CСЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[001] Согласно настоящей заявке испрашивается приоритет в соответствии с заявкой на патент Китая № 201910841159.9, озаглавленной «ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ» («PYRIMIDINE COMPOUND AND PREPARATION METHOD thereof»), и заявкой на патент Китая № 201910846545.7, озаглавленной «ПРОИЗВОДНЫЕ ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ» («Pyrimidine Derivatives and Use thereof»), обе из которых поданы 6 сентября 2019 г. в Китайское национальное управление по интеллектуальной собственности, полное раскрытие каждой из которых включено в настоящий документ посредством их ссылок.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
[002] Настоящее изобретение относится к области медицинской химии. В частности, настоящее изобретение относится к пиримидиновым соединениям и, более конкретно, настоящее изобретение относится к пиримидиновым соединениям, способам их получения и их применению в получении лекарственных средств.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[003] Аутотаксин (сокращенно АТХ) представляет собой секретируемый гликопротеин, обладающий фосфодиэстеразной (PDE) активностью, и является представителем семейства внеклеточных пирофосфатаз/фосфодиэстераз (ENPP). Таким образом, аутотаксин также называют ENPP2. ATX также обладает активностью лизофосфолипазы D (LysoPLD) и может гидролизовать лизофосфатидилхолин (LPC) в лизофосфатидную кислоту (LPA), обладающую биологической активностью. LPA является внутриклеточным липидным медиатором, влияющим на многие биологические и биохимические процессы.
[004] Исследования показали, что при патологических состояниях уровень LPA может быть снижен путем ингибирования ATX, тем самым обеспечивая терапевтические преимущества для неудовлетворенных клинических потребностей. Связанные с этим заболевания включают в себя рак, хоминг лимфоцитов, хроническое воспаление, невропатическую боль, фиброз, тромбоз, холестатический зуд или фиброзные заболевания, которые индуцируются, опосредуются и/или распространяются посредством повышенного уровня LPA и/или активации ATX.
[005] Положительная регуляция сигнального пути ATX-LPA может наблюдаться при различных воспалительных состояниях. Например, провоспалительные эффекты LPA включают в себя дегрануляцию тучных клеток, сокращение гладкомышечных клеток и высвобождение цитокинов из дендритных клеток. В качестве проявления общей роли LPA в воспалении положительная регуляция сигнального пути ATX-LPA наблюдалась на модели воздушного мешка с каррагинаном у мышей, которая используется для разработки противовоспалительных препаратов, включая в себя ингибиторы циклооксигеназы для лечения артрита (Hidenobu Kanda, Rebecca Newton, Russell Klein et al., Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs[J] Nature Immunology. 2008, 9(4):415-423.). Кроме того, снижение содержания LPA в плазме и в воздушном мешке наблюдалось в модели воздушного мешка с каррагинаном у крыс с использованием ингибитора АТХ, что подтверждает роль АТХ как основного источника LPA при воспалении. В качестве другой общей роли LPA в воспалительных заболеваниях был подтвержден «синергетический эффект» между LPA и хемокинами миграции лимфоцитов. Высокий уровень экспрессии ATX может быть обнаружен в местах хронического воспаления. Подтверждено, что хоминг Т-клеток в лимфатические ткани ингибируется внутривенным введением инактивированного АТХ, что может быть достигнуто за счет конкуренции с эндогенным АТХ и оказания доминирующего отрицательного эффекта. В некоторых случаях АТХ способствует проникновению лимфоцитов в лимфоидные органы. Следовательно, ингибиторы АТХ могут блокировать миграцию лимфоцитов во вторичные лимфоидные органы и оказывать положительные эффекты при аутоиммунных заболеваниях.
[006] Было подтверждено, что при ревматоидном артрите экспрессия АТХ увеличивается в синовиальных фибробластах у пациентов с ревматоидным артритом (RA), а устранение экспрессии АТХ в интерстициальных клетках (включая в себя синовиальные фибробласты) приводит к ослаблению симптомов в модели ревматоидного артрита на мышах. Таким образом, роль аутотаксина при ревматоидном артрите полностью установлена.
[007] LPA также может положительно регулировать белки, связанные с болью, через один из своих гомологичных рецепторов, LPA1. Целенаправленное ингибирование ATX-опосредованного биосинтеза LPA может служить механизмом предотвращения невропатической боли, вызванной повреждением нервов, такой как боль, связанная с остеоартритом. Было замечено, что ингибиторы аутотаксина снижают содержание LPA и PGE2, а также облегчают воспалительную боль. В ходе исследований также было показано, что целенаправленное ингибирование опосредованного АТХ биосинтеза LPA может быть новым механизмом предотвращения невропатической боли, вызванной повреждением нервов.
[008] После стихания воспаления и устранения повреждения ткани ткань, как правило, восстанавливается до своего исходного состояния. Когда в этом нет необходимости, чрезмерное и неконтролируемое восстановление тканей может привести к состоянию, общепринято называемому фиброзом. Фиброз характеризуется избыточным отложением компонентов внеклеточного матрикса и чрезмерным ростом фибробластов. Фиброз может возникать во всех тканях, но особенно часто он встречается в органах, которые часто подвергаются химическим и биологическим повреждениям, включая в себя легкие, кожу, пищеварительный тракт, почки и печень. Фиброз часто серьезно вредит нормальным функциям органов.
[009] В некоторых случаях LPA стимулирует пролиферацию звездчатых клеток печени, ингибируя синтез ДНК в гепатоцитах. Уровень LPA и активность АТХ в сыворотке повышены у пациентов с хроническим гепатитом С. В крови кроликов с различными поражениями печени концентрация LPA в плазме и активность АТХ в сыворотке относительно выше при фиброзе печени, индуцированном четыреххлористым углеродом. Концентрация LPA в плазме и активность АТХ в сыворотке увеличиваются с увеличением тяжести различных повреждений печени.
[0010] Легочный фиброз представляет собой конечную стадию большой группы заболеваний легких, характеризующихся пролиферацией фибробластов и накоплением большого количества внеклеточного матрикса, сопровождающихся воспалительным повреждением и разрушением структуры ткани, т.е. структурной аномалией (образование рубцов), что вызвано аномальной репарацией после повреждения нормальных тканей альвеол. Когда повреждение легких связано с различными причинами, интерстициальные ткани выделяют коллаген для восстановления. Если они восстанавливаются чрезмерно, т.е. чрезмерно пролиферируют фибробласты и накапливается большое количество внеклеточного матрикса, разовьется легочный фиброз.
[0011] Сигнал LPA способствует фиброзу эпителиальных клеток, эндотелиальных клеток и фибробластов, в частности, через рецептор LPA1: генетическая делеция этого рецептора уменьшает апоптоз эпителиальных клеток, пропотевание жидкости через сосуды и накопление фибробластов в модели легочного фиброза.
[0012] Идиопатический легочный фиброз (IPF) представляет собой хроническую, прогрессирующую и фиброзную интерстициальную пневмонию неизвестной этиологии, характеризующуюся диффузным альвеолитом и альвеолярными структурными нарушениями и, как правило, проявляющуюся как обычная интерстициальная пневмония при визуализации и патологической гистологии. По мере прогрессирования заболевания возникает легочный фиброз, легочная ткань пациента утолщается и твердеет, в результате чего образуются стойкие рубцы или образование сотообразного легкого пациента, также ярко именуемого «сотовым легким» или «легкое-люффа». Это хроническое прогрессирующее заболевание вызывает необратимое и постоянное снижение функции легких. 50% пациентов могут иметь среднюю продолжительность жизни всего 2,8 года с момента постановки диагноза. Таким образом, идиопатический легочный фиброз также называют «опухолеподобным заболеванием». В настоящее время существующие медикаментозные методы лечения характеризуются такими проблемами, как множество побочных реакций, плохие терапевтические эффекты, а немедикаментозные методы лечения в основном включают в себя трансплантацию легких, но трансплантация органов является дорогостоящей и ограниченной по ресурсам, а также сопровождается определенными клиническими рисками.
[0013] Имеются данные, демонстрирующие, что пролиферация и сокращение фибробластов и секреция внеклеточного матрикса, стимулируемые LPA, способствуют фиброзной пролиферации при других заболеваниях дыхательных путей, таких как перибронхиальный фиброз, присутствующий при хроническом бронхите и интерстициальном заболевании легких, а также при тяжелой бронхиальной астме. LPA играет роль в фиброзном интерстициальном заболевании легких и облитерирующем бронхиолите, при которых увеличивается количество как коллагена, так и миофибробластов. Исследования, относящиеся к идиопатическому легочному фиброзу (IPF), показали, что уровень LPA в жидкости бронхоальвеолярного лаважа пациентов повышен. Дальнейшие исследования нокаута LPA1 и ингибиторов показали, что LPA играет ключевую роль в процессе легочного фиброза, что дополняется исследованием с использованием клеточно-специфических мышей с нокаутом, лишенных ATX в бронхиальных эпителиальных клетках и макрофагах. Было показано, что эти мыши были менее чувствительны к модели легочного фиброза. Роль LPA при других фиброзных заболеваниях (почечных и кожных) основана на аналогичных наблюдениях. Роль LPA в ремоделировании легких связана с эффектами LPA как на фибробласты легких (через LPA1), так и на эпителиальные клетки (через LPA2). Было показано, что LPA2 играет ключевую роль в активации TGFβ в эпителиальных клетках в случае фиброзных нарушений. Роли LPA в ремоделировании и фиброзе связаны с COPD, IPF и бронхиальной астмой, а ремоделирование легких в долгосрочной перспективе будет ограничивать функцию легких. Наконец, что касается заболеваний легких, ATX является одним из трех основных локусов количественных признаков, которые, по-видимому, связаны с различиями в функции легких у мышей.
[0014] В ходе исследования установлено, что концентрация LPA увеличивается в плазме и асците у пациентов с раком яичников на ранних и поздних стадиях. Повышенный уровень LPA и изменения в экспрессии и ответе рецепторов LPA могут быть одной из причин возникновения, прогрессирования или исхода рака яичников. LPA также связан с раком предстательной железы, раком молочной железы, меланомой, раком головы и шеи, раком кишечника, раком головного мозга и раком щитовидной железы. LPA участвует в пролиферации и инвазии опухолевых клеток в соседние ткани, что приводит к метастазированию. Эти биологические и патобиологические процессы инициируются активацией рецепторов, связанных с G-белком, с помощью LPA. Уровень LPA можно снизить путем ингибирования ферментов, участвующих в биосинтезе LPA, таких как ATX, для лечения пациентов с опухолями.
[0015] В процессе ангиогенеза АТХ и другие ангиогенные факторы вместе приводят к ангиогенезу. Опухоль может питаться за счет ангиогенеза во время роста опухоли. Соответственно, важной отправной точкой для лечения рака и опухолей является ингибирование ангиогенеза. В патентной заявке WO 2014202458A1 перечисляются эффекты передачи сигналов ATX-LPA при различных патофизиологических состояниях, таких как пролиферативные заболевания, невропатическая боль, воспаление, аутоиммунные заболевания, фиброз, отслеживание лимфоцитов в лимфатических узлах, ожирение, диабет или образование кровеносных сосудов у эмбрионов.
[0016] В настоящее время достигнуты определенные успехи в лечении рака, фиброзных заболеваний, пролиферативных заболеваний, воспалительных заболеваний, аутоиммунных заболеваний, респираторных заболеваний, сердечно-сосудистых заболеваний, нейродегенеративных заболеваний, дерматологических нарушений и/или заболеваний, связанных с аномальным ангиогенезом, но их все равно недостаточно. В настоящее время продаваемые терапевтические лекарственные средства для лечения IPF включают в себя пирфенидон и нинтеданиб. Пирфенидон может вызывать поражение печени (например, печеночную недостаточность, желтуху), реакции гиперчувствительности (такие как отек лица, отек гортани, одышка, свистящее дыхание и т.д.) и тяжелые желудочно-кишечные реакции, а анализы фотогенотоксичности показали, что он может вызывать структурную аномалию хромосом и может вызвать рак кожи при воздействии света. Нинтеданиб вызывает побочные реакции, такие как диарея, тошнота и боль в области живота, частота желудочно-кишечных реакций может достигать 50%, а их распространенные побочные реакции включают в себя потерю массы тела, потерю аппетита, повреждение печени, кровотечение и т.д. Среди пациентов, принимавших пирфенидон и нинтеданиб, вероятность прекращения лечения из-за серьезных нежелательных явлений составила 20,9% и 26,3% соответственно (Toby M Maher, et al. Rationale, design and objectives of two phase III, randomised, placebocontrolled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2)[J]. BMJ Open Respiratory Research. 2019, 21;6(1).). Качество жизни пациентов с IPF будет серьезно снижено, и ни пирфенидон, ни нинтеданиб не могут улучшить качество жизни пациентов в ходе клинических испытаний. Хотя оба этих лекарственных средства могут улучшить общие результаты, они могут только отсрочить течение болезни, но не могут вызывать обратное развитие легочного фиброза. В связи с этим пациентам с тяжелым специфическим легочным фиброзом он может не принести пользы. GLPG-1690, как один из препаратов для лечения IPF, который в настоящее время находится в стадии разработки и быстро прогрессирует, проявляет тенденцию к обращению вспять течения заболевания, но имеет проблемы низкой ферментативной активности, большой дозировкой клинического препарата и несоблюдения пациентом режима приема лекарственного средства. Поэтому существующие в настоящее время методы лечения все еще неудовлетворительны. По-прежнему существует большое количество пациентов, которые нуждаются в новых методах лечения с более высокой активностью и большей эффективностью, чтобы в большей степени замедлить течение болезни или даже обратить ее вспять, улучшить соблюдение пациентом режима приема лекарственных средств и позволить большему количеству пациентов с идиопатическим легочным фиброзом извлечь из этого пользу.
[0017] Принимая во внимание вышеизложенное, согласно настоящему раскрытию на основе предшествующего уровня техники разработано соединение, представленное формулой (I), для получения ингибитора АТХ с новой структурой, лучшими фармакокинетическими свойствами, лучшей фармакодинамикой и сильной лекарственной способностью, для эффективного лечения заболеваний и состояний, связанных с ATX, включая в себя без ограничения рак, метаболические заболевания, заболевания почек, заболевания печени, фиброзные заболевания, интерстициальные заболевания легких, легочный фиброз, фиброз печени, пролиферативные заболевания, воспалительные заболевания, боль, связанную с остеоартритом боль, аутоиммунные заболевания, респираторные заболевания, сердечно-сосудистые заболевания, нейродегенеративные заболевания, дерматологические нарушения и/или заболевания, связанные с аномальным ангиогенезом.
КРАТКОЕ РАСКРЫТИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0018] Настоящее раскрытие направлено на решение по меньшей мере одной из вышеуказанных технических проблем или по меньшей мере на предоставление применимого бизнес-варианта.
[0019] Согласно одному аспекту настоящего раскрытия настоящее раскрытие относится к соединению, которое представляет собой соединение, представленное формулой (I), или таутомер, стереоизомер, гидрат, сольват, фармацевтически приемлемую соль или пролекарственное средство соединения, представленного формулой (I):
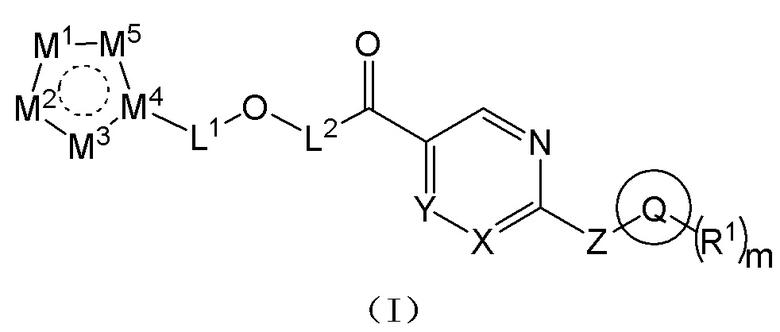
где Q выбран из C3-C10 циклоалкила, трех-десятичленной гетероциклической группы, C6-C10 арила или пяти-десятичленного гетероарила; причем C3-C10 циклоалкил, трех-десятичленная гетероциклическая группа, C6-C10 арил и пяти-десятичленный гетероарил необязательно замещены m группами R1, m представляет собой целое число, выбранное из 0-9;
каждая из m групп R1 независимо выбрана из водорода, галогена, -CN, -OH, -SH, -NO2, C1-C10 алкила, C3-C10 циклоалкила или C1-C10 алкокси;
X и Y независимо выбраны из -N= или -C(R2)=;
Z выбран из -O-, -S- или -N(R3)-;
L1 выбран из одинарной связи или 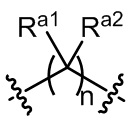 ;
;
L2 выбран из одинарной связи или 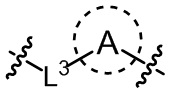 ;
;
L3 выбран из одинарной связи или 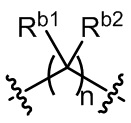 ;
;
Ra1, Ra2, Rb1 и Rb2 каждый независимо выбран из водорода, галогена, циано, -OH или C1-C3 алкила;
 выбран из C3-C8 циклоалкила, C4-C8 гетероциклоалкила или семи-восьмичленной N-спироциклической группы, и C3-C8 циклоалкил, C4-C8 гетероциклоалкил и семи-восьмичленная N-спироциклическая группа необязательно замещены по меньшей мере одним Rc;
выбран из C3-C8 циклоалкила, C4-C8 гетероциклоалкила или семи-восьмичленной N-спироциклической группы, и C3-C8 циклоалкил, C4-C8 гетероциклоалкил и семи-восьмичленная N-спироциклическая группа необязательно замещены по меньшей мере одним Rc;
каждый из по меньшей мере одного Rc независимо выбран из -H, галогена, -OH, -CN или C1-C3 алкила;
n представляет собой целое число, выбранное из 0-3;
M1, M2, M3, M4 и M5 каждый независимо выбран из -N=, -N(R4)- или -C(R5)=, по меньшей мере один из M1, M2, M3, M4 или M5 представляет собой -N= или -N(R4)-, и по меньшей мере один из M1, M2, M3, M4 или M5 представляет собой -C(R5)=;
R2 и R5 каждый независимо выбран из водорода, галогена, -CN, -OH, -SH или C1-C3 алкила;
R3 и R4 каждый независимо выбран из водорода или C1-C3 алкила; и
соединение не включает в себя ни одно из следующих соединений или их энантиомеров или стереоизомеров:
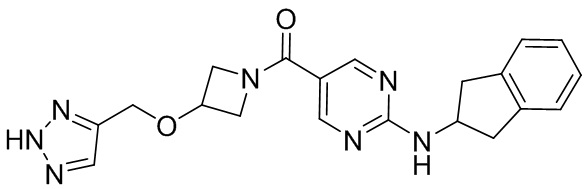
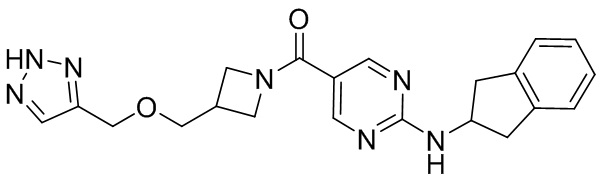
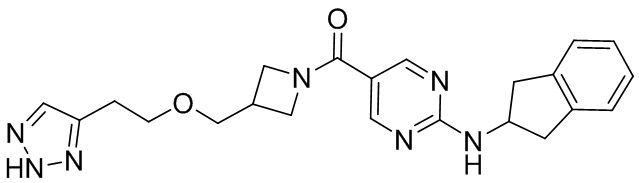
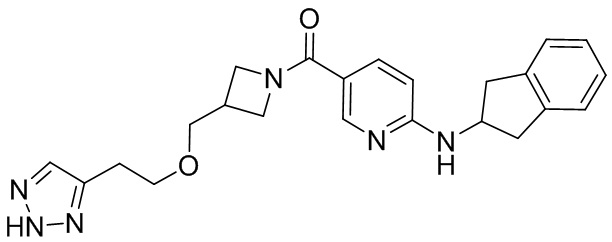
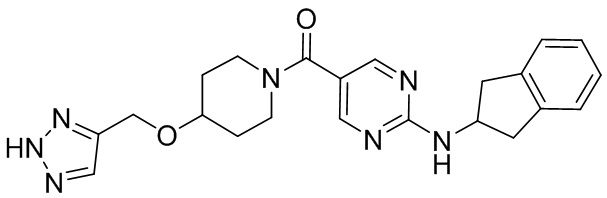
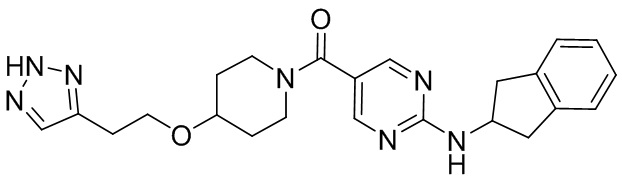
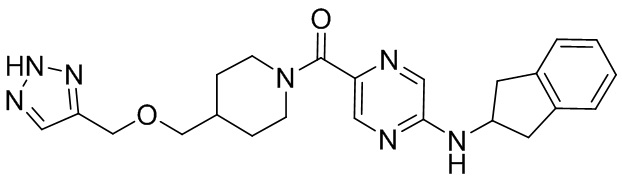
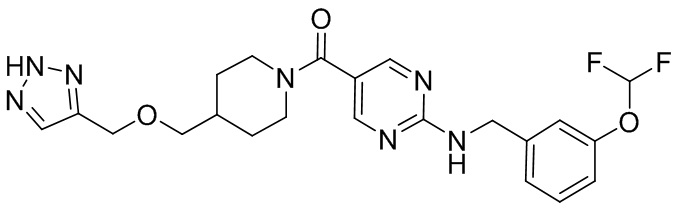
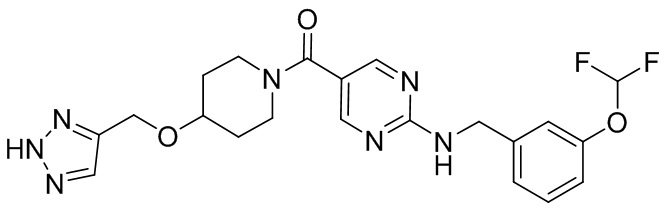
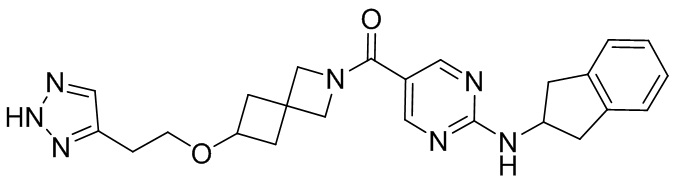
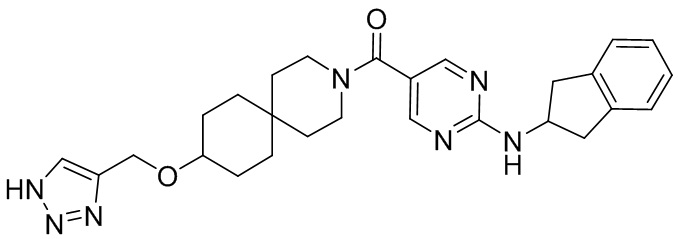
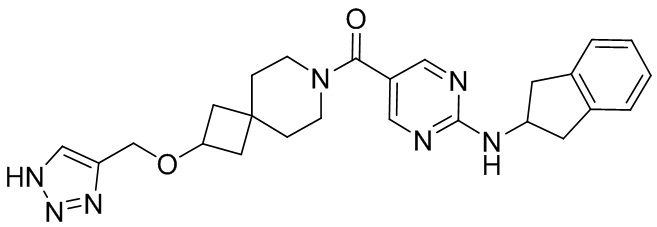
 .
.
[0020] Авторы настоящего изобретения синтезировали новое соединение, представленное формулой (I), и обнаружили, что это соединение или его фармацевтически приемлемая соль, таутомер, стереоизомер, гидрат, сольват или пролекарственное средство оказывает значительный ингибирующий эффект на фермент АТХ и проявляет превосходную стабильность метаболизма в печени и метаболизируется медленнее в организме человека и характеризуется более высокой степенью воздействия.
[0021] Согласно иллюстративным вариантам осуществления настоящего раскрытия в соединении, представленном формулой (I), Q выбран из циклопропила, циклобутила, циклопентила, циклогексила, азетидинила, пирролидинила, тетрагидрофуранила, пиперидинила, пиперазинила, морфолинила, фенила, инденила, нафтила, пирролила, пиразолила, имидазолила, триазолила, тетразолила, фурила, тиенила, тиазолила, оксазолила, пиридинила, пиримидинила, пиразинила, пиридазинила, бензимидазолила, индолила или хинолинила; m равно 0, 1, 2 или 3; и M1, M2 и M3 все выбраны из -N= или -N(R4)-, и каждый из M4 и M5 представляет собой -C(R5)=.
[0022] Согласно вариантам осуществления настоящего раскрытия каждая из m групп R1 независимо выбрана из водорода, галогена или C1-C3 алкила, и предпочтительно R1 выбран из H, F, Cl, метила или этила; и предпочтительно R1 выбран из H, F, Cl, метила или этила;
необязательно, когда Q представляет собой инденил, инденил необязательно не замещен R1 или замещен одной или двумя группами R1;
необязательно, когда X представляет собой -N=, Y представляет собой -C(R2)=; или когда X представляет собой -C(R2)=, Y представляет собой -N=;
необязательно, Z выбран из -O-, -S- или -N(R3)-; когда Z представляет собой -N(R3)-, R3 представляет собой водород или C1-C3 алкил;
необязательно, когда L1 представляет собой , n равно 1 или 2;
необязательно, когда L1 представляет собой , Ra1 и Ra2 каждый независимо выбран из водорода, F, Cl, -CN, метила или этила;
необязательно, когда L1 представляет собой , Ra1 и Ra2 каждый независимо выбран из водорода, F, Cl, -CN, метила или этила, и по меньшей мере один Ra1 представляет собой водород;
необязательно, когда L3 представляет собой , n равно 1 или 2;
необязательно, когда L3 представляет собой , Rb1 и Rb2 каждый независимо выбран из водорода, -F, -Cl, -CN, метила или этила;
необязательно, когда L3 представляет собой , Rb1 и Rb2 каждый независимо выбран из водорода, -F, -Cl, -CN, метила или этила, и по меньшей мере один Rb1 представляет собой водород;
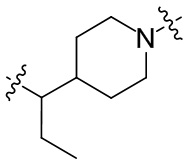
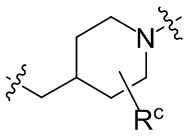
необязательно, когда указанный L2 представляет собой , выбран из следующих групп, незамещенных или необязательно замещенных по меньшей мере одним Rc: пиперидинил или семи-восьмичленная N-спироциклическая группа;
необязательно, когда указанный L2 представляет собой , представляет собой пиперидинил, незамещенный или необязательно замещенный по меньшей мере одним Rc, указанный L3 и N в пиперидиниле независимо находятся в орто-, мета- или пара-положении по отношению друг к другу;
необязательно, когда указанный L2 представляет собой , представляет собой пиперидинил, незамещенный или необязательно замещенный по меньшей мере одним Rc, по меньшей мере один Rc и N в пиперидиниле независимо находятся в орто- или метаположении по отношению друг к другу;
необязательно, когда указанный L2 представляет собой , представляет собой пиперидинил, незамещенный или необязательно замещенный по меньшей мере одним Rc, по меньшей мере один Rc выбран из -H, -F, -Cl, метила или этила;
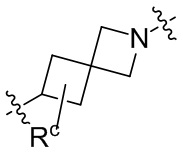
необязательно, когда указанный L2 представляет собой , и L3 представляет собой одинарную связь, выбран из семи-восьмичленных N-спироциклических групп, незамещенных или необязательно замещенных по меньшей мере одним Rc;
необязательно, когда указанный выбран из семи-восьмичленных N-спироциклических групп, незамещенных или необязательно замещенных по меньшей мере одним Rc, N-спироциклическая группа выбрана из  или
или  ; и
; и
необязательно M1, M2 и M3 каждый выбран из -N= или -N(R4)-, M4 и M5 оба представляют собой -C(R5)=, R4 представляет собой водород, и R5 представляет собой водород.
[0023] Согласно иллюстративным вариантам осуществления настоящего раскрытия соединение, представленное формулой (I), представляет собой соединение, представленное следующей формулой (I-0):
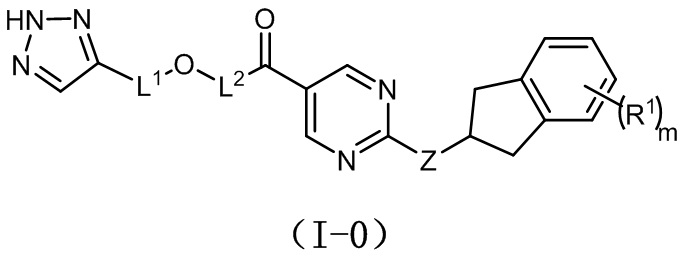
в которой R1 представляет собой -F или метил;
необязательно m равно 0, 1 или 2;
необязательно Z представляет собой -NH-;
необязательно L1 выбран из 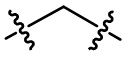 или
или 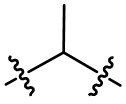 ;
;
необязательно L2 выбран из 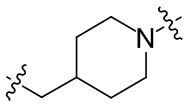 ,
, 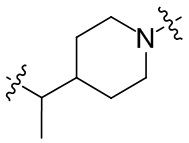 ,
,  ,
,  ,
,  или
или 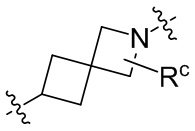 , где Rc выбран из -H, -F, -Cl, метила или этила.
, где Rc выбран из -H, -F, -Cl, метила или этила.
[0024] Согласно другому аспекту настоящее раскрытие относится к соединению, представленному формулой (I-1), или таутомеру, стереоизомеру, гидрату, сольвату, фармацевтически приемлемой соли или пролекарственному средству соединения, представленного формулой (I-1):
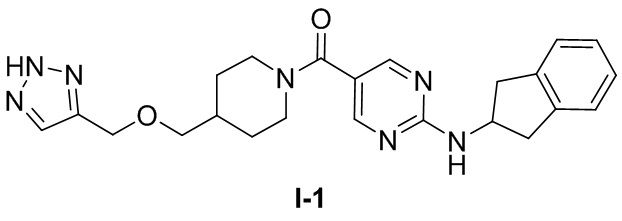 .
.
[0025] Согласно другому аспекту настоящее раскрытие относится к соединению, представленному формулой (I-9), или таутомеру, стереоизомеру, гидрату, сольвату, фармацевтически приемлемой соли или пролекарственному средству соединения, представленного формулой (I-9):
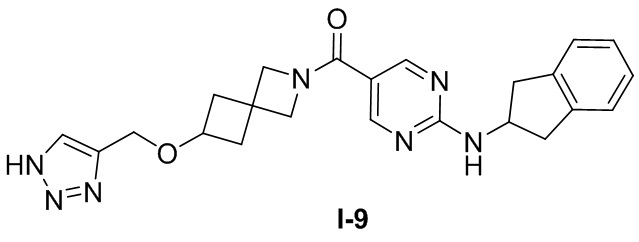 .
.
[0026] Согласно вариантам осуществления настоящего раскрытия соединение, представленное формулой (I), представляет собой любое из следующих соединений:
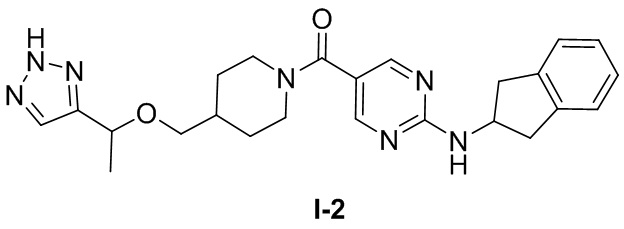
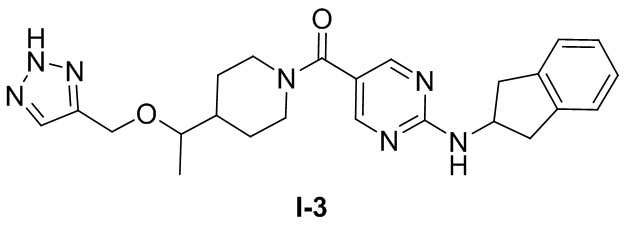
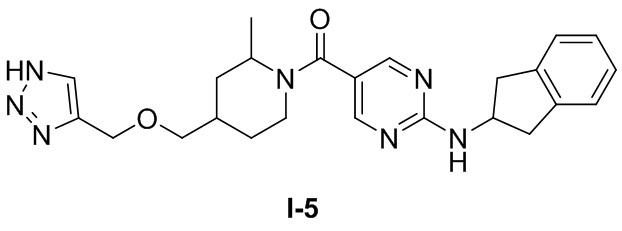
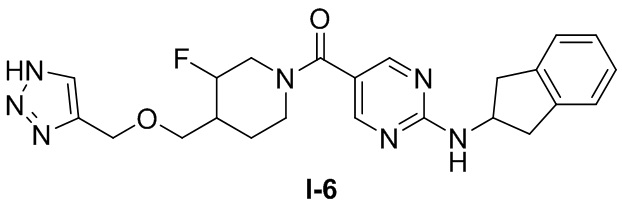
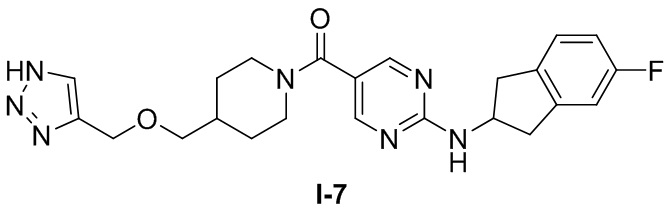
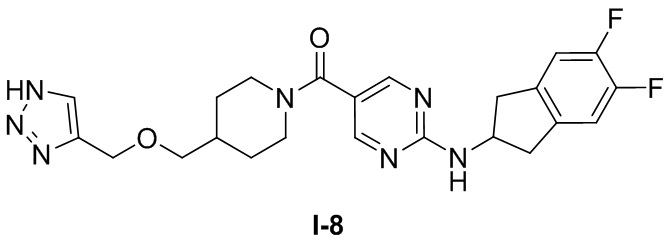
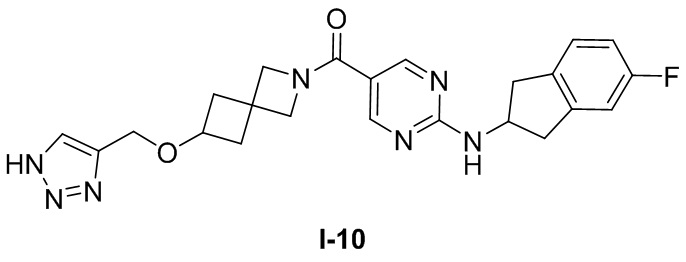 .
.
[0027] Соединения согласно настоящему раскрытию могут характеризоваться таутомерией. Настоящее раскрытие включает в себя все таутомерные формы соединений, либо они находятся в равновесном состоянии, либо одна форма является доминирующей, настоящее раскрытие включает в себя соответствующие таутомерные формы.
[0028] Согласно другому аспекту настоящего раскрытия настоящее раскрытие относится к фармацевтической композиции, которая включает в себя эффективную дозу соединения, представленного формулой (I).
[0029] Согласно другому аспекту настоящего раскрытия настоящее раскрытие относится к применению соединения, представленного формулой (I), или его таутомера, стереоизомера, гидрата, сольвата, фармацевтически приемлемой соли или пролекарственного средства в производстве лекарственного средства для лечения заболевания, связанного с АТХ.
[0030] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, выбрано из рака, метаболических заболеваний, заболеваний почек, заболеваний печени, фиброзных заболеваний, интерстициальных заболеваний легких, пролиферативных заболеваний, воспалительных заболеваний, боли, аутоиммунных заболеваний, респираторных заболеваний, сердечно-сосудистых заболеваний, нейродегенеративных заболеваний, дерматологических нарушений и/или заболеваний, связанных с аномальным ангиогенезом.
[0031] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, выбрано из интерстициальных заболеваний легких, легочного фиброза, фиброза печени или фиброза почек, и предпочтительно заболевание, связанное с АТХ, представляет собой идиопатический легочный фиброз. Согласно вариантам осуществления настоящего раскрытия соединения согласно настоящему раскрытию обладают значительными преимуществами при лечении легочного фиброза, особенно идиопатического легочного фиброза
[0032] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, представляет собой метаболическое заболевание и предпочтительно выбрано из диабета II типа или неалкогольного стеатогепатита. Согласно вариантам осуществления настоящего раскрытия соединения согласно настоящему раскрытию обладают значительными преимуществами при лечении метаболического заболевания, особенно диабета II типа и неалкогольного стеатогепатита.
[0033] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, выбрано из невропатической боли или воспалительной боли и предпочтительно заболевание, связанное с АТХ, представляет собой боль, связанную с остеоартритом. Согласно вариантам осуществления настоящего раскрытия соединения согласно настоящему раскрытию обладают значительными преимуществами при лечении боли, связанной с остеоартритом.
[0034] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, представляет собой рак. Согласно вариантам осуществления настоящего раскрытия соединения согласно настоящему раскрытию обладают значительными преимуществами при лечении рака.
[0035] Согласно другому аспекту настоящего раскрытия настоящее раскрытие относится к способу лечения заболевания, связанного с АТХ, и способ включает в себя: введение пациенту, страдающему заболеванием, связанным с АТХ, фармацевтической композиции, содержащей эффективную дозу соединения, представленного формулой (I).
[0036] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, выбрано из рака, метаболических заболеваний, заболеваний почек, заболеваний печени, фиброзных заболеваний, интерстициальных заболеваний легких, пролиферативных заболеваний, воспалительных заболеваний, боли, аутоиммунных заболеваний, респираторных заболеваний, сердечно-сосудистых заболеваний, нейродегенеративных заболеваний, дерматологических нарушений и/или заболеваний, связанных с аномальным ангиогенезом.
[0037] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, выбрано из интерстициальных заболеваний легких, легочного фиброза, фиброза печени или фиброза почек и предпочтительно заболевание, связанное с АТХ, представляет собой идиопатический легочный фиброз.
[0038] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, представляет собой метаболическое заболевание и предпочтительно выбрано из диабета II типа или неалкогольного стеатогепатита.
[0039] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, выбрано из невропатической боли или воспалительной боли и предпочтительно заболевание, связанное с АТХ, представляет собой боль, связанную с остеоартритом.
[0040] Согласно вариантам осуществления настоящего раскрытия заболевание, связанное с АТХ, представляет собой рак.
[0041] Определения и пояснения терминов
[0042] Если не указано иное, определения групп и терминов, приведенные в настоящем описании и формуле изобретения, включают в себя фактические определения, иллюстративные определения, предпочтительные определения, определения, записанные в таблицах, и определения конкретных соединений в примерах и т.д., которые могут быть произвольно объединены и интегрированы друг с другом. Определения групп и структуры соединений, которые объединяются и интегрируются, должны подпадать под объем настоящего раскрытия.
[0043] Если не указано иное, все научные и технологические термины согласно настоящему раскрытию имеют те же значения, которые обычно понимаются специалистами в области техники, к которой относятся объекты формулы изобретения. Если не указано иное, все выданные патенты, патентные заявки и опубликованные документы, цитируемые в настоящем раскрытии, полностью включены в настоящее раскрытие посредством ссылки. Если термин имеет несколько определений в настоящем раскрытии, определения в этом разделе имеют преимущественную силу.
[0044] Если не указано иное, используются общепринятые методы в технической области предшествующего уровня техники, такие как масс-спектрометрия, ЯМР, спектроскопия в ИК и УФ/видимой части спектра, а также фармакологические методы. Если не предоставлены конкретные определения, термины, используемые в настоящем описании, относящиеся к аналитической химии, химии органического синтеза, фармацевтике и медицинской химии, известны в настоящей области техники. Стандартные методы можно использовать при химическом синтезе, химическом анализе, получении, составлении и доставке лекарственных средств, а также при лечении пациентов. Например, реакцию и очистку можно проводить в соответствии с инструкциями производителя по применению набора или способами, известными в соответствующей области техники или в описании настоящего изобретения. Как правило, вышеупомянутые методики и способы могут быть реализованы в соответствии с описаниями в ряде сводных и более конкретных документов, цитируемых и обсуждаемых в настоящем раскрытии, в соответствии с общепринятыми способами, хорошо известными в соответствующей области техники. В настоящем описании группы и их заместители могут быть выбраны специалистами в настоящей области техники для получения стабильных структурных частей и соединений. Когда заместитель описывается обычной химической формулой, написанной слева направо, заместитель также включает в себя химически эквивалентный заместитель, полученный, когда структурная формула написана справа налево. Например, CH2O эквивалентен OCH2.
[0045] Когда числовой диапазон, описанный в настоящем описании и пунктах формулы настоящего изобретения, понимается как «целые числа», его следует понимать как запись двух конечных точек диапазона и всех целых чисел в диапазоне. Например, под «целым числом от 1 до 6» следует понимать запись целых чисел 1, 2, 3, 4, 5 и 6. Под «целым числом от 0 до 9» следует понимать запись целых чисел 0, 1, 2, 3, 4, 5, 6, 7, 8 и 9. Когда числовой диапазон понимается как «числовое число», следует понимать как запись двух конечных точек диапазона, соответствующих целых чисел в диапазоне и соответствующих десятичных дробей в диапазоне. Например, под «числом от 1 до 10» следует понимать не только запись соответствующих целых чисел 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10, но также по меньшей мере запись сумм каждого из целых чисел и 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8 и 0,9 соответственно.
[0046] Термин «фармацевтически приемлемый» относится к соединениям, материалам, композициям и/или лекарственным формам, подходящим для применения в контакте с тканями человека и животных без избыточной токсичности, раздражения, аллергических реакций или других проблем или осложнений в рамках надежного медицинского заключения и соответствуя разумному соотношению польза/риск.
[0047] Термин «фармацевтически приемлемая соль» относится к фармацевтически приемлемой соли нетоксичной кислоты или основания, включая в себя соли неорганических кислот и оснований, а также органических кислот и оснований. Соли, полученные из неорганических оснований, включают в себя, но не ограничиваются ими, соли металлов, образованные Al, Ca, Li, Mg, K, Na и Zn. Соли, полученные из органических оснований, включают в себя, но не ограничиваются ими, соли первичных, вторичных или третичных аминов, включая в себя органические соли, образованные природными замещенными или незамещенными аминами, циклическими аминами и основными ионообменными смолами, например, органические соли, образованные аммонием, изопропиламином, триметиламином, диэтиламином, триэтиламином, трипропиламином, диэтаноламином, этаноламином, диметилэтаноламином, 2-диметиламиноэтанолом, 2-диэтиламиноэтанолом, дициклогексиламином, кофеином, прокаином, холином, бетаином, бенетамин пенициллином, этилендиамином, глюкозамином, метилглюкамином, теобромином, триэтаноламином, трометамолом, пурином, пиперазином, пиперидином, N-этилпиперидином или полиаминовой смолой. Соли, полученные из неорганических и органических кислот, включают в себя, но не ограничиваются ими, органические соли, образованные следующим: серная кислота, фосфорная кислота, азотная кислота, бромистоводородная кислота, соляная кислота, муравьиная кислота, уксусная кислота, пропионовая кислота, бензолсульфокислота, бензойная кислота, фенилуксусная кислота, салициловая кислота, альгиновая кислота, антраниловая кислота, камфорная кислота, лимонная кислота, винилсульфоновая кислота, муравьиная кислота, фумаровая кислота, фурановая кислота, глюконовая кислота, глюкуроновая кислота, глутаминовая кислота, гликолевая кислота, изетионовая кислота, молочная кислота, малеиновая кислота, яблочная кислота, миндальная кислота, муциновая кислота, памоевая кислота, пантотеновая кислота, стеариновая кислота, янтарная кислота, сульфаниловая кислота, винная кислота, п-толуолсульфокислота, малоновая кислота, 2-гидроксипропионовая кислота, щавелевая кислота, гликолевая кислота, глюкуроновая кислота, галактуроновая кислота, лимонная кислота, лизин, аргинин, аспарагиновая кислота, коричная кислота, п-толуолсульфокислота, метансульфокислота, этансульфокислота, трифторметансульфокислота и т.д.
[0048] В дополнение к фармацевтически приемлемым солям в настоящем раскрытии могут быть приняты другие соли, и они могут служить промежуточными продуктами при очистке соединений или при получении других фармацевтически приемлемых солей или могут использоваться для идентификации, определения характеристик или очистки соединений согласно настоящему раскрытию.
[0049] Термин «стереоизомер» относится к изомеру, образованному различным пространственным расположением атомов в молекуле, включая в себя цис- и транс-изомеры, энантиомеры, диастереомеры и конформационные изомеры. Определения и правила стереохимии, используемые в настоящем раскрытие информации, как правило, следуют “S.P.Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. and Wilen, S., “Stereochemistry of Organic Compounds”, John Wiley & Sons, Inc., New York, 1994”.
[0050] В зависимости от выбранных исходных материалов и способов соединения согласно настоящему раскрытию могут существовать в форме одного из возможных изомеров или их смесей, например, в виде чистых оптических изомеров, или в виде смеси изомеров, таких как рацемические и диастереомерные смеси, в зависимости от числа асимметрических атомов углерода. При описании оптически активных соединений префиксы D и L или R и S используются для обозначения абсолютных конфигураций молекулы по отношению к одному или нескольким хиральным центрам. Префиксы D и L или (+) и (-) являются символами, используемыми для обозначения вращения плоскополяризованного света, вызванного соединением, где (-) или L указывает на то, что соединение является левовращающим, а префикс (+) или D указывает, что соединение является правовращающим. Для данной химической структуры эти стереоизомеры идентичны, за исключением того, что эти стереоизомеры являются зеркальным отображением друг друга. Конкретные стереоизомеры можно назвать энантиомерами, а смесь таких изомеров, как правило, называют энантиомерной смесью. Смесь энантиомеров в соотношении 50:50 называется рацемической смесью или рацематом, что может иметь место, когда в химической реакции или процессе отсутствует стереоселективность или стереоспецифичность. Многие геометрические изомеры олефинов, двойные связи C=N и т.д. также могут быть включены в соединения, описанные в настоящем документе, и все такие стабильные изомеры рассматриваются в настоящем раскрытии. Когда описанные в настоящем документе соединения содержат олефиновые двойные связи, если не указано иное, такие двойные связи включают в себя геометрические изомеры E и Z. Если соединение содержит дизамещенную циклоалкильную группу, заместитель циклоалкильной группы может иметь цис- или транс-конфигурацию.
[0051] Когда связь с хиральным атомом углерода в формуле согласно настоящему раскрытию изображена прямой линией, следует понимать, что две конфигурации (R) и (S) хирального атома углерода и обе образующиеся энантиомерно чистые соединения и смеси входят в объем, определяемый общей формулой. Схематическое представление рацемата или чистого энантиомерного соединения в настоящем документе взято из Maehr, J. Chem. Ed. 1985, 62: 114-120. Если не указано иное, клиновидная связь и пунктирная связь используются для представления абсолютной конфигурации стереоцентра.
[0052] Оптически активные (R)- или (S)-изомеры могут быть получены с использованием хиральных синтонов или хиральных препаратов или они могут быть разделены с использованием общепринятых способов. Соединения согласно настоящему раскрытию, содержащие асимметрично замещенные атомы углерода, могут быть разделены в оптически активной форме или в рацемической форме. Разделение рацемической смеси соединения можно проводить любым из множества способов, известных в настоящей области техники. Например, способы включают в себя фракционную перекристаллизацию с использованием хиральных разделяющих кислот, которые представляют собой оптически активные солеобразующие органические кислоты. Например, подходящими разделяющими средствами для фракционной перекристаллизации являются оптически активные кислоты, такие как винная кислота, диацетилвинная кислота, дибензоилвинная кислота, миндальная кислота, яблочная кислота, молочная кислота или различные оптически активные камфорсульфокислоты, такие как D и L формы β-камфорсульфоновой кислоты. Другие разделяющие средства, подходящие для фракционной кристаллизации, включают в себя α-метилбензиламин в стереоизомерно чистой форме (например, формы S и R или диастереомерно чистая форма), 2-фенилглицинол, норэфедрин, эфедрин, N-метилэфедрин, циклогексилэтиламин, 1,2-диаминоциклогексан и т.д. Разделение рацемической смеси можно проводить также путем элюирования колонки, заполненной оптически активным разделяющим средством (например, динитробензоилфенилглицином). Также можно использовать высокоэффективную жидкостную хроматографию (ВЭЖХ) или сверхкритическую жидкостную хроматографию (СЖХ). Конкретный метод, условия элюирования и хроматографические колонки могут быть выбраны специалистами в настоящей области техники в соответствии со структурами соединений и экспериментальными результатами. Кроме того, оптически чистые исходные материалы или ресредства с известной конфигурацией также можно использовать для получения любых энантиомеров или диастереомеров соединений, описанных в настоящем раскрытии, посредством стереоорганического синтеза.
[0053] Термин «таутомер» относится к изомеру функциональной группы, образующемуся в результате быстрого перемещения атома между двумя положениями в молекуле. Соединение согласно настоящему раскрытию может проявлять таутомерию. Таутомерные соединения могут присутствовать в двух или более взаимопревращающихся формах. Прототропный таутомер возникает в результате переноса ковалентно связанных атомов водорода между двумя атомами. Таутомер, как правило, существует в равновесной форме. При попытке разделить одиночный таутомер, как правило, получается смесь, физические и химические свойства которой согласуются со смесью соединений. Положение равновесия зависит от внутримолекулярных химических свойств. Например, для многих алифатических альдегидов и кетонов, таких как ацетальдегид, преобладает кетоновая форма; а для фенолов преобладает енольная форма. Все таутомерные формы соединений включены в настоящее раскрытие.
[0054] В примерах согласно настоящему раскрытию протоны могут занимать два или более положения циклической формы гетероциклической кольцевой системы, например, 1H- и 3H-имидазол, 1H-, 2H- и 4H-1,2,4-триазол, 1H- и 2H-изоиндол и 1H- и 2H-пиразол. Таутомерные формы могут находиться в равновесии или стерически фиксироваться в одной форме путем соответствующего замещения, например:
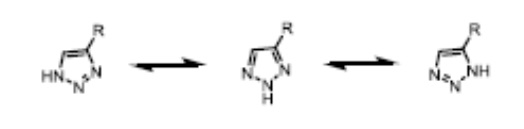
[0055] Из-за резонанса атом водорода на атоме азота триазола может быть расположен на любом из трех атомов азота триазола, поэтому их названия различны, но эти три формы фактически представляют собой одно и то же соединение.
[0056] Термин «фармацевтическая композиция» относится к смеси одного или нескольких соединений, описанных в настоящем документе, или их физиологически/фармацевтически приемлемых солей или пролекарственных средств и других химических компонентов. Другими химическими компонентами могут быть, например, физиологически/фармацевтически приемлемые носители и вспомогательные вещества. Фармацевтическая композиция направлена на облегчение введения соединения в организм.
[0057] Что касается лекарственных средств или фармакологически активных средств, термин «эффективная доза», «эффективное количество» или «терапевтически эффективное количество» относится к достаточному количеству лекарственного средства или средства, которое нетоксично, но может достигать требуемого эффекта. Для пероральной лекарственной формы согласно настоящему раскрытию «эффективное количество» одного активного вещества в композиции относится к количеству, необходимому для достижения требуемого эффекта при комбинации с другим активным веществом в композиции. Эффективное количество может варьироваться от человека к человеку, в зависимости от возраста и общего состояния субъектов, а также от конкретного активного вещества. Подходящее эффективное количество в конкретном случае может быть определено специалистами в настоящей области техники согласно рутинным экспериментам.
[0058] Термин «активный ингредиент», «терапевтическое средство», «активное вещество» или «активное средство» относится к химическому соединению, которое может эффективно лечить целевое нарушение, заболевание или состояние.
[0059] Термин «сольват» относится к соединению согласно настоящему раскрытию или его соли, включая в себя стехиометрический или нестехиометрический растворитель, связанный посредством межмолекулярной нековалентной силы. Когда растворителем является вода, сольват представляет собой гидрат.
[0060] Термин «пролекарственное средство» может быть преобразован в соединение согласно настоящему раскрытию, обладающее биологической активностью в физиологических условиях или посредством сольволиза. Пролекарственное средство согласно настоящему раскрытию получают путем модификации функциональных групп в соединении, и модификацию можно удалить с помощью общепринятых операций или in vivo, чтобы получить исходное соединение. Пролекарственное средство включает в себя соединение, которое образуется путем присоединения фрагмента к гидроксильной группе или аминогруппе в соединении согласно настоящему раскрытию. Когда пролекарственное средство соединения согласно настоящему раскрытию вводят млекопитающему, пролекарственное средство диссоциирует с образованием свободной гидроксильной или аминогруппы.
[0061] Соединение согласно настоящему раскрытию может содержать не встречающееся в природе соотношение атомных изотопов на одном или нескольких атомах, составляющих соединение. Например, соединение может быть помечено радиоактивным изотопом, таким как дейтерий (2Н), тритий (3Н), иод-125 (125I) или С-14 (14С). Превращения всех изотопных составов соединений согласно настоящему раскрытию, радиоактивных или нет, включены в объем настоящего раскрытия.
[0062] Термин «вспомогательное вещество» относится к фармацевтически приемлемому инертному ингредиенту. Примеры «вспомогательного вещества» включают в себя без ограничения связующие вещества, разрыхлители, смазывающие вещества, вещества, способствующие скольжению, стабилизаторы, наполнители, разбавители и т.д. Вспомогательные вещества могут улучшать рабочие свойства фармацевтического состава, делая состав более подходящим для прямого прессования за счет повышения текучести и/или адгезии. Примерами типичных «фармацевтически приемлемых носителей», подходящих для вышеуказанных составов, являются: сахара, такие как лактоза, сахароза, маннит и сорбит; крахмалы, такие как кукурузный крахмал, крахмал тапиоки и картофельный крахмал; целлюлоза и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и метилцеллюлоза; фосфаты кальция, такие как дикальцийфосфат и трикальцийфосфат; сульфат натрия; сульфат кальция; поливинилпирролидон; поливиниловый спирт; стеариновая кислота; соли щелочноземельных металлов и стеариновой кислоты, такие как стеарат магния и стеарат кальция; стеариновая кислота; растительные масла, такие как арахисовое масло, хлопковое масло, кунжутное масло, оливковое масло и кукурузное масло; неионогенные, катионогенные и анионные поверхностно-активные вещества; полимеры этиленгликоля; жирные спирты; и гидролизованные твердые вещества зерна и другие нетоксичные совместимые наполнители, связующие вещества, разрыхлители, буферы, консерванты, антиоксиданты, смазывающие вещества, красители и другие вспомогательные вещества, обычно используемые в лекарственных составах.
[0063] Термин «C1-C10 алкил» следует понимать как обозначающий неразветвленную или разветвленную насыщенную одновалентную углеводородную группу, содержащую 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода. Алкильная группа представляет собой, например, метил, этил, пропил, бутил, пентил, гексил, изопропил, изобутил, втор-бутил, трет-бутил, изоамил, 2-метилбутил, 1-метилбутил, 1-этилпропил, 1,2-диметилпропил, неопентил, 1,1-диметилпропил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 2-этилбутил, 1-этилбутил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 2,3-диметилбутил, 1,3-диметилбутил или 1,2-диметилбутил и т.д., или их изомеры. В частности, алкильная группа содержит 1, 2, 3, 4, 5 или 6 атомов углерода («C1-C6 алкил»), такая как метил, этил, пропил, бутил, изопропил, изобутил, втор-бутил, трет-бутил, и более конкретно, алкильная группа содержит 1, 2 или 3 атома углерода («C1-C3 алкил»), такая как метил, этил, н-пропил или изопропил.
[0064] Термин «C3-C10 циклоалкил» следует понимать как обозначающий насыщенное моноциклическое или бициклическое одновалентное углеводородное кольцо, содержащее 3-10 атомов углерода, включая в себя конденсированную или мостиковую полициклическую систему, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил или циклодецил, или бициклические углеводородные группы, такие как декагидронафталиновое кольцо.
[0065] Термин «трех-десятичленная гетероциклическая группа» следует понимать как обозначающий насыщенное, ненасыщенное или частично насыщенное моноциклическое, бициклическое или трициклическое кольцо, содержащее 3-10 атомов, в котором 1, 2, 3, 4 или 5 атомов кольца выбраны из N, O или S, и они могут быть соединены атомом углерода или азота, если не указано иное. Группа -CH2- в гетероциклической группе селективно заменена на -C(O)-. Если не указано иное, атом азота или атом серы в кольце селективно окисляется с образованием N-оксида или S-оксида, или атом азота в кольце селективно кватернизуется. -NH в кольце селективно замещен ацетилом, формилом, метилом или метилсульфонилом. Кольцо селективно замещено одним или несколькими атомами галогена. Следует понимать, что когда общее количество атомов S и атомов О в гетероциклической группе превышает 1, эти гетероатомы не являются смежными друг с другом. Если гетероциклическая группа является бициклической или трициклической, то по меньшей мере одно кольцо может селективно представлять собой гетероароматическое кольцо или ароматическое кольцо при условии, что по меньшей мере одно кольцо не является гетероароматическим. Если гетероциклическая группа является моноциклической, она не должна быть ароматической. Примеры гетероциклических групп включают в себя без ограничения пиперидинил, N-ацетилпиперидинил, N-метилпиперидинил, N-формилпиперазинил, N-метансульфонилпиперазинил, гомопиперазинил, пиперазинил, азетидинил, оксетанил, морфолинил, тетрагидроизохинолинил, тетрагидрохинолинил, индолинил, тетрагидропиранил, дигидро-2H-пиранил, тетрагидрофуранил, тетрагидротиопиранил, тетрагидротиопиран-1-оксид, тетрагидротиопиран-1,1-диоксид, 1H-пиридин-2-он и 2,5-диоксоимидазолидинил.
[0066] Термин «C1-C10 алкокси» следует понимать как -O-( C1-C10 алкил), где «C1-C10 алкил» имеет определение, описанное выше.
[0067] Термин «C6-C10 арильная» группа следует понимать как одновалентное ароматическое или частично ароматическое моноциклическое, бициклическое или трициклическое углеводородное кольцо, содержащее 6-10 атомов углерода, особенно кольцо, содержащее 6 атомов углерода («C6 арил»), такое как фенил; или бифенил, или кольцо, содержащее 9 атомов углерода («C9 арил»), такое как инданил или инденил, или кольцо, содержащее 10 атомов углерода («C10 арил»), такое как тетрагидронафтил, дигидронафтил или нафтил. Когда указанный C6-C10 арил является замещенным, он может быть монозамещенным или многократно замещенным. Кроме того, место замещения не ограничено, например, можно использовать орто-, пара- или метазамещение.
[0068] Термин «C6-C10 арилокси» следует понимать как -O-( C6-C10 арил), где C6-C10 арил имеет приведенное выше определение.
[0069] Термин «пяти-десятичленный гетероарил» следует понимать как моноциклическую, бициклическую или трициклическую одновалентную ароматическую кольцевую группу, содержащую 5-10 атомов кольца и содержащую 1-5 гетероатомов, независимо выбранных из N, O или S. Например, «пяти-четырнадцатичленный гетероарил» следует понимать как моноциклическую, бициклическую или трициклическую одновалентную ароматическую кольцевую группу, содержащую 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 атомов кольца, особенно 5 или 6 или 9 или 10 атомов углерода, и содержащую 1-5, предпочтительно 1-3 гетероатомов, выбранных из N, O или S; и дополнительно, в каждом случае, она может быть бензоконденсированной. В частности, гетероарильная группа выбрана из группы, состоящей из тиенила, фурила, пирролила, оксазолила, тиазолила, имидазолила, пиразолила, изоксазолила, изотиазолила, оксадиазолила, триазолила, тиадиазолила и т.д. и их бензоконденсированных производных, таких как бензофуранил, бензотиенил, бензоксазолил, бензизоксазолил, бензимидазолил, бензотриазолил, индазолил, индолил, изоиндолил, и т.д.; или пиридинила, пиридазинила, пиримидинила, пиразинила, триазинила и т.д., и их бензопроизводных, таких как хинолинил, хиназолинил, изохинолинил, и т.д.; или азоцинила, индолизинила, пуринила и т.д. и их бензопроизводных; или цинолинила, фталазинила, хиназолинила, хиноксалинила, нафтиридинила, птеридила, карбазолила, акридинила, феназинила, фенотиазинила, феноксазинила и т.д.
[0070] Термин «спироциклическая» относится к полициклической группе, которая имеет один общий атом углерода (называемый спироатомом) между пяти-двадцатичленными моноциклическими кольцами. Спироциклическая группа может включать в себя одну или несколько двойных связей, но ни одно из колец не содержит полностью сопряженной π-электронной системы. Кольца предпочтительно являются шести-четырнадцатичленными, более предпочтительно семи-восьмичленными. На основании количества общих спироатомов между кольцами спироциклоалкильную группу можно классифицировать как моноспироциклоалкильную группу, биспироциклоалкильную группу или полиспироциклоалкильную группу, и моноспироциклоалкильная группа и биспироциклоалкильная группа являются предпочтительными. Более предпочтительно спироциклоалкил представляет собой 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспироциклоалкил. Примеры спироциклоалкила включают в себя, но не ограничиваются ими:
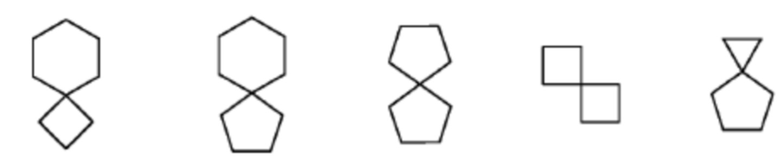 .
.
[0071] Спироциклоалкильная группа, в которой моноспироциклоалкил и гетероциклоалкил имеют общий спироатом, также включена, и ее примеры включают в себя, но не ограничиваются ими:
 .
.
[0072] Термин «галогенированная группа» или «галоген» относится к фтору, хлору, брому и иоду.
[0073] «Галогенированный алкил» относится к насыщенной алифатической углеводородной группе с разветвленной и неразветвленной цепью, содержащей определенное число атомов углерода и замещенной одним или несколькими атомами галогена (например, -CvFw, где v=от 1 до 3, w=от 1 до (2v+1)). Примеры галогенированного алкила включают в себя без ограничения трифторметил, трихлорметил, пентафторэтил, пентахлорэтил, 2,2,2-трифторэтил, гептафторпропил и гептахлорпропил.
Благоприятные эффекты
[0074] Согласно конкретным примерам согласно настоящему раскрытию соединение, представленное формулой (I) согласно настоящему раскрытию, или его фармацевтически приемлемая соль, таутомер, стереоизомер, гидрат, сольват или пролекарственное средство, оказывает значительный ингибирующий эффект на фермент АТХ.
[0075] В соответствии с конкретными примерами согласно настоящему раскрытию соединение согласно настоящему раскрытию обладает хорошим ингибирующим эффектом на фермент АТХ, и соединение согласно настоящему раскрытию проявляет превосходную стабильность метаболизма в печени, медленнее метаболизируется в организме человека и характеризуется более высокой степенью воздействия.
[0076] Дополнительные аспекты и преимущества настоящего раскрытия будут частично приведены в последующем описании, а часть из них станет очевидна из следующего описания или может быть понята посредством реализации настоящего раскрытия.
описание вариантов осуществления
[0077] Решения согласно настоящему раскрытию будут объяснены ниже в сочетании с примерами. Специалистам в настоящей области техники понятно, что следующие примеры используются просто для иллюстрации настоящего раскрытия и не должны рассматриваться как ограничивающие объем настоящего раскрытия. Примеры без указания конкретных методов или условий должны быть реализованы в соответствии с технологией или условиями, описанными в литературе в соответствующей области техники, или в соответствии с инструкциями к продукту. Все ресредства или инструменты представляют собой общепринятые продукты, коммерчески доступные, если их производители не указаны.
[0078] Варианты осуществления согласно настоящему раскрытию относятся к соединениям, представленным формулой (I), или их таутомерам, стереоизомерам, гидратам, сольватам, фармацевтически приемлемым солям или пролекарственным средствам; способам и промежуточным соединениям для получения соединений, представленных формулой (I), или их таутомеров, стереоизомеров, гидратов, сольватов, фармацевтически приемлемых солей или пролекарственных средств; фармацевтическим композициям; и применениям соединений и фармацевтических композиций согласно настоящему раскрытию в производстве лекарственных средств.
[0079] Растворители в реакциях, используемые на стадиях реакции согласно настоящему раскрытию, конкретно не ограничены, и любой растворитель, который может растворять исходные материалы до определенной степени и не ингибирует реакцию, должен быть включен в настоящее раскрытие. Кроме того, многие подобные модификации, эквивалентные замены, комбинации растворителей, эквивалентные растворителям, описанным в настоящем раскрытии, и различные соотношения комбинаций растворителей считаются включенными в объем настоящего раскрытия.
[0080] Структуры соединений идентифицируют с помощью ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Единица сдвига ЯМР составляет 10-6 (м.д.). Сдвиг ЯМР описан в единицах 10-6 (м.д.). Растворителем для измерения ЯМР является дейтерированный диметилсульфоксид, дейтерированный хлороформ, дейтерированный метанол и т.д., а внутренний стандарт представляет собой тетраметилсилан (TMS).
[0081] Измерения методом жидкостной хроматографии с масс-спектрометрией (ЖХ-МС) проводили с использованием масс-спектрометра Waters Acquity H-класса Uplc-QDA и контролировали с помощью хроматографической колонки ACQUITY UPLC BEH C18, 2,1×50 мм, 1,7 мкм. Условия градиентного элюирования: при скорости потока 1,0 мл/мин, 95-5% растворителя А1 и 5-95% растворителя В1, затем 95% В1 и 5% А1 в течение 0,5 мин, где процентное содержание представляет собой процент от объема определенного растворителя в общем объеме растворителя. Среди них растворитель A1 представлял собой 0,1% муравьиной кислоты в водном растворе, а растворитель B1 представлял собой 0,1% муравьиной кислоты в растворе ацетонитрила. Процент представляет собой объемный процент растворенного вещества в растворе.
[0082] Сокращения в настоящем раскрытии определяются следующим образом:
[0083] DIPEA: также называется DIEA, диизопропилэтиламин, т.е. N,N-диизопропилэтиламин
[0084] DMF: N,N-диметилформамид
[0085] Et3N: триэтиламин
[0086] MeOH: метанол
[0087] н.: эквивалентная концентрация, например, 2 н. соляная кислота означает раствор соляной кислоты с концентрацией 2 моль/л.
[0088] NADPH: восстановленный кофермент II
[0089] NaH: гидрид натрия
[0090] NMM: N-метилморфолин
[0091] NMP: N-метилпирролидон
[0092] СЖХ: сверхкритическая жидкостная хроматография
[0093] T3P: ангидрид пропилфосфоновой кислоты, т.е. 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфин-2,4,6-триоксид или ангидрид 1-пропилфосфорной кислоты
[0094] THF: тетрагидрофуран
[0095] TMSN3: азидотриметилсилан
[0096] IC50: Полуингибирующая концентрация, которая относится к концентрации, при которой достигается половина максимального ингибирующего эффекта
[0097] Если не указано иное, соединения, приведенные в качестве примеров в настоящем документе, названы и пронумерованы с помощью ChemBioDraw Ultra 13.0.
[0098] Контрольное соединение 1, используемое в следующих испытаниях согласно настоящему раскрытию, имеет структуру:
Контрольное соединение 1.
[0099] Контрольное соединение синтезировали в соответствии с патентной заявкой WO2014110000A1.
[01] Контрольное соединение 2 используемое в следующих испытаниях согласно настоящему раскрытию, имеет структуру:
Контрольное соединение 2
[02] Контрольное соединение 2 синтезировали в соответствии с патентной заявкой WO2014139882A1.
[03] Известно, что контрольные соединения 1 и 2 обладают хорошим ингибирующим эффектом в отношении фермента АТХ.
Пример получения 1: Получение промежуточного соединения A
[04] 2-((2,3-Дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновая кислота (промежуточное соединение A)
[05] Схема синтеза промежуточного соединения A проиллюстрирована ниже:
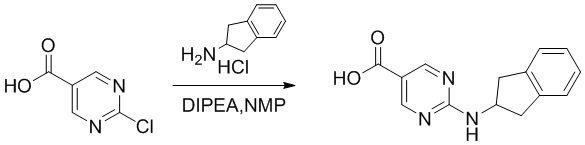
[06] 2-Хлорпиримидин-5-карбоновую кислоту (2 г, 12,61 ммоль) растворяли в N-метилпирролидоне (10 мл) и добавляли гидрохлорид 2-аминоиндана (2,57 г, 15,14 ммоль) и N,N-диизопропилэтиламин (8,15 г, 63,1 ммоль). Смесь нагревали до 100°C и проводили реакцию в течение 24 часов. Реакционный раствор упаривали под масляным насосом для удаления растворителя, к остатку добавляли этилацетат (30 мл) для диспергирования и фильтрации, фильтровальный осадок промывали водой (30 мл), фильтровали и сушили на воздухе при 50°C в течение 3 часов с получением серого твердого вещества, т.е. 2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновой кислоты (2,4 г, выход 74,5%). Промежуточное соединение A, используемое в следующих примерах, можно получить в соответствии с приведенной выше схемой синтеза и способом получения.
[07] LC-MS m/z: 256,2[M+H]+
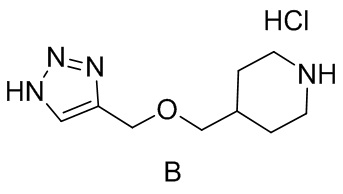
Пример получения 2: Получение промежуточного соединения B
[08] Гидрохлорид 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидина (промежуточное соединение B)
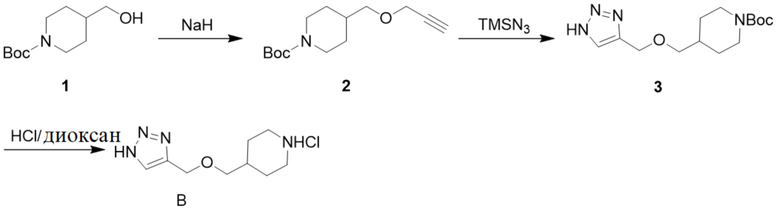
[09] Схема синтеза промежуточного соединения B проиллюстрирована ниже:
 .
.
[010] Стадия 1: Синтез трет-бутил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (2)
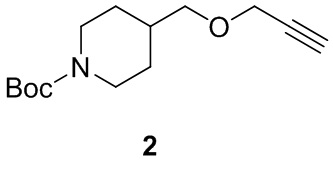
[011] трет-Бутил-4-(гидроксиметил)пиперидин-1-карбоксилат (150 г, 697 ммоль) добавляли к THF (1000 мл) при 0° C. Затем порциями добавляли гидрид натрия (33,4 г, 836 ммоль, чистота 60%), перемешивали при комнатной температуре в течение 1 ч и затем охлаждали до 0°C. Медленно по каплям добавляли 3-бромпропин (104 г, 871 ммоль) и перемешивали при комнатной температуре в течение 8 часов после завершения добавления. После завершения реакции реакционный раствор выливали в насыщенный водный раствор хлорида аммония (1000 мл) и экстрагировали этилацетатом (300 мл×3) и органические слои объединяли с получением неочищенного продукта. Неочищенный продукт разделяли и очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.)=20:1) с получением бесцветного маслянистого соединения, т.е. трет-бутил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (135 г, выход 76%).
[012] Стадия 2: Синтез трет-бутил-4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-карбоксилата (3)
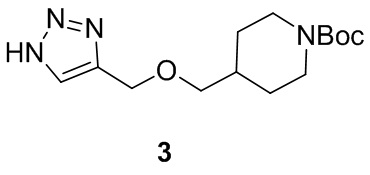
[013] Субстрат, трет-бутил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилат (60 г, 237 ммоль), растворяли в смешанном растворителе DMF/MeOH (400 мл /100 мл) и добавляли иодид меди (I) (2,26 г, 11,84 ммоль). После охлаждения смеси до 0°C медленно по каплям добавляли азидотриметилсилан (40,9 г, 355 ммоль) и после завершения добавления реакционную смесь медленно нагревали до 100°C и перемешивали в течение 18 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры и разбавляли MeOH и затем отфильтровывали осадок. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт разделяли и очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.)=5:1) с получением желтого маслянистого соединения, т.е. трет-бутил-4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-карбоксилата (55 г, выход 78%).
[014] LC-MS, M/Z: 297,1 [M+H]+
[015] Стадия 3: Синтез гидрохлорида 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидина (промежуточное соединение B)
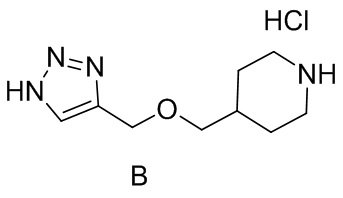
[016] Субстрат, трет-бутил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилат (55 г, 186 ммоль), растворяли в 1,4-диоксане (90 мл). Затем при перемешивании медленно по каплям добавляли раствор хлористого водорода в 1,4-диоксане (93 мл, 371 ммоль, 4 M) и затем перемешивали при комнатной температуре в течение 2 часов. После завершения реакции реакционный раствор разбавляли трет-бутилметиловым эфиром (200 мл) и хорошо перемешивали в течение 2 часов. Образовавшееся твердое вещество быстро отсасывали с получением гидрохлорида 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидина (42 г, выход 84%).
[017] LC-MS, M/Z: 197,1 [M+H]+
Пример 1: Получение целевого соединения I-1
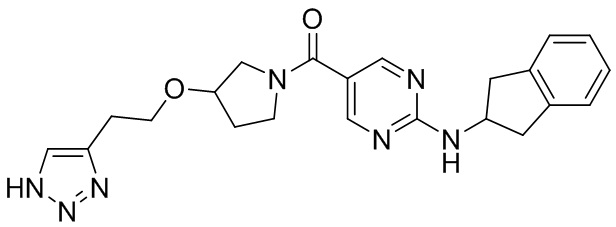
[018] (4-(((2H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-1)
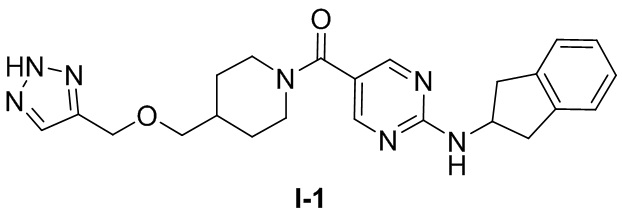
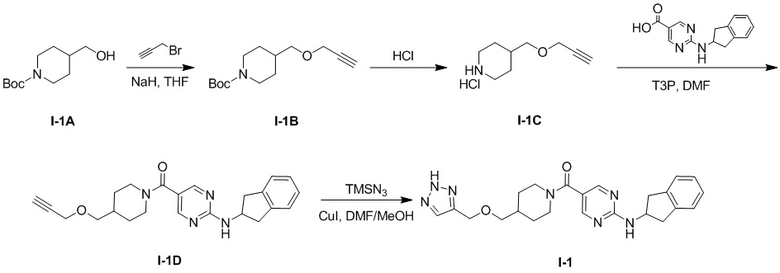
[019] Схема синтеза целевого соединения I-1 проиллюстрирована ниже:
 .
.
[020] Стадия 1: Синтез трет-бутил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (I-1B)
[021] трет-Бутил-4-(гидроксиметил)пиперидин-1-карбоксилат (I-1A) (2 г, 9,29 ммоль) растворяли в тетрагидрофуране (20 мл) и охлаждали до 0° C. Затем добавляли гидрид натрия (409 мг, 10,22 ммоль, 60%) с последующим добавлением по каплям 3-бромпропина (1,66 г, 14 ммоль) и после завершения добавления смесь подвергали реакции при комнатной температуре в течение 18 часов. Реакцию гасили водой (50 мл) и экстрагировали этилацетатом (50 мл×3) и органические фазы объединяли и сушили безводным сульфатом натрия с последующим фильтрованием и концентрированием. Остаток разделяли и очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.) =100: 1) с получением желтой жидкости, т.е. трет-бутил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (I-1B) (1,6 г, выход 68%).
[022] Стадия 2: Синтез гидрохлорида 4-((проп-2-ин-1-илокси)метил)пиперидина (I-1C)
[023] Раствор хлористого водорода в диоксане (20 мл) добавляли в трет-бутил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилат (I-1B) (1,6 г, 6,32 ммоль) и перемешивали при комнатной температуре для реакции в течение 2 часов. Растворитель удаляли при пониженном давлении с получением неочищенного продукта, который непосредственно использовали на следующей стадии реакции без очистки.
[024] Стадия 3: Синтез 2-(2,3-дигидро-1H-инден-2-иламино)пиримидин-5-карбоновой кислоты
[025] 2-Хлорпиримидин-5-карбоновую кислоту (2 г, 12,61 ммоль) растворяли в N-метилпирролидоне (10 мл) и добавляли гидрохлорид 2-аминоиндана (2,57 г, 15,14 ммоль) и N,N-диизопропилэтиламин (8,15 г, 63,1 ммоль). Смесь нагревали до 100°C и проводили реакцию в течение 24 часов. Реакционный раствор упаривали под масляным насосом для удаления растворителя, к остатку добавляли этилацетат (30 мл) для диспергирования и фильтрации и фильтровальный осадок промывали водой (30 мл), фильтровали и сушили на воздухе при 50°C в течение 3 часов с получением серого твердого вещества, т.е. 2-(2,3-дигидро-1H-инден-2-иламино)пиримидин-5-карбоновой кислоты (2,4 г, выход 74,5%).
[026] LC-MS, M/Z (ESI): 256,2 (M+H).
[027] Стадия 4: Синтез (2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)(4-((проп-2-ин-1-илокси)метил)пиперидин-1-ил)метанона (I-1D)
[028] N,N-диметилформамид (10 мл), N,N-диизопропилэтиламин (1,02 г, 7,91 ммоль) и 2-(2,3-дигидро-1H-инден-2-иламино)пиримидин-5-карбоновую кислоту (424 мг, 1,66 ммоль) добавляли в неочищенный продукт, полученный на предыдущей стадии, т.е. гидрохлорид 4-((проп-2-ин-1-илокси)метил)пиперидина (I-1C) (300 мг, 1,58 ммоль). Реакционный раствор охлаждали до приблизительно 0oC и по каплям добавляли 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид (1,31 г, 2,06 ммоль, 50% раствор N,N-диметилформамида). После завершения добавления смесь подвергали реакции при комнатной температуре в течение 16 часов. Реакционный раствор гасили добавлением воды (60 мл) и экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток разделяли и очищали на пластине с силикагелем (петролейный эфир: этилацетат) (об./об.)=5:1) с получением белого твердого вещества, т.е. (2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)(4-((проп-2-ин-1-илокси)метил)пиперидин-1-ил)метанона (I-1D) (400 мг, выход 64,8%).
[029] LC-MS, M/Z (ESI): 391,4 (M+1).
[030] Стадия 5: Синтез (4-(((2H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-1)
[031] В защитной атмосфере азота (2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)(4-((проп-2-ин-1-илокси)метил)пиперидин-1-ил)метанон (I-1D) (200 мг, 0,512 ммоль) растворяли в N,N-диметилформамиде (4 мл) и метаноле (2 мл). Добавляли L-аскорбат натрия (203 мг, 1,024 ммоль) и затем добавляли азидотриметилсилан (590 мг, 5,12 ммоль) и пентагидрат сульфата меди (32,7 мг, 0,205 ммоль). Смесь нагревали до 90°C и проводили реакцию в течение 4 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли воду (40 мл) и экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток разделяли и очищали на пластине с силикагелем (этилацетат: метанол (об./об.)=10:1, водный аммиак) с получением белого твердого вещества, т.е. (4-(((2H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-1) (19 мг, выход 8,56%).
[032] 1H ЯМР (400 МГц, DMSO-d6): δ8,37 (s, 2H), 7,96 (d, 1H), 7,82 (s, 1H), 7,25-7,10 (m, 4H), 4,75-4,60 (m, 1H), 4,54 (s, 2H), 4,05 (m, 2H), 3,32-3,20 (m, 4H), 2,97-2,85 (m, 4H), 1,83 (m, 1H), 1,75-1,60 (m, 2H), 1,2-1,05 (m, 2H).
[033] LC-MS, M/Z (ESI): 434,4 (M+1).
Пример 2: Получение целевого соединения I-2
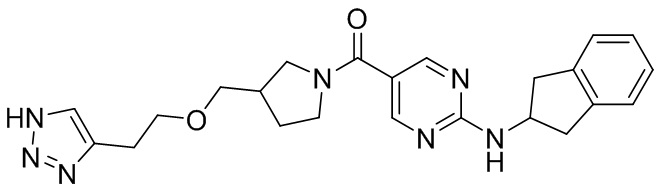
[034] (4-((1-(2H-1,2,3-триазол-4-ил)этокси)метил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-2)
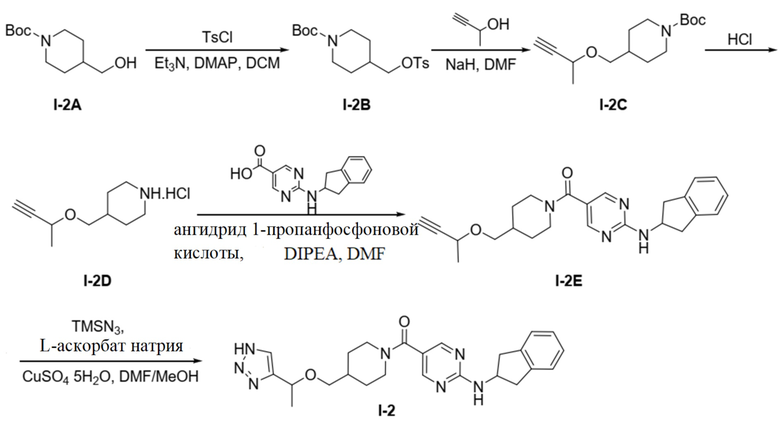
[035] Схема синтеза целевого соединения I-2 проиллюстрирована ниже:
 .
.
[036] Стадия 1: Синтез трет-бутил-4-((тозилокси)метил)пиперидин-1-карбоксилата (I-2B)
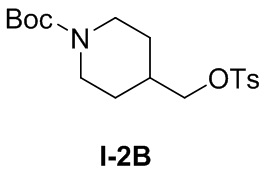
[037] трет-Бутил-4-(гидроксиметил)пиперидин-1-карбоксилат (1,5 г, 6,97 ммоль), триэтиламин (1,41 г, 13,93 ммоль), 4-диметиламинопиридин (0,426 г, 3,48 ммоль) и дихлорметан (20 мл) добавляли в одногорлую колбу и растворяли и добавляли п-толуолсульфонилхлорид (1,73 г, 9,06 ммоль) при 0°C. Смесь перемешивали и подвергали реакции при комнатной температуре в течение 12 часов, ТСХ (петролейный эфир: этилацетат (об./об.) =5:1) показала, что реакция завершена, проводили концентрирование и остаток очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.) = 5:1) с получением бесцветного твердого вещества, трет-бутил-4-((тозилокси)метил)пиперидин-1-карбоксилата (1 г, 2,71 ммоль, выход 38,8%).
[038] Стадия 2: Синтез трет-бутил-4-((бут-3-ин-2-илокси)метил)пиперидин-1-карбоксилата (I-2C)
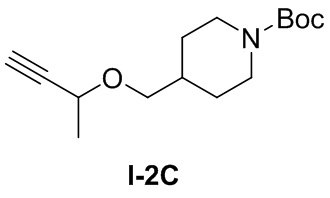
[039] В одногорлую колбу добавляли бут-3-ин-2-ол (0,285 г, 4,06 ммоль) и растворяли с помощью N,N-диметилформамида (10 мл). Добавляли 60% гидрид натрия (0,162 г, 4,06 ммоль). Смесь перемешивали при комнатной температуре в течение 0,5 часов добавляли и трет-бутил-4-((тозилокси)метил)пиперидин-1-карбоксилат (1 г, 2,71 ммоль) и перемешивали для реакции при 80°C в течение 12 часов. ТСХ (петролейный эфир: этилацетат (об./об.) =3:1) показала, что реакция завершена, для гашения реакции добавляли метанол (10 мл) и проводили концентрирование. Остаток очищали с использованием колонки с силикагелем (петролейный эфир: этилацетат (об./об.) = 10:1) с получением бесцветного маслянистого продукта, трет-бутил-4-((бут-3-ин-2-илокси)метил)пиперидин-1-карбоксилата (0,25 г, 0,935 ммоль, выход 34,5%).
[040] Стадия 3: Синтез гидрохлорида 4-((бут-3-ин-2-илокси)метил)пиперидина (I-2D)
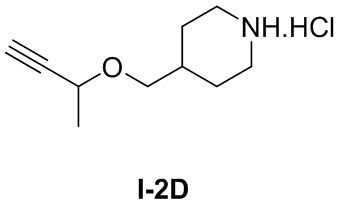
[041] трет-Бутил-4-((бут-3-ин-2-илокси)метил)пиперидин-1-карбоксилат (0,25 г, 0,935 ммоль) и хлористый водород в 1,4-диоксане (2,5 M, 2 мл) добавляли в одногорлую колбу, перемешивали при комнатной температуре в течение 3 часов и концентрировали досуха с получением бесцветного твердого неочищенного продукта, т.е. гидрохлорида 4-((бут-3-ин-2-илокси)метил)пиперидина (0,204 г).
[042] Стадия 4: Синтез (4-((бут-3-ин-2-илокси)метил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-2E)
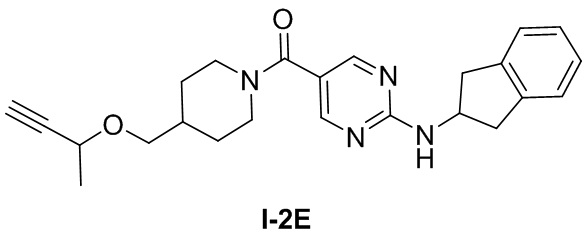
[043] Гидрохлорид 4-((бут-3-ин-2-илокси)метил)пиперидина (204 мг, 1,0 ммоль), 2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновую кислоту (142 мг, 0,555 ммоль), диизопропилэтиламин (0,717 г, 5,55 ммоль) и N,N-диметилформамид (4 мл) добавляли в одногорлую колбу, растворяли при комнатной температуре и затем охлаждали до 0°C. Добавляли по каплям 50% раствор ангидрида 1-пропанфосфоновой кислоты/N,N-диметилформамида (0,529 г, 0,832 ммоль) и подвергали реакции при комнатной температуре в течение ночи после завершения добавления по каплям. ТСХ (метанол: дихлорметан (об./об.) = 1:10) показала, что реакция завершена, для разбавления добавляли воду (10 мл) и водную фазу экстрагировали дихлорметаном (50 мл×2). Органические фазы объединяли и промывали насыщенным раствором соли (50 мл×2) для разделения жидкостей. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали и остаток очищали на колонке с силикагелем (метанол: дихлорметан (об./об.) = 1:10) с получением белого твердого вещества, (4-((бут-3-ин-2-илокси)метил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (200 мг, 0,494 ммоль, выход 89%).
[044] LC-MS m/z: 405,51[M+H]+
[045] Стадия 5: Синтез (4-((1-(2H-1,2,3-триазол-4-ил)этокси)метил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (целевое соединение I-2)
[046] Натриевую соль L(+)аскорбиновой кислоты (196 мг, 1,0 ммоль), (4-((бут-3-ин-2-илокси)метил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (200 мг, 0,494 ммоль), азидотриметилсилан (0,171 г, 1,48 ммоль), пентагидрат сульфата меди (25 мг, 0,099 ммоль), N,N-диметилформамид (4 мл) и метанол (0,4 мл) добавляли в одногорлую колбу и нагревали до 90°C для реакции в течение 2 часов. ЖХ-МС показала, что большая часть исходных материалов прореагировала. Реакционный раствор охлаждали до комнатной температуры, добавляли насыщенный раствор соли (10 мл) и экстрагировали этилацетатом (50 мл×2). Органические фазы дважды промывали насыщенным раствором соли (20 мл×2), сушили над безводным сульфатом натрия, фильтровали и концентрировали и остаток разделяли препаративной хроматографией с получением белого твердого вещества, (4-((1-(2H-1,2,3-триазол-4-ил)этокси)метил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (целевое соединение I-2, 30 мг, 0,067 ммоль, выход 13,6%).
[047] 1H ЯМР (400 МГц, DMSO-d6) δ8,33 (s, 2H), 7,92-7,93 (d, 1H), 7,74 (s, 1H), 7,16-7,19 (m, 2H), 7,09-7,13 (m, 2H), 4,56-4,64 (m, 2H), 3,09-3,25 (m, 5H), 2,85-2,90(q, 4H), 1,63-1,74 (m, 3H), 1,38-1,40 (d, 3H),1,03-1,12 (m, 3H).
[048] LC-MS m/z: 448,54[M+H]+
Пример 3: Получение целевого соединения I-3
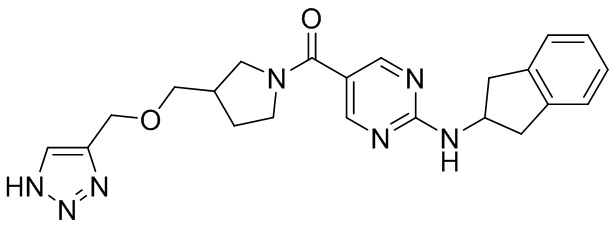
[049] (4-(1-((1H-1,2,3-триазол-4-ил)метокси)этил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-3)
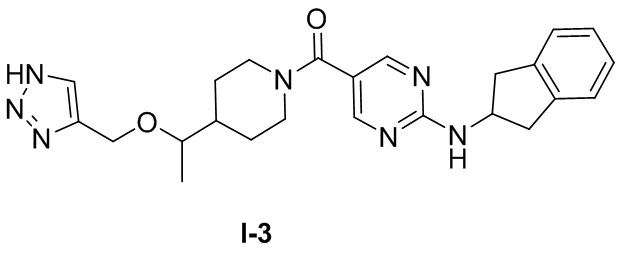 , т.е.
, т.е.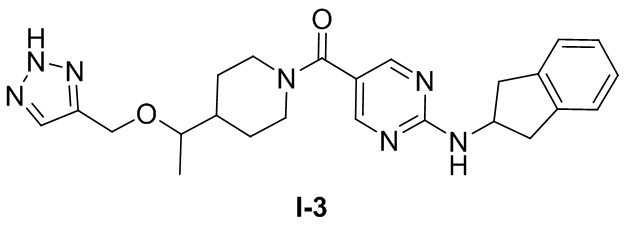
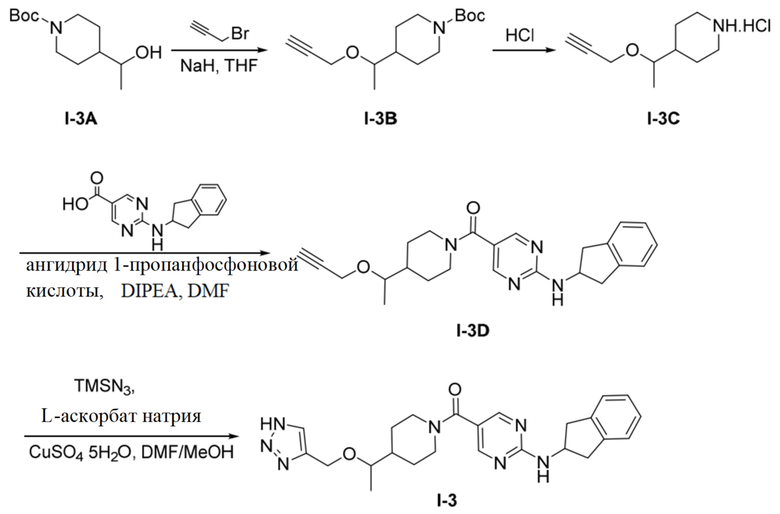
[050] Схема синтеза целевого соединения I-3 проиллюстрирована ниже:
 .
.
[051] Стадия 1: Синтез трет-бутил-4-(1-(проп-2-ин-1-илокси)этил)пиперидин-1-карбоксилата (I-3B)
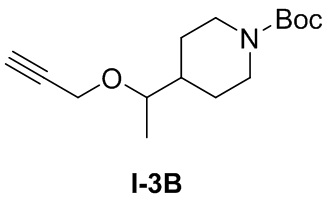
[052] трет-Бутил-4-(1-гидроксиэтил)пиперидин-1-карбоксилат (2,18 г, 5,34 ммоль) добавляли в одногорлую колбу и растворяли тетрагидрофураном (10 мл) и при комнатной температуре добавляли 60% гидрид натрия (0,131 г, 3,27 ммоль). Смесь перемешивали при комнатной температуре в течение 0,5 часов, добавляли бромпропин (0,778 г, 6,54 ммоль) и перемешивали при комнатной температуре в течение 12 часов. ТСХ (петролейный эфир: этилацетат (об./об.) = 3:1) показала, что реакция завершена. Реакцию гасили добавлением метанола (10 мл) с последующим концентрированием. Остаток очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.) = 3:1) с получением бесцветного маслянистого продукта, трет-бутил-4-(1-(проп-2-ин-1-илокси)этил)пиперидин-1-карбоксилата (300 мг, 1,12 ммоль, выход 51,5%).
[053] Стадия 2: Синтез гидрохлорида 4-(1-(проп-2-ин-1-илокси)этил)пиперидина (I-3C)
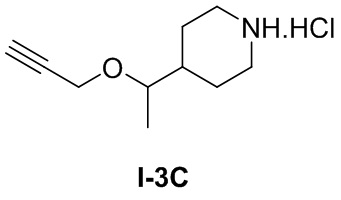
[054] трет-Бутил-4-(1-(проп-2-ин-1-илокси)этил)пиперидин-1-карбоксилат (300 мг, 1,12 ммоль) и раствор хлористого водорода/1,4-диоксан (2,5M, 2 мл) добавляли в одногорлую колбу, перемешивали при комнатной температуре в течение 3 часов и концентрировали досуха с получением белого твердого неочищенного продукта, т.е. гидрохлорида 4-(1-(проп-2-ин-1-илокси)этил)пиперидина (0,22 г).
[055] Стадия 3: Синтез (2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)(4-(1-(проп-2-ин-1-илокси)этил)пиперидин-1-ил)метанона (I-3D)
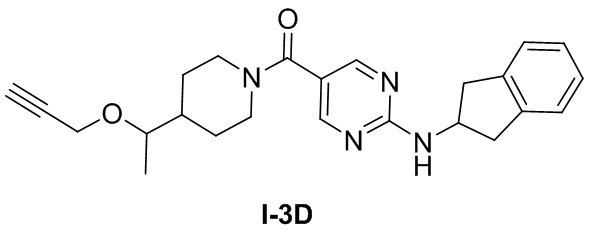
[056] Неочищенный продукт, т.е. гидрохлорид 4-(1-(проп-2-ин-1-илокси)этил)пиперидина (0,22 г, 1,08 ммоль), 2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновую кислоту (153 мг, 0,56 ммоль), диизопропилэтиламин (0,774 г, 5,99 ммоль) и N,N-диметилформамид (4 мл) добавляли в одногорлую колбу, растворяли при комнатной температуре и затем охлаждали до 0°C. Добавляли по каплям 50% раствор ангидрида 1-пропанфосфоновой кислоты/N,N-диметилформамида (0,572 г, 0,90 ммоль) и подвергали реакции в течение ночи при комнатной температуре после завершения добавления по каплям. ТСХ (метанол: дихлорметан (об./об.) = 1:10) показала, что реакция завершена. Реакционный раствор разбавляли водой (10 мл) и водную фазу экстрагировали дихлорметаном (50 мл×2). Органические фазы объединяли и промывали насыщенной солью водой (50 мл×2) для разделения жидкостей. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали и остаток очищали на колонке с силикагелем (метанол: дихлорметан (об./об.) = 1:10) с получением белого твердого вещества, т.е. (2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)(4-(1-(проп-2-ин-1-илокси)этил)пиперидин-1-ил)метанона (150 мг, 0,37 ммоль, выход 61,9%).
[057] LC-MS m/z: 405,51[M+H]+
[058] Стадия 4: Синтез (4-(1-((1H-1,2,3-триазол-4-ил)метокси)этил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (целевое соединение I-3)
[059] Натриевую соль L(+)аскорбиновой кислоты (147 мг, 0,74 ммоль), (2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)(4-(1-(проп-2-ин-1-илокси)этил)пиперидин-1-ил)метанон (150 мг, 0,37 ммоль), азидотриметилсилан (0,13 г, 1,12 ммоль), пентагидрат сульфата меди (19 мг, 0,074 ммоль), N,N-диметилформамид (4 мл) и метанол (0,4 мл) добавляли в одногорлую колбу и нагревали до 90°C для реакции в течение 2 часов. ЖХ-МС показала, что большая часть исходных материалов прореагировала. Реакционный раствор охлаждали до комнатной температуры, добавляли насыщенную солью воду (10 мл) и экстрагировали этилацетатом (50 мл×2). Органические фазы промывали насыщенной солью водой дважды (20 мл×2), сушили над безводным сульфатом натрия, фильтровали и концентрировали и остаток разделяли препаративной хроматографией с получением не совсем белого твердого вещества, (4-(1-((1H-1,2,3-триазол-4-ил)метокси)этил)пиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (57 мг, 0,127 ммоль, выход 34,3%).
[060] 1H ЯМР (400 МГц, DMSO-d6) δ8,33 (s, 2H), 7,92-7,93 (d, 1H), 7,77 (s, 1H), 7,16-7,19 (m, 2H), 7,10-7,13 (m, 2H), 4,57-4,66(m, 2H), 4,42-4,46 (d, 2H), 3,31-3,30 (q, 4H), 2,85-2,91 (q, 3H), 1,53-1,75 (m, 4H), 1,05-1,30 (m, 6H).
[061] LC-MS m/z: 448,54[M+H]+
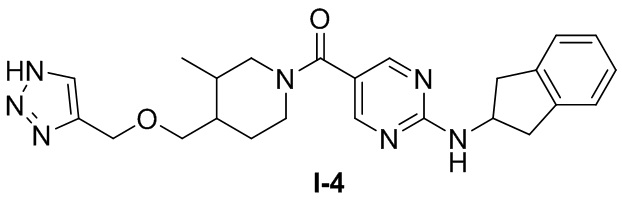
Пример 4: Получение целевого соединения I-4
[062] (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-метилпиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-4)
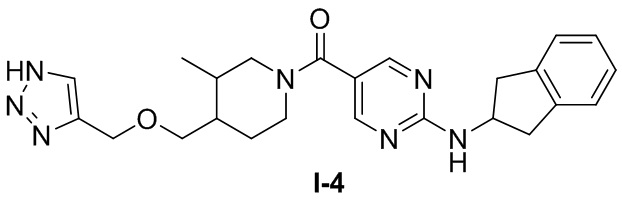
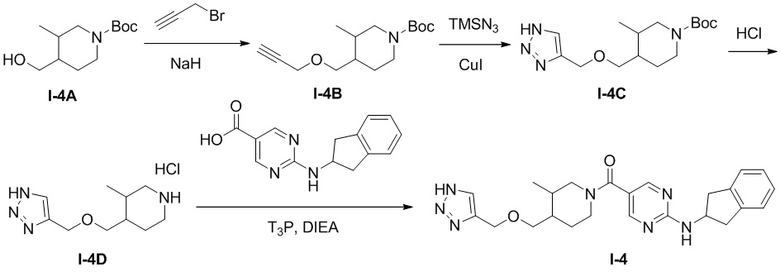
[063] Схема синтеза целевого соединения I-4 проиллюстрирована ниже:
 .
.
[064] Стадия 1: Синтез трет-бутил 3-метил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (I-4B)
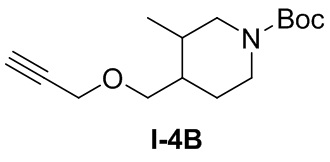
[065] трет-Бутил-4-(1-гидроксиметил)-3-метилпиперидин-1-карбоксилат (I-4A) (1,2 г, 5,23 ммоль) растворяли в сухом тетрагидрофуране (15 мл) и охлаждали до 0-5°С. Добавляли гидрид натрия (60% чистота, 0,209 г, 5,23 ммоль) и температуру реакционного раствора доводили до комнатной и перемешивали в течение 30 мин. Добавляли 3-бромпропин (0,934 г, 7,85 ммоль) и после завершения добавления реакцию продолжали при комнатной температуре в течение 20 часов. Реакцию гасили добавлением насыщенного раствора хлорида аммония (50 мл) и экстрагировали этилацетатом (30 мл×3). Органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.)=50:1) с получением желтого маслянистого вещества, трет-бутил 3-метил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (I-4B) (450 мг, выход 32,3%)
[066] 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-метилпиперидин-1-карбоксилат (I-4C)
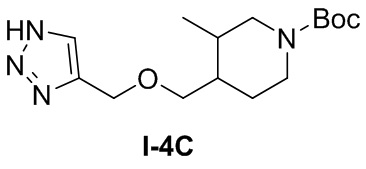
[067] трет-Бутил 3-метил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилат (I-4B) (450 мг, 1,683 ммоль) растворяли в N,N-диметилформамиде (5 мл) и метанол (0,5 мл) и добавляли иодид меди (96 мг, 0,505 ммоль). Смесь охлаждали до 0-5°C, добавляли по каплям азидотриметилсилан (291 мг, 2,52 ммоль) и после завершения добавления по каплям замену атмосферы азота проводили три раза и затем смесь нагревали до 95°C и проводили реакцию в течение 18 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали и остаток очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.) = 1: 1) с получением светло-желтого маслянистого вещества, трет-бутил-4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-метилпиперидин-1-карбоксилата (I-4C) (95 мг, выход 18,2%).
[068] Стадия 3: Синтез гидрохлорида 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-метилпиперидина (I-4D)
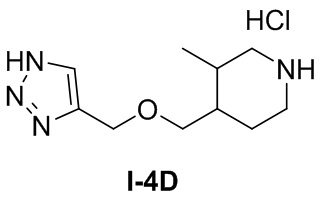
4 M[069] раствор хлористого водорода в 1,4-диоксане (2 мл, 8 ммоль) добавляли в трет-бутил-4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-метилпиперидин-1-карбоксилат (I-4C) (95 мг, 0,306 ммоль) и подвергали реакции при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали и неочищенный продукт использовали непосредственно на следующей стадии реакции.
[070] Стадия 4: Синтез (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-метилпиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-4)
[071] N,N-диметилформамид (5 мл), 2-((2,3-дигидро-1H-индан-2-ил)амино)пиримидин-5-карбоновую кислоту (79 мг, 0,308 ммоль) и N,N-диизопропилэтиламин (199 мг, 1,54 ммоль) добавляли в неочищенный продукт, полученный на предыдущей стадии, т.е. гидрохлорид 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-метилпиперидина (I-4D). Реакционный раствор охлаждали до приблизительно 0°С и добавляли по каплям T3P (294 мг, 0,462 ммоль, 50% раствор N,N-диметилформамида). После завершения добавления по каплям смесь подвергали реакции при комнатной температуре в течение 4 часов. Реакцию гасили добавлением воды (20 мл) в реакционный раствор, реакционный раствор экстрагировали дихлорметаном (15 мл×3) и органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха. Остаток разделяли и очищали на пластине с силикагелем (этилацетат: метанол (об./об.)=10 :1) с получением белого твердого вещества, (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-метилпиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-4) (21 мг, выход 15,2%).
[072] 1H ЯМР (400 МГц, DMSO-d6): δ 8,33 (s, 2H), 7,94-7,92 (d, 1H), 7,80 (s, 1H), 7,21-7,18 (m, 2H), 7,14-7,11 (m, 2H), 4,65-4,48 (m, 3H), 3,28-3,20 (m, 2H), 2,99-2,86 (m, 3H), 2,43-2,37 (m, 1H), 2,01-1,89 (m, 2H), 1,43-1,32 (m, 3H), 121-1,20 (m, 1H), 0,94-0,93 (d, 3H), 0,71-0,70 (d, 2H).
[073] LC-MS, M/Z: 448,3 [M+H]+.
Пример 5: Получение целевого соединения I-5
[074] (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-2-метилпиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-5)
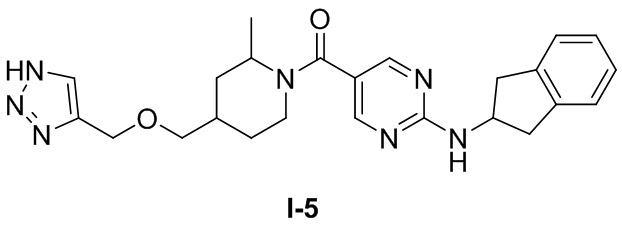
[075] Схема синтеза целевого соединения I-5 проиллюстрирована ниже:
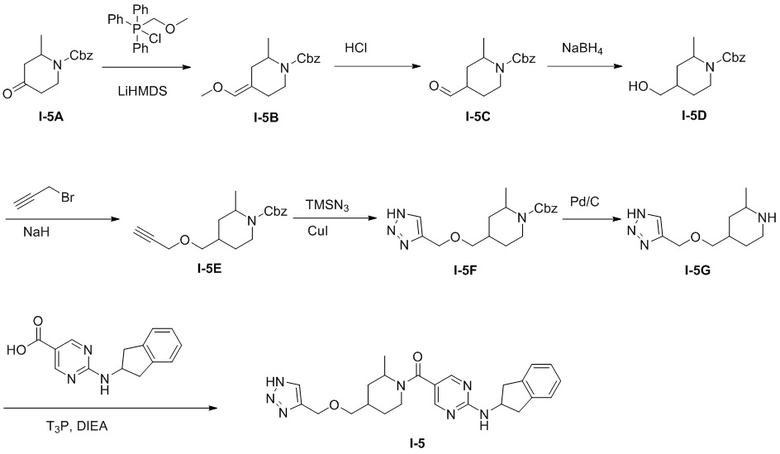 .
.
[076] Стадия 1: Синтез бензил-(Z)-4-(метоксиметилен)-2-метилпиперидин-1-карбоксилата (I-5B)
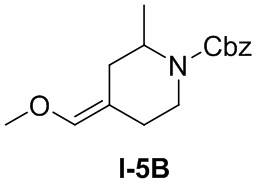
[077] В защитной атмосфере азота хлор(метоксиметил)трифенилфосфин (5,53 г, 16,13 ммоль) растворяли в сухом тетрагидрофуране (15 мл), смесь охлаждали до 0°С и по каплям добавляли раствор бис(триметилсилил)амида лития (1 M, 16,13 мл, 16,13 ммоль), после чего температуру поддерживали на уровне 0℃ для реакции в течение 30 мин. Затем по каплям добавляли тетрагидрофурановый раствор (10 мл) бензил-2-метил-4-оксопиперидин-1-карбоксилата (3 г, 12,13 ммоль) и после добавления реакцию продолжали при 0-5°С в течение 1 часа. Реакцию гасили добавлением насыщенного раствора хлорида аммония (50 мл) и реакционный раствор экстрагировали этилацетатом (50 мл×3), органические фазы объединяли и сушили над безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали колоночной хроматографией (петролейный эфир: этилацетат (об./об.)=50:1) с получением светло-желтого маслянистого вещества, бензил-(Z)-4-(метоксиметилен)-2-метилпиперидин-1-карбоксилата (I-5B) ( 3,37 г, выход 101%).
[078] Стадия 2: Синтез бензил-4-формил-2-метилпиперидин-1-карбоксилата (I-5C)
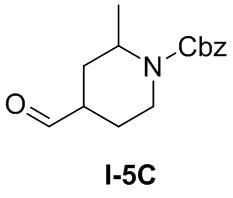
[079] Бензил-(Z)-4-(метоксиметилен)-2-метилпиперидин-1-карбоксилат (I-5B) (3,37 г, 12,24 ммоль) растворяли в ацетоне (20 мл), добавляли концентрированную соляную кислоту (0,245 г, 2,448 ммоль) и смесь нагревали до 50°C и проводили реакцию в течение 20 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли насыщенный раствор бикарбоната натрия для доведения рН выше 8. Реакционный раствор экстрагировали этилацетатом (10 мл×3) и органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали с получением желтого маслянистого вещества, бензил-4-формил-2-метилпиперидин-1-карбоксилата (I-5C) (3,08 г, выход 96 %).
[080] Стадия 3: Синтез бензил-4-(гидроксиметил)-2-метилпиперидин-1-карбоксилата (I-5D)
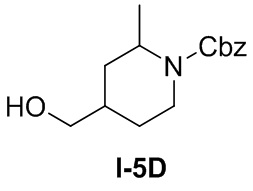
[081] Бензил-4-формил-2-метилпиперидин-1-карбоксилат (I-5C) (3,08 г, 11,79 ммоль) растворяли в метаноле (30 мл). Затем порциями добавляли боргидрид натрия (0,892 г, 23,57 ммоль) и после завершения добавления смесь перемешивали при комнатной температуре и проводили реакцию в течение 18 часов. Затем реакцию гасили добавлением воды (50 мл). Реакционный раствор экстрагировали этилацетатом (30 мл×3), органические фазы объединяли, сушили над безводным сульфатом натрия, концентрировали и разделяли и очищали колоночной хроматографией (петролейный эфир: этилацетат (об./об.)=3:1) с получением бледно-желтого маслянистого вещества, бензил-4-(гидроксиметил)-2-метилпиперидин-1-карбоксилата (I-5D) (2,44 г, выход 79%).
[082] Стадия 4: Синтез бензил-2-метил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (I-5E)
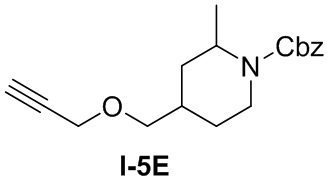
[083] Бензил-4-(гидроксиметил)-2-метилпиперидин-1-карбоксилат (I-5D) (2,44 г, 9,27 ммоль) растворяли в сухом тетрагидрофуране (25 мл) и охлаждали до приблизительно 0°С. Добавляли гидрид натрия (60% чистота, 0,371 г, 9,27 ммоль) и температуру реакционного раствора доводили до комнатной для реакции в течение 30 мин. По каплям добавляли 3-бромпропин (1,653 г, 13,90 ммоль) и после завершения добавления смесь подвергали реакции при комнатной температуре в течение 20 часов. Реакцию гасили добавлением насыщенного раствора хлорида аммония (50 мл) и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали колоночной хроматографией (петролейный эфир: этилацетат (об./об.)=10:1) с получением желтой жидкости, бензил-2-метил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (I-5E) (2,48 г, выход 89%).
[084] Стадия 5: Синтез бензил-4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-2-метилпиперидин-1-карбоксилата (I-5F)
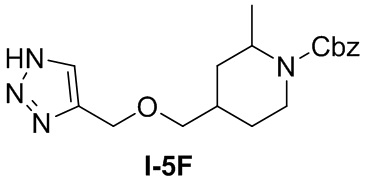
[085] Бензил-2-метил-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилат (I-5E) (1,2 г, 3,98 ммоль) растворяли в N,N-диметилформамиде (10 мл) и метаноле (1 мл), добавляли иодид меди (0,227 г, 1,195 ммоль) и смесь охлаждали до приблизительно 0°C. По каплям добавляли азидотриметилсилан (0,688 г, 5,97 ммоль), и после этого замену атмосферы азота проводили три раза, температуру повышали до 95°C для реакции в течение 20 часов. Реакционный раствор концентрировали и остаток разделяли и очищали колоночной хроматографией (петролейный эфир: этилацетат (об./об.) = 1: 1) с получением коричневого маслянистого вещества, бензил-4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-2-метилпиперидин-1-карбоксилата (I-5F) (0,45 г, выход 32,8%).
[086] Стадия 6: Синтез 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-2-метилпиперидина (I-5G)
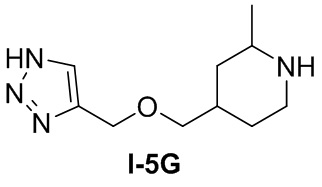
[087] Бензил-4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-2-метилпиперидин-1-карбоксилат (I-5F) (450 мг, 1,307 ммоль) растворяли в метаноле (10 мл), добавляли сухой палладий на углероде (10%, 278 мг, 0,261 ммоль) и замену атмосферы водорода проводили три раза. Смесь подвергали реакции при комнатной температуре под защитой атмосферы водорода в течение 48 часов, затем фильтровали через диатомит и концентрировали с получением неочищенного продукта непосредственно для следующей стадии реакции.
[088] Стадия 7: Синтез (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-2-метилпиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-5)
[089] N,N-диметилформамид (10 мл), 2-((2,3-дигидро-1H-индан-2-ил)амино)пиримидин-5-карбоновую кислоту (334 мг, 1,308 ммоль) и N,N-диизопропилэтиламин (507 мг, 3,92 ммоль) добавляли в неочищенный продукт, полученный на предыдущей стадии, т.е. 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-2-метилпиперидин (I-5G). Реакционный раствор охлаждали до приблизительно 0℃ и по каплям добавляли T3P (1,248 г, 1,962 ммоль, 50% раствор N,N-диметилформамида). После завершения добавления смесь подвергали реакции при комнатной температуре в течение 4 часов. Реакцию гасили добавлением воды (50 мл) к реакционному раствору и реакционный раствор экстрагировали этилацетатом (30 мл×3). Органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха. Остаток разделяли и очищали на пластине с силикагелем (этилацетат: метанол (об./об.)=10: 1) с получением белого твердого вещества, (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-2-метилпиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-5) (48 мг, выход 8,2%).
[090] 1H ЯМР (400 МГц, DMSO-d6) δ 8,33 (s, 2H), 7,93-7,91 (d, 1H), 7,80 (s, 1H), 7,21-7,17 (m, 2H), 7,14-7,10 (m, 2H), 4,67-4,58 (m, 1H), 4,51 (s, 2H), 3,29-3,20 (m, 7H), 2,92-2,86 (dd, 2H), 2,03 (s, 1H), 1,68-1,53 (dd, 2H), 1,38-1,04 (m, 5H).
[091] LC-MS, M/Z: 448,3 [M+H]+.
Пример 6: Получение целевого соединения I-6
[092] (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-фторпиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-6)
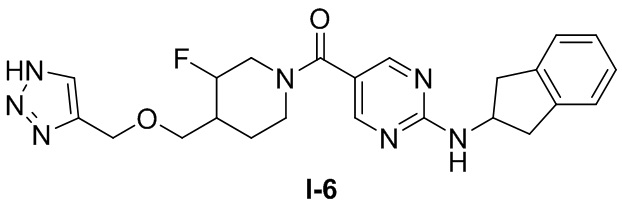
[093] Схема синтеза целевого соединения I-6 проиллюстрирована ниже:
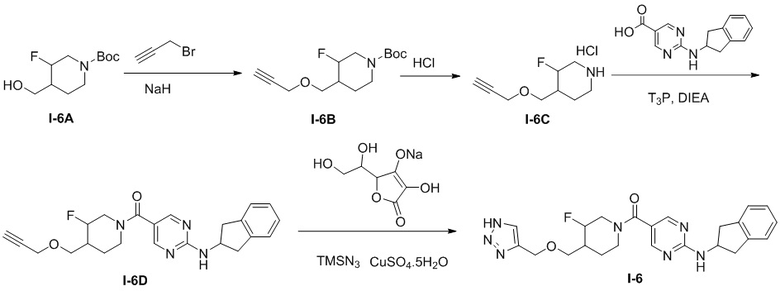 .
.
[094] Стадия 1: Синтез трет-бутил-3-фтор-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (I-6B)
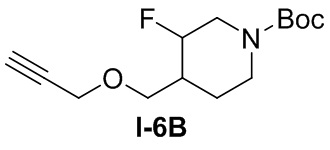
[095] трет-Бутил-3-фтор-4-(гидроксиметил)пиперидин-1-карбоксилат (I-6А) (500 мг, 2,143 ммоль) растворяли в сухом тетрагидрофуране (5 мл) и охлаждали до приблизительно 0℃, добавляли гидрид натрия (60% чистота, 86 мг, 2,143 ммоль) и температуру смеси восстанавливали до комнатной температуры для реакции в течение 30 мин. По каплям добавляли 3-бромпропин (382 мг, 3,22 ммоль) и после этого реакцию продолжали при комнатной температуре в течение 18 часов. Реакцию гасили добавлением насыщенного раствора хлорида аммония (30 мл) к реакционному раствору и реакционный раствор экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали колоночной хроматографией (петролейный эфир: этилацетат (об./об.)=20: 1) с получением желтого маслянистого вещества, трет-бутил-3-фтор-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилата (I-6B) (450 мг, выход 77%).
[096] Стадия 2: Синтез гидрохлорида 3-фтор-4-((проп-2-ин-1-илокси)метил)пиперидина (I-6C)
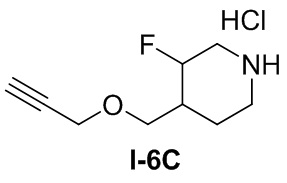
4 M[097] раствор хлористого водорода в 1,4-диоксане (5 мл, 20 ммоль) добавляли в трет-бутил-3-фтор-4-((проп-2-ин-1-илокси)метил)пиперидин-1-карбоксилат (I-6B) (200 мг, 0,737 ммоль) и подвергали реакции при комнатной температуре в течение 1 ч. Реакционный раствор концентрировали и неочищенный продукт напрямую подавали на следующую стадию реакции.
[098] Стадия 3: Синтез (2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)(3-фтор-4-((проп-2-ин-1-илокси)метил)пиперидин-1-ил)метанона (I-6D)
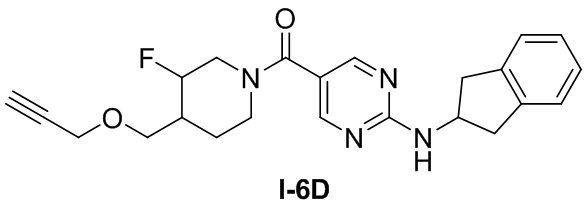
[099] N,N-диметилформамид (5 мл), 2-((2,3-дигидро-1H-индан-2-ил)амино)пиримидин-5-карбоновую кислоту (188 мг, 0,737 ммоль) и N,N-диизопропилэтиламин (476 мг, 3,68 ммоль) добавляли в неочищенный продукт, полученный на предыдущей стадии, т.е. гидрохлорид 3-фтор-4-((проп-2-ин-1-илокси)метил)пиперидина (I-6C). Реакционный раствор охлаждали до приблизительно 0°C. По каплям добавляли T3P (703 мг, 1,105 ммоль, 50% раствор N,N-диметилформамида). После завершения добавления по каплям смесь подвергали реакции при комнатной температуре в течение 4 часов. Реакцию гасили добавлением воды (50 мл) к реакционному раствору и реакционный раствор экстрагировали этилацетатом (30 мл×3). Органические фазы сушили безводным сульфатом натрия, фильтровали и концентрировали досуха. Остаток разделяли и очищали колоночной хроматографией (петролейный эфир: этилацетат (об./об.) = 1:2) с получением бесцветного маслянистого вещества, (2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)(3-фтор-4-((проп-2-ин-1-илокси)метил)пиперидин-1-ил)метанона (I-6D) (250 мг, выход 83%).
[0100] LC-MS, M/Z: 409,3 [M+H]+.
[0101] Стадия 4: Синтез (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-фторпиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-6)
[0102] (2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)(3-фтор-4-((проп-2-ин-1-илокси)метил)пиперидин-1-ил)метанон (I-6D) (230 мг, 0,563 ммоль) растворяли в N,N-диметилформамиде (5 мл) и метаноле (2 мл). Последовательно добавляли натриевую соль аскорбиновой кислоты (223 мг, 1,126 ммоль), пентагидрат сульфата меди (28,1 мг, 0,113 ммоль) и азидотриметилсилан (324 мг, 2,82 ммоль), замену атмосферы азота проводили три раза, реакционный раствор нагревали до 95°С и проводили реакцию в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли воду (50 мл) и экстрагировали дихлорметаном (30 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали. Остаток очищали на пластине с силикагелем (этилацетат: метанол (об./об.)=20:1) с получением белого твердого вещества, (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)-3-фторпиперидин-1-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-6) (140 мг, выход 55,1%).
[0103] 1H ЯМР (400 МГц, DMSO-d6) δ 8,37 (s, 2H), 8,02-8,00 (d, 1H), 7,84 (s, 1H), 7,24-7,20 (m, 2H), 7,17-7,13 (m, 2H), 4,88 (s, 0,5H), 4,76 (s, 0,5H), 4,69-4,64 (m, 1H), 4,58 (s, 2H), 3,48-3,44(t, 4H), 3,29-3,23 (m, 4H), 2,95-2,89 (dd, 2H), 2,16-2,00 (m, 1H), 1,59-1,55 (m, 1H), 1,42-1,35 (m, 1H).
[0104] LC-MS, M/Z: 452,3 [M+H]+.
Пример 7: Получение целевого соединения I-7
[0105] (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-ил)(2-((5-фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-7)
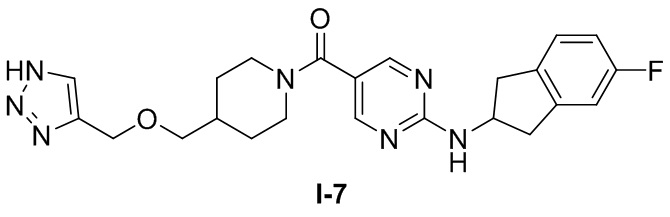
[0106] Схема синтеза целевого соединения I-7 проиллюстрирована ниже:
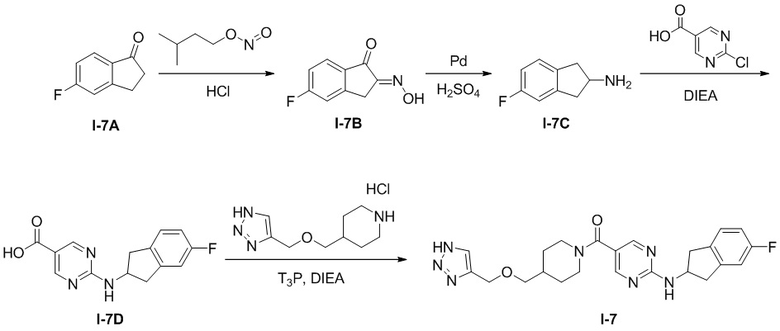 .
.
[0107] Стадия 1: Синтез (E)-5-фтор-2-(гидроксиимино)-2,3-дигидро-1H-инден-1-она (I-7B)
[0108] 5-фтор-2,3-дигидро-1H-инден-1-он (I-7A) (5 г, 33,3 ммоль) растворяли в метаноле (50 мл), реакционный раствор нагревали до 40°С, добавляли изопентилнитрит (6,6 мл, 5,74 ммоль) и по каплям добавляли концентрированную соляную кислоту (5 г, 50,1 ммоль). После этого смесь подвергали реакции при 40-50°С в течение 30 мин. Твердое вещество осаждали, фильтровали и сушили с получением белого твердого вещества, (E)-5-фтор-2-(гидроксиимино)-2,3-дигидро-1H-инден-1-она (I-7B) (4,2 г, выход 70,4%).
[0109] LC-MS, M/Z: 180,1 [M+H]+.
[0110] Стадия 2: Синтез 5-фтор-2,3-дигидро-1H-инден-2-амина (I-7C)
[0111] (E)-5-фтор-2-(гидроксиимино)-2,3-дигидро-1H-инден-1-он (I-7B) (3,1 г, 17,30 мл) растворяли в уксусной кислоте (90 мл), добавляли 10% сухой палладий на угле (5,52 г, 5,19 ммоль) и концентрированную серную кислоту (6,06 г, 60,6 ммоль) и трижды проводили замену атмосферы водорода. Под защитой атмосферы водорода реакционный раствор нагревали до 60°С и проводили реакцию в течение 20 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали через диатомит, фильтровальный осадок промывали водой (20 мл), фильтрат концентрировали. К остатку добавляли 50% раствор гидроксида натрия для доведения pH до значения выше 11 и реакционный раствор экстрагировали хлороформом (30 мл×5). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали колоночной хроматографией (дихлорметан: метанол (об./об.)=50:1) с получением темно-коричневого маслянистого вещества, 5-фтор-2,3-дигидро-1H-инден-2-амина (I-7C) (1 г, выход 38,2%).
[0112] LC-MS, M/Z: 152,2 [M+H]+.
[0113] Стадия 3: Синтез 2-((5-фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновой кислоты (I-7D)
[0114] 5-Фтор-2,3-дигидро-1H-инден-2-амин (I-7C) (524 мг, 3,47 ммоль) и 2-хлорпиримидин-5-карбоновую кислоту (500 мг, 3,15 ммоль) растворяли в N-метилпирролидоне (5 мл) и добавляли N,N-диизопропилэтиламин (2,038 г, 15,77 ммоль). Смесь нагревали до 100oC и проводили реакцию в течение 20 часов. Затем реакционный раствор охлаждали и концентрировали путем удаления растворителя и к остатку добавляли изопропилацетат (5 мл), превращали в пульпу и фильтровали. Фильтровальный осадок промывали водой (10 мл) и сушили с получением серого твердого вещества, 2-((5-фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновой кислоты (I-7D) (650 мг, выход 75%).
[0115] LC-MS, M/Z: 274,2 [M+H]+.
[0116] Стадия 4: Синтез (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-ил)(2-((5-фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-7)
[0117] 2-((5-фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновую кислоту (I-7D) (200 мг, 0,732 ммоль) и гидрохлорид 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидина (170 мг, 0,732 ммоль) растворяли в сухом N,N-диметилформамиде (5 мл) и добавляли N,N-диизопропилэтиламин (473 мг, 3,66 ммоль). Реакционный раствор охлаждали до приблизительно 0oC, по каплям добавляли T3P (699 мг, 1,098 ммоль, 50% раствор N,N-диметилформамида). После завершения добавления по каплям смесь подвергали реакции при комнатной температуре в течение 3 часов. Реакцию гасили добавлением воды (20 мл) и реакционный раствор экстрагировали дихлорметаном (30 мл×3). Органические фазы сушили безводным сульфатом натрия, фильтровали и концентрировали досуха. Остаток разделяли и очищали на пластине с силикагелем (этилацетат: метанол (об./об.)=10:1) с получением белого твердого вещества, (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-ил)(2-((5-фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-7) (146 мг, выход 44,2%).
[0118] 1H ЯМР (400 МГц, DMSO-d6) δ 8,36 (s, 2H), 7,98-7,96 (d, 1H), 7,81 (bs, 1H), 7,23-7,20 (m, 1H), 7,06-7,03 (dd, 1H), 6,97-6,92 (m, 1H), 4,71-4,62 (m, 1H), 4,53 (s, 2H), 3,29-3,18 (m, 6H), 2,94-2,83 (m, 4H), 1,86-1,79 (m, 1H), 1,70-1,67 (d, 2H), 1,23-1,07 (m, 2H).
[0119] LC-MS, M/Z: 452,4 [M+H]+.
Пример 8: Получение целевого соединения I-8
[0120] (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-ил)(2-((5,6-дифтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-8)
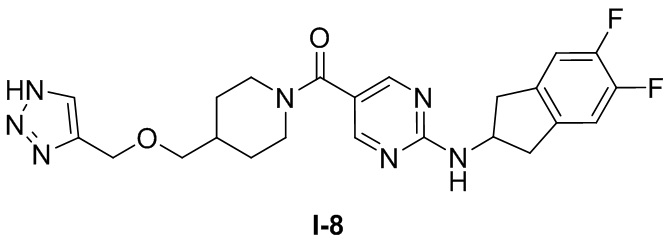
[0121] Схема синтеза целевого соединения I-8 проиллюстрирована ниже:
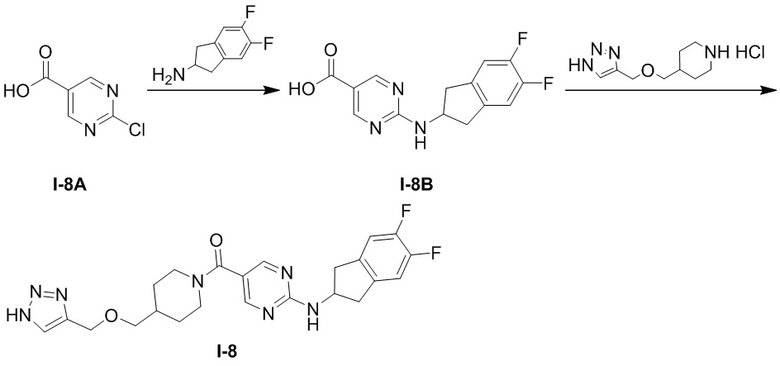 .
.
[0122] Стадия 1: Синтез 2-((5,6-дифтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновой кислоты (I-8B)
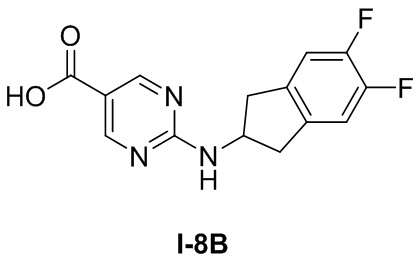
[0123] 5,6-Дифтор-2,3-дигидро-1H-инден-2-амин (150 мг, 0,887 ммоль), 2-хлорпиримидин-5-карбоновую кислоту (141 мг, 0,887 ммоль) и DIEA (229 мг, 1,773 ммоль) добавляли к NMP (2 мл). Систему нагревали до 95°С и перемешивали в течение ночи. После завершения реакции реакционный раствор концентрировали досуха и превращали в пульпу добавлением изопропилацетата и затем фильтровали с получением продукта, 2-((5,6-дифтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновой кислоты (I-8B) (200 мг, выход 77 %).
[0124] Стадия 2: Синтез (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-ил)(2-((5,6-дифтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-8)
[0125] Гидрохлорид 4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидина (162 мг, 0,824 ммоль), 2-((5,6-дифтор-2,3-дигидро-1H-индан-2-ил)амино)пиримидин-5-карбоновую кислоту (200 мг, 0,687 ммоль) и DIEA (133 мг, 1,030 ммоль) последовательно добавляли к раствору DMF (3 мл). По каплям добавляли T3P (437 мг, 0,687 ммоль, 50% раствор DMF) на бане со льдом и после завершения добавления по каплям смесь подвергали реакции в течение ночи при комнатной температуре. После завершения реакции реакционный раствор концентрировали путем удаления DMF, добавляли 10 мл водного раствора бикарбоната натрия, экстрагировали дихлорметаном (10 мл×3), сушили, концентрировали и разделяли на пластине для получения с получением бледно-желтого твердого вещества, (4-(((1H-1,2,3-триазол-4-ил)метокси)метил)пиперидин-1-ил)(2-((5,6-дифтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-8) (70 мг, выход 21,7%).
[0126] LC-MS, M/Z: 470,3 [M+H]+
[0127] 1H ЯМР (400 МГц, DMSO-d6) δ 8,32 (s, 2H), 7,94 (s, 1H), 7,22 (s, 2H), 4,72-4,62 (m, 3H), 3,99 (b, 1H), 3,18 (m, 4H), 2,85 (m, 4H), 1,75-1,61 (m, 3H), 1,16-1,10 (m, 4H).
Пример 9: Получение целевого соединения I-9
[0128] (6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-9)
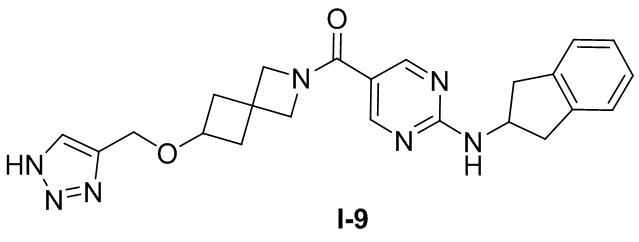
[0129] Схема синтеза целевого соединения I-9 проиллюстрирована ниже:
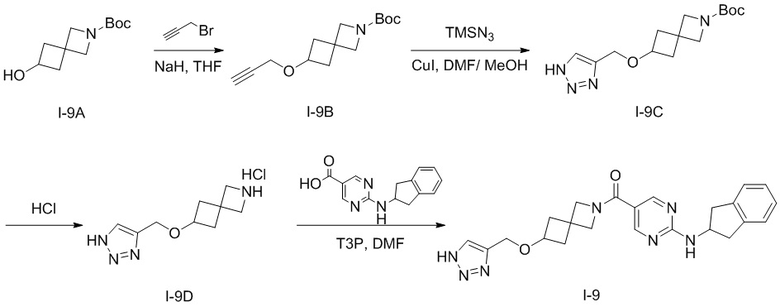 .
.
[0130] Стадия 1: Синтез трет-бутил-6-(проп-2-ин-1-илокси)-2-азаспиро[3.3]гептан-2-карбоксилата (I-9B)
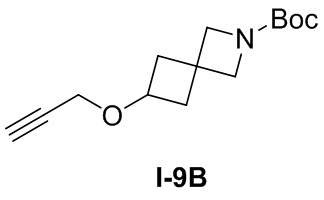
[0131] Гидрид натрия (11,25 г, 281 ммоль) растворяли в тетрагидрофуране (400 мл) и охлаждали до 0-5°C. Добавляли трет-бутил-6-гидрокси-2-азаспиро[трет-бутил][3.3]гептан-2-карбоксилат (I-9A) (50 г, 234 ммоль) и затем температуру смеси восстанавливали до комнатной температуры для реакции в течение 1 часа. По каплям добавляли 3-бромпропин (41,8 г, 352 ммоль) и затем реакцию продолжали при 25-30°C в течение 20 часов. Реакцию гасили добавлением насыщенного раствора хлорида аммония (1L) к реакционному раствору и реакционный раствор разделяли. Водную фазу экстрагировали этилацетатом (500 мл×2) и органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.) = 10: 1) с получением желтого маслянистого вещества, трет-бутил-6-(проп-2-ин-1-илокси)-2-азаспиро[3.3]гептан-2-карбоксилата (I-9B) (60 г, выход 102%).
[0132] Стадия 2: Синтез трет-бутил-6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-карбоксилата (I-9C)
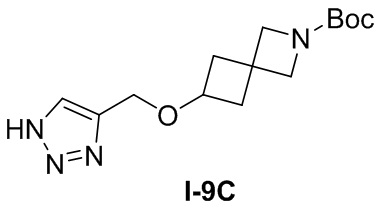
[0133] трет-Бутил-6-(проп-2-ин-1-илокси)-2-азаспиро[3.3]гептан-2-карбоксилат (I-9B) (40 г, 159 ммоль) и иодид меди (9,09 г, 47,7 ммоль) растворяли в N,N-диметилформамиде (400 мл) и метаноле (40 мл). Азидотриметилсилан (27,5 г, 239 ммоль) по каплям добавляли к реакционному раствору. В реакционной системе замену атмосферы азота проводили три раза. Реакционный раствор нагревали до 95°С и проводили реакцию в течение 18 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли воду (1 л) и экстрагировали этилацетатом (500 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.) = 2: 1) с получением бесцветного маслянистого вещества, трет-бутил-6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-карбоксилата (I-9C) (33,7 г, выход 71,9%).
[0134] Стадия 3: Синтез гидрохлорида 6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептана (I-9D)
[0135] трет-Бутил-6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-карбоксилат (I-9C) (25 г, 85 ммоль) растворяли в 1,4-диоксане (150 мл), добавляли раствор хлористого водорода в диоксане (42,5 мл, 170 ммоль, 4 M) и смесь перемешивали для реакции при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении с получением неочищенного продукта и неочищенный продукт непосредственно использовали на следующей стадии реакции без очистки.
[0136] Стадия 4: Синтез (6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-9)
[0137] N,N-диметилформамид (300 мл), N,N-диизопропилэтиламин (50,6 г, 392 ммоль) и 2-(2,3-дигидро-1H-инден-2-иламино)пиримидин-5-карбоновую кислоту (20 г, 78 ммоль) добавляли в неочищенный продукт, полученный на предыдущей стадии, т.е. гидрохлорид 6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептана (I-9D) (19,52 г, 85 ммоль). Реакционный раствор охлаждали до приблизительно 0°C. По каплям добавляли 2,4,6-трипропил-1,3,5,2,4,6-триоксотрифосфат-2,4,6-триоксид (74,8 г, 118 ммоль, 50% раствор N,N-диметилформамида) и затем смесь подвергали реакции при 25-30oC в течение 20 часов. Реакцию гасили добавлением воды (10 мл) к реакционному раствору и реакционный раствор концентрировали досуха, затем превращали в пульпу добавлением воды (200 мл) и метанола (20 мл). После фильтрации твердое вещество дважды перерабатывали в горячую пульпу путем добавления изопропилацетата и метанола ((об./об.) =10: 1) и затем сушили на воздухе при 50°C в течение 20 часов с получением белого твердого вещества, (6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-ил)(2-((2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-9) (7,2 г, выход 21,3%).
[0138] 1H ЯМР (400 МГц, DMSO-d6): δ8,53-8,49 (d, 2H), 8,09-8,07 (d, 1H), 7,78 (s, 1H), 7,19-7,16 (m, 2H), 7,13-7,10 (m, 2H), 4,67-4,59 (m, 1H), 4,40 (s, 3H), 4,34-4,29 (d, 1H), 3,97-3,91 (m, 3H), 3,29-3,19 (dd, 2H), 2,90-2,85 (dd, 2H), 2,47-2,41(m,2H), 2,04-1,99 (m, 2H).
[0139] LC-MS, M/Z (ESI): 432,4 (M+H).
Пример 10: Получение целевого соединения I-10
[0140] (6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-ил)(2-((5-фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанон (целевое соединение I-10)
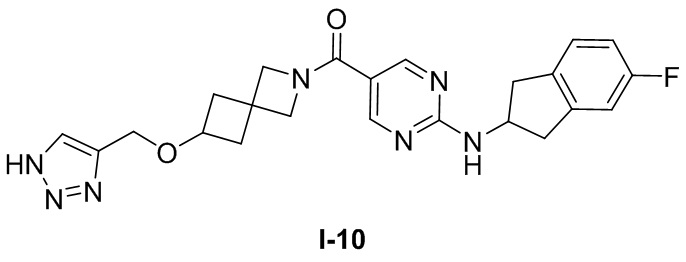
[0141] Схема синтеза целевого соединения I-10 проиллюстрирована ниже:
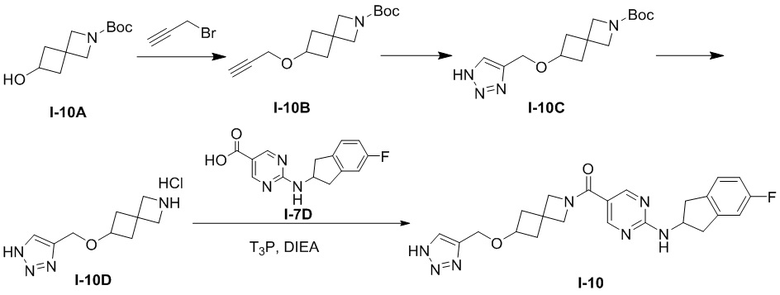 .
.
[0142] Стадия 1: Синтез трет-бутил-6-(проп-2-ин-1-илокси)-2-азаспиро[3.3]гептан-2-карбоксилата (I-10B)
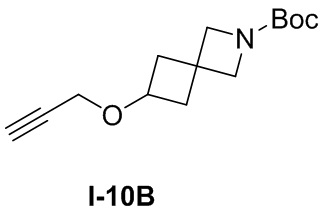
[0143] Гидрид натрия (60% чистота, 11,25 г, 281 ммоль) растворяли в тетрагидрофуране (400 мл) и охлаждали до 0-5°C. Добавляли трет-бутил-6-гидрокси-2-азаспиро[3.3]гептан-2-карбоксилат (I-10A) (50 г, 234 ммоль) и затем температуру смеси восстанавливали до комнатной температуры и смесь проводили реакцию в течение 1 часа. По каплям добавляли 3-бромпропин (41,8 г, 352 ммоль) и после этого реакцию продолжали при 25-30°C в течение 20 часов. Реакцию гасили добавлением насыщенного раствора хлорида аммония (1 л) к реакционному раствору с последующим разделением жидкости. Водную фазу экстрагировали этилацетатом (500 мл×2) и органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.)=10: 1) с получением желтого маслянистого вещества, трет-бутил-6-(проп-2-ин-1-илокси)-2-азаспиро[3.3]гептан-2-карбоксилата (I-10B) (60 г, выход 102%).
[0144] Стадия 2: Синтез трет-бутил-6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-карбоксилата (I-10C)
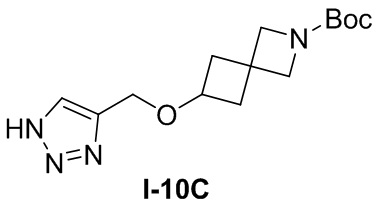
[0145] трет-Бутил-6-(проп-2-ин-1-илокси)-2-азаспиро[3.3]гептан-2-карбоксилат (I-10B) (40 г, 159 ммоль) и иодид меди (9,09 г, 47,7 ммоль) растворяли в N,N-диметилформамиде (400 мл) и метаноле (40 мл) и азидотриметилсилан (27,5 г, 239 ммоль) добавляли по каплям к реакционному раствору. В реакционной системе замену атмосферы азота проводили три раза, реакционный раствор нагревали до 95oC для реакции в течение 18 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли воду (1 л) и экстрагировали этилацетатом (500 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали. Остаток разделяли и очищали на колонке с силикагелем (петролейный эфир: этилацетат (об./об.) = 2: 1) с получением бесцветного маслянистого вещества, трет-бутил-6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-карбоксилата (I-10C) (33,7 г, выход 71,9% ).
[0146] Стадия 3: Синтез гидрохлорида 6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептана (I-10D)
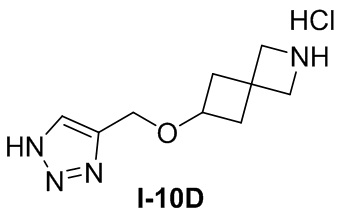
[0147] трет-Бутил-6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-карбоксилат (I-10C) (25 г, 85 ммоль) растворяли в 1,4-диоксане (150 мл) и добавляли 4 M раствор хлористого водорода в 1,4-диоксане (42,5 мл, 170 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов и растворитель удаляли при пониженном давлении с получением неочищенного продукта. Неочищенный продукт использовали непосредственно на следующей стадии реакции без очистки.
[0148] Стадия 4: Синтез (6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-ил)(2-((5-фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-10)
[0149] 2-((5-Фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-карбоновую кислоту (I-7D) (278 мг, 1,019 ммоль) и гидрохлорид 6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептана (I-10D) (235 мг, 1,019 ммоль) растворяли в высушенном N,N-диметилформамиде (5 мл) и добавляли N,N-диизопропилэтиламин (658 мг, 5,09 ммоль). Реакционный раствор охлаждали до приблизительно 0°C. По каплям добавляли T3P (972 мг, 1,528 ммоль, 50% раствор N,N-диметилформамида). После завершения добавления по каплям смесь подвергали реакции при комнатной температуре в течение 3 часов. Реакцию гасили добавлением воды (20 мл) в реакционный раствор. Реакционный раствор экстрагировали дихлорметаном (30 мл×3). Органические фазы сушили безводным сульфатом натрия, фильтровали и концентрировали досуха. Остаток разделяли и очищали на пластине с силикагелем (этилацетат: метанол (об./об.)=10:1) с получением белого твердого вещества, (6-((1H-1,2,3-триазол-4-ил)метокси)-2-азаспиро[3.3]гептан-2-ил)(2-((5-фтор-2,3-дигидро-1H-инден-2-ил)амино)пиримидин-5-ил)метанона (I-10) (125,3 мг, выход 27,4%).
[0150] 1H ЯМР (400 МГц, DMSO-d6) δ 8,56-8,53 (d, 2H), 8,15-8,13 (d, 1H), 7,81(bs, 1H), 7,25-7,21 (m, 1H), 7,07-7,04 (m, 1H), 6,99-6,94 (m, 1H), 4,73-4,64 (m, 1H), 4,44 (s, 2H), 4,38-4,33 (d, 2H),4,06-3,96 (m, 3H), 3,30-3,20 (m, 2H), 2,95-2,84 (m, 2H), 2,48-2,45 (m, 2H), 2,08-2,00 (m, 2H).
[0151] LC-MS, M/Z: 450,4 [M+H]+.
Примеры испытаний биологической активности и связанных с ней свойств
Пример испытания 1: Испытание на ингибирование ферментативной активности аутотаксина (АТХ)
[0152] Ингибирующие эффекты соединений на фермент аутотаксин определяли с использованием набора для скрининга ингибиторов аутотаксина (Cayman, 700580). Сначала исследуемое соединение получали в виде 10 мМ исходного раствора в растворителе DMSO, а затем исходный раствор разбавляли DMSO до 8 градиентов концентрации. Затем 8 концентраций разбавляли буфером для анализа аутотаксина (1×), поставляемым в наборе, до 19× рабочих растворов соединения (содержание DMSO составляло 1,9%). Реагент для анализа аутотаксина (10×) извлекали и разбавляли в 10 раз буфером для анализа аутотаксина (1×). Субстрат аутотаксина извлекали, растворяли путем добавления 1,2 мл буфера для анализа аутотаксина (1×), равномерно перемешивали и оставляли стоять при комнатной температуре. В 96-луночном планшете 150 мкл буфера для анализа аутотаксина (1×), 10 мкл приготовленного и разведенного 19× рабочего раствора соединения, 10 мкл реагента для анализа аутотаксина (1×) и 20 мкл растворенного субстрата аутотаксина добавляли в каждую лунку для каждой концентрации и гомогенно смешивали. 96-луночный планшет встряхивали в шейкере с постоянной температурой при 37°С и инкубировали в темноте в течение 30 минут, затем планшет вынимали и помещали на устройство для считывания микропланшетов для определения OD405. Экспериментальные результаты вводили в программное обеспечение GraphPad Prism, и IC50 каждого соединения рассчитывали путем аппроксимации.
[0153] В соответствии с описанными выше экспериментальными способами определяли ингибирующие эффекты контрольного соединения 1 и соединений согласно настоящему раскрытию на активность фермента АТХ. Результаты приведены в таблице 1:
[0154] [Таблица 1] Результаты ингибирующих эффектов исследуемых соединений на активность фермента АТХ
[0155] Экспериментальные результаты показали, что значение IC50 соединений согласно настоящему раскрытию аналогично или ниже, чем у контрольного соединения 1, что указывает на превосходные ингибирующие активности соединений согласно настоящему раскрытию в отношении фермента АТХ. Кроме того, ингибирующая активность соединений I-1 и от I-5 до I-10 в отношении фермента АТХ выше, чем у контрольного соединения 1.
Пример испытания 2: Испытание на ингибирование активности фермента АТХ в плазме человека
[0156] Цельную кровь брали у здоровых добровольцев и предохраняли от свертывания с помощью гепарина. Пробирки для сбора крови центрифугировали при 3000 об/мин в течение 10 мин, а плазму отбирали и хранили при -80°C для использования.
[0157] Соединение серийно разбавляли DMSO в соответствии с общепринятыми требованиями к концентрации, затем 3 мкл каждого из разведенных растворов соединения добавляли в 96-луночный планшет и 147 мкл PBS добавляли в каждую лунку, содержащую 3 мкл раствор соединения. После гомогенного перемешивания 50 мкл смеси отбирали и добавляли в новый 96-луночный планшет. Плазму человека извлекали из холодильника с температурой -80°С и размораживали при быстром встряхивании на водяной бане с температурой 37°С. Отбирали 50 мкл плазмы человека и добавляли в 96-луночный планшет, содержащий 50 мкл разведенного соединения (конечная система содержала 1% DMSO). Группа без соединения была установлена как положительная группа. 96-луночный планшет встряхивали, однородно перемешивали и инкубировали при 37°С в течение 3 часов. Также предусмотрена группа холостой пробы, и плазму группы холостой пробы хранили при -80°C. Группа холостой пробы предназначена для определения исходной концентрации эндогенного LPA.
[0158] После завершения инкубации группу холостой пробы оттаивали на льду и переносили в инкубационный планшет. Избыток ацетонитрила, содержащего внутренний стандарт LPA17:0, добавляли в инкубационный планшет для осаждения белка плазмы. После центрифугирования вихревым способом супернатант отбирали и разбавляли, и с помощью масс-спектрометрии ЖХ-МС/МС определяли площадь пика LPA18:2 и площадь пика внутреннего стандарта LPA17:0.
[0159] Рассчитывали отношение площади пика LPA18:2 к площади пика внутреннего стандарта LPA17:0 и рассчитывали степень ингибирования образования LPA18:2 по следующей формуле:
[0160] Степень ингибирования (%) = 100 – (группа соединения с различными концентрациями – группа холостой пробы)/(положительная группа – группа холостой пробы)*100
[0161] В соответствии со значениями степени ингибирования различных концентраций соединения рассчитывали значение IC50 соединения для ингибирования активности фермента АТХ в плазме человека.
[0162] Ингибирующий эффект на активность фермента АТХ в плазме человека контрольного соединения 1, контрольного соединения 2 и соединений согласно настоящему раскрытию испытывали в соответствии с описанным выше экспериментальным способом, и результаты приведены в таблице 2.
[0163] [Таблица 2] Результаты ингибирующих эффектов исследуемых соединений на активность фермента АТХ в плазме человека
[0164] Известно, что контрольные соединения 1 и 2 обладают хорошей ингибирующей активностью в отношении фермента АТХ. Экспериментальные результаты в таблице 2 показывают, что соединения согласно настоящему раскрытию обладают хорошим ингибирующим эффектом на фермент АТХ в плазме человека, особенно соединения I-1, I-9 и I-10 обладают более сильным ингибирующим эффектом на фермент АТХ в плазме человека, чем контрольные соединения 1 и 2.
Пример испытания 3: Испытание на ингибирование активности фермента АТХ в плазме крысы
[0165] Цельную кровь собирали у крыс и предохраняли от свертывания с помощью гепарина. Пробирки для сбора крови центрифугировали при 3000 об/мин в течение 15 минут, а плазму помещали на лед для использования.
[0166] Соединение серийно разбавляли DMSO:30% ACN=1:19 в соответствии с общепринятыми требованиями к концентрации, а затем помещали на лед. 147 мкл плазмы, которую помещали на лед, добавляли в 96-луночный планшет, а затем добавляли по 3 мкл каждого из градиентных растворов соединения (конечная система содержала 1‰ DMSO). Группу, не содержащую ни одного из соединений, устанавливали в качестве положительной группы. 96-луночный планшет однородно перемешивали и инкубировали при 37°С в течение 3 часов. Кроме того, предусмотрена группа холостой пробы. В начале инкубации к плазме группы холостой пробы добавляли избыток ацетонитрила, содержащего внутренний стандарт LPA17:0, для осаждения белка плазмы, встряхивали для перемешивания и хранили при 4°C. Группа холостой пробы предусмотрена для определения исходной концентрации эндогенного LPA.
[0167] После завершения инкубации избыток ацетонитрила, содержащего внутренний стандарт LPA17:0, добавляли в инкубационный планшет для осаждения белка плазмы, вышеупомянутую группу холостой пробы, в которой белки были осаждены, переносили в инкубационный планшет, после центрифугирования вихревым способом супернатант отбирали и разбавляли. Площадь пика LPA18:2 и площадь пика внутреннего стандарта LPA17:0 определяли с помощью масс-спектрометрии ЖХ-МС/МС.
[0168] Рассчитывали отношение площади пика LPA18:2 к площади пика внутреннего стандарта LPA17:0 и рассчитывали степень ингибирования образования LPA18:2 по следующей формуле:
[0169] Степень ингибирования (%) = 100 – (группа соединения с различными концентрациями – группа холостой пробы)/(положительная группа – группа холостой пробы)*100
[0170] В соответствии со значениями степени ингибирования различных концентраций соединения рассчитывали значение IC50 соединения для ингибирования активности фермента АТХ в плазме крыс.
[0171] Ингибирующий эффект на активность фермента АТХ в плазме крыс контрольного соединения 1, контрольного соединения 2 и соединений согласно настоящему раскрытию испытывали в соответствии с описанным выше экспериментальным способом, и результаты приведены в таблице 3.
[0172] [Таблица 3] Результаты ингибирующих эффектов исследуемых соединений на активность фермента АТХ в плазме крыс
[0173] Экспериментальные результаты показывают, что соединения согласно настоящему раскрытию обладают хорошей ингибирующей активностью в отношении фермента АТХ в плазме крыс, особенно соединения I-1, I-5, I-9 и I-10 обладают более высокой ингибирующей активностью в отношении фермента АТХ в плазме крыс, чем контрольные соединения 1 и 2.
Пример испытания 4: Испытание на стабильность микросом печени человека
[0174] Испытание на стабильность микросом печени человека проводили путем инкубации соединения и микросом печени человека in vitro для обнаружения. Сначала получали исследуемое соединение в виде 10 мМ исходного раствора в растворителе DMSO, а затем соединение разбавляли до 0,5 мМ ацетонитрилом. Микросомы печени человека (Corning) разбавляли PBS и готовили в виде раствора микросом с буфером, который использовали для разбавления 0,5 мМ раствора соединения до рабочего раствора. В рабочем растворе концентрация соединения составляла 1,5 мкМ, а концентрация микросом печени человека — 0,75 мг/мл. Брали многолуночный планшет с глубокими лунками, в каждую лунку последовательно добавляли 30 мкл рабочего раствора и 15 мкл предварительно подогретого раствора NADPH (6 мМ) для инициации реакции, реакцию инкубировали при 37°С. На 0, 5, 15, 30 и 45 мин инкубации в соответствующие лунки добавляли по 135 мкл ацетонитрила для прекращения реакции. После того как реакцию останавливали ацетонитрилом в последний момент времени 45 мин, многолуночный планшет с глубокими лунками встряхивали вихревым способом в течение 10 мин (600 об/мин), а затем центрифугировали в течение 15 мин. После центрифугирования супернатант собирали и добавляли к нему очищенную воду в соотношении 1:1 для проведения обнаружения ЖХ-МС/МС. Соответственно, получали отношение площади пика соединения к площади пика внутреннего стандарта в каждый момент времени, и отношения площадей пиков соединения через 5, 15, 30 и 45 минут сравнивали с отношением площадей пиков через 0 мин для расчета оставшегося процента соединения в каждый момент времени. T1/2 рассчитывали с помощью Excel.
[0175] Стабильность контрольного соединения 1, контрольного соединения 2 и соединений согласно настоящему раскрытию в микросомах печени человека испытывали в соответствии с описанным выше экспериментальным способом, и результаты приведены в таблице 4.
[0176] [Таблица 4] Результаты испытания на стабильность микросом печени человека
[0177] По сравнению с контрольным соединением 1 и контрольным соединением 2, соединения согласно настоящему раскрытию проявляли лучшую метаболическую стабильность в печени, они более медленно метаболизировались в организме человека и характеризовались более высокой степенью воздействия. T1/2 соединений согласно настоящему раскрытию лучше, чем у контрольных соединений. Соответственно, клиническая дозировка и частота введения могут быть снижены, токсические и побочные эффекты клинического введения могут быть снижены, а соблюдение предписанного режима терапии может быть улучшено.
Пример испытания 5: Обнаружение ингибирующего эффекта соединений на hERG с использованием полностью автоматической электрофизиологической системы для локальной фиксации потенциала QPatch
[0178] Полностью автоматическую электрофизиологическую систему для локальной фиксации потенциала QPatch использовали для обнаружения ингибирующего эффекта соединений на hERG. Клетки, использованные в этом испытании, представляли собой клеточную линию СНО, трансфицированную кДНК hERG и стабильно экспрессирующую каналы hERG (предоставлены Sophion Bioscience, Дания), и количество клеточных пассажей составляло P24. Клетки культивировали в среде, содержащей следующие компоненты (все приобретены у Invitrogen): среда Хэма F12, инактивированная фетальная бычья сыворотка (10% (об./об.)), гигромицин В (100 мкг/мл) и генетицин (100 мкг /мл). Клетки CHO hERG выращивали в чашке Петри, содержащей указанную выше среду, и культивировали в инкубаторе при 37°С и содержании 5% СО2.
[0179] Получали внеклеточную жидкость (2 мМ CaCl2, 1 мМ MgCl2, 4 мМ KCl, 145 мМ NaCl, 10 мМ глюкозы, 10 мМ HEPES, рН: приблизительно 7,4, осмотическое давление: приблизительно 305 мОсм) и внутриклеточную жидкость (5,374 мМ CaCl2, 1,75 мМ MgCl2, 120 мМ KCl, 10 мМ HEPES, 5 мМ EGTA, 4 мМ Na-ATP, рН приблизительно 7,25, осмотическое давление приблизительно 295 мОсм).
[0180] Исследуемое соединение готовили в виде 10 мМ исходного раствора в растворителе DMSO, соединение разбавляли до 3 мМ, 1 мМ, 0,3 мМ и 0,1 мМ DMSO, а затем соединение разбавляли до 30 мкМ, 10 мкМ, 3 мкМ, 1 мкМ, 0,3 мкМ и 0,1 мкМ с помощью внеклеточной жидкости, так что, за исключением того, что конечная концентрация DMSO в 30 мкМ соединении составляла 0,3%, конечная концентрация DMSO в растворах соединения всех других концентраций составляла 0,1%.
[0181] Клетки CHO hERG после расщепления и ресуспендирования добавляли в полностью автоматическую систему QPatch (Sophion, Дания) и подвергали испытанию в соответствии со следующей предварительно установленной процедурой.
[0182] После достижения состояния конфигурации разорванной целой клетки на начальной стадии ток всей клетки регистрировали при комнатной температуре (приблизительно 25°C) в течение по меньшей мере 120 секунд для достижения стабильности. Для испытания отбирали стабильные клетки. В течение всего испытания фиксацию потенциала клеток проводили при напряжении -80 мВ, напряжение фиксации потенциала клеток деполяризовали до +20 мВ для активации калиевых каналов hERG, а через 2,5 секунды напряжение фиксации потенциала клеток составляло -50 мВ для устранения инактивации и генерации направленного наружу следового тока. Пиковый следовой ток использовали как значение тока hERG. Описанный выше режим напряжения прикладывали к клеткам для электрофизиологического испытания каждые 15 секунд. К клеткам добавляли внутриклеточную жидкость, содержащую 0,1% диметилсульфоксида (растворитель), для установления исходного уровня, а затем позволяли току стабилизироваться в течение 3 мин. После добавления раствора соединения клетки выдерживали в среде для испытания до тех пор, пока действие соединения не достигало стационарного состояния или до достижения 4 мин. В экспериментах испытания с различными градиентами концентрации соединения соединение добавляли к клеткам с фиксированным потенциалом от низкой до высокой концентрации. После окончания испытания соединения клетки промывали внеклеточной жидкостью до тех пор, пока ток не возвращался в стабильное состояние. Данные испытаний проанализировали с помощью программного обеспечения для анализа Qpatch, предоставленного Sophion, Excel, Graphpad Prism и т.д.
[0183] В соответствии с описанными выше экспериментальными способами определяли ингибирующие эффекты контрольного соединения 1 и соединений согласно настоящему раскрытию на hERG. Результаты приведены в таблице 5:
[0184] [Таблица 5] Результаты ингибирующих эффектов соединений на hERG
[0185] По сравнению с контрольным соединением 1 соединения согласно настоящему раскрытию проявляют более слабый ингибирующий эффект на hERG. Принимая во внимание значение IC50 соединений для ингибирования активности фермента АТХ, это указывает на то, что соединения согласно настоящему раскрытию демонстрируют лучшее окно безопасности для ингибирования hERG и обладают значительными преимуществами в отношении безопасности для сердца.
Пример испытания 6: Фармакокинетическое испытание на крысах
[0186] Для фармакокинетического испытания на крысах использовали 3 самцов крыс SD (180-240 г), которых не кормили в течение ночи. Крысам проводили пероральное введение через желудочный зонд (10 мг/кг). Кровь собирали перед введением и через 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 8 ч и 24 ч после введения. Образцы крови центрифугировали при 8000 об/мин при 4°С в течение 6 мин, собирали плазму и хранили при -20°С. Плазму в каждый момент времени брали и добавляли раствор ацетонитрила, содержащий внутренний стандарт в 3-5-кратном количестве, встряхивали вихревым способом и перемешивали в течение 1 мин и центрифугировали при 13000 об/мин при 4°С в течение 10 мин. Супернатант собирали, добавляли воду в 3-кратном количестве и перемешивали. Соответствующее количество смеси брали для анализа ЖХ-МС/МС. Основные фармакокинетические параметры анализировали с использованием программного обеспечения WinNonlin 7.0 с помощью некомпартментной модели.
[0187] [Таблица 6] Результаты фармакокинетического испытания на крысах
(нг/мл)
(ч)
(ч*нг/мл)
[0188] Экспериментальные результаты показывают, что по сравнению с контрольными соединениями 1 и 2 соединения согласно настоящему раскрытию проявляют лучшие фармакокинетические свойства.
Пример испытания 7: Фармакокинетическое испытание на мышах
[0189] Для фармакокинетического испытания на мышах использовали самцов мышей CD-1 (от 20 до 25 г), которых не кормили в течение ночи. Мышам проводили пероральное введение через желудочный зонд (10 мг/кг). Кровь собирали перед введением и через 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 8 ч и 24 ч после введения. Образцы крови центрифугировали при 8000 об/мин при 4°С в течение 6 мин, собирали плазму и хранили при -20°С. Плазму в каждый момент времени брали и добавляли раствор ацетонитрила, содержащий внутренний стандарт в 3-5-кратном количестве, встряхивали вихревым способом и перемешивали в течение 1 мин и центрифугировали при 13000 об/мин при 4°С в течение 10 мин. Супернатант собирали, добавляли воду в 3-кратном количестве и перемешивали. Соответствующее количество смеси брали для анализа ЖХ-МС/МС. Основные фармакокинетические параметры анализировали с использованием программного обеспечения WinNonlin 7.0 с помощью некомпартментной модели.
[0190] [Таблица 7] Результаты фармакокинетического испытания на мышах
(нг/мл)
(ч)
(ч*нг/мл)
[0191] Экспериментальные результаты показывают, что по сравнению с контрольным соединением 1 соединения согласно настоящему раскрытию проявляют лучшие фармакокинетические свойства.
Пример испытания 8: Фармакокинетическое испытание на собаках
[0192] Для фармакокинетического испытания на собаках использовали 3 самцов биглей (от 8 до 10 кг), которых не кормили в течение ночи. Собакам проводили пероральное введение через желудочный зонд (5 мг/кг). В остальном операция аналогична фармакокинетическому испытанию на крысах.
[0193] [Таблица 8] Результаты фармакокинетического испытания на собаках
(нг/мл)
(ч)
(ч*нг/мл)
[0194] Результаты испытаний показывают, что по сравнению с контрольными соединениями 1 и 2 соединения согласно настоящему раскрытию проявляют лучшие фармакокинетические свойства.
Пример испытания 9: Испытание на термодинамическую растворимость
[0195] Приготовили следующие растворы: фосфатный буфер (PBS, pH 7,4), раствор FeSSIF (pH 5,8, содержащий 10 мМ таурохолата натрия, 2 мМ лецитина, 81,65 мМ гидроксида натрия, 125,5 мМ хлорида натрия, 0,8 мМ олеата натрия, 5 мМ глицерилмоноолеата, 55,02 мМ малеиновой кислоты) и раствор FaSSGF (рН 1,6, 1 л раствора, содержащего 80 мкМ таурохолата натрия, 20 мкМ лецитина, 0,1 г пепсина и 34,2 мМ хлорида натрия).
[0196] Соединение точно взвешивали и добавляли приготовленный фосфатный буфер (pH 7,4), раствор FeSSIF (pH 5,8) и раствор FaSSGF (pH 1,6) для получения раствора с концентрацией 4 мг/мл, который встряхивали при 1000 об/мин в течение 1 часа, а затем инкубировали в течение ночи при комнатной температуре. После инкубации раствор центрифугировали при 12000 об/мин в течение 10 мин для удаления нерастворившихся частиц, а супернатант переносили в новую центрифужную пробирку. Супернатант соответствующим образом разбавляли, затем добавляли раствор ацетонитрила, содержащий внутренний стандарт, и количественно определяли с использованием стандартной кривой, полученной с той же матрицей.
[0197] [Таблица 9] Результаты испытания на термодинамическую растворимость
[0198] Результаты испытаний показывают, что по сравнению с контрольным соединением термодинамическая растворимость соединений согласно настоящему раскрытию в искусственном желудочном соке, искусственном кишечном соке и нейтральных условиях значительно улучшилась. Соответственно, ожидается, что кишечная абсорбция соединений в организме человека значительно улучшится, а степень воздействия при пероральном введении будет выше, так что доза при клиническом введении может быть снижена, а соблюдение пациентом предписанного режима терапии может быть улучшено.
[0199] Хотя варианты осуществления настоящего раскрытия проиллюстрированы и описаны выше, можно понять, что упомянутые выше варианты осуществления являются иллюстративными и не должны рассматриваться как ограничивающие настоящее раскрытие. Специалисты в настоящей области техники могут вносить изменения, модификации, замены и вариации на основе вышеупомянутых вариантов осуществления в рамках объема настоящего раскрытия.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ПИРРОЛОПИРИМИДИНА И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2780254C1 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
| КОМПОЗИЦИИ ТЕТРАГИДРОХИНОЛИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНА ВЕТ | 2014 |
|
RU2727169C2 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ЧЕЛОВЕЧЕСКОЙ ФОСФАТИДИЛИНОЗИТ 3-КИНАЗЫ ДЕЛЬТА | 2013 |
|
RU2658006C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТРИАЗОЛОПИРАЗИНА И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2643361C2 |
| 5-ЗАМЕЩЕННЫЕ ИНДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2008 |
|
RU2487873C2 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛ-2,7-ДИАЗАСПИРО[3.5]НОНАНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ИЛИ ОБРАТНЫХ АГОНИСТОВ ГРЕЛИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2524341C2 |
| МОДУЛЯТОРЫ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2797323C2 |
| ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИНГИБИТОРЫ JAK | 2012 |
|
RU2632870C2 |
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
Изобретение относится к соединению формулы (I), его таутомеру или фармацевтически приемлемой соли, которые обладают ингибирующей активностью к аутотаксину (АТХ). В формуле (I) Q представляет инданил, необязательно замещенный m группами R1, m представляет собой целое число, выбранное из 0-8, каждая из m групп R1 независимо выбрана из водорода или галогена; X выбран из -N=; Y выбран из -C(R2)=; Z выбран из -N(R3)-; L1 выбран из 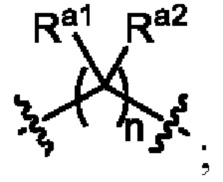 L2 выбран из
L2 выбран из 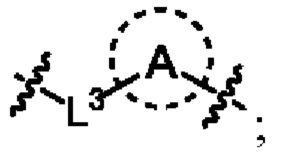 L3 выбран из одинарной связи или
L3 выбран из одинарной связи или 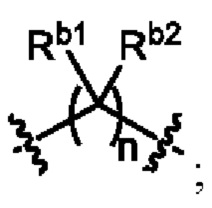 Ra1 и Ra2 каждый выбран из водорода или С1-С3 алкила; Rb1 и Rb2 независимо выбраны из водорода;
Ra1 и Ra2 каждый выбран из водорода или С1-С3 алкила; Rb1 и Rb2 независимо выбраны из водорода;  выбран из одинарной связи, С4-С8 гетероциклоалкила или семи-восьмичленной N-спироциклической группы, где С4-8 гетероциклоалкил и семи-восьмичленная N-спироциклическая группа необязательно замещены одним Rc, выбранным из галогена или C1-С3 алкила, и 1 атом кольца С4-С8 гетероциклоалкила выбран из N; n представляет собой целое число, выбранное из 0-3; М1, М2 и М3 независимо выбраны из -N= или -N(R4)-; М4 выбран из С; М5 выбран из -C(R5)=; R2 и R5 каждый выбран из водорода; R3 и R4 каждый выбран из водорода. Изобретение также относится к соединениям формул I-1 и I-9 и фармацевтической композиции, содержащей эффективную дозу соединения формулы (I) и обладающей свойством ингибирования аутотаксина. 4 н. и 4 з.п. ф-лы, 9 табл., 19 пр.
выбран из одинарной связи, С4-С8 гетероциклоалкила или семи-восьмичленной N-спироциклической группы, где С4-8 гетероциклоалкил и семи-восьмичленная N-спироциклическая группа необязательно замещены одним Rc, выбранным из галогена или C1-С3 алкила, и 1 атом кольца С4-С8 гетероциклоалкила выбран из N; n представляет собой целое число, выбранное из 0-3; М1, М2 и М3 независимо выбраны из -N= или -N(R4)-; М4 выбран из С; М5 выбран из -C(R5)=; R2 и R5 каждый выбран из водорода; R3 и R4 каждый выбран из водорода. Изобретение также относится к соединениям формул I-1 и I-9 и фармацевтической композиции, содержащей эффективную дозу соединения формулы (I) и обладающей свойством ингибирования аутотаксина. 4 н. и 4 з.п. ф-лы, 9 табл., 19 пр.
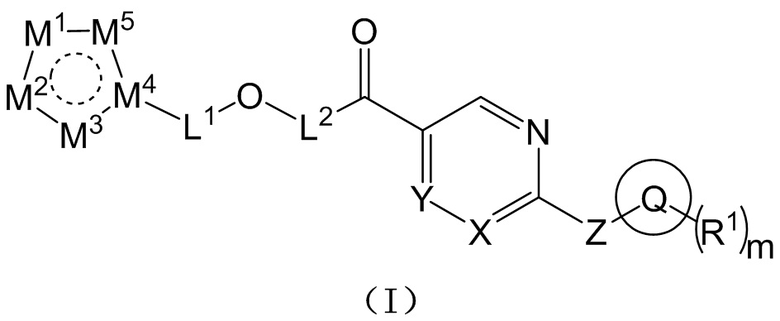
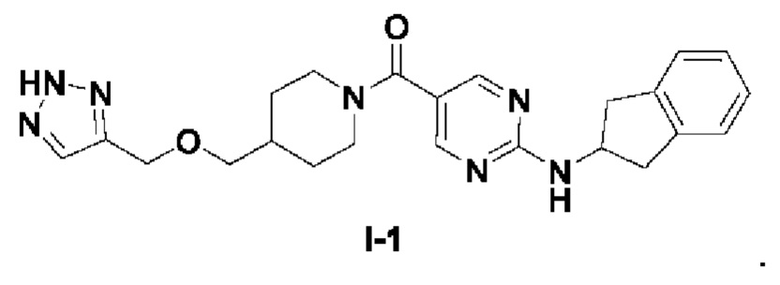
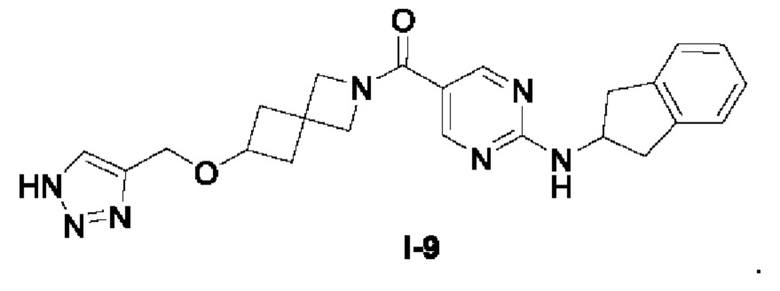
1. Соединение, причем соединение представляет собой соединение, представленное формулой (I), или представляет собой таутомер или фармацевтически приемлемую соль соединения, представленного формулой (I):
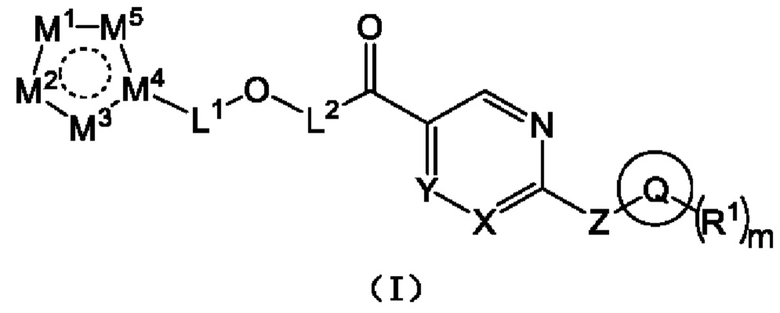 ,
,
где Q представляет собой инданил, необязательно замещенный m группами R1, m представляет собой целое число, выбранное из 0-8;
каждая из m групп R1 независимо выбрана из водорода или галогена;
X выбран из -N=;
Y выбран из -C(R2)=;
Z выбран из -N(R3)-;
L1 выбран из 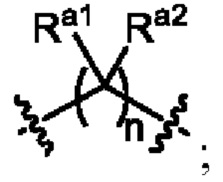
L2 выбран из 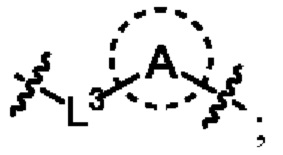
L3 выбран из одинарной связи или 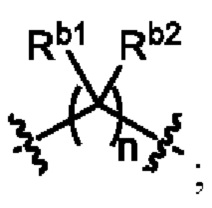
Ra1 и Ra2 выбраны из водорода или С1-С3 алкила;
Rb1 и Rb2 каждый выбран из водорода;
 выбран из одинарной связи, С4-С8 гетероциклоалкила или семи-восьмичленной N-спироциклической группы, где С4-8 гетероциклоалкил и семи-восьмичленная N-спироциклическая группа необязательно замещены одним Rc, выбранным из галогена или C1-С3 алкила, и 1 атом кольца С4-С8 гетероциклоалкила выбран из N;
выбран из одинарной связи, С4-С8 гетероциклоалкила или семи-восьмичленной N-спироциклической группы, где С4-8 гетероциклоалкил и семи-восьмичленная N-спироциклическая группа необязательно замещены одним Rc, выбранным из галогена или C1-С3 алкила, и 1 атом кольца С4-С8 гетероциклоалкила выбран из N;
n представляет собой целое число, выбранное из 0-3;
М1, М2 и М3 независимо выбраны из -N= или -N(R4)-;
М4 выбран из С;
М5 выбран из -C(R5)=;
R2 и R5 каждый выбран из водорода;
R3 и R4 каждый выбран из водорода; и
соединение не содержит ни одного из следующих соединений или их энантиомеров или стереоизомеров:
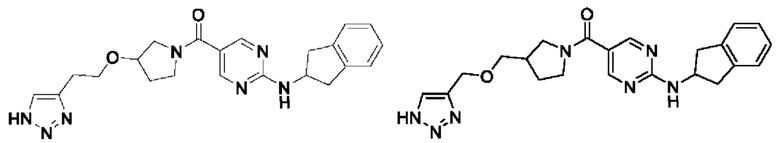
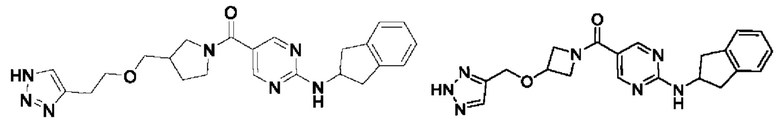
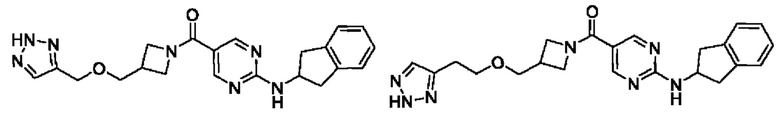
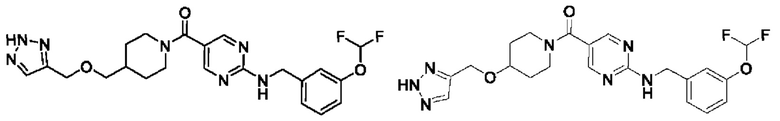
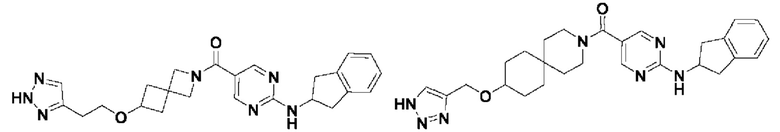
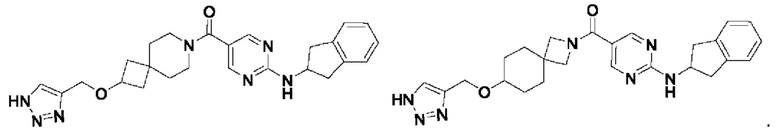
2. Соединение по п. 1, где в соединении, представленном формулой (I), m равно 0, 1, 2 или 3.
3. Соединение по п. 1, где
R1 выбран из Н, F или Cl;
необязательно n равно 1 или 2;
необязательно Ra1 и Ra2 независимо выбран из водорода, метила или этила;
необязательно Ra1 и Ra2 независимо выбран из водорода, метила или этила, и по меньшей мере один Ra1 представляет собой водород;
необязательно  выбран из пиперидинила, необязательно замещенного одним Rc, или семи-восьмичленной N-спироциклической группы;
выбран из пиперидинила, необязательно замещенного одним Rc, или семи-восьмичленной N-спироциклической группы;
необязательно  представляет собой пиперидинил, необязательно замещенный одним Rc, указанный L3 и N в пиперидиниле независимо находятся в орто-, мета- или пара-положении по отношению друг к другу;
представляет собой пиперидинил, необязательно замещенный одним Rc, указанный L3 и N в пиперидиниле независимо находятся в орто-, мета- или пара-положении по отношению друг к другу;
необязательно  представляет собой пиперидинил, необязательно замещенный одним Rc, Rc и N в пиперидиниле независимо находятся в орто- или мета-положении по отношению друг к другу;
представляет собой пиперидинил, необязательно замещенный одним Rc, Rc и N в пиперидиниле независимо находятся в орто- или мета-положении по отношению друг к другу;
необязательно  представляет собой пиперидинил, необязательно замещенный одним Rc, по меньшей мере один Rc выбран из -F, -Cl, метила или этила;
представляет собой пиперидинил, необязательно замещенный одним Rc, по меньшей мере один Rc выбран из -F, -Cl, метила или этила;
необязательно L3 представляет собой одинарную связь, и  выбран из семи-восьмичленной N-спироциклической группы;
выбран из семи-восьмичленной N-спироциклической группы;
необязательно указанный  выбран из семи-восьмичленной N-спироциклической группы, выбранной из
выбран из семи-восьмичленной N-спироциклической группы, выбранной из 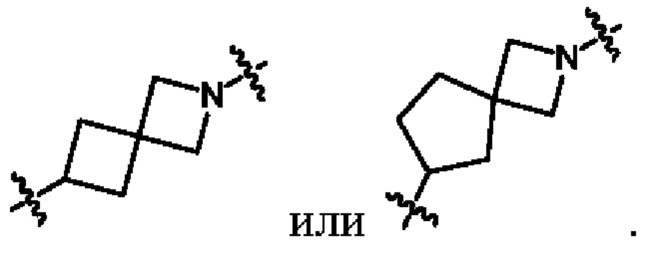
4. Соединение по п. 1, причем соединение, представленное формулой (I), представляет собой соединение, представленное следующей формулой (I-0):
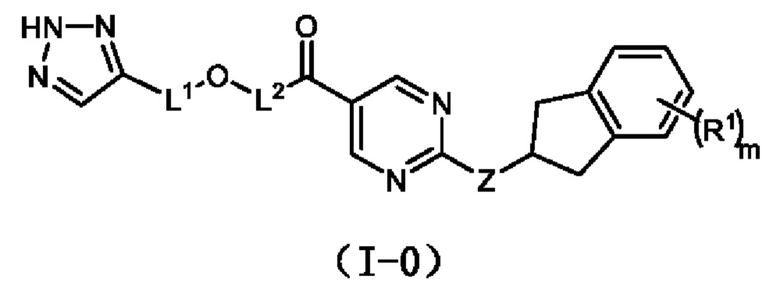 ,
,
где R1 представляет собой -F; необязательно m равно 0, 1 или 2;
необязательно L1 выбран из 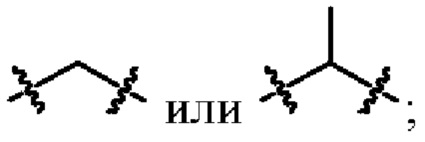
необязательно L2 выбран из 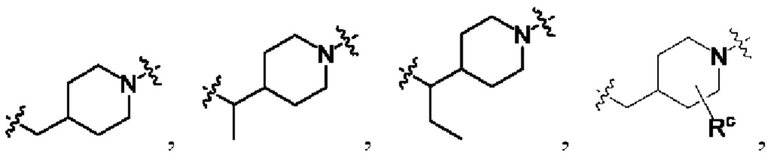
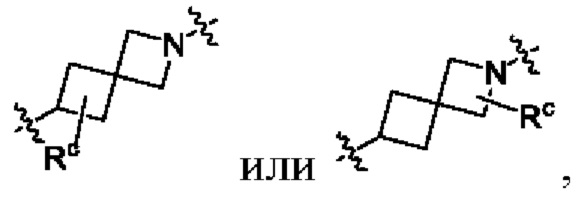 где Rc выбран из -F, -Cl, метила или этила.
где Rc выбран из -F, -Cl, метила или этила.
5. Соединение, представленное формулой (I-1), или таутомер, или фармацевтически приемлемая соль соединения, представленного формулой (I-1):
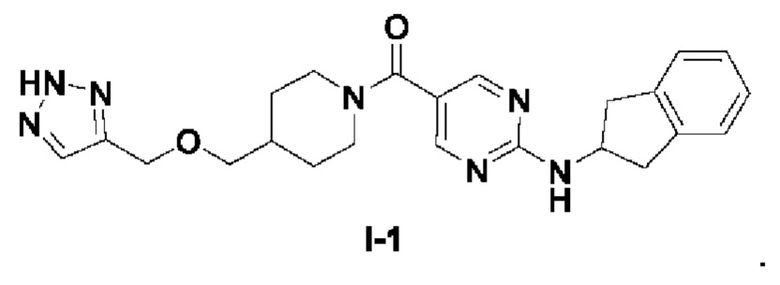
6. Соединение, представленное формулой (I-9), или таутомер, или фармацевтически приемлемая соль соединения, представленного формулой (I-9):
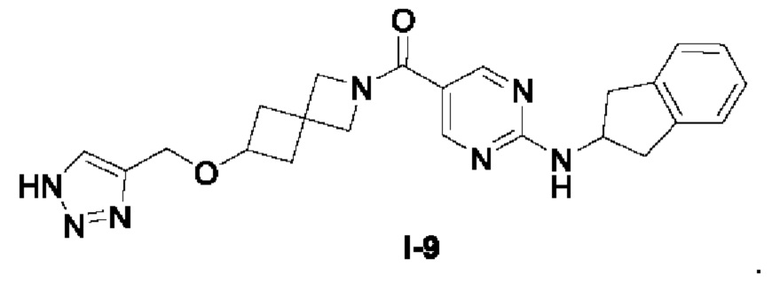
7. Соединение по п. 1, причем соединение, представленное формулой (I), представляет собой любое из следующих соединений:
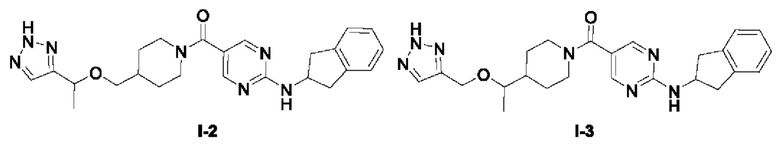
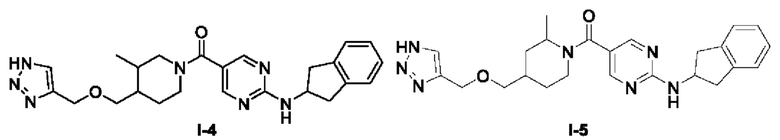
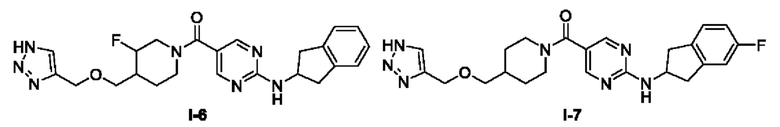
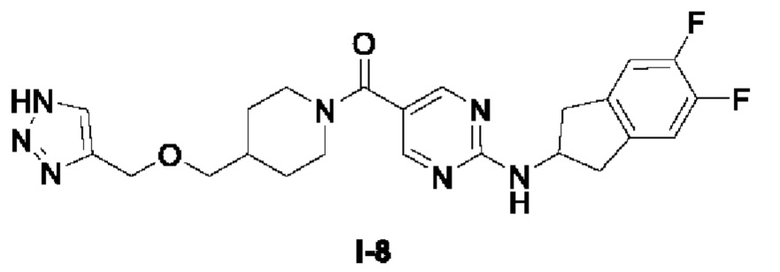
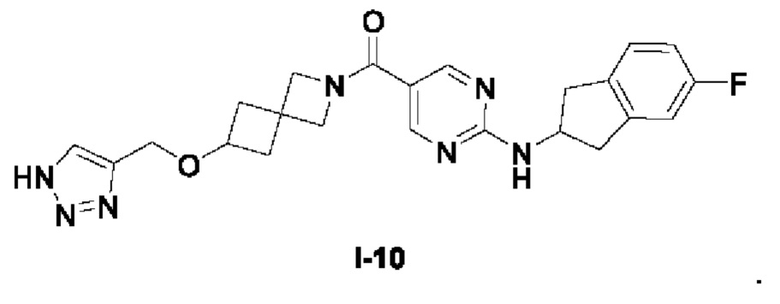
8. Фармацевтическая композиция, обладающая свойством ингибирования аутотаксина и содержащая эффективную дозу соединения по любому из пп. 1-7.
| База данных PubСhem [онлайн] NCBI, PubСhem CID 56010207 от 25.01.2012 доступно из Интернет: https://pubchem.ncbi.nlm.nih.gov/compound/56010207 | |||
| CN 103958481 A, 30.07.2014 | |||
| CN 106220572 A, 14.12.2016 | |||
| CN 106103446 A, 09.11.2016 | |||
| US 2019225611 A1, 25.07.2019 | |||
| Приспособление для профилирования дорожного полотна | 1932 |
|
SU28509A1 |

