Область техники
Настоящее изобретение относится к реагентам, в частности к алкин-содержащим амидофосфитам, полученным из 1,3-диолов, содержащих в своей структуре фрагмент 2,2-бис(гидроксиметил)уксусной кислоты, которые используются для введения алкиновой группы, необходимой для функционализации или мечения синтетических олигонуклеотидов по методу «клик-химии».
Предшествующий уровень техники
Для функционализации или мечения синтетических олигонуклеотидов удобно использовать реагенты, синтезированные на основе 1,3-диолов, которые могут применяться в любом коммерческом синтезаторе олигонуклеотидов так же, как стандартные амидофосфиты нуклеозидов и/или CPG-реагенты. При вставке в олигонуклеотиды реагентов на основе 1,3-диолов, сохраняется природное межнуклеотидное расстояние в 3 атома углерода между фосфатными группами, что позитивно сказывается на дальнейшей их химической модификации.
Амидофосфитные реагенты на основе 1,3-диолов могут быть использованы для многократного мечения и модификации синтетических олигонуклеотидов, то есть могут быть включены несколько раз, причем в произвольных позициях синтетического нуклеотида (подряд или рандомизировано). Они позволяют маркировать и модифицировать синтетические олигонуклеотиды в любом положении (3', 5' или внутри).
Наличие алкиновой группы в реагентах на основе 1,3-диолов позволяет по реакции «клик-химии» вводить любую флуоресцентную метку (I. K. Astakhova, J. Wengel, Chem. Eur. J. 2013, 19, 1112; M. Taskova, K. Astakhova, Bioconjugate Chem.2019, 30, 3007) или радиоактивную метку (J. Flagothier, G. Kaisin, F. Mercier, D. Thonon, N. Teller, J. Wouters, A. Luxen, Appl. Radiat. Isot., 2012, 70, 1549; F. Mercier, J. Paris, G. Kaisin, D. Thonon, J. Flagothier, N. Teller, C. Lemaire, A. Luxen, Bioconjugate Chem. 2011,22, 108), тем самым получая широкий набор маркеров. Реакцию введения красителя или метки по алкиновой связи можно осуществить, как до, так и после включения реагента в олигонуклеотид (N. Klöcker, F.P. Weissenboeck, A. Rentmeister, Chem. Soc. Rev., 2020, 49, 8749). Такие молекулы, как пептиды (M. Jezowska, D. Honcharenko, A. Ghidini, R. Strömberg, M. Honcharenko, Bioconjugate Chem. 2016, 27, 2620), олигонуклеотиды (S.S. Pujari, P. Leonard, F. Seela, J. Org. Chem. 2014, 79, 4423), углеводы (G. Pourceau, A. Meyer, J. J. Vasseur, F. Morvan, J. Org. Chem. 2009, 74, 1218) и биотин (K. Nakamoto, Y. Akao, Y. Ueno, Bioorg. Med. Chem. Lett. 2018, 28, 2906), также могут быть присоединены по алкиновой группе.
Известно несколько подходов по получению на основе 1,3-диолов амидофосфитных реагентов, содержащих алкиновую группу. Первые, и до сих пор наиболее распространенные работы, связаны непосредственно с модификацией нуклеозидов путём введения линкера с алкиновой группой по 2'-положению(M. Grøtli, M. Douglas, R. Eritja, B.S. Sproat, Tetrahedron, 1998, 54, 5899; S.S. Pujari, P. Leonard, F. Seela, J. Org. Chem. 2014, 79, 4423; H. K. Walter, B. Olshausen, U. Schepers, H.A. Wagenknecht, Beilstein J. Org. Chem. 2017,13,127). В последнее время для этих целей широко используются восстановленные производные 4-гидрокси-L-пролина (J. Flagothier, G. Kaisin, F. Mercier, D. Thonon, N. Teller, J. Wouters, A. Luxen, Appl. Radit. Isot., 2012, 70, 1549; Yu.V. Martynenko-Makaev, V.V. Udodova, O. L. Sharko, V.V. Shmanai, Russ. J. Gen. Chem., 2018, 88, 452; P.K. Yuen, S.A. Green, J. Ashby, K.T. Lay, A. Santra, X. Chen, M.P. Horvath, S.S. David, ACS Chem. Biol. 2019, 14, 27). В литературе описан способ синтеза подобных амидофосфитных реагентов из глюкозы в 6 стадий (M. Taskova, K. Astakhova, Bioconjugate Chem. 2019, 30, 3007) или возможность их получения из серинола (патентная заявка US 20110077389 A1 от 31.03.2011).
Недостатками данных способов являются трудоемкость синтеза, заключающаяся в многостадийности, необходимости применения значительного количества стадий по введению и снятию защитных групп или использование исходных соединений, которые сами по себе могут быть крайне редкими и дорогостоящими продуктами.
Краткое описание настоящего изобретения
Целью настоящего изобретения является предоставление нового алкин-содержащего амидофосфита, предназначенного для функционализации синтетических олигонуклеотидов методом азид-алкинового циклоприсоединения («клик-химии»), а также способа его получения.
Указанная цель была достигнута путём создания нового алкин-содержащего амидофосфита, а также путём разработки простого и удобного способа его получения с использованием простых и доступных реагентов.
В одном аспекте настоящее изобретение предоставляет алкин-содержащий амидофосфит, предназначенный для функционализации синтетических олигонуклеотидов методом азид-алкинового циклоприсоединения, имеющий следующую общую структуру:
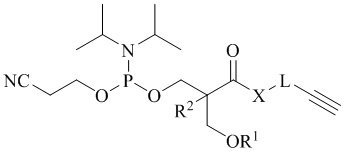
где R1 представляет собой защитную группу, стабильную в условиях синтеза олигонуклеотидов, R2 представляет собой водород, метил или этил, X представляет собой -О- или -NH-, и L представляет собой спейсерную группу. В одном из вариантов настоящего изобретения защитная группа, стабильная в условиях синтеза олигонуклеотидов, выбрана из 4,4'-диметокситритильной (DMTr), 4-монометокситритильной (MMTr), 9-фенилксантен-9-ильной (Px), тритильной (Tr), триметокситритильной (TMTr), карбонатной и силильной групп.
В одном из вариантов настоящего изобретения L представляет собой представляет собой спейсерную группу -(CH2)-n, где n = 1, 2 или 3; или -(CH2CH2O)nCH2, где n = 1 или 2.
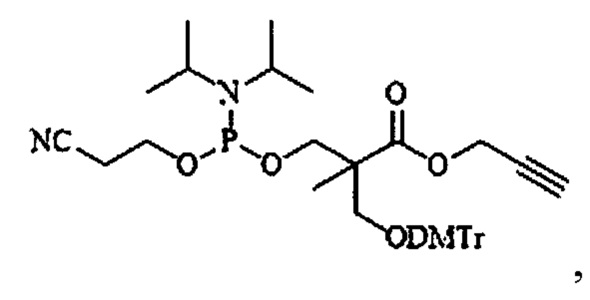
В одном из конкретных вариантов настоящего изобретения указанный алкин-содержащий амидофосфит выбран из группы, включающей соединения, имеющие следующие структурные формулы:
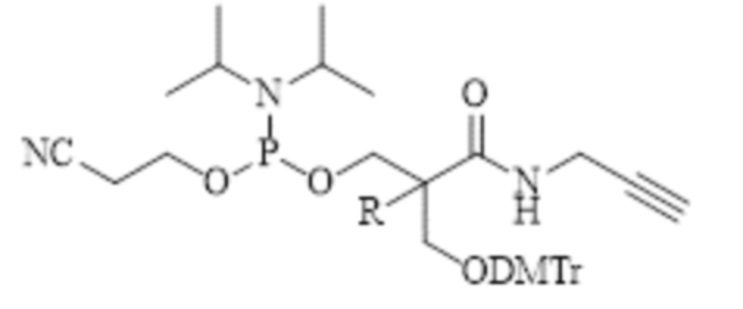 , где R представляет собой метил или этил;
, где R представляет собой метил или этил;
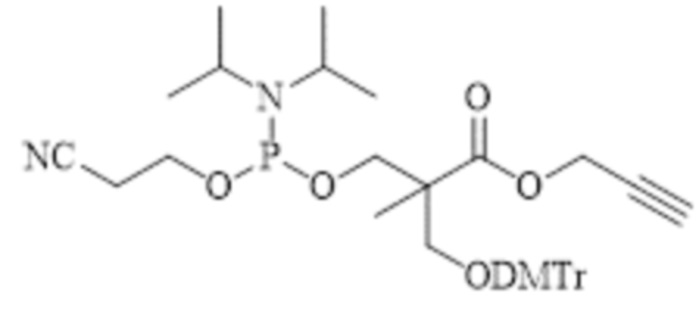 , и
, и
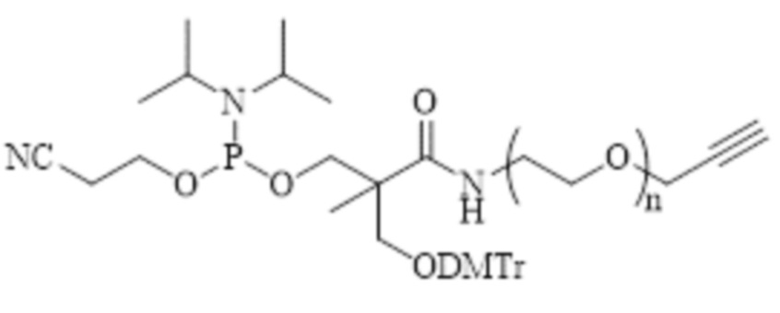 , где n = 1 или 2.
, где n = 1 или 2.
В другом аспекте настоящее изобретение предоставляет способ получения указанного алкин-содержащего амидофосфита, включающий стадии:
1) взаимодействия 2,2-бис(гидроксиметил)карбоновой кислоты с соединением формулы HC≡C(CH2)n-NH2, где n=1, 2 или 3, или с соединением формулы HC≡CCH2(OCH2CH2)n-NH2
2) где n = 1 или 2, посредством активации карбоксильной группы в присутствии N-гидроксисукцинимида и дициклогексилкарбодиимида в случае, когда Х представляет собой -NH-; или с соединением формулы HC≡C(CH2)nBr, где n=1, 2 или 3 в присутствии КОН в случае, когда Х представляет собой -О-;
2) взаимодействия соединения, полученного на стадии 1, с субэквимолярным количеством реагента для защиты одной гидроксильной группы соединения, полученного на стадии 1; и
3) взаимодействия соединения, полученного на стадии 2, с цианоэтил-N,N-диизопропилхлорамидофосфитом.
В одном из вариантов настоящего изобретения в качестве 2,2-бис(гидроксиметил)карбоновой кислоты используют 2,2-бис(гидроксиметил)пропионовую кислоту или 2,2-бис(гидроксиметил)масляную кислоту.
В одном из вариантов настоящего изобретения соединением формулы HC≡C(CH2)n-NH2 является пропаргиламин.
В одном из вариантов настоящего изобретения соединением формулы HC≡CCH2(OCH2CH2)n-NH2 является 2-(2-пропинилокси)этиламин или пропаргил-PEG2-амин.
В одном из вариантов настоящего изобретения на стадии 1 для активации карбоксильной группы используют N-гидроксисукцинимид и дициклогексилкарбодиимид в эквимолярных количествах.
В одном из вариантов настоящего изобретения соединением формулы HC≡C(CH2)nBr является пропаргилбромид.
В одном из вариантов настоящего изобретения в качестве реагента для защиты гидроксильной группы на стадии 2 используют 4,4'-диметокситритилхлорид (DMTrCl), 4-монометокситритилхлорид (MMTrCl), 9-фенилксантен-9-илхлорид, тритилхлорид (TrCl), триметокситритилхлорид (TMTrCl), карбонаты или силилхлориды.
Алкин-содержащие амидофосфиты согласно настоящему изобретению могут быть использованы в химии нуклеиновых кислот для до или пост-синтетического введения флуоресцентных красителей и гасителей флуоресценции, биотина и других функциональных молекул в состав синтетических олигонуклеотидов (ДНК или РНК) с целью дальнейшего использования таких фрагментов в качестве гибридизационных зондов в полимеразной цепной реакции в реальном времени, для секвенирования ДНК и РНК, для in vivo и ex vivo гибридизации, для конъюгации биологических макромолекул за счет образования комплексов биотин-стрептавидин и др.
Настоящее изобретение позволяет преодолеть недостатки, встречающиеся в решениях, известных из уровня техники, путем упрощения и удешевления процессов получения реагентов, пригодных для прямой модификации или мечения олигонуклеотидов, при этом указанные реагенты можно легко включать в олигонуклеотиды в процессе их автоматического синтеза вместо любого нуклеозида, как 5'- или 3'-концевого, так и внутреннего.
Подробное описание настоящего изобретения
Настоящее изобретение предоставляет алкин-содержащий амидофосфит, предназначенный для функционализации синтетических олигонуклеотидов методом азид-алкинового циклоприсоединения, имеющий следующую структурную формулу:
,
где R1 представляет собой защитную группу, выбранную из 4,4'-диметокситритильной (DMTr), 4-монометокситритильной (MMTr) или 9-фенилксантен-9-ильной (Px) групп, R2 представляет собой водород, метил или этил, X представляет собой -О- или -NH-, и L представляет собой спейсерную группу.
Выбор кислот из ряда, содержащего во 2 положении два гидроксиметильных заместителя, обусловлен их доступностью. Исходные соединения, содержащие фрагмент 2,2-бис(гидроксиметил)уксусной кислоты обладают универсальной структурой, позволяющей всего в три стадии получать целевые продукты. В частности, подобные производные не требуют использования лишних стадий по присоединению и снятию защитных групп, наличие карбоксильной группы позволяет в одну стадию вводить алкиновую группу на заданном расстоянии от нее за счет спейсерной группы (L) нужной структуры или размера, обладающей необходимой гидрофобностью или гидрофильностью. В свою очередь, наличие алкиновой группы позволяет в дальнейшем по реакции «клик-химии» вводить в состав синтетического олигонуклеотида любую целевую молекулу (флуоресцентный краситель, гаситель флуоресценции, биотин, радиоактивную метку и др.) в виде соответствующего азидного производного.
Структура и размер указанной спейсерной группы (L) может варьировать в зависимости от целей, которые необходимо достигнуть, и задач, которые необходимо решить, с использованием алкин-содержащего олигонуклеотида. Специалист в данной области техники может легко подобрать структуру и размер указанной спейсерной группы (L), исходя из поставленных целей.
Конкретными вариантами спейсерной группы (L) могут быть группы -(CH2)-n, где n = 1, 2 или 3, или -(CH2CH2O)nCH2-, где n = 1 или 2, но не ограничиваются ими.
Группы, используемые для защиты гидроксильной группы в рамках настоящего изобретения, включают любые группы, хорошо известные специалистам в области автоматического синтеза олигонуклеотидов, например, 4,4'-диметокситритильную (DMTr), 4-монометокситритильную (MMTr), 9-фенилксантен-9-ильную (пиксильную, Px) тритильную (Tr), триметокситритильную (TMTr), карбонатную или силильную группы. Предпочтительно использовать защитные группы, которые быстро и количественно удаляются в процессе автоматического синтеза олигонуклеотидов обработкой ди- или трифторуксусной кислотой в безводном дихлорметане, например,
4,4'-диметокситритильную (DMTr), 4-монометокситритильную (MMTr), 9-фенилксантен-9-ильную (пиксильную, Px) группы. Наиболее предпочтительной и традиционно используемой защитной группой является DMTr-группа.
Конкретными примерами алкин-содержащего амидофосфита согласно настоящему изобретению являются соединения, имеющие следующие структурные формулы:
, где R представляет собой метил или этил;
, и
, где n = 1 или 2.
Следует понимать, что указанные соединения приведены исключительно в иллюстративных целях и ими не ограничиваются рамки настоящего изобретения, определяемые формулой изобретения. Специалист в данной области техники на основании настоящего описания легко может получить другие варианты соединений, подпадающих под общую структурную формулу соединения, указанную выше.
Наличие алкиновой группы в соединениях согласно настоящему изобретению, полученных на основе 1,3-диолов, позволяет по реакции «клик-химии» вводить любую метку, например, флуоресцентную метку (I. K. Astakhova, J. Wengel, Chem. Eur. J. 2013, 19, 1112; M. Taskova, K. Astakhova, Bioconjugate Chem. 2019, 30, 3007) или радиоактивную метку (J. Flagothier, G. Kaisin, F. Mercier, D. Thonon, N. Teller, J. Wouters, A. Luxen, Appl. Radiat. Isot., 2012, 70, 1549; F. Mercier, J. Paris, G. Kaisin, D. Thonon, J. Flagothier, N. Teller, C. Lemaire, A. Luxen, Bioconjugate Chem. 2011,22, 108), тем самым получая широкий набор маркеров. Реакцию введения красителя или метки по алкиновой связи можно осуществить, как до, так и после включения реагента в олигонуклеотид (N. Klöcker, F.P. Weissenboeck, A. Rentmeister, Chem. Soc. Rev., 2020, 49, 8749). Такие молекулы, как пептиды (M. Jezowska, D. Honcharenko, A. Ghidini, R. Strömberg, M. Honcharenko, Bioconjugate Chem. 2016, 27, 2620), олигонуклеотиды (S.S. Pujari, P. Leonard, F. Seela, J. Org. Chem. 2014, 79, 4423), углеводы (G. Pourceau, A. Meyer, J. J. Vasseur, F. Morvan, J Org. Chem. 2009, 74, 1218) и биотин (K. Nakamoto, Y. Akao, Y. Ueno, Bioorg. Med. Chem. Lett. 2018, 28, 2906), также могут быть присоединены к олигонуклеотиду, содержащему алкиновую группу.
Также настоящее изобретение предоставляет способ получения указанного алкин-содержащего амидофосфита, включающий стадии:
1) взаимодействия 2,2-бис(гидроксиметил)карбоновой кислоты с соединением формулы HC≡C(CH2)n-NH2, где n=1, 2 или 3, или с соединением формулы HC≡CCH2(OCH2CH2)n-NH2, где n = 1 или 2, посредством активации карбоксильной группы в присутствии N-гидроксисукцинимида и дициклогексилкарбодиимида в случае, когда Х представляет собой -NH-; или с соединением формулы HC≡C(CH2)nBr, где n=1, 2 или 3 в присутствии КОН в случае, когда Х представляет собой -О-;
2) взаимодействия соединения, полученного на стадии 1, с субэквимолярным количеством реагента для защиты одной гидроксильной группы соединения, полученного на стадии1;
3) взаимодействия соединения, полученного на стадии 2, с цианоэтил-N,N-диизопропилхлорамидофосфитом.
В качестве 2,2-бис(гидроксиметил)карбоновой кислоты можно использовать такие кислоты, как 2,2-бис(гидроксиметил)пропионовая кислота или 2,2-бис(гидроксиметил)масляная кислота, но список кислот, которые возможно использовать в способе согласно настоящему изобретению, не ограничивается ими.
В одном из вариантов настоящего изобретения соединением формулы HC≡C(CH2)n-NH2 является пропаргиламин. В другом варианте настоящего изобретения соединением формулы HC≡CCH2(OCH2CH2)n-NH2 является 2-(2-пропинилокси)этиламин или пропаргил-PEG2-амин.
Для активации карбоксильной группы для последующего присоединения соединения, содержащего аминогруппу, используют любые методы, известные специалистам в данной области техники, в частности, метод активации карбоксильной группы смесью N-гидроксисукцинимида и дициклогексилкарбодиимида в эквимолярных количествах.
В одном из вариантов настоящего изобретения соединением формулы HC≡C(CH2)nBr является пропаргилбромид. В случае использования соединений, содержащих бром, реакцию присоединения проводят в присутствии КОН в полярном органическом растворителе, например, в растворе ДМФА.
Далее для защиты одной из гидроксильных групп соединения, полученного на первой стадии синтеза, используют 4,4'-диметокситритилхлорид (DMTrCl),
4-монометокситритилхлорид (MMTrCl), 9-фенилксантен-9-илхлорид, тритилхлорид(TrCl), триметокситритилхлорид (TMTrCl), карбонаты или силилхлориды. Предпочтительным и наиболее традиционным является использование 4,4'-диметокситритилхлорида (DMTrCl).
Вторую оставшуюся гидроксильную группу соединения, полученного на второй стадии синтеза, функционализируют амидофосфитом. Предпочтительным и наиболее традиционным является использование цианоэтил-N,N-диизопропилхлорамидофосфита.
Далее в качества вариантов способа получения алкин-содержащих амидофосфитов согласно настоящему изобретению приведены три схемы их синтеза.
Схема 1
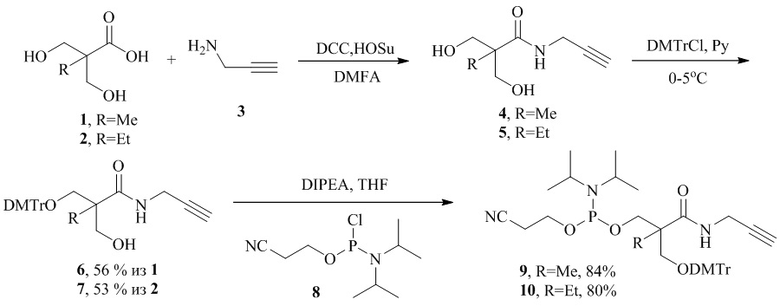
На первом этапе 2,2-бис(гидроксиметил)пропионовую кислоту 1 или 2,2-бис(гидроксиметил)масляную кислоту 2 сразу функционализируют по карбоксильной группе пропаргиламином 3 через стадию активации карбоксильной группы в присутствии эквимолярных количеств N-гидроксисукцинимида и дициклогексилкарбодиимида в растворе диметилформамида (ДМФА). Последующую селективную защиту одной первичной гидроксильной группы в пропаргиламиде 2,2-бис-(гидроксиметил)пропионовой кислоты 4 или 2,2-бис(гидроксиметил)масляной кислоты 5 проводят действием 0,75 эквивалентов 4,4'-диметокситритилхлорида (DMTrCl) в пиридине при температуре 0-5°С с получением монозамещенных продуктов 6 и 7 соответственно. Заключительную активацию второй гидроксильной группы осуществляют обработкой продуктов 6 и 7 амидофосфитом 8. В результате получают целевые реагенты 9 и 10 с общим выходом 47% и 42% по трем стадиям соответственно.
Схема 2
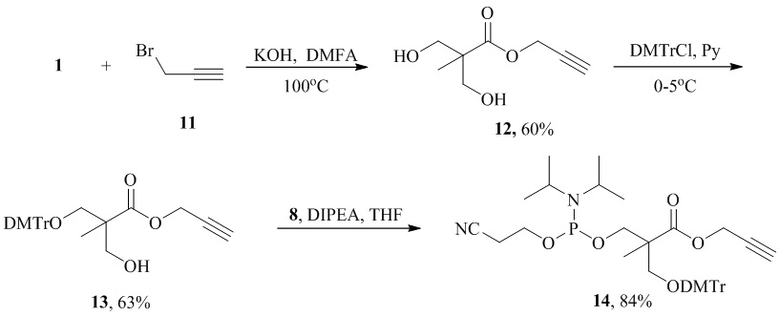
При получении реагента 14, содержащего сложноэфирную функциональную группу, этерификацию 2,2-бис(гидроксиметил)пропионовой кислоты 1 проводят по известной методике с использованием пропаргилбромида 11 в присутствии KOH в растворе ДМФА. Последующие стадии постепенного блокирования первичных гидроксильных групп с применением DMTrCl и амидофосфита 8 осуществляют аналогично, как и в случаях с амидными производными. Общий выход продукта 14 составляет 32%.
Схема 3
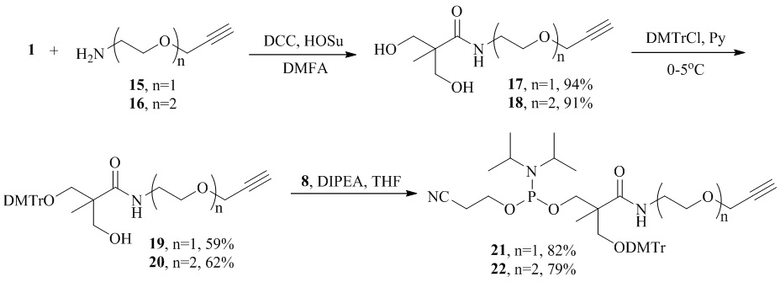
Получение амидофосфитных производных 21 и 22, содержащих более длинную линейную спейсерную группу, осуществляют с использованием
2-(2-пропинилокси)этиламина 15, пропаргил-PEG2-амина 16 и
2,2-бис(гидроксиметил)пропионовой кислоты 1. При синтезе целевых реагентов 21 и 22 применяют аналогичные методы амидирования карбоксильной группы, селективной защиты и активации первичных гидроксильных групп, как в случае получения соединения 9 из кислоты 1. В этих примерах общий выход продуктов 21 и 22 составил 45%.
Все предложенные в настоящем изобретении реагенты, содержащие алкиновую группу, могут быть получены с любой защитной группой, стабильной в условиях синтеза олигонуклеотидов. Вместо DMTr группы могут быть использованы другие триарилметильные группы, например, тритильная Tr, метокситритильная MMTr, триметокситритильная TMTr, а также 9-фенилксантен-9-ильные, карбонатные или силильные защитные группы. Способ синтеза таких реагентов может быть осуществлен в три стадии с высоким выходом аналогично описанному. Для всех этих защитных групп хорошо отработаны методы их введения и удаления с гидроксильных групп и показаны возможности использования подобных реагентов в олигнуклеотидном синтезе (H. Seliger, Protection of 5′-Hydroxy Functions of Nucleosides, Current Protocols in Nucleic Acid Chemistry, 2000, 2.3.1-2.3.34).
Таким образом, преимуществами предлагаемого способа получения амидофосфитных реагентов, содержащих алкиновую группу, по сравнению с описанными, являются простота, меньшая трудоёмкость, практичность и экономичность достигаемые за счёт использования простых, недорогих коммерчески доступных исходных соединений.
Далее приведены примеры вариантов осуществления настоящего изобретения. Указанные иллюстративные примеры приведены исключительно для целей разъяснения сути настоящего изобретения и не ограничивают каким-либо образом рамки настоящего изобретения, определяемые формулой настоящего изобретения.
Примеры
Пример 1. Синтез амидофосфитного реагента 9 (R1=DMTr, R2=Me, X=NH, L=CH2).
Синтез пропаргиламида 2,2-бис(гидроксиметил)пропионовой кислоты ( 4 ).
К раствору (10,0 г, 74,55 ммоль) 2,2-бис-(гидроксиметил)пропионовой кислоты 1 в 100 мл безводного ДМФА при комнатной температуре добавляли (15,4 г, 74,55 ммоль) дициклогексилкарбодиимида (DCC) и (8,63 г, 74,9 ммоль) N-гидроксисукцинимида (HOSu). Реакционную массу перемешивали в течение 3 ч при комнатной температуре. Выпавший осадок отфильтровывали, осадок промывали ДМФА. В маточный раствор при перемешивании добавляли (10,25 мл, 160,0 ммоль) пропаргиламина 3. Реакционную массу перемешивали в течение 5 мин. Растворитель упаривали при пониженном давлении. Получали 26,0 г концентрата пропаргиламида 2,2-бис-(гидроксиметил)-пропионовой кислоты 4, чистота продукта - 45-49%.
Синтез 3-[бис(4-метоксифенил)(фенил)метокси]-2-(гидроксиметил)-2-метил-N-(проп-2-ин-1-ил)пропанамида ( 6 ).
К охлажденному до 0°С раствору (26,0 г, 74,55 ммоль) концентрата пропаргиламида 2,2-бис(гидроксиметил)пропионовой кислоты 4 в 400 мл безводного пиридина при перешивании порциями в течение 4-х часов добавляли (18,9 г, 55,9 ммоль) 4,4'-диметокситритилхлорида (DMTrCl). Реакционную массу перемешивали в течение 24 ч, далее добавляли 20 мл сухого МеОН и дополнительно раствор перемешивали в течение 30 мин. Растворитель упаривали, остаток растворяли в 300 мл хлороформа. Раствор промывали последовательно насыщенным раствором NaHCO3 (~200 мл) и насыщенным раствором NaСl (~200 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход продукта - 14,82 г (56% из 1).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,45 (dd, J = 8,4, 1,3 Гц, 2H, Ph-2), 7,30-7,37 (m, 6H, Ph-3, PhOMe-2), 7,24 (tt, J = 6,9, 1,3 Гц, 1H, Ph-4), 6,87 (ddd, J = 9,0, 3,2, 1,9 Гц, 4H, PhOMe-3), 4,11 (ddd, J = 17,6, 5,4, 2,6 Гц, 1H, CH2N), 4,01 (ddd, J = 17,6, 4,9, 2,6 Гц, 1H, CH2N), 3,81 (s, 6H, ОCH3), 3,66 (d, J = 11,3 Гц, 1H, CH2OH), 3,62 (d, J = 11,3 Гц, 1H, CH2OH), 3,35 (d, J = 9,3 Гц, 1H, CH2ODMTr), 3,28 (br.s, 1H, OH), 3,23 (d, J = 9,3 Гц, 1H, CH2ODMTr), 2,20 (t, J = 2,6 Гц, 1H, CH≡), 1,19 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ 176,04 (CO), 158,83, 158,81 (4-PhOMe), 144,46 (1-Ph), 135,45, 135,21 (1-PhOMe), 130,17 (2-PhOMe), 128,23 (2-Ph), 128,08 (3-Ph), 127,17 (4-Ph), 113,53, 113,52 (3-PhOMe), 87,11 (C-Ph), 79,69 (C≡), 71,74 (CH≡), 68,04 (CH2OTr), 67,57 (CH2OH), 55,38 (OCH3), 47,50 (CCO), 29,23 (CH2N), 18,86 (CH3).
Синтез 2-{[бис(4-метоксифенил)(фенил)метокси]метил}-2-[(проп-2-ин-1-ил)карбамоил]пропил (2-цианоэтил) диизопропиламидофосфита ( 9 ).
К раствору (5,25 г, 11,08 ммоль) диметокситритилового эфира 6 в 180 мл тетрагидрофурана (ТГФ) при перешивании добавляли (4,67 мл, 26,82 ммоль) N,N-диизопропилэтиламина (DIPEA) и по каплям (4,44 мл, 18,83 ммоль) 2-цианоэтил-N,N-диизопропилхлорамидофосфита 8. Реакционную массу перемешивали в течение 1 часа при комнатной температуре. Далее осадок отфильтровывали, промывали небольшим количеством ТГФ. Органический слой упаривали, остаток растворяли в 100 мл хлороформа. Раствор 1 раз промывали фосфатным буфером pH=7,0 (~100 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход продукта - 6,29 г (84%).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,44-7,48 (m, 2H, Ph-2), 7,33-7,37 (m, 4H, PhOMe-2), 7,29-7,33 (m, 2H, Ph-3), 7,21-7,26 (m, 1H, Ph-4), 6,84-6,88 (m, 4H, PhOMe-3), 3,99-4,04 (m, 2H, CH2N), 3,81 (s, 6H, OCH3), 3,66-3,81 (m, 4H, CH2O), 3,56 (h, J = 6,8 Гц, 1H, CHMe2), 3,31 (d, J = 9,2 Гц, 1H, CH2ODMTr), 3,26 (d, J = 9,2 Гц, 1H, CH2ODMTr), 2,53-2,59 (m, 2H, CH2CN), 2,17 (t, J = 2,6 Гц, 1H, CH≡), 1,27 (s, 3H, CH3), 1,18 (d, J = 6,8 Гц, 6H, CH3), 1,14 (d, J = 6,8 Гц, 3H, CH3), 1,12 (d, J = 6,8 Гц, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ 174,40, 174,31 (CO); 158,71 (4-PhOMe); 144,76, 144,73 (1-Ph); 135,76, 135,73, 135,66, 135,62 (1-PhOMe); 130,27, 130,25 (2-PhOMe); 128,22 (2-Ph); 128,06 (3-Ph); 127,02 (4-Ph); 117,75, 117,72 (CN); 113,37 (3-PhOMe); 86,60, 86,56 (CPh); 79,89, 79,84 (C≡); 71,57, 71,54 (CH≡); 67,19 (d, J = 7,4 Гц, CCH2OP), 67,06(d, J = 7,4 Гц, CCH2OP); 65,95, 65,72 (CH2ODMTr); 58,51 (d, J = 3,1 Гц, CH2OP), 58,65 (d, J = 3,5 Гц, CH2OP); 55,37 (OCH3); 48,16 (d, J = 7,8 Гц, CCO), 48,06 (d, J = 7,9 Гц, CCO); 43,31, 43,30, 43,41, 43,39 (CHCH3); 29,27, 29,25 (CH2N), 24,78, 24,75, 24,73, 24,69 (CHCH3); 20,54, 20,49 (CH2CN); 18,86, 18,75 (CH3). 31P-ЯМР (202 МГц, CDCl3) δ 148,61, 148,51.
Пример 2. Синтез амидофосфитного реагента 10 (R1=DMTr, R2=Et, X=NH, L=CH2).
Синтез пропаргиламида 2,2-бис(гидроксиметил)масляной кислоты ( 5 ).
К раствору (3,0 г, 20,25 ммоль) 2,2-бис-(гидроксиметил)масляной кислоты 2 в 30 мл безводного ДМФА при комнатной температуре добавляли (4,2 г, 20,25 ммоль) DCC и (2,34 г, 20,35 ммоль) HOSu. Реакционную массу перемешивали в течение 3 ч при комнатной температуре. Выпавший осадок отфильтровывали, осадок промывали ДМФА. В маточный раствор при перемешивании добавляли (2,78 мл, 43,5 ммоль) пропаргиламина 3. Реакционную массу перемешивали в течение 5 мин. Растворитель упаривали при пониженном давлении. Получали 7,8 г концентрата пропаргиламида 2,2-бис-(гидроксиметил)масляной кислоты 5, чистота продукта - 45-49%.
Синтез 2-{[бис(4-метоксифенил)(фенил)метокси]метил}-2-(гидроксиметил)-N-(проп-2-ин-1-ил)бутанамида ( 7 ).
К охлажденному до 0°С раствору (7,8 г, 20,25 ммоль) концентрата пропаргиламида 2,2-бис(гидроксиметил)масляной кислоты 5 в 150 мл безводного пиридина при перешивании порциями в течение 4-х часов добавляли (5,15 г, 15,2 ммоль) DMTrCl. Реакционную массу перемешивали в течение 24 ч, далее добавляли 10 мл сухого МеОН и дополнительно раствор перемешивали в течение 30 мин. Растворитель упаривали, остаток растворяли в 150 мл хлороформа. Раствор 1 раз промывали насыщенным раствором NaHCO3 (~150 мл), 1 раз насыщенным раствором NaСl (~150 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход продукта - 3,90 г (53% из 2).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,45 (dd, J = 8,4, 1,3 Гц, 2H, Ph-2), 7,30-7,37 (m, 6H, Ph-3, PhOMe-2), 7,24 (tt, J = 6,9, 1,3 Гц, 1H, Ph-4), 6,87 (ddd, J = 9,0, 3,2, 1,9 Гц, 4H, PhOMe-3), 4,11 (ddd, J = 17,6, 5,4, 2,6 Гц, 1H, CH2N), 4,01 (ddd, J = 17,6, 4,9, 2,6 Гц, 1H, CH2N), 3,81 (s, 6H, ОCH3), 3,67 (d, J = 11,3 Гц, 1H, CH2OH), 3,61 (d, J = 11,3 Гц, 1H, CH2OH),3,35 (d, J = 9,3 Гц, 1H, CH2ODMTr), 3,35 (br.s, 1H, OH), 3,23 (d, J = 9,3 Гц, 1H, CH2ODMTr), 2,19 (t, J = 2,6 Гц, 1H, CH≡), 1,81 (q, J = 7,5 Гц, 2H, CH2CH3), 0,90 (t, J = 7,4 Гц, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ 176,10 (CO), 158,83, 158,81 (4-PhOMe), 144,46 (1-Ph), 135,45, 135,21 (1-PhOMe), 130,17 (2-PhOMe), 128,23 (2-Ph), 128,08 (3-Ph), 127,17 (4-Ph), 113,53, 113,52 (3-PhOMe), 87,11 (C-Ph), 79,72 (C≡), 71,76 (CH≡), 67,97 (CH2ODMTr), 67,62 (CH2OH), 55,38 (OCH3), 51,90 (CCO), 29,23 (CH2N), 21,22 (CH2) 8,80 (CH3).
Синтез 2-{[бис(4-метоксифенил)(фенил)метокси]метил}-2-[(проп-2-ин-1-ил)-карбамоил]бутил (2-цианоэтил) диизопропиламидофосфита ( 10 ).
К раствору (3,90 г, 8,00 ммоль) диметокситритилового эфира 7 в 100 мл ТГФ при перешивании добавляли (3,37 мл, 19,36 ммоль) DIPEA и по каплям (3,03 мл, 13,60 ммоль) 2-цианоэтил-N,N-диизопропилхлорамидофосфита 8. Реакционную массу перемешивали в течение 1 часа при комнатной температуре. Далее осадок отфильтровывали, промывали небольшим количеством ТГФ. Органический слой упаривали, остаток растворяли в 100 мл хлороформа. Раствор 1 раз промывали фосфатным буфером pH=7,0 (~100 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход продукта - 4,40 г (80%).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,44-7,48 (m, 2H, Ph-2), 7,33-7,37 (m, 4H, PhOMe-2), 7,29-7,33 (m, 2H, Ph-3), 7,21-7,26 (m, 1H, Ph-4), 6,84-6,88 (m, 4H, PhOMe-3), 3,99-4,04 (m, 2H, CH2N), 3,81 (s, 6H, OCH3), 3,65-3,80 (m, 4H, CH2O), 3,56 (h, J = 6,8 Гц, 2H, CHMe2), 3,31 (d, J = 9,2 Гц, 1H, CH2ODMTr), 3,24 (d, J = 9,2 Гц, 1H, CH2ODMTr), 2,53-2,59 (m, 2H, CH2CN), 2,17 (t, J = 2,6 Гц, 1H, CH≡), 1,81 (q, J = 7,5 Гц, 2H, CH2CH3), 1,18 (d, J = 6,8 Гц, 6H, CH3), 1,14 (d, J = 6,8 Гц, 3H, CH3), 1,12 (d, J = 6,8 Гц, 3H, CH3),0,90 (t, J = 7,4 Гц, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ 174,40, 174,31 (CO); 158,71 (4-PhOMe); 144,76, 144,73 (1-Ph); 135,76, 135,73, 135,66, 135,62 (1-PhOMe); 130,27, 130,25 (2-PhOMe); 128,22 (2-Ph); 128,06 (3-Ph); 127,02 (4-Ph); 117,75, 117,72 (CN); 113,37 (3-PhOMe); 86,60, 86,56 (CPh); 79,89, 79,84 (C≡); 71,57, 71,54 (CH≡); 67,15 (d, J = 7,4 Гц, CCH2OP), 67,08(d, J = 7,4 Гц, CCH2OP); 65,95, 65,72 (CH2OTr); 58,51 (d, J = 3,1 Гц, CH2OP), 58,66 (d, J = 3,5 Гц, CH2OP); 55,37 (OCH3); 51,96 (d, J = 7,8 Гц, CCO), 51,86 (d, J = 7,9 Гц, CCO); 43,31, 43,30, 43,41, 43,39 (CHCH3); 29,29, 29,27 (CH2N), 24,78, 24,75, 24,73, 24,69 (CHCH3); 21,22, 21,09 (CH2); 20,54, 20,49 (CH2CN); 8,75, 8,80 (CH3).
Пример 3. Синтез амидофосфитного реагента 14 (R1=DMTr, R2=Me, X=O, L=CH2).
Синтез пропаргилового эфира 2,2-бис(гидроксиметил)пропионовой кислоты ( 12 ).
К раствору (1,8 г, 13,4 ммоль) 2,2-бис-(гидроксиметил)пропионовой кислоты 1 в 10 мл безводного ДМФА добавляли (0,82 г, 14,6 ммоль) KOH. Реакционную массу перемешивали в течение 2 ч при 100°С, после раствор охлаждали до 45°С и добавляли (2,02 г, 13,7 ммоль) пропаргилбромида 11. Реакционную массу перешивали 72 ч при 45°С, выпавший осадок отфильтровывали, растворитель упаривали при пониженном давлении. Полученный остаток растворяли в 5 мл воды и экстрагировали хлороформом (5×20мл). Органический слой сушили над MgSO4, упаривали. Получили 1,39 г (60%) пропаргилового эфира 2,2-бис-(гидроксиметил)пропионовой кислоты 12.
1H-ЯМР (CDCl3, 500 МГц) δ: 4,73 (d, J = 2,5 Гц, 2H, СH2O), 3,88 (d, J = 11,4 Гц, 2H, СH2OH), 3,70 (d, J = 11,4 Гц, 2H, СH2OH), 2,51-2,80 (br s, 2H, OH), 2,49 (t,J= 2,5 Гц, 1H, CH≡), 1,09 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ 175,23 (CO), 77,47 (C≡), 75,37 (CH≡),67,33 (CH2OH), 52,56 (CH2O), 49,49 (CCO), 17,12 (CH3).
Синтез проп-2-ин-1-ил3-[бис(4-метоксифенил)(фенил)метокси]-2-(гидроксиметил)-2-метилпропионата ( 13 ).
К охлажденному до 0°С раствору (1,3 г, 7,55 ммоль) пропаргилового эфира 2,2-бис(гидроксиметил)пропионовой кислоты 12 в 25 мл безводного пиридина при перешивании порциями в течение 4-х часов добавляли (1,9 г, 5,66 ммоль) DMTrCl. Реакционную массу перемешивали в течение 24 ч, далее добавляли 3 мл сухого МеОН и дополнительно раствор перемешивали в течение 30 мин. Растворитель упаривали, остаток растворяли в 50 мл хлороформа. Раствор промывали 1 раз насыщенным раствором NaHCO3 (~50 мл), 1 раз насыщенным раствором NaСl (~50 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход продукта - 2,26 г (63%).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,45 (dd, J = 8,4, 1,3 Гц, 2H, Ph-2), 7,30-7,37 (m, 6H, Ph-3, PhOMe-2), 7,24 (tt, J = 6,9, 1,3 Гц, 1H, Ph-4), 6,87 (ddd, J = 9,0, 3,2, 1,9 Гц, 4H, PhOMe-3), 4,70 (d, J = 2,5 Гц, 2H, CH2O), 3,80 (s, 6H, ОCH3), 3,66 (d, J = 10,8 Гц, 1H, CH2OH), 3,60 (d, J = 10,8 Гц, 1H, CH2OH),3,33 (d, J = 8,8 Гц, 1H, CH2ODMTr), 3,20 (d, J = 8,8 Гц, 1H, CH2ODMTr), 2,50 (t, J = 2,5 Гц, 1H, CH≡), 1,15 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ 173,55 (CO), 158,83, 158,81 (4-PhOMe), 144,46 (1-Ph), 135,45, 135,21 (1-PhOMe), 130,17 (2-PhOMe), 128,23 (2-Ph), 128,08 (3-Ph), 127,17 (4-Ph), 113,53, 113,52 (3-PhOMe), 87,11 (CPh), 77,49 (C≡), 74,43 (CH≡), 68,17 (CH2ODMTr), 67,39 (CH2OH), 55,38 (OCH3), 52,52 (CH2O), 48,30 (CCO), 18,07 (CH3).
Синтез проп-2-ин-1-ил3-[бис(4-метоксифенил)(фенил)метокси]-2-({[(2-цианоэтокси)(диизопропиламино)фосфанил]окси}метил)-2-метилпропионата ( 14 ).
К раствору (2,20 г, 4,63 ммоль) диметокситритилового эфира 13 в 100 мл ТГФ при перешивании добавляли (1,61 мл, 9,26 ммоль) DIPEA и по каплям (1,75 мл, 7,87 ммоль) 2-цианоэтил-N,N-диизопропилхлорамидофосфита 8. Реакционную массу перемешивали в течение 1 часа при комнатной температуре. Далее осадок отфильтровывали, промывали небольшим количеством ТГФ. Органический слой упаривали, остаток растворяли в 50 мл хлороформа. Раствор 1 раз промывали фосфатным буфером pH=7,0 (~50 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход продукта - 2,63 г (84%).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,44-7,48 (m, 2H, Ph-2), 7,33-7,37 (m, 4H, PhOMe-2), 7,29-7,33 (m, 2H, Ph-3), 7,21-7,26 (m, 1H, Ph-4), 6,84-6,88 (m, 4H, PhOMe-3), 4,65-4,70 (m, 2H, CH2O), 3,80 (s, 6H, OCH3), 3,68-3,84 (m, 4H, CH2O),3,56 (h, J = 6,8 Гц, 2H, CHMe2),3,33 (d, J = 8,9 Гц, 1H, CH2ODMTr), 3,21 (d, J = 8,9 Гц, 1H, CH2ODMTr), 2,53-2,59 (m, 2H, CH2CN), 2,47 (t, J = 2,5 Гц, 1H, CH≡), 1,18 (d, J = 6,8 Гц, 6H, CH3), 1,14 (d, J = 6,8 Гц, 3H, CH3), 1,12 (d, J = 6,8 Гц, 3H, CH3),1,10 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ 171,25, 171,16 (CO); 158,71 (4-PhOMe); 144,76, 144,73 (1-Ph); 135,76, 135,73, 135,66, 135,62 (1-PhOMe); 130,27, 130,25 (2-PhOMe); 128,22 (2-Ph); 128,06 (3-Ph); 127,02 (4-Ph); 117,75, 117,72 (CN); 113,37 (3-PhOMe); 86,60, 86,56 (CPh); 77,59, 77,54 (C≡); 74,20, 74,17 (CH≡);67,21 (d, J = 7,4 Гц, CCH2OP), 67,08(d, J = 7,4 Гц, CCH2OP); 65,99, 65,77 (CH2ODMTr); 58,51 (d, J = 3,1 Гц, CH2OP), 58,65 (d, J = 3,5 Гц, CH2OP); 55,37 (OCH3); 48,86 (d, J = 7,8 Гц, CCO), 48,76 (d, J = 7,9 Гц, CCO); 43,31, 43,30, 43,41, 43,39 (CHCH3); 29,27, 29,25 (CH2N), 24,78, 24,75, 24,73, 24,69 (CHCH3); 20,54, 20,49 (CH2CN); 18,12, 18,01 (CH3).
Пример 4. Синтез амидофосфитного реагента 17 (R1=DMTr, R2=Me, X=NH, L=CH2CH2OCH2).
Синтез 3‐гидрокси-2-(гидроксиметил)-2-метил-N-[2-(проп‐2-ин-1-илокси)этил]-пропанамида ( 17 ).
К раствору (1,0 г, 7,45 ммоль) 2,2-бис-(гидроксиметил)пропионовой кислоты 1 в 10 мл безводного ДМФА при комнатной температуре добавляли (1,54 г, 7,45 ммоль) DCC и (0,86 г, 7,49 ммоль) HOSu. Реакционную массу перемешивали в течение 3 ч при комнатной температуре. Выпавший осадок отфильтровывали, осадок промывали ДМФА. В маточный раствор при перемешивании добавляли (1,49 г, 15,0 ммоль) 2-(проп-2-ин-1-илокси)этан-1-амина 15. Реакционную массу перемешивали в течение 5 мин. Растворитель упаривали при пониженном давлении, продукт выделяли колоночной хроматографией на SiO2. Выход продукта - 1,5 г (94%).
1H-ЯМР (CDCl3, 500 МГц) δ: 4,90 (br.s, 1H, NH), 4,13 (d, J = 2,4 Гц, 2H, CH2C≡), 3,86 (d, J = 11,4 Гц, 2H, СH2OH),3,70 (d, J = 11,4 Гц, 2H, СH2OH),3,56 (t, J = 5,1 Гц, 2H, CH2O), 3,29-3,33 (m, 2H, CH2N), 2,60-2,80 (brs, 2H, OH), 2,42 (t, J = 2,4 Гц, 1H, CH≡), 1,10 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ:175,98 (CO), 80,02 (C≡), 75,93 (CH≡),69,61 (CH2O), 67,29 (2CH2OH), 58,80 (CH2C≡), 47,40 (CCO),41,04 (CH2N), 18,84 (CH3).
Синтез 3-[бис(4-метоксифенил)(фенил)метокси]-2-(гидроксиметил)-2-метил-N-[2-(проп-2-ин-1-илокси)этил]пропанамида ( 19 ).
К охлажденному до 0°С раствору (1,0 г, 4,65 ммоль) амида 17 в 25 мл безводного пиридина при перешивании порциями в течение 4-х часов добавляли (1,18 г, 3,49 ммоль) DMTrCl. Реакционную массу перемешивали в течение 24 ч, далее добавляли 2 мл МеОН и дополнительно раствор перемешивали в течение 30 мин. Растворитель упаривали, остаток растворяли в 50 мл хлороформа. Раствор 1 раз промывали насыщенным раствором NaСl (~10 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход диметокситритилового эфира 19 - 1,07 г (59%).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,46 (dd, J = 8,4, 1,3 Гц, 2H, Ph-2), 7,30-7,37 (m, 6H, Ph-3, PhOMe-2), 7,21-7,26 (m, 1H, Ph-4), 6,87 (ddd, J = 9,0, 3,2, 1,9 Гц, 4H, PhOMe-3), 4,13 (d, J = 2,4 Гц, 2H, CH2C≡), 3,82 (s, 6H, ОCH3), 3,70 (d, J = 11,3 Гц, 1H, СH2OH), 3,61 (d, J = 11,3 Гц, 1H, СH2OH), 3,56 (t, J = 5,1 Гц, 2H, CH2O), 3,29-3,33 (m, 3H, CH2N, CH2ODMTr), 3,23 (d, J = 9,2 Гц, 1H, CH2ODMTr), 2,42 (t, J = 2,4 Гц, 1H, CH≡), 1,10 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ: 176,06 (CO), 158,82, 158,80 (4-PhOMe), 144,47 (1-Ph), 135,45, 135,22 (1-PhOMe), 130,15 (2-PhOMe), 128,25 (2-Ph), 128,08 (3-Ph), 127,17 (4-Ph), 113,52, 113,51 (3-PhOMe), 87,09 (CPh), 80,00 (C≡), 75,96 (CH≡), 69,63 (CH2O), 68,08 (CH2ODMTr), 67,57 (CH2OH), 58,75 (CH2C≡), 55,37 (OCH3), 47,41 (CCO), 41,06 (CH2N), 18,83 (CH3).
Синтез 2-{[бис(4-метоксифенил)(фенил)метокси]метил}-2-метил-3-оксо-3-{2-[2-(проп-2-ин-1-илокси)этил]амино}пропил (2-цианоэтил) диизопропиламидофосфита( 21 ).
К раствору (1,0 г, 1,93 ммоль) диметокситритилового эфира 19 в 30 мл ТГФ при перешивании добавляли (0,82 мл, 4,70 ммоль) DIPEA и по каплям (0,73 мл, 3,27 ммоль) 2-цианоэтил-N,N-диизопропилхлорамидофосфита 8. Реакционную массу перемешивали в течение 1 часа при комнатной температуре. Далее осадок отфильтровывали, промывали небольшим количеством ТГФ. Органический слой упаривали, остаток растворяли в 30 мл хлороформа. Раствор 1 раз промывали фосфатным буфером pH=7,0 (~10 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход продукта 1,14 г (82%).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,44-7,48 (m, 2H, Ph-2), 7,33-7,37 (m, 4H, PhOMe-2), 7,29-7,33 (m, 2H, Ph-3), 7,21-7,26 (m, 1H, Ph-4), 6,84-6,88 (m, 4H, PhOMe-3), 4,13 (d, J = 2,4 Гц, 2H, CH2C≡), 3,82 (s, 6H, ОCH3), 3,66-3,81 (m, 4H, CH2O), 3,53-3,59 (m, 4H, CHMe2, CH2O), 3,29-3,33 (m, 3H, CH2N, CH2ODMTr), 3,25 (d, J = 9,3 Гц, 1H, CH2ODMTr), 2,53-2,59 (m, 2H, CH2CN), 2,40 (t, J = 2,4 Гц, 1H, ≡CH), 1,18 (d, J = 6,8 Гц, 6H, CH3), 1,14 (d, J = 6,8 Гц, 6H, CH3), 1,10 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ: 174,44, 174,35 (CO); 158,71 (4-PhOMe); 144,76, 144,73 (1-Ph); 135,76, 135,73, 135,66, 135,62 (1-PhOMe); 130,27, 130,25 (2-PhOMe); 128,22 (2-Ph); 128,06 (3-Ph); 127,02 (4-Ph); 117,75, 117,72 (CN); 113,37 (3-PhOMe); 86,60, 86,56 (CPh); 80,12, 80,10 (C≡); 75,91, 75,89 (CH≡); 69,63, 69,60 (CH2O); 65,84, 65,62 (CH2ODMTr); 67,19 (d, J = 7,4 Гц, CCH2OP), 67,06 (d, J = 7,4 Гц, CCH2OP); 58,78, 58,75 (CH2C≡); 58,51 (d, J = 3,0 Гц, CH2OP), 58,65 (d, J = 3,5 Гц, CH2OP); 55,37 (OCH3); 48,14 (d, J = 7,8 Гц, CCO), 48,04 (d, J = 7,9 Гц, CCO); 43,31, 43,30, 43,41, 43,39 (CHCH3); 41,08, 41,06 (CH2N); 24,78, 24,75, 24,73, 24,69 (CHCH3); 20,54, 20,49 (CH2CN); 18,85, 18,73 (CH3).
Пример 5. Синтез амидофосфитного реагента 22 (R1=DMTr, R2=Me, X=NH, L=CH2CH2OCH2CH2OCH2).
Синтез 3‐гидрокси-2-(гидроксиметил)-2-метил-N-{2-[2-(проп‐2-ин-1-илокси)-этокси]этил}пропанамида ( 18 ).
К раствору (1,0 г, 7,45 ммоль) 2,2-бис-(гидроксиметил)пропионовой кислоты 1 в 10 мл безводного ДМФА при комнатной температуре добавляли (1,54 г, 7,45 ммоль) DCC и (0,86 г, 7,49 ммоль) HOSu. Реакционную массу перемешивали в течение 3 ч при комнатной температуре. Выпавший осадок отфильтровывали, осадок промывали ДМФА. В маточный раствор при перемешивании добавляли (2,15 г, 15,0 ммоль) 3-[2-(2-аминоэтокси)этокси]проп-1-ина 16. Реакционную массу перемешивали в течение 5 мин. Растворитель упаривали при пониженном давлении, продукт выделяли колоночной хроматографией на SiO2. Выход продукта - 1,76 г (91%).
1H-ЯМР (CDCl3, 500 МГц) δ: 4,96 (s, 1H,NH), 4,19 (d, J=2,5 Гц, 2H, CH2C≡), 3,88 (d, J = 11,4 Гц, 2H, СH2OH), 3,70-3,66 (m, 4H,CH2O, СH2OH), 3,65-3,60 (m, 2H,CH2O), 3,53 (t, J = 5,4 Гц, 2H,CH2O), 3,29-3,33 (m, 2H,CH2N), 2,60-2,80 (brs, 2H, OH), 2,43 (t, J = 2,4 Гц, 1H,CH≡), 1,09 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ: 176,04 (CO), 79,20 (C≡), 74,74 (CH≡), 70,28, 70,11, 69,03 (CH2O), 67,33 (2CH2OH), 58,39 (CH2C≡),47,50 (CCO), 40,30 (CH2N), 18,86 (CH3).
Синтез 3-[бис(4-метоксифенил)(фенил)метокси]-2-(гидроксиметил)-2-метил-N-{2-[2-(проп‐2-ин-1-илокси)этокси]этил}пропанамида ( 20 ).
К охлажденному до 0°С раствору (1,0 г, 3,86 ммоль) амида 18 в 20 мл безводного пиридина при перешивании порциями в течение 4-х часов добавляли (0,98 г, 2,89 ммоль) DMTrCl. Реакционную массу перемешивали в течение 24 ч, далее добавляли 2 мл сухого МеОН и дополнительно раствор перемешивали в течение 30 мин. Растворитель упаривали, остаток растворяли в 50 мл хлороформа. Раствор 1 раз промывали насыщенным раствором NaСl (~10 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход продукта - 1,01 г (62%).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,47 (dd, J = 8,4, 1,3 Гц, 2H, Ph-2), 7,30-7,37 (m, 6H, Ph-3, PhOMe-2), 7,21-7,26 (m, 1H, Ph-4), 6,87 (ddd, J = 9,0, 3,2, 1,9 Гц, 4H, PhOMe-3), 4,19 (d, J = 2,5 Гц, 2H, CH2C≡), 3,81 (s, 6H, ОCH3), 3,70-3,66 (m, 3H, СH2OH, CH2O), 3,65-3,60 (m, 3H, СH2OH, CH2O), 3,53 (t, J = 5,4 Гц, 2H, CH2O), 3,29-3,35 (m, 3H, CH2N, CH2ODMTr), 3,23 (d, J = 9,3 Гц, 1H, CH2ODMTr), 2,43 (t, J = 2,4 Гц, 1H, CH≡), 1,10 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ: 176,06 (CO), 158,81, 158,79 (4-PhOMe), 144,47 (1-Ph), 135,46, 135,22 (1-PhOMe), 130,15 (2-PhOMe), 128,25 (2-Ph), 128,08 (3-Ph), 127,17 (4-Ph), 113,52, 113,50 (3-PhOMe), 87,10 (C-Ph), 79,22 (C≡), 74,78 (CH≡), 70,26, 70,14, 69,01 (CH2O), 68,12 (CH2ODMTr), 67,50 (CH2OH), 58,36 (CH2C≡), 55,36 (OCH3), 47,47 (CCO), 40,38 (CH2N), 18,85 (CH3).
Синтез 2-{[бис(4-метоксифенил)(фенил)метокси]methyl}-2-метил-3-оксо-3-({2-[2-(проп-2-ин-1-илокси)этокси]этил}амино)пропил (2-цианоэтил) диизопропилфосфор-амидита ( 22 ).
К раствору (1,0 г, 1,78 ммоль) диметокситритилового эфира 20 в 30 мл ТГФ при перешивании добавляли (0,75 мл, 4,30 ммоль) DIPEA и по каплям (0,68 мл, 3,03 ммоль) 2-цианоэтил-N,N-диизопропилхлорамидофосфита 8. Реакционную массу перемешивали в течение 1 часа при комнатной температуре. Далее осадок отфильтровывали, промывали небольшим количеством ТГФ. Органический слой упаривали, остаток растворяли в 30 мл хлороформа. Раствор 1 раз промывали фосфатным буфером pH=7,0 (~10 мл). Органический слой сушили над Na2SO4, упаривали и выделяли колоночной хроматографией на SiO2. Выход продукта - 1,07 г (79%).
1H-ЯМР (CDCl3, 500 МГц) δ: 7,44-7,48 (m, 2H, Ph-2), 7,33-7,37 (m, 4H, PhOMe-2), 7,29-7,33 (m, 2H, Ph-3), 7,21-7,26 (m, 1H, Ph-4), 6,84-6,88 (m, 4H, PhOMe-3), 4,19 (d, J = 2,5 Гц, 2H, CH2C≡), 3,80 (s, 6H, ОCH3), 3,80-3,66 (m, 6H, CH2O), 3,65-3,60 (m, 2H, CH2O), 3,53-3,59 (m, 4H, CHMe2, CH2O), 3,29-3,35 (m, 3H, CH2N, CH2ODMTr), 3,24 (d, J = 9,3 Гц, 1H, CH2ODMTr), 2,53-2,59 (m, 2H, CH2CN), 2,41 (t, J = 2,4 Гц, 1H, CH≡), 1,18 (d, J = 6,8 Гц, 6H, CH3), 1,14 (d, J = 6,8 Гц, 6H, CH3), 1,12 (s, 3H, CH3). 13С-ЯМР (126 МГц, CDCl3) δ: 174,35, 174,27 (CO); 158,69 (4-PhOMe); 144,78, 144,75 (1-Ph); 135,75, 135,73, 135,65, 135,62 (1-PhOMe); 130,27, 130,25 (2-PhOMe); 128,22 (2-Ph); 128,06 (3-Ph); 127,03 (4-Ph); 117,73, 117,71 (CN); 113,37 (3-PhOMe); 86,60, 86,56 (CPh); 79,20, 79,18 (C≡); 74,82, 74,80 (CH≡); 71,06, 71,04, 70,34, 70,31, 69,01, 68,98 (CH2O);65,90, 65,70 (CH2ODMTr); 67,23 (d, J = 7,4 Гц, CCH2OP), 67,11 (d, J = 7,4 Гц, CCH2OP); 58,55 (d, J = 3,1 Гц, CH2OP), 58,69 (d, J = 3,5 Гц, CH2OP); 58,37, 58,35 (CH2C≡); 55,38 (OCH3);48,13 (d, J = 7,8 Гц, CCO), 48,03 (d, J = 7,8 Гц, CCO); 43,29, 43,30, 43,40, 43,38 (CHCH3); 40,38, 40,36 (CH2N);24,76, 24,73, 24,71, 24,67 (CHCH3); 20,54, 20,49 (CH2CN); 18,84, 18,72 (CH3).
Хотя настоящее изобретение описано в деталях выше, для специалиста в данной области техники очевидно, что могут быть сделаны изменения и произведены эквивалентные замены, и такие изменения и замены не выходят за рамки настоящего изобретения, которые определяются приложенной формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Четвертичные аммониевые соединения на основе 3-гидроксипиридина, обладающие антибактериальной активностью | 2021 |
|
RU2778507C1 |
| Соединения фторхинолонового ряда на основе производных пиридоксина, обладающие антибактериальными свойствами | 2019 |
|
RU2713932C1 |
| МОДУЛЯТОРЫ ИНТЕГРИРОВАННОГО СИГНАЛЬНОГО ПУТИ СТРЕССА | 2017 |
|
RU2769327C2 |
| АНТРАЦИКЛИНОВЫЕ ГЛИКОЗИДЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ДИИОДОПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1991 |
|
RU2100366C1 |
| Бис-аммониевые соединения на основе пиридоксина, обладающие антибактериальными и антимикотическими свойствами | 2020 |
|
RU2731999C1 |
| ИОДИРОВАННЫЕ АРИЛЬНЫЕ СОЕДИНЕНИЯ И ДИАГНОСТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2145955C1 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2525392C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2466729C2 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2014 |
|
RU2678327C2 |
| ПРОТИВОВОСПАЛИТЕЛЬНЫЕ СРЕДСТВА | 2004 |
|
RU2365585C2 |
Группа изобретений относится к области органической химии и химии нуклеиновых кислот и предназначена для функционализации синтетических олигонуклеотидов методом азид-алкинового циклоприсоединения («клик-химии»). Раскрывается соединение, имеющее структурную формулу, где R1 представляет собой защитную группу, стабильную в условиях синтеза олигонуклеотидов, выбранную из 4,4'-диметокситритильной (DMTr), 4-монометокситритильной (MMTr), тритильной (Tr), триметокситритильной (TMTr), 9-фенилксантен-9-ильной (Px), карбонатной или силильной групп, R2 представляет собой водород, метил или этил, X представляет собой -О- или -NH-, и L представляет собой спейсерную группу -(CH2)-n, где n = 1, 2 или 3; или -(CH2CH2O)nCH2-, где n = 1 или 2. Также раскрыт способ получения вышеуказанных амидофосфитных реагентов, включающий функционализацию карбоксильной группы линкерами, содержащими алкиновую группу, с последующей селективной защитой одной гидроксильной группы и фосфитилированием второй гидроксильной группы с получением целевых реагентов с общим выходом 32-47%. Группа изобретений позволяет модифицировать синтетические олигонуклеотиды в любом положении (3', 5' или внутри) и при их вставке сохраняется природное межнуклеотидное расстояние в 3 атома углерода между фосфатными группами. 2 н. и 7 з.п. ф-лы, 5 пр.
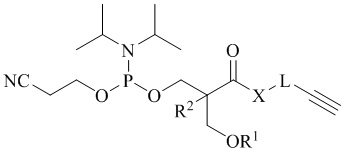
1. Соединение, имеющее структурную формулу
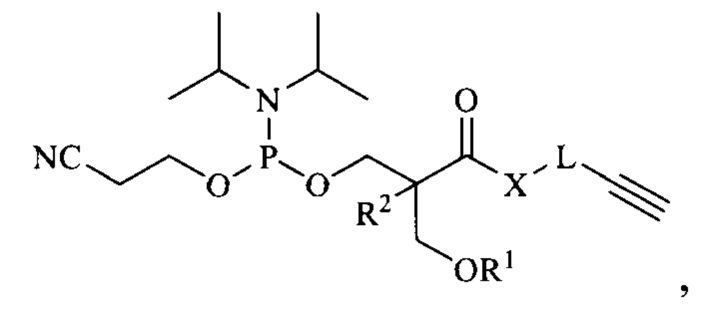
где R1 представляет собой защитную группу, стабильную в условиях синтеза олигонуклеотидов, выбранную из 4,4'-диметокситритильной (DMTr), 4-монометокситритильной (MMTr), тритильной (Tr), триметокситритильной (TMTr), 9-фенилксантен-9-ильной (Рх), карбонатной или силильной групп, R2 представляет собой водород, метил или этил, X представляет собой -О- или -NH-, и L представляет собой спейсерную группу -(СН2)-n, где n=1, 2 или 3; или -(СН2СН2О)nCH2-, где n=1 или 2.
2. Соединение по п. 1, выбранное из группы, включающей соединения, имеющие следующие структурные формулы:

где R представляет собой метил или этил;
 и
и
где n=1 или 2.
3. Способ получения соединения по п. 1, включающий стадии:
1) взаимодействия 2,2-бис(гидроксиметил)карбоновой кислоты с соединением формулы HC≡C(CH2)n-NH2, где n=1, 2 или 3, или с соединением формулы HC≡CCH2(OCH2CH2)n-NH2, где n=1 или 2, посредством активации карбоксильной группы в присутствии N-гидроксисукцинимида и дициклогексилкарбодиимида в случае, когда X представляет собой -ΝΗ-; или с соединением формулы HC≡C(CH2)nBr, где n=1, 2 или 3, в присутствии КОН в случае, когда X представляет собой -О-;
2) взаимодействия соединения, полученного на стадии 1, с субэквимолярным количеством реагента для защиты одной гидроксильной группы соединения, полученного на стадии 1;
3) взаимодействия соединения, полученного на стадии 2, с цианоэтил-Ν,Ν-диизопропилхлорамидофосфитом.
4. Способ по п. 3, отличающийся тем, что в качестве 2,2-бис(гидроксиметил)карбоновой кислоты используют 2,2-бис(гидроксиметил)пропионовую кислоту или 2,2-бис(гидроксиметил)масляную кислоту.
5. Способ по п. 3, отличающийся тем, что соединением формулы HC≡C(CH2)n-NH2 является пропаргиламин.
6. Способ по п. 3, отличающийся тем, что соединением формулы НС≡ССН2(ОСН2СН2)n-NH2 является 2-(2-пропинилокси)этиламин или пропаргил-PEG2-амин.
7. Способ по п. 3, отличающийся тем, что на стадии 1 при активации карбоксильной группы используют N-гидроксисукцинимид и дициклогексилкарбодиимид в эквимолярных количествах.
8. Способ по п. 3, отличающийся тем, что соединением формулы НС≡С(СН2)nBr является пропаргилбромид.
9. Способ по п. 3, отличающийся тем, что в качестве реагента для защиты гидроксильной группы на стадии 2 используют 4,4'-диметокситритилхлорид (DMTrCl), 4-монометокситритилхлорид (MMTrCl), 9-фенилксантен-9-илхлорид, тритилхлорид(TrCl), триметокситритилхлорид (TMTrCl), карбонаты или силилхлориды.
| СПОСОБ ПОЛУЧЕНИЯ АМИДОФОСФИТНОГО МОНОМЕРА АХИРАЛЬНОЙ НЕНУКЛЕОТИДНОЙ ВСТАВКИ ДЛЯ МОДИФИКАЦИИ ОЛИГОНУКЛЕОТИДОВ | 2011 |
|
RU2460721C1 |
| RU 2014134380 A, 20.03.2016 | |||
| WO 2002079216 A, 10.10.2002 | |||
| WO 2004063208 A1, 24.07.2004 | |||
| IHARA TOSHIHIRO et al | |||
| Synthesis of the Amidite Reagent to Built Bipyridine Units into DNA Backbone | |||
| Heterocycles, 2005, 65 (2), p | |||
| ПРИСПОСОБЛЕНИЕ ДЛЯ ПОДАЧИ УГЛЯ В ТЕНДЕР ПАРОВОЗА | 1920 |
|
SU293A1 |

