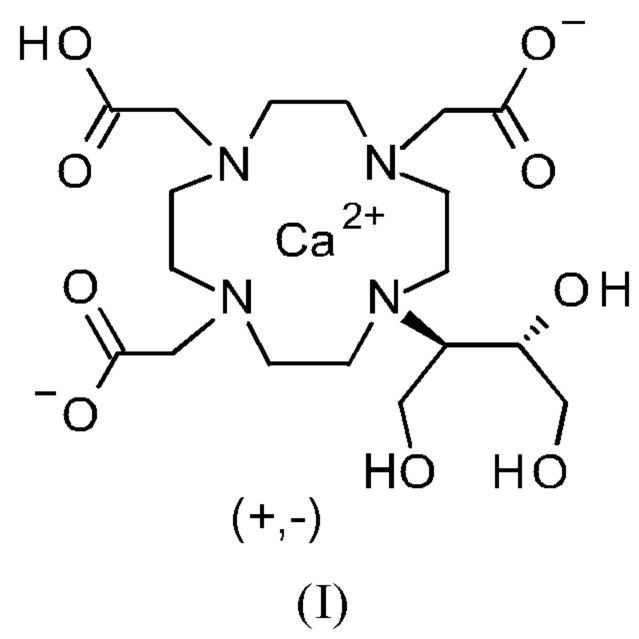
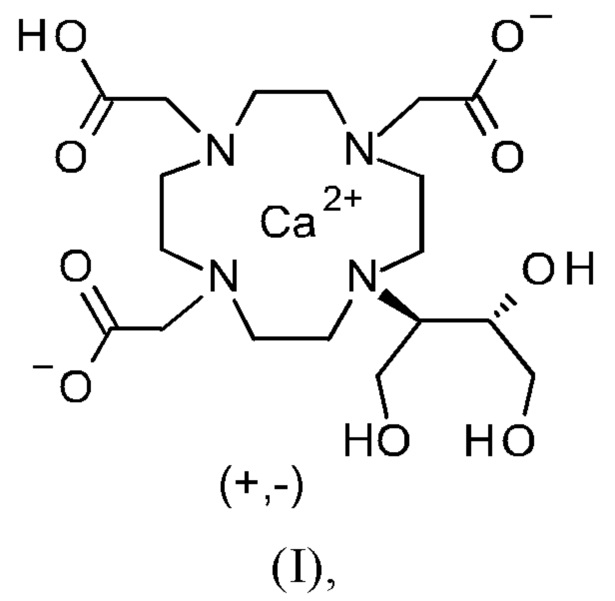
Изобретение относится к способу получения кристаллической формы модификации А кальциевого комплекса дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты (калкобутрол) формулы (I)
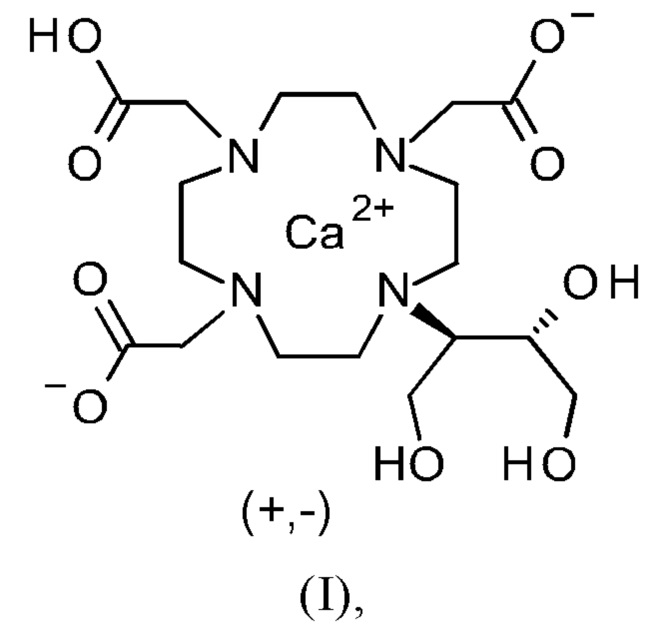
и применению кристаллической модификации А кальциевого комплекса дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты (калкобутрол) формулы (I) для получения галенов препаратов гадобутрола.
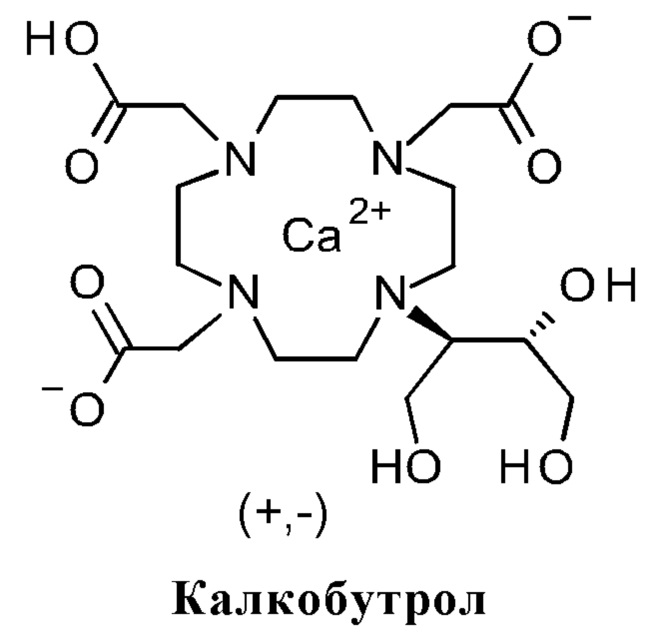
Калкобутрол представляет собой добавку в галенов препаратах гадобутрола и имеет задачу предотвращения высвобождения гадолиния в препарате (растворы). Производство калкобутрола высокой чистоты описано в WO 2011/054827 A1 (Bayer AG) и в WO 2016/043462 А2 (ST PHARM CO., LTD.):
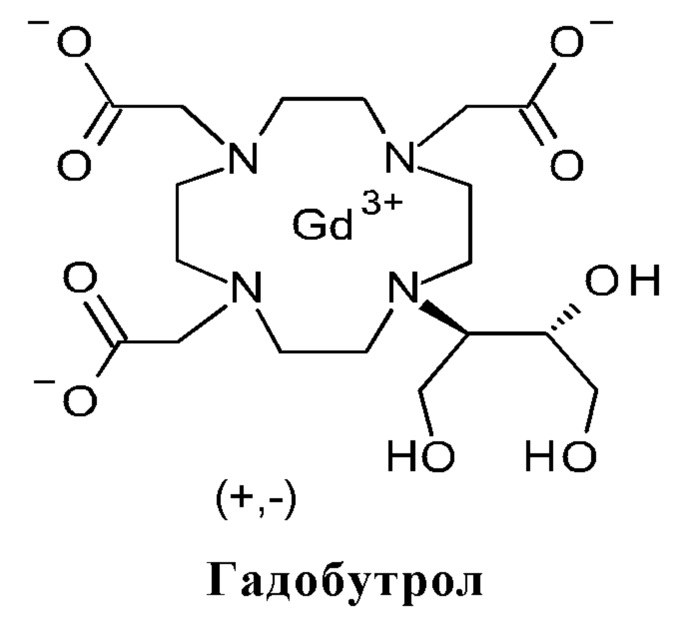
Гадобутрол представляет собой гадолиний-содержащее контрастное средство для ядерной спиновой томографии и с 2000 был одобрен в Германии как Gadovist® в обозначении "Contrast enhancement in cranial and spinal magnetic resonance tomography (MRT)" (EP 0448191 B1, EP 0643705 B1, EP 0986548 B1, EP 0596586 B1 и Патент CA 1341176). Производство гадобутрола высокой чистоты описано в патентной заявке WO 2012/143355 А1. Он представляет собой неионный комплекс, включающий гадолиний(III) и макроциклический лиганд дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты (бутрол).
Гадовист продают в виде 1 молярного водного раствора, который состоит из следующих компонентов в препарате: гадобутрол, натриевая соль калкобутрола, трометамол, соляная кислота и вода для инъекций.
С наиболее гадолиний-содержащим контрастным средством, было найдено выгодным применять избыток гадолиниевого комплексообразующего лиганда в форме кальциевого комплекса в препарате, ЕР 0 270 483 В2. По сути это определяет задачу предотвращения высвобождения гадолиния в препарате (например, при многолетнем хранении или перекомплексообразовании с инородными ионами из стекла).
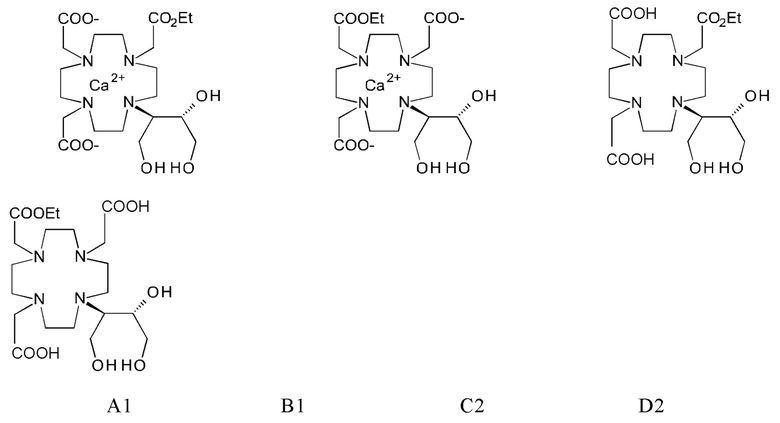
Синтезы кальциевого комплекса (калкобутрола) описаны в Inorg. Chem 1997, 36, 6086-6093. Для этого, проводят комплексообразование бутролового лиганда в воде с карбонатом кальция, водный раствор затем высушивают сублимацией и оставшийся порошок упаривают в виде суспензии в 26-кратном этаноле, это не способ кристаллизации, но скорее горячее экстракционное перемешивание суспензии. Тем не менее, описанный способ там не приводит к высокой чистоте требуемой ведомствами, во время упаривания в чистом этаноле имеет место возникновение новых примесей до значительной наблюдаемой степени, с которыми также будут, помимо других примесей, также два этиловых сложных эфира А1 и В1, и сложные эфиры лиганда С2 и D2:
Может быть получен только материал с чистотой приблизительно 94% (100% способ, ВЭЖХ), так как эти два этиловых сложных эфира также кристаллизуются в чистом этаноле. У лиганда, полученного из синтеза гадобутрола (бутрол), нет необходимой высокой чистоты для него, чтобы быть непосредственно преобразованным в кальциевый комплекс. Из-за его высоко цвитерионной природы, дальнейшая очистка лиганда трудная и дорогостоящая.
Разложение гадолиниевых комплексов щавелевой кислотой с добавлением минеральной кислоты (главным образом соляная кислота) описан в литературе, смотрите, например, Inorganica Chimica Acta 249 (1996), 191-199. Здесь производство лиганда высокой чистоты обрисовано в общих чертах, где продукт, наконец, перемешивается в метаноле при комнатной температуре, чтобы, таким образом, создать лиганд высокой чистоты для определения константы устойчивости. Однако способ, описанный там, не подходит для увеличения масштаба, а также не раскрывает производство, выделение и очистку калкобутрола. Таким образом, в US PS 5595714 раскрыто, что, с одной стороны, гадолиний, а также свободные лиганды, могут быть восстановлены из гадолиний-содержащего контрастного средства путем разложения комплекса с щавелевой кислотой/соляной кислотой. Тем не менее, использование способа получения кальциевых солей не упомянуто в рамках этого документа.
В то время как нейтральный гадолиниевый комплекс (гадобутрол) может быть очищен на ионообменных смолах, и в заключении может быть получен с высокой чистотой (>>99%) очень эффективной кристаллизацией, это не возможно с калкобутролом из-за дополнительной кислотной функции. Очистка комплекса не имела успеха, поскольку, даже препаративной ВЭЖХ, примеси, очень близко подходящие к главному пику, не могли быть удалены.
Цель данного изобретения состоит в том, чтобы воспроизводимо получить очень чистый калкобутрол со стабильной, определяемой полиморфной формой. Трудность во всех способах очистки по существу состоит, с одной стороны, в воспроизводимом получении высокой чистоты, а также стехиометрии 1:1 Са:лиганд. Калкобутрол стабилен только при нейтральных условиях и во время любой операции по очистке, либо это хроматография или обработка ионообменной смолой, всегда теряет значительные пропорции кальция через разложение комплекса.
С данным изобретением был найден очень эффективный способ, который позволяет выполнить вышеупомянутые требования.
В описании патента WO 2011/054827 A1 (Bayer AG), неожиданно было найдено, что эффективное производство возможно, начиная с гадобутрола высокой чистоты, как например, описано в WO 2012/143355 А1. Гадолиний удаляют из комплекса гадобутрола разложением комплекса, таким образом, получают лиганд очень высокой чистоты, а затем связывают в комплекс с ионами кальция 2+. В WO 2011/054827 A1 (Bayer AG) кристаллизация из водного этанола описана как пример, где кристаллизация была выполнена из водного этанола, что дает на выходе очень чистый калкобутрол.
Сейчас неожиданно было найдено, что водный эквивалент этанольного раствора в диапазоне должен находиться ≥9% и ≤11%, чтобы воспроизводимо получить одну определенную модификацию (целевая модификация А). Это отличается от способов, опубликованных прежде (Inorganic Chemistry 1997, 36, 6086-6093 и WO 2011/054827 A1), в которых не были предприняты специальные попытки, чтобы регулировать содержание воды во время процесса кристаллизации. Перегонка сырья до неопределенного целевого количества применяемых бутроловых лигандов (как описано в предшествующем уровне техники) представляет собой, в особенности, только грубое ориентировочное значение для дополнительного улучшения устойчивости к изменениям способа, и не может, вне всяких сомнений, соперничать с надежным технологически контролем для воды; таким образом, эта величина может относительно просто быть приведена в желаемый диапазон 9-11% путем дополнительного добавления этанола или воды. Более того, было преимущественно найдено, что если после комплексообразования с карбонатом кальция была обеспечена стехиометрия 1:1 в Са:бутрол, ее снова проверяют с помощью технологического контроля, и необязательно дополнительно откорректируют путем добавления малых количеств карбоната кальция или бутрола, таким образом, чтобы точно получить стехиометрию 1:1. Неожиданно, здесь было замечено, что даже наименьшие отклонения от этой стехиометрии влияют на чистоту и полученную полиморфную форму. Воспроизводимое получение имеет успех только с новым изобретательным способом. Это, с одной стороны, важно, поскольку, среди найденных четырех полиморфных форм (модификаций) А, В, С и D, только форма А (модификация А) имеет хорошую стабильность при хранении, в то время как В, С и D в большой степени имеют тенденцию быть гигроскопическими, что вызывает значительные проблемы в производстве фармацевтического препарата (Gadovist®). Сильная гигроскопичность всегда является проблемой в фармацевтике во время хранения и взвешивании оптовых количеств.
Упомянутые четыре полиморфные формы представляют собой полиморфные модификации А, В, С и D. Они проявляют свойства, показанные в Таблице 1:
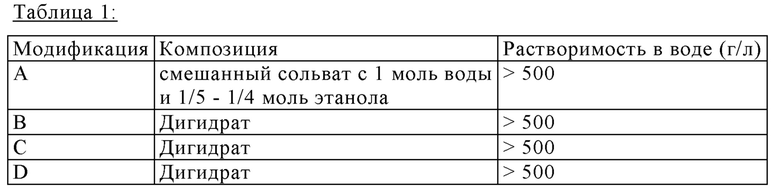
На хранении при условиях высокой атмосферной влажности все четыре модификации преобразовываются в аморфное вещество. Модификации В, С и D могут, например, быть получены кристаллизацией из воды с увеличенными водными эквивалентами >12%.
Посредством нового процесса стало возможно решить проблему гигроскопичности, в которой кристаллизацию осуществляют из водного этанола, в то же время поддерживая водный эквивалент между 9% и 11%. Неожиданно, было найдено, что, работая в водных эквивалентах 9-11% содержание этиловых сложных эфиров A1, B1, C2 и D2, описанных выше, уменьшается значительно, так как во-первых, этерификация сильно ингибируется и во-вторых эти сложные эфиры лучше растворимы в водном этаноле, чем в чистом этаноле, что относительно неожиданно для специалиста в данной области техники. В конечном продукте эти сложные эфиры больше не будут определены (ниже предела обнаружения способа).
При практическом применении бутрол связывается в комплекс с карбонатом кальция предпочтительно при 20-30°С. В этом диапазоне температур подавляется чрезмерное вспенивание. Стехиометрию затем анализируют при помощи технологического контроля для Са и бутрола, и необязательно регулируют до точно 1:1 путем добавления корректирующего количества карбоната кальция или бутрола. Затем смесь концентрируют, как описано в WO 2011/054827 A1 (Bayer AG), т.е. отгоняют воду под вакуумом, но до определенного конечного объема, основанного на 7-8 кратном количестве примененного карбоната кальция. После этого, приблизительно 26-кратное количество этанола (например, также денатурированный МЕК = метилэтилкетон) отмеряют на протяжении 60-70 минут при температуре кипения, и после охлаждения до 20°С определяют водный эквивалент при помощи технологического контроля. Путем добавления корректирующего количества этанола или метанола, водный эквивалент регулируют до тех пор, пока он не ляжет в пределы целевого коридора 9-11%. Выгодно, установлена величина приблизительно 10%. Затем, смесь нагревают в течение 3 часов с обратным холодильником. Эта процедура также позволяет применение нормального коммерческого спирта, денатурированного например, толуолом, метилэтилкетоном, гексаном или тиофеном.
При водных эквивалентах <9%, примеси также уже кристаллизуются, так, что некоторые партии выпадают из спецификации. Кроме этого, в полиморфе А затем возникают аморфные части до увеличенной степени. При водных эквивалентах >11%, по общему признанию получены очень чистые продукты, но относительно резко падают выходы, поскольку растворимость калкобутрола слишком высока, и для кристаллизации возникают препятствия, кроме того, предпочтительно наблюдается формирование полиморфных форм В, С и D. При применении водного эквивалента в диапазоне 9-11%, с одной стороны, есть очень хороший выход, что очень интересно с экономической точки зрения, и с другой стороны, качество полиморфа (модификация А) очень высокое. Работа с водными эквивалентами в диапазоне 9-11% гарантирует воспроизводимую, надежную и масштабную операцию процесса, которая теперь может быть расширена до нужных масштабов желаемым образом. Новый процесс очень прост в управлении, поскольку он требует только измерения водного эквивалента путем простого технологического контроля. Этот технологический контроль, например, может проводиться путем титрования Карла Фишера или также другим сопоставимым методом. Ход операции существенно не затрагивается технологическим контролем, поскольку относительно быстро может быть определен результат.
Дополнительное преимущество нового процесса согласно изобретению в уже упомянутом воспроизводимом получении определенного полиморфа (для характеристики полиморфных форм, смотрите примеры).
Изобретение существенным образом включает способ получения кальциевого комплекса дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты (калкобутрол), где он связывается в комплекс с ионами кальция2+ в воде, и затем кристаллизуется из этанола, где содержание воды (водный эквивалент) (Карл Фишер) выгодно лежит в диапазоне 9-11%, чтобы получить желаемый целевой полиморф (модификацию А).
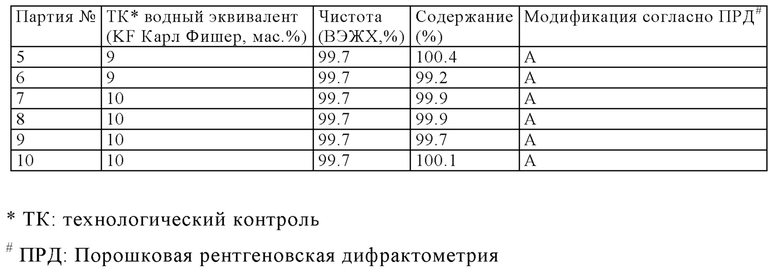
Таблица 2 показывает содержание воды и этанола трех обычных партий получения (24, 25, 26), для которых кристаллическую форму определяли ПРД (Порошковая рентгеновская дифрактометрия). Эти три партии получены в полиморфной формой А (модификация А). Таблица 2 также показывает свойства двух дополнительных партий, которые были получены согласно опубликованным процедурам (Пример 5: Inorganic Chemistry 1997, 36, 6086-6093 и Пример 6: WO 2011/054827 А1).
Партии согласно Примерам 5 и 6 характеризуются их значительно низким содержанием воды и их чрезмерно низким содержанием этанола, обе из которых ясно указывают, что эти партии не соответствуют полиморфной форме А (Модификация А). Эти результаты подтверждены сравнением соответствующих спектров ПРД, которые показывают полностью различные отраженные рентгенограммы.
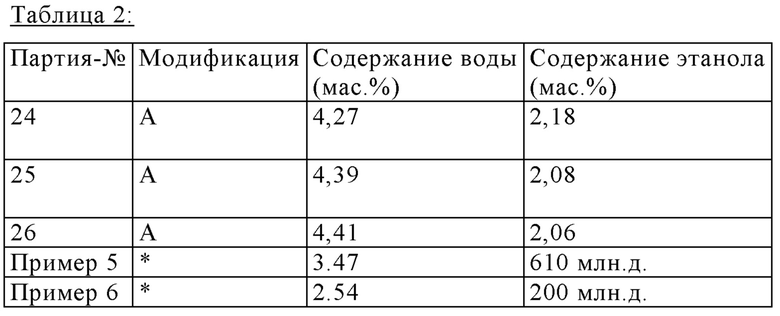
*) модификация, которая отличается от модификации А
Для сравнения применяют теоретические содержания воды и этанола для модификации А:
Модификация А = калкобутрол * 1 H2O * 1/5 этанол
Точнее говоря, изобретение также включает технологические параметры для кристаллизации кальциевого комплекса дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты, где он сперва связывается в комплекс, применяя очень умеренные условия, с ионами кальция2+ при 20-25°С (это важное отличие от предшествующего уровня техники, где комплексообразование проводят при существенно более высоких температурах (80-90°С)), после завершения реакции стехиометрию в Са:бутрол регулируют при помощи технологического контроля и необязательно путем устранения недостатков, таким как добавление карбоната кальция или бутрола, затем продукт кристаллизуют из водного этанола, предпочтительно при содержании воды (водный эквивалент) 9-11%, и затем после выделения сушат под вакуумом.
В качестве подходящих источников ионов кальция2+ для комплексообразования, были найдены карбонат кальция, оксид кальция или гидроксид кальция. Это комплексообразование предпочтительно проводят в водном растворе при различных температурах от 20-90°С. Тем не менее, с карбонатом кальция, комплексообразование может уже быть произведено особенно мягко при 20-30°С.
Конечное преобразование в калкобутрол проводят комплексообразованием бутрола со стехиометрическим количеством карбоната кальция в воде. Тем не менее, также могут применяться оксид кальция (СаО) или гидроксид кальция Са(ОН)2. Предпочтительно применяют карбонат кальция (СаСО3).
Для удаления частиц и уменьшения микробов, смесь обрабатывают активированным углем, и отфильтровывают его. Фильтрат существенно концентрируют под вакуумом и путем добавления этанола проводят технологический контроль воды, необязательно повторно регулируют смесь и затем подвергают кристаллизации. Для этого его нагревают с обратным холодильником, и наконец охлаждают. Выпавший кристаллический продукт отфильтровывают и затем промывают небольшим количеством этанола. Затем его сушат (до постоянной массы) в вакуумной камере.
Калкобутрол модификации А, полученный таким образом, характеризуется очень высоким качеством. Продукт бесцветно растворим в воде и имеет чистоту >99.0%, с производственными партиями в производстве чистота обычно составляет 99.7% (чистота 100% способом, ВЭЖХ). Полный процесс, начинающийся от гадобутрола до калкобутрола, характеризуется высокой воспроизводимостью и эффективностью. Полный выход (полных двух этапов), таким образом, очень хороший. Продукт стабильный при хранении и может применяться в фармацевтике для производства препаратов раствора Гадовист. Натриевую соль калкобутрола создают in situ путем добавления стехиометрического количества раствора гидроксида натрия. Растворы Гадовиста, полученные таким образом, являются стабильными при хранении в течение нескольких лет, и гарантируют безопасность, что свободный токсичный гадолиний никогда не попадет в раствор.
Таким образом, возможно встретить желание органов и фармацевтов, чтобы недорого предоставить калкобутрол высокой чистоты и определенной полиморфной формой (модификация А), который непосредственно подходит для последующей обработки и производства Гадовиста.
Производственный процесс, как описано и заявлено в формуле изобретения здесь, приводит к стабильному и однородному Калкобутролу полиморфной формы А (модификация А) надежным способом. Раньше потенциальных различных форм нужно было избегать, чтобы выполнить увеличивающиеся обязательные требования и требования правил организации производства и контроля качества лекарственных средств.
Различные полиморфные формы даже для фармацевтического наполнителя включают риск незначительных нерастворимых твердых остатков, которые на самом деле наблюдались в прошлом. Чтобы избежать этого риска для парентерального применения должен применяться только однородный полностью растворимый полиморф.
Кроме этого, различные полиморфные формы приводят также к различным ПРД и инфракрасным спектрам, которых нужно избежать, чтобы иметь ясное соответствие идентичности, которая является существенным тестом на фармацевтическое применение.
Хотя структура калкобутрола уже известна, полиморфная форма А уникальна для применения в парентеральных фармацевтических продуктах и описана здесь подробно впервые.
Изобретение также включает применение полиморфа А кальциевого комплекса дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты для получения обычных коммерческих галенов препаратов гадобутрола.
Объект данного изобретения представляет собой соединение формулы (I) в кристаллической форме модификации А
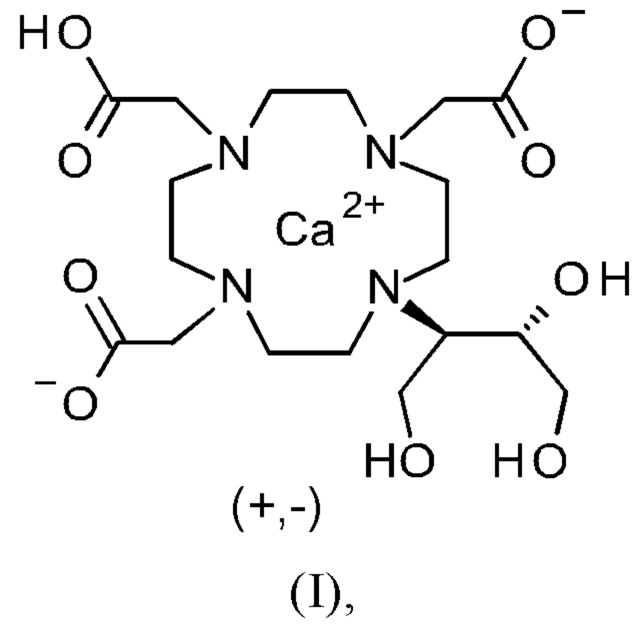
в которой диаграмма рентгеновской порошковой дифрактометрии соединения показывает максимум пика угла 2 тета при 7.6°, 9.1°, 11.1°, 11.3°, 11.9° и 12.3° .
Дополнительным объектом данного изобретения является соединение формулы (I) в кристаллической форме модификации А, где оно представляет собой моногидрат с 2.0-2.5 мас.% этанола.
Дополнительным объектом данного изобретения является соединение формулы (I) в кристаллической форме модификации А, где оно представляет собой моногидрат с 2.0-2.2 мас.% этанола.
Дополнительным объектом данного изобретения является соединение формулы (I) в кристаллической форме модификации А, где оно представляет собой моногидрат с 2.0-2.5 мас.% этанола и где диаграмма рентгеновской порошковой дифрактометрии соединения показывает максимум пика угла 2 тета при 7.6°, 9.1°, 11.1°, 11.3°, 11.9° и 12.3° .
Дополнительным объектом данного изобретения является соединение формулы (I) в кристаллической форме модификации А, где оно представляет собой моногидрат с 2.0-2.2 мас.% этанола и где диаграмма рентгеновской порошковой дифрактометрии соединения показывает максимум пика угла 2 тета при 7.6°, 9.1°, 11.1°, 11.3°, 11.9° и 12.3° .
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, в котором подвергают разложению гадолиниевый комплекс дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты (гадобутрол), осажденную соль гадолиния удаляют, затем раствор со свободными лигандами связывают кислотной ионообменной смолой, затем элюируют водным основным раствором, затем связывают в комплекс ионами кальция2+, затем стехиометрию Са:бутрол доводят до 1:1 с помощью технологического контроля, затем кристаллизуют из водного раствора этанола с содержанием воды 9-11 мас.% с помощью технологического контроля для определения воды и продукт затем высушивают и таким образом соединение формулы (I) выделяют в кристаллической форме модификации А.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, в котором гадолиниевый комплекс дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты (гадобутрол) со щавелевой кислотой в воде подвергают разложению теплом, осажденный оксалат гадолиния фильтруют, затем свободный лиганд связывают с кислотной ионообменной смолой, затем элюируют водным раствором аммиака, после концентрирования раствор связывают в комплекс с ионами кальция2+, затем с помощью технологического контроля стехиометрию Са:бутрол доводят до 1:1, затем нагревают с обратным холодильником из водного раствора этанола с содержанием воды 9-11 мас.% воды, сушат под вакуумом после выделения, и, таким образом, соединение формулы (I) выделяют в кристаллической форме модификации А.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, где для комплексообразования применяют карбонат кальция, оксид кальция или гидроксид кальция.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, где для комплексообразования применяют карбонат кальция.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, который отличается тем, что для комплексообразования применяют карбонат кальция и что комплексообразование проводят в температурном диапазоне ≥0°С и ≤50°С.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, который отличается тем, что для комплексообразования применяют карбонат кальция и что комплексообразование проводят в температурном диапазоне ≥10°С и ≤40°С.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, который отличается тем, что для комплексообразования применяют карбонат кальция и что комплексообразование проводят в температурном диапазоне ≥15°С и ≤35°С.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, который отличается тем, что для комплексообразования применяют карбонат кальция и что комплексообразование проводят в температурном диапазоне ≥20°С и ≤30°С.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, который отличается тем, что для комплексообразования применяют карбонат кальция и что комплексообразование проводят в температурном диапазоне ≥20°С и ≤25°С.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, в котором стехиометрию в кальций:бутрол доводят до 1:1.
Дополнительным объектом данного изобретения является способ получения соединения формулы (I) в кристаллической форме модификации А, где при помощи технологического контроля водный эквивалент во время кристаллизации устанавливается в диапазоне между 9 и 11%.
Дополнительным объектом данного изобретения является соединение формулы (I) в кристаллической форме модификации А, где соединение получают способом, упомянутым как объект данного изобретения.
Дополнительным объектом данного изобретения является соединение формулы (I) в кристаллической форме модификации А, где чистота составляет ≥99.0%.
Дополнительным объектом данного изобретения является соединение формулы (I) в кристаллической форме модификации А, где чистота составляет ≥99.6%.
Дополнительным объектом данного изобретения является соединение формулы (I) в кристаллической форме модификации А, где чистота составляет ≥99.7%.
Дополнительным объектом данного изобретения является применение соединения формулы (I) в кристаллической форме модификации А для получения галенов препаратов гадобутрола.
Следующие примеры служат, чтобы описать объект изобретения.
Экспериментальная часть
Пример 1
Общая процедура получения для калкобутрола с помощью технологического контроля для воды во время кристаллизации
2 кг гадобутрола вместе с 0.8 кг дигидрата щавелевой кислоты суспендировали в 14 л воды и перемешивали в течение, по меньшей мере, 3 часов при 80°С (обычно 3-5 часа). Этому давали возможность охладиться до 20°С и дополнительно перемешивали в течение 60 минут при 20°. Осажденный оксалат гадолиния отфильтровывали и ополаскивали дважды каждый раз 3 л воды (~ 20 л элюата ионообменной смолы). Ионообменную колонку, заполненную катионообменной смолой Amberlite 252 С промывали водой до тех пор пока элюат не достигал проводимости <10 мкСм/см (целевая рН>4.5 и <5.5). В этом случае обнаруживали рН 4.73 и проводимость 4.42 мкСм/см. Вышеуказанный элюат ионообменной смолы (приблизительно 20 л) подавали на ионообменную колонку (скорость подачи 250-350 мл/мин). Ее затем промывали 15 л воды до тех пор пока проводимость элюата не составила <5 мкСм/см (измеренные значения составляли рН 4.48 и 4.99 мкСм/см). Ионообменную колонку затем медленно промывали 1.4-1.7% водным раствором аммиака (применяли 1.4% раствор). Собирали фракции элюата, содержащие продукт (рН 11.01). Элюат концентрировали под вакуумом при 64.4°С до тех пор пока раствор не достиг плотности 1.07 г/мл. Получали 7.906 кг раствора с рН 3.8. Предварительно отбирали 127.13 г образца для того, чтобы определить рН, плотность и содержание бутролового лиганда. После взятия образца, осталось 7.7795 кг раствора с содержанием бутролового лиганда 18.99%) (измерения относительно внешнего стандарта), который соответствует массе 1.477 кг бутрола. Для комплексообразования с кальцием добавляли 328 г карбоната кальция и затем ополаскивали 1019.7 г воды. Его затем оставляли перемешиваться в течение 60 минут при 23°С (во время которых растворялся карбонат кальция). Следующим проводили технологический контроль для избытка кальция или избытка бутрола. Избыток кальция связывали в комплекс дополнительным добавлением соответствующего количества лиганда, аналогично, в случае избытка бутролового лиганда добавляли соответствующее количество карбоната кальция, до тех пор пока стехиометрия Са:бутрол не составила 1:1. После этого, 120.03 г добавляли активированного угля NORIT SX PLUS, 20 μs и промывали 91.6 г воды. Все это перемешивали в течение 60 минут при 23°С, затем активированный уголь отфильтровывали и промывали 0.4 л воды. Фильтрат фильтровали через стерильный фильтр Sartopore 2 mini cartrige 0.2 мкм (получали 9.3537 кг раствора). Затем воду отгоняли при 75 мбар и 70°С воды приблизительно до 7-8-крат в пересчете на количество применяемого карбоната кальция (применяли 328 г карбоната кальция, это означает, что дистилляцию выполняли до конечного количества от 2296 г до 2624 г). Таким образом, отгоняли 6755.5 г воды, которая соответствует количеству конечного объема раствора калкобутрола приблизительно 7.9-крат в пересчете на карбонат кальция. Смесь нагревали до появления конденсата и отмеряли в нее 8556.9 г этанола, МЕК (метилэтилкетон)-денатурированного на протяжении 60 минут (приблизительно 26-кратное количество в пересчете на количество применяемого карбоната кальция). Смесь давали возможность охладиться до 23°С и проводили технологический контроль водного эквивалента согласно Карлу Фишеру. Водный эквивалент в этой точке должен составлять ≥9 и ≤11 мас.%, и по возможности истинное значение должно находиться около 10 мас.% (в любом случае тем не менее в этом диапазоне 9-11 мас.%). Если значение отклоняется от этого окна нужно добавлять воду (в случае <9 мас.%), или этанол, МЕК-денатурированный (в случае >11 мас.%) соответственно. Устанавливали содержание воды 9.24 мас.% и продолжали обработку. Для этого, смесь нагревали в течение 3 часов с обратным холодильником, затем охлаждали (градиент) на протяжении 14 часов до 20°С и кристаллизованную смесь перемешивали в течение дополнительных 60 минут. Осажденный продукт кристаллизации отфильтровывали, затем промывали в 2 порциях с общим количеством 812 г этанола, МЕК-денатурированного. Осадок помещали на штатив в стерильной комнате и затем высушивали (до постоянной массы, 20-85 ч). После высушивания получали 1264.9 г продукта, который соответствует размеру обычной производственной партии.
Пример 2
Получение калкобутрола в техническом масштабе
Следующая таблица представляет результаты получения калкобутрола, который производили согласно общей процедуре аналогично примеру 1. Средний размер партии составлял 1.0-1.2 кг. Таблица 3 показывает высокую чистоту и высокие содержания, которые получили, когда водный эквивалент устанавливали между 9 и 11 мас.%. 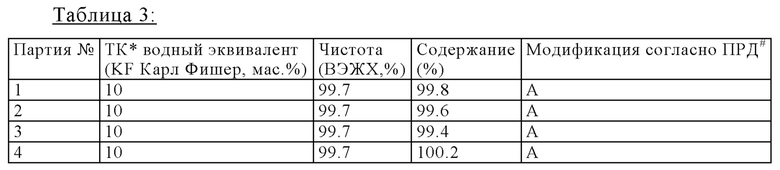
Пример 3
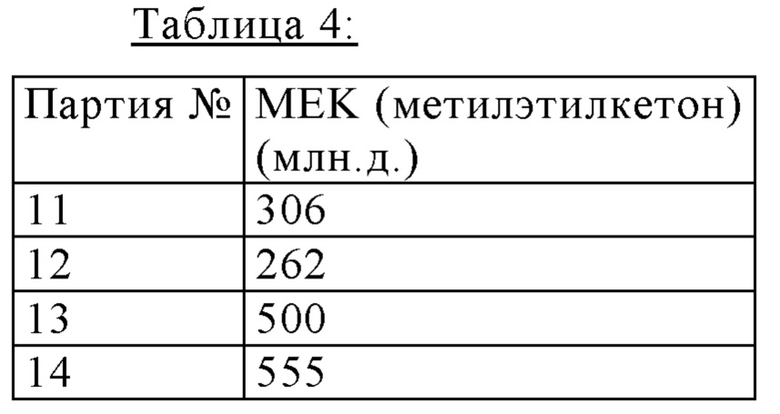
Содержания остатка растворителей при применении денатурированного этанола с метилэтилкетоном
Партии, описанные здесь, получали как описанно выше, и применяли денатурированный этанол с метилэтилкетоном. Таблица 4 показывает, что наблюдается граница остатка растворителей ≤5,000 млн.д., требуемая ICH (International Conference on Harmonisation) Guideline.
Пример 4
Полиморфизм
Полиморфы В, С и D характеризуются высоким содержанием воды и могут быть получены из полиморфа А (модификация А) путем добавления воды (≥12 мас.%) во время процесса кристаллизации. Таблица 5 показывает свойства этих полиморфов, которые получили в малом масштабе, в лаборатории. 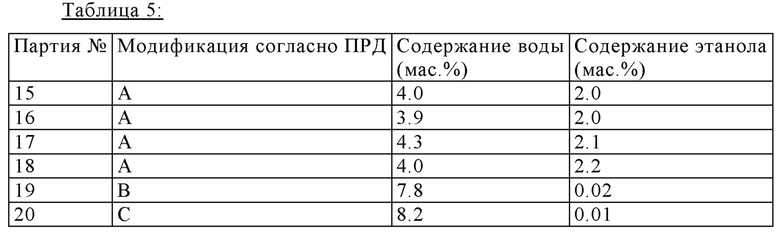
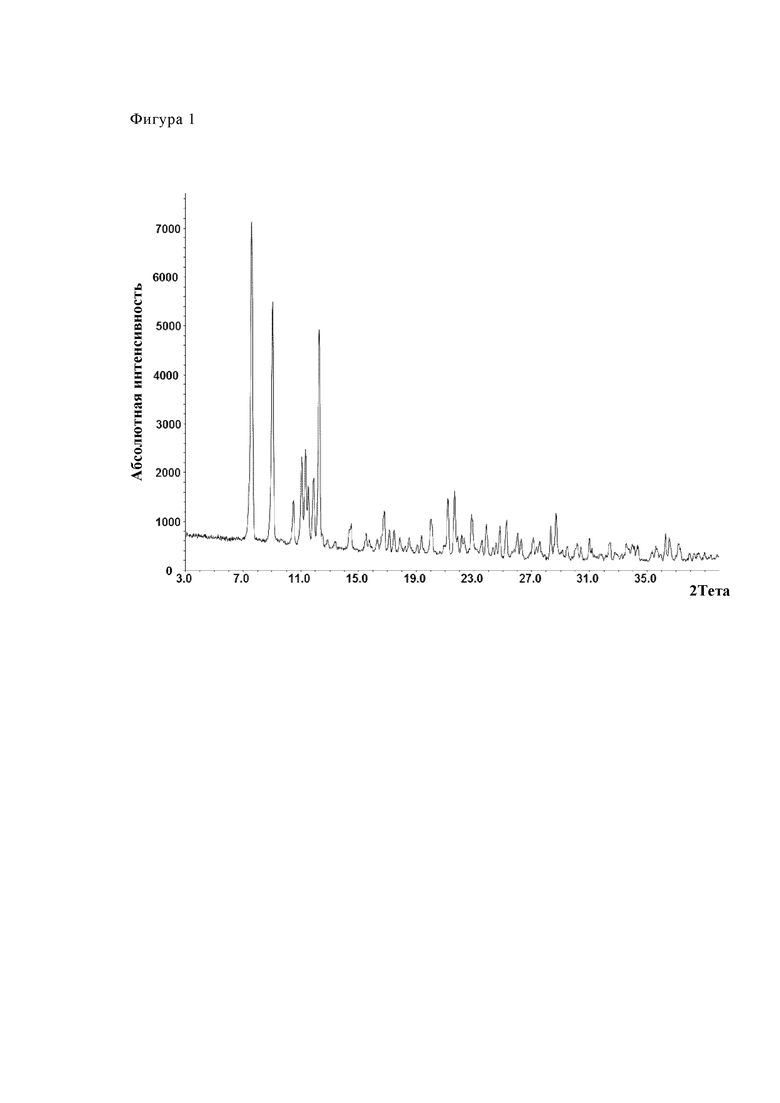
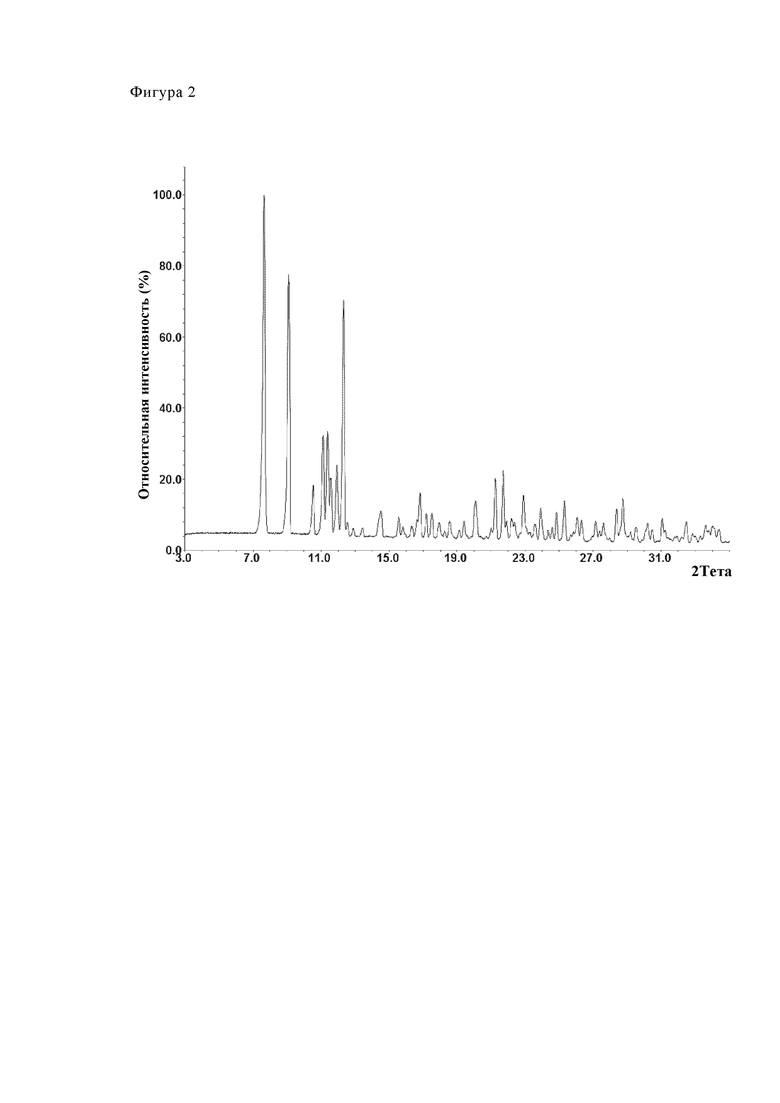
Фиг. 1 показывает диаграмму рентгеновской порошковой дифрактометрии модификации А из партии №16.
Таблица 6 показывает путем примера три типичных получения партий (24, 25, 26), в которых полиморфную форму определяли с помощью ПРД. Во всех партиях воспроизводимо получали модификацию А.
В дополнение, таблица 6 показывает аналитические данные для двух партий, которые были получены согласно процедурам, описанным в предшествующем уровне техники (Примеры 5 и 6, процедуры для получения и изолирования как описано ниже). Эти партии характеризуются их значительно низким содержанием воды и их чрезмерно низким содержанием этанола, оба из которых ясно указывают, что эти партии не соответствуют полиморфной форме А (Модификация А). Эти результаты подтверждены сравнением соответствующих спектров ПРД (см ниже).
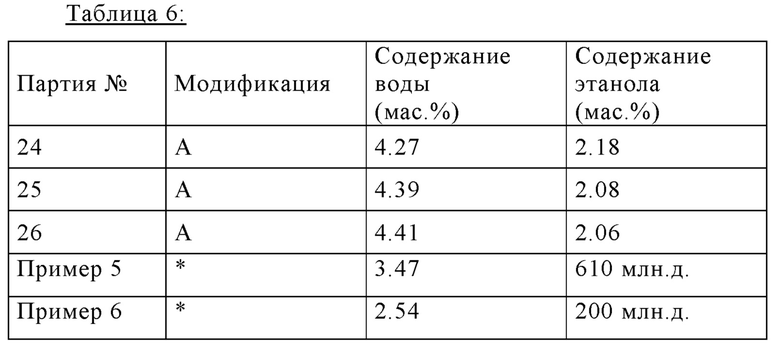
*) модификация, которая отличается от модификации А
В качестве сравнения применяют теоретические содержания воды и этанола для модификации А:
Модификация А = калкобутрол * 1 H2O * 1/5 этанол
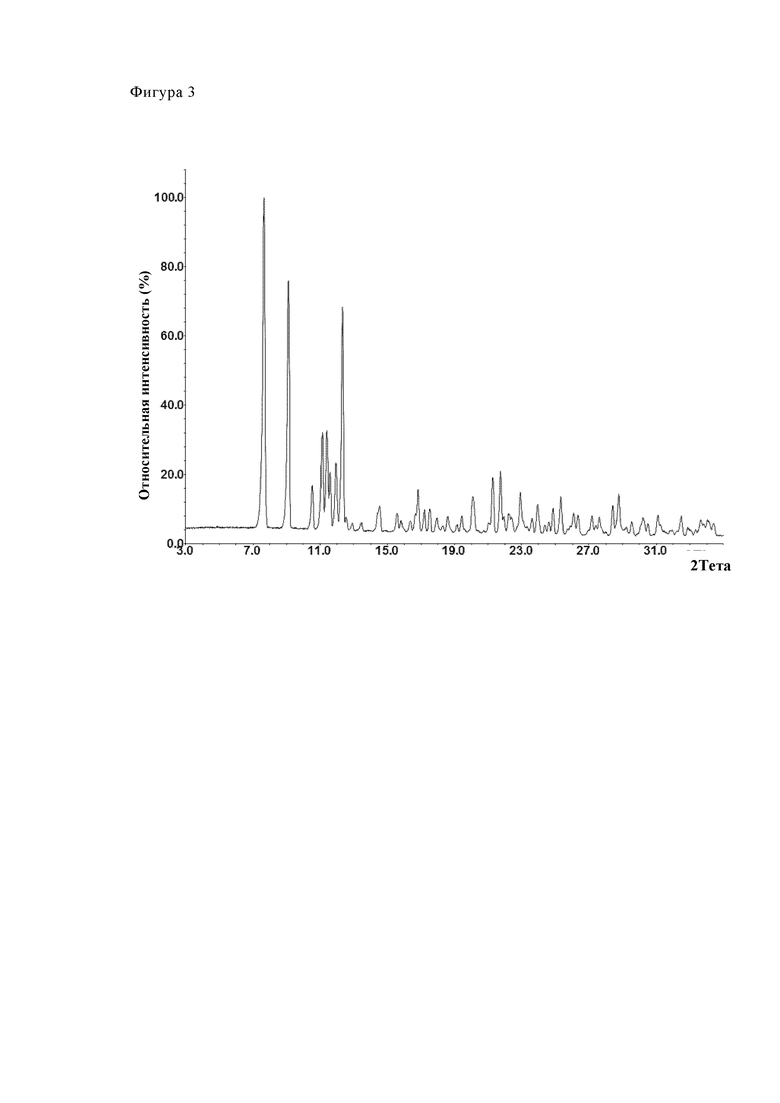
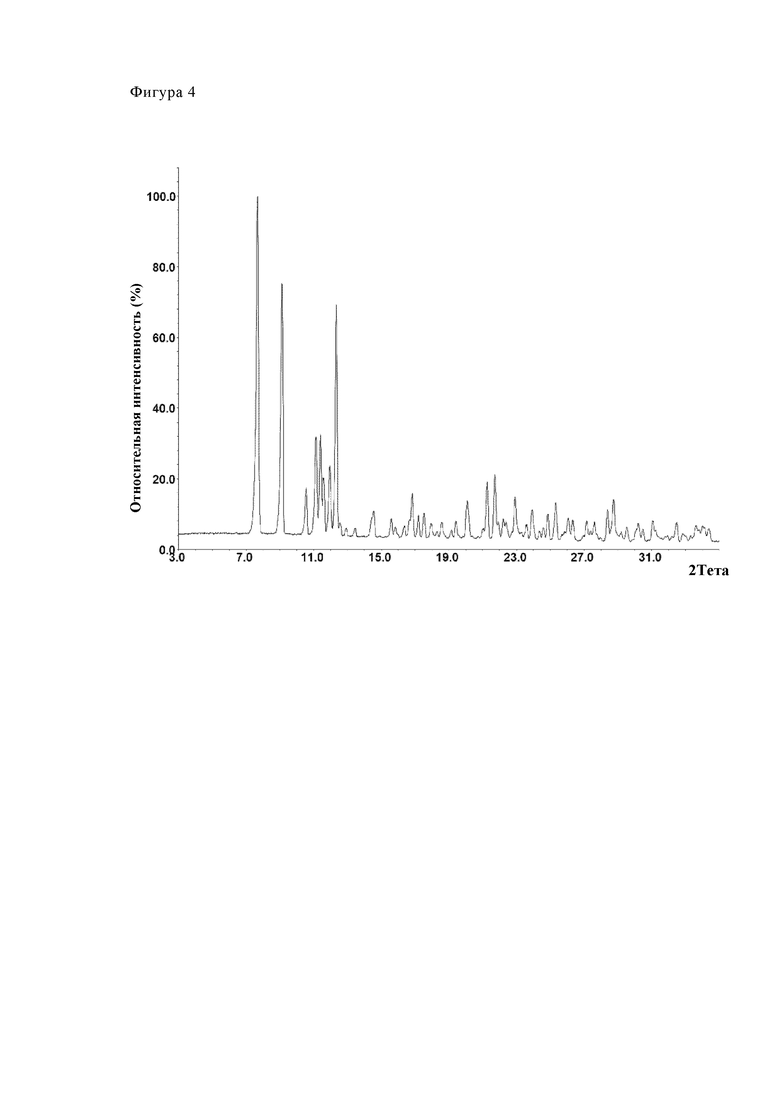
Фиг. 2 (Партия №24), 3 (Партия №25) и 4 (Партия №26) показывают путем примера три диаграммы дифрактометрии. Партии соответствуют форме А (модификация А).
Таблица 7 показывает влияние водного эквивалента на модификацию, полученную при кристаллизации.
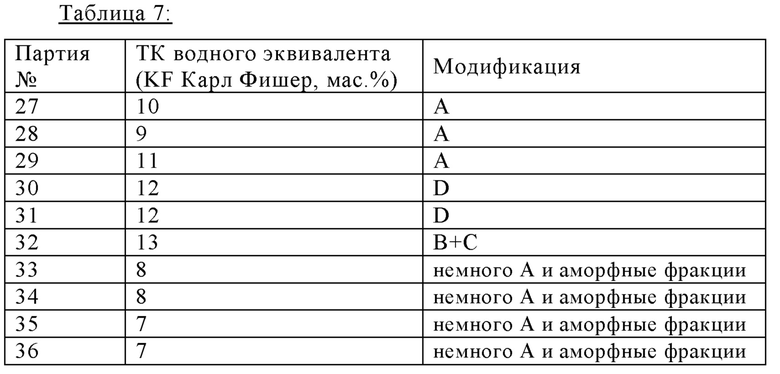
Партии 33-36: партии, при получении которых водный эквивалент регулировали до 7-8%, являются гигроскопическими, так как они содержат более высокую пропорцию аморфного вещества.
Порошковая рентгеновская дифрактометрия калкобутрола (ПРД)
Таблица 8 показывает величины 2θ дифракционных пиков калкобутрола модификации А (партии 16, 24, 25, 26). Максимумы пика найдены при углах 2θ 7.6°, 9.1°, 11.1°, 11.3°, 11.9° и 12.3°.


Установка прибора для ПРД:
Сравнение с предшествующим уровнем техники
Следующие примеры демонстрируют, что полиморфная форма как получаемая заявленным способом данного изобретения отличается от полиморфной формы материала, который может быть получен при помощи опубликованных процедур. Для этой цели готовили две партии калкобутрола согласно опубликованным процедурам, и соответствующие спектры ПРД сравнивали со спектрами ПРД, полученными из партии получения Калкобутрола, полученного согласно изобретению. Синтез калкобутрола согласно опубликованным процедурам проводили, начиная с водного раствора Бутрола (обычно 17-22%), который доступен в больших объемах.
Процедура общего получения для приготовления водного раствора Бутролового лиганда 2 кг гадобутрола вместе с 0.8 кг дигидрата щавелевой кислоты суспендировали в 14 л воды и перемешивали в течение, по меньшей мере, 3 часов при 80°С (обычно 3-5 часов). Всему этому дали возможность охладиться до 20°С и дополнительно перемешивали в течение 60 минут при 20°С. Осажденный оксалат гадолиния отфильтровывали и промывали дважды 3 л воды каждый раз (~ 20 л элюата ионообменной смолы). Ионообменную колонку, наполненную катионообменной смолой Amberlite 252 С промывали водой до тех пор, пока не достигли проводимости <10 мкСм/см (целевая рН >4.5 и <5.5). В этом случае находили рН 4.73 и проводимость 4.42 мкСм/см. Вышеуказанный элюат ионообменной смолы (приблизительно 20 л) подавали на ионообменную колонку (скорость подачи 250-350 мл/мин). Все это затем промывали 15 л воды до тех пор, пока проводимость элюата не стала <5 мкСм/см (измеренные значение составили рН 4.48 и 4.99 мкСм/см). Ионообменную колонку затем медленно промывали 1.4-1.7% водным раствором аммиака (применяли 1.4% раствор). Собирали фракции элюата, содержащие продукт (рН 11.01). Элюат концентрировали под вакуумом при 64.4°С до тех пор, пока раствор не достиг плотности 1.07 г/мл. Получали 7.906 кг раствора с рН 3.8. Предварительно отбирали 127.13 г образца для того, чтобы определить рН, плотность и содержание бутролового лиганда. После взятия образца осталось 7.7795 кг раствора с содержанием бутролового лиганда 18.99% (измерения относительно внешнего стандарта), которое соответствует массе 1.477 кг бутрола.
Для следующих экспериментов водный раствор бутролового лиганда получали подобным образом.
Пример 5
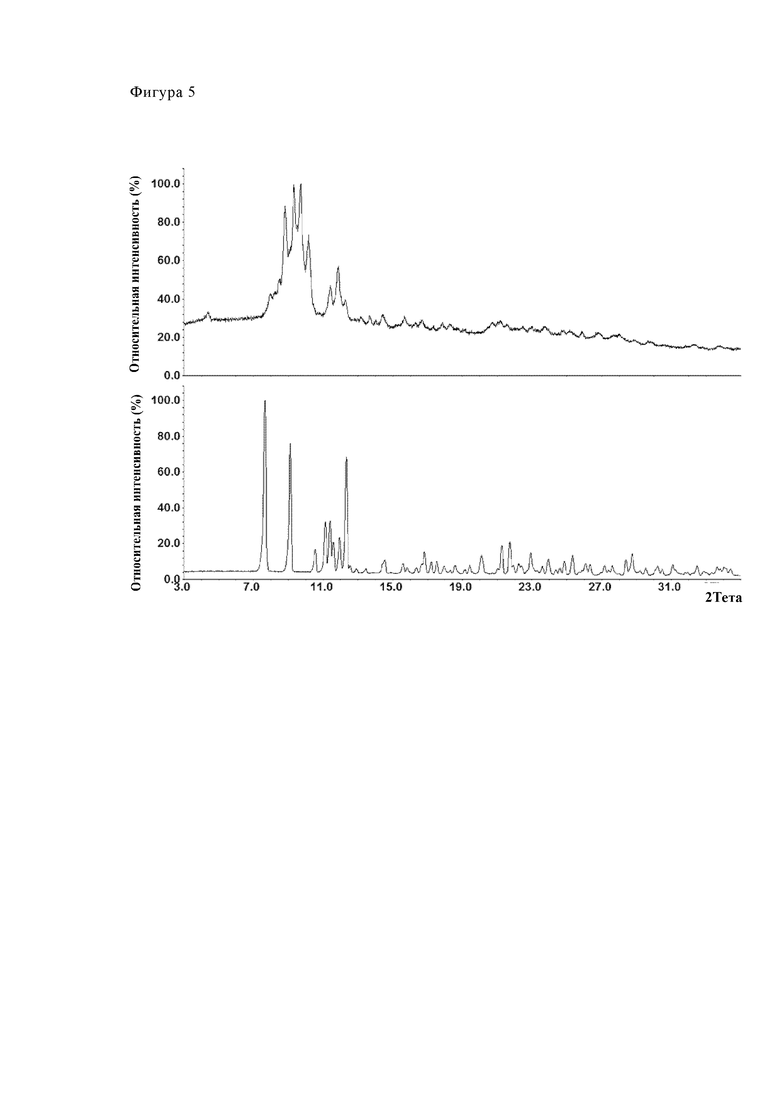
Получение калкобутрола согласно Inorganic Chemistry 1997, 36, 6086-6093:
245.52 г 18% водного раствора бутрола (112 ммоль, полученного как описано выше) разбавили 330 мл воды при комнатной температуре. Затем добавляли 10.99 г (112 ммоль) карбоната кальция и раствор нагревали при 80°С в течение 2 ч. Его охлаждали до 22.6°С и раствор фильтровали, а остаток на фильтре промывали 20 мл воды. Фильтрат высушивали вымораживанием. Высушенный вымораживанием порошок (55.52 г) суспендировали в 1315 мл этанола (денатурированный метилэтилкетоном) и перемешивали при 50°С в течение 1 ч. Для выделения продукта реакции суспензию охлаждали до 0°С и продукт изолировали путем фильтрования. Продукт промывали 395 мл этанола (денатурированный метилэтилкетоном) и затем высушивали под вакуумом до тех пор, пока масса не стала постоянной (при 60°С).
Выход: получали 52.62 г (96.2%-й) мелкодисперсного белого порошка.
Данные анализа:
Для соответствующего спектра ПРД см. Фиг. 5.
Пример 6
Получение калкобутрола согласно WO 2011/054827 А1:
245.54 г 18% водного раствора бутрола (50.45 г = 112 ммоль, полученного как описано в пример 1), разбавили 208 мл воды при комнатной температуре. Затем добавляли 11.34 г (112 ммоль) карбоната кальция и раствор нагревали при 90°С в течение приблизительно 3 ч. Его охлаждали до комнатной температуры (+20°С). После этого добавляли 5.06 г свежепромытого угля (Norit SX Plus) и смесь перемешивали в течение 1 ч при комнатной температуре. Раствор фильтровали и остаток на фильтре (уголь) промывали 3 раза, каждый 50 мл воды. Фильтрат упаривали дистилляцией под вакуумом при 80°С до определенного объема, который соответствует 1.4 раза исходного бутролового лиганда (1.4 раза: 1.4×50.45 массы бутролового лиганда = 70.6 мл). Добавляли 505 мл этанола (денатурированный метилэтилкетоном) и смесь нагревали в течение 3 ч с обратным холодильником. Для выделения продукта реакции суспензию охлаждали до 20°С и продукт отделяли фильтрованием. Продукт промывали два раза, каждый 50 мл этанола (денатурированный метилэтилкетоном) и затем сушили под вакуумом до тех пор, пока масса не станет постоянной (при 70°С).
Выход: получали 49.15 г (89.9%-й) мелкодисперсного белого порошка.
Данные анализа:
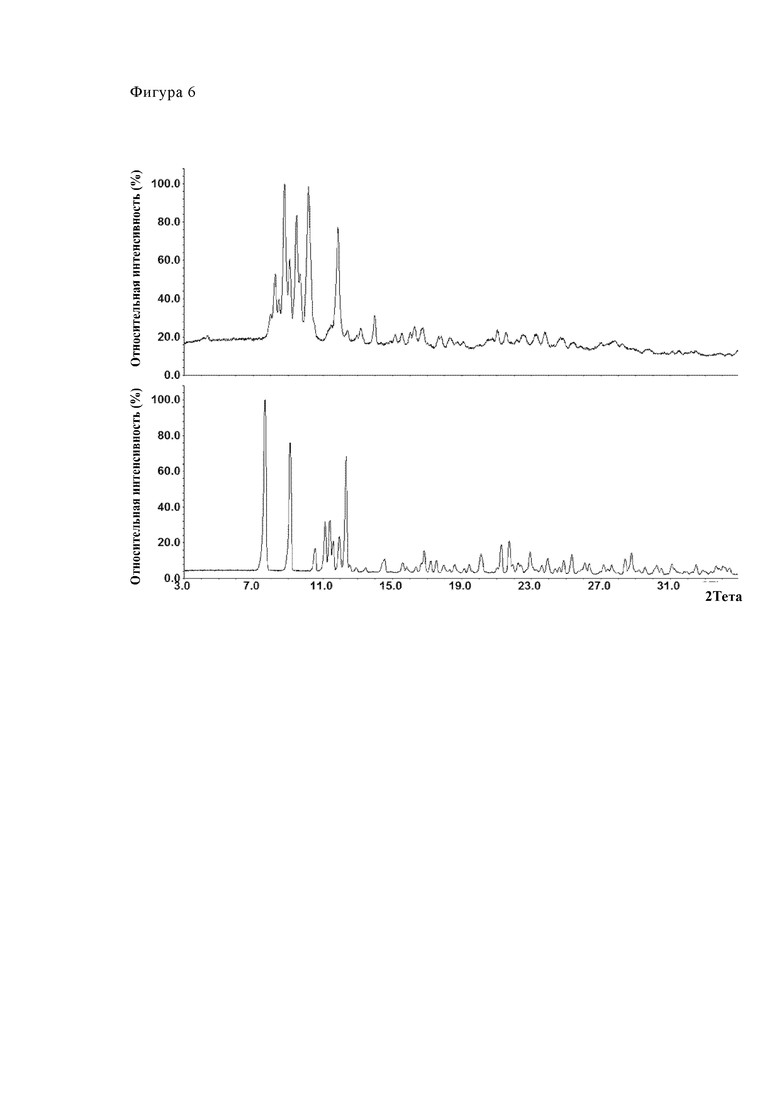
Для соответствующего спектра ПРД см. Фиг. 6.
Пример 7
Гигроскопические свойства
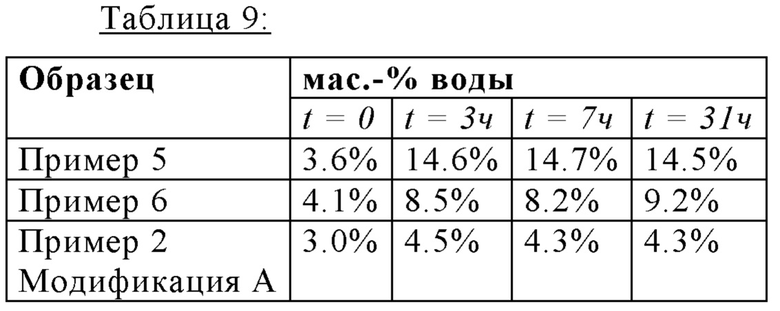
Определяли гигроскопические свойства трех разных образцов калкобутрола, один образец получали способом, описанным в Inorganic Chemistry 1997, 36, 6086-6093 (Пример 5), один образец получали методом, описанным в WO2011/054827A1 (Пример 6), и один образец получали способом согласно данного изобретения (Пример 2).
2 г каждого образца подвергали воздуху окружающей среды (приблизительно 20°С, приблизительно 60% отн. влажности). Содержание воды измеряли при помощи титрованием по Карлу Фишеру после начального действия (t=0), через три часа (t=3ч), семи часов (t=7ч), и тридцать одного часа (t=31ч) после начального действия.
Таблица 9 показывает увеличение мас.% воды разных образцов в течение времени. Образцы Калкобутрола, полученные согласно способам, описанным в предшествующим уровнем техники, являются намного более гигроскопичными, чем Калкобутрол, полученный согласно способу данного изобретения.
Описание Фигур
Фиг. 1 показывает диаграмму рентгеновской порошковой дифрактометрии модификации А из партии №16.
Фиг. 2 показывает путем примера диаграмму дифрактометрии партии № 24. Партия соответствует модификации А.
Фиг. 3 показывает путем примера диаграмму дифрактометрии партии № 25. Партия соответствует модификации А.
Фиг. 4 показывает путем примера диаграмму дифрактометрии партии № 26. Партия соответствует модификации А.
Фиг. 5 показывает диаграмму рентгеновской порошковой дифрактометрии примера 5 (верхняя диаграмма) в сравнении с диаграммой рентгеновской порошковой дифрактометрии партии, соответствующей модификации А (нижняя диаграмма). Это сравнение ясно демонстрирует, что полиморфная форма, соответствующая примеру 5, которая была получена согласно процедуре, описанной в Inorganic Chemistry 1997, 36, 6086-6093, отличается от модификации А как получают заявленным способом.
Фиг. 6 показывает диаграмму рентгеновской порошковой дифрактометрии примера 6 (верхняя диаграмма) в сравнении с диаграммой рентгеновской порошковой дифрактометрии партии, соответствующей модификации А (нижняя диаграмма). Это сравнение ясно демонстрирует, что полиморфная форма, соответствующая примеру 6, которая была получена согласно процедуре, описанной в WO 2011/054827 A1, отличается от модификации А как получают заявленным способом.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КАЛЬКОБУТРОЛА | 2019 |
|
RU2779668C1 |
| ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ГАДОБУТРОЛА И СПОСОБ ПОЛУЧЕНИЯ ГАДОБУТРОЛА С ИСПОЛЬЗОВАНИЕМ ТАКОГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2018 |
|
RU2740901C1 |
| ПОЛУЧЕНИЕ ЗАЩИЩЕННОГО DO3A | 2020 |
|
RU2828009C1 |
| ДИАСТЕРЕОИЗОМЕРНО ОБОГАЩЕННЫЙ КОМПЛЕКС ГАДОЛИНИЯ И ХЕЛАТИРУЮЩЕГО ЛИГАНДА НА ОСНОВЕ PCTA, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ОЧИСТКИ | 2020 |
|
RU2806027C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ КОМПЛЕКСЫ МЕТАЛЛОВ РЯДА ЛАНТАНОИДОВ | 2015 |
|
RU2707070C2 |
| СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2002 |
|
RU2305096C2 |
| Составы металлических комплексов | 2015 |
|
RU2710360C2 |
| СПОСОБ ОЧИСТКИ ПОЛИАМИНОКАРБОКСИЛАТОВ | 2012 |
|
RU2621896C2 |
| ДИАСТЕРЕОИЗОМЕРНО ОБОГАЩЕННЫЙ КОМПЛЕКС ГАДОЛИНИЯ И ХЕЛАТИРУЮЩЕГО ЛИГАНДА НА ОСНОВЕ PCTA И СПОСОБ СИНТЕЗА | 2020 |
|
RU2810975C2 |
| КОМПЛЕКСЫ КАСКАДНЫХ ПОЛИМЕРОВ, СОДЕРЖАЩЕЕ ИХ ДИАГНОСТИЧЕСКОЕ КОНТРАСТНОЕ СРЕДСТВО И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1996 |
|
RU2197495C2 |
Изобретение относится к способу получения соединения формулы (I) в кристаллической форме модификации А, которое представляет собой моногидрат с 2.0-2.5 мас.% этанола и диаграмма рентгеновской порошковой дифрактометрии соединения показывает максимум пика угла 2 тета при 7.6°, 9.1°, 11.1°, 11.3°, 11.9° и 12.3°. Способ заключается в том, что подвергают разложению гадолиниевый комплекс дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты (гадобутрол), осажденную соль гадолиния удаляют, затем раствор с свободными лигандами связывают с кислотной ионообменной смолой, затем элюируют водным основным раствором, затем связывают в комплекс с ионами кальция2+, затем стехиометрию Са:бутрол доводят до 1:1, затем осуществляют кристаллизацию из водного этанола при содержании воды 9-11 мас.% и затем продукт сушат и, таким образом, выделяют соединение формулы (I). Технический результат – разработан способ получения кристаллической формы модификации А соединения формулы (I) с высокой чистотой, которую применяют при получении Гадовиста. 2 н. и 2 з.п. ф-лы, 6 ил., 9 табл., 7 пр.
1. Способ получения соединения формулы (I) в кристаллической форме модификации А
которое отличается тем, что оно представляет собой моногидрат с 2.0-2.5 мас.% этанола и что диаграмма рентгеновской порошковой дифрактометрии соединения показывает максимум пика угла 2 тета при 7.6°, 9.1°, 11.1°, 11.3°, 11.9° и 12.3°, который отличается тем, что подвергают разложению гадолиниевый комплекс дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты (гадобутрол), осажденную соль гадолиния удаляют, затем раствор с свободными лигандами связывают с кислотной ионообменной смолой, затем элюируют водным основным раствором, затем связывают в комплекс с ионами кальция2+, затем стехиометрию Са:бутрол доводят до 1:1, затем осуществляют кристаллизацию из водного этанола при содержании воды 9-11 мас.% и затем продукт сушат и, таким образом, выделяют соединение формулы (I).
2. Способ получения соединения формулы (I) в кристаллической форме модификации А
которое отличается тем, что оно представляет собой моногидрат с 2.0-2.5 мас.% этанола и что диаграмма рентгеновской порошковой дифрактометрии соединения показывает максимум пика угла 2 тета при 7.6°, 9.1°, 11.1°, 11.3°, 11.9° и 12.3°, который отличается тем, что гадолиниевый комплекс дигидрокси-гидрокси-метилпропил-тетраазациклододекан-триуксусной кислоты (гадобутрол) подвергают разложению щавелевой кислотой в воде при нагревании, осажденный оксалат гадолиния отфильтровывают, затем свободный лиганд связывают с кислотной ионообменной смолой, затем элюируют водным раствором аммиака, и связывают в комплекс с ионами кальция2+ после концентрирования раствора, затем стехиометрию Са:бутрол доводят до 1:1, затем его нагревают с обратным холодильником из водного этанола при содержании воды 9-11 мас.% воды, затем охлаждают, после его выделения сушат, и таким образом выделяют соединение формулы (I).
3. Способ получения соединения формулы (I) по п. 1 или 2, который отличается тем, что для комплексообразования применяют карбонат кальция.
4. Способ получения соединения формулы (I) по пп. 1, 2 или 3, который отличается тем, что для комплексообразования применяют карбонат кальция и что комплексообразование проводят в температурном диапазоне ≥20°С и ≤25°С.
| WO 2011054827 A1, 12.05.2011 | |||
| RU 2003120513 A, 27.12.2004 | |||
| MINO R.CAIRA, Crystalline Polymorphism of Organic Compounds, 1998, p.163-208 | |||
| Sherry L.Morissette et al.: "High-throughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids", ADVANCED DRUG DELIVERY REVIEWS, 2004, v.56, pp.275-300 | |||
| Приспособление для регулировки ветряного двигателя с вращаемыми вокруг своих осей цилиндрами | 1927 |
|
SU10198A1 |

