ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям, селективно ингибирующим киназу JAK1. Настоящее изобретение также относится к фармацевтическим композициям, содержащим одно или более соединений в качестве активного ингредиента, и к применению соединений в лечении связанных с JAK1 расстройств, например респираторных состояний, таких как астма или COPD.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Янус-киназы (JAK) представляют собой семейство внутриклеточных нерецепторных тирозинкиназ, которые передают опосредованные цитокинами сигналы сигнальным путем JAK-STAT. После связывания цитокинов с их рецепторами рецепторы олигомеризуются для привлечения JAK-киназ, которые ассоциируются с цитоплазматическими хвостами рецепторов и облегчают трансфосфорилирование и активацию тирозиновых остатков на JAK-киназе. Фосфорилированные JAK-киназы связывают и активируют различные белки, являющиеся сигнальными трансдукторами и активаторами транскрипции (STAT), которые затем димеризуются и транслоцируются в ядро для активации транскрипции цитокинотвечающих генов.
Семейство JAK включает в себя JAK1, JAK2, JAK3 и TYK2. JAK1 важна для передачи сигналов определенных цитокинов типа I и типа II и играет решающую роль в инициировании ответных реакций множества основных семейств цитокиновых рецепторов. Например, JAK1 взаимодействует с общей гамма-цепью (γс) цитокиновых рецепторов типа I, вызывая сигналы рецепторов семейства IL-2 (например, IL-2R, IL-7R, IL-9R и IL-15R), рецепторов семейства IL-4 (например, IL-4R и IL-13R) и рецепторов семейства gp130 (например, IL-6R, IL-11R, LIF-R CNTF-R и рецептор нейротрофина-1). JAK1 также важна для передачи сигнала интерферонов типа I (IFN-α/β), интерферона типа II (IFN-γ) и членов семейства IL-10 через цитокиновые рецепторы типа II. Было продемонстрировано, что JAK1 связана с такими расстройствами, как рак, аутоиммунные заболевания, отторжение трансплантата и воспаление.
Учитывая, что члены семейства JAK играют разные роли, существует терапевтический потенциал нацеливания их селективно. Однако разработка селективных ингибиторов JAK1 была сложной задачей, и соединения, идентифицированные как селективные ингибиторы JAK1, демонстрируют лишь крайне низкую селективность в отношении JAK1 (Menet et al., Future Med Chem (2015) 7:203-35). Следовательно, существует потребность в разработке высокоэффективных и селективных ингибиторов JAK1 для лечения связанных с JAK1 расстройств, например астмы или COPD, без реальных или предполагаемых побочных эффектов, связанных с нецелевой активностью, таких как анемия.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте настоящего изобретения предложено соединение, представленное формулой (I):
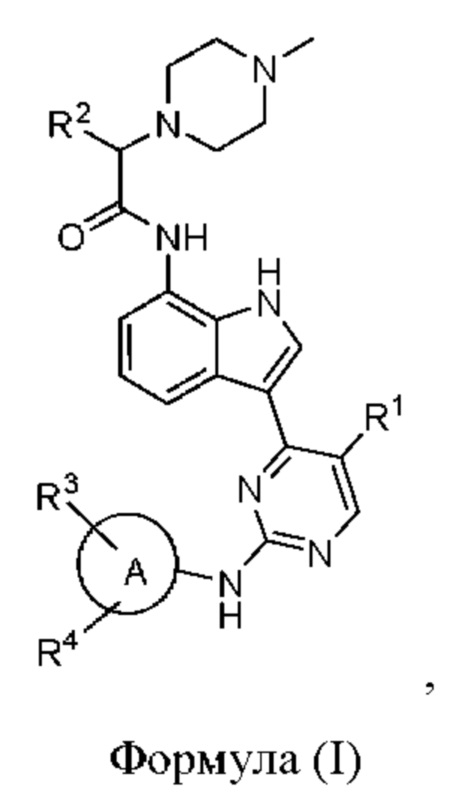
или его фармацевтически приемлемая соль, где кольцо A, R1, R2, R3, R4 такие, как определено в данном документе.
В другом аспекте настоящего изобретения предложено соединение, представленное формулой (Ia):
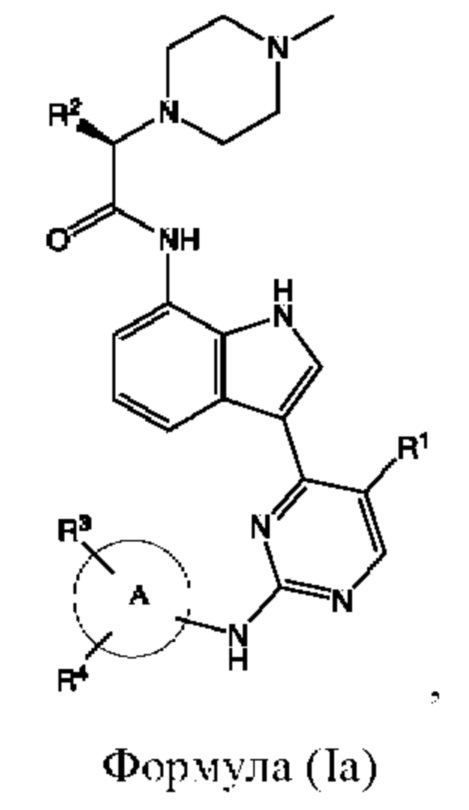
или его фармацевтически приемлемая соль, где кольцо A, R1, R2, R3, R4 такие, как определено в данном документе.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая одно или более соединений формулы (I), формулы (Ia) или их фармацевтически приемлемых солей в качестве активного ингредиента.
В другом аспекте настоящего изобретения также предложено соединение формулы (I), формулы (Ia) или их фармацевтически приемлемая соль, или фармацевтическая композиция одного или более из вышеуказанных для применения в ингибировании киназы JAK-1.
В еще одном другом аспекте настоящего изобретения предложено применение соединений формулы (I), формулы (Ia) или их фармацевтически приемлемых солей или фармацевтической композиции одного или более из вышеуказанных в изготовлении лекарственного средства для ингибирования киназы JAK-1 у субъекта.
В другом аспекте настоящего изобретения предложен способ ингибирования киназы JAK-1 путем использования одного или более соединений формулы (I), формулы (Ia) или их фармацевтически приемлемых солей или фармацевтической композиции одного или более из вышеуказанных.
В другом аспекте настоящего изобретения предложен способ лечения связанного с JAK-1 расстройства (например, респираторного заболевания, такого как астма или COPD) путем использования соединений формулы (I), формулы (Ia) или их фармацевтически приемлемых солей или фармацевтической композиции одного или более из вышеуказанных. В дополнительном аспекте настоящего изобретения предложено соединение формулы (I), формулы (Ia) или его фармацевтически приемлемая соль в комбинации со вторым терапевтическим агентом, предпочтительно противовоспалительным агентом.
В другом аспекте настоящего изобретения предложено совместное применение соединения формулы (I), формулы (Ia) или его фармацевтически приемлемой соли и второго терапевтического агента, предпочтительно противовоспалительного агента.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения
В одном аспекте настоящего изобретения предложено соединение формулы (I):
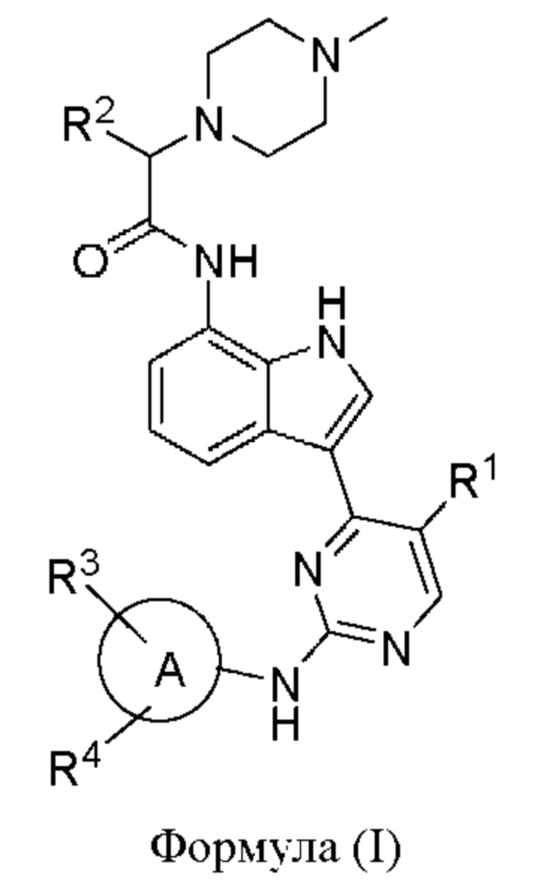
или его фармацевтически приемлемая соль,
где
кольцо А представляет собой моноциклический гетероарил или насыщенное или ненасыщенное 8-10-членное бициклическое кольцо, имеющее 0-5 кольцевых гетероатомов, выбранных из атомов кислорода, серы и азота, где одна или более чем одна образующая кольцо группа -CH2- гетероарила или бициклического кольца может быть заменена группой -С(О)-;
R1 представляет собой водород, галоген, гидроксил, амино, циано или С1-3алкил;
R2 представляет собой водород, С1-12алкил или С1-12алкоксил, возможно моно- или мультизамещенный галогеном, гидроксилом, амино, циано или С1-12алкоксилом;
каждый из R3 и R4 независимо отсутствует или представляет собой галоген, гидроксил, С1-6алкил, карбоксил, С1-6алкоксил, С1-6алкоксикарбонил, -NRaRb, -C(O)NRaRb, сульфинил, C1-6алкилсульфинил, сульфонил, С1-6алкилсульфонил, сульфоноксил, сульфоксиминил, C1-6алкилсульфоксиминил, сульфонимидоил, S-(С1-6алкил)сульфонимидоил, N-(С1-6алкил)сульфонимидоил, N, S-(С1-6алкил)2сульфонимидоил, фосфиноил, С1-6алкилфосфиноил, (С1-6алкил)2фосфиноил, C1-6алкилфосфонил, 3-10-членный насыщенный или ненасыщенный карбоциклил, 3-10-членный насыщенный или ненасыщенный гетероциклил, который возможно может быть моно- или независимо мультизамещен галогеном, гидроксилом, С1-6алкилом, С1-6алкоксилом, C1-6карбоксилом, С1-6алкоксикарбонилом, -NRaRb, -C(O)NRaRb, сульфонилом, C1-6алкилсульфонилом, карбамоилом, N-(С1-6алкил)карбамоилом или N,N-(С1-6алкил)2карбамоилом, фосфиноилом, C1-6алкилфосфиноилом, (С1-6алкил)2фосфиноилом, где одна или более чем одна образующая кольцо группа -CH2- карбоциклила или гетероциклила может быть заменена группой -С(О)-;
где каждый из Ra и Rb независимо выбран из водорода, C1-6алкила, C1-6алкилкарбонила, который возможно может быть моно- или мультизамещен галогеном, гидроксилом или C1-6алкокси.
В некоторых воплощениях предложенные соединения имеют структуру формулы (Ia)
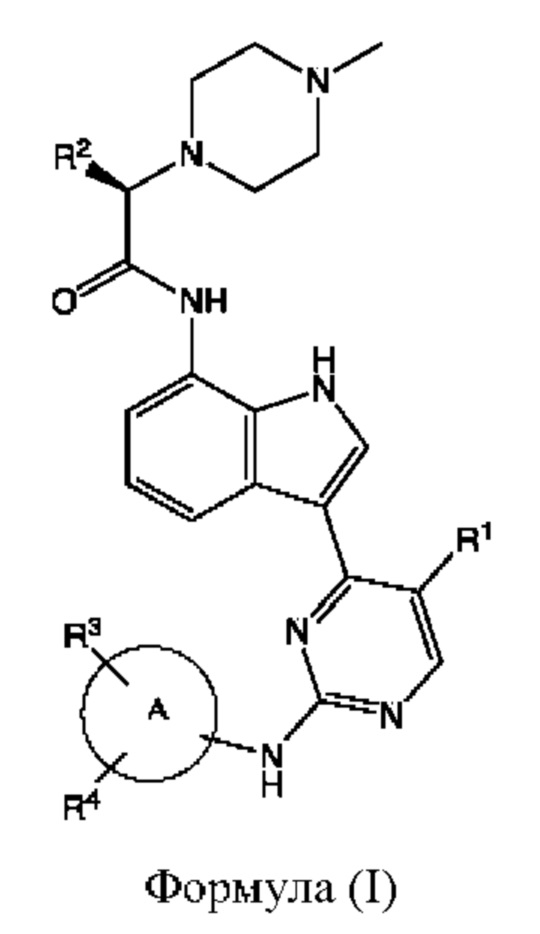
или его фармацевтически приемлемая соль, где
кольцо А представляет собой моно циклический гетероарил или насыщенное или ненасыщенное 8-10-членное бициклическое кольцо, имеющее 0-5 кольцевых гетероатомов, выбранных из атомов кислорода, серы и азота, где одна или более чем одна образующая кольцо группа -СН2- гетероарила или бициклического кольца может быть заменена группой -С(О)-;
R1 представляет собой водород, галоген, гидроксил, амино, циано или С1-3алкил;
R2 представляет собой водород, С1-12алкил или С1-12алкоксил, возможно моно- или мультизамещенный галогеном, гидроксилом, амино, циано или С1-12алкоксилом;
каждый из R3 и R4 независимо отсутствует или представляет собой галоген, гидроксил, С1-6алкил, карбоксил, С1-6алкоксил, С1-6алкоксикарбонил, -NRaRb, -C(O)NRaRb, сульфинил, C1-6алкилсульфинил, сульфонил, C1-6алкилсульфонил, сульфоноксил, сульфоксиминил, C1-6алкилсульфоксиминил, сульфонимидоил, S-(С1-6алкил)сульфонимидоил, N-(С1- 6алкил)сульфонимидоил, N, S-(С1-6алкил)2сульфонимидоил, фосфиноил, С1-6алкилфосфиноил, (С1-6алкил)2фосфиноил, C1-6алкилфосфонил, 3-10-членный насыщенный или ненасыщенный карбоциклил, 3-10-членный насыщенный или ненасыщенный гетероциклил, который возможно может быть моно- или независимо мультизамещен галогеном, гидроксилом, С1-6алкилом, С1-6алкоксилом, C1-6карбоксилом, С1-6алкоксикарбонилом, -NRaRb, -C(O)NRaRb, сульфонилом, C1-6алкилсульфонилом, карбамоилом,
N-(С1-6алкил)карбамоилом или N,N-(С1-6алкил)2карбамоилом, фосфиноилом, C1-6алкилфосфиноилом, (С1-6алкил)2фосфиноилом, где одна или более чем одна образующая кольцо группа -CH2- карбоциклила или гетероциклила может быть заменена группой -С(О)-;
где каждый из Ra и Rb независимо выбран из водорода, С1-6алкила, C1-6алкилкарбонила, который возможно может быть моно- или независимо мультизамещен галогеном, гидроксилом или С1-6алкокси.
В некоторых воплощениях кольцо А представляет собой конденсированное с фенилом или пиридинилом бициклическое гетероарильное кольцо, имеющее 0-5 кольцевых гетероатомов, выбранных из атомов кислорода, серы и азота, где одна или более чем одна образующая кольцо группа -CH2- бициклического кольца может быть заменена группой -С(О)-.
В некоторых воплощениях кольцо А выбрано из группы, состоящей из
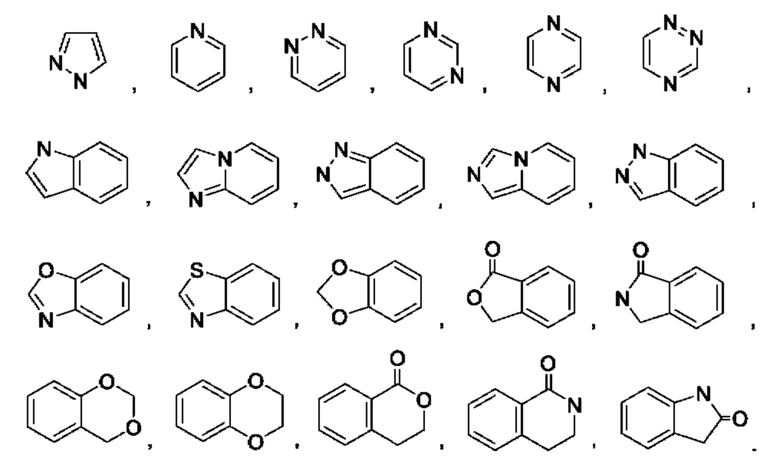
В некоторых воплощениях кольцо А представляет собой моноциклический гетероарил, выбранный из пиразолила, пиридинила, пиридазинила, пиримидинила, пиразинила или триазинила. В некоторых воплощениях кольцо А представляет собой пиримидинил.
В некоторых воплощениях кольцо А выбрано из пиримидин-3-ила, пиримидин-4-ила, 1Н-пиразоло[4,3-b]пиридин-6-ила, 6-(оксазол-2-ил)пиридин-3-ила, 1H-пиразол-4-ила, бензо[d]тиазол-5-ила.
В некоторых воплощениях R1 представляет собой галоген, выбранный из брома, фтора, хлора и йода. В некоторых воплощениях R1 представляет собой фтор.
В некоторых воплощениях R2 представляет собой С1-6алкил, возможно моно- или мультизамещенный C1-6алкоксилом. В некоторых воплощениях R2 представляет собой R2 представляет собой C1-3алкил, возможно моно- или мультизамещенный С1-3алкоксилом. В некоторых воплощениях R2 представляет собой метоксиметил.
В некоторых воплощениях каждый из R3 и R4 независимо отсутствует или представляет собой C1-6алкил, C1-6алкоксил, карбоксил, С1-6алкоксикарбонил, -C(O)NRaRb, который возможно может быть моно- или независимо мультизамещен галогеном, гидроксилом, C1-6алкилом, С1-6алкоксилом, C1-6алкил-карбоксилом, С1-6алкоксикарбонилом, -NRaRb, -C(O)NRaRb, сульфонилом, C1-6алкилсульфонилом, карбамоилом, N-(C1-6алкил)карбамоилом или N,N-(C1-6алкил)2карбамоилом.
В некоторых воплощениях по меньшей мере один из R3 и R4 отсутствует.
В некоторых воплощениях ни R3, ни R4 не отсутствует, и указанный R3 или R4 находится в орто-положении. В некоторых воплощениях ни R3, ни R4 не отсутствует, и указанный R3 или R4 находится в мета-положении.
В некоторых воплощениях каждый из R3 и R4 независимо отсутствует или выбран из C1-6алкила, C1-6алкоксикарбонила, возможно замещенных гидроксилом или C1-6алкоксикарбонилом.
В некоторых воплощениях каждый из R3 и R4 независимо отсутствует или выбран из карбоксила, гидроксила, карбамоила, амино, метила, метоксила, этоксила, метоксиметила, метоксиэтоксила, гидроксиметила, гидроксиэтила, гидроксибутила, гидроксиметоксила, гидроксиэтоксила, карбамоилметоксила, метилкарбамоила, гидроксиацетамидо, (гидроксиэтил)карбамоила, метилкарбамоилметоксила, диметилкарбамоилэтоксила, карбоксиметоксила, метоксикарбонила, этоксикарбонила, изопропоксикарбонила, трет-бутоксикарбонила, метоксикарбонилметила, метоксикарбонилэтила, этоксикарбонилметила, метоксикарбонилметоксила, метиламино, диметиламино, диметиламиноэтила, диметиламиноэтоксикарбонила, диметиламинометила, пропионамидо, метилкарбонил амино, диметиламиноэтоксикарбонила, фосфиноила, метилфосфиноила, диметилфосфиноила, сульфонила, метилсульфонила, S-метил-сульфонимидоила, N,S-диметил-сульфонимидоила, диметилсульфоксиминила, метилсульфоноксила, оксетанила, оксетанил-2-она, азетидин-2-ила, азетидин-3-ил-2-она, метилазетидин-3-ил-2-она, тетрагидрофуран-3-ила или тетрагидропиран-4-ила.
В некоторых воплощениях каждый из R3 и R4 выбран из гидроксиметила, метоксиметила, гидроксиацетамидо или пропионамидо.
В некоторых воплощениях, когда кольцо А представляет собой пиразолил, тогда ни R3, ни R4 не является C1-3алкилом или C1-3алкоксилом.
В некоторых воплощениях R1 представляет собой фтор; R2 представляет собой метоксиметил; кольцо А выбрано из пиримидин-3-ила, пиримидин-4-ила, 1Н-пиразоло[4,3-b]пиридин-6-ила, 6-(оксазол-2-ил)пиридин-3-ила, 1Н-пиразол-4-ила и бензо[d]тиазол-5-ила; каждый из R3 и R4 выбран из гидроксиметила, метоксиметила, гидроксиацетамидо и пропионамидо.
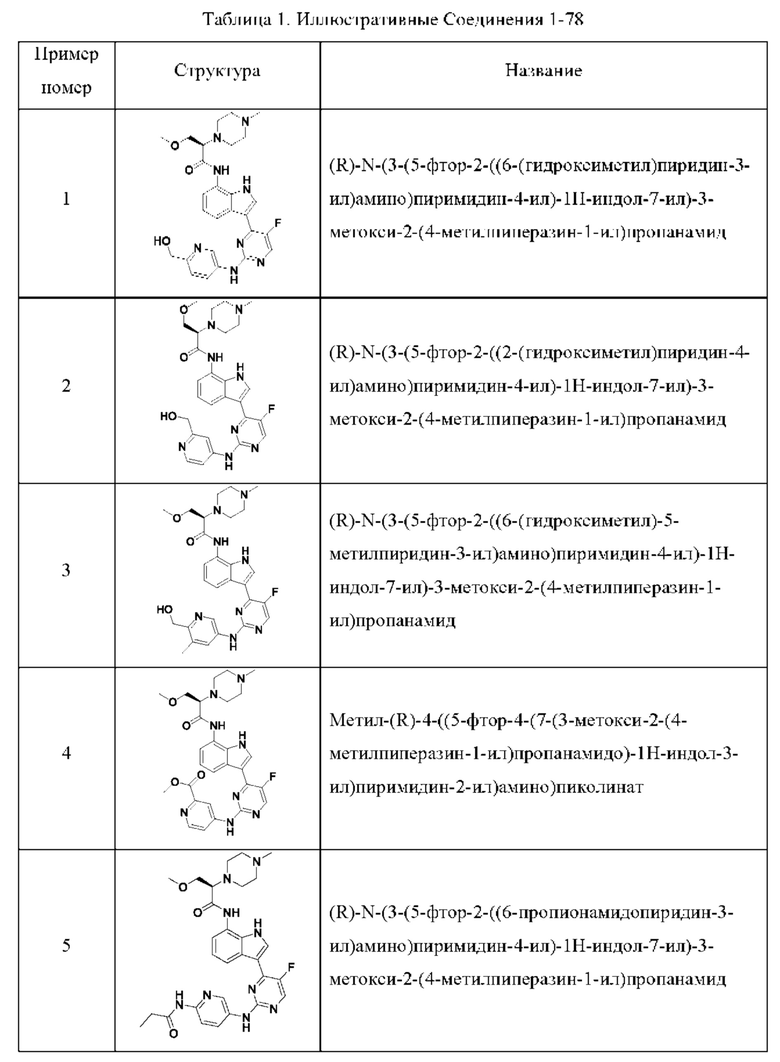
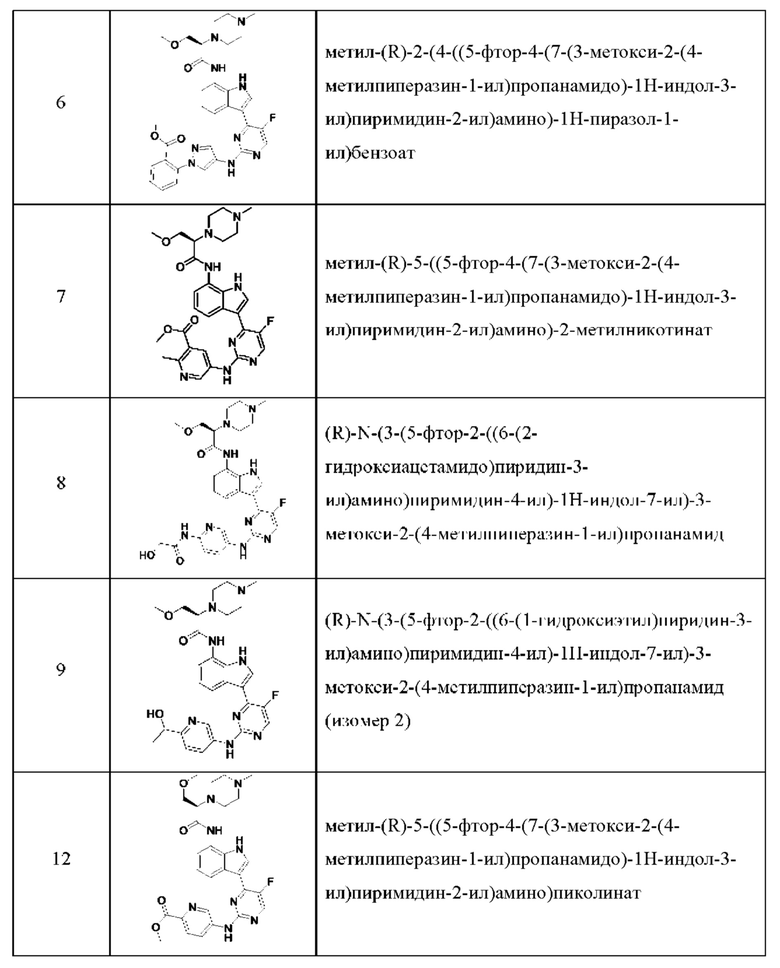
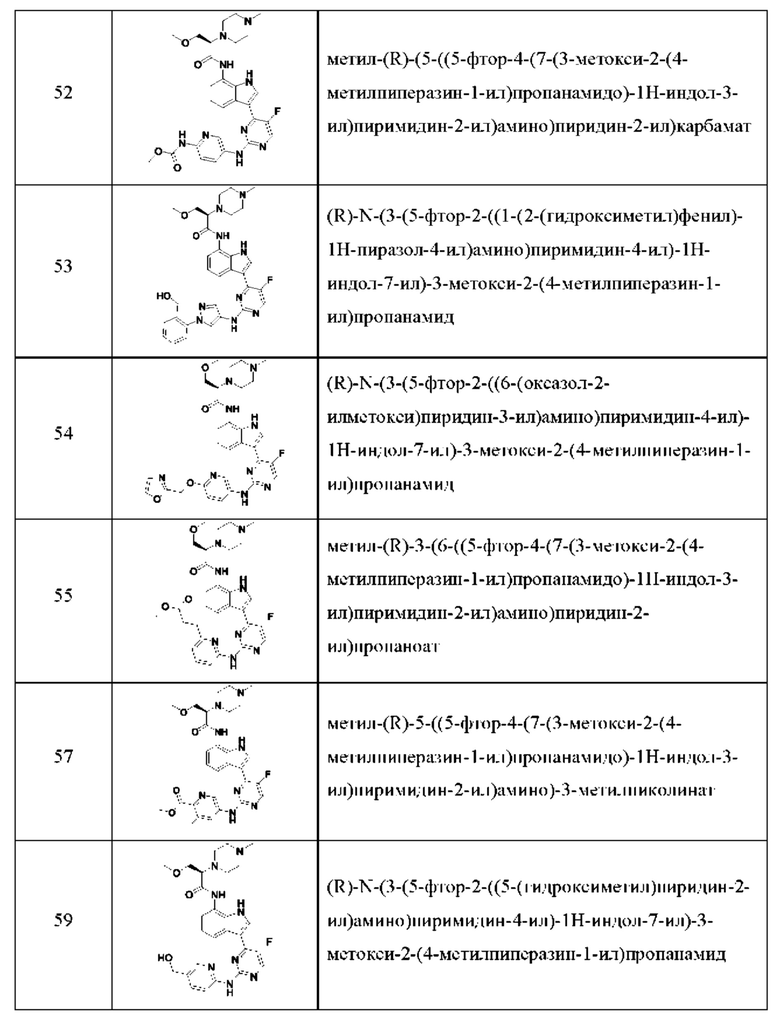
Иллюстративные соединения 1-78 формулы (I) приведены в Таблице 1 ниже.









Понятно, что некоторые признаки настоящего изобретения, которые для ясности описаны в контексте отдельных воплощений, также могут быть предусмотрены в комбинации в единственном воплощении. И наоборот, различные признаки настоящего изобретения, которые для ясности описаны в контексте единственного воплощения, могут быть также предусмотрены отдельно или в любой подходящей их субкомбинации.
В разных местах настоящего описания изобретения описаны линкерные заместители. В тех случаях, когда структура четко требует линкерную группу, тогда понятно, что переменные Маркуша, перечисленные для этой группы, являются линкерными группами. Например, если структура требует линкерную группу, и в определении группы Маркуша для этой переменной указан "алкил", то понятно, что "алкил" представляет собой линкерную алкиленовую группу.
Использованный в данном документе термин "замещенный", когда он относится к химической группе, означает, что химическая группа имеет один или более атомов водорода, который(ые) удален(ы) и заменен(ы) заместителем(ями). Использованный в данном документе термин "заместитель" имеет обычное значение, известное в данной области, и относится к химической группировке, которая ковалентно присоединена к родительской группе или, если это подходит, конденсирована с родительской группой. Использованный в данном документе термин "возможно замещенный" или "возможно…замещенный" означает, что химическая группа может не иметь заместителей (т.е. является незамещенной) или может иметь один или более заместителей (т.е. является замещенной). Следует иметь в виду, что замещение по данному атому ограничено валентностью.
Использованный в данном документе термин "Ci-j" указывает диапазон количества атомов углерода, где i и j являются целыми числами, и этот диапазон количества атомов углерода включает конечные точки (т.е. i и j) и каждое целое число между ними, и где j больше, чем i. Например, С1-6 указывает диапазон от одного до шести атомов углерода, включая один атом углерода, два атома углерода, три атома углерода, четыре атома углерода, пять атомов углерода и шесть атомов углерода. В некоторых воплощениях термин "С1-12" указывает от 1 до 12, включая от 1 до 10, от 1 до 8, от 1 до 6, от 1 до 5, от 1 до 4, от 1 до 3 или от 1 до 2 атомов углерода.
Использованный в данном документе термин "алкил", как часть другого термина или использованный независимо, относится к насыщенной или ненасыщенной углеводородной цепи, причем последняя может быть дополнительно подразделена на углеводородную цепь, имеющую по меньшей мере одну двойную или тройную связь (алкенил или алкинил). В некоторых воплощениях алкил относится к насыщенной углеводородной цепи. Углеводородная цепь, упомянутая выше, может быть прямоцепочечной или разветвленной. Термин "Ci-jалкил" относится к алкилу, имеющему от i до j атомов углерода. Примеры насыщенной алкильной группы включают, но без ограничения, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, трет-бутил; высшие гомологи, такие как 2-метил-1-бутил, н-пентил, 3-пентил, н-гексил, 1,2,2-триметилпропил и т.п. Примеры ненасыщенных алкильных групп включают, но без ограничения, этенил, н-пропенил, изопропенил, н-бутенил, трет-бутенил, этинил, пропин-1-ил, пропин-2-ил и т.п. Примеры "С1-6алкила" включают, но без ограничения, метил, этил, пропил, изопропил, н-бутил, изобутил и трет-бутил. Примеры "С1-3алкила" включают, но без ограничения, метил, этил, пропил и изопропил.
Когда "алкил" представляет собой линкерную алкиленовую группу, примеры алкиленовых групп включают, но без ограничения, метилен, 1,1-этилен, 1,2-этилен, 1,1-пропилен, 1,2-пропилен, 1,3-пропилен, 2,2-пропилен, трет-бутанилен и т.п.
Использованный в данном документе термин "амино" относится к группе формулы "-NH2".
Использованный в данном документе термин "карбамоил" относится к аминокарбонильной группе (т.е. NH2-C(=O)-).
Использованный в данном документе термин "циано" относится к группе формулы "-C≡N".
Использованные в данном документе термины "галоген" и "галогено" относятся к фтору, хлору, брому или йоду.
Использованный в данном документе термин "гидроксил" относится к группе формулы "-ОН".
Использованный в данном документе термин "сульфинил" относится к группе формулы "-S(=O)-".
Использованный в данном документе термин "сульфонил" относится к группе формулы "-S(=O)2-".
Использованный в данном документе термин "сульфоноксил" относится к группе формулы "-O-(S(=O)2H)".
Использованный в данном документе термин "сульфоксиминил" относится к группе формулы "-N=S=O".
Использованный в данном документе термин "сульфонимидоил" относится к группе формулы "-S(=O)(=NH)-".
Использованный в данном документе термин "фосфиноил" относится к группе формулы "-P(=O)H3".
Использованный в данном документе термин "фосфонил" относится к группе формулы "-Р(=O)(-ОН)2".
Использованный в данном документе термин "алкокси", как часть другого термина или использованный независимо, относится к группе формулы -О-алкил.
Термин "Ci-jалкокси" означает, что алкильная группировка группы алкокси имеет от i до j атомов углерода. Примеры групп алкокси включают, но без ограничения, метоксил, этоксил, пропоксил (например, н-пропокси и изопропокси), трет-бутокси и т.п. Примерами "С1-12алкоксила" являются метоксил, этоксил и пропоксил.
Использованный в данном документе термин "гидроксиС1-12алкил", относится к группе формулы "-С1-12алкил-ОН", где алкильная группировка группы имеет от 1 до 12 атомов углерода, и одна или более гидроксильных групп могут быть связаны с любыми атомами углерода в алкильной группировке. В некоторых воплощениях "Ci-jалкил-ОН" имеет одну гидроксильную группу. Примерами группы "С1-12алкил-ОН" являются гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил и 1-гидроксиизопропил.
Использованный в данном документе термин "Ci-jгалогеналкил" относится к Ci-jалкильной группе, замещенной (моно- или мультизамещенной) галогеном. Примерами "С1-12галогеналкила" являются фторметил, дифторметил, трифторметил, фторэтил, дифторэтил, трифторэтил, хлорэтил и бромизопропил. Примером "дифторэтила" является 1,1-дифторэтил. Примерами "трифторэтила" являются 2,2,2-трифторэтил и 1,2,2-трифторэтил.
Примерами "Ci-jгалогеналкоксила" являются фторметоксил, дифторметоксил, или трифторметоксил. Примерами "трифторэтокси" являются 2,2,2-трифторэтокси и 1,2,2-трифторэтокси.
Примерами "N-(С1-12алкил)амино" являются метиламино и этиламино.
Примерами "N-(С1-12галогеналкил)амино" являются фторметиламино, дифторметиламино, трифторметиламино, 2-хлорэтиламино и 1-бромизопропиламино.
Использованный в данном документе термин "C1-6алкоксикарбонил" относится к группе формулы "С1-6алкил-O-С(O)-".
Примерами "C1-6алкилсульфинила" являются метилсульфинил, этилсульфинил, и пропилсульфинил.
Примерами "C1-6алкилсульфонила" являются метилсульфонил и этилсульфонил.
Примерами "C1-6алкилсульфоксиминила" являются метилсульфоксиминил и этилсульфоксиминил.
Примерами "S-(С1-6алкил)сульфонимидоила" являются S-метилсульфоксимидоил и S-этилсульфоксимидоил.
Примерами "N-(С1-6алкил)сульфонимидоила" являются N-метилсульфоксимидоил и N-этилсульфоксимидоил.
Примерами "N,S-(С1-6алкил)2сульфонимидоила" являются N,S-диметил-сульфонимидоил, N-метил-S-этил-сульфонимидоил и N-этил-S-метил-сульфонимидоил.
Примерами "C1-6алкилфосфиноила" являются метилфосфиноил и этилфосфиноил.
Примерами "(С1-6алкил)2фосфиноила" являются диметилфосфиноил и диэтилфосфиноил.
Примерами "С1-6алкилфосфонила" являются метилфосфонил и этилфосфонил.
Использованный в данном документе термин "Ci-jалканоил" относится к Ci-jалкилкарбонилу.
Примерами "С1-12алканоила" являются пропионил и ацетил.
Примерами "С1-12алканоиламино" являются формамидо, ацетамидо и пропионамидо.
Примером"С1-12алканоилокси" является ацетокси.
Примерами "С1-12алкоксикарбонила" являются метоксикарбонил, этоксикарбонил, н- и трет-бутоксикарбонил.
Примерами "N-(С1-12алкил)карбамоила" являются метилкарбамоил и этилкарбамоил.
Примерами "N,N-(С1-12алкил)2карбамоила" являются диметилкарбамоил и метилэтилкарбамоил.
Примерами "N,N-(С1-12алкил)2амино" являются ди-(N-метил)амино, ди-(N-этил)амино и N-этил-N-метиламино.
Использованный в данном документе термин "арил" или "ароматический", как часть другого термина или использованный независимо, относится к кольцевой системе с перемежающимися двойными и одинарными связями между атомами, образующими кольца. В настоящем изобретении термин "арил" или "ароматический" также охватывает псевдоароматический. Термин "псевдоароматический" относится к кольцевой системе, которая не является строго ароматической, но которая стабилизирована за счет делокализации электронов и ведет себя аналогично ароматическим кольцам. Арильная или ароматическая группа может иметь монокольцо или поликольца. Примеры арильных групп включают, но без ограничения, фенил, нафтил, тетрагидронафтил, инданил и т.п.
Использованный в данном документе термин "гетероарил" в данном документе относится к арилу, который содержит по меньшей мере один образующий кольцо гетероатом, выбранный из О, S, N, Р и т.п. Гетероарил включает, но без ограничения, фурил, тиенил, пиридинил, триазинил, пиридил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, индолизинил, индолил, изо индол ил, индолинил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,4-оксадиазол-5-он, 1,2,3-триазолил, 1,3,4-тиадиазолил, пиридазинил, пиримидинил, пиразинил, хиназолинил, изохиназолинил, 1,3,5-триазинил, 1Н-тиено[2,3-с]пиразолил, тиено[2,3-b]фурил, 3Н-индолил, бензо[b]фуранил, бензо[b]тиофенил, 1Н-индазолил, бензимидазолил, тетразолил, уридинил и цитозинил.
Использованный в данном документе термин "карбоциклил", как часть другого термина или использованный независимо, относится к любому кольцу, включая моноциклическое(ие) кольцо(а) или полициклическое(ие) кольцо(а) (например, имеющие 2 или 3 конденсированных, мостиковых или спироколец), в котором все кольцевые атомы являются атомами углерода, и которое содержит по меньшей мере три образующих кольцо атома углерода. В некоторых воплощениях карбоциклил может содержать от 3 до 12 образующих кольцо атомов углерода (т.е. 3-12 членных атомов углерода), от 3 до 10 образующих кольцо атомов углерода, от 3 до 9 образующих кольцо атомов углерода или от 4 до 8 образующих кольцо атомов углерода. Карбоциклильные группы могут быть насыщенными, частично ненасыщенными или полностью ненасыщенными. В некоторых воплощениях карбоциклильная группа может представлять собой насыщенную циклическую алкильную группу. В некоторых воплощениях карбоциклильная группа может представлять собой ненасыщенную циклическую алкильную группу, которая содержит по меньшей мере одну двойную связь в ее кольцевой системе. В некоторых воплощениях ненасыщенная карбоциклильная группа может содержать одно или более ароматических колец. В некоторых воплощениях одна или более чем одна образующая кольцо группа -СН2- насыщенного или ненасыщенного карбоциклила может быть заменена группой -С(О)-.
В некоторых воплощениях карбоциклильная группа представляет собой моноциклическую алкильную группу. В некоторых воплощениях карбо цикл ильная группа представляет собой насыщенную моноциклическую алкильную группу. Примеры моноциклических насыщенных или ненасыщенных карбо циклильных групп включают, но без ограничения, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептатриенил и т.п.
Использованный в данном документе термин "спиро" кольца относится к кольцевым системам, имеющим два кольца, соединенные посредством одного единственного общего атома; термин "конденсированные" кольца относится к кольцевым системам, имеющим два кольца, имеющих два общих соседних атома; и термин "мостиковые" кольца относится к кольцевым системам с двумя кольцами, имеющими три или более общих атомов.
3-12-, 3-10- или 5-6-"членный насыщенный или ненасыщенный карбоциклил" представляет собой насыщенную, частично ненасыщенную или полностью ненасыщенную моно- или полициклическую кольцевую систему, имеющую от 3 до 12, от 3 до 10 или от 5 до 6 образующих кольцо атомов углерода соответственно, где одна или более чем одна образующая кольцо группа -CH2- возможно может быть заменена группой -С(О)-.
Примерами "3-12-членного насыщенного или ненасыщенного карбоциклила" являются С3-4циклоалкил, циклогексил, циклогексенил, циклопентил, фенил, нафтил и бицикло[1.1.1]пентан-1-ил. Примерами "С3-4циклоалкила" являются циклопропил и циклобутил. Примерами "5-6-членного насыщенного или ненасыщенного карбоциклила" являются циклопентил и фенил.
Использованный в данном документе термин "гетероциклил" относится к карбоциклильной группе, где один или более (например, 1, 2 или 3) кольцевых атомов заменены гетероатомами, которые включают, но без ограничения, О, S, N, Р и т.п. В некоторых воплощениях гетероциклил представляет собой насыщенный гетероциклил. В некоторых воплощениях гетероциклил представляет собой ненасыщенный гетероциклил, имеющий одну или более двойных связей в его кольцевой системе. В некоторых воплощениях гетероциклил представляет собой частично ненасыщенный гетероциклил. В некоторых воплощениях гетероциклил представляет собой полностью ненасыщенный гетероциклил. В некоторых воплощениях ненасыщенная гетероциклильная группа может содержать одно или более ароматических колец. В некоторых воплощениях одна или более чем одна образующая кольцо группа -СН2-гетероциклила возможно может быть заменена группой -С(О)-, -S-, -S(O)- или S(O)2-. В некоторых воплощениях, где гетероциклил содержит атом серы в его кольцевой системе, указанный образующий кольцо атом серы возможно может быть окислен с образованием S-оксидов. В некоторых воплощениях гетероциклил связан с другой частью соединения через его образующий кольцо атом углерода. В некоторых воплощениях гетероциклил связан с другой частью соединения через его образующий кольцо атом азота.
В некоторых воплощениях 3-12-членный насыщенный или ненасыщенный моно- или полициклический гетероциклил имеет 1, 2, или 3 гетероатома, выбранные из N, О или S.
3-12-, 3-10- или 5-6-"членный насыщенный или ненасыщенный гетероциклил" представляет собой насыщенную, частично ненасыщенную или полностью ненасыщенную моно- или полициклическую кольцевую (например, имеющую 2 или 3 конденсированных, мостиковых или спироколец) систему, имеющую от 3 до 12, от 3 до 10 или от 5 до 6 образующих кольцо атомов соответственно, из которых по меньшей мере один образующий кольцо атом выбран из атома азота, серы или кислорода, который может, если конкретно не указано иное, быть связан с другой частью соединения через ее образующий кольцо атом углерода или азота, где одна или более чем одна образующая кольцо группа -СН2- насыщенного или ненасыщенного гетероциклила может быть заменена группой -С(О)-, -S-, -S(O)- или -S(O)2-, и где, когда гетероциклил содержит атом серы в его кольцевой системе, тогда указанный кольцевой атом серы возможно может быть окислен с образованием S-оксидов.
Иллюстративные моноциклические гетероциклильные группы включают, но без ограничения, оксетанил, пиранил, 1,1-диоксотиетанилпирролидил, тетрагидрофурил, тетрагидротиенил, пирролил, фуранил, тиенил, пиразолил, имидазолил, триазолил, оксазолил, тиазолил, пиперидил, пиперидил, пиперазинил, морфолинил, пиридинил, пиразинил, пиримидинил, пиридазинил, триазинил, пиридонил, пиримидонил, пиразинонил, пиримидонил, пиридазонил, триазинонил и т.п.
Примеры спирогетероциклила включают, но без ограничения, спиропиранил, спирооксазинил и т.п. Примеры конденсированного гетероциклила включают, но без ограничения, конденсированное с фенилом кольцо или конденсированное с пиридинилом кольцо, такое как хинолинил, изохинолинил, хиноксалинил, хинолизинил, хиназолинил, азаиндолизинил, птеридинил, хроменил, изохроменил, индолил, изоиндолил, индолизинил, индазолил, пуринил, бензофуранил, изобензофуранил, бензимидазолил, бензотиенил, бензотиазолил, карбазолил, феназинил, фенотиазинил, фенантридинил, имидазо[1,2-а]пиридинил, [1,2,4]триазоло[4,3-а]пиридинил, [1,2,3]триазоло[4,3-а]пиридинил и т.п. Примеры мостиковых гетероциклилов включают, но без ограничения, морфанил, гексаметилентетраминил, 8-аза-бицикло[3.2.1]октан, 1-аза-бицикло[2.2.2]октан, 1,4-диазабицикло[2.2.2]октан (DABCO) и т.п.
Примерами "насыщенного или ненасыщенного 8-10-членного бициклического кольца" являются индолил, индазолил, бензо[d]тиазол-5-ил, 2-оксоиндолин-6-ил, бензо[d]тиазол-5-ил, бензо[d]тиазол-6-ил, 1-оксоизохроман-6-ил, 1Н-пиразоло[4,3-b]пиридин-6-ил, 1-оксо-1,2,3,4-тетрагидроизохинолин-7-ил, 1-оксоизохроман-7-ил, бензо[d]оксазол-6-ил, 1Н-бензо[d]имидазол-6-ил, имидазо[1,5-а]пиридин-6-ил, бензо[d]оксазол-5-ил.
"Соединение" по настоящему изобретению охватывает все стереоизомеры, геометрические изомеры и таутомеры изображенных структур, если конкретно не указано иное.
Термин "стереоизомер" относится к любым различным стереоизомерным конфигурациям (например, энантиомерам, диастереомерам и рацематам) асимметрических соединений (например, тем, которые имеют один или более асимметрически замещенных атомов углерода или "асимметрических центров"). Соединения по настоящему изобретению, которые содержат асимметрические центры, могут быть выделены в оптически активных (энантиомеры или диастереомеры) или оптически неактивных (рацемических) формах. Термин "энантиомер" охватывает пары стереоизомеров, которые представляют собой зеркальные отражения друг друга, не накладывающиеся друг на друга. Смесь 1:1 пары энантиомеров является "рацемической смесью". Термины "диастереомеры" или "диастереоизомеры" охватывают стереоизомеры, которые имеют по меньшей мере два асимметрических атома, но которые не являются зеркальными отражениями друг друга. Некоторые соединения, содержащие один или более асимметрических центров, могут приводить к энантиомерам, диастереомерам или другим стереоизомерным формам, которые могут быть определены в терминах абсолютной конфигурации как (R)- или (S)- по каждому асимметрическому центру согласно R-S системе Кана-Ингольда-Прелога. Разделенные соединения, чья абсолютная конфигурация не известна, могут быть обозначены с использованием термина "или" по асимметрическому центру. Способы получения оптически активных форм из рацемических смесей известны в данной области, такие как разделение методом ВЭЖХ или посредством стереоселективного синтеза.
Термины "геометрические изомеры" или "цис- и транс-изомеры" относятся к соединениям с одинаковой формулой, но их функциональные группы вращаются в разной ориентации в трехмерном пространстве.
Термин "таутомеры" включает прототропные таутомеры, которые представляют собой состояния протонирования соединений, имеющих одинаковую формулу и общий заряд. Примеры прототропных таутомеров включают, но без ограничения, кетон-енольные пары, амид-имидные кислотные пары, лактам-лактимные пары, енамин-иминные пары и кольцевые формы, где протон может занимать два или более положений гетероциклической системы, например, 1H- и 3Н-имидазол, 1Н-, 2Н- и 4Н-1,2,4-триазол, 1Н- и 2Н-изоиндол и 1H- и 2Н-пиразол. Таутомеры могут находиться в равновесии или могут быть стерически заблокированы в одной форме соответствующим замещением. Соединения по настоящему изобретению, идентифицированные по названию или структуре как одна конкретная таутомерная форма, включают другие таутомерные формы, если конкретно не указано иное.
"Соединение" по настоящему изобретению также охватывает все изотопы атомов в соединениях. Изотопы атомов включают атомы, имеющие одинаковый атомный номер, но разные массовые числа. Например, если конкретно не указано иное, атомы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора, брома или йода в "соединении" по настоящему изобретению включают также их изотопы, такие как, но без ограничения, 1Н, 2Н, 3Н, 11C, 12С, 13С, 14С, 14N, 15N, 16О, 17O, 18O, 31Р, 32Р, 32S, 33S, 34S, 36S, 17F, 19F, 35Cl, 37Cl, 79Br, 81Br, 127I и 131I. В некоторых воплощениях водород включает протий, дейтерий и тритий. В некоторых воплощениях термин "замещенный дейтерием" или "дейтерием замещенный" использован для замены другой изоформы водорода (например, протия) в химической группе дейтерием. В некоторых воплощениях углерод включает в себя 12С и 13С. В некоторых воплощениях "соединение" по настоящему изобретению охватывает только изотопы водорода в соединении. В некоторых воплощениях "соединение" по настоящему изобретению охватывает только изотопы атомов, распространенных в природе.
Следует также иметь в виду, что "соединение" по настоящему изобретению может существовать в сольватированных, а также в несольватированных формах, таких как, например, гидратированные формы, твердые формы, и настоящее изобретение охватывает все такие сольвированные и несольватированные формы.
Следует также иметь в виду, что "соединение" по настоящему изобретению может существовать в формах фармацевтически приемлемых солей.
Использованный в данном документе термин "фармацевтически приемлемый" относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые в рамках обоснованного медицинского суждения являются подходящими для контактирования с тканями людей и животных без излишней токсичности, раздражения, аллергических реакций или других проблем или осложнений, и которые соизмеримы с разумным соотношением польза/риск. В некоторых воплощениях соединения, вещества, композиции и/или лекарственные формы, которые являются фармацевтически приемлемыми, относятся к тем из них, которые одобрены регулирующим органом (таким как Управление по контролю качества пищевых продуктов и лекарственных средств США, Управление по контролю качества пищевых продуктов и лекарственных средств Китая или Европейское агентство по лекарственным средствам) или перечислены в общепризнанных фармакопеях (таких как Фармакопея США, Фармакопея Китая или Европейская фармакопея) для применения у животных и, более конкретно, у людей.
Использованные в данном документе "фармацевтически приемлемые соли" относятся к производным соединений по настоящему изобретению, где родительское соединение модифицировано путем превращения существующей кислотной группировки (например, карбоксила и т.п.) или основной группировки (например, аминной, щелочной и т.п.) в его солевую форму. Во многих случаях соединения по настоящему изобретению способны образовывать соли присоединения кислоты и/или основания в силу присутствия амино и/или карбоксильных групп или групп, подобных им. Фармацевтически приемлемые соли представляют собой соли присоединения кислоты и/или основания, которые сохраняют биологическую эффективность и свойства родительского соединения и которые обычно не являются биологически или иным образом нежелательными. Подходящие фармацевтически приемлемые соли соединения по настоящему изобретению включают, например, соль присоединения кислоты, которая может быть получена из, например неорганической кислоты (например, соляной, бромистоводородной, серной, азотной, фосфорной кислоты и т.п.) или органической кислоты (например, муравьиной, уксусной, пропионовой, гликолевой, оксалиновой, малеиновой, малоновой, янтарной, фумаровой, винной, тримезиновой, лимонной, молочной, фенилуксусной, бензойной, миндальной, метансульфоновой, нападисиловой, этансульфоновой, толуолсульфоновой, трифторуксусной, салициловой, сульфосалициловой кислоты и т.п.). В некоторых воплощениях фармацевтически приемлемая соль соединения по настоящему изобретению представляет собой соль муравьиной кислоты. В некоторых воплощениях фармацевтически приемлемая соль соединения по настоящему изобретению представляет собой соль TFA.
Подходящие фармацевтически приемлемые соли соединения по настоящему изобретению также включают, например, соль присоединения основания, которая может быть получена, например, из неорганических оснований (например, натриевые, калиевые, аммониевые соли и гидроксидные, карбонатные, бикарбонатные соли металлов из групп I-XII Периодической таблицы, таких как кальций, магний, железо, серебро, цинк, медь и т.п.) или органических оснований (например, первичных, вторичных и третичных аминов, замещенных аминов, в том числе встречающихся в природе замещенных аминов, циклических аминов, основных ионообменных смол и т.п.). Некоторые органические амины включают, без ограничения, изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин. Специалисты в данной области поймут, что присоединение кислот или оснований для образования солей присоединения кислоты/основания, иных, чем те, которые представлены в примерах, также могут быть возможны. Перечни дополнительных подходящих солей можно найти, например, в "Remington's Pharmaceutical Sciences", 20th ed., Mack Publishing Company, Easton, Pa., (1985); и в "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). В некоторых воплощениях подходящей фармацевтически приемлемой солью соединения по настоящему изобретению является соль присоединения неорганического основания.
Настоящее изобретение также охватывает активные промежуточные соединения, активные метаболиты и пролекарства соединений по настоящему изобретению. Использованный в данном документе термин "активное промежуточное соединение" относится к промежуточному в процессе синтеза соединению, которое проявляет такую же или по существу такую же биологическую активность, как и конечное синтезированное соединение.
Использованный в данном документе "активный метаболит" относится к расщепленному или конечному продукту соединения по настоящему изобретению или его соли или пролекарству, образовавшемуся в результате метаболизма или биотрансформации в организме животного или человека, который проявляет такую же или по существу такую же биологическую активность, как и конкретно указанное соединение. Такие метаболиты могут образовываться в результате, например, окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, деэтерификации, ферментативного расщепления и т.п. введенного соединения или его соли или пролекарства.
Использованные в данном документе "пролекарства" относятся к любым соединениям или конъюгатам, которые высвобождают активное родительское лекарственное средство при введении субъекту-животному или человеку. Пролекарства могут быть получены путем модификации функциональных групп, присутствующих в соединениях, таким образом, чтобы модификации отщеплялись либо в результате рутинных манипуляциях, либо in vivo от родительских соединениях. Пролекарства включают соединения, где гидроксильная, амино, сульфгидрильная или карбоксильная группа связана с любой группой, которая при введении субъекту-млекопитающему отщепляется с образованием свободной гидроксильной, амино, сульфгидрильной или карбоксильной группы соответственно. Примеры пролекарств включают, без ограничения, ацетатные, формиатные и бензоатные производные функциональных спиртовых и аминогрупп в соединениях по настоящему изобретению. Получение и применение пролекарств рассматривается в Т. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems", Vol. 14 of A.C.S. Symposium Series, и в Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, которые оба во всей их полноте включены в данный документе посредством ссылки.
В данном документе раскрыты новые соединения или фармацевтически приемлемые соли, которые могут селективно ингибировать JAK1. Кроме того, эти соединения могут быть, в частности, эффективными для лечения респираторных состояний, когда они адаптированы для введения ингаляцией. И эти соединения обладают определенными преимущественными свойствами, например превосходными ингибирующими свойствами, хорошими фармакокинетическими профилями, включая скорость захвата/всасывания, низкий прогнозируемый клиренс у человека и т.д. Они также могут обладать благоприятными профилями токсичности и/или благоприятными метаболическими или фармакокинетическими профилями по сравнению с известными ингибиторами JAK1.
Способы синтеза
Предусмотренный в данном документе синтез соединений, включая их соли, сложные эфиры, гидраты или сольваты или стереоизомеры, иллюстрируется на схемах синтеза в примерах. Предусмотренные в данном документе соединения могут быть получены с использованием любых известных способов органического синтеза и могут быть синтезированы в соответствии с любым из многочисленных возможных путей синтеза, и поэтому эти схемы являются только иллюстративными и не служат ограничением других возможных способов, которые могут быть использованы для получения предусмотренных в данном документе соединений. Кроме того, стадии на Схемах указаны для лучшей иллюстрации, и они могут быть изменены, если это целесообразно. Воплощения соединений в примерах были синтезированы в Китае в целях исследования и потенциального представления в регулирующие органы.
Реакции для получения соединений по настоящему изобретению могут быть осуществлены в подходящих растворителях, которые легко могут быть выбраны специалистом в области органического синтеза. Подходящими растворителями могут быть растворители, по существу неспособные вступать в реакции с исходными веществами (реагентами), промежуточными соединениями или продуктами при температурах, при которых проводят реакции, например при температурах, которые могут находиться в диапазоне от температуры замерзания растворителя до температуры кипения растворителя. Данную реакцию можно проводить в одном растворителе или в смеси растворителей. Специалист в данной области может выбрать подходящие растворители для конкретной реакционной стадии в зависимости от конкретной реакционной стадии.
Получение соединений по настоящему изобретению может включать в себя защиту и снятие защиты с различных химических групп. Необходимость в защите и снятии защиты и выбор подходящих защитных групп легко может определить специалист в данной области. Химию защитных групп можно найти, например, в Т. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999), который во всей его полноте включен в данный документ посредством ссылки.
Реакции можно контролировать любым подходящим методом, известным в данной области. Например, образование продукта можно контролировать спектроскопическими методами, такими как спектроскопия ядерного магнитного резонанса (например, 1Н или 13С), инфракрасная спектроскопия, спектрофотометрия (например, в УФ-видимой области), масс-спектроскопия, или хроматографическими методами, такими как высокоэффективная жидкостная хроматография (ВЭЖХ), жидкостная хроматография/масс-спектроскопия (ЖХ-МС) или тонкослойная хроматография (ТСХ). Соединения могут быть очищены специалистом в данной области различными методами, включая высокоэффективную жидкостную хроматографию (ВЭЖХ) ("Preparative LC-MS Purification: Improved Compound Specific Method Optimization" Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883, который во всей его полноте включен в данный документ посредством ссылки) и нормальнофазную хроматографию на диоксиде кремния.
Сокращения, использованные в данном документе, определены следующим образом: "1 ×" или "× 1" означает однократно, "2 ×" или "× 2" означает дважды, "3 ×" или "× 3" означает трижды, "4 ×" или "× 4" означает четыре раза, "5 ×" или "× 5" означает пять раз, "°С" означает градусы Цельсия, "экв." означает эквивалент или эквиваленты, "г" означает грамм или граммы, "мг" означает миллиграмм или миллиграммы, "л" означает литр или литры, "мл" означает миллилитр или миллилитры, "мкл" означает микролитр или микролитры, "н." означает нормальный, "М" означает молярный, "ммоль" означает миллимоль или миллимоли, "мин" означает минута или минуты, "ч" означает час или часы, "к.т." означает комнатная температура, "атм" означает атмосфера, "фунт/кв.дюйм" означает фунты на квадратный дюйм, "конц." означает концентрированный, "насыщ." означает насыщенный, "МС" или "масс-спек" означает масс-спектрометрия, "ИЭР" означает масс-спектроскопия с ионизацией электрораспылением, "ЖХ-МС" означает жидкостная хроматография/масс-спектрометрия, "ЖХВД" означает жидкостная хроматография высокого давления, "ОФ" означает с обращенной фазой, "ТСХ" означает тонкослойная хроматография, "ИВ" означает исходное вещество, "ЯМР" означает спектроскопия ядерного магнитного резонанса, "1Н" означает протон, "δ" означает дельта, "s" означает синглет, "d" означает дублет, "t" означает триплет, "q" означает квартет, "m" означает мультплет, "br" означает уширенный, и "Гц" означает герц, "α", "β", "R", "S", "Е" и "Z" являются стереохимическим обозначениями, известными специалисту в данной области.
Фармацевтическая композиция
Согласно настоящему изобретению предложены фармацевтические композиции, содержащие по меньшей мере одно соединение по настоящему изобретению. В некоторых воплощениях фармацевтическая композиция содержит более чем одно соединение по настоящему изобретению. В некоторых воплощениях фармацевтическая композиция содержит одно или более чем одно соединение по настоящему изобретению и фармацевтически приемлемый носитель.
Фармацевтически приемлемые носители представляют собой традиционные медицинские носители, которые могут быть получены способом, общеизвестным в данной области. В некоторых воплощениях соединение по настоящему изобретению или его фармацевтически приемлемая соль может быть смешано(а) с фармацевтически приемлемым носителем с получением фармацевтической композиции.
Термин "фармацевтически приемлемый носитель" в данном документе относится к фармацевтически приемлемому веществу, композиции или носителю, такому как жидкий или твердый наполнитель, разбавитель, эксципиент, растворитель или инкапсулирующее вещество, участвующему в переносе или транспорте предложенного в данном документе соединения из одного местоположения, жидкости организма, ткани, органа (внутреннего или внешнего) или части тела в другое местоположение, жидкость организма, ткань, орган или часть тела. Фармацевтически приемлемые носители могут представлять собой носители, разбавители, эксципиенты или другие вещества, которые могут быть использованы для контактирования с тканями животного без излишней токсичности или неблагоприятных эффектов. Иллюстративные фармацевтически приемлемые носители включают сахара, крахмалы, целлюлозы, солод, трагакант, желатин, раствор Рингера, альгиновую кислоту, изотонический физиологический раствор, буферные агенты и т.п. Фармацевтически приемлемый носитель, который может быть использован в настоящем изобретении, включает носители, общеизвестные в данной области, такие как те, которые описаны в "Remington Pharmaceutical Sciences" Mack Pub. Co., New Jersey (1991), который включен в данное описание посредством ссылки.
Некоторые примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) эксципиенты, такие как масло какао и суппозиторные воски; (9) масла, такие как арахисовое масло, хлопковое масло, подсолнечное масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический физиологический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатные буферные растворы; и (21) другие нетоксичные совместимые вещества, используемые в фармацевтических препаратах, такие как ацетон.
Фармацевтические композиции могут содержать фармацевтически приемлемые вспомогательные вещества, требуемые для приближения к физиологическим условиям, такие как агенты, регулирующие рН, и буферные агенты, агенты, регулирующие токсичность, и т.п., например ацетат натрия, хлорид натрия, хлорид калия, хлорид кальция, лактат натрия и т.п.
Форма фармацевтической композиции зависит от ряда критериев, включающих, но без ограничения, путь введения, степень заболевания или дозу, подлежащую введению. Фармацевтические композиции могут быть приготовлены для перорального, назального, ректального, подкожного, внутривенного или внутримышечного введения. Например, лекарственные формы для назального введения могут быть для удобства приготовлены в виде аэрозолей, растворов, капель, гелей или сухих порошков; лекарственные формы для интраназального введения могут быть приготовлены в виде жидкой композиции. В соответствии с желаемым путем введения фармацевтические композиции могут быть приготовлены в форме таблеток, капсул, пилюль, драже, порошков, гранул, саше, облаток, пастилок, суспензий, эмульсий, растворов, сиропов, аэрозолей (в виде твердого вещества или в жидкой среде), спрея, мази, пасты, крема, лосьона, геля, пластыря, ингалируемого препарата или суппозитория.
Для композиций, подходящих и/или адаптированных для введения ингаляцией, предпочтительно, чтобы активное вещество было в форме измельченных частиц, и более предпочтительно чтобы измельченная форма была получена или могла быть получена микронизацией. Предпочтительный размер частиц измельченного (например, микронизированного) соединения, или соли, или сольвата определяется значением D50 примерно от 0,5 до примерно 10 микрон (например, при измерении с использованием лазерной дифракции). Лекарственные формы для ингаляционного введения для удобства могут быть приготовлены в виде аэрозолей или сухих порошков.
Аэрозольные композиции для ингаляционного введения могут содержать раствор или тонкодисперсную суспензию активного вещества в фармацевтически приемлемом водном или неводном растворителе. Аэрозольные композиции могут быть представлены в однодозовых или многодозовых количествах в стерильной форме в запечатанном контейнере, который может принимать форму картриджа или заправки для использования с распыляющим устройством или ингалятором. Альтернативно, запечатанный контейнер может представлять собой унитарное дозирующее устройство, такое как однодозовый назальный ингалятор или дозатор аэрозоля, снабженный мерным клапаном (ингалятор отмеренной дозы), который предназначен для утилизации после исчерпания содержимого контейнера.
Если лекарственная форма содержит аэрозольный дозатор, такой как ингалятор отмеренной дозы под давлением (pMDI), который высвобождает отмеренную дозу при каждом срабатывании, то она предпочтительно содержит подходящий пропеллент под давлением, такой как сжатый воздух, диоксид углерода или органический пропеллент, такой как гидрофторалканы (HFA), также известные как гидрофторуглерод (HFC). Подходящие пропелленты HFC включают 1,1,1,2,3,3,3-гептафторпропан (HFA 227) и 1,1,1,2-тетрафторэтан (HFA 134а). Аэрозольные лекарственные формы могут также принимать форму насоса-распылителя. Аэрозоль под давлением может содержать раствор или суспензию активного соединения. Для этого может потребоваться включение в состав дополнительных эксципиентов, например сорастворителей и/или поверхностно-активных веществ для улучшения дисперсионных характеристик и гомогенности суспензионных композиций. Композиции в форме раствора могут также требовать добавления сорастворителей, таких как этанол. Другие эксципиенты-модификаторы также могут быть включены в состав для улучшения, например, стабильности, и/или вкуса, и/или характеристик массы тонких частиц (количество и/или профиль) композиции. Композиция может содержать другие фармацевтически приемлемые эксципиенты для ингаляционного применения, такие как этанол, олеиновая кислота, поливинилпирролидон и т.п.
pMDI типично имеет два компонента. Во-первых, имеется компонент баллончик, в котором частицы лекарственного средства хранятся под давлением в форме суспензии или раствора. Во-вторых, имеется компонент резервуар, используемый для хранения и приведения в действие баллончика. Типично баллончик будет содержать в себе множество доз композиции, хотя также возможны однодозовые баллончики. Компонент баллончик типично включает в себя выпускной клапан, из которого содержимое баллончика может быть выгружено. Аэрозольное лекарственное средство дозируется из pMDI в результате приложения усилия к компоненту баллончику, чтобы вдавить его в компонент резервуар, тем самым открывая выходное отверстие клапана и обеспечивая транспорт частиц лекарственного средства из выходного отверстия клапана через компонент резервуар и выпуск из выходного отверстия резервуара. После выпуска из баллончика частицы лекарственного средства "распыляются", образуя аэрозоль. Предполагается, что пациент координирует выпуск аэрозоля лекарственного средства его или ее вдохом, так что частицы лекарственного средства захватываются аспираторным потоком воздуха пациента и поступают в легкие.
Предпочтительно, сухая порошковая ингалируемая композиция содержит сухую порошковую смесь соединения формулы I или его фармацевтически приемлемой соли (предпочтительно в измельченной форме, например в микронизированной форме), порошковую основу, такую как лактоза, глюкоза, трегалоза, маннит или крахмал, и возможно модификатор характеристик, такой как L-лейцин или другая аминокислота, и/или металлические соли стеариновой кислоты, такие как стеарат магния или кальция. Лактоза предпочтительно представляет собой гидрат лактозы, например моногидрат лактозы, и/или предпочтительно лактозу сорта для ингаляций, и/или лактозу высшего сорта. Предпочтительно, размер частиц лактозы определяют как 90% или более (по массе или по объему) частиц лактозы менее чем 1000 микрон (микрометров) (например, 10-1000 микрон, например 30-1000 микрон) в диаметре и/или 50% или более частиц лактозы менее 500 микрон (например, 10-500 микрон) в диаметре. Более предпочтительно, размер частиц лактозы определяют как 90% или более частиц лактозы менее 300 микрон (например, 10-300 микрон, например 50-300 микрон) в диаметре, и/или 50% или более частиц лактозы менее 100 микрон в диаметре. Возможно, размер частиц лактозы определяют как 90% или более частиц лактозы менее 100-200 микрон в диаметре, и/или 50% или более частиц лактозы менее 40-70 микрон в диаметре. Предпочтительно, чтобы от примерно 3 до примерно 30% (например, примерно 10%) (по массе или по объему) частиц были менее 50 микрон или менее 20 микрон в диаметре. Например, без ограничения, подходящей лактозой сорта для ингаляций является лактоза Е9334 (10% тонкодисперсных частиц).
Возможно, сухая порошковая ингалируемая композиция может быть введена во множество запечатанных дозовых контейнеров (например, содержащих сухую порошковую композицию), расположенных продольно в полоске или ленте внутри ингаляционного устройства. Контейнер разрывается и открывается по требованию, и доза сухой порошковой композиции может быть введена ингаляцией с помощью такого устройства, как устройство DISKUS (GlaxoSmithKline). Другие сухие порошковые ингаляторы известны специалистам в данной области, и многие такие устройства имеются в продаже, причем репрезентативные устройства включают Aerolizer (Novartis), Airmax (WAX), ClickHaler (Innovata Biomed), Diskhaler (GlaxoSmithKline), Accuhaler (GlaxoSmithKline), Easyhaler (Orion Pharma), Eclipse (Aventis), FlowCaps (Hovione), Handihaler (Boehrmger Ingelheim), Pulvinal (Chiesi), Rotahaler (GlaxoSmithKline), SkyeHaler или Certihaler (SkyePharma), Twisthaler (Schering-Plough), Turbuhaler (AstraZeneca), Ultrahaler (Aventis) и т.п. Могут быть приготовлены также фармацевтические композиции, обеспечивающие быстрое, длительное или отсроченное высвобождение активного ингредиента после введения пациенту путем использования методик, известных в данной области. В некоторых воплощениях фармацевтическая композиция приготовлена в форме длительного высвобождения. Использованный в данном документе термин "форма длительного высвобождения" относится к высвобождению активного агента из фармацевтической композицию таким образом, что становится доступным для биовсасывания у субъекта, прежде всего в желудочно-кишечном тракте субъекта, в течение пролонгированного периода времени (пролонгированное высвобождение) или в определенном месте (контролируемое высвобождение). В некоторых воплощениях пролонгированный период времени может составлять от примерно 1 часа до 24 часов, от 2 часов до 12 часов, от 3 часов до 8 часов, от 4 часов до 6 часов, от 1 до 2 суток или более. В некоторых воплощениях пролонгированный период времени составляет по меньшей мере примерно 4 часа, по меньшей мере примерно 8 часов, по меньшей мере примерно 12 часов или по меньшей мере примерно 24 часа. Фармацевтическая композиция может быть приготовлена в форме таблетки. Например, скорость высвобождения активного агента можно не только контролировать за счет растворения активного агента в желудочно-кишечной жидкости и последующей диффузии из таблетки или пилюли независимо от рН, но также может находиться под влиянием физических процессов разрыхления и разрушения таблетки. В некоторых воплощениях полимерные материалы, которые раскрыты в "Medical Applications of Controlled Release," Langer and Wise (eds.). CRC Pres., Boca Raton, Florida (1974); "Controlled Drug Bioavailability," Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, 1983, J MacromolSci. Rev. Macromol Chem. 23:61; см. также Levy et al., 1985, Science 228:190; During et al., 1989. Ann. Neurol. 25:351; Howard et al., 1989. J. Neurosurg. 71:105. могут быть использованы для длительного высвобождения. Вышеуказанные источники информации во всей их полноте включены в данное описание изобретения посредством ссылки.
В некоторых воплощениях фармацевтические композиции содержат от примерно 0,0001 мг до примерно 100 мг соединения по настоящему изобретению (например, от примерно 0,0001 мг до примерно 10 мг, от примерно 0,001 мг до примерно 10 мг, от примерно 0,01 мг до примерно 10 мг, от примерно 0,1 мг до примерно 10 мг, от примерно 0,1 мг до примерно 5 мг, от примерно 0,1 мг до примерно 4 мг, от примерно 0,1 мг до примерно 3 мг, от примерно 0,1 мг до примерно 2 мг, от примерно 0,1 мг до примерно 1 мг, от примерно 0,1 мг до примерно 0,5 мг, от примерно 1 мг до примерно 10 мг, от примерно 1 мг до примерно 5 мг, от примерно 5 мг до примерно 10 мг, от примерно 5 мг до примерно 20 мг, от примерно 5 мг до примерно 30 мг, от примерно 5 мг до примерно 40 мг, от примерно 5 мг до примерно 50 мг, от примерно 10 мг до примерно 100 мг, от примерно 20 мг до примерно 100 мг, от примерно 30 мг до примерно 100 мг, от примерно 40 мг до примерно 100 мг, от примерно 50 мг до примерно 100 мг). Подходящие дозировки на субъекта в сутки могут составлять от примерно 0,1 мг до примерно 10 мг, предпочтительно от примерно 0,1 мг до примерно 5 мг, от примерно 5 мг до примерно 10 мг или от примерно 1 мг до примерно 5 мг.
В некоторых воплощениях фармацевтические композиции могут быть приготовлены в стандартной лекарственной форме, причем каждая лекарственная форма содержит от примерно 0.0001 мг до примерно 10 мг, от примерно 0,001 мг до примерно 10 мг, от примерно 0,01 мг до примерно 10 мг, от примерно 0,1 мг до примерно 10 мг, от примерно 0,1 мг до примерно 5 мг, от примерно 0,1 мг до примерно 4 мг, от примерно 0,1 мг до примерно 3 мг, от примерно 0,1 мг до примерно 2 мг, от примерно 0,1 мг до примерно 1 мг, от примерно 0,1 мг до примерно 0,5 мг, от примерно 1 мг до примерно 10 мг, от примерно 5 мг до примерно 10 мг, от примерно 5 мг до примерно 20 мг, от примерно 5 мг до примерно 30 мг, от примерно 5 мг до примерно 40 мг, от примерно 5 мг до примерно 50 мг, от примерно 10 мг до примерно 100 мг, от примерно 20 мг до примерно 100 мг, от примерно 30 мг до примерно 100 мг, от примерно 40 мг до примерно 100 мг, от примерно 50 мг до примерно 100 мг соединения по настоящему изобретению. Термин "стандартные лекарственные формы" относится к физически дискретным единицам, подходящим в качестве унитарных дозировок для субъектов-людей и других млекопитающих, причем каждая единица содержит предопределенное количество активного вещества, рассчитанное таким образом, чтобы оно вызывало желаемый терапевтический эффект, совместно с подходящим фармацевтическим носителем.
В некоторых воплощениях фармацевтические композиции содержат одно или более соединений по настоящему изобретению в качестве первого активного ингредиента и дополнительно содержат второй активный ингредиент. Второй активный ингредиент может представлять собой любой противовоспалительный или антигиперпролиферативный агент, который полезен для лечения связанных с JAK1 расстройств (например, астмы или COPD).
Примеры таких антигиперпролиферативных агентов можно найти в Cancer Principles and Practice of Oncology by V.T. Devita and S. Hellman (editors), 6th edition (Feb. 15, 2001), Lippincott Williams & Wilkins Publishers. Специалист в данной области также способен распознать, какие комбинации агентов будут полезны, основываясь на конкретных характеристиках вовлеченных лекарственных средств и видов рака.
Примеры противовоспалительных агентов включают, но без ограничения, (1) ингибиторы TNF-α, такие как ремикейд и энбрел); (2) неселективные ингибиторы СОХ-I/COX-2 (такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенаминовая кислота, индометацин, сулиндак, апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин); (3) ингибиторы СОХ-2 (такие как мелоксикам, целекоксиб, рофекоксиб, вальдекоксиб и эторикоксиб); (4) другие агенты для лечения ревматоидного артрита, включающие метотрексат в низкой дозе, лефуномид, цикдесонид, гидроксихлороквин, d-пеницилламин, ауранофин или парентерал или пероральное золото; (5) ингибитор биосинтеза лейкотриенов, ингибитор 5-липоксидазы (5-LO) или антагонист 5-липоксидаза-активирующего белка (FLAP), такой как зилейтон; (6) антагонист рецептора LTD4, такой как зафирлукаст, монтелукаст и пранлукаст; (7) ингибитор PDE4, такой как рофлумиласт; (8) антагонисты антигистаминового рецептора H1; такие как цетиризин, лоратадин, дезлоратадин, фексофенадин, астемизол, азеластин и хлорфенирамин; (9) агонист α1- и α2-адреноцерепторов вазоконстрикторный симпатомиметический агент, такой как пропилгекседрин, фенилэфрин, фенилпропаноламин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид и этилнорэпинефрина гидрохлорид; (10) антихолинергические агенты, такие как ипратропия бромид, тиотропия бромид, окситропия бромид, аклидиния бромид, гликопирролат, пирензепин и телензепин; (11) агонисты β-адренорецептора, такие как метапротеренол, изпротеренол, изпреналин, албутерол, сальбутамол, формотерол, сальметерол, тербуталин, орципреналин, бутолтерола мезилат и пирбутерол, или метилксантанины, включающие теофиллин и аминофиллин, натрия кромогликат; (12) миметик инсулиноподобного фактора роста типа I (IGF-1); (13) ингалируемый глюкокортикоид с пониженными системными побочными эффектами, такой как преднизон, преднизолон, флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат, цикдесонид и мометазона фуроат.
Предпочтительно, эта комбинация предназначена для лечения и/или профилактики астмы, COPD или аллергического ринита. Репрезентативными примерами такой комбинации являются соединение формулы I или его фармацевтически приемлемая соль в комбинации с компонентами Advair (сальметерола ксинафоат и флутиказона пропионат), Symbicort (будесонид и формотерола фумарат), или Dulera (мометазона фуроат и формотерола фумарат), сальметерол или его фармацевтически приемлемая соль (например, сальметерола ксинофоат) или флутиказона пропионат.
Способ лечения
Согласно настоящему изобретению предложен способ лечения связанных с JAK1 расстройств, включающий введение субъекту эффективного количества одного или более соединений, их фармацевтически приемлемых солей или фармацевтической композиции по настоящему изобретению.
Согласно настоящему изобретению также предложен способ лечения связанных с JAK1 расстройств. В некоторых воплощениях способ включает в себя введение субъекту эффективного количества одного или более соединений, их фармацевтически приемлемых солей или фармацевтической композиции по настоящему изобретению.
Использованный в данном документе термин "связанных с JAK1 расстройств" относится к заболеваниям, чье начало или развитие или и то, и другое связано с экспрессией или активностью JAK1. Примеры включают, но без ограничения, респираторные состояния, аутоиммунные заболевания, гиперпролиферативное расстройство (например, рак) и другие заболевания.
Связанные с JAK1 расстройства включают, но без ограничения, (1) респираторные состояния, такие как астма, бронхит, бронхоэктаз, силикоз, пневмокониоз, острый респираторный дисстресс-синдром, хроническую эозинофильную пневмонию и хроническое обструктивное заболевание легких (COPD); (2) аутоиммунные заболевания, такие как псориаз, склеродерма, ревматоидный артрит, псориатический артрит, ювенильный артрит, миелофиброз, болезнь Кастлемана, волчанковский нефрит, системную красную волчанку, синдром Шегрена, рассеянный склероз, воспалительное заболевание кишечника, болезнь Бехчета, тяжелую миастению, сахарный диабет типа 1, иммуноглобулиновую нефропатию, аутоиммунные тиреоидные заболевания; и (3) гиперпролиферативное расстройство, такое как рак, например лейкоз, глиобластому, меланому, хондросаркому, холангио карциному, остеосаркому, лимфому, рак легкого, аденому, миелому, печеночноклеточную карциному, адренокортикальную карциному, рак поджелудочной железы, рак молочной железы, рак мочевого пузыря, рак предстательной железы, рак печени, рак желудка, рак ободочной кишки, колоректальный рак, рак яичника, рак шейки матки, рак головного мозга, рак пищевода, рак кости, рак яичка, рак кости, рак почки, мезотелиому, нейробластому, рак щитовидной железы, рак в области головы и шеи, рак пищевода, рак глаза, рак предстательной железы, назофарингиальный рак или рак ротовой полости.
Использованные в данном документе термины "лечение", "лечить" и "проведение лечения" относятся к реверсированию, облегчению, задержке начала или торможению прогрессирования заболевания или расстройства или одного или более его симптомов, как описано в данном документе. В некоторых воплощениях лечение может быть назначено после развития одного или более симптомов. В других воплощениях лечение может быть назначено в отсутствие симптомов. Например, лечение может быть назначено восприимчивому индивидууму до начала симптомов (например, в свете истории симптомов и/или в свете генетических факторов или других факторов восприимчивости). Лечение может быть также продолжено после исчезновения симптомов, например чтобы предупредить или отсрочить их повторное появление.
В некоторых воплощениях предложенные одно или более соединений, их фармацевтически приемлемых солей или фармацевтическую композицию вводят парентеральным путем или непарентеральным путем. В некоторых воплощениях одно или более соединений, фармацевтически приемлемых солей, гидратов, сольватов или их стереоизомеров или фармацевтическую композицию вводят перорально, энтерально, трансбуккально, назально, интраназально, трансмукозально, эпидермально, трансдермально, дермально, офтальмически, пульмонарно, сублингвально, ректально, вагинально, местно, подкожно, внутривенно, внутримышечно, интраартериально, интратекально, интракапсулярно, интраорбитально, интракардиально, интрадермально, интраперитонеально, транстрахеально, субкутикулярно, интраартикулярно, субкапсулярно, субарахноидально, интраспинально или интрастернально.
Соединения, предложенные в данном документе, можно вводить в чистой форме, в комбинации с другими активными ингредиентами или в форме фармацевтической композиции по настоящему изобретению. В некоторых воплощениях соединения, предложенные в данном документе, можно вводить нуждающемуся субъекту параллельно или последовательно в комбинации с одним или более противораковым или противовоспалительным агентом(ами), известным в данной области. Индивидуальные соединения таких комбинаций можно вводить либо последовательно, либо одновременно в отдельных или объединенных фармацевтических композициях. Предпочтительно, индивидуальные соединения будут вводить одновременно в объединенной фармацевтической композиции. Соответствующие дозы известных терапевтических агентов легко определит специалист в данной области.
В некоторых воплощениях введение продолжают один раз в сутки, дважды в сутки, три раза в сутки или один раз каждые двое суток, один раз каждые трое суток, один раз каждые четверо суток, один раз каждые пять суток, один раз каждые шесть суток, один раз в неделю.
В некоторых воплощениях одно или более соединений, их фармацевтически приемлемых солей или фармацевтическую композицию, предложенные в данном документе, вводят перорально. Для перорального введения подходящей является любая доза, которая достигает желаемой цели. В некоторых воплощениях подходящие суточные дозировки составляют примерно 0,001-100 мг, предпочтительно от 0,1 мг до 5 г, более предпочтительно от 5 мг до 1 г, более предпочтительно от 10 мг до 500 мг, и введение проводят один раз в сутки, дважды в сутки, три раза в сутки, каждые сутки или 3-5 суток в неделю. В некоторых воплощениях доза одного или более соединений, их фармацевтически приемлемых солей или фармацевтической композиции, предложенных в данном документе, колеблется между примерно 0,0001 мг, предпочтительно 0,001 мг, 0,01 мг, 0,1 мг, 0,2 мг, 0,3 мг, 0,4 мг, 0,5 мг, 0,6 мг, 0,7 мг, 0,8 мг, 0,9 мг, 1 мг, 2 мг, 3 мг, 4 мг, 5 мг, 6 мг, 7 мг, 8 мг, 9 мг, 10 мг в сутки.
Применение соединений
В некоторых воплощениях согласно настоящему изобретению предложено применение соединений, их фармацевтически приемлемых солей или фармацевтической композиции по настоящему изобретению в изготовлении лекарственных средств для лечения связанных с JAK1 расстройств. В некоторых воплощениях связанные с JAK1 расстройства включают в себя виды рака.
Соединения и фармацевтические композиции по настоящему изобретению можно применять в предупреждении или лечении начала или развития любых связанных с JAK1 расстройств (экспрессия или активность) у млекопитающих, в частности у людей.
В такой ситуации согласно настоящему изобретению также предложен способ скрининга пациента, подходящего для лечения соединениями или фармацевтической композицией по настоящему изобретению, одними или совместно с другими ингредиентами (например, вторым активным ингредиентом, например противовоспалительным или противораковым агентом). Этот способ включает в себя секвенирование образцов тканей пациентов и обнаружение накопления JAK1 у пациента.
ПРИМЕРЫ
Нижеследующее дополнительно объясняет общие способы по настоящему изобретению. Соединения по настоящему изобретению могут быть получены способами, известными в данной области. Нижеследующее иллюстрирует подробно способы получения предпочтительных соединений по настоящему изобретению. Однако это не означает, что они ограничивают способы получения соединений по настоящему изобретению.
ПРИМЕРЫ СИНТЕЗА
Синтез соединений, предложенных в данном документе, включая их фармацевтически приемлемые соли, иллюстрируются на схемах синтеза и в примерах. Соединения, предложенные в данном документе, могут быть получены с использованием методов органического синтеза и могут быть синтезированы любым из возможных многочисленных путей синтеза, и поэтому эти схемы являются только иллюстративными и не служат ограничением других возможных способов, которые могут быть использованы для получения предложенных соединений. Дополнительно, стадии на Схемах даны для лучшей иллюстрации и могут быть изменены, если это целесообразно. Воплощения соединений в примерах были синтезированы в целях исследования и потенциального представления в регулирующие органы.
Реакции для получения соединений по настоящему изобретению могут быть проведены в подходящих растворителях, которые легко сможет выбрать специалист в области органического синтеза. Подходящие растворители по существу могут не вступать в реакции с исходными веществами (реагентами), промежуточными соединениями или продуктами при температурах, при которых проводят реакции, например при температурах, которые могут колебаться от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может быть проведена в одном растворителе или в смеси более чем одного растворителя. Специалист в данной области сможет выбрать подходящие растворители для конкретной реакционной стадии в зависимости от конкретной реакционной стадии.
Получение соединений по настоящему изобретению может включать в себя защиту и снятие защиты с различных химических. Необходимость в защите и снятии защиты и выбор подходящих защитных групп легко определит специалист в данной области. Химию защитных групп можно найти, например, в Т. W. Greene and P. G. М. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999), который во всей его полноте включен в данное описание посредством ссылки.
Реакции можно контролировать любым подходящим методом, известным в данной области. Например, образование продукта можно контролировать спектроскопическими методами, такими как спектроскопия ядерного магнитного резонанса (например, 1H или 13С), инфракрасная спектроскопия, спектрофотометрия (например, в Уф-видимой области), масс-спектрометрия, или хроматографическими методами, такими как высокоэффективная жидкостная хроматография (ВЭЖХ), жидкостная хроматография-масс-спектрометрия (ЖХ-МС) или тонкослойная хроматография (ТСХ). Соединения могут быть очищены специалистами в данной области различными методами, включая высокоэффективную жидкостную хроматографию (ВЭЖХ) ("Preparative LC-MS Purification: Improved Compound Specific Method Optimization" Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883, который во всей его полноте включен в данное описание посредством ссылки) и нормально-фазовую хроматографию на диоксиде кремния.
Структуры соединений в примерах охарактеризованы ядерным магнитным резонансом (ЯМР) или/и жидкостной хроматографией/масс-спектромерией (ЖХ-МС). ЯМР химический сдвиг (δ) приводится в единицах 10-6 (м.д. (миллионные доли)). 1H-ЯМР спектры регистрируют в диметилсульфоксиде-d6 (DMSO-d6), или CDCl3, или CD3OD, или D2O, или ацетоне-d6, или CD3CN (от Innochem, или Sigma-Aldrich, или Cambridge Isotope Lab., Inc.) на спектрометрах Bruker AVANCE NMR (300 МГц или 400 МГц) с использованием ICON-NMR (под контролем программы TopSpin) с тетраметилсиланом в качестве внутреннего стандарта.
Измерение методом МС проводят с использованием масс-спектрометра Shimadzu 2020 с источником электрораспыления в режиме регистрации положительных и отрицательных ионов.
Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводят на системах Shimadzu LC-20AD, или системах Shimadzu LC-20ADXR, или системах Shimadzu LC-30AD с использованием колонки Shim-pack XR-ODS С18 (3,0 х 50 мм, 2,2 мкм), или колонки Ascentis Express С18 (2,1 × 50 мм, 2,7 мкм), или колонки Agilent Poroshell HPH-C18 (3,0 × 50 мм, 2,7 мкм).
Тонкослойную хроматографию проводят с использованием силикагелевых пластин Sinopharm Chemical Reagent Beijing Co., Ltd. и Xinnuo Chemical. Используют силикагелевые пластины для тонкослойной хроматография (ТСХ) 175-225 мкм. Для разделения и очистки продуктов методом ТСХ используют силикагелевые пластины 1,0 мм.
Для очистки используется хроматографическая колонка с силикагелем в качестве носителя (100-200, 200-300 или 300-400 меш, производимый компанией Rushanshi Shangbang Xincailiao Co., Ltd. или Rushan Taiyang Desiccant Co., Ltd. И т.д.), или флэш-колонка (обращенно-фазовая колонка С18 20-45 мкм, производимая Agela Technologies) во флэш-системе Agela Technologies. Размеры колонок корректируются в зависимости от количества соединения.
Известные исходные вещества согласно настоящему изобретению могут быть синтезированы с использованием способов или согласно способам, известным в данной области, или могут быть приобретены у Alfa Aesar, TCI, Sigma-Aldrich, Bepharm, Bide pharmatech, PharmaBlock, Еnamine, Innochem и JW&Y PharmLab и т.д.
Если конкретно не указано иное, реакции все проводят в атмосфере аргона или азота. В атмосфере аргона или азота означает, что реакционную колбу соединяют с баллоном аргона или азота объемом примерно 1 л. Гидрирование обычно проводят под давлением. Если конкретно не указано иное, реакционная температура в примерах представляет собой температуру окружающей среды, которая равна 10°С-30°С. Протекание реакций контролируют методом ТСХ или/и ЖХ-МС. Системы элюентов, использованных для реакционных смесей, включают систему дихлорметан-метанол и систему петролейный эфир-этилацетат. Объемные соотношения растворителей регулируют в соответствии с разной полярностью соединений.
Система элюирования колоночной хроматографии, используемая для очистки соединений, и система элюентов ТСХ включают систему дихлорметан-метанол и систему петролейный эфир-этилацетат. Объемные соотношения растворителей корректируют в соответствии с разной полярностью соединений. Небольшое количество щелочных или кислотных агентов (0,1%-1%), таких как муравьиная кислота, или уксусная кислота, или TFA, или аммиак, может быть добавлено для корректировки.
Сокращения для химических соединений, использованных в синтезе предложенных соединений, перечислены ниже:

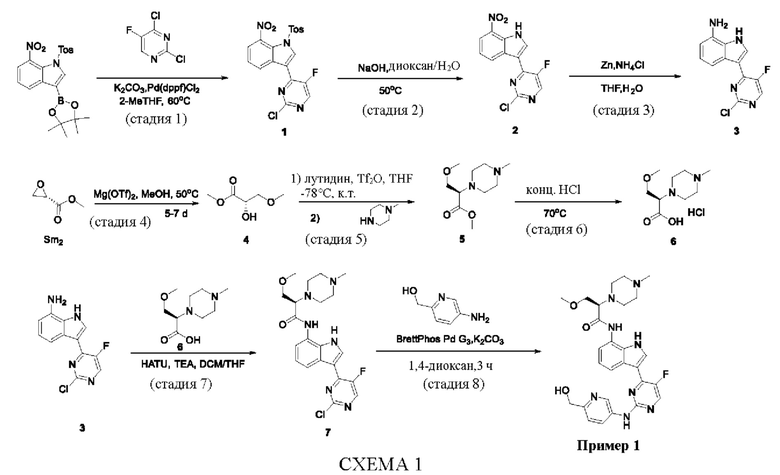
Пример 1
Получение (R)-N-(3-(5-Фтор-2-((6-(гидроксиметил)пиридин-3-ил)амино)-пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
Стадия 1. 3-(2-Хлор-5-фторпиримидин-4-ил)-7-нитро-1-тозил-1Н-индол
В раствор 1-(4-метилбензолсульфонил)-7-нитро-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индола (20,00 г, 45,219 ммоль, 1,00 экв.) и 2,4-дихлор-5-фторпиримидина (9,81 г, 58,785 ммоль, 1,30 экв.) в 2-метилтетрагидрофуране (400,00 мл) и воде (4,0 мл) добавляли K2CO3 (18,69 г, 135,205 ммоль, 2,99 экв.) и Pd(dppf)Cl2.CH2Cl2 (2,95 г, 3,618 ммоль, 0,08 экв.). После перемешивания в течение 15 ч при 60°С в атмосфере азота продукт осаждали добавлением воды (300 мл). Осажденное твердое вещество собирали фильтрованием и промывали РЕ (1 х 40 мл). Полученное твердое вещество сушили под инфракрасным светом с получением 3-(2-хлор-5-фторпиримидин-4-ил)-1-(4-метилбензолсульфонил)-7-нитро-1Н-индола (16 г, 79,19%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 447,1. 1H-ЯМР (300 МГц, DMSO-d6) δ 2.40 (3Н, s), 7.50 (2H, d), 7.68 (1H, t), 7.98 (3H, dd), 8.72-8.85 (2H, m), 9.03 (1H, d).
Стадия 2. 3-(2-Хлор-5-фторпиримидин-4-ил)-7-нитро-1Н-индол
В раствор 3-(2-хлор-5-фторпиримидин-4-ил)-1-(4-метилбензолсульфонил)-7-нитро-1H-индола (7,00 г, 15,666 ммоль, 1,00 экв.) в 1,4-диоксане (210,00 мл) добавляли NaOH (6,27 г, 156,66 ммоль, 10,0 экв.) в воде (105 мл). После перемешивания в течение 5 ч при 60°С смесь подкисляли до рН 6 2М HCl. Осажденное твердое вещество собирали фильтрованием и промывали РЕ (1 × 30 мл). Это привело к получению 3-(2-хлор-5-фторпиримидин-4-ил)-7-нитро-1Н-индола (4,1 г, 89,43%) в виде темно-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 293,0. 1H-ЯМР (300 МГц, DMSO-d6) δ 7.53 (1H, t), 8.13-8.40 (2H, m), 8.83 (1H, d), 8.98 (1H, d), 12.82 (1H, s).
Стадия 3. 3-(2-Хлор-5-фторпиримидин-4-ил)-1Н-индол-7-амин
В раствор 3-(2-хлор-5-фторпиримидин-4-ил)-7-нитро-1Н-индола (10,00 г, 34,171 ммоль, 1,00 экв.) в THF (400,00 мл) добавляли порошок цинка (17,9 г, 273,4 ммоль, 8,0 экв.). Затем в смесь добавляли NH4Cl (18,3 г, 341,7 ммоль, 10,0 экв.) в воде (100,00 мл). После перемешивания в течение 15 ч при комнатной температуре полученную смесь фильтровали, и осадок на фильтре промывали ЕА (3 × 20 мл). Фильтрат концентрировали при пониженном давлении с получением 3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-амина (5 г, 55,71%) в виде красновато-коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 263,1. 1h-ямр (300 МГц, DMSO-d6) δ 5.30 (2H, s), 6.48 (1H, dd), 6.96 (1H, t), 7.76 (1H, d), 8.27 (1H, t), 8.62 (1H, d), 11.84 (1H, s).
Стадия 4. (2S)-2-Гидрокси-3-метоксипропаноат
Смесь метил-(2S)-оксиран-2-карбоксилата (20,00 г, 195,907 ммоль, 1,00 экв.) и дитрифторметансульфоната магния (18,95 г, 58,772 ммоль, 0,30 экв.) в МеОН (500 мл) перемешивали в течение 3 суток при 50°С в атмосфере азота. Смесь оставляли охлаждаться до комнатной температуры и концентрировали при пониженном давлении. Остаток растворяли в DCM (350 мл) и промывали 1 х 300 мл воды. Водный слой экстрагировали смесью CH2Cl2/MeOH (10/1) (5 × 200 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (1:1), с получением метил-(2S)-2-гидрокси-3-метоксипропаноата (20,6 г, 78,39%) в виде бесцветного масла. 1H-ЯМР (300 МГц, DMSO-d6) δ 3.41 (3Н, s), 3.63-3.78 (2H, m), 3.83 (3Н, s), 4.33 (1H, t), 5.56 (1H, d).
Стадия 5. (R)-3-Метокси-2-(4-метилпиперазин-1-ил)пропаноат
В перемешиваемый раствор метил-(2S)-2-гидрокси-3-метоксипропаноата (8,00 г, 59,643 ммоль, 1,00 экв.) и 2,6-лутидина (9,73 мл, 90,761 ммоль, 1,4 экв.) в DCM (150,00 мл) добавляли трифторметансульфонил-трифторметансульфонат (21,88 г, 77,536 ммоль, 1,3 экв.) по каплям при -78°С в атмосфере азота. Полученную смесь перемешивали в течение 1 ч при комнатной температуре в атмосфере азота. В вышеуказанную смесь добавляли 1-метилпиперазин (12,55 г, 125,293 ммоль, 2,10 экв.) по каплям в течение 10 мин при 0°С. Полученную смесь перемешивали в течение еще 15 ч при комнатной температуре. Реакционную смесь гасили добавлением воды (150 мл) при комнатной температуре и экстрагировали CH2Cl2 (3 × 150 мл). Объединенные органические слои сушили над безводным MgSO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (от 10:1 до 0:1), с получением метил-(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропаноата (12 г, 93,03%) в виде коричневого масла. ЖХ-МС (ПЭР): m/z [M+H]+ = 217,3. 1H ЯМР (300 МГц, DMSO-d6) δ 2.35 (3H, s), 2.57 (4H, s), 2.73 (4H, t), 3.37 (3H, s), 3.40-3.52 (1H, m), 3.65 (1H, dd), 3.69-3.79 (4H. m).
Стадия 6. (R)-3-Метокси-2-(4-метилпиперазин-1-ил)пропановая кислота
Раствор метил-(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропаноата (10,00 г, 46,236 ммоль, 1,00 экв.) в конц. HCl (37,97 мл, 1041,355 ммоль, 10,00 экв., 37%) перемешивали в течение 30 ч при 70°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток растворяли в iPrOH (150 мл). Полученную смесь концентрировали под вакуумом, снова растворяли и концентрировали 3 раза с получением (R)-3-метокси-2-(4-метилпиперазин-1-ил)пропановой кислоты гидрохлорида (11 г, 99,66%), который напрямую использовали на следующей стадии. ЖХ-МС (ИЭР): m/z [M+H]+ = 203,1.
Стадия 7. (R)-N-(3-(2-Хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид
В перемешиваемую смесь (R)-3-метокси-2-(4-метилпиперазин-1-ил)пропановой кислоты дигидрохлорида (17,29 г, 62,816 ммоль, 1,50 экв.), HATU (16,72 г, 43,972 ммоль, 1,05 экв.) и 3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-амина (11,00 г, 41,878 ммоль, 1,00 экв.) в DCM (280,00 мл) и THF (140,00 мл) добавляли TEA (23,28 мл, 230,097 ммоль, 4,00 экв.) по каплям при 0°С в атмосфере азота. Полученную смесь перемешивали в течение 2 ч при 25°С в атмосфере азота. Реакционную смесь гасили добавлением насыщ. NaHCO3 (водн.) (150 мл). Полученную смесь экстрагировали CH2Cl2 (2 × 150 мл). Объединенные органические слои сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью CH2Cl2/MeOH (15:1). Неочищенный продукт промывали смесью гексан/EtOAc (3:1) с получением (2R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (7,8 г, 41,68%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 447,3. 1H-ЯМР (300 МГц, DMSO-d6) δ 1.25 (3H, s), 2.46 (3H, s), 2.70-2.90 (8H, m), 3.54-3.91 (3H, m), 7.25 (1H, t), 7.57 (1H, dd), 8.28-8.52 (2H, m), 8.73 (1H, d), 9.99 (1H, s), 11.81 (1H, s).
Стадия 8 (R)-N-[3-(5-Фтор-2-[[6-(гидроксиметил)пиридин-3-ил]амино]-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 1)
В сосуд вместимостью 40 мл добавляли (2R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (200,00 мг, 0,448 ммоль, 1,00 экв.) и (5-аминопиридин-2-ил)метанол (83,33 мг, 0,671 ммоль, 1,50 экв.), BrettPhos Pd G3 (40,57 мг, 0,045 ммоль, 0,1 экв.), K2CO3 (123,70 мг, 0,895 ммоль, 2 экв.) в 1,4-диоксане (15,00 мл) при комнатной температуре. Затем смесь перемешивали при 70°С в атмосфере азота в течение 3 ч. Полученную смесь разбавляли водой (20 мл) и экстрагировали EtOAc (3 × 20 мл). Объединенные органические слои промывали рассолом (3 х 10 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 25% В до 40% В за 7 мин; 254/220 нм; Rt: 5,77 мин) с получением (R)-N-[3-(5-фтор-2-[[6-(гидроксиметил)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил) пропанамида (30 мг, 12,54%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 535,4. 1H-ЯМР (300 МГц, DMSO-d6) δ 2.14 (3H, s), 2.36 (4H, s), 2.63 (2H, s), 2.73 (2H, s), 3.30 (3H, s), 3.49-3.86 (1H, t), 3.67 (1H, dd), 3.79 (1H, dd), 4.52 (2H, d), 5.28 (1H, t), 7.13 (1H, t), 7.39 (1H, d), 7.53 (1H, d), 8.22 (2H, dd), 8.49 (2H, dd), 8.78 (1H, d), 9.65 (1H, s), 9.86 (1,H s), 11.47 (1H, s).
Следующие указанные в таблице соединения синтезированы способом, аналогичным способу, описанному в Примере 1.
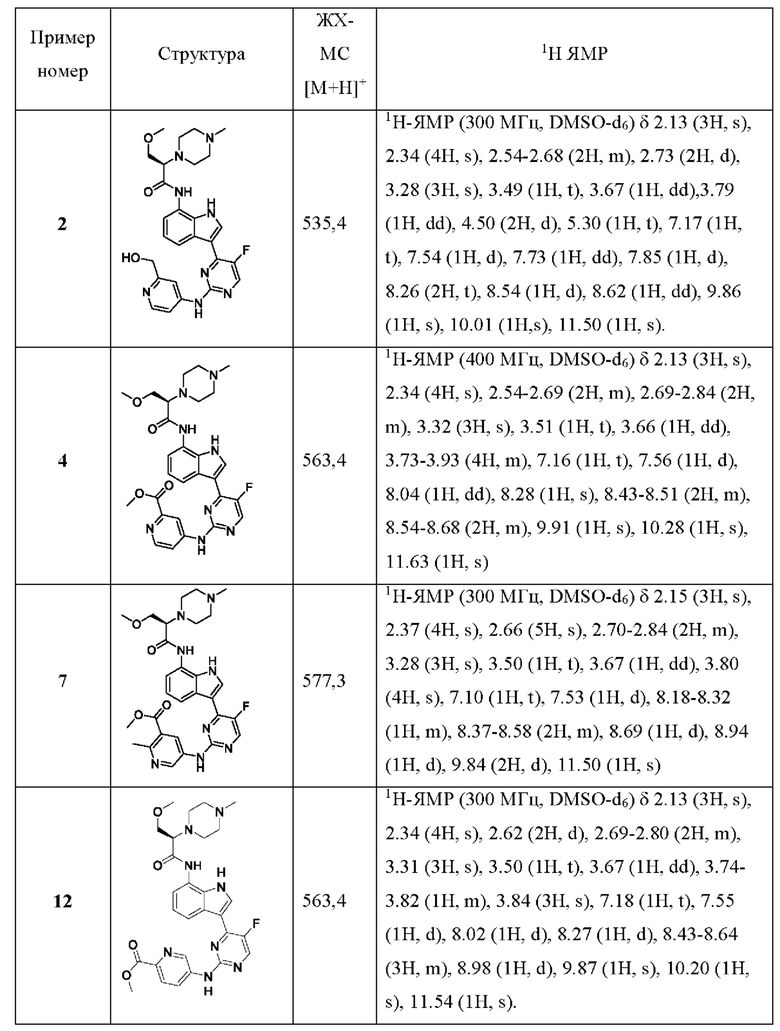
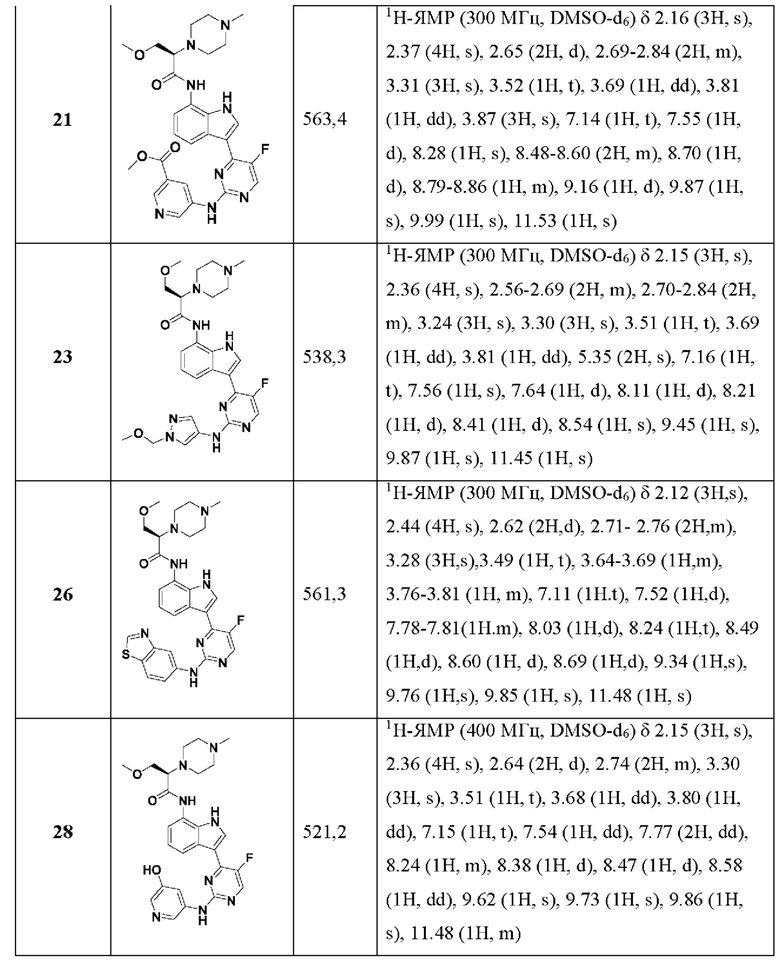

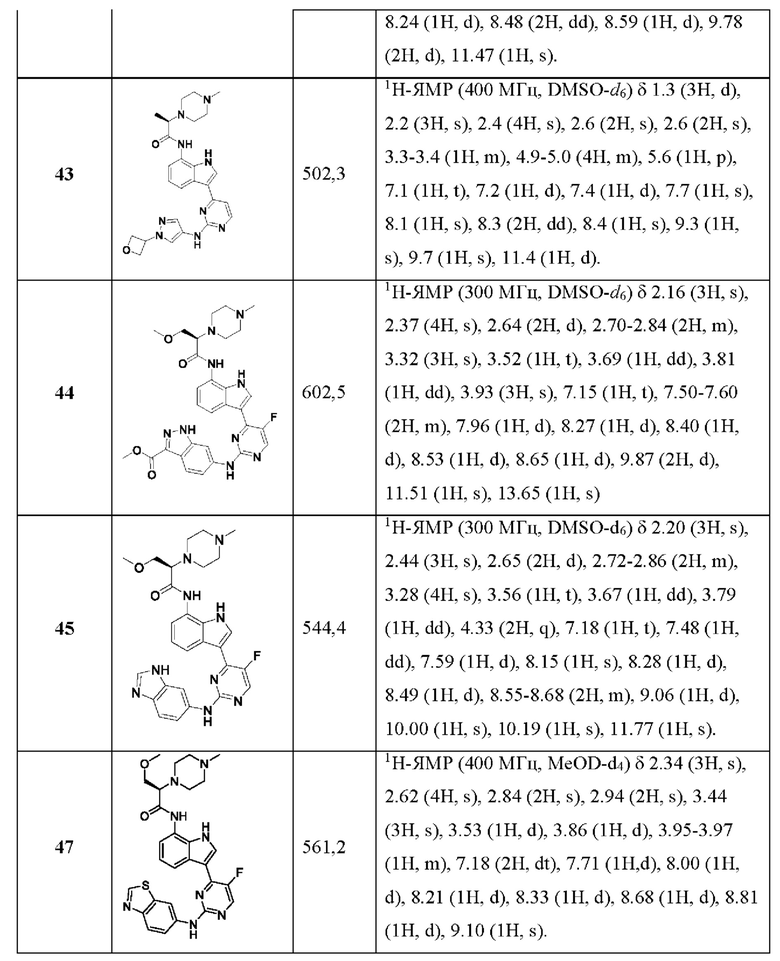
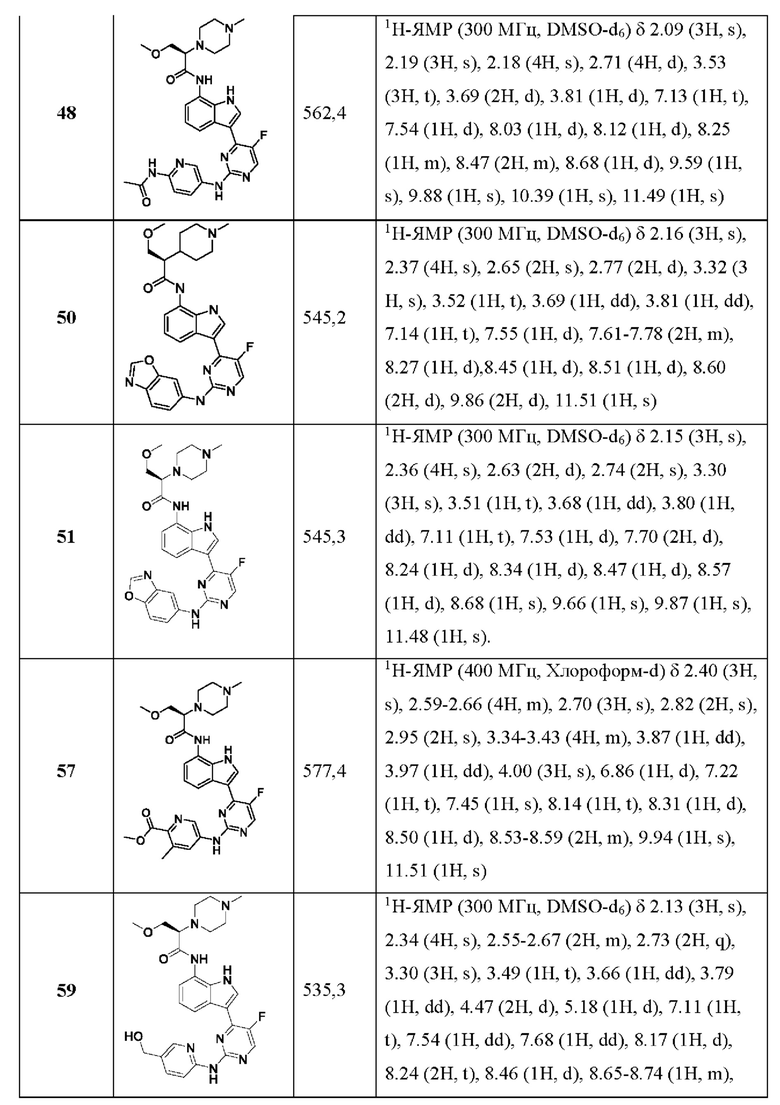
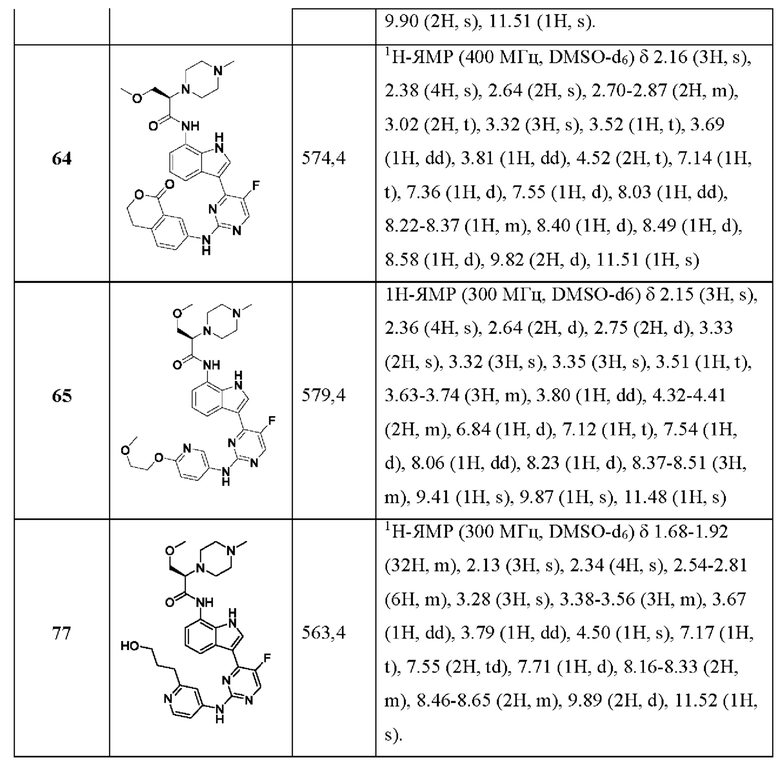
Пример 3
Получение (R)-N-(3-(5-фтор-2-((6-(гидроксиметил)-5-метилпиридин-3-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
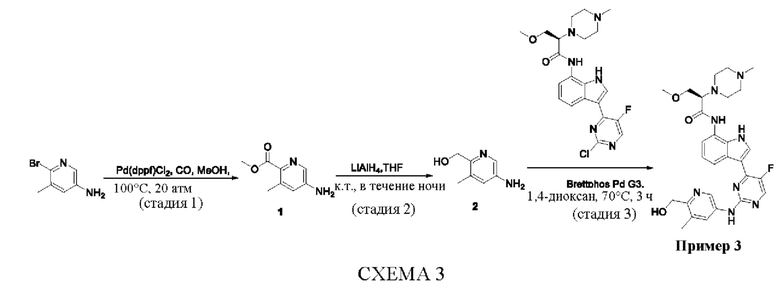
Стадия 1. Метил-5-амино-3-метилпиколинат
Смесь 6-бром-5-метилпиридин-3-амина (2000,00 мг, 10,693 ммоль, 1,00 экв.) и Pd(dppf)Cl2 (1564,80 мг, 2,139 ммоль, 0,20 экв.) в МеОН (20,00 мл) перемешивали в течение в течение ночи при 100°С в атмосфере моноксида углерода при 20 атм. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/MeOH 20:1) с получением метил-5-амино-3-метилпиридин-2-карбоксилата (280 мг, 15,76%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 167,3.
Стадия 2. (5-Амино-3-метилпиридин-2-ил)метанол
Смесь метил-5-амино-3-метилпиридин-2-карбоксилата (200,00 мг, 1,204 ммоль, 1,00 экв.) и LiAlH (137,03 мг, 3,610 ммоль, 3,00 экв.) в THF (20,00 мл) перемешивали в течение в течение ночи при комнатной температуре в воздушной атмосфере. Полученную смесь фильтровали, и осадок на фильтре промывали THF (2 × 5 мл). Фильтрат концентрировали под вакуумом. Неочищенный продукт напрямую использовали на следующей стадии без дополнительной очистки. ЖХ-МС (ИЭР): m/z [M+H]+ = 139,3.
Стадия 3. (R)-N-[3-(5-Фтор-2-[[6-(гидроксиметил)-5-метилпиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (пр. 3)
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (200,00 мг, 0,448 ммоль, 1,00 экв.) и (5-амино-3-метилпиридин-2-ил)метанола (92,75 мг, 0,671 ммоль, 1,50 экв.) в диоксане (20,00 мл) добавляли BrettPhos Pd G3 (81,13 мг, 0,090 ммоль, 0,20 экв.) и Cs2CO3 (437,43 мг, 1,343 ммоль, 3,00 экв.) порциями при 70°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт (50 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 18 В до 38 В за 7 мин; 254,220 нм; RT1: 6,80) с получением (R)-N-[3-(5-фтор-2-[[6-(гидроксиметил)-5-метилпиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (15 мг; 6,11%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 549,4. 1H-ЯМР (400 Гц, Метанол-d4) δ 2.33 (3Н, s), 2.43 (3Н, s), 2.60 (4H, s), 2.83 (2H, s), 2.92 (2H, s), 3.43 (3Н, s), 3.51 (1H, t), 3.85 (1H, dd), 3.94 (1H, dd), 4.72 (2H, s), 7.16-7.23 (2H, m), 8.17 (1H, d), 8.20-8.24 (1H, m), 8.29 (1H, d), 8.62 (2H, dd).
Пример 5
Получение (R-N-(3-[5-фтор-2-[(6-пропанамидопиридин-3-ил)амино]пиримидин-4-ил]-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида

Стадия 1. N-(5-Нитропиридин-2-ил)пропанамид
В сосуд вместимостью 40 мл добавляли 5-нитропиридин-2-амин (800,00 мг, 5,751 ммоль, 1,00 экв.), пропаноилхлорид (691,67 мг, 7,476 ммоль, 1,30 экв.), TEA (1454,78 мг, 14,377 ммоль, 2,5 экв.) и DCM (20,00 мл) при комнатной температуре. Затем смесь перемешивали при 0°С в атмосфере азота в течение 3 ч. Полученную смесь экстрагировали EtOAc (3 × 20 мл). Объединенные органические слои промывали рассолом (3 × 10 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении с получением N-(5-нитропиридин-2-ил)пропанамида (145 мг, 12,92%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 196,0.
Стадия 2. N-(5-Аминопиридин-2-ил)пропанамид
В сосуд вместимость 100 мл добавляли N-(5-нитропиридин-2-ил)пропанамид (100,00 мг, 0,512 ммоль, 1,00 экв.), Pd/C (5,45 мг, 0,051 ммоль, 0,10 экв.) и МеОН (15,00 мл) при комнатной температуре. Затем смесь перемешивали при 0°С в атмосфере Н2 в течение 3 ч. Полученную смесь фильтровали, и осадок на фильтре промывали DCM (3 × 20 мл). Фильтрат концентрировали при пониженном давлении с получением N-(5-аминопиридин-2-ил)пропанамида (35 мг, 41,35%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 166,2.
Стадия 3. (R)-N-(3-[5-Фтор-2-[(6-пропанамидопиридин-3-ил)амино]пиримидин-4-ил]-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 5)
В сосуд вместимостью 40 мл добавляли (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (180,00 мг, 0,403 ммоль, 1,00 экв.) и N-(5-аминопиридин-2-ил)пропанамид (99,80 мг, 0,604 ммоль, 1,50 экв.), BrettPhos Pd G3 (36,51 мг, 0,040 ммоль, 0,1 экв.), K2CO3 (111,33 мг, 0,806 ммоль, 2 экв.) и диоксан (20,00 мл) при комнатной температуре. Затем смесь перемешивали при 70°С в атмосфере азота в течение 3 ч. Полученную смесь экстрагировали EtOAc (3 × 20 мл). Объединенные органические слои промывали рассолом (3 × 10 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05%NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 25% В до 40% В за 7 мин; 254/220 нм; Rt: 5,77 мин) с получением (R)-N-(3-[5-фтор-2-[(6-пропанамидопиридин-3-ил)амино]пиримидин-4-ил]-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (30 мг, 12,94%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 576,4. 1H-ЯМР (300 МГц, DMSO-d6) δ 1.07 (3Н, t), 2.13 (3Н, s), 2.37 (6H, dd), 2.54-2.66 (2H, m), 2.73 (2H, q), 3.32 (3Н, s), 3.49 (1H, t), 3.66 (1H, dd), 3.79 (1H, dd), 7.11 (1H, t), 7.52 (1H, d), 7.97-8.17 (2H, m), 8.22 (1H, d), 8.45 (2,H dd), 8.66 (1H, d), 9.54 (1H, s), 9.85 (1H, s), 10.27 (1H, s), 11.47 (1H, s).
Пример 6
Получение метил-2-[4-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиразол-1-ил]бензоата
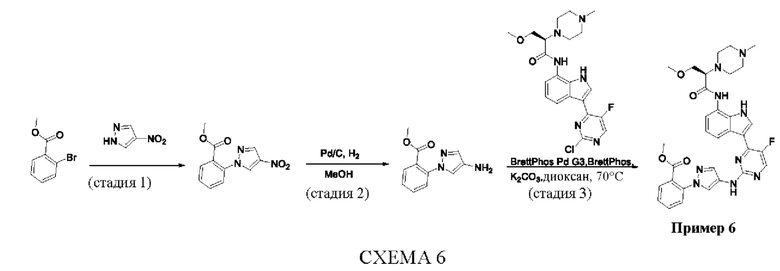
Стадия 1. Метил-2-(4-нитропиразол-1-ил)бензоат
В смесь метил-2-бромбензоата (7,61 г, 35,374 ммоль, 2,00 экв.) и 4-нитропиразола (2,00 г, 17,687 ммоль, 1,00 экв.) в диоксане (30,00 мл) добавляли Cs2CO3 (17288,54 мг, 53,062 ммоль, 3,00 экв.), (1S,2S)-N1,N2-диметилциклогексан-1,2-диамин (1509,56 мг, 10,612 ммоль, 0,60 экв.) и CuI (1347,41 мг, 7,075 ммоль, 0,40 экв.). После перемешивания в течение в течение ночи при 110°С в атмосфере азота полученную смесь фильтровали, и осадок на фильтре промывали DCM (3 × 20 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (3:1), с получением метил-2-(4-нитропиразол-1-ил)бензоата (410 мг, 9,38%) в виде белого твердого вещества. 1H ЯМР (300 МГц, CDCl3-d1) δ 3.79 (3Н, s), 7.50-7.53 (1H, m), 7.60-7.64 (1H, m), 7.67-7.73 (1H, m), 8.00-8.02 (1H, m), 8.26 (1H, s), 8.41-8.47 (1H, m).
Стадия 2. Метил-2-(4-аминопиразол-1-ил)бензоат
В круглодонную колбу на 50 мл добавляли метил-2-(4-нитропиразол-1-ил)бензоат (410,00 мг, 1,659 ммоль, 1,00 экв.) и Pd/C (353,00 мг, 3,317 ммоль, 2,00 экв.) в МеОН (25,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение 2 ч при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, и осадок на фильтре промывали МеОН (3 × 10 мл). Фильтрат концентрировали при пониженном давлении. Это привело к получению метил-2-(4-аминопиразол-1-ил)бензоата (360 мг, 79,3%) в виде черного масла. ЖХ-МС (ИЭР): m/z [M+H]+ = 218,2 1H ЯМР (400 МГц, CDCl3-d) δ 3.79 (3Н, s), 7.32-7.49 (4H, m), 7.53-7.57 (1H, m), 7.73-7.76 (1H, m).
Стадия 3. Метил-2-[4-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиразол-1-ил]бензоат (Пр. 6)
В смесь метил 2-(4-аминопиразол-1-ил)бензоата (94,78 мг, 0,436 ммоль, 1,5 экв.) и (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (130,00 мг, 0,291 ммоль, 1,00 экв.) в диоксане (10,00 мл) добавляли BrettPhos Pd G3 (26,37 мг, 0,029 ммоль, 0,10 экв.), BrettPhos (15,61 мг, 0,029 ммоль, 0,10 экв.) и Cs2CO3 (284,33 мг, 0,873 ммоль, 3,00 экв.). После перемешивания в течение 2 ч при 80°С в атмосфере азота полученную смесь концентрировали при пониженном давлении. Неочищенный продукт (50 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: CHIRALPAK IC-3, 4,6 х 50 мм, 3 мкм; подвижная фаза A: (Hex:DCM = 3:1)(0,1% DEA):EtOH = 50:50, скорость потока: 1,5 мл/мин) с получением метил-2-[4-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиразол-1-ил]бензоата (Пр. 6) (7 мг, 3,80%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 628,3 1h-ямр (300 МГц, DMSO-d6) δ 2.17 (3H, s), 2.38 (4H, s), 2.65 (4H, s), 2.75 (3H, s), 3.49-3.54 (1H, m), 3.67 (3H, s), 3.71 (1H, d), 3.78-3.84 (1H, m), 7.13 (1H, s), 7.42-7.58 (2H, m), 7.68 (3H, d), 7.83 (1H, s), 8.23 (1H, s), 8.39 (1H, s), 8.46 (1H, d), 8.47-8.48 (1H, m), 9.61 (1H, s), 9.87 (1H, s), 11.46 (1H, s).
Пример 8
Получение (R)-N-(3-(5-фтор-2-((6-(2-гидроксиацетамидо)пиридин-3-ил)амино)-пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида

Стадия 1. Получение 2-((5-нитропиридин-2-ил)амино)-2-оксоэтилацетата
В перемешиваемую смесь 5-нитропиридин-2-амина (500,00 мг, 3,594 ммоль, 1,00 экв.) и TEA (909,24 мг, 8,985 ммоль, 2,50 экв.) в DCM (20,00 мл) добавляли 2-хлор-2-оксоэтилацетат (736,07 мг, 5,391 ммоль, 1,50 экв.) по каплям при комнатной температуре в атмосфере азота. Полученную смесь фильтровали, и осадок на фильтре промывали DCM (3 × 10 мл). Фильтрат концентрировали при пониженном давлении. Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/MeOH=10:1) с получением [(5-нитропиридин-2-ил)карбамоил]метилацетата (300 мг, 34,90%) в виде коричнево-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 240,3.
Стадия 2. Получение 2-((5-аминопиридин-2-ил)амино)-2-оксоэтилацетата
В перемешиваемую смесь [(5-нитропиридин-2-ил)карбамоил]метилацетата (300,00 мг, 1,254 ммоль, 1,00 экв.) и Pd/C (26,70 мг, 0,251 ммоль, 0,20 экв.) в МеОН (20,00 мл) при комнатной температуре в атмосфере Н2. Полученную смесь фильтровали, и осадок на фильтре промывали МеОН (3 × 10 мл). Фильтрат концентрировали при пониженном давлении с получением [(5-аминопиридин-2-ил)карбамоил]метилацетата (250 мг, 95,28%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+H]+ = 210,3.
Стадия 3. Получение N-(5-аминопиридин-2-ил)-2-гидроксиацетамида
В перемешиваемую смесь [(5-аминопиридин-2-ил)карбамоил]метилацетата (250,00 мг, 1,195 ммоль, 1,00 экв.) и LiOH.H2O (250,73 мг, 5,975 ммоль, 5,00 экв.) в THF (18,00 мл) и воде (6,00 мл) порциями при комнатной температуре в атмосфере азота. Полученную смесь фильтровали, осадок на фильтре промывали DCM (3 × 20 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН=10:1) с получением N-(5-аминопиридин-2-ил)-2-гидроксиацетамида (100 мг, 35,13%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 168,1.
Стадия 4. Получение (R)-N-(3-(5-фтор-2-((5-гидрокси-6-(гидроксиметил)-пиридин-3-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 8)
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-гидрокси-2-(4-метилпиперазин-1-ил)пропанамида (150,00 мг, 0,347 ммоль, 1,00 экв.) и N-(5-аминопиридин-2-ил)-2-гидроксиацетамида (86,89 мг, 0,520 ммоль, 1,50 экв.) в диоксане (15,00 мл) добавляли BrettPhos Pd G3 (31,41 мг, 0,035 ммоль, 0,10 экв.), K2CO3 (95,78 мг, 0,693 ммоль, 2,00 экв.) и BrettPhos (37,20 мг, 0,069 ммоль, 0,20 экв.) порциями при 70°С в атмосфере азота. Полученную смесь фильтровали, и осадок на фильтре промывали DCM (3 × 20 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/MeOH=10:1) с получением неочищенного продукта (100 мг), который очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅Н2О), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 31% В до 45% В за 7 мин; 254/220 нм; Rt: 6,30 мин) с получением (R)-N-[3-(5-фтор-2-[[6-(2-гидроксиацетамидо)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (25,1 мг, 12,45%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 578.4. 1h-ямр (300 МГц, DMSO-d6) δ 2.16 (3H. s), 2.37 (4H, s). 2.64 (2H, d), 2.75 (2H, d), 3.30 (3H, s), 3.51 (1H, t), 3.69 (1H, dd), 3.81 (1H, dd), 4.05 (2H, d), 5.75 (1H, t), 7.14 (1H, t), 7.54 (1H, d), 8.07 (1H, d), 8.14-8.30 (2H, m), 8.40-8.57 (2H, m), 8.64-8.76 (1H, m), 9.56 (1H, s), 9.64 (1H, s), 9.87 (1H, s), 11.49 (1H, s).
Пример 9/29
Получение (R)-N-(3-(5-фтор-2-((6-(1-гидроксиэтил)пиридин-3-ил)амино)-пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 9 в виде изомера 2 и Пр. 29 в виде изомера 1)
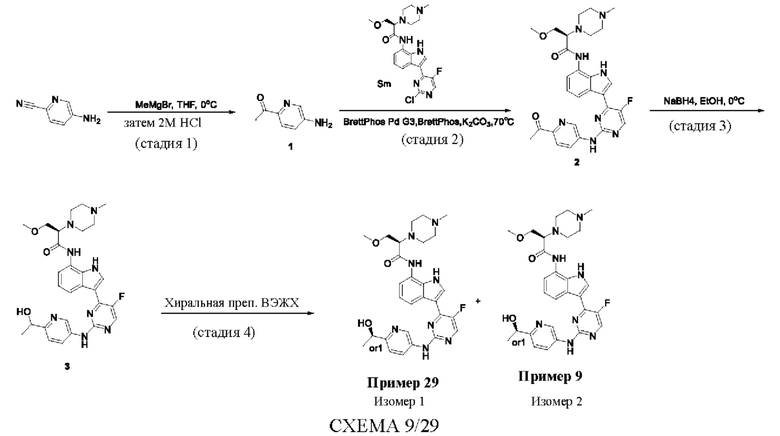
Стадия 1. 1-(5-Аминопиридин-2-ил)этан-1-он
В перемешиваемый раствор 5-аминопиридин-2-карбонитрила (800 мг, 6,716 ммоль, 1,00 экв.) в THF (35,00 мл) добавляли бром(метил)магний (7,83 мл, 23,490 ммоль, 3,50 экв.) по каплям при 0°С в атмосфере азота. Полученную смесь перемешивали в течение 2 ч при 0°С в атмосфере азота. Реакционную смесь гасили 2 М HCl (водн.) при 0°С. Полученную смесь перемешивали в течение 4 ч при комнатной температуре. Смесь подщелачивали до рН 8 насыщенным NaHCO3 (водн.). Полученную смесь экстрагировали EtOAc (3 × 20 мл). Объединенные органические слои промывали рассолом (1 х 50 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (1:1), с получением 1-(5-аминопиридин-2-ил)этанона (550 мг, 60,15%) в виде светло-коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 137.1. 1H-ЯМР (300 МГц, Хлороформ-d) δ 2.66 (3Н, s), 4.15 (2H, d), 7.01 (1H, dd), 7.93 (1H, d), 8.08 (1H, d).
Стадия 2. (R)-N-(3-(2-((6-Ацетилпиридин-3-ил)амино)-5-фторпиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид
Смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (220,00 мг, 0,492 ммоль, 1,00 экв.), BrettPhos Pd G3 (44,62 мг, 0,049 ммоль, 0,10 экв.), BrettPhos (26,42 мг, 0,049 ммоль, 0,10 экв.), K2CO3 (136,07 мг, 0,985 ммоль, 2,00 экв.) и 1-(5-аминопиридин-2-ил)этанона (100,54 мг, 0,738 ммоль, 1,50 экв.) в 1,4-диоксане (10,00 мл) перемешивали в течение 3 ч при 80°С в атмосфере азота. Полученную смесь фильтровали, и осадок на фильтре промывали CH2Cl2 (2 × 5 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/MeOH 8:1) с получением (R)-N-(3-[2-[(6-ацетилпиридин-3-ил)амино]-5-фторпиримидин-4-ил]-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (200 мг, 74,33%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 547,5.
Стадия 3. (R)-N-(3-(5-Фтор-2-((6-(1-гидроксиэтил)пиридин-3-ил)амино)-пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид
В перемешиваемый раствор (R)-N-(3-[2-[(6-ацетилпиридин-3-ил)амино]-5-фторпиримидин-4-ил]-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (200,00 мг, 0,366 ммоль, 1,00 экв.) в МеОН (10,00 мл) добавляли NaBH4 (41,53 мг, 1,098 ммоль, 3,00 экв.) порциями при 0°С в атмосфере азота. Полученную смесь перемешивали в течение 1 ч при 0°С в атмосфере азота. Реакционную смесь гасили добавлением смеси вода/лед. Полученную смесь экстрагировали CH2Cl2 (3 × 15 мл). Объединенные органические слои сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Неочищенный продукт (180 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 23 В до 43 В за 7 мин) с получением (R)-N-[3-(5-фтор-2-[[6-(1-гидроксиэтил)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (120 мг, 59,78%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 549,0.
Стадия 4. (R)-N-(3-(5-Фтор-2-((6-(1-гидроксиэтил)пиридин-3-ил)амино)-пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 29/9)
Неочищенный продукт (100 мг) очищали хиральной препаративной ВЭЖХ в нижеследующих условиях (колонка: CHIRALPAK IC, 2 × 25 см, 5 мкм; подвижная фаза A: Hex:DCM = 1:1(10 мМ NH3-МЕОН)-ВЭЖХ, подвижная фаза В: IPA--ВЭЖХ; скорость потока: 20 мл/мин; градиент: от 20 В до 20 В за 19 мин; 254/220 нм; RT1: 14,362; RT2: 16,774; впрыскиваемый объем: 0,3 мл; число прогонов: 10) с получением (R)-N-[3-[5-фтор-2-([6-[(1R)-1-гидроксиэтил]пиридин-3-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 29) (изомер 1, 40 мг, 40,00%) в виде белого твердого вещества ЖХ-МС m/z (ИЭР), [М+Н]+ = 549,4. 1H-ЯМР (400 МГц, DMSO-d6) δ 1.38 (3Н, d), 2.15 (3H, s), 2.36 (4H, s), 2.63 (2H, s), 2.68-2.84 (2H, m), 3.30 (3H, s), 3.51 (1H, t), 3.68 (1H, dd), 3.80 (1H, dd), 4.60-4.78 (1H, m), 5.23 (1H, d), 7.14 (1H, t), 7.44 (1H, d), 7.54 (1H, d), 8.09-8.29 (2H, m), 8.45 (1H, d), 8.53 (1H, d), 8.78 (1H, s), 9.63 (1H, s), 9.86 (1H, s), 11.48 (1H, s).
(R)-N-[3-[5-Фтор-2-([6-[1-гидроксиэтил]пиридин-3-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 9) (изомер 2, 45 мг, 44,55%) в виде белого твердого вещества, ЖХ-МС (ИЭР): m/z [M+H]+ = 549,4. 1H-ЯМР (400 МГц, DMSO-d6) δ 1.38 (3Н, d), 2.15 (3Н, s), 2.36 (4H, s), 2.63 (2H, s), 2.68-2.84 (2H, m), 3.30 (3Н, s), 3.51 (1H, t), 3.68 (1H, dd), 3.80 (1H, dd), 4.60-4.78 (1H, m), 5.23 (1H, d), 7.14 (1H, t), 7.44 (1H, d), 7.54 (1H, d), 8.09-8.29 (2H, m), 8.45 (1H, d), 8.53 (1H, d), 8.78 (1H, s), 9.63 (1H, s), 9.86 (1H, s), 11.48 (1H, s).
Пример 13
Получение (R)-N-[3-[5-фтор-2-(1Н-индазол-6-иламино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
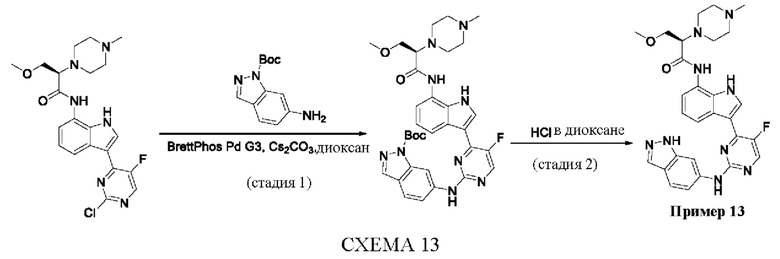
Стадия 1. трет-Бутил-6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]индазол-1-карбоксилат
Раствор (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (150,00 мг, 0,336 ммоль, 1,00 экв.) и трет-бутил-6-аминоиндазол-1-карбоксилата (117,44 мг, 0,503 ммоль, 1,50 экв.), BrettPhos Pd G3 (30,43 мг, 0,034 ммоль, 0,10 экв.), BrettPhos (18,02 мг, 0,034 ммоль, 0,10 экв.), Cs2CO3 (218,72 мг, 0,671 ммоль, 2,00 экв.) в диоксане (5,00 мл) перемешивали в течение 2 ч при 100°С в атмосфере азота. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью CHCl3/МеОН (12:1), с получением трет-бутил-6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]индазол-1-карбоксилата (120 мг, 55,54%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 644,6
Стадия 2. (R)-N-[3-[5-Фтор-2-(1Н-индазол-6-иламино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 13)
В перемешиваемый раствор трет-бутил-6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]индазол-1-карбоксилата (110,00 мг, 0,171 ммоль, 1,00 экв.) в HCl (газ) в 1,4-диоксане (10 мл) при комнатной температуре в атмосфере азота. Полученную смесь концентрировали под вакуумом. Неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 19 х 250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 25 мл/мин; градиент: от 31 В до 40 В за 10 мин; 254,220 нм; RT1: 9,87) с получением (R)-N-[3-[5-фтор-2-(1Н-индазол-6-иламино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (30 мг, 32,30%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 544,3. 1H-ЯМР (400 МГц, DMSO-d6) δ 2.15 (3Н, s), 2.36 (4H, s), 2.64 (2Н, d), 2.75 (2Н, q), 3.30 (3H, s), 3.51 (1H, t), 3.68 (1H, dd), 3.80 (1H, dd), 7.13 (1H, t), 7.37 (1H, dd), 7.54 (1H, dd), 7.64 (1H, d), 7.94 (1H, d), 8.25 (2H, dd), 8.49 (1H, d), 8.64 (1H, m), 9.67 (1H, s), 9.87 (1H, s), 11.47 (1H, m), 12.78 (1H, s).
Пример 14
Получение метил-2-([5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]окси)ацетата
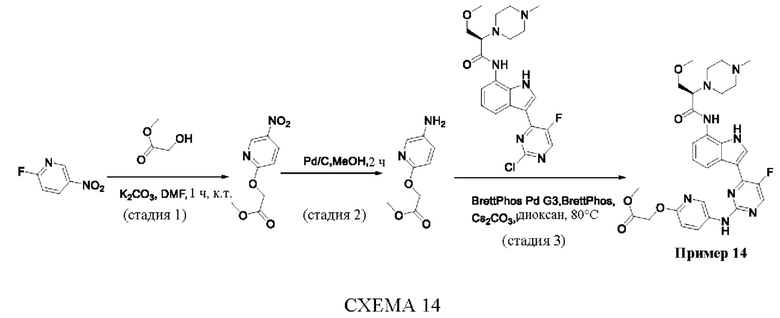
Стадия 1. Метил-2-[(5-нитропиридин-2-ил)окси]ацетат
В перемешиваемый раствор 2-фтор-5-нитропиридина (300,00 мг, 2,11 ммоль, 1,00 экв.) и метил-2-гидроксиацетата (380,4 мг, 4,22 ммоль, 2,00 экв.) в DMF (20,00 мл) добавляли K2CO3 (583,6 мг, 4,22 ммоль, 2,00 экв.) порциями при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 1 ч при комнатной температуре в атмосфере N2. Полученную смесь разбавляли водой (100 мл) и экстрагировали EtOAc (3 × 100 мл). Объединенные органические слои промывали рассолом (1 × 30 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (РЕ/EtOAc 3:1) с получением метил-2-[(5-нитропиридин-2-ил)окси]ацетата (200 мг, 26,79%) в виде светло-коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 213,2.
Стадия 2. Метил-2-[(5-аминопиридин-2-ил)окси]ацетат
Смесь метил-2-[(5-нитропиридин-2-ил)окси]ацетата (200,00 мг, 1 экв.) и Pd/C (30,00 мг) в МеОН (20,00 мл) перемешивали в течение 2 ч при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Это привело к получению метил-2-[(5-аминопиридин-2-ил)окси] ацетата (150 мг, 87,34%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 1833.
Стадия 3. Метил-2-([5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]окси)ацетат (пр. 14)
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (140,00 мг, 0,313 ммоль, 1,00 экв.) и метил-2-[(5-аминопиридин-2-ил)окси]ацетата (114,14 мг, 0,627 ммоль, 2 экв.) в диоксане (20,00 мл) добавляли BrettPhos Pd G3 (42,60 мг, 0,047 ммоль, 0,15 экв.) и BrettPhos (25,22 мг, 0,047 ммоль, 0,15 экв.), Cs2CO3 (21,87 мг, 0,067 ммоль, 3,00 экв.) при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 ч при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт (30 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 38 В до 48 В за 7 мин; 254;220 нм; RT1: 5,93) с получением метил-2-([5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]окси)ацетата (21 мг, 11,31%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 593,3. 1H-ЯМР (300 МГц, MeOD-d4) δ 2.34 (3Н, s), 2.62 (4H, s), 2.84 (2H, s), 2.93 (2H, s), 3.43 (3H, s), 3.52 (1H, t), 3.78 (3H, s), 3.85 (1H, d), 3.94 (1H, d), 4.93 (2H, s), 6.91 (1H, d), 7.17 (2H, m), 8.07 (1H, d), 8.17 (1H, d), 8.23 (1H, d), 8.38 (1H, m), 8.53 (1H, d).
Пример 15
Получение метил-(R)-3-(4-((5-фтор-4-(7-(3-метокси-2-(4-метилпиперазин-1-ил)-пропанамидо)-1Н-индол-3-ил)пиримидин-2-ил)амино)пиридин-2-ил)пропаноата
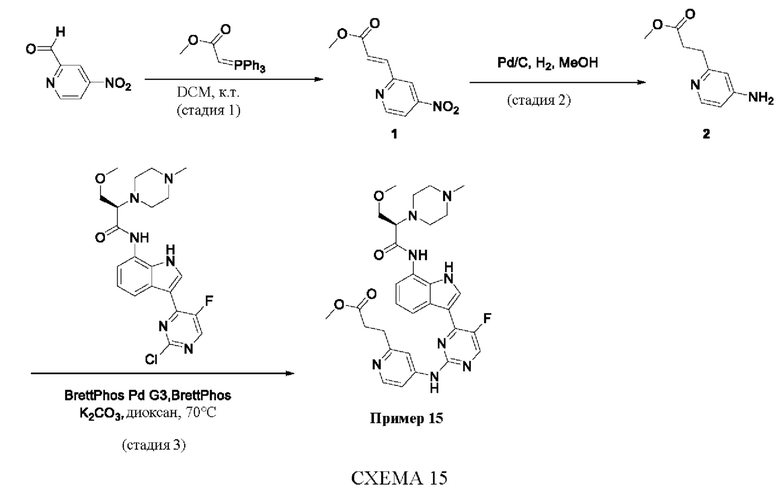
Стадия 1. Метил-3-(4-нитропиридин-2-ил)акрилат
В перемешиваемый раствор 4-нитропиридин-2-карбальдегида (0,50 г, 3,287 ммоль, 1,00 экв.) и метил-2-(трифенил-лямбда5-фосфанилиден)ацетата (1,65 г, 4,935 ммоль, 1,50 экв.) в DCM (10,00 мл) при комнатной температуре в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/MeOH 15:1) с получением метил-(3-(4-нитропиридин-2-ил)проп-2-еноата (450 мг, 65,76%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 209,2.
Стадия 2. Получение метил-3-(4-аминопиридин-2-ил)пропаноата
Смесь метил-3-(4-нитропиридин-2-ил)проп-2-еноата (200,00 мг, 0,961 ммоль, 1,00 экв.) и Pd/C (20,45 мг, 0,192 ммоль, 0,20 экв.) в МеОН (15,00 мл) перемешивали при комнатной температуре в атмосфере Н2 в течение 1 ч. Полученную смесь фильтровали, и осадок на фильтре промывали МеОН (3 × 10 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1) с получением метил-3-(4-аминопиридин-2-ил)пропаноата (100 мг, 57,76%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [M+H]+ = 181,2.
Стадия 3. Метил-(R)-3-(4-((5-фтор-4-(7-(3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо)-1Н-индол-3-ил)пиримидин-2-ил)амино)пиридин-2-ил)пропаноат (Пр. 15)
В смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (130,00 мг, 0,291 ммоль, 1,00 экв.) и метил-3-(4-аминопиридин-2-ил)пропаноата (78,63 мг, 0,436 ммоль, 1,50 экв.) в диоксане (5,00 мл) добавляли BrettPhos Pd G3 (26,37 мг, 0,029 ммоль, 0,10 экв.), BrettPhos (31,23 мг, 0,058 ммоль, 0,20 экв.) и K2CO3 (80,40 мг, 0,582 ммоль, 2,00 экв.) при к.т. в атмосфере азота. Полученную смесь перемешивали при 70°С в течение 2 ч в атмосфере N2. Полученную смесь фильтровали, и осадок на фильтре промывали DCM (3 × 20 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/MeOH 10:1) с получением неочищенного продукта (100 мг), который очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 х 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 31% В до 45% В за 7 мин; 254;220 нм; Rt: 6,30 мин) с получением метил-3-[4-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метил-пиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]пропаноата (40,8 мг, 23,13%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 591,4 1h-ямр (300 МГц, DMSO-d6) 2.16 (3Н, s), 2.38 (4H, s), 2.65 (2H, d), 2.76 (4H, t), 2.96 (2H, t), 3.32 (3Н, d), 3.53 (1H, d), 3.61(3H, s), 3.69 (1H, dd), 3.81 (1H, dd), 7.20 (1H, t), 7.57 (2H, dd), 7.79 (1H, d), 8.23-8.31 (2H, m), 8.53-8.63 (2H, m), 9.89 (1H, s), 9.98 (1H, s), 11.55 (1H, s).
Пример 16
Получение (R)-N-[3-(5-фтор-2-[[6-(2-гидроксиэтокси)пиридин-3-ил]амино]-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
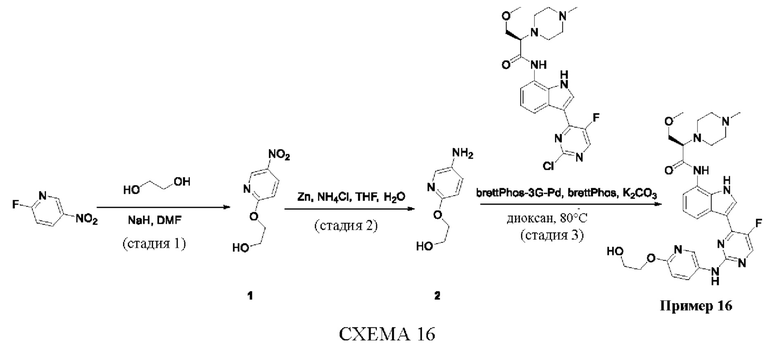
Стадия 1. 2-[(5-Нитропиридин-2-ил)окси]этанол
Смесь 2-фтор-5-нитропиридина (1,50 г, 10,557 ммоль, 1,00 экв.), этилен гликоля (0,98 г, 15,835 ммоль, 1,50 экв.) и NaH (0,63 г, 15,730 ммоль, 1,49 экв., 60%) в DMF (20,00 мл, 258,435 ммоль, 24,48 экв.) перемешивали в течение 2 ч при 0°С в атмосфере азота. Полученную смесь разбавляли H2O (100 мл) и экстрагировали ЕА (3 × 100 мл), и объединенные органические слои промывали рассолом (2 × 20 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (PE/EtOAc 1:1) с получением 2-[(5-нитропиридин-2-ил)окси]этанола (1,78 г, 91,56%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=185,2. 1H-ЯМР (300 МГц, DMSO-d6) 5 3.75 (2Н, q), 4.31-4.51 (2Н, m), 4.92 (1H, t), 7.04 (1H, dd), 8.48 (1H, dd), 9.08 (1H, d).
Стадия 2. 2-[(5-Аминопиридин-2-ил)окси]этанол
Смесь 2-[(5-нитропиридин-2-ил)окси]этанола (200,00 мг, 1,086 ммоль, 1,00 экв.), Zn (710,38 мг, 10,861 ммоль, 10,00 экв.) и NH4Cl (580,95 мг, 10,861 ммоль, 10,00 экв.) в THF (4,00 мл) и H2O (2,00 мл) перемешивали в течение 4 ч при комнатной температуре в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (5 мл). Фильтрат концентрировали при пониженном давлении. Это привело к получению 2-[(5-аминопиридин-2-ил)окси]этанола (150 мг, 89,59%) в виде желтого масла. ЖХ-МС (ИЭР): m/z [М+Н]+=155,2.
Стадия 3. (R)-N-[3-(5-Фтор-2-[[6-(2-гидроксиэтокси)пиридин-3-ил]амино]-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 16)
Смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (150,00 мг, 0,336 ммоль, 1,00 экв.), 2-[(5-аминопиридин-2-ил)окси]этанола (62,09 мг, 0,403 ммоль, 1,20 экв.), BrettPhos Pd G3 (60,85 мг, 0,067 ммоль, 0,20 экв.), BrettPhos (36,03 мг, 0,067 ммоль, 0,20 экв.) и K2СО3 (115,97 мг, 0,839 ммоль, 2,50 экв.) в диоксане (3,00 мл) перемешивали в течение ночи при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1). Неочищенный продукт (200 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 21 В до 41 В за 7 мин; 254; 220 нм; RT1: 6,98) с получением (R)-N-[3-(5-фтор-2-[[6-(2-гидроксиэтокси)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (110 мг, 58,04%) в виде белого твердого вещества. Этот неочищенный продукт ((R)-N-[3-(5-фтор-2-[[6-(2-гидроксиэтокси)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (110,00 мг)) очищали препаративной хиральной ВЭЖХ в нижеследующих условиях (колонка: CHIRAL ART Cellulose-SB, 4,6 × 100 мм, 3 мкм; подвижная фаза A: Hex (0,1% DEA):EtOH=50:50, подвижная фаза В; скорость потока: 1 мл/мин; градиент: от 0 В до 0 В) с получением (R)-N-[3-(5-фтор-2-[[6-(2-гидроксиэтокси)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метил-пиперазин-1-ил)пропанамида (53,07 мг, 48,25%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=565,4. 1H-ЯМР (300 МГц, DMSO-d6) δ 2.16 (3Н, s), 2.37 (4Н, s), 2.54-2.66 (2Н, m), 2.75 (2Н, q), 3.32 (3Н, s), 3.51 (1H, t), 3.65-3.90 (4Н, m), 4.26 (2Н, dd), 4.83 (1Н, t), 6.82 (1H, d), 7.12 (1H, t), 7.53 (1H, dd), 8.05 (1Н, dd), 8.17-8.31 (1H, m), 8.33-8.55 (3Н, m), 9.40 (1H, s), 9.86 (1H, s), 11.47 (1H, s).
Пример 17.
Получение (R)-N-(3-(5-фтор-2-((3-метил-1Н-индазол-6-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
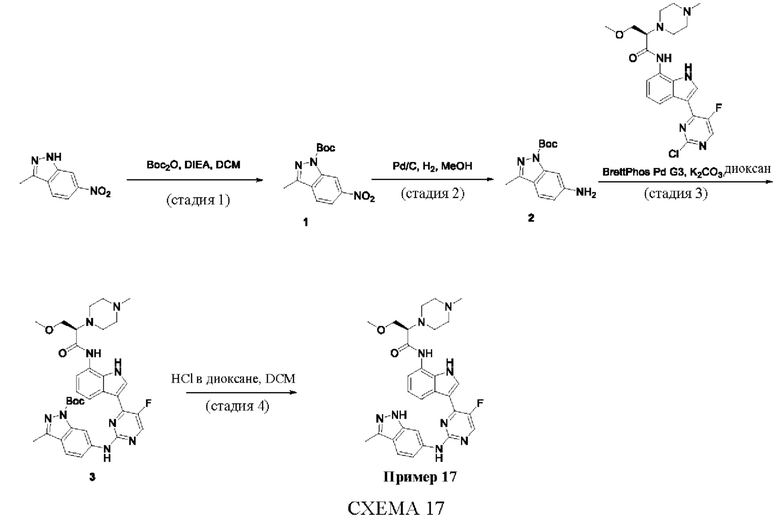
Стадия 1. трет-Бутил-3-метил-6-нитро-1Н-индазол-1-карбоксилат
Раствор 3-метил-6-нитро-1H-индазола (500,00 мг, 2,822 ммоль, 1,00 экв.) и Вос20 (923,92 мг, 4,233 ммоль, 1,50 экв.), DIEA (729,52 мг, 5,645 ммоль, 2,00 экв.) в DCM (10,00 мл) перемешивали в течение в течение ночи при комнатной температуре в атмосфере азота. Полученную смесь гасили водой (10 мл) и экстрагировали CH3Cl (20 мл × 3). Объединенные органические слои промывали рассолом (10 мл × 3) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (5:1), с получением трет-бутил-3-метил-6-нитроиндазол-1-карбоксилата (550 мг, 70.28%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=278,3.
Стадия 2. трет-Бутил-6-амино-3-метилиндазол-1-карбоксилат
Раствор трет-бутил-3-метил-6-нитроиндазол-1-карбоксилата (540,00 мг, 1,947 ммоль, 1,00 экв.) и Pd/C (20,73 мг, 0,195 ммоль, 0,10 экв.) в МеОН (10,00 мл) перемешивали в течение 3 ч при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (10 мл × 3). Фильтрат концентрировали при пониженном давлении с получением трет-бутил-6-амино-3-метилиндазол-1-карбоксилата (400 мг, 83,05%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=248,1
Стадия 3. трет-Бутил-6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил) пропанамидо]-1Н-индол-3-ил1пиримидин-2-ил)амино]-3-метилиндазол-1-карбоксилат
Раствор трет-бутил-6-амино-3-метилиндазол-1-карбоксилата (124,50 мг, 0,503 ммоль, 1,50 экв.) и (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (150,00 мг, 0,336 ммоль, 1,00 экв.), BrettPhos Pd G3 (30,43 мг, 0,034 ммоль, 0,10 экв.), K2СО3 (92,77 мг, 0,671 ммоль, 2,00 экв.) в диоксане (4,00 мл) перемешивали в течение 2 ч при 70°С в атмосфере азота. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью CH2Cl2/МеОН (7:1), с получением трет-бутил-6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]-3-метилиндазол-1-карбоксилата (140 мг, 63,42%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=658,6.
Стадия 4. (R)-N-(3-[5-Фтор-2-[(3-метил-1Н-индазол-6-ил)амино]пиримидин-4-ил]-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 17)
Раствор трет-бутил-6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]-3-метилиндазол-1-карбоксилата (140,00 мг, 0,213 ммоль, 1,00 экв.) и HCl (газ) в 1,4-диоксане (2,00 мл) в DCM (2,00 мл) перемешивали в течение 3 ч при комнатной температуре в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 30 В до 50 В за 7 мин; 254, 220 нм; RT1: 6,63) с получением (R)-N-(3-[5-фтор-2-[(3-метил-1Н-индазол-6-ил)амино]пиримидин-4-ил]-1Н-индол-7-ил)-3-метокси-2-(4-метил-пиперазин-1-ил)пропанамида (90 мг, 75,83%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=558,3 1H-ЯМР (300 МГц, DMSO-d6) δ 2.14 (3Н, s), 2.35 (4Н, s), 2.44 (3H, s), 2.62 (2Н, m), 2.74 (2Н, m), 3.28 (3H, s), 3.50 (1H, t), 3.67 (1H, dd), 3.79 (1Н, dd), 7.12 (1H, t), 7.32 (1H, dd), 7.54 (2Н, m), 8.14 (1Н, d), 8.23 (1Н, m), 8.47 (1H, d), 8.62 (1H, dd), 9.63 (1H, s), 9.86 (1H, s), 11.47 (1H, s), 12.33 (1H, s).
Пример 18
Получение (R)-N-[3-(5-фтор-2-[[1-(оксан-4-ил)пиразол-4-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
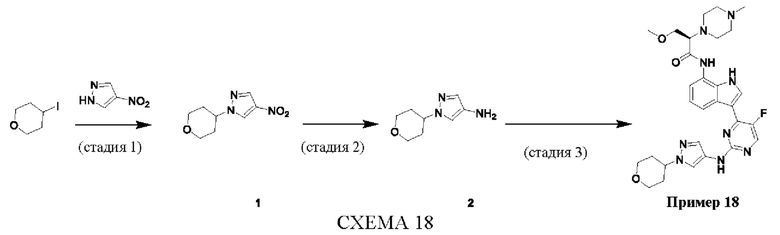
Стадия 1. 4-Нитро-1-(оксан-4-ил)пиразол
В перемешиваемую смесь 4-йодоксана (2,06 г, 9,728 ммоль, 1,10 экв.) и 4-нитропиразола (1,00 г, 8,844 ммоль, 1,00 экв.) в DMF (13,33 мл, 182,397 ммоль, 19,48 экв.) добавляли Cs2CO3 (8,64 г, 26,531 ммоль, 3,00 экв.) при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 суток при 80°С в воздушной атмосфере. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 20:1) с получением неочищенного твердого вещества. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью CH2Cl2/МеОН (20:1), с получением 4-нитро-1-(оксан-4-ил)пиразола (343 мг, 19,28%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ 1.98-2.01 (4Н, m), 3.43-3.49 (2Н, m), 3.95-3.99 (2Н, m), 4.48-4.56 (1H, m), 8.29 (1Н, s), 8.96 (1H, s).
Стадия 2. 1-(Оксан-4-ил)пиразол-4-амин
В круглодонную колбу на 100 мл добавляли 4-нитро-1-(оксан-4-ил)пиразол (315,00 мг, 1,597 ммоль, 1,00 экв.) и Pd/C (3399,93 мг, 31,948 ммоль, 20,00 экв.) в МеОН (20,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение в течение ночи при 120°С в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (3 × 10 мл). Фильтрат концентрировали при пониженном давлении. Это привело к получению 1-(оксан-4-ил)пиразол-4-амина (200 мг, 67,39%) в виде красного твердого вещества. ЖХ-МС (ПЭР): m/z [М+Н]+=168,2. 1Н ЯМР (300 МГц, DMSO-d6) δ 1.71-1.92 (4Н, m), 3.22-3.53 (2Н, m), 3.75 (2Н, s), 3.87-3.89 (1H, m), 3.91-3.97 (1H, m), 4.11-4.18 (1H, m), 6.89 (1H, d), 7.05 (1H, d).
Стадия 3. (R)-N-[3-(5-Фтор-2-[[1-(оксан-4-ил)пиразол-4-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил1-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 18)
В раствор 1-(оксан-4-ил)пиразол-4-амина (101,02 мг, 0,604 ммоль, 1,50 экв.) и (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (180,00 мг, 0,403 ммоль, 1,00 экв.) в диоксане (5 мл) добавляли BrettPhos (6,01 мг, 0,011 ммоль, 0,10 экв.), Cs2CO3(393,69 мг, 1,208 ммоль, 3,00 экв.) и BrettPhos Pd G3 (36,51 мг, 0,040 ммоль, 0,10 экв.). После перемешивания в течение 2 ч при 80°С в атмосфере азота полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 7:1). Неочищенный продукт (105 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 31% В до 43% В за 7 мин; 254; 220 нм; Rt: 6,75 мин). Неочищенный продукт (80 мг) очищали препаративной хиральной ВЭЖХ в нижеследующих условиях (колонка: CHIRAL ART Cellulose-SB, 4,6 × 100 мм, 3 мкм; подвижная фаза A: MtBE (0,l%DEA):EtOH=95:5, подвижная фаза В; скорость потока: 1 мл/мин; градиент: от 0 В до 0 В) с получением (R)-N-[3-(5-фтор-2-[[1-(оксан-4-ил)пиразол-4-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (37 мг, 15,74%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=578,4. 1H-ЯМР (400 МГц, DMSO-d6) δ 1.65-1.83 (4Н, m), 1.95 (3H, s), 2.16 (4Н, s), 2.40-2.47 (2Н, m), 2.52-2.59 (2Н, m), 3.21-3.35 (3H, m), 3.32 (3H, s), 3.47-3.51 (1Н, m), 3.58-3.62 (1H, m), 3.77 (2H, d), 4.14-4.19 (1H, m), 6.92-6.95 (1H, m), 7.33-7.35 (2H, m), 7.80 (1H, s), 7.97-8.02 (1H, m), 8.18 (1H, d), 8.29 (1H, s), 9.11 (1H, s), 9.66 (1H, s), 11.23 (1H, s).
Пример 19
Получение (R)-N-(3-(2-((1Н-пиразоло[4,3-b]пиридин-6-ил)амино)-5-фторпиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
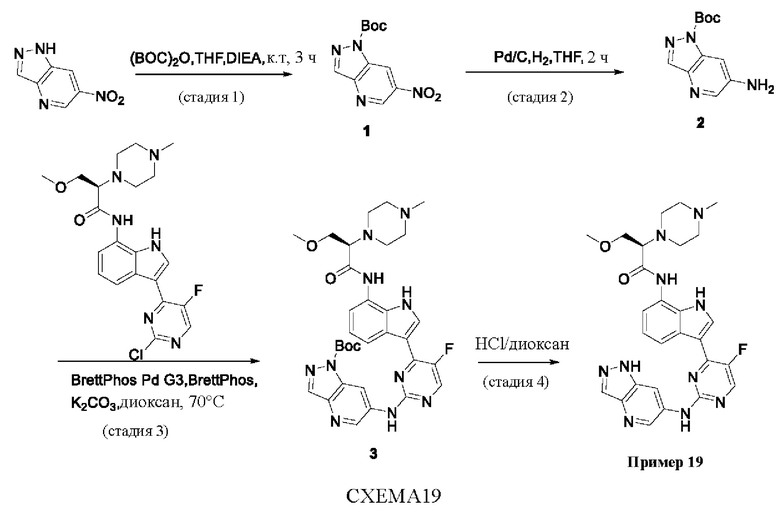
Стадия 1. трет-Бутил-6-нитро-1H-пиразоло[4,3-b]пиридин-1-карбоксилат
В перемешиваемую смесь 6-нитро-1Н-пиразоло[4,3-b]пиридина (300,00 мг, 1,828 ммоль, 1,00 экв.) и (Вос)2O (598,40 мг, 2,742 ммоль, 1,50 экв.) в THF (40,00 мл) добавляли DIEA (708,73 мг, 5,484 ммоль, 3,00 экв.) порциями при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 3 ч при комнатной температуре в воздушной атмосфере. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (РЕ/EtOAc 2:1) с получением трет-бутил-6-нитропиразоло[4,3-b]пиридин-1-карбоксилата (310 мг,64.18%) в виде желтого твердого вещества. ЖХ-МС (ПЭР): m/z [М+Н]+=265,0
Стадия 2. трет-Бутил-6-амино-1Н-пиразоло[4,3-b]пиридин-1-карбоксилат
Смесь трет-бутил-6-нитропиразоло[4,3-b]пиридин-1-карбоксилата (290,00 мг, 1,097 ммоль, 1,00 экв.) и Pd/C (23,36 мг, 0,219 ммоль, 0,20 экв.) в THF (30,00 мл) перемешивали в течение в течение ночи при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, и осадок на фильтре промывали МеОН (3 × 10 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН=12:1) с получением трет-бутил-6-аминопиразоло[4,3-b]пиридин-1-карбоксилата (200 мг, 77,79%) в виде желтого твердого вещества ЖХ-МС (ПЭР): m/z [М+Н]+=235,1.
Стадия 3. трет-Бутил-6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиразоло[4,3-b]пиридин-1-карбоксилат
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (200,00 мг, 0,448 ммоль, 1,00 экв.) и трет-бутил-6-аминопиразоло[4,3-b]пиридин-1-карбоксилата (157,25 мг, 0,671 ммоль, 1,50 экв.) в диоксане (30,00 мл) добавляли Brettphos Pd G3 (81,13 мг, 0,090 ммоль, 0,20 экв.) и K2СО3 (123,70 мг, 0,895 ммоль, 2,00 экв.) порциями при 70°С в атмосфере азота. Полученную смесь перемешивали в течение 2 ч при 70°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН=10:1) с получением трет-бутил-6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиразоло[4,3-b]пиридин-1-карбоксилата (150 мг, 51,99%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=645,3.
Стадия 4. (R)-N-[3-(5-Фтор-2-[1Н-пиразоло[4,3-b]пиридин-6-иламино]-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 19)
Смесь трет-бутил-6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиразоло[4,3-b]пиридин-1-карбоксилата (130,00 мг, 0,202 ммоль, 1,00 экв.) и HCl (газ) в 1,4-диоксане (7,35 мг, 0,202 ммоль, 1,00 экв.) в DCM (20,00 мл) перемешивали в течение 3 ч при комнатной температуре в воздушной атмосфере. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт (80 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 22 В до 42 В за 7 мин; 254/220 нм; RT1: 8,52) с получением (R)-N-[3-(5-фтор-2-[1Н-пиразоло[4,3-b]пиридин-6-иламино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (30 мг, 27,32%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=545,4. 1H ЯМР (400 МГц, DMSO-d6) δ 2.15 (3H, s), 2.36 (4Н, s), 2.63 (2Н, s), 2.75 (2Н, d), 3.32 (3H, s), 3.52 (1H, t), 3.68 (1H, dd), 3.80 (1H, dd), 7.15 (1H, t), 7.55 (1H, d), 8.16 (1H, s), 8.27 (1H, d), 8.54 (1H, d), 8.56-8.66 (2Н, m), 8.72 (1H, d), 9.92 (2Н, d), 11.54 (1H, s), 13.01 (1H, s)
Пример 20
Получение (R)-N-(3-(5-фтор-2-((6-(2-(метиламино)этокси)пиридин-3-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
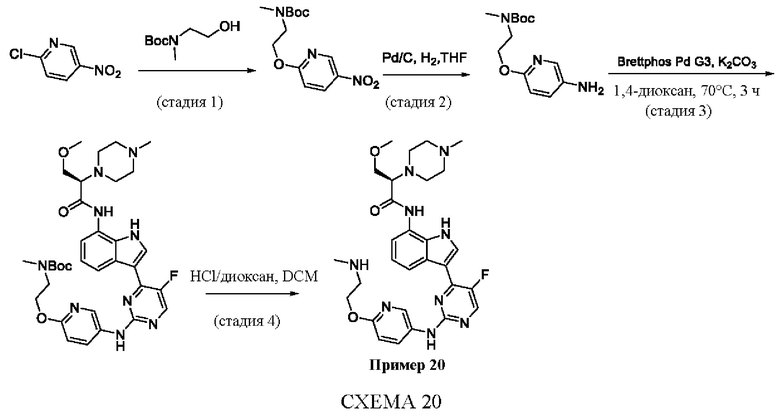
Стадия 1. трет-Бутил-N-метил-N-[2-[(5-нитропиридин-2-ил)окси]этил]карбамат
В перемешиваемую смесь 2-хлор-5-нитропиридина (200,00 мг, 1,262 ммоль, 1,00 экв.) и трет-бутил-N-(2-гидроксиэтил)-N-метилкарбамата (331,58 мг, 1,892 ммоль, 1,50 экв.) в DMF (20,00 мл) добавляли NaH (30,27 мг, 1,262 ммоль, 1,00 экв.) порциями при комнатной температуре в воздушной атмосфере. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (РЕ/EtOAc=1:1) с получением трет-бутил-N-метил-N-[2-[(5-нитропиридин-2-ил)окси]этил]карбамата (300 мг, 79,99%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=298,1.
Стадия 2. трет-Бутил-N-[2-[(5-аминопиридин-2-ил)окси]этил]-N-метилкарбамат
Смесь трет-бутил-N-метил-N-[2-[(5-нитропиридин-2-ил)окси]этил]карбамата (200,00 мг, 0,673 ммоль, 1,00 экв.) и Pd/C (71,59 мг, 0,673 ммоль, 1,00 экв.) в THF (20,00 мл) перемешивали в течение 2 ч при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением трет-бутил-N-[2-[(5-аминопиридин-2-ил)окси]этил]-N-метилкарбамата (150 мг, 83,41%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=268,1.
Стадия 3. трет-Бутил-N-[2-([5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]окси)этил]-N-метил карбамат
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (200,00 мг, 0,448 ммоль, 1,00 экв.) и трет-бутил-N-[2-[(5-аминопиридин-2-ил)окси]этил]-N-метилкарбамата (239,27 мг, 0,895 ммоль, 2,00 экв.) в диоксане (20,00 мл) добавляли BrettPhos Pd G3 (81,13 мг, 0,089 ммоль, 0,20 экв.) и K2СО3 (123,70 мг, 0,895 ммоль, 2 экв.) порциями при 70°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН=10:1) с получением трет-бутил-N-[2-([5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]окси)этил]-N-метилкарбамата (50 мг, 16,48%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [M+Na]+=700,3.
Стадия 4. (R)-N-[3-[5-Фтор-2-([6-[2-(метиламино)этокси]пиридин-3-ил]амино)-пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 20)
Смесь трет-бутил-N-[2-([5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]окси)этил]-N-метил-карбамата (50,00 мг, 0,074 ммоль, 1,00 экв.) и HCl (газ) в 1,4-диоксане (8,07 мг, 0,221 ммоль, 3,00 экв.) в DCM (10,00 мл) перемешивали в течение 2 ч при комнатной температуре в воздушной атмосфере. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт (30 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 21 В до 41 В за 7 мин; RT1: 7,03) с получением (R)-N-[3-[5-фтор-2-([6-[2-(метиламино)этокси]пиридин-3-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (5 мг,11,73%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=578,4. 1Н-ЯМР (400 МГц, Метанол-d4) δ 2.29 (3H, s), 2.48 (3H, s), 2.56 (4Н, s), 2.70-2.84 (2Н, m), 2.84-2.95 (2Н, m), 2.95-3.07 (2Н, m), 3.40 (3H, s), 3.47 (1H, t), 3.74- 3.98 (2Н, m), 4.33-4.45 (2Н, m), 6.83 (1H, dd), 7.05-7.18 (2Н, m), 8.02 (1H, dd), 8.11 (1H, d), 8.18 (1H, d), 8.37 (1H, dd), 8.49 (1H, dd).
Пример 22
Получение (R)-N-(3-(5-фтор-2-((6-(оксазол-2-ил)пиридин-3-ил)амино)-пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
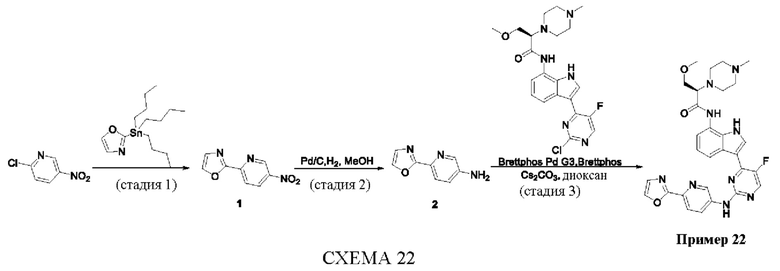
Стадия 1. Получение 2-(5-нитропиридин-2-ил)оксазола
Смесь пиридина, 2-хлор-5-нитро- (100,00 мг, 0,631 ммоль, 1,00 экв.), Pd(PPh3)4 (72,89 мг, 0,063 ммоль, 0,1 экв.) и 2-(трибутилстаннил)-1,3-оксазола (293,65 мг, 0,820 ммоль, 1,30 экв.) в диоксане (6,00 мл) перемешивали в течение 16 ч при 110°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали препаративной ТСХ (РЕ/EtOAc=5:1) с получением 5-нитро-2-(1,3-оксазол-2-ил)пиридина (10 мг, 8.29%) в виде светло-желтого твердого вещества. 1H-ЯМР (300 МГц, DMSO-d6) δ 7.61 (1H, d), 8.35-8.37 (1H, m), 8.47 (1Н, d), 8.75 -8.77 (1H, m), 9.49-9.51(1Н, m).
Стадия 2. Получение 6-(оксазол-2-ил)пиридин-3-амина
Смесь 5-нитро-2-(1,3-оксазол-2-ил)пиридина (200,00 мг, 1,046 ммоль, 1,00 экв.) и Pd/C (200,43 мг, 1,883 ммоль, 1,80 экв.) в МеОН (50,00 мл) перемешивали в течение 2 ч при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (2 × 10 мл). Фильтрат концентрировали при пониженном давлении. Это дало 6-(1,3-оксазол-2-ил)пиридин-3-амин (160 мг, 94,88%) в виде светло-желтого масла. ЖХ-МС (ИЭР): m/z [М+Н]+=162,2. 1Н-ЯМР (300 МГц, DMSO-d6) δ 5.91 (2Н, s), 7.00-7.03 (1H, m), 7.28 (1Н, d), 7.76 (1H, d), 8.00 (1H, d), 8.10 (1H, d).
Стадия 3. Получение (R)-N-(3-(5-фтор-2-((6-(оксазол-2-ил)пиридин-3-ил)амино) пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 22)
Смесь 6-(1,3-оксазол-2-ил)пиридин-3-амина (51,93 мг, 0,322 ммоль, 1,2 экв.), (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (120,00 мг, 0,269 ммоль, 1,00 экв.), K2СО3 (111,33 мг, 0,806 ммоль, 3,00 экв.), BrettPhos (28,83 мг, 0,054 ммоль, 0,20 экв.) и BrettPhos Pd G3 (24,34 мг, 0,027 ммоль, 0,10 экв.) в диоксане (20,00 мл) перемешивали в течение 2 ч при 70°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью CH2Cl2/МеОН (12:1), с получением неочищенного твердого вещества. Этот неочищенный продукт (90 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 30 В до 50 В за 7 мин; RT1: 6,20) с получением (R)-N-[3-(5-фтор-2-[[6-(1,3-оксазол-2-ил)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (65 мг) в виде белого твердого вещества. Неочищенный продукт (65 мг) очищали препаративной хиральной ВЭЖХ в нижеследующих условиях (колонка: CHIRALPAK IC-3, 4,6 × 50 мм, 3 мкм; подвижная фаза А: МТВЕ (0,1% DEA):MeOH=60:40, скорость потока: 1 мл/мин) с получением (R)-N-[3-(5-фтор-2-[[6-(1,3-оксазол-2-ил)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (52 мг, 33,88%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=572,4. 1H-ЯМР (300 МГц, MeOD-d4) δ 2.37 (3H, s), 2.67 (4Н, s), 2.89 (4Н, d), 3.42 (3H, s), 3.52 (1H, t), 3.79-3.98 (2Н, m), 7.15-7.26 (2Н, m), 7.34 (1H, d), 7.99-8.09 (2Н, m), 8.16 (1H, d), 8.33 (1H, d), 8.53 (1H, dd), 8.68 (1H, dd), 8.99 (1H, d).
Пример 24
Получение (R)-N-(3-(2-((6-(1Н-имидазол-1-ил)пиридин-3-ил)амино)-5-фторпиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 24)
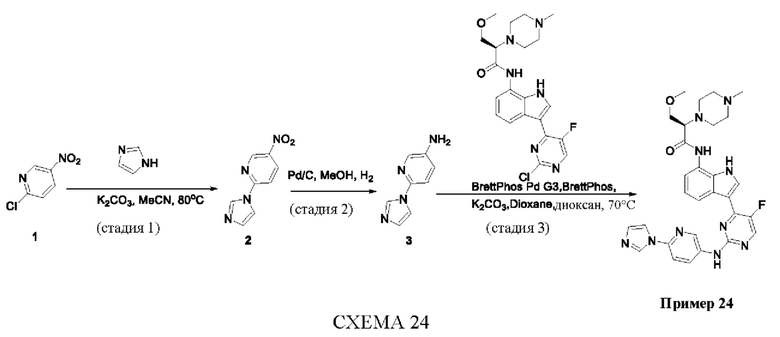
Стадия 1. Получение 2-(1Н-имидазол-1-ил)-5-нитропиридина
Смесь 2-хлор-5-нитро-пиридина (500,00 мг, 3,154 ммоль, 1,00 экв.), K2СО3 (1089,67 мг, 7,884 ммоль, 2,50 экв.) и имидазола (429,41 мг, 6,308 ммоль, 2,00 экв.) в MeCN (20,00 мл) перемешивали в течение 2 ч при 80°С в атмосфере азота. Выпавшее в осадок твердое вещество собирали фильтрованием и промывали MeCN (3 × 10 мл) с получением 2-(имидазол-1-ил)-5-нитропиридина (375 мг, 60,46%) в виде коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=191,0
Стадия 2. Получение 6-(1Н-имидазол-1-ил)пиридин-3-амина
Смесь 2-(имидазол-1-ил)-5-нитропиридина (180,00 мг, 0,947 ммоль, 1,00 экв.) и Pd/C (50,37 мг, 0,473 ммоль, 0,50 экв.) в МеОН (15,00 мл) перемешивали при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали DCM (3×10 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1) с получением 6-(имидазол-1-ил)пиридин-3-амина (120 мг, 79,15%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=161,2.
Стадия 3. (R)-N-(3-(2-((6-(1Н-Имидазол-1-ил)пиридин-3-ил)амино)-5-фторпиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 24)
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (150,00 мг, 0,336 ммоль, 1,00 экв.) и 6-(имидазол-1-ил)пиридин-3-амина (80,64 мг, 0,503 ммоль, 1,50 экв.) в диоксане (20,00 мл) добавляли BrettPhos Pd G3 (60,85 мг, 0,067 ммоль, 0,20 экв.), BrettPhos (54,05 мг, 0,101 ммоль, 0,30 экв.) и K2СО3 (115,97 мг, 0,839 ммоль, 2,50 экв.). Смесь перемешивали при 80°С в атмосфере азота. Полученную смесь фильтровали, осадок на фильтре промывали DCM (3 × 20 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl3/МеОН 10:1) с получением неочищенного продукта (100 мг), который очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 31% В до 45% В за 7 мин; 254, 220 нм; Rt: 6,30 мин) с получением (R)-N-[3-(5-фтор-2-[[6-(имидазол-1-ил)пиридин-3-ил]амино]пиримидин-4-ил)-1H-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (60,8 мг, 31,75%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=571,4 1H-ЯМР (300 МГц, DMSO-d6) δ 2.16 (3H, s), 2.37 (4Н, s), 2.59-2.69 (2Н, m), 2.71-2.82 (2Н, m), 3.30 (3H, s), 3.51 (1H, t), 3.69 (1Н, dd), 3.81 (1H, dd), 7.12 (1Н, t), 7.19 (1H, t), 7.55 (1Н, d), 7.78 (1H, d), 7.91 (1H, t), 8.27 (1H, d), 8.42 (1H, dd), 8.47 (1Н, m), 8.50 (1H, d), 8.56 (1H, d), 8.80-8.89 (1H, m), 9.87 (2Н, d), 11.50 (1H, s)
Пример 25
Получение (R)-N-[3-(5-фтор-2-[[5-(3-гидроксипропил)пиридин-3-ил]амино]-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
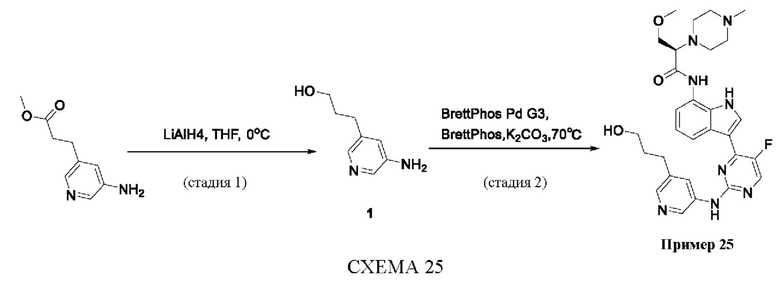
Стадия 1. 3-(5-Аминопиридин-3-ил)пропан-1-ол
В перемешиваемую смесь LiAlH4 (44,23 мг, 1,165 ммоль, 3 экв.) в THF (1 мл) добавляли метил-3-(5-аминопиридин-3-ил)пропаноат (70,00 мг, 0,388 ммоль, 1,00 экв.) в THF (20.0 мл) по каплям при 0°С. Полученную смесь перемешивали в течение 30 мин при 0°С. Целевой продукт можно было обнаружить методом ЖХ-МС. Реакционную смесь гасили добавлением Na2SO4⋅10H2O. Полученную смесь фильтровали, и осадок на фильтре промывали этилацетатом (3×5 мл). Фильтрат концентрировали при пониженном давлении с получением 3-(5-аминопиридин-3-ил)пропан-1-ола (56 мг, 94,72%) в виде красновато-коричневого масла. ЖХ-МС (ИЭР): m/z [М+Н]+=153,3.
Стадия 2. (R)-N-[3-(5-Фтор-2-[[5-(3-гидроксипропил)пиридин-3-ил]амино]-пиримидин-4-ил)-1Н-индол-7-ил1-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 25)
В смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (150,00 мг, 0,336 ммоль, 1,00 экв.) и 3-(5-аминопиридин-3-ил)пропан-1-ола (66,41 мг, 0,436 ммоль, 1,30 экв.) в диоксане (20,0 мл) добавляли BrettPhos (36,03 мг, 0,067 ммоль, 0,20 экв.), BrettPhos Pd G3 (60,85 мг, 0,067 ммоль, 0,20 экв.) и K2СО3 (92,77 мг, 0,671 ммоль, 2,00 экв.). После перемешивания в течение 2 ч при 80°С в атмосфере азота остаток очищали ТСХ (CH2Cl2/МеОН=5:1) с получением неочищенного твердого вещества. Этот неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 19 В до 39 В за 7 мин; RT1: 6,53) с получением (R)-N-[3-(5-фтор-2-[[5-(3-гидроксипропил)пиридин-3-ил]амино]-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (16 мг, 8,47%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=563,4. 1Н-ЯМР (300 МГц, DMSO-d6) δ 1.60-1.82 (2Н, m), 2.13 (3H, s), 2.34 (4Н, s), 2.61 (4Н, q), 2.67-2.81 (2Н, m), 3.28 (3H, s), 3.41 (2Н, q), 3.49 (1H, t), 3.67 (1H, dd), 3.79 (1H, dd), 4.48 (1H, t), 7.13 (1Н, t), 7.52 (1Н, d), 8.03 (1H, d), 8.11 (1H, t), 8.23 (1H, d), 8.38-8.56 (2Н, m), 8.70 (1H, d), 9.63 (1H, s), 9.85 (1H, s), 11.47 (1H, s).
Пример 30/33
Получение (R)-N-[3-[5-фтор-2-([1-[оксолан-3-ил]пиразол-4-ил]амино)-пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 30 в виде изомера 1 и Пр. 33 в виде изомера 2)
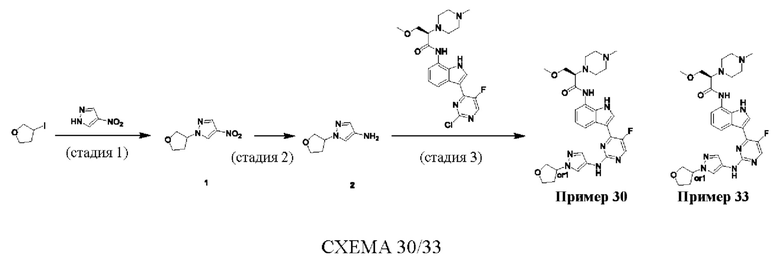
Стадия 1. 4-Нитро-1-(оксолан-3-ил)пиразол
В сосуд вместимостью 40 мл добавляли 3-йодоксолан (665 мг, 3,36 ммоль, 1,00 экв.) и 4-нитропиразол (380 мг, 3,36 ммоль, 1,00 экв.) в DMF (20,00 мл) при комнатной температуре. Конечную реакционную смесь перемешивали в течение в течение ночи при 80°С. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1) с получением 4-нитро-1-(оксолан-3-ил)пиразола (600 мг, 59,02%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=184,3. 1Н-ЯМР (300 МГц, MeOD-d4) δ 2.36-2.39 (1H, m), 2.52 (1H, dtd), 3.91-3.94 (1H, m), 4.00-4.11 (2Н, m), 4.06-4.19 (1H, m), 5.08-5.12 (1H, m), 8.13 (1H, s), 8.57-8.63 (1H, m).
Стадия 2. 1-(Оксолан-3-ил)пиразол-4-амин
Смесь 4-нитро-1-(оксолан-3-ил)пиразола (600 мг, 3,27 ммоль, 1,00 экв.) и Pd/C (0,03 г, 0,327 ммоль, 0,10 экв.) в МеОН (20,00 мл) перемешивали в течение 1 ч при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (2 × 10 мл). Фильтрат концентрировали при пониженном давлении. Это привело к получению 1-(оксолан-3-ил)пиразол-4-амина (500 мг, 92,67%) в виде пурпурного масла. ЖХ-МС (ИЭР): m/z [М+Н]+=154,1. 1Н-ЯМР (300 МГц, DMSO-d6) δ 2.05-2.21 (1H, m), 2.23-2.28 (1H, m), 3.58-4.04 (6Н, m), 4.74-4.82 (1H, m), 6.91 (1H, d), 7.03 (1H, d).
Стадия 3. (R)-N-[3-[5-Фтор-2-([1-[оксолан-3-ил]пиразол-4-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 30 и Пр. 33)
В раствор 1-(оксолан-3-ил)пиразол-4-амина (102,83 мг, 0,671 ммоль, 1,50 экв.) и (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метил-пиперазин-1-ил)пропанамида (200,00 мг, 0,448 ммоль, 1,00 экв.) в диоксане (20,00 мл) добавляли BrettPhos (24,02 мг, 0,045 ммоль, 0,10 экв.), BrettPhos Pd G3 (40,57 мг, 0,045 ммоль, 0,10 экв.) и Cs2CO3 (437,43 мг, 1,343 ммоль, 3,00 экв.). После перемешивания в течение 3 ч при 80°С в атмосфере азота полученную смесь концентрировали при пониженном давлении. Неочищенный продукт (40 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: CHIRAL ART Cellulose-SB, 4,6 × 100 мм, 3 мкм; подвижная фаза A: (Hex:DCM=5:1) (0,1% DEA):IPA=85:15, подвижная фаза В; скорость потока: 1 мл/мин; градиент: от 0 В до 0 В) с получением (R)-N-[3-[5-фтор-2-([1-[оксолан-3-ил]пиразол-4-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 33) (11 мг, 4,32%) ЖХ-МС (ИЭР): m/z [М+Н]+=564,4; 1Н-ЯМР (300 МГц, DMSO-d6) δ 2.32 (5Н, s), 2.54-2.82 (8Н, m), 3.30 (3H, s), 3.59 (1H, s), 3.56-3.74 (1H, m), 3.75-4.04 (2Н, m), 3.83-4.00 (3H, m), 4.98 (1H, s), 7.12-7.17 (1H, m), 7.56 (2Н, d), 7.99 (1H, s), 8.19 (1H, s), 8.39 (2Н, d), 9.34 (1H, s), 9.94 (1H, s), 11.52 (1H, s); и (R)-N-[3-[5-фтор-2-([1-[оксолан-3-ил]пиразол-4-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 30) (7 мг, 13,86%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=564,4; 1Н-ЯМР (300 МГц, DMSO-d6) δ 1.24 (3H, s), 1.95-2.06 (1H, m), 2.16 (3H, s), 2.25 (1H, s), 2.28-2.47 (4Н, m), 2.64 (2Н, d), 2.75 (2Н, d), 3.51 (1H, t), 3.65-3.69 (1H, m), 3.74-3.87 (2Н, m), 3.84-4.04 (3H, m), 4.95-5.03 (1H, m), 7.11-7.17 (1H, m), 7.53 (2Н, d), 7.99 (1H, s), 8.18-8.20 (1H, m), 8.38-8.39 (1H, m), 8.49 (1H, s), 9.34 (1H, s), 9.85 (1H, s), 11.42 (1H, s).
Пример 34
Получение (R)-N-[3-(5-фтор-2-[[6-(гидроксиметил)-5-метоксипиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
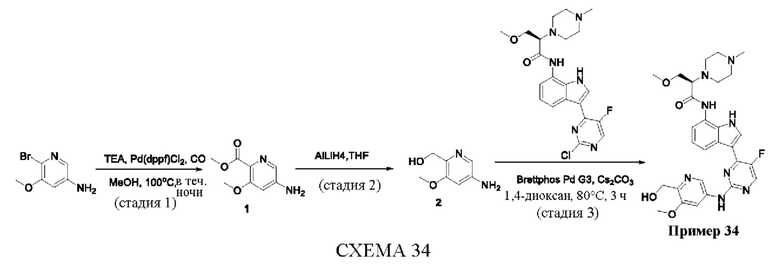
Стадия 1. Метил-5-амино-3-метоксипиридин-2-карбоксилат
В перемешиваемую смесь 6-бром-5-метоксипиридин-3-амина (1000,00 мг, 4,925 ммоль, 1,00 экв.) и TEA (996,75 мг, 9,850 ммоль, 2,00 экв.) в МеОН (100,00 мл) добавляли Pd(dppf)Cl2 (720,75 мг, 0,985 ммоль, 0,20 экв.) Полученную смесь перемешивали при 100°С в атмосфере монооксида углерода. Полученную смесь перемешивали в течение в течение ночи при 100°С в атмосфере монооксида углерода. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 20:1) с получением метил-5-амино-3-метоксипиридин-2-карбоксилата (700 мг, 78,02%) в виде светло-коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=183,2
Стадия 2. (5-Амино-3-метоксипиридин-2-ил)метанол
Смесь метил-5-амино-3-метоксипиридин-2-карбоксилата (300,00 мг, 1,647 ммоль, 1,00 экв.) и LiAlH4 (187,50 мг, 4,940 ммоль, 3,00 экв.) в THF (30,00 мл) перемешивали в течение в течение ночи при комнатной температуре в воздушной атмосфере. Реакционную смесь гасили смесью вода/лед при комнатной температуре. Полученную смесь фильтровали, осадок на фильтре промывали THF (3 × 10 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт напрямую использовали на следующей стадии без дополнительной очистки с получением (5-амино-3-метоксипиридин-2-ил)метанола (200 мг, 78,78%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=155,3.
Стадия 3. (R)-N-[3-(5-Фтор-2-[[6-(гидроксиметил)-5-метоксипиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1H-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (120,00 мг, 0,269 ммоль, 1,00 экв.) и (5-амино-3-метоксипиридин-2-ил)метанола (82,79 мг, 0,537 ммоль, 2,00 экв.) в диоксане (20,00 мл) добавляли Cs2CO3 (262,46 мг, 0,806 ммоль, 3,00 экв.) и BrettPhos Pd G3 (48,68 мг, 0,054 ммоль, 0,20 экв.) порциями при 80°С в атмосфере азота. Полученную смесь перемешивали в течение 2 ч при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт (80 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 26 В до 36 В за 7 мин; 254;220 нм; RT1: 7,28) с получением (R)-N-[3-(5-фтор-2-[[6-(гидроксиметил)-5-метоксипиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (10 мг, 6,60%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=565,4. 1H-ЯМР (400 МГц, DMSO-d6) δ 2.13 (3H, s), 2.34 (4Н, s), 2.54-2.67 (2Н, m), 2.73 (2Н, d), 3.28 (3H, s), 3.49 (1Н, t), 3.66 (1Н, dd), 3.72-3.85 (4Н, m), 4.48 (2Н, d), 4.73 (1H, t), 7.13 (1H, t), 7.53 (1H, dd), 7.93 (1H, d), 8.24 (1H, d), 8.39-8.58 (3H, m), 9.78 (2Н, d), 11.43 (1H, s)
Пример 36
Получение этил-5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)-пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-карбоксилата
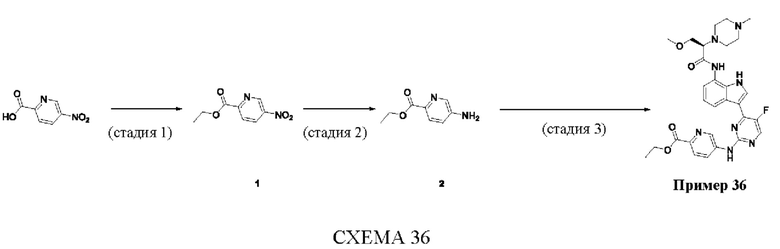
Стадия 1. Этил-5-нитропиридин-2-карбоксилат
В перемешиваемый раствор 5-нитропиридин-2-карбоновой кислоты (700,00 мг, 4,164 ммоль, 1,00 экв.) в EtOH (20,00 мл) добавляли SOCl2 (1,01 мл, 7,480 ммоль, 3,00 экв.) по каплям при 0°С в воздушной атмосфере. Полученную смесь перемешивали в течение 2 ч при 80°С в воздушной атмосфере. Полученную смесь концентрировали при пониженном давлении. Реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (50 мл) при комнатной температуре. Смесь экстрагировали EtOAc (2 × 25 мл). Объединенные органические слои промывали рассолом (1 × 20 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Это привело к получению этил-5-нитропиридин-2-карбоксилата (600 мг, 72,72%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=197,2. 1H-ЯМР (300 МГц, MeOD-d4) δ1.40-1.47 (3H, m), 4.44-4.52 (2Н, m), 8.33-8.38 (1H, m), 8.74-8.79 (1H, m), 9.43-9.46 (1H, m).
Стадия 2. Этил-5-аминопиридин-2-карбоксилат
Смесь этил-5-нитропиридин-2-карбоксилата (400,00 мг, 2,039 ммоль, 1,00 экв.) и Pd/C (434,01 мг, 4,078 ммоль, 2,00 экв.) в МеОН (25,00 мл) перемешивали при комнатной температуре в атмосфере водорода в течение 1 ч. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (3 × 15 мл). Фильтрат концентрировали при пониженном давлении. Это привело к получению этил-5-аминопиридин-2-карбоксилата (312 мг, 91,15%) в виде серого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=167,3. 1H-ЯМР (300 МГц, DMSO-d6) δ1.25 (3H, t), 4.17-4.31 (2Н, m), 6.21 (2Н, s), 6.89-6.93 (1H, m), 7.72 (1H, d), 7.96 (1H, d).
Стадия 3. Этил-5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)-пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-карбоксилат (Пр. 36)
В раствор этил-5-аминопиридин-2-карбоксилата (55,78 мг, 0,336 ммоль, 1,50 экв.) и (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метил-пиперазин-1 -ил)пропанамида (100,00 мг, 0,224 ммоль, 1,00 экв.) в диоксане (10,00 мл) добавляли BrettPhos (12,01 мг, 0,022 ммоль, 0,10 экв.), Cs2CO3 (218,72 мг, 0,671 ммоль, 3,00 экв.) и BrettPhos Pd G3 (20,28 мг, 0,022 ммоль, 0,10 экв.). После перемешивания в течение 2 ч при 80°С в атмосфере азота полученную смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью CH2Cl2/МеОН (20:3). Неочищенный продукт (100 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 30 В до 50 В за 7 мин; 254;220 нм; RT1: 7,43) с получением этил-5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-карбоксилата (20 мг, 15,35%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=577,5. 1Н-ЯМР (300 МГц, DMSO-d6) δ 1.29-1.34 (3H, ш), 2.13 (3H, s), 2.34 (4Н, s), 2.62 (2Н, s), 3.47-3.52 (2Н, m), 3.32 (3H, s), 3.64-3.69 (1H, m), 3.76-3.81 (2Н, m), 4.29- 4.34 (2Н, m), 7.15-7.20 (1H, m), 7.54 (1H, d), 8.02 (1H, d), 8.27 (1H, s), 8.45-8.62 (3H, m), 8.97 (1H, d), 9.87 (1H, s), 10.19 (1H, s), 11.53 (1H, s).
Пример 39
Получение (R)-N-(3-(5-фтор-2-((6-(2-(метиламино)-2-оксоэтил)пиридин-3-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
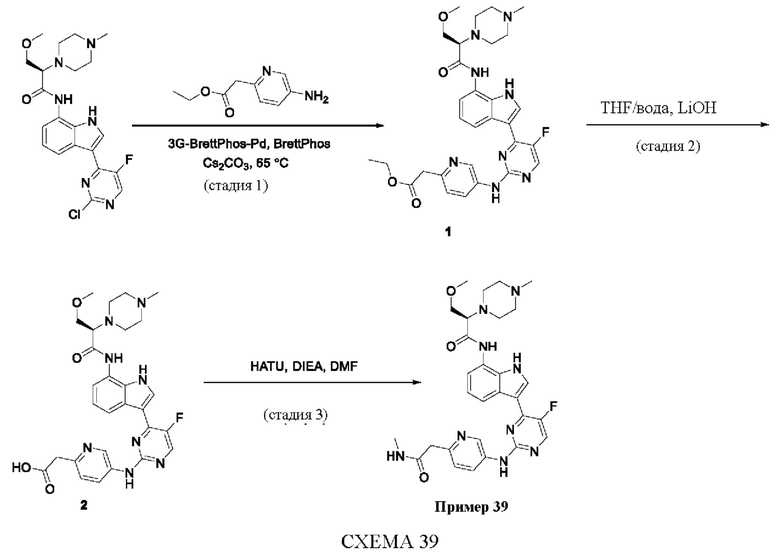
Стадия 1. Этил-(R)-2-(5-((5-фтор-4-(7-(3-метокси-2-(4-метилпиперазин-1-ил) пропанамидо)-1Н-индол-3-ил)пиримидин-2-ил)амино)пиридин-2-ил)ацетат
В сосуд вместимостью 40 мл добавляли этил-2-(5-аминопиридин-2-ил)ацетат (72,58 мг, 0,403 ммоль, 1,20 экв.) и (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (150,00 мг, 0,336 ммоль, 1,00 экв.), BrettPhos (18,02 мг, 0,034 ммоль, 0,10 экв.), BrettPhos Palladacycle (26,81 мг, 0,034 ммоль, 0,10 экв.), Cs2CO3 (218,72 мг, 0,671 ммоль, 2,00 экв.) в диоксане (10,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение 2 ч при 80°С в атмосфере азота. Полученную смесь фильтровали, осадок на фильтре промывали DCM (2 × 10 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 200:15) с получением этил-2-[5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]ацетата (120 мг, 60,6%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=591,3.
Стадия 2. (R)-2-(5-((5-Фтор-4-(7-(3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо)-1Н-индол-3-ил)пиримидин-2-ил)амино)пиридин-2-ил)уксусная кислота
В сосуд вместимостью 40 мл добавляли этил-2-[5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]ацетат (140,00 мг, 0,237 ммоль, 1,00 экв.) в THF (3,00 мл) и LiOH (56,76 мг, 2,370 ммоль, 10,00 экв.) в воде (0.50 мл) при комнатной температуре. Полученную смесь перемешивали в течение 3 ч при комнатной температуре в воздушной атмосфере. Реакционную смесь подкисляли раствором HCl (1 М), затем выпаривали с получением неочищенного твердого вещества. Это неочищенное твердое вещество напрямую использовали на следующей стадии без очистки. ЖХ-МС (ИЭР): m/z [М+Н]+=563,4.
Стадия 3. (R)-N-(3-(5-Фтор-2-((6-(2-(метиламино)-2-оксоэтил)пиридин-3-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 39)
В сосуд вместимостью 8 мл добавляли [5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]уксусную кислоту (80 мг, 0,142 ммоль, 1,00 экв.) и метиламин (0,36 мл, 0,720 ммоль, 5,06 экв.), HATU (108,13 мг, 0,284 ммоль, 2,00 экв.), Et3N (43,17 мг, 0,427 ммоль, 3,00 экв.) в DMF (2,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение 2 ч при комнатной температуре в воздушной атмосфере. Полученную смесь разбавляли водой (10 мл). Водный слой экстрагировали CH2Cl2 (3 ×10 мл). Объединенный органический слой сушили над безводным Na2SO4, фильтровали и выпаривали с получением желтого твердого вещества. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 8:1) с получением желтого твердого вещества. Неочищенный продукт (40 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 27 В до 37 В за 7 мин; 254;220 нм; RT1: 5,17) с получением (R)-N-[3-[5-фтор-2-([6-[(метилкарбамоил)метил]пиридин-3-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (10 мг, 12,22%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=576,3. 1H-ЯМР (400 МГц, DMSO-d6) δ2.16 (3H, s), 2.38 (4Н, s), 2.61 (5Н, d), 2.76 (2Н, t), 3.30 (3H, s), 3.54 (3H, d), 3.69 (1H, dd), 3.81 (1H, dd), 7.16 (1Н, t), 7.28 (1Н, d), 7.55 (1H, d), 7.96 (1Н, q), 8.16 (1Н, dd), 8.22-8.29 (1H, m), 8.46 (1H, d), 8.54 (1Н, d), 8.79 (1H, d), 9.65 (1H, s), 9.88 (1Н, s), 11.50 (1H, d)
Пример 40
Получение (R)-N-[3-(5-фтор-2-[[6-(1,3-оксазол-5-ил)пиридин-3-ил]амино]-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
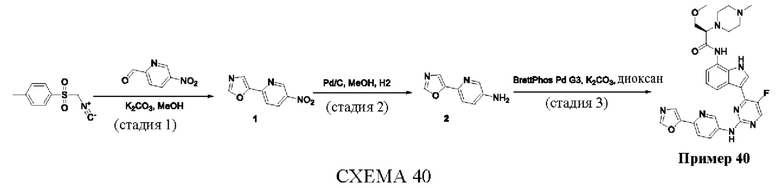
Стадия 1. 5-Нитро-2-(1,3-оксазол-5-ил)пиридин
Смесь TosMIC (1,00 г, 5,122 ммоль, 1,00 экв.) и 5-нитропиридин-2-карбальдегида (779,09 мг, 5,122 ммоль, 1,00 экв.), K2СО3 (1061,81 мг, 7,683 ммоль, 1,50 экв.) в МеОН (20,00 мл) перемешивали в течение 5 ч при 75°С в атмосфере азота. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (5:1), с получением 5-нитро-2-(1,3-оксазол-5-ил)пиридина (500 мг, 51,07%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=192,2.
Стадия 2. 6-(1,3-Оксазол-5-ил)пиридин-3-амин
Смесь 5-нитро-2-(1,3-оксазол-5-ил)пиридина (250,00 мг, 1,308 ммоль, 1,00 экв.) и Pd/C (27,84 мг, 0,262 ммоль, 0,20 экв.) в МеОН (10,00 мл) перемешивали в течение 3 ч при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (10 мл × 3). Фильтрат концентрировали при пониженном давлении с получением 6-(1,3-оксазол-5-ил)пиридин-3-амина (180 мг, 85,39%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=162,3.
Стадия 3. (R)-N-[3-(5-Фтор-2-[[6-(1,3-оксазол-5-ил)пиридин-3-ил1амино1-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 40)
Смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (120,00 мг, 0,269 ммоль, 1,00 экв.) и 6-(1,3-оксазол-5-ил)пиридин-3-амина (64,91 мг, 0,403 ммоль, 1,50 экв.), BrettPhos Pd G3 (24,34 мг, 0,027 ммоль, 0,10 экв.), K2СО3 (74,22 мг, 0,537 ммоль, 2,00 экв.) в диоксане (4,00 мл) перемешивали в течение 2 ч при 70°С в атмосфере азота. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН=15:1) с получением неочищенного продукта. Этот неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 19 × 250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 25 мл/мин; градиент: от 32 В до 52 В за 7 мин; RT1: 6,40) с получением (R)-N-[3-(5-фтор-2-[[6-(1,3-оксазол-5-ил)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (100 мг, 65,15%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=572,2 1Н-ЯМР (400 МГц, DMSO-d6) δ 2.15 (3H, s), 2.36 (4Н, s), 2.64 (2Н, d), 2.76 (2Н, m), 3.30 (3H, s), 3.51 (1Н, t), 3.68 (1H, dd), 3.80 (1H, dd), 7.17 (1H, t), 7.56 (1H, d), 7.65 (1H, s), 7.73 (1H, d), 8.27 (1H, d), 8.44 (1H, dd), 8.47 (1H, s), 8.50 (1H, d), 8.57 (1H, d), 8.96 (1H, d), 9.89 (1H, s), 9.95 (1H, s), 11.54 (1H, s).
Пример 41
Получение (R)-N-[3-[5-фтор-2-(1Н-индол-5-иламино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
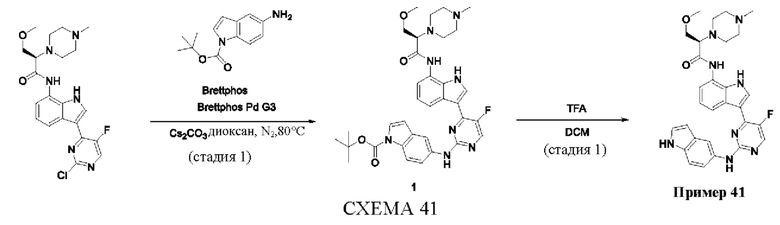
Стадия 1. трет-Бутил-5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо 1-1Н-индол-3-ил1пиримидин-2-ил)амино1индол-1-карбоксилат
В раствор (R)-Н-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (180,00 мг, 0,403 ммоль, 1,00 экв.) и трет-бутил-5-аминоиндол-1-карбоксилата (121,62 мг, 0,524 ммоль, 1,3 экв.) в диоксане (10,0 мл) добавляли BrettPhos (43,24 мг, 0,081 ммоль, 0,2 экв.) и BrettPhos Pd G3 (73,02 мг, 0,081 ммоль, 0,2 экв.) и Cs2CO3 (262,46 мг, 0,806 ммоль, 2 экв.). После перемешивания в течение 16 ч при 80°С в атмосфере азота. Остаток очищали ТСХ (CH2Cl2/МеОН 8:1) с получением трет-бутил-5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]индол-1-карбоксилата (130 мг, 50,22%) в виде красновато-коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=643,4.
Стадия 2. (R)N-[3-[5-фтор-2-(1Н-индол-5-иламино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 41).
В перемешиваемый раствор трет-бутил-5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метил-пиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]индол-1-карбоксилата (130,00 мг, 0,202 ммоль, 1,00 экв.) в DCM (6,0 мл) добавляли TFA (2,00 мл, 26,926 ммоль, 133,13 экв.). Полученную смесь перемешивали в течение 2 ч при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. Смесь подщелачивали до рН 8 насыщенным NaHCO3 (водн.). Полученную смесь экстрагировали CH2Cl2 (8 × 30 мл), и объединенные органические слои сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении с получением (R)-N-[3-[5-фтор-2-(1Н-индол-5-иламино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (54 мг, 49,20%) в виде красновато-коричневого масла. Этот неочищенный продукт (54 мг) очищали препаративной хиральной ВЭЖХ в нижеследующих условиях (колонка: CHIRAL ART Cellulose-SB, 2 × 25 см, 5 мкм; подвижная фаза А: МТВЕ (10 мМ NH3-МЕОН)--ВЭЖХ, подвижная фаза В: EtOH-ВЭЖХ; скорость потока: 20 мл/мин; градиент: от 10 В до 10 В за 12 мин; 220/254 нм; RT1: 8,928; RT2: 10,344; впрыскиваемый объем: 0,6 мл; число прогонов: 20) с получением (R)-N-[3-[5-фтор-2-(1Н-индол-5-иламино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (32,96 мг, 72,30%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=543,3. 1H-ЯМР (300 МГц, DMSO-d6) 5 2.14 (3H, s), 2.35 (4Н, s), 2.63 (2Н, d), 2.73 (2Н, s), 3.29 (3H, s), 3.50 (1H, m), 3.68 (1Н, dd), 3.80 (1Н, dd), 6.36 (1H, t), 7.02 (1H, t), 7.23-7.42 (3H, m), 7.51 (1H, d), 8.01 (1H, s), 8.21 (1H, d), 8.38 (1Н, d), 8.55 (1H, d), 9.22 (1H, s), 9.85 (1H, s), 10.95 (1Н, s), 11.43 (1H, s).
Пример 42
Получение (R)-N-(3-(5-фтор-2-((1-оксоизохроман-6-ил)амино)пиримидин-4-ил)-1H-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
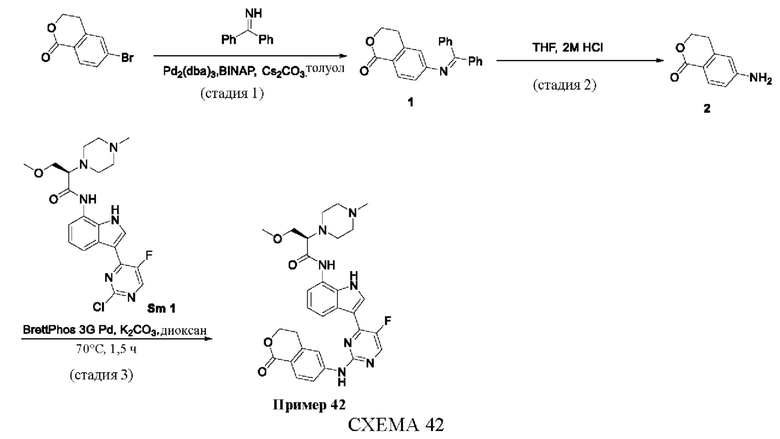
Стадия 1. 6-((Дифенилметилен)амино)изохроман-1-он
В сосуд вместимостью 40 мл добавляли 6-бром-3,4-дигидро-2-бензопиран-1-он (500,00 мг, 2,202 ммоль, 1,00 экв.) и бензолметанимин, ?-фенил- (518,83 мг, 2,863 ммоль, 1,30 экв.), Pd2(dba)3 (201,65 мг, 0,220 ммоль, 0,10 экв.), BIN АР (274,24 мг, 0,440 ммоль, 0,20 экв.), Cs2CO3 (1434,97 мг, 4,404 ммоль, 2,00 экв.) в толуоле (20,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение 2 ч при 90°С в атмосфере азота. Реакционную смесь оставляли охлаждаться до к.т., твердое вещество отфильтровывали, осадок на фильтре промывали МеОН (10 мл), и фильтрат концентрировали при пониженном давлении. Остаток очищали ТСХ (ЕА:РЕ=1:3) с получением 6-[(дифенилметилиден)амино]-3,4-дигидро-2-бензопиран-1-она (458 мг, 63,53%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=328,2.
Стадия 2. 6-Аминоизохроман-1-он
В круглодонную колбу на 50 мл добавляли 6-[(дифенилметилиден)амино]-3,4-дигидро-2-бензопиран-1-он (458,00 мг, 1,399 ммоль, 1,00 экв.) в THF (10 мл), и раствор HCl (2 М) в воде (5 мл) добавляли в вышеуказанный раствор при комнатной температуре. Полученную смесь перемешивали в течение 1 ч при комнатной температуре в воздушной атмосфере. Смесь подщелачивали до рН 8 насыщенным NaHCO3 (водн.). Водный слой экстрагировали CH2Cl2 (3 × 20 мл). Объединенные органические слои сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/MeOH 20:1) с получением 6-амино-3,4-дигидро-2-бензопиран-1-она (112 мг, 49,06%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=164,1
Стадия 3. (R)-N-(3-(5-Фтор-2-((1-оксоизохроман-6-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 42)
В сосуд вместимостью 40 мл добавляли 6-амино-3,4-дигидро-2-бензопиран-1-он (35,05 мг, 0,215 ммоль, 1,20 экв.) и (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (80,00 мг, 0,179 ммоль, 1,00 экв.), BrettPhos Pd G3 (16,23 мг, 0,018 ммоль, 0,10 экв.), K2СО3 (74,22 мг, 0,537 ммоль, 3,00 экв.) в диоксане (2,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение 2 ч при 70°С в атмосфере азота. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (2 × 10 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1) с получением желтого твердого вещества. Неочищенный продукт (40 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 31 В до 51 В за 7 мин; 254;220 нм; RT1: 6,77) с получением (R)-N-(3-[5-фтор-2-[(1-оксо-3,4-дигидро-2-бензопиран-6-ил)амино]пиримидин-4-ил]-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (10 мг, 9,74%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=574,4 1Н-ЯМР (400 МГц, MeOD-d4) 2.35 (3H, s), 2.63 (4Н, s), 2.84 (2Н, s), 2.94 (2Н, s), 3.07 (2Н, t), 3.43 (3H, s), 3.53 (1Н, t), 3.85 (1H, dd), 3.94 (1Н, dd), 4.56 (2Н, t), 7.21 (2Н, d), 7.67 (1H, dd), 7.95 (1H, d), 8.02 (1H, d), 8.19 (1H, d), 8.35 (1H, d), 8.69 (1H, q).
Пример 46
Получение (R)-N-[3-(5-фтор-2-[[6-(гидроксиметил)пиридин-2-ил]амино]-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида

Стадия 1. Метил-2-(5-бромпиридин-2-ил)-2-(N-гидроксиимино)ацетат
Смесь метил-2-(5-бромпиридин-2-ил)ацетата (3,00 г, 13,040 ммоль, 1,00 экв.) в АсОН (15,00 мл) перемешивали в течение 30 мин при 0°С в воздушной атмосфере. В вышеуказанную смесь добавляли раствор NaNO2 (0,90 г, 13,040 ммоль, 1,00 экв.) в воде (2 мл) по каплям в течение 1 мин при комнатной температуре. Полученную смесь перемешивали в течение еще 1 ч при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. Полученную смесь экстрагировали EtOAc (2 × 20 мл). Объединенные органические слои промывали рассолом (1 × 20 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Это привело к получению метил-2-(5-бромпиридин-2-ил)-2-(N-гидроксиимино)ацетата (3 г, 87,92%) в виде розового твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=260,9.
Стадия 2. Метил-2-амино-2-(5-бромпиридин-2-ил)ацетат
В круглодонную колбу на 250 мл добавляли метил-2-(5-бромпиридин-2-ил)-2-(N-гидроксиимино)ацетат (5,00 г, 19,301 ммоль, 1,00 экв.), Zn (3,16 г, 48,252 ммоль, 2,50 экв.), муравьиную кислоту (20,00 мл, 530,142 ммоль, 27,47 экв.), МеОН (20,00 мл, 493,978 ммоль, 25,59 экв.) и H2O (20,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение в течение ночи при комнатной температуре в воздушной атмосфере. Полученную смесь концентрировали при пониженном давлении. Остаток нейтрализовали до рН 7 насыщенным NaHCO3 (водн.). Полученную смесь экстрагировали EtOAc (3×15 мл). Объединенные органические слои промывали рассолом (1 × 20 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Это привело к получению метил-2-амино-2-(5-бромпиридин-2-ил)ацетата (6 г, 60,89%) в виде черного масла. Этот неочищенный продукт использовали на следующей стадии без другой очистки. ЖХ-МС (ИЭР): m/z [М+Н]+=244,9.
Стадия 3. Метил-6-бромимидазо[1,5-а]пиридин-1-карбоксилат В круглодонную колбу на 250 мл добавляли метил-2-амино-2-(5-бромпиридин-2-ил)ацетат (5,00 г, 20,402 ммоль, 1,00 экв.) и (диметоксиметил)диметиламин (2,67 г, 22,442 ммоль, 1,10 экв.) в толуоле (50 мл) при комнатной температуре. Полученную смесь перемешивали в течение в течение ночи при 110°С в воздушной атмосфере. Полученную смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (1:1), с получением метил-6-бромимидазо[1,5-а]пиридин-1-карбоксилата (3,962 г, 74,61%) в виде темно-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=254,9.
Стадия 4. Метил-6-Г(дифенилметилиден)амино1имидазо[1,5-а]пиридин-1-карбоксилат
В раствор метил-6-бромимидазо[1,5-а]пиридин-1-карбоксилата (3,00 г, 11,761 ммоль, 1,00 экв.) и дифенилметанимина (3,20 г, 17,642 ммоль, 1,50 экв.) в толуоле (25,00 мл) добавляли Pd2(dba)3 (1,08 г, 1,176 ммоль, 0,10 экв.), BINAP (1,46 г, 2,352 ммоль, 0,20 экв.) и Cs2CO3 (11,50 г, 35,284 ммоль, 3,00 экв.). После перемешивания в течение 2 ч при 90°С в атмосфере азота полученную смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (5:1), с получением метил-6-[(дифенилметилиден)амино]имидазо[1,5-а]пиридин-1-карбоксилата (1,9 г, 40,00%) в виде темно-желтого твердого вещества. 1H-ЯМР (300 МГц, CDCl3-d1) δ 1.18-1.32 (0Н, m), 3.95 (3H, s), 6.66-6.70 (1H, m), 7.04-7.22 (3H, m), 7.34 (1H, s), 7.28-7.40 (2Н, m), 7.40-7.48 (1H, m), 7.44-7.59 (3H, m), 7.72-7.86 (2Н, m), 7.94 (2Н, d).
Стадия 5. Метил-6-аминоимидазо[1.5-а]пиридин-1-карбоксилат
В круглодонную колбу на 50 мл добавляли метил-6-[(дифенилметилиден)-амино]имидазо[1,5-а]пиридин-1-карбоксилат (1,80 г, 5,065 ммоль, 1,00 экв.), HCl (2М) (2,00 мл) и THF (20,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение 1 ч при комнатной температуре в воздушной атмосфере. Полученную смесь концентрировали под вакуумом. Остаток нейтрализовали до рН 7 насыщенным NaHCO3 (водн.). Полученную смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (3:1), с получением метил-6-аминоимидазо[1,5-а]пиридин-1-карбоксилата (731 мг, 73,23%) в виде темно-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=192,2.
Стадия 6. [6-Аминоимидазо[1,5-а]пиридин-1-ил]метанол
В сосуд вместимостью 40 мл добавляли метил-6-аминоимидазо[1,5-а]пиридин-1-карбоксилат (200,00 мг, 1,046 ммоль, 1,00 экв.) и LiAlH4 (119,11 мг, 3,138 ммоль, 3 экв.) в THF (15,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение 5 ч при 65°С в воздушной атмосфере. Реакционную смесь гасили добавлением NaOH (120 мг в 1 мл) при комнатной температуре. Полученную смесь фильтровали, осадок на фильтре промывали DCM (3×8 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CHCl3/МеОН 10:1) с получением [6-аминоимидазо[1,5-а]пиридин-1-ил]метанола (53 мг, 42,34%) в виде черного масла. Этот неочищенный продукт использовали на следующей стадии без другой очистки. ЖХ-МС (ИЭР): m/z [М+Н]+=164,0
Стадия 7. (R)-N-[3-(5-Фтор-2-[[1-(гидроксиметил)имидазо[1,5-а]пиридин-6-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 46)
В раствор (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (100,00 мг, 0,224 ммоль, 1,00 экв.) и [6-аминоимидазо[1,5-а]пиридин-1-ил]метанола (36,51 мг, 0,224 ммоль, 1,00 экв.) в диоксане (10,00 мл) добавляли BrettPhos (12,01 мг, 0,022 ммоль, 0,10 экв.), BrettPhos Pd G3 (20,28 мг, 0,022 ммоль, 0,10 экв.) и K2СО3 (61,85 мг, 0,448 ммоль, 2,00 экв.). После перемешивания в течение 2 ч при 80°С в атмосфере азота полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (DCM:MEOH 10:1). Неочищенный продукт (20 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 19 В до 39 В за 7 мин; 254/220 нм; RT1: 6,47) с получением (R)-N-[3-(5-фтор-2-[[1-(гидроксиметил)имидазо[1,5-а]пиридин-6-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (7 мг, 5,29%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=574,5 1Н-ЯМР (300 МГц, DMSO-d6) δ2.15 (3H, s), 2.37 (4Н, s), 2.55-2.85 (2Н, m), 3.30 (2Н, s), 3.32 (3H, s), 3.49-3.53 (1Н, m), 3.66-3.71 (1H, m), 3.78 (1H, d), 4.67 (2H, d), 4.89-4.93 (1H, m), 6.97 (1H, d), 7.10-7.15 (1H, m), 7.54 (1H, d), 7.60 (1H, d), 8.22 (2H, d), 8.49 (1H, d), 8.56 (1H, d), 9.06 (1H, s), 9.48 (1H, s), 9.87 (1H, s), 11.49 (1H, s).
Пример 52
Получение метил-(R)-(5-((5-фтор-4-(7-(3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо)-1Н-индол-3-ил)пиримидин-2-ил)амино)пиридин-2-ил)карбамата
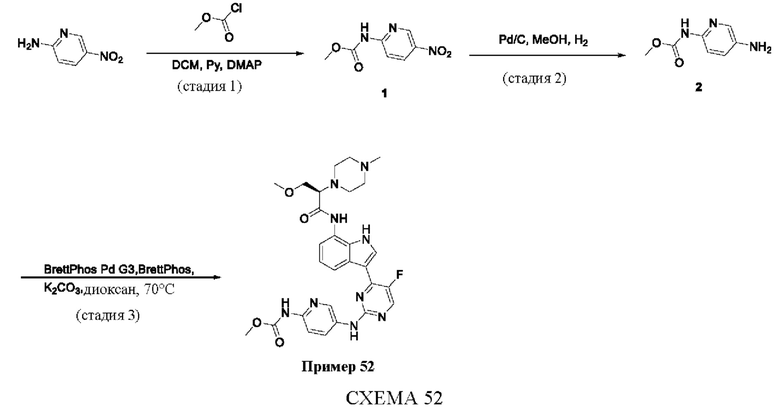
Стадия 1. Метил-(5-нитропиридин-2-ил)карбамат
В перемешиваемый раствор 5-нитро-2-пиридинамина (500,00 мг, 3,594 ммоль, 1,00 экв.), DMAP (87,82 мг, 0,719 ммоль, 0,20 экв.) и пиридина (852,90 мг, 10,783 ммоль, 3,00 экв.) в DCM (25,00 мл) добавляли метилхлорформиат (679,23 мг, 7,188 ммоль, 2,00 экв.) по каплям при 0°С в атмосфере азота. Полученную смесь перемешивали в течение 13 ч при 30°С в атмосфере азота. Выпавшее в осадок твердое вещество собирали фильтрованием и промывали CH2Cl2 (1×3 мл) с получением метил-N-(5-нитропиридин-2-ил)карбамата (300 мг, 42,34%) (неочищенного) в виде коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=198,2.
Стадия 2. Метил-(5-аминопиридин-2-ил)карбамат
Смесь метил-N-(5-нитропиридин-2-ил)карбамата (250,00 мг, 1,268 ммоль, 1,00 экв.) и Pd/C (161,94 мг, 1,522 ммоль, 2,00 экв.) в МеОН (15,00 мл) перемешивали в течение 2 ч при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (2 × 10 мл). Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН=20:1) с получением метил-N-(5-аминопиридин-2-ил)карбамата (89 мг, 41,98%) в виде не совсем белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=168,2.
Стадия 3. Метил-(R)-(5-((5-фтор-4-(7-(3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо)-1Н-индол-3-ил)пиримидин-2-ил)амино)пиридин-2-ил)карбамат (Пр. 52)
Смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (110,00 мг, 0,246 ммоль, 1,00 экв.), RuPhos Palladacycle Gen.3 (20,59 мг, 0,025 ммоль, 0,10 экв.), RuPhos (11,49 мг, 0,025 ммоль, 0,10 экв.), K2СО3 (68,03 мг, 0,492 ммоль, 2,00 экв.) и метил-N-(5-аминопиридин-2-ил)карбамата (61,72 мг, 0,369 ммоль, 1,50 экв.) в 1,4-диоксане (8,00 мл) перемешивали в течение 2 ч при 70°С в атмосфере азота. Полученную смесь фильтровали, осадок на фильтре промывали CH2Cl2 (2×5 мл). Полученную смесь концентрировали под вакуумом. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 8:1) с получением неочищенного продукта (110 мг), который очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 28 В до 48 В за 7 мин; 254;220 нм; RT1: 5,82) с получением метил-N-[5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-l-ил)пропанамидо]-lH-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]карбамата (65 мг, 45,72%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=578,4. 1Н-ЯМР (400 МГц, DMSO-d6) δ 2.15 (3H, s), 2.35 (4Н, s), 2.60-2.68 (2Н, m), 2.74 (2Н, s), 3.30 (3H, s), 3.51 (1Н, t), 3.68 (4Н, s), 3.80 (1H, dd), 7.12 (1H, t), 7.53 (1H, d), 7.77 (1H, d), 8.14 (1H, dd), 8.23 (1H, d), 8.43 (1H, d), 8.50 (1H, d), 8.60 (1H, d), 9.53 (1H, s), 9.86 (1H, s), 9.99 (1H, s), 11.48 (1H, s).
Пример 53
Получение (R)-Н-[3-[5-фтор-2-([1-[2-(гидроксиметил)фенил]пиразол-4-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
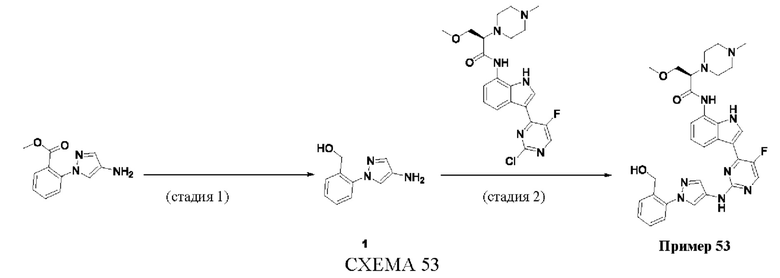
Стадия 1. [2-(4-Аминопиразол-1-ил)фенил1 метанол
В круглодонную колбу на 50 мл добавляли метил-2-(4-аминопиразол-1-ил)бензоат (350,00 мг, 1,611 ммоль, 1,00 экв.) и LiAlH4 (183,46 мг, 4,834 ммоль, 3,00 экв.) в THF (20,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение 1 ч при комнатной температуре в воздушной атмосфере. Реакционную смесь гасили добавлением NaOH при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без другой очистки. ЖХ-МС (ИЭР): m/z [М+Н]+=190,3.
Стадия 2. (R)-N-[3-[5-Фтор-2-([1-[2-(гидроксиметил)фенил]пиразол-4-ил]амино)-пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 53)
В раствор (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1H-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (50 мг, 0,112 ммоль, 1,00 экв.) и [2-(4-аминопиразол-1-ил)фенил]метанола (31,75 мг, 0,168 ммоль, 1,50 экв.) в диоксане (5,00 мл) добавляли BrettPhos Pd G3 (10,14 мг, 0,011 ммоль, 0,10 экв.), BrettPhos (6,01 мг, 0,011 ммоль, 0,10 экв.) и Cs2CO3 (109,36 мг, 0,336 ммоль, 3,00 экв.). После перемешивания в течение 2 ч при 80°С в атмосфере азота полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1). Неочищенный продукт (60 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 29 В до 49 В за 7 мин; 254;220 нм; RT1: 6,22). Неочищенный продукт (30 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: CHIRALPAK IC-3, 4,6 × 50 мм, 3 мкм; подвижная фаза A:(Hex:DCM=3:l)(0.1%DEA):EtOH=50:50, подвижная фаза В; скорость потока: 1 мл/мин; градиент: от 0 В до 0 В) с получением (R)-N-[3-[5-фтор-2-([1-[2-(гидроксиметил)фенил]пиразол-4-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (7 мг, 10,43%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=600,3. 1Н-ЯМР (300 МГц, DMSO-d6) δ 2.24 (3H, s), 2.49 (4Н, s), 2.68 (2Н, s), 2.78 (2Н, s), 3.30 (3H, s), 3.53 (1Н, t), 3.63-3.83 (2Н, m), 4.51 (2Н, d), 5.25-5.27 (1Н, m), 7.11 (1Н, s), 7.43 (3H, d), 7.52 (1Н, d), 7.66 (1H, s), 7.85 (1H, s), 8.21 (1H, s), 8.31 (1H, s), 8.42 (2Н, d), 9.53 (1H, s), 9.87 (1H, s), 11.45 (1H, s).
Пример 54
Получение (R)-N-[3-(5-фтор-2-[[6-(1,3-оксазол-2-илметокси)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
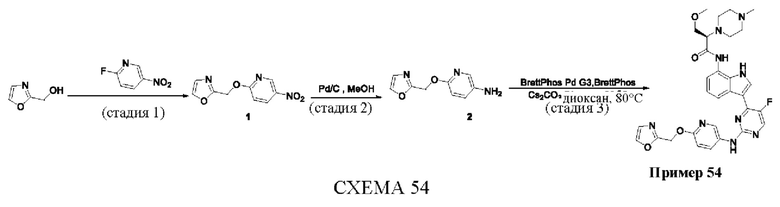
Стадия 1. 5-Нитро-2-(1,3-оксазол-2-илметокси)пиридин
В перемешиваемую смесь 1,3-оксазол-2-илметанола (500,00 мг, 5,046 ммоль, 1,00 экв.) и NaH (157,42 мг, 6,560 ммоль, 1,30 экв.) при 0°С в DMF (20,00 мл) добавляли 2-фтор-5-нитропиридин (716,98 мг, 5,046 ммоль, 1,00 экв.) по каплям при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 ч при комнатной температуре в воздушной атмосфере. Полученную смесь разбавляли водой (150 мл) и экстрагировали EtOAc (3 × 200 мл). Объединенные органические слои промывали рассолом (3 × 50 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Это привело к получению 5-нитро-2-(1,3-оксазол-2-илметокси)пиридина (900 мг, 80,64%) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=222,2. 1Н-ЯМР (300 МГц, MeOD-d4) δ 5.63 (2Н, s), 7.08 (1H, dd), 7.22 (1H, d), 7.97 (1H, d), 8.52 (1H, dd), 9.07 (1H, dd).
Стадия 2. 6-(1,3-Оксазол-2-илметокси)пиридин-3-амин
Смесь 5-нитро-2-(1,3-оксазол-2-илметокси)пиридина (500,00 мг) и Pd/C (20,00 мг) в МеОН (30,00 мл) перемешивали при комнатной температуре в атмосфере водорода в течение 1 ч. Полученную смесь фильтровали, и осадок на фильтре промывали метанолом (3 × 100 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1) с получением 6-(1,3-оксазол-2-илметокси)пиридин-3-амина (420 мг, 97,2%) в виде коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=192,2. 1Н ЯМР (300 МГц, MeOD-d4) δ 5.34 (2Н, s), 6.70 (1H, dd), 7.17 (3H, m), 7.61 (1Н, dd), 7.92 (2Н, d).
Стадия 3. (R)-N-[3-(5-Фтор-2-[[6-(1,3-оксазол-2-илметокси)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 54)
В перемешиваемый раствор (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (120,00 мг, 0,269 ммоль, 1,00 экв.) и 6-(1,3-оксазол-2-илметокси)пиридин-3-амина (102,67 мг, 0,537 ммоль, 2,00 экв.) в диоксане (20,00 мл) добавляли BrettPhos Pd G3 (36,51 мг, 0,040 ммоль, 0,15 экв.) и BrettPhos (21,62 мг, 0,040 ммоль, 0,15 экв.) и K2СО3 (111,33 мг, 0,806 ммоль, 3,00 экв.) при комнатной температуре в атмосфере азота. Полученную смесь перемешивали в течение 2 ч при 80°С в атмосфере азота. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1) с получением неочищенного твердого вещества. Этот неочищенный продукт (100 мг) очищали хиральной препаративной ВЭЖХ в нижеследующих условиях (колонка: CHIRAL ART Cellulose-SB, 4,6 × 100 мм, 3 мкм; подвижная фаза A: MtBE (0,1% DEA):EtOH=90:10, подвижная фаза В; скорость потока: 1 мл/мин; градиент: от 0 В до 0 В) с получением (R)-N-[3-(5-фтор-2-[[6-(1,3-оксазол-2-илметокси)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (71.9 мг, 44.06%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+=602,4. 1Н ЯМР (300 МГц, DMSO-d6) δ 2.16 (3H, s), 2.37 (4Н, s), 2.63 (2Н, m), 2.76 (2Н, m), 3.51 (3H, t), 3.69 (2Н, dd), 3.81 (1H, dd), 5.44 (2Н, s), 6.93 (1H, d), 7.13 (1H, t), 7.26 (1H, d), 7.54 (1H, dd), 8.12 (2Н, m), 8.24 (1H, d), 8.42 (1H, d), 8.48 (2Н, m), 9.48 (1H, s), 9.86 (1H, s), 11.47 (1H, s).
Пример 55
Получение метил-(R)-3-(6-((5-фтор-4-(7-(3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо)-1Н-индол-3-ил)пиримидин-2-ил)амино)пиридин-2-ил)пропаноата
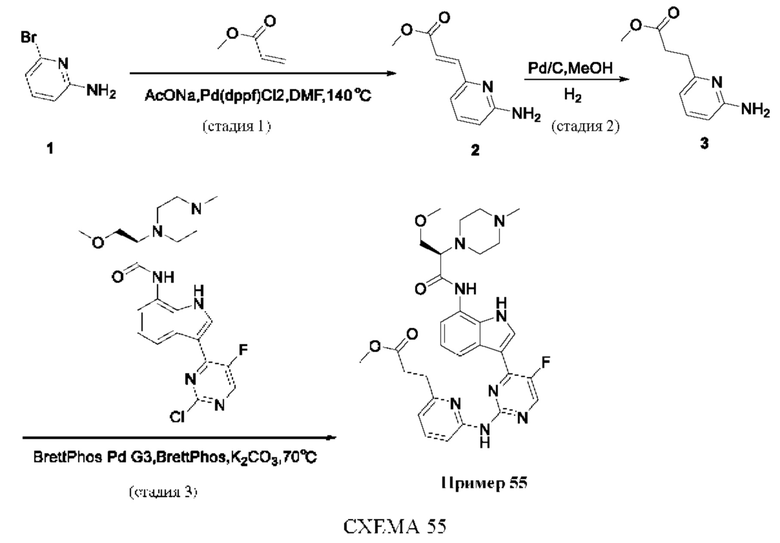
Стадия 1. Получение метил-3-(6-аминопиридин-2-ил)акрилата
Смесь метилакрилата (0,75 г, 8,712 ммоль, 1,51 экв.) и 6-бромпиридин-2-амина (1,00 г, 5,780 ммоль, 1,00 экв.) в DMF (20,00 мл), AcONa (0,95 г, 11,581 ммоль, 2,00 экв.) и Pd(dppf)Cl2 (0,42 г, 0,574 ммоль, 0,10 экв.) перемешивали при 140°С в атмосфере азота. Полученную смесь экстрагировали CH2Cl2 (3 × 20 мл). Объединенные органические слои промывали водой (3 × 50 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1) с получением метил-3-(6-аминопиридин-2-ил)проп-2-еноата (450 мг, 40,02%) в виде желтого твердого вещества. [М+Н]+=179,0.
Стадия 2. Метил-3-(б-аминопиридин-2-ил)пропаноат
Смесь метил-3-(6-аминопиридин-2-ил)проп-2-еноата (80 мг, 0,449 ммоль, 1,00 экв.) и Pd/C (9,56 мг, 0,090 ммоль, 0,20 экв.) в МеОН (8,00 мл) перемешивали при комнатной температуре в атмосфере водорода в течение 1 ч. Полученную смесь фильтровали, и осадок на фильтре промывали МеОН (3 × 10 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1) с получением метил-3-(6-аминопиридин-2-ил)пропаноата (135 мг, 64,54%) в виде желтого твердого вещества. [М+Н]+=181,1.
Стадия 3. Получение метил-(R)-3-(6-((5-фтор-4-(7-(3-метокси-2-(4-метил-пиперазин-1-ил)пропанамидо)-1Н-индол-3-ил)пиримидин-2-ил)амино)пиридин-2-ил)пропаноата (Пр. 55)
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (150,00 мг, 0,336 ммоль, 1,00 экв.) и метил-3-(6-аминопиридин-2-ил)пропаноата (90,73 мг, 0,503 ммоль, 1,50 экв.) в диоксане (5,00 мл) добавляли BrettPhos Pd G3 (45,64 мг, 0,050 ммоль, 0,15 экв.), K2СО3 (92,77 мг, 0,671 ммоль, 2,00 экв.) и BrettPhos (36,03 мг, 0,067 ммоль, 0,20 экв.). Полученную смесь перемешивали при 70°С в атмосфере азота. Полученную смесь фильтровали, и осадок на фильтре промывали DCM (3 × 20 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1) с получением неочищенного продукта (100 мг), который очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD C18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 31% В до 45% В за 7 мин; 254;220 нм; Rt: 6,30 мин) с получением метил-3-[6-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метил-пиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]пропаноата (33,8 мг, 16,71%) в виде не совсем белого твердого вещества. [М+Н]+=591,4. 1H-ЯМР (300 МГц, DMSO-d6) δ 2.16 (3H, s), 2.37 (4Н, s), 2.64 (2Н, d), 2.80 (4Н, dd), 2.97 (2Н, t), 3.30 (3H, s), 3.51 (1H, t), 3.61 (3H, s), 3.69 (1Н, dd), 3.81 (1Н, dd), 6.89 (1Н, d), 7.15 (1H, t), 7.54 (1H, d), 7.60-7.72 (1H, m), 8.07 (1H, d), 8.27 (1Н, s), 8.50 (1H, d), 8.69-8.78 (1Н, m), 9.84 (2Н, d), 11.48 (1H, s)
Пример 60
Получение (R)-N-(3-(5-фтор-2-((6-(2-гидроксиэтил)пиридин-3-ил)амино)-пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр.60)

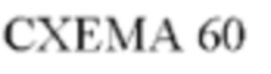
Стадия 1. 2-(5-Аминопиридин-2-ил)этан-1-ол
В круглодонную колбу на 50 мл добавляли LiAlH4 (189,55 мг, 4,994 ммоль, 3,00 экв.) в THF (13 мл) при комнатной температуре. Раствор этил-2-(5-аминопиридин-2-ил)ацетата (300,00 мг, 1,665 ммоль, 1,00 экв.) в THF (7 мл) добавляли в вышеуказанную смесь при 0°С. Полученную смесь перемешивали в течение 0,5 ч при 0°С в воздушной атмосфере. Реакционную смесь гасили добавлением воды (0,2 мл) при комнатной температуре и затем 15% NaOH (0,6 мл), воды (0,2 мл). Полученную смесь сушили над безводным MgSO4, твердое вещество отфильтровывали, и фильтрат выпаривали с получением 2-(5-аминопиридин-2-ил)этанола (200 мг, 86,95%) в виде желтого твердого вещества. 1H-ЯМР (400 МГц, CDCl3) δ 2.91 (2Н, t), 3.95-4.03 (2Н, m), 6.91-7.00 (2Н, m), 8.00 (1H, t)
Стадия 2. (R)-N-(3-(5-Фтор-2-((6-(2-гидроксиэтил)пиридин-3-ил)амино)-пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 60)
В сосуд вместимостью 40 мл добавляли (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (100,00 мг, 0,224 ммоль, 1,00 экв.), 2-(5-аминопиридин-2-ил)этанол (37,10 мг, 0,269 ммоль, 1,20 экв.), BrettPhos (12,01 мг, 0,022 ммоль, 0,10 экв.), BrettPhos Pd G3 (20,28 мг, 0,022 ммоль, 0,10 экв.) и Cs2CO3 (218,72 мг, 0,671 ммоль, 3,00 экв.) в диоксане (20 мл) при комнатной температуре. Полученную смесь перемешивали в течение 1,5 ч при 80°С. Твердое вещество отфильтровывали, и осадок на фильтре промывали МеОН (2×10 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 7:1) с получением неочищенного твердого вещества. Этот неочищенный продукт (80 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30×150 мм, 5 мкм; подвижная фаза А:; подвижная фаза В; скорость потока: 60 мл/мин; градиент: % В; 254, 220 нм; RT1: 7,25) с получением (R)-N-[3-(5-фтор-2-[[6-(2-гидроксиэтил)пиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метил-пиперазин-1-ил)пропанамида (25 мг, 20,37%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 549,3 1Н-ЯМР (400 МГц, DMSO-d6) δ 2.15 (3Н, s), 2.35 (4Н, s), 2.58-2.66 (2Н, m), 2.75 (2Н, dt), 2.85 (2Н, t), 3.30 (3Н, s), 3.51 (1Н, t), 3.64-3.84 (4Н, m), 4.64 (1Н, t), 7.08-7.27 (2Н, m), 7.55 (1Н, dd), 8.12 (1Н, dd), 8.24 (1Н, d), 8.44 (1Н, d), 8.50-8.56 (1Н, m), 8.78 (1H, dd), 9.59 (1H, s), 9.88 (1H, s), 11.47 (1H, s).
Пример 61
Получение (R)-N-(3-(5-фтор-2-((4-(гидроксиметил)-1Н-индазол-6-ил)амино)-пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
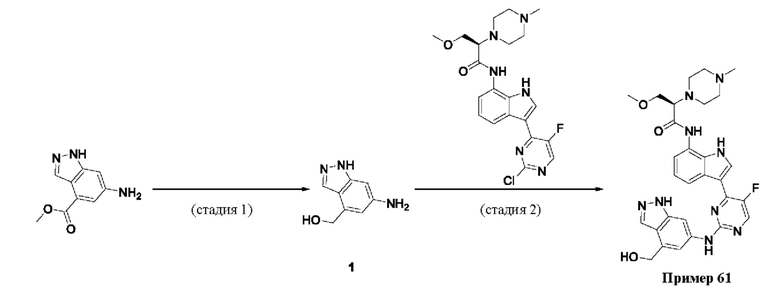
СХЕМА 61
Стадия 1. Получение (6-амино-1Н-индазол-4-ил)метанола
В перемешиваемую смесь метил-6-амино-1Н-индазол-4-карбоксилата (300,00 мг, 1,569 ммоль, 1,00 экв.) в THF (5,00 мл) добавляли LiAlH4 (178,66 мг, 4,707 ммоль, 3,00 экв.) порциями при 0°С. Полученную смесь перемешивали в течение 1 ч при 70°С. Реакционную смесь гасили добавлением воды (0,08 мл) и NaOH (0,08 мл, 15%) при 0°С. Полученную смесь фильтровали, и осадок на фильтре промывали THF (3×10 мл). Фильтрат концентрировали при пониженном давлении. Это дало (6-амино-1H-индазол-4-ил)метанол (100 мг, 39,06%) в виде светло-желтого масла. ЖХ-МС (ИЭР): m/z [М+Н]+ = 164,2.
Стадия 2. Получение (R)-N-(3-(5-фтор-2-((4-(гидроксиметил)-1Н-индазол-6-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 61)
Смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (50,00 мг, 0,112 ммоль, 1,00 экв.), (6-амино-1Н-индазол-4-ил)метанола (21,91 мг, 0,134 ммоль, 1,20 экв.), K2CO3 (46,39 мг, 0,336 ммоль, 3,00 экв.), BrettPhos (12,01 мг, 0,022 ммоль, 0,20 экв.) и BrettPhos Pd G3 (10,14 мг, 0,011 ммоль, 0,10 экв.) в диоксане (10,00 мл) перемешивали в течение 2 ч при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 12:1) с получением (R)-N-[3-(5-фтор-2-[[4-(гидроксиметил)-1Н-индазол-6-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (20 мг, неочищенный) в виде светло-желтого твердого вещества. Этот неочищенный продукт (20 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 21 В до 41 В за 7 мин; 254/220 нм; RT1: 5,65) с получением (R)-N-[3-(5-фтор-2-[[4-(гидроксиметил)-1Н-индазол-6-ил]амино]-пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (2,5 мг, 3,90%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 574,4. 1Н-ЯМР (300 МГц, MeOD-d4) δ 2.31 (3Н, s), 2.58 (4Н, s), 2.86 (4Н, d), 3.41 (3Н, s), 3.49 (1Н, t), 3.75-3.98 (2Н, m), 7.04-7.22 (2Н, m), 7.30 (1Н, d), 8.07-8.19 (3Н, m), 8.29 (1Н, d), 8.67 (1Н, dd)
Пример 66
Получение (R)-N-[3-(5-фтор-2-[[6-(2-гидроксиэтил)-5-метоксипиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
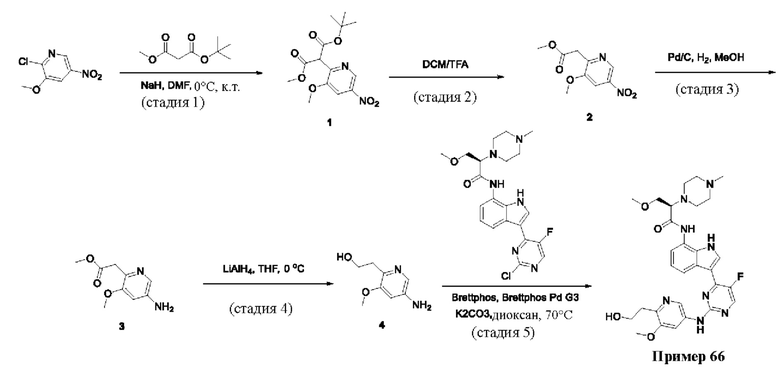
СХЕМА 66
Стадия 1. 1-трет-Бутил-3-метил-2-(3-метокси-5-нитропиридин-2-ил)-пропандиоат
Раствор 2-хлор-3-метокси-5-нитропиридина (1,00 г, 5,303 ммоль, 1,00 экв.) в DMF (100.0 мл) обрабатывали NaH (0,32 г, 13,258 ммоль, 2,50 экв.) при 0°С. Раствор перемешивали в течение 10 мин при комнатной температуре. В вышеуказанную смесь добавляли 1-трет-бутил-3-метил-пропандиоат (1,52 г, 8,750 ммоль, 1,65 экв.) по каплям при 0°С. Полученную смесь перемешивали в течение 15 ч при комнатной температуре. Полученную смесь гасили водой (30 мл) и экстрагировали EtOAc (3×35 мл). Объединенные органические слои промывали рассолом (1×30 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (РЕ/EtOAc 5:1) с получением 1-трет-бутил-3-метил-2-(3-метокси-5-нитропиридин-2-ил)пропандиоата (1,46 г, 84,37%) в виде красновато-коричневого масла. ЖХ-МС (ПЭР): m/z [М+Н]+ = 327,3. 1Н-ЯМР (300 МГц, Хлороформ-d) δ 1.50 (9Н, s), 3.82 (3Н, s), 3.98 (3Н, s), 5.09 (1Н, s), 7.94 (1Н, d), 9.02 (1Н, d).
Стадия 2. Метил-2-(3-метокси-5-нитропиридин-2-ил)ацетат
В перемешиваемый раствор 1-трет-бутил-3-метил-2-(3-метокси-5-нитропиридин-2-ил)пропандиоата (1,40 г, 4,290 ммоль, 1,00 экв.) в DCM (20,0 мл) добавляли TFA (6,00 мл, 80,778 ммоль, 18,83 экв.). Полученную смесь перемешивали в течение 18 ч при 25°С. Полученную смесь концентрировали при пониженном давлении. Смесь подщелачивали до рН 8 насыщенным NaHCO3 (водн.). Полученную смесь экстрагировали CH2Cl2 (3×80 мл). Объединенный органический слой сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении с получением метил-2-(3-метокси-5-нитропиридин-2-ил)ацетата (0,88 г, 90,68%) в виде красновато-коричневого масла. ЖХ-МС (ИЭР): m/z [М+Н]+ = 227,2. 1Н-ЯМР (300 МГц, Хлороформ-d) δ 3.74 (3Н, s), 3.98 (3Н, s), 4.00 (2Н, s), 7.92 (1Н, d), 9.01 (1Н, d).
Стадия 3. Метил-2-(5-амино-3-метоксипиридин-2-ил)ацетат
В раствор метил-2-(3-метокси-5-нитропиридин-2-ил)ацетата (840,00 мг, 3,714 ммоль, 1,00 экв.) в МеОН (50 мл) добавляли Pd/C (10%, 79,04 мг) в атмосфере азота в круглодонной колбе на 250 мл. Смесь гидрировали при комнатной температуре в течение 1 ч в атмосфере водорода, используя водородный баллон. Смесь фильтровали через фильтр из целита, и фильтрат концентрировали при пониженном давлении с получением метил-2-(5-амино-3-метоксипиридин-2-ил)ацетата (445 мг, 61,07%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 197,2.
Стадия 4. 2-(5-Амино-3-метоксипиридин-2-ил)этанол
В перемешиваемый раствор LiAlH4 (203,11 мг, 5,352 ммоль, 3,00 экв.) в THF (10 мл) добавляли метил-2-(5-амино-3-метоксипиридин-2-ил)ацетат (350,00 мг, 1,784 ммоль, 1,00 экв.) в THF (20 мл) по каплям при 0°С. Полученную смесь перемешивали в течение 30 мин при 0°С. Реакционную смесь гасили добавлением Na2SO4⋅10H2O. Полученную смесь фильтровали, и осадок на фильтре промывали этилацетатом (3×5 мл). Фильтрат концентрировали при пониженном давлении с получением 2-(5-амино-3-метоксипиридин-2-ил)этанола (243 мг, 80,99%) в виде светло-оранжевого твердого вещества. ЖХ-МС (ПЭР): m/z [М+Н]+ = 169,0.
Стадия 5. (R)-N-[3-(5-фтор-2-[[6-(2-гидроксиэтил)-5-метоксипиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 66)
В раствор (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (130,00 мг, 0,291 ммоль, 1,00 экв.) и 2-(5-амино-3-метоксипиридин-2-ил)этанола (63,60 мг, 0,378 ммоль, 1,3 экв.) в диоксане (10,0 мл) добавляли BrettPhos (31,23 мг, 0,058 ммоль, 0,20 экв.) и BrettPhos Pd G3 (52,74 мг, 0,058 ммоль, 0,20 экв.) и K2CO3 (80,40 мг, 0,582 ммоль, 2,00 экв.). После перемешивания в течение 2 ч при 70°С в атмосфере азота. Остаток очищали ТСХ (CH2Cl2/МеОН 8:1) с получением (R)-N-[3-(5-фтор-2-[[6-(2-гидроксиэтил)-5-метоксипиридин-3-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (34,35 мг, 20,41%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 579,4. 1H-ЯМР (300 МГц, DMSO-d6) δ 2.13 (3Н, s), 2.22-2.44 (4Н, m), 2.54-2.80 (4Н, m), 2.86 (2Н, t), 3.28 (3Н, s), 3.49 (1Н, t), 3.59-3.70 (3Н, m), 3.72-3.84 (4Н, m), 4.57 (1Н, t), 7.11 (1Н, t), 7.52 (1Н, d), 7.85 (1Н, d), 8.23 (1Н, d), 8.30-8.64 (3Н, m), 9.60 (1Н, s), 9.86 (1Н, s), 11.47 (1Н, s).
Пример 67
Получение (R)-N-(3-(5-фтор-2-((1-(3-(гидроксиметил)фенил)-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
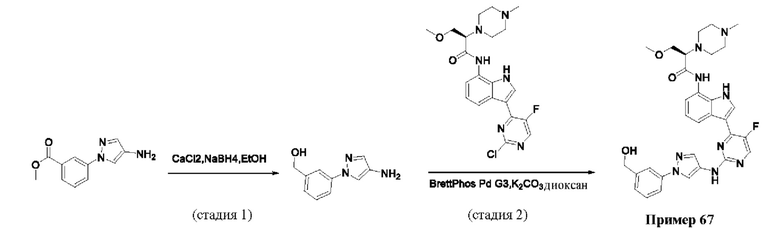
СХЕМА 67
Стадия 1. (3-(4-Амино-1Н-пиразол-1-ил)фенил)метанол
В сосуд вместимостью 40 мл добавляли метил-3-(4-аминопиразол-1-ил)бензоат (130,00 мг, 0,598 ммоль, 1,00 экв.), CaCl2 (99,63 мг, 0,898 ммоль, 1,50 экв.), NaBH4 (67,92 мг, 1,795 ммоль, 3 экв.) и EtOH (15,00 мл) при комнатной температуре, и эту реакционную смесь перемешивали при 0°С в течение 3 ч. Полученную смесь экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали рассолом (3×10 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 25% В до 40% В за 7 мин; 254/220 нм; Rt: 5,77 мин) с получением [3-(4-аминопиразол-1-ил)фенил]метанола (80 мг, 70,65%) в виде белого твердого вещества. ЖХ-МС (ПЭР): m/z [М+Н]+ = 190,3.
Стадия 2. (R)-N-[3-[5-Фтор-2-([1-[3-(гидроксиметил)фенил]пиразол-4-ил]амино)-пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 67)
В сосуд вместимостью 40 мл добавляли (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (100,00 мг, 0,224 ммоль, 1,00 экв.) и [3-(4-аминопиразол-1-ил)фенил]метанол (63,51 мг, 0,336 ммоль, 1,50 экв.), BrettPhos Pd G3 (20,28 мг, 0,022 ммоль, 0,1 экв.), K2CO3 (61,85 мг, 0,448 ммоль, 2 экв.), диоксан (15,00 мл) при комнатной температуре. Затем смесь перемешивали при 70°С в атмосфере азота в течение 3 ч, и ЖХ-МС показала, что все в порядке. Полученную смесь разбавляли водой (10 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали рассолом (3×10 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 25% В до 40% В за 7 мин) с получением (R)-N-[3-[5-фтор-2-([1-[3-(гидроксиметил)фенил]пиразол-4-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (25 мг, 18,63%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 600,4. 1H-ЯМР (300 МГц, DMSO-d6) δ 2.14 (3Н, s), 2.35 (4Н, s), 2.56-2.68 (2Н, m), 2.74 (2Н, q), 3.30 (3Н, s), 3.50 (1Н, t), 3.67 (1Н, dd), 3.79 (1H, dd), 4.56 (2H, d), 5.30 (1H, d), 7.11 (1H, t), 7.21 (1H, d), 7.41 (1H, t), 7.57 (2H, dd), 7.72 (1H, t), 7.82 (1H, s), 8.17-8.25 (1H, m), 8.43-8.63 (3H, m), 9.60 (1H, s), 9.87 (1H, s), 11.46 (1H, s).
Пример 68
Получение (R)-N-(3-(5-фтор-2-((5-(2-(метиламино)-2-оксоэтокси)пиридин-3-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
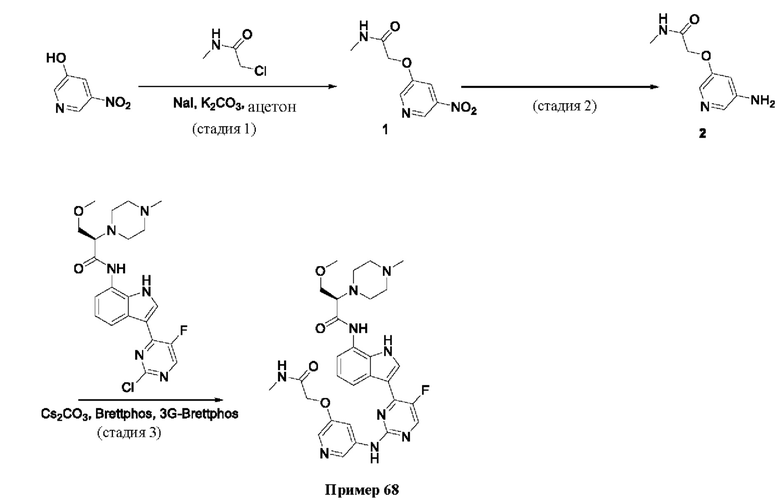
СХЕМА 68
Стадия 1. N-Метил-2-((5-нитропиридин-3-ил)окси)ацетамид
Смесь 5-нитропиридин-3-ола (70,00 мг, 0,500 ммоль, 1,00 экв.), NaI (7,49 мг, 0,050 ммоль, 0,10 экв.), 2-хлор-N-метил-ацетамида (80,60 мг, 0,749 ммоль, 1,50 экв.) и K2CO3 (138,11 мг, 0,999 ммоль, 2,00 экв.) в пропан-2-оне (5,00 мл) перемешивали в течение 2 ч при 65°С в воздушной атмосфере. Полученную смесь концентрировали под вакуумом. Неочищенный продукт подвергали перекристаллизации из смеси EtOAc/РЕ с получением N-метил-2-[(5-нитропиридин-3-ил)окси]ацетамида (525 мг, 69,66%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 212,0. 1Н-ЯМР (400 МГц, CDCl3) δ 2.98 (3Н, d), 4.66 (2Н, s), 8.02 (1Н, t), 8.70 (1Н, d), 9.17 (1Н, d).
Стадия 2. 2-((5-Аминопиридин-3-ил)окси)-N-метилацетамид
В перемешиваемый раствор N-метил-2-[(5-нитропиридин-3-ил)окси]ацетамида (240,00 мг, 1,136 ммоль, 1,00 экв.) в МеОН (20,00 мл) добавляли Pd/C (120,94 мг, 1,136 ммоль, 1,00 экв.). Полученную смесь перемешивали в течение 4 ч при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, и осадок на фильтре промывали МеОН (3×20 мл). Фильтрат концентрировали при пониженном давлении с получением 2-[(5-аминопиридин-3-ил)окси]-N-метилацетамида (201 мг, 97,61%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 182,2.
Стадия 3. (R)-N-(3-(5-Фтор-2-((5-(2-(метиламино)-2-оксоэтокси)пиридин-3-ил)амино)пиримидин-4-ил)-1Н-индол-7-ил)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 68)
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (150,00 мг, 0,336 ммоль, 1,00 экв.) и 2-[(5-аминопиридин-3-ил)окси]-N-метилацетамида (121,63 мг, 0,671 ммоль, 2,00 экв.) в диоксане (2,00 мл) добавляли Brettphos (36,03 мг, 0,067 ммоль, 0,20 экв.) и BrettPhos Pd G3 (60,85 мг, 0,067 ммоль, 0,20 экв.), Cs2CO3 (328,07 мг, 1,007 ммоль, 3,00 экв.). Полученную смесь перемешивали в течение 2 ч при 80°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 8:1) с получением неочищенного продукта. Этот неочищенный продукт (150 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 29 В до 31 В за 7 мин; 254;220 нм; RT1: 5,85) с получением твердого вещества. Неочищенный продукт (80 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: CHIRAL ART Cellulose-SB, 2×25 см, 5 мкм; подвижная фаза А: Нех(8 ммоль/л NH3⋅МеОН)--ВЭЖХ, подвижная фаза В: EtOH--ВЭЖХ; скорость потока: 20 мл/мин; градиент: от 50 В до 50 В за 15 мин; 254/220 нм; RT1: 8,698; RT2: 11,463; впрыскиваемый объем: 0,85 мл; число прогонов: 4) с получением (R)-N-[3-[5-фтор-2-([5-[(метилкарбамоил)метокси]пиридин-3-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (40 мг, 20,14%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 592,3. 1Н-ЯМР (300 МГц, DMSO-d6) δ 2.12 (3Н, s), 2.34 (4Н, s), 2.64-2.7Ц5Н, m), 2.72-2.75 (2H, m), 3.27 (3Н, s), 3.49 (1Н, t), 3.64-3.69 (1H, m), 3.76-3.81 (1H, m), 4.51 (2H, s), 7.14 1H. t), 7.53 (1H, d), 7.91-7.96 (2H, m), 8.06 (1H, d), 8.25 (1H, s), 8.47 (1H, d), 8.56 (2H, t), 9.76(1H, s),9.86 (1H, s), 11.50 (1H, s).
Пример 69
Получение метил-(R)-N-[3-[5-фтор-2-([6-[2-(гидроксиметил)фенил]пиридин-3-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 69)
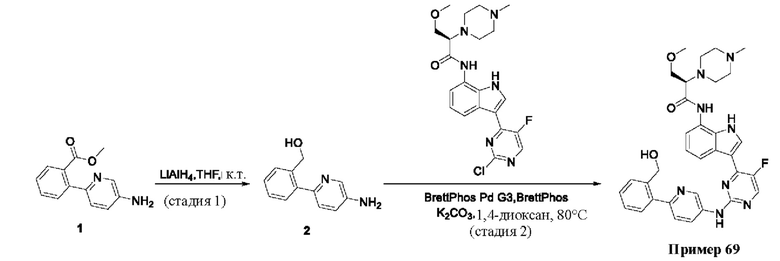
СХЕМА 69
Стадия 1. [2-(5-Аминопиридин-2-ил)фенил]метанол
В перемешиваемый раствор метил-2-(5-аминопиридин-2-ил)бензоата (400,00 мг, 1,752 ммоль, 1,00 экв.) в THF (20,00 мл) добавляли LiAlH4 (266,05 мг, 7,010 ммоль, 4,00 экв.) порциями при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 1 ч при комнатной температуре в воздушной атмосфере. Реакционную смесь гасили добавлением воды (0,3 мл) при 0°С. Смесь подщелачивали до рН 7 NaOH (266 мг). Полученную смесь фильтровали, и осадок на фильтре промывали CH2Cl2 (3×30 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 20:1) с получением [2-(5-аминопиридин-2-ил)фенил]метанола (135 мг, 38,47%) в виде красного твердого вещества. ЖХ-МС (ПЭР): m/z [М+Н]+ = 201,2.
Стадия 2. (R)-N-[3-[5-Фтор-2-([6-[2-(гидроксиметил)фенил]пиридин-3-ил]амино)-пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (Пр. 69)
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (120,00 мг, 0,269 ммоль, 1,00 экв.) и 3-(5-аминопиридин-2-ил)-2-метилпента-2,4-диен-1-ола (102,17 мг, 0,537 ммоль, 2,00 экв.) в диоксане (20,00 мл) добавляли BrettPhos Pd G3 (36,51 мг, 0,040 ммоль, 0,15 экв.), BrettPhos (21,62 мг, 0,040 ммоль, 0,15 экв.) и K2CO3 (111,33 мг, 0,806 ммоль, 3,00 экв.) при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 ч при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1). Неочищенный продукт (100 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: YMC-Actus Triart С18 30×250, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 52 В до 72 В за 7 мин; 254;220 нм; RT1: 6,05) с получением (R)-N-[3-[5-фтор-2-([6-[2-(гидроксиметил)фенил]пиридин-3-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (52,3 мг, 31,89%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 611,4. 1H-ЯМР (300 МГц, DMSO-d6) δ 2.13 (3Н, s), 2.34 (4Н, s), 2.63 (2Н, s), 2.74 (2Н, s), 3.49 (3Н, t), 3.67 (1Н, dd), 3.79 (2Н, dd), 4.55 (2Н, d), 5.45 (1Н, t), 7.17 (1Н, t), 7.37 (2Н, m), 7.55 (4Н, m), 8.26 (1Н, d), 8.32 (1Н, dd), 8.50 (1Н, d), 8.56 (1Н, d), 9.02 (1Н, d), 9.85 (2Н, d), 11.49 (1Н, s).
Пример 74
Получение (R)-N-[3-(5-фтор-2-[[1-(1-метилпиперидин-4-ил)-1Н-пиразол-4-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
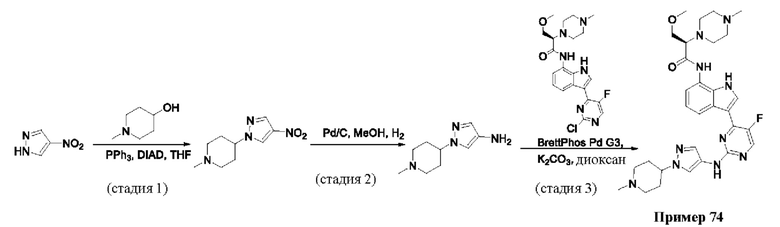
СХЕМА 74
Стадия 1. 1-Метил-4-(4-нитропиразол-1-ил)пиперидин
В перемешиваемую смесь 4-нитропиразола (30,00 мг, 0,265 ммоль, 1,00 экв.) и 1-метилпиперидин-4-ола (91,67 мг, 0,796 ммоль, 3,00 экв.) в THF (2,00 мл) добавляли PPh3 (208,76 мг, 0,796 ммоль, 3,00 экв.) и DIAD (160,94 мг, 0,796 ммоль, 3,00 экв.) порциями при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 ч при 70°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 1:1) с получением 1-метил-4-(4-нитропиразол-1-ил) пиперидина (10,33 мг, 18,52%) в виде коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 211,2.
Стадия 2. 1-(1-Метилпиперидин-4-ил)пиразол-4-амин
В перемешиваемую смесь 1-метил-4-(4-нитропиразол-1-ил)пиперидина (500,00 мг) и Pd/C (20,00 мг) в МеОН (20,00 мл) порциями при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 1 ч при комнатной температуре в атмосфере H2. Полученную смесь фильтровали, и осадок на фильтре промывали МеОН (3×30 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 1:1) с получением 1-(1-метилпиперидин-4-ил)пиразол-4-амина (333 мг) в виде красновато-коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 181,3.
Стадия 3. (R)-N-[3-(5-фтор-2-[[1-(1-метилпиперидин-4-ил)пиразол-4-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид Пр. 74)
В перемешиваемую смесь (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (120,00 мг, 0,269 ммоль, 1,00 экв.) и 1-(1-метилпиперидин-4-ил)пиразол-4-амина (72,60 мг, 0,403 ммоль, 1,5 экв.) в диоксане (20,00 мл) добавляли BrettPhos Pd G3 (36,51 мг, 0,040 ммоль, 0,15 экв.), BrettPhos (21,62 мг, 0,040 ммоль, 0,15 экв.) и K2CO3 (111,33 мг, 0,806 ммоль, 3 экв.) порциями при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 ч при 70°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 10:1). Неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 37 В до 57 В за 7 мин; RT1: 6,03) с получением (R)-N-[3-(5-фтор-2-[[1-(1-метилпиперидин-4-ил)пиразол-4-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (24 мг,15,13%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 591,4. 1Н ЯМР (300 МГц, DMSO-d6) δ 1.99 (6Н, m), 2.18 (6Н, d), 2.36 (4Н, s), 2.63 (2Н, m), 2.74 (1Н, s), 2.77 (1Н, d), 2.86 (2Н, d), 3.30 (3Н, s), 3.51 (1Н, t), 3.69 (1Н, dd), 3.81 (1Н, dd), 4.06 (1Н, dq), 7.13 (1Н, t), 7.53 (2Н, m), 7.98 (1Н, s), 8.20 (1Н, s), 8.38 (1Н, d), 8.40 (1Н, s), 9.30 (1Н, s), 9.86 (1Н, s), 11.43 (1Н, s).
Пример 75
Получение (R)-N-[3-[5-фтор-2-([2-[2-(гидроксиметил)фенил]пиридин-4-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
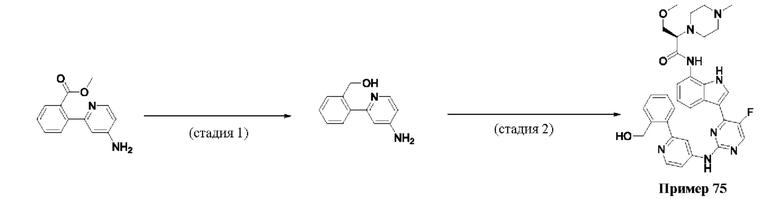
СХЕМА 75
Стадия 1. [2-(4-Аминопиридин-2-ил)фенил]метанол
В круглодонную колбу на 50 мл добавляли метил-2-(4-аминопиридин-2-ил)бензоат (200,00 мг, 0,876 ммоль, 1,00 экв.) и LiAlH4 (133,03 мг, 3,505 ммоль, 4,00 экв.) в THF (10,00 мл) при комнатной температуре. Полученную смесь перемешивали в течение в течение ночи при 70°С в воздушной атмосфере. Реакционную смесь гасили добавлением NaOH (133 мг в воде) при 5°С. Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН 10:1 с TEA) с получением [2-(4-аминопиридин-2-ил)фенил]метанола (70 мг, 29,12%) в виде черного масла. ЖХ-МС (ПЭР): m/z [М+Н]+ = 201,0.
Стадия 2. (R)-N-[3-[5-Фтор-2-([2-[2-(гидроксиметил)фенил]пиридин-4-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 75)
В раствор [2-(4-аминопиридин-2-ил)фенил]метанола (67,21 мг, 0,336 ммоль, 1,50 экв.) и (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (100 мг, 0,224 ммоль, 1,00 экв.) в диоксане (10,00 мл) добавляли BrettPhos Pd G3 (20,28 мг, 0,022 ммоль, 0,10 экв.), BrettPhos (12,01 мг, 0,022 ммоль, 0,10 экв.) и K2CO3 (61,85 мг, 0,448 ммоль, 2,00 экв.). После перемешивания в течение 2 ч при 70°С в атмосфере азота полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (PE/EtOAc 3:1) с получением неочищенного твердого вещества. Это неочищенное твердое вещество очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: YMC-Actus Triart С18, 30×250, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: A CN; скорость потока: 60 мл/мин; градиент: от 52 В до 72 В за 7 мин; 254,220 нм; RT1: 6,05) с получением (R)-N-[3-[5-фтор-2-([2-[2-(гидроксиметил)фенил]пиридин-4-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (25 мг, 18,11%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 611,3. 1Н-ЯМР (400 МГц, DMSO-d6) δ 2.15 (3Н, s), 2.36 (4Н, s), 2.75 (4Н, s), 3.28 (3Н, s), 3.50 (1Н, t), 3.69 (1Н, dd), 3.76-3.84 (1Н, m), 4.50 (2Н, d), 5.62 (1Н, t), 7.08 (1Н, t), 7.35 (1Н, t), 7.42 (1Н, t), 7.50 (2Н, dd), 7.58 (1Н, d), 7.79-7.85 (1Н, m), 8.05 (1Н, d), 8.27 (1Н, s), 8.44 (1Н, d), 8.52-8.60 (2Н, m), 9.86 (1Н, s), 10.14 (1Н, s), 11.51 (1Н, s). Пример 76
Получение (R)-N-[3-(2-[[6-(аминометил)пиридин-3-ил]амино]-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1 -ил)пропанамида
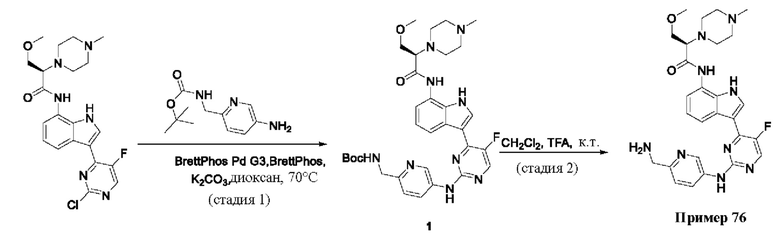
СХЕМА 76
Стадия 1. трет-Бутил-N-([5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]метил)карбамат
В перемешиваемый раствор/смесь (R)-Н-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (150,00 мг, 0,336 ммоль, 1,00 экв.) и трет-бутил-N-[(5-аминопиридин-2-ил)метил]карбамата (149,88 мг, 0,671 ммоль, 2 экв.) в диоксане (20,00 мл) добавляли BrettPhos Pd G3 (45,64 мг, 0,050 ммоль, 0,15 экв.), BrettPhos (27,02 мг, 0,050 ммоль, 0,15 экв.) и K2CO3 (139,16 мг, 1,007 ммоль, 3 экв.) порциями при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 ч при 80°С в атмосфере азота. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 10:1) с получением трет-бутил-N-([5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]метил)карбамата (150 мг, 70,52%) в виде коричневого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 634,4.
Стадия 2. (R)-N-[3-[5-Фтор-2-([6-[2-(гидроксиметил)фенил]пиридин-3-ил]амино)пиримидин-4-ил]-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (Пр. 76)
В перемешиваемый раствор трет-бутил-N-([5-[(5-фтор-4-[7-[(R)-3-метокси-2-(4-метилпиперазин-1-ил)пропанамидо]-1Н-индол-3-ил]пиримидин-2-ил)амино]пиридин-2-ил]метил)карбамата (100,00 мг) в CH2Cl2 (3,00 мл) и TFA (10,00 мл) по каплям при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 1 ч при комнатной температуре в воздушной атмосфере. Полученную смесь экстрагировали CH2Cl2 (3 × 30 мл). Объединенные органические слои промывали рассолом (1×30 мл) и сушили над безводным Na2SO4. После фильтрования фильтрат концентрировали при пониженном давлении. Неочищенный продукт (80 мг) очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30 × 150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 17 В до 37 В за 7 мин; 254/220 нм; RT1: 6,58) с получением (R)-N-[3-(2-[[6-(аминометил)пиридин-3-ил]амино]-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (24,1 мг) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 534,2. 1Н-ЯМР (300 МГц, MeOD-d4) δ 2.33 (3Н, s), 2.61 (4Н, s), 2.85 (2Н, s), 2.93 (2Н, s), 3.44 (3Н, s), 3.52 (1Н, t), 3.90 (1Н, m), 3.95 (3Н, s), 7.21 (2Н, m), 7.42 (1Н, d), 8.19 (1Н, d), 8.32 (2Н, q), 8.65 (1Н, m), 8.88 (1Н, d).
Пример 78
Получение (R)-N-[3-(5-фтор-2-[[2-(гидроксиметил)-6-метилпиридин-4-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида
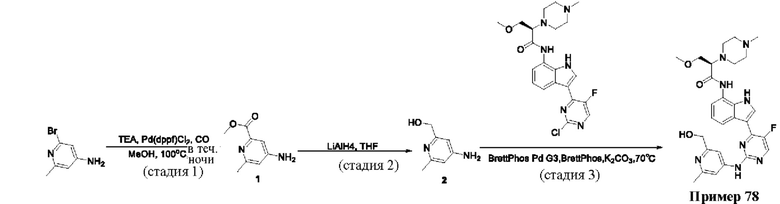
СХЕМА 78
Стадия 1. Метил-4-амино-6-метилпиридин-2-карбоксилат
В реактор высокого давления на 250 мл добавляли 2-бром-6-метилпиридин-4-амин (1.00 г, 5,346 ммоль, 1,00 экв.), Pd(dppf)Cl2 CH2Cl2 (436,61 мг, 0,535 ммоль, 0,10 экв.) и TEA (1,623 г, 16,039 ммоль, 3,00 экв.) в МеОН (50,00 мл) под давлением 20 атм СО (газ) при 100°С в течение 6 ч. Целевой продукт можно было обнаружить методом ЖХ-МС. Полученную смесь концентрировали под вакуумом. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью РЕ/EtOAc (1:1), с получением метил-4-амино-6-метилпиридин-2-карбоксилата (500 мг, 56,28%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 167,3.
Стадия 2. (4-Амино-6-метилпиридин-2-ил)метанол
В герметичную пробирку на 40 мл добавляли метил-4-амино-6-метилпиридин-2-карбоксилат (332,00 мг, 1,998 ммоль, 1,00 экв.) и LiAlH4 (151,65 мг, 3,996 ммоль, 2,00 экв.) в THF (15,00 мл) при 0°С, затем смесь перемешивали при комнатной температуре в течение 1 ч. Целевой продукт можно было обнаружить методом ЖХ-МС. Реакционную смесь гасили добавлением воды (1 мл) при 0°С. Выпавшее в осадок твердое вещество собирали фильтрованием и промывали МеОН (2×50 мл). Полученную смесь концентрировали при пониженном давлении. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 10:1) с получением (4-амино-6-метилпиридин-2-ил)метанола (210 мг, 76,08%) в виде желтого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 139,2.
Стадия 3. (R)-N-[3-(5-Фтор-2-[[2-(гидроксиметил)-6-метилпиридин-4-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид Пр. 78)
В герметичную пробирку на 40 мл добавляли (R)-N-[3-(2-хлор-5-фторпиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамид (120,00 мг, 0,269 ммоль, 1,00 экв.), (4-амино-6-метилпиридин-2-ил)метанол (74,20 мг, 0,537 ммоль, 2,00 экв.), BrettPhos (14,41 мг, 0,027 ммоль, 0,1 экв.), BrettPhos Pd G3 (24,34 мг, 0,027 ммоль, 0,1 экв.) и K2CO3 (74,22 мг, 0,537 ммоль, 2 экв.) в диоксане (8,00 мл) при 80°С.Целевой продукт можно было обнаружить методом ЖХ-МС. Полученную смесь концентрировали под вакуумом. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 10:1) с получением неочищенного твердого вещества. Этот неочищенный продукт очищали препаративной ВЭЖХ в нижеследующих условиях (колонка: XBridge Prep OBD С18 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 34 В до 54 В за 7 мин, RT1: 5,90) с получением (R)-N-[3-(5-фтор-2-[[2-(гидроксиметил)-6-метилпиридин-4-ил]амино]пиримидин-4-ил)-1Н-индол-7-ил]-3-метокси-2-(4-метилпиперазин-1-ил)пропанамида (65 мг, 44,12%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z [М+Н]+ = 549,4. 1Н-ЯМР (300 МГц, DMSO-d6) δ 2.13 (3Н, s), 2.34 (4Н, s), 2.37 (3Н, s), 2.56-2.66 (2Н, m), 2.69-2.79 (2Н, m), 3.28 (3Н, s), 3.49 (1Н, t), 3.67 (1Н, dd), 3.79 (1Н, dd), 4.45 (2Н, d), 5.24 (1Н, t), 7.17 (1Н, t), 7.49-7.60 (2Н, m), 7.70 (1Н, d), 8.26 (1Н, d), 8.53 (1Н, d), 8.59 (1Н, dd), 9.89 (2Н, d), 11.51 (1Н, s).
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Иллюстративные соединения, раскрытые в данном документе, были охарактеризованы в одном или более нижеследующих биологических анализов.
Пример 79: Ферментативный анализ и клеточный анализ p-STAT6
Рекомбинантные JAK1, JAK2, JAK3 и TYK2 были приобретены у Carna Biosciences. Эффективность ингибирования соединений против JAK1, JAK2, JAK3 и TYK2 оценивали с использованием анализа Lance Ultra Kinase Assay.
Кратко, рекомбинантные киназы предварительно инкубировали в присутствии или в отсутствие соединения при комнатной температуре в течение 15 минут. Реакцию инициировали добавлением 5 мМ АТР (аденозинтрифосфат) и пептидного субстрата, который может быть фосфорилирован киназами в результате реакции. После 60 минут инкубирования реакцию останавливали добавлением смеси детекционных реагентов, содержащей EDTA. Флуоресценцию измеряли при 615 нм и 665 нм соответственно при длине волны возбуждения 320 нм. Вычисленное соотношение сигналов при 665 нм/615 нм пропорционально активности киназы. Концентрацию соединения, вызывающего 50%-ное ингибирование соответственной киназы (IC50), вычисляли, используя четырехпараметрическую логистическую модель подгонки с использованием XLfit.
Чтобы детектировать фосфорилированный STAT6 (сигнальный белок и активатор транскрипции 6 (pSTAT6)), клетки ТНР-1 харвестировали центрифугированием при 250g в течение 5 минут и ресуспендировали в среде для анализа (RPMI1640+10% FBS (фетальная бычья сыворотка)) до 2×105 клеток на лунку. Тестируемые соединения наносили на аналитические планшеты в последовательных разведениях от 1 мкМ до 0,3 нМ в DMSO. Клетки ТНР-1 инкубировали с соединениями в последовательных разведениях в течение 60 мин при комнатной температуре с последующей стимуляцией интерлейкина (IL-13, 10 нг/мл) в течение 30 минут, фиксировали в буфере Cytofix (BD Biosciences) и пермеабилизовали в 90% метаноле на льду. РЕ анти-pSTAT6 (BD Biosciences) антитела окрашивали в течение 60 мин при комнатной температуре, после чего анализировали методом проточной цитометрии. В анализе соединения были, таким образом, разбавлены, и кривые доза-ответ для ингибирования сигнала определяют IC50 для соединений.
Ингибирующая активность протестированных соединений по отношению к киназам JAK1, JAK2, JAK3, TYK2 и к фосфорилированию STAT6 показана в Таблице 2 ниже. Соотношения селективности в отношении JAK1/JAK2 для всех протестированных соединений составляют свыше 10 (вплоть до 1000 или больше) в по результатам (JAK2 IC50/JAKI IC50). Об ингибировании фосфорилирования STAT6, подтверждающего релевантность сигнального пути JAK-STAT при воспалении дыхательных путей, сообщалось в предшествующем уровне техники. Было доказано также, что соединения, которые продемонстрировали мощную ингибирующую активность в отношении JAK1, являются эффективными в ингибировании фосфорилирования STAT6.
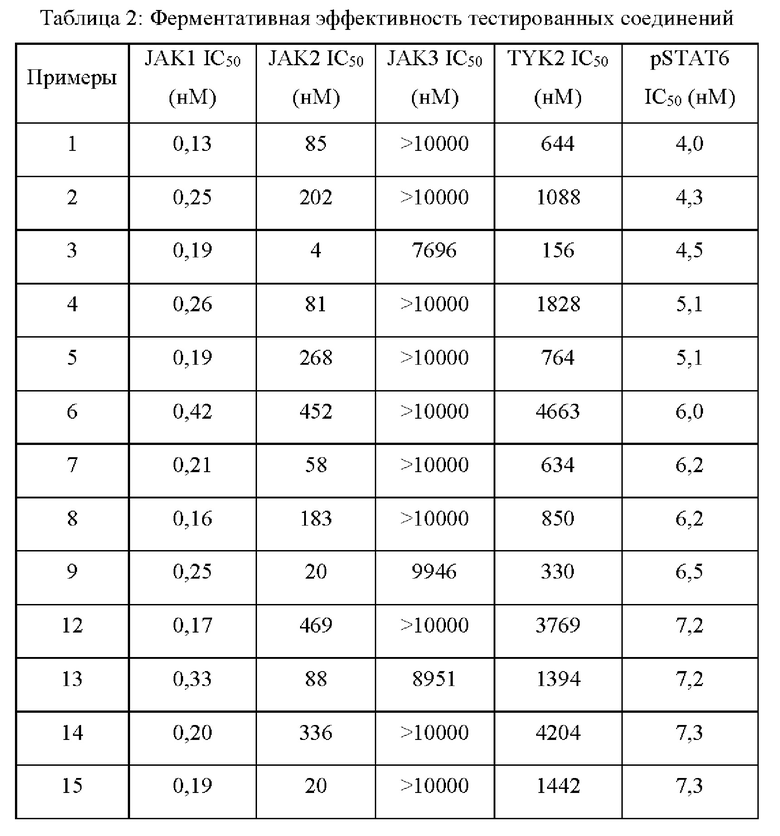
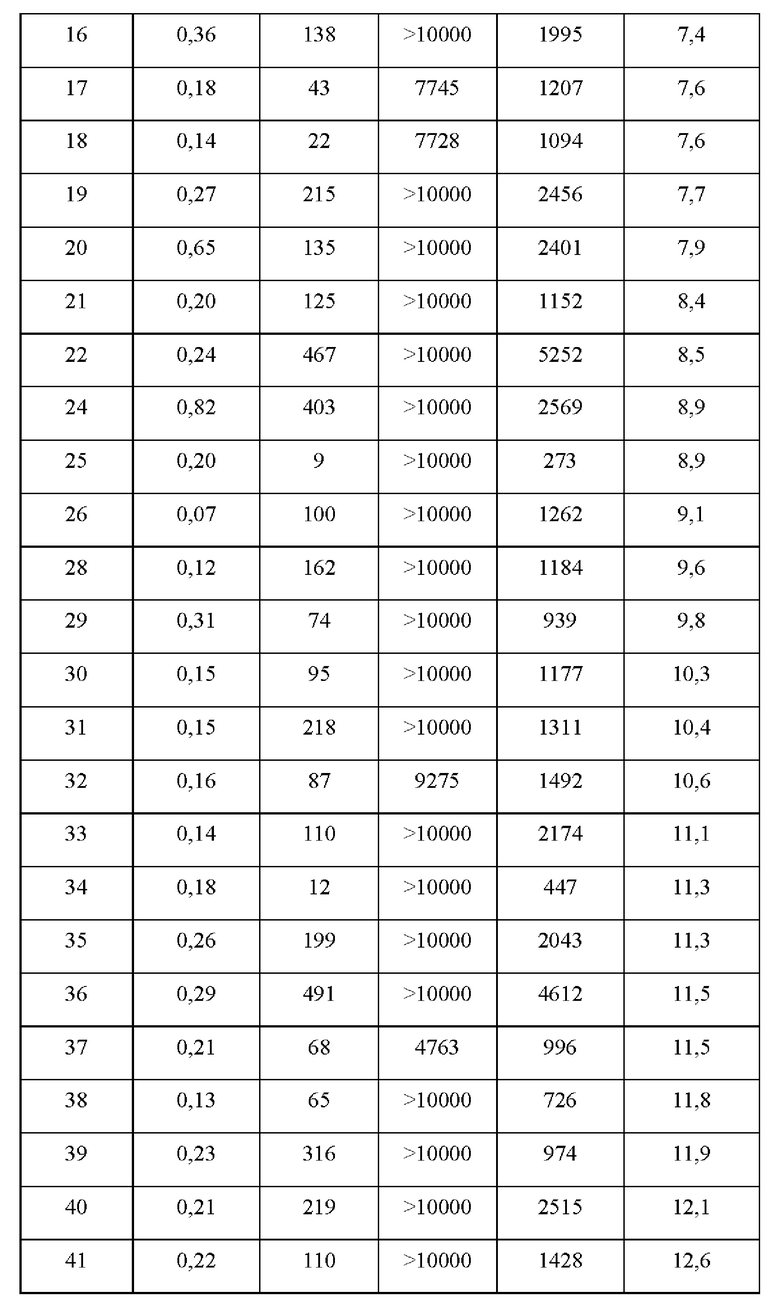

Пример 80: Метаболическая стабильность в гепатоцитах крысы и микросоме печени человека
Гепатоциты крыс мужского пола и микросомы печени человека были получены от коммерческих поставщиков (например, BioreclamationIVT) и хранились при -150°С до использования.
Для анализа метаболической стабильности с крысиными гептатоцитами виалы с криоконсервированными гепатоцитами или микросомами извлекали из хранилища и обеспечивали, чтобы виалы оставались при криогенных температурах. 1 мкМ каждого тестируемого соединения (в ацетонитриле; 0,01% DMSO) инкубировали с 250 мкл клеток гепатоцитов (1×106 клеток/мл) в 96-луночном планшете с глубокими лунками. Реакцию останавливали в разные моменты времени (0, 0,5, 5, 15, 30, 45, 60, 80, 100 и 120 минут) добавлением 3 объемов охлажденного ацетонитрила к 20 мкл реакционной смеси и центрифугировали при 4°С в течение 15 мин. 40 мкл надосадочной жидкости разбавляли до 200 мкл чистой водой и анализировали методом ЖХ-МС/МС.
Для анализа метаболической стабильности с микромами печени человека 1 мкМ каждого тестируемого соединения инкубировали с 1 мг/мл микросом (объединенные HLM (микросомы печени человека) с белковым конусом 20 мг/мл) при 37°С в 250 мкл буфера (100 мМ фосфатный буфер, рН-7,4), содержащего 1 мМ раствора NADPH. 20 мкл инкубационной смеси гасили 5 объемами охлажденного ацетонитрила в разные моменты времени 0, 0,5, 5, 10, 15, 20 и 30 мин в свежем 96-луночном планшете. Гашеный планшет центрифугировали при 4000 об/мин в течение 15 мин. 40 мкл надосадочной жидкости разбавляли до 200 мкл чистой водой и анализировали методом ЖХ-МС/МС.
Клиренс гепатоцитов оценивали in vitro, основываясь на определении периода полувыведения (T1/2) соединений, исчезновения из их начальных концентраций. Вычисляли отношение площади пика каждого соединения (тестируемого или контроля) к ИВ (исходное вещество). Строили кривую зависимости Ln (% контроля) от времени инкубирования (мин), и вычисляли тангенс угла наклона линии линейной аппроксимации. Константу скорости элиминации лекарственного средства k (мин-1), Т1/2 (мин) и in vitro собственный клиренс CLint (мкл/мин/Е6) вычисляли согласно следующим уравнениям:
k = -тангенс угла наклона
Т1/2 = 0,693/k
CLint = k/Chep
где Chep (клетки × мкл-1) означает концентрацию клеток в инкубационной системе.
Данные представлены ниже в Таблице 3.
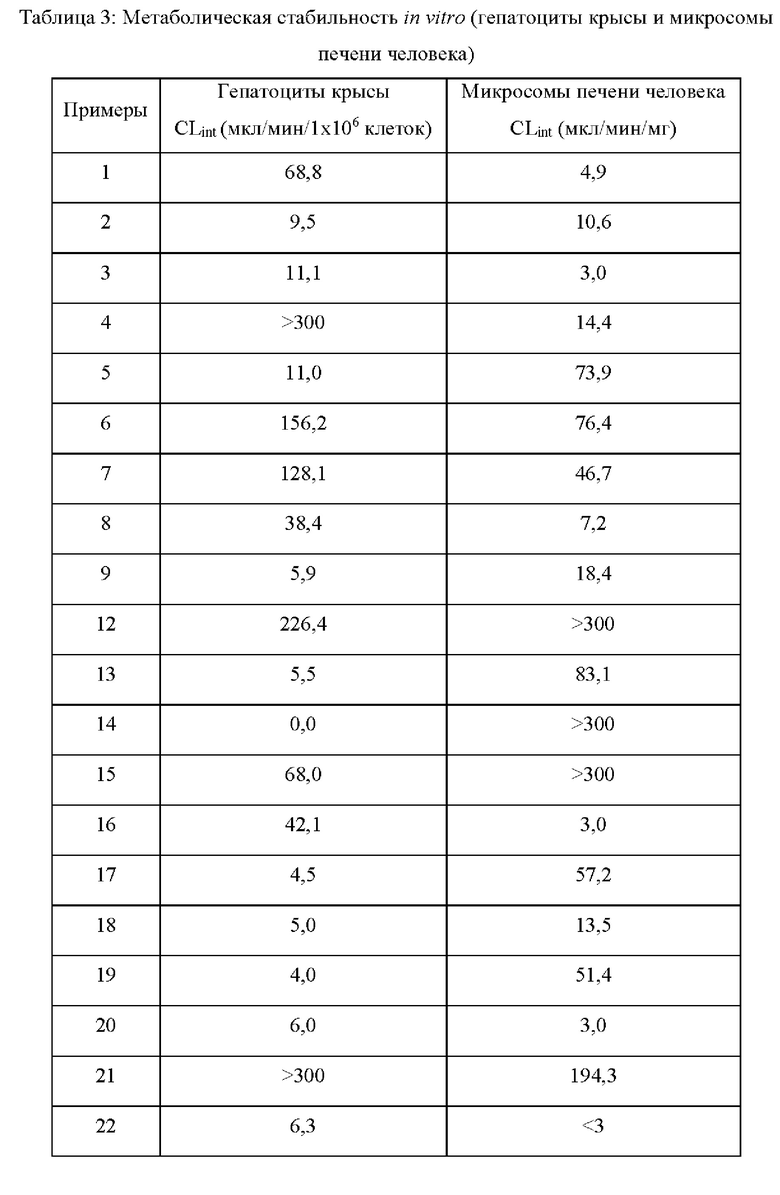
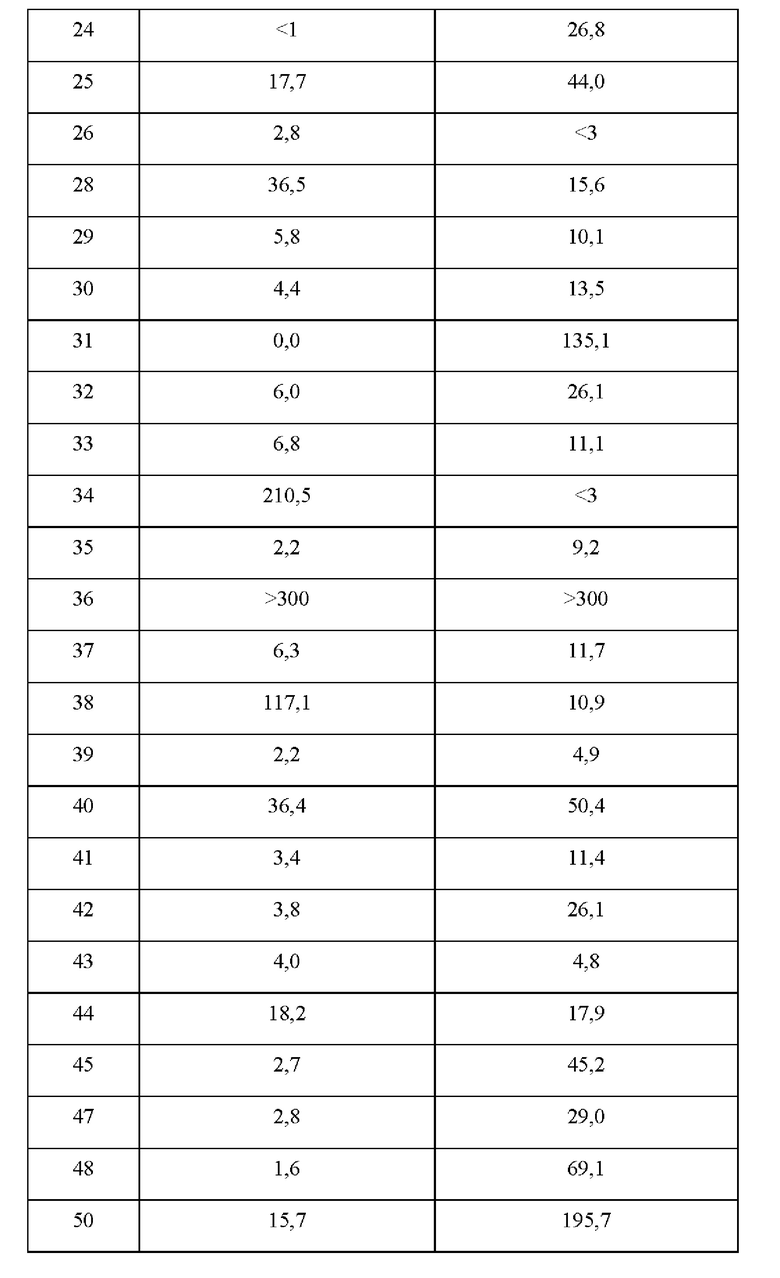

Пример 81: Фармакокинетика в плазме крови и легком мыши
Легочную PK (фармакокинетика) соединений тестировали посредством интратрахеальной (IT) истилляции у самцов-мышей CD1. Уровни тестируемых соединений в плазме крови и легком и их соотношения определяли следующим образом. Тестируемые соединения дозировали кассетно в виде композиции 0,4 мг/мл суспензии 0,5% НРМС (гидроксипропилметилцеллюлоза), 0,1% Tween 80 в физиологическом растворе. Животного анестезировали с использованием 5% изофлурана в течение 5 мин, открывали его рот и выводили наружу язык, свет фокусировали на шее мыши и локализовали трахею, и шприц вставляли в трахею, пока трахея находилась в открытом состоянии, и тестируемые соединения впрыскивали в трахею. В разные моменты времени (обычно 5 мин, 1, 4, 24 часа) после введения дозы брали приблизительно по 0,250 мл образцов крови посредством пункции сердца, и интактные легкие иссекали из мышей. Каждый образец крови переносили в пластиковую центифужную микропробирку, содержащую K2EDTA. Образцы крови затем центрифугировали (центрифуга Eppendorf, 5804R) в течение 4 минут при приблизительно 12000 об/мин при 4°С для сбора плазмы. Перед сбором тканей мыши должны быть полностью обескровлены. Образцы легких собирали в принятые моменты времени, и цельное легкое взвешивали и гомогенизировали. Концентрации тестируемых соединений в образцах плазмы и легких анализировали методом ЖХ-МС/МС. Для фармакокинетических вычислений будет использовано WinNonlin (PhoenixTM) или другое подобное программное обеспечение. Протестированные соединения продемонстрировали концентрацию в легком на один-два порядка величины больше, чем концентрация в плазме у мыши.
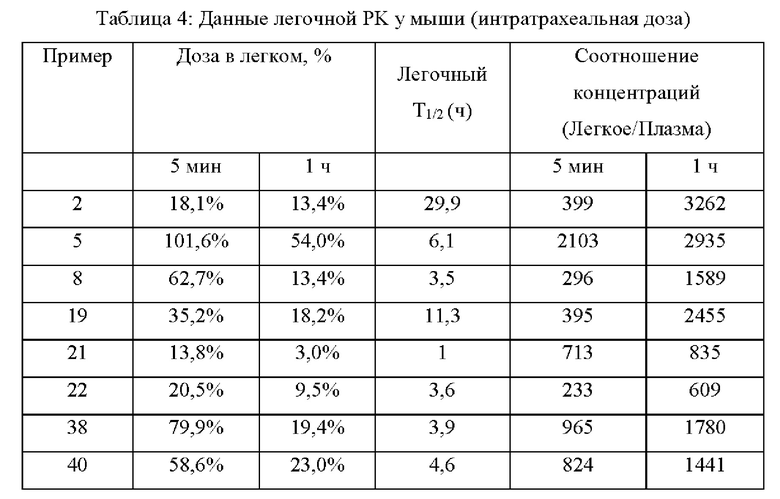
Пример 82: Мышиная модель вызванного альтернарией эозинофильного воспаления легкого
Эозинофилия дыхательных путей является отличительной чертой астмы человека. Alternaria alternata (альтернария) представляет собой грибковый аэроаллерген, который может усугублять астму у людей и вызывает эозинофильное воспаление в легких мышей (Havaux et al. Clin Exp Immunol. 2005, 139(2): 179-88). Ha мышах было продемонстрировано, что альтернария опосредованно активирует тканевые резидентные врожденные лимфоидные клетки типа 2 в легком, которые реагируют на (например, IL-2 и IL-7) и высвобождают JAK-зависимые цитокины (например IL-5 и IL-13) и координируют эозинофильное воспаление (Bartemes et al. J. Immunol. 2012, 188(3): 1503-13.
В исследовании используют самцов мышей С57 в возрасте от семи до девяти недель от Taconic. В день исследования животных слегка анестезируют изофлураном и вводят им либо носитель, либо тестируемое соединение (0,1-1,0 мг/мл, 50 мкл общий объем за несколько вдохов) посредством орофарингеальной аспирации. После введения дозы животных размещают в лежачем положении на боку и осуществляют наблюдение в отношении полного выхода из анестезии, после чего возвращают в домашнюю клетку. Один час спустя животных еще раз ненадолго анестезируют и вводят им либо носитель, либо экстракт альтернарии (в сумме 200 мкг экстракта, общий объем 50 мл) посредством орофарингиальной апирации, после чего осуществляют наблюдение в отношении полного выхода из анестезии и возвращают в домашнюю клетку. Через сорок восемь часов после введения альтернарии собирают жидкость бронхоальвеолярного лаважа (BALF) и подсчитывают эозинофилы в BALF с использованием системы Advia 120 Hematology System (Siemens).
Иллюстративные соединения, раскрытые в данном документе, тестируют в этом анализе с альтернарией. Об активности в этой модели свидетельствует снижение уровня эозинофилов, присутствующих через сорок восемь часов в BALF животных, которым вводили соединения, по сравнению с контрольными животными, которым вводили носитель, которые были аллергизированы альтернарией. Данные выражают в процентах ингибирования ответной реакции эозинофилов в BALF на введение носителя, аллергизацию альтернарией. Для вычисления процента ингибирования количество эозинофилов в BALF для каждого условия преобразуют в процент от среднего количества эозинофилов в BALF при введении носителя, аллергизации альтернарией и вычитают из ста процентов. Тестируемые соединения демонстрируют ингибирование вызванных альтернарией эозинофилов в BALF.
Хотя настоящее изобретение конкретно показано и описано со ссылкой на конкретные воплощения (некоторые из которых являются предпочтительными воплощениями), специалисту должно быть понятно, что различные изменения по форме и деталям могут быть сделаны без отклонения от замысла и объема настоящего изобретения, как оно раскрыто в данном документе.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| МОДУЛЯТОРЫ MAS-СВЯЗАННОГО РЕЦЕПТОРА G-БЕЛКА X2 И СВЯЗАННЫЕ С НИМИ ПРОДУКТЫ И СПОСОБЫ | 2021 |
|
RU2841536C1 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2760554C1 |
| ПРОИЗВОДНОЕ ПИРИМИДИН-4,6-ДИАМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2834353C2 |
| НОВЫЕ АМИНОКИСЛОТНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2747673C2 |
| ИНГИБИТОРЫ HPK1 И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2839132C2 |
| Соединения и способы для ингибирования JAK | 2016 |
|
RU2760359C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| СОЕДИНЕНИЯ ПИРИМИДИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2807277C2 |
Группа изобретений относится к области органической химии и включает соединение формулы (I), его фармацевтически приемлемые соли, фармацевтическую композицию и комбинацию, его содержащие, способ ингибирования JAK1 и способ лечения. В формуле (I) кольцо А выбрано из группы, указанной в формуле изобретения, R1 представляет собой водород, галоген, гидроксил, амино, циано или C1-3алкил; R2 представляет собой водород, С1-12алкил или С1-12алкоксил, возможно моно- или мультизамещенный галогеном, гидроксилом, амино, циано или С1-12алкоксилом; каждый из R3 и R4 независимо отсутствует или представляет собой галоген, гидроксил, C1-6алкил, карбоксил, C1-6алкоксил, C1-6алкоксикарбонил, -NRaRb, -C(O)NRaRb, 3-10-членный насыщенный или ненасыщенный карбоциклил, 3-10-членный насыщенный или ненасыщенный гетероциклил, имеющий 1, 2 или 3 гетероатома, выбранных из N, О или S, который возможно может быть моно- или независимо мультизамещен галогеном, гидроксилом, С1-6алкилом, C1-6алкоксилом, С1-6карбоксилом, С1-6алкоксикарбонилом, -NRaRb, -C(O)NRaRb, где одна или более чем одна образующая кольцо группа-CH2- карбоциклила или гетероциклила, имеющего 1, 2 или 3 гетероатома, выбранных из N, О или S, может быть заменена группой -С(О)-; где каждый из Ra и Rb независимо выбран из водорода, С1-6алкила, C1-6алкилкарбонила, который возможно может быть моно- или независимо мультизамещен галогеном, гидроксилом или C1-6алкокси. Соединения формулы (I) обладают ингибирующей активностью в отношении JAK1. 5 н. и 13 з.п. ф-лы, 8 табл., 82 пр.
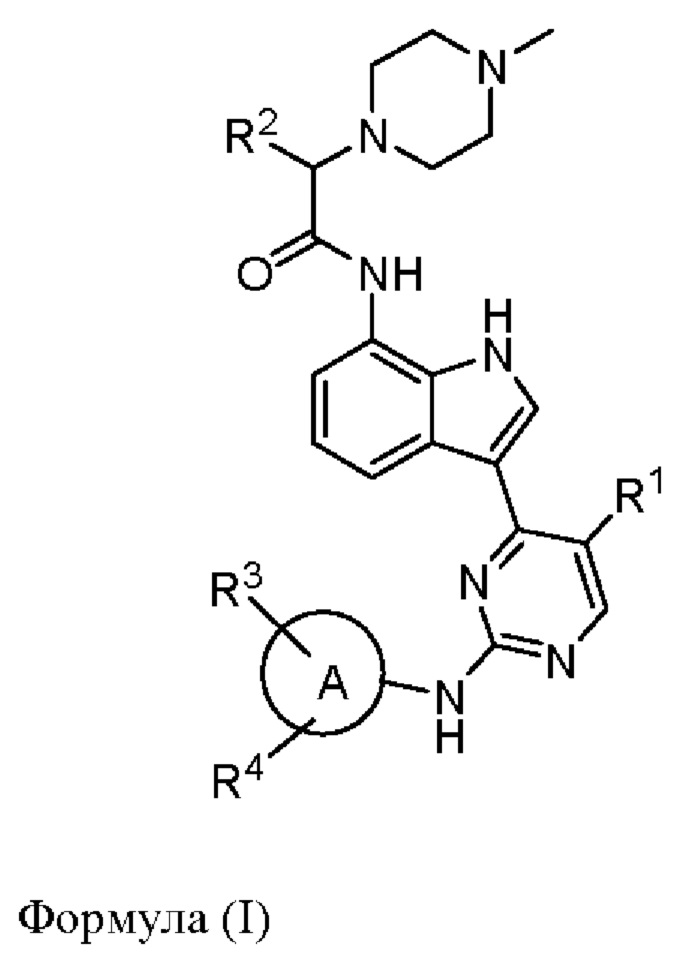
1. Соединение формулы (I):
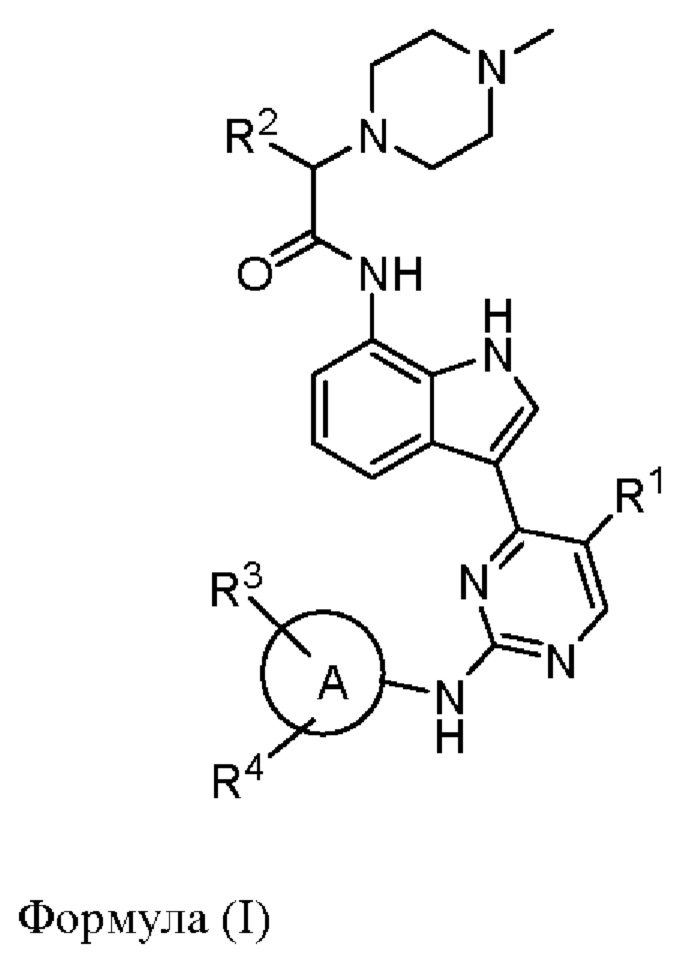
или его фармацевтически приемлемая соль,
где
кольцо А выбрано из группы, состоящей из:
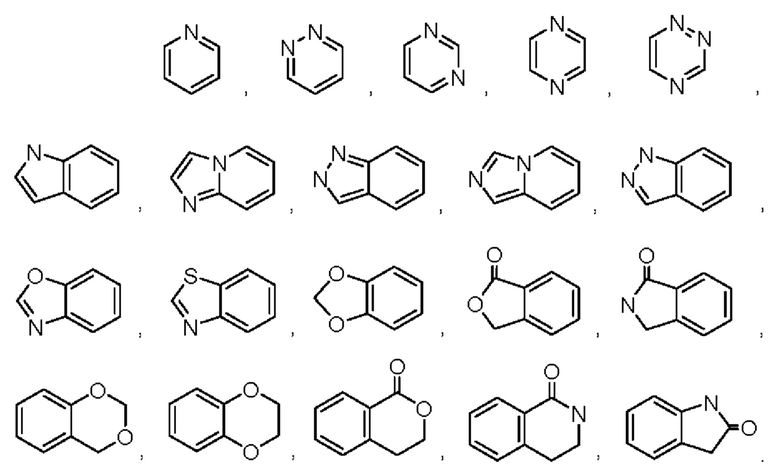
R1 представляет собой водород, галоген, гидроксил, амино, циано или C1-3алкил;
R2 представляет собой водород, С1-12алкил или С1-12алкоксил, возможно моно- или мультизамещенный галогеном, гидроксилом, амино, циано или С1-12алкоксилом;
каждый из R3 и R4 независимо отсутствует или представляет собой галоген, гидроксил, C1-6алкил, карбоксил, C1-6алкоксил, C1-6алкоксикарбонил, -NRaRb, -C(O)NRaRb, 3-10-членный насыщенный или ненасыщенный карбоциклил, 3-10-членный насыщенный или ненасыщенный гетероциклил, имеющий 1, 2 или 3 гетероатома, выбранных из N, О или S, который возможно может быть моно- или независимо мультизамещен галогеном, гидроксилом, С1-6алкилом, C1-6алкоксилом, С1-6карбоксилом, С1-6алкоксикарбонилом, -NRaRb, -C(O)NRaRb, где одна или более чем одна образующая кольцо группа-CH2- карбоциклила или гетероциклила, имеющего 1, 2 или 3 гетероатома, выбранных из N, О или S, может быть заменена группой -С(О)-;
где каждый из Ra и Rb независимо выбран из водорода, С1-6алкила, C1-6алкилкарбонила, который возможно может быть моно- или независимо мультизамещен галогеном, гидроксилом или C1-6алкокси.
2. Соединение по п. 1, имеющее структуру формулы (Ia)

3. Соединение по п. 1, где кольцо А представляет собой моноциклический гетероарил, выбранный из пиридинила, пиридазинила, пиримидинила, пиразинила или триазинила.
4. Соединение по п. 1, где каждый из R3 и R4 независимо отсутствует или представляет собой С1-6алкил, С1-6алкоксил, карбоксил, С1-6алкоксикарбонил, -C(O)NRaRb, который возможно может быть моно- или независимо мультизамещен галогеном, гидроксилом, C1-6алкилом, С1-6алкоксилом, C1-6алкоксикарбонилом, -NRaRb, -C(O)NRaRb.
5. Соединение по п. 1, где по меньшей мере один из R3 и R4 отсутствует.
6. Соединение по п. 1, где ни R3, ни R4 не отсутствует, и указанный R3 или R4 находится в орто-положении.
7. Соединение по п. 1, где каждый из R3 и R4 независимо отсутствует или выбран из C1-6алкила, C1-6алкоксикарбонила, возможно замещенных гидроксилом или C1-6 алкоксикарбонилом.
8. Соединение по п. 1, где каждый из R3 и R4 независимо отсутствует или представляет собой карбоксил, гидроксил, карбамоил, амино, метил, метоксил, этоксил, метоксиметил, метоксиэтоксил, гидроксиметил, гидроксиэтил, гидроксибутил, гидроксиметоксил, гидроксиэтоксил, карбамоилметоксил, метилкарбамоил, гидроксиацетамидо, (гидроксиэтил)карбамоил, метилкарбамоилметоксил, диметилкарбамоилэтоксил, карбоксиметоксил, метоксикарбонил, этоксикарбонил, изопропоксикарбонил, трет-бутоксикарбонил, метоксикарбонилметил, метоксикарбонилэтил, этоксикарбонилметил, метоксикарбонилметоксил, метиламино, диметиламино, диметиламиноэтил, диметиламиноэтоксикарбонил, диметиламинометил, пропионамидо, метилкарбониламино, диметиламиноэтоксикарбонил, оксетанил, оксетанил-2-он, азетидин-2-ил, азетидин-3-ил-2-он, метилазетидин-3-ил-2-он, тетрагидрофуран-3-ил или тетрагидропиран-4-ил.
9. Соединение по п. 1, где каждый из R3 и R4 независимо отсутствует или представляет собой метил, метоксикарбонил или гидроксиметил.
10. Соединение по п. 1, которое выбрано из группы, состоящей из
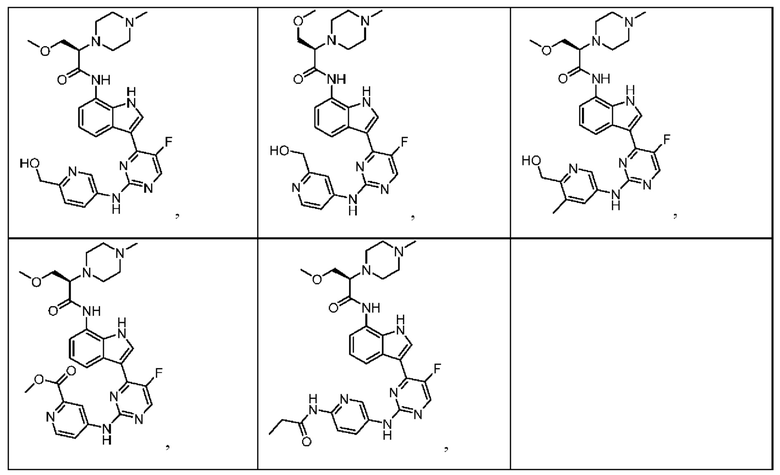
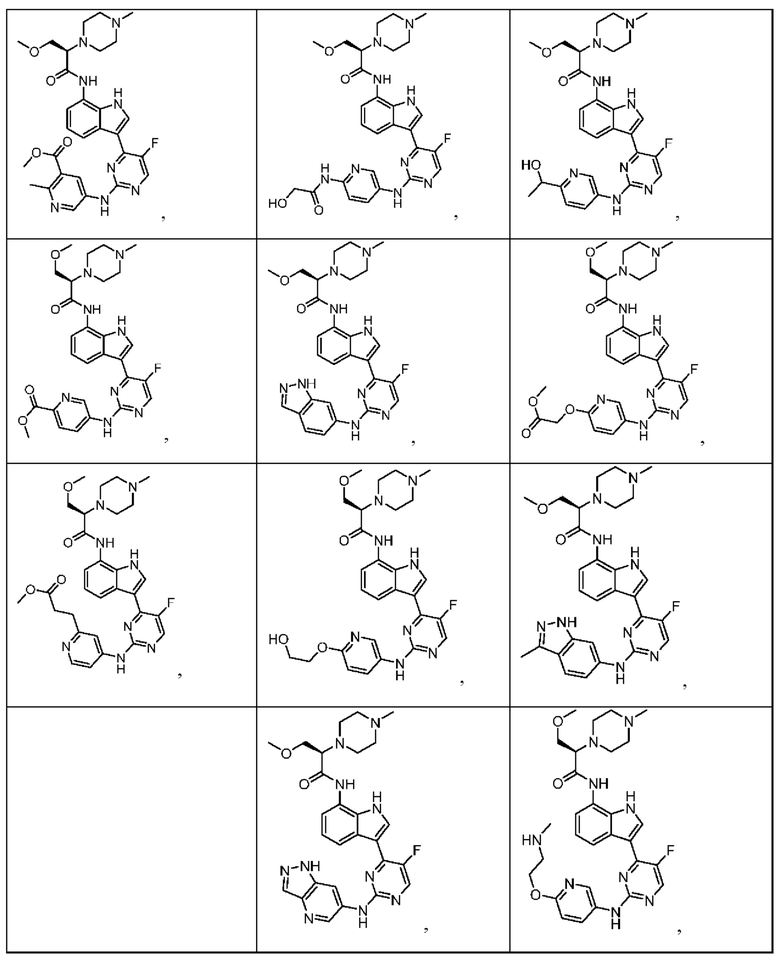

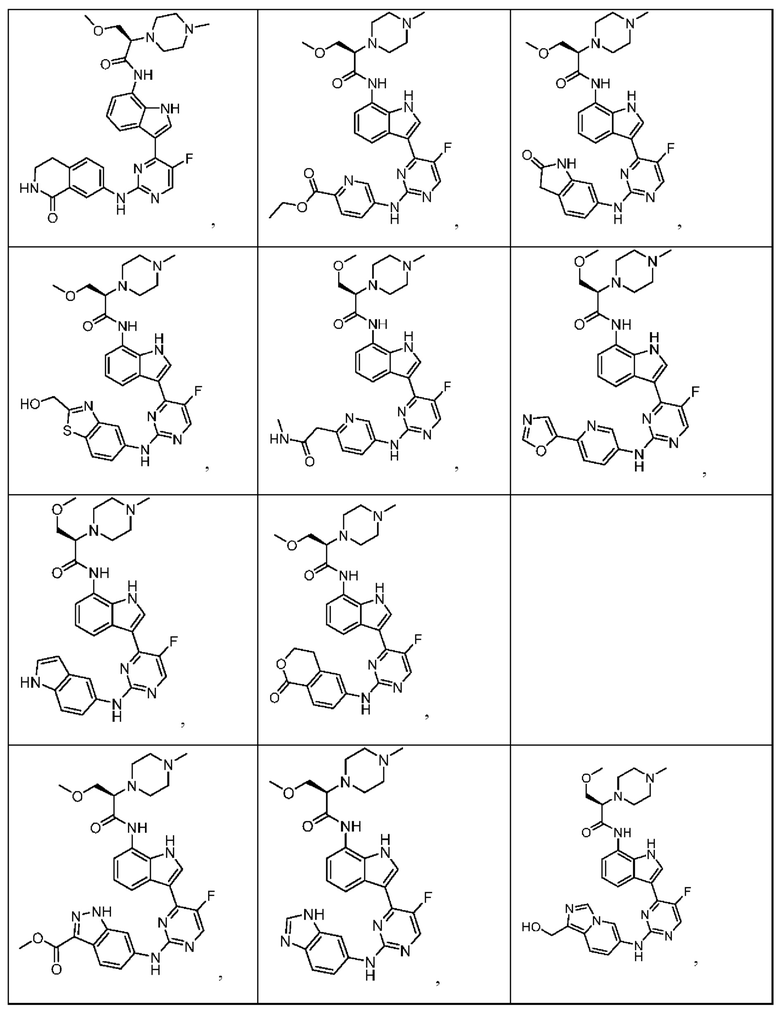
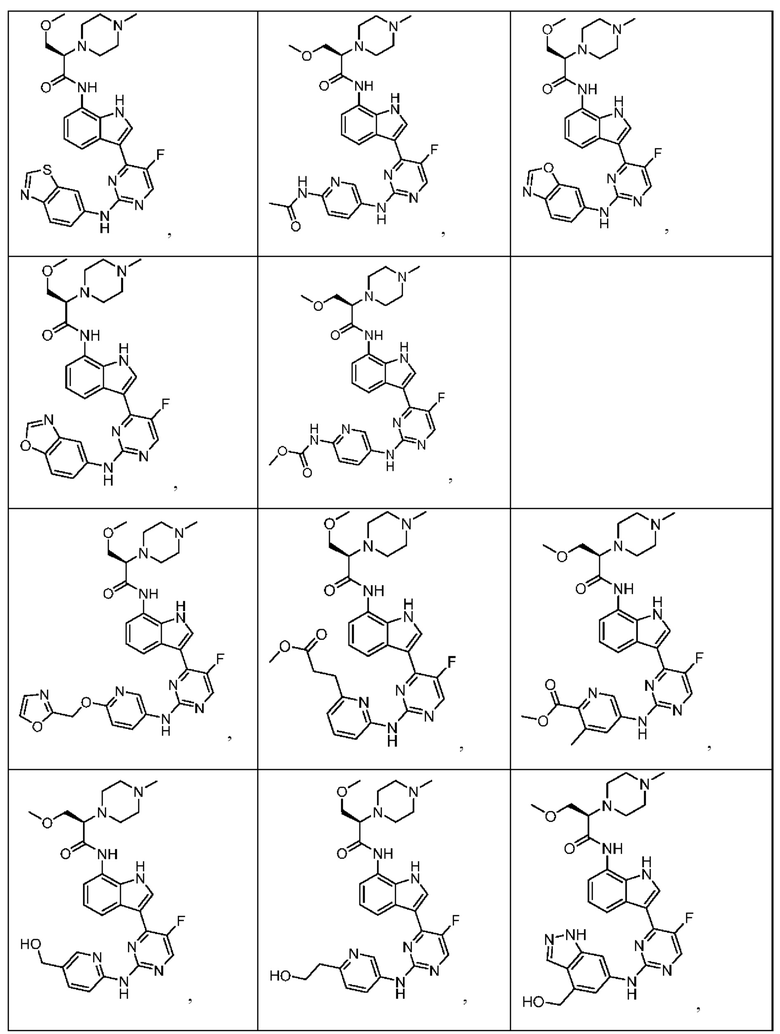

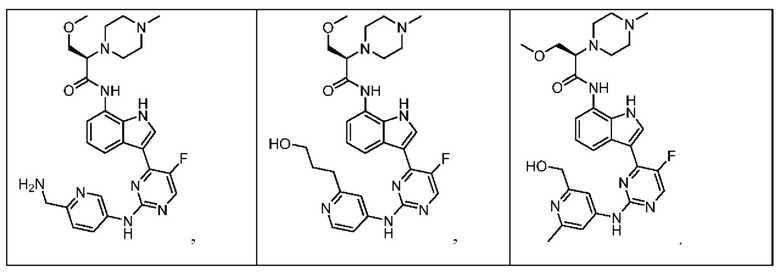
11. Фармацевтическая композиция для ингибирования JAK1 (Янус-киназа 1), содержащая эффективное количество одного или более соединений формулы (I) или их фармацевтически приемлемых солей по любому из пп. 1-10 в качестве активного ингредиента и фармацевтически приемлемый разбавитель, эксципиент или носитель.
12. Фармацевтическая композиция по п. 11, которая приготовлена в виде препарата для ингаляции.
13. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-10 для применения в качестве лекарственного средства для ингибирования JAK1.
14. Способ ингибирования JAK1 путем использования эффективного количества одного или более соединений, их фармацевтически приемлемых солей по любому из пп. 1-10 или фармацевтической композиции по п. 11 или 12.
15. Способ лечения связанных с JAK1 расстройств у субъекта, включающий введение субъекту эффективного количества одного или более соединений или их фармацевтически приемлемых солей по любому из пп. 1-10 или фармацевтической композиции по п. 11 или 12.
16. Способ по п. 15, где субъектом является теплокровное животное, такое как человек.
17. Способ по п. 16, где связанное с JAK1 расстройство представляет собой респираторное состояние, такое как астма или COPD (хроническое обструктивное заболевание легких).
18. Фармацевтическая комбинация, обладающая ингибирующей активностью в отношении JAK1, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль по любому из пп. 1-10 в комбинации со вторым терапевтическим агентом, выбранным из противовоспалительного агента или антигиперпролиферативного агента.
| WO 2017050938 А1, 30.03.2017 | |||
| WO 2018134213 A1, 26.07.2018 | |||
| Neil P | |||
| Ggrimster et al., Discovery and optimization of a novel series of highly selective JAK1 kinase inhibitors | |||
| Journal of medicinal chemistry, 2018, vol | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| Горизонтальный ветряный двигатель | 1926 |
|
SU5235A1 |
| Хлеборезка | 1928 |
|
SU20777A1 |

