Настоящее изобретение относится к объектам, охарактеризованным в формуле изобретения, а именно к макроциклическим комплексам металлов, а также к их применению для получения конъюгатов с биомолекулами. Подобные конъюгаты пригодны для получения контрастных веществ, предназначенных для использования при ЯМР-диагностике и рентгенодиагностике, а также средств, предназначенных для радиотерапии.
Предпосылкой целенаправленной и эффективной терапии является, как очевидно, точность постановки диагноза. За прошедшие годы именно в области диагностики был достигнут значительный прогресс, о чем свидетельствуют, например, современные методы ЯМР-диагностики, которая позволяет с высокой точностью избирательно визуализировать практически любую анатомическую деталь. Однако во многих случаях соответствующие структуры становятся визуально различимы только при применении контрастных веществ. Помимо этого существует также возможность придания контрастным веществам таких свойств, благодаря которым они приобретают способность избирательно накапливаться в требуемых структурах-мишенях. Благодаря этому удается повысить точность визуализации определенных структур организма при одновременном снижении дозы контрастного вещества, в которой его требуется вводить в организм пациента для достижения необходимого эффекта.
Для применения в качестве контрастных веществ при ЯМР-диагностике пригодны хелатные комплексы парамагнитных металлов. Теоретические и практические аспекты применения хелатных соединений гадолиния(III) в качестве контрастных веществ при ЯМР-диагностике подробно рассматриваются в обзорной статье Р.Caravan и др., опубликованной в Chem. Rev.99, 1999, cc.2293-2352.
Интенсивность изображения при исследованиях методом протонного ЯМР определяется в основном протонами воды. Она зависит от времени ядерной релаксации. Комплексы парамагнитных переходных металлов и лантаноидов сокращают время релаксации соседних протонов за счет диполярных взаимодействий. Получение изображений при диагностике методом протонного ЯМР обеспечивается не за счет непосредственного, а за счет косвенного детектирования парамагнитных контрастных веществ на основе того факта, что они способны изменять время релаксации соседних протонов, таких как протоны воды. Предпочтительными для применения при ЯМР-диагностике парамагнитными катионами металлов являются благодаря их высокому магнитному моменту и высокой релаксационной эффективности катионы Gd3+, Fe3+, Mn2+.
Важной физической величиной, описывающей релаксационные свойства протонов, является время их продольной релаксации T1. Ткани, для которых характерно короткое время релаксации T1, в целом дают изображения более высокой интенсивности по сравнению с тканями, для которых характерно более длительное время релаксации. Если для определенного парамагнитного иона построить график зависимости величины, обратной его измеренному времени релаксации T1, от его концентрации с, то такой график будет иметь вид прямой с наклоном R. Подобный наклон называют также релаксационностью, которая является мерой, характеризующей способность соответствующего парамагнитного иона сокращать время релаксации соседних протонов.
Равным образом в области биологических и медицинских исследований уже достаточно давно известно применение радиофармацевтических препаратов в диагностических и терапевтических целях. Подобные радиофармацевтические препараты используются главным образом для визуализации определенных структур организма, таких, например, как скелет, различные органы или ткани. Радиоактивные средства для возможности их применения в диагностических целях должны обладать способностью после их введения в организм пациента специфически накапливаться непосредственно в тех структурах организма, которые требуется исследовать. Такие локально накапливающиеся в организме радиоактивные средства можно после их введения обнаруживать, регистрировать с помощью графопостроителя или сцинтиграфировать с использованием соответствующих детекторов, таких, например, как сцинтилляционные камеры или иные пригодные для этой цели средства и методы регистрации. По распределению и относительной интенсивности детектированного излучения, испускаемого радиоактивным средством, можно определить место его нахождения в исследуемой структуре и таким путем визуализировать наличие аномалий в структурах организма и в выполняемых ими функциях, патологические изменения и т.д.
Аналогичным образом радиофармацевтические препараты можно использовать и в качестве терапевтических средств для облучения определенных больных тканей или областей организма. Для подобного лечения требуется получение радиоактивных терапевтических средств, способных накапливаться в определенных структурах, органах или тканях.
Требуемые ионы по причине их отчасти сравнительно высокой токсичности обычно вводят в организм не в виде их водорастворимых солей, а в виде хелатных комплексов. Такие хелатные комплексы могут практически в неизменном виде выводиться из организма. Чем меньше размеры присутствующих в растворе комплексов, тем меньше их момент инерции и тем выше скорость их вращения в растворе (Tumbling Motion Time). Соответственно чем выше скорость вращения комплекса, тем ниже его релаксационность. Тем самым релаксационность возрастает с увеличением молекулярной массы всего комплекса. Высокую молекулярную массу комплексов можно обеспечить за счет их связывания с макромолекулами. Эффективное контрастное вещество для ЯМР-диагностики должно помимо прочего обладать высоким показателем релаксационности.
Конъюгаты из Gd-ДТПУ (диэтилентриаминопентауксусная кислота) с альбумином описаны, например, у M.D.Ogan и др. в Invest. Radiol. 22, 1987, cc.665-671, а также у U.Schmiedl и др. в Radiology 162, 1987, cc.205-210. В WO 95/31444 описаны конъюгаты из макроциклических комплексов металлов и биомолекул. Для повышения избирательности контрастных веществ в WO 01/08712 было предложено контрастное вещество, которое содержит по меньшей мере два представляющих собой хелатные соединения металлов фрагмента в качестве улучшающих качество изображения групп и по меньшей мере два "связывающихся с мишенью фрагмента", обеспечивающих в организме присоединение молекулы контрастного вещества к требуемой молекуле-мишени или органу-мишени.
Согласно WO 97/02051 крупные молекулы контрастных веществ с высокой молекулярной массой получают путем встраивания макроциклических комплексов металлов в каскадные полимеры.
В ЕР-А 0565930 описаны производные тетраазациклододекантетрауксусной кислоты, которые обладают высокой стабильностью и хорошей растворимостью из-за отсутствия у них заряда и которые пригодны для присоединения к биомолекулам.
Описанное выше присоединение макроциклических комплексов металлов к биомолекулам позволяет повысить не только релаксационность контрастного вещества, но и его избирательность. Чем выше релаксационность контрастного вещества, тем в меньшем количестве его требуется вводить в организм пациента и тем выше контрастность получаемого изображения. По этой причине желательно далее получение контрастных веществ для ЯМР-диагностики с максимально высокой релаксационностью.
В соответствии с этим в основу настоящего изобретения была положена задача предложить более эффективные контрастные вещества для ЯМР-диагностики и рентгенодиагностики, а также средства для радиотерапии. Такие контрастные вещества для ЯМР-диагностики должны обладать прежде всего максимально высокой релаксационностью и способностью с максимально высокой избирательностью накапливаться в требуемом месте в организме.
Согласно изобретению неожиданно было установлено, что указанную задачу можно решить за счет присоединения к 1,4,7,10-тетраазациклододекановому макроциклу особых лигандов. Новые качественные свойства предлагаемых в изобретении соединений проявляются при их присоединении к биомолекулам. За счет присоединения к макроциклу особых лигандов достигается высокая релаксационность получаемого в результате контрастного вещества и помимо этого обеспечивается возможность точного ее регулирования с учетом цели применения предлагаемых в изобретении соединений.
В соответствии с этим настоящее изобретение относится к соединениям формулы I

в которой
Z обозначает атом водорода или по меньшей мере два Z представляют собой эквивалент иона металла,
В обозначает атом водорода или С1-С4алкильный остаток,
R обозначает атом водорода или прямой, разветвленный либо циклический насыщенный либо ненасыщенный C1-С10алкильный или арильный остаток, который необязательно замещен карбоксильной группой, группой -SO3Н или группой -РО3Н2, при этом алкильная цепь C1-С10алкильного остатка необязательно содержит арильную группу и/или 1-2 атома кислорода,
при условии, что остатки В и R оба одновременно не обозначают атомы водорода,
А обозначает прямую либо разветвленную, насыщенную либо ненасыщенную C1-С30углеводородную цепь, которая необязательно содержит 1-5 атомов кислорода, 1-5 атомов азота и/или 1-5 -NR'-остатков, где R' имеет указанные для R, но выбираемые независимо от него значения, и необязательно замещена 1-3 карбоксильными группами, 1-3 группами -SO3Н, 1-3 группами -РО3Н2 и/или 1-3 атомами галогена и в которой 1-3 атома углерода необязательно присутствуют в виде карбонильных групп, при этом такая цепь или ее часть может быть замкнута в кольцо, и которая далее имеет такую структуру, что Х по меньшей мере через 3 атома связан с атомом азота, к которому присоединен остаток А, и
Х представляет собой группу, способную вступать в реакцию с биомолекулой, включая соли этого соединения, и к его применению для получения конъюгата с биомолекулой.
Соответствующее макроциклическое соединение, у которого каждый из четырех атомов азота макроциклической структуры замещен группой -СН(CO2Н)СН2СН2CO2Н, описано у Р.Caravan и др., Chem. Rev.99, 1999, cc.2293-2352. Однако возможное применение этого соединения для получения конъюгатов с биомолекулами в этой публикации не рассматривается. В WO 97/02051 описаны макроциклические соединения, в которых А представляет собой остаток -CH(R4)-CO-NR2-U6- и которые используются в качестве промежуточных соединений для получения каскадных полимеров. В ЕР-А-0565930 описаны макроциклические соединения, в которых А представляет собой остаток -CH(R3)-C(O)-NH-(CH2)1-6-NH-D-. При этом о повышении релаксационности таких соединений за счет использования определенных заместителей в этой публикации ничего не говорится. В соответствии с этим подобные соединения не подпадают под определение соединения формулы I, указанное в п.1 формулы изобретения, и исключены из объема настоящего изобретения.
Под "алкильным остатком" в контексте настоящего изобретения подразумевается, если не указано иное, насыщенный или ненасыщенный, неразветвленный или разветвленный либо циклический алкильный остаток с указанным числом атомов углерода. Если такой остаток может содержать другие группы или атомы, то в этом случае в контексте настоящего изобретения подразумевается, что подобные другие группы или атомы могут присутствовать в дополнение к уже имеющимся у остатка атомам и могут находиться в любом его положении, включая концевые положения.
Под "арилом" в контексте настоящего изобретения предпочтительно подразумеваются фенил, бисфенил, пиридил, фуранил, пирролил и имидазолил. Особо предпочтительным при этом является фенил.
Под "углеводородной цепью", которая полностью или частично может быть замкнута в кольцо, в контексте настоящего изобретения предпочтительно подразумевается углеводородная цепь, такая как алкильная цепь, которая, например, может содержать алифатическое или ароматическое, необязательно гетероциклическое, 5- или 6-членное кольцо (например, фенил(ен), пиридил(ен) или циклогексил(ен)) либо состоять из него.
В предлагаемом в изобретении соединении формулы I три из четырех атомов азота макроциклического кольца замещены необязательно замещенными уксуснокислыми, соответственно метилкарбоксилатными остатками. Эти остатки способствуют координации, соответственно компенсации заряда координированного иона металла. Поэтому Z обозначает либо атом водорода, либо эквивалент иона металла.
Уксуснокислые, соответственно метилкарбоксилатные остатки у трех из атомов азота макроциклического кольца дополнительно могут быть замещены заместителем R. Помимо этого макроциклическое кольцо может быть замещено по четырем его атомам углерода дополнительным заместителем В. Характерная особенность предлагаемых в изобретении соединений заключается в том, что оба заместителя В и R одновременно не могут обозначать атомы водорода, т.е. макроциклическое кольцо должно быть замещено такими дополнительными заместителями непосредственно по его кольцевым атомам и/или по уксуснокислым, соответственно метилкарбоксилатным заместителям его атомов азота. За счет соответствующего выбора подобных дополнительных заместителей обеспечивается целенаправленное точное регулирование релаксационности полученного с применением предлагаемого в изобретении соединения контрастного вещества.
Заместитель В может представлять собой атом водорода или С1-С4алкильный остаток. Предпочтительными С1-С4алкильными остатками являются метил, этил и изопропил.
Если в предлагаемых в изобретении соединениях формулы I В представляет собой атом водорода, то R обозначает прямой, разветвленный и/или циклический, насыщенный либо ненасыщенный C1-С10алкильный (предпочтительно С5-С10алкильный) или арильный остаток, который необязательно замещен карбоксильной группой, группой -SO3Н или группой -РО3Н2, при этом алкильная цепь C1-С10алкильного остатка необязательно содержит арильную группу и/или 1-2 атома кислорода. В качестве алкильных остатков предпочтительны неразветвленные или разветвленные, предпочтительно насыщенные C1-С10- и прежде всего С1-С4алкильные остатки, такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил и трет-бутил, а также циклогексил. В другом варианте предпочтительны неразветвленные, разветвленные или циклические, предпочтительно насыщенные С5-С10алкильные остатки, такие как пентил, гексил, циклогексил, гептил, октил, нонил и децил. C1-С10алкильный остаток, значение которого может иметь заместитель R, необязательно может быть замещен карбоксильной группой, группой -SO3Н или группой -РО3Н2. В качестве предпочтительных примеров подобных замещенных алкильных групп можно назвать -СН2-СООН и -С(СН3)2-СООН. Помимо этого алкильная цепь C1-С10алкильного остатка может содержать арильную группу и/или 1-2 атома кислорода. Эти арильная группа и атомы кислорода могут находиться в любом положении в алкильной цепи. Арильная группа может, кроме того, находиться также в концевом положении в алкильной цепи и образовывать совместно с атомом кислорода арилоксигруппу. Наиболее предпочтительной арильной группой является прежде всего фенильная группа.
Предпочтительной алкильной цепью в качестве значения заместителя R, которая необязательно содержит арильную группу и 1-2 атома кислорода, является остаток формулы -(СН2)m-(O)n-(фенилен)р-Y, где m обозначает целое число от 1 до 5, n обозначает 0 или 1, p обозначает 0 или 1, a Y обозначает атом водорода, метоксиостаток, карбоксильную группу, -SO3Н или -РО3Н2. Заместитель Y находится при этом предпочтительно в пора-положении.
Арильный остаток в качестве значения заместителя R предпочтительно представляет собой фенильный остаток, который необязательно замещен карбоксильной группой, группой -SO3Н или группой -РО3Н2.
В том случае, когда В обозначает атом водорода, R предпочтительно представляет собой изопропил, изобутил, трет-бутил, неразветвленный либо разветвленный С5-С10алкильный остаток, циклогексил, -СН2-СООН, -С(СН3)2-СООН, фенильный остаток или остаток формулы -(CH2)m-(O)n-(фенилен)р-Y, где m обозначает целое число от 1 до 5, n обозначает 0 или 1, p обозначает 0 или 1, a Y обозначает атом водорода, метоксиостаток, карбоксильную группу, -SO3Н или -РО3Н2, наиболее предпочтительно R представляет собой изопропил, циклогексил или фенил.
Замещенное макроциклическое кольцо соединения формулы I может быть присоединено к биомолекуле посредством группы X, способной вступать в реакцию с биомолекулой, через спейсер А.
При этом такой спейсер А представляет собой прямую либо разветвленную, насыщенную либо ненасыщенную C1-С30углеводородную цепь, которая необязательно содержит 1-5 атомов кислорода, 1-5 атомов азота и/или 1-5 -NR'-остатков, где R' имеет указанные выше для R, но выбираемые независимо от него значения, и необязательно замещена 1-3 карбоксильными группами, 1-3 группами -SO3Н, 1-3 группами -РО3Н2 и/или 1-3 атомами галогена и в которой 1-3 атома углерода необязательно присутствуют в виде карбонильных групп, при этом такая цепь или ее часть может быть замкнута в кольцо, и которая далее имеет такую структуру, что Х по меньшей мере через 3 атома связан с атомом азота, к которому присоединен остаток А.
Подобный спейсер должен иметь по меньшей мере три, предпочтительно по меньшей мере четыре, атома, расположенных в цепь между атомом азота макроциклического кольца и заместителем X. Под такой цепью атомов при этом подразумевается также кратчайшее соединение между атомом азота макроциклического кольца и заместителем Х в том числе и через кольцо. При подобном трактовании в качестве спейсера с цепью из четырех атомов могла бы рассматриваться, например, пара-фениленовая группа, а в качестве спейсера с цепью из трех атомов - мета-фениленовая группа. При определении длины такой цепи атомов подсчитывается общее количество атомов вне зависимости от того, является ли каждый из них атомом углерода, азота или кислорода. Заместители у этих атомов или боковые цепи не относятся к числу образующих эту цепь атомов.
Для -А-Х предпочтительно выбирать отличное от заместителей -CH(R)-CO2Z значение.
Спейсер А предпочтительно представлять в виде остатка A'-U, где А' присоединен к атому азота макроциклического кольца, a U присоединен к X. При этом А' предпочтительно обозначает
а)связь,
б) группу -CH(СО2Н)-,
в) группу формулы

в которой Q обозначает атом водорода, C1-С10алкильный остаток, который необязательно замещен карбоксильной группой, или арильный остаток, который необязательно замещен карбоксильной группой, С1-С15алкоксигруппой, арилоксигруппой или атомом галогена, a R' имеет указанные для R, но выбираемые независимо от него значения, или
г) группу формулы
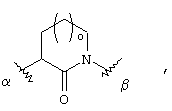
в которой о обозначает 0 или 1, а кольцо необязательно аннелировано с бензольным кольцом, которое при его наличии может быть замещено метоксигруппой, карбоксильной группой, группой -SO3Н или группой -РО3Н2.
В представленных выше в п.п. в) и г) группах их обозначенные символом  положения присоединены к смежным группам, при этом α-положение связано с атомом азота макроциклического кольца, а β-положение связано с U.
положения присоединены к смежным группам, при этом α-положение связано с атомом азота макроциклического кольца, а β-положение связано с U.
В группе формулы
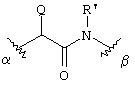
Q предпочтительно обозначает линейный либо разветвленный C1-С10-, прежде всего С1-С4алкильный остаток, такой как метил, этил или изопропил, или циклогексильный остаток. Эти остатки необязательно могут быть замещены карбоксильной группой, при этом предпочтителен карбоксиметильный остаток. Предпочтительным арильным остатком в качестве значения заместителя Q является фенил. Такой арильный остаток может быть замещен карбоксильной группой, С1-С15алкоксигруппой, арилоксигруппой, прежде всего феноксигруппой, или атомом галогена, например фтором, хлором, бромом или иодом, но прежде всего фтором или хлором. Если арильным остатком является фенильный остаток, то он предпочтительно замещен в пара-положении одной из указанных групп. Наиболее предпочтительными группами в качестве значений заместителя Q являются метил, фенил и n-додеканоксифенил.
R' имеет указанные выше для заместителя R значения, которые, однако, могут выбираться независимо от значений этого заместителя R. Наиболее предпочтительно R' обозначает атом водорода.
Предпочтительно А' обозначает связь, -СН(CO2Н)-, -С(СН3)Н-СО-NH-, -С(фенил)Н-СО-NH-, -С(n-додеканоксифенил)Н-СО-NH-,
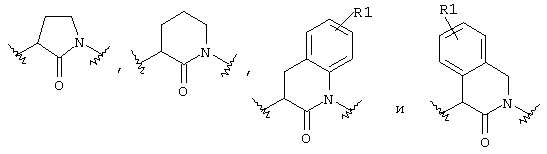
где R1 обозначает -ОСН3, -CO2H, -SO3H или -РО3Н2.
Если спейсер А представить в виде остатка A'-U, где А' имеет указанное выше значение, то U предпочтительно обозначает прямую либо разветвленную, насыщенную либо ненасыщенную C1-С30углеводородную цепь, которая необязательно содержит 1-3 атома кислорода, 1-3 атома азота и/или 1-3 -NR''-остатка, где R'' имеет указанные выше для R, но выбираемые независимо от него значения, и в которой 1-3 атома углерода необязательно присутствуют в виде карбонильных групп, при этом такая цепь или ее часть может быть замкнута в кольцо. Наиболее предпочтительно U обозначает арильный остаток или С1-С20алкильный остаток (предпочтительно линейный или по меньшей мере частично циклический и насыщенный), который необязательно содержит 1-3 атома кислорода, 1-3 -NR''-остатка, 1-2 фениленовых остатка и/или пиридиленовый остаток и в котором 1-3 атома углерода необязательно присутствуют в виде карбонильных групп и который далее необязательно замещен арильным остатком (например, фенилом). А' и U совместно должны образовывать такую структуру, чтобы Х по меньшей мере через три атома был связан с атомом азота, к которому присоединен А'. Такая цепь из по меньшей мере трех атомов рассмотрена выше при раскрытии значений заместителя А.
Предпочтительным арильным остатком в качестве значения заместителя U является фенильный остаток. Предпочтительным С1-С20алкильным остатком в качестве значения заместителя U является линейный, насыщенный C1-С10алкильный остаток, циклогексильный остаток или циклогексил-С1-С5алкильный остаток. Алкильные фрагменты в этих остатках необязательно могут быть прерваны 1 атомом кислорода, 1 фениленовым остатком и/или 1 пиридиленовым остатком или могут содержать -СО-NR''-остаток либо могут быть замещены фенилом. К предпочтительным значениям заместителя U относятся -СН2-, -(СН2)5-, -(СН2)10-, -фенилен-О-СН2-, -фенилен-O-(СН2)3, -фенилен-O-(СН2)10-, -СН2-фенилен-, -циклогексилен-О-СН2-, -фенилен-, -С(фенил)Н-, -СН2-пиридилен-О-СН2-, -СН2-пиридилен- и -СН2-СО-NH-СН2-СН2-. В указанных выше в качестве предпочтительных значений заместителя U группах фениленовые группы предпочтительно замещены в пара-положении, а пиридиленовые группы предпочтительно представляют собой пирид-2,5-иленовые или пирид-2,4-иленовые группы.
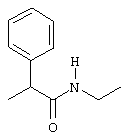
Предпочтительными группами в качестве значений спейсера А являются следующие:

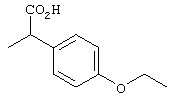
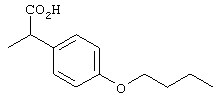

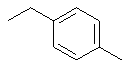



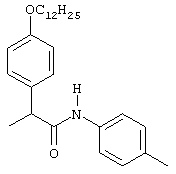







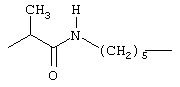

Через спейсер А в соединениях формулы I группа Х присоединена к макроциклическому кольцу. Под такой группой Х подразумевается группа, способная вступать в реакцию с биомолекулой. В качестве примеров подобной группы можно назвать карбоксил (-СООН), активированный карбоксил, аминогруппу (-NH2), изоцианат (-NCO), изотиоцианат (-NCS), гидразин (-NHNH2), семикарбазид (-NHCONHNH2), тиосемикарбазид (-NHCSNHNH2), хлорацетамид (-NHCOCH2Cl), бромацетамид (-NHCOCH2Br), иодацетамид (-NHCOCH2I), ациламиногруппу, такую как ацетиламиногруппа (-NHCOCH3), смешанные ангидриды, азид, гидроксид, сульфонилхлорид, карбодиимид или группу формулы

где Hal обозначает атом галогена.
Под активированной карбоксильной группой выше подразумеваются такие карбоксильные группы, которые дериватизированы таким образом, что они облегчают реакцию с биомолекулой. Конкретные группы, которые могут использоваться для подобного активирования, хорошо известны в данной области, и в этом отношении можно сослаться на публикацию М. и А.Bodanszky, "The Practice of Peptide Synthesis", изд-во Springer Verlag, 1984. В качестве примеров при этом можно назвать аддукты карбоновой кислоты с карбодиимидами или активированных сложных эфиров, таких как гидроксибензотриазоловый эфир. Наиболее предпочтительно активированную карбоксильную группу в качестве значения заместителя Х выбирать из


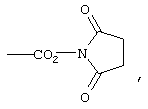
 и
и 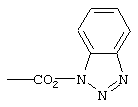
Z в формуле I обозначает атом водорода или эквивалент иона металла. Ион какого именно металла должен присутствовать в предлагаемом в изобретении соединении в образующем комплекс виде, зависит от предусматриваемого применения полученных с использованием предлагаемых в изобретении соединений конъюгатов с биомолекулой. Соответствующие конъюгаты могут использоваться, например, при ЯМР-диагностике, рентгенодиагностике и радиотерапии, а также при нейтронозахватной терапии. Наиболее предпочтительно использовать такие конъюгаты в качестве контрастных веществ при ЯМР-диагностике.
Комплексные соединения для ЯМР-диагностики можно получать методом, описанным в ЕР 71564, ЕР 130934 и DE-OS 3401052. С этой целью оксид или соль (например, хлорид, нитрат, ацетат, карбонат или сульфат) соответствующего металла растворяют или суспендируют в воде и/или низшем спирте (таком как метанол, этанол или изопропанол) и подвергают взаимодействию с раствором или суспензией эквивалентного количества предлагаемого в изобретении комплексообразователя.
Если такие комплексообразователи предусматривается использовать для получения рентгенодиагностических или радиотерапевтических средств, то комплексные соединения можно получать из комплексообразователей по методам, описанным в "Radiotracers for Medical Applications", т.I, изд-во CRC-Press, Boca Raton, Florida.
Предлагаемые в изобретении соединения могут использоваться в следующих целях:
1. для ЯМР-диагностики в виде их комплексов с ионами парамагнитных элементов с порядковыми номерами 21-29, 42, 44 и 58-70, при этом в качестве примера приемлемых ионов можно назвать ионы хрома(III), железа(II), кобальта(II), никеля(II), меди(II), празеодима(III), неодима(III), самария(III) и иттербия(III), а наиболее предпочтительны для ЯМР-диагностики благодаря их высокому магнитному моменту ионы гадолиния(III), тербия(III), диспрозия(III), гольмия(III), эрбия(III), марганца(II) и железа(III);
2. для рентгенодиагностики и радиотерапии в виде их комплексов с радиоизотопами элементов с порядковыми номерами 26, 27, 29, 31, 32, 37-39, 43, 46, 47, 49, 61, 62, 64, 67, 70, 71, 75, 77, 82 и 83.
Предлагаемые в изобретении соединения и прежде всего их конъюгаты с биомолекулами отвечают самым различным требованиям, которыми определяется их пригодность для применения в качестве контрастных веществ в ЯМР-томографии. Так, в частности, эти соединения, соответственно их конъюгаты с биомолекулами позволяют за счет увеличения интенсивности сигнала после их перорального или парентерального введения повысить информативность изображения, полученного с помощью ЯМР-томографа.
Помимо этого такие соединения, соответственно их конъюгаты с биомолекулами обладают высокой эффективностью, которая необходима для снижения концентрации вводимых в организм чужеродных веществ до минимально возможного уровня, и вместе с тем обладают хорошей переносимостью, которая необходима для сохранения неинвазивного характера исследований.
Благодаря хорошей растворимости предлагаемых в изобретении соединений и их конъюгатов с биомолекулами в воде и их малой осмомолярности появляется возможность получать на их основе высококонцентрированные растворы, что позволяет поддерживать объемную перегрузку системы кровообращения в допустимых пределах и компенсировать разбавление таких растворов жидкостями организма, т.е. водорастворимость средств для ЯМР-диагностики должна в 100-1000 раз превышать водорастворимость средств для ЯМР-спектроскопии. Предлагаемые в изобретении соединения обладают далее не только высокой стабильностью in vitro, но и проявляют неожиданно высокую стабильность in vivo, благодаря чему высвобождение или обмен не ковалентно связанных в комплексах ионов, которые по своей природе являются токсичными, происходит лишь крайне медленно в течение промежутка времени, за который новые контрастные вещества полностью выводятся из организма.
Предлагаемые в изобретении комплексные соединения могут, кроме того, эффективно применяться в качестве реагентов с магнитной восприимчивостью и в качестве реагентов сдвига в ЯМР-спектроскопии in vivo.
Предлагаемые в изобретении соединения и их конъюгаты с биомолекулами благодаря их оптимальным радиоактивным свойствам и высокой стабильности содержащихся в них комплексных соединений пригодны также для применения в качестве рентгенодиагностических и радиотерапевтических средств. Более подробно применение таких средств и их дозировка описаны, например, в публикации "Radiotracers for Medical Applications", изд-во CRC-Press, Boca Raton, Florida, 1983, а также в Eur. J. Nucl. Med. 17, 1990, cc.346-364, и в Chem. Rev.93, 1993, cc.1137-1156.
Для применения при однофотонной эмиссионной компьютерной томографии (ОФЭКТ) пригодны комплексы с изотопами 111In и 99mTc.
В качестве примера другого метода визуализации, основанного на применении радиоизотопов, можно назвать позитронно-эмиссионную томографию, где используются испускающие протоны изотопы, такие, например, как 43Sc, 44Sc, 52Fe, 55Co, 68Ga, 64Cu, 86Y и 94mTc (W.D.Heiss и М.Е.Phelps, Positron Emission Tomography of Brain, изд-во Springer Verlag, Berlin, Heidelberg, New York, 1983).
Предлагаемые в изобретении соединения и их конъюгаты с биомолекулами могут, как неожиданно было установлено, применяться также для дифференциации злокачественных и доброкачественных опухолей в областях без гематоэнцефалического барьера.
Предлагаемые в изобретении соединения и их конъюгаты с биомолекулами отличаются также тем, что они полностью выводятся из организма, проявляя тем самым хорошую переносимость.
Поскольку предлагаемые в изобретении соединения и прежде всего их конъюгаты с биомолекулами накапливаются в злокачественных опухолях (отсутствие диффузии в здоровые ткани, но высокая проницаемость через опухолевые сосуды), их можно также применять в качестве вспомогательных средств при лучевой терапии злокачественных опухолей. Отличие лучевой терапии от соответствующей диагностики состоит лишь в количестве и типе используемого изотопа. Целью при этом является разрушение опухолевых клеток под воздействием высокоэнергетического (мощного) коротковолнового излучения с предельно малым радиусом действия. При этом используется взаимодействие содержащихся в комплексных соединениях металлов (таких, например, как железо или гадолиний) с ионизирующим излучением (например, с рентгеновскими лучами) или с нейтронным излучением. Благодаря этому эффекту удается значительно повысить локальную дозу облучения в том месте, где находится комплекс металла (например, в опухолях). Для обеспечения такой же дозы облучения в злокачественной ткани применение подобных комплексов металлов позволяет существенно снизить дозу облучения здоровых тканей и предотвратить тем самым нежелательные для пациентов побочные действия. Поэтому предлагаемые в изобретении конъюгаты с комплексами металлов пригодны также для применения в качестве радиосенсибилизирующих веществ при лучевой терапии злокачественных опухолей (например, за счет использования эффектов Мессбауэра или при нейтронозахватной терапии). В качестве примера приемлемых испускающих β-излучение ионов можно назвать 46Sc, 47Sc, 48Sc, 72Ga, 73Ga, 90Y, 67Cu, 109Pd, 111Ag, 149Pm, 153Sm, 166Ho, 177Lu, 186Re и 188Re. Предпочтительны при этом ионы 90Y, 177Lu, 72Ga, 153Sm и 67Cu. В качестве приемлемых испускающих α-излучение ионов с малым периодом полураспада можно назвать, например, 211At, 211Bi, 212Bi, 213Bi и 214Bi, предпочтителен из которых 212Bi. Приемлемым испускающим фотоны и электроны ионом является 158Gd, который можно получить из 157Gd путем захвата нейтронов.
Если предлагаемое в изобретении соединение или его конъюгат с биомолекулой предназначены для применения при лучевой терапии в соответствии с методикой, предложенной R.L.Mills и др. (Nature, т.336, 1988, с.787), то центральный ион должен быть производным мессбауэровского изотопа, такого, например, как 57Fe или 151Eu.
Возможно еще присутствующие свободные карбоксигруппы нейтрализуют с помощью неорганических оснований, например гидроксидов, карбонатов или бикарбонатов натрия, калия, лития, магния или кальция, и/или органических оснований, таких, в частности, как первичные, вторичные и третичные амины, например этаноламин, морфолин, глюкамин, N-метил- и N,N-диметилглюкамин, а также основных аминокислот, таких, например, как лизин, аргинин и орнитин, либо амидов исходно нейтральных или кислых аминокислот.
Для получения нейтральных комплексных соединений требуемое основание можно, например, добавлять к кислым солям комплексов в водном растворе или суспензии в таком количестве, при котором достигается точка нейтральности. Полученный раствор можно затем досуха концентрировать в вакууме. Образовавшиеся нормальные соли часто предпочтительно осаждать добавлением смешивающихся с водой растворителей, таких, например, как низшие спирты (метанол, этанол, изопропанол и другие), низшие кетоны (ацетон и другие), полярные простые эфиры (тетрагидрофуран, диоксан, 1,2-диметоксиэтан и другие), с получением таким путем легко выделяемых и хорошо поддающихся очистке кристаллизатов. Было установлено, что соответствующее основание предпочтительно добавлять к реакционной смеси уже в процессе комплексообразования, что позволяет на одну сократить количество стадий в способе получения предлагаемых в изобретении соединений.
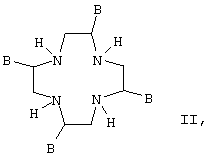
Предлагаемые в изобретении соединения формулы I можно получать известными методами. Так, например, соединения формулы I можно получать способом, в соответствии с которым соединение формулы II
в которой В имеет указанные выше значения, необязательно после введения защитных групп для атомов азота, подвергают взаимодействию с молекулами Nu-A-X' и Nu-CH(R)-CO2Z', где А и R имеют указанные выше значения, Nu обозначает нуклеофоб, X' соответствует заместителю Х или защищенной форме заместителя X, который имеет указанные выше значения, a Z' обозначает атом водорода, эквивалент иона металла, предпочтительно щелочного или щелочноземельного металла, прежде всего натрия или калия, или защитную группу для карбоксила. После этого из полученного соединения можно удалить возможно присутствующие в нем защитные группы и затем по известному методу подвергнуть его взаимодействию по меньшей мере с одним оксидом или по меньшей мере с одной солью требуемого металла. В последующем в полученных таким путем комплексах еще присутствующие в них кислые атомы водорода можно при необходимости полностью или частично заместить на катионы неорганических и/или органических оснований, аминокислот или амидов аминокислот.
Ниже более подробно рассмотрены три предпочтительных варианта способа получения предлагаемых в изобретении соединений.
В первом варианте сначала незамещенный по атомам азота макроцикл подвергают взаимодействию с защищенным фрагментом А-Х'. При этом группа А несет нуклеофоб в качестве уходящей группы. Благодаря стехиометрическому контролю реакции один из четырех атомов азота в макроцикле реагирует с группой А с отщеплением уходящей группы. Таким путем получают монофункционализованный макроцикл, содержащий остаток Х в защищенной форме (X'). На второй реакционной стадии каждый из трех оставшихся нуклеофильных атомов азота макроцикла подвергают взаимодействию с защищенной карбоновой кислотой, которая в α-положении по отношению к карбоксильной группе несет нуклеофоб. После отщепления защитных групп от функциональных карбоксильных групп добавляют оксид металла или соль металла с получением комплекса из парамагнитного иона металла и хелатного лиганда. Ниже этот вариант способа проиллюстрирован на реакционной схеме, на которой остатки в формулах имеют указанные выше значения:
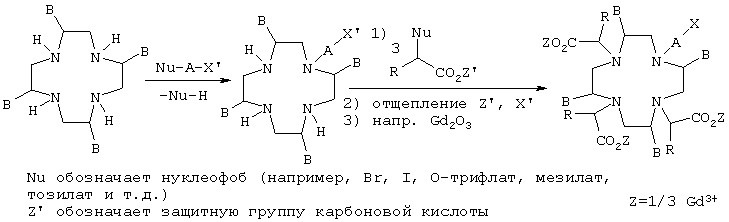
Во втором варианте в качестве эдукта используют макроцикл, который уже несет на трех из четырех его атомов азота соответствующие защитные группы SG. Такими защитными группами в этом случае могут служить, например, трет-бутилоксикарбонил (t-ВОС), COCF3, карбобензоксигруппа (Cbo) или флуоренилметоксикарбонил (FMOC) и т.д. При наличии подобных защитных групп нуклеофильным остается только один из четырех атомов азота, который может реагировать с молекулой А-Х', которая аналогично описанному выше варианту несет нуклеофоб Nu. После соединения обеих молекул с удалением уходящей группы отщепляют три защитных группы от атомов азота. За этой стадией следует дериватизация с помощью производных карбоновых кислот, уже описанная выше для предыдущего варианта. Ниже этот второй вариант способа проиллюстрирован на реакционной схеме, на которой остатки в формулах имеют указанные выше значения:
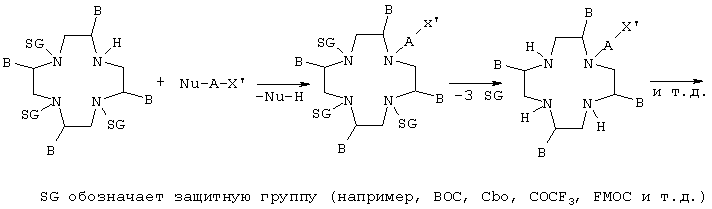
В третьем варианте сначала один из четырех атомов азота макроцикла блокируют соответствующей защитной группой SG. В качестве примера пригодных для применения в этих целях защитных групп можно назвать формил, бензил, Вос-тритил и т.д. После этого проводят взаимодействие по трем оставшимся нуклеофильным атомам азота с соответствующим образом защищенными производными карбоновых кислот, несущими в α-положении соответствующий нуклеофоб. Затем отщепляют введенную на первой стадии защитную группу SG у первого атома азота и дериватизируют молекулой А-Х', которая в свою очередь также несет нуклеофоб. Ниже этот третий вариант способа проиллюстрирован на реакционной схеме, на которой остатки в формулах имеют указанные выше значения:

Нуклеофобом предпочтительно служат следующие остатки: Cl, Br, I, O-трифлат, мезилат и тозилат.
Реакцию проводят в смеси воды и органических растворителей, таких как изопропанол, этанол, метанол, бутанол, диоксан, тетрагидрофуран, диметилформамид, диметилацетамид, формамид или дихлорметан. Предпочтительны при этом тройные смеси из воды, изопропанола и дихлорметана.
Реакцию проводят при температуре в интервале от -10 до 100°С, предпочтительно от 0 до 30°С.
Указанные выше группы можно защищать различными, известными специалисту методами. Использование подобных защитных групп поясняется ниже на примере некоторых вариантов, которыми, однако, не ограничиваются возможные методы синтеза предлагаемых в изобретении соединений.
В качестве кислотозащитных групп могут использоваться C1-С6алкильные, С6-С10арильные и С6-С10-Ar(С1-С4)алкильные группы, а также триалкилсилильные группы. Предпочтительны при этом метильная, этильная, пропильная, изопропильная, н-бутильная, изобутильная и трет-бутильная группы.
Такие кислотозащитные группы отщепляют известными специалистам методами, например путем гидролиза, гидрогенолиза, щелочного омыления сложных эфиров щелочами в водно-спиртовом растворе при температуре от 0 до 50°С, кислотного омыления минеральными кислотами либо в случае сложных трет-бутиловых эфиров - с помощью трифторуксусной кислоты.
Для введения и последующего удаления NH-защитных групп могут использоваться самые разнообразные подходы. Так, в частности, N-трифторацетильное производное отщепляют карбонатом калия или натрия в воде (Н.Newman, J. Org. Chem., 30, 1965, с.287; M.A.Schwartz и др., J. Am. Chem. Soc., 95 G12, 1973) либо в простейшем случае отщепляют обработкой раствором аммиака (М.Imazama и F.Eckstein, J. Org. Chem. 44, 1979, с.2039). Равным образом в мягких условиях следует отщеплять трет-бутилоксикарбонильное производное, для чего вполне достаточно перемешивания с трифторуксусной кислотой (B.F.Lundt и др., J. Org. Chem. 43, 1978, с.2285). Большое число NH-защитных групп можно отщеплять путем гидрогенолиза или восстановления. Так, например, N-бензильную группу целесообразно отщеплять под действием водорода в присутствии палладия на угле (W.H.Hartung и R.Rimonoff, Org. Reactions VII, 1953, с.262), что в равной степени относится также к тритильной группе (L.Zervas и др., J. Am. Chem. Soc. 78, 1956, с.1359) и к бензилоксикарбонильной группе (М.Bergmann и L.Zervas, Ber. 65, 1932, с.1192).
Активированные сложные эфиры вышеописанных соединений получают известными специалистам методами. В случае изотиоцианатов или α-галогенацетатов соответствующие концевые аминовые предшественники по известным из литературы методам подвергают взаимодействию с тиофосгеном или галогенидами 2-галоуксусной кислоты. Возможно также взаимодействие с соответствующим образом дериватизированными сложными эфирами N-гидроксисукцинимида, как, например,

(где Hal обозначает галоген).
В целом для этой цели могут использоваться все обычные методы активирования карбоновых кислот, известные из уровня техники. Молекулу Nu-A-X' предпочтительно синтезировать независимо на начальной стадии. Такую молекулу, когда она содержит амидную группу, получают, например, взаимодействием активированной карбоновой кислоты с амином. При этом карбоновую кислоту активируют традиционными методами. В качестве примера приемлемых активирующих агентов можно назвать дициклогексилкарбодиимид (ДЦК), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (ЭДК), гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (БОФ) и гексафторфосфат O-(бензотриазол-1-ил)-1,1,3,3-тетраметилурония (ГБТУ), предпочтителен среди которых ДЦК. Помимо этого можно также добавлять O-нуклеофильные катализаторы, такие, например, как N-гидроксисукцинимид (N-ГС) или N-гидроксибензотриазол.
Если группа Х представляет собой функциональную карбоксильную группу, то ее можно использовать в защищенной форме (например, в виде сложного бензилового эфира), и в этом случае отщеплять защитную группу в последующем можно путем гидрогенолиза.
Такую функциональную карбоксильную группу для ее присоединения к соответствующей функциональной группе соответствующей биомолекулы сначала, как правило, необходимо активировать. С этой целью предпочтительно получать промежуточные активированные сложные эфиры, которые затем атакуются нуклеофильной группой биомолекулы. В результате между биомолекулой и предлагаемым в изобретении соединением формулы I образуется ковалентная связь. К числу предпочтительных активированных сложных эфиров относятся сложные эфиры N-гидроксисукцинимида, сложные эфиры паранитрофенола или сложные эфиры пентафторфенола. Если группа Х должна присоединяться к биомолекуле в виде изотиоцианата, то предпочтительно сначала использовать концевой амин, который при необходимости можно защитить приемлемой защитной группой. Пригодные для применения в этих целях защитные группы известны из химии пептидов. После отщепления защитной группы взаимодействием первичного концевого амина с тиофосгеном можно образовать изотиоцианат. К нему затем можно присоединить нуклеофильные группы биомолекулы.
В одном из вариантов группа Х может представлять собой имид малеиновой кислоты, который, например, может избирательно реагировать с функциональными тиольными группами биомолекулы.
В другом варианте группой Х является нуклеофил (NH2, SH), который атакует соответствующую функциональную группу биомолекулы (активированный сложный эфир, имид малеиновой кислоты и т.д.). Самые разнообразные функционализованные имидами малеиновой кислоты биомолекулы являются коммерчески доступными продуктами.
Настоящее изобретение относится, кроме того, к применению описанных выше соединений формулы I для получения конъюгатов с биомолекулой.
Процесс синтеза конъюгатов обычно состоит в первоначальном получении дериватизированного и функционализованного хелатного комплекса, который затем присоединяют к биомолекуле. Однако в другом варианте при использовании синтезированных биомолекул предлагаемый в изобретении хелатный комплекс можно также встраивать в биомолекулу в процессе ее синтеза. Так, например, подобное встраивание хелатного комплекса может происходить в ходе последовательного синтеза олигопептидов в роботизированном синтезаторе. В дополнение к этому в предлагаемое в изобретении соединение при необходимости можно вводить обычно применяемые при синтезе соответствующей биомолекулы защитные группы. В последующем эти защитные группы вновь отщепляют в ходе выполнения обычных алгоритмов синтеза в синтезаторе.
Под "биомолекулой" в контексте настоящего изобретения подразумевается любая молекула, которая встречается в естественных условиях, например, в организме, либо которую можно получить путем синтеза со структурой, которая аналогична структуре встречающейся в естественных условиях молекулы. Под это же определение подпадают, кроме того, и те молекулы, которые способны взаимодействовать с биологической, например встречающейся в организме, молекулой либо со встречающейся в нем структурой, что приводит к накоплению, например, конъюгатов в определенных местах организма, где требуется обеспечить их присутствие. Под "организмом" в контексте настоящего изобретения подразумевается любой растительный организм или организм животного, предпочтительно организм животного и прежде всего человека.
К биомолекулам относятся прежде всего встречающиеся в живых организмах молекулы, которые в качестве продуктов эволюционного отбора выполняют в организме за счет упорядоченных и сложных взаимодействий особые задачи и образуют основу его жизненных, соответственно биологических функций (обмен веществ, смена форм, размножение, поддержание энергетического баланса). Обычно в биомолекулах простые структурные элементы (аминокислоты, нуклеиновые основания, моносахариды, жирные кислоты и т.д.) образуют более крупные молекулы (белки, нуклеиновые кислоты, полисахариды, липиды и т.д.). Соответствующие макромолекулы называют также биополимерами.
В предпочтительном варианте биомолекула может иметь, например, полипептидный скелет из аминокислот с боковыми цепями, которые могут вступать в реакцию с реакционноспособной группой Х предлагаемых в изобретении соединений формулы I. Такими боковыми цепями являются например, карбоксильные группы остатков аспарагиновой кислоты и глутаминовой кислоты, аминогруппы остатков лизина, ароматические группы остатков тирозина и гистидина и сульфгидрильные группы остатков цистеина.
Обзорную информацию о биомолекулах с самыми разнообразными их примерами можно найти в работе "Chemie der Biomoleküle", которая издана при Техническом университете г.Грац (Н Berthold и др., Институт органической химии (Institut für Organische Chemie), Технический университет г.Грац (TU-Graz), 2001) и с которой можно также ознакомиться в сети Интернет на сайте по адресу www.orgc.tu-graz.ac.at. Содержание этой публикации включено в настоящее описание в качестве ссылки.
Для образования конъюгатов с предлагаемыми в изобретении соединениями наиболее пригодны следующие биомолекулы: биополимеры, белки, такие как белки, выполняющие биологическую функцию, сывороточный белок человека (СБЧ), бычий сывороточный альбумин (БСА) и т.д., белки и пептиды, накапливающиеся в определенных местах в организме (например, на рецепторах, на клеточных мембранах, в каналах и т.д.), расщепляемые протеазами пептиды, пептиды с искусственными точками запрограммированного разрыва (например, лабильные сложные эфиры, амиды и т.д.), пептиды, расщепляемые под действием металлопротеаз, пептиды с фоторасщепляемыми линкерами, пептиды с отщепляемыми под действием окислителей (оксидаз) группами, пептиды с природными и не встречающимися в естественных условиях аминокислотами, гликопротеиды (гликопептиды), сигнальные белки, антивирусные белки и апоктоз, синтетически модифицированные биополимеры, такие как дериватизированные линкерами биополимеры, модифицированные металлопротеазы, дериватизированная оксидаза и т.д., углеводы (моно- и полисахариды), такие как дериватизированные сахара, расщепляемые в организме сахара, циклодекстрин и его производные, аминосахара, хитозан, полисульфаты и производные сиаловой кислоты, антитела, такие как моноклональные антитела, фрагменты антител, поликлональные антитела, "миниантитела", одиночные цепи (в том числе и такие, которые соединены линкерами в многократно повторяющиеся фрагменты), эритроциты и другие компоненты крови, раковые маркеры (например, САА) и вещества клеточной адгезии (например, антиген Льюис Х и антитела к видоизмененному антигену Льюис X), фрагменты ДНК и РНК, такие как дериватизированные ДНК и РНК (например, ДНК и РНК, выявленные методом SELEX), синтетические РНК и ДНК (в том числе и с не встречающимися в естественных условиях основаниями), агглютинины земляного ореха (PNA) (фирма Hoechst) и антисмысловые (олигонуклеотидные) последовательности, β-аминокислоты (фирма Seebach), векторные амины для встраивания в клетку, биогенные амины, фармацевтические препараты, онкологические препараты, синтетические полимеры, направленные на биологическую мишень (например, рецептор), стероиды (природные и модифицированные), простагландины, таксол и его производные, эндотелины, алкалоиды, фолиевая кислота и ее производные, биоактивные липиды, жиры, эфиры жирных кислот, синтетически модифицированные моно-, ди- и триглицериды, липосомы, дериватизированные на поверхности, мицеллы из природных жирных кислот или из перфторалкильных соединений, порфирины, тексафрины, расширенные порфирины, цитохромы, ингибиторы, нейрамидазы, нейропептиды, иммуномодуляторы, такие как FK 506, САРЕ и глиотоксин, эндогликозидазы, субстраты, активируемые ферментами, такими как калмодулин-киназа, казеин-киназа II, глютатион-3-трансфераза, гепариназа, матриксные металлопротеазы, инсулин-β-рецепторная киназа, UDPглюкоза-4-эпимераза, фукозидазы, G-белки, галактозидазы, гликозидазы, гликозилтрансферазы и ксилозидаза, антибиотики, витамины и их аналоги, гормоны, ДНК-интеркаляторы, нуклеозиды, нуклеотиды, лектины, витамин В12, антиген Льюис Х и родственные ему антигены, псорален, диентриеновые антибиотики, карбациклин, VEGF-фактор (васкулярный эндотелиальный фактор роста), соматостатин и его производные, производные биотина, антигормоны, опухолеспецифичные белки и синтетические материалы, полимеры, накапливающиеся в областях организма с кислой или щелочной средой (рН-регулируемое распределение), миоглобины, апомиоглобины и т.д., пептиды-нейромедиаторы, факторы некроза опухоли, пептиды, накапливающиеся в воспаленной ткани, реагенты на белки плазмы крови, белки-переносчики анионов и катионов, сложные полиэфиры (например, молочной кислоты), полиамиды и полифосфаты.
Большинство из перечисленных выше биомолекул являются коммерчески доступными продуктами, выпускаемыми, например, фирмами Merck, Alderich, Sigma, Calibochem или Bachem.
Помимо этого к биомелекулам можно также отнести и использовать в предусмотренных изобретением целях все описанные в WO 96/23526 и WO 01/08712 "связывающиеся с белками плазмы группы", соответственно "связывающиеся с мишенью группы". Содержание обеих этих публикаций тем самым включено в настоящее описание в качестве ссылки.
Количество предлагаемых в изобретении соединений формулы I из расчета на одну биомолекулу в принципе может быть любым, предпочтительно, однако, молекулярное соотношение от 0,1:1 до 10:1, прежде всего от 0,5:1 до 7:1.
Помимо этого предлагаемые в изобретении соединения пригодны для конъюгации со всеми теми молекулами, которые в соответствии с уровнем техники подвергают взаимодействию с флуоресцентными красителями, например, для определения их местоположения в клетке с помощью эпифлуоресцентной микроскопии. Предлагаемые в изобретении соединения можно также конъюгировать в принципе с любыми медикаментами, что позволяет после введения медикамента отслеживать его перемещение в организме ЯМР-методами. Помимо этого конъюгаты из предлагаемых в изобретении соединений и биомолекул могут дополнительно содержать другие молекулы, конъюгированные с биомолекулами. Таким образом, в понятие "биомолекула" в контексте настоящего изобретения включены все молекулы, которые встречаются в биологических системах, и все молекулы, которые являются биосовместимыми.
Получаемые с использованием предлагаемых в изобретении соединений конъюгаты предпочтительно использовать в качестве контрастных веществ при ЯМР-диагностике. Поэтому такие конъюгаты должны быть водорастворимыми. Если получаемые с использованием предлагаемых в изобретении соединений конъюгаты предназначены для применения в качестве контрастных веществ при ЯМР-исследованиях, то их предпочтительно вводить в организм в дозе, составляющей от 0,0001 до 5 ммолей/кг веса тела, наиболее предпочтительно от 0,005 до 0,5 ммоля/кг веса тела. Более детально применение подобного рода контрастных веществ рассматривается, например, у H.-J.Weinmann и др., Am. J. of Roentgenology 142, 1984, с.619. Благодаря неожиданно высокой релаксационности предлагаемых в изобретении соединений при одновременно высокой их специфичности в отношении мишени полученные с использованием этих соединений конъюгаты можно использовать в исключительно низкой дозировке, например для обнаружения опухолей.
Подробно применение радиотерапевтических средств описано, например, у R.W.Kozak и др., TIBTEC, октябрь 1986, с.262 (см. также Bioconjugate Chem. 12, 2001, cc.7-34).
Ниже настоящее изобретение более подробно поясняется на примерах, которые, однако, не ограничивают его объем.
Примеры
Пример 1
а) 10-[4-(бензилоксикарбонил)-1-метил-2-оксо-3-азабутил]-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 27,9 г (162,2 ммоля) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 25 г (81,1 ммоля) бензилового эфира 2-бромпропионилглицина (пример 1е в WO 98/24774) и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: хлороформ/метанол/25%-ный водный аммиак в соотношении 10:5:1).
Полученный таким путем 1-[4-(бензилоксикарбонил)-1-метил-2-оксо-3-азабутил]-1,4,7,10-тетраазациклододекан (19,6 г, 50 ммолей, 62% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull., 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 32,0 г (73% от теории) бесцветного кристаллического порошка.
б) 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
26,3 г (30 ммолей) соединения, указанного в заголовке примера 1а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 15,7 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
10,4 г (20 ммолей) описанного в примере 1б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 10,1 г (69% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,3%.
Пример 2
а) 10-[4-(бензилоксикарбонил)-1-метил-2-оксо-3-азабутил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
19,6 г (50 ммолей) описанного в примере 1а в качестве промежуточного продукта 1-[4-(бензилоксикарбонил)-1-метил-2-оксо-3-азабутил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 68,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)изовалериановой кислоты (Walker и др., Tetrahedron 53(43), 1997, с.14591) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 33,7 г (70% от теории) бесцветного кристаллического порошка.
б) 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
28,9 г (30 ммолей) соединения, указанного в заголовке примера 2а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 18,0 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
12,0 г (20 ммолей) описанного в примере 2б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 12,0 г (72% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 9,1%.
Пример 3
а) 10-[4-(бензилоксикарбонил)-1-метил-2-оксо-3-азабутил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
19,6 г (50 ммолей) описанного в примере 1а в качестве промежуточного продукта 1-[4-(бензилоксикарбонил)-1-метил-2-оксо-3-азабутил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 76,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)-2-циклогексилуксусной кислоты (Qabar и др., Tetrahedron Letters 39(33), 1998, с.5895) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 41,1 г (76% от теории) бесцветного кристаллического порошка.
б) 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
32,5 г (30 ммолей) соединения, указанного в заголовке примера 3а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 22,0 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
14,4 г (20 ммолей) описанного в примере 3б лиганда растворяют в 150 мл воды и 150 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 8 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и упаривают досуха. Остаток растворяют в муравьиной кислоте и затем при добавлении дихлорметана многократно упаривают досуха, после чего сушат в вакууме до постоянства массы.
Выход: 12,4 г (65% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,0%.
Пример 4
а) 10-[4-(трет-бутоксикарбонил)-1-фенил-2-оксо-3-азабутил]-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 27,9 г (162,2 ммоля) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 26,6 г (81,1 ммоля) трет-бутилового эфира N-[2-бром-2-фенилацетил] глицина (пример 6а в WO 98/24775) и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: хлороформ/метанол/25%-ный водный аммиак в соотношении 10:5:1). Полученный таким путем 1-[4-(трет-бутоксикарбонил)-1-фенил-2-оксо-3-азабутил]-1,4,7,10-тетраазациклододекан (21,0 г, 50 ммолей, 62% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull. 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 34,0 г (75% от теории) бесцветного кристаллического порошка.
б) 10-(4-(трет-бутоксикарбонил-1-фенил-2-оксо-3-азабутил)-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
27,2 г (30 ммолей) соединения, указанного в заголовке примера 4а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 17,5 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-(4-карбокси-1-фенил-2-оксо-3-азабутил)-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
11,6 г (20 ммолей) описанного в примере 4б трет-бутилового эфира растворяют в минимальном количестве трифторуксусной кислоты и перемешивают в течение 15 мин при комнатной температуре. После добавления 250 мл диэтилового эфира реакционную смесь перемешивают в течение 2 ч, после чего осадок отделяют вакуум-фильтрацией и сушат в вакууме.
Полученный таким путем свободный лиганд растворяют в 200 мл воды и 80 мл изопропанола, значение рН с помощью разбавленного аммиака устанавливают на 7 и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 11,6 г (72% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 9,0%.
Пример 5
а) Метиловый эфир 4-(этоксикарбонилметокси)фенилуксусной кислоты
10 г (60,2 ммоля) метилового эфира гидроксифенилуксусной кислоты (фирма Aldrich) растворяют в 75 мл ацетона. Далее добавляют 18,4 г (133 ммоля) твердого карбоната калия, в течение 15 мин при нагревании с обратным холодильником по каплям добавляют 17,8 мл (123 ммоля) этилового эфира бромуксусной кислоты, выдерживают при этой же температуре в течение последующих 4 ч и перемешивают в течение ночи при комнатной температуре. После этого осадок отфильтровывают, раствор упаривают досуха и хроматографируют на силикагеле (гексан/этилацетат в соотношении 3:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 14,6 г (96% от теории).
б) Метиловый эфир α-бром-4-(этоксикарбонилметокси)фенилуксусной кислоты
13,5 г (53,5 ммоля) соединения, указанного в заголовке примера 5а, растворяют в 75 мл четыреххлористого углерода. Далее добавляют 9,52 г (53,5 ммоля) N-бромсукцинимида и 48 мг дибензоилпероксида, в течение 5 ч кипятят с обратным холодильником и перемешивают в течение ночи при комнатной температуре. Суспензию дважды промывают раствором гидрокарбоната натрия и однократно водой, органическую фазу сушат сульфатом магния, отфильтровывают осушитель и фильтрат досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (гексан/этилацетат в соотношении 3:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 15,4 г (87% от теории).
в) 10-[α-(4-(этоксикарбонилметокси)фенил)метоксикарбонилметил]-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 27,9 г (162,2 ммоля) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 26,9 г (81,1 ммоля) описанного в предыдущем примере 5б бромзамещенного соединения и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол/триэтиламин в соотношении 10:5:0,1). Полученный таким путем 1-[α-(4-(этоксикарбонилметокси)фенил)метоксикарбонилметил]-1,4,7,10-тетраазациклододекан (21,1 г, 50 ммолей, 62% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull. 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 34,1 г (75% от теории) бесцветного кристаллического порошка.
г) 10-[α-(4-(этоксикарбонилметокси)фенил)метоксикарбонилметил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
27,3 г (30 ммолей) соединения, указанного в заголовке примера 5в, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 19,3 г (количественный) бесцветного порошка.
д) Gd-комплекс 10-[α-(4-карбоксиметоксифенил)карбоксиметил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
13,3 г (20 ммолей) соединения, указанного в заголовке примера 5г, растворяют в 250 мл 2 н. раствора едкого натра и 250 мл тетрагидрофурана и перемешивают в течение 5 дней при 40°С. После этого значение pH водной фазы устанавливают на 7 с помощью Amberlite IR-120® (Н+-форма), добавляют 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение pH с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (H+-форма). Кислый элюат сушат вымораживанием.
Выход: 8,6 г (61% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 9,3%.
Пример 6
а) Метиловый эфир 4-(этоксикарбонилпропокси)фенилуксусной кислоты
10 г (60,2 ммоля) метилового эфира гидроксифенилуксусной кислоты (фирма Aldrich) растворяют в 75 мл ацетона. Далее добавляют 18,4 г (133 ммоля) твердого карбоната калия, в течение 15 мин при нагревании с обратным холодильником по каплям добавляют 17,8 мл (123 ммоля) этилового эфира 4-броммасляной кислоты, выдерживают при этой же температуре в течение последующих 4 ч и перемешивают в течение ночи при комнатной температуре. После этого осадок отфильтровывают, раствор упаривают досуха и хроматографируют на силикагеле (гексан/этилацетат в соотношении 3:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 16,4 г (97% от теории).
б) Метиловый эфир α-бром-[4-(этоксикарбонилпропокси)фенил]уксусной кислоты
15,0 г (53,5 ммоля) соединения, указанного в заголовке примера 6а, растворяют в 75 мл четыреххлористого углерода. Далее добавляют 9,52 г (53,5 ммоля) N-бромсукцинимида и 48 мг дибензоилпероксида, в течение 5 ч кипятят с обратным холодильником и перемешивают в течение ночи при комнатной температуре. Суспензию дважды промывают раствором гидрокарбоната натрия и однократно водой, органическую фазу сушат сульфатом магния, отфильтровывают осушитель и фильтрат досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (гексан/этилацетат в соотношении 3:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 15,9 г (83% от теории).
в) 10-[α-(4-(этоксикарбонилпропокси)фенил)метоксикарбонилметил]-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 27,9 г (162,2 ммоля) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 29,1 г (81,1 ммоля) описанного в предыдущем примере 6б бромзамещенного соединения и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол/триэтиламин в соотношении 10:5:0,1). Полученный таким путем 1-[α-(4-(этоксикарбонилпропокси)фенил)метоксикарбонилметил]-1,4,7,10-тетраазациклододекан (22,5 г, 50 ммолей, 62% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull. 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 30,5 г (65% от теории) бесцветного кристаллического порошка.
г) 10-[α-(4-(этоксикарбонилпропокси)фенил)метоксикарбонилметил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
28,1 г (30 ммолей) соединения, указанного в заголовке примера 6в, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 20,0 г (количественный) бесцветного порошка.
д) Gd-комплекс 10-[α-(4-карбоксипропоксифенил)карбоксиметил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
13,3 г (20 ммолей) соединения, указанного в заголовке примера 6г, растворяют в 250 мл 2 н. раствора едкого натра и 250 мл тетрагидрофурана и перемешивают в течение 5 дней при 40°С. После этого значение рН водной фазы устанавливают на 7 с помощью Amberlite IR-120® (Н+-форма), добавляют 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 9,3 г (55% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,0%.
Пример 7
а) Метиловый эфир 4-(этоксикарбонилдецилокси)фенилуксусной кислоты
10 г (60,2 ммоля) метилового эфира гидроксифенилуксусной кислоты (фирма Aldrich) растворяют в 75 мл ацетона. Далее добавляют 18,4 г (133 ммоля) твердого карбоната калия, после чего по каплям добавляют 36,1 г (123 ммоля) этилового эфира ω-бромундекановой кислоты в 50 мл ацетона, в течение 8 ч кипятят с обратным холодильником и перемешивают в течение ночи при комнатной температуре. Нерастворившиеся компоненты отфильтровывают, раствор упаривают досуха и хроматографируют на силикагеле (гексан/этилацетат в соотношении 3:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 20,3 г (89% от теории).
б) Метиловый эфир α-бром[4-(этоксикарбонилдецилокси)фенил]уксусной кислоты
20,2 г (53,5 ммоля) соединения, указанного в заголовке примера 7а, растворяют в 75 мл четыреххлористого углерода. Далее добавляют 9,52 г (53,5 ммоля) N-бромсукцинимида и 48 мг дибензоилпероксида, в течение 5 ч кипятят с обратным холодильником и перемешивают в течение ночи при комнатной температуре. Суспензию дважды промывают раствором гидрокарбоната натрия и однократно водой, органическую фазу сушат сульфатом магния, отфильтровывают осушитель и фильтрат досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (гексан/этилацетат в соотношении 3:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 21,0 г (86% от теории).
в) 10-[α-(4-(этоксикарбонилдецилокси)фенил)метоксикарбонилметил]-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 27,9 г (162,2 ммоля) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 37,1 г (81,1 ммоля) описанного в предыдущем примере 7б бромзамещенного соединения и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол/триэтиламин в соотношении 10:5:0,1). Полученный таким путем 1-[α-(4-(этоксикарбонилдецилокси)фенил)метоксикарбонилметил]-1,4,7,10-тетраазациклододекан (27,4 г, 50 ммолей, 62% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull. 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 33,6 г (65% от теории) бесцветного кристаллического порошка.
г) 10-[α-(4-(этоксикарбонилдецилокси)фенил)метоксикарбонилметил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
31,1 г (30 ммолей) соединения, указанного в заголовке примера 7в, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 23,0 г (количественный) бесцветного порошка.
д) Gd-комплекс 10-[α-(4-карбоксидецилоксифенил)карбоксиметил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
15,3 г (20 ммолей) соединения, указанного в заголовке примера 7г, растворяют в 250 мл 2 н. раствора едкого натра и 250 мл тетрагидрофурана и перемешивают в течение 5 дней при 40°С. После этого значение pH водной фазы устанавливают на 7 с помощью Amberlite IR-120® (Н+-форма), добавляют 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 11,5 г (60% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,5%.
Пример 8
а) 10-(n-метоксикарбонилбензил)-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 27,9 г (162,2 ммоля) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 18,6 г (81,1 ммоля) метилового эфира 4-бромметилбензойной кислоты (фирма Aldrich) в 150 мл хлороформа и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: метанол/25%-ный водный аммиак в соотношении 8:1). Полученный таким путем 1-(n-метоксикарбонилбензил)-1,4,7,10-тетраазациклододекан (21,6 г, 67,3 ммоля, 83% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull. 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 41,8 г (77% от теории) бесцветного кристаллического порошка.
б) 10-(n-карбоксибензил)-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
24,2 г (30 ммолей) соединения, указанного в заголовке примера 8а, растворяют в 400 мл метанола, смешивают со 100 мл 15 н. раствора едкого натра, в течение 6 ч кипятят с обратным холодильником и перемешивают в течение ночи при комнатной температуре. После упаривания в вакууме остаток растворяют в 200 мл воды и добавлением катионита IR-120® (Н+-форма) значение рН устанавливают на 7. После этого ионит отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток без последующего определения его характеристик используют в реакции комплексообразования.
Система растворителей для тонкослойной хроматографии: н-бутанол/водн. аммиак/этанол/вода в соотношении 12:6:3:3.
Выход: 16 г.
в) Gd-комплекс 10-(n-карбоксибензил)-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
11 г (20 ммолей) описанного в примере 8б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 8,9 г (61% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,2%.
Пример 9
а) 10-(n-метоксикарбонилбензил)-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
21,6 г (67,3 ммоля) описанного в примере 8а в качестве промежуточного продукта 1-(n-метооксикарбонилбензил)-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 85,1 г (0,25 моля) бензилового эфира 2-(трифторметансульфонилокси)изовалериановой кислоты (Walker и др., Tetrahedron 53(43), 1997, с.14591) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 48,5 г (81% от теории) бесцветного кристаллического порошка.
б) 10-(n-карбоксибензил)-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
26,7 г (30 ммолей) соединения, указанного в заголовке примера 9а, растворяют в 400 мл метанола, смешивают с 100 мл 15 н. раствора едкого натра, в течение 6 ч кипятят с обратным холодильником и перемешивают в течение ночи при комнатной температуре. После упаривания в вакууме остаток растворяют в 200 мл воды и добавлением катионита IR-120® (Н+-форма) значение рН устанавливают на 7. После этого ионит отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток без последующего определения его характеристик используют в реакции комплексообразования.
Система растворителей для тонкослойной хроматографии: н-бутанол/водн. аммиак/этанол/вода в соотношении 12:6:3:3.
Выход: 19 г.
в) Gd-комплекс 10-(n-карбоксибензил)-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
12,6 г (20 ммолей) описанного в примере 9б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (H+-форма). Кислый элюат сушат вымораживанием.
Выход: 10,9 г (65% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 9,0%.
Пример 10
а) 10-(n-метоксикарбонилбензил)-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазапиклододекан
21,6 г (67,3 ммоля) описанного в примере 8а в качестве промежуточного продукта 1-(n-метоксикарбонилбензил)-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 95,1 г (0,25 моля) бензилового эфира 2-(трифторметансульфонилокси)-2-циклогексилуксусной кислоты (Qabar и др., Tetrahedron Letters, 39(33), 1998, с.5895) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 48,3 г (71% от теории) бесцветного кристаллического порошка.
б) 10-(n-карбоксибензил)-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
30,3 г (30 ммолей) соединения, указанного в заголовке примера 10а, растворяют в 400 мл метанола, смешивают с 100 мл 15 н. раствора едкого натра, в течение 6 ч кипятят с обратным холодильником и перемешивают в течение ночи при комнатной температуре. После упаривания в вакууме остаток растворяют в 200 мл воды и добавлением катионита IR-120® (Н+-форма) значение рН устанавливают на 7. После этого ионит отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток без последующего определения его характеристик используют в реакции комплексообразования.
Система растворителей для тонкослойной хроматографии: н-бутанол/водн. аммиак/этанол/вода в соотношении 12:6:3:3.
Выход: 22,5 г.
в) Gd-комплекс 10-(n-карбоксибензил)-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
15,0 г (20 ммолей) описанного в примере 10б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и упаривают досуха. Остаток растворяют в муравьиной кислоте и затем при добавлении дихлорметана многократно упаривают досуха, после чего сушат в вакууме до постоянства массы.
Выход: 11,9 г (63% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,0%.
Пример 11
а) 10-(n-метоксикарбонилбензил)-1,4,7-α,α',α''-трифенил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
21,6 г (67,3 ммоля) описанного в примере 8а в качестве промежуточного продукта 1-(n-метоксикарбонилбензил)-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 93,6 г (0,25 моля) бензилового эфира 2-(трифторметансульфонилокси)-2-фенилуксусной кислоты (Qabar и др., Tetrahedron Letters, 39(33), 1998, с.5895) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 50,8 г (76% от теории) бесцветного кристаллического порошка.
б) 10-(n-карбоксибензил)-1,4,7-α,α',α''-трифенил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
29,8 г (30 ммолей) соединения, указанного в заголовке примера 11a, растворяют в 400 мл метанола, смешивают с 100 мл 15 н. раствора едкого натра, в течение 6 ч кипятят с обратным холодильником и перемешивают в течение ночи при комнатной температуре. После упаривания в вакууме остаток растворяют в 200 мл воды и добавлением катионита IR-120® (Н+-форма) значение рН устанавливают на 7. После этого ионит отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток без последующего определения его характеристик используют в реакции комплексообразования.
Система растворителей для тонкослойной хроматографии: н-бутанол/водн. аммиак/этанол/вода в соотношении 12:6:3:3.
Выход: 22,0 г.
в) Gd-комплекс 10-(n-карбоксибензил)-1,4,7-α,α',α''-трифенил-1,4,7-трис-(карбоксиметил)-1,4,7,10-тетраазациклододекана
14,6 г (20 ммолей) описанного в примере 11б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и упаривают досуха. Остаток растворяют в муравьиной кислоте и затем при добавлении дихлорметана многократно упаривают досуха, после чего сушат в вакууме до постоянства массы.
Выход: 13,1 г (70% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,1%.
Пример 12
а) 10-[4-(трет-бутоксикарбонил)-1-фенил-2-оксо-3-азабутил]-1,4,7-α,α',α''-трифенил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 27,9 г (162,2 ммоля) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 26,6 г (81,1 ммоля) трет-бутилового эфира N-[2-бром-2-фенилацетил]глицина (пример 6а в WO 98/24775) и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: хлороформ/метанол/25%-ный водный аммиак в соотношении 10:5:1). Полученный таким путем 1-[4-(трет-бутоксикарбонил)-1-фенил-2-оксо-3-азабутил]-1,4,7,10-тетраазациклододекан (21,0 г, 50 ммолей, 62% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 74,9 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)-2-фенилуксусной кислоты (Qabar и др., Tetrahedron Letters 39(33), 1998, с.5895) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 30:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 37,7 г (69% от теории) бесцветного кристаллического порошка.
б) 10-(4-(трет-бутоксикарбонил-1-фенил-2-оксо-3-азабутил)-1,4,7-α,α',α''-трифенил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
32,8 г (30 ммолей) соединения, указанного в заголовке примера 12а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 24,8 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-(4-карбокси-1-фенил-2-оксо-3-азабутил)-1,4,7-α,α',α''-трифенил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
16,4 г (20 ммолей) описанного в примере 12б трет-бутилового эфира растворяют в минимальном количестве трифторуксусной кислоты и перемешивают в течение 15 мин при комнатной температуре. После добавления 250 мл диэтилового эфира перемешивают в течение 2 ч, осадок отделяют вакуум-фильтрацией и сушат в вакууме. Полученный таким путем свободный лиганд растворяют в 200 мл воды и 80 мл изопропанола, значение рН с помощью разбавленного аммиака устанавливают на 7 и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 25:15:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (H+-форма). Кислый элюат сушат вымораживанием.
Выход: 11,7 г (59% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,5%.
Пример 13
а) 10-[4-(бензилоксикарбонил)-2-оксо-3-азабутил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 34,4 г (0,2 моля) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 23,2 г (81,1 ммоля) бензилового эфира 2-бромацетилглицина (Teger-Nilsson и др., WO 93/11152, с.38) и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: хлороформ/метанол/25%-ный водный аммиак в соотношении 10:5:1). Полученный таким путем 1-[4-(бензилоксикарбонил)-2-оксо-3-азабутил]-1,4,7,10-тетраазациклододекан (19,6 г, 50 ммолей, 62% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 68,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)изовалериановой кислоты (Walker и др., Tetrahedron 53(43), 1997, с.14591) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 37,0 г (78% от теории) бесцветного кристаллического порошка.
б) 10-(4-карбокси-2-оксо-3-азабутил)-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
28,4 г (30 ммолей) соединения, указанного в заголовке примера 13а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 17,7 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-(4-карбокси-2-оксо-3-азабутил)-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
11,8 г (20 ммолей) описанного в примере 13б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 12,1 г (75% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,0%.
Пример 14
а) 10-[4-(бензилоксикарбонил)-2-оксо-3-азабутил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
18,9 г (50 ммолей) описанного в примере 13а в качестве промежуточного продукта 1-[4-(бензилоксикарбонил)-2-оксо-3-азабутил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 76,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)-2-циклогексилуксусной кислоты (Qabar и др., Tetrahedron Letters 39(33), 1998, с.5895) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 38,5 г (72% от теории) бесцветного кристаллического порошка.
б) 10-(4-карбокси-2-оксо-3-азабутил)-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
32,1 г (30 ммолей) соединения, указанного в заголовке примера 14а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 21,2 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-(4-карбокси-2-оксо-3-азабутил)-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
14,2 г (20 ммолей) описанного в примере 14б лиганда растворяют в 150 мл воды и 150 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 8 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и упаривают досуха. Остаток растворяют в муравьиной кислоте и затем при добавлении дихлорметана многократно упаривают досуха, после чего сушат в вакууме до постоянства массы.
Выход: 13,5 г (71% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 9,0%.
Пример 15
а) Три-трет-бутиловый эфир 10-[4-(бензилоксикарбонил)-1-метил-2-оксо-3-азабутил]-2,5,8,11-тетраметил-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты, натрийбромидный комплекс
К 1,14 г (5 ммолей) 2,5,8,11-тетраметил-1,4,7,10-тетраазациклододекана (Petrov и др., DE 19608307; Ranganathan и др., WO 95/31444), растворенного в 10 мл хлороформа, добавляют 0,50 г (1,67 ммоля) бензилового эфира 2-бромпропионилглицина (пример 1е в WO 98/24774) и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: хлороформ/метанол/25%-ный водный аммиак в соотношении 10:5:1). К полученному таким путем 1-[4-(бензилоксикарбонил)-1-метил-2-оксо-3-азабутил]-2,5,8,11-тетраметил-1,4,7,10-тетраазациклододекану (0,70 г, 1,27 ммоля, 76% от теории) и 541 мг (5,1 ммоля) карбоната натрия в 5 мл ацетонитрила добавляют 822 мг (4,2 ммоля) трет-бутилового эфира бромуксусной кислоты и перемешивают в течение 12 ч при 60°С. После этого охлаждают до 0°С и соли отфильтровывают. Фильтрат упаривают досуха и остаток хроматографируют на силикагеле (элюент: метиленхлорид/метанол в соотношении 20:1).
Выход: 964 мг (85% от теории) бесцветного твердого вещества.
б) Три-трет-бутиловый эфир 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-2,5,8,11-тетраметил-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты (натрийбромидный комплекс)
893 мг (1,0 ммоль) соединения, указанного в заголовке примера 15а, растворяют в 10 мл изопропанола и на кончике шпателя добавляют палладиевый катализатор (10%-ный Pd/C). После этого гидрируют в течение ночи при комнатной температуре. Затем катализатор отфильтровывают и фильтрат упаривают досуха. Остаток перекристаллизовывают из диоксана.
Выход: 562 мг (70% от теории) кристаллического твердого вещества.
в) Гадолиниевый комплекс 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-2,5,8,11-тетраметил-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты
803 мг (1,0 ммоль) соединения, указанного в заголовке примера 15б, растворяют в 5 мл трифторуксусной кислоты и перемешивают в течение 3 ч при комнатной температуре. После этого упаривают досуха, остаток растворяют в 300 мл воды и раствор вносят в колонку, заполненную Reillex® 425 PVP. Затем элюируют водой. Содержащие продукт фракции объединяют и упаривают досуха (446 мг, 0,84 ммоля) и вновь растворяют в 4 мл воды. Далее добавляют 152 мг (0,42 ммоля) оксида гадолиния, нагревают до 90°С и выдерживают при этой температуре в течение 3 ч. После этого упаривают досуха (в вакууме) и остаток перекристаллизовывают из 90%-ного водного этанола. Кристаллы отделяют вакуум-фильтрацией, однократно промывают этанолом, а затем ацетоном и в завершение диметиловым эфиром и сушат в вакуумной печи при 130°С (в течение 24 ч).
Выход: 469 мг (65% от теории) бесцветного кристаллического порошка.
Содержание воды: 5%.
Пример 16
Gd-комплекс 10-[8-(N-малеимидо)-1-метил-2,5-диоксо-3,6-диазаоктил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
2,27 г (3 ммоля) описанного в примере 2 комплекса Gd-кислота растворяют в 15 мл ДМФ, при охлаждении льдом смешивают с 380 мг (3,3 ммоля) N-гидроксисукцинимида и 681 мг (3,3 ммоля) дициклогексилкарбодиимида и в течение 1 ч предварительно активируют в ледяной бане. После этого добавляют смесь из 839 мг (3,3 ммоля) трифторацетатной соли N-(2-аминоэтил)малеимида (Arano и др., J. Med. Chem. 39, 1996, с.3458) и 0,7 мл (4 ммоля) N,N-диизопропилэтиламина в 10 мл ДМФ и перемешивают в течение ночи при комнатной температуре. Затем реакционную смесь вновь охлаждают в ледяной бане, фильтруют и фильтрат досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 1:1).
Выход: 997 мг (35% от теории).
Содержание воды (с использованием реактива Карла Фишера): 7,5%.
Пример 17
Gd-комплекс 10-[8-(N-малеимидо)-1-метил-2,5-диоксо-3,6-диазаоктил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
2,63 г (3 ммоля) описанного в примере 3 комплекса Gd-кислота растворяют в 15 мл ДМФ, при охлаждении льдом смешивают с 380 мг (3,3 ммоля) N-гидроксисукцинимида и 681 мг (3,3 ммоля) дициклогексилкарбодиимида и в течение 1 ч предварительно активируют в ледяной бане. После этого добавляют смесь из 839 мг (3,3 ммоля) трифторацетатной соли N-(2-аминоэтил)малеимида (Arano и др., J. Med. Chem. 39, 1996, с.3458) и 0,7 мл (4 ммоля) N,N-диизопропилэтиламина в 10 мл ДМФ и перемешивают в течение ночи при комнатной температуре. Затем реакционную смесь вновь охлаждают в ледяной бане, фильтруют и фильтрат досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 1:1).
Выход: 1,24 г (39% от теории).
Содержание воды (с использованием реактива Карла Фишера): 6,0%.
Пример 18
а) Бензиловый эфир (3-бром-2-оксопирролидин-1-ил)уксусной кислоты
67,7 г (0,2 моля) тозилата бензилового эфира глицина и 61,2 мл (0,44 моля) триэтиламина растворяют в 200 мл метиленхлорида, при 0°С в течение 45 мин по каплям добавляют к раствору 52,9 г (0,2 моля) хлорангидрида 2,4-диброммасляной кислоты (Gramain и др., Synth. Commun. 27, 1997, с.1827) в 200 мл метиленхлорида и перемешивают в течение 18 ч при комнатной температуре. Затем реакционную смесь при 0°С по каплям добавляют к раствору 400 мл водного 32%-ного гидроксида натрия и 2 г гидрокарбоната тетрабутиламмония (примерно в течение 15 мин) и перемешивают в течение 30 мин. После этого фазы разделяют и водную фазу трижды экстрагируют дихлорметаном порциями по 200 мл. Органические фазы сушат над сульфатом натрия, раствор упаривают досуха и хроматографируют на силикагеле (метиленхлорид). Содержащие продукт фракции объединяют и упаривают.
Выход: 29,3 г (47% от теории).
б) 10-[1-(бензилоксикарбонилметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 28,7 г (165,8 ммоля) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 20,7 г (66,3 ммоля) бензилового эфира (3-бром-2-оксопирролидин-1-ил)уксусной кислоты и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: хлороформ/метанол/25%-ный водный аммиак в соотношении 10:5:1). Полученный таким путем 1-[1-(бензилоксикарбонилметил)-2-оксопирролидин-3-ил]-1,4,7,10-тетраазациклододекан (20,9 г, 51,8 ммоля, 78% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull. 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 32,7 г (71% от теории) бесцветного кристаллического порошка.
в) 10-[1-(карбоксиметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис-(карбоксиметил)-1,4,7,10-тетраазациклододекан
26,7 г (30 ммолей) соединения, указанного в заголовке примера 18б, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 15,8 г (количественный) бесцветного порошка.
г) Gd-комплекс 10-[1-(карбоксиметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
10,6 г (20 ммолей) описанного в примере 18в лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 9,7 г (67% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,3%.
Пример 19
а) 10-[1-(бензилоксикарбонилметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
20,2 г (50 ммолей) описанного в примере 18б в качестве промежуточного продукта 1-[1-(бензилоксикарбонилметил)-2-оксопирролидин-3-ил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 68,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)изовалериановой кислоты (Walker и др., Tetrahedron 53(43), 1997, с.14591) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 34,1 г (70% от теории) бесцветного кристаллического порошка.
б) 10-[1-(карбоксиметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
29,2 г (30 ммолей) соединения, указанного в заголовке примера 19а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 18,4 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-[1-(карбоксиметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
12,3 г (20 ммолей) описанного в примере 19б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 11,9 г (75% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,2%.
Аналогичным путем с применением 12,3 г (20 ммолей) описанного в примере 19б лиганда и 3,73 г (10 ммолей) оксида диспрозия вместо оксида гадолиния получают Dy-комплекс 10-[1-(карбоксиметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана.
Выход: 11,4 г (71% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,0%.
Пример 20
а) 10-[1-(бензилоксикарбонилметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
20,2 г (50 ммолей) описанного в примере 18б в качестве промежуточного продукта 1-[1-(бензилоксикарбонилметил)-2-оксопирролидин-3-ил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 76,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)-2-циклогексилуксусной кислоты (Qabar и др., Tetrahedron Letters 39(33), 1998, с.5895) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 37,2 г (68% от теории) бесцветного кристаллического порошка.
б) 10-[1-(карбоксиметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
32,8 г (30 ммолей) соединения, указанного в заголовке примера 20а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 22,0 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-[1-(карбоксиметил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
14,6 г (20 ммолей) описанного в примере 20б лиганда растворяют в 150 мл воды и 150 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 8 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и упаривают досуха. Остаток растворяют в муравьиной кислоте и затем при добавлении дихлорметана многократно упаривают досуха, после чего сушат в вакууме до постоянства массы.
Выход: 12,1 г (65% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,0%.
Пример 21
а) Бензиловый эфир (3-бром-2-оксопирролидин-1-ил)бензойной кислоты
45,5 г (0,2 моля) бензилового эфира 4-аминобензойной кислоты и 30,6 мл (0,22 моля) триэтиламина растворяют в 200 мл метиленхлорида, при 0°С в течение 45 мин по каплям добавляют к раствору 52,9 г (0,2 моля) хлорангидрида 2,4-диброммасляной кислоты (Gramain и др. Synth. Commun. 27, 1997, с.1827) в 200 мл метиленхлорида и перемешивают в течение 18 ч при комнатной температуре. Затем реакционную смесь при 0°С по каплям добавляют к раствору 400 мл водного 32%-ного гидроксида натрия и 2 г гидрокарбоната тетрабутиламмония (примерно в течение 15 мин) и перемешивают в течение 30 мин. После этого фазы разделяют и водную фазу трижды экстрагируют дихлорметаном порциями по 200 мл. Органические фазы сушат над сульфатом натрия, раствор упаривают досуха и хроматографируют на силикагеле (метиленхлорид). Содержащие продукт фракции объединяют и упаривают.
Выход: 38,2 г (51% от теории).
б) 10-[1-(4-бензилоксикарбонилфенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 31,2 г (180 ммолей) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 26,9 г (71,9 ммоля) бензилового эфира (3-бром-2-оксопирролидин-1-ил)бензойной кислоты и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: хлороформ/метанол/25%-ный водный аммиак в соотношении 10:5:1). Полученный таким путем 1-[1-(4-бензилоксикарбонилфенил)-2-оксопирролидин-3-ил]-1,4,7,10-тетраазациклододекан (26,1 г, 56,1 ммоля, 78% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull. 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 36,3 г (68% от теории) бесцветного кристаллического порошка.
в) 10-[1-(4-карбоксифенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
28,6 г (30 ммолей) соединения, указанного в заголовке примера 21б, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 17,7 г (количественный) бесцветного порошка.
г) Gd-комплекс 10-[1-(4-карбоксифенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
11,8 г (20 ммолей) описанного в примере 21в лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (H+-форма). Кислый элюат сушат вымораживанием.
Выход: 11,1 г (71% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,5%.
Пример 22
а) 10-[1-(4-бензилоксикарбонилфенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
23,3 г (50 ммолей) описанного в примере 21б в качестве промежуточного продукта 1-[1-(4-бензилоксикарбонилфенил)-2-оксопирролидин-3-ил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 68,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)изовалериановой кислоты (Walker и др., Tetrahedron 53(43), 1997, с.14591) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 35,3 г (68% от теории) бесцветного кристаллического порошка.
б) 10-[1-(4-карбоксифенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
31,1 г (30 ммолей) соединения, указанного в заголовке примера 22а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 20,2 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-[1-(4-карбоксифенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
13,5 г (20 ммолей) описанного в примере 22б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 12,4 г (72% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,8%.
Аналогичным путем с применением 13,5 г (20 ммолей) описанного в примере 22б лиганда и 3,73 г (10 ммолей) оксида диспрозия вместо оксида гадолиния получают Dy-комплекс 10-[1-(4-карбоксифенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана.
Выход: 13,0 г (75% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,5%.
Пример 23
a) 10-[1-(4-бензилоксикарбонилфенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
23,3 г (50 ммолей) описанного в примере 21б в качестве промежуточного продукта 1-[1-(4-бензилоксикарбонилфенил)-2-оксопирролидин-3-ил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 76,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)-2-циклогексилуксусной кислоты (Qabar и др., Tetrahedron Letters 39(33), 1998, с.5895) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 41,1 г (71% от теории) бесцветного кристаллического порошка.
б) 10-[1-(4-карбоксифенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
34,7 г (30 ммолей) соединения, указанного в заголовке примера 23а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 23,8 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-[1-(4-карбоксифенил)-2-оксопирролидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
15,9 г (20 ммолей) описанного в примере 23б лиганда растворяют в 150 мл воды и 150 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 8 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и упаривают досуха. Остаток растворяют в муравьиной кислоте и затем при добавлении дихлорметана многократно упаривают досуха, после чего сушат в вакууме до постоянства массы.
Выход: 12,9 г (65% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,0%.
Пример 24
а) Бензиловый эфир (3-бром-2-оксопиперидин-1-ил)уксусной кислоты
67,7 г (0,2 моля) тозилата бензилового эфира глицина и 61,2 мл (0,44 моля) триэтиламина растворяют в 200 мл метиленхлорида, при 0°С в течение 45 мин по каплям добавляют к раствору 55,7 г (0,2 моля) хлорангидрида 2,5-дибромвалериановой кислоты (Okawara и др. Chem. Pharm. Bull. 30, 1982, с.1225) в 200 мл метиленхлорида и перемешивают в течение 18 ч при комнатной температуре. Затем реакционную смесь при 0°С по каплям добавляют к раствору 400 мл водного 32%-ного гидроксида натрия и 2 г гидрокарбоната тетрабутиламмония (примерно в течение 15 мин) и перемешивают в течение 30 мин. После этого фазы разделяют и водную фазу трижды экстрагируют дихлорметаном порциями по 200 мл. Органические фазы сушат над сульфатом натрия, раствор упаривают досуха и хроматографируют на силикагеле (метиленхлорид). Содержащие продукт фракции объединяют и упаривают.
Выход: 33,2 г (51% от теории).
б) 10-[1-(бензилоксикарбонилметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 30,3 г (175 ммолей) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 18,9 г (58 ммолей) бензилового эфира (3-бром-2-оксопиперидин-1-ил)уксусной кислоты и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: хлороформ/метанол/25%-ный водный аммиак в соотношении 10:5:1). Полученный таким путем 1-[1-(бензилоксикарбонилметил)-2-оксопиперидин-3-ил]-1,4,7,10-тетраазациклододекан (20,3 г, 48,6 ммоля, 84% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull. 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 32,5 г (74% от теории) бесцветного кристаллического порошка.
в) 10-[1-(карбоксиметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
27,1 г (30 ммолей) соединения, указанного в заголовке примера 24б, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 16,3 г (количественный) бесцветного порошка.
г) Gd-комплекс 10-[1-(карбоксиметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
10,9 г (20 ммолей) описанного в примере 24в лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 9,6 г (65% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,2%.
Пример 25
а) 10-[1-(бензилоксикарбонилметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
20,9 г (50 ммолей) описанного в примере 24б в качестве промежуточного продукта 1-[1-(бензилоксикарбонилметил)-2-оксопиперидин-3-ил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 68,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)изовалериановой кислоты (Walker и др., Tetrahedron 53(43), 1997, с.14591) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 36,2 г (73% от теории) бесцветного кристаллического порошка.
б) 10-[1-(карбоксиметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
29,6 г (30 ммолей) соединения, указанного в заголовке примера 25а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 18,8 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-[1-(карбоксиметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
12,6 г (20 ммолей) описанного в примере 25б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 11,7 г (71% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 8,1%.
Аналогичным путем с применением 12,6 г (20 ммолей) описанного в примере 25б лиганда и 3,73 г (10 ммолей) оксида диспрозия вместо оксида гадолиния получают Dy-комплекс 10-[1-(карбоксиметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана.
Выход: 10,8 г (66% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,6%.
Пример 26
а) 10-[1-(бензилоксикарбонилметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
20,9 г (50 ммолей) описанного в примере 24б в качестве промежуточного продукта 1-[1-(бензилоксикарбонилметил)-2-оксопиперидин-3-ил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 76,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)-2-циклогексилуксусной кислоты (Qabar и др., Tetrahedron Letters 39(33), 1998, с.5895) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 39,8 г (72% от теории) бесцветного кристаллического порошка.
б) 10-[1-(карбоксиметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
33,3 г (30 ммолей) соединения, указанного в заголовке примера 26а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 22,4 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-[1-(карбоксиметил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
14,9 г (20 ммолей) описанного в примере 26б лиганда растворяют в 150 мл воды и 150 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 8 ч кипятят с обратным холодильником. По завершении комплексообразования значение рН с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и упаривают досуха. Остаток растворяют в муравьиной кислоте и затем при добавлении дихлорметана многократно упаривают досуха, после чего сушат в вакууме до постоянства массы.
Выход: 12,9 г (68% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,6%.
Пример 27
а) Бензиловый эфир (3-бром-2-оксопиперидин-1-ил)бензойной кислоты
45,5 г (0,2 моля) бензилового эфира 4-аминобензойной кислоты и 30,6 мл (0,22 моля) триэтиламина растворяют в 200 мл метиленхлорида, при 0°С в течение 45 мин по каплям добавляют к раствору 55,3 г (0,2 моля) хлорангидрида 2,5-дибромвалериановой кислоты (Okawara и др., Chem. Pharm. Bull. 30, 1982, с.1225) в 200 мл метиленхлорида и перемешивают в течение 18 ч при комнатной температуре. Затем реакционную смесь при 0°С по каплям добавляют к раствору 400 мл водного 32%-ного гидроксида натрия и 2 г гидрокарбоната тетрабутиламмония (примерно в течение 15 мин) и перемешивают в течение 30 мин. После этого фазы разделяют и водную фазу трижды экстрагируют дихлорметаном порциями по 200 мл. Органические фазы сушат над сульфатом натрия, раствор упаривают досуха и хроматографируют на силикагеле (метиленхлорид). Содержащие продукт фракции объединяют и упаривают.
Выход: 38,8 г (50% от теории).
б) 10-[1-(4-бензилоксикарбонилфенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 31,2 г (180 ммолей) 1,4,7,10-тетраазациклододекана, растворенного в 300 мл хлороформа, добавляют 26,6 г (68,5 ммоля) бензилового эфира (3-бром-2-оксопиперидин-1-ил)бензойной кислоты и перемешивают в течение ночи при комнатной температуре. Далее добавляют 250 мл воды, органическую фазу отделяют и дополнительно дважды промывают ее водой порциями по 200 мл. Затем органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: хлороформ/метанол/25%-ный водный аммиак в соотношении 10:5:1). Полученный таким путем 1-[1-(4-бензилоксикарбонилфенил)-2-оксопиперидин-3-ил]-1,4,7,10-тетраазациклододекан (27,6 г, 57,5 ммоля, 84% от теории) и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 62,45 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)пропионовой кислоты (Kitazaki и др., Chem. Pharm. Bull. 47(3), 1999, с.360) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 39,4 г (71% от теории) бесцветного кристаллического порошка.
в) 10-[1-(4-карбоксифенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
29,0 г (30 ммолей) соединения, указанного в заголовке примера 27б, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 18,1 г (количественный) бесцветного порошка.
г) Gd-комплекс 10-[1-(4-карбоксифенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-триметил-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
12,1 г (20 ммолей) описанного в примере 27в лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение pH с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 11,4 г (72% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,1%.
Пример 28
а) 10-[1-(4-бензилоксикарбонилфенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
24,0 г (50 ммолей) описанного в примере 27б в качестве промежуточного продукта 1-[1-(4-бензилоксикарбонилфенил)-2-оксопиперидин-3-ил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметана добавляют к 68,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)изовалериановой кислоты (Walker и др., Tetrahedron 53(43), 1997, с.14591) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 37,8 г (72% от теории) бесцветного кристаллического порошка.
б) 10-[1-(4-карбоксифенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
31,5 г (30 ммолей) соединения, указанного в заголовке примера 28а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 20,7 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-[1-(4-карбоксифенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
13,8 г (20 ммолей) описанного в примере 28б лиганда растворяют в 200 мл воды и 80 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 3 ч кипятят с обратным холодильником. По завершении комплексообразования значение pH с помощью аммиака устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и пропускают через катионообменную колонку IR-120® (Н+-форма). Кислый элюат сушат вымораживанием.
Выход: 12,0 г (68% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,5%.
Аналогичным путем с применением 13,8 г (20 ммолей) описанного в примере 28б лиганда и 3,73 г (10 ммолей) оксида диспрозия вместо оксида гадолиния получают Dy-комплекс 10-[1-(4-карбоксифенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(изопропил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана.
Выход: 12,4 г (70% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,5%.
Пример 29
а) 10-[1-(4-бензилоксикарбонилфенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(бензилоксикарбонилметил)-1,4,7,10-тетраазациклододекан
24,0 г (50 ммолей) описанного в примере 27б в качестве промежуточного продукта 1-[1-(4-бензилоксикарбонилфенил)-2-оксопиперидин-3-ил]-1,4,7,10-тетраазациклододекана и 60 мл (0,35 моля) N-этилдиизопропиламина в 200 мл дихлорметан добавляют к 76,1 г (0,2 моля) бензилового эфира 2-(трифторметансульфонилокси)-2-циклогексилуксусной кислоты (Qabar и др., Tetrahedron Letters 39(33), 1998, с.5895) в 400 мл дихлорметана и перемешивают в течение 6 ч при нагревании с обратным холодильником, а затем в течение ночи при комнатной температуре. После этого трижды экстрагируют водой порциями по 500 мл, органическую фазу сушат над сульфатом магния и упаривают досуха. Остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1). Содержащие продукт фракции объединяют и упаривают.
Выход: 40,9 г (70% от теории) бесцветного кристаллического порошка.
б) 10-[1-(4-карбоксифенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
35,1 г (30 ммолей) соединения, указанного в заголовке примера 29а, растворяют в 400 мл изопропанола, смешивают с 40 мл воды и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют в течение 8 ч при 50°С. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 24,3 г (количественный) бесцветного порошка.
в) Gd-комплекс 10-[1-(4-карбоксифенил)-2-оксопиперидин-3-ил]-1,4,7-α,α',α''-трис(циклогексил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
16,2 г (20 ммолей) описанного в примере 29б лиганда растворяют в 150 мл воды и 150 мл изопропанола и подкисляют добавлением 5 мл уксусной кислоты. Далее добавляют 3,6 г (10 ммолей) оксида гадолиния и в течение 8 ч кипятят с обратным холодильником. По завершении комплексообразования значение pH с помощью аммиака вновь устанавливают на 7,4 и хроматографируют на силикагеле (элюент: дихлорметан/метанол/аммиак в соотношении 20:20:1). Содержащие продукт фракции объединяют и упаривают досуха. Остаток растворяют в муравьиной кислоте и затем при добавлении дихлорметана многократно упаривают досуха, после чего сушат в вакууме до постоянства массы.
Выход: 13,6 г (68% от теории) бесцветного порошка.
Содержание воды (с использованием реактива Карла Фишера): 7,5%.
Примеры 30-90
В примерах 30-90 описаны конъюгаты рассмотренных выше гадолиниевых комплексов с биомолекулами. Эти конъюгаты получали в соответствии с описанными ниже общими методиками I-IV. Полученные конъюгаты и их характеристики представлены в таблице 1. При этом сокращение "ОМ" означает общую методику, по которой получали соответствующий конъюгат, сокращение "АКТГ" означает адренокортикотропный гормон, а сокращение "RP-18" (от англ. "reversed phase") означает стационарную хроматографическую обращенную фазу. Число комплексов, приходящееся на одну биомолекулу, определяли с помощью ИСП (атомно-эмиссионная спектроскопия с индуктивно-связанной плазмой).
Общая методика (ОМ) I: Альбумин-амидные конъюгаты
3 ммоля комплекса Gd-кислота растворяют в 15 мл ДМФ, при охлаждении льдом смешивают с 380 мг (3,3 ммоля) N-гидроксисукцинимида и 681 мг дициклогексилкарбодиимида и в течение 1 ч предварительно активируют в ледяной бане. Реакционную смесь, содержащую активированный сложный эфир, в течение 30 мин по каплям добавляют к раствору 16,75 г (0,25 ммоля) бычьего сывороточного альбумина (БСА) в 150 мл фосфатного буфера (рН 7,4) и перемешивают в течение 2 ч при комнатной температуре. После этого реакционный раствор фильтруют, фильтрат пропускают через ультрафильтрационную мембрану AMICON® YM30 (предельная проницаемость 30000 Да), ретентат (вещество, не прошедшее через фильтрационную мембрану) хроматографируют на колонке Sephadex® G50 и содержащие продукт фракции сушат вымораживанием.
Общая методика (ОМ) II: Альбумин-малеимидные конъюгаты
0,0438 ммоля комплекса Gd-малеимид в 1 мл ДМФ добавляют к 0,84 г (0,0125 ммоля) бычьего сывороточного альбумина (БСА), растворенного в 15 мл фосфатного буфера (рН 7,4), и перемешивают в течение часа при комнатной температуре. Реакционный раствор фильтруют, фильтрат пропускают через ультрафильтрационную мембрану AMICON® YM30 (предельная проницаемость 30000 Да), ретентат хроматографируют на колонке Sephadex® G50 и содержащие продукт фракции сушат вымораживанием.
Общая методика (ОМ) III: Получение амидных конъюгатов
3 ммоля комплекса Gd-кислота растворяют в 15 мл ДМФ, при охлаждении льдом смешивают с 380 мг (3,3 ммоля) N-гидроксисукцинимида и 681 мг дициклогексилкарбодиимида и в течение 1 ч предварительно активируют в ледяной бане. Реакционную смесь, содержащую активированный сложный эфир, по каплям добавляют к раствору 2,5 ммоля аминового компонента в 15-150 мл ДМФ и перемешивают в течение ночи при комнатной температуре. Реакционный раствор фильтруют и хроматографируют на силикагеле.
Общая методика (ОМ) IV: Получение малеимид-SH-конъюгатов
3 ммоля комплекса Gd-малеимид в 15 мл ДМФ по каплям добавляют к 2,5 ммоля SH-компонента в 15-150 мл ДМФ и перемешивают в течение часа при комнатной температуре. Реакционный раствор хроматографируют на силикагеле.
Пример 91
В этом примере показатели релаксационности конъюгатов из примеров 30-38 сравнивали с показателями релаксационности двух сравнительных веществ. В качестве таких сравнительных веществ использовали Gd-ДТПУ (1) формулы:
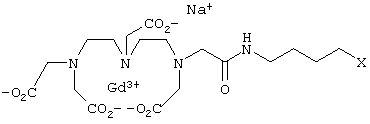
и Gd-GlyMeДОТА (2) формулы:
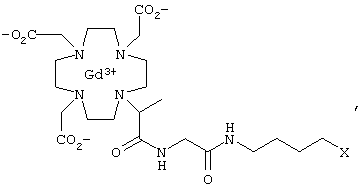
каждое из которых подвергали взаимодействию с бычьим сывороточным альбумином (БСА).
Измерения проводили в каждом случае в водном растворе и в плазме при +37°С и частоте 20 МГц. Полученные результаты приведены в таблице 2, при этом указанные значения релаксационности на моль гадолиния рассчитывали на основе измеренных значений.
Полученные в этом примере данные свидетельствуют о том, что полученные с предлагаемыми в изобретении соединениями конъюгаты несмотря на наличие у них небольшого числа атомов гадолиния, приходящихся на одну биомолекулу, неожиданно обладают более высокой релаксационностью в сопоставлении со сравнительными веществами. Так, в частности, присоединение к макроциклическому кольцу особых лигандов позволило значительно повысить релаксационность конъюгатов в сопоставлении со сравнительным веществом 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ КОМПЛЕКСЫ С ОСТАТКАМИ САХАРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2280644C2 |
| ПРИМЕНЕНИЕ ПЕРФТОРАЛКИЛСОДЕРЖАЩИХ КОМПЛЕКСОВ МЕТАЛЛОВ В КАЧЕСТВЕ КОНТРАСТНЫХ ВЕЩЕСТВ В МАГНИТНО-РЕЗОНАНСНОЙ ТОМОГРАФИИ ДЛЯ ВИЗУАЛИЗАЦИИ БЛЯШЕК, ОПУХОЛЕЙ И НЕКРОЗОВ | 2001 |
|
RU2290206C2 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ УКАЗАННЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2242477C2 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ КОПЛЕКСЫ С ПОЛЯРНЫМИ ОСТАТКАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО | 2001 |
|
RU2289579C2 |
| ХЕЛАТЫ МЕТАЛЛОВ, ИМЕЮЩИЕ ПЕРФТОРИРОВАННЫЙ ПЭГ РАДИКАЛ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2470014C2 |
| КАСКАДНЫЕ ПОЛИМЕРНЫЕ КОМПЛЕКСЫ, ИСХОДНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2166501C2 |
| КОМПЛЕКСЫ КАСКАДНЫХ ПОЛИМЕРОВ, СОДЕРЖАЩЕЕ ИХ ДИАГНОСТИЧЕСКОЕ КОНТРАСТНОЕ СРЕДСТВО И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1996 |
|
RU2197495C2 |
| КОНЪЮГАТЫ, ПОЛУЧЕННЫЕ ИЗ КОМПЛЕКСОВ МЕТАЛЛОВ И ОЛИГОНУКЛЕОТИДОВ, СРЕДСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТЫ, ИХ ИСПОЛЬЗОВАНИЕ В РАДИОДИАГНОСТИКЕ, А ТАКЖЕ СПОСОБ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2165771C2 |
| СОЕДИНЕНИЯ ДЛЯ РАЗДЕЛЕНИЯ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ И S-, P-, D-МЕТАЛЛОВ, СПОСОБЫ РАЗДЕЛЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2783526C2 |
| НОВЫЕ ХЕЛАТНЫЕ СОЕДИНЕНИЯ ГАДОЛИНИЯ С ВЫСОКОЙ РЕЛАКСИВНОСТЬЮ ДЛЯ ПРИМЕНЕНИЯ В МАГНИТНО-РЕЗОНАНСНОЙ ВИЗУАЛИЗАЦИИ | 2017 |
|
RU2755181C2 |
Описаны макроциклические комплексы металлов и их получение, а также их применение для получения конъюгатов с биомолекулами. Подобные конъюгаты пригодны для применения в качестве контрастных веществ при ЯМР-диагностике и рентгенодиагностике, а также для радиотерапии. За счет присоединения к макроциклам особых лигандов достигается высокая релаксационность и обеспечивается возможность точного ее регулирования. 2 н. и 10 з.п. ф-лы, 2 табл.

в которой
Z обозначает атом водорода или по меньшей мере два Z представляют собой эквивалент иона радиоактивного или парамагнитного металла,
В обозначает атом водорода или С1-С4алкильный остаток,
R обозначает атом водорода или неразветвленный, разветвленный либо циклический насыщенный либо ненасыщенный С1-С10алкильный или фенильный остаток, который необязательно замещен карбоксильной группой, при этом алкильная цепь C1-С10алкильного остатка необязательно содержит фенил и/или 1-2 атома кислорода,
при условии, что остатки В и R оба одновременно не обозначают атомы водорода,
А обозначает неразветвленную либо разветвленную, насыщенную либо ненасыщенную C1-С30углеводородную цепь, которая необязательно содержит 1-5 атомов кислорода, 1-5 атомов азота и/или 1-5 -NR'-остатков, где R' имеет указанные для R, но выбираемые независимо от него значения, и необязательно замещена 1-3 карбоксильными группами, и в которой 1-3 атома углерода необязательно присутствуют в виде карбонильных групп, при этом такая цепь или ее часть может быть замкнута в кольцо, и которая далее имеет такую структуру, что Х по меньшей мере через 3 атома связан с атомом азота, к которому присоединен остаток А, и Х представляет собой группу, способную вступать в реакцию с биомолекулой,
а также их соли,
при условии, что
а) если В представляет собой атом водорода, а R представляют собой группу -СН2СН2CO2Н, то А-Х совместно не обозначают группу -СН(CO2Н)СН2СН2CO2Н,
б) если В представляет собой атом водорода, а R представляет собой метильный или этильный остаток, который необязательно замещен карбоксигруппой, то А не обозначает остаток -CH(R4)-CO-NR2-U6-, где R2 представляет собой атом водорода, метильный или этильный остаток, который необязательно замещен 1 карбоксигруппой, R4 представляет собой неразветвленную, разветвленную, насыщенную либо ненасыщенную C1-С30алкильную цепь, которая необязательно прервана 1-10 атомами кислорода, 1 фениленовой группой, 1 фениленоксигруппой и/или необязательно замещена 1-5 гидроксигруппами, 1-3 карбоксигруппами, 1 фенильной группой, и U6 представляет собой необязательно содержащую 1-5 иминогрупп, 1-3 фениленовые группы, 1-3 фениленоксигруппы, 1-3 фенилениминогруппы, 1-5 амидных групп, 1-2 гидразидные группы, 1-5 карбонильных групп, 1-5 этиленоксигрупп, 1 мочевинную группу, 1 тиомочевинную группу, 1-2 карбоксиалкилиминогруппы, 1-2 сложноэфирные группы, 1-10 атомов кислорода, 1-5 атомов серы и/или 1-5 атомов азота и/или необязательно замещенную 1-5 гидроксигруппами, 1-2 меркаптогруппами, 1-5 оксогруппами, 1-5 тиоксогруппами, 1-3 карбоксигруппами, 1-5 карбоксиалкильными группами, 1-5 сложноэфирными группами и/или 1-3 аминогруппами неразветвленную, разветвленную, насыщенную либо ненасыщенную С1-С20алкиленовую группу, при этом необязательно присутствующие фениленовые группы могут быть замещены 1-2 карбоксигруппами, 1-2 сульфоновыми группами или 1-2 гидроксигруппами, и,
в) если В представляет собой атом водорода, a R представляет собой С1-С4алкильный остаток, то А не обозначает остаток формулы
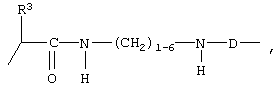
где R3 представляет собой атом водорода или С1-С4алкильный остаток, D представляет собой насыщенную либо ненасыщенную, неразветвленную либо разветвленную С1-С4алкиленовую группу, которая необязательно может быть прервана или замещена карбонильной группой, и D связан с X.
а) связь,
б) группу -СН(CO2Н)-,
в) группу формулы

в которой Q обозначает атом водорода, С1-С10алкильный остаток, который необязательно замещен карбоксильной группой, или фенильный остаток, который необязательно замещен карбоксильной группой, C1-С15алкоксигруппой, фенилоксигруппой, а R' имеет указанные для R в п.1, но выбираемые независимо от него значения, или
г) группу формулы

в которой о обозначает 0 или 1, а кольцо необязательно аннелировано с бензольным кольцом, которое при его наличии может быть замещено метоксигруппой или карбоксильной группой, при этом в представленных в подпунктах в) и г) группах их обозначенные символом  положения присоединены к смежным группам, α-положение связано с атомом азота макроциклического кольца, а β-положение связано с U,
положения присоединены к смежным группам, α-положение связано с атомом азота макроциклического кольца, а β-положение связано с U,
U обозначает неразветвленную либо разветвленную, насыщенную либо ненасыщенную C1-С30углеводородную цепь, которая необязательно содержит 1-3 атома кислорода, 1-3 атома азота и/или 1-3 -NR''-остатка, где R'' имеет указанные для R в п.1, но выбираемые независимо от него значения, и в которой 1-3 атома углерода необязательно присутствуют в виде карбонильных групп, при этом такая цепь или ее часть может быть замкнута в кольцо, при условии, что А' и U совместно образуют такую структуру, что Х по меньшей мере через три атома связан с атомом азота, к которому присоединен А'.

выбрана из -C(CH3)H-CO-NH-, -С(фенил)Н-СО-NH- и -С(n-додеканоксифенил)Н-СО-NH-.

выбрана из

где R1 обозначает -ОСН3, -СО2Н.

где Hal обозначает атом галогена.


 и
и 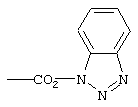
в которой Z, В, R, А и Х имеют указанные в п.1 значения, при условии, что В и R одновременно не обозначают атомы водорода и, если В представляет собой атом водорода, а R представляет собой С1-С4алкильный остаток, то А не обозначает остаток формулы
где R3 представляет собой атом водорода или С1-С4алкильный остаток, D представляет собой насыщенную либо ненасыщенную, неразветвленную либо разветвленную С1-С4алкиленовую группу, которая необязательно может быть прервана или замещена карбонильной группой, и D связан с X, заключающийся в том, что соединение формулы II

в которой В имеет указанные в п.1 значения, необязательно после введения защитных групп для атомов азота, подвергают взаимодействию с Nu-A-X' и Nu-CH(R)-CO2Z', где А и R имеют указанные в п.1 значения, Nu обозначает нуклеофоб, X' соответствует заместителю Х или защищенной форме заместителя X, который имеет указанные в п.1 значения, a Z' обозначает атом водорода, эквивалент иона радиоактивного или парамагнитного металла или защитную группу для карбоксила, после чего удаляют возможно присутствующие защитные группы, а затем при необходимости по известному методу подвергают взаимодействию по меньшей мере с одним оксидом или по меньшей мере с одной солью требуемого металла.
| US 5006643 А, 09.04.1991 | |||
| WO 9801476 A, 23.02.1989 | |||
| US 5972307 A, 26.10.1999 | |||
| Присадка к смазочным маслам | 1974 |
|
SU565930A1 |

