Область техники, к которой относится изобретение
Настоящее изобретение касается способа извлечения йода из водных растворов, содержащих органический йод, в частности йодсодержащие ароматические соединения.
Уровень техники
Большинство коммерческих рентгенографических контрастных агентов, используемых в настоящее время (например, включая йопамидол и йомепрол от Bracco Imaging) характеризуются содержанием трийодированного ароматического кольца.
Способ изготовления этих контрастных агентов и их интермедиатов генерирует маточные растворы и сточные воды, содержащие йод, который, в зависимости от способа, может присутствовать в неорганической форме в разных степенях окисления (т.е. в виде свободных ионов или внедренный в неорганические соединения) или в органической форме (т.е. внедренный в органические соединения).
В последние годы извлечение йода становится все более и более важным по экономическим и экологическим причинам; это действительно дорогой исходный материал, и его выбросы подвергаются очень строгим ограничениям.
В перспективе, выполнение очистки сточных вод, обеспечивающей высокие выходы извлечения органического и неорганического йода и дающей по существу свободные от йода стоки, является основным вопросом для промышленных изготовителей йодированных контрастных агентов.
Ключевым этапом эффективного извлечения йода является удаление органического йода, внедренного в йодсодержащие ароматические соединения, обычно йодированные контрастные агенты и их интермедиаты, и его превращение в молекулярный йод, который затем извлекают.
ЕР0106934 раскрывает способ минерализации для превращения йода, содержащегося в органических соединениях, в йодид в присутствии ионов меди или тонкодиспергированной меди. Извлечение йода затем выполняют путем подкисления минерализованного раствора, окисления образованного йодида и извлечения элементарного йода.
ЕР1133346 предлагает улучшение вышеописанного способа, которое содержит выполнение термического концентрирования маточного раствора или сточных вод во время минерализации, позволяя удалять органические растворители, и очистку концентрированного раствора путем нанофильтрации перед этапом окисления.
СN103508421 раскрывает альтернативную процедуру извлечения йода, в которой превращение (а именно щелочное восстановление) йода, содержащегося в органических соединениях, в йодид-ионы выполняют при дефлегмации в щелочных условиях (рН, по меньшей мере, 12) в присутствии восстановителя, выбранного из порошка меди, порошка магния, порошка цинка, порошка железа и их смесей. Затем воздействуют композитным окислителем Н2О2–FеСl3, превращая йодид-ионы в молекулярный йод, который затем отделяют фильтрацией.
СN105460896 раскрывает многостадийный способ извлечения йода из сточных вод, в котором йод, содержащийся в органических соединениях, превращают в молекулярный йод или йодид-ионы в кислотных условиях, используя окислительную смесь, выбранную из С/Fе2+ и Fе/Fе3+ (окислительное превращение).
Сущность изобретения
Настоящее изобретение, в общем, касается способа обработки водных растворов, содержащих йодсодержащие (кратко "йодированные") ароматические соединения, в котором йод, содержащийся в упомянутых ароматических соединениях (далее "органический йод"), прямо превращается в молекулярный йод и извлекается.
Более конкретно, настоящее изобретение касается способа извлечения молекулярного йода из водного раствора, содержащего йодсодержащее ароматическое соединение и йодсодержащее неорганическое соединение, где упомянутый способ включает этапы:
А) превращения йода упомянутого йодсодержащего ароматического соединения в молекулярный йод; и
В) сбора упомянутого молекулярного йода,
где рН водного раствора, содержащего йодсодержащее ароматическое соединение, подвергающееся превращению на этапе А, составляет 1 или меньше.
Еще более конкретно настоящее изобретение касается вышеописанного способа, в котором рН водного раствора составляет 0,5 или меньше, например от 0,01 до 0,3.
Температура реакции предпочтительно составляет, по меньшей мере, 80°С или выше, более предпочтительно, по меньшей мере, 90°С или выше, еще более предпочтительно 100°С или выше; данная температура предпочтительно меньше чем 300°С и более предпочтительно меньше чем 200°С. В частности, превращение на этапе А выполняют путем нагрева водного раствора при температуре от 80°С до 300°С, предпочтительно от 90°С до 200°С и более предпочтительно от 100°С до 150°С.
Согласно способу данного изобретения превращение на этапе А йода, содержащегося в ароматическом соединении, в молекулярный йод эффективно протекает в отсутствие катализатора.
Данное превращение предпочтительно выполняют в присутствии неорганических анионов, например, включая галогениды, сульфаты или нитраты. Предпочтительные водные растворы (обрабатываемые) дополнительно содержат йодид-ионы.
В одном варианте осуществления данное изобретение касается способа обработки и извлечения йода из водного раствора, содержащего йодированные ароматические соединения, в котором йод, внедренный в эти соединения, превращается в молекулярный йод при рН меньше чем 1 в отсутствие катализатора.
В предпочтительном варианте осуществления данное изобретение касается способа обработки и извлечения йода из водных растворов промышленных способов изготовления радиографических контрастных агентов или их йодированных интермедиатов.
В одном варианте осуществления данный способ включает этапы:
А) превращения йода, содержащегося в йодированных ароматических соединениях, в молекулярный йод при рН меньше чем 1;
А´) необязательного окисления йодид-ионов в молекулярный йод;
В) сбора образованного молекулярного йода; и
С) необязательного извлечения остаточного йода.
Подробное описание изобретения
Если не указано иное, в настоящем описании и формуле изобретения термин:
– "йодсодержащее ароматическое соединение" относится к ароматическим соединениям, содержащим, по меньшей мере, один атом йода, соединенный с атомом углерода в нем;
– "органический йод" относится к йоду, содержащемуся в органических, предпочтительно ароматических, соединениях, обычно ковалентно присоединенному к атому углерода в них;
– "йодсодержащее неорганическое соединение" относится к йоду, содержащемуся в неорганических соединениях, таких как неорганические кислоты или соли, включая йодид-ионы;
– "йодид" или "йодид-ионы" относится к йоду со степенью окисления –1, т.е. в виде иона I–, который может объединяться с протоном или с любым другим положительным противоионом, обычно в диссоциированной форме в водном растворе;
– "неорганический йод" относится к йоду в разных степенях окисления, включая йод, содержащийся в неорганических соединениях, таких как неорганические кислоты или соли (например, йодноватая кислота или йодат).
Данное изобретение касается, в целом, способа обработки и извлечения молекулярного йода из водного раствора, содержащего йодсодержащее ароматическое соединение и йодсодержащее неорганическое соединение, где упомянутый способ включает этапы:
А) превращения йода упомянутого йодсодержащего ароматического соединения в молекулярный йод; и
В) сбора упомянутого молекулярного йода
где рН водного раствора, содержащего йодированное ароматическое соединение, подвергающееся превращению на этапе А, составляет 1 или меньше.
Этап А данного способа выполняют в кислотных условиях, как обсуждается выше.
Подходящие условия включают в себя рН ниже 1 и предпочтительно ниже 0,5. Более предпочтительно, этап А способа выполняют с водными растворами, имеющими рН ниже 0,3, например, приблизительно 0. Если необходимо, рН обрабатываемого раствора можно надлежащим образом доводить до этих величин рН, например, путем добавления (сильной) кислоты, например, выбранной из НСl, Н3РО4 и 50% Н2SО4.
Реакцию предпочтительно проводят при температуре выше 80°С; например, от 80°С до 300°С и, более предпочтительно, от 90°С до 200°С.
Более предпочтительно, этап А способа выполняют при температуре в интервале от 100°С до 150°С, например, приблизительно 140°С–150°С.
В особенно предпочтительном варианте осуществления этап А способа содержит выполнение превращения йода, содержащегося в ароматическом соединении, непосредственно в молекулярный йод, которое выполняют путем нагрева водных растворов, имеющих рН ниже 1 и предпочтительно ниже 0,5, при температуре выше 90°С и предпочтительно от 100°С до 150°С, например, приблизительно 140°С–150°С.
Прямое превращение йода, содержащегося в ароматических соединениях, молекулярный йод является, в частности, реакцией гидродейодирования.
Термин "гидродейодирование" (или "протодейодирование") содержит в себе реакцию, в которой атом йода, соединенный с атомом углерода ароматического кольца, заменяется атомом водорода.
Водные растворы, подвергаемые гидродейодированию, включают в себя йодсодержащее ароматическое соединение. Концентрация ароматических соединений в растворе предпочтительно составляет, по меньшей мере, 0,01 мг/г и предпочтительно находится в интервале, например, от 0,1 мг/г до 20 мг/г.
В предпочтительном варианте осуществления водный раствор, подвергаемый гидродейодированию, содержит йодсодержащие ароматические соединения и йодид-ионы.
Подходящие примеры этих растворов включают в себя, например, водные растворы, поступающие из промышленных способов изготовления йодированных контрастных агентов, особенно неионных рентгеновских контрастных агентов, которые обычно имеют рН ниже 1 и содержат:
– йодиды и, возможно, молекулярный йод;
– йодированные ароматические соединения.
Эти водные растворы могут дополнительно содержать:
– неорганический йод, иной, чем йодиды или молекулярный йод;
– нейодированные органические соединения;
– нейодированные неорганические анионы, такие как хлориды, сульфаты, фосфаты и так далее.
Предпочтительно, массовое отношение йодид : органический йод (т.е. массовое количество йодид-ионов относительно массового количества йода, внедренного в ароматические соединения) в растворе составляет, по меньшей мере, 0,5 или выше.
Более предпочтительно, данное отношение составляет, по меньшей мере, 1 или выше, и еще более предпочтительно, по меньшей мере, 2 или выше, например, до 10.
В вышеуказанных предпочтительных условиях может быть получено почти полное гидродейодирование йодированных ароматических соединений, позволяя почти количественное превращение органического йода (в обработанных растворах) в молекулярный йод, который можно легко собирать на этапе В данного способа.
Существенно, что согласно способу данного изобретения это превращение достигается в отсутствие катализатора дегалогенирования или превращения.
В одном варианте осуществления реакцию гидродейодирования на этапе А выполняют при атмосферном давлении и температурах ниже 100°С.
В альтернативном варианте осуществления этап А выполняют при более высоких температурах, например выше 100°С. В этом случае применяют подходящее оборудование, позволяющее достигать температур реакции приблизительно 140°С–150°С, например, содержащее бак, в котором верхний конец присоединен внешним фланцем к корпусу бака, позволяя выполнять реакцию гидродейодирования при давлении выше атмосферного давления, ускоряя завершение реакции гидродейодирования. В зависимости от температуры реакции рабочее давление может меняться, например, от 1 атм до 85 атм, предпочтительно от 1 атм до 15 атм, более предпочтительно от 1 атм до 5 атм, например, приблизительно 3,5 атм.
Обычно время реакции, позволяющее полное превращение йодсодержащего ароматического соединения в растворах в молекулярный йод, будет зависеть от содержания йодид-ионов и условий гидродейодирования, например, включая температуру реакции и рН раствора.
Например, когда гидродейодирование выполняют в водных растворах, имеющих высокое содержание йодид-ионов, например, по меньшей мере, равное (по массе) содержанию органического йода, и рН ниже 0,5, например приблизительно от 0 до 0,3, при температуре выше чем 100°С и предпочтительно от 140°С до 150°С, превращение органического йода в молекулярный йод, по меньшей мере, 90% (мас./мас.), более предпочтительно 95% (мас./мас.) и наиболее предпочтительно 97% (мас./мас.), и даже больше (относительно количества йода, содержащегося в ароматических соединениях исходного начального раствора) достигается меньше чем за 3 часа, например, за время в интервале от 20 мин до 120 мин и предпочтительно от 20 мин до 60 мин.
Когда альтернативно этап А способа выполняют при меньшей температуре, например приблизительно 100°С, аналогичная конверсия достигается за время, например, от 20 до 50 часов, предпочтительно от 20 до 30 часов и более предпочтительно приблизительно 25 часов.
После (или во время) осуществления гидродейодирования на этапе А оставшийся водный раствор обрабатывают на этапе В, собирая образованный молекулярный йод, возможно после окисления остаточного неорганического йода на этапе А´.
В одном варианте осуществления молекулярный йод, образованный во время этапа А данного способа, прямо удаляют (по меньшей мере, частично) возгонкой из реакционной среды (например, непрерывным способом) с образованием паров чистого йода, которые затем собираются, например, в отдельном устройстве на этапе В способа. Альтернативно, молекулярный йод, полученный во время этапа А, может оставаться в водном растворе внутри реактора до завершения реакции, где он осаждается (например, после охлаждения) и удаляется фильтрованием.
В одном альтернативном варианте осуществления после этапа А способ дополнительно содержит возможный этап А´, включающий окисление возможных остаточных йодид-ионов в молекулярный йод.
Возможный этап А´ может преимущественно выполняться, чтобы обрабатывать водные растворы, богатые йодидами, например, в которых отношение йодиды : органический йод (в растворе) составляет выше 0,5, предпочтительно выше 1.
Предпочтительно, окисление на этапе А´ выполняют в кислотных условиях, например путем добавления окислителя в кислотные растворы, полученные из этапа А.
В одном варианте осуществления окислитель добавляют в кислотные растворы, собранные из этапа А, превращая возможные остаточные йодид-ионы в растворах в молекулярный йод.
Альтернативно, окислитель можно добавлять непосредственно в реактор, в котором происходит гидродегалогенирование, когда оно заканчивается.
Подходящие окислители выбирают, например, из группы, состоящей из гипохлорита, хлората, хлора (газ), нитрита и пероксида водорода, последний особенно предпочтителен.
Окислитель, например пероксид водорода (Н2О2), добавляют, например порциями, в кислотный раствор, собранный из этапа А, за время от 30 от 120 мин и предпочтительно от 50 до 70 мин, например приблизительно 60 мин, пока редокс потенциал смеси не достигает 600 мВ (смотри например, D.R. Lide, CRC Handbook of Chemistry and Physics, ed. 83rd, 2002 CRC Press LLC, от стр.8–21 до стр.8–23).
В одном варианте осуществления Н2О2 добавляют в раствор, собранный из этапа А, предварительно охлажденный, например, до интервала температур ниже 50°С, предпочтительно от 20°С до 40°С и более предпочтительно от 20°С до 30°С, например, при комнатной температуре, промотируя осаждение молекулярного йода, образованного путем окисления йодидов, который затем собирают фильтрацией на этапе В способа.
В предпочтительном варианте осуществления этап А´ способа содержит порционное добавление Н2О2 непосредственно в кислотные растворы из этапа А, поддерживаемые при перемешивании в том же реакторе при температуре приблизительно 150°С.
Когда работают при этой температуре, молекулярный йод, образованный на этапе А и возможно на этапе А´, надлежащим образом удаляют из горячих растворов путем возгонки с образованием паров чистого йода, которые затем собирают, например, в отдельном устройстве согласно этапу В способа.
В частности, в одном варианте осуществления этап В способа содержит удаление йода, образованного во время этапов А и/или А´ способа, путем возгонки, например в потоке паров, и сбор сублимированного йода в отдельных контейнерах. Альтернативно, особенно когда этап А выполняют в закрытом реакторе под давлением, йод, полученный во время этапа А, может оставаться в таком реакторе до возможного начала этапа А´.
Например, на этапе В пары, содержащие йод, превращаются и собираются в адсорбционной жидкости, например органическом растворителе и, предпочтительно, в щелочи, например 30% гидроксиде натрия (NаОН), в которой I2 диспропорционирует на йодат (I+5) и йодид (I–1) ионы. Подкисление щелочного раствора, например, 50% серной кислотой (Н2SО4), ведет затем к осаждению чистого молекулярного йода (I2), который собирают фильтрацией и возвращают в способ изготовления желаемого радиографического контрастного агента.
После каждого из этапов А, А´ или В получают водный раствор с переменными (обычно уменьшается от А до В) количествами остаточного органического, неорганического и/или молекулярного йода.
В одном варианте осуществления способ данного изобретения дополнительно содержит дополнительный этап С для извлечения возможных остаточных следов органического йода, неорганического йода и/или молекулярного йода. Остаточные следы органического йода могут происходить, например, из неполного дейодирования на этапе А способа, например, выполняемого при некоторой температуре и в течение времени, большем, чем возможное, или с недостаточно кислыми отходами, или бедными йодидами. Остаточные следы молекулярного йода могут также происходить от неполного извлечения на этапе В, например, выполняемого осаждением и фильтрацией из водного раствора, в котором молекулярный йод имеет некоторую (пониженную) растворимость, или вызываться неполной возгонкой из водного раствора.
Этап С, например, выполняют путем подачи водного раствора, образующегося на этапе В, т.е. того, который остается после удаления и сбора йода, образовавшегося на этапах А и А´, в одну или несколько колонок, заполненных поглотителем, например, углеродом или ХАD смолами. Остаточные следы органического, неорганического и/или молекулярного йода сначала закрепляются на колонках, а затем десорбируются, например, путем использования подходящих растворителей или оснований, давая концентрированный раствор, который может возвращаться на этап А данного способа или, альтернативно, может обрабатываться согласно известным процедурам, например, описанным в ЕР0106934.
Более конкретно, этап С вышеописанного способа предпочтительно содержит:
i) подачу водного раствора из любого из вышеуказанных этапов А, А´ или В в колонку или набор последовательных колонок, содержащих поглотитель, предпочтительно выбранный из углерода или ХАD смолы, прикрепление остаточных, не разложившихся йодсодержащих ароматических соединений, йодсодержащих неорганических соединений и/или молекулярного йода; и затем выпуск использованных элюатов, которые по существу свободны от йода;
ii) десорбцию йодсодержащих ароматических соединений, йодсодержащих неорганических соединений и/или молекулярного йода, например, путем обработки колонки растворителем или основанием (например, 30% NаОН), получая концентрированный основный раствор, содержащий упомянутые йодсодержащие ароматические соединения, йодсодержащие неорганические соединения и/или молекулярный йод;
iii) возврат данного основного раствора на этап А способа, возможно после подкисления до указанных величин; или, альтернативно;
iv) воздействие минерализации на основные растворы, собранные на этапе ii), например, как описано в ЕР0106934, с превращением органического йода в йодиды, которые возвращаются на этап А или этап А´ способа, или прямо окисляются в молекулярный йод, который затем собирают фильтрацией или возгонкой, как упоминается выше.
Молекулярный йод (I2) из этапа ii) выше обычно диспропорционирует на йодат (I+5) и йодид (I–1) ионы.
Соответственно, в одном варианте осуществления способ данного изобретения содержит:
А) гидродейодирование йодированных ароматических соединений для превращения йода, содержащегося в упомянутых ароматических соединениях, в молекулярный йод;
А´) окисление возможных остаточных неорганических йодсодержащих соединений (например, йодидов) в молекулярный йод;
В) удаление и сбор образованного йода, например, путем осаждения или возгонки, и
С) извлечение остаточных йодсодержащих соединений или молекулярного йода.
Вышеописанный способ, современный в любом его варианте, имеет общее применение и позволяет эффективно обрабатывать растворы (особенно промышленные растворы), содержащие йод в разных формах (например, органический йод в смеси с йодидами, молекулярным и/или неорганическим йодом, как описано выше). В частности, данный способ обеспечивает по существу количественное извлечение полного содержания йода, независимо от концентрации каждой формы йода, со сбором отходов, по существу свободных от йода, которые можно выпускать. При этом, в кислотных условиях этапа А способа и в присутствии йодидов, если присутствуют, возможные следы неорганического йода в степени окисления выше 1, если присутствуют, обычно могут превращаться в молекулярный йод.
Следует заметить, что этап С поглощения/десорбции позволяет эффективно извлекать органический йод, присутствующий в разбавленных растворах, без применения какой–либо концентрирующей обработки, такой как термическое концентрирование. Такое термическое концентрирование требует большой энергии, его устранение особенно выгодно в способах, применяемых в промышленном масштабе.
В частности, способ данного изобретения позволяет эффективно обрабатывать сильнокислые растворы, например, прямо собираемые из способа промышленного производства интермедиатов радиографических агентов, которые обычно имеют высокое содержание йодидов, способствуя превращению органического йода в растворах в молекулярный йод прямо на этапе А с, по существу, количественным извлечением всего йода с этапов А и А´ способа, как показано, например, тестами примеров 1 и 9.
В предпочтительном варианте осуществления данный способ содержит:
А) превращение йода, содержащегося в ароматическом соединении, прямо в молекулярный йод при рН меньше чем 1;
А´) окисление возможных остаточных йодид-ионов в молекулярный йод путем добавления окислителя в раствор из этапа А;
В) сбор молекулярного йода, образованного на этапах А и/или А´; и
С) извлечение возможных остаточных йодсодержащих ароматических соединений и/или молекулярного йода.
На практике, вышеописанный способ может выполняться, как обсуждается выше, путем осуществления этапов от А до С в предложенном порядке, где гидродейодирование йодированных ароматических соединений согласно этапу А предшествует окислению остаточных йодидов, выполняемому согласно этапу А´, возможно непосредственно в том же реакторе, с образованием молекулярного йода, который собирают согласно этапу В, и затем оставшийся раствор возможно подвергают извлечению возможных остатков органического йода и/или молекулярного йода согласно этапу С из предложенной процедуры.
Считается, что возможные варианты вышеописанного исполнения, в которых этапы от А´ до С выполняют в разных последовательностях, или которые не включают один или оба из возможных этапов А´ и С, содержатся в пределах объема настоящего изобретения.
Например, в одном варианте осуществления, в особенности, когда обрабатываемый водный раствор имеет низкую концентрацию йодид-ионов (например, когда отношение йодиды : органический йод в растворе меньше чем 0,5), способ данного изобретения может выполняться без включения этапа А´. В этом случае, этап С, содержащий извлечение любого остаточного органического или молекулярного йода, выполняют прямо после того, как извлечение йода, образованного на этапе А способа, выполняют (позднее) согласно этапу В.
В другом варианте осуществления, например, когда реакции этапа А или А´ выполняют в открытом реакторе, этап В может начинаться до соответствующего окончания этих этапов.
В другом варианте осуществления, этап С способа может предшествовать этапу А´, удаляя остаточный органический йод из раствора, полученного на этапе А. В этом случае, например, последующее окисление йодидов может удобно выполняться при меньших температурах, например, при комнатной температуре, приводя к осаждению молекулярного йода, который отфильтровывают и возможно дополнительно очищают согласно известным процедурам.
В предпочтительном варианте осуществления способ данного изобретения находит эффективное применение в обработке жидкостей, собранных из промышленных способов для получения рентгеноконтрастных агентов и их йодированных интермедиатов, включая йодированные или частично йодированные производные бензольных моно– или дикарбоновых кислот или аминобензольных моно– или дикарбоновых кислот (например, моно–, ди– и трийодированные производные аминобензолкарбоновых кислот).
Эти способы получения включают в себя, в виде общего этапа, йодирование ароматического субстрата, обычно 5–амино–1,3–бензолдикарбоновой кислоты, например, выполняемые, как описано в WО96/37458.
Альтернативно, способы изготовления радиографических контрастных агентов могут содержать йодирование N, N´–бис–амидов 5–амино–1,3–бензолдикарбоновой кислоты, например, соединений с формулой (I)
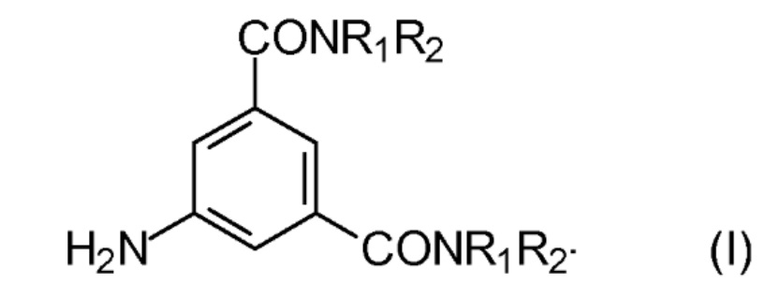
где
R1 обозначает С1–С3 алкил, замещенный одной или несколькими гидроксильными группами; и
R2 обозначает Н или является таким же, как R1,
которое выполняют в кислотных условиях и в присутствии NаIСl2 или КIСl2, например, как описано в GВ 14720250 или ЕР 1337505.
Предпочтительно, в формуле (I) выше R1 является пропандиолом, например, 1,3–пропандиолом, а R2 является Н.
Маточные растворы, прямо собираемые из вышеописанных способов йодирования, имеют оптимальные кислотные условия, а именно рН ниже 1 и обычно ниже 0,5, значительные количества непрореагировавших йодидов и йодированные или частично йодированные ароматические соединения (например, моно–, ди– и трийодированные производные 5–амино–1,3–бензолдикарбоновых кислот).
Эти растворы могут эффективно обрабатываться способом данного изобретения, не требуя какого–либо термического концентрирования или добавления кислот или возможных дополнительных реагентов, давая общий выход извлечения йода, например, выше 95%, предпочтительно от 96 до 100%, более предпочтительно от 97 до 100% и наиболее предпочтительно от 98 до 100%, например, от 98 до 99%, прямо на этапе В.
Интересно, что, когда эти промышленные растворы обрабатывают способом данного изобретения для извлечения йода, почти количественное превращение органического йода (в обрабатываемых растворах) в молекулярный йод, например, выше 95% и предпочтительно 97%, преимущественно получают непосредственно на этапе А способа, делая излишним и необязательным дополнительный этап С, которого, таким образом, можно избежать.
В особенно предпочтительном варианте осуществления данное изобретение касается способа обработки и извлечения йода из растворов, прямо собираемых из промышленных способов йодирования для изготовления радиографических интермедиатов, выбранных из группы, состоящей из 5–амино–2,4,6–трийод–1,3–бензолдикарбоновой кислоты и ее 1,3–бензолкарбоксамидов с вышеуказанной формулой (I).
Наиболее предпочтительно, данный способ содержит извлечение и возврат йода из водных растворов, собранных из промышленного способа изготовления 5–амино–2,4,6–трийод–1,3–бензолдикарбоновой кислоты.
Дополнительные подробности, касающиеся способа йодирования настоящего изобретения, представлены в следующем экспериментальном разделе с единственной целью лучше проиллюстрировать настоящее изобретение без представления каких–либо его ограничений.
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
Следующие примеры выполняли либо с промышленными растворами, либо с их модельными составами, содержащими органический йод в виде моно–, ди– и трийодированных производных 5–амино–1,3–бензолдикарбоновой кислоты или 1,3–бензолдикарбоксамидов, и йодиды.
Пример 1: гидродейодирование+окисление
Этап А: ГИДРОДЕЙОДИРОВАНИЕ
1,000 кг промышленных маточных растворов, полученных из способа йодирования, например описанного в WО96/37458, с рН<0,1, содержащих 10,28 г йодидов (определено с помощью аргентометрического титрования) и 2,00 горганического йода (определено с помощью аргентометрического титрования после разрушения органического йода обработкой Zn/NаОН (смотри, например, йодный анализ, представленный для IOPAMIDOL от Europen Pharmacopeia Ed. 9.0, pag. 2795–2797) в виде моно–, ди– и трийодированных производных 5–амино–1,3–бензолдикарбоновой кислоты, загружали в оболочечный реактор, оборудованный механической мешалкой и термометром и соединенный с ловушкой Дрекселя, заполненной 15% раствором гидроксида натрия.
Данную смесь нагревали до 100°С и оставляли при перемешивании в течении приблизительно 24 ч: гидродейодирование протекало с образованием пурпурных паров молекулярного йода, которые собирали путем поглощения в ловушке, содержащей гидроксид натрия.
В конце концентрация остаточного органического йода была 0,06 г/кг, соответствуя прямой конверсии органического йода в молекулярный йод, равной 97,0%.
Йод, собранный в ловушке, соответствовал приблизительно 31,6% от начального полного содержания йода.
Этап А´: ОКИСЛЕНИЕ
Затем добавляли 5% водный пероксид водорода, поддерживая смесь при 100°С; добавление выполняли за 0,75 ч до редокс потенциала 600 мВ: йодиды окислялись до элементарного (молекулярного) йода, который возгонялся, образуя пары, которые собирали путем поглощения в ловушке, содержащей гидроксид натрия.
Полученную смесь дополнительно перемешивали при той же температуре в течение 1 ч, завершая реакцию и возгонку йода, и затем охлаждали до комнатной температуры за 80 мин.
Использованный раствор (0,926 кг) содержал остаточное количество йодидов (0,08 г) и органического йода (0,055 г).
В конце этапа А´ йод, собранный в ловушке, соответствовал общему выходу извлечения 98,8%.
Пример 2: гидродейодирование+окисление+обработка на угольных колонках
Этап А: ГИДРОДЕЙОДИРОВАНИЕ
3,750 кг водного раствора с рН=0,24, содержащего 5,48 г/кг йодидов и 2,04 г/кг органического йода по существу в виде моно–, ди– и трийодированных производных 5–амино–1,3–бензолдикарбоновой кислоты, загружали в оболочечный реактор, оборудованный механической мешалкой и термометром и соединенный с ловушкой Дрекселя, заполненной 15% раствором гидроксида натрия. Данную смесь нагревали до 100°С и оставляли при перемешивании в течении приблизительно 24,5 ч. Гидродейодирование протекало с образованием пурпурных паров молекулярного йода, которые собирали путем поглощения в ловушке, содержащей гидроксид натрия. В конце концентрация остаточного органического йода была 0,24 г/кг, соответствуя прямой конверсии органического йода в молекулярный йод, равной 88,2%.
Йод, собранный в ловушке, соответствовал приблизительно 48% от начального полного содержания йода.
Этап А´: ОКИСЛЕНИЕ
Затем добавляли 5% водный пероксид водорода, поддерживая смесь при 100°С; добавление выполняли за 1 ч до достижения редокс-потенциала смеси 600 мВ: йодиды окислялись, образуя пары молекулярного йода, которые собирали путем поглощения в ловушке, содержащей гидроксид натрия. Полученную смесь дополнительно перемешивали при той же температуре в течение 1 ч, завершая реакцию и возгонку йода, и затем охлаждали до комнатной температуры за 60 мин. Использованный раствор (3,459 кг) содержал остаточное количество йодидов (0,09 г/кг) и органического йода (0,24 г/кг).
В конце этапа А´ йод, собранный в ловушке, соответствовал общему выходу извлечения 96,0%.
Этап С: ОБРАБОТКА НА УГОЛЬНОЙ КОЛОНКЕ
13,55 кг использованного раствора готовили, повторяя процедуры этапа А и этапа А´ (йодиды/молекулярный йод 0,12 г/кг; органический йод 0,20 г/кг).
Данный раствор загружали в угольную колонку, заряженную 30,5 г сухого угля (эквивалентно слою 80 мл мокрого угля). Операцию загрузки выполняли при скорости потока 3 об/ч и 60°С, нагревая и раствор, и уголь.
Элюирование контролировали с помощью УФ, используя детектор Кнауэра (λ=335 нм), и подачу останавливали, когда поглощение превышало 0,03 мкА.
Приблизительно 85 мг органического йода и 24 мг йодидов/молекулярного йода поглощалось на 1 угля. 80 мл гидроксида натрия и 320 мл воды загружали в угольную колонку при 60°С при скорости потока 2 об/ч, чтобы извлечь адсорбированный йод.
Полученный раствор (412 г) имел содержания органического йода и йодидов соответственно 5,24 г/кг и 0,56 г/кг, и его можно было обрабатывать согласно предшествующему уровню техники, описанному в ЕР106934, или возвращать на этап А.
Общий выход извлечения йода, считая этапы А, А´ и С, был 98,2%.
После этапа десорбции слой угля можно было возвращать и повторно использовать.
Пример 3: гидродейодирование+окисление
Этап А: ГИДРОДЕЙОДИРОВАНИЕ
3,790 кг водного раствора с рН=0,28, содержащего 6,66 г/кг йодидов и 1,51 г/кг органического йода, главным образом в виде моно–, ди– и трийодированных производных 5–амино–1,3–бензолдикарбоновой кислоты, загружали в оболочечный реактор, оборудованный механической мешалкой и термометром и соединенный с ловушкой Дрекселя, заполненной 15% раствором гидроксида натрия.
Данную смесь нагревали до 100°С и оставляли при перемешивании в течении приблизительно 25 ч: гидродейодирование протекало с образованием пурпурных паров молекулярного йода, которые собирали путем поглощения в ловушке, содержащей гидроксид натрия.
В конце концентрация остаточного органического йода была 0,16 г/кг, соответствуя прямой конверсии органического йода в молекулярный йод, равной 89,4%.
Йод, собранный в ловушке, соответствовал приблизительно 33% от начального полного содержания йода.
Этап А´: ОКИСЛЕНИЕ
Затем добавляли 5% водный пероксид водорода, поддерживая смесь при 100°С; добавление выполняли за 1 ч до достижения редокс-потенциала смеси 600 мВ: йодиды окислялись, образуя пары молекулярного йода, которые собирали путем поглощения в ловушке, содержащей гидроксид натрия.
Полученную смесь дополнительно перемешивали при той же температуре в течение 1 ч, завершая реакцию и возгонку йода, и затем охлаждали до комнатной температуры за 60 мин.
Использованный раствор (3,563 кг) содержал остаточное количество йодидов/молекулярного йода (0,07 г/кг) и органического йода (0,15 г/кг).
В конце этапа А´ йод, собранный в ловушке, соответствовал общему выходу извлечения 97,4%.
Использованный раствор затем обрабатывали углем согласно процедуре этапа С, представленной в примере 1, доводя общий выход извлечения йода до 98,5%.
Пример 4: гидродейодирование при 140°С
Эксперимент выполняли в замкнутом реакторе, проверяя влияние на конверсию температуры выше чем 100°С, приблизительно 140°С.
45 г раствора с рН=0,27, содержащего йодиды (3,93 мг/г) и органический йод (1,11 мг/г) поровну помещали в 10 мл закрытые реакторы, поддерживая при перемешивании и нагреве до 140°С.
После 90 мин реакция гидродейодирования завершалась, и остаточный органический йод был равен 0,9% от начального органического йода, соответствуя выходу превращения >99%.
Пример 5: гидродейодирование при 140°С с добавлением кислоты
Эксперимент выполняли, чтобы оценить влияние рН на кинетику гидродейодирования.
28,4 г такого же раствора, как в примере 4, содержащего йодиды (3,93 мг/г) и органический йод (0,94 мг/г), дополнительно подкисляли добавлением 37% соляной кислоты (6,6 г).
Полученный раствор поровну помещали в 10 мл закрытые реакторы и нагревали до 140°С при перемешивании.
После 30 мин реакция гидродейодирования завершалась, и остаточный органический йод был равен 0,5% от начального органического йода.
Сравнение результатов примеров 4 и 5 показывает, что меньшие величины рН приводят в увеличению кинетики реакции.
Пример 6: гидродейодирование при 80°С
Данный тест выполняли, чтобы выяснить влияние субоптимальных температур гидродейодирования.
0,5 кг водных растворов с рН=0,21, содержащих йодиды (3,99 мг/г водных растворов) и органический йод (1,24 мг/г, в виде моно–, ди– и трийодированных производных 5–амино–1,3–бензолдикарбоновой кислоты), загружали в оболочечный реактор, нагревали до 80°С и оставляли при перемешивании в течении приблизительно 55 ч. Гидродейодирование протекало с образованием пурпурных паров молекулярного йода, которые собирали путем поглощения в растворе 15% гидроксида натрия, приводя к снижению концентрации органического йода до измеренной величины 0,78 мг/г, соответствующей конверсии органического йода, равной 37%.
Использованный раствор затем подвергали окислению йодидов и обработке угольным поглощением, выполняемым, как описано в примере 1, приводя к общему выходу извлечения йода 97%.
Пример 7: гидродейодирование+окисление при 100°С
Этап А: ГИДРОДЕЙОДИРОВАНИЕ
952,29 г водного раствора, содержащего соляную кислоту (рН=0,10), йодиды (1,40 мг/г) и органический йод (1,45 мг/г), главным образом в виде моно–, ди– и трийодированных производных 5–амино–1,3–бензолдикарбоновой кислоты, загружали в оболочечный реактор, оборудованный механической мешалкой и термометром и соединенный с ловушкой Дрекселя, заполненной 15% раствором гидроксида натрия.
Данную смесь нагревали до 100°С и оставляли при перемешивании в течении приблизительно 25,5 ч: гидродейодирование протекало с образованием пурпурных паров молекулярного йода, которые собирали путем поглощения в ловушке, содержащей гидроксид натрия.
В конце концентрация остаточного органического йода была 0,50 мг/г, соответствуя прямой конверсии органического йода в молекулярный йод, равной 65,7%.
Йод, собранный в ловушке, соответствовал приблизительно 67,8% от начального полного содержания йода.
Этап А´: ОКИСЛЕНИЕ
Затем добавляли 5% водный пероксид водорода, поддерживая смесь при 100°С; добавление выполняли за 0,5 ч до достижения редокс-потенциала смеси 600 мВ: йодиды окислялись, образуя пары молекулярного йода, которые собирали путем поглощения в ловушке, содержащей гидроксид натрия.
Полученную смесь дополнительно перемешивали при той же температуре в течение 1,5 ч, завершая реакцию и возгонку йода, и затем охлаждали до комнатной температуры за 60 мин.
Использованный раствор (866,95 г) содержал только остаточный органический йод (0,48 мг/г).
В конце этапа А´ йод, собранный в ловушке, соответствовал общему выходу извлечения 84,7%.
Использованный раствор затем обрабатывали углем, как представлено в примере 2, достигая общего выхода более 96,5%.
Пример 8: частичное гидродейодирование+окисление+угольные колонки
Данный тест включает частичное гидродейодирование, предположительно выполненное совместно с этапом окисления, чтобы определить эффективность этапа С) в компенсации любых субоптимальных условий гидродейодирования.
ЧАСТИЧНОЕ ГИДРОДЕЙОДИРОВАНИЕ И ОКИСЛЕНИЕ
3,700 кг маточного раствора, полученного из способа йодирования, например, описанного в WО96/37458, с рН<0,1, содержащего 37,08 гйодидов и 7,32 горганического йода (главным образом в виде моно–, ди– и трийодированных производных 5–амино–1,3–бензолдикарбоновой кислоты), загружали в оболочечный реактор, оборудованный механической мешалкой и термометром и соединенный с ловушкой Дрекселя, заполненной 15% раствором гидроксида натрия.
Раствор нагревали до 100°С за приблизительно 1 ч и затем добавляли 5% водный пероксид водорода (приблизительно 95 г), поддерживая смесь при 100°С; добавление выполняли за 1 ч до достижения редокс-потенциала смеси 600 мВ: молекулярный йод, образующийся во время реакции, возгонялся, и пары собирали путем поглощения в ловушке.
Полученную смесь дополнительно перемешивали при той же температуре в течение 1 ч, завершая реакцию и возгонку йода, и затем охлаждали до комнатной температуры за 80 мин.
Использованный раствор (3,419 кг) содержал остаточное количество йодидов (0,53 г) и органического йода (3,49 г).
В указанных условиях органический йод частично разлагался по механизму гидродейодирования, и йод, собранный в ловушке, соответствовал выходу извлечения 88,9%.
ОБРАБОТКА НА УГОЛЬНОЙ КОЛОНКЕ
9,78 кг раствора готовили, повторяя предыдущую реакцию (все йодиды/молекулярный йод 0,29 г/кг; органический йод 1,39 г/кг).
Данный раствор загружали в две угольных колонки, расположенные последовательно (Col1 и Col2); каждую колонку заряжали 38 г сухого угля (эквивалентно слою 100 мл мокрого угля).
Загрузку выполняли при 20–25°С при скорости потока 3 об/ч и останавливали, когда колонка Col2 начинала выделять йодированные органические соединения (точка проскока). Элюат (9,673 кг) из Col2 содержал остаточное количество йодидов (0,18 г/кг) и органического йода (0,04 г/кг), соответствуя общему извлечению йода 98,5%.
Пример 9: гидродейодирование при 140°С+окисление
Этап А: ГИДРОДЕЙОДИРОВАНИЕ
52,0 г маточного раствора, полученного из способа йодирования, например, описанного в WО96/37458, с рН=0,06, содержащего йодиды (9,86 г/кг) и органический йод (1,92 г/кг), главным образом в виде моно–, ди– и трийодированных производных 5–амино–1,3–бензолдикарбоновой кислоты, загружали равными порциями в 20 мл закрытые реакторы и нагревали до 140°С при перемешивании.
После 30 мин концентрация остаточного органического йода была 0,04 г/кг, соответствуя 2,1% от начального органического йода.
Этап А´: ОКИСЛЕНИЕ
Этап А´ выполняли, как описано в примере 1. В конце этапа А´ йод, собранный в ловушке, соответствовал общему выходу извлечения 99,0%.
Пример 10: гидродейодирование при 100°С
725 г маточного раствора, полученного от йодирования 5–амино–N, N´–бис[2–гидрокси–1–(гидроксиметил)этил]–1,3–бензолдикарбоксамида, выполняемого согласно WО96/737458, содержащего 3,68 горганического йода и 3,49 гйодидов, нагревали до 100°С за 30 мин и поддерживали при перемешивании при этой температуре в течение 47 ч.
В конце остаточный органический и молекулярный йод составляли 0,30 г и 0,20 г соответственно, соответствуя общему извлечению йода 93%. Никакие остаточные йодиды не присутствовали в растворе.
Использованный раствор затем обрабатывали поглощением на угольной колонке согласно процедуре, например, представленной в примере 2, достигая общего выхода извлечения выше 97%.
Пример 11: гидродейодирование при 140°С
Данный тест представляет пример способа согласно данному изобретению, который не включает в себя этапа А´.
30 г водного раствора, подкисленного до рН=0,1 серной кислотой, содержащего йодиды (0,73 мг/г) и органический йод (1,46 мг/г), главным образом в виде 5–амино–2,4,6–трийод–1,3–бензолдикарюоновой кислоты, нагревали до 140°С при перемешивании. После 6 часов остаточный органический йод был равен 30,1% от начального органического йода.
Полученный раствор обрабатывали углем согласно процедуре, представленной в примере 2, достигая общего выхода извлечения 96%.
Изобретение может быть использовано при очистке сточных вод, содержащих рентгенографические контрастные агенты. Способ извлечения молекулярного йода из водного раствора, содержащего йодсодержащее ароматическое соединение и йодсодержащее неорганическое соединение, которое содержит йодид-ионы, включает этапы, на которых превращают йод упомянутого йодсодержащего ароматического соединения в молекулярный йод и собирают упомянутый молекулярный йод. При этом рН водного раствора, содержащего йодсодержащее ароматическое соединение, подвергающееся превращению, составляет 1 или меньше, а массовое отношение между йодид-ионами и йодом, содержащимся в ароматическом соединении в водном растворе, составляет, по меньшей мере, 0,5 или выше. Изобретение позволяет обеспечить высокую степень извлечения йода из сточных вод. 18 з.п. ф-лы, 11 пр.
1. Способ извлечения молекулярного йода из водного раствора, содержащего йодсодержащее ароматическое соединение и йодсодержащее неорганическое соединение, которое содержит йодид-ионы, причем упомянутый способ включает этапы, где
А) превращают йод упомянутого йодсодержащего ароматического соединения в молекулярный йод; и
В) собирают упомянутый молекулярный йод,
где рН водного раствора, содержащего йодсодержащее ароматическое соединение, подвергающееся превращению на этапе А, составляет 1 или меньше, и где массовое отношение между йодид-ионами и йодом, содержащимся в ароматическом соединении в водном растворе, составляет, по меньшей мере, 0,5 или выше.
2. Способ по п. 1, в котором рН водного раствора составляет 0,5 или меньше.
3. Способ по п. 1 или 2, в котором этап А выполняют в отсутствие катализатора.
4. Способ по любому из пп. 1-3, в котором превращение на этапе А выполняют путем нагрева водного раствора до температуры от 80°С до 300°С.
5. Способ по п. 4, в котором температура составляет от 100°С до 150°С.
6. Способ по п. 1, в котором упомянутое массовое отношение составляет, по меньшей мере, 1 или выше.
7. Способ по любому из пп. 1-6, дополнительно включающий этап А´, на котором добавляют окислитель к раствору, собранному на этапе А, и превращают возможные остаточные йодид-ионы в молекулярный йод.
8. Способ по любому из пп. 1-7, в котором на этапе В удаляют молекулярный йод, образовавшийся на этапах А и/или А´ данного способа, возгонкой и собирают его в отдельном устройстве.
9. Способ по любому из пп. 1-8, дополнительно включающий этап С, на котором извлекают возможное остаточное йодсодержащее ароматическое соединение и/или молекулярный йод.
10. Способ по п. 9, в котором на этапе С:
i) подают водный раствор с любого из этапов А, А´ или В в колонку, содержащую материал-поглотитель, закрепляющий остаточные, не разложившиеся йодсодержащие ароматические соединения, йодсодержащие неорганические соединения и/или молекулярный йод;
ii) десорбируют йодсодержащие ароматические соединения, йодсодержащие неорганические соединения и молекулярный йод путем обработки колонки основанием, с получением концентрированного основного раствора, содержащего упомянутые йодсодержащие ароматические соединения, йодсодержащие неорганические соединения и/или молекулярный йод.
11. Способ по п. 10, в котором упомянутый основный раствор, необязательно после подкисления, возвращают на этап А способа.
12. Способ по п. 10, в котором упомянутый основный раствор подвергают процедуре минерализации для превращения йодсодержащих ароматических соединений в йодиды.
13. Способ по п. 12, в котором упомянутые йодиды возвращают на этап А или А´ способа.
14. Способ по любому из предыдущих пунктов, в котором водный раствор для этапа А собирают непосредственно из промышленных процессов приготовления рентгеноконтрастных агентов или их йодсодержащих интермедиатов.
15. Способ по п. 14, в котором упомянутый водный раствор содержит йодированные производные бензол(моно- или ди-)карбоновых кислот или аминобензол(моно- или ди-)карбоновых кислот.
16. Способ по п. 14, в котором осуществляют йодирование радиографических интермедиатов, выбранных из группы, состоящей из: 5-амино-1,3-бензолдикарбоновой кислоты и ее N,N´-бисамидов формулы (I)
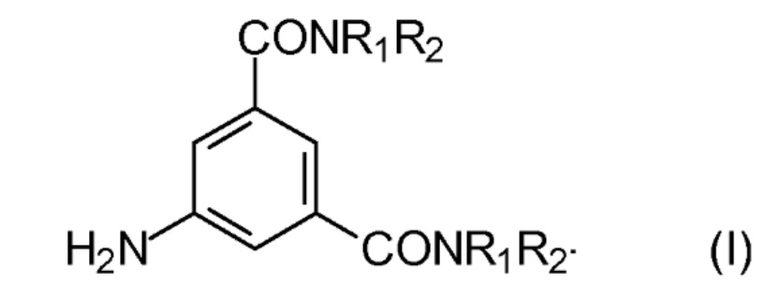
где
R1 обозначает С1-С3 алкил, замещенный одной или несколькими гидроксильными группами; и
R2 обозначает Н или является таким же, как R1.
17. Способ по п. 16, в котором интермедиатом является 5-амино-1,3-бензолдикарбоновая кислота.
18. Способ по любому из пп. 14-17, в котором на этапе А способа достигают конверсии йода, содержащегося в ароматических соединениях, в молекулярный йод, по меньшей мере, 95% (мас./мас.).
19. Способ по любому из предыдущих пунктов, включающий следующие этапы:
А) превращение йода, содержащегося в ароматическом соединении, непосредственно в молекулярный йод при рН меньше чем 1;
А´) окисление возможных остаточных йодид-ионов в молекулярный йод путем добавления окислителя в раствор с этапа А;
В) сбор молекулярного йода, образованного на этапах А и/или А´; и
С) извлечение возможных остаточных йодсодержащих ароматических соединений и/или молекулярного йода.
| DE 19702814 A1, 06.08.1998 | |||
| US 4461711 A, 24.07.1984 | |||
| CN 103508421 A, 15.01.2014 | |||
| КОЛОННА СОРБЦИИ ЙОДА ИЗ РАСТВОРОВ | 2002 |
|
RU2233792C2 |
| WO 9637458 A1, 28.11.1996 | |||
| Шумозащитное вентиляционное окно | 1983 |
|
SU1337505A1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ ЙОДА ИЗ РАСТВОРОВ | 2001 |
|
RU2207976C2 |
| CN 105460896 A, 06.04.2016. | |||

