Изобретение относится к усовершенствованиям в области контрастных средств, в частности к йодированным рентгеноконтрастным веществам.
Контрастные средства могут вводиться в ходе медицинских процедур визуализации, например при получении рентгеновского и магнитно-резонансного и ультразвукового изображения, для усиления контрастности изображения объекта исследования, обычно тела человека и животного. Достигаемая усиленная контрастность дает возможность более четко наблюдать или идентифицировать различные органы, типы тканей или пространственные перегородки организма. При рентгеновском исследовании контрастные вещества действуют путем изменения характеристик поглощения рентгеновских лучей теми участками тела, в которых они распространяются.
Однако очевидно, что пригодность соединений для использования в качестве контрастных веществ зависит от их токсичности, их диагностической эффективности и прочих неблагоприятных воздействий, оказываемых на пациента, которому их вводят, и от легкости их хранения и легкости введения.
Поскольку такие контрастные вещества обычно используют скорее в целях диагностики, чем для достижения определенного терапевтического эффекта, то для создания новых контрастных веществ в основном требуется получить вещества, обладающие возможно низким воздействием на различные биологические механизмы клеток или организма, что должно, как правило, приводить к более низкой токсичности в отношении животных и к менее вредным клиническим влияниям.
Токсичность и вредные биологические влияния контрастных средств складываются из совместного действия компонентов среды, например растворителя или носителя, а также контрастного соединения или его компонентов (например, ионов, где соединение существует в ионной форме) и метаболитов.
Установлены следующие основные факторы, вносящие вклад в токсичность и вредные воздействия контрастных средств:
- хемотоксичность контрастного агента,
- способность контрастного вещества к осмосу (осмоляльность),
- ионная структура (или ее отсутствие) контрастного вещества.
Например, в коронарной ангиографии инъекция контрастного вещества в сердечно-сосудистую систему связана с рядом серьезных воздействий на сердечную функцию. Эти воздействия достаточно тяжелые, что налагает ограничения на применение в ангиографии некоторых контрастных веществ.
В этой процедуре на короткий период времени вместо крови контрастное вещество протекает через сердечно-сосудистую систему и различия в химической и физико-химической природе контрастного вещества и крови приводят к тому, что эти временные замены могут вызывать нежелательные эффекты, например аритмию, Q Т-пролонгацию и особенно ослабление сократительной способности сердца и возникновение фибрилляции желудочков.
Проведены многочисленные исследования этих негативных воздействий на функцию сердца при введении контрастного вещества в сердечно-сосудистую систему, например, при ангиографии, и испытан широкий спектр способов снижения или устранения этих эффектов.
Ранее вводившиеся рентгеноконтрастные вещества, основанные на солях трийодфенилкарбоксилата, связаны в частности с осмотоксичными эффектами, происходящими от гипертоничности (повышенного осмотического давления) инъецируемого контрастного вещества.
Гипертоничность вызывает осмотические эффекты, такие как отток воды из эритроцитов, эндотелиальных клеток и мышечных клеток сердца и кровяных сосудов. Потеря воды делает эритроциты жесткими и повышенное осмотическое давление, хемотоксичность и неоптимальная ионная структура обеспечивают по отдельности или вместе ослабление сократительной способности мышечных клеток и вызывают расширение мелких кровеносных сосудов и приводят в результате к снижению кровяного давления.
Проблеме осмотоксичности была адресована разработка неионных трийодфенильных мономеров, таких как иохексол (iohexol), позволяющих достигать тех же самых контрастно-эффективных концентраций йода со значительно сниженными сопутствующими эффектами осмотоксичности.
Разработки в направлении снижения осмотоксичности привели к созданию неионных бис-(трийодфения)-димеров, таких как йодиксанол (iodixanol), которые снижают связанные с осмотоксичностью проблемы, позволяя при этом достигать контрастно-эффективных концентраций йода гипотоническими растворами.
Способность достигать контрастно-эффективных концентраций йода без доведения растворов до изотонических уровней (около 300 мосм/кг H2О) дополнительно создает возможность влияния на вклад в токсичность, вызванную ионным дисбалансом, путем включения различных катионов плазмы, как обсуждается, например, в WO-90/01194, WO-91/13636 of Nycomed Imaging AS.
Однако рентгеноконтрастные вещества при таких высоких промышленных концентрациях йода, как приблизительно 300 мг I /мл, обладают относительно высокими вязкостями, находящимися в интервале примерно 15-60 мП при температуре окружающей среды, при этом димерные контрастные вещества обычно более вязкие, чем мономерные контрастные вещества. При таких вязкостях возникают проблемы введения контрастных веществ, требующие использования игл со сравнительно большим отверстием или применения высокого давления, которые особенно ярко выражены в педиатрической радиографии и в радиографических методиках, требующих быстроты, введения болюсов, например в ангиографии.
Данное изобретение посвящено проблемам вязкости, возникающим в связи с материалами, известными из уровня техники, и таким образом рассматриваемое в этом аспекте изобретение предлагает йодированные арильные соединения, полезные в качестве рентгеноконтрастных соединений, формулы I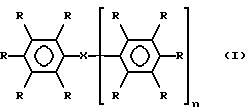
где n равно 0 или 1, и когда n равно 1, каждый из С6R5 фрагментов может быть одинаковым или различным; каждая группа R обозначает атом водорода, атом йода или гидрофильный радикал М или М1, две или три не смежных R-группы в каждом C6R5-фрагменте представляют собой йод и по крайней мере одна, а предпочтительно две или три R-группы в каждом C6R5-фрагменте представляют М или М1; X обозначает связь или группу, обеспечивающую цепь из 1-7 атомов, например цепь из 1, 2, 3 или 4 атомов, связывает два С6X5-фрагмента, или когда n равно 0, X обозначает группу R; каждый М независимо обозначает неионный гидрофильный радикал и каждый М1 независимо обозначает С1-4-алкильную группу, замещенную по крайней мере одной гидроксильной группой и необязательно связанную с фенильным циклом посредством карбонильной, сульфоновой или сульфоксидной группы, по меньшей мере одна R-группа, предпочтительно по крайней мере две R-группы и особенно предпочтительно - по меньшей мере одна R-группа в каждом из C6R5-фрагментов, обозначает М1 радикал; при условии, что когда n равно нулю, либо присутствует по крайней мере одна M1-группа, отличная от гидроксиметильной или 1,2-дигидроксиэтильной (или необязательно отличная от гидроксиэтильной) группы, либо если присутствует одна гидроксиметильная или 1,2-дигидроксиэтильная М1-группа (и необязательно любая гидроксиэтильная группа), то также присутствует по меньшей мере одна связанная с азотом М-группа, содержащая гидроксилированную алкильную (предпочтительно С1-4-алкильную группу) и их изомеры, особенно стереоизомеры и ротамеры.
Найдено, что соединения по изобретению обладают преимущественно низкой вязкостью в водном растворе (таблица 1); предполагается, что причиной тому служит наличие М1-групп на фенильных фрагментах, асимметричность соединения и для димерных соединений - природа связующего X (особенно когда X обозначает связующую группу длиной менее чем в 5 углеродных атомов).
Так, например, все испытанные водорастворимые мономерные соединения по изобретению обладают меньшей вязкостью, чем иохексол. Кроме того, соединения формулы I, введенные в количестве, превышающем эффективное, не вызывают какого-либо токсического действия (таблица 2).
Соединения формулы 1 предпочтительно являются асимметричными. Для мономерных соединений (где n = 0) низкая вязкость может достигаться за счет асимметричного замещения фенильного цикла. Для димеров это может быть достигнуто использованием группы X, образующей цепь с 2 или более асимметричными атомами, и/или выбором неидентичных C6R5-групп, например неидентичного замещения йодфенильных концевых групп. Асимметричные молекулы предпочтительны, так как найдено, что они обладают лучшей растворимостью в воде.
Такое неидентичное замещение фенильных концевых групп, радикалов С6R5, можно достигнуть, используя различное число и/или различные положения йодного замещения и/или различное число, положения или виды М- или М1-замещений. Для максимального йодного содержания предпочтительны трийодфенильные концевые группы, например группы формулы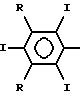
и в них две R-группы могут быть одинаковыми или различными, хотя предпочтительно, чтобы обе обозначали М- или М1-группы.
Когда фенильная концевая группа замещена йодом, предпочтительно она имеет формулу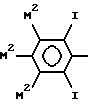
или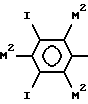
(где каждый из М2 может быть одинаковым или различным и обозначает M1- или М-группу, причем по меньшей мере один на каждом кольце представляет М1).
Обычно, димеры дийодфенил-дийодфенил менее предпочтительны, чем дийодфенил-трийодфенил- или трийодфенил-трийодфенил-димеры, главным образом из-за их низкого йодного содержания, то есть 4 атома йода на молекулу димера, вместо 5 или 6 атомов. Действительно, трийодфенил-трийодфенил-димеры особенно предпочтительны благодаря их большему йодному насыщению.
Для мономеров опять же предпочтительны трийодфенильные соединения.
Солюбизирующие группы М могут представлять собой любые неионизирующие группы, обычно используемые для изменения растворимости в воде. Подходящие группы включают, например, линейные или разветвленные С1-10-алкильные группы, предпочтительно С1-5-группы, необязательно с одной или более CH2- или CH-группами, замененными атомами кислорода или азота, и необязательно - замещенные одной или более группами, выбранными из: оксо, гидрокси, амино, карбоксильного производного и оксо, замещенной серы или фосфора. Конкретные примеры включают полигидроксиалкил, гидроксиалкоксиалкил и гидроксиполиалкоксиалкил и такие группы, присоединенные к фенильной группе посредством амидной связи такие, как гидроксиалкиламинокарбонил, N-алкилгидроксиалкиламинокарбонил и бис-гидроксиалкиламинокарбонил.
Из этих групп предпочтительными являются группы, содержащие 1, 2, 3, 4, 5 или 6, особенно 1, 2 или 3 гидроксигруппы, например
- CONH-CH2CH2OH
- CONH-СН2CHOHCH2ОН
- CONH-CH(CH2OH)2
-CON(CH2CH2OH)2
равно как другие группы, такие как
-CONH2
-CONHCH3
-OCOCH3
-N(COCH3)H
-N(COCH3)С1-3-алкил
-N(COCH3)-моно, бис- или тригидрокси С1-4-алкил
-N(COCH2OH) -моно, бис- или тригидрокси С1-4-алкил
-N(COCH2OH)2
-CON(CH2CHOHCH2OH) (CH2CH2ОН)
-CONH-C(CH2OH)3 и
-CONH-CH(CH2OH)(CHOHCH2ОН).
В основном каждая М-группа предпочтительно включает полигидрокси С1-4-алкильную группу, такую как 1,3-дигидроксипроп-2-ил или 2,3-дигидроксипроп-1-ил.
Могут также быть использованы другие М-группы, такие как общеприняты в области трийодфенильных рентгеноконтрастных веществ, и введение М-групп в йодфенильные структуры можно осуществлять общепринятыми способами.
В основном М-группы предпочтительно включают С1-4-алкильные группы, замещенные 1, 2, 3 или 4 гидроксильными группами (например, гидроксиметил, 2-гидроксиэтил, 2,3-бисгидроксипропил, 1,3-бисгидроксипроп-2-ил, 2, 3, 4-тригидроксибутил и 1, 2, 4- тригидроксибут-2-ил), необязательно присоединенные к фенильному кольцу посредством СО, SO или SO2 группы (например, COCH2ОН или SO2CH2ОН).
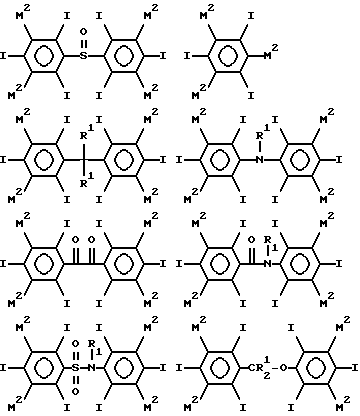
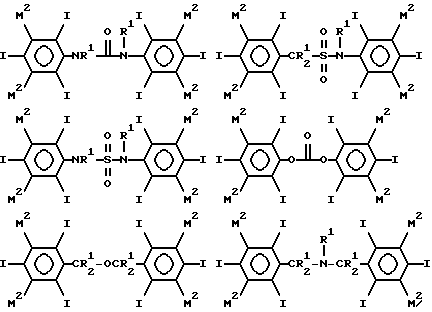
В димерных соединениях по данному изобретению связующая группа X обозначает обычно связь или 1-7, например 1-, 2-, 3- или 4-членную цепь, включающую атомы углерода, азота, кислорода или серы, например:
простая химическая связь,
О, S, N или C - одноатомная цепь,
NN, NC, NS, СС или CO двухатомная цепь,
или NCN, OCN, CNC, OCO, NSN, CSN, COC, OCC или CCC трехатомная цепь,
например:
атом кислорода или группа NR1, СО, SO2 или CR2 1;
группа COCO, CONR1, COCR2 1, SOCR2 1, SO2NR1, CR2 1CR2 1, CR2 1NR1 или CR2 1O;
группа NR1CONR1, OCONR1, CONR1CO, CONR1CR2 1, OCOO, CR2 1OCR2 1, OCR2 1CO, CR2 1CONR1, CR2 1CR2 1CR2 1, COCR1R1CO, CR2 1NR1CR2 1, CR2 1SO2NR1, CR2 1OCO или NR1SO2NR1;
где R1 обозначает водород или C1-6-алкильную или алкоксигруппу, необязательно замещенную гидрокси, алкокси, окса или оксо (например, полигидроксиалкил, формил, ацетил, гидроксил, алкокси или гидроксиалкокси) и там, где R1 присоединена к атому углерода, она может также обозначать гидроксильную группу.
Когда X обеспечивает 4-7-атомную связь, могут быть использованы обычные связующие группы, такие как, например, предложены Justesa в WO- 93/10078 или Bracco в US-A-4348377 и WO-94/ 14478.
В основном такие связующие группы включают необязательно аза- или оксазамещенные алкиленовые цепи, необязательно несущие R1 заместители, особенно желательны такие группы, заканчивающиеся иминовым атомом азота или более предпочтительно карбонильным углеродным атомом, предпочтительно относящимся к иминокарбонильным функциональностям в цепи. Особенно предпочтительны цепи, содержащие гидроксильные группы, такие как присутствуют в йодиксаноле.
Примерами таких цепей служат NCCN, NCCCN, CNCCCNC и CNCCN, например
-NR1COCONR1-
-NR1COCR2 1CONR1-
-NR1CR2 1CR1OHCR2 1NR1-
-CONR1CR2 1CONR1- и
-N (COR1)CR2 1CR1OHN(COR1) -,
например, как присутствует в иотролане, иофратоле, иоксаглиновой кислоте и йодиксаноле или в других соединениях, приведенных в WO-94/14478.
Преимущественно, в димерных соединениях X-группа несимметричная. Это может быть достигнуто введением асимметричных заместителей в симметричную цепь (например, N-C-N замещенная до NHCONR1) или выбором асимметричной цепи (например, OCN замещенной до OCONR1). В частности, желательно, чтобы связующая группа X была полярной, а также гидрофильной.
Таким образом примеры предпочтительных структур по изобретению включают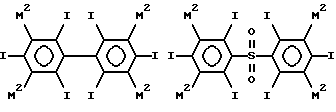
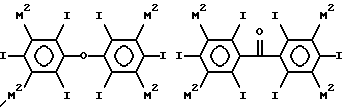
где каждый из М2 обозначает М1 или М, по крайней мере один М2 в каждом соединении (и предпочтительно, в каждом кольце) обозначает M1, особенно когда по крайней мере один М2 обозначает C1-4-алкильную группу, замещенную 1, 2, 3 или 4 гидроксильными группами (например, гидроксиметил, 2-гидроксиэтил, 2,3- бисгидрокси-пропил 1,3-бисгидроксипроп-2-ил, 2,3,4- тригидроксибутил и 1, 2, 4-тригидроксибут-2-ил), необязательно связанную с фенильным кольцом посредством СО, SO или SO2 группы (например, COCH2ОН или SO2CH2ОН), например гидроксиалкильную или гидроксиалкилкарбонильную группу, в частности гидроксиметильную, гидроксиметилкарбонильную, 2-гидроксиэтильную или 2-гидроксиэтилкарбонильную группу, и где R1 обозначает водород, гидроксил, гидроксиалкил (например, 2-гидроксиэтил), ацетил или гидроксиалкилкарбонил.
Конкретными предпочтительными соединениями являются соединения формул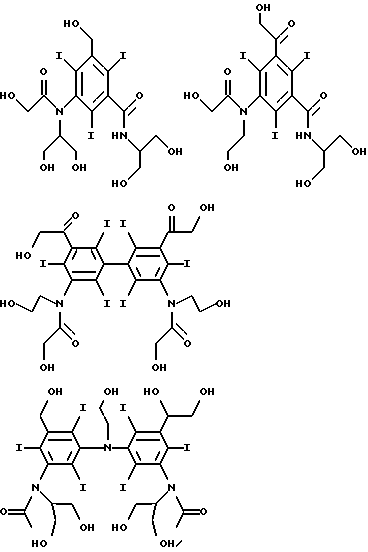
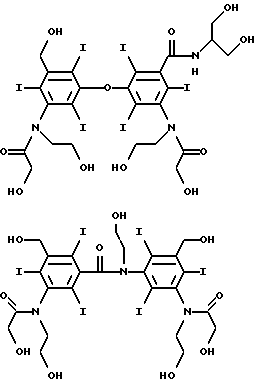
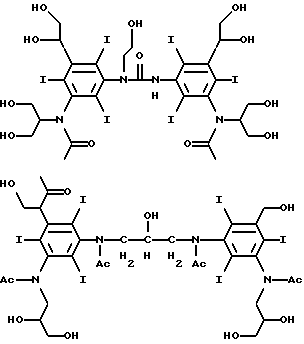
Соединения по данному изобретению обычно могут быть получены в две или три стадии: (a) получения димера (где необходимо), (b) йодирование фенильных групп и (c) замещение фенильных групп и/или необязательно связующих групп солюбилизирующими группами.
Хотя теоретически стадии (a), (b) и (c) могут быть выполнены в любом порядке, в большинстве случаев предпочтительно выполнять стадию получения димера перед стадией йодирования, и из экономических соображений предпочтительно выполнять стадию йодирования последней в синтезе, так как это дает возможность снижения йодных потерь. Стадия получения димера может сама по себе быть многостадийным процессом с первоначальным взаимодействием соответствующего активированного связующего звена с одним из мономеров до того, как образующийся конъюгатсвязующее звено-мономер вступает во взаимодействие со вторым мономером. Или же иначе получение димера можно осуществлять взаимодействием аналогично или совместно замещенных мономеров, сопровождающимся конъюгацией мономеров, приводящей к образованию димеров.
При желании связующая группа X может быть получена модификацией, например замещением, окислением или восстановлением предшествующего связующего звена, например, в предшествующем мономере.
Для мономерных соединений, особенно тех, где замещение цикла асимметричное, обычно эффективное введение йода перед или после частичного замещения фенильного кольца R-группами.
Во всех случаях могут быть использованы обычные синтетические пути, хорошо известные из литературы (например, способы, аналогичные тем, что использованы и описаны для получения соединений в WO-94/14478).
Соединения по данному изобретению могут быть использованы в качестве рентгеноконтрастных средств и с этой целью они могут быть сформулированы с общепринятыми носителями или эксципиентами с получением диагностических контрастных веществ.
Таким образом, исходя из другого аспекта изобретение предлагает диагностические композиции, включающие соединение формулы I (определенной выше) совместно с по крайней мере одним физиологически приемлемым носителем или эксципиентом, например в водном растворе в воде для инъекций необязательно вместе с ионами плазмы или растворенным кислородом.
Композиции контрастных веществ по изобретению могут быть приготовлены в концентрациях, готовых к применению, либо в форме концентрата, предназначенного для разбавления перед применением. Обычно композиции в готовой для применения форме должны иметь концентрации йода не ниже 100 мг I/мл, предпочтительно по крайней мере 150 мг I/мл, при этом обычно предпочтительны концентрации по меньшей мере 300 мг I/мл, например 320-400 мг I/мл. Чем выше концентрация йода, тем выше диагностическая ценность, но при этом выше вязкость раствора и осмотические способности. Обычно максимальная йодная концентрация для данного соединения определяется его растворимостью и согласно верхним приемлемым пределам вязкости и осмоляльности.
Для контрастного вещества, вводимого путем инъекции, верхний желательный предел вязкости раствора при температуре окружающей среды (20oC) составляет 30 мП; однако вязкость до 50 или даже до 60 мП может быть приемлема, хотя ее применение в педиатрической радиографии обычно противопоказано. Для контрастных веществ, вводимых болюсной инъекцией, например в ангиографических процедурах, следует учитывать осмотические эффекты и предпочтительно осмоляльность должна быть ниже 1 осм/кг H2О, в основном ниже 850 мосм/кг H2О, в частности 50 или ниже, желательно в пределах 10 мосм изотоничности (около 300 мосм/кг H2О).
Для соединений по изобретению такие требования относительно вязкости, осмоляльности и йодных концентраций легко могут быть удовлетворены. Действительно, эффективные йодные концентрации могут быть достигнуты с гипотоническими растворами. Таким образом, может быть желательно доводить изотоничность раствора добавлением катионов плазмы с целью снижения вклада в токсичность, обусловленную эффектами ионного дисбаланса, сопровождающими болюсную инъекцию. Такие катионы желательно включать в пределах, предложенных в WO-90/01194 и WO-91/13636.
Предпочтительные содержания катионов плазмы для контрастных веществ по изобретению, особенно контрастных веществ для ангиографии, следующие:
натрий 2-100, главным образом 15-75, в частности 20-70, более конкретно 25-35 мМ,
кальций до 3,0, предпочтительно 0,05-1,6, желательно 0,1- 1,2, в частности 0,15-0,7 мМ,
калий до 2, предпочтительно 0,2-1,5, желательно 0,3-1,2, в частности 0,4-0,9 мМ,
магний до 0,8, предпочтительно 0,05-0,6 желательно 0,1-0,5, в частности 0,1-0,25 мМ.
В целом или частично катионы плазмы могут рассматриваться как противоионы в ионных контрастных веществах.
Обычно они предусматриваются в основном в форме солей с физиологически приемлемыми противоионами, например с хлоридом, сульфатом, фосфатом, гидрокарбонатом и так далее, особенно желательно их использование с анионами плазмы.
Кроме катионов плазмы контрастные вещества могут содержать другие противоионы, когда димер является ионным, и такие противоионы предпочтительно, конечно, должны быть физиологически приемлемыми. Примеры таких ионов включают ионы щелочных или щелочно-земельных металлов, аммония, меглумина, этаноламина, диэтаноламина, хлорида, фосфата и гидрокарбоната. Могут также быть использованы другие общепринятые для фармацевтических составов противоионы. Кроме того, композиции могут содержать другие компоненты, общепринятые для рентгеноконтрастных веществ, например буферы и так далее.
Приведенные в тексте источники приведены для ссылки.
Изобретение дополнительно описывается приведенными следующими далее неограничивающими примерами.
Пример 1. 1,3,5-Трийод-2, 4-ди(1,2,3-тригидрокси- 1-пропил)-6-(3-гидрокси-1 -пропен-1-ил)бензол.
a. 1, 3, 5-Трийод-2, 4, 6-триметилбензол.
Йод (19,0 г, 75 ммоль) растворяют в четыреххлористом углероде (75 мл). Добавляют мезитилен (7,0 мл, 50 ммоль) и бис(трифторацетокси)фенилйодид (35,5 г, 82 ммоль) и раствор перемешивают при температуре окружающей среды в течение 2 часов. Собранный и отфильтрованный осадок промывают охлажденным четыреххлористым углеродом и сушат. Выход: 20,5 г (82%).
1H-ЯМР (CDCl3): 3,31 (с).
b. 1,3,5-Трийод-2,4, 6-триацетоксиметилбензол.
Трийодмезитилен (19,5 г, 39 ммоль) добавляют к ледяной уксусной кислоте (200 мл), содержащей уксусный ангидрид (400 мл) и концентрированную серную кислоту (40 мл). Затем доставляют небольшими порциями за 3 часа твердый перманганат калия (24,6 г, 156 ммоль). После перемешивания в течение 16 часов растворитель упаривают и добавляют воду (200 мл). Водную суспензию экстрагируют дихлорметаном (250 мл) и органическую фазу промывают водой (3 х 50 мл), сушат (MgSO4) и упаривают. Твердый остаток суспендируют в ацетоне и белый кристаллический продукт собирают и фильтруют. Выход: 9,3 г (35%).
1H-ЯМР (CDCl3): 5,66 (с, 6H), 2,20 (с, 9H).
c. 1, 3, 5-Трийод-2, 4, 6-тригидроксиметилбензол.
1, 3, 5-Трийод-2, 4, 6-триацетоксиметилбензол (9,3 г, 13,8 ммоль) суспендируют в метаноле (120 мл) и добавляют К2СО3 (0,32 г, 2,3 ммоль). Смесь перемешивают при температуре окружающей среды в течение 16 часов и после нейтрализации раствора 2 М водной HCl органический растворитель упаривают. Остаток суспендируют в воде и собранный фильтрацией белый твердый продукт промывают водой, метанолом и диэтиловым эфиром. Выход: 7,1 г (94%).
1H-ЯМР (ДМСО-d6): 5,08 (с, 6H), 3,35 (ушир. с, 3H).
d. 1, 3, 5-Трийодбензол-2, 4, 6-триальдегид.
1, 3, 5-Трийод-2,4,6-тригидроксиметилбензол (4,5 г, 8,2 ммоль) растворяют в ДМСО (80 мл). Добавляют триэтиламин (51,7 мл, 371 ммоль) и пиридин SO3 (11,8 г, 74,2 ммоль) и смесь перемешивают в течение 2 часов. Две фазы разделяют и составляющую нижний слой фазу охлаждают до 0oC, выливают в воду (150 мл) и перемешивают 30 мин при 0oC. Собранный фильтрацией белый твердый продукт промывают водой и сушат. Выход: 3,0 г (67%).
1H-ЯМР (ДМСО-d6): 9,64 (с).
е. Метиловый эфир 1, 3, 5-трийод-бензол-2, 4, 6-трис (2-проп- 1-еновой кислоты).
Гидрид натрия (194 мг 80% в минеральном масле, 6,5 ммоль) растворяют в ДМСО (13 мл) и добавляют триэтилфосфоноацетат (1,16 мл, 5,80 ммоль). После перемешивания раствора в течение 30 мин добавляют 1, 3, 5-трийодбензол-2, 4, 6-триальдегид (700 мг, 1,30 ммоль) и реакционную смесь перемешивают 16 часов. Затем добавляют воду (200 мл) и доводят pH до 1 с помощью 2 М водной HCl. Суспензию экстрагируют дихлорметаном (2х200 мл) и объединенные органические фазы промывают водой (3х50 мл), сушат (MgSO4) и упаривают. Очистка колоночной хроматографией (силикагель CH2Cl2:метанол 99:1) дает чистый продукт в виде белого твердого вещества. Выход: 426 мг (44%).
1H-ЯМР (CDCl3): 7,48 (д. 3H, J=16,2 Гц), 5,95 (д, 3H, J=16,2 Гц), 4,30 (кв, 6H, J=7,2 Гц), 1,36 (т, 9H, J=7,2 Гц).
f. 1,3,5-Трийод-2,4,6-трис (1-гидроксипроп-ен-3-ил) бензол.
Метиловый эфир 1, 3, 5-трийод-бензол-2, 4, 6-трис (2-проп-1-еновой кислоты) (650 мг, 0,87 ммоль) растворяют в толуоле (10 мл) и добавляют диизобутилалюмогидрид (5,44 мл 1,2 М раствора в толуоле) при 0oC. После перемешивания в течение 40 минут при 0oC раствор выливают в метанол (50 мл) и образовавшуюся суспензию перемешивают еще 45 минут. Твердые продукты отфильтровывают и раствор упаривают, получая белый твердый остаток, который чистят, растирая с диэтиловым эфиром. Выход: 510 мг (94%).
1H-ЯМР (СD3ОD): 6,44 (д. 3H, J=16,0 Гц), 5,57 (дв. т. 3H, Jд=16,0 Гц, Jт=5,8 Гц), 4,23 (м, 6H).
g. 1,3,5-Трийод-2,4,6-трис (1-ацетоксипроп-ен-3-ил) бензол.
1,3,5-Трийод-2,4,6-трис (1-гидроксипроп-ен-3-ил) бензол (560 мг, 0,90 ммоль) растворяют в смеси пиридина (8 мл) и уксусного ангидрида (8 мл). После перемешивания при температуре окружающей среды в течение 16 часов растворитель упаривают и остаток чистят препаративной ЖХВР (RP-18, CH3CN: H2O 80:20). Выход: 310 мг (46%).
1H-ЯМР (CDCl3): 6,46 (дв. т. 3H, Jд=16,2 Гц, Jт=1,6 Гц), 5,68 (дв. т. 3H, Jд=16,2 Гц, Jт=5,6 Гц), 4,83 (дв. т. 6H, J1=5,6 Гц, J2=1,6 Гц), 2,12 (с, 9H).
13C-ЯМР (CDCl3): 170,6, 146,3, 140,9, 131,5, 98,7, 63,5, 20,9.
h. 1,3,5-Трийод-2,4-ди (1,2,3-тригидрокси-1-пропил) -6-(3-гидрокси-1-пропен-1-ил)бензол.
1,3,5-Трийод-2,4,6- трис(1-ацетоксипроп-ен-3-ил) бензол (100 мг, 0,133 ммоль) растворяют в муравьиной кислоте (5 мл), содержащей перекись водорода (0,054 мл). Смесь перемешивают при температуре окружающей среды 21 час и растворитель упаривают. Добавляют метанол (5 мл) и впоследствии твердый К2СО3 (195 мг) и после перемешивания в течение 1 часа растворитель упаривают. Продукт чистят препаративной ЖХВР (CH3CN:H2О 3:97).
1H-ЯМР (D2О): 6,45 (д. 1H, J=16,0 Гц), 5,40-5,55 (м, 1H), 4,54-4,90 (м, 11H), 4,23-4,31 (м, 2H), 3,62-3,91 (м, 4H).
MC(ESP): 692(М+).
Пример 2. 1,3,5-Трийод-2,4,6-три (1,2,3-тригидрокси-1- пропил)бензол.
1,3,5-Трийод-2,4,6- трис-(1-ацетоксипроп-ен-3-ил) бензол (100 мг, 0,133 ммоль, из примера 1g) растворяют в муравьиной кислоте (5 мл), содержащей перекись водорода (0,081 мл). Смесь перемешивают 40 часов при комнатной температуре и растворитель упаривают. Добавляют метанол (5 мл) и впоследствии твердый К2CO3 (195 мг) и после перемешивания в течение 1 часа растворитель упаривают. Продукт чистят препаративной ЖХВР (CH3С:H2О 3:97).
1H-ЯМР (СD3ОD): 4,57-4,94 (м, 15H), 3,62-3,99 (м, 6H).
МС (ESP): 744 (М+18).
Пример 3. N-Ацетил-3-гидроксиметил-5-(2, 3- дигидроксипропил-аминокарбонил)-N-(2, 3-дигидроксипропил)-2, 4, 6- трийоданилин.
а. Метиловый эфир 1-гидроксиметил-3-нитро-5-бензойной кислоты.
Монометиловый эфир 1-нитроизофталевой кислоты (22,5 г, 100 ммоль) растворяют в сухом ТГФ (675 мл) и добавляют BF3•Et2O (25,2 мл, 200 ммоль). Затем порциями за 1 час добавляют NaBH4 (5,1 г, 135 ммоль). После дополнительного перемешивания в течение 2 часов медленно добавляют этанол (20 мл) и впоследствии воду (200 мл) и диэтиловый эфир (400 мл). Фазы разделяют и водную фазу экстрагируют один раз диэтиловым эфиром (100 мл). Объединенные органические фазы промывают насыщенным водным раствором NaHCO3, сушат (Na2SO4) и упаривают. Выход: 20 г (96%). ЖХВР-анализ показывает > 95% чистоту продукта.
1H-ЯМР (CDCl3): 8,72 (с, 1H), 8,42 (с, 1H), 8,32 (с, 1H), 4,86 (с, 2H), 3,97 (с, 3H), 2,37 (ушир. с, 1H).
b. 1-Гидроксиметил-3-нитро-5-(2, 3- дигидроксипропиламинокарбонил)бензол.
Сложный метиловый эфир из примера 3а (20,5 г, 97 ммоль) смешивают с 2,3-дигидроксипропиламином (9,6 г, 106 ммоль) и смесь нагревают до 90oC. Через 45 минут давление снижают до 200 мм и нагревание продолжают в течение 2 часов. Сырой продукт >95% чистоты по ЖХВР-анализу используют в следующей стадии без дополнительной очистки. Выход: 22,8 г (87%).
1H-ЯМР (СD3ОD): 8,57 (с, 1H), 8,38 (с, 1H), 8,19 (с, 1H), 4,77 (с, 2H), 3,81-3,88 (м, 1H), 3,39-3,63 (м, 4H).
с. 3-Гидроксиметил-5-(2, 3-дигидроксипропиламинокарбонил)- анилин.
1-Гидроксиметил-3-нитро-5- (2,3-дигидроксипропиламинокарбонил) бензол (12,0 г, 44,4 ммоль) гидрируют в метаноле (150 мл) при 60 фунт/кв. дюйм (4,218 кг/см2) H2, используя Pd/C (10%, 100 мг) в качестве катализатора. Катализатор удаляют фильтрацией и остаток упаривают. Добавление метанола (10 мл) вызывает осаждение продукта в виде белого твердого вещества, которое отфильтровывают и сушат. Выход: 6,6 г (62%).
1H-ЯМР (СD3ОD): 7,05-7,09 (м, 1H), 6,98-7,03 (м, 1H), 6,83- 6,87 (м, 1H), 4,53 (с, 2H), 3,77-3,85 (м, 1H), 3,8-3,59 (м, 4H), 3,32-3,42 (м, 1H).
МС (ESP, m/e): 241 ([M+1]+, 100%).
d. 3-Гидроксиметил-5-(2, 3-дигидроксипропиламинокарбонил)-2, 4, 6-трийоданилин.
3-Гидроксиметил-5- (2, 3-дигидроксипропиламинокарбонил) -анилин (500 мг, 2,1 ммоль) растворяют в воде (175 мл) и добавляют водный раствор KICl2 (70%, вес/вес) порциями по 0,1 мл за 8 часов. Общее количество добавленного раствора KICl2 составляет 1,0 мл. После общего реакционного времени 6 часов, раствор экстрагируют этилацетатом (1000 мл), который отделяют и промывают водным раствором Na2S2O3 (0,2 М, 100 мл). Упаривание с последующей очисткой препаративной ЖХВР дает 432 мг (33%) чистого продукта.
1H-ЯМР (СD2ОD): 5,10 (с, 2H), 3,90-3,98 (м, 1H), 3,72 (тр. д, J1=0,7 Гц, J2= 4,2 Гц, J= 11,4 Гц, 1H), 3,60 (дв. д, J1=6,0 Гц, J2=11,4 Гц, 1H), 3,49 (тр. д, J1=1,2 Гц, J2=6,0 Гц, J3=13,5 Гц, 1H), 3,37 (тр. д, J1=1,2 Гц, J2= 6,1 Гц, J3 =13,2 Гц, 1H), 2,62 (с, 1H), 2,28 и 2,34 (2с, 2H).
МС (ESP, m/e): 618 (М+100%), 640 ([M+Na]+, 55%).
e. N-ацетил-3-ацетоксиметил-5-(2, 3-диацетоксипропиламинокарбонил)- 2,4,6-трийоданилин.
3-Гидроксиметил-5-(2, 3-дигидроксипропиламинокарбонил) -2,4,6-трийоданилин (3,3 г, 5,3 ммоль) суспендируют в ледяной уксусной кислоте (12 мл), содержащей уксусный ангидрид (48 мл) и концентрированную серную кислоту (0,08 мл). Смесь перемешивают при 60oC 3 часа, дают охладиться до комнатной температуры и добавляют CH2Cl2 (100 мл) и воду (100 мл). Органическую фазу промывают водой (3х50 мл) и насыщенным водным раствором NaHCO3 (2х50 мл). После высушивания (MgSO4) и упаривания остаток растворяют в смеси CH2Cl2 и метанола (9:1) и фильтруют через тонкий слой силикагеля и упаривают. Выход: 3,0 г (71%).
1H-ЯМР (CDCl3): 8,27-8,32 (м, 1H), 5,51 (с, 2H), 5,18-5,22 (м, 1H), 4,17-4,42 (м, 2H), 3,67-3,84 (м, 1H), 3,41-3,60 (м, 1H), 2,56 (с, 3H), 2,04-2,14 (8c, 12H).
МС (ESP, m/e): 786 (М+, 100%), 809 ([M+Na]+, 81%).
f. N-Ацетил-3-гидроксиметил-5-(2, 3- дигидроксипропиламинокарбонил)-N-(2, 3-дигидроксипропил)-2, 4, 6-трийоданилин.
N-Ацетил-3-ацетоксиметил-5- (2, 3- диацетоксипропиламинокарбонил)-2,4,6-трийоданилин (1,0, 1,27 ммоль) суспендируют в смеси метанола (6 мл) и воды (30 мл) и рН доводят до 12,0, используя 2 М водный раствор. После перемешивания в течение 1 часа добавляют 1-бром-2, З-пропандиол (0,99 г, 6,4 ммоль) и pH доводят до 11,6, используя 2 М водный раствор HCl. Вновь добавляют 1-бром-2, 3-пропандиол (0,99 г, 6,4 ммоль) через 16 и 18 часов и после 24 часов pH доводят до 6,5, используя 2 М водный раствор HCl. После упаривания остаток чистят препаративной ЖХВР. Выход: 0,373 г (40%).
1H-ЯМР (D2О): 5,20 (с, 2H), 3,23-3,99 (м, 12H), 1,79 (2c, 3H).
MC (ESP, m/e): 734 (M+, 60%), 756 ([M+Na]+, 100%).
Пример 4. N, N1-бис (гидроксиацетил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
а. 3, 5-Диаминобензиловый спирт.
Раствор 3, 5-динитробензилового спирта (2,20 г, 11,1 ммоль) в метаноле (90 мл) гидрируют в гидрогенизаторе Парра при 60 фунт/кв. дюйм в присутствии Pd/C-катализатора (10%, 100 мг). Раствор фильтруют и растворитель удаляют упариванием. Сырой продукт используют без очистки в следующей стадии.
1H-ЯМР (CDCl3): 6,12-6,14 (м, 2H), 5,96-5,98 (м, 1H), 4,51 (с, 2H), 3,60 (ушир. с, 4H).
b. 3,5-Диамино-2, 4, 6-трийодбензиловый спирт.
Сырой продукт из предыдущего примера растворяют в смеси метанола (310 мл) и воды (60 мл) и pH доводят до 1,5, используя 4 М водный раствор HCl. Добавляют по каплям раствор KICl2 (70%, 11,2 г) при такой скорости, что между каждым добавлением происходит исчезновение окраски. После дополнительного перемешивания в течение 5 минут осадок отфильтровывают и промывают водой (3х50 мл), диэтиловым эфиром (3х50 мл) и сушат. Выход: 4,50 г (80%).
1H-ЯМР (ДМСО-d6): 5,20 (с, 4H), 4,81-4,94 (м, 3H).
с. 3, 5-Диамино-2, 4, 6-трийодбензилацетат.
3, 5-Диамино-2, 4, 6-трийодбензиловый спирт (4,42 г, 8,56 ммоль) растворяют в смеси пиридина (50 мл) и уксусного ангидрида (2,5 мл) и смесь перемешивают при комнатной температуре 16 часов. Растворители упаривают и остаток промывают диэтиловым эфиром (3х50 мл), водой (3х50 мл) и сушат. Выход: 4,52 г (95%).
1H-ЯМР (ДМСО-d6): 5,35 [с, 2H), 5,28 (с, 4H), 2,04 (с, 3H).
1C-ЯМР (ДМСО-d6): 170,5, 148,1, 139,0, 78,0, 73,6, 70,0, 20,8.
d. 3, 5-Бис (ацетоксиацетиламино)-2, 4, 6-трийодбензилацетат.
3, 5-Диамино-2, 4, 6-трийодбензилацетат (5,58 г, 10 ммоль) смешивают с ацетоксиацетилхлоридом (3,22 мл, 30 ммоль) и диметилацетамидом (50 мл) и смесь перемешивают 17 часов. Добавляют диэтиловый эфир (600 мл) и через 20 минут осадок собирают, промывают водой (3х50 мл) и сушат. Перекристаллизация из ацетонитрила дает 3,3 г (44%) чистого продукта.
1H-ЯМР (ДМСО-d6): 10,27 и 10,19 (2c, 2: 1,2Н), 5,51 (с, 2H), 4,66 (с, 3H), 2,12 (с, 3H), 2,06 (с, 3H), 2,13 (с, 3H).
е. N, N' -Бис(гидроксиацетил)-3, 5-диамино-2, 4, 6- трийодбензиловый спирт.
3, 5-Бис (ацетоксиацетиламино)-2, 4, 6-трийодбензилацетат (152 мг, 0,2 ммоль) растворяют в смеси метанола (6 мл) и 1 М водного NaOH (2 мл) и раствор перемешивают 2 часа при комнатной температуре. После нейтрализации 1 М HCl, растворители удаляют упариванием и продукт чистят препаративной ЖХВР. Выход: 105 мг (83%).
МС (ESP, m/e): 655 ([M+Na]+, 100%).
Пример 5. N,N1 -Бис (гидроксиацетил)-N-(2-гидроксиэтил)-3, 5- диамино-2, 4, 6-трийодбензиловый спирт.
a. N, N1 -Бис (ацетоксиацетил)-N-(2-ацетоксиэтил)-3, 5- диамино- 2, 4, 6-трийодбензилацетат.
3,3-Бис (ацетоксиацетиламино)-2, 4, 6-трийодбензилацетат (2,15 г, 2,84 ммоль) растворяют в смеси ДМСО (5 мл) и диметилацетамида (5 мл), содержащей Cs2CO3 (1,0 г, 3,07 ммоль) и 2-бромэтилацетат (0,31 мл, 2,84 мл). Смесь перемешивают при комнатной температуре 48 часов, добавляют диэтиловый эфир (100 мл) и водный NaH2PO4-буфер и органическую фазу промывают водой и сушат. Очистка препаративной ЖХВР дает 520 мг (22%) продукта.
1H-ЯМР (CDCl3): 7,86 (с, 1H), 5,66 (с, 2H), 4,80 (с, 2H), 3,81-4,44 (м, 6H), 2,13-2,27 (м, 12H).
МС (ESP, m/e): 845 ([M+1]+, 100%), 866 ([M+Na]+, 24%).
b. N,N1 -Бис(гидроксиацетил)-N-(2-гидроксиэтил)-3, 5-диамино- 2, 4, 6-трийодбензиловый спирт.
N, N1 -Бис(ацетоксиацетил)-N-(2-aцeтoкcиэтил)-3,5-диамино -2, 4, 6- трийодбензилацетат (0,50 г, 0,59 ммоль) растворяют в смеси метанола (5 мл), воды (5 мл) и водного 1 М NaOH (1 мл). Раствор перемешивают 2 часа, pH доводят до 2, используя водную HCl и продукт чистят препаративной ЖХВР. Выход: 240 мг (67%).
1H-ЯМР (ДМСО-d6): 9,85 (ушир. с, 1H), 5,77 (ушир. с, 1H), 4,79-5,26 (м, 3H), 3,20-3,71 (м, 6H).
МС (ESP, m/e): 676 (М+, 57%), 698 ([M+Na]+, 100%).
Пимер 6. N,N1- Бис(гидроксиацетил)-N,N1 -бис(2-гидроксиэтил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
3,5-Бис (ацетоксиацетиламино)-2, 4, 6- трийодбензилацетат (190 мг, 0,25 ммоль), полученный по примеру 4, растворяют в диметилацетамиде (5 мл) в атмосфере аргона. Добавляют 2-бромэтилацетат (0,22 мл, 2,0 ммоль) и К2СО3 (138 мг, 1,0 ммоль). Через 16 часов добавляют ДМСО (1,5 мл), 2-бромэтилацетат (0,22 мл, 2,0 ммоль) и К2СО3 (138 мг, 1,0 ммоль) и суспензию дополнительно перемешивают 24 часа. Добавляют водный NaH2PO4 и полученный раствор экстрагируют эфиром (3х25 мл). Объединенные органические фазы промывают водой (4х20 мл) и затем сушат (MgSO4). Растворитель удаляют упариванием и остаток, бесцветное масло, растворяют в смеси метанола (3 мл) и 1 М водного NaOH (3 мл). Раствор перемешивают 1 час pH доводят до 6, используя водную HCl, и растворители удаляют упариванием. Очистка препаративной ЖХВР дает 60 мг (33%) продукта.
МС (ESP, m/e): 720 (М+, 100%), 742 ([M+Na]+, 36%).
Пример 7. N,N1 -Бис (гидроксиацетил)-N-(2, 3- дигидроксипропил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
a. N,N1- Бис(ацетоксиацетил)-N-[(2,2-диметил-1,3-диоксолан-4- ил)-метил] -3, 5-диамино-2, 4, 6-трийодбензилацетат.
3,5-Бис (ацетоксиацетиламино)-2, 4, 6-трийодбензилацетат (90 мг, 0,12 ммоль), полученный по примеру 4, растворяют в смеси диметилацетамида (3 мл) и ДМСО (3 мл). Добавляют 4-бром-метил-2,2- диметил-1,3-диоксолан (0,097 г, 0,5 ммоль) и (0,10 г, 0,3 ммоль) и раствор перемешивают при комнатной температуре 48 часов. Добавляют водный NaH2PO4 и раствор экстрагируют диэтиловым эфиром (3х25 мл). Объединенные органические фазы промывают водой (3х15 мл), сушат (MgSO4) и упаривают. Очистка препаративной ЖХВР дает 36 мг (35%) чистого продукта.
1H-ЯМР (ДМСО-d6): 10,30 (ушир. с, 1H), 5,53 (с, 2H), 3,61-4,68 (м, 9H), 2,13 (с, 3H), 2,07 (с, 3H), 2,06 (с, 3H), 1,17-1,34 (м, 6H).
МС (ESP, m/e): 894 ([M+Na]+, 100%), 910 ([M+K]+, 11%).
b. N,N1- Бис (гидроксиацетил)-N-[2, 3-дигидроксипропил)-3, 5- диамино-2, 4, 6-трийодбензиловый спирт.
N, N1-Бис(ацетоксиацетил)-N-(2, 2-диметил-1, 3-диоксолан-4-ил)- метил-3, 5-диамино-2, 4, 6-трийодбензилацетат (36 мг, 0,042 ммоль) растворяют в смеси метанола (3 мл) и воды (4 мл) и pH доводят до 12, используя 1 М водный раствор NaOH. После перемешивания в течение 2 часов pH доводят до 1, используя 1 М водную HCl, и перемешивание продолжают 16 часов. Раствор нейтрализуют водным NaH2PO4-буфером, растворители удаляют упариванием и остаток чистят препаративной ЖХВР, получая 24 мг (81%) чистого продукта.
МС (ESP, m/e): 704 (М+, 100%), 726 ([M+Na]+, 34%).
Пример 8. N, N1- Бис(2-гидроксипропионил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
a. N, N1- Бис(2-ацетоксипропионил)-3, 5-диамино-2, 4, 6- трийодбензилацетат.
3, 5-Диамино-2, 4, 6-трийодбензилацетат (2,79 г, 5,0 ммоль) растворяют в диметилацетамиде (25 мл) и охлаждают до 0oC. Добавляют по каплям 2-ацетоксипропионилхлорид (3,73 г, 25 ммоль) и смесь перемешивают при комнатной температуре 17 часов. Растворители упаривают и остаток чистят флэш-хроматографией на силикагеле, используя смесь CH2Cl2 и ацетонитрила (5:1) в качестве элюента. Выход: 2,21 г (56%).
1H-ЯМР (ДМСО-d6): 10,20- 10,23 (м, 2H), 5,52 (с, 2H), 5,21-5,24 (м, 2H), 2,06-2,13 (м, 9H), 1,51 (д, J=6,9 Гц, 6H).
b. N,N1- Бис(2-гидроксипропионил)-3, 5-диамино-2, 4, 6- трийодбензиловый спирт.
N, N1- Бис(2-ацетоксипропионил) -3, 5-диамино-2, 4, 6-трийодбензилацетат (0,16 г, 0,2 ммоль) растворяют в смеси метанола (5 мл) и воды (5 мл) и pH доводят до 12, используя 1 М водный раствор NaOH. После перемешивания в течение 15 часов раствор нейтрализуют 1 М HCl и растворители удаляют упариванием. Очистка препаративной ЖХВР дает 61 мг (46%) чистого продукта.
МС (ESP, m/e): 660 (М+, 5%), 682 ([M+Na]+, 100%), 698 ([M+K]+, 17%).
Пример 9. N,N1- Бис(2-гидроксипропионил)-N-(2- гидроксиэтил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
a. N, N1- Бис(2-ацетоксипропионил)-N- (2-ацетоксиэтил) -3, 5-диамино-2, 4, 6-трийодбензилацетат.
N, N1- Бис(2-ацетоксипропионил)-3, 5-диамино-2, 4, 6-трийодбензилацетат (393 мг, 0,50 ммоль) растворяют в смеси диметилацетамида (4 мл) и ДМСО (4 мл), содержащей 2-бромэтилацетат (0,083 г, 0,50 ммоль) и Cs2СО3 (244 мг, 0,75 ммоль). Смесь перемешивают 17 часов, добавляют воды (20 мл) и смесь экстрагируют диэтиловым эфиром (3х25 мл). Объединенные органические фазы промывают водой (3х20 мл), сушат (MgSO4) и упаривают. Остаток чистят препаративной ЖХВР, получая 80 мг (18%) чистого продукта.
1H-ЯМР (СD3ОD): 5,72-5,82 (м, 2H), 5,20-5,42 (м, 2H), 3,55-4,48 (м, 4H), 1,90-2,24 (м, 12H), 1,66 (д, J=7,1 Гц, 6H).
MC(ESP, m/e): 1004 ([M+Cs]+, 100%).
b. N,N1- Бис(2-гидроксипропионил)-N- (2-гидроксиэтил-3, 5- диамино-2, 4, 6-трийодбензиловый спирт.
N,N1 -Бис(2-ацетоксипропионил) -N- (2-ацетоксиэтил) -3, 5- диамино-2, 4, 6-трийодбензилацетат (120 мг, 0,14 ммоль) растворяют в смеси воды (7 мл) и метанола (7 мл) и pH доводят до 12, используя 1 М водный раствор NaOH. Смесь перемешивают 2 часа, pH доводят до 7 с помощью водной HCl и растворители упаривают. Продукт чистят препаративной ЖХВР. Выход: 70 мг (72%).
1H-ЯМР (СD3ОD): 5,27-5,34 (м, 2H), 4,31-4,41 (м, 1H), 3,82- 4,12 (м, 4H), 3,55-3,73 (м, 1H), 1,51-1,60 (м, 3H), 1,23-1,32 (м, 3H).
MC (ESP, m/e): 726 ([M+Na]+, 100%).
Пример 10. N,N1- Бис(2-гидроксипропионил)-N,N1 -бис (2-гидроксиэтил-3,5- диамино-2,4,6- трийодбензиловый спирт.
а. N,N1 -Бис (2-ацетоксипропионил)-N,N1 -бис (2- ацетоксиэтил)- 3, 5-диамино-2, 4, 6-трийодбензилацетат.
N, N1 -Бис(2-ацетоксипропионил) -3, 5-диамино-2, 4, 6-трийодбензилацетат (197 мг, 0,25 ммоль) растворяют в смеси диметилацетамида (5 мл) и ДМСО (1,5 мл), содержащей 2-бромэтилацетат (0,11 мл, 1,0 ммоль) и Cs2CO3 (162 мг, 0,50 ммоль). Смесь перемешивают 67 часов, добавляют воду и смесь экстрагируют диэтиловым эфиром (2х75 мл). Объединенные органические фазы промывают водой (5х75 мл), сушат (Na2SO4) и упаривают. Остаток чистят препаративной ЖХВР, получая 35 мг (15%) чистого продукта.
1H-ЯМР (ДМСО-d6): 5,49-5,73 (м, 2H), 4,97-5,22 (м, 2H), 3,49-4,00 (м, 6H), 1,86-12,08 (м, 15H), 1,09-1,58 (м, 6H).
b. N,N1 -Бис(2-гидроксипропионил)-N,N1 -бис (2- гидроксиэтил)- 3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
N, N1 -Бис (2-ацетоксипропионил)-N,N1 - бис (2-ацетоксиэтил-3, 5-диамино-2, 4, 6-трийодбензилацетат (175 мг, 0,18 ммоль) растворяют в смеси метанола (8 мл) и воды (8 мл) и pH доводят до 12 с помощью 1 М водного NaOH. После перемешивания в течение 3 часов раствор нейтрализуют водной HCl. Очистка препаративной ЖХВР дает 50 мг (37%) чистого продукта.
1H-ЯМР (СD3ОD): 5,26-5,38 (м, 2H), 3,44-4,08 (м, 6H), 1,32- 1,59 (м, 6H).
МС (ESP, m/e): 770 ([M+Na]+, 100%).
Пимер 11. N,N1 -Бис(2-гидроксипропионил)-N- (2,3- дигидроксипропил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
a. N, N1 -Бис(ацетоксипропионил)-N-[(2,2-диметил-1, 3-диоксолан-4-ил)-метил]-3, 5-диамино-2, 4, 6-трийодбензилацетат.
N, N1-Бис (2-ацетоксипропионил)-3, 5-диамино-2, 4, 6-трийодбензилацетат (393 мг, 0,50 ммоль) растворяют в смеси ДМСО (4 мл) и диметилацетамида (4 мл), содержащей Cs2CO3 (1,80 г, 5,52 ммоль) и 4-бромметил-2, 2-диметил-1, 3-диоксолан (1,0 мл). Смесь перемешивают 7 дней и затем обрабатывают аналогично примеру 7а. Очистка препаративной ЖХВР дает 115 мг (26%) чистого продукта.
1H-ЯМР (СD3ОD): 5,61-5,75 (м, 2H), 5,03-5,44 (м, 2H), 3,47- 4,55 (м, 6H), 1,98-2,23 (м, 9H), 1,30-1,71 (м, 12H).
МС (ESP, m/e): 922 ([M+Na]+, 100%).
b. N,N1- Бис(2-гидроксипропионил)-N-(2, 3- дигидроксипропил)- 3,5-диамино-2, 4, 6-трийодбензиловый спирт.
N, N1 -Бис(ацетоксипропионил)-N-[(2, 2- диметил-1, 3-диоксолан-4-ил)-метил]-3, 5-диамино-2, 4, 6- трийодбензилацетат (115 мг, 0,13 ммоль) растворяют в смеси метанола (8 мл) и воды (8 мл) и pH доводят до 12 с помощью водного 1 M NaOH. Через 2,5 часа pH доводят до 1 с помощью 2 М водной HCl. После перемешивания в течение 17 часов раствор нейтрализуют водным NaH2PO4-буфером и растворители удаляют упариванием. Очистка препаративной ЖХВР дает 65 мг (69%) чистого продукта.
МС (ESP, m/e): 756 ([M+Na]+, 100%).
Пример 12. N, N1 -Бис(2, 3-дигидроксипропионил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
а. N,N1 -Бис(2, 2-диметил-1, 3-диоксолан-4-карбонил)-3, 5-диамино-2, 4, 6- трийодбензилацетат.
3, 5-Диамино-2, 4, 6-трийодбензилацетат (3,54 г, 6,3 ммоль) и хлорангидрид 2,2-диметил-1,3-диоксолан-4-карбоновой кислоты (3,13 г, 19 ммоль) растворяют в диметилацетамиде (40 мл) и раствор перемешивают 3,5 часа. Растворитель удаляют упариванием и остаток обрабатывают водным раствором NaHCO3. Кристаллический остаток отфильтровывают, промывают водой и сушат. Очистка флэш-хроматографией с использованием смеси CH2Cl2 и CH3CN (3:1) в качестве элюента дает 1,90 г (37%) чистого продукта.
1H-ЯМР (ДМСО-d6): 9,93-10,02 (м, 2H), 5,30 (с, 2H), 4,58 (т, J=6,2 Гц, 1H), 4,29 (т, J=7,3 Гц, 1H), 4,10 (т, J=6,1 Гц, 1H), 2,06 (с, 3H), 1,54 (с, 3H), 1,38 (с, 3H).
МС (ESP, m/e): 902 ([М+диметилацетамид]+, 100%).
b. N, N1 -Бис(2, 3-дигидроксипропионил)-3, 5-диамино-2, 4, 6- трийодбензиловый спирт.
N, N1 -Бис(2, 2-диметил-1, 3-диоксолан-4-карбонил)-3, 5-диамино-2, 4, 6- трийодбензилацетат (1,25 г, 1,55 ммоль) растворяют в смеси воды (50 мл), метанола (25 мл) и концентрированной HCl (0,5 мл). После перемешивания в течение 4 часов раствор нейтрализуют водным NaH2PO4 и растворители удаляют упариванием. Остаток растворяют в воде (10 мл) и pH доводят до 12 с помощью водного NaOH. Через 30 минут раствор снова нейтрализуют и растворитель упаривают. Продукт чистят препаративной ЖХВР. Выход: 483 мг (45%).
1H-ЯМР: 9,65-9,83 (м, 2H), 5,77 (с, 2H), 5,20 (с, 1H), 4,95-5,03 (м, 2H), 4,81 (с, 2H), 4,00-4,08 (м, 2H), 3,72-3,82 (м, 2H), 3,50-3,63 (м, 2H).
МС (ESP, m/e): 692 (М+, 62%), 714 ([M+Na]+, 100%).
Пример 13. N, N1 -Бис(2, 3-дигидроксипропионил)-N-(2- гидроксиэтил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
N, N1 -Бис(2, 2-диметил-1, 3-диоксолан-4-карбонил)-3, 5- диамино-2, 4, 6-трийодбензилацетат (204 мг, 0,25 ммоль) растворяют в смеси диметилацетамида (4 мл) и ДМСО (2,5 мл), содержащей Cs3CO3 (650 мг, 2,0 ммоль) и 2-бромэтилацетат (0,035 мл, 0,31 ммоль). После перемешивания в течение 1 недели добавляют диэтиловый эфир (150 мл) и NaH2PO4-буфер (100 мл), органическую фазу отделяют и водную фазу экстрагируют диэтиловым эфиром (150 мл). Затем объединенные органические фазы промывают водой (6х100 мл), сушат (Na2SO4) и упаривают. Остаток растворяют в смеси метанола (20 мл) и воды (20 мл) и pH доводят до 12 с помощью водного NaOH. После перемешивания в течение 1 часа pH доводят до 1,5 с помощью концентрированной HCl и перемешивают дополнительно 16 часов. Раствор нейтрализуют водным NaH2PO4 и растворители упаривают. Препаративная ЖХВР дает 55 мг (30%) чистого продукта.
МС (ESP, m/e): 736 (М+, 28%), 758 ([M+Na]+, 100%).
Пример 14. N,N1 -Бис(2, 3-дигидроксипропионил)-N,N1 -бис-(2- гидроксиэтил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
N, N1 -Бис (2, 2-диметил-1, 3-диоксолан-4-карбонил)-3, 5-диамино-2, 4, 6-трийодбензилацетат (204 мг, 0,25 ммоль) растворяют в смеси диметилацетамида (5 мл) и ДМСО (1,5 мл), содержащей К2СО3 (276 мг, 2,0 ммоль) и 2-бромэтилацетат (0,44 мл, 4,0 ммоль). После перемешивания в течение 48 часов добавляют водный NaH2PO4-буфер и смесь экстрагируют диэтиловым эфиром (2х150 мл). Объединенные органические фазы промывают водой (6х100 мл), сушат (Na2SO4) и упаривают. Твердый остаток растворяют в смеси метанола (12 мл) и воды (12 мл) и pH доводят до 12 с помощью водного NaOH. После перемешивания в течение 18 часов раствор подкисливают концентрированной HCl (0,70 мл) и перемешивание продолжают 3 часа. Раствор нейтрализуют и растворители упаривают. Продукт чистят препаративной ЖХВР. Выход: 98 мг (50%).
МС (ESP, m/e): 802 ([M+Na]+, 100%).
Пример 15. N, N1 -Бис (2, 3-дигидроксипропионил)-N-(2, 3- дигидроксипропил)-3, 5-диамино-2, 4, 6-трийодбензиловый спирт.
N, N1 -Бис (2, 2-диметил-1, 3-диоксолан-4-карбонил)-3, 5- диамино-2, 4, 6-трийодбензилацетат (408 мг, 0,50 ммоль) растворяют в смеси диметилацетамида (4 мл) и ДМСО (4 мл), содержащей Cs2SO3 (1,80 г, 5,52 ммоль) и хлорангидрид 2, 2-диметил-1,3-диоксолан-4-карбоновой кислоты (2,0 мл). После перемешивания в течение 8 дней добавляют водный NaH2PO4 (100 мл) и смесь экстрагируют диэтиловым эфиром (2х150 мл). Объединенные органические фазы промывают водой (6х100 мл), сушат (Na2SO4) и упаривают. Остаток растворяют в смеси метанола (10 мл) и воды (10 мл) и pH доводят до 12 с помощью водного NaOH. После перемешивания в течение 2 часов добавляют концентрированную HCl (1,0 мл) и перемешивание продолжают 16 часов. После нейтрализации растворители упаривают и остаток чистят препаративной ЖХВР. Выход: 149 мг (39%).
МС (ESP, m/e): 766 (М+, 60%), 788 ([M+Na]+, 100%).
Пример 16. Бис-[3-гидроксиметил-5-(2, 3-дигидроксипропиламинокарбонил)- 2, 4, 6-трийодбензоламид] щавелевой кислоты.
а. 3-Ацетоксиметил-5-(2, 3-диацетоксипропиламинокарбонил)-2, 4, 6-трийоданилин.
3-Гидроксиметил-5-(2, 3-дигидроксипропиламинокарбонил) -2, 4, 6- трийоданилин (1,89 г, 3,06 ммоль), полученный в примере 3d, растворяют в смеси уксусного ангидрида (5 мл) и пиридина (5 мл). Смесь перемешивают при комнатной температуре 24 часа, добавляют CH2Cl2 (100 мл) и раствор промывают водой (3х25 мл), насыщенным водным раствором NaHCO3, сушат (Na2SO4) и упаривают. Продукт чистят флэш-хроматографией на силикагеле, используя смесь CH2Cl2 и метанола (98:2) в качестве элюента. Выход: 1,30 г (57%).
1H-ЯМР (CDCl3): 6,10-6,25 (м, 1H), 5,48 (с, 2H), 5,20-5,28 (м, 1H), 4,22-4,43 (м, 2H), 3,53-3,89 (м, 2H), 2,06-2,13 (м, 9H).
МС (ESP, m/e): 744 (М+, 100%).
b. Бис-[3-гидроксиметил-5-(2, 3- дигидроксипропиламинокарбонил)-2, 4, 6-трийодбензоламид] щавелевой кислоты.
3-Ацетоксиметил-5- (2, 3-диацетоксипропиламинокарбонил)- 2, 4, 6-трийоданилин (100 мг, 0,134 ммоль) растворяют в диоксане (1,0 мл) и раствор нагревают до 90oC. Добавляют оксалилхлорид (0,096 ммоль) и смесь перемешивают при 78oC в течение 17 часов. После охлаждения до комнатной температуры добавляют воду (1,0 мл) и pH доводят до 12 с помощью водного NaOH. После перемешивания в течение 4 часов раствор нейтрализуют, растворители упаривают и остаток чистят препаративной ЖХВР. Выход: 14 мг (16%).
1H-ЯМР (СD3ОD): 5,24 (с, 2H), 3,38-4,03 (м, 10H).
МС (ESP, m/e): 1290 (М+, 33%), 1312 ([M+Na]+,100%).
Пример 17. Бис-[3-гидроксиметил-5-(2, 3-дигидроксипропиламинокарбонил)- 2,4,6-трийодбензоламид малоновой кислоты.
3-Ацетоксиметил-5- (2,3-диацетоксипропиламинокарбонил) -2, 4, 6-трийоданилин (100 мг, 0,134 ммоль), полученный по примеру 16а, растворяют в диоксане (1,0 мл) и добавляют малонилхлорид (0,097 ммоль). Смесь перемешивают при 90oC в течение 2 часов и раствору дают охладиться до комнатной температуры. Добавляют воду (1 мл) и pH доводят до 12 с помощью водного NaOH. После перемешивания при 60oC в течение 18 часов раствор нейтрализуют и растворитель упаривают. Продукт чистят препаративной ЖХВР. Выход: 34 мг (39%).
МС (ESP, m/e): 1304 (М+, 68%), 1326 ([M+Na]+, 100%).
Пример 18. N,N1 -Диацетил-N,N1 -бис [3-гидроксиметил-5-(2, 3- дигидроксипропиламинокарбонил)-2, 4, 6-трийодфенил]-1, 3-диамино-2- гидроксипропан.
N-Ацетил-3-ацетоксиметил-5-(2, 3-ацетоксипропиламинокарбонил)-2, 4, 6- трийоданилин (300 мг, 0,38 ммоль), полученный по примеру 3e, растворяют в смеси воды (1,2 мл) и метанола (0,2 мл) и pH доводят до 12 с помощью водного NaOH. Добавляют эпихлоргидрин (0,28 ммоль) и смесь перемешивают при комнатной температуре 65 часов. Раствор нейтрализуют и продукт выделяют препаративной ЖХВР. Выход: 60 мг (20%).
МС (ESP, m/e): 1398 ([M+Na]+, 100%).
Пример 19. N-3-Гидроксиметил-5- (2,3-дигидроксипропиламинокарбонил) -2,4,6-трийодфенил -N'[3,5-бис(2,3-дигидроксипропиламинокарбонил)-2, 4, 6-трийодфенил] карбамид.
а. N-[3-ацетоксиметил-5-(2, 3-диацетоксипропиламинокарбонил)- 2, 4, 6-трийодфенил] -N1 [3, 5-бис (2, 3-диацетоксипропиламинокарбонил) -2, 4, б-трийодфенил] карбамид.
3, 5-Бис (2, 3-диацетоксипропиламинокарбонил)-2, 4, 6-трийоданилин (260 мг, 0,30 ммоль) растворяют в диоксане (1,0 мл) и добавляют раствор фосгена в толуоле (1,93 М, 1,8 мл). Сосуд плотно запечатывают и затем нагревают до 60oC в течение 17 часов. После охлаждения до комнатной температуры растворитель отгоняют при пониженном давлении. Добавляют диоксан (3 мл) и снова отгоняют. Эту процедуру повторяют дважды. Добавляют диоксан (1 мл) и впоследствии 3-ацетоксиметил-5-(2, 3-диацетоксипропиламинокарбонил) -2, 4, 6-трийоданилин (0,245 г, 0,31 ммоль), полученный по примеру 16a и Hg (OCOCF3)2 (20 мг). Смесь перемешивают 16 часов при комнатной температуре, растворитель упаривают и остаток чистят препаративной ЖХВР. Выход: 0,192 г (39%).
МС (ESP, m/e): 1643 (М+, 100%), 1665 (М+Па+, 34%).
b. N-[3- Гидроксиметил-5-(2, 3-дигидроксипропиламинокарбонил)- 2, 4, 6-трийодфенил] -N1 [3, 5-бис (2, 3-дигидроксипропиламинокарбонил)- 2, 4, 6-трийодфенил] карбамид.
Продукт примера 19a растворяют в смеси метанола (5 мл) и воды (5 мл) и pH доводят до 12, используя 2 М водный раствор NaOH. После перемешивания в течение 2 часов pH доводят до 6,5, используя водную HCl и растворители упаривают. Продукт чистят, используя препаративную ЖХВР. Выход: 68 мг (44%).
МС (ESP, m/e): 1349 (М+, 15%), 1372 ([M+Na]+, 100%).
Пример 20. N- Гидроксиацетил-3-(1, 2-дигидроксиэтил)-5-(2, 3- дигидроксипропиламинокарбонил)-2, 4, 6-трийоданилин.
а. Метиловый эфир 3-нитро-5-(2-триметилсилилвинил)бензойной кислоты.
Смесь метилового эфира 3-йод-5-нитробензойной кислоты (307 мг, 1,0 ммоль), Pd(OAc)2 (67 мг, 0,30 ммоль), трифенилфосфина (0,032 г, 0,60 ммоль), AgNO3 (170 мг, 1,0 ммоль), триэтиламина (0,167 мл, 1,2 ммоль) и винилтриметилсилана (0,309 мл, 2,0 ммоль) и винилтриметилсилана (0,309 мл, 2,0 ммоль) растворяют в ацетонитриле (10 мл) и раствор нагревают до 60oC в закрытом сосуде в течение 48 часов. Осадившиеся соли отфильтровывают и фильтрат упаривают. Остаток хроматографируют на силикагеле, используя смесь этилацетата и гептана (1:11) в качестве элюента. Выход: 210 мг (75%).
1H-ЯМР (CDCl3): 8,70-8,73 (м, 1H), 8,36-8,47 (м, 2H), 6,93 (д, J=19,2 Гц, 1H), 6,75 (д, J=19,2 Гц, 1H), 3,99 (с, 3H), 0,20 (с, 9H).
МС (APci, m/e): 279 (М+, 100%).
b. Метиловый эфир 3-нитро-5-винилбензойной кислоты.
Метиловый эфир 3-нитро-5-(2- триметилсилилвинил)-бензойной кислоты (2,44 г, 8,71 ммоль) растворяют в ацетонитриле (150 мл), раствор нагревают до температуры кипения с обратным холодильником и газообразный HCl барботируют через раствор до исчезновения исходного соединения в соответствии с ЖХВР-анализом. Раствору дают охладиться и растворитель удаляют упариванием. Чистота остатка соответствует > 95% по ЖХВР, и продукт используют без дальнейшей очистки. Выход: 2,02 г (89%).
1H-ЯМР (CD3CN): 8,64 (с, 1H), 8,54 (с, 1H), 8,45 (с, 1H), 6,96 (дд, J= 10,8 Гц, J=17,4 Гц, 1H), 6,11 (д, J=17,4 Гц, 1H), 5,59 (д, J=10,8 Гц, 1H), 4,00 (с, 3H).
с. Метиловый эфир 3-нитро-5-(1, 2-дигидроксиэтил)бензойной кислоты.
Метиловый эфир 3-нитро-5-винилбензойной кислоты (2,02 г, 9,76 ммоль) растворяют в смеси ацетона и воды (200 мл, 9:1), и после охлаждения до 0oC добавляют OsO4 (60 мг, 0,24 ммоль) и впоследствии N-метилморфолин-N-оксид (2,34 г, 20,0 ммоль). После перемешивания в течение 46 часов при комнатной температуре добавляют водный раствор Na2S2O5 (3,7 г) в воде (150 мл) и раствор подкисливают разбавленной водной HCl. Объем раствора снижают до 150 мл путем упаривания и остаток экстрагируют этилацетатом (3х100 мл). Объединенные органические фазы упаривают и остаток чистят колоночной хроматографией на силикагеле, используя этилацетат в качестве элюента. Выход: 1,60 г (60%).
1H-ЯМР (CD3CN): 8,62-8,66 (м, 1H), 8,44-8,48 (м, 1H), 8,36- 8,40 (м, 1H), 4,88-4,94 (м, 1H), 3,98 (с, 3H), 3,60-3,79 (м, 4H).
d. 1-(2, 3-Дигидроксипропиламинокарбонил)-3-нитро-5-(1, 2- дигидроксиэтил)бензол.
Метиловый эфир 3-нитро-5- (1, 2-дигидроксиэтил)бензойной кислоты (0,40 г, 1,69 ммоль) и 2,3-дигидроксипропиламин (0,17 г, 1,86 ммоль) растворяют в метаноле (2 мл) и раствор перемешивают при 75oC в течение 1 часа. Затем давление снижают до 200 мм Hg и перемешивание продолжают при 95oC в течение 2 часов. Сырую реакционную смесь чистят препаративной ЖХВР. Выход: 0,40 г (78%).
МС (ESP, m/e): 299 ([М-1]+, 100%).
е. 3-(2, 3-дигидроксипропиламинокарбонил)-5-(1, 2-дигидроксиэтил) анилин.
1-(2, 3-Дигидроксипропиламинокарбонил)-3-нитро-5-(1, 2- дигидроксиэтил) бензол (0,40 г, 1,32 ммоль) растворяют в смеси метанола (40 мл) и воды (20 мл). Раствор гидрируют при 60 фунт/кв. дюйм, используя Pd/С-катализатор (10%, 50 мг). Раствор фильтруют через цеолит и растворители удаляют упариванием. По ЖХВР-анализу чистота продукта соответствует > 95% и соединение используют без дополнительной очистки.
МС (ESP, m/e): 271 ([M+1]+, 100%), 293 ([M+Na]+, 45%).
f. 3-(2, 3-Дигидроксипропиламинокарбонил)-5-(1, 2- дигидрокси- этил)-2, 4, 6-трийоданилин.
3-(2, 3-Дигидроксипропиламинокарбонил)-5-(1, 2-дигидрокси-этил)анилин (0,37 г, 1,35 ммоль) растворяют в смеси метанола (30 мл) и воды (90 мл). Добавляют KICl2 (1,37 г, 4,05 ммоль) и раствор перемешивают при 35oC 24 часа. Добавляют дополнительное количество KICl2 (1,0 ммоль) и перемешивание продолжают при 60oC в течение 72 часов. Добавляют водный раствор Na2S2O5 (1,0 г в 50 мл) и растворители удаляют упариванием. Очистка препаративной ЖХВР дает 87 мг (10%) чистого продукта.
1H-ЯМР (СD3ОD): 8,60 (м, 1H), 5,38-5,47 (м, 1H), 3,96-4,26 (м, 3H), 3,30-3,84 (м, 10H).
МС (ESP, m/e): 648 (М+, 15%), 670 ([M+Na]+, 100%).
g. N-Гидpoкcиaцeтил-3-(2, 3-дигидроксипропиламинокарбонил)- 5-(1, 2-дигидроксиэтил)-2, 4, 6-трийоданилин.
3-(2, 3-Дигидроксипропиламинокарбонил)-5-(1, 2-дигидрокси-этил) -2, 4, 6- трийоданилин (0,059 г, 0,091 ммоль) смешивают с ацетоксиацетилхлоридом (1,0 мл), содержащим N,N-диметилацетамид (0,4 мл) и смесь перемешивают при 60oC 48 часов. Смеси дают охладиться до комнатной температуры, добавляют воду и растворители удаляют упариванием. Остаток растворяют в смеси метанола (10 мл) и воды (5 мл) и добавляют водный раствор NaOH (5 М, 1 мл). Раствор перемешивают при комнатной температуре 1 час, раствор нейтрализуют водной HCl и растворители упаривают. Очистка ЖХВР дает чистый продукт.
МС (ESP, m/e): 706 (М+, 100%).
Пример 21. 3,5- Ди(гидроксиацетиламино)-2, 4, 6- трийодацетофенон.
a. 1, 3-Диамино-5-(1-гидроксиэтил)бензол.
3, 5-Динитроацетофенон (2,02 г, 9,5 ммоль), полученный по методике (Y. Nagase et al., Macromol.Chem. Rapid Comm. (1990), 11, 185-191) растворяют в метаноле (100 мл) и гидрируют при 60 фунт/кв. дюйм, используя Pd/C-катализатор (5%, 100%). Катализатор отфильтровывают и растворитель удаляют упариванием. Продукт используют без очистки в следующей стадии. Выход: 1,22 г (84%).
1H-ЯМР (CDCl3): 6,20 (д, J=2,0 Гц, 2H), 6,08 (т, J=2,0 Гц, 1H), 4,90 (ушир. с, 4H), 4,62 (кв. J=7,0 Гц, 1H), 3,37 (д, J=7,0 Гц, 3H).
МС (ESP, m/e): 151 ([М-1]+, 100%).
b. 1, 3-Диамино-2, 4, 6-трийодацетофенон.
1, 3-Диамино-5-(1-гидроксиэтил) бензол (1,18 г, 7,72 ммоль) растворяют в смеси метанола и воды (5:1, 168 мл), содержащей 1 М водную HCl (16 мл). Добавляют быстро водный раствор KICl2 (7,31 г, 30,9 ммоль) и после перемешивания в течение 50 минут твердый продукт отфильтровывают, промывают водой и сушат. Продукт чистят ТСХ и анализируют методом 1H-ЯМР. Выход: 3,62 г (89%).
1H-ЯМР (CDCl3): 4,86 (ушир. с, 4H), 2,62 (с, 3H).
13C-ЯМР (CDCl3): 204,7, 151,1, 146,8, 28,7.
МС (APci, m/e): 528 (M+, 100%).
с. 1, 3-Ди (ацетоксиацетиламино)-2, 4, 6-трийодацетофенон.
1, 3-Диамино-2, 4, 6-трийодацетофенон (1,8 г, 3,41 ммоль) растворяют в диметилацетамиде (15 мл), содержащем ацетоксиацетилхлорид (1,1 мл, 10,2 ммоль) и раствор перемешивают 65 часов при комнатной температуре. Растворители удаляют упариванием и остаток чистят препаративной ЖХВР. Выход: 1,69 г (68%).
1H-ЯМР (ДМСО-d6): 10,13-10,27 (м, 2H), 4,65 (с, 4H), 2,56 (с, 3H), 2,12 (с, 6H).
МС (ESP, m/e): 750 ([M+Na]+, 100%), 766 ([M+K]+, 26%).
d. 1, 3-Ди(гидроксиацетиламино)-2, 4, 6-трийодацетофенон.
1, 3-Ди (ацетоксиацетиламино)-2, 4, 6-трийодацетофенон (0,171 г, 0,23 ммоль) растворяют в смеси метанола (30 мл) и воды (5 мл), содержащей 2 М водный NaOH (3 мл). Раствор перемешивают в течение 90 минут и затем нейтрализуют, используя сильнокислую катионообменную смолу. Растворители удаляют упариванием и остаток чистят препаративной ЖХВР. Выход: 98 мг (54%).
МС (ESP, m/e): 644 (М+, 100%), 666 ([M+Na]+, 95%).
Пример 22. 3,5-Ди (гидроксиацетиламино)-1-гидроксиацетил-2,4,6- трийодбензол.
a. 3, 5-Ди (ацетоксиацетиламино)-1-бромацетил-2, 4, 6-трийодбензол.
1,3-Ди (ацетоксиацетиламино)-2,4,6- трийодацетофенон (0,20 г, 0,279 ммоль) растворяют в ледяной уксусной кислоте и добавляют бром (0,044 г, 0,28 ммоль). Реакционную смесь перемешивают 2,5 часа при 75oC и затем дают охладиться. Растворители удаляют упариванием и остаток используют непосредственно на следующей стадии.
МС (ESP, m/e): 806 (М+, 100%), 808 (М+, 98%).
b. 3, 5-Ди(ацетоксиацетиламино)-1-ацетоксиацетил-2, 4, 6- трийодбензол.
3, 5-Ди(ацетоксиацетиламино)-1-бромацетил-2, 4, 6-трийодбензол (10 мг, 0,12 ммоль) превращают в соответствующий ацетат нагреванием до 110oC в ледяной уксусной кислоте (5 мл), содержащей ацетат натрия (1 ммоль) и AgOCOCF3 (0,11 г, 0,5 ммоль) в течение 16 часов. Продукт чистят препаративной ЖХВР. Выход не определен.
МС (ESP, m/e): 786 (М+, 100%).
с. 3, 5-Ди (гидроксиацетиламино)-1-гидроксиацетил-2, 4, 6- трийодбензол.
Гидролиз 3, 5-Ди (ацетоксиацетиламино)-1-ацетоксиацетил-2, 4, 6-трийодбензола проводят аналогично примеру 4e. Сырой продукт чистят препаративной ЖХВР. Выход не определен.
МС (ESP, m/e): 687 ([М+HCOOH]+, 100%).
Пример 23. 3,5-Ди (гидроксиацетиламино)-1-(1, 2-дигидроксиэтил)- 2, 4, 6-трийодбензол.
а. 3,5-Динитрофенилэтанол.
3,5-Динитроацетофенон (3,27 г, 0,0156 ммоль) растворяют в смеси абсолютного этанола (75 мл) и ТГФ (37,5 мл) и смесь охлаждают до -10oC. Добавляют NaBH4 (0,30 г, 7,9 ммоль) и смесь перемешивают 1 час при -10oC. Добавляют воду (80 мл) и этилацетат, фазы разделяют и органическую фазу промывают водой (80 мл) и сушат (Na2SO4). Растворители удаляют упариванием и остаток чистят хроматографически на нейтральной окиси алюминия, используя смесь пентана и этилацетата (1:1) в качестве элюента. Выход: 2,52 г (76%).
1H-ЯМР (CDCl3): 8,95 (т, J=2,0 Гц, 1H), 8,60 (д, J=2,0 Гц, 1H), 8,59 (д, J=2,0 Гц, 1H), 5,15 (кв, J=7,5 Гц, 1H), 1,61 (д, J=7,5 Гц, 3H).
b. 3,5-Динитростирол.
3, 5-Динитрофенилэтанол (1,0 г, 4,7 ммоль) смешивают с P2О5 (1,0 г, 0,71 ммоль) и перемешивают смесь, нагревая до 100oC. Через 3 часа смеси дают охладиться до комнатной температуры и добавляют воду (0,4 мл), pH доводят до 9, используя 1 М водный NaOH и экстрагируют диэтиловым эфиром (2х25 мл). Объединенные органические фазы сушат (Na2SO4) и упаривают. Сырой продукт используют без дополнительной очистки на следующей стадии.
1H-ЯМР (CDCl3): 8,92 (т, J=2,0 Гц, 1H), 8,56 (д, J=2,0 Гц, 2H), 6,86 (дд, J=18,4 Гц, J=10,9 Гц 1H), 6,08 (д, J=18,0 Гц, 1H), 5,67 (д, J=10,9 Гц, 1H).
с. 1-(1, 2-Дигидроксиэтил)-3, 5-динитробензол.
3, 5-Динитростирол (0,50 г, 2,58 ммоль) растворяют в смеси ацетона и воды (8: 1, 70 мл) и раствор охлаждают до 0oC. Добавляют OsO4 (0,046 г, 0,18 ммоль) и NMO (0,60 г, 5,15 ммоль) и раствор перемешивают при комнатной температуре 16 часов. Добавляют раствор Na2S2O5 (1,5 г) в воде (120 мл) и органический растворитель удаляют упариванием. Водную фазу экстрагируют этилацетатом (2х70 мл) и объединенные органические фазы сушат (Na2SO4) и упаривают. Продукт чистят препаративной ЖХВР. Выход: 0,44 г (75%).
1H-ЯМР (CD3CN): 8,87 (т, J=2,0 Гц, 1H), 8,63 (т, J=2,0 Гц, 2H), 4,98 (т, J=6,0 Гц, 1H), 3,64-3,79 (м, 2H), 2,34 (с, 2H).
MC (ESP-, m/e): 227 [M-, 50%), 197 ([M-CH2O]-, 100%).
d. 1- (1, 2-Дигидроксиэтил)-3, 5-диаминобензол.
1- (1, 2-Дигидроксиэтил)-3, 5-динитробензол (0,10 г, 0,44 ммоль) растворяют в метаноле (35 мл) и гидрирование проводят при 60 фунт/кв. дюйм, используя Pd/С- катализатор (10%, 50 мг). Катализатор отфильтровывают и растворитель упаривают. Выход: 0,074 г (100%).
1H-ЯМР (СD3ОD): 6,14 (д, J=2,0 Гц, 2H), 6,06 (т, J=2,0 Гц, 1H), 4,98 (ушир. с, 6H), 4,43-4,50 (м, 1H), 3,52-3,57 (м, 2H).
MC (ESP, m/e): 170 [М+, 100%), 210 ([M+K]+, 18%).
e. 1-(1, 2-Дигидроксиэтил)-3, 5-диамино-2, 4, 6-трийодбензол.
1- (1, 2-Дигидроксиэтил)-3, 5-диаминобензол (0,0584 г, 0,242 ммоль) растворяют в смеси метанола (5 мл) и водный 2М HCl (1,2 мл) и добавляют одной порцией раствор KICl2 (70% в воде, 0,97 ммоль). После перемешивания в течение 20 минут при комнатной температуре добавляют 10% водный раствор NaHSO3 (0,2 мл), растворители удаляют упариванием и остаток чистят, препаративной ЖХВР. Выход: 31,4 мг (24%).
1H-ЯМР (СD3ОD): 5,56-5,63 (м, 1H), 4,03-4,12 (м, 1H), 3,79-3,87 (м, 1H), 5,06 (ушир. с, 4H).
f. 1- (1, 2-Дигидроксиэтил)-3, 5-ди (гидроксиацетиламино)-2, 4, 6-трийодбензол.
1-(1, 2-Дигидроксиэтил)-3, 5-диамино-2, 4, 6-трийодбензол ацилируют ацетоксиацетилхлоридом, используя, например, методику примера 4d. Сырой продукт гидролизуют затем аналогично примеру 4e, получая конечный продукт. Очистку сырого продукта проводят, используя препаративную ЖХВР.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИОРАЗЛАГАЕМЫЕ НЕСШИТЫЕ ПОЛИМЕРЫ | 1993 |
|
RU2114865C1 |
| КОНТРАСТНЫЕ АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 1993 |
|
RU2122432C1 |
| НОВЫЕ СОЕДИНЕНИЯ - АНТАГОНИСТЫ РЕЦЕПТОРА НЕЙРОКИНИНА 1 | 2013 |
|
RU2631319C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ КАТЕХОЛА И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1997 |
|
RU2180898C2 |
| ПИРАЗОЛПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2735522C2 |
| ИНГИБИТОРЫ ДВУХ САЙТОВ СВЯЗЫВАНИЯ АЦЕТИЛХОЛИНЭСТЕРАЗЫ ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ АЛЬЦГЕЙМЕРА | 2003 |
|
RU2325379C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2838028C2 |
| ПРОИЗВОДНЫЕ ВИТАМИНА-D, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ТЕРАПЕВТИЧЕСКИЙ АГЕНТ, СПОСОБ ЛЕЧЕНИЯ | 2001 |
|
RU2230738C9 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |


Описываются новые йодированные арильные соединения общей формулы I, где значения R, X, n указаны в п. 1 формулы с низкой вязкостью, полезные в качестве рентгеноконтрастных соединений. Описывается также диагностическая композиция на основе соединений формулы I. 2 с. и 14 з.п. ф-лы, 2 табл. 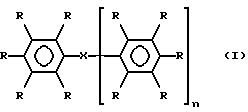
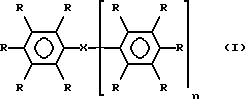
где n = 0 или 1, при этом, когда n = 1, каждый из C6R5 фрагментов может быть одинаковым или различным; каждая группа R обозначает атом водорода или иода или гидрофильный радикал M или M1; две или три не смежных R группы в каждом C6R5 фрагменте представляют собой иод и по меньшей мере одна R группа в каждом C6R5 фрагменте представляет M или M1; X обозначает связь или цепь из 1 - 4 атомов, связывающих два C6R5 фрагмента и включающих атомы углерода, азота или кислорода, или, когда n = 0, X обозначает группу R; каждый M независимо обозначает неионный гидрофильный радикал, выбранный из группы, состоящей из C1-6алкила, необязательно с одной или более CH2 или CH группами, замененными атомами кислорода или азота и необязательно замещенными одной или более группами оксо, гидрокси, амино, карбокси; каждый M1 независимо означает C1-4-алкильную группу, замещенную по меньшей мере одной гидроксильной группой и необязательно связанную с фенильным кольцом посредством карбонильной группы, причем по меньшей мере одна R группа является M1 радикалом; при условии, что, когда n = 0, либо присутствует по меньшей мере одна M1 группа, отличная от гидроксиметильной или 1,2-дигидроксиэтильной группы, либо, если присутствует одна гидроксиметильная или 1,2-дигидроксиэтильная M1 группа, то по меньшей мере одна связанная с азотом гидроксилированная алкильная группа, содержащая M группу, также присутствует; и их изомеры.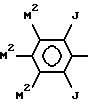
где каждый M2 независимо обозначает группу M или M1.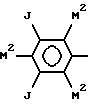
где каждый M2 независимо обозначает группу M или M1.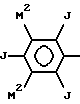
где каждый M2 независимо обозначает группу M или M1.
CR2 1OCO, где R1 обозначает водород или C1-6-алкил или алкоксигруппу, необязательно замещенную гидрокси, алкокси, окса или оксо и, когда эта группа R1 присоединена к углеродному атому, она может также быть гидроксильной группой.
| WO 9414478 А, 1994 | |||
| WO 9113636 А, 1991 | |||
| Конвейер для транспортирования штучных грузов | 1974 |
|
SU576255A1 |
| US 3794729 А, 1974 | |||
| Способ получения замещенных амидов или их солей | 1977 |
|
SU793387A3 |
| Способ получения бис-(3,5-дикарбамоил-2,4,6-трийоданилидов) дикарбоновых кислот | 1977 |
|
SU917696A3 |
