Область техники, к которой относится изобретение
Настоящее изобретение касается способа получения производных 7H-пирроло[2,3-d]пиримидина, которые могут применяться в качестве ингибитора Янус-киназы (JAK), их сокристаллы, способа получения указанных сокристаллов и способ очистки производных 7H-пирроло[2,3-d]пиримидина с использованием сокристаллов.
JAK является представителем семейства протеин-тирозин киназ цитоплазмы и включает, например, JAK1, JAK2, JAK3 и TYK2.
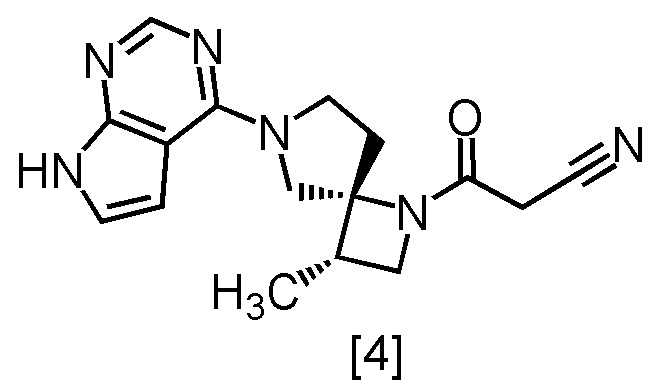
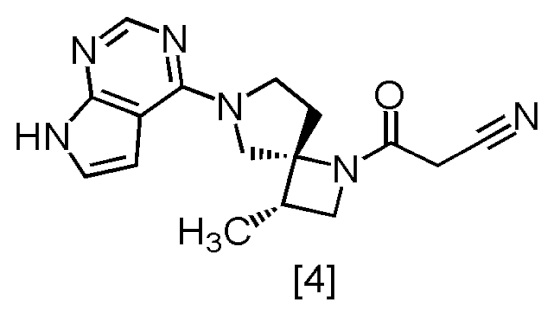
В патентной литературе 1 описано соединение A (3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрил: в настоящем тексте именуется также соединение [4]), которое может применяться в качестве ингибитора JAK.
Список процитированной литературы
Патентная литература
Патентная литература 1: WO 2011/013785 реферат
Краткое описание изобретения
В настоящем изобретении описаны способы получения производных 7H-пирроло[2,3-d]пиримидина, которые могут применяться в качестве ингибитора JAK, их сокристаллы, способы получения указанных сокристаллов и способы получения или очистки производных 7H-пирроло[2,3-d]пиримидина с использованием сокристаллов.
Настоящее изобретение включает следующий вариант осуществления:
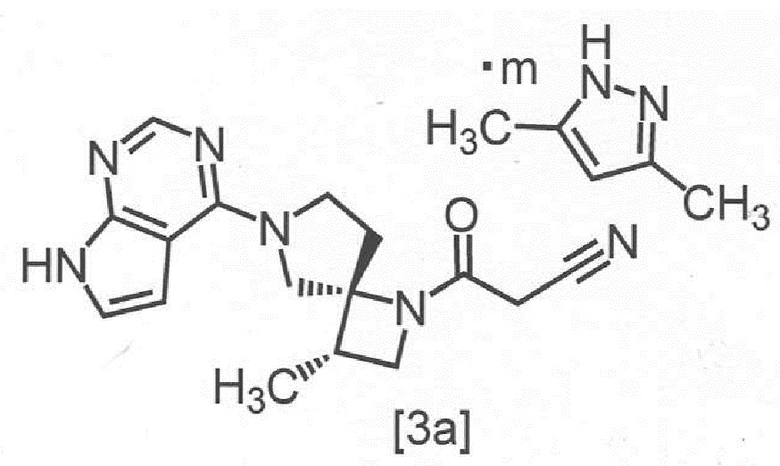
Сокристалл 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрила с 3,5-диметилпиразолом.
Краткое описание чертежей
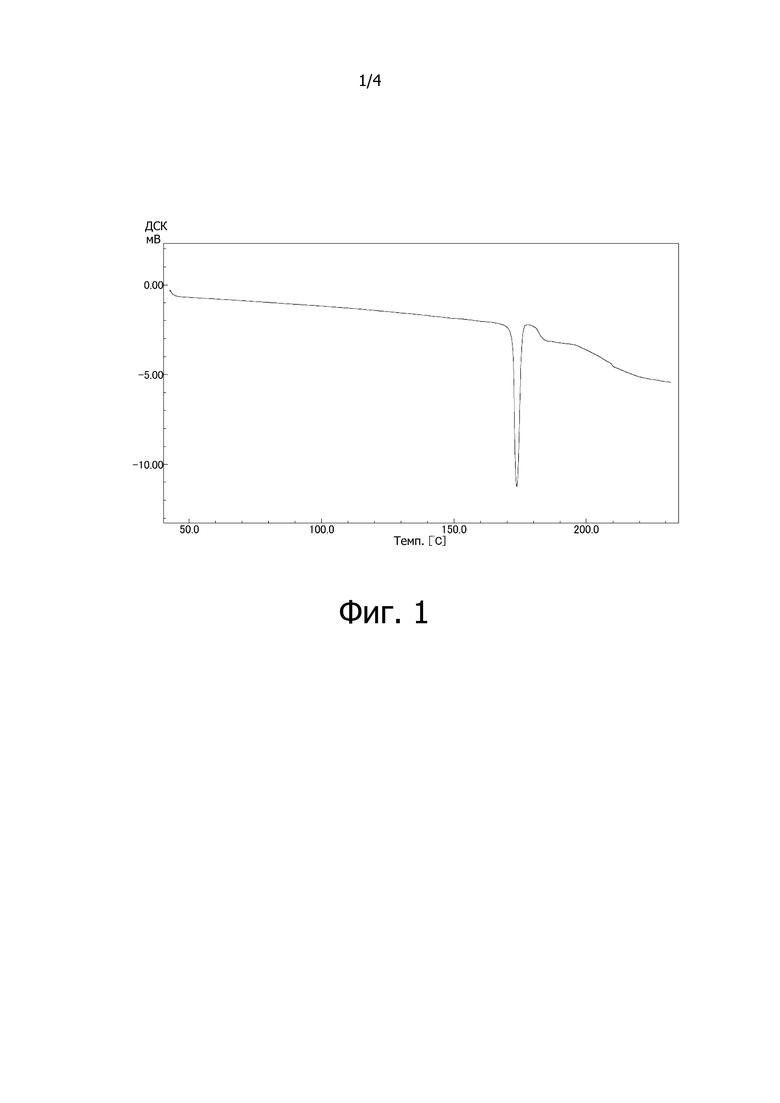
На фиг. 1 изображена кривая, построенная по результатам дифференциальной сканирующей калориметрии (ДСК) для сокристалла (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) в качестве затравочного кристалла.
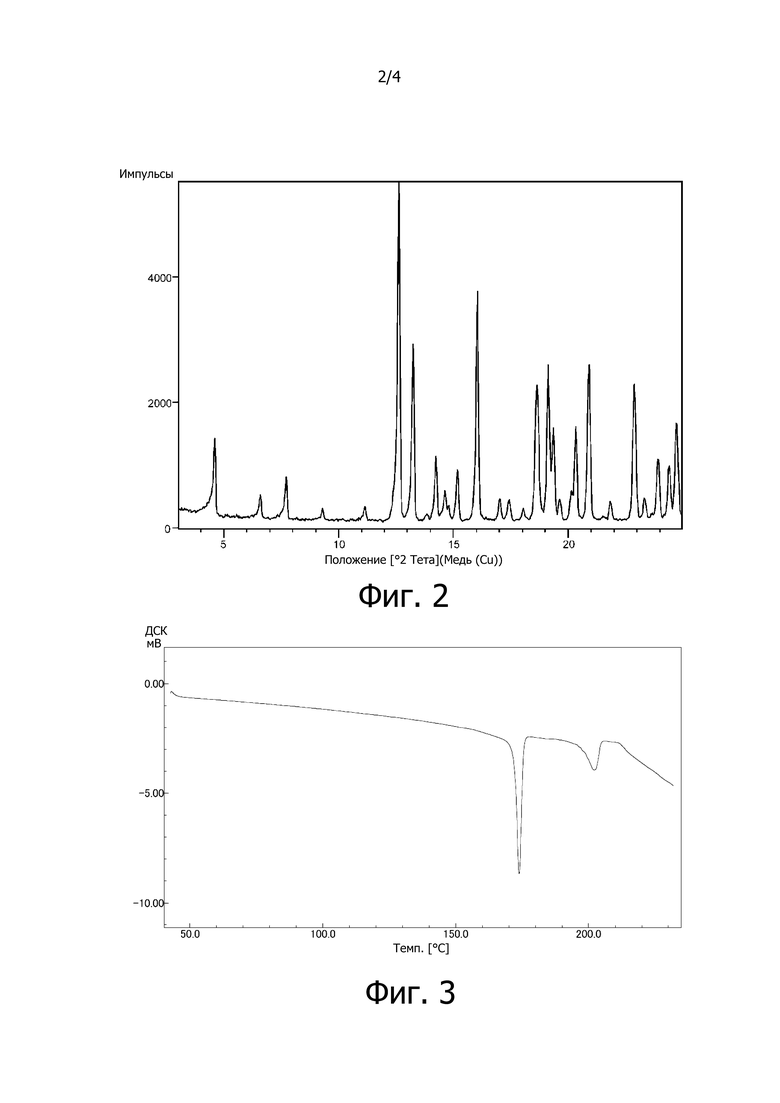
На фиг. 2 изображена диаграмма порошковой рентгеновской дифракции сокристалла (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) в качестве затравочного кристалла. Интенсивность дифракционной картины (имп/с: количество импульсов в секунду) приведено по вертикальной оси, а дифракционный угол 2θ (°) показан по горизонтальной оси.
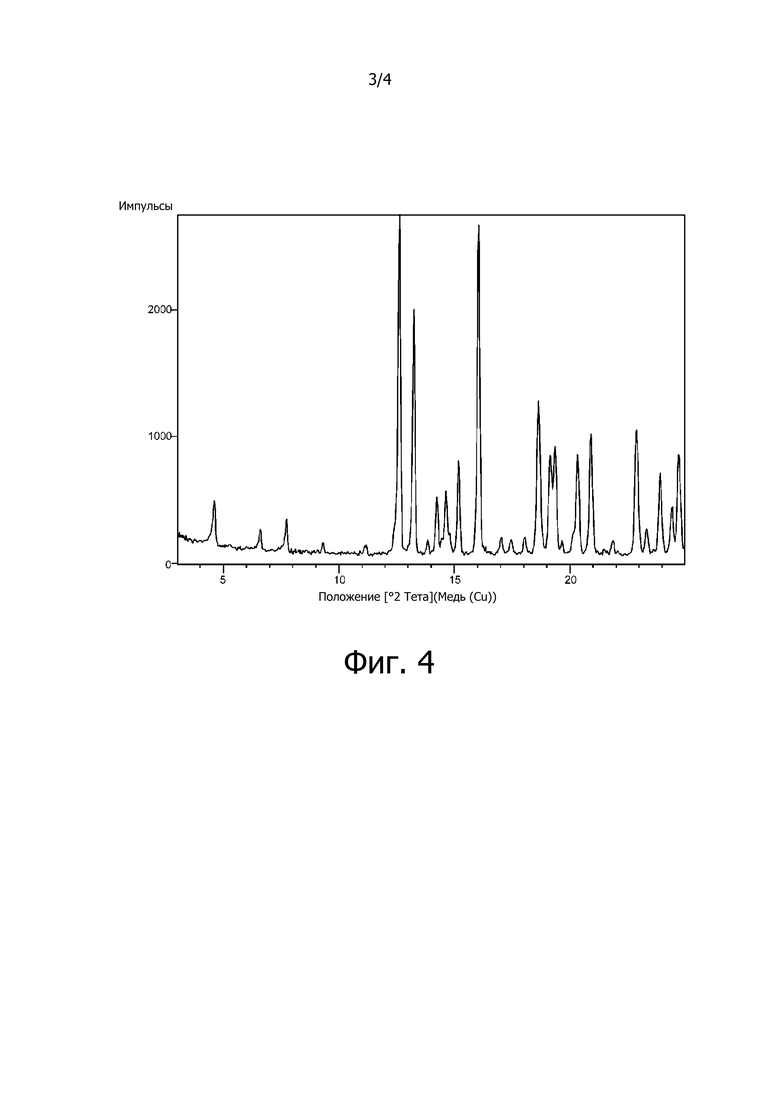
На фиг. 3 изображена кривая, построенная по результатам дифференциальной сканирующей калориметрии (ДСК) для сокристалла (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение).
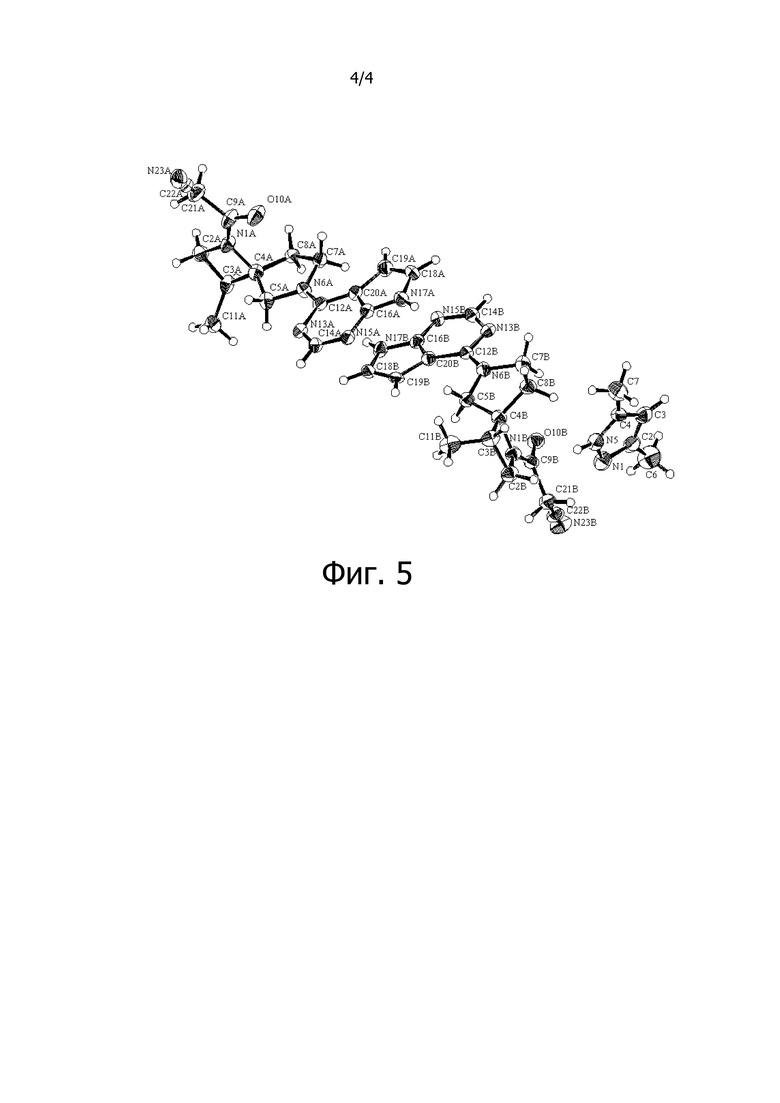
На фиг. 4 изображена диаграмма порошковой рентгеновской дифракции сокристалла (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение). Интенсивность дифракционной картины (имп/с: количество импульсов в секунду) приведено по вертикальной оси, а дифракционный угол 2θ (°) показан по горизонтальной оси.
На фиг. 5 приведено ORTEP изображение для сокристалла (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение).
Описание вариантов осуществления
Применяющиеся в настоящем тексте термины имеют указанные ниже значения.
В способе получения соединения, имеющего формулу [4]
или его соли, фраза "использование сокристалла 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрила с 3,5-диметилпиразолом (например, соединение [3a])" означает любой из следующих вариантов осуществления:
(1) Выделение соединения, имеющего формулу [4], в форме сокристалла (например, соединение [3a]) из реакционной смеси; и
(2) Добавление сокристалла (например, соединение [3a]), полученного заранее, в качестве затравочного кристалла в реакционную смесь, с последующим выделением соединения, имеющего формулу [4], в форме сокристалла (например, соединение [3a]) из реакционной смеси.
В описанном способе получения соединения, имеющего формулу [4], или его соли, соединение, имеющее формулу [4], или его соль можно получить из сокристалла (например, соединения [3a]), выделенного согласно указанным выше пунктам (1) или (2).
В способе очистки соединения, имеющего формулу [4]
или его соли, фраза "использование сокристалла 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрила с 3,5-диметилпиразолом (например, соединение [3a])" означает любой из следующих вариантов осуществления:
(1) Выделение соединения, имеющего формулу [4], в форме сокристалла (например, соединение [3a]) из реакционной смеси;
(2) Превращение сырого соединения, имеющего формулу [4], или его соли в сокристалл (например, соединение [3a]) с последующим выделением соединения, имеющего формулу [4], в форме сокристалла (например, соединение [3a]);
(3) Добавление сокристалла (например, соединения [3a]), полученного заранее, в качестве затравочного кристалла в реакционную смесь, с последующим выделением соединения, имеющего формулу [4], в форме сокристалла (например, соединение [3a]) из реакционной смеси; и
(4) Превращение сырого соединения, имеющего формулу [4], или его соли в сокристалл (например, соединение [3a]) с добавлением сокристалла (например, соединения [3a]), полученного заранее, в качестве затравочного кристалла, с последующим выделением соединения, имеющего формулу [4], в форме сокристалла (например, соединения [3a]).
В способе очистки соединения, имеющего формулу [4], или его соли, очищенное соединение, имеющее формулу [4], или его соль можно получить в ходе стадии, включающей кристаллизацию после растворения сокристалла (например, соединения [3a]), выделенного согласно любым из указанных выше пунктов (1) - (4).
Соединение, имеющее формулу [4], в настоящем тексте может именоваться также соединение [4].
Соли соединений могут представлять собой любые соли, если такие соли могут быть сформированы с соединением по настоящему изобретению, и включают, например, соли с неорганическими кислотами, соли с органическими кислотами, соли с неорганическими основаниями, соли с органическими основаниями, соли с аминокислотами.
Неорганические кислоты включают, например, соляную кислоту, азотную кислоту, серную кислоту, фосфорную кислоту, бромистоводородную кислоту.
Органические кислоты включают, например, щавелевую кислоту, малоновую кислоту, малеиновую кислоту, лимонную кислоту, фумаровую кислоту, терефталевую кислоту, молочную кислоту, яблочную кислоту, янтарную кислоту, винную кислоту, уксусную кислоту, трифторуксусную кислоту, глюконовую кислоту, аскорбиновую кислоту, метансульфокислоту, бензолсульфокислоту, п-толуолсульфокислоту, 10-камфорасульфокислоту.
Соли с неорганическими основаниями включают, например, натриевые соли, калиевые соли, кальциевые соли, магниевые соли, аммонийные соли.
Органические основания включают, например, метиламин, диэтиламин, триметиламин, триэтиламин, этаноламин, диэтаноламин, триэтаноламин, этилендиамин, трис(гидроксиметил)метиламин, дициклогексиламин, N,N-дибензилэтилендиамин, гуанидин, пиридин, пиколин, холин, цинхонин, меглумин.
Аминокислоты включают, например, лизин, аргинин, аспарагиновую кислоту, глутаминовую кислоту.
Согласно известным методикам, соединение по настоящему изобретению можно вводить в реакцию с неорганическими основаниями, органическими основаниями, неорганическими кислотами, органическими кислотами или аминокислотами, получая соли соединений по настоящему изобретению.
Соединение или его соль по настоящему изобретению может существовать в виде сольвата.
Сольват представляет собой соединение, в котором молекула растворителя координируется с соединением или его солью по настоящему изобретению, и включает гидрат. Предпочтительным сольватом является фармацевтически приемлемый сольват, и он включает, например, гидрат, этанолят, сольват с ДМСО, 1-пропанолят, 2-пропанолят, сольват с хлороформом, сольват с диоксаном, сольват с анизолом, сольват с ацетоном, сольват с этиленгликолем или сольват с диметилацетамидом, соединения или его соли по настоящему изобретению.
Сольват соединения или его соли по настоящему изобретению можно получить согласно известным методикам
Соединение по настоящему изобретению может существовать в виде таутомера. В этом случае, соединение по настоящему изобретению может существовать в виде единственного таутомера или в виде смеси отдельных таутомеров.
Соединение по настоящему изобретению может содержать двойную связь углерод-углерод. В этом случае, соединение по настоящему изобретению может существовать в виде E-формы, Z-формы или смеси E-формы и Z-формы.
Соединение по настоящему изобретению может существовать в виде стереоизомера, являющегося цис/транс изомером. В этом случае, соединение по настоящему изобретению может существовать в виде цис-формы, транс-формы или смеси цис-формы и транс-формы.
Соединение по настоящему изобретению может содержать один или больше асимметрических атомов углерода. В этом случае, соединение по настоящему изобретению может существовать в виде единственного энантиомера, единственного диастереомера, смеси энантиомеров или смеси диастереомеров.
Соединение по настоящему изобретению может существовать в виде атропоизомера. В этом случае, соединение по настоящему изобретению может существовать в виде единственного атропоизомера или смеси отдельных атропоизомеров.
Соединение по настоящему изобретению может одновременно содержать несколько структурных элементов, вызывающих появление указанных изомеров. Соединение по настоящему изобретению может содержать такие изомеры в любых соотношениях.
При отсутствии других указаний, таких как подписи и т.п., приведенные в настоящем тексте формулы, химические структуры и названия соединений без указания их стереохимии включают все упомянутые выше изомеры, которые могут существовать.
Химическая связь, изображенная волнистой линией, означает, что соединение представляет собой смесь стереоизомеров или любой из стереоизомеров. Например, соединение, имеющее формулу [10]:
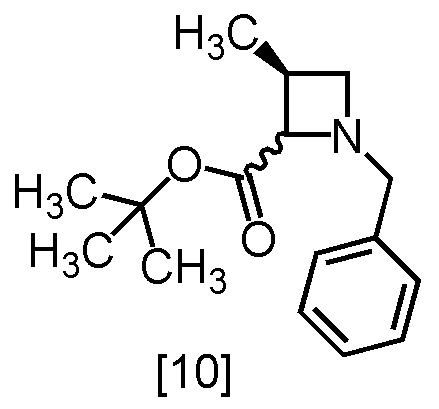
представляет собой смесь соединений, имеющих формулы [10-1] и [10-2]:
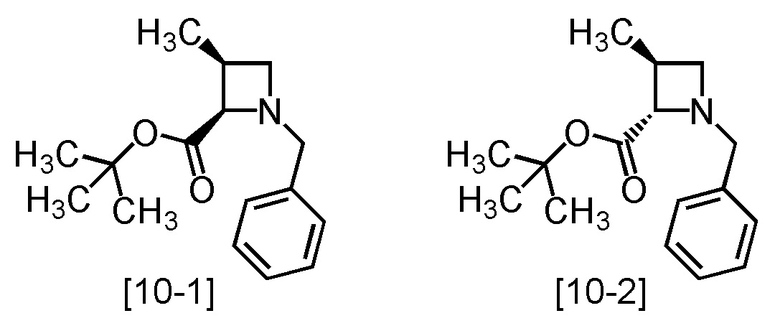
или любое из этих соединений.
Диастереомерную смесь можно разделить на отдельные диастереомеры общеизвестным способом, таким как хроматография или кристаллизация. Каждый диастереомер можно также получить с применением стереохимически индивидуального исходного соединения или методом синтеза с применением стереоселективной реакции.
Разделение смеси энантиомеров на отдельные энантиомеры можно провести с помощью хорошо известных в данной области методов.
Например, в стандартном методе, таком как дробная кристаллизация или хроматография, можно выделить диастереомер с более высоким соотношением изомеров или практически чистый диастереомер из смеси диастереомеров, которая образуется при реакции смеси энантиомеров с хиральным вспомогательным веществом, представляющим собой практически чистый энантиомер. Выделенный диастереомер можно превратить в целевой энантиомер посредством удаления вспомогательного хирального фрагмента реакцией отщепления.
Целевой энантиомер можно также получить прямым разделением смеси энантиомеров методом хроматографии с применением хиральной твердой фазы, хорошо известной в данной области.
Альтернативно, целевой энантиомер можно также получить при использовании практически чистого оптически активного исходного вещества или стереоселективным синтезом с использованием хирального вспомогательного вещества или асимметрического катализатора с прохиральным синтетическим интермедиатом, т.е. с применением асимметрической индукции.
Абсолютную конфигурацию можно определить методом рентгеноструктурного анализа кристаллического финального продукта или промежуточного продукта в синтезе. При необходимости абсолютную конфигурацию можно определить с использованием кристаллического финального продукта или промежуточного продукта, модифицированного реагентом, имеющим асимметрический центр с известной стереоконфигурацией. В настоящем тексте конфигурацию определяли методом рентгеноструктурного анализа кристаллического хлорформиата соединения [4].
Соединение по настоящему изобретению может быть кристаллическим или аморфным.
Соединение по настоящему изобретению может иметь изотопную метку, включая 3H, 14C, 35S.
Сокристалл 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрила с 3,5-диметилпиразолом в настоящем изобретении предпочтительно представляет собой сокристалл 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрила с 3,5-диметилпиразолом с мольным соотношением в диапазоне от 2:0,8 до 2:1, Более предпочтительным мольным соотношением является 2:1,
Сокристалл, имеющий формулу [3a]
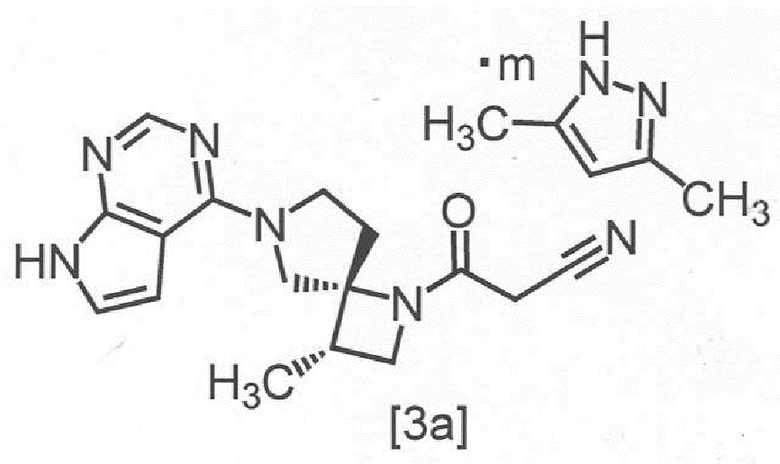 ,
,
где m представляет собой любое число от 0,4 до 0,5, в настоящем изобретении предпочтительно представляет собой сокристалл, где m равно 0,5.
В другом предпочтительном варианте осуществления, сокристалл, имеющий формулу [3a], представляет собой сокристалл, где m равно от 0,40 до 0,48, от 0,40 до 0,46, от 0,40 до 0,44, от 0,40 до 0,42, от 0,42 до 0,50, от 0,44 до 0,50, от 0,46 до 0,50, от 0,48 до 0,50, от 0,42 до 0,44, от 0,44 до 0,46, или от 0,46 до 0,48.
Соединение, имеющее формулу [4]
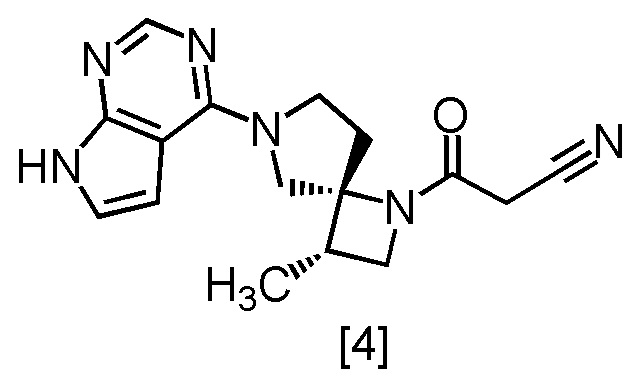
или его соль в настоящем изобретении предпочтительно представляет собой свободную форму соединения, имеющего формулу [4].
Способы получения сокристалла по настоящему изобретению, или соединения или его соли по настоящему изобретению, или его сольвата проиллюстрированы ниже.
На каждой стадии реакцию можно проводить в растворителе.
Соединение, получаемое на каждой стадии, при необходимости можно выделять и очищать известным способом, таким как перегонка, перекристаллизация, колоночная хроматография, или можно опционально использовать на последующей стадии без выделения или очистки.
Комнатная температура в настоящем тексте означает условия, когда температура не контролируется, и в одном варианте осуществления включает диапазон от 1°C до 40°C. Температура реакции может включать указанную температуру ± 5°C, предпочтительно ±2°C.
Пример способа получения сокристалла по настоящему изобретению, или соединения или его соли по настоящему изобретению, или его сольвата показан на приведенной ниже Схеме. В частности, приведена схема синтеза через соединение [3a].
На приведенной схеме, m представляет собой любое число от 0,4 до 0,5.
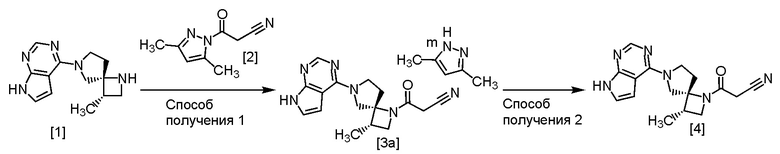
Ниже дано подробное описание способов, изображенных на приведенной выше схеме.
[Способ получения 1] Получение сокристалла, имеющего формулу [3a]
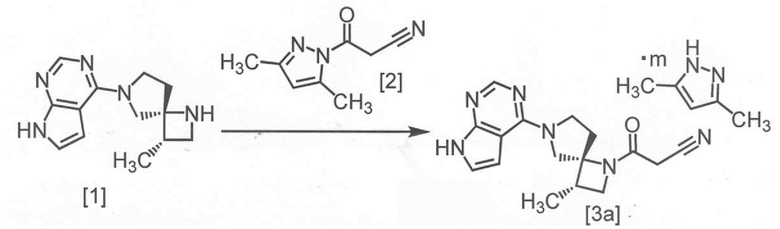
На приведенной схеме, m представляет собой любое число от 0,4 до 0,5.
Сокристалл, имеющий формулу [3a], можно получить конденсацией соединения, имеющего формулу [1], с 1-цианоацетил-3,5-диметил-1H-пиразолом (DPCN) [2]. Соединение, имеющее формулу [1], может находиться в форме соли, и образование солевой формы из свободной формы или образование свободной формы из соли можно осуществить по любой из методик, известных в данной области техники.
Предпочтительным растворителем является ацетонитрил.
DPCN [2] можно применять, например, в количестве от 0,95 до 1,2 эквивалентов относительно соединения, имеющего формулу [1], предпочтительно 1,1 ± 0,05 эквивалентов. Другим предпочтительным вариантом является 1,0 ± 0,05 эквивалентов.
Температура реакции находится в диапазоне, например, от комнатной температуры до 80°C, предпочтительно от 70°C до 80°C.
Время реакции составляет, например, от 0,5 часов до 12 часов, предпочтительно от 0,5 часов до 6 часов.
Символ "m" в сокристалле, имеющем формулу [3a], может представлять собой любое число от 0,4 до 0,5, в зависимости от реакции, условий отфильтровывания сокристаллов или сушки.
Соединение, имеющее формулу [3a], может представлять собой, например, кристалл, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один (например, по меньшей мере 1, 2 или 3) пик со значением дифракционного угла (2θ) = 4,6° ± 0,2°, 18,6° ± 0,2° или 20,9° ± 0,2°, при анализе с применением CuKα излучения.
Предпочтительно, соединение, имеющее формулу [3a], может представлять собой кристалл, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один (например, по меньшей мере 1, 2 или 3) пик со значением дифракционного угла (2θ) = 4,6° ± 0,1°, 18,6° ± 0,1° или 20,9° ± 0,1°, при анализе с применением CuKα излучения.
Более предпочтительно, соединение, имеющее формулу [3a], может представлять собой кристалл, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один (например, по меньшей мере 1, 2 или 3) пик со значением дифракционного угла (2θ) = 4,6° ± 0,06°, 18,6° ± 0,06° или 20,9° ± 0,06°, при анализе с применением CuKα излучения.
Кроме того, соединение, имеющее формулу [3a], может также представлять собой, например, кристалл, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один (например, по меньшей мере 1, 2, 3, 4, или 5) пик со значением дифракционного угла (2θ) = 4,6° ± 0,2°, 12,6° ± 0,2°, 16,1° ± 0,2°, 18,6° ± 0,2° или 20,9° ± 0,2°, при анализе с применением CuKα излучения.
Предпочтительно, соединение, имеющее формулу [3a], может также представлять собой кристалл, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один (например, по меньшей мере 1, 2, 3, 4, или 5) пик со значением дифракционного угла (2θ) = 4,6° ± 0,1°, 12,6° ± 0,1°, 16,1° ± 0,1°, 18,6° ± 0,1° или 20,9° ± 0,1°, при анализе с применением CuKα излучения.
Более предпочтительно, соединение, имеющее формулу [3a], может также представлять собой кристалл, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один (например, по меньшей мере 1, 2, 3, 4, или 5) пик со значением дифракционного угла (2θ) = 4,6° ± 0,06°, 12,6° ± 0,06°, 16,1° ± 0,06°, 18,6° ± 0,06° или 20,9° ± 0,06°, при анализе с применением CuKα излучения.
Сокристалл, имеющий формулу [3a], представляет собой сокристалл, показывающий экстраполированную температуру начала перехода 172 ± 5°C, при анализе методом дифференциальной сканирующей калориметрии.
Предпочтительный сокристалл, имеющий формулу [3a], представляет собой сокристалл, показывающий экстраполированную температуру начала перехода 172 ± 3°C, при анализе методом дифференциальной сканирующей калориметрии.
Более предпочтительный сокристалл, имеющий формулу [3a], представляет собой сокристалл, показывающий экстраполированную температуру начала перехода 172 ± 1°C, при анализе методом дифференциальной сканирующей калориметрии.
Сокристалл, имеющий формулу [3a], представляет собой сокристалл, имеющий эндотермический пик при 173 ± 5°C, при анализе методом дифференциальной сканирующей калориметрии.
Предпочтительный сокристалл, имеющий формулу [3a], представляет собой сокристалл, имеющий эндотермический пик при 173 ± 3°C, при анализе методом дифференциальной сканирующей калориметрии.
Более предпочтительный сокристалл, имеющий формулу [3a], представляет собой сокристалл, имеющий эндотермический пик при 173 ± 1°C, при анализе методом дифференциальной сканирующей калориметрии.
[Способ получения 2] Получение (очистка) соединения, имеющего формулу [4]
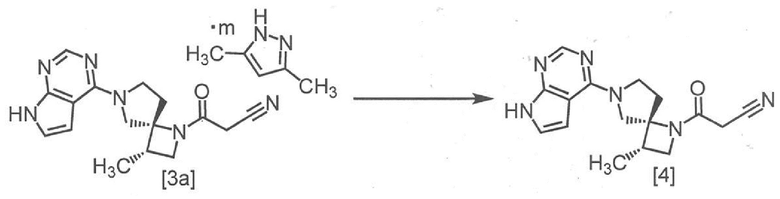
На приведенной схеме, m имеет указанное выше значение.
Соединение, имеющее формулу [4], можно получить кристаллизацией после растворения соединения, имеющего формулу [3a]. Получение (очистку) можно осуществить посредством добавления 2,6-ди-трет-бутил-4-метилфенола (BHT) во время кристаллизации.
Примеры растворителя для кристаллизации включают, например, 1-бутанол и 1-пропанол. Предпочтительным растворителем является 1-бутанол. Растворитель можно применять, например, в количестве от 8,0-кратного до 20-кратного относительно веса соединения, имеющего формулу [3a], предпочтительно 8,5 ± 0,5 –кратное количество.
Температура для растворения соединения [3a] в растворителе для кристаллизации находится в диапазоне, например, от 100°C до 117°C, предпочтительно 110°C ± 5°C.
Время прохождения кристаллизации составляет, например, от 15 часов до 48 часов, предпочтительно от 18 часов до 24 часов.
Способ получения сокристалла по настоящему изобретению, или соединение или его соль по настоящему изобретению, или его сольват могут, например, иметь следующее преимущество над Препаратом 6 из Патентной Литературы 1:
(1) Процедуры по выделению и очистке посредством экстракции и колоночной хроматографии на силикагеле могут быть необязательны благодаря высокой стабильности сокристалла, который можно напрямую выделять из реакционной смеси. Соединение A (соединение [4]) можно получать с высокой химической чистотой.
Варианты осуществления настоящего изобретения включают следующее:
Пункт 1, Сокристалл 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрила с 3,5-диметилпиразолом.
Пункт 2. Сокристалл по пункту 1, имеющий структуру, отвечающую формуле [3a]
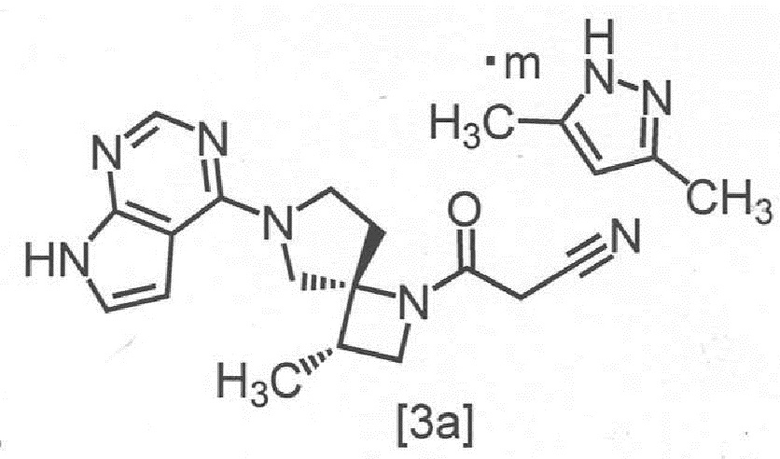 ,
,
где m представляет собой любое число от 0,4 до 0,5.
Пункт 3. Сокристалл по пункту 2, где m равно 0,5.
Пункт 4. Сокристалл по любому из пунктов 1-3, показывающий экстраполированную температуру начала перехода 172 ± 5°C при анализе методом дифференциальной сканирующей калориметрии.
Пункт 5. Сокристалл по любому из пунктов 1 - 4, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один пик со значением дифракционного угла (2θ) = 4,6° ± 0,2°, 18,6° ± 0,2° или 20,9° ± 0,2°, при анализе с применением CuKα излучения.
Пункт 6. Сокристалл по любому из пунктов 1 - 4, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один пик со значением дифракционного угла (2θ) = 4,6° ± 0,2°, 12,6° ± 0,2°, 16,1° ± 0,2°, 18,6° ± 0,2° или 20,9° ± 0,2°, при анализе с применением CuKα излучения.
Пункт 7. Способ получения соединения, имеющего формулу [4]
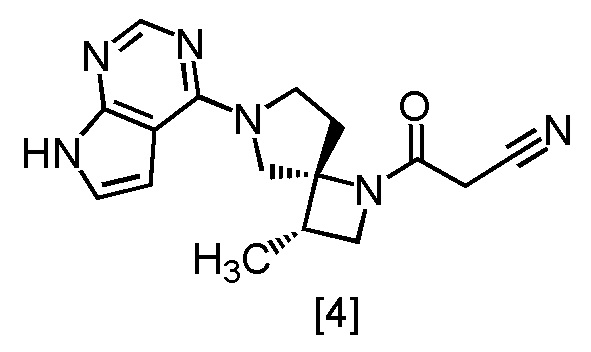
или его соли с использованием сокристалла по любому из пунктов 1 - 6.
Пункт 8: Способ по п. 7, дополнительно включающий стадию реакции соединения, имеющего формулу [1]
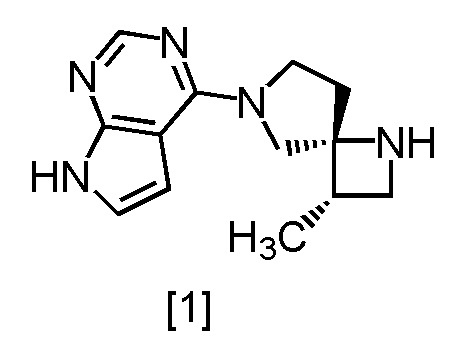
или его соли с соединением, имеющим формулу [2]
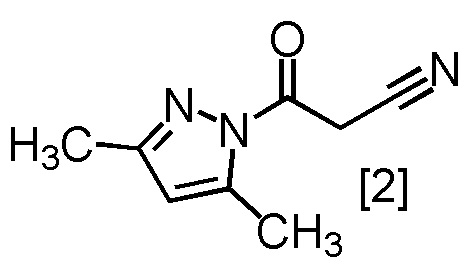
с получением соединения, имеющего формулу [4], или его соли.
Пункт 9: Способ очистки соединения, имеющего формулу [4]
или его соли с использованием сокристалла по любому из пунктов 1 - 6.
Пункт 10, Способ по п. 9, дополнительно включающий стадию реакции соединения, имеющего формулу [1]
или его соли с соединением, имеющим формулу [2]
с получением соединения, имеющего формулу [4], или его соли.
Пункт 11, Способ получения сокристалла, имеющего структуру, отвечающую формуле [3a]
 ,
,
где m представляет собой любое число от 0,4 до 0,5, включающий стадию реакции соединения, имеющего формулу [1]
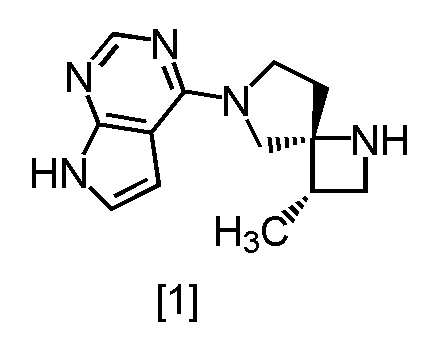
или его соли с соединением, имеющим формулу [2]
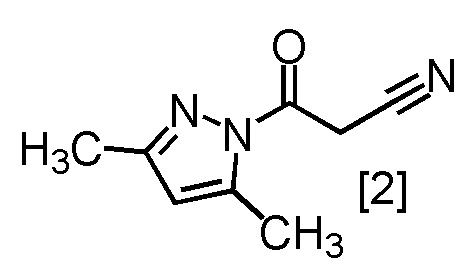
с получением сокристалла, имеющего формулу [3a].
Пункт 12. Способ получения сокристалла, имеющего структуру, отвечающую формуле [3a]
,
где m представляет собой любое число от 0,4 до 0,5, включающий стадию реакции соединения, имеющего формулу [4]
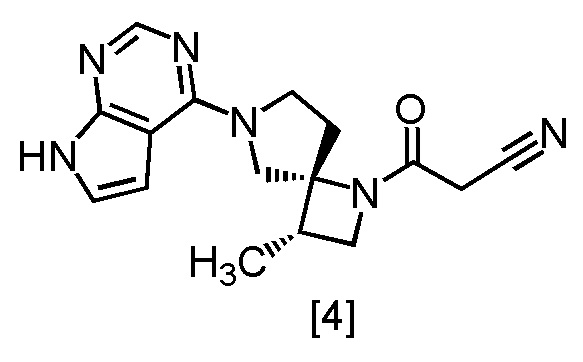
или его соли с соединением, имеющим формулу [5]
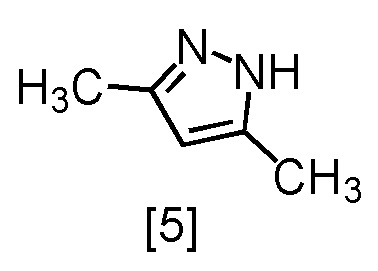
с получением сокристалла, имеющего формулу [3a].
Пункт 13. Способ по п. 11 или 12, где m равно 0,5.
Пункт 14. Способ по любому из пп. 11 - 13, где сокристалл, имеющий формулу [3a], показывает экстраполированную температуру начала перехода 172 ± 5°C при анализе методом дифференциальной сканирующей калориметрии.
Пункт 15. Способ по любому из пп. 11 - 14, где сокристалл, имеющий формулу [3a], имеет спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один пик со значением дифракционного угла (2θ) = 4,6° ± 0,2°, 18,6° ± 0,2° или 20,9° ± 0,2°, при анализе с применением CuKα излучения.
Пункт 16. Способ по любому из пп. 11 - 14, где сокристалл, имеющий формулу [3a], имеет спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один пик со значением дифракционного угла (2θ) = 4,6° ± 0,2°, 12,6° ± 0,2°, 16,1° ± 0,2°, 18,6° ± 0,2° или 20,9° ± 0,2°, при анализе с применением CuKα излучения.
Пункт 17. Соединение, имеющее формулу [4]
или его соль, которое получено или может быть получено способом по п. 7 или 8.
Пункт 18. Сокристалл, имеющий формулу [3a]
,
,
где m представляет собой любое число от 0,4 до 0,5, которое получено или может быть получено способом по п. 11 или 12.
Пункт 19. Сокристалл по пункту 18, где m равно 0,5.
Примеры
Частные способы получения сокристалла по настоящему изобретению, или соединения или его соли по настоящему изобретению, или его сольвата проиллюстрированы в приведенных ниже примерах. Однако настоящее изобретение не ограничивается только приведенными ниже примерами.
На стадиях кристаллизации в получении (очистке) соединения A (соединение [4]) (пример 3), получении соединения [6] (пример 4 Стадия 4) и получении соединения [20] (пример 14) применяли затравочные кристаллы для облегчения кристаллизации. Кристаллы этих соединений можно получить согласно методикам, описанным в примерах, даже без использования затравочных кристаллов.
Ниже приведены расшифровки сокращений, использованных в настоящем тексте.
SR-MDOP: 4-[(3S,4R)-3-метил-1,6-диазаспиро[3,4]-октан-6-ил]-7H-пирроло[2,3-d]пиримидин
Соединение A (соединение [4]): 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрил
S-BAPO: (S)-2-(бензиламино)пропан-1-ол
S-BBMO: трет-бутил (S)-N-бензил-N-(1-гидроксипропан-2-ил)глицинат
R-BCAB: трет-бутил (R)-N-бензил-N-(2-хлорпропил)глицинат
S-MABB: трет-бутил (3S)-1-бензил-3-метилазетидин-2-карбоксилат
S-MABB-HC: трет-бутил (3S)-1-бензил-3-метилазетидин-2-карбоксилат гидрохлорид
S-MACB-HC: трет-бутил (3S)-3-метилазетидин-2-карбоксилат гидрохлорид
S-ZMAB: 1-бензил 2-(трет-бутил) (3S)-3-метилазетидин-1,2-дикарбоксилат
RS-ZMBB: 1-бензил 2-(трет-бутил)(2R,3S)-2-(2-(трет-бутокси)-2-оксоэтил)-3-метилазетидин-1,2-дикарбоксилат
RS-ZMAA: (2R,3S)-1-((бензилокси)карбонил)-2-(карбоксиметил)-3-метилазетидин-2-карбоновая кислота
RS-ZMAA-DN·2H2O: динатрия (2R,3S)-1-((бензилокси)карбонил)-2-(карбоксиметил)-3-метилазетидин-2-карбоксилат дигидрат
RS-ZMOO: бензил (2R,3S)-2-(2-гидроксиэтил)-2-(гидроксиметил)-3-метилазетидин-1-карбоксилат
RS-ZMSS: бензил (2R,3S)-3-метил-2-(2-((метилсульфонил)окси)этил)-2-(((метилсульфонил)окси)метил)азетидин-1-карбоксилат
SR-ZMDB: бензил (3S,4R)-6-бензил-3-метил-1,6-диазаспиро[3,4]октан-1-карбоксилат
SR-MDOZ: бензил (3S,4R)-3-метил-1,6-диазаспиро[3,4]октан-1-карбоксилат
SR-MDOZ-OX: бензил (3S,4R)-3-метил-1,6-диазаспиро[3,4]октан-1-карбоксилат оксалат
SR-MDPZ: бензил-(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-карбоксилат
BHT: 2,6-ди-трет-бутил-4-метилфенол
DPCN: 1-цианоацетил-3,5-диметил-1H-пиразол
CPPY: 4-хлор-7H-пирроло[2,3-d]пиримидин
TBBA: трет-бутиловый эфир бромуксусной кислоты
PTFE: политетрафторэтилен
Применявшиеся в примерах приборы и условия снятия спектров описаны ниже.
1H-ЯМР спектры регистрировали в CDCl3, ДМСО-d6 или оксиде дейтерия с применением триметилсилана в качестве внутреннего стандарта, и все значения δ указаны в миллионных долях (м.д.). Спектры регистрировали на ЯМР-приборе с рабочей частотой 400 МГц, если не указано иное.
Символы в примерах имеют указанные ниже значения.
с: синглет
д: дублет
т: триплет
кв: квартет
дд: дублет дублетовд.кв: дублет квартетов
ддд: дублет дублетов дублетов
ушир.с: уширенный синглет
м: мультиплет
J: константа спин-спинового взаимодействия
Диаграммы порошковой рентгеновской дифракции образцов были получены методом рентгеновской дифракции на порошках.
Прибор: X'Pert Pro (SPECTRIS)
Условия проведения анализа:
Антикатод: Cu
Ток и напряжение в рентгеновской трубке: 45 кВ, 40 мА
Скорость вращения образца: каждые 1 сек.
Щель Соллера для падающего луча: 0,02 рад
Щель вертикальной дивергенции для падающего луча: 15 мм
Щель расходимости для падающего луча: автом., ширина излучения 15 мм
Щель рассеивания для падающего луча: 1°
Фильтр отраженного луча: никелевый фильтр
Щель Соллера для отраженного луча: 0,02 рад
Щель расходимости для отраженного луча: автом., ширина излучения 15 мм
Детектор: X'Celerator
Режим детектора: сканирование
Эффективная ширина детектора: 2,122°
Ось сканирования: Гонио.
Режим сканирования: непрерывный
Диапазон сканирования: 3° - 60°
Время единичного шага: 10 сек.
Вес.% углерода, водорода и азота в образцах определяли элементным анализом.
Содержание ионов в образце определяли как среднее значение из трех измерений для образца.
Прибор: ионообменный хроматограф LC-20 (Shimadzu Corporation)
Условия анализа: Детектор электропроводности SHIMADZU CDD-10A VP
Колонка для анализа анионов SHIMADZU SHIM-PAC IC-A3
Колонка для анализа катионов SHIMADZU SHIM-PAC IC-C1
Содержание воды в образце определяли методом титрования по Карлу Фишеру.
Прибор: кулонометрический титратор для определения содержания воды CA-06 (Mitsubishi Chemical Corporation)
Условия анализа: Количество образца: около 20 мг
Реагент: анодный раствор Aquamicron AX (API Corporation)
Катодный раствор Aquamicron CXU (API Corporation)
Пример 1. Получение сокристалла (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) (Затравочный кристалл)
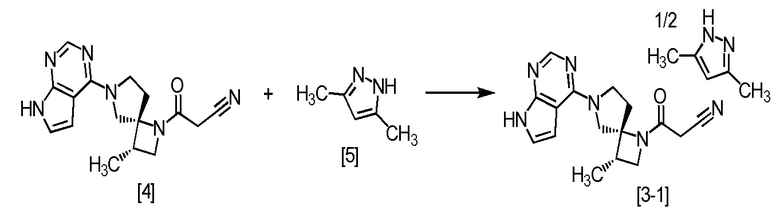
К соединению A (соединение [4]) (70,0 г, 226 ммоль) и 3,5-диметилпиразолу [5] (21,7 г, 226 ммоль) добавляли ацетонитрил (490 мл) в атмосфере азота, и смесь растворяли при нагревании до 80°C. Смесь перемешивали при 65°C в течение 2 часов. После выпадения кристаллического осадка, смесь медленно охлаждали до комнатной температуры. Смесь перемешивали при охлаждении на ледяной бане 2 часа, выпавший осадок собирали на фильтре и промывали ледяным ацетонитрилом (140 мл). Полученное влажное твердое вещество сушили при пониженном давлении, получая сокристалл (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) (75,3 г, 210 ммоль) с выходом 93,1%.
Регистрировали ЯМР-спектр, проводили элементный анализ и дифференциальную сканирующую калориметрию для синтезированного сокристалла соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение). 1H-ЯМР (ДМСО-d6) δ: 11,98 (ушир.с, 0,5H), 11,59 (ушир.с, 1H), 8,08 (с, 1H), 7,11 (дд, 1H, J = 3,5, 2,2 Гц), 6,58 (дд, 1H, J = 3,5, 1,4 Гц), 5,73 (с, 0,5H), 4,16 (т, 1H, J = 8,3 Гц), 4,09-3,93 (м, 3H), 3,84-3,74 (м, 1H), 3,70 (д, 1H, J = 19,0 Гц), 3,65 (д, 1H, J = 19,0 Гц), 3,58 (дд, 1H, J = 8,2, 5,9 Гц), 2,70-2,58 (м, 2H), 2,22-2,12 (м, 1H), 2,12 (с, 3H), 1,12 (д, 3H, J = 7,2 Гц).
Элементный анализ: C 61,9 вес.%, H 6,1 вес.%, N 27,2 вес.% (Теоретическое значение C 62,0 вес.%, H 6,2 вес.%, N 27,4 вес.%).
Дифференциальная сканирующая калориметрия:
Анализ проводили на дифференциальном сканирующем калориметре DSC-60A (производство Shimadzu Corporation) при скорости повышения температуры 5°C/мин (герметично закрытая алюминиевая ячейка). Полученная в эксперименте кривая ДСК изображена на фиг. 1, Энтальпия эндотермических пиков в кривой ДСК составила 100,26 Дж/г, температура начала эндотермического перехода 173,66°C, и экстраполированная температура начала перехода составила 172,36°C. Полученный спектр изображен на фиг. 1.
Дифракционный угол 2θ и интенсивность дифракционной картины определяли методом порошковой рентгеновской дифракции для сокристалла соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение). Полученный спектр изображен на фиг. 2.
Соответствующие пики на фиг. 2 имеют характеристики, приведенные ниже в таблице.
[2θ (°)]
интенсивность
[%]
картины
[имп/с]
Пример 2. Получение сокристалла (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение)
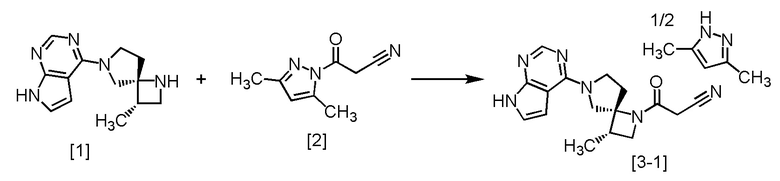
К SR-MDOP [1] (800 г, 3,29 моль) добавляли ацетонитрил (8,0 л) в атмосфере азота, и затем в смесь по каплям добавляли раствор DPCN [2] (563 г, 3,45 моль) в ацетонитриле (4,8 л) при 75°C. Использованную капельную воронку промывали ацетонитрилом (0,8 л), и промывные растворы добавляли в реакционную смесь. После перемешивания реакционной смеси при 75°C в течение 1,5 часов, смесь упаривали при пониженном давлении до объема 8,0 л. В остаток добавляли при 65°C сокристалл (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) (80 мг), синтезированный в примере 1, После перемешивания при 65°C в течение 2 часов, смесь перемешивали 2 часа при охлаждении на ледяной бане. Выпавший твердый осадок собирали на фильтре и промывали ледяным ацетонитрилом (2,4 л). Полученное влажное твердое вещество сушили при пониженном давлении, получая сокристалл (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) (1070 г, 2,99 моль) с выходом 90,8%.
Регистрировали ЯМР-спектр, проводили элементный анализ и дифференциальную сканирующую калориметрию для синтезированного сокристалла соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение).
1H-ЯМР (ДМСО-d6) δ: 11,99 (ушир.с, 0,5H), 11,59 (ушир.с, 1H), 8,11 (с, 1H), 7,11 (с, 1H), 6,58 (д, 1H, J = 3,0 Гц), 5,73 (с, 0,5H), 4,16 (т, 1H, J = 8,4 Гц), 4,10-3,92 (м, 3H), 3,85-3,74 (м, 1H), 3,70 (д, 1H, J = 19,1 Гц), 3,65 (д, 1H, J = 19,1 Гц), 3,57 (дд, 1H, J = 7,9, 6,1 Гц), 2,70-2,58 (м, 2H), 2,22-2,14 (м, 1H), 2,12 (с, 3H), 1,12 (д, 3H, J = 6,9 Гц).
Элементный анализ: C 62,0 вес.%, H 6,2 вес.%, N 27,2 вес.% (Теоретическое значение C 62,0 вес.%, H 6,2 вес.%, N 27,4 вес.%)
Дифференциальная сканирующая калориметрия:
Анализ проводили на дифференциальном сканирующем калориметре DSC-60A (производство Shimadzu Corporation) при скорости повышения температуры 5°C/мин (герметично закрытая алюминиевая ячейка). Полученная в эксперименте кривая ДСК изображена на фиг. 3. Энтальпия эндотермических пиков в кривой ДСК составила 78,02 Дж/г, температура начала эндотермического перехода 173,81°C, и экстраполированная температура начала перехода составила 172,025°C. Полученный спектр изображен на фиг. 3.
Дифракционный угол 2θ и интенсивность дифракционной картины определяли методом порошковой рентгеновской дифракции для синтезированного сокристалла соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение). Полученный спектр изображен на фиг. 4.
Соответствующие пики на фиг. 4 имеют характеристики, приведенные ниже в таблице.
[2θ (°)]
[%]
[имп/с]
Сокристаллы, в которых мольное соотношение соединения A (соединение [4]) и 3,5-диметилпиразола находилось в диапазоне от 2:0,842 до 2:0,864, в частности 2:0,842, 2:0,848, 2:0,856, 2:0,862 и 2:0,864, получали по методике, аналогичной описанной в примере 2.
Пример 3. Получение (очистка) соединения A (соединение [4])
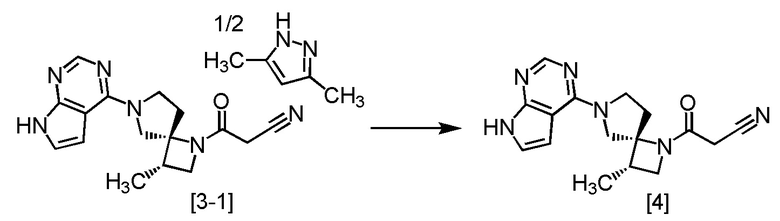
Сокристалл (соединение [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) (2,00 кг, 5,88 моль), BHT (60 г) и 1-бутанол (16 л) смешивали в атмосфере азота и растворяли при 110°C. После охлаждения смеси до 85°C, добавляли в смесь заранее приготовленный кристалл (200 мг) соединения A (соединение [4]). После перемешивания при 85°C в течение 2 часов, смесь медленно охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение 3 часов. Выпавший твердый осадок собирали на фильтре и последовательно промывали 1-бутанолом (4 л) и этилацетатом (4 л). Полученный влажный твердый продукт сушили при пониженном давлении с получением соединения A (соединение [4]) (1,63 кг, 5,27 моль) с выходом 94,4%.
Регистрировали спектры ЯМР и МС для соединения A (соединение [4]), которое было синтезировано по описанной выше методике.
1H-ЯМР (ДМСО-d6) δ: 11,58 (ушир.с, 1H), 8,08 (с, 1H), 7,11 (дд, 1H, J = 3,5, 2,3 Гц), 6,58 (дд, 1H, J = 3,5, 1,6 Гц), 4,16 (т, 1H, J = 8,4 Гц), 4,10-3,94 (м, 3H), 3,84-3,74 (м, 1H), 3,70 (д, 1H, J = 19,0 Гц), 3,65 (д, 1H, J = 18,7 Гц), 3,58 (дд, 1H, J = 8,2, 5,9 Гц), 2,70-2,59 (м, 2H), 2,23-2,12 (м, 1H), 1,12 (д, 3H, J = 7,2 Гц).
МС: m/z = 311 [M+H]+
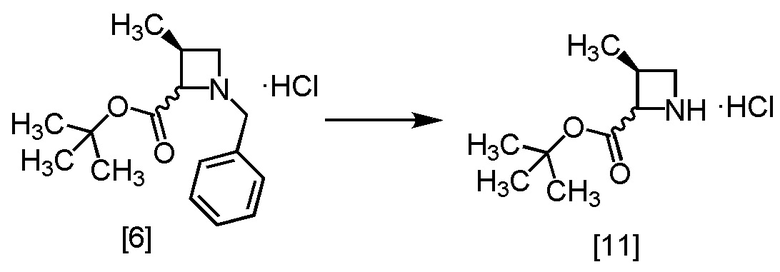
Пример 4. Получение S-MABB-HC (соединение [6])
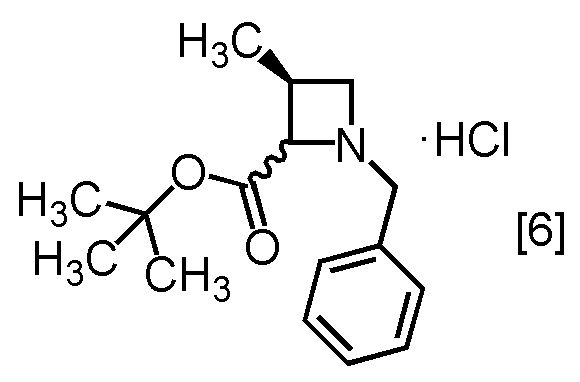
Стадия 1
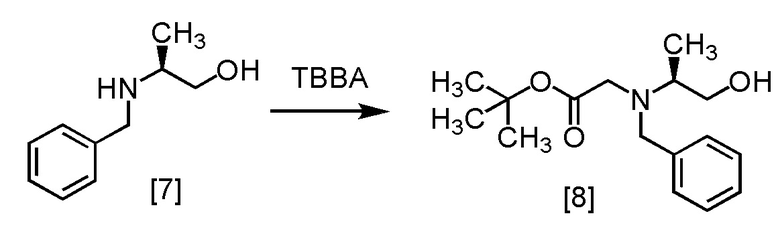
S-BAPO [7] (35,0 г, 212 ммоль) добавляли в воду (175 мл) при комнатной температуре в атмосфере азота. В полученную суспензию добавляли толуол (53 мл) и карбонат калия (32,2 г, 233 ммоль) при комнатной температуре. В полученный раствор по каплям добавляли TBBA (434,4 г, 223 ммоль) при комнатной температуре, затем использованную капельную воронку промывали толуолом (17 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при 65°C в течение 21 часов, затем охлаждали до комнатной температуры. Добавляли в реакционную смесь толуол (105 мл), затем смесь перемешивали, органический слой отделяли. Органический слой промывали водой (175 мл), водный слой отделяли и удаляли растворитель из органического слоя в вакууме. Толуол (105 мл) добавляли в остаток, и полученный толуольный раствор упаривали. Эту операцию повторяли еще два раза, получая толуольный раствор S-BBMO [8] (74,0 г, 212 ммоль теоретически). Полученный толуольный раствор S-BBMO [8] использовали на следующей стадии, считая выход равным 100%.
Сырой продукт S-BBMO [8], который был синтезирован по описанной выше методике, упаривали и сушили, затем регистрировали спектры ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 7,36-7,13 (5H, м), 4,26 (1H, дд, J = 6,8, 3,9 Гц), 3,72 (2H, дд, J = 14,2, 6,8 Гц), 3,47-3,38 (1H, м), 3,30-3,08 (3H, м), 2,79 (1H, секстет, J = 6,8 Гц), 1,35 (9H, с), 0,96 (3H, д, J = 6,8 Гц).
МС: m/z = 280 [M+H]+
Стадия 2
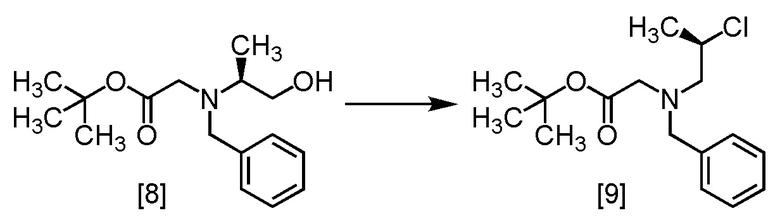
В толуольный раствор S-BBMO [8] (74,0 г, 212 ммоль) добавляли толуол (200 мл), тетрагидрофуран (35 мл) и затем триэтиламин (25,7 г, 254 ммоль) при комнатной температуре в атмосфере азота. В полученную смесь по каплям добавляли метансульфонилхлорид (26,7 г, 233 ммоль) при 0°C, затем использованную капельную воронку промывали толуолом (10 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем при 65°C в течение 22 часов, и охлаждали до комнатной температуры. Добавляли в реакционную смесь водный раствор бикарбоната натрия (105 мл) и перемешивали, органический слой отделяли. Органический слой промывали водой (105 мл), отделяли водный слой и затем удаляли из органического слоя растворитель в вакууме. Добавляли в остаток толуол (105 мл), и полученный толуольный раствор упаривали. Эту операцию повторяли еще два раза, получая толуольный раствор R-BCAB [9] (75,3 г, 212 ммоль теоретически). Полученный толуольный раствор R-BCAB использовали на следующей стадии, считая выход равным 100%.
Сырой продукт R-BCAB, который был синтезирован по описанной выше методике, упаривали и сушили, затем регистрировали спектры ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 7,28-7,11 (5H, м), 4,24-4,11 (1H, м), 3,80 (2H, д, J = 3,6 Гц), 3,24 (2H, д, J = 3,6 Гц), 2,98-2,78 (2H, м), 1,46-1,37 (12H, м).
МС: m/z = 298 [M+H]+
Стадия 3
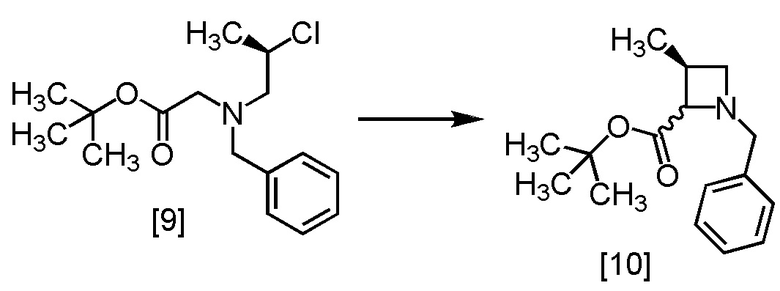
В толуольный раствор R-BCAB [9] (75,3 г, 212 ммоль) добавляли тетрагидрофуран (88,0 мл) и 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинон (42,0 мл) при комнатной температуре в атмосфере азота. В полученный раствор по каплям добавляли раствор лития бис(триметилсилил)амид/тетрагидрофуран (195 мл, 233 ммоль) при 0°C, затем использованную капельную воронку промывали тетрагидрофураном (17,0 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при 0°C в течение 1 часа, затем нагревали до комнатной температуры. Затем добавляли в реакционную смесь воду (175 мл) и толуол (175 мл), смесь перемешивали, отделяли органический слой. Полученный органический слой промывали водным раствором хлорида аммония (175 мл) и затем водой (175 мл), и растворитель удаляли из органического слоя в вакууме. Этилацетат (175 мл) добавляли в остаток, и полученный этилацетатный раствор упаривали. Эту операцию повторяли еще два раза, получая этилацетатный раствор S-MABB [10] (66,5 г, 212 ммоль теоретически). Полученный этилацетатный раствор S-MABB использовали на следующей стадии, считая выход равным 100%.
Сырой продукт S-MABB [10], который был синтезирован по описанной выше методике, упаривали и сушили, затем регистрировали спектры ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 7,28-7,25 (10H, м), 3,75 (1H, д, J = 12,7 Гц), 3,68 (1H, д, J = 1,4 Гц), 3,66 (1H, д, J = 6,7 Гц), 3,46 (2H, д, J = 12,7 Гц), 3,30-3,17 (2H, м), 2,95 (1H, дд, J = 6,2, 1,2 Гц), 2,77 (1H, дд, J = 6,1, 2,2 Гц), 2,65-2,55 (1H, м), 2,48-2,40 (2H, м), 1,35 (9H, с), 1,35 (9H, с), 1,12 (3H, д, J = 7,2 Гц), 1,09 (3H, д, J = 6,2 Гц).
МС: m/z = 262 [M+H]+
Содержание Cl (йонная хроматография): 11,9% (11,9% теоретически)
Стадия 4
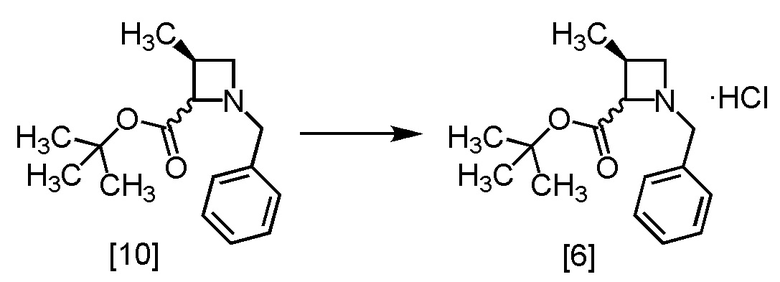
В полученный этилацетатный раствор S-MABB [10] (66,5 г, 212 ммоль теоретически) добавляли этилацетат (175 мл) и активированный уголь (3,5 г) в атмосфере азота, и затем смесь перемешивали при комнатной температуре в течение 2 часов. Активированный уголь удаляли фильтрованием, и осадок на фильтре промывали этилацетатом (175 мл). Промывные растворы добавляли в фильтрат. В раствор добавляли кристалл S-MABB-HC (17,5 мг), который был получен по описанной в настоящем тексте методике, при 0°C, и затем прикапывали 4M раствор хлороводорода в этилацетате (53,0 мл, 212 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 17 часов, затем выпавший твердый осадок собирали на фильтре и промывали этилацетатом (70 мл). Полученный влажный твердый продукт сушили в вакууме, получая S-MABB-HC [6] (48,3 г, 162 ммоль, выход: 76,4%).
Регистрировали спектры ЯМР, МС и определяли содержание Cl для S-MABB-HC [6], который был синтезирован по описанной выше методике.
1H-ЯМР (ДМСО-d6) δ: 11,08 (1H, ушир.с), 10,94 (1H, ушир.с), 7,52-7,42 (10H, м), 5,34 (1H, т, J = 8,4 Гц), 4,90 (1H, ушир.с), 4,45-4,10 (5H, м), 3,92-3,49 (3H, ушир.м), 3,10-2,73 (2H, ушир.м), 1,35 (9H, с), 1,29 (9H, с), 1,24 (3H, д, J = 6,7 Гц), 1,17 (3H, д, J = 7,4 Гц).
МС: m/z = 262 [M+H-HCl]+
Пример 5. Получение S-MACB-HC (соединение [11])
В раствор S-MABB-HC [6] (5,0 г, 16,8 ммоль) в метаноле (15,0 мл) добавляли 5% палладий на угле (производство Kawaken Fine Chemicals Co., Ltd., PH type, 54,1% содержание воды, 1,0 г) при комнатной температуре в атмосфере азота. Реакционный сосуд заполняли водородом, реакционную смесь перемешивали под давлением водорода 0,4 МПа при комнатной температуре в течение 12 часов, заменяли водород в реакционном сосуде на азот, и затем 5% палладий на угле удаляли фильтрованием. Реакционный сосуд и 5% палладий на угле промывали метанолом (10 мл). Промывные растворы добавляли в фильтрат, получая метанольный раствор S-MACB-HC [11] (24,8 г, 16,8 ммоль теоретически). Полученный метанольный раствор S-MACB-HC использовали на следующей стадии, считая выход равным 100%.
Сырой продукт S-MACB-HC, который был синтезирован по описанной выше методике, упаривали и сушили, затем регистрировали спектры ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 9,60 (ушир.с, 1H), 4,97 (д, 1H, J = 9,2 Гц), 4,61 (д, 1H, J = 8,4 Гц), 4,01 (дд, 1H, J = 10,0, 8,4 Гц), 3,78-3,74 (м, 1H), 3,54 (дд, 1H, J = 9,6, 8,4 Гц), 3,35 (дд, 1H, J = 10,0, 6,0 Гц), 3,15-3,03 (м, 1H), 3,00-2,88 (м, 1H), 1,49 (с, 9H), 1,47 (с, 9H), 1,22 (д, 3H, J = 6,8 Гц), 1,14 (д, 3H, J = 7,2 Гц).
МС: m/z = 172 [M+H]+ (свободная форма)
Пример 6. Получение S-ZMAB (соединение [12])
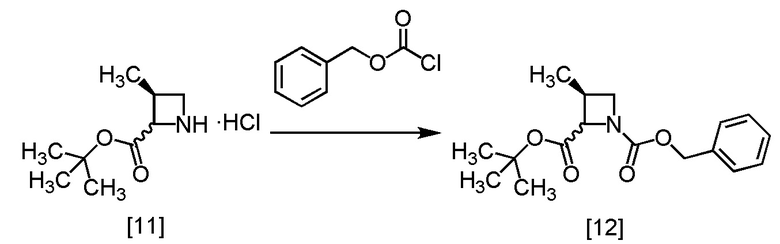
В метанольный раствор S-MACB-HC [11] (24,8 г, 16,8 ммоль теоретически) по каплям добавляли N,N-диизопропилэтиламин (4,8 г, 36,9 ммоль) при комнатной температуре в атмосфере азота, затем использованную капельную воронку промывали тетрагидрофураном (2,5 мл), и промывные растворы добавляли в реакционную смесь. В результирующую реакционную смесь по каплям добавляли бензилхлорформиат (3,0 г, 17,6 ммоль) при 0°C, затем использованную капельную воронку промывали тетрагидрофураном (2,5 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при 0°C в течение 1 часа, и растворитель удаляли в вакууме. Толуол (25,0 мл) и водный раствор лимонной кислоты (25,0 мл) добавляли в остаток, затем смесь перемешивали и отделяли органический слой. Полученный органический слой промывали водным раствором бикарбоната натрия (25,0 мл) и затем водой (25,0 мл), и удаляли растворитель из органического слоя в вакууме. Толуол (15,0 мл) добавляли в остаток, и полученный толуольный раствор упаривали. Эту операцию повторяли еще раз, получая толуольный раствор S-ZMAB [12] (6,9 г, 16,8 ммоль теоретически). Полученный толуольный раствор S-ZMAB использовали на следующей стадии, считая выход равным 100%.
Сырой продукт S-ZMAB, который был синтезирован по описанной выше методике, упаривали и сушили, затем регистрировали спектры ЯМР и МС.
1H-ЯМР (CDCl3) δ: 7,38-7,28 (м, 10H), 5,16-5,04 (м, 4H), 4,60 (д, 1H, J = 9,2 Гц), 4,18-4,12 (м, 2H), 4,04 (т, 1H, J = 8,6 Гц), 3,66 (дд, 1H, J = 7,6, 7,2 Гц), 3,50 (дд, 1H, J = 8,0, 5,2 Гц), 3,05-2,94 (м, 1H), 2,60-2,50 (м, 1H), 1,43 (ушир.с, 18H), 1,33 (д, 3H, J = 6,5 Гц), 1,15 (д, 3H, J = 7,2 Гц).
МС: m/z = 328 [M+Na]+
Пример 7. Получение RS-ZMBB (соединение [13])
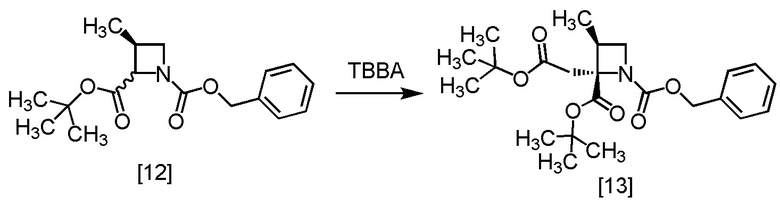
В толуольный раствор S-ZMAB [12] (6,9 г, 16,8 ммоль) добавляли тетрагидрофуран (15,0 мл) при комнатной температуре в атмосфере азота. По каплям добавляли раствор лития бис(триметилсилил)амид/тетрагидрофуран (14,7 мл, 17,6 ммоль) в толуольный раствор при -70°C. Использованную капельную воронку промывали тетрагидрофураном (2,5 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при -70°C в течение 6 часов, и затем по каплям добавляли в реакционную смесь раствор TBBA (3,4 г, 17,6 ммоль) в тетрагидрофуране (2,5 мл) при -70°C. Использованную капельную воронку промывали тетрагидрофураном (2,5 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при -70°C в течение 1 часа, затем нагревали до комнатной температуры. В реакционную смесь добавляли водный раствор хлорида аммония (25 мл) и толуол (25 мл), затем смесь перемешивали, отделяли органический слой. Полученный органический слой промывали водным раствором лимонной кислоты (25 мл x 2), водным раствором бикарбоната натрия (25 мл) и водой (25 мл), затем растворитель удаляли из органического слоя в вакууме. Ацетонитрил (15 мл) добавляли в остаток, и полученный ацетонитрильный раствор упаривали. Эту операцию повторяли еще два раза. Добавляли в остаток ацетонитрил (15 мл) и активированный уголь (0,25 г), смесь перемешивали при комнатной температуре в течение 2 часов. Активированный уголь удаляли фильтрованием, реакционную колбу и осадок на фильтре промывали ацетонитрилом (10 мл). Промывные растворы добавляли в фильтрат, затем фильтрат упаривали в вакууме, получая ацетонитрильный раствор RS-ZMBB [13] (13,2 г, 16,8 ммоль теоретически). Полученный ацетонитрильный раствор RS-ZMBB использовали на следующей стадии, считая выход равным 100%.
Сырой продукт RS-ZMBB, который был синтезирован по описанной выше методике, упаривали и сушили, затем регистрировали спектры ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 7,38-7,29 (м, 5H), 5,09-4,96 (м, 2H), 3,91 (т, 0,4H, J = 8,0 Гц), 3,79 (т, 0,6H, J = 8,0 Гц), 3,55 (т, 0,4H, J = 7,2 Гц), 3,46 (т, 0,6H, J = 7,5 Гц), 3,14-3,04 (м, 1H), 2,83-2,72 (м, 2H), 1,38 (ушир.с, 9H), 1,37 (ушир.с, 3,6H), 1,34 (ушир.с, 5,4H), 1,12-1,09 (м, 3H).
МС: m/z = 420 [M+H]+
Пример 8. Получение RS-ZMAA-DN·2H2O (соединение [14])
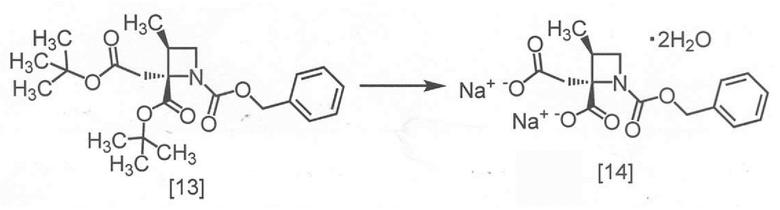
В полученный ацетонитрильный раствор RS-ZMBB [13] (13,2 г, 16,8 ммоль теоретически) добавляли ацетонитрил (15 мл) при комнатной температуре в атмосфере азота. Моногидрат п-толуолсульфокислоты (6,4 г, 33,6 ммоль) добавляли в раствор при комнатной температуре. Реакционную смесь перемешивали при 50°C в течение 12 часов, затем охлаждали до комнатной температуры и по каплям добавляли воду (7,5 мл) в реакционную смесь. Реакционную смесь охлаждали до 0°C, затем по каплям добавляли 4 моль/л водный раствор гидроксида натрия (17,6 мл, 70,5 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 1 часа, по каплям добавляли ацетонитрил (75 мл) при комнатной температуре, и реакционную смесь перемешивали в течение 3 часов. Выпавший твердый осадок собирали на фильтре и промывали смесью ацетонитрил : вода = 4 : 1 (10 мл) и затем ацетонитрилом (10 мл). Полученный влажный твердый продукт сушили в вакууме, получая RS-ZMAA-DN·2H2O [14] (5,2 г, 13,4 ммоль, выход: 85,4%).
Регистрировали спектры ЯМР и МС, определяли содержание Na и содержание воды для RS-ZMAA-DN·2H2O, который был получен по описанной выше методике.
1H-ЯМР (ДМСО-d6) δ: 7,32-7,22 (м, 5H), 4,97 (д, 1H, J = 12,7 Гц), 4,84 (д, 1H, J = 12,7 Гц), 3,79 (т, 1H, J = 8,0 Гц), 3,29 (д, 1H, J = 14,8 Гц), 3,16-3,12 (м, 1H), 2,17-2,09 (м, 2H), 1,07 (д, 3H, J = 6,9 Гц).
МС: m/z = 352 [M+H]+ (безводный)
Содержание Na (ионная хроматография): 13,3% (после корректировки на содержание воды) (13,1% теоретически)
Содержание воды (метод Карла Фишера): 9,8% (9,3% теоретически)
Пример 9. Получение RS-ZMAA (соединение [15])
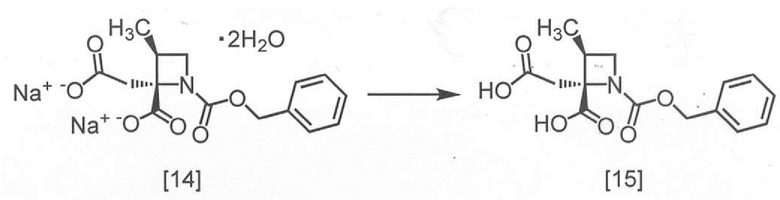
В 1 моль/л соляную кислоту (180 мл) добавляли RS-ZMAA-DN·2H2O [14] (30 г, 77,5 ммоль) и ацетонитрил (60 мл), и смесь перемешивали при комнатной температуре в течение примерно 15 минут. Этилацетат (240 мл) добавляли в реакционную смесь, затем смесь перемешивали и отделяли органический слой. Органический слой промывали 10%-ным раствором хлорида натрия (60 мл x 2). Органический слой перемешивали с сульфатом магния (6 г), сульфат магния удаляли фильтрованием, и осадок на фильтре промывали этилацетатом (60 мл). Фильтрат и промывные растворы объединяли, и растворитель удаляли в вакууме. Тетрагидрофуран (240 мл) добавляли в остаток, и полученный тетрагидрофурановый раствор упаривали. Эту операцию повторяли еще два раза. Тетрагидрофуран (60 мл) добавляли в остаток, получая тетрагидрофурановый раствор RS-ZMAA [15]. Полученный тетрагидрофурановый раствор RS-ZMAA использовали на следующей стадии, считая выход равным 100%.
Регистрировали спектры ЯМР и МС для RS-ZMAA, который был получен по описанной выше методике.
1H-ЯМР (ДМСО-D6) δ: 7,35-7,28 (м, 5H), 5,06-4,94 (м, 2H), 3,86 (дт, 1H, J = 48,4, 7,9 Гц), 3,50 (дт, 1H, J = 37,9, 7,4 Гц), 3,16-3,02 (ушир.м, 1H), 2,91-2,77 (ушир.м, 2H), 1,08 (д, 3H, J = 6,9 Гц).
МС: m/z = 308 [M+H]+
Пример 10. Получение RS-ZMOO (соединение [16])
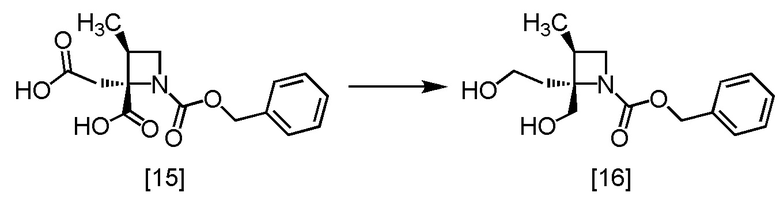
В тетрагидрофурановый раствор RS-ZMAA [15] (25,8 ммоль теоретически) добавляли тетрагидрофуран (50 мл) в атмосфере азота. Комплексный эфират трехфтористого бора (4,40 г) по каплям добавляли при температуре от 0°C до 5°C. Использованную капельную воронку промывали тетрагидрофураном (5 мл), и промывные растворы добавляли в реакционную смесь. В реакционную смесь по каплям добавляли 1,2 моль/л раствор комплекса боран-тетрагидрофуран (43,0 мл) при температуре от 0°C до 5°C, и смесь перемешивали при температуре от 0°C до 5°C в течение примерно 30 минут, затем дополнительно перемешивали при комнатной температуре в течение ночи. В реакционную смесь по каплям добавляли 1,2 моль/л раствор комплекса боран-тетрагидрофуран (21,1 мл) при температуре от 0°C до 5°C, и затем реакционную смесь перемешивали при комнатной температуре в течение ночи. После перемешивания по каплям добавляли в реакционную смесь воду (40 мл) при температуре от 0°C до 15°C. В реакционную смесь добавляли бикарбонат натрия (5,42 г) при температуре от 0°C до 15°C. Оставшийся в емкости бикарбонат натрия смывали водой (10 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем добавляли толуол (50 мл), и реакционную смесь перемешивали дополнительно. Органический слой отделяли. Полученный органический слой промывали 10%-ным раствором хлорида натрия (20 мл x 1), смесью (x3) 5%-ного водного раствора бикарбоната натрия (20 мл) и 10%-ного раствора хлорида натрия (20 мл), смесью (x1) 5%-ного водного раствора гидросульфата калия (10 мл) и 10%-ного раствора хлорида натрия (10 мл), и затем 10%-ным раствором хлорида натрия (20 мл x 2). Органический слой перемешивали с сульфатом магния (8,9 г), сульфат магния удаляли фильтрованием, и осадок на фильтре промывали толуолом (20 мл). Промывные растворы добавляли в фильтрат, и фильтрат упаривали в вакууме. В остаток от упаривания добавляли толуол (80 мл). Раствор упаривали в вакууме и добавляли толуол (15 мл), получая толуольный раствор RS-ZMOO [16]. Полученный толуольный раствор RS-ZMOO использовали на следующей стадии, считая выход равным 100%.
Регистрировали спектры ЯМР и МС для RS-ZMOO, который был получен по описанной выше методике.
1H-ЯМР (CDCl3) δ: 7,39-7,30 (м, 5H), 5,10 (с, 2H), 4,15-4,01 (ушир.м, 2H), 3,83-3,73 (ушир.м, 3H), 3,48 (дд, 1H, J = 8,3, 6,4 Гц), 2,59-2,50 (ушир.м, 1H), 2,46-2,40 (ушир.м, 1H), 2,07-1,99 (м, 1H), 1,14 (д, 3H, J = 7,2 Гц).
МС: m/z = 280 [M+H]+
Пример 11. Получение RS-ZMSS (соединение [17])
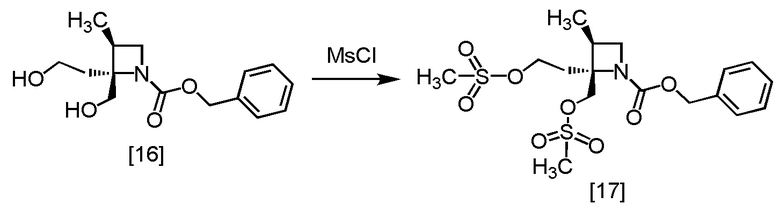
В толуольный раствор RS-ZMOO [16] (23,7 ммоль теоретически) добавляли толуол (55 мл) в атмосфере азота, по каплям добавляли триэтиламин (5,27 г) при температуре от -10°C до 10°C, использованную капельную воронку промывали толуолом (1,8 мл), и промывные растворы добавляли в реакционную смесь. В полученную реакционную смесь по каплям добавляли метансульфонилхлорид (5,69 г) при температуре от -10°C до 10°C, затем использованную капельную воронку промывали толуолом (1,8 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при температуре от 0°C до 10°C в течение примерно 2 часов, затем по каплям добавляли воду (28 мл) при температуре от 0°C до 20°C. Реакционную смесь перемешивали при температуре от 0°C до 20°C в течение примерно 30 минут, и отделяли органический слой. Полученный органический слой промывали два раза 10%-ным раствором хлорида натрия (18 мл). Органический слой перемешивали с сульфатом магния (2,75 г), сульфат магния удаляли фильтрованием, и осадок на фильтре промывали толуолом (18 мл). Промывные растворы добавляли в фильтрат и удаляли растворитель из фильтрата в вакууме. В остаток от упаривания добавляли толуол до 18 мл, получая толуольный раствор RS-ZMSS [17]. Полученный толуольный раствор RS-ZMSS использовали на следующей стадии, считая выход равным 100%.
Регистрировали спектры ЯМР и МС для RS-ZMSS, который был получен по описанной выше методике.
1H-ЯМР (ДМСО-D6) δ: 7,37-7,27 (ушир.м, 5H), 5,10-4,98 (м, 2H), 4,58-4,22 (ушир.м, 4H), 3,84 (дт, 1H, J = 45,6, 8,1 Гц), 3,48-3,33 (ушир.м, 1H), 3,17-3,10 (м, 6H), 2,81-2,74 (ушир.м, 1H), 2,22-2,12 (м, 2H).
МС: m/z = 436 [M+H]+
Пример 12. Получение SR-ZMDB (соединение [18])
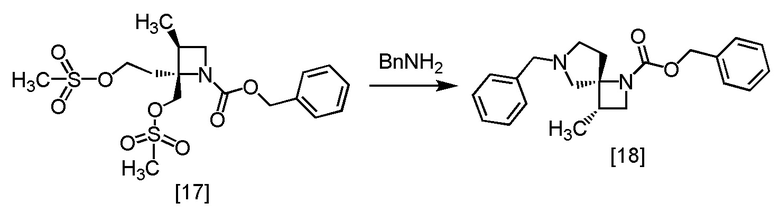
В толуольный раствор RS-ZMSS [17] (23,7 ммоль теоретически) добавляли толуол (55 мл) в атмосфере азота, по каплям добавляли бензиламин (17,8 г) при комнатной температуре, использованную капельную воронку промывали толуолом (9,2 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при комнатной температуре в течение примерно 1 часа, при температуре от 55°C до 65°C в течение примерно 3 часов, и затем при температуре от 70°C до 80°C в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры, по каплям добавляли 10%-ный раствор NaCl (28 мл), и реакционную смесь перемешивали при комнатной температуре в течение примерно 30 минут. Толуол (37 мл) добавляли в реакционную смесь, смесь перемешивали и отделяли органический слой. Полученный органический слой промывали смесью (x 2) 10%-ного раствора хлорида натрия (18 мл) и уксусной кислоты (2,84 г), затем 10%-ным раствором хлорида натрия (11 мл x 1). Растворитель из органического слоя удаляли в вакууме до половины объема и добавляли в остаток от упаривания уксусный ангидрид (1,45 г) при комнатной температуре. Смесь перемешивали в течение примерно 3 часов. В реакционную смесь добавляли по каплям раствор гидросульфата калия (3,87 г) и воду (92 мл) при комнатной температуре. Реакционную смесь перемешивали, затем отделяли водный слой. Полученный водный слой промывали толуолом (18 мл), и толуолом (73 мл), затем добавляли в водный слой бикарбонат натрия (6,56 г) при комнатной температуре, и смесь перемешивали. Органический слой отделяли и промывали 10%-ным раствором хлорида натрия (11 мл). Органический слой перемешивали с сульфатом магния (2,75 г) и удаляли сульфат магния фильтрованием. Осадок на фильтре промывали толуолом (18 мл), промывные растворы добавляли в фильтрат, затем фильтрат упаривали в вакууме. Добавляли в остаток от упаривания толуол (44 мл), получая толуольный раствор SR-ZMDB [18]. Полученный толуольный раствор SR-ZMDB использовали на следующей стадии, считая выход равным 100%.
1H-ЯМР (CDCl3) δ: 7,35-7,20 (м, 10H), 5,08 (д, 2H, J = 23,6 Гц), 3,94 (кв, 1H, J = 7,9 Гц), 3,73-3,42 (ушир.м, 2H), 3,30-3,23 (м, 1H), 3,05 (дд, 1H, J = 19,7, 9,5 Гц), 2,79 (дт, 1H, J = 69,6, 6,1 Гц), 2,57-2,32 (ушир.м, 4H), 1,96-1,89 (м, 1H), 1,09 (д, 3H, J = 6,9 Гц).
МС: m/z = 351 [M+H]+
Пример 13. Получение SR-MDOZ (соединение [19])
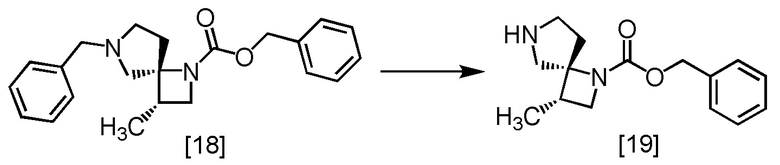
В раствор 1-хлорэтилхлорформиата (3,72 г) и толуола (28 мл) по каплям добавляли раствор SR-ZMDB [18] в толуоле (соответствует 23,7 ммоль) в атмосфере азота в диапазоне температур от 0°C до 10°C. Капельную воронку промывали толуолом (4,6 мл), и промывные растворы добавляли в реакционную смесь. В реакционную смесь добавляли триэтиламин (718 мг) в диапазоне температур от 0°C до 10°C, и смесь перемешивали в диапазоне температур от 15°C до 25°C в течение примерно 2 часов. Затем добавляли метиловый спирт (46 мл), и смесь перемешивали в диапазоне температур от 50°C до 60°C еще примерно 2 часа. Растворитель удаляли из реакционной смеси при пониженном давлении, так чтобы остаток составлял примерно 37 мл или меньше. В остаток от упаривания по каплям добавляли 2 моль/л раствор соляной кислоты (46 мл) в диапазоне температур от 15°C до 20°C, и смесь перемешивали. Затем отделяли водный слой. Полученный водный слой промывали толуолом (28 мл) два раза. В водный слой добавляли 20%-ный раствор хлорида натрия (46 мл) и тетрагидрофуран (92 мл), и затем по каплям добавляли 8 моль/л водный раствор гидроксида натрия (18 мл) в диапазоне температур от 0°C до 10°C. Органический слой отделяли и промывали 20%-ным раствором хлорида натрия (18 мл) два раза, затем растворитель из органического слоя удаляли при пониженном давлении. Процедуру с добавлением тетрагидрофурана (92 мл) в остаток от упаривания и упариванием полученной смеси при пониженном давлении повторяли еще два раза. Остаток от упаривания растворяли в тетрагидрофуране (92 мл) и добавляли сульфат магния (2,75 г). Смесь перемешивали и отфильтровывали сульфат магния. Отфильтрованный остаток промывали тетрагидрофураном (28 мл), фильтрат и промывные растворы объединяли и удаляли растворитель при пониженном давлении. Остаток от упаривания разводили тетрагидрофураном примерно до 20 мл, получая раствор SR-MDOZ [19] в тетрагидрофуране (чистое количество: 4,01 г, 15,4 моль) с выходом 65,0%.
SR-MDOZ, который был синтезирован по описанной выше методике, упаривали и сушили, затем регистрировали спектры ЯМР и МС.
1H-ЯМР (CDCl3) δ: 7,37-7,28 (м, 5H), 5,08 (дд, 2H, J = 16,8, 12,8 Гц), 4,00 (дд, 1H, J = 17,1, 8,3 Гц), 3,40-3,31 (м, 1H), 3,24 (д, 1H, J = 12,7 Гц), 3,00 (дд, 1H, J = 54,9, 12,4 Гц), 2,87-2,57 (м, 3H), 2,47-2,27 (м, 1H), 1,91-1,80 (м, 1H), 1,14 (д, 3H, J = 7,2 Гц).
МС: m/z = 261 [M+H]+
Пример 14. Получение SR-MDOZ-OX (соединение [20])
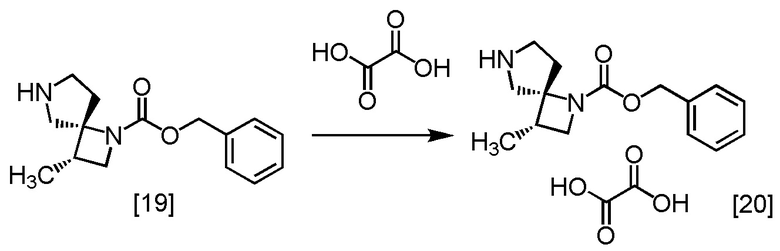
Щавелевую кислоту (761 мг) растворяли в тетрагидрофуране (40 мл) в атмосфере азота, затем по каплям добавляли раствор SR-MDOZ [19] в тетрагидрофуране (соответствует 3,84 ммоль) при комнатной температуре. В раствор добавляли при комнатной температуре кристалл SR-MDOZ-OX (1 мг), который был получен заранее по методике, аналогичной описанной выше. Смесь перемешивали при комнатной температуре в течение примерно 3,5 часов, при этом выпадал кристаллический осадок. В полученную суспензию по каплям добавляли раствор SR-MDOZ в тетрагидрофуране (3,84 ммоль) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение примерно 1 часа. Суспензию нагревали и перемешивали при температуре от 50°C до 60°C в течение примерно 2 часов, и затем перемешивали при комнатной температуре в течение ночи. Смесь фильтровали, влажные кристаллы промывали тетрагидрофураном (10 мл) и сушили при пониженном давлении, получая SR-MDOZ-OX [20] (2,32 г, 6,62 моль) с выходом 86,2%.
Регистрировали спектры ЯМР и МС и проводили элементный анализ для SR-MDOZ-OX, который был синтезирован по описанной выше методике.
1H-ЯМР (ДМСО-D6) δ: 7,37-7,30 (м, 5H), 5,15-5,01 (м, 2H), 3,92 (дт, 1H, J = 43,5, 8,4 Гц), 3,48-3,12 (ушир.м, 5H), 2,67-2,56 (м, 1H), 2,46-2,35 (м, 1H), 2,12-2,05 (м, 1H), 1,13 (д, 3H, J = 6,9 Гц).
МС: m/z = 261 [M+H]+
Элементный анализ: C 58,4 вес.%, H 6,4 вес.%, N 7,9% вес.% (Теоретическое значение C 58,3 вес.%, H 6,3 вес.%, N 8,0 вес.%).
Пример 15. Получение SR-MDPZ (соединение [21])
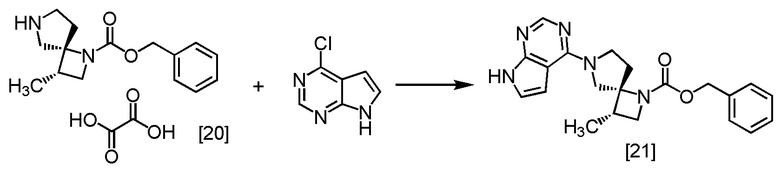
К SR-MDOZ-OX [20] (12,0 г, 34,2 ммоль) добавляли этанол (36 мл) в атмосфере азота, и затем последовательно добавляли воду (72 мл), CPPY (5,36 г, 34,9 ммоль) и K3PO4 (21,8 г, 103 ммоль). Реакционную смесь перемешивали при 80°C в течение 5 часов, затем охлаждали до 40°C. Добавляли толуол (120 мл) при 40°C и отделяли органический слой. Полученный органический слой промывали 20%-ным водным раствором карбоната калия (48 мл), затем промывали водой (48 мл) два раза. Растворитель из органического слоя удаляли при пониженном давлении. Добавляли трет-бутанол (60 мл) в остаток от упаривания. Операцию упаривания повторяли три раза. В остаток от упаривания добавляли трет-бутанол (36 мл) и получали раствор SR-MDPZ [21] в трет-бутаноле (61,1 г, соответствует 34,2 ммоль). Полученный раствор SR-MDPZ в трет-бутаноле использовали на следующей стадии, считая выход равным 100%.
SR-MDPZ, который был синтезирован по описанной выше методике, получали в виде твердого вещества после обработки смесью этилацетата и н-гептана, для него регистрировали спектры ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 11,59 (ушир.с, 1H), 8,08 (с, 1H), 7,41-7,26 (ушир.м, 3H), 7,22-7,08 (ушир.м, 3H), 6,64-6,51 (ушир.м, 1H), 5,07-4,91 (ушир.м, 2H), 4,09-3,67 (ушир.м, 5H), 3,47-3,32 (ушир.м, 1H), 2,67-2,55 (ушир.м, 2H), 2,21-2,15 (ушир.м, 1H), 1,11 (д, 3H, J = 6,9 Гц).
МС: m/z = 378 [M+H]+
Пример 16. Получение SR-MDOP (соединение [1])
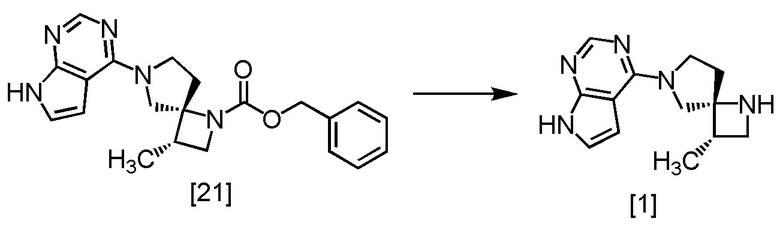
В раствор SR-MDPZ [21] в трет-бутаноле (соответствует 34,2 ммоль) в атмосфере азота добавляли формиат аммония (10,8 г, 171 ммоль), воду (60 мл) и 10% палладий на угле (производство Kawaken Fine Chemicals Co., Ltd., type M, 52,6% содержание воды, 1,20 г). Реакционную смесь перемешивали при 40°C в течение 13 часов и затем охлаждали до комнатной температуры. Нерастворенные частицы отфильтровывали. Реакционный сосуд и осадок на фильтре промывали трет-бутанолом (24 мл), и в промывные растворы и фильтрат добавляли 8M водный раствор гидроксида натрия (25,7 мл, 205 ммоль) и хлорида натрия (13,2 г). Реакционную смесь перемешивали при 50°C в течение 2 часов, затем добавляли толуол (84 мл) при комнатной температуре и отделяли органический слой. Полученный органический слой промывали 20%-ным раствором хлорида натрия (60 мл) и добавляли безводный сульфат натрия. Смесь перемешивали, затем сульфат натрия отфильтровывали. Осадок на фильтре промывали смесью (48 мл) толуол:трет-бутанол = 1:1, Фильтрат и промывные растворы объединяли и удаляли растворитель при пониженном давлении. В остаток от упаривания добавляли толуол (60 мл), и смесь перемешивали при 50°C в течение 2 часов. Затем растворитель удаляли при пониженном давлении. В остаток от упаривания добавляли толуол (60 мл), и полученную смесь упаривали. В остаток от упаривания добавляли толуол (48 мл), смесь перемешивали при комнатной температуре в течение 1 часа и затем при охлаждении на ледяной бане 1 час. Выпавший твердый осадок отфильтровывали и промывали толуолом (24 мл). Полученный влажный твердый продукт сушили при пониженном давлении, получая SR-MDOP [1] (7,07 г, 29,1 ммоль) с выходом 84,8%.
Регистрировали спектры ЯМР и МС для SR-MDOP, который был синтезирован по описанной выше методике.
1H-ЯМР (ДМСО-d6) δ: 11,57 (ушир.с, 1H), 8,07 (с, 1H), 7,10 (д, 1H, J = 3,2 Гц), 6,58 (д, 1H, J = 3,2 Гц), 3,92-3,59 (ушир.м, 4H), 3,49 (дд, 1H, J = 8,3, 7,2 Гц), 2,93 (дд, 1H, J = 7,2, 6,1 Гц), 2,61-2,53 (м, 2H), 2,12-2,01 (ушир.м, 2H), 1,10 (д, 3H, J = 6,9 Гц).
МС: m/z = 244 [M+H]+
Пример 17. Рентгеноструктурный анализ монокристалла
Монокристалл сокристалла (соединения [3-1]) соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) получали и анализировали методом рентгеноструктурного анализа.
Метод получения монокристалла
К сокристаллу соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) (10 мг) добавляли ацетонитрил (1 мл), и смесь нагревали до 70°C в течение 2 часов. Полученный раствор отфильтровывали через мембранный фильтр (PTFE, 13 мкм). Фильтрат оставляли при комнатной температуре на 4 дня, получая монокристалл.
Рентгеноструктурный анализ проводили на приборе Beamline BL2S1 в исследовательском центре Aichi Synchrotron Radiation Center.
Условия проведения анализа
Длина волны: 0,74998 Å
Размер луча: 100 мкм
Длина камеры: 90 мм
Смещение: 70 мм (вертикальное направление)
Угол отклонения: 2°
Диапазон углов измерения: 180°
Температура съемки: 100K (-173,15°C)
Результаты, проанализированные указанной ниже программой анализа данных, приведены в следующей далее таблице
Программа анализа данных
Регистрация данных, обработка данных дифракции: XDS
Анализ структуры кристалла: SHELX97
Уточнение структуры: Метод наименьших квадратов в полноматричном приближении по F2
4 (3,5-диметилпиразол)
1 (3,5-диметилпиразол)
[I > 2σ1]
Исследованный монокристалл представляет собой сокристалл, имеющий водородную связь между соединением A (соединение [4]) и 3,5-диметилпиразолом. ORTEP изображение результирующего сокристалла соединения A (соединение [4]) с 3,5-диметилпиразолом (2:1, мольное соотношение) показано на фиг. 5.
Промышленная применимость
Сокристалл соединения A (соединение [4]) с 3,5-диметилпиразолом (например, соединение [3a]) по настоящему изобретению можно применять для получения соединения A (соединение [4]). В настоящем изобретении описан способ стабильного получения этого сокристалла с хорошей химической чистотой. В настоящем изобретении также описан способ стабильного получения соединения A (соединение [4]) с хорошей химической чистотой. Кроме того, способ получения по настоящему изобретению можно применять для промышленного производства в большом количестве, поскольку сокристалл можно выделять напрямую из реакционной смеси.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА ЯНУС-КИНАЗЫ | 2017 |
|
RU2838992C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 7Н-ПИРРОЛО[2,3-D] ПИРИМИДИНА И ЕГО ИНТЕРМЕДИАТА | 2016 |
|
RU2755618C2 |
| ИНГИБИТОРЫ КИНАЗ, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И ДРУГИХ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2482112C2 |
| НОВЫЙ СПОСОБ СИНТЕЗА СОЕДИНЕНИЯ ФЕНОКСИДИАМИНОПИРИМИДИНА | 2019 |
|
RU2805661C2 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| СУЛЬФОНАМИДНЫЕ ПЕРИ-ЗАМЕЩЕННЫЕ БИЦИКЛЫ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ПОРАЖЕНИЯ АРТЕРИЙ | 2005 |
|
RU2403240C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ АРИЛКОНДЕНСИРОВАННЫХ АЗАПОЛИЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2282619C9 |
| БЕНЗОКСАЗИН-ОКСАЗОЛИДИНОНОВОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ АЗОТСОДЕРЖАЩИМ ГЕТЕРОЦИКЛОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2744784C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| РАПП-ПОДДЕРЖИВАЮЩИЕ ТИАЗОЛИДИНДИОНЫ И ИХ КОМБИНАЦИИ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2010 |
|
RU2570424C2 |
Изобретение относится к области органической химии, а именно к сокристаллу 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрила с 3,5-диметилпиразолом формулы [3а], где m представляет собой любое число от 0,4 до 0,5. Также изобретение относится к способу получения соединения формулы [4] путем использования соединения формулы [3а]. Технический результат: разработан способ получения соединения формулы [4] известного ингибитора ВТК из сокристалла формулы [3а], отличающийся высокой чистотой целевого соединения и возможностью масштабирования для промышленного производства. 2 н. и 4 з.п. ф-лы, 5 ил., 16 пр.
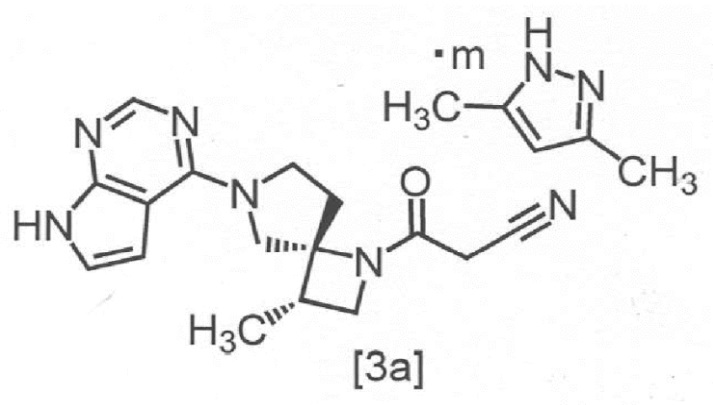
1. Сокристалл 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3,4]октан-1-ил]-3-оксопропаннитрила с 3,5-диметилпиразолом, имеющий структуру, отвечающую формуле [3a]
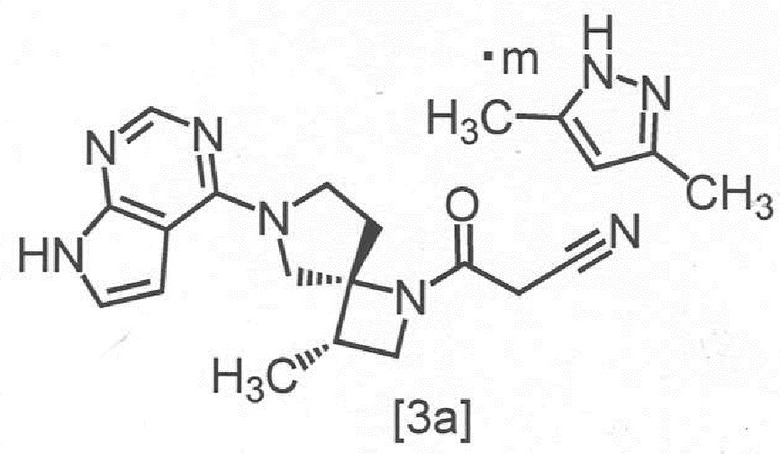 ,
,
где m представляет собой любое число от 0,4 до 0,5.
2. Сокристалл по п. 1, где m равно 0,5.
3. Сокристалл по п. 1 или 2, показывающий экстраполированную температуру начала перехода 172±5°C при анализе методом дифференциальной сканирующей калориметрии.
4. Сокристалл по любому из пп. 1-3, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один пик со значением дифракционного угла (2θ)=4,6°±0,2°, 18,6°±0,2° или 20,9°±0,2°, при анализе с применением CuKα излучения.
5. Сокристалл по любому из пп. 1-3, имеющий спектр порошковой рентгеновской дифракции, содержащий по меньшей мере один пик со значением дифракционного угла (2θ)=4,6°±0,2°, 12,6°±0,2°, 16,1°±0,2°, 18,6°±0,2° или 20,9°±0,2°, при анализе с применением CuKα излучения.
6. Способ получения соединения, имеющего формулу [4]
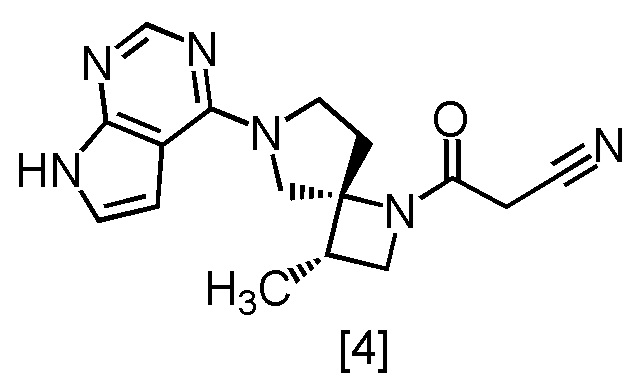 ,
,
путем реакции соединения, имеющего формулу [1]
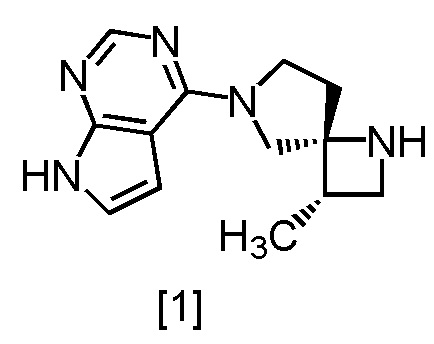 ,
,
с соединением, имеющим формулу [2]
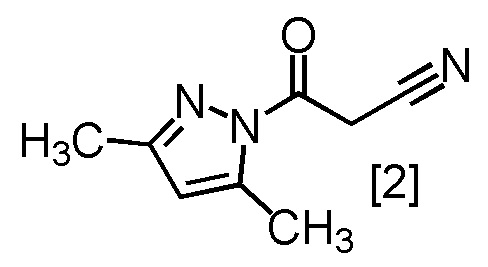 ,
,
которое находится в количестве от 0.95 до 1.2 эквивалентов относительно соединения формулы [1], при температуре от 70°С до 80°С в растворителе, включающем ацетонитрил, с получением сокристалла по любому из пп. 1-5, и
растворение сокристалла и кристаллизация соединения формулы [4] при температуре от 100°С до 117°С в растворителе, включающем 1-бутанол или 1-пропанол, где количество указанного растворителя составляет 8.5±0.5–кратное количество относительно веса сокристалла.
| EP 3181570, 30.07.2010 | |||
| LOPEZ C | |||
| et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Crystal Growth & Design, 12.2007, 7(6), с.1176-1184 | |||
| YADAV AV et al | |||
| Co-Crystals: A Novel Approach to Modify Physicochemical Properties of Active Pharmaceutical Ingredients | |||
| Indian Journal | |||

