Область техники, к которой относится изобретение
Заявленная группа изобретений относится к способу получения новой аморфной формы N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3Н-индол-3-илиден)метил]-2,4-диметил-1Н-пиррол-3-карбоксамида малата (международное непатентованное название - сунитиниб), полученному продукту и его применению в фармацевтических композициях, которые могут быть использованы для лечения иммунологических и/или онкологических заболеваний.
Уровень техники
Из многочисленных источников хорошо известно, что соединение сунитиниб (N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1Н-пиррол-3-карбоксамид) способно одновременно ингибировать рецепторы различных тирозинкиназ (РТК), участвующих в процессе роста опухолей, патологического ангиогенеза и образования метастазов. Проявляет ингибирующую активность в отношении многих киназ (>80 киназ). Было показано, что он является мощным ингибитором рецепторов тромбоцитарного фактора роста (PDGRFα, PDGRFβ), рецепторов фактора роста сосудистого эндотелия (VEGRF1, VEGRF2, VEGRF3), рецептора фактора стволовых клеток (KIT), рецептора Fms-подобной тирозинкиназы-3 (FLT), рецептора колониестимулирующего фактора (CSF-1R) и рецептора нейротрофического глиального фактора (RET). Активность основного метаболита была сходной с таковой у сунитиниба.
Сунитиниб ингибирует фосфорилирование многих РТК (PDGRFα, VEGRF2, KIT) в ксенографтах опухолей, экспрессирующих целевые РТК in vivo и демонстрирует подавление роста опухоли или ее регрессию и/или подавление метастазов на экспериментальных моделях различных опухолей. Сунитиниб демонстрирует способность ингибировать рост опухолевых клеток, экспрессирующих дерегулированные целевые РТК (PDGRF, RET или KIT) in vitro и PDGRFβ-, VEGRF2- зависимый ангиогенез in vivo.
Метаболизм сунитиниба осуществляет в основном изофермент CYP3A4, фермент цитохрома Р450, в результате чего образуется основной активный метаболит, который далее метаболизируется тем же изоферментом CYP3A4. Доля активного метаболита составляет 23-37% от величины AUC. Сунитиниб выводится в основном кишечником (61%); через почки в виде препарата и его метаболитов выводится примерно 16% от введенной дозы. Сунитиниб и его главный активный метаболит являются основными веществами, обнаруженными в плазме, моче и фекалиях с радиоактивностью соответственно 91,5%, 86,4%, 73,8%. Меньшие метаболиты обнаруживаются в моче и фекалиях, но не обнаруживаются в плазме. После однократного перорального приема здоровыми добровольцами время полувыведения сунитиниба и его активного метаболита составляет 40-60 и 80-110 часов, соответственно. При повторном ежедневном применении происходит 3-4-кратное накопление сунитиниба и 7-10- кратное накопление его основного метаболита. Равновесные концентрации сунитиниба и его основного активного метаболита достигаются через 10-14 дней [Инструкция по медицинскому применению препарата «Сутент» ЛСР-002516/07-240215, зарегистрированная в Государственном реестре лекарственных средств, далее ИМП препарата «Сутент»].
Известно противоопухолевое средство, ингибитор протеин-тирозин киназы «Сутент», выпускаемое в виде твердых желатиновых капсул с дозировкой 12,5 мг; 25 мг; 50 мг. Согласно инструкции по применению капсула дозировкой 12,5 мг содержит следующие компоненты:
- Сунитиниба малат - 16,7 мг, эквивалентно 12,5 мг сунитиниба;
- Маннитол - 80,0 мг;
- Кроскармеллоза натрия - 6,6 мг;
- Повидон - 5,6 мг;
- Магния стеарат - 1,1 мг.
Согласно ИМП препарата «Сутент» применяется при:
- Гастроинтестинальных стромальных опухолях при отсутствии эффекта от терапии иматинибом вследствие резистентности или непереносимости;
- Распространенном и/или метастатическом почечноклеточном раке у пациентов, не получавших ранее специфического лечения;
- Распространенном и/или метастатическом почечноклеточном раке при отсутствии эффекта от лечения цитокинами;
- Нерезектабельных или метастатических высокодифференцированных нейроэндокринных опухолях поджелудочной железы у взрослых с прогрессированным заболеванием.
Аморфное состояние вещества отличается от кристаллического отсутствием дальнего порядка взаимного расположения молекул, более высокой внутренней энергией и межмолекулярным расстоянием. Способность химических соединений существовать в нескольких аморфных формах называют полиаморфизмом. В термодинамически строгом смысле под этим следует понимать возможное существование двух аморфных фаз, между которыми имеется четкий фазовый переход первого рода. Однако зачастую новая аморфная фаза не подходит под это определение. Так, стекловидные материалы находятся в термодинамически неравновесном состоянии, но при этом могут оставаться стабильными в течение длительного времени при температурах ниже точки стеклования. Хэнкок и др. предложили для таких случаев термин «псевдополиаморфизм» [J. Pharm. Pharmacol. 2002, 54 (8), 1151-2], который не прижился глубоко в научной литературе. Чтобы избежать путаницы в терминологии, далее по тексту мы будем считать аморфные формы одного вещества разными, если эти формы отличаются своими свойствами.
Аморфное состояние вещества характеризуется более высокой свободной энергией Гиббса. Из этого следует, что оно обладает более высокой растворимостью и скоростью растворения по сравнению с кристаллическим состоянием, что приводит к увеличению биодоступности лекарственного средства. С другой стороны, из-за избыточной энтропии, энтальпии и свободной энергии аморфное состояние вещества нестабильно, и характеризуется склонностью к спонтанной кристаллизации. Таким образом, обнаружение новых аморфных форм лекарственных соединений, обладающих улучшенными фармакологическими, физико-химическими и технологическими свойствами, является актуальной задачей современной науки, а создание таких форм представляет собой важное техническое достижение.
Аморфные формы органических соединений обычно характеризуют такими физическими методами, как рентгеновская дифракция, дифференциальная сканирующая калориметрия, инфракрасная спектроскопия, рамановская спектроскопия, терагерцовая спектроскопия, спектроскопия твердофазного ядерного магнитного резонанса и другими.
Хорошо известно, что свойства аморфного вещества могут зависеть от способа, которым оно было получено. Существенное отличие в физических свойствах было обнаружено для α-аморфной, β-аморфной, α-кристаллической и β-кристаллической форм азелнидипина. При помощи ИК спектроскопии было установлено, что указанные кристаллические и аморфные формы отличаются характером и прочностью водородных связей [Shuang Du, et al. RSC Adv., 2018, 8, 32756-32764]. В другом исследовании было показано, что аморфный симвастатин, полученный методом криоизмельчения, обладает более низкой стабильностью, чем аморфный симвастатин, приготовленный путем переохлаждения расплава [K.A. Graeser, С.J. Strachan, J. Е. Patterson, K.С. Gordon and Т. Rades, Physicochemical properties and stability of two differently prepared amorphous forms of simvastatin, Cryst. Growth Des. 8, 2008, 128-135]. Аморфная форма индометацина, полученная методом криоизмельчения, является наименее стабильной по сравнению с аморфными формами, полученными при помощи быстрого охлаждения и распылительной сушки [Karmwar et al., Int. J. Pharm. 417, 2011, 94-100]. Таким образом, для описания разных аморфных форм химических соединений возможно использовать как признаки, относящиеся непосредственно к веществу, так и признаки способа их получения, включающие последовательность технологических стадий и режимов их проведения.
В патентной заявке WO 2009156837 А2 заявлены аморфная форма основания и аморфная форма сунитиниба малата, а также способы их получения. Аморфная форма сунитиниба малата охарактеризована спектральными методами: дифрактограмма представляет собой характерное широкое гало, а в ИК-спектре присутствуют следующие сигналы: 3417, 1680, 1576, 1516, 1479, 1443, 1403, 1330, 1302, 1260, 1233, 1200, 1147, 1109, 1050, 667, 587 см-1. Способ получения аморфной формы сунитиниба малата включает в себя растворение кристаллической формы в растворителе, перемешивание полученной суспензии и удаление растворителя. Удаление растворителя подразумевает под собой фильтрование, выпаривание под вакуумом, распылительную сушку или лиофилизацию. В качестве растворителя выступает N-метил-2-пирролидон.
В международной патентной заявке WO 2010039798 А2 описаны аморфные композиции сунитиниба малата и вспомогательных веществ, среди которых заявлены целлюлоза, поливинилпирролидон, циклодекстрины, дисахариды и их производные.
В международной патентной заявке WO 2013160916 А1 заявлена аморфная твердая дисперсия, содержащая фармацевтически приемлемый носитель. В качестве носителей заявлены такие полимеры, как коповидон, этилцеллюлоза, Span 20, гидроксипропилметилцеллюлоза, полиэтиленгликоль или Soluplus. Процесс получения твердой дисперсии включает в себя смешивание сунитиниба малата и полимера в воде и лиофилизацию полученной суспензии. Аморфные твердые дисперсии на основе сунитиниба малата являются стабильными, воспроизводимыми и негигроскопичными.
В патентной заявке Китая CN 106974890 А раскрывается способ получения твердых дисперсий, содержащих аморфный сунитиниба малат. Заявлено, что такие твердые дисперсии обладают улучшенной стабильностью и дисперсностью, увеличенной скоростью растворения сунитиниба малата. Способ получения отличается своей доступностью, низкой стоимостью, а также может применяться в промышленности. Сунитиниб малат, L-яблочную кислоту и соответствующий полимер растворяют в подходящем растворителе, нагревают, перемешивают, затем удаляют растворитель в вакууме. В результате получают аморфную твердую дисперсию сунитиниба малата и подходящего полимера. Полимер выбирается из следующего списка: гидроксипропилцеллюлоза, липосомы, сополимер метакриловой кислоты, поливинил ацетат, гидроксипропилметилцеллюлоза, полиакриловая смола, карбоксиметилцеллюлоза, фталат карбоксиметилцеллюлозы, поликарбоксиэтилен, карбоксилактон, поливиниловый спирт, хитозан, коллаген и т.д.
Раскрытие изобретения
Изобретение относится к способу получения стабильной аморфной формы N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3Н-индол-3-илиден)метил]-2,4-диметил-1Н-пиррол-3-карбоксамида малата (международное непатентованное название - сунитиниб; активная фармацевтическая субстанция известна как сунитиниба малат), полученному указанным способом продукту (аморфной форме сунитиниба малата) и его применению в фармацевтических композициях, которые могут быть использованы для лечения онкологических и иммунологических заболеваний.
Сунитиниба малат характеризуется следующей структурной формулой:
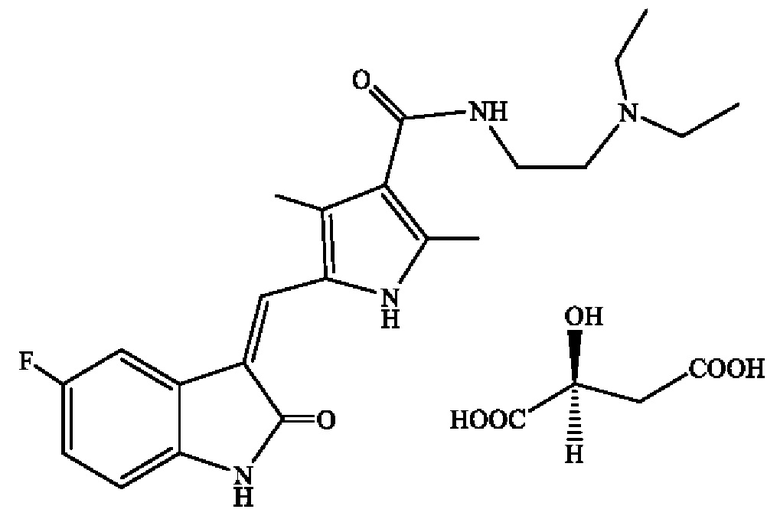
Брутто-формулой: C22H27FN4O2 * С4Н6О5
Молекулярной массой: 532,56 (сунитиниба малат).
Под малатом здесь и далее подразумевается кислотно-аддитивная соль L-яблочной кислоты.
Заявленная группа изобретений направлена на решение актуальной задачи расширения арсенала технических средств определенного назначения. Данная задача решается путем создания новой стабильной слабо гигроскопичной аморфной формы N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3Н-индол-3-илиден)метил]-2,4-диметил-1Н-пиррол-3-карбоксамид малата (сунитиниба малата), а также ее применению в фармацевтических композициях для лечения онкологических и иммунологических заболеваний.
Техническим результатом заявленной группы изобретений является уменьшенное содержание примесей, а также неожиданные обнаруженные свойства, такие как улучшенная стабильность, биодоступность и терапевтическая эффективность полученной заявленным способом новой аморфной формы сунитиниба малата.
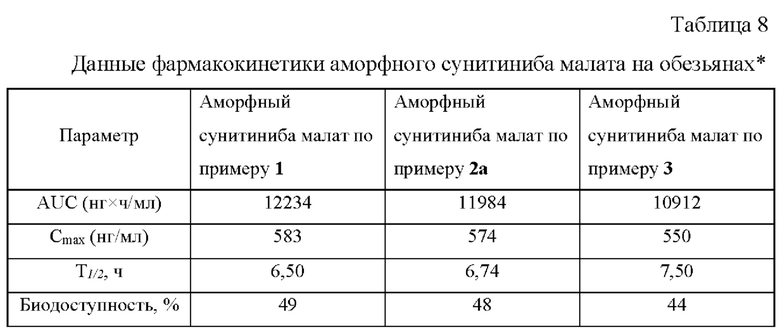
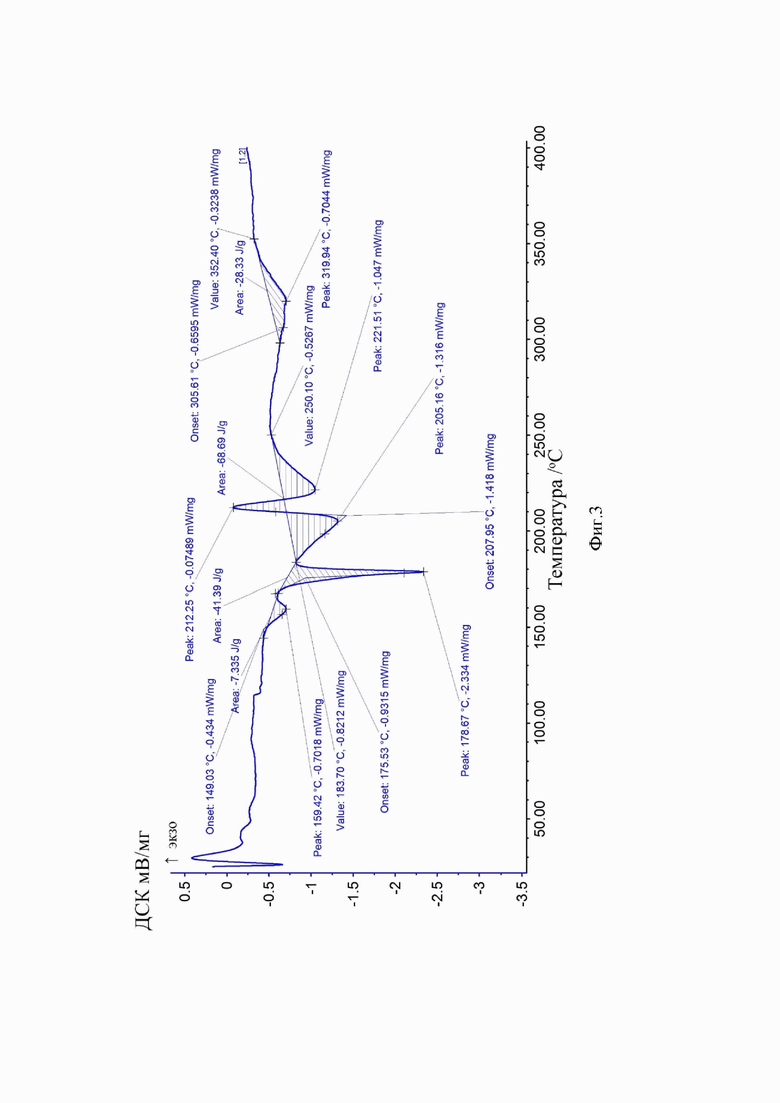
Аморфная форма сунитиниба малата по изобретению характеризуется широким гало в спектре порошковой рентгеновской дифракции (Фиг. 1). Для образца аморфного сунитиниба малата, полученного по примеру 1, в условиях дифференциальной сканирующей калориметрии при скорости нагрева 10°С/мин, наблюдаются следующие тепловые эффекты: 149,03° (эндо), 175,53° (плавление, эндо), 183,70° (эндо), 207,95° (экзо). Температура плавления 175,5°С является одной из существенных характеристик предлагаемой новой аморфной формы, которая отличается от известных значений для данного химического соединения. Поскольку координаты экстремума на кривой ДСК существенно зависят от конструкции прибора и условий проведения эксперимента, во всех случаях в качестве температуры фазового перехода принималась температура, соответствующая точке пересечения экстраполированной в область пика базовой линии графика с касательной к точке перегиба на левом плече кривой (Tonset).
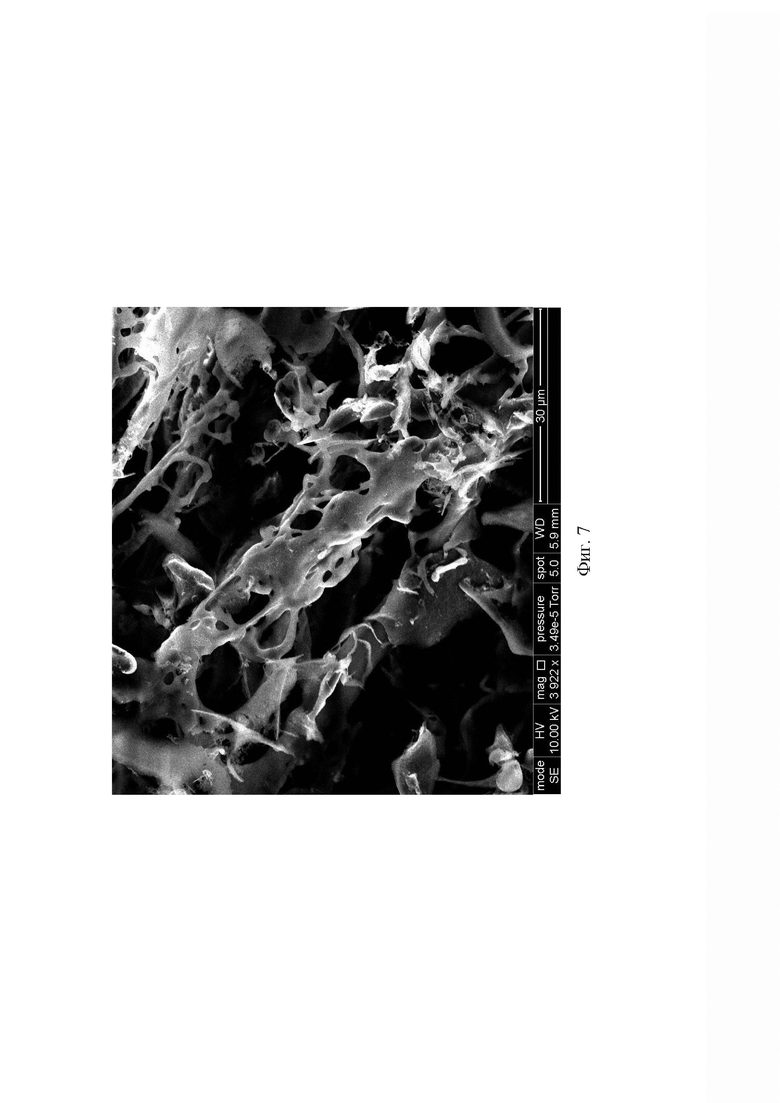
Микрофотография новой аморфной формы сунитиниба малата демонстрирует развитую микроструктуру и значительную удельную поверхность. При этом микроструктура аморфной формы хорошо воспроизводилась (Фиг. 6 и 7).
Наиболее близким аналогом (прототипом) аморфной формы по изобретению можно считать единственную известную аморфную форму сунитиниба малата, описанную в международной патентной заявке WO 2009156837 А2. Для получения аморфной формы по WO 2009156837 используют высококипящий растворитель, что приводит к трудностям его удаления. Это является проблемой, которая часто встречается в фармацевтической технологии, особенно при переходе от лабораторных объемов к промышленным. Зачастую ее не удается преодолеть путем увеличения времени или температуры сушки, поскольку такое решение приводит к частичной деградации основного вещества.
В предпочтительном варианте осуществления способа получения новой аморфной формы сунитиниба малата, ее получают сплавлением кристаллического N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида малата (сунитиниба малата), D-фруктозы и мочевины в массовом соотношении 1:2:3 с последующим прибавлением воды при перемешивании, и выделении продукта путем фильтрования и промывания избытком воды, после чего высушивают под вакуумом до постоянной массы. Преимуществом данного варианта является неиспользование органических растворителей, что обеспечивает их отсутствие в готовом продукте, а также повышает пожарную безопасность производства и, вследствие этого, снижает технические требования к производственному помещению.
В дополнительном варианте осуществления способа получения новой аморфной формы сунитиниба малата, ее получают из сунитиниба малата, синтезируемого при взаимодействии 5-[(Z)-5-фтор-1,2-дигидро-2-оксо-3Н-индол-3-илиден)метил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты и N,N-диэтил-1,2-этилендиамина в среде N,N-диметилформамида с последующей обработкой L-яблочной кислотой в среде ацетона. Полученный таким образом сунитиниба малат сплавляют с D-фруктозой и мочевиной в массовом соотношении 1:2:3 с последующим прибавлением воды при перемешивании, и выделении продукта путем фильтрования и промывания избытком воды, после чего высушивают под вакуумом до постоянной массы.
Высокая чистота аморфного сунитиниба малата по изобретению исключает возможность проявления примесями и остаточными органическими растворителями «пластифицирующего» эффекта. Более низкая гигроскопичность и повышенная стабильность аморфной формы сунитиниба малата по настоящему изобретению в сравнении с известной аморфной формой продемонстрирована соответствующими исследованиями (примеры 7 и 8).
Испытания на животных показывают, что аморфная форма сунитиниба малата по изобретению, характеризующаяся указанными выше параметрами и полученная предложенными в настоящем изобретении способами, обладает улучшенной биологической доступностью и терапевтической эффективностью при лечении опухолевых заболеваний, чем известная аморфная форма (примеры 9 и 10).
Краткое описание чертежей
Для пояснения сущности заявляемого технического решения к описанию приложены Фигуры 1-8:
На Фиг. 1 приведен спектр порошковой рентгеновской дифракции аморфной формы сунитиниба малата, полученной по примеру 1.
На Фиг. 2 приведен спектр порошковой рентгеновской дифракции исходной кристаллической формы сунитиниба малата.
На Фиг. 3 приведена дериватограмма аморфной формы сунитиниба малата по примеру 1.
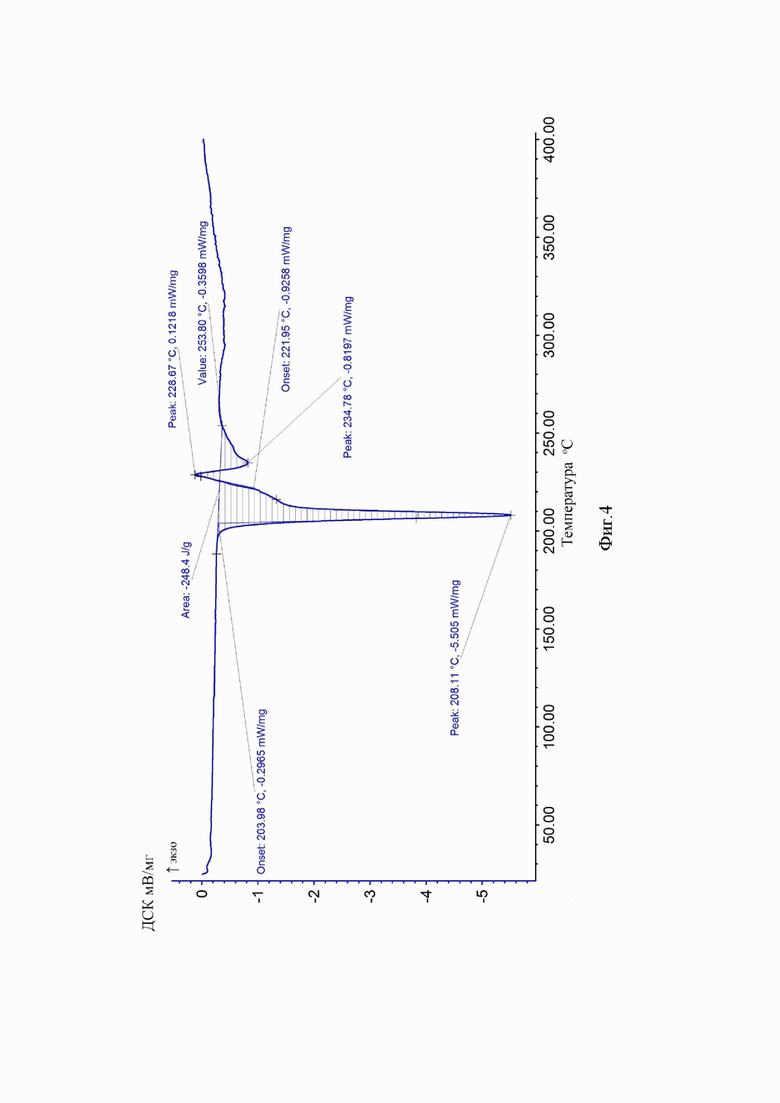
На Фиг. 4 приведена дериватограмма исходной кристаллической формы сунитиниба малата.
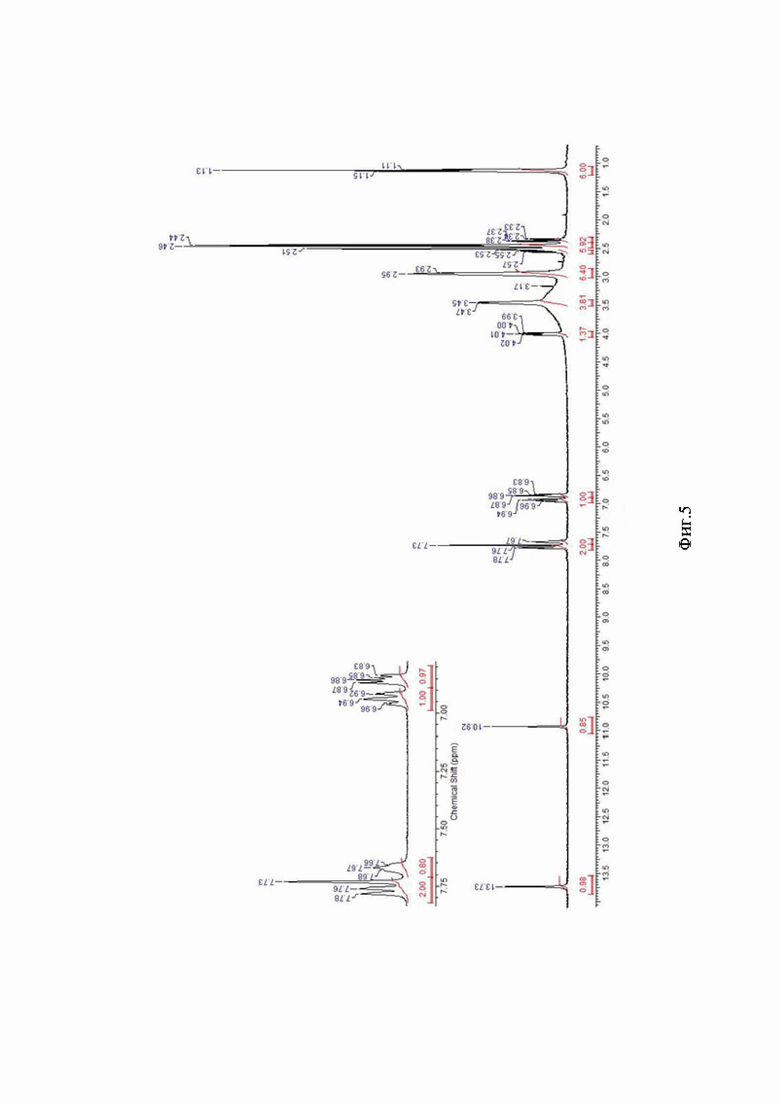
На Фиг. 5 приведен спектр 1Н ЯМР основания сунитиниба по примеру 2а (ДМСО-D6, 400,13 МГц).
На Фиг. 6 представлена микрофотография аморфной формы сунитиниба малата по примеру 1.
На Фиг. 7 представлена микрофотография аморфной формы сунитиниба малата по примеру 2а.
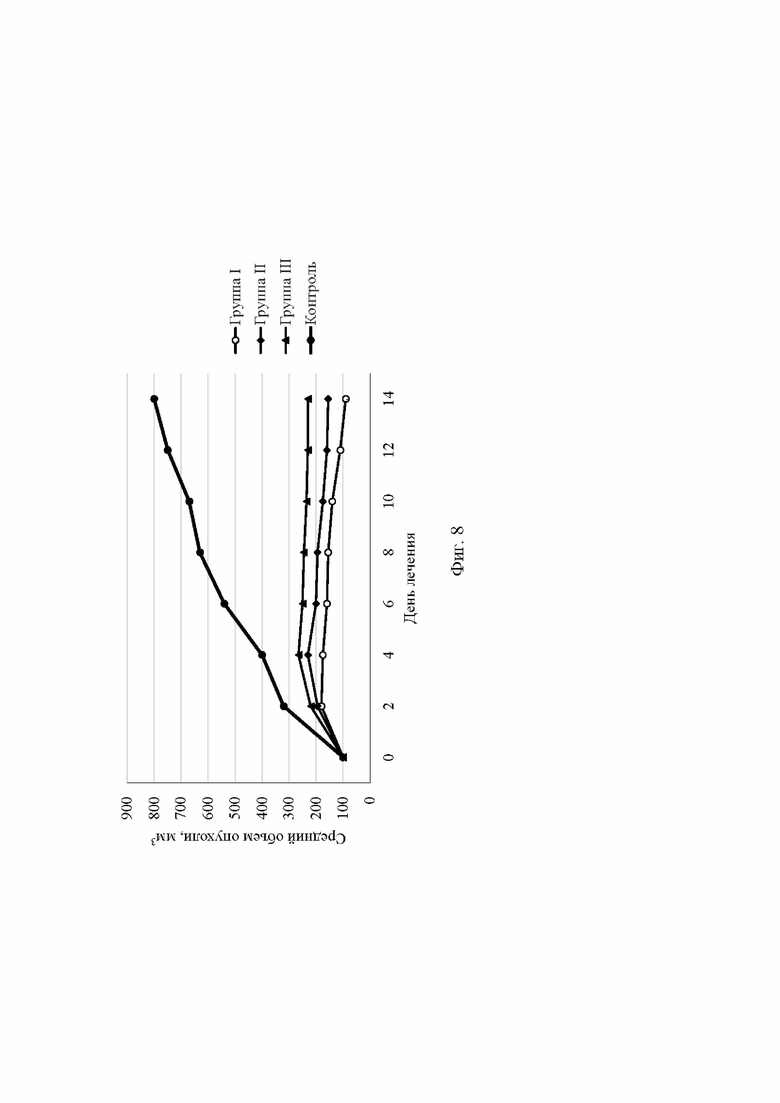
На Фиг. 8 приведен график с результатами изучения фармакотерапевтической активности (группа I - аморфный сунитиниба малат по примеру 1, группа II - аморфный сунитиниба малат по примеру 2а, группа III - аморфный сунитиниба малат по примеру 3, контрольная группа - 30% раствор Captisol).
Осуществление изобретения
Физико-химический анализ сунитиниба малата был осуществлен методом ядерной магнитной спектроскопии 1Н ЯМР, ИК-спектроскопии, ВЭЖХ, ГЖХ и рентгенофазовым анализом. Спектры ЯМР были зарегистрированы в насыщенном растворе в дейтерированном диметилсульфоксиде (ДМСО-D6) на ЯМР-спектрометре высокого разрешения VXR-400 фирмы "VARIANT" (США) на рабочей частоте 400,13 МГц. Инфракрасный спектр субстанции регистрировали по методу многократно нарушенного полного внутреннего отражения (МНПВО) с преобразованием Фурье в области от 4000 до 580 см-1 на приборе Shimadzu IRAffinity-1s. Рентгенофазовый анализ (РФА) проводили на дифрактометре Rigaku D/MAX-2500 (Rigaku, Япония) на CuKα излучении (λ=1,54056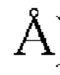 ). Содержание воды анализировалось на автоматическом титраторе C20D, Mettler Toledo (Швейцария). Термоаналитические исследования проводили на приборе NETZSCH DSC 204 F1. Измерительную систему калибровали согласно норме ISO 11357-1 по параметрам фазовых переходов стандартных веществ (С6Н12, Hg, бензойная кислота, Ga, KNO3, In, Sn, Bi, CsCl, чистота 99.99%). Систематическая ошибка температурной калибровки (определена по In) составляет 0,1°. Образцы тестировали в стандартных алюминиевых ячейках (V=56 мм3, d=6 мм), завальцованных крышкой с отверстием (отношение площади дна ячейки к площади отверстия составляло порядка 40) в потоке (40 мл/мин) азота (ВЧ) в интервале температур 20-80°С при скорости нагревания 10°/мин. Экспериментальные данные обрабатывали с помощью пакета анализа NETZSCH Proteus Analysis согласно стандарту, ISO/CD 11358. Микрофотографии получены на сканирующем (растровом) электронном микроскопе "QUANTA 650 FEG" с автоэмиссионным катодом. Условия съемки приведены в подписи к микрофотографии.
). Содержание воды анализировалось на автоматическом титраторе C20D, Mettler Toledo (Швейцария). Термоаналитические исследования проводили на приборе NETZSCH DSC 204 F1. Измерительную систему калибровали согласно норме ISO 11357-1 по параметрам фазовых переходов стандартных веществ (С6Н12, Hg, бензойная кислота, Ga, KNO3, In, Sn, Bi, CsCl, чистота 99.99%). Систематическая ошибка температурной калибровки (определена по In) составляет 0,1°. Образцы тестировали в стандартных алюминиевых ячейках (V=56 мм3, d=6 мм), завальцованных крышкой с отверстием (отношение площади дна ячейки к площади отверстия составляло порядка 40) в потоке (40 мл/мин) азота (ВЧ) в интервале температур 20-80°С при скорости нагревания 10°/мин. Экспериментальные данные обрабатывали с помощью пакета анализа NETZSCH Proteus Analysis согласно стандарту, ISO/CD 11358. Микрофотографии получены на сканирующем (растровом) электронном микроскопе "QUANTA 650 FEG" с автоэмиссионным катодом. Условия съемки приведены в подписи к микрофотографии.
Ввиду высокой светочуствительности, все растворы, содержащие сунитиниба малат, защищают от воздействия света использованием посуды из темного стекла и обертыванием фольгой. Пробоподготовку для исследований методом ВЭЖХ следует проводить в помещении, освещенном осветительными приборами с красным светофильтром.
Возможность осуществления заявленной группы изобретений иллюстрируется следующими примерами, но не ограничивается только ими.
Пример 1. Получение аморфного N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3Н-индол-3-илиден)метил]-2,4-диметил-1Н-пиррол-3-карбоксамид малата.
В трехгорлую круглодонную колбу объемом 1 л, оснащенную механической якорной мешалкой, загрузили 15,00 г кристаллического сунитиниба малата (производство Hyper Chem, Китай), 30,00 г D-фруктозы и 45,00 г мочевины. Смесь нагревали до температуры 60°С при медленном перемешивании, при этом происходило сплавление компонентов. В полученный темного цвета расплав прибавили 0,7 л воды и интенсивно перемешивали в течение 30 мин. Выделившийся осадок сунитиниба малата отфильтровали на стеклянном пористом фильтре Шотта (S4), повторно суспендировали в 0,7 л воды очищенной и перемешивали в течение 1 часа при температуре около 20°С. Осадок отфильтровали на стеклянном пористом фильтре Шотта (S4), промыли на фильтре 3×100 мл воды, и высушили под вакуумом до постоянной массы при температуре не выше 40°С. Получили 14,10 г. Выход 94%. ESI-MS: m/z=400 (М+Н+). Образец полученного вещества полностью рентгеноаморфен по данным порошковой рентгеновской дифракции (Фиг. 1). Отсутствие значимых уровней адсорбционной воды подтверждено кулонометрическим титрованием по К. Фишеру (ГФ XIII, Т. 1, стр. 719).
Пример 2а. Получение аморфного N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3Н-индол-3-илиден)метил]-2,4-диметил-1Н-пиррол-3-карбоксамид малата.
100,00 г 5-[(Z)-5-фтор-1,2-дигидро-2-оксо-3Н-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоновой кислоты в 1,2 л N,N-диметилформамида смешивают с 68,94 г 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида (EDC⋅HCl) и 40,22 г N-гидроксисукцинимида. Реакционную смесь перемешивают в течение 14 часов. Затем прибавляют 40,25 г N,N-диэтил-1,2-этилендиамина. Полученный раствор перемешивают в течение 12 часов и выливают в 3,0 л очищенной охлажденной до 5°С воды. Осадок отфильтровывают на стеклянном пористом фильтре Шотта (S3) и промывают 500 мл 5% р-ром Na2CO3, затем 3×500 мл водой, высушивают на воздухе. Полученное основание сунитиниба переносят в 1,0 л ацетона, нагретого до 40°С, добавляют 50,00 г L-яблочной кислоты, перемешивают в течение 12 часов. Осадок отфильтровывают на стеклянном пористом фильтре Шотта (S3), промывают очищенной водой (3×500 мл) и высушивают под вакуумом до постоянной массы при температуре 40°С.
Аморфный сунитиниба малат получают по методике из примера 1 с использованием полученного осадка вместо готового кристаллического сунитиниба малата и пропорциональным увеличением загрузки всех компонентов. Выход реакции составил 133,00 г (75%). 1Н ЯМР-спектр представлен на Фиг. 5.
Пример 2b. Получение аморфного N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3Н-индол-3-илиден)метил]-2,4-диметил-1Н-пиррол-3-карбоксамид малата.
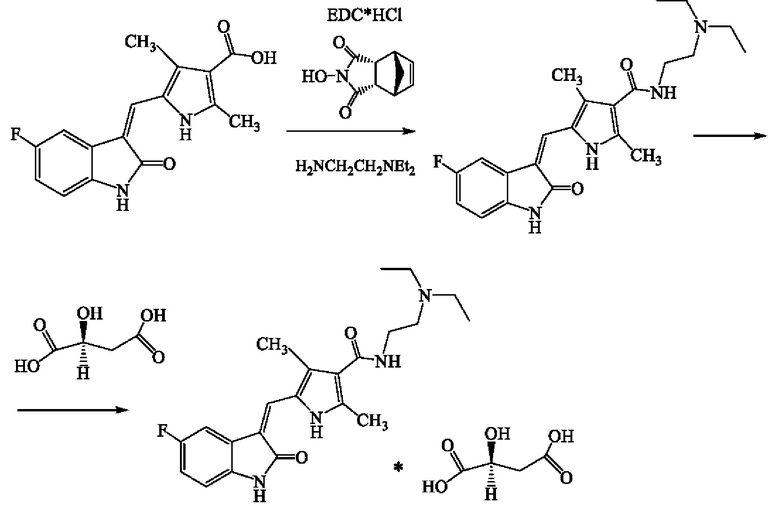
1,00 г 5-[(Z)-5-фтор-l,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты в 20 мл N,N-диметилформамида смешивают с 0,69 г 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида и 0,66 г имида N-гидрокси-5-норборнен-2,3-дикарбоновой кислоты. Реакционную смесь перемешивают в течение 14 часов. Затем прибавляют 0,43 г N,N-диэтил-1,2-этилендиамина. Полученный раствор перемешивают в течение 12 часов и выливают в 50 мл очищенной охлажденной до 5°С воды. Осадок отфильтровывают на стеклянном пористом фильтре Шотта (S3) и промывают 20 мл 5% р-ром Na2CO3, 3×20 мл водой, высушивают на воздухе. Полученное основание сунитиниба переносят в 50 мл ацетона, нагретого до 40°С, добавляют 0,5 г L-яблочной кислоты, перемешивают в течение 12 часов. Осадок отфильтровывают на стеклянном пористом фильтре Шотта (S3), промывают очищенной водой (3×20 мл) и высушивают под вакуумом до постоянной массы при температуре 40°С.
Аморфный сунитиниба малат получают по методике из примера 1 с использованием полученного осадка вместо готового кристаллического сунитиниба малата и пропорциональным уменьшением загрузки всех компонентов. Выход реакции составил 1,27 г (72%).
Пример 3. Получение аморфного сунитиниба малата известным способом (по международной заявке WO 2009156837 A2, пример 2).
1,0 г кристаллического сунитиниба малата растворяют в 10 мл N-метил-2-пирролидона при нагревании. Перемешивание продолжают в течение 24 часов при комнатной температуре. Затем N-метил-2-пирролидон удаляют под вакуумом при температуре 40°С до постоянной массы. Т.пл. 198°С. Рентгенограмма полученного аморфного сунитиниба малата совпадает с рентгенограммой, представленной в патентной заявке WO 2009156837 А2. Выход составил 0,98 г (98%).
Пример 4. Определение остаточных органических растворителей в образцах аморфного сунитиниба малата.
Для образцов аморфного сунитиниба малата по изобретению остаточные органические растворители определяют методом газовой хроматографии в соответствии с ГФ РФ (ОФС.1.1.0008.15 «Остаточные органические растворители», ОФС.1.2.1.2.0004.15 «Газовая хроматография», ОФС.1.2.1.2.0001.15 «Хроматография») на газовом хроматографе с программированием температуры, снабженном пламенно-ионизационным детектором GC-2010 Plus, Shimadzu (Япония) и автоматическим устройством для анализа равновесной паровой фазы типа «Headspace», АОС-5000 Plus, Shimadzu (Швейцария).
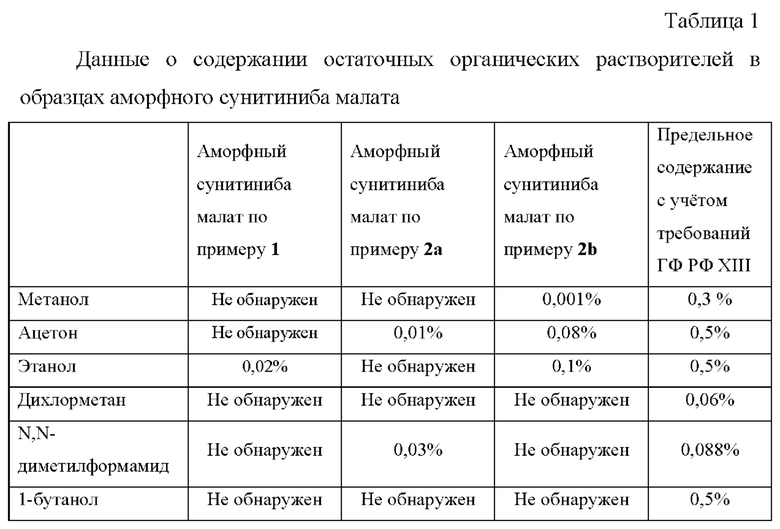
Хроматографические условия:
- капиллярная кварцевая колонка размером 30 м × 0,32 мм, заполненная сорбентом (6%-цианопропилфенил) - 94% диметилполисилоксан, толщина неподвижной фазы 1,8 мкм (типа ZB-624, кат. №: 7HM-G005-31, «Phenomenex», США или аналогичная);
- температура колонки - градиент:
45°С (5 мин) → 20°С/мин → 190°С (3 мин);
- температура инжектора - 200°С;
- детектор - пламенно-ионизационный (ПИД);
- скорость подачи воздуха для ПИД - 450 мл/мин;
- скорость подачи водорода для ПИД - 45 мл/мин;
- температура детектора - 250°С;
- газ-носитель - азот;
- скорость газа-носителя - 1 мл/мин;
- время хроматографирования - 15,25 мин.
Пример 5. Получение готового лекарственного средства в форме твердых желатиновых капсул, содержащих аморфный сунитиниба малат 12,5 мг.
Отвешивают на весах и просеивают в индивидуальные маркированные контейнеры следующие компоненты:
- Сунитиниба малат, полученный по примерам 1, 2а, 2b, 3 - 1550,09±31 г;
- Маннитол (Parteck М 200, производства Merck, Германия) - 7425,60±74,25 г;
- Кроскармеллоза натрия (Solutab А, производства Blanver, Бразилия) - 612,61±6,1 г;
- Повидон (Kollidon 30, производства BasF SE, Германия)- 519,80±5,2 г;
- Магния стеарат (NutriMag STv производства CALMAGS GmbH, Германия) - 102,1±1,0 г.
Для просева стеарата магния используют сита с размером ячеек 0,200±0,0083 мм, для остальных компонентов - 0,400±0,015 мм.
В смеситель - гранулятор загружают аморфный сунитиниба малат, маннитол, кроскармеллозу натрия, повидон и перемешивают массу в течение 10-20 мин со скоростью 16-22 об/мин. Далее загружают магния стеарат и перемешивают 3-5 мин на скорости 16-22 об/мин. Полученную капсульную смесь вручную выгружают из бункера смесителя в промаркированный тарированный контейнер. Капсульную массу фасуют в твердые желатиновые капсулы №4 производства Capsulgel на автоматической капсулонаполняющей машине. Содержимое капсул - порошок желтого цвета массой 12,5 мг ± 5,0% (от 11,88 мг до 13,13 мг).
Пример 6. Исследование стабильности образцов аморфного сунитиниба малата.
Стабильность образцов аморфного сунитиниба малата, полученных по примерам 1, 2а, 2b была подтверждена отсутствием пиков на порошковой дифрактограмме после двух месяцев хранения при температуре 25±2°С и относительной влажности 60±5%. Для образца известной аморфной формы сунитиниба малата по примеру 3 после хранения при тех же условиях на порошковой дифрактограмме наблюдалось появление рефлексов, превышающих уровень шума, на фоне сохраняющегося широкого гало.
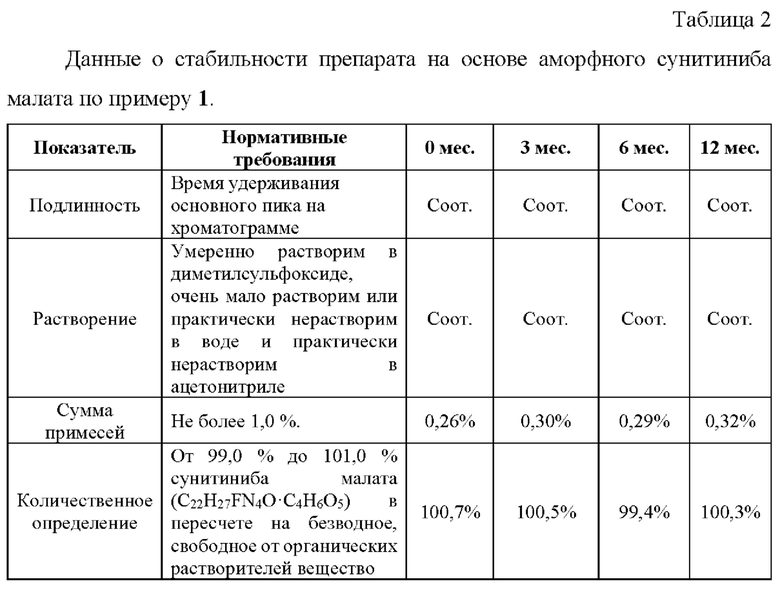
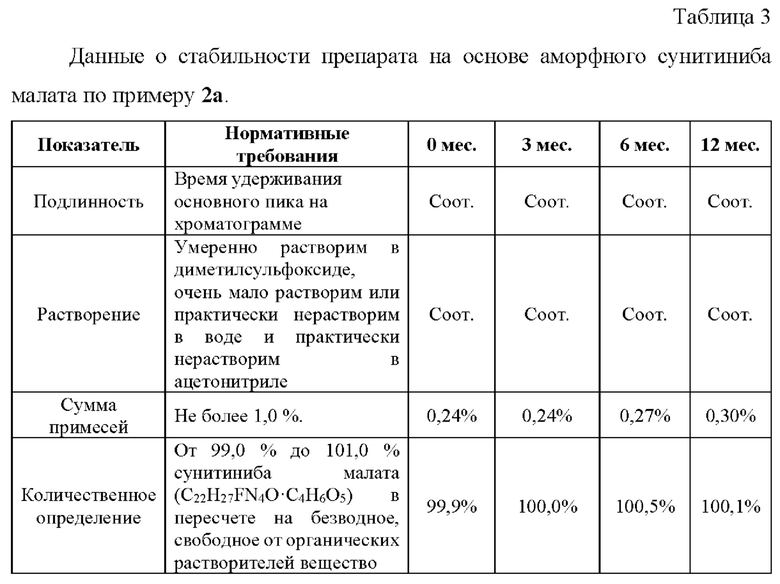
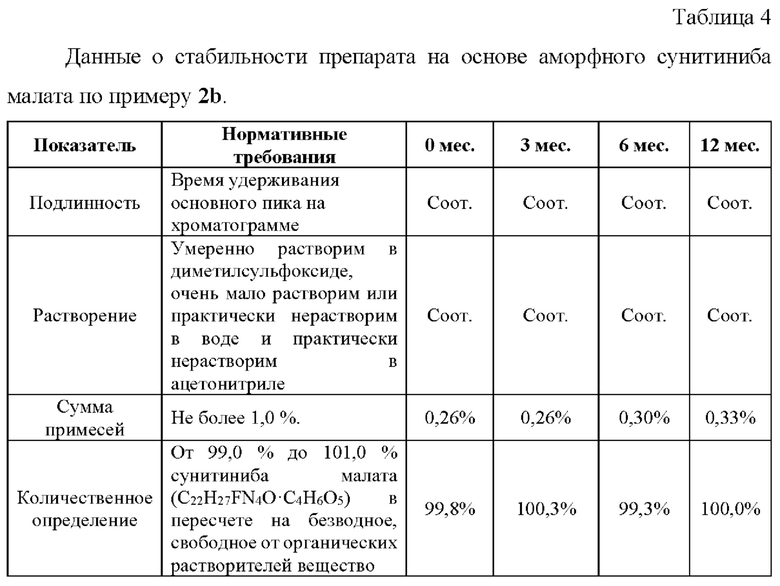
Пример 7. Ускоренные испытания стабильности препаратов, содержащих аморфный сунитиниба малат, 12,5 мг.
Ускоренные испытания стабильности препарата проводили в течение 6 месяцев при температуре 40±2°С и относительной влажности 75±5%. На основании результатов изучения стабильности подтвержден срок годности лекарственного препарата в течение 24 месяцев.
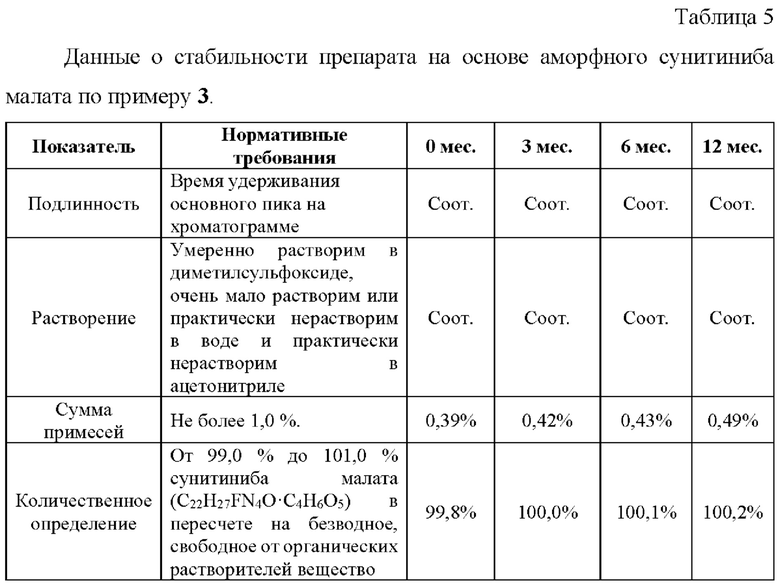
Пример 8. Исследование гигроскопичности образцов аморфного сунитиниба малата.
Оценка гигроскопичности образцов аморфного сунитиниба малата, полученных по примерам 1, 2а, 2b, 3 производилась в соответствии с Европейской Фармакопеей [Characters section in monographs. European Pharmacopoeia 6, version 6.8, Section 5.11 ed.2010] в условиях относительной влажности 80±2% при 25°С в течение 24 ч.
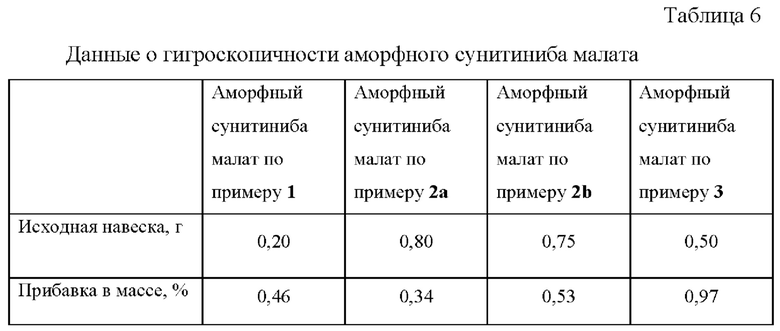
В соответствии с критериями Европейской Фармакопеи - прибавка в массе образца <2% и ≥0,2%, все исследованные образцы сунитиниба малата следует классифицировать как слабо гигроскопичные.
Пример 9а. Исследование фармакокинетики аморфного сунитиниба малата in vivo.
Эксперимент проводился на крысах Спрег-Доули весом 180-200 г. Животные содержались в стандартных лабораторных условиях при температуре 22 - 25°С и влажности 45±5%, имели свободный доступ к еде и воде. Забор крови проводился через сонную артерию.
Животные были разделены на 3 группы по 12 особей в каждой. Первая группа получала разовую дозу аморфного сунитиниба малата, полученного по примеру 1, вторая группа - аморфного сунитиниба малата, полученного по примеру 2а. Третьей группе вводился аморфный сунитиниба малат, полученный по примеру 3.
Субстанция на основе сунитиниба малата в дозировке 10 мг/кг вводилась через желудочный зонд в виде суспензии, содержащей сунитиниба малат, 0,5% карбоксиметилцеллюлозы, 0,9% хлорида натрия, 0,4% полисорбата 80 и 0,9% бензилового спирта. Образцы крови (0,2 мл) отбирались через 5, 15, и 30 мин, и 1, 3, 6, 9 и 24 ч и помещались в капилляры с гепарином. Плазма центрифугировалась при комнатной температуре в течение 1,5 мин при 4200 об/мин и хранилась при температуре -80°С.
Для внутривенного введения использовали водный раствор сунитиниба малата (2,0 мг/мл) в цитратном буфере с рН=3,5, содержащем полисорбат 80, полиэтиленгликоль 300.
При внутривенном введении субстанции сунитиниба малата (дозировка 8 мг/кг) были получены следующие результаты AUCB/B=7430 нгхч/мл, t1/2=2,5 ч.
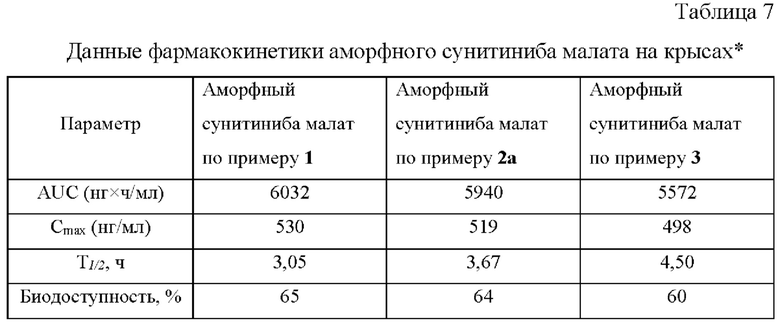
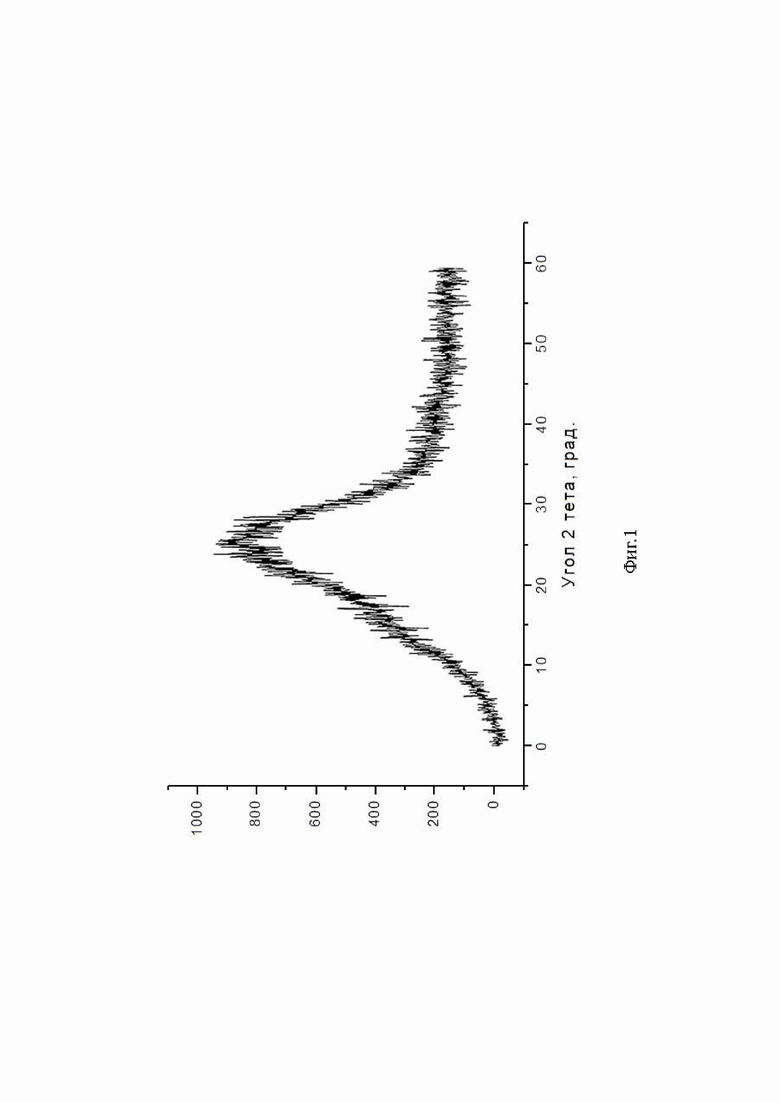
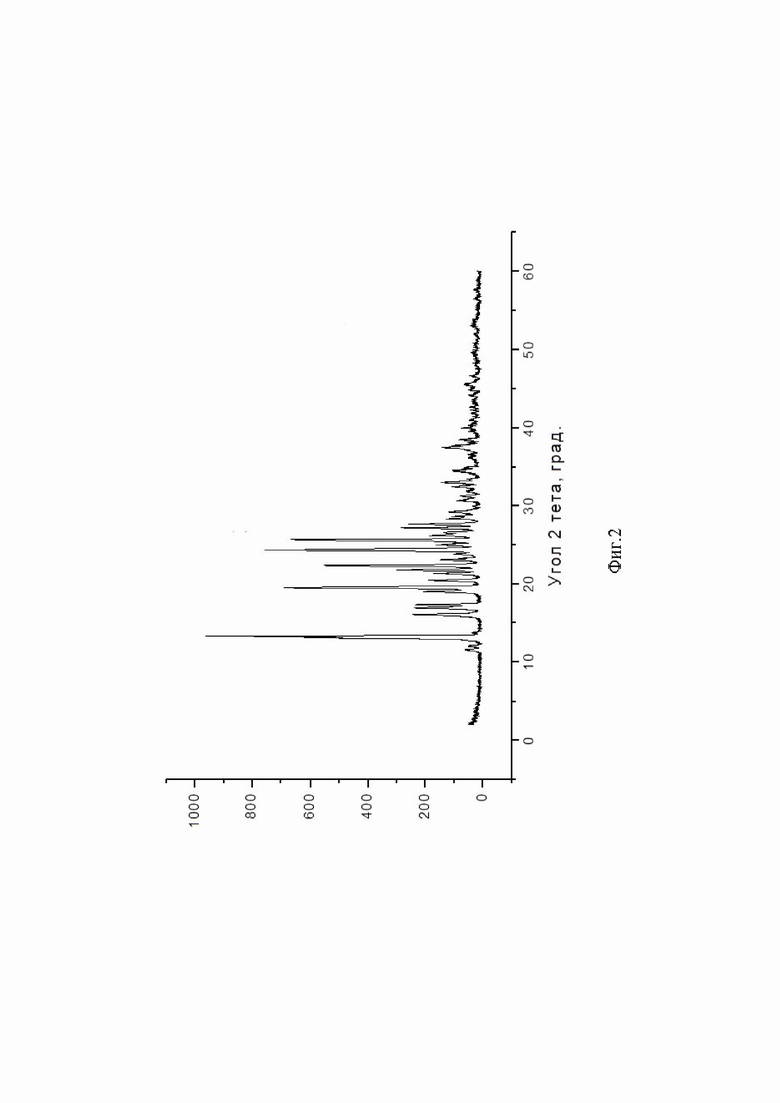
Биодоступность рассчитывали по следующей формуле:
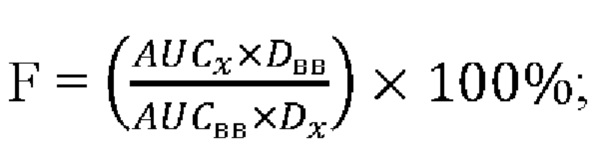
где AUCx - площадь под кривой концентрация - время после перорального введения;
AUCв/в - площадь под кривой концентрация - время после внутривенного введения;
Dx - дозировка препарата для перорального введения;
Dв/в - дозировка препарата для внутривенного введения.
*Различия можно считать статистически достоверными с учетом уровня значимости р≤0,05.
Пример 9b. Исследование фармакокинетики аморфного сунитиниба малата in vivo.
Эксперимент проводился на обезьянах рода Cynomolgus в возрасте 4-6 недель. Животные содержались в стандартных лабораторных условиях при температуре 23 - 25°С, влажность 45±5%. Забор крови проводился из подкожной вены.
Животные были разделены на 3 группы по 12 особей в каждой. Первая группа получала разовую дозу аморфного сунитиниба малата, полученного по примеру 1, вторая группа - аморфного сунитиниба малата, полученного по примеру 2а. Третьей группе вводился аморфный сунитиниба малат, полученный по примеру 3.
Субстанция на основе сунитиниба малата в дозировке 50 мг/кг вводилась через желудочный зонд в виде суспензии, содержащей сунитиниба малат, 0,5% натриевой соли карбоксиметилцеллюлозы, 0,9% хлорида натрия, 0,4% Tween 80 и 0,9% бензилового спирта. Образцы крови (0,4 мл) отбирались через 0,5, 1, 1,5, 2, 3, 6, 9 и 24 ч и помещались в капилляры с гепарином. Плазма центрифугировалась при комнатной температуре в течение 1,5 мин при 4200 об/мин и хранилась при температуре -80°С. Для внутривенного введения использовали водный раствор сунитиниба малата (2,0 мг/мл) в цитратном буфере с рН=3,5, содержащем полисорбат 80, полиэтиленгликоль 300.
При внутривенном введении (дозировка 3 мг/кг) были получены следующие параметры AUCв/в=1488 нгхч/мл, t1/2=7,7 ч.
Биодоступность рассчитывали по следующей формуле:
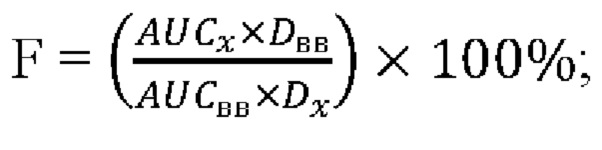
где AUCx - площадь под кривой концентрация - время после перорального введения;
AUCв/в - площадь под кривой концентрация - время после внутривенного введения;
Dx - дозировка препарата для перорального введения;
Dв/в - дозировка препарата для внутривенного введения.
*Различия можно считать статистически достоверными с учетом уровня значимости р≤0,05.
Пример 10. Исследование терапевтической эффективности аморфного сунитиниба малата.
Терапевтическая эффективность сунитиниба малата изучалась на ксенографтной модели почечноклеточной карциномы (HCC-PDX). HCC-PDX модели были созданы из опухолевой ткани методом резекции. Части опухолевой ткани ксенографта (~5×106 клеток) имплантировались в печень мыши с помощью иглы. Лечение сунитинибом малатом начинали примерно через 7 дней, когда объем опухоли достигал 100 мм3. Размер опухоли измерялся раз в два дня, используя штангенциркуль. Объем опухоли рассчитывался, исходя из формулы (L×W2)/2, где L - наибольший диаметр опухоли, W - наименьший диаметр. Мышей разделяли на 5 групп, по 12 особей в каждой. Группа I получала лечение аморфным сунитинибом малатом, полученным по примеру 1, группа II - аморфным сунитинибом малатом, полученным по примеру 2а, группа III - аморфным сунитинибом малатом, полученным по примеру 3. Контрольной группе вводили 200 мкл 30% раствора Captisol.
Суспензию сунитиниба малата вводили через желудочно-кишечный зонд. Сунитиниба малат в дозировке 40 мг/кг растворяли в 30% растворе Captisol.
Для исследования применялись иммунодефицитные мыши-самки, безволосые альбиносы, в возрасте 9-10 недель. Животные содержались при температуре 23°С, имели свободный доступ к еде и воде. При лечении сунитинибом малатом потеря массы тела мышей составила не более 10%.
Наибольший терапевтический эффект наблюдался в группах I и II, где лечение проводилось аморфным сунитинибом малатом, полученным по примерам 1 и 2а (см. Фиг. 8).

Настоящее изобретение относится к способу получения аморфной формы N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида малата. Способ включает следующие стадии: (а) сплавление кристаллического N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида малата, D-фруктозы и мочевины в массовом соотношении 1:2:3, при температуре 60°С при перемешивании; (b) прибавление к полученному расплаву воды; (c) перемешивание полученной смеси; (d) фильтрование осадка; (e) приготовление суспензии осадка в воде; (f) перемешивание полученной суспензии при температуре 20°С; (g) фильтрование осадка; (h) промывание осадка водой на фильтре; (i) высушивание осадка до постоянной массы под вакуумом при температуре 40°С. Технический результат – получение аморфной формы N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида малата, содержащей уменьшенное количество примесей. 1 з.п. ф-лы, 8 ил, 8 табл., 12 пр.
1. Способ получения аморфной формы N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида малата, отличающийся тем, что он включает следующие стадии:
a) сплавление кристаллического N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида малата, D-фруктозы и мочевины в массовом соотношении 1:2:3 при температуре 60°С при перемешивании;
b) прибавление к полученному расплаву воды;
c) перемешивание полученной смеси;
d) фильтрование осадка;
e) приготовление суспензии осадка в воде;
f) перемешивание полученной суспензии при температуре 20°С;
g) фильтрование осадка;
h) промывание осадка водой на фильтре;
i) высушивание осадка до постоянной массы под вакуумом при температуре 40°С.
2. Способ по п.1, отличающийся тем, что перед сплавлением N-[2-(диэтиламино)этил]-5-[(Z)-(5-фтор-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида малата, D-фруктозы и мочевины осуществляют следующие стадии:
a) взаимодействие 5-[(Z)-5-фтор-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоновой кислоты, N-гидроксиимидного соединения, 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида в присутствии N,N-диэтил-1,2-этилендиамина в среде N,N-диметилформамида при перемешивании;
b) смешение реакционной массы с водой;
c) фильтрование осадка;
d) промывание осадка раствором бикарбоната натрия и водой;
e) высушивание полученного осадка;
f) растворение осадка в ацетоне, нагретом до 40°С;
g) прибавление к полученному раствору L-яблочной кислоты;
h) перемешивание полученной смеси;
i) фильтрование осадка;
j) промывание осадка водой;
k) высушивание осадка под вакуумом до постоянной массы при температуре 40°С.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| CN 109503555 A, 22.03.2019 | |||
| Вентиль-отвод для сгущающихся жидкостей | 1927 |
|
SU5996A1 |
| Sun, L., Liang, C., Shirazian, S., Zhou, Y., Miller, T., Cui, J., et al.: " Discovery of 5-[5-Fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic Acid (2-Diethylaminoethyl)amide, a Novel Tyrosine | |||

