Область техники, к которой относится изобретение
Настоящее изобретение относится к области фармацевтики, а конкретнее – оно касается бензоксазин-оксазолидиноновых соединений, замещенных азотсодержащим гетероциклом, имеющих формулу (I), способа их получения, фармацевтической композиции, содержащей указанные соединения в качестве действующих веществ, и их применения в производстве лекарственного средства для лечения и/или профилактики инфекционного заболевания, вызванного Mycobacterium tuberculosis.
Предшествующий уровень техники
Туберкулез (ТБ) представляет собой хроническую болезнь со смертельным исходом, вызванную Mycobacterium tuberculosis, а также серьезное инфекционное заболевание, угрожающее здоровью человека и приводящее к смерти. Туберкулез, наряду со СПИД, является одной из главных причин смерти во всем мире. Согласно отчету (Global tuberculosis report 2015) Всемирной Организации Здравоохранения (ВОЗ), оценочно в 2014 году в мире страдали от туберкулеза 9.6 миллиона человек, включая 5.4 миллиона мужчин, 3.2 миллиона женщин и 1 миллион детей. В 2014 году от туберкулеза умерло 1.5 миллиона человек (1.1 миллион ВИЧ-отрицательных и 0.4 миллиона ВИЧ-положительных пациентов), включая 890000 мужчин, 480000 женщин и 140000 детей.
Химическая терапия является главным методом лечения туберкулеза. Применение стрептомицина в 1944 году открыло новую эру противотуберкулезных препаратов. Разработка изониазида, рифампицина и пиразинамида сократила длительность лечения туберкулеза до шести месяцев, открыв эру "короткой химиотерапии". Однако пациентам трудно привыкнуть к регулярному приему лекарств из-за возникновения побочных эффектов от длительной комбинированной терапии. Кроме того, большинство лекарственных средств было разработано в пятидесятые и шестидесятые годы прошлого столетия, а длительное широкое и неправильное применение лекарственных средств вызвало развитие лекарственно-устойчивых бактерий и появление туберкулеза с множественной лекарственной устойчивостью (MDR-TB), туберкулеза с широкой лекарственной устойчивостью (XDR-TB), туберкулеза с тотальной лекарственной устойчивостью (TDR-TB), которые требуют применения дорогих и более токсичных противотуберкулезных лекарственных средств второй линии и даже третьей линии.
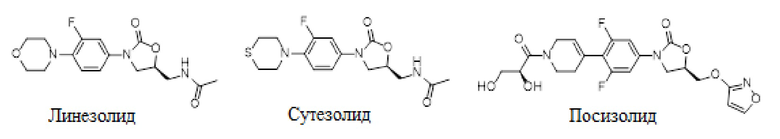
В сравнении с существующими противотуберкулезными лекарственными средствами, оксазолидиноны имеют новую химическую структуру и уникальный механизм действия. Линезолид, одобренный FDA в США в 2000 году, использовали для лечения грамположительных бактериальных инфекций с наибольшей продолжительностью периода лечения 28 дней. Линезолид применяют в клинике для лечения туберкулеза по незарегистрированным показаниям, потому что его механизм действия отличается от противотуберкулезных лекарственных средств, использующихся в настоящий момент в клинике. Линезолид показывает преимущество в лечении сложных случаев MDR-TB/XRD-TB. Однако, длительный период лечения туберкулеза (6 месяцев или больше) и серьезные побочные эффекты сильно ограничивают применение линезолида в лечении туберкулеза (Linezolid in the treatment of drug-resistant tuberculosis: the challenge of its narrow therapeutic index. Expert Review of Anti-infective Therapy. 2016, 14 (10):901-915). Токсичность по отношению к крови (лейкоцитопения, гемоцитопения, тромбоцитопения и анемия) является симптомом токсичности в отношении костного мозга, среди прочих побочных эффектов, и ее связывают с ингибированием синтеза белка митохондрий (MPS) (Inhibition of Mammalian Mitochondrial Protein Synthesis by Oxazolidinones. Antimicrobial Agents and Chemotherapy, 2006, 50(6): 2042-2049). Сутезолид, находящийся на II фазе клинических испытаний, показал более высокую противотуберкулезную активность in vitro, чем линезолид, а также более высокую безопасность в клинических испытаниях (без гемоцитопении и периферической нейропатии). Однако, у некоторых пациентов в клинических испытаниях наблюдалось повышение уровня ALT, поэтому есть опасения относительно гепатотоксичности сутезолида. Посизолид (AZD5847), разработанный компанией AstraZeneca, перешел во II стадию клинических испытаний, однако испытания были прерваны из-за проблем с безопасностью и эффективностью.
В свете описанных выше ситуаций, в настоящее время существует потребность в исследовании и разработке новых оксазолидинонов с новыми структурами, более высокой противотуберкулезной активностью и меньшими побочными эффектами.
В международной заявке на патент WO2011/147259 A1, поданной 1 декабря 2011 года, описаны соединения, имеющие приведенную ниже общую формулу (IV), которые были использованы для лечения инфекционных заболеваний, в особенности инфекционных заболеваний, вызванных бактериями с множественной лекарственной устойчивостью, выбранными из группы, состоящей из Enterococcus, Staphylococcus aureus, Staphylococcus epidermidis, и pneumococcus:
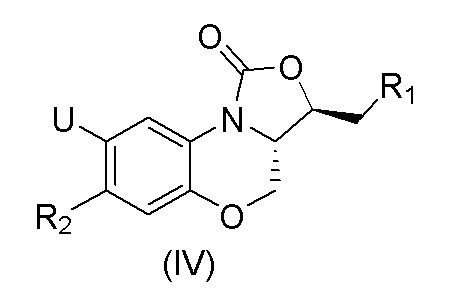
где U представляет собой H или F, R1 представляет собой 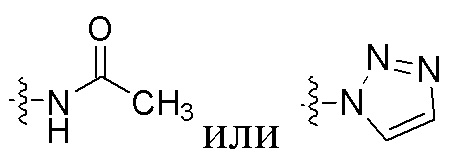 , R2 представляет собой фенильную группу или 5-членную или 6-членную ароматическую или неароматическую гетероциклическую группу.
, R2 представляет собой фенильную группу или 5-членную или 6-членную ароматическую или неароматическую гетероциклическую группу.
В патенте CN102260277 B, выданном 24 июля 2013 года, описаны соединения, имеющие формулу (IV), где R2 представляет собой фенильную группу или 5-членную или 6-членную ароматическую гетероциклическую группу.
В документе WO2011/147259 A1, бактерии с множественной лекарственной устойчивостью, выбранные из группы, состоящей из Enterococcus, Staphylococcus aureus, Staphylococcus epidermidis и pneumococcus, относятся к фирмикутам (Firmicutes). Однако, Mycobacterium tuberculosis относится к актинобактериям (Actinobacteria). Таксономически они отличаются. Нет сведений, что соединения, имеющие формулу (IV) из документа WO2011/147259 A1, обладают активностью в лечении Mycobacterium tuberculosis.
Кроме того, в другом документе (J. Med. Chem. 2011, 54, 7493–7502) описано следующее соединение:
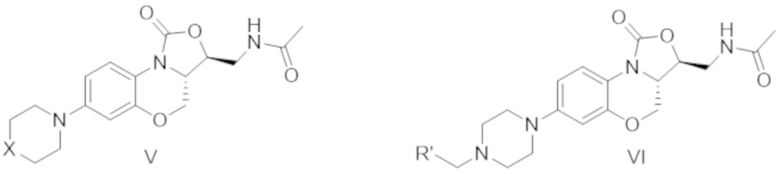
где X представляет собой N или O, R' представляет собой 3-нитрофенил, 2-нитрофенил, 4-пиридинил, 3-пиридинил, 2-пиридинил, 2-фуранил, 3-фуранил и 2-нитро-5-фуранил. В этом документе описана их активность против Staphylococcus aureus, метициллин-резистентного Staphylococcus aureus, метициллин-резистентного Staphylococcus epidermis, пенициллин-резистентного Streptococcus pneumoniae и enterococcus. Нет сведений, что соединение из указанного документа обладает активностью в лечении Mycobacterium tuberculosis.
Краткое описание изобретения
Задачей настоящего изобретения является разработка бензоксазин-оксазолидиноновых соединений, замещенных азотсодержащим гетероциклом, имеющих новую структуру, низкую токсичность и высокую активность против Mycobacterium tuberculosis, а также прекрасные фармакокинетические свойства. Авторы настоящего изобретения обнаружили, что указанные соединения обладают высокой активностью против Mycobacterium tuberculosis и низкой цитотоксичностью, в особенности соединения по настоящему изобретению оказывают очень слабое ингибирующее действие на синтез белков митохондрий, что может сильно уменьшить токсичность в отношении костного мозга. Таким образом, в настоящем изобретении описан класс бензоксазин-оксазолидиноновых соединений с новой структурой, высокой противотуберкулезной активностью, высокой безопасностью и прекрасными фармакокинетическими свойствами, которые можно применять для лечения туберкулеза. Это легло в основу настоящего изобретения.
В первом аспекте настоящего изобретения описано соединение, имеющее формулу (I), или его изомеры или его фармацевтически приемлемая соль,
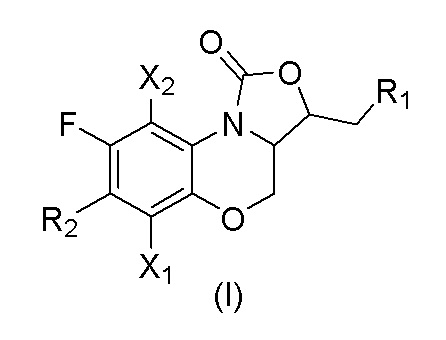
где
X1 и X2 каждый независимо выбран из H или F;
R1 представляет собой -OR3, -NHR3, -NHCOR3, -NHCSR3, -NHSO2R3, -NHCOOR3, -NHCSOR3, -NHCONHR3, -NHCSNHR3, замещенный или незамещенный 5-6-членный гетероарил;
R3 независимо выбран из группы, состоящей из атома водорода, замещенного или незамещенного C1-C4 алкила, замещенного или незамещенного 3-6-членного циклоалкила, замещенной или незамещенной 3-6-членной гетероциклической группы, замещенного или незамещенного 5-6-членного гетероарила, замещенного или незамещенного фенила;
указанный замещенный или незамещенный 5-6-членный гетероарил в R1 или R3 и замещенная или незамещенная 3-6-членная гетероциклическая группа в R3 содержат по меньшей мере один гетероатом, выбранный из N, O или S;
заместители в R1 или R3 каждый независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы;
R2 представляет собой замещенный или незамещенный 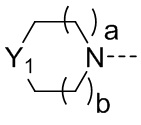 , замещенный или незамещенный
, замещенный или незамещенный 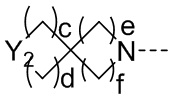 , замещенный или незамещенный
, замещенный или незамещенный  или замещенный или незамещенный
или замещенный или незамещенный  ;
;
Y1 представляет собой -S-, -S(=O)-, -S(O2)-, -C(HF)-, -C(F2)- или -C(=O)-;
Y2 представляет собой -O-, -S-, -S(=O)-, -S(O2)-, -C(HF)-, -C(F2)- или -C(=O)-;
a и b каждый равны 0, 1 или 2;
c и d каждый равны 0, 1 или 2, и c и d не могут быть равны 0 одновременно;
e и f каждый равны 1 или 2;
заместители в R2 независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы.
В предпочтительном варианте осуществления, в настоящем изобретении описано соединение, имеющее формулу (II), или его изомеры или его фармацевтически приемлемая соль,
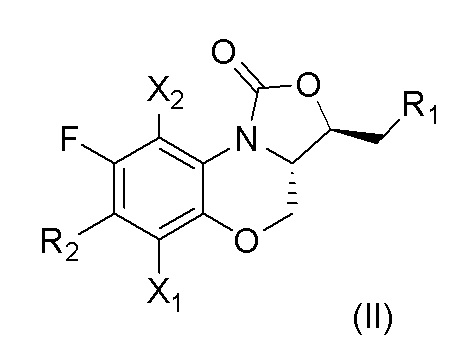
где
X1, X2, R1 и R2 имеют такие же значения, как описано в первом аспекте.
В другом предпочтительном варианте осуществления, в настоящем изобретении описано соединение, имеющее формулу (III), или его изомеры или его фармацевтически приемлемая соль,
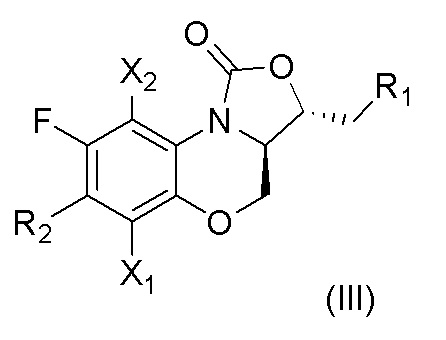
где
X1, X2, R1 и R2 имеют такие же значения, как описано в первом аспекте.
В другом предпочтительном варианте осуществления, в настоящем изобретении описано соединение, имеющее формулу (II) или (III), или его изомеры или его фармацевтически приемлемая соль,
где
X1 и X2 каждый представляют собой H;
R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3 или замещенный или незамещенный 5-6-членный гетероарил;
R3 независимо выбран из группы, состоящей из атома водорода, замещенного или незамещенного C1-C4 алкила, замещенного или незамещенного 3-6-членного циклоалкила, замещенной или незамещенной 3-6-членной гетероциклической группы, замещенного или незамещенного 5-6-членного гетероарила;
указанный замещенный или незамещенный 5-6-членный гетероарил в R1 или R3 и замещенная или незамещенная 3-6-членная гетероциклическая группа в R3 содержат по меньшей мере один гетероатом, выбранный из N, O или S;
заместители в R1 или R3 независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы;
R2 представляет собой замещенный или незамещенный  , замещенный или незамещенный
, замещенный или незамещенный 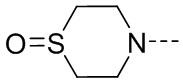 , замещенный или незамещенный
, замещенный или незамещенный 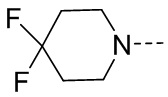 , замещенный или незамещенный
, замещенный или незамещенный 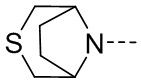 , замещенный или незамещенный
, замещенный или незамещенный 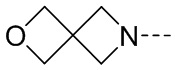 , замещенный или незамещенный
, замещенный или незамещенный  или замещенный или незамещенный
или замещенный или незамещенный  ;
;
заместители в R2 независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы.
В одном аспекте, соединение, имеющее формулу (II), выбрано из формулы (II-A),
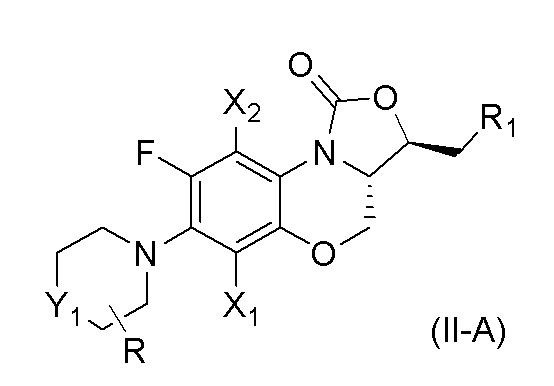
где
X1 и X2 каждый представляют собой H;
Y1 представляет собой S, S=O, CF2, SO2;
R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3 или замещенный или незамещенный 5-6-членный гетероарил;
R3 независимо выбран из группы, состоящей из атома водорода, замещенного или незамещенного C1-C4 алкила, замещенного или незамещенного 3-6-членного циклоалкила, замещенной или незамещенной 3-6-членной гетероциклической группы, замещенного или незамещенного 5-6-членного гетероарила;
указанный замещенный или незамещенный 5-6-членный гетероарил в R1 или R3 и замещенная или незамещенная 3-6-членная гетероциклическая группа в R3 содержат по меньшей мере один гетероатом, выбранный из N, O или S;
заместители в R1 или R3 независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы;
R представляет собой один или больше заместителей, одинаковых или разных, каждый из которых независимо выбран из группы, состоящей из H, F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы.
В другом аспекте, соединение, имеющее формулу (II), выбрано из формулы (II-B),
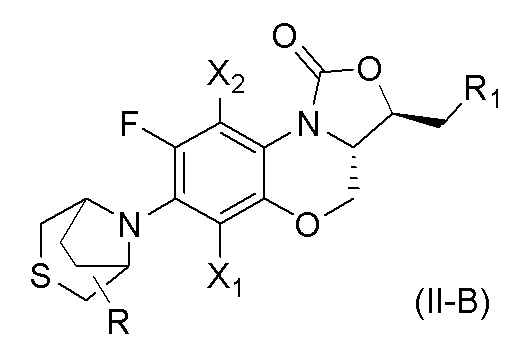
где
X1 и X2 каждый представляют собой H;
R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3 или замещенный или незамещенный 5-6-членный гетероарил;
R3 независимо выбран из группы, состоящей из атома водорода, замещенного или незамещенного C1-C4 алкила, замещенного или незамещенного 3-6-членного циклоалкила, замещенной или незамещенной 3-6-членной гетероциклической группы, замещенного или незамещенного 5-6-членного гетероарила;
указанный замещенный или незамещенный 5-6-членный гетероарил в R1 или R3 и замещенная или незамещенная 3-6-членная гетероциклическая группа в R3 содержат по меньшей мере один гетероатом, выбранный из N, O или S;
заместители в R1 или R3 независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы;
R представляет собой один или больше заместителей, одинаковых или разных, каждый из которых независимо выбран из группы, состоящей из H, F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы.
В другом аспекте, соединение, имеющее формулу (II), выбрано из формулы (II-C),
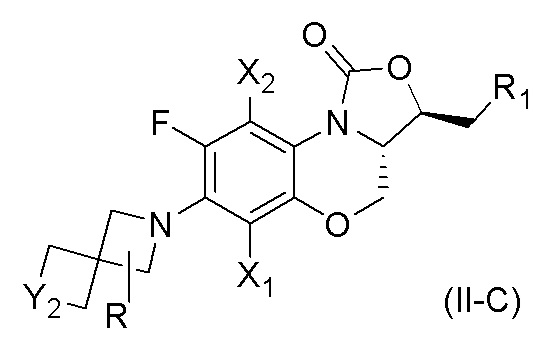
X1 и X2 каждый представляют собой H;
Y2 представляет собой O или S;
R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3 или замещенный или незамещенный 5-6-членный гетероарил;
R3 независимо выбран из группы, состоящей из атома водорода, замещенного или незамещенного C1-C4 алкила, замещенного или незамещенного 3-6-членного циклоалкила, замещенной или незамещенной 3-6-членной гетероциклической группы, замещенного или незамещенного 5-6-членного гетероарила;
указанный замещенный или незамещенный 5-6-членный гетероарил в R1 или R3 и замещенная или незамещенная 3-6-членная гетероциклическая группа в R3 содержат по меньшей мере один гетероатом, выбранный из N, O или S;
заместители в R1 или R3 независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы;
R представляет собой один или больше заместителей, одинаковых или разных, каждый из которых независимо выбран из группы, состоящей из H, F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы.
В одном варианте осуществления, соединение, имеющее формулу (III), выбрано из формулы (III-A),
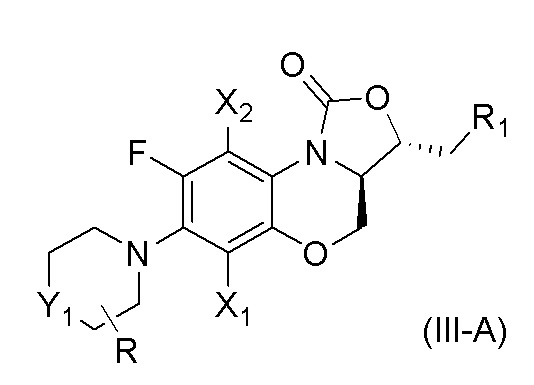
где
X1 и X2 каждый представляют собой H;
Y1 представляет собой S, S=O, CF2, SO2;
R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3, замещенный или незамещенный 5-6-членный гетероарил;
R3 независимо выбран из группы, состоящей из атома водорода, замещенного или незамещенного C1-C4 алкила, замещенного или незамещенного 3-6-членного циклоалкила, замещенной или незамещенной 3-6-членной гетероциклической группы, замещенного или незамещенного 5-6-членного гетероарила;
указанный замещенный или незамещенный 5-6-членный гетероарил в R1 или R3 и замещенная или незамещенная 3-6-членная гетероциклическая группа в R3 содержат по меньшей мере один гетероатом, выбранный из N, O или S;
заместители в R1 или R3 независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы;
R представляет собой один или больше заместителей, одинаковых или разных, каждый из которых независимо выбран из группы, состоящей из H, F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы.
В другом варианте осуществления, соединение, имеющее формулу (III), выбрано из формулы (III-B),
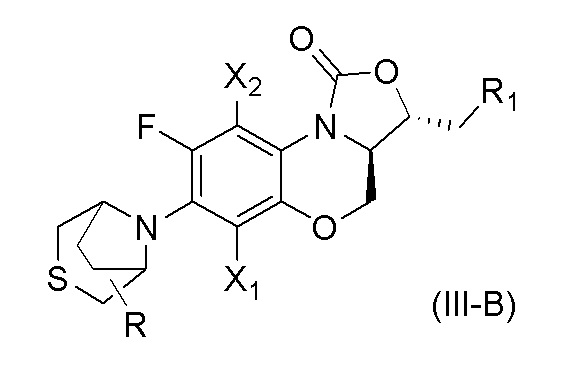
где
X1 и X2 каждый представляют собой H;
R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3 или замещенный или незамещенный 5-6-членный гетероарил;
R3 независимо выбран из группы, состоящей из атома водорода, замещенного или незамещенного C1-C4 алкила, замещенного или незамещенного 3-6-членного циклоалкила, замещенной или незамещенной 3-6-членной гетероциклической группы, замещенного или незамещенного 5-6-членного гетероарила;
указанный замещенный или незамещенный 5-6-членный гетероарил в R1 или R3 и замещенная или незамещенная 3-6-членная гетероциклическая группа в R3 содержат по меньшей мере один гетероатом, выбранный из N, O или S;
заместители в R1 или R3 независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы;
R представляет собой один или больше заместителей, одинаковых или разных, каждый из которых независимо выбран из группы, состоящей из H, F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы.
В другом варианте осуществления, соединение, имеющее формулу (III), выбрано из формулы (III-C),
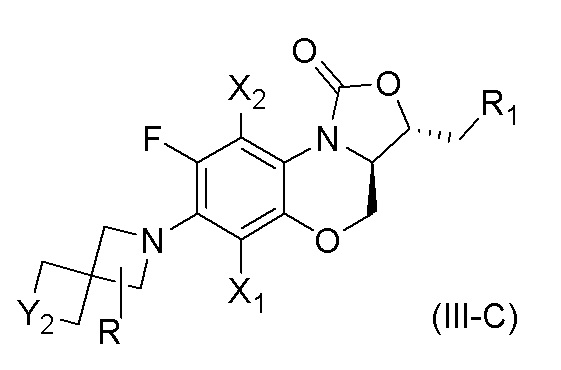
где
X1 и X2 каждый представляют собой H;
Y2 представляет собой O или S;
R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3, замещенный или незамещенный 5-6-членный гетероарил;
R3 независимо выбран из группы, состоящей из атома водорода, замещенного или незамещенного C1-C4 алкила, замещенного или незамещенного 3-6-членного циклоалкила, замещенной или незамещенной 3-6-членной гетероциклической группы, замещенного или незамещенного 5-6-членного гетероарила;
указанный замещенный или незамещенный 5-6-членный гетероарил в R1 или R3 и замещенная или незамещенная 3-6-членная гетероциклическая группа в R3 содержат по меньшей мере один гетероатом, выбранный из N, O или S;
заместители в R1 или R3 независимо выбраны из группы, состоящей из F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы;
R представляет собой один или больше заместителей, одинаковых или разных, каждый из которых независимо выбран из группы, состоящей из H, F, Cl, Br, гидроксила, амино-группы, нитро-группы, циано-группы, трифторметила, трифторметокси-группы, C1-C3 алкила, галогенированного C1-C3 алкила, C1-C3 алкокси-группы и C1-C3 алкиламино-группы.
Предпочтительно,
X1 представляет собой H;
X2 представляет собой H;
R1 представляет собой амино-группу, метиламино-группу, 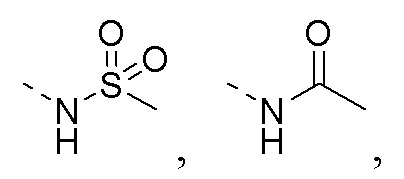
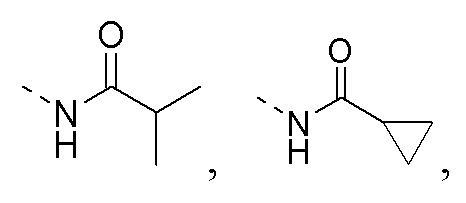
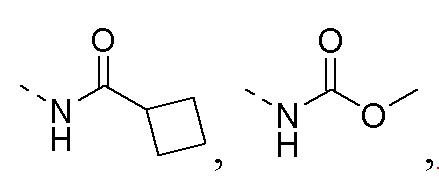
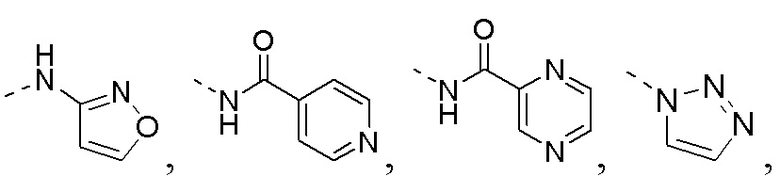
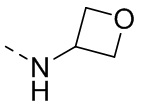 или
или 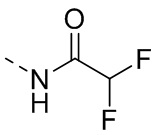 ;
;
R2 представляет собой , , , , , 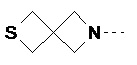 ,
, 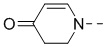 или .
или .
Фармацевтически приемлемая соль соединения по настоящему изобретению представляет собой соль, образованную, соответственно, со следующими кислотами: хлористоводородная кислота, п-толуолсульфокислота, винная кислота, малеиновая кислота, молочная кислота, метансульфокислота, серная кислота, фосфорная кислота, лимонная кислота, уксусная кислота или трифторуксусная кислота; предпочтительно хлористоводородная кислота, п-толуолсульфокислота или трифторуксусная кислота.
В первом аспекте, соединение по настоящему изобретению относится к целевым соединениям (представленным формулой или систематическим названием) в примерах, или их стереоизомерам или их фармацевтически приемлемым солям.
Предпочтительными соединениями согласно первому аспекту, являются следующие:
Соединение 1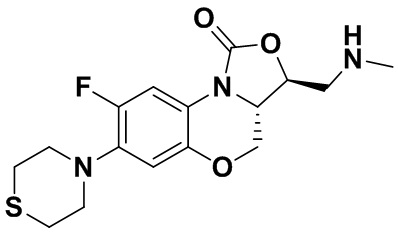
Соединение 2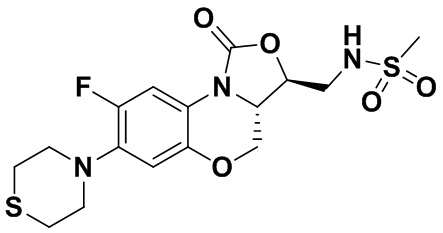
Соединение 3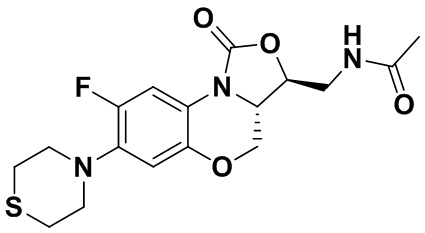
Соединение 4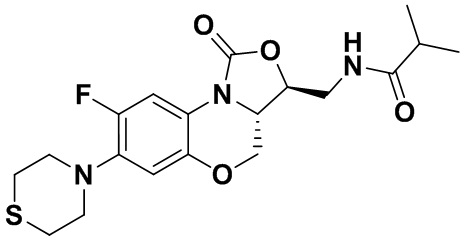
Соединение 5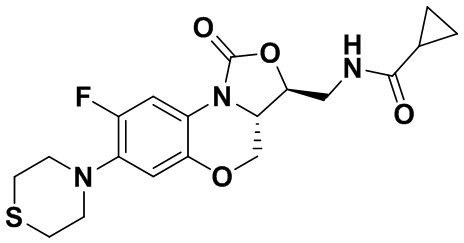
Соединение 6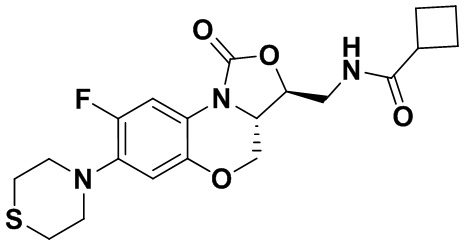
Соединение 7
Соединение 8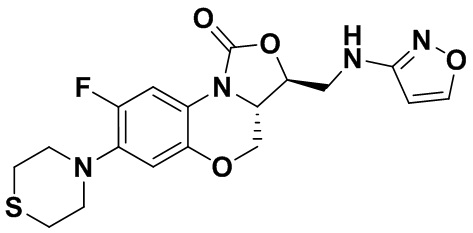
Соединение 9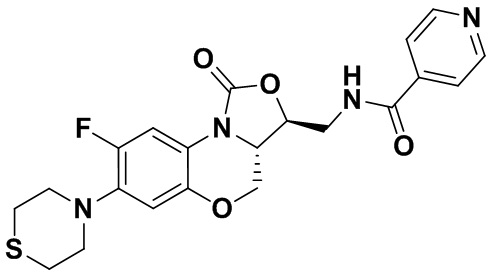
Соединение 10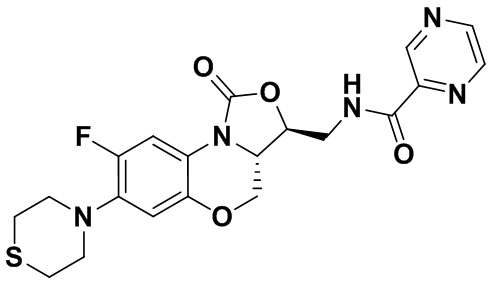 Соединение 11
Соединение 11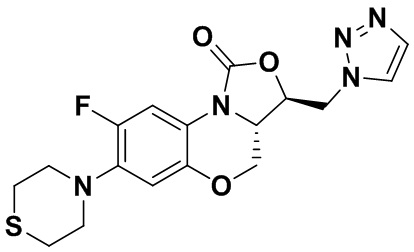
Соединение 12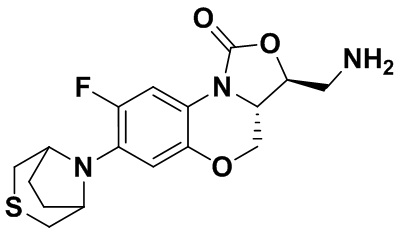
Соединение 13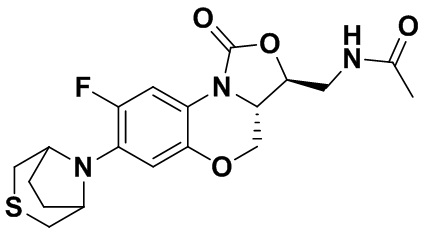
Соединение 14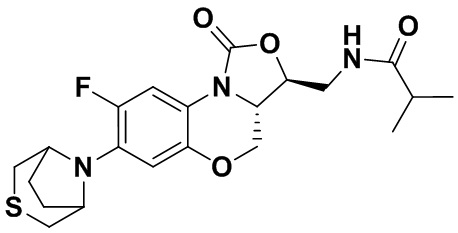 Соединение 15
Соединение 15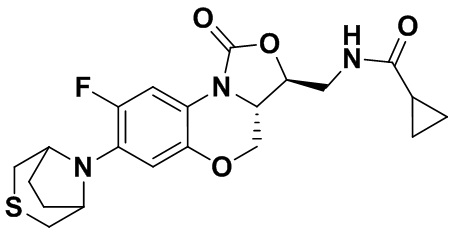
Соединение 16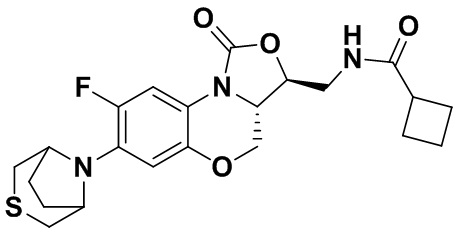
Соединение 17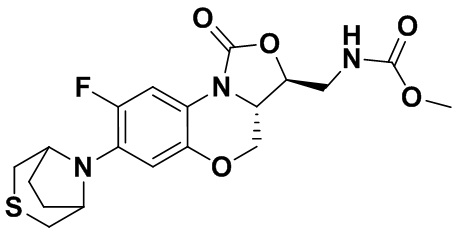
Соединение 18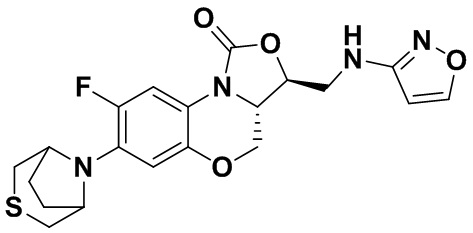
Соединение 19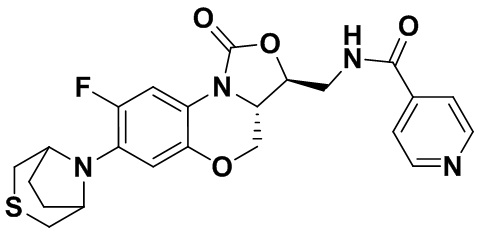
Соединение 20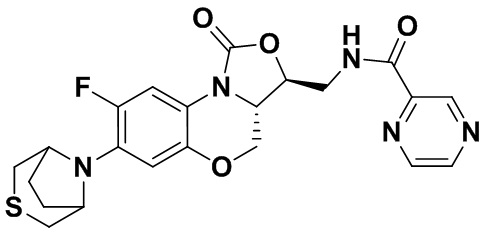
Соединение 21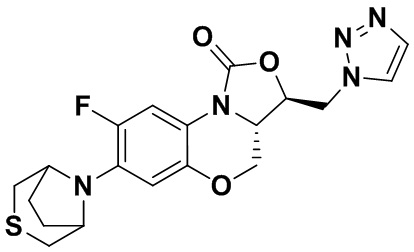
Соединение 22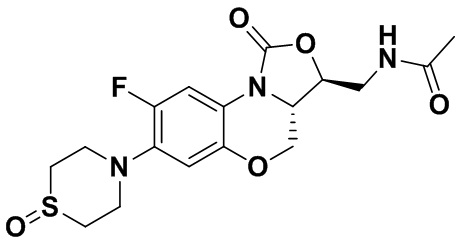
Соединение 23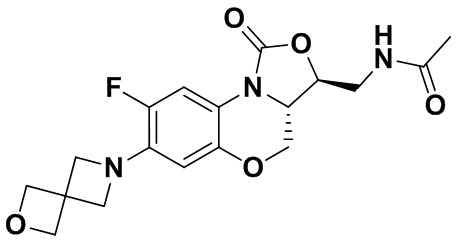
Соединение 24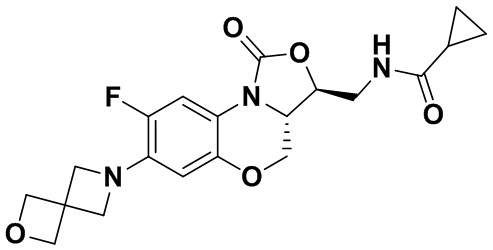
Соединение 25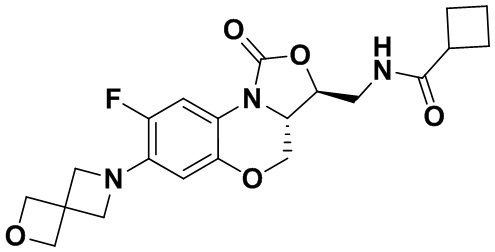
Соединение 26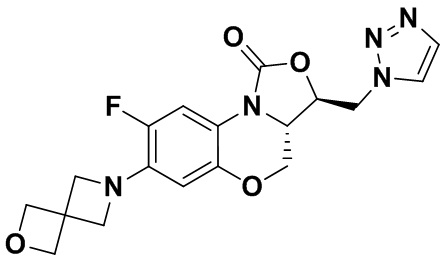
Соединение 27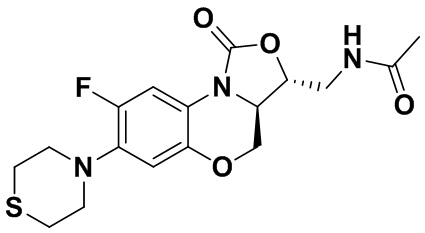
Соединение 28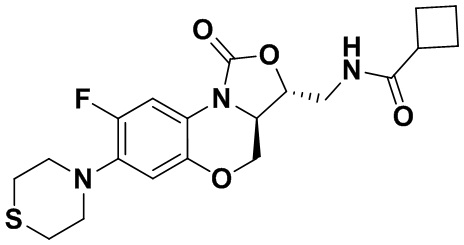
Соединение 29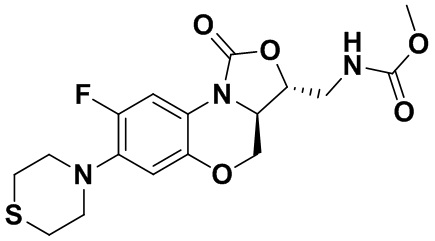
Соединение 30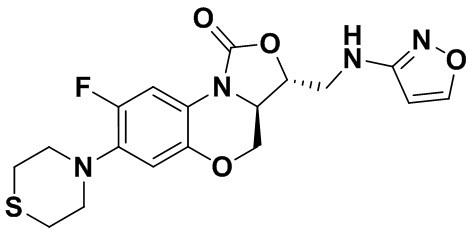
Соединение 31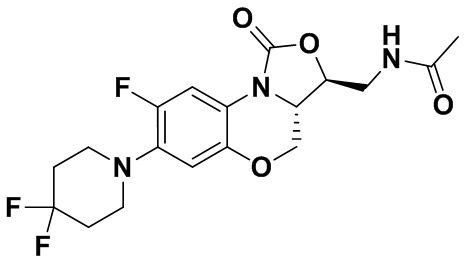
Соединение 32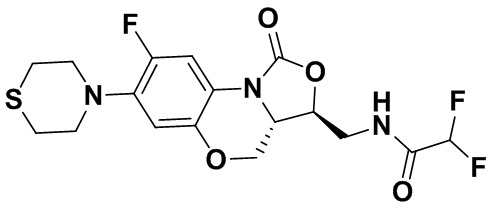
Соединение 33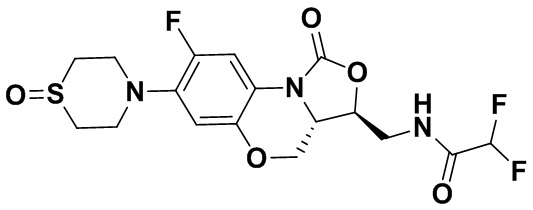
Соединение 34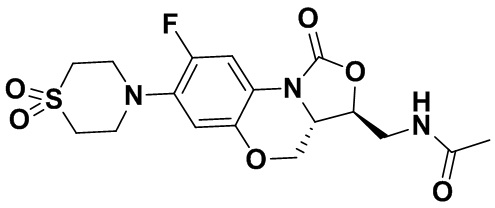
Соединение 35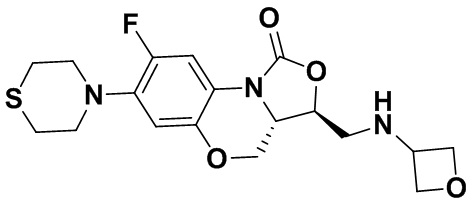
Соединение 36
Во втором аспекте настоящего изобретения описан способ получения соединений, описанных в первом аспекте, который включает следующие стадии:
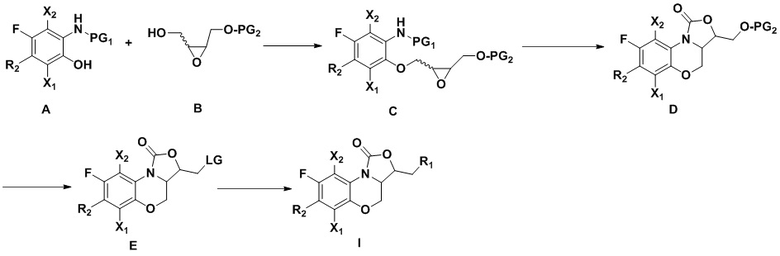
Соединение A реагирует с соединением B по реакции Мицунобу (см. книгу “Modern Organic Reactions”, chemical industry press, Yuefei Hu, Guoqiang Lin ed., первое издание 2008 года, volume 3, 187-244), с получением соединения C. Циклизация соединения C в присутствии литий-содержащего основания дает соединение D. Защитную группу PG2 с гидроксила в соединении D удаляют, получая гидроксильный продукт (см. книгу “Protection Group in Organic Synthesis”, East China University of Science and Technology press, перевод East China University of Science and Technology of organic chemistry, October 2004, первое издание), и затем полученный гидроксильный продукт превращают в соединение E, содержащее уходящую группу LG (например, галоген, псевдогалоген). Соединения, имеющие формулу (I), можно синтезировать из соединения E согласно известному в области оксазолидинонов методу синтеза.
Конкретнее,
1) Соединение A получают согласно следующей схеме:
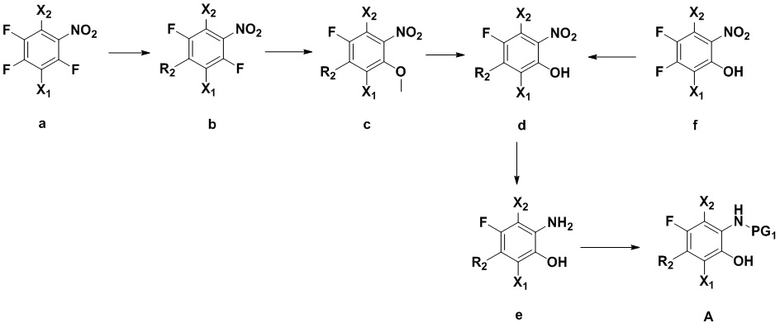
Соединение a реагирует с азотсодержащим R2H в щелочных условиях (например, в присутствии органического основания: триэтиламин, диизопропилэтиламин, DBU, или неорганического основания: бикарбонат натрия, карбонат цезия, карбонат калия), в подходящем растворителе (например, тетрагидрофуран, ацетонитрил, ДМФА, NMP, вода), при температуре от -10°C до 20°C, с получением соединения b.
Соединение b реагирует с метилатом натрия в метаноле с получением соединения c.
Соединение c реагирует с деметилирующими реагентами (например, HBr, HI, BBr3 или LiCl) в подходящем растворителе (например, дихлорметан или ДМФА) с получением соединения d.
Соединение e получают из соединения d в определенных условиях реакции (например, никель Ренея-H2, Pd/C-H2, порошок цинка – уксусная кислота, порошок цинка – формиат аммония или порошок железа – соляная кислота).
Соединение e реагирует с хлорформиатами (такими как метилхлорформиат, этилхлорформиат, трет-бутилхлорформиат или бензилхлорформиат) или ангидридом (например, (Boc)2O) в присутствии основания (такого как карбонат натрия, бикарбонат натрия, карбонат калия, триэтиламин или диизопропилэтиламин) в растворителе (таком как тетрагидрофуран, вода или смесь обоих растворителей) при температуре от 0°C до 30°C, с получением соединения A, содержащего амино-защитную группу PG1.
Соединение d можно получить путем смешивания соединения f и азотсодержащего R2H в щелочных условиях (например, в присутствии органического основания: триэтиламин, диизопропилэтиламин, DBU, N-метилморфолин, или неорганического основания: бикарбонат натрия, карбонат цезия, карбонат калия), при температуре от -10°C до 20°C, в подходящем растворителе (например, тетрагидрофуран, ацетонитрил, ДМФА, NMP или вода);
2) Защитная группа PG2 в соединении B включает силиловые эфиры (например, трет-бутилдиметилсилиловый эфир или трет-бутилдифенилсилиловый эфир), бензиловые эфиры (п-бромбензиловый эфир, п-метоксибензиловый эфир или тритиловый эфир), бензоаты (например, бензоаты или п-нитробензоаты), и рацемическое соединение B можно получить окислением 3-хлорпероксибензойной кислотой. Оптически активное соединение B можно получить реакцией эпоксидирования по Шарплессу.
3) Соединение C можно получить путем смешивания соединения A и соединения B в растворителях (например, дихлорметан, толуол, тетрагидрофуран или трет-бутил-метиловый эфир) в присутствии фосфиновых реагентов (например, трифенилфосфин или трибутилфосфин) и азодикарбонатных соединений (например, диэтилазодикарбонат или диизопропилазодикарбонат) или азодикарбонамиды (например, 1,1'-(азодикарбонил)-дипиперидин) по реакции Мицунобу.
4) Соединение D можно получить циклизацией соединения C в растворителях (например, тетрагидрофуран или трет-бутил-метиловый эфир) в присутствии литий-содержащих оснований (например, трет-бутоксилитий, бутиллитий, LiHMDS, LDA).
5) Удаление защитной группы из соединения D подходящими методами (например, силил-эфирная защитная группа может быть удалена фторидом тетрабутиламмония или кислотой, бензил-эфирная защитная группа может быть удалена гидрированием или кислотой, и бензоатная защитная группа может быть удалена неорганическим основанием) дает спиртовой интермедиат, и дальнейшее превращение спирта в уходящую группу LG (например, хлор, бром, иод или псевдогалоген) дает соединение, имеющее формулу E.
6) В соответствии с предшествующим уровнем техники, соединение E можно превратить в соединение, имеющее формулу (I), с различными группами R1;
заменяя соединение B на 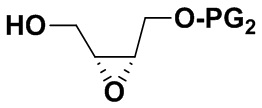 в описанных выше стадиях синтеза, можно получить соответствующее соединение, имеющее формулу (II);
в описанных выше стадиях синтеза, можно получить соответствующее соединение, имеющее формулу (II);
заменяя соединение B на 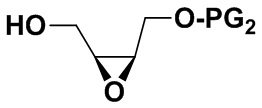 в описанных выше стадиях синтеза, можно получить соответствующее соединение, имеющее формулу (III);
в описанных выше стадиях синтеза, можно получить соответствующее соединение, имеющее формулу (III);
где значения X1, X2, R1 и R2 такие же, как описано в первом аспекте настоящего изобретения.
В третьем аспекте настоящего изобретения описана фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по первому аспекту, его изомеров или его фармацевтически приемлемой соли, и один или больше фармацевтически приемлемый адъювант.
В четвертом аспекте настоящего изобретения описано применение соединения по первому аспекту, его изомеров или его фармацевтически приемлемой соли по первому аспекту, или фармацевтической композиции по третьему аспекту в производстве лекарственного средства для лечения и/или профилактики микробных инфекций, вызванных Mycobacterium tuberculosis.
Отличительный признак любого аспекта или любого из аспектов настоящего изобретения может быть также применен к любому другому аспекту или любому из других аспектов, при условии, что они не противоречат друг другу, и, разумеется, когда они имеют отношение друг к другу, соответствующие отличительные признаки могут быть соответствующим образом модифицированы. В настоящем изобретении, например, в выражении “любой из первых аспектов настоящего изобретения” термин “любой” означает любой из подаспектов первого аспекта настоящего изобретения и имеет аналогичное значение при использовании сходным образом в других аспектах.
Подробное описание изобретения
Различные аспекты и отличительные признаки настоящего изобретения подробнее описаны ниже.
Все литературные источники, упомянутые в настоящем изобретении, включены в текст настоящего изобретения посредством ссылки во всей их полноте. Если содержание в приведенных ссылках не соответствует настоящему изобретению, то приоритетным считается настоящее изобретение. Кроме того, различные термины и обороты, использованные в настоящем изобретении, имеют значения, общеизвестные квалифицированным специалистам в данной области. Тем не менее, термины и обороты подробно описаны и разъяснены в настоящем изобретении. Если упомянутые термины и обороты расходятся в своих значениях с общепринятыми, то приоритетными являются определения, данные в настоящем изобретении. Далее приведены определения различных терминов, используемых в настоящем изобретении, которые применимы к терминам, применяющимся в тексте настоящей заявки, если иное не оговорено особо.
Соединения по настоящему изобретению содержат асимметрические центры. Соединения, содержащие асимметрично замещенные атомы, в настоящем изобретении можно разделить на оптически активные или рацемические формы. Квалифицированным специалистам в данной области известно, как получить оптически активные формы, например, методом разделения рацемических форм или синтезом из оптически активного исходного вещества. Если иное не оговорено особо, настоящее изобретение включает все оптические изомеры, диастереомеры и рацемические формы. Способ получения соединений по настоящему изобретению и их интермедиатов составляет часть настоящего изобретения. Все таутомеры соединений по настоящему изобретению также составляют часть настоящего изобретения.
Термин "стереоизомеры" относится к соединениям, которые имеют одинаковое химическое строение, но отличаются расположением атомов или групп атомов в пространстве. Стереоизомеры включают энантиомеры, диастереомеры, конформационные изомеры (ротамеры), геометрические изомеры, атропоизомеры и т.д.
Термин "хиральные молекулы" означает молекулы, которые не могут быть совмещены со своим зеркальным отображением, в то время как термин “ахиральные молекулы” означает молекулы, которые могут быть совмещены со своим зеркальным отображением.
Термин “энантиомеры” относится к двум стереоизомерам соединения, которые являются несовмещаемыми зеркальными отображениями друг друга.
Термин “диастереомер” относится к стереоизомерам с двумя или больше хиральными центрами, молекулы которых не являются зеркальными отображениями друг друга. Диастереомеры имеют различающиеся физические свойства, например, температуру плавления, температуру кипения, спектральные характеристики и реакционную способность. Смесь диастереомеров можно разделить аналитическими методами высокого разрешения, такими как электрофорез и хроматография, такая как ВЭЖХ.
Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активных соединений используют такие префиксы, как D и L, или R и S, для обозначения абсолютной конфигурации хиральных центров молекулы. Префиксы D и L, или (+) и (-) применяют для обозначения знака вращения соединением плоскополяризованного света, где (-) или L означает, что соединение является левовращающим. Соединение с префиксом (+) или D является правовращающим. Конкретный стереоизомер может именоваться энантиомером, и смесь таких стереоизомеров именуют энантиомерной смесью. Смесь энантиомеров с соотношением 50:50 называют рацемической смесью или рацематом, что может иметь место в том случае, когда в химической реакции или в процессе нет стереоселективности или стереоспецифичности.
Любой асимметрический атом (например, атом углерода или атом Н) в соединении(-ях) по настоящему изобретению может присутствовать в рацемической форме или в энантиомерно обогащенной форме, например, может иметь место (R)-, (S)-, (R,S)-, (с,R)-, (R,R)- или (с,S)- конфигурация. В некоторых вариантах осуществления, каждый асимметрический атом имеет по меньшей мере 50% энантиомерный избыток, по меньшей мере 60% энантиомерный избыток, по меньшей мере 70% энантиомерный избыток, по меньшей мере 80% энантиомерный избыток, по меньшей мере 90% энантиомерный избыток, по меньшей мере 95% энантиомерный избыток, или по меньшей мере 99% энантиомерный избыток, с превалированием (R)- или (S)-конфигурации.
В зависимости от выбора исходных соединений и методик, соединения могут присутствовать в форме одного из возможных стереоизомеров или их смесей, например, рацематов и смесей диастереоизомеров, в зависимости от количества асимметрических атомов углерода. Оптически активные изомеры можно получить с использованием хиральных синтонов или хиральных реагентов, или путем расщепления с применением общеизвестных методов.
Полученную смесь любых стереоизомеров можно разделить на чистые или практически чистые геометрические изомеры, энантиомеры, диастереомеры, например, хроматографией и/или дробной кристаллизацией, в зависимости от различий в физико-химических свойствах компонентов.
Термин "замещенный" означает замещение одного или больше атомов водорода в структуре определенными группами-заместителями, при условии, что валентность конкретного атома сохраняется нормальной и что получаемое после замещения соединение является устойчивым. Если не указано иное, каждое способное к замещению положение в группе может быть замещено опциональным заместителем. Если больше одного положения в структуре может быть замещено одним или более заместителями, выбранными из определенной группы, каждое положение может быть замещено одинаковыми или разными заместителями. Описанные в настоящем тексте заместители включают (но не ограничиваются только ими) атом водорода, атом дейтерия, оксо-группу (= O), галоген, циано-группу, нитро-группу, гидроксил, меркапто-группу, амино-группу (-NH2), ароматическую амино-группу, аминоалкил, алкил, алкилтио-группу, гидроксилалкил, галогеналкил, циклоалкил, гетероциклил, арил, гетероарил, -C(=O)Ra, -ORb, -COORb, -SO2Rb, -NRcRd, -CONRcRd, -SO2NRcRd, -C(NRcRd), где Ra, Rb, Rc и Rd каждый независимо представляют собой атом водорода, циано-группу, амино-группу, алкиламино-группу, ароматическую амино-группу, алкилтио-группу, алкокси-группу, арилокси-группу, гидроксил, меркапто-группу, алкил, галогеналкил, циклоалкил, гетероциклил, арил, гетероарил, алкилсульфонил, аминосульфонил, гидроксилалкил, аминоалкилкарбонил или алкилкарбонил.
Количество атомов углерода в различных углеводород-содержащих фрагментах выражается префиксом, указывающим минимальное и максимальное число атомов углерода в соответствующем фрагменте. Ci-Cj означает фрагмент, содержащий от числа "i" (включая i) до числа "j" (включая j) атомов углерода. Таким образом, например, C1-C4 алкил означает алкил, содержащий от 1 до 4 (включая 1 и 4) атомов углерода, в частности метил, этил, C3 алкил и C4 алкил.
При использовании в настоящем тексте, термин "алкил" означает алкил с указанным числом атомов углерода, который представляет собой линейный или разветвленный алкил, и может включать его подгруппы. Например, термин "C1-C4 алкил" охватывает поддиапазоны, представленные C1-C3 алкилом, C1-C2 алкилом, C2-C4 алкилом, C3-C4 алкилом и т.д., и частные варианты групп, такие как метил, этил, н-пропил, изопропил. Термины "алкокси" и "алкиламино" являются общепринятыми и означают алкильные группы, присоединенные к остальной части молекулы через атом кислорода или аминогруппу, где алкильные группы такие, как описано в настоящем тексте. Алкокси-группы включают (но не ограничиваются только ими) метокси-, этокси-, изопропокси-, н-пропокси-группу и т.д. Алкиламино-группа включает (но не ограничивается только ими) метиламино-группу, этиламино-группу, изопропиламино-группу, н-пропиламино-группу и т.п.
Термин “галогеналкил" означает, что алкил замещен один или больше галогеновыми атомами, включая (но не ограничиваясь только ими) трифторметил, дифторметил и т.п.
При использовании в настоящем тексте, термины "галоген", "атом галогена" и "галогенированный" означают фтор (F), хлор (Cl), бром (Br) или иод (I).
При использовании в настоящем тексте, термин “псевдогалоген” означает сульфонилокси-группу, в частности трифторметилсульфонилокси-группу, п-метилфенилсульфонилокси-группу, метилсульфонилокси-группу и п-нитрофенилсульфонилокси-группу.
При использовании в настоящем тексте, термин "циклоалкил" означает циклический алкил с указанным числом атомов углерода, а также включает его поддиапазоны, например, термин "3-6-членный циклоалкил" означает поддиапазон групп, представленный 3-5-членным циклоалкилом, 4-6-членным циклоалкилом и т.п., и частные варианты групп, такие как циклопропил и циклобутил, циклопентил и циклогексил.
При использовании в настоящем тексте, термин "гетероциклил" означает циклическую гетероалкильную группу с указанным числом атомов в цикле, включая группы с одним циклом или конденсированными циклами. В кольце содержится от 4 до 10 атомов, один или два из которых выбраны из гетероатомов – азота, кислорода и серы, а остальные атомы в цикле представляют собой углерод. Эти кольца могут также содержать одну или больше двойных связей, но они не содержат полностью сопряженной π-электронной системы. Гетероциклические группы включают (но не ограничиваются только ими) оксетанил, азетидинил, пирролидинил, пиразолидинил, дигидротиенил, 1.3-диоксоланил, дитиоланил, тетрагидропиранил, тетрагидротиопиранил, пиперидинил, 1,2-дигидропиридил, морфолинил, тиоморфолинил, гексагидропиримидил, пиперазинил, гомопиперазинил, 1,3-бензоксазинил, оксазолидинил, гомопиридинил и т.п.
При использовании в настоящем тексте, термин "гетероарил" означает ароматические группы, содержащие в цикле от 1 до 3 гетероатомов, выбранных из кислорода, серы и азота, а остальные атомы в цикле представляют собой углерод. Пример "5-6-членного гетероарила" включает 5-членный гетероарил и 6-членный гетероарил. 5-членный гетероарил включает (но не ограничивается только ими) имидазолил, фуранил, тиенил, триазолил, тетразолил, пиразолил (например, 2-пиразолил), тиазолил, оксазолил и изоксазолил. 6-членный гетероарил включает пиридил, пиридазинил, пиримидинил, пиразинил или 1,3,5-триазинил. В некоторых вариантах осуществления, гетероарил представляет собой триазолил, пиразинил, изоксазолил или пиридинил.
При использовании в настоящем тексте, термин "кольцо" означает замещенный или незамещенный циклоалкил, замещенный или незамещенный гетероциклил, замещенный или незамещенный арил, или замещенный или незамещенный гетероарил. Указанное кольцо включает конденсированное кольцо. Число атомов в кольце обычно определяется как число в цикле. Например, "3-6-членное кольцо" означает циклическое расположение 3 - 6 атомов в кольце.
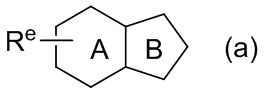
При использовании в настоящем тексте, кольцевая система, образованная заместителем Re, который одной связью соединен с центральным циклом, означает, что один или больше одинаковых или разных заместителей Re могут являться заместителями в любом способном к замещению положении кольца. Например, формула (a) показывает, что любое способное к замещению положение в кольце A или B может быть замещено одним или больше заместителями Re.
Термин "уходящая группа" или "LG" имеет общеизвестное значение, связанное с синтетической органической химией, а именно атомы или группы, которые могут быть заменены нуклеофильными группами, включая галогены, алифатический или ароматический сульфонилоксил, такие как атом хлора, атом брома, атом иода, метилсульфонилокси-группа, толилсульфонилокси-группа, трифторметилсульфонилокси-группа и т.п.
"Защитная группа" или “PG” означает заместитель, который обычно применяется для блокировки или защиты определенной функциональной группы при проведении реакции по другой функциональной группе, присутствующей в соединении. Например, "амино-защитная группа" означает заместитель, присоединенный к аминогруппе для блокировки или защиты аминной функциональности в соединении. Подходящие амино-защитные группы включают трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz), метоксикарбонил и этоксикарбонил. Аналогичным образом, "гидроксил-защитная группа" означает заместитель в гидроксильной группе, который блокирует или защищает гидроксильную функциональную группу.
При использовании в настоящем тексте, термин "эффективное количество" означает количество лекарственного средства, которое можно применять субъекту для достижения желаемого лечения в отношении описанных в настоящем тексте заболеваний или симптомов.
При использовании в настоящем тексте, термин "фармацевтически приемлемая", например, при описании "фармацевтически приемлемой соли", означает, что соль не только физиологически приемлема для субъекта, но и представляет собой синтетическое вещество, имеющее ценность для фармацевтики, такое как соль, сформированная в качестве интермедиатов для хирального разделения, и хотя в этом случае соль интермедиата нельзя напрямую давать субъектам, ее можно использовать для получения финального продукта по настоящему изобретению.
При использовании в настоящем тексте, термин "фармацевтическая композиция" может также означать "композицию", которую можно применять для лечения у субъектов, в особенности у млекопитающих, заболеваний или симптомов, описанных в настоящем тексте.
"Лечение" заболеваний включает:
(1) предотвращение заболевания, предотвращение появления клинических симптомов заболевания у млекопитающих, входящих в контакт или восприимчивых к заболеванию, но без наличия или проявления его симптомов,
(2) подавление заболевания, т.е. предотвращение или уменьшение развития заболевания или его клинических симптомов.
(3) облегчение заболевания, то есть вызывание восстановления от заболевания или его клинических симптомов.
"Терапевтически эффективное количество" означает количество соединения, достаточное для лечения заболевания при введении млекопитающим. Терапевтически эффективное количество варьируется в зависимости от соединения, заболевания, подвергающегося лечению, и степени его тяжести, а также от возраста, веса тела и пола млекопитающего. Терапевтически эффективное количество означает также любое количество соединения, достаточное для достижения желаемого положительного эффекта, который включает предотвращение заболевания, подавление заболевания или облегчение заболевания, как описано выше в пунктах (1) – (3). Например, количество соединения может составлять 0.1-250 мг/кг, или предпочтительно 0.5-100 мг/кг, или более предпочтительно 1-50 мг/кг, или еще более предпочтительно 2-20 мг/кг. Предпочтительно, указанное количество соединения вводят млекопитающим два раза в сутки. Более предпочтительно, указанное количество соединения вводят млекопитающим один раз в сутки. Более предпочтительно, указанное количество соединения вводят млекопитающим один раз неделю или два раза в неделю.
При использовании в настоящем тексте, термин "заболевание и/или симптомы" относится к физическому состоянию субъекта, которое релевантно заболеванию и/или симптомам, описанным в настоящем изобретении. Например, заболевания и/или симптомы, описанные в настоящем изобретении, относятся к инфекционным заболеваниям, вызванным туберкулезной бациллой.
При использовании в настоящем тексте, термин "субъект" может относиться к пациенту или другим животным, в частности к млекопитающему, такому как человек, собака, обезьяна, крупный рогатый скот, лошадь и т.д., которые принимают соединение, имеющее формулу (I), или его фармацевтическую композицию для лечения описанного в настоящем тексте заболевания или симптомов.
С другой стороны, настоящее изобретение относится также к фармацевтической композиции, в которой описанные в настоящем тексте соединения используются в качестве действующих веществ. Фармацевтическую композицию можно приготовить в соответствии с известными в данной области методами. Любой препарат, подходящий для человека или животного, можно приготовить путем комбинирования соединения по настоящему изобретению с одним или больше фармацевтически приемлемыми твердыми или жидкими вспомогательными веществами и/или адъювантами.
Соединения по настоящему изобретению или содержащие их фармацевтические композиции можно вводить в виде однократных дозированных форм перорально или парентерально, например, перорально, внутривенно, внутримышечно, подкожно, назально или через слизистую рта, глаза, легкие и органы дыхания, кожу, вагину, прямую кишку и т.п.
Дозированные формы могут представлять собой жидкие дозированные формы, твердые дозированные формы или полутвердые дозированные формы. Жидкие дозированные формы могут представлять собой раствор (включая истинный раствор и коллоидный раствор), эмульсию (включая эмульсию типа вода-в-масле, масло-в-воде и гетерогенную эмульсию), суспензию, инъекции (включая водную инъекцию, порошок для инъекции и инфузии), глазные капли, назальные капли, лосьон и мазь, и т.д. Твердый препарат может представлять собой таблетку (включая обычную таблетку, таблетку с кишечно-растворимым покрытием, буккальную таблетку, диспергируемую таблетку, жевательную таблетку, шипучую таблетку, распадающуюся во рту таблетку), капсулы (включая твердые капсулы, мягкие капсулы, капсулы с кишечно-растворимым покрытием), гранулы, порошки, пеллеты, пилюли, капли, суппозитории, мембраны, чрезкожные пластыри, аэрозоль, спреи и т.п. Полутвердые препараты могут представлять собой мазь, желатин, пасту и т.п.
Соединение по настоящему изобретению можно вводить в состав традиционных препаратов, препаратов с замедленным высвобождением, препаратов с контролируемым высвобождением, таргетированных препаратов и различных систем доставки частиц.
Для введения соединения по настоящему изобретению в состав таблеток можно применять широко используемые разнообразные вспомогательные вещества, известные в данной области техники, включая разбавители, адгезивы, увлажняющие агенты, разрыхлители, лубриканты и сорастворители. Разбавители могут представлять собой крахмал, декстрин, сахарозу, глюкозу, лактозу, маннит, сорбит, ксилит, микрокристаллическую целлюлозу, сульфат кальция, гидрофосфат кальция, карбонат кальция и т.д. Увлажняющие агенты могут представлять собой воду, этанол, изопропанол и т.д. Адгезивы могут представлять собой крахмальный сироп, декстрин, сироп, мед, раствор глюкозы, микрокристаллическую целлюлозу, гуммиарабик, желатин, натрия карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, этилцеллюлозу, акриловый полимер, карбомер, поливинилпирролидон, полиэтиленгликоль и т.д. Разрыхлители могут представлять собой крахмал, микрокристаллическую целлюлозу, низкозамещенную гидроксипропилцеллюлозу, сшитый поливинилпирролидон, сшитую натрия карбоксиметилцеллюлозу, натрия карбоксиметилкрахмал, бикарбонат натрия и лимонную кислоту, сложный эфир полиоксиэтиленсорбита и жирной кислоты, лаурилсульфонат натрия и т.д. Лубриканты и сорастворители могут представлять собой порошок талька, диоксид кремния, стеарат, винную кислоту, жидкий парафин, полиэтиленгликоль и т.д.
Таблетки можно также дополнительно превращать в таблетки с покрытием, например, в таблетки, покрытые сахаром, таблетки с пленочным покрытием, таблетки с кишечнорастворимым покрытием, или в двуслойные или многослойные таблетки.
Для заключения лекарственного средства в капсулу, соединение по настоящему изобретению в качестве действующего вещества можно смешивать с разбавителями и сорастворителями, и смесь помещать в твердую капсулу или мягкую капсулу. Соединение по настоящему изобретению в качестве действующего вещества можно также смешивать в виде гранул или пеллет с разбавителями, адгезивами и разрыхлителями, и затем заполнять полученной смесью твердые капсулы или мягкие капсулы. Различные разбавители, адгезивы, увлажняющие агенты, разрыхлители и сорастворители, применяемые для приготовления таблеток с описанными в настоящем тексте соединениями, можно также использовать для приготовления капсул с описанными в настоящем тексте соединениями.
Для введения соединения по настоящему изобретению в состав раствора для инъекций, в качестве растворителя можно применять воду, этанол, изопропанол, пропиленгликоль или их смесь, а также использовать приемлемое количество обычно применяемых в данной области техники солюбилизаторов, сорастворителей, рН регуляторов и регуляторов осмотического давления. Солюбилизаторы или сорастворители могут представлять собой полоксамер, лецитин, гидроксипропил-бета-циклодекстрин и т.д. рН регуляторы могут представлять собой фосфат, ацетат, соляную кислоту, гидроксид натрия и т.д. Регуляторы осмотического давления могут представлять собой хлорид натрия, маннит, глюкозу, фосфат, ацетат и т.д. Если готовят лиофилизованный порошок для приготовления инъекционного раствора, в качестве подложки можно применять маннит и глюкозу.
Кроме того, при необходимости можно добавлять в фармацевтические композиции красители, консерванты, отдушки, ароматизаторы или другие добавки.
Для достижения цели медицинского применения и усиления терапевтического эффекта, соединение или фармацевтическую композицию по настоящему изобретению можно вводить любым известным методом.
Соединения или композиции по настоящему изобретению можно вводить отдельно или в комбинации с другими терапевтическими лекарственными средствами или средствами для снятия симптомов. Когда соединение по настоящему изобретению демонстрирует синергетический эффект с другими терапевтическими средствами, его дозировку следует подстраивать в соответствии с конкретной ситуацией.
Положительный технический эффект
Авторы настоящего изобретения обнаружили, что соединения по настоящему изобретению обладают очень высокой противотуберкулезной активностью in vitro. Минимальная подавляющая концентрация (MIC) для семи соединений против Mycobacterium tuberculosis in vitro составляет менее 0.1 мкг/мл, что очевидно лучше, чем у линезолида и приближается или превосходит противотуберкулезную активность сутезолида. Кроме того, они показывают хорошие параметры безопасности для клеток Vero при низкой цитотоксичности (IC50 выше 30 мкг/мл). Следует отметить, что ингибирование синтеза белков митохондрий у соединений по настоящему изобретению намного слабее, чем у позитивного контроля – таких лекарств, как линезолид и сутезолид, что говорит об их большей безопасности. Соединения по настоящему изобретению демонстрируют не только высокую противотуберкулезную активность и высокую безопасность in vitro, но также прекрасные фармакокинетические характеристики и прекрасную противотуберкулезную активность in vivo. В настоящем изобретении описан класс бензоксазин-оксазолидиноновых соединений, имеющих новую структуру, высокую активность, низкую токсичность и прекрасные фармакокинетические характеристики, которые можно применять для лечения и профилактики инфекционных заболеваний, вызванных Mycobacterium tuberculosis, и даже сензитивными или лекарственно-устойчивыми Mycobacterium tuberculosis.
В патенте WO2011/147259 A1 от 1 декабря 2011 года описаны соединения, имеющие формулу (IV), для лечения инфекционных заболеваний, в особенности инфекционных заболеваний, вызванных бактериями с множественной лекарственной устойчивостью, при этом указанные инфекционные заболевания вызваны бактериями с множественной лекарственной устойчивостью, включая Enterococcus, Staphylococcus aureus, Staphylococcus epidermidis и Pneumococcus:
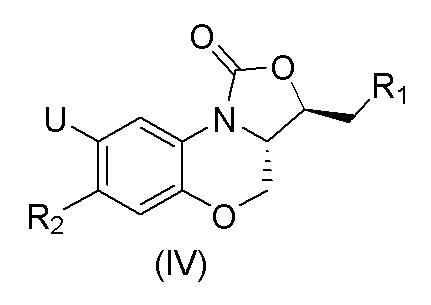
где U представляет собой H или F, R1 представляет собой 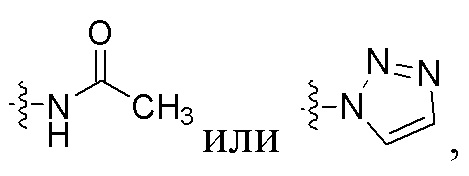 R2 представляет собой фенильную группу или 5-членную или 6-членную ароматическую или неароматическую гетероциклическую группу.
R2 представляет собой фенильную группу или 5-членную или 6-членную ароматическую или неароматическую гетероциклическую группу.
В патенте CN102260277 B, выданном 24 июля 2013 года, описаны соединения, имеющие формулу (IV), где R2 представляет собой фенильную группу или 5-членную или 6-членную ароматическую гетероциклическую группу.
Кроме того, в другом документе (J. Med. Chem. 2011, 54, 7493–7502) описано следующее соединение:
где X представляет собой N или O, R' представляет собой 3-нитрофенил, 2-нитрофенил, 4-пиридинил, 3-пиридинил, 2-пиридинил, 2-фуранил, 3-фуранил и 2-нитро-5-фуранил. В этом документе описана их активность против Staphylococcus aureus, метициллин-резистентного Staphylococcus aureus, метициллин-резистентного Staphylococcus epidermis, пенициллин-резистентного Streptococcus pneumoniae и enterococcus. Нет сведений, что соединение из указанного документа обладает активностью в лечении Mycobacterium tuberculosis.
Авторы настоящего изобретения синтезировали соединение, имеющее формулу (VII), т.е. соединение, в котором X представляет собой атом кислорода, как показано в формуле (V), согласно известным схемам синтеза (J. Med. Chem. 2011, 54, 7493-7502), и определили их противотуберкулезную активность in vitro и подавление синтеза белков митохондрий.
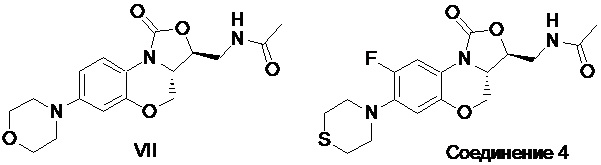
Для соединения, имеющего формулу (VII), значение MIC против Mycobacterium tuberculosis и значение IC50 для подавления синтеза белков митохондрий составляют 1.546 мкг/мл и 35.82 мкM, соответственно. Однако для соединения 4 по настоящему изобретению значение MIC составляет 0.044 мкг/мл, а его противотуберкулезная активность значительно выше, чем у соединения, имеющего формулу (VII) (MIC = 1.546 мкг/мл). В то же время, для соединения 4 эффект подавления синтеза белков митохондрий очевидно меньше, чем для соединения, имеющего формулу (VII), что говорит о его большей безопасности. Противотуберкулезная активность соединения, имеющего формулу (VII), ниже, чем у соединения по настоящему изобретению, и активность в отношении лекарственно-устойчивого туберкулеза у соединения, имеющего формулу (VII), ниже, чем у соединения по настоящему изобретению. Кроме того, у соединения по настоящему изобретению эффект подавления синтеза белков митохондрий слабее, чем у соединения, имеющего формулу (VII), и поэтому соединение по настоящему изобретению имеет лучший профиль безопасности.
Примеры
Настоящее изобретение более подробно описано с помощью приведенных ниже вариантов осуществления, которые никоим образом не ограничивают объем настоящего изобретения. Настоящее изобретение было подробно описано, также были описаны частные варианты его осуществления. Для квалифицированных специалистов в данной области очевидно, что можно проводить различные изменения и улучшения в частных вариантах осуществления настоящего изобретения, не выходя за рамки сути и объема изобретения.
Во всех приведенных ниже примерах применяли стандартные операции и методы очистки, известные квалифицированным специалистам в данной области. Если иное не указано особо, все значения температуры приведены в °C (градусы Цельсия). Структуры соединений определяли методом ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Температуры плавления (т.пл.) приведены в °C и нескорректированы.
Примеры синтеза
Структуру соединений определяли методом 1H-ЯМР или масс-спектрометрии. Значения химсдвига (δ) в спектрах протонного магнитного резонанса (1H-ЯМР) приведены в миллионных долях (м.д.). Значения констант спин-спинового взаимодействия (J) приведены в Герцах (Гц). ЯМР спектры записывали на ЯМР-спектрометре Mercury-400 или Brucker-500, используя CDCl3 или ДМСО-d6 в качестве растворителей, а тетраметилсилан (ТМС) в качестве референса.
Температуру плавления определяли на приборе для определения температуры плавления Yanaco M.P-500D, изготовленном в Японии, температура нескорректирована.
Масс-спектры высокого разрешения записывали на масс-спектрометре LC/MSD Agilent 1100 series.
Электронные весы – японские Yanaco LY-300.
Для колоночной хроматографии использовали силикагель 200-300 меш в качестве неподвижной фазы.
Безводные растворители получали стандартными методами. Другие реагенты коммерчески доступны с аналитической чистотой.
В настоящем тексте применяются перечисленные ниже аббревиатуры:
ADDP = 1,1'-(азодикарбонил)-дипиперидин.
ДМФА = N, N-диметилформамид.
EDCI = 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид.
HOBt = 1-гидроксибензотриазол.
TPP = трифенилфосфин.
Примеры синтеза
Пример получения 1
Получение ((2R,3S)-3-(((трет-бутилдиметилсилил)окси)метил)оксиран-2- ил)метанола (Интермедиат 1)
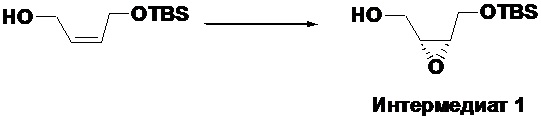
В трехгорлую колбу объемом 500 мл помещали молекулярные сита 4A (7.2 г) и безводный дихлорметан (180 мл). Смесь охлаждали до -20°C в атмосфере аргона. Добавляли D-(-)-диэтилтартрат (7.8 мл, 45.7 ммоль), затем тетраизопропанолят титана (12 мл, 39.8 ммоль), и реакционная смесь становилась желтой. После перемешивания в течение 0.5 часа, добавляли в смесь (Z)-4-((трет-бутилдиметилсилил)окси)бут-2-ен-1-ол (12 г, 59.4 ммоль) и перемешивали 1 час при той же температуре. Добавляли раствор трет-бутилгидропероксида (в толуоле, 5M, 28.5 мл, 142 ммоль) и перемешивали в течение ночи при той же температуре. После завершения реакции согласно данным ТСХ-мониторинга, добавляли раствор винной кислоты (10%, 192 мл), содержащий FeSO4·7H2O (23.4 г, 84 ммоль), и перемешивали при 0°C в течение 5 часов, затем реакционную смесь оставляли расслаиваться. Смесь фильтровали, органическую фазу отделяли, водную фазу экстрагировали дихлорметаном один раз. Органические фазы объединяли и упаривали, получая масло. Растворяли полученное масло в 100 мл диэтилового эфира, добавляли по каплям 1н. раствор гидроксида натрия (50 мл) при охлаждении на ледяной бане, перемешивали в течение 10 минут, затем отделяли эфирный слой и промывали его последовательно водой и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали, получая бледно-желтое масло. После колоночной хроматографии (петролейный эфир/этилацетат = 9/1) получали 8.1 г интермедиата 1 в виде светло-желтого масла, выход: 62.8%.
Пример получения 2
Получение ((2S,3R)-3-(((трет-бутилдиметилсилил)окси)метил)оксиран-2- ил)метанола (Интермедиат 2)
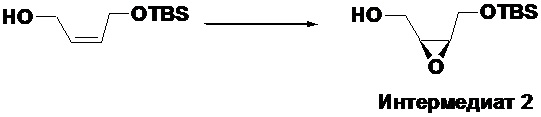
Методика такая же, как в случае получения интермедиата 1, за исключением того, что L-(+)-диэтилтартрат был использован вместо D-(-)-диэтилтартрата, с получением 3.0 г интермедиата 2 в виде светло-желтого масла, выход: 55.6%.
Пример получения 3
Получение ((2R,3S)-3-((тритилокси)метил)оксиран-2-ил)метанола (Интермедиат 3)
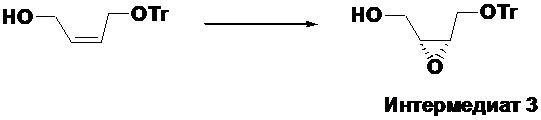
В четырехгорлую колбу объемом 1 л помещали молекулярные сита 4А (12 г) и безводный дихлорметан (330 мл). Смесь охлаждали до -40°C в атмосфере аргона. Добавляли D-(-)-диэтилтартрат (13.6 мл, 79.2 ммоль), затем тетраизопропанолят титана, и реакционная смесь становилась желтой. После перемешивания в течение 0.5 часа, добавляли в смесь раствор (Z)-4-(тритилокси)бут-2-ен-1-ола (26.1 г, 79.2 ммоль) в дихлорметане (120 мл), перемешивали в течение 2 часов при той же температуре и затем в течение ночи при -20°C. Добавляли раствор трет-бутилгидропероксида (в толуоле, 5M, 28.5 мл, 142 ммоль) и перемешивали в течение ночи при той же температуре. После завершения реакции согласно данным ТСХ-мониторинга, добавляли раствор винной кислоты (10%, 200 мл), содержащий FeSO4·7H2O (30 г) и перемешивали при 0°C в течение 1 часа, затем реакционную смесь оставляли расслаиваться. Органическую фазу отделяли, водную фазу экстрагировали дихлорметаном один раз. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия два раза, фильтровали и упаривали, получая твердое вещество, которое растирали в н-гексане, получая 30 г не совсем белого твердого вещества, затем перекристаллизовывали его из смеси петролейный эфир/этилацетат, получая 15 г интермедиата 3 в виде не совсем белого твердого вещества с выходом 57.5%.
Пример 1
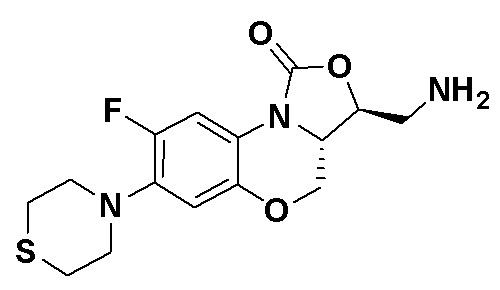
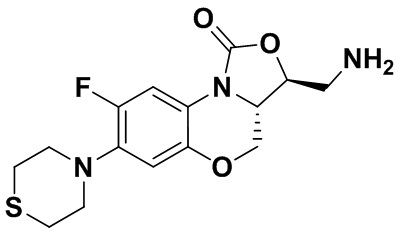
(3S,3aS)-3-(аминометил)-8-фтор-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-1-он (соединение 1)
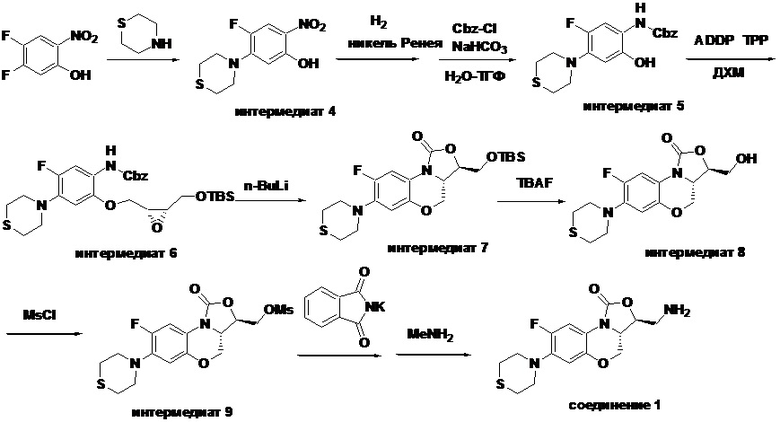
Стадия 1: Получение 4-фтор-2-нитро-5-тиоморфолинофенола (интермедиат 4)
В раствор 4,5-дифтор-2-нитрофенола (1.75 г, 10 ммоль) в ацетонитриле (20 мл) добавляли N-метилморфолин (1.5 мл), затем тиоморфолин (11 мл, 11 ммоль). Реакционную смесь нагревали при 80°C в течение 3 часов. После охлаждения добавляли воду (20 мл), получая твердый осадок, который отфильтровывали и промывали водой, сушили под инфракрасной лампой, получая 2.53 г интермедиата 4 в виде оранжевого твердого вещества с выходом 98.1%.
1H-ЯМР (400 МГц, CDCl3) δ: 11.20 (с, 1H), 7.64 (д, J = 12.4 H, 1H), 5.82 (д, J =7.6 Гц, 1H), 4.24 (с, 4H), 3.43 (с, 4H). LC-MS (ESI): m/z [M+H]+: 271.0744.
Стадия 2: Получение бензил(5-фтор-2-гидрокси-4-тиоморфолинофенил) карбамата (интермедиат 5)
Интермедиат 4 (4 г, 15.5 ммоль) суспендировали в смеси этанола и тетрагидрофурана (1:1, 40 мл). Никель Ренея (1 г) добавляли и гидрировали при среднем давлении 2 часа. Реакционную смесь фильтровали в колбу, содержащую бикарбонат натрия (2.6 г, 31 ммоль) и воду (10 мл), в защитной атмосфере аргона. Добавляли по каплям бензилхлорформиат (1.95 мл, 14.4 ммоль) при охлаждении на ледяной бане и перемешивали в течение 20 минут при той же температуре. Органическую фазу промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая красное твердое вещество. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/этилацетат = 7/3, получая интермедиат 5 (4.5 г, 80.4%) в виде розового твердого вещества.
LC-MS (ESI): m/z [M+H]+: 363.1905.
Стадия 3: Получение бензил (2-(((2R,3S)-3-(((трет-бутилдиметилсилил)окси) метил)оксиран-2-ил)метокси)-5-фтор-4-тиоморфолинофенил)карбамата (интермедиат 6)
В двугорлую колбу объемом 50 мл добавляли интермедиат 5 (1 г, 2.76 ммоль), интермедиат 1 (0.9 г, 4.14 ммоль), трифенилфосфин (1.45 г, 5.52 ммоль) и безводный дихлорметан (20 мл), и затем добавляли в четыре порции ADDP (1.39 г, 5.52 ммоль). После завершения реакции согласно данным ТСХ-мониторинга, добавляли н-гексан для разбавления, реакционную смесь фильтровали, и фильтрат упаривали. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/этилацетат = 92/8, получая светло-желтое масло, которое затвердевало при комнатной температуре с получением интермедиата 6 (1.2 г, 77.4%).
1H-ЯМР (400 МГц, CDCl3) δ: 7.92 (д, J = 12.4 Гц, 1H), 7.44-7.31 (м, 5H), 7.19 (ушир.с, 1H), 6.59 (ушир.с, 1H), 5.20 (с, 2H), 4.31 (дд, J = 11.6, 3.2 Гц, 1H), 4.04 (дд, J = 11.6, 7.2 Гц, 1H), 3.91-3.79 (м, 2H), 3.40-3.20 (м, 6H), 2.81 (ушир.с, 4H), 0.90 (с, 9H), 0.10 (с, 3H), 0.09 (с, 3H). LC-MS (ESI): m/z [M+H]+: 563.2954.
Стадия 4: Получение (3R,3aS)-3-(((трет-бутилдиметилсилил)окси)метил)-8- фтор-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 7)
В раствор интермедиата 6 (1.7 г, 3.02 ммоль) в безводном тетрагидрофуране (30 мл) в атмосфере аргона при -78°C по каплям добавляли n-BuLi (1.6 М раствор в н-гексане 2 мл, 3.3 ммоль). После добавления полученную смесь перемешивали в течение 1.5 часов при той же температуре, затем нагревали до комнатной температуры и перемешивали в течение ночи. Насыщенный раствор хлорида аммония (2 мл) добавляли для остановки реакции. Растворитель упаривали и добавляли этилацетат и воду. Органическую фазу отделяли, и водную фазу снова экстрагировали этилацетатом. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме, получая светло-розовое твердое вещество. Полученный остаток очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/дихлорметан/ этилацетат = 80/10/10, получая интермедиат 7 (1.29 г, 94.2%) в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.73 (д, J = 12.8 Гц, 1H), 6.61 (д, J = 6.8 Гц, 1H), 4.42 (дд, J = 10.4, 3.2 Гц, 1H), 4.28-4.22 (м, 1H), 4.09-4.02 (м, 1H), 3.96-3.81 (м, 3H), 3.28 (м, 4H), 2.82 (т, J = 4.8 Гц, 4H), 0.90 (с, 9H), 0.11 (с, 3H), 0.10 (с, 3H). LC-MS (ESI): m/z [M+H]+455.2620.
Стадия 5: Получение (3R,3aS)-8-фтор-3-(гидроксиметил)-7-тиоморфолино-3a,4- дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 8)
В раствор интермедиата 7 (1.2 г, 2.64 ммоль) в ТГФ (10 мл) в колбе объемом 50 мл, помещенной в ледяную баню, добавляли фторид тетрабутиламмония (3.2 мл, 3.2 ммоль, 1M раствор в тетрагидрофуране). После перемешивания в течение 0.5 часа, при добавлении воды выпадал осадок. Смесь фильтровали и осадок на фильтре промывали водой, сушили, получая интермедиат 8 (0.70 г, 78.3%) в виде не совсем белого твердого вещества.
Стадия 6: Получение ((3R,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро- 1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил метансульфоната (интермедиат 9)
В раствор интермедиата 8 (1.0 г, 2.94 ммоль) в дихлорметане (20 мл) в двугорлой колбе объемом 50 мл, охлажденный до 0°C на бане с ледяной водой, добавляли триэтиламин (1.2 мл, 8.8 ммоль) и затем метансульфонилхлорид (0.34 мл, 4.4 ммоль). Реакционную смесь упаривали, получая твердый остаток. В полученный остаток добавляли воду и фильтровали, получая интермедиат 9 (1.11 г, 90.2%) в виде светло-красного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.70 (дд, J = 12.8, 1.0 Гц, 1H), 6.61 (д, J = 7.9 Гц, 1H), 4.57-4.46 (м, 4H), 4.13-4.05 (м, 1H), 3.87 (т, J = 10.2 Гц, 1H), 3.35-3.22 (м, 4H), 3.14 (с, 3H), 2.82 (т, J = 4.8 Гц, 4H). LC-MS (ESI): m/z [M+H]+ 419.1083.
Стадия 7: Получение (3S,3aS)-3-(аминометил)-8-фтор-7-тиоморфолино-3a,4- дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (соединение 1)
Интермедиат 9 (1.1 г, 2.63 ммоль) растворяли в ДМФА (20 мл). Добавляли фталимид калия (1.09 г, 5.88 ммоль) и смесь оставляли для протекания реакции при 80°C на 5 часов. Затем добавляли после охлаждения ледяную воду (20 мл). Выпадал твердый осадок, который отфильтровывали, промывали и сушили, получая светло-розовое твердое вещество. Этот твердый остаток помещали в герметично закрываемую пробирку и добавляли раствор метиламина в метаноле (5 мл). Реакционную смесь нагревали при 80°C в течение 4 часа. После охлаждения добавляли воду для разбавления, и смесь экстрагировали этилацетатом 3 раза. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле с получением соединения 1 (250 мг, 25.0%) в виде светло-желтого твердого вещества. Т.пл.: 155-157oC.
1H-ЯМР (400 МГц, CDCl3) δ: 7.75 (д, J = 13.0 Гц, 1H), 6.55 (д, J = 7.8 Гц, 1H), 4.45 (дд, J = 10.4, 3.0 Гц, 1H), 4.29-4.23 (м, 1H), 4.07-4.00 (м, 1H), 3.86 (т, J = 10.2 Гц, 1H), 3.32-3.20 (м, 4H), 3.19-3.04 (м, 2H), 2.83-2.76 (м, 4H), 1.41 (ушир.с, 2H). HR-MS (ESI): m/z [M+H]+ вычислено для C15H19FN3O3S: 340.1126; найдено: 340.1111.
Пример 2
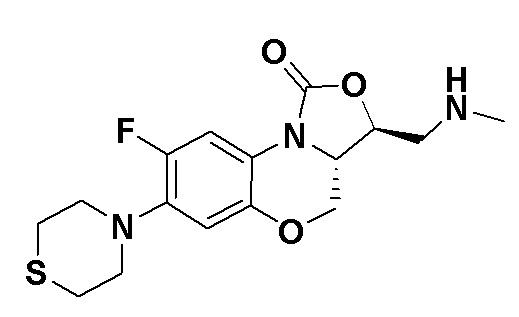
(3S,3aS)-8-фтор-3-((метиламино)метил)-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-он (соединение 2)
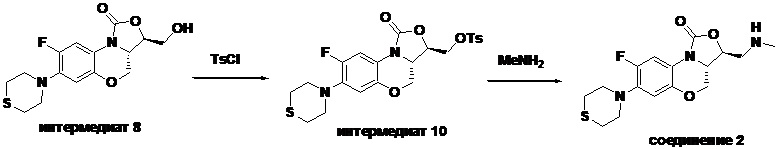
Стадия 1: Получение ((3R,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро- 1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил-4-метил бензолсульфоната (интермедиат 10)
Интермедиат 8 (1.2 г, 3.53 ммоль) помещали в двугорлую колбу объемом 50 мл и растворяли в дихлорметане (30 мл). Температуру понижали до 0°C на бане с ледяной водой, добавляли триэтиламин (0.99 мл, 7.06 ммоль) и DMAP (50 мг) и затем порциями добавляли п-метилбензолсульфонил хлорид (0.81 г, 4.24 ммоль). После перемешивания в течение 1 часа при той же температуре, реакционную смесь разбавляли дихлорметаном, промывали последовательно водой, 10%-ным раствором лимонной кислоты, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали. Фильтрат упаривали при пониженном давлении и очищали методом колоночной хроматографии на силикагеле (дихлорметан/этилацетат = 80/20), получая интермедиат 10 (1.59 г, 85.0%) в виде белого твердого вещества.
Стадия 2: Получение (3S,3aS)-8-фтор-3-((метиламино)метил)-7-тиоморфолино- 3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (соединение 2)
Интермедиат 10 (0.090 г, 0.17 ммоль) помещали в герметично закрываемую пробирку, добавляли метанольный раствор (3 мл) метиламина и тетрагидрофуран (3 мл), нагревали при 100°C в течение 1 часа. Реакционную смесь упаривали и очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол/гидроксид аммония = 100/2/1), получая соединение 2 (30 мг, 50.0%) в виде не совсем белого твердого вещества. Т.пл.: 155-156°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.75 (д, J = 13.0 Гц, 1H), 6.54 (д, J = 7.9 Гц, 1H), 6.03 (ушир.с, 1H), 4.44 (дд, J = 10.4, 3.0 Гц, 1H), 4.35 (дд, J = 12.2, 5.4 Гц, 1H), 4.08-3.98 (м, 1H), 3.84 (т, J = 10.2 Гц, 1H), 3.33-3.20 (м, 4H), 3.04-2.91 (м, 2H), 2.83-2.75 (м, 4H), 2.51 (с, 3H). HR-MS (ESI): m/z [M+H]+ вычислено для C16H21FN3O3S: 354.1282; найдено: 354.1275.
Пример 3
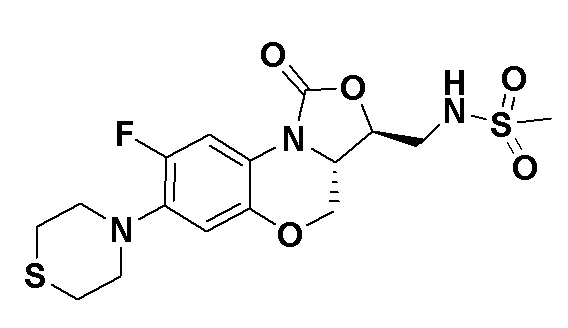
N-(((3S,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)метансульфонамид (соединение 3)
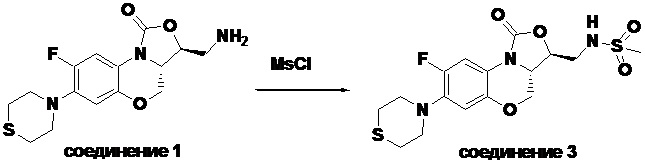
В раствор соединения 1 (0.070 г, 0.175 ммоль) в дихлорметане (4 мл) добавляли триэтиламин (0.037 мл, 0.26 ммоль) при охлаждении на ледяной бане. Добавляли метилсульфонилхлорид (0.016 мл, 0.21 ммоль). После завершения реакции согласно данным ТСХ-мониторинга, растворитель упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99/1), получая соединение 3 (43 мг, 51.8%) в виде не совсем белого твердого вещества. Т.пл.: 234-235°C.
1H-ЯМР (400 МГц, ДМСО-d6) δ: 7.61-7.51 (м, 2H), 6.68 (д, J = 8.2 Гц, 1H), 4.55-4.47 (м, 2H), 4.04-3.94 (м, 2H), 3.50-3.35 (м, 2H), 3.17 (ушир.с, 4H), 2.96 (с, 3H), 2.77-2.69 (м, 4H). HR-MS (ESI): m/z [M+H]+ вычислено для C16H21FN3O5S2: 418.0901; найдено: 418.0894.
Пример 4
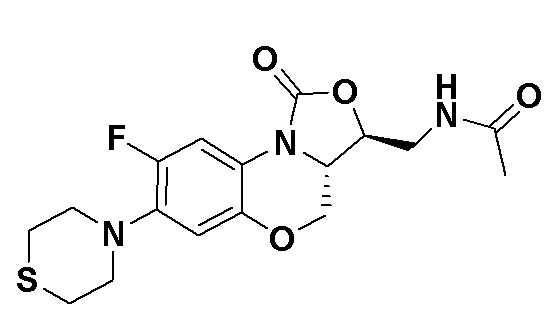
N-(((3S,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамид (соединение 4)
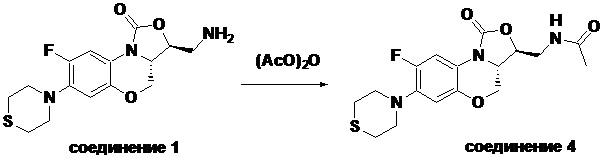
В раствор соединения 1 (0.18 г, 0.53 ммоль) в дихлорметане (6 мл) добавляли пиридин (0.086 мл, 1.06 ммоль), охлажденный на ледяной бане. Добавляли уксусный ангидрид (0.076 мл, 0.8 ммоль) и перемешивали смесь в течение 40 минут. Растворитель упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99/1), получая соединение 4 (128 мг, 63.4%) в виде не совсем белого твердого вещества. Т.пл.: 190-192°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.76 (д, J = 13.0 Гц, 1H), 6.83 (ушир.с, 1H), 5.96 (т, J = 6.0 Гц, 1H), 4.51 (дд, J = 10.4, 2.8 Гц, 1H), 4.43-4.37 (м, 1H), 3.95-3.88 (м, 1H), 3.82 (т, J = 10.2 Гц, 1H), 3.79-3.63 (м, 2H), 3.39-3.31 (м, 4H), 2.89 (ушир.с, 4H), 2.05 (с, 3H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H21FN3O4S: 382.1231; найдено: 382.1222.
Пример 5
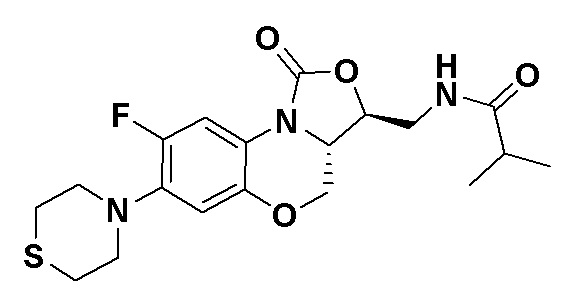
N-(((3S,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)изобутирамид (соединение 5)
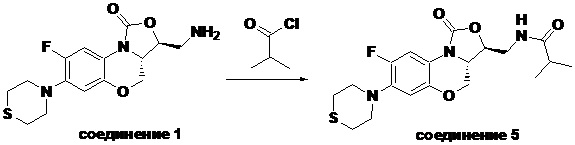
В раствор соединения 1 (0.055 г, 0.16 ммоль) в дихлорметане (6 мл) добавляли триэтиламин (0.034 мл, 0.24 ммоль), при охлаждении на ледяной бане. Добавляли изобутирилхлорид (0.020 мл, 0.18 ммоль) и полученную смесь перемешивали в течение 30 минут. Растворитель упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99/1), получая соединение 5 (38 мг, 58.5%) в виде не совсем белого твердого вещества. Т.пл.: 193-195°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.76 (д, J = 13.0 Гц, 1H), 7.01 (ушир.с, 1H), 6.06 (т, J = 5.4 Гц, 1H), 4.51 (дд, J = 10.4, 2.6 Гц, 1H), 4.45-4.36 (м, 1H), 4.00-3.87 (м, 1H), 3.86-3.62 (м, 3H), 3.41 (ушир.с, 4H), 2.94 (ушир.с, 4H), 2.49-2.36 (м, 1H), 1.17 (д, J = 4.0 Гц, 3H), 1.16 (д, J = 4.0 Гц, 3H). HR-MS (ESI): m/z [M+H]+ вычислено для C19H25FN3O4S: 410.1544; найдено: 410.1528.
Пример 6
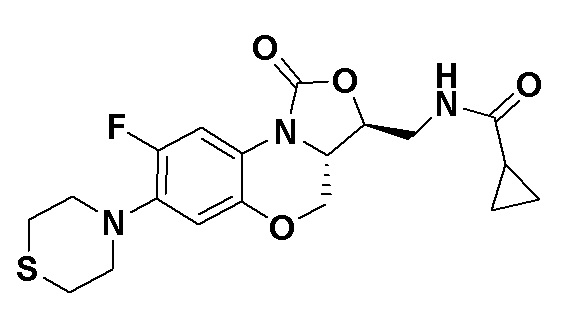
N-(((3S,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)циклопропанкарбоксамид (соединение 6)
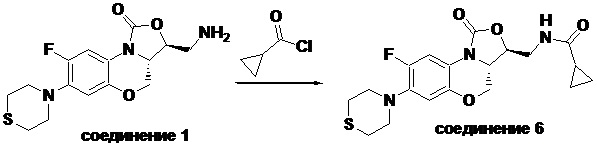
В раствор соединения 1 (0.10 г, 0.30 ммоль) в тетрагидрофуране (7 мл) добавляли триэтиламин (0.13 мл, 0.9 ммоль) при охлаждении на ледяной бане. Добавляли циклопропанкарбонилхлорид (0.035 мл, 0.39 ммоль) и полученную смесь перемешивали в течение 15 минут. Растворитель упаривали. Полученный остаток разбавляли дихлорметаном и промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99/1), получая соединение 6 (86 мг, 70.5%) в виде не совсем белого твердого вещества. Т.пл. 209-211°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.74 (д, J = 12.8 Гц, 1H), 6.60 (д, J = 7.6 Гц, 1H), 6.16 (т, J = 6.0 Гц, 1H), 4.48 (дд, J = 10.2, 2.8 Гц, 1H), 4.43-4.36 (м, 1H), 3.95-3.88 (м, 1H), 3.82 (т, J = 10.2 Гц, 1H), 3.79–3.65 (м, 2H), 3.32–3.23 (м, 4H), 2.86–2.76 (м, 4H), 1.45–1.36 (м, 1H), 1.01–0.94 (м, 2H), 0.85-0.77 (м, 2H). HR-MS (ESI): m/z [M+H]+ вычислено для C19H23FN3O4S: 408.1388; найдено: 408.1379.
Пример 7
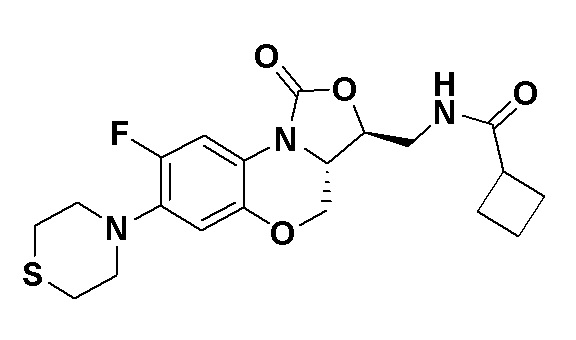
N-(((3S,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)циклобутанкарбоксамид (соединение 7)
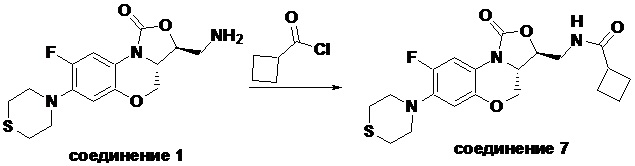
В раствор соединения 1 (0.098 г, 0.29 ммоль) в дихлорметане (3 мл) добавляли триэтиламин (0.081 мл, 0.58 ммоль), охлажденный на ледяной бане. Добавляли циклобутанкарбонилхлорид (0.037 мл, 0.37 ммоль), и полученную смесь перемешивали в течение 2 часов. Полученную смесь разбавляли дихлорметаном и промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99/1), получая соединение 7 (30 мг, 24.8%) в виде не совсем белого твердого вещества. Т.пл. 176-178°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.71 (д, J = 12.8 Гц, 1H), 6.63 (д, J = 7.4 Гц, 1H), 5.83 (т, J = 6.0 Гц, 1H), 4.50 (дд, J = 10.2, 2.8 Гц, 1H), 4.42-4.36 (м, 1H), 3.94-3.86 (м, 1H), 3.82 (т, J = 10.2 Гц, 1H), 3.78-3.71 (м, 1H), 3.71-3.62 (м, 1H), 3.36-3.22 (м, 4H), 3.11-2.97 (м, 1H), 2.89-2.76 (м, 4H), 2.34-2.11 (м, 4H), 2.07-1.81 (м, 2H). HR-MS (ESI): m/z [M+H]+ вычислено для C20H25FN3O4S: 422.1544; найдено: 422.1534.
Пример 8
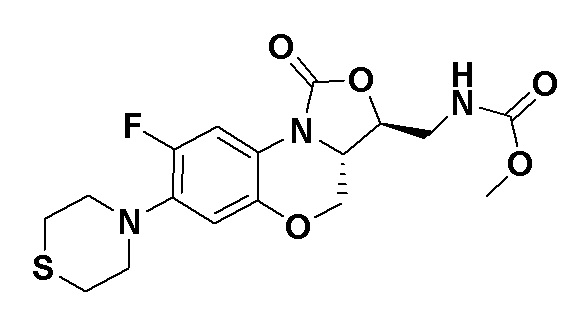
Метил (((3S,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)карбамат (соединение 8)
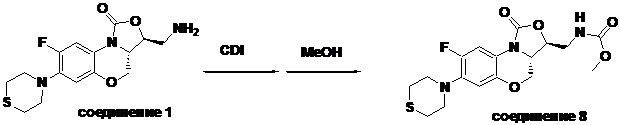
В раствор соединения 1 (0.10 г, 0.29 ммоль) в тетрагидрофуране (9 мл) добавляли 1,1'-карбонилдиимидазол (CDI, 0.49 г, 3 ммоль), и смесь перемешивали при комнатной температуре в течение 50 минут. Добавляли безводный метанол (3 мл), и полученную смесь перемешивали в течение ночи при комнатной температуре. Растворитель упаривали, и остаток разбавляли дихлорметаном. Полученную смесь промывали насыщенным раствором хлорида аммония, насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/ этилацетат = 60/40), получая соединение 8 (77 мг, 64.7%) в виде белого твердого вещества. Т.пл. 155-156°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.74 (д, J = 12.8 Гц, 1H), 6.66 (д, J = 5.8 Гц, 1H), 5.16 (ушир.с, 1H), 4.49 (дд, J = 10.4, 3.0 Гц, 1H), 4.41-4.35 (м, 1H), 3.99-3.91 (м, 1H), 3.84 (т, J = 10.2 Гц, 1H), 3.70 (с, 3H), 3.67-3.58 (м, 2H), 3.26-3.24 (м, 4H), 2.83 (т, J = 4.8 Гц, 4H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H21FN3O5S: 398.1180; найдено: 398.1172.
Пример 9
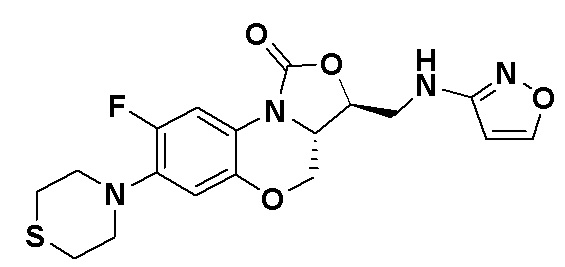
(3S,3aS)-8-фтор-3-((изоксазол-3-иламино)метил)-7-тиоморфолино-3a,4-дигидро- 1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-он (соединение 9)
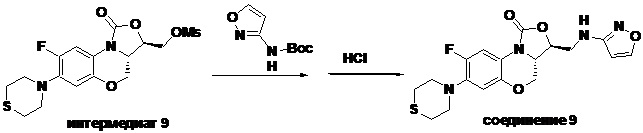
В раствор N-Boc-3-аминоизоксазола (0.057 г, 0.31 ммоль) в безводном ДМФА (2 мл), охлажденный ледяной бане, добавляли NaH (60%, 15 мг, 0.34 ммоль). После перемешивания в течение 5 минут, добавляли интермедиат 9 (0.13 г, 0.31 ммоль) и оставляли для прохождения реакции при 70°C на 2 часа. После охлаждения добавляли по каплям ледяную воду (10 мл), смесь экстрагировали дихлорметаном два раза, и органические фазы объединяли. Органическую фазу промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 85/15), получая 141 мг масла с выходом 89.8%.
В раствор описанного выше масла в этилацетате (2 мл) добавляли метанольный раствор хлороводорода (5н., 4 мл) и перемешивали при комнатной температуре в течение 30 минут. Растворитель упаривали, добавляли воду (3 мл), и значение pH доводили до щелочного с помощью насыщенного раствора бикарбоната натрия. Выпадал твердый осадок, который отфильтровывали. Осадок промывали на фильтре водой до нейтрального рН, сушили, получая соединение 9 (94 мг, 75.2%) в виде белого твердого вещества. Т.пл. 148-150°C.
1H-ЯМР (400 МГц, CDCl3) δ: 8.08 (д, J = 1.8 Гц, 1H), 7.73 (д, J = 12.8 Гц, 1H), 6.55 (д, J = 7.8 Гц, 1H), 5.88 (д, J = 1.8 Гц, 1H), 4.61–4.54 (м, 1H), 4.51 (дд, J = 10.4, 3.0 Гц, 1H), 4.35 (т, J = 6.4 Гц, 1H), 4.05-3.98 (м, 1H), 3.86 (т, J = 10.2 Гц, 1H), 3.82-3.74 (м, 1H), 3.74-3.65 (м, 1H), 3.31-3.20 (м, 4H), 2.79 (т, J = 5.2 Гц, 4H). HR-MS (ESI): m/z [M+H]+ вычислено для C18H20FN4O4S: 407.1184; найдено: 407.1174.
Пример 10
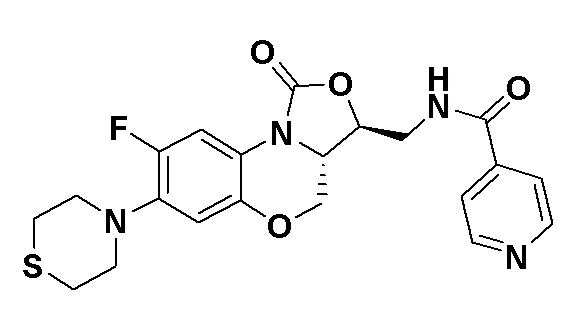
N-(((3S,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)изоникотинамид (соединение 10)
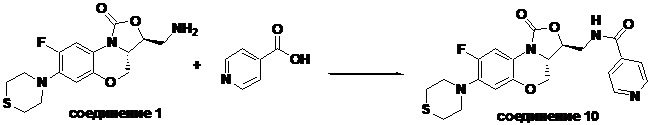
Соединение 1 (60 мг, 0.18 ммоль), изоникотиновую кислоту (26 мг, 0.21 ммоль), EDCI (40 мг, 0.21 ммоль), HOBt (28 мг, 0.21 ммоль) и триэтиламин (0.050 мл, 0.35 ммоль) добавляли в колбу объемом 5 мл. Добавляли ДМФА (2 мл), и смесь перемешивали в течение ночи при комнатной температуре. При добавлении ледяной воды выпадал твердый осадок, который отделяли фильтрованием. Полученное твердое вещество растворяли в дихлорметане и очищали методом колоночной хроматографии на силикагеле (200-300 меш) (дихлорметан/этилацетат/метанол = 50/50/1), получая соединение 10 (46 мг, 59.0%) в виде белого твердого вещества. Т.пл. 135-137°C.
1H-ЯМР (400 МГц, CDCl3) δ: 8.77 (д, J = 4.4 Гц, 2H), 7.69 (д, J = 5.6 Гц, 2H), 7.65 (д, J = 12.8 Гц, 1H), 7.35 (т, J = 5.8 Гц, 1H), 6.55 (д, J = 7.8 Гц, 1H), 4.61-4.49 (м, 2H), 4.03-3.92 (м, 2H), 3.93-3.82 (м, 2H), 3.33-3.18 (м, 4H), 2.8-2.75 (м, 4H). HR-MS (ESI): m/z [M+H]+ вычислено для C21H22FN4O4S: 445.1340; найдено: 445.1324.
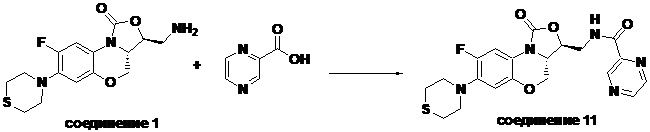
Пример 11
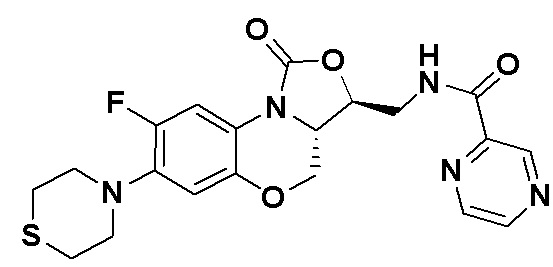
N-(((3S,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)пиразин-2-карбоксамид (соединение 11)
Соединение 1 (60 мг, 0.18 ммоль), 2-пиразинкарбоновую кислоту (25 мг, 0.21 ммоль), EDCI (40 мг, 0.21 ммоль), HOBt (28 мг, 0.21 ммоль) и триэтиламин (0.050 мл, 0.35 ммоль) добавляли в колбу объемом 5 мл. Добавляли ДМФА (2 мл), и смесь перемешивали в течение ночи при комнатной температуре. При добавлении ледяной воды выпадал твердый осадок, который отделяли фильтрованием, растворяли в дихлорметане и очищали методом колоночной хроматографии на силикагеле (200-300 меш) (дихлорметан/этилацетат/метанол = 50/50/1), получая соединение 11 (41 мг, 51.9%) в виде бледно-желтого твердого вещества. Т.пл. 212-214°C.
1H-ЯМР (400 МГц, CDCl3) δ: 9.38 (д, J = 1.2 Гц, 1H), 8.80 (д, J = 2.4 Гц, 1H), 8.57 (дд, J = 2.2, 1.4 Гц, 1H), 8.27 (т, J = 6.0 Гц, 1H), 7.73 (д, J = 12.8 Гц, 1H), 6.68 (ушир.с, 1H), 4.60-4.46 (м, 2H), 4.08-3.81 (м, 4H), 3.37-3.22 (м, 4H), 2.83 (ушир.с, 4H). HR-MS (ESI): m/z [M+H]+ вычислено для C20H21FN5O4S: 446.1298; найдено: 445.1276.
Пример 12
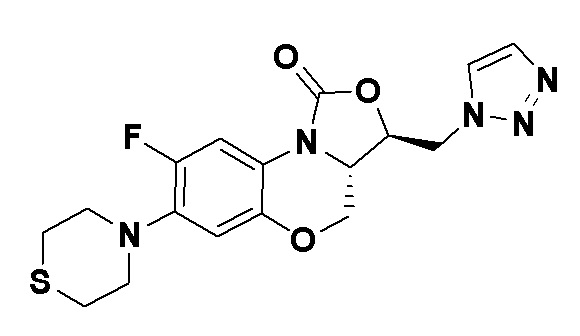
(3S,3aS)-3-((1H-1,2,3-Триазол-1-ил)метил)-8-фтор-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-он (соединение 12)
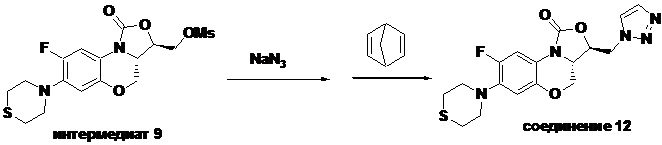
Интермедиат 9 (418 мг, 1 ммоль) растворяли в ДМФА (10 мл). Добавляли в раствор азид натрия (130 мг, 2 ммоль). Реакционную смесь нагревали при 80°C в течение 2.5 часов и затем охлаждали до комнатной температуры. После добавления ледяной воды (10 мл) выпадал твердый осадок, который отфильтровывали, промывали водой и сушили под инфракрасной лампой, получая 351 мг не совсем белого твердого вещества с выходом 96.2%.
LC-MS (ESI): m/z [M+H]+: 366.8012.
Полученное как описано выше твердое вещество (0.11 г, 0.3 ммоль) растворяли в 1,4-диоксане (3 мл) и добавляли дициклогептадиен (0.31 мл, 3 ммоль). Реакционную смесь кипятили 5 часов и упаривали растворитель. Полученный остаток очищали методом колоночной хроматографии на силикагеле (200-300 меш) (дихлорметан/метанол = 99/1), получая соединение 12 (83 мг, 70.9%) в виде не совсем белого твердого вещества. Т.пл. 223-225°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.80-7.77 (м, 2H), 7.64 (д, J = 12.8 Гц, 1H), 6.64 (д, J = 6.6 Гц, 1H), 4.84 (д, J = 4.8 Гц, 2H), 4.72-4.66 (м, 1H), 4.46 (дд, J = 10.6, 3.0 Гц, 1H), 4.08-4.00 (м, 1H), 3.84 (т, J = 10.2 Гц, 1H), 3.35-3.23 (м, 4H), 2.83 (т, J = 4.4 Гц, 4H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H19FN5O3S: 392.1187; найдено: 392.1178.
Пример 13
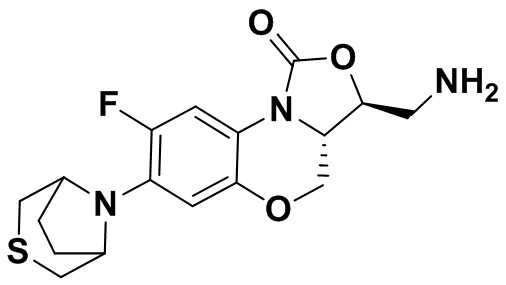
(3S,3aS)-3-(аминометил)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8-фтор- 3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-он (соединение 13)

Стадия 1: Получение 5-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-4-фтор- 2-нитрофенола (интермедиат 11)
В раствор 4,5-дифтор-2-нитрофенола (3.55 г, 20 ммоль) в ацетонитриле (40 мл) добавляли N-метилморфолин (6.7 мл, 60 ммоль), затем (1R, 5S)-3-тиа-8-азабицикло[3.2.1] октан гидроиодид (5.14 г, 20 ммоль), реакцию оставляли при 70°C на 8 часов перед тем как охладить. После добавления воды (30 мл) выпадал твердый осадок, который отфильтровывали, промывали водой и сушили под ультрафиолетовой лампой, получая 4.3 г оранжевого твердого интермедиата 11 с выходом 75.7%.
1H-ЯМР (400 МГц, CDCl3) δ: 11.05 (с, 1H), 7.74 (д, J = 14.6 Гц, 1H), 6.29 (д, J = 7.6 Гц, 1H), 4.72 (с, 2H), 3.28 (дд, J = 13.2, 1.8 Гц, 2H), 2.20 (м, 6H).
Стадия 2: Получение бензил (4-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-5- фтор-2-гидроксифенил)карбамата (интермедиат 12)
Интермедиат 11 (4 г, 14.1 ммоль) суспендировали в смеси этанола и тетрагидрофурана (1:1, 40 мл). Добавляли никель Ренея (1 г) и гидрировали при среднем давлении 3 часа. В реакционную смесь добавляли тетрагидрофуран (20 мл) и фильтровали в колбу, содержащую бикарбонат натрия (2.37 г, 28 ммоль) и воду (20 мл), в защитной атмосфере аргона. Добавляли бензилхлорформиат (1.9 мл, 14.1 ммоль) по каплям при охлаждении на ледяной бане и перемешивали в течение 20 минут при той же температуре. Растворитель упаривали и добавляли воду. Полученную смесь экстрагировали этилацетатом два раза. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая красное твердое вещество. Полученный остаток очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/этилацетат = 7/3, получая интермедиат 12 (4.16 г, 81.6%) в виде фиолетового твердого вещества.
Стадия 3: Получение бензил (4-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-5- фтор-2-(((2R,3S)-3-((тритилокси)метил)оксиран-2-ил)метокси)фенил)карбамата (интермедиат 13)
В трехгорлую колбу объемом 250 мл добавляли интермедиат 12 (3.62 г, 10 ммоль), интермедиат 1 (4.5 г, 13 ммоль), трифенилфосфин (5.2 г, 20 ммоль) и безводный дихлорметан (100 мл), затем в два приема добавляли ADDP (5 г, 20 ммоль). После завершения реакции согласно данным ТСХ-мониторинга, добавляли н-гексан для разбавления, смесь фильтровали, и фильтрат упаривали. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/ дихлорметан /этилацетат = 80/10/10, получая интермедиат 13 (2.3 г, 33.3%) в виде не совсем белой твердой пены.
1H-ЯМР (400 МГц, CDCl3) δ: 7.84 (д, J = 13.6 Гц, 1H), 7.47-7.19 (м, 20H), 7.07 (с, 1H), 6.34 (д, J = 7.8 Гц, 1H), 5.18 (с, 2H), 4.27 (ушир.с, 2H), 4.18 (д, J = 11.8 Гц, 1H), 3.83-3.74 (м, 1H), 3.43 (дд, J = 10.6, 5.4 Гц, 1H), 3.36-3.23 (м, 4H), 3.14 (дд, J = 10.4, 4.8 Гц, 1H), 2.17-1.93 (м, 3H).
Стадия 4: Получение (3R,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8- фтор-3-((тритилокси)метил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 14)
В раствор интермедиата 13 (2.1 г, 3 ммоль) в безводном тетрагидрофуране (33 мл) в атмосфере аргона при -78°C по каплям добавляли n-BuLi (1.6 М раствор в н-гексане 2 мл, 3.3 ммоль). После добавления полученную смесь перемешивали 1 час при той же температуре, затем нагревали до комнатной температуры и перемешивали в течение ночи. Добавляли насыщенный раствор хлорида аммония (2 мл) для остановки реакции. Растворитель упаривали и при перемешивании добавляли воду. Смесь перемешивали и фильтровали. Осадок на фильтре промывали водой до нейтрального рН, сушили и промывали н-гексаном два раза, получая интермедиат 14 (1.7 г, 94.4%) в виде светло-фиолетового твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.70 (д, J = 14.2 Гц, 1H), 7.47-7.41 (м, 6H), 7.36-7.23 (м, 9H), 6.40 (д, J = 8.0 Гц, 1H), 4.38-4.31 (м, 3H), 4.27-4.20 (м, 1H), 4.01-3.93 (м, 1H), 3.81 (т, J = 10.2 Гц, 1H), 3.48 (дд, J = 4.8, 3.6 Гц, 2H), 3.38-3.28 (м, 2H), 2.21-1.99 (м, 6H).
Стадия 5: Получение (3R,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8- фтор-3-(гидроксиметил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 15)
В раствор интермедиата 14 (1.64 г, 2.7 ммоль) в дихлорметане (25 мл) в колбе объемом 50 мл добавляли трифторуксусную кислоту (2.5 мл, 32.4 ммоль) при охлаждении на ледяной бане и перемешивали в течение ночи при комнатной температуре. Реакционную смесь доводили до слабощелочного значения рН 4н. раствором гидроксида натрия и экстрагировали два раза дихлорметаном. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая твердое вещество. Полученный твердый продукт очищали методом колоночной хроматографии на силикагеле (200-300 меш) (дихлорметан/метанол = 98/2), получая интермедиат 15 (0.83 г, 84.0%) в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.71 (д, J = 14.2 Гц, 1H), 6.43 (д, J = 8.0 Гц, 1H), 4.44 (дд, J = 10.5, 3.0 Гц, 1H), 4.41-4.31 (м, 3H), 4.18-4.10 (м, 1H), 4.02 (дд, J = 12.4, 3.8 Гц, 1H), 3.93-3.82 (м, 2H), 3.41-3.31 (м, 2H), 2.22-2.02 (м, 7H). LC-MS (ESI): m/z [M+H]+: 367.1288.
Стадия 6: Получение ((3R,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8- фтор-1-оксо-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил-4- метил бензолсульфоната (интермедиат 16)
В раствор интермедиата 15 (0.79 г, 2.16 ммоль) и DMAP (80 мг) в дихлорметане (20 мл) в двугорлой колбе объемом 50 мл добавляли триэтиламин (0.46 мл, 3.24 ммоль). Полученную смесь охлаждали до 0°C на бане с ледяной водой и порциями добавляли п-метилбензолсульфонилхлорид (0.49 г, 2.59 ммоль). Реакционную смесь перемешивали в течение 3 часов при той же температуре и разбавляли дихлорметаном. Реакционную смесь промывали последовательно водой, 10%-ным раствором лимонной кислоты и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая интермедиат 16 (1.07 г, 95.5%) в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.81 (дд, J = 6.8, 1.6 Гц, 2H), 7.62 (д, J = 14.0 Гц, 1H), 7.40 (дд, J = 8.0, 0.4 Гц, 2H), 6.41 (д, J = 7.8 Гц, 1H), 4.48-4.30 (м, 5H), 4.26 (дд, J = 11.0, 5.8 Гц, 1H), 4.06-4.00 (м, 1H), 3.84 (т, J = 10.2 Гц, 1H), 3.34 (т, J = 11.4 Гц, 1H), 2.48 (с, 3H), 2.21-1.99 (м, 6H). LC-MS (ESI): m/z [M+H]+: 521.1164.
Стадия 7: Получение (3S,3aS)-3-(аминометил)-7-((1R,5S)-3-тиа-8-азабицикло [3.2.1]октан-8-ил)-8-фтор-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (соединение 13)
В раствор интермедиата 16 (0.8 г, 1.54 ммоль) в тетрагидрофуране (24 мл) в герметично закрытой пробирке добавляли гидроксид аммония (15 мл). Реакционную смесь нагревали при 100°C в течение 7 часов. После охлаждения тетрагидрофуран упаривали, и водную фазу экстрагировали дихлорметаном три раза. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая светло-желтое твердое вещество. Полученный остаток очищали методом колоночной хроматографии на силикагеле (200-300 меш) (дихлорметан/метанол/ гидроксид аммония =100/2/1), получая соединение 13 (464 мг, 82.6%) в виде не совсем белого твердого вещества. Т.пл. 168-169°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.70 (д, J = 14.2 Гц, 1H), 6.41 (д, J = 8.0 Гц, 1H), 4.43 (дд, J = 10.4, 3.0 Гц, 1H), 4.39-4.31 (м, 2H), 4.29-4.22 (м, 1H), 4.07-3.99 (м, 1H), 3.87 (т, J = 10.2 Гц, 1H), 3.38-3.28 (м, 2H), 3.19-3.04 (м, 2H), 2.23-2.00 (м, 6H), 1.39-1.22 (ушир.с, 2H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H21FN3O3S: 366.1282; найдено: 366.1265.
Пример 14
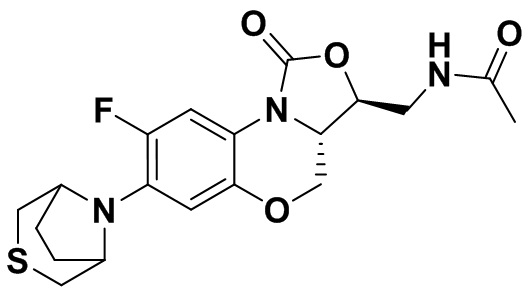
N-(((3S,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8-фтор-1-оксо-3a,4- дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамид (соединение 14)
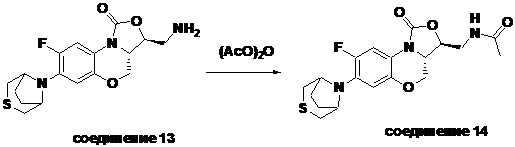
В раствор соединения 13 (0.062 г, 0.17 ммоль) в дихлорметане (4 мл) добавляли пиридин (0.042 мл, 0.52 ммоль) при охлаждении на ледяной бане. Добавляли уксусный ангидрид (0.024 мл, 0.26 ммоль) и затем полученную смесь перемешивали в течение 1.5 часов. Растворитель упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол=98.5/1.5), получая соединение 14 (43 мг, 62.3%) в виде белого твердого вещества. Т.пл.: 235-236°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.67 (д, J = 14.2 Гц, 1H), 6.42 (д, J = 8.0 Гц, 1H), 6.14 (т, J = 6.0 Гц, 1H), 4.48 (дд, J = 10.0, 2.6 Гц, 1H), 4.43-4.31 (м, 3H), 3.95-3.88 (м, 1H), 3.84 (т, J = 10.0 Гц, 1H), 3.78-3.63 (м, 2H), 3.39-3.29 (м, 2H), 2.21-1.99 (с, 9H). HR-MS (ESI): m/z [M+H]+ вычислено для C19H23FN3O4S: 408.1388; найдено: 408.1368.
Пример 15
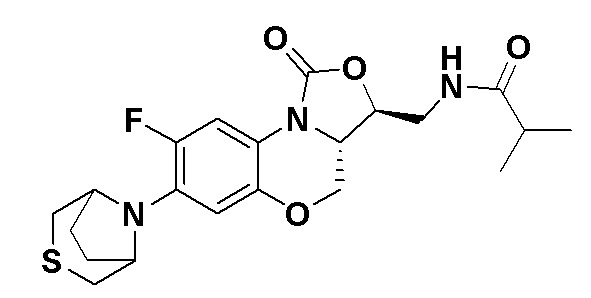
N-(((3S,3aS)-8-фтор-1-оксо-7-тиоморфолил-3a,4-дигидро-1H,3H-бензо[b]оксазоло [3,4-d][1,4]оксазин-3-ил)метил)изобутирамид (соединение 15)
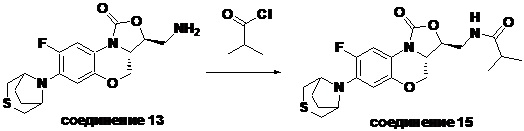
В раствор соединения 13 (0.062 г, 0.17 ммоль) в дихлорметане (6 мл) добавляли триэтиламин (0.036 мл, 0.26 ммоль) при охлаждении на ледяной бане. Добавляли изобутирилхлорид (0.022 мл, 0.21 ммоль) и полученную смесь перемешивали в течение 1 часа. Растворитель упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол=99.5/0.5), получая соединение 15 (54 мг, 73.0%) в виде белого твердого вещества. Т.пл.: 164-165°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.65 (д, J = 13.2 Гц, 1H), 6.41 (ушир.с, 1H), 6.06 (т, J = 6.0 Гц, 1H), 4.47 (дд, J = 10.0, 2.6 Гц, 1H), 4.43-4.28 (м, 3H), 3.97-3.79 (м, 2H), 3.79-3.61 (м, 2H), 3.34 (т, J = 11.4 Гц, 2H), 2.49-2.36 (м, 1H), 2.21-1.99 (м, 6H), 1.17 (д, J = 2.8 Гц, 3H), 1.16 (д, J = 2.4 Гц, 3H). HR-MS (ESI): m/z [M+H]+ вычислено для C21H27FN3O4S: 436.1701; найдено: 436.1680.
Пример 16
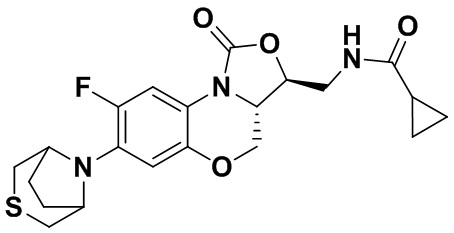
N-(((3S,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8-фтор-1-оксо-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)циклопропан- карбоксамид (соединение 16)
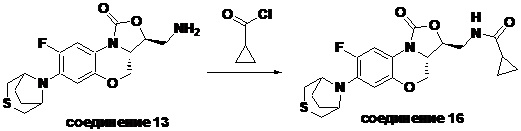
В раствор соединения 13 (0.062 г, 0.17 ммоль) в дихлорметане (6 мл) добавляли триэтиламин (0.036 мл, 0.26 ммоль) при охлаждении на ледяной бане. Добавляли циклопропанкарбонилхлорид (0.019 мл, 0.21 ммоль) и затем полученную смесь перемешивали в течение 1 часа. Растворитель упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол=99.5/0.5), получая соединение 16 (57 мг, 77.0%) в виде не совсем белого твердого вещества. Т.пл. 169-171°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.69 (д, J = 14.2 Гц, 1H), 6.41 (д, J = 8.0 Гц, 1H), 6.23 (т, J = 6.0 Гц, 1H), 4.46 (дд, J = 10.0, 2.6 Гц, 1H), 4.43-4.28 (м, 3H), 3.96-3.63 (м, 4H), 3.39-3.25 (м, 2H), 2.23-1.95 (м, 6H), 1.47-1.35 (м, 1H), 1.02-0.92 (м, 2H), 0.84-0.74 (м, 2H). HR-MS (ESI): m/z [M+H]+ вычислено для C21H25FN3O4S: 434.1544; найдено: 434.1525.
Пример 17
N-(((3S,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8-фтор-1-оксо-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)циклобутан- карбоксамид (соединение 17)
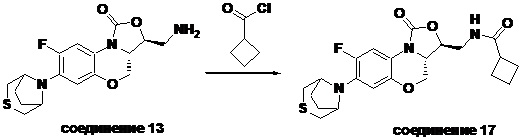
В раствор соединения 13 (0.062 г, 0.17 ммоль) в дихлорметане (6 мл) добавляли триэтиламин (0.036 мл, 0.26 ммоль) при охлаждении на ледяной бане. Добавляли циклобутанкарбонилхлорид (0.020 мл, 0.21 ммоль) и полученную смесь перемешивали в течение 1 часа. Полученную смесь упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99.5/0.5), получая соединение 17 (65 мг, 85.5%) в виде белого твердого вещества. Т.пл. 195-196°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.66 (д, J = 14.0 Гц, 1H), 6.41 (д, J = 7.6 Гц, 1H), 5.87 (т, J = 6.2 Гц, 1H), 4.47 (дд, J = 10.0, 2.6 Гц, 1H), 4.43-4.28 (м, 3H), 3.97-3.79 (м, 2H), 3.79-3.61 (м, 2H), 3.34 (т, J = 11.0 Гц, 2H), 3.14-2.95 (м, 1H), 2.35-1.80 (м, 12H). HR-MS (ESI): m/z [M+H]+ вычислено для C22H27FN3O4S: 448.1701; найдено: 448.1683.
Пример 18
Метил (((3S,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8-фтор-1-оксо- 3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)карбамат (соединение 18)
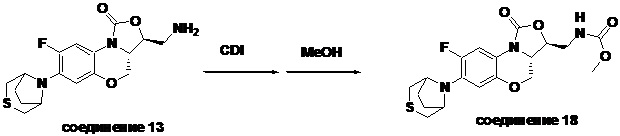
В раствор соединения 13 (0.062 г, 0.17 ммоль) в дихлорметане (4 мл) добавляли 1,1'-карбонилдиимидазол (CDI, 0.41 г, 2.55 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли безводный метанол (2 мл) и перемешивали в течение ночи при комнатной температуре. В смесь добавляли воду и дихлорметан. Полученную смесь промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 50/50), получая соединение 18 (25 мг, 34.7%) в виде не совсем белого твердого вещества. Т.пл. 149-151°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.69 (д, J = 14.0 Гц, 1H), 6.44 (д, J = 7.4 Гц, 1H), 5.19 (с, 1H), 4.47 (дд, J = 10.2, 2.6 Гц, 1H), 4.43-4.31 (м, 3H), 3.95 (ушир.с, 1H), 3.85 (т, J = 10.2 Гц, 1H), 3.71 (с, 3H), 3.67-3.56 (м, 2H), 4.43-3.32 (м, 2H), 2.22-2.00 (м, 6H). HR-MS (ESI): m/z [M+H]+ вычислено для C19H23FN3O5S: 424.1337; найдено: 424.1326.
Пример 19
(3S,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8-фтор-3-((изоксазол-3-иламино)метил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-он (соединение 19)
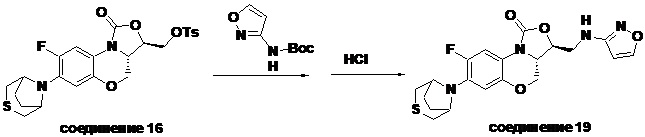
В раствор N-Boc-3-аминоизоксазола (0.041 г, 0.22 ммоль) в безводном ДМФА (2 мл), охлаждаемый в бане с ледяной водой, добавляли NaH (60%, 11 мг, 0.26 ммоль). После перемешивания в течение 10 минут, добавляли интермедиат 16 (0.11 г, 0.22 ммоль) и оставляли для прохождения реакции при 70°C на 1.5 часа. После охлаждения добавляли воду (10 мл), и смесь экстрагировали этилацетатом два раза. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 85/15), получая светло-розовое масло.
В раствор описанного выше масла в дихлорметане (2 мл) добавляли метанольный раствор хлороводорода (5н., 4 мл) и перемешивали при комнатной температуре в течение ночи. Растворитель упаривали, добавляли воду (3 мл), и значение pH доводили до щелочного с помощью насыщенного раствора бикарбоната натрия. Выпадал твердый осадок, его отфильтровывали. Осадок на фильтре промывали водой до нейтрального рН и сушили, получая соединение 19 (65 мг, 68.4% за две стадии) в виде светло-розового твердого вещества. Т.пл. 180-182°C.
1H-ЯМР (400 МГц, CDCl3) δ: 8.08 (с, 1H), 7.70 (д, J = 14.0 Гц, 1H), 6.45(д, J = 7.8 Гц, 1H), 5.89 (с, 1H), 4.58 (ушир.с, 1H), 4.53-4.46 (м, 1H), 4.42-4.31 (ушир.с, 2H), 4.06-3.98 (м, 1H), 3.87 (т, J = 10.0 Гц, 1H), 3.81-3.65 (м, 2H), 3.44-3.34 (м, 2H), 2.22-2.02 (м, 6H). HR-MS (ESI): m/z [M+H]+ вычислено для C20H22FN4O4S: 433.1340; найдено: 433.1321.
Пример 20
N-(((3S,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8-фтор-1-оксо-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)изоникотинамид (соединение 20)
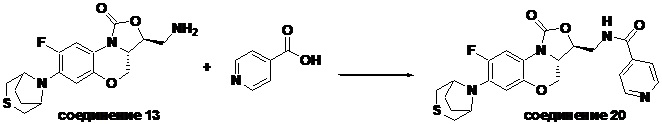
Соединение 13 (62 мг, 0.17 ммоль), изоникотиновую кислоту (25 мг, 0.2 ммоль), EDCI (38 мг, 0.2 ммоль), HOBt (27 мг, 0.2 ммоль) и триэтиламин (0.072 мл, 0.51 ммоль) помещали в колбу объемом 5 мл. Добавляли ДМФА (2 мл) и перемешивали в течение ночи при комнатной температуре. При добавлении ледяной воды выпадал твердый осадок, который отделяли фильтрованием. Полученное твердое вещество растворяли в дихлорметане и очищали методом колоночной хроматографии на силикагеле (200-300 меш) (дихлорметан/метанол/гидроксид аммония=100/1.5/1), получая соединение 20 (54 мг, 67.5%) в виде белого твердого вещества. Т.пл. 149-150°C.
1H-ЯМР (400 МГц, CDCl3) δ: 8.78 (дд, J = 4.4, 1.6 Гц, 2H), 7.68 (дд, J = 4.4, 1.6 Гц, 2H), 7.63 (д, J = 14.2 Гц, 1H), 7.17 (д, J = 6.0 Гц, 1H), 6.41 (д, J = 8.0 Гц, 1H), 4.58-4.48 (м, 2H), 4.35 (ушир.с, 2H), 4.04-3.95 (м, 2H), 3.94-3.82 (м, 1H), 3.36-3.27 (м, 2H), 2.21-2.00 (м, 6H). HR-MS (ESI): m/z [M+H]+ вычислено для C23H24FN4O4S: 471.1497; найдено: 471.1479.
Пример 21
N-(((3S,3aS)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1]октан-8-ил)-8-фтор-1-оксо-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)пиразин-2-карбоксамид (соединение 21)
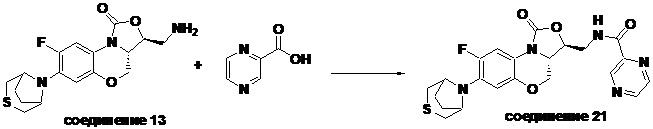
Соединение 13 (62 мг, 0.17 ммоль), 2-пиразинкарбоновую кислоту (25 мг, 0.2 ммоль), EDCI (38 мг, 0.2 ммоль), HOBt (27 мг, 0.2 ммоль) и триэтиламин (0.072 мл, 0.51 ммоль) помещали в колбу объемом 5 мл. Добавляли ДМФА (2 мл) и полученную смесь перемешивали в течение ночи при комнатной температуре. При добавлении ледяной воды выпадал твердый осадок, который отделяли фильтрованием и растворяли в дихлорметане, затем очищали методом колоночной хроматографии на силикагеле (200-300 меш) (дихлорметан/метанол = 99/1), получая соединение 21 (64 мг, 80.0%) в виде светло-желтого твердого вещества. Т.пл. >250°C.
1H-ЯМР (400 МГц, CDCl3) δ: 9.39 (с, 1H), 8.80 (д, J = 2.4 Гц, 1H), 8.58-8.55 (м, 1H), 8.28 (т, J = 6.2 Гц, 1H), 7.67 (дд, J = 4.4, 1.6 Гц, 1H), 6.41 (д, J = 7.2 Гц, 1H), 4.56-4.46 (м, 2H), 4.34 (ушир.с, 2H), 4.05-3.83 (м, 4H), 3.38-3.26 (м, 2H), 2.20-1.99 (м, 6H). HR-MS (ESI): m/z [M+H]+ вычислено для C22H23FN5O4S: 472.1449; найдено: 472.1430.
Пример 22
(3S,3aS)-3-((1H-1,2,3-триазол-1-ил)метил)-7-((1R,5S)-3-тиа-8-азабицикло[3.2.1] октан-8-ил)-8-фтор-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-он (соединение 22)
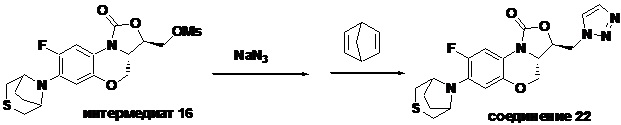
Интермедиат 16 (135 мг, 0.26 ммоль) растворяли в ДМФА (5 мл). Азид натрия (34 мг, 0.4 ммоль) добавляли в раствор. Реакционную смесь нагревали при 80°C в течение ночи и охлаждали до комнатной температуры. После добавления ледяной воды (10 мл) выпадал твердый осадок, который фильтровали, промывали водой, и сушили под инфракрасной лампой, получая не совсем белое твердое вещество (100 мг, 98.0%).
Полученное твердое вещество растворяли в 1,4-диоксане (3 мл) и добавляли дициклогептадиен (0.26 мл, 2.6 ммоль). Реакционную смесь кипятили в течение ночи и упаривали растворитель. Полученный остаток очищали методом колоночной хроматографии на силикагеле (200-300 меш) (дихлорметан/метанол = 99/1), получая соединение 22 (70 мг, 65.4%) в виде не совсем белого твердого вещества. Т.пл. 225-227°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.79 (д, J = 0.8 Гц, 1H), 7.78 (д, J = 1.2 Гц, 1H), 7.58 (д, J = 14.0 Гц, 1H), 6.39 (д, J = 8.0 Гц, 1H), 4.86-4.81 (м, 2H), 4.72-4.66 (м, 1H), 4.44 (дд, J = 10.4, 3.0 Гц, 1H), 4.39-4.30 (м, 2H), 4.07-3.99 (м, 1H), 3.86 (т, J = 10.2 Гц, 1H), 3.37-3.26 (м, 2H), 2.20-2.00 (м, 6H). HR-MS (ESI): m/z [M+H]+ вычислено для C19H21FN5O3S: 418.1344; найдено: 418.1324.
Пример 23
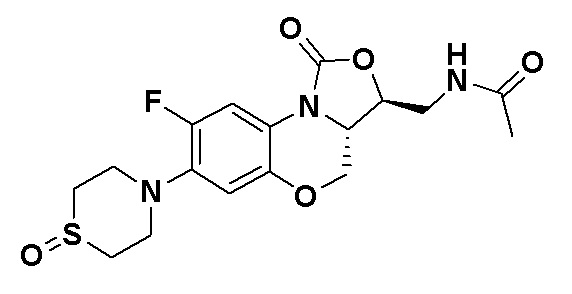
N-(((3S,3aS)-8-фтор-1-оксо-7-(1-оксидотиоморфолино)-3a,4-дигидро-1H,3H- бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамид (соединение 23)
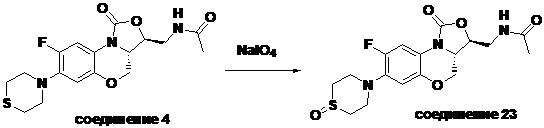
Смесь соединения 4 (0.1 г, 0.26 ммоль) и периодата натрия (0.11 г, 0.52 ммоль) помещали в одногорлую колбу объемом 25 мл. Добавляли метанол (4 мл) и воду (1.5 мл), и полученную смесь перемешивали в течение ночи при комнатной температуре. Растворитель упаривали, и остаток растворяли в метаноле. Нерастворенное вещество отфильтровывали, и фильтрат очищали методом колоночной хроматографии на силикагеле (этилацетат/метанол = 96/4), получая соединение 23 (74 мг, 71.8%) в виде белого твердого вещества. Т.пл. 214-216°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.76 (д, J = 12.8 Гц, 1H), 6.68 (д, J = 7.8 Гц, 1H), 6.04 (т, J = 6.0 Гц, 1H), 4.52 (дд, J = 10.4, 3.0 Гц, 1H), 4.43-4.37 (м, 1H), 3.96-3.89 (м, 1H), 3.83 (т, J = 10.2 Гц, 1H), 3.79-3.63 (м, 4H), 3.31-3.20 (м, 2H), 3.06-2.93 (м, 4H), 2.05 (с, 3H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H21FN3O5S: 398.1180; найдено: 398.1162.
Пример 24
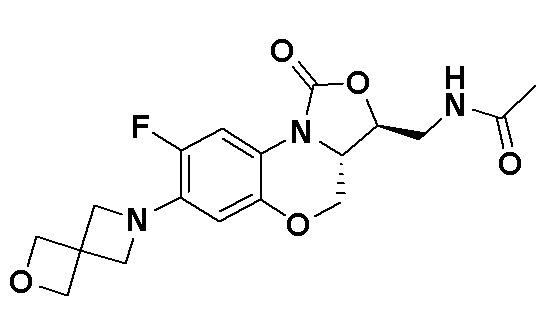
N-(((3S,3aS)-8-фтор-1-оксо-7-(2-окса-6-азаспиро[3.3]гептан-6-ил)-3a,4-дигидро- 1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамид (соединение 24)
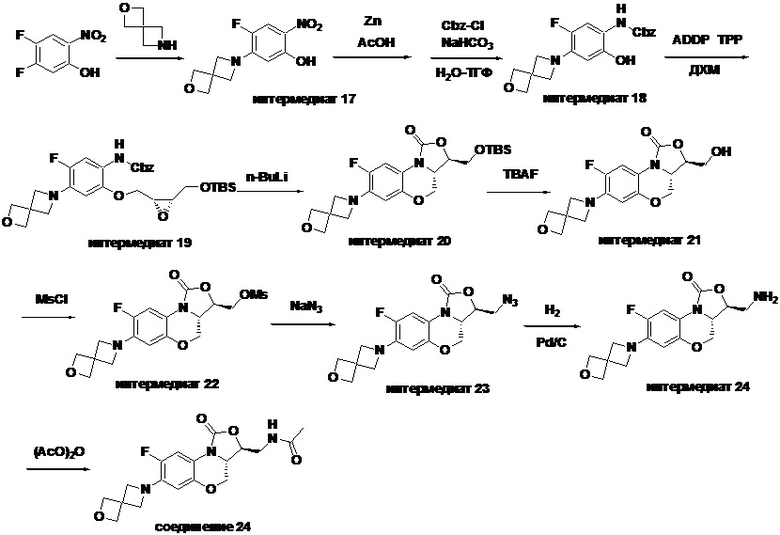
Стадия 1: Получение 4-фтор-2-нитро-5-(2-окса-6-азаспиро[3.3]гептан-6- ил)фенола (интермедиат 17)
В раствор 4,5-дифтор-2-нитрофенола (1.2 г, 6.9 ммоль) в ацетонитриле (5 мл) добавляли N-метилморфолин (1.5 мл), затем 2-окса-6-азаспиро[3.3]гептан (1 г, 10 ммоль), и реакционную смесь выдерживали при 80°C в течение 3 часов перед тем как охладить. После добавления воды (20 мл) выпадал твердый осадок, который отфильтровывали, промывали водой и сушили под инфракрасной лампой, получая интермедиат 17 (2.53 г, 77.7%) в виде желтого твердого вещества.
Стадия 2: Получение бензил (5-фтор-2-гидрокси-4-(2-окса-6-азаспиро[3.3]гептан- 6-ил)фенил)карбамата (интермедиат 18)
В суспензию интермедиата 17 (3.8 г, 15 ммоль) в тетрагидрофуране (80 мл) добавляли активную цинковую пыль (3.9 г, 60 ммоль) в атмосфере аргона. Добавляли по каплям уксусную кислоту (4.3 мл, 75 ммоль) и поддерживали температуру на уровне 30-40°C. После завершения реакции согласно данным ТСХ-мониторинга, реакционную смесь фильтровали в колбу, содержащую бикарбонат натрия (3.8 г, 45 ммоль) и воду (40 мл), в атмосфере аргона. Добавляли бензилхлорформиат (2 мл, 15 ммоль) при охлаждении на ледяной бане и перемешивали в течение 20 минут при той же температуре. Растворитель упаривали, добавляли воду, и смесь экстрагировали этилацетатом три раза. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая красное твердое вещество. Полученный твердый продукт очищали методом колоночной хроматографии на силикагеле (дихлорметан/этилацетат = 80/20), получая интермедиат 18 (1.5 г, 27.8%) в виде красного твердого вещества.
Стадия 3: Получение бензил (2-(((2R,3S)-3-(((трет-бутилдиметилсилил)окси) метил)оксиран-2-ил)метокси)-5-фтор-4-(2-окса-6-азаспиро[3.3]гептан-6-ил)фенил) карбамата (интермедиат 19)
В двугорлую колбу объемом 50 мл добавляли интермедиат 18 (1 г, 2.8 ммоль), интермедиат 1 (0.8 г, 3.64 ммоль), трифенилфосфин (1.47 г, 5.6 ммоль) и безводный дихлорметан (20 мл), и затем добавляли в два приема ADDP (1.4 г, 5.6 ммоль). После завершения реакции согласно данным ТСХ-мониторинга, для разбавления добавляли н-гексан, реакционную смесь фильтровали, и фильтрат упаривали. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/этилацетат = 80/20, получая интермедиат 19 (0.7 г, 44.9%) в виде светло-желтого масла, которое затвердевало при комнатной температуре.
LC-MS (ESI): m/z [M+H]+559.8687.
Стадия 4: Получение (3R,3aS)-3-(((трет-бутилдиметилсилил)окси)метил)-8- фтор-7-(2-окса-6-азаспиро[3.3]гептан-6-ил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло [3,4-d][1,4]оксазин-1-она (интермедиат 20)
В раствор интермедиата 19 (0.93 г, 1.67 ммоль) в безводном тетрагидрофуране (18 мл) в атмосфере аргона при -78°C по каплям добавляли n-BuLi (1.6 М раствор в н-гексане 1.2 мл, 1.8 ммоль). После добавления полученную смесь перемешивали в течение 1 часа при той же температуре, затем нагревали до комнатной температуры и перемешивали в течение ночи. Добавляли насыщенный раствор хлорида аммония (2 мл) для остановки реакции. Растворитель упаривали и добавляли этилацетат и воду. Органическую фазу отделяли и водную фазу снова экстрагировали этилацетатом. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 75/25), получая интермедиат 20 (0.67 г, 89.2%) в виде светло-желтой твердой пены.
1H-ЯМР (400 МГц, CDCl3) δ: 7.62 (дд, J = 12.8, 0.4 Гц, 1H), 6.03 (д, J = 8.2 Гц, 1H), 4.81 (с, 4H), 4.37 (дд, J = 10.4, 3.2 Гц, 1H), 4.22 (м, 1H), 4.02 (м, 5H), 3.93-3.78 (м, 3H), 0.88 (с, 9H), 0.09 (2s, 6H). LC-MS (ESI): m/z [M+H]+ 451.8929.
Стадия 5: Получение (3R,3aS)-8-фтор-3-(гидроксиметил)-7-(2-окса-6-азаспиро[3.3] гептан-6-ил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 21)
В раствор интермедиата 20 (0.64 г, 1.4 ммоль) в тетрагидрофуране (10 мл) при охлаждении на ледяной бане добавляли фторид тетрабутиламмония (1.7 мл, 1.7 ммоль, 1M в тетрагидрофуране). После перемешивания в течение 0.5 часа, большую часть растворителя упаривали, и при добавлении воды выпадал твердый осадок. Полученный твердый продукт отфильтровывали, и осадок на фильтре промывали водой, сушили, получая интермедиат 21 (0.41 г, 86.6%) в виде не совсем белого твердого вещества.
Стадия 6: Получение ((3R,3aS)-8-фтор-1-оксо-7-(2-окса-6-азаспиро[3.3]гептан-6- ил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил метансульфоната (интермедиат 22)
В раствор интермедиата 21 (0.4 г, 1.2 ммоль) в дихлорметане (10 мл), охлажденный до 0°C на бане с ледяной водой, добавляли N-метилморфолин (0.26 мл, 2.4 ммоль) и затем добавляли метансульфонилхлорид (0.11 мл, 1.4 ммоль). Реакционную смесь перемешивали в течение 5.5 часов при комнатной температуре и упаривали, получая твердое вещество. Полученный остаток разбавляли водой и фильтровали, получая интермедиат 22 (0.49 г, 99.4%) в виде светло-желтого твердого вещества.
LC-MS (ESI): m/z [M+H]+ 415.7708.
Стадия 7: Получение (3S,3aS)-3-(азидометил)-8-фтор-7-(2-окса-6-азаспиро[3.3] гептан-6-ил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 23)
В раствор интермедиата 22 (384 мг, 0.93 ммоль) в ДМФА (10 мл) добавляли азид натрия (120 мг, 1.86 ммоль). Реакционную смесь нагревали при 70°C в течение 3 часов и охлаждали до комнатной температуры. После добавления ледяной воды (10 мл) выпадал твердый осадок, который фильтровали, промывали водой и сушили, получая интермедиат 23 (285 мг, 84.8%) в виде не совсем белого твердого вещества.
LC-MS (ESI): m/z [M+H]+362.7897.
Стадия 8: Получение N-(((3S,3aS)-8-фтор-1-оксо-7-(2-окса-6-азаспиро[3.3]гептан- 6-ил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамида (соединение 24)
В раствор интермедиата 23 (70 мг, 0.19 ммоль) в тетрагидрофуране (5 мл) добавляли Pd/C (10%, 10 мг). Реакционную смесь выдерживали в атмосфере водорода 2 часа. Реакционную смесь, содержащую интермедиат 24, фильтровали в колбу объемом 25 мл. Добавляли пиридин (0.031 мл, 0.38 ммоль). После охлаждения реакционной смеси на ледяной бане, добавляли уксусный ангидрид (0.029 мл, 0.3 ммоль) по каплям, и полученную смесь перемешивали в течение ночи при комнатной температуре. Смесь разбавляли дихлорметаном. Органическую фазу промывали последовательно 0.5н. раствором соляной кислоты и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали, упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 98/2), получая соединение 24 (42 мг, 58.3%) в виде не совсем белого твердого вещества. Т.пл. 105-107°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.63 (д, J = 12.8 Гц, 1H), 6.05 (д, J = 8.2 Гц, 1H), 5.96 (т, J = 5.8 Гц, 1H), 4.82 (с, 4H), 4.50-4.43 (м, 1H), 4.39-4.33 (м, 1H), 4.06 (д, J = 2.0 Гц, 4H), 3.92-3.77 (м, 2H), 3.76-3.61 (м, 2H), 2.04 (с, 3H). HR-MS (ESI): m/z [M+H]+ вычислено для C18H21FN3O5: 378.1460; найдено: 378.1449.
Пример 25
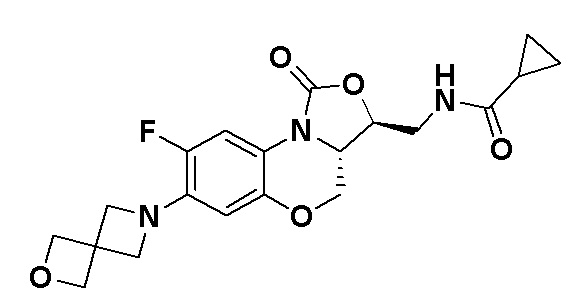
N-(((3S,3aS)-8-фтор-1-оксо-7-(2-окса-6-азаспиро[3.3]гептан-6-ил)-3a,4-дигидро- 1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)циклопропанкарбоксамид (соединение 25)
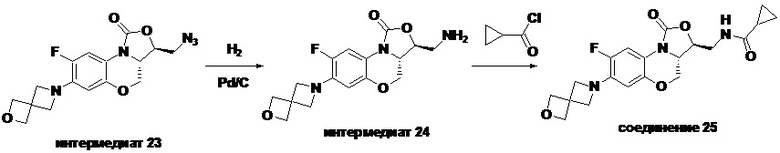
В раствор интермедиата 23 (70 мг, 0.19 ммоль) в тетрагидрофуране (5 мл) добавляли Pd/C (10%, 10 мг). Реакционную смесь выдерживали в атмосфере водорода 3 часа. Реакционную смесь, содержащую интермедиат 24, фильтровали в колбу объемом 25 мл. Добавляли триэтиламин (0.054 мл, 0.38 ммоль). После охлаждения реакционной смеси на ледяной бане, добавляли по каплям циклопропанкарбонилхлорид (0.025 мл, 0.27 ммоль), и полученную смесь перемешивали в течение 1 часа при комнатной температуре. Растворитель упаривали, остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99/1), получая соединение 25 (45 мг, 58.4%) в виде не совсем белого твердого вещества. Т.пл. 181-183°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.65 (д, J = 12.8 Гц, 1H), 6.10 (т, J = 6.0 Гц, 1H), 6.04 (д, J = 8.2 Гц, 1H), 4.82 (с, 4H), 4.45 (дд, J = 10.0, 2.8 Гц, 1H), 4.40-4.34 (м, 1H), 4.05 (д, J = 2.0 Гц, 4H), 3.93-3.72 (м, 3H), 3.72-3.63 (м, 1H), 1.45-1.35 (м, 1H), 1.02-0.95 (м, 2H), 0.84-0.76 (м, 2H). HR-MS (ESI): m/z [M+H]+ вычислено для C20H23FN3O5: 404.1616; найдено: 4040.1608.
Пример 26
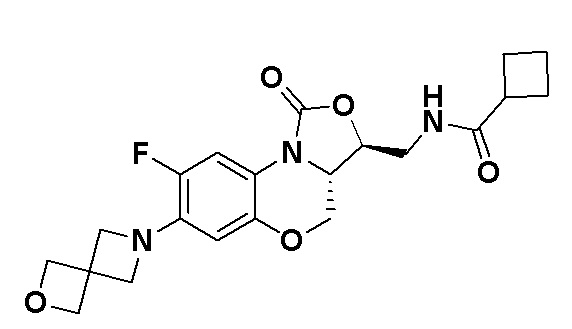
N-(((3S,3aS)-8-фтор-1-оксо-7-(2-окса-6-азаспиро[3.3]гептан-6-ил)-3a,4-дигидро- 1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)циклобутанкарбоксамид (соединение 26)
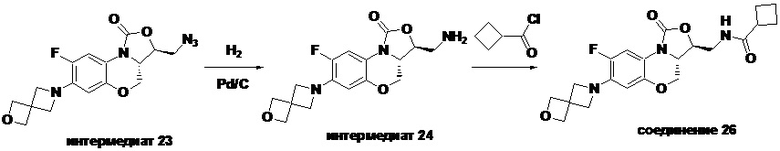
В раствор интермедиата 23 (70 мг, 0.19 ммоль) в тетрагидрофуране (5 мл) добавляли Pd/C (10%, 10 мг). Реакционную смесь выдерживали в атмосфере водорода 3 часа. Реакционную смесь, содержащую интермедиат 24, фильтровали в колбу объемом 25 мл. Добавляли триэтиламин (0.054 мл, 0.38 ммоль). После охлаждения реакционной смеси на ледяной бане, добавляли по каплям циклобутанкарбонилхлорид (0.026 мл, 0.27 ммоль), и полученную смесь перемешивали в течение 1 часа при комнатной температуре. Растворитель упаривали, и остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99/1), получая соединение 26 (44 мг, 55.7%) в виде не совсем белого твердого вещества. Т.пл. 118-120°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.60 (д, J = 12.8 Гц, 1H), 6.03 (д, J = 8.2 Гц, 1H), 5.81 (т, J = 6.2 Гц, 1H), 4.81 (с, 4H), 4.44 (дд, J = 10.0, 2.6 Гц, 1H), 4.38-4.32 (м, 1H), 4.04 (д, J = 2.0 Гц, 4H), 3.91-3.76 (м, 2H), 3.76-3.68 (м, 1H), 3.67-3.59 (м, 1H), 3.08-2.95 (м, 1H), 2.32-2.10 (м, 4H), 2.05-1.78 (м, 2H). HR-MS (ESI): m/z [M+H]+ вычислено для C21H25FN3O5: 418.1773; найдено: 418.1762.
Пример 27
(3S,3aS)-3-((1H-1,2,3-Триазол-1-ил)метил)-8-фтор-7-(2-окса-6-азаспиро[3.3] гептан-6-ил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-он (соединение 27)
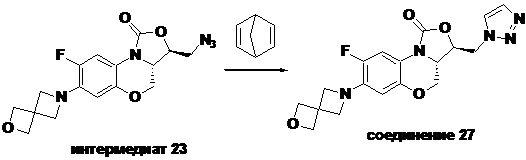
В раствор интермедиата 23 (70 мг, 0.19 ммоль) в 1,4-диоксане (3 мл) добавляли дициклогептадиен (0.19 мл, 1.9 ммоль). Смесь кипятили в течение ночи. Растворитель упаривали, и остаток очищали методом колоночной хроматографии на силикагеле (200-300 меш) (дихлорметан/метанол = 98/2), получая соединение 27 (42 мг, 56.8%) в виде не совсем белого твердого вещества. Т.пл. 219-221°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.79-7.78 (д, J = 0.8 Гц, 1H), 7.78-7.77 (д, J = 0.8 Гц, 1H), 7.53 (д, J = 12.8 Гц, 1H), 6.03 (д, J = 8.2 Гц, 1H), 4.85-4.80 (м, 6H), 4.69-4.63 (м, 1H), 4.42 (дд, J = 10.4, 3.0 Гц, 1H), 4.05 (д, J = 2.0 Гц, 4H), 4.04-3.97 (м, 1H), 3.82 (т, J = 10.2 Гц, 1H). HR-MS (ESI): m/z [M+H]+ вычислено для C18H19FN5O4: 388.1416; найдено: 388.1405.
Пример 28
N-(((3R,3aR)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамид (соединение 28)
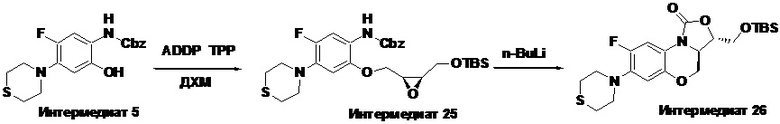
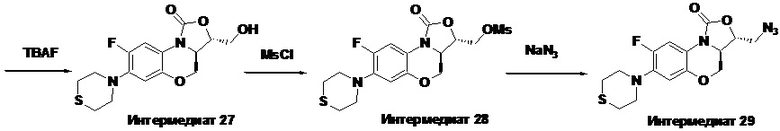
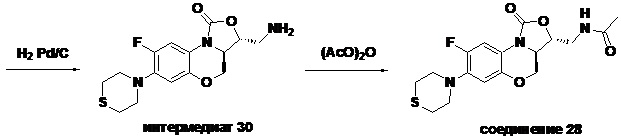
Стадия 1: Получение бензил (2-(((2S,3R)-3-(((трет-бутилдиметилсилил)окси) метил)оксиран-2-ил)метокси)-5-фтор-4-тиоморфолинофенил)карбамата (интермедиат 25)
В двугорлую колбу объемом 50 мл добавляли интермедиат 5 (1 г, 2.76 ммоль), интермедиат 2 (0.78 г, 3.59 ммоль), трифенилфосфин (1.45 г, 5.52 ммоль) и безводный дихлорметан (20 мл), затем в два приема добавляли ADDP (1.39 г, 5.52 ммоль). После завершения реакции согласно данным ТСХ-мониторинга, добавляли н-гексан для разбавления, смесь фильтровали, и фильтрат упаривали. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/этилацетат = 90/10, получая интермедиат 25 (1.14 г, 73.5%) в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.93 (д, J = 12.8 Гц, 1H), 7.44-7.31 (м, 5H), 7.20 (ушир.с, 1H), 6.65 (ушир.с, 1H), 5.20 (с, 2H), 4.32 (дд, J = 11.4, 3.0 Гц, 1H), 4.05 (дд, J = 11.4, 7.0 Гц, 1H), 3.91-3.79 (м, 2H), 3.40-3.20 (м, 6H), 2.83 (ушир.с, 4H), 0.90 (с, 9H), 0.10 (с, 3H), 0.09 (с, 3H). LC-MS (ESI): m/z [M+H]+ 563.8978.
Стадия 2: Получение (3S,3aR)-3-(((трет-бутилдиметилсилил)окси)метил)-8-фтор- 7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 26)
В раствор интермедиата 25 (1.07 г, 1.9 ммоль) в безводном тетрагидрофуране (20 мл) в атмосфере аргона при -78°C по каплям добавляли n-BuLi (1.6 М раствор в н-гексане 1.3 мл, 2.1 ммоль). После добавления полученную смесь перемешивали в течение 1.5 часов при той же температуре, затем нагревали до комнатной температуры и перемешивали в течение ночи. Добавляли насыщенный раствор хлорида аммония (2 мл) для остановки реакции. Растворитель упаривали и добавляли этилацетат и воду. Органическую фазу отделяли, водную фазу снова экстрагировали этилацетатом. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме, получая светло-фиолетовое твердое вещество. Полученный остаток растирали в н-гексане и фильтровали, получая интермедиат 26 (0.79 г, 91.5%) в виде светло-фиолетового твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.73 (д, J = 13.0 Гц, 1H), 6.62 (д, J = 6.6 Гц, 1H), 4.42 (дд, J = 10.4, 3.2 Гц, 1H), 4.28-4.22 (м, 1H), 4.09-4.02 (м, 1H), 3.97-3.79 (м, 3H), 3.28 (м, 4H), 2.82 (т, J = 4.8 Гц, 4H), 0.90 (с, 9H), 0.11 (с, 3H), 0.10 (с, 3H). LC-MS (ESI): m/z [M+H]+ 455.9834.
Стадия 3: Получение (3S,3aR)-8-фтор-3-(гидроксиметил)-7-тиоморфолино-3a,4- дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 27)
В раствор интермедиата 26 (0.75 г, 1.65 ммоль) в ТГФ (10 мл), помещенный в ледяную баню, добавляли фторид тетрабутиламмония (2 мл, 2 ммоль, 1M раствор в тетрагидрофуране). После перемешивания в течение 1 часа упаривали большую часть растворителя, и при добавлении воды выпадал твердый осадок. Твердый продукт отфильтровывали и промывали осадок на фильтре водой, сушили, получая светло-фиолетовое твердое вещество, которое растирали в смеси н-гексана и диэтилового эфира (1:1) и фильтровали, получая интермедиат 27 (0.54 г, 95.7%) в виде не совсем белого твердого вещества.
Стадия 4: Получение ((3S,3aR)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро- 1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метилметансульфоната (интермедиат 28)
В суспензию интермедиата 27 (0.52 г, 1.54 ммоль) в дихлорметане (8 мл) добавляли N-метилморфолин (0.34 мл, 3.08 ммоль) и охлаждали до 0°C на бане с ледяной водой. Добавляли метансульфонилхлорид (0.18 мл, 2.3 ммоль), постепенно растворялся нерастворенный осадок, и затем снова выпадал осадок в ходе добавления. После завершения реакции согласно данным ТСХ-мониторинга, растворитель упаривали досуха, получая твердое вещество, к которому добавляли воду и фильтровали, получая интермедиат 28 (0.63 г, 97.5%) в виде светло-розового твердого вещества.
LC-MS (ESI): m/z [M+H]+ 419.7741.
Стадия 5: Получение (3R,3aR)-3-(азидометил)-8-фтор-7-тиоморфолино-3a,4- дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 29)
В раствор интермедиата 28 (490 мг, 1.17 ммоль) в ДМФА (12 мл) добавляли азид натрия (152 мг, 2.34 ммоль). Реакционную смесь нагревали при 80°C в течение 3 часов и охлаждали до комнатной температуры. Добавляли ледяную воду (10 мл), выпадал твердый осадок, который отфильтровывали, промывали водой и сушили, получая интермедиат 29 (400 мг, 93.7%) в виде светло-розового твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.75 (дд, J = 12.8, 1.8 Гц, 1H), 6.64 (д, J = 7.0 Гц, 1H), 4.49-4.42 (м, 1H), 4.40-4.32 (м, 1H), 4.05-3.96 (м, 1H), 3.88-3.80 (м, 1H), 3.79-3.66 (м, 2H), 3.35-3.21 (м, 4H), 2.83 (ушир.с, 4H).
Стадия 6: Получение N-(((3R,3aR)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро- 1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамида (соединение 28)
В раствор интермедиата 29 (120 мг, 0.33 ммоль) в тетрагидрофуране (6 мл) добавляли Pd/C (10%, 20 мг). Реакционную смесь выдерживали в атмосфере водорода 7 часов. Реакционную смесь, содержащую интермедиат 29, фильтровали в колбу объемом 25 мл. Добавляли пиридин (0.053 мл, 0.66 ммоль). После охлаждения реакционной смеси в ледяной бане, добавляли по каплям уксусный ангидрид (0.047 мл, 0.5 ммоль), и полученную смесь перемешивали в течение 1 часа. Смесь разбавляли дихлорметаном. Органическую фазу промывали 0.5н. водным раствором соляной кислоты и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали, упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 98/2), получая соединение 28 (86 мг, 68.2%) в виде не совсем белого твердого вещества. Т.пл. 210-212°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.73 (д, J = 13.0 Гц, 1H), 6.74 (ушир.с, 1H), 6.27 (ушир.с, 1H), 4.52 (дд, J = 10.2, 2.8 Гц, 1H), 4.45-4.37 (м, 1H), 3.97-3.88 (м, 1H), 3.83 (т, J = 10.2 Гц, 1H), 3.77-3.64 (м, 2H), 3.32 (ушир.с, 4H), 2.86 (ушир.с, 4H), 2.06 (с, 3H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H21FN3O4S: 382.1231; найдено: 382.1224.
Пример 29
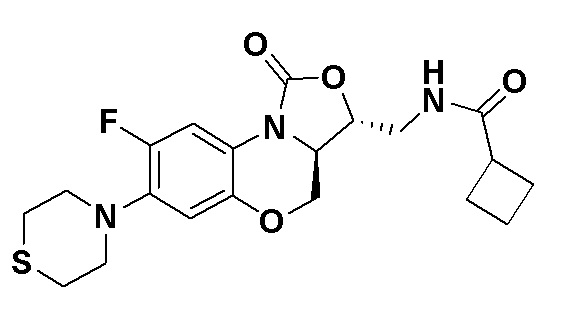
N-(((3R,3aR)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)циклобутан карбоксамид (соединение 29)
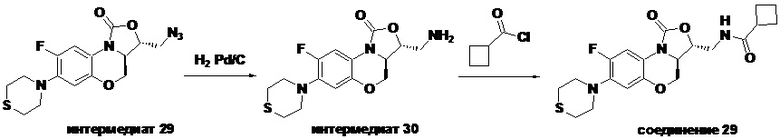
В раствор интермедиата 29 (120 мг, 0.33 ммоль) в тетрагидрофуране (6 мл) добавляли Pd/C (10%, 20 мг). Реакционную смесь выдерживали в атмосфере водорода 7 часов. Реакционную смесь, содержащую интермедиат 30, фильтровали в колбу объемом 25 мл. Добавляли пиридин (0.053 мл, 0.66 ммоль). После охлаждения реакционной смеси в ледяной бане, добавляли по каплям циклобутанкарбонилхлорид (0.042 мл, 0.43 ммоль), и полученную смесь перемешивали в течение 1 часа при комнатной температуре. Смесь разбавляли дихлорметаном, и промывали водой и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали, упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99/1), получая соединение 29 (92 мг, 66.2%) в виде белого твердого вещества. Т.пл. 183-185°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.71 (д, J = 12.8 Гц, 1H), 6.65 (д, J = 7.2 Гц, 1H), 5.86 (т, J = 6.0 Гц, 1H), 4.50 (дд, J = 10.2, 2.8 Гц, 1H), 4.42-4.35 (м, 1H), 3.95-3.87 (м, 1H), 3.82 (т, J = 10.2 Гц, 1H), 3.79-3.62 (м, 2H), 3.37-3.23 (м, 4H), 3.11-2.98 (м, 1H), 2.89-2.77 (м, 4H), 2.34-2.11 (м, 4H), 2.06-1.81 (м, 2H). HR-MS (ESI): m/z [M+H]+ вычислено для C20H25FN3O4S: 422.1544; найдено: 422.1534.
Пример 30
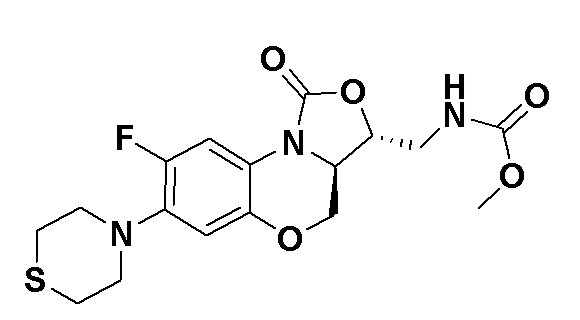
Метил (((3R,3aR)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b] оксазоло[3,4-d][1,4]оксазин-3-ил)метил)карбамат (соединение 30)
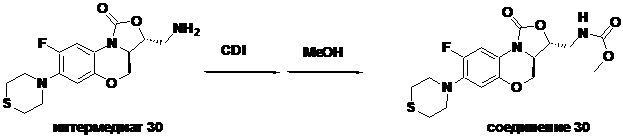
В раствор интермедиата 30 (0.11 г, 0.33 ммоль) в тетрагидрофуране (9 мл) добавляли 1,1'-карбонилдиимидазол (CDI, 0.80 г, 5 ммоль) и перемешивали при комнатной температуре в течение 50 минут. Добавляли безводный метанол (3 мл) и перемешивали в течение ночи при комнатной температуре. Растворитель упаривали и разбавляли смесь дихлорметаном. Полученную смесь промывали насыщенным раствором хлорида аммония и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали досуха. Полученный остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 60/40), получая соединение 30 (60 мг, 45.8%) в виде не совсем белого твердого вещества. Т.пл. 158-160°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.74 (д, J = 12.8 Гц, 1H), 6.66 (д, J = 6.0 Гц, 1H), 5.12 (ушир.с, 1H), 4.49 (дд, J = 10.4, 3.0 Гц, 1H), 4.41-4.34 (м, 1H), 4.00-3.91 (м, 1H), 3.84 (т, J = 10.2 Гц, 1H), 3.69 (с, 3H), 3.68-3.56 (м, 2H), 3.38-3.23 (м, 4H), 2.83 (т, J = 4.8 Гц, 4H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H21FN3O5S: 398.1180; найдено: 398.1176.
Пример 31
(3R,3aR)-8-фтор-3-((изоксазол-3-иламино)метил)-7-тиоморфолино-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-он (соединение 31)
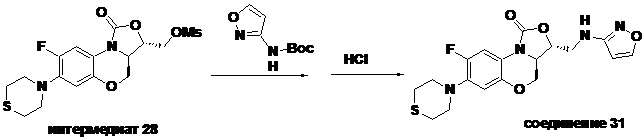
В раствор N-Boc-3-аминоизоксазола (0.048 г, 0.26 ммоль) в безводном ДМФА (2 мл) при охлаждении на ледяной бане добавляли NaH (60%, 12 мг, 0.29 ммоль). После перемешивания в течение 10 минут, добавляли интермедиат 28 (0.13 г, 0.31 ммоль) и оставляли для прохождения реакции при 70°C на 3 часа. После охлаждения добавляли ледяную воду (10 мл). Смесь экстрагировали дихлорметаном два раза. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 85/15), получая масло (80 мг, 60.6%).
В раствор полученного как описано выше масла в этилацетате (2 мл) добавляли метанольный раствор хлороводорода (5н., 4 мл), перемешивали при комнатной температуре в течение 30 минут. Растворитель упаривали, добавляли воду (3 мл), и значение pH доводили до щелочного с помощью насыщенного раствора бикарбоната натрия. Выпадал твердый осадок, его отфильтровывали. Осадок на фильтре промывали водой до нейтрального рН, сушили, получая соединение 31 (53 мг, 82.8%) в виде не совсем белого твердого вещества. Т.пл. 149-150°C.
1H-ЯМР (400 МГц, CDCl3) δ: 8.10-8.06 (м, 1H), 7.76 (дд, J = 12.8, 4.4 Гц, 1H), 6.69 (с, 1H), 5.95-5.80 (м, 1H), 4.67-4.43 (м, 2H), 4.34 (с, 1H), 4.01 (д, J = 3.0 Гц, 1H), 3.94-3.61 (м, 3H), 3.31 (с, 4H), 2.84 (с, 4H). HR-MS (ESI): m/z [M+H]+ вычислено для C18H20FN4O4S: 407.1184; найдено: 407.1175.
Пример 32
N-(((3S,3aS)-7-(4,4-Дифторпиперидин-1-ил)-8-фтор-1-оксо-3a,4-дигидро-1H,3H- бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамид (соединение 32)
Стадия 1. Получение 5-(4,4-дифторпиперидин-1-ил)-4-фтор-2-нитрофенола (интермедиат 31)
В раствор 4,5-дифтор-2-нитрофенола (2.8 г, 16 ммоль) в ацетонитриле (15 мл) добавляли N-метилморфолин (4 мл) и 4,4-дифторпиперидин гидрохлорид (3.5 г, 22 ммоль). Реакционную смесь выдерживали при 80°C в течение 4 часов. После охлаждения добавляли воду (15 мл), реакционную смесь оставляли на ночь, она расслаивалась, после этого смесь фильтровали. Полученный твердый продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир/дихлорметан = 80/20), получая интермедиат 31 (3 г, 68.2%) в виде желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 10.83 (с, 1H), 7.75 (д, J = 13.2 Гц, 1H), 6.47 (д, J = 7.7 Гц, 1H), 3.47 (т, J= 5.6 Гц, 4H), 2.22-2.07 (м, 4H).
Стадия 2. Получение бензил (4-(4,4-дифторпиперидин-1-ил)-5-фтор-2- гидроксифенил)карбамата (интермедиат 32)
В раствор интермедиата 31 (3 г, 10.87 ммоль) в тетрагидрофуране (30 мл) добавляли Никель Ренея (1 г). Реакционную смесь гидрировали при среднем давлении 2 часа. Реакционную смесь фильтровали в колбу, содержащую бикарбонат натрия (1.6 г, 19.2 ммоль) и воду (20 мл), в защитной атмосфере аргона. Добавляли по каплям бензил хлорформиат (1.48 мл, 10.87 ммоль) при охлаждении на ледяной бане и перемешивали в течение 20 минут при той же температуре. Растворитель упаривали и добавляли воду. Полученную смесь экстрагировали этилацетатом два раза. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая красное твердое вещество. Полученный остаток очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/этилацетат = 80/20, получая интермедиат 32 (4.7 г, 77.0%) в виде светло-розового твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.49-7.30 (м, 5H), 7.01 (ушир.с, 1H), 6.87-6.55 (м, 2H), 5.22 (с, 2H), 3.18 (ушир.с, 4H), 2.17 (с, 4H). LC-MS (ESI): m/z [M+H]+: 381.2007.
Стадия 3. Получение бензил (4-(4,4-дифторпиперидин-1-ил)-5-фтор-2-(((2R,3S)-3- ((тритилокси)метил)оксиран-2-ил)метокси)фенил)карбамата (интермедиат 33)
В трехгорлую колбу объемом 100 мл добавляли интермедиат 32 (1.6 г, 4.2 ммоль), интермедиат 3 (1.87 г, 5.4 ммоль), трифенилфосфин (2.2 г, 8.4 ммоль) и безводный дихлорметан (30 мл), затем в три приема добавляли ADDP (2.1 г, 8.4 ммоль). После завершения реакции согласно данным ТСХ-мониторинга, добавляли н-гексан для разбавления, смесь фильтровали, и фильтрат упаривали. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/ дихлорметан /этилацетат = 80/10/10, получая интермедиат 33 (2.2 г, 76.3%) в виде светло-желтого масла.
Стадия 4. Получение (3R,3aS)-7-(4,4-дифторпиперидин-1-ил)-8-фтор-3- ((тритилокси)метил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 34)
В раствор интермедиата 33 (4.5 г, 6.36 ммоль) в безводном тетрагидрофуране (65 мл) в атмосфере аргона при -78°C добавляли н-BuLi (1.6 М раствор в н-гексане, 4.7 мл, 7.63 ммоль) по каплям. После добавления полученную смесь перемешивали в течение 1.5 часов при той же температуре, затем нагревали до комнатной температуры и перемешивали в течение ночи. Насыщенный раствор хлорида аммония (2 мл) добавляли для остановки реакции. Растворитель упаривали и добавляли дихлорметан и воду. Органическую фазу отделяли, и водную фазу экстрагировали дихлорметаном еще раз. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле, элюируя смесью петролейный эфир/дихлорметан/этилацетат = 80/10/10, получая интермедиат 34 (2.97 г, 77.7%) в виде светло-розового твердого вещества.
Стадия 5. Получение (3R,3aS)-7-(4,4-дифторпиперидин-1-ил)-8-фтор-3- (гидроксиметил)-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-она (интермедиат 35)
В раствор интермедиата 34 (2.9 г, 4.83 ммоль) в дихлорметане (50 мл) добавляли трифторуксусную кислоту (5 мл) при охлаждении на ледяной бане, и перемешивали в течение ночи при комнатной температуре. Реакционную смесь доводили до щелочного pH раствором бикарбоната натрия и экстрагировали дихлорметаном три раза. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 98/2), получая интермедиат 35 (1.5 г, 88.2%) в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7.78 (д, J = 13.0 Гц, 1H), 6.75 (д, J = 7.6 Гц, 1H), 4.47 (дд, J = 10.6, 3.2 Гц, 1H), 4.37-4.33 (м, 1H), 4.18-4.11 (м, 1H), 4.03 (дд, J = 12.4, 3.8 Гц, 1H), 3.92-3.83 (м, 2H), 3.28-3.13 (м, 4H), 2.28-2.14 (м, 4H).
Стадия 6. Получение N-(((3S,3aS)-7-(4,4-дифторпиперидин-1-ил)-8-фтор-1-оксо- 3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамида (соединение 32)
Раствор интермедиата 35 (0.4 г, 1.12 ммоль) в дихлорметане (15 мл) охлаждали до 0°C на бане с ледяной водой. Добавляли триэтиламин (0.24 мл, 1.68 ммоль) и затем порциями добавляли п-метилбензолсульфонилхлорид (0.26 г, 1.34 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Смесь промывали последовательно водой и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая интермедиат 36 в виде твердой пены, которую напрямую использовали в следующей стадии без дополнительной очистки.
Полученное как описано выше твердое вещество растворяли в тетрагидрофуране (8 мл) и добавляли гидроксид аммония (6 мл). Реакционную смесь нагревали в герметично закрытой пробирке 5 часов при 100°C и затем охлаждали. Тетрагидрофуран упаривали и добавляли воду. Смесь экстрагировали этилацетатом три раза. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали, получая твердое вещество (330 мг, 82.5% выход за две стадии).
В суспензию описанного выше твердого вещества (112 мг, 0.31 ммоль) в дихлорметане (5 мл) добавляли пиридин (0.050 мл, 0.62 ммоль), затем уксусный ангидрид (0.039 мл, 0.41 ммоль). Смесь перемешивали в течение 1 часа при комнатной температуре и разбавляли дихлорметаном. Смесь промывали водой, 0.5н. водным раствором соляной кислоты и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 98.5/1.5), получая соединение 32 (95 мг, 76.6%) в виде светло-желтого твердого вещества. Т.пл. 230-232°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.74 (д, J = 13.0 Гц, 1H), 6.63 (д, J = 7.8 Гц, 1H), 6.03 (т, J = 6.2 Гц, 1H), 4.51 (дд, J = 10.2, 2.8 Гц, 1H), 4.43-4.36 (м, 1H), 3.95-3.88 (м, 1H), 3.83 (т, J = 10.2 Гц, 1H), 3.78-3.63 (м, 2H), 3.23-3.09 (м, 4H), 2.23-2.10 (м, 4H), 2.05 (с, 3H). HR-MS (ESI): m/z [M+H]+ вычислено для C18H21F3N3O4: 400.1479; найдено: 400.1458.
Пример 33
2,2-Дифтор-N-(((3S,3aS)-8-фтор-1-оксо-7-тиоморфолино-3a,4-дигидро-1H,3H- бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамид (соединение 33)
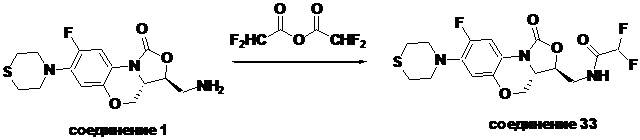
В раствор соединения 1 (174 мг, 0.52 ммоль) в дихлорметане (8 мл) добавляли пиридин (0.084 мл, 1.04 ммоль). После охлаждения на ледяной бане добавляли по каплям дифторуксусный ангидрид (0.070 мл, 0.56 ммоль), и затем смесь перемешивали при комнатной температуре в течение 40 минут. Полученную смесь упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 99/1), получая соединение 33 (181 мг, 83.4%) в виде не совсем белого твердого вещества. Т.пл. 200-202°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.71 (д, J = 12.8 Гц, 1H), 6.92 (ушир.с, 1H), 6.65 (с, 1H), 5.95 (т, J = 54.0 Гц, 1H), 4.51 (дд, J = 10.0, 2.4 Гц, 1H), 4.47-4.40 (м, 1H), 3.95-3.81 (м, 3H), 3.77-3.68 (м, 1H), 3.36-3.23 (м, 4H), 2.89-2.73 (м, 4H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H19F3N3O4S: 418.1043; найдено: 418.1039.
Пример 34
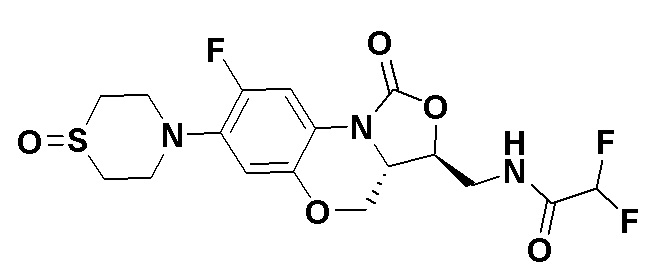
2,2-Дифтор-N-(((3S,3aS)-8-фтор-7-(1-оксидотиоморфолино)-1-оксо-3a,4-дигидро-1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамид (соединение 34)
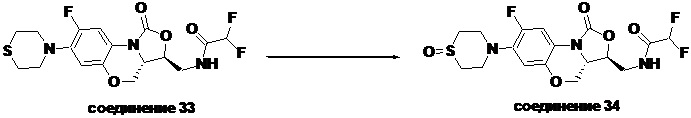
Соединение 33 (0.11 г, 0.26 ммоль) и периодат натрия (0.068 г, 0.32 ммоль) помещали в колбу объемом 25 мл, добавляли метанол (1 мл) и воду (0.7 мл). Полученную смесь перемешивали в течение ночи при комнатной температуре и упаривали. Полученный остаток растворяли в метаноле и отфильтровывали нерастворенный осадок. Фильтрат очищали методом колоночной хроматографии на силикагеле (этилацетат/метанол = 97/3), получая соединение 34 (55 мг, 48.7%) в виде белого твердого вещества. Т.пл.: 190-192°С.
1H-ЯМР (500 МГц, ДМСО-d6) δ: 9.18 (т, J = 5.4 Гц, 1H), 7.60 (д, J = 13.2 Гц, 1H), 6.78 (д, J = 8.2 Гц, 1H), 6.28 (т, J = 53.6 Гц, 1H), 4.61-4.46 (м, 2H), 4.04-3.92 (м, 2H), 3.65 (т, J = 5.2 Гц, 2H), 3.49 (т, J = 12.0 Гц, 2H), 3.24-3.12 (м, 2H), 3.01 (т, J = 12.2 Гц, 2H), 2.89-2.77 (м, 2H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H19F3N3O5S: 434.0992; найдено: 434.0991.
Пример 35
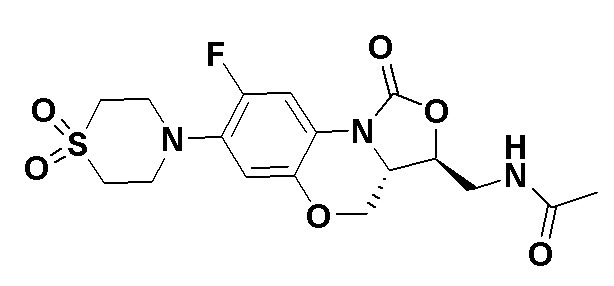
N-(((3S,3aS)-7-(1,1-диоксидотиоморфолино)-8-фтор-1-оксо-3a,4-дигидро-1H,3H- бензо[b]оксазоло[3,4-d][1,4]оксазин-3-ил)метил)ацетамид (соединение 35)
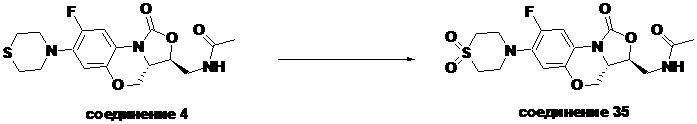
Соединение 4 (0.5 г, 1.3 ммоль) и периодат натрия (0.41 г, 1.95 ммоль) помещали в колбу объемом 25 мл. Добавляли метанол (20 мл) и воду (2 мл). Смесь перемешивали в течение ночи при комнатной температуре и затем упаривали на водяной бане при 50oC. Полученный остаток растворяли в метаноле и отфильтровывали нерастворенный осадок. Фильтрат очищали методом колоночной хроматографии на силикагеле (этилацетат/метанол = 96/3), получая соединение 35 (102 мг, 29.0%) в виде белого твердого вещества. Т.пл. 226-228°C.
1H-ЯМР (400 МГц, CDCl3) δ: 7.77 (д, J = 12.6 Гц, 1H), 6.60 (д, J = 7.8 Гц, 1H), 6.04 (т, J = 6.0 Гц, 1H), 4.52 (дд, J = 10.4, 3.0 Гц, 1H), 4.44-4.37 (м, 1H), 3.96-3.89 (м, 1H), 3.83 (т, J = 10.2 Гц, 1H), 3.79-3.63 (м, 2H), 3.59-3.52 (м, 4H), 3.23-3.16 (м, 4H), 2.05 (с, 3H). HR-MS (ESI): m/z [M+H]+ вычислено для C17H21FN3O6S: 414.1130; найдено: 414.1126.
Пример 36
(3S,3aS)-8-фтор-3-((оксетан-3-иламино)метил)-7-тиоморфолино-3a,4-дигидро- 1H,3H-бензо[b]оксазоло[3,4-d][1,4]оксазин-1-он (соединение 36)
Соединение 1 (0.1 г, 0.29 ммоль), 3-оксетанон (0.031 г, 0.44 ммоль) и триацетоксиборгидрид натрия (184 мг, 0.87 ммоль) добавляли в колбу объемом 25 мл. Добавляли в смесь дихлорметан (4 мл) и каплю уксусной кислоты и перемешивали в течение ночи при комнатной температуре. Добавляли насыщенный раствор бикарбоната натрия (2 мл), смесь интенсивно перемешивали 5 минут и разбавляли дихлорметаном. Органическую фазу отделяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали, и упаривали, получая коричневое масло. Полученный остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол/гидроксид аммония = 100/2/1), получая соединение 36 (31 мг, 25.8%) в виде не совсем белого твердого вещества. Т.пл.: 162-164°C.
1H-ЯМР (400 МГц, CDCl3) δ 7.75 (д, J = 12.8 Гц, 1H), 6.55 (д, J = 7.8 Гц, 1H), 4.84 (дд, J = 7.4 Гц, 6.4 Гц, 2H), 4.54-4.35 (м, 3H), 4.35-4.28 (м, 1H), 4.10-3.97 (м, 2H), 3.85 (т, J = 10.4 Гц, 1H), 3.32-3.21 (м, 4H), 3.04 (дд, J = 13.0, 4.4 Гц, 1H), 2.94 (дд, J = 13.0, 5.4 Гц, 1H), 2.83-2.76 (м, 4H), 2.05 (с, 1H). HR-MS (ESI): m/z [M+H]+ вычислено для C18H23FN3O4S: 396.1388; найдено: 396.1390.
Тестирование биологической активности
Пример 1. Тест на противотуберкулезную активность in vitro
Метод: для определения противотуберкулезной активности in vitro применяли количественный анализ в микропланшете с использованием красителя Alamar Blue (MABA).
Принцип проведения эксперимента: Alamar Blue можно применять как редокс-индикатор при добавлении в культуральную среду. Цвет меняется с синего на красный, показывая поглощение молекул кислорода тестируемыми микроорганизмами. Изменения цвета красителя Alamar Blue можно измерить фотометром с длиной волны испускаемого света 590 нм.
Методика эксперимента: В стерильном 96-луночном планшете (Falcon 3072; Becton Dickinson, Lincoln Park, N.J.), тестируемое соединение растворяли в ДМСО, готовя начальный раствор с концентрацией 5 мг/мл. В лунку с наивысшей концентрацией добавляли 199 мкл среды 7H9 и 1 мкл начального раствора соединения. После перемешивания смесь распределяли в остальные лунки поочередно, используя метод двукратного разбавления. Финальная концентрация соединения составляла 25, 12.5, 6.25, 3.125, 1.56, 0.78, 0.39, 0.2, 0.1, 0.05, 0.025 мкг/мл. Суспензию Mycobacterium tuberculosis H37Rv, культивируемую в течение 2-3 недель, добавляли в среду 7H9, содержащую 0.05% Tween 80 и 10% ADC. Полученную суспензию инкубировали при 37°C в течение 1-2 недель. Когда мутность составляла McFarland 1 (эквивалентно 107 КОЕ/мл), 100 мкл данной суспензии добавляли в каждую лунку, получая финальную концентрацию суспензии 106 КОЕ/мл после разбавления 1:20. На каждом планшете оставляли две лунки контроля роста без добавления противомикробного агента, и 96-луночный микропланшет инкубировали при 37°C. Через 7 дней лунки контроля роста инкубировали со смесью 20 мкл 10×Alamar Blue и 50 мкл 5% Tween80 в течение 24 часов. Во время 24-часового инкубирования при 37oC, если цвет менялся с синего на розовый, то указанное выше количество смеси Alamar Blue и Tween80 добавляли в каждую лунку с исследуемым лекарственным средством. Цвет каждой лунки замеряли после 24 часов инкубирования при 37°C. Значение флуоресценции определяли на считывающем устройстве для микропланшетов при 590 нм, и затем вычисляли значение MIC90.
Таблица 1. In vitro противотуберкулезная активность соединений по настоящему изобретению
(мкг/мл)
(мкг/мл)
По данным в Таблице 1 видно, что соединения по настоящему изобретению обладают прекрасной анти-Mycobacterium tuberculosis активностью in vitro.
Пример 2. Тест цитотоксичности
Метод: MTT-тест
Принцип проведения эксперимента: Активные клетки восстанавливают 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия бромид (торговое название: thiazole blue)/MTT [3-(4,5-диметилтиазо-2-ил)-2,5-дифенилтетразолия бромид] до нерастворимого синего формазана с помощью митохондриальной дегидрогеназы (например, сукцинатдегидрогеназы). После растворения смеси в ДМСО, активность клеток можно измерить по степени превращения, которая коррелирует с числом живых клеток.
Методика эксперимента: 1. Приготовление суспензии клеток. Клетки Vero, выращенные до стадии логарифмического роста, обрабатывали 0.25% трипсином в течение 2-3 минут, затем лизирующий раствор удаляли и добавляли надлежащее количество культуральной среды. После перемешивания проводили подсчет на 20 мкл смеси под микроскопом с гематологическим счетчиком, и готовили суспензию с надлежащей концентрацией клеток для дальнейшего использования. В это время готовили раствор MTT (5 г/л) в PBS (фосфатно-солевой буферный раствор) и фильтровали для удаления бактерий, для дальнейшего использования. 2. Приготовление раствора лекарственного соединения и тест цитотоксичности. Тестируемое соединение растворяли в ДМСО и разбавляли в 50 раз культуральной средой, получая наивысшую концентрацию соединения. Затем полученный раствор разбавляли культуральной средой в соотношении 1:3 в 96-луночном микропланшете. Для каждого соединения готовили 6 концентраций, наивысшая концентрация составляла 64 мкг/мл, и каждую концентрацию распределяли в шесть параллельных лунок в количестве 50 мкг/лунку. Приготовленную суспензию клеток распределяли в 96-луночный микропланшет в количестве 50 мкл/лунку с концентрацией клеток 4 x 105 клеток/мл. В то же время оставляли контрольную лунку без лекарственного вещества и пустую контрольную лунку с культуральной средой. После 48 часов инкубирования добавляли MTT в количестве 10 мкл/лунку и продолжали инкубировать 4 часа. Затем осторожно удаляли культуральную среду, добавляли 100 мкл ДМСО в каждую лунку, и планшет встряхивали до полного растворения гранул формазана. Измеряли оптическую плотность (OD570) методом ELISA при 570 нм. 3. Обработка результатов. Процент ингибирования клеток (%)= [(значение OD570 в контрольной группе - значение OD570 в тестируемой группе) / (значение OD570 в контрольной группе - значение OD570 в пустой группе)]×100%. Строили кривую зависимости доза-ответ с помощью программы Origin 7.0 и вычисляли концентрацию различных соединений, обеспечивающую 50% ингибирование клеток (IC50).
Таблица 2. Цитотоксичность соединений по настоящему изобретению
(мкг/мл)
(мкг/мл)
Из данных Таблицы 2 видно, что цитотоксичность соединений по настоящему изобретению низкая, а это значит, что эти соединения очень безопасны.
Пример 3. Активность ингибирования синтеза белков митохондрий
Эксперимент проводили согласно литературным данным (Antimicrobial Agents and Chemotherapy, 2006, 50 (6), 2042-2049).
Таблица 3. Активность ингибирования синтеза белков митохондрий соединениями по настоящему изобретению
(мкг/мл)
(мкг/мл)
Согласно данным в Таблице 3, ингибирующее действие соединений по настоящему изобретению на синтез белков митохондрий очень слабое, что обеспечивает значительное преимущество над линезолидом и сутезолидом, поскольку вероятность токсичности в отношении костного мозга, вызванной соединениями по настоящему изобретению, очень низкая.
4. In vitro активность соединений против лекарственно-устойчивого туберкулеза
Примененный частный экспериментальный метод относится к методу скрининга из Примера 1 настоящего изобретения на штамме H37Rv.
Таблица 4. In vitro активность соединений по настоящему изобретению против лекарственно-устойчивого туберкулеза
Согласно данным в Таблице 4, соединения по настоящему изобретению обладают высокой противотуберкулезной активностью против клинически изолированных 12525 штаммов (рифампицин- и изониазид-резистентные штаммы) и превосходят соединение VII. Кроме того, соединение 4 и соединение 28 показали еще более высокую противотуберкулезную активность против линезолид-резистентного штамма, чем соединение VII. В особенности соединение 4 очень эффективно против линезолид-резистентного штамма.
Хотя выше были проиллюстрированы и описаны примеры реализации настоящего изобретения, следует понимать, что описанные выше примеры являются иллюстративными и не могут рассматриваться как ограничения настоящего изобретения, и что квалифицированные специалисты в данной области могут изменять, заменять и модифицировать описанные выше примеры в рамках объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| 5-ФТОРПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ФУНГИЦИДОВ | 2009 |
|
RU2522430C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2010 |
|
RU2619463C2 |
| ФЕНИЛАТНОЕ ПРОИЗВОДНОЕ, ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2744975C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ИЛИ ПРЕДОТВРАЩЕНИИ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2741496C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2808166C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2684103C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНИЛАНТРАЦИКЛИНА | 2014 |
|
RU2666356C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕКОТОРЫХ ЛЕЙКОЗОВ | 2019 |
|
RU2804709C2 |
| FGFR И ЕГО ИНГИБИТОР МУТАЦИЙ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2811207C1 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
Изобретение относится к области органической химии, а именно к соединению формулы (I) или к его изомерам, где X1 и X2 каждый представляет собой H; R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3, незамещенный 5-членный гетероарил, причем незамещенный 5-членный гетероарил в R1 содержит три атома N; R3 независимо выбран из атома водорода, замещенного или незамещенного C1-C4 алкила, незамещенного 3-6-членного циклоалкила, незамещенной 4-членной гетероциклической группы, незамещенного 5-6-членного гетероарила; причем указанная незамещенная 4-членная гетероциклическая группа и указанный незамещенный 5-6-членный гетероарил в R3 содержат от одного до трех гетероатомов, выбранных из N или O; заместители в R3 каждый независимо выбран из F, C1-C3 алкила; R2 представляет собой незамещенный  , незамещенный
, незамещенный 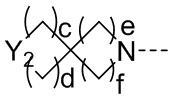 , незамещенный
, незамещенный  ; Y1 представляет собой -S-, -S(=O)-, -S(O2)-, -C(F2)-; Y2 представляет собой -O-; a, b, c, d, e и f каждый равны 1. Также изобретение относится к способу получения соединения формулы (I), фармацевтической композиции на основе соединения формулы (I) и его применению. Технический результат: получены новые гетероциклические соединения, полезные для лечения и/или профилактики микробной инфекции, вызванной Mycobacterium tuberculosis. 4 н. и 9 з.п. ф-лы, 4 табл., 39 пр.
; Y1 представляет собой -S-, -S(=O)-, -S(O2)-, -C(F2)-; Y2 представляет собой -O-; a, b, c, d, e и f каждый равны 1. Также изобретение относится к способу получения соединения формулы (I), фармацевтической композиции на основе соединения формулы (I) и его применению. Технический результат: получены новые гетероциклические соединения, полезные для лечения и/или профилактики микробной инфекции, вызванной Mycobacterium tuberculosis. 4 н. и 9 з.п. ф-лы, 4 табл., 39 пр.
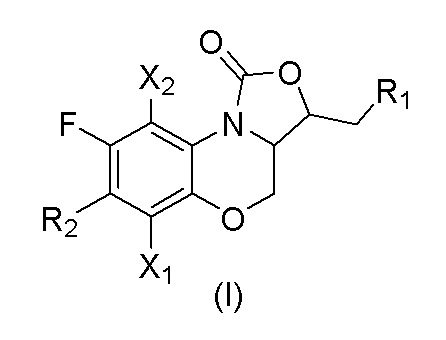
1. Соединение, имеющее формулу (I) или его изомеры
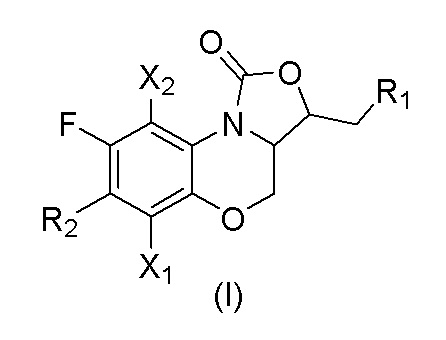
где
X1 и X2 каждый представляет собой H;
R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3, незамещенный 5-членный гетероарил, причем незамещенный 5-членный гетероарил в R1 содержит три атома N;
R3 независимо выбран из атома водорода, замещенного или незамещенного C1-C4 алкила, незамещенного 3-6-членного циклоалкила, незамещенной 4-членной гетероциклической группы, незамещенного 5-6-членного гетероарила;
причем указанная незамещенная 4-членная гетероциклическая группа и указанный незамещенный 5-6-членный гетероарил в R3 содержат от одного до трех гетероатомов, выбранных из N или O;
заместители в R3 каждый независимо выбран из F, C1-C3 алкила;
R2 представляет собой незамещенный 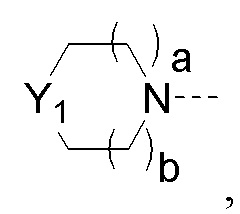 незамещенный
незамещенный 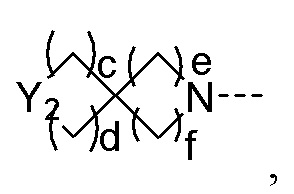 незамещенный
незамещенный 
Y1 представляет собой -S-, -S(=O)-, -S(O2)-, -C(F2)-;
Y2 представляет собой -O-;
a и b каждый равны 1;
c и d каждый равны 1;
e и f каждый равны 1.
2. Соединение или его изомеры по п.1, где указанное соединение имеет формулу (II),
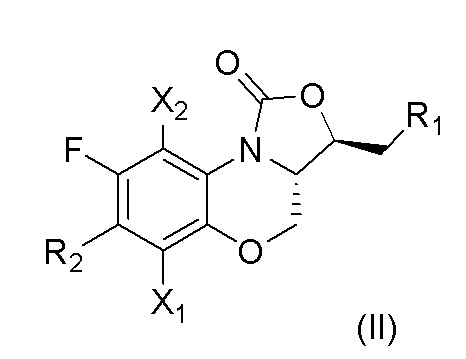
где
X1, X2, R1 и R2 имеют такие же значения, как в п.1.
3. Соединение или его изомеры по п.1, где указанное соединение имеет формулу (III),
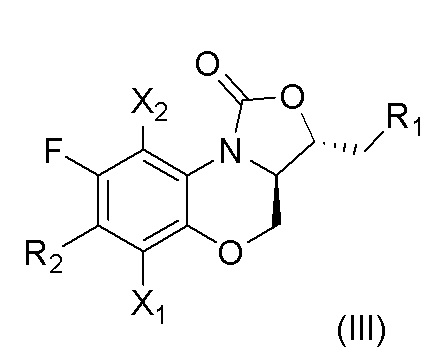
где
X1, X2, R1 и R2 имеют такие же значения, как в п.1.
4. Соединение или его изомеры по п.2, где указанное соединение имеет формулу (II-A),
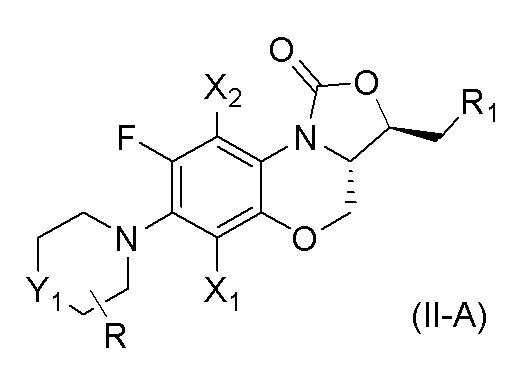
X1 и X2 каждый представляют собой H;
Y1 представляет собой S, S=O, CF2, SO2;
R1 представляет собой -NHR3, -NHCOR3, -NHSO2R3, -NHCOOR3 или незамещенный 5-членный гетероарил; причем незамещенный 5-членный гетероарил в R1 содержит три атома N;
R3 независимо выбран из атома водорода, замещенного или незамещенного C1-C4 алкила, незамещенного 3-6-членного циклоалкила, незамещенной 4-членной гетероциклической группы, незамещенного 5-6-членного гетероарила;
указанная незамещенная 4-членная гетероциклическая группа и указанный незамещенный 5-6-членный гетероарил в R3 содержат от одного до трех гетероатомов, выбранных из N или O;
заместители в R3 независимо выбраны из F, C1-C3 алкила;
R представляет собой один H.
5. Соединение или его изомеры по п.2, где указанное соединение имеет формулу (II-B),
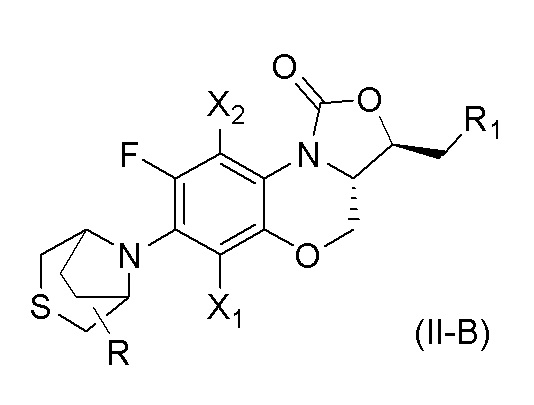
X1 и X2 каждый представляют собой H;
R1 представляет собой -NHR3, -NHCOR3, -NHCOOR3 или незамещенный 5-членный гетероарил;
R3 независимо выбран из атома водорода, незамещенного C1-C4 алкила, незамещенного 3-6-членного циклоалкила, незамещенного 5-6-членного гетероарила;
указанный незамещенный 5-6-членный гетероарил в R3 содержит от одного до тех гетероатомов, выбранных из N или O;
R представляет собой H.
6. Соединение или его изомеры по п.2, где указанное соединение имеет формулу (II-C),
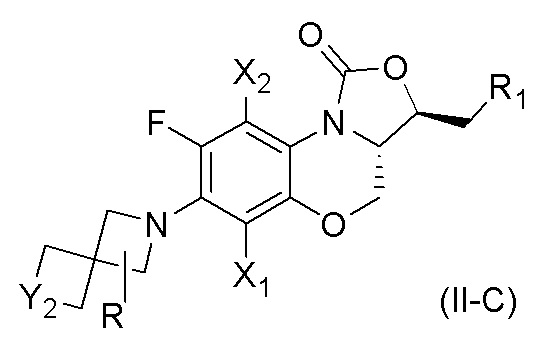
X1 и X2 каждый представляют собой H;
Y2 представляет собой O;
R1 представляет собой -NHCOR3 или незамещенный 5-членный гетероарил, причем незамещенный 5-членный гетероарил в R1 содержит три атома N;
R3 независимо выбран из незамещенного C1-C4 алкила, незамещенного 3-6-членного циклоалкила;
R представляет собой H.
7. Соединение или его изомеры по п.3, где указанное соединение имеет формулу (III-A),
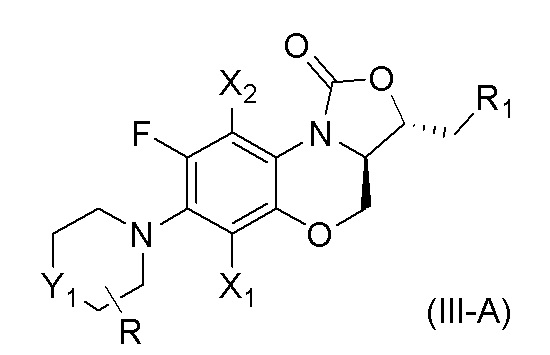
X1 и X2 каждый представляют собой H;
Y1 представляет собой S;
R1 представляет собой -NHR3, -NHCOR3, -NHCOOR3;
R3 независимо выбран из незамещенного C1-C4 алкила, незамещенного 3-6-членного циклоалкила, незамещенного 5-членного гетероарила;
указанный незамещенный 5-членный гетероарил в R3 содержит от одного до трех гетероатомов, выбранных из N или O;
R представляет собой H.
8. Соединение или его изомеры по любому из пп.1-7, где
X1 представляет собой H;
X2 представляет собой H;
R1 представляет собой -NH2, -NHCH3, 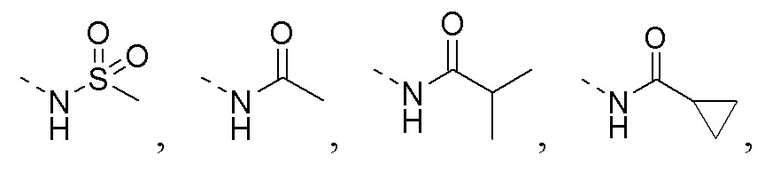
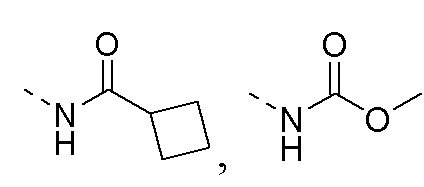 ,
, 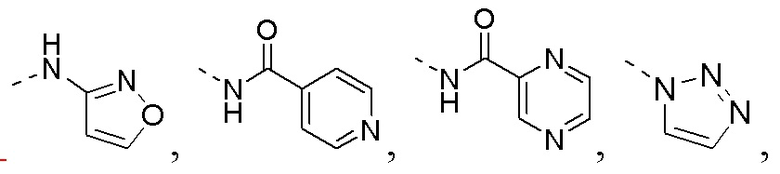
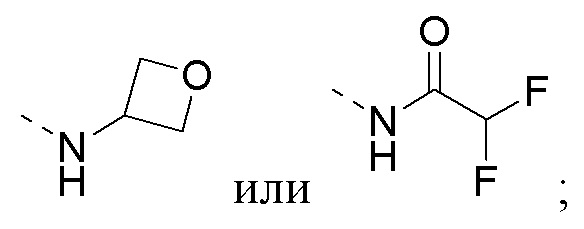
R2 представляет собой 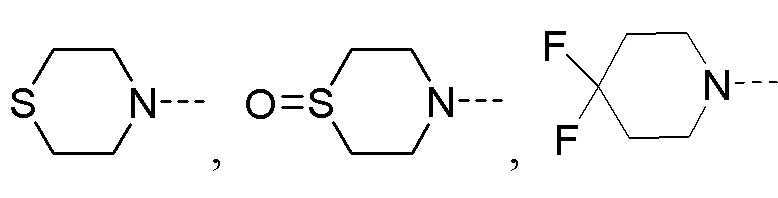 ,
, 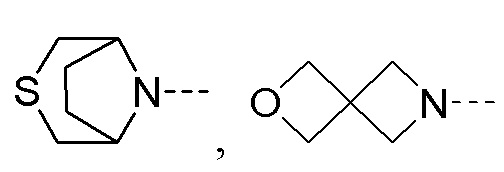 или
или 
9. Соединение или его изомеры по п.1, где соединение, имеющее общую формулу (I), выбрано из следующих соединений:
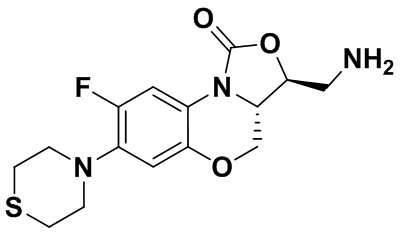
Соединение 1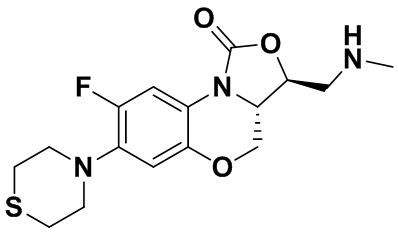
Соединение 2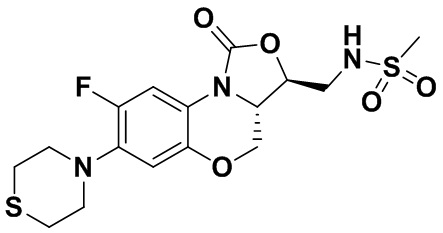
Соединение 3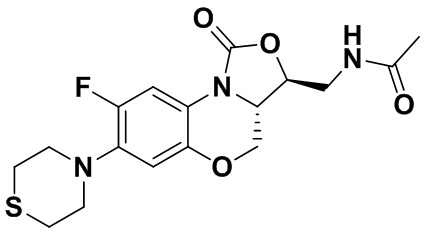
Соединение 4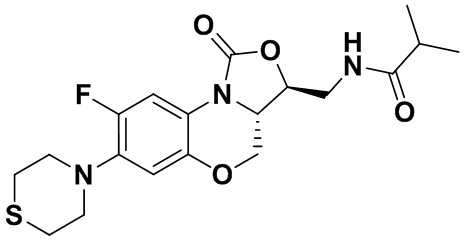
Соединение 5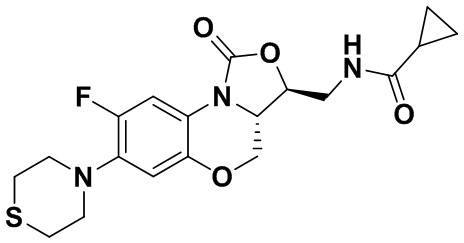
Соединение 6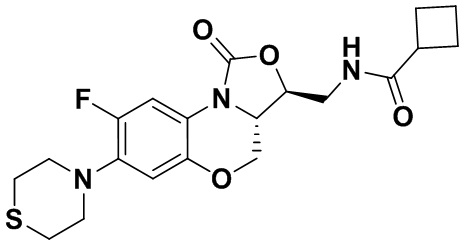
Соединение 7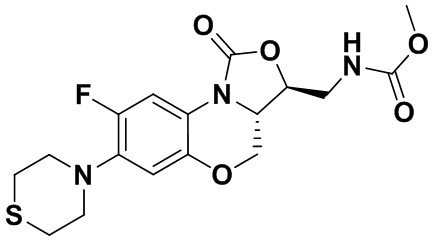
Соединение 8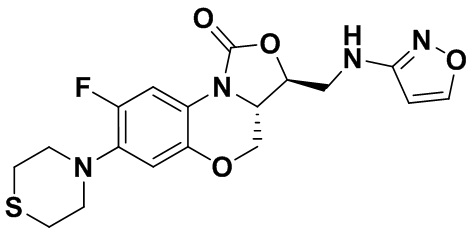
Соединение 9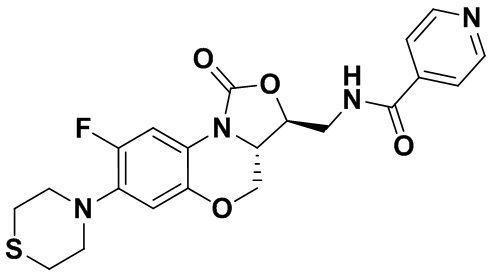
Соединение 10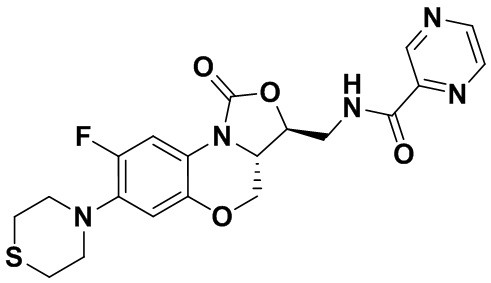 Соединение 11
Соединение 11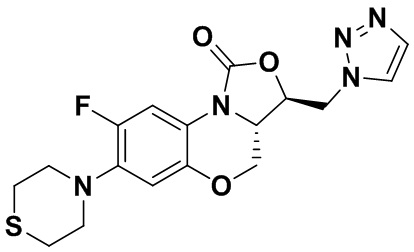
Соединение 12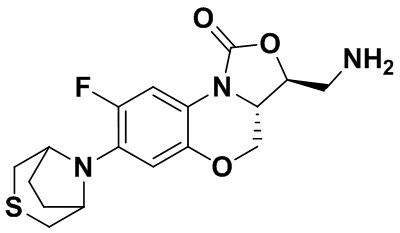
Соединение 13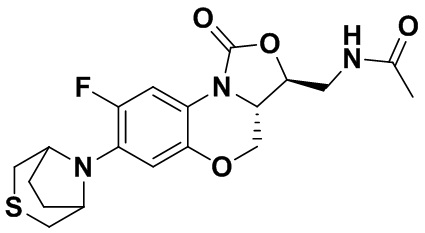
Соединение 14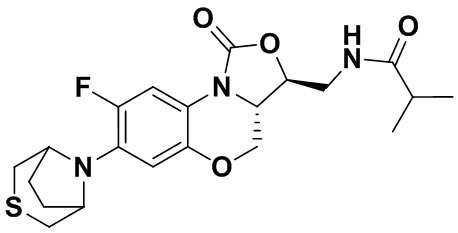 ССоединение 15
ССоединение 15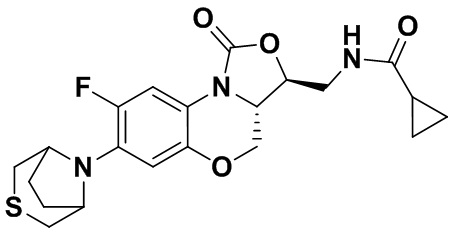
Соединение 16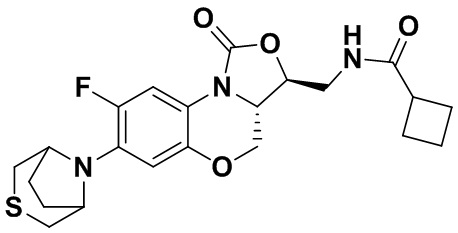
Соединение 17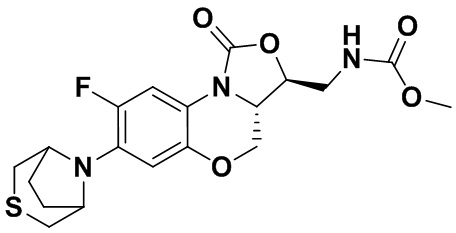
Соединение 18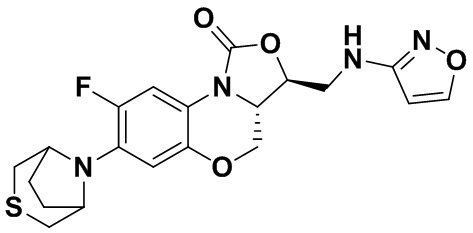
Соединение 19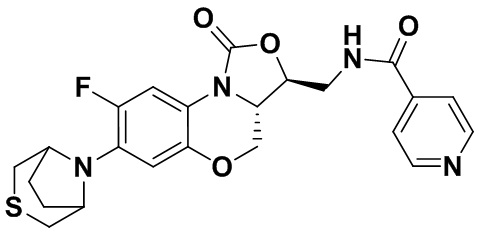
Соединение 20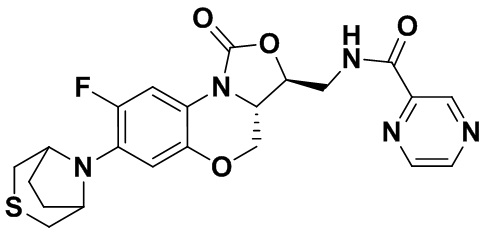
Соединение 21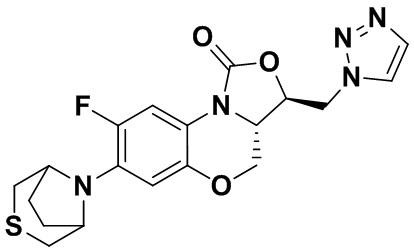
Соединение 22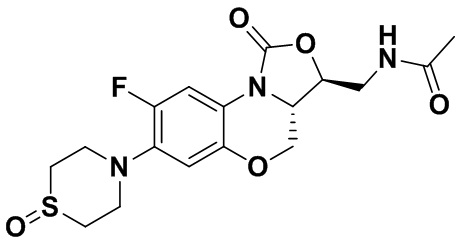
Соединение 23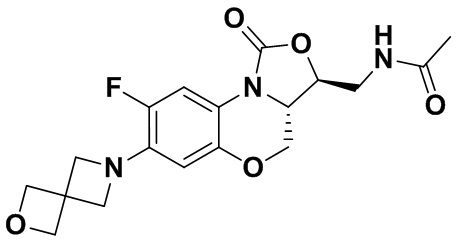
Соединение 24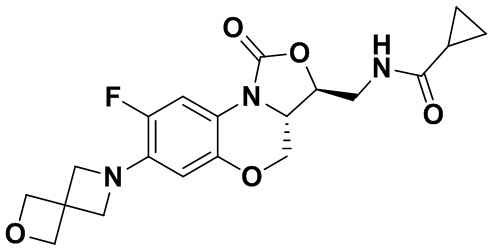
Соединение 25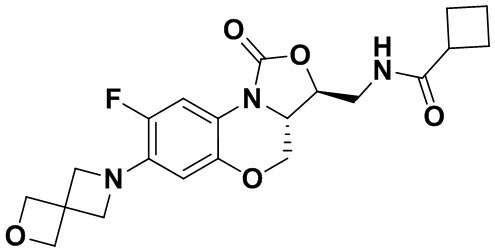
Соединение 26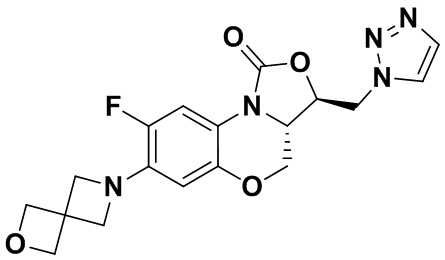
Соединение 27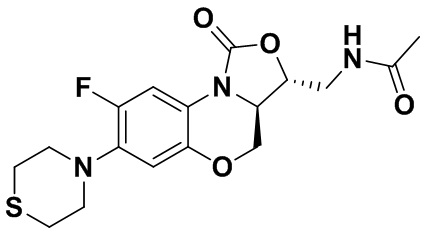
Соединение 28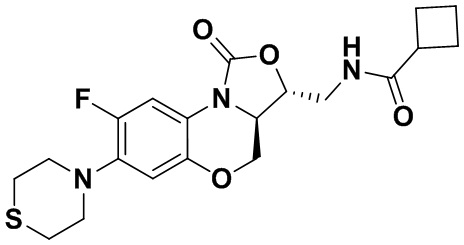
Соединение 29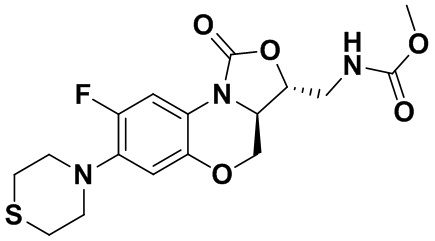
Соединение 30
Соединение 31
Соединение 32
Соединение 33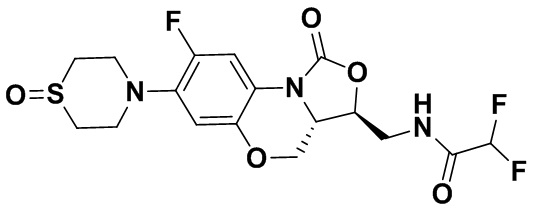
Соединение 34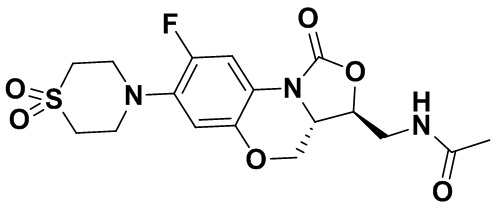
Соединение 35
Соединение 36
10. Способ получения соединений по любому из пп. 1-9, который включает следующие стадии:
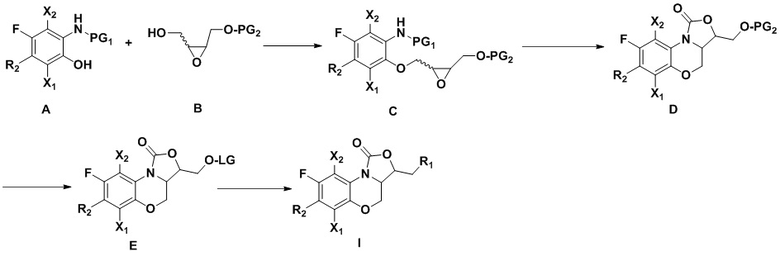
соединение A реагирует с соединением B по реакции Мицунобу с получением соединения C, затем циклизация соединения C в щелочных условиях дает соединение D, затем удаляют защитную группу в соединении D, и превращают соединение D в соединение E, которое содержит уходящую группу, и соединение E подвергают серии превращений функциональной группы с получением соответствующего соединения, имеющего формулу (I);
причем реакцию Мицунобу при получении соединения C проводят в растворителе в присутствии реагента и азодикарбонатных соединений или азодикарбонамида, указанным растворителем является дихлорметан, толуол, тетрагидрофуран или трет-бутилметиловый эфир, указанным реагентом является трифенилфосфин или трибутилфосфин, указанным азодикарбонатом является диэтилазодикарбонат или диизопропилазодикарбонат, указанный азодикарбонамид представляет собой 1,1'- (азодикарбонил) дипиперидин;
циклизацию при получении соединения D проводят в растворителе в присутствии литий-содержащих оснований, указанным растворителем является тетрагидрофуран или трет-бутилметиловый эфир, указанными литий-содержащими основаниями являются трет-бутоксилитий, бутиллитий, LiHMDS, LDA;
в способах удаления защитной группы используют фторид тетрабутиламмония или кислоту, когда –O-PG2 представляет собой силил-эфирную защитную группу, гидрирование или кислоту используют, когда –O-PG2 представляет собой бензил-эфирную защитную группу, указанным растворителем является тетрагидрофуран;
замена B на 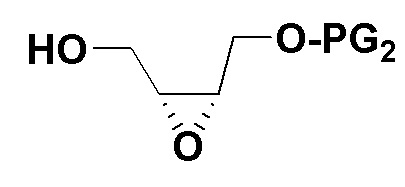 в описанных выше стадиях синтеза дает соответствующее соединение, имеющее формулу (II);
в описанных выше стадиях синтеза дает соответствующее соединение, имеющее формулу (II);
замена B на 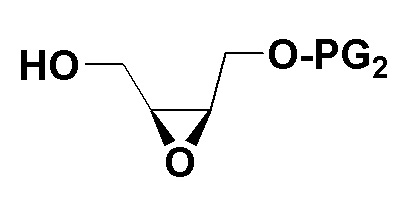 в описанных выше стадиях синтеза дает соответствующее соединение, имеющее формулу (III);
в описанных выше стадиях синтеза дает соответствующее соединение, имеющее формулу (III);
где LG в формуле (Е) выбрана из галогена или псевдогалогена и указанный галоген выбран из F, Cl, Br или I, указанный псевдогалоген выбран из трифторметилсульфонилокси-группы, п-метилфенилсульфонилокси-группы, метилсульфонилокси-группы и п-нитрофенилсульфонилокси-группы; PG1 представляет собой защитную группу для амино-группы и выбрана из трет-бутоксикарбонил-группы, бензилоксикарбонил-группы и этоксикарбонил-группы; PG2 представляет собой защитную группу для гидроксила и выбрана из силилового эфира и бензилового эфира; X1, X2, R1 и R2 имеют те же значения, которые указаны в любом из пп. 1-8.
11. Способ по п. 10, в котором силиловый эфир выбран из трет-бутилдиметилсилилового эфира или трет-бутилдифенилсилилового эфира и бензиловый эфир выбран из п-бромбензилового эфира, п-метоксибензилового эфира или тритилового эфира.
12. Фармацевтическая композиция для лечения и/или профилактики микробной инфекции, вызванной Mycobacterium tuberculosis, содержащая терапевтически и/или профилактически эффективное количество соединения или его изомеров по любому из пп. 1-9, и один или больше фармацевтически приемлемый адъювант.
13. Применение соединения или его изомеров по любому из пп. 1-9 или фармацевтической композиции по п. 12 для изготовления лекарственного средства для лечения и/или профилактики микробных инфекций, вызванных Mycobacterium tuberculosis.
| ЛЕКАРСТВЕННЫЕ СРЕДСТВА, СВЯЗАННЫЕ С ДИПЕПТИДАМИ | 2009 |
|
RU2578591C2 |
| CN 105593232 A, 18.05.2016 | |||
| Xin, Q | |||
| и др.: "Design, Synthesis, and Structure-Activity Relationship Studies of Highly Potent Novel Benzoxazinyl-Oxazolidinone Antibacterial Agents", Journal of Medicinal Chemistry, 2011, 54(21), с.7493-7502 (doi:10.1021/jm200614t ) | |||
| НИТРОИМИДАЗООКСАЗИНОВЫЕ И НИТРОИМИДАЗООКСАЗОЛЬНЫЕ АНАЛОГИ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2540860C2 |

