[ОБЛАСТЬ ТЕХНИКИ]
[0001]
Настоящее изобретение относится к способу получения карбонилгалогенида, эффективному с точки зрения количества применяемого галогенированного метана.
УРОВЕНЬ ТЕХНИКИ
[0002]
Карбонилгалогенид, такой как фосген, очень важен как синтетический промежуточный продукт различных соединений и сырьевой материал. Например, карбонатное соединение обычно получают из фосгена и спиртового соединения.
[0003]
Однако фосген очень токсичен. Например, фосген легко вступает в реакцию с водой с образованием хлористого водорода и имеет историю применения в качестве ядовитого газа. Как правило, фосген получают в результате газофазной реакции с выделением большого количества тепла между безводным газообразным хлором и монооксидом углерода высокой чистоты в присутствии катализатора на основе активированного угля. Угарный газ, применяемый в данной реакции, также токсичен. Основной способ производства фосгена не сильно изменился с 1920-х годов. Данный способ производства фосгена требует дорогостоящего крупномасштабного оборудования. Всесторонние гарантии безопасности при проектировании установок необходимы в связи с высокой токсичностью фосгена, и это приводит к увеличению производственных затрат.
[0004]
Изобретатели настоящего изобретения разработали способ получения галогена и/или карбонилгалогенида облучением галогенированного углеводорода светом в присутствии кислорода (патентный документ 1). Данный способ является безопасным, так как карбонилгалогенид, полученный данным способом, может быть непосредственно введен в реакционноспособное соединение, являющееся субстратом, такое как аминовое соединение и спиртовое соединение. Кроме того, карбонилгалогенид, не израсходованный в реакции, можно собрать, чтобы он не просочился наружу, с помощью ловушки. Например, изобретатели также разработали способ получения галогенированного сложного эфира карбоновой кислоты путем облучения светом смеси, содержащей галогенированный углеводород и спирт, в присутствии кислорода (патентный документ 2). Кроме того, изобретатели также разработали способ получения карбонатного производного путем облучения светом композиции, состоящей из галогенированного углеводорода, соединения, содержащего нуклеофильную функциональную группу, и основания в присутствии кислорода (патентный документ 3 и патентный документ 4).
[0005]
Когда карбонилгалогенид получают вышеописанными способами, остается большое количество галогенированного углеводорода по сравнению с полученным карбонилгалогенидом. Галогенированные углеводороды не могут быть легко утилизированы и нуждаются в очистке и повторном применении из-за высокой нагрузки на окружающую среду.
[0006]
Давно известно, что карбонилгалогенид разлагается под действием света. Например, патентный документ 5 описывает способ разложения фосгена с целью его удаления фоторазложением путем облучения ультрафиолетовым светом до треххлористого бора, содержащего фосген в качестве примеси. В непатентном документе 1 также описано, что фосген разлагается при облучении светом.
ДОКУМЕНТЫ ПРЕДШЕСТВУЮЩЕГО УРОВНЯ ТЕХНИКИ
ПАТЕНТНЫЙ ДОКУМЕНТ
[0007]
Патентный документ 1: JP 2013-181028 А
Патентный документ 2: WO 2015/156245
Патентный документ 3: WO 2018/211952
Патентный документ 4: WO 2018/211953
Патентный документ 5: US 4,405,423 В
НЕПАТЕНТНЫЙ ДОКУМЕНТ
[0008]
Непатентный документ 1: С.W. Mostgomery et al., J. Am. Chem. Soc., 1934, 56, 5, стр. 1089-1092
ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
ПРОБЛЕМЫ, КОТОРЫЕ РЕШАЕТ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
[0009]
Изобретатели настоящего изобретения разработали способ получения карбонилгалогенид облучением галогенированного углеводорода светом, как описано выше, и эффективность реакции полученного карбонилгалогенида и спиртового соединения или подобных является высокой. Но выход израсходованного галогенированного углеводорода является низким, так как применяют большое количество галогенированного углеводорода.
Таким образом, цель настоящего изобретения заключается в обеспечении способа получения карбонилгалогенид, эффективного в отношении применяемого галогенированного метана.
СПОСОБЫ РЕШЕНИЯ ПРОБЛЕМ
[0010]
Изобретатели настоящего изобретения повторили интенсивные исследования, чтобы решить вышеописанные проблемы. Например, изобретатели предполагали, что если испаряющийся галогенированный метан облучать высокоэнергетическим светом, то галогенированный метан может эффективно фоторазлагаться, но образующийся карбонилгалогенид также может быстро фоторазлагаться в газовой фазе. С одной стороны, изобретатели исследовали различные условия реакции; в результате изобретатели завершили настоящее изобретение, обнаружив, что карбонилгалогенид неожиданно можно получить с высоким выходом путем облучения высокоэнергетическим светом испаренного текущего галогенированного метана.
Настоящее изобретение описано далее.
[0011]
[1] Способ получения карбонилгалогенид, включающий стадии:
получения смешанного газа, содержащего кислород и галогенированный метан, содержащий одну или более галогеновых групп, выбранных из группы, состоящей из хлора, брома и йода, и
пропускания потока смешанного газа и облучения высокоэнергетическим светом протекающего смешанного газа.
[2] Способ по [1] выше, где кратчайшее расстояние от источника высокоэнергетического света до протекающей газовой смеси составляет 1 м или меньше.
[3] Способ по [1] или [2] выше, где продолжительность облучения высокоэнергетическим светом протекающей газовой смеси составляет 1 секунду или больше и 10000 секунд или меньше.
[4] Способ по любому из [1]-[3] выше, где температура при облучении высокоэнергетическим светом протекающей газовой смеси составляет 40°С и больше и 200°С или меньше.
[5] Способ получения фторированного карбонатного соединения, включающий стадии:
получения карбонилгалогенида способом по любому из [1]-[4] выше и
реакции фторированного спиртового соединения и карбонилгалогенида,
где молярное отношение фторированного спиртового соединения к галогенированному метану регулируют равным 1 или более.
[6] Способ получения нефторированного карбонатного соединения, включающий стадии:
получения карбонилгалогенида способом по любому из [1]-[4] выше и
реакции нефторированного спиртового соединения и карбонилгалогенид,
где молярное отношение нефторированного спиртового соединения к галогенированному метану регулируют равным 1 или более.
[7] Способ получения соединения фторированного сложного эфира галогенированной муравьиной кислоты, включающий стадии:
получения карбонилгалогенида способом по любому из [1]-[4] выше и
реакции фторированного спиртового соединения и карбонилгалогенида,
где молярное отношение фторированного спиртового соединения к галогенированному метану регулируют равным меньшим чем 1.
[8] Способ получения соединения нефторированного сложного эфира галогенированной муравьиной кислоты, включающий стадии:
получения карбонилгалогенида способом по любому из [1]-[4] выше и
реакции нефторированного спиртового соединения и карбонилгалогенида,
где молярное отношение нефторированного спиртового соединения к галогенированному метану регулируют равным меньше чем 1.
[9] Способ получения изоцианатного соединения, включающий стадии:
получения карбонилгалогенида способом по любому из [1]-[4] выше и
реакции первичного аминосоединения и карбонилгалогенида,
где молярное отношение первичного аминосоединения к галогенированному метану регулируют равным меньше чем 1.
[10] Способ получения N-карбоксильного ангидрида аминокислоты, включающий стадии:
получения карбонилгалогенида способом по любому из выше [1]-[4] выше и
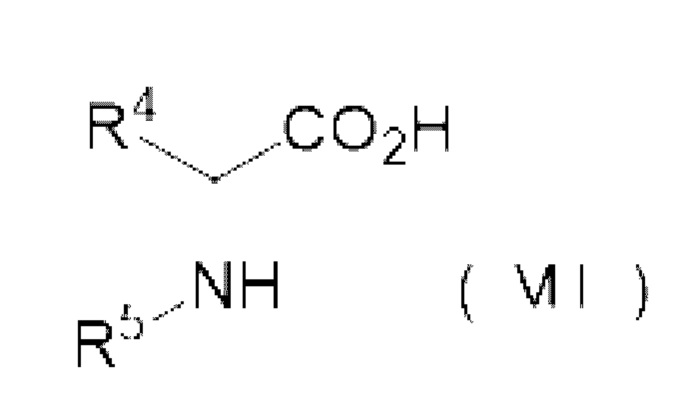
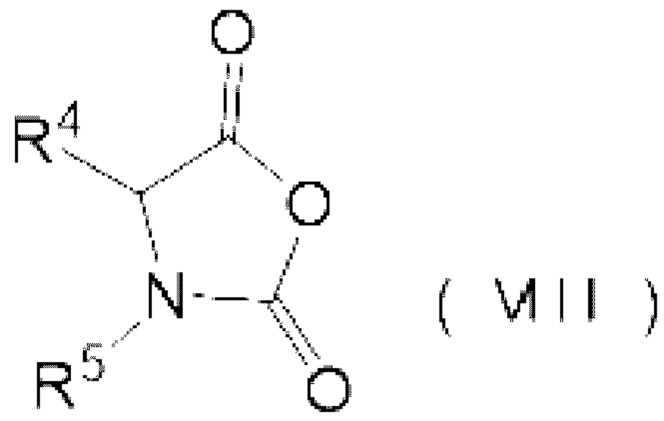
реакции аминокислотного соединения, представленного следующей формулой (VII), и карбонилгалогенида,
где N-карбоксильный ангидрид аминокислоты представлен следующей формулой (VIII):
[0012]
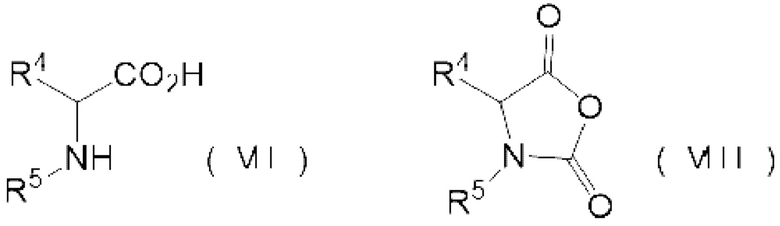
[0013] где
R4 представляет собой группу боковой цепи аминокислоты, где реакционная группа защищена,
R5 представляет собой Н или Р1-[-NH-CHR6-C(=O)-]1-, где R6 представляет собой группу боковой цепи аминокислоты, где реакционная группа защищена, Р1 представляет собой защитную группу аминогруппы, 1 представляет собой целое 1 или более, и когда 1 представляет собой целое 2 или более, множество R6 могут быть одинаковыми или отличными друг от друга.
[11] Способ получения реагента Вильсмейера,
где реагент Вильсмейера представляет собой соль, представленную следующей формулой (X):
[0014]
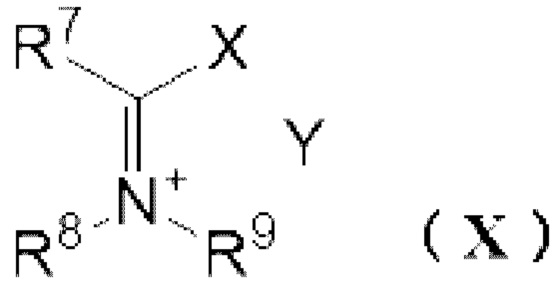
[0015] где
R7 представляет собой атом водорода, С1-6 алкильную группу или необязательно замещенную C6-12 ароматическую углеводородную группу,
R8 и R9 независимо представляют собой С1-6 алкильную группу или необязательно замещенную C6-12 ароматическую углеводородную группу, или R8 и R9 могут образовывать 4 или более и 7 или менее членную кольцевую структуру вместе друг с другом,
X представляет собой атом галогена, выбранный из группы, состоящей из хлора, брома или йода,
Y- представляет собой противоион,
включающий стадии:
получения карбонилгалогенида способом по любому из [1]-[4] выше, и
реакцию карбонилгалогенида и амидного соединения, представленного следующей формулой (IX):
[0016]
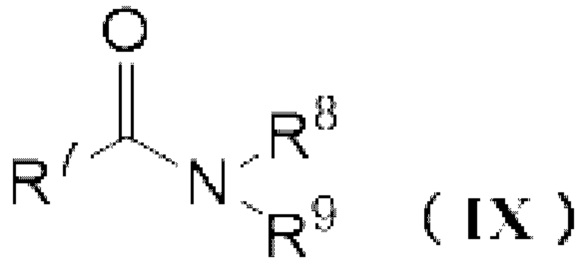
[0017]
где R7-R9 имеют значения как выше.
ЭФФЕКТ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0018]
Применение и обработка галогенированного метана ограничена из-за сильного воздействия на окружающую среду. С одной стороны, карбонилгалогенид можно эффективно получить из применяемого галогенированного метана, и галогенированный метан можно эффективно применять настоящим изобретением. Таким образом, настоящее изобретение является очень пригодным в промышленном отношении в качестве технологии, которая обеспечивает эффективное применение галогенированного метана и эффективное производство карбонилгалогенида, такого как фосген.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0019]
Фигура 1 представляет собой схематическое изображение, показывающее один пример строения реакционной системы, пригодной в способе настоящего изобретения.
Фигура 2 представляет собой схематическое изображение, показывающее один пример строения реакционной системы, пригодной в способе настоящего изобретения.
Фигура 3 представляет собой схематическое изображение, показывающее один пример строения реакционной системы, пригодной в способе настоящего изобретения.
Фигура 4 представляет собой схематическое изображение, показывающее один пример строения реакционной системы, пригодной в способе настоящего изобретения.
Фигура 5 представляет собой схематическое изображение, показывающее один пример строения реакционной системы, пригодной в способе настоящего изобретения.
Фигура 6 представляет собой схематическое изображение, показывающее один пример строения реакционной системы, пригодной в способе настоящего изобретения.
СПОСОБ ОСУЩЕСТВЛЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0020]
Способ настоящего изобретения описан далее поэтапно, и настоящее изобретение не ограничивается следующими конкретными примерами.
[0021]
1. Стадия получения смешанного газа
Смешанный газ, содержащий кислород и галогенированный метан, содержащий один или более атомов галогена, выбранных из группы, состоящей из хлора, брома или йода, в данной стадии.
[0022]
Галогенированный метан, применяемый в настоящем изобретении, обозначает метан, который содержит один или более атомов галогена, выбранных из группы, состоящей из хлора, брома или йода. Галогенированный метан может разлагаться кислородом и высокоэнергетичным светом, образуя карбонилгалогенид.
[0023]
Галогенированный метан может разлагаться кислородом и высокоэнергетичным светом, как описано выше, и играет роль, аналогичную карбонилгалогениду в настоящем изобретении. Галогенированный метан предпочтительно представляет собой полигалогенированный метан, содержащий 2 или более атомов галогена, и предпочтительно пергалогенированный метан, в котором все из атомов водорода замещены атомами галогена.
[0024]
Пример конкретного галогенированного метана включает галогенированный метан, такой как дихлорметан, хлороформ, дибромметан, бромоформ, йодметан и дийодметан.
[0025]
Галогенированный метан можно подходящим образом выбрать в зависимости от целевой химической реакции и требуемого продукта. Можно применять только один тип галогенированного метана, или два или более типов галогенированных метанов можно применять в комбинации. В зависимости от получаемого целевого соединения применяют только один вид галогенированного метана. Галогенированный метан, содержащий хлорную группу, является предпочтительным среди галогенированных метанов с точки зрения испарения и стоимости.
[0026]
Обычный продукт из галогенированного метана может содержать стабилизирующий агент, такой как спирт, для ингибирования разложения галогенированного метана. Поскольку галогенированный метан подвергается окислительному фоторазложению в настоящем изобретении, стабилизирующий агент можно удалять из галогенированного метана, который будут применять. Когда применяют галогенированный метан, из которого удален стабилизирующий агент, возможно более эффективное разложение галогенированного метана. Например, можно применять высокоэнергетический свет, энергия которого относительно невелика, и Продолжительность облучения высокоэнергетическим светом может быть уменьшено. Способ удаления стабилизирующего агента из галогенированного метана специально не ограничен. Например, галогенированный метан можно промыть водой для удаления водорастворимого стабилизирующего агента, и затем высушить.
[0027]
Хлороформ, который применяют в качестве растворителя общего назначения и является недорогим, можно применять в качестве галогенированного метана, применяемого в способе настоящего изобретения. Например, галогенированный метан который когда-то применяли в качестве растворителя, может регенерировать для повторного применения. Предпочтительно, чтобы данный примененный галогенированный метан был до некоторой степени очищен для применения, так как при содержании большого количества примеси и воды реакция может ингибироваться. Например, предпочтительно, чтобы вода и водорастворимые примеси удалялись путем промывки водой, а затем галогенированный метан сушился над безводным сульфатом натрия, безводным сульфатом магния или подобными. Излишняя очистка, при которой снижается производительность, не требуется, так как реакция может протекать даже при содержании воды около 1% по массе. Содержание воды составляет приблизительно 0,5% по массе или меньше, более предпочтительно 0,2% по массе или меньше, и даже предпочтительно 0,1% по массе или меньше. Содержание воды составляет предпочтительно предел обнаружения или менее, или 0% по массе. Вышеописанный повторно применяемый галогенированный метан может содержать продукт разложения галогенированного метана.
[0028]
В частности, когда галогенированный метан не является жидкостью при атмосферной температуре и атмосферном давлении или трудно испаряется, можно применять растворитель в добавление к галогенированному метану. Данный растворитель может ускорить разложение галогенированного метана. Кроме того, растворитель может ингибировать разложение карбонилгалогенида, образующееся при разложении галогенированного метана. Предпочтительно, чтобы растворитель мог подходящим образом растворять галогенированный метан и не препятствовал разложению галогенированного метана. Пример растворителя включает кетоновый растворитель, такой как ацетон, метилэтилкетон, метилизобутилкетон и циклогексанон; сложноэфирный растворитель, такой как этилацетат; алифатический углеводородный растворитель, такой как н-гексан; ароматический углеводородный растворитель, такой как бензол, толуол, ксилол и бензонитрил; эфирный растворитель, такой как диэтиловый эфир, тетрагидрофуран и диоксан; и нитриловый растворитель, такой как ацетонитрил.
[0029]
Источником кислорода может быть газ, содержащий кислород, например, можно применять воздух или очищенный кислород. Очищенный кислород можно смешивать с применяемым инертным газом, таким как азот и аргон. Предпочтительнее применять воздух с точки зрения стоимости и простоты. Содержание кислорода в кислородсодержащем газе, применяемом в качестве источника кислорода, составляет приблизительно 15 об. % или более и приблизительно 100 об. % или менее с точки зрения высокой эффективности разложения галогенированного метана при облучении высокоэнергетическим светом. Предпочтительно применять по существу только кислород кроме неизбежной примеси. Содержание кислорода может быть соответствующим образом определено в зависимости от типа галогенированного метана или подобных. Например, когда в качестве галогенированного метана применяют хлорметан, такой как дихлорметан и хлороформ, содержание кислорода составляет приблизительно 15 об. % или более и 100 об. % или менее. При применении бромметана, такого как дибромметан и бромоформ, содержание кислорода составляет 90 об. % или более и 100 об. % или менее. Даже когда применяют кислород с содержанием кислорода 100 об. %, содержание кислорода можно регулировать в вышеописанном диапазоне путем регулирования количества кислорода, подаваемого в реакционную систему.
[0030]
В качестве источника кислорода можно применять осушенный воздух. Поскольку даже воздух, содержащий водяной пар, не может чрезмерно ингибировать реакцию, воздух можно применять без регулирования содержания водяного пара. Концентрация кислорода в воздухе составляет приблизительно 21 об. %, и концентрация кислорода в источнике кислорода также может быть доведена до 20±5 об. %. Концентрация предпочтительно составляет 20±2 об. %. Когда в качестве источника кислорода применяют воздух, компоненты воздуха, отличные от кислорода, могут чрезмерно поглощать высокоэнергетический свет и снижать концентрацию образующихся карбонилгалогенидов; в результате может быть ингибировано разложение образующихся карбонилгалогенидов.
[0031]
Смешанный газ, содержащий газообразный галогенированный метан и кислород, получают на данной стадии. Условия получения смешанного газа конкретно не ограничены. Например, галогенированный метан подают в нагреватель 3 шприцевым насосом 1 с заданным расходом для испарения галогенированного метана путем нагревания до температуры кипения и выше, и заданный расход кислорода и испаряемого галогенированного метана смешивают с помощью регулятора массового расхода 2 для получения смешанный газ, как показано на рисунках 1, 2, 4-6.
[0032]
Альтернативно, температуру бани реактора для фотореакции 12, оснащенного источником света 11 и баней 13, предварительно устанавливают на температуру кипения или выше галогенированного метана, и затем галогенированный метан подают в реактор для фотореакции 12 для испарения, как показано на рисунке 3. Подаваемый галогенированный метан можно перемешивать, применяя мешальник 14, для ускорения испарения галогенированного метана. Галогенированный метан испаряется, и кислородсодержащий газ подают в газовую фазу реактора для фотореакции 12 при заданном расходе для получения смешанного газа, содержащего галогенированный метан и кислород в реакторе для фотореакции 12
[0033]
Соотношения испаренного галогенированного метана и кислорода в смешанном газе можно соответствующим образом регулировать при условии, что можно успешно получать карбонилгалогенид. Например, соотношение расхода кислорода в кислородсодержащем газе к расходу галогенированного метана в смешанном газе может быть доведено до 0,1 или более и 10 или менее. При соотношении 0,1 или более галогенированный метан может в достаточной степени подвергнуться окислительному фоторазложению. Когда отношение составляет 10 или меньше, можно в достаточной степени предотвратить дальнейшее окислительное фоторазложение полученного карбонилгалогенида. Соотношение предпочтительно составляет 0,2 или более, более предпочтительно 0,4 или более, даже более предпочтительно 0,5 более или более предпочтительно 8 или менее, более предпочтительно 6 или менее. В частности, при соотношении 0,5 или более образование побочного продукта и нарушение работы реакционной системы из-за побочного продукта могут быть более эффективно подавлены.
[0034]
Когда кислородсодержащий газ подают в газовую фазу, содержащую испаренный галогенированный метан в реакционной системе, показанной как рисунок 3, предпочтительно применяют достаточное количество кислорода, которое вызывает окислительное фоторазложение галогенированного метана. Например, расход кислорода в 1 минуту на 1 моль галогенированного метана может быть доведен до 0,1 л или более и 100 л или менее. Соотношение предпочтительно составляет 1 л или более, более предпочтительно 5 л или более, и даже более предпочтительно 10 л или более.
[0035]
2. Стадия окислительного фоторазложения
Смешанный газ, содержащий галогенированный метан и кислород, течет, и высокоэнергетическим светом облучают текущий смешанный газ в газовой фазе для получения карбонилгалогенида окислительным фоторазложением галогенированного метана на данной стадии.
[0036]
Высокоэнергетический свет, излучаемый в протекающий смешанный газ, предпочтительно включает коротковолновый свет и более предпочтительно включает ультрафиолетовый свет. Свет, включающий свет с длиной волны 180 нм или более и 500 нм или менее, и свет, включающий свет с пиковой длиной волны 180 нм или более и 500 нм или менее, являются предпочтительными. Длина волны высокоэнергетического света может быть соответствующим образом определена, она составляет более предпочтительно 400 нм или менее и даже более предпочтительно 300 нм или менее, и свет, пиковая длина волны которого входит в вышеуказанные диапазоны, также является предпочтительным.
Когда облучаемый свет включает свет, длина волны которого входит в указанные выше диапазоны, галогенированный метан может эффективно подвергаться окислительному фоторазложению. Например, можно применять свет, включающий УФ-В с длиной волны 280 нм или более и 315 нм или менее, и/или УФ-С с длиной волны 180 нм или более и 280 нм или менее, предпочтительно применять свет, включающий УФ-С с длиной волны 180 нм или более и 280 нм или меньше, и также предпочтителен свет, пиковая длина волны которого включена в данные диапазоны.
[0037]
Поскольку газообразный галогенированный метан подвергается окислительному фоторазложению в настоящем изобретении, даже высокоэнергетический свет, обладающий относительно низкой энергией, может окислительно фоторазлагать галогенированный метан. В частности, при применении галогенированного метана, который не содержит стабилизирующего агента, даже высокоэнергетический свет, обладающий относительно низкой энергией, может подвергать галогенированный метан окислительному фоторазложению. Примером высокоэнергетического света, имеющего относительно низкую энергию, является свет, пиковая длина волны которого входит в диапазон длин волн видимого света. Диапазон длин волн видимого света может составлять 350 нм или более и 830 нм или менее, и предпочтительно составляет 360 нм или более, более предпочтительно 380 нм или более, даже более предпочтительно 400 нм или более, предпочтительно 800 нм или менее, более предпочтительно 780 нм или менее, даже более предпочтительно 500 нм или менее.
[0038]
Средства облучения светом конкретно не ограничены при условии, что свет, имеющий вышеописанную длину волны, может излучаться данными средствами. Пример источника света с данным диапазоном длин волн включает солнечный свет, ртутную лампу низкого давления, ртутную лампу среднего давления, ртутную лампу высокого давления, ртутную лампу сверхвысокого давления, химическую лампу, лампу черного света, металлогалогенную лампу и светодиодную лампу. Предпочтительно применять ртутную лампу низкого давления с точки зрения эффективности реакции и стоимости.
[0039]
Параметр, такой как интенсивность света, может быть соответствующим образом определен в зависимости от галогенированного метана или подобных. Например, интенсивность света при кратчайшем расстоянии текущего смешанного газа от источника света определяют в зависимости от масштаба производства и длина волны облучающего света и предпочтительно составляет 1 мВт/см2 или более и 200 мВт/см2 или менее. Например, когда длина волны облучающего света является относительно короткой, интенсивность света более предпочтительно составляет 100 мВт/см2 или менее или 50 мВт/см2 или менее и даже более предпочтительно 20 мВт/см2 или менее или 10 мВт/см2 или менее. Когда длина волны облучающего света является относительно длинной, интенсивность света более предпочтительно составляет 10 мВт/см2 или более и 20 мВт/см2 или более и может составлять 50 мВт/см2 или более или 100 мВт/см2 или более. Кратчайшее расстояние между источником света и текущим смешанным газом предпочтительно составляет 1 м или менее, более предпочтительно 50 см или менее, и даже более предпочтительно 10 см или менее или 5 см или менее. Нижний предел кратчайшего расстояния конкретно не ограничен и может составлять 0 см. Другими словами, источник света помещается в протекающий смешанный газ.
[0040]
Условия облучения высокоэнергетическим светом текущего смешанного газа конкретно не ограничены. Например, проточное устройство для фотореакции 4 сконструировано путем размещения одной или более реакционных трубок вокруг источника света, или проточное устройство для фотореакции 4 может иметь вход для газа и выход для газа на обоих концах, и в него вставлен источник света. Смешанный газ можно подавать через проточное устройство для фотореакции 4, как показано на рисунках 1, 2 и 4. Реакционную трубку можно спирально обернута вокруг источника света для эффективного облучения высокоэнергетическим светом смешанного газа в проточном устройстве для фотореакции 4. Проточное устройство для фотореакции 4 может быть оборудован средствами подогрева для поддержания газового состояния галогенированного метана. Пример данного средства нагрева включает горячую ванну, в которую может быть погружена часть или все проточное устройство для фотореакции 4, и нагреватель, который может нагревать часть или всю внешнюю часть проточного устройства для фотореакции 4. Альтернативно, получают реактор для фотореакции 12, содержащий источник света 11 внутри, галогенированный метан испаряется в реакторе для фотореакции 12, и высокоэнергетический свет может излучаться из источника света 11 с подачей кислородсодержащего газа в реактор для фотореакции 12, как показано на рисунке 3. Кроме того, испаренный галогенированный метан и кислородсодержащий газ могут подаваться в реактор для фотореакции 12, как показано на рисунках 5 и 6.
[0041]
Испаренный галогенированный метан может подвергаться окислительному фоторазложению до карбонилгалогенида кислородом и высокоэнергетическим светом. Также известно, что карбонилгалогенид разлагается высокоэнергетическим светом. Таким образом, важно регулировать условия облучения высокоэнергетическим светом, чтобы чрезмерно не разлагать образующийся карбонилгалогенид.
[0042]
Например, продолжительность облучения описанным выше высокоэнергетическим светом описанного выше текущего смешанного газа можно регулировать в зависимости от длины волны облучения светом и температуры реакции и составляет предпочтительно 1 секунду или более и 2000 секунд или менее.
Продолжительность облучения высокоэнергетическим светом соответствует времени удерживания текущего смешанного газа в реакторе для фотореакции, в котором высокоэнергетический свет непрерывно облучает текущий смешанный газ. Когда продолжительность составляет 1 секунду или более, испаренный галогенированный метан может более надежно подвергаться окислительному фоторазложению. Когда продолжительность составляет 2000 секунд или менее, избыточное разложение образовавшегося карбонилгалогенида может ингибироваться более надежно. Продолжительность предпочтительно составляет 5 секунд или более, более предпочтительно 10 секунд или более, даже более предпочтительно 20 секунд или более или 30 секунд или более, и предпочтительно 1500 секунд или менее, 1000 секунд или менее, 500 секунд или менее или 300 секунд или менее, более предпочтительно 100 секунд или менее, даже более предпочтительно 60 секунд или менее или 50 секунд или менее. Когда применяют галогенированный метан, который не содержит стабилизирующего агента, разложение образовавшегося карбонилгалогенида можно дополнительно ингибировать, применяя свет с относительно большой длиной волны. Продолжительность светового облучения можно регулировать в пределах 1 секунды или более и 10000 секунд или менее в данном случае. Продолжительность светового облучения составляет предпочтительно 5000 секунд или менее и более предпочтительно 1000 секунд или менее с точки зрения эффективности производства.
[0043]
Когда продолжительность облучения высокоэнергетическим светом является более длинной, галогенированный метан может разлагаться более эффективно, но образующийся карбонилгалогенид может подвергаться дальнейшему окислительному фоторазложению. С одной стороны, когда концентрацию кислорода в смешанном газе регулируют так, чтобы она была низкой, галогенированный метан может подвергаться окислительному фоторазложению с подавлением последующего окислительного фоторазложения карбонилгалогенида. Например, когда концентрацию кислорода в смешанном газе регулируют равной 15±5 об. % и предпочтительно 15±2 об. %, продолжительность облучения светом смешанного газа можно регулировать равной 50 секунд или более, 100 секунд или более, 150 секунд или более, 200 секунд или более, 500 секунд или более или 1000 секунд или более.
[0044]
Расход текущего смешанного газа в реакторе для фотореакции для облучения высокоэнергетическим светом текущего смешанного газа определяют с учетом внутреннего объема реактора для фотореакции. Например, когда внутренний объем реактора для фотореакции является большим, продолжительность пребывания смешанного газа имеет тенденцию к увеличению, и, таким образом, скорость потока предпочтительно регулировать так, чтобы она увеличивалась. С одной стороны, когда внутренний объем реактора для фотореакции является небольшим, скорость потока смешанного газа предпочтительно регулировать так, чтобы она уменьшалась. В частности, поскольку внутренний объем реактора для фотореакции (л)/расход текущего смешанный газ (л/с) соответствует времени пребывания (с) текущего смешанного газа, расход текущего смешанного газа можно определить с учетом требуемого времени пребывания и внутреннего объема реактора для фотореакции. Расход текущего смешанного газа можно считать таким же, как расход кислородсодержащего газа в варианте осуществления, показанном на рисунке 3.
[0045]
Линейная скорость текущего смешанного газа в реакторе для фотореакции можно регулировать равным 0,001 м/мин или более и 100 м/мин или менее. Когда линейная скорость составляет 0,001 м/мин или более, фоторазложение карбонилгалогенида, полученного из галогенированного метана, реакцией в газовой фазе может ингибироваться более более надежно. Когда линейная скорость составляет 100 м/мин или менее, продолжительность превращения галогенированного метана в карбонилгалогенид может быть обеспечена более надежно. Линейную скорость можно рассчитать делением скорости потока текущего смешанного газа, проходящего через реактор для фотореакции на площадь поперечного сечения реактора для фотореакции. Когда площадь поперечного сечения реактора для фотореакции не является постоянной, площадь поперечного сечения можно считать средней величиной площадей поперечных сечений реактора для фотореакции в направлении движения текущего смешанного газа. Среднее значение можно рассчитать, разделив внутренний объем реактора для фотореакции на длину реактора для фотореакции в направлении движения текущего смешанного газа. Линейная скорость составляет предпочтительно 0,01 м/мин или более, и предпочтительно 50 м/мин или менее или 20 м/мин или менее, более предпочтительно 10 м/мин или менее или 5 м/мин или менее, даже более предпочтительно 1 м/мин или менее или 0,5 м/мин или менее.
[0046]
Температуру при облучении высокоэнергетическим светом испаряющийся галогенированного метана можно соответствующим образом отрегулировать при условии, что можно поддерживать испарение галогенированного метана и ингибировать чрезмерное разложение образовавшегося карбонилгалогенида. Например, хотя температура кипения дихлорметана составляет 40°С, а температура кипения хлороформа составляет 61,2°С при атмосферном давлении, газообразное состояние галогенированного метана можно поддерживать даже при температуре ниже температуры кипения путем смешивания газообразного галогенированного метана с кислородом или воздухом. Например, температуру можно регулировать равной 35°С или больше и 250°С или меньше. Температура предпочтительно составляет 40°С или больше или 50°С или больше, более предпочтительно 70°С или больше или 80°С или больше, даже более предпочтительно 85°С или больше, и предпочтительно 200°С или меньше, более предпочтительно 150°С или меньше, даже более предпочтительно 120°С или меньше. Температуру можно регулировать температурой испаренного галогенированного метана и/или температурой кислородсодержащего газа, подаваемого в реакционную емкость. Реакционную емкость можно нагревать, применяя теплоноситель для поддержания температуры смешанного газа в реакционной емкости.
[0047]
Когда высокоэнергетическим светом облучают галогенированный метан, смешанный газ, содержащий галогенированный метан и кислород, может находиться под давлением, но может находиться под давлением до такой степени, что по меньшей мере смешанный газ может пройти через реакционную емкость. Кроме того, производительность может быть повышена за счет повышения давления смешанного газа. Манометрическое давление смешанного газа в реакционной емкости можно регулировать до 0 МПа изб. или более и 2 МПа изб. или менее и оно предпочтительно составляет 1 МПа изб. или менее и более предпочтительно 0,5 МПа изб. или менее.
[0048]
Галогенированный метан подвергается окислительному фоторазложению и можно получить карбонилгалогенид [Х-С(=O)-Х, где X представляет собой один или более атомов галогена, выбранных из группы, состоящей из хлора, брома или йода]. Карбонилгалогенидоподобное соединение, которое играет аналогичную роль карбонилгалогениду, можно также получить в добавление к карбонилгалогениду. Карбонилгалогенидоподобное соединение включено в карбонилгалогенид настоящего изобретения. Репрезентативные реакции, в которых применяют карбонилгалогенид, описаны ниже.
[0049]
3. стадия после реакции - получение карбонатного соединения
Карбонатное соединение можно получить реакцией карбонилгалогенида и спиртового соединения.
[0050]
Условия реакции специально не ограничены. Например, газ, содержащий полученный карбонилгалогенид, можно продувать в композицию, содержащую спиртовое соединение в реакционной емкости 6, как показано на рисунке 1. соединения могут вступать в реакцию в змеевиковом реакционном устройстве, как показано на рисунке 2 и рисунке 4. Кроме того, спиртовое соединение подают в реакционное устройство с регулируемой температурой змеевика 9, и карбонилгалогенид и спиртовое соединение могут вступать в реакцию в змеевиковом реакционном устройстве, как показано на рисунке 2 и рисунке 4.
Спиртовое соединение можно испарить, регулируя температуру змеевикового реакционного устройства, чтобы в данном случае карбонилгалогенид и спиртовое соединение реагировали в газовой фазе. Кроме того, карбонилгалогенид, образующийся в реакторе для фотореакции 12, выталкивается из реактора для фотореакции 12 подаваемым кислородосодержащим газом и продувается в композицию, содержащую спиртовое соединение в реакционной емкости 16, как показано на рисунках 3, 5 и 6. Охлаждающий конденсатор 15 можно установить между реактором для фотореакции 12 и реакционной емкостью 16, как показано на рисунке 3. Температура охлаждающего конденсатора предпочтительно регулируется таким образом, чтобы образовавшийся карбонилгалогенид мог пройти через него. Например, поскольку температура кипения фосгена среди карбонилгалогенидов составляет 8,2°С, температура охлаждающего конденсатора 15 предпочтительно регулируют равной 10°С или больше в случае фосгена.
[0051]
Спиртовое соединение обозначает органическое соединение, содержащее гидроксильную группу, и может быть представлено одновалентным спиртовым соединением, представленным следующей формулой (I), или двухвалентным спиртовым соединением, представленным следующей формулой (II). В настоящем изобретении далее, соединение, представленное формулой х, сокращено обозначают как "соединение х" в некоторых случаях. Например, моновалетное спиртовое соединение, представленное формулой (I), сокращено обозначают как "моновалетное спиртовое соединение (I)" в некоторых случаях

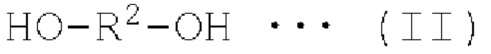
где R1 представляет собой моновалентную органическую группу, и R2 представляет собой двухвалентную органическую группу.
[0052]
Органическая группа конкретно не ограничена при условии, что органическая группа является неактивной в реакции данной стадии и представлена необязательно замещенной С1-10 алифатической углеводородной группой, необязательно замещенной С6-12 ароматической углеводородной группой, необязательно замещенной гетероарильной группой, органической группой, полученной соединением 2 или более и 5 или менее необязательно замещенных С1-10 алифатических углеводородных групп и необязательно замещенных С6-12 ароматических углеводородных групп, и органической группой, полученной соединением 2 или более и 5 или менее необязательно замещенных С1-10 алифатических углеводородных групп и необязательно замещенных гетероарильных групп.
[0053]
Пример С1-10 алифатической углеводородной группы включает С1-10 алифатическую углеводородную группу, С3-10 циклическую алифатическую углеводородную группу и органическую группу, образованную соединением 2 или более и 5 или менее С1-10 алифатических углеводородных групп и С3-10 циклических алифатических углеводородных групп.
[0054]
"С1-10 алифатическая углеводородная группа" обозначает линейную или разветвленную насыщенную или ненасыщенную алифатическую углеводородную группу, содержащую количество атомов углерода 1 или более и 10 или менее. Пример моновалентной С1-10 алифатической углеводородной группы включает С1-10 алкильную группу, С2-10 алкенильную группу и С2-10 алкинильную группу.
[0055]
Пример С1-10 алкильной группы включает метил, этил, н-пропил, изопропил, н-бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил, 2,2-лиметилэтил, н-пентил, н-гексил, 2-гексил, 3-гексил, 4-метил-2-пентил, н-гептил, н-октил и н-децил. C1-10 алкильная группа предпочтительно представляет собой С2-8 алкильную группу и более предпочтительно С4-6 алкильную группу.
[0056]
Пример С2-10 алкенильной группы включает этенил (винил), 1-пропенил, 2-пропенил (аллил), бутенил, гексенил, октенил и деценил. С2-10 алкенильная группа предпочтительно представляет собой С2-8 алкенильную группу и более предпочтительно С4-6 алкенильную группу.
[0057]
Пример С2-10 алкинильной группы включает этинил, пропинил, бутинил, гексинил, октинил и пентадецинил. С2-10 алкинильная группа предпочтительно представляет собой С2-8 алкинильную группу и более предпочтительно С2-6 алкинильную группу.
[0058]
"С3-10 циклическая алифатическая углеводородная группа" обозначает циклическую насыщенную или ненасыщенную алифатическую углеводородную группу, содержащую количество атомов углерода 1 или более и 10 или менее. Пример моновалентной С3-10 циклической алифатической углеводородной группы включает С3-10 циклоалкильную группу, С4-10 циклоалкенильную группу и С4-10 циклоалкинильную группу.
[0059]
Пример органической группы, образованной соединением 2 или более и 5 или менее С1-10 алифатических углеводородных групп и С3-10 циклических алифатических углеводородных групп включает С3-10 моновалентную циклическую алифатическую углеводородную группу - С1-10 двухвалентную алифатическую углеводородную группу, и С1-10 моновалентную алифатическую углеводородную группу - С3-10 двухвалентную циклическую алифатическую углеводородную группу - С1-10 двухвалентную алифатическую углеводородную группу.
[0060]
"С6-12 ароматическая углеводородная группа" обозначает ароматическую углеводородную группу, содержащую количество атомов углерода 6 или более и 12 или менее. Пример моновалентной С6-12 ароматической углеводородной группы включает фенил, инденил, нафтил и бифенил, и фенил является предпочтительным.
[0061]
"Гетероарильная группа" обозначает 5-членную ароматическую гетероциклическую группу, 6-членную ароматическую гетероциклическую группу и конденсированную кольцевую ароматическую гетероциклическую группу, содержащую по меньшей мере один гетероатом, такой как атом азота, атом кислорода и атом серы. Пример гетероарильной группы включает моновалентную 5-членную ароматическую гетероциклическую группу, такую как пирролил, имидазолил, пиразолил, тиенил, фурил, оксазолил, изоксазолил, тиазолил, изотиазолил и тиадиазолил; моновалентную 6-членную ароматическую гетероциклическую группу, такую как пиридинил, пиразинил, пиримидинил и пиридазинил; и конденсированную кольцевую ароматическую гетероциклическую группу, такую как индолил, изоиндолил, хинолинил, изохинолинил, бензофуранил, изобензофуранил и хроменил.
[0062]
Пример "органической группы, образованной соединением 2 или более и 5 или менее С1-10 алифатических углеводородных групп и С6-12 ароматических углеводородных групп" включает С6-12 ароматическую углеводородную группу - С1-10 алифатическую углеводородную группу, С1-10 алифатической углеводородную группу - С6-12 ароматическую углеводородную группу, С1-10 алифатическую углеводородную группу - С6-12 ароматическую углеводородную группу - С1-10 алифатическую углеводородную группу и С6-12 ароматическую углеводородную группу - С1-10 алифатическую углеводородную группу - С6-12 ароматическую углеводородную группу. Пример "органической группы, образованной соединением 2 или более и 5 или менее С1-10 алифатических углеводородных групп и гетероарильных групп" включает гетероарильную группу - С1-10 алифатическую углеводородную группу, С1-10 алифатическую углеводородную группу - гетероарильную группу, С1-10 алифатическую углеводородную группу - гетероарильную группу - С1-10 алифатическую углеводородную группу, и гетероарильную группу - С1-10 алифатической углеводородную группу - гетероарильную группу.
[0063]
Пример замещающей группы, которую С1-10 алифатическая углеводородная группа может необязательно содержать, включает одну или более замещающих групп, выбранных из группы, состоящей из атома галогена, нитрогруппы и цианогруппы, и атом галогена является предпочтительным. Пример замещающей группы, которую С6-12 ароматическая углеводородная группа и гетероарильная группа могут необязательно содержать, включает одну или более замещающих групп, выбранных из группы, состоящей из С1-6 алкильной группы, С1-6 алкоксигруппы, атома галогена, нитрогруппы и цианогруппы, и атом галогена является предпочтительным. Пример атома галогена включает фтор, хлор, бром и йод, и фтор является предпочтительным.
[0064]
Спиртовое соединение можно классифицировать как фторированное спиртовое соединение и нефторированное спиртовое соединение. Фторированное спиртовое соединение обязательно содержит фторгруппу в качестве замещающей группы, и нефторированное спиртовое соединение не замещено фторгруппой. Атом галогена, который нефторированное спиртовое соединение может необязательно содержать, представляет собой один или более из атомов галогена, выбранных из хлора, брома и йода. Группу "Rx", содержащую фтор в качестве замещающей группы, можно описать как "RFx".
[0065]
"С1-6 Алкильная группа" обозначает линейную или разветвленную моновалентную насыщенную алифатической углеводородную группу, содержащую количество атомов углерода 1 или более и 6 или менее. Пример С1-6 алкильной группы включает метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил и н-гексил. С1-6 алкильная группа предпочтительно представляет собой C1-4 алкильную группу, более предпочтительно C1-2 алкильную группу и даже более предпочтительно метил.
[0066]
"С1-6 алкоксигруппа" обозначает линейную или разветвленную моновалентную насыщенную алифатическую углеводородную оксигруппу, содержащую количество атомов углерода 1 или более и 6 или менее. Пример С1-6 алкоксигруппы включает метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси, н-пентокси и н-гексокси. С1-6 алкоксигруппа предпочтительно представляет собой C1-4 алкоксигруппу, более предпочтительно C1-2 алкоксигруппу и даже более предпочтительно метокси.
[0067]
Моновалентное спиртовое соединение (I) может представлять собой фторированное спиртовое соединение. Пример моновалентного фторированного спиртового соединения (I) включает фторированный этанол, такой как дифторэтанол и трифторэтанол; фторированный пропанол, такой как монофторпропанол, дифторпропанол, трифторпропанол, тетрафторпропанол, пентафторпропанол и гексафторпропанол.
[0068]
Пример двухвалентной органической группы включает двухвалентные органические группы, полученный из примеров моновалентной органической группы. Например, двухвалентная органическая группа, полученная из С1-10 алкильной группы, С2-10 алкенильной группы и С2-10 алкинильной группы в качестве моновалентной органической группы, представляет собой С1-10 ал канди ильную группу, С2-10 алкендиильную группу и С2-10 алкиндиильную группу.
[0069]
Двухвалентная органическая группа может представлять собой двухвалентную (поли)алкиленгликольную группу: - [-O-R2-]n-, где R2 представляет собой C1-8 алкандиильную группу, и n представляет собой целое 1 или более и 50 или менее.
[0070]
Кроме того, пример двухвалентного спиртового соединения (II) включает следующее двухвалентное спиртовое соединение (II-1):
[0071]
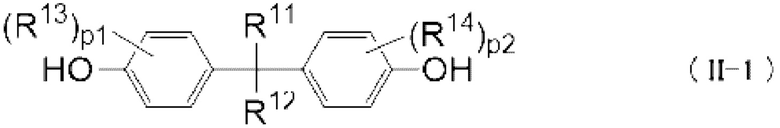
[0072] Где
R11 и R12 независимо представляют собой Н, C1-6 алкильную группу, C1-6 фторалкильную группу или С6-12 ароматическую углеводородную группу, или образуют С3-6 циклоалкильную группу, необязательно содержащую C1-6 алкил в качестве заместителя вместе друг с другом, R13 и R14 независимо представляют собой Н, C1-6 алкильную группу или С6-12 ароматическую углеводородную группу, и когда p1 или р2 представляет собой целое 2 или более, множество R11 или R12 являются одинаковыми или отличными друг от друга,
p1 и р2 независимо представляют собой целые 0 или более и 4 или менее.
[0073]
Пример конкретного двухвалентного нефторированного спиртового соединения (II-1) включают 2,2-бис(4-гидроксифенил)пропан, 1,1-бис(4-гидроксифенил)-1-фенилэтан, 2,2-бис(4-гидроксифенил)гексафторпропан, 2,2-бис(4-гидроксифенил)бутан, бис(4-гидроксифенил)дифенилметан, 2,2-бис(3-метил-4-гидроксифенил)пропан, 1,1-бис(4-гидроксифенил)этан, бис(4-гидроксифенил)метан и 2,2-бис(4-гидрокси-3-изопропилфенил)пропан, и 2,2-бис(4-гидроксифенил)пропан и т.д. Бисфенол А является предпочтительным.
[0074]
Двухвалентное спиртовое соединение (II) может представлять собой фторированное спиртовое соединение. Пример данного двухвалентного фторированного спиртового соединения (II) включают фторированный этиленгликоль; фторированный пропиленгликоль, такой как монофторпропиленгликоль и дифторпропиленгликоль; фторированный бутандиол, такой как монофторбутандиол, дифторбутандиол, трифторбутандиол и тетрафторбутандиол; фторированный пентандиол, такой как монофторпентандиол, дифторпентандиол, трифторпентандиол, тетрафторпентандиол, пентафторпентандиол и гексафторпентандиол; фторированный гександиол, такой как монофторгександиол, дифторгександиол, трифторгександиол, тетрафторгександиол, пентафторгександиол, гексафторгександиол, гептафторгександиол и октафторгександиол; фторированный гептандиол, такой как монофторгептандиол, дифторгептандиол, трифторгептандиол, тетрафторгептандиол, пентафторгептандиол, гексафторгептандиол, гептафторгептандиол, октафторгептандиол, нонафторгептандиол и декафторгептандиол; фторированный октандиол, такой как монофтороктандиол, дифтороктандиол, трифтороктандиол, тетрафтороктандиол, пентафтороктандиол, гексафтороктандиол, гептафтороктандиол, октафтороктандиол, нонафтороктандиол, декафтороктандиол, ундекафтороктандиол и додекафтороктандиол; фторированный нонандиол, такой как монофторнонандиол, дифторнонандиол, трифторнонандиол, тетрафторнонандиол, пентафторнонандиол, гексафторнонандиол, гептафторнонандиол, октафторнонандиол, нонафторнонандиол, декафторнонандиол, ундекафторнонандиол, додекафторнонандиол, тридекафторнонандиол и тетрадекафторнонандиол; фторированный декандиол, такой как монофтордекандиол, дифтордекандиол, трифтордекандиол, тетрафтордекандиол, пентафтордекандиол, гексафтордекандиол, гептафтордекандиол, октафтордекандиол, нонафтордекандиол, декафтордекандиол, ундекафтордекандиол, додекафтордекандиол, тридекафтордекандиол, тетрадекафтордекандиол, пентадекафтордекандиол и гексадекафтордекандиол; фторированный полиэтиленгликоль, такой как фторированный диэтиленгликоль, фторированный триэтиленгликоль, фторированный тетраэтиленгликоль, фторированный пентаэтиленгликоль и фторированный гексаэтиленгликоль.
[0075]
Применяемое количество спиртового соединения можно подходящим образом отрегулировать при условии, что реакция успешно протекает. Например, можно применять 1 или более молярное отношение двухвалентного спиртового соединения к получаемому карбонилгалогениду, и можно применять 2 или более молярное отношение моновалентного спиртового соединения к получаемому карбонилгалогениду. Карбонатное соединение можно более эффективно получить, применяя избыточное количество спиртового соединения. Поскольку выход карбонилгалогенида по отношению к применяемому галогенированному метану не является постоянным, молярное отношение двухвалентного спиртового соединения к галогенированному метану предпочтительно регулируют равным 1 или более, и молярное отношение моновалентного спиртового соединения к галогенированному метану предпочтительно регулируют равным 2 или более. Молярное отношение двухвалентного спирта предпочтительно составляет 1,5 или более, более предпочтительно 2 или более, и предпочтительно 10 или менее, более предпочтительно 5 или менее. Молярное отношение моновалентного спирта предпочтительно составляет 2 или более, более предпочтительно 4 или более, и предпочтительно 20 или менее, более предпочтительно 10 или менее.
[0076]
Основание можно применять для ускорения реакции карбонилгалогенида и спиртового соединения. Основание классифицируют как неорганическое основание и органическое основание. Пример неорганического основания включает карбонатную соль щелочного металла, такую как карбонат лития, карбонат натрия, карбонат калия и карбонат цезия; карбонатную соль металла 2 группы, такую как карбонат магния, карбонат кальция и карбонат бария; гидрокарбонатную соль щелочного металла, такую как гидрокарбонат лития, гидрокарбонат натрия, гидрокарбонат калия и гидрокарбонат цезия; гидроксид щелочного металла, такой как гидроксид лития, гидроксид натрия и гидроксид калия; гидроксид металла 2 группы, такой как гидроксид магния и гидроксид кальция; фторидную соль щелочного металла, такую как фторид лития, фторид натрия, фторид калия и фторид цезия. Карбонатная соль или гидрокарбонатная соль щелочного металла или металла 2 группы является предпочтительной из-за низкой влагопоглощающей способности и низкой текучести, и более предпочтительна карбонатная соль щелочного металла. Например, три(С1-4 алкил)амин, такой как триметиламин, триэтиламин и диизопропилэтиламин; трет-бутоксид щелочного металла, такой как трет-бутоксид натрия и трет-бутоксид калия; ненуклеофильное органическое основание, такое как диазабициклоундецен, диизопропиламид лития, тетраметилпиперидин лития, lf4-диазабицикло[2,2,2]октан (DABCO), 1,5,7-триазабицикло[4,4,0]дец-5-ен (TBD), 7-метил-1,5,7-триазабицикло[4,4,0]дец-5-ен (MTBD), 1,8-диазабицикло[5,4,0]ундец-7-ен (ДБУ), 1,5-диазабицикло[4,3,0]нон-5-ен (ДБН), 1,1,3,3-тетраметилгуанидин (ТМГ) и N-метилморфолин можно применять в качестве органического основания в связи с низкой реакционной способности с продуктом фотореакции тетрагалогенэтилена. Также можно применять слабонуклеофильные органические основания, такие как пиридин и лутидин.
[0077]
Галогеноводород, такой как хлороводород, образуется как побочный продукт в ходе окислительного фоторазложения галогенированного метана и реакции карбонилгалогенида и спиртового соединения. Основание является пригодным для захвата данного галогеноводорода. Но когда применяют реакционную трубку небольшого диаметра, такую как змеевиковое реакционное устройство, показанное на рисунке 2 и рисунке 4, может осаждаться соль галогеноводорода и основания и, таким образом, в некоторых случаях реакционная трубка может засориться. В данном случае применяют основание, соль которого с галогеноводородом представляет собой ионную жидкость. Пример основания включает органическое основание, такое как производное имидазола, например, 1-метилимидазол. Кроме того, можно применять основание, гидрохлоридная соль которого имеет относительно низкую температуру плавления, такое как пиридин.
[0078]
Применяемое количество основания можно подходящим образом регулировать при условии, что реакция протекает успешно и, например, применяемое количество на 1 ммоль галогенированного метана можно регулировать равным 1 моль или более и 10 моль или менее.
[0079]
Например, основание можно добавлять к спиртовому соединению, или основание можно непрерывно подавать к спиртовому соединению.
[0080]
При реакции карбонилгалогенида и спиртового соединения можно применять растворитель. Например, растворитель можно добавить к композиции, содержащей спиртовое соединение. Пример растворителя включает кетоновый растворитель, такой как ацетон, метилэтилкетон, метилизобутилкетон и циклогексанон; сложноэфирный растворитель, такой как этилацетат; алифатический углеводородной растворитель, такой как н-гексан; ароматический углеводородной растворитель, такой как бензол, толуол, ксилол и бензонитрил; эфирный растворитель, такой как диэтиловый эфир, тетрагидрофуран и диоксан; нитриловый растворитель, такой как ацетонитрил; и галогенированный углеводородной растворитель, такой как дихлорметан и хлороформ.
[0081]
Температура реакции карбонилгалогенида и спиртового соединения конкретно не ограничена, и ее можно подходящим образом регулировать. Например, температуру можно регулировать равной 0°С или больше и 250°С или меньше. Температура более предпочтительно составляет 10°С или больше, даже более предпочтительно 20°С или больше, и более предпочтительно 200°С или меньше или 150°С или меньше, даже более предпочтительно 100°С или меньше или 80°С или меньше. Когда основание не применяют, или основание применяют для дополнительного ускорения реакции, температуру можно регулировать так, чтобы она была относительно высокой, например, 50°С или больше или 100°С или больше.
[0082]
Продолжительность реакции карбонилгалогенида и спиртового соединения конкретно не ограничена, и ее можно подходящим образом регулировать. Например, продолжительность составляет предпочтительно 0,5 часов или более и 50 часов или менее. Продолжительность реакции более предпочтительно составляет 1 час или более, даже более предпочтительно 5 часов или более, и более предпочтительно 30 час или менее, даже более предпочтительно 20 часов или менее. Например, даже после окончания получения карбонилгалогенида реакционную смесь можно непрерывно перемешивать до тех пор, пока не будет подтверждено потребление спиртового соединения.
[0083]
Когда применяют моновалентное спиртовое соединение (I), цепочечное карбонатное соединение, представленное следующей формулой (III), получают реакцией карбонилгалогенида и спиртового соединения. Когда применяют двухвалентное спиртовое соединение (II), получают поликарбонатное соединение, содержащее звено, представленное следующей формулой (IV-1), или циклической карбонатное соединение, представленное следующей формулой (IV-2). Когда применяют двухвалентное спиртовое соединение (II), получают ли поликарбонатное соединение (IV-1) или циклической карбонатное соединение (IV-2), и его получаемая доля в основном зависит от расстояния между двумя гидроксильными группами и гибкости химической структуры двухвалентного спиртового соединения (II) и может быть специально определено предварительным экспериментом или подобными.
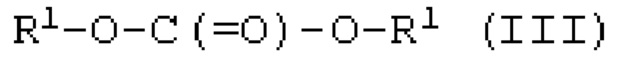
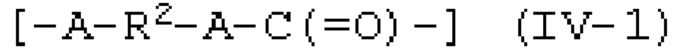
[0084]
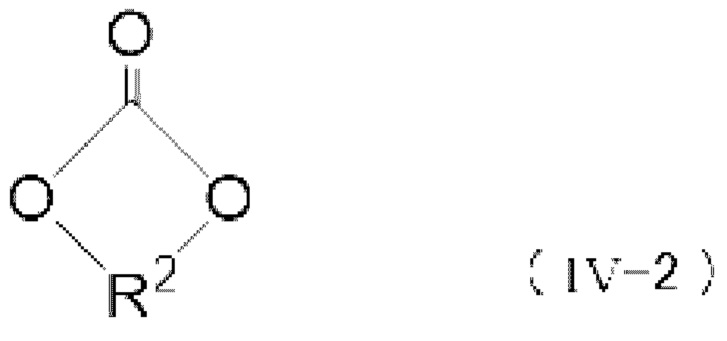
[0085]
4. Стадия после реакции - получение галогенированного эфира муравьиной кислоты
Галогенированный эфир муравьиной кислоты можно получить регулированием молярного отношения спиртового соединения к галогенированному метану равным меньше чем 1 без применения основания в описанном выше способе получения карбонатного соединения. Молярное отношение предпочтительно составляет 0, 9 или менее и более предпочтительно 0,8 или менее. Описанное выше моновалентное спиртовое соединение (I) можно применять в качестве спиртового соединения. Фторированный эфир галогенированной муравьиной кислоты можно получить из фторированного моновалентного спиртового соединения (I), и нефторированный эфир галогенированной муравьиной кислоты можно получить из нефторированного моновалентного спиртового соединения (I).
[0086]
5. стадия после реакции - получение изоцианатного соединения
Изоцианатное соединения можно получить реакцией карбонилгалогенида и первичного аминосоединения. Изоцианатное соединение является пригодным в качестве исходного соединения карбаматного соединения, уретанового соединения или подобных. Первичное аминосоединения можно применять вместо спиртового соединения в описанном выше способе получения карбонатного соединения за исключением следующих моментов в качестве варианта осуществления реакции.
[0087]
Первичное аминосоединение конкретно не ограничено при условии, что соединение содержит 1 или более аминогрупп (-NH2 групп). Пример первичного аминосоединения включает первичное аминосоединение (V): R3-(NH2)m, где R3 представляет собой м-валентную органическую группу, и m равно целому 1 или более и 6 или менее, предпочтительно 5 или менее, 4 или менее или 3 или менее, более предпочтительно 1 или 2, и даже более предпочтительно 2.
[0088]
Пример моновалентной органической группы среди органической группы R3 включает ту же группу как моновалентная органическая группа R1 в описанном выше способе получения карбонатного соединения. Пример двухвалентной органической группы включает ту же группу как двухвалентная органическая группа R2. Пример трех или более валентной органической группы включает трех или более валентную органическую группу, полученную из примеров моновалентной органической группы R1. Например, трехвалентная органическую группу, полученная из С1-10 алкильной группы, С2-10 алкенильной группы и С2-10 алкинильной группы в качестве моновалентной органической группы, представляет собой C1-10 алкантриильную группу, С2-10 алкентриильную группу и С2-10 алкинтриильную группу.
[0089]
Изоцианатное соединение (VI): R3-(N=C=O)m можно получить реакцией карбонилгалогенида и первичного аминового соединения (V). Полученный R3-(N=C=O)m может вступать в реакцию с первичным аминовым соединения (V) с образованием соединения на основе мочевины: R3-[NH-C(=O)-NH-R3]m, и предпочтительно ингибировать реакцию, в которой молярное отношение первичного аминового соединения (V) к галогенированному метану регулируют равным 1 или менее, в качестве первичного аминового соединения (V) применяют соль, или не применяют основание. Кроме того, изоцианатное соединение можно эффективно получить при следующем условии: полученный карбонилгалогенид растворяют в растворителе для получения раствора карбонилгалогенида, а молярное отношение карбонилгалогенида к первичному аминовому соединению (V) поддерживают равным более 1 путем добавления первичного аминового соединения (V) или его раствора к раствору карбонилгалогенида.
[0090]
Когда целевое соединение представляет собой изоцианатное соединение, молярное отношение первичного аминового соединения (V) к полученному карбонилгалогениду предпочтительно регулируют равным 1 или менее. Поскольку в некоторых случаях может быть трудно предсказать точное количество образующегося карбонилгалогенида, отношение первичного аминового соединения (V) к применяемому галогенированному метану регулируют меньшим ниже 1. Молярное отношение предпочтительно составляет 0,5 или менее, более предпочтительно 0,2 или менее, и предпочтительно 0,001 или более, более предпочтительно 0,05 или более. Когда целевое соединение представляет собой соединение на основе мочевины, соотношение предпочтительно составляет 2 или более, более предпочтительно 4 или более, и предпочтительно 20 или менее, более предпочтительно 15 или менее.
[0091]
Когда целевое соединение представляет собой изоцианатное соединение, применяют соль в качестве первичного аминового соединения (V), поскольку изоцианатное соединение практически не реагирует с солью амина. Например, данная соль включает соль неорганической кислоты, такую как гидрохлоридная соль, гидробромидная соль, гидройодидная соль, сульфатная соль, нитратная соль, перхлоратная соль и фосфатная соль; и органическую соль, такую как оксалатная соль, малонатная соль, малеатная соль, фумарат, лактатная соль, малатная соль, цитратная соль, тартратная соль, бензоатная соль, трифторацетатная соль, ацетатная соль, метансульфонатная соль, п-толуолсульфонатная соль и трифторметансульфонатная соль
[0092]
Температура реакции карбонилгалогенида и первичного аминосоединения предпочтительно регулируют меньшей чем температура реакции со спиртовым соединением для того чтобы поддерживать жидкое состояние карбонилгалогенида. Например, температуру предпочтительно регулируют равным 15°С или меньше, и она предпочтительно составляет 10°С или меньше, более предпочтительно 5°С или меньше и даже более предпочтительно 2°С или меньше. Нижний предел температуры конкретно не ограничен, и температура предпочтительно составляет -80°С или больше и более предпочтительно -20°С или больше или -15°С или больше.
[0093]
Когда целевое соединение представляет собой изоцианатное соединение и применяют основание, основание предпочтительно представляет собой 1 или более оснований, выбранных из гетероциклического ароматического амина и ненуклеофильного сильного основания. Гетероциклический ароматический амин обозначает соединение, которое содержит по меньшей мере одно гетерокольцо и по меньшей мере одну аминовую функциональную группу, отличную -NH2. Пример гетероциклического ароматического амина включает пиридин и его производное, такое как пиридин, α-пиколин, β-пиколин, γ-пиколин, 2,3-лутидин, 2,4-лутидин, 2,6-лутидин, 3,5-лутидин, 2-хлорпиридин, 3-хлорпиридин, 4-хлорпиридин, 2,4,6-триметилпиридин и 4-диметиламинопиридин.
[0094]
«Ненуклеофильное органическое основание» означает основание, в котором нуклеофильность неподеленной электронной пары атома азота является низкой из-за стерических затруднений, но основность которого является высокой. Пример ненуклеофильного органического основания включает триэтиламин, N,N-диизопропилэтиламин, трипропиламин, триизопропиламин, трибутиламин, трипентиламин, тригексиламин, тригептиламин, триоктиламин, тридециламин, тридодециламин, трифениламин, трибензиламин, N,N-диизопропилэтиламин, 1,5,7-триазабицикло[4,4,0]дец-5-ен (TBD), 7-метил-1,5,7-триазабицикло[4,4,0]дец-5-ен (MTBD), 1,8-диазабицикло[5,4,0]ундец-7-ен (DBU), 1,5-диазабицикло[4,3,0]нон-5-ен (DBN) и 1,1,3,3-тетраметилгуанин (TMG). Кроме того, можно применять основание, у которого относительно высокая основность. Например, TBD (pKBH+: 25,98), MTBD (pKBH+: 25,44), DBU (pKBH+: 24,33), DBN (pKBH+: 23,89) и TMG (pKBH+: 23,30) можно применять в качестве основания, чья основность (pKBH+) в ацетонитриле равна 20 или более.
[0095]
Кроме того, разнообразные органические амины, такие как триметиламин, диметилэтиламин, диэтилметиламин, N-этил-N-метилбутиламин и 1-метилпирролидон можно применять в качестве основания.
[0096]
Когда целевое соединение представляет собой соединение на основе мочевины, молярное отношение первичного аминосоединения к галогенированному метану или образующемуся карбонилгалогениду предпочтительно регулируют равным более 1. Молярное отношение предпочтительно составляет 1,5 или более и более предпочтительно 2 или более.
[0097]
6. Стадия после реакции - получение NCA
N-карбоксиангидрид аминокислоты (VIII) (NCA) можно получить, применяя аминокислотное соединение (VII) вместо спиртового соединения в описанном выше способе получения карбонатное соединение.
[0098]
[0099] Где
R4 представляет собой группу боковой цепи аминокислоты, где реакционная группа защищена,
R5 представляет собой Н или Р1-[-NH-CHR6-C(=O)-]1-, где R6 представляет собой группу боковой цепи аминокислоты, где реакционная группа защищена, Р1 представляет собой защитную группу аминогруппы, 1 представляет собой целое 1 или более, и когда 1 представляет собой целое 2 или более, множество R6 могут быть одинаковыми или отличными друг от друга.
[0100]
7. Стадия после реакции - получение реагента Вильсмейера
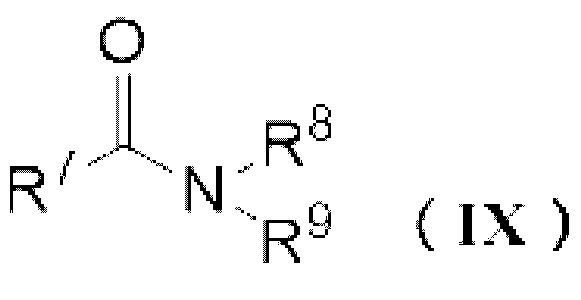
Реагент Вильсмейера (X) можно получить реакцией карбонилгалогенида и амидного соединения (IX). Реагент Вильсмейера можно получить аналогично описанному выше способу получения карбонатного соединения за исключением того, что применяют амидное соединение (IX) вместо спиртового соединения и основание не применяют.
[0101]
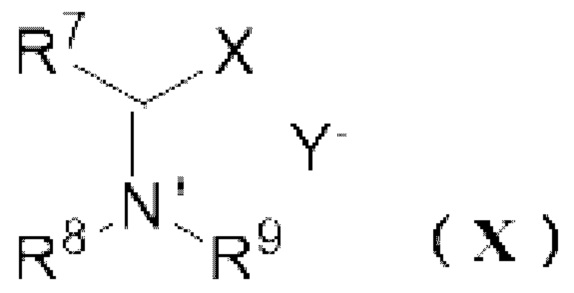
[0102]
где
R7 представляет собой атом водорода, C1-6 алкильную группу или необязательно замещенную С6-12 ароматическую углеводородную группу,
R8 и R9 независимо представляют собой C1-6 алкильную группу или необязательно замещенную C6-12 ароматическую углеводородную группу, или R8 и R9 могут образовывать 4 или более и 7 или менее-членную кольцевую структуру вместе друг с другом,
X представляет собой атом галогена, выбранный из группы, состоящей из хлора, брома или йода,
Y- представляет собой противоион.
[0103]
Замещающая группа, которая может необязательно содержать С6-12 ароматическую углеводородную группу, конкретно не ограничена при условии, что замещающая группа не ингибирует реакцию настоящего изобретения, и примерами являются 1 или более замещающих групп, выбранных из группы, состоящей из C1-6 алкильной группы, C1-6 алкоксигруппы, атома галогена, нитрогруппы и цианогруппы. Количество замещающих групп конкретно не ограничено при условии, что С6-12 ароматическая углеводородная группа может быть замещена и может составлять 1 или более и 5 или менее. Количество предпочтительно составляет 3 или менее, более предпочтительно 2 или менее, и даже более предпочтительно 1. Когда количество замещающих групп равно 2 или более, замещающие группы могут быть одинаковыми или отличными друг от друга.
[0104]
Пример 4 или более и 7 или менее-членной кольцевой структуры, которая образована R8, R9 и атомом азота вместе друг с другом, включает пирролидильную группу, пиперидильную группу и морфолино группу.
[0105]
Пример конкретного амидного соединения (IX) включает N,N-диметилформамид (ДМФА), N,N-диметилацетамид (DMA), N-метил-N-фенилформамид, N-метилпирролидон (NMP), 1,3-диметилмидазолидинон (DMI), тетраметилмочевину, тетраэтилмочевину и тетрабутилмочевину, и ДМФА является предпочтительным по универсальности и стоимости.
[0106]
Y- в формуле (X) конкретно не ограничен и примером является хлорид-ион, бромид-ион и йодид-ион.
[0107]
Применяемое количество амидного соединения можно подходящим образом регулировать при условии, что реакция протекает успешно. Например, применяемое количество на 1 мл галогенированного метана можно регулировать равным 0,1 моль или более и 100 моль или менее.
[0108]
Когда реагируют карбонилгалогенид и амидное соединение, можно применять растворитель. Например, растворитель смешивают с композицией, содержащей амидное соединение. Пример растворителя включает кетоновый растворитель, такой как ацетон, метилэтилкетон, метилизобутилкетон и циклогексанон; сложноэфирный растворитель, такой как этилацетат; алифатический углеводородной растворитель, такой как н-гексан; ароматический углеводородной растворитель, такой как бензол, толуол, ксилол и бензонитрил; эфирный растворитель, такой как диэтиловый эфир, тетрагидрофуран и диоксан; и нитриловый растворитель, такой как ацетонитрил
[0109]
Температура реакции карбонилгалогенида и амидного соединения конкретно не ограничена, и ее можно подходящим образом регулировать. Например, температуру можно регулировать равной 0°С или больше и 120°С или меньше. Температура более предпочтительно составляет 10°С или больше, даже более предпочтительно 20°С или больше, и более предпочтительно 100°С или меньше, даже более предпочтительно 80°С или меньше или 50°С или меньше.
[0110]
Продолжительность реакции карбонилгалогенида и амидного соединения конкретно не ограничена, и ее можно подходящим образом регулировать. Например, продолжительность предпочтительно составляет 0,5 часов или более и 50 часов или менее. Продолжительность реакции более предпочтительно составляет 1 час или более, даже более предпочтительно 5 часов или более, и более предпочтительно 30 час или менее, даже более предпочтительно 20 часов или менее. Например, даже после завершения получения карбонилгалогенида реакционную смесь можно непрерывно перемешивать до тех пор, пока не будет подтверждено потребление амидного соединения.
[0111]
Реакция Вильсмейера-Хаака с применением реагента Вильсмейера позволяет формилировать или кетонировать ароматическое соединение, содержащее активную группу. Кроме того, было известно, что карбоксильная группа соединения карбоновой кислоты можно превратить в галоформильную группу реагентом Вильсмейера. Кроме того, эфир муравьиной кислоты можно получить взаимодействием реагента Вильсмейера и соединения, содержащего гидроксильную группу.
[0112]
Ароматическое соединение, содержащее активную группу, обозначает ароматическое соединение, активированное замещающей группой или подобное. Ароматическое соединение далее в настоящем изобретении описывают как "активное ароматическое соединение". Например, гидрокси группа и амино группа, такая как алкиламиногруппа, замещенная алкильной группой, сильно активирует ароматическое соединение. Кроме того, алкилкарбониламиногруппа (-N(C=O)R), алкилкарбонилоксигруппа (-O(C=O)R), эфирная группа (-OR), алкильная группа (-R) (R представляет собой алкильную группу и предпочтительно представляет собой С1-6 алкильную группу) и ароматическая группа активируют ароматическое соединение. Замещающие группы далее называют активирующими группами. Кроме того, соединение, созданное конденсацией ароматических колец и из которых удлиняется сопряженная система, такое как антрацен, также активируется и может быть подвергнуто формилированию или кетонизации реагентом Вильсмейера. Пи-электрон в активированной части может вступать в электрофильную реакцию с реагентом Вильсмейера, подвергаясь формилированию или кетонизации.
[0113]
Активное ароматическое соединение конкретно не ограничено при условии, что активное ароматическое соединение активируется и может подвергаться формилированию или кетонизации реагентом Вильсмейера. Пример активного ароматического соединения включает С6-10 ароматический углеводород, такой как бензол и нафталин, которые могут быть замещены описанной выше активирующей группой; конденсированный ароматический углеводород, такой как фенантрен и антрацен, которые могут быть замещены описанной выше активирующей группой; 5-членный гетероарил, такой как пиррол, имидазол, пиразол, тиофен, фуран, оксазол, изоксазол, тиазол, изотиазол и тиадиазол, которые могут быть замещены описанной выше активирующей группой; 6-членный гетероарил, такой как пиридин, пиразин, пиримидин и пиридазин, которые могут быть замещены описанной выше активирующей группой; конденсированный гетероарил, такой как индол, изоиндол, хинолин, изохинолин, бензофуран, изобензофуран и хромен, которые могут быть замещены описанной выше активирующей группой. Хотя не сообщалось, что незамещенные фуран и тиофен подвергаются формилированию или кетонизации по обычной реакции Вильсмайера-Хаака, формилирование или кетонизация углерода, соседнего с гетероэлементом, является возможным способом настоящего изобретения.
[0114]
Ароматическое соединение, содержащее активирующую группу, соединение на основе карбоновой кислоты и соединение, содержащее гидроксильную группу, в качестве соединения-субстрата описанной выше реакции можно добавлять в реакционную смесь после того, как газ, содержащий карбонилгалогенид, подают к композиции, содержащей амидное соединение, или добавлять в реакционную смесь до или во время подачи карбонилгалогенидсодержащего газа к композиции, содержащей амидное соединение.
[0115]
Применяемое количество ароматического соединения, содержащего активирующую группу, соединения на основе карбоновой кислоты и соединения, содержащего гидроксильную группу, можно регулировать, и можно регулировать, например, равным в 0,1 раза или более на моль и в 1,0 раз или менее на моль амидного соединения.
[0116]
Реагент Вильсмейера является пригодным для получения галогенида карбоновой кислоты из соединения на основе карбоновой кислоты. Реагент Вильсмейера становится амидным соединением после того, как Реагент Вильсмейера галогенирует соединение на основе карбоновой кислоты. Сложноэфирное соединение можно получить реакцией полученного галогенида карбоновой кислоты со спиртовым соединением, и ангидрид карбоновой кислоты можно получить реакцией полученного галогенида карбоновой кислоты с карбоновой кислотой. Когда соединение на основе карбоновой кислоты и основание применяют вместо амидного соединения, соединение на основе карбоновой кислоты может быть анионизировано основанием, и затем анионизированное карбоксилатное соединение может быть непосредственно превращено в галогенид карбоновой кислоты карбонилгалогенидом. Данный галогенид карбоновой кислоты можно также применять для получения сложноэфирного соединения и ангидрида карбоновой кислоты.
[0117]
8. стадия дополнительной обработки
Поскольку многие карбонилгалогениды являются токсичными, образующийся карбонилгалогенид не вытекает из реакционной системы. Например, предпочтительно подавать газовую фазу, выходящую из реакционной емкости для реакции образовавшегося карбонилгалогенида, в спиртовую ловушку, и газовая фаза, выходящая из спиртовой ловушки, далее подавалась в ловушку с щелочью, как показано на рисунках 1-6. Спиртовую ловушку можно охлаждать при условии, что применяемый спирт не коагулирует. Например, спиртовую ловушку можно охлаждать до -80°С или больше и 50°С или меньше. Кроме того, например, водный раствор гидроксида натрия и насыщенный водный раствор гидрокарбоната натрия можно применять для щелочной ловушки.
[0118]
Когда соединение, полученное из карбонилгалогенида, является относительно нестабильным соединением, таким как изоцианатное соединение, то после реакции карбонилгалогенида в реакционную смесь можно добавить реакционноспособный субстрат. Когда соединение, полученное из карбонилгалогенида, является относительно стабильным соединением, таким как карбонатное соединение, целевое соединение может быть очищено из реакционной смеси. Например, к реакционной смеси добавляют воду и нерастворимый в воде органический растворитель, такой как хлороформы, водную фазу и органическую фазу разделяют, органическую фазу сушат над безводным сульфатом натрия или безводным сульфатом магния и затем концентрируют при пониженном давлении, и целевое соединение можно очищать хроматографией или подобным.
[0119]
В настоящей заявке испрашивается преимущество даты приоритета заявки на патент Японии №2021-21001, поданной 12 февраля 2021 г. Все содержание заявки на патент Японии №2021-21001, поданной 12 февраля 2021 г., включено с помощью ссылки в настоящее изобретение.
ПРИМЕРЫ
[0120]
Далее примеры описаны для более конкретной демонстрации настоящего изобретения, но настоящее изобретение никоим образом не ограничено примерами, и примеры могут быть надлежащим образом модифицированы для осуществления в пределах диапазона, который адаптируется к содержанию настоящего описания. Данный модифицированный пример также включен в объем настоящего изобретения.
[0121]
Пример 1: получение фосгена
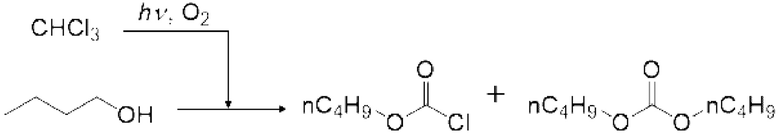
Фотореакцию в газовой фазе проводили с применением системы фотореакции, схематически представленной на рисунке 1. В частности, ртутную лампу низкого давления («SUV40D» производства SEN Light, 40 Вт, ϕ22,3×380 мм, длина волны: 185-600 нм, пик длина волны: 184,9 нм и 253,7 нм) помещали в кожух из кварцевого стекла диаметром 30 мм × длиной 320 мм, и 12 кварцевых трубок с внутренним диаметром 2,1 мм и длиной 320 мм и объемом 1,033 мл помещали вокруг кожуха из кварцевого стекла для получения цилиндрического проточного устройства для фотореакции 4. Общий объем, содержащий соединенные части цилиндрического проточного устройства для фотореакции 4, составлял 13,3 мл. Интенсивность освещения светом с длиной волны 185 нм в положении 5 мм от центра ртутной лампы низкого давления составляла 3,93 мВт/см2, и интенсивность освещения светом с длиной волны 254 нм составляла 11,02 мВт/см2.
Жидкий хлороформ подавали в тефлоновую трубку с внутренним диаметром 1 мм при расходе, указанном в таблице 1, с помощью шприцевого насоса 1 и испаряли с помощью нагревателя, нагретого до температуры, указанной в таблице 1. Испаренный хлороформ смешивали с газообразным кислородом, расход которого регулировали регулятором массового расхода 2, смешанный газ подавали в проточное устройство для фотореакции 4. Давление внутри проточного устройства для фотореакции регулировали с помощью регулятора обратного давления 5, присоединенного к выходу проточного устройства для фотореакции. Впрыскиваемый хлороформ испарялся и смешивался с газообразным кислородом, смешанный газ подавали при температуре нагревателя. Каждая максимальная скорость газового потока испаряемых хлороформа и газообразного кислорода, линейная скорость и время пребывания смешанного газа рассчитывали по таблице 1.
Газ, полученный окислительным фоторазложением смешанного газа газообразных хлороформа и кислорода, подавали к достаточному количеству перемешиваемого 1-бутанола в присоединенной реакционной емкости 6 при атмосферной температуре в течение от 2 до 6 часов. Непрореагировавший газ улавливали 1-бутанолом в присоединенном сосуде-ловушке 7, и оставшийся непрореагировавший газ очищали путем подачи в присоединенную щелочную ловушку, чтобы токсичный газ не вытекал наружу.
Кроме того, был проведен аналогичный эксперимент, за исключением того, что применяли воздушный насос («Nisso air pump Silent β-60» производства Marukan), и воздух, высушенный адсорбентом («Silica Gel, Medium Granular, Blue» производства FUJIFILM Wako Pure Chemical), применяли вместо кислорода.
После реакции реакционные смеси в реакционной емкости 6 и сосуде-ловушке 7 анализировали с помощью 1Н ЯМР для измерения степеней конверсии в образующиеся эфир хлормуравьиной кислоты и карбонат, и количество образовавшегося фосгена рассчитывали из суммарных степеней конверсии. Результат представлен в таблице 1. Выход фосгена равен выходу применяемого хлороформа в таблице 1.
[0122]
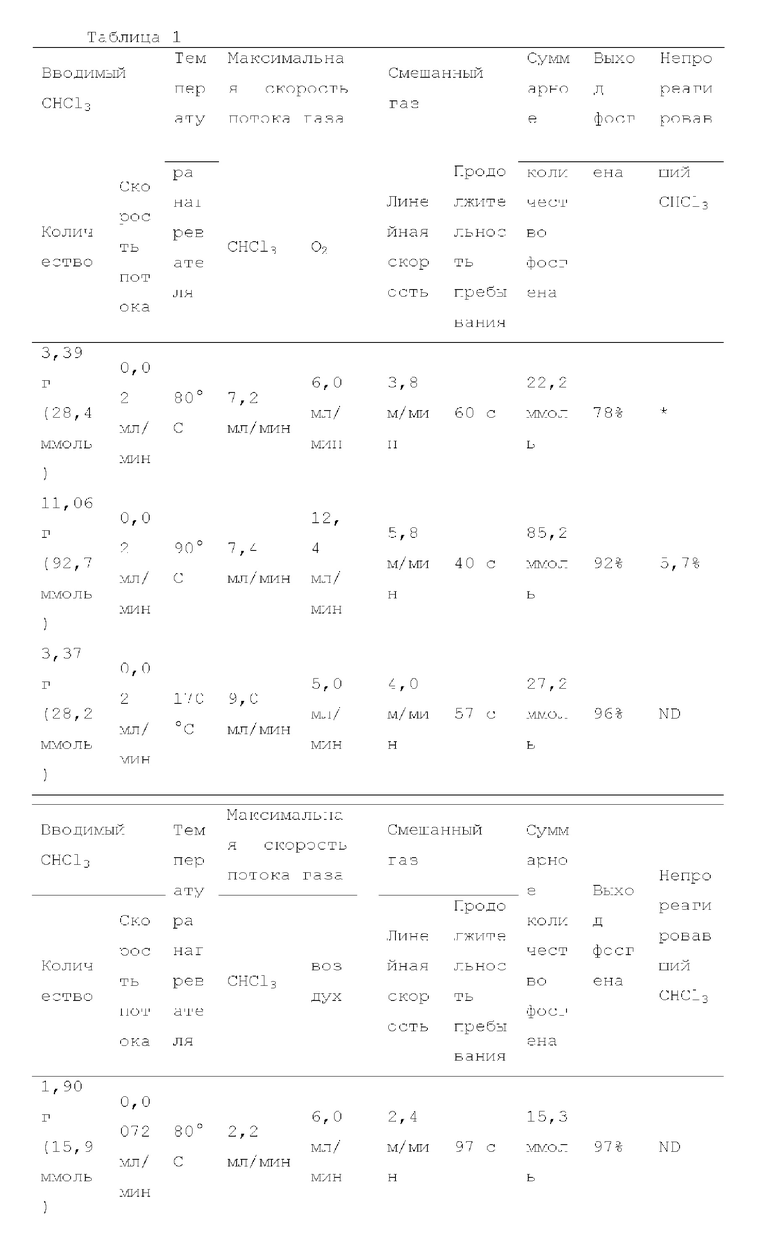

[0123]
Проточную фотореакцию в газовой фазе проводили нагреванием хлороформа при 80°С с применением змеевикового нагревателя для испарения и смешивания испарившегося хлороформа с газообразным кислородом; в результате из хлороформа удалось получить фосген с коэффициентом конверсии 78%, как описанный выше результат.
Температура змеевикового нагревателя дополнительно повышали до 90°С и увеличивали расход кислорода; в результате удалось получить фосген из хлороформа со степенью конверсии 92% и непрореагировавшим хлороформом 5,7%. Остальные приблизительно 2% могут составлять гексахлорэтан, образующийся при фоторазложении хлороформа в качестве побочного продукта и продуктов фоторазложения фосгена, таких как CO2, СО и Cl2.
Когда температуру змеевикового нагревателя дополнительно повышают до 170°С, выход увеличивается.
При применении осушенного воздуха вместо кислорода можно было получать фосген с высоким выходом, но выделенное количество уменьшалось по сравнению со случаем применения кислорода. При увеличении количества вводимого воздуха количество непрореагировавшего хлороформа несколько увеличивалось.
[0124]
Пример 2: эксперимент с расходом газа
Исследовали, изменился ли выход фосгена или нет от времени облучения светом аналогично примеру 1, за исключением того, что были изменены скорости газовых потоков хлороформа и кислорода и, таким образом, была изменена продолжительность пребывания в проточном устройстве для фотореакции. Результат показан в таблице 2.
[0125]

[0126]
Установлено, что количество непрореагировавшего хлороформа уменьшается, и количество образующегося фосгена уменьшается с увеличением продолжительности пребывания в проточном устройстве для фотореакции, как описанный выше результат. Образующийся фосген может разлагаться, но резко не уменьшается и описанный выше выход фосгена является достаточно практичным. Таким образом, система настоящего изобретения может иметь функцию, которая ингибирует фоторазложение образующегося фосгена. Например, поскольку газообразный кислород имеет полосу поглощения света в диапазоне от 100 до 250 нм, избыточный высокоэнергетический свет может поглощаться кислородом и, таким образом, может подавляться разложение образовавшегося фосгена.
[0127]
Пример 3: получение изоцианата проточной световой системой
Раствор (20 мл) гидрохлорида гексиламина (1,38 г, 10 ммоль) в 1,1,2,2-тетрахлорэтане применяли вместо 1-бутанола в условиях примера 1, в которых применяли 11,06 г (92,7 ммоль) хлороформа, и температуру змеевикового нагревателя устанавливали равной 90°С, раствор нагревали до 100°С и перемешивали, и газ, образующийся в проточном устройстве для фотореакции, подавали в раствор. Дихлорметан добавляли к реакционной смеси в качестве внутреннего стандарта после реакции, и смесь анализировали с помощью 1Н ЯМР; в результате было подтверждено, что гексилизоцианат был получен в виде целевого соединения с выходом 83%.
[0128]
Пример 4: получение изоцианата проточной световой системой
Газ, полученный проточным устройством для фотореакции фотореакционной системы, схематически показанной на рисунке 1 в условиях, показанных в таблице 3, продували в хлороформеиную суспензию (30 мл) дигидрохлорида 2,2,3,3,4,4,5,5-октафторгексан-1, 6-диамина (0,34 г, 1,0 ммоль) в течение 3,5 часов, и затем смесь перемешивали при 0°С. В реакционную смесь вводили 2,6-лутидин (0,58 мл, 5 ммоль) в течение 1 часа, применяя шприцевой насос. Смесь нагревали до 50°С и перемешивали в течение 1 часа.
Реакционную смесь анализировали 1Н ЯМР после реакции; как результат, подтверждали, что целевое соединение получали с выходом >99% относительно исходного соединение, диаминового соединения.
[0129]
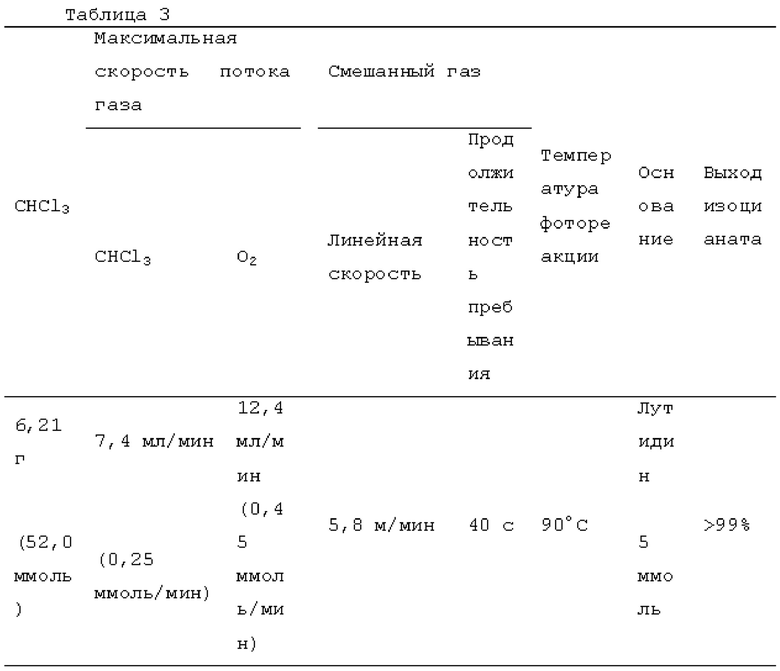
[0130]
Пример 5: Получение карбоната проточной световой системой
Газ, полученный проточным устройством для фотореакции фотореакционной системы, схематически показанной на рисунке 1, в условиях, показанных в таблице 4, подавали в перемешиваемый спирт, содержащий пиридин или 1-метилимидазол (NMI) в качестве основания в реакционной емкости 6 при атмосферной температуре. В ловушку 7 добавляли 1-бутанол для захвата газа, вытекающего из реакционной емкости 6.
После реакции, 1,1,2,2-тетрахлорэтан добавляли к реакционной смеси в качестве внутреннего стандартного материала, и смесь анализировали 1Н ЯМР; как результат, подтверждали, что карбонаты получали в качестве целевого соединения с высокими выходами.
С одной стороны, выход фосгена, рассчитанный по карбонату, полученному в реакционной емкости 6, и эфира хлоромуравьиной кислоты и карбоната, полученного в ловушке 7, был относительно низким в некоторых случаях. Пиридин мог вероятно вызывать разложение фосгена.
[0131]


 inn
inn
[0132]
Пример 6: Получение поликарбоната проточной световой системой
(1) гомогенная полимеризация
Смешанный газ испаренного хлороформа и кислорода подавали в проточное устройство для фотореакции для фотореакции при 90°С в условиях, приведенных в таблице 5, с применением фотореакционной системы, схематически показанной на рисунке 1. Газ, полученный в проточном устройстве для фотореакции, подавали в перемешиваемый раствор, полученный смешиванием бисфенола А, пиридина (4 мл, 50 ммоль) и хлороформа в реакционной емкости 6. Затем реакционную смесь перемешивают при 50°С в течение 1 часа. Далее к реакционной смеси добавляли метанол (150 мл), полученный осадок отфильтровывали и сушили в вакууме с получением белого целевого соединения.
[0133]


[0134]
Полученный поликарбонат анализировали гель-проникающей хроматографией (ГПХ) для определения молекулярной массы. Результат показан в таблице 6.
[0135]
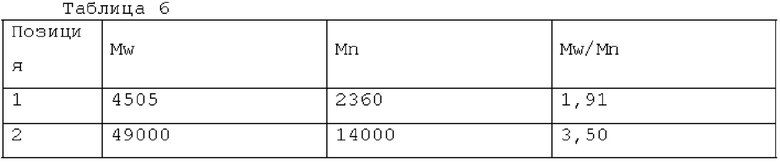
[0136]
(2) гомогенная полимеризация
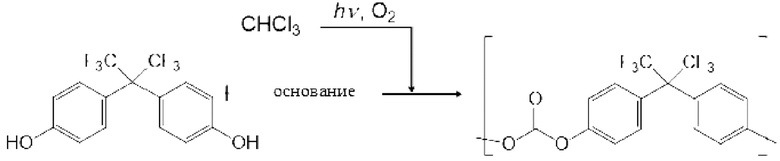
Смешанный газ испаренного хлороформа и кислорода подавали в проточное устройство для фотореакции для фотореакции при 90°С в условиях, приведенных в таблице 7, с применением фотореакционной системы, схематически показанной на рисунке 1. Газ, полученный в проточном устройстве для фотореакции, подавали в перемешиваемый раствор, полученный смешиванием бисфенола AF (BPAF), пиридина и хлороформа в реакционной емкости 6. Затем реакционную смесь перемешивали при 50°С в течение 1 часа.
Далее к реакционной смеси добавляли метанол (150 мл), полученный осадок отфильтровывали и сушили в вакууме с получением белого целевого соединения.
[0137]


[0138]
Полученный поликарбонат анализировали гель-проникающей хроматографией (ГПХ) для определения молекулярной массы. Результат показан в таблице 8.
[0139]

[0140]
(3) Межфазная полимеризация
Смешанный газ испаренного хлороформа и кислорода подавали в проточное устройство для фотореакции для фотореакции при 90°С в условиях, показанных в таблице 9 с применением фотореакционной системы, схематически показанной на рисунке 1. Газ, полученный в проточном устройстве для фотореакции, подавали в перемешиваемый раствор, полученный смешиванием бисфенола А, 17% по массе водного раствора гидроксида натрия и дихлорметана в реакционной емкости 6.
Затем, воду и дихлорметан к реакционной смеси, и верхний слой и нижний слой были разделяли. Нижний слой сушили над безводным сульфатом натрия, смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Метанол добавляли к остатку, и образовавшийся осадок отфильтровывали и сушили в вакууме для получения желто-белого целевого соединения (выход относительно хлороформа: 19%, выход относительно ВРА: 84%).
[0141]


[0142]
Полученный поликарбонат анализировали гель-проникающей хроматографией (ГПХ) для определения молекулярной массы. Результат показан в таблице 10.
[0143]

[0144]
Пример 7: Получение поликарбоната проточной световой системой
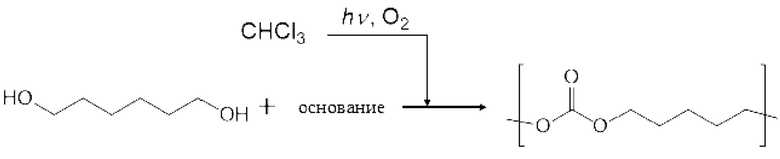
Смешанный газ испаренного хлороформа и кислорода подавали в проточное устройство для фотореакции для фотореакции при 90°С в условиях, показанных в таблице 11, с применением фотореакционной системы, схематически показанной на рисунке 1. Газ, полученный в проточном устройстве для фотореакции, подавали в перемешиваемый раствор, полученный смешиванием 1,6-гександиола (9,45 г, 80 ммоль), пиридина (32 мл, 400 ммоль) и дихлорметана (40 мл) в реакционной емкости 6.
Затем, 10% по массе хлористоводородную кислоту добавляли к реакционной смеси, и верхний слой и нижний слой были разделяли. Нижний слой сушили над безводным сульфатом натрия, и смесь фильтровали. Растворитель отгоняли из фильтрата при пониженном давлении, и остаток сушили в вакууме при 50°С в течение 2 часов для получения целевого белого твердого вещества цвета (выход относительно хлороформа: 58%, выход относительно исходного диола: 73%).
[0145]

[0146]
Полученный поликарбонат анализировали гель-проникающей хроматографией (ГПХ) для определения молекулярной массы. Результат показан в таблице 12.
[0147]

[0148]
Пример 8: Получение соединения на основе мочевины проточной световой системой
Газ, полученный проточным устройством для фотореакции, применяя 11,46 г (94,3 ммоль) хлороформа, подавали в перемешиваемый раствор, полученный смешиванием анилина (42,3 г, 460 ммоль) и дихлорметана (50 мл) вместо 1-бутанола при стандартной температуре в реакционной емкости 6 в условиях примера 1, для которого температура змеевикового нагревателя составляла 90°С.
Затем, воду добавляли к реакционной смеси, и образовавшийся осадок получали фильтрованием. Осадок промывали и затем сушили в вакууме при 50°С в течение 3 часов для получения белого целевого твердого соединения (выход относительно хлороформа: 93%). Соединение на основе мочевины можно получить реакцией фосгена и анилина, получая изоцианатное соединение, которое реагирует с анилином.
Фосген, применяемый для получения дифенилмочевины, и непрореагировавший фосген, уловленный спиртовой ловушкой (ловушка 7), объединяли для расчета степени конверсии из хлороформа в фосген как 98%.
[0149]
Пример 9: Получение N-карбоксильного ангидрида аминокислоты проточной световой системой
(1) N-карбоксильный ангидрид L-фенилаланина
Газ, полученный проточным устройством для фотореакции, применяя 5,7 г (47,7 ммоль) хлороформа, подавали в перемешиваемую суспензию, полученные смешиванием L-фенилаланина (1,65 г, 10 ммоль), хлороформа (20 мл) и ацетонитрила (15 мл) вместо 1-бутанола при 70°С в реакционной емкости 6 в условиях примера 1, для которого температура змеевикового нагревателя составляла 90°С.
Затем, непрореагировавшее сырье отделяли фильтрованием, и фильтрат концентрировали при пониженном давлении с получением целевого соединения (выход относительно исходного L-фенилаланина: 61%).
[0150]
(2) Получение N-карбоксильного ангидрида аминокислоты, применяя воздух
Газофазную фотореакцию проводили с применением фотореакционной системы, схематически показанной на рисунке 1. Жидкий хлороформ подавались в тефлоновую трубку с внутренним диаметром 1 мм при расходе 838 мкл/мин (0,1 ммоль/мин) и смешивали с воздухом, расход которого устанавливали равным 7 мл/мин с помощью воздушного насоса и регулятора массового расхода, и смешанный газ подавали в проточное устройство для фотореакции при 80°С. Введенный хлороформ испарялся и смешивался с воздухом, и смешанный газ протекал. Соответствующую максимальную газовая скорость потока испаренного хлороформа и газообразного кислорода, линейную скорость и продолжительность пребывания смешанного газа рассчитывали, как в таблице 13.
Газ, полученный окислительным фоторазложением смешанного газа газообразного хлороформа и воздуха, подавали в перемешиваемый раствор ТГФ, содержащий L-аминокислоту (10 ммоль) в присоединенной реакционной емкости 6 при 60°С в течение 3 часов. Непрореагировавший газ обрабатывали путем подачи в дополнительно подключенную щелочную ловушку, чтобы токсичный газ не вытекал наружу. Затем, полученный раствор промывали водой и подвергали экстракции дихлорметаном. Органический слой сушили над безводным сульфатом натрия, растворитель отгоняли при пониженном давлении. Белое твердое целевое соединение получали перекристаллизацией из остатка, применяя диэтиловый эфир и гексан. Результат показан в таблице 13.
[0151]
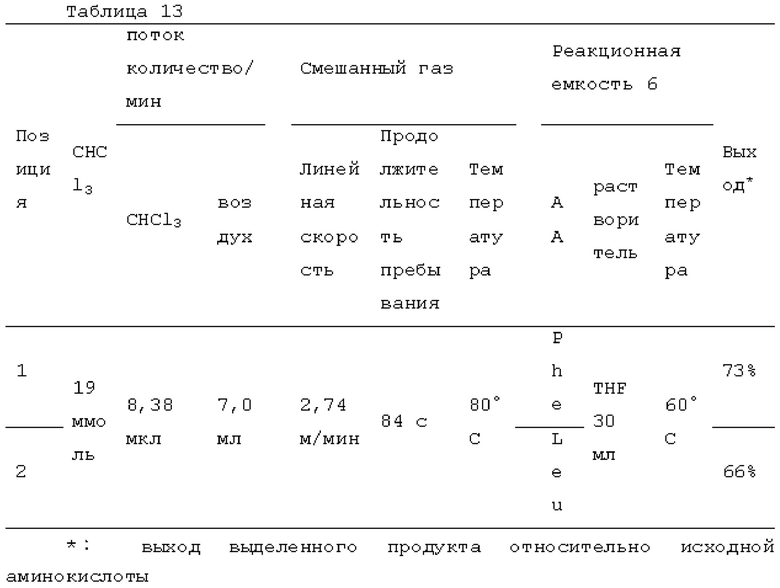
[0152]
Пример 10: Получение карбонилдиимидазола проточной световой системой
Хлороформ (2,4 мл, 30 ммоль) применяли и газ, полученный в проточном устройстве для фотореакции подавали в перемешиваемый раствор ТГФ имидазола (5,11 г, 75 ммоль) вместо 1-бутанола при 0°С в условиях примера 1, в которых температуру змеевикового нагревателя устанавливали равной 90°С.
Затем, осадок гидрохлоридной соли имидазола, полученный в качестве побочного продукта при протекании реакции, отделяли фильтрованием, и фильтрат концентрировали при пониженном давлении, получая целевое соединение (выход относительно хлороформа: 74%).
[0153]
Пример 11: Получение поликарбоната проточной световой системой
Хлороформ (3,14 г, 26,3 ммоль) применяли и паз, полученный в проточном устройстве для фотореакции, подавали в перемешиваемый раствор, полученный смешиванием 4,4'-циклододецилиденбисфенола (BisP-CDE) (3,52 г, 10 ммоль), пиридина (4 мл, 50 ммоль) и хлороформа (40 мл) вместо 1-бутанола при 30°С в течение 125 минут в условии примера 1, в котором температуру змеевикового нагревателя устанавливали равной 90°С.
Затем, метанол (150 мл) добавляли к реакционной смеси, и образовавшийся осадок отфильтровывали и сушили в вакууме для получения белого поликарбоната (выход относительно BisP-CDE: >99%).
Полученный поликарбонат анализировали гель-проникающей хроматографией (ГПХ) для определения молекулярной массы. Результат показан в таблице 14.
[0154]

[0155]
Пример 12: Непрерывное поточное получение проточной световой системой
Шприцевой насос 8 для ввода реакционноспособного субстрата и терморегулируемый змеевиковый реактор 9 с внутренним диаметром 1,0 мм, длиной 2830 мм и объемом 2,22 мл дополнительно подключали к проточной фотореакционной системе рисунка 1 для непрерывной реакции, как показано на рисунке 2. В газообразный фосген, образующийся в проточной системе для фотореакции, из шприцевого насоса 8 дополнительно вводили 1-бутанол для подачи реакционного субстрата с расходом 0,052 мл/мин для реакции в змеевиковом реакторе 9, для которого температуру устанавливали равной 0-100°С в условиях, приведенных в таблице 12. Двугорлую колбу охлаждали до 0°С и присоединяли к выходу змеевика реактора в качестве емкости для извлечения 10 для получения продукта. Непрореагировавший разложившийся газ улавливали в дополнительно присоединенной спиртовой ловушке в качестве ловушки-сосуда 7, и отходящий газ обрабатывали в щелочной ловушке, чтобы он не вытекал наружу.
Выходы эфира хлормуравьиной кислоты и карбоната, образующегося в сосуде-уловителе 10 и сосуде-ловушке 7, рассчитывали по 1Н ЯМР спектрам путем сравнения 50 ммоль 1,1,2,2-тетрахлорэтана в качестве внутреннего стандартного материала, и количество полученного фосгена оценивали по его общему количеству. Результат показан в таблице 15.
[0156]


[0157]
Когда 1-бутанол в качестве алифатического спирта вводили из шприцевого насоса 8 для введения реакционного субстрата, эфир хлормуравьиной кислоты и карбонат можно получить с выходом приблизительно 90% относительно хлороформа, введенного из шприцевого насоса 1, как результат, показанный в таблице 15. Эффективность реакции была высокой при реакции в змеевиковом реакторе, поскольку спирт, вводимый при высокой температуре, может испаряться, реагируя с фосгеном в газовой фазе. Как результат, фосген не был обнаружен на выходе из змеевикового реактора, и таким образом можно добиться того, что величина обнаружения фосгена на выходе из системы была равна нулю на лабораторном уровне.
Реакция в газовой фазе при высокой температуре в змеевиковом реакторе становится возможной при разработке реакционной системы с непрерывным потоком, как описанный выше результат.
Кроме того, вводили 2-пропанол (IPA); как результат, целевой эфир хлормуравьиной кислоты можно получить с выходом 74%, хотя 2-пропанол обычно не реагирует с фосгеном при комнатной температуре.
[0158]
Пример 13: Получение эфира хлоромуравьиной кислоты проточной световой системой
Температуру змеевикового нагревателя в проточной системе с облучением светом, схематически показанной на рисунке 2, устанавливали равной 90°С, газ после окислительного фоторазложения, полученный из 7,4 мл/мин газообразного хлороформа и 12,4 мл/мин газообразного кислорода, смешивали со спиртом, введенным шприцевым насосом, и смешанный газ подавали в ПТФЭ трубчатом реакторе при 30°С для реакции в потоке. Продукт получали присоединенной двугорлой колбе, которую охлаждали до 0°С. Непрореагировавший газ подавали в дополнительно присоединенную щелочную ловушку, чтобы он не просачивался наружу.
После реакции, реакционную смесь анализировали 1Н ЯМР; как результат, целевой эфир хлормуравьиной кислоты можно получить с выходами, показанными в таблице 16.
[0159]
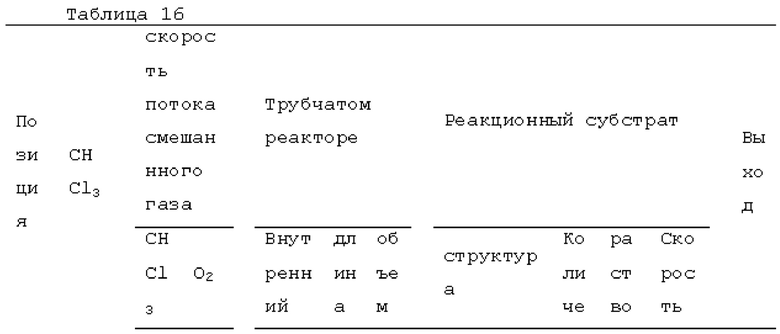
[0160]
Пример 14: Получение этиленкарбоната
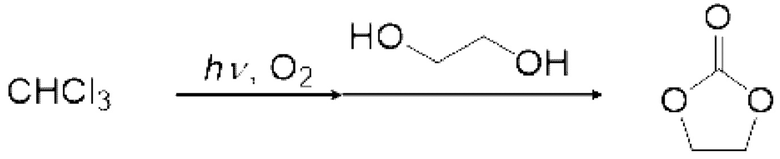
Этиленгликоль (1,62 мл, 29,0 ммоль) дополнительно вводили в газообразный фосген, полученный из хлороформа (всего 2,37 мл, 29,4 ммоль) с помощью шприцевого насоса с расходом 0,014 мл/мин в условиях, аналогичных примеру 12, в течение 2 часов, чтобы они реагировали в змеевиковом реакторе, который нагревали до 200°С. Двугорлую колбу, охлажденную до 0°С, присоединяли к выходу змеевикового реактора в качестве емкости для извлечения 10 для сбора продукта. Непрореагировавший разложившийся газ улавливали в дополнительно присоединенной спиртовой ловушке в качестве ловушки-сосуда 7, и отходящий газ очищали щелочной ловушкой. Фосген не обнаружен на стадии спиртовой ловушки
К полученной реакционной смеси 1,1,2,2-тетрахлорэтан добавляли в качестве внутреннего стандартного материала. Смесь анализировали 1Н ЯМР; как результат, подтверждали, что этиленкарбонат получали в качестве целевого соединения с выходом 54% относительно применяемых хлороформа и этиленгликоля.
[0161]
Пример 15: получение в непрерывном потоке, применяя NMI проточной световой системой
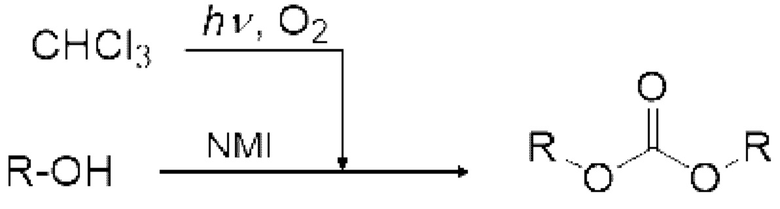
Если органическое основание вводят в качестве катализатора в змеевиковый реактор в проточной реакционной системе с непрерывным потоком, показанной на рисунке 2, по ходу реакции образуется HCl, который реагирует с органическим основанием с образованием соли. Соль может вызвать закупорку трубы реактора. Изобретателям настоящего изобретения пришла в голову идея выбрать органическое основание, для которого соль с HCl не осаждается, иными словами, для которого соль представляет собой ионную жидкость в качестве жидкой соли в системе настоящего изобретения.
[0162]
(1) Получение бис-1,1,1,3,3,3-гексафтор-1-пропилкарбонат
Смешанную жидкость гексафторизопропанола (HFIP) (14,8 мл, 188 ммоль) и 1-метилимидазола (NMI) (7,9 мл, 75 ммоль) вводили в газообразный фосген, полученный непрерывной проточной системой фотореакции, из шприцевого насоса 8 для введения реакционного субстрата при расходе 0,125 мл/мин в течение в сумме 3 часов условии, аналогичном примеру 12, за исключением того, что змеевиковый реактор с регулируемой температурой заменяли на реактор с внутренним диаметром 2,4 мм, длиной 2830 мм и объемом 12,8 мл. Смесь подавали в змеевиковый реактор 9, нагретый до 100°С, для проведения реакции. Продукт собирали в емкости для извлечения 10-1, который был соединен с выходом змеевика реактора, и емкости для извлечения 10-2. Температуру емкости для извлечения 10-1 устанавливали равной 100°С, чтобы можно было достаточно поддерживать жидкое состояние соли NMI и HCl и извлекать целевой карбонат. С одной стороны, температура емкости для извлечения 10-2 устанавливали равной 0°С, так как возможна утечка целевого карбоната из емкости для извлечения 10-1. Газ, выходящий из емкости для извлечения 10-2, улавливался дополнительно присоединенной спиртовой ловушкой, по мере того как емкость-уловитель 7 и отработанный газ очищались щелочной ловушкой, но на стадии спиртовой ловушки фосген не обнаруживали.
Собранные продукты в улавливающей емкости 10-1 и улавливающей емкости 10-2 объединяли. Смесь разделяли на два слоя. Нижний слой промывали 1,4 моль/л хлороводородной кислотой (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением целевого соединения в виде бесцветной и прозрачной жидкости (выход относительно примененного HFIP: 61%).
[0163]
(2) Получение бис-2,2,3,3-тетрафторпропилкарбоната
Смешанную жидкость гексафторизопропанола (HFIP) (8,86 мл, 100 ммоль) и 1-метилимидазола (NMI) (12 мл, 150 ммоль) вводили в газообразный фосген, полученный непрерывной проточной системой фотореакции, из шприцевого насоса 8 для введения реакционного субстрата при расходе 0,12 мл/мин в течение в сумме 3 часов условии, аналогичном примеру 12, за исключением того, что змеевиковый реактор с регулируемой температурой заменяли на реактор с внутренним диаметром 2,4 мм, длиной 2830 мм и объемом 12,8 мл. Смесь подавали в змеевиковый реактор 9, нагретый до 100°С, для проведения реакции. Продукт собирали в емкости для извлечения 10-1, который был соединен с выходом змеевика реактора, и емкости для извлечения 10-2. Емкость для извлечения 10-1 нагревали до 100°С, и емкости для извлечения 10-2 охлаждали до 0°С. Непрореагировавший разложившийся газ улавливали дополнительно присоединенной спиртовой ловушкой в качестве сосуда ловушки 7, и отходящий газ очищали щелочной ловушкой, но на стадии спиртовой ловушки фосген не обнаруживали.
К полученному раствору реакционного образца 1,1,2,2-тетрахлорэтан добавляли в качестве внутреннего стандартного материала. Смесь анализировали 1Н ЯМР; как результат, подтверждали, что целевое соединение получали с выходом 70% относительно примененного спирта.
Сырье удаляли из полученного раствора образца перегонкой. Раствор образца промывали 1,4 моль/л HCl (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением целевого соединения в виде бесцветной и прозрачной жидкости (выход относительно примененного 2,2,3,3-тетрафтор-1-пропанола: 44%).
[0164]
(3) Получение бис-2,2,2-трифторэтилкарбоната
Смешанную жидкость 2,2,2-трифторэтанола (10 г, 100 ммоль) и НМИ (12 мл, 150 ммоль) вводили в газообразный фосген, полученный непрерывной проточной системой фотореакции, из шприцевого насоса 8 для введения реакционного субстрата при расходе 0,11 мл/мин в течение в сумме 3 часов условии, аналогичном примеру 12, за исключением того, что змеевиковый реактор с регулируемой температурой заменяли на реактор с внутренним диаметром 2,4 мм, длиной 2830 мм и объемом 12,8 мл. Смесь подавали в змеевиковый реактор 9, нагретый до 100°С, для проведения реакции. Продукт собирали в емкости для извлечения 10-1, соединенном с выходом змеевика реактора, и емкости для извлечения 10-2. Емкость для извлечения 10-1 нагревали до 100°С, и емкость для извлечения 10-2 охлаждали до 0°С. Непрореагировавший разложившийся газ улавливали дополнительно присоединенной спиртовой ловушкой в качестве сосуда ловушки 7, и отходящий газ очищали щелочной ловушкой, но на стадии спиртовой ловушки фосген не обнаруживали.
К полученной реакционной смеси 1,1,2,2-тетрахлорэтан добавляли в качестве внутреннего стандартного материала. Смесь анализировали 1Н ЯМР; как результат, подтверждали, что целевое соединение получали с выходом 60% относительно примененного спирта.
Исходное соединение удаляли из полученного раствора образца перегонкой. Полученный таким образом остаток промывали 3 М соляной кислотой (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением целевого соединения в виде бесцветной и прозрачной жидкости (выделенное количество: 6,43 г, выход относительно примененного 2,2,2-трифторэтанола: 44%).
[0165]
(4) Получение дифенилкарбоната
Смешанную жидкость фенола (18,8 мл, 200 ммоль) и NMI (24 мл, 300 ммоль) вводили в газообразный фосген, полученный непрерывной проточной системой фотореакции, из шприцевого насоса 8 для введения в реакционный субстрат при расходе 0,23 мл/мин в течение в сумме 3 часов условии, аналогичном примеру 12, за исключением того, что змеевиковый реактор с регулируемой температурой заменяли на реактор с внутренним диаметром 2,4 мм, длиной 2830 мм и объемом 12,8 мл. Смесь подавали в змеевиковый реактор 9, нагретый до 115°С, для проведения реакции. Продукт собирали в емкости для извлечения 10, соединенном с выходом змеевикового реактора и нагретом до 100°С. Непрореагировавший разложившийся газ улавливали дополнительно присоединенной спиртовой ловушкой в качестве сосуда ловушки 7, и отработанный газ очищали щелочной ловушкой, но фосген не обнаруживали на стадии спиртовой ловушки.
К полученной реакционной смеси, 1,1,2,2-тетрахлорэтан добавляли в качестве внутреннего стандартного материала. Смесь анализировали 1Н ЯМР; как результат, подтверждали, что целевое соединение получали с выходом 72% относительно примененного фенола.
Дихлорметан и 1,4 моль/л HCl добавляли к полученному раствору образца, и верхний слой и нижний слой разделяли. Нижний слой сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли при пониженном давлении из фильтрата. Остаток сушили в вакууме в условиях нагревания, применяя стеклянную трубку, получая белое целевое твердое соединение (выделенный выход относительно примененного хлороформа: 56%).
[0166]


[0167]
В качестве описанного выше результата было обнаружено, что даже когда основание, образующее ионную жидкость с гидрохлоридом, полученным в качестве побочного продукта, такое как 1-метилимидазол, образуется в змеевиковом реакторе, змеевик не засоряется, и непрерывная реакция может протекать успешно.
[0168]
Пример 16: непрерывное проточное получение проточной системой, применяя пиридин
Смешанную жидкость фенола (9,41 г, 100 ммоль) и пиридина (12 мл, 150 ммоль) вводили в газообразный фосген, полученный непрерывной проточной системой фотореакции, из шприцевого насоса 8 для введения реакционного субстрата при расходе 0,11 мл/мин в течение в сумме 3 часов в условии, аналогичном примеру 15. Смесь подавали в змеевиковый реактор 9, нагретый до 160°С, для реакции. Продукт собирали в емкости для извлечения 10-1, соединенном с выходом змеевика реактора, и емкости для извлечения 10-2. Температуру емкости для извлечения 10-1 устанавливали равной 155°С, чтобы можно было в достаточной степени поддерживать жидкое состояние соли пиридина и HCl и можно было получить целевой карбонат. С одной стороны, температуру емкости для извлечения 10-2 устанавливали равной -5°С, так как возможна утечка целевого карбоната из емкости для извлечения 10-1. Газ, выходящий из емкости для извлечения 10-2, улавливался дополнительно присоединенной спиртовой ловушкой в качестве емкости-ловушки 7, и отходящий газ очищали щелочной ловушкой, но фосген на стадии спиртовой ловушки не обнаруживали.
Как результат, засорение гидрохлоридной солью пиридина не обнаруживали в змеевиковом реакторе, и, таким образом, полученная гидрохлоридная соль пиридина могла быть растворена или расплавлена для вытекания.
К полученной смеси, 1,1,2,2-тетрахлорэтан добавляли в качестве внутреннего стандартного материала. Смеси анализировали 1Н ЯМР; как результат, подтвердили, что был получен целевой дифенилкарбонат (выход отнсительно примененного фенола: 65%, выход относительно примененного хлороформа: 72%).
[0169]
Пример 17: непрерывное проточное получение проточной системой, применяя растворитель
Если органическое основание вводят в качестве катализатора в змеевиковый реактор в проточной реакционной системе с непрерывным потоком, показанной на рисунке 2, по ходу реакции образуется HCl, которая реагирует с органическим основанием с образованием соли. Соль может вызвать закупорку трубы реактора. Изобретателям настоящего изобретения пришла в голову идея применять растворитель для растворения образующейся соли HCl в системе настоящего изобретения.
Смешанную жидкость, полученную растворением фенола (1,88 г, 20 ммоль) и пиридина (6,4 мл, 80 ммоль) в хлороформе (16 мл), вводили в газообразный фосген, полученный непрерывной проточной фотореакционной системой, из шприцевого насоса 8 для введение реакционного субстрата при расходе 0,2 мл/мин в течение в сумме 2 часов в условии, аналогичном примеру 15. Смесь подавали в змеевиковый реактор 9 для проведения реакции при комнатной температуре. Продукт собирали в двугорлую колбу в качестве емкости для извлечения 10, подсоединенной к выходу змеевикового реактора. Отработанный газ очищали щелочной ловушкой в качестве ловушки 7.
Как результат, поскольку отложение гидрохлорида пиридина не обнаруживали в змеевиковом реакторе, таким образом полученная гидрохлоридная соль пиридина могла быть растворена в хлороформе для вытекания.
К полученной реакционной смеси 1,1,2,2-тетрахлорэтан добавляли в качестве внутреннего стандартного материала. Смесь анализировали 1Н ЯМР; как результат, подтверждали, что получали целевой дифенилкарбонат (выход относительно примененного фенола: 90%).
Затем, полученный раствор пробы промывали 1 М соляной кислотой и водой, экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток подвергали перегонке при пониженном давлении с применением сушильного шкафа для стеклянных трубок с получением белого твердого дифенилкарбоната из фракции 100-155°С (выделенное количество: 1,79 г, выход: 84%).
[0170]
Пример 18: непрерывное проточное получение проточной системой, применяя растворитель
Смешанную жидкость, полученную растворением 2,2,3,3-тетрафтор-1-пропанола (1,96 мл, 20 ммоль) и пиридина (6,4 мл, 80 ммоль) в 16 мл хлороформа, вводили в газообразный фосген, полученный непрерывной проточной системой для фотореакции, из шприцевого насоса 8 для ввода реакционного субстрата при расходе 0,2 мл/мин в течение в сумме 2 часов в условии, аналогичном примеру 15. Смесь подавали в змеевиковый реактор 9 для проведения реакции при комнатной температуре. Продукт собирали в двугорлую колбу в качестве емкости для извлечения 10, присоединенной к выходу змеевикового реактора. Отработанный газ очищали щелочной ловушкой в качестве ловушки 7.
К полученной реакционной смеси 1,1,2,2-тетрахлорэтан добавляли в качестве внутреннего стандартного материала. Смесь анализировали 1Н ЯМР; как результат, подтверждали, что получали целевой бис(2,2,3,3-тетрафторпропил)карбонат (выход относительно примененного спирта: 94%).
Затем, полученный раствор образца промывали 1 М хлороводородной кислотой и водой, экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, и получали целевого соединения в виде бесцветной жидкости из фракций 60-70°С перегонкой при пониженном давлении, применяя диафрагменный насос (выделенное количество: 2,09 г, выход: 72%).
[0171]
Пример 19: Непрерывное проточное получение проточной системой, применяя растворитель
Смешанную жидкость, полученную растворением HFIP (2,31 мл, 20 ммоль) и пиридина (6, 4 мл, 80 ммоль) в хлороформе (16 мл), вводили в газообразный фосген, полученный непрерывной проточной системой для фотореакции, из шприцевого насоса 8 для ввода реакционного субстрата при расходе 0,2 мл/мин в течение в сумме 2 часов в условии, аналогичном примеру 15. Смесь подавали в змеевиковый реактор 9 для проведения реакции при комнатной температуре. Продукт собирали в двугорлую колбу в качестве емкости для извлечения 10, присоединенной к выходу змеевикового реактора. Отработанный газ очищали щелочной ловушкой в качестве ловушки 7. К полученной реакционной смеси, 1,1,2,2-тетрахлорэтан добавляли в качестве внутреннего стандартного материала. Смесь анализировали 1Н ЯМР; как результат, подтверждали, что получали целевой бис(1,1,1,3,3,3-гексафторпропан-2-ил)карбонат (выход относительно примененного спирта: 61%).
Затем, полученный раствор образца промывали 1 М соляной кислотой и водой, экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, и получали целевое соединение в виде бесцветной жидкости из фракций 65-75°С перегонкой (выделенное количество: 1,45 г, выход: 40%).
[0172]
Пример 20: Получение карбоната газовым способом
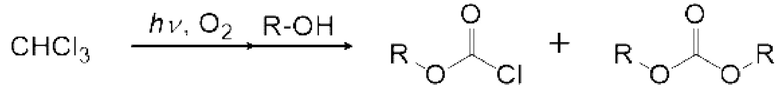
Фотореакционную систему конструировали помещением рубашки из кварцевого стекла диаметром 30 мм в цилиндрическую реакционную емкость диаметром 42 мм и объемом 100 мл и последующей установкой ртутной лампы низкого давления ("UVL20PH-6" производства SEN Light, 20 Вт, ϕ24 мм × 120 мм) в оболочку из кварцевого стекла, как схематически показано на рисунке 3. Свет, излучаемый ртутной лампой низкого давления, содержал УФ-С с длиной волны 185 нм и длиной волны 254 нм, и интенсивность освещения светом с длиной волны 185 нм в положении 5 мм от центра лампы до реакционной смеси составляла 2,00-2,81 мВт/см2, и интенсивность освещения светом с длиной волны 254 нм составляла 5,60-8,09 мВт/см2. Цилиндрическую реакционную емкость 12 снабжали охлаждающим конденсатором 15, который применяли для селективной транспортировки образующегося низкокипящего газового компонента и охлаждали до 10°С, и двугорлой колбой в качестве реакционной емкости 16, снабженной подключенным к ней охлаждающим холодильником 15, охлаждаемым до -10°С. Охлаждающий холодильник 15 дополнительно соединяли с двугорлой грушевидной колбой со спиртом и сосудом-ловушкой с водным раствором щелочи.
Температура бани реактора для фотореакции 12 устанавливали равной температуре, показанной в таблице 18, и затем жидкий хлороформ подавали реактор для фотореакции из ПТФЭ трубки с внутренним диаметром 1 мм при скорости потока, указанным в таблице 18, применяя шприцевой насос. Хлороформ перемешивали для ускорения испарения. Затем, кислород подавали в газовую фазу реакционной емкости при скорости 0,1 мл/мин с другого направления для получения смешанного газа хлороформа и кислорода в реакторе для фотореакции 12, и ее облучали светом с помощью ртутной лампы низкого давления. Газ, полученный окислительным фоторазложением смешанного газа, подавали в перемешиваемый 1-гексанол (30 мл, 239 ммоль) в присоединенной двугорлой грушевидной колбах при комнатной температуре. Непрореагировавший газ улавливали присоединенной далее ловушкой с 1-гексанолом в качестве ловушки, и отходящий газ из ловушки очищался щелочной ловушкой, чтобы токсичный газ не вытекал наружу.
Выходы эфира хлормуравьиной кислоты и карбоната, получаемых в реакционной емкости 16 и сосуде-ловушке, оценивали по 1Н ЯМР спектрам, и количество образующегося фосгена определяли исходя из их суммарного количества. Результат показаны в таблице 18.
[0173]
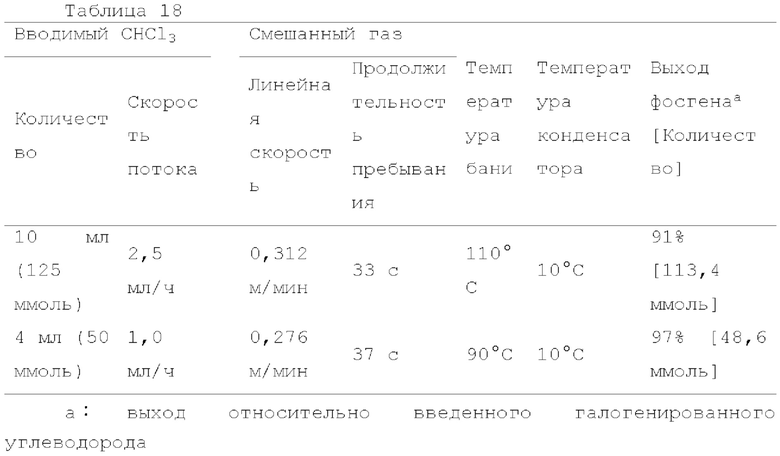
[0174]
Фотореакцию в газовой фазе испаренного хлороформа и газообразного кислорода проводили непрерывным введением хлороформа в реактор для фотореакции, нагретый до температуры кипения или выше хлороформа, извне с помощью шприцевого насоса; как результат, хлороформ может относительно быстро разлагаться с образованием фосгена, как показано в таблице 8.
Газообразный фосген, полученный с высокой степенью конверсии 97% или больше, не содержал заметных побочных продуктов и подвергался реакции со спиртом с образованием эфира хлормуравьиной кислоты или карбоната высокой чистоты. Небольшое количество побочного продукта представляло собой монооксид углерода и диоксид углерода.
[0175]
Пример 21: Получение реагента Вильсмейера фотореакцией в газовой фазе
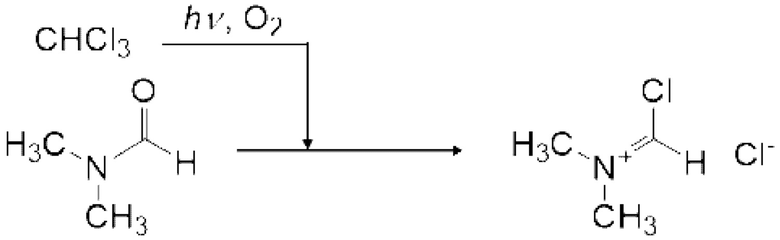
Газ окислительного фоторазложения, полученный в сумме из 6,83 г (57,2 ммоль) хлороформа с применением фотореакционной системы, схематически показанной на рисунке 1, подавали в перемешиваемый N,N-диметилформамид (ДМФА) (5,5 мл, 70 ммоль) при атмосферной температуре в присоединенную двугорлую грушевидную колбу в течение 4 часов, аналогично примеру 1. Непрореагировавший газ подавали в дополнительно присоединенную спиртовую ловушку и щелочную ловушку, которые обрабатывались так, чтобы газ не вытекал наружу.
Реакционную смесь анализировали 1Н ЯМР после реакции; как результат, подтвердили, что реагент Вильсмейера можно получить с выходом 51% или более относительно примененному хлороформу. Фактическое выделенное количество может быть больше полученного, так как реагент Вильсмейера вступает в реакцию с влагой воздуха, разлагаясь до исходного амида. Результат показан в таблице 19.
[0176]

[0177]
Хлороформ нагревали до 90°С, применяя змеевиковый нагреватель, для испарения, и испарившийся хлороформ смешивали с газообразным кислородом для проточной фотореакции в газовой фазе; как результат, реагент Вильсмейера можно получить с выходом более 51% из ДМФА и фосгена, полученного из хлороформа в газовой фазе.
[0178]
Пример 22: Получение реагента Вильсмейера и хлорангидрида кислоты фотореакцией в газовой фазе
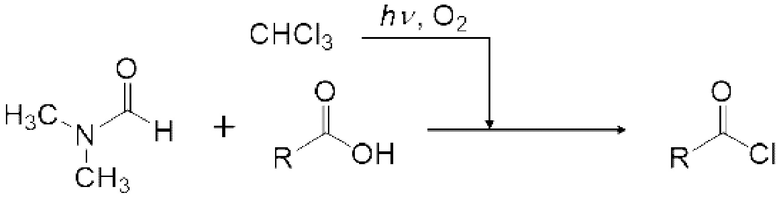
Газ окислительного фоторазложения, полученный из газообразного хлороформа при 7,4 мл/мин и газообразного кислорода при 12,4 мл/мин подавали в перемешиваемый раствор хлороформа (100 мл), полученный растворением бензойной кислоты или пропионовой кислоты и ДМФ в подсоединенной двугорлой грушевидной колбе при 30°С в течение 3-5 часов с применением проточной фотореакционной системы аналогично примеру 21. Непрореагировавший газ подавали в дополнительно подсоединенную спиртовую ловушку и щелочную ловушку, чтобы газ не просочился наружу. Реакционную смесь анализировали 1Н ЯМР после реакции; как результат, целевой хлорангидрид карбоновой кислоты можно получить, как показано в таблице 20.
[0179]


[0180]
Как результат, представленный в таблице 20, было установлено, что степень конверсии карбоновой кислоты в хлорангидрид карбоновой кислоты становится тем выше, чем больше количество хлороформа и отношение ДМФА к карбоновой кислоте.
[0181]
Пример 23: Получение реагента Вильсмейера и эфира муравьиной кислоты фотореакцией в газовой фазе
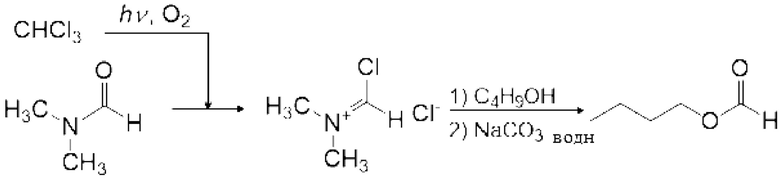
Газ окислительного фоторазложения, полученный из смешанного газа газообразного хлороформа при 7,4 мл/мин и газообразного кислорода при 12,4 мл/мин из 5,82 г (48,8 ммоль) хлороформа, подавали в перемешиваемый ДМФА (8,0 мл, 100 ммоль) в подсоединенной двугорлой грушевидной колбе при атмосферной температуре в течение 3 часов с применением проточной фотореакционной системы аналогично примеру 21.
Непрореагировавший газ подавали в дополнительно присоединенную спиртовую ловушку и щелочную ловушку, чтобы газ не просочился наружу.
К реакционной смеси добавляли 1-бутанол (4,57 мл, 50 ммоль). Смесь перемешивали при атмосферной температуре в течение 1,5 часов. Затем к ней добавляли насыщенный водный раствор карбоната натрия (50 мл) для гидролиза. Добавляли к ней дихлорметан, и разделяли органическую фазу и водную фазу. Органическую фазу сушили над безводным сульфатом натрия и затем фильтровали.
Полученный фильтрат анализировали 1Н ЯМР; как результат, подтвердили, что целевой эфир муравьиной кислоты получали с выходом 60% относительно примененного хлороформа. Результат показан в таблице 21.
[0182]
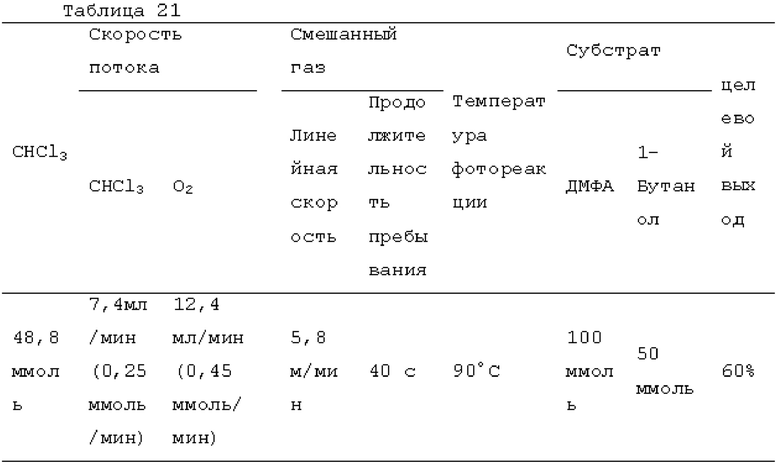
[0183]
Пример 24: Получение реагента Вильсмейера и реакция формилирование фотореакцией в газовой фазе
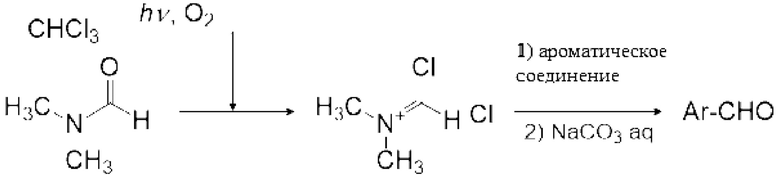
Газ окислительного фоторазложения, полученный из смешанного газа газообразного хлороформа при 7,4 мл/мин и газообразного кислорода при 12,4 мл/мин из 5,37 г (45,0 ммоль) хлороформа подавали в перемешиваемый ДМФА (3,9 мл, 50 ммоль) в подсоединенной двугорлой грушевидной колбе при атмосферной температуре в течение 3 часов с применением проточной системы фотореакции аналогично примеру 21. Непрореагировавший газ подавали в дополнительно присоединенные спиртовую ловушку и щелочную ловушку, чтобы газ не вытекал наружу.
К реакционной смеси, 1-метилпиррол или 2-метилфуран (50 ммоль) добавляли в качестве ароматического соединения. Смесь перемешивали при 70°С в течение 2 часов. Затем, добавляли к ней насыщенный водный раствор карбоната натрия (100 мл) для гидролиза. Добавляли к ней воду и дихлорметан, и разделяли органическую фазу и водную фазу. Органическую фазу сушили над безводным сульфатом натрия и затем фильтровали. Растворитель удаляли из фильтрата испарителем, и целевое соединение получали очисткой из полученного темно-зеленого масла колоночной хроматографией с выделенным выходом 98% или 74%. Результат показан в таблице 22.
[0184]

[0185]
Пример 25: Получение эфира ангидрида карбоновой кислоты
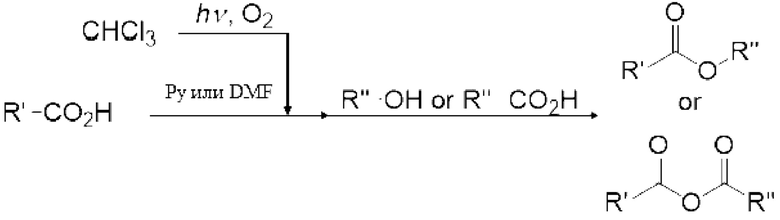
Температуру змеевикового нагревателя световой проточной системы, схематически показанной на рисунке 2, устанавливали равной 90°С. Газ окислительного фоторазложения, полученный из газообразного хлороформа (0,02 мл/мин) и газообразного кислорода (10 мл/мин), и раствор, вводимый из шприцевого насоса, содержащий карбоновую кислоту и 5 краткое количество по молям пиридина или 1 кратное количество по молям ДМФА, смешивали, применяя смеситель Т-типа, и смесь подавали в ПТФЭ трубчатый реактор с внутренним диаметром 2,4 мм, длиной 2830 мм и объемом 12,8 мл для проточной реакции. Продукт подавали в перемешиваемый хлороформный раствор спирта или карбоновой кислоты в двугорлой колбе. Непрореагировавший газ подавали в дополнительно присоединенную щелочную ловушку, чтобы он не просачивался наружу.
Реакционную смесь анализировали 1Н ЯМР после реакции; как результат, эфир или ангидрид карбоновой кислоты получали с выходом, показанным в таблице 23.
Кроме того, реакционную смесь промывали водой и экстрагировали дихлорметаном. Экстракционную жидкость сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Далее, целевое соединение очищали перегонкой при пониженном давлении или колоночной хроматографией. В таблице «ВА» представляет собой бензойную кислоту, «4-FBA» представляет собой 4-фторбензойную кислоту, «РА» представляет собой пропионовую кислоту, и «Ру» представляет собой пиридин.
[0186]
[0187]
Сложный эфир или ангидрид карбоновой кислоты можно получить прямым взаимодействием продукта разложения хлороформа с карбоновой кислотой или получением реагента Вильсмейера из продукта разложения хлороформа и ДМФА и реакцией реагента Вильсмейера с карбоновой кислотой с получением хлорангидрида карбоновой кислоты, и дальнейшей реакцией хлорангидрида карбоновой кислоты со спиртом или карбоновой кислотой в змеевиковом реакторе, как описано выше.
[0188]
Пример 26: Получение амида
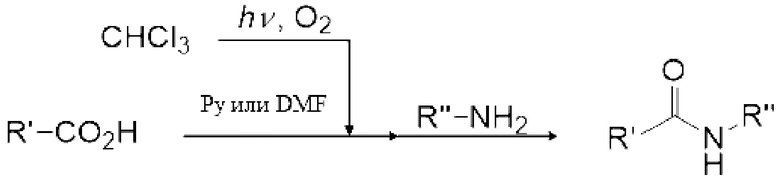
Температуру змеевикового нагревателя световой проточной системы, схематически показанной на рисунке 2, устанавливали равной 90°С. Газ окислительного фоторазложения, полученный из газообразного хлороформа (2,4 мл, 30 ммоль, 0,02 мл/мин) и газообразного кислорода (10 мл/мин), и раствор, вводимый из шприцевого насоса, содержащий карбоновую кислоту и 5 краткое количество по молям пиридина или 1 кратное количество по молям ДМФА, смешивали, применяя смеситель Т-типа, и смесь подавали в ПТФЭ трубчатый реактор с внутренним диаметром 2,4 мм, длиной 2830 мм и объемом 12,8 мл для проточной реакции. Продукт подавали в перемешиваемый раствор амина в двугорлой колбе. Непрореагировавший газ подавали в дополнительно присоединенную щелочную ловушку, чтобы он не просачивался наружу.
Реакционную смесь анализировали 1Н ЯМР после реакции; как результат, амид получали с выходом, показанным в таблице 24.
Кроме того, реакционную смесь промывали водой, и проводили экстракцию, применяя дихлорметан. Экстракционную жидкость сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Далее, целевое соединение очищали перекристаллизацией при необходимости. В таблице "cHex-NH2" представляет собой циклогексилламин.
[0189]
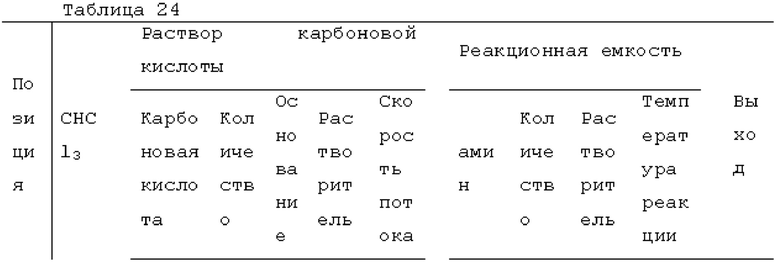
[0190]
Амид можно получить непосредственной реакцией продукта разложения хлороформа с карбоновой кислотой или получением реагента Вильсмейера из продукта разложения хлороформа и ДМФА и реакцией реагента Вильсмейера с карбоновой кислотой с получением хлорангидрида карбоновой кислоты и дальнейшей реакцией хлорангидрида карбоновой кислоты с амином в змеевиковом реакторе, как описано выше.
[0191]
Пример 27: формилирование ароматического соединения реагентом Вильсмейера
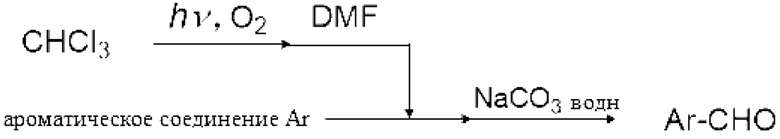
(1) Формилирование 1-метилпиррола
Шприцевой насос 8-1 для введения реакционного субстрата и трубчатый реактор 17 с внутренним диаметром 2,4 мм, длиной 182 мм и объемом 0, 8 мл подключали к проточной фотореакционной системе рисунка 1 для непрерывной реакции, и шприцевой насос 8-2 для введения реакционного субстрата и регулируемый по температуре змеевиковый реактор 9, имеющий внутренний диаметр 2,4 мм, длину 994 мм и объем 4,5 мл, дополнительно подключали для последующей непрерывной реакции, как показано на рисунке 4.
Смешанную жидкость ДМФА (2,32 мл, 30 ммоль) и хлороформа (7,8 мл) вводили в газообразный фосген, полученный проточной фотореакционной системой, из шприцевого насоса 8-1 для введения реакционного субстрата при расходе 3,26 мл/ч аналогично примеру 1, и смесь подавали в трубчатом реакторе 17 для реакции при комнатной температуре. Смешанную жидкость 1-метилпиррола (2,66 мл, 30 ммоль) и дихлорметана (7,0 мл) далее вводили из другого шприцевого насоса 8-2 для введения реакционного субстрата при расходе 3,2 мл/ч в течение в сумме 3 часов. Смесь подавали в змеевиковый реактор 9 для реакции при комнатной температуре.
Продукт вводили в насыщенный водный раствор карбоната натрия (50 мл) в двугорлой колбе в качестве емкости для извлечения 10, соединенной с выходом змеевикового реактора, и смеси перемешивали при температуре бани 0°С. Дихлорметан и воду добавляли к реакционной смеси, и разделяли органическую фазу и водную фазу. Органическую фазу сушили над безводным сульфатом натрия и затем фильтровали. Ацетон (0,74 мл, 10 ммоль) добавляли к полученной высушенной органической фазе в качестве внутреннего стандартного, и смесь анализировали 1Н ЯМР; как результат, подтвердили, что в качестве целевого соединения был получен 1-метил-1Н-пирролкарбоальдегид (выход относительно примененного 2-метилпиррола и ДМФА: 53%).
[0192]
(2) Формилирование 2-метилфурана
Смешанную жидкость ДМФА (2,32 мл, 30 ммоль) и хлороформа (7,8 мл) вводили в газообразный фосген, полученный проточной фотореакционной системой, из шприцевого насоса 8-1 для введения реакционного субстрата при расходе 3,26 мл/ч аналогично примеру 27 (1), и смесь подавали в трубчатом реакторе 17 для реакции при комнатной температуре. Смешанную жидкость 2-метилфурана (2,32 мл, 30 ммоль) и дихлорметана (7,0 мл) далее вводили из другого шприцевого насоса 8-2 для введения реакционного субстрата при расходе 3,2 мл/ч в течение в сумме 3 часов. Смесь подавали в змеевиковый реактор 9 для реакции при комнатной температуре.
Продукт вводили в насыщенный водный раствор карбоната натрия (50 мл) в двугорлой колбе в качестве емкости для извлечения 10, соединенной с выходом змеевикового реактора, и смеси перемешивали при температуре бани 0°С. Дихлорметан и воду добавляли к реакционной смеси, и разделяли органическую фазу и водную фазу. Органическую фазу сушили над безводным сульфатом натрия и затем фильтровали. Ацетон (0,74 мл, 10 ммоль) добавляли к полученной высушенной органической фазе в качестве внутреннего стандартного, и смесь анализировали 1Н ЯМР; как результат, подтверждали, что 5-метил-2-фуральдегид получали в качестве целевого соединения (степень конверсии относительно примененного 2-метилфурана и ДМФА: 95%).
Растворитель отгоняли из описанной выше высушенной органической фазы, и полученный остаток подвергали колоночной хроматографии на силикагеле (элюент: дихлорметан) для выделения целевого соединения (выход относительно примененного 2-метилфурана и ДМФА: 80%).
[0193]
Пример 28: отбор газофазного реактора для фотореакции
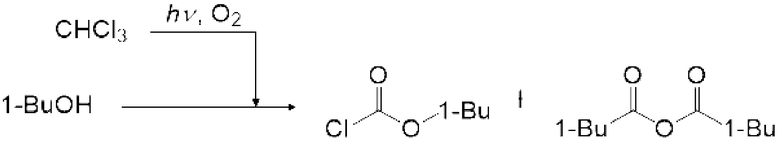
Газофазную фотореакцию проводили с применением фотореакционной системы, схематически показанной на рисунке 5. В частности, реакционная система была сконструирована помещением кожуха из кварцевого стекла в цилиндрическую реакционную емкость, и затем помещением ртутной лампы низкого давления ("SUL-20P" производства SEN Light, 20 Вт, длина светоизлучающей части: 130 мм, длина волны: 185-600 нм, пиковая длина волны: 184,9 нм и 253,7 нм) в кожухе из кварцевого стекла. Размеры цилиндрической реакционной емкости в качестве внешней трубы и кожуха из кварцевого стекла в качестве внутренней трубы приведены в таблице 25.
Жидкий хлороформ и высушенный воздух 20°С подавали в нагретый до 90°С змеевиковый нагреватель 3 при расходе, указанном в таблице 25, для перемешивания и нагрева, и подавали в реактор для фотореакции 12 в течение 2 часов. Температуру нагревателя 13 для нагревания реактора для фотореакции устанавливали равной 100°С.
Газ, выделившийся из реактора для фотореакции 12, подавали в 1-бутанол в реакционной емкости 16-1, и газ, выделившийся из реакционной емкости 16-1, далее подавали в 1-бутанол в реакционной емкости 16-2. В реакционной емкости 16-1 и в реакционной емкости 16-2 соответственно добавлялось 1,5-2,0 кратное количество по молям 1-бутанола относительно примененного хлороформа. Газ, выходящий из реакционной емкости 16-2, далее подавался в щелочную ловушку, чтобы токсичный газ не вытекал наружу.
К реакционным смесям в реакционной емкости 16-1 и реакционной емкости 16-2, 1,2-дихлорэтан добавляли в качестве внутреннего стандарта после реакции, и смесь анализировали 1Н ЯМР для определения выделенных количеств и выходов полученных эфира хлормуравьиной кислоты и ангидрида карбоновой кислоты. Количество образовавшегося фосгена рассчитывали исходя из его общего количества. Результат показан в таблице 25. Выход фосгена в таблице 25 представляет собой выход относительно примененного хлороформа.
[0194]


[0195]
Когда объем реактора для фотореакции относительно невелик, количество вводимого хлороформа должно быть небольшим, чтобы поддерживать высокий выход фосгена и уменьшать количество непрореагировавшего хлороформа за счет короткого времени пребывания смешанного газа, как результат, показанный в таблице 25.
Таким образом, объем реактора для фотореакции делают большим. Как результат, вводимое количество становилось больше, и выделенное количество фосгена могло быть больше, поскольку продолжительность пребывания смешанного газа становилась большой. Но количество непрореагировавшего хлороформа было немного увеличено, так как могла быть область, которая не была достаточно облучено высокоэнергетическим светом.
[0196]
Пример 29: отбор газофазного реактора для фотореакции
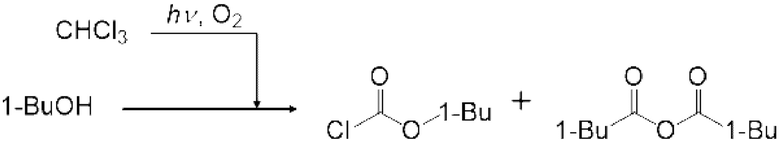
Газофазную фотореакцию проводили с применением фотореакционной системы, схематически показанной на рисунке 5. В частности, реакционную систему конструировали помещением кожуха из кварцевого стекла ϕ30×1,5t и длинной 550 мм в цилиндрическую реакционную емкость ϕ60×2,4t и длинной 550 мм, и затем помещением ртутной лампы низкого давления ("SUL-400" производства SEN Light, 40 Вт, длина светоизлучающей части: 380 мм, длина волны: 185-600 нм, пиковая длина волны: 184,9 нм и 253, 7 нм) в кожухе из кварцевого стекла. Эффективный объем реактора для фотореакции составлял 976 мл.
Жидкий хлороформ и кислород 20°С подавали в нагретый до 150°С змеевиковый нагреватель 3 при расходе, указанном в таблице 26, с применением шприцевого насоса 1 и регулятора массового расхода 2 («MODEL8500MC» производства KOFLOC) для смешивания и нагревания, и подавали в реактор для фотореакции 12 в течение 2 часов. Температуру нагревателя 13 для нагрева реакционной емкости устанавливали равной 100°С
Газ, выделившийся из реактора для фотореакции 12, подавали в 1-бутанол в реакционной емкости 16-1, и газ, выделившийся из реакционной емкости 16-1, далее подавали в 1-бутанол в реакционной емкости 16-2. К применяемому хлороформу добавляли соответственно 1,5-2,0 кратного количества по молям 1-бутанола в реакционной емкости 16-1 и реакционной емкости 16-2. Газ, выходящий из реакционной емкости 16-2, далее подавали в щелочную ловушку, чтобы токсичный газ не вытекал наружу.
После реакции, 1,2-дихлорэтан добавляли в качестве внутреннего стандарта в реакционные смеси в реакционной емкости 16-1 и реакционной емкости 16-2, и смесь анализировали 1Н ЯМР для определения выделенных количеств и выходов полученных эфира хлормуравьиной кислоты и ангидрида карбоновой кислоты. Количество синтезированного фосгена рассчитывали на основе его общего количества. Результат показан в таблице 26. Выход фосгена в таблице 26 является выходом относительно примененного хлороформа.
[0197]
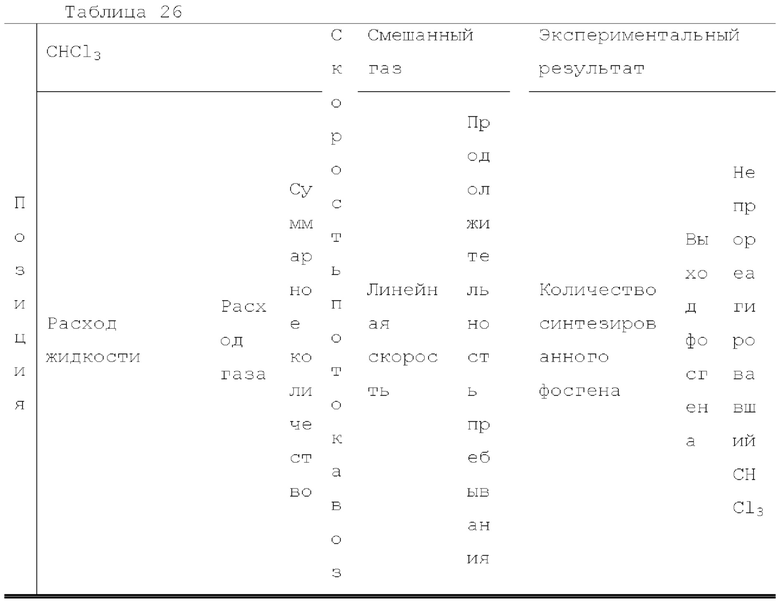

[0198]
Эффективность разложения хлороформа можно дополнительно улучшить за счет применения длинного источника света и, таким образом, облучения сильным высокоэнергетическим светом хлороформа/кислородсодержащего газа в течение длительного времени.
[0199]
Пример 30: выбор источника кислорода
Эффективность синтеза фосгена тестировали аналогично примеру 29, за исключением того, что условия реакции были изменены, как в таблице 27. Воздух применяли в качестве источника кислорода, воздух сушили с применением влагопоглотителя («силикагель, средний гранулированный, синий» производства FUJIFILM Wako Pure Chemical) в позиции 1, позиции 4 и позиции 5, воздух с влажностью 60-80% во время дождя применяли непосредственно в позиции 2, и воздух, полученный из резервуара для осушенного воздуха, применяли в позиции 3. Результат показан в таблице 27.
[0200]



[0201]
Как результат, представленный в таблице 27, было установлено, что некоторое количество воды не становится проблемой, так как эффективность разложения хлороформа и выход фосгена не снижаются даже в том случае, когда воздух применяли без осушения (позиция 2).
Когда расход кислорода был меньше расхода хлороформа (позиция 1 и позиция 4), непрореагировавшего хлороформа было мало, и выход фосгена относительно хлороформа был высоким.
Хотя расход кислорода был меньше, чем расход хлороформа, соотношение непрореагировавшего хлороформа было относительно большим в позиции 5. Причина может заключаться в том, что расход хлороформ и воздуха был большим. Но эффективность позиции 5 была высокой, так как количество синтезированного фосгена было очень большим, и выход фосгена также был относительно высоким в случае позиции 5.
[0202]
Пример 31: выбор газофазного реактора для фотореакции
Фотореакцию в газовой фазе проводили, применяя систему для фотореакции, сконструированную соединением двух газофазных реакторов для фотореакции последовательно, как схематически показано на рисунке 6. В частности, реактор для фотореакции 12-1 был сконструирован вставкой кожуха из кварцевого стекла ϕ30×1,5t в цилиндрическую реакционную емкость ϕ80×2,5t или ϕ140×2,5t, и последующей установкой ртутной лампы низкого давления ("SUL-20P" производства SEN Light, 20 Вт, длина светоизлучающей части: 130 мм, длина волны: 18-600 нм, пиковая длина волны: 184,9 нм и 253,7 нм) в кожух из кварцевого стекла. Также реактор для фотореакции 12-2 был сконструирован помещением кожуха из кварцевого стекла ϕ30×1,5t в цилиндрическую реакционную емкость ϕ60×2,5t, и последующей установкой ртутной лампы низкого давления в кожух из кварцевого стекла. Реакционная система сконструирована соединением реактора для фотореакции 12-1 и реактора для фотореакции 12-2.
Жидкие хлороформ и кислород 20°С подавали в нагретый до 90°С змеевиковый нагреватель 3 при расходе, указанном в таблице 28, для смешения и нагревания, и подавали в реактор для фотореакции 12-1 в течение 2 часов. Температуру нагревателя 13 для нагрева реактора для фотореакции 12 устанавливали равной 100°С.
Газ, выделившийся из реактора для фотореакции 12-2, подавали в 1-бутанол в реакционной емкости 16-1, и выделившийся из реакционной емкости газ 16-1 далее подавали в 1-бутанол в реакционной емкости 16-2. К применяемому хлороформу добавляли соответственно 1,5-2,0 кратное количество по молям 1-бутанола в реакционную емкость 16-1 и реакционную емкость 16-2. Газ, выходящий из реакционной емкости 16-2, далее подавали в щелочную ловушку, чтобы токсичный газ не вытекал наружу.
После реакции, 1,2-дихлорэтан добавляли в качестве внутреннего стандарта в реакционных смесях в реакционной емкости 16-1 и реакционной емкости 16-2, и смеси анализировали 1Н ЯМР, определяя выделенные количества и выходы образовавшихся эфира хлормуравьиной кислоты и ангидрида карбоновой кислоты. Количество образовавшегося фосгена рассчитывали, исходя из его суммарного количества. Результат показан в таблице 28. Выход фосгена в таблице 28 представляет собой выход относительно примененного хлороформа.
[0203]
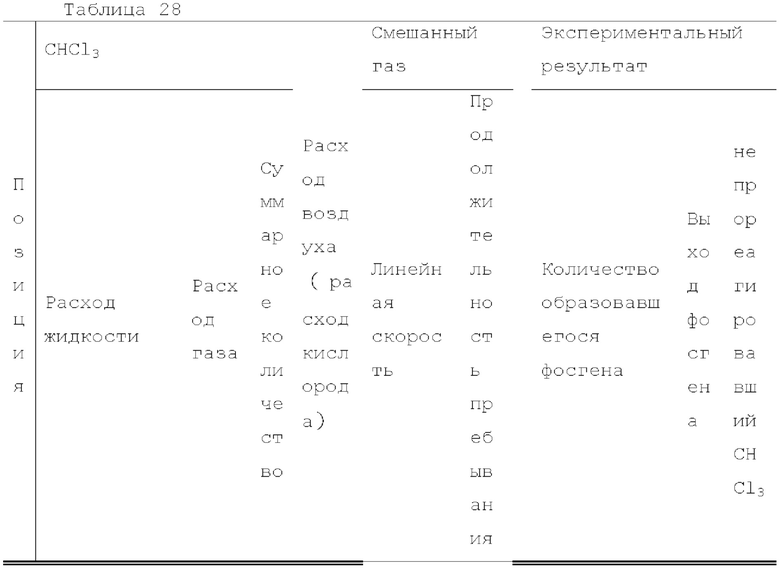
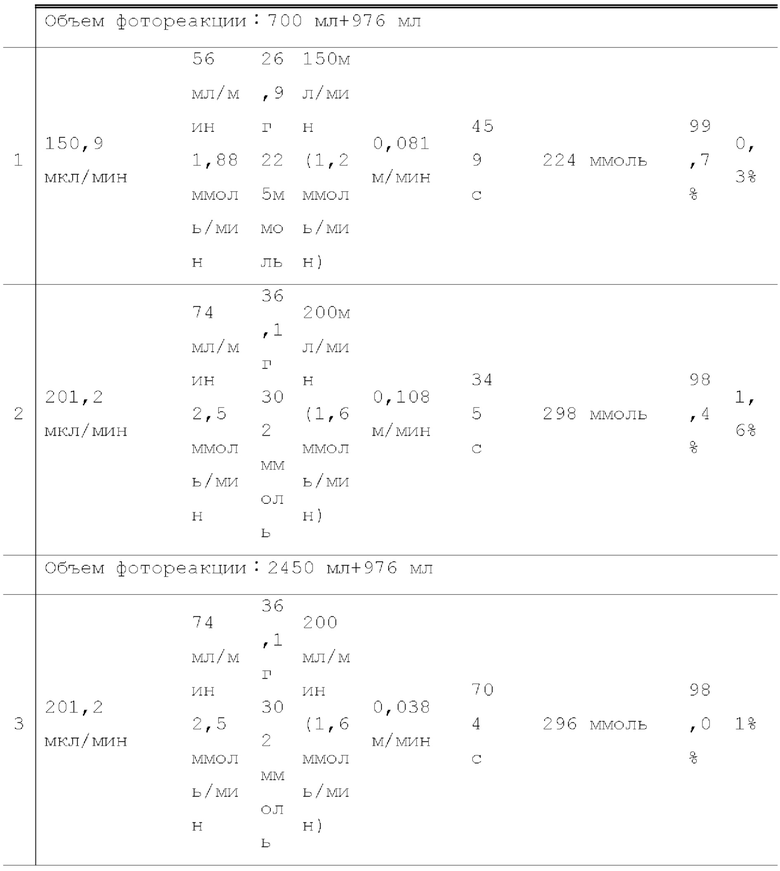
[0204]
Например, сравнивая позицию 1 примера 30 с позицией 2 примера 31, можно утверждать, что степень конверсии хлороформа, количество фосгена и выход фосгена можно повысить соединением двух реакторов для фотореакции. Кроме того, когда смешанный газ проходит через тонкую трубку, соединяющую первый реактор для фотореакции 12-1 и второй реактор для фотореакции 12-2, непрореагировавший испарившийся хлороформ и кислород смешиваются более гомогенно и, таким образом, может эффективно протекать окислительное фоторазложение хлороформа во втором реакторе для фотореакции 12-2.
[0205]
Пример 32: выбор газофазного реактора для фотореакции
Фотореакцию в газовой фазе проводили с применением фотореакционной системы, схематически показанной на рисунке 6. В частности, реактор для фотореакции 12-1 сконструировали помещением кожуха из кварцевого стекла ϕ30×1,5t в цилиндрическую реакционную емкость ϕ80×2,4t и последующей установкой ртутной лампы низкого давления ("SUV-40D" производства SEN Light, 40 Вт, длина светоизлучающая часть: 380 мм, длина волны: 185-600 нм, пиковая длина волны: 184,9 нм и 253,7 нм) в кожух из кварцевого стекла. Также реактор для фотореакции 12-2 сконструировали помещением кожуха из кварцевого стекла (p30xl,5t в цилиндрическую реакционную емкость ϕ80×2,5t и последующей установкой ртутной лампы низкого давления ("SUL-20P" производства SEN Light, 20 Вт, длина светоизлучающей части: 130 мм, длина волны: 185-600 нм, пиковая длина волны: 184,9 нм и 253,7 нм) в кожух из кварцевого стекла. Реакционная система сконструирована соединением реактора для фотореакции 12-1 и реактора для фотореакции 12-2.
Жидкий хлороформы и осушенный воздух 20°С подавали в нагретый до 150°С змеевиковый нагреватель 3 при расходе, указанном в таблице 29, для смешения и нагревания, и подавали в реактор для фотореакции 12-1 в течение 2 часов. Температуру нагревателя для нагрева реактора для фотореакции 12 устанавливали равной 100°С.
Газ, выделившийся из реактора для фотореакции 12, подавали в 1-бутанол в реакционной емкости 16-1, и выделившийся из реакционной емкости газ 16-1 далее подавали в 1-бутанол в реакционной емкости 16-2. К применяемому хлороформу добавляли соответственно 1,5-2,0 кратное количество по молям 1-бутанола в реакционной емкости 16-1 и реакционной емкости 16-2. Газ, выходящий из реакционной емкости 16-2, далее подавали в щелочную ловушку, чтобы токсичный газ не вытекал наружу.
После реакции, 1,2-дихлорэтан добавляли в качестве внутреннего стандарта в реакционные смеси в реакционной емкости 16-1 и реакционная емкость 16-2, и смеси анализировали 1Н ЯМР, определяя выделенные количества и выходы образовавшихся эфира хлормуравьиной кислоты и ангидрида карбоновой кислоты. Количество образовавшегося фосгена рассчитывали, исходя из его суммарного количества. Результат показан в таблице 29. Выход фосгена в таблице 29 представляет собой выход относительно примененного хлороформа.
[0206]

[0207]
Как результат, представленный в таблице 29, было установлено, что степень конверсии хлороформа, количество и выход фосгена заметно увеличиваются при смешивании хлороформа и воздуха, содержащего кислород, при молярном отношении приблизительно 0, 55 относительно хлороформа и применении смешанного газа в реакции фоторазложения. Кроме того, воздух может препятствовать дальнейшему разложению образовавшегося фосгена, поглощая чрезмерно высокоэнергетический свет, особенно ультрафиолетовый свет.
[0208]
Пример 33
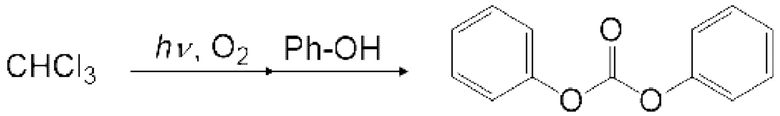
Фотореакцию в газовой фазе проводили, применяя систему, аналогичную фотореакционной системе, схематически показанной на рисунке 5. В частности, фотореакционную систему сконструировали помещением кожуха из кварцевого стекла диаметром 30 мм в цилиндрическую реакционную емкость диаметром 60 мм и объемом 1360 мл, и затем помещением ртутной лампы низкого давления ("SUV40 D" производства SEN Light, 40 Вт, ϕ22,3×380 мм, длина волны: 185-600 нм, пиковая длина волны: 184,9 нм и 253, 7 нм) в кожух из кварцевого стекла. Облучаемый свет содержал УФ-С с длиной волны 185 нм и длиной волны 254 нм, и интенсивность освещения светом с длиной волны 185 нм в положении 5 мм от центра лампы к реакционной смеси составляла 2,00-2,81 мВт/см2, и интенсивность освещения светом с длиной волны 254 нм составляла 5,60-8,09 мВт/см2. Общий объем цилиндрического проточного устройства для фотореакции 12 составлял 976 мл.
Жидкий хлороформ (450 ммоль) подавали в ПТФЭ трубку с внутренним диаметром 1 мм шприцевым насосом 1 при расходе 0,2 мл/мин (2,5 ммоль/мин), смешивали с воздухом, расход которого устанавливали равным 200 мл/мин воздушным насосом и регулятором массового расхода 2, и испаряли нагревателем 3, нагретым до 60°С, подавая в описанное выше проточное устройство для фотореакции 4. Смешанный газ снова нагревали в проточном реактора для фотореакции нагревателем 13, присоединенным к днищу проточного устройства для фотореакции. Температуру нагревателя устанавливали равной 100°С. Линейную скорость смешанного газа в фотореакционном устройстве можно было оценить в 0,18 м/мин, и продолжительность пребывания можно было оценить в 188 секунд.
Пиридин вводили в перемешиваемый фенол (900 ммоль) в реакционной емкости 16, применяя шприцевой насос при расходе 0,52 мл/мин (6,5 ммоль/мин), и газ, полученный окислительным фоторазложением смешанного газа хлороформа и воздуха, подавали в него при 0°С в течение 3 часов. Непрореагировавший газ подавали в дополнительно присоединенную щелочную ловушку, чтобы он не просачивался наружу.
Затем, реакционную смесь в реакционной емкости 16 промывали 1 М хлороводородной кислотой и водой, и экстракцию проводили дихлорметаном. Полученный экстракт сушили над безводным сульфатом натрия, растворитель отгоняли при пониженном давлении с получением белого твердого дифенилкарбоната (выделенное количество: 70,9 г, выход: 74%).
[0209]
Пример 34
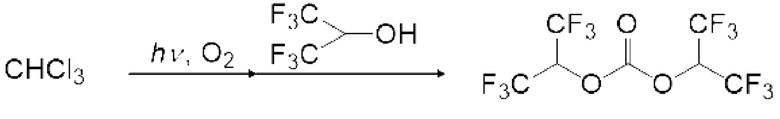
Фотореакцию в газовой фазе проводили, применяя фотореакционную систему, применяемую в примере 33. Жидкий хлороформ (450 ммоль) подавали в ПТФЭ трубку с внутренним диаметром 1 мм шприцевым насосом 1 при расходе 0,2 мл/мин (2,5 ммоль/мин), смешивали с воздухом, расход которого устанавливали равным 200 мл/мин воздушным насосом и регулятором массового расхода 2, и испаряли нагревателем 3, нагретым до 60°С, для подачи в проточное устройство для фотореакции 4. Смешанный газ снова нагревали в проточном устройстве для фотореакции 4 нагревателем 13, присоединенным к днищу проточного устройства для фотореакции. Температуру нагревателя устанавливали равной 100°С. Линейная скорость смешанного газа может быть оценена в 0,18 м/мин, и продолжительность пребывания может быть оценена в 188 секунд.
Пиридин добавляли к перемешиваемому хлороформному раствору HFIP (900 ммоль) в реакционной емкости 16, применяя шприцевой насос при расходе 0,52 мл/мин (6,5 ммоль/мин), и газ, полученный окислительным фоторазложением смешанного газа хлороформа и воздуха, подавали в него при 0°С в течение 3 часов. Непрореагировавший газ подавали в дополнительно присоединенную щелочную ловушку для обработки, чтобы токсичный газ не вытекал наружу.
Затем реакционную смесь в реакционной емкости 16 промывали 1 М хлористоводородной кислотой и водой и анализировали с помощью 1Н ЯМР. Как результат, подтвердили, что бис(гексафторизопропил)карбонат (БГФУ) получали с выходом 85%.
Кроме того, промытую реакционную смесь сушили над безводным сульфатом натрия, и бесцветную жидкость БГФУ выделяли перегонкой (выделенное количество: 88,0 г, 384 ммоль, выход относительно HFIP: 54%).
[0210]
Пример 35
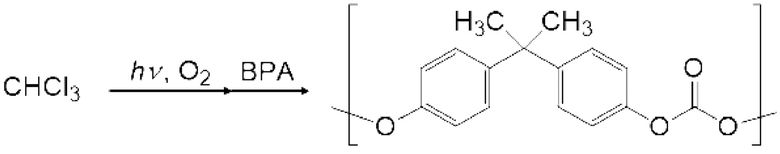
Испаренный хлороформ подвергали окислительному фоторазложению, применяя высокоэнергетический свет, аналогично примеру 34.
Газ, полученный окислительным фоторазложением смешанного газа хлороформа и воздуха, подавали в перемешиваемый хлороформный раствор, содержащий бисфенол А (ВРА) (405 ммоль) и пиридин (2025 ммоль), в реакционную емкость 16 при 0°С в течение 3 часов. Непрореагировавший газ подавали в дополнительно присоединенную щелочную ловушку для обработки, чтобы токсичный газ не вытекал наружу.
Затем, реакционную смесь в реакционной емкости 16 промывали 1 М хлороводородной кислотой и водой, экстракцию проводили дихлорметаном. Полученный экстракт сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении с получением белого твердого поликарбоната (выделенное количество: 102 г, выход: 95%).
Полученный поликарбонат анализировали гель-проникающей хроматографией (ГПХ) для определения молекулярной массы. Результат показан в таблице 30.
[0211]

[0212]
Пример 36
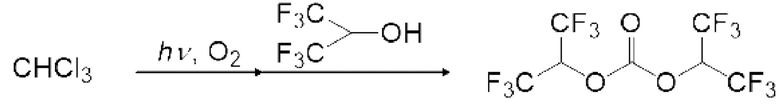
Фотореакцию в газовой фазе проводили, применяя систему, аналогичную фотореакционной системе, схематически показанной на рисунке 5. В частности, фотореакционную систему сконструировали вставлением кожуха из кварцевого стекла диаметром 30 мм в цилиндрическую реакционную емкость диаметром 140 мм и объемом 2575 мл, и последующей установкой ртутной лампы низкого давления ("UVL20PH-6" производства SEN Light, 20 Вт, ϕ24 мм × 120 мм, длина волны: 185-600 нм, пиковая длина волны: 184,9 нм и 253,7 нм) в кожух из кварцевого стекла. Свет, излучаемый ртутной лампой низкого давления, содержал УФ-С с длиной волны 185 нм и длиной волны 254 нм, и интенсивность освещения светом с длиной волны 185 нм в положении на расстоянии 5 мм от центра лампы к реакционной смеси составляла 2,00-2,81 мВт/см2, и интенсивность освещения светом с длиной волны 254 нм составляла 5,60-8,09 мВт/см2. Общий объем цилиндрического проточного устройства для фотореакции 12 составлял 2450 мл.
Жидкий хлороформ (900 ммоль) подавали в ПТФЭ трубку с внутренним диаметром 1 мм шприцевым насосом 1 при расходе 0,4 мл/мин (5,0 ммоль/мин), смешивали с воздухом, расход которого устанавливали равным 200 мл/мин воздушным насосом и регулятором массового расхода 2, и испаряли нагревателем 3, нагретым до 60°С, подавали в описанное выше проточное устройство для фотореакции 12. Смешанный газ снова нагревали в проточном реактора для фотореакции нагревателем 13, присоединенным к днищу проточного устройства для фотореакции. Температуру нагревателя устанавливали равной 100°С. Линейную скорость смешанного газа в устройстве для фотореакции можно было оценить в 0,046 м/мин, и продолжительность пребывания можно было оценить в 235 секунд.
Газ, полученный окислительным фоторазложением смешанного газа хлороформа и воздуха подавали в перемешиваемый хлороформный раствор, содержащий HFIP (1200 ммоль) и пиридин (1800 ммоль), в реакционную емкость 16 при 0°С в течение 3 часов. Непрореагировавший газ подавали в дополнительно присоединенную щелочную ловушку для обработки, чтобы токсичный газ не вытекал наружу.
Затем, реакционную смесь в реакционной емкости 16 промывали 1 М хлористоводородной кислотой и водой, и анализировали 1Н ЯМР. Как результат, подтверждали, что образовывался бис(гексафторизопропил)карбонат (вьделенное количество: 588 ммоль, выход: 98%).
[0213]
Пример 37: Разложение хлороформа видимым светом
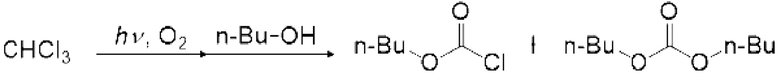
Хлороформный продукт (500 мл), который содержал этанол в качестве стабилизирующего агента, промывали дистиллированной водой, и затем хлороформный слой и водный слой разделяли. Данную процедуру повторяли 3 раза. Хлороформный слой сушили над безводным сульфатом натрия, и безводный сульфат натрия удаляли фильтрованием. Добавляли гидрид кальция (1 г), смешивали и перемешивали при 30°С в течение ночи. Далее, хлороформ, который не содержал стабилизирующего агента, получали перегонкой.
Фотореакцию в газовой фазе проводили, применяя систему, аналогичную фотореакционной системе, схематически показанной на рисунке 5. В частности, фотореакционную систему сконструировали помещением кожуха из кварцевого стекла ϕ30×1,5t в цилиндрическую реакционную емкость ϕ140×2,5t, в которую помещали ртутная лампа низкого давления ("UVL20PH-6" производства SEN Light, 20 Вт, ϕ24 мм × 120 мм, длина волны: 185-600 нм, пиковая длина волны: 184,9 нм и 253,7 нм) в кожухе из кварцевого стекла, и далее помещали светодиодную лампу 365 нм ("LKI-152" производства Polarstar, 30 Вт, светоизлучающая часть: 250×150 мм, пиковая длина волны: 365 нм) на расстоянии приблизительно 1 см от внешней стороны цилиндрической реакционной емкости. Эффективный объем внутренней части устройства для фотореакции 12 составлял 2,5 л. Интенсивность освещения светом на расстоянии 5 мм от применяемой светодиодной лампы составляла 34-38 мВт/см2.
Жидкий хлороформ, из которого стабилизирующий агент удаляли, как описано выше, и кислород 20°С подавали в змеевиковый нагреватель 3, нагретый до 110°С, применяя шприцевой насос 1 и регулятор массового расхода 2 ("MODEL8500MC" производства KOFLOC) при расходе, показанном в таблице 31, для смешения и нагревания. Смешанный газ подавали в реактор для фотореакции 12 в течение 2 часов. Температуру нагревателя 13 для нагревания реакционного устройства устанавливали равной 100°С.
Облучали видимым светом смешанный газ в устройстве для фотореакции 12 с применением только светодиодной лампы, или облучали видимым светом с помощью светодиодной лампы в течение 5 минут и затем дополнительно облучали ультрафиолетовым светом с помощью ртутной лампы низкого давления в течение 1 минуты и снова облучали только видимым светом.
Газ, выделившийся из реактора для фотореакции, подавали в 1-бутанол в реакционной емкости 16-1, и газ, выделившийся из реакционной емкости 16-1, далее подавали в 1-бутанол в реакционной емкости 16-2. В реакционной емкости 16-1 и реакционной емкости 16-2 к примененному хлороформу соответственно добавляли 2,0 кратное количество по молям 1-бутанола. Газ, выходящий из реакционной емкости 16-2, далее подавали в щелочную ловушку для очистки от утечки ядовитого газа наружу.
После реакции, 1,2-дихлорэтан добавляли в качестве внутреннего стандарта в реакционные смеси в реакционной емкости 16-1 и реакционной емкости 16-2, и смесь анализировали 1Н ЯМР, определяя выделенные количества и выходы синтезированных эфира хлормуравьиной кислоты и ангидрида карбоновой кислоты. Количество образовавшегося фосгена рассчитывали, исходя из его суммарного количества. Результат показан в таблице 31. Выход фосгена в таблице 31 представляет собой выход относительно примененного хлороформа.
[0214]
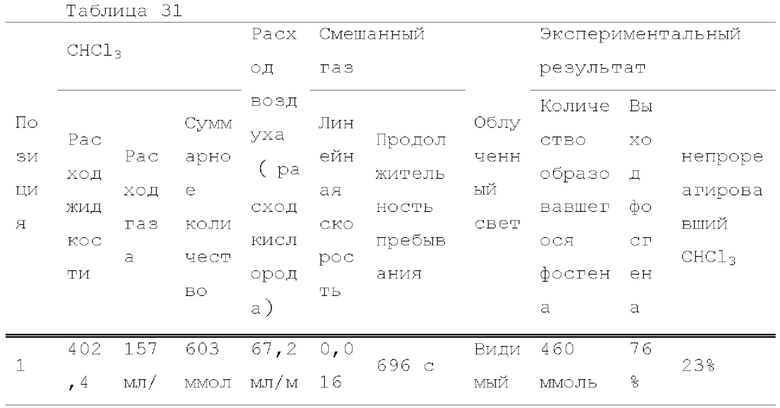
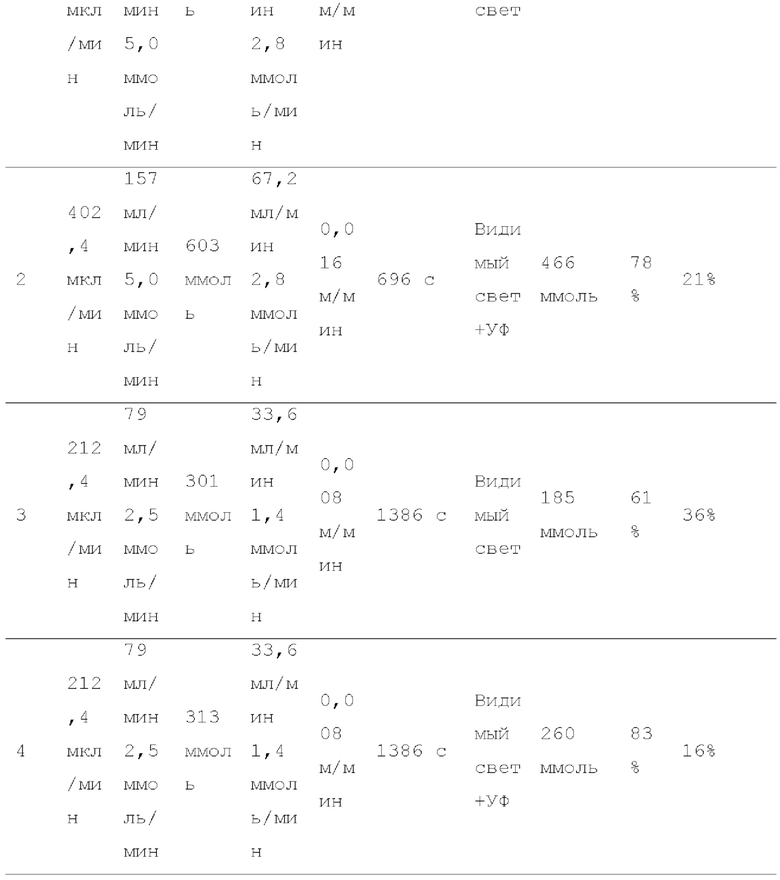
[0215]
При применении хлороформа, который не содержал стабилизирующего агента, количество непрореагировавшего хлороформа было несколько увеличено, но хлороформ мог разлагаться, и фосген можно было получить даже под действием видимого света, как показано в таблице 31. Причина, почему разлагался хлороформ и образовывался фосген при облучении светом, энергия которого была относительно низкой, может заключаться в том, что хлороформ не содержали стабилизирующего агента, и реакцию окислительного фоторазложения проводили в газовой фазе.
Кроме того, выделенное количество и выход повышались, и количество непрореагировавшего хлороформа уменьшалось при кратковременном облучении ультрафиолетовым светом в добавление к видимому свету на ранней стадии реакции окислительного фоторазложения. Причина может заключаться в том, что связь С-Cl расщеплялась высокоэнергетическим ультрафиолетовым светом и затем реакция окислительного фоторазложения хлороформа протекала по радикально-цепному механизму даже при применении только видимого света.
ОБЪЯСНЕНИЕ ПОЗИЦИЙ
[0216]
1: Шприцевой насос, 2: регулятор массового расхода, 3: нагреватель, 4: проточное устройство для фотореакции, 5: регулятор обратного давления, 6: реакционная емкость, 7: емкость-ловушка, 8: шприцевой насос для введения реакционного субстрат, 9: змеевиковое реакционное устройство, 10: емкость для извлечения, 11: источник света, 12: устройство для фотореакции, 13: баня, 14: мешальник, 15: охлаждающий конденсатор, 16: реакционная емкость, 17: трубчатый реактор.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО ПРОИЗВОДНОГО | 2019 |
|
RU2798090C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО ПРОИЗВОДНОГО | 2018 |
|
RU2771748C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИКАРБОНАТА | 2019 |
|
RU2798088C2 |
| СИНТЕЗ УГЛЕВОДОРОДОВ | 2004 |
|
RU2366642C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО НИТРИЛЬНОГО СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО ЭФИРА | 2019 |
|
RU2804510C2 |
| СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА | 2019 |
|
RU2719176C1 |
| ПРОИЗВОДНОЕ БЕНЗОЛА, ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛОМ, И ГЕРБИЦИД | 1997 |
|
RU2162849C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 1,1,1,3,3-ПЕНТАФТОРПРОПАНА, СПОСОБ ПОЛУЧЕНИЯ 1,1,1,3,3-ПЕНТАФТОР-2-ГАЛОГЕНО-3-ХЛОРПРОПАНА, СПОСОБ ПОЛУЧЕНИЯ 1,1,1,2,3,3-ГЕКСАХЛОРПРОПЕНА, СПОСОБ ПОЛУЧЕНИЯ 1,1,1,3,3-ПЕНТАФТОР-2,3-ДИХЛОРПРОПАНА | 1993 |
|
RU2114813C1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО НИТРИЛЬНОГО СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО СЛОЖНОГО ЭФИРА | 2017 |
|
RU2739444C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГАЛОИДИРОВАННЫХ 1,3-ДИОКСОЛАНОВ И ГАЛОИДИРОВАННЫЕ 1,3-ДИОКСОЛАНЫ | 1991 |
|
RU2039055C1 |
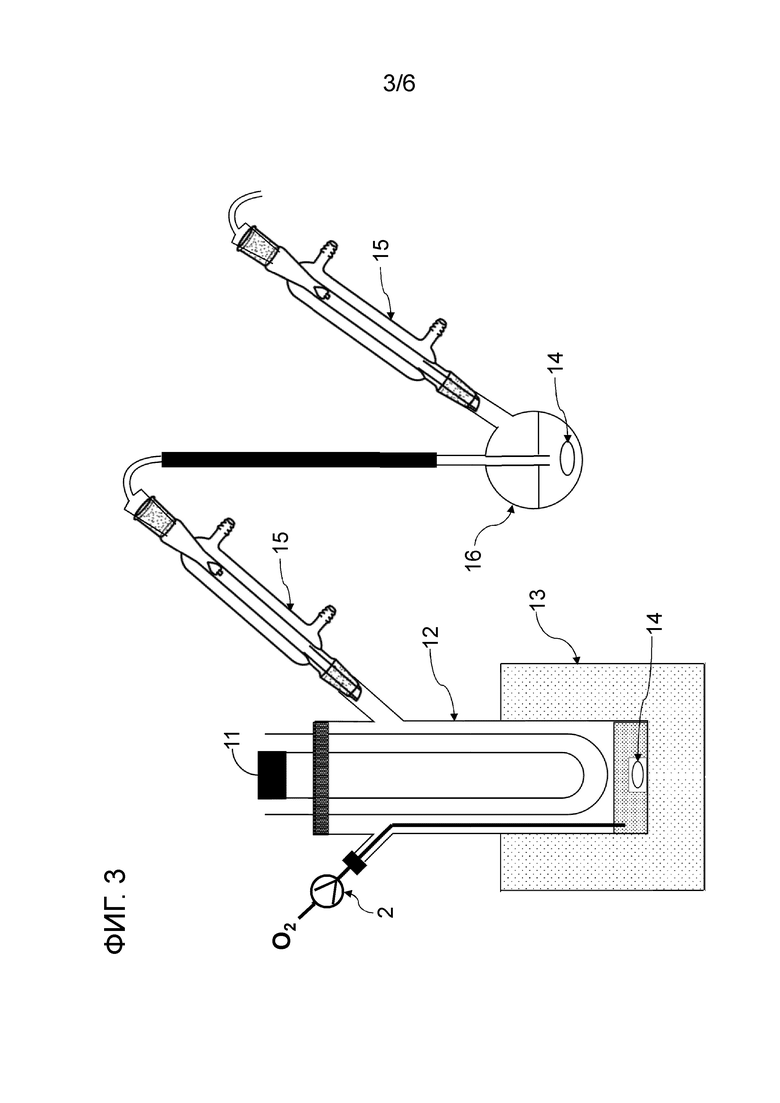
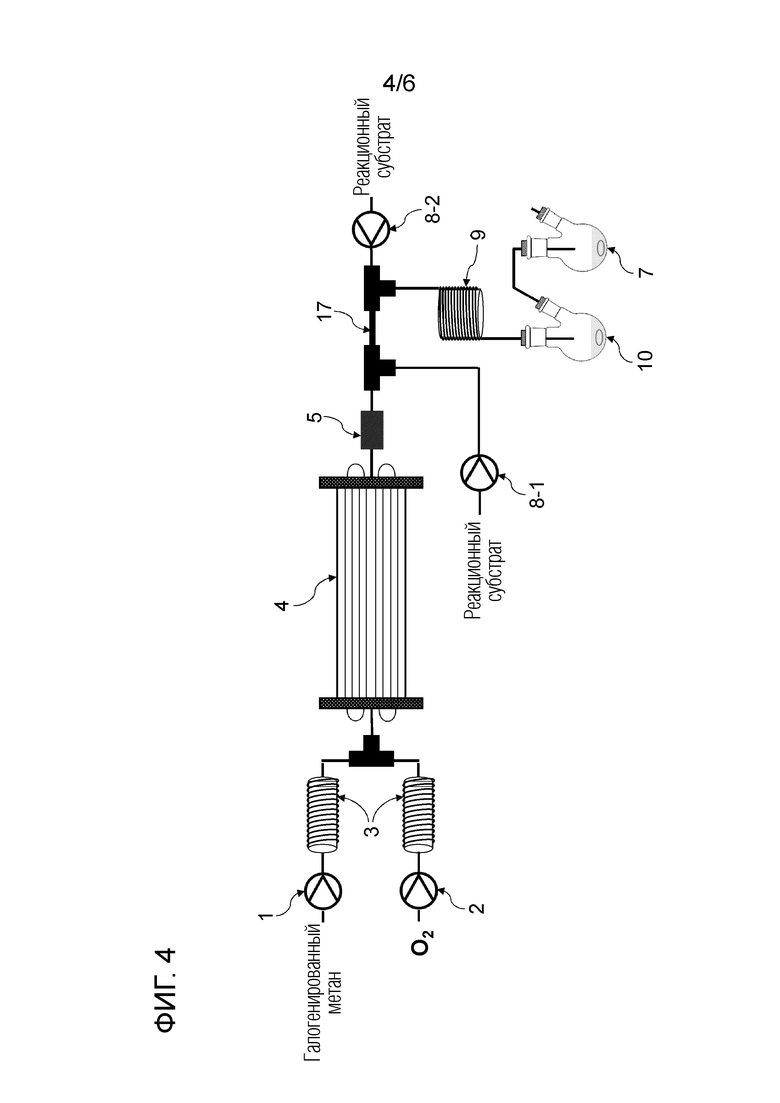
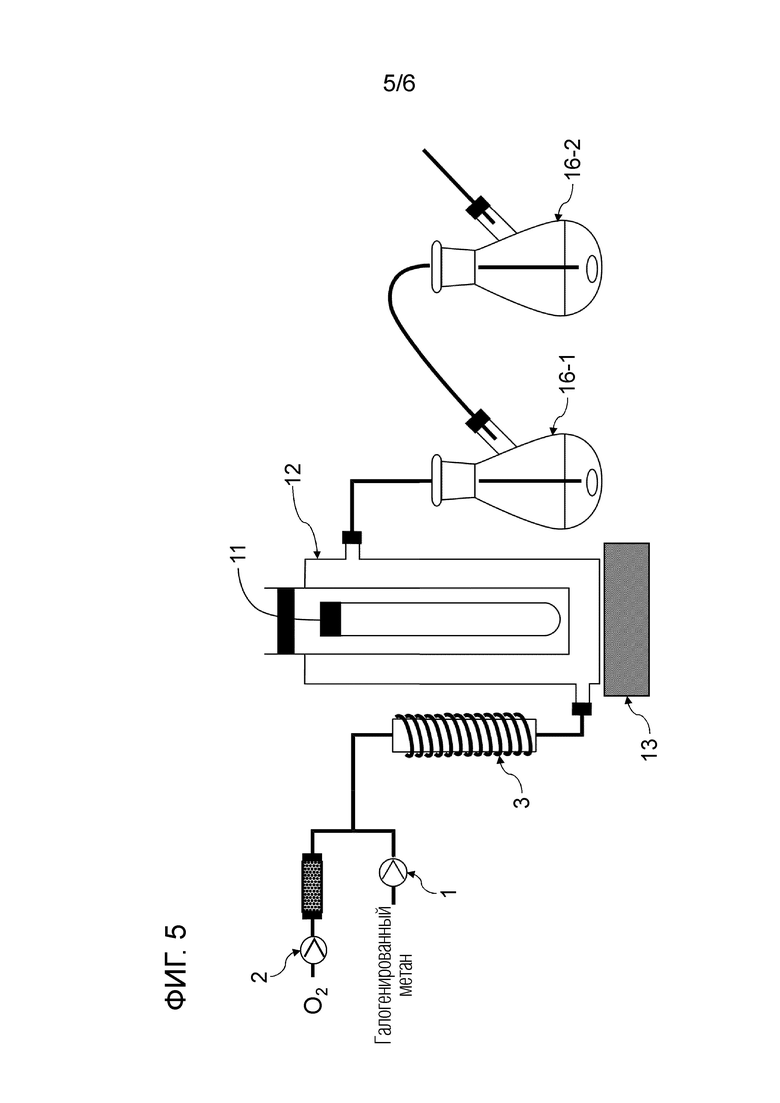
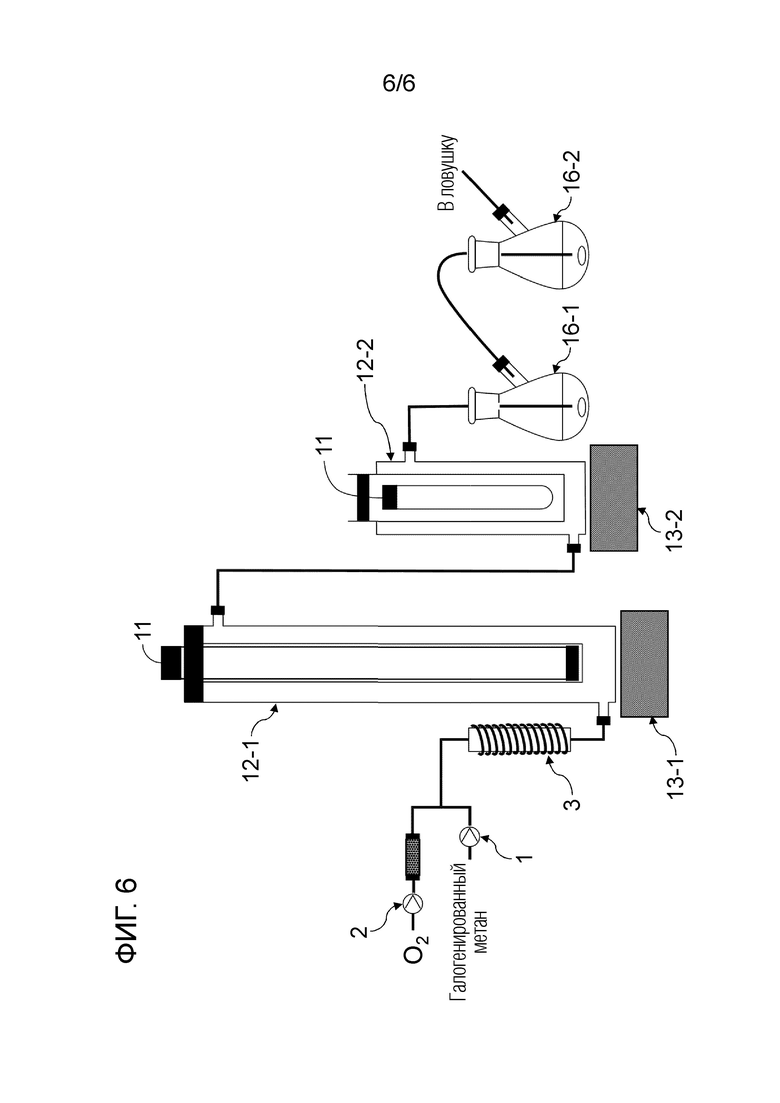
Настоящая группа изобретений относится к области органической химии, конкретно к методу синтеза карбонилгалогенида, а также его производных, включая фторированное и нефторированное карбонатное соединение, фторированный и нефторированный сложный эфир галогенированной муравьиной кислоты, изоцианатное соединение, N-карбоксильный ангидрид аминокислоты и реагент Вильсмейера. Способ получения карбонилгалогенида заключается в получении смешанного газа, содержащего кислород и галогенированный метан, содержащий два или более атомов галогена, выбранного из Cl и Br, и облучении текущего смешанного газа высокоэнергетическим светом. Технический результат заключается в обеспечении эффективным способом получения карбонилгалогенида из галогенированного метана. 8 н. и 3 з.п. ф-лы, 6 ил., 31 табл., 37 пр.
1. Способ получения карбонилгалогенида, включающий стадии:
получения смешанного газа, содержащего кислород и галогенированный метан, содержащий два или более атомов галогена, выбранного из хлора и брома, и
подачи смешанного газа и облучения высокоэнергетическим светом текущий смешанный газ.
2. Способ по п. 1, где кратчайшее расстояние от источника высокоэнергетического света до протекающей газовой смеси составляет 1 м или меньше.
3. Способ по п. 1 или 2, где продолжительность облучения высокоэнергетическим светом протекающей газовой смеси составляет 1 секунду или больше и 10000 секунд или меньше.
4. Способ по любому из пп. 1-3, где температура при облучении высокоэнергетическим светом протекающей газовой смеси составляет 40°С и больше и 200°С или меньше.
5. Способ получения фторированного карбонатного соединения, включающий стадии:
получения карбонилгалогенида способом по любому из пп. 1-4, и
реакции фторированного спиртового соединения и карбонилгалогенида,
где молярное отношение фторированного спиртового соединения к галогенированному метану регулируют равным 1 или более.
6. Способ получения нефторированного карбонатного соединения, включающий стадии:
получения карбонилгалогенида способом по любому из пп. 1-4 и
реакции нефторированного спиртового соединения и карбонилгалогенида,
где молярное отношение нефторированного спиртового соединения к галогенированному метану регулируют равным 1 или более.
7. Способ получения соединения фторированного сложного эфира галогенированной муравьиной кислоты, включающий стадии:
получения карбонилгалогенида способом по любому из пп. 1-4 и
реакции фторированного спиртового соединения и карбонилгалогенида,
где молярное отношение фторированного спиртового соединения к галогенированному метану регулируют равным меньше чем 1.
8. Способ получения соединения нефторированного сложного эфира галогенированной муравьиной кислоты, включающий стадии:
получения карбонилгалогенида способом по любому из пп. 1-4 и
реакции нефторированного спиртового соединения и карбонилгалогенида,
где молярное отношение нефторированного спиртового соединения к галогенированному метану регулируют равным меньше чем 1.
9. Способ получения изоцианатного соединения, включающий стадии:
получения карбонилгалогенида способом по любому из пп. 1-4, и
реакции первичного аминосоединения и карбонилгалогенида,
где молярное отношение первичного аминосоединения к галогенированному метану регулируют равным меньше чем 1.
10. Способ получения N-карбоксильного ангидрида аминокислоты, включающий стадии:
получения карбонилгалогенида способом по любому из пп. 1-4, и
реакции аминокислотного соединения, представленного следующей формулой (VII), и карбонилгалогенида,
где N-карбоксильный ангидрид аминокислоты представлен следующей формулой (VIII):
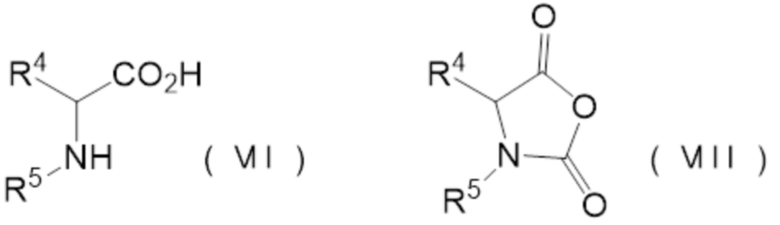
где R4 представляет собой группу боковой цепи аминокислоты,
R5 представляет собой H.
11. Способ получения реагента Вильсмейера,
где реагент Вильсмейера представляет собой соль, представленную следующей формулой (X):
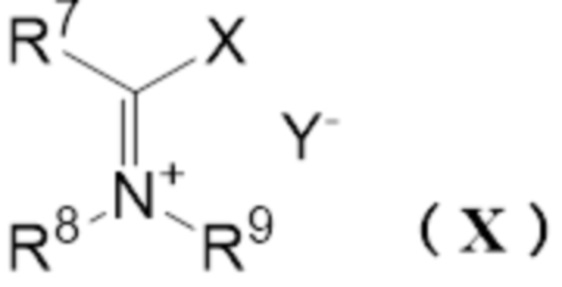
где R7 представляет собой атом водорода,
R8 и R9 независимо представляют собой C1-6 алкильную группу,
X представляет собой атом галогена, выбранный из группы, состоящей из хлора и брома,
Y- представляет собой хлорид-ион или бромид-ион,
включающий стадии:
получения карбонилгалогенида способом по любому из пп. 1-4 и
реакции карбонилгалогенида и амидного соединения, представленного следующей формулой (IX):
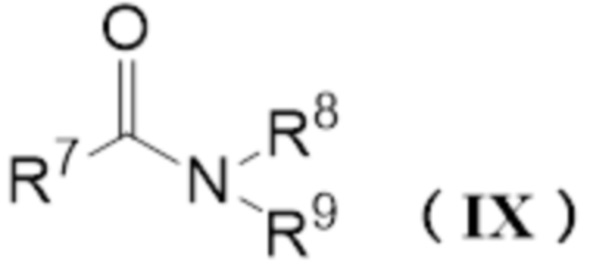
где R7-R9 имеют значения как выше.
| JP 2013181028 A, 12.09.2013 | |||
| WO 2018211953 A1, 22.11.2018 | |||
| WO 2015156245 A1, 15.10.2015 | |||
| JP 2020083765 A, 04.06.2020 | |||
| WO 2020100971 A, 22.05.2020 | |||
| WO 2020100977 A1, 22.05.2020 | |||
| СПОСОБ ПОЛУЧЕНИЯ ФОСГЕНА | 1992 |
|
RU2042618C1 |

