ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001]
Настоящее изобретение относится к способу получения карбонатного производного безопасным и эффективным образом.
УРОВЕНЬ ТЕХНИКИ
[0002]
Линейный карбонат среди карбонатных производных традиционно используется в качестве растворителя или тому подобного. В частности, в последнее время увеличивается объем производства линейного карбоната в качестве неводного растворителя для электролита литий–ионной вторичной батареи. Поликарбонат получают реакцией угольной кислоты с бисфенольным соединением и широко используют в качестве конструкционного пластика с превосходной прозрачностью и ударопрочностью. Мочевинная смола широко используется в качестве материала для клея и посуды. Полидитиокарбонат допустим в качестве стабильного оптического материала со слабым окрашиванием.
[0003]
Карбонатное производное обычно получают из фосгена и соединения, содержащего нуклеофильную функциональную группу. Фосген является очень токсичным. Например, фосген легко вступает в реакцию с водой с образованием хлористого водорода и имеет историю использования в качестве отравляющего газа. Кроме того, карбонатное производное получают с помощью реакции монооксида углерода, спирта и кислорода, но этот способ имеет проблему, заключающуюся в том, что токсичный монооксид углерода должен использоваться под высоким давлением. Соответственно, были изучены различные безопасные способы получения сложного карбонатного эфира и поликарбоната.
[0004]
Например, способ получения целевого карбонатного производного путем подвергания сложного карбонатного эфира реакции переэтерификации в присутствии катализатора описан в патентном документе 1. Этот способ, однако, не дает принципиального решения, поскольку в данном способе остается проблема получения карбонатного производного в качестве исходного соединения. Данный способ также имеет проблемы использования дорогостоящего катализатора и обратной реакции и побочной реакции из–за остаточного катализатора.
[0005]
Способ получения карбонатного производного из эпоксисоединения и диоксида углерода в присутствии катализатора описан в патентном документе 2. В этом способе нет необходимости использовать фосген и монооксид углерода. Но этот способ не подходит для массового промышленного производства, поскольку необходимо использовать дорогостоящий катализатор и доводить давление углекислого газа до высоких уровней.
[0006]
Автор настоящего изобретения разработал способ получения галогенированного сложного карбонатного эфира, подвергая галогенированный углеводород и спирт окислительной фотореакции (патентный документ 3) и способ получения галогенированного сложного эфира муравьиной кислоты, который включает стадии получения смеси фосгена путем облучения светом хлороформа в присутствии кислорода и реакции спирта со смесью без выделения фосгена (патентный документ 4).
ДОКУМЕНТЫ ИЗВЕСТНОГО УРОВНЯ ТЕХНИКИ ПАТЕНТНЫЕ ДОКУМЕНТЫ
[0007]
Патентный документ 1: JP H7–10811 A
Патентный документ 2: JP 2001–129397 A
Патентный документ 3: WO 2014/171367
Патентный документ 4: JP 2013–181028 A
НЕПАТЕНТНЫЕ ДОКУМЕНТЫ
[0008]
Непатентный документ 1: OKUMA Seiichi et al., The journal of the Japan Society for Analytical Chemistry, Vol. 24, pp. 385–387 (1975)
Непатентный документ 2: TSURUGI Jitsuo et al., Journal of the Society of Rubber Science and Technology, Japan, Vol. 43, Number 5, pp. 337–346 (1970)
Непатентный документ 3: Jerzy Herbich et al., J. Photochem. Photobiol. A: Chem., 80, pp. 157–160 (1994)
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
ЗАДАЧИ, РЕШАЕМЫЕ ИЗОБРЕТЕНИЕМ
[0009]
Как правило, фосген используется для получения карбонатного производного, как описано выше. Даже при способе производства без использования фосгена возникает проблема, связанная с необходимостью использования другого токсичного соединения или дорогостоящего катализатора или использования фосгена для получения исходного соединения.
С учетом вышеизложенного, целью настоящего изобретения является предложить способ получения карбонатного производного безопасным и эффективным образом.
СРЕДСТВА ДЛЯ РЕШЕНИЯ ЗАДАЧ
[0010]
Автор настоящего изобретения провел обширные исследования для решения указанных выше проблем. В результате автор изобретения осуществил настоящее изобретение, обнаружив, что карбонатное производное можно получить безопасным и эффективным образом, подвергая галогенированное углеводородное соединение и специфическое соединение, содержащее нуклеофильную функциональную группу, фотореакции в присутствии кислорода и специфического основания. Поскольку общеизвестно, что органическое основание образует пигмент путем фотореакции, играет роль антиоксиданта для захвата радикала и гасит флуоресценцию соединения с помощью такого механизма, как перенос электронов, и пиридин распадается на глутаконовый альдегид или тому подобное под воздействием ультрафиолета (непатентные документы 1–3), предполагается, что органическое основание является неблагоприятным для фотореакции, как в изобретениях автора, описанных в патентном документе 3 и патентном документе 4. С другой стороны, очень удивительно, что карбонатное производное эффективно образуется путем фотореакции в присутствии специфического основания.
Далее описывается настоящее изобретение.
[0011]
[1] Способ получения карбонатного производного, включающий
облучение светом композиции, содержащей C1–4 галогенированный углеводород, имеющий один или более видов атомов галогена, выбранных из группы, состоящей из атома хлора, атома брома и атома йода, соединение, содержащее нуклеофильную функциональную группу, и основание, в присутствии кислорода,
где соединение, содержащее нуклеофильную функциональную группу, представлено следующей формулой (i), и карбонатное производное представляет собой линейное карбонатное производное, представленное следующей формулой (I), или
соединение, содержащее нуклеофильную функциональную группу, представлено следующей формулой (ii), и карбонатное производное представляет собой поликарбонатное производное, содержащее фрагмент, представленный следующей формулой (II–1), или циклическое карбонатное производное, представленное следующей формулой (II–2), и
где основание представляет собой одно или более оснований, выбранных из группы, в основном состоящей из гетероциклического ароматического амина, ненуклеофильного сильного основания и неорганического основания.
(i) R1–A–H
(ii) H–A–R2–A–H
(I) R1–A–C(=O)–A–R1
(II–1) [–A–R2–A–C(=O)–]

где
A представляет собой O, S или NR3, где R3 представляет собой H или C1–4 алкильную группу, или R3 образует азотсодержащую гетероциклическую группу с R1 и N,
R1 представляет собой C6–14 арильную группу, C4–14 гетероарильную группу или C2–24 алкилполиоксиалкиленовую группу,
R2 представляет собой C2–10 алкиленовую группу, C6–14 ариленовую группу, C4–14 гетероариленовую группу или C2–24 полиоксиалкиленовую группу.
[0012]
[2] Способ получения в соответствии с указанным выше [1], в котором C1–4 галогенированный углеводород представляет собой C1–4 полигалогенированный углеводород.
[0013]
[3] Способ получения в соответствии с указанным выше [1], в котором C1–4 галогенированный углеводород представляет собой хлороформ.
[0014]
[4] Способ получения по любому из вышеуказанных [1] – [3], в котором гетероциклический ароматический амин представляет собой пиридин, пиколин или лутидин.
[0015]
[5] Способ получения по любому из вышеуказанных [1] – [4], в котором ненуклеофильное сильное основание представляет собой 1,5,7–триазабицикло[4.4.0]дец–5–ен, 7–метил–1,5,7–триазабицикло[4.4.0]дец–5–ен, 1,8–диазабицикло[5.4.0]ундец–7–ен, 1,5–диазабицикло[4.3.0]нон–5–ен или 1,1,3,3–тетраметилгуанидин.
[0016]
[6] Способ получения по любому из вышеуказанных [1] – [5], в котором неорганическое основание представляет собой гидроксид щелочного металла, гидрокарбонат щелочного металла или карбонат щелочного металла.
[0017]
[7] Способ получения по любому из вышеуказанных [1] – [6], в котором использовали 0,001–кратное или более и 1–кратное или менее молярное количество соединения, содержащего нуклеофильную функциональную группу, относительно С1–4 галогенированного углеводорода.
[0018]
[8] Способ получения по любому из вышеуказанных [1] – [7], в котором использовали 1,5–кратное или более и 10–кратное или менее молярное количество основания относительно соединения, содержащего нуклеофильную функциональную группу.
[0019]
[9] Способ получения по любому из вышеуказанных [1] – [8], в котором длина волны света, облучающего композицию, составляет 180 нм или более и 500 нм или менее.
ЭФФЕКТ ИЗОБРЕТЕНИЯ
[0020]
В способе настоящего изобретения не требуется, чтобы в качестве исходного соединения использовался дорогостоящий катализатор и чрезвычайно токсичное соединение, такое как фосген и монооксид углерода. В связи с этим, способ настоящего изобретения является промышленно очень ценным в качестве технологии получения подходящего карбонатного производного безопасным и эффективным образом.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0021]
На фиг.1 представлено схематическое изображение для демонстрации примера реакционного устройства, используемого в способе настоящего изобретения.
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
[0022]
В способе получения карбонатного производного по настоящему изобретению облучают светом композицию, содержащую C1–4 галогенированный углеводород, имеющий один или более видов атомов галогена, выбранных из группы, состоящей из атома хлора, атома брома и атома йода, соединение, содержащее нуклеофильную функциональную группу, и специфическое основание, в присутствии кислорода.
[0023]
1. С1–4 галогенированный углеводород
В реакции настоящего изобретения C1–4 галогенированный углеводород может разлагаться вследствие облучения светом и воздействия кислорода на галогенированное карбонильное или галогенированное карбонилоподобное соединение и реагировать с соединением, содержащим гидроксильную группу, с образованием карбонатного производного. Даже если образуется токсичный галогенированный карбонил, этот галогенированный карбонил немедленно реагирует с соединением, содержащим гидроксильную группу, вследствие чрезвычайно высокой реакционной способности. В результате, не происходит утечки галогенированного карбонила из реакционной смеси или, если утечка галогенированного карбонила происходит, – объем утечки является небольшим. Например, на транспортировку фосгена наложены строгие ограничения, поскольку фосген среди галогенированных карбонилов является очень токсичным; с другой стороны, С1–4 галогенированный углеводород, конечно, не так опасен. Реакция по настоящему изобретению, возможно, может не быть опосредована галогенированным карбонилом или галогенированным карбонилоподобным соединением, поскольку реакция протекает даже в присутствии водного раствора неорганического основания, как описано ниже.
[0024]
С1–4 галогенированный углеводород, который является жидким при атмосферной температуре и атмосферном давлении, используется в качестве органического растворителя или тому подобного в большом количестве, но при попадании в атмосферу вызывает загрязнение окружающей среды, такое как загрязнение воздуха и разрушение озонового слоя. Настоящее изобретение представляет собой технологию получения полезного соединения путем фотолиза C1–4 галогенированного углеводорода и вносит значительный вклад как в промышленность, так и в экологию.
[0025]
С1–4 галогенированный углеводород представляет собой алкан, алкен или алкин, которые имеют число атомов углерода 1 или более и 4 или менее, и который замещен одним или более атомами галогена, выбранными из группы, состоящей из атома хлора, атома брома и атома йода. Как описано выше, C1–4 галогенированный углеводород может разлагаться при облучении светом и под воздействием кислорода и может действовать аналогично галогенированному карбонилу в настоящем изобретении. С1–4 галогенированный углеводород предпочтительно представляет собой С1–2 галогенированный углеводород и более предпочтительно – галогенированный метан. Когда число атомов углерода составляет 2 или более и 4 или менее, C1–4 галогенированный углеводород предпочтительно представляет собой алкен или алкин, имеющий одну или более ненасыщенных связей для более легкого разложения. В дополнение к этому, предпочтительно, чтобы C1–4 галогенированный углеводород имел два или более из описанных выше атомов галогена. Кроме того, С1–4 полигалогенированный углеводород, имеющий два или более из описанных выше атомов галогена на одном и том же атоме углерода, является предпочтительным, даже если вышеописанный атом галогена переносится при разложении.
[0026]
В качестве специфического С1–4 галогенированного углеводорода С1–4 галогенированный алкан, С2–4 галогенированный алкен или С2–4 галогенированный алкин является предпочтительным, галогенированный метан, галогенированный этен или галогенированный ацетилен является более предпочтительным с точки зрения легкости образования галогенированного карбонилоподобного соединения, полигалогенированный метан, полигалогенированный этен или полигалогенированный ацетилен, имеющий два или более из описанных выше атомов галогена, является особенно предпочтительным, и полигалогенированный метан является наиболее предпочтительным. Пример C1–4 галогенированного углеводорода включает галогенированный метан, такой как дихлорметан, хлороформ, дибромметан, бромоформ, йодметан и дийодметан; галогенированный этан, такой как 1,1,2–трихлорэтан, 1,1,1–трихлорэтан, 1,1,2,2–тетрахлорэтан и 1,1,1,2–тетрахлорэтан; галогенированный пропан, такой как 1,1,1,3–тетрахлорпропан; пергалогенированный алкан, такой как тетрахлорметан, тетрабромметан, тетрайодметан, гексахлорэтан и гексабромэтан; пергалогенированный этен, такой как 1,1,2,2–тетрахлорэтен и 1,1,2,2–тетрабромэтен.
[0027]
С1–4 галогенированный углеводород может быть соответствующим образом выбран в зависимости от целевой реакции и желаемого продукта. Один C1–4 галогенированный углеводород может быть использован отдельно, или два или более C1–4 галогенированных углеводорода могут использоваться в комбинации. Предпочтительно, чтобы только один С1–4 галогенированный углеводород использовался в зависимости от целевого соединения. С1–4 галогенированный углеводород, имеющий группу хлора, является предпочтительным.
[0028]
C1–4 галогенированный углеводород, используемый в способе настоящего изобретения, может представлять собой C1–4 галогенированный углеводород, который однажды использовался в качестве, например, растворителя. Предпочтительно, чтобы такой использованный C1–4 галогенированный углеводород был очищен в некоторой степени для использования, поскольку, если содержится большое количество примесей и воды, реакция может быть ингибирована. Например, предпочтительно, чтобы вода и водорастворимые примеси были удалены путем промывки водой, и затем С1–4 галогенированный углеводород сушился с помощью безводного сульфата натрия, безводного сульфата магния или тому подобного. Чрезмерно большая очистка, в результате которой снижается производительность, не нужна, поскольку даже при наличии воды реакция может протекать. Содержание воды предпочтительно составляет 0,5 об.% или менее, более предпочтительно 0,2 об.% или менее и еще более предпочтительно 0,1 об.% или менее. C1–4 галогенированный углеводород для повторного использования может содержать продукт разложения С1–4 галогенированного углеводорода.
[0029]
2. Соединение, содержащее нуклеофильную функциональную группу
«Соединение, содержащее нуклеофильную функциональную группу» в настоящем изобретении представляет собой соединение, которое имеет нуклеофильную функциональную группу, содержащую нуклеофильный атом кислорода, атом серы и/или атом азота, и которое представлено формулой (i) или формулой (ii). В ряде случаев это соединение сокращенно называется, соответственно, «соединение (i), содержащее нуклеофильную функциональную группу» или «соединение (ii), содержащее нуклеофильную функциональную группу». Соединение, содержащее нуклеофильное функциональное группу, используемое в настоящем изобретении, не имеет атома фтора в качестве замещающей группы; и в результате, карбонатное производное, полученное способом настоящего изобретения, также не имеет атома фтора в качестве замещающей группы. Специфическое соединение, содержащее нуклеофильную функциональную группу, используется в настоящем изобретении; поэтому реакция продолжается до тех пор, пока не будет получено карбонатное производное.
[0030]
Когда соединение (i), содержащее нуклеофильную функциональную группу, используется в настоящем изобретении, полученное карбонатное производное представляет собой линейный карбонат, представленный формулой (I), который в ряде случаев сокращенно называется «линейный карбонат (I)». Когда используется соединение, содержащее гидроксильную группу (ii), полученное карбонатное производное представляет собой поликарбонатное производное, содержащее фрагмент, представленный формулой (II–1), которое в ряде случаев сокращенно называется «поликарбонатное производное (II–1)», или циклическое карбонатное производное, представленное формулой (II–2), которое в ряде случаев сокращенно называется «циклическое карбонатное производное (II–2)».
[0031]
Соединение (i), содержащее нуклеофильную функциональную группу, и соединение (ii), содержащее нуклеофильную функциональную группу, используемые в способе получения настоящего изобретения в качестве исходного соединения, и линейное карбонатное производное (I), поликарбонатное производное (II–1) и циклическое карбонатное производное (II–2) в качестве целевого соединения описаны ниже.
(i) R1–A–H
(ii) H–A–R2–A–H
(I) R1–A–C(=O)–A–R1
(II–1) [–A–R2–A–C(=O)–]
[0032]
[0033]
где
A представляет собой O, S или NR3, где R3 представляет собой H или C1–4 алкильную группу, или R3 образует азотсодержащую гетероциклическую группу с R1 и N,
R1 представляет собой C6–14 арильную группу, C4–14 гетероарильную группу или C2–24 алкилполиоксиалкиленовую группу,
R2 представляет собой C2–10 алкиленовую группу, C6–14 ариленовую группу, C4–14 гетероариленовую группу или C2–24 полиоксиалкиленовую группу.
[0034]
В данном описании галогенированный углеводород, имеющий 1 или более и 4 или менее атомов углерода, называется «C1–4 галогенированный углеводород». Другие группы и соединения описываются аналогичным образом.
[0035]
R3 в соединении (i), содержащем нуклеофильную функциональную группу, предпочтительно представляет собой H. Азотсодержащая гетероциклическая группа, образованная R1, R3 и N, может представлять собой неароматическую азотсодержащую гетероциклическую группу или ароматическую азотсодержащую гетероциклическую группу. Пример неароматической азотсодержащей гетероциклической группы включает пирролидинил и пиперидинил. Пример ароматической азотсодержащей гетероциклической группы включает пирролил, имидазолил и пиразол.
[0036]
Атом водорода в C6–14 арильной группе может быть замещен атомом хлора, атомом брома, атомом йода или C1–8 алкильной группой.
[0037]
С4–14 гетероарильная группа означает ароматическую гетероциклическую группу, имеющую один или более атомов азота, атомов кислорода или атомов серы. Примером гетероциклической группы является пятичленная гетероарильная группа, такая как пирролил, имидазолил, пиразолил, тиенил, фурил, оксазолил, изооксазолил, тиазолил, изотиазолил и тиадиазол; шестичленная гетероарильная группа, такая как пиридинил, пиразинил, пиримидинил и пиридазинил; конденсированная ароматическая гетероциклическая группа, такая как индолил, изоиндолил, хинолинил, изохинолинил, бензофуранил, изобензофуранил и хроменил, предпочтительно C4–14 гетероарильная группа, содержащая атом азота, и более предпочтительно пиридинил.
[0038]
C2–24 алкилполиоксиалкиленовая группа предпочтительно представляет собой группу, представленную формулой: –(QHO)mRH где QH представляет собой –CH2–, –CH2CH2–, –CH2CH2CH2–, –CH2CH(CH3)– или –CH2CH2CH2CH2–, RH представляет собой –CH3 или –CH2CH3, m является целым числом, равным 1 или более и 20 или менее. Когда m равно 2 или более, QH может состоять только из одного типа или может состоять из нескольких типов. Когда QH может состоять из нескольких типов, расположение нескольких типов QH может быть случайным или блочным.
[0039]
C2–24 полиоксиалкиленовая группа предпочтительно представляет собой группу, представленную формулой: –(QHO)mQH–.
[0040]
С2–10 алкиленовая группа может быть линейной, разветвленной или циклической. C2–10 алкиленовая группа предпочтительно представляет собой C2–6 алкиленовую группу, и более предпочтительно C2–4 алкиленовую группу. C2–10 алкиленовая группа предпочтительно представляет собой этиленовую группу, необязательно замещенную одной или двумя C1–4 алкильными группами, более предпочтительно – этиленовую группу, необязательно замещенную одной или двумя C1–2 алкильными группами, и еще более предпочтительно этиленовую группу, необязательно замещенную одной или двумя метильными группами, с точки зрения легкости получения циклического карбоната. Вышеописанная этиленовая группа, замещенная алкильной группой, также может быть описана как 1,2–алкиленовая группа.
[0041]
C6–14 ариленовая группа, C4–14 гетероариленовая группа и C2–24 полиоксиалкиленовая группа представляют собой соответственно двухвалентные органические группы, соответствующие C6–14 арильной группе, C4–14 гетероарильной группе и C2–24 алкилполиоксиалкиленовой группе.
[0042]
Пример соединения (i), содержащего нуклеофильную функциональную группу, включает соединение (i), содержащее гидроксильную группу, соединение (i), содержащее тиольную группу, и соединение (i), содержащее аминогруппу. Пример соединения (i), содержащего гидроксильную группу, включает фенол и его производное, в том числе фенол, 2–хлорфенол, 3–хлорфенол, 4–хлорфенол, 2–бромфенол, 3–бромфенол, 4–бромфенол, 2–метилфенол, 3–метилфенол и 4–метилфенол; C3–10 циклоалканол, как например, циклогексанол; бензиловый спирт и его производное, в том числе бензиловый спирт и 2,6–бензиловый спирт; простой моно(C1–4 алкиловый) эфир алкиленгликоля, такой как простой монометиловый эфир этиленгликоля и простой монометиловый эфир пропиленгликоля; простой моно(C1–4 алкиловый) эфир олигоалкиленгликоля, такой как простой монометиловый эфир диэтиленгликоля, простой монометиловый эфир триэтиленгликоля и простой монометиловый эфир тетраэтиленгликоля.
[0043]
Пример соединения (i), содержащего тиольную группу, включает тиофенол и его производное, в том числе тиофенол, 2–хлортиофенол, 3–хлортиофенол, 4–хлортиофенол, 2–бромтиофенол, 3–бромтиофенол, 4–бромтиофенол, 2–метилтиофенол, 3–метилтиофенол и 4–метилтиофенол; C3–10 циклоалкантиол, такой как циклогексантиол; бензилмеркаптан и его производное, в том числе бензилмеркаптан, 2–хлорбензилмеркаптан, 4–хлорбензилмеркаптан и 4–метоксибензилмеркаптан; простой моно(C1–4–алкиловый) тиоэфир 1,2–этандитиола, такой как HSCH2CH2SCH3, HSCH2CH(CH3)SCH3 и HSCH(CH3)CH2SCH3; простой моно(C1–4–алкиловый) тиоэфир олиго(1,2–этандитиол)алкиленгликоля, такой как простой монометиловый тиоэфир ди(1,2–этандитиола), простой монометиловый тиоэфир три(1,2–этандитиола) и простой монометиловый тиоэфир тетра(1,2–этандитиола).
[0044]
Пример соединения (i), содержащего аминогруппу, включает анилин и его производное, в том числе анилин, 2–хлоранилин, 3–хлоранилин, 4–хлоранилин, 2–броманилин, 3–броманилин, 4–броманилин, 2–метиланилин, 3–метиланилин и 4–метиланилин; C3–10 циклоалкиламин, как например, циклогексиламин; гетероциклический амин, такой как пиперазин и пиперидин; бензиловый спирт и его производное, такое как бензиламин, 4–(аминометил)бензонитрил, 2–хлорбензиламин, 3–хлорбензиламин, 4–хлорбензиламин, 2–бромбензиламин, 3–бромбензиламин, 4–бромбензиламин и 4–трет–бутилбензиламин; простой моно(C1–4 алкиловый) эфир алкиленгликоля, такой как N–метилэтилендиамин, N, N–диметилэтилендиамин, N–метилпропилендиамин и N, N–диметилпропилендиамин; N–моно(C1–4 алкил)oлигoэтилендиамин или N, N–ди(C1–4 алкил)oлигoэтилендиамин, такой как N–метилдиэтилентриамин, N, N–диметилдиэтилентриамин, N–метилтриэтилентетрамин, N, N–диметилтриэтилентетрамин, N–метилтетраэтиленпентамин и N, N–диметилтетраэтиленпентамин.
[0045]
Только одно соединение (i), содержащее нуклеофильную функциональную группу, может использоваться отдельно, или же два или более соединения (i), содержащие нуклеофильную функциональную группу, могут использоваться в сочетании. Например, при использовании двух соединений (i), содержащих нуклеофильные функциональные группы, можно синтезировать асимметричное линейное карбонатное производное. Однако предпочтительно, чтобы использовалось только одно соединение (i), содержащее нуклеофильную функциональную группу.
[0046]
Соединение (ii), содержащее нуклеофильную функциональную группу, предпочтительно представляет собой соединения, представленные следующими формулами (ii–1) и (ii–2).
(ii–1) H–A–R21–A–H
(ii–2) H–A–R22–R23–R24–A–H
где
А имеет указанное выше значение,
R21 представляет собой C2–10 алкиленовую группу, C6–14 ариленовую группу или C4–14 гетероариленовую группу,
R22 и R24 независимо представляют собой C6–14 ариленовую группу или C4–14 гетероариленовую группу,
R23 представляет собой C1–10 алкиленовую группу.
[0047]
Когда соединение (ii), содержащее гидроксигруппу, используется в качестве исходного соединения, может быть получено поликарбонатное производное (II–1) или циклическое карбонатное производное (II–2). В частности, когда число атомов углерода в главной цепи в R2 равно 2 или 3, и стабильная структура, такая как пятичленное кольцо и шестичленное кольцо образуется с карбонатной сложноэфирной группой (–O–C(=O)–O–), карбонатной сложнодитиоэфирной группой (–S–C(=O)–S–) или карбамидной группой (–NH–C(=O)–NH–), – в основном образуется циклическое карбонатное производное. В частности, когда C2–10 алкиленовая группа, C6–14 ариленовая группа и C4–14 гетероариленовая группа в соединении (ii–1), содержащем нуклеофильную функциональную группу, представляют собой, соответственно, 1,2–C2–10 алкиленовую группу, 1,2–C6–14 ариленовую группу и 1,2–C4–14 гетероариленовую группу, – в основном образуется циклический карбонат. Когда число атомов углерода в основной цепи в R2 составляет 4 или более, предпочтительно образуется химически более стабильное соединение между циклическим карбонатным производным и поликарбонатным производным в зависимости от условий реакции или тому подобного.
[0048]
Пример 1,2–ариленовой группы включает 1,2–нафталинилен, 1,8–нафталинилен и 2,3–нафталинилен, имеющие следующую структуру в дополнение к 1,2–фениленовой группе и 1,2–бифенилену. То же самое относится к 1,2–(С2–10 алкиленовой) группе и 1,2–(С4–14 гетероариленовой) группе.
[0049]
[0050]
Пример соединения (ii), содержащего нуклеофильную функциональную группу, включает соединение (ii), содержащее гидроксильную группу, соединение (ii), содержащее тиольную группу, и соединение (ii), содержащее аминогруппу. Пример соединения (ii), содержащего гидроксильную группу, включает соединение гликоля, такое как 1,2–пропандиол, 1,2–этандиол и 1,4–бутандиол; дигидроксибензольное соединение, такое как катехол и резорцин; дигидроксигетероарильное соединение, такое как 4,6–дигидрокси–2–метилпиримидин и 3,6–дигидрокси–4–метилпиридазин; бисфенольное соединение, такое как бисфенол A, бисфенол AP, бисфенол B, бисфенол BP, бисфенол E, бисфенол F, бисфенол TMC и бисфенол Z.
[0051]
Например, при использовании бисфенола А в качестве соединения (ii), содержащего гидроксильную группу, получают сложный поликарбонатный эфир, представленный следующей формулой.
[0052]

[0053]
Пример соединения (ii), содержащего тиольную группу, включает C1–4 алкилендитиольное соединение, такое как 1,2–пропандитиол, 1,2–этандитиол и 1,4–бутандитиол; бензолдитиольное соединение, такое как 1,2–бензолдитиол и 1,3–бензолдитиол; гетероарилдитиольное соединение, такое как 2–метилпиримидин–4,6–дитиол и 4–метилпиридазин–3,6–дитиол; бистиофенoльное соединение, такое как 4,4’–тиобисбензолтиол, 2,2–бис(4–меркаптофенил)пропан, 1,1–бис(4–меркаптофенил)–1–фенилэтан, 2,2–бис(4–меркаптофенил)бутан, бис(4–меркаптофенил)дифенилметан, 1,1–бис(4–меркаптофенил)этан, бис(4–меркаптофенил)метан, 1,1–бис(4–меркаптофенил)–3,3,5–триметилциклогексан и 1,1–бис(4–меркаптофенил)циклогексан.
[0054]
Пример соединения (ii), содержащего аминогруппу, включает C1–4 алкилендиаминовое соединение, такое как 1,2–пропилендиамин, 1,3–пропилендиамин, 1,2–этилендиамин и 1,4–бутилендиамин; фенилендиаминовое соединение, такое как 1,2–фенилендиамин и 1,4–фенилендиамин; гетероарилдитиольное соединение, такое как 4,6–диамино–2–метилпиримидин и 3,6–диамино–4–метилпиридазин; бисаминобензольное соединение, такое как 2,2–бис(4–аминофенил)пропан, 1,1–бис(4–аминофенил)–1–фенилэтан, 2,2–бис(4–аминофенил)бутан, бис(4–аминофенил)дифенилметан, 1,1–бис(4–аминофенил)этан, бис(4–аминофенил)метан, 1,1–бис(4–аминофенил)–3,3,5–триметилциклогексан и 1,1–бис(4–аминофенил)циклогексан.
[0055]
Использование C1–4 галогенированного углеводорода и соединения, содержащего нуклеофильную функциональную группу, не имеет особых ограничений при условии, что реакция протекает, и может быть получен целевой продукт, и например, даже когда используется 1–кратное молярное количество соединения, содержащего нуклеофильную функциональную группу, относительно числа моль С1–4 галогенированного углеводорода, реакция протекает. Молярное отношение соединения, содержащего нуклеофильную функциональную группу, к С1–4 галогенированному углеводороду, т.е. [соединение, содержащее нуклеофильную функциональную группу] / [С1–4 галогенированный углеводород], предпочтительно составляет 0,001 или более и 1 или менее с точки зрения эффективности реакции и времени реакции. Молярное отношение более предпочтительно составляет 0,01 или более, еще более предпочтительно 0,1 или более, и более предпочтительно 0,8 или менее, еще более предпочтительно 0,5 или менее. Когда молярное отношение является слишком большим, количество соединения, содержащего нуклеофильную функциональную группу, становится пропорционально большим, и количество непрореагировавшего соединения, содержащего нуклеофильную функциональную группу, увеличивается. С другой стороны, когда молярное отношение является слишком малым, количество непрореагировавшего С1–4 галогенированного углеводорода увеличивается, и может быть утечка галогенированного карбонила из реакционной системы. Когда C1–4 галогенированный углеводород является жидким при обычной температуре и обычном давлении и может использоваться в качестве растворителя, отношение соединения, содержащего нуклеофильную функциональную группу, к C1–4 галогенированному углеводороду может составлять 1 мг/мл или более и 500 мг/мл или менее.
[0056]
3. Основание
Одно или более оснований, выбранных из группы, в основном состоящей из гетероциклического ароматического амина, ненуклеофильного сильного основания и неорганического основания, используются в способе настоящего изобретения. Реакция может продолжаться до тех пор, пока поликарбонатное производное образуется с помощью основания.
[0057]
Гетероциклический ароматический амин означает соединение, имеющее по меньшей мере одно гетероциклическое кольцо и по меньшей мере одну функциональную аминогруппу. Пример такого гетероциклического ароматического амина включает пиридин и его производное, в том числе пиридин, α–пиколин, β–пиколин, γ–пиколин, 2,3–лутидин, 2,4–лутидин, 2,6–лутидин, 3,5–лутидин, 2–хлорпиридин, 3–хлорпиридин и 4–хлорпиридин.
[0058]
«Ненуклеофильное сильное основание» означает основание, в котором нуклеофильность неподеленной пары на атоме азота является слабой из–за стерической обструкции, и основность которого (pKBH+) в ацетонитриле составляет 20 или более. Пример такого ненуклеофильного сильного основания включает 1,5,7–триазабицикло[4.4.0]дец–5–ен (TBD, pKBH+: 25,98), 7–метил–1,5,7–триазабицикло[4.4.0]дец–5–ен (MTBD, pKBH+: 25,44), 1,8–диазабицикло[5.4.0]ундец–7–ен (DBU, pKBH+: 24,33), 1,5–диазабицикло[4.3.0]нон–5–ен (DBN, pKBH+: 23,89) и 1,1,3,3–тетраметилгуанидин (TMG, pKBH+: 23,30).
[0059]
Пример неорганического основания включает гидроксид щелочного металла, такой как гидроксид лития, гидроксид натрия и гидроксид калия; гидроксид щелочноземельного металла, такой как гидроксид кальция; карбонат щелочного металла, такой как карбонат натрия и карбонат калия; карбонат щелочноземельного металла, такой как карбонат кальция; гидрокарбонат щелочного металла, такой как гидрокарбонат натрия.
[0060]
Неорганическое основание может быть измельчено непосредственно перед использованием для добавления в реакционную смесь, или предпочтительно добавляют его водный раствор. Концентрация водного раствора неорганического основания может быть соответствующим образом скорректирована и, например, может быть доведена до 0,05 г/мл или более и 2 г/мл или менее. Водный раствор неорганического основания используется для разложения фосгена. В частности, фосген разлагается водой на углекислый газ и хлористый водород, и хлористый водород может быть нейтрализован неорганическим основанием. Поскольку автор изобретения полагал, что реакция настоящего изобретения протекает через фосген, примечательным является тот факт, что реакция настоящего изобретения протекает даже тогда, когда используется водный раствор неорганического основания, как в описанном ниже примере. Кроме того, поскольку реакция настоящего изобретения протекает даже при использовании водного раствора неорганического основания, реакция может протекать без прохождения через фосген.
[0061]
Одно основание может быть использовано отдельно, или два или более основания могут использоваться в комбинации.
[0062]
Использование основания может быть соответствующим образом скорректировано при условии, что реакция успешно продолжается, и например, отношение к соединению, содержащему нуклеофильную функциональную группу, может быть доведено в молях до 1,5–кратного или более и 10–кратного или менее. Когда используется больше основания, – выход обычно становится больше; следовательно, отношение в молях предпочтительно 2,0–кратное или более, более предпочтительно 3,0–кратное или более и еще более предпочтительно 4,0–кратное или более.
[0063]
4. Условия реакции
Способ настоящего изобретения включает стадию облучения светом композиции, содержащей C1–4 галогенированный углеводород, соединение, содержащее нуклеофильную функциональную группу, и основание, в присутствии кислорода.
[0064]
Способ смешивания С1–4 галогенированного углеводорода, соединения, содержащего нуклеофильную функциональную группу, и основания не имеет особых ограничений. Например, полное количество каждого соединения может быть предварительно смешано в реакционном сосуде, соединения могут добавляться несколькими порциями или непрерывно добавляться с любой скоростью. Когда одно или оба из С1–4 галогенированного углеводорода и соединения, содержащего нуклеофильную функциональную группу, не являются жидкими при обычной температуре и обычном давлении, можно использовать растворитель, который может надлежащим образом растворять исходные соединения и который не ингибирует реакцию настоящего изобретения. Пример такого растворителя включает алифатический углеводородный растворитель, такой как н–гексан; ароматический углеводородный растворитель, такой как бензол, толуол, ксилол и хлорбензол; простой эфирный растворитель, такой как простой диэтиловый эфир, тетрагидрофуран и диоксан; и нитрильный растворитель, такой как ацетонитрил.
[0065]
Источником кислорода может быть газ, содержащий кислород, и, например, может использоваться воздух или очищенный кислород. Очищенный кислород может быть смешан с инертным газом, таким как азот и аргон, для использования. Предпочтительно использовать воздух с точки зрения стоимости и легкости. Содержание кислорода в воздухе, используемом в качестве источника кислорода, предпочтительно составляет примерно 15 об.% или более и примерно 100 об.% или менее с точки зрения высокой эффективности разложения C1–4 галогенированного углеводорода при облучении светом. Содержание кислорода может быть подходящим образом определено в зависимости от вида C1–4 галогенированного углеводорода или тому подобного. Например, когда в качестве С1–4 галогенированного углеводорода используется C1–4 хлоруглеводородное соединение, такое как дихлорметан, хлороформ и тетрахлорэтилен, содержание кислорода предпочтительно составляет 15 об.% или более и 100 об.% или менее. Когда используется C1–4 бромуглеводородное соединение, такое как дибромметан и бромоформ, содержание кислорода предпочтительно составляет 90 об.% или более и 100 об.% или менее. Даже когда используется кислород или содержание кислорода составляет 100 об.%, содержание кислорода можно регулировать в вышеописанном диапазоне путем корректировки скорости потока кислорода в реакционную систему. Способ подачи газа, содержащего кислород, не имеет особых ограничений, и газ может подаваться в реакционную систему из кислородного баллона, снабженного регулятором расхода, или из устройства, генерирующего кислород.
[0066]
Термин «в присутствии кислорода» означает любое одно из состояний, в котором описанное выше каждое соединение контактирует с кислородом, и состояние, в котором присутствует кислород в описанной выше композиции. Реакцию настоящего изобретения можно проводить в потоке газа, содержащего кислород, но предпочтительно подавать газ, содержащий кислород, в композицию путем барботирования с точки зрения высокого выхода продукта.
[0067]
Количество кислородсодержащего газа может быть надлежащим образом определено в зависимости от количества C1–4 галогенированного углеводорода или формы реакционного сосуда. Например, количество газа, подаваемого в реакционный сосуд за 1 мин относительно C1–4 галогенированного углеводорода в реакционном сосуде, предпочтительно в 5 или более раз больше по объему. Отношение более предпочтительно в 25 или более раз больше по объему, и еще более предпочтительно – в 50 или более раз больше по объему. Верхний предел отношения не имеет особых ограничений, и отношение предпочтительно в 500 или менее раз больше по объему, более предпочтительно в 250 или менее раз больше по объему и еще более предпочтительно в 150 или менее раз больше по объему. Количество кислорода, подаваемого в реакционный сосуд за 1 мин, относительно С1–4 углеводородного соединения в реакционном сосуде может быть в 5 или более раз больше по объему и в 25 или менее раз больше по объему. Когда количество газа является слишком большим, C1–4 углеводородное соединение может улетучиваться, но когда это количество является слишком малым, может быть затруднено развитие реакции.
[0068]
Свет, облучающий композицию, предпочтительно представляет собой свет, содержащий коротковолновый свет, более предпочтительно свет, содержащий ультрафиолетовый свет, и предпочтительно свет, содержащий свет, в частности имеющий длину волны 180 нм или более и 500 нм или менее. Длина волны света может быть соответствующим образом определена в зависимости от вида С1–4 галогенированного углеводорода и составляет более предпочтительно 400 нм или менее и еще более предпочтительно 300 нм или менее. Когда облучающий свет содержит свет описанного выше диапазона длин волн, С1–4 галогенированный углеводород подвергается окислительному фотохимическому разложению эффективным образом.
[0069]
Средство для облучения светом не имеет особых ограничений при условии, что с помощью этого средства может излучаться свет описанной выше длины волны. Пример источника света, имеющего такой диапазон длин волн, включает солнечный свет, ртутную лампу низкого давления, ртутную лампу среднего давления, ртутную лампу высокого давления, ртутную лампу сверхвысокого давления, химическую лампу, лампу черного света, металлогалогенную лампу и светодиодную лампу. Ртутная лампа низкого давления предпочтительно используется с точки зрения эффективности реакции и стоимости.
[0070]
Условия, такие как интенсивность облучающего света, время облучения или тому подобное, могут быть соответствующим образом определены в зависимости от вида и количества используемых исходных соединений, и например, интенсивность облучающего света предпочтительно составляет 10 мкВт/см2 или более и 500 мкВт/см2 или менее. Интенсивность облучающего света более предпочтительно составляет 100 мкВт/см2 или менее, и еще более предпочтительно 40 мкВт/см2 или менее. Время облучения предпочтительно составляет 0,5 ч или более и 10 ч или менее, более предпочтительно 1 ч или более и 6 ч или менее, и еще более предпочтительно 2 ч или более и 4 ч или менее. Способ облучения светом также не имеет особых ограничений, и могут быть выбраны любые способы. Например, облучение светом может быть непрерывным с начала реакции и до завершения реакции, периоды облучения светом и его прекращения могут поочередно повторяться, и облучение светом может происходить с момента начала реакции только в течение заданного времени. Предпочтительно непрерывное облучение светом с начала реакции до завершения реакции.
[0071]
Температура во время реакции не имеет особых ограничений и может быть соответствующим образом отрегулирована, и, например, может быть доведена до 0°C или выше и до 50°C или ниже. Температура более предпочтительно составляет 10°С или выше, еще более предпочтительно 20°С или выше и более предпочтительно 40°С или ниже, еще более предпочтительно 30°С или ниже.
[0072] Примером реакционного устройства, используемого в способе получения настоящего изобретения, является реакционный сосуд, снабженный средством облучения светом. Реакционное устройство может быть снабжено перемешивающим устройством и средством регулирования температуры. Один из вариантов осуществления реакционного устройства, используемого в способе получения настоящего изобретения, показан на фиг.1. Реакционное устройство, показанное на фиг.1, имеет средство 1 облучения светом в трубчатом реакционном сосуде 6. Описанные выше исходные соединения добавляют в трубчатый реакционный сосуд 6 и облучают светом с использованием средства 1 облучения светом, в то время как газ, содержащий кислород, подается в трубчатый реакционный сосуд 6 или газ, содержащий кислород, вдувается в композицию, чтобы вызвать барботирование (не показано на фигуре) для осуществления реакции. Когда средство 1 облучения светом покрыто рубашкой 2 или тому подобным, предпочтительно, чтобы рубашка состояла из материала, через который проникает коротковолновый свет. Облучение светом может происходить снаружи реакционного сосуда. В таком случае реакционный сосуд состоит из материала, через который проникает коротковолновый свет. Материал, через который проникает коротковолновый свет, не имеет особых ограничений при условии, что эффект настоящего изобретения не ингибируется, и предпочтительно представляет собой кварцевое стекло.
[0073]
Продукт, полученный в результате реакции, может быть очищен общеизвестным способом. Пример такого способа очистки включает перегонку, удаление исходных соединений при пониженном давлении, колоночную хроматографию, разделение жидкостей, экстракцию, промывку и перекристаллизацию.
[0074]
Когда соединение, содержащее гидроксильную группу, соединение, содержащее тиольную группу, или соединение, содержащее аминогруппу, используется в качестве исходного соединения, содержащего нуклеофильную функциональную группу, получают карбонатное производное, имеющее, соответственно, карбонатную сложноэфирную группу (–O–C(=O)–O–), карбонатную сложнодитиоэфирную группу (–S–C(=O)–S–) или мочевинную группу (–NH–C(=O)–NH–). Когда соединение, содержащее гидроксильную группу, и соединение, содержащее аминогруппу, используются в комбинации, получают карбонатное производное, имеющее уретановую группу (–O–C(=O)–NH–). Когда соединение, содержащее тиольную группу, и соединение, содержащее аминогруппу, используются в комбинации, получают карбонатное производное, имеющее тиоуретановую группу (–S–C(=O)–NH–).
[0075]
Линейное карбонатное производное (I), полученное способом настоящего изобретения, может быть использовано в качестве неводного растворителя или тому подобного. Например, линейный карбонат (I) может использоваться в качестве электролитного растворителя литий–ионной вторичной батареи. Поликарбонат (II) может использоваться в качестве превосходного конструкционного пластика.
[0076]
Настоящая заявка испрашивает преимущество на дату подачи над японской патентной заявкой № 2017–97681, поданной 16 мая 2017. Все содержание японской патентной заявки № 2017–97681, поданной 16 мая 2017, включено в настоящее описание посредством ссылки.
ПРИМЕРЫ
[0077]
Далее настоящее изобретение будет описано более подробно с помощью примеров. Однако настоящее изобретение никоим образом не ограничивается следующими примерами, и можно осуществить настоящее изобретение в соответствии с данными примерами с дополнительными соответствующими изменениями в пределах вышеприведенных описаний и последующих описаний. Такой измененный вариант осуществления также входит в технический объем настоящего изобретения.
[0078]
Сравнительный пример 1: Синтез диметилкарбоната
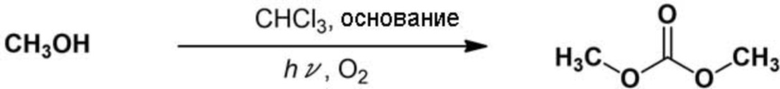
Рубашку из кварцевого стекла диаметром 30 мм вставляли в трубчатый реакционный сосуд, имеющий диаметр 42 мм и объем 100 мл, и ртутную лампу низкого давления («UVL20PH–6» производства SEN Light, 20 Вт, Ø24 × 120 мм) далее вставляли в рубашку из кварцевого стекла для сбора реакционной системы. Схематическое изображение реакционной системы показано на фиг.1. В реакционный сосуд добавляли очищенный хлороформ (20 мл), метанол (0,405 мл, 10 ммоль) и 5–кратное количество моль пиридина (4,03 мл) относительно количества метанола. Смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Реакционную смесь анализировали с помощью 1H–ЯМР через 3 ч; в результате было подтверждено, что выход диметилкарбоната в качестве целевого соединения составлял почти 1,6%.
[0079]
Сравнительный пример 2
Реакцию проводили аналогично описанному выше сравнительному примеру 1, за исключением того, что вместо метанола использовали этанол. Не подтверждалось, что реакция протекала даже спустя 3 ч. Принимая во внимание данный результат, наряду с результатом сравнительного примера 1, становится очевидно, что способ настоящего изобретения трудно применить к одновалентному спирту.
[0080]
Пример 1: Синтез дифенилкарбоната

(1) Использование пиридина в качестве основания
Реакцию проводили аналогично описанному выше сравнительному примеру 1, за исключением того, что вместо метанола использовали фенол (0,94 г, 10 ммоль), используемое количество пиридина в расчете на моли доводили до 3,5–кратного количества относительно фенола, и время реакции составляло 2 ч. После реакции к реакционной смеси добавляли воду и дихлорметан, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия, и затем растворитель отгоняли при пониженном давлении. Полученное таким образом твердое вещество перекристаллизовывали с использованием дихлорметана и н–гексана с получением дифенилкарбоната в качестве белого твердого целевого соединения (выход выделенного продукта: 61%).
[0081]
(2) Использование пиридина в качестве основания
Дифенилкарбонат в виде белого твердого целевого соединения получали (выход выделенного продукта: более 99%) аналогично описанному выше примеру 1(1), за исключением того, что использовали 5–кратное молярное количество пиридина относительно фенола, и время реакции составляло 1 ч.
[0082]
(3) Использование 2,6–лутидина в качестве основания
Дифенилкарбонат в качестве целевого соединения получали (выход выделенного продукта: 60%) аналогично описанному выше примеру 1(1), за исключением того, что использовали 2,6–лутидин вместо пиридина, и время реакции составляло 1 ч.
[0083]
(4) Использование четыреххлористого углерода
Реакцию проводили аналогично описанному выше примеру 1(1), за исключением того, что вместо хлороформа использовали четыреххлористый углерод (25 мл) и 5–кратное молярное количество пиридина относительно фенола. После реакции к реакционной смеси добавляли воду и дихлорметан, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия, и затем растворитель отгоняли при пониженном давлении. Полученное таким образом твердое вещество перекристаллизовывали с использованием дихлорметана и н–гексана с получением дифенилкарбоната в качестве белого твердого целевого соединения (выход выделенного продукта: 69%).
[0084]
Сравнительный пример 3
Реакцию проводили аналогично описанному выше примеру 1(2), за исключением того, что вместо пиридина использовали триэтиламин. Однако было выделено лишь небольшое количество черного смолистого вещества, и дифенилкарбонат в качестве целевого соединения выделить не удалось. Как только что было описано, когда в качестве органического основания использовали триэтиламин, дифенилкарбонат получить не удавалось; с другой стороны дифенилкарбонат мог быть получен с выходом более 99% только за счет замены органического основания с триэтиламина на пиридин.
[0085]
Пример 2: Синтез бис(пентахлорфенил)карбоната
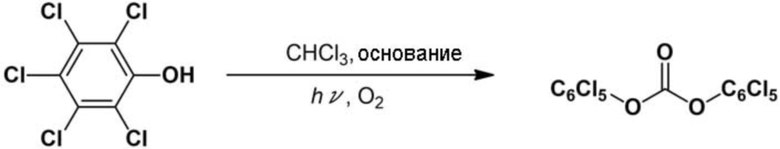
Реакцию проводили аналогично описанному выше сравнительному примеру 1, за исключением того, что вместо метанола использовали пентaхлорфенол (1,13 г, 5 ммоль). После реакции к суспендированной реакционной смеси добавляли метанол с образованием белого твердого вещества. Образовавшееся белое твердое вещество собирали фильтрованием с отсасыванием в качестве целевого соединения, бис(пентахлорфенил)карбоната (выход выделенного продукта: 72%).
[0086]
Пример 3: Синтез 1,3–бензодиоксол–2–она
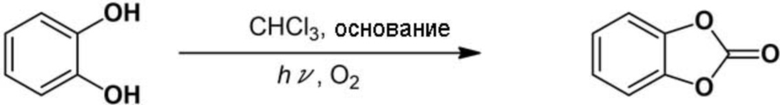
Очищенный хлороформ (20 мл), катехол (1,1 г, 10 ммоль) и 5–кратное молярное количество пиридина (4,03 мл) относительно катехола добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Через 2 ч к реакционной смеси добавляли воду и дихлорметан, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия, и затем растворитель отгоняли при пониженном давлении. Полученное таким образом твердое вещество перекристаллизовывали с использованием дихлорметана и н–гексана с получением целевого 1,3–бензодиоксол–2–она (выход выделенного продукта: более 99%).
[0087]
Пример 4: Синтез этиленкарбоната
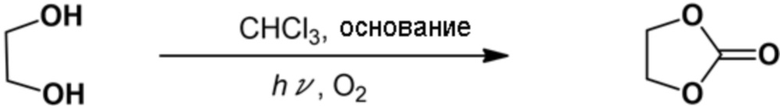
Реакцию проводили аналогично описанному выше примеру 3, за исключением того, что вместо катехина использовали этиленгликоль (0,28 мл, 10 ммоль). После реакции реакционную смесь анализировали с помощью 1Н–ЯМР для подтверждения образования целевого этиленкарбоната (выход выделенного продукта: 44%).
[0088]
Пример 5: Синтез поликарбоната бисфенола А

Очищенный хлороформ (20 мл), бисфенол А (2,28 г, 10 ммоль) и 5–кратное молярное количество пиридина (4,03 мл) относительно бисфенола А добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Поскольку перемешивающий стержень не мог вращаться из–за повышенной вязкости реакционной смеси через 40 мин, к реакционной смеси добавляли метанол (30 мл), облучали ультразвуковыми волнами и реакционную смесь фильтровали путем отсасывания. Полученное твердое вещество промывали метанолом и затем сушили под вакуумом с получением белого твердого вещества. Белое твердое вещество анализировали с помощью 1H–ЯМР; в результате было подтверждено, что целевой поликарбонат бисфенола А может быть получен с выходом более 99%.
Полученный поликарбонат бисфенола А анализировали методом гель–проникающей хроматографии (ГПХ) в следующих условиях с определением молекулярной массы. Результаты показаны в таблице 1.
Устройство: устройство высокоэффективной ГПХ («HLC–8320GPC» производства Tosoh Corporation)
Колонка: Колонка для сверхвысокомолекулярных соединений («TSKgel GMHHR–H×2» производства Tosoh Corporation)
Подвижная фаза: хлороформ; Скорость потока: 1,0 мл/мин
Температура печи: 40°C; Концентрация: 0,3% масс./об.
Впрыскиваемое количество: 100 мкл
Стандарт молекулярной массы: полистирол
Детектор: RI
[0089]
Таблица 1.
[0090]
По результату, представленному в таблице 1, было обнаружено, что сложный поликарбонатный эфир, синтезированный способом настоящего изобретения, имеет достаточно высокую молекулярную массу, и молекулярно–массовое распределение является относительно узким.
[0091]
Пример 6: Синтез карбоната бис(простого монометилового эфира триэтиленгликоля)

Реакцию проводили аналогично описанному выше сравнительному примеру 1, за исключением того, что вместо метанола использовали простой монометиловый эфир триэтиленгликоля (1,64 г, 10 ммоль). После реакции к реакционной смеси добавляли воду и смешанный растворитель дихлорметан:этилацетат=1:1, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении с получением карбоната бис(простого монометилового эфира триэтиленгликоля) в виде коричневого маслянистого целевого соединения (выход выделенного продукта: более 99%).
[0092]
Пример 7: Синтез поликарбоната тетраэтиленгликоля

Реакцию проводили аналогично описанному выше сравнительному примеру 1, за исключением того, что вместо метанола использовали тетраэтиленгликоль (1,50 г, 10 ммоль), и время реакции составляло 2 ч. После реакции к реакционной смеси добавляли воду и этилацетат, и органическую фазу и водную фазу разделяли. Органическую фазу трижды промывали соленой водой. Органическую фазу сушили с помощью безводного сульфата натрия, и затем концентрировали при пониженном давлении с получением поликарбоната тетраэтиленгликоля в виде коричневого маслянистого целевого соединения (выход выделенного продукта: более 99%).
1H–NMR (400 МГц, CDCl3) δ 4,28 (т, J=4,8 Гц, –CO2CH2–), 3,73 (т, J=4,8 Гц, –CH2–), 3,68–3,63 (м, –CH2–);
FAB–MS: m/z 519, 739, 958;
ИК (KBr): 2955, 2891, 1740, 1459, 1396, 1354, 1271, 1100, 1029, 950, 864, 791 см–1
[0093]
Пример 8: Фотоиндуцированная сополимеризация бисфенола А и гексаметилендиамина

Очищенный хлороформ (30 мл), бисфенол А (0,685 г, 3 ммоль), гексаметилендиамин (0,412 г, 3 ммоль) и водный раствор гидроксида натрия (20 мл, 100 ммоль) добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Через 2 ч воду и дихлорметан добавляли в реакционную смесь. Образовавшийся осадок получали фильтрованием, промывали метанолом и сушили под вакуумом при 70°С. Органическую фазу и водную фазу фильтрата разделяли. Органическую фазу концентрировали при пониженном давлении. Полученный таким образом остаток промывали метанолом и затем сушили под вакуумом при 70°С с получением светло–оранжевого порошка (выход: 39%). Полученный порошок анализировали с помощью 1H–ЯМР и ИК, в результате чего было подтверждено образование целевого сополимера.
Как описано выше, даже при использовании водного раствора неорганического основания может быть получено карбонатное производное. Поскольку для разложения фосгена использовали водный раствор неорганического основания, то вышеописанный экспериментальный результат никоим образом не ожидается, и считается, что реакция по настоящему изобретению может протекать без прохождения через фосген.
Поскольку первоначально полученный осадок был нерастворим в растворителе, и порошок, полученный из фильтрата, был растворим в ДМСО или тому подобном, молекулярные массы обоих порошков могут быть различными.
[0094]
Пример 9: Синтез бисфенилкарбоната

Очищенный хлороформ (20 мл), фенол (0,941 г, 10 ммоль) и водный раствор гидроксида натрия (20 мл, 100 ммоль) добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Через 3 ч воду и дихлорметан добавляли в реакционную смесь. Органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении при 70°С с получением светло–оранжевого твердого вещества (выход: 55%). Полученное твердое вещество анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0095]
Пример 10: Синтез дициклогексилкарбоната

Светло–желтую жидкость получали (выход: 13%) аналогично описанному выше примеру 9, за исключением того, что вместо фенола использовали циклогексанол (1,06 мл, 10 ммоль). Полученную жидкость анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0096]
Пример 11: Синтез бис(4–трет–бутилфенил)карбоната

Очищенный хлороформ (20 мл), 4–трет–бутилфенол (1,53 г, 10 ммоль), водный раствор карбоната натрия (20 мл, 50 ммоль) и пиридин (0,202 мл, 5 ммоль) добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Через 3 ч к реакционной смеси добавляли хлороформ и воду, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении при 70°С. Полученный таким образом остаток перекристаллизовывали с получением светло–оранжевого порошка (выход: 57,0%). Полученный порошок анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0097]
Пример 12: Синтез бис(4–метоксифенил)карбоната

Коричневое твердое вещество получали (выход: 60%) аналогично описанному выше примеру 9, за исключением того, что вместо фенола использовали 4–метоксифенол (10 ммоль) и использовали 30 мл хлороформа. Полученное твердое вещество анализировали с помощью 1H–ЯМР и ИК, в результате чего было подтверждено образование целевого соединения.
[0098]
Пример 13: Синтез бис(4–нитрофенил)карбоната
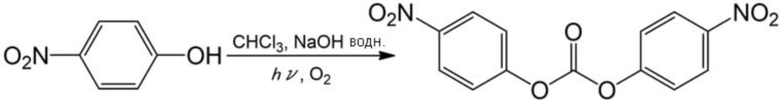
Белый порошок получали (выход: 5%) аналогично описанному выше примеру 9, за исключением того, что вместо фенола использовали 4–нитрофенол (1,391 г, 10 ммоль) и использовали 30 мл хлороформа, и время реакции составляло 2 ч. Полученный порошок анализировали с помощью 1H–ЯМР и ИК, в результате чего было подтверждено образование целевого соединения.
[0099]
Пример 14: Синтез поликарбоната бисфенола А

Очищенный хлороформ (20 мл), бисфенол А (1,14 г, 5 ммоль) и водный раствор гидроксида натрия (100 ммоль, 20 мл) добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Через 2 ч органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении. Затем туда добавляли хлороформ и метанол и растворитель удаляли декантацией, остаток сушили при 70°С при пониженном давлении с получением белого твердого вещества (выход: 79%). Полученное твердое вещество анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
Молекулярную массу полученного поликарбоната бисфенола А определяли в условиях, аналогичных описанному выше примеру 5. Результаты показаны в таблице 2.
[0100]
Таблица 2
[0101]
По результату, представленному в таблице 2, было обнаружено, что сложный поликарбонатный эфир, синтезированный способом настоящего изобретения, имеет достаточно высокую молекулярную массу, и молекулярно–массовое распределение является относительно узким.
[0102]
Пример 15: Синтез дигексилкарбоната

Реакцию проводили аналогично описанному выше примеру 9, за исключением того, что вместо фенола использовали 1–гексанол (1,25 мл, 10 ммоль). Реакционную смесь сушили с помощью безводного сульфата натрия. К ней добавляли дихлорметан (0,64 мл, 10 ммоль) в качестве внутреннего стандарта, и реакционную смесь непосредственно анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения (выход: >99%).
[0103]
Пример 16: Синтез дипентилкарбоната

Реакцию проводили аналогично описанному выше примеру 9, за исключением того, что вместо фенола использовали 1–пентанол (10 ммоль). Реакционную смесь сушили с помощью безводного сульфата натрия. К ней добавляли дихлорметан (0,64 мл, 10 ммоль) в качестве внутреннего стандарта, и реакционную смесь непосредственно анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения (выход: 12%).
[0104]
Пример 17: Синтез 1,3–дифенилмочевины

Очищенный хлороформ (20 мл), анилин (0,93 г, 10 ммоль) и водный раствор гидроксида натрия (NaOH: 4 г, 20 мл) добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Через 2 ч к реакционной смеси добавляли дихлорметан и воду, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении. Полученное таким образом черное твердое вещество перекристаллизовывали с использованием дихлорметана и н–гексана с получением черного порошка (выход по массе: 0,13 г, выход: 12%). Полученное твердое вещество анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0105]
Пример 18: Синтез 1,3–дициклогексилмочевины
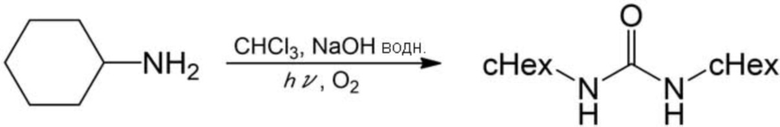
Получали белый порошок (выход по массе: 0,69 г, выход: 62%) аналогично описанному выше примеру 17, за исключением того, что вместо анилина использовали циклогексиламин (1,17 мл, 10 ммоль), время реакции составляло 3 ч, и после реакции осадок, образованный добавлением гексана и воды, получали фильтрованием и сушили под вакуумом. Полученный порошок анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0106]
Пример 19: Синтез 1,3–дибензилмочевины

Получали светло–коричневый порошок в качестве целевого соединения (выход по массе: 0,78 г, выход: 65%) аналогично описанному выше примеру 17, за исключением того, что вместо анилина использовали бензиламин (1,07 г, 10 ммоль), время реакции составляло 5 ч, и после реакции к реакционной смеси добавляли гексан и воду, чтобы получить осадок путем фильтрования, и осадок сушили под вакуумом. Полученный порошок анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0107]
Пример 20: Синтез 1,3–дигексилмочевины

Получали белый порошок (выход по массе: 0,58 г, выход: 51%) аналогично описанному выше примеру 17, за исключением того, что вместо анилина использовали 1–гексиламин (1,01 г, 10 ммоль), температура реакции составляла 10°С, и время реакции составляло 3 ч. Полученный порошок анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0108]
Пример 21: Синтез 1,3–дигексилмочевины

Получали желтые кристаллы (выход по массе: 0,08 г, выход: 14%) аналогично описанному выше примеру 17, за исключением того, что вместо анилина использовали гидрохлорид этиламина (0,82 г, 10 ммоль), температура реакции была 10°С, время реакции составляло 5 ч, и после реакции вместо дихлорметана использовали этилацетат. Полученные кристаллы анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0109]
Пример 22: Синтез 1,3–дипиперидинилмочевины
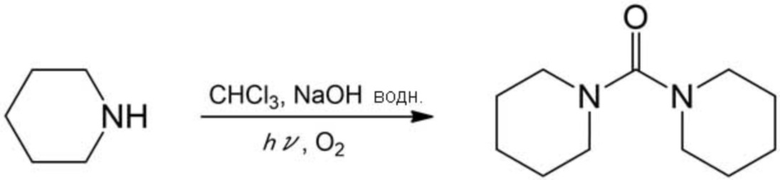
Получали желтые кристаллы (выход по массе: 0,38 г, выход: 38%) аналогично описанному выше примеру 17, за исключением того, что вместо анилина использовали пиперидин (0,85 г, 10 ммоль), время реакции составляло 3 ч, и целевое соединение очищали, используя короткую колонку с силикагелем (элюент: дихлорметан). Полученные кристаллы анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0110]
Пример 23: Фотоиндуцированная сополимеризация бисфенола А и гексаметилендиамина
Реакцию проводили при 20°С в течение 2 ч аналогично описанному выше примеру 8, за исключением того, что вместо водного раствора гидроксида натрия использовали диазабициклоундецен (60 ммоль). Затем реакцию дополнительно проводили при 50°С в течение 15 мин. После реакции добавляли воду и реакционную смесь оставляли стоять в течение ночи. Затем органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении. Остаток промывали гексаном и сушили при 70°С при пониженном давлении. После того, как остаток дополнительно промывали дихлорметаном и гексаном, остаток сушили при пониженном давлении с получением светло–оранжевого порошка (выход: > 99%). Полученный порошок анализировали с помощью 1H–ЯМР и ИК, в результате чего было подтверждено образование целевого сополимера.
[0111]
Пример 24: Синтез 1,3–дифенилмочевины

Очищенный хлороформ (20 мл), анилин (0,93 г, 10 ммоль) и пиридин (4,01 мл, 50 ммоль) добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Через 2 ч к реакционной смеси добавляли дихлорметан и воду, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении. Полученный таким образом остаток растворяли в дихлорметане и этилацетате и анилиновую сажу удаляли пропусканием раствора через колонку с оксидом алюминия. Жидкость, очищенную колонкой, концентрировали при пониженном давлении и остаток перекристаллизовывали, используя этилацетат и гексан, с получением светло–коричневых игольчатых кристаллов (выход по массе: 0,54 г, выход: 51%). Полученные кристаллы анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0112]
Пример 25: Синтез 1,3–дифенилмочевины
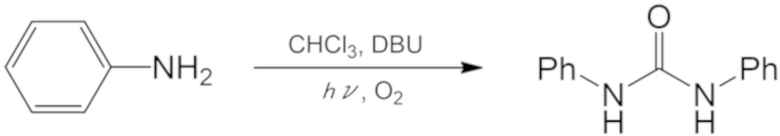
Реакцию осуществляли при 20°С в течение 2 ч аналогично описанному выше примеру 24, за исключением того, что вместо пиридина использовали диазабициклоундецен (7,48 мл, 50 ммоль). После реакции к реакционной смеси добавляли дихлорметан и воду, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении. Полученный таким образом остаток растворяли в THF и примесь удаляли пропусканием раствора через колонку с оксидом алюминия. Жидкость, очищенную колонкой, концентрировали при пониженном давлении и остаток перекристаллизовывали, используя дихлорметан и гексан, с получением светло–оранжевых кристаллов (выход по массе: 0,44 г, выход: 38%).
[0113]
Пример 26: Синтез 1,3–дициклогексилмочевины

Реакцию осуществляли при 20°С в течение 4 ч аналогично описанному выше примеру 24, за исключением того, что вместо анилина использовали циклогексиламин (1,17 мл, 10 ммоль). После реакции добавляли дихлорметан и воду, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении. Остаток перекристаллизовывали, используя дихлорметан и гексан, с получением светло–коричневых кристаллов (выход по массе: 0,16 г, выход: 14%). Полученные кристаллы анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения.
[0114]
Пример 27: Синтез полимочевины
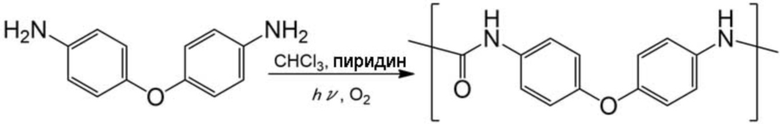
Очищенный хлороформ (20 мл), простой 4,4’–диаминодифениловый эфир (0,50 г, 2,5 ммоль) и пиридин (1,0 мл, 12,5 ммоль) добавляли в описанный выше реакционный сосуд, и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. Через 1,5 ч к реакционной смеси добавляли дихлорметан и воду, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении. Полученный таким образом остаток промывали метанолом с получением коричневого порошка (выход по массе: 0,14 г, выход: 25%). Полученные кристаллы анализировали с помощью 1H–ЯМР и ИК, в результате чего было подтверждено образование целевого соединения.
[0115]
Пример 28: Синтез карбонилдиимидазола

Очищенный хлороформ (20 мл), имидазол (0,68 г, 10 ммоль) и 2,6–лутидин (5,79 мл, 50 ммоль) добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин при 20°С, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления. После прекращения облучения реакцию проводили при 50°С в течение 30 мин. Дихлорметан (5 мл) добавляли к реакционной смеси в качестве внутреннего стандарта и реакционную смесь анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения с выходом 38%.
[0116]
Пример 29: Синтез S, S’–дифенилдитиокарбоната

Очищенный хлороформ (20 мл), тиофенол (1,03 мл, 10 ммоль) и водный раствор гидроксида натрия (NaOH: 4 г, 20 ммоль) добавляли в описанный выше реакционный сосуд и смесь перемешивали для смешивания. Газообразный кислород вдували в перемешиваемую реакционную смесь при скорости потока 0,5 л/мин, чтобы вызвать барботирование, и облучали светом от ртутной лампы низкого давления для осуществления реакции при 20°С в течение 2 ч. Затем к реакционной смеси добавляли дихлорметан и воду, и органическую фазу и водную фазу разделяли. Органическую фазу сушили с помощью безводного сульфата натрия и затем концентрировали при пониженном давлении с получением коричневой жидкости. Полученную коричневую жидкость анализировали с помощью 1H–ЯМР, в результате чего было подтверждено образование целевого соединения с выходом 20%.
ОПИСАНИЕ ССЫЛОЧНЫХ ПОЗИЦИЙ
[0117]
1: средство облучения светом,
2: рубашка,
3: водяная ванна,
4: перемешивающий стержень,
5: теплоноситель или охлаждающая среда,
6. трубчатый реакционный сосуд

Изобретение относится к способу получения карбонатного производного, включающему облучение светом композиции, содержащей галогенированный метан, имеющий один или более видов атомов галогена, выбранных из группы, состоящей из атома хлора, атома брома и атома йода, соединение, содержащее нуклеофильную функциональную группу, и основание в присутствии кислорода, где соединение, содержащее нуклеофильную функциональную группу, представлено следующей формулой (i) и карбонатное производное представляет собой линейное карбонатное производное, представленное следующей формулой (I), или соединение, содержащее нуклеофильную функциональную группу, представленное следующей формулой (ii), бисфенолом A, бисфенолом AP, бисфенолом B, бисфенолом BP, бисфенолом TMC и бисфенолом Z, и карбонатное производное представляет собой поликарбонатное производное, содержащее фрагмент, представленный следующей формулой (II–1), сложный поликарбонатный эфир бисфенола A, бисфенола AP, бисфенола B, бисфенола BP, бисфенола TMC или бисфенола Z, или циклическое карбонатное производное, представленное следующей формулой (II–2), и где основание представляет собой одно или более оснований, выбранных из группы, состоящей из гетероциклического ароматического амина, ненуклеофильного сильного основания и неорганического основания, где гетероциклический ароматический амин представляет собой пиридин или производное пиридина, выбранное из группы, состоящей из α-пиколина, β-пиколина, γ-пиколина, 2,3-лутидина, 2,4-лутидина, 2,6-лутидина, 3,5-лутидин, 2-хлорпиридин, 3-хлорпиридин и 4-хлорпиридин, где ненуклеофильное сильное основание представляет собой 1,5,7–триазабицикло[4.4.0]дец–5–ен, 7–метил–1,5,7–триазабицикло[4.4.0]дец–5–ен, 1,8–диазабицикло[5.4.0]ундец–7–ен, 1,5–диазабицикло[4.3.0]нон–5–ен или 1,1,3,3–тетраметилгуанидин и где неорганическое основание представляет собой гидроксид щелочного металла, карбонат щелочного металла или гидрокарбонат щелочного металла (значения для групп в структурных формулах приведены в описании). Целью настоящего изобретения является предложить способ получения карбонатного производного безопасным и эффективным образом. 5 з.п. ф-лы, 29 пр., 2 табл., 1 ил.
(i) R1–A–H
(ii) H–A–R2–A–H
(I) R1–A–C(=O)–A–R1
(II–1) [–A–R2–A–C(=O)–]
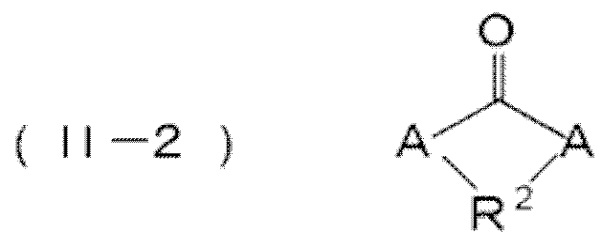
1. Способ получения карбонатного производного, включающий
облучение светом композиции, содержащей галогенированный метан, имеющий один или более видов атомов галогена, выбранных из группы, состоящей из атома хлора, атома брома и атома йода, соединение, содержащее нуклеофильную функциональную группу, и основание в присутствии кислорода,
где соединение, содержащее нуклеофильную функциональную группу, представлено следующей формулой (i), и карбонатное производное представляет собой линейное карбонатное производное, представленное следующей формулой (I), или
соединение, содержащее нуклеофильную функциональную группу, представленное следующей формулой (ii), бисфенолом A, бисфенолом AP, бисфенолом B, бисфенолом BP, бисфенолом TMC и бисфенолом Z, и карбонатное производное представляет собой поликарбонатное производное, содержащее фрагмент, представленный следующей формулой (II–1), сложный поликарбонатный эфир бисфенола A, бисфенола AP, бисфенола B, бисфенола BP, бисфенола TMC или бисфенола Z, или циклическое карбонатное производное, представленное следующей формулой (II–2), и
где основание представляет собой одно или более оснований, выбранных из группы, состоящей из гетероциклического ароматического амина, ненуклеофильного сильного основания и неорганического основания,
где гетероциклический ароматический амин представляет собой пиридин или производное пиридина, выбранное из группы, состоящей из α-пиколина, β-пиколина, γ-пиколина, 2,3-лутидина, 2,4-лутидина, 2,6-лутидина, 3,5-лутидин, 2-хлорпиридин, 3-хлорпиридин и 4-хлорпиридин,
где ненуклеофильное сильное основание представляет собой 1,5,7–триазабицикло[4.4.0]дец–5–ен, 7–метил–1,5,7–триазабицикло[4.4.0]дец–5–ен, 1,8–диазабицикло[5.4.0]ундец–7–ен, 1,5–диазабицикло[4.3.0]нон–5–ен или 1,1,3,3–тетраметилгуанидин, и
где неорганическое основание представляет собой гидроксид щелочного металла, карбонат щелочного металла или гидрокарбонат щелочного металла,
(i) R1–A–H,
(ii) H–A–R2–A–H,
(I) R1–A–C(=O)–A–R1,
(II–1) [–A–R2–A–C(=O)–],
 ,
,
где
A представляет собой O, S или NR3, где R3 представляет собой H или C1–4 алкильную группу или R3 образует азотсодержащую гетероциклическую группу с R1 и N,
R1 представляет собой C6–14 арильную группу, C4–14 гетероарильную группу или C2–24 алкилполиоксиалкиленовую группу,
R2 представляет собой C2–10 алкиленовую группу, C6–14 ариленовую группу, C4–14 гетероариленовую группу или C2–24 полиоксиалкиленовую группу.
2. Способ получения по п.1, в котором галогенированный метан представляет собой хлороформ.
3. Способ получения по любому из пп.1,2, в котором гетероциклический ароматический амин представляет собой пиридин, пиколин или лутидин.
4. Способ получения по любому из пп.1–3, в котором использовали 0,001–кратное или более и 1–кратное или менее молярное количество соединения, содержащего нуклеофильную функциональную группу, относительно галогенированного метана.
5. Способ получения по любому из пп.1–4, в котором использовали 1,5–кратное или более и 10–кратное или менее молярное количество основания относительно соединения, содержащего нуклеофильную функциональную группу.
6. Способ получения по любому из пп.1–5, в котором длина волны света, облучающего композицию, составляет 180 нм или более и 500 нм или менее.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| JP 2013181028 A, 12.09.2003 | |||
| В.Н | |||
| Коваленко "ЦИКЛОПРОПАНОЛЬНЫЙ ПОДХОД К ДИФФЕРЕНЦИАЦИИ ФУНКЦИОНАЛЬНЫХ ГРУПП ЯБЛОЧНОЙ КИСЛОТЫ" | |||

