Область изобретения
Настоящее изобретение принадлежит к области медицины и относится к оксаспиропроизводному, способу его получения и его применения в лекарственных средствах. В частности, настоящее изобретение относится к оксаспиропроизводному, представленному формулой (I), способу его получения и к фармацевтической композиции, содержащей указанное производное, и его применению в качестве агониста рецептора MOR (μ-опиодиный рецептор), и к его применению при получении лекарственного средства для лечения и/или профилактики боли и связанных с болью заболеваний.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Опиоидный рецептор представляет собой важный рецептор, сопряженный с G-белком (GPCR), и является мишенью комбинированных эндогенных опиоидных пептидов и опиоидных лекарственных средств. Активированные опиоидные рецепторы играют регуляторную роль в иммунитете нервной системы и эндокринной системы. Опиоидные лекарственные средства являются сильнейшими и традиционно применяемыми анальгетиками центрального действия. Эндогенные опиоидные пептиды представляют собой опиоидоподобные активные вещества, встречающиеся в природе у млекопитающих. Известные в настоящее время эндогенные опиоидные пептиды можно примерно разделить на энкефалин, эндорфин, динорфин и ноцицептин (Pharmacol Rev 2007; 59: 88-123). В центральной нервной системе находятся соответствующие опиоидные рецепторы, т.е. μ (MOR), δ (DOR – δ-опиоидный рецептор), κ (KOR - κ-опиоидный рецептор) рецепторы и т.п. Обнаружено, что сила обезболивающего действия эндогенных опиоидных пептидов в основном зависит от уровня экспрессии опиоидных рецепторов. Опиоидные рецепторы являются мишенями обезболивающего действия опиоидных лекарственных средств и эндогенных опиоидных пептидов. Zadina et al. обнаружили, что рецептор MOR обладает самой сильной связывающей способностью по отношению к морфиновому пептиду 1 (360 пМ). Это значение в 4000 раз превышает связывающую способность рецептора DOR по отношению к морфиновому пептиду 1 и в 15 000 раз превышает связывающую способность рецептора KOR по отношению к морфиновому пептиду 1. Рецептор MOR является важнейшим опиоидным рецептором, опосредующим обезболивающее действие (Science, 2001, 293: 311-315; Biochem Biophys Res Commun 235:567-570; Life Sci 61:PL409-PL415).
Современные исследования показывают, что GPCR опосредует и регулирует физиологические функции в основном посредством двух метаболических путей: пути G-белка и пути β-аррестина. Активация пути передачи сигнала G-белка может происходить за счет связывания традиционного агониста GPCR и рецептора и включает систему вторичных посредников, таких как ион кальция, аденилциклаза (АЦ), митоген-активируемые протеинкиназы (MAPK) и т.п. В то время как активация метаболического пути β-аррестина осуществляется в основном посредством β-аррестин-смещенного лиганда. Отклик β-аррестина, опосредованный GPCR, в основном включает три аспекта: 1) β-аррестин в качестве отрицательного регулятора взаимодействует с киназой рецепторов, сопряженных с G-белком (GRK), вызывая таким образом десенсибилизацию рецептора в GPCR и блокируя преобразование сигнала G-белка; 2) β-аррестин в качестве каркасного белка рекрутирует эндоцитозный белок и индуцирует эндоцитоз GPCR; 3) β-аррестин в качестве адаптерного белка образует комплекс с нижележащими сигнальными молекулами GPCR и активирует молекулы преобразования сигнала, такие как MAPK, протеинтирозинкиназы Src и Akt и т.д., независимым от G-белка образом. Различия стимуляции лигандов при передаче сигнала G-белка и/или при передаче сигнала β-аррестина в конечном счете определяют лиганд-специфичные клеточные биологические эффекты GPCR.
MOR является мишенью опиоидных анальгетических лекарственных средств, таких как эндогенный энкефалин и морфин. В ранних исследованиях продемонстрировано, что эндогенный энкефалин и опиоидное лекарственное средство эторфин способны оказывать агонистическое действие на G-белок и вызывать эндоцитоз рецептора, при этом морфин совсем не способен вызывать эндоцитоз рецептора. Это связано с тем, что морфин обладает слишком слабой способностью к агонистическому действию на фосфорилирование MOR, и на мембране рекрутируются лишь следовые количества β-аррестина (Zhang et al., Proc Natl Acad Sci USA, 1998, 95 (12): 7157-7162). Осуществление этими лигандами физиологических функций полностью опосредовано через метаболический путь передачи сигнала G-белка, а не через метаболический путь β-аррестина. Исследование показало, что после инъекции морфина мышам «нокаутным» по β-аррестину-2 обезболивающее действие, опосредованное сигналом G-белка, является более сильным и продолжительным (Bohn et al., Science, 1999). Можно предположить, что в случае более сильного отрицательного смещения таких лигандов по отношению к β-аррестину, они способны даже избежать опосредованной β-аррестином десенсибилизации рецептора, что, таким образом, приведет к более продолжительной передаче сигнала G-белка и более эффективным обезболивающим действиям.
Заявки на патенты, в которых раскрыты агонисты MOR, включают публикации международных заявок на патенты WO2014022733, WO2008009415, WO2009018169, WO2012129495, WO2001049650, WO2002020481, WO2010051476 и WO2013087589 и т.п.
Длительное применение опиоидных лекарственных средств приводит к побочным эффектам, таким как толерантность (привыкание), угнетение дыхания и запор. Было продемонстрировано, что указанные побочные эффекты тесно связаны с функцией β-аррестина. Для того, чтобы снизить побочные эффекты опиоидных лекарственных средств, лекарственные средства могут быть разработаны на основе лиганда MOR с отрицательным смещением по отношению к β-аррестину, тем самым уменьшая опосредованные β-аррестином побочные эффекты и усиливая терапевтический эффект. При исследовании оксо-спиро-производных по настоящему изобретению, применяемых в качестве MOR-селективных лекарственных средств, компанией Trevena Inc. обнаружено, что при нахождении заместителя в бензильном положении арила, активность производных снижается (J. Med. Chem. 2013, 56, 8019-8031), но после проведения серии исследований автором настоящего изобретения установлено, что оксоспиропроизводные обладают высокой активностью в случае замыкании кольца бензильного положения, при этом также значимо повышался Emax (максимальный эффект лекарства), а также значительно улучшалась экспрессия hERG (human Ether-à-go-go-Related Gene), и в дальнейших исследованиях показано, что соединение с единственной конфигурацией обладает более высокой селективностью к MOR.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы (I-A) или к его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, или к их фармацевтически приемлемой соли:
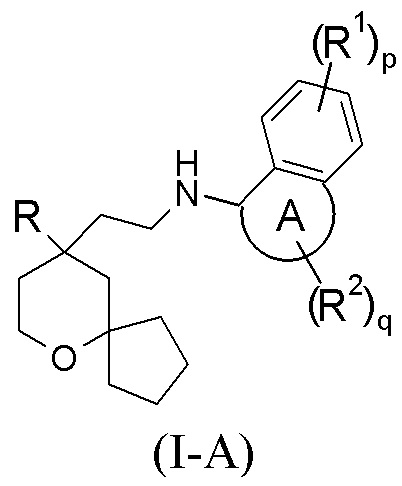
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль,
где:
кольцо A выбрано из группы, состоящей из циклоалкила и гетероциклила;
R выбран из группы, состоящей из арила и гетероарила, где каждый из арила и гетероарила необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, галогеналкила, атома галогена, амино, нитро, циано, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, –OR3, –C(O)R3, –C(O)OR3, –S(O)mR3 и –NR4R5;
каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкокси, галогеналкила, атома галогена, амино, нитро, гидрокси, циано, циклоалкила, гетероциклила, арила, гетероарила, –OR3, –C(O)R3, –C(O)OR3, –S(O)mR3 и –NR4R5, где каждый из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, галогеналкила, атома галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
каждый R2 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкокси, галогеналкила, атома галогена, амино, нитро, гидрокси, циано, оксо, алкенила, циклоалкила, гетероциклила, арила, гетероарила, –OR3, –C(O)R3, –C(O)OR3, –S(O)mR3 и –NR4R5, где каждый из алкила, алкокси, алкенила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или более групп, выбранных из группы, состоящей из дейтерия, алкила, галогеналкила, атома галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
или два R2 вместе образуют циклоалкил или гетероциклил, где каждый из циклоалкила или гетероциклила необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, галогеналкила, атома галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R3 выбран из группы, состоящей из атома водорода, алкила, дейтерированного алкила, амино, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкокси, гидроксиалкила, гидрокси, амино, алкоксикарбонила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, гидрокси, амино, алкоксикарбонила, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
p и q каждое независимо представляет собой 0, 1, 2, 3 или 4; и
m представляет собой 0, 1 или 2.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I-A) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль представляет собой соединение формулы (I):
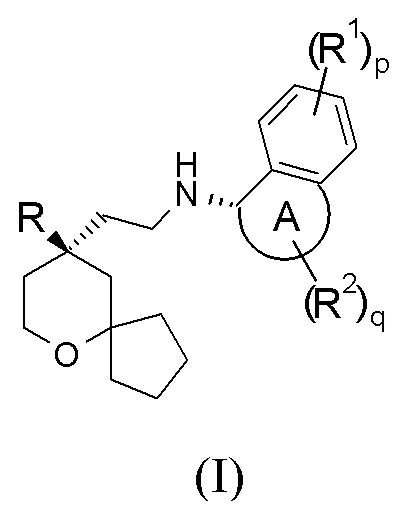
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль,
где:
кольцо A, R, R1, R2, p и q являются такими, как определено в формуле (I-A).
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или формулы (I-A) или их таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли кольцо A выбрано из группы, состоящей из 5–6-членного гетероциклила и 5–6-членного циклоалкила.
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или формулы (I-A), или их таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли R представляет собой пиридил.
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или формулы (I-A), или их таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода и атома галогена.
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или формулы (I-A), или их таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли каждый R2 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, оксо, алкокси, гидрокси, атома галогена и –OR3, где каждый из алкила и алкокси необязательно замещен одной или более групп, выбранных из группы, состоящей из дейтерия, алкила, атома галогена, гидрокси, амино, алкоксикарбонила, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила; R3 выбран из группы, состоящей из атома водорода, алкила и циклоалкила, где алкил необязательно замещен атомом галогена или циклоалкилом.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I-A) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль представляет собой соединение формулы (II-A):
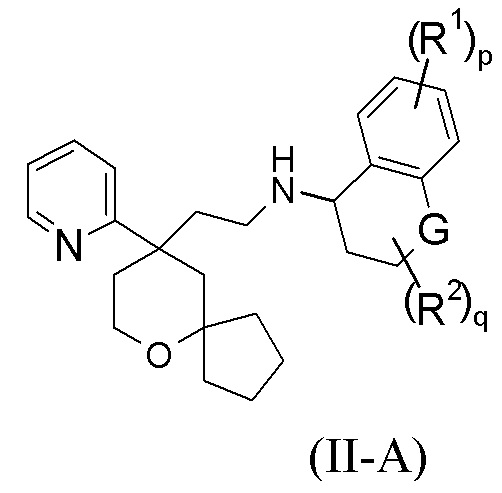
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль,
где:
G выбран из группы, состоящей из связи, CRaRb, C=O, NR4 и атома кислорода;
Ra и Rb каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкокси, галогеналкила, атома галогена, амино, нитро, гидрокси, циано, циклоалкила, гетероциклила, арила, гетероарила, –OR3, –C(O)R3, –C(O)OR3, –S(O)mR3 и -NR4R5, где каждый из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, галогеналкила, атома галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила, предпочтительно гидрокси или –OR3;
или Ra и Rb вместе образуют циклоалкил или гетероциклил, где каждый из циклоалкила или гетероциклила необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, галогеналкила, атома галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R1–R5, p, m и q являются такими, как определено в формуле (I-A).
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (II-A) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль представляет собой соединение формулы (II-B):
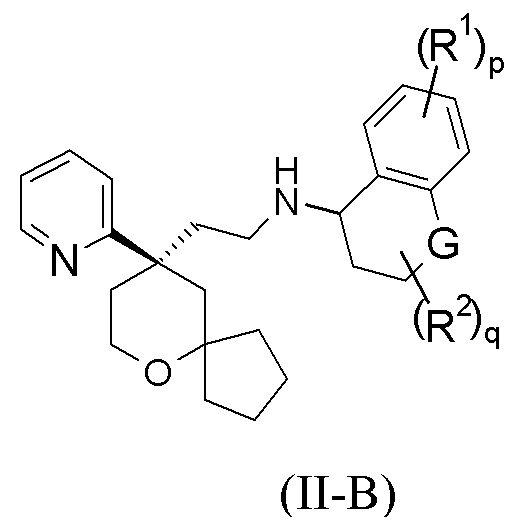
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль,
где:
G выбран из группы, состоящей из связи, CRaRb, C=O, NR4 и атома кислорода; и
R1, R2, R4, Ra, Rb, p и q являются такими, как определено в формуле (II-A).
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (II-A) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль представляет собой соединение формулы (II):
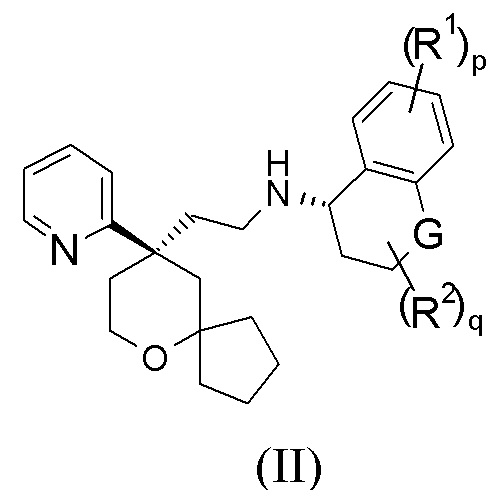
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль,
где:
G выбран из группы, состоящей из связи, CRaRb, C=O, NR4 и атома кислорода; и
Ra, Rb, R1, R2, R4, p и q являются такими, как определено в формуле (II-A).
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (II-A) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль представляет собой соединение формулы (IV-A):
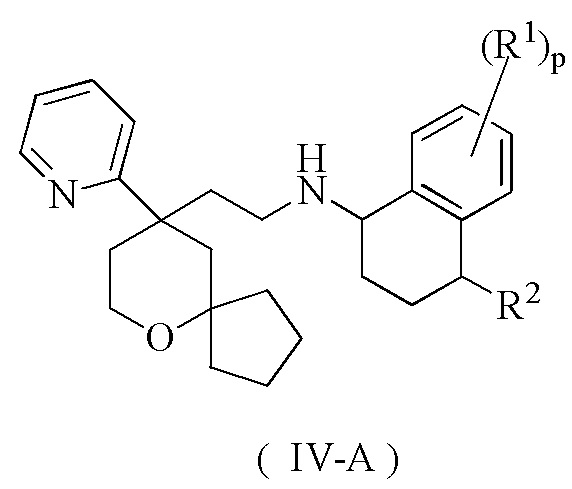
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль,
где:
R1, R2 и p являются такими, как определено в формуле (II-A).
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль представляет собой соединение формулы (IV):
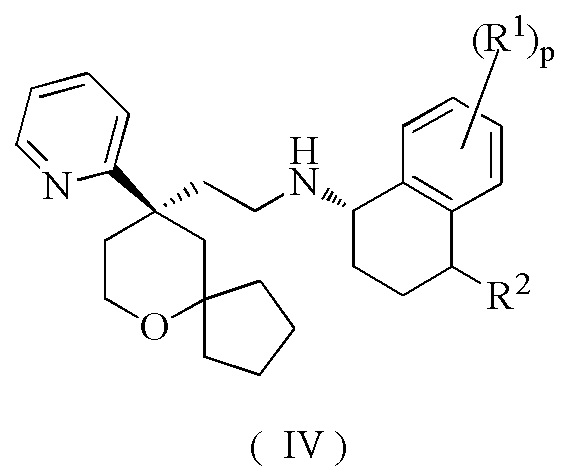
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль,
где:
R1, R2 и p являются такими, как определено в формуле (II).
Типичные соединения формулы (I-A) включают, но не ограничиваются указанными:
№









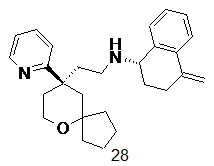
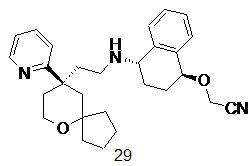
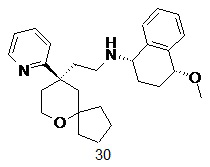
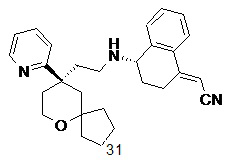
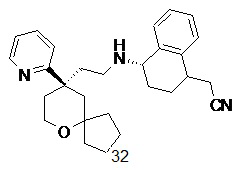
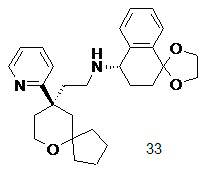

или их таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль,
В другом аспекте настоящее изобретение также относится к способу получения соединения формулы (I-A) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, включающему стадию:
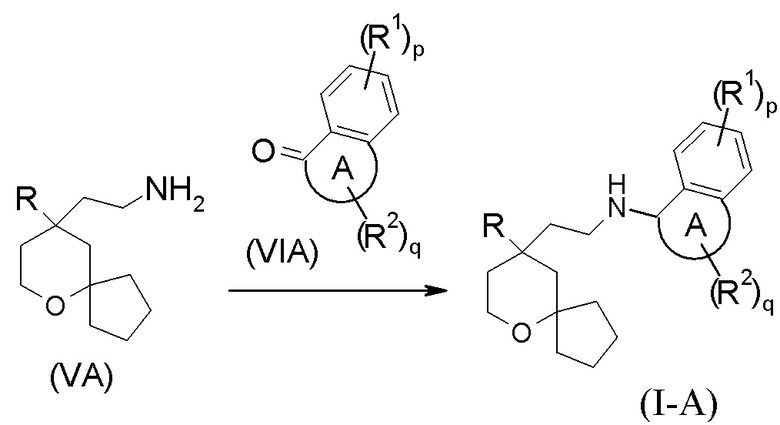
взаимодействия соединения формулы (VA) или его гидрохлорида с соединением формулы (VIA) посредством восстановительного аминирования с получением соединения формулы (I-A);
где:
кольцо A, R, R1, R2, p и q являются такими, как определено в формуле (I-A).
В другом аспекте настоящее изобретение также относится к способу получения соединения формулы (I-A) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, включающему стадию:
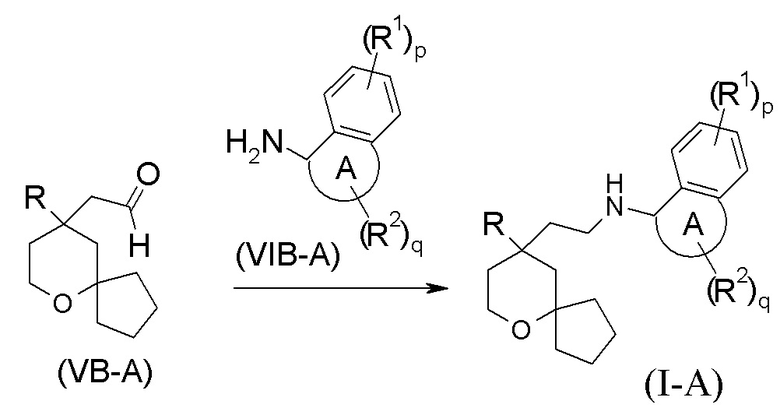
взаимодействия соединения формулы (VB-A) с соединением формулы (VIB-A) или его гидрохлоридом посредством восстановительного аминирования с получением соединения формулы (I-A);
где:
кольцо A, R, R1, R2, p и q являются такими, как определено в формуле (I-A).
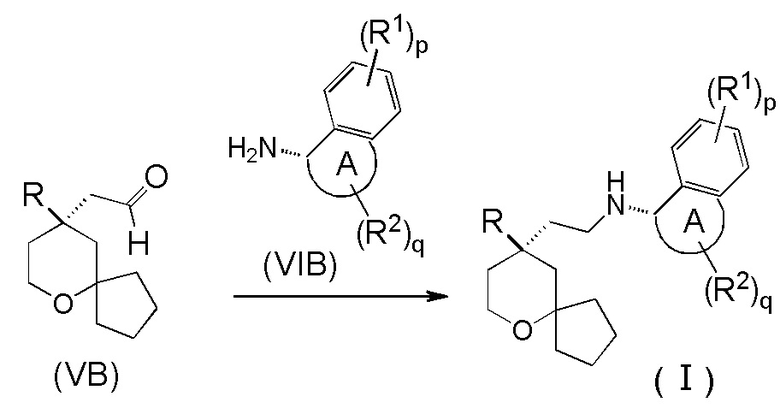
В другом аспекте настоящее изобретение также относится к способу получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, включающему стадию:
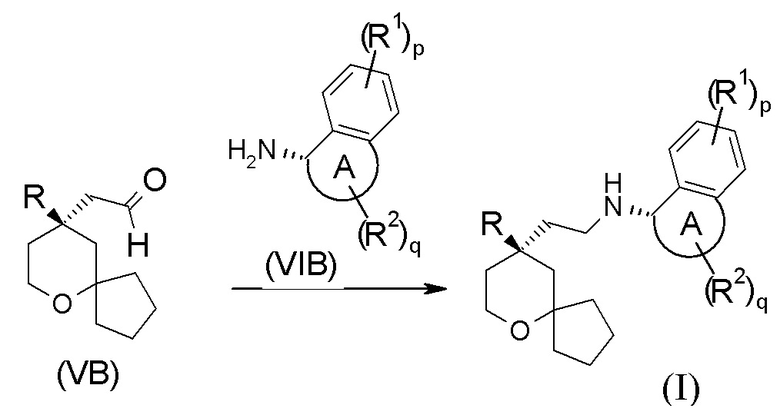
взаимодействия соединения формулы (VB) с соединением формулы (VIB) или его гидрохлоридом посредством восстановительного аминирования с получением соединения формулы (I);
где:
кольцо A, R, R1, R2, p и q являются такими, как определено в формуле (I).
В другом аспекте настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения каждой из указанных выше формул или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Настоящее изобретение также относится к способу получения указанной выше композиции, включающему стадию смешивания соединения, представленного каждой из формул, или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли с одним или более фармацевтически приемлемым носителем, разбавителем или эксципиентом.
Настоящее изобретение дополнительно относится к применению соединения каждой из формул, или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, или содержащей их фармацевтической композиции при получении лекарственного средства для агонизации или антагонизации рецептора MOR.
Настоящее изобретение дополнительно относится к применению соединения каждой из формул, в частности формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, или содержащей их фармацевтической композиции при получении лекарственного средства для профилактики и/или лечения опосредованного агонистом MOR и связанного с ним заболевания, где заболевание выбрано из группы, состоящей из боли, иммунной дисфункции, воспаления, эзофагеального рефлюкса, неврологических и психиатрических расстройств, заболеваний мочевыводящих путей и репродуктивной системы, сердечно-сосудистого заболевания и респираторных заболеваний.
Настоящее изобретение дополнительно относится к применению соединения каждой из формул, в частности формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, или содержащей их фармацевтической композиции при получении лекарственного средства для профилактики и/или лечения боли и связанных с болью заболеваний у млекопитающих, где боль может представлять собой послеоперационную боль, боль, вызванную раком, нейропатическую боль, травматическую боль и воспалительную боль и т. д., где рак выбран из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака предстательной железы, рака яичника, опухоли фаллопиевых труб, опухоли яичника, гемофилии и лейкоза.
Настоящее изобретение также относится к способу профилактики или лечения опосредованного агонистом MOR и связанного с ним заболевания, включающему введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения каждой из формул, в частности формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли. Этот способ показывает выраженную эффективность и меньшие побочные эффекты. При этом заболевание выбрано из группы, состоящей из боли, иммунной дисфункции, воспаления, эзофагеального рефлюкса, неврологических и психиатрических расстройств, заболеваний мочевыводящих путей и репродуктивной системы, сердечно-сосудистых заболеваний и респираторных заболеваний; предпочтительно боли.
В другом аспекте настоящее изобретение также относится к способу профилактики или лечения боли и связанных с болью заболеваний у млекопитающих, включающему стадию введения нуждающемуся в этом пациенту терапевтически эффективного количества соединения каждой из формул, в частности формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли. Этот способ обладает выраженной эффективностью и меньшими побочными эффектами. При этом боль может представлять собой послеоперационную боль, боль, вызванную раком, нейропатическую боль, травматическую боль и воспалительную боль; где рак выбран из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака предстательной железы, рака яичника, опухоли фаллопиевых труб, опухоли яичника, гемофилии и лейкоза.
Настоящее изобретение отноится к соединению каждой из формул, в частности формулы (I), или к его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, или их фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в качестве лекарственного средства для лечения иммунной дисфункции, воспаления, эзофагеального рефлюкса, неврологических и психиатрических расстройств, заболеваний мочевыводящих путей и репродуктивной системы, злоупотребления лекарственным средством и алкоголем, гастрита и диареи, сердечно-сосудистых заболеваний, респираторных заболеваний и кашля.
Фармацевтические композиции, содержащие активную фармацевтическую субстанцию, могут иметь форму, приемлемую для перорального введения, например, таблетки, леденца, лепешки, водной или масляной суспензии, диспергируемого порошка или гранулы, эмульсии, твердой или мягкой капсулы, либо сиропа или эликсира. Пероральные композиции могут быть получены в соответствии с любым способом, известным в данной области техники для получения фармацевтических композиций. Такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из подсластителей, корригентов, красителей и консервантов, для того чтобы обеспечить фармацевтической лекарственной форме приятный вид и вкус. Таблетка содержит активную фармацевтическую субстанцию в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, подходящими для производства таблетки.
Пероральные лекарственные формы могут быть предложены в виде твердых желатиновых капсул, в которых активная фармацевтическая субстанция смешана с инертным твердым разбавителем, таким как карбонат кальция, фосфат кальция или каолин, или в виде мягких желатиновых капсул, в которых активная фармацевтическая субстанция смешана с водорастворимым носителем, таким как полиэтиленгликоль, или с масляной средой, такой как арахисовое масло, жидкий парафин или оливковое масло.
Водная суспензия содержит активную фармацевтическую субстанцию в смеси с эксципиентами, приемлемыми для производства водной суспензии.
Активная фармацевтическая субстанция в смеси с диспергирующими или смачивающими агентами, суспендирующим агентом или с одним, или более консервантом может быть получена в виде диспергируемого порошка или гранулы, приемлемых для получения водной суспензии путем добавления воды. Примеры приемлемых диспергирующих или смачивающих агентов и суспендирующих агентов уже упомянуты выше. Могут быть также добавлены дополнительные эксципиенты, такие как подсластители, корригенты и красящие вещества. Эти композиции можно консервировать путем добавления антиоксиданта, такого как аскорбиновая кислота.
Настоящая фармацевтическая композиция также может иметь форму эмульсии масло-в-воде.
Фармацевтическая композиция по настоящему изобретению может иметь форму стерильного водного раствора. Приемлемыми средами, которые переносят лекарственное вещество, и растворителями, которые можно применять, являются вода, раствор Рингера и изотонический раствор хлорида натрия.
Фармацевтическая композиция по настоящему изобретению может иметь форму стерильной инъекционной водной или масляной суспензии для внутримышечного и подкожного введения.
Специалистам в данной области техники хорошо известно, что дозировка лекарственного средства зависит от ряда факторов, включающих, но не ограничивающихся указанными, следующие факторы: активность конкретного соединения, возраст пациента, массу тела пациента, общее состояние здоровья пациента, поведение пациента, рацион питания пациента, время введения, путь введения, скорость выведения, комбинацию лекарственного средства и т.п. В дополнение к этому наилучшее лечение, подразумевающее способ лечения, суточную дозу соединения формулы (I) или тип его фармацевтически приемлемой соли, может быть подтверждено традиционными терапевтическими схемами.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, термины, используемые в описании и в формуле изобретения, имеют описанные ниже значения.
«Алкил» относится к насыщенной алифатической углеводородной группе, включающей от C1 до C20 прямоцепочечные и разветвленные группы, предпочтительно алкил, содержащий от 1 до 12 атомов углерода, и более предпочтительно алкил, содержащий от 1 до 6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, содержащий от 1 до 6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкильная группа может быть замещенной или незамещенной. В случае замещения группа(-ы) заместителя(-ей) может(-гут) быть присоединена(-ы) в любой доступной точке присоединения. Группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Алкенил» относится к алкилу, такому, как определено выше, содержащему по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную двойную связь, например, к этенилу, 1-пропенилу, 2-пропенилу, 1-, 2- или 3-бутенилу и т. п. Алкенильная группа может быть замещенной или незамещенной. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио и гетероциклического алкилтио.
«Циклоалкил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, содержащей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 6 атомов углерода и наиболее предпочтительно от 5 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т. п. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
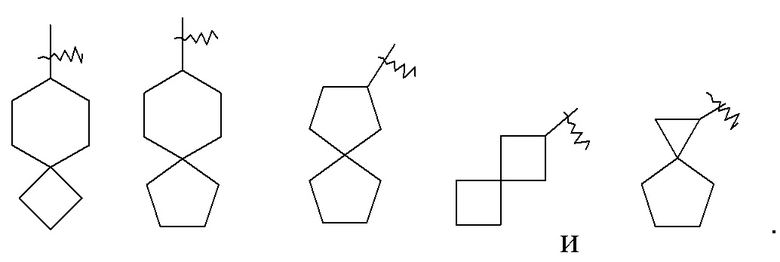
«Спироциклоалкил» относится к 5–20-членной полициклической группе, в которой кольца соединены посредством одного общего атома углерода (называемого спироатомом), где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной системы пи-электронов, предпочтительно к 6–14-членному спироциклоалкилу и более предпочтительно к 7–10-членному спироциклоалкилу. В соответствии с числом общих спироатомов между кольцами спироциклоалкил можно разделить на моноспироциклоалкил, диспироциклоалкил или полиспироциклоалкил и предпочтительно моноспироциклоалкил или диспироциклоалкил, и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспироциклоалкил. Неограничивающие примеры спироциклоалкила включают:
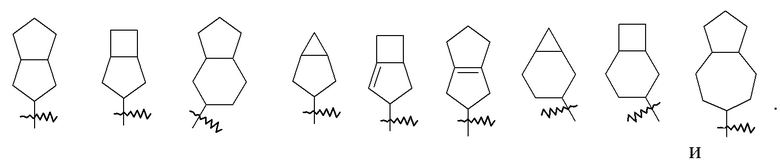
«Конденсированный циклоалкил» относится к 5–20-членной полностью углеродной полициклической группе, где каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной системы пи-электронов, предпочтительно к 6–14-членному конденсированному циклоалкилу и более предпочтительно к 7–10-членному конденсированному циклоалкилу. В соответствии с числом колец-членов конденсированный циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, предпочтительно бициклический или трициклический конденсированный циклоалкил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:
«Мостиковый циклоалкил» относится к 5–20-членной полностью углеродной полициклической группе, где каждые два кольца в системе имеют два общих не соединенных друг с другом атомов углерода, где кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной системы пи-электронов, предпочтительно к 6–14-членному мостиковому циклоалкилу и более предпочтительно к 7–10-членному мостиковому циклоалкилу. В соответствии с числом колец-членов мостиковый циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, предпочтительно бициклический, трициклический или тетрациклический мостиковый циклоалкил и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостикового циклоалкила включают:
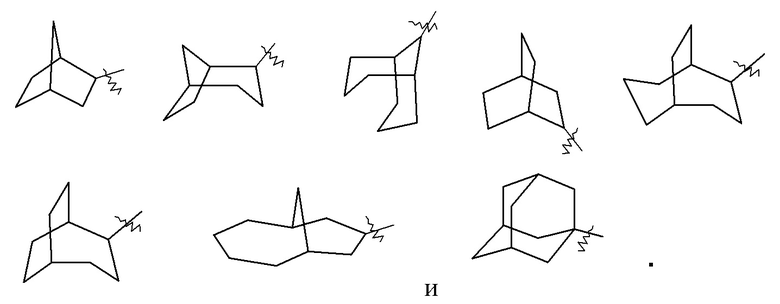
Кольцо циклоалкила может быть конденсировано с кольцом арила, гетероарила или гетероциклила, где кольцом, связанным с исходной структурой, является циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и т. п., предпочтительно бензоциклопентил, тетрагидронафтил. Циклоалкил может быть необязательно замещенным или незамещенным. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Гетероциклил» относится к 3–20-членной насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более гетероатомов, выбранных из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2), в качестве кольцевых атомов, но за исключением –O–O–, –O–S– или –S–S– в кольце, где остальными кольцевыми атомами являются атомы углерода. Предпочтительно гетероциклил содержит от 3 до 12 атомов, где от 1 до 4 атомов представляют собой гетероатомы, более предпочтительно от 3 до 8 атомов, где от 1 до 3 атомов представляют собой гетероатомы, и наиболее предпочтительно от 5 до 6 атомов, где от 1 до 2 или от 1 до 3 атомов представляют собой гетероатомы. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиенил, дигидроимидазолил, дигидрофуранил, дигидропиразолил, дигидропирролил, пиперидил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.п, предпочтительно тетрагидропиранил, пиперидил или пирролидинил. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
«Спирогетероциклил» относится к 5–20-членному полициклическому гетероциклилу, кольца которого соединены посредством одного общего атома (называемого спироатомом), где кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, а остальными кольцевыми атомами являются атомы углерода, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной системы пи-электронов, предпочтительно к 6–14-членному спирогетероциклилу и более предпочтительно к 7–10-членному спирогетероциклилу. В соответствии с числом общих спироатомов между кольцами спирогетероциклил можно разделить на моноспирогетероциклил, диспирогетероциклил или полиспирогетероциклил, и предпочтительно моноспирогетероциклил или диспирогетероциклил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-гетероциклил. Неограничивающие примеры спирогетероциклила включают:
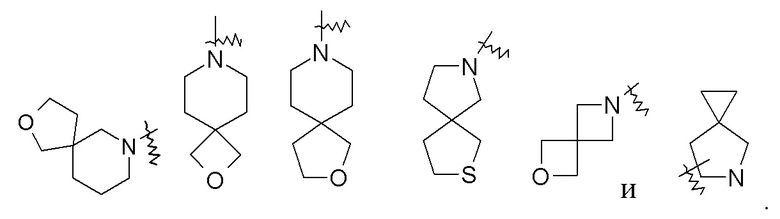
«Конденсированный гетероциклил» относится к 5–20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую пару соседних атомов с другим кольцом, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной системы пи-электронов, и где кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, а остальными кольцевыми атомами являются атомы углерода; предпочтительно 6–14-членный конденсированный гетероциклил и более предпочтительно 7–10-членный конденсированный гетероциклил. В соответствии с числом колец-членов конденсированный гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, предпочтительно бициклический или трициклический конденсированный гетероциклил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:
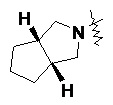
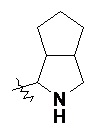




 и
и  .
.
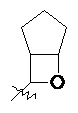
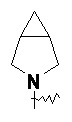
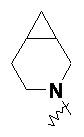
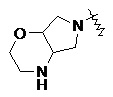
«Мостиковый гетероциклил» относится к 5–14-членной полициклической гетероциклильной группе, где каждые два кольца в системе имеют два общих не соединенных друг с другом атома, где кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной системы пи-электронов, и где кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, а остальными кольцевыми атомами являются атомы углерода; предпочтительно 6–14-членный мостиковый гетероциклил и более предпочтительно 7–10-членный мостиковый гетероциклил. В соответствии с числом колец-членов мостиковый гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, предпочтительно бициклический, трициклический или тетрациклический мостиковый гетероциклил и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостикового гетероциклила включают:
 .
.
Гетероциклильное кольцо может быть конденсировано с кольцом арила, гетероарила или циклоалкила, где кольцом, связанным с исходной структурой, является гетероциклил. Неограничивающие примеры включают:

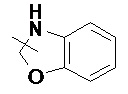
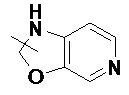 и
и 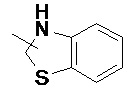 и т.д.
и т.д.
Гетероциклил может быть необязательно замещенным или незамещенным. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Арил» относится к 6–14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т. е. каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом в системе), имеющему полностью конъюгированную систему пи-электронов, предпочтительно к 6–10-членному арилу и более предпочтительно к 5–6-членному арилу, например, к фенилу и нафтилу. Арильное кольцо может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, где кольцом, связанным с исходной структурой, является арильное кольцо. Неограничивающие примеры включают:
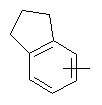
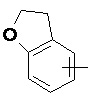
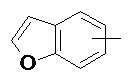
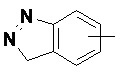


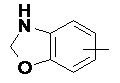
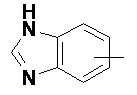
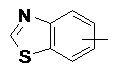
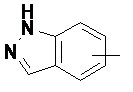
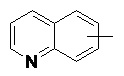
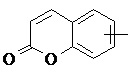 и
и 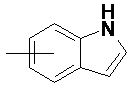 .
.
Арил может быть необязательно замещенным или незамещенным. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила и циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси, алкоксикарбонила.
«Гетероарил» относится к 5–14-членной гетероароматической системе, содержащей от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, S и N в качестве кольцевых атомов, предпочтительно к 5–10-членному гетероарилу, имеющему от 1 до 3 гетероатомов, и более предпочтительно к 5- или 6-членному гетероарилу, имеющему от 1 до 2 гетероатомов, например к имидазолилу, фурилу, тиенилу, тиазолилу, пиразолилу, оксазолилу, пирролилу, тетразолилу, пиридилу, пиримидинилу, тиадиазолилу, пиразинилу и т.п., предпочтительно к имидазолилу, пиразолилу, пиримидинилу или тиазолилу и более предпочтительно к пиразолилу. Гетероарильное кольцо может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, где кольцом, связанным с исходной структурой, является гетероарильное кольцо. Неограничивающие примеры включают:
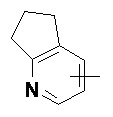
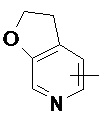
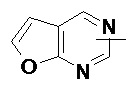

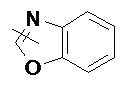

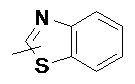
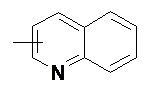 и
и  .
.
Гетероарил может быть необязательно замещенным или незамещенным. В случае замещения группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси, алкоксикарбонила.
«Алкокси» относится к группе -O-(алкил) или -O-(незамещенный циклоалкил), где алкил является таким, как определено выше. Неограничивающие примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и т. п. Алкокси может быть необязательно замещенным или незамещенным. В случае замещения заместитель предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси, алкоксикарбонила.
«Дейтерированный алкил» относится к алкилу, замещенному атомом(-ами) дейтерия, где алкил является таким, как определено выше.
«Гидроксиалкил» относится к алкилу, замещенному гидроксигруппой(-ами), где алкил является таким, как определено выше.
«Гидрокси» относится к группе –OH.
«Атом галогена» относится к атому фтора, хлора, брома или йода.
«Амино» относится к группе –NH2.
«Циано» относится к группе –CN.
«Нитро» относится к группе –NO2.
«Карбокси» относится к группе –C(O)OH.
«Алкоксикарбонил» относится к группе –C(O)O(алкил) или к группе –C(O)O(циклоалкил), где алкил и циклоалкил являются такими, как определено выше.
«Ацилгалогенид» относится к соединению, содержащему группу –C(O)-атом галогена.
Все выражения «X выбран из группы, состоящей из A, B или C», «X выбран из группы, состоящей из A, B и C», «X представляет собой A, B или C», «X представляет собой A, B и C» и т. п. имеют одно и то же значение. Это означает, что X может представлять собой любое одно или более из A, B и C. «Необязательный» или «необязательно» означает, что описанное впоследствии событие или обстоятельство может произойти, но необязательно произойдет, и это описание включает ситуацию, в которой это событие или обстоятельство происходит или не происходит. Например, «гетероциклическая группа необязательно замещена алкилом» означает, что алкильная группа может присутствовать, но необязательно присутствует, и это описание включает ситуацию, в которой гетероциклическая группа замещена алкилом, и в которой гетероциклическая группа не замещена алкилом.
«Замещенный» относится к одному или более атомов водорода в группе, предпочтительно вплоть до 5, более предпочтительно от 1 до 3 атомов водорода, которые независимо замещены соответствующим количеством заместителей. Без пояснений понятно, что заместители могут существовать только в их химически возможном положении. Специалист в данной области техники способен определить, возможно или невозможно замещение, с помощью экспериментов или теоретически, не прилагая слишком больших усилий. Например, комбинация амино- или гидрокси-группы, имеющей свободные атомы водорода, и атомов углерода, имеющих ненасыщенные двойные связи (такие как олефиновые связи), может быть нестабильной.
«Фармацевтическая композиция» относится к смеси одного или более соединений в соответствии с настоящим изобретением или их физиологически/фармацевтически приемлемых солей, или пролекарств и других химических компонентов, а также других компонентов, таких как физиологически/фармацевтически приемлемые носители и эксципиенты. Цель фармацевтической композиции состоит в том, чтобы способствовать введению соединения в организм, обеспечить условия абсорбции активной фармацевтической субстанции и, следовательно, проявления им биологической активности.
«Фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которая является безопасной и эффективной для млекопитающих и обладает желаемой биологической активностью.
СПОСОБ СИНТЕЗА СОЕДИНЕНИЯ ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Для достижения цели настоящего изобретения применены следующие технические решения.
Способ получения соединения формулы (I-A) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли включает следующую стадию:
Схема 1

взаимодействия соединения формулы (VA) или его гидрохлорида с соединением формулы (VIA) посредством восстановительного аминирования с получением соединения формулы (I-A);
где:
кольцо A, R, R1, R2, p и q являются такими, как определено в формуле (I-A).
Соединение формулы (I-A) по настоящему изобретению может быть также получено следующим образом:
Схема 2

взаимодействием соединения формулы (VB-A) с соединением формулы (VIB-A) или его гидрохлоридом посредством восстановительного аминирования с получением соединения формулы (I-A);
где:
кольцо A, R, R1, R2, p и q являются такими, как определено в формуле (I-A).
В другом аспекте настоящее изобретение также относится к способу получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, включающему стадию:
Схема 1
взаимодействия соединения формулы (VB) с соединением формулы (VIB) или его гидрохлоридом посредством восстановительного аминирования с получением соединения формулы (I);
где:
кольцо A, R, R1, R2, p и q являются такими, как определено в формуле (I).
ПРЕДПОЧТИТЕЛЬНЫЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение будет описано со ссылкой на приведенные ниже примены, но эти примеры не следует рассматривать как ограничивающие объем изобретения.
Примеры:
Структуры соединений идентифицируют с помощью ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Химические сдвиги (δ) ЯМР приведены в 10-6 (млн-1). ЯМР определяют на устройстве Bruker AVANCE-400. Растворителями для определения являются дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), и внутренним стандартом является тетраметилсилан (ТМС).
МС проводят с помощью масс-спектрометра FINNIGAN LCQAd (ионизация электрораспылением (ИЭР)) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводят на спектрометре, сопряженном с жидкостным хроматографом высокого давления, Agilent 1200DAD (хроматографическая колонка Sunfire C18 150 на 4,6 мм), и на спектрометре, сопряженном с жидкостным хроматографом высокого давления, Waters 2695-2996 (хроматографическая колонка Gimini C18 150 на 4,6 мм).
Средние скорости ингибирования киназы и значения IC50 (концентрация полумаксимального ингибирования) определяют с помощью набора реагентов для твердофазного иммуносорбентного ферментного анализа (ИФА) ELISA NovoStar (BMG Co., Германия).
Для тонкослойной гель-хроматографии на силикагеле (ТСХ) используют селикагелевую пластину Yantai Huanghai HSGF254 или Qingdao GF254. Размеры пластины для силикагеля, используемой в ТСХ, составляет от 0,15 до 0,2 мм, а размеры пластины силикагеля, используемой в очистке продукта, составляет от 0,4 до 0,5 мм.
В качестве носителя для колоночной хроматографии используют силикагель Yantai Huanghai от 200 до 300 меш.
Известные исходные материалы настоящего изобретения могут быть получены традиционными способами синтеза, известными в данной области техники, или могут быть приобретены у компаний ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc. или Dari chemical Company и т. д.
Если не указано иное, реакции проводят в атмосфере азота или в атмосфере аргона.
Термин «атмосфера азота» или «атмосфера аргона» означает, что реакционная колба оборудована баллоном азота или аргона емкостью 1 л.
Термин «атмосфера водорода» означает, что реакционная колба оборудована баллоном водорода емкостью 1 л.
Реакции гидрогенизации под давлением проводят с помощью аппарата Парра 3916EKX для гидрогенизации и генератора водорода QL-500 или аппарата для гидрогенизации HC2-SS.
В реакциях гидрогенизации в реакционной системе обычно создают вакуум и заполняют ее водородом, и описанную выше операцию повторяют три раза.
В микроволновой реакции используют микроволновой реактор CEM Discover-S 908860.
Если не указано иное, раствор, используемый в реакциях, относится к водному раствору.
Если не указано иное, температура реакционной смеси при проведении реакций относится к комнатной температуре в диапазоне от 20 до 30 °C.
Ход реакции отслеживают с помощью тонкослойной хроматографии (ТСХ), и система растворителей для проявления включает: A: систему дихлорметана и метанола, B: систему н-гексана и этилацетата, C: систему дихлорметана и ацетона. Соотношение объема растворителя можно регулировать в зависимости от полярности соединений.
Элюирующая система для очистки соединений с помощью колоночной хроматографии и тонкослойной хроматографии включает: A: систему дихлорметана и метанола, B: систему н-гексана и этилацетата, C: систему дихлорметана и ацетона. Соотношение объема растворителя можно регулировать в зависимости от полярности соединений, и в некоторых случаях можно добавить небольшое количество щелочного реагента, такого как триэтиламин, или кислого реагента, такого как уксусная кислота.
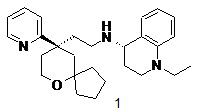
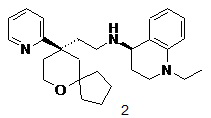
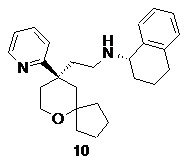
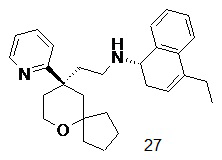
Примеры 1, 2:
(S)-1-этил-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4- тетрагидрохинолин-4-амин 1
(R)-1-этил-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4- тетрагидрохинолин-4-амин 2
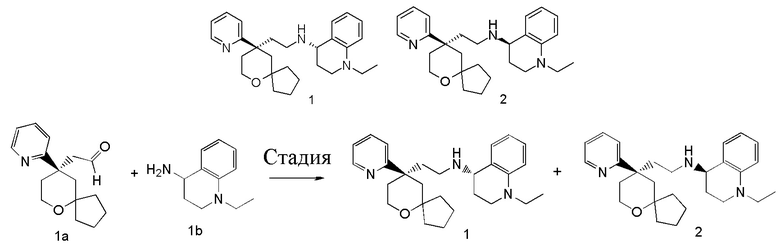
(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 1a (294 мг, 1,135 ммоль, полученный способом, раскрытым в заявке на патент WO2012129495) и 1-этил-1,2,3,4-тетрагидрохинолин-4-амин 1b (200 мг, 1,135 ммоль, полученный способом, раскрытым в заявке на патент WO2014078454) растворяли в 15 мл дихлорметана, и смесь перемешивали в течение 1 часа. Затем добавляли триацетоксиборгидрид натрия (1,203 г, 5,675 ммоль), и полученную в результате смесь перемешивали в течение 16 часов. Добавляли 20 мл воды, и реакционный раствор экстрагировали дихлорметаном (3 раза по 20 мл). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью высокоэффективной жидкостной хроматографии с получением соединения, указанного в заголовке, 1-этил-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидрохинолин-1-амина, которое впоследствии подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Superchiral S-AD (Chiralway), внутренний диаметр 2 см, 25 см, 5 мкм; подвижная фаза: CO2:метанол:диэтаноламин составляет 75:25:0,05, скорость потока: 50 г/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений 1 (98 мг, коричневое масло) и 2 (95 мг, желтое твердое вещество), указанных в заголовке.
Пример 1:
МС m/z (ИЭР): 420,3 [M+1];
Анализ с помощью хиральной ВЭЖХ: время удерживания 4,028 мин, хиральная чистота: 99,7 % (хроматографическая колонка: Superchiral S-AD (Chiralway), внутренний диаметр 0,46 см, 15 см, 5 мкм; подвижная фаза: CO2:метанол:диэтаноламин составляет 75:25:0,05 (об./об./об.))
1H ЯМР (400 МГц, ДМСО-d6) δ 8,54 (s, 1H), 7,72 (s, 1H), 7,45 (d, 1H), 7,20 (s, 1H), 6,95 (s, 1H), 6,78 (d, 1H), 6,52 (d, 1H), 6,37 (s, 1H), 3,60 (br, 2H), 3,18-3,43 (m, 3H), 2,99 (m, 1H), 2,33-2,45 (m, 3H), 1,77-1,99 (m, 3H), 1,19-1,60 (m, 12H), 1,00-1,06 (m, 4 H), 0,63 (m, 1H).
Пример 2:
МС m/z (ИЭР): 420,3 [M+1];
Анализ с помощью хиральной ВЭЖХ: время удерживания 3,725 мин, хиральная чистота: 99,8 % (хроматографическая колонка: Superchiral S-AD (Chiralway), внутренний диаметр 0,46 см, 15 см, 5 мкм; подвижная фаза: CO2: метанол:диэтаноламин составляет 75:25:0,05 (об./об./об.))
1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (s, 1H), 7,72 (s, 1H), 7,46 (d, 1H), 7,20 (s, 1H), 6,97 (s, 1H), 6,85 (d, 1H), 6,54 (d, 1H), 6,40 (s, 1H), 3,61 (br, 2H), 3,17-3,25 (m, 3H), 3,00-3,01 (m, 1H), 2,33-2,46 (m, 3H), 1,78-1,97 (m, 3H), 1,24-1,65 (m, 12H), 1,01-1,06 (m, 4 H), 0,61 (m, 1H).
Пример 3:
(1R,2R)-1-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-2,3-дигидро-1H-инден-2-ол 3
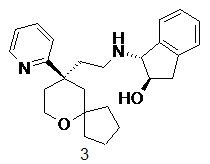
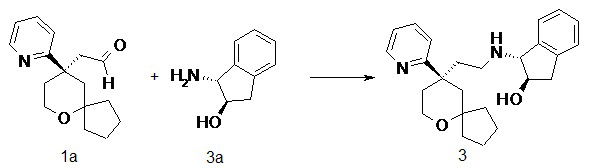
Соединение 1a (50 мг, 0,193 ммоль) и (1R,2R)-1-амино-2,3-дигидро-1H-инден-2-ол 3a (31,6 мг, 0,212 ммоль, полученный способом, раскрытым в заявке на патент WO2010148191), растворяли в 15 мл дихлорметана, добавляли необходимое количество метанола для усиления растворимости. Полученную в результате смесь перемешивали в течение 1 часа при комнатной температуре, затем добавляли триацетоксиборгидрид натрия (200 мг, 0,965 ммоль). После перемешивания в течение 16 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 3, указанного в заголовке, в виде белого твердого вещества (50 мг, выход 66 %).
МС m/z (ИЭР): 393,5 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 8,51 (d, 1H), 7,73-7,66 (m, 1H), 7,37 (d, 1H), 7,28-7,20 (m, 3H), 7,19-7,12 (m, 2H), 4,75 (d, 1H), 4,61 (d, 1H), 3,82-3,71 (m, 4H), 3,41-3,31 (m, 2H), 2,30-2,89 (m, 2H), 2,41-2,25 (m, 2H), 1,96-1,90 (m, 2H), 1,85-1,61 (m, 4H), 1,61-1,25 (m, 6H).
Пример 4:
(1R,2R)-2-метокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-2,3-дигидро-1H-инден-1-амин 4
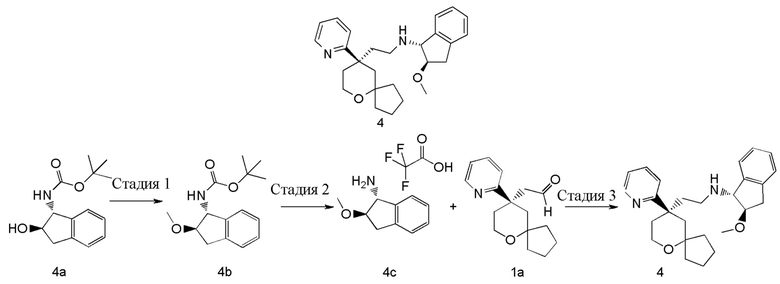
Стадия 1
трет-Бутил-((1R,2R)-2-метокси-2,3-дигидро-1H-инден-1-ил)карбамат 4b
трет-Бутил-((1R,2R)-2-гидрокси-2,3-дигидро-1H-инден-1-ил)карбамат 4a (350 мг, 1,34 ммоль, полученный хорошо известным способом, раскрытым в публикации Angewandte Chemie-International Edition, 2012, 51(34), 8495-8499) растворяли в 15 мл дихлорметана, затем добавляли оксид серебра (930 мг, 4,02 ммоль), иодметан (0,25 мл, 4,02 ммоль) и небольшое количество активированных молекулярных сит 4Å. Полученную в результате смесь перемешивали в течение 16 часов при комнатной температуре, фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой B с получением соединения 4b, указанного в заголовке, в виде белого твердого вещества (200 мг, выход 57 %).
МС m/z (ИЭР): 208,2 [M-56+1]
Стадия 2
(1R,2R)-2-метокси-2,3-дигидро-1H-инден-1-амина 2,2,2-трифторацетат 4c
Соединение 4b (60 мг, 0,228 ммоль) растворяли в 5 мл дихлорметана, затем добавляли 0,5 мл трифторуксусной кислоты. После перемешивания в течение 2 часов реакционный раствор концентрировали при пониженном давлении с получением неочищенного соединения 4c, указанного в заголовке, в виде желтого масла (66 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 164,2 [M+1]
Стадия 3
(1R,2R)-2-метокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-2,3-дигидро-1H-инден-1-амин 4
Соединение 1a (50 мг, 0,193 ммоль) и неочищенное соединение 4c (66 мг, 0,228 ммоль) растворяли в 15 мл дихлорметана. Полученную в результате смесь перемешивали в течение 30 минут при комнатной температуре, затем добавляли триацетоксиборгидрид натрия (200 мг, 0,965 ммоль). После перемешивания в течение 16 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 4, указанного в заголовке, в виде светло-желтого масла (25 мг, выход 32 %).
МС m/z (ИЭР): 407,3 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 8,55 (d, 1H), 7,71 (d, 1H), 7,58 (d, 1H), 7,40 (d, 1H), 7,28 (d, 1H), 7,25-7,10 (m, 3H), 4,39 (d, 1H), 4,26 (d, 1H), 3,82-3,70 (m, 5H), 3,30 (s, 3H), 2,88-2,30 (m, 2H), 2,40-2,26 (m, 2H), 1,96-1,91 (m, 2H), 1,85-1,62 (m, 4H), 1,61-1,24 (m, 6H).
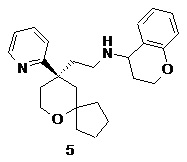
Пример 5:
N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)хроман-4-амин
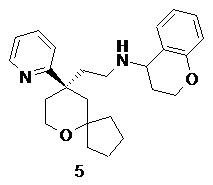
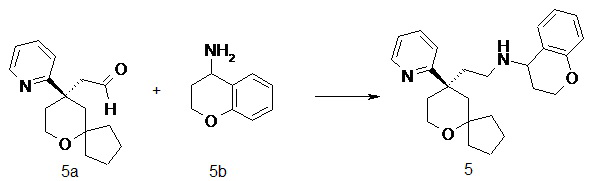
(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (20 мг, 0.08 ммоль, полученный способом, раскрытым в заявке на патент WO2012129495) и хроман-4-амин 5b (23 мг, 0,15 ммоль, полученный способом, раскрытым в публикации Bioorganic & Medicinal Chemistry Letters, 2011, 21(5), 1338-1341) растворяли в 10 мл дихлорметана, и смесь перемешивали в течение 2 часов. Затем добавляли триацетоксиборгидрид натрия (65 мг, 0,31 ммоль), и полученную в результате смесь перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 5, указанного в заголовке, в виде желтого масла (6 мг, выход 20 %).
МС m/z (ИЭР): 393,5 [M+1]
1H ЯМР (400 МГц, метанол-d4) δ 8,55 (s, 1H), 7,78 (t, 1H), 7,52 (d, 1H), 7,27 (d, 1H), 7,01-7,12 (m, 2H), 6,66-6,85 (m, 2H), 4,05-4,23 (m, 2H), 3,71-3,86 (m, 2H), 3,59-3,69 (m, 1H), 2,51-2,65 (m, 2H), 2,37-2,47 (m, 1H), 1,98-2,17 (m, 2H), 1,84-1,96 (m, 2H), 1,37-1,83 (m, 9H), 1,24-1,35 (m, 1H), 1,05-1,17 (m, 1H), 0,65-0,71 (m, 1H).
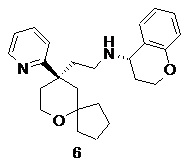
Пример 6:
(S)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)хроман-4-амин


(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (80 мг, 0,31 ммоль) и (S)-хроман-4-амина гидрохлорид 6a (86 мг, 0,46 ммоль, полученный способом, раскрытым в публикации ACS Catalysis, 3(4), 555-559; 2013) растворяли в 10 мл смеси дихлорметана и метанола (об./об составляет 5:1), и смесь перемешивали в течение 1 часа. Затем добавляли триацетоксиборгидрид натрия (263 мг, 1,24 ммоль), и полученную в результате смесь перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 6, указанного в заголовке, в виде белого вязкого твердого вещества (36 мг, выход 32,1 %).
МС m/z (ИЭР): 393,5 [M+1]
1H ЯМР (400 МГц, метанол-d4) δ 8,55 (d, 1H), 7,80-7,76 (m, 1H), 7,53 (d, 1H), 7,26-7,25 (m, 1H), 7,05-7,01 (m, 2H), 6,78-6,70 (m, 2H), 4,17-4,10 (m, 2H), 3,79-3,63 (m, 3H), 2,56-2,42 (m, 3H), 2,19-2,10 (m, 2H), 1,92-1,82 (m, 2H), 1,80-1,44 (m, 12H).
Пример 7:
(R)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)хроман-4-амин

(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (80 мг, 0,31 ммоль, полученный способом, раскрытым в заявке на патент WO2012129495), (R)-хроман-4-амина гидрохлорид 7a (115 мг, 0,62 ммоль, полученный способом, раскрытым в публикации European Journal of Organic Chemistry, 2014(31), 7034-7038, 2014) и триацетоксиборгидрид натрия (197 мг, 0,93 ммоль) растворяли в 10 мл смеси дихлорметана и метанола (об./об. составляет 5:1), и смесь перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 7, указанного в заголовке, в виде светло-желтого масла (30 мг, выход 24,8 %).
МС m/z (ИЭР): 393,5 [M+1]
1H ЯМР (400 МГц, метанол-d4) δ 8,63 (d, 1H), 7,93 (t, 1H), 7,64 (d, 1H), 7,39 (t, 1H), 7,29 (t, 1H), 7,19 (d, 1H), 6,81-6,97 (m, 2H), 4,25-4.35 (m, 1H), 4,14-4,24 (m, 1H), 3,79 (d, 2H), 2,47-2,65 (m, 3H), 2,13-2,32 (m, 3H), 1,87-2,03 (m, 2H), 1,72-1,85 (m, 2H), 1,40-1,71 (m, 5H), 1,25-1,35 (m, 2H), 1,06-1,15 (m, 1H), 0,66-0,75 (m, 1H).
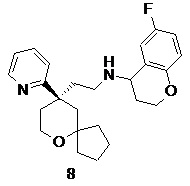
Пример 8:
6-фтор-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)хроман-4-амин

(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (30 мг, 0,12 ммоль, полученный способом, раскрытым в заявке на патент WO2012129495) и 6-фторхроман-4-амин 8a (39 мг, 0,23 ммоль, полученный способом, раскрытым в публикации Bioorganic & Medicinal Chemistry Letters, 2011, 21(5), 1338-1341) растворяли в 20 мл дихлорметана, затем добавляли триацетоксиборгидрид натрия (74 мг, 0,35 ммоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 8, указанного в заголовке, в виде светло-желтого твердого вещества (10 мг, выход 20,4 %).
МС m/z (ИЭР): 411,2 [M+1]
1H ЯМР (400 МГц, CDCl3) δ 8,56 (d, 1H), 7,67-7,64 (m, 1H), 7,34-7,31 (m, 1H), 7,16-7,14 (m, 1H), 6,84-6,74 (m, 2H), 6,73-6,7 (m, 1H), 4,02-4,08 (m, 2H), 3,78-3,75 (m, 3H), 2,66-2,12 (m, 6H), 2,1-1,59 (m, 9H), 1,35-1,18 (m, 4H).
Пример 9:
(R)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин
(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (35 мг, 0,14 ммоль, полученный способом, раскрытым в заявке на патент WO2012129495) и (R)-1,2,3,4-тетрагидронафталин-1-амин 9a (40 мг, 0,27 ммоль, полученный способом, раскрытым в публикации Angewandte Chemie-International Edition, 45(28), 4641-4644, 2006) растворяли в 5 мл дихлорметана. Полученную в результате смесь перемешивали в течение 1 часа, затем добавляли триацетоксиборгидрид натрия (144 мг, 0,68 ммоль). После перемешивания в течение 1 часа реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 9, указанного в заголовке, в виде желтого твердого вещества (15 мг, выход 27,5 %).
МС m/z (ИЭР): 391,2 [M+1]
1H ЯМР (400 МГц, CDCl3) δ 8,57 (d, 1H), 7,65 (t, 1H), 7,32 (d, 1H), 7,16 (d, 1H), 7,11-7,07 (m, 3H), 7,05 (d, 1H), 3,77 (d, 2H), 3,60-3,57 (br, 1H), 2,73-2,70 (m, 3H), 2,45 (d, 1H), 2,34 (d, 1H), 2,15-2,08 (m, 1H), 2,05-2,02 (m, 1H), 1,91 (d, 1H), 1,75 -1,70 (m, 12H), 1,50-1,44 (m, 3H).
Пример 10:
(S)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин

(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (20 мг, 0,14 ммоль, полученный способом, раскрытым в заявке на патент WO2012129495) и (S)-1,2,3,4-тетрагидронафталин-1-амин 10a (50 мг, 0,272 ммоль, полученный способом, раскрытым в публикации Angewandte Chemie-International Edition, 45(28), 4641-4644, 2006) растворяли в 20 мл дихлорметана. Полученную в результате смесь перемешивали в течение 1 часа, затем добавляли триацетоксиборгидрид натрия (144 мг, 0,68 ммоль). После перемешивания в течение 1 часа реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 10, указанного в заголовке, в виде желтого твердого вещества (15 мг, выход 28,3 %).
МС m/z (ИЭР): 391,2 [M+1]
1H ЯМР (400 МГц, метанол-d4) δ 8,77 (d, 1H), 8,28 (t, 1H), 7,92 (d, 1H), 7,71 (t, 1H), 7,33-7,19 (m, 4H), 4,38 (t, 1H), 3,80-3,74 (m, 2H), 3,23-3,11 (m, 1H), 3,08-2,98 (m, 1H), 2,87-2,82 (m, 2H), 2,56-2,48 (m, 3H), 2,26-2,04 (m, 5H), 1,85-1,81 (m, 3H), 1,56-1,32 (m, 5H), 1,34-1,31 (m, 1H), 0,82-0,79 (m, 1H).
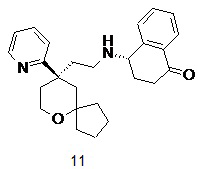
Пример 11:
(S)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-3,4-дигидронафталин-1(2H)-он

Стадия 1
(S)-трет-бутил-(1,2,3,4-тетрагидронафталин-1-ил)карбамат 11a
(S)-1,2,3,4-тетрагидронафталин-1-амин 10a (3 г, 20,41 ммоль, полученный способом, раскрытым в публикации Angewandte Chemie-International Edition, 45(28), 4641-4644, 2006) растворяли в 100 мл дихлорметана, затем добавляли триэтиламин (5,7 мл, 40,82 ммоль) и ди-трет-бутилдикарбонат (4,9 г, 22,45 ммоль). После перемешивания в течение 12 часов реакционный раствор последовательно промывали водой (100 мл) и насыщенным раствором бикарбоната натрия (100 мл). Органическую фазу высушивали над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении с получением неочищенного соединения 11a (5,6 г) в виде светло-желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 248,3 [M+1]
Стадия 2
(S)-трет-бутил-(4-оксо-1,2,3,4-тетрагидронафталин-1-ил)карбамат 11b
Неочищенный (S)-трет-бутил-(1,2,3,4-тетрагидронафталин-1-ил)карбамат 11a (5,6 г, 20,41 ммоль) растворяли в 90 мл смеси ацетона и воды (об./об. составляет 2:1), затем медленно добавляли сульфат магния (5,5 г, 45,66 ммоль) и перманганат калия (7,22 г, 45,66 ммоль) при перемешивании. Реакционную систему перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой B с получением соединения 11b, указанного в заголовке, в виде беловатого твердого вещества (3,1 мг, выход 52 %).
МС m/z (ИЭР): 262,3 [M+1]
Стадия 3
(S)-4-амино-3,4-дигидронафталин-1(2H)-он 11c
(S)-трет-бутил-(4-оксо-1,2,3,4-тетрагидронафталин-1-ил)карбамат 11b (1 г, 3,83 ммоль) растворяли в 20 мл дихлорметана, затем добавляли 8 мл 4M раствора хлорида водорода в 1,4-диоксане. После перемешивания в течение 2 часов реакционный раствор концентрировали при пониженном давлении, к полученному в результате остатку добавляли 10 мл этанола и по каплям добавляли 30% водный раствор аммиака для доведения значения pH до 8. Смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 11c, указанного в заголовке, в виде зеленого вязкого вещества (400 мг, выход 64,8 %).
МС m/z (ИЭР): 162,3 [M+1]
Стадия 4
(S)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-3,4-дигидронафталин-1(2H)-он 11
(S)-4-амино-3,4-дигидронафталин-1(2H)-он 11c (200 мг, 1,24 ммоль) и (R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (268 мг, 1,04 ммоль, полученный способом, раскрытым в заявке на патент WO2012129495) растворяли в 20 мл дихлорметана, и смесь перемешивали в течение 1 часа, затем добавляли триацетоксиборгидрид натрия (1,1 г, 5,18 ммоль). После перемешивания в течение 2 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 11, указанного в заголовке, в виде белого твердого вещества (136 мг, выход 32,4 %).
МС m/z (ИЭР): 405,6 [M+1]
1H ЯМР (400 МГц, метанол-d4) δ 8,73 (d, 1H), 8,15-8,09 (m, 2H), 7,83 (d, 1H), 7,81-7,69 (m, 3H), 7,47 (d, 1H), 4,45 (t, 1H), 3,77-3,74 (m, 2H), 3,03-2,98 (m, 1H), 2,75-2,68 (m, 3H), 2,51-2,44 (m, 5H), 2,05-2,01 (m, 2H), 1,57-1,48 (m, 7H), 1,20-1,05 (m, 1H), 0,80-0,77 (m, 1H).
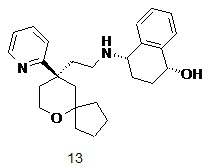
Пример 12 и пример 13:
(1S,4S)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-1,2,3,4-тетрагидронафталин-1-ол 12
(1R,4S)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-1,2,3,4-тетрагидронафталин-1-ол 13
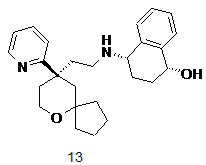
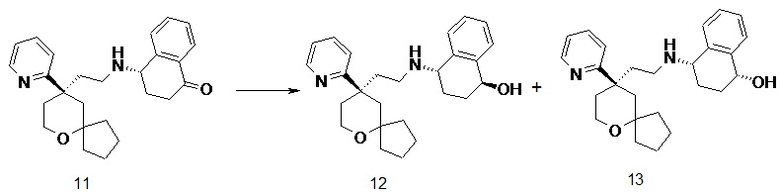
(S)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-3,4-дигидронафталин-1(2H)-он 11 (50 мг, 0,12 ммоль) растворяли в 10 мл дихлорметана, добавляли по каплям 0,29 мл 1M раствора гидроксида диизобутилалюминия при -78 oC, и смесь перемешивали в течение 2 часов при -78 oC. Добавляли 5 мл метанола для гашения реакции. Реакционный раствор подогревали до комнатной температуры и концентрировали при пониженном давлении. Полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением указанных в заголовке соединений 12 (18 мг, выход 35,3 %) в виде беловатого вязкого твердого вещества и 13 (20 мг, выход 39,2 %) в виде беловатого вязкого твердого вещества.
12: МС m/z (ИЭР): 407,6 [M+1],
1H ЯМР (400МГц, CDCl3) δ 8,51 (d, 1H), 7,50 (t, 1H), 7,36 (d, 1H), 7,33-7,30 (m, 3H), 7,21-7,18 (m, 2H), 4,83 (t, 1H), 4,25 (t, 1H), 3,81-3,75 (m, 2H), 2,85-2,83 (m, 1H), 2,36-2,30 (m, 5H), 1,98-1,80 (m, 2H) , 1,78-1,60 (m, 9H), 1,48-1,25 (m, 5H).
13: МС m/z (ИЭР): 407,6 [M+1],
1H ЯМР (400МГц, CDCl3) δ 8,51 (d, 1H), 7,50 (t, 1H), 7,36 (d, 1H), 7,33-7,30 (m, 3H), 7,21-7,18 (m, 2H), 4,83 (t, 1H), 4,25 (t, 1H), 3,81-3,75 (m, 2H), 2,85-2,83 (m, 1H), 2,36-2,30 (m, 5H), 1,98-1,80 (m, 2H), 1,78-1,60 (m, 9H), 1,48-1,25 (m, 5H).
Пример 14:
(1S,4S)-4-метокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин
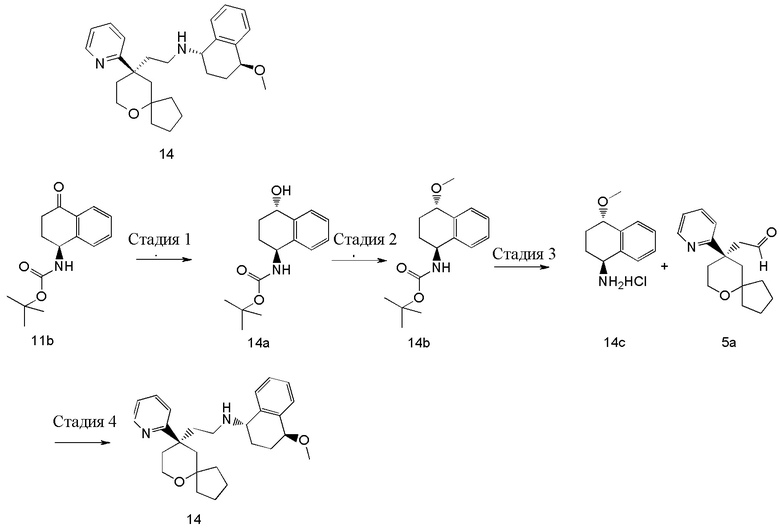
Стадия 1
трет-Бутил-((1S,4S)-4-гидрокси-1,2,3,4-тетрагидронафталин-1-ил)карбамат 14a
(S)-трет-бутил-(4-оксо-1,2,3,4-тетрагидронафталин-1-ил)карбамат 11b (100 мг, 0,883 ммоль) растворяли в 5 мл толуола, реакционную смесь охлаждали до 0 oC, добавляли (R)-2-метил-CBS-оксазаборолидин (0,1 мл, 0,076 ммоль) и перемешивали в течение 5 минут. Затем добавляли боран метилсульфид (0,88 мл, 0,76 ммоль) и реакционную смесь перемешивали в течение 2 часов. Реакционную смесь гасили добавлением 50 мл насыщенного раствора хлорида натрия и экстрагировали этилацетатом (3 раза по 30 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 14a, указанного в заголовке, в виде белого твердого вещества (60 мг, выход 60 %).
МС m/z (ИЭР): 208,3 [M-55]
Стадия 2
трет-Бутил-((1S,4S)-4-метокси-1,2,3,4-тетрагидронафталин-1-ил)карбамат 14b
Неочищенное соединение 14a (30 мг, 0,11 ммоль) растворяли в 4 мл дихлорметана, затем добавляли оксид серебра (76 мг, 0,33 ммоль) и метилиодид (62 мг, 0,44 ммоль). После перемешивания в течение 48 часов реакционный раствор фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения 14b, указанного в заголовке, в виде желтого масла (30 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 278,4 [M+1].
Стадия 3
(1S,4S)-4-метокси-1,2,3,4-тетрагидронафталин-1-амина гидрохлорид 14c
Неочищенное соединение 14b (30 мг, 0,11 ммоль) растворяли в 0,5 мл дихлорметана, затем добавляли 1 мл 4M раствора хлорида водорода в 1,4-диоксане. Реакционную смесь перемешивали в течение 2,5 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного соединения 14c, указанного в заголовке, в виде белого твердого вещества (24 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 178,4 [M+1].
Стадия 4
(1S,4S)-4-метокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 14
Соединение 5a (29 мг, 0,11 ммоль), неочищенное соединение 14c (24 мг, 0,11 ммоль) и сульфат натрия растворяли в 4 мл метанола, и смесь перемешивали в течение 12 часов. Затем добавляли боргидрид натрия (8 мг, 0,22 ммоль), и смесь перемешивали в течение 15 минут. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 14, указанного в заголовке, в виде белого твердого вещества (4 мг, выход 8,7 %).
МС m/z (ИЭР): 407,6 [M+1]
1H ЯМР (400 МГц, CDCl3) δ 8,56 (d, 1H), 7,66 (t, 1H), 7,33 (d, 1H), 7,15 (d, 1H), 7,08-7,06 (m, 3H), 7,04 (d, 1H), 3,76 (d, 2H), 3,61-3,58 (br, 1H), 3,41 (s, 3H), 2,74-2,72 (m, 3H), 2,46 (d, 1H), 2,32 (d, 1H), 2,13-2,08 (m, 1H), 2,03-2,00 (m, 1H), 1,90 (d, 1H), 1,75 -1,72 (m, 11H), 1,51-1,46 (m, 3H).
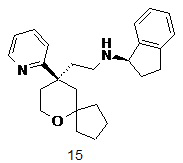
Пример 15:
(R)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-2,3-дигидро-1H-инден-1-амин
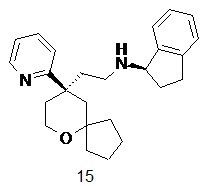

(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (20 мг, 0,08 ммоль) и (R)-2,3-дигидро-1H-инден-1-амина гидрохлорид 15a (27 мг, 0,16 ммоль, полученный способом, раскрытым в публикации Synthesis, (14), 2283-2287, 2008) растворяли в 10 мл дихлорметана, и смесь перемешивали в течение 2 часов, затем добавляли триацетоксиборгидрид натрия (51 мг, 0,24 ммоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 15, указанного в заголовке, в виде желтого масла (5 мг, выход 16,7 %).
МС m/z (ИЭР): 377,5 [M+1]
1H ЯМР (400 МГц, метанол-d4) δ 8,62 (d, 1H), 7,91 (t, 1H), 7,60 (d, 1H), 7,37 (s, 4H), 7,35 (d, 1H), 4,64-4,70 (m, 1H), 3,76 (d, 2H), 2,91-3,15 (m, 2H), 2,41-2,60 (m, 4H), 1,85-2,11 (m, 4H), 1,70-1,81 (m, 2H), 1,41-1,69 (m, 5H), 1,31-1,39 (m, 1H), 1,10-1,20 (m, 1H), 0,71-0,80 (m, 1H).
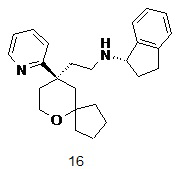
Пример 16:
(S)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-2,3-дигидро-1H-инден-1-амин


(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (20 мг, 0,08 ммоль) и (S)-2,3-дигидро-1H-инден-1-амина гидрохлорид 16a (26 мг, 0,15 ммоль, полученный способом, раскрытым в публикации Tetrahedron Asymmetry, 14(22), 3479-3485; 2003) растворяли в 10 мл дихлорметана, и смесь перемешивали в течение 2 часов, затем добавляли триацетоксиборгидрид натрия (49 мг, 0,23 ммоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 16, указанного в заголовке, в виде желтого масла (5 мг, выход 17 %).
МС m/z (ИЭР): 377,5 [M+1]
1H ЯМР (400 МГц, метанол-d4) δ 8,63 (d, 1H), 7,90 (t, 1H), 7,60 (d, 1H), 7,38 (s, 4H), 7,35 (d, 1H), 4,65-4,70 (m, 1H), 3,76 (d, 2H), 2,90-3,16 (m, 2H), 2,40-2,60 (m, 4H), 1,85-2,10 (m, 4H), 1,70-1,80 (m, 2H), 1,40-1,69 (m, 5H), 1,30-1,39 (m, 1H), 1,10-1,20 (m, 1H), 0,70-0,80 (m, 1H).
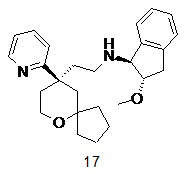
Пример 17:
(1S,2S)-2-метокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-2,3-дигидро-1H-инден-1-амин

Стадия 1
(1S,2S)-2-метокси-2,3-дигидро-1H-инден-1-амина трифторацетат 17b
трет-Бутил-((1S,2S)-2-метокси-2,3-дигидро-1H-инден-1-ил)карбамат 17a (110 мг, 0,42 ммоль, получен способом, раскрытым в заявке на патент WO2008080015) растворяли в 5 мл дихлорметана, затем добавляли 1 мл трифторуксусной кислоты. После перемешивания в течение 2 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой A с получением неочищенного соединения 17b, указанного в заголовке, в виде желтого масла (70 мг, выход 60,3 %).
МС m/z (ИЭР): 164,1 [M+1].
Стадия 2
(1S,2S)-2-метокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-2,3-дигидро-1H-инден-1-амин 17
(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (25 мг, 0,96 ммоль) и (1S,2S)-2-метокси-2,3-дигидро-1H-инден-1-амина трифторацетат 17b (54 мг, 0,19 ммоль) растворяли в 10 мл дихлорметана, затем добавляли триацетоксиборгидрид натрия (61 мг, 0,29 ммоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 17, указанного в заголовке, в виде желтого масла (5 мг, выход 25,5 %).
МС m/z (ИЭР): 407,6 [M+1]
1H ЯМР (400 МГц, CDCl3) δ 8,58 (d, 1H), 7,71 (t, 1H), 7,57 (d, 1H), 7,40 (d, 1H), 7,29 (d, 1H), 7,16-7,24 (m, 3H), 3,77 (d, 3H), 3,31 (s, 3H), 2,87-3,05 (m, 2H), 2,24-2,50 (m, 4H), 2,14-2,24 (m, 1H), 1,61-1,84 (m, 4H), 1,35-1,51 (m, 5H), 1,24-1,35 (m, 2H), 1,11-1,20 (m, 1H), 0,65-0,75 (m, 1H).
Пример 18:
(1S,2S)-1-((2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-2,3-дигидро-1H-инден-2-ол

Стадия 1
2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 18b
2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетонитрил 18a (500 мг, 1,95 ммоль, полученный способом, раскрытым в заявке на патент WO2012129495) растворяли в 20 мл толуола, медленно добавляли по каплям 4,2 мл 1 M раствора диизобутилалюмогидрида при -78 °C, и реакционную смесь перемешивали в течение 1,5 часов. Затем добавляли 18 мл 2 M раствора соляной кислоты, и смесь перемешивали в течение 30 минут. Добавляли по каплям 5 M раствор гидроксида натрия до доведения значения pH реакционного раствора до 9–10. Смесь подогревали до комнатной температуры и экстрагировали этилацетатом (3 раза по 30 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой A с получением соединения 18b, указанного в заголовке, в виде желтого масла (270 мг, выход 53,4 %).
МС m/z (ИЭР): 260,5 [M+1].
Стадия 2
(1S,2S)-1-((2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-2,3-дигидро-1H-инден-2-ол 18
Соединение 18b (20 мг, 0,08 ммоль) и (1S,2S)-1-амино-2,3-дигидро-1H-инден-2-ол 18c (23 мг, 0,15 ммоль, полученный способом, раскрытым в публикации Advanced Synthesis & Catalysis, 350(14+15), 2250-2260; 2008) растворяли в 15 мл смеси дихлорметана и метанола (об./об. составляет 5:1), смесь перемешивали в течение 2 часов, затем добавляли триацетоксиборгидрид натрия (49 мг, 0,23 ммоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 18, указанного в заголовке, в виде желтого масла (10 мг, выход 33 %).
МС m/z (ИЭР): 393,5 [M+1]
1H ЯМР (400 МГц, CDCl3) δ 8,50 (d, 1H), 7,70 (t, 1H), 7,37 (d, 1H), 7,20-7,26 (m, 3H), 7,11-7,19 (m, 2H), 3,76 (d, 3H), 3,36 (d, 1H), 2,88-3,05 (m, 2H), 2,25-2,50 (m, 4H), 2,15-2,24 (m, 1H), 1,60-1,84 (m, 4H), 1,36-1,51 (m, 5H), 1,25-1,35 (m, 2H), 1,10-1,20 (m, 1H), 0,65-0,75 (m, 1H).
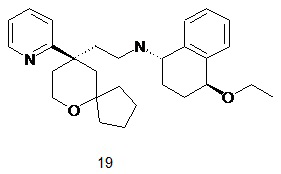
Пример 19:
(1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин
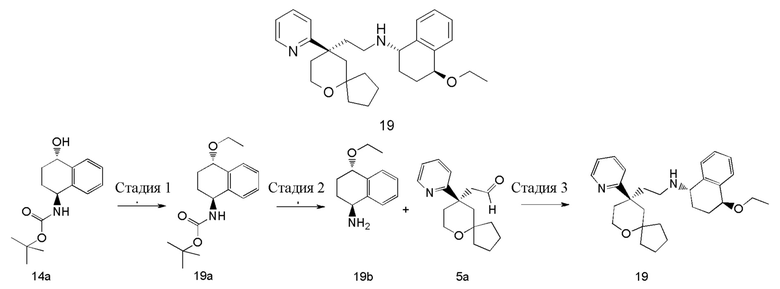
Стадия 1
трет-бутил-((1S,4S)-4-этокси-1,2,3,4-тетрагидронафталин-1-ил)карбамат 19a
Неочищенное соединение трет-бутил-((1S)-4-гидрокси-1,2,3,4-тетрагидронафталин -1-ил) карбамат 14a (850 мг, 3,23 ммоль), оксид серебра (76 мг, 0,33 ммоль) и иодэтан (1,3 мл, 16,15 ммоль) растворяли в 30 мл дихлорметана, и смесь перемешивали в течение 48 часов. Реакционный раствор фильтровали, и фильтрат концентрировали при пониженном давлении с получением неочищенного соединения 19a (800 мг) в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 236,1 [M-55].
Стадия 2
(1S,4S)-4-этокси-1,2,3,4-тетрагидронафталин-1-амин 19b
Неочищенное соединение 19a (698 мг, 2,4 ммоль) растворяли в 4 мл дихлорметана, затем добавляли 8 мл 4M раствора хлорида водорода в 1,4-диоксане. После перемешивания в течение 2 часов реакционный раствор концентрировали при пониженном давлении, растирали с этилацетатом (30 мл) и фильтровали. Фильтрационный кек растворяли в 20 мл смеси дихлорметана и метанола (об./об. составляет 5:1). Добавляли насыщенный раствор бикарбоната натрия для доведения занчения pH реакционного раствора до 7–8. Реакционный раствор концентрировали при пониженном давлении, промывали смесью дихлорметана и метанола (об./об. составляет 5:1) (2 раза по 30 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения 19b, указанного в заголовке, в виде желтой жидкости (310 мг), которую использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 191,1 [M+1].
Стадия 3
(1S,4S)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 19
(R)-2-(9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)ацетальдегид 5a (500 мг, 1,85 ммоль) и неочищенное соединение 19b (310 мг, 1,85 ммоль) растворяли в 30 мл дихлорметана, и смесь перемешивали в течение 40 минут, затем добавляли триацетоксиборгидрид натрия (980 мг, 4,63 ммоль). После перемешивания в течение 2 часов реакционный раствор последовательно промывали насыщенным раствором бикарбоната натрия (3 раза по 30 мл) и насыщенным раствором хлорида натрия (3 раза по 30 мл). Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 19, указанного в заголовке, в виде желтого вязкого твердого вещества (280 мг, выход 35 %).
МС m/z (ИЭР): 435,3 [M+1]
1H ЯМР (400 МГц, CDCl3) δ 9,74 (d, 1H), 9,58 (d, 1H), 8,94 (d, 1H), 8,37 (d, 1H), 7,94 (d, 1H), 7,67 (d, 1H), 7,52 (d, 1H), 7,47 (t, 1H), 4,46-4,49 (m, 1H), 4,30-4,33 (m, 1H), 3,84-3,87 (m, 1H), 3,66-3,70 (m, 2H), 3,53-3,56 (m, 2H), 2,82-2,85 (d, 2H), 2,67 (s, 2H), 2,39-2,41 (m, 4H), 2,30-2,33 (m, 4H), 1,85 (s, 2H), 1,48-1,52 (m, 6H), 1,27 (m, 3H).
Пример 20:
(1S,4S)-4-(циклопропилметокси)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин
Стадия 1
трет-Бутил-((S)-4-оксо-1,2,3,4-тетрагидронафталин-1-ил)(2-((R)-9-(пиридин-2-ил)- 6-оксаспиро[4.5]декан-9-ил)этил)карбамат 20a
Соединение 11 (220 мг, 0,54 ммоль), ди-трет-бутилдикарбонат (173 мг, 0,82 ммоль) и триэтиламин (0,15 мл, 1,08 ммоль) растворяли в 20 мл дихлорметана. После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографией с элюирующей системой A с получением соединения 20a, указанного в заголовке, в виде светло-желтого вязкого твердого вещества (100 мг, выход 37 %).
МС m/z (ИЭР): 505,3 [M+1].
Стадия 2
трет-Бутил-((1S,4S)-4-гидрокси-1,2,3,4-тетрагидронафталин-1-ил)(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)карбамат 20b
Соединение 20a (100 мг, 0,2 ммоль) и 1M (R)-2-метил-CBS-оксаборолидин (0,04 мл, 0,4 ммоль) растворяли в 10 мл толуола, реакционную смесь охлаждали до 0 °C, затем добавляли 2 M раствор боран метилсульфида а (0,02 мл, 0,4 ммоль). Реакционную смесь подогревали до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь гасили добавлением 10 мл насыщенного раствора хлорида натрия и экстрагировали этилацетатом (3 раза по 10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 10 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 20b, указанного в заголовке, в виде белого твердого вещества (10 мг, выход 10 %).
МС m/z (ИЭР): 507,3 [M+1].
Стадия 3
трет-Бутил-((1S,4S)-4-(циклопропилметокси)-1,2,3,4-тетрагидронафталин-1-ил)(2-((R)-9- (пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)карбамат 20c
Соединение 20b (10 мг, 0,02 ммоль) растворяли в 5 мл N,N-диметилформамида, затем добавляли гидрид натрия (2,2 мг, 0,06 ммоль). Смесь перемешивали в течение 30 минут, затем добавляли циклопропилметилбромид (6,7 мг, 0,05 ммоль). После перемешивания в течение 3 часов, реакционную смесь гасили добавлением 20 мл воды и экстрагировали этилацетатом (3 раза по 10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 10 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения 20c, указанного в заголовке, в виде белого твердого вещества (5 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 561,0 [M+1].
Стадия 4
(1S,4S)-4-(циклопропилметокси)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 20d
Неочищенное соединение 20c (5 мг, 0,0089 ммоль) растворяли в 5 мл дихлорметана, затем добавляли 0,1 мл 4M раствора соляной кислоты в 1,4-диоксане. После перемешивания в течение 2 часов, реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 20d, указанного в заголовке, в виде белого твердого вещества (3 мг, выход 73,2 %).
МС m/z (ИЭР): 461,3 [M+1]
1H ЯМР (400 МГц, CD3OD) δ 8,59 (d, 1H), 7,84-7,81 (m, 1H), 7,55 (d, 1H), 7,53 (d, 1H), 7,47-7,40 (m, 1H), 7,39-7,29 (m, 2H), 7,25 (d, 1H), 4,48-4,46 (m, 1H), 4,28-4,25 (m, 1H), 3,77-3,75 (m, 2H), 3,45-3,43 (m, 2H), 3,35-3,30 (m, 2H), 2,93-2,92 (m, 1H), 2,53-2,50 (m, 2H), 2,49-2,48 (m, 1H), 2,25-2,13 (m, 2H), 1,95-1,31 (m, 11H), 1,10-1,08 (m, 2H), 0,76-0,73 (m, 1H), 0,55-0,53 (m,2H),0,25-0,23 (m,2H).
Пример 21:
(1S,4S)-4-(2-фторэтокси)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин
Стадия 1
трет-Бутил-((1S,4S)-4-(2-фторэтокси)-1,2,3,4-тетрагидронафталин-1-ил)(2-((R)- 9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)карбамат 21a
20b (45 мг, 0,088 ммоль) растворяли в 5 мл N,N-диметилформамида, затем добавляли гидрид натрия (20 мг, 0,44 ммоль). Смесь перемешивали в течение 20 минут, затем добавляли 1-бром-2-фторэтан (23 мг, 0,176 ммоль). После перемешивания в течение 16 часов, реакционную смесь гасили добавлением 5 мл воды и экстрагировали этилацетатом (3 раза по 10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 21a, указанного в заголовке, в виде желтого масла (30 мг, выход 61,1 %).
МС m/z (ИЭР): 553,4 [M+1].
Стадия 2
(1S,4S)-4-(2-фторэтокси)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 21
Соединение 21a (30 мг, 0,543 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 0,3 мл 4M раствора соляной кислоты в 1,4-диоксане. Смесь перемешивали в течение 1 часа, затем добавляли 10 мг карбоната натрия. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 21, указанного в заголовке, в виде белого вязкого вещества (10 мг, выход 40,7 %).
МС m/z (ИЭР): 453,4 [M+1]
1H ЯМР (400 МГц, CDCl3) δ 9,76 (d, 1H), 9,61 (d, 1H), 8,89 (d, 1H), 8,34 (d, 1H), 7,94 (d, 1H), 7,69 (d, 1H), 7,58 (d, 1H), 7,44 (t, 1H), 4,43-4,49 (m, 2H), 4,28-4,33 (m, 2H), 3,81-3,87 (m, 1H), 3,61-3,71 (m, 2H), 3,51-3,56 (m, 2H), 2,81-2,89 (d, 2H), 2,67 (s, 2H), 2,39-2,43 (m, 4H), 2,30-2,36 (m, 4H), 1,85 (s, 2H), 1,48-1,61 (m, 6H).
Примеры 22, 23:
(1S,4S)-4-(метоксиметил)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 22
(1S,4R)-4-(метоксиметил)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 23

Стадия 1
(S)-трет-бутил-(4-метилен-1,2,3,4-тетрагидронафталин-1-ил)карбамат 22a
Бромид метилтрифенилфосфония (2,95 г, 11,5 ммоль) растворяли в 20 мл тетрагидрофурана. Реакционную смесь охлаждали до 0 °C, добавляли трет-бутоксид калия (1,29 г, 11,5 ммоль) и перемешивали в течение 30 минут. Затем добавляли 11b (1 г, 7,66 ммоль), реакционную смесь подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении и растворяли в метаноле. Полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой B с получением соединения 22a, указанного в заголовке, в виде белого твердого вещества (200 мг, выход 22,2 %).
МС m/z (ИЭР): 204,2 [M-55].
Стадия 2
трет-бутил-((1S)-4-(гидроксиметил)-1,2,3,4-тетрагидронафталин-1-ил)карбамат 22b
22a (780 мг, 3 ммоль) растворяли в 20 мл тетрагидрофурана, реакционную смесь охлаждали до 0 °C, затем добавляли 6 мл 1 M раствора борана в тетрагидрофуране. Реакционный раствор перемешивали в течение 5 часов. Добавляли 12 мл 3 M раствора гидроксида натрия, и смесь перемешивали в течение 30 минут. После добавления 12 мл 30% пероксида водорода, реакционный раствор подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении и экстрагировали дихлорметаном. Органические фазы объединяли, концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 22b, указанного в заголовке, в виде белого твердого вещества (740 мг, выход 89,2 %).
МС m/z (ИЭР): 222,1 [M-55].
Стадия 3
трет-бутил ((1S)-4-(метоксиметил)-1,2,3,4-тетрагидронафталин-1-ил)карбамат 22c
22b (200 мг, 0,72 ммоль) растворяли в 10 мл тетрагидрофурана, затем добавляли гидрид натрия (60 мг, 1,4 ммоль), и реакционную смесь перемешивали в течение 1 часа. Затем добавляли иодметан (123 мг, 0,86 ммоль), и реакционную смесь перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении и растворяли в метаноле. Полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой B с получением соединения 22c, указанного в заголовке, в виде белого твердого вещества (20 мг, выход 9,5 %).
МС m/z (ИЭР): 236,2 [M-55].
Стадия 4
(1S)-4-(метоксиметил)-1,2,3,4-тетрагидронафталин-1-амин 22d
Неочищенное соединение 22c (20 мг, 0,07 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 10 мл 4M раствора соляной кислоты в 1,4-диоксане. После перемешивания в течение 2 часов реакционный раствор концентрировали при пониженном давлении с получением неочищенного соединения 22d, указанного в заголовке, в виде желтого масла (13 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 192,2 [M-55].
Стадия 5
(1S,4S)-4-(метоксиметил)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 22
5a (30 мг, 0,116 ммоль) и неочищенное соединение 22d (22 мг, 0,116 ммоль) растворяли в 20 мл смеси дихлорметана и метанола (об./об. составляет 1:1), затем добавляли цианоборгидрид натрия (15 мг, 0,23 ммоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 22, указанного в заголовке, в виде белого твердого вещества (10 мг, выход 20 %) и соединения 23, указанного в заголовке, в виде белого твердого вещества (8 мг, выход 16 %).
МС m/z (ИЭР): 435,3 [M+1]
Пример 22:
1H ЯМР (400 МГц, CDCl3) δ 8,26-8,25 (d, 1H), 7,76-7,72 (t, 1H), 7,42-7,40 (d, 1H), 7,34-7,29 (m, 3H), 7,29-7,27 (m, 1H), 7,18-7,16 (m, 1H), 4,16 (s, 1H), 3,75-3,70 (m, 2H), 3,47-3,45 (m, 2H), 3,41 (s, 3H), 3,35-3,33 (m, 1H), 3,18-3,17 (m, 1H), 2,80-2,70 (m, 1H), 2,4-2,33 (m, 1H), 2,28-1,95 (m, 7H), 1,81-1,62 (m, 5H), 1,59-1,51 (m, 1H), 1,46-1,20 (m, 4H), 1,22-1,1 (m,1H).
Пример 23:
1H ЯМР (400 МГц, CDCl3) δ 8,47-8,46 (d, 1H), 7,72-7,68 (t, 1H), 7,39-7,37 (d, 1H), 7,37-7,33 (m, 1H), 7,24-7,21 (m, 1H), 7,15-7,05 (m, 2H), 6,93-6,91 (d, 1H), 3,94 (s, 1H), 3,68-3,60 (m, 2H), 3,59-3,57 (m, 2H), 3,22 (s, 3H), 3,22-3,19 (m, 1H), 2,71-2,70 (m, 1H), 2,34-2,30 (m, 5H), 2,28-2,25 (m, 1H), 1,84-1,81(m, 1H), 1,81-1,71 (m, 5H), 1,69-1,51 (m, 2H), 1,45-1,4 (m, 2H), 1,32-1,26 (m,2H), 1,23-1,15 (m, 1H), 1,1-0,95 (m, 1H).
Пример 24:
(S)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-3',4'-дигидро-2'H-спиро[[1,3]дитиолан-2,1'-нафталин]-4'-амин 24
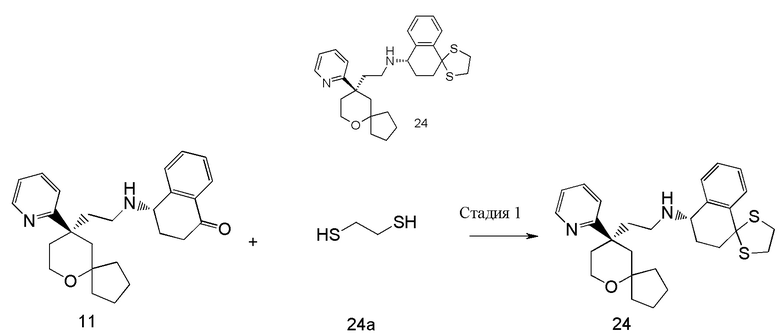
Соединение 11 (35 мг, 0,0865 ммоль), этан-1,2-дитиол 24a (82 мг, 0,865 ммоль) и пиридиния пара-толуолсульфонат (240 мг, 0,952 ммоль) растворяли в 15 мл толуола, реакционную смесь подогревали до 110 °C и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 24, указанного в заголовке, в виде светло-желтого твердого вещества (40 мг, выход 96 %).
МС m/z (ИЭР): 481,2 [M+1]
1H ЯМР (400 МГц, CD3OD): δ 8,60 (d, 1H), 7,60 (t, 1H), 7,25-7,31 (m, 2H), 7,15-7,20 (m, 4H), 4,26-4,30 (m, 1H), 3,76 (d, 2H), 2,81-3,01 (m, 4H), 2,41-2,60 (m, 2H), 2,21-2,30 (m, 2H), 1,86-2,13 (m, 4H), 1,70-1,81 (m, 2H), 1,41-1,69 (m, 5H), 1,31-1,39 (m, 2H), 1,10-1,20 (m, 2H), 0,71-0,80 (m, 2H).
Пример 25:
(1S,4R)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4- тетрагидронафталин-1-амин 25
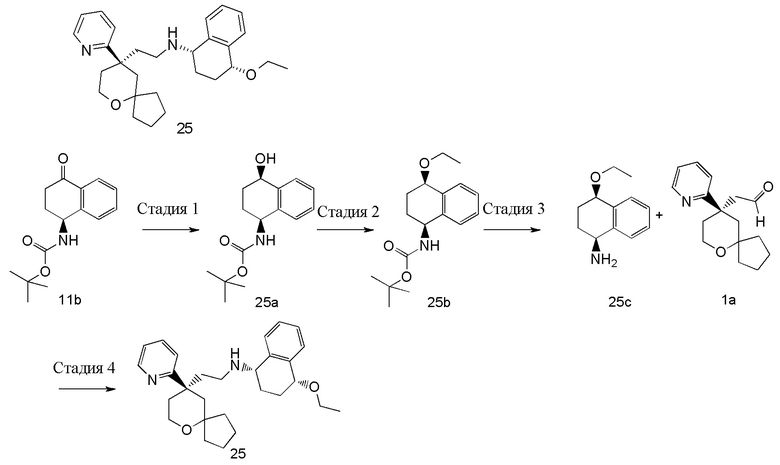
Стадия 1
трет-Бутил-((1S,4R)-4-гидрокси-1,2,3,4-тетрагидронафталин-1-ил)карбамат 25a
(S)-2-метил-CBS-оксазаборолидин (221,8 мг, 0,8 ммоль) растворяли в 140 мл тетрагидрофурана, затем добавляли боран триметилсульфид а (2,4 мл, 48 ммоль) в атмосфере аргона. Реакционную смесь подогревали до 30 °C, затем добавляли по каплям 80 мл предварительно полученного раствора 11b (10,5 г, 40 ммоль) в тетрагидрофуране в течение 30 минут. Реакционную смесь перемешивали в течение 1 часа при 30 °C. Добавляли 100 мл метанола при 15 °C и перемешивали в течение 1 часа для гашения реакционной смеси. Реакционный раствор концентрировали при пониженном давлении. Добавляли 200 мл этилацетата и 5 г активированного углерода. Смесь перемешивали в течение 30 минут при микрокипении и фильтровали. Фильтрационный кек промывали этилацетатом (3 раза по 100 мл). Фильтрат концентрировали при пониженном давлении с получением соединения 25a, указанного в заголовке, в виде бесцветного масла (10,5 г).
МС m/z (ИЭР): 264,4 [M+1].
Стадии 2–4
(1S,4R)-4-этокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 25
В соответствии с путем синтеза примера 19; исходное вещество 14a заменяли на 25a, соответственно, было получено соединение 25, указанное в заголовке, в виде светло-красного масла (7 г).
МС m/z (ИЭР): 435,5 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 0,62 (dt, 1H), 0,92-1,03 (m, 1H), 1,12 (t, 3H), 1,34 (td, 2H), 1,41-1,69 (m, 9H), 1,79 (d, 1H), 1,82-1,92 (m, 2H), 2,02 (td, 1H), 2,26-2,38 (m, 2H), 2,43 (d, 1H), 3,37-3,48 (m, 2H), 3,52-3,66 (m, 3H), 4,25 (t, 1H), 7,11-7,16 (m, 2H), 7,16-7,20 (m, 1H), 7,21-7,28 (m, 2H), 7,45 (d, 1H), 7,71 (td, 1H), 8,52 (dd, 1H).
Пример 26:
(1S,4S)-4-(этокси-d5)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дец-9-ил)этил) -1,2,3,4-тетрагидронафталин-1-амин 26
Стадия 1
трет-Бутил-((1S,4S)-4-(этокси-d5)-1,2,3,4-тетрагидронафталин-1-ил)карбамат 26a
Соединение 14a (3,3 г, 12,5 ммоль) растворяли в 50 мл N,N-диметилформамида, затем добавляли активированные молекулярные сита. После этого реакционный раствор охлаждали до 0 °C, добавляли гидроксид натрия (0,75 г, 18,75 ммоль) в атмосфере аргона. Реакционную смесь перемешивали в течение 0,5 часов при 0 °C. Затем добавляли дейтерированный иодэтан-d5 (0,8 мл, 10 ммоль), и реакционную смесь герметично закрывали на 16 часов при 0 °C. После завершения реакции реакционный раствор наливали в смесь 50 мл воды, 50 мл н-гексана и 5 мл этилацетата, перемешивали в течение 10 минут и фильтровали. Нерастворимые вещества удаляли. Фильтрат разделяли на две фазы, и водную фазу экстрагировали смесью н-гексана и этилацетата (об./об. составляет 10:1) (2 раза по 33 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (2 раза по 30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью устройства для препаративной флэш-хроматографии CombiFlash с элюирующей системой B с получением соединения 26a, указанного в заголовке, в виде белого твердого вещества (1,89 г, выход 64 %).
МС m/z (ИЭР): 241,4 [M-56+1].
Стадия 2
(1S,4S)-4-(этокси-d5)-1,2,3,4-тетрагидронафталин-1-амин 26b
Добавляли 8 мл 4M раствора хлорида водорода в 1,4-диоксане к соединению 26a (1,89 г, 6,38 ммоль). Реакционный раствор перемешивали в течение 1 часа и концентрировали при пониженном давлении. Добавляли 30 мл этилацетата, и смесь концентрировали при пониженном давлении. К полученному в результате остатку добавляли 1 мл насыщенного раствора карбоната натрия, и смесь перемешивали в течение 5 минут. Добавляли 30 мл этилацетата, 2 г твердого карбоната натрия и 10 г сульфата натрия, реакционный раствор перемешивали в течение 30 минут до исчезновения мутности раствора. Смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 26b (1,21 мг, светло-коричневая жидкость), которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 197,4 [M+1].
Стадия 3
(1S,4S)-4-(этокси-d5)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]дец-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 26
Соединение 1a (1,37 г, 5,31 ммоль) и неочищенное соединение 26b (1,21 г, 6,16 ммоль) растворяли в 50 мл дихлорэтана, добавляли каплю уксусной кислоты, и реакционную смесь перемешивали в течение 1 часа. Добавляли триацетоксиборгидрид натрия (2,81 г, 13,27 ммоль), и реакционную смесь перемешивали в течение 16 часов. К реакционному раствору добавляли 10 мл насыщенного раствора карбоната натрия и перемешивали в течение 5 минут. Добавляли последовательно 10 мл 15% раствора гидроксида натрия, 30 мл воды, 30 мл дихлорметана, и смесь перемешивали в течение 5 минут. Две фазы разделяли, и водную фазу экстрагировали дихлорметаном (2 раза по 50 мл). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой A с получением соединения 26, указанного в заголовке, в виде светло-желтой жидкости (1,7 г, выход 73 %).
МС m/z (ИЭР): 440,5 [M+1]
1H ЯМР (400 МГц, CDCl3) δ 0,70 (dt, 1H), 1,09 - 1,16 (m, 1H), 1,45 - 1,55 (m, 4H), 1,62 - 1,84 (m, 6H), 1,86 - 2,04 (m, 4H), 2,23 (td, 1H) 2,34 (dd, 1H), 2,44 (dd, 1H), 2,53 (td, 1H), 3,68 (br. s., 1H), 3,72 - 3,81 (m, 2H), 4,34 (t, 1H), 7,11 (ddd, 1 H), 7,17 (t, 2H), 7,18 - 7,23 (m, 1 H), 7,31 (t, 2 H), 7,62 (td, 1 H), 8,55 (dd, 1H).
Пример 27:
(S)-4-этил-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2-дигидронафталин-1-амин 27
Стадия 1
(S,E)-трет-бутил-(4-этилиден-1,2,3,4-тетрагидронафталин-1-ил)карбамат 27a
Бромид этилтрифенилфосфония (2,1 г, 5,75 ммоль) растворяли в 20 мл тетрагидрофурана. Трет-бутоксид калия (643 мг, 5,75 ммоль) добавляли в ледяной бане, и реакционную смесь перемешивали в течение 30 минут в ледяной бане. Добавляли по каплям предварительно полученный раствор 11b (1 г, 3,83 ммоль) в тетрагидрофуране, и смесь перемешивали в течение 16 часов при 25 °C. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой B с получением соединения 27a, указанного в заголовке, в виде светло-желтого масла (530 мг, выход 51 %).
Стадия 2
(S)-4-этил-1,2-дигидронафталин-1-амина гидрохлорид 27b
Соединение 27a (273 мг, 1 ммоль) растворяли в 5 мл дихлорметана, затем добавляли 2 мл 4M раствора хлорида водорода в 1,4-диоксане. После перемешивания в течение 1 часа реакционный раствор концентрировали при пониженном давлении с получением неочищенного соединения 27b, указанного в заголовке, в виде коричневого масла (173 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
(S)-4-этил-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2-дигидронафталин-1-амин 27
Соединение 1a (150 мг, 0,58 ммоль) и неочищенное соединение 27b (158 мг, 0,58 ммоль) растворяли в 30 мл смеси дихлорэтана и метанола (об./об. составляет 10:1), затем добавляли триацетоксиборгидрид натрия (369 мг, 1,74 ммоль). После перемешивания в течение 16 часов реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 27, указанного в заголовке, в виде светло-желтого твердого вещества (40 мг, выход 17 %).
МС m/z (ИЭР): 417,2 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 8,58 (d, 1H), 7,83-7,78 (m, 1H), 7,53-7,48 (m, 3H), 7,33-7,29 (m, 2H), 7,21 (d, 1H), 5,84 (t, 1H), 4,25 (t, 1H), 3,73-3,72 (m, 3H), 3,41-3,31 (m, 2H), 2,81-2,80 (m, 2H), 2,41-2,25 (m, 3H), 1,96-1,90 (m, 3H), 1,85-1,61 (m, 8H), 1,25 (t, 3H), 1,23-1,21 (m, 1H), 0,68-0,65 (m, 1H).
Пример 28:
(S)-4-метилен-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4- тетрагидронафталин-1-амин 28
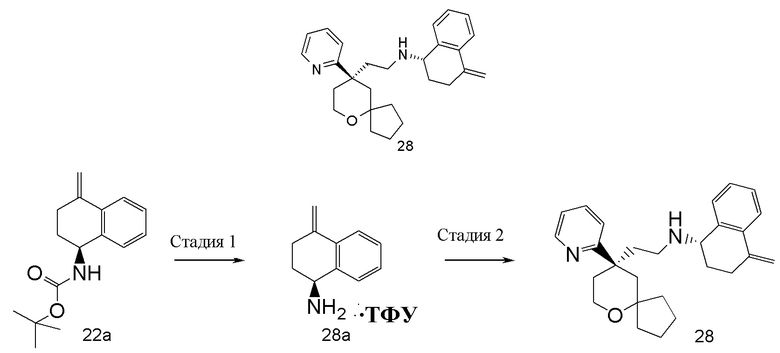
В соответствии с путем синтеза примера 17; исходное вещество 17a заменяли на 22a, соответственно, было получено соединение 28, указанное в заголовке, в виде коричневого твердого вещества (20 мг).
МС m/z (ИЭР): 403,5 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 8,50 (d, 1H), 7,72 (t, 1H), 7,68-7,21 (m, 6H), 5, 95 (d, 1H), 4,09 (d, 1H), 3,71-3,69 (m, 3H), 3,01-2,80 (m, 2H), 2,67-2,63 (m, 2H), 2,11 (d, 1H), 1,74-1,21 (m, 14H), 0,99-0,98 (m, 1H), 0,45-0,42 (m, 1H).
Пример 29:
2-(((1S,4S)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-1,2,3,4-тетрагидронафталин-1-ил)окси)ацетонитрил 29

Стадия 1
трет-Бутил-((1S,4S)-4-(цианометокси)-1,2,3,4-тетрагидронафталин-1-ил)(2-((R)-9- (пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)карбамат 29a
Соединение 20b (40 мг, 0,08 ммоль) растворяли в 10 мл тетрагидрофурана, затем последовательно добавляли трет-бутоксид калия (45 мг, 0,4 ммоль) и бромацетонитрил (20 мг, 0,16 ммоль), и реакционную смесь перемешивали в течение 16 часов. Добавляли 20 мл воды и 20 мл этилацетата и перемешивали. Смесь оставляли для выдерживания и разделения и экстрагировали этилацетатом (2 раза по 30 мл). Органические фазы объединяли и концентрировали при пониженном давлении с получением неочищенного соединения 29a, указанного в заголовке, в виде масла (50 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
2-(((1S,4S)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-1,2,3,4-тетрагидронафталин-1-ил)окси)ацетонитрил 29
Неочищенное соединение 29a (50 мг, 0,1 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 0,1 мл 4 M раствора хлорида водорода в диоксане. Реакционную смесь перемешивали в течение 0,5 часа. Добавляли водный раствор аммиака, пока реакционный раствор не становился щелочным. Смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 29, указанного в заголовке, в виде белого воска (10 мг, выход 8 %).
МС m/z (ИЭР): 446,3 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 8,54 (d, 1H), 7,75-7,72 (m, 1H), 7,43 (d, 1H), 7,37-7,32 (m, 2H), 7,28-7,15 (m, 3H), 4,67 (d, 1H), 4,40 (d, 2H), 4,31 (d, 1H), 3,97 (d, 1H), 3,63-3,51 (m, 2H), 2,41-2,25 (m, 2H), 2,16-2,06 (m, 2H), 2,04-1,87 (m, 2H), 1,86-1,72 (m, 4H), 1,62-1,21 (m, 8H), 1,04-0,94 (m, 1H), 0,68-0,61 (m, 1H).
Пример 30
(1S,4R)-4-метокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 30

Стадия 1
трет-Бутил-((1S,4R)-4-гидрокси-1,2,3,4-тетрагидронафталин-1-ил)(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)карбамат 30a
Соединение 13 (46 мг, 0,11 ммоль), ди-трет-бутилдикарбонат (27 мг, 0,121 ммоль) и триэтиламин (23 мг, 0,22 ммоль) растворяли в 15 мл дихлорметана, и реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 30a, указанного в заголовке, в виде белого твердого вещества (46 мг, выход 82 %).
МС m/z (ИЭР): 507,3 [M+1].
Стадия 2
трет-Бутил-((1S,4R)-4-метокси-1,2,3,4-тетрагидронафталин-1-ил)(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)карбамат 30b
Соединение 30a (46 мг, 0,091 ммоль) растворяли в 10 мл тетрагидрофурана, затем добавляли гидрид натрия (8 мг, 0,182 ммоль). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Добавляли иодметан (16 мг, 0,11 ммоль), и реакционную смесь перемешивали в течение 16 ч при комнатной температуре. Добавляли 50 мл воды и 50 мл этилацетата, и две фазы разделяли. Органическую фазу концентрировали при пониженном давлении с получением неочищенного соединения 30b, указанного в заголовке, в виде коричневого твердого вещества (47 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 521,3 [M+1].
Стадия 4
(1S,4R)-4-метокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4- тетрагидронафталин-1-амин 30
Неочищенное соединение 30b (47 мг, 0,091 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 0,1 мл 4 M раствора хлорида водорода в 1,4-диоксане, и реакционную смесь перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли этанол, и доводили значение pH до 8, используя водный раствор аммиака. Смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 30, указанного в заголовке, в виде желтого вязкого вещества (36 мг, выход 95 %).
МС m/z (ИЭР): 421,3 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 8,55 (d, 1H), 7,75-7,72 (m, 1H), 7,46 (d, 1H), 7,37-7,32 (m, 2H), 7,28-7,15 (m, 3H), 4,67 (d, 1H), 4,30 (d, 1H), 3,97 (d, 1H), 3,64-3,50 (m, 2H), 3,35 (s, 3H), 2,41-2,26 (m, 2H), 2,16-2,06 (m, 2H), 2,04-1,87 (m, 2H), 1,86-1,72 (m, 4H), 1,62-1,21 (m, 8H), 1,04-0,94 (m, 1H), 0,68-0,61 (m, 1H).
Пример 31:
2-((S,E)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-3,4- дигидронафталин-1(2H)-илиден)ацетонитрил 31
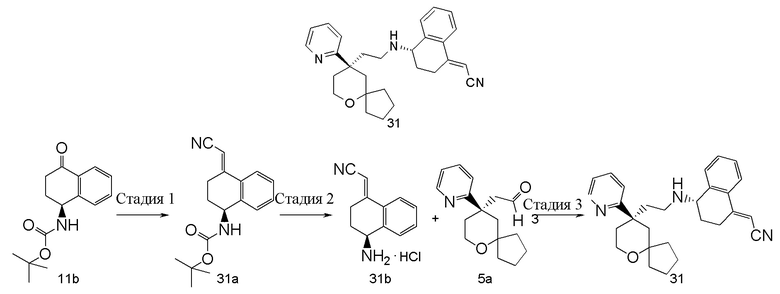
Стадия 1
(S,E)-трет-бутил-(4-(цианометилен)-1,2,3,4-тетрагидронафталин-1-ил)карбамат 31a
Диэтилцианометилфосфонат (200 мг, 0,76 ммоль) растворяли в 20 мл тетрагидрофурана. Добавляли гидрид натрия (61 мг, 1,52 ммоль) в ледяной бане, и реакционную смесь перемешивали в течение 30 минут в ледяной бане. Добавляли по каплям предварительно полученный раствор соединения 11b (200 мг, 0,76 ммоль) в тетрагидрофуране, и смесь перемешивали в течение 16 часов при 25 °C. Реакционный раствор наливали в ледяную воду и экстрагировали этилацетатом три раза. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой B с получением соединения 31a, указанного в заголовке, в виде бесцветного вязкого вещества (150 мг, выход 69 %).
МС m/z (ИЭР): 285,1 [M+1].
Стадия 2
(S,E)-2-(4-амино-3,4-дигидронафталин-1(2H)-илиден)ацетонитрила гидрохлорид 31b
Соединение 31a (150 мг, 0,52 ммоль) растворяли в 5 мл дихлорметана, затем добавляли 2 мл 1M раствора соляной кислоты в 1,4-диоксане. Реакционную смесь перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного соединения 31b, указанного в заголовке, в виде белого твердого вещества (110 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
2-((S,E)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-3,4-дигидронафталин-1(2H)-илиден)ацетонитрил 31
Соединение 5a (100 мг, 0,39 ммоль), и неочищенное соединение 31b (85 мг, 0,39 ммоль) растворяли в 10 мл смеси дихлорэтана и метанола (об./об. составляет 10:1), затем добавляли триацетоксиборгидрид натрия (165 мг, 0,78 ммоль), и реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 31, указанного в заголовке, в виде светло-желтого вязкого вещества (30 мг, выход 18 %).
МС m/z (ИЭР): 428,0 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 8,57 (d, 1H), 7,86-7,78 (m, 1H), 7,76-7,74 (m, 1H), 7,39-7,22 (m, 3H), 7,26-7,23 (m, 2H), 6,36-6,35 (m,1H), 3,65-3,54 (m, 3H), 2,90-2,60 (m, 2H), 2,42-2,37 (m, 3H), 2,03-1,90 (m, 4H), 1,82-1,78 (m, 2H), 1,51-1,24 (m, 10H).
Пример 32:
2-((4S)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-1,2,3,4-тетрагидронафталин-1-ил)ацетонитрил 32
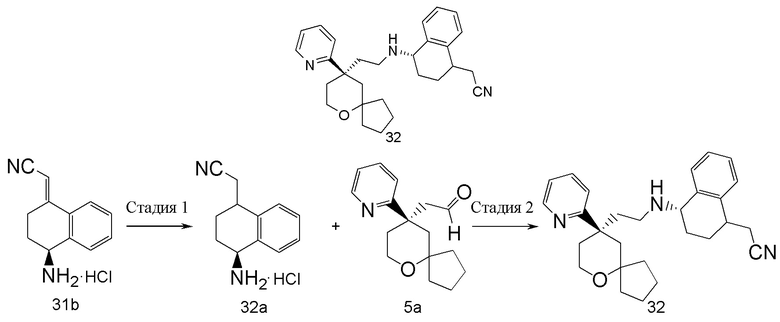
Стадия 1
2-((4S)-4-амино-1,2,3,4-тетрагидронафталин-1-ил)ацетонитрила гидрохлорид 32a
Соединение 31b (50 мг, 0,227 ммоль) растворяли в 5 мл этанола, затем добавляли 5 мг Pd/C, и реакционную систему продували водородом три раза. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре в атмосфере водорода. Нерастворимые вещества удаляли фильтрованием, и фильтрат концентрировали при пониженном давлении с получением неочищенного соединения 32a, указанного в заголовке, в виде бесцветного вязкого вещества (45 мг), которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
2-((4S)-4-((2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)амино)-1,2,3,4-тетрагидронафталин-1-ил)ацетонитрил 32
Соединение 5a (53 мг, 0,2 ммоль), и неочищенное соединение 32b (25 мг, 0,2 ммоль) растворяли в 10 мл смеси дихлорэтана и метанола (об./об. составляет 10:1), затем добавляли триацетоксиборгидрид натрия (80 мг, 0,4 ммоль), и реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный в результате остаток очищали с помощью тонкослойной хроматографии с элюирующей системой A с получением соединения 32, указанного в заголовке, в виде светло-желтого вязкого вещества (5 мг, выход 5,8 %).
МС m/z (ИЭР): 430,3 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 8,55 (d, 1H), 7,85-7,78 (m, 1H), 7,75-7,72 (m, 1H), 7,35-7,20 (m, 3H), 7,25-7,21 (m, 2H), 3,75-3,60 (m, 3H), 2,95-2,80 (m, 2H), 2,70-2,65 (m, 4H), 2,41-2,30 (m, 4H), 1,95-1,89 (m, 4H), 1,85-1,60 (m, 4H), 1,55-1,21 (m, 6H).
Пример 33:
(S)-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-3,4-дигидро-2H-спиро[[1,3]диоксолан-2,1'-нафталин]-4'-амин 33
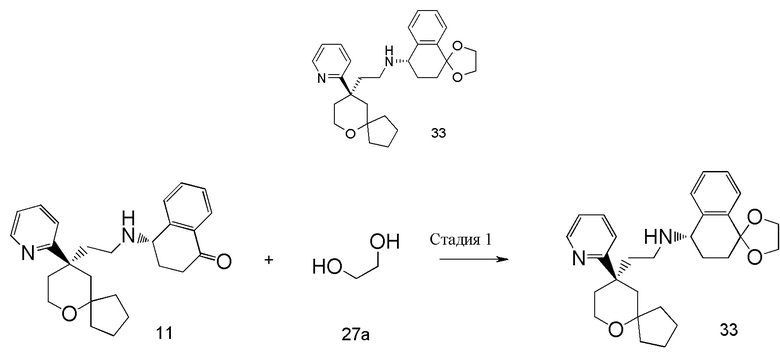
В соответствии с путем синтеза примера 27; исходное вещество 2 заменяли на 11, соответственно, было получено соединение 33, указанное в заголовке, в виде желтого масла (5 мг).
МС m/z (ИЭР): 449,0 [M+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 8,58 (d, 1H), 7,81 (s, 1H), 7,77-7,70 (m, 1H), 7,51 (d, 1H), 7,27-7,11 (m, 4H), 3,85 (s, 1H), 3,66-3,50 (m, 5H), 3,51-3,42 (m, 1H), 3,42-3,33 (m, 1H), 2,48-2,35 (m, 2H), 2,38-2,32 (m, 1H), 2,20-2,08 (m, 2H), 2,01-1,88 (m, 2H), 1,85-1,75 (m, 3H), 1,71-1,31 (m, 8H), 1,00-0,96 (m, 1H), 0,70-0,62 (m, 1H).
Пример 34:
(1S,4S)-4-пропокси-N-(2-((R)-9-(пиридин-2-ил)-6-оксаспиро[4.5]декан-9-ил)этил)-1,2,3,4-тетрагидронафталин-1-амин 34

В соответствии с путем синтеза примера 21; исходное вещество 1-бром-2-фторэтан заменяли на иодпропан, соответственно, было получено соединение 34, указанное в заголовке, в виде желтого твердого вещества (8 мг).
МС m/z (ИЭР): 449,3 [M+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (d, 1H), 7,75-7,70 (m, 1H), 7,45 (d, 1H), 7,37-7,33 (m, 2H), 7,28-7,17 (m, 3H), 4,65 (d, 1H), 4,32 (d, 1H), 3,98 (d, 1H), 3,64-3,52 (m, 3H), 3,49-3,40 (m, 1H), 2,62-2,52 (m, 1H), 2,41-2,27 (m, 2H), 2,16-2,06 (m, 1H), 2,04-1,87 (m, 2H), 1,86-1,71 (m, 5H), 1,67 (d, 1H), 1,60-1,20 (m, 8H), 1,13 (t, 3H), 1,03-0,95 (m, 1H), 0,68-0,60 (m, 1H).
БИОЛОГИЧЕСКИЙ АНАЛИЗ
Далее настоящее изобретение будет описано со ссылкой на приведенные ниже экспериментальные примеры, но эти примеры не следует рассматривать как ограничивающие объем изобретения.
Экспериментальный пример 1:
1. Цель эксперимента
Цель данного эксперимента состоит в определении агонистического действия соединений по настоящему изобретению на MOR, KOR, DOR и в оценке активности in vitro соединений в соответствии со значениями EC50 (полумаксимальная эффективная концентрация) и Emax.
2. Исследование активности MOR
2.1 Цель эксперимента
Соединения по настоящему изобретению могут активировать μ-опиоидные рецепторы (MOR). Активированные MOR могут регулировать внутриклеточный уровень цАМФ (Циклический аденозинмонофосфат), и цАМФ проникает в ядро и связывается с областью CRE (– цАМФ-ответный элемент) гена-репортера люциферазы, инициируя таким образом экспрессию гена-репортера. Люцифераза может взаимодействовать со своим субстратом с испусканием флуоресценции, и измеряемые сигналы флуоресценции отражают агонистическую активность соединений.
2.2 Способ эксперимента
Активность исследуемых в примере соединений при агонизации MOR и влияние на нижележащий уровень цАМФ исследовали следующим способом.
2.1.1 Материалы эксперимента
2.2.2 Методика эксперимента
1) Получение моноклональных линий клеток HEK293/MOR/CRE
Клетки HEK293 трансфицировали векторами MOR/pcDNA3.1 (+) и CRE/pGL4.29. В культуральную среду добавляли G418 и гигромицин и проводили скрининг моноклональных линий клеток HEK293/MOR/CRE в 96-луночном планшете для культур клеток.
2) Агонистическое действие соединений из примеров на MOR
Моноклональные клетки HEK293/MOR/CRE культивировали в среде DMEM/высокое содержание глюкозы (10% ФБС, 1 мг/мл G418, 200 мкг/мл гигромицина, смешивали до однородного состояния) и пересевали раз в 3 суток. В день эксперимента получали суспензию клеток со свежей клеточной средой, добавляли в 96-луночный планшет (BD, #356692) в концентрации 20 000 клеток/лунка и инкубировали в 5% CO2 при 37 °C. На вторые сутки соединение растворяли в чистом ДМСО в концентрации 20 мМ, затем вводили в ДМСО до получения первой концентрации 4 мМ и разбавляли с 10-кратным градиентом концентраций до 6 концентраций. В лунку для холостой пробы и контрольную лунку добавляли 90 мкл ДМСО; затем по 2,5 мкл растворов соединений, приготовленных в ДМСО, с градиентом концентраций, вводили в 97,5 мкл свежей среды для культуры клеток, содержащей 5 мкМ форсколина; 10 мкл приготовленного раствора соединения добавляли в лунки планшета для культуры клеток с получением конечной концентрации соединения 10000, 1000, 100, 10, 1, 0,1, 0,01 нМ, и планшет инкубировали при 37 °C в 5% CO2 в течение 5 часов. В каждую лунку 96-луночного планшета для культуры клеток добавляли 100 мкл раствора для анализа люциферазы (Promega, # E6110). Планшет помещали в темное место при комнатной температуре на 10–15 минут, продували и аспирировали 10 раз и переносили пипеткой по 100 мкл в лунки белого 96-луночного планшета. Значения сигнала хемилюминесценции регистрировали на считывающем устройстве для микропланшетов (PE, Victor3) и проводили статистическую обработку полученных данных с помощью программного обеспечения.
2.3 Результаты эксперимента
Активность соединения по настоящему изобретению при агонизации MOR и влиянию на нижележащий уровень цАМФ определяли с помощью описанного выше эксперимента, и значение EC50 представлено в таблице 1.1. Emax представляет собой максимальное действие соединения из примера по активации MOR и влиянию на метаболический путь передачи сигнала цАМФ (максимальное действие TRV-130 равно 100 %).
3. Определение активности KOR и DOR
3.1 Цель эксперимента
Цель эксперимента состоит в определении активности соединений по настоящему изобретению при агонизации рецепторов KOR и DOR и влиянию на нижележащие уровни цАМФ
3.2 Методика эксперимента
90 мкл клеток HEK293/KOR/CRE или HEK293/DOR/CRE (кДНК CRE приобретена у компании Promega, номер продукта E8471, кДНК KOR и кДНК DOR была сконструирована компанией, выполнившей изобретение) высевали в 96-луночный планшет при плотности 1×104 клеток/лунка, затем клетки инкубировали в течение ночи при 37 °C в 5% CO2. Лекарственное средство получали в виде 20 мМ исходного раствора, который впоследствии разбавляли 100% ДМСО до 200-кратного градиента концентрации, а затем разбавляли 20-кратной средой DMEM/высокое содержание глюкозы (SH30243.01B, Hyclone). Планшет для культуры клеток, засеянный в первые сутки, извлекали, и в каждую лунку добавляли 10 мкл разбавленного лекарственного средства или контрольного раствора (0,5% ДМСО). Планшет осторожно встяхивали и помещали в инкубатор при 37 °C, 5% CO2 в течение 4 часов. Наконец, в каждую лунку добавляли 100 мкл реагента для обнаружения ONE-Glo (E6120, Promega) и помещали планшет в условия комнатной температуры на 5 минут. Значение поглощения измеряли с помощью модели считывающего устройства для микропланшетов с холодным светом (PE, Victor3). Значение EC50 соединения рассчитывали с помощью программного обеспечения Graphpad Prism в соответствии с каждой концентрацией соединения и соответствующего значения сигнала. Emax представляет собой максимальное действие соединения на изменения уровня цАМФ.
3.3 Результаты эксперимента
Активность соединения по настоящему изобретению при агонизации рецепторов KOR или DOR и влиянию на нижележащий уровень цАМФ определяли с помощью описанного выше эксперимента, и значение EC50 представлено в таблице 1.2. Emax представляет собой максимальное действие соединения из примера, влияющее на метаболический путь передачи сигнала цАМФ (максимальное действие морфина равно 100 %).
Таблица 1.1. Значения EC50 и Emax соединения по настоящему изобретению при агонизации рецептора MOR и влиянию на нижележащий уровень цАМФ
Таблица 1.2. Значения EC50 и Emax соединения по настоящему изобретению при агонизации рецепторов KOR и DOR и влиянию на нижележащий уровень цАМФ
Вывод: Агонистическая активность соединений по настоящему изобретению в отношении рецептора KOR или рецептора DOR является очевидно слабой; и соединения по настоящему изобретению обладают высокой селективностью к рецептору MOR.
Экспериментальный пример 2:
1. Цель эксперимента
Цель эксперимента состоит в определении активности соединения по настоящему изобретению при активации метаболического пути передачи сигнала β-аррестина рецептором MOR.
2. Способ эксперимента
2.1 Методика эксперимента
90 мкл клеток CHO-K1 OPRM1 β-Arrestin (93-0213C2, DiscoveRX) засевали в 96-луночный планшет при плотности 1×104 клеток/лунка, затем клетки инкубировали в течение ночи при 37 °C, при 5% CO2. Лекарственное средство получали в виде 20 мМ исходного раствора, который впоследствии разбавляли 100% ДМСО до 200-кратного градиента концентрации, а затем разбавляли 20-кратной средой для посева клеток AssayComplete™ Cell Plating 2 Reagent (93-0563R2B, DiscoveRX). Планшет для культуры клеток, засеянный в первые сутки, извлекали, и в каждую лунку добавляли 10 мкл разбавленного лекарственного средства или контрольного раствора (0,5% ДМСО). Планшет осторожно встряхивали и помещали в инкубатор при 37 °C, 5% CO2 в течение 90 минут. Наконец, в каждую лунку добавляли 50 мкл реагента для обнаружения (93-0001, DiscoveRX) и помещали планшет в условия комнатной температуры на 60 минут. Значение поглощения измеряли с помощью модели считывающего устройства для микропланшетов с холодным светом (PE, Victor3). Значение EC50 соединения рассчитывали с помощью программного обеспечения Graphpad Prism в соответствии с каждой концентрацией соединения и соответствующего значения сигнала.
2.2 Результаты эксперимента
Активность соединения по настоящему изобретению при активации метаболического пути передачи сигнала β-аррестина определяли с помощью описанного выше анализа, и значение EC50 представлено в таблице 2. Emax представляет собой максимальное действие соединения из примера, влияющее на метаболический путь передачи сигнала β-аррестина (максимальное действие морфина равно 100 %).
Таблица 2: Значение EC50 соединения по настоящему изобретению, влияющее на метаболический путь передачи сигнала β-аррестина
Вывод: Соединения по настоящему изобретению оказывают незначительное действие на метаболический путь передачи сигнала β-аррестина.
Экспериментальный пример 3:
1. Цель эксперимента
Блокирующее действие соединения по настоящему изобретению и положительного контрольного соединения TRV-130 (Journal of Pharmacology and Experimental Therapeutics, Volume 344, Issue 3, Pages 708-717, 2013) на ток калиевых каналов hERG исследовали на стабильной линии клеток, трансфицированных геном калиевого канала hERG, с использованием автоматического метода точечной фиксации потенциалов (пэтч-кламп).
2. Способ эксперимента
2.1 Материалы и устройства для эксперимента
2.1.1 Материалы эксперимента
2.1.2 Устройства
2.2 Экспериментальная методика автоматической точечной фиксации потенциала
Стабилизированные клетки HEK293-hERG пересевали при плотности 1:4 в среде MEM/EBSS (Earle's Balanced Salt Solution - сбалансированный раствор соли Эрла) (10 % ФБС, 400 мкг/мл G418, 1% раствора незаменимых аминокислот MEM (100-кратный раствор), 1% раствор пирувата натрия) в течение 48–72 часов, затем проводили эксперимент по автоматической точечной фиксации потенциалов. В день эксперимента клетки диссоциировали 0,25% трипсином, затем клетки собирали центрифугированием и ресуспендировали во внеклеточной жидкости (140 мМ NaCl, 4 мМ KCl, 1 мМ MgCl2, 2 мМ CaCl2, 5 мМ D-глюкозы моногидрат, 10 мМ буферный раствор ГЭПЭС, pH 7,4, 298 мОсмоль) с получением суспензии клеток. Суспензию клеток помещали в клеточный банк устройства Patchliner, клетки наносили на микрочип (NPC-16) с помощью регулятора отрицательного давления устройства Patchliner, и за счет отрицательного давления отдельные клетки притягивались к небольшому отверстию микрочипа. После формирования цельноклеточной модели устройство будет генерировать ток hERG в соответствии с заданной программой тока и напряжения hERG, после чего устройство может автоматически осуществлять перфузию соединения от низкой концентрации к высокой концентрации. HEAK Patchmaster, усилитель точечной фиксации потенциала HEAK EPC10 (Nanion), программное обеспечение Pathliner и программное обеспечение для анализа данных поставлялись компанией Pathcontrol HT. Программное обеспечение использовали для анализа тока соединений при различных концентрациях и тока холостого контрольного раствора.
2.3 Результаты эксперимента
Блокирующее действие соединения по настоящему изобретению на ток калиевого канала hERG определяли с помощью описанного выше эксперимента, и значения IC50 представлено в таблице 3.
Таблица 3: IC50 соединений по настоящему изобретению в отношении блокирования тока калиевых каналов hERG
Вывод: Соединения по настоящему изобретению оказывают более слабое ингибиторное действие на hERG по сравнению с положительным контролем, и различие является статистически значимым.
Настоящее изобретение относится к области органической химии, а именно к соединению формулы (I):
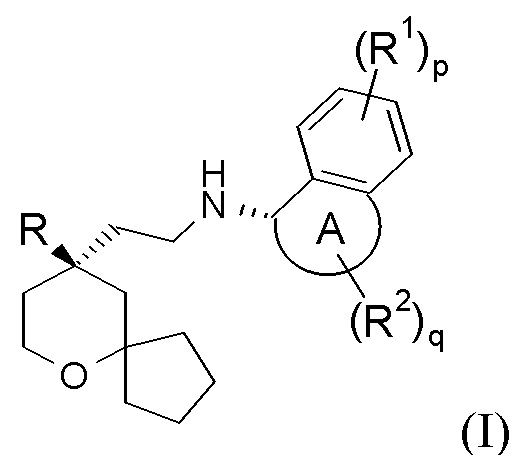
или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, или их фармацевтически приемлемой соли. В соединение формулы (I) кольцо A выбрано из группы, состоящей из C3-6 циклоалкила и 5-6-членного гетероциклила, содержащего один атом азота или один атом кислорода; R выбран из незамещенного 5-6-членного гетероарила, содержащего один атом азота; каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода и атома галогена; каждый R2 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, C1-6 алкила, C1-6 алкокси, гидроксила, оксо, =CH2 и =CH-CN, где каждый из C1-6 алкил и C1-6 алкокси необязательно замещены одной или более групп, выбранных из группы, состоящей из дейтерия, C1-6 алкила, атома галогена, циано, C1-6 алкокси и C3-6 циклоалкила; или два R2 вместе образуют незамещенный 5-6-членный гетероциклил, содержащий два атома серы или два атома кислорода; p и q каждое независимо представляет собой 0, 1 или 2. Также предложен способ получения соединения формулы (I), фармацевтическая композиция и применение соединения (I). Технический результат: предложено оксаспиропроизводное соединение общей формулы (I), которое может быть использовано в качестве агониста рецептора MOR (μ-опиоидный рецептор). 4 н. и 10 з.п. ф-лы, 8 табл., 31 пр.
1. Соединение формулы (I):
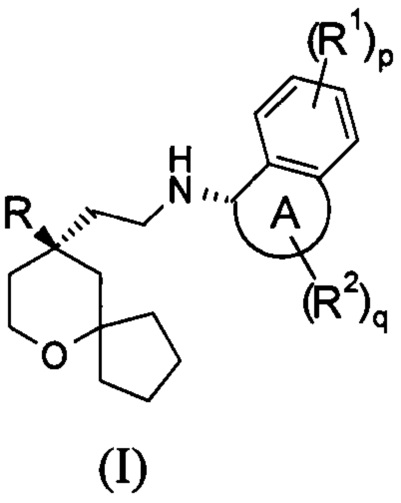
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль,
где:
кольцо A выбрано из группы, состоящей из C3-6 циклоалкила и 5-6-членного гетероциклила, содержащего один атом азота или один атом кислорода;
R выбран из незамещенного 5-6-членного гетероарила, содержащего один атом азота;
каждый R1 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода и атома галогена;
каждый R2 идентичен другому или отличается от него, и каждый независимо выбран из группы, состоящей из атома водорода, C1-6 алкила, C1-6 алкокси, гидроксил, оксо, =CH2 и =CH-CN, где каждый из C1-6 алкил и C1-6 алкокси необязательно замещены одной или более групп, выбранных из группы, состоящей из дейтерия, C1-6 алкила, атома галогена, циано, C1-6 алкокси и C3-6 циклоалкила;
или два R2 вместе образуют незамещенный 5-6-членный гетероциклил, содержащий два атома серы или два атома кислорода;
p и q каждое независимо представляет собой 0, 1 или 2.
2. Соединение формулы (I) по п. 1, где кольцо A выбрано из группы, состоящей из 5-6-членного гетероциклила, содержащего один атом азота или один атом кислорода, и 5-6-членного циклоалкила.
3. Соединение формулы (I) по п. 1 или 2, где R представляет собой пиридил.
4. Соединение формулы (I) по п. 1, представляющее собой соединение формулы (II):

или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль,
где:
G выбран из группы, состоящей из связи, CRaRb, C=O, NR4 и атома кислорода;
Ra и Rb каждый независимо выбран из группы, состоящей из атома водорода, C1-6 алкила, C1-6 алкокси и гидроксила, где каждый из C1-6 алкил и C1-6 алкокси необязательно замещены одной или более групп, выбранных из группы, состоящей из атома галогена, циано, C1-6 алкокси и C3-6 циклоалкила;
или Ra и Rb вместе образуют незамещенный 5-6-членный гетероциклил, содержащий два атома серы или два атома кислорода;
R4 выбран из группы, состоящей из атома водорода и C1-6 алкила; и
R1-R2, p и q являются такими, как определено в п. 1.
5. Соединение формулы (I) по п. 1, где R1 представляет собой атом водорода.
6. Соединение формулы (I) по п. 1, где R2 выбран из группы, состоящей из атома водорода, C1-6 алкила, оксо, C1-6 алкокси, гидроксила, где каждый из C1-6 алкил и C1-6 алкокси необязательно замещен одной или более групп, выбранных из группы, состоящей из дейтерия, C1-6 алкила, атома галогена, циано, C1-6 алкокси и C3-6 циклоалкила.
7. Соединение формулы (I) по п. 6, где R2 представляет собой C1-6 алкил, где C1-6 алкил необязательно замещен C1-6 алкокси или C3-6 циклоалкилом.
8. Соединение формулы (I) по п. 4, представляющее собой соединение формулы (IV):

или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль,
где
R1, R2 и р являются такими, как определено в п. 1.
9. Соединение формулы (I) по п. 1, выбранное из группы, состоящей из:

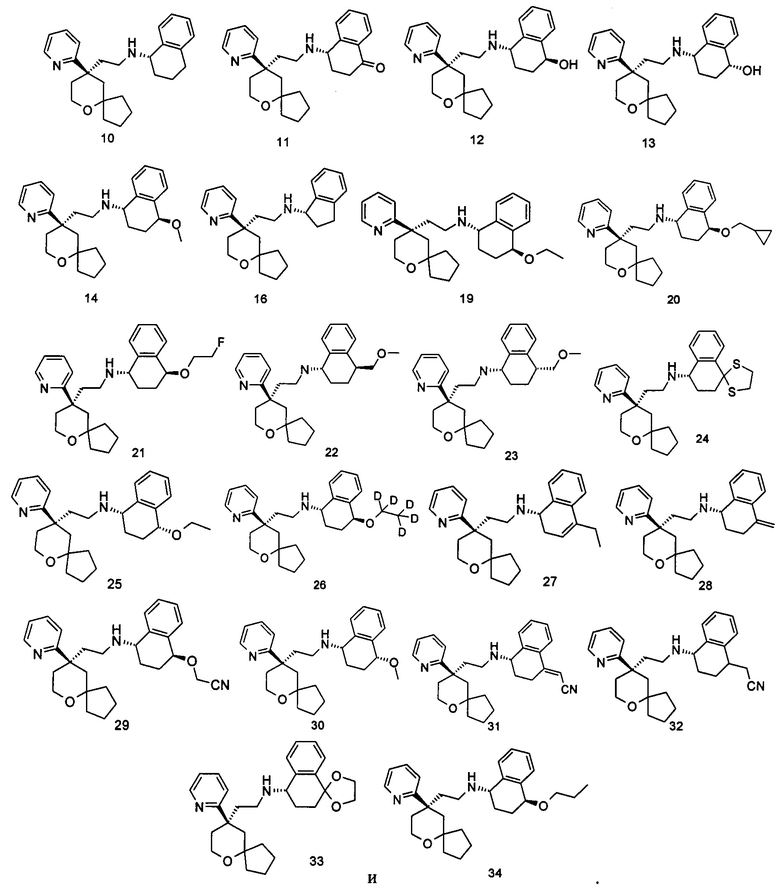
10. Способ получения соединения формулы (I) по п. 1, включающий стадию:

взаимодействия соединения формулы (VB) с соединением формулы (VIB) или его гидрохлоридом посредством восстановительного аминирования с получением соединения формулы (I);
где:
кольцо A, R, R1, R2, р и q являются такими, как определено в п. 1.
11. Фармацевтическая композиция, обладающая свойством агониста MOR (μ-опиоидный рецептор), содержащая терапевтически эффективное количество соединения формулы (I) по любому из пп. 1-9 и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
12. Применение соединения формулы (I) по любому из пп. 1-9 или фармацевтической композиции по п. 11 для получения лекарственного средства для профилактики и/или лечения заболевания, опосредованного агонистом рецептора MOR или связанного с ним, где указанное заболевание выбрано из группы, состоящей из боли, иммунной дисфункции, воспаления, эзофагеального рефлюкса, неврологических и психиатрических расстройств, заболеваний мочевыводящих путей и репродуктивной системы, сердечно-сосудистых заболеваний и респираторных заболеваний.
13. Применение по п. 12, где боль выбрана из группы, состоящей из послеоперационной боли, боли, вызванной раком, нейропатической боли, травматической боли и воспалительной боли.
14. Применение по п. 13, где рак выбран из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака предстательной железы, рака яичника, опухоли фаллопиевых труб, опухоли яичника, гемофилии и лейкоза.
| WO 2010051476 A1, 06.05.2010 | |||
| CN 103702561 A, 02.04.2014 | |||
| US 2015246904 A1, 03.09.2015 | |||
| ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2007 |
|
RU2459806C2 |

