снижает выход целевого продукта.
П р и м е р 21. К 50 мл 0,2 М раствора Mg1,8ГПК-6 (10-2 моль) при перемешивании в токе CO2 при 50оС прикапывают за 110 мин 15 мл петролейного эфира, в которых растворено 0,144 г (9 ˙ 10-4 моль) фракции нафтолов. После реакции выделяют 0,166 г (75%) продукта I и 0,041 г (26%) смолы. Соотношение субстрата и катализатора 1:11. Зола, полученная сожжением смолы (0,002 г), являлась V2O5, что указывало, что катализаторы с высоким содержанием ванадия в молекуле ГПК при работе теряют часть ванадия и потому непригодны для практики.
П р и м е р 22. К 50 мл 0,2 молярного раствора Mg0,6ГПК-2 (10-2моля) при перемешивании в токе CO2 при 52оС прикапывают за 90 мин 10 мл петролейного эфира с 0,158 г (10-3 моль) фракции нафтолов. После реакции получают 0,069 г (40%) соединения I и 0,015 г смолы (62%). Этот пример показывает, что каталитические свойства Mg0,6ГПК-2 близки к таковым катализаторам Mg0,9ГПК-1 (пример 19) и Mg0,3ГПК-1 (пример 20).
П р и м е р 23. К 80 мл 0,5 м раствора ГПК-2, содержащего 4 ˙ 10-2моль гетерополикислоты (ГПК-2 = H5PMo10V2O40) при перемешивании в токе CO2 при 70оС прикапывают за 120 мин 120 мл гексанового раствора, содержащего 1,6 г (0,009 моль) смеси нафтолов того же состава, что в примерах 11-12б с одновременной отгонкой растворителя. В опыте молярное соотношение субстрата и катализатора равно 4:44. После реакции продукт в виде твердого оранжевого остатка отфильтрован от водного раствора катализатора и перегнан с водяным паром. Выделено 1,05 г 2-метил-1,4-нафтохинона (68%) и 0,464 г смолы (29%).
Этот пример показывает, что верхний предел температур реакции, обеспечивающий приемлемые избирательности катализатора достигает 70оС. Кроме того, часть побочных продуктов реакции (100-68-29 = 3%) оказывается водорастворимой, переходит из органического слоя в водный - к катализатору и сгорает при регенерации катализатора.
Таким образом, данный способ позволяет существенно увеличить выход целевого продукта - до 85% (против 64% по прототипу) и упростить технологию процесса за счет упрощения стадии выделения целевого продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТИЛ-1,4-НАФТОХИНОНА | 1997 |
|
RU2142935C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТИЛ-1,4 НАФТОХИНОНА И КАТАЛИЗАТОР ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2000 |
|
RU2162837C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТИЛ-1,4-НАФТОХИНОНА | 1992 |
|
RU2061669C1 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕСИ 2-МЕТИЛ-1-НАФТОЛА И 2,4-ДИМЕТИЛ-1-НАФТОЛА | 1991 |
|
RU2027694C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРАЗИНОВ, СОДЕРЖАЩИХ О-ГИДРОКСИФЕНИЛЬНУЮ ГРУППУ | 1991 |
|
RU2026295C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТИЛ-1,4-НАФТОХИНОНА | 2005 |
|
RU2278106C1 |
| СПОСОБ НИТРОВАНИЯ НАФТАЛИНА | 1993 |
|
RU2079482C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,4,4А,9А-ТЕТРАГИДРОАНТРАХИНОНА | 1995 |
|
RU2095341C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5,5'-БИАЦЕНАФТЕНИЛА | 1993 |
|
RU2061667C1 |
| СПОСОБ ПОЛУЧЕНИЯ КУБОВЫХ КРАСИТЕЛЕЙ И ПИГМЕНТОВ, СОДЕРЖАЩИХ ПЕРИЛЕНОВЫЙ ФРАГМЕНТ | 1997 |
|
RU2128200C1 |


Использование: в качестве витамина К. Сущность изобретения: 2-метил-1,4-нафтохинон, т.пл. 105-106°С. Реагент 1: 2-метил-1-нафтол или его смесь с 2,4-диметил-1-нафтолом. Реагент 2 : кислород. Условия реакции: двухфазная система, катализатор формулы H(3+n)PMo(12-n)VnO40, где n = 1-6, концентрация катализатора 0,1-0,5 моль/л, 20 - 70°С, инертный газ. 1 з.п. ф-лы, 1 табл.
H(3+n) RMo(12-n) VnO40,
где N = 1 - 6,
отличающийся тем, что, с целью увеличения выхода целевого продукта и упрощения технологии процесса, в качестве производного нафталина используют 2-метил-1-нафтол или его смесь с 2,4-диметил-1-нафтолом, а процесс проводят в двухфазной системе, в которой катализатор входит в состав водной фазы в количестве, соответствующем его концентрации 0,1 - 0,5 моль/л, а производное нафталина - в состав органической фазы, не смешивающей с водной фазой, при интенсивном перемешивании фаз при 20 - 70oС в атмосфере инертного газа в течение 0,5 - 3 ч с последующим выделением целевого продукта из органической фазы и окислением водной фазы, содержащей восстановленную форму катализатора, кислородом или кислородсодержащим газом.
Mea Н(3+n-a) Р Mo(12-n) VnO40,
где Me = Na+, K+, Mg2+, Ca2+, Zn2+;
n = 1 - 6;
a определяется валентностью металлов.
| Способ получения 2-метил-1,4-нафтохинона | 1983 |
|
SU1121255A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
