ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к области биомедицины, в частности, настоящее изобретение относится к производным пирролопиримидина и, конкретнее, настоящее изобретение относится к производным пирролопиримидина, способам получения производных пирролопиримидина и применению производных пирролопиримидина в изготовлении лекарственных средств.
УРОВЕНЬ ТЕХНИКИ
[0002] Аутотаксин (сокращенно называемый АТХ) представляет собой секретируемый гликопротеин с активностью фосфодиэстеразы (PDE) и является представителем семейства внеклеточных пирофосфатаз/фосфодиэстераз (ENPP). Таким образом, аутотаксин также называется ENPP2. АТХ также обладает активностью лизофосфолипазы D (LysoPLD) и может гидролизовать лизофосфатидилхолин (LPC) до лизофосфатидной кислоты (LPA) с биологической активностью. LPA представляет собой внутриклеточный липидный медиатор, который влияет на многие биологические и биохимические процессы.
[0003] Исследования показали, что в патологических условиях уровень LPA можно понижать за счет ингибирования АТХ, обеспечивая тем самым терапевтическую пользу для неудовлетворенных клинических потребностей, включая рак, хоминг лимфоцитов, хроническое воспаление, невропатическую боль, фиброз, тромбоз и холестатический зуд, или фиброзирующие заболевания, которые индуцируются, опосредуются и/или распространяются за счет повышения уровня LPA и/или активации АТХ.
[0004] Повышенную регуляцию сигнального пути ATX-LPA можно наблюдать в различных воспалительных условиях. Например, провоспалительные эффекты LPA включают дегрануляцию тучных клеток, сокращение гладкомышечных клеток и высвобождение цитокинов из дендритных клеток. Как проявление его общей роли в воспалении, повышенную регуляцию сигнального пути ATX-LPA наблюдали в мышиной модели воздушного мешка с каррагенаном, которая является общепринятой для разработки противовоспалительных лекарственных средств, включая ингибиторы циклооксигеназы для артрита (Hidenobu Kanda, Rebecca Newton, Russell Klein et at, Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs[J] Nature Immunology. 2008, 9(4):415-423.). Кроме того, снижение количества LPA в плазме и в воздушном мешке наблюдали в крысиной модели воздушного мешка с каррагенаном с применением ингибитора АТХ, которая подтверждает роль АТХ как основного источника LPA во время воспаления. В качестве другой общей роли в воспалительных заболеваниях был подтвержден «синергетический эффект» между LPA и хемокинами, обуславливающими миграцию лимфоцитов. В участках хронического воспаления можно обнаружить высокую экспрессию АТХ. Было подтверждено, что хоминг Т-клеток в лимфатических тканях можно ингибировать путем внутривенной инъекции инактивированного АТХ, что можно обеспечить посредством конкуренции с эндогенным АТХ и оказания доминантного негативного эффекта. В некоторых случаях АТХ облегчает попадание лимфоцитов в лимфоидные органы. Следовательно, ингибиторы АТХ могут блокировать миграцию лимфоцитов во вторичные лимфоидные органы и полезны при аутоиммунных заболеваниях.
[0005] Было подтверждено, что при ревматоидном артрите экспрессия АТХ повышена в синовиальных фибробластах от пациентов с ревматоидным артритом (РА), а устранение экспрессии АТХ в мезенхимальных клетках (включая синовиальные фибробласты) приводит к ослаблению симптомов в мышиной модели ревматоидного артрита. Следовательно, роль аутотаксина при ревматоидном артрите была полностью установлена.
[0006] LPA также может осуществлять повышающую регуляцию связанных с болью белков посредством LPA1, который является одним из его гомологичных рецепторов. Нацеленное ингибирование против АТХ-опосредованного биосинтеза LPA может служить механизмом предотвращения невропатической боли, вызванной повреждением нерва, такой как боль, связанная с остеоартритом. Было установлено, что ингибиторы аутотаксина снижают количество LPA и PGE2 и также облегчают воспалительную боль. Также посредством исследований было показано, что нацеленное ингибирование против АТХ-опосредованного биосинтеза LPA может быть новым механизмом для предотвращения невропатической боли, вызванной повреждением нерва.
[0007] После того, как воспаление проходит, а повреждение ткани исправляется, ткань обычно восстанавливается до своего исходного состояния. Избыточное или неконтролируемое восстановление ткани может приводить к тому, что обычно называют фиброзом. Фиброз характеризуется избыточным отложением компонентов внеклеточного матрикса и избыточным ростом фибробластов. Фиброз может возникать во всех тканях, но в особенности распространен в органах, которые часто подвергаются химическому и биологическому повреждению, включая легкие, кожу, пищеварительный тракт, почки и печень. Фиброз часто серьезно вредит нормальному функционированию органов.
[0008] В некоторых случаях LPA стимулирует пролиферацию звездчатых клеток печени, в то же время ингибируя синтез ДНК в гепатоцитах. Уровень LPA и сывороточная активность АТХ повышены у пациентов с хроническим гепатитом С. В крови кроликов с разными поражениями печени плазменная концентрация LPA и сывороточная активность АТХ относительно выше при индуцированном черыреххлористым углеродом фиброзе печени. Плазменная концентрация LPA и сывороточная активность АТХ повышаются с тяжестью разных поражений печени.
[0009] Легочный фиброз представляет собой изменение терминальной стадии большой группы заболеваний легких, которые характеризуются пролиферацией фибробластов и накоплением большого количества внеклеточного матрикса, что сопровождается воспалительным повреждением и разрушение структуры ткани, т.е. структурной аномалией (рубцеванием), вызванной аномальным восстановлением после повреждения нормальных альвеолярных тканей. Когда повреждение легких обусловлено различными причинами, интерстициальные ткани будут секретировать коллаген для восстановления. Если восстановление происходит в избытке, т.е. наблюдается избыточная пролиферация фибробластов и накопление внеклеточного матрикса, будет развиваться легочный фиброз.
[0010] Сигнал LPA имеет эффект стимуляции фиброза на эпителиальных клетках, эндотелиальных клетках и фибробластах, в частности, посредством рецептора LPA1: генетическая делеция этого рецептора снижает апоптоз эпителиальных клеток, пропотевание сосудов и накопление фибробластов в модели легочного фиброза.
[0011] Идиопатический легочный фиброз (ИЛФ) представляет собой хроническую, прогрессирующую и фиброзирующую пневмонию с неизвестной этиологией, характеризуемую диффузным альвеолитом и альвеолярными структурными нарушениями, как правило, проявляющуюся как обычная интерстициальная пневмония при визуализации и патологической гистологии. По мере того, как заболевание прогрессирует, ткань легких пациента становится толще и жестче, приводя к появлению перманентных рубцов, или же легкие пациента выглядят как соты, что наглядно называется «сотовым легким» или «люфовидным легким». Хроническое прогрессирующее заболевание приводит к необратимому и непрерывному снижению легочной функции. 50% пациентов могут иметь среднее время выживаемости, составляющее всего 2,8 года с момента постановки диагноза. Таким образом, идиопатический легочный фиброз также называется «опухолеподобным заболеванием». В настоящее время существующие виды лекарственной терапии имеют проблемы, такие как большое количество нежелательных реакций, плохой терапевтический эффект; а нелекарственная терапия включает, главным образом, трансплантацию легких, но органная трансплантация является дорогостоящей и ограниченной по ресурсам, а также сопровождается определенным клиническим риском.
[0012] Существует свидетельство, демонстрирующее, что пролиферация и сокращение фибробластов и секреция внеклеточного матрикса, стимулируемые LPA, способствуют фиброзирующей пролиферации при других заболеваниях дыхательных путей, таких как хронический бронхит и интерстициальное заболевание легких, а также перибронхиальный фиброз при тяжелой астме. LPA играет роль при фиброзирующем интерстициальном заболевании легких и облитерирующем бронхиолите, при которых повышены как коллаген, так и миофибробласты. Исследования, относящиеся к идиопатическому легочному фиброзу (ИЛФ), показали, что уровень LPA в жидкости бронхоальвеолярного лаважа повышен. Дальнейшие исследования нокаута и ингибиторов LPA1 показали, что LPA играет ключевую роль в процессе легочного фиброза, что дополняет исследование на мышах с клеточно-специфическим нокаутом, у которых отсутствует АТХ в клетках бронхиального эпителия и макрофагах. Было показано, что эти мыши были менее чувствительны в модели легочного фиброза. Роль LPA в других фиброзирующих заболеваниях (почек и кожи) основана на схожих наблюдениях. Роль LPA в ремоделировании легких связана с эффектами LPA на легочные фибробласты (посредством LPA1) и эпителиальные клетки (посредством LPA2). Было показано, что LPA2 играет поворотную роль в активации TGFβ в эпителиальных клетках в случае фиброзирующих нарушений. Роли LPA в ремоделировании и фиброзе связаны с ХОБЛ, ИЛФ и астмой, а ремоделирование легких, как отдаленный результат, будет ограничивать легочную функцию. И наконец, если сфокусироваться на легочных заболеваниях, АТХ является одним из трех основных локусов количественных признаков, которые, вероятно, связаны с разницей в легочной функции у мышей.
[0013] В исследовании обнаружили, что концентрация LPA повышена в плазме и асцитах у пациентов с раком яичника на ранних или поздних стадиях. Повышенный уровень LPA и изменения экспрессии и ответа рецепторов LPA могут быть одной из причин начала, прогрессирования или исхода рака яичника. LPA также связана с раком предстательной железы, раком молочной железы, меланомным раком, раком головы и шеи, раком кишечника, раком головного мозга и раком щитовидной железы. LPA участвует в пролиферации и инвазии опухолевых клеток в смежные ткани, что приводит к метастазированию. Эти биологические и патобиологические процессы инициируются активацией сопряженных с протеином G рецепторов LPA. Уровень LPA можно снизить путем ингибирования ферментов, участвующих в биосинтезе LPA, таких как АТХ, для лечения опухолевых пациентов.
[0014] В процессе ангиогенеза АТХ и другие ангиогенные факторы вместе приводят к ангиогенезу. Во время опухолевого роста опухоль может подпитываться за счет ангиогенеза. Соответственно, важной стартовой точкой для лечения рака и опухолей является ингибирование ангиогенеза.
[0015] В патентной заявке WO 2014202458 A1 перечислены эффекты сигнализации ATX-LPA в разных патофизиологических условиях, таких как пролиферативные заболевания, невропатическая боль, воспаление, аутоиммунные заболевания, фиброз, мечение лимфоцитов в лимфатических узлах, ожирение, диабет или эмбриональное образование кровеносных сосудов.
[0016] В настоящее время был достигнут определенный прогресс в лечении рака, фиброзирующих заболеваний, пролиферативных заболеваний, воспалительных заболеваний, аутоиммунных заболеваний, респираторных заболеваний, сердечно-сосудистых заболеваний, нейродегенеративных заболеваний, дерматологических нарушений и/или связанных с аномальным ангиогенезом заболеваний, но его все еще недостаточно. Имеющиеся на сегодня на рынке терапевтические лекарственные средства для ИЛФ включают пирфенидон и нинтеданиб. Пирфенидон может приводить к повреждению печени (такому как печеночная недостаточность, желтуха), реакциям гиперчувствительности (таким как отечность лица, отек гортани, одышка, свистящее дыхание и т.д.) и тяжелым реакциям со стороны желудочно-кишечного тракта, а анализ фотогенотоксичности продемонстрировал, что он может приводить к структурной аномалии хромосом и может приводить к раку кожи при воздействии света. Нинтеданиб характеризуется такими нежелательными явлениями, как диарея, тошнота и боль в животе, частота реакций со стороны желудочно-кишечного тракта может достигать 50%, а распространенные нежелательные явления включают потерю массы, потерю аппетита, повреждение печени и кровотечение и т.д. Среди пациентов, которые принимают пирфенидон и нинтеданиб вероятность прекращения лечения вследствие серьезных нежелательных явлений составляла 20,9% и 26,3%, соответственно (Toby М Maher, et al. Rationale, design and objectives of two phase III, randomised, placebocontrolled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2)[TJ. BMJ Open Respiratory Research. 2019, 21; 6(1).). Качество жизни пациентов с ИЛФ сильно ухудшается, при этом в клинических исследованиях ни пирфенидон ни нинтеданиб не смогли улучшить качество жизни этих пациентов. Хотя оба из этих лекарственных средств могут улучшить общие результаты, они могут лишь замедлить течение заболевания, но не могут обратить легочный фиброз. В связи с этим польза от них для пациентов с тяжелым специфическим легочным фиброзом может отсутствовать. GLPG-1690, как одно из лекарственных средств для лечения ИЛФ, которое сейчас находится на стадии разработки и демонстрирует быстрый прогресс, проявляет тенденцию к обращению течения заболевания, но имеет проблемы, связанные с низкой ферментативной активностью, большой дозировкой клинического лекарственного средства и плохим соблюдением приема лекарственного средства.
[0017] Следовательно, на данный момент существующие варианты терапии все еще остаются неудовлетворительными. Все еще существует большое количество пациентов, нуждающихся в новых вариантах лечения с большей активностью и лучшей эффективностью, которые могут замедлять течение заболевания в большей степени или даже обращать течение заболевания, улучшать соблюдение приема лекарственного средства и обеспечивать пользу для большего числа пациентов с идиопатический легочным фиброзом.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0018] Цель настоящего изобретения состоит в предоставлении соединения, которое может эффективно ингибировать АТХ и которое можно использовать в качестве улучшения или замещения существующих лекарственных средств или ингибиторов АТХ.
[0019] В первом аспекте настоящего изобретения предложено соединение, представленное формулой I, или мезомер, рацемат, таутомер, стереоизомер, гидрат, сольват, соль или пролекарство соединения, представленного формулой I,
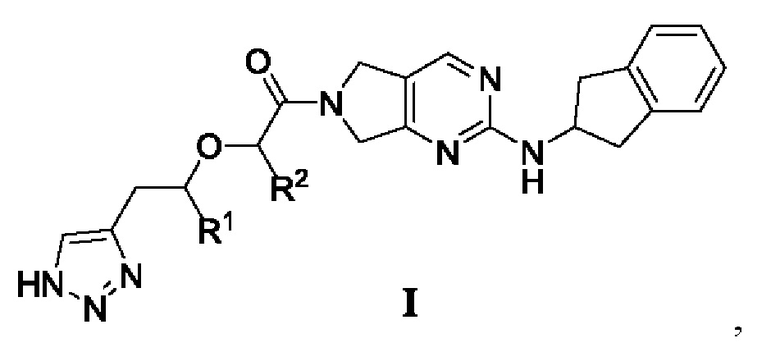
где
R1 и R2 каждый независимо выбраны из -Н или -СН3,
при условии, что:
R1 и R2 одновременно не представляют собой -Н; или
R1 и R2 одновременно не представляют собой -СН3.
[0020] В соответствии с вариантами осуществления настоящего изобретения вышеуказанное соединение может дополнительно иметь по меньшей мере один из следующих технических признаков.
[0021] В соответствии с вариантом осуществления настоящего изобретения R1 представляет собой -Н, a R2 представляет собой -СН3.
[0022] В соответствии с вариантом осуществления настоящего изобретения R1 представляет собой -СН3, a R2 представляет собой -Н.
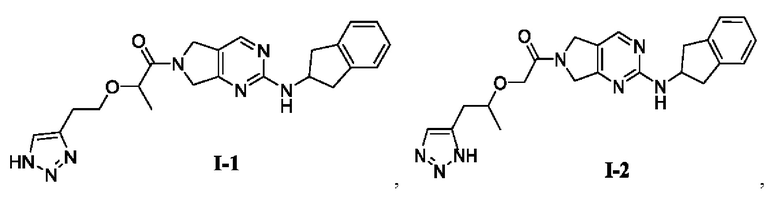
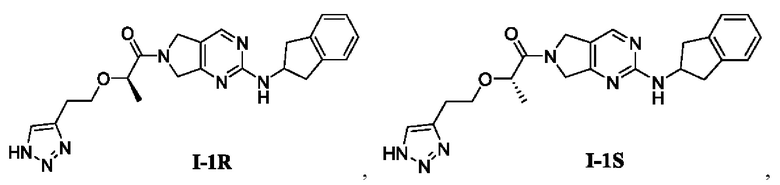
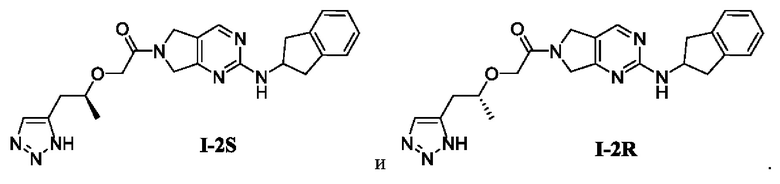
[0023] В соответствии с вариантом осуществления настоящего изобретения соединение представляет собой одно из следующих соединений; или соединение представляет собой мезомер, рацемат, таутомер, стереоизомер, гидрат, сольват, соль или пролекарство одного из следующих соединений:
[0024] В соответствии с вариантом осуществления настоящего изобретения соль включает фармацевтически приемлемые соли и представляет собой соль по меньшей мере одной кислоты, выбранной из серной кислоты, фосфорной кислоты, азотной кислоты, бромистоводородной кислоты, хлористоводородной кислоты, муравьиной кислоты, уксусной кислоты, пропионовой кислоты, бензолсульфоновой кислоты, бензойной кислоты, фенилуксусной кислоты, салициловой кислоты, альгиновой кислоты, антраниловой кислоты, камфорной кислоты, лимонной кислоты, винилсульфоновой кислоты, муравьиной кислоты, фумаровой кислоты, пирослизевой кислоты, глюконовой кислоты, глюкуроновой кислоты, глутаминовой кислоты, гликолевой кислоты, изэтионовой кислоты, молочной кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, слизевой кислоты, памовой кислоты, пантотеновой кислоты, стеариновой кислоты, янтарной кислоты, сульфаниловой кислоты, винной кислоты, n-толуолсульфоновой кислоты, малоновой кислоты, 2-гидроксипропионовой кислоты, щавелевой кислоты, гликолевой кислоты, глюкуроновой кислоты, галактуроновой кислоты, лимонной кислоты, лизина, аргинина, асперагиновой кислоты, коричной кислоты, n-толуолсульфоновой кислоты, метансульфоновой кислоты, этансульфоновой кислоты или трифторметансульфоновой кислоты. Для специалистов в данной области техники понятно, что помимо фармацевтически приемлемых солей в настоящем изобретении также можно использовать другие соли, действующие как промежуточные соединения при очистке соединений или при получении других фармацевтически приемлемых солей, или для идентификации, изучения характеристик или очистки соединений по настоящему изобретению.
[0025] Во втором аспекте настоящего изобретения предложена фармацевтическая композиция. В соответствии с вариантами осуществления настоящего изобретения фармацевтическая композиция содержит вышеуказанное соединение в качестве активного ингредиента.
[0026] В третьем аспекте настоящего изобретения предложено применение вышеуказанного соединения или вышеуказанной фармацевтической композиции в производстве лекарственного средства для лечения или предотвращения связанных с АТХ заболеваний.
[0027] В соответствии с вариантами осуществления настоящего изобретения применение может дополнительно включать по меньшей мере один из следующих технических признаков.
[0028] В соответствии с вариантом осуществления настоящего изобретения связанное с АТХ заболевание включает по меньшей мере одно, выбранное из рака, метаболического заболевания, заболевания почек, заболевания печени, фиброзирующего заболевания, интерстициального заболевания легких, пролиферативного заболевания, воспалительного заболевания, боли, аутоиммунного заболевания, респираторного заболевания, сердечно-сосудистого заболевания, нейродегенеративных заболеваний, дерматологического нарушения и/или связанного с аномальным ангиогенезом заболевания.
[0029] В соответствии с вариантом осуществления настоящего изобретения связанное с АТХ заболевание включает по меньшей мере одно, выбранное из интерстициального заболевания легких, легочного фиброза, печеночного фиброза или почечного фиброза.
[0030] В соответствии с вариантом осуществления настоящего изобретения связанное с АТХ заболевание включает идиопатический легочный фиброз.
[0031] В соответствии с вариантом осуществления настоящего изобретения связанное с АТХ заболевание включает диабет типа II и неалкогольный стеатогепатит.
[0032] В соответствии с вариантом осуществления настоящего изобретения связанное с АТХ заболевание включает невропатическую боль и воспалительную боль.
[0033] В соответствии с вариантом осуществления настоящего изобретения связанное с АТХ заболевание включает боль, связанную с остеоартритом.
[0034] В четвертом аспекте настоящего изобретения предложена лекарственная комбинация. В соответствии с вариантом осуществления настоящего изобретения лекарственная комбинация содержит вышеуказанное соединение или вышеуказанную фармацевтическую композицию; и дополнительное лекарственное средство для лечения или предотвращения связанных с АТХ заболеваний.
[0035] В соответствии с вариантом осуществления настоящего изобретения соединение или фармацевтическую композицию по настоящему изобретению можно использовать, чтобы предоставить нуждающимся в этом пациентам лучшие и более эффективные клинические лекарственные средства или схемы. В соответствии с вариантом осуществления в настоящем изобретении предложена серия ингибиторов АТХ с новыми структурами, лучшими фармакокинетическими свойствами, лучшей эффективностью и хорошей лекарственной ориентированностью, способных обеспечивать эффективное лечение связанных с АТХ заболеваний или нарушений.
[0036] Настоящее изобретение дополнительно относится к способу лечения связанных с АТХ заболеваний. Этот способ включает введение пациенту терапевтически эффективной дозы фармацевтического состава, содержащего соединение по настоящему изобретению или его фармацевтически приемлемую соль.
[0037] Определения и пояснения терминов
[0038] Если не указано иное, определения групп и термины, описанные в тексте и формуле изобретения, включают действительные определения, типовые определения, предпочтительные определения, определения, записанные в таблицах, и определения конкретных соединений в примерах и т.д., которые можно произвольным образом комбинировать и интегрировать друг с другом. Комбинируемые и интегрируемые группы определений и структуры соединений должны входить в объем настоящего изобретения.
[0039] Термин «фармацевтически приемлемый» означает соединения, материалы, композиции и/или дозированные формы, которые подходят для применения в контакте с тканями людей и животных без чрезмерных токсичности, раздражения, аллергических реакций или других проблем или осложнений с надлежащей медицинской точки зрения, и соответствуют разумному соотношению польза/риск.
[0040] Термин «фармацевтически приемлемая соль» относится к фармацевтически приемлемой соли нетоксичной кислоты или основания, включая соли неорганических кислот и оснований, а также органических кислот и оснований. Соли, полученные из неорганических оснований включают, но не ограничиваются этим, соли металлов, образуемые Al, Са, Li, Mg, K, Na и Zn. Соли, полученные из органических оснований, включают, но не ограничиваются этим, соли первичных, вторичных или третичных аминов, включая органические соли, образуемые природными замещенными или незамещенными аминами, циклическими аминами и основными ионообменными смолами, например, органические соли, образуемые аммонием, изопропиламином, триметиламином, диэтиламином, триэтиламином, трипропиламином, диэтаноламином, этаноламином, диметилэтаноламином, 2-диметиламиноэтанолом, 2-диэтиламиноэтанолом, дициклогексил амином, кофеином, прокаином, холином, бетаином, бенетамин пенициллином, этиле ндиамином, глюкозамином, метилглюкамином, теобромином, триэтаноламином, трометамином, пурином, пиперазином, пиперидином, N-этилпиперидином или полиаминовой смолой. Соли, полученные из неорганических и органических кислот, включают, но не ограничиваются этим, органические соли, образуемые серной кислотой, фосфорной кислотой, азотной кислотой, бромистоводородной кислотой, хлористоводородной кислотой, муравьиной кислотой, уксусной кислотой и т.д.
[0041] Помимо фармацевтически приемлемых солей в настоящем изобретении также можно применять другие соли, которые могут действовать как промежуточные соединения при очистке соединений или при получении других фармацевтически приемлемых солей, или которые можно использовать для идентификации, изучения характеристик или очистки соединений по настоящему изобретению.
[0042] Термин «стереоизомер» относится к изомеру, получаемому за счет разного пространственного упорядочения атомов в молекуле. Определения и правила стереохимии, используемые в настоящем изобретении, в целом соответствуют "McGraw-Hill Dictionary of Chemical Terms (1984)", S.P. Parker, Ed., McGraw-Hill Book Company, New York; and "Stereochemistry of Organic Compounds", Eliel, E. and Wilen, S., John Wiley & Sons, Inc., New York, 1994. Соединение по настоящему изобретению может содержать центр асимметрии или хиральный центр, и, таким образом, могут существовать разные стереоизомерные формы. Все стереоизомерные формы соединения по настоящему изобретению, включая, но не ограничиваясь этим, диастереоизомеры, энантиомеры, атропизомеры, геометрические (или конформационные) изомеры и их смеси, такие как рацемические смеси, входят в объем настоящего изобретения.
[0043] Многие органические соединения существуют в оптически активных формах, т.е. они способны вращать плоскость плоскополяризованного света. При описании оптически активных соединений приставки D и L, или R и S, используют для обозначения абсолютной конфигурации молекулы относительно ее хирального центра. Приставки D и L, или (+) и (-) представляют собой символы, используемые для уточнения вращения плоскополяризованного света, обусловленного соединением, где (-) или L указывает на то, что соединение является левовращающим, а приставка (+) или D указывает на то, что соединение является правовращающим. Для данной химической структуры эти стереоизомеры идентичны, за исключением того, что эти стереоизомеры являются зеркальными отображениями друг друга. Определенные стереоизомеры могут называться энантиомерами, а смесь таких изомеров называется энантиомерной смесью. Смесь энантиомеров в соотношении 50:50 называется рацемической смесью или рацематом, которые могут возникать в тех случаях, когда в химической реакции или химическом процессе отсутствует стереоселективность или стереоспецифичность.
[0044] В соответствии с выбором исходным материалов и методов соединение по настоящему изобретению может существовать в форме одного из возможных изомеров или их смеси, например, в виде чистого оптического изомера или в виде смеси изомеров, такой как рацемическая смесь изомеров и диастереоизомерная смесь, в зависимости от числа асимметричных атомов углерода. Оптически активные (R)- или (S)- изомеры можно получать, используя хиральные синтоны или хиральные препараты, или разделять, используя традиционные методики. Если соединение содержит двойную связь, заместители могут находиться в Е- или Z-конфигурации; если соединение содержит двузамещенный циклоалкил, заместитель циклоалкила может иметь цис- или транс-конформацию.
[0045] Когда связь с хиральным атомом углерода в формуле по настоящему изобретению изображена прямой линией, это следует понимать как то, что обе конфигурации (R) и (S) хирального атома углерода и как получаемое в результате энантиомерно чистое соединение, так и смесь включены в объем, определяемый общей формулой. В данном документе схематическое представление рацемата или энантиомерно чистого соединения взято из Maehr, J. Chem. Ed. 1985, 62: 114-120. Если не указано иное, клиновидная связь и изображенная пунктиром связь используются для представления абсолютной конфигурации стереоцентра.
[0046] Соединения по настоящему изобретению, содержащие асимметрически замещенные атомы углерода, можно выделять в оптически активной форме или в рацемической форме. Разделение рацемической смеси соединения можно осуществлять любым из ряда способов, известных в данной области техники. Например, эти способы включают фракционную рекристаллизацию с использованием хиральных расщепляющих кислот, которые представляют собой оптически активные солеобразующие органические кислоты. Например, подходящими расщепляющими агентами для фракционной рекристаллизации являются оптически активные кислоты, такие как винная кислота, диацетилвинная кислота, дибензоилвинная кислота, миндальная кислоты, яблочная кислота, молочная кислота или различные оптически активные камфорсульфоновые кислоты, такие как D и L формы β-камфорсульфоновой кислоты. Другие расщепляющие агенты для фракционной рекристаллизации включают α-метилбензиламин в чистой стереоизомерной форме (например, S и R формах или чистой диастереомерной форме), 2-фенилглицинол, норэфедрин, эфедрин, N-метилэфедрин, циклогексилэтиламин, 1,2-диаминоциклогексан и т.д. Разделение рацемической смеси также можно осуществлять путем элюирования колонки, наполненной оптически активным расщепляющим агентом (например, динитробензоилфенилглицином). Также можно применять высокоэффективную жидкостную хроматографию (ВЭЖХ) или сверхкритическую жидкостную хроматографию (СЖХ). Специалисты в данной области техники смогут выбрать конкретные способ, условия элюирования и хроматографические колонки в соответствии со структурами соединений и экспериментальными результатами. Кроме того, также можно использовать чистые оптически активные исходные материалы или реагенты с известной конфигурацией для получения любых энантиомеров или диастереомеров соединений, описанных в настоящем изобретении, путем стереоорганического синтеза.
[0047] Многие геометрические изомеры олефинов, двойные связи C=N и т.п. также могут присутствовать в соединениях, описанных в данном документе, и все такие стабильные изомеры рассматриваются в настоящем изобретении. Когда описанное в данном документе соединение содержит этиленовую двойную связь, такая двойная связь включает Е- и Z-геометрические изомеры, если не указано иное.
[0048] Термин «таутомер» относится к изомеру функциональной группы, получаемому в результате быстрого перемещения атома между двумя положениями в молекуле. Соединение по настоящему изобретению может проявлять таутомеризм. Таутомерные соединения могут находится в двух или более взаимно преобразуемых формах. Прототропный таутомер образуется в результате переноса ковалентно связанных атомов водорода между двумя атомами. Таутомер в общем случае существует в равновесной форме. При попытке выделения одного таутомера обычно образуется смесь, физические и химические свойства которой согласуются со смесью соединений. Положение равновесия зависит от внутримолекулярных химических свойств. Например, для многих алифатических альдегидов и кетонов, таких как ацетальдегид, доминантным является кетонный тип; а для фенолов доминантным является енольный тип. Все таутомерные формы соединений включены в настоящее изобретение.
[0049] В примерах настоящего изобретения протоны могут занимать два или более положений циклической формы гетероциклической кольцевой системы, например, 1Н- и 3Н-имидазол, 1Н-, 2Н- и 4Н-1,2,4-триазол, 1H- и 2Н-изоиндол и 1H- и 2Н-пиразол. Таутомерные формы могут находиться в равновесии или быть стерически зафиксированы в одной форме посредством подходящей замены, например,
[0050] 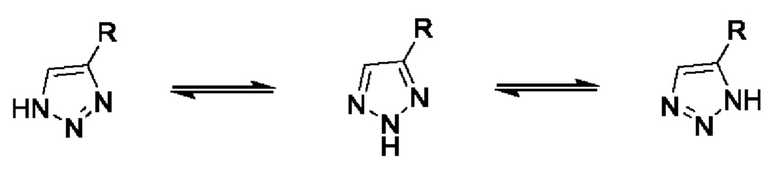
[0051] Вследствие резонанса атом водорода на атоме азота может быть расположен на любом из трех атомов азота триазола, таким образом, их названия различаются, но фактически эти три формы представляют одно и то же соединение.
[0052] Как пример, соединение, представленное следующей формулой, может существовать в форме следующих таутомеров, которые все входят в объем соединения по настоящему изобретению:
[0053] 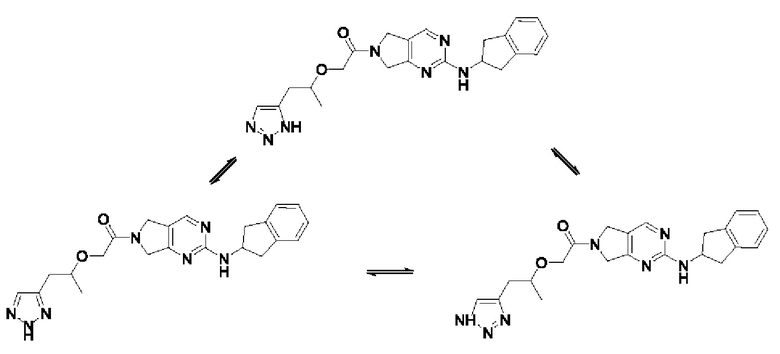
[0054] Термин «фармацевтическая композиция» относится к смеси одного или более соединений, описанных в данном документе, или их физиологически/фармацевтически приемлемых солей или пролекарств, и других химических компонентов. Другие химические компоненты могут представлять собой, например, физиологически/фармацевтически приемлемые носители и эксципиенты. Целью фармацевтической композиции является облегчение введения соединения в организм.
[0055] Термин «сольват» относится к соединению по настоящему изобретению или его соли, содержащим стехиометрический или нестехиометрический растворитель, связанный посредством межмолекулярных нековалентных сил. Когда растворителем является вода, сольват называется гидратом.
[0056] Термин «пролекарство» можно преобразовать в соединение по настоящему изобретению, имеющее биологическую активность в физиологических условиях, или посредством сольволиза. Пролекарство по настоящему изобретению получают путем модификации функциональных групп в соединении, а модифицирующий фрагмент можно удалять посредством традиционных операций или in vivo, так, чтобы получить родительское соединение. Пролекарство включает соединение, образованное путем соединения фрагмента с гидроксильной группой или аминогруппой в соединении по настоящему изобретению. Когда пролекарство соединения по настоящему изобретению вводят субъекту-млекопитающему происходит диссоциация пролекарства с образованием свободной гидроксильной или аминогруппы.
[0057] Соединение по настоящему изобретению может содержать неприродное соотношение атомных изотопов на одном или более атомах, составляющих соединение. Например, соединение может быть помечено радиоактивным изотопом, таким как тритий (3Н), йод-125 (125I) или С-14 (14С). Трансформация всех изотопных композиций соединений по настоящему изобретению, радиоактивных или нет, включена в объем настоящего изобретения.
[0058] Термин «эксципиент» относится к фармацевтически приемлемому инертному ингредиенту. Примеры «эксципиентов» включают, но не ограничиваются этим, связующие вещества, разрыхлители, смазывающие вещества, глиданты, стабилизаторы, наполнители, разбавители и т.п. Эксципиенты могут улучшать свойства в терминах обработки фармацевтического состава, т.е. позволять составу быть более стабильным в случае прямого прессования за счет повышения текучести и/или адгезии. Примерами типичных «фармацевтически приемлемых носителей», подходящих для вышеуказанных составов, являются сахара, такие как лактоза, сахароза, маннит и сорбит; крахмалы, такие как кукурузный крахмал, крахмал тапиоки и картофельный крахмал; целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и метилцеллюлоза; фосфаты кальция, такие как дикальцийфосфат и трикальцийфосфат; сульфат натрия; сульфат кальция; поливинилпирролидон; поливиниловый спирт; стеариновая кислота; соли стеариновой кислоты и щелочноземельных металлов, такие как стеарат магния и стеарат кальция; стеариновая кислота; растительные масла, такие как арахисовое масло, хлопковое масло, кунжутное масло, оливковое масло и кукурузное масло; неионные, катионные и анионные поверхностно-активные вещества; полимеры этиле нгликоля; жирные спирты; и гидролизаты твердых составляющих злаковых и другие нетоксичные совместимые наполнители, связующие вещества, разрыхлители, буферы, консерванты, антиоксиданты, смазывающие вещества, красители и другие эксципиенты, обычно используемые в фармацевтических составах.
[0059] В соответствии с вариантом осуществления настоящего изобретения соединения по настоящему изобретению и/или их композиция могут эффективно ингибировать активность фермента АТХ, обладают преимуществами лучшей печеночной метаболической стабильности и сердечной безопасности и имеют лучшие фармакокинетические свойства, более высокую воздействующую дозу in vivo, большее T1/2, меньшие дозировку и частоту введения и лучшее соблюдение приема, за счет чего имеют широкие перспективы применения в производстве лекарственных средств для лечения связанных с АТХ заболеваний.
[0060] Дополнительные аспекты и преимущества настоящего изобретения будут частично приведены в следующем описании, а часть из них станут очевидными из следующего описания или могут стать понятными посредством реализации настоящего изобретения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0061] На Фиг. 1 приведена диаграмма, иллюстрирующая изменение массы тела животного после введения в соответствии с вариантом осуществления настоящего изобретения; и
[0062] На Фиг. 2 приведена диаграмма, иллюстрирующая изменения концентрации TGFβ1 в легочной ткани и жидкости бронхоальвеолярного лаважа после введения в соответствии с вариантом осуществления настоящего изобретения.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0063] Решения по настоящему изобретению будут объяснены ниже в сочетании с примерами. Специалистам в данной области техники будет понятно, что следующие примеры используются просто для иллюстрации настоящего изобретения, и их не следует воспринимать как ограничивающие объем настоящего изобретения. Методики и условия, которые явным образом не указаны, представляют собой методики или условия, описанные в литературе известного уровня техники, или соответствуют инструкциям к продукту. В случае, когда не указан производитель, реагенты или инструменты все представляют собой традиционные продукты, являющиеся коммерчески доступными.
[0064] Если не указано иное, структуры соединений по настоящему изобретению определены методом ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Единица ЯМР-сдвига составляет 10-6 (м.д.). Растворителем для ЯМР-измерений является дейтерированный диметилсульфоксид, дейтерированный хлороформ, дейтерированный метанол и т.д., а внутренним стандартом является тетраметилсилан (ТМС).
[0065] Сокращения в настоящем изобретении определены следующим образом:
[0066] водн.: водный раствор
[0067] диоксан: 1,4-диоксан
[0068] ДМФА: N,N-диметилформамид
[0069] Т3Р: раствор пропилфосфонового ангидрида, т.е. 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфин-2,4,6-триоксид
[0070] Н: эквивалентная концентрация, например, 1 Н хлористоводородная кислота обозначает 1 моль/л раствор хлористоводородной кислоты
[0071] NMM: N-метилморфолин
[0072] ДИПЭА: диизопропилэтиламин, т.е. N,N-диизопропилэтиламин
[0073] ВЭЖХ: высокоэффективная жидкостная хроматография
[0074] СЖХ: сверхкритическая жидкостная хроматография
[0075] ДМСО: диметилсульфоксид
[0076] NADPH: восстановленный кофермент II
[0077] ГЭПЭС: (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновоя кислота)
[0078] ЭГТА: этиленгликоль-бис(2-аминоэтилэфир)-N,N,N',N'-тетрауксусная кислота
[0079] IC50: Полуингибирующая концентрация, которая относится к концентрации, при которой достигается половина максимального ингибирующего эффекта.
[0080] Если не указано иное, соединения, проиллюстрированные в данном документе, названы и пронумерованы с помощью ChemBioDraw Ultra 13.0.
[0081] Пример 1: Получение целевого соединения I-1
[0082] 2-(2-(1H-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-он (целевое соединение I-1)
[0083] 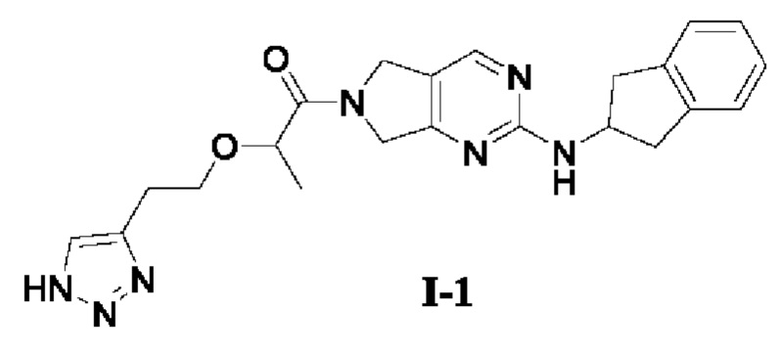
[0084] Схема синтеза целевого соединения I-1 проиллюстрирована ниже:
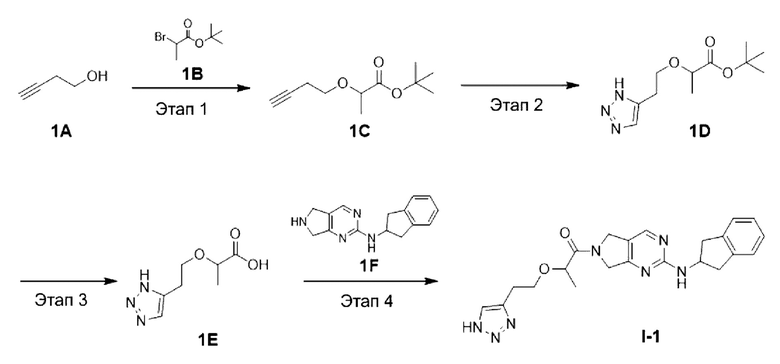
[0085] Этап 1: Синтез трет-бутил 2-(бут-3-ин-1-илокси)пропаноата (1С)
[0086] 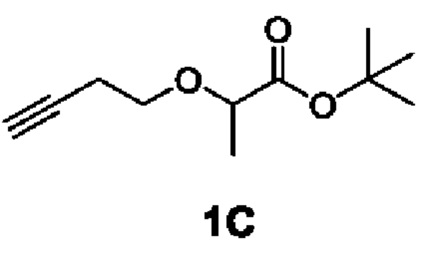
[0087] При температуре 0°С 3-бутин-1-ол (15 г, 214 ммоль) добавляли в дихлорметан (300 мл), а затем последовательно добавляли тетрабутиламмония гидросульфат (7,27 г, 21,40 ммоль), гидроксид натрия (300 мл, 3,57 ммоль, 40% водн.) и трет-бутил 2-бромпропионат (49,2 г, 235 ммоль). Затем смесь медленно нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разводили водой (300 мл), а затем экстрагировали дихлорметаном (200 мл × 3). Органические слои объединяли с получением неочищенного продукта, которые разделяли и очищали с помощью силикагелевой колонки (петролейный эфир) с получение бесцветного маслянистого соединения, трет-бутил 2-(бут-3-ин-1-илокси)пропаноата (1С) (35 г, выход 82%).
[0088] Этап 2: Синтез трет-бутил 2-(2-(1Н-1,2,3-триазол-5-ил)этокси)пропаноата (1D)
[0089] 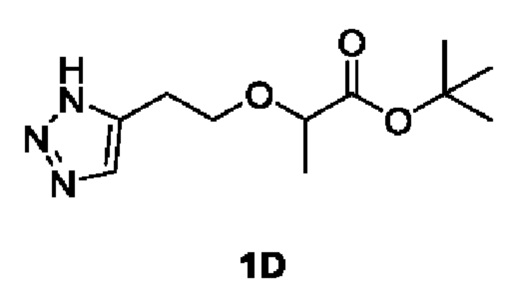
[0090] В условиях азотной защиты трет-бутил 2-(бут-3-ин-1-илокси)пропаноат (50 г, 252 ммоль) и йодид меди (I) (2,4 г, 12,6 ммоль) добавляли в перемешанный раствор N,N-диметилформамида (300 мл)/метанола (30 мл) с последующим добавлением триметилсилилазида (43,6 г, 378 ммоль) при 0°С. Смесь перемешивали при 90°С в течение 18 ч, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разделяли и очищали с помощью силикагелевой колонки (петролейный эфир: этил ацетат (об./об.) = от 10:1 до 5:1) с получением указанного в заголовке соединения трет-бутил 2-(2-(1Н-1,2,3-триазол-5-ил)этокси)пропаноата (1D) (40 г, 166 ммоль, выход 65,7%).
[0091] ЖХ-МС, M/Z (ИЭР): 242,2 (М+1)
[0092] Этап 3: Синтез 2-(2-(1Н-1,2,3-триазол-5-ил)этокси)пропановой кислоты (1Е)
[0093] 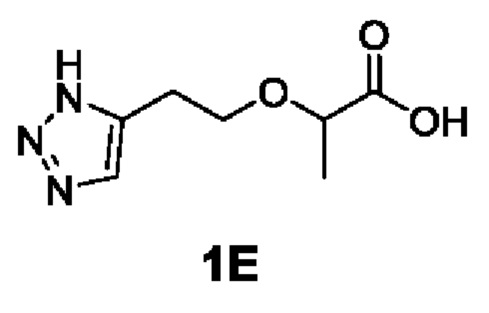
[0094] При 0°С трифторуксусную кислоту (150 мл) добавляли в раствор трет-бутил 2-(2-(1Н-1,2,3-триазол-5-ил)этокси)пропаноата (50 г, 207 ммоль) в дихлорметане (150 мл), а потом перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали с получением неочищенного продукта 2-(2-(1Н-1,2,3-триазол-5-ил)этокси)пропановой кислоты трифторацетата (62 г, 207 ммоль, выход 100%), который непосредственно использовали на следующем этапе реакции.
[0095] Этап 4: Синтез 2-(2-(1Н- 1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-она (соединение I-1)
[0096] 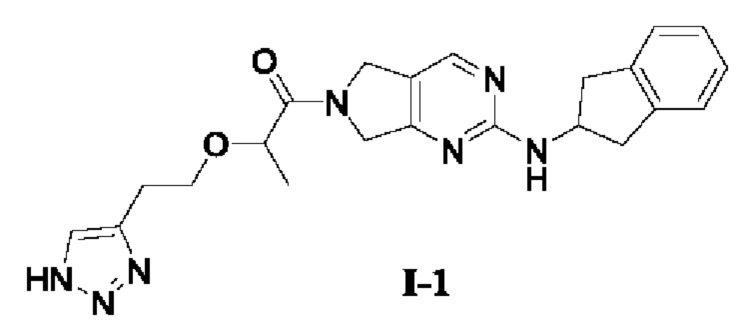
[0097] При комнатной температуре N-(2,3-дигидро-1Н-инден-2-ил)-6,7-дигидро-5Н-пирроло[3,4-d]пиримидин-2-амина гидрохлорид (его синтез описан в патентной заявке WO 2014110000 A1) (7 г, 21,5 ммоль) добавляли в ДМФ (40 мл) с последующим последовательным добавлением 2-(2-(1Н-1,2,3-триазол-4-ил)этокси)пропановой кислоты трифторацетата (9,66 г, 32,3 ммоль) и ДИЭА (27,8 г, 215 ммоль). 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфин-2,4,6-триоксид (10,3 г, 32,3 ммоль, 50% раствор ДМФ) добавляли при 0°С. Затем смесь медленно нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 12 ч. Завершение реакции определяли с помощью ТСХ, реакционный раствор, перемешивая, вливали в дистиллированную воду (150 мл), а осажденное твердое вещество разбивали с ацетонитрилом (50 мл) и фильтровали с получением твердого соединения 2-(2-(1H-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-она (8,1 г, выход 90%).
[0098] 1H ЯМР (400 МГц, CDCl3) δ 8,17 (м, 1H), 7,45 (с, 1H), 7,19-7,09 (м, 4Н), 5,53 (с, 1H), 4,73-4,51 (м, 5Н), 4,18-4,15 (м, 1H), 3,73-3,68 (м, 2Н), 3,53-3,29 (м, 2Н), 3,03-2,77 (м, 4Н), 1,19-1,15 (м, 3Н).
[0099] ЖХ-МС, M/Z (ИЭР): 420,3 (М+1).
[00100] Пример 2: Получение целевого соединения I-1R
[00101] (R)-2-(2-(1Н-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-он (целевое соединение I-1R)
[00102] 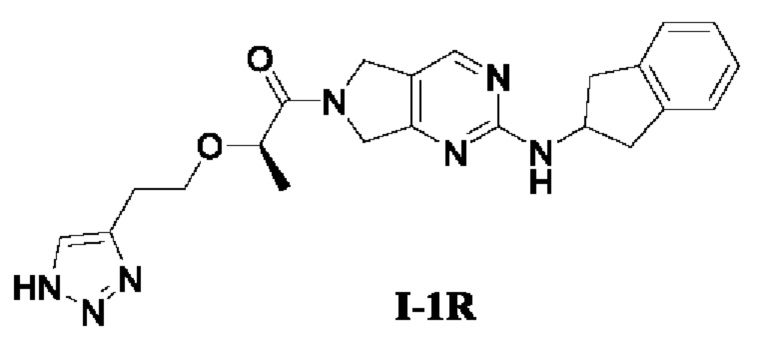
[00103] Схема синтеза целевого соединения I-1R проиллюстрирована ниже:
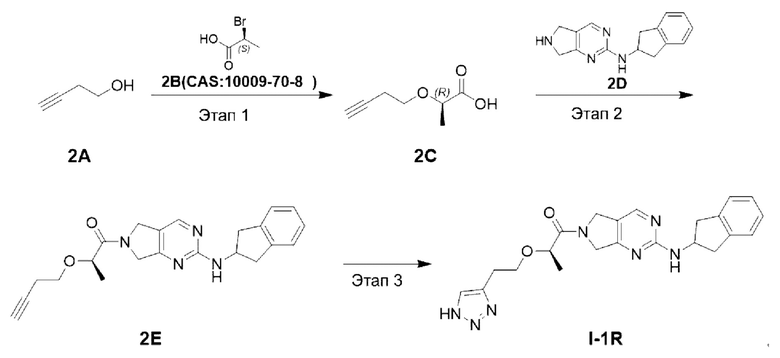
[00104] Этап 1: Синтез (R)-2-(бут-3-ин-1-илокси)пропановой кислоты (2С)
[00105] 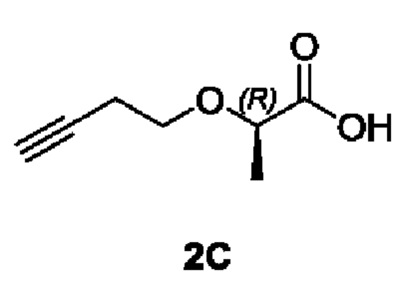
[00106] 3-бутиновый-1 спирт (350 мг, 5 ммоль) добавляли в ДМФ (5 мл), охлаждали до 0°С с последующим добавлением NaH (400 мг, 10 ммоль, 60%), перемешиванием в течение 30 мин. Затем добавляли исходный материал (S)-2-бромпропановую кислоту (700 мг, 4,5 ммоль) с последующим перемешиванием при 0°С в течение 5 ч. Затем в реакционный раствор добавляли воду (10 мл) при 0°С и доводили рН до 1-2 с помощью хлористоводородной кислоты (1 Н). Реакционный раствор экстрагировали этилацетатом (20 мл × 3); а органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (петролейный эфир : этилацетат (об./об.) = 1:1) с получением указанного в заголовке соединения в виде светло-желтой жидкости (R)-2-(бут-3-ин-1-илокси)пропановой кислоты (002С) (390 мг, выход 60%).
[00107] Этап 2: Синтез (R)-2-(бут-3-ин-1-илокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-она (2Е)
[00108] 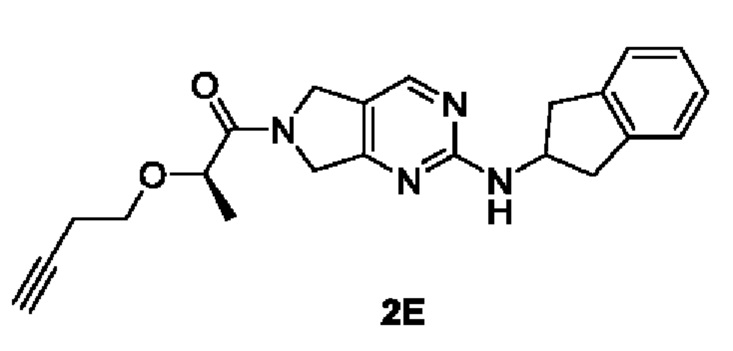
[00109] Исходный материал (R)-2-(бут-3-ин-1-илокси)пропановую кислоту (56 мг, 4 ммоль) добавляли в ДМФ (2 мл) при комнатной температуре с последующим последовательным добавлением исходных материалов Н-(2,3-дигидро-1Н-инден-2-ил)-6,7-дигидро-5Н-пирроло[3,4-d]пиримидин-2-амина (100 мг, 4 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфата (180 мг, 4,4 ммоль) и триэтиламина (82 мг, 8 ммоль). Затем реакционный раствор нагревали до 50°С и перемешивали в течение 15 ч. Реакционный раствор охлаждали до комнатной температуры, добавляли воду (6 мл) и экстрагировали этилацетатом (5 мл × 3). Органические фазы объединяли, сушили безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (дихлорметан : метанол (об./об.) = 10:1) с получением указанного в заголовке соединения, которое представляло собой желтое твердое вещество (R)-2-(бут-3-ин-1-илокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-она (80 мг, 53,9%).
[00110] Этап 3: Синтез (R)-2-(2-(1Н-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-она (соединение I-1R)
[00111] 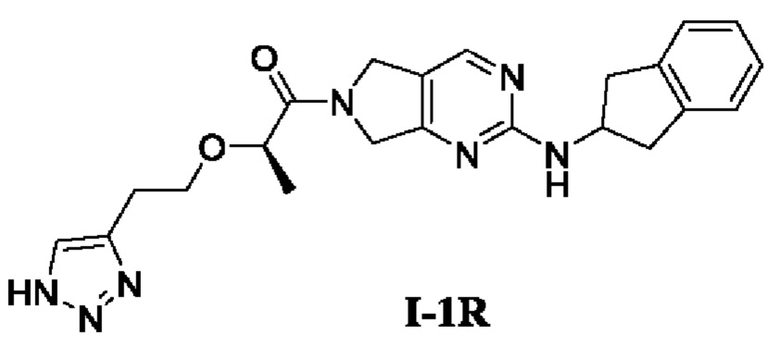
[00112] При комнатной температуре исходный материал (R)-2-(бут-3-ин-1-илокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-он (80 мг, 0,21 ммоль) добавляли в ДМФ (2 мл) и метанол (0,5 мл) с последующим последовательным добавлением триметилсилилазида (37 мг, 0,32 ммоль) и йодида меди (5 мг, 0,025 ммоль) в условиях азотной защиты. Реакционный раствор нагревали до 80°С и перемешивали в течение 15 ч. Затем реакционный раствор охлаждали до комнатной температуры, добавляли воду (8 мл) и экстрагировали этилацетатом (5 мл × 3). Органические фазы объединяли, сушили безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (дихлорметан : метанол (об./об.) = 10:1) с получением указанного в заголовке соединения, желтого твердого вещества (R)-2-(2-(1Н-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-она (8 мг, 8,9%).
[00113] 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,31 (д, 1H), 7,61 (с, 1H), 7,57 (с, 1Н), 7,22 (м, 2Н), 7,16 (м, 2Н), 4,69-4,61 (м, 4Н), 4,55-4,53 (м, 1H), 4,30-4,28 (м, 1H), 3,68-3,64 (м, 2Н), 3,33-3,17 (м, 2Н), 2,91-2,85 (м, 4Н), 1,28-1,19 (м, 3Н).
[00114] ЖХ-МС, M/Z (ИЭР): 420,2 (М+1)
[00115] Пример 3: Получение целевого соединения I-1R и целевого соединения I-1S
[00116] (R)-2-(2-(1Н-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-он (целевое соединение I-1R)
[00117] (S)-2-(2-(1Н-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-он (целевое соединение I-1S)
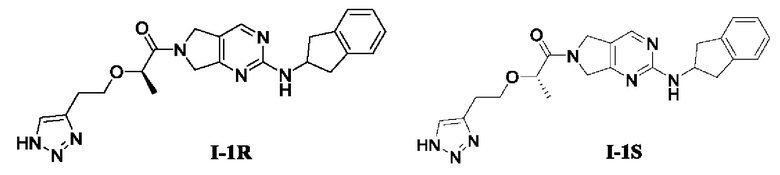
[00118] Целевые соединения получали с помощью СЖХ-разделения.
[00119] Рацемат
2-(2-(1H-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-она (10,1 г, 24,1 ммоль) разделяли с помощью СЖХ в следующих условиях разделения: тип колонки: Chiralpak IC-350 * 4,6 мм, 3 мкм; подвижная фаза: подвижная фаза А представляла собой СО2, подвижная фаза В представляла собой 40% этанол (содержащий 0,05% диэтиламина); градиентное элюирование: 40% этанол в СО2 (содержащий 0,05% диэтиламина); скорость потока: 3 мл/мин; длина волны: 220 нм; температура колонки: 35°С; обратное давление: 100 бар. Были получены один изомер (R)-2-(2-(1Н-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-она (целевое соединение I-1R) (3,51 г, 100% э. и., выход 34,7%) и один изомер (S)-2-(2-(1Н-1,2,3-триазол-4-ил)этокси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)пропан-1-она (целевое соединение I-1S) (3,8 г, 97% э.п., выход 37,6%).
[00120] Посредством сравнения с помощью хиральной ВЭЖХ определили, что при СЖХ-разделении были получены две абсолютные конфигурации соединения.
[00121] Целевое соединение I-1R:
[00122] 1H ЯМР (400 МГц, ДМСО-d6) δ 14,7 (шир. с, 1H), 8,32 (шир. с, 0,6Н), 8,27 (шир. с, 0,4Н), 7,67 (шир. с, 1H), 7,55 (т, J=12,0 Гц, 1H), 7,21-7,13 (м, 4Н), 4,72-4,59 (м, 3Н), 4,55-4,49 (м, 1H), 4,45-4,44 (м, 1Н), 4,33-4,27 (м, 1H), 3,70-3,64 (м, 2Н), 3,28-3,18 (м, 2Н), 2,93-2,85 (м, 4Н), 1,27 (дд, J=8,0, 4,0 Гц, 3Н).
[00123] ЖХ-МС, M/Z (ИЭР): 420,3 (М+1).
[00124] Время удержания: 2,82 мин
[00125] Целевое соединение I-1S:
[00126] 1Н ЯМР (400 МГц, ДМСО-d6) δ 14,5 (шир. с, 1Н), 8,32 (шир. с, 0,6Н), 8,27 (шир. с, 0,4Н), 7,63 (шир. с, 1Н), 7,55 (т, J=12,0 Гц, 1Н), 7,22-7,13 (м, 4Н), 4,72-4,59 (м, 3Н), 4,54-4,49 (м, 1H), 4,45-4,44 (м, 1Н), 4,33-4,27 (м, 1H), 3,68-3,64 (м, 2Н), 3,32-3,22 (м, 2Н), 2,92-2,87 (м, 4Н), 1,27 (дд, J=8,0, 4,0 Гц, 3Н).
[00127] ЖХ-МС, M/Z (ИЭР): 420,3 (М+1).
[00128] Время удержания: 4,48 мин
[00129] Пример 4: Получение целевого соединения I-2
[00130] 2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-он (целевое соединение I-2)
[00131] 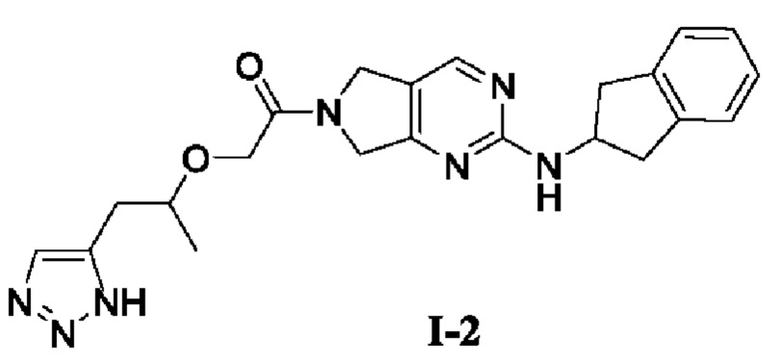
[00132] Схема синтеза целевого соединения I-2 проиллюстрирована ниже:
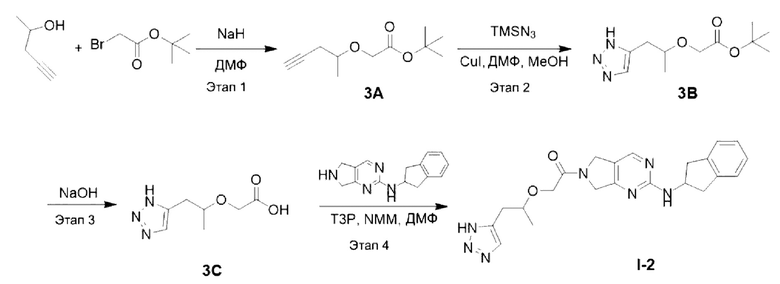
[00133] Этап 1: Синтез трет-бутил 2-(пент-4-ин-2-илокси)ацетата (3А)
[00134] 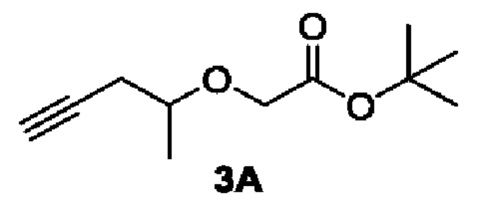
[00135] Исходный материал 4-пентин-2-ол (84,0 г, 1,0 моль) добавляли в 1000 мл сухого тетрагидрофурана и охлаждали до 0°С, добавляли 60% NAH (80,0 г, 2,0 моль), перемешивали смесь в течение 30 мин, добавляли исходный материал т-бутил бромацетат (234,0 г, 1,2 моль) при 0°С, полученную в результате смесь естественным образом нагревали до комнатной температуры и перемешивали в течение 16 ч. В реакционный раствор добавляли воду (2000 мл) при 0°С, рН доводили до 1-2 с помощью 1 Н хлористоводородной кислоты и экстрагировали этилацетатом (2000 мл × 3). Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (петролейный эфир : этилацетат (об./об.) = 3:1) с получением указанного в заголовке соединения в виде светло-желтой жидкости, трет-бутил 2-(пент-4-ин-2-илокси)ацетата (3А) (180 г, выход 65%).
[00136] Этап 2: Синтез трет-бутил 2-((1-(1H-1,2,3-триазол-5-ил)пропан-2-ил)окси)ацетата (3В)
[00137] 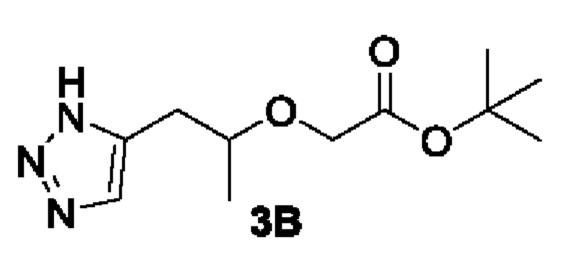
[00138] Исходный материал трет-бутил 2-(пент-4-ин-2-илокси)ацетат (40,0 г, 0,2 моль) добавляли в 400 мл ДМФ и 50 мл метанола при комнатной температуре. В условиях азотной защиты добавляли триметилсилилазид (34,6 г, 0,3 моль) и йодид меди (8 г, 0,4 моль), соответственно. Затем реакционный раствор нагревали до 90°С и перемешивали в течение 15 ч. Реакционный раствор охлаждали до комнатной температуры, затем добавляли воду (1000 мл) и экстрагировали этилацетатом (1200 мл × 3). Органические фазы объединяли, сушили безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (петролейный эфир : этилацетат (об./об.) = 1:1) с получением указанного в заголовке соединения, желтой жидкости трет-бутил 2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)ацетата (26,0 г, 53%).
[00139] Этап 3: Синтез 2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)уксусной кислоты (3С)
[00140] 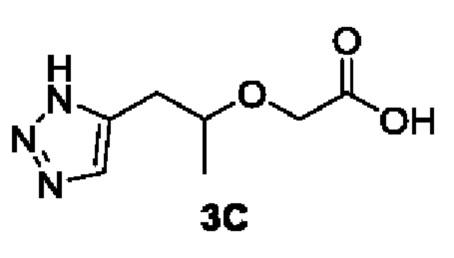
[00141] При комнатной температуре исходный материал трет-бутил 2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)ацетат (26,0 г, 0,11 моль) добавляли в 300 мл ДМФ и 100 мл воды, а затем добавляли NaOH (8,6 г, 0,22 моль). Смесь перемешивали при комнатной температуре в течение 40 ч и затем добавляли воду (300 мл). Реакционный раствор экстрагировали этилацетатом (150 мл × 3), а водную фазу концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (дихлорметан : метанол (об./об.) = 5:1) с получением указанного в заголовке соединения, желтой жидкости 2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)уксусной кислоты (20 г, 85%).
[00142] Этап 4: Синтез 2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-она (целевое соединение I-2)
[00143] 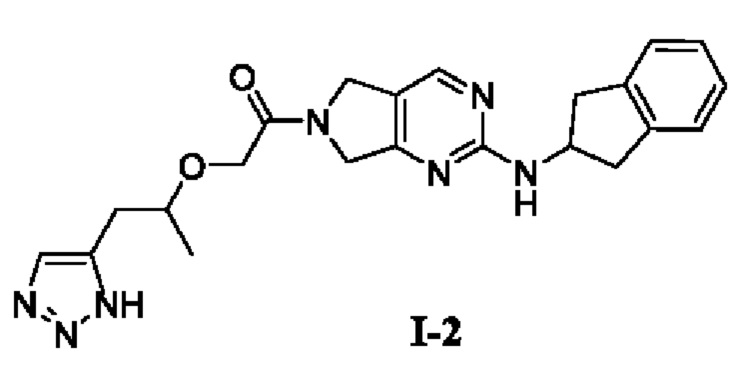
[00144] Исходный материал 2-((1-(1H-1,2,3-триазол-5-ил)пропан-2-ил)окси)уксусную кислоту (20 г, 0,11 моль) добавляли в 400 мл ДМФ при комнатной температуре. N-(2,3-дигидро-1H-инден-2-ил)-6,7-дигидро-5Н-пирроло[3,4-d] пиримидин-2-амин (его синтез описан в патентной заявке WO 201411000 A1) (12 г, 47,6 ммоль), Т3Р (45,4 г, 71,4 ммоль, 50% раствор этилацетата) и NMM (24 г, 238 ммоль) добавляли при 0°С. Смесь естественным образом нагревали до комнатной температуры и затем перемешивали в течение 16 ч. Реакционный раствор фильтровали. В фильтрат добавляли воду (300 мл) и экстрагировали этилацетатом (1500 мл × 3). Органические фазы объединяли, сушили безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (дихлорметан : метанол (об./об.) = 10:1) с получением указанного в заголовке соединения, желтого твердого вещества 2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-она (19,3 г, 92%).
[00145] 1H ЯМР (400 МГц, ДМСО-d6) δ 8,30 (д, 1H), 7,64 (шир., 1Н), 7,57 (т, 1H), 7,22-7,20 (м, 2Н), 7,16-7,12 (м, 2Н), 4,65-4,59 (м, 3Н), 4,52 (с, 1H), 4,42 (с, 1H), 4,25-4,17 (м, 2Н), 3,87-3,81 (м, 1H), 3,27-3,21 (м, 2Н), 2,90-2,85 (м, 4Н), 1,19 (т, 3Н).
[00146] ЖХ-МС, M/Z (ИЭР): 420,2 (М+1)
[00147] Пример 5: Получение целевого соединения I-2S
[00148] (S)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-он (целевое соединение I-2 S)
[00149] 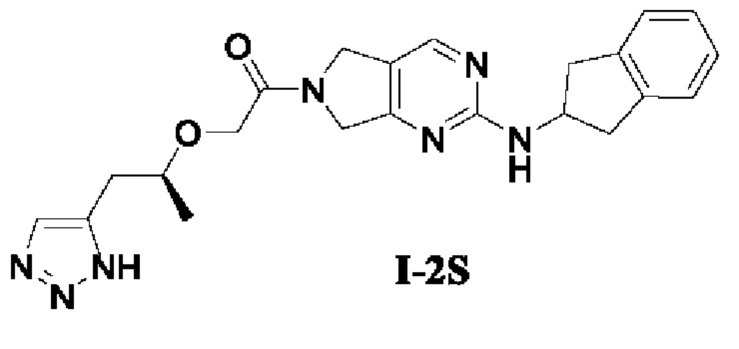
[00150] Схема синтеза целевого соединения I-2S проиллюстрирована ниже:
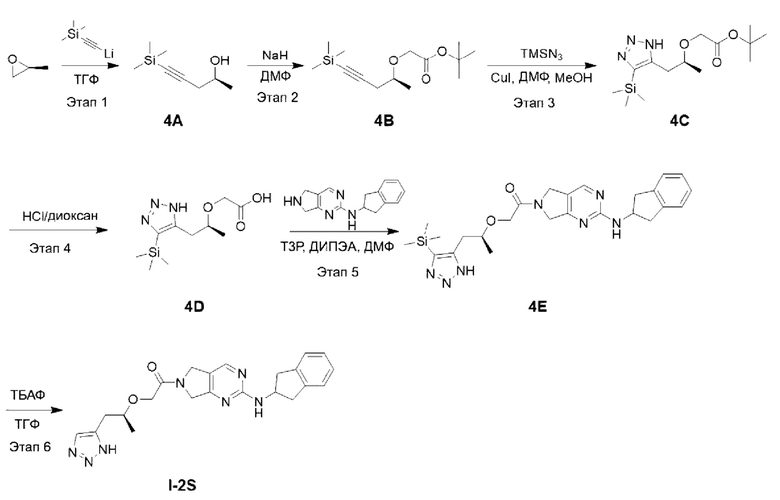
[00151] Этап 1: Синтез (S)-5-(триметилсилил)пент-4-ин-2-ола (4А)
[00152] 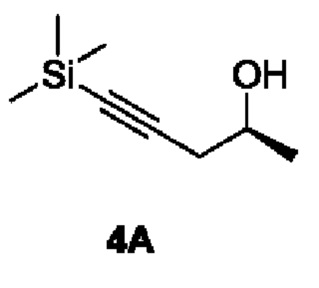
[00153] Раствор тетрагидрофурана и (триметилсилил)ацетилида лития (0,5 М, 52 мл) добавляли в трехгорлую колбу и охлаждали до -70°С в условиях азотной защиты с последующим добавлением раствора тетрагидрофурана и трифторида бора (50%, 2,4 мл) и медленного по каплям добавления (S)-пропиленоксида (1,5 г). После покапельного добавления смесь перемешивали, поддерживая температуру, в течение 1 часа, а затем добавляли насыщенный водный раствор хлорида аммония (10 мл), чтобы погасить реакцию. После нагревания до комнатной температуры раствор разделяли на органическую фазу и водную фазу. Органическую фазу сушили и концентрировали с получением неочищенного продукта (2,0 г), который непосредственно использовали на следующем этапе реакции.
[00154] Этап 2: Синтез трет-бутил (S)-2-((5-(триметилсилил)пент-4-ин-2-ил)окси)ацетата (4В)
[00155] 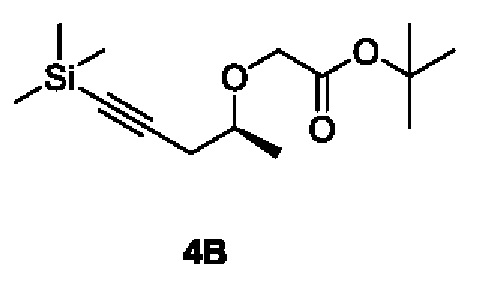
[00156] Исходный материал (S)-5-(триметилсилил)пент-4-ин-2-ол (2,0 г, 12,8 ммоль) добавляли в 10 мл сухого тетрагидрофурана, охлаждали до 0°С, доводили 60% NaH (0,49 г, 12,8 ммоль), перемешивали в течение 30 минут, а затем добавляли исходный материал трет-бутил 2-бромацетат (3,0 г, 15,4 ммоль) при 0°С. Смесь естественным образом нагревали до комнатной температуры и перемешивали в течение 16 ч. В реакционный раствор добавляли воду (20 мл) при 0°С, рН доводили до 12 с помощью 1 H хлористоводородной кислоты и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (петролейный эфир : этилацетат (об./об.) = 3:1) с получением указанного в заголовке соединения, светло-желтой жидкости трет-бутил (S)-2-((5-(триметилсилил)пент-4-ин-2-ил)окси)ацетата (4В) (2,1 г, выход 60,7%).
[00157] Этап 3: Синтез трет-бутил (S)-2-((1-(4-(триметилсилил)-1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)ацетата (4С)
[00158] 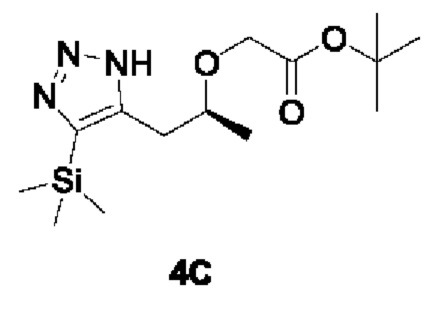
[00159] При комнатной температуре исходный материал трет-бутил (S)-2-((5-(триметилсилил)пент-4-ин-2-ил)окси)ацетат (1,0 г, 3,7 ммоль) добавляли в 10 мл ДМФ и 5 мл метанола с последующим добавлением триметилсилилазида (0,64 г, 5,5 ммоль) и йодида меди (0,14 г, 0,74 ммоль) в условиях азотной защиты, соответственно. Реакционный раствор нагревали до 90°С и перемешивали в течение 15 ч. Реакционный раствор охлаждали до комнатной температуры, добавляли воду (50 мл) и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, сушили безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (петролейный эфир : этилацетат (об./об.) = 1:1) с получением указанного в заголовке соединения, желтой жидкости трет-бутил (S)-2-((1-(4-(триметилсилил)-1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)ацетата (0,6 г, 51,8%).
[00160] Этап 4: Синтез (S)-2-((1-(4-(триметилсилил)-1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)уксусной кислоты (4D)
[00161] 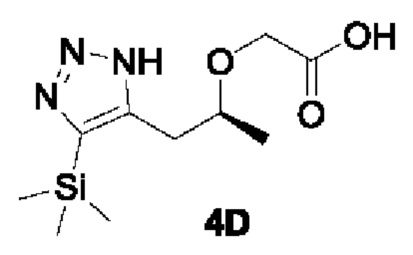
[00162] При комнатной температуре исходный материал трет-бутил (S)-2-((1-(4-(триметилсилил)-1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)ацетат (0,6 г, 1,9 ммоль) добавляли в раствор хлорида водорода и 1,4-диоксана (4 М, 10 мл), перемешивали при комнатной температуре в течение 2 ч и концентрировали до сухости с получением неочищенного продукта (S)-2-((1-(4-(триметилсилил)-1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)уксусной кислоты (0,5 г, 100%), который непосредственно использовали на следующем этапе реакции.
[00163] Этап 5: Синтез (S)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)-2-((1-(4-(триметилсилил)-1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)этан-1-она (4Е)
[00164] 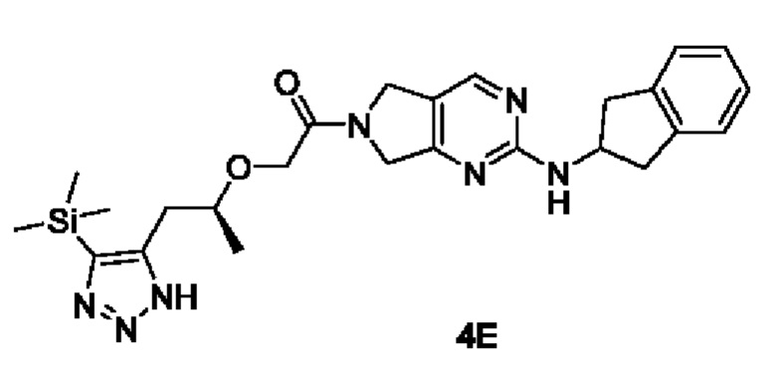
[00165] Исходный материал (S)-2-((1-(4-(триметилсилил)-1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)уксусную кислоту (0,5 г, 1,9 ммоль) добавляли в 4 мл ДМФ с последующим добавлением N-(2,3-дигидро-1H-инден-2-ил)-6,7-дигидро-5Н-пирроло[3,4-d]пиримидин-2-амина (0,7 г, 2,1 ммоль), Т3Р (1,5 г, 2,3 ммоль, 50% раствор ДМФ) и диизопропилэтиламина (0,5 г, 3,8 ммоль) при 0°С. Смесь естественным образом нагревали до комнатной температуры и перемешивали в течение 16 ч. Реакционный раствор фильтровали, а в фильтрат добавляли воду (30 мл) и экстрагировали этилацетатом (15 мл × 3). Органические фазы объединяли, сушили безводным сульфатом натрия и концентрировали с получением неочищенного продукта (S)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)-2-((1-(4-(триметилсилил)-1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)этан-1-она (0,3 г).
[00166] Этап 6: Синтез (S)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-она (целевое соединение I-2S)
[00167] 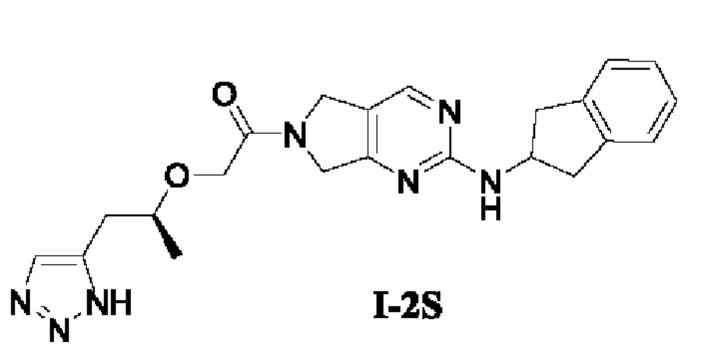
[00168] Неочищенный продукт (S)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)-2-((1-(4-(триметилсилил)-1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)этан-1-он (0,3 г) добавляли в тетрагидрофуран (10 мл) с последующим добавлением тригидрата фторида тетрабутиламмония (0,4 г, 1,2 ммоль). Смесь перемешивали при комнатной температуре в течение 5 ч, добавляли воду (20 мл) и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, сушили безводным сульфатом натрия и концентрировали. Остаток разделяли с помощью препаративной хроматографии и лиофилизировали с получением (S)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-она (68,3 мг, выход за два этапа 8,4%).
[00169] 1H ЯМР (400 МГц, ДМСО-d6) δ 8,30 (д, 1H), 7,64 (шир., 1H), 7,57 (т, 1H), 7,22-7,20 (м, 2Н), 7,16-7,12 (м, 2Н), 4,65-4,59 (м, 3Н), 4,52 (с, 1H), 4,42 (с, 1H), 4,25-4,17 (м, 2Н), 3,87-3,81 (м, 1Н), 3,27-3,21 (м, 2Н), 2,90-2,85 (м, 4Н), 1,19 (т, 3Н).
[00170] ЖХ-МС, M/Z (ИЭР): 420,4 (М+1)
[00171] Пример 6: Получение целевого соединения I-2R
[00172] (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-он (целевое соединение I-2R)
[00173] Этап 1: Синтез (R)-5-(триметилсилил)пент-4-ин-2-ола (5А)
[00174] 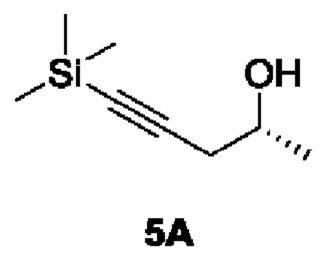
[00175] В условиях азотной защиты триметилсилилацетилен (51,7 г) и диэтиловый эфир (600 мл) добавляли в трехгорлую колбу, охлаждали до -78°С с последующим медленным по каплям добавлением н-бутиллития (2,5 М, 217 мл). После завершения покапельного добавления проводили реакцию смеси в течение 1 часа, поддерживая температуру на -78°C, затем добавляли раствор тетрагидрофурана и трифторида бора (50%, 30 мл) и медленно по каплям добавляли (R)-пропиленоксид (30 г). После завершения покапельного добавления в течение 1 часа поддерживали температуру, перемешивая, и добавляли насыщенный водный раствор бикарбоната натрия (300 мл), чтобы погасить реакцию. После нагревания до комнатной температуры реакционный раствор разделяли на слои, а органическую фазу сушили, смешивали с силикагелем и затем разделяли и очищали с помощью силикагелевой колонки (петролейный эфир : этилацетат (об./об.) = 10:1) с получением продукта в виде светло-желтой жидкости соединения (R)-5-(триметилсилил)пент-4-ин-2-ола (34 г, выход 42,1%).
[00176] Этап 2: Синтез трет-бутил (R)-2-((5-(триметилсилил)пент-4-ин-2-ил)окси)ацетата (5В)
[00177] 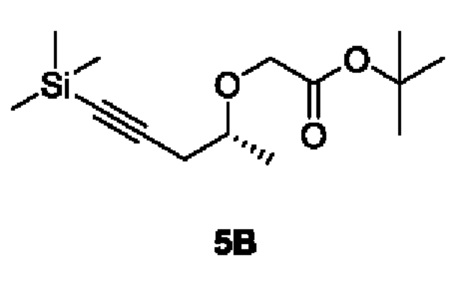
[00178] Исходный материал (R)-5-(триметилсилил)пент-4-ин-2-ол (34 г, 218 ммоль) добавляли в 340 мл сухого тетрагидрофурана и охлаждали до 0°С, с последующим добавлением 60% NaH (10,44 г, 261 ммоль), перемешиванием в течение 30 мин и добавлением исходного материала трет-бутил 2-бромацетата (46,7 г, 239 ммоль) при 0°С.Смесь естественным образом нагревали до комнатной температуры и перемешивали в течение 16 ч. При 0°С в реакционный раствор добавляли метанол (20 мл), смешивали с силикагелем, концентрировали и разделяли и очищали с помощью силикагелевой колонки (петролейный эфир : этилацетат (об./об.) = 10:1) с получением светло-желтой жидкости соединения трет-бутил (R)-2-((5-(триметилсилил)пент-4-ин-2-ил)окси)ацетата (5В) (50 г, выход 85%).
[00179] Этап 3: Синтез трет-бутил (R)-2-(пент-4-ин-2-илокси)ацетата (5С)
[00180] 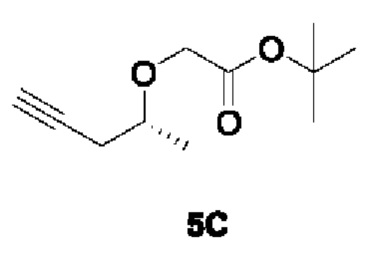
[00181] При комнатной температуре исходный материал трет-бутил (R)-2-((5-(триметилсилил)пент-4-ин-2-ил)окси)ацетата (50 г, 185 ммоль) добавляли в 500 мл тетрагидрофурана, а затем добавляли фторид тетрабутиламмония (53,2 г, 203 ммоль). Реакцию смеси проводили при комнатной температуре в течение 15 ч, смешивали с силикагелем и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (петролейный эфир : этилацетат (об./об.) = 10:1) с получением указанного в заголовке желтого жидкого соединения трет-бутил (R)-2-(пент-4-ин-2-илокси)ацетата (27 г, 73,7%).
[00182] Этап 4: Синтез трет-бутил (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)ацетата (5D)
[00183] 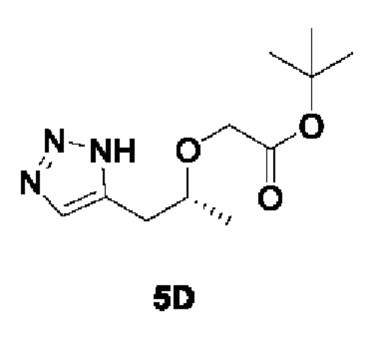
[00184] Исходный материал трет-бутил (R)-2-(пент-4-ин-2-илокси)ацетат (27 г, 136 ммоль) добавляли в 150 мл ДМФ и 20 мл метанола при комнатной температуре. Затем в условиях азотной защиты добавляли триметилсилилазид (23,53 г, 204 ммоль) и йодид меди (2,08 г, 10,89 ммоль), соответственно. Реакционный раствор нагревали до 90°С и перемешивали в течение 15 ч. Затем реакционный раствор охлаждали до 40°С, концентрировали до сухости, разводили дихлорметаном, смешивали с силикагелем и концентрировали. Остаток разделяли и очищали с помощью силикагелевой колонки (петролейный эфир : этилацетат (об./об.) = 1:1) с получением желтого маслянистого соединения трет-бутил (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)ацетата (14 г, 42,6%).
[00185] Этап 5: Синтез (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)уксусной кислоты (5Е)
[00186] 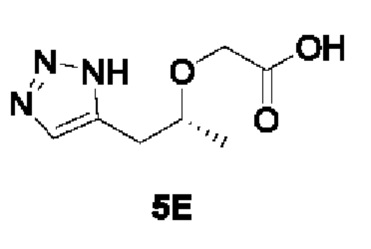
[00187] Исходный материал трет-бутил (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)ацетат (14 г, 58 ммоль) добавляли в раствор хлорида водорода и 1,4-диоксана (4 моль/л, 70 мл) при комнатной температуре, перемешивали при комнатной температуре в течение 16 ч и фильтровали. Твердое вещество промывали метил-трет-бутиловым эфиром и сушили с получением белого твердого вещества (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)уксусной кислоты (9,2 г, 86%).
[00188] Этап 6: Синтез (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-она (целевое соединение I-2R)
[00189] 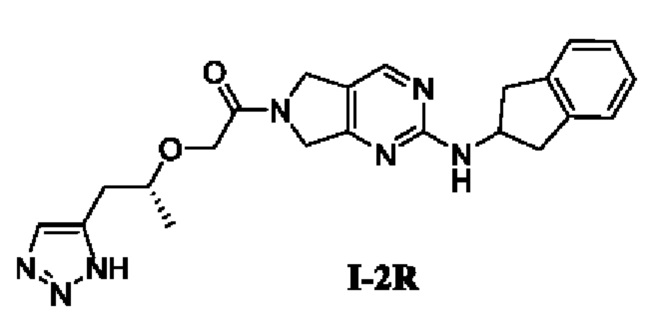
[00190] Исходные материалы (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)уксусную кислоты (9,41 г, 42,5 ммоль) и N-(2,3-дигидро-1H-инден-2-ил)-6,7-дигидро-5Н-пирроло[3,4-d]пиримидин-2-амин (9,2 г, 28,3 ммоль) добавляли в 1000 мл ДМФ с последующим добавлением Т3Р (50% раствор ДМФ) (27 г, 42,5 ммоль) и диизопропилэтиламина (21,95 г, 170 ммоль) при 0°С. Смесь естественным образом нагревали до комнатной температуры и перемешивали в течение 16 ч. Реакционный раствор фильтровали, в фильтрат добавляли воду (3 мл) и концентрировали до сухости. Остаток разделяли и очищали с помощью силикагелевой колонки (дихлорметан : метанол (об./об.) = 10:1) с получением 12 г неочищенного продукта. Неочищенный продукт разбивали со 120 мл изопропилацетата в течение 10 ч, фильтровали и сушили с получением (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-она (7,8 г, выход 65,7%).
[00191] 1H ЯМР (400 МГц, ДМСО-d6) δ 8,30 (д, 1H), 7,64 (шир., 1Н), 7,57 (т, 1H), 7,22-7,20 (м, 2Н), 7,16-7,12 (м, 2Н), 4,65-4,59 (м, 3Н), 4,52 (с, 1Н), 4,42 (с, 1H), 4,25-4,17 (м, 2Н), 3,87-3,81 (м, 1H), 3,27-3,21 (м, 2Н), 2,90-2,85 (м, 4Н), 1,19 (т, 3Н).
[00192] ЖХ-МС, M/Z (ИЭР): 420,4 (М+1)
[00193] Пример 7: Получение целевого соединения I-2S и целевого соединения I-2R
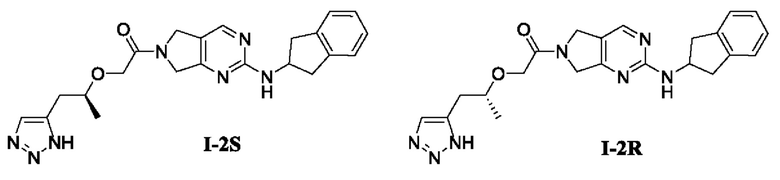
[00194] Целевые соединения получали с помощью ВЭЖХ-разделения.
[00195] Рацемат
2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амин о)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-она (22 г, 52,5 ммоль) разделяли с помощью ВЭЖХ в следующих условиях разделения: тип колонки: Chiralpak ID (ID00CD-QG003) 4,6 мм, В. О. × 15 см л; подвижная фаза: 100% метанол; скорость потока: 1,0 мл/мин; длина волны: 254 нм; температура колонки: 35°C; обратное давление: 10 МПа. Соединения было получены в одной единственной конфигурации и записаны как пик 1 (7,0 г, 100% э. и., выход 63,6%) и пик 2 (9,9 г, 97% э. и., выход 90,0%), соответственно.
[00196] Путем сравнения времени удержания по данным СЖХ-анализа определили, что при ВЭЖХ-разделении были получены две абсолютные конфигурации соединения.
[00197] Целевое соединение I-2S:
[00198] СЖХ-анализ: тип колонки: Chiralpak AY-3 50 × 4,6 мм В. Д., 3 мкм; подвижная фаза: этанол (содержащий 0,05% диэтиламина); градиентное элюирование: 60% этанол в CO2 (содержащий 0,05% диэтиламина); скорость потока: 3,0 мл/мин; длина волны: 254 нм; температура колонки: 35°C; обратное давление: 100 бар;
[00199] Время удержания: 0,964 мин.
[00200] Целевое соединение I-2R:
[00201] СЖХ-анализ: тип колонки: Chiralpak AY-3 50 × 4,6 мм В. Д., 3 мкм; подвижная фаза: этанол (содержащий 0,05% диэтиламина); градиентное элюирование: 60% этанол в СО2 (содержащий 0,05% диэтиламина); скорость потока: 3,0 мл/мин; длина волны: 254 нм; температура колонки: 35°C; обратное давление: 100 бар;
[00202] Время удержания: 2,118 мин.
[00203] По данным того же СЖХ-анализа время удержания для пика 1 составляло 1,006 мин, а время удержания для пика 1 составляло 2,205 мин. Посредством сравнения времени удержания определили, что пик 1 соответствует соединению I-2S, т.е. (S)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-ону, а пик 2 соответствует соединению I-2R, т.е. (R)-2-((1-(1Н-1,2,3-триазол-5-ил)пропан-2-ил)окси)-1-(2-((2,3-дигидро-1Н-инден-2-ил)амино)-5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)этан-1-ону.
[00204] Пример 8: Контрольное соединение и его получение
[00205] Контрольное соединение синтезировали в соответствии с патентной заявкой WO 2014110000 A1.
[00206] Контрольное соединение в тестовых примерах ниже представляет собой соединение, упоминаемое в примере 8.
[00207] Тестовый пример 1: Анализ ингибирования активности фермента аутотаксина (АТХ)
[00208] Ингибирующую активность соединений в отношении фермента аутотаксина выявляли, используя аналитический набор для скрининга ингибиторов аутотаксина (Cayman, 700580). Сначала тестовое соединение получали в виде 10 мМ маточного раствора в растворителе ДМСО, а затем маточный раствор разводили ДМСО до 8 градиентных концентраций. После этого 8 концентраций разводили аутотаксиновым аналитическим буфером (1×), предоставленном в наборе, до 19× рабочих растворов соединения (содержание ДМСО составляло 1,9%). Доставали аутотаксиновый аналитический реагент (10×) и разводили в 10 раз аутотаксиновым аналитическим буфером (1×). Доставали аутотаксиновый субстрат, растворяли путем добавления 1,2 мл аутотаксинового аналитического буфера (1×), равномерно смешивали и отстаивали при комнатной температуре. В 96-луночном планшете 150 мкл аутотаксинового аналитического буфера (1×), 10 мкл полученных и разведенных 19× рабочих растворов соединения, 10 мкл аутотаксинового аналитического реагента (1×) и 20 мкл растворенного аутотаксинового субстрата добавляли в каждую из лунок для каждой концентрации и равномерно смешивали. 96-Луночный планшет встряхивали в шейкере с постоянной температурой при 37°С и инкубировали в темноте в течение 30 мин, затем планшет доставали и помещали в микропланшетный ридер для считывания ОП405. Экспериментальные результаты вводили в программное обеспечение GraphPad Prism и рассчитывали IC50 каждого соединения путем аппроксимации.
[00209] 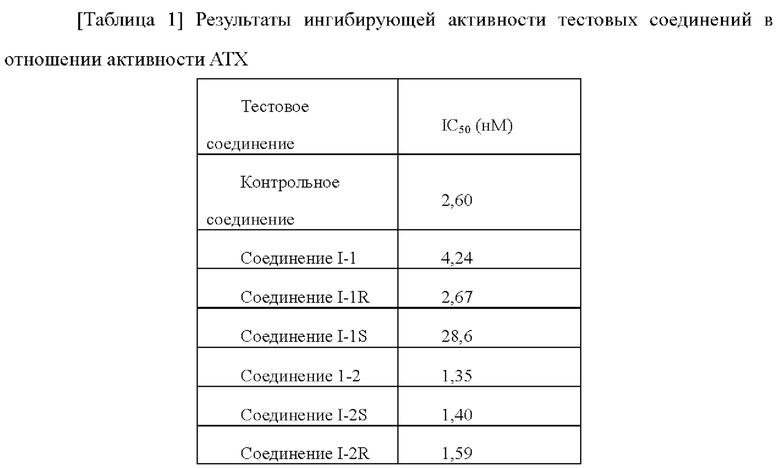
[00210] Экспериментальные результаты показали, что соединения по настоящему изобретению имеют хорошую ингибирующую активность в отношении фермента АТХ и могут эффективно ингибировать активность фермента АТХ.
[00211] Тестовый пример 2: Тест на стабильность с микросомами печени человека
[00212] Тест на стабильность с микросомами печени человека проводили путем инкубации соединения и микросом печени человека in vitro. Сначала тестовое соединение получали в виде 10 мМ маточного раствора в растворителе ДМСО, а затем соединение разводили до 0,5 мМ ацетонитрилом. Микросомы печени человека (Corning) разводили ФСБ и получали в виде раствора микросомы/буфер, который использовали для разведения 0,5 мМ соединения до рабочего раствора. В рабочем растворе концентрация соединения составляла 1,5 мкМ, а концентрация микросом печени человека составляла 0,75 мг/мл. Брали многолуночный планшет с глубокими лунками, в каждую лунку последовательно добавляли 30 мкл рабочего раствора и 15 мкл предварительно нагретого раствора NADPH (6 мМ) для инициации реакции и инкубировали реакционную смесь при 37°С. Через 0, 5, 15, 30 и 45 мин инкубации в соответствующие лунки добавляли по 135 мкл ацетонитрила для терминации реакции. После терминации реакции ацетонитрилом в последний момент времени 45 мин планшет с глубокими лунками вортексно перемешивали в течение 10 мин (600 об/мин), а затем центрифугировали в течение 15 мин. После центрифугирования собирали супернатант и добавляли очищенную воду в соотношении 1:1 для проведения обнаружения методом ЖХ-МС/МС. Соответственно, в каждый момент времени получали соотношение площади пика соединения и площади пика внутреннего стандарта, а соотношения площади пика соединения в моменты 5, 15, 30 и 45 мин сравнивали с соотношением площади пика в момент 0 мин, чтобы рассчитать оставшееся процентное содержание соединения в каждый момент времени. T1/2 рассчитывали, используя Excel.
[00213] 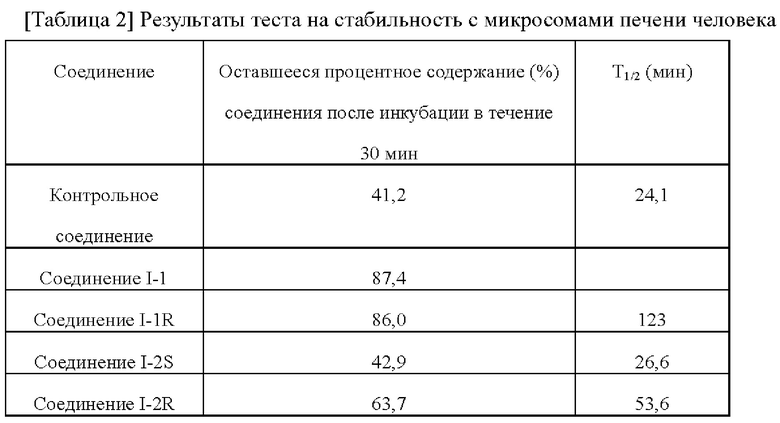
[00214] По сравнению с контрольным соединением соединения по настоящему изобретению проявляли лучшую печеночную метаболическую стабильность, метаболизировались медленнее в организме человека и имели большее воздействие. Т1/2 стабильности в микросомах печени соединений по настоящему изобретению лучше, чем для контрольного соединения и даже может превышать значение для контрольного соединения в два раза. Соответственно, клиническую дозировку и частоту введения можно уменьшить, снизить токсические и побочные эффекты клинического введения и улучшить клиническое соблюдение приема.
[00215] Тестовый пример 3: Выявление ингибирующего эффекта соединений в отношении hERG с использованием полностью автоматического электрофизиологического метода фиксации потенциала QPatch
[00216] Полностью автоматический электрофизиологический метод фиксации потенциала QPatch использовали для выявления ингибирующего эффекта соединений в отношении hERG. Клетками, используемыми в этом тесте, была линия клеток СНО, трансфицированных кДНК hERG и стабильно экспрессирующих каналы hERG (предоставленная Sophion Bioscience, Дания), а номер пассажа был Р24. Клетки культивировали в среде, содержащей следующие компоненты (все приобретены у Invitrogen): среда Хэма F12, инактивированная фетальная бычья сыворотка (10% (об./об.)), гигромицин В (100 мкг/мл) и генетицин (100 мкг/мл). Клетки СНО hERG выращивали в чашке Петри, содержащей вышеуказанную среду, и культивировали в инкубаторе при 37°С, содержащем 5% CO2.
[00217] Готовили внеклеточную жидкость (2 мМ CaCl2, 1 мМ MgCl2, 4 мМ KCl, 145 мМ NaCl, 10 мМ Glucose, 10 мМ ГЭПЭС, рН: около 7,4, осмотическое давление: около 305 мОсм) и внутриклеточную жидкость (5,374 мМ CaCl2, 1,75 мМ MgCl2, 120 мМ KCl, 10 мМ ГЭПЭС, 5 мМ ЭГТА, 4 мМ Na-АТФ, рН около 7,25, осмотическое давление около 295 мОсм).
[00218] Подлежащее тестированию соединение получали в виде 10 мМ маточного раствора в растворителе ДМСО, разводили соединение до 3 мМ, 1 мМ, 0,3 мМ и 0,1 мМ с помощью ДМСО, а затем разводили соединение до 30 мкМ, 10 мкМ, 3 мкМ, 1 мкМ, 0,3 мкМ и 0,1 мкМ внеклеточной жидкость так, что за исключением того, что конечная концентрация ДМСО в 30 мкМ растворе соединения составляла 0,3%, конечная концентрация ДМСО в растворах соединения всех других концентраций составляла 0,1%.
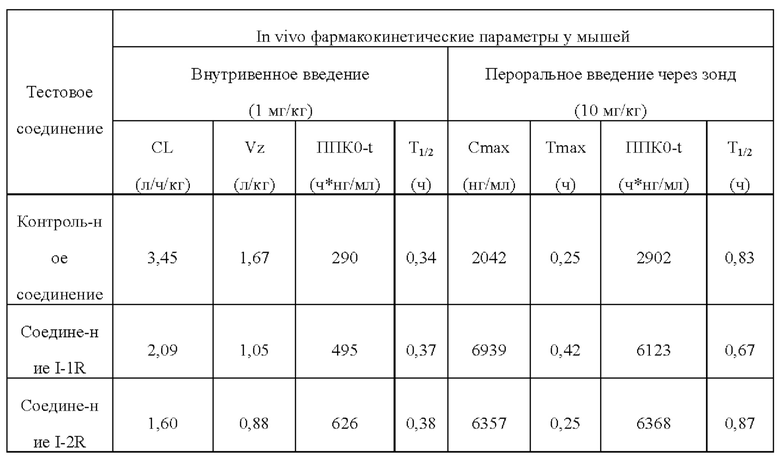
[00219] После расщепления и ресуспендирования клетки СНО hERG добавляли в полностью автоматическую систему QPatch (Sophion, Дания) и проводили тест в соответствии со следующей процедурой.
[00220] После достижения конфигурационного состояния разорванных цельных клеток на исходной стадии записывали цельноклеточный ток при комнатной температуре (около 25°С), и для обеспечения стабильности запись клеток проводили в течение по меньшей мере 120 секунд. Для теста отбирали стабильные клетки. Во время цельного теста фиксацию потенциала клеток проводили при -80 мВ, напряжение при фиксации потенциала клеток было деполяризовано до +20 мВ, чтобы активировать калиевые каналы hERG, а через 2,5 секунды напряжение при фиксации потенциала клеток составляло -50 мВ, чтобы устранить инактивацию и создать направленный наружу следовой ток. Пиковый следовой ток использовали в качестве значения тока hERG. Описанный выше режим подачи напряжения применяли к клетками для электрофизиологических тестов каждые 15 секунд. Внутриклеточную жидкость, содержащую 0,1% диметилсульфоксида (растворитель) добавляли в клетки, чтобы установить базовую линию, а затем давали току установиться в течение 3 мин. После добавления раствора соединения клетки держали в тестовом окружении до тех пор, пока эффект соединения не достигал стационарного состояния или до 4 мин. В тестовых экспериментах с разными концентрационными градиентами соединения соединение добавляли в фиксированные клетки в концентрации от низкой до высокой. После окончания теста соединения клетки промывали внеклеточной жидкостью до тех пор, пока ток не возвращался к стабильному состоянию.
[00221] Данные теста анализировали с помощью аналитического программного обеспечения Qpatch, предоставленного Sophion, Excel и Graphpad Prism.
[00222] 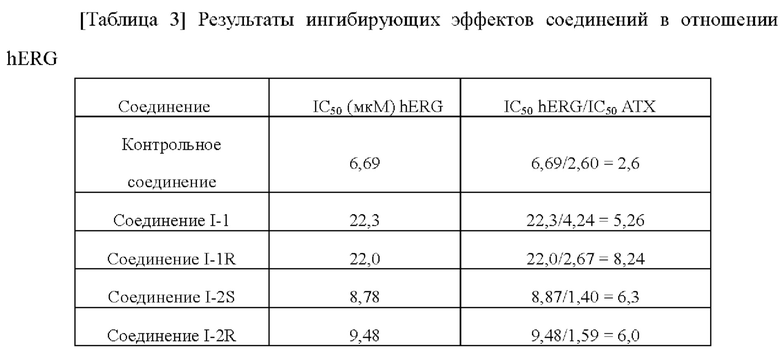
[00223] По сравнению с контрольным соединением соединения по настоящему изобретению проявляют более слабую ингибирующую активность в отношении hERG. С учетом значения IC50 соединений, указывающие на ингибирование активности фермента ATX, соединения по настоящему изобретению демонстрируют благоприятное окно безопасности в отношении ингибирования hERG и имеют значительные преимущества в отношении кардиологической безопасности.
[00224] Тестовый пример 4: Тест термодинамической растворимости
[00225] Получали фосфатно-солевой буферный раствор (ФСБ, рН 7,4), раствор FeSSIF (рН 5,8, содержащий 10 мМ таурохолата натрия, 2 мМ лецитина, 81,65 мМ гидроксида натрия, 125,5 мМ хлорида натрия, 0,8 мМ олеата натрия, 5 мМ глицерил моноолеата, 55,02 мМ малеиновой кислоты) и раствор FaSSGF (рН 1,6, 1 л раствор, содержащий 80 мкМ таурохолата натрия, 20 мкМ лецитина, 0,1 г пепсина и 34,2 мМ хлорида натрия).
[00226] Соединение аккуратно взвешивали и добавляли полученный фосфатный буфер (рН 7,4), раствор FeSSIF (рН 5,8) и раствор FaSSGF (рН 1,6) для получения раствора с концентрацией 4 мг/мл, который встряхивали при 1000 об/мин в течение 1 часа, а затем инкубировали в течение носи при комнатной температуре. После инкубации раствор центрифугировали при 12000 об/мин в течение 10 мин для удаления нерастворенных частиц, а супернатант переносили в новую центрифужную пробирку. Супернатант надлежащим образом разводили, затем добавляли раствор ацетонитрила, содержащий внутренний стандарт, и проводили количественное определение, используя стандартную кривую, полученную с той же матрицей.
[00227] 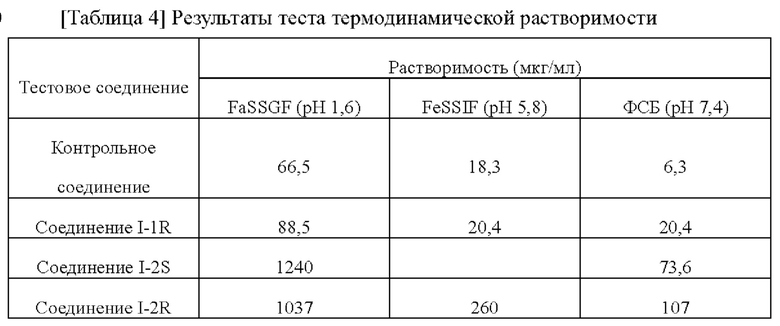
[00228] Экспериментальные результаты показывают, что растворимость контрольного соединения является относительно слабой и, таким образом, предполагается, что всасывание в желудочно-кишечном тракте будет относительно слабым, что не способствует разработке лекарственных средств для перорального введения. По сравнению с контрольным соединением термодинамическая растворимость соединений по настоящему изобретению в стимулированном желудочном соке, стимулированном кишечном соке и в нейтральных условиях существенно улучшена. Соответственно, ожидается, что кишечное всасывание в организме человека будет значительно улучшено, а воздействие при пероральном введении будет больше так, чтобы можно было уменьшить клиническую вводимую дозу и улучшить клиническое соблюдение приема.
[00229] Тестовый пример 5: Фармакокинетический тест
[00230] Для in vivo фармакокинетического теста на крысах использовали 6 самцов крыс SD, 180-240 г, не принимавших пищи в течение ночи. Трем крысам соединение вводили перорально через зонд (10 мг/кг), а кровь брали до введения и через 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 8 ч и 24 ч после введения. 3 другим крысам соединение вводили внутривенно (1 мг/кг), а кровь брали до введения и через 5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 8 ч и 24 ч после введения. Образцы крови центрифугировали при 8000 об/мин при 4°С в течение 6 мин, а плазму собирали и хранили при -20°С. В каждый момент времени брали плазму и добавляли раствор ацетонитрила, содержащий внутренний стандарт в 3-5-кратном количестве, перемешивали на вортексе и смешивали в течение 1 мин, центрифугировали при 13000 об/мин при 4°С в течение 10 мин. Супернатант собирали, добавляли и смешивали с водой в 3-кратном количестве. Надлежащее количество смеси брали для анализа методом ЖХ-МС/МС. Основные фармакокинетические параметры анализировали, используя программное обеспечение WinNonlin 7.0 с некомпартментной моделью.
[00231] Для in vivo фармакокинетического теста на мышах использовали 18 самцов мышей ICR, 20 25 г, не принимавших пищи в течение ночи. Из них 9 мышам соединение вводили перорально через зонд (10 мг/кг), брали кровь 3 мышей в каждый момент сбора крови, и кровь 9 мышей брали поочередно. Другим 9 мышам соединение вводили внутривенно (1 мг/кг), брали кровь 3 мышей в каждый момент сбора крови, и кровь 9 мышей брали поочередно. Остальные операции были такими же, как и в случае фармакокинетических тестов на крысах.
[00232] 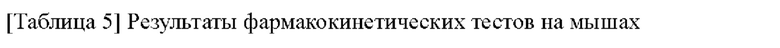
[00233] 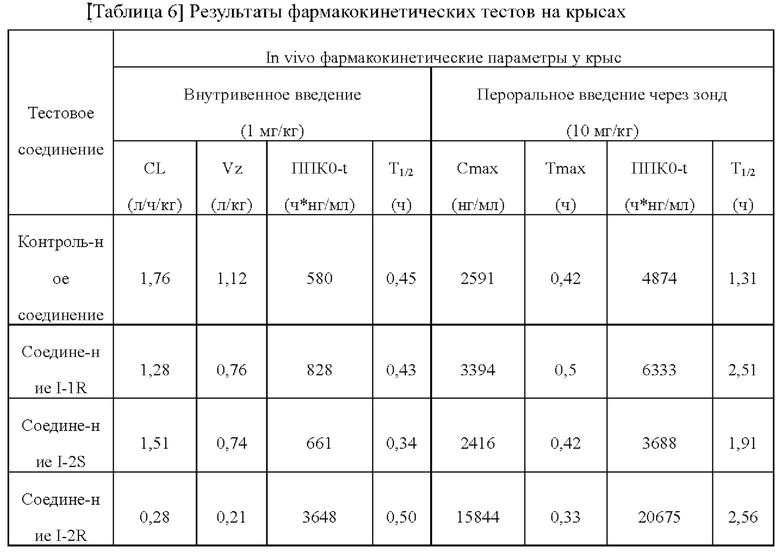
[00234] Экспериментальные результаты показывают, что по сравнению с контрольным соединением соединения по настоящему изобретению проявляли лучшие фармакокинетические свойства. В особенности у крыс, скорость выведения (CL) соединения I-2R по настоящему изобретению намного ниже, составляя около 1/4 от контрольного соединения, что говорит о том, что это соединение является относительно стабильным в организме, а его пероральные Cmax и ППК0-t могут достигать значение, в 6,1 раза и 4,2 раза больших, чем у контрольного соединения, соответственно.
[00235] Тестовый пример 6: Тест ингибирования активности фермента АТХ в плазме человека
[00236] цельную кровь брали у здоровых добровольцев и проводили антикоагуляцию гепарином. Пробирки для сбора крови центрифугировали при 3000 об/мин в течение 10 мин, отбирали плазму и хранили при -80°С для использования.
[00237] Соединение серийно разводили ДМСО в соответствии с традиционными концентрационными требованиями, а затем в 96-луночный планшет добавляли 3 мкл соединения, а в каждую лунку, содержащую 3 мкл соединения, добавляли 147 мкл ФСБ. После равномерного смешивания отбирали 50 мкл смеси и добавляли в новый 96-луночный планшет. Человеческую плазму доставали из морозильной камеры при -80°С и размораживали посредством быстрого встряхивания на водяной бане при 37°С. Брали 50 мкл человеческой плазмы и добавляли в 96-луночный планшет, содержащий 50 мкл разведенного соединения (конечная система соответствовала 1% ДМСО). Группа без соединения была назначена группой положительного контроля. 96-луночный планшет встряхивали и равномерно смешивали и инкубировали при 37°С в течение 3 ч. Также была предоставлена холостая группа, а плазму холостой группы хранили при -80°С. Холостая группа предоставлена для определения базовой концентрации эндогенной LPA.
[00238] После завершения инкубации холостую группу размораживали на льду и переносили в планшет для инкубации. Избыток ацетонитрила, содержащего внутренний стандарт LPA17:0, добавляли в планшет для инкубации для осаждения белка плазмы. После вортексного центрифугирования отбирали и разводили супернатант, а площадь пика LPA18: 2 и площадь пика внутреннего стандарта LPA17: 0 определяли, используя масс-спектрометрию ЖХ-МС/МС.
[00239] Рассчитывали соотношение площади пика LPA18: 2 и площади пика внутреннего стандарта LPA17: 0, а уровень ингибирования образования LPA18: 2 рассчитывали в соответствии со следующей формулой:
[00240] Уровень ингибирования (%) = 100-(группа соединения с разной концентрацией - холостая группа)/(группа положительного контроля - холостая группа)*100
[00241] В соответствии с уровнями ингибирования для разных концентраций соединения рассчитывали значение IC50, которое относится к ингибированию активности фермента АТХ в плазме человека.
[00242] 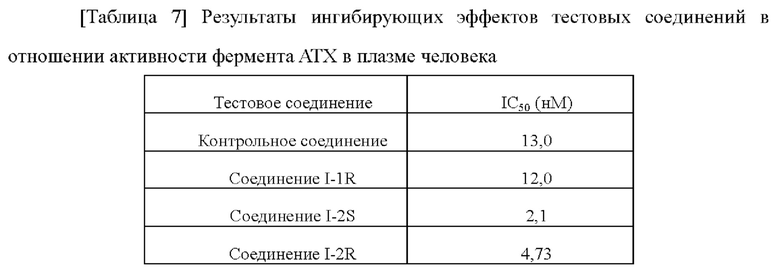
[00243] Экспериментальные данные показывают, что соединения по настоящему изобретению имеют хорошую ингибирующую активность в отношении фермента АТХ в плазме человека, могут эффективно ингибировать активность фермента АТХ и существенно лучше контрольного соединения.
[00244] Тестовый пример 7: Модель индуцированного блеомицином ИЛФ у крыс
[00245] Используя самцов крыс BN, 180-240 г, индуцировали модель идиопатического легочного фиброза (модель ИЛФ) дозой 5 Е/кг блеомицина. После установления модели животных случайным образом делили на группы, включая контрольную группу растворителя, группу GLPG-1690 (соединение галапагосской клинической фазы III), контрольную группу соединения, группу соединения I-2S и группу соединения I-2R. На вторые сутки после установления модели животным дважды в сутки проводили пероральное введение через зонд, при этом группе введения каждый раз вводили дозу 30 мг/кг, а контрольной группе растворителя вводили пустой растворитель, и продолжали введение в течение 21 суток.
[00246] Во время введения массу тела измеряли каждые трое суток. На 21 сутки введения получали альвеолярный лаваж через 2 ч после первого ведения, подсчитывали воспалительные клетки в жидкости лаважа и выявляли связанные биомаркеры в супернатанте жидкости лаважа. После лаважа фиксировали левые легкие крыс и окрашивали трихромно по Массону для оценки фиброзной патологии. Оставшиеся доли легких подвергали криоконсервации. Брали супернатант жидкости альвеолярного лаважа и свежезамороженную ткань легких трех групп соединений и проводили выявление в отношении содержания белка TGF-βl и количества общего белка, используя ELISA, и рассчитывали количество TGF-β1 на миллиграмм общего белка.
[00247] Экспериментальные результаты показывают, что снижение массы животных из групп соединения I-2S и соединения I-2R было существенно меньшим, чем в группе контрольного соединения, а соединения по настоящему изобретению имеют лучшую безопасность (результаты проиллюстрированы на Фиг. 1); содержание TGF-β1 в супернатанте жидкости альвеолярного лаважа и свежезамороженной ткани легких группы соединения I-2R существенно ниже, чем в контрольной группе растворителя, и, таким образом, соединения по настоящему изобретению имеют существенный противофиброзный эффект (результаты приведены на Фиг. 2).
[00248] В данном тексте описания со ссылкой на термины «вариант осуществления», «некоторые варианты осуществления», «примеры», «конкретные примеры» или «некоторые примеры» и т.д. означают конкретные признаки, структуры, материалы или характеристики, описанные в сочетании с вариантом осуществления или примером, включены по меньшей мере в один вариант осуществления или пример по настоящему изобретению. В данном тексте вышеприведенные термины являются иллюстративными и необязательно относятся к одному и тому же варианту осуществления или примеру. Кроме того, описанные конкретные признаки, структуры, материалы или характеристики можно комбинировать подходящим образом в любом одном или более вариантах осуществления или примерах. Дополнительно, специалисты в данной области техники могут комбинировать разные варианты осуществления или примеры и признаки разных вариантов осуществления или примеров без противоречий между ними.
[00249] Хотя варианты осуществления настоящего изобретения проиллюстрированы и описаны выше, можно понять, что вышеупомянутые варианты осуществления являются иллюстративными, и их не следует воспринимать как ограничения настоящего изобретения. Специалисты в данной области техники могут делать изменения, модификации, замены и вариации на основании вышеупомянутых вариантов осуществления в рамках объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2790017C1 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
| БИЦИКЛИЧЕСКИЕ КЕТОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2797922C2 |
| СП0СОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ДИГИДРОИНДЕНАМИДА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИИ, СОДЕРЖАЩИЕ ДАННЫЕ СОЕДИНЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2009 |
|
RU2528408C2 |
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛ-2,7-ДИАЗАСПИРО[3.5]НОНАНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ИЛИ ОБРАТНЫХ АГОНИСТОВ ГРЕЛИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2524341C2 |
| НОВЫЕ АКТИВАТОРЫ РАСТВОРИМОЙ ГУАНИЛАТЦИКЛАЗЫ И ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗЫ С ДВОЙНЫМ МЕХАНИЗМОМ ДЕЙСТВИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2758373C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
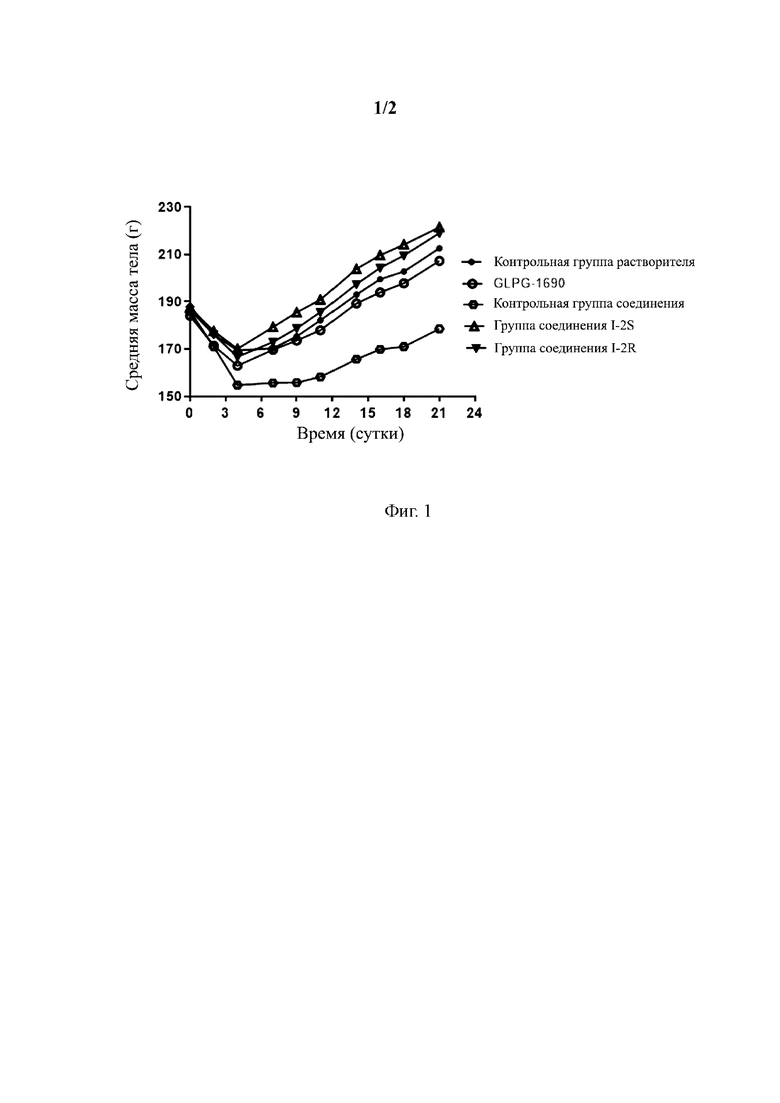
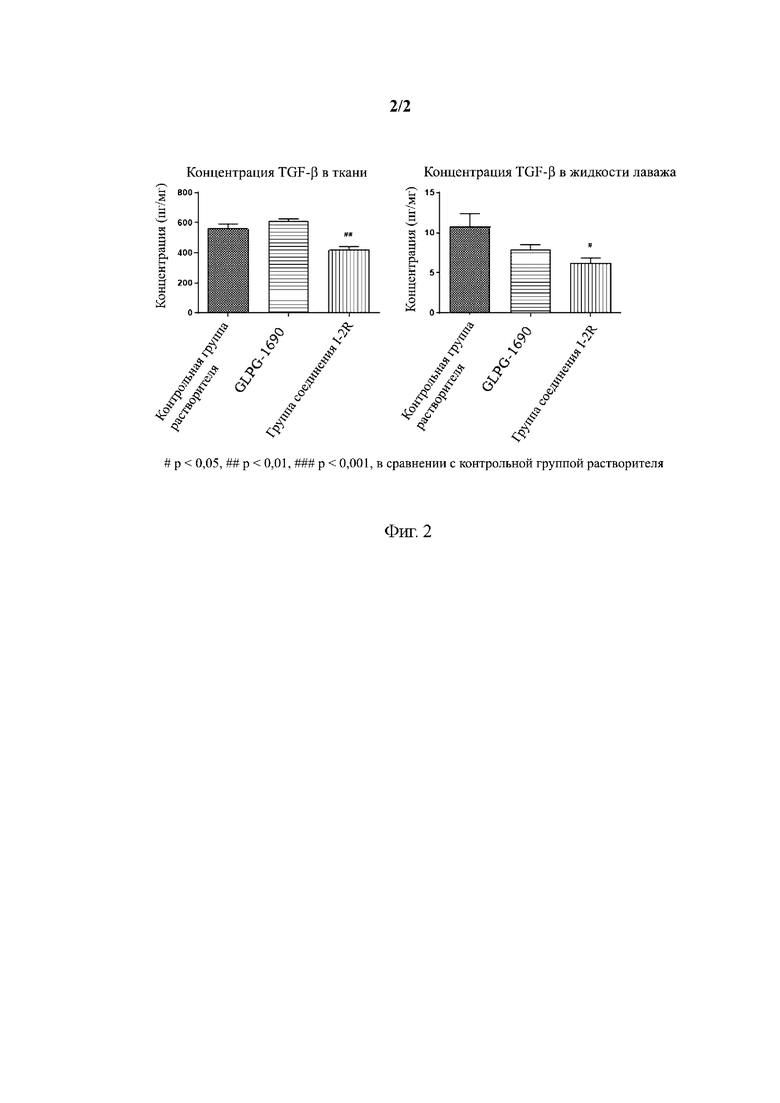
Изобретение относится к соединению или к его таутомеру или стереоизомеру, представленному формулой I, где R1 и R2 каждый независимо выбран из -Н или -СН3 при условии, что R1 и R2 одновременно не представляют собой -Н; или R1 и R2 одновременно не представляют собой -СН3. Технический результат – получено новое соединение, которое может найти применение в медицине в производстве лекарственного средства для лечения или предупреждения связанных с АТХ заболеваний. 2 н. и 9 з.п. ф-лы, 2 ил., 7 табл., 8 пр.
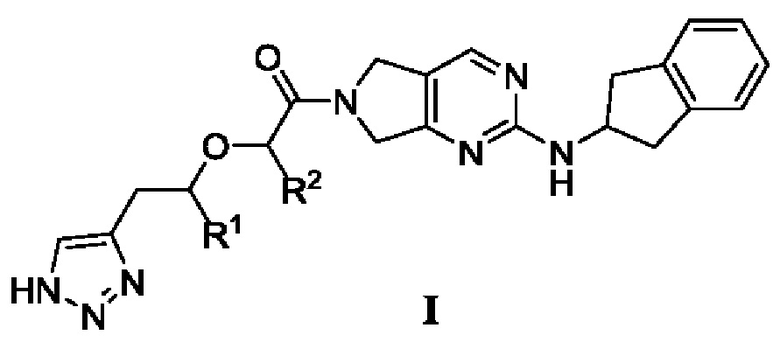
1. Соединение, являющееся соединением, представленным формулой I, или являющееся таутомером или стереоизомером соединения, представленного формулой I
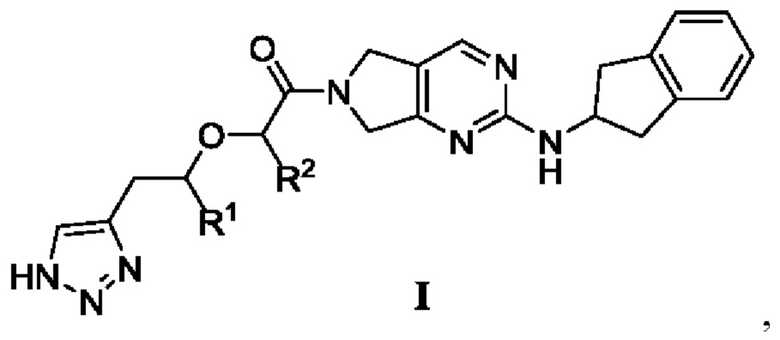
где
R1 и R2 каждый независимо выбран из -Н или -СН3 при условии, что:
R1 и R2 одновременно не представляют собой -Н или
R1 и R2 одновременно не представляют собой -СН3.
2. Соединение по п. 1, где R1 представляет собой -Н и R2 представляет собой -СН3.
3. Соединение по п. 1, где R1 представляет собой -СН3 и R2 представляет собой -Н.
4. Соединение по п. 1, отличающееся тем, что соединение представляет собой одно из следующих соединений или таутомер или стереоизомер одного из следующих соединений
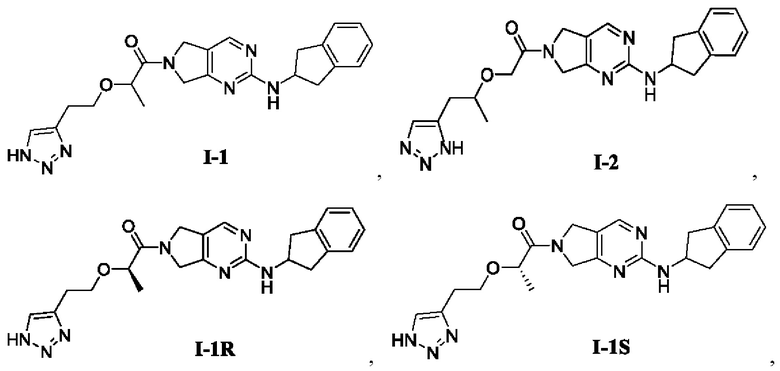
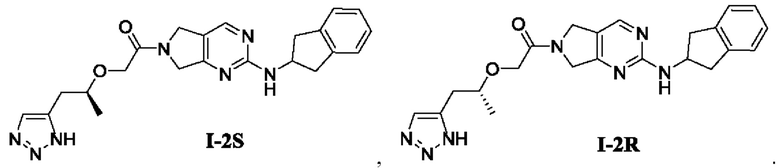
5. Применение соединения по любому из пп. 1-4 в производстве лекарственного средства для лечения или предупреждения связанных с АТХ заболеваний.
6. Применение по п. 5, отличающееся тем, что связанные с АТХ заболевания включают по меньшей мере одно, выбранное из рака, метаболического заболевания, заболевания почек, заболевания печени, фиброзирующего заболевания, интерстициального заболевания легких, пролиферативного заболевания, воспалительного заболевания, боли, аутоиммунного заболевания, респираторного заболевания, сердечно-сосудистого заболевания, нейродегенеративных заболеваний, дерматологического нарушения и/или связанного с аномальным ангиогенезом заболевания.
7. Применение по п. 5, отличающееся тем, что связанные с АТХ заболевания включают по меньшей мере одно, выбранное из интерстицильного заболевания легких, легочного фиброза, печеночного фиброза и почечного фиброза.
8. Применение по п. 5, отличающееся тем, что связанные с АТХ заболевания включают идиопатический легочный фиброз.
9. Применение по п. 5, отличающееся тем, что связанные с АТХ заболевания включают диабет II типа и неалкогольный стеатогепатит.
10. Применение по п. 5, отличающееся тем, что связанные с АТХ заболевания включают невропатическую боль и воспалительную боль.
11. Применение по п. 5, отличающееся тем, что связанные с АТХ заболевания включают связанную с остеоартритом боль.
| Способ сушки свекловичной стружки | 1931 |
|
SU25106A1 |
| JONES S.B | |||
| ET AL | |||
| ACS Medicinal Chemistry Letters, vol | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Радиатор | 1925 |
|
SU857A1 |
| WO 2014143583 A1, 18.09.2014 | |||
| Способ приготовления бумаги для мульчирования посевов | 1930 |
|
SU28364A1 |

