Настоящее изобретение относится к способу синтеза полипептида, содержащего предварительно определенную аминокислотную последовательность. Способ по настоящему изобретению включает циклы связывания для связывания С-конца аминокислотного структурного блока, защищенного по N-концу, с незащищенной N-концевой аминогруппой аминокислотной цепи, где по меньшей мере один цикл связывания включает стадию (а) связывания, стадию (b) кэппирования и стадию (с) удаления защитной группы.
Настоящее изобретение дополнительно относится к композиции, содержащей 0,5-5% об./об. уксусного ангидрида и 0,2-2% об./об. диизопропилэтиламина, а также к ее применению в качестве кэппирующего реагента для ацетилирования незащищенной аминогруппы в ходе синтеза полипептида.
Разработанные способы твердофазного синтеза пептидов предусматривают связывание посредством линкера предварительно определенной С-концевой аминокислоты синтезируемой аминокислотной цепи с полимерным носителем. Используемая для присоединения аминокислота представляет собой аминокислотный структурный блок с защищенной на N-конце аминогруппой, при этом указанная защитная группа представляет собой временно связанную группу Fmoc. После успешного присоединения защитную группу Fmoc отщепляют и осуществляют присоединение следующего Fmoc-защищенного аминокислотного структурного блока к свободной функциональной аминогруппе. После синтеза требуемой аминокислотной цепи ее отщепляют от твердой фазы. На фигуре 1 представлен обзор описываемого подхода.
Поскольку реакция сочетания не всегда завершается, оставшиеся свободные аминогруппы можно ацетилировать уксусным ангидридом (см. фигуру 1). Такую реакцию называют кэппированием. Целью кэппирования является предотвращение появления примесей (N-1) (т. е. продуктов, в которых отсутствует аминокислотный структурный блок в одном положении), которые может быть трудно отделить от требуемого конечного продукта.
В литературе описаны многочисленные кэппирующие смеси для твердофазного синтеза пептидов. В работе Fields et al. (PNAS 85, 1384-1388, 1988) используют уксусный ангидрид и диизопропилэтиламин (DIPEA) в метиленхлориде для синтеза грамицидина A. Сначала добавляют 1 эквивалент DIPEA в 20 мл метиленхлорида. Через пять минут добавляют 4 эквивалента уксусного ангидрида. Общее время инкубации составляет 20 минут. В работе Eritja et al. (Tetrahetron 43, 2675-2680, 1987) описывается применение приблизительно 9% уксусного ангидрида и приблизительно 16% DIPEA в DMF в сочетании со временем инкубации, составляющим 30 минут, для процесса кэппирования. В работе Echner et al. раскрыта смесь уксусного ангидрида, пиридина и метиленхлорида для процесса кэппирования (Liebigs Ann. Chem. 1988, 1095-1097).
Твердофазный синтез ликсисенатида (также известного как AVE0010 или ZP-10), описанный в WO 01/04156 A1, которая включена в данный документ посредством ссылки, включает связывание отдельных Fmoc-защищенных аминокислотных структурных блоков одним и тем же способом, без какого-либо процесса кэппирования.
Ликсисенатид имеет последовательность дез-Pro36-эксендин-4(1-39)-Lys6-NH2. Данное вещество раскрыто в WO 01/04156, SEQ ID NO: 93 (см. SEQ ID NO:1 и фигуру 2 настоящей заявки). Эксендины представляют собой группу пептидов, которые могут снижать концентрацию глюкозы в крови. Эксендины обладают определенной схожестью с последовательностью GLP-1(7-36) (53%, Göke et al., J. Biol. Chem. 268, 19650-55). При взаимодействии с эксендиновыми рецепторами эксендин-3 и эксендин-4 стимулируют повышение внутриклеточного синтеза цАМФ в ациноцитах поджелудочной железы морских свинок (Raufman, 1996, Reg. Peptides 61:1-18). В отличие от эксендина-4 эксендин-3 оказывает свое действие путем увеличения высвобождения амилазы в ациноцитах поджелудочной железы. Эксендины выполняют роль антагонистов GLP-1.
Глюкагон-подобный пептид 1 (GLP-1) представляет собой эндокринный гормон, который усиливает инсулиновый ответ после перорального приема глюкозы или жира. Обычно GLP-1 снижает концентрации глюкагона, замедляет опорожнение желудка, стимулирует биосинтез (про-)инсулина, повышает чувствительность к инсулину и стимулирует инсулин-независимый биосинтез гликогена (Holst (1999), Curr. Med. Chem. 6:1005, Nauck et al. (1997), Exp. Clin. Endocrinol. Diabetes 105:187, Lopez-Delgado et al. (1998), Endocrinology 139:2811). Человеческий GLP-1 имеет 37 аминокислотных остатков (Heinrich et al., Endocrinol. 115:2176 (1984), Uttenthal et al., J. Clin. Endocrinol. Metabol. (1985), 61:472). К активным фрагментам GLP-1 относятся GLP-1(7-36) и GLP-1(7-37).
Было высказано предположение о том, что эксендин-3, эксендин-4 и агонисты эксендина можно применять для лечения сахарного диабета и для предупреждения развития гипергликемии, поскольку они уменьшают опорожнение и моторику желудка (US 5424286 и WO 98/0535 A1).
Аналоги эксендина можно охарактеризовать с помощью аминокислотных замен и/или С-концевых усечений нативной последовательности эксендина-4. Такие аналоги эксендина описаны в WO 99/07404, WO 99/25727 и WO 99/25728.
Авторами настоящего изобретения было обнаружено, что неочищенный продукт стандартного твердофазного синтеза ликсисенатида с Fmoc-защищенными аминокислотными структурными блоками характеризуется повышенными количествами определенных ацетилированных ошибочных последовательностей по сравнению с другими ацетилированными ошибочными последовательностями. Эти неожиданно выраженные примеси представляют собой, в частности, Ac(20-44), Ac(17-44), Ac(13-44), Ac(10-44) и Ac(4-44).
Задача, подлежащая решению с помощью настоящего изобретения, заключается в уменьшении количества ацетилированных ошибочных последовательностей, встречающихся при пептидном синтезе, и, следовательно, в увеличении выхода продукта пептидного синтеза. В частности, следует уменьшить количество тех ацетилированных ошибочных последовательностей, которые в повышенном количестве содержатся в неочищенном продукте.
Было обнаружено, что на стадии кэппирования твердофазного синтеза ликсисенатида защитная группа Fmoc нежелательно отщепляется от некоторых только что связанных аминокислотных структурных блоков. Такое нежелательное отщепление было обнаружено только при кэппировании в определенных аминокислотных положениях во время цикла синтеза. Пять из этих положений представляют собой следующие:
• кэппирование после связывания Arg(20),
• кэппирование после связывания Glu(17),
• кэппирование после связывания Gln(13),
• кэппирование после связывания Leu(10),
• кэппирование после связывания Gly(4).
Таким образом, дополнительная проблема, подлежащая решению с помощью настоящей заявки, заключается в обеспечении условий кэппирования, при которых нежелательные ацетилированные продукты более не будут присутствовать в таких повышенных количествах.
Неожиданно было обнаружено, что применение более мягких условий кэппирования приводит к более низким концентрациям побочных продуктов в неочищенном продукте или даже к их полному устранению. Путем уменьшения или полного удаления побочных продуктов Ac(17-44), Ac(13-44) и Ac(10-44), хроматографические пики которых близки к пикам предполагаемого продукта, значительно облегчается очистка предполагаемого продукта из неочищенного продукта. Чем меньше получают смешанных фракций, тем выше выход очищенного ликсисенатида. Выход дополнительно увеличивается, поскольку за счет мягких условий кэппирования минимизируется концентрация побочных продуктов Ас(20-44) и Ас(4-44), пики которых находятся еще дальше от пика предполагаемого продукта.
Один аспект настоящего изобретения относится к способу синтеза полипептида, содержащего предварительно определенную аминокислотную последовательность, при этом способ включает циклы связывания аминокислотных структурных блоков с аминокислотной цепью, при этом указанные аминокислотные структурные блоки содержат незащищенную С-концевую карбоксильную группу и защищенную N-концевую аминогруппу, и при этом указанная аминокислотная цепь содержит незащищенную N-концевую аминогруппу, при этом по меньшей мере один цикл связывания включает следующие стадии:
(a) связывание С-конца аминокислотного структурного блока с незащищенной N-концевой аминогруппой аминокислотной цепи, так чтобы между аминокислотной цепью и аминокислотным структурным блоком образовалась амидная связь,
(b) приведение в контакт продукта, полученного на стадии (а), с кэппирующим реагентом, содержащим кэппирующее соединение, при этом кэппирующее соединение связывается с незащищенной N-концевой аминогруппой аминокислотной цепи, с которой на стадии (а) не был связан ни один структурный блок, и
(с) удаление защитной группы с N-концевой аминогруппы аминокислотного структурного блока.
В настоящем изобретении термины "кэппирующий реагент", "кэппирующая композиция" или "кэппирующая смесь" используют взаимозаменяемо. Реагент можно получить перед стадией (b) или же компоненты кэппирующего реагента добавляют в ходе стадии (b).
Аминокислотная цепь, кэппированная на стадии (b), не способна связывать дополнительный аминокислотный структурный блок, так что удлинение цепи этой молекулы прекращается.
Во время синтеза полипептида циклы связывания аминокислотных структурных блоков проводят таким образом, чтобы аминокислотная цепь полипептида образует структурный блок за структурным блоком. Для стадии связывания можно применять современные методики, в частности твердофазный синтез на основе Fmoc-защищенных аминокислотных структурных блоков. Можно использовать все виды твердых фаз, подходящих для твердофазного синтеза пептидов. В частности, можно использовать твердую фазу, содержащую смолу. Смола может представлять собой смолу Ринка (амидную смолу Ринка) или смолу Tentagel®. В предпочтительном аспекте смола, выступающая в качестве твердой фазы, представляет собой смолу Ринка или амидную смолу Ринка.
Циклы, включающие стадии (а), (b) и (c) по настоящему изобретению, можно повторять один или несколько раз. Для каждого подлежащего связыванию аминокислотного структурного блока проводят один цикл. Соответствующий аминокислотный структурный блок независимо выбирают для каждого цикла в зависимости от предварительно определенной аминокислотной последовательности. Можно использовать все виды аминокислотных структурных блоков, которые описаны в данном документе.
Способ по настоящему изобретению позволяет комбинировать циклы связывания, которые являются одинаковыми или разными по отношению к условиям стадии кэппирования, таким как температура кэппирования, состав кэппирующего реагента или/и продолжительность кэппирования. В одном аспекте все циклы связывания по настоящему изобретению проводят в одинаковых условиях кэппирования. В другом аспекте по меньшей мере одна стадия кэппирования отличается от других стадий кэппирования по настоящему изобретению, например, измененной температурой кэппирования, составом кэппирующего реагента или/и продолжительностью кэппирования. В одном аспекте способ по настоящему изобретению включает более одного условия стадии кэппирования, например, два, три, четыре или пять различных условий стадии кэппирования на протяжении всего синтеза полипептида.
В одном аспекте способ по настоящему изобретению включает по меньшей мере один цикл связывания, по меньшей мере два цикла связывания, по меньшей мере три цикла связывания, по меньшей мере четыре цикла связывания или по меньшей мере пять циклов связывания, включающих стадию кэппирования (b).
Кэппирующее соединение предпочтительно выбирают из группы, состоящей из уксусного ангидрида (CAS 108-24-7), гомологов уксусного ангидрида, бензоилхлорида (CAS 98-88-4), N-(бензилоксикарбонилокси)сукцинимида (CAS 13139-17-8), бензилхлорформиата (CAS 501-53-1), сложных эфиров хлормуравьиной кислоты, 1-ацетилимидазола (CAS 2466-76-4), ди-трет-бутилдикарбоната (CAS 24424-99-5) и N-(трет-бутоксикарбонилокси)сукцинимида (CAS 13139-12-3). В предпочтительном аспекте кэппирующее соединение выбирают из уксусного ангидрида и гомологов уксусного ангидрида, и предпочтительно оно представляет собой уксусный ангидрид.
В одном аспекте кэппирующий реагент содержит уксусный ангидрид в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация уксусного ангидрида составляет 1-3% объем/объем, более предпочтительно 2% объем/объем.
В другом аспекте кэппирующий реагент содержит гомологи уксусного ангидрида в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация гомологов уксусного ангидрида составляет 1-3% об./об., более предпочтительно - 2% об./об.
В другом аспекте кэппирующий реагент содержит бензоилхлорид в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация бензоилхлорида составляет 1-3% об./об., более предпочтительно - 2% об./об.
В другом аспекте кэппирующий реагент содержит N-(бензилоксикарбонилокси)сукцинимид в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация N-(бензилоксикарбонилокси)сукцинимида составляет 1-3% об./об., более предпочтительно - 2% об./об.
В другом аспекте кэппирующий реагент содержит бензилхлорформиат в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация бензилхлорформиата составляет 1-3% об./об., более предпочтительно - 2% об./об.
В другом аспекте кэппирующий реагент содержит сложные эфиры хлормуравьиной кислоты в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация сложных эфиров хлормуравьиной кислоты составляет 1-3% об./об., более предпочтительно - 2% об./об.
В другом аспекте кэппирующий реагент содержит 1-ацетилимидазол в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация 1-ацетилимидазола составляет 1-3% об./об., более предпочтительно - 2% об./об.
В другом аспекте кэппирующий реагент содержит сложные эфиры ди-трет-бутилдикарбоната в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация ди-трет-бутилдикарбоната составляет 1-3% об./об., более предпочтительно - 2% об./об.
В другом аспекте кэппирующий реагент содержит сложные эфиры N-(трет-бутоксикарбонилокси)сукцинимида в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация N-(трет-бутоксикарбонилокси)сукцинимида составляет 1-3% об./об., более предпочтительно - 2% об./об.
Кэппирующий реагент может содержать диизопропилэтиламин (также называемый DIPEA, N-этилдиизопропиламином или N, N-диизопропилэтиламином). В одном аспекте кэппирующий реагент содержит диизопропилэтиламин, при этом концентрация диизопропилэтиламина может составлять 0,2-2% об./об. и предпочтительно составляет 0,5-2% об./об. Предпочтительная концентрация диизопропилэтиламина составляет 1% объем/объем.
В одном аспекте кэппирующая композиция содержит диизопропилэтиламин и уксусный ангидрид, при этом концентрация диизопропилэтиламина может составлять 0,2-2% объем/объем и предпочтительно составляет 0,5-2% объем/объем, а концентрация уксусного ангидрида может составлять 0,5-5% объем/объем и предпочтительно составляет 1-3% объем/объем.
В одном аспекте кэппирующая композиция или кэппирующий реагент содержит диизопропилэтиламин в концентрации, составляющей приблизительно 1% об./об. и уксусный ангидрид в концентрации, составляющей приблизительно 2% об./об.
В способе по настоящему изобретению N-концевая аминогруппа аминокислотного структурного блока предпочтительно представляет собой лабильную по основанию защитную группу, более предпочтительно Fmoc.
Растворитель, используемый на стадии кэппирования, предпочтительно представляет собой полярный безводный растворитель, такой как ацетонитрил, диметилсульфоксид (DMSO), метанол, метиленхлорид, N, N-диметилформамид (DMA), N, N-диметилформамид (DMF), N-метилпирролидон или их смеси. В предпочтительном аспекте растворитель, используемый на стадии кэппирования, представляет собой DMF.
В настоящем изобретении термин "приблизительно" или "примерно" означает диапазон ±10%, ±5% или ±1%.
Реакцию кэппирования (b) по настоящему изобретению можно осуществлять при комнатной температуре. Комнатная температура, в соответствии с настоящим изобретением, относится к температуре приблизительно 15-25°C, температуре в диапазоне приблизительно 20-23°C, температуре в диапазоне приблизительно 19-21°C или температуре, составляющей приблизительно 20°C.
В способе по настоящему изобретению стадию (b) предпочтительно проводят в течение 5-15 мин., предпочтительно в течение 10 мин.
В предпочтительном аспекте стадию (b) проводят в течение 10 мин. с кэппирующим реагентом, содержащим 2% об./об. уксусного ангидрида и 1% об./об. DIPEA.
В другом предпочтительном аспекте стадию (b) проводят в течение 10 мин с кэппирующим реагентом, содержащим 2% об./об. уксусного ангидрида и 1% об./об. DIPEA в DMF, в положениях Arg(20), Glu(17), Gln(13), Leu(10) или/и Gly(4) последовательности ликсисенатида или эксендина-4.
В еще одном предпочтительном аспекте стадию (b) проводят в течение 10 мин с кэппирующим реагентом, содержащим 2% об./об. уксусного ангидрида и 1% об./об. DIPEA в DMF, в положениях Arg(20), Glu(17), Gln(13), Leu(10) или/и Gly(4) последовательности ликсисенатида или эксендина-4 при комнатной температуре, как описано в данном документе.
В одном аспекте способ по настоящему изобретению может включать по меньшей мере один цикл связывания, по меньшей мере два цикла связывания, по меньшей мере три цикла связывания, по меньшей мере четыре цикла связывания или по меньшей мере пять циклов связывания без стадии кэппирования, в частности в положениях, отличных от Arg(20), Glu(17), Gln(13), Leu(10) или/и Gly(4), последовательности ликсисенатида или эксендина-4.
В другом аспекте способ по настоящему изобретению включает кэппирование согласно стадии (b) после связывания аминокислотного структурного блока Arg(20), Glu(17), Gln(13), Leu(10) или/и Gly(4) последовательности ликсисенатида или эксендина-4, в частности, в течение приблизительно 10 мин. с кэппирующим реагентом, содержащим 2% об./об. уксусного ангидрида и 1% об./об. диизопропилэтиламина. Стадию (b) можно проводить после связывания остатка Arg, Glu, Gln, Leu или/и Gly на стадии (a), в частности, Arg(20), Glu(17), Gln(13), Leu(10) или/и Gly(4) последовательности ликсисенатида или эксендина-4. Предпочтительно в других аминокислотных положениях стадию кэппирования не проводят или/и кэппирование проводят в течение 20 мин. с 10% уксусным ангидридом и 5% об./об. DIPEA в DMF. В частности, кэппирование проводят при комнатной температуре, как описано в данном документе.
В еще одном аспекте настоящего изобретения способ по настоящему изобретению включает кэппирование согласно стадии (b) после связывания всех аминокислотных структурных блоков последовательности ликсисенатида или эксендина-4, в частности, в течение приблизительно 10 мин. с кэппирующим реагентом, содержащим 2% об./об. уксусного ангидрида и 1% об./об. диизопропилэтиламина. Стадию (b) можно проводить после связывания всех аминокислотных остатков последовательности ликсисенатида или эксендина-4 на стадии (а). На отдельных стадиях связывания аминокислот кэппирование можно не проводить. Стадию (b) можно проводить после связывания по меньшей мере 30 или по меньшей мере 35 аминокислотных остатков последовательности ликсисенатида или эксендина-4 на стадии (а).
В одном аспекте способ по настоящему изобретению может включать по меньшей мере один цикл связывания без стадии кэппирования (b) и по меньшей мере один цикл связывания, включающий стадию кэппирования (b), как описано в данном документе. В одном аспекте способ по настоящему изобретению может включать по меньшей мере один цикл связывания, по меньшей мере два цикла связывания, по меньшей мере три цикла связывания, по меньшей мере четыре цикла связывания или по меньшей мере пять циклов связывания без стадии кэппирования (b) и по меньшей мере один цикл связывания, по меньшей мере два цикла связывания, по меньшей мере три цикла связывания, по меньшей мере четыре цикла связывания или по меньшей мере пять циклов связывания, включающих стадию кэппирования (b), как описано в данном документе. В частности, в положениях, отличных от Arg(20), Glu(17), Gln(13), Leu(10) или/и Gly(4) последовательности ликсисенатида или эксендина-4, кэппирование не проводят.
В одном аспекте способ по настоящему изобретению может включать по меньшей мере один цикл связывания, по меньшей мере два цикла связывания, по меньшей мере три цикла связывания, по меньшей мере четыре цикла связывания или по меньшей мере пять циклов связывания, включающих стадии (а), (b') и (c), при этом стадию кэппирования (b') проводят в условиях, отличных от условий стадии кэппирования (b). Цикл связывания, включающий стадию (b'), в частности, применяют для связывания аминокислотного структурного блока, отличного от Arg(20), Glu(17), Gln(13), Leu(10) или/и Gly(4) последовательности ликсисенатида или эксендина-4. В одном аспекте стадия (b') может предусматривать применение кэппирующего реагента, содержащего приблизительно 10% об./об. уксусного ангидрида и приблизительно 5% об./об. диизопропилэтиламина в DMF, при этом ее проводят, например, в течение приблизительно 20 мин.
В одном аспекте способ по настоящему изобретению может включать по меньшей мере один цикл связывания, включающий стадию кэппирования (b) по настоящему изобретению, и по меньшей мере один цикл связывания, включающий стадию кэппирования (b’), как описано в данном документе. В одном аспекте способ по настоящему изобретению может включать по меньшей мере один цикл связывания, по меньшей мере два цикла связывания, по меньшей мере три цикла связывания, по меньшей мере четыре цикла связывания или по меньшей мере пять циклов связывания, включающих стадию кэппирования (b) по настоящему изобретению, и по меньшей мере один цикл связывания, по меньшей мере два цикла связывания, по меньшей мере три цикла связывания, по меньшей мере четыре цикла связывания или по меньшей мере пять циклов связывания, включающих стадию кэппирования (b’), как описано в данном документе.
Для связывания аминокислотного структурного блока, как описано в данном документе, включающего связывание более одной аминокислоты за один отдельный цикл, предпочтительно используют цикл связывания, включающий стадию (b'), например, для связывания дипептидов, таких как Pro-Pro или His-Gly с применением аминокислотных структурных блоков Fmoc-Pro-Pro-OH и Fmoc-His (Trt) -Gly-OH соответственно.
Способы проведения стадии связывания согласно стадии (а) и стадии удаления защитной группы согласно стадии (с) известны специалистам в данной области техники. Пептидный синтез предпочтительно проводят в форме твердофазного синтеза. В предпочтительном аспекте циклы присоединения осуществляют в направлении от C-конца к N-концу синтезируемой последовательности. Специалисту в данной области техники известны условия реакции для стадий (а) и (с), используемые при твердофазном пептидном синтезе в направлении от С-конца к N-концу с применением аминокислотных структурных блоков.
Аминокислотный структурный блок по настоящему изобретению представляет собой соединение, которое обеспечивает удлинение синтезируемой аминокислотной цепи на одну или несколько аминокислот за один цикл пептидного синтеза. В предпочтительном аспекте аминокислотный структурный блок по настоящему изобретению обеспечивает удлинение синтезируемой аминокислотной цепи на 1, 2, 3 или 4 аминокислоты. В конкретном предпочтительном аспекте аминокислотный структурный блок по настоящему изобретению обеспечивает удлинение синтезируемой аминокислотной цепи на одну или две аминокислоты.
Аминокислотный структурный блок по настоящему изобретению предпочтительно содержит одну аминокислоту (структурный блок из одной аминокислоты) или олигонуклеотид, содержащий 2, 3, 4 или больше аминокислот. В предпочтительном аспекте аминокислотный структурный блок по настоящему изобретению содержит одну аминокислоту или пептид, содержащий две аминокислоты, такие как, например, Pro-Pro или His-Gly. Аминокислоты из аминокислотного структурного блока, содержащего более одной аминокислоты, предпочтительно соединяются пептидными связями. Особенно предпочтительными аминокислотными структурными блоками, содержащими две аминокислоты, являются Fmoc-Pro-Pro-OH и Fmoc-His(Trt)-Gly-OH.
Было обнаружено, что применение Fmoc-His(Trt)-Gly-OH вместо аминокислотных структурных блоков His и Gly в положениях 1 и 2 в синтезе ликсисенатида и эксендина-4 позволяет предотвратить образование нежелательного дез-Gly(2)-ликсисенатида. Более того, у полученного ликсисенатида не наблюдали увеличенные значения D-His, которые являются следствием рацемизации.
Fmoc-His(Trt)-Gly-OH можно получить, например, посредством способа, предусматривающего стадии:
i) проведения реакции между Fmoc-His(Trt)-OH и тозилатом H-Gly-OBzl и
ii) отщепления бензильной группы от продукта, полученного на стадии i),
с получением Fmoc-His(Trt)-Gly-OH.
Иллюстративные условия реакции изложены в примере 3.
Аминокислотные структурные блоки по настоящему изобретению могут содержать модификации, подходящие для избирательного удлинения аминокислотной цепи только в требуемых положениях. Изменения аминокислотного структурного блока можно осуществлять на N-конце, на C-конце или/и на боковых цепях аминокислот.
Для защиты N-концевой функциональной аминогруппы в аминокислотном структурном блоке (т. е. аминогруппы, которая после успешного присоединения представляет собой N-конец аминоцепи) можно использовать все виды защитных групп, обычно применяемых для синтеза пептидов, особенно для твердофазного синтеза полипептидов. Специалисту в данной области техники известны такие виды подходящих временных защитных групп. В предпочтительном аспекте можно применять защитные группы, которые являются нестабильными в щелочной среде. В предпочтительном аспекте N-концевую аминогруппу в аминокислотном структурном блоке защищают посредством защитной группы Fmoc.
С-концевую карбоксильную группу из аминокислотного структурного блока предпочтительно оставляют незащищенной.
Аминокислотный структурный блок по настоящему изобретению может содержать, независимо один от другого, D-аминокислоты и глицин, L-аминокислоты и глицин или/и их комбинации. В предпочтительном аспекте аминокислоты в аминокислотном структурном блоке по настоящему изобретению выбраны независимо друг от друга из L-аминокислот и глицина. В предпочтительном аспекте аминокислоты могут быть выбраны из α-аминокислот. В дополнительном аспекте аминокислоты могут быть выбраны из встречающихся в природе аминокислот, таких как аминокислоты, встречающиеся в природе в полипептидах. В другом аспекте аминокислотный структурный блок по настоящему изобретению может содержать искусственные аминокислоты, такие как Met(O) (метионинсульфоксид или метионинсульфон), Trp(O2) (N-формилкинуренин) или/и изо-Asp (β-аспартат или изоаспартат). В еще одном дополнительном предпочтительном аспекте аминокислоты выбраны из Ser, Thr, Trp, Lys, Ala, Asn, Asp, Val, Met, Phe, Ile, Pro, Arg, Glu, Gln, Leu, каждая из которых, в частности, представлена в D-форме или каждая из которых представлена в L-форме, и Gly. В особенно предпочтительном аспекте аминокислотный структурный блок по настоящему изобретению содержит аминокислоты, выбранные из Arg, Glu, Gln, Leu, каждая из которых, в частности, представлена в D-форме или каждая из которых представлена в L-форме, и Gly.
В частности, аминокислоты выбраны независимо друг от друга, например, независимо из Ser, Thr, Trp, Lys, Ala, Asn, Asp, Val, Met, Phe, Ile, Pro, Arg, Glu, Gln, Leu, каждая из которых, в частности, представлена в D-форме или каждая из которых представлена в L-форме, и Gly.
В одном аспекте по меньшей мере одну боковую цепь аминокислотного структурного блока по настоящему изобретению можно защитить дополнительной защитной группой. Дополнительная защитная группа является предпочтительно ортогональной по отношению к N-концевой защитной группе. Подходящие защитные группы для указанных боковых цепей известны специалисту в данной области техники. Примерами подходящих защитных групп являются, например, Trt, Boc, Bzl, Pdf, tBu и OtBu, которые можно использовать для защиты определенных боковых цепей. Специалист в данной области техники осведомлен, какую боковую цепь нужно защищать и каким типом защитной группы. В одном аспекте можно использовать аминокислотные структурные блоки, которые упомянуты в примере 1.4. В случае, когда аминокислотный структурный блок содержит больше одной боковой цепи, одну или несколько данных боковых цепей можно защитить посредством защитных групп, независимо выбранных из подходящих защитных групп, которые известны специалисту в данной области техники.
Синтезируемым полипептидом может быть любой возможный пептид с предварительно определенной последовательностью. В предпочтительном аспекте синтезируемым полипептидом является агонист GLP-1. Полипептидом может быть агонист GLP-1, при этом агонист GLP-1 выбран из группы, состоящей из GLP-1 и его аналогов и производных, эксендина-3 и его аналогов и производных, эксендина-4 и его аналогов и производных. В предпочтительном аспекте полипептид выбран из группы, состоящей из эксендина-4 и ликсисенатида. Наиболее предпочтительным является ликсисенатид. В дополнительном предпочтительном аспекте полипептид выбран из албиглутида, дулаглутида и семаглутида.
Эксендин-3, аналоги и производные эксендина-3, эксендин-4 и аналоги и производные эксендина-4 описаны в WO 01/04156, WO 98/30231, US 5424286, EP 99610043.4 и WO 2004/005342. Эти документы включены в данный документ посредством ссылки. Эксендин-3, эксендин-4 и их аналоги и производные, описанные в данных документах, можно синтезировать посредством способа по настоящему изобретению, в то же время после завершения синтеза можно осуществить дополнительные модификации.
Ликсисенатид (SEQ ID NO: 1, фигура 2), эксендин-4 (SEQ ID NO: 2, фигура 2) и эксендин-3 (SEQ ID NO: 3, фигура 2) имеют высокую степень идентичности последовательности. Последовательности ликсисенатида и эксендина-4 идентичны в положениях 1-37. Последовательность 1-39 эксендина-4 идентична эксендину-3 в положениях 37 из 39 (94%) (J. Biol. Chem. 267, 1992, 7402-7405). Положения в последовательности приведены в данном документе применительно к последовательности ликсисенатида или эксендина-4. На основе данных последовательностей специалист в данной области техники сможет легко определить соответствующие положения в других последовательностях.
Аналоги и производные эксендина-3 или/и эксендина-4, в частности, содержат модифицированную аминокислотную последовательность. В одном аспекте аминокислотную последовательность модифицируют путем удаления одной или нескольких аминокислот (например дез-Pro36, дез-Pro37, дез-Asp28, дез-Met(O14) в эксендине-4 и соответствующих положений в эксендине-3). В одном аспекте можно заменить одну или несколько аминокислот (например, Met(O14), Trp(O2)25, изо-Asp28, Asp28, Pro38 в эксендине-4 и в соответствующих положениях в эксендине-3), при этом можно ввести встречающиеся в природе или искусственные аминокислоты, такие как, например, Met(O) (метионинсульфоксид или метионинсульфон), Trp(O2) (N-формилкинуренин) или/и изо-Asp (β-аспартат или изоаспартат). За один цикл синтеза в последовательность можно легко встроить искусственные аминокислоты с применением соответствующих аминокислотных структурных блоков.
В одном аспекте C-конец или/и N-конец полипептида можно модифицировать, например, путем добавления таких последовательностей, как -(Lys)-, -(Lys)2-, -(Lys)3-, -(Lys)4-, -(Lys)5-, -(Lys)6- и -Asn-(Glu)5-. В предпочтительном аспекте дополнительные аминокислотные последовательности представляют собой, например, -(Lys)4-, -(Lys)5-, -(Lys)6- и -Asn-(Glu)5-. С-концевая карбоксильная группа предпочтительно представляет собой кислую аминогруппу (-NH2). Необязательно, модификацию C-конца или/и N-конца осуществляют на отдельной стадии после завершения циклов синтеза способа по настоящему изобретению.
После завершения циклов синтеза способа по настоящему изобретению на дополнительной стадии необязательно можно получить фармацевтически приемлемые соли синтезированных полипептидов. Способы получения фармацевтически приемлемых солей полипептидов известны специалисту в данной области техники. Предпочтительными фармацевтически приемлемыми солями являются, например, ацетатные соли.
В одном аспекте агонист GLP-1 предпочтительно выбирают из группы, состоящей из эксендина-4, его аналогов и производных и его фармацевтически приемлемых солей.
Еще один предпочтительный агонист GLP-1 представляет собой аналог эксендина-4, выбранный из группы, состоящей из:
H-дез-Pro36-эксендин-4-Lys6-NH2,
H-дез-(Pro36,37)-эксендин-4-Lys4-NH2,
H-дез-(Pro36,37)-эксендин-4-Lys5-NH2 и их фармацевтически приемлемых солей.
Еще один предпочтительный агонист GLP-1 представляет собой аналог эксендина-4, выбранный из группы, состоящей из:
дез-Pro36[Asp28]эксендина-4 (1-39),
дез-Pro36[изо-Asp28]эксендина-4 (1-39),
дез-Pro36[Met(O)14,Asp28]эксендина-4 (1-39),
дез-Pro36[Met(O)14,изо-Asp28]эксендина-4 (1-39),
дез-Pro36[Trp(O2)25,Asp28]эксендина-2 (1-39),
дез-Pro36[Trp(O2)25,изо-Asp28]эксендина-2 (1-39),
дез-Pro36[Met(O)14Trp(O2)25,Asp28]эксендина-4 (1-39),
дез-Pro36[Met(O)14Trp(O2)25, изо-Asp28]эксендина-4(1-39) и их фармацевтически приемлемых солей.
Еще один предпочтительный агонист GLP-1 представляет собой аналог эксендина-4, выбранный из описанных выше групп, дополнительно модифицированный пептидом -Lys6-NH2 на C-конце.
Еще один предпочтительный агонист GLP-1 представляет собой аналог эксендина-4, выбранный из группы, состоящей из:
H-(Lys)6-дез-Pro36[Asp28]эксендин-4(1-39)-Lys6-NH2, дез-Asp28Pro36,Pro37,Pro38эксендин-4(1-39)-NH2,
H-(Lys)6-дез-Pro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5дез-Pro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-NH2,
дез-Pro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дез-Pro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-дез-Pro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дез-Pro36[Trp(O2)25,Asp28]эксендин-4(1-39)-Lys6-NH2,
H-дез-Asp28 Pro36,Pro37,Pro38[Trp(O2)25]эксендин-4(1-39)-NH2,
H-(Lys)6-дез-Pro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дез-Pro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-NH2,
дез-Pro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дез-Pro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-дез-Pro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дез-Pro36[Met(O)14,Asp28]эксендин-4(1-39)-Lys6-NH2,
desMet(O)14,Asp28,Pro36,Pro37,Pro38-эксендин-4(1-39)-NH2,
H-(Lys)6-дез-Pro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дез-Pro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-NH2,
дез-Pro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дез-Pro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-Lys6-NH2,
H-Asn-(Glu)5-дез-Pro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дез-Pro36[Met(O)14, Trp(O2)25,Asp28]эксендин-4(1-39)-Lys6-NH2,
дез-Asp28Pro36,Pro37,Pro38[Met(O)14, Trp(O2)25]эксендин-4(1-39)-NH2,
H-(Lys)6-дез-Pro36,Pro37,Pro38[Met(O)14, Trp(O2)25,Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дез-Pro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-NH2,
дез-Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дез-Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-дез-Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2 и их фармацевтически приемлемых солей.
В дополнительном аспекте предпочтительный агонист GLP-1 выбран из группы, состоящей из GLP-1 (в частности, GLP-1(7-36)-амида, SEQ ID NO: 4), Arg34,Lys26(Nε(γ-глутамил(Nαгексадеканоил)))GLP-1(7-37) (лираглутида), албиглутида, дулаглутида, семаглутида и их фармацевтически приемлемых солей. В частности, предпочтительный агонист GLP-1 выбран из группы, состоящей из албиглутида, дулаглутида, семаглутида и их фармацевтически приемлемых солей.
Еще один предпочтительный агонист GLP-1 представляет собой ликсисенатид (SEQ ID NO: 1), а также его фармацевтически приемлемые соли.
В одном аспекте синтезируемый полипептид предпочтительно представляет собой ликсисенатид или эксендин-4, при этом после связывания аминокислотного структурного блока Arg(20), Glu (17), Gln(13), Leu(10) или/и Gly(4) проводят стадию (b) в течение приблизительно 10 мин. с кэппирующим реагентом, содержащим 2% об./об. уксусного ангидрида и 1% об./об. диизопропилэтиламина.
В одном аспекте способ по настоящему изобретению включает синтез полипептидов в форме твердофазного синтеза. Способ по настоящему изобретению необязательно включает дополнительную стадию:
(d) отщепления полипептида, связанного с твердой фазой.
Стадию (d), в частности, проводят после завершения синтеза аминокислотной цепи. Стадию (d) можно проводить путем отщепления от твердой фазы полипептида, связанного с твердой фазой, которое предусматривает приведение твердой фазы, с которой связан полипептид, в контакт с композицией, фактически состоящей из трифторуксусной кислоты и 1,2-этандитиола, при температуре в диапазоне от приблизительно 23°C до приблизительно 29°C.
Отщепление можно проводить с помощью композиции, содержащей трифторуксусную кислоту в количестве от приблизительно 95 до приблизительно 99% об./об.
Отщепление также можно проводить с помощью композиции, содержащей 1,2-этандитиол в количестве от приблизительно 1 до приблизительно 5% об./об.
Предпочтительно, отщепление проводят с помощью композиции, которая в основном состоит из трифторуксусной кислоты в количестве, составляющем приблизительно 97% об./об., и 1,2-этандитиола в количестве, составляющем приблизительно 3% об./об.
Отщепление можно проводить путем приведения композиции в контакт с твердой фазой, с которой связан полипептид, при температуре от приблизительно 25°C до приблизительно 27°C.
Предпочтительно, отщепление проводят путем приведения композиции в контакт с твердой фазой, с которой связан полипептид, при температуре приблизительно 26°C.
Наиболее предпочтительно, отщепление проводят с помощью композиции, которая в основном состоит из трифторуксусной кислоты в количестве, составляющем приблизительно 97% об./об., и 1,2-этандитиола в количестве, составляющем приблизительно 3% об./об., при температуре приблизительно 26°C.
Наиболее предпочтительно, отщепление проводят с помощью композиции, которая в основном состоит из трифторуксусной кислоты в количестве, составляющем приблизительно 97% об./об., и 1,2-этандитиола в количестве, составляющем приблизительно 3% об./об., при температуре приблизительно 26°C в течение 4 часов.
Предпочтительный аспект настоящего изобретения относится к способу твердофазного синтеза полипептида, содержащего предварительно определенную аминокислотную последовательность, при этом способ включает циклы связывания аминокислотных структурных блоков с аминокислотной цепью,
где указанные аминокислотные структурные блоки содержат незащищенную C-концевую карбоксильную группу и защищенную N-концевую аминогруппу,
и где указанная аминокислотная цепь содержит незащищенную N-концевую аминогруппу,
при этом по меньшей мере один цикл связывания включает следующие стадии:
(a) связывание С-конца аминокислотного структурного блока с незащищенной N-концевой аминогруппой аминокислотной цепи, так чтобы между аминокислотной цепью и аминокислотным структурным блоком образовалась амидная связь,
(b) приведение продукта, полученного на стадии (а), в контакт с кэппирующим реагентом, содержащим уксусный ангидрид, при этом уксусный ангидрид связывается с незащищенной N-концевой аминогруппой аминокислотной цепи, с которой на стадии (а) не был связан ни один структурный блок, и
(с) удаление защитной группы с N-концевой аминогруппы аминокислотного структурного блока,
где кэппирующий реагент содержит 0,5-5% об./об. уксусного ангидрида и 0,2-2% об./об. диизопропилэтиламина.
Следующий аспект настоящего изобретения относится к композиции, содержащей 0,5-5% об./об. уксусного ангидрида и 0,2-2% об./об. диизопропилэтиламина в DMF. В предпочтительном аспекте композиция по настоящему изобретению содержит 1-3% об./об. уксусного ангидрида и 0-5-2% об./об. диизопропилэтиламина в DMF, предпочтительно - приблизительно 2% об./об. уксусного ангидрида и приблизительно 1% об./об. диизопропилэтиламина в DMF.
Еще одним аспектом настоящего изобретения предусмотрена композиция, содержащая диизопропилэтиламин в концентрации, составляющей приблизительно 1% об./об. и уксусный ангидрид в концентрации, составляющей приблизительно 2% об./об.
Еще одним аспектом настоящего изобретения предусмотрена композиция, содержащая гомолог уксусного ангидрида в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация гомолога уксусного ангидрида составляет 1-3% об./об., более предпочтительно приблизительно 2% об./об. Композиция может содержать DIPEA, как описано в данном документе.
Еще один аспект настоящего изобретения относится к композиции, содержащей бензоилхлорид в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация бензоилхлорида составляет 1-3% об./об., более предпочтительно приблизительно 2% об./об. Композиция может содержать DIPEA, как описано в данном документе.
Еще один аспект настоящего изобретения относится к композиции, содержащей N-(бензилоксикарбонилокси)сукцинимид в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация N-(бензилоксикарбонилокси)сукцинимида составляет 1-3% об./об., более предпочтительно приблизительно 2% об./об. Композиция может содержать DIPEA, как описано в данном документе.
Еще один аспект настоящего изобретения относится к композиции, содержащей бензилхлорформиат в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация бензилхлорформиата составляет 1-3% об./об., более предпочтительно приблизительно 2% об./об. Композиция может содержать DIPEA, как описано в данном документе.
Еще один аспект настоящего изобретения относится к композиции, содержащей сложный эфир хлормуравьиной кислоты в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация сложного эфира хлормуравьиной кислоты составляет 1-3% об./об., более предпочтительно приблизительно 2% об./об. Композиция может содержать DIPEA, как описано в данном документе.
Еще один аспект настоящего изобретения относится к композиции, содержащей 1-ацетилимидазол в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация 1-ацетилимидазола составляет 1-3% об./об., более предпочтительно приблизительно 2% об./об. Композиция может содержать DIPEA, как описано в данном документе.
Еще один аспект настоящего изобретения относится к композиции, содержащей ди-трет-бутилдикарбонат в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация ди-трет-бутилдикарбоната составляет 1-3% об./об., более предпочтительно приблизительно 2% об./об. Композиция может содержать DIPEA, как описано в данном документе.
Еще один аспект настоящего изобретения относится к композиции, содержащей N-(трет-бутоксикарбонилокси)сукцинимид в концентрации, составляющей 0,5-5% об./об. В предпочтительном аспекте концентрация N-(трет-бутоксикарбонилокси)сукцинимида составляет 1-3% об./об., более предпочтительно приблизительно 2% об./об. Композиция может содержать DIPEA, как описано в данном документе.
Композицию по настоящему изобретению можно применять для кэпирования свободных аминогрупп в синтезе полипептида, в частности, в синтезе полипептида, описанного в данном документе. Более того, композицию по настоящему изобретению можно применять для ацетилирования свободной аминогруппы со связанным углеродом, как описано в данном документе.
В предпочтительном аспекте композицию по настоящему изобретению применяют на стадии (b) способа по настоящему изобретению, например, в течение периода, составляющего 10 мин., после стадии связывания (a) в положениях Arg(20), Glu(17), Gln(13), Leu(10) или/и Gly(4) ликсисенатида, эксендина-3 или эксендина-4 или соответствующих положениях другого аналога GLP-1.
Еще одним аспектом настоящего изобретения является применение композиции, описанной в данном документе, для ацетилирования незащищенной аминогруппы в ходе синтеза полипептида.
Сокращения
Ac(N1-N2): ацетилированный на N-конце фрагмент полипептида от положения N1 до N2.
H(N1-N2) или (N1-N2): фрагмент полипептида от положения N1 до N2, содержащий свободную, N-концевую функциональную аминогруппу.
Fmoc(N1-N2): фрагмент полипептида от положения N1 до N2, содержащий защищенную N-концевую функциональную аминогруппу, где защитной группой является Fmoc.
(N-1)-примесь: относится к образованию непредусмотренного пептида в ходе синтеза пептида, у которого в определенном положении отсутствует структурный блок. Если предусмотренный синтезированный полипептид имеет длину N, примесь имеет длину N-1. Появление примесей (N-1) предотвращают путем кэппирования.
Fmoc флуоренилметилхлороформиат
Boc трет-бутоксикарбонил
Bzl бензил
Pbf 2,2,5,7,8-пентаметилдигидробензофуран-5-сульфонил
tBu трет-бутил
OtBu O-трет-бутил
Trt тритил
DIPE простой диизопропиловый эфир
Настоящее изобретение дополнительно охарактеризовано приведенными ниже фигурами и примерами.
Фигуры
Фигура 1: твердофазный синтез пептидов.
Фигура 2: последовательность ликсисенатида (SEQ ID NO: 1), эксендина-4 (SEQ ID NO: 2), эксендина-3 (SEQ ID NO: 3) и GLP-1(GLP-1(7-36)-амида, SEQ ID NO: 4).
Фигура 3: образование ацетилированных ошибочных последовательностей в ходе синтеза ликсисенатида. Присоединение Fmoc-Arg(20)-OH и последующее кэппирование/отщепление Fmoc. Следует отметить, что из синтеза было исключено положение 21 (Leu). (1) Fmoc-(22-44)+Arg, (2) (22-44)+Arg, (3) Ac(22-44)+Arg, (4) Fmoc-(22-44)+Arg+Val. По этим данным видно, что в ходе стадии кэппирования уж образовались ацетилированные фрагменты, однако ацетилировано неправильное положение [Ас(22-24)+Arg уже присутствует в ходе кэппирования Arg].
Фигура 4: образование ацетилированных ошибочных последовательностей в ходе синтеза ликсисенатида. Присоединение Fmoc-Gln(13)-OH и последующее кэппирование/отщепление Fmoc. (1) Ac(14-44), (2) Fmoc(13-44), (3) Ac(13-44), (4) (13-44), (5) (14-44). По этим данным видно, что в ходе стадии кэппирования уже образовались ацетилированные фрагменты, однако ацетилировано неправильное положение (Ac(13-44)).
Фигура 5: образование ацетилированных ошибочных последовательностей в ходе синтеза ликсисенатида. Присоединение Fmoc-Lys(12)-OH и последующее кэппирование/отщепление Fmoc. (1) Ac(13-44), (2) Fmoc(12-44), (3) Ac(12-44), (4) (12-44). По этим данным видно, что в ходе стадии кэппирования уже образовались ацетилированные фрагменты, однако ацетилировано неправильное положение (Ac(12-44)).
Фигура 6: результаты сравнения синтеза ликсисенатида с использованием способа кэппирования по настоящему изобретению (B) с кэппированием с использованием 10% уксусного ангидрида и 5% объем/объем DIPEA в DMF в течение 20 мин (A), полученные посредством HPLC-хроматографии. (C) Наложение HPLC-хроматограмм (A) и (B).
Фигура 7: HPLC ликсисенатида (неочищенный продукт). Красным: нежелательные ацетилированные побочные продукты.
Фигура 8: образование Ac(36-44) в зависимости от кэппирующей смеси и температуры.
Фигура 9: образование Ac(23-44) в зависимости от кэппирующей смеси и температуры.
Фигура 10: образование Ac(21-44) в зависимости от кэппирующей смеси и температуры.
Фигура 11: образование Ac(19-44) в зависимости от кэппирующей смеси и температуры.
Фигура 12: образование Ac(18-44) в зависимости от кэппирующей смеси и температуры.
Фигура 13: образование Ac(15-44) в зависимости от кэппирующей смеси и температуры.
Фигура 14: образование Ac(12-44) в зависимости от кэппирующей смеси и температуры.
Фигура 15: образование Ac(8-44) в зависимости от кэппирующей смеси и температуры.
Фигура 16: образование Ac(6-44) в зависимости от кэппирующей смеси и температуры.
Фигура 17: результаты сравнения содержания Ac(X-44) при кэппировании в 9 различных положениях в ходе синтеза ликсисенатида при 15°C, комнатной температуре (RT) и 30°C.
Фигура 18: результаты сравнения содержания Ac[(X-1)-44] при кэппировании в 9 различных положениях в ходе синтеза ликсисенатида при 15°C, комнатной температуре (RT) и 30°C.
Фигура 19: результаты сравнения содержания Ac(X-44) при кэппировании при различных условиях или без кэппирования в 9 различных положениях в ходе синтеза ликсисенатида при различных условиях кэппирования.
Фигура 20: результаты сравнения содержания Ac[(X-1)-44] при кэппировании при различных условиях или без кэппирования в 9 различных положениях в ходе синтеза ликсисенатида при различных условиях кэппирования.
Пример 1
Синтез ликсисенатида
Активное вещество ликсисенатид представляет собой амид полипептида, состоящий из 44 аминокислот; при этом ацетат выполняет функцию противоиона.
В однобуквенном коде аминокислотная последовательность ликсисенатида выглядит следующим образом:
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-l-E-W-L-K-N-G-G-P-S-S-G-A-P-P-S-K-K-K-K-K-K-NH2
Пептидную цепь синтезировали посредством линейного твердофазного синтеза, начиная от C-конца Lys-44.
Способ синтеза представляет собой твердофазный пептидный синтеза с использованием Fmoc, в котором для получения амида пептида использовали амидную смолу Ринка. Реакции осуществляли в DMF при комнатной температуре. Между реакциями периодически проводили промывки, преимущественно посредством DMF, при этом одну из промежуточных стадий промывки проводили с использованием изопропанола.
Синтез ликсисенатида на полимерной подложке можно разделить на следующие стадии:
• присоединение первой Fmoc-аминокислоты (Fmoc-Lys(Boc)-OH) со смолой Ринка;
• кэппирование не вступившей в реакцию аминогруппы;
• отщепление временной защитной группы Fmoc;
• присоединение дополнительных Fmoc-аминокислот или Fmoc-дипептидов;
• кэппирование не вступившей в реакцию аминогруппы;
• конечное отщепление Fmoc;
• отщепление ликсисенатида от смолы и одновременное удаление защитных групп на боковых цепях.
Цикл синтеза проиллюстрирован на фигуре 1.
1.1 Присоединение первой Fmoc-аминокислоты (Fmoc-Lys(Boc)-OH) со смолой Ринка
До начала синтеза амидную смолу Ринка обрабатывали DMF, чтобы она набухла. Набуханию ее подвергали на протяжении 2-15 ч. После этого от амидной смолы Ринка отщепляли временную защитную группу Fmoc с использованием 25% пиперидина в DMF. Данное отщепление проводили дважды; время отщепления составляло 5 минут и 20 минут. После отщепления Fmoc смолу повторно промывали DMF и однократно изопропанолом.
Присоединение первой Fmoc-аминокислоты, Fmoc-Lys(Boc)-OH, осуществляли в избытке, составлявшем 2,4 экв., с целью загрузки смолы. Гидрат HOBt, HBTU и DIPEA выступали в качестве конденсирующих реагентов. Время присоединения составляло 60-120 мин.
Для полной загрузки смолы Ринка посредством Fmoc-Lys(Boc)-OH проводили дополнительную загрузку с помощью конденсирующих реагентов - гидрата HOBt и DIC. Время присоединения составляло 6-18 ч. В ходе осуществления стадии 1.1 смесь перемешивали. Затем осуществляли кэппирование.
1.2 Кэппирование не вступившей в реакцию аминогруппы
Следствием неполной загрузки смолы было то, что на смоле еще оставались не вступившие в реакцию аминогруппы. Их инактивировали, а значит делали недоступными для дальнейшего присоединения, путем добавления смеси уксусный ангидрид/DIPEA/DMF (10:5:85). Кэппирующую смесь оставляли на смоле в течение 20 минут при перемешивании. Ацилировали оставшуюся свободную аминогруппу. После этого смолу повторно промывали DMF и однократно изопропанолом.
Способ кэппирования по настоящему изобретению в по меньшей мере 5 положениях в ходе синтеза ликсисенатида описан в примерах 4 и 5.
1.3. Отщепление временной защитной группы Fmoc
Временную защитную группу Fmoc отщепляли с использованием 25% пиперидина в DMF. Данное отщепление проводили дважды; время отщепления составляло 5 минут и 20 минут. После отщепления Fmoc смолу повторно промывали DMF и однократно изопропанолом.
1.4 Присоединение дополнительных Fmoc-аминокислот или Fmoc-дипептидов
Следующую Fmoc-аминокислоту присоединяли к незащищенной аминогруппе, присутствующей на смоле. Присоединение осуществляли в DMF в разных эквивалентах. Время присоединения составляло от 2 ч до 18 ч. В качестве конденсирующих реагентов использовали HOBt/DIC, а также HBTU/DIPEA.
В качестве Fmoc-аминокислот использовали следующие производные:
• Fmoc-Lys(Boc)-OH
• Fmoc-Ser(tBu)-OH
• Fmoc-Pro-OH
• Fmoc-Ala-OH x H2O
• Fmoc-Gly-OH
• Fmoc-Asn(Trt)-OH
• Fmoc-Leu-OH
• Fmoc-Trp(Boc)-OH
• Fmoc-Glu(OtBu)-OH x H2O
• Fmoc-lle-OH
• Fmoc-Phe-OH
• Fmoc-Arg(Pbf)-OH
• Fmoc-Val-OH
• Fmoc-Met-OH
• Fmoc-Gln(Trt)-OH
• Fmoc-Asp(OtBu)-OH
• Fmoc-Thr(tBu)-OH
• Fmoc-His(Trt)-OH
Альтернативно можно было использовать Fmoc-дипептиды (способ по настоящему изобретению):
• Fmoc-Pro-Pro-OH (CAS 129223-22-9)
• Fmoc-Ala-Pro-OH (CAS 186023-44-9)
• Fmoc-Ser(tBu)-Gly-OH (CAS 113247-80-6)
• Fmoc-Gly-Pro-OH (CAS 212651-48-4)
• Fmoc-Gly-Gly-OH (CAS 35665-38-4)
• Fmoc-Asn(Trt)-Gly-OH (от Bachem B-3630)
• Fmoc-Glu(OtBu)-Gly-OH (CAS 866044-63-5)
• Fmoc-His(Trt)-Gly-OH
Если присоединение оказывалось неполным в соответствии с тестом Кайзера (E. Kaiser et al, Anal. Biochem. 34, 1970, 595), тогда можно провести дополнительное присоединение. С этой целью Fmoc-аминокислоту повторно присоединяли с использованием HBTU/DIPEA/гидрата HOBt.
1.5 Кэппирование не вступившей в реакцию аминогруппы
См. описание в разделе 1.2.
1.6 Конечное отщепление Fmoc
Конечное отщепление Fmoc осуществляли так, как описано в пункте 1.3. В завершении смолу повторно промывали простым диизопропиловым эфиром и высушивали при пониженном давлении.
1.7 Отщепление ликсисенатида от смолы и одновременное удаление защитных групп на боковых цепях
Отщепление ликсисенатида от смолы Ринка осуществляли так, как это описано в примере 6.
1.8 Синтез ликсисенатида с применением дипептидов по настоящему изобретению
Присоединение первой Fmoc-Lys(Boc)-OH к смоле осуществляли с использованием HBTU/DIPEA/гидрата HOBt. После присоединения первой аминокислоты Fmoc-Lys(Boc)-OH к свободному амину амидной смолы Ринка дальнейшие стадии способа проводили в длительно повторяющемся цикле (также см. стадии 1.3-1.6):
• отщепление Fmoc;
• присоединение;
• дополнительное присоединение, если это необходимо;
• кэппирование;
• отщепление N-концевой группы Fmoc после присоединения конечного аминокислотного звена.
Стандартные Fmoc-защищенные аминокислоты присоединяли к DIC/HOBt, при этом избыток аминокислот и конденсирующих реагентов составлял от 2 до 4 эквивалентов.
В положениях Pro(36) и Pro(37) вместо двух аминокислотных производных Fmoc-Pro-OH осуществляли присоединение дипептида Fmoc-Pro-Pro-OH с использованием HBTU/DIPEA.
В положении Pro(31) присоединение осуществляли с использованием HBTU/DIPEA/гидрата HOBt.
В положениях His(1) и Gly(2) вместо аминокислотных производных Fmoc-His(Boc)-OH и Fmoc-Gly-OH осуществляли присоединение дипептида Fmoc-His(Trt)-Gly-OH.
После стадий присоединения осуществляли кэппирование, в каждом случае с использованием Ac2O/DIPEA, как это описано в примерах 4 и 5.
Отщепление Fmoc осуществляли с использованием 25% пиперидина в DMF, в каждом случае последовательно, начиная с 5-минутного времени реакции и продолжая 20-40-минутным временем реакции.
Степень завершенности присоединения проверяли в тесте Кайзера.
После последнего присоединения и последнего отщепления группы Fmoc смолу промывали сначала несколько раз DMF, затем изопропанолом, а в завершении простым диизопропиловым эфиром, и затем ее сушили при 35°C при пониженном давлении.
Отщепление неочищенного пептида от смолы осуществляли в трифторуксусной кислоте с такими акцепторами, как 1,2-этандитиол.
Неочищенный пептид подвергали очистке в двухстадийной HPLC, при этом в качестве твердой фазы выступал силикагель C18 RP. На первой стадии очистки применяли буферную систему с ацетонитрилом/водой и 0,1% TFA; на второй стадии применяли буферную систему с ацетонитрилом/водой и AcOH. После концентрирования объединенных растворов чистый пептид получали посредством лиофилизации.
Использование 3500 г амидной смолы Ринка с загрузкой 0,3 ммоль/г (т. е. 1,05 мольную партию) давало 9970 г пептида, связанного со смолой. Из него получали 4636 г неочищенного пептида.
После очистки из них получали 576 г чистого пептида. MS: 4855,5 (моноизотопная молярная масса); полученная: 4855,6. Секвенирование аминокислотной последовательности: получена правильная последовательность. Анализ: 89,0% (как есть).
1.9 Синтез ликсисенатида без применения дипептидов
Пептидную цепь синтезировали посредством линейного твердофазного синтеза, начиная от C-конца Lys-44.
Стандартные Fmoc-защищенные аминокислоты присоединяли к DIC/HOBt, при этом избыток аминокислот и конденсирующих реагентов составлял от 2 до 4 эквивалентов.
В положениях Pro(37), Pro(36), Pro(31) присоединение осуществляли с использованием HBTU/DIPEA/гидрата HOBt.
После каждого присоединения проводили кэппирование с использованием Ac2O/DIPEA. Отщепление Fmoc осуществляли с использованием 25% пиперидина в DMF, в каждом случае последовательно, начиная с 5-минутного времени реакции и продолжая 20-минутным временем реакции.
Степень завершенности присоединения проверяли в тесте Кайзера. После последнего присоединения и последнего отщепления группы Fmoc смолу промывали сначала несколько раз DMF, затем изопропанолом, а в завершении простым диизопропиловым эфиром, и затем ее сушили при 35°C при пониженном давлении.
Отщепление неочищенного пептида от смолы осуществляли в трифторуксусной кислоте с такими акцепторами, как 1,2-этандитиол, тиоанизол, фенол и вода.
Неочищенный пептид подвергали очистке в двухстадийной HPLC, при этом в качестве твердой фазы выступал силикагель C18 RP. После концентрирования объединенных растворов чистый пептид получали посредством лиофилизации. В таблице 1 представлены результаты сравнения содержания рацемизированного D-His-ликсисенатида и содержания некоторых примесей в чистом пептиде при синтезе с использованием и без использования дипептидов.
Таблица 1
Результаты сравнения синтезов ликсисенатида с использованием и без использования дипептидов.
ликсисенатид
дез-Pro(36)-
ликсисенатид
ди-Pro(36)-
ликсисенатид
по настоящему изобретению
По этим данным видно, что применение дипептида Fmoc-His (Trt)-GIy-OH давало ликсисенатид, который не содержал повышенных количеств D-His, возникающих в результате рацемизации. Более того, при использовании Fmoc-His(Trt)-GIy-OH дез-Gly(2)-ликсисенатид больше не выявляли. Кроме того, не были выявлены и пептиды N-1 и N+1 рядом с положением Pro(36) и Pro(37) в цепи (например, дез-Pro(36)-ликсисенатид или ди-Pro(36)ликсисенатид).
Пример 2
Синтез, очистка и определение характеристик эксендина-4 (по настоящему изобретению)
Активное вещество эксендин-4 представляет собой амид полипептида, состоящий из 39 аминокислот; при этом ацетат выполняет функцию противоиона.
В однобуквенном коде аминокислотная последовательность выглядит следующим образом:
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-I-E-W-L-K-N-G-G-P-S-S-G-A-P-P-P-S-NH2
MW 4186,66 г/моль; MW (моноизотопная) = 4184,03 г/моль.
Синтез эксендина-4 осуществляли так же, как это описано в синтезе ликсисенатида, в соответствии с указанной выше последовательностью. В положениях 1 и 2 присоединение осуществляли за один цикл с Fmoc-His(Trt)-Gly-OH. В положениях 37 и 38 присоединение осуществляли за один цикл с Fmoc-Pro-Pro-OH. В других положениях присоединение осуществляли с Fmoc-аминокислотами (звеньями моноаминокислот).
Использование 26,666 г амидной смолы Ринка с загрузкой 0,42 ммоль/г (т. е. партия с 11,2 ммоль) давало 74 г пептида, связанного со смолой. Из этого количества отбирали 65 г пептида, связанного со смолой, для отщепления и получали 28 г неочищенного пептида. Для очистки из этого количества брали 21,3 г неочищенного пептида и получали 4,01 г чистого пептида. MS: 4184,03 (моноизотопная молярная масса); полученная: 4185,1[M+H]. Чистота 98,25 FI%.
Использование дипептидов подтверждало результаты, полученные для ликсисенатида. Применение дипептида Fmoc-His(Trt)-GIy-OH давало эксендин-4, который не содержал повышенных количеств D-His, возникающих в результате рацемизации. Более того, при использовании Fmoc-His(Trt)-GIy-OH дез-Gly(2)-эксендин-4 больше не выявляли. Кроме того, не были выявлены и пептиды N-1 и N+1 рядом с положением Pro(36) и Pro(37) в цепи (например, дезPro(36)-эксендин-4 или диPro(36)эксендин-4)ликсисенатид).
Пример 3
Синтез Fmoc-His(Trt)-Gly-OH
3.1 Fmoc-His(Trt)-Gly-OBzl
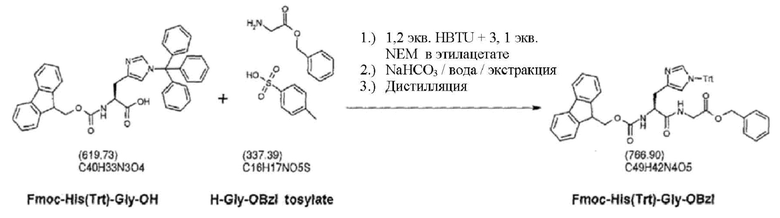
40 г Fmoc-His(Trt)-OH растворяли вместе с 32,7 г тозилата H-Gly-OBzl и 29,37 г HBTU в 400 мл этилацетата. Затем добавляли 33,32 мл N-этилморфолина. Реакционную смесь перемешивали в течение 4 ч при 30°C. Затем трижды экстрагировали с использованием каждый раз 256 г 8% раствора бикарбоната натрия, а затем осуществляли однократное промывание с использованием 250 мл воды. Половину полученного в результате раствора в этилацетате выпаривали и дополнительно обрабатывали на следующей стадии.
3.2 Fmoc-His(Trt)-Gly-OH

К этилацетатной фазе добавляли THF и метанол с тем, чтобы образовывалась 5:2:2 (вес/вес/вес) смесь THF/этилацетат/MeOH. После этого добавляли 10 г палладия на углеродном катализаторе (5%) и данную смесь подвергали гидрогенизации при 30°C и под давлением водорода, равным 1,1 бар, в течение 2,5 ч. Затем катализатор отфильтровывали и полученный в результате раствор выпаривали до начала образования осадка. После этого осуществляли перемешивание в течение 1 ч и оставляли раствор отстаиваться при комнатной температуре на 4 дня. Продукт отфильтровывали и после этого экстрагировали путем перемешивания в 2-бутаноне при 80°C в течение 4 ч. Выход: 32,9 г Fmoc-His(Trt)-Gly-OH (75%).
Пример 4
Ацетилированные ошибочных последовательностей в ходе синтеза ликсисенатида
4.1 Определение содержания ацетилированных ошибочных последовательностей в ходе синтеза ликсисенатида
В HPLC-профиле неочищенного продукта ликсисенатида можно встретить некоторые ацетилированные ошибочные последовательности. Обычно они возникают из-за кэппирования не вступивших в реакцию аминогрупп на смоле. С помощью кэппирования достигается отсутствие примесей (N-1), которые лишь незначительно отличаются от целевого продукта, и, следовательно, их трудно удалить при очистке.
Степень завершенности, а также кинетику связывания в выбранных положениях контролировали с помощью реакции расщепления Эдмана. Отбирали образец смолы, полученный в результате синтеза ликсисенатида, и отщепляли от него группу Fmoc. Затем данный образец смолы подвергали расщеплению по Эдману, и, таким образом, можно было определить соотношение присоединенной аминокислоты и (N-1) аминокислоты, из чего можно непосредственно сделать вывод о конечном результате присоединения. Результаты расщепления по Эдману (таблица 2) демонстрировали высокие значения присоединения. Эти значения были настолько высоки, что они не могли пояснить таких количеств ацетилированных ошибочных последовательностей (данные HPLC в таблице 2). Это означает, что должен был существовать альтернативный способ образования данных побочных продуктов. Разъяснение по данному вопросу будет описано в следующих разделах.
Ac(5-44)
Ac(4-44)
Gly(4)
Glu(3)
99,1-99,8%
98,2-99,4%
Таблица 2: конечные данные присоединения и содержания ацетилированных фрагментов в ходе синтеза ликсисенатида. Сравнивали процентное содержание ацетилированных ошибочных последовательностей согласно данным HPLC и данным по Эдману (совместное элюирование Ас(6-44), Ас(5-44) и Ас(4-44)).
4.2 Образование ацетилированных ошибочных последовательностей
Для исследования в цикле синтеза точек, в которых образуются ацетилированные ошибочные последовательности, в ходе цикла присоединения отбирали образцы смолы и отщепляли пептид и исследовали его с помощью LC-MS. Данные исследования проводили в положениях присоединения Fmoc-Arg(20)-OH и присоединения Fmoc-Gln(13)-OH.
При присоединении Fmoc-Arg(20)-OH к пептиду, связанному с твердой фазой, с частичной последовательностью ликсисенатида H(22-24) отбирали образцы после периодов присоединения, составляющих 1 ч, 2 ч, 4 ч, 8 ч и 24 ч, а также после кэппирования, последующего отщепления Fmoc и присоединении валина(19). На фигуре 3 видно, что ошибочная последовательность Ac(22-44)+Arg впервые возникала на стадии кэппирования (3,1%). Следовательно, в ходе кэппирования небольшая часть группы Fmoc отщеплялась (утрачивалась) и сразу же ацилировалась. Для пояснения обозначения Ac(22-24)+Arg следует отметить, что положение 21 (Leu) исключили из синтеза.
Тот же эксперимент проводили для присоединения Fmoc-Gln(13)-OH в ходе синтеза ликсисенатида (фигура 4). В данном случае ошибочную последовательность Ac(13-44) впервые выявили (4,6%) при отщеплении Fmoc после связывания и кэппирования глутамина(13). В остальном цикле синтеза после присоединения Fmoc-Lys(12)-OH можно было увидеть, что в ходе кэппирования также образовывался Ac(12-44) (4,1%) (см. фигуру 5).
Эксперимент продемонстрировал, что необходим поиск условий кэппирования, при которых предотвращается нежелательное образование ацетилированной ошибочной последовательности с N-й аминокислотой (последней присоединенной) без снижения кэппирующей способности используемой смеси до такой степени, чтобы потенциальная примесь (N-1) больше не подвергалась кэппированию.
4.3 Варианты условий кэппирования
Изучали варианты присоединения Fmoc-Arg(20)-OH, Fmoc-Leu(10)-OH, Fmoc-Gly(4)-OH и Fmoc-Thr(5)-OH. Сравнивали различные условия кэппирования.
Варьировали условия кэппирования в ходе лабораторного синтеза ликсисенатида. Особое внимание уделяли содержанию нежелательного Ас(N-44) и требуемого Ас([N-1]-44). Тестируемые условия были следующими:
• 10% уксусный ангидрид/5% DIPEA в DMF в течение 20 минут;
• 10% уксусный ангидрид/5% DIPEA в DMF в течение 10 минут;
• 2% уксусный ангидрид/1% DIPEA в DMF в течение 20 минут;
• 2% уксусный ангидрид/1% DIPEA в DMF в течение 10 минут.
Исследования проводили в положениях Arg(20), Leu(10), Thr(5) и Gly(4). Результаты собраны в таблицах 3-6.
Данные также сравнивали с результатом GMP-синтеза ликсисенатида ("GMP-кэппирование" в таблицах 3-6). Условия кэппирования соответствовали условиям 10% уксусного ангидрида/5% DIPEA в DMF. Время контакта смолы с кэппирующей смесью в GMP-партии было на 7-8 минут дольше и, следовательно, составило 27-28 минут. Причиной этому было то, что на откачку кэппирующей смеси затрачивалось больше времени.
4.3.1 Присоединение в положении Arg(20)
Осуществляли присоединение Fmoc-Arg(Pbf)-OH к Leu(21). На тех же цепях, на которых не происходило присоединение (продукт H(21-44)), при последующем кэппировании образовывался продукт Ac(21-44). Оба продукта Ac(20-44) и H(20-44) образовывались, если во время кэппирования группа Fmoc нежелательным образом отщеплялась (образование H(20-44)) и происходило ацетилирование (образование Ac(2044)).
Из таблицы 3 видно, что степень образования нежелательных продуктов H(20-44) и Ac(20-44) зависела как от времени кэппирования, так и от количества уксусного ангидрида и DIPEA (см. столбец Ac(20-44)%). Наивысшее процентное значение можно увидеть при GMP-кэппировании. Наименьшее содержание Ас(20-44) обнаруживали в условиях "2% уксусный ангидрид/1% DIPEA в DMF в течение 10 минут".
Кэппирующая способность различных кэппирующих смесей (и, следовательно, исходное назначение) примерно была одинаковой (см. столбец Ас(21-44)), т. е. все кэппирующие смеси превращали H(21-44)). Смесь "2% уксусный ангидрид/1% DIPEA в DMF в течение 10 минут" также выполняла свое назначение по исключению примесей (N-1).
Таблица 3: результаты присоединения Fmoc-Arg(Pbf)-OH в положении 20. В таблице показано содержание ацетилированных и неацетилированных фрагментов в зависимости от условий кэппирования. Результаты получили с помощью LC-MS. Данные сравнивали с результатами GMP-синтеза ("GMP-кэппирование").
4.3.2 Связывание в положениях Leu(10), Gly(4) и Thr(5)
Результаты для Leu(10) приведены в таблице 4 и подтверждают результаты, полученные для положения Arg(20). Содержание нежелательных продуктов Ac(10-44) и H(10-44), которые образовывались в ходе кэппирования свободных аминогрупп продукта H(11-44), было наиболее низким в условиях "2% уксусный ангидрид, 1% DIPEA в течение 10 минут". Кэппирующую способность сравнивали у различных кэппирующих смесей.
Таблица 4: результаты связывания Fmoc-Leu-OH в положении 10. В таблице показано содержание ацетилированных и неацетилированных фрагментов в зависимости от условий кэппирования. Результаты получили с помощью LC-MS.
Также в случае связывания Gly(4) содержание нежелательных продуктов Ас(4-44) зависело от кэппирующей смеси и времени реакции. Кэппирующая способность у различных смесей была одинаковой (таблица 5).
Таблица 5: результаты присоединения Fmoc-Gly-OH в положении 4. В таблице показано содержание ацетилированных и неацетилированных фрагментов в зависимости от условий кэппирования. Результаты получили с помощью LC-MS.
Помимо положений Arg(20), Leu(10) и Gly(4) также исследовали положение Thr(5). В отличие от трех предыдущих положений содержание нежелательного продукта Ас(N-44) (Ас(5-44) в положении 5) было примерно одинаковым при различных условиях кэппирования. Тем не менее, в данном случае кэппирующая способность различных смесей также была сопоставимой (таблица 6).
Таблица 6: результаты присоединения Fmoc-Thr(tBu)-OH в положении 5. В таблице показано содержание ацетилированных и неацетилированных фрагментов в зависимости от условий кэппирования. Результаты получили с помощью LC-MS.
4.3.3 Выводы
В положениях Arg(20), Leu(10) и Gly(4) смесь для умеренного кэппирования (2% уксусный ангидрид/1% DIPEA в DMF в течение 10 минут) была достаточной эффективной для того, чтобы сохранялся требуемый эффект устранения (N-1) примесей при ацилировании. Тем не менее, в этих трех случаях соответствующее образование Ас(20-44), Ас(10-44) и Ас(4-44) зависело от времени кэппирования, а также от кэппирующей смеси. Это не применимо к положению Thr(5).
Пример 5
Синтез ликсисенатида
Пример относится к синтезу ликсисенатида (см. SEQ ID NO: 1). В начале синтеза линкер, связанный с твердой фазой, несет защитную группу Fmoc. Осуществляли присоединение отдельных аминокислотных звеньев, начиная от С-конца (положения 44) в направлении N-конца в циклах присоединения, которые состояли из стадий:
• отщепления Fmoc;
• присоединения Fmoc-защищенного аминокислотного звена и
• кэппирования.
В положениях Arg(20), Glu(17), Gln(13), Leu(10) и Gly(4) использовали способ кэппирования по настоящему изобретению (2% уксусный ангидрид/1% DIPEA в DMF в течение 10 минут). Для этих положений указания относительно цикла присоединения описаны ниже. В других положениях кэппирование осуществляли с использованием 10% уксусного ангидрида/5% DIPEA в DMF в течение 20 минут. В качестве примера данное кэппирование описано в положении Thr(5). Способ кэппирования по настоящему изобретению предусматривает более мягкие условия.
Размер партии составил 1050 ммоль смолы Ринка.
5.1. Присоединение Fmoc-Arg(Pbf)-OH в положении 20
5.1.1 Отщепление Fmoc
В реактор вносили 7 л DMF, а затем смесь 7,9 л пиперидина в 16,6 л DMF. Данную смесь перемешивали в течение 5 минут, затем фильтровали с откачкой. Данный процесс повторяли и осуществляли перемешивание в течение 30 минут; затем снова проводили фильтрацию с откачкой. После отщепления Fmoc смолу промывали 7 раз в следующей последовательности: DMF (31,1 л), DMF (31,1 л), изопропанол (31,1 л), DMF (31,1 л), DMF (8 л), DMF (31,1 л), DMF (31,1 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.1.2 Присоединение Fmoc-Arg(Pbf)-OH
В реактор вносили 21 л DMF. Затем добавляли навеску 2,125 кг FmocArg(Pbf)-OH и добавляли 5,3 л DMF. После полного растворения данный раствор выливали в реактор с последующим внесением раствора 502 г гидрата гидроксибензотриазола (гидрата HOBt) в 2,2 л DMF. И в завершении, в реактор вносили 413 г N, N-диизопропилкарбодиимида (DIC). Время присоединения составило 6-18 ч. После присоединения растворитель отфильтровывали из смолы путем откачки и без остановки осуществляли кэппирование.
5.1.3 Кэппирование (по настоящему изобретению)
В реактор заливали 26,3 л DMF. Одновременно 1,2 л DMF, 0,53 л уксусного ангидрида и 0,26 л диизопропилэтиламина (DIPEA) смешивали в 2-литровом сосуде Шотта и добавляли к смоле в реакторе. Содержимое реактора перемешивали в течение 10 минут, затем проводили фильтрацию с откачкой. После кэппирования смолу промывали 5 раз в следующей последовательности: DMF (24 л), изопропанол (31,1 л), DMF (8 л), DMF (31,5 л), DMF (31,5 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.2. Присоединение гидрата Fmoc-Glu(OtBu)-OH в положении 17
5.2.1 Отщепление Fmoc
В реактор вносили 7 л DMF, а затем смесь 7,9 л пиперидина в 16,6 л DMF. Данную смесь перемешивали в течение 5 минут, затем фильтровали с откачкой. Данный процесс повторяли и осуществляли перемешивание в течение 30 мин; затем снова проводили фильтрацию с аспирацией. После отщепления Fmoc смолу промывали 7 раз в следующей последовательности: DMF (31,1 л), DMF (31,1 л), изопропанол (31,1 л), DMF (31,1 л), DMF (8 л), DMF (31,1 л), DMF (31,1 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.2.2 Присоединение гидрата Fmoc-Glu(OtBu)-OH
В реактор вносили 21 л DMF. Затем добавляли навеску 1,453 кг гидрата FmocGlu(OtBu)-OH и добавляли 5,3 л DMF. После полного растворения данный раствор выливали в реактор с последующим внесением раствора 502 г гидрата гидроксибензотриазола (гидрата HOBt) в 2,2 л DMF. И в завершении, в реактор вносили 413 г N, N-диизопропилкарбодиимида (DIC). Время присоединения составило 6-18 ч. После присоединения растворитель отфильтровывали из смолы путем откачки и без остановки осуществляли кэппирование.
5.2.3 Кэппирование (по настоящему изобретению)
В реактор заливали 26,3 л DMF. Одновременно 1,2 л DMF, 0,53 л уксусного ангидрида и 0,26 л диизопропилэтиламина (DIPEA) смешивали в 2-литровом сосуде Шотта и добавляли к смоле в реакторе. Содержимое реактора перемешивали в течение 10 минут, затем проводили фильтрацию с откачкой. После кэппирования смолу промывали 5 раз в следующей последовательности: DMF (24 л), изопропанол (31,1 л), DMF (8 л), DMF (31,5 л), DMF (31,5 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.3 Присоединение Fmoc-Gln(Trt)-OH в положении 13
5.3.1 Отщепление Fmoc
В реактор вносили 7 л DMF, а затем смесь 7,9 л пиперидина в 16,6 л DMF. Данную смесь перемешивали в течение 5 минут, затем фильтровали с откачкой. Данный процесс повторяли и осуществляли перемешивание в течение 35 минут; затем снова проводили фильтрацию с откачкой. После отщепления Fmoc смолу промывали 7 раз в следующей последовательности: DMF (31,1 л), DMF (31,1 л), изопропанол (31,1 л), DMF (31,1 л), DMF (8 л), DMF (31,1 л), DMF (31,1 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.3.2 Присоединение Fmoc-Gln(Trt)-OH
В реактор вносили 21 л DMF. Затем добавляли навеску 2,001 кг FmocGln(Trt)-OH и добавляли 5,3 л DMF. После полного растворения данный раствор выливали в реактор с последующим внесением раствора 502 г гидрата гидроксибензотриазола (гидрата HOBt) в 2,2 л DMF. И в завершении, в реактор вносили 413 г N, N-диизопропилкарбодиимида (DIC). Время присоединения составило 6-18 ч. После присоединения растворитель отфильтровывали из смолы путем откачки и без остановки осуществляли кэппирование.
5.3.3 Кэппирование (по настоящему изобретению)
В реактор заливали 26,3 л DMF. Одновременно 1,2 л DMF, 0,53 л уксусного ангидрида и 0,26 л диизопропилэтиламина (DIPEA) смешивали в 2-литровом сосуде Шотта и добавляли к смоле в реакторе. Содержимое реактора перемешивали в течение 10 минут, затем проводили фильтрацию с откачкой. После кэппирования смолу промывали 5 раз в следующей последовательности: DMF (24 л), изопропанол (31,1 л), DMF (8 л), DMF (31,5 л), DMF (31,5 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.4 Присоединение Fmoc-Leu-OH в положении 10
5.4.1 Отщепление Fmoc
В реактор вносили 7 л DMF, а затем смесь 7,9 л пиперидина в 16,6 л DMF. Данную смесь перемешивали в течение 5 минут, затем фильтровали с откачкой. Данный процесс повторяли и осуществляли перемешивание в течение 35 минут; затем снова проводили фильтрацию с откачкой. После отщепления Fmoc смолу промывали 7 раз в следующей последовательности: DMF (31,1 л), DMF (31,1 л), изопропанол (31,1 л), DMF (31,1 л), DMF (8 л), DMF (31,1 л), DMF (31,1 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.4.2 Присоединение Fmoc-Leu-OH
В реактор вносили 21 л DMF. Затем добавляли навеску 1,158 кг Fmoc-Leu-OH и добавляли 5,3 л DMF. После полного растворения данный раствор выливали в реактор с последующим внесением раствора 502 г гидрата гидроксибензотриазола (гидрата HOBt) в 2,2 л DMF. И в завершении, в реактор вносили 413 г N, N-диизопропилкарбодиимида (DIC). Время присоединения составило 6-18 ч. После присоединения растворитель отфильтровывали из смолы путем откачки и без остановки осуществляли кэппирование.
5.4.3 Кэппирование (по настоящему изобретению)
В реактор заливали 26,3 л DMF. Одновременно 1,2 л DMF, 0,53 л уксусного ангидрида и 0,26 л диизопропилэтиламина (DIPEA) смешивали в 2-литровом сосуде Шотта и добавляли к смоле в реакторе. Содержимое реактора перемешивали в течение 10 минут, затем проводили фильтрацию с откачкой. После кэппирования смолу промывали 5 раз в следующей последовательности: DMF (24 л), изопропанол (31,1 л), DMF (8 л), DMF (31,5 л), DMF (31,5 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.5 Связывание Fmoc-Gly-OH в положении 4
5.5.1 Отщепление Fmoc
В реактор вносили 7 л DMF, а затем смесь 7,9 л пиперидина в 16,6 л DMF. Данную смесь перемешивали в течение 5 минут, затем фильтровали с откачкой. Данный процесс повторяли и осуществляли перемешивание в течение 35 минут; затем снова проводили фильтрацию с откачкой. После отщепления Fmoc смолу промывали 7 раз в следующей последовательности: DMF (31,1 л), DMF (31,1 л), изопропанол (31,1 л), DMF (31,1 л), DMF (8 л), DMF (31,1 л), DMF (31,1 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.5.2 Присоединение Fmoc-Gly-OH
В реактор вносили 21 л DMF. Затем добавляли навеску 1,217 кг Fmoc-Gly-OH и добавляли 5,3 л DMF. После полного растворения данный раствор выливали в реактор с последующим внесением раствора 627 г гидрата гидроксибензотриазола (гидрата HOBt) в 2,2 л DMF. И в завершении, в реактор вносили 517 г N, N-диизопропилкарбодиимида (DIC). Время присоединения составило 6-18 ч. После присоединения растворитель отфильтровывали из смолы путем откачки и без остановки осуществляли кэппирование.
5.5.3 Кэппирование (по настоящему изобретению)
В реактор заливали 26,3 л DMF. Одновременно 1,2 л DMF, 0,53 л уксусного ангидрида и 0,26 л диизопропилэтиламина (DIPEA) смешивали в 2-литровом сосуде Шотта и добавляли к смоле в реакторе. Содержимое реактора перемешивали в течение 10 минут, затем проводили фильтрацию с откачкой. После кэппирования смолу промывали 5 раз в следующей последовательности: DMF (24 л), изопропанол (31,1 л), DMF (8 л), DMF (31,5 л), DMF (31,5 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.6 Присоединение Fmoc-Thr(tBu)-OH в положении 5
5.6.1 Отщепление Fmoc
В реактор вносили 7 л DMF, а затем смесь 7,9 л пиперидина в 16,6 л DMF. Данную смесь перемешивали в течение 5 минут, затем фильтровали с откачкой. Данный процесс повторяли и осуществляли перемешивание в течение 35 минут; затем снова проводили фильтрацию с откачкой. После отщепления Fmoc смолу промывали 7 раз в следующей последовательности: DMF (31,1 л), DMF (31,1 л), изопропанол (31,1 л), DMF (31,1 л), DMF (8 л), DMF (31,1 л), DMF (31,1 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.6.2 Присоединение Fmoc-Thr(tBu)-OH
В реактор вносили 21 л DMF. Затем добавляли навеску 1,628 кг FmocThr(tBu)-OH и добавляли 5,3 л DMF. После полного растворения данный раствор выливали в реактор с последующим внесением раствора 627 г гидрата гидроксибензотриазола (гидрата HOBt) в 2,2 л DMF. И в завершении, в реактор вносили 517 г N, N-диизопропилкарбодиимида (DIC). Время присоединения составило 6-18 ч. После присоединения растворитель отфильтровывали из смолы путем откачки и без остановки осуществляли кэппирование.
5.6.3 Кэппирование
В реактор заливали 10,5 л DMF. Одновременно 15,8 л DMF, 3,2 л уксусного ангидрида и 1,6 л диизопропилэтиламина (DIPEA) смешивали в сосуде для перемешивания и добавляли к смоле в реакторе. Содержимое реактора перемешивали в течение 20 минут, затем проводили фильтрацию с откачкой. После кэппирования смолу промывали 5 раз в следующей последовательности: DMF (24 л), изопропанол (31,1 л), DMT (8 л), DMF (31,5 л), DMF (31,5 л). При этом реактор каждый раз заполняли соответствующим промывающим растворителем, затем осуществляли перемешивание в течение 3 минут и снова проводили фильтрацию с откачкой.
5.7 Результаты
На фигуре 6 показана HPLC-хроматограмма неочищенного продукта синтеза ликсисенатида при помощи способа кэппирования по настоящему изобретению в положениях Arg(20), Glu(17), Gln(13), Leu(10) и Gly(4) и с кэппированием на других стадиях присоединения, как описано в разделе 5.6.3. Указаны пики, соответствующие примесями ацетил(20-44), ацетил(17-44), ацетил(13-44), ацетил(10-44) и ацетил(4-44)/ацетил(6-44).
5.8 Сравнение
Проводили стадии кэппирования со всеми вариантами присоединения, как это описано в разделе 5.6.3, что приводило к увеличению образования нежелательных ошибочных последовательностей Ac(20-44), Ac(17-44), Ac(13-44), Ac(10-44) и Ас(4-44)/Ас(6-44). HPLC-хроматограмма неочищенного ликсисенатида по результатам данного теста показана на фигуре 6А.
На фигуре 6B показана HPLC-хроматограмма неочищенного ликсисенатида, синтезированного посредством способа кэппирования по настоящему изобретению в положениях Arg(20), Glu(17), Gln(13), Leu(10) и Gly(4).
На фигуре 6C показано наложение HPLC-хроматограмм из фигур 6A и B. Очевидно, что синтез ликсисенатида посредством способа кэппирования по настоящему изобретению в периодическом режиме приводил к отчетливому снижению количества ошибочных последовательностей Ac(20-44), Ас(17-44), Ас(13-44), Ас(10-44) и Ас(4-44)/Ас(6-44).
При использовании смеси для более мягкого кэппирования (2% уксусного ангидрида/1% DIPEA в DMF в течение 10 минут) можно было снизить количество ацетилированных ошибочных последовательностей Ac(20-44), Ac(17-44), Ac(13-44), Ас(10-44) и Ас(4-44) в неочищенном продукте ликсисенатида или устранить их из него. Поскольку неочищенный продукт ликсисенатида, который был получен путем кэппирования по настоящему изобретению, содержал ацетилированные побочные продукты Ас(17-44), Ас(13-44) и Ас(10-44), в частности, в значительно сниженных количествах, очистка ликсисенатида упрощалась. Как результат, объединение фракций после первого цикла препаративной хроматографии с ликсисенатидом давало больше фракций, которые соответствовали техническим критериям и поэтому не подлежали отбраковке. Это приводило к увеличению выхода.
Пример 6
Кэппирование в 9 конкретных положениях при синтезе ликсисенатида
Как рассмотрено в примере 5, использование "мягких" условий кэппирования при синтезе ликсисенатида в положениях Arg(20), Glu(17), Gln(13), Leu(10) или/и Gly(4) улучшило профиль нежелательных побочных продуктов.
В данном примере описано влияние условий кэппирования на образование ацетилированных и неацетилированных побочных продуктов. Варьировали температуру (15°C, комнатная температура [20°C-23°C], 30°C), длительность кэппирования и ингредиенты кэппирующей композиции:
• без кэппирования,
• мягкие условия кэппирования: 10 мин кэппирования с 2% уксусным ангидридом и 1% DIPEA (диизопропилэтиламина),
• "нормальные" условия кэппирования: 20 мин кэппирования с 10% уксусным ангидридом и 5% DIPEA,
• 40 мин кэппирования с 10% уксусным ангидридом и 5% DIPEA.
Условия кэппирования по настоящему изобретению являются "мягкими условиями". Данные условия использовали в примере 5. Обнаружили, что данные условия являются предпочтительными.
В 9 положениях, выбранных в данном примере, получали ацетилированные последовательности с кэппированием (N-1) положения (фигура 7). Кроме того, на стадии кэппирования может происходить нежелательное удаление группы Fmoc в аминокислотном структурном блоке. Незащищенную аминогруппу можно ацетилировать кэппирующим реагентом или кэппирующей композицией. В таком случае улучшенные условия кэппирования могут позволить избежать нежелательного отщепления группы Fmoc.
6.1 Кэппирование в положении 36/35 после присоединения дипептидного структурного блока Pro-Pro (36-44)
Пептид Fmoc-(36-44)-AVE0010 получали путем твердофазного синтеза. Смолу сушили разделенной на 4 порции. Каждую порцию подвергали одной из четырех описанных выше процедур кэппирования при комнатной температуре (20°C - 23°C). Образцы высушивали и отщепляли пептид от смолы. Данную процедуру повторяли, при этом кэппирование проводили при 15°C или при 30°C.
Всего получали 12 образцов пептидов. 12 образцов пептидов анализировали с помощью LCMS. Показатели молекулярной массы определяли исходя из TIC (полного ионного тока). Определяли показатели молекулярной массы у следующих соединений:
Таблица 7
В таблице 8 показано содержание продуктов, полученных после присоединения Fmoc-ProPro и последующего кэппирования (% от общего содержания пептидов).
Результаты описаны на фигуре 8. Соединения (36-44), (38-44) и Ac(38-44) не были обнаружены или были обнаружены в небольших количествах. Во всех случаях количество нежелательного продукта Ac(36-44) возрастало с увеличением концентрации кэппирующей смеси и продолжительности кэппирования. Количество данного продукта увеличивалось с повышением температуры.
6.2 Кэппирование в положении 23 после присоединения структурного блока Ile, (23-44)
Синтез Fmoc(23-44) проводили так, как описано в разделе 6.1. Эксперименты при 15°C/30°C и при комнатной температуре проводили с разными партиями.
В таблице 9 показано содержание продуктов, полученных после присоединения Fmoc-Ile и последующего кэппирования (% от общего содержания пептидов).
Результаты описаны на фигуре 9. В зависимости от реагента для кэппирования при RT и 30°C увеличивалось содержание нежелательного соединения Ac(23-44). "Нормальное" кэппирование при 20°C давало в результате 0,26% Ac(23-44). Продление кэппирования (40 мин вместо 20 мин) оказывало отрицательное влияние на содержание Ac(23-44).
Образование целевого продукта Ac(24-44) не зависело от кэппирующей композиции.
6.3 Кэппирование в положении 21 после связывания структурного блока Leu (21-44)
Синтез Fmoc(21-44) проводили так, как описано в разделе 6.1. Эксперименты при 15°C/30°C и при комнатной температуре проводили с разными партиями.
В таблице 10 показано содержание продуктов, полученных после присоединения Fmoc-Leu и последующего кэппирования (% от общего содержания пептидов).
Результаты описаны на фигуре 10. Содержание нежелательного соединения Ac(21-44) было наибольшим при "40 мин, 10% Ac2O, 5% DIPEA" при 15°C, RT и 30°C. Содержание соединения Ac(21-44) возрастало с увеличением температуры.
Образование целевого соединения Ac(22-44) не зависело от кэппирующей композиции. Данное соединение образовывалось даже без кэппирования.
6.4 Кэппирование в положении 19 после связывания структурного блока Val (19-44)
Синтез Fmoc(19-44) проводили так, как описано в разделе 6.1. Эксперименты при 15°C/30°C и при комнатной температуре проводили с разными партиями.
В таблице 11 показано содержание продуктов, полученных после присоединения Fmoc-Val и последующего кэппирования (% от общего содержания пептидов).
Результаты описаны на фигуре 11. Содержание соединения Ac(19-44) было наибольшим при "40 мин, 10% Ac2O, 5% DIPEA" при 15°C, RT и 30°C. Содержание соединения Ac(19-44) возрастало с увеличением температуры.
Образование целевого соединения Ac(20-44) возрастало с увеличением концентрации кэппирующей композиции. Содержание нежелательного продукта (20-44) снижалось с увеличением концентрации кэппирующей композиции.
6.5 Кэппирование в положении 18 после присоединения структурного блока Ala (18-44)
Синтез Fmoc(18-44) проводили так, как описано в разделе 6.1. Эксперименты при 15°C/30°C и при комнатной температуре проводили с разными партиями.
В таблице 12 показано содержание продуктов, полученных после присоединения Fmoc-Ala и последующего кэппирования (% от общего содержания пептидов).
Результаты описаны на фигуре 12. Содержание нежелательного соединения Ac(18-44) было наибольшим при "40 мин, 10% Ac2O, 5% DIPEA" при 15°C и 30°C. Содержание соединения Ac(18-44) возрастало с увеличением температуры от 15°C до 30°C.
Образование целевого соединения Ac(19-44) возрастало при 15°C и 30°C с увеличением концентрации кэппирующей композиции.
6.6 Кэппирование в положении 15 после присоединения структурного блока Glu (15-44)
Синтез Fmoc(15-44) проводили так, как описано в разделе 6.1. Эксперименты при 15°C/30°C и при комнатной температуре проводили с разными партиями.
В таблице 13 показано содержание продуктов, полученных после присоединения Fmoc-Glu и последующего кэппирования (% от общего содержания пептидов).
Результаты описаны на фигуре 13. Содержание нежелательного соединения Ac(15-44) было наибольшим при "40 мин, 10% Ac2O, 5% DIPEA" при 15°C, RT и 30°C. Содержание соединения Ac(15-44) возрастало с увеличением температуры.
Образование целевого соединения Ac(16-44) не зависело от кэппирующей композиции. Данное соединение образовывалось даже без кэппирования.
6.7 Кэппирование в положении 12 после присоединения структурного блока Lys (12-44)
Синтез Fmoc(12-44) проводили так, как описано в разделе 6.1. Эксперименты при 15°C/30°C и при комнатной температуре проводили с разными партиями.
В таблице 14 показано содержание продуктов, полученных после присоединения Fmoc-Lys и последующего кэппирования (% от общего содержания пептидов).
Результаты описаны на фигуре 14. Содержание нежелательного соединения Ac(12-44) было наибольшим при "40 мин, 10% Ac2O, 5% DIPEA" при 15°C, RT и 30°C. Содержание соединения Ac(12-44) возрастало с увеличением температуры.
Образование целевого соединения Ac(13-44) не зависело от кэппирующей композиции. Данное соединение образовывалось даже без кэппирования.
6.8 Кэппирование в положении 8 после присоединения структурного блока Ser (8-44)
Синтез Fmoc(8-44) проводили так, как описано в разделе 6.1. Эксперименты при 15°C, RT и 30°C проводили с одной и той же партией.
В таблице 15 показано содержание продуктов, полученных после присоединения Fmoc-Ser связывания и последующего кэппирования (% от общего содержания пептидов).
Результаты описаны на фигуре 15. Содержание нежелательного соединения Ac(8-44) было наибольшим при "40 мин, 10% Ac2O, 5% DIPEA" при 15°C, RT и 30°C. Содержание соединения Ac(8-44) возрастало с увеличением температуры.
6.9 Кэппирование в положении 6 после присоединения структурного блока Phe (6-44)
Синтез Fmoc(86-44) проводили так, как описано в разделе 6.1. Эксперименты при 15°C, RT и 30°C проводили с одной и той же партией.
В таблице 16 показано содержание продуктов, полученных после присоединения Fmoc-Phe и последующего кэппирования (% от общего содержания пептидов).
Результаты описаны на фигуре 16. Содержание нежелательного соединения Ac(6-44) было наибольшим при "40 мин, 10% Ac2O, 5% DIPEA" при 15°C, RT и 30°C. Содержание соединения Ac(6-44) возрастало с увеличением температуры.
Образование целевого соединения Ac(7-44) не зависело от кэппирующей композиции. Данное соединение образовывалось даже без кэппирования.
Температура оказывала лишь незначительное влияние на образование целевого соединения Ac(7-44). Содержание нежелательного продукта (7-44) снижалось с увеличением концентрации кэппирующей композиции.
6.10 Выводы
Нежелательное образование соединения Ac(X-44) в значительной степени зависело от продолжительности кэппирования, кэппирующей композиции и температуры кэппирования. При увеличении продолжительности кэппирования, повышении температуры кэппирования и повышенном содержании уксусного ангидрида и DIPEA в кэппирующей композиции возрастало содержание нежелательного соединения Ac(X-44).
6.11 Кэппирование при "нормальных" условиях в зависимости от температуры
На фигурах 17 и 18 обобщены данные, полученные при кэппировании при различных температурах в 9 положениях в ходе синтеза ликсисенатида при "нормальных" условиях "20 мин, 10% Ac2O, 5% DIPEA", как описано в данном примере.
На фигуре 17 показаны результаты сравнения GMP-кэппирования Ac(X-44) в зависимости от температуры реакции. Значения, приведенные для условий 15°C и 30°C, являются положительными и отрицательными отклонениями от значений для условия "комнатная температура" (серая область).
Образование нежелательного продукта Ac(X-44) составило ≤0,5% в 5 из 9 положений, в 3 положениях от 0,5% до 1% и лишь в одном положении >1%. Значительное увеличение наблюдали при 30°C, тогда как при 15°C образование Ac(X-44) несколько уменьшалось.
Это означает, что GMP-кэппирование "20 мин, 10% Ac2O, 5% DIPEA" можно осуществлять в различных положениях в диапазоне от 15°C до комнатной температуры, которая может составлять 20-23°C.
На фигуре 18 показаны результаты сравнения GMP-кэппирования Ac[(X-1)-44] в зависимости от температуры реакции. Значения, приведенные для условий 15°C и 30°C, являются положительными и отрицательными отклонениями от значений для условия "комнатная температура" (серая область).
Что касается требуемого образования соединений Ac[(X-1)-44] при RT, то отклонение при 15°C составило от +0,23 до -0,25%. При 30°C отклонение составило от +0,29 до -0,33%. Таким образом, образование целевого продукта кэппирования Ac[(X-1)-44] в меньшей степени зависело от температуры, чем нежелательное образование Ac(X-44).
При 15°C и 30°C наблюдали отрицательные отклонения содержания целевого соединения Ac[(X-1)-44] по сравнению с кэппированием при комнатной температуре. Это означает, что кэппирование при "нормальных" условиях следует проводить при комнатной температуре.
6.12 Кэппирование с различными кэппирующими композициями при комнатной температуре.
На фигурах 19 и 20 обобщены данные, полученные при кэппировании с разными кэппирующими композициями при комнатной температуре в 9 положениях при синтезе ликсисенатида, как описано в данном примере.
На фигуре 19 показаны результаты сравнения содержания Ac(X-44) в зависимости от кэппирующей композиции при комнатной температуре. Значения, приведенные для условий "без кэппирования", "мягкие условия" и "40 мин", являются положительными и отрицательными отклонениями от значений при "нормальном кэппировании" (серая область).
Наибольшим образование нежелательного продукта Ac(X-44) было при условиях "20 мин, 10% Ac2O, 5% DIPEA" и "40 мин, 10% Ac2O, 5% DIPEA". Образование Ac(X-44) при "нормальных" условиях (40 мин, 10% Ac2O, 5% DIPEA) составило от 0,2% до 1,12%. Значительное снижение наблюдали при мягких условиях кэппирования.
На фигуре 20 показаны результаты сравнения содержания Ac[(X-1)-44] в зависимости от кэппирующей композиции при комнатной температуре. Значения, приведенные для "без кэппирования", "мягкие условия" и "40 мин", являются положительными и отрицательными отклонениями от значений "нормальное кэппирование" (серая область).
Образование целевого продукта Ac[(X-1)-44] при условиях "без кэппирования", "мягкие условия" и "40 мин" составляло в пределах от -0,14% до +0,16% в сравнении с "нормальными" условиями.
В частности, при "мягких" условиях (10 мин, 2% Ac2O, 1% DIPEA) по настоящему изобретению можно осуществить достаточное кэппирование при синтезе ликсисенатида.
Таким образом, мягкие условия кэппирования, в частности кэппирование в течение 10 мин с 2% Ac2O и 1% DIPEA в растворителе, являются предпочтительными в твердофазном синтезе ликсисенатида, который описан в данном документе.
Если кэппирование отсутствует после присоединения в определенных аминокислотных положениях, то в ходе очистки может быть затруднительным удаление нежелательных побочных продуктов, содержащих неполную аминокислотную последовательность и присутствующих в небольшом количестве.
Пример 7
Отщепление ликсисенатида от твердой фазы
Данный пример относится к отщеплению по настоящему изобретению ликсисенатида от твердой фазы. Брали твердую фазу (смолу Ринка), с которой был связан пептид ликсисенатид. Пептид синтезировали на смоле путем постадийного присоединения аминокислотных звеньев.
В качестве сравнительного теста проводили отщепление согласно известному из уровня техники способу (King et al., Int. J. Peptide Protein Res. 1990, 36: 255-266).
Способ отщепления по настоящему изобретению отличается от способа, известного из предшествующего уровня техники, следующими изменениями:
• температура реакции от 20°C до 26°C
• количество компонентов в отщепляющей смеси уменьшено с 5 до 2 компонентов в сочетании с увеличением соотношения смолы и используемой смеси для расщепления.
a) 0,5 г фенола
b) 0,5 мл тиоанизола
c) 0,25 мл 1‚2-этандитиола
d) 0,5 мл воды
e) 8,25 мл TFA
("смесь Кинга")
смоле"]:
a) 0,25 мл 1‚2-этандитиола
b) 8,25 мл TFA
Профильтрованную смолу добавляли к TFA (10 мл на г смолы), перемешивали в течение 1 ч и отфильтровывали смолу.
Профильтрованную смолу добавляли к TFA (10 мл на г смолы), перемешивали в течение 1 ч и отфильтровывали смолу.
Таблица 17: результаты сравнения способа отщепления по настоящему изобретению и способа отщепления, известного из предшествующего уровня техники. Отличия выделены жирным шрифтом/подчеркнуты.
За счет использования отщепления по настоящему изобретению по сравнению с отщеплением, известным из предшествующего уровня техники, стало возможным увеличение показателей выхода неочищенного ликсисенатида на примерно 5% (с 20% до 25%) с незначительным изменением профиля примесей.
Способ из данного примера является подходящим для увеличения масштаба до полупромышленного и промышленного.
В таблице 18 обобщены результаты, полученные в сравнительном способе (см. таблицу 17). Использовали три разные партии: 2E002, 2B008 и 2B006 ликсисенатида, связанного со смолой (1-44). Средние значения и стандартное отклонение рассчитывали отдельно для каждой партии. Сравнение между разными условиями отщепления должно проводиться в тестах с использованием той же партии ликсисенатида, связанного со смолой (1-44). В различных партиях на выход мог оказывать влияние твердофазный синтез. Если не указано иное, в качестве исходного материала использовали 10 г ликсисенатида, связанного со смолой (1-44).
Таблица 18: содержание ликсисенатида в смоле и выход ликсисенатида после отщепления ликсисенатида от смолы в стандартных условиях (сравнительный способ, см. таблицу 17).
7.1 Выход после отщепления в зависимости от температуры отщепления от 20°C до 35°C
Отщепление ликсисенатида от смолы (1-44) проводили в стандартных условиях (сравнительный способ, см. таблицу 17) в течение 4 ч.
[°C]
Таблица 19: выход после отщепления в зависимости от температуры
Результаты: выход ликсисенатида после отщепления в стандартных условиях повышался с увеличением температуры до оптимальной, составляющей приблизительно 26°C. Повышение температуры от 23°C до 26°C неожиданно приводило к значительному увеличению выхода.
7.2 Выход после отщепления в зависимости от продолжительности отщепления
Отщепление ликсисенатида от смолы (1-44) проводили в стандартных условиях (сравнительный способ, см. таблицу 17) при 20°C.
[°C]
Таблица 20: выход после отщепления в зависимости от продолжительности отщепления
Результаты: выход ликсисенатида возрастал с увеличением продолжительности отщепления. Максимальный выход достигался через приблизительно 8 ч отщепления.
7.3 Выход после отщепления в зависимости от температуры при продолжительности отщепления 12 ч
Отщепление ликсисенатида от смолы (1-44) проводили в стандартных условиях (сравнительный способ, см. таблицу 17) в течение 4 ч.
[°C]
Таблица 21: выход после отщепления в зависимости от температуры при продолжительности отщепления 12 ч
Результаты: выход увеличивался при продолжительности отщепления 12 ч, если повышали температуру реакции. Максимальный выход получали при 26°C, как описано в примере 7.1 для 4-часового отщепления. Тесты 70586-044 (4 ч, 26°C, пример 7) и 70586-045 (12 ч, 26°C) давали аналогичные показатели выхода (14,8% в сравнении с 14,0%).
7.4 Выход после отщепления в зависимости от температуры отщепления не более 20°C
Отщепление ликсисенатида от смолы (1-44) проводили в стандартных условиях (сравнительный способ, см. таблицу 17) в течение 4 ч.
[°C]
Таблица 22: выход после отщепления в зависимости от температуры отщепления не более 20°C
Результаты: в случае отщепления при температуре ниже 20°C требовалась большая продолжительность, как и ожидалось, для достижения выхода, получаемого при отщеплении при 20°C в течение 4 ч (стандартные условия, сравнительный способ, таблица 17).
7.5 Модифицированная смесь для отщепления
Стандартный способ предусматривает применение смеси для отщепления, содержащей пять компонентов: фенол, тиоанизол, 1,2-этандитиол, воду и TFA. Предметом данного примера были упрощенные смеси для отщепления с исключением от одного до трех из тиоанизола, фенола и воды. Определяли выход ликсисенатида после отщепления ликсисенатида от смолы (1-44). Смесь "без модификации" описана в таблице 17, "сравнительный способ".
Таблица 23: модифицированная смесь для отщепления
Результаты: отсутствие одного или нескольких компонентов приводило к увеличенному выходу за исключением теста 71002-010 (исключение тиоанизола).
Упрощенная отщепляющая смесь (смесь для отщепления) обладала рядом преимуществ:
(a) упрощение определения аналитических показателей и контроля качества,
(b) снижение затрат,
(c) упрощение манипуляций в производственном цикле.
7.6 Содержание TFA и 1,2-этандитиола в смеси для отщепления
Начиная с теста 71002-042, исследовали влияние соотношения TFA:1,2-этандитиол на выход после отщепления:
TFA и 1,2-этандитиола на г "пептида, связанного со смолой"
Таблица 24: разное соотношение TFA и 1,2-этандитиола в смеси для отщепления
Результаты: увеличение содержания 1,2-этандитиола приводило к значительному снижению выхода ликсисенатида. Было выявлено, что соотношение TFA:1,2-этандитиол 8,25:0,25 обеспечивает наибольший выход (партия 2E002). Этот результат был подтвержден в экспериментах с использованием партии 2B008.
7.7 Объем смеси для отщепления
Изучали влияние объема (и, следовательно, концентрации) смеси для отщепления.
Таблица 25: выход после отщепления в зависимости от объема смеси для отщепления
Результаты: уменьшение до 30% включительно не оказывало влияния на выход после отщепления. Более крупные уменьшения объема приводили к сниженному выходу.
7.8 Обеспечение набухания "пептида, связанного со смолой" посредством coрастворителя (толуола или CH2Cl2) перед отщеплением
Обоснованием для данного эксперимента было обнаружение того, что отщепление ликсисенатида от смолы может приводить к повышению температуры на 5-8°C включительно, что может приводить к образованию нежелательных побочных продуктов и потенциально может отрицательно влиять на стабильность и, следовательно, выход после отщепления. Обеспечение набухания "пептида, связанного со смолой" в органическом растворителе могло уменьшить экзотермический эффект, а значит могло увеличить выход.
Таблица 26: выход после отщепления в зависимости от присутствия сорастворителя. * Общий объем TFA и толуола/CH2Cl2 сохраняли постоянным.
Результаты: обеспечение набухания при помощи органического сорастворителя не повышало выход после отщепления.
7.9 Концентрация в присутствии сорастворителя
Присутствие сорастворителя с более высокой температурой кипения, чем у TFA, и в котором ликсисенатид не растворим, может увеличивать выход после отщепления от смолы, поскольку в ходе отгонки TFA из фильтрата присутствие сорастворителя может приводить к осаждению ликсисенатида, а значит может предотвращать деградацию ликсисенатида в ходе отщепления в смеси Кинга.
Таблица 27: выход после отщепления в зависимости от присутствия сорастворителя в ходе отгонки TFA.
Результаты: присутствие толуола при отгонке фильтрата после отщепления ликсисенатида от смолы давало незначительно повышенный выход.
7.10 Оптимизированная процедура отщепления по настоящему изобретению
Исходя из описанных выше результатов, полученных в данном примере, выбрали и протестировали следующие оптимизированные условия отщепления:
(a) температура реакции 26°C,
(b) смесь для отщепления из TFA и 1,2-этандитиола. Смесь содержала приблизительно 97% TFA и приблизительно 3% 1,2-этандитиола. Использовали следующие количества: 8,25 мл/г "пептида, связанного со смолой" в случае TFA и 0,25 мл/г "пептида, связанного со смолой" в случае 1,2-этандитиола.
Выход после отщепления у данной смеси по сравнению со стандартной сравнительной смесью тестировали в партиях 2E002 и 2B008.
Таблица 28: оптимизированная процедура отщепления по настоящему изобретению
Результаты: в обеих партиях выход увеличивался на приблизительно 5%, что свидетельствовало о значительном улучшении отщепления пептида от твердой фазы при использовании способа по настоящему изобретению.
7.11 Второе (последующее) отщепление
После отщепления с применением сравнительной смеси (смеси Кинга) или смеси для отщепления по настоящему изобретению проводили второе (последующее) отщепление (см. таблицу 17).
Проводили первое отщепление, получали фильтрат. К смеси TFA-влажная смола добавляли TFA. После 1 ч перемешивания фильтровали смолу. Фильтраты объединяли и концентрировали.
Исследовали влияние второго последующего отщепления на выход ликсисенатида.
Таблица 29: влияние второго (последующего) отщепления на выход ликсисенатида. EDT: 1,2-этандитиол.
Результаты: последующее отщепление приводило к повышению выхода лишь на приблизительно 0,7%. Данное повышение связано со значительным ростом затрат на исходные материалы (TFA) и дополнительные усилия по удалению TFA из пептидного продукта. Необходимо учитывать, что при объединении фильтратов из первой и второй стадии отщепления значительно увеличивалось количество TFA.
Авторы пришли к выводу, что ввиду небольшого повышения выхода исключение второго отщепления приводит к снижению затрат, а также упрощается манипуляция в ходе производственного процесса. Уменьшаются количества TFA, поэтому упрощается удаление TFA.
7.12 Аналитические показатели
Получали две партии: 71001-016 (сравнительная партия, отщепление с использованием смеси Кинга в соответствии со стандартным способом) и 71001-013 (отщепление ликсисенатида в соответствии с настоящим изобретением).
Таблица 30: аналитические показатели
Результаты: при анализе партий наблюдали практически идентичную чистоту. Содержание в полученной партии по настоящему изобретению было несколько сниженным. В партии по настоящему изобретению выход по весу увеличивался, что давало повышенный выход.
7.13 Выводы
Способ отщепления по настоящему изобретению обладает следующими преимуществами:
(a) увеличение выхода ликсисенатида на приблизительно 5%, что дает снижение затрат и повышение производительности,
(b) в смеси для отщепления присутствуют лишь два компонента (по сравнению с пятью компонентами в сравнительной смеси Кинга), поэтому улучшен аналитический контроль качества и снижены затраты,
(c) отсутствие второй стадии отщепления дает снижение затрат и облегчает выполнение технологических операций в ходе производственного процесса. Уменьшаются количества TFA, поэтому упрощается удаление TFA.
Предметом настоящего изобретения также являются следующие аспекты:
1. Способ синтеза полипептида, содержащего предварительно определенную аминокислотную последовательность, при этом способ включает циклы связывания аминокислотных структурных блоков с аминокислотной цепью,
где указанные аминокислотные структурные блоки содержат незащищенную C-концевую карбоксильную группу и защищенную N-концевую аминогруппу,
и где указанная аминокислотная цепь содержит незащищенную N-концевую аминогруппу,
при этом по меньшей мере один цикл связывания включает следующие стадии:
(a) связывание С-конца аминокислотного структурного блока с незащищенной N-концевой аминогруппой аминокислотной цепи, так чтобы между аминокислотной цепью и аминокислотным структурным блоком образовалась амидная связь,
(b) приведение в контакт продукта, полученного на стадии (а), с кэппирующим реагентом, содержащим кэппирующее соединение, при этом кэппирующее соединение связывается с незащищенной N-концевой аминогруппой аминокислотной цепи, с которой на стадии (а) не был связан ни один структурный блок, и
(с) удаление защитной группы с N-концевой аминогруппы аминокислотного структурного блока.
2. Способ по пункту 1, где кэппирующее соединение выбрано из группы, состоящей из уксусного ангидрида (CAS 108-24-7), гомологов уксусного ангидрида, бензоилхлорида (CAS 98-88-4), N-(бензилоксикарбонилокси)сукцинимида (CAS 13139-17-8), бензилхлорформиата (CAS 501-53-1), сложных эфиров хлормуравьиной кислоты, 1-ацетилимидазола (CAS 2466-76-4), ди-трет-бутилдикарбоната (CAS 24424-99-5) и N-(трет-бутоксикарбонилокси)сукцинимида (CAS 13139-12-3).
3. Способ по пункту 1 или 2, где кэппирующий реагент 0,5-5% об./об. уксусного ангидрида.
4. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит 1-3% об./об. уксусного ангидрида.
5. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит 2% об./об. уксусного ангидрида.
6. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит 0,2-2% об./об. диизопропилэтиламина.
7. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит 0,5-2% об./об. диизопропилэтиламина.
8. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит 1% об./об. диизопропилэтиламина.
9. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит 1% об./об. диизопропилэтиламина и 2% об./об. уксусного ангидрида.
10. Способ по любому из предыдущих пунктов, где реагент для кэппирования содержит DMF.
11. Способ по любому из предыдущих пунктов, где стадию (b) проводят при температуре 15-25°C.
12. Способ по любому из предыдущих пунктов, где стадию (b) проводят в течение 5-15 мин.
13. Способ по любому из предыдущих пунктов, где аминокислотный структурный блок содержит α-аминокислоту.
14. Способ по любому из предыдущих пунктов, где аминокислотный структурный блок выбран из Ser, Thr, Trp, Lys, Ala, Asn, Asp, Val, Met, Phe, Ile, Pro, Arg, Glu, Gln, Leu и Gly.
15. Способ по любому из предыдущих пунктов, где аминокислотный структурный блок выбран из Arg, Glu, Gln, Leu и Gly.
16. Способ по любому из предыдущих пунктов, где боковая цепь аминокислотного структурного блока содержит защитную группу, которая является ортогональной по отношению к N-концевой защитной группе аминокислотного структурного блока.
17. Способ по любому из предыдущих пунктов, включающий твердофазный синтез.
18. Способ по любому из предыдущих пунктов, где N-концевая аминогруппа в аминокислотном структурном блоке защищена лабильной по основанию защитной группой.
19. Способ по любому из предыдущих пунктов, где N-концевая аминогруппа в аминокислотном структурном блоке защищена посредством Fmoc.
20. Способ по любому из предыдущих пунктов, где полипептид представляет собой агонист GLP-1.
21. Способ по любому из предыдущих пунктов, где полипептид выбран из GLP-1, его аналогов и производных, эксендина-3, его аналогов и производных и эксендина-4, его аналогов и производных.
22. Способ по любому из пунктов 1-21, где полипептид выбран из эксендина-4 и ликсисенатида.
23. Способ по любому из пунктов 1-22, где полипептид представляет собой ликсисенатид.
24. Способ по любому из пунктов 1-21, где полипептид выбран из албиглутида, дулаглутида и семаглутида.
25. Способ по пункту 1, где полипептид представляет собой ликсисенатид или эксендин-4, при этом после связывания аминокислотного структурного блока Arg(20), Glu (17), Gln(13), Leu(10) или/и Gly(4) проводят стадию (b) в течение приблизительно 10 мин. с кэппирующим реагентом, содержащим 2% об./об. уксусного ангидрида и 1% об./об. диизопропилэтиламина.
26. Композиция, отличающаяся тем, что она содержит 0,5-5% об./об. уксусного ангидрида и 0,2-2% об./об. диизопропилэтиламина в DMF.
27. Композиция по пункту 26, содержащая 1% об./об. диизопропилэтиламина и 2% об./об. уксусного ангидрида.
28. Применение композиции по пункту 26 или 27 для ацетилирования незащищенной аминогруппы в ходе синтеза полипептида.
29. Применение композиции по пункту 28, где полипептид представляет собой ликсисенатид.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Sanofi-Aventis Deutschland GmbH
<120> Кэппирование незащищенных аминогрупп при синтезе пептидов
<130> 65946PEP
<160> 4
<170> PatentIn версия 3.5
<210> 1
<211> 44
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> ликсисенатид
<400> 1
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Asn Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Ser Lys Lys Lys Lys Lys Lys
35 40
<210> 2
<211> 39
<212> БЕЛОК
<213> Heloderma suspectum
<220>
<223> эксендин-4
<400> 2
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Asn Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Pro Ser
35
<210> 3
<211> 39
<212> БЕЛОК
<213> Heloderma horridum
<220>
<223> эксендин-3
<400> 3
His Ser Asp Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Asn Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Pro Ser
35
<210> 4
<211> 30
<212> БЕЛОК
<213> Человек
<220>
<223> GLP-1(7-36)
<400> 4
His Ala Glu Gly Thr Phe Thr Ser Asp Val Ser Ser Tyr Leu Glu Gly
1 5 10 15
Gln Ala Ala Lys Glu Phe Ile Ala Trp Leu Val Lys Gly Arg
20 25 30
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОТЩЕПЛЕНИЯ ПЕПТИДОВ, СВЯЗАННЫХ С ТВЕРДОЙ ФАЗОЙ, ОТ ТВЕРДОЙ ФАЗЫ | 2019 |
|
RU2771712C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТ ЭКСЕНДИН-ЛИНКЕР | 2011 |
|
RU2593774C2 |
| СТАБИЛИЗИРОВАННЫЕ СОЕДИНЕНИЯ ЭКСЕНДИНА-4 | 2003 |
|
RU2376314C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ИНСУЛИНА С ЧРЕЗВЫЧАЙНО ЗАМЕДЛЕННЫМ ПРОФИЛЕМ ВРЕМЯ/ДЕЙСТВИЕ | 2009 |
|
RU2524423C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ИНСУЛИНА С СИЛЬНО ЗАМЕДЛЕННЫМ ПРОФИЛЕМ ВРЕМЯ/ДЕЙСТВИЕ | 2009 |
|
RU2529952C2 |
| ПРЕПАРАТ ИНСУЛИНА, СОДЕРЖАЩИЙ МЕТИОНИН | 2010 |
|
RU2540485C2 |
| УСТРОЙСТВО ДОСТАВКИ ЛЕКАРСТВЕННОГО ВЕЩЕСТВА И ДЕРЖАТЕЛЬ КАРТРИДЖА ДЛЯ УСТРОЙСТВА ДОСТАВКИ ЛЕКАРСТВЕННОГО ВЕЩЕСТВА | 2012 |
|
RU2605422C2 |
| МЕДИЦИНСКОЕ УСТРОЙСТВО, СОДЕРЖАЩЕЕ ОСВЕТИТЕЛЬНОЕ ПРИСПОСОБЛЕНИЕ | 2012 |
|
RU2627635C2 |
| ПРИВОДНОЙ МЕХАНИЗМ ДЛЯ УСТРОЙСТВА ДОСТАВКИ ЛЕКАРСТВЕННОГО ВЕЩЕСТВА И УСТРОЙСТВО ДОСТАВКИ ЛЕКАРСТВЕННОГО ВЕЩЕСТВА | 2011 |
|
RU2577243C2 |
| ПРИВОДНОЙ МЕХАНИЗМ ДЛЯ УСТРОЙСТВА ДОСТАВКИ ЛЕКАРСТВЕННОГО ВЕЩЕСТВА И УСТРОЙСТВО ДОСТАВКИ ЛЕКАРСТВЕННОГО ВЕЩЕСТВА | 2011 |
|
RU2577451C2 |
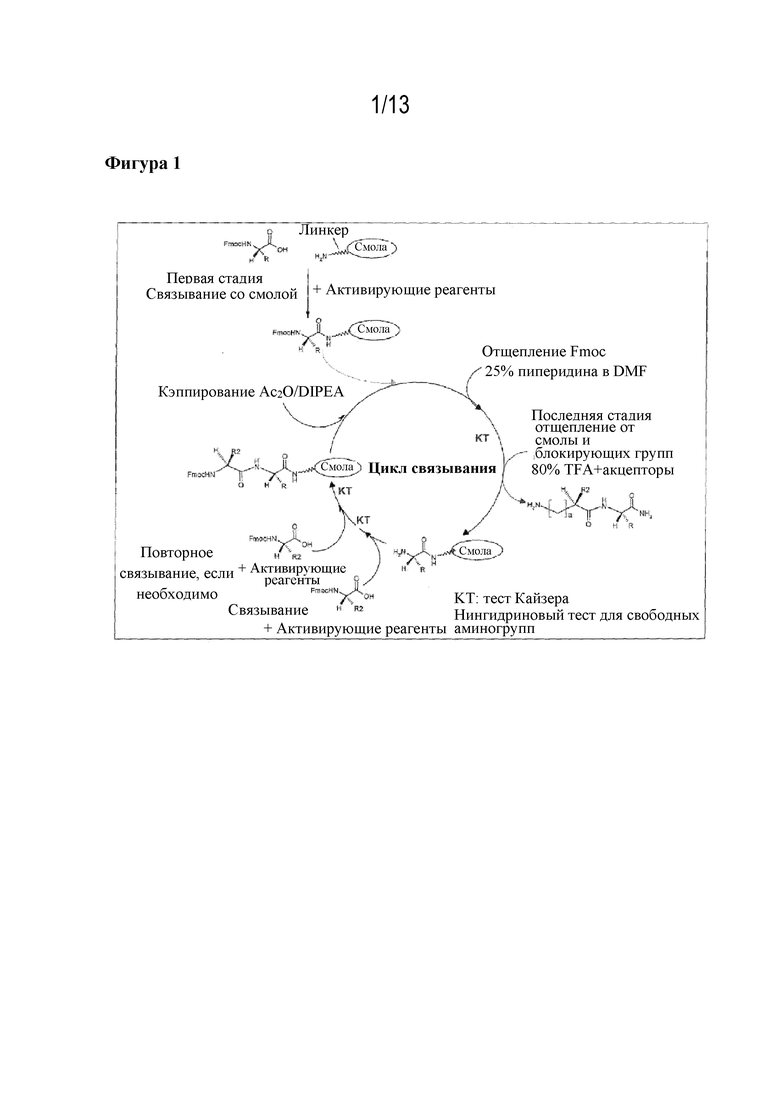


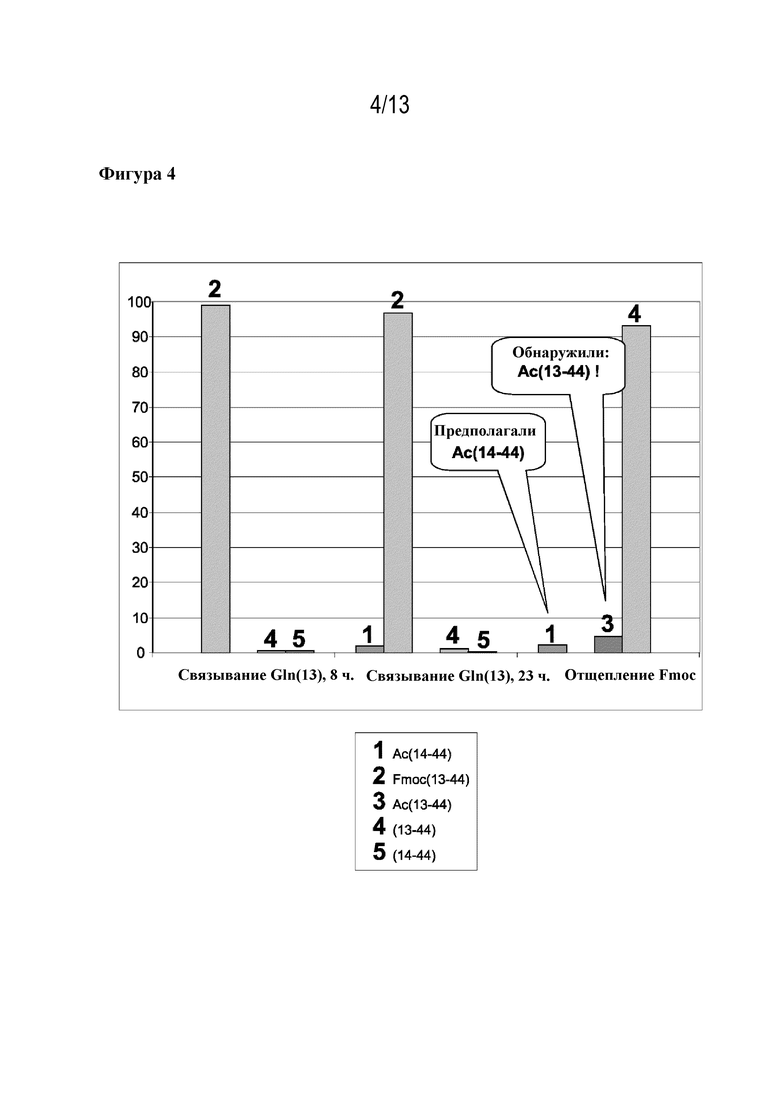
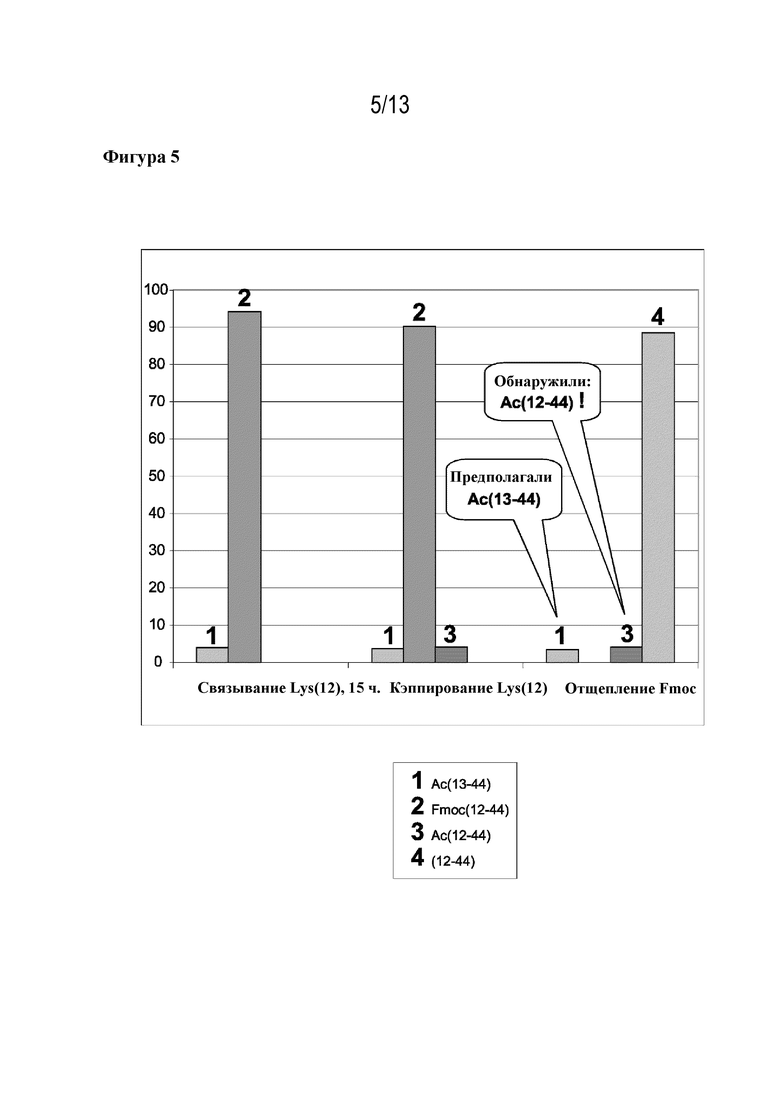
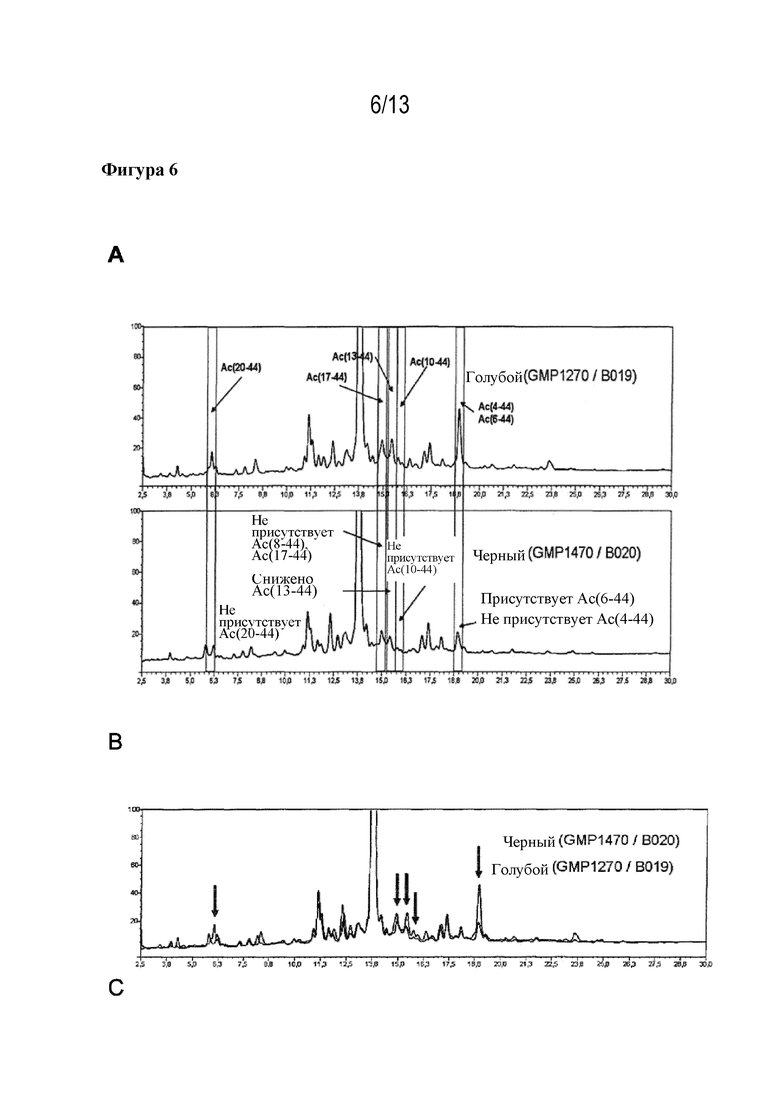
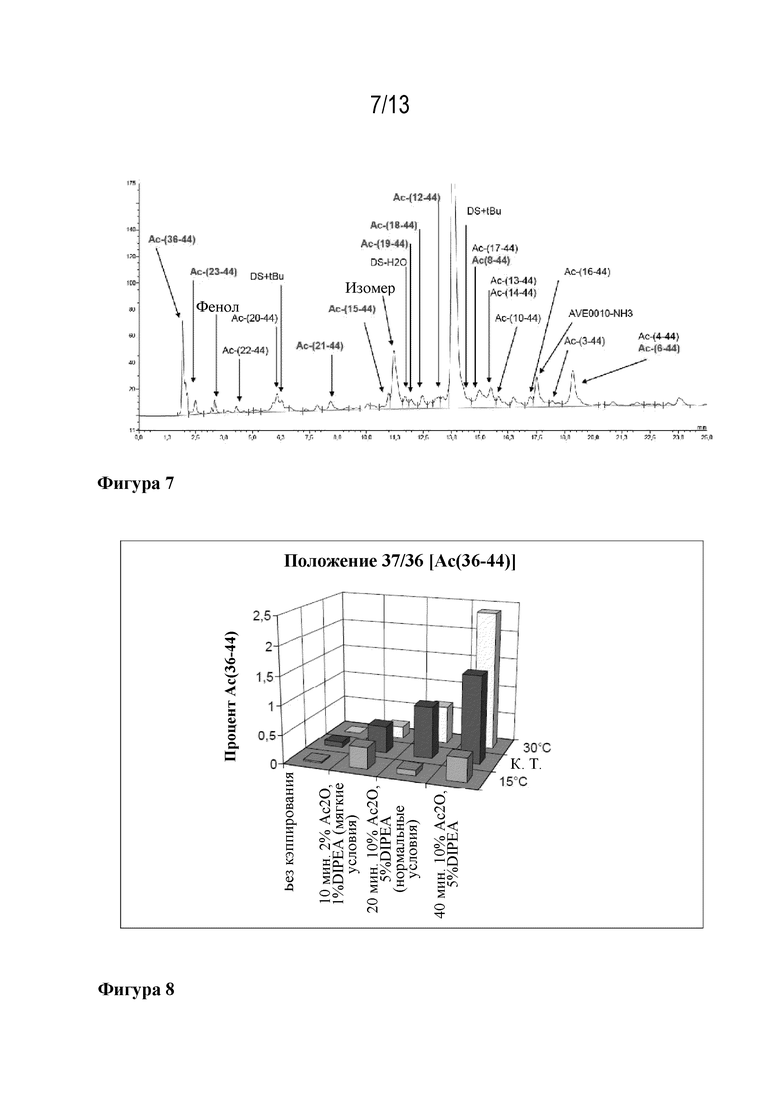
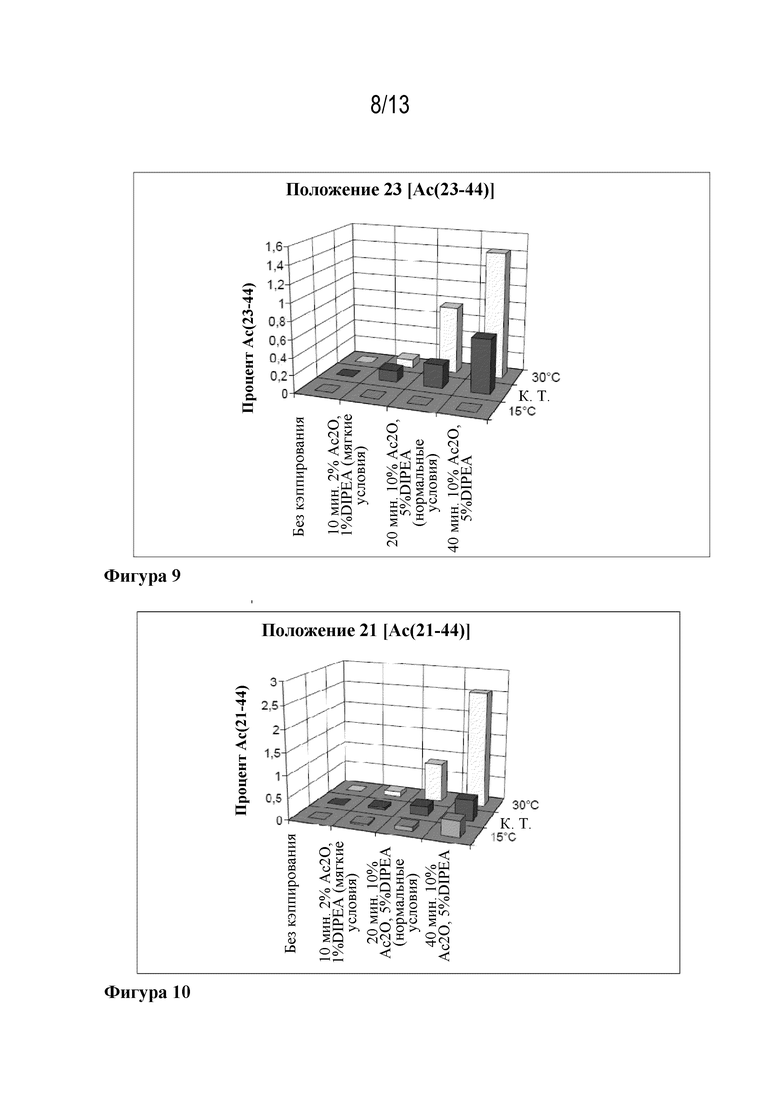
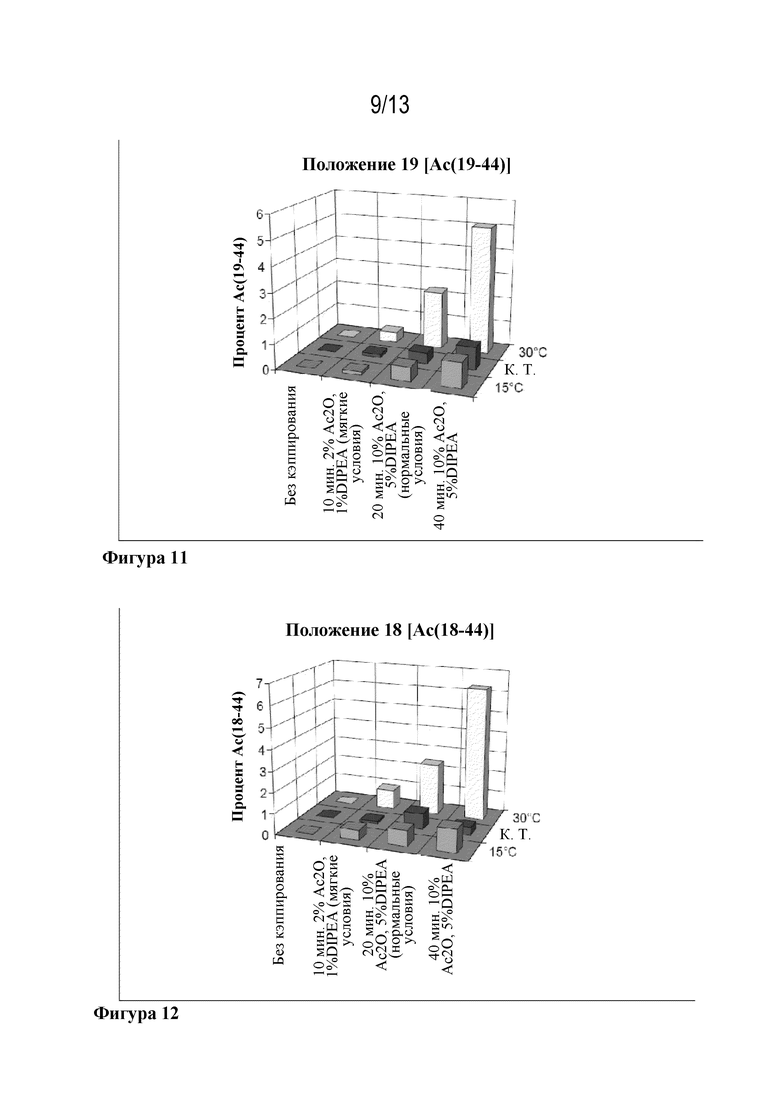
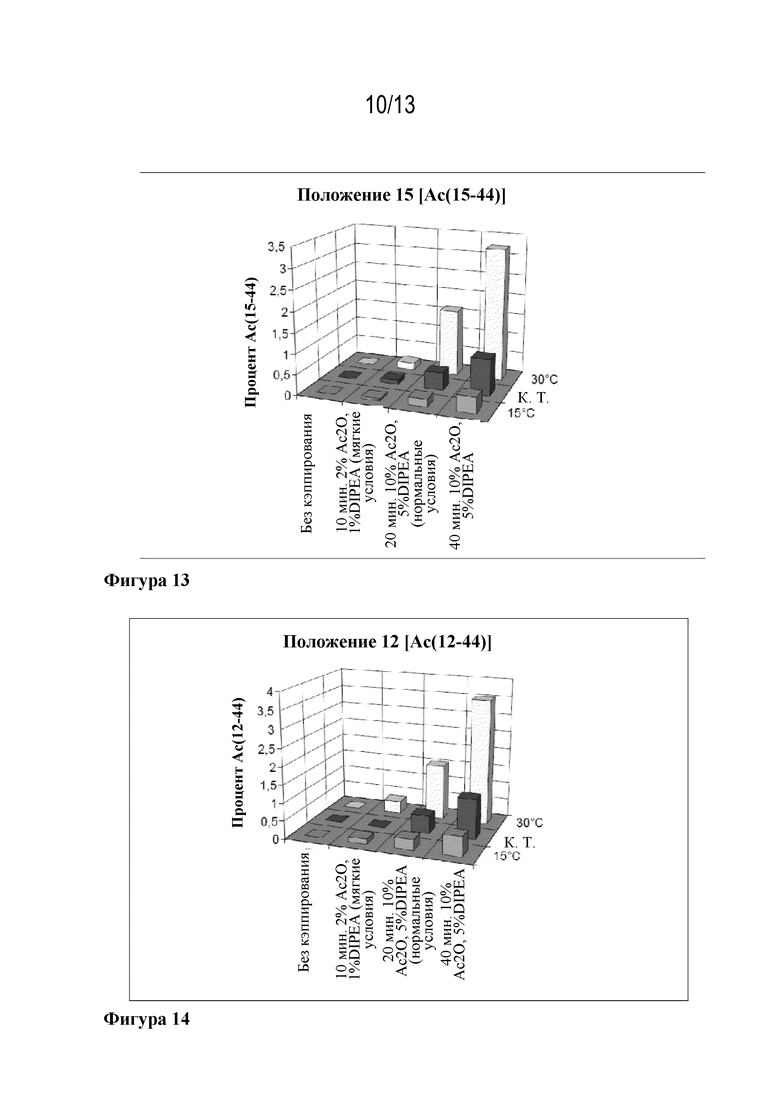
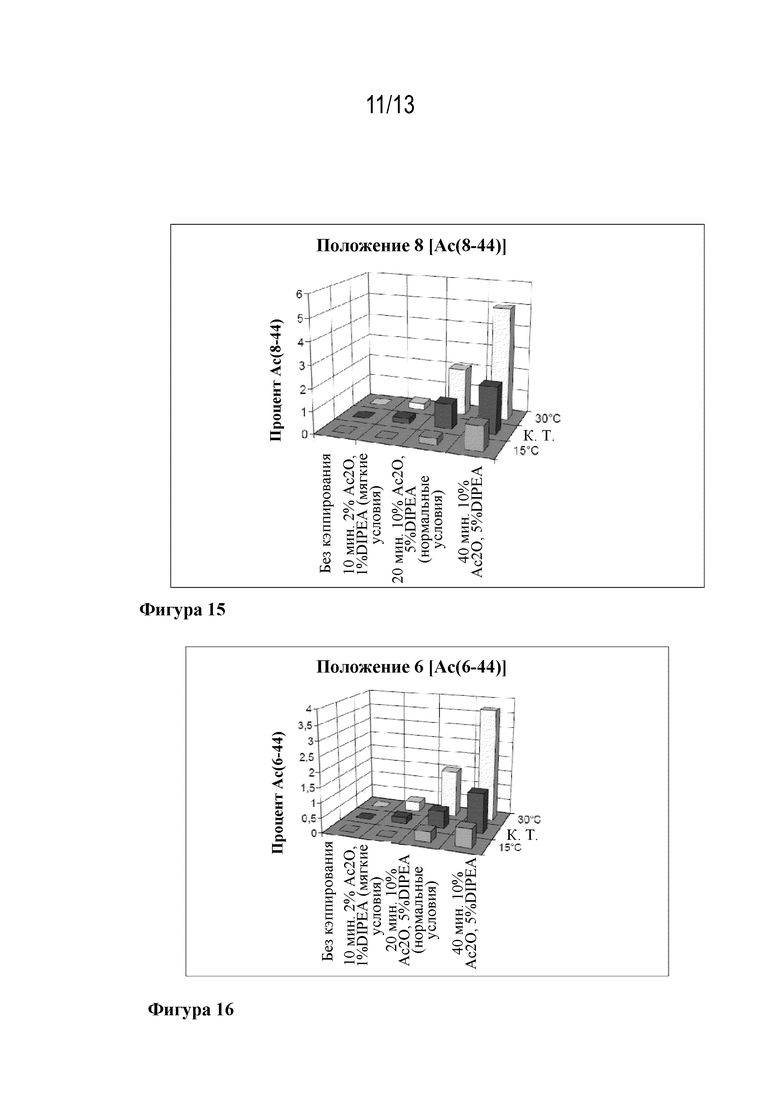
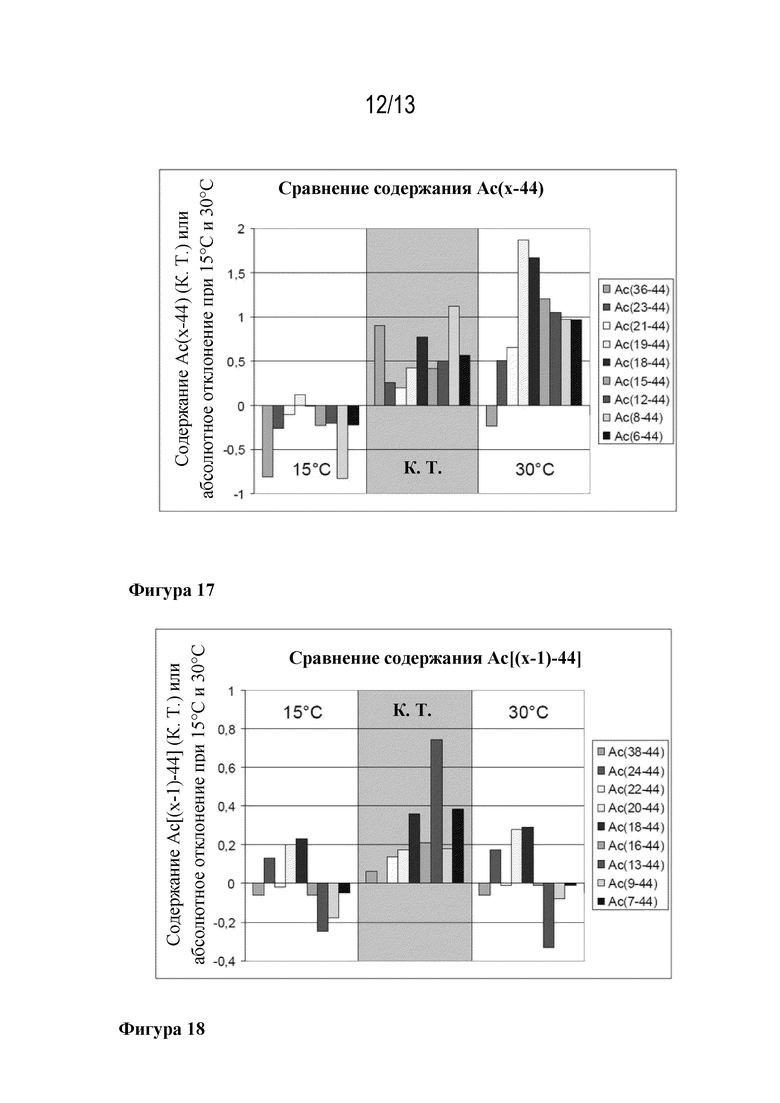
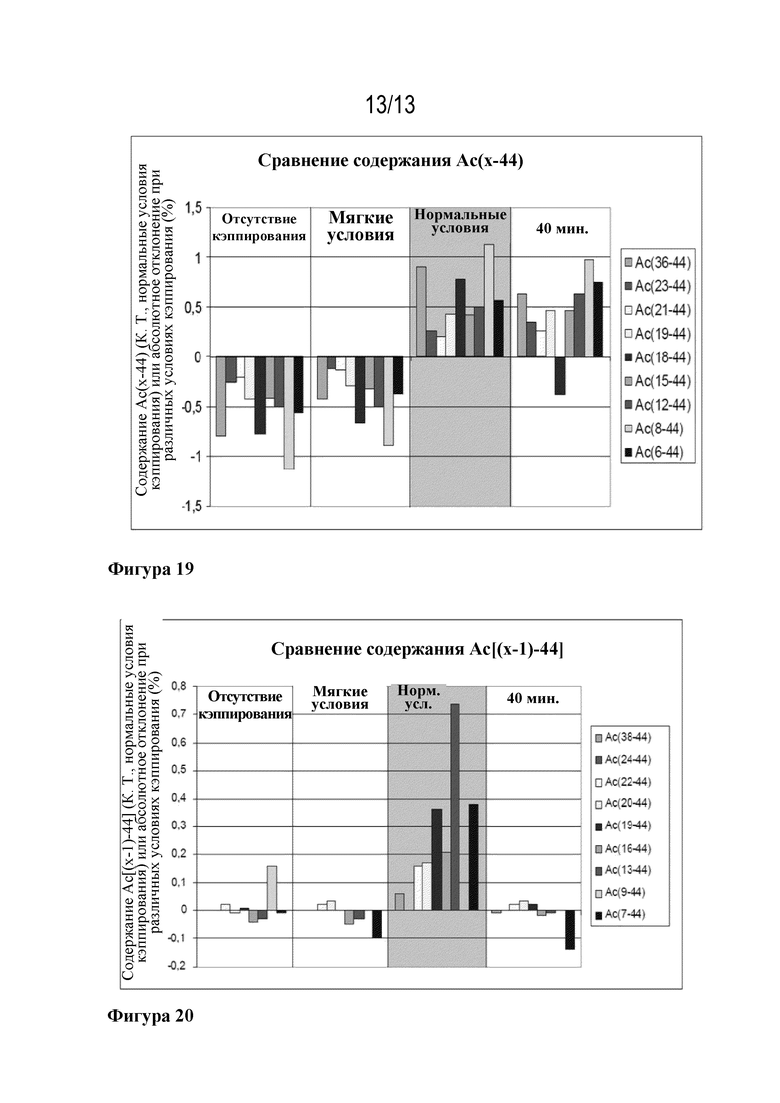
Изобретение относится к способу твердофазного синтеза полипептида, содержащего предварительно определенную аминокислотную последовательность, при этом способ включает циклы связывания аминокислотных структурных блоков с аминокислотной цепью, где указанные аминокислотные структурные блоки содержат незащищенную C-концевую карбоксильную группу и защищенную N-концевую аминогруппу, и где указанная аминокислотная цепь содержит незащищенную N-концевую аминогруппу, при этом по меньшей мере один цикл связывания включает следующие стадии: (a) связывание С-конца аминокислотного структурного блока с незащищенной N-концевой аминогруппой аминокислотной цепи, так чтобы между аминокислотной цепью и аминокислотным структурным блоком образовалась амидная связь, (b) приведение продукта, полученного на стадии (а), в контакт с кэппирующим реагентом, содержащим кэппирующее соединение, при этом кэппирующее соединение связывается с незащищенной N-концевой аминогруппой аминокислотной цепи, с которой на стадии (а) не был связан ни один структурный блок, и (c) удаление защитной группы с N-концевой аминогруппы аминокислотного структурного блока, где кэппирующий реагент содержит 0,5-5% об./об. уксусного ангидрида и 0,2-2% об./об. диизопропилэтиламина, где полипептид представляет собой агонист GLP-1, выбранный из GLP-1, эксендина-3, эксендина-4 и аналогов эксендина-4. Способ позволяет уменьшить количества ацетилированных ошибочных последовательностей, встречающихся при пептидном синтезе, и увеличить выход продукта пептидного синтеза. 2 н. и 12 з.п. ф-лы, 20 ил., 30 табл., 7 пр.
1. Способ твердофазного синтеза полипептида, содержащего предварительно определенную аминокислотную последовательность, при этом способ включает циклы связывания аминокислотных структурных блоков с аминокислотной цепью,
где указанные аминокислотные структурные блоки содержат незащищенную C-концевую карбоксильную группу и защищенную N-концевую аминогруппу,
и где указанная аминокислотная цепь содержит незащищенную N-концевую аминогруппу,
при этом по меньшей мере один цикл связывания включает следующие стадии:
(a) связывание С-конца аминокислотного структурного блока с незащищенной N-концевой аминогруппой аминокислотной цепи, так чтобы между аминокислотной цепью и аминокислотным структурным блоком образовалась амидная связь,
(b) приведение продукта, полученного на стадии (а), в контакт с кэппирующим реагентом, содержащим кэппирующее соединение, при этом кэппирующее соединение связывается с незащищенной N-концевой аминогруппой аминокислотной цепи, с которой на стадии (а) не был связан ни один структурный блок, и
(c) удаление защитной группы с N-концевой аминогруппы аминокислотного структурного блока,
где кэппирующий реагент содержит 0,5-5% об./об. уксусного ангидрида и 0,2-2% об./об. диизопропилэтиламина,
где полипептид представляет собой агонист GLP-1, выбранный из GLP-1, эксендина-3, эксендина-4 и аналогов эксендина-4, выбранных из группы, состоящей из
H-дезPro36-эксендин-4-Lys6-NH2,
H-дез(Pro36,37)-эксендин-4-Lys4-NH2,
H-дез(Pro36,37)-эксендин-4-Lys5-NH2,
дезPro36[Asp28]эксендин-4 (1-39),
дезPro36[IsoAsp28]эксендин-4 (1-39),
дезPro36[Met(O)14,Asp28]эксендин-4 (1-39),
дезPro36[Met(O)14,IsoAsp28]эксендин-4 (1-39),
дезPro36[Trp(O2)25,Asp28]эксендин-4 (1-39),
дезPro36[Trp(O2)25,IsoAsp28]эксендин-4 (1-39),
дезPro36[Met(O)14Trp(O2)25,Asp28]эксендин-4 (1-39),
дезPro36[Met(O)14Trp(O2)25,IsoAsp28]эксендин-4(1-39)
дезPro36[Asp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[IsoAsp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Met(O)14,Asp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Met(O)14,IsoAsp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Trp(O2)25,Asp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Trp(O2)25,IsoAsp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Met(O)14Trp(O2)25,Asp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Met(O)14Trp(O2)25,IsoAsp28]эксендин-4(1-39)-Lys6-NH2,
H-(Lys)6-дезPro36[Asp28]эксендин-4(1-39)-Lys6-NH2,
дезAsp28Pro36,Pro37,Pro38эксендин-4(1-39)-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-NH2,
дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36[Trp(O2)25,Asp28]эксендин-4(1-39)-Lys6-NH2,
H-дезAsp28 Pro36,Pro37,Pro38[Trp(O2)25]эксендин-4(1-39)-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-NH2,
дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36[Met(O)14,Asp28]эксендин-4(1-39)-Lys6-NH2,
дезMet(O)14 Asp28 Pro36, Pro37, Pro38эксендин-4(1-39)-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-NH2,
дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-Lys6-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-Lys6-NH2,
дезAsp28Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25]эксендин-4(1-39)-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-NH2,
дезPro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2, и их фармацевтически приемлемых солей.
2. Способ по п. 1 или 2, где кэппирующий реагент содержит 1-3% об./об. уксусного ангидрида.
3. Способ по п. 1 или 2, где кэппирующий реагент содержит 2% об./об. уксусного ангидрида.
4. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит 0,5-2% об./об. диизопропилэтиламина.
5. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит 1% об./об. диизопропилэтиламина.
6. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит 1% об./об. диизопропилэтиламина и 2% об./об. уксусного ангидрида.
7. Способ по любому из предыдущих пунктов, где кэппирующий реагент содержит DMF.
8. Способ по любому из предыдущих пунктов, где стадию (b) проводят при температуре 15-25°C.
9. Способ по любому из предыдущих пунктов, где стадию (b) проводят в течение 5-15 мин.
10. Способ по любому из предыдущих пунктов, где аминокислотный структурный блок содержит α-аминокислоту.
11. Способ по любому из предыдущих пунктов, где аминокислотный структурный блок выбран из Arg, Glu, Gln, Leu и Gly.
12. Способ по любому из предыдущих пунктов, где полипептид выбран из эксендина-4, ликсисенатида, албиглутида, дулаглутида и семаглутида.
13. Применение композиции для ацетилирования незащищенной аминогруппы в ходе твердофазного синтеза полипептида, при этом указанная композиция содержит 0,5-5% об./об. уксусного ангидрида и 0,2-2% об./об. диизопропилэтиламина в DMF, где полипептид представляет собой агонист, выбранный из GLP-1, эксендина-3, эксендина-4 и аналогов эксендина-4, выбранных из группы, состоящей из
H-дезPro36-эксендин-4-Lys6-NH2,
H-дез(Pro36,37)-эксендин-4-Lys4-NH2,
H-дез(Pro36,37)-эксендин-4-Lys5-NH2,
дезPro36[Asp28]эксендин-4 (1-39),
дезPro36[IsoAsp28]эксендин-4 (1-39),
дезPro36[Met(O)14,Asp28]эксендин-4 (1-39),
дезPro36[Met(O)14,IsoAsp28]эксендин-4 (1-39),
дезPro36[Trp(O2)25,Asp28]эксендин-4 (1-39),
дезPro36[Trp(O2)25,IsoAsp28]эксендин-4 (1-39),
дезPro36[Met(O)14Trp(O2)25,Asp28]эксендин-4 (1-39),
дезPro36[Met(O)14Trp(O2)25,IsoAsp28]эксендин-4(1-39)
дезPro36[Asp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[IsoAsp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Met(O)14,Asp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Met(O)14,IsoAsp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Trp(O2)25,Asp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Trp(O2)25,IsoAsp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Met(O)14Trp(O2)25,Asp28]эксендин-4 (1-39)-Lys6-NH2,
дезPro36[Met(O)14Trp(O2)25,IsoAsp28]эксендин-4(1-39)-Lys6-NH2,
H-(Lys)6-дезPro36[Asp28]эксендин-4(1-39)-Lys6-NH2,
дезAsp28Pro36,Pro37,Pro38эксендин-4(1-39)-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-NH2,
дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36[Trp(O2)25,Asp28]эксендин-4(1-39)-Lys6-NH2,
H-дезAsp28 Pro36,Pro37,Pro38[Trp(O2)25]эксендин-4(1-39)-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-NH2,
дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36[Met(O)14,Asp28]эксендин-4(1-39)-Lys6-NH2,
дезMet(O)14 Asp28 Pro36, Pro37, Pro38эксендин-4(1-39)-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-NH2,
дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-Lys6-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-Lys6-NH2,
дезAsp28Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25]эксендин-4(1-39)-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Met(O)14,Asp28]эксендин-4(1-39)-NH2,
дезPro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-дезPro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-дезPro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]эксендин-4(1-39)-(Lys)6-NH2, и их фармацевтически приемлемых солей.
14. Применение по п. 13, где композиция содержит 1% об./об. диизопропилэтиламина и 2% об./об. уксусного ангидрида в DMF.
| US 20130289241 A1, 31.10.2013 | |||
| WO 2017162650 A1, 28.09.2017 | |||
| WO 2014072916 A1, 15.05.2014 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА ЭКСЕНАТИДА | 2011 |
|
RU2458066C1 |
| US 7431685 B2, 07.10.2008 | |||
| Установка для непрерывного стыкования,анкеровки и резки арматурных стержней | 1983 |
|
SU1107969A2 |

