Область техники, к которой относится изобретение
Настоящее изобретение относится к кристаллическим формам ингибитора Янус-киназы (JAK) 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила (Соединение A), а также к их композициям, способам их получения, способам их применения и способам количественного анализа.
Уровень техники
Ингибиторы Янус-киназы (JAK) представляют интерес для лечения различных заболеваний, включая аутоиммунные заболевания, воспалительные заболевания и рак. К настоящему моменту два ингибитора JAK получили одобрение Управления США по контролю за лекарствами и пищевыми продуктами (FDA). Руксолитиниб был одобрен для лечения первичного миелофиброза и истинной полицитемии (PV), а тофацитиниб был одобрен для лечения ревматоидного артрита. В литературе описаны другие и ингибиторы JAK. Соединение 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрил (Соединение A) (см. структуру ниже) является примером спироциклического ингибитора JAK, описанного в публикации заявки на патент США 2011/0136778 и международной заявке PCT/JP2016/070046.
[Хим. 1]
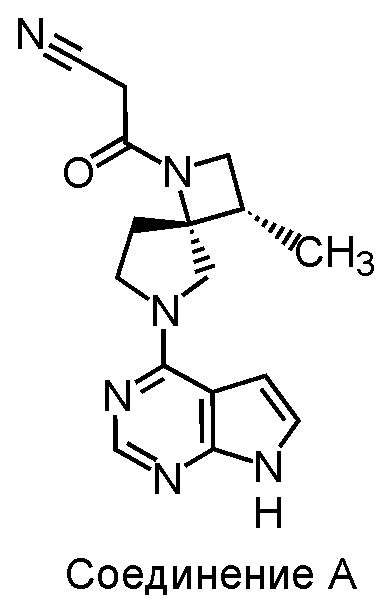
Лекарственные соединения, такие как, например, Соединение A, обычно комбинируют с другими фармацевтически приемлемыми ингредиентами для формирования композиций, подходящих для введения пациентам. Для твердых препаратов часто требуется, чтобы лекарственное соединение имело надлежащие характеристики в твердом состоянии, такие как устойчивость к нагреву и влажности, легкость обращения и другие характеристики, которые облегчают приготовление твердых дозированных форм. Соответственно, имеется постоянная потребность в твердых формах существующих лекарственных молекул. Описанные здесь кристаллические формы Соединения A направлены на решение этой задачи.
Краткое описание сущности изобретения
Настоящее изобретение относится, среди прочего, к кристаллическим Формам β и γ 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила (Соединение A).
Настоящее изобретение относится также к композициям, содержащим Форму α вместе с Формой β или Формой γ, или с ними обеими.
Настоящее изобретение относится также к фармацевтическим композициям, содержащим одну или больше кристаллических форм или композиций по настоящему изобретению.
Настоящее изобретение относится также к способам получения кристаллических форм по настоящему изобретению.
Настоящее изобретение относится также к способам оценки количества кристаллических форм по настоящему изобретению.
Настоящее изобретение относится также к способам ингибирования Янус-киназы, включающим приведение кристаллической формы по настоящему изобретению в контакт с Янус-киназой.
Настоящее изобретение относится также к способу лечения или предотвращения заболевания у пациента, включающему введение пациенту терапевтически эффективного количества кристаллической формы или композиции по настоящему изобретению.
Настоящее изобретение относится также к кристаллической форме или композиции по настоящему изобретению для применения в профилактике или терапии.
Настоящее изобретение относится также к применению кристаллической формы или композиции по настоящему изобретению для приготовления лекарственного средства для применения в терапии или профилактике.
Краткое описание чертежей
На Фиг. 1 приведен спектр порошковой рентгеновской дифракции, соответствующий Форме α.
На Фиг. 2 приведен спектр порошковой рентгеновской дифракции, соответствующий Форме β.
На Фиг. 3 приведен спектр порошковой рентгеновской дифракции, соответствующий Форме γ.
На Фиг. 4 приведена термограмма ДСК, соответствующая Форме α.
На Фиг. 5 приведена термограмма ДСК, соответствующая Форме β.
На Фиг. 6 приведена термограмма ДСК, соответствующая Форме γ.
На Фиг. 7 приведены данные термогравиметрического дифференциально-термического анализа (ТГ-ДТА), соответствующие Форме α.
На Фиг. 8 приведены данные термогравиметрического дифференциально-термического анализа (ТГ-ДТА), соответствующие Форме β.
На Фиг. 9 приведены данные термогравиметрического дифференциально-термического анализа (ТГ-ДТА), соответствующие Форме γ.
На Фиг. 10 приведены данные твердотельной 13C ЯМР-спектроскопии, соответствующие Форме α.
На Фиг. 11 приведены данные твердотельной 13C ЯМР-спектроскопии, соответствующие Форме β.
На Фиг. 12 приведены данные твердотельной 13C ЯМР-спектроскопии, соответствующие Форме γ.
На Фиг. 13 изображены наложенные спектры порошковой рентгеновской дифракции, полученные для стандартного образца Соединения А, содержащего 0, 2, 5 и 10% Формы β (см. Пример 4).
На Фиг. 14 изображены наложенные спектры порошковой рентгеновской дифракции, полученные для стандартного образца Соединения А, содержащего 0, 1, 2, 5 и 10% Формы γ (см. Пример 4).
На Фиг. 15 изображены (снизу вверх) спектры порошковой рентгеновской дифракции из Примера 6 для использовавшейся Формы α Соединения A, использовавшейся Формы β Соединения A, смеси 1:1 по массе Форм α и β до перемешивания, и кристаллов после перемешивания.
На Фиг. 16 изображены спектры порошковой рентгеновской дифракции из Примера 7 для использовавшейся Формы α Соединения A, использовавшейся Формы γ Соединения A, смеси 1:1 по массе Форм α и γ до перемешивания, и кристаллов после перемешивания.
На Фиг. 17 изображены спектры порошковой рентгеновской дифракции из Примера 8 для использовавшейся Формы γ Соединения A, использовавшейся Формы β Соединения A, смеси 1:1 по массе Форм γ и β до перемешивания, кристаллов, полученных из формамида, кристаллов из N,N-диметилформамида, и кристаллов из диметилсульфоксида.
Варианты осуществления изобретения
В настоящем изобретении описаны, среди прочего, кристаллические формы 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила, которые являются ингибитором Янус-киназы (JAK) и могут применяться для лечения различных заболеваний, связанных с повышением количества рецепторов Янус-киназы или их сверхэкспрессией. В некоторых вариантах осуществления кристаллические формы по настоящему изобретению ингибируют JAK3. В некоторых вариантах осуществления кристаллические формы по настоящему изобретению ингибируют JAK2.
Одним из аспектов настоящего изобретения является следующее.
[Пункт 1] Кристаллическая форма 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила, имеющая Форму β.
[Пункт 2] Кристаллическая форма по Пункту 1, имеющая спектр порошковой рентгеновской дифракции, содержащий характеристический пик со значением 2θ (°) примерно 11,8.
[Пункт 3] Кристаллическая форма по Пункту 1, имеющая спектр порошковой рентгеновской дифракции, содержащий два или больше характеристических пиков со значениями 2θ (°), выбранными из примерно 10,5, примерно 11,8, примерно 19,3 и примерно 22,0.
[Пункт 4] Кристаллическая форма по Пункту 1, имеющая спектр порошковой рентгеновской дифракции, содержащий три или больше характеристических пиков со значениями 2θ (°), выбранными из примерно 7,8, примерно 10,5, примерно 11,8, примерно 13,4, примерно 13,9, примерно 17,8, примерно 19,3, примерно 22,0, примерно 23,6 и примерно 28,0.
[Пункт 5] Кристаллическая форма по любому из Пунктов 1-4, имеющая ДСК-термограмму, отличающуюся наличием эндотермического пика примерно при 186°C.
[Пункт 6] Кристаллическая форма по любому из Пунктов 1-5, имеющая твердотельный 13C ЯМР спектр, который отличается наличием по меньшей мере одного пика со значением химсдвига примерно 165,1 м.д.
[Пункт 7] Кристаллическая форма по любому из Пунктов 1-5, имеющая твердотельный 13C ЯМР спектр, который отличается наличием по меньшей мере 5 пиков, выбранных из пиков со значениями химсдвигов примерно 16,5, примерно 25,8, примерно 26,5, примерно 33,1, примерно 34,8, примерно 36,7, примерно 38,8, примерно 48,2, примерно 53,4, примерно 77,7, примерно 79,5, примерно 101,2, примерно 102,6, примерно 117,5, примерно 120,6, примерно 151,1, примерно 154,3 и примерно 165,1 м.д.
[Пункт 8] Кристаллическая форма по любому из Пунктов 1-7, имеющая пространственную группу P1 со следующими параметрами элементарной ячейки:
Таблица 1
[Пункт 9] Кристаллическая форма по любому из Пунктов 1-8, имеющая чистоту по меньшей мере примерно 50%.
[Пункт 10] Кристаллическая форма по любому из Пунктов 1-8, имеющая чистоту по меньшей мере примерно 75%.
[Пункт 11] Кристаллическая форма по любому из Пунктов 1-8, имеющая чистоту по меньшей мере примерно 85%.
[Пункт 12] Кристаллическая форма по любому из Пунктов 1-8, имеющая чистоту по меньшей мере примерно 90%.
[Пункт 13] Кристаллическая форма по любому из Пунктов 1-8, имеющая чистоту по меньшей мере примерно 95%.
[Пункт 14] Кристаллическая форма 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила, имеющая Форму γ.
[Пункт 15] Кристаллическая форма по Пункту 14, имеющая спектр порошковой рентгеновской дифракции, содержащий характеристический пик со значением 2θ (°) примерно 16,5.
[Пункт 16] Кристаллическая форма по Пункту 14, имеющая спектр порошковой рентгеновской дифракции, содержащий два или больше характеристических пиков со значениями 2θ (°), выбранными из примерно 16,5, примерно 17,7, примерно 21,4, примерно 21,8 и примерно 23,1.
[Пункт 17] Кристаллическая форма по Пункту 14, имеющая спектр порошковой рентгеновской дифракции, содержащий три или больше характеристических пиков со значениями 2θ (°), выбранными из примерно 7,7, примерно 10,6, примерно 13,3, примерно 13,9, примерно 15,5, примерно 16,5, примерно 17,7, примерно 17,9, примерно 19,0, примерно 21,4, примерно 21,8, примерно 23,1, примерно 23,7 и примерно 28,1.
[Пункт 18] Кристаллическая форма по любому из Пунктов 14-17, имеющая ДСК-термограмму, отличающуюся наличием эндотермического пика примерно при 196°C.
[Пункт 19] Кристаллическая форма по любому из Пунктов 14-18, имеющая твердотельный 13C ЯМР спектр, который отличается наличием по меньшей мере одного пика со значением химсдвига примерно 162,9 м.д.
[Пункт 20] Кристаллическая форма по любому из Пунктов 14-19, имеющая твердотельный 13C ЯМР спектр, который отличается наличием по меньшей мере 5 пиков, выбранных из пиков со значениями химсдвигов примерно 16,9, примерно 26,5, примерно 32,9, примерно 36,4, примерно 48,1, примерно 53,7, примерно 78,6, примерно 102,6, примерно 116,4, примерно 117,9, примерно 121,5, примерно 151,8, примерно 154,6 и примерно 162,9 м.д.
[Пункт 21] Кристаллическая форма по любому из Пунктов 14-20, имеющая пространственную группу P21 со следующими параметрами элементарной ячейки:
Таблица 2
[Пункт 22] Кристаллическая форма по любому из Пунктов 14-21, имеющая чистоту по меньшей мере примерно 50%.
[Пункт 23] Кристаллическая форма по любому из Пунктов 14-21, имеющая чистоту по меньшей мере примерно 75%.
[Пункт 24] Кристаллическая форма по любому из Пунктов 14-21, имеющая чистоту по меньшей мере примерно 85%.
[Пункт 25] Кристаллическая форма по любому из Пунктов 14-21, имеющая чистоту по меньшей мере примерно 90%.
[Пункт 26] Кристаллическая форма по любому из Пунктов 14-21, имеющая чистоту по меньшей мере примерно 95%.
[Пункт 27] Способ получения кристаллической формы по любому из Пунктов 1-13, включающий кристаллизацию 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила из растворителя.
[Пункт 28] Способ по Пункту 27, в котором растворитель содержит 1-бутанол и ацетонитрил.
[Пункт 29] Способ по Пункту 28, в котором соотношение 1-бутанола к ацетонитрилу составляет примерно 1:3 (об/об).
[Пункт 30] Кристаллическая форма β, полученная способом по любому из Пунктов 27-29.
[Пункт 31] Способ получения кристаллической формы по любому из Пунктов 14-26, включающий превращение Формы α 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила в Форму γ.
[Пункт 32] Способ по Пункту 31, включающий перемешивание Формы α 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила в диметилформамиде (ДМФА).
[Пункт 33] Способ по Пункту 31, включающий перемешивание Формы α 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила в формамиде.
[Пункт 34] Кристаллическая форма γ, полученная способом по любому из Пунктов 31-33.
[Пункт 35] Композиция кристаллического 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила, содержащая Форму α и Форму β.
[Пункт 36] Композиция по Пункту 35, состоящая, главным образом, из Формы α и Формы β.
[Пункт 37] Композиция по Пункту 35 или 36, в которой Форма β присутствует в количестве от примерно 1 до примерно 50% мас./мас. относительно Формы α.
[Пункт 38] Композиция по Пункту 35 или 36, в которой Форма β присутствует в количестве от примерно 1 до примерно 20% мас./мас. относительно Формы α.
[Пункт 39] Композиция по Пункту 35 или 36, в которой Форма β присутствует в количестве от примерно 1 до примерно 10% мас./мас. относительно Формы α.
[Пункт 40] Композиция по Пункту 35 или 36, в которой Форма β присутствует в количестве от примерно 1 до примерно 5% мас./мас. относительно Формы α.
[Пункт 41] Композиция по любому из Пунктов 35-40, дополнительно содержащая Форму γ.
[Пункт 42] Композиция кристаллического 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила, содержащая Форму β и Форму γ.
[Пункт 43] Композиция по Пункту 42, состоящая, главным образом, из Формы β и Формы γ.
[Пункт 44] Композиция по Пункту 42 или 43, в которой Форма γ присутствует в количестве от примерно 1 до примерно 50% мас./мас. относительно Формы β.
[Пункт 45] Композиция по Пункту 42 или 43, в которой Форма γ присутствует в количестве от примерно 1 до примерно 20% мас./мас. относительно Формы β.
[Пункт 46] Композиция по Пункту 42 или 43, в которой Форма γ присутствует в количестве от примерно 1 до примерно 10% мас./мас. относительно Формы β.
[Пункт 47] Композиция по Пункту 42 или 43, в которой Форма γ присутствует в количестве от примерно 1 до примерно 5% мас./мас. относительно Формы β.
[Пункт 48] Композиция по любому из Пунктов 42-47, дополнительно содержащая Форму α.
[Пункт 49] Фармацевтическая композиция, содержащая кристаллическую форму или композицию по любому из Пунктов 1-48 и фармацевтически приемлемый носитель.
[Пункт 50] Фармацевтическая композиция по Пункту 49, дополнительно содержащая второе терапевтическое средство.
[Пункт 51] Фармацевтическая композиция по Пункту 49 или 50, которая подходит для перорального, парентерального, легочного, местного или наружного применения.
[Пункт 52] Фармацевтическая композиция по Пункту 49 или 50, которая подходит для наружного применения.
[Пункт 53] Фармацевтическая композиция по Пункту 49 или 50 в форме таблетки, капсулы, пилюли, порошка или мази.
[Пункт 54] Фармацевтическая композиция по Пункту 49 или 50 в форме порошка, подходящего для разведения в жидкости для внутривенного, внутримышечного или подкожного введения.
[Пункт 55] Фармацевтическая композиция по Пункту 49 или 50, содержащая белый мягкий парафин, твердый парафин, сквален или их смесь.
[Пункт 56] Способ ингибирования Янус-киназы, включающий приведение Янус-киназы в контакт с кристаллической формой или композицией по любому из Пунктов 1-55.
[Пункт 57] Способ по Пункту 56, в котором Янус-киназа представляет собой Янус-киназу 3 (JAK3).
[Пункт 58] Способ по Пункту 56, в котором Янус-киназа представляет собой Янус-киназу 2 (JAK2).
[Пункт 59] Способ лечения или профилактики заболевания, выбранного из отторжения органа после пересадки, реакции трансплантат-против-хозяина после пересадки, аутоиммунного заболевания, аллергических заболеваний и хронического миелопролиферативного заболевания, включающий введение млекопитающему терапевтически эффективного количества кристаллической формы или композиции по любому из Пунктов 1-55.
[Пункт 60] Способ лечения или профилактики ревматоидного артрита, псориаза, очаговой алопеции, сухости глаз, атопического дерматита, экземы или экземы рук, включающий введение млекопитающему терапевтически эффективного количества кристаллической формы или композиции по любому из Пунктов 1-55.
[Пункт 61] Кристаллическая форма или композиция по любому из Пунктов 1-55 для применения в качестве фармацевтически активного ингредиента.
[Пункт 62] Кристаллическая форма или композиция по любому из Пунктов 1-55 для применения в лечении или профилактике отторжения органа после пересадки, реакции трансплантат-против-хозяина после пересадки, аутоиммунного заболевания, аллергических заболеваний или хронического миелопролиферативного заболевания.
[Пункт 63] Кристаллическая форма по любому из Пунктов 1-55 для применения в лечении или профилактике ревматоидного артрита, псориаза, очаговой алопеции, сухости глаз, атопического дерматита, экземы или экземы рук.
[Пункт 64] Способ измерения количества Формы β, присутствующей при получении Формы α 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила, включающий измерение площади пика в спектре порошковой рентгеновской дифракции, являющегося характеристическим для Формы β, и сравнение площади этого пика со стандартом.
[Пункт 65] Способ по Пункту 64, в котором пик, характеристический для Формы β, находится примерно при 10,6° 2-тета.
[Пункт 66] Способ измерения количества Формы γ, присутствующей при получении Формы α 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила, включающий измерение площади пика в спектре порошковой рентгеновской дифракции, являющегося характеристическим для Формы γ, и сравнение площади этого пика со стандартом.
[Пункт 67] Способ по Пункту 66, в котором пик, характеристический для Формы γ, находится примерно при 16,6° 2-тета.
При использовании в настоящем документе “кристаллическая форма” означает определенную конфигурацию кристаллической решетки кристаллического соединения. Разные кристаллические формы одного и того же соединения обычно имеют разные кристаллические решетки (например, размер элементарной ячейки) и разные физические свойства, связанные с различием в кристаллических решетках, и, в некоторых случаях, имеют разное содержание воды или растворителя. Разные кристаллические решетки могут быть идентифицированы методами исследования твердого тела, такими как порошковая рентгеновская дифракция. Другие методы исследования, такие как дифференциальная сканирующая калориметрия (ДСК), термогравиметрический анализ (ТГА), динамическая сорбция пара (ДСП), твердотельный ЯМР и т.п., дополнительно помогают идентифицировать кристаллическую форму, а также помогают оценить ее стабильность и содержание воды/растворителя.
Разные кристаллические формы одного соединения могут включать как безводные формы этого соединения, так и сольватированные/гидратированные формы этого соединения, где каждая из безводных форм и сольватированных/гидратированных форм отличается от других различием в спектрах порошковой рентгеновской дифракции, указывая тем самым на различие в кристаллических решетках. В некоторых случаях одна кристаллическая форма (например, идентифицированная по уникальному спектру порошковой рентгеновской дифракции) может иметь разное содержание воды или растворителя, при этом решетка остается практически неизменной (как и спектр порошковой рентгеновской дифракции), несмотря на вариации в содержании воды и/или растворителя.
Спектр порошковой рентгеновской дифракции, состоящий из отражений (пиков), обычно рассматривается, как отпечаток пальцев данной кристаллической формы. Хорошо известно, что относительные интенсивности пиков в спектре порошковой рентгеновской дифракции могут варьироваться в зависимости, среди прочего, от методики приготовления образца, распределения кристаллов по размерам, методики закрепления образца и применяемого для анализа прибора. В некоторых случаях в зависимости от типа прибора или его настроек (например, применяется Ni фильтр или нет) могут наблюдаться новые пики или могут исчезать существующие пики. Кроме того, различия в приборах и другие факторы могут влиять на значения 2-тета. Так, значения пиков, такие как приведенные в настоящем документе, могут варьироваться на ±0,2°, ±0,1° или ±0,04° (2-тета), и термин “по существу” при использовании в контексте порошковой рентгеновской дифракции в настоящем документе охватывает перечисленные выше отклонения.
Аналогичным образом, значения температур при обсуждении результатов ДСК, ТГА или других термических экспериментов могут варьироваться примерно на ±3°C в зависимости от прибора, его настроек, подготовки образца и т.д. Таким образом, значения температуры, такие как приведенные в настоящем документе, могут варьироваться на ±3°C, и описанная в настоящем тексте кристаллическая форма, имеющая ДСК термограмму или другую термограмму, “по существу” такую же, как на любой из фигур, охватывает такие отклонения.
Кроме того, значения химических сдвигов могут варьироваться до ±0,2 м.д. в 13C ЯМР-спектрах, и термин “по существу” при использовании в контексте данных ЯМР в настоящем тексте охватывает такие отклонения.
Форма β
В настоящем изобретении описана кристаллическая форма 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила (Соединение A), имеющая Форму β. Кристаллическая форма, соответствующая Форме β, является безводной, что доказывается, например, данными ДСК и ТГ-ДТА (см. Пример 2). В некоторых вариантах осуществления Форма β имеет спектр порошковой рентгеновской дифракции, содержащий характеристический пик со значением 2θ (°) примерно 11,8. В других вариантах осуществления Форма β имеет спектр порошковой рентгеновской дифракции, содержащий два или больше характеристических пиков со значениями 2θ (°), выбранными из примерно 10,5, примерно 11,8, примерно 19,3 и примерно 22,0. В других вариантах осуществления Форма β имеет спектр порошковой рентгеновской дифракции, содержащий три или больше характеристических пиков со значениями 2θ (°), выбранными из примерно 7,8, примерно 10,5, примерно 11,8, примерно 13,4, примерно 13,9, примерно 17,8, примерно 19,3, примерно 22,0, примерно 23,6 и примерно 28,0. В других вариантах осуществления Форма β имеет спектр порошковой рентгеновской дифракции, содержащий три или больше характеристических пиков со значениями 2θ (°), выбранными из пиков, перечисленных в Таблице 6 (см. Пример 2). В других вариантах осуществления Форма β имеет спектр порошковой рентгеновской дифракции, по существу такой, как на Фиг. 2.
В некоторых вариантах осуществления Форма β имеет ДСК термограмму, которая отличается эндотермическим пиком примерно при 186°C. В некоторых вариантах осуществления Форма β имеет ДСК термограмму, которая отличается экстраполированной температурой начала перехода примерно 185°C. В других вариантах осуществления Форма β имеет ДСК термограмму, по существу такую как на Фиг. 5.
В некоторых вариантах осуществления Форма β имеет твердотельный 13C ЯМР спектр, который отличается наличием по меньшей мере одного пика со значением химсдвига примерно 165,1 м.д. В других вариантах осуществления Форма β имеет твердотельный 13C ЯМР спектр, который отличается наличием по меньшей мере 5 пиков, выбранных из пиков со значениями химсдвигов примерно 16,5, примерно 25,8, примерно 26,5, примерно 33,1, примерно 34,8, примерно 36,7, примерно 38,8, примерно 48,2, примерно 53,4, примерно 77,7, примерно 79,5, примерно 101,2, примерно 102,6, примерно 117,5, примерно 120,6, примерно 151,1, примерно 154,3 и примерно 165,1 м.д. В других вариантах осуществления Форма β имеет твердотельный 13C ЯМР спектр, по существу такой, как изображенный на Фиг. 11.
В некоторых вариантах осуществления Форма β имеет пространственную группу P1 со следующими параметрами элементарной ячейки:
Таблица 3
согласно данным рентгеноструктурного анализа монокристалла (см. Пример 5).
Форма β может иметь степень чистоты по меньшей мере примерно 50%, по меньшей мере примерно 75%, по меньшей мере примерно 85%, по меньшей мере примерно 90% или по меньшей мере примерно 95%. В некоторых вариантах осуществления Форма β по существу чистая.
Форму β можно получить кристаллизацией 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила (Соединение A) из растворителя. Например, Соединение A (например, в виде Формы α) можно комбинировать с растворителем, получая раствор или суспензию, из которой получают кристаллы Формы β. В некоторых вариантах осуществления растворитель включает нитрил, такой как ацетонитрил, или смесь 1-бутанола и ацетонитрил. В некоторых вариантах осуществления растворитель содержит примерно 1:3 (об/об) 1-бутанола и ацетонитрила, соответственно. В некоторых вариантах осуществления Соединение A можно комбинировать с растворителем и нагревать до температуры между примерно 30 и примерно 50°C. В некоторых вариантах осуществления смесь можно нагревать до температуры между примерно 35 и примерно 45°C, или примерно до 40°C. После нагревания смесь можно охладить, например примерно до комнатной температуры (например, примерно 23°C), получая кристаллический продукт - Форму β.
В настоящем изобретении также описана Форма β, полученная любым из описанных выше способов.
Форма γ
В настоящем изобретении также описана кристаллическая форма 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила (Соединение A), имеющая Форму γ. Кристаллическая форма, соответствующая Форме γ, является безводной, что доказывается, например, данными ДСК и ТГ-ДТА (см. Пример 3). В некоторых вариантах осуществления Форма γ имеет спектр порошковой рентгеновской дифракции, содержащий характеристический пик со значением 2θ (°) примерно 16,5. В других вариантах осуществления Форма γ имеет спектр порошковой рентгеновской дифракции, содержащий два или больше характеристических пиков со значениями 2θ (°), выбранными из примерно 16,5, примерно 17,7, примерно 21,4, примерно 21,8 и примерно 23,1. В других вариантах осуществления Форма γ имеет спектр порошковой рентгеновской дифракции, содержащий три или больше характеристических пиков со значениями 2θ (°), выбранными из примерно 7,7, примерно 10,6, примерно 13,3, примерно 13,9, примерно 15,2, примерно 16,5, примерно 17,7, примерно 17,9, примерно 19,0, примерно 21,4, примерно 21,8, примерно 23,1, примерно 23,7 и примерно 28,1. В других вариантах осуществления Форма γ имеет спектр порошковой рентгеновской дифракции, содержащий три или больше характеристических пиков со значениями 2θ (°), выбранными из пиков, перечисленных в Таблице 7 (см. Пример 3). В других вариантах осуществления Форма γ имеет спектр порошковой рентгеновской дифракции, по существу такой, как на Фиг. 3.
В некоторых вариантах осуществления Форма γ имеет ДСК термограмму, которая отличается эндотермическим пиком примерно при 196°C. В некоторых вариантах осуществления Форма γ имеет ДСК термограмму, которая отличается экстраполированной температурой начала перехода примерно 196°C. В других вариантах осуществления Форма γ имеет ДСК термограмму, по существу такую, как на Фиг. 6.
В некоторых вариантах осуществления Форма γ имеет твердотельный 13C ЯМР спектр, который отличается наличием по меньшей мере одного пика со значением химсдвига примерно 162,9 м.д. В других вариантах осуществления Форма γ имеет твердотельный 13C ЯМР спектр, который отличается наличием по меньшей мере 5 пиков, выбранных из пиков со значениями химсдвигов примерно 16,9, примерно 26,5, примерно 32,9, примерно 36,4, примерно 48,1, примерно 53,7, примерно 78,6, примерно 102,6, примерно 116,4, примерно 117,9, примерно 121,5, примерно 151,8, примерно 154,6 и примерно 162,9 м.д. В других вариантах осуществления Форма γ имеет твердотельный 13C ЯМР спектр, по существу такой, как изображенный на Фиг. 11.
В некоторых вариантах осуществления Форма γ имеет пространственную группу P21 со следующими параметрами элементарной ячейки:
Таблица 4
согласно данным рентгеноструктурного анализа монокристалла (см. Пример 5).
Форма γ может иметь степень чистоты по меньшей мере примерно 50%, по меньшей мере примерно 75%, по меньшей мере примерно 85%, по меньшей мере примерно 90% или по меньшей мере примерно 95%. В некоторых вариантах осуществления Форма γ по существу чистая.
Форму γ можно получить превращением Формы α 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила в Форму γ. Это превращение можно проводить, например, перемешиванием Формы α в диметилформамиде (ДМФА). Перемешивание можно проводить, например, при комнатной температуре. В альтернативном варианте Форму γ можно получить путем объединения Соединения A с формамидом и добавления затравочного кристалла Формы γ. Такое получение можно проводить при комнатной температуре и необязательно в инертной атмосфере, такой как азот.
В настоящем изобретении также описана Форма γ, полученная любым из описанных выше способов.
Композиции
Помимо Форм β и γ, Соединение A можно получить в виде безводной кристаллической Формы α, которая описана в Примере 1. Соответственно, в настоящем изобретении описаны смеси двух или больше из Форм α, β, и γ Соединения A.
В некоторых вариантах осуществления в настоящем изобретении описана композиция кристаллического 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила (Соединение A), содержащая Форму α и Форму β. В некоторых вариантах осуществления Форма β присутствует в количестве от примерно 1 до примерно 50% мас./мас. относительно Формы α. В других вариантах осуществления Форма β присутствует в количестве от примерно 1 до примерно 20% мас./мас. относительно Формы α. В других вариантах осуществления Форма β присутствует в количестве от примерно 1 до примерно 10% мас./мас. относительно Формы α. В других вариантах осуществления Форма β присутствует в количестве от примерно 1 до примерно 5% мас./мас. относительно Формы α. В некоторых вариантах осуществления композиция содержит Форму α и Форму β и по существу не содержит других кристаллических форм Соединения A. В других вариантах осуществления композиция, содержащая Форму α и Форму β, дополнительно содержит Форму γ. В некоторых вариантах осуществления композиция состоит практически только из Формы α и Формы β.
Кроме того, в настоящем изобретении описана композиция кристаллического 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрил (Соединение A), содержащая Форму β и Форму γ. В некоторых вариантах осуществления Форма γ присутствует в количестве от примерно 1 до примерно 50% мас./мас. относительно Формы β. В других вариантах осуществления Форма γ присутствует в количестве от примерно 1 до примерно 20% мас./мас. относительно Формы β. В других вариантах осуществления Форма γ присутствует в количестве от примерно 1 до примерно 10% мас./мас. относительно Формы β. В других вариантах осуществления Форма γ присутствует в количестве от примерно 1 до примерно 5% мас./мас. относительно Формы β. В некоторых вариантах осуществления композиция содержит Форму β и Форму γ и по существу не содержит других кристаллических форм Соединения A. В других вариантах осуществления композиция, содержащая Форму β и Форму γ, дополнительно содержит Форму α. В некоторых вариантах осуществления композиция состоит практически только из Формы β и Формы γ.
В настоящем изобретении также описаны композиции, содержащие Форму β или Форму γ и одно или больше других веществ. В некоторых вариантах осуществления композиция содержит по меньшей мере примерно 50%, по меньшей мере примерно 70%, по меньшей мере примерно 80%, по меньшей мере примерно 90%, по меньшей мере примерно 95%, по меньшей мере примерно 97%, по меньшей мере примерно 98% или по меньшей мере примерно 99% по массе Формы β или Формы γ.
Аналитические методы
В настоящем изобретении описан способ определения количества Формы β, присутствующей при получении Формы α, включающий измерение площади пика в спектре порошковой рентгеновской дифракции, являющегося характеристическим для Формы β, и сравнение площади этого пика со стандартом. В некоторых вариантах осуществления пик, характеристический для Формы β, находится примерно при 10,6° 2-тета. Стандартом может служить стандартная кривая, вычисленная из площадей пиков известных количеств Формы β в Форме α. Аналогичным образом, в настоящем изобретении описан способ измерения количества Формы γ, присутствующей при получении Формы α, включающий измерение площади пика в спектре порошковой рентгеновской дифракции, являющегося характеристическим для Формы γ, и сравнение площади этого пика со стандартом. В некоторых вариантах осуществления пик, характеристический для Формы γ, находится примерно при 16,6° 2-тета. Стандартом может служить стандартная кривая, вычисленная из площадей пиков известных количеств Формы γ в Форме α.
Фармацевтические композиции и применение
Кристаллические формы по настоящему изобретению можно ввести в состав фармацевтических композиций, которые содержат кристаллическую форму по настоящему изобретению или композицию по настоящему изобретению вместе с по меньшей мере одним фармацевтически приемлемым носителем (или вспомогательным веществом). В одном варианте осуществления фармацевтическая композиция подходит для перорального, парентерального, легочного, местного или наружного применения. В некоторых вариантах осуществления фармацевтическая композиция имеет форму препарата для перорального введения, такого как таблетка, капсула, гранула, порошок, литая таблетка, сироп, эмульсия, суспензия, или препарата для парентерального введения, такого как препарат для наружного применения, суппозиторий, инъекция, капли, препарат для назального введения, препарат для легочного введения. В некоторых вариантах осуществления фармацевтическая композиция подходит для наружного применения, например имеет вид мази.
Фармацевтические композиции по настоящему изобретению можно приготовить смешиванием одной или более кристаллических форм по настоящему изобретению или композиции по настоящему изобретению с по меньшей мере одним или больше фармацевтически приемлемыми носителями в подходящих количествах согласно методикам, известным в области медицинских препаратов. Количество Соединения A в фармацевтической композиции зависит от дозированных форм, дозировок и т.д., и может составлять, например, примерно от 0,1 до 100 мас.% от массы композиции. Фармацевтические композиции по настоящему изобретению могут быть, например, в форме таблетки, капсулы, пилюли, порошка или мази. В некоторых вариантах осуществления фармацевтические композиции по настоящему изобретению могут быть в форме порошка, подходящего для разведения в жидкости для внутривенного (IV), внутримышечного (IM) или подкожного (SC) введения. Кроме того, фармацевтические композиции по настоящему изобретению могут дополнительно содержать второе терапевтическое средство.
Термин “фармацевтически приемлемый носитель” включает различные традиционные органические или неорганические носители для фармацевтических соединений, например разбавитель, разрыхляющее вещество, связующее вещество, добавку для придания текучести и лубрикант для твердых препаратов, растворитель, солюбилизатор, суспендирующий агент, регулятор тоничности, буферную добавку и успокаивающее вещество для жидких препаратов, и основу, эмульгатор, увлажнитель, стабилизатор, стабилизирующее вещество, диспергатор, пластификатор, регулятор рН, стимулятор абсорбции, гелеобразователь, антисептик, наполнитель, противовоспалительное вещество, солюбилизатор и суспендирующий агент для полутвердых препаратов. Кроме того, при необходимости можно применять такие добавки, как консервант, антиоксидант, краситель и подсластитель.
Примеры разбавителей включают лактозу, сахарозу, D-маннит, D-сорбит, кукурузный крахмал, декстрин, микрокристаллическую целлюлозу, кристаллическую целлюлозу, кармеллозу, кармеллозу кальция, натрия карбоксиметилкрахмал, низкозамещенную гидроксипропилцеллюлозу, аравийскую камедь и т.д.
Примеры разрыхлителей включают кармеллозу, кармеллозу кальция, кармеллозу натрия, натрия карбоксиметилкрахмал, кросскармеллозу натрия, кросповидон, низкозамещенную гидроксипропилцеллюлозу, гидроксипропилцеллюлозу, кристаллическую целлюлозу и т.д.
Примеры связующих веществ включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, повидон, кристаллическую целлюлозу, сахарозу, декстрин, крахмал, желатин, кармеллозу натрия, аравийскую камедь и т.д.
Примеры добавок для придания текучести включают легкую безводную кремневую кислоту, стеарат магния и т.д.
Примеры лубрикантов включают стеарат магния, стеарат кальция, тальк и т.д.
Примеры растворителей включают очищенную воду, этанол, пропиленгликоль, макрогол, кунжутное масло, кукурузное масло, оливковое масло и т.д.
Примеры солюбилизаторов включают пропиленгликоль, D-маннит, бензилбензоат, этанол, триэтаноламин, карбонат натрия, цитрат натрия и т.д.
Примеры суспендирующих агентов включают бензалконий хлорид, кармеллозу, гидроксипропилцеллюлозу, пропиленгликоль, повидон, метилцеллюлозу, глицерил моностеарат и т.д.
Примеры регуляторов тоничности включают глюкозу, D-сорбит, хлорид натрия, D-маннит и т.д.
Примеры буферных добавок включают гидрофосфат натрия, ацетат натрия, карбонат натрия, цитрат натрия и т.д.
Примеры успокаивающих веществ включают бензиловый спирт и т.д.
Примеры консервантов включают этилпараоксибензоат, хлорбутанол, бензиловый спирт, дегидроацетат натрия, сорбиновую кислоту и т.д.
Примеры антиоксидантов включают сульфит натрия, аскорбиновую кислоту и т.д.
Примеры красителей включают пищевые красители (например, пищевой краситель красный № 2 или 3, пищевой краситель желтый №4 или 5, и т.д.), β-каротин и т.д.
Примеры подсластителей включают сахарин натрия, дикалия глицирризинат, аспартам и т.д.
Фармацевтические композиции по настоящему изобретению можно перорально или парентерально (например, местно, ректально, внутривенно и т.д.) вводить пациенту, такому как отличные от человека млекопитающие (например, мыши, крысы, грызуны, морские свинки, кролики, кошки, собаки, свиньи, коровы, лошади, овцы, обезьяны и т.д.) или люди, в терапевтически эффективном количестве для лечения или предотвращения заболевания. Термин “млекопитающее” включает и человека, и отличных от человека млекопитающих. Доза фармацевтической композиции зависит от субъектов, заболеваний, патологических состояний, дозированной формы, способа введения. Доза для перорального введения взрослым пациентам (вес тела примерно 60 кг), страдающим от отторжения органа после пересадки, реакции трансплантат-против-хозяина после пересадки, аутоиммунного заболевания или аллергического заболевания и т.д., может составлять, например, примерно от 1 мг до 1 грамма в сутки. Всю дозу можно вводить за один прием или в виде нескольких отдельных доз.
Для наружных фармацевтических композиций носитель (или разбавитель) может представлять собой белый мягкий парафин, твердый парафин, сквален или их комбинацию. Белый мягкий парафин, твердый парафин и сквален можно комбинировать в соотношении 70-90 мас.%, 5-10 мас.% и 5-20 мас.%, соответственно. Пример такого препарата содержит Соединение A, белый мягкий парафин, 5±2 мас.% твердого парафина и 10±2 мас.% сквалена.
Наружное средство можно наносить, например, путем простого нанесения, размазывания или опрыскивания, в зависимости от дозированной формы и т.д. Количество наносимого наружного средства на пораженную площадь можно выбрать в зависимости от содержания действующего вещества и т.д., и наружное средство можно наносить, например, однократно или в несколько приемов в течение дня. В некоторых вариантах осуществления нанесение осуществляют один раз в сутки или два раза в сутки.
Фармацевтические композиции можно приготовить согласно общепринятым в фармацевтике методикам, таким как, например, методики, описанные в работе Remingtons Pharmaceutical Sciences, 17-е издание под редакцией Alfonoso R. Gennaro, Mack Publishing Company, Easton, PA (1985), которая в полном объеме включена в настоящий документ посредством ссылки.
Кристаллические формы по настоящему изобретению можно применять в способе ингибирования Янус-киназы, таком как ингибирование JAK3, JAK2 или обеих, включающем контакт кристаллической формы по настоящему изобретению с ингибитором Янус-киназы. Контакт может происходить in vitro или in vivo.
Кристаллические формы или композиции по настоящему изобретению можно применять в качестве действующего вещества в способе лечения или предотвращения одного или более перечисленных ниже заболеваний у пациента:
(a) отторжение органа после пересадки или реакция трансплантат-против-хозяина после пересадки;
(b) аутоиммунные заболевания, включая ревматоидный артрит, псориаз, псориатический артрит, рассеянный склероз, язвенный колит, болезнь Крона, системная красная волчанка, диабет I типа, миастения гравис, болезнь Кастлемена, ювенильный идиопатический артрит, сухость глаз; и
(c) аллергические заболевания, включая астму, атопический дерматит, ринит.
В некоторых вариантах осуществления кристаллические формы по настоящему изобретению можно применять в качестве действующего вещества в терапевтическом или профилактическом средстве против ревматоидного артрита, псориаза, очаговой алопеции, сухости глаз, атопического дерматита, экземы или экземы рук.
Кристаллические формы по настоящему изобретению можно применять в качестве действующего вещества в терапевтическом или профилактическом средстве против хронических миелопролиферативных заболеваний, включая истинную полицитемию, первичный миелофиброз, идиопатическую тромбоцитемию и т.д.
Термин “терапевтически эффективное количество” соединения при использовании в настоящем тексте означает количество, достаточное для излечения, ослабления или частичной остановки клинических проявлений определенного заболевания и его осложнений. Количество, достаточное для решения такой задачи, называют “терапевтически эффективным количеством”. Эффективное количество для каждой цели зависит от степени тяжести заболевания или поражения, а также от веса тела и общего состояния здоровья субъекта.
Термин “лечение” при использовании в настоящем документе включает ослабление симптома, предотвращение развития осложнения, поддержание ремисии, предотвращение обострения и предотвращение повторного появления. Термин “лечение” может также включать замедление прогрессирования заболевания, нарушения или патологического состояния, ослабление или избавление от симптомов и осложнений, и/или лечение или полное устранение заболевания, нарушения или патологического состояния.
Термин “лечение” может также означать уход за пациентом с целью борьбы с заболеванием, нарушением или патологического состоянием.
Термин “предотвращение” означает подавление появления симптома.
Комбинированная терапия
Кристаллические формы по настоящему изобретению можно вводить пациенту в комбинации с терапевтически эффективным количеством одного или более дополнительных терапевтических средств. Кристаллическую форму по настоящему изобретению можно вводить одновременно (например, совместно) с дополнительным терапевтическим средством (например, в виде единой дозированной формы или в виде отдельных дозированных форм). Аналогичным образом, кристаллическую форму по настоящему изобретению и дополнительное терапевтическое средство можно вводить пациенту последовательно. Например, дополнительное терапевтическое средство вводят тогда, когда кристаллическая форма по настоящему изобретению уже проявляет свое терапевтическое действие, или наоборот.
Ниже приведены некоторые примеры с целью более эффективного понимания описанного в настоящем документе изобретения. Следует понимать, что эти примеры приведены исключительно в иллюстративных целях и никоим образом не ограничивают объем настоящего изобретения.
Примеры
Пример 1
Получение 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила (Соединение A) и описание кристаллической Формы α.
Получение Соединения A
Соединение A получали методом синтеза, представленным ниже и описанным в PCT/JP2016/070046 (Примеры 1-15). На стадиях кристаллизации применяли затравочный кристалл, но можно получить кристаллы каждого соединения согласно описанным методикам и без применения затравочного кристалла.
Применяющиеся в настоящем тексте аббревиатуры имеют следующие значения:
S-BAPO: (S)-2-(бензиламино)пропан-1-ол
S-BBMO: трет-бутил (S)-N-бензил-N-(1-гидроксипропан-2-ил)глицинат
R-BCAB: трет-бутил (R)-N-бензил-N-(2-хлорпропил)глицинат
S-MABB: трет-бутил (3S)-1-бензил-3-метилазетидин-2-карбоксилат
S-MABB-HC: трет-бутил (3S)-1-бензил-3-метилазетидин-2-карбоксилат гидрохлорид
S-MACB-HC: трет-бутил (3S)-3-метилазетидин-2-карбоксилат гидрохлорид
S-ZMAB: 1-бензил 2-(трет-бутил) (3S)-3-метилазетидин-1,2-дикарбоксилат
RS-ZMBB: 1-бензил 2-(трет-бутил) (2R,3S)-2-(2-(трет-бутокси)-2-оксоэтил)-3-метилазетидин-1,2-дикарбоксилат
RS-ZMAA: (2R,3S)-1-((бензилокси)карбонил)-2-(карбоксиметил)-3-метилазетидин-2-карбоновая кислота
RS-ZMAA-DN⋅2H2O: динатрия (2R,3S)-1-((бензилокси)карбонил)-2-(карбоксиметил)-3-метилазетидин-2-карбоксилат дигидрат
RS-ZMOO: бензил (2R,3S)-2-(2-гидроксиэтил)-2-(гидроксиметил)-3-метилазетидин-1-карбоксилат
RS-ZMSS: бензил (2R,3S)-3-метил-2-(2-((метилсульфонил)окси)этил)-2-(((метилсульфонил)окси)метил)азетидин-1-карбоксилат
SR-ZMDB: бензил (3S,4R)-6-бензил-3-метил-1,6-диазаспиро[3.4]октан-1-карбоксилат
SR-MDOZ: бензил (3S,4R)-3-метил-1,6-диазаспиро[3.4]октан-1-карбоксилат
SR-MDOZ-OX: бензил (3S,4R)-3-метил-1,6-диазаспиро[3.4]октан-1-карбоксилат оксалат
SR-MDPZ: бензил (3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-карбоксилат
SR-MDOP: 4-[(3S,4R)-3-метил-1,6-диазаспиро[3.4]-октан-6-ил]-7H-пирроло[2,3-d]пиримидин
Соединение A: 3-[(3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил]-3-оксопропаннитрил
CPPY: 4-хлор-7H-пирроло[2,3-d]пиримидин
DPCN: 1-цианоацетил-3,5-диметил-1H-пиразол
TBBA: трет-бутиловый эфир бромуксусной кислоты
ТГФ: тетрагидрофуран.
Применявшиеся приборы и условия регистрации спектров указаны ниже.
1H-ЯМР спектры регистрировали в CDCl3 или ДМСО-d6 с применением тетраметилсилана в качестве внутреннего стандарта, и все значения δ указаны в миллионных долях (м.д.). Спектры регистрировали на ЯМР-приборе с частотой 400 МГц, если не указано иное.
Символы в Примерах имеют указанные ниже значения.
с: синглет
д: дублет
т: триплет
кв: квартет
дд: дублет дублетов
ддд: дублет дублетов дублетов
ушир.с: уширенный синглет
м: мультиплет
J: константа спин-спинового взаимодействия
Содержание ионов в образце определяли, как среднее по трем измерениям для образца.
Прибор: Ионообменный хроматограф LC-20 (Shimadzu Corporation)
Условия анализа: Детектор электропроводности SHIMADZU CDD-10A VP
Колонка для анализа анионов SHIMADZU SHIM-PAC IC-A3
Колонка для анализа катионов SHIMADZU SHIM-PAC IC-C1
Содержание воды в образце определяли методом титрования по Карлу Фишеру.
Прибор: Титратор для определения содержания воды CA-06 (Mitsubishi Chemical)
Условия анализа:
Количество образца: около 20 мг
Реагент: анодный раствор Aquamicron AX (API Corporation)
Катодный раствор Aquamicron CXU (API Corporation)
Массовые проценты углерода, водорода и азота в образцах определяли элементным анализом.
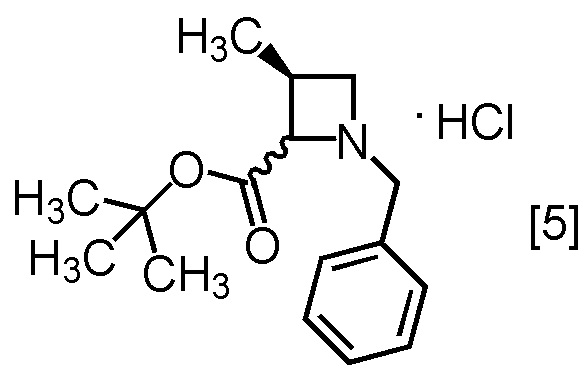
Стадия A. Получение S-MABB-HC (Соединение [5])
[Хим. 2]
Стадия 1
[Хим. 3]
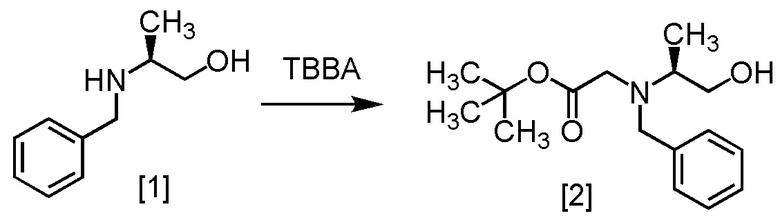
S-BAPO [1] (35,0 г, 212 ммоль) добавляли в воду (175 мл) при комнатной температуре в атмосфере азота. В полученную суспензию добавляли толуол (53 мл) и карбонат калия (32,2 г, 233 ммоль) при комнатной температуре. В полученный раствор по каплям добавляли TBBA (434,4 г, 223 ммоль) при комнатной температуре, затем использованную капельную воронку промывали толуолом (17 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при 65°C в течение 21 часов, затем охлаждали до комнатной температуры. Добавляли в реакционную смесь толуол (105 мл), затем смесь перемешивали, органический слой отделяли. Органический слой промывали водой (175 мл), водный слой отделяли и удаляли растворитель из органического слоя в вакууме. Толуол (105 мл) добавляли в остаток, и полученный толуольный раствор упаривали. Эту операцию повторяли еще два раза, получая толуольный раствор S-BBMO [2] (74,0 г, 212 ммоль теоретически). Полученный толуольный раствор S-BBMO использовали на следующей стадии, считая выход равным 100%.
Сырой продукт S-BBMO, который был получен по описанной выше методике, упаривали досуха и затем исследовали методами ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 7,36-7,13 (5H, м), 4,26 (1H, дд, J = 6,8, 3,9 Гц), 3,72 (2H, дд, J = 14,2, 6,8 Гц), 3,47-3,38 (1H, м), 3,30-3,08 (3H, м), 2,79 (1H, секстет, J = 6,8 Гц), 1,35 (9H, с), 0,96 (3H, д, J = 6,8 Гц).
МС: m/z = 280 [M+H]+
Стадия 2
[Хим. 4]
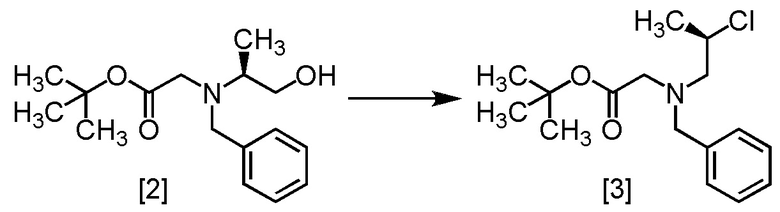
В толуольный раствор S-BBMO [2] (74,0 г, 212 ммоль) добавляли толуол (200 мл), тетрагидрофуран (35 мл) и триэтиламин (25,7 г, 254 ммоль) при комнатной температуре в атмосфере азота. В полученную смесь по каплям добавляли метансульфонилхлорид (26,7 г, 233 ммоль) при 0°C, затем использованную капельную воронку промывали толуолом (10 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем при 65°C в течение 22 часов и охлаждали до комнатной температуры. Добавляли в реакционную смесь водный раствор бикарбоната натрия (105 мл) и смесь перемешивали, отделяли органический слой. Органический слой промывали водой (105 мл), водный слой отделяли и удаляли растворитель из органического слоя в вакууме. Толуол (105 мл) добавляли в остаток, и толуольный раствор упаривали. Эту операцию повторяли еще два раза, получая толуольный раствор R-BCAB [3] (75,3 г, 212 ммоль теоретически). Полученный толуольный раствор R-BCAB использовали на следующей стадии, считая выход равным 100%.
Сырой продукт R-BCAB, который был получен по описанной выше методике, упаривали досуха и затем исследовали методами ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 7,28-7,11 (5H, м), 4,24-4,11 (1H, м), 3,80 (2H, д, J = 3,6 Гц), 3,24 (2H, д, J = 3,6 Гц), 2,98-2,78 (2H, м), 1,46-1,37 (12H, м).
МС: m/z = 298 [M+H]+
Стадия 3
[Хим. 5]
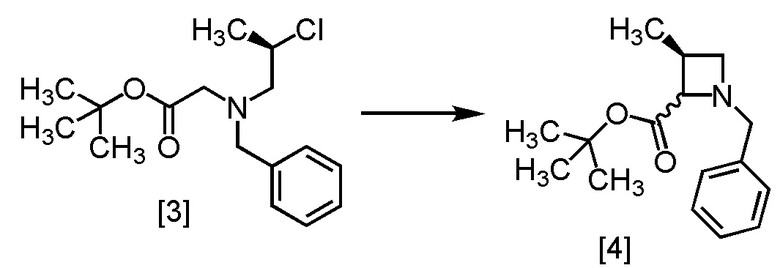
В толуольный раствор R-BCAB [3] (75,3 г, 212 ммоль) в атмосфере азота добавляли тетрагидрофуран (88,0 мл) и 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинон (42,0 мл) при комнатной температуре. В полученный раствор по каплям добавляли раствор лития бис(триметилсилил)амид/тетрагидрофуран (195 мл, 233 ммоль) при 0°C, затем использованную капельную воронку промывали тетрагидрофураном (17,0 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при 0°C в течение 1 часа, затем нагревали до комнатной температуры. Добавляли в реакционную смесь воду (175 мл) и толуол (175 мл), затем смесь перемешивали, органический слой отделяли. Полученный органический слой промывали водным раствором хлорида аммония (175 мл) и затем водой (175 мл), и удаляли растворитель из органического слоя в вакууме. Этилацетат (175 мл) добавляли в остаток и упаривали полученный этилацетатный раствор. Эту операцию повторяли еще два раза, получая этилацетатный раствор S-MABB [4] (66,5 г, 212 ммоль теоретически). Полученный этилацетатный раствор S-MABB использовали на следующей стадии, считая выход равным 100%.
Сырой продукт S-MABB, который был получен по описанной выше методике, упаривали досуха и затем исследовали методами ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 7,28-7,25 (10H, м), 3,75 (1H, д, J = 12,7 Гц), 3,68 (1H, д, J = 1,4 Гц), 3,66 (1H, д, J = 6,7 Гц), 3,46 (2H, д, J = 12,7 Гц), 3,30-3,17 (2H, м), 2,95 (1H, дд, J = 6,2, 1,2 Гц), 2,77 (1H, дд, J = 6,1, 2,2 Гц), 2,65-2,55 (1H, м), 2,48-2,40 (2H, м), 1,35 (9H, с), 1,35 (9H, с), 1,12 (3H, д, J = 7,2 Гц), 1,09 (3H, д, J = 6,2 Гц).
МС: m/z = 262 [M+H]+
Стадия 4
[Хим. 6]
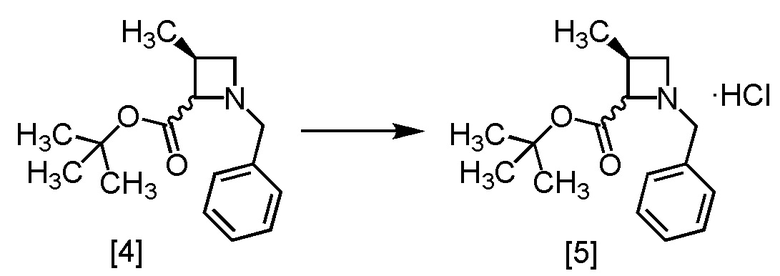
В полученный этилацетатный раствор S-MABB [4] (66,5 г, 212 ммоль теоретически) добавляли этилацетат (175 мл) и активированный уголь (3,5 г) в атмосфере азота, затем смесь перемешивали при комнатной температуре в течение 2 часов. Активированный уголь удаляли фильтрованием, осадок на фильтре промывали этилацетатом (175 мл). Промывные растворы добавляли в фильтрат. В раствор при 0°C добавляли кристалл S-MABB-HC (17,5 мг), который был получен по описанной в настоящем тексте методике, затем прикапывали 4M раствор хлороводорода в этилацетате (53,0 мл, 212 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 17 часов, выпавший твердый осадок собирали на фильтре и промывали этилацетатом (70 мл). Полученный влажный твердый продукт сушили в вакууме, получая S-MABB-HC [5] (48,3 г, 162 ммоль, выход: 76,4%).
S-MABB-HC, который был получен по описанной выше методике, исследовали методом ЯМР, МС и определяли содержание Cl.
1H-ЯМР (ДМСО-d6) δ: 11,08 (1H, ушир.с), 10,94 (1H, ушир.с), 7,52-7,42 (10H, м), 5,34 (1H, т, J = 8,4 Гц), 4,90 (1H, ушир.с), 4,45-4,10 (5H, м), 3,92-3,49 (3H, ушир.м), 3,10-2,73 (2H, ушир.м), 1,35 (9H, с), 1,29 (9H, с), 1,24 (3H, д, J = 6,7 Гц), 1,17 (3H, д, J = 7,4 Гц).
МС: m/z = 262 [M+H-HCl]+
Содержание Cl (ионообменная хроматография): 11,9% (теоретически: 11,9%).
Стадия B. Получение S-MACB-HC (Соединение [6])
[Хим. 7]
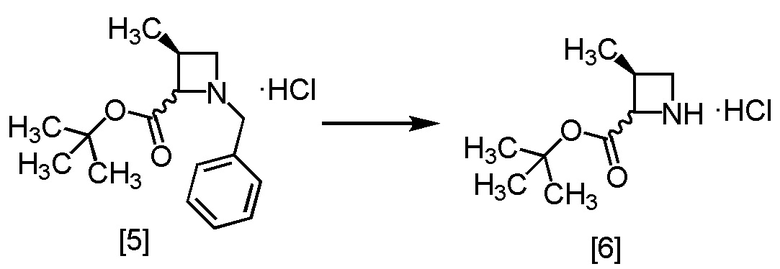
В раствор S-MABB-HC [5] (5,0 г, 16,8 ммоль) в метаноле (15,0 мл) при комнатной температуре добавляли 5% палладий на угле (производство Kawaken Fine Chemicals Co., Ltd., PH type, 54,1% содержание воды 1,0 г) в атмосфере азота. Реакционный сосуд заполняли водородом, смесь перемешивали под давлением водорода 0,4 МПа при комнатной температуре в течение 12 часов, заменяли водород в реакционном сосуде на азот, и затем 5% палладий на угле удаляли фильтрованием. Реакционный сосуд и 5% палладий на угле промывали метанолом (10 мл). Промывные растворы добавляли в фильтрат, получая метанольный раствор S-MACB-HC [6] (24,8 г, 16,8 ммоль теоретически). Полученный метанольный раствор S-MACB-HC использовали на следующей стадии, считая выход равным 100%.
Сырой продукт S-MACB-HC, который был получен по описанной выше методике, упаривали досуха и исследовали методами ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 9,60 (ушир.с, 1H), 4,97 (д, 1H, J = 9,2 Гц), 4,61 (д, 1H, J = 8,4 Гц), 4,01 (дд, 1H, J = 10,0, 8,4 Гц), 3,78-3,74 (м, 1H), 3,54 (дд, 1H, J = 9,6, 8,4 Гц), 3,35 (дд, 1H, J = 10,0, 6,0 Гц), 3,15-3,03 (м, 1H), 3,00-2,88 (м, 1H), 1,49 (с, 9H), 1,47 (с, 9H), 1,22 (д, 3H, J = 6,8 Гц), 1,14 (д, 3H, J = 7,2 Гц).
МС: m/z = 172 [M+H]+ (свободная форма).
Стадия C. Получение S-ZMAB (Соединение [7])
[Хим. 8]
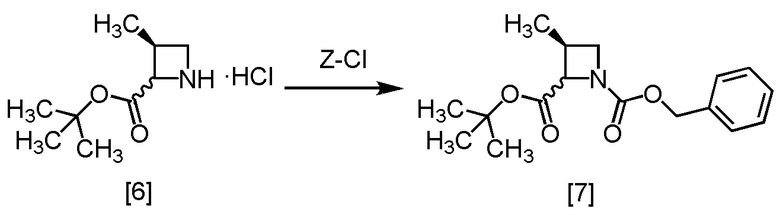
В метанольный раствор S-MACB-HC [6] (24,8 г, 16,8 ммоль теоретически) по каплям добавляли N,N-диизопропилэтиламин (4,8 г, 36,9 ммоль) при комнатной температуре в атмосфере азота, затем использованную капельную воронку промывали тетрагидрофураном (2,5 мл), и промывные растворы добавляли в реакционную смесь. В результирующую реакционную смесь по каплям добавляли бензилхлорформиат (3,0 г, 17,6 ммоль) при 0°C, затем использованную капельную воронку промывали тетрагидрофураном (2,5 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при 0°C в течение 1 часа, затем растворитель удаляли в вакууме. Добавляли в остаток толуол (25,0 мл) и водный раствор лимонной кислоты (25,0 мл), смесь перемешивали, органический слой отделяли. Полученный органический слой промывали водным раствором бикарбоната натрия (25,0 мл) и затем водой (25,0 мл), и растворитель удаляли из органического слоя в вакууме. Толуол (15,0 мл) добавляли в остаток, и полученный толуольный раствор упаривали. Эту операцию повторяли еще раз, получая толуольный раствор S-ZMAB [7] (6,9 г, 16,8 ммоль теоретически). Полученный толуольный раствор S-ZMAB использовали на следующей стадии, считая выход равным 100%.
Сырой продукт S-ZMAB, который был получен по описанной выше методике, упаривали досуха и затем исследовали методами ЯМР и МС.
1H-ЯМР (CDCl3) δ: 7,38-7,28 (м, 10H), 5,16-5,04 (м, 4H), 4,60 (д, 1H, J = 9,2 Гц), 4,18-4,12 (м, 2H), 4,04 (т, 1H, J = 8,6 Гц), 3,66 (дд, 1H, J = 7,6, 7,2 Гц), 3,50 (дд, 1H, J = 8,0, 5,2 Гц), 3,05-2,94 (м, 1H), 2,60-2,50 (м, 1H), 1,43 (ушир.с, 18H), 1,33 (д, 3H, J = 6,5 Гц), 1,15 (д, 3H, J = 7,2 Гц).
МС: m/z = 328 [M+Na]+.
Стадия D. Получение RS-ZMBB (Соединение [8])
[Хим. 9]
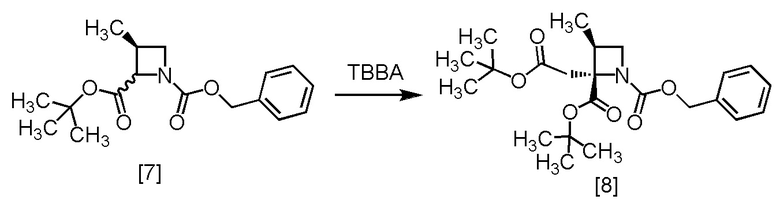
В толуольный раствор S-ZMAB [7] (6,9 г, 16,8 ммоль) добавляли тетрагидрофуран (15,0 мл) при комнатной температуре в атмосфере азота. Раствор лития бис(триметилсилил)амид/тетрагидрофуран (14,7 мл, 17,6 ммоль) по каплям добавляли в толуольный раствор при -70°C. Использованную капельную воронку промывали тетрагидрофураном (2,5 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при -70°C в течение 6 часов, затем по каплям добавляли в реакционную смесь раствор TBBA (3,4 г, 17,6 ммоль) в тетрагидрофуране (2,5 мл) при -70°C. Использованную капельную воронку промывали тетрагидрофураном (2,5 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при -70°C в течение 1 часа, затем нагревали до комнатной температуры. В реакционную смесь добавляли водный раствор хлорида аммония (25 мл) и толуол (25 мл), смесь перемешивали, органический слой отделяли. Полученный органический слой промывали водным раствором лимонной кислоты (25 мл, x 2), водным раствором бикарбоната натрия (25 мл) и водой (25 мл), затем растворитель удаляли из органического слоя в вакууме. Ацетонитрил (15 мл) добавляли в остаток, и полученный ацетонитрильный раствор упаривали. Эту операцию повторяли еще два раза. Ацетонитрил (15 мл) и активированный уголь (0,25 г) добавляли в остаток, смесь перемешивали при комнатной температуре в течение 2 часов. Активированный уголь удаляли фильтрованием, реакционную колбу и осадок на фильтре промывали ацетонитрилом (10 мл). Промывные растворы добавляли в фильтрат и упаривали в вакууме, получая ацетонитрильный раствор RS-ZMBB [8] (13,2 г, 16,8 ммоль теоретически). Полученный ацетонитрильный раствор RS-ZMBB использовали на следующей стадии, считая выход равным 100%.
Сырой продукт RS-ZMBB, который был получен по описанной выше методике, упаривали досуха и затем исследовали методами ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 7,38-7,29 (м, 5H), 5,09-4,96 (м, 2H), 3,91 (т, 0,4H, J = 8,0 Гц), 3,79 (т, 0,6H, J = 8,0 Гц), 3,55 (т, 0,4H, J = 7,2 Гц), 3,46 (т, 0,6H, J = 7,5 Гц), 3,14-3,04 (м, 1H), 2,83-2,72 (м, 2H), 1,38 (ушир.с, 9H), 1,37 (ушир.с, 3,6H), 1,34 (ушир.с, 5,4H), 1,12-1,09 (м, 3H).
МС: m/z = 420 [M+H]+.
Стадия E. Получение RS-ZMAA-DN,2H2O (Соединение [9])
[Хим. 10]
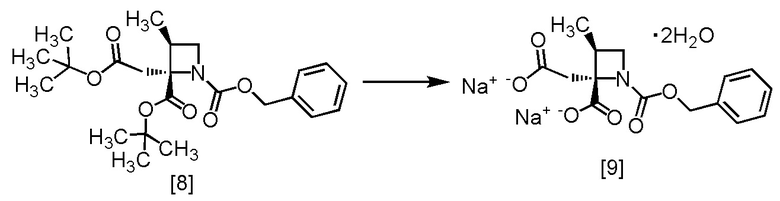
В полученный ацетонитрильный раствор RS-ZMBB [8] (13,2 г, 16,8 ммоль теоретически) добавляли ацетонитрил (15 мл) при комнатной температуре в атмосфере азота. Добавляли в раствор моногидрат п-толуолсульфокислоты (6,4 г, 33,6 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 50°C в течение 12 часов, затем охлаждали до комнатной температуры и по каплям добавляли в реакционную смесь воду (7,5 мл). Реакционную смесь охлаждали до 0°C и по каплям добавляли 4 моль/л водный раствор гидроксида натрия (17,6 мл, 70,5 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 1 часа, по каплям добавляли ацетонитрил (75 мл) при комнатной температуре, и смесь перемешивали в течение 3 часов. Выпавший твердый осадок собирали на фильтре и промывали смесью ацетонитрил: вода = 4:1 (10 мл), и затем ацетонитрилом (10 мл). Полученный влажный твердый продукт сушили в вакууме, получая RS-ZMAA-DN,2H2O [9] (5,2 г, 13,4 ммоль, выход: 85,4%).
RS-ZMAA-DN,2H2O, который был получен по описанной выше методике, анализировали методами ЯМР, МС, определяли содержание Na и содержание воды.
1H-ЯМР (ДМСО-d6) δ: 7,32-7,22 (м, 5H), 4,97 (д, 1H, J = 12,7 Гц), 4,84 (д, 1H, J = 12,7 Гц), 3,79 (т, 1H, J = 8,0 Гц), 3,29 (д, 1H, J = 14,8 Гц), 3,16-3,12 (м, 1H), 2,17-2,09 (м, 2H), 1,07 (д, 3H, J = 6,9 Гц).
МС: m/z = 352 [M+H]+ (безводный).
Содержание Na (ионообменная хроматография): 13,3% (после корректировки на содержание воды) (13,1% теоретически).
Содержание воды (метод Карла Фишера): 9,8% (9,3% теоретически).
Стадия F. Получение RS-ZMAA (Соединение [10])
[Хим. 11]
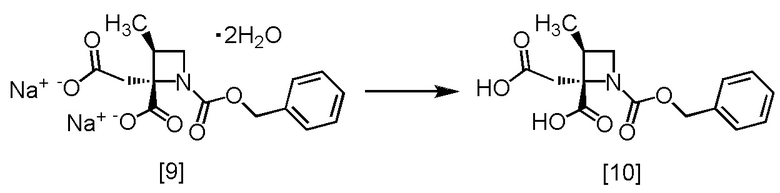
К 1 моль/л соляной кислоте (180 мл) добавляли RS-ZMAA-DN,2H2O [9] (30 г, 77,5 ммоль) и ацетонитрил (60 мл), и смесь перемешивали при комнатной температуре в течение примерно 15 минут. Добавляли в реакционную смесь этилацетат (240 мл), смесь перемешивали, органический слой отделяли. Органический слой промывали 10%-ным раствором хлорида натрия (60 мл x 2). Органический слой перемешивали с сульфатом магния (6 г), удаляли сульфат магния фильтрованием и промывали осадок на фильтре этилацетатом (60 мл). Фильтрат и промывные растворы объединяли, растворитель удаляли в вакууме. Тетрагидрофуран (240 мл) добавляли в остаток, и полученный тетрагидрофурановый раствор упаривали. Эту операцию повторяли еще два раза. Тетрагидрофуран (60 мл) добавляли в остаток, получая тетрагидрофурановый раствор RS-ZMAA [10]. Полученный тетрагидрофурановый раствор RS-ZMAA использовали на следующей стадии, считая выход равным 100%.
RS-ZMAA, который был получен по описанной выше методике, исследовали методами ЯМР и МС.
1H-ЯМР (ДМСО-D6) δ: 7,35-7,28 (м, 5H), 5,06-4,94 (м, 2H), 3,86 (дт, 1H, J = 48,4, 7,9 Гц), 3,50 (дт, 1H, J = 37,9, 7,4 Гц), 3,16-3,02 (ушир.м, 1H), 2,91-2,77 (ушир.м, 2H), 1,08 (д, 3H, J = 6,9 Гц).
МС: m/z = 308 [M+H]+.
Стадия G. Получение RS-ZMOO (Соединение [11])
[Хим. 12]
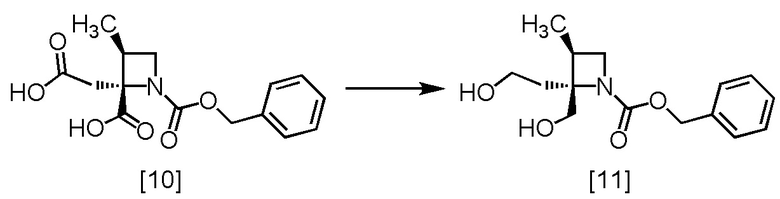
В тетрагидрофурановый раствор RS-ZMAA [10] (25,8 ммоль теоретически) добавляли тетрагидрофуран (50 мл) в атмосфере азота. Комплексный эфират трехфтористого бора (4,40 г) добавляли по каплям при температуре от 0°C до 5°C. Использованную капельную воронку промывали тетрагидрофураном (5 мл), и промывные растворы добавляли в реакционную смесь. В реакционную смесь по каплям добавляли 1,2 моль/л раствор комплекса боран-тетрагидрофуран (43,0 мл) при температуре от 0°C до 5°C, реакционную смесь перемешивали при 0°C до 5°C в течение примерно 30 минут и затем дополнительно перемешивали при комнатной температуре в течение ночи. В реакционную смесь по каплям добавляли 1,2 моль/л раствор комплекса боран-тетрагидрофуран (21,1 мл) при температуре от 0°C до 5°C, и реакционную смесь перемешивали при комнатной температуре в течение ночи. После перемешивания по каплям добавляли в реакционную смесь воду (40 мл) при температуре от 0°C до 15°C. В реакционную смесь добавляли бикарбонат натрия (5,42 г) при температуре от 0°C до 15°C. Оставшийся в емкости бикарбонат натрия смывали водой (10 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем добавляли толуол (50 мл), и реакционную смесь перемешивали дополнительно. Органический слой отделяли. Полученный органический слой промывали 10%-ным раствором хлорида натрия (20 мл x 1), смесью (x 3) 5%-ного водного раствора бикарбоната натрия (20 мл) и 10%-ного раствора хлорида натрия (20 мл), смесью (x 1) 5%-ного водного раствора гидросульфата калия (10 мл) и 10%-ного раствора хлорида натрия (10 мл), и затем 10%-ным раствором хлорида натрия (20 мл x 2). Органический слой перемешивали с сульфатом магния (8,9 г), сульфат магния удаляли фильтрованием, осадок на фильтре промывали толуолом (20 мл). Промывные растворы добавляли в фильтрат, и фильтрат упаривали в вакууме. В остаток от упаривания добавляли толуол (80 мл). Раствор упаривали в вакууме и добавляли толуол (15 мл), получая толуольный раствор RS-ZMOO [11]. Полученный толуольный раствор RS-ZMOO использовали на следующей стадии, считая выход равным 100%.
RS-ZMOO, который был получен по описанной выше методике, исследовали методами ЯМР и МС.
1H-ЯМР (CDCl3) δ: 7,39-7,30 (м, 5H), 5,10 (с, 2H), 4,15-4,01 (ушир.м, 2H), 3,83-3,73 (ушир.м, 3H), 3,48 (дд, 1H, J = 8,3, 6,4 Гц), 2,59-2,50 (ушир.м, 1H), 2,46-2,40 (ушир.м, 1H), 2,07-1,99 (м, 1H), 1,14 (д, 3H, J = 7,2 Гц).
МС: m/z = 280 [M+H]+.
Стадия H. Получение RS-ZMSS (Соединение [12])
[Хим. 13]
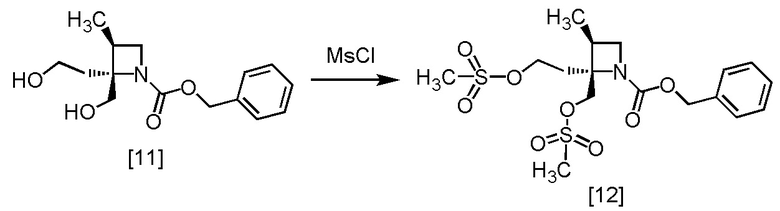
В толуольный раствор RS-ZMOO [11] (23,7 ммоль теоретически) добавляли толуол (55 мл) в атмосфере азота. По каплям добавляли триэтиламин (5,27 г) при температуре от -10°C до 10°C, использованную капельную воронку промывали толуолом (1,8 мл), и промывные растворы добавляли в реакционную смесь. В полученную реакционную смесь по каплям добавляли метансульфонилхлорид (5,69 г) при температуре от -10°C до 10°C, затем использованную капельную воронку промывали толуолом (1,8 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при температуре от 0°C до 10°C в течение примерно 2 часов, затем по каплям добавляли воду (28 мл) при температуре от 0°C до 20°C. Реакционную смесь перемешивали при температуре от 0°C до 20°C в течение примерно 30 минут и отделяли органический слой. Полученный органический слой промывали два раза 10%-ным раствором хлорида натрия (18 мл). Органический слой перемешивали с сульфатом магния (2,75 г), сульфат магния удаляли фильтрованием, и осадок на фильтре промывали толуолом (18 мл). Промывные растворы добавляли в фильтрат, затем растворитель удаляли из фильтрата в вакууме. В остаток от упаривания добавляли толуол до 18 мл, получая толуольный раствор RS-ZMSS [12]. Полученный толуольный раствор RS-ZMSS использовали на следующей стадии, считая выход равным 100%.
RS-ZMSS, который был получен по описанной выше методике, анализировали методами ЯМР и МС.
1H-ЯМР (ДМСО-D6) δ: 7,37-7,27 (ушир.м, 5H), 5,10-4,98 (м, 2H), 4,58-4,22 (ушир.м, 4H), 3,84 (дт, 1H, J = 45,6, 8,1 Гц), 3,48-3,33 (ушир.м, 1H), 3,17-3,10 (м, 6H), 2,81-2,74 (ушир.м, 1H), 2,22-2,12 (м, 2H).
МС: m/z = 436 [M+H]+.
Стадия I. Получение SR-ZMDB (Соединение [13])
[Хим. 14]
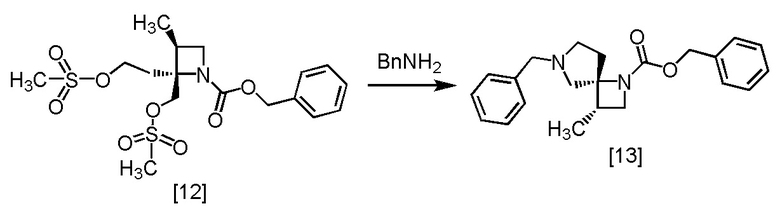
В толуольный раствор RS-ZMSS [12] (23,7 ммоль теоретически) добавляли толуол (55 мл) в атмосфере азота. По каплям добавляли бензиламин (17,8 г) при комнатной температуре, использованную капельную воронку промывали толуолом (9,2 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при комнатной температуре в течение примерно 1 часа при температуре от 55°C до 65°C в течение примерно 3 часов и затем при температуре от 70°C до 80°C в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры по каплям добавляли 10%-ный раствор NaCl (28 мл), и реакционную смесь перемешивали при комнатной температуре примерно 30 минут. Добавляли в реакционную смесь толуол (37 мл), смесь перемешивали, органический слой отделяли. Полученный органический слой промывали смесью (x 2) 10%-ного раствора хлорида натрия (18 мл) и уксусной кислоты (2,84 г), и затем 10%-ным раствором хлорида натрия (11 мл, x 1). Растворитель из органического слоя удаляли в вакууме до половины объема и добавляли в остаток от упаривания уксусный ангидрид (1,45 г) при комнатной температуре. Смесь перемешивали в течение примерно 3 часов. В реакционную смесь добавляли по каплям раствор гидросульфата калия (3,87 г) и воду (92 мл) при комнатной температуре. Реакционную смесь перемешивали, водный слой отделяли. Полученный водный слой промывали толуолом (18 мл) и добавляли в водный слой толуол (73 мл) и затем бикарбонат натрия (6,56 г) при комнатной температуре, смесь перемешивали. Органический слой отделяли и промывали 10%-ным раствором хлорида натрия (11 мл). Органический слой перемешивали с сульфатом магния (2,75 г), сульфат магния удаляли фильтрованием. Осадок на фильтре промывали толуолом (18 мл), и промывные растворы добавляли в фильтрат, затем фильтрат упаривали в вакууме. Толуол (44 мл) добавляли в остаток от упаривания, получая толуольный раствор SR-ZMDB [13]. Полученный толуольный раствор SR-ZMDB использовали на следующей стадии, считая выход равным 100%.
1H-ЯМР (CDCl3) δ: 7,35-7,20 (м, 10H), 5,08 (д, 2H, J = 23,6 Гц), 3,94 (кв, 1H, J = 7,9 Гц), 3,73-3,42 (ушир.м, 2H), 3,30-3,23 (м, 1H), 3,05 (дд, 1H, J = 19,7, 9,5 Гц), 2,79 (дт, 1H, J = 69,6, 6,1 Гц), 2,57-2,32 (ушир.м, 4H), 1,96-1,89 (м, 1H), 1,09 (д, 3H, J = 6,9 Гц).
МС: m/z = 351 [M+H]+.
Стадия J. Получение SR-MDOZ (Соединение [14])
[Хим. 15]
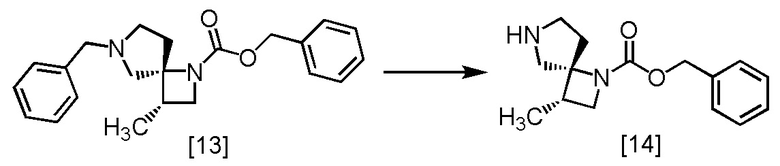
В раствор 1-хлорэтил хлорформиата (3,72 г) в толуоле (28 мл) по каплям добавляли толуольный раствор SR-ZMDB [13] (23,7 ммоль теоретически) при температуре от 0°C до 10°C в атмосфере азота, затем использованную капельную воронку промывали толуолом (4,6 мл), и промывные растворы добавляли в реакционную смесь. В реакционную смесь добавляли триэтиламин (718 мг) при температуре от 0°C до 10°C, и реакционную смесь перемешивали при температуре от 15°C до 25°C в течение примерно 2 часов. Затем добавляли в реакционную смесь метиловый спирт (46 мл), и смесь перемешивали при температуре от 50°C до 60°C в течение еще примерно 2 часов. Растворитель из реакционной смеси удаляли в вакууме до объема меньше примерно 37 мл. В остаток от упаривания по каплям добавляли 2 моль/л соляную кислоту (46 мл) при температуре от 15°C до 20°C, смесь перемешивали и отделяли водный слой. Полученный водный слой промывали толуолом (28 мл, x 2). В водный слой добавляли 20%-ный раствор хлорида натрия (46 мл) и тетрагидрофуран (92 мл), затем по каплям добавляли 8 моль/л водный раствор гидроксида натрия (18 мл) при температуре от 0°C до 10°C. Органический слой отделяли, промывали 20%-ным раствором хлорида натрия (18 мл, x 2), и затем растворитель из органического слоя удаляли в вакууме. В остаток от упаривания добавляли тетрагидрофуран (92 мл), и раствор упаривали в вакууме. Эту операцию повторяли еще раз. Остаток от упаривания растворяли в тетрагидрофуране (92 мл). Раствор перемешивали с сульфатом магния (2,75 г) и удаляли сульфат магния фильтрованием. Осадок на фильтре промывали тетрагидрофураном (28 мл), промывные растворы добавляли в фильтрат, и фильтрат упаривали в вакууме. Объем остатка от упаривания доводили до примерно 20 мл тетрагидрофураном, получая тетрагидрофурановый раствор SR-MDOZ [14] (чистый вес: 4,01 г, 15,4 моль, выход: 65,0%).
SR-MDOZ, который был получен по описанной выше методике, упаривали досуха и затем исследовали методами ЯМР и МС.
1H-ЯМР (CDCl3) δ: 7,37-7,28 (м, 5H), 5,08 (дд, 2H, J = 16,8, 12,8 Гц), 4,00 (дд, 1H, J = 17,1, 8,3 Гц), 3,40-3,31 (м, 1H), 3,24 (д, 1H, J = 12,7 Гц), 3,00 (дд, 1H, J = 54,9, 12,4 Гц), 2,87-2,57 (м, 3H), 2,47-2,27 (м, 1H), 1,91-1,80 (м, 1H), 1,14 (д, 3H, J = 7,2 Гц).
МС: m/z = 261 [M+H]+.
Стадия K. Получение SR-MDOZ-OX (Соединение [15])
[Хим. 16]
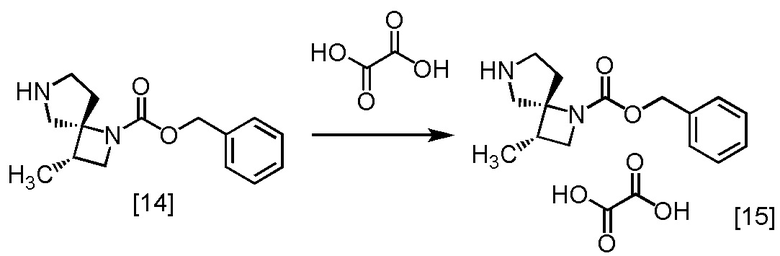
В атмосфере азота растворяли щавелевую кислоту (761 мг) в тетрагидрофуране (40 мл), и по каплям добавляли тетрагидрофурановый раствор SR-MDOZ [14] (3,84 ммоль теоретически) в полученный раствор щавелевой кислоты при комнатной температуре. В раствор при комнатной температуре добавляли кристалл SR-MDOZ-OX (1 мг), который был получен по описанной в настоящем тексте методике, и смесь перемешивали при комнатной температуре в течение примерно 3,5 часов, наблюдая выпадение кристаллического осадка. В полученную суспензию по каплям добавляли тетрагидрофурановый раствор SR-MDOZ (3,84 ммоль) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение примерно 1 часа. Суспензию нагревали и перемешивали при температуре от 50°C до 60°C в течение примерно 2 часов, и затем перемешивали при комнатной температуре в течение ночи. Суспензию фильтровали и промывали влажные кристаллы на фильтре тетрагидрофураном (10 мл), сушили в вакууме, получая SR-MDOZ-OX [15] (2,32 г, 6,62 моль, выход: 86,2%).
SR-MDOZ-OX, который был получен по описанной выше методике, анализировали методами ЯМР, МС, и проводили элементный анализ.
1H-ЯМР (ДМСО-D6) δ: 7,37-7,30 (м, 5H), 5,15-5,01 (м, 2H), 3,92 (дт, 1H, J = 43,5, 8,4 Гц), 3,48-3,12 (ушир.м, 5H), 2,67-2,56 (м, 1H), 2,46-2,35 (м, 1H), 2,12-2,05 (м, 1H), 1,13 (д, 3H, J = 6,9 Гц).
МС: m/z = 261 [M+H]+
Элементный анализ: C 58,4 мас.%, H 6,4 мас.%, N 7,9 мас.% (теоретически: C 58,3 мас.%, H 6,3 мас.%, N 8,0 мас.%).
Стадия L. Получение SR-MDPZ (Соединение [16])
[Хим. 17]
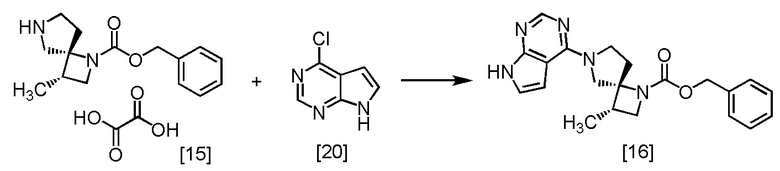
К SR-MDOZ-OX [15] (12,0 г, 34,2 ммоль) добавляли этанол (36 мл), воду (72 мл), CPPY [20] (5,36 г, 34,9 ммоль) и затем K3PO4 (21,8 г, 103 ммоль) в атмосфере азота. Реакционную смесь перемешивали при 80°C в течение 5 часов, затем охлаждали до 40°C. Добавляли толуол (120 мл) при 40°C и отделяли органический слой. Полученный органический слой промывали 20%-ным водным раствором карбоната калия (48 мл), затем два раза промывали водой (48 мл). Растворитель из органического слоя удаляли в вакууме. Добавляли в остаток трет-бутанол (60 мл) и упаривали полученный трет-бутанольный раствор. Эту операцию повторяли еще два раза. Добавляли в остаток от упаривания трет-бутанол (36 мл), получая раствор SR-MDPZ [16] в трет-бутаноле (61,1 г, 34,2 ммоль теоретически). Полученный трет-бутанольный раствор SR-MDPZ использовали на следующей стадии, считая выход равным 100%.
SR-MDPZ, который был получен по описанной выше методике, выделяли в виде твердого вещества из смеси этилацетата и н-гептана, и затем исследовали методами ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 11,59 (ушир.с, 1H), 8,08 (с, 1H), 7,41-7,26 (ушир.м, 3H), 7,22-7,08 (ушир.м, 3H), 6,64-6,51 (ушир.м, 1H), 5,07-4,91 (ушир.м, 2H), 4,09-3,67 (ушир.м, 5H), 3,47-3,32 (ушир.м, 1H), 2,67-2,55 (ушир.м, 2H), 2,21-2,15 (ушир.м, 1H), 1,11 (д, 3H, J = 6,9 Гц).
МС: m/z = 378 [M+H]+
Стадия M. Получение SR-MDOP (Соединение [17])
[Хим. 18]
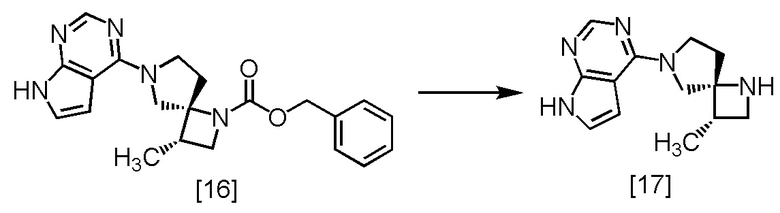
В раствор SR-MDPZ [16] в трет-бутаноле (34,2 ммоль теоретически) добавляли формиат аммония (10,8 г, 171 ммоль), воду (60 мл), и 10% палладий на угле (производство Kawaken Fine Chemicals Co., Ltd., M type, 52,6% содержание воды, 1,20 г) в атмосфере азота. Реакционную смесь перемешивали при 40°C в течение 13 часов, затем охлаждали до комнатной температуры, и выпавший осадок удаляли фильтрованием. Реакционный сосуд и осадок на фильтре промывали трет-бутанолом (24 мл), промывные растворы добавляли в фильтрат и добавляли в фильтрат 8M водный раствор гидроксида натрия (25,7 мл, 205 ммоль) и хлорид натрия (13,2 г). Реакционную смесь перемешивали при 50°C в течение 2 часов, затем добавляли при комнатной температуре толуол (84 мл) и отделяли органический слой. Полученный органический слой промывали 20%-ным раствором хлорида натрия (60 мл), перемешивали с безводным сульфатом натрия, затем сульфат натрия удаляли фильтрованием. Осадок на фильтре промывали смесью толуол : трет-бутанол = 1 : 1 (48 мл), промывные растворы добавляли в фильтрат, и фильтрат упаривали в вакууме. В остаток от упаривания добавляли толуол (60 мл), и полученный раствор перемешивали при 50°C в течение 2 часов, затем растворитель удаляли в вакууме. В остаток от упаривания снова добавляли толуол (60 мл) и упаривали полученный раствор. В остаток от упаривания добавляли толуол (48 мл), и раствор перемешивали при комнатной температуре в течение 1 часа, затем при охлаждении льдом 1 час. Выпавший твердый осадок собирали на фильтре и промывали толуолом (24 мл). Полученный влажный твердый продукт сушили в вакууме, получая SR-MDOP [17] (7,07 г, 29,1 ммоль, выход: 84,8%).
SR-MDOP, который был получен по описанной выше методике, исследовали методами ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 11,57 (ушир.с, 1H), 8,07 (с, 1H), 7,10 (д, 1H, J = 3,2 Гц), 6,58 (д, 1H, J = 3,2 Гц), 3,92-3,59 (ушир.м, 4H), 3,49 (дд, 1H, J = 8,3, 7,2 Гц), 2,93 (дд, 1H, J = 7,2, 6,1 Гц), 2,61-2,53 (м, 2H), 2,12-2,01 (ушир.м, 2H), 1,10 (д, 3H, J = 6,9 Гц).
МС: m/z = 244 [M+H]+.
Стадия N. Получение Соединения A моно-этанолята (Соединение [18])
[Хим. 19]
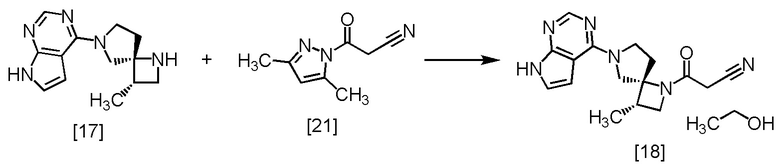
В атмосфере азота добавляли ацетонитрил (60 мл) и триэтиламин (416 мг, 4,11 ммоль) к SR-MDOP [17] (5,00 г, 20,5 ммоль), и по каплям добавляли в полученный раствор DPCN [21] (3,69 г, 22,6 ммоль) в ацетонитриле (35 мл) при 45°C, затем использованную капельную воронку промывали ацетонитрилом (5,0 мл), и промывные растворы добавляли в реакционную смесь. Реакционную смесь перемешивали при 45°C в течение 3 часов, затем охлаждали до комнатной температуры. Добавляли в реакционную смесь 5%-ного водного раствора бикарбоната натрия (25 мл), 10%-ного раствора хлорида натрия (25 мл) и этилацетата (50 мл), затем смесь перемешивали и отделяли органический слой. Растворитель из органического слоя удаляли в вакууме. Тетрагидрофуран (50 мл) добавляли в остаток, и полученный тетрагидрофурановый раствор упаривали. Эту операцию повторяли еще три раза. В остаток от упаривания добавляли тетрагидрофуран (50 мл) и воду, доводя содержание воды до 5,5%. Выпавший осадок удаляли фильтрованием. Реакционный сосуд и осадок на фильтре промывали тетрагидрофураном (15 мл), промывные растворы добавляли в фильтрат и удаляли из фильтрата растворитель в вакууме. В остаток от упаривания добавляли этанол (50 мл) и кристалл Соединения A (5,1 мг), который получали способом, описанным далее в Примере 15. Смесь перемешивали при комнатной температуре в течение 1 часа и упаривали в вакууме. В остаток добавляли этанол (50 мл), и раствор снова упаривали. В остаток от упаривания добавляли этанол (15 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. Выпавший твердый осадок собирали на фильтре и промывали этанолом (20 мл). Полученный влажный твердый продукт сушили в вакууме, получая Соединения A моно-этанолят [18] (6,26 г, 17,6 ммоль, выход: 85,5%).
Соединения A моно-этанолят, который был получен по описанной выше методике, анализировали методами ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 11,59 (ушир.с, 1H), 8,08 (с, 1H), 7,11 (дд, 1H, J = 3,5, 2,3 Гц), 6,58 (дд, 1H, J = 3,5, 1,8 Гц), 4,34 (т, 1H, J = 5,1 Гц), 4,16 (т, 1H, J = 8,3 Гц), 4,09-3,92 (м, 3H), 3,84-3,73 (м, 1H), 3,71 (д, 1H, J = 19,0 Гц), 3,65 (д, 1H, J = 19,0 Гц), 3,58 (дд, 1H, J = 8,2, 5,9 Гц), 3,44 (дкв, 2H, J = 6,7, 5,1 Гц), 2,69-2,60 (м, 2H), 2,23-2,13 (ушир.м, 1H), 1,12 (д, 3H, J = 7,1 Гц), 1,06 (т, 3H, J = 6,7 Гц).
МС: m/z = 311 [M+H]+
Стадия O. Очистка Соединения A (Соединение [19])
[Хим. 20]
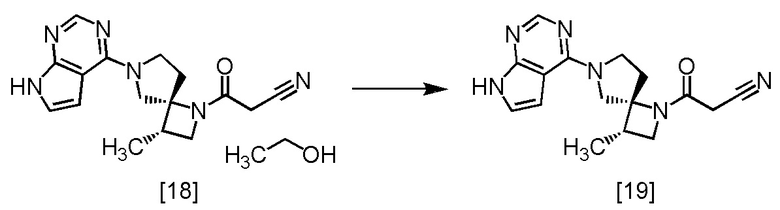
Соединения A моно-этанолят [18] (4,00 г, 11,2 ммоль) и н-бутанол (32 мл) смешивали в атмосфере азота и растворяли смесь при 110°C. Смесь охлаждали до 85°C, и добавляли кристалл Соединения A (4,0 мг), который был получен по описанной в настоящем тексте методике, затем смесь перемешивали при 85°C в течение 2 часов, при 75°C в течение 1 часа и при комнатной температуре в течение 16 часов. Выпавший твердый осадок собирали на фильтре и промывали н-бутанолом (8,0 мл) и затем этилацетатом (8,0 мл). Полученный влажный твердый продукт сушили в вакууме с получением соединения A [19] (3,18 г, 10,2 ммоль, выход: 91,3%).
Соединение A, которое было получено по описанной выше методике, анализировали методами ЯМР и МС.
1H-ЯМР (ДМСО-d6) δ: 11,59 (ушир.с, 1H), 8,08 (с, 1H), 7,11 (дд, 1H, J = 3,5, 2,5 Гц), 6,58 (дд, 1H, J = 3,5, 1,8 Гц), 4,16 (т, 1H, J = 8,3 Гц), 4,09-3,93 (м, 3H), 3,84-3,73 (м, 1H), 3,71 (д, 1H, J = 19,0 Гц), 3,65 (д, 1H, J = 19,0 Гц), 3,58 (дд, 1H, J = 8,2, 5,9 Гц), 2,69-2,59 (м, 2H), 2,23-2,13 (м, 1H), 1,12 (д, 3H, J = 7,2 Гц).
МС: m/z = 311 [M+H]+
Используя Соединение A, которое получали аналогичным методом, проводили рентгеноструктурный анализ монокристалла.
(1) Получение монокристалла
К 10 мг Соединения A в широкогорлом сосуде LaPha ROBO Vial® объемом 2,0 мл добавляли 0,5 мл хлороформа. Закрывали сосуд крышкой, Соединение A полностью растворялось. Для медленного испарения растворителя делали отверстие иглой от шприца TERUMO® в септе, вмонтированной в крышку, и сосуд оставляли стоять при комнатной температуре. Полученный монокристалл использовали для проведения рентгеноструктурного анализа.
(2) Прибор
Источник излучения: SPring-8 BL32B2
Детектор: дифрактометр Rigaku R-AXIS V diffractometer
(3) Метод измерения
Монокристалл подвергали излучению 0,71068 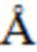 , регистрируя дифракцию рентгеновских лучей
, регистрируя дифракцию рентгеновских лучей
(4) Анализ
Используя эффект аномального рассеяния рентгеновских лучей на атоме хлора в полученном хлороформенном сольвате Соединения A, абсолютную конфигурацию Соединения A определяли как (3S,4R). Основываясь на установленной абсолютной конфигурации Соединения A, идентифицировали абсолютную конфигурацию каждого промежуточного продукта в синтезе.
Описание Формы α
Порошковая рентгеновская дифракция
Форму α характеризовали методом порошковой рентгеновской дифракции на приборе Spectris Co., Ltd. PANalytical instrument Model Number: X’Pert Pro со следующими параметрами:
Число шагов:7
Размер стадии: 1,000
Диапазон:6
Спектр порошковой рентгеновской дифракции для Формы α показан на Фиг. 1, и полученные данные приведены ниже в Таблице 5.
[2θ (°)]
[%]
[имп/с]
Дифференциальная сканирующая калориметрия (ДСК)Данные ДСК измеряли на приборе TA Instruments Model Number Q2000 со следующими параметрами:
Размер образца: примерно 3 мг
Ячейка: герметично закрытая алюминиевая ячейка
Диапазон: 25-230°C
Скорость нагрева: 2°C/мин в токе азота.
ДСК термограмма для Формы α приведена на Фиг. 4, в ней наблюдается единственный эндотермический пик при 188°C с началом при 185°C.
Термогравиметрический дифференциальный термический анализ (ТГ-ДТА)
Данные ТГ-ДТА измеряли на приборе Mettler Toledo Model Number: TGA/SDTA851e/SF со следующими параметрами:
Образец: примерно 5 мг
Диапазон: 25-250°C
Скорость нагрева: 5°C/мин в токе азота.
Экстраполированная температура начала перехода составила 186,29°C, не наблюдалось потери веса в диапазоне от 25 до 220°C. ТГ-ДТА данные для Формы α показаны на Фиг. 7.
Твердотельный 13C ЯМР
Данные твердотельного 13C ЯМР регистрировали на приборе Bruker BioSpin Corporation AVANCE III 400 со следующими параметрами:
Датчик: 4 мм CP/MAS
Температура регистрации спектра: комнатная температура
Стандарт: глицин (Внешний стандарт: слабопольному пику присвоено значение 176,03 м.д.)
Ядро: 13C (100,6228303 МГц)
Скорость вращения: 14,3 кГц
Время задержки: 5 секунд
Число сканов: 3072
Режим датчика: CP/MAS
Спектр твердотельного 13C ЯМР для Формы α изображен на Фиг. 10, и содержит пики: 14,8, 17,0, 24,5, 27,2, 35,0, 35,9, 38,6, 48,8, 49,5, 53,6, 78,3, 101,4, 103,0, 116,2, 118,0, 122,0, 122,7, 151,2, 154,8, 162,4 и 163,3 м.д.
Пример 2
Получение и описание Формы β Соединения A
Получение
Соединение A (3992,3 г, кристаллическая форма α), предварительно пропущенное через сито с размером ячейки 500 мкм, размалывали на мельнице Spiral Jet Mill 100AS (производство Hosokawa Micron Corporation) в указанных ниже условиях, получая размолотый продукт (3473,6 г), при этом Соединение A (361,0 г) налипало на сопловое кольцо.
Условия размалывания
Применяющийся газ: азот
Сопло размалывателя: 1,5 мм в диаметре x 4
Сопло эжектора: 1,3 мм в диаметре
Давление воздуха размалывателя: 0,50-0,60 МПа
Давление воздуха эжектора: 0,50-0,60 МПа
Скорость подачи Соединения A: 8 г/мин
Соединение A (50,0 г, 161 ммоль), налипшее на сопловое кольцо, добавляли в смесь 1-бутанол/ацетонитрил (1:3 об/об) (250 мл), и смесь перемешивали при 40°C в течение 10 дней. Образовавшуюся суспензию охлаждали до комнатной температуры и дополнительно перемешивали при комнатной температуре в течение 24 часов. Выпавшее в осадок кристаллическое вещество собирали на фильтре. Полученный кристаллический продукт промывали этилацетатом (50 мл) два раза и сушили при пониженном давлении при 40°C, получая Соединение A, кристаллическая форма β (35,5 г, выход 71%).
Описание
Порошковая рентгеновская дифракция
Спектр порошковой рентгеновской дифракции для Формы β показан на Фиг. 2, и полученные данные приведены ниже в Таблице 6. Прибор и параметры эксперимента такие же, как описано выше в Примере 1 для исследования Формы α.
[2θ (°)]
[%]
[имп/с]
Дифференциальная сканирующая калориметрия (ДСК)
Данные ДСК измеряли на приборе TA Instruments Model Number Q2000 со следующими параметрами:
Размер образца: примерно 3 мг
Ячейка: герметично закрытая алюминиевая ячейка
Диапазон: 25-230°C
Скорость нагрева: 2°C/мин в токе азота.
ДСК термограмма для Формы β приведена на Фиг. 5, в ней наблюдается единственный эндотермический пик при 187°C с началом при 185°C.
Термогравиметрический дифференциальный термический анализ (ТГ-ДТА)
Данные ТГ-ДТА получали, как описано выше в Примере 1, за исключением того, что диапазон измерений для Формы β составлял от 25 до 230°C. Эндотермический пик был детектирован при 188,59°C, экстраполированная температура начала перехода составила 186,68°C, не наблюдалось потери веса в диапазоне от 25 до 220°C. Данные для Формы β показаны на Фиг. 8.
Твердотельный 13C ЯМР
Данные твердотельного 13C ЯМР для Формы β получали как описано выше в Примере 1. 13C ЯМР спектр приведен на Фиг. 11 и содержит пики: 16,5, 25,8, 26,5, 33,1, 34,8, 36,7, 38,8, 48,2, 53,4, 77,7, 79,5, 101,2, 102,6, 117,5, 120,6, 151,1, 154,3 и 166,1 м.д.
Пример 3
Получение и описание Формы γ Соединения A
Получение 1
Соединение A (2491,5 г, кристаллическая форма α), предварительно пропущенное через сито с размером ячейки 500 мкм, размалывали на мельнице Spiral Jet Mill 100AS (производство Hosokawa Micron Corporation) в указанных ниже условиях, получая размолотый продукт (2106,5 г), при этом Соединение A (247,9 г) налипало на сопловое кольцо.
Условия размалывания
Применяющийся газ: азот
Сопло размалывателя: 1,5 мм в диаметре x 4
Сопло эжектора: 1,3 мм в диаметре
Давление воздуха размалывателя: 0,50-0,60 МПа
Давление воздуха эжектора: 0,50-0,60 МПа
Скорость подачи Соединения A: 4 г/мин
Соединение A (100,0 г, 322 ммоль), налипшее на сопловое кольцо, добавляли в формамид (330 мл) в атмосфере азота и добавляли в смесь затравочный кристалл (1,00 г Кристаллической формы γ, которая может также содержать некоторое количество Формы β Соединения A). Смесь перемешивали при комнатной температуре в течение 30 дней. Кристаллический осадок собирали на фильтре и промывали этилацетатом (150 мл) два раза. Полученное влажное кристаллическое вещество добавляли в этилацетат (300 мл) в атмосфере азота, и смесь перемешивали при комнатной температуре в течение 30 минут. Кристаллический осадок собирали на фильтре, промывали этилацетатом (150 мл) два раза и сушили при пониженном давлении при 40°C, получая Соединение A, кристаллическая форма γ (74,1 г, выход 74%).
Получение 2
Соединение A, Форма α (содержащее небольшое, но не определенное точно количество Формы β) (100 мг, 0,322 ммоль) добавляли в N,N-диметилформамид (0,2 мл), и смесь перемешивали при комнатной температуре в течение 6 дней. Кристаллический осадок отделяли фильтрованием. Полученное влажное кристаллическое вещество сушили при пониженном давлении при комнатной температуре в течение 18 часов, получая соединение A, Форма γ (13,7 мг, выход 13,7%).
Описание
Порошковая рентгеновская дифракция
Спектр порошковой рентгеновской дифракции для Формы γ показан на Фиг. 3, и полученные данные приведены ниже в Таблице 7. Прибор и параметры эксперимента такие же, как описано выше в Примере 1 для исследования Формы α.
[2θ (°)]
[%]
[имп/с]
Дифференциальная сканирующая калориметрия (ДСК) Данные ДСК измеряли на приборе TA Instruments Model Number Q2000 со следующими параметрами:
Размер образца: примерно 3 мг
Ячейка: герметично закрытая алюминиевая ячейка
Диапазон: 25-230°C
Скорость нагрева: 2°C/мин в токе азота.
ДСК термограмма для Формы γ приведена на Фиг. 6, в ней наблюдается единственный эндотермический пик при 196°C с началом при 196°C.
Термогравиметрический дифференциальный термический анализ (ТГ-ДТА)
Данные ТГ-ДТА получали как описано выше в Примере 1. Эндотермический пик был детектирован при 198,68°C, экстраполированная температура начала перехода составила 197,38°C, не наблюдалось потери веса в диапазоне от 25 до 220°C. Данные для Формы γ показаны на Фиг. 9.
Твердотельный 13C ЯМР
Данные твердотельного 13C ЯМР для Формы γ получали как описано выше в Примере 1. 13C ЯМР спектр приведен на Фиг. 12 и содержит пики: 16,9, 26,5, 32,9, 36,4, 48,1, 53,7, 78,6, 102,6, 116,4, 117,9, 121,5, 151,8, 154,6 и 162,9 м.д.
Пример 4
Количественная оценка содержания Форм β и γ в образцах Формы α
Производственные партии Соединения A, Форма α, а также препарат в виде мази, содержащей Соединение A, Форму α, тестировали на присутствие Форм β и γ. Используя метод порошковой рентгеновской дифракции можно количественно определить содержание Форм β и γ с точностью до 1 мас.%.
Партии Соединения A, Форма α, которые использовались для анализа, приведены в Таблице 8.
Партии мази, содержащей Форму α Соединения A (3%), которые использовались для анализа, приведены в Таблице 9. Отн.вл. означает относительную влажность. M означает длительность в месяцах.
Содержание Форм β и γ количественно оценивали в указанных выше партиях и мазях с использованием порошкового рентгеновского дифрактометра PANalytical X’Pert PRO со следующими рабочими параметрами:
(Product No. 9430 018 18251)
Держатель
(Product No. 9430 018 18401)
Тест на Форму β
Приготовление тестового образца (Соединение A, Форма α, лекарственная субстанция)
Часть тестируемого образца Соединения A измельчали в агатовой ступке в мелкий порошок. Примерно 10 мг закрепляли в держателе образца.
Приготовление тестируемого образца (мазь)
Примерно 1 грамм тестируемого образца мази суспендировали в 80 мл н-гексана. Колбу закрывали пробкой и интенсивно встряхивали 30 секунд. Полученную смесь отфильтровывали при отсасывании на фильтровальной бумаге для количественного анализа (5B), промывали порциями по 5 мл н-гексана пять раз. Примерно 10 мг осадка закрепляли в держателе образца.
Получение стандартного образца
К Соединению A, Партия U (Форма α), добавляли партию P-3273-58 (Форма β) с получением финальных концентраций 2, 5 и 10 мас./мас.%, и смешивали в ступке. Примерно 10 мг продукта закрепляли в держателе образца.
Методика
Анализ методом порошковой рентгеновской дифракции проводили для тестируемых образцов и стандартных образцов согласно установкам прибора и параметрам регистрации данных, указанным выше (см. X-ray Powder Diffraction Method, General Tests, Processes and Apparatus, The Japanese Pharmacopoeia, 17-е издание, 1 апреля 2016 г., английское издание, стр. 79-83). Площади дифракционного пика (10,6°), полученные для стандартных образцов и тестируемого образца, определяли автоматическим интегрированием или вручную. Количество (%) Формы β в тестируемом образце вычисляли с помощью калибровочной кривой, которую строили по площадям пиков стандартных образцов.
Спектры порошковой рентгеновской дифракции, полученные для стандартного образца, содержащего 0%, 2%, 5% и 10% мас./мас. Формы β, показаны на Фиг. 13.
Тест на Форму γ
Приготовление тестового образца (Соединение A, Форма α, лекарственная субстанция)
Часть тестируемого образца Соединения A измельчали в агатовой ступке в мелкий порошок. Примерно 10 мг закрепляли в держателе образца.
Приготовление тестируемого образца (мазь)
Примерно 1 грамм тестируемого образца мази суспендировали в 80 мл н-гексана. Колбу закрывали пробкой и интенсивно встряхивали 30 секунд. Полученную смесь отфильтровывали при отсасывании на фильтровальной бумаге для количественного анализа (5B), промывали порциями по 5 мл н-гексана пять раз. Примерно 10 мг осадка закрепляли в держателе образца.
Получение стандартного образца
К Партии U (Форма α) добавляли Соединение A Партия YKF56-28G (Форма γ) с получением финальных концентраций 1, 2 и 5 мас./мас.%, и смешивали в ступке. Примерно 10 мг продукта закрепляли в держателе образца.
Методика
Анализ методом порошковой рентгеновской дифракции проводили для тестируемых образцов и стандартных образцов согласно установкам прибора и параметрам регистрации данных, указанным выше (см. X-ray Powder Diffraction Method, General Tests, Processes and Apparatus, The Japanese Pharmacopoeia, 17-е издание, 1 апреля 2016 г., английское издание, стр. 79-83). Площади дифракционного пика (16,6°), полученные для стандартных образцов и тестируемого образца, определяли автоматическим интегрированием или вручную. Если площадь пика в тестируемом образце была не больше, чем в 1%-ном стандартном образце, значение фиксировали как «не больше 1%». Если площадь пика в тестируемом образце была больше, чем в 1%-ном стандартном образце, и не больше, чем в 2%-ном стандартном образце, значение фиксировали как «не больше 2%». Если площадь пика в тестируемом образце была больше, чем в 2%-ном стандартном образце, и не больше, чем в 5%-ном стандартном образце, значение фиксировали как «не больше 5%».
Спектры порошковой рентгеновской дифракции, полученные для стандартного образца, содержащего 0%, 1%, 2% и 5% мас./мас. Формы γ, показаны на Фиг. 14.
Результаты
Результаты количественного определения содержания сведены в Таблицу 10. Соединение A, фармацевтическая субстанция Партия P, JTE-052 Мазь Партия 223-1A и Партия 239-1A, фармацевтическая субстанция Производственная Партия P - все содержали измеримые количества Формы β.
Пример 5
Рентгеноструктурный анализ монокристалла
Рентгеновские структуры монокристаллов были определены для Форм α, β и γ согласно указанным ниже параметрам. Структурные данные приведены в Таблице 11.
Для Формы α рентгеновскую структуру монокристалла определяли с применением детектора Rayonix MX225HE и источника излучения SPring-8 BL41XU с длиной волны излучателя 0,71068 Å.
Для Форм β и γ рентгеновскую структуру монокристаллов определяли с применением детектора DECTRIS PILATUS3 6M и источника излучения SPring-8 BL41XU с длиной волны излучателя 0,71068 Å.
(0,8-1,3)
Пример 6
Сравнительный эксперимент между Формами α и β Соединения A
Формы α и β Соединения A смешивали в соотношении 1:1 по массе, и в полученную смесь (80 мг) добавляли 1-бутанол (800 мкл). Полученную суспензию перемешивали при комнатной температуре в течение 12 дней. Осадок отфильтровывали при той же температуре и затем сушили при пониженном давлении при комнатной температуре в течение 19 часов. Дифракционные углы 2θ и интенсивность дифракционных пиков определяли методом порошковой рентгеновской дифракции для использовавшихся Форм α и β Соединения A, для смеси 1:1 по массе Форм α и β Соединения A до перемешивания, и для полученных кристаллов. Прибор и параметры регистрации спектров в этом случае такие же, как описанные выше для исследования Формы α в Примере 1. Полученные спектры показаны на Фиг. 15. На этой Фигуре изображены спектры использовавшейся Формы α Соединения A, использовавшейся Формы β Соединения A, смеси 1:1 по массе Форм α и β Соединения A до перемешивания, и полученных после перемешивания кристаллов, снизу вверх.
Сравнивали соотношение интенсивностей дифракционных пиков до и после перемешивания: пика с дифракционным углом 2θ 10,2° для Формы α и пика с дифракционным углом 2θ 10,6° для Формы β. Как показано в приведенной ниже таблице, соотношение интенсивностей дифракционного пика Формы β и пика Формы α после перемешивания увеличивалось по сравнению с соотношением до перемешивания.
Таблица 12
(Форма α) [имп/с]
(Форма β) [имп/с]
Пример 7
Сравнительный эксперимент между Формами α и γ Соединения A
Формы α и γ Соединения A смешивали в соотношении 1:1 по массе, и в полученную смесь (100 мг) добавляли 1-бутанол (1 мл). Полученную суспензию перемешивали при комнатной температуре в течение 11 дней. Осадок отфильтровывали при той же температуре. Дифракционные углы 2θ и интенсивность дифракционных пиков определяли методом порошковой рентгеновской дифракции для использовавшихся Форм α и γ Соединения A, для смеси 1:1 по массе Форм α и γ Соединения A до перемешивания, и для полученных кристаллов. Прибор и параметры регистрации спектров в этом случае такие же, как описанные выше для исследования Формы α в Примере 1. Полученные спектры показаны на Фиг. 16. На этой Фигуре изображены спектры использовавшейся Формы α Соединения A, использовавшейся Формы γ Соединения A, смеси 1:1 по массе Форм α и β Соединения A до перемешивания, и полученных после перемешивания кристаллов, снизу вверх.
Сравнивали соотношение интенсивностей дифракционных пиков до и после перемешивания: пика с дифракционным углом 2θ 10,2° для Формы α и пика с дифракционным углом 2θ 10,7° для Формы γ. Как показано в приведенной ниже таблице, соотношение интенсивностей дифракционного пика Формы γ и пика Формы α после перемешивания увеличивалось по сравнению с соотношением до перемешивания.
Таблица 13
(Форма α) [имп/с]
(Форма γ) [имп/с]
Пример 8
Сравнительный эксперимент между Формами β и γ Соединения A
(1) Формы β и γ Соединения A смешивали в соотношении 1:1 по массе, и в полученную смесь (150 мг) добавляли формамид (1 мл). Полученную суспензию перемешивали при комнатной температуре 1 день. Осадок отфильтровывали при той же температуре.
(2) Формы β и γ Соединения A смешивали в соотношении 1:1 по массе, и в полученную смесь (150 мг) добавляли N,N-диметилформамид (1 мл). Полученную суспензию перемешивали при комнатной температуре 1 день. Осадок отфильтровывали при той же температуре.
(3) Формы β и γ Соединения A смешивали в соотношении 1:1 по массе, и в полученную смесь (800 мг) добавляли диметилсульфоксид (1 мл). Полученную суспензию перемешивали при комнатной температуре 1 день. Осадок отфильтровывали при той же температуре.
Дифракционные углы 2θ и интенсивность дифракционных пиков определяли методом порошковой рентгеновской дифракции для использовавшихся Форм β и γ Соединения A, смеси 1:1 по массе Форм β и γ Соединения A до перемешивания, и для кристаллов, полученных, как описано выше в (1), (2) и (3). Прибор и параметры регистрации спектров в этом случае такие же, как описанные выше для исследования Формы α в Примере 1. Полученные спектры показаны на Фиг. 17. На этой Фигуре изображены спектры использовавшейся Формы γ Соединения A, использовавшейся Формы β Соединения A, смеси 1:1 по массе Форм β и γ Соединения A до перемешивания, и кристаллов, полученных после перемешивания в формамиде, в N,N-диметилформамиде и в диметилсульфоксиде, снизу вверх.
Согласно результатам для описанных выше (1), (2) и (3), смеси кристаллических форм формировали единственную кристаллическую форму γ.
Пример 9
Тестирование растворимости
Каждый образец (т.е. Формы α, β и γ Соединения A) отвешивали в стеклянный сосуд объемом 4 мл. В сосуд добавляли тестовый раствор (3 мл) и смесь суспендировали. Примерно 150 мг образца отвешивали для тестового раствора, представляющего собой жидкость №1 для проведения теста растворимости по Японской Фармакопее (JP 1), и примерно 15 мг образца отвешивали для тестового раствора, представляющего собой жидкость №2 для проведения теста растворимости по Японской Фармакопее (JP 2), и для воды. Каждую смесь встряхивали на шейкере с точным поддержанием заданной температуры (производство TAITEC) при 20°C в течение 3 часов. После встряхивания отфильтровывали надосадочный раствор через политетрафторэтиленовый дисковый фильтр (Millex-LG; производство Millipore) с размером частиц 0,2 мкм и диаметром 4 мм. Дальнейший анализ проводили методом высокоэффективной жидкостной хроматографии (ВЭЖХ). Полученные результаты показаны в приведенной ниже таблице.
Таблица 14
(безводная форма)
Жидкость JP 2 (Японская Фармакопея): Смешать один объем воды с одним объемом фосфатно-солевого буферного раствора с pH = 6,8.
Форма α показала самую высокую растворимость в каждом из протестированных растворителей, а Форма γ показала самую низкую растворимость в каждом из протестированных растворителей.
Пример 10
Препараты
Примеры препаратов, содержащих Соединение A (например, в виде Формы α, Формы β, Формы γ или смеси любых из перечисленных), приведены ниже, и они не являются ограничивающими.
Препарат 1 (Приготовление капсулы)
1) Соединение A 30 мг
2) Микрокристаллическая целлюлоза 10 мг
3) Лактоза 19 мг
4) Стеарат магния 1 мг
1), 2), 3) и 4) смешивают для заполнения в желатиновую капсулу.
Препарат 2 (Приготовление таблетки)
1) Соединение A 10 г
2) Лактоза 50 г
3) Кукурузный крахмал 15 г
4) Кармеллоза кальция 44 г
5) Стеарат магния 1 г
Все количество 1), 2) и 3) и 30 грамм 4) объединяли с водой, сушили в вакууме и затем гранулировали. Полученные гранулы смешивали с 14 граммами 4) и 1 граммом 5), и таблетировали на таблеточном прессе. Таким образом получали 1000 таблеток, каждая из которых содержала 10 мг Соединения A.
Различные модификации настоящего изобретения, в дополнение к описанным в настоящем документе, будут очевидны специалистам в рассматриваемой области техники из приведенного выше описания. Такие модификации также входят в объем настоящего изобретения. Каждая литературная ссылка, включая все процитированные в настоящей заявке патенты, заявки на патенты и публикации в журналах, включена в настоящий текст в полном объеме посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 7Н-ПИРРОЛО[2,3-d]ПИРИМИДИНА И ИХ СОКРИСТАЛЛОВ | 2017 |
|
RU2779212C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 7Н-ПИРРОЛО[2,3-D] ПИРИМИДИНА И ЕГО ИНТЕРМЕДИАТА | 2016 |
|
RU2755618C2 |
| ГИДРОКСИАЛКИЛТИАДИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2737892C1 |
| СОЛЬ ПРОИЗВОДНОГО ПИРРОЛИДИН-3-ИЛ-УКСУСНОЙ КИСЛОТЫ И ЕЕ КРИСТАЛЛЫ | 2014 |
|
RU2640047C2 |
| СОЛИ И ТВЕРДЫЕ ФОРМЫ МОНОБАКТАМНОГО АНТИБИОТИКА | 2016 |
|
RU2754180C2 |
| АМИНОВАЯ СОЛЬ И ЕЕ КРИСТАЛЛЫ | 2013 |
|
RU2658823C2 |
| КРИСТАЛЛ ПРОИЗВОДНОГО 1,3,5-ТРИАЗИНА ИЛИ ЕГО СОЛЬВАТА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2837449C1 |
| СОЛИ ПРОИЗВОДНОГО ИНДАЗОЛА И ИХ КРИСТАЛЛЫ | 2017 |
|
RU2747399C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2801220C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2838718C2 |
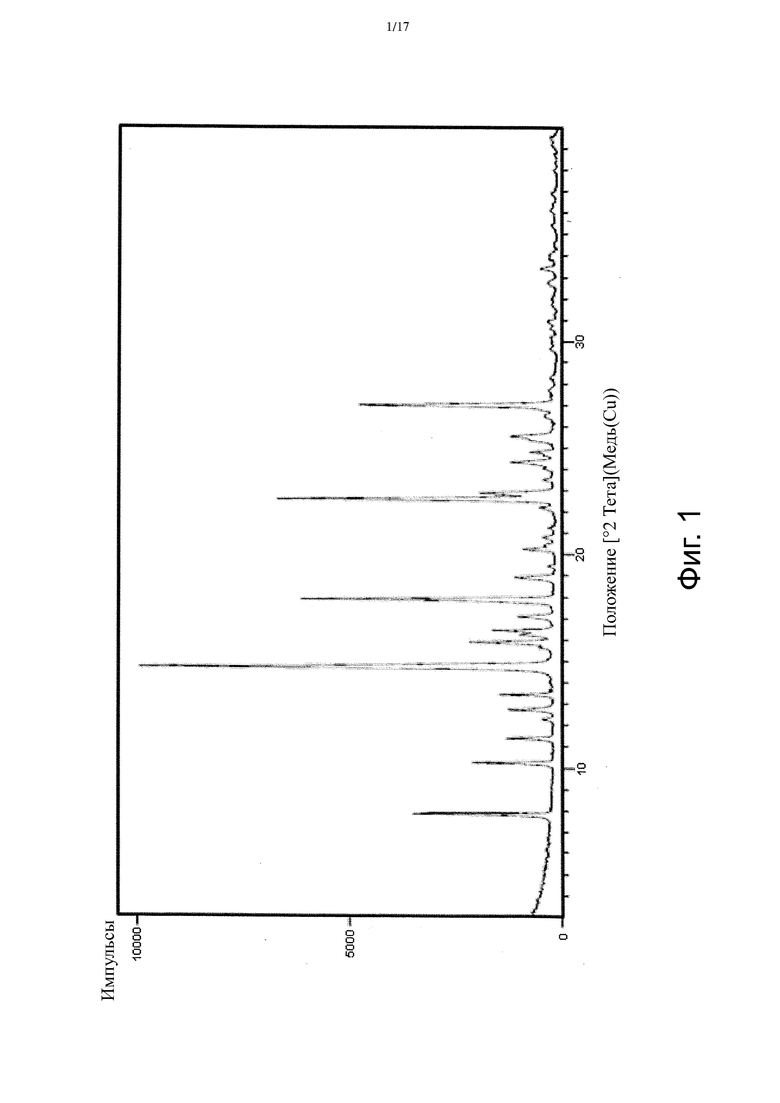
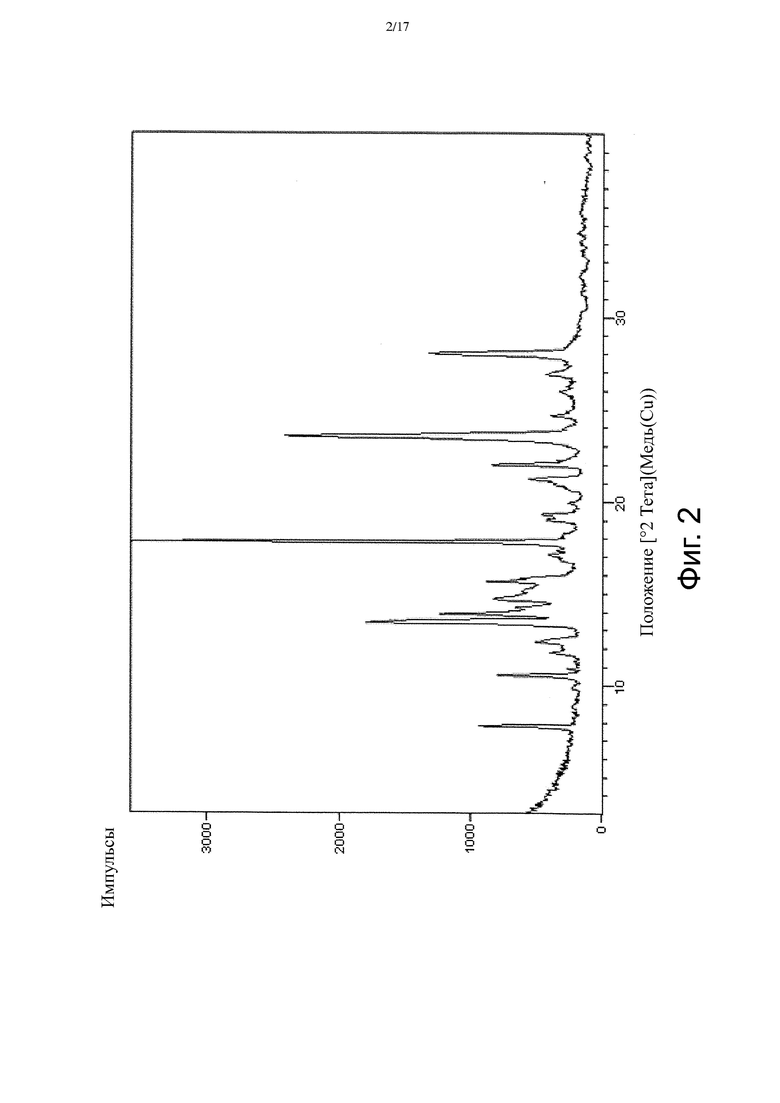
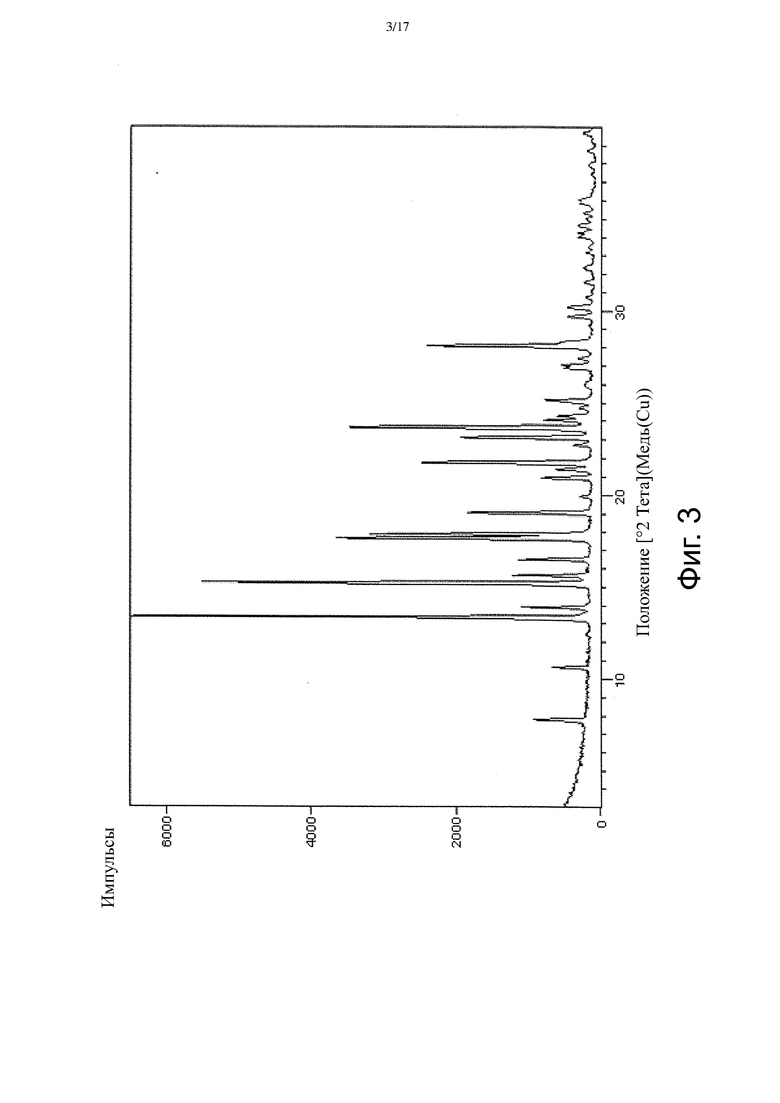
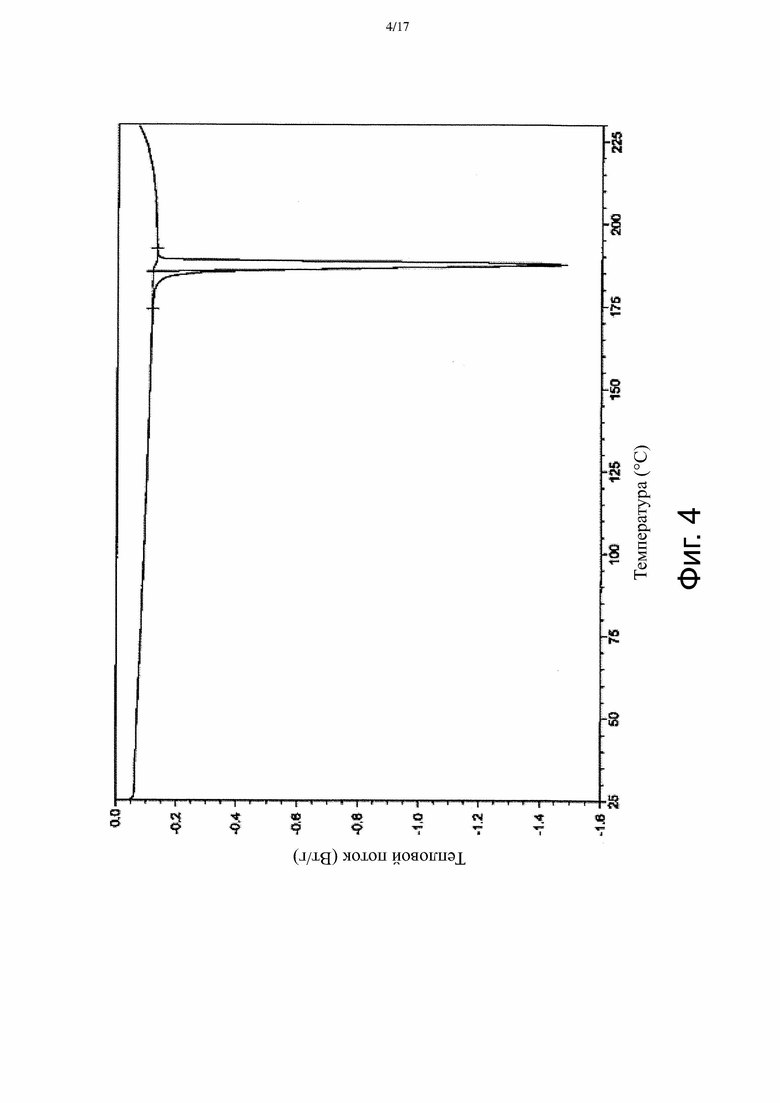
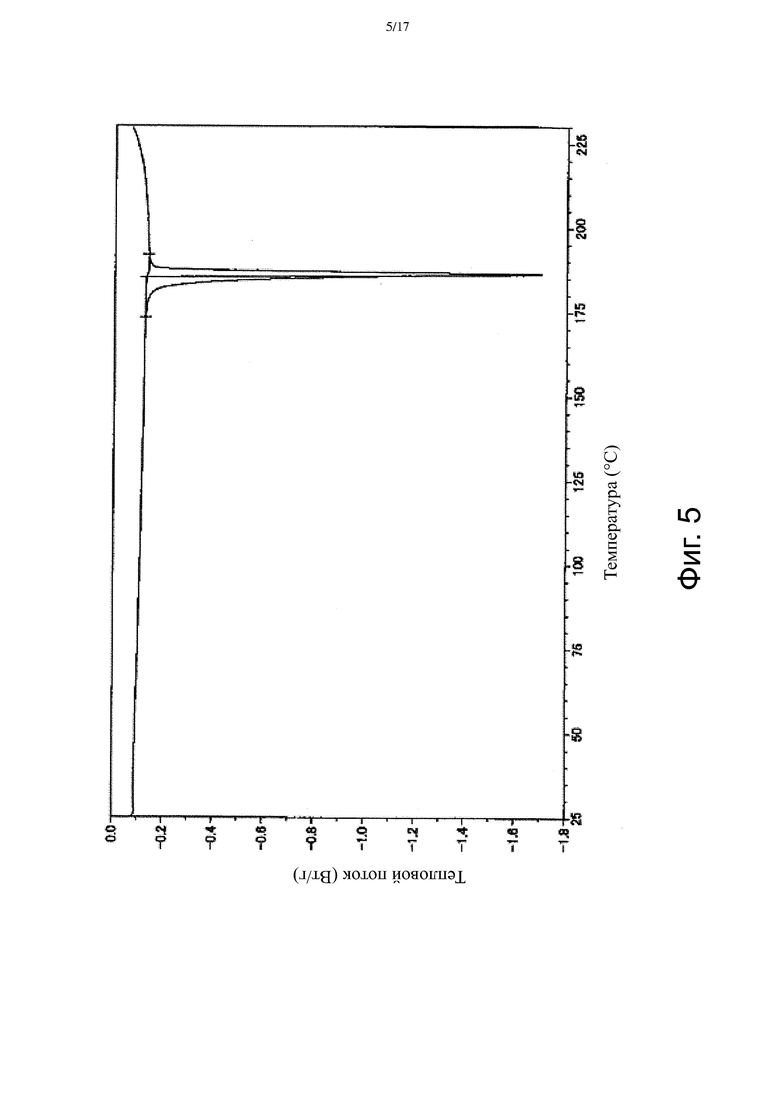
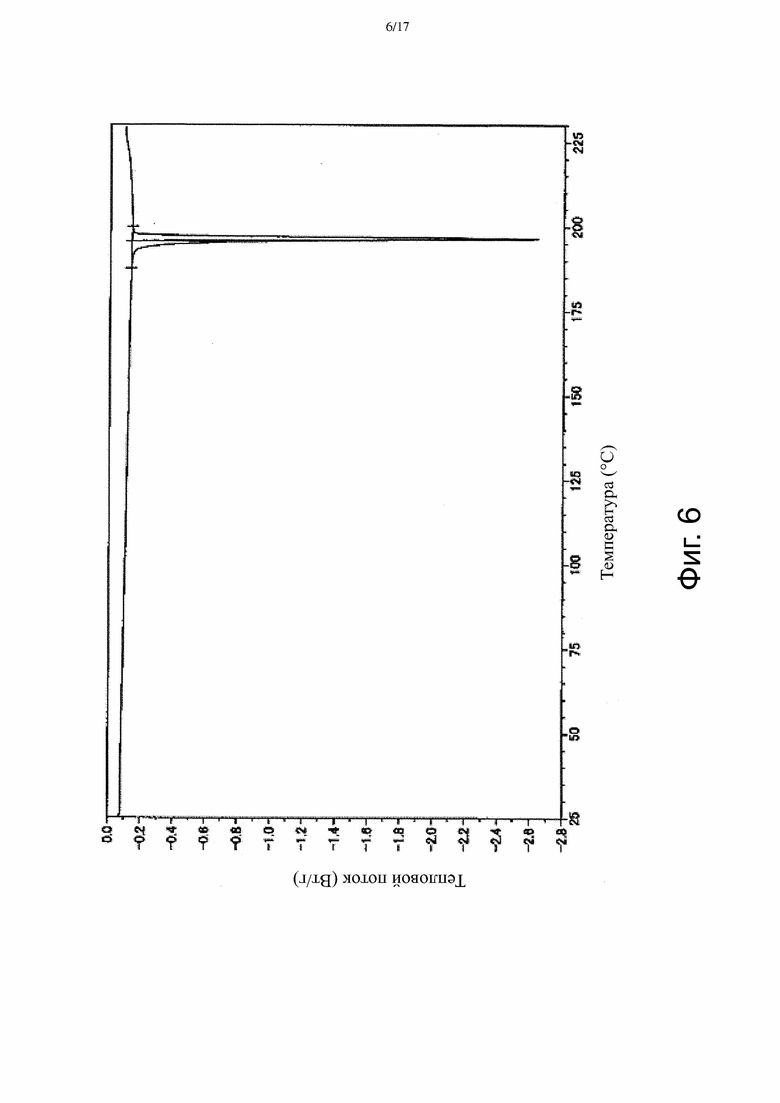
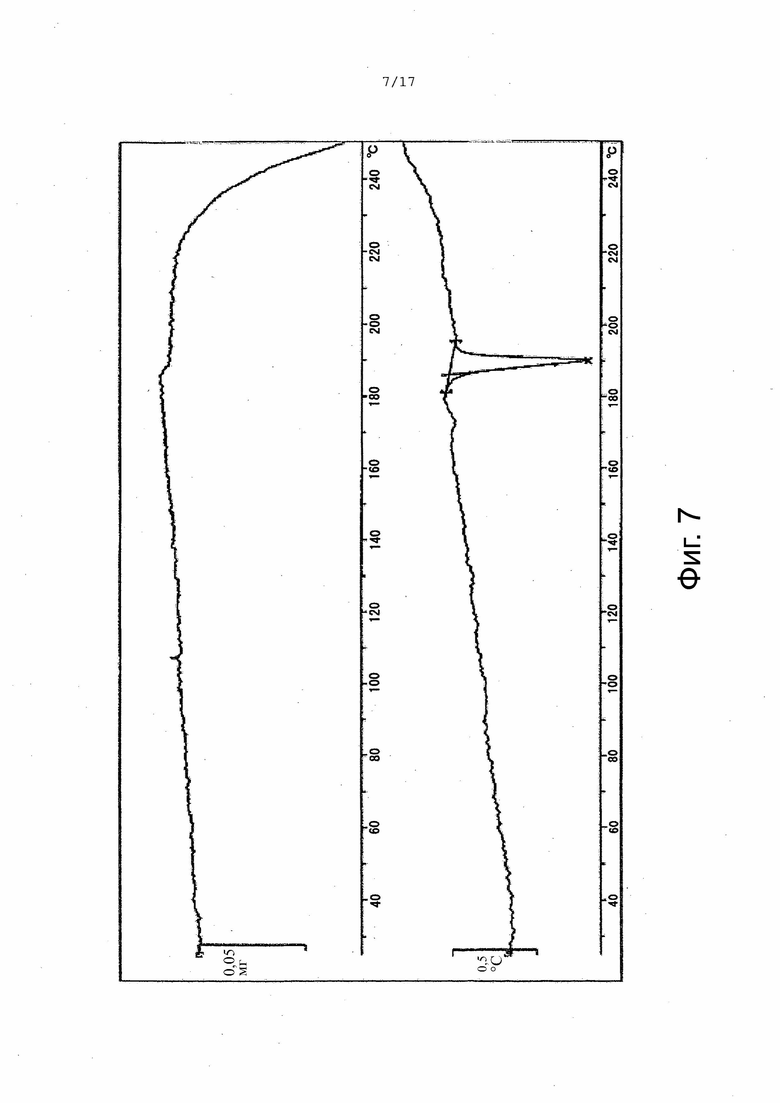
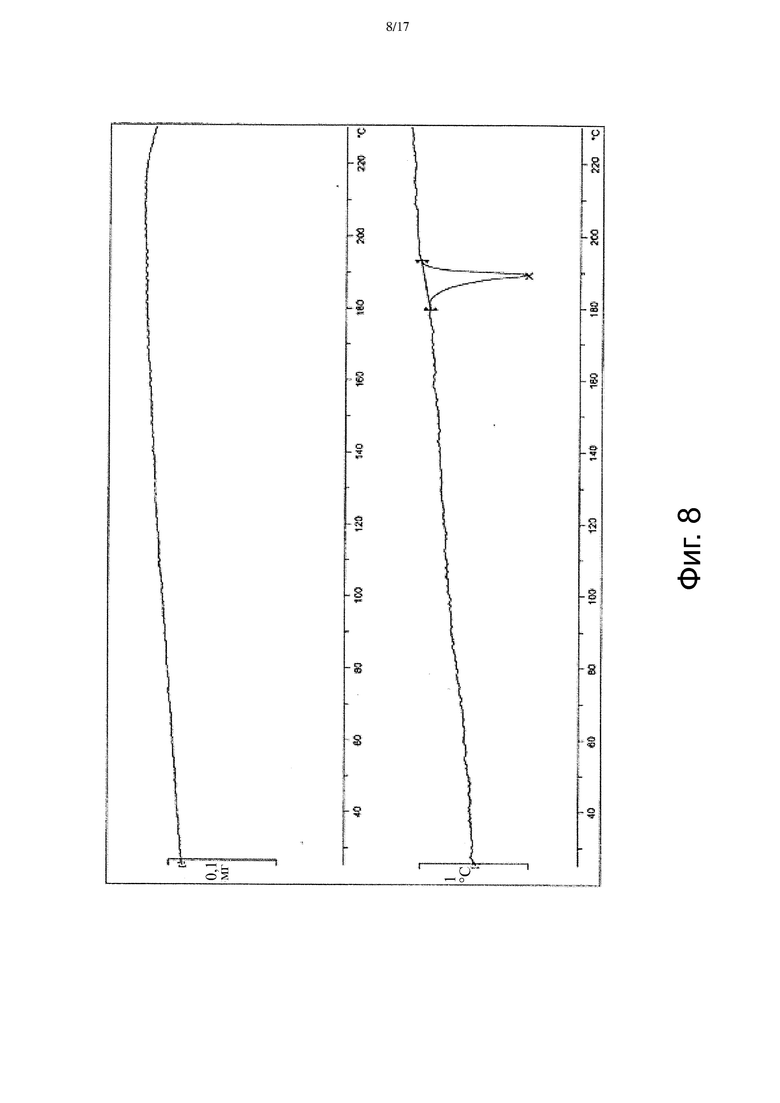
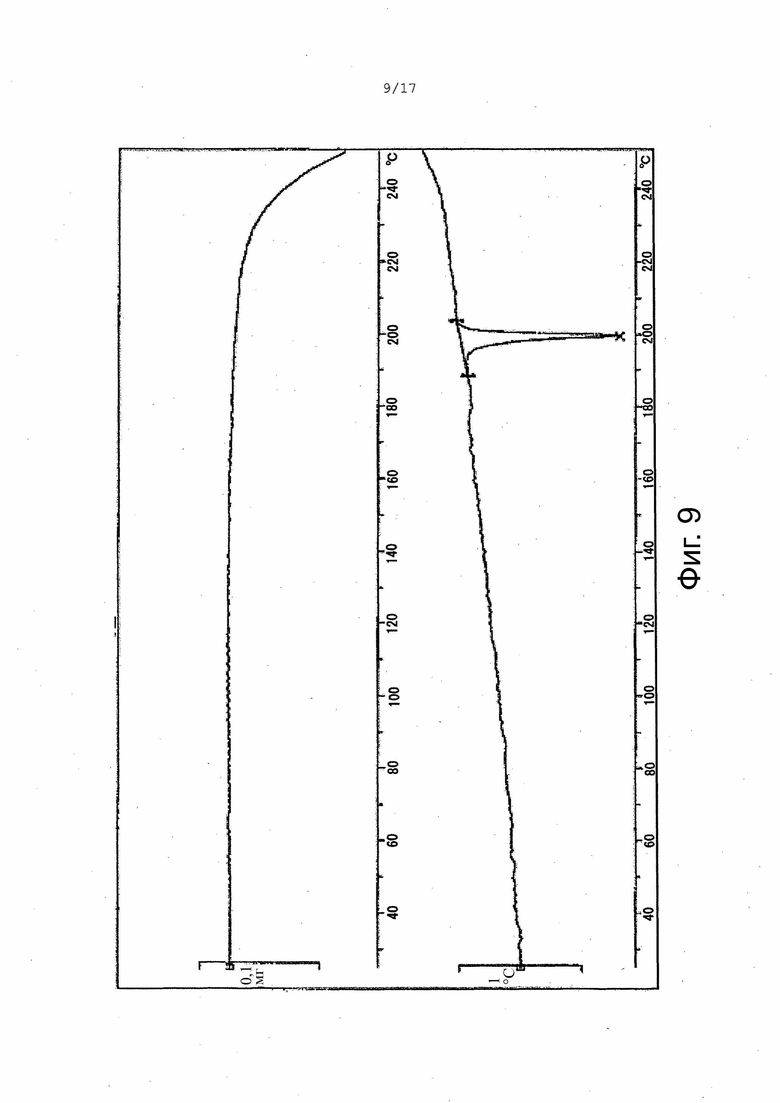
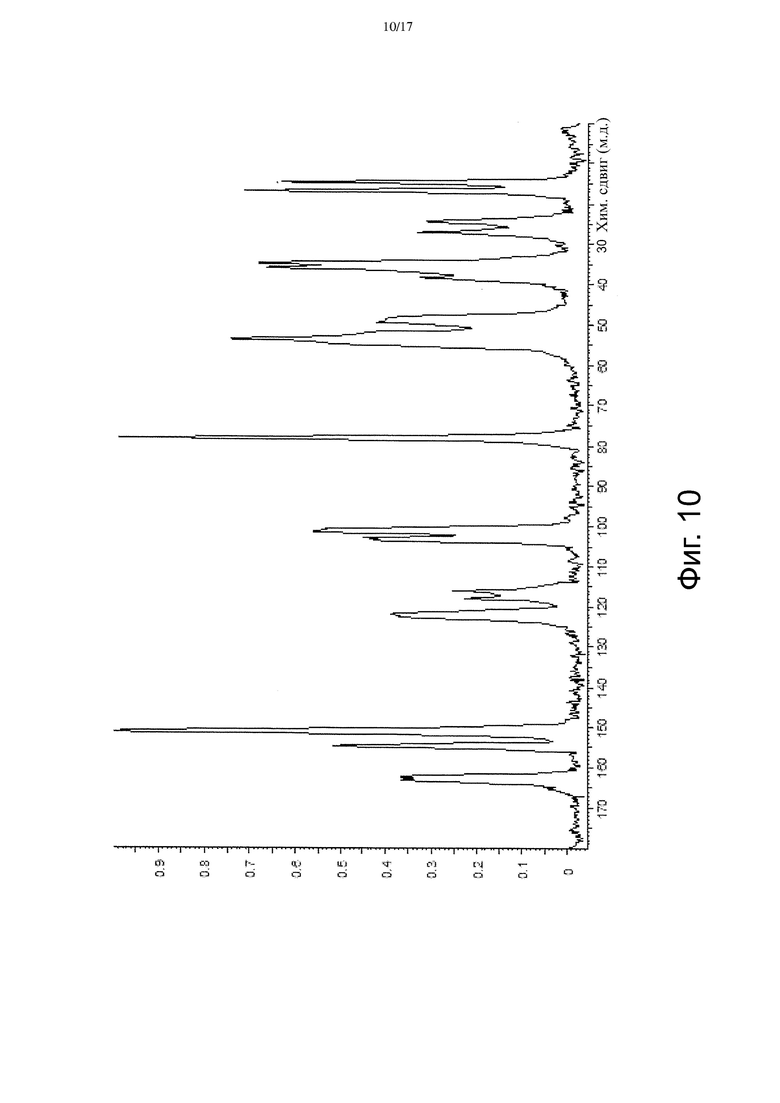
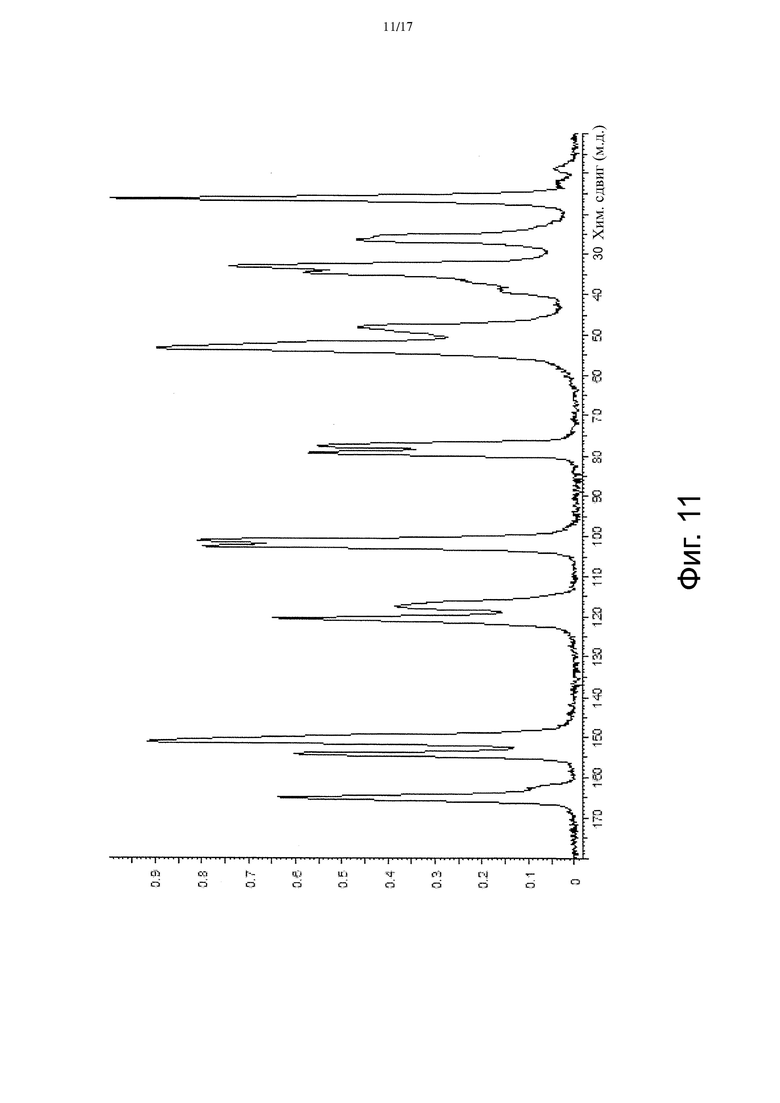
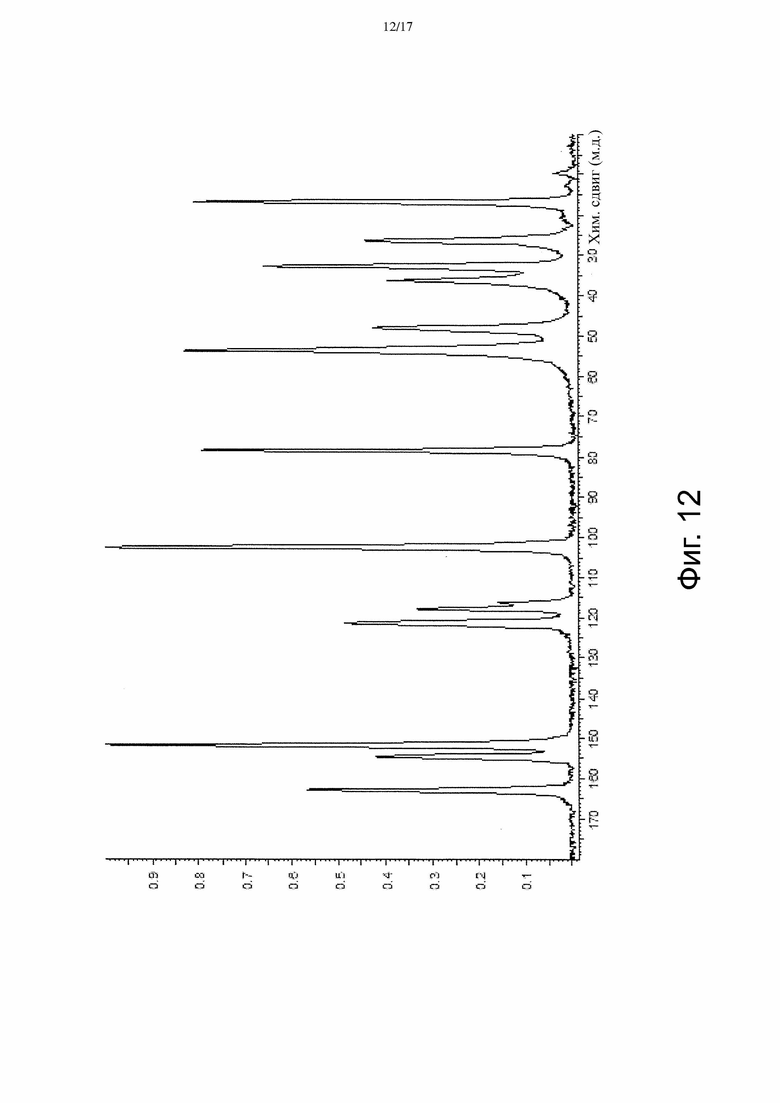
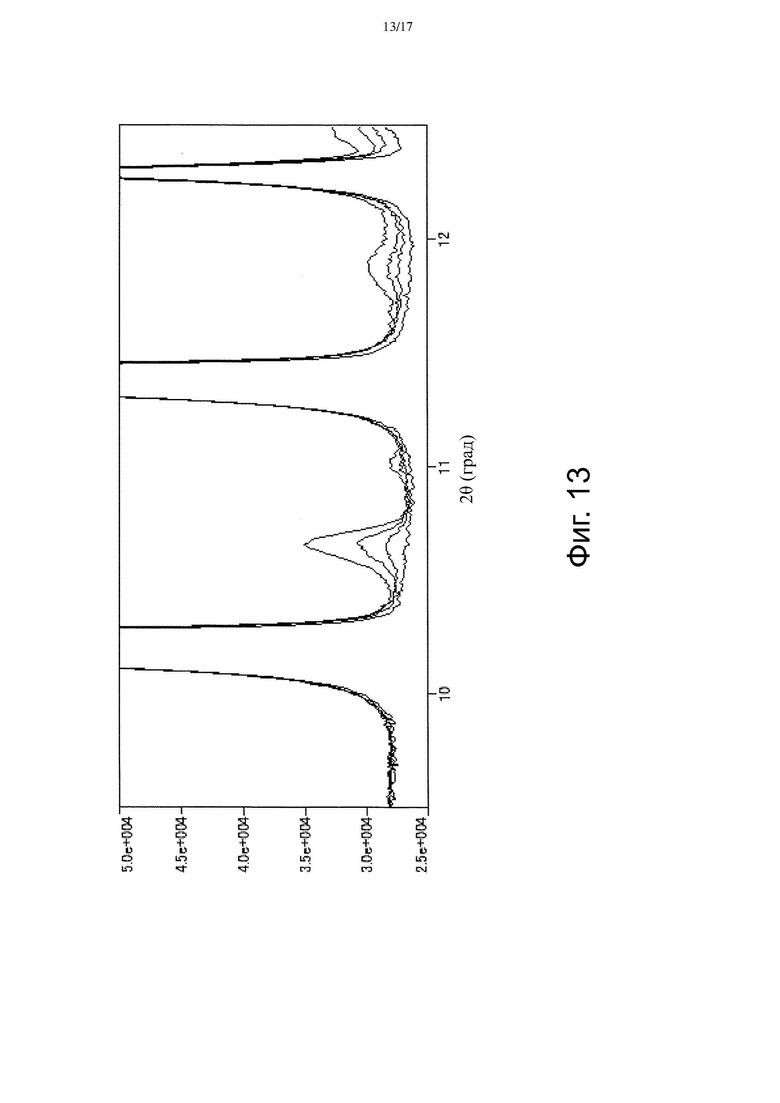
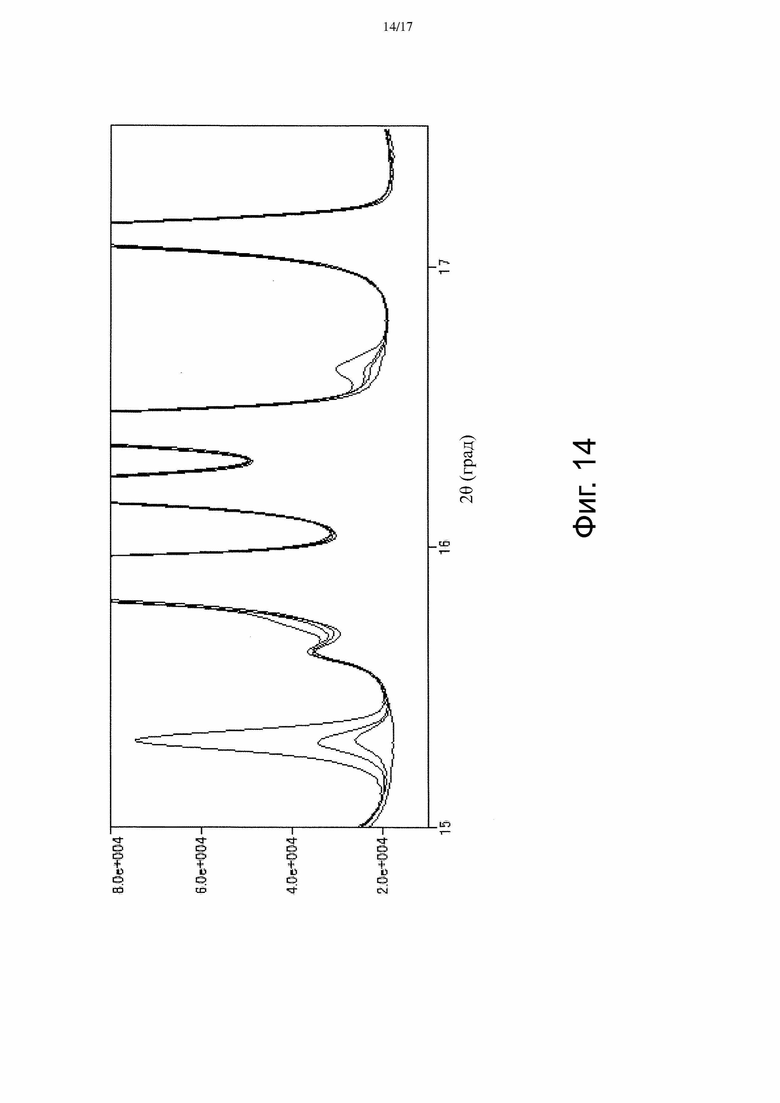
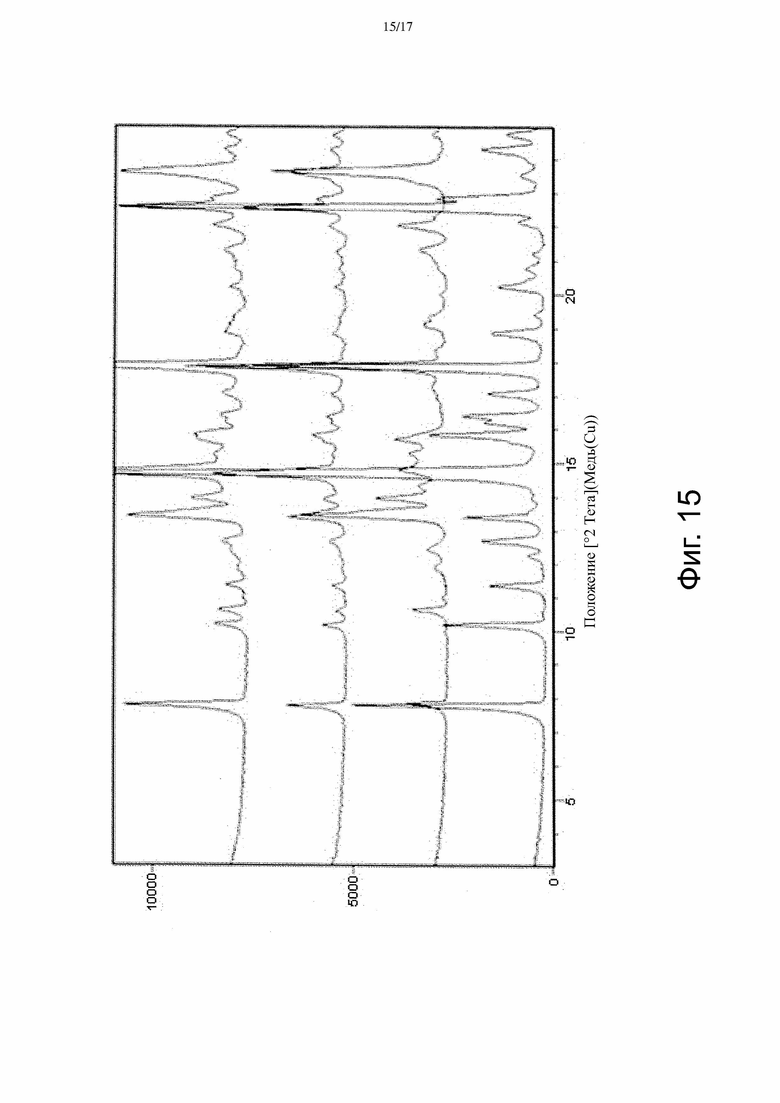
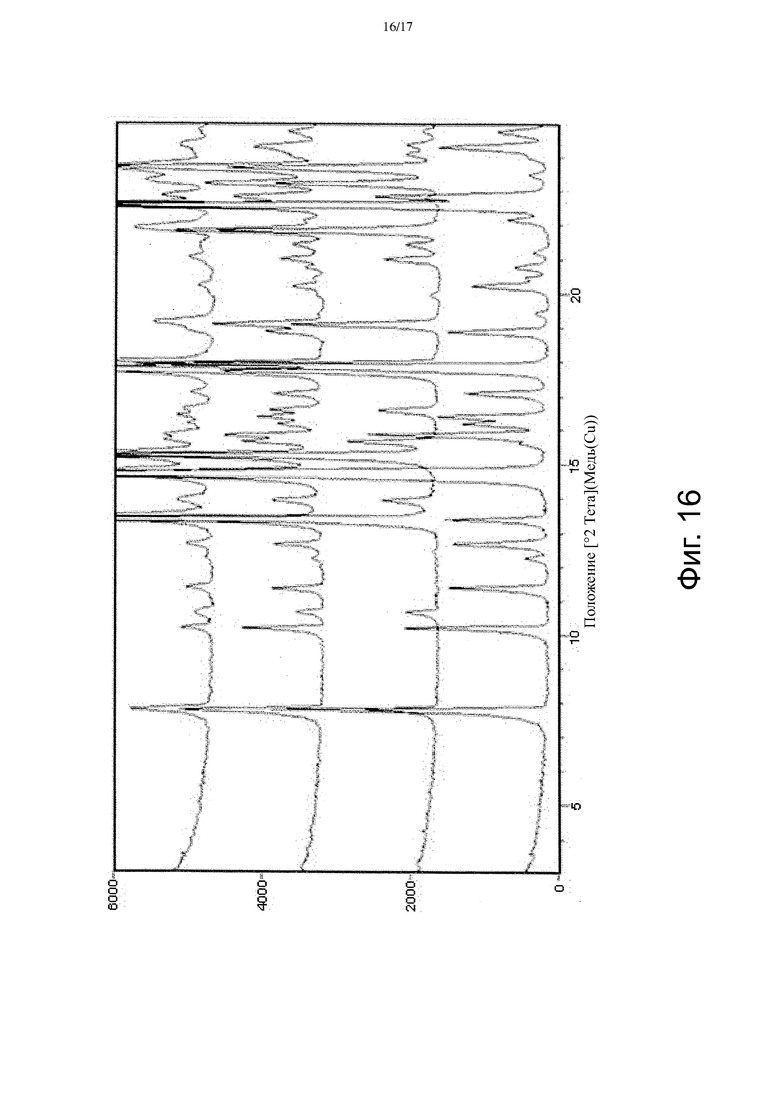

Изобретение относится к способу получения кристаллической формы 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила, имеющей Форму β, имеющей спектр порошковой рентгеновской дифракции, содержащий характеристические пики со значениями 2θ (°): 10,5±0.2, 11,8±0.2, 19,3±0.2 и 22,0±0.2. Способ осуществляют путем кристаллизации 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила из растворителя, в котором растворитель содержит 1:3 (об/об) 1-бутанола и ацетонитрила соответственно. При этом смесь соединения и растворителя перемешивают при 40°С в течение 10 дней, с последующим охлаждением до комнатной температуры. Технический результат – получение стабильной кристаллической формы 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила. 17 ил., 14 табл., 10 пр.
Способ получения кристаллической формы 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила, имеющей Форму β, имеющей спектр порошковой рентгеновской дифракции, содержащий характеристические пики со значениями 2θ (°): 10,5±0.2, 11,8±0.2, 19,3±0.2 и 22,0±0.2, включающий кристаллизацию 3-((3S,4R)-3-метил-6-(7H-пирроло[2,3-d]пиримидин-4-ил)-1,6-диазаспиро[3.4]октан-1-ил)-3-оксопропаннитрила из растворителя, в котором растворитель содержит 1:3 (об/об) 1-бутанола и ацетонитрила соответственно, при этом смесь соединения и растворителя перемешивают при 40°С в течение 10 дней, с последующим охлаждением до комнатной температуры.
| US 2011136778 A1, 09.06.2011 | |||
| MINO R | |||
| CAIRA | |||
| "Crystalline Polymorphism of Organic Compounds", TOPICS IN CURRENT CHEMISTRY, 1998, vol.198, pp.163-208 | |||
| Anamika Sharma и др | |||
| "Polymorphism in pharmaceutical compounds", январь 2011, конференция Advancement and futuristic trends in material science" с.39-48 | |||
| "Polymorphism: in the pharmaceutical |

