Область изобретения
Настоящее изобретение относится к новому способу получения соединения феноксидиаминопиримидина или его фармацевтически приемлемой соли, которые потенциально являются пригодными для потенциального лечения заболеваний, ассоциированных пуринергическими рецепторами P2X, например, респираторных и связанных с болью заболеваний, состояний и нарушений.
УРОВЕНЬ ТЕХНИКИ, К КОТОРОМУ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Субъединицы рецептора P2X находятся на афферентных волокнах в уротелии мочевого пузыря грызунов и человека. Существуют данные, указывающие на то, что АТФ может высвобождаться из эпителиальных/эндотелиальных клеток мочевого пузыря или других полых органов в результате растяжения (Burnstock (1999) J. Anatomy 194:335-342; и Ferguson et al. (1997) J. Physiol. 505:503-511). АТФ, высвободившийся таким образом, может играть роль в передаче информации в чувствительные нейроны, находящиеся в субэпителиальных компонентах, например, субуротелиальной собственной пластинке (Namasivayam, et al. (1999) BJU Intl. 84:854-860). Рецепторы P2X исследованы в ряде нейронов, включая чувствительные, симпатические, парасимпатические и центральные нейроны (Zhong, et al. (1998) Br. J. Pharmacol. 125:771-781). Эти исследования указывают на то, что пуринергические рецепторы могут играть роль в афферентной нейротрансмиссии из мочевого пузыря, и что модуляторы рецепторов P2X потенциально являются пригодными для лечения нарушений мочевого пузыря и других мочеполовых заболеваний или состояний.
Недавние данные также указывают на роль эндогенного АТФ и пуринергических рецепторов в ноцицептивных ответах у мышей (Tsuda, et al. (1999) Br. J. Pharmacol. 128:1497-1504). Было показано, что АТФ-индуцируемая активация рецепторов P2X на нервных окончаниях ганглия заднего корешка стимулирует высвобождение глутамата - ключевого нейротрансмиттера, вовлеченного в ноцицептивную передачу сигнала (Gu and MacDermott, Nature 389:749-753 (1997)). Рецепторы P2X3 идентифицированы на ноцицептивных нейронах в пульпе зуба (Cook et al., Nature 387:505-508 (1997)). Таким образом, АТФ, высвободившийся из поврежденных клеток, может вызывать боль посредством активации P2X3 и/или P2X2/3, включая рецепторы на ноцицептивных чувствительных нервных окончаниях. Это согласуется с индукцией боли посредством внутрикожного введения АТФ в модели основания волдыря (Bleehen, Br J Pharmacol 62:573-577 (1978)). В моделях на животных было показано, что антагонисты P2X являются болеутоляющими (Driessen and Starke, Naunyn Schmiedebergs Arch Pharmacol 350:618-625 (1994)). Эти данные указывают на то, что P2X2 и P2X3 вовлечены в ноцицепцию, и что модуляторы рецепторов P2X потенциально пригодны в качестве обезболивающих средств.
Другие исследователи показали, что рецепторы P2X3 экспрессируются в толстом кишечнике человека и экспрессируются на более высоких уровнях в воспаленной толстой кишке, чем в нормальной толстой кишке (Y. Yiangou et al, Neuroeastroenterol Mot (2001) 13:365-69). Другие исследователи предположили, что рецептор P2X3 вовлечен в детекцию растяжения или внутрипросветного давления в кишечнике и инициацию рефлекторных сокращений (X. Bian et al., J Physiol (2003) 551.1:309-22), и связали это с колитом (G. Wynn et al., Am J Physiol Gastrointest Liver Physiol (2004) 287:G647-57); Inge Brouns et al. (Am J Respir Cell Mol Biol (2000) 23:52-61) обнаружили, что рецепторы P2X3 экспрессируются в нейроэпителиальных тельцах легких (NEB), что подразумевает вовлечение рецептора в трансмиссию боли в легком. Исследователи также предположили, что рецепторы P2X2 и P2X3 вовлечены в детекцию pO2 в NEB легких (W. Rong et al., J Neurosci (2003) 23(36):11315-21).
Таким образом, существует потребность в усовершенствованных способах получения соединений, которые являются эффективными модуляторами рецепторов P2X, включая рецепторы P2X3 и P2X2/3.
В патентах США номер 7858632 и 7741484 описана методология и способы синтеза, которые можно использовать для получения производных феноксидиаминопиримидина. Однако известные в настоящее время способы имеют различные недостатки, включающие сложную химию и/или низкий выход. Остается потребность в усовершенствованных способах синтеза для получения производных феноксидиаминопиримидина с упрощенной химией и/или более высоким выходом.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
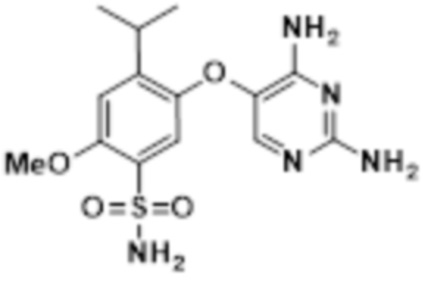
В настоящем описании описан новый способ получения соединения A, соединения феноксидиаминопиримидина следующей формулы, или его фармацевтически приемлемой соли:
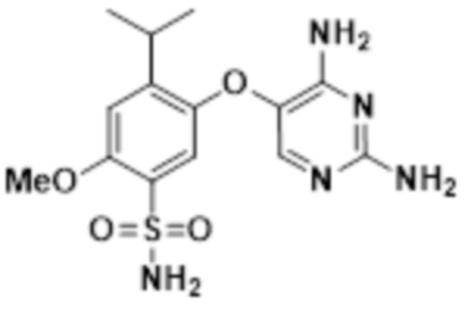 Соединение A.
Соединение A.
Также в настоящем описании описаны различные соли и сольваты соединения A.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
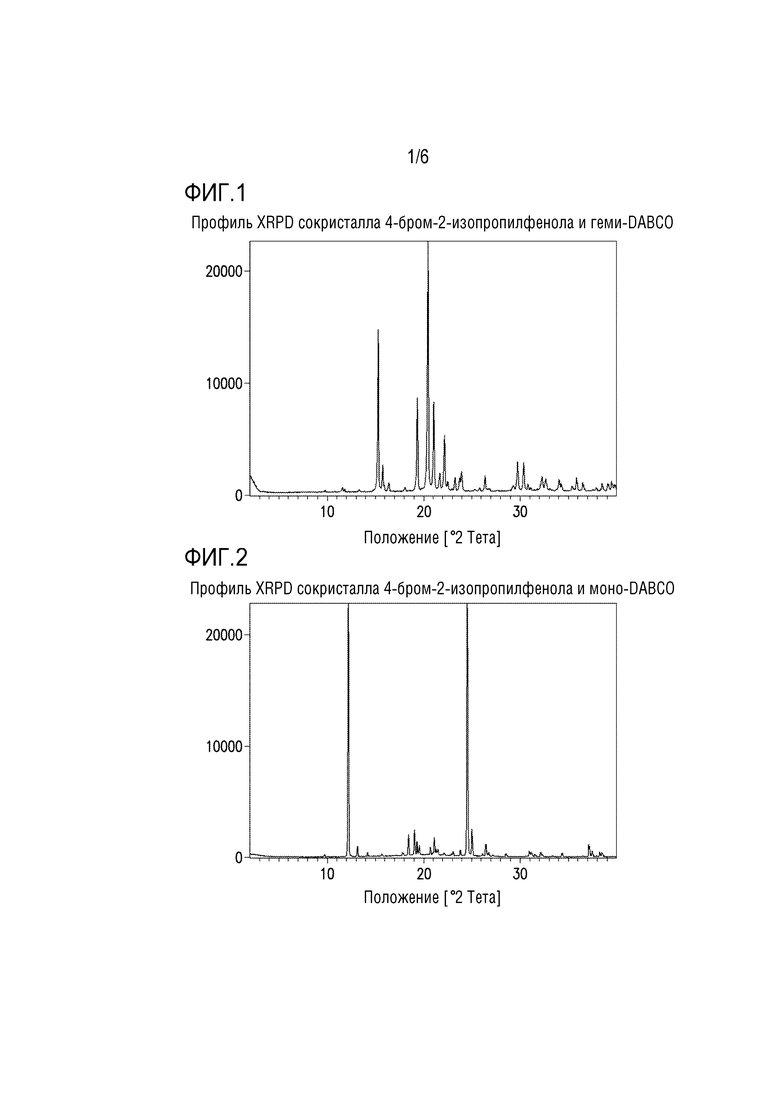
Фигура 1. Профиль XRPD сокристалла 4-бром-2-изопропилфенола и геми-DABCO.
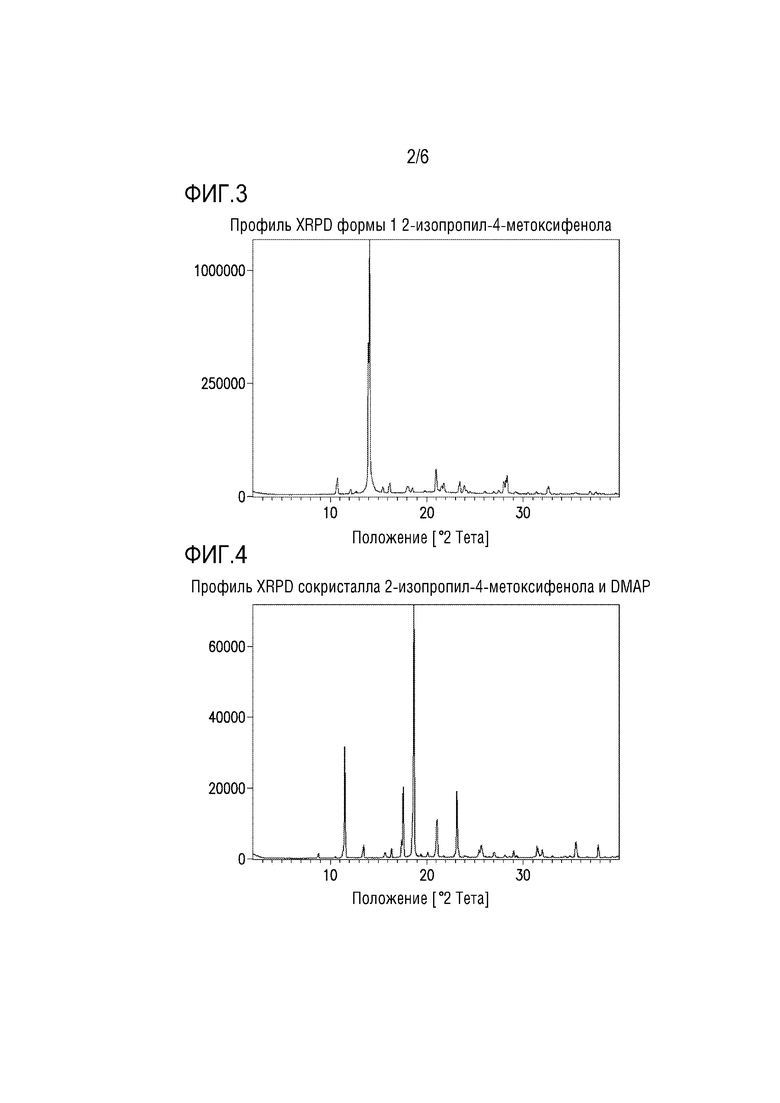
Фигура 2. Профиль XRPD сокристалла 4-бром-2-изопропилфенола и моно-DABCO.
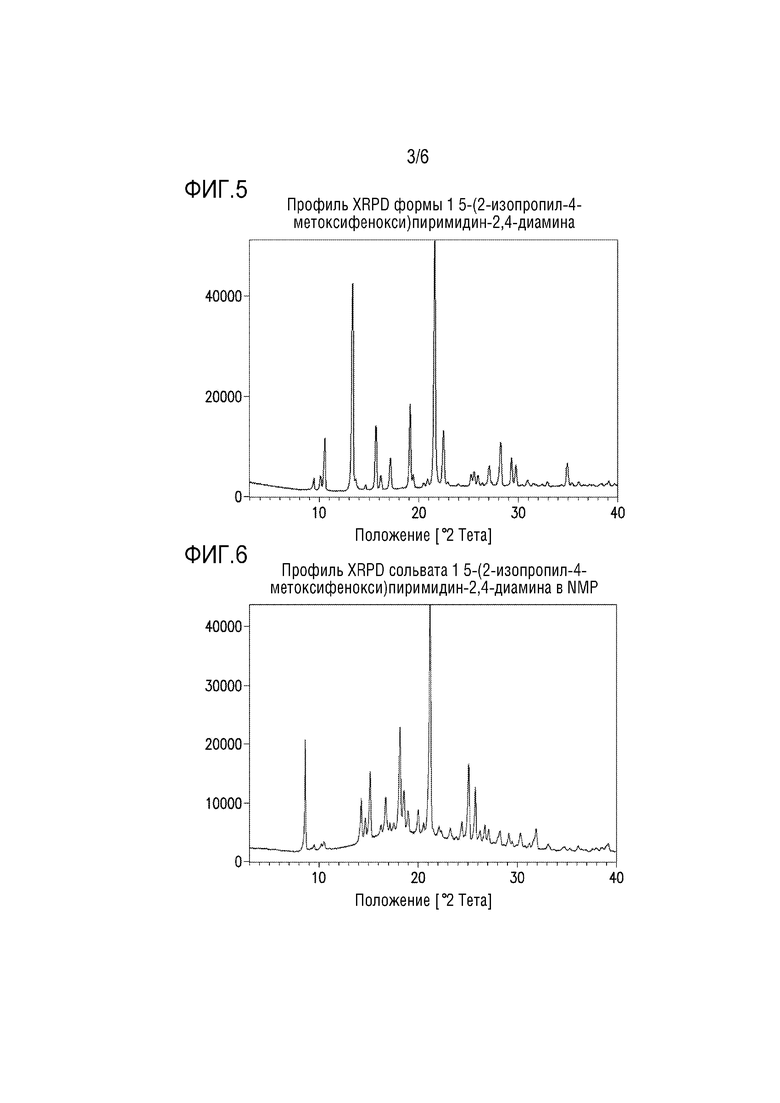
Фигура 3. Профиль XRPD формы 1 2-изопропил-4-метоксифенола.
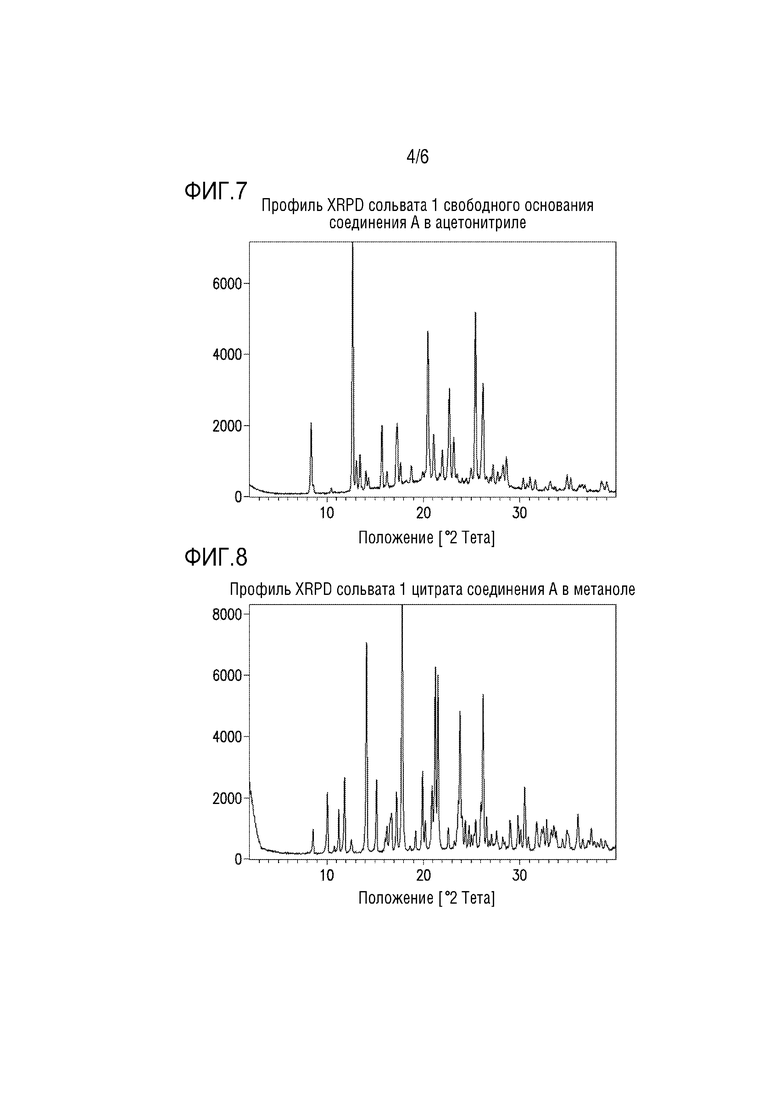
Фигура 4. Профиль XRPD сокристалла 2-изопропил-4-метоксифенола и DMAP.
Фигура 5. Профиль XRPD формы 1 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина.
Фигура 6. Профиль XRPD сольвата 1 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина в NMP.
Фигура 7. Профиль XRPD сольвата 1 свободного основания соединения A в ацетонитриле.
Фигура 8. Профиль XRPD сольвата 1 цитрата соединения A в метаноле.
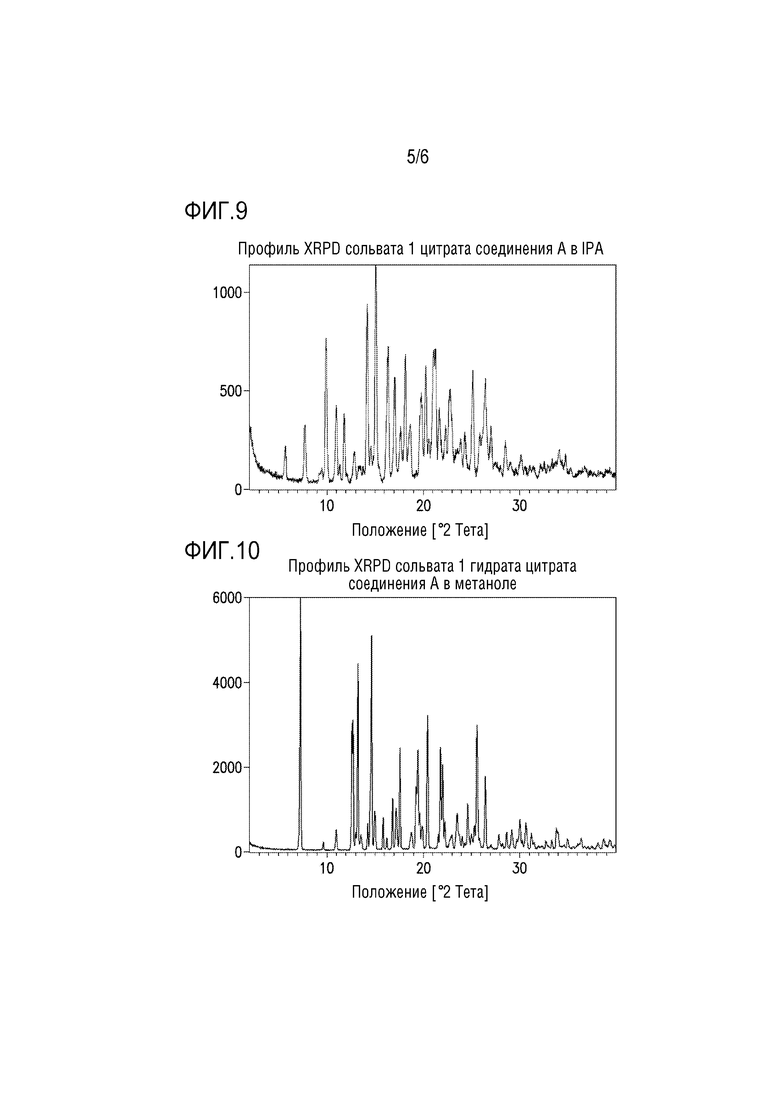
Фигура 9. Профиль XRPD сольвата 1 цитрата соединения A в IPA.
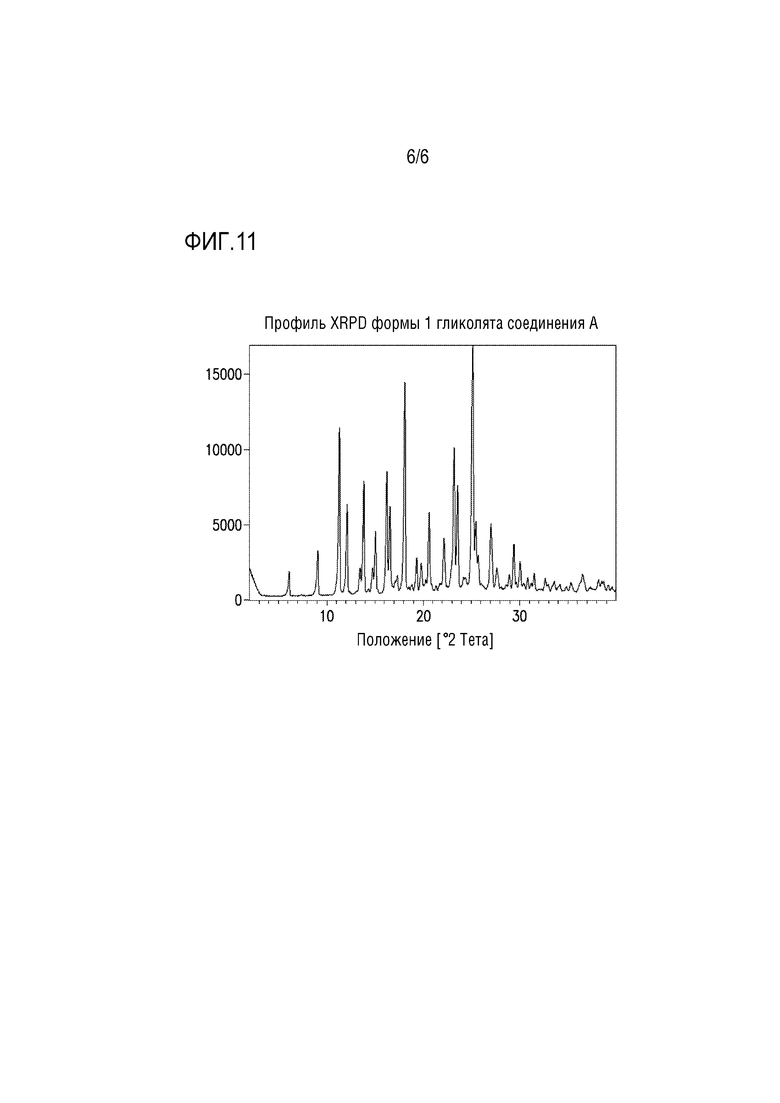
Фигура 10. Профиль XRPD сольвата 1 гидрата цитрата соединения A в метаноле.
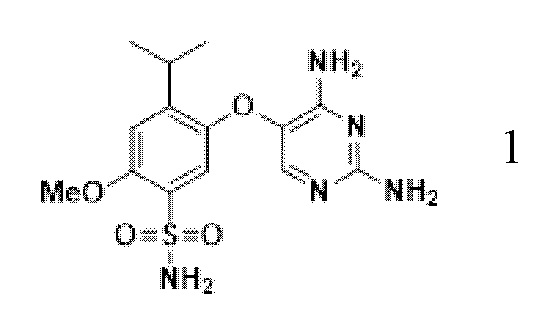
Фигура 11. Профиль XRPD формы 1 гликолята соединения A.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Способ получения свободного основания соединения A или его фармацевтически приемлемой соли с упрощенной химией и более высоким выходом описан в настоящем описании.
В одном варианте осуществления цитрат соединения A получают способом, включающим синтез и выделение сокристалла 4-бром-2-изопропилфенола и DABCO, синтез 2-изопропил-4-метоксифенола и 2-(2-изопропил-4-метоксифенокси)ацетонитрила, синтез и выделение 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина, синтез свободного основания соединения A, 5-((2,4-диаминопиримидин-5-ил)окси)-4-изопропил-2-метоксибензолсульфонамида, и конечное конвертирование свободного основания соединения A в соответствующий цитрат, как показано на схеме 1.
Схема 1
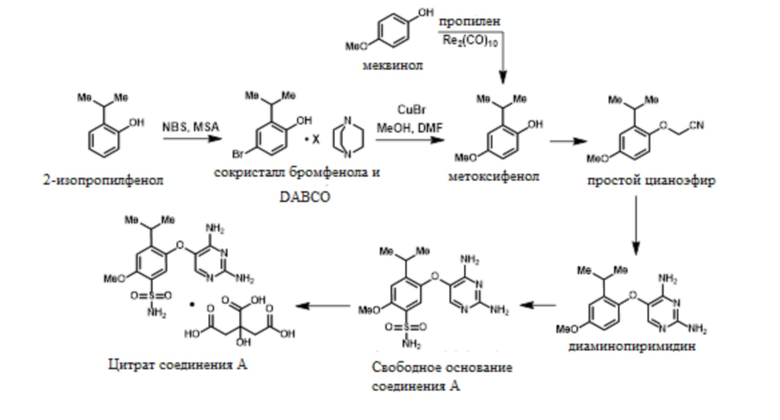
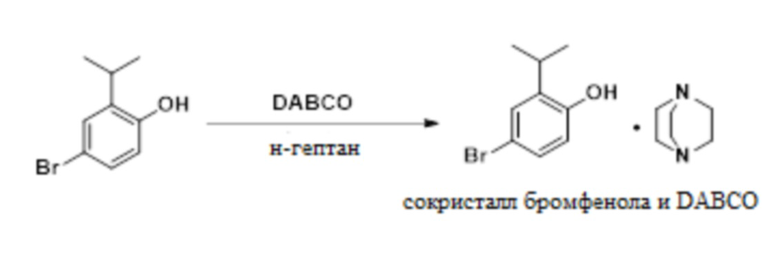
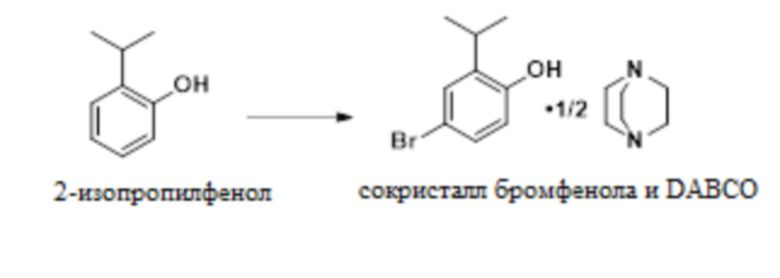
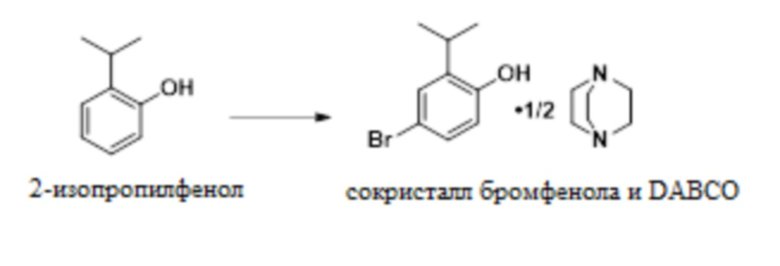
В одном варианте осуществления 2-изопропилфенол обрабатывают NBS в присутствии MSA с образованием 4-бром-2-изопропилфенола. Это соединение обрабатывают DABCO с получением твердого сокристалла 4-бром-2-изопропилфенола с DABCO.
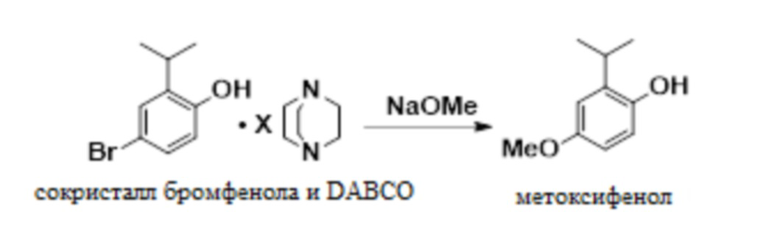
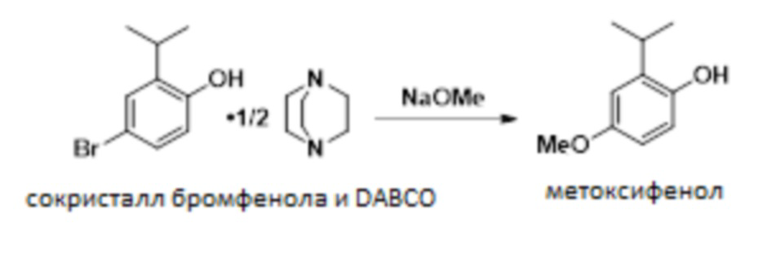
В одном варианте осуществления сокристалл 4-бром-2-изопропилфенола и DABCO конвертируют в 2-изопропил-4-метоксифенол в присутствии бромида меди(I), DMF и метоксида натрия. В одном варианте осуществления как DMF, так и DABCO требуются в этой реакции для предотвращения димеризации продукта.
В альтернативном варианте осуществления 2-изопропил-4-метоксифенол синтезируют из меквинола в присутствии дирения декарбонила и пропилена при высоких температурах и давлениях.
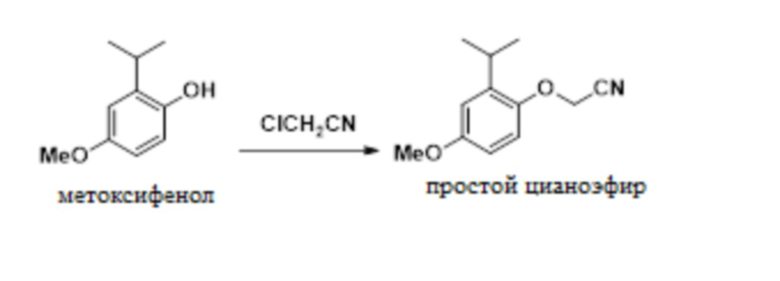
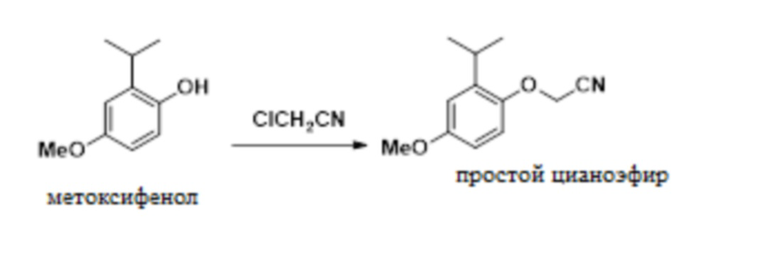
В одном варианте осуществления 2-изопропил-4-метоксифенол обрабатывают алкилирующим средством с получением желаемого 2-(2-изопропил-4-метоксифенокси)ацетонитрила. В одном варианте осуществления эту реакцию проводят с карбонатом калия в ацетонитриле. В другом варианте осуществления эту реакцию проводят с гидроксидом натрия в смеси толуола и NMP. Эти условия реакции позволяют полное конвертирование фенола в 2-(2-изопропил-4-метоксифенокси)ацетонитрил, в то время как предшествующие способы были не способны обеспечивать полное конвертирование фенола в 2-(2-изопропил-4-метоксифенокси)ацетонитрил.
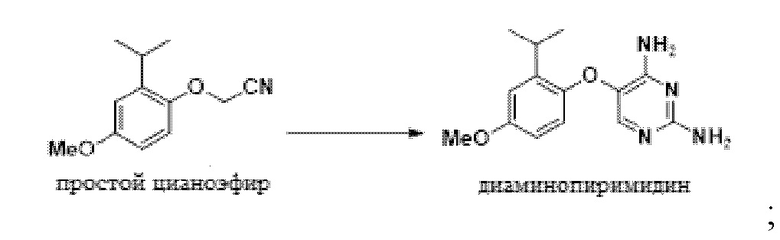
В одном варианте осуществления 2-(2-изопропил-4-метоксифенокси)ацетонитрил обрабатывают этилформиатом в NMP, и эту смесь добавляют к раствору трет-бутоксида калия в NMP. Этот порядок добавления позволяет образование желаемой связи; в то время как добавление реагентов в альтернативном порядке может приводить к димеризации исходного материала в виде цианометилового эфира и образованию монооксида углерода. Полученный энолят обрабатывают гуанидином с получением 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина.
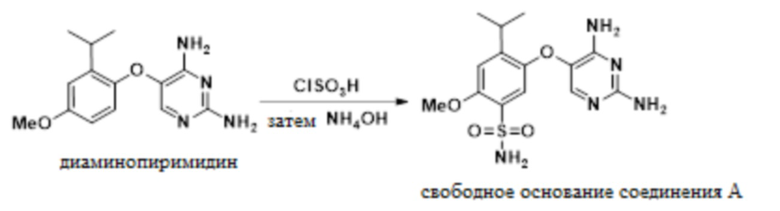
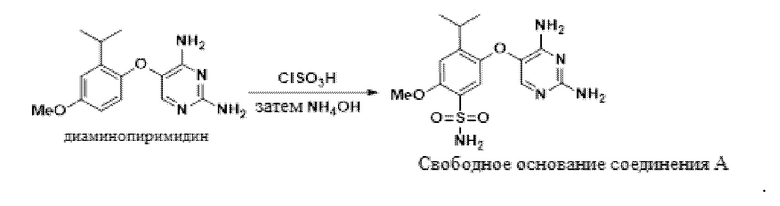
В одном варианте осуществления 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин обрабатывают хлорсульфоновой кислотой в присутствии ацетонитрила с получением сульфонилхлоридного промежуточного соединения. В ацетонитриле эта реакция является однородной, и ее можно прямо гасить водным гидроксидом аммония с получением свободного основания соединения A, 5-((2,4-диаминопиримидин-5-ил)окси)-4-изопропил-2-метоксибензолсульфонамида.
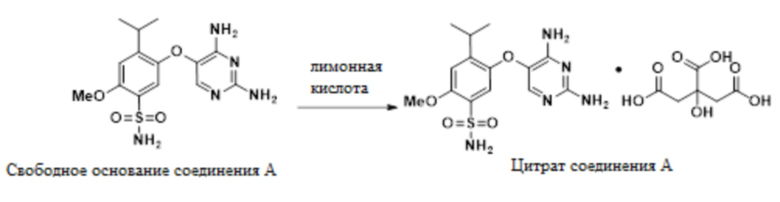
В одном варианте осуществления свободное основание соединения A, 5-((2,4-диаминопиримидин-5-ил)окси)-4-изопропил-2-метоксибензолсульфонамид, обрабатывают гликолевой кислотой в метаноле с образованием гликолята. Добавление лимонной кислоты конвертирует гликолят в цитрат в смеси метанола и изопропанола с образованием цитрата соединения A. Этот новый подход солевого обмена для выделения позволяет получение желаемой формы и контроль примесей.
Методика согласно схеме 1 обеспечивает несколько важных преимуществ. Синтез промежуточного соединения 2-изопропил-5-метоксифенола проводят за одну или две стадии вместо последовательности множества стадий, требуемой для предшествующих способов синтеза. Желаемое алкилирование проводят безопасным контролируемым способом без предельных экзотерм. 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин синтезируют непосредственно из 2-(2-изопропил-4-метоксифенокси)ацетонитрила без каких-либо промежуточных соединений или выделений, и без использования анилинов, которые могут усложнять очистку желаемого соединения. Реакцию хлорсульфонилирования проводят в ацетонитриле для того, чтобы прямо получить сульфонилхлорид без необходимости в оксихлориде фосфора. Свободное основание 5-((2,4-диаминопиримидин-5-ил)окси)-4-изопропил-2-метоксибензолсульфонамид выделяют из смеси воды и ацетонитрила, что позволяет легкую кристаллизацию нескольких форм желаемого продукта. Этот способ обеспечивает упрощенную химию, более высокий выход и использует стандартное оборудование для производства, тем самым обеспечивая производительный, экологичный и портативный процесс производства.
В одном варианте осуществления данный способ включает стадию конвертирования 2-изопропилфенола в сокристалл 4-бром-2-изопропилфенола и DABCO:
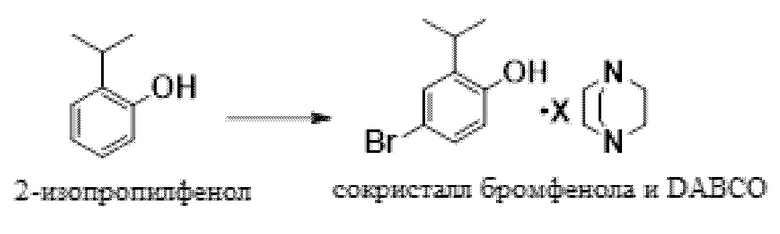
В одном варианте осуществления указанное выше конвертирование проводят в присутствии NBS, MSA и растворителя, такого как MeCN. В другом варианте осуществления можно использовать пероксид водорода и бромистоводородную кислоту. В другом варианте осуществления используют трибромид пиридиния. В другом варианте осуществления можно использовать другие бромирующие реагенты.
В одном варианте осуществления описанное выше конвертирование проводят при температуре приблизительно от -5°C до 10°C, или более конкретно приблизительно от -5°C до 0°C.
В одном варианте осуществления сокристалл 4-бром-2-изопропилфенола и DABCO выделяют в качестве сокристалла 4-бром-2-изопропилфенола и геми-DABCO. В другом варианте осуществления сокристалл выделяют в качестве сокристалла 4-бром-2-изопропилфенола и моно-DABCO.
В одном варианте осуществления процесс получения сокристалла 4-бром-2-изопропилфенола и геми-DABCO включает следующую стадию:
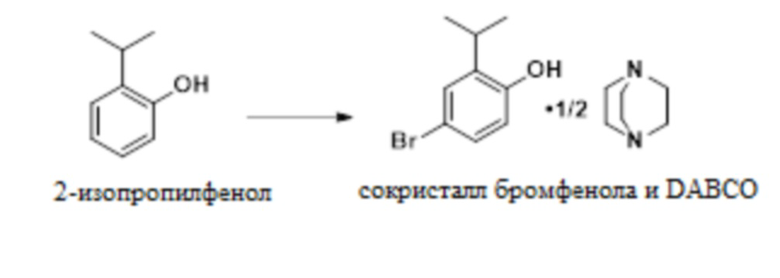 .
.
В одном варианте осуществления данный способ дополнительно включает стадию реакции сокристалла 4-бром-2-изопропилфенола и DABCO с NaOMe с получением 2-изопропил-4-метоксифенола:
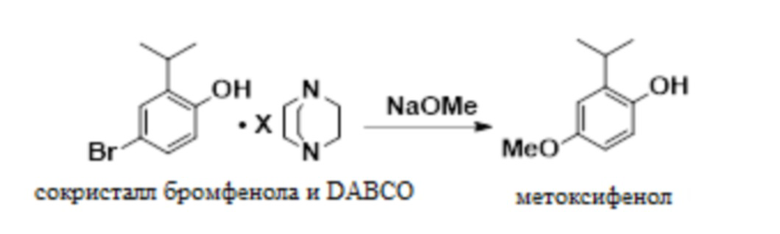 .
.
В одном варианте осуществления указанную выше реакцию проводят в присутствии бромида меди (I) в качестве катализатора. В других вариантах осуществления в качестве катализатора можно использовать бромид меди (II), йодид меди (I) и другие соли меди (I) или меди (II).
В одном варианте осуществления сокристалл 4-бром-2-изопропилфенола и DABCO загружают в качестве сокристалла 4-бром-2-изопропилфенола и геми-DABCO. В другом варианте осуществления сокристалл загружают в качестве сокристалла 4-бром-2-изопропилфенола и моно-DABCO.
В одном варианте осуществления реакцию проводят в присутствии MeOH и DMF. В других вариантах осуществления в качестве растворителя можно использовать диметилацетамид, диоксан или DMSO.
В одном варианте осуществления 2-изопропил-4-метоксифенол не является выделенным. В другом варианте осуществления 2-изопропил-4-метоксифенол выделяют в качестве ангидрата. В следующем варианте осуществления 2-изопропил-4-метоксифенол выделяют в качестве сокристалла с DMAP.
В одном варианте осуществления данный способ дополнительно включает стадию реакции 2-изопропил-4-метоксифенола с ClCH2CN с образованием 2-(2-изопропил-4-метоксифенокси)ацетонитрила, как показано ниже:
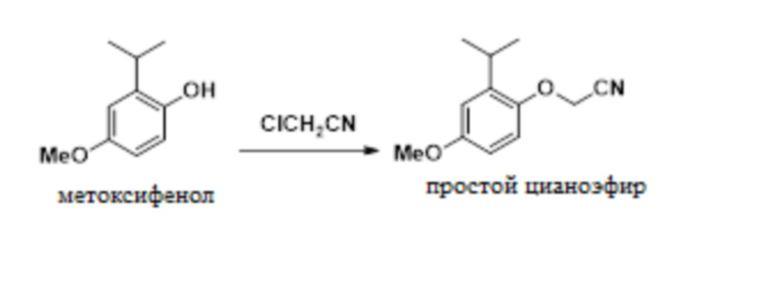 .
.
В одном варианте осуществления описанную выше реакцию проводят в присутствии NMP и толуола. В другом варианте осуществления реакцию проводят в присутствии ацетонитрила. В другом варианте осуществления можно использовать другие полярные апротонные растворители.
В одном варианте осуществления описанную выше реакцию проводят в присутствии основания. В одном варианте осуществления основание представляет собой NaOH. В другом варианте осуществления основание представляет собой карбонат калия. В другом варианте осуществления можно использовать другие гидроксидные или карбонатные основания.
В одном варианте осуществления в качестве алкилирующего реагента используют хлорацетонитрил. В другом варианте осуществления можно использовать бромацетонитрил, йодацетонитрил и другие 2-замещенные ацетонитрилы.
В одном варианте осуществления указанную выше реакцию проводят при температуре от приблизительно 0°C до приблизительно 10°C. В другом варианте осуществления указанную выше реакцию проводят при температуре от приблизительно 25°C до приблизительно 35°C.
В одном варианте осуществления данный способ дополнительно включает стадию конвертирования соединения простого цианоэфира, имеющего приведенную ниже формулу, в 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин:
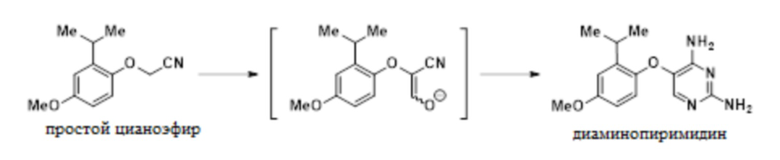 .
.
В одном варианте осуществления указанное выше конвертирование проводят, сначала получая раствор 2-(2-изопропил-4-метоксифенокси)ацетонитрила с этилформиатом в толуоле с получением промежуточного соединения 3-гидрокси-2-(2-изопропил-4-метоксифенокси)акрилонитрилинолята. В других вариантах осуществления эти соединения растворяют в NMP, DMI или других полярных апротонных растворителях.
В одном варианте осуществления раствор, содержащий промежуточное соединение-энолят 3-гидрокси-2-(2-изопропил-4-метоксифенокси)акрилoнитрил смешивают с источником гуанидина непосредственно без выделения энолята.
В одном варианте осуществления источником гуанидина является гуанидина гидрохлорид. В другом варианте осуществления можно использовать другие солевые формы гуанидина, включая карбонат гуанидина и гидросульфат гуанидина.
В одном варианте осуществления соли аммония присутствуют в ходе добавления гуанидина.
В одном варианте осуществления толуол отгоняют из реакционной смеси перед кристаллизацией. В других вариантах осуществления толуол не удаляют перед кристаллизацией.
В одном варианте осуществления в качестве растворителя используют N-метилпирролидинон. В другом варианте осуществления используют другие полярные апротонные растворители.
В одном варианте осуществления указанную выше реакцию между смесью и гуанидином-HCl проводят при температуре приблизительно от 80°C до 98°C. В другом варианте осуществления температура реакции составляет приблизительно от 95°C до 110°C.
В одном варианте осуществления 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин выделяют в качестве ангидрата. В другом варианте осуществления 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин выделяют в качестве сольвата в NMP. 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин также образует сольваты с ацетоном, DMA или DMI.
В одном варианте осуществления данный способ дополнительно включает стадию реакции 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина с ClSO3H, а затем с аммиаком, с получением свободного основания соединения A:
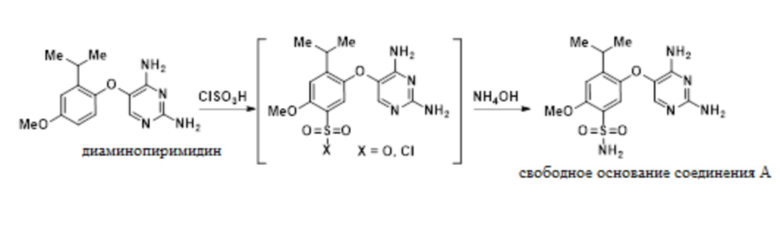
В одном варианте осуществления аммиак подают в качестве гидроксида аммония. В другом варианте осуществления можно использовать другие соли аммония. В другом варианте осуществления используют аммиак в органических растворителях.
В одном варианте осуществления указанную выше реакцию между 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамином и ClSO3H проводят в присутствии MeCN.
В одном варианте осуществления указанную выше реакцию между 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамином и ClSO3H проводят при температуре от приблизительно 25°C до приблизительно 35°C. В другом варианте осуществления температура реакции составляет от приблизительно 35°C до приблизительно 45°C. В другом варианте осуществления температура реакции составляет от приблизительно 45°C до приблизительно 70°C.
В одном варианте осуществления в ходе конвертирования сульфонилхлорида в соединение A присутствует метанол. В другом варианте осуществления в ходе конвертирования сульфонилхлорида в соединение A присутствует вода.
В одном варианте осуществления свободное основание соединения A получают в качестве ангидрата. В другом варианте осуществления свободное основание соединения A получают в качестве сольвата в ацетонитриле. Свободное основание соединения A также образует сольваты с DMSO, DMAc, DMF, ацетоном, NMP, IPA, метанолом, THF, MEK, сульфоланом или NMP/IPA.
В одном варианте осуществления свободное основание соединения A в указанных выше реакциях получают с выходом более 80%. В другом варианте осуществления выход составляет более 85%. В другом варианте осуществления выход превышает 90%.
В одном варианте осуществления данный способ дополнительно включает стадию конвертирования свободного основания соединения A в цитрат соединения A.

В одном варианте осуществления конвертирование проводят в присутствии MeOH и гликолевой кислоты. В другом варианте осуществления в ходе конвертирования также присутствует IPA.
В одном варианте осуществления цитрат выделяют в качестве ангидрата. В другом варианте осуществления цитрат выделяют в качестве сольвата в метаноле или изопропаноле. Цитрат также образует сольваты с метанолом/водой, этанолом/водой, IPA/водой, THF или NMP.
В одном варианте осуществления гликолят выделяют в качестве ангидрата, а затем конвертируют в цитрат. Гликолят также образует сольваты или гидраты с метанолом, IPA, трет-амиловым спиртом, водой или комбинацией этих растворителей, и образует десольватированные/дегидратированные формы из этих сольватов/гидратов.
В одном варианте осуществления указанное выше конвертирование имеет выход более 85%. В другом варианте осуществления выход составляет более 90%. В другом варианте осуществления выход составляет приблизительно 93%.
В одном варианте осуществления способ получения цитрата соединения A включает следующие стадии:
(a) конвертирование 2-изопропилфенола в сокристалл 4-бром-2-изопропилфенола и DABCO:
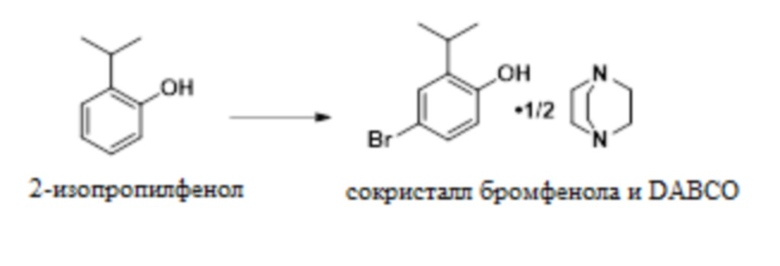 ;
;
(b) реакция полученного сокристалла 4-бром-2-изопропилфенола и DABCO с NaOMe с образованием 2-изопропил-5-метоксифенола:
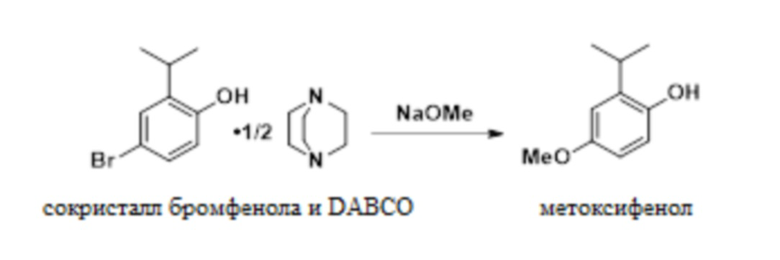 ;
;
(c) реакция 2-изопропил-5-метоксифенола с ClCH2CN с получением 2-(2-изопропил-4-метоксифенокси)ацетонитрила:
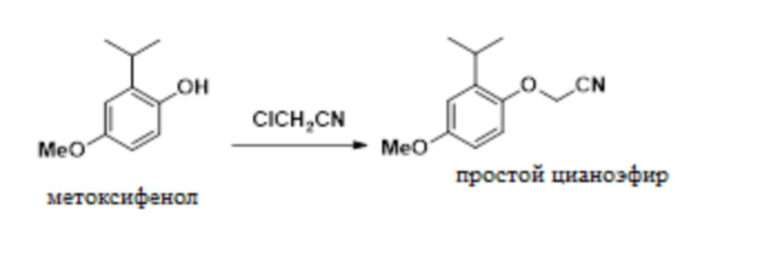 ;
;
(d) конвертирование 2-(2-изопропил-4-метоксифенокси)ацетонитрила в 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин:
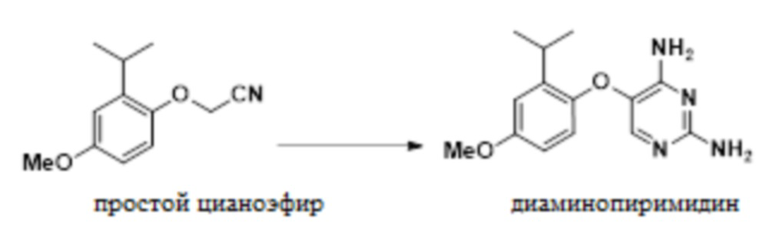 ;
;
(e) реакция 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина с ClSO3H, а затем с NH4OH с получением свободного основания соединения A, 5-((2,4-диаминопиримидин-5-ил)окси)-4-изопропил-2-метоксибензолсульфонамида:
 ; и
; и
(f) конвертирование свободного основания соединения A, 5-((2,4-диаминопиримидин-5-ил)окси)-4-изопропил-2-метоксибензолсульфонамида, в цитрат соединения A в присутствии гликолевой кислоты и MeOH:
 .
.
В одном варианте осуществления общий выход цитрата соединения A из 2-изопропилфенола составляет более 50%. В другом варианте осуществления общий выход составляет более 55%. В другом варианте осуществления общий выход составляет более 60%.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приведенные ниже примеры предоставлены только для иллюстрации, а не для ограничения каким-либо образом. Сокращенные обозначения, использованные в настоящем описании и в описании, представляют собой сокращенные обозначения, общепринятые в данной области или следующие.
°C: градусы Цельсия
DABCO: 1,4-диазабицикло[2.2.2]октан
DMA: диметиламин
DMAP: диметиламинопиридин
DMF: N, N-диметилформамид
DMI: 1,3-диметил-2-имидазолидинон
EtOAc: этилацетат
EtOH: этанол
г: граммы
ч: час(ы)
IPA: изопропиловый спирт
кг: килограммы
л: литр
LC: жидкостная хроматография
MeOH: метанол
MSA: метансульфоновая кислота
MeCN: ацетонитрил
MEK: метилэтилкетон
мин: минуты
мл: миллилитр(ы)
моль: количество моль
Н: нормальный
NBS: N-бромсукцинимид
нМ: наномолярный
NMP: N-метил-2-пирролидон
К.т. или к.т.: комнатная температура
насыщ.: насыщенный
THF: тетрагидрофуран
масс или масс.: масса
XRPD: порошковая рентгенодифракция
В одном варианте осуществления свободное основание соединения A и его цитрат получают способом, включающим синтез и выделение сокристалла 4-бром-2-изопропилфенола и DABCO, синтез 2-изопропилфенила, 2-изопропил-5-метоксифенола и 2-(2-изопропил-4-метоксифенокси)ацетонитрила, синтез и выделение 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина, синтез свободного основания соединения A и конечное конвертирование свободного основания соединения A в соответствующий цитрат, как проиллюстрировано ниже.
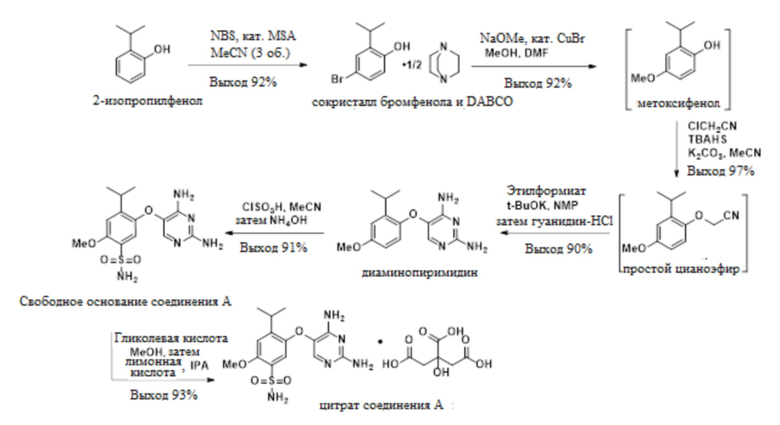
Стадия 1. Получение сокристалла 4-бром-2-изопропилфенола и DABCO
Следующий сокристалл 4-бром-2-изопропилфенола и геми-DABCO получают с чистотой более 99% и выходом приблизительно 85-92% посредством следующего процесса:
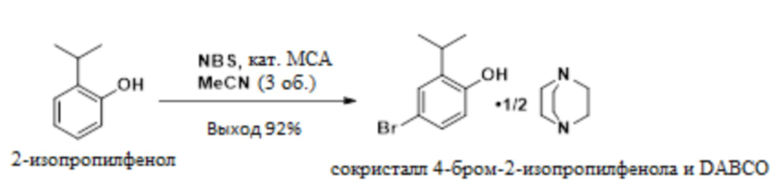
К раствору 2-изопропилфенола (75,0 г, 550 ммоль) в ацетонитриле (225 мл) добавляли MSA (0,520 г, 5,41 ммоль). Смесь охлаждали до -10°C и порционно добавляли NBS (98,01 г, 550 ммоль) при поддержании внутренней температуры ниже 10°C. Реакционную смесь выдерживали в течение от 30 мин до 1 ч, а затем нагревали до 20°C, разбавляли водой (450 мл) и экстрагировали толуолом (225 мл). Органический слой последовательно промывали 9 масс.% фосфорной кислотой (150 мл) и 5 масс.% NaCl (150 мл). Органические слои концентрировали приблизительно до 150 мл и отфильтровывали в чистый реактор. Смесь нагревали до 30-40°C и добавляли н-гептан (28,5 мл), а затем DABCO (30,89 г, 275 ммоль). В смесь вносили затравку (затравка может быть синтезирована из предшествующей партии этой методики, проведенной без затравки) сокристалла 4-бром-2-изопропилфенола и геми-DABCO (75 мг, 0,277 ммоль), разбавляли 52,5 мл н-гептана и перемешивали в течение 1 ч. Взвесь охлаждали до 20°C в течение 1 ч и добавляли 370 мл н-гептана в течение 2 ч. Взвесь охлаждали до 5°C в течение 2 ч, выдерживали в течение 2 ч, фильтровали и промывали н-гептаном (2×75 мл). Твердое вещество сушили при 20-25°C в вакууме с получением сокристалла 4-бром-2-изопропилфенола и геми-DABCO (134,8 г, 90%) в качестве твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ 7,20 (д, J=2,5 Гц, 1H), 7,13 (дд, J=8,5, 2,6 Гц, 2H), 6,73 (д, J=8,5 Гц, 2H), 3,16 (гепт, J=6,9 Гц, 2H), 2,60 (с, 12H), 1,14 (д, J=6,9 Гц, 12H).
Кристаллизация на стадии 1 дает сокристалл 4-бром-2-изопропилфенола и геми-DABCO, сокристалл бромфенола и моно-DABCO или смесь сокристалла бромфенола и геми-DABCO и сокристалла бромфенола и моно-DABCO. Профиль XRPD для сокристалла бромфенола и геми-DABCO представлен на фиг.1.
Сокристалл бромфенола и моно-DABCO можно получать по следующей методике:
В колбу с магнитной мешалкой помещали DABCO (1,7 г, 15 ммоль), фенол (2,5 г, 15 ммоль) и 2 мл н-гептана. Полученную взвесь перемешивали при 23°C в течение ночи. Затем взвесь фильтровали и полученный влажный остаток промывали 2 мл н-гептана, имевшего температуру 5°C. Остаток сушили в вакууме с продуванием азотом с получением сокристалла 4-бром-2-изопропилфенола и моно-DABCO (2,9 г, выход 70%) в виде твердого вещества. 1H-ЯМР (500 МГц, DMSO-d6) δ 9,65 (с, 1H), 7,20 (с, 1H), 7,14 (д, J=8,5 Гц, 1H), 6,74 (д, J=8,5 Гц, 1H), 3,17 (гепт, J=6,8 Гц, 1H), 2,61 (с, 12H), 1,15 (д, J=6,9 Гц, 6H).
Профиль XRPD для сокристалла бромфенола и моно-DABCO представлен на фиг.2.
Стадия 2a. Получение 2-изопропил-4-метоксифенола
2-изопропил-4-метоксифенол, представленный ниже, получают с выходом приблизительно 92% посредством следующего процесса:
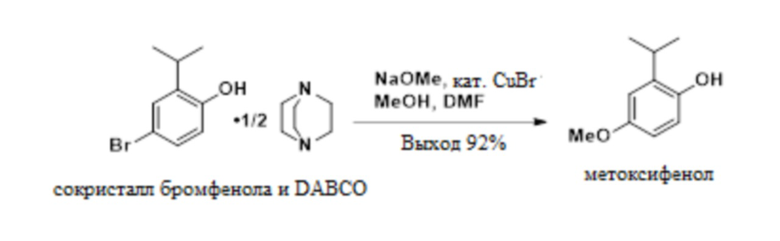
К раствору сокристалла 4-бром-2-изопропилфенола и геми-DABCO (120 г, 442 ммоль) в 25 масс.% метоксиде натрия в метаноле (430 г) добавляли 60 мл DMF. Раствор продували азотом под давлением, к смеси добавляли бромид меди (I) (3,23 г, 22,5 ммоль), и реакционную смесь нагревали до температуры кипения с обратным холодильником в течение 12-16 ч. Реакционную смесь охлаждали до 0-5°C и гасили 6 M HCl до тех пор, пока pH раствора не составлял менее 5. Взвесь разбавляли 492 мл толуола и 720 мл воды с получением однородного раствора со слоем остатков между слоями. Водный слой удаляли в отходы. Органический слой фильтровали для удаления слоя остатков и промывали 240 мл воды с получением 2-изопропил-4-метоксифенола (491 г, 13,3 масс.%, выход 89%) в качестве раствора в толуоле. 1H-ЯМР (500 МГц, DMSO-d6) δ 8,73 (с, 1H), 6,68 (д, J=8,6 Гц, 1H), 6,66 (д, J=3,0 Гц, 1H), 6,55 (дд, J=8,6, 3,1 Гц, 1H), 3,65 (с, 3H), 3,17 (гепт, J=6,9 Гц, 1H), 1,14 (д, J=6,9 Гц, 6H).
Стадия 2b. Получение 2-изопропил-4-метоксифенола
Альтернативно метоксифенол получают посредством следующего процесса:

В емкость высокого давления помещали 400 мл безводного толуола, Re2(CO)10 (3,16 г, 4,84 ммоль) и меквинол (100 г, 806 ммоль) при к.т. Затем емкость дегазировали пропиленом и в нее загружали пропилен (85,0 г, 2,02 моль). Емкость закрывали и нагревали до 170°C. Внутреннее давление было измерено на уровне приблизительно 250 фунт./кв. дюйм. Реакционную смесь перемешивали в этих условиях в течение 72 ч. Затем емкости позволяли остыть до 23°C. Внутреннее давление осторожно снижали до 1 атмосферы, и раствор в толуоле был проанализирован с результатом 91%, и его использовали непосредственно на следующей стадии или выделяли в качестве твердого вещества.
Стадия 2a/2b обеспечивает безводную форму 1 2-изопропил-4-метоксифенола. Профиль XRPD формы 1 метоксифенола представлен на фиг.3.
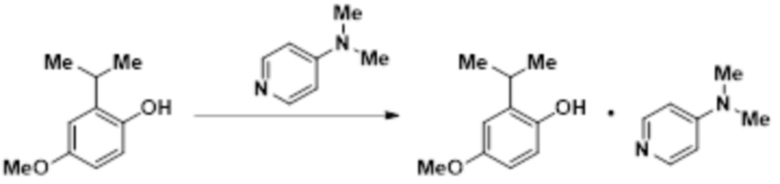
В другом варианте осуществления продукт выделяли в качестве сокристалла с DMAP:
 .
.
В колбу с магнитной мешалкой помещали DMAP (3,67 г, 30,1 ммоль), 2,5 мл толуола и 2-изопропил-4-метоксифенол (5,00 г, 30,1 ммоль). Реакционную смесь перемешивали при к.т. в течение 5 мин, и образовывался однородный раствор. Затем реакционную смесь охлаждали до 5°C. Медленно добавляли десять мл н-гептана в течение 20 мин. Полученную взвесь перемешивали при 5°C в течение ночи. Взвесь фильтровали и полученный влажный остаток промывали 3 мл н-гептаном, имевшим температуру 5°C. Остаток сушили в вакууме с продуванием азотом с получением сокристалла 2-изопропил-4-метоксифенола и DMAP (7,01 г, 81%) в виде твердого вещества. 1H-ЯМР (500 МГц, DMSO-d6) δ 8,78 (с, 1H), 8,10 (д, J=6,1 Гц, 2H), 6,71-6,65 (м, 2H), 6,57 (дд, J=11,3, 6,0 Гц, 3H), 3,66 (с, 3H), 3,17 (гепт, J=6,8 Гц, 1H), 2,95 (с, 6H), 1,14 (д, J=6,9 Гц, 6H).
Кристаллизация обеспечивает безводный кристалл 2-изопропил-4-метоксифенола с DMAP. Профиль XRPD сокристалла 2-изопропил-4-метоксифенола и DMAP представлен на фиг.4.
Стадия 3a. Получение простого цианоэфира, 2-(2-изопропил-4-метоксифенокси)ацетонитрила
Простой цианоэфир получают с выходом приблизительно 95% посредством следующего процесса:
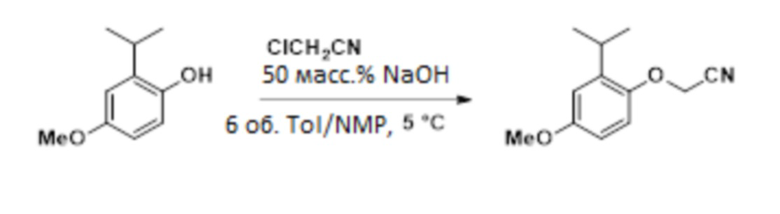
12-15 масс.% раствор 2-изопропил-4-метоксифенола (314,3 г, 12 масс.%, 226,8 ммоль) концентрировали до более чем 50 масс.% 2-изопропил-4-метоксифенола в толуоле в вакууме при 40-50°C. К раствору добавляли 189 мл NMP и смесь охлаждали до 5°C. К смеси последовательно добавляли гидроксид натрия (27,2 г, 50 масс.% в воде, 340 ммоль) и хлорацетонитрил (36 г, 340 ммоль) при поддержании внутренней температуры ниже 10°C. Реакционную смесь выдерживали в течение 2 ч, а затем разбавляли 150 мл толуола и 226 мл воды при поддержании температуры ниже 10°C. Смесь нагревали до 20-25°C, слои разделяли и органический слой промывали 75 мл 20 масс.% NaCl (водн.). Органический слой фильтровали с получением 2-(2-изопропил-4-метоксифенокси)ацетонитрила (56,8 г, 74,6 масс.%) в виде раствора в толуоле. Фильтр промывали NMP с получением дополнительного 2-(2-изопропил-4-метоксифенокси)ацетонитрила (27,1 г, 5,0 масс.%) в виде раствора в NMP. Совокупный выход составлял приблизительно 94%. 1H-ЯМР (500 МГц, DMSO-d6) δ 7,05 (д, J=8,8 Гц, 1H), 6,81 (д, J=3,0 Гц, 1H), 6,78 (дд, J=8,8, 3,1 Гц, 1H), 5,11 (с, 2H), 3,73 (с, 3H), 3,20 (гепт, J=6,9 Гц, 1H), 1,17 (д, J=6,9 Гц, 6H).
Стадия 3b. Получение простого цианоэфира, 2-(2-изопропил-4-метоксифенокси)ацетонитрила
Альтернативно простой цианоэфир, представленный ниже, получали с выходом приблизительно 92% посредством следующего процесса:
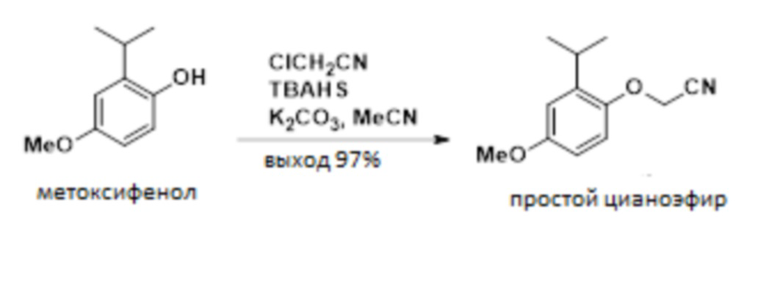
Раствор 2-изопропил-4-метоксифенола в толуоле (491 г, 13,3 масс.%, 393 ммоль) концентрировали и растворитель заменяли на ацетонитрил в вакууме при 40-50°C. В отдельную емкость добавляли карбонат калия (164,5 г, 1190 ммоль) и гидросульфат тетрабутиламмония (1,5 г, 4,42 ммоль), и емкость продували газообразным азотом под давлением. В реакционную емкость последовательно добавляли раствор фенола в ацетонитриле и хлорацетонитриле. Емкость нагревали до 40°C и выдерживали в течение 4 ч. Смеси позволяли остыть до 25°C и разбавляли 326 мл воды. Слои разделяли и органический слой промывали 130 мл 10 масс.% NaCl. Проводили смену растворителя на толуол в вакууме, и органический слой фильтровали через две кассеты 16D Cuno #5. Органический слой концентрировали с получением 2-(2-изопропил-4-метоксифенокси)ацетонитрила в толуоле (128,2 г, 58 масс.%, выход 92%).
Стадия 4. Получение диаминопиримидина, 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина
Диаминопиримидин получают с выходом приблизительно 90% посредством следующего процесса:
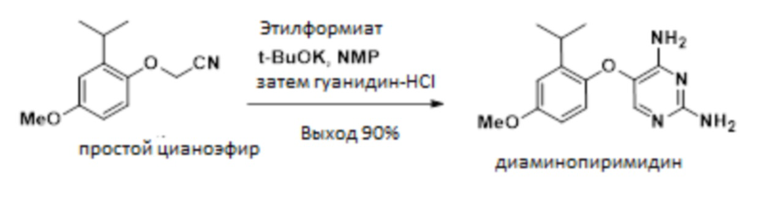
Раствор трет-бутоксида калия (44,8 г, 0399 ммоль) в NMP (180 мл) охлаждали до -10°C. К основному раствору добавляли раствор 2-(2-изопропил-4-метоксифенокси)ацетонитрила, простого цианоэфира, (59,3 г, 61,4 масс.%, 177 ммоль) в толуоле и этилформиате (26,3 г, 355 ммоль) при поддержании внутренней температуры между -12°C и -8°C. После выдерживания в течение 3 ч к смеси добавляли гидрохлорид гуанидина (136 г, 1420 ммоль) и реакционную смесь нагревали до 115°C в течение 6 ч. Смеси позволяли остыть до 90°C, разбавляли 200 мл воды и выдерживали до тех пор пока реакционная смесь не становилась однородной (приблизительно 30-45 мин). После растворения всех твердых веществ к реактору применяли вакуум (400 мм рт. ст.) для удаления толуола. Вакуум отсоединяли и раствору позволяли остыть до 85°C. Добавляли затравку 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина (49,8 мг) (затравку можно синтезировать способом, описанным в патенте США 7741484), раствор выдерживали в течение 2 ч, добавляли 200 мл воды и партии позволяли остыть до 20°C в течение 6 ч. Взвесь выдерживали в течение 10 ч при 20°C, фильтровали, промывали смесью вода:NMP 2:1 (3×100 мл) и водой (3×100 мл), и сушили в вакууме при 50°C с получением указанного в заголовке соединения (42,2 г, 88%) в виде твердого вещества. 1H-ЯМР (500 МГц, DMSO-d6) δ 7,23 (с, 1H), 6,83 (д, J=3,0 Гц, 1H), 6,70 (дд, J=8,9, 3,0 Гц, 1H), 6,63 (д, J=8,8 Гц, 1H), 6,32 (с, 2H), 5,75 (с, 2H), 3,71 (с, 3H), 3,28 (гепт, J=6,9 Гц, 1H), 1,20 (д, J=6,9 Гц, 6H); 13C-ЯМР (126 МГц, DMSO-d6) δ 159,7, 157,2, 155,1, 148,4, 144,2, 139,0, 130,4, 116,9, 112,5, 111,3, 55,4, 26,57, 22,83.
Кристаллизация на стадии 4 обеспечивает форму 1 безводного 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина. Профиль XRPD для формы 1 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина представлен на фиг.5.
В одном варианте осуществления сольват 1 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина в NMP получают посредством добавления избыточного количества формы 1 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина в NMP в закрытую емкость для получения суспензии. Суспензию перемешивают при к.т. до завершения перехода формы. Кристаллы сольвата 1 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина в NMP можно собирать фильтрацией и сразу измерять посредством XRPD для предотвращения десольватации. Профиль XRPD для сольвата 1 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина в NMP представлен на фиг.6.
Стадия 5. Получение свободного основания соединения A
Свободное основание соединения A получают с выходом приблизительно 91% посредством процесса, включающего стадии:
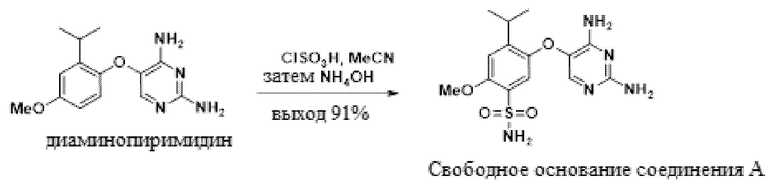
К суспензии 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина, диаминопиримидина, (47,0 г, 171 ммоль) в 141 мл ацетонитрила при -10°C добавляли хлорсульфоновую кислоту (63,1 мл, 942 ммоль) при поддержания внутренней температуры ниже 25°C. Раствор выдерживали в течение 1 ч при 25°C, а затем нагревали до 45°C в течение 12 ч. Раствору позволяли остыть до 20°C и добавляли к раствору 235 мл гидроксида аммония и 71 мл ацетонитрила при -10°C при поддержании внутренней температуры ниже 15°C. Взвесь выдерживали при 10°C в течение 1 ч, нагревали до 25°C и выдерживали в течение 1 ч. Взвесь разбавляли 564 мл воды и 188 мл 50 масс.% гидроксида натрия с получением однородного раствора, который нагревали до 35°C в течение 2 ч. Раствору позволяли остыть до 22°C и pH раствора доводили до 12,9 посредством 2 M раствора лимонной кислоты. В раствор вносили затравку свободного основания соединения A (470 мг, 1,19 ммоль) (затравку можно синтезировать способом, описанным в патенте США 7741484), выдерживали в течение 2 ч, подкисляли до pH 10,5-11,3 2 M раствором лимонной кислоты в течение 5-10 ч, а затем выдерживали в течение 2 ч. Взвесь фильтровали, полученный остаток промывали смесью вода:ацетонитрил 90:10 (2×118 мл) и водой (2×235 мл), и сушили при 55°C в вакууме с получением свободного основания соединения A (50,9 г, 91%) в виде твердого вещества. 1H-ЯМР (500 МГц, DMSO-d6) δ 7,36 (с, 1H), 7,07 (с, 1H), 7,05-6,89 (м, 3H), 6,37 (с, 2H), 5,85 (с, 2H), 3,89 (с, 3H), 3,41 (гепт, J=6,6 Гц, 1H), 1,27 (д, J=6,8 Гц, 6H).
Кристаллизация на стадии 5 обеспечивает форму 1 свободного основания соединения A.
В одном варианте осуществления сольват 1 свободного основания соединения A в ацетонитриле можно получать добавлением избыточного количества свободной формы 1 соединения A в ацетонитрил в закрытой емкости для получения суспензии. Суспензию перемешивают при 50°C до завершения перехода формы. Кристаллы сольвата 1 свободного основания соединения A в ацетонитриле можно собирать фильтрацией и сразу измерять посредством XRPD для предотвращения десольватации. Профиль XRPD сольвата 1 свободного основания соединения A в ацетонитриле представлен на фиг.7.
Стадия 6a. Получение цитрата соединения A
Цитрат соединения A получали посредством процесса, включающего стадии:
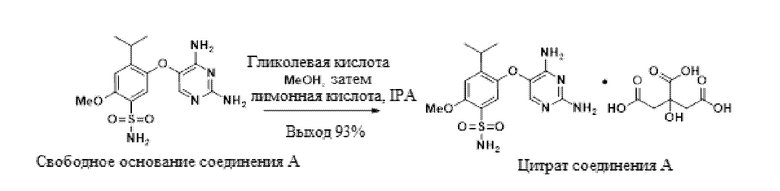
Свободное основание соединения A (30,0 г, 84,9 ммоль) и гликолевую кислоту (22,6 г, 297 ммоль) добавляли к метанолу (360 мл). Раствор нагревали до 60°C, выдерживали в течение 1 ч и фильтровали через 0,6-мкм фильтр в чистую емкость. Раствор лимонной кислоты (32,6 г, 170 ммоль) в 2-пропаноле (180 мл) при к.т. фильтровали через 0,6-мкм фильтр в раствор метанола в течение 30 мин при поддержании температуры раствора в метаноле между 58-62°C. В раствор вносили затравку цитрата соединения A (450 мг, 0,825 ммоль) (затравку можно синтезировать способом, описанным в патентной заявке номер PCT/US17/66562), выдерживали в течение 1 ч и разбавляли 180 мл 2-пропанола в течение 3 ч при поддержании температуры между 58-62°C. Взвесь охлаждали до 50°C в течение 3 ч. Взвесь фильтровали при 50°C, промывали смесью метанол:2-пропанол 1:1 (120 мл) и 2-пропанолом (120 мл) при 50°C, и сушили в вакууме при 35°C с получением цитрата соединения A (45,1 г, 97%) в виде твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ 10,89 (с, 3H), 7,33 (с, 1H), 7,10 (с, 1H), 7,07 (с, 3H), 7,04 (с, 2H), 6,44 (с, 2H), 3,91 (с, 3H), 3,34 (гепт, J=6,7 Гц, 1H), 2,69 (д, J=15,3 Гц, 2H), 2,60 (д, J=15,3 Гц, 2H), 1,26 (д, J=6,9 Гц, 6H).
Стадия 6b. Альтернативный способ получения цитрата соединения A
Альтернативно цитрат соединения A получают способом, включающим стадии:

К суспензии цитрата соединения A (4,5 г, 8,25 ммоль) в метаноле (72 мл) и 2-пропаноле (36 мл) при 50°C добавляли одновременно через отдельные 0,6-мкм фильтры раствор свободного основания соединения A (30,0 г, 84,9 ммоль) и гликолевой кислоты (22,6 г, 297 ммоль) в 360 мл метанола при 50°C и раствор лимонной кислоты (19,5 г, 101 ммоль) в 180 мл 2-пропанола при 25°C в течение 8 ч при поддержании температуры затравочного раствора 60°C. После завершения одновременного добавления к взвеси добавляли лимонную кислоту (13,2 г, 68,7 ммоль) в 180 мл 2-пропанола в течение 8 ч при поддержании температуры 60°C. Взвеси позволяли остыть до 50°C и выдерживали в течение 1 ч, фильтровали при 50°C, промывали смесью метанол:2-пропанол 1:1 (2×120 мл) и 2-пропанолом (120 мл), и сушили в вакууме при 35°C с получением цитрата соединения A (45,1 г, 88%) в виде твердого вещества.
Кристаллизация на стадии 6a/6b обеспечивает форму 1 безводного цитрата соединения A.
В другом варианте осуществления сольват 1 цитрата соединения A в метаноле можно получать через насыщенный раствор формы 1 цитрата соединения A в метаноле при 50°C. Раствор подвергают охлаждению естественным образом до температуры окружающей среды или упаривают при температуре окружающей среды до получения кристаллов сольвата 1 цитрата соединения A в метаноле. Профиль XRPD для сольвата 1 цитрата соединения A в метаноле представлен на фиг.8.
В другом варианте осуществления сольват 1 цитрата соединения A в IPA можно получать путем добавления избыточного количества сольвата 1 цитрата соединения A в метаноле к IPA в закрытой емкости для получения суспензии. Суспензию перемешивают при 5°C или ниже до завершения перехода формы. Кристаллы сольвата 1 цитрата соединения A в IPA можно собирать фильтрацией и сразу измерять посредством XRPD для предотвращения десольватации. Профиль XRPD для сольвата 1 цитрата соединения A в IPA представлен на фиг.9.
В другом варианте осуществления сольвата 1 гидрата цитрата соединения A в метаноле 1 можно получать добавлением избыточного количества формы 1 цитрата соединения A в смесь метанол:вода 3:1 (об./об.) в закрытой емкости для получения суспензии. Суспензию перемешивают при к.т. до завершения перехода формы. Кристаллы сольвата 1 гидрата цитрата соединения A в метаноле можно собирать фильтрацией и сразу измерять посредством XRPD для предотвращения десольватации. Профиль XRPD для сольвата 1 гидрата цитрата соединения A в метаноле представлен на фиг.10.
В другом варианте осуществления форму 1 безводного гликолята соединения A можно получать добавлением избыточного количества формы 1 свободного основания соединения A и 4-5 экв гликолевой кислоты в смесь метанол:IPA 3:1 (об./об.) в закрытую емкость для получения суспензии. Суспензию перемешивают при 50°C до завершения перехода формы. Кристаллы формы 1 гликолята соединения A можно собирать фильтрацией при 50°C, промывать горячим IPA при 50°C и сушить в вакууме при 50°C. Профиль XRPD формы 1 гликолята соединения A представлен на фиг.11.
В то время как в вышеуказанном описании описаны принципы настоящего изобретения, причем примеры предоставлены только для иллюстрации, применение настоящего изобретения на практике охватывает все типичные изменения, адаптации и/или модификации, которые входят в объем прилагаемой ниже формулы изобретения. Все публикации, патенты и патентные заявки, цитированные в настоящем описании, включены в описание в качестве ссылок в полном объеме.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ ОСТРОГО, ХРОНИЧЕСКОГО И ПОДОСТРОГО КАШЛЯ И НЕПРЕОДОЛИМОГО ЖЕЛАНИЯ ОТКАШЛЯТЬСЯ | 2014 |
|
RU2650118C2 |
| СУЛЬФОНАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ НАТРИЕВЫХ КАНАЛОВ | 2014 |
|
RU2680401C2 |
| ПРОИЗВОДНОЕ ХИНАЗОЛИНОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЯ | 2017 |
|
RU2730500C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ЗАМЕЩЕННЫХ ПИРРОЛО (2,3-α)ПИРИМИДИНОВ | 1993 |
|
RU2127274C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 7Н-ПИРРОЛО[2,3-d]ПИРИМИДИНА И ИХ СОКРИСТАЛЛОВ | 2017 |
|
RU2779212C2 |
| АНАЛОГ ПИРИДО[1,2-A]ПИРИМИДОНА, ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2753696C2 |
| ПРОЛЕКАРСТВА ПИРИДОНАМИДОВ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ НАТРИЕВЫХ КАНАЛОВ | 2014 |
|
RU2692766C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ATR КИНАЗЫ (ВАРИАНТЫ) | 2014 |
|
RU2720408C2 |
| ДЕЙТЕРИРОВАННЫЕ ДИАМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ТАКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2632907C2 |
Изобретение относится к органической химии. Предложены варианты получения свободного основания соединения A формулы 1, включающие стадию конвертирования 2-изопропилфенола в сокристалл 4-бром-2-изопропилфенола и геми-DABCO, дополнительно включающие стадию конвертирования сокристалла 4-бром-2-изопропилфенола и геми-DABCO в соединение метоксифенола, дополнительно включающие стадию реакции 2-изопропил-4-метоксифенола с ClCH2CN с получением соединения простого цианоэфира, 2-(2-изопропил-4-метоксифенокси)ацетонитрила. Технический результат - разработка простого способа получения свободного основания соединения А с высоким выходом. 2 н. и 9 з.п. ф-лы, 11 ил., 1 пр.
1. Способ получения свободного основания соединения A, имеющего следующую формулу:
 ,
,
включающий стадию конвертирования 2-изопропилфенола в сокристалл 4-бром-2-изопропилфенола и геми-DABCO, как показано ниже:
 ;
;
дополнительно включающий стадию конвертирования сокристалла 4-бром-2-изопропилфенола и геми-DABCO в соединение метоксифенола, представляющего собой 2-изопропил-4-метоксифенол, как показано ниже:
 ;
;
дополнительно включающий стадию реакции 2-изопропил-4-метоксифенола с ClCH2CN с получением соединения простого цианоэфира, 2-(2-изопропил-4-метоксифенокси)ацетонитрила, как показано ниже:
 ;
;
дополнительно включающий стадию конвертирования соединения простого цианоэфира, 2-(2-изопропил-4-метоксифенокси)ацетонитрила, в соединение диаминопиримидина, 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин:
 и
и
дополнительно включающий стадию реакции 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина с ClSO3H, а затем с NH4OH с получением свободного основания соединения A, 5-((2,4-диаминопиримидин-5-ил)окси)-4-изопропил-2-метоксибензолсульфонамида, как показано ниже:
2. Способ по п.1, где конвертирование в сокристалл 4-бром-2-изопропилфенола и геми-DABCO проводят в присутствии NBS и MSA.
3. Способ по п.1, где конвертирование в 2-изопропил-4-метоксифенол проводят в присутствии NaOMe и с CuBr в качестве катализатора.
4. Способ по п.3, где конвертирование проводят в присутствии MeOH и DMF.
5. Способ по п.1, где реакцию для получения соединения простого цианоэфира проводят при температуре от приблизительно 20°C до приблизительно 60°C.
6. Способ по п.1, где конвертирование в 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин проводят в присутствии трет-бутоксида калия в NMP и соединение простого цианоэфира находится в растворе в этилформиате и толуоле.
7. Способ по п.6, где конвертирование в 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин проводят дополнительно в присутствии гидрохлорида гуанидина.
8. Способ по п.1, где реакцию между 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамином и ClSO3H проводят в присутствии MeCN.
9. Способ получения свободного основания соединения A, включающий следующие стадии:
(a) конвертирование 2-изопропилфенола в сокристалл 4-бром-2-изопропилфенола и геми-DABCO:
 ;
;
(b) конвертирование сокристалла 4-бром-2-изопрофилфенола и геми-DABCO в 2-изопропил-4-метоксифенол, показанный ниже, в присутствии NaOMe:
 ;
;
(c) реакция 2-изопропил-5-метоксифенола с ClCH2CN с получением 2-(2-изопропил-4-метоксифенокси)ацетонитрила:
 ;
;
(d) конвертирование 2-(2-изопропил-4-метоксифенокси)ацетонитрила в 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамин:
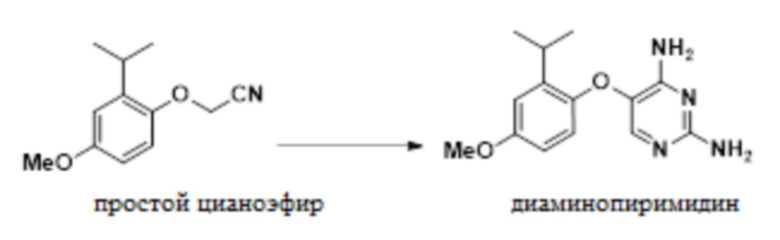 ; и
; и
(e) реакция 5-(2-изопропил-4-метоксифенокси)пиримидин-2,4-диамина с ClSO3H, а затем с NH4OH с получением свободного основания соединения A, 5-((2,4-диаминопиримидин-5-ил)окси)-4-изопропил-2-метоксибензолсульфонамида:
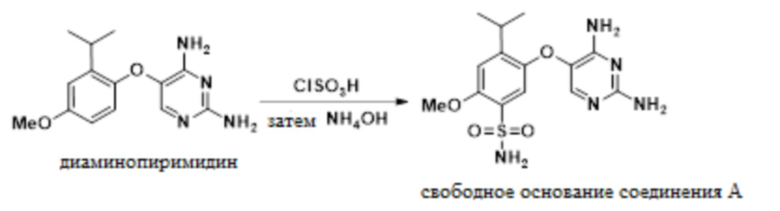 .
.
10. Способ по п.9, дополнительно включающий конвертирование свободного основания соединения A, в цитрат соединения A в присутствии гликолевой кислоты и MeOH:
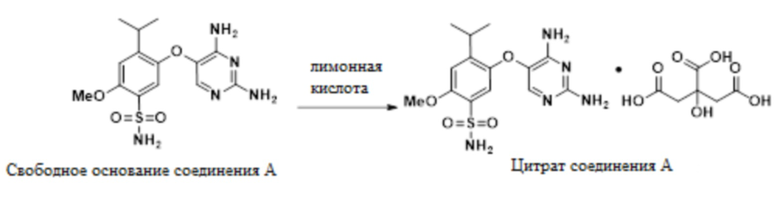 .
.
11. Способ по п.10, где общий выход цитрата соединения A из 2-изопропилфенола превышает 50%.
| WO 2008040652 A1, 10.04.2008 | |||
| US 20170326142 A1, 16.11.2017 | |||
| MARTA DABROS и др | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Angewandte Chemie (International ed | |||
| in English), 2007, 46(22), с.4132-4135, | |||
