Область техники
Изобретение относится к медицине, а именно к фармакологии, и касается новых химических соединений, обладающих высокой эффективностью в ингибировании P-гликопротеина, которые могут быть использованы в терапии заболеваний центральной нервной системы, инфекционных, аллергических, аутоиммунных, онкологических и прочих заболеваний. В частности, данные химические соединения могут быть использованы для подавления множественной лекарственной устойчивости у больных, длительно получающих различные лекарственные препараты.
Уровень техники
P-гликопротеин (P-gp) принадлежит к семейству ABC-переносчиков (ATP-binding cassette (ABC) transporters) - белков, осуществляющих ATФ-зависимый транспорт веществ через мембрану клетки против градиента концентраций [Higgins, C.F., Nature, 2007. 446(7137): p. 749-57]. Вследствие широкой субстратной специфичности, P-гликопротеин человека участвует в экспорте лекарственных веществ из клеток, что приводит к возникновению множественной лекарственной устойчивости [Borst, P. and R.O. Elferink, Annu Rev Biochem, 2002. 71: p. 537-92]. Множественная лекарственная устойчивость - это невосприимчивость клеток одновременно к целому ряду химиотерапевтических препаратов разного химического строения. Кроме того, поскольку специфические ABC-переносчики экспрессируются в патогенных микроорганизмах, обеспечивая резистентность к антибиотикам, то ингибирование бактериальных ABC-переносчиков может повысить эффективность антимикробной химиотерапии [Ouellette, M., D. Legare, and B. Papadopoulou, J Mol Microbiol Biotechnol, 2001. 3(2): p. 201-6]. P-gp активно экспрессируется в эндотелии капилляров головного мозга и играет ключевую роль в регулировании проницаемости гемато-энцефалического барьера для лекарств и ксенотоксинов [Schinkel, A.H., Adv Drug Deliv Rev, 1999. 36(2-3): p. 179-194].
Активное изучение P-гликопротеина продолжается более 30 лет и, по-видимому, P-гликопротеин является наиболее хорошо изученным белком из семейства ABC-переносчиков. За это время было найдено большее количество различных синтетических и природных соединений, способных ингибировать P-гликопротеин, приводя к повышению проникновения лекарственных веществ в клетку и подавлению множественной лекарственной устойчивости [Shukla, S., C.P. Wu, and S.V. Ambudkar, Expert Opin Drug Metab Toxicol, 2008. 4(2): p. 205-23].
К настоящему моменту известно три поколения ингибиторов P-гликопротеина: большинство ингибиторов P-gp первого поколения были разработаны до 1990 года и обладали собственной фармакологической активностью (например, Верапамил [US20020165120 (2002)], Циклоспорин [US200710060635A1 (2007), US20090088393 (2009), US200510288222A1 (2005)], Хинидин, Резерпин и т.д.), однако возможность их использования для подавления множественной лекарственной устойчивости была ограниченна высокой токсичностью этих соединений. Для устранения проблем, связанных с токсичностью соединений первого поколения, были предложены ингибиторы P-gp второго поколения (например, D-изомер Верапамила, который обладал существенно меньшей кардиотоксичностью по сравнению с рацематом) [Krishna, R. and L.D. Mayer, Eur J Pharm Sci, 2000. 11(4): p. 265-83]. В то же время, ингибиторы P-gp второго поколения обладали низкой селективностью относительно других белков из семейства ABC-переносчиков и за счет этого в ряде случаев приводили к развитию серьезных побочных эффектов [Thomas, H. and H.M. Coley, Cancer Control, 2003. 10(2): p. 159-65; Boesch, D., et al., Cancer Res, 1991. 51(16): p. 4226-33]. Интенсивные поиски новых, более специфичных и малотоксичных ингибиторов P-gp, привели к разработке третьего поколения соединений. Ингибиторы P-gp третьего поколения (Элакридар [Van Tellingen, O.Z., NL, Methods and Means for the Treatment of Cancer. 2009: United States; Kawamura, K., et al., Nucl Med Biol, 2009. 36(3): p. 239-46], Тариквидар [Pajeva, I.K. and M. Wiese, AAPS J, 2009. 11(3): p. 435-44], Зосиквидар [Sato, W., et al., Cancer Res, 1991. 51(9): p. 2420-4]) существенно превосходят ингибиторы первого и второго поколения, как по своей аффинности относительно P-гликопротеина, так и по селективности относительно других ABC-переносчиков. В то же время, клинические испытания большинства ингибиторов P-gp третьего поколения были прекращены или по причине недостаточной эффективности, или из-за серьезных побочных эффектов.
Многие природные соединения, такие как куркумин и флавоноиды (например, Кемпферол и Кверцетин), способны ингибировать P-гликопротеин и подавлять множественную лекарственную устойчивость [Limtrakul, P., O. Khantamat, and K. Pintha, J Chemother, 2005. 17(1): p. 86-95]. Ближайшим аналогом соединений данного изобретения является Куркумин I. Куркумин (смесь куркуминоидов, выделенных из порошка куркумы), широко использующийся в качестве специи, красителя и лечебного средства в восточной медицине, обладает ингибирующей активностью относительно P-гликопротеина [Chearwae, W., et al., Biochem Pharmacol, 2004. 68(10): p. 2043-52]. Куркумин I (присутствующий в смеси в концентрации >70%) является наиболее активным ингибитором P-gp. В то же время, Куркумин I имеет ряд недостатков - сравнительно низкая аффинность к P-gp (по различным данным от 1 до 10 uM), низкая биодоступность при пероральном введении и быстрый метаболизм.
Таким образом, создание новых соединений, способных ингибировать P-гликопротеин, обладающих хорошими фармакокинетическими параметрами и не вызывающих серьезных побочных эффектов, является практически важной задачей.
Данное изобретение касается новой группы химических соединений, обладающих повышенной эффективностью в ингибировании Р-гликопротеина, а также высокой аффинностью к P-гликопротеину и высокой биодотупностью, и перспективных для подавления множественной лекарственной устойчивости, вызванной активностью P-гликопротеина.
Раскрытие изобретения
Задачей (техническим результатом) настоящего изобретения является разработка новых химических соединений, обладающих высокой эффективностью в ингибировании Р-гликопротеина, а также обладающих высокой биодоступностью и аффинностью к Р-гликопротеину, и перспективных для применения в терапии заболеваний, в механизм развития которых вовлечен P-гликопротеин, в частности, путем подавления множественной лекарственной устойчивости или для повышения всасывания или распределения лекарственного препарата. Такие заболевания могут представлять собой заболевания центральной нервной системы, инфекционные, аллергические, аутоиммунные, онкологические заболевания.
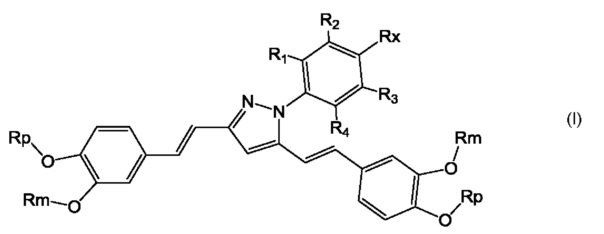
Указанный технический результат достигается путем получения соединений общей формулы (I) или их фармацевтически приемлемых солей, гидратов или сольватов:

где:
R1, R2, R3, R4 - выбираются независимо и представляют собой H, галоген, OH,
C1-6-алкокси, NO2, -C(=S)NHRA, -C(=O)ORA, -S(=O)2ORA, -C(=S)ORA, -S(=O)2NHRA, -C(=O)NHRA, -S(=O)2NRARB или -C(=O)NRARB;
RX - выбран из группы -C(=S)NHRA, -C(=O)ORA, -S(=O)2ORB, -C(=S)ORB,
-S(=O)2NHRA, -C(=O)NHRA, -S(=O)2NRARB, -C(=O)NRARB, 5-6-членный частично или полностью ненасыщенный гетероцикл, содержащий 1-2 атома N;
RA - выбирается независимо и представляет собой C1-6-алкил, C1-8-алкокси, частично или полностью галогенированный С1-8-алкил или С5-6-циклоалкил;
RB - выбирается независимо и представляет собой H, C1-8-алкил, C1-8-алкокси или частично или полностью галогенированный С1-8-алкил;
альтернативно, в случаях, когда любой из R1, R2, R3, R4, RX представляет собой
-S(=O)2NRARB или -C(=O)NRARB, RA и RB, совместно с атомом азота, к которому они присоединены, могут образовывать насыщенный цикл, содержащий от 5 до 6 атомов;
Rp - выбирается независимо и представляет собой С1-6-алкил, частично или полностью галогенированный С1-6-алкил;
Rm - выбирается независимо и представляет собой H, С1-6-алкил, частично или полностью галогенированный С1-8-алкил;
альтернативно, соседние группы Rp и Rm совместно с двумя атомами кислорода, к которым они присоединены, и двумя атомами углерода бензольного кольца могут образовывать неароматический цикл, содержащий от 5 до 6 атомов.
Химические соединения, составляющие предмет настоящего изобретения, характеризуются концентрацией полумаксимального ингибирования P-гликопротеина в диапазоне от 0.1 до 5000 nM.
Предпочтительные варианты воплощения изобретения
Отдельный класс соединений, представляющих интерес, включает соединения формулы (I), в которых:
R1, R2, R3, R4 - выбираются независимо и представляют собой H, галоген, NO2;
RX - представляет собой -C(=S)NHRA, -C(=O)ORA, -C(=S)ORB, -C(=O)NHRA,
-S(=O)2NRARB, -C(=O)NRARB или 5-6-членный частично или полностью ненасыщенный гетероцикл, содержащий 1-2 атома N;
RA - выбирается независимо и представляет собой C1-6-алкил, C1-8-алкокси, частично или полностью галогенированный С1-8-алкил;
RB - выбирается независимо и представляет собой H, C1-8-алкил, C1-8-алкокси, частично или полностью галогенированный С1-8-алкил;
альтернативно, в случаях, когда RX представляет собой -C(=O)NRARB, RA и RB совместно с атомом азота, к которому они присоединены, могут образовывать насыщенный цикл, содержащий от 5 до 6 атомов;
Rp - выбирается независимо и представляет собой CH3, частично или полностью галогенированный С1-6-алкил;
Rm - выбирается независимо и представляет собой H, CH3, частично или полностью галогенированный С1-8-алкил.
Другой отдельный класс соединений, представляющих интерес, включает соединения формулы (I), в которых:
R1, R2, R3, R4 - выбираются независимо и представляют собой H, галоген, NO2;
RX - представляет собой -C(=S)NHRA, -C(=O)ORA, -C(=S)ORB, -C(=O)NHRA,
-S(=O)2NRARB, -C(=O)NRARB, 5-6-членный частично или полностью ненасыщенный гетероцикл, содержащий 1-2 атома N;
RA - выбирается независимо и представляет собой C1-6-алкил, C1-8-алкокси, частично или полностью галогенированный С1-8-алкил;
RB - выбирается независимо и представляет собой H, C1-8-алкил, C1-8-алкокси, частично или полностью галогенированный С1-8-алкил;
альтернативно, в случаях, когда RX представляет собой -C(=O)NRARB, RA и RB совместно с атомом азота, к которому они присоединены, могут образовывать насыщенный цикл, содержащий от 5 до 6 атомов;
Rp - выбирается независимо и представляет собой CH3, частично или полностью галогенированный С1-6-алкил;
Rm - выбирается независимо и представляет собой H, CH3, частично или полностью галогенированный С1-8-алкил,
при этом соседние группы Rp и Rm совместно с двумя атомами кислорода, к которым они присоединены, и двумя атомами углерода бензольного кольца образуют неароматический цикл, содержащий от 5 до 6 атомов.
Иллюстративные примеры отдельного подкласса соединений, представляющего интерес, включают соединения по формуле (I), в которых Rm представляют CH3:
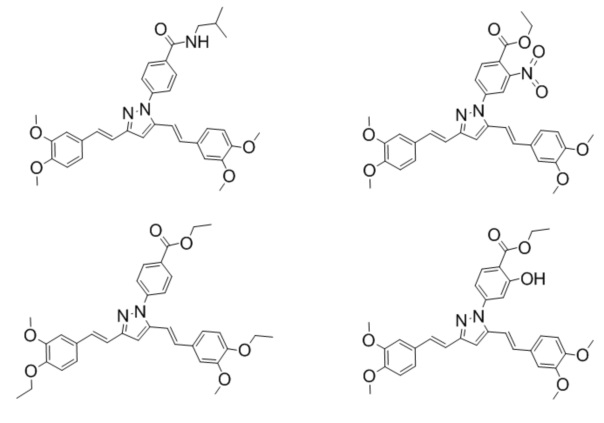
Иллюстративные примеры отдельного подкласса соединений, представляющего интерес, включают соединения формулы (I), в которых Rp и Rm совместно с двумя атомами кислорода, к которым они присоединены, и двумя атомом углерода бензольного кольца образуют неароматический цикл:

Иллюстративные примеры отдельного подкласса соединений, представляющего интерес, включают соединения по формуле (I), в которых Rp и/или Rm представляют собой частично или полностью галогенированный С1-6-алкил:

Иллюстративные примеры отдельного подкласса соединений, представляющего интерес, включают соединения формулы (I), в которых R1 и/или R2 и/или R3 и/или R4 представляет собой H или галоген: 
Иллюстративные примеры отдельного подкласса соединений, представляющего интерес, включают соединения формулы (I), в которых RX представляет собой -C(=O)ORA,
-C(=O)NHRA, -C(=O)NRARB, то есть замещенный эфир или амид карбоновой кислоты:

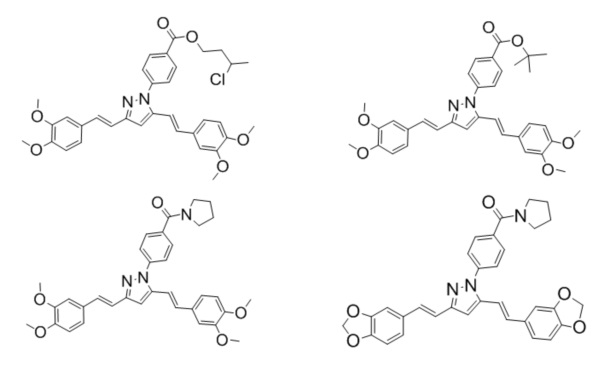

Иллюстративные примеры отдельного подкласса соединений, представляющего интерес, включают соединения формулы (I), в которых RX представляет собой
-C(=S)NHRA, -C(=S)ORB, то есть замещенный эфир или амид тио-карбоновой кислоты. Иллюстративными примерами этого класса являются следующие соединения:

Иллюстративные примеры отдельного подкласса соединений, представляющего интерес, включают соединения формулы (I), в которых RX представляет собой замещенный имидазол, имидазолил или сульфонамид. Иллюстративными примерами этого класса являются следующие соединения:


Конкретные примеры соединений, представленные в описании, должны рассматриваться как иллюстративные, а не как ограничительные. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
Данное изобретение также относится к применению соединений по изобретению в качестве ингибиторов P-гликопротеина.
Настоящее изобретение также относится к применению соединений, являющихся предметом изобретения, для получения фармацевтической композиции для лечения заболеваний, связанных с активностью P-гликопротеина.
В частности, такие фармацевтические композиции могут быть использованы для подавления множественной лекарственной устойчивости, вызванной активностью Р-гликопротеина.
Кроме того, изобретением предусматривается применение соединений по изобретению для получения фармацевтической композиции, которая может быть использована для повышения биодоступности различных пероральных лекарственных средств, обладающих низкой кишечной адсорбцией, за счет подавления активности Р-гликопротеина в клетках пищеварительного тракта человека или животных.
Также изобретение предполагает применение соединений, являющихся предметом изобретения, для получения фармацевтической композиции для повышения концентрации лекарственных средств, например, противоэпилептических лекарственных средств, в клетках головного мозга человека или животных.
Изобретение также предполагает применение соединений, являющихся предметом изобретения, для получения фармацевтической композиции для повышения концентрации цитостатических и/или цитотоксических лекарственных средств в опухолевых клетках. В частности, такие цитостатические или цитотоксические лекарственные средства могут представлять собой доксорубицин, даунорубицин, эпирубицин, идарубицин, тенипозид, метотрексат, пеметрексед, этопозид.
Изобретение также охватывает фармацевтические композиции, содержащие соединения настоящего изобретения, включая соединения любого из описанных классов или подклассов, в том числе любой из формул, описанных выше, в терапевтически эффективном количестве, в совокупности с, по крайней мере, одним терапевтически приемлемым носителем, растворителим и/или наполнителем.
Помимо этого, изобретение предусматривает фармацевтические композиции, описанные выше, которые дополнительно включают химиотерапевтическое или фармакотерапевтическое средство.
В частности, химиотерапевтическое средство может представлять собой этопозид, доксорубицин, винбластин, тикарциллин, карбпенициллин, мезлоциллин, азлоциллин, цефтобипрол, цефтаролин, цефтолозан, метотрексат и другие.
Фармакотерапевтическое средство, в частности, может представлять собой дигоксин, дексаметазон и другие.
Изобретение также включает получение соединений по формуле (I) или любых других соединений настоящего изобретения.
Определения
Следующие определения применяются в данном документе, если иное не указано явно. Кроме того, если не указано иное, все вхождения функциональных групп выбираются независимо.
Термин «алкил» сам по себе, или как часть другого заместителя, относится к насыщенным углеводородным группам с прямой или разветвленной цепью, включая углеводородные группы, имеющие указанное число атомов углерода (то есть С1-6 подразумевает от одного до шести атомов углерода). Например, «алкил» может означать метил, этил, н-пропил, изопропил, циклопропил, бутил, изобутил, втор-бутил, циклобутил, трет-бутил, циклобутил, пентил, циклопентил, трет-пентил, изопентил, гексил, изогексил, циклогексил и т.д. В качестве иллюстрации, замещенные алкильные группы включают, но не ограничиваются, следующими группами: фторметил, дифторметил, трифторметил, 2-фторэтил, 3-фторпропил, гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил и т.д.
Термин «алкокси» относится к алкильным группам, соответствующим определению, приведенному выше, и которые присоединяются к молекуле посредством мостикового атома кислорода. Например, термин «C1-8алкокси» означает -O-алкил, где алкильная группа содержит от 1 до 8 атомов углерода в виде линейной (неразветвленной) или разветвленной цепи или в виде цикла. В качестве иллюстрации алкокси группы включают, но не ограничиваются, следующими группами: метокси, этокси, н-пропокси, н-бутокси, трет-бутокси, аллилокси, циклобутокси и т.д.
Термин «галоген» сам по себе или в части другого термина относится к атому фтора, хлора, брома или йода.
Термин «галогенированный алкил» включает разветвленные и линейные насыщенные углеводородные цепи, в которых один или несколько атомов водорода замещены на галоген. Примеры галогенированных алкильных групп включают, но не ограничиваются, следующие группы: дифторметил, трифторметил, трихлорметил, пентафторэтил, 1,1,1,3,3,3-гексафтор-2-метилпропан-2-ил и т.п.
Термин «циклоалкил» обозначает алициклическую насыщенную группу, имеющую указанное число атомов углерода.
Термин «неароматический цикл» относится к насыщенным или частично ненасыщенным циклам.
Термин «гетероцикл» («гетероциклический»), в контексте изобретения, означает циклическую систему, имеющую от 5 до 6 атомов, в состав которых, наряду с углеродом, входят и атомы других элементов. Такими гетероатомами согласно изобретению могут быть N или O. Термин «гетероцикл» относится к насыщенным, частично ненасыщенным или ароматическим циклам (гетероарилам). Степень насыщенности гетероцикла, а также количество гетероатомов указываются отдельно.
Данное изобретение содержит только такие комбинации заместителей и производных, которые образуют стабильное или химически возможное соединение. Стабильным или химически возможным соединением называется такое соединение, стабильности которого достаточно для его синтеза и аналитического детектирования. Предпочтительные соединения данного изобретения являются достаточно стабильными и не разлагаются при температуре до 40°C в отсутствие химически активных условий, в течение, по крайней мере, одной недели.
Осуществление изобретения
Соединения, являющиеся предметом настоящего изобретения, могут быть получены с использованием различных общеизвестных синтетических методик, в том числе с использованием описанных ниже. Синтез химических соединений, являющихся предметом настоящего изобретения, может быть проведен из коммерчески доступных исходных реагентов или исходных реагентов, которые могут быть получены по методикам, описанным в работах, известных из уровня техники (Вейганд - Хильгетаг. Методы эксперимента в органической химии. Под ред. проф. Н. Н. Суворова. М., Химия, 1968, «Синтезы гетероциклических соединений» вып. 1-16 Ереван 1956-1987; "Синтезы органических препаратов" ч.1-12 М. 1949-1964; «Синтезы органических соединений с изотопами водорода» Мэррей А., Уильямс Д.Л. Москва, Издательство ИЛ, 1961;). Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза.
Общая схема конечной стадии получения соединений по изобретению
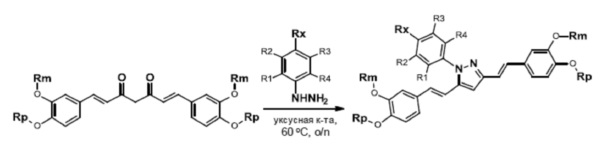
Все соединения, являющиеся предметом данного изобретения, могут быть получены на основании изложенных в описании синтетических подходов, примеров экспериментальных методик и общеизвестных методик и материалов.
Примеры синтеза соединений по изобретению
Синтез промежуточных соединений
Синтез Куркуминоида I (CUR I)
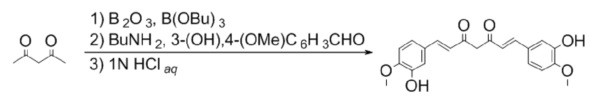
К раствору 350 мг (5.0 ммоль) ангидрида борной кислоты в 60 мл безводного этилацетата прибавляют 1.0 мл (10 ммоль) ацетилацетона и перемешивают реакционную смесь в течение 30 минут при 40°С, после прибавляют 3,04 г (20 ммоль) изо-ванилина и 10.8 мл (40 ммоль) трибутилбората. Реакционную смесь перемешивают 30 минут при 50°С после чего по каплям, в течение 15 минут прибавляют 0.4 мл (5 ммоль) н-бутиламина в 5 мл безводного этилацетата. Реакционную смесь нагревают до 70°С и перемешивают при этой температуре 10 часов, добавляют 30 мл 1N водной соляной кислоты и перемешивают при 60°С еще 2 часа, охлаждают, органический слой отделяют, а водный экстрагируют этилацетатом (3х50 мл). Объединенные органические фазы сушат над безводным сульфатом натрия и концентрируют в вакууме. Остаток перекристаллизовывают из метанола и сушат. Получают: 957 мг (52%) продукта.
Синтез Куркуминоида II (CUR II)
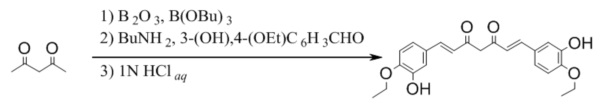
К раствору 350 мг (5.0 ммоль) ангидрида борной кислоты в 60 мл безводного этилацетата прибавляют 1.0 мл (10 ммоль) ацетилацетона и перемешивают реакционную смесь в течение 30 минут при 40°С, после прибавляют 3.32 г (20 ммоль) 4-этокси-3-гидроксибензальдегида и 10.8 мл (40 ммоль) трибутилбората. Реакционную смесь перемешивают 30 минут при 50°С после чего по каплям, в течение 15 минут прибавляют 0.4 мл (5 ммоль) н-бутиламина в 5 мл безводного этилацетата. Реакционную смесь нагревают до 70°С и перемешивают при этой температуре 10 часов, добавляют 30 мл 1N водной соляной кислоты и перемешивают при 60°С еще 2 часа, охлаждают, органический слой отделяют, а водный экстрагируют этилацетатом (3×50 мл). Объединенные органические фазы сушат над безводным сульфатом натрия и концентрируют в вакууме. Остаток перекристаллизовывают из метанола и сушат. Получают: 524 мг (34%)продукта.
Синтез Диметиликуркумина (DMCUR)
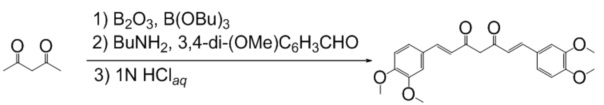
К раствору 350 мг (5.0 ммоль) ангидрида борной кислоты в 60 мл безводного этилацетата прибавляют 1.0 мл (10 ммоль) ацетилацетона и перемешивают реакционную смесь в течение 30 минут при 40°С, после прибавляют 3.32 г (20 ммоль) 3,4-диметоксибензальдегида и 10.8 мл (40 ммоль) трибутилбората. Реакционную смесь перемешивают 30 минут при 50°С после чего по каплям, в течение 15 минут прибавляют 0.4 мл (5 ммоль) н-бутиламина в 5 мл безводного этилацетата. Реакционную смесь нагревают до 70°С и перемешивают при этой температуре 10 часов, добавляют 30 мл 1N водной соляной кислоты и перемешивают при 60°С еще 2 часа, охлаждают, органический слой отделяют, а водный экстрагируют этилацетатом (3×50 мл). Объединенные органические фазы сушат над безводным сульфатом натрия и концентрируют в вакууме. Остаток перекристаллизовывают из метанола и сушат. Получают: 1090 мг (55%)продукта.
Синтез Куркуминоида III (PICUR)

К раствору 350 мг (5.0 ммоль) ангидрида борной кислоты в 60 мл безводного этилацетата прибавляют 1.0 мл (10 ммоль) ацетилацетона и перемешивают реакционную смесь в течение 30 минут при 40°С, после прибавляют 3.00 г (20 ммоль) гелиотропина и 10.8 мл (40 ммоль) трибутилбората. Реакционную смесь перемешивают 30 минут при 50°С после чего по каплям, в течение 15 минут прибавляют 0.4 мл (5 ммоль) н-бутиламина в 5 мл безводного этилацетата. Реакционную смесь нагревают до 70°С и перемешивают при этой температуре 10 часов, добавляют 30 мл 1N водной соляной кислоты и перемешивают при 60°С еще 2 часа, охлаждают, органический слой отделяют, а водный экстрагируют этилацетатом (3×50 мл). Объединенные органические фазы сушат над безводным сульфатом натрия и концентрируют в вакууме. Остаток перекристаллизовывают из метанола и сушат. Получают: 673 мг (37%) продукта.
Синтез соединений по изобретению
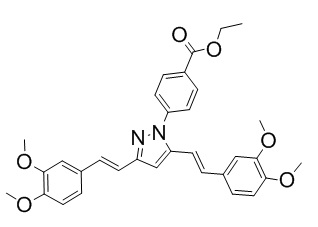
Синтез этил 4-(3,5-бис((E)-3-гидрокси-4-метоксистирил)-1H-пиразол-1-ил)бензоата

К раствору 368 мг (1 ммоль) Куркуминоида I в 10 мл ледяной уксусной кислоты прибавляют 540 мг (3 ммоль) арилгидразина в виде раствора в 2 мл ледяной уксусной кислоты. Реакционную смесь нагревают при перемешивании до 65-70°С, прибавляют 10 мг безводного ацетата натрия и выдерживают при заданной температуре до окончания реакции. Затем, реакционную смесь охлаждают и выливают в 100 мл воды со льдом, выпавший осадок отфильтровывают, сушат и разделяют хроматографически дополнительно перекристаллизовывая целевую фракцию из смеси дихлорметан/гексан. Получают: 307 мг (60%) продукта.
Синтез этил 4-(3,5-бис((E)-3-гидрокси-4-метоксистирил)-1H-пиразол-1-ил)-2-фторбензоата

К раствору 368 мг (1 ммоль) Куркуминоида I в 10 мл ледяной уксусной кислоты прибавляют 594 мг (3 ммоль) в виде раствора в 2 мл ледяной уксусной кислоты. Реакционную смесь нагревают при перемешивании до 65-70°С и прибавляют 10 мг безводного ацетата натрия и выдерживают при заданной температуре до окончания реакции. Затем, реакционную смесь охлаждают и выливают в 100 мл воды со льдом, выпавший осадок отфильтровывают, сушат и разделяют хроматографически дополнительно перекристаллизовывая целевую фракцию из смеси дихлорметан/гексан. Получают: 264 мг (50%) продукта.
Синтез этил 4-(3,5-бис((E)-3-гидрокси-4-етоксистирил)-1H-пиразол-1-ил)бензоата

К раствору 396 мг (1 ммоль) Куркуминоида I в 10 мл ледяной уксусной кислоты прибавляют 540 мг (3 ммоль) в виде раствора в 2 мл ледяной уксусной кислоты. Реакционную смесь нагревают при перемешивании до 65-70°С, прибавляют 1 каплю триэтиламина и выдерживают при заданной температуре до окончания реакции. Затем, реакционную смесь охлаждают, выливают в 100 мл воды со льдом, выпавший осадок отфильтровывают, сушат и разделяют хроматографически дополнительно перекристаллизовывая целевую фракцию из смеси дихлорметан/гексан. Получают 154 мг (34%) продукта.
Синтез 2,2,2-трифторэтил 4-(3,5-бис((E)-4-гидрокси-3-метоксистирил)-1H-пиразол-1-ил)бензоата
Получение 2,2,2-трифторэтил-4-аминобензоатa

К раствору 740 мг (3.01 ммоль) 2,2,2,-трифторэтил-4-нитробензоата (6) в 40 мл этанола прибавляют 1.32 мл (13 ммоль) циклогексена и 200 мг 10% палладия на угле. Реакционную смесь кипятят в атмосфере аргона в течение 15 часов, охлаждают, фильтруют через слой целита, растворитель удаляют в вакууме, остаток перекристаллизовывают из смеси дихлорметан/гексан. Получают:572 мг (87%) продукта.
Получение 2,2,2-трифторэтил 4-гидразинобензоата
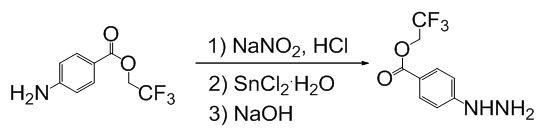
К раствору 4.38 г (20 ммоль) 2,2,2-трифторэтилового эфира п-аминобензойной в 30 мл концентрированной соляной кислоты при -10°С прибавляют 370 мг нитрита натрия в 4 мл воды поддерживая температуру ниже -10°С. Реакционную смесь перемешивают 1.5 часа при заданной температуре и прибавляют раствор 2.74 г (14.2 ммоль) дигидрата хлорида олова (II) в 10 мл концентрированной соляной кислоты, таки образом, чтобы температура реакционной смеси не поднималась выше -10°С и перемешивают еще 2 часа при -10°С, а затем 2 часа при комнатной температуре. Осадок отфильтровывают, растворяют в минимальном количестве горячей воды и добавляют 4N водный раствор NaOH до pH 9. Полученную смесь экстрагируют диэтиловым эфиром (3×50 мл), органические фазы объединяют, промывают насыщенным раствором хлорида натрия (3×10 мл), растворитель удаляют, остаток сушат в вакууме. Получают: 2.48 г (53%) технического продукта.
Синтез 2,2,2-трифторэтил 4-(3,5-бис((E)-3-гидрокси-4-метоксистирил)-1H-пиразол-1-ил)бензоата
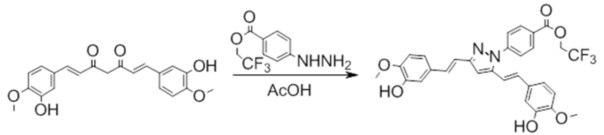
К раствору 368 мг (1 ммоль) Куркуминоида I в 10 мл ледяной уксусной кислоты прибавляют 702 мг (3 ммоль) в виде раствора в 3 мл ледяной уксусной кислоты. Реакционную смесь нагревают при перемешивании до 65-70°С, прибавляют 1 каплю триэтиламина и выдерживают при заданной температуре до окончания реакции. Затем, реакционную смесь охлаждают, выливают в 100 мл воды со льдом, выпавший осадок отфильтровывают, сушат и разделяют хроматографически дополнительно перкристаллизовывая целевую фракцию из смеси дихлорметан/гексан. Получают: 198 мг (35%) продукта.
Синтез этил 4-(3,5-бис((E)-3-гидрокси-4-етоксистирил)-1H-пиразол-1-ил)-2-фторбензоата
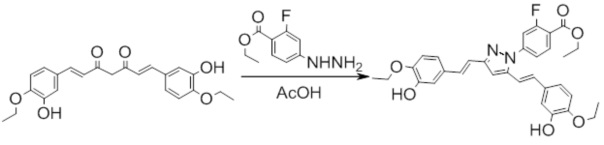
К раствору 396 мг (1 ммоль) в 10 мл ледяной уксусной кислоты прибавляют 594 мг (3 ммоль) в виде раствора в 2 мл ледяной уксусной кислоты. Реакционную смесь нагревают при перемешивании до 65-70°С, прибавляют 1 каплю триэтиламина и выдерживают при заданной температуре до окончания реакции. Затем, реакционную смесь охлаждают и выливают в 100 мл воды со льдом, выпавший осадок отфильтровывают, сушат и разделяют хроматографически дополнительно перекристаллизовывая целевую фракцию из смеси дихлорметан/гексан. Получают 117 мг (25%) продукта.
Синтез этил 4-(3,5-бис((E)-3,4-диметоксистирил)-1H-пиразол-1-ил)-2-нитробензоата
Получение этил 4-гидразино-2-нитробензоат

К суспензии 4.20 г (20 ммоль) этил 4-гидразино-2-нитробензоат в 30 мл концентрированной соляной кислоты при -10°С прибавляют 370 мг нитрита натрия в 4 мл воды, поддерживая температуру ниже -10°С. Реакционную смесь перемешивают 1.5 часа при заданной температуре, охлаждают до -35°С и,поддерживая данную температуру, прибавляют раствор 2.74 г (14.2 ммоль) дигидрата хлорида олова (II) в 10 мл концентрированной соляной кислоты, перемешивают один час при -30°С, а затем добавляют 1N водный раствор NaOH до pH 9. Полученную смесь экстрагируют диэтиловым эфиром (3×50 мл), органические фазы объединяют, промывают насыщенным раствором хлорида натрия (3×10 мл), растворитель удаляют, остаток сушат в вакууме и разделяют хроматографически используя смесь этилацетат/дихлорметан 19:1 Получают: 676 мг (15%) продукта.
Получение 4-(3,5-бис((E)-3,4-диметоксистирил)-1H-пиразол-1-ил)-2-нитробензоата
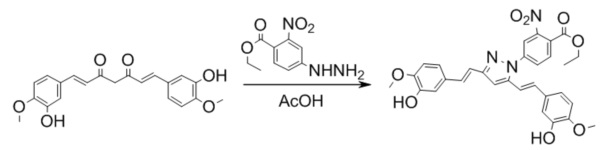
К раствору 368 мг (1 ммоль) диметилкуркуминоида I в 10 мл ледяной уксусной кислоты прибавляют 675 мг (3 ммоль) в виде раствора в 3 мл ледяной уксусной кислоты. Реакционную смесь нагревают при перемешивании до 65-70°С, прибавляют 1 каплю триэтиламина и выдерживают при заданной температуре до окончания реакции. Затем, реакционную смесь охлаждают, выливают в 100 мл воды со льдом, выпавший осадок отфильтровывают, сушат и разделяют хроматографически дополнительно перкристаллизовывая целевую фракцию из смеси дихлорметан/гексан. Получают: 167 мг (30%) продукта.
Синтез 4-(3,5-бис((E)-3,4-диметоксистирил)-1H-пиразол-1-ил)-N-изопропилбензамида

К раствору 368 мг (1 ммоль) в 10 мл ледяной уксусной кислоты прибавляют 675 (3 ммоль) в виде раствора в 1 мл ледяной уксусной кислоты. Реакционную смесь нагревают при перемешивании до 65-70°С, прибавляют 1 каплю триэтиламина и выдерживают при заданной температуре до окончания реакции. Затем, реакционную смесь охлаждают, выливают в 100 мл воды со льдом, выпавший осадок отфильтровывают, сушат и разделяют хроматографически дополнительно перкристаллизовывая целевую фракцию из смеси дихлорметан/гексан. Получают: 258 мг (44%) получение.
Синтез 4-(3,5-бис((E)-3,4-диметоксистирил)-1H-пиразол-1-ил)-N-изобутилбензамида

К суспензии 512.5 мг (1 ммоль) кислоты в 5 мл безводного ДМФА прибавляют 390 мкл (2.2 ммоль) диизопропилэтиламина, перемешивают в течение 30 минут и прибавляют 570 мг (1.5 ммоль) HATU. Реакционную смесь перемешивают 30 минут при 40°С, прибавляют 150 мкл (1.5 ммоль) диизопропилэтиламина и перемешивают при заданной температуре в течение 12 часов, охлаждают, выливают в 50 мл воды, осадок отфильтровывают, промывают на фильтре водой, сушат и разделяют хроматографически используя в качестве элюента систему гексан:дихлорметан нарастающей полярности. Получают: 380 мг (67%) продукта.
Применение химических соединений по изобретению
Применение соединений по медицинским показаниям
Соединения, описанные в данном изобретении, могут применяться для подавления множественной лекарственной устойчивости (MDR) у больных, длительно получающих различные лекарственные препараты. В частности, соединения, описанные в данном изобретении, способны ингибировать белок семейства ABC-переносчиков P-гликопротеин. Кроме того показано, что применение ряда соединений, составляющих настоящее изобретение, совместно с цитостатическими лекарственными препаратами (например, доксорубицином) приводит к усилению действия цитостатических препаратов. Соединения, описанные в данном изобретении, также могут применяться в лечении инфекционных заболеваний, при которых патогенный микроорганизм обладает множественной лекарственной устойчивостью (MDR), особенно P-гликопротеин-опосредованной MDR, в качестве примеров можно привести формы малярии (малярийного плазмодия), туберкулеза, лейшманиоза и амебной дизентерии. Введение соединений, описанных в настоящем изобретении, совместно (раздельно, одновременно или последовательно) с препаратом для лечения инфекционных заболеваний способно потенцировать эффект последнего. Соединения, описанные в данном изобретении, также могут быть использованы для повышения всасывания или распределения лекарственного препарата. Этот способ применения включает введение пациенту, раздельно, одновременно или последовательно, одного из соединений настоящего изобретения и упомянутого лекарственного препарата. В частности, этот способ может быть использован для повышения проникновения лекарственного препарата в центральную нервную систему. Например, соединения по настоящему изобретению могут быть использованы в способе облегчения доставки лекарственных средств через гематоэнцефалический барьер.
Таким образом, мы предполагаем, что использование ингибиторов P-гликопротеина, составляющих предмет настоящего изобретения, в сочетании с текущими или новыми средствами химиотерапии против различных онкологических заболеваний или в сочетании с противоинфекционными лекарственными средствами, для терапии инфекционных заболеваний, позволит достигнуть существенной и длительной ремиссии.
Способ терапевтического применения соединений
Предмет данного изобретения включает также введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения по изобретению. «Терапевтически эффективным количеством» называется такое количество соединения, которое необходимо для подавления множественной лекарственной устойчивости. Кроме того, применение терапевтически эффективного количества соединения по изобретению совместно с цитостатическими лекарственными препаратами должно приводить к детектируемому уничтожению раковых клеток или ингибированию их роста или скорости распространения по организму, размера или количества опухолей, или других характеристик онкологического заболевания. Точное требуемое количество может меняться от субъекта к субъекту в зависимости от вида, возраста и общего состояния пациента, тяжести заболевания, особенностей противоракового агента, методики введения препарата, комбинированного лечения с другими препаратами и т.п. Соединение, или фармацевтическая композиция, содержащая соединение, может быть введена в организм пациента в любом количестве и любым путем введения, эффективным для подавления множественной лекарственной устойчивости. Количество соединения, которое будет эффективным в лечении или профилактике конкретного расстройства или состояния, зависит, в частности, от хорошо известных факторов, влияющих на эффективную дозировку препаратов. Кроме того, опционально могут применяться измерения in vitro или in vivo для определения оптимального дозового диапазона. Грубым путем определения эффективной дозы может стать экстраполяция кривых доза - отклик, которые будут зависеть от модели тестирования in vitro или на животных. Точный уровень дозировки, определяемый лечащим врачом, зависит от хорошо известных факторов, включающих способ введения препарата, а также возраста, массы тела, пола и общего состояния здоровья пациента; характера, тяжести и клинического состояния заболевания; использования (или неиспользования) сопутствующей терапии; а также характера и степени генетических изменений в клетках пациента.
Фармацевтически приемлемые производные соединений
Соединения данного изобретения могут существовать в свободной форме в процессе обработки, или, если требуется, в виде фармацевтически приемлемой соли или другого производного. Используемый здесь термин «фармацевтически приемлемые соли» относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Примерами фармацевтически приемлемых нетоксичных солей могут служить соли, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная, лимонная или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, метансульфонат (мезилат), 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, полуфумарат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат (тозилат), ундеканат, валериат и подобные.
Под «сольватом» понимается ассоциация или комплекс одного или нескольких молекул растворителя и соединения по изобретению. Примеры растворителей, образующих сольваты, включают, но ими не ограничиваются, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Термин «гидрат» относится к комплексу, где молекулой растворителя является вода.
Фармацевтические композиции
Изобретение также относится к фармацевтическим композициям, которые содержат, по меньшей мере, одно из описанных здесь соединений (или пролекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, растворителей и/или наполнителей. Данные композиции также могут содержать один или несколько дополнительных терапевтических агентов. Кроме того, соединение данного изобретения может быть введено пациенту, нуждающемуся в соответствующей терапии, в комбинации с одним или более других терапевтических средств.
Фармацевтические композиции, заявляемые в данном изобретении, содержат соединения данного изобретения совместно с фармацевтически приемлемыми носителями, которые могут включать в себя любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазочные материалы и т.д., подходящие для конкретной формы дозирования. За исключением таких случаев, когда среда обычных носителей несовместима с соединением изобретения, например, при появлении любых нежелательных биологических эффектов и иных нежелательных взаимодействий с любым другим компонентом (компонентами) фармацевтической композиции, использование таких композиций находится в рамках данного изобретения. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно- и олигосахаридами, а также их производными; солодом, желатином; тальком; эксципиентами, такими как: какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Лекарственные формы
Предметом данного изобретения являются также лекарственные формы - класс фармацевтических композиций, состав которых оптимизирован для определенного пути введения в организм в терапевтически эффективной дозе. Лекарственные композиции данного изобретения могут быть введены в организм орально, местно, ректально, внутриглазным способом, пульмональным, например, в виде ингаляционного спрея, или внутрисосудистым способом, интраназально, интраперитонально, подкожно, внутримышечно, интрастернально, трансдермально, а также инфузионным способом, в рекомендованных дозировках.
Лекарственная форма данного изобретения может содержать соединение описанной здесь формулы или его фармацевтически приемлемую соль, сольват или гидрат и любой фармацевтически приемлемый носитель, адъювант или растворитель. Термин «фармацевтически приемлемый носитель или адъювант» означает носитель или адъювант, который может быть введен в организм пациента совместно с соединением, составляющем суть данного изобретения, и который не разрушает фармакологической активности этого соединения, и является нетоксичным при введении в дозах, достаточных для доставки терапевтического количества соединения.
Лекарственные формы данного изобретения могут содержать составы, полученные методами использования липосом или микрокапсуляционные методы, методами приготовления наноформ препарата, и прочие примеры, известные в фармацевтике.
Характеристика биологической активности соединений
Изучение влияние соединений по изобретению на накопление доксорубицина в клетках с множественной лекарственной устойчивостью
Биологическая активность соединений, являющихся предметом настоящего изобретения, была изучена различными методами. Например, было исследовано ингибирование Р-гликопротеина по накоплению субстрата Р-гликопротеина (доксорубицина) внутри опухолевой клетки. Для изучения накопления доксорубицина в клетках с множественной лекарственной устойчивостью и влияния соединений, являющихся предметом настоящего изобретения на этот процесс, клетки сублинии К562/Dox рассевали в 6-луночные планшеты. Добавляли доксорубицин (1 мкМ, 30 мин.) в отсутствие или в присутствии соединений (каждое в концентрации 5 мкМ; выбор концентрации определялся диапазоном токсических концентраций, установленном в МТТ-тесте). Клетки отмывали холодным физиологическим буфером, суспендировали в том же буфере на льду (для предотвращения вытекания доксорубицина) и анализировали флуоресценцию клеток. О флуоресценции клеток (следовательно, о накоплении доксорубицина) судили по величине среднего канала флуоресценции (mean fluorescence channel) в FL1 или FL2, определяемого на оси абсцисс прибора. Все эксперименты проведены в 2-3 повторностях.
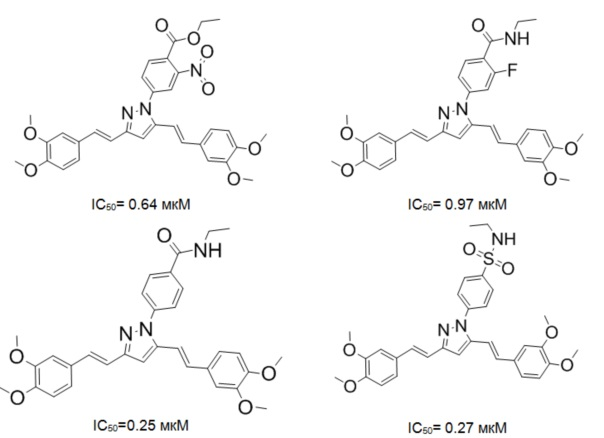
Иллюстративные примеры соединений (увеличивающих накопление доксорубицина в 1.2-10 раз), продемонстрировавших активность в отношении ингибирование Р-гликопротеина по накоплению субстрата Р-гликопротеина (доксорубицина) внутри опухолевой клетки: 

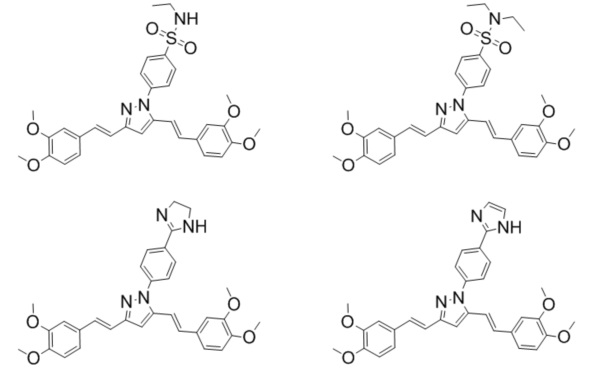

Изучение способности соединений по изобретению ингибировать Р-гликопротеин
Эффективность ингибиторов Р-гликопротеина исследовалась путем определения способности соединений усиливать цитотоксичность доксорубицина (и других цитостатических препаратов) при совместной инкубации с резистентными клетками и оценка гибели колориметрическим методом. Клетки сублинии К562/Dox рассевали на 96-луночные планшеты (Costar, США) (104 клеток в 190 мкл культуральной среды). В лунки вносили куркумин или соединения по изобретению из серийных разведений стоковых растворов. Контролем служили лунки с растворителем (ДМСО). Объем раствора испытуемого соединения не превышал 5% объема культуральной среды в лунках. Каждую концентрацию препарата изучали в 3-х повторностях. Клетки инкубировали при 37°С, 5% СО2 в течение 72 часов. По окончании инкубации в лунки вносили 20 мкл (50 мкг) тетразолиевой соли (3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Р-тетразолий, внутренняя соль), и планшеты помещали в СО2-инкубатор на 2 часа. О жизнеспособности клеток судили по цветной реакции, развивающейся при восстановлении тетразолиевой соли в формазан дегидрогеназами митохондрий. Окраску регистрировали на спектрофотометре (LKB, Швеция) при длине волны возбуждения 540 нм. Оптическую плотность в лунках, где клетки инкубировались только с растворителем (контроль), принимали за 100%. Показатели оптической плотности в лунках с каждой концентрацией испытуемых препаратов усредняли и вычисляли процент выживших клеток при той или иной концентрации исследуемого препарата.



Примеры фармацевтических композиций
Соединения, описанные в данном изобретении, могут быть использованы для профилактики и лечения заболеваний человека, например, в виде следующих составов (в приведенных ниже примерах под «Соединением» понимается активный ингредиент):
Данные составы могут быть приготовлены в соответствии со стандартными фармацевтическими методиками. Таблетки (I)-(II) могут быть покрыты кишечнорастворимой оболочкой с использованием, например, фталата ацетата целлюлозы. Аэрозольный состав (I) может быть использован в сочетании со стандартными диспенсерами; в качестве суспендирующего агента вместо триолеата сорбитана и соевого лецитина может быть использован моноолеат сорбитана, полуолеат сорбитана, полисорбат 80, олеат полиглицерина или олеиновая кислота.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКОЕ АМИДНОЕ СОЕДИНЕНИЕ, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2823875C1 |
| НОВЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ (ВАРИАНТЫ) И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2550346C2 |
| СОЕДИНЕНИЕ, НАЦЕЛЕННОЕ НА БЕЛОК И ЕГО ДЕГРАДАЦИЮ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2829459C1 |
| ИНГИБИТОРЫ ТИПА ErbB | 2006 |
|
RU2592703C9 |
| ПРОИЗВОДНЫЕ N4-ФЕНИЛХИНАЗОЛИН-4-АМИНА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ ТИПА ERBB ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2006 |
|
RU2428421C2 |
| ЗАМЕЩЕННЫЕ ДИГИДРОТИЕНОПИРИМИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2018 |
|
RU2817698C2 |
| Производные бензо[d]изоксазола и их применение | 2016 |
|
RU2638155C1 |
| СОЕДИНЕНИЯ 2,3-ДИГИДРОХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NaV1.8 | 2020 |
|
RU2833870C2 |
| Ингибиторы альдостеронсинтазы (CYP11B2) человека | 2022 |
|
RU2811745C1 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
Изобретение относится к медицине, а именно к фармакологии, и касается новых химических соединений общей формулы (I) или их фармацевтически приемлемых солей, которые являются ингибиторами P-гликопротеина. В формуле (I) R1, R2, R3, R4 независимо представляют собой Н, галоген, NO2; Rx - выбран из группы -C(=S)NHRA, -C(=O)ORA, -S(=O)2NRARB, -C(=O)NRARB, 5-членного частично или полностью ненасыщенного гетероцикла, содержащего 2 атома N; RA выбирается независимо и представляет собой C1-6-алкил, частично или полностью галогенированный C1-8-алкил; RB выбирается независимо и представляет собой Н, С1-8-алкил или частично или полностью галогенированный C1-8-алкил; альтернативно, в случае, когда Rx представляет собой -C(=O)NRARB, RA и RB совместно с атомом азота, к которому они присоединены, могут образовывать насыщенный цикл, содержащий 5 атомов; Rp выбирается независимо и представляет собой C1-6-алкил, частично или полностью галогенированный C1-6-алкил; Rm выбирается независимо и представляет собой Н, C1-6-алкил, частично или полностью галогенированный C1-8-алкил; альтернативно соседние группы Rp и Rm совместно с двумя атомами кислорода, к которым они присоединены, и двумя атомами углерода бензольного кольца могут образовывать неароматический цикл, содержащий от 5 до 6 атомов. Данные соединения могут быть использованы в терапии заболеваний центральной нервной системы, инфекционных, аллергических, аутоиммунных, онкологических и прочих заболеваний. В частности, данные химические соединения могут быть использованы для подавления множественной лекарственной устойчивости у больных, длительно получающих различные лекарственные препараты. Кроме того, изобретение относится к применению описанных выше соединений и фармацевтической композиции их содержащей. 4 н. и 5 з.п. ф-лы.
 (I)
(I)
1. Соединение формулы (I)
или его фармацевтически приемлемая соль, где:
R1, R2, R3, R4 - выбираются независимо и представляют собой Н, галоген, NO2;
Rx - выбран из группы -C(=S)NHRA, -C(=O)ORA, -S(=O)2NRARB, -C(=O)NRARB, 5-членного частично или полностью ненасыщенного гетероцикла, содержащего 2 атома N;
RA - выбирается независимо и представляет собой C1-6-алкил, частично или полностью галогенированный C1-8-алкил;
RB - выбирается независимо и представляет собой Н, С1-8-алкил или частично или полностью галогенированный C1-8-алкил;
альтернативно, в случаях, когда любой из Rx представляет собой
-C(=O)NRARB, RA и RB совместно с атомом азота, к которому они присоединены, могут образовывать насыщенный цикл, содержащий 5 атомов;
Rp - выбирается независимо и представляет собой C1-6-алкил, частично или полностью галогенированный C1-6-алкил;
Rm - выбирается независимо и представляет собой Н, C1-6-алкил, частично или полностью галогенированный C1-8-алкил;
альтернативно соседние группы Rp и Rm совместно с двумя атомами кислорода, к которым они присоединены, и двумя атомами углерода бензольного кольца могут образовывать неароматический цикл, содержащий от 5 до 6 атомов.
2. Соединение по п.1, в котором
R1, R2, R3, R4 - выбираются независимо и представляют собой Н, галоген, NO2;
Rx - представляет собой -C(=S)NHRA, -C(=O)ORA,
-S(=O)2NRARB, -C(=O)NRARB или 5-членный частично или полностью ненасыщенный гетероцикл, содержащий 2 атома N;
RA - выбирается независимо и представляет собой С1-6-алкил, частично или полностью галогенированный C1-8-алкил;
RB - выбирается независимо и представляет собой Н, С1-8-алкил, частично или полностью галогенированный C1-8-алкил;
альтернативно, в случаях, когда Rx представляет собой -C(=O)NRARB, RA и RB совместно с атомом азота, к которому они присоединены, могут образовывать ненасыщенный цикл, содержащий 5 атомов;
Rp - выбирается независимо и представляет собой СН3, частично или полностью галогенированный C1-6-алкил;
Rm - выбирается независимо и представляет собой Н, СН3, частично или полностью галогенированный C1-8-алкил.
3. Соединение по п.1, выбранное из группы и представляющее собой:


4. Применение соединения по п. 1 в качестве ингибитора Р-гликопротеина.
5. Применение соединения по п. 1 для получения фармацевтической композиции для лечения заболеваний, связанных с активностью Р-гликопротеина.
6. Применение по п. 5, в котором заболевание характеризуется множественной лекарственной устойчивостью, вызванной активностью Р-гликопротеина.
7. Фармацевтическая композиция для лечения заболеваний, связанных с активностью Р-гликопротеина, характеризующаяся тем, что она содержит эффективное количество соединения по п. 1 и фармацевтически приемлемый носитель, растворитель и/или наполнитель.
8. Фармацевтическая композиция по п. 7, которая дополнительно содержит цитостатическое средство.
9. Фармацевтическая композиция по п. 8, в которой цитостатическое средство представляет собой доксорубицин, винбластин или метотрексат.
| KUMAR Denesh et al | |||
| "Isolation, synthesis and pharmacological evaluation of some novel curcumin derivatives as anticancer agents", Journal of Medicinal Plants Research, 2012, vol.6 (14), pp.2880-2884 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| RU 2013150083 A, 20.05.2011 | |||
| ANAND Preetha et al | |||
| " Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature", 2008, 76, pp.1590-1611. | |||

