[0001] Данная заявка испрашивает приоритет по предварительной заявке США 62/550137, поданной 25 августа 2017 года, содержание которой включено в данный документ в полном объеме посредством ссылки.
[0002] Все патенты, патентные заявки и публикации, процитированные в данном документе, включены в данный документ в полном объеме с помощью ссылки. Раскрытие данных публикаций во всей их полноте включено в данную заявку посредством ссылки для более полного описания данного уровня техники, известного специалистам в данной области техники на дату изобретения, описанного и заявленного в данном документе.
[0003] Данное раскрытие сущности изобретения содержит данные, которые подлежат защите авторских прав. Владелец авторских прав не возражает против факсимильного воспроизведения кем-либо патентного документа или раскрытия сущности изобретения, как это указано в патенте США или записях патентного ведомства США по патентам и товарным знакам, но в остальном сохраняет за собой любые и все авторские права.
ГОСУДАРСТВЕННЫЕ КАПИТАЛОВЛОЖЕНИЯ
[0004] Данное изобретение было выполнено при государственной поддержке в рамках гранта №Р30 GM103340, присужденного Национальным институтом здравоохранения. Таким образом правительство имеет определенные права на данное изобретение.
ОБЛАСТЬ ТЕХНИКИ
[0005] В данном изобретении предложены композиции, способы и наборы, которые могут быть использованы для лечения или уменьшения боли.
УРОВЕНЬ ТЕХНИКИ
[0006] Для многих типов боли (например, общая головная боль, остеоартрит) ацетаминофен (АрАР, N-ацетил-пара-аминофенол) обладает такой же активностью и эффективностью, что и ацетилсалициловая кислота (аспирин). Тем не менее с точки зрения безопасности АрАР имеет риск, особенно для пациента с нарушением функции печени. Передозировка (непреднамеренная или намеренная) или использовании у пациентов с нарушением функции печени является наиболее распространенной причиной фульминантной печеночной недостаточности в странах западного мира (Bernal, William, et al. "Acute Liver failure." The Lancet 376.9736 (2010): 190-201). У данных пациентов острая фульминантная печеночная недостаточность представляет собой быстрое развитие печеночной дисфункции, переходящей в энцефалопатию, коагулопатию и прогрессирующую полиорганную недостаточность.
[0007] Передозировка АрАР является основной причиной звонков в Токсикологические центры по всей территории США с более чем 100 000 ежегодных вызовов и является основной причиной более чем 56 000 посещений отделения неотложной помощи, и 2600 госпитализаций ежегодно, что привело к приблизительно 458 смертельным случаям из-за острой печеночной недостаточности в 2014 году (Mercola, FDA Finally Changes Prescription Recommendations for High-Dose ApAP, 2014).
[0008] Считается, что токсичность АрАР опосредуется токсичным метаболитом N-ацетилбензохинонимином (NAPQI), который истощает печеночный и почечный глутатион, цитопротективный эндогенный метаболит (Mason, R.P. и V. Fischer). Federation proceedings. Vol. 45. No. 10. 1986.; Mitchell et al., 1983). Гепатотоксичность с АрАР может возникать в дозах, только в 4-8 раз превышающих максимальную рекомендованную обезболивающую дозу (Neuberger et al., 1980); нефротоксичность редко наблюдается в клинических случаях. Фармацевтические комбинации, которые содержат АрАР и обезболивающее соединение центрального действия, могут быть даже более опасными, чем отдельно АрАР. При повторном использовании данные комбинации требуют более высоких доз для получения того же обезболивающего эффекта из-за увеличения толерантности. По мере того как доза комбинации увеличивается, чтобы компенсировать обезболивающего толерантность, безопасность препарата снижается, так как более высокие дозы компонента АрАР увеличивают гепатотоксичность.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0009] В данном изобретении предлагаются обезболивающие соединения для лечения боли.
[0010] В некоторых вариантах реализации обезболивающее соединение включает соединение формулы (I):
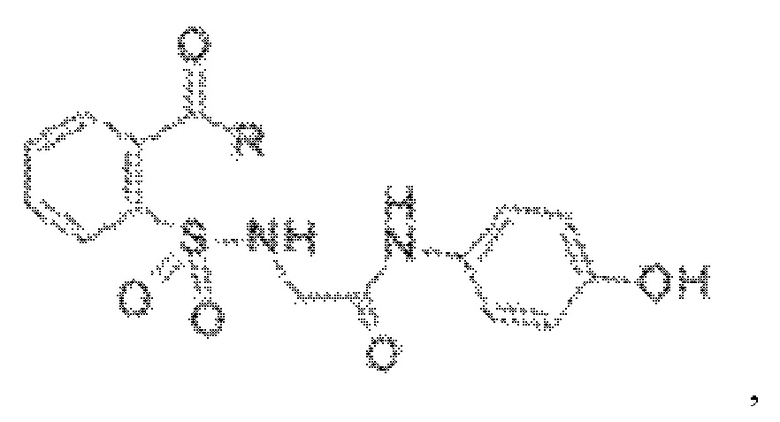
где R включает NH2, N(CH3)2, NHCH3, N(CH2CH3)2, N2(CH2)4CH2C6H5, NH(CH2)2C6H5, NHCH2C6H5, NO(CH2)4, NHCH2CH2CH2CH3, NHCH2C6H4CH3, NHCH2C6H3C12, NHCH2C6H5CH3, NHCH2C6H5C1, NHCH2C6H5NO2, NHC5H4, NHCH2C(CH3)2, NHC(CH3)2, NHCH2CH2C6H3(OH)2, NHCH2C6H4N, NHCH2C6H3NCH3, NHCH2CH2C4H4N, N(CH3)CH2CH2OH, NHCH2CH(OH)CH2NH2; или его фармацевтически приемлемую соль.
[0011] В некоторых вариантах реализации обезболивающее соединение включает соединение формулы (II):
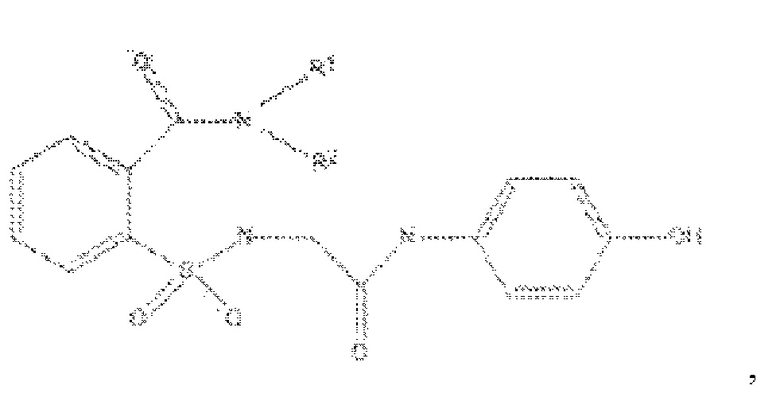
где R1 представляет собой Н, ОН, алкильную группу, галогеналкильную группу, галогенбензильную группу, фенильную группу, -О-(алкил), -О-(галогеналкил), -О-(галогенбензил), -О-(фенил), алкилфенил, галогеналкилфенил, алкилгалогенбензол, алкилнитробензол, -О-(алкилфенил), -O-(галогеналкил)фенил, циклоалкановую группу и где R выбран из группы, состоящей из Н и алкильной группы; или его фармацевтически приемлемую соль. В некоторых вариантах реализации R1 включает Н, СН3, (СН2)2С6Н5, СН2С6Н5, СН2СН2СН2СН3, CH2C6H3Cl2, СН2С6Н5СН3, CH2C6H5Cl, CH2C5H5NO2, С5Н4, СН2С(СН3)2, С(СН3)2, СН2СН2С6Н3(ОН)2, CH2C6H4N, CH2C6H3NCH3, CH2CH2C4H4N, СН2СН2ОН или CH2CH(OH)CH2NH2; и где R2 выбран из группы, состоящей из Н и СН3, или его фармацевтически приемлемую соль.
[0012] В некоторых вариантах реализации алкил может включать CNH2N-1, например, где N равно 1-10.
[0013] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
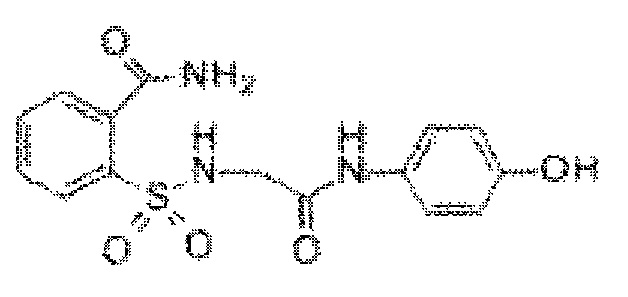
[0014] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:

[0015] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
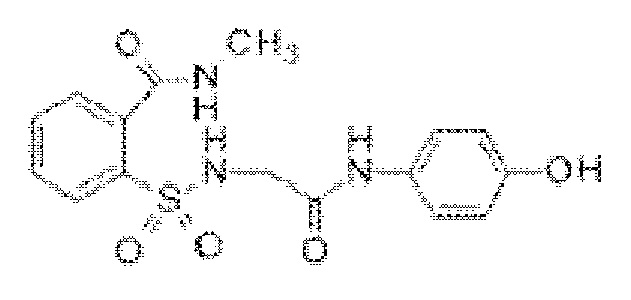
[0016] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
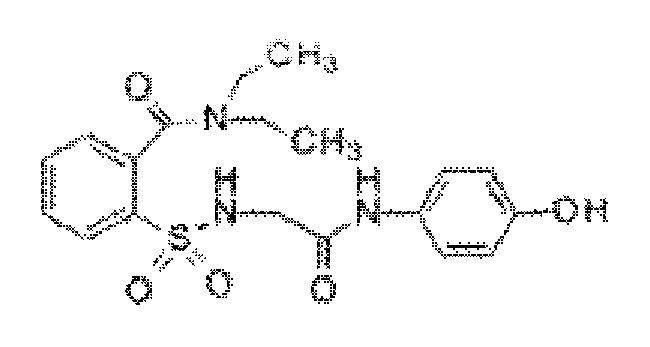
[0017] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
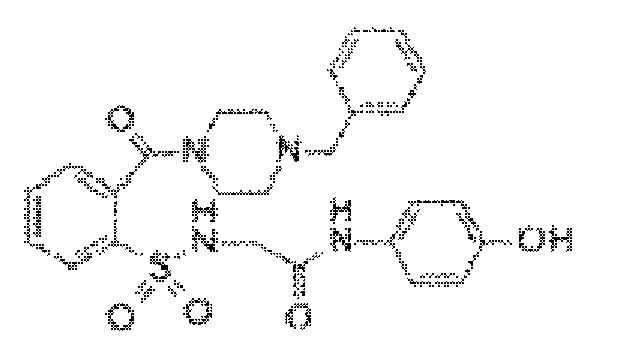
[0018] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
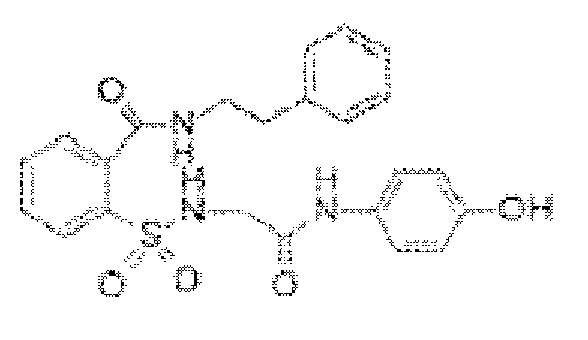
[0019] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
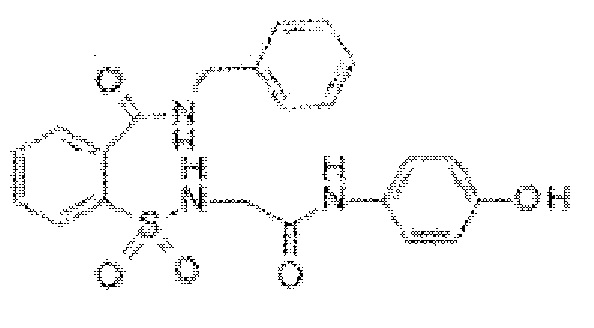
[0020] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
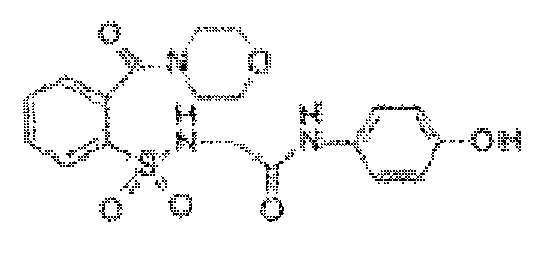
[0021] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
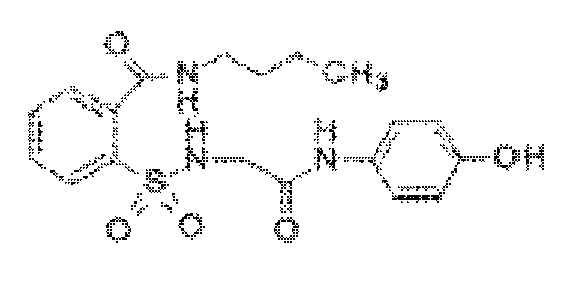
[0022] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
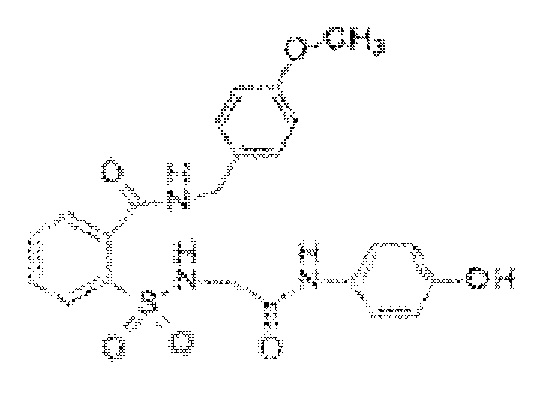
[0023] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
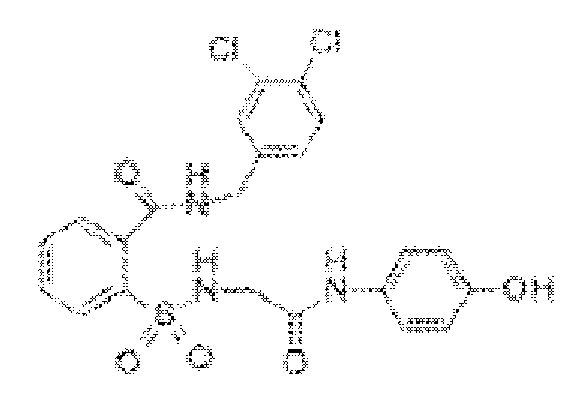
[0024] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
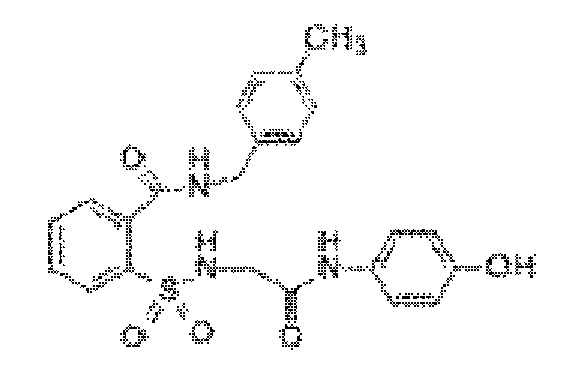
[0025] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
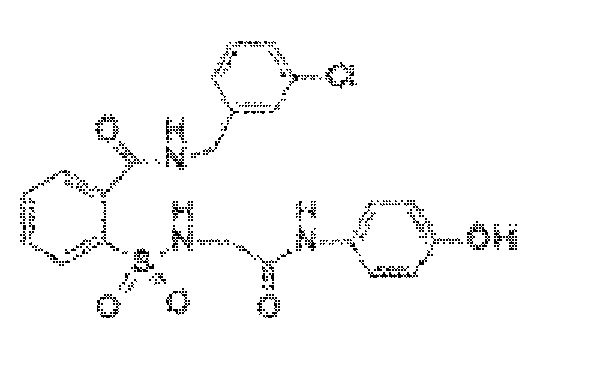
[0026] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
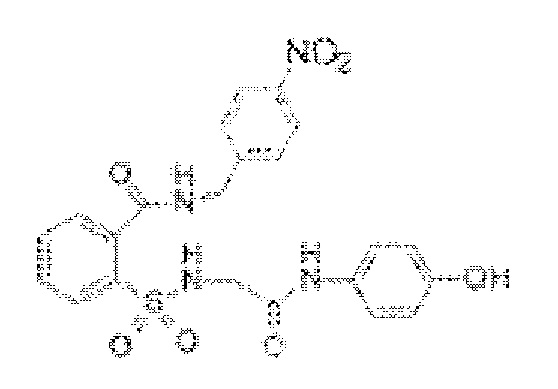
[0027] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
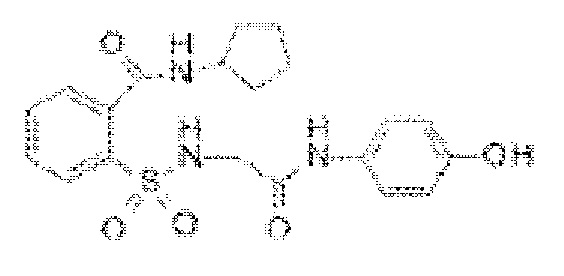
[0028] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
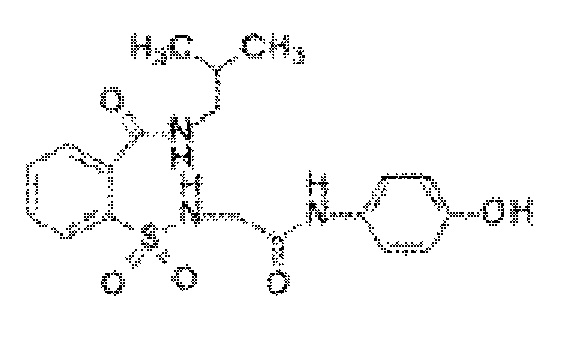
[0029] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
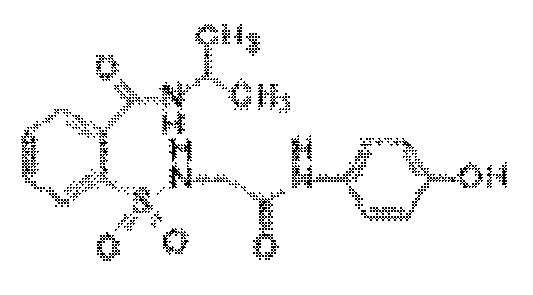
[0030] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
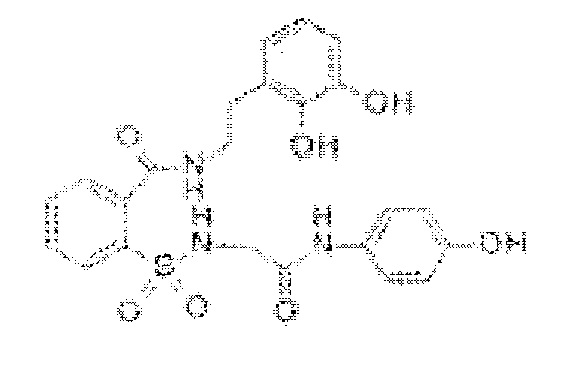
[0031] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
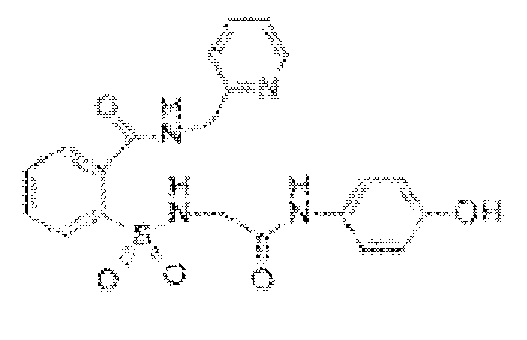
[0032] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
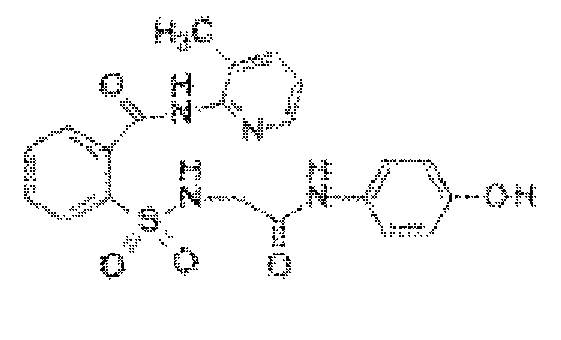
[0033] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
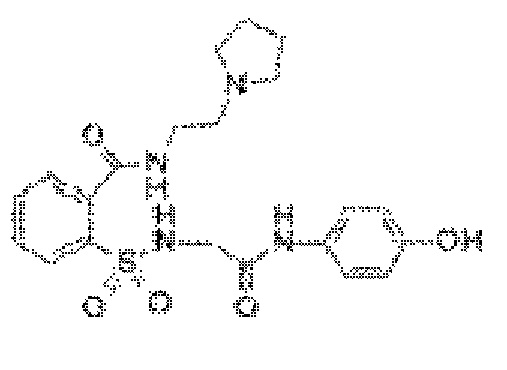
[0034] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
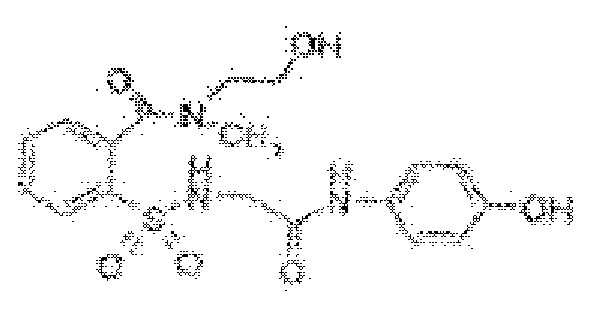
[0035] В некоторых вариантах реализации обезболивающее соединение имеет следующую химическую структуру:
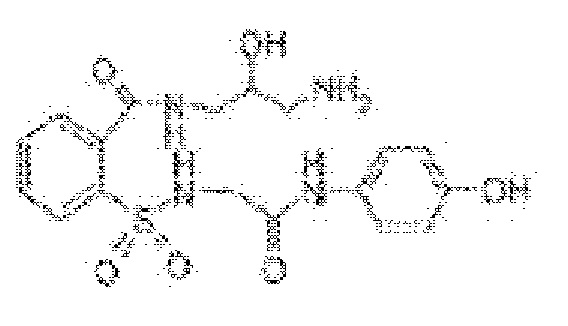
[0036] В некоторых вариантах реализации обезболивающее соединение имеет пониженный риск гепатотоксичности при введении субъекту in vivo. Например, композиция может снижать риск гепатотоксичности по меньшей мере на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45% или 50%.
[0037] В некоторых вариантах реализации обезболивающее соединение проявляет обезболивающий эффект, сравнимый с АрАР, при введении субъекту in vivo.
[0038] В некоторых вариантах реализации обезболивающее соединение представляет собой ненаркотическое обезболивающее средство.
[0039] В некоторых вариантах реализации обезболивающее соединение проявляет жаропонижающий эффект.
[0040] В некоторых вариантах реализации композиция не метаболизируется до NAPQI.
[0041] В некоторых вариантах реализации обезболивающее соединение имеет пониженный риск гепатотоксичности, проявляет обезболивающий эффект, сравнимый с АрАР, является ненаркотическим, проявляет жаропонижающий эффект и не метаболизируется до NAPQI при введении субъекту in vivo.
[0042] В данном изобретении дополнительно предлагаются фармацевтические композиции, содержащие обезболивающее соединение, как описано в данном документе, и второй активный ингредиент, такой как опиоид или нестероидное противовоспалительное лекарственное средство (НСПВ). Неограничивающие примеры таких опиоидов включают кодеин, фентанил, гидрокодон, гидрокодон/АрАР, гидроморфон, меперидин, метадон, морфин, оксикодон, оксикодон и АрАР, оксикодон и налоксон. Неограничивающие примеры НСПВ включают аспирин, целекоксиб, диклофенак, дифлунисал, этодолак, ибупрофен, индометацин, кетопрофен, кеторолак, набуметон, напроксен, оксапроз, пироксикам, салсалат, сулиндак и толметин.
[0043] В данном изобретении также предложен способ лечения боли у субъекта.
[0044] В данном изобретении дополнительно предложен способ облегчения боли у субъекта.
[0045] Кроме того, в данном изобретении предложен способ предотвращения боли у субъекта, уменьшения частоты возникновения боли у субъекта, задержки развития боли у субъекта, предотвращения развития боли у субъекта и/или временного облегчения боли у субъекта.
[0046] Неограничивающие примеры такой боли включают острую боль, хроническую боль, невропатическую боль, ноцицептивную боль, послеоперационную боль, боль в глазах, зубную боль и/или боль у животных. В некоторых вариантах реализации невропатическая боль включает послеоперационную боль, невропатическую боль, зубную боль, глазную боль, боль при артрите, пост- и/или травматическую боль, или их комбинацию.
[0047] В некоторых вариантах реализации способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества обезболивающего соединения или композиции, как описано в данном документе. Например, терапевтически эффективное количество обезболивающего соединения или композиции, вводимых субъекту, может включать дозу от около 10 мкМ до около 10 мМ или дозу от около 50 мкМ до около 1 мМ.
[0048] В некоторых вариантах реализации обезболивающее соединение или композицию вводят субъекту в однократной дозе, такой как болюс. В других вариантах реализации соединение вводят с интервалами около 4 ч, 12 ч или 24 ч. В еще других вариантах реализации соединение вводят непрерывно, например, с помощью капельной в/в инфузии.
[0049] В некоторых вариантах реализации композиция может быть введена перорально, например, в виде пилюли, таблетки, водного раствора или капсулы; парентерально, например, в виде внутривенной или внутримышечной инъекции; трансдермально, например, в виде крема, лосьона или пластыря; или назально, например, в виде спрея. В других вариантах реализации композиция может быть введена подкожно, внутрилегочно, местно, интравитреально, трансмукозально, ректально и интраназально.
[0050] В некоторых вариантах реализации композиция или обезболивающее соединение, как описано в данном документе, может быть введена субъекту вместе с терапевтически эффективным количеством второго активного ингредиента, такого как опиоид и/или НСПВ. Второй активный ингредиент может быть введен до, одновременно с или после введения композиции или обезболивающего соединения, как описано в данном документе.
[0051] В данном изобретении также предложен медицинский набор для лечения боли. В вариантах реализации набор содержит напечатанные инструкции для введения соединения субъекту, страдающему от боли, и обезболивающее соединение или композиции, как описано в данном документе.
[0052] Другие цели и преимущества данного изобретения станут очевидными из последующего описания.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0053] На Фиг. 1 продемонстрировано обезболивающее действие новых соединений SRP6D, R сравнима с АрАР в мышиной модели in vivo с использованием двух анализов боли. А) Тест корчей, вызванный абдоминальным введением уксусной кислоты. Количество растяжений (корчей) брюшной полости, вызванных инъекцией уксусной кислоты, n=7, р<0,05. SRP6D (n=5, р=0,022) и С) SRP6R (n=5, р=0,008) показывают уровень обезболивания по сравнению только с контролем/носителем. В) Тест отдергивания хвоста. Процент максимального обезболивания для каждой мыши рассчитывали по формуле:
Процент максимального обезболивания для каждой мыши рассчитывали по формуле. Процентное обезболивание = 100*{[(Латентность отдергивания хвоста после инъекции лекарственного средства)-(Латентность отдергивания хвоста базовой линии)]:[(12 сек Время отключения)-(Базовая латентность)]}. Данные выражены как среднее ± СОИ, n=10.
[0054] На Фиг. 2 представлены анализы гепатотоксичности первичных гепатоцитов человека (hHEP), которые показывают пониженную токсичность для новых соединений SRP6D, R, по сравнению с АрАР и производными сахарина АрАР первого поколения, SCP-1 и SCP-1M. Например, испытанными дозами были 500 мкМ АрАР, SCP1, SCP1M, SRP6D и SRP6R.
[0055] На Фиг. 3 представлены анализы гепатотоксичности первичных гепатоцитов человека (hHEP), выявляющие сниженную токсичность для новых соединений SRP6D и R по сравнению с АрАР и производными сахарина АрАР первого поколения, SCP-1 и SCP-1М. А) Высвобождение лактатдегидрогеназы (LDH) увеличивается, и В) Количество восстановленного глутатиона (GSR) уменьшается в hHEP в зависимости от времени и дозы для АрАР, но не для SRP6D и R. Дозы, протестированные в (А) и (В), представляли собой 500 мкМ (5 мМ) и 1000 мкМ (1 мМ), С) для SRP6D и R отмечается заметное снижение в пробах функции печени по сравнению с АрАР, наибольшим из которых является ALT. Испытанная дозировка составляла 600 мг/кг.
[0056] На Фиг. 4 представлен жаропонижающий эффект соединений SRP. Графики изменения температуры демонстрируют сопоставимый жаропонижающий эффект с АрАР для A) SRP6D и В) SRP6R на модели LPS-индуцированной лихорадки у мышей. Отмечено, что через 2, 8 и 10 ч жаропонижающий эффект похож на АрАР, SRP6D и SRP6R. Вызванная дрожжами искусственная лихорадка демонстрирует аналогичные жаропонижающие эффекты АрАР, С) SRP6D и D) SRP6R, n=10 на группу. Е) Пирогенная доза пекарских дрожжей (15% дрожжей 0,1 мл/10 г массы тела; контрольная группа получала внутрибрюшинную инъекцию носителя (0,9% физиологический раствор), внутрибрюшинно и через 4 ч регистрировали температуру, после чего лекарственные средства вводили перорально животным с лихорадкой, принадлежащим к группам лечения - (соединения АрАР и SRP при 300 мг/кг массы тела). Через два часа после инъекции по ректальной температуре определяли общее изменение температуры тела. Данные представлены как среднее ± СОИ, n=10.
[0057] На Фиг. 5 представлен эффективный метаболизм цитохромом Р450 для SRP6D и SRP6R в различных изоферментах Р450. Красная хеш-метка демонстрирует эффект АрАР, а зеленая хеш-метка обозначает первое поколение, производное сахарина АрАР. Верхние панели представляют активность ферментов в относительных единицах флуоресценции, а нижние панели представляют процентную активность ферментов.
[6058] На Фиг. 6 представлены стандартные кривые для проб функции печени (LFT) для соединений SRP
[0059] На Фиг. 7 представлены анализы функции печени. (АЛТ, ACT и ALP) проводили после введения мышам-самцам CDI 600 мг/кг соединений - соединений SRP и АРАР посредством п/о (перорально) (кормление через желудочный зонд). Проводили анализы сыворотки, собранной у мышей, которым инъецировали соединения или носитель, в течение ночи (15 ч). После введения лекарственного средства мышам ad libitum давали воду и пищу. Результаты показали повышенные уровни активности ферментов печени для АРАР, тогда как другие соединения были аналогичны носителю, уровни LFT для соединений SRP не достигали уровней для АРАР.
[0060] На Фиг. 8 представлены химические варианты реализации данного изобретения.
[0061] На Фиг. 9 представлен функциональный анализ для определения уровней креатинина в крови после введения мышам-самцам CD1 600 мг/кг (масса тела) соединений - соединений SRP и АРАР посредством п/о (перорально) (кормление через желудочный зонд). Проводили анализы сыворотки, собранной у мышей, которым инъецировали соединения или носитель, в течение ночи (15 ч). После введения лекарственного средства мышам давали воду и пищу ad libitum. Результаты показали повышенный уровень креатинина для АРАР, тогда как для SRP6D эффект был схож с таковым для носителя. Уровни LFT для других соединений SRP не достигли уровней LFT для АРАР.
[0062] На Фиг. 10 продемострирована гистограмма, показывающая распределение рассчитанных значений logP более 3000 лекарственных средств, представленных на рынке.
[0063] На Фиг. 11 представлена столбчатая диаграмма, показывающая результаты газовой хроматографии, использованной для обнаружения токсичного метаболита N-ацетилбензохинонимина (NAPQI). Пик при 5,738.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0064] Сокращения и определения
[0065] Подробное описание одного или более вариантов реализации данного изобретения предоставлено в данном документе. Однако следует понимать, что данное изобретение может быть воплощено в различных формах. Следовательно, конкретные подробности, раскрытые в данном документе, должны интерпретироваться не как ограничивающие, а скорее, как основание для формулы изобретения и как репрезентативная основа для обучения специалиста в данной области техники использованию данного изобретения любым подходящим способом.
[0066] Формы единственного числа включают множественное число, если контекст явно не предписывает иное. Использование термина в единственном числе при использовании в сочетании с термином «содержащий» в формуле изобретения и/или описании может означать «один», но также согласуется со значением «один или более», «по меньшей мере один» и «один или более чем один».
[0067] В каком бы месте не использовались какие-либо из фраз «например», «такие как», «включая» и тому подобное, в этом месте следует подразумевать фразу «и без ограничения», если явно не указано иное. Аналогично, «пример», «примерный» и т.п. понимаются как не ограничивающие.
[0068] Термин «по существу» допускает отклонения от характеристики, которые не оказывают негативного влияния на намеченную цель. Описательные термины понимаются как измененные термином «по существу», даже если слово «по существу» не указано явно.
[0069] Термины «содержащий» и «включающий» и «имеющий» (и аналогично «содержит», «включает», «имеет») и тому подобное используются взаимозаменяемо и имеют одинаковый смысл. В частности, каждый из терминов определен в соответствии с общим определением в патентном праве США «содержащий» и поэтому интерпретируется как открытый термин, означающий «по меньшей мере следующее», а также интерпретируется, не исключая дополнительные признаки, ограничения, аспекты и т.д. Таким образом, например, «способ, включающий стадии a, b и с», означает, что способ включает по меньшей мере стадии a, b и с. Везде, где используются термины в единственном числе, подразумевается «один или более», если только такая интерпретация не бессмысленна по контексту.
[0070] Термин «около», в контексте данного документа, может относиться к приблизительно, примерно, вокруг или в области. Когда термин «около» используется в сочетании с числовым диапазоном, он изменяет этот диапазон, расширяя границы выше и ниже указанных числовых значений. В общем, термин «около» используется в данном документе для изменения числового значения выше и ниже заявленного значения на 20 процентов вверх или вниз (выше или ниже).
[0071] Обезболивающие соединения
[0072] Аспекты данного изобретения направлены на обезболивающие соединения и композиции, содержащие обезболивающие соединения. Термин «обезболивающее соединение» или «обезболивание может относиться к агенту, который уменьшает, облегчает, снижает, избавляет или полностью удаляет боль в области тела субъекта.
[0073] Кроме того, аспекты данного изобретения направлены на соединения и/или композиции, которые проявляют жаропонижающий эффект. Например, обезболивающие соединения, как описано в данном документе, могут считаться «жаропонижающими» или «жаропонижающими соединениями», которые могут относиться к соединению или композиции, которая обладает способностью снижать температуру тела субъекта, например, до физиологически нормальных уровней, когда субъект имеет аномально высокую температуру тела (например, лихорадка). Жаропонижающие соединения, например, описанные в данном документе, также могут блокировать начало лихорадки.
[0074] Варианты реализации данного изобретения демонстрируют обезболивающие и/или жаропонижающие свойства при отсутствии или снижении уровней гепатотоксичности. Термин «гепатотоксичность» может относиться к химическому или медикаментозно индуцированному поражению печени. Медикаментозное поражение или повреждение печени является причиной острого или хронического заболевания печени. Гепатотоксичность может быть вызвана определенными лекарственными средствами при приеме в передозировке или иногда даже при введении в терапевтических пределах.
[0075] АрАР обычно хорошо переносится в предписанной дозе, но передозировка является частой причиной медикаментозно индуцированных заболеваний печени и острой печеночной недостаточности. Повреждение печени происходит не из-за самого препарата, а из-за токсичного метаболита (N-ацетил-р-бензохинонимин (NAPQI)), который вырабатывается ферментами цитохрома Р-450 в печени. В нормальных условиях, этот метаболит детоксифицируется путем конъюгирования с глутатионом во 2 фазе реакции. При передозировке образуется большое количество NAPQI, которое подавляет процесс детоксикации и приводит к повреждению клеток печени. Оксид азота также играет роль в индукции токсичности. На риск повреждения печени влияют несколько факторов, таких как принимаемая доза, одновременный прием алкоголя или других лекарственных средств и/или интервал между пероральным приемом препарата и антидота. Доза, токсичная для печени, варьируется и зависит от конкретного человека, и часто считается ниже у хронических алкоголиков. Измерение показателей крови имеет важное значение при оценке прогноза, при этом более высокие уровни показателей прогнозируют неутешительный прогноз. Те, у кого острая печеночная недостаточность только развивается, все еще могут восстановиться спонтанно, но также может потребоваться трансплантация, если присутствуют плохие прогностические признаки, такие как энцефалопатия или коагулопатия.
[0076] В некоторых вариантах реализации обезболивающее соединение или композиция, содержащая его, имеет пониженный риск гепатотоксичности, например, по сравнению с АрАР, при введении субъекту in vivo. Например, композиция может снизить риск гепатотоксичности по меньшей мере на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45% или 50%.
[0077] В некоторых вариантах реализации обезболивающее соединение имеет формулу (I):
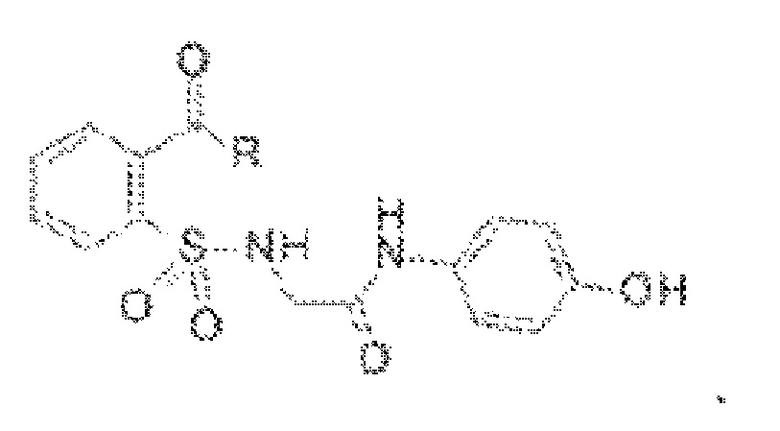
[0078] В других вариантах реализации R включает NH2, N(CH3)2, NHCH3, N(СН2СН3)2, N2(CH2)4CH2C6H5, NH(CH2)2C6H5, NHCH2C6H5, NO(CH2)4, NHCH2CH2CH2CH3, NHCH2C6H4CH3, NHCH2C6H3C12, NHCH2C6H5CH3, NHCH2C6H5C1, NHCH2C6H5NO2, NHC5H4, NHCH2C(CH3)2, NHC(CH3)2, NHCH2CH2C6H3(OH)2, NHCH2C6H4N, NHCH2C6H3NCH3, NHCH2CH2C4H4N, N(CH3)CH2CH2OH, NHCH2CH(OH)CH2NH2; или его фармацевтически приемлемую соль.
[0079] В некоторых вариантах реализации обезболивающее соединение имеет формулу (II);
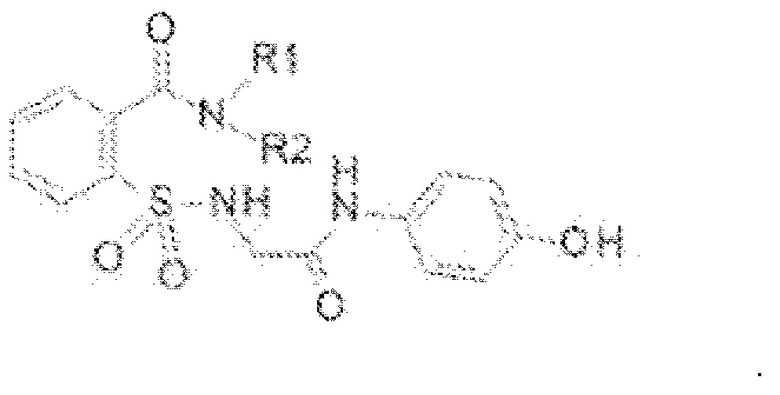
[0080] В некоторых вариантах реализации R1 может представлять собой Н, ОН, алкильную группу, галогеновую группу, галогеналкильную группу, галогенбензильную группу, фенильную группу, -О-(алкил), -О-(галогеналкил), -О-(фенил), -О-(галогенбензил), алкилфенил, галогеналкилфенил, алкилгалогенбензол, алкилнитробензол, -О-(алкилфенил), -O-(галогеналкил)фенил, циклоалкановую группу. В дополнительных вариантах реализации алкильная группа включает углеродные цепи С1-С2, углеродные цепи С1-С3, углеродные цепи С1-С4, углеродные цепи С1-С5, углеродные цепи С1-С6, углеродные цепи С1-С7, углеродные цепи С1-С8, углеродные цепи С1-С9, углеродные цепи С1-С10. В других вариантах реализации алкильная группа может представлять собой С1-С4 углеродные цепи. В некоторых вариантах реализации галогеном может быть F, Br, Cl. В некоторых вариантах реализации циклоалкановая группа может представлять собой 5-членное кольцо. В некоторых вариантах реализации циклоалкановая группа может представлять собой 6-членное кольцо. В некоторых вариантах реализации обезболивающее соединение, описанное в данном документе, представляет собой фармацевтически приемлемую соль.
[0081] В еще других вариантах реализации R2 может представлять собой Н, ОН, алкильную группу, галогеновую группу, галогеналкильную группу, галогенбензильную группу, фенильную группу, -О-(алкил), -О-(галогеналкил), -О-(фенил), -О-(галогенбензил), алкилфенил, галогеналкилфенил, алкилгалогенбензол, алкилнитробензол, -О-(алкилфенил), -O-(галогеналкил)-фенил, циклоалкановую группу. В дополнительных вариантах реализации алкильная группа включает углеродные цепи С1-С2, углеродные цепи С1-С3, углеродные цепи С1-С4, углеродные цепи С1-С5, углеродные цепи С1-С6, углеродные цепи С1-С7, углеродные цепи С1-С8, углеродные цепи С1-С9, углеродные цепи С1-С10. В других вариантах реализации алкильная группа может представлять собой С1-С4 углеродные цепи. В некоторых вариантах реализации галогеном может быть F, Br, Cl. В некоторых вариантах реализации циклоалкановая группа может представлять собой 5-членное кольцо. В некоторых вариантах реализации циклоалкановая группа может представлять собой 6-членное кольцо. В некоторых вариантах реализации обезболивающее средство, описанное в данном документе, представляет собой фармацевтически приемлемую соль.
[0082] В других вариантах реализации R1 включает Н, СН3, (СН2)2С6Н5, СН2С6Н5, СН2СН2СН2СН3, CH2C6H3Cl2, СН2С6Н5СН3, CH2C6H5Cl, CH2C5H5NO2, С5Н4, СН2С(СН3)2, С(СН3)2, СН2СН2С6Н3(ОН)2, CH2C6H4N, CH2C6H3NCH3, CH2CH2C4H4N, СН2СН2ОН или CH2CH(OH)CH2NH2; и R2 выбран из группы, состоящей из Н и СН3, или его фармацевтически приемлемую соль.
[0083] Неограничивающие примеры обезболивающих соединений по данному изобретению приведены в Таблице 1 ниже. При желании такие соединения также могут быть представлены в виде их солей.
[0084] ТАБЛИЦА 1:

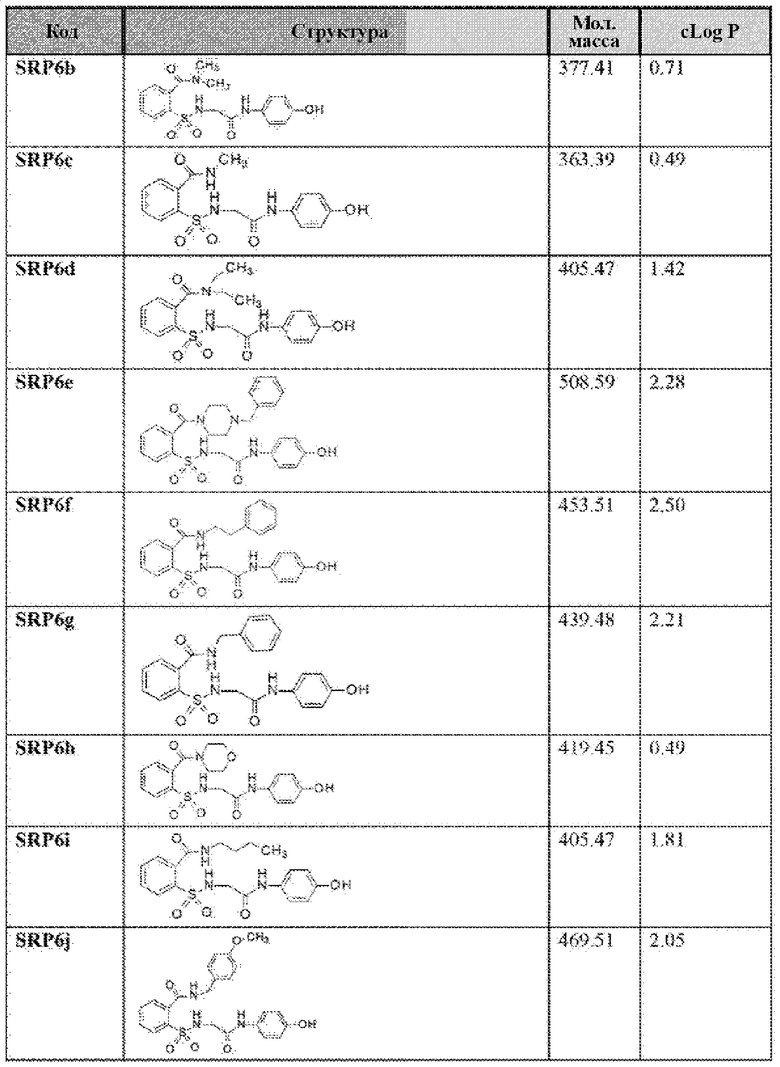
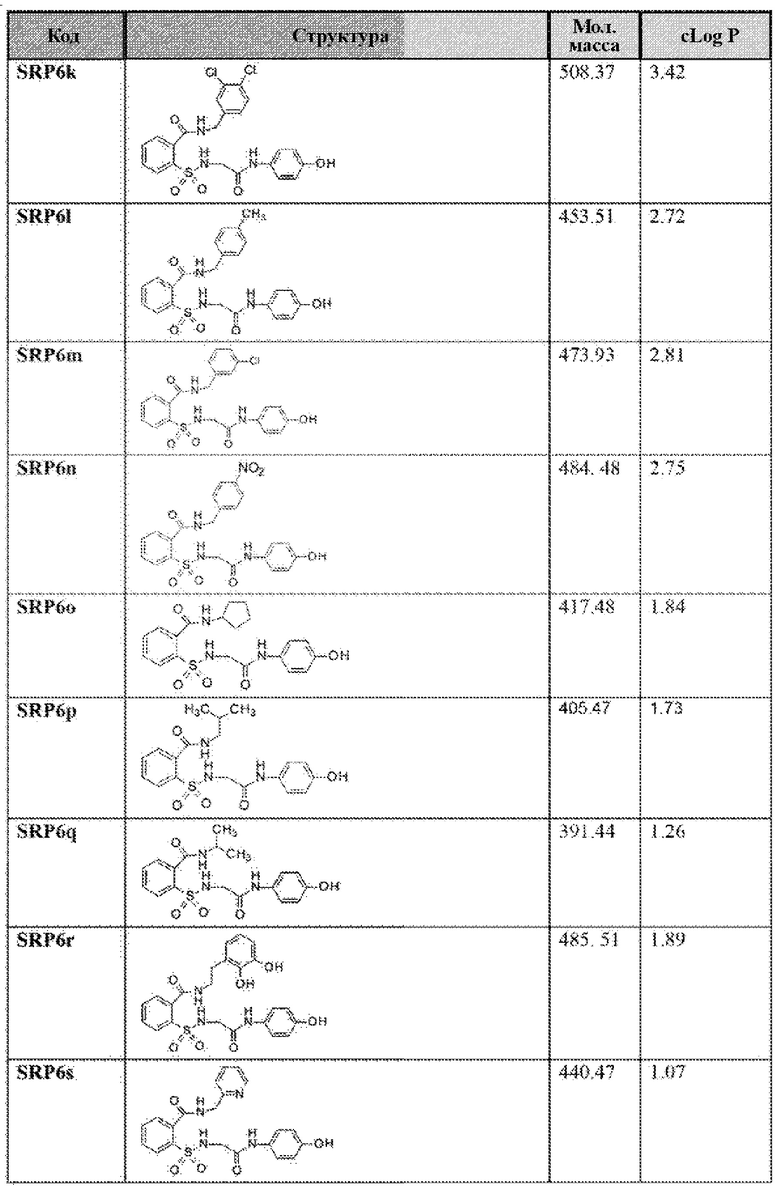
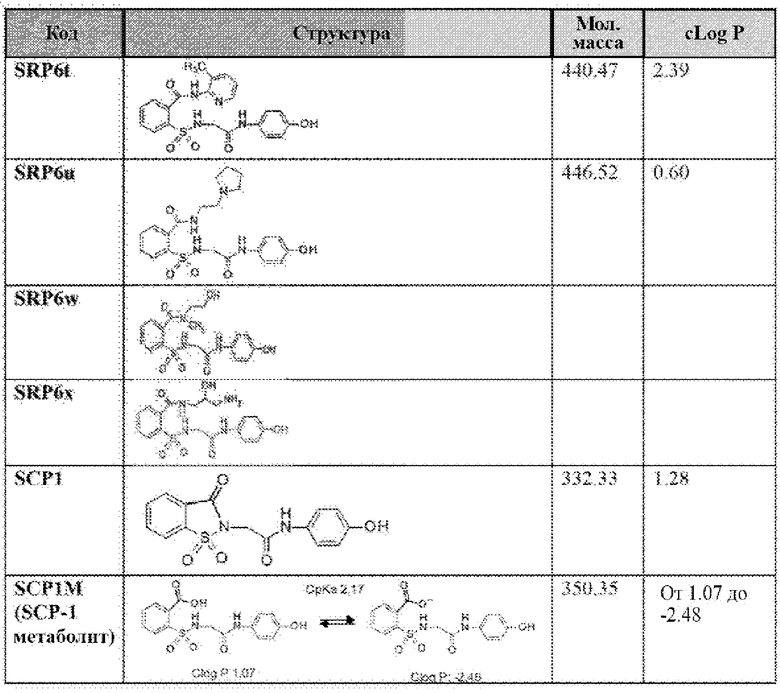
[0085] Значение logP соединения является мерой гидрофильности соединения. Значение logP соединения, которое представляет собой логарифм его коэффициента распределения между н-октанолом и водой log(Cоктанол/Cвода), является мерой гидрофильности соединения.
Как правило, низкая растворимость приводит к плохой абсорбции. Низкие гидрофильности характеризуются высоким значением logP. Высокие значения logP могут указывать на плохую абсорбцию или проникновение. Низкие расчетные значения Log Р (cLog Р) могут указывать на соединения с более коротким периодом полураспада и плохой абсорбцией. Низкие гидрофильности и, следовательно, высокие значения logP вызывают плохую абсорбцию или проникновение. Не будучи связанным теорией, для понимания разумной вероятности того, что соединения были хорошо абсорбированы, их значение logP не должно превышать 5,0. Иллюстративные варианты реализации включают соединения с промежуточными значениями cLog Р, такими как около 1,2, 1,4, 1,6, 1,8, 2,0, 2,2, 2,4, 2,6, 2,8 или 3. В некоторых вариантах реализации значение cLog Р соединения составляет около 2,0. Варианты реализации также могут содержать соединение со значением logP не более чем около 5,0. Распределение рассчитанных значений logP для более 3000 препаратов, представленных на рынке показано на Фиг. 10 [источник: http://wvw.openmolecules.org/properties/properties.html].
[0086] Боль
[0087] Варианты реализации могут использоваться для лечения или ослабления боли, неограничивающие примеры которой включают послеоперационную, невропатическую, стоматологическую, офтальмологическую, артритическую, пост- и/или травматическую боль.
[0088] Термин «боль» может относиться ко всем типам боли. Например, этот термин может относиться к острым и хроническим болям, таким как невропатическая боль и постхирургическая/послеоперационная боль, хроническая боль в пояснице, офтальмологическая боль, боль в суставах, посттравматическая боль, травматическая боль, кластерные головные боли, невралгия герпеса, фантомная боль в конечностях, центральная боль, зубная боль, опиоид-резистентная боль, висцеральная боль, хирургическая боль, боль при травмах костей, боль во время схваток и родов, боль в результате ожогов, включая солнечные ожоги, послеродовые боли, мигрень, ангинозную боль и боль в мочеполовом тракте, включая цистит. Термин может также относиться к ноцицептивной боли или ноцицепции, такой как соматическая боль (нормальный нервный ответ на болевой раздражитель). Боль также может относиться к боли, которая временно классифицируется как, например, хроническая боль и острая боль; боль, которая классифицируется по степени тяжести, например слабая, умеренная или сильная; и боль, которая является симптомом или результатом болезненного состояния или синдрома, например воспалительная боль, боль при раке, боль при СПИДе, артропатия, мигрень, невралгия тройничного нерва, ишемия сердца и диабетическая невропатия (см., например, Harrison's Principles of Internal Medicine pp. 93-98 (Wilson et al., eds., 12th ed. 1991); Williams et al., J. of Medicinal Chem. 42: 1481-1485 (1999), каждый из которых включен в данное описание в полном объеме в качестве ссылки).
[0089] Термин «нейропатическая боль» (НБ) может относиться к типу хронической боли, которая часто развивается после повреждения или заболевания нервной или периферической ткани. Нейропатическая боль (НБ) может развиваться с постоянными, спонтанными, пароксизмальными и болевыми компонентами боли. Такие НБ почти всегда связаны с нарушениями чувствительности кожи при формах аллодинии (ощущение боли от раздражителей, которые обычно не являются болезненными), гипералгезии (повышение чувствительности к обычно болевым раздражителям) и дизестезии (неприятные ненормальные ощущения). Хотя знания о механизмах нейропатической боли значительно продвинулись, удовлетворительные способы лечения НБ были недостижимыми.
[0090] Нейропатическая боль согласно данному раскрытию может быть разделена на «периферическую» (берущую начало в периферической нервной системе) и «центральную» (берущую начало в головном или спинном мозге).
[0091] Известно, что признаки невропатической боли отличаются от общего, ноцицептивного типа боли. Ноцицептивный тип боли может относиться к хронической или острой боли, связанной с болевым раздражителем. Большинство моделей на животных, используемых для изучения боли и ее лечения, основаны на ноцицептивном типе боли, например, модели отдергивания хвоста или горячей пластины. Невропатическая боль может быть вызвана безвредными стимулами и реагировать на некоторые лекарственные средства гораздо меньше, чем ноцицептивная боль. Например, опиоиды редко оказывают обезболивающее действие на нейропатическую боль, тогда как опиоиды успешно оказывают обезболивающее действие на ноцицептивную боль. Невропатическая боль может возникать в результате травмы периферического нерва (например, ампутация), инфекции (например, постгерпетическая невралгия), инфаркта или метаболического нарушения (например, диабетическая невралгия). Для лечения невропатической боли необходимы новые стратегии лечения.
[0092] Термин «зубная боль» может относиться к боли, ощущаемой в области рта, например, десны, зубов и/или челюсти. Боль в зубах может указывать на проблему со здоровьем полости рта, например, заболевание десен, разрушение зубов или расстройство ВНЧС, хотя боль также может быть вызвана состояниями, которые не относятся к зубам, таким как инфекции пазух или ушей, или проблемы с сердцем.
[0093] В большинстве случаев зубная боль может быть вызвана или быть результатом кариеса. В случае, когда полость становится больше, она начинает раздражать пульпу, которая является центром зуба, который содержит нервы и кровеносные сосуды. Пульпа зуба также может быть раздраженной, когда к зубу прикасаются или он контактирует с холодной, горячей или очень сладкой пищей и напитками. В более поздних случаях разрушения зуба, разрушение эмали и дентина (средний слой зуба) может позволить бактериям проникать в пульпу, что может привести к инфекции и привести к абсцессу зуба. Всякий раз, когда пульпа становится раздраженной, ее нервы посылают сигналы в мозг, вызывая боль. Хотя боль может со временем исчезнуть без какого-либо лечения, состояние будет продолжать ухудшаться, и боль может возобновиться, если ткани и кости, окружающие пораженный зуб, будут инфицированы.
[0094] Гингивит также может быть причиной зубной боли. Мягкие ткани десен могут воспаляться из-за образования зубного налета вдоль линии десен. В результате десны ослабляются и отделяются от зубов, образуя глубокие карманы пространства между деснами и зубами. Бактерии проникают в эти карманы, вызывая отеки, кровотечение и боль. В тяжелых случаях, когда бактерии растворяют кости, окружающие корни зубов, может произойти потеря зубов и костей. В случае, когда корни зубов становятся открытыми из-за удаляющихся десен или потери костной массы, может возникнуть чувствительность зубов. Нервные окончания, содержащиеся в нижней части зуба, реагируют на определенные раздражители, такие как холодный воздух, еда или напитки, вызывая зубную боль.
[0095] Зубная боль также может возникать в области челюсти и может быть вызвана, например, мышечным напряжением. Мышцы, контролирующие височно-нижнечелюстной сустав (ВНЧС), могут вызывать спазм и боль. Это часто случается у пациентов с нестабильным прикусом, отсутствующими или неправильно выровненными зубами.
[0096] Дополнительные оральные симптомы, которые могут быть связаны с зубной болью, зависят от ее причины, неограничивающие примеры которой включают чувствительность к определенным раздражителям (например, холод, тепло, воздух, прикус, жевание), рыхлые зубы, плохое дыхание (неприятный запах изо рта), красные и/или опухшие десны, кровоточащие десны, опущенные десны, затруднения при открытии или закрытии рта, треск при открывании челюсти, неприятные выделения и/или гной вблизи источника боли. Кроме того, симптомы в других областях тела могут появляться наряду с зубной болью, неограничивающими примерами которой являются жар, головные боли и трудности с глотанием или дыханием.
[0097] Боль в зубах может быть вызвана множеством медицинских состояний, неограничивающими примерами которых являются разрушение зубов, заболевания десен, инородные тела, расстройства височно-нижнечелюстного сустава (ВНЧС) и/или скрежетание зубами во время сна (бруксизм). Другие причины зубной боли включают прорезывание зубов, например, у детей, или вколачивание зуба; трещиноватые, поврежденные или разбитые зубы; открытый корень зуба; луночковый постэкстракционный альвеолит (осложнение после удаления зуба), травма головы или зубов; неправильный прикус; недавнее лечение зубов; и/или «метамфетаминовый рот» (вызванный применением метамфетамина). Кроме того, зубная боль также может быть результатом состояния других частей тела, такого как ушная инфекция, инфекция пазух, мигрени, проблемы с сердцем (например, боль, которая усиливается при физической нагрузке), неврологические состояния (например, невралгия тройничного нерва), синдром жжения рта и/или дисфункция слюнных желез.
[0098] Термин «офтальмологическая боль» или «боль в глазах», также известная как офтальмалгия, может относиться к одной из двух категорий: глазная боль, возникающая на поверхности глаза, и/или орбитальная боль, возникающая в глазу.
[0099] Боль в глазах, возникающая на поверхности, может представлять собой ощущение царапин, жжения или зуда. Поверхностная боль может быть вызвана раздражением от постороннего предмета, инфекцией или травмой.
[00100] Боль в глазах, которая возникает глубже в глазу, может быть ноющей, зудящей, колющей или пульсирующей.
[00101] Боль в глазах может сопровождаться потерей зрения.
[00102] Офтальмологическая боль, возникающая на поверхности глаза, может быть вызвана, например, посторонним предметом, конъюнктивитом, раздражением контактной линзой, истиранием роговицы, травмой, химическими ожогами и внезапными ожогами глаза, блефаритом и/или острым воспалением железы хряща века или сальной железы.
[00103] Офтальмологическая боль, возникающая в глазу (например, боль в глазнице), может быть вызвана глаукомой, невритом зрительного нерва, синуситом, мигренью, травмой, притом и/или воспалением глаза.
[00104] Термин «боль при артрите» может относиться к любой боли, возникающей анатомически из суставов и соседних костей, и не костных тканей. Любая боль при артрите может быть излечена с помощью данного изобретения, включая, без ограничения, любую боль, возникающую в результате аутоиммунного, инфекционного, воспалительного, пролиферативного, регенеративного или дегенеративного процесса, затрагивающая суставы животного или человека. Как таковая, подходящая боль, которую можно лечить с помощью данного изобретения, включает боль от ревматоидного артрита или остеоартрита.
[00105] Термин «послеоперационная боль» (взаимозаменяемо обозначаемая как «постинцизионная» или «посттравматическая боль») может относиться к боли, возникающей в результате внешней травмы, такой как порез, прокол, разрез, разрыв или рана в ткани индивида (включая травму, которая возникает в результате всех хирургических процедур, как инвазивных, так и неинвазивных).
[00106] В некоторых вариантах реализации послеоперационная боль представляет собой внутреннюю или внешнюю (включая периферическую) боль, и при этом рана, порез, травма, слеза или разрез могут возникать случайно (как при травматической ране) или преднамеренно (как при хирургическом вмешательстве).
[00107] Послеоперационная боль, в контексте данного документа, включает аллодинию (то есть повышенный ответ (т.е. вредное восприятие) на обычно безвредное воздействие) и гипералгезию (то есть повышенный ответ на обычно вредный или неприятное воздействие), который, в свою очередь, может быть термическим или механическим (тактильным) по своей природе. В некоторых вариантах реализации послеоперационная боль характеризуется термической чувствительностью, механической чувствительностью и/или болью в состоянии покоя. В некоторых вариантах реализации послеоперационная боль включает механически индуцированную боль или боль в состоянии покоя. В других вариантах реализации послеоперационная боль включает боль в состоянии покоя.
[00108] Фармацевтические комбинации
[00109] Варианты реализации включают структурные аналоги молекул АрАР, которые функционируют как нетоксичные, не вызывающие привыкания, обезболивающие средства. Такие соединения могут быть компонентом фармацевтических комбинаций для лечения или ослабления боли.
[00110] Фармацевтические комбинации по данному изобретению содержат обезболивающие соединения, как описано в данном документе, такие как SRP6D и SRP6R, в смеси с обезболивающим соединением, как описано в данном документе, вместе с фармацевтически приемлемым носителем, приготовленным в соответствии с традиционными фармацевтическими способами. Согласно данному изобретению фармацевтически приемлемый носитель может содержать любые растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические и замедляющие абсорбцию агенты и тому подобное, совместимые с фармацевтическим способом введением. Использование таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области техники. Неограничивающие примеры фармацевтически приемлемых носителей включают твердые или жидкие наполнители, разбавители и инкапсулирующие вещества, включая, но не ограничиваясь этим, лактозу, декстрозу, сахарозу, сорбит, маннит, ксилит, эритрит, мальтит, крахмалы, аравийскую камедь, альгинат, желатин, фосфат кальция, силикат кальция, целлюлозу, метилцеллюлозу, микрокристаллическую целлюлозу, поливинилпирролидон, воду, метилбензоат, пропилбензоат, тальк, стеарат магния и минеральное масло. Количество носителя, используемого вместе с комбинацией, является достаточным для обеспечения практического количества материала на стандартную дозу обезболивающего соединения.
[00111] Использование таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области техники. Можно использовать любой обычный носитель или агент, который совместим с активным соединением. Дополнительные активные соединения также могут быть включены в композиции.
[00112] Фармацевтически приемлемые носители для перорального введения включают сахара, крахмалы, целлюлозу и ее производные, солод, желатин, тальк, сульфат кальция, растительные масла, синтетические масла, полиолы, альгиновую кислоту, фосфатные буферные растворы, эмульгаторы, изотонический солевой раствор и апирогенную воду. Фармацевтически приемлемые носители для парентерального введения включают изотонический солевой раствор, пропиленгликоль, этилолеат, пирролидон, водный этанол, кунжутное масло, кукурузное масло и их комбинации.
[00113] Могут быть использованы различные пероральные лекарственные формы, неограничивающие примеры которых включают твердые формы, такие как таблетки, капсулы, гранулы, суппозитории и/или порошки. Таблетки могут быть спрессованы, таблетки для растирания, покрыты энтеросолюбильной оболочкой, покрыты сахаром, покрыты пленочной оболочкой или многократно спрессованы, содержат подходящие связующие, смазывающие вещества, разбавители, разрыхлители, красители, ароматизаторы, агенты, индуцирующие поток, и плавящие агенты. Жидкие пероральные лекарственные формы включают водные растворы, эмульсии, суспензии, сиропы, аэрозоли и/или восстановленные растворы, и/или суспензии. Композиция может быть альтернативно составлена для наружного местного применения или в форме стерильного раствора для инъекций.
[00114] Фармацевтически эффективные комбинации могут быть предоставлены в виде композиции, содержащей от 0,1 до 2000 мг/кг обезболивающего соединения, как описано в данном документе, такого как SRP6D и SRP6R. Например, фармацевтически эффективные комбинации могут быть предоставлены в виде композиции, содержащей около 0,1 мг/кг, 1 мг/кг, 10 мг/кг, 20 мг/кг, 30 мг/кг, 40 мг/кг, 50 мг/кг, 60 мг/кг, 70 мг/кг, 80 мг/кг, 90 мг/кг, 100 мг/кг, 125 мг/кг, 150 мг/кг, 175 мг/кг, 200 мг/кг, 225 мг/кг, 250 мг/кг, 275 мг/кг, 300 мг/кг 325 мг/кг, 350 мг/кг, 375 мг/кг, 400 мг/кг, 425 мг/кг, 450 мг/кг, 475 мг/кг, 500 мг/кг, 525 мг/кг, 550 мг/кг, 575 мг/кг, 600 мг/кг, 625 мг/кг, 650 мг/кг, 675 мг/кг, 700 мг/кг, 725 мг/кг, 750 мг/кг, 775 мг/кг, 800 мг/кг, 825 мг/кг, 850 мг/кг, 875 мг/кг, 900 мг/кг, 925 мг/кг, 950 мг/кг, 975 мг/кг, 1000 мг/кг, 1100 мг/кг, 1200 мг/кг, 1300 мг/кг, 1400 мг/кг, 1500 мг/кг, 1600 мг/кг, 1700 мг/кг, 1800 мг/кг, 1900 мг/кг, 2000 мг/кг обезболивающего средства. Полезные фармацевтически эффективные комбинации могут содержать от около 300 мг/кг до около 1000 мг/кг обезболивающего соединения, как описано в данном документе, такого как SRP6D и SRP6R. Например, варианты реализации, как описано в данном документе, могут содержать около 300 мг/кг обезболивающего соединения.
[00115] Фармацевтически эффективные комбинации, такие как пилюля или таблетка, могут содержать от 0,1 до 2000 мг обезболивающего соединения, как описано в данном документе, такого как SRP6D и SRP6R. Например, фармацевтически эффективные комбинации могут содержать около 0,1 мг, 1 мг, 10 мг, 20 мг, 30 мг, 40 мг, 50 мг, 60 мг, 70 мг, 80 мг, 90 мг, 100 мг, 125 мг, 150 мг, 175 мг, 200 мг, 225 мг, 250 мг, 275 мг, 300 мг, 325 мг, 350 мг, 375 мг, 400 мг, 425 мг, 450 мг, 475 мг, 500 мг, 525 мг, 550 мг, 575 мг, 600 мг, 625 мг, 650 мг, 675 мг, 700 мг, 725 мг, 750 мг, 775 мг, 800 мг, 825 мг, 850 мг, 875 мг, 900 мг, 925 мг, 950 мг, 975 мг, 1000 мг, 1100 мг, 1200 мг, 1300 мг, 1400 мг, 1500 мг, 1600 мг, 1700 мг, 1800 мг, 1900 мг, 2000 мг обезболивающего соединения. Полезные фармацевтически эффективные комбинации могут содержать от около 300 мг до около 1000 мг обезболивающего соединения, как описано в данном документе, такого как SRP6D и SRP6R. Например, варианты реализации, как описано в данном документе, могут содержать около 300 мг обезболивающего соединения.
[00116] Данное изобретение также включает образование фармацевтически приемлемых стабильных солей соединений, как описано в данном документе, таких как SRF6D и SRF6R, с металлами или аминами. Неограничивающие примеры металлов, используемых в качестве катионов, включают щелочные металлы, такие как Na+ или K+, и щелочноземельные металлы, такие как Mg2+ и Са2+. Неограничивающие примеры аминов включают N,N-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, N-метилглюкамин и прокаин.
[00117] Фармацевтическая композиция по данному изобретению составлена так, чтобы она была совместима с предполагаемым путем введения. Примеры путей введения включают парентеральное, например, внутривенное, внутрикожное, подкожное, пероральное (например, ингаляция), трансдермальное (местное), транс-слизистое и ректальное введение. Растворы или суспензии, используемые для парентерального, внутрикожного или подкожного применения, могут содержать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетатные, цитратные или фосфатные буферы, и агенты для регулирования тоничности, такие как хлорид натрия или декстроза, рН можно регулировать кислотами или основаниями, такими как соляная кислота или гидроксид натрия. Препарат для парентерального введения может быть заключен в ампулы, одноразовые шприцы или многодозовые флаконы, изготовленные из стекла или пластика.
[00118] В качестве примерного варианта реализации фармацевтические комбинации по данному изобретению могут быть введены перорально, либо в форме таблеток, содержащих наполнители, такие как крахмал или лактоза, либо в капсулах, либо отдельно, либо в смеси со вспомогательными веществами, либо в форме сиропов или суспензии, содержащие красители или ароматизаторы. Они также могут быть введены парентерально, например, внутримышечно, внутривенно или подкожно. При парентеральном введении они могут быть использованы в форме стерильного водного раствора, который может содержать другие растворенные вещества, такие как, например, любая соль или глюкоза, для того, чтобы раствор был изотоническим.
[00119] Соединения по данному изобретению могут быть введены субъекту для лечения боли, например, перорально, либо в желатиновых капсулах, либо в виде таблеток для рассасывания. Для перорального терапевтического введения указанные соединения могут быть смешаны со вспомогательными веществами и использоваться в форме таблеток для рассасывания, таблеток, капсул, эликсиров, суспензий, сиропов, пластинок, жевательной резинки и тому подобного. Эти препараты могут содержать по меньшей мере 0,5% активного соединения, но могут варьироваться в зависимости от каждой формы, в частности от 4% до 75% приблизительно от массы каждой единицы. Количество активного соединения в таких композициях должно быть таким, которое необходимо для получения соответствующей дозировки. Например, композиции и препараты, описанные в данном документе, могут быть приготовлены таким образом, что каждая единица пероральной дозы может содержать от 0,1 мг до 300 мг активного соединения.
[00120] При парентеральном терапевтическом введении активные соединения по данному изобретению могут быть включены в раствор или суспензию. Такие препараты, например, могут содержать по меньшей мере 0,1% активного соединения, но количество может варьироваться приблизительно от 0,5% до 50% от массы препарата. Например, такие препараты составляют около 0,1%, 0,5%, 1%, 5%, 10%, 15%, 25%, 30%, 35%, 40%, 45%, 50% от массы препарата. Количество активного соединения в таких композициях должно быть таким, которое необходимо для получения соответствующей дозировки. Композиции и препараты, описанные в данном документе, могут быть приготовлены таким образом, что каждая парентеральная единица дозировки может содержать от 0,01 до 1000 мг, например, от около 0,5 мг до 100 мг, например, активного соединения. В то время как внутримышечное введение может быть осуществлено в одной дозе или разделено на несколько доз, например, три дозы, внутривенное введение может включать капельное устройство для введения дозы с помощью внутривенного вливания. Парентеральное введение может осуществляться с помощью ампул, одноразовых шприцев или многодозовых флаконов, изготовленных из стекла или пластика.
[00121] Фармацевтические композиции, подходящие для инъекционного применения, могут содержать стерильные водные растворы (в случае водорастворимости) или дисперсии и стерильные порошки для немедленного приготовления стерильных инъекционных растворов или дисперсий. Для внутривенного введения подходящие носители могут включать физиологический солевой раствор, бактериостатическую воду, Cremophor ЕМ™ (BASF, Парсиппани, Нью-Джерси) или фосфатно-солевой буферный раствор (PBS). В вариантах реализации композиция может быть стерильной и должна быть жидкой до такой степени, чтобы была возможность легко осуществить шприцевание. Он может быть стабильным в условиях производства и хранения, и может быть защищен от загрязнения микроорганизмами, такими как бактерии и грибы. Носителем может быть растворитель или дисперсионная среда, содержащая, например, воду, этанол, фармацевтически приемлемый полиол, такой как глицерин, пропиленгликоль, жидкий полиэтеиленгликоль и их подходящие смеси. Надлежащая текучесть может поддерживаться, например, путем использования покрытия, такого как лецитин, путем поддержания требуемого размера частиц в случае дисперсии и путем использования поверхностно-активных веществ. Предотвращение действия микроорганизмов может быть достигнуто с помощью различных антибактериальных и противогрибковых агентов, например, парабенов, хлорбутанола, фенола, аскорбиновой кислоты и тимеросала. Во многих случаях может быть полезно включить в композицию изотонические агенты, например, сахара, полиспирты, такие как маннит, сорбит, хлорид натрия. Пролонгированная абсорбция инъецируемых композиций может происходить путем включения в композицию агента, который задерживает абсорбцию, например, моносте арата алюминия и желатина.
[00122] Стерильные растворы для инъекций могут быть приготовлены путем включения соединения в необходимом количестве в подходящем растворителе с одним или комбинацией ингредиентов, перечисленных в данном документе, по мере необходимости, с последующей стерилизованной фильтрацией. Обычно дисперсии готовят путем включения активного соединения в стерильный носитель, который содержит основную дисперсионную среду и требуемые другие ингредиенты из перечисленных в данном документе. В случае стерильных порошков для приготовления стерильных растворов для инъекций примерами полезных способов приготовления являются: вакуумная сушка и сублимационная сушка, которая дает порошок активного ингредиента плюс любой дополнительный желаемый ингредиент из его предварительно стерильно отфильтрованного раствора.
[00123] Пероральные композиции обычно включают инертный разбавитель или пищевой носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. С целью перорального терапевтического введения активное соединение может быть включено со вспомогательными веществами и использоваться в форме таблеток, троше или капсул. Композиции для ухода за полостью рта также могут быть приготовлены с использованием жидкого носителя для использования в качестве жидкости для полоскания рта, при этом соединение в желаемой жидкости вводится перорально и взбивается, откашливается или проглатывается.
[00124] Фармацевтически приемлемые связующие агенты и/или адъюванты могут быть включены в состав композиции. Таблетки, пилюли, капсулы, таблетки и тому подобное могут содержать любой из следующих ингредиентов или соединений аналогичной природы: связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; вспомогательное вещество, такое как крахмал или лактоза, дезинтегрирующий агент, такой как альгиновая кислота, примогель или комкрахмал; смазывающее вещество, такое как стеарат магния или стероты; глидант, такой как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или ароматизатор, такой как мята перечная, метилсалицилат или апельсиновый ароматизатор.
[00125] Системное введение также может осуществляться трансмукозальным или трансдермальным путем. Для трансмукозального или трансдермального введения в препарате используются пенетранты, подходящие для проникающего барьера. Такие пенетранты, как правило, известны в данной области техники и включают, например, для трансмукозального введения, детергенты, соли желчных кислот и производные фузидиевой кислоты. Введение через слизистую оболочку может быть достигнуто путем использования назальных спреев или суппозиториев. Для трансдермального введения активные соединения составляют в виде мазей. Маски, гели или кремы, общеизвестные в данной области техники.
[00126] Соединения по данному изобретению могут быть введены субъекту для лечения боли в виде одной дозы или в виде нескольких доз в течение определенного периода времени. Кроме того, соединение может быть введено с интервалами около 4 ч, 8 ч, 12 ч, 24 ч или дольше. Например, таблетка может быть введена субъекту до начала боли, чтобы предотвратить боль, или множество таблеток может быть введена в течение периода времени, чтобы облегчить боль в течение указанного периода.
[00127] По необходимости, могут быть изменения, которые будут зависеть от массы и состояния субъекта, поддающегося лечению, и от конкретного выбранного пути введения.
[00128] Способы лечения
[00129] Варианты реализации могут использоваться для лечения или ослабления боли, неограничивающие примеры которых включают послеоперационную, невропатическую, стоматологическую, офтальмологическую, артритическую, пост- и/или травматическую боль.
[00130] В вариантах реализации соединение, как описано в данном документе, такое как SRP6D или SRP6R, используют в качестве единственного физически активного соединения при лечении невропатической боли без второго активного агента, такого как аналог ГАМК, например, Габапентин (Нейронтин).
[00131] В других вариантах реализации композиции, как описано в данном документе, могут быть введены субъекту одновременно и/или в комбинации со вторым активным ингредиентом, таким как опиоид или нестероидное противовоспалительное лекарственное средство (НСПВ). Опиоидные препараты работают путем связывания с опиоидными рецепторами в головном и спинном мозге. Неограничивающие примеры таких опиоидов включают кодеин, фентанил, гидрокодон, гидрокодон/АрАР, гидроморфон, меперидин, метадон, морфин, оксикодон, оксикодон и АрАР, оксикодон и налоксон. Нестероидные противовоспалительные препараты (НСПВ) блокируют ферменты ЦОГ и снижают уровень простагландинов по всему организму. Как следствие, продолжающееся воспаление, боль и жар уменьшаются. Неограничивающие примеры НСПВ включают: аспирин, целекоксиб, диклофенак, дифлунисал, этодолак, ибупрофен, индометацин, кетопрофен, кеторолак, набуметон, напроксен, оксапрозин, пироксикам, сальсалат, сулиндак и толметин.
[00132] Соединения, как описано в данном документе, например, SRD6R и SRP6D, могут быть включены в фармацевтические композиции, подходящие для введения. Такие композиции могут содержать соединение, как описано в данном документе, и фармацевтически приемлемый носитель. Таким образом, в некоторых вариантах реализации соединения по данному изобретению присутствуют в фармацевтической композиции.
[00133] Например, фармацевтические композиции, содержащие соединение, как описано в данном документе, могут быть использованы для предотвращения и/или лечения боли, например, терапевтически эффективное количество SRP6D и SRP6R в смеси с фармацевтически приемлемым носителем или наполнителем. Например, терапевтически эффективное количество SRP6D может быть введено субъекту для предотвращения возникновения боли или предотвращения усиление выраженности боли.
[00134] Термин «лечение» может относиться к подходу для получения полезных или желаемых клинических результатов, например, улучшения или облегчения любого аспекта боли, такой как острая, хроническая, воспалительная, невропатическая или послеоперационная боль. Благоприятные или желательные клинические результаты включают, но не ограничиваются этим, одно или более из следующих: включая уменьшение тяжести, ослабление одного или более симптомов, связанных с болью, включая любой аспект боли (такой как сокращение продолжительности боли и/или уменьшение болевой чувствительности или ощущения).
[00135] Термин «уменьшение» боли или одного, или более симптомов боли может означать уменьшение или улучшение одного, или более симптомов боли по сравнению с не введением композиции, как описано в данном документе, такой как SRP6D и SRP6R. Термин «улучшение» также может включать укорочение или уменьшение продолжительности симптома, например, терапевтически эффективное количество SRP6D или SRP6R может быть введено субъекту, страдающему от боли, чтобы ослабить или уменьшить боль.
[00136] Термин «облегчение» или «облегчать» может относиться к облегчению или уменьшению тяжести симптома, состояния или расстройства. Например, можно сказать, что лечение, такое как SRP6D или SRP6R, которое уменьшает тяжесть боли у субъекта, облегчает боль. Например, терапевтически эффективное количество SRP6D может быть введено субъекту, страдающему от боли, при этом степень боли уменьшается. Понятно, что при определенных обстоятельствах лечение может облегчить симптом или состояние без лечения лежащего в основе расстройства. В определенных аспектах этот термин может быть синонимом слова «паллиативное лечение».
[00137] Варианты реализации могут использоваться для уменьшения частоты возникновения боли или задержки, неограничивающие примеры включают послеоперационную, невропатическую, стоматологическую, офтальмологическую, артритическую, пост- и/или травматическую боль. Термин «уменьшение частоты» боли может относиться к любому из снижения тяжести (которое может включать уменьшение потребности и/или количества (например, воздействия) других лекарственных средств и/или методов лечения, обычно применяемых для этих состояний), длительности и/или частоты (включая, например, задержку или увеличение времени до повторного появления боли у человека). Как понятно специалистам в данной области техники, индивидуумы могут варьировать в зависимости от их реакции на лечение, и, например, «способ уменьшения частоты возникновения боли у индивидуума» отражает введение композиций, как описано в данном документе, таких как SRP6D и SRP6R, исходя из разумных ожиданий, что такое введение может вызвать соответствующее изменение в уровне заболеваемости у данного конкретного лица.
[00138] Термин «задержка» развития боли может относиться к отсрочке, затруднению, замедлению, стабилизации и/или отсрочке прогрессирования боли. Эта задержка может быть различной продолжительности, в зависимости от истории болезни и/или лиц, которые поддаются лечению. Как очевидно для специалиста в данной области техники, достаточная или значительная задержка может, по сути, включать профилактику, поскольку у индивидуума не возникает боль. Способ, который «задерживает» развитие симптома, представляет собой способ, который уменьшает вероятность развития симптома в заданном временном интервале и/или уменьшает степень симптомов в заданном временном интервале по сравнению с неиспользованием указанного способа.
[00139] Термины «развитие» или «прогрессирование» боли могут относиться к начальному проявлению и/или последующему прогрессированию расстройства. Развитие боли можно обнаружить и оценить с использованием стандартных клинических методов, хорошо известных в данной области техники. Тем не менее, развитие также относится к прогрессу, который может быть не обнаруживаемым. Для целей данного изобретения развитие или прогрессирование относится к биологическому течению симптомов. Термин «развитие» включает возникновение, повторение и начало. Используемый в данном документе термин «начало» или «возникновение» боли включает начальное начало и/или рецидив. Например, варианты реализации, как описано в данном документе, могут использоваться для предотвращения развития боли или предотвращения прогрессирования боли.
[00140] Варианты реализации могут использоваться для смягчения боли, неограничивающие примеры которых включают послеоперационную, невропатическую, стоматологическую, офтальмологическую, артритическую, пост- и/или травматическую боль.
[00141] Термин «смягчение» боли или одного, или более симптомов боли могут относиться к уменьшению степени одного или более нежелательных клинических проявлений боли у индивидуума или группы лиц, которые поддаются лечению композицией, как описано в данном документе, такой как SRP6D и SRP6R.
[00142] Варианты реализации включают введение субъекту эффективного количества композиции, как описано в данном документе, такой как SRP6D и SRP6R, для лечения боли.
[00143] Термин «эффективное количество», «достаточное количество» или «терапевтически эффективное количество» может относиться к количеству, достаточному для достижения полезных или желательных клинических результатов, включая удаление или уменьшение болевого ощущения. Для целей данного изобретения эффективное количество композиции, как описано в данном документе, например SRP6D и SRP6R, может включать количество, достаточное для лечения, ослабления, уменьшения интенсивности или предотвращения боли любого рода, включая острую, хроническую, воспалительную, невропатическая или послеоперационная боль. В некоторых вариантах реализации эффективное количество композиций, как описано в данном документе, может модулировать порог чувствительности к внешним раздражителям до уровня, сопоставимого с тем, который наблюдается у здоровых субъектов. В других вариантах реализации этот уровень не сопоставим с уровнем, наблюдаемым у здоровых субъектов, но он ниже по сравнению с отсутствием комбинированной терапии.
[00144] Конкретные композиции, как описано в данном документе, такие как SRF6D и SRP6R, могут вводиться субъекту любыми подходящими способами, такими как пероральное, внутривенное, парентеральное, подкожное, внутрилегочное, местное, интравитреальное, дермальное, трансмукозальное, ректальное и интраназальное введение. Парентеральные инфузии включают внутримышечное, внутривенное, внутриартериальное или внутрибрюшинное введение. Соединения также можно вводить трансдермально, например, в форме подкожного имплантата с медленным высвобождением или в виде трансдермального пластыря. Их также можно вводить путем ингаляции. Хотя прямое пероральное введение может вызывать некоторую потерю желаемой активности, например, обезболивающего действия, обезболивающие лекарственные средства могут быть упакованы таким образом, чтобы защитить активный(ые) ингредиент(ы) от переваривания путем использования энтеросолюбильных покрытий, капсул или других способов, применяемых в данной области техники
[00145] Фармацевтические продукты с контролируемым высвобождением имеют общую цель улучшения лекарственной терапии по сравнению с тем, которого достигают их аналоги с неконтролируемым высвобождением. Использование оптимально разработанного препарата с контролируемым высвобождением в лечении характеризуется минимальным количеством лекарственного вещества, используемого для лечения или контроля состояния в течение минимального промежутка времени. Преимущества составов с контролируемым высвобождением включают расширенную активность лекарственного средства, сниженную частоту дозирования и улучшенное соблюдение пациентом режима и схемы применения состава. Кроме того, композиции с контролируемым высвобождением могут использоваться для воздействия на время начала действия или других характеристик, таких как уровни лекарственного средства в крови, и, таким образом, могут влиять на возникновение побочных (например, неблагоприятных) эффектов.
[00146] Большинство составов с контролируемым высвобождением предназначены для первоначального высвобождения количества лекарственного средства (активного ингредиента), которое быстро производит желаемый терапевтический эффект, и постепенного и непрерывного высвобождения других количеств лекарственного средства для поддержания этого уровня терапевтического или профилактического эффекта в течение более длительного периода времени. Чтобы поддерживать указанный постоянный уровень лекарственного средства в организме, лекарственное средство должно высвобождаться из лекарственной формы со скоростью, которая будет замещать количество лекарственного средства, которое метаболизируется и выводится из организма. Контролируемое высвобождение активного ингредиента может стимулироваться различными условиями, включая, но не ограничиваясь ими, рН, температуру, ферменты, воду или другие физиологические условия или соединения.
[00147] Растворы или суспензии, используемые для парентерального, внутрикожного или подкожного применения, могут включать, например, следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; противовоспалительные агенты; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатообразующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулирования тонуса, такие как хлорид натрия или декстроза. рН можно регулировать кислотами или основаниями, такими как хлористоводородная кислота или гидроксид натрия.
[00148] Композиции, как описано в данном документе, такие как SRP6D и SRP6R, могут вводиться субъекту один раз (например, в виде однократной инъекции или применения). Хотя период введения по меньшей мере одного соединения для предотвращения боли варьируется в зависимости от вида, а также от природы и тяжести состояния, которое необходимо предотвратить или лечить, соединение можно вводить людям в течение короткого или длительного периода, т.е. в течение 1 недели и до 1 года. Например, введение может осуществляться один или два раза в день субъекту, нуждающемуся в этом, в течение периода времени, такого как одна неделя или один месяц.
[00149] Дозировка может варьироваться в зависимости от известных факторов, таких как фармакодинамические характеристики активного ингредиента и его способ и путь введения; время введения активного ингредиента; возраст, пол, здоровье и масса пациента; характер и степень выраженности симптомов; вид сопутствующего лечения, частота лечения и желаемый эффект; и скорость выведения.
[00150] Терапевтически эффективная доза может зависеть от ряда факторов, известных специалистам в данной области техники. Доза(ы) может варьироваться, например, в зависимости от индивидуальности, размера и состояния субъекта или образца, подвергаемого лечению, дополнительно в зависимости от пути введения композиции, если это применимо, и эффекта, который практикующий доктор желает достичь. Эти количества могут быть легко определены специалистом в данной области техники. [00151] В некоторых вариантах реализации терапевтически эффективное количество составляет по меньшей мере около 0,1 мг/кг массы тела, по меньшей мере около 0,25 мг/кг массы тела, по меньшей мере около 0,5 мг/кг массы тела, по меньшей мере около 0,75 мг/кг массы тела, по меньшей мере около 1 мг/кг массы тела, по меньшей мере около 2 мг/кг массы тела, по меньшей мере около 3 мг/кг массы тела, по меньшей мере около 4 мг/кг массы тела, по меньшей мере около 5 мг/кг массы тела, по меньшей мере около 6 мг/кг массы тела, по меньшей мере около 7 мг/кг массы тела, по меньшей мере около 8 мг/кг массы тела, по меньшей мере около 9 мг/кг массы тела, по меньшей мере около 10 мг/кг массы тела, по меньшей мере около 15 мг/кг массы тела, по меньшей мере около 20 мг/кг массы тела, по меньшей мере около 25 мг/кг массы тела, по меньшей мере около 30 мг/кг массы тела, по меньшей мере около 40 мг/кг массы тела, по меньшей мере около 50 мг/кг массы тела, по меньшей мере около 75 мг/кг массы тела, по меньшей мере около 100 мг/кг массы тела, по меньшей мере около 200 мг кг массы тела, по меньшей мере около 250 мг кг массы тела, по меньшей мере около 300 мг кг массы тела, по меньшей мере около 3500 мг/кг массы тела, по меньшей мере около 400 мг/кг массы тела, по меньшей мере около 450 мг/кг массы тела, по меньшей мере около 500 мг/кг массы тела, по меньшей мере около 550 мг/кг массы тела, по меньшей мере около 600 мг/кг массы тела, по меньшей мере около 650 мг/кг массы тела, по меньшей мере около 700 мг/кг массы тела, по меньшей мере около 750 мг/кг массы тела, по меньшей мере около 800 мг/кг массы тела, по меньшей мере около 900 мг/кг массы тела или по меньшей мере около 1000 мг/кг массы тела.
[00152] Терапевтически эффективная доза может зависеть от ряда факторов, известных специалистам в данной области техники. Доза(ы) может варьироваться, например, в зависимости от индивидуальности, размера и состояния субъекта или образца, подвергаемого лечению, дополнительно в зависимости от пути введения композиции, если это применимо, и эффекта, который практикующий врач желает достичь. Эти количества могут быть легко определены специалистом в данной области техники.
[00153] В одном варианте реализации рекомендуемый диапазон суточной дозы соединения, как описано в данном документе, для обезболивания, как описано в данном документе, находится в диапазоне от около 1 мг/кг массы тела до около 10 г/кг массы тела, например, от около 5 мг/кг массы тела до около 5 г/кг массы тела, или, например, от около 10 мг/кг массы тела до около 2 г/кг массы тела активного ингредиента для лечения указанного заболевания, и средняя разовая доза от около 0,5 мг до около 1 мг, около 5 мг, около 10 мг, около 50 мг, около 100 мг, около 250 мг, около 500 мг, около 1 г, около 2 г и около 3 г, может быть обычно введена. Суточная доза для введения людям для лечения или ослабления боли может находиться в диапазоне от около 1 мг/кг до около 300 мг/кг.
[00154] Соединение, как описано в данном документе, например, SRP6D или SRP6R, или композиция, содержащая его, могут быть введены субъекту один раз (например, в виде однократной инъекции или введения). Альтернативно, введение может осуществляться один или два раза в день субъекту, нуждающемуся в этом, в течение периода от около 2 до около 28 дней, или от около 7 до около 10 дней, или от около 7 до около 15 дней. Соединение также можно вводить субъекту один или два раза в день в течение 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 раз в год или их комбинации.
[00155] Разовые единичные дозированные формы по данному изобретению подходят для перорального, слизистого (например, назального, сублингвального, вагинального, буккального или ректального), парентерального (например, подкожного, внутривенного, болюсного введения, внутримышечного или внутриартериального), местного (например, глазные капли или другие офтальмологические препараты), трансдермального (например, крем, лосьон или дермальный спрей) или чрезкожного введения пациенту. Примеры лекарственных форм включают, но не ограничиваются ими: таблетки; капсулы, такие как мягкие эластичные желатиновые капсулы; облатки; троше; таблетки для рассасывания; дисперсии; суппозитории; порошки; аэрозоли (например, назальные спреи или ингаляторы); гели; жидкие лекарственные формы, подходящие для перорального или слизистого введения пациенту, включая суспензии (например, водные или неводные жидкие суспензии или растворы, эмульсии масло-в-воде или жидкие эмульсии вода-в-масле), растворы и эликсиры; жидкие лекарственные формы, подходящие для парентерального введения пациенту; глазные капли или другие офтальмологические препараты, подходящие для местного применения; и стерильные твердые вещества (например, кристаллические или аморфные твердые вещества), которые могут быть восстановлены для обеспечения жидких лекарственных форм для парентерального введения субъекту.
[00156] Состав, форма и тип лекарственных форм по данному изобретению обычно будут варьироваться в зависимости от их применения. Кроме того, дозировка может варьироваться в зависимости от известных факторов, таких как фармакодинамические характеристики активного ингредиента и его способ, и путь введения; время введения активного ингредиента; возраст, пол, здоровье и масса пациента; характер и степень выраженности симптомов; вид сопутствующего лечения, частота лечения и желаемый эффект; и скорость выведения.
[00157] Например, лекарственная форма, используемая при лечении острого заболевания, может содержать большие количества одного или более активных агентов, которые она содержит, чем лекарственная форма, используемая при хроническом лечении того же заболевания. Аналогично, парентеральная лекарственная форма может содержать меньшие количества одного или более активных агентов, которые она содержит, чем пероральная лекарственная форма, используемая для лечения того же заболевания. Эти и другие способы, которыми конкретные лекарственные формы, охватываемые настоящим документом, будут отличаться друг от друга, будут очевидны для специалистов в данной области техники. Смотрите, например, Remington's Pharmaceutical Sciences, 18th., Mack Publishing, Easton Pa. (1990).
[00158] Любое из описанных в данном документе терапевтических применений может быть применено к любому субъекту, нуждающемуся в такой терапии, включая, например, млекопитающее, такое как мышь, крыса, собака, кошка, корова, лошадь, кролик, обезьяна, свинья, овца, коза или человек. В некоторых вариантах реализации субъект представляет собой мышь, крысу, свинью или человека. В некоторых вариантах субъектом является мышь. В некоторых вариантах субъектом является крыса. В некоторых вариантах субъектом является свинья. В некоторых вариантах субъектом является человек.
[00159] Медицинские наборы
[00160] Термины «набор» или «медицинский набор» по данному изобретению включают лекарственную форму соединения по данному изобретению, такого как SRP6D или SRP6R, или его фармацевтически приемлемую соль, сольват, гидрат, стереоизомер, пролекарство или клатрат. Набор также может включать как SRP6D, так и SRP6R, либо в комбинации, например, в одной таблетке, либо отдельно, например, в двух таблетках.
[00161] Наборы могут дополнительно содержать дополнительные активные агенты, например, опиоиды или нестероидные противовоспалительные средства, примеры которых описаны в данном документе. Например, опиоид может быть предоставлен в наборе, описанном в данном документе, в дозе ниже, чем та, которая используется в настоящее время субъектом, чтобы снизить общее потребление опиоидов организмом и вредные эффекты, связанные с длительным употреблением опиоидов. Наборы по данному изобретению могут дополнительно содержать устройства, которые используются для введения активного ингредиента. Примеры таких устройств включают, но не ограничиваются ими, шприцы, капельные пакеты, пластыри и ингаляторы. Наборы могут также содержать инструкции для введения соединения субъекту.
[00162] Наборы по данному изобретению могут дополнительно содержать фармацевтически приемлемые носители, которые можно использовать для введения одного или более активных ингредиентов. Например, если активный ингредиент предоставляется в твердой форме, которая должна быть восстановлена для парентерального введения, набор может содержать герметичный контейнер с подходящим носителем, в котором активный ингредиент может быть растворен с образованием стерильного раствора без частиц, который подходит для парентерального введения. Примеры фармацевтически приемлемых носителей включают, но не ограничиваются ими: Вода для инъекций USP; водные носители, такие как, но не ограничиваясь ими, инъекция хлорида натрия, инъекция раствора Рингера, инъекция декстрозы, инъекция декстрозы и хлорида натрия и инъекция лактированного раствора Рингера; смешивающиеся с водой носители, такие как, но не ограничиваясь ими, этиловый спирт, полиэтиленгликоль и полипропиленгликоль; и неводные носители, такие как, но не ограничиваясь ими, кукурузное масло, хлопковое масло, арахисовое масло, кунжутное масло, этилолеат, изопропилмиристат и бензилбензоат.
ПРИМЕРЫ
[00163] Ниже предоставлены примеры для облегчения более полного понимания данного изобретения. Следующие примеры иллюстрируют примерные способы воссоздания и практического применения данного изобретения. Однако объем данного изобретения не ограничен конкретными вариантами реализации, раскрытыми в данных примерах, которые приведены только в целях иллюстрации, поскольку могут быть использованы альтернативные способы для получения аналогичных результатов.
ПРИМЕР 1
[00164] Открытие аналогов АрАР, сохраняющих обезболивающий эффект и имеющих минимальную гепатотоксичность
[00165] Хотя АрАР является одним из наиболее часто используемых лекарственных средств в мире, гепатотоксичность представляет собой наиболее значительный риск, и передозировка им или его применение у пациентов с нарушенной функцией печени является наиболее частой причиной молниеносной печеночной недостаточности. Окисление АрАР до метаболита N-ацетил-п-бензохинонимина (NAPQI) является вероятным механизмом гепатотоксичности. Ранее мы синтезировали аналог АрАР, содержащий гетероциклический фрагмент, соединенный с фрагментом п-ациламинофенола. Аналоги метаболита этого аналога АрАР были синтезированы (SRP 6D и R) для дальнейшего улучшения профиля безопасности этих новых химических веществ с сохраненным обезболивающим эффектом. Данные новые соединения имеют обезболивающий эффект, сравнимый с АрАР, с сохраненным жаропонижающим эффектом на мышиной модели, в то же время демонстрируя пониженную гепатотоксичность в отношении гепатоцитов человека (hHEP). По сравнению с АрАР, SRP 6D и R приводили к снижению уровня лактатдегидрогеназы и увеличению уровня восстановленного глутатиона в hHEP, демонстрировали благоприятный метаболизм цитохромом Р450 и имели снижение тестов функции печени на мышиной модели in vivo. Учитывая широкое использование АрАР в качестве самостоятельного лекарственного средства и в различных комбинированных составах, эти доклинические данные устанавливают новый набор соединений для разработки, которые сохраняют обезболивающий эффект, но заметно снижают гепатотоксичность.
[00166] Введение
[00167] Ацетаминофен, АрАР, также известный как парацетамол, представляет собой N-ацетил-пара-аминофенол (АрАР), наиболее распространенный среди находящихся в свободной продаже обезболивающих лекарственных средств, используемых во всем мире [2]. Химическая структура АрАР, н-ацетил-п-аминофенола и метаболита АрАР N-арахидоноилфеноламина (АМ404) представлена ниже:
[00168]
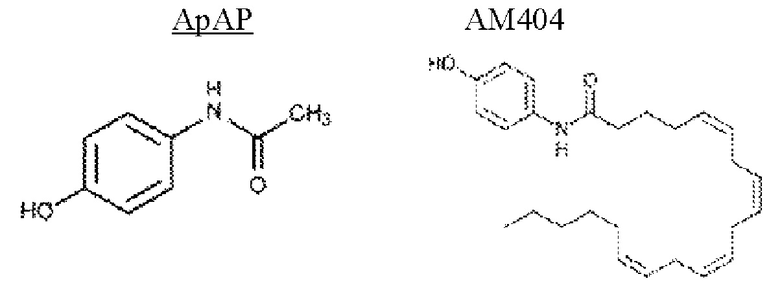
[00169] Анилиновое обезболивающее средство было синтезировано в 1878 году в поисках более безопасного производного ацетанилида, лишенного побочных эффектов, как метгемоглобинемия, которая в свою очередь вызывает цианоз, имеющего обезболивающую и жаропонижающую активность, и был кратко внедрен в клиническую практику в 1887 году [3]. Тем не менее, он не был широко принят в качестве обезболивающего лекарственного средства до 1950-х годов после того, как Броди и Аксельрод продемонстрировали, что АрАР был основным метаболитом аналинов, ацетанилида и фенацетина [4], лишенным метгемоглобинемии и нефропатии, как побочных эффектов, повторно вводя АрАР.
[00170] Несмотря на то, что АрАР использовался в течение нескольких десятилетий, он имеет довольно узкий терапевтический индекс, и при передозировке возникают значительные побочные эффекты, в первую очередь гепатотоксичность. Гепатотоксичность была главной оговоркой при использовании АрАР, главным образом из-за случайной или преднамеренной передозировки, являющейся наиболее распространенной причиной молниеносной печеночной недостаточности в США [5] и в западном мире [2], требующей агрессивной поддерживающей интенсивной терапии, и, в редких случаях, трансплантация печени [6]. Случаи гепатотоксичности АрАР также могут наблюдаться у пациентов с нарушенной функцией печени, не принимающих большие дозы АрАР. Гепатотоксичность возникает в результате окисления АрАР до соответствующего N-ацетил-п-бензохинонимина (NAPQI) [2,7] через цитохром Р450, что приводит к истощению глутатиона, митохондриальной дисфункции и окислительному стрессу.
[00171] Несмотря на эту долгую историю, механизм действия АрАР все еще неясен, и это было проблемой для разработки более безопасных обезболивающих и жаропонижающих аналогов. Отличен от нестероидных противовоспалительных препаратов (НСПВ), которые, в основном, обладают противовоспалительным и только умеренным обезболивающим и жаропонижающим эффектом за счет ингибирования циклооксигеназ (ЦОГ-1, -2) независимо от того, является ли АрАР ингибитором ЦОГ или нет, АрАР не имеет заметных клинических противовоспалительных функций. Некоторые полагают, что он обладает способностью выступать в роли ингибитора ЦОГ, восстанавливая катион радикал протопорфирина в пероксидазном сайте простагландин Н2 синтаз (ЦОГ-ферменты) [8], тем самым снижая простагландиновый ответ за гипертермию [9].
[00172] Открытие в 2005 году метаболита АрАР N-арахидоноилфеноламина (АМ404, Фиг. 1) в виде амида, образованного из 4-аминофенола и арахидоновой кислоты жирной амидгидролазой в головном и спинном мозге [10], позволяет предположить, что АрАР может оказывать свое действие воздействует через активацию рецептора капсаицина/TRPV 1 (переходной рецепторный потенциальный канал, подсемейство V, член 1) [11] и/или системы рецепторов каннабиноида СВ1 [12]. Эти предполагаемые механизмы действия привели к разработке аналогов АрАР, включая адамантильный аналог АрАР [13], который нацелен на ионный канал TRPA1 (член 1 подсемейства А переходного рецепторного потенциального канала). Другая используемая стратегия заключалась в том, чтобы воздействовать на каннабиноидные рецепторы СВ1 и СВ2 через модификацию основного метаболита АрАР, АМ404, путем размещения анандамидной цепи вместо ацетамидогруппы [14].
[00173] Мы выбрали другой подход, заключающийся в создании новых аналогов АрАР, которые не метаболизируются до NAPQI и, следовательно, приводят к минимальной гепатотоксичности. Это было достигнуто путем замены метильной группы на сахарин и различные его производные и аналоги, получив 2-(1,1-диоксидо-3-оксо-1,2-бензизотиазол-2(3Н)-ил)-N-(4-гидроксифенил)алканкарбоксамиды с гетероциклическим фрагментом, связанным с фрагментом п-ациламинофенола (SCP-1) [15]. Эта модификация также значительно улучшает растворимость в воде и сохраняет обезболивающий эффект [16]. В данном документе мы опишем синтез новых обезболивающих средств, 2-[(4-гидроксифенилкарбамоил)метил]сульфамоил-н,н-диэтилбензамида (SRP-6D) и н-[2-(2,3-дигидроксифенил)-этил-2-[(4-гидроксифенилкарбамоил)метил]сульфамоилбензамида (SRP-6R, Фиг. 1), которые являются аналогами метаболита SCP-1 (SCP-1M), проявляющего обезболивающую активность, благоприятный профиль метаболизма cytP450 и минимальную гепатотоксичность.
[00174] Способы
[00175] Синтез производных сахарина
[00176] Получение исходного вещества 5 и производных сахарина 7
[00177] 2-Хлор-N-(4-гидроксифенил)ацетамид 3 синтезировали ацилированием 4-аминофенола 1 2-хлоруксусным ангидридом, используя катализатор NaHSO4, нанесенный на силикагель (NaHSO4 SiO2), в CH2Cl2 при комнатной температуре. Соединение 3 было получено с выходом 75% (Схема 1).
[00178] Получение соединения 5 проводили с использованием процедуры, описанной в Trudell et al. [17]. Таким образом, 2-хлор-N-(4-гидроксифенил)ацетамид 3 и натриевую соль сахарина 4 нагревали с обратным холодильником в ДМ ФА с каталитическим количеством NaI. Производное сахарина 5 получали осаждением льдом/водой и кристаллизовали из смеси этанол/вода, с получением соединения 5 (Схема 1),
[00179] Реакция между соединением 5 и аминами 6 приводит к раскрытию гетероциклического кольца сахарина с получением желаемых N-замещенных амидов (Схема 2). Данные реакции проводили в основном в водном растворе до полного исчезновения 5 согласно ТСХ
[00180] Получение производных сахарина
[00181] NaHSO4 SiO2 катализатор. К раствору 4,14 г (0,03 моль) NaHSO4 H2O в 20 мл воды добавляли 10 г силикагеля (для колоночной хроматографии, 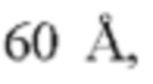 200-400 меш). Смесь перемешивали в течение 15 мин при комнатной температуре, а затем осторожно нагревали на роторном испарителе, пока не было получено свободно текучее белое твердое вещество. Катализатор дополнительно высушивали в вакууме в течение по меньшей мере 48 ч перед использованием.
200-400 меш). Смесь перемешивали в течение 15 мин при комнатной температуре, а затем осторожно нагревали на роторном испарителе, пока не было получено свободно текучее белое твердое вещество. Катализатор дополнительно высушивали в вакууме в течение по меньшей мере 48 ч перед использованием.
[00182] 2-Хлор-N-(4-гидроксифенил)ацетамид, 3. К смеси 4-аминофенола 1 (4,36 г, 40 ммоль) и 2-хлоруксусного ангидрида 2 (8,2 г, 48 ммоль) в CH2Cl2 (200 мл) добавляли NaHSO4 SiO2 (4 г). Смесь нагревали до 35°С в течение 2,5 ч и реакцию контролировали с помощью ТСХ. Осадок растворяли в этаноле и смесь фильтровали. Фильтрат концентрировали и остаток промывали водой, получая 3 (5,74 г, 75%) в виде белого твердого вещества: т.пл. 142-144°С; 1H ЯМР (300 МГц, ДМСО-d6) δ 4,18 (с, 2Н, CH2), 6,71 (д, 2Н, J=8,8 Гц, H-1,5), 7,36 (д, 2Н, J=8,9 Гц, H-2,4), 9,27 (с, 1Н, OH), 10,03 (с, 1Н, NH) м.д. 13С ЯМР (75 МГц, ДМСО-d6) δ 44,0, 115,6, 121,6, 130,5, 154,2, 164,3 м.д.
[00183] 2-(1,1-Диоксо-1,2-дигидробензо[d]изотиазол-3-он-2-ил)-N-(4-гидроксифенил)ацетамид, 5. 2-Хлор-N-(4 гидроксифенил)ацетамид 3 (5 г, 27 ммоль) и соль моногидрата натрия сахарина 4 (7,25 г, 32,4 ммоль) смешивали вместе в присутствии NaI (0,015 г, 2,30 ммоль) в ДМФА (15 мл). Смесь кипятили с обратным холодильником (160°С) в течение 2 ч, охлаждали до 25°С и выливали в холодную воду до тех пор, пока не образовывался дополнительный осадок для вытеснения ДМФА. Липкий белый осадок собирали вакуумной фильтрацией и оставляли сушиться на воздухе в течение 90 мин. Полученный остаток растворяли в 50% этаноле-воде и кристаллизовали, получая 6,72 г 5 в виде белых кристаллов, выход 75%: т.пл. 204-207°С; 1Н ЯМР (300 МГц, ДМСО-d6) δ 4,52 (с, 2Н, CH2), 6,71 (д, 2Н, J=8,5 Гц, H-1,5), 7,34 (д, 2Н, J=8,5 Гц, H-2,4), 8,05 (м, 2Н, 2H-аром.), 8,15 (д, 1Н, J=7,1 Гц, H-аром.), 8,35 (д, 1H, J=7,4 Гц, H-аром.), 9,26 (с, 1H, OH), 10,06 (с, 1H, NH) м.д. 13С ЯМР (75 МГц, ДМСО-d6) δ 40,6, 115,2, 121,0, 121,8, 125,2 126,5, 130,1, 135,5 135,9, 136,0, 153,7, 158,8 162,6 м.д.
[00184] 2-[(4-Гидроксифенилкарбамоил)метил]сульфамоил-N-метилбензамид (7а). OSA-6c
[00185] К водному раствору метиламина (10 мл, 40%, 119 ммоль) добавляли соединение 5 (0,6 г, 1,8 ммоль). Смесь перемешивали в течение 35 мин и сушили в вакууме, получая белое твердое вещество. Полученное соединение очищали колоночной флэш-хроматографией (этилацетат/гексан 9:1), получая 0,59 г, 89%: т.пл. 93-95°С; 1Н ЯМР (300 МГц, ДМСО-d6) δ 2,80 (д, 3Н, J=4,5 Гц, СН3), 3,67 (с, 2Н, СН2), 6,64 (д, 2Н, J=8,8 Гц, H-1,5), 7,20 (д, 2Н, J=8,8 Гц, H-2,4), 7,49 (с, 1H, NH), 7,55 (д, J=7,2 Гц, H-аром.), 7,65 (м, 2Н, аром.), 7,86 (д, J=7,4 Гц, H-аром.), 8,69 (с, 1H, J=4,5 Гц, NH), 9,20 (с, 1H, NH), 9,76 (с, 1H, OH) м.д. 13С ЯМР (75 МГц, ДМСО-d6) δ 26,7, 39,1, 39,4, 39,7, 39,9, 40,2, 40,5, 40,8, 46,3, 115,4, 129,0, 121,4, 129,6, 130,3, 130,4, 133,2, 136,2, 137,2, 153,9, 165,8, 169,2 м.д.
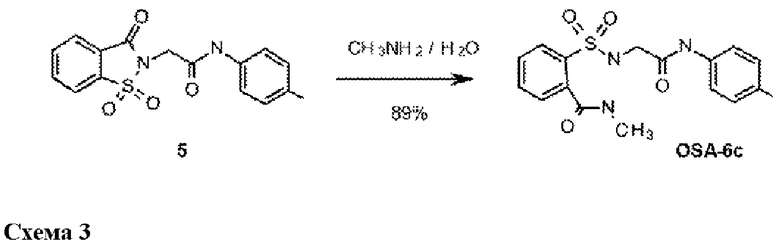
[00186] 2-[(4-Гидроксифенилкарбамоил)метил]сульфамоил-N,N-диэтилбензамид (7b). OSA-6d
[00187] К раствору диэтиламина (0,657 г, 0,93 мл, 9 ммоль) в воде (15 мл) добавляли соединение 5 (1 г, 3 ммоль). Смесь перемешивали в течение 2 ч при комнатной температуре. Полученное твердое вещество сушили при пониженном давлении и очищали посредством колоночной флэш-хроматографии (этилацетат/EtOH 6: 4), получая белое твердое вещество (0,960 г, 76%): т.пл. 187-189°С; 1H ЯМР (300 МГц, ДМСО-d6) δ 1,19 (т, J=7,2 Гц, 6Н, 2CH3), 2,93 (кв., J=7,2 Гц, 4Н, 2CH2), 3,55 (с, 2Н, СН2), 6,63 (д, J=8,7 Гц, 2Н, H-аром.), 7,23 (д, 2Н, J=8,7 Гц, H-аром.), 7,40 (т, J=7,2 Гц, 1H), 7,52 (т, J=7,1 Гц, 1H, H-аром.), 7,60 (д, J=7,2 Гц, 1H, H-аром.), 7,73 (д, J=7,5 Гц, 1H, H-аром.), 8,93 (уш. с, 1H, NH), 9,20 (с, 1H, NH 9,99 (с, 1Н, OH) м.д. 13С ЯМР (75 МГц, ДМСО-d6) δ 11,1, 41,3, 46,4, 114,8, 120,8, 127,1, 127,2, 129,9, 130,0, 132,0, 135,3, 141,6, 153,2, 165,4 м.д.
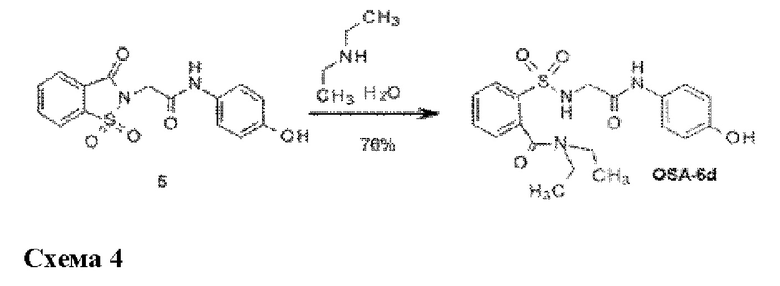
[00188] 2-[(4-Гидроксифенилкарбамоил)метил]сульфамоил-N-(2-пирролидин-1-илэтил)бензамид (7с). OSA-6U
[00189] К раствору 1-(2-аминоэтил)пирролидина (0,684 г, 0,76 мл, 6 ммоль) в ацетонитриле (15 мл) добавляли соединение 5 (0,941 г, 2,8 ммоль). Смесь перемешивали в течение 1 ч при комнатной температуре. Полученное твердое вещество отфильтровывали и очищали колоночной флэш-хроматографией (этилацетат/EtOH 9:1), получая белое твердое вещество (1,1 г, 80%): т.пл. 141-142°С. 1H ЯМР (300 МГц, ДМСО-d6) δ 1,71 (с, 4Н), 2,54 (с, 4Н), 2,64 (с, 2Н), 3,43 (с, 2Н), 3,85 (с, 2Н), 6,59 (д, J=7,8 Гц, 2Н), 6,95 (д, 2Н, J=7,7 Гц, 2Н), 7,44 (дд, J=7,4 Гц, 2Н), 7,56 (т, J=7,5 Гц, 1H), 7,80 (д, J=7,7 Гц, 1H), 9,21 (с, 1H), 8,81 (с, 1H), 9,87 (с, 1H) м.д. 13С ЯМР (75 МГц, ДМСО-d6) 523,6, 38,2, 46,0, 53,4, 54,5, 115,7, 122,2, 129,5, 129,8, 133,2, 137,2, 137,5, 154,5, 166,9, 167,8 м.д.
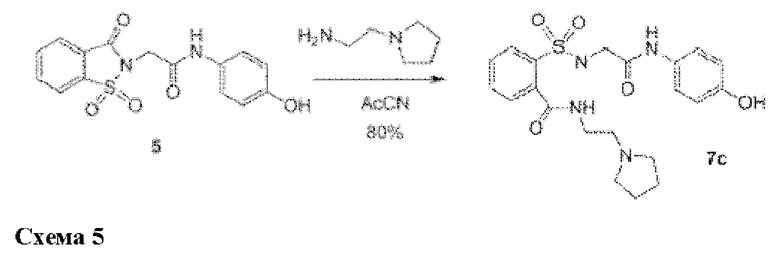
[00190] N-[2-(2,3-дигидроксифенил-1)-этил-2-[(4-гидроксифенилкарбамоил)метил]сульфамоилбензамид (7d), OSA-6r
[00191] Дофамин гидрохлорид (0,381 г, 2 ммоль), Na2CO3 (0,215 г, 2 ммоль) и соединение 5 (0,333 г, 1 ммоль) добавляли к 10 мл EtOH и нагревали до 65°С. Смесь перемешивали в течение 18 ч. Полученное твердое вещество отфильтровывали и очищали посредством колоночной флэш-хроматографии (этилацетат/EtOH 9:1), получая соединение 7d (0,350 г, 71%): т.пл. 87-89°С; 1Н ЯМР (300 МГц, ДМСО-d6) δ 2,68 (т, J=7,4 Гц, 2Н, CH2), 3,40 (т, J=6,8 Гц, 2Н, CH2), 3,68 (д, J=5,8 Гц, 2Н, CH2), 6,50 (дд, J=8,0, 1,5 Гц, 1H, H-аром.), 6,70-6,58 (м, 4Н), 7,21 (д, J=8,7 Гц, 2Н, H-аром), 7,50-7,39 (м, 2Н, Н-аром., NH), 7,63 (дкв, J=7,5, 6,8 Гц, 2Н, Н-аром), 7,87 (дд, J=7,3, 1,0 Гц, 1H, Н-аром.), 8,65 (с, 1H, ОН), 8,77 (с, 1Н, ОН), 8,82 (т, J=5,6 Гц, 2Н, NH), 9,18 (с, 1H, NH), 9,75 (с, 1H, ОН) м.д. 13С ЯМР (75 МГц, ДМСО-d6) δ 35,0, 42,1, 46,7, 115,7, 116,2, 116,7 120,0, 121,6, 129,2, 129,8, 130,5, 130,6, 130,7, 133,4, 136,4, 137,4, 144,2, 145,7, 154,1, 165,9, 168, 8 м.д.
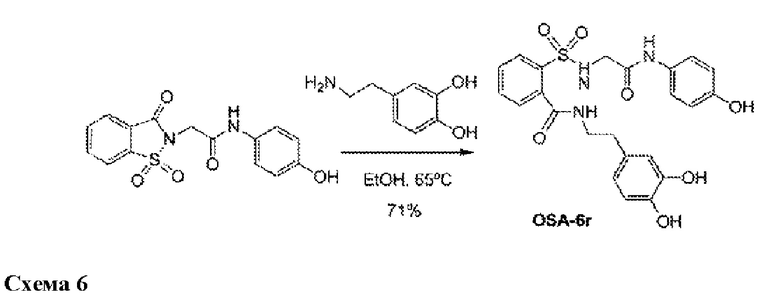
[00192] Дополнительные аналоги описанных выше новых соединений были получены аналогично в результате реакции соединения 5 с различными реагентами посредством процессов, аналогичных описанным выше в отношении избранных аналогов, описанных в данном документе новых обезболивающих лекарственных средств. В некоторых вариантах реализации соединение 5 смешивали с желаемым реагентом в растворе в воде, этаноле, метаноле, ацетонитриле, тетрагидрофуране, водном метиламине или любых других органических или неорганических растворителей, известных в данной области техники, в зависимости от ситуации.
[00193] Потенциальные реагенты включают NH3, NH(CH3)2, NH2CH3, NH(CH2CH3)2, N2(CH2)4HCH2C6H5, NH2(CH2)2C6H5, NH2CH2C6H5, NHO(CH2)4, NH2CH2CH2CH2CH3, NH2CH2C6H4CH3, NH2CH2C6H3Cl2, NH2CH2C6H5CH3, NH2CH2C6H5Cl, NH2CH2C6H5NO2, NH2C5H4, NH2C5H4, NH2CH2C(CH3)2, NH2C(CH3)2, NH2CH2CH2C6H3(OH)2, NH2CH2C6H4N, NH2CH2C6H3NCH3, NH2CH2CH2C4H4N, NH(CH3)CH2CH2OH, NH2CH2CH(OH)CH2NH2, или их фармацевтически приемлемые соли.
[00194] Аналоги, полученные в соответствии со способами синтеза, описанными в данном документе, включают композиции, имеющие следующие химические формулы:
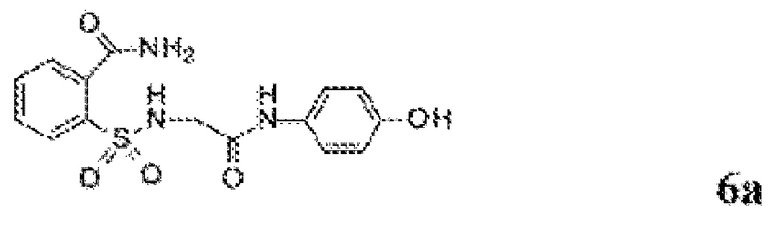
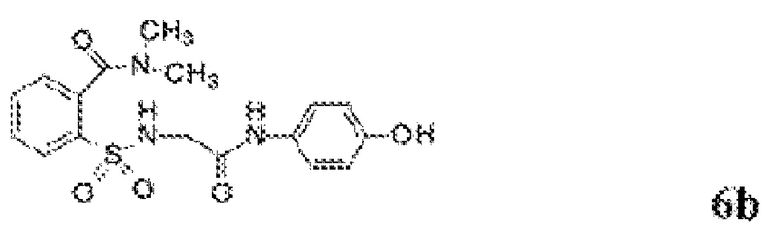
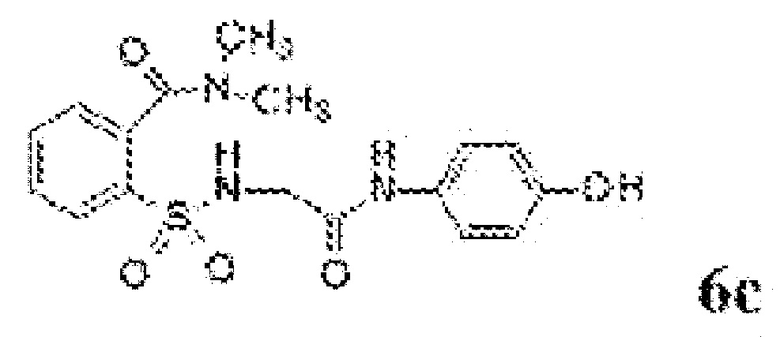
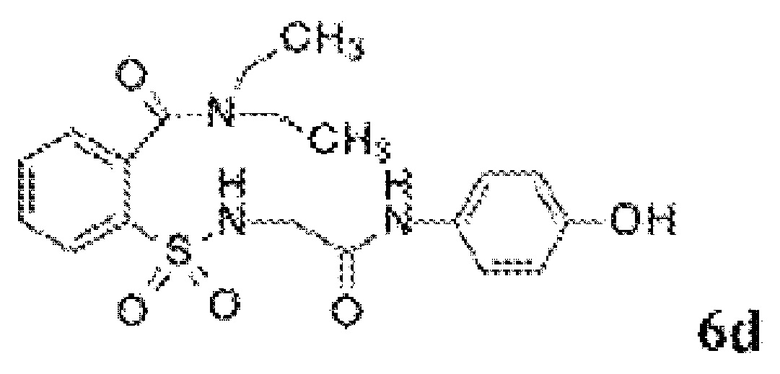
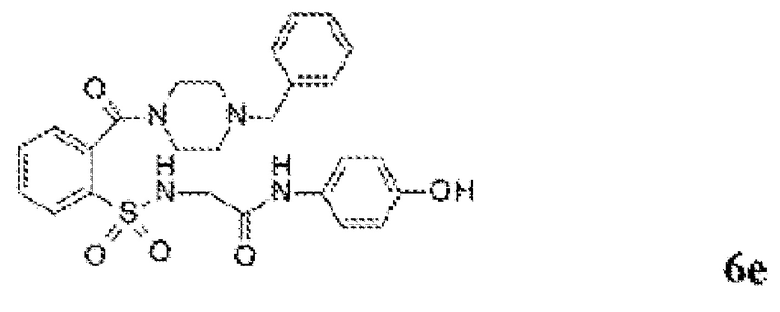
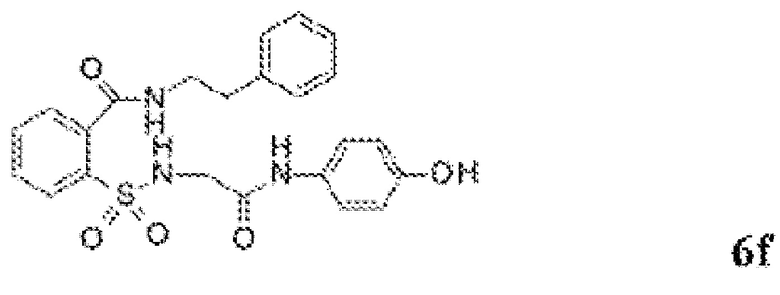
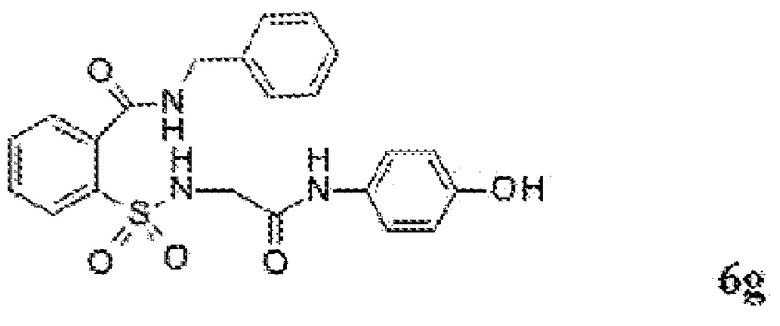
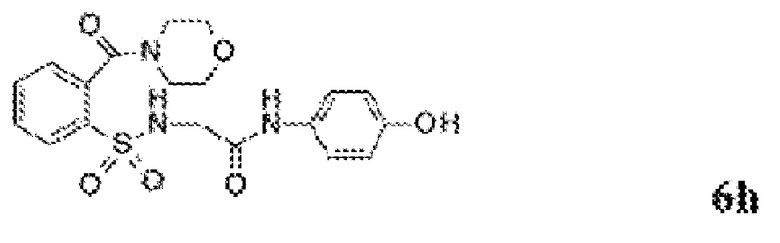
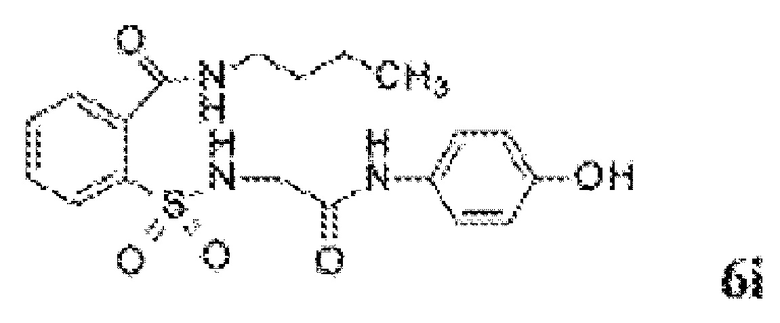
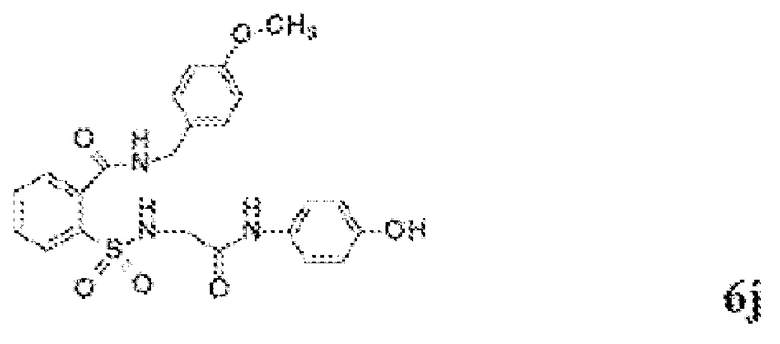
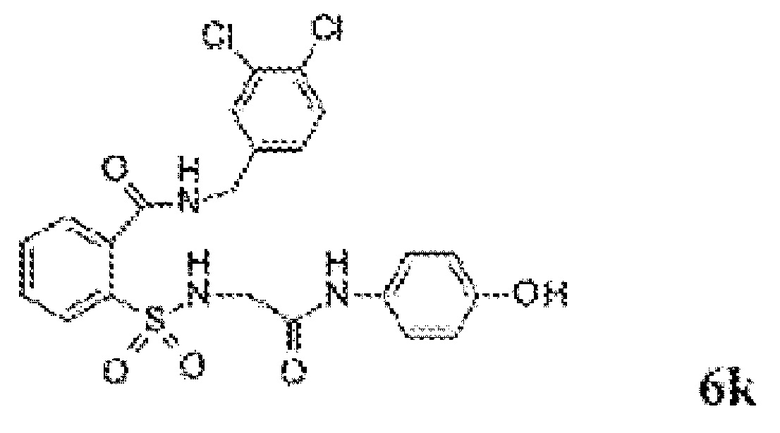
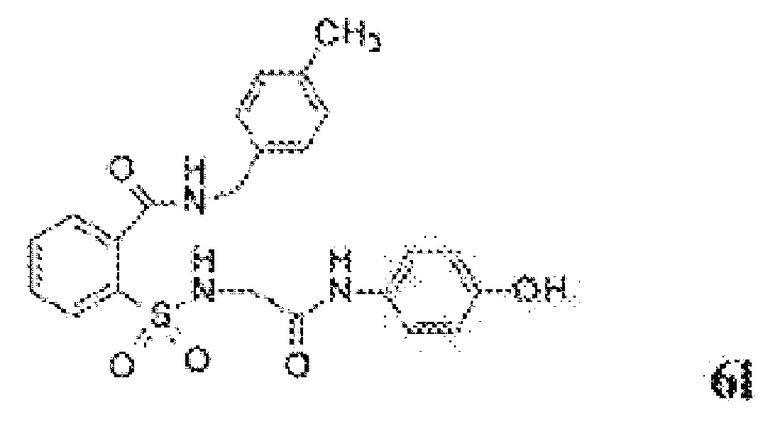
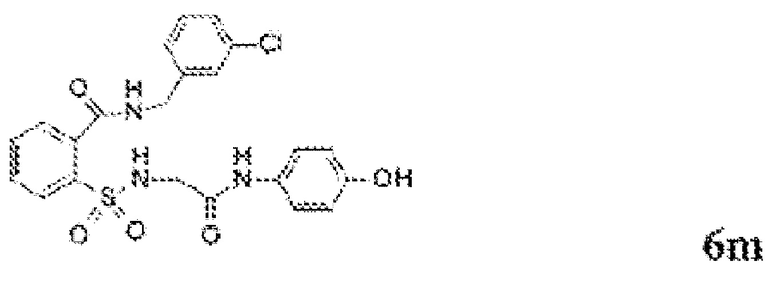
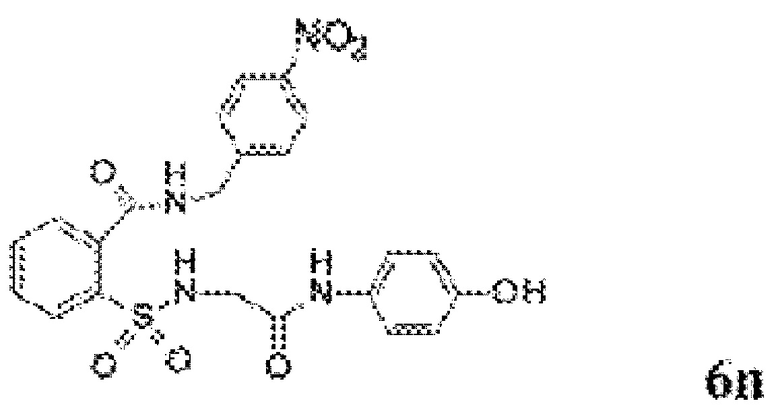
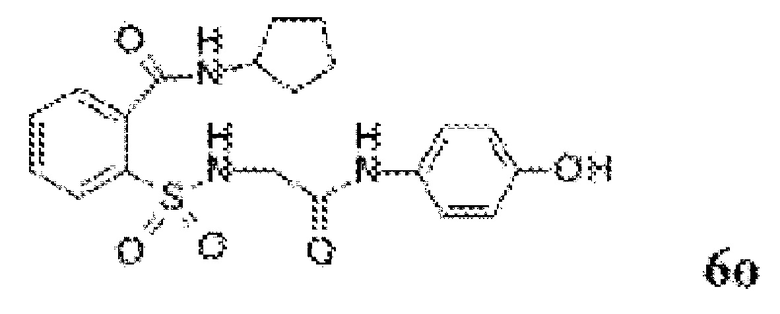
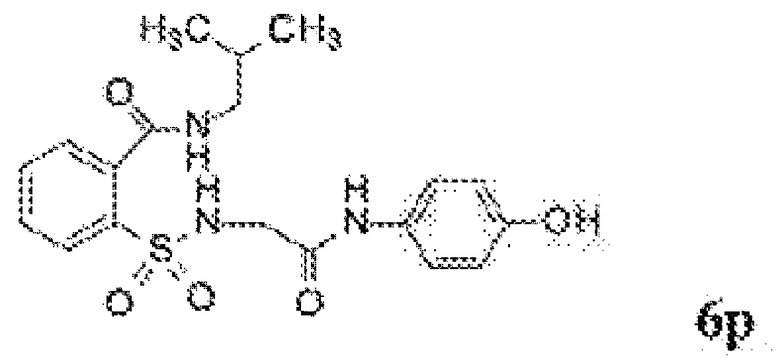
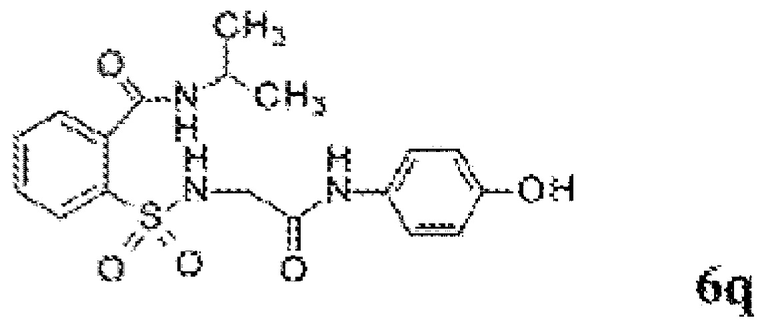
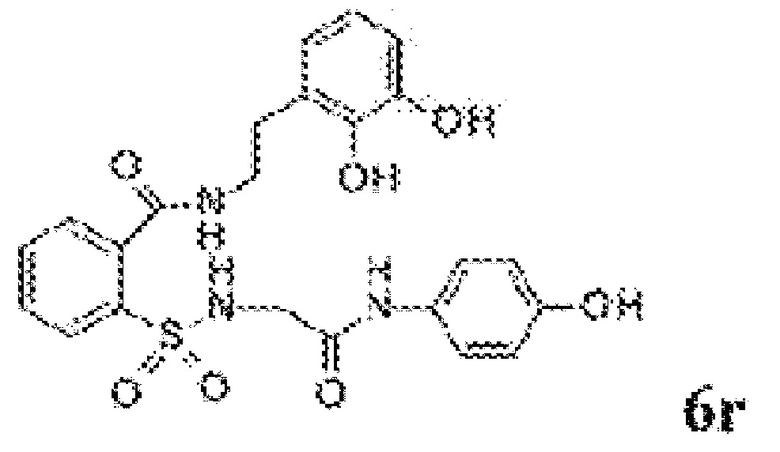
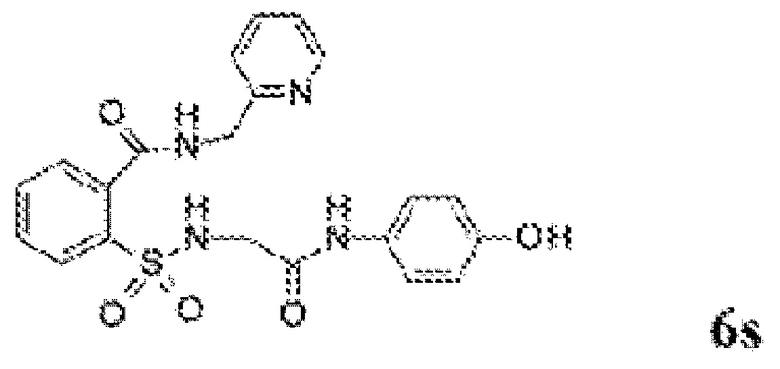
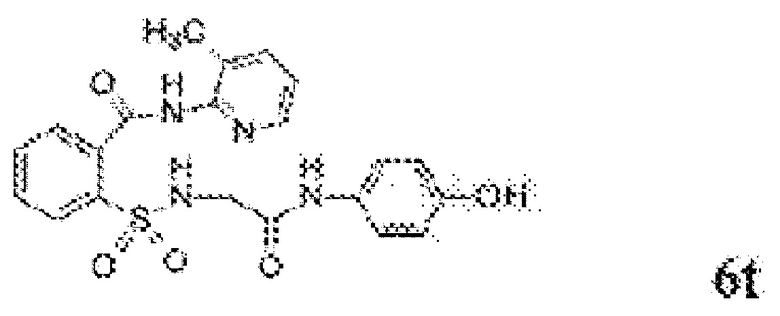
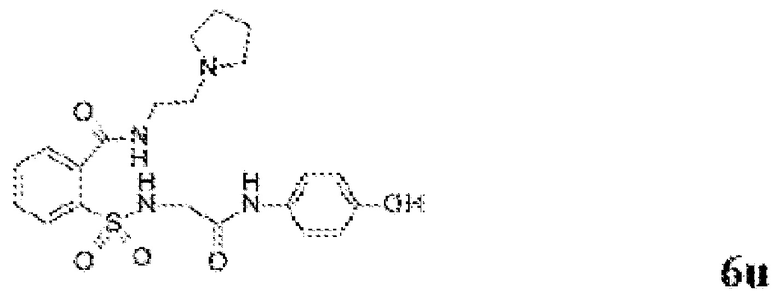
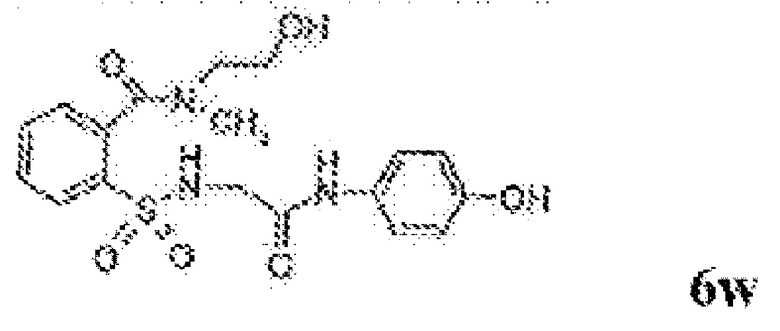
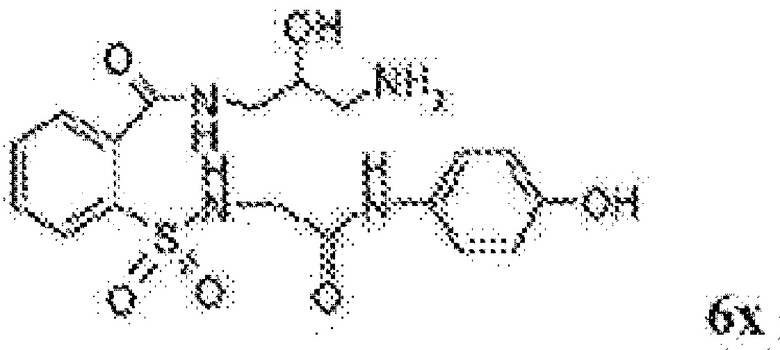
[00195] Анализы на обезболивающий эффект
[00196] Два различных анализа были использованы для количественной оценки обезболивающего действия соединений. Все тесты были выполнены на самцах мышей CD1. Соединения SCP-1, SCP-1M и SRP-6D, R. (South Rampart Pharmaceuticals, Новый Орлеан, Луизиана) или ARAP (Sigma, Сэнт Луис, Миссури) вводили в концентрации 75 мг/кг после суспендирования в носителе (0,9% физиологический раствор). В некоторых вариантах реализации носитель может содержать агент К, 0,2%, Bio-serve или Labrafil 1944 (Готтефозе, Франция). Соединение или носитель вводили перорально с помощью лаважа под кратковременной анестезией галотаном животным в течение ночи.
[00197] Анализ корчей индуцированных введением уксусной кислоты в брюшную полость. Сокращение мышц живота и растяжение задних конечностей являются ответом на внутрибрюшинную (ip) инъекцию раствора уксусной кислоты, как описано Hendershot and Forsaith, 1959. В этой модели висцеральной боли абдоминальные сокращения (корчи) у мышей индуцируются внутрибрюшинной инъекцией 0,4% уксусной кислоты в дозе 10 мл/кг через 25 мин после введения препарата. Количество корчей подсчитывается для 10 мм, начиная с 5 мм после введения уксусной кислоты. Все животные (самцы мышей CD1) голодали в течение ночи (15 ч) перед тестированием, и соединения вводили перорально животным, принадлежащим к группам обработки, соединениям АрАР и SRP в количестве 75 мг/кг массы тела. Данные выражены как среднее +/-СОИ, n=7.
[00198] Анализ отдергивания хвоста. Обезболивающий эффект препаратов определяли с помощью времени реакции (латентности) мышей на термостимуляцию кончика хвоста. Все животные (самцы мышей CD1) голодали в течение ночи (15 ч), и их базовая латентность отдергивания хвоста (сек) регистрировалась с использованием измерителя обезболивания хвоста IITS Tail Flick. Хвост каждой мыши подвергался воздействию сфокусированного луча света, и латентность отдергивания хвоста с пути стимула регистрировалась в электронном виде с использованием фотоэлемента измерителя обезболивания IITC Tail Flick. Интенсивность стимула была отрегулирована таким образом, чтобы исходные задержки составляли 3-6 сек. После измерения исходной латентности, лекарства вводили перорально животным, принадлежащим к группам лечения - (соединения АрАР и SRP при 600 мг/кг массы тела); контрольная группа получала носитель (только 0,9% физиологический раствор). Через 30 мин после введения снова регистрировали задержку отдергивания хвоста, чтобы определить общее изменение задержки. Процент максимального обезболивания для каждой мыши рассчитывали по формуле. Процентное обезболивание=100*{[(Латентность отдергивания хвоста после инъекции лекарственного средства)-(Латентность отдергивания хвоста базовой линии):(12 сек Время отключения)-(Базовая латентность)]}. Данные выражены как среднее ± СОИ, n=10.
[00199] Жаропонижающий эффект
[00200] Жаропонижающий эффект соединений оценивали с использованием гипертермии, вызванной пекарскими дрожжами. Все животные (самцы мышей CD1) голодали в течение ночи (15 ч), и их базовая температура регистрировалась с использованием зонда ректального термометра Коул-Палмера. Затем им вводили внутрибрюшинно пирогенную дозу пекарских дрожжей (1,5% дрожжей, 0,1 мл/10 г массы тела), в то время как контрольная группа и группа носителя получали внутрибрюшинную инъекцию носителя (0,9% физиологический раствор). Температуру снова регистрировали через 4 ч, после чего соединения вводили перорально лихорадочным животным, принадлежащим к группам лечения - (соединения АрАР и SRP при 300 мг/кг массы тела). Через два ч после инъекции ректальные температуры регистрировали еще раз, чтобы определить общее изменение температуры тела. Процентное изменение температуры тела рассчитывается по формуле. Процентное изменение = [(Общее изменение температуры тела)/(Базовая температура) * 100]. Данные выражены как среднее ± СОИ, n=10.
[00201] Клетки/Клеточные линии
[00202] Клеточная линия гепатоцитов (клетки HEPG-2) и первичные гепатоциты человека (hHEP). Клетки HepaRG, полученные от Thermo Fisher Scientific (Invitrogen), представляют собой терминально дифференцированные печеночные клетки, которые получены из линии клеток-предшественников печени, которая сохраняет характеристики первичных гепатоцитов человека. Клетки HepaRG выращивали и поддерживали в среде ЕМЕМ, содержащей NEAA (не незаменимые аминокислоты), дополненной 10% эмбриональной бычьей сывороткой (FBS), и инкубировали при 37°С/5% CO2. hHEP, приобретенный у SCKISUI Xenotech, был получен от белого 61 летнего мужчины-донора мужского пола, который не употреблял алкоголь и не курил. hHEP выращивали в НСМ (Clonetics, Уолкерсвиль, Мэриленд) и поддерживали в НММ (Clonetics, Уолкерсвиль, Мэриленд) при 37°С/5% СО2, культуры (80% слияния) клеток HepaRG и hHEP, растущие в 6 и 24 луночных планшетах, соответственно, выдерживали 6-8 ч в бессывороточной среде (ЕМЕМ, 0,5% FBS для HEPG-2 и НММ для первичных гепатоцитов) перед добавлением обезболивающих средств. Клетки, лишенные сыворотки, обрабатывали АРАР, SRP-6D, R, SCP-1, SCP-1M или контрольным носителем в течение 6-8 ч при 37°С.
[00203] Тест на гепатотоксичность для опосредованного АрАР (АРАР) повреждения печени (A1L1) в гепатоцитах
[00204] Анализ LDH. С использованием набора для анализа цитотоксичности Pierce LDH от Thermo Scientific клетки инкубировали в присутствии различных лекарственных соединений с последующим сбором супернатанта среды. Высвобождение LDH измеряли в 96-луночных планшетах. Поглощение измеряли при 490 нм и 680 нм, и конечным результатом было поглощение, наблюдаемое при 680 нм, вычитаемое из поглощения, наблюдаемого при 490 нм (А490 нм - А 680 нм). GSH Анализ. С помощью реагента ThiolTracker Violet Glutathione Detection от Molecular Probes (Invitrogen) после инкубации гепатоцитов в присутствии различных соединений инкубационную среду удаляли, клетки промывали кондиционированной средой D-PBS с последующей инкубацией с предварительно нагретым красителем ThiolTracker Violet (рабочий раствор, приготовленный в соответствии с инструкциями производителя) в течение 30 мин. Флуоресценцию измеряли на следующих длинах волн: возбуждение (404 нм) и излучение (526 нм). Окончательный результат выражали в относительных единицах флуоресценции (ОЕФ), что указывает на клеточный уровень восстановленного глутатиона (GSH) в интактных клетках.
[00205] Анализ печеночного перехода. Уровень аланин аминотрансферазы (АЛТ), аспартат аминотрансферазы (ACT) и щелочной фосфатазы (ALP) определяли после введения мышам CD1 600 мг/кг соединений - АрАР и SRP D и R перорально через желудочный зонд. Анализы проводили с сывороткой, собранной у мышей, которым инъецировали соединения или носитель, после голодания в течение ночи (15 ч). После введения лекарственного средства мышам ad libitum давали воду и пищу.
[00206] Профиль метаболизма цитохромом Р450
[00207] Набор для скрининга VIVIDCYP450 (Life Technologies. In vitro gen/Thermo Fisher Scientific) использовали для высокопроизводительного скрининга in vitro. В данном документе каждое соединение было смешано с основной предварительной смесью, содержащей CYP450 BACULOSOMES (которые представляют собой микросомы, полученные из клеток насекомых, экспрессирующих специфический изофермент Р450 человека), реагент и систему регенерации, которая содержала глюкозо-6-фосфат и глюкозо-6-фосфат дегидрогеназу. Смесь предварительно инкубировали при комнатной температуре в течение 20 мин. После этого добавляли каждый субстрат, специфичный для фермента CYP, и NADP' и смесь инкубировали при комнатной температуре в течение 30 минут. Реакцию останавливали добавлением 0,5 М трис-основания. Активность CYP оценивали путем измерения флуоресценции флуоресцентного метаболита, генерируемого из каждого субстрата, специфичного для фермента CYP.
[00208] Статистика. Изменения порогов отмены или латентных периодов, вызванных лекарственным средством, сначала анализировали односторонним ANOVA. Сравнения между эффектами различных лекарств были затем подвергнуты t-тестам для непарных средств. Значение р<0,05 считалось значимым.
[00200] Определение метаболитов: Для обнаружения NAPQI использовали газовую хроматографию (Фиг. 11). Не будучи связанными теорией, токсичность АрАР опосредуется токсичным метаболитом N-ацетил-бензохинонимином (NAPQI), который истощает печеночный и почечный глутатион, цитопротективный эндогенный метаболит. Газовая хроматография демонстрирует заметно более низкие уровни метаболизма до NAPQI для отдельных соединений SRP по сравнению с АрАР, что объясняет снижение гепатотоксичности.
[00210] Двухмесячные мыши CD-I голодали в течение ночи. На следующее утро после легкой анестезии галотаном им вводили перорально 5а, 3 ммоль/кг в Твин 20 (носитель). Их помещали в метаболические клетки Nalgene (по две мыши на клетку) с водой ad libitum. Питание было предоставлено через 6 ч после введения дозы. Моча была собрана в пластиковый контейнер, который содержался во льду в течение 24-чого периода сбора. Ее хранили при 20°С до использования. Во время инъекции ВЭЖХ аликвоты центрифугировали в микроцентрифуге при 6000 об/мин, 15°С в течение 10 минут, фильтровали с помощью нейлоновых фильтров (0,45 мкм) и сразу использовали или лиофилизировали.
[00211] В анализе ВЭЖХ-МС вводили объем 20 мкл. Когда использовали лиофилизированные образцы, их растворяли в смеси ацетонитрил-0,1% раствор ацетата аммония, рН 7 (50:50, об./об.). ВЭЖХ-МС анализы проводили на приборе Agilent 1100. Аналитическая колонка представляла собой колонку Luna 150 ⋅ 4,6 мм, С18 (5 мкм) Phenomenex. Подвижная фаза автоматически дегазировалась электронной системой дегазирования. Перед анализом колонку уравновешивали и использовали градиентную программу для анализа образцов. Скорость потока поддерживалась на уровне 1,5 мл/мин, а колонка поддерживалась при 45°С.
[00212] Результаты
100213] Молекулярная масса соединения и рассчитанный Log Р (cLog Р). Низкие значения Clog P означают продукты с более коротким периодом полураспада, но более высокие значения cLog Р указывают на потенциальные трудности с поглощением соединения. Следовательно, промежуточные значения около 2, вероятно, желательны, особенно для соединений, способных преодолевать гематоэнцефалический барьер.
[00214]
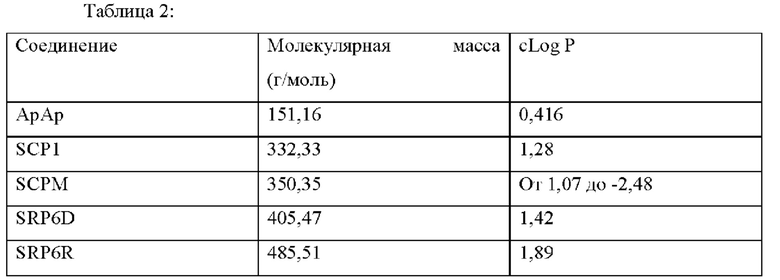
[00215] В нашем проекте было синтезировано 21 соединение, чтобы найти новые химические аналоги метаболита гетероциклического фрагмента, связанного с п-ациламинофенольным фрагментом АрАР, но мы сфокусируемся на двух (SRP 6D и R), которые показали обезболивающий эффект, сравнимый с АрАР, минимальную гепатотоксичность, жаропонижающий эффект и благоприятный метаболизм цитохромом Р450. Анализ 21 соединения, которое было синтезировано, выявил некоторые с низкими значениями cLogP, ~ 0,5, что указывает на вероятную плохую биодоступность при использовании в качестве пероральных препаратов. Тем не менее, SRP6D (Clog Р: 1,42) и SRP6R (Clog Р: 1,89) сопоставимы с АрАР (Clog Р 0,91) и SCP-1 (Clog Р 1,28), Табл. Обратите внимание, что SCP-1M имеет интервал Clog Р, который варьируется от 1,07 до -2,48 в зависимости от рН среды, поскольку он имеет поляризуемую ионизируемую группу.
[00216] В двух различных моделях обезболивающего эффекта у мышей in vivo, корчей, вызванных абдоминальным введением уксусной кислоты, и тестом отдергивания хвоста, SRP6D и R сравнимы с АрАР (Фиг. 1). Количество корчей, вызванных инъекцией уксусной кислоты, составило 15,7±1,2 (n=3) для SRP6D (n=7, р<0,02) и 9,5±4, для SRP6R (n=7, р<0,008; Фиг. 1А) по сравнению с 42,3±7,2 только для транспортного средства. Вторая серия экспериментов с n=7 демонстрирует обезболивающую активность различных соединений SRP, которая похожа на АрАР, но ниже, чем контроль (необработанные данные для Фиг. 1А).
[00217] Таблица 3. Обезболивающая активность соединений SRP по сравнению с АрАР, оцененная с помощью анализа корчей, вызванных абдоминальным введением уксусной кислоты
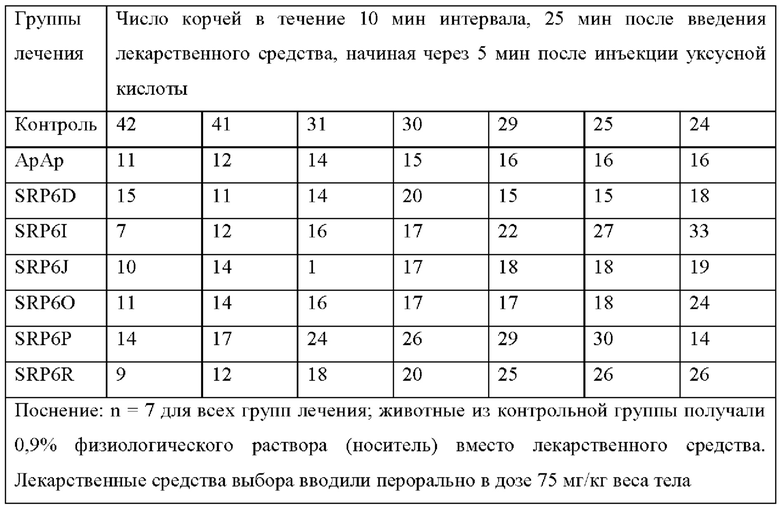
[00218] Тест отдергивания хвоста демонстрировал сопоставимую значительное обезболивание для SRP6D, R относительно АрАР и отмечал улучшенную задержку по сравнению только с носителем (Фиг. 1B).
[00219] SRP6D и SRP6R сохраняют жаропонижающий эффект, сравнимый с АрАР (Фиг. 4). Жаропонижающий эффект определяли с использованием двух разных мышиных анализов. Температурные кривые демонстрируют сопоставимый жаропонижающий эффект относительно АрАР для SRP6D и SRP6R на модели LPS-индуцированной лихорадки у мышей (Фиг. 4.А, В). Обратите внимание, что через 2 ч, 8 ч и 10 ч, жаропонижающий эффект сопоставим с АрАР, SRP6D и SRP6R. Модель жаропонижающего эффекта, вызванная пекарскими дрожжами, продемонстрировала сходные жаропонижающие эффекты АрАР и SRP6D и SRF6R (Фиг. 4С, D). Сопоставимое действие жаропонижающих средств на гипертермию, вызванную пекарскими дрожжами, отмечено на Фиг. 4Е для АрАР и SRP6D, и SRP6R.
[00220] Затем определяли профиль гепатотоксичности для соединений SRP6D и R по сравнению с АрАР и производными сахарина АрАР первого поколения, SCP-1 и SCP-1 М, как в HepaRG, так и в hHEP. Снижение токсичности было отмечено для SRP6D и R по сравнению с АрАР: высвобождение лактатдегидрогеназы (LDH) последовательно уменьшалось, а количество восстановленного глутатиона (GSH) увеличивалось для SRP6D и R, тогда как АрАР приводил к увеличению высвобождения LDH (Фиг. 3) и истощению GSH (Фиг. 2) в зависимости от времени и дозы. В печени мыши и гепатоцитах человека (HepaRG и hHEP) при эквивалентных терапевтических дозах для человека наблюдалось дозозависимое и зависящее от времени влияние снижения гепатотоксичности через снижение LDH и увеличение выделения GSH с прогрессирующими клиническими признаками повреждения печени для SRP6D и R, но не АрАР. Отмечено значительное снижение функциональных проб печени для SRP6D и R, по сравнению с АрАР, наибольшим из которых является АЛТ. Отмечены повышенные уровни активности ферментов АЛТ, ACT и АР для АРАР, тогда как SRP6D и R были аналогичны контролю (только носитель, Фиг. 3С).
[00221] Наконец, благоприятный метаболизм цитохромом Р450 для SRP6D и SRP6R был отмечен в различных изоформах цитохрома Р450, включая CYP3A4, CYP2D6 и CYP2E1. SRP6D и SRP6R ингибируют активность CYP2E1 и CYP3A4 только на -25% по сравнению с 50% для АрАР и имеют незначительный ингибирующий эффект для CYP2IM (Фиг. 5).
[00222] Обсуждение
[00223] Мы показали минимальную гепатотоксичность для двух новых соединений, которые являются аналогами метаболита сахаринового производного АрАР. Используя две хорошо известные линии клеток печени [18] hHEP и HepaRG в качестве клеточных моделей печени in vitro, мы показали последовательное снижение высвобождения LDH и повышенное снижение выделения глутатиона (GSH). Эти клетки обладают самой высокой прогностической способностью к острой печеночной недостаточности, вызванной АрАР [19]. Снижение гепатотоксичности было дополнительно подтверждено на модели in vivo. Поскольку механизмы гепатотоксичности АрАР одинаковы как у людей, так и у мышей [20], мы выбрали мышей для изучения клинических признаков повреждения печени (LFT).
[00224] В анализах токсичности in vitro использовались hHEP и HepaRG, поскольку они являются более воспроизводимыми для токсикологических исследований. Несмотря на врожденные ограничения первичных клеточных культур, включая ограниченную доступность и короткую продолжительность жизни, hHEPs являются лучшими клетками, доступными для изучения гепатотоксичности in vitro [21]. Однако при использовании первичной клеточной культуры существуют внутренние трудности, и недавний токсигеномный анализ показал, что HepaRGs, клеточная линия печени человека, полученная из гепатоцеллюлярной карциномы, экспрессирует пять специфичных генов при сходных уровнях hHEP [19] и демонстрирует лучший фенотип взрослых гепатоцитов из всех доступных клеточных линий печени [18].
[00225] Несмотря на широкое применение в качестве безрецептурного обезболивающего лекарственного средства во всем мире, основным недостатком АрАР является его дозозависимая гепатотоксичность, терапевтический индекс которого еще более сужается у лиц с нарушенным резервом печени. Тем не менее, случаи непреднамеренной или преднамеренной передозировки не могут быть распознаны в этот короткий период времени, и АрАР остается наиболее распространенной причиной острой молниеносной печеночной недостаточности в Соединенных Штатах [5], обычно после случайного приема больших количеств или потребления более 3-4 г в день у пациентов с нарушениями функции печени. АрАР доступен в виде одноразового безрецептурного лекарственного препарата (ОТС) и в сочетании с другими безрецептурными лекарственными средствами, включая противоотечные, а также в виде рецептурных АрАР-опиоид составов. АрАР, вызывающий ALP, который вероятно, возникает в непреднамеренных случаях, когда люди принимают внутрь АрАР, не зная о том, что АрАР присутствует в этих различных составах. В США ежегодно около 30 000 пациентов поступают в больницы для лечения гепатотоксичности АрАР [22]. Хотя большинство пациентов испытывают только легкую заболеваемость, такую как гепатит, холестаз и кратковременное повышение уровня печеночных трансаминаз, острая печеночная недостаточность развивается у пациентов без лечения, принимающих большие дозы, и может прогрессировать до судорог, комы и смерти, если не будет своевременно распознана и подвергнута лечению. N-ацетилцистеин (NAC) может предотвратить повреждение АрАР-печени, предоставляя цистеин для восстановления GSH, если он вводится в течение 12 ч после проглатывания АрАР.
[00226] Другое применение этой технологии может заключаться в том, чтобы обуздать крупную эпидемию опиоидов в Соединенных Штатах. В 2016 году смертность от передозировки наркотиков достигла пика в > 65000 случаев, главным образом из-за опиоидных болеутоляющих и героина. Травмы на рабочем месте могут быть причиной многих из этих случаев, потому что есть свидетельства того, что назначенные оральные наркотики являются вероятным источником, и две из самых больших концентраций смертельных случаев передозировки находятся в Аппалачах и на юго-западе США (CDC 2016).
[00227] NAPQI. АРАР-индуцированная гепатотоксичность связана с образованием электрофильного реактивного метаболита NAPQI, который детоксифицируется путем конъюгации с восстановленным глутатионом (GSH). GSH является важным клеточным антиоксидантом в печени, и истощение GSH, вероятно, является важным событием при АРАР-индуцированном остром повреждении печени, хотя этот механизм все еще недостаточно изучен [23]. АрАР метаболизируется ферментами CYP, главным образом CYP2E1 и CYP3A до NAPQI. Однако после токсической дозы истощение GSH сопровождается образованием активных форм кислорода и азота, что приводит к проницаемости митохондрий и гибели гепатоцитов [24]. Не будучи связанными теорией, механизм, с помощью которого эти соединения являются минимально гепатологическими, может заключаться в том, что они не генерируют NAPQI.
[00228] Ссылки в примерах
[1] W. Gamal, Р. Tresk.es, К. Samuel, G;J Sullivan. R. Siller, V. Srsen, K. Morgan, A. Bryans, A. Kozlowska, A, Koulovasilopoulos, I. Undenvood, S. Smith, J. Del-Pozo S Moss, A.I. Thompson, N.C. Henderson, P.C. Hayes, J.N. Plevris, P.O. Bagnaninchi, LJ. Nelson, Low-dose acetaminophen induces early disruption of cell-cell tight junctions in human hepatic cells and mouse liver. Scientific reports, 7 (2017) 37541.
[2] K. Brune, B. Renner, G. Tiegs, Acetaminophen/paracetamol: A history of errors, failures and false decisions, European journal of pain, 19 (2015) 953-965.
[3] A. Bertolini, A. Ferrari, A. Ottani, S. Guetzoni, R. Tacchi, S. Leone, Paracetamol: new vistas of an old drug, CHS Drug Rev, 12 (2006) 250-275.
[4] B.B. Brodie, J. Axelrod, The fate of acetanilide in man. The Journal of pharmacology and experimental therapeutics, 94 (1948) 29-38.
[5] A.M. Larson, J. Poison, R.J. Fontana, T.J. Davern, E. Lalani, L.S. Hynan, J.S. Reisch, F.V. Schiodt, G. Ostapowicz, A.O. Shakil, W.M. Lee, G. Acute Liver Failure Study, Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study, Hepatology, 42 (2005) 1364-1372.
[6] A. Reuben. H. Tillman, R.J. Fontana, T. Davern, B.McGuire, R.T. Stravitz, V. Durkalski, A.M. Larson, I.Lion, O.Fix, M.Schilsky, T.McCashland, J.E. Hay, N. Murray, O.S. Shaikh, D. Ganger, A. Zaman, S.B. Han, R.T. Chung, A. Smith, R. Brown, J. Crippin, M.E. Harrison, D. Koch, S. Munoz, K.R. Reddy, L. Rossaro, R. Satyanarayana, T.Hassanein, A.J. Hanje, J. Olson, R. Subramanian, C. Karvellas, B. Hameed, A.H. Sherker, P. Robuck, W.M. Lee, Outcomes in Adults With Acute Liver Failure Between 1998 and 2013: An Observational Cohort Study, Annals of internal medicine, 164 (2016) 724-732.
[7] J.G Bessems, N.P. Vermeulen, Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches, Crit Rev Toxicol, 31 (2001) 55-138.
[8] M. Ouellet, M.D. Percival, Mechanism of acetaminophen inhibition of cyclooxygenase isoforms. Archives of biochemistry and biophysics, 387 (2001) 273-280.
[9] R.J. Flower, J.R. Vane, Inhibition of prostaglandin synthetase in brain explains the antipyretic activity of paracetamol (4-acetamidophenol), Nature, 240 (1972) 410-411.
[10] E.D. Hogestalt, B.A. Jonsson, A. Ermund, D.A. Andersson, H. Bjork, J.P. Alexander, B.F. Cravatt, A.I. Basbaum, P.M. Zygmunt, Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via laity acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system, The Journal of biological chemistry, 280 (2005) 31405-31412.
[11] C. Mallet, D.A. Barriere, A. Ennund, B.A. Jonsson, A. Eschaller, P.M. Zygmunt, E.D. Hogestatt, TRPV1 in brain is involved in acetaminophen-induced antinociception, PloS one, 5 (2010).
[12] M. Dani, L Guindon, C. Lambert, P. Beaulieu, The local antinociceptive effects of paracetamol in neuropathic pain are mediated by cannabinoid receptors, European journal of pharmacology, 573 (2007) 214-215.
[13] N. Fresno, R. Perez-Fernandez, C. Goicoechea, I. Alkorta, A. Fernandez-Carvajal, R. de la Torre-Martinez, S. Quirce, A. Ferrer-Montiel, M.I. Martin, P. Goya, J. Elguero, Adamanlyl analogues of paracetamol as potent analgesic drugs via inhibition of TRPA1, PloS one, 9 (2014) el13841.
[14] C. Sinning, B. Watzer, O. Coste, R.M. Nosing, I. Ott, A. Ligresti, V. Di Matzo, P. Imming. New analgesics synthetically derived from the paracetamol metabolite N-(4-hydroxyphenyl)-(5Z,8Z,11Z,14Z)-icosatetra-5,8,11,14-enamide. Journal of medicinal chemistry, 51 (2008) 7800-7805.
[15] A.L. Vaccarino, D. Paul, P.K. Mukherjee, E.B. Rodriguez de Turco, V.L. Marcheselli, L. Xu, M.L. Trudell, J.M. Minguez, M.P. Matia, C. Sunkel, J. Alvarez-Builla, N.G. Bazan, Synthesis and in vivo evaluation, of non-hepatotoxic acetaminophen analogs, Bioorg Med Chem. 15 (2007) 2206-2215.
[16] J.G Cui, X. Zhang, Y.H. Zhao, C. Chen, N. Bazan, Allodynia. and hyperalgesia suppression by a novel analgesic in experimental neuropathic pain. Biochemical and biophysical research communications, 350 (2006) 358-363.
[17] L. Miao, L. Xu, K.W. Narducy, M.L, Trudell, First Multi-gram Preparation SCP-123, A Novel Water Soluble Analgesic, Org Process Res Dev, 13 (2009) 820.
[18] M. Santoh, S. Sanoh,Y. Ohtsuki, Y. Ejiri, Y. Kotake, S. Ohta, Acetaminophen analog N-acetyl-m-aminophenol, but not its reactive metabolite, N-acetyl-p-benzoquinone imine induces CYP3A activity via inhibition of protein degradation, Biochemical and biophysical research communications, 486 (2017) 639-644.
[19] R.M. Rodrigues, A. Heymans, V. De Вое, A. Sachinidts, U. Chaudhari, O. Govaere, T. Roskams, T. Vanhaecke, V. Rogiers, J. De Kock, Toxicogenomics-based prediction of acetaminophen-induced liver injury using human hepatic cell systems, Toxicology letters, 240 (2016) 50-59.
[20] M.R. McGill, C.D. Williams, Y. Xie, A. Ramachandran, H. Jaeschke, Acetanminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity, Toxicology and applied pharmacology, 264(2012)387-394.
[21] E.L. LeCluyse, R.P. Witek, M.E. Andersen, M.J. Powers, Organotypic liver culture models: meeting current challenges in toxicity testing, Crit Rev Toxicol, 42 (2012) 501-548.
[22] M. Blieden, L.C. Paramore, D. Shah, R. Ben-Joseph, A perspective on the epidemiology of acetaminophen exposure and toxicity in the United States, Expert Rev Clin Pharmacol, 7 (2014) 341-348.
[23] M.A. Abdelmegeed, K.H. Moon, C. Chen, F.J. Gonzalez, B.J. Song, Role of cytochrome P450 2E1 in protein nitration and ubiquitin-mediated degradation during acetaminophen toxicity. Biochemical pharmacology, 79 (2010) 57-66.
[24] J.A. Hinson, A.B. Reid, S.S. McCullough, L.P. James, Acetaminophen-induced hepatotoxicity: rote of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition, Drug Metab Rev, 36 (2004) 805-822.
[00229]
ЭКВИВАЛЕНТЫ
[00230] Специалисты в данной области техники распознают или смогут установить, используя не более чем обычные эксперименты, многочисленные эквиваленты конкретных веществ и описанных процедур в данном описании. Такие эквиваленты считаются находящимися в рамках данного изобретения и охватываются следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ N-АЦЕТИЛЦИСТЕИНА И СПОСОБЫ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ТОКСИЧНОСТИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2002 |
|
RU2291689C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПОЛИКИСТОЗНОЙ БОЛЕЗНИ ПОЧЕК | 2016 |
|
RU2742300C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛЕНИЯ И ИШЕМИЧЕСКОГО ПОРАЖЕНИЯ | 2012 |
|
RU2638807C2 |
| ЛЕЧЕНИЕ ДЕМИЕЛИНИЗИРУЮЩИХ ЗАБОЛЕВАНИЙ | 2019 |
|
RU2805061C2 |
| ЛЕЧЕНИЕ НЕВРОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ МЕТАЛЛОПОРФИРИНАМИ | 2012 |
|
RU2623207C2 |
| Лечение боли | 2009 |
|
RU2630969C2 |
| ГЛЮКУРОНИДИРОВАННЫЙ АЦЕТАМИНОФЕН КАК МАРКЕР РАССТРОЙСТВ ПЕЧЕНИ | 2009 |
|
RU2555331C2 |
| ДЕЗОКСИНУКЛЕОЗИДНАЯ ТЕРАПИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ НЕСБАЛАНСИРОВАННЫМИ ПУЛАМИ НУКЛЕОТИДОВ, В ТОМ ЧИСЛЕ СИНДРОМОВ ИСТОЩЕНИЯ МИТОХОНДРИАЛЬНОЙ ДНК | 2016 |
|
RU2827432C2 |
| КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ АНТИТЕЛА К CD38 И КАРФИЛЗОМИБ | 2014 |
|
RU2670127C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ T-КЛЕТОЧНОЙ ЛИМФОМЫ КОЖИ | 2014 |
|
RU2669941C2 |
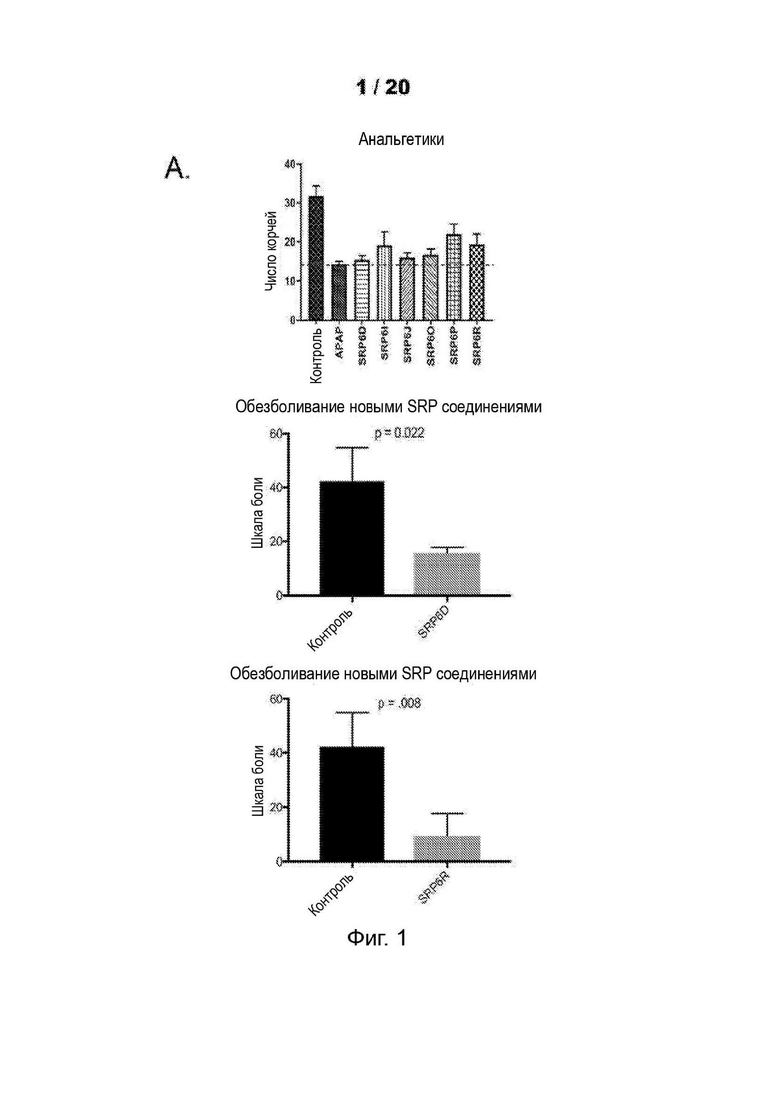
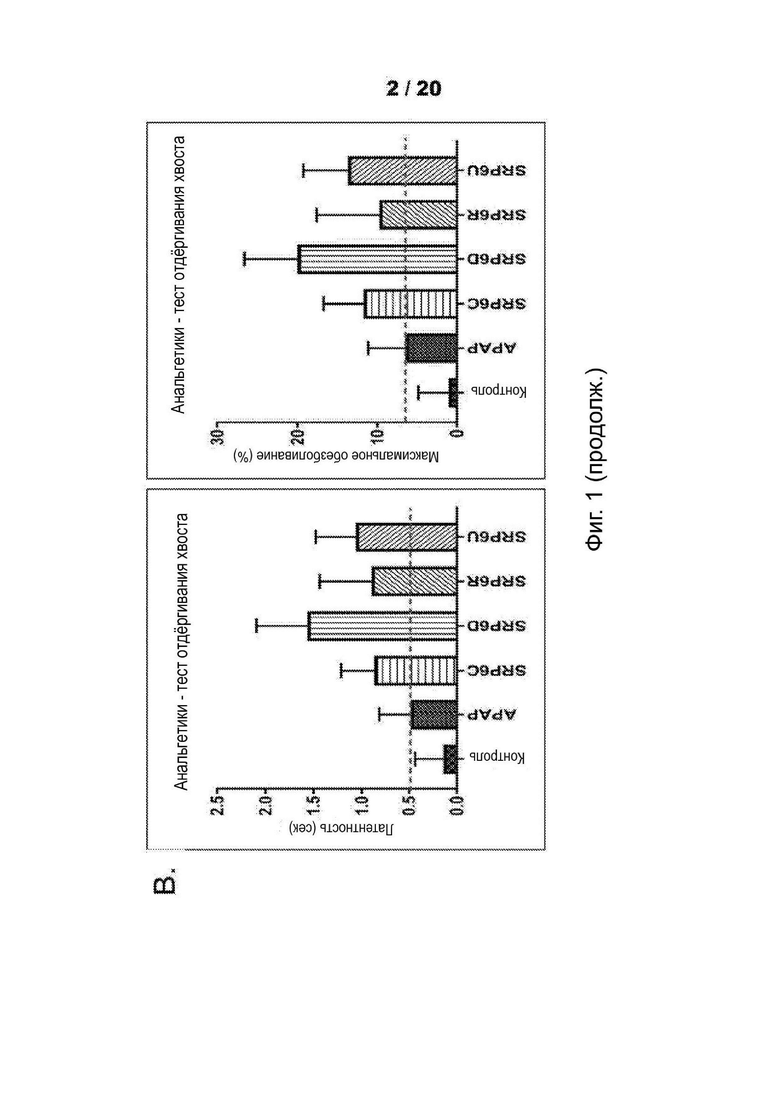
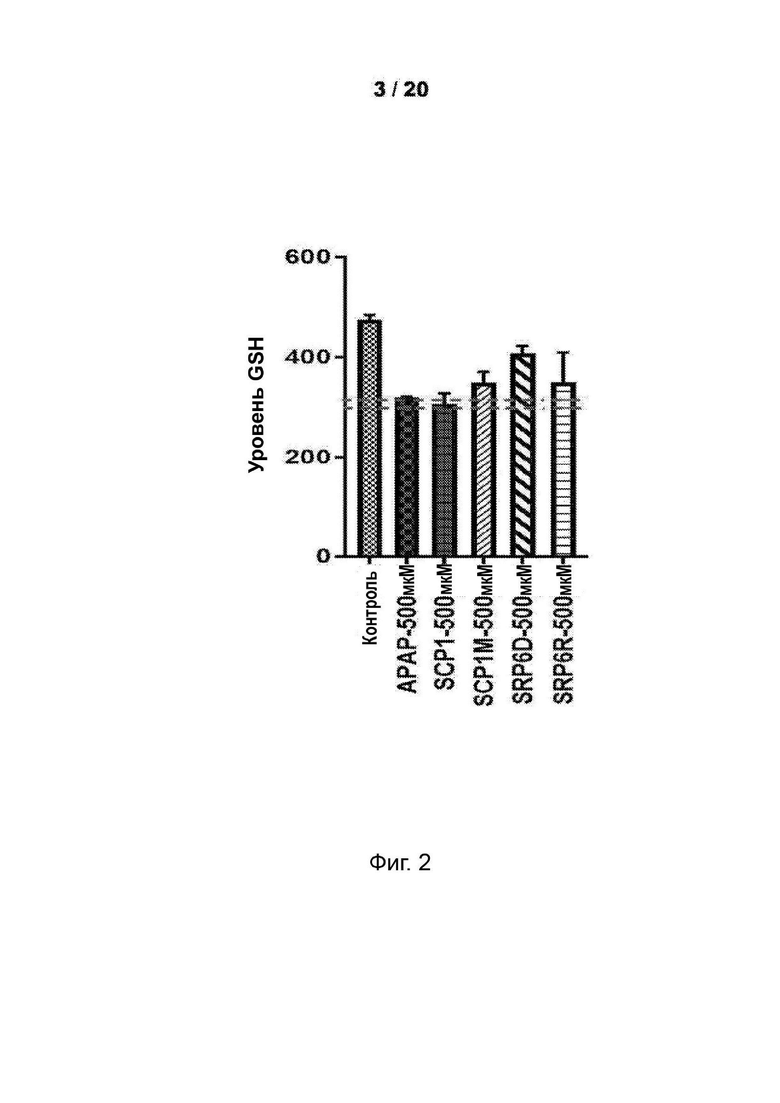
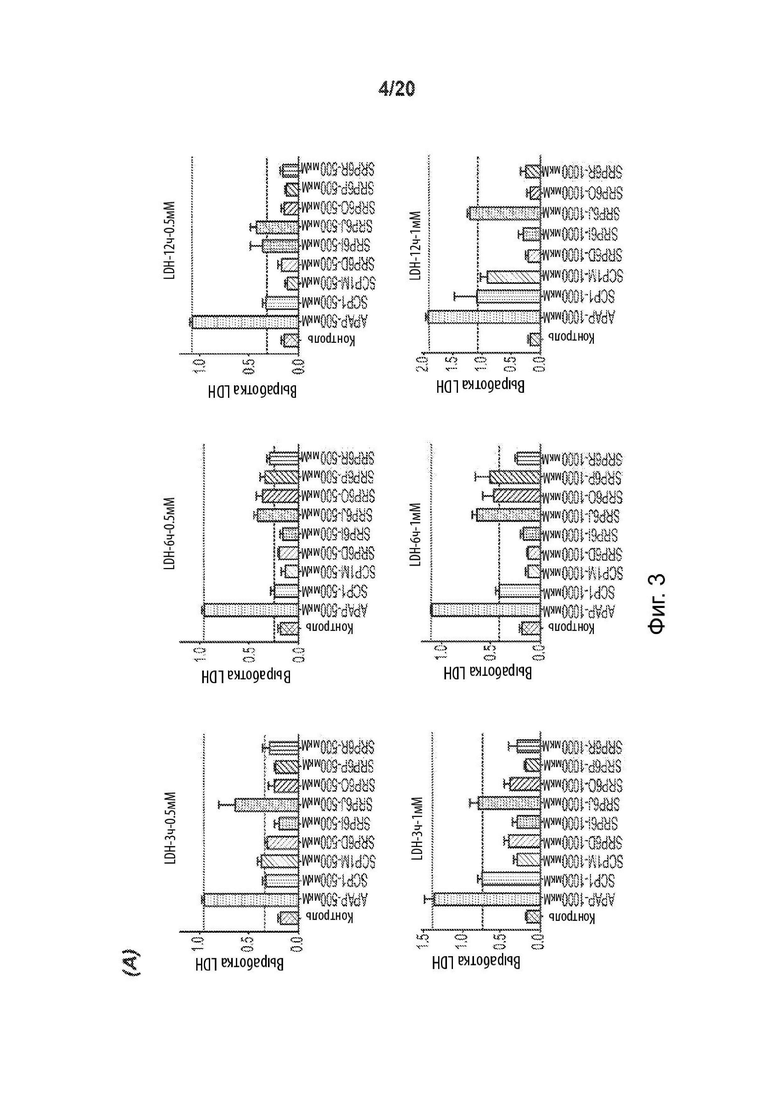
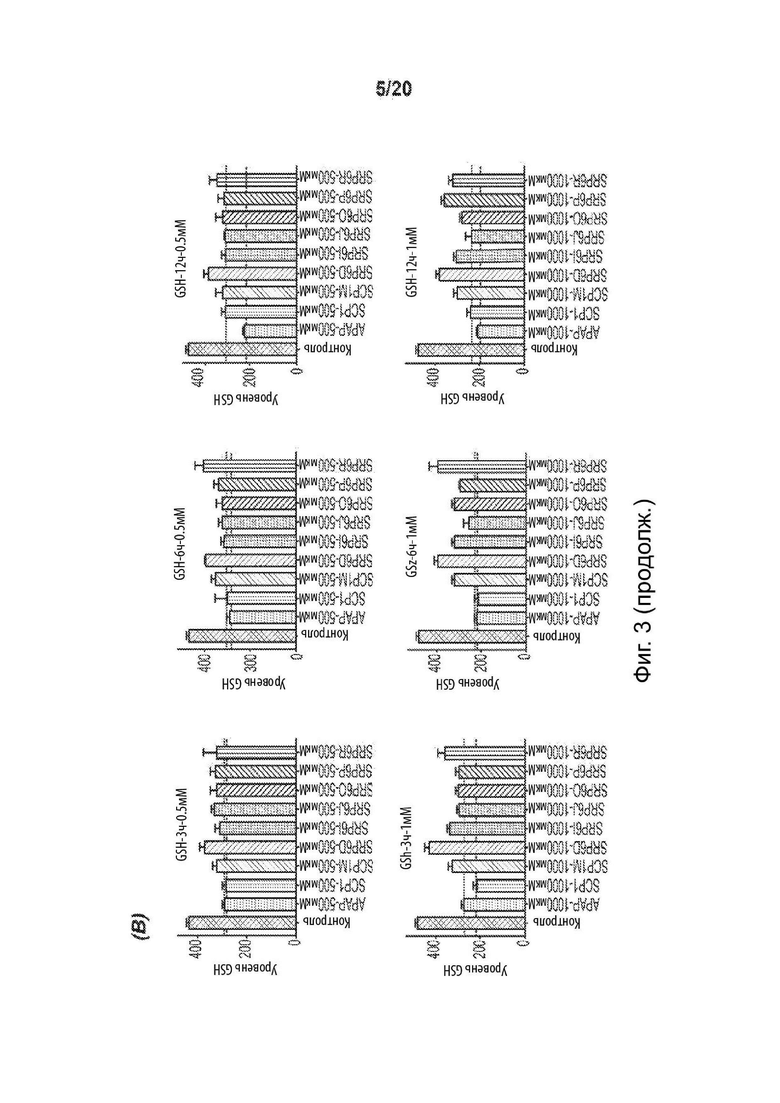
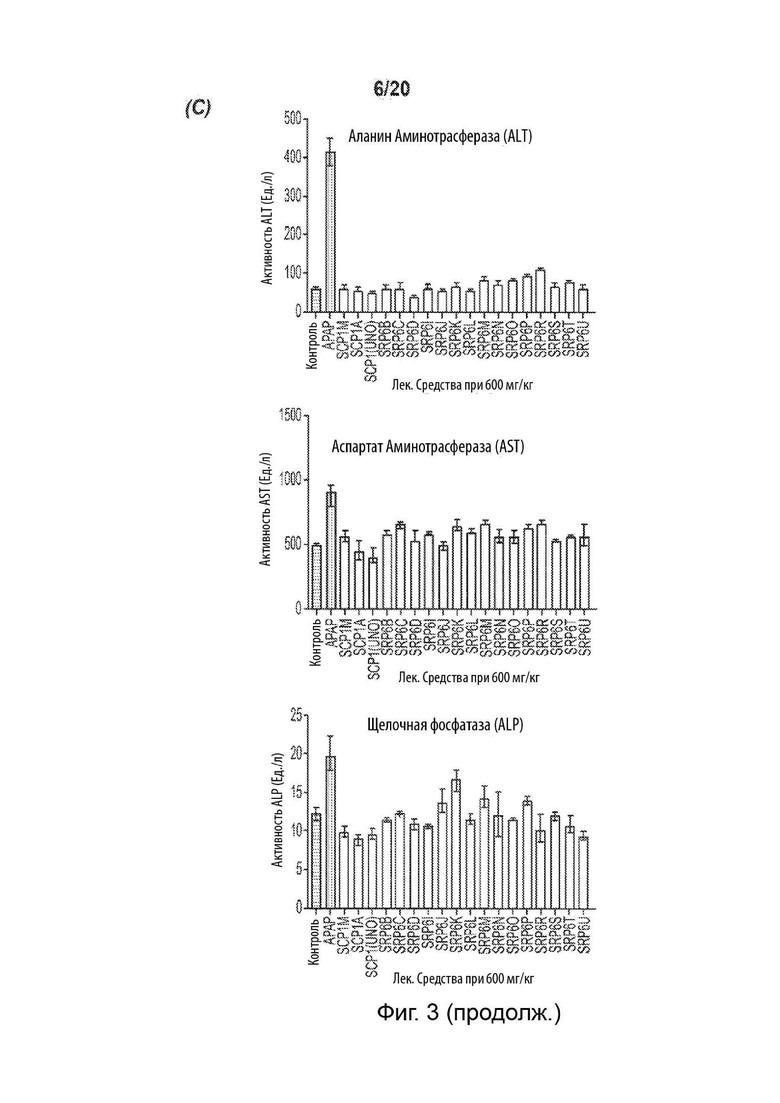
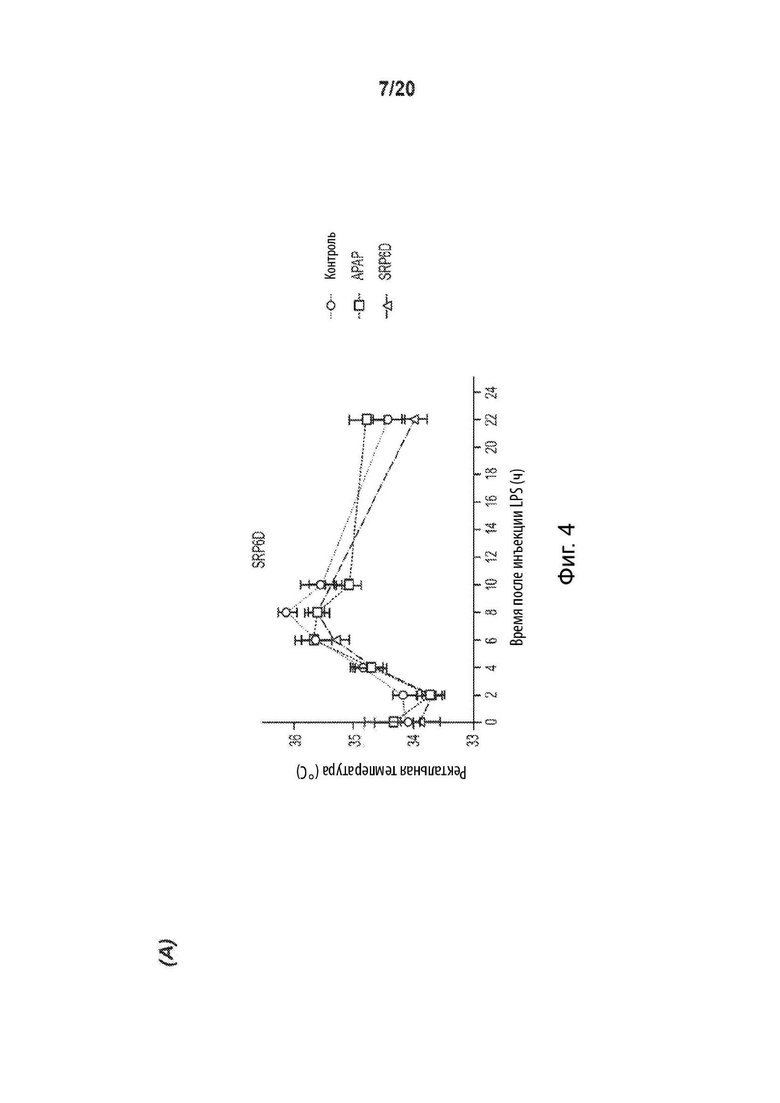
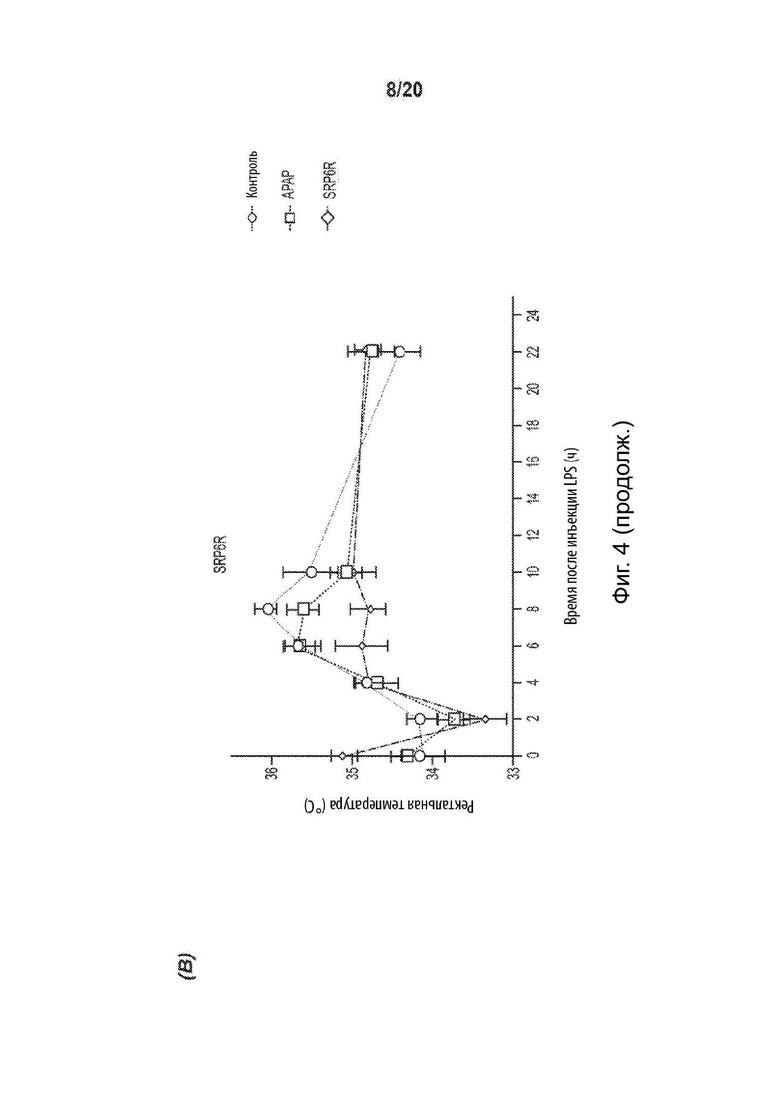
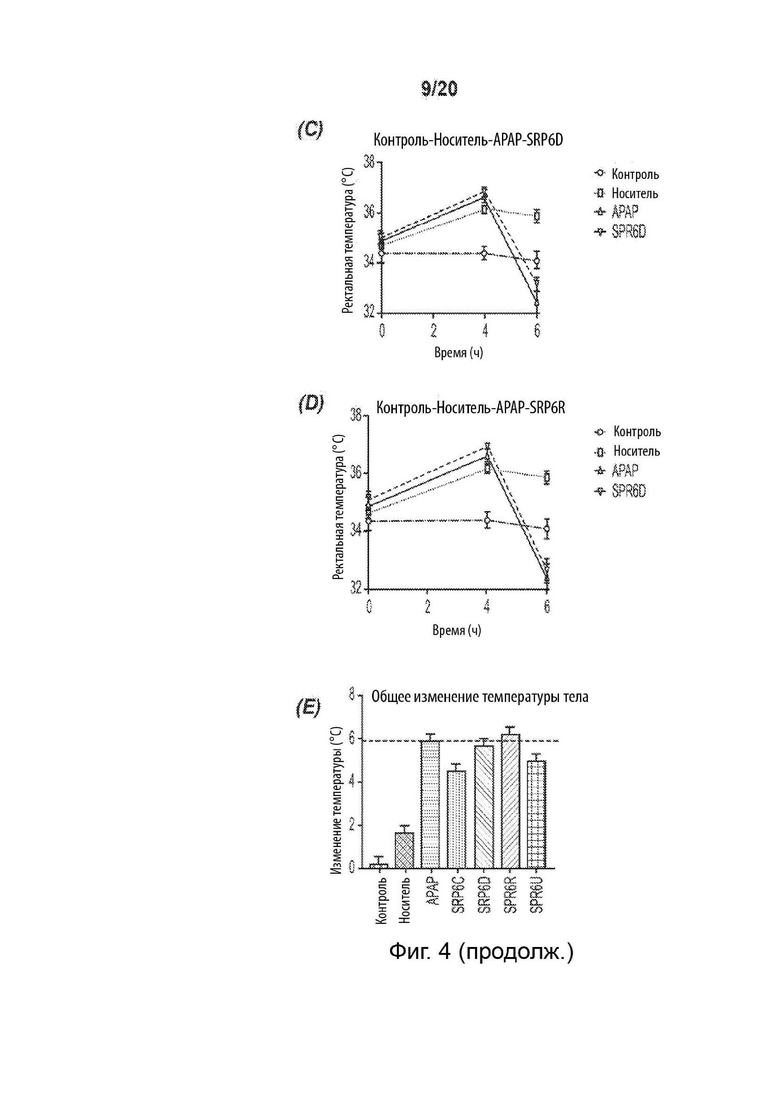
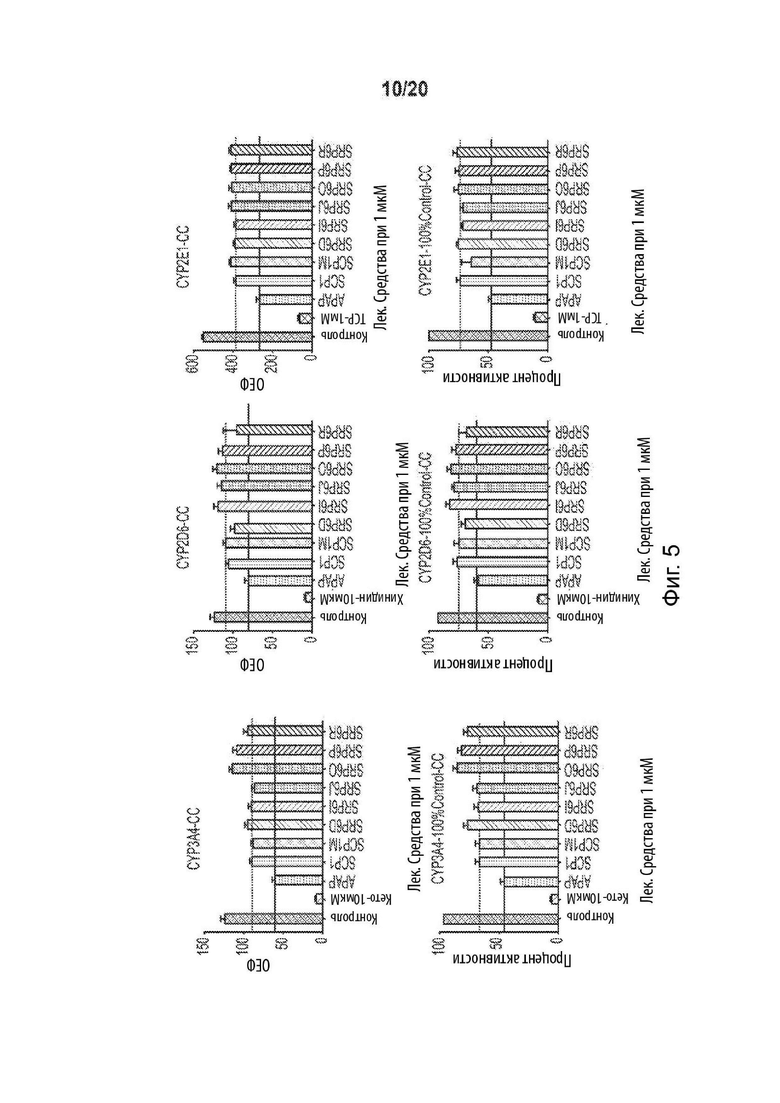
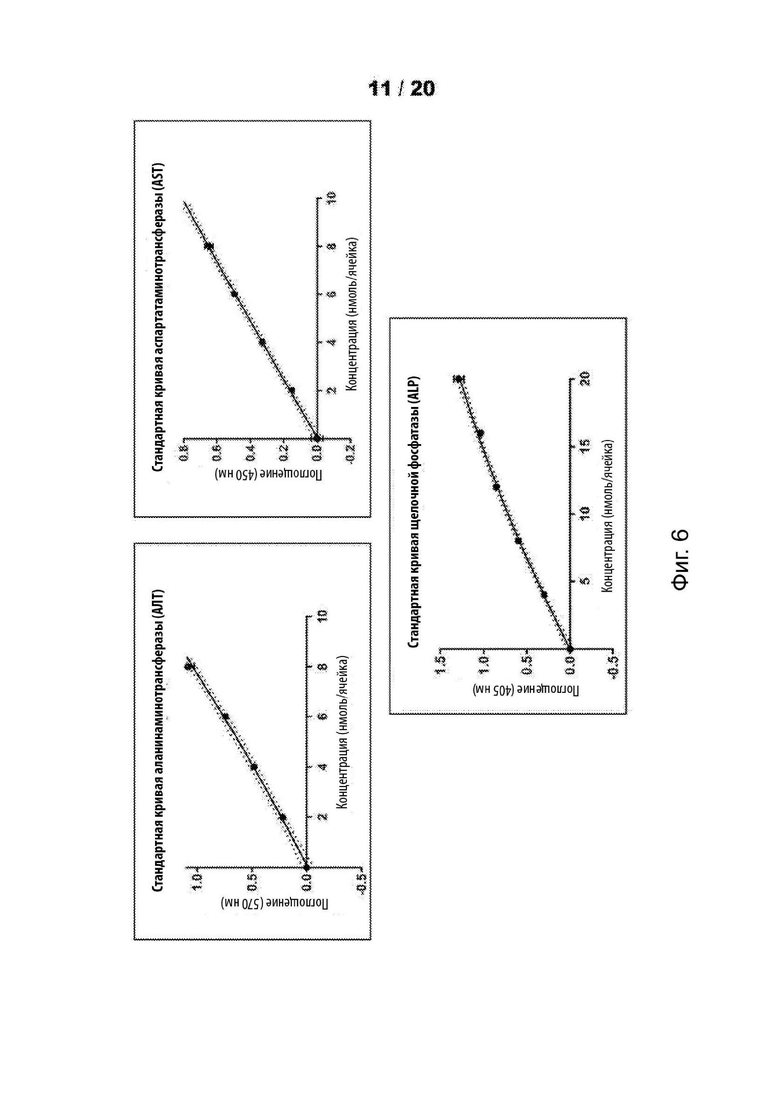
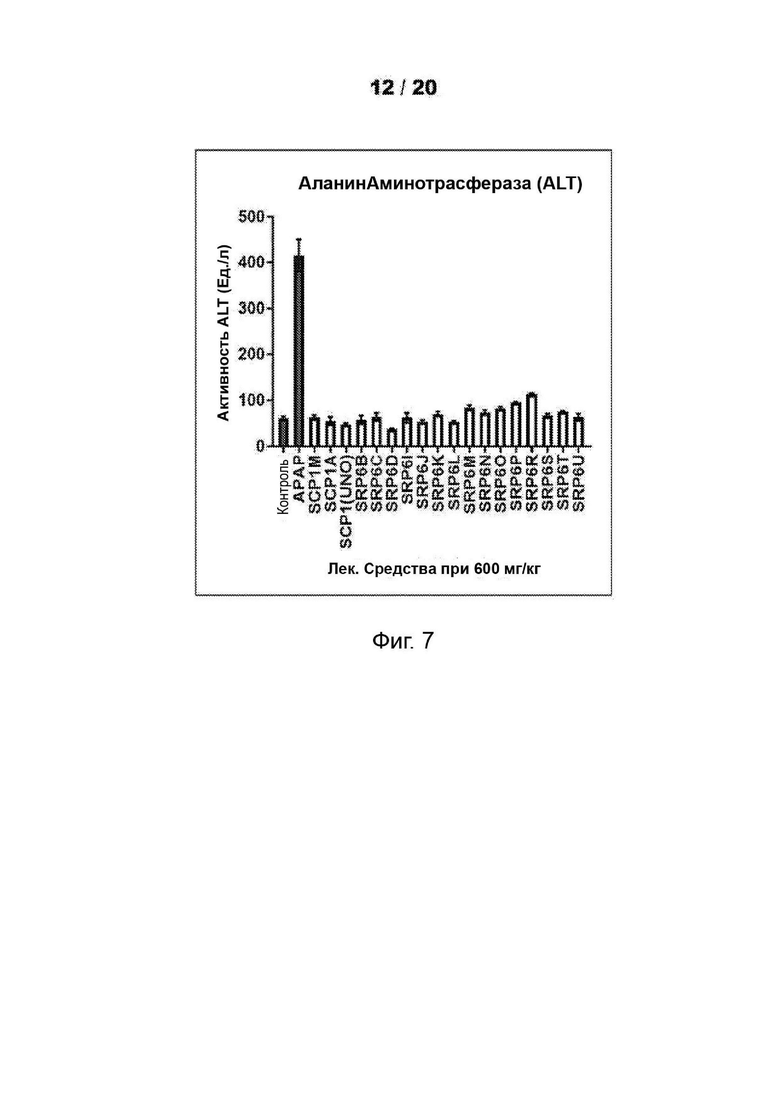
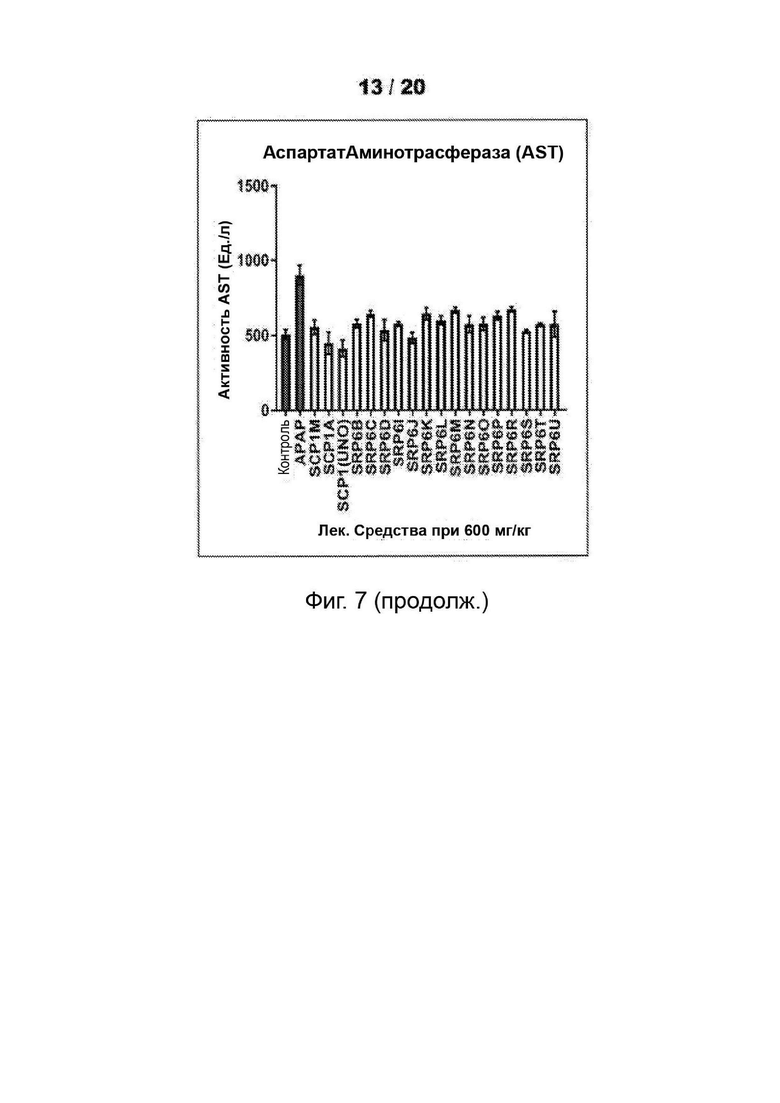
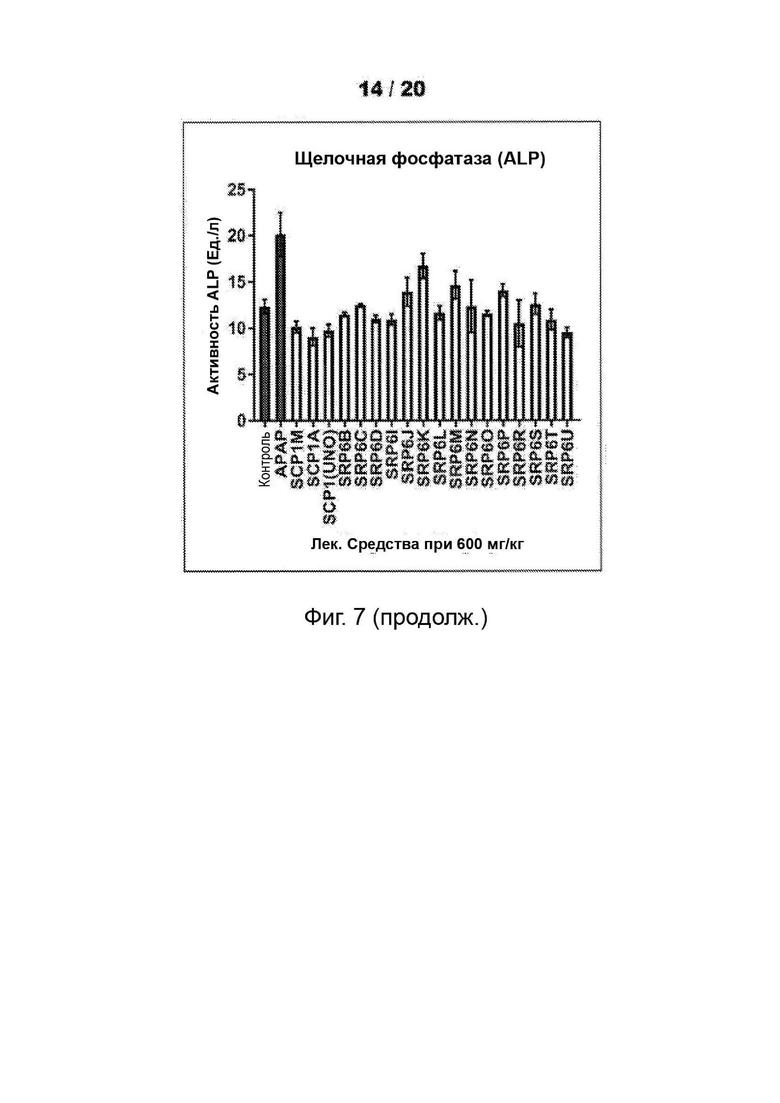
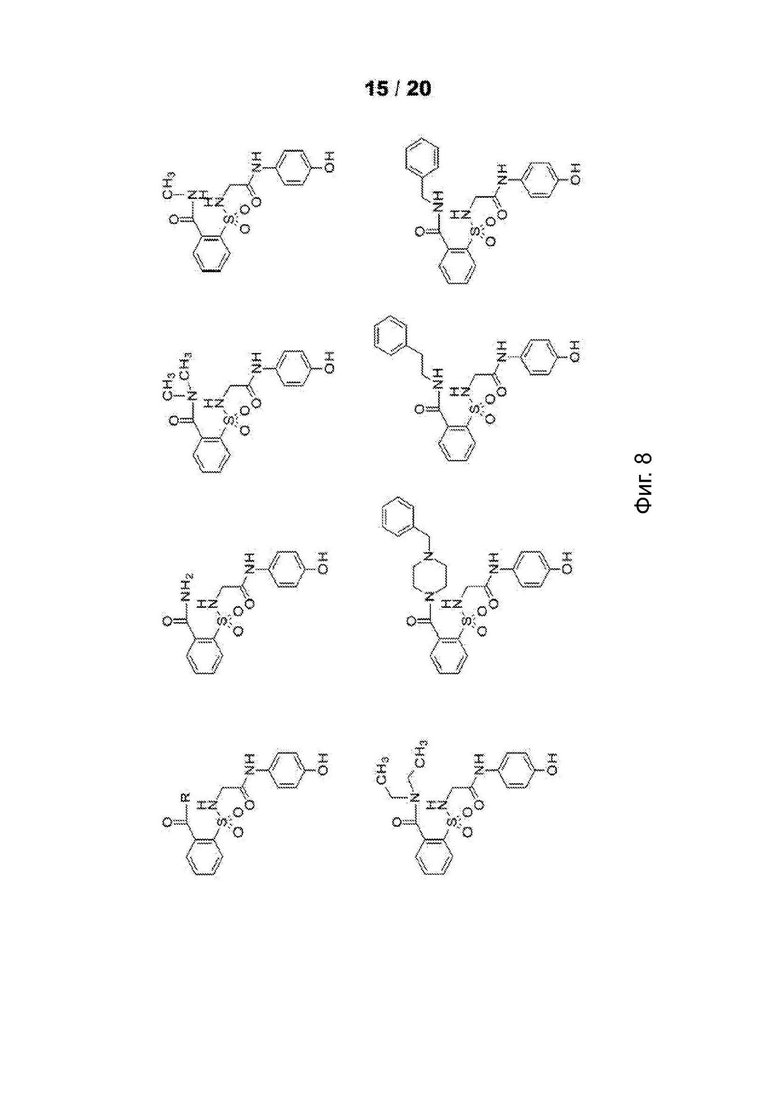
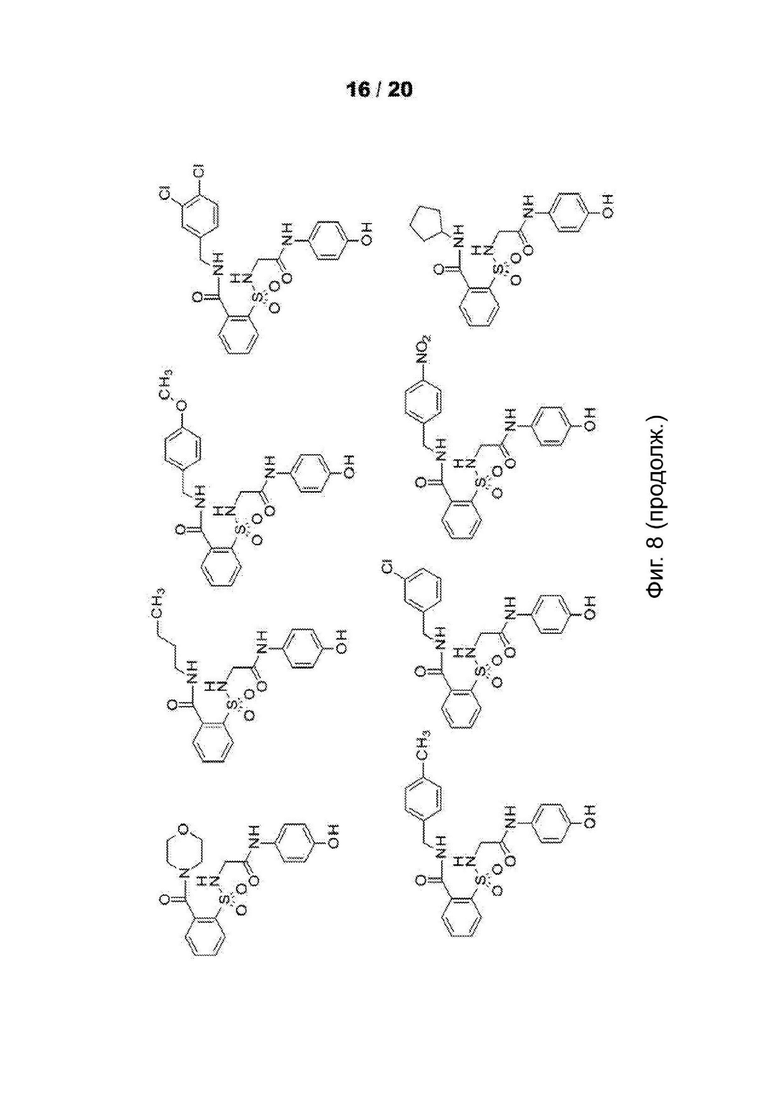
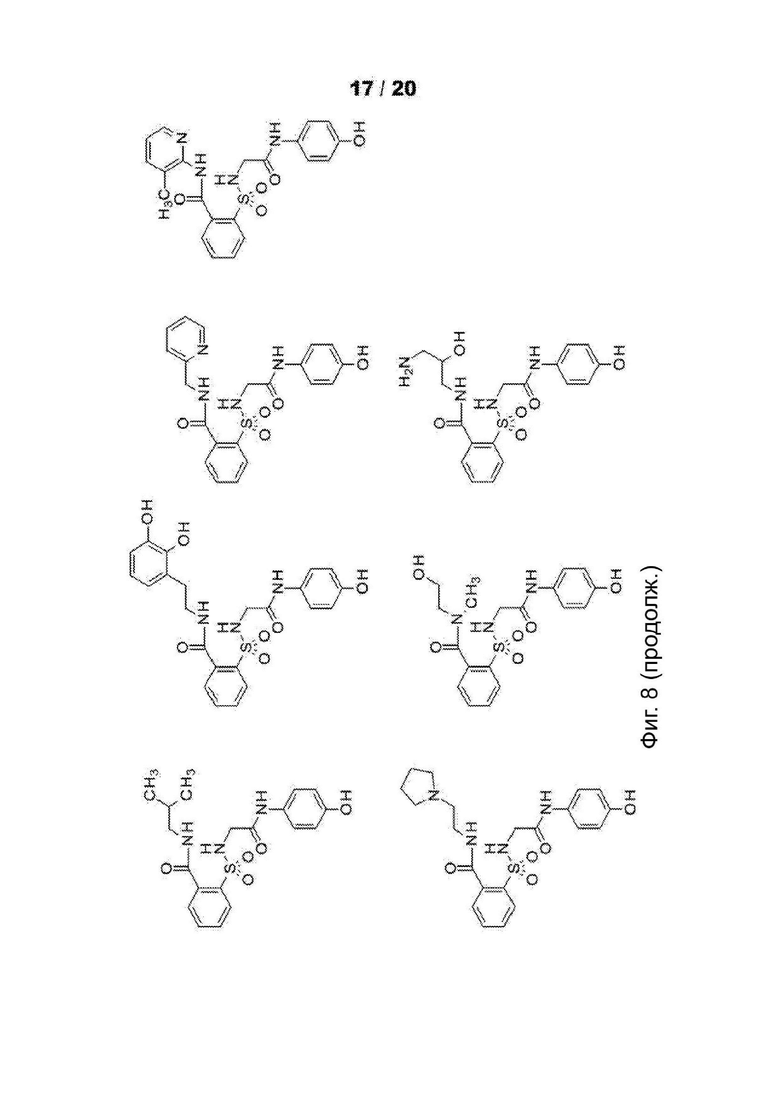
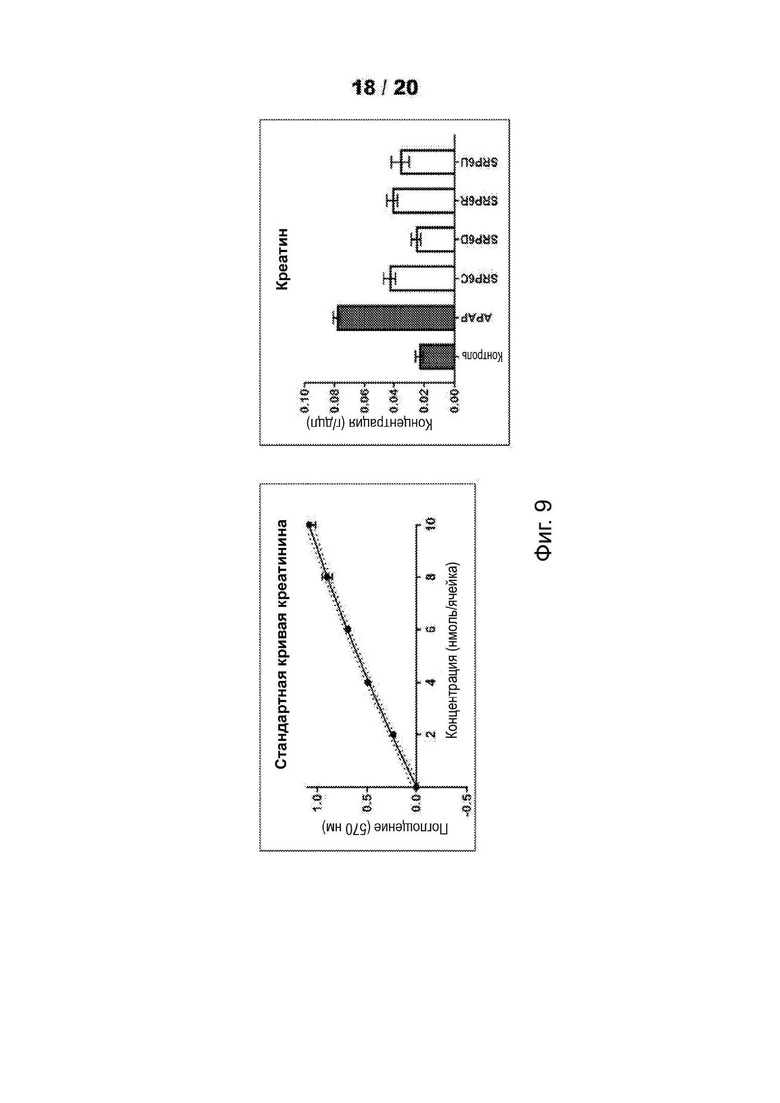
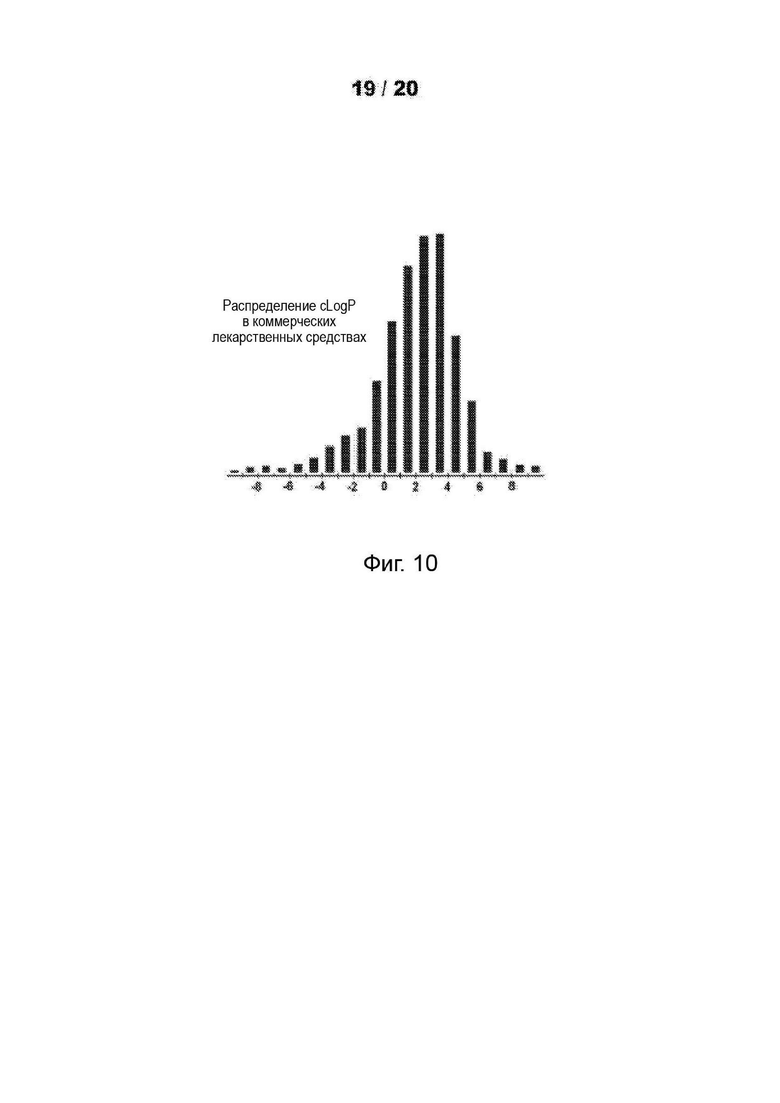
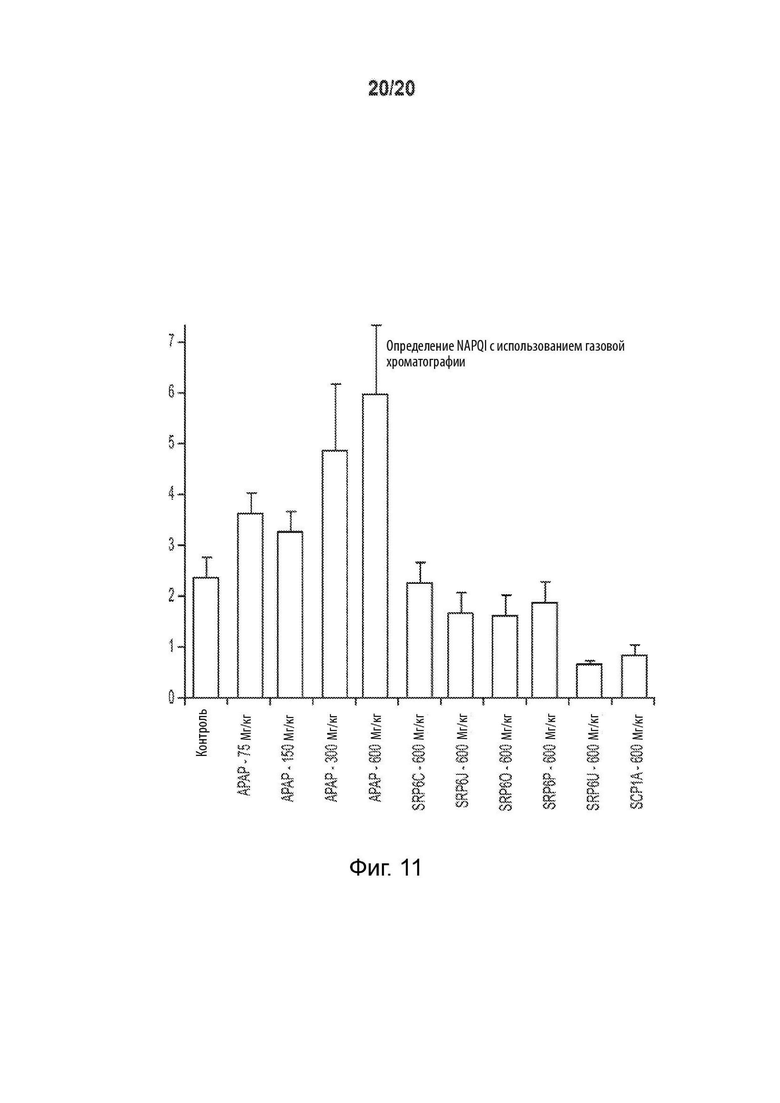
Изобретение относится к области органической химии, а именно к соединению формулы (I), где R выбран из группы, состоящей из NH2, N(CH3)2, NHCH3, N(CH2CH3)2, NH(CH2)2C6H5, NHCH2C6H5, NHCH2CH2CH2CH3, NHCH2C6H4ОСH3, NHCH2C6H3Cl2, NHCH2C6H4CH3, NHCH2C6H4Cl, NHCH2C6H4NO2, NH-циклопентил, NHCH2CН(CH3)2, NHCH(CH3)2, NHCH2CH2C6H3(OH)2, NHCH2CH2-пирролидинил, N(CH3)CH2CH2OH, NHCH2CH(OH)CH2NH2; или его фармацевтически приемлемая соль. Также изобретение относится к соединению формулы (II), значения радикалов указаны в формуле изобретения, к способу облегчения или предотвращения висцеральной и ноцицептивной боли. Технический результат: эффективное облегчение боли производным общей формулы (I) и (II). 11 н. и 9 з.п. ф-лы, 3 табл., 1 пр., 11 ил.
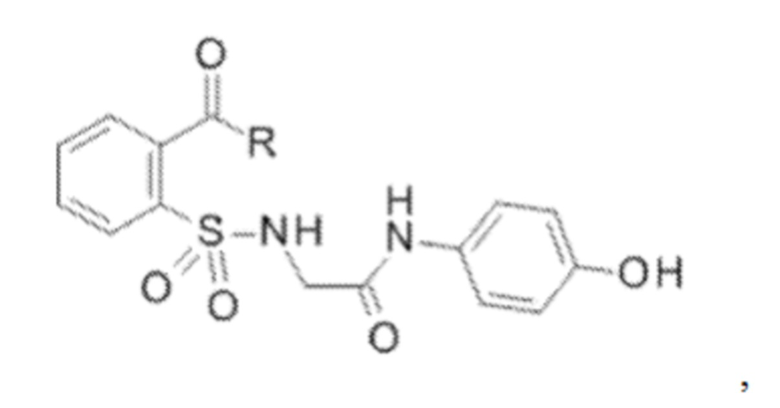 (I)
(I)
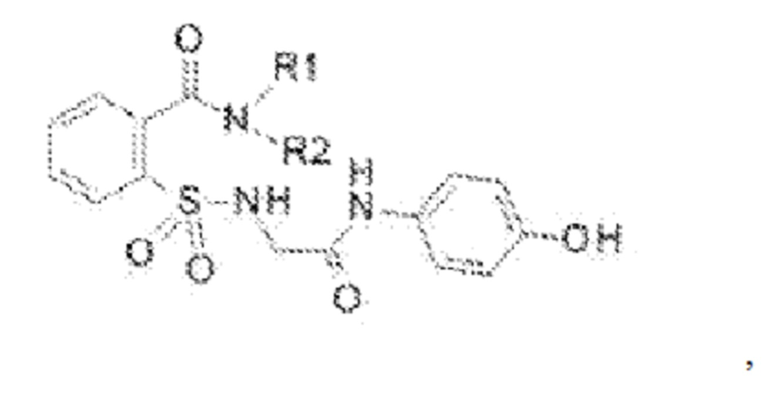 (II)
(II)
1. Обезболивающее соединение формулы (I):
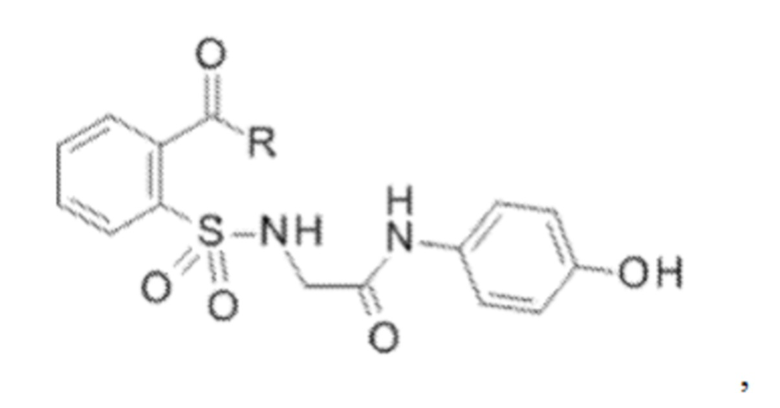
где R выбран из группы, состоящей из NH2, N(CH3)2, NHCH3, N(CH2CH3)2, NH(CH2)2C6H5, NHCH2C6H5, NHCH2CH2CH2CH3, NHCH2C6H4ОСH3, NHCH2C6H3Cl2, NHCH2C6H4CH3, NHCH2C6H4Cl, NHCH2C6H4NO2, NH-циклопентил, NHCH2CН(CH3)2, NHCН(CH3)2, NHCH2CH2C6H3(OH)2, NHCH2CH2-пирролидинил, N(CH3)CH2CH2OH, NHCH2CH(OH)CH2NH2; или его фармацевтически приемлемая соль.
2. Обезболивающее соединение Формулы (II):
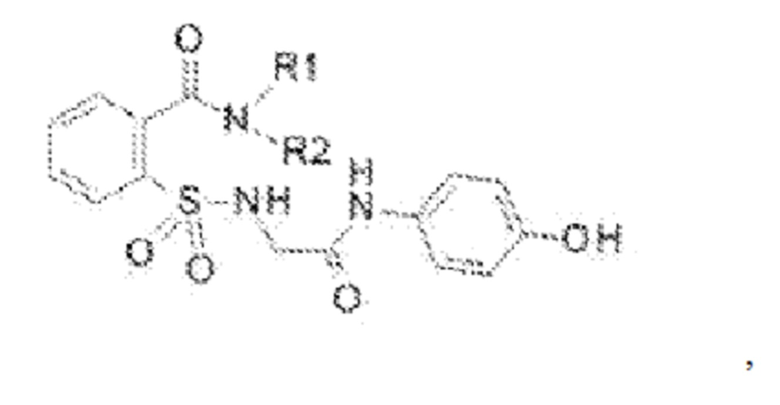
где R1 выбран из группы, состоящей из Н, С1-С4алкила, фенильной группы, С1-С3алкилфенила, CH2C6H3Cl2, CH2C6H5Cl, CH2C6H5NO2, CH2C6H4CH3, CH2CH2C6H3(OH)2, CH2CH2-пирролидинила, CH2CH2OH, CH2CH(OH)CH2NH2 и циклопентила, и где R2 выбран из группы, состоящей из Н и С1-С2алкила, или его фармацевтически приемлемая соль.
3. Обезболивающее соединение по п. 2, где R1 выбран из группы, состоящей из H, CH3, CH2CH3, (CH2)2C6H5, CH2C6H5, CH2CH2CH2CH3, CH2C6H3Cl2, CH2C6H4Cl, CH2C6H5NO2, циклопентила, CH2C(CH3)2, CH(CH3)2; и где R2 выбран из группы, состоящей из Н и СН3, или его фармацевтически приемлемая соль.
4. Обезболивающее соединение по п. 1, имеющее химическую структуру, выбранную из группы, состоящей из:
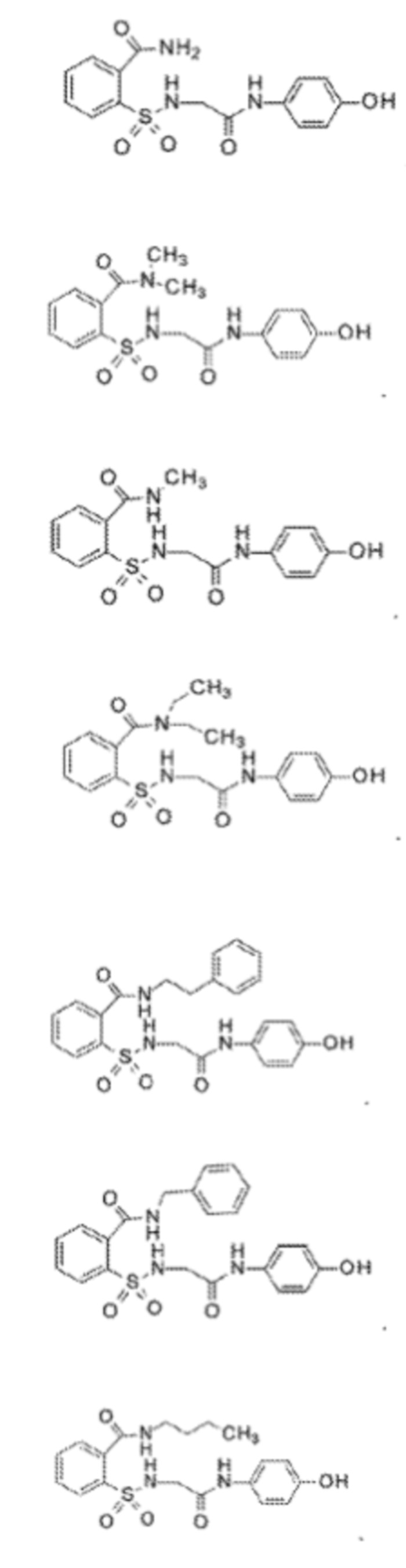
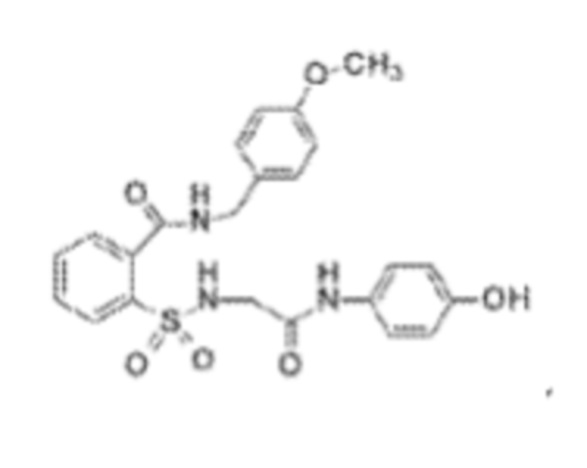
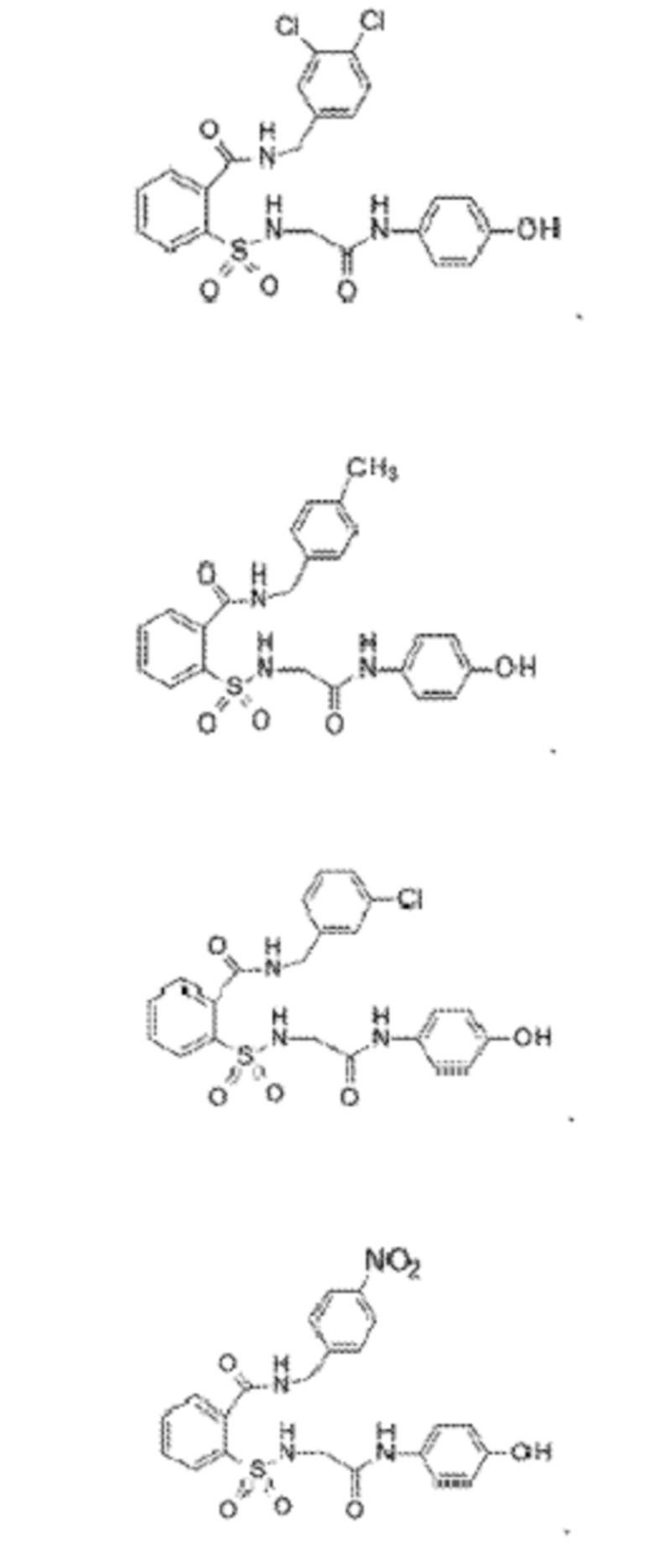
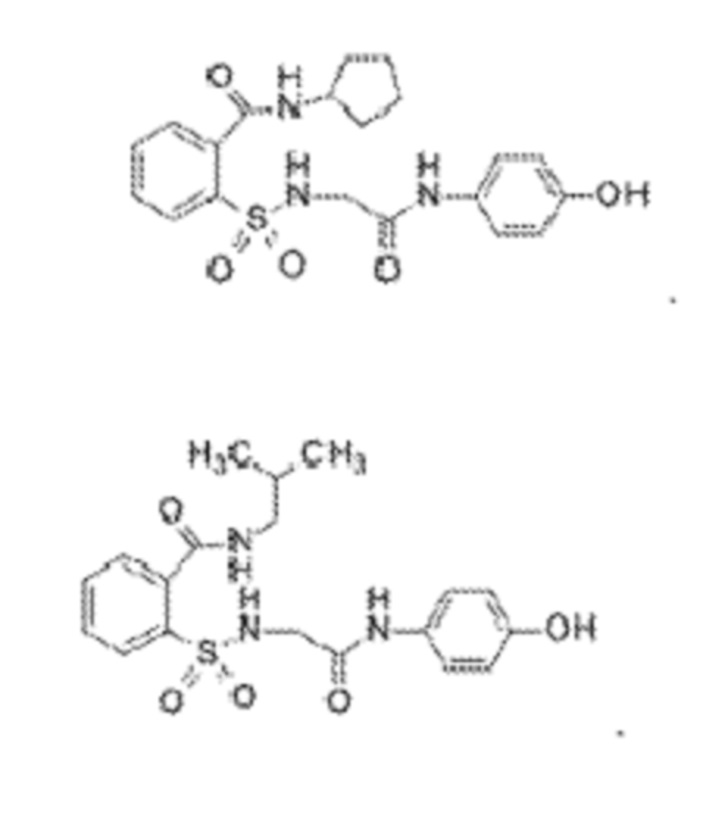
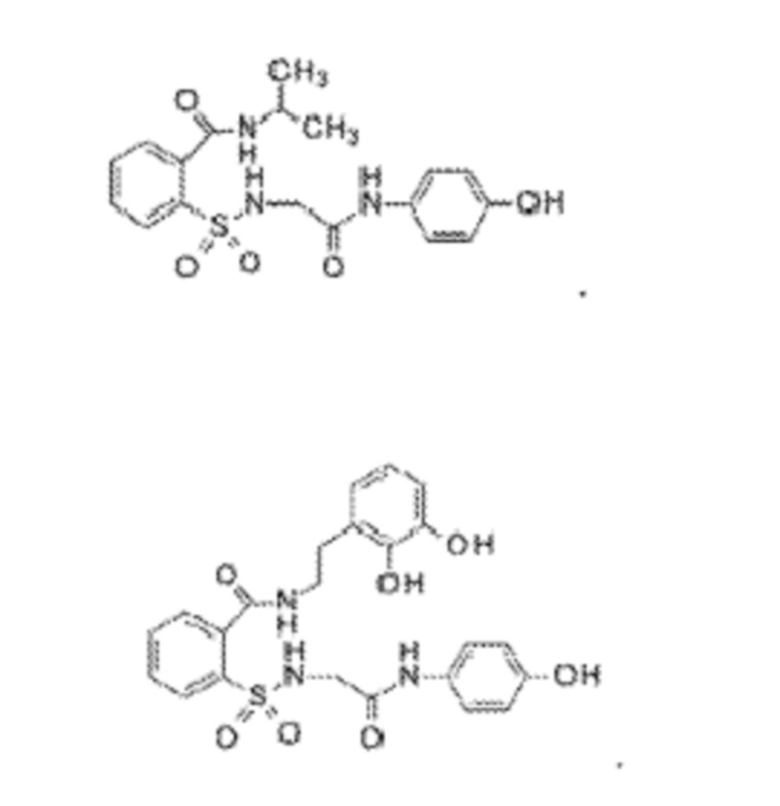
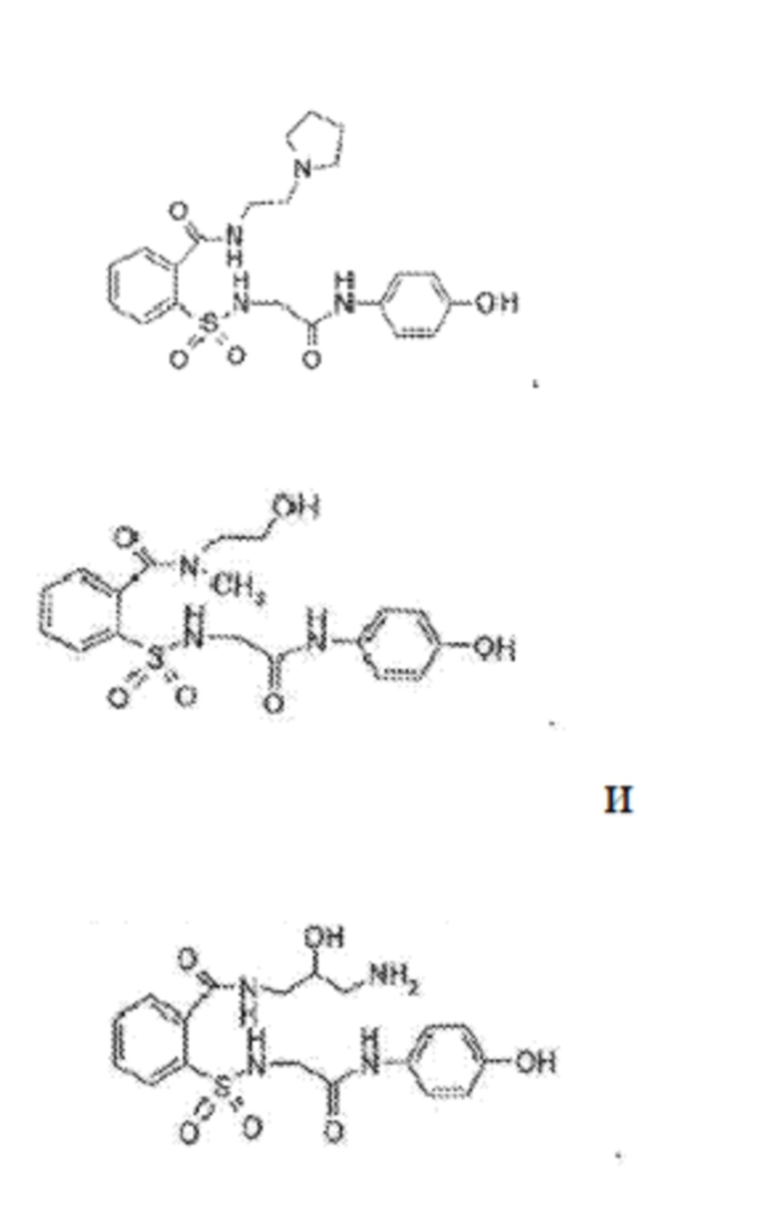
5. Способ облегчения висцеральной и ноцицептивной боли у субъекта, включающий введение указанному субъекту, страдающему от такой боли, терапевтически эффективного количества обезболивающего соединения по любому из пп. 1, 2.
6. Способ предотвращения висцеральной и ноцицептивной боли у субъекта, включающий введение указанному субъекту, страдающему от такой боли, терапевтически эффективного количества обезболивающего соединения по любому из пп. 1, 2.
7. Способ ослабления висцеральной и ноцицептивной боли у субъекта, включающий введение указанному субъекту, страдающему от такой боли, терапевтически эффективного количества обезболивающего соединения по любому из пп. 1, 2.
8. Способ уменьшения частоты возникновения висцеральной и ноцицептивной боли у субъекта, включающий введение указанному субъекту, страдающему от такой боли, терапевтически эффективного количества обезболивающего соединения по любому из пп. 1, 2.
9. Способ задержки развития висцеральной и ноцицептивной боли у субъекта, включающий введение указанному субъекту, страдающему от такой боли, терапевтически эффективного количества обезболивающего соединения по любому из пп. 1, 2.
10. Способ предотвращения развития висцеральной и ноцицептивной боли у субъекта, включающий введение указанному субъекту, страдающему от такой боли, терапевтически эффективного количества обезболивающего соединения по любому из пп. 1, 2.
11. Способ облегчения висцеральной и ноцицептивной боли у субъекта, включающий введение указанному субъекту, страдающему от такой боли, терапевтически эффективного количества обезболивающего соединения по любому из пп. 1, 2.
12. Способ по любому из пп. 5-11, где терапевтически эффективное количество включает дозу от около 10 мкМ до около 10 мМ соединения, вводимого субъекту.
13. Способ по п. 12, где терапевтически эффективное количество включает дозу от около 50 мкМ до около 1 мМ соединения, вводимого субъекту.
14. Способ по любому из пп. 5-11, где соединение вводят в разовой дозе.
15. Способ по любому из пп. 5-11, где соединение вводят с интервалами около 4 ч, 12 ч или 24 ч.
16. Способ по любому из пп. 5-11, где соединение вводят перорально, парентерально, трансдермально или назально.
17. Способ по любому из пп. 5-11, где соединение вводят в форме пилюли, капсулы, крема, спрея, лосьона или водного раствора.
18. Способ по любому из пп. 5-11, дополнительно включающий введение субъекту одновременно или последовательно терапевтически эффективного количества опиоида и/или НСПВ.
19. Обезболивающая фармацевтическая композиция, содержащая терапевтически эффективное количество обезболивающего соединения по п. 1 или 2 и фармацевтически приемлемый носитель.
20. Медицинский набор для лечения висцеральной и ноцицептивной боли, содержащий напечатанные инструкции по введению соединения субъекту, страдающему от висцеральной и ноцицептивной боли; и обезболивающее соединение по любому из пп. 1, 2 или фармацевтическую композицию по п. 19.
| US 6806291 B1, 19.10.2004 | |||
| Miao, L | |||
| Устройство для разметки подлежащих сортированию и резанию лесных материалов | 1922 |
|
SU123A1 |
| D | |||
| Reddy, Y | |||
| и др.: "A Facile and Green Synthesis of Novel Imide and Amidic Acid Derivatives of Phenacetin as Potential Analgesic and Anti-Pyretic Agents", Letters in | |||

