Настоящее изобретение относится к соединению (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамиду, и в частности к новым физическим формам указанного соединения, способу получения указанного соединения и промежуточным соединениям для применения в указанном способе и новым составам, содержащим указанное соединение, а также к терапевтическим применениям указанного соединения.
УРОВЕНЬ ТЕХНИКИ
Передача сигнала MAPK и роль ERK1/2
Киназы, регулируемые внеклеточными сигналами (ERK1/2), представляют собой повсеместно экспрессируемые серин-треониновые киназы, которые являются одним из ключевых компонентов пути передачи сигнала митоген-активируемых протеинкиназ (MAPK). Путь MAPK представляет собой эволюционно консервативный путь передачи сигнала в клетке, который регулирует различные процессы в клетке, включая прохождение клеточного цикла, миграцию, выживание, дифференцировку, метаболизм, пролиферацию клеток и транскрипцию в них. Путь передачи сигнала ERK/MAPK отвечает на внеклеточные стимулы от рецепторных тирозинкиназ (RTK) клеточной поверхности. После активации RTK ГТФ-азы семейства RAS (K-RAS, N-RAS и H-RAS) переходят из неактивного ГДФ-связанного состояние в активное ГТФ-связанное состояние. Активированная RAS фосфорилирует и таким образом активирует RAF (A-RAF, B-RAF и C-RAF), которая, в свою очередь, фосфорилирует и активирует киназу с двойной специфичностью МЕК (MEK1/2). Затем активированная MEK фосфорилирует и активирует ERK1/2. После активации ERK1/2 активирует несколько субстратов в ядре и цитоплазме. В настоящее время известно более 200 субстратов ERK1/2, которые включают факторы транскрипции, киназы, фосфатазы и белки цитоскелета (Roskoski, Pharmacol. Res. 2012; 66: 105-143).
Идентифицирован ряд изозимов ERK (ERK1, ERK2, ERK3/4, ERK5, ERK7), однако интенсивнее всего изучаются изозимы ERK1 и ERK2: см. R. Roberts, J. Exp. Pharm., The extracellular signal-regulated kinase (ERK) pathway: a potential therapeutic target in hypertension 2012: 4, 77-83, и Cargnello с соавт., Microbiol. & Mol. Biol. Rev., Activation and Function of the MAPKs and Their Substrates, the MAPK-Activiated Protein Kinases 2011, 50-83.
Повышающая регуляция передачи сигнала ERK1/2 при раке
При раке часто наблюдается повышающая активация ERK1/2, обусловленная активирующими мутациями в вышележащих компонентах пути MAPK. Приблизительно 30% раковых заболеваний у человека характеризуются мутациями, активирующими RAS (Roberts, Der, Oncogene. 2007; 26: 3291-3310). Чаще всего мутации содержатся в изоформе K-RAS, которая мутирована в 22% всех опухолей. Мутации KRAS особенно часто встречаются при аденокарценоме поджелудочной железы, (70-90%), немлекоклеточной карциноме (10-20%) и раке толстой и прямой кишки (25-35%) (Neuzillet с соавт., 2014. Pharmacol. Ther. 141; 160-171). Мутации N-RAS и H-RAS присутствуют при 8% и 3% раков, соответственно (Prior с соавт., Cancer Res. 2012; 72 (10); 2457-2467). Важно, что активирующие мутации N-RAS отмечаются в 15-20% случаев меланомы. Кроме того, активирующие мутации B-RAF присутствуют в 8% всех опухолей и особенно часто встречаются при меланоме (50-60%), папиллярном раке щитовидной железы (40-60%), раке толстой и прямой кишки (5-10%) и немелкоклеточном раке легких (3-5%) (Neuzillet с соавт., 2014. Pharmacol. Ther. 141; 160-171). В дополнение к возникновению активирующих мутаций RAS и RAF, повышающая регуляция пути передачи сигнала MAPK при раке может быть обусловлена гиперэкспрессией или активацией в результате мутации вышележащих рецепторных тирозинкиназ, таких как EGFR (рецептор эпителиального фактора роста) (Lynch с соавт., N Engl J Med. 2004; 350: 2129-2139), HER2 (Stephens с соавт., Nature. 2004; 431: 525-526) и FGFR (Ahmed с соавт., Biochim. Biophys. Acta Mol. Cell. Res. 2012; 1823: 850-860).
Существует несколько механизмов, за счет которых аберрантная передача сигнала ERK1/2 может вносить вклад в прогрессирование рака. После активации ERK1/2 фосфорилирует и активирует широкий диапазон факторов транскрипции, участвующих в стимуляции пролиферации и дифференцировки клеток, таких как с-Fos (Murphy с соавт., Nat. Cell Biol. 2002: 4 (8):556-64) и ELK-1 (Gille с соавт., EMBO J. 1995; 14 (5):951-62). Также известно, что передача сигнала ERK1/2 стимулирует прохождение клеточного цикла за счет нескольких механизмов, включающих индукцию циклинов D-типа и подавление ингибитора циклин-зависимой киназы p27KIP1 (Kawada с соавт., Oncogene. 1997; 15: 629-637, Lavoie с соавт., J. Biol. Chem. 1996; 271: 20608-20616). Кроме-того, передача сигнала ERK1/2 может стимулировать выживание клеток путем регуляции ряда белков апоптоза. Примеры таких механизмов включают ERK1/2-зависимое подавления белков BIM1 и BAD проапоптотического семейства BCL-2 (She с соавт., J. Biol Chem. 2002; 277: 24039-24048. Ley с соавт., J. Biol. Chem. 2003; 278: 18811-18816) и ERK1/2-зависимую стабилизацию анти-апоптотических белков, таких как MCL-1 (Domina с соавт., Oncogene. 2004; 23: 5301-5315).
Роль ERK1/2 в устойчивости к ингибиторам MAPK
В многочисленных доклинических исследованиях было продемонстрировано, что ингибирование пути MAPK подавляет рост линий раковых клеток, несущих мутации B-Raf или Ras (Friday & Adjei, Clin. Cancer Res. 2008; 14: 342-346). Ингибиторы RAF вемурафениб и дабрафениб и ингибитор МЕКтраметиниб клинически одобрены для лечения меланомы с мутацией BRAF. Эти агенты вызывают глубинные противоопухолевые ответы у большинства пациентов, но продолжительность этих ответов невелика из-за возникновения приобретенной лекарственной устойчивости (Chapman с соавт., N. Engl. J. Med. 2011; 364 2507-2516. Hauschild с соавт., Lancet. 2012; 380: 358-365. Solit и Rosen, N Engl J Med. 2011; 364 (8): 772-774. Flaherty с соавт., N. Engl. J. Med. 2012; 367: 1694-1703). Выявлено несколько механизмов приобретенной устойчивости к ингибиторам B-RAF. Эти механизмы включают повышающую регуляцию или активацию альтернативных активаторов МЕК, таких как C-RAF или СОТ1 (Villanueva с соавт., Cancer Cell. 2010; 18:683-95. Johannessen с соавт., Nature. 2010; 468: 968-72), повышающую регуляцию передачи сигнала RTK или NRAS (Nazarian с соавт., Nature. 2010; 468:973-7) и возникновение мутаций, приводящих к активации MEK (Wagle с соавт., J Clin Oncol. 2011; 29:3085-96). Механизмы устойчивости к ингибиторам MEK включают возникновение мутаций MEK, которые снижают связывание лекарственного средства или повышают собственную активность MEK (Emery с соавт., Proc Natl. Acad. Sci. 2009; 106: 20411-20416. Wang с соавт., Cancer Res. 2011; 71: 5535-5545), а также амплификацию BRAF или KRAS (Little с соавт., Biochem Soc. Trans. 2012; 40(1): 73-8). Общей чертой механизмов устойчивости к ингибиторам RAF или MEK является реактивация передачи сигнала ERK1/2, которая обуславливает пролиферацию и выживание клеток в присутствии ингибиторов. На основании этого наблюдения было выдвинуто предположение о том, что прямое ингибирование ERK1/2 может быть эффективным терапевтическим подходом к преодолению приобретенной устойчивости к ингибиторам RAF или MEK. Доклинические исследования свидетельствуют в пользу того, что ингибирование ERK1/2 позволяет преодолеть приобретенную устойчивость к ингибиторам RAF или MEK (Hatzivassiliou с соавт., Mol Cancer Ther. 2012; 11(5):1143-54. Morris с соавт., Cancer Discov. 2013; 3(7):742-50).
Дополнительные заболевания
В дополнение к онкологическим заболеваниям, абнормальная передача сигнала ERK1/2 также отмечалась при других заболеваниях, включая сердечно-сосудистые заболевания (Muslin, Clin. Sci. 2008; 115: 203-218), болезнь Альцгеймера (Giovannini с соавт., Neuroscience. 2008; 153: 618-633), поликистозную болезнь почек (Omori с соавт., J Am Soc Nephrol. 2006; 17:1604-1614), астму (Duan с соавт., J Immunol. 2004; 172: 7053-7059) и эмфизему (Mercer с соавт., J. Biol. Chem. 2004; 279: 17690-17696).
(2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид
В нашей более ранней международной патентной заявке номер PCT/IB2016/001507 (содержание которой включено сюда посредством ссылки) раскрыт класс соединений бензолактама в качестве ингибиторов ERK2. Одним из конкретно раскрытых в указанной заявке соединений является соединение (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(S3)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид, имеющее формулу (1):

Получение указанного соединения описано в Примере 685 заявки PCT/IB2016/001507 и включает реакцию соединения формулы (2):
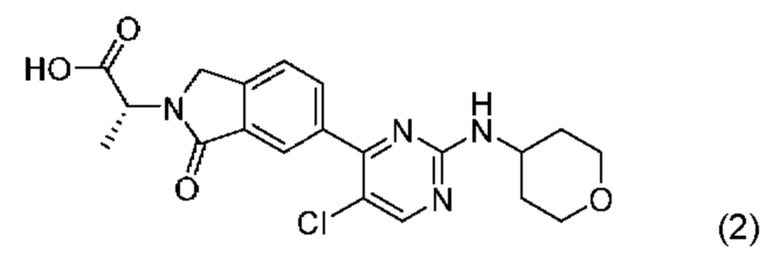
с соединением, описанным формулой (3):
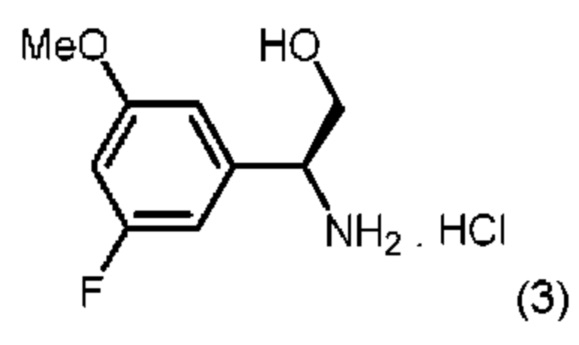
в диметилформамиде (ДМФА) и триэтиламине в присутствии ускорителя образования амидной связи O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (ТБТУ).
В Примере 685 раскрыто, что после обработки и частичной хроматографической очистки на диоксиде кремния элюат из хроматографической колонки упаривают с получением стекла, которое затем перетирают с эфиром, получая соединение (1) в виде аморфного твердого вещества.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В Примере 685 заявки PCT/IB2016/001507 соединение (1) получают в аморфной форме. Однако в настоящее время было обнаружено, что может быть получена кристаллическая форма соединения формулы (1). Соответственно, в первом аспекте изобретение относится к соединению формулы (1), в по существу кристаллической форме.
В настоящем изобретении также предложены новые способы получения соединения формулы (1) и синтетических промежуточных соединений, а также новые составы, содержащие соединение формулы (1).
КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЕДИНЕНИЯ ФОРМУЛЫ (1)
В первом аспекте согласно настоящему изобретению предложен (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид, имеющий формулу (1):
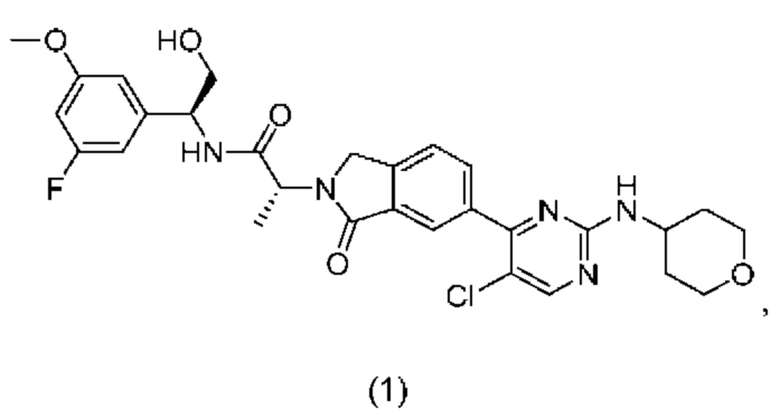
или его таутомерная форма, в по существу кристаллической форме.
Несмотря на то, что соединение формулы (1) может образовывать соли, ссылки на соединение в кристаллической форме являются ссылками на свободное основание.
Ссылки на соединение формулы (1), где это допускается контекстом, включают в свои рамки все сольваты, таутомеры и их изотопные варианты.
В твердом аморфном состоянии трехмерная структура, которая обычно существует в кристаллической форме, не существует, и положения молекул относительно друг друга в аморфной форме по существу случайны, см. в качестве примера Hancock et al. J. Pharm. Sci. (1997), 86, 1.
Термин «по существу кристаллический» относится к формам соединения формулы (1), в которых оно является кристаллическим на 50%-100%. В этом интервале соединение формулы (1) может быть по меньшей мере на 55% кристаллическим, или по меньшей мере на 60% кристаллическим, или по меньшей мере на 70% кристаллическим, или по меньшей мере на 80% кристаллическим, или по меньшей мере на 90% кристаллическим, или по меньшей мере на 95% кристаллическим, или по меньшей мере на 98% кристаллическим, или по меньшей мере на 99% кристаллическим, или по меньшей мере на 99,5% кристаллическим, или по меньшей мере на 99,9% кристаллическим, например, на 100% кристаллическим.
В частном варианте реализации кристаллическая форма является кристаллической на 95%-100%, например, по меньшей мере на 98% кристаллической, или по меньшей мере на 99% кристаллической, или по меньшей мере на 99,5% кристаллической, или по меньшей мере на 99,6% кристаллической, или по меньшей мере на 99,7% кристаллической, или по меньшей мере на 99,8% кристаллической, или по меньшей мере на 99,9% кристаллической, например, на 100% кристаллической.
Кристаллические формы соединения согласно настоящему изобретению могут быть сольватированы (например, гидратированы) или не сольватированы (например, безводны).
В одном варианте реализации соединение является безводным.
Используемый в настоящем описании термин «безводный» не исключает возможности присутствия некоторого количества воды на соединении или в соединении (например, в кристалле соединения). Например, некоторое количество воды может присутствовать на поверхности соединения (например, кристалла соединения), или незначительные количества могут присутствовать в теле соединения (например, в кристалле). Как правило, безводная форма содержит менее 0,4 молекул воды на молекулу соединения и более предпочтительно содержит менее 0,1 молекулы воды на молекулу соединения, например, 0 молекул воды.
В другом варианте реализации соединение сольватировано, например, гидратировано. Когда кристаллические формы гидратированы, они могут содержать, например, до трех молекул кристаллизационной воды, чаще всего до двух молекул воды, например, одна молекула воды или две молекулы воды. Также могут быть получены нестехиометрические гидраты, в которых количество присутствующих молекул воды меньше единицы или иначе не является целым числом. Например, там, где присутствует менее одной молекулы воды, может присутствовать, например, 0,4, или 0,5, или 0,6, или 0,7, или 0,8, или 0,9 молекул воды на молекулу соединения (1).
В одном варианте реализации кристаллическая форма соединения (1) представляет собой моногидрат, а кристаллическая форма содержит одну молекулу кристаллизационной воды.
Другие сольваты включают в себя алкоголяты, такие как этилаты и изопропилаты.
Описанные здесь кристаллические формы, их кристаллы и их кристаллическая структура образуют дополнительные аспекты настоящего изобретения.
Кристаллы и их кристаллические структуры могут быть охарактеризованы с использование ряда методов, включая порошковую рентгеновскую дифракцию (ПРД), дифракцию рентгеновских лучей на монокристаллах, дифференциальную сканирующую калориметрию (ДСК) и термогравиметрический анализ (ТГА). Поведение кристаллов в условиях различной влажности может быть проанализировано с помощью гравиметрических исследований сорбции пара (таких как динамическая сорбция пара).
Кристаллическая структура соединения может быть проанализирована методом твердофазной порошковой рентгеновской дифракции (ПРД). ПРД может быть проведена в соответствии с традиционным методами, такими как описано в настоящем документе (см. пример 4А) и во введении книги Introduction to X-ray Powder Diffraction, Roy Jenkins and Robert L. Snyder (John Wiley & Sons, New York, 1996). Наличие определенных пиков (в отличие от случайного фонового шума) на дифрактограмме ПРД указывает на то, что соединение имеет степень кристалличности.
Дифрактограмма порошковой рентгеновской дифракции соединения характеризуется параметрами угла дифракции (26) и межплоскостного расстояния (d) спектра дифракции рентгеновских лучей. Они связаны уравнением Брэгга, nλ=2d Sin θ, (где n=1; λ = длина волны рентгеновского излучения; d = межплоскостное расстояние; и θ = угол дифракции). Здесь межплоскостные расстояния, угол дифракции и общая картина важны для идентификации кристалла при порошковой рентгеновской дифракции по характеристическим данным. Относительную интенсивность не следует строго интерпретировать, поскольку она может варьироваться в зависимости от направления роста кристаллов, размеров частиц и условий измерения. Кроме того, под углами дифракции обычно понимают углы, которые совпадают в пределах 2θ±0,2°.
К настоящему времени идентифицированы две конкретные кристаллические формы свободного основания соединения (1), и они упоминаются в настоящем описании как «форма А» и «форма В». Из указанных двух форм форма В, по-видимому, является наиболее стабильной. Характеристические данные как для формы А, так и для формы В представлены в разделе «Примеры» ниже.
Обе формы А и В кристаллического (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида согласно настоящему изобретению охарактеризованы с помощью ПРД (см. примеры 3 и 4 и фигуры 1 и 2).
В каждом случае результаты порошковой рентгеновской дифракции могут быть выражены через угол дифракции (2θ), межплоскостное расстояние (d) и относительные интенсивности пиков на дифрактограмме.
Форма А
Первую кристаллическую форму (форму А) свободного основания соединения (1) можно получить диспропорционированием из раствора соли соляной кислоты соединения (1) в смеси растворителей вода:пропанол в соотношении 3:1, как описано в Примере 3А ниже. Дифрактограмма ПРД для формы А показана на фигуре 1. В процессе диспропорционирования кислота диссоциирует от свободного основания и остается в растворе, оставляя суспензию свободного основания соединения (1).
Кристаллическая форма А также может быть получена путем суспендирования аморфной соли соляной кислоты соединения (1) в воде при 70°С в течение длительного периода (например, 96 часов) и дальнейшей фильтрации кристаллического материала.
Форма В
Вторая кристаллическая форма (форма В) свободного основания также может быть получена путем диспропорционирования кислых солей (таких как гидрохлорид, сульфат или гидробромид) путем длительного перемешивания водной суспензии соли в очищенной воде, но при более низкой температуре, чем та, которая используется для образования кристаллической формы А выше. Таким образом, форму В можно получить, как описано в Примерах 3B-3D ниже, путем суспендирования аморфной кислой соли минеральных кислот (такой как соль гидрохлорида, сульфата или гидробромида) соединения (1) в очищенной воде при температуре 18-23°С, затем перемешивания смеси при 45-50°С в течение 20 часов с последующим дополнительным перемешиванием при 30-35°С в течение 96 часов и дальнейшей фильтрации кристаллов формы В. Порошковая дифрактограмма в формы В показана на фигуре. 2.
Диспропорционированию аморфной соли кислотного остатка соединения (1) в кристаллическую форму В может способствовать добавление к реакционной смеси спиртового сорастворителя, такого как изопропанол.
В другом способе получения кристаллической формы В соединение (1) в форме аморфного свободного основания может быть суспендировано в воде, которая может быть нестабилизрована или стабилизирована буферным раствором при рН от приблизительно 2 до рН 7, а затем перемешана при слегка повышенной температуре (например, 30°С) в течение периода времени (например, до шести дней, например, приблизительно пяти дней), достаточного для превращения аморфного соединения (1) в кристаллическую форму В.
Дифрактограмма рентгеновской дифракции кристаллической формы В соединения (1) демонстрирует пики наибольшей интенсивности при углах дифракции, указанных в таблице А, т.е. 14,0°, 20,6°, 24,0° и 24,2° (±0,2°).
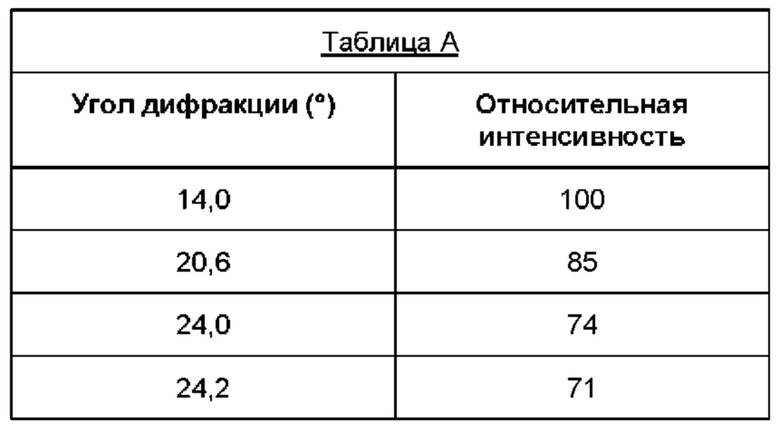
Соответственно, в другом варианте реализации настоящего изобретения предложена по существу кристаллическая форма (форма В) (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, дифрактограмма порошковой рентгеновской дифракции которого характеризуется наличием основных пиков при углах дифракции (29) 14,0 0 и/или 20,6 0 и/или 24,0 0 и/или 24,2 0 (±0,2°).
В одном варианте реализации дифрактограмма порошковой рентгеновской дифракции характеризуется наличием по меньшей мере одного пика под углом дифракции, выбранным из 14,0°, 20,6°, 24,0° и 24,2° (±0,2°).
Так, например, в одном варианте реализации настоящего изобретения предложена по существу кристаллическая форма (форму В) соединения (1), дифрактограмма порошковой рентгеновской дифракции которого характеризуется наличием основного пика при угле дифракции 14.0° (±0.2°).
В другом варианте реализации настоящего изобретения предложена по существу кристаллическая форма (форма В) соединения (1), дифрактограмма порошковой рентгеновской дифракции которого характеризуется наличием основного пика при угле дифракции 20.6° (±0.2°).
В еще одном варианте реализации настоящего изобретения предложена по существу кристаллическая форма (форма В) соединения (1), дифрактограмма порошковой рентгеновской дифракции которой характеризуется наличием основного пика при угле дифракции 24.0° (±0.2°).
В еще одном варианте реализации настоящего изобретения предложена по существу кристаллическая форма (форма В) соединения (1), дифрактограмма порошковой рентгеновской дифракции которой характеризуется наличием основного пика при угле дифракции 24.2° (±0.2°).
В других вариантах реализации по существу кристаллическая форма (форма В) соединения (1) имеет дифра кто грамму порошковой рентгеновской дифракции, характеризующуюся наличием основных пиков на двух или более, например, трех или более, и, в частности, четырех углах дифракции, выбранные из 14.0°, 20.6°, 24.0° и 24.2° (±0.2°).
Дифрактограмма порошковой рентгеновской дифракции формы В соединения (1) также может иметь меньшие пики, присутствующие при углах дифракции, указанных в таблице В, т.е. 8,8, 13,0, 13,8, 14,4, 17,3, 19,3, 21,3 и 28,7 (±0,2°).
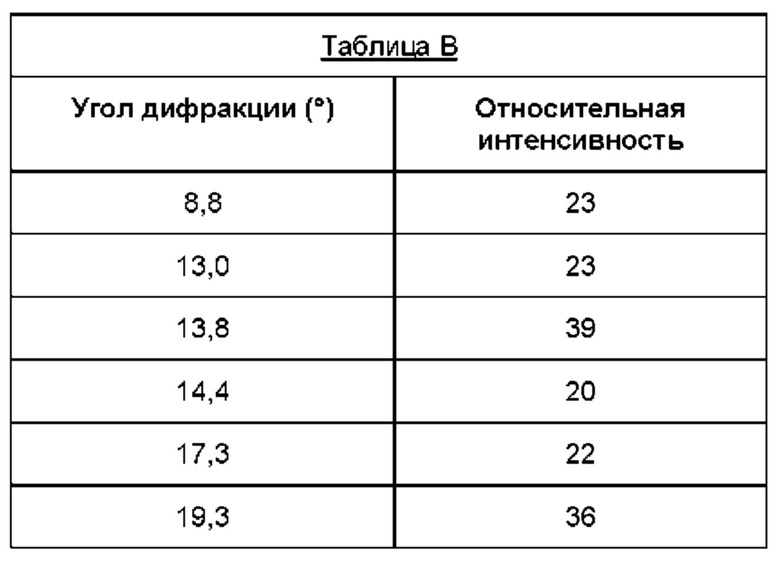
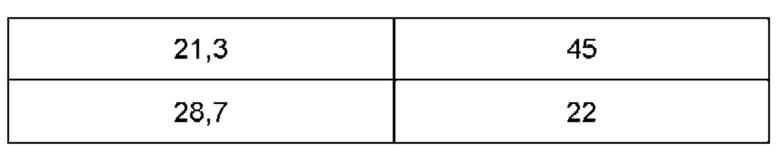
Следовательно, изобретение также обеспечивает по существу кристаллическую форму (форму В) соединения (1), дифрактограмма порошковой рентгеновской дифракции которого характеризуется наличием основных пиков при углах дифракции 14,0° и/или, 20,6 0 и/или 24,0 0 и/или 24,2 0 (±0,2°) как определено выше, и, необязательно, один или несколько дополнительных пиков при углах дифракции, выбранных из 8,8°, 13,0°, 13,8°, 14,4°, 17,3°, 19,3°, 21,3 0 и/или 28,7 0 (±0,2°).
Водном варианте реализации по существу кристаллическая форма (форма В) соединения (1) имеет дифра кто грамму порошковой рентгеновской дифракции, характеризующуюся наличием основных пиков при углах дифракции 14,0 0 и/или 20,6 0 и/или 24,0 0 и/или 24,2 0 (±0,2°); и, необязательно, один или несколько дополнительных пиков при углах дифракции 13,8 0 и/или 19,3 0 и/или 21,3 0 (±0,2°).
В конкретном варианте реализации по существу кристаллическая форма (форма В) соединения (1) имеет порошковая рентгенограмму, характеризующуюся наличием основных пиков при углах дифракции 14,0°, 20,6°, 24,0°, 24,2°, 13,8°, 19,3° и 21,3 0 (±0,2°).
В другом конкретном варианте реализации по существу кристаллическая форма (форма В) соединения (1) имеет дифра кто грамму порошковой рентгеновской дифракции, характеризующуюся наличием основных пиков при углах дифракции 14,0°, 20,6°, 24,0°, 24,2°, 8,8°, 13,0°, 13,8°, 14,4°, 17,3°, 19,3°, 21,3 0 и 28,7 0 (±0,2°).
Дифрактограмма порошковой рентгеновской дифракции может дополнительно характеризоваться наличием дополнительных пиков при углах дифракции (29) (±0,2°), указанных в таблице С.
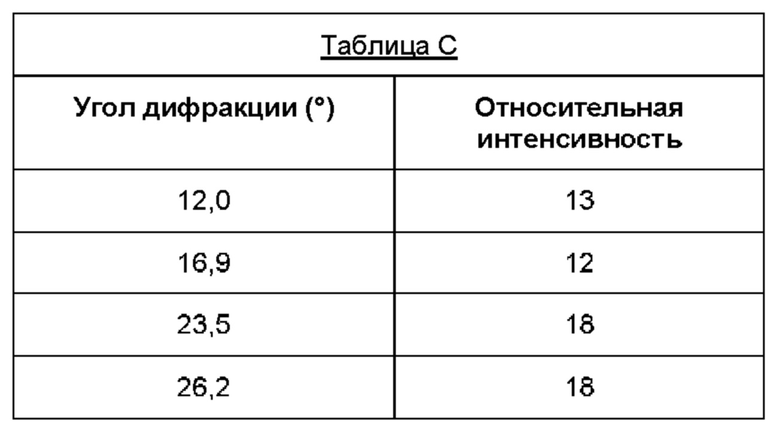
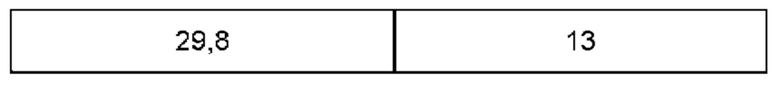
В настоящем изобретении также предложена по существу кристаллическая форма (форма В) соединения (1), которая демонстрирует пики при тех же углах дифракции, что и пики на дифрактограмме порошковой рентгеновской дифракции, показанной на фигуре 2. Предпочтительно пики имеют туже относительную интенсивность, что и пики на фигуре 2.
В предпочтительном варианте реализации в настоящем изобретении предложена по существу кристаллическая форма (форма В) соединения (1), имеющая дифрактограмму порошковой рентгеновской дифракции, по существу такую, как показано на фигуре 2.
Кристаллическая форма согласно настоящему изобретению также может быть охарактеризована с помощью дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма В соединения (1) была проанализирована с помощью ДСК и было обнаружено, что она демонстрирует эндотермический эффект с начальной температурой от 100°С до 110°С (в частности, от 101°С до 108°С) и пиком между 110°С и 125°С (в частности, между 111°С и 114°С) как показано на фигуре 3 прилагаемых чертежей. Это событие связано с выделением воды.
Соответственно, изобретение обеспечивает по существу кристаллическую форму (форму В) соединения (1), которая демонстрирует эндотермический эффект с начальной температурой от 100°С до 110°С (в частности, от 101°С до 108°С) при воздействии ДСК. Изобретение также обеспечивает по существу кристаллическую форму (форму В) соединения (1), которая демонстрирует эндотермический эффект с пиком между 110°С и 125°С (в частности, между 111°С и 113°С).
Кристаллическая форма (форма В) согласно настоящему изобретению также может быть охарактеризована с помощью термогравиметрического анализа (ТГА).
По существу кристаллическая форма В соединения (1) была проанализирована с помощью ТГА и демонстрирует переход потери массы с начальной температурой от 85°С до 95°С, например 90,86°С, который завершается при температуре от 110°С до 130°С, например 120°С (см. фигуру 4). Потеря массы соответствует выделению воды.
На основании данных ДСК и ТГА считают, что по существу кристаллическая форма В соединения (1), описанного выше, представляет собой моногидрат. Это также было подтверждено исследованиями дифракции рентгеновских лучей на монокристалле (см. Пример 4Е ниже).
В кристаллической по существу форме В соединения (1) может преобладать одна монокристаллическая форма, хотя другие кристаллические формы могут присутствовать в незначительных и предпочтительно незначительных количествах.
В предпочтительном варианте реализации изобретение относится к соединению (1) в практически кристаллической форме (форма В), содержащей одну кристаллическую форму, обладающую свойствами ПРД, описанными выше, и не более 5 масс. % любых других кристаллических форм соединения.
Предпочтительно, монокристаллическая форма (форма В) сопровождается менее чем 4%, или менее чем 3%, или менее чем 2% других кристаллических форм, и, в частности, содержит менее или равную приблизительно 1% по массе других кристаллических форм. Более предпочтительно, чтобы монокристаллическая форма сопровождалась менее чем 0,9%, или менее чем 0,8%, или менее чем 0,7%, или менее чем 0,6%, или менее чем 0,5%, или менее чем 0,4%, или менее чем 0,3% или менее 0,2%, или менее 0,1%, или менее 0,05%, или менее 0,01% по массе других кристаллических форм, например, 0% по массе других кристаллических форм.
Как видно из предыдущих абзацев, кристаллическая форма (форма В) соединения (1) может характеризоваться рядом различных физико-химических параметров. Соответственно, в одном конкретном варианте реализации изобретение обеспечивает по существу кристаллическую форму (форму В) соединения (1), которая характеризуется любым одним или несколькими (в любой комбинации) или всеми из следующих параметров, а именно, что она:
(a) имеет дифра кто грамму порошковой рентгеновской дифракции, характеризующуюся наличием основных пиков при углах дифракции (29) и интенсивностях, указанных в таблице А и, необязательно, в таблице В; и, дополнительно, необязательно, где дифрактограмма порошковой рентгеновской дифракции характеризуется наличием основных пиков при углах дифракции (29) и интенсивностях, указанных в таблице С; и/или
(b) демонстрирует пики при тех же углах дифракции, что и углы дифрактограммы порошковой рентгеновской дифракции, показанной на фигуре 2, и необязательно, где пики имеют те же относительные интенсивности, что и пики на фигуре 2; и/или
(c) имеет дифрактограмму порошковой рентгеновской дифракции, по существу такую, как показано на фигуре 2; и/или
(d) демонстрирует эндотермический пик между 100 и 115°С при воздействии ДСК; и/или
(е) демонстрирует потерю массы между 85 и 130°С (например, 90-120°С) при проведении термогравиметрического анализа (ТГА).
Способы получения кристаллических форм свободного основания соединения (1)
В настоящем изобретении также предложены способы получения кристаллических форм свободного основания соединения (1).
Соответственно, в другом аспекте настоящего изобретения предложен способ получения по существу кристаллической формы свободного основания соединения (1), включающий:
(i) получение водной суспензии кислотно-аддитивной соли соединения (1) и перемешивание суспензии при температуре от 25°С до 75°С в течение периода времени, достаточного для того, чтобы обеспечить диспропорционирование указанной кислотно-аддитивной соли, образование кристаллической формы свободного основания соединения (1) и после этого выделение кристаллической формы; или
(ii) получение водной суспензии аморфной формы свободного основания соединения (1), причем указанная водная суспензия не обработана буфером или обработана буфером до рН от 1,75 до 7,25, и перемешивание водной суспензии при температуре от 25°С до 55°С в течение периода времени, достаточного для того, чтобы обеспечить превращение аморфной формы свободного основания соединения (1) в кристаллическую форму соединения (1), и после этого выделение кристаллической формы.
В варианте способа (i) выбор температуры и времени перемешивания будет влиять на то, образуется ли кристаллическая форма А или форма В. Так, например, способ может быть проведен при более высокой температуре, например при 65-75°С (и более конкретно приблизительно при 70°С), для того чтобы получить форму А. И наоборот, указанный способ может быть проведен при более низкой температуре, например, при 25-55°С, с получением кристаллической формы В.
Образование кристаллической формы может быть облегчено добавлением в технологическую смесь спирта, такого как изопропанол.
Исходный материал для варианта способа (i) представляет собой кислотно-аддитивную соль соединения (1). Указанная кислотно-аддитивная соль может быть аморфной.
Кислотно-аддитивная соль соединения (1) может быть, например, солью минеральной кислоты, такой как гидрохлоридная, гидробромидная или сульфатная соль. Способы получения таких солей будут хорошо известны специалистам в данной области техники. Указанная соль может быть получена путем добавления свободного основания соединения к раствору противоиона в растворителе. Растворитель может быть полярным протонным растворителем, таким как 2-пропанол или метанол, и может содержать полярный апротонный сорастворитель, такой как дихлорметан.
Вариант способа (ii) обычно приводит к образованию формы В.
В варианте способа (ii) водная суспензия может быть обработана буфером или не обработана буфером. Например, она может быть либо не обработана буфером или обработана буфером до рН 2, 5 или 7. Водную суспензию перемешивают при слабом нагревании, например, при температуре от приблизительно 25 до приблизительно 35°С, например, приблизительно при 30°С. Водную суспензию перемешивают в течение периода времени, достаточного для того, чтобы обеспечить превращение аморфной формы свободного основания соединения (1) в кристаллическую форму свободного основания соединения (1). Как правило, ее перемешивают в течение по меньшей мере одного дня, чаще в течение по меньшей мере двух дней или по меньшей мере трех дней и в одном из вариантов реализации перемешивают в течение пяти дней.
При альтернативном наборе условий в варианте способа (ii) указанный процесс может быть проведен в течение более короткого периода времени (например, по меньшей мере в течение 12 часов или по меньшей мере 15 часов; например, 20 часов) при более высокой температуре (например, при 45-55°С, в частности при приблизительно 50°С). Затравочный кристалл либо формы А, либо формы В могут быть успешно добавлены с целью способствовать образованию кристаллической формы В.
Аморфные соли соединения Формулы (1)
Был получен ряд аморфных солей соединений формулы (1) (см. пример 2). Соответственно, в дополнительном аспекте согласно настоящему изобретению предложена соль соединения формулы (1) в аморфной форме.
Указанная соль может быть гидрохлоридом, сульфатом, нападизилатом (нафталин-1,5-дисульфонатом), эдисилатом (этандисульфонатом), тозилатом (п-толуолсульфонатом), мезилатом (метансульфонатом), напсилатом (2-нафталинсульфонатом), безилатом (бензолсульфонатом), изетионатом (2-гидроксиэтансульфонатом), эсилатом (этансульфонатом) или гидробромид ной солью соединения формулы (1).
Один набор солей состоит из аморфных гидрохлоридных, сульфатных, гид роб ром ид ных или нападизилатных солей соединения формулы (1).
В других вариантах реализации настоящего изобретения предложены:
• аморфная гидрохлоридная соль соединения формулы (1);
• аморфная сульфатная соль соединения формулы (1);
• аморфная гидробромидная соль соединения формулы (1); и
• аморфная нападизилатная соль соединения формулы (1).
Указанные аморфные соли могут быть получены взаимодействием формы свободного основания соединения с подходящей кислотой в органическом растворителе, предпочтительно в полярном апротонном растворителе (например, в изопропилацетате) или в смеси полярного апротонного растворителя и полярного протонного растворителя (например, смеси изопропилацетата и 2-пропанола).
Аморфные соли согласно настоящему изобретению могут быть предоставлены в форме частиц со среднемассовым диаметром в диапазоне от 1 мкм до 100 мкм.
Указанные частицы могут иметь среднемассовый диаметр в диапазоне от 2 мкм до 50 мкм, например, от 2 мкм до 25 мкм или от 2 мкм до 10 мкм. Частицы могут быть введены перорально (обычно в виде перорально вводимого состава, необязательно содержащего один или несколько фармацевтически приемлемых вспомогательных веществ) или другими способами, например, путем ингаляции. Когда частицы предназначены для введения путем ингаляции, частицы обычно имеют среднемассовый диаметр от 1 мкм до 10 мкм или от 1 мкм до 5 мкм.
Размеры частиц могут быть установлены методами анализа изображений, методами лазерной дифракции или методами просеивания.
Указанные частицы могут быть получены либо посредством способов механической микронизации, либо посредством способов разделения фаз на основе раствора. Примеры способов механической микронизации включают методы измельчения.
Основанные на растворах методы обычно включают в себя применение жидкостей, сжатых газов, околокритических жидкостей или сверхкритических флюидов в качестве растворителей или криогенных сред для быстрого замораживания. Эти методы включают разделение фаз растворителя и фармацевтического соединения путем выпаривания, расширения, замораживания или изменения состава растворителя.
Частицы могут быть получены лиофилизацией. В качестве альтернативы, частицы могут быть получены путем распылительной сушки. Соответственно, в одном из вариантов реализации аморфные соли согласно настоящему изобретению высушены распылением.
Аморфные соли могут быть применены в качестве терапевтических агентов или могут быть применены в качестве промежуточных соединений при получении кристаллических форм свободного основания соединения формулы (1), описанного выше.
СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ ФОРМУЛЫ (1)
Соединение (1) может быть получено посредством последовательных стадий способа, показанных на схеме 1 ниже.
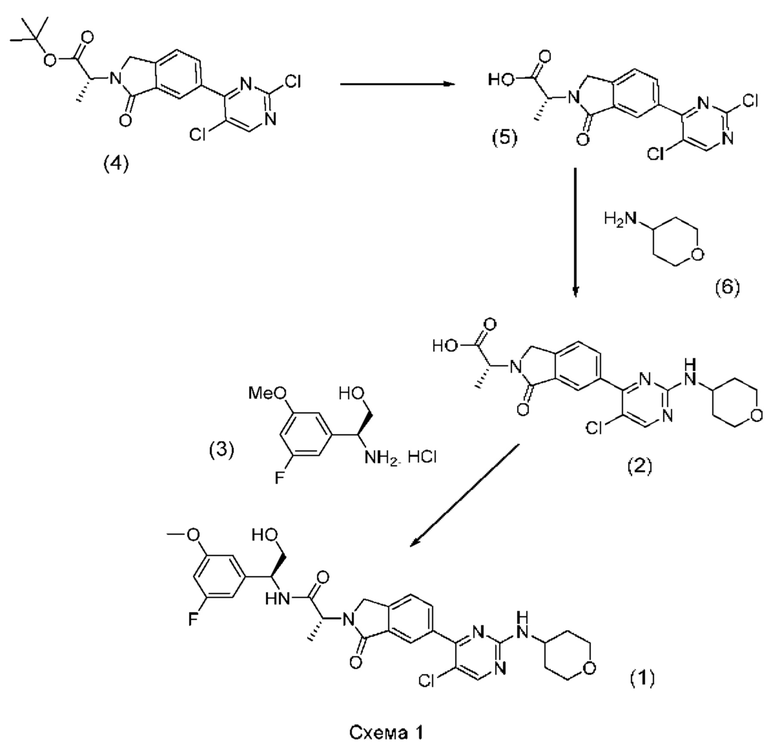
Реакция промежуточного соединения (2) с промежуточным соединением (3) с получением соединения (1) описана в примере 685 нашей предыдущей заявки PCT/IB2016/001507. Реакцию проводят в диметилформамиде (ДМФА) и триметиламине в присутствии промотора образования амидной связи O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (TBTU). В PCT/IB2016/001507 промежуточное соединение (2) получают гидролизом соответствующего трет-бутилсложного эфира, который получают реакцией промежуточного соединения (4) с амином (6).
Согласно настоящему изобретению был внесен ряд модификаций в способ, описанный в PCT/IB2016/001507.
Во-первых, были модифицированы условия способа для конечной стадии, реакции промежуточных соединений (2) и (3). Так, вместо применения реагента сочетания TBTU были применены альтернативные реагенты сочетания, такие как N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (HATU) и 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCI). Кроме того, наряду с триэтиламином были применены альтернативные основания, такие как диизопропилэтиламин (DIPEA) и 4-диметиламинопиридин (DMAP).
Во-вторых, вместо взаимодействия промежуточного соединения (4) с амином (6) и последующего гидролиза фрагмента трет-бутилсложного эфира с образованием соединения (2), как описано в PCT/IB2016/001507, фрагмент трет-бутилсложного эфира в промежуточном соединении (4) сначала гидролизуют с образованием карбоновой кислоты (5), которая затем реагирует с амином (6) с образованием соединения (2).
В соответствии с дополнительным аспектом согласно настоящему изобретению предложен способ получения соединения формулы (1), включающий проведение реакции соединения формулы (2) с соединением формулы (3):
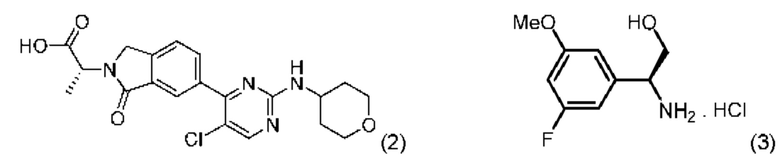
в апротонном растворителе в присутствии третичного аминного основания и промотирующего образование амидной связи агента, причем указанный промотирующий образование амидной связи агент выбран из N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфата (HATU) и 1-этил-3-(3-диметиламинопропил)карбодиимида (EDCI).
В конкретном варианте реализации указанный промотирующий образование амидной связи агент представляет собой N,N',N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (HATU).
Примеры третичных аминных оснований для применения в указанном способе представляют собой диизопропилэтиламин (DIPEA), 4-диметиламинопиридин (DMAP) и триэтиламин, а также их смеси.
В конкретном варианте реализации третичное аминное основание представляет собой диизопропилэтиламин (DIPEA).
Примеры апротонных растворителей представляют собой дихлорметан, этилацетат и диметилформамид.
В конкретном варианте реализации указанный апротонный растворитель представляет собой дихлорметан.
В предпочтительном варианте реализации предложен способ получения соединения формулы (1), включающий реакцию соединения формулы (2) с соединением формулы (3) в апротонном растворителе, представляющем собой дихлорметан, в присутствии третичного аминного основания, которое представляет собой диизопропилэтиламин (DIPEA), и промотирующего образование амидной связи агента, представляющего собой N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (HATU).
Реакцию между соединениями (2) и (3) обычно проводят без внешнего нагревания и она может, например, быть проведена при температуре не выше 25°С. Так, например, после того, как реагенты были смешаны с получением реакционной смеси, указанную реакционную смесь можно перемешивать при температуре в диапазоне 15-25°С до завершения реакции.
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (1), определенного в настоящем описании, включающий:
а) проведение реакции соединения формулы (5):

с соединением формулы (6)
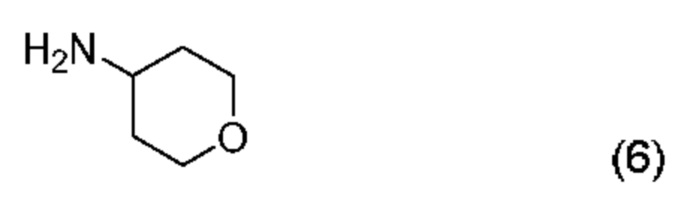
с получением соединения формулы (2):

и
b) проведение реакции соединения формулы (2) с соединением формулы (3):
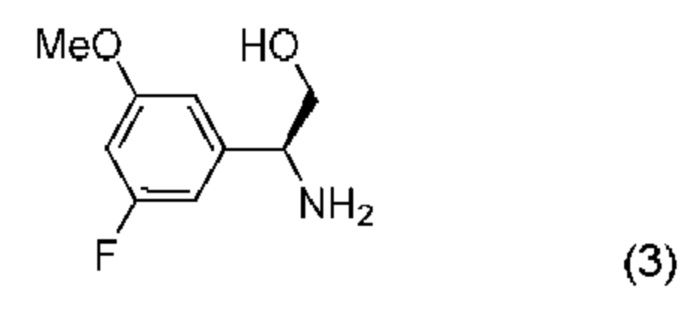
с получением соединения формулы (1) и после этого необязательное получение его соли или кристаллической формы.
Стадию (а) обычно проводят в полярном апротонном растворителе, таком как 1-метил-2-пирролидион (НМП). Указанную реакцию можно проводить при повышенных температурах; например, при температуре, превышающей 60°С, чаще превышающей 70°С, в частности при температуре в диапазоне 75-95°С (например, от 80 до 95°С).
Стадию (а) проводят в присутствии основания, которое может представлять собой неорганическое основание, такое как карбонат щелочного металла, например, карбонат калия.
Ход реакции между соединениями формул (5) и (6) можно контролировать с целью установления степени протекания реакции. Например, реакцию можно контролировать до тех пор, пока остаточное количество соединения формулы (5) не станет меньше желаемого уровня (например, менее 1 мол.% от его исходного количества). Время реакции на стадии (а) обычно составляет от 1 до 8 часов, например от 2 до 7 часов, и обычно от 4 до 6 часов.
Стадию (b) проводят в условиях, описанных выше для реакции между соединениями (2) и (3) с получением соединения (1).
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (2), включающий:
а) проведение реакции соединения формулы (5):
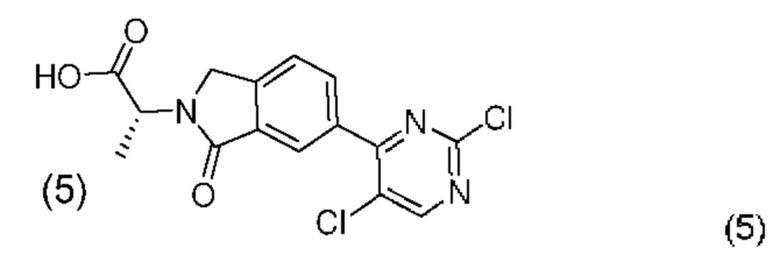
с соединением формулы (6)
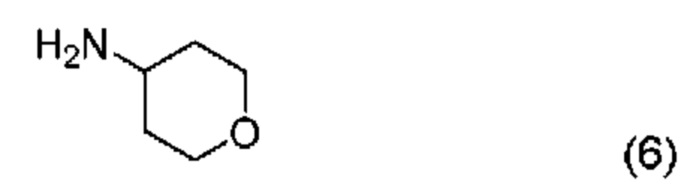
с получением соединения формулы (2):

Указанная реакция может быть проведена в условиях, описанных для стадии (b) выше.
Соединение (5) может быть получено гидролизом трет-бутилового сложного эфира соединения формулы (4):

например, с применением неорганической кислоты, такой как концентрированная соляная кислота. Реакции гидролиза может способствовать мягкое нагревание, например, до температуры в диапазоне 30-45°С и, более типично, в диапазоне от 35 до 40°С. Может быть применен сорастворитель, который может представлять собой углеводородный или хлорированный углеводородный растворитель. В одном случае такой сорастворитель представляет собой толуол.
Соединение (4) может быть получено способами, описанными в PCT/IB2016/001507; см., например, пример получения 94 в указанной заявке.
КОМПОЗИЦИИ, СОДЕРЖАЩИЕ СОЕДИНЕНИЕ ФОРМУЛЫ (1)
Соединение формулы (1) имеет относительно низкую растворимость в воде. В настоящем изобретении, таким образом, предложены композиции соединения (1), в которых содержится основной носитель, отличный от воды. Такие композиции подходят для перорального введения.
Было обнаружено, что соединение (1) имеет хорошую растворимость в ряде неводных растворителей. Соответственно, в настоящем изобретении предложена фармацевтическая композиция, содержащая соединение формулы (1) и носитель, выбранный из:
- С2-4 спирта;
- простого полиэфирного соединения;
- сложных моноэфиров жирных кислот с длиной цепи от С8 до C18 с глицерином или пропиленгликолем;
- ди- или триглицеридов жирных кислот с длиной цепи от С8 до С10; и их смесей.
Фармацевтическая композиция может принимать форму раствора соединения (1) в носителе.
В дополнительном аспекте согласно настоящему изобретению предложен способ получения фармацевтической композиции, содержащей соединение формулы (1), включающий диспергирование соединения формулы (1) в носителе, выбранном из:
- С2-4 спирта;
- простого полиэфирного соединения;
- сложных моноэфиров жирных кислот с длиной цепи от С8 до C18 с глицерином или пропиленгликолем;
- ди- или триглицеридов жирных кислот с длиной цепи от С8 до С10; и их смесей.
Обычно соединение формулы (1) диспергируют в носителе с получением раствора или суспензии. В одном из вариантов реализации соединение формулы (1) суспендируют в носителе. В другом варианте реализации соединение формулы (1) растворяют в носителе с получением суспензии.
Носитель может содержать одноатомный или многоатомный С2-4 спирт, предпочтительно С2-3 спирт, например, этанол или про пилен гликоль.
Когда носитель содержит простое полиэфирное соединение, указанное простое полиэфирное соединение может представлять собой полиэтиленгликоль (ПЭГ). Полиэтиленгликоль может иметь среднюю молекулярную массу от 200 до 10000 г/моль, например, от 300 до 8000 г/моль. В одном из вариантов реализации полиэтиленгликоль имеет среднюю молекулярную массу приблизительно от 200 до 400 г/моль, например, от 300 до 450 г/моль.
В зависимости от природы и относительных количеств компонентов носителя указанные композиции могут быть жидкими, полутвердыми или твердыми. Например, при применении ПЭГ с более высокой молекулярной массой, вязкость композиции может быть увеличена до такой степени, что ее можно рассматривать как «полутвердую» или твердую, тогда как ПЭГ с более низкой молекулярной массой могут приводить к образованию жидких композиций. Ссылки на растворы в контексте таких композиций включают твердые растворы, а также жидкие (или полутвердые) растворы.
В качестве альтернативы или дополнительно носитель может содержать каприловую или каприновую кислоту и моно-, ди- и три-сложные эфиры каприловой или каприновой кислот.Примеры таких сложных эфиров включают монокаприлат пропиленгликоля, монокаприлат глицерина, дикаприлат глицерина, трикаприлат глицерина, монокапрат глицерина, дикапрат глицерина и трикапрат глицерина. Каприлокапроил макрогол-8 глицериды (Labrasol® ALF) представляют собой коммерчески доступный носитель, который содержит смесь моно-, ди- и триглицериновых сложных эфиров каприловой и каприновой кислот, а также моно- и дисложные эфиры полиэтиленгликолей со средней молекулярной молекулярной массой от 200 до 400 г/моль.
В другом альтернативном варианте указанный носитель может содержать моноглицерид жирной кислоты с более длинной цепью, такой как линолевая кислота или олеиновая кислота. Примеры таких носителей включают монолинолеат глицерина (Maisine СС™) и моноолеаты глицерина (тип 40, Peceol™).
В одном из вариантов реализации изобретения носитель выбран из этанола, пропиленгликоля, полиэтиленгликоля и их смесей. Например, указанный носитель может содержать комбинацию пропиленгликоля и этанола, такую как комбинацию пропиленгликоля и этанола в отношении от 50:50 до 90:10% масс./масс. (например, пропиленгликоль и этанол в отношении 75:25 или 85:15% масс./масс.). В одном из вариантов реализации носитель содержит комбинацию пропиленгликоля и этанола в отношении от 75:25 до 90:10% масс./масс.
В другом варианте реализации носитель выбран из этанола, полиэтиленгликоля 400 (ПЭГ 400) и пропиленгликоля, а также их смесей.
Композиция обычно позволяет вводить соединение (1) перорально с получением общей суточной дозы до 1,2 г в сутки. В композициях согласно настоящему изобретению концентрация соединения (1) в носителе может находиться в диапазоне от 10 мг/мл до 130 мг/мл, например от 40 мг/мл до 125 мг/мл и, более конкретно, от 110 мг/мл до 125 мг/мл. Концентрация 120 мг/мл позволяет вводить дозу 1,2 г соединения (1) в 10 мл композиции.
В качестве альтернативы композиция может содержаться в капсуле. Подходящие капсулы для доставки композиций, описанных в настоящем документе, включают твердые или мягкие желатиновые капсулы. В одном из вариантов реализации композиция содержится в мягкой желатиновой капсуле. В контексте настоящего описания термин «желатин» относится не только к капсулам, изготовленным из желатина как такового, но также к капсулам, изготовленным из нежелатиновых эквивалентов, таких как пуллулан, или модифицированные целлюлозы, такие как гидроксипропилметилцеллюлоза.
Композиция также может содержать одно или несколько поверхностно-активных веществ, способствующих растворимости соединения (1) в выбранном носителе (носителях). Поверхностно-активные вещества также могут ингибировать осаждение соединения формулы (1) при разведении композиции в желудочно-кишечном тракте.
Поверхностно-активные вещества обычно представляют собой неионные поверхностно-активные вещества.
Неионным поверхностно-активным веществом может быть, например, сложный эфир полиола, сложный эфир полиоксиэтилена или полоксамер.
В одном из вариантов реализации указанное поверхностно-активное вещество представляет собой токоферол полиэтиленгликоля (TPG), такой как D-α-токоферол полиэтиленгликоль сукцинат (TPGS), имеющий формулу:
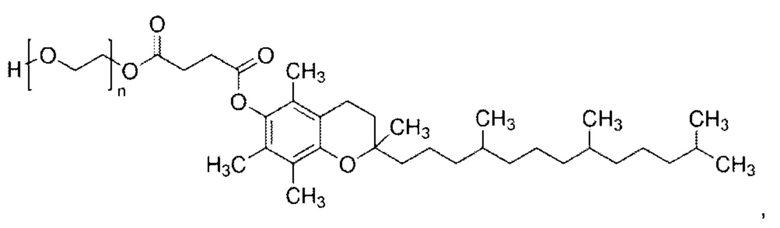
где среднее значение n находится в области от приблизительно 10 до приблизительно 30, более типично в диапазоне от приблизительно 15 до приблизительно 27; например, в диапазоне от приблизительно 20 до 25. Один конкретный TPGS представляет собой α-токоферол полиэтиленгликоль 1000 сукцинат (приблизительная средняя молекулярная масса 1513), где полиоксиэтиленовый фрагмент [-O-СН2-СН2]n имеет молекулярную массу приблизительно 1000 (например, от 950 до 1050), и среднее значение n составляет приблизительно 22. Примеры сложных эфиров полиолов включают сложные эфиры гликоля и глицерина и производные сорбитана.
Сложные эфиры жирных кислот и сорбита (обычно называемые Spans (спанами)) и их этоксилированные производные (обычно называемые Tweens (твинами)) включают сорбитан монолаурат (Span 20), сорбитан монопальмитат (Span 40), сорбитан моностеарат (Span 60), сорбитан моноолеат (Span 80), сорбитан тристеарат (Span 65), сорбитан триолеат (Span 8), полиоксиэтилен (20) сорбитан монолаурат (Tween 20), полиоксиэтилен (20) сорбитан монопальмитат (Tween 40), полиоксиэтилен (20) сорбитан моностеарат (Tween 60), полиоксиэтилен (20) сорбитан моноолеат (Tween 80), полиоксиэтилен (20) сорбитан тристеарат (Tween 65) и полиоксиэтилен (20) сорбитан триолеат (Tween 85).
Другие конкретные примеры поверхностно-активных веществ включают полиоксил 40 гидрированное касторовое масло (Cremophor RH 40, Kolliphor® RH40), полиоксил 35 касторовое масло (Cremophor EL, Kolliphor® EL), полисорбат 80 (Tween 80), Gelucire 44/14 (глицериды лауроил макрогола-32), Solutol HS-15 (гидроксистеарат макрогола 15) и Labrasol® ALF (глицериды каприлокапроил макрогола-8). В одном из вариантов реализаци поверхностно-активное вещество представляет собой Cremophor RH 40.
В одном из вариантов реализации композиция содержит соединение формулы (1), этанол и токоферол полиэтиленгликоль (TPG), например D-α-токоферол полиэтиленгликоль 1000 сукцинат (TPGS). Этанол и TPG могут присутствовать в отношении от 20:80 этанол:ТРС до 60:40 этанол:ТРС, например, от 30:70 этанол:ТРО до 50:50 этанол:ТРС
В другом варианте реализации композиция содержит, в дополнение к соединению формулы (1), носитель, содержащий:
(i) про пилен гликоль;
(ii) неионное поверхностно-активное вещество (такое как Cremophor RH40 и токоферол полиэтиленгликоль); и необязательно
(iii) этанол.
В другом варианте реализации композиция содержит, в дополнение к соединению формулы (1), носитель, содержащий:
(i) про пилен гликоль;
(ii) неионное поверхностно-активное вещество на основе сложного эфира полиоксиэтилена; и необязательно
(iii) этанол.
Указанное неионогенное поверхностно-активное вещество на основе сложного эфира полиоксиэтилена может представлять собой, например, токоферол полиэтиленгликоль (TPG), такой как D-α-токоферол полиэтиленгликоль сукцинат (TPGS), как определено в настоящем описании выше.
В одном конкретном варианте реализации указанные композиции не содержат этанол (iii).
В другом конкретном варианте реализации указанные композиции содержат этанол (iii).
В другом конкретном варианте реализации носитель содержит пропиленгликоль и токоферол полиэтиленгликоль (TPG). В этом варианте реализации указанный носитель может содержать TPGS в массовом отношении от 1:2 до 10:1 пропиленгликоль:ТРС5 и может необязательно дополнительно содержать этанол в массовом отношении от 1:10 до 2:1 этанол:пропиленгликоль (например, D-a-токоферол полиэтиленгликоль 1000 сукцинат (TPGS)) в массовом отношении от 1:2 до 5:1 пропиленгликоль:токоферол полиэтиленгликоль; и необязательно дополнительно содержать этанол в массовом отношении этанол:пропиленгликоль от 1:10 до 2:1.
Композиции, содержащие соединение формулы (1), пропиленгликоль, неионное поверхностно-активное вещество на основе полиоксиэтиленового сложного эфира; и необязательно этанол, могут легко быть заключены в капсулу, например, в твердую желатиновую или мягкую желатиновую капсулу.
Соединение формулы (1) на стадии а) может быть в кристаллической форме. В одном из вариантов реализации соединение формулы (1) на стадии а) находится в кристаллической форме, как описано в настоящем описании.
Низкая растворимость в воде фармацевтических соединений может быть улучшена путем уменьшения размера твердых частиц соединений. При уменьшении размера частиц фармацевтического соединения площадь поверхности, доступная для сольватации, увеличивается.
Соответственно, в дополнительном аспекте изобретения предложено соединение формулы (1) в форме частиц, имеющих средне массовый диаметр от 1 мкм до 100 мкм.
Указанные частицы могут иметь среднемассовый диаметр от 2 мкм до 50 мкм, например, от 2 мка до 25 мкм или от 2 мкм до 10 мкм. Частицы можно вводить перорально (обычно в виде перорально вводимого состава, необязательно содержащего один или несколько фармацевтически приемлемых вспомогательных веществ) или другими способами, например, путем ингаляции. Когда частицы предназначены для введения путем ингаляции, частицы обычно имеют среднемассовый диаметр от 1 мкм до 10 мкм или от 1 мкм до 5 мкм.
Размеры частиц могут быть установлены способами анализа изображений, способами лазерной дифракции или методами просеивания.
Указанные частицы могут быть получены либо посредством способов механической микронизации, либо посредством способов разделения фаз на основе раствора. Примеры способов механической микронизации включают методы измельчения.
Основанные на растворах методы обычно включают применение жидкостей, сжатых газов, околокритических жидкостей или сверхкритических флюидов в качестве растворителей или криогенных сред для быстрого замораживания. Эти методы включают разделение фаз растворителя и фармацевтического соединения путем испарения, расширения, замораживания или изменения композиции растворителя.
Частицы могут быть получены лиофилизацией. В качестве альтернативы, частицы могут быть сформированы распылительной сушкой.
ОПРЕДЕЛЕНИЯ
Соединение формулы (1) может быть упомянуто в настоящей заявке под его химическим названием или, для удобства, как «соединение», «соединение формулы (1)», «соединение (1)» или «соединение согласно настоящему изобретению». Каждый из этих синонимов относится к соединению, изображенному в формуле (1) выше и имеющему химическое название (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид.
Термин «среднемассовый диаметр», используемый в настоящем описании для определения размеров частиц, определен как диаметр, при котором 50% частиц по массе имеют больший диаметр и 50% меньший диаметр. Указанный диаметр относится к эквивалентному сферическому диаметру, который для несферической частицы равен диаметру сферической частицы, имеющей такой же объем, как у несферической частицы.
Под ERK1/2 мы подразумеваем либо один из, либо оба изозима ERK1 и ERK2 киназ, регулируемых внеклеточными сигналами (ERK).
«Удельная активность» представляет собой меру активности лекарственного средства в терминах количества, требующегося для достижения эффекта заданной интенсивности. Высокоэффективный лекарственный препарат вызывает более сильный ответ при низких концентрациях. Удельная активность пропорциональна сродству и эффективности. Аффинность - это способность лекарственного средства связываться с ферментом. Эффективность - это взаимосвязь между способностью связывать мишень и способностью инициировать ответ на молекулярном, клеточном, тканевом или системном уровне.
Термин «ингибитор» относится к ингибитору фермента, который представляет собой тип лиганда или лекарственного средства, которое блокирует или ослабляет биологические реакции, опосредуемые ERK1/2. Ингибиторы осуществляют свое действие путем связывания с активным сайтом или с аллостерическими сайтами ферментов или могут взаимодействовать с уникальными сайтами связывания, которые обычно не участвуют в биологической регуляции активности фермента. Ингибирование может осуществляться прямо или косвенно и может быть опосредовано любым механизмом и на любом физиологическом уровне. В результате ингибирование лигандами или лекарственными средствами может при различных обстоятельствах проявляться функционально по-разному. Ингибирующая активность может быть обратимой или необратимой в зависимости от долговечности комплекса ингибитор-фермент, который, в свою очередь, зависит от природы связывания ингибитора и фермента.
Термин «лечение», использующийся в настоящем документе в контексте лечения состояния, т.е. состояния, нарушения или заболевания, обычно относится к лечению и терапии человека или животного (например, в ветеринарных применениях), в которых достигаются некоторый желаемый терапевтический эффект, например, ингибирование прогрессирования состояния, и включает снижение скорости прогрессирования, остановку прогрессирования, улучшение состояния, снижение или уменьшение по мере одного симптома, связанного с состоянием или вызванного состоянием, которое подвергается лечению, и лечение этого состояния. Например, лечением может быть уменьшение одного или нескольких симптомов нарушения или полное искоренение нарушения.
Термин «профилактика» (т.е. применение соединений в качестве профилактической меры), используемый в настоящем документе в контексте лечения состояния, т.е. состояния, нарушения или заболевания, относится обычно к профилактике или предотвращению для человека или животного (например, в ветеринарных применениях), в которых достигается некоторый желаемый профилактический эффект, например, предотвращение возникновения заболевания или защита от заболевания. Профилактика включает полное и общее блокирование всех симптомов нарушения в течение неопределенного периода времени, простое замедление наступления одного или нескольких симптомов заболевания или уменьшение вероятности возникновения заболевания и не включает улучшение состояния, уменьшение или облегчение по крайней мере одного симптома, связанного с состоянием или вызванного им, подлежащего лечению, и лечение этого состояния.
Указания на профилактику или лечение болезненного состояния или состояния, такого как рак, включают облегчение или уменьшение заболеваемости, например, раком.
В настоящем тексте термин «опосредуемый», используемый, например, в сочетании с ERK1/2, как описано в настоящем документе (и применяемый, например, к различным физиологическим процессам, заболеваниям, патологическим состояниям, состояниям, терапиям, лечениям или вмешательствам), предназначен для ограниченного использования так, что различные процессы, заболевания, патологические состояния, состояния, лечения и вмешательства, к которым применяется этот термин, относятся к тем, в которых указанный белок играет биологическую роль. В случаях, когда этот термин применяется к заболеванию, патологическому состоянию или состоянию, биологическая роль белка может быть прямой или косвенной и может быть необходимой и/или достаточной для проявления симптомов заболевания, патологического состояния или состояния (или его этиология или прогрессирование). Соответственно, функция белка (и в частности аберрантные уровни функционирования, например, повышенная экспрессия или пониженная экспрессия) необязательно должна быть проксимальной причиной заболевания, патологического состояния или состояния: скорее предполагается, что опосредуемые заболевания, патологические состояния или состояния включают те, которые имеют многофакторную этиологию и сложное прогрессирование, в которых рассматриваемый белок участвует лишь частично. В тех случаях, когда этот термин применяется для лечения, профилактики или вмешательства, действие белка может быть прямым или косвенным и может быть необходимым и/или достаточным для лечения, профилактики или результата вмешательства. Соответственно, болезненное состояние или состояние, опосредуемое белком, включает развитие устойчивости к любому конкретному лекарственному препарату или лечению.
Комбинации настоящего изобретения могут давать терапевтически эффективный результат по отношению к терапевтическому результату отдельных соединений/агентов при их раздельном введении.
Термин «эффективный» включает такие благоприятные эффекты, как аддитивность, синергизм, снижение побочных эффектов, снижение токсичности, увеличение времени до прогрессирования заболевания, увеличение времени выживания, сенсибилизация или обратная сенсибилизация одного агента к другому или улучшение скорости ответа. Преимущественно эффективный результат может позволить снижать дозы каждого или любого из компонентов, которые будут вводиться пациенту, тем самым снижая токсичность химиотерапии, одновременно получая и/или поддерживая тот же терапевтический результат.«Синергетический» результат в данном контексте относится к терапевтическому результату, создаваемому комбинацией, который больше, чем сумма терапевтических эффектов агентов комбинации при их индивидуальном ведении. «Аддитивный» результат в данном контексте относится к терапевтическому результату, создаваемому комбинацией, который больше, чем терапевтический результат любого из агентов комбинации при индивидуальном введении. Термин «скорость ответа», использующийся в настоящем документе, относится, в случае солидной опухоли, к степени уменьшения размера опухоли в определенный момент времени, например, 12 недель. Так, например, 50%-ная скорость ответа означает уменьшение размера опухоли на 50%. Ссылки в настоящем документе на «клинический ответ» относятся к частоте ответов 50% или более. «Частичный ответ» определяется в настоящем документе как показатель отклика менее 50% при условии, что он больше 0%.
В настоящем тексте термин «комбинация», применяемая к двум или более соединениям и/или агентам, предназначен для определения материала, в котором находятся вместе два или более агентов. Термины «комбинированное» и «комбинирование» в этом контексте должны интерпретироваться соответствующим образом.
Объединение двух или более соединений/агентов в комбинации может быть физическим или нефизическим. Примеры физически объединенных комбинированных соединений/агентов включают:
• композиции (например, единые составы), содержащие два или более соединений/агентов в смеси (например, в одной и той же единице лекарственной формы),
• композиции, содержащие материал, в котором два или более соединения/агента являются химически/физико-химически связанными (например, поперечными сшивками, в виде молекулярного агломерата или связывания с фрагментами общего носителя),
• композиции, содержащие материал, в котором два или более соединений/агентов химически/физико-химически совместно упакованы (например, расположенными на липидных везикулах, частицах (например, микро- или наночастицах) или каплях эмульсии или внутри них),
• фармацевтические наборы, фармацевтические упаковки или упаковки для пациентов, в которых два или более соединений/агентов совместно упакованы или совместно представлены (например, как часть массива единичных доз),
Примеры нефизически объединенных комбинированных соединений/агентов включают:
• материал (например, неединый состав), включающий по меньшей мере одно из двух или более соединений/агентов вместе с инструкциями для экстемпорального объединения по мере одного соединения с образованием физической композиции двух или более соединений/агентов,
• материал (например, неединый состав), включающий по меньшей мере одно из двух или более соединений/агентов вместе с инструкциями для комбинированной терапии с применением двух или более соединений/агентов,
• материал, включающий по меньшей мере одно из двух или более соединений/агентов вместе с инструкциями для введения популяции пациентов, в которой другое (другие) из двух или более соединений/агентов были введены (или вводятся),
• материал, содержащий по меньшей мере одно из двух или более соединений/агентов в количестве или в форме, которая специально адаптирована для применения в комбинации с другим (другими) веществами двух или более соединений/агентов.
В настоящем документе термин «комбинированная терапия» предназначен для определения видов терапии, которые включают применение комбинации двух или более соединений/агентов (как определено выше). Таким образом, указания на «комбинированную терапию», «комбинации» и применение соединений/агентов «в комбинации» в этой заявке могут относиться к соединениям/агентам, которые вводят как часть одного и того же режима лечения. Таким образом, дозирование каждого из двух или более соединений/агентов может различаться: они могут вводиться в одно и то же время или в разное время. Поэтому следует понимать, что соединения/агенты комбинации можно вводить последовательно (например, до или после) или одновременно, либо в одном фармацевтическом составе (т.е. вместе), либо в разных фармацевтических составах (т.е., отдельно). Одновременное введение в одном составе определяется как единый состав, тогда как одновременное введение в разных фармацевтических составах представляет собой неединый состав. Дозирование каждого из двух или более соединений/агентов в комбинированной терапии может также различаться в отношении путей введения.
В настоящем тексте термин «фармацевтический набор» определяет массив из одной или нескольких единичных доз фармацевтической композиции вместе с дозирующими средствами (например, измерительным устройством) и/или средствами доставки (например, ингалятор или шприц), при этом все из них, необязательно, содержатся в общей внешней упаковке. В фармацевтических наборах, содержащих комбинацию из двух или более соединений/агентов, отдельные соединения/агенты могут иметь унитарные или неединые составы. Единичная доза (дозы) могут содержаться в блистерной упаковке. Фармацевтический набор может дополнительно содержать инструкцию по применению.
В настоящем тексте термин «фармацевтическая упаковка» определяет массив одной или нескольких единичных доз фармацевтической композиции, необязательно содержащихся в общей внешней упаковке. В фармацевтических упаковках, содержащих комбинацию из двух или более соединений/агентов, отдельные соединения/агенты могут иметь унитарные или неединые составы. Единичная доза (дозы) может содержаться в блистерной упаковке. Фармацевтическая упаковка может дополнительно содержать инструкцию по применению.
Соли, сольваты, таутомеры и изотопы
Ссылка на соединение формулы (1) включает ионные формы, соли, сольваты, таутомеры и их изотопные варианты, если в контексте не указано иное.
Соли
Соединение формулы (1) может существовать в форме солей и, в частности, кислотно-аддитивных солей. Все такие соли входят в объем настоящего изобретения, и ссылки на соединение формулы (1) включают солевые формы соединения, если в контексте не указано иное.
Соли соединения (1) могут быть синтезированы из соединения (1) традиционными химическими способами, такими как способы, описанные в Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 pages, August 2002. Обычно такие соли могут быть получены реакцией формы свободного основания соединения с подходящей кислотой в воде или в органическом растворителе или в смеси того и другого; обычно применяют неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Кислотно-аддитивные соли (моно- или дисоли) могут быть образованы с широким спектром кислот, как неорганических, так и органических. Примеры кислотно-аддитивных солей включают моно- или дисоли, образованные кислотой, выбранной из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетамидобензойной, масляной, (+)-камфорной, камфор-сульфоновой, (+)-(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламовой, додецилсульфоновой, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, галогенводородной кислоты (например, бромистоводородной, соляной, йодистоводородной), изэтиновой, молочной (например, (+)-L-молочной, (±)-DL-молочной), лактобионовой, малеиновой, яблочной, (-) -L-малоновой, малоновой, (±)-DL-миндальной, метансульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой, 1-гидрокси-2-нафталевой, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памоевой, фосфорной, пропионовой, пирувиновой, L-пироглутаминовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, (+)-L-винной, тиоциановой, п-толуолсульфоновой, ундециленовой и валериановой кислот, ацилированных аминокислот и катионообменных смол.
Одна конкретная группа солей состоит из солей, образованных из уксусной, соляной, йодоводородной, фосфорной, азотной, серной, лимонной, молочной, янтарной, малеиновой, яблочной, изиотионовой, фумаровой, бензолсульфоновой, толуолсульфоновой, метансульфоновой (мэзилат), этансульфоновой, нафталинсульфоновой, валериановой, уксусной, пропионовой, бутановой, малоновой, глюкуроновой и лактобионовой кислот. Одной конкретной солью является соль гидрохлорида.
Если соединение является анионным или имеет функциональную группу, которая может быть анионной (например, СООН может быть СОО-), то соль может быть образована с органическими или неорганическими основаниями, образующими подходящий катион. Примеры подходящих неорганических катионов включают ионы щелочных металлов, такие как Li+, Na+ и K+, катионы щелочноземельных металлов, такие как Са2+ и Mg2+, и другие катионы, такие как Al3+ или Zn+, но не ограничиваются ими. Примеры подходящих органических катионов включают ионы аммония (т.е., NH4+) и замещенные ионы аммония (например, NH3R+, NH2R2+, NHR3+, NR4+), но не ограничиваются ими. Примерами некоторых подходящих замещенных ионов аммония являются те, которые получены из метиламина, этиламина, диэтиламина, пропиламина, дициклогексиламина, триэтиламина, бутиламина, этилендиамина, этаноламина, диэтаноламина, пиперазина, бензиламина, фенилбензиламина, холина, меглумина и тромэтамина, а также аминокислот, таких как лизин и аргинин. Пример обычного четвертичного аммония представляет собой N(СН3)4+.
Когда соединения формулы (0) содержат аминную функциональную группу, они могут образовывать соли четвертичного аммония, например, путем взаимодействия с алкилирующим агентом в соответствии с методами, хорошо известными специалисту. Такие соединения четвертичного аммония находятся в рамках Формулы (0).
Соединения согласно настоящему изобретению могут существовать в виде моно- или ди-солей в зависимости от рКа кислоты, из которой образуется соль.
Солевые формы соединений настоящего изобретения обычно являются фармацевтически приемлемыми солями, и примеры фармацевтически приемлемых солей обсуждаются в Berge и др., 1977, «Pharmaceutically Acceptable Salts» J. Pharm. Sci., т. 66, с. 1-19. Однако соли, которые не являются фармацевтически приемлемыми, также могут быть получены в виде промежуточных форм, которые затем могут быть превращены в фармацевтически приемлемые соли. Такие не фармацевтически приемлемые соли, которые могут быть полезными, образуются, например, в соединениях при очистке или разделении соединений согласно настоящему изобретению, которые также являются частью настоящего изобретения.
Геометрические изомеры и таутомеры
Соединение формулы (1) может существовать в ряде различных таутомерных форм, и ссылки на соединение формулы (1) включают все такие формы, если в контексте не указано иное. Во избежание сомнений, когда указанное соединение может существовать в одной из таутомерных форм и только одна определенным образом описана или показана, все другие, тем не менее, охвачены формулой (1).
Для характеристики конкретных стереохимических форм используется обозначение в виде «пунктирных» или «клиновидных» линий для указания стереохимии, например, как проиллюстрировано двумя молекулами, изображенными ниже.


Оптические изомеры могут быть охарактеризованы и идентифицированы по их оптической активности (т.е., как + и - изомеры или d и I изомеры), или они могут быть охарактеризованы в терминах их абсолютной стереохимии с использованием номенклатуры «R и S», разработанной Каном, Ингольдом и Прелогом (Cahn, Ingold и Prelog), см. Advanced Organic Chemistry, Jerry March, 4-е издание, John Wiley & Sons, Нью-Йорк, 1992, стр. 109-114, и см. также Cahn, Ingold & Prelog, Angew. Chem. Int. Ed. Engl., 1966, 5, 385-415.
Оптические изомеры могут быть разделены рядом методов, включая хиральную хроматографию (хроматография на хиральной подложке), и такие методы хорошо известны специалисту в данной области техники.
В качестве альтернативы хиральной хроматографии оптические изомеры могут быть разделены путем образования диастереомерных солей с хиральными кислотами, такими как (+)-винная кислота, (-)-пироглутаминовая кислота, (-)-дитолуол-L-винная кислота, (+)-миндальная кислота, (-)-яблочная кислота, и (-)-камфорсульфоновая, разделения диастереомеров предпочтительной кристаллизацией, а затем диссоциацией солей, в результате чего получают индивидуальный энантиомер свободного основания. Аналогично, оптические изомеры кислотных соединений могут быть разделены путем образования диастереомерных солей с хиральными аминами, такими как бруцин, цинхонидин, хинин и т.д.
Кроме того, энантиомерное разделение может быть осуществлено путем ковалентного связывания энантиомерно чистого хирального вспомогательного вещества с соединением и последующего разделения диастереоизомеров с использованием обычных методов, таких как хроматография. Затем расщепляют указанную ковалентную связь для получения соответствующего энантиомерно чистого продукта. Например, оптические изомеры хиральных соединений, содержащих свободную гидроксильную группу, можно разделить, образуя кислотные сложные эфиры Мошера, затем разделяя полученные диастереомеры хроматографией, затем расщепляя эфир для получения свободной гидроксильной группы.
Если в соединениях в настоящей заявке показана определенная стереохимическая конфигурация, это может означать, что в этой стереохимической форме присутствует по меньшей мере 55% (например, по меньшей мере 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95%) соединения в отличие от других изомерных форм соединения. В одном общем варианте реализации 99% или более (например, по существу, все количество) от общего количества соединения формулы (1) присутствует в изображенной стереохимической конфигурации.
Изотопные варианты
Ссылки в настоящем описании на соединение (1) включают все его фармацевтически приемлемые изотопно-меченные варианты, где один или более атомов заменены атомами, имеющими тот же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, обычно встречающихся в природе.
Примеры изотопов, пригодных для включения в соединение согласно настоящему изобретению, включают изотопы водорода, такие как 2Н (D) и 3Н (Т), углерода, такие как 11С, 13С и 14С, хлора, такие как 36Cl, фтора, такие как 18F, азота, такие как 13N и 15N, и кислорода, такие как 15O, 17O и 18O.
Некоторые изотопно-меченные соединения формулы (1), например содержащие радиоактивный изотоп, пригодны в исследованиях распределения лекарственного средства и/или субстрата в тканях. Соединение формулы (1) также может иметь ценные диагностические свойства в том смысле, что они могут быть применены для обнаружения или идентификации образования комплекса между меченым соединением и другими молекулами, пептидами, белками, ферментами или рецепторами. Способы обнаружения или идентификации могут применять соединения, которые помечены мечеными агентами, такими как радиоизотопами, ферментами, флуоресцентными веществами, светящимися веществами (например, люминолом, производными люминола, люциферином, экворином и люциферазой) и т.д. Радиоактивные изотопы тритий, т.е. 3Н (Т), и углерод-14, то есть 14С, являются особенно подходящими для этой цели ввиду простоты их внедрения и готовых средств обнаружения.
Замена более тяжелыми изотопами, такими как дейтерий, т.е. 2Н (D), может дать определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенным периодом полувыведения in vivo или сниженными требованиями к дозировке, и, следовательно, может быть предпочтительна в некоторых обстоятельствах. В частности, каждая ссылка на водород в настоящей заявке должна быть истолкована как охватывающая 1Н и 2Н, независимо от того, определен ли водород явно или водород присутствует неявно, чтобы насытить валентность соответствующего атома (в частности, углерода).
Замена позитрон-излучающими изотопами, такими как 11С, 18F, 15O и 13N, может быть полезна в исследованиях позитронно-эмиссионной топографии (ПЭТ) для изучения степени занятости мишени.
Изотопно-меченные соединения формулы (1), как правило, могут быть получены традиционными методами, известными специалистам в данной области техники, или способами, аналогичными тем, которые описаны в прилагаемых «Примерах» и «Примерах получения», с применением подходящих изотопно-меченных реагентов вместо немаркированных ранее применяемых реагентов.
Комплексы
Формула (1) также включает в свой объем комплексы соединения (например, комплексы включения или клатраты с такими соединениями, как циклодекстрины или комплексы с металлами). Комплексы включения, клатраты и комплексы металлов могут быть получены способами, хорошо известными специалисту в данной области техники.
Биологические свойства
Предполагается, что соединение (1) будет пригодно для примененения в медицине или в терапии.
Соединение согласно настоящему изобретению представляет собой ингибитор ERK1/2 и будет пригодно для применения для профилактики или лечения заболеваний и состояний, описанных в настоящем документе, например заболеваний и состояний, обсуждаемых ниже, и заболеваний и состояний, описанных в вышеуказанном разделе «Уровень техники», в которых играет роль ERK1/2. Помимо этого, соединение согласно настоящему изобретению будет пригодно для применения для преотвращения или лечения заболеваний или состояний, опосредованных ERK1/2, например заболеваний или состояний, таких как рак, при которых активность ERK1/2 необходима или имеет повышающую регуляцию в результате активации мутаций в вышестоящих компонентах (таких как RAS, K-RAS, NRAS и RAF) пути МАРК.
Указания на предотвращение или профилактику или лечение заболевания или состояния, такого как рак, включают в свой объем ослабление или уменьшение частоты возникновения заболевания или состояния. Таким образом, например, предполагается, что соединение согласно настоящему изобретению будет пригодно для применения для ослабления или снижения частоты рака.
Приведенные ниже ссылки на соединение формулы (1), включают кристаллические формы соединения формулы (1), описанного в настоящем документе, и соединение формулы (1), полученное в соответствии со способами, описанными в настоящем описании.
Соответственно, в других вариантах реализации настоящего изобретения предложены:
• Соединение формулы (1) для применения в медицине.
• Соединение формулы (1) для применения для предотвращения или лечения опосредованного ERK1/2 заболевания или состояния.
• Применение соединения формулы (1) для получения лекарственного средства для предотвращения или лечения опосредованного ERK1/2 заболевания или состояния.
• Способ предотвращения или лечения опосредованного ERK1/2 заболевания или состояния у субъекта (например, субъекта-млекопитающего, такого как человек, нуждающегося в этом), включающий введение субъекту терапевтически эффективного количества соединения формулы (1).
• Соединение формулы (1) для применения для ослабления или уменьшения частоты возникновения опосредованных ERK1/2 заболеваний или состояний.
• Применение соединения формулы (1) для получения лекарственного средства для ослабления или уменьшения частоты возникновения опосредованного ERK1/2 заболевания или состояния.
• Способ ослабления или уменьшения частоты возникновения опосредованного ERK1/2 заболевания или состояния у субъекта (например, субъекта-млекопитающего, такого как человек, нуждающегося в этом), который включает введение субъекту терапевтически эффективного количества соединение формулы
(1)-
Более конкретно, соединение (1) представляет собой ингибитор ERK1/2. Например, соединение согласно настоящему изобретению обладает ингибирующей активностью в отношении ERK1 или ERK2, и в частности, в отношении ERK1/2.
Соединение-ингибитор ERK формулы (1) способно связываться с ERK1/2 и проявлять активность в отношении ERK1/2. Водном из вариантов реализации соединение-ингибитор формулы (1) проявляет селективность в отношении ERK1/2 по сравнению с другими членами семейства киназ и может быть способно связываться и/или проявлять предпочтительную ингибирующую способность в отношении ERK1 и/или ERK2 по сравнению со связыванием и/или проявлением ингибирующей способности в отношении других членов указанного семейства киназ.
Функция ERK1/2 контроля клеточной передачи сигналов также участвует во многих заболеваниях, включая расстройства, связанные с накоплением клеток (например, рак, аутоиммунные нарушения, воспаление и рестеноз), расстройства, при которых чрезмерный апоптоз приводит к потере клеток (например, инсульт, сердечная недостаточность), нейродегенерация, такая как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, боковой амиотрофический склероз, СПИД, ишемия (инсульт, инфаркт миокарда) и остеопороз или лечение аутоиммунных заболеваний, таких как рассеянный склероз (PC).
Опосредованное ERK1/2 заболевание или состояние, которое упоминается в любом из вышеупомянутых вариантов реализации, может представлять собой любое одно или несколько из указанных выше заболеваний и нарушений.
Таким образом, также предусматривается, что соединение согласно настоящему изобретению, определенное в настоящем описании, может быть пригодно для применения для лечения других состояний, таких как воспаление, гепатит, язвенный колит, гастрит, аутоиммунитет, воспаление, рестеноз, инсульт, сердечная недостаточность, нейродегенеративные состояния, такие как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, миотоническая дистрофия и боковой амиотрофический склероз, СПИД, ишемия, такая как травматическое повреждение головного мозга, повреждение спинного мозга, церебральная ишемия, церебральное ишемическое/реперфузионное (И/Р) повреждение, острое и хроническое ишемическое повреждение ЦНС, инсульт или инфаркт миокарда, дегенеративные заболевания опорно-двигательного аппарата, такие как остеопороз, аутоиммунные заболевания, такие как рассеянный склероз (PC) и диабет I типа, а также глазные заболевания, такие как дегенерация сетчатки, которые возникают в результате утраты контроля над запрограммированной клеточной смертью.
Вследствие своей аффинности по отношению к ERK1/2 соединение согласно настоящему изобретению будет пригодно для применения для обеспечения средств, контролирующих передачу сигналов клетками. Таким образом, ожидается, что указанное соединение может доказать востребованность для лечения или предотвращения пролиферативных нарушений, таких как раковые заболевания.
Соответственно, в других вариантах реализации настоящего изобретения предложено:
• Соединение формулы (1) для применения для предотвращения или лечения пролиферативных нарушений, таких как раковые заболевания.
• Применение соединения формулы (1) для получения лекарственного средства для предотвращения или лечения пролиферативных нарушений, таких как раковые заболевания.
• Способ предотвращения или лечения пролиферативных нарушений, таких как раковые заболевания у субъекта (например, субъекта-млекопитающего, такого как человек, нуждающегося в этом), включающий введение субъекту терапевтически эффективного количества соединения формулы (1).
• Соединение формулы (1) для применения для ослабления или уменьшения частоты возникновения пролиферативных нарушений, таких как раковые заболевания.
• Применение соединения формулы (1) для получения лекарственного средства для ослабления или уменьшения частоты возникновения пролиферативных нарушений, таких как раковые заболевания.
• Способ ослабления или уменьшения частоты возникновения пролиферативных нарушений, таких как раковые заболевания, у субъекта (например, субъекта-млекопитающего, такого как человек, нуждающегося в этом), который включает введение субъекту терапевтически эффективного количества соединения формулы (1)-
Примеры раковых заболеваний (и их доброкачественных аналогов), которые можно лечить (или ингибировать), включают, но не ограничиваются ими, опухоли эпителиального происхождения (аденомы и карциномы различных типов, включая аденокарциномы, плоскоклеточный рак, карциномы переходных клеток и другие карциномы), такие как карциномы мочевого пузыря и мочевыводящих путей, молочной железы, желудочно-кишечного тракта (в том числе пищевода, желудка (гастрический), тонкой кишки, толстой кишки, прямой кишки и заднего прохода), печени (гепатоцеллюлярная карцинома), желчного пузыря и желчевыводящей системы, экзокринной поджелудочной железы, почек, легких (например, аденокарциномы, мелкоклеточный рак легкого, немелкокпеточный рак легкого, бронхоальвеолярный рак и мезотелиомы), головы и шеи (например, рак языка, буккальной полости, гортани, глотки, носоглотки, миндалины, слюнных желез, носовой раковины и околоносовых пазух), яичника, маточных труб, брюшины, влагалища, вульвы, полового члена, шейки матки, миометрия, эндометрия, щитовидной железы (например, фолликулярный рак щитовидной железы), надпочечников, простаты, кожи и придаточных клеткок (например, меланома, базально-клеточный рак, плоскоклеточный рак, кератоакантома, диспластический наевус); гематологические злокачественные заболевания (например, лейкозы, лимфомы) и предзлокачественные гематологические нарушения и нарушения пограничной злокачественности, включая гематологические злокачественные заболевания и связанные состояния лимфоидной линии (например, острый лимфоцитарный лейкоз [ОЛЛ], хронический лимфолейкоз [ХЛЛ], В-клеточную лимфому, такую как диффузная крупноклеточная В-клеточная лимфома [ДВКЛ], фолликулярную лимфому, лимфому Беркитта, мантийноклеточную лимфому, Т---клеточные лимфомы и лейкозы, лимфомы из клеток-натуральных киллеров [NK], лимфомы Ходжкина, волосатоклеточный лейкоз, моноклональную гаммапатию неопределенной значимости, плазмацитому, множественную миелому и посттрансплантационные лимфопролиферативные расстройства), а также гематологические злокачественные заболевания и связанные состояния миелоидной линии (например, острый миелогенный лейкоз [ОМЛ], хронический миелогенный лейкоз [ХМЛ], хронический миеломоноцитарный лейкоз [ХММЛ], гиперэозинофильный синдром, миелопролиферативные нарушения, такие как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативный синдром, миелодиспластический синдром и промиелоцитарный лейкоз); опухоли мезенхимального происхождения, например саркомы мягких тканей, кости или хряща, такие как остеосаркомы, фибросаркомы, хондросаркомы, рабдомиосаркомы, лейомиосаркомы, липосаркомы, ангиосаркомы, саркому Капоши, саркому Юинга, синовиальные саркомы, эпителиоидные саркомы, желудочно-кишечные стромальные опухоли, доброкачественную и злокачественную гистиоцитомы и выбухающую дерматофибросаркому; опухоли, происходящие из клеток нервного гребня, включая меланоцитарные опухоли (например, злокачественную меланому или увеальную меланому), опухоли периферических и черепных нервов, периферические нейробластические опухоли (например, нейробластому), эмбриональные опухоли ЦНС, параганглиому; опухоли центральной или периферической нервной системы (например, астроцитомы, глиомы и глиобластомы, менингиомы, эпендимомы, опухоли шишковидной железы и шванномы); эндокринные опухоли (например, опухоли гипофиза, опухоли надпочечников, опухоли островковых клеток, опухоли околощитовидной железы, карциноидные опухоли и медуллярную карциному, рак щитовидной железы); опухоли глаза и придаточного аппарата (например, ретинобластому); опухоли половых клеток и трофобластов (например, тератомы, семиномы, дисгерминомы, доброкачественную гестационную трофобластическую болезнь и хориокарциномы); а также детские и эмбриональные опухоли (например, медуллобластому, нейробластому, опухоль Вильмса и примитивные нейроэктодермальные опухоли); или синдромы, врожденные или иные, которые делают пациента восприимчивым к злокачественному заболеванию (например, пигментную ксеродерму). Дополнительные примеры раковых заболеваний (и их доброкачественных аналогов), которые можно лечить (или ингибировать), включают, но не ограничиваются ими, опухоли яичек и головного мозга (например, невромы).
Таким образом, в фармацевтических композициях, применениях или способах согласно настоящему изобретению для лечения заболевания или состояния, включающего аномальный рост клеток (то есть неконтролируемый и/или быстрый рост клеток), заболевание или состояние, включающее аномальный рост клеток, в одном из вариантов реализации представляет собой рак.
В одном из вариантов реализаци гематологическое злокачественное заболевание представляет собой лейкоз. В другом варианте реализации гематологическое злокачественное заболевание представляет собой лимфому. В одном из вариантов реализации соединение согласно настоящему изобретению предназначено для профилактики или лечения лейкоза, такого как острый или хронический лейкоз, в частности острый миелоидный лейкоз (ОМЛ), острый лимфоцитарный лейкоз (ОЛЛ), хронический лимфоцитарный лейкоз (ХЛЛ) или хронический миелоидный лейкоз (ХМЛ). В одном из вариантов реализации соединение согласно настоящему изобретению предназначено для применения для профилактики или лечения лимфомы, такой как острая или хроническая лимфома, в частности лимфома Беркитта, лимфома Ходжкина, неходжкинская лимфома или диффузная крупноклеточная В-клеточная лимфома. Водном из вариантов реализации соединение согласно настоящему изобретению предназначено для профилактики или лечения острого миелоидного лейкоза (ОМЛ) или острого лимфоцитарного лейкоза (ОЛЛ). В одном из вариантов реализации указанный рак представляет собой ОМЛ. В другом варианте реализации указанный рак представляет собой ХЛЛ.
Многие заболевания характеризуются стойким и нерегулируемым ангиогенезом. Хронические пролиферативные заболевания часто сопровождаются сильным ангиогенезом, который может способствовать воспалительному и/или пролиферативному состоянию или поддерживать его, либо который ведет к разрушению ткани через инвазивную пролиферацию кровеносных сосудов. Было обнаружено, что рост опухоли и метастазирование зависят от ангиогенеза. Соединение согласно настоящему изобретению может таким образом быть пригодно для предотвращения и препятствования инициации ангиогенеза опухоли. В частности, соединение согласно настоящему изобретению может быть пригодно для лечения метастазирования и метастатических раковых заболеваний.
Метастазирование или метастазирующее заболевание представляет собой распространение заболевания от одного органа или части к другому несмежному органу или части. Раковые заболевания, которые могут быть подвергнуты действию соединения согласно настоящему изобретению, включают первичные опухоли (т.е. раковые клетки в месте возникновения), локальную инвазию (раковые клетки, которые проникают и инфильтрируют в окружающие нормальные ткани в локальной области) и метастатические (или вторичные) опухоли, то есть опухоли, которые образовались из злокачественных клеток циркулирующих через кровоток (гематогенное распространение) или через лимфатические узлы, или через полости тела (транскеломное) в другие участки и ткани организма.
В указанных выше вариантах реализации конкретные раковые заболевания включают гепатоцеллюлярную карциному, меланому, раковые заболевания пищевода, почки, толстой кишки, толстой и прямой кишки, молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичников, предстательной железы и легкого, например, мезотелиомы или аденокарциномы легкого.
Другая подгруппа раковых заболеваний состоит из меланомы, раковых заболеваний почки, толстой кишки, легких, молочной железы, яичников и предстательной железы.
Другая подгруппа раковых заболеваний состоит из рака поджелудочной железы.
Другая подгруппа раковых заболеваний состоит из лейкоза, такого как острый и хронический лейкоз, острый миелоидный лейкоз (ОМЛ) и хронический лимфоцитарный лейкоз (ХЛЛ).
Еще одна подгруппа раковых заболеваний состоит из мезотелиомы, включая злокачественную мезотелиому брюшины или злокачественную мезотелиому плевры.
Определенные раковые заболевания устойчивы к лечению конкретными лекарственными средствами. Это может быть связано с типом опухоли (наиболее распространенные эпителиальные злокачественные заболевания по своей природе являются хеморезистентными), или резистентность может возникать спонтанно по мере прогрессирования заболевания или в результате лечения. В связи с этим ссылки на мезотелиому включают мезотелиому с устойчивостью к ядам топоизомеразы, алкилирующим агентам, антитубулинам, антифолатам, платиновым соединениям и лучевой терапии, в частности мезотелиому, устойчивую к цисплатину. Аналогичным образом, ссылки на множественную миелому включают чувствительную к бортезомибу множественную миелому или трудноподдающуюся лечению множественную миелому, а ссылки на хронический миелогенный лейкоз включают чувствительный к имитанибу хронический миелогенный лейкоз и рефрактерный хронический миелогенный лейкоз. В этой связи ссылки на рак предстательной железы включают раковые заболевания предстательной железы с резистентностью к анти-андрогенной терапии, в частности абиратерону или энзалутамиду, или кастрационно-резистентный рак предстательной железы. Ссылки на меланому включают меланомы, которые резистентны к лечению ингибиторами BRAF и/или МЕК.
Указанные раковые заболевания могут представлять собой раковые заболевания, которые чувствительны к ингибированию ERK1 или ERK2, или наиболее конкретно ERK1/2.
Кроме того, предполагается, что соединение согласно настоящему изобретению будет особенно пригодно для лечения или предотвращении раковых заболеваний тех типов, которые связаны с или характеризуются наличием повышенной передачи сигналов Ras, BRAF и/или МЕК.
Повышенные уровни передачи сигналов Ras, BRAF или МЕК обнаруживают при многих раковых заболеваниях и они связаны с плохим прогнозом. Помимо этого, раковые заболевания с активирующими Ras, BRAF или МЕК мутациями также могут быть чувствительными кингибитору ERK1/2. Повышенные уровни передачи сигналов Ras, BRAF или МЕК и мутаций в Ras, BRAF или МЕК могут быть идентифицированы с помощью методов, кратко изложенных в настоящем описании. Является ли конкретный рак тем раком, который чувствителен к ингибированию ERK1/2, может быть определено способом, который изложен в разделе «Способы диагностики».
Еще одна подгруппа раковых заболеваний состоит из NRas меланомы и NRas ОМЛ.
Другая подгруппа раковых заболеваний состоит из KRas рака легкого, KRas рака поджелудочной железы и KRas рака толстой и прямой кишки (CRC).
Другая подгруппа раковых заболеваний состоит из BRAF рака толстой и прямой кишки (CRC), BRAF рака легких и BRAF меланомы.
В других вариантах реализации настоящего изобретения предложено:
• Соединение формулы (1) для применения для предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной МЕК.
• Применение соединения формулы (1) для получения лекарственного средства для предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной МЕК.
• Способ предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной МЕК у субъекта (например, субъекта-млекопитающего, такого как человек, нуждающегося в этом), который включает введение субъекту терапевтически эффективного количества соединения формулы (1).
• Соединение формулы (1) для применения для ослабления или уменьшения частоты возникновения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной МЕК.
• Применение соединения формулы (1) для получения лекарственного средства для ослабления или уменьшения частоты возникновения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной МЕК.
• Способ ослабления или уменьшения частоты возникновения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной МЕК у субъекта (например, субъекта-млекопитающего, такого как человек, нуждающегося в этом), включающий введение субъекту терапевтически эффективного количества соединения формулы (1).
• Соединение формулы (1) для применения для лечения (или уменьшения частоты возникновения) рака, выбранного из NRas меланомы и NRas ОМЛ.
• Соединение формулы (1) для применения для лечения (или уменьшения частоты возникновения) рака, выбранного из KRas рака легких, KRas рака поджелудочной железы и KRas рака толстой и прямой кишки (CRC).
• Соединение формулы (1) для применения для лечения (или уменьшения частоты возникновения) рака, выбранного из BRAF рака тоетой и прямой кишки (CRC), BRAF рака легких и BRAF меланомы.
• Соединение формулы (1) для применения для лечения (или уменьшения частоты возникновения) рака, представляющего собой BRAF меланому.
• Применение соединения формулы (1) для получения лекарственного средства для предотвращения или лечения рака, как определено в настоящем описании.
• Способ лечения (или уменьшения частоты возникновения) рака у субъекта (например, субъекта-млекопитающего, такого как человек), который включает введение субъекту терапевтически эффективного количества соединения формулы (1).
• Соединение формулы (1) для применения для лечения заболевания или состояния, как описано в настоящем описании, в частности рака.
• Применение соединения формулы (1) для получения лекарственного средства для лечения заболевания или состояния, как описано в настоящем описании, в частности рака.
• Способ предотвращения или лечения заболевания или состояния, как описано в настоящем описании, в частности рака, у субъекта (например, субъекта-млекопитающего, такого как человек, нуждающегося в этом), который включает введение субъекту терапевтически эффективного количества соединения формулы (1).
• Соединение формулы (1) для применения для ослабления или уменьшения частоты возникновения заболевания или состояния, как описано в настоящем описании, в частности рака.
• Применение соединения формулы (1) для получения лекарственного средства для ослабления или уменьшения частоты возникновения заболевания или состояния, как описано в настоящем описании, в частности рака.
• Способ ослабления или уменьшения частоты возникновения заболевания или состояния, как описано в настоящем описании, в частности рака, у субъекта (например, субъекта-млекопитающего, такого как человек, нуждающегося в этом), который включает введение субъекту терапевтически эффективного количества соединения формулы (1).
Соединение (1) также может быть пригодно для лечения роста опухолей, патогенеза, устойчивости к химио- и лучевой терапии путем сенсибилизации клеток к химиотерапии и в качестве антиметастатического агента.
Терапевтические противораковые вмешательства всех типов неизбежно увеличивают стрессы, налагаемые на опухолевые клетки-мишени. При смягчении вредных эффектов таких стрессов ERK1/2 непосредственно вовлекается в противодействие эффектам противораковых лекарств и схем лечения. Таким образом, ингибиторы ERK1/2 представляют собой класс химиотерапевтических средств с потенциалом для: (i) сенсибилизации злокачественных клеток к противораковым лекарственным средствам и/или разновидностям лечения; (ii) облегчения или уменьшения частоты возникновения резистентности к противораковым лекарственным средствам и/или разновидностям лечения; (iii) изменения резистентности к противораковым лекарственным средствам и/или разновидностям лечения; (iv) потенцирование активности противораковых лекарственных средств и/или разновидностей лечения; (v) задержки или предотвращения появления устойчивости к противораковым лекарственным средствам и/или разновидностям лечения.
Как следствие его ингибирования по отношению к ERK1/2, указанное соединение будет пригодно для обеспечения средств, контролирующих передачу сигналов клетками. Таким образом, также предполагается, что соединение согласно настоящему изобретению может быть пригодно для применения для лечения других состояний, таких как воспалительные расстройства, таких как гепатит, язвенный колит и гастрит; ней роде генеративных состояний, таких как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, миотонической дистрофии и бокового амиотрофического склероза; СПИДа, ишемии, такой какрестеноз, травматического повреждения головного мозга, повреждения спинного мозга, церебральной ишемии, церебрального ишемического/реперфузионного (И/Р) повреждения, острого и хронического ишемического повреждения ЦНС, инсульта или инфаркта миокарда; дегенеративных заболеваний опорно-двигательного аппарата, таких как остеопороз; аутоиммунных заболеваний, таких как рассеянный склероз (PC) и диабет I типа, а также заболеваний глаз, таких как дегенерация сетчатки.
Аффинность соединения согласно настоящему изобретению в качестве ингибитора ERK1/2 может быть измерена с применением биологических и биофизических анализов, изложенных в приведенных в настоящем описании примерах.
Способы диагностики
Перед введением соединения формулы (1) субъект (например, пациент) может быть подвергнут скринингу для определения того, представляет ли собой заболевание или состояние, от которого страдает или может страдать пациент, именно то, которое может быть восприимчиво к лечению соединением, демонстрирующим ингибирование ERK1/2. Термин «пациент» включает людей и ветеринарных пациентов.
Например, можно исследовать биологический образец, взятый у пациента, для определения того, является ли состояние или заболевание, такое как рак, которым пациент страдает или может страдать, состоянием или заболеванием, характеризующимся генетическим нарушением или анормальной экспрессией белка, которые приводят к повышению уровней передачи сигнала ERK1/2 или к сенсибилизации какого-либо пути к нормальной функции ERK1/2 или к повышающей регуляции какого-либо биохимического пути «ниже» активации ERK1/2.
Примеры таких нарушений, которые приводят к активации или сенсибилизации пути ERK1/2, включают активирующие мутации в какой-либо изоформе Ras, такой как KRAS, или в BRAF, как обсуждается в разделе «УРОВЕНЬ ТЕХНИКИ».
Мутации Ras были выявлены с клеточных линиях и первичных опухолях, включая следующие, но не ограничиваясь ими: меланома, рак толстой и прямой кишки, немел коклеточный рак легких и раковые заболевания поджелудочной железы, предстательной железы, щитовидной железы, мочевыводящих путей и верхних дыхательных путей (Cancer Res. 2012; 72: 2457-2467).
Термин «повышение» (повышающая регуляция) включает повышенную экспрессию или сверхэкспрессию, включая амплификацию гена (т.е., несколько копий гена), цитогенетические аберрации и повышенную экспрессию, обусловленную влиянием на транскрипцию, или повышенную передачу сигнала за счет активации ERK1/2. Соответственно, пациент может быть подвергнут диагностическому тестированию для детектирования маркерного признака повышающей регуляции ERK1/2. Термин «диагностика» включает скрининг.Под маркером понимают, например, измерение состава ДНК для идентификации присутствия мутаций Ras (например, KRAS) или BRAF. Термин «маркер» включает также маркеры, которые являются признаком повышающей регуляции ERK1/2, включая уровни белка, состояние белка и уровни мРНК указанных белков. Амплификация гена включает более 7 копий, а также увеличение количества копий в диапазоне от 2 до 7.
Диагностические тесты для детектирования мутаций KRAS и BRAF описаны в источнике de Castro с соавт.Br. J. Cancer. 2012, 10 июля 10;107(2):345-51. doi: 10.1038/bjc.2012.259. Epub 2012, 19 июня, «А comparison of three methods for detecting KRAS mutations in formalin-fixed colorectal cancer specimens.*; и Gonzalez с coaem., Br J Dermatol. 2013, Apr; 168(4): 700-7. doi: 10.1111/bjd.12248, «BRAF mutation testing algorithm for vemurafeniob treatment in melanoma: recommendations from an expert paneb и источниках, цитируемых в настоящем документе.
Управление по контролю в сфере лекарств и пищевых продуктов (FDA) одобрило ряд диагностических тестов для мутаций BRAF, и подробное описание этих тестов можно найти на веб-сайте FDA. Примеры таких диагностических тестов включают тест на мутации 4800 BRAF V600 от Cobas, вспомогательный тест для продукта Roche вемурафениба тест на BRAF от THxID, вспомогательные тесты для продуктов тацинлар (дабрафениб) и мекинист (траметиниб).
Диагностические тесты и скрининговые исследования обычно проводят на биологическом образце (т.е., ткани организма или физиологических жидкостях), выбранном из биоптатов опухоли, образцов крови (выделение и обогащение слущенных опухолевых клеток), цереброспинальной жидкости, плазмы, сыворотки, слюны, биопсий стула, мокроты, аналитических образцов хромосом, плевральной жидкости, перитонеальной жидкости, соскобов щеки, биопсии кожи и мочи.
Способы идентификации и анализа цитогенетических аберраций, амплификации гена, мутаций и повышающей регуляции белков известны специалистам в данной области. Клиническое исследование большинства генетических вариантов может включать стандартные методы, такие как аллель-специфическая полимеразная цепная реакция (ПЦР), полимеразно-цепная реакция с обратной транскриптазой (ОТ-ПЦР), анализ последовательностей ДНК с применением традиционных методов по Сенгеру или методов секвенирования следующего поколения, дидезокси-секвенирование по Сенгеру, пиросеквенирование, мультиплексная лигазозависимая амплификация зондов (MLPA) или методика ПЦР ARMS (амплификация рефрактерной мутационной системы), но не ограничивается ими. Клинические исследования на число копий генов и структурные варианты генов могут включать стандартные методы, такие как секвенирование РНК (RNAseq), тесты высокого разрешения NanoString, основанные на гибридизации РНК, с реагентами nCounter или гибридизация in-situ, флуоресцентная гибридизация in situ (FISH), но не ограничиваются ими. Более новые методики секвенирования следующего поколения (NGS), такие как массовое параллельное секвенирование, дают возможность полноэкзомного или пол но геном но го секвенирования.
При скрининге методом ОТ-ПЦР, уровень мРНК в опухоли оценивают, создавая кДНК-копию матричной РНК с последующей амплификацией этой кДНК методом ПЦР. Методы ПЦР-амплификации, выбор праймеров и условий для амплификации, известны специалистам в данной области. Манипуляции с нуклеиновыми кислотами и ПЦР проводят с применением стандартных методов, описанных, например, в Ausubel, F.M. ссоавт., изд. (2004) Current Protocols in Molecular Biology, John Wiley & Sons Inc., или Innis, M.A. с соавт., изд. (1990) PCR Protocols: a guide to methods and applications, Academic Press, San Diego. Реакции и манипуляции с методиками на основе нуклеиновых кислот описаны также в источнике Sambrook с соавт., (2001), 3е изд., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press. В альтернативном варианте можно применять коммерчески доступные набора для ОТ-ПЦР (например, Roche Molecular Biochemicals), или процедуры, описанные в патентах США 4 666 828, 4 683 202, 4 801 531, 5 192 659, 5 272 057, 5 882 864 и 6 218 529, которые включены в настоящий документ посредством ссылки. Примером методик на основе гибридизации in-situ для оценки экспрессии мРНК является флуоресцентная гибридизация in-situ (FISH) (см. Angerer (1987) Meth. Enzymol., 152: 649).
В целом, гибридизация in situ включает следующие основные этапы: (1) фиксация ткани для анализа, (2) обработка образца перед гибридизацией для повышения доступности целевой нуклеиновой кислоты и для снижения неспецифического связывания, (3) гибридизация смеси нуклеиновых кислот с нуклеиновой кислотой в ткани или какой-либо биологическое структуре ткани, (4) промывки после гибридизации для удаления фрагментов нуклеиновых кислот, не связавшихся при гибридизации, и (5) детектирование гибридизованных фрагментов нуклеиновой кислоты. Зонды, применяемые в таких приложениях обычно мечены, например, радиоизотопами или флуоресцентными репортерами. Конкретные зонды являются достаточно длинными, например, от приблизительно 50, 100 или 200 нукпеотидов до приблизительно 1000 или более нуклеотидов, для того, чтобы обеспечивать специфическую гибридизацию с целевой нуклеиновой кислотой или кислотами в жестких условиях. Стандартные методы осуществления FISH описаны в источниках Ausubel, F.M. с соавт., изд. (2004) Current Protocols in Molecular Biology, John Wiley & Sons Inc и Fluorescence In Situ Hybridization: Technical Overview, John M. S. Bartlett, в Molecular Diagnosis of Cancer, Methods and Protocols, 2e изд., ISBN: 1-59259-760-2, Март 2004, стр. 077-088, Серия: Methods in Molecular Medicine.
Методы исследования экспрессии генов описаны в источнике (DePrimo с соавт.(2003), ВМС Cancer, 3:3). Вкратце, применяют следующий протокол: синтезируют двухцепочечную кДНК с тотальной РНК с применением (dT)24-оголимера для инициации синтеза первой цепи кДНК, например, с полиаденилированной мРНК, а затем синтезируют вторую цепь кДНЕ с применением случайных гексамерных праймеров. Эту двухцепочечную кДНКзатем используют в качестве матрицы для in vitro транскрипции кРНК с применением биотинилированных рибонуклеотидов. кРНК подвергают гимическому фрагментированию с применением протоколов, описанных компанией Affymetrix (Santa Clara, СА, США), а затем гибридизуют в течение ночи на матрицах генома человека или ген-специфичными олигонуклеотидными зондами на матрицах геном человека (Human Genome Array). В альтернативном варианте можно применять матрицы однонуклеотидных полиморфизмов (SNP), представляющих собой тип ДНК-матриц, для детектирования полиморфизмов в популяции.
В альтернативном варианте можно исследовать белковые продукты, экспрессируемые с мРНК, методами иммуногистохимии или иммунофлуоресценции образцов опухоли, твердофазного иммуноанализа на микротитрационных планшетах, Вестерн-блоттинга, капилларного электрофореза, 2-мерного гель-электрофореза на полиакриламидном гене с додецилсульфатом натрия, ИФА в формате ELISA, поточной цитометрии и других методов, известных в области детектирования конкретных белков. Способы детектирования могут включать применение сайт-специфичных антител. Специалисту будет понятно, что в настоящем случае можно применять хорошо известные методики для детектирования повышающей регуляции ERK1/2, детектирования вариантов или мутантов ERK1/2 или детектирования амплификации 11q22.
Абнормальные уровни белков, таких как ERK1/2, можно измерять с применением стандартных анализов на белки, описанных в настоящем документе. Повышенные уровни или сверхэкспрессию также можно детектировать в образце ткани, например, опухолевой ткани, путем измерения уровней белка с применением теста, например, предлагаемого компанией Chemicon International. Представляющий интерес белок можно подвергнуть иммунопреципитации из лизированного образца и измерить его уровни. Аналитические метода включают также применение маркеров.
Сверхэкспрессия ERK может быть измерена путем биопсии опухоли. Способы оценки изменений количества копий гена включают методики, рутинно применяемые в цитогенетических лабораториях, такие как MLPA (мультиплексная лигазозависимая амплификация зондов) - метод мультиплексной ПЦР для детектирования аберрантного числа копий, или другие ПЦР-методики, которые позволяют детектировать амплификацию, увеличение числа копий и делецию генов.
В соответствующих случаях можно также применять нефункциональные анализы, например, измерение циркулирующих лейкозных клеток у пациента с раком, для оценки ответа на провокацию ингибитором.
Таким образом, все эти методы также могут быть применены для идентификации опухолей, особенно подходящих для лечения соединением согласно настоящему изобретению.
Соответственно, в дополнительных вариантах реализации настоящего изобретения предложено:
• Соединение формулы (1) для применения для лечения или профилактики (или для применения для ослабления или уменьшения частоты возникновения) заболевания или состояния у пациента, который был подвергнут скринингу и был определен как страдающий от заболевания или состояния, либо подверженный риску заболевания или состояния, которое может быть восприимчиво к лечению соединением, проявляющим способность к ингибированию ERK1/2 (т.е. ингибитором ERK1/2).
• Применение соединения формулы (1) для получения лекарственного средства для лечения или профилактики (для применения для ослабления или уменьшения частоты возникновения) заболевания или состояния у пациента, который был подвергнут скринингу и был определен как страдающий от заболевания или состояния, либо подверженный риску заболевания или состояния, которое может быть восприимчиво к лечению соединением, проявляющим способность к ингибированию ERK1/2 (т.е. ингибитором ERK1/2).
• Способ лечения или профилактики (для применения для ослабления или уменьшения частоты возникновения) заболевания или состояния у пациента, который был подвергнут скринингу и был определен как страдающий от заболевания или состояния, либо подверженный риску заболевания или состояния, которое может быть восприимчиво к лечению соединением, проявляющим способность к ингибированию ERK1/2 (т.е. ингибитором ERK1/2), причем этот способ включает введение субъекту терапевтически эффективного количества соединения формулы (1).
Другой аспект согласно настоящему изобретению включает соединение согласно настоящему изобретению для применения в профилактике или лечении рака у пациента, выбранного из субпопуляции, обладающей сверхэкспрессией или активирующей мутацией в сигнальном пути ERK1/2 (например, Ras, BRAF или МЕК), Соответственно, в дополнительных вариантах реализации настоящего изобретения предложено:
• Соединение формулы (1) для применения для лечения или профилактики (или для ослабления или уменьшения частоты возникновения) рака у пациента, выбранного из субпопуляции, обладающей избыточной экспрессией или активирующей мутацией в сигнальном пути ERK1/2, например Ras (например, KRAS), BRAF или МЕК.
• Применение соединения формулы (1) для получения лекарственного средства для лечения или профилактики (для применения для ослабления или уменьшения частоты возникновения) рака у пациента, выбранного из субпопуляции, обладающей сверхэкспрессией или активирующей мутацией в сигнальном пути ERK1/2 Ras (например, KRAS), BRAF или МЕК.
• Способ лечения или профилактики (для применения для ослабления или уменьшения частоты возникновения) рака у пациента, выбранного из субпопуляции, обладающей сверхэкспрессией или активирующей мутацией в сигнальном пути ERK1/2 Ras (например, KRAS), BRAF или МЕК, который включает введение субъекту терапевтически эффективного количества соединения формулы (1).
• Способ диагностики и лечения заболевания или состояния, опосредованного ERK1/2, включающий (i) скрининг пациента для определения того, представляет собой ли заболевание или расстройство, от которого пациент страдает или может страдать, то заболевание, которое восприимчиво к лечению соединением, имеющим сродство к ERK1/2; и (ii) если показано, что заболевание или расстройство пациента является восприимчивым, после этого указанному пациенту вводят соединение формулы (1).
Фармацевтические составы
Соединение (1), полученное с помощью нового способа согласно настоящему изобретению, и новые кристаллические формы соединения (1) могут быть введены субъекту самостоятельно, но более типично они представлены в виде фармацевтической композиции (например, состава).
Таким образом, в другом варианте реализации настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (1) и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество и, необязательно, другие терапевтические или профилактические агенты, как описано в настоящем документе.
В настоящем изобретении дополнительно предложены способы получения фармацевтической композиции, включающие приведение в ассоциировнное состояние с соединением формулы (1) (например, примешивание) по меньшей мере одного указанного фармацевтически приемлемого вспомогательного вещества и, необязательно, других терапевтических или профилактических агентов, как описано в настоящем документе.
Фармацевтически приемлемое вспомогательное вещество (вещества) может быть выбрано из, например, носителей (например, твердых, жидких или полутвердых носителей), адъювантов, разбавителей, наполнителей или объемооразующих агентов, гранулирующих агентов, покровных веществ, агентов, контролирующих высвобождение, связующих, разрыхлителей, смазывающих агентов, консервантов, антиоксидантов, буферных веществ, суспендирующих веществ, загустителей, вкусоароматических веществ, подсластителей, агентов, маскирующих вкус, стабилизаторов или любых других вспомогательных веществ, традиционно применяемых в фармацевтических композициях. Примеры вспомогательных веществ для различных типов фармацевтических композиций описаны ниже более подробно.
Термин «фармацевтически приемлемый» в настоящем документе относится к соединениям, материалам, композициям и/или лекарственным формам, которые в рамках обоснованного медицинского суждения подходят для применения в контакте с тканями субъекта (например, субъекта-человека) и при этом не вызывают излишнюю токсичность, раздражение, аллергическую реакцию, или другие проблемы или осложнения, и характеризуются разумным соотношением риск/польза. Каждое вспомогательное вещество также должно быть «приемлемым» в смысле совместимости с другими ингредиентами состава.
Фармацевтические композиции, содержащие соединение (1), могут быть приготовлены в соответствии с известными методами, см., например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Истон, Пенсильвания, США.
Фармацевтические композиции могут быть представлены в любой форме, подходящей для перорального, парентерального, топического, интраназального введения, введения в бронхи, подъязычного, глазного введения, введения в ухо, ректального, внутривагинального или трансдермального введения. В тех случаях, когда композиции предназначены для парентерального введения, они могут быть выполнены с возможностью внутривенного, внутрибрюшинного, подкожного введения или доставки непосредственно в орган или ткань -мишень путем инъекции, инфузии или с применением других средств доставки. Доставка может осуществляться путем болюсной инъекции, кратковременной инфузии или более продолжительной инфузии, и может осуществляться посредством пассивной доставки или с применением подходящего инфузионного насоса или шприцевого насоса.
Фармацевтические составы, пригодные для парентерального введения включают водные и неводные стерилные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатики, сорастворители, поверхностно-активные вещества, смеси органических растворителей, комплексообразователей на основе циклодекстрина, эмульгаторов (для образованя и стабилизации составов в форме эмульсий), компонентов липосом для образования липосом, гелеобразующих полимеров для образования полимерных гелей, криопротекторов и комбинаций агентов для, среди прочего, стабилизации активного ингредиента в растворимой форме и придания составу изотоничности относительно крови предполагаемого реципиента. Фармацевтические составы для парентерального введения также могут иметь форму водных или неводных суспензий, которые могут включать суспендирующие вещества и загустители (R.G. Strickly, Solubilizing Excipients in oral and injectable formulations, Pharmaceutical Research, т.21(2) 2004, p 201-230).
Составы могут быть представлены в однодозовых или многодозовых контейнерах, например, запаянных ампулах, склянках и предзаполненных шприцах, и могут храниться в сублимированной (после сушки замораживанием, лиофилизированной) форме, к которой достаточно добавить стерильных жидкий носитель, например, например, воду для инъекций, непосредственно перед применением. В одном варианте реализации предложенный состав представляет собой активный фармацевтический ингредиент (например, в сублимированной или другой мелкоизмельченной форме) во флаконе для последующего восстановления с применением подходящего разбавителя.
Фармацевтический состав может быть получен лиофилизацией соединения формулы (1). Лиофилизация относится к процедуре сублимационной сушки композиции. Поэтому сублимационная сушка и лиофилизация применяются здесь как синонимы. Лиофилизация представляет собой способ дегидратации, при котором содержащий растворитель субстрат замораживают и затем подвергают действию вакуума, так что растворитель удаляется сублимацией, то есть прямым переводом из твердого замороженного состояния в газообразное состояние. Лиофилизация обычно включает три стадии; стадию замораживания; стадию первичной сушки; и стадию вторичной сушки. Во время стадии замораживания субстрат замораживают до температуры значительно ниже его температуры плавления. На последующей стадии первичной сушки замороженный субстрат подвергают воздействию вакуума, что позволяет удалять растворитель сублимацией. Большая часть растворителя удаляется из субстрата на этой стадии. Однако небольшое количество растворителя может оставаться связанным с субстратом или адсорбированным на субстрате в конце стадии первичной сушки. Чтобы удалить этот остаточный растворитель, вакуум поддерживают, но частично высушенный субстрат нагревают до температуры, при которой он больше не замерзает.Таким образом, остаточный растворитель удаляют испарением.
Процедуру сублимационной сушки можно проводить в аппарате для лиофилизации, конструкция которого может быть полностью традиционной. Аппарат для лиофилизации обычно имеет камеру, в которую могут быть помещены содержащие раствор для лиофилизации контейнеры для осуществления сублимационной сушки. Камера обычно соединена с источником вакуума (например, вакуумным насосом), чтобы дать возможность снизить давление в камере. Устройство также может иметь средства для замораживания или нагревания содержимого камеры.
Фармацевтический состав также может быть получен распылительной сушкой соединения формулы (1). Распылительная сушка представляет собой способ получения частиц микронного размера, в котором раствор или суспензию субстрата в растворителе тонко распыляют с образованием дисперсии капелек раствора или суспензии, а затем подвергают нагреву и/или частичному вакууму, так что растворитель удаляется испарением. Сначала субстрат растворяют или суспендируют в подходящем растворителе, а затем раствор пропускают через атомизатор или распылительное сопло в сушильную камеру. Нагретый газ (например, воздух или азот) также может впрыскиваться в сушильную камеру для контакта с распыленным сырьем или сушильная камера может подвергаться частичному вакууму, чтобы инициировать испарение растворителя. Твердые частицы субстрата образуются по мере испарения растворителя и могут быть извлечены. Размер образующихся в результате частиц в порошке можно изменить, выбрав подходящий атомизатор или распылительное сопло.
Процедуру распылительной сушки можно проводить с помощью распылительной сушилки, конструкция которой может быть полностью традиционной. Распылительная сушилка обычно имеет атомизатор или распылительное сопло, которые распыляют раствор в мелкодисперсные брызги (обычно диаметром менее 500 мкм). Мелкодисперсные брызги направляют в сушильную камеру. Сушильная камера обычно также имеет впускное отверстие для приема нагретого газа (такого как воздух или азот) и, необязательно, также соединена с источником вакуума (например, вакуумным насосом), чтобы дать возможность снизить давление внутри камеры. Сушильная камера также может иметь выход, через который могут быть собраны твердые частицы, образовавшиеся в результате испарения растворителя из распыляемых капелек.
Экстемпоральные растворы и суспензии для инъекций могут быть приготовлены из стерильных порошков, гранул и таблеток.
Фармацевтические композиции согласно настоящему изобретению для парентеральных инъекций могут также содержать фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для восстановления с получением стерильных растворов или дисперсий для инъекций непосредственно перед применением. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или основ включают воду, этанол, многоатомные спирты (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.), карбоксиметилцеллюлозу и их подходящие смеси, растительные масла (такие как оливковое масло, подсолнечное масло, сафлоровое масло или кукурузное масло), и подходящие для инъекций сложные органические эфиры, такие как этилолеат.Подходящую текучесть можно поддерживать, например, путем применения обволакивающих материалов (или загустителей), таких как лецитин, путем поддержания необходимого размера частиц в случае дисперсий и путем применения поверхностно-активных веществ.
Композиции согласно настоящему изобретению могут также содержать адъюванты, такие как консерванты, смачивающие вещества, эмульгаторы и диспергирующие агенты. Предотвратить воздействие микроорганизмов можно путем добавления различных антибактериальных и противогрибковых агентов, например, парабена, хлорбутанола, фенола, сорбиновой кислоты и т.п. Также может быть желательно добавлять агенты, регулирующие тоничность, такие как сахара, хлорид натрия и т.п. Фармацевтические формы с пролонгированным всасыванием и инъекционные формы можно получить путем добавления агентов, которые замедляют всасывание, таких как моностеарат алюминия и желатин.
В одном из вариантов реализации согласно настоящему изобретению указанная фармацевтическая композиция находится в форме, подходящей для внутривенного введения, например, путем инъекции или инфузии. Для внутривенного введения указанный раствор может быть дан дозой как есть или может быть инжектирован в инфузионный пакет (содержащий фармацевтически приемлемое вспомогательное вещество, такое как 0,9% физиологический раствор или 5% декстроза) перед введением.
В другом варианте реализации фармацевтическая композиция находится в форме, подходящей для подкожного (п/к) введения.
Фармацевтические лекарственные формы, подходящие для перорального введения, включают таблетки (с покрытием или без), капсулы (в твердой или мягкой оболочке), каплеты, пилюли, леденцы, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, подъязычные таблетки, пастилки или пластыри, такие как буккальные пластыри.
Так, композиции для таблеток могут содержать единицу дозы активного соединения совместно с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например: лактоза, сахароза, сорбит или маннит, и/или несахарный разбавитель, такой как карбонат натрия, фосфат кальция, карбонат кальция или соответствующие производные целлюлозы, такие как микрокристаллическая целлюлоза (МСС), метилцеллюлоза, этил целлюлоза, гид роксипропил метилцеллюлоза, и крахмалы, такие как кукурузный крахмал. Таблетки могут также содержать такие стандартные ингредиенты как связующие и гранулирующие агенты, такие как поливинилпирролидон, разрыхлители (например, набухающие поперечно-сшитые полимеры, такие как поперечно-сшитая карбоксиметилцеллюлоза), смазывающие агенты (например, стеараты), консерванты (например, парабены), антиоксиданты (например, ВНТ (бутилгидрокситолуол)), буферные вещества (например, фосфатные или цитратные буферы) и шипучие вещества, такие как цитрат/бикарбонатные смеси. Такие вспомогательные вещества хорошо известны, и необходимости подробно обсуждать их в настоящем документе нет.
Таблетки могут быть выполнены с возможностью высвобождения лекарственного средства либо после контакта с желудочным соком (таблетки с мгновенным высвобождением) либо контролируемого высвобождения (таблетки с контролируемым высвобождением) в течение продолжительного периода времени или в конкретной области ЖКТ.
Составы в капсулах могут принадлежать к типу жестких или мягких желатиновых капсул и могут содержать активный компонент в твердой, полутвердой или жидкой форме. Желатиновые капсулы могут быть выполнены из животного желатина или его синтетических или растительных аналогов.
Твердые лекарственные формы (например, таблетки, капсулы и т.д.) могут иметь покрытие или не иметь покрытия. Покрытия могут действовать либо как защитная пленка (например, полимерная, восковая или лаковая) или в качестве средства для контроля высвобождения лекарственного средства или могут служить для эстетических целей или целей идентификации. Покрытие (например, полимер типа Eudragit™) может быть выполнен с возможностью высвобождения активного компонента в желаемом положении желудочно-кишечного тракта. Соответственно, покрытие может быть выбрано таким образом, чтобы разрушаться при определенных условиях рН в желудочно-кишечном тракте, обеспечивая таким образом селективное высвобождение соединения в желудке или в подвздошной кишке, двенадцетиперстной кишке, толстой кишке или тонкой кишке.
Вместо покрытия или в дополнение к нему, лекарственное средство может быть представлено в твердой матрице, содержащей агент для контроля высвобождения, например, агент, замедляющий высвобождение, который может применяться для контролируемого высвобождения соединения в желудочно-кишечном тракте. В альтернативном варианте лекарственное средство может быть представлено в полимерном покрытии, в покрытии из полиметакрилатного полимера, которое можно применять для селективного высвобождения соединения в условиях различной кислотности или щелочности в желудочно-кишечном тракте. В альтернативном варианте материал матрицы или покрытие, замедляющее высвобождение, могут иметь форму эродируемого полимера (например, полимерного малеинового ангидрида), который по существу непрерывно эродирует по мере продвижения лекарственного препарата по желудочно-кишечному тракту. В другой альтернативе может быть предусмотрено покрытие, разлагающиеся под действием микрофлоры кишечника. В качестве еще одной альтернативы активное соединение может быть включено в систему доставки, которая обеспечивает осмотический контроль высвобождения соединения. Составы с осмотическим высвобождением или с замедленным высвобождением (например, составы на основе ионообменных смол) могут быть получены в соответствии со способами, хорошо известными специалистам в данной области.
Соединение формулы (1) может быть приготовлено в составе с переносящей средой и введено в форме наночастиц. Наночастицы увеличивают площадь поверхности, способствуя абсорбции соединения, и дают возможность прямого проникновения в клетку. Системы доставки лекарств на основе наночастиц описаны в «Nanoparticle Technology for Drug Delivery*, под редакцией Ram В Gupta и Uday В. Kompella, Informa Healthcare, ISBN 9781574448573, опубликованной 13 марта 2006 года. Наночастицы для доставки лекарств также описаны в J. Control. Release, 2003, 91 (1-2), 167-172 и в Sinha и др., Mol. Cancer Ther. 1 August (2006) 5, 1909.
Указанные фармацевтические композиции могут содержать от приблизительно 1% (масс/масс.) до приблизительно 95% активного ингредиента и от 99% (масс/масс.) до 5% (масс/масс.) фармацевтически приемлемого вспомогательного вещества или комбинации вспомогательных веществ. Более конкретно, композиции могут включать от приблизительно 20% (масс/масс.) до приблизительно 90% (масс/масс.) активного ингредиента и от 80% (масс./масс.) до 10% фармацевтического вспомогательного вещества или комбинации вспомогательных веществ.
Фармацевтические композиции согласно настоящему изобретению могут быть, например, в форме единичной дозы, такой как ампулы, виалы, суппозитории, драже, таблетки или капсулы или предварительно заполненные шприцы.
Фармацевтически приемлемое вспомогательное вещество (вещества) могут быть выбраны в соответствии с необходимой физической формой состава и могут, например, быть выбраны из разбавителей (например, твердых разбавителей, таких как наполнители или объемооразующие агенты, и жидких разбавителей, таких как растворители и сорастворители), разрыхлителей, буферных веществ, смазывающих веществ, веществ, улучшающих текучесть, агентов, контролирующих высвобождение (например, замедляющие или откладывающие высвобождение полимеры или воски), связующие, гранулирующие агенты, пигменты, пластификаторы, антиоксиданты, консерванты, вкусоароматические вещества, вкусомаскирующие агенты, агенты, регулирующие тоничность, и покровные вещества.
Специалист сможет выбрать подходящие количестве ингредиентов для применения в составах. Например, таблетки и капсулы обычно содержат 0-20% разрыхлителей, 0-5% смазывающих веществ, 0-5% веществ, улучшающих текучесть, и/или 0-99% (масс./масс.) наполнителей/объемооразующих агентов (в зависимости от дозы лекарственного средства). Они могут также содержать 0-10% (масс/масс.) полимерных связующих, 0-5% (масс/масс.) антиоксидантов, 0-5% (масс/масс.) пигментов. Таблетки с медленным высвобождением будут дополнительно содержать 0-99% (масс/масс.) полимеров (в зависимости от дозы). Пленочные покрытия на таблетках или капсулах обычно содержат 0-10% (масс/масс.) замедляющих высвобождение (например, откладывающих) полимеров, 0-3% (масс/масс.) пигментов, и/или 0-2% (масс/масс.) пластификаторов.
Составы для парентерального введения обычно содержат 0-20% (масс/масс.) буферов, 0-50% (масс/масс.) сорастворителей, и/или 0-99% (масс/масс.) воды для инъекций (WFI) (в зависимости от дозы и того, лиофилизирован ли состав). Составы для внутримышечных депо могут также содержать 0-99% (масс/масс.) масел.
Фармацевтические композиции для перорального введения могут быть получены путем объединения активного ингредиента с твердыми носителями, при желании гранулирования полученной смеси и переработки смеси, где это желательно или необходимо, после добавления соответствующих вспомогательных веществ, в таблетки, ядра драже или капсулы. Также возможно вводить их в полимерную или восковую матрицу, которая обеспечивает возможность диффундирования или высвобождения дозированных количества активных ингредиентов.
Соединения согласно настоящему изобретению можно также включать в составы в форму твердых дисперсий. Твердые дисперсии представляют собой гомогенные, исключительно тонкие дисперсные фазы из одного или более твердых веществ. Хорошо известно применение твердых растворы (молекулярно-дисперсных систем), одного из типов твердых дисперсий, в фармацевтике (см. (Chiou, Riegelman, J. Pharm. Sci., 60, 1281-1300 (1971)), они полезны для повышения скоростей растворения и повышения биодоступности слабо растворимых в воде лекарственных средств.
Согласно настоящему изобретению также предложены твердые лекарственные формы, содержащий твердый раствор, описанный выше. Твердые лекарственные формы включают таблетки, капсулы и жевательные таблетки, диспергируемые или шипучие таблетки, известные вспомогательные вещества можно смешивать с твердым раствором с получением желаемой лекарственной формы. Например, капсула может содержать твердый раствор, смешанный с (а) разрыхлителем и смазывающим веществом, или (b) разрыхлителем, смазывающим веществом и поверхностно-активным веществом. Кроме того, капсула может содержать наполнитель, такой как лактоза или микрокристаллическая целлюлоза. Таблетка может содержать твердый раствор, смешанный с по меньшей мере одним разрыхлителем, смазывающим веществом, поверхностно-активным веществом, наполнителем и скользящим веществом. Жевательная таблетка может содержать твердый раствор, смешанный с наполнителем, смазывающим веществом, и, при желании, дополнительным подсластителем (таким как искусственный подсластитель) и подходящими вкусоароматическими веществами. Твердые растворы могут также быть получены путем распыления растворов лекарственного средства и подходящего полимера на инертные носители, такие как сахарные крупинки ('нон па релей'). Затем этими крупинками можно наполнить капсулы или спрессовать их в таблетки.
Фармацевтические составы могут предоставляться пациенту в «упаковках для пациента», содержащих полный курс лечения в одной упаковке, обычно в блистерной упаковке. Упаковки для пациентов предпочтительнее обычных методов выписывания лекарств, в которых фармацевт выделяет количество, предназначенное для пациента, из общего количества, поскольку пациент имеет постоянный доступ к вкладышу, содержащемуся в упаковке для пациента, который обычно отсутствует при выдачи препарата фармацевтом. Было показано, что включение в упаковку вкладыша улучшает приверженность пациента (комплаентность) указаниям врача.
Композиции для топического применения и назальной доставки включают мази, кремы, спреи, пластыри, гели, жидкие капли и вставки (например, внутриглазные вставки (вкладыши)). Такие композиции могут быть изготовлены известными способами.
Примеры составов для ректального и внутривагинального введения включают пессарии и суппозитории, которые могут быть изготовлены, например, из формуемого или воскового материала, содержащего активное соединение, приданием формы. Растворы активного соединения можно также применять для ректального введения.
Композиции для введения путем ингаляции могут иметь форму композиций в виде порошков или жидкостей для ингаляций, и их можно вводить в стандартной форме с применением порошковых ингаляторов или аэрозольных устройств. Такие устройства хорошо известны. Для введения путем ингаляции, составы в форме порошка обычно содержат активное соединение с инертным твердым разбавителем в форме порошка, таким как лактоза.
Соединение формулы (1), как правило, будет представлено в форме с однократной дозировкой, и такая форма обычно будет содержать достаточное количество соединения для обеспечения желаемого уровня биологической активности. Например, состав может содержать от 1 нанограмма до 2 граммов активного ингредиента, например от 1 нанограмма до 2 миллиграммов активного ингредиента. Внутри этого диапазона конкретные поддиапазоны соединения составляют от 0,1 миллиграмма до 2 граммов активного ингредиента (в более типичных случаях от 10 миллиграммов до 1 грамма, например, от 50 миллиграммов до 500 миллиграммов) или от 1 микрограмма до 20 миллиграммов (например, от 1 микрограмма до 10 миллиграммов, например, от 0,1 миллиграмма до 2 миллиграммов активного ингредиента).
Для пероральных композиций форма однократной дозировки может содержать от 1 миллиграмма до 2 граммов, более типично от 10 милиграммов до 1 грамма, например от 50 миллиграммов до 1 грамма, например от 100 милиграммов до 1 грамма активного вещества.
Соединение согласно настоящему изобретению будет введено нуждающемуся в этом пациенту (например, человеку или пациенту-животному) в количестве, достаточном для достижения желаемого терапевтического эффекта.
Способы лечения
Соединение формулы (1) может быть применено для профилактики или лечения ряда заболеваний и состояний, опосредованных ERK1/2. Примеры таких заболеваний и состояний изложены выше.
Соединение обычно вводят субъекту, нуждающемуся в таком введении, например человеку или пациенту-животному, в частности человеку.
Указанное соединение обычно вводят в количествах, которые терапевтически или профилактически пригодны для применения и которые обычно нетоксичны. Однако в определенных ситуациях (например, в случае угрожающих жизни заболеваний) преимущества введения соединения формулы (1) могут перевешивать недостатки любых токсических или побочных эффектов, и в этом случае можно считать желательным вводить соединение в количествах, которые основаны на степени токсичности.
Соединение можно вводить в течение длительного периода времени для поддержания полезных терапевтических эффектов или можно вводить только в течение короткого периода времени. В качестве альтернативы, его можно вводить непрерывным образом или таким образом, который обеспечивает прерывистое дозирование (например, пульсирующим образом).
Типичная суточная доза соединения формулы (1) может находиться в диапазоне от 100 пикограммов до 100 миллиграммов на килограмм массы тела, более типично от 5 нанограммов до 25 миллиграммов на килограмм массы тела, и более обычно от 10 нанограммов до 15 миллиграммов на килограмм (например, от 10 нанограммов до 10 миллиграммов и более типично от 1 микрограмма на килограмм до 20 миллиграммов на килограмм, например, от 1 микрограмма до 10 миллиграммов на килограмм) массы тела, хотя при необходимости можно вводить более высокие или более низкие дозы. Соединение формулы (1) и его подформулы можно вводить на суточной основе или на основе повторений каждые 2, или 3, или 4, или 5, или 6, или 7, или 10, или 14, или 21, или 28 дней например.
Соединение согласно настоящему изобретению можно вводить перорально в диапазоне доз, например, от 1 до 1500 мг, от 2 до 800 мг или от 5 до 500 мг, например, от 2 до 200 мг или от 10 до 1000 мг, конкретные примеры доз включают 10, 20, 50 и 80 мг. Соединение может быть введено однократно или несколько раз каждый день для получения желаемого терапевтического эффекта. Соединение можно вводить непрерывно (т.е. принимать каждый день без перерыва в течение всего курса лечения). В качестве альтернативы, соединение можно вводить прерывисто (т.е. непрерывно принимать в течение определенного периода, такого как неделя, затем прерывать на период, такой как неделя, и затем непрерывно принимать в течение другого периода, такого как неделя, и так далее в течение всего курса лечения). Примеры курсов лечения, включающих прерывистое введение, включают схемы, в которых введение осуществляется циклами одной недели использования, одной недели перерыва; или двух недель использования, одной недели перерыва; или трех недель использования, одной недели перерыва; или двух недель использования, двух недель перерыва; или четырех недель использования на две недели перерыва; или одной недели использования на три недели перерыва - для одного или более циклов, например, 2, 3, 4, 5, 6, 7, 8, 9 или 10 или более циклов. Это лечение с перерывами может также быть основано на количестве дней, а не на полной неделе. Например, лечение может включать ежедневное дозирование в течение от 1 до 6 дней, отсутствие дозирования в течение от 1 до 6 дней с повторением этой схемы по ходу протокола лечения. Количество дней (или недель), в течение которых соединения согласно настоящему изобретению не дозируются, необязательно должно равняться количеству дней (или недель), в которые соединения согласно настоящему изобретению дозируются.
В одной конкретной схеме дозирования пациенту будут давать инфузию соединения формулы (1) в течение одного часа ежедневно на срок до десяти дней, в частности до пяти дней в течение одной недели, и лечение повторяют с желаемым интервалом, таким как две - четыре недели, в частности каждые три недели.
Соединение согласно настоящему изобретению также можно вводить болюсом или непрерывной инфузией. Соединение согласно настоящему изобретению можно вводить ежедневно один раз каждую неделю или один раз каждые две недели или один раз каждые три недели или один раз каждые четыре недели в течение цикла лечения. При ежедневном введении в течение цикла лечения это ежедневное дозирование может быть прерывистым на протяжении нескольких недель цикла лечения: например, дозирование в течение недели (или количества дней), отсутствие дозирования в течение недели (или количества дней), с повторяющимся во время цикла лечения шаблоном.
В одной из схем дозирования пациенту дают инфузию соединения формулы (1) в течение периодов один час ежедневно в течение 5 дней, и лечение повторяют каждые три недели.
В другой конкретной схеме дозирования пациенту дают инфузию в течение от 30 минут до 1 часа с последующими поддерживающими инфузиями переменной продолжительности, например от 1 до 5 часов, например 3 часа.
В дополнительной конкретной схеме дозирования пациенту дают непрерывную инфузию в течение периода от 12 часов до 5 дней, в частности непрерывную инфузию от 24 часов до 72 часов.
В конечном счете, однако, количество вводимого соединения и тип используемой композиции будут соразмерны природе заболевания или подлежащего лечению физиологического состояния и будут определены врачом.
Было обнаружено, что ингибиторы ERK1/2 можно применять в качестве единственного агента или в комбинации с другими противораковыми агентами. Например, можно получить преимущество от комбинирования ингибитора, который подавляет передачу сигналов ERK, с другим агентом, который действует через другую точку в каскаде сигнальной трансдукции или с помощью другого механизма регулирования роста клеток, таким образом подвергая лечению две характерные особенности развития рака. Комбинированные эксперименты могут быть выполнены, например, как описано в Chou ТС, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regulat 1984; 22: 27-55.
Соединение согласно настоящему изобретению можно вводить в качестве единственного терапевтического агента или его можно вводить в комбинированной терапии с одним или более другими соединениями (или разновидностями терапии) для лечения конкретного заболевания, например, неопластического заболевания, такого как рак, как определено выше в настоящем документе. Для лечения вышеуказанных состояний соединение согласно настоящему изобретению может преимущественно применяться в комбинации с одним или более другими лечебными средствами, более конкретно, с другими противораковыми агентами или адъювантами (поддерживающими терапевтическими агентами) в терапии рака. Примеры других терапевтических агентов или видов лечения, которые можно вводить вместе (или параллельно, или через разные промежутки времени) с соединением формулы (1), включают, но не ограничиваются ими:
• ингибиторы топоизомеразы I
• антиметаболиты
• Агенты, воздействующие на тубулин
• ДНК-связывающие агенты о и ингибиторы топоизомеразы II
• Алкилирующие агенты
• Моноклональные антитела
• Антигормоны
• Ингибиторы передачи сигнала
• Ингибиторы убиквитин-протеасомного пути
• Иммуно-терапевтические средства
• Регуляторы гибели клеток
• Ингибиторы ДНК-метил-трансферазы
• Цитокины и ретиноиды
• Терапевтические средства, нацеленные на хроматин,
• Радиотерапия и
• другие терапевтические и профилактические средства.
Конкретные примеры противораковых агентов или адъювантов (или их солей) включают следующие, но не ограничиваются ими: любые один или более агентов, выбранных их групп (i)-(xlviii) и необязательно группы (xlix) и или (I), приведенных ниже:
(i) Соединения платины, например, цисплатин (необязательно в комбинации с амифостином), карбоплатин или оксалиплатин,
(ii) Соединения класса таксанов, например, паклитаксел, частицы паклитаксела, связанного с белком (Abraxane™), доцетаксел, кабазитаксел или ларотаксел,
(iii) ингибиторы топоизомеразы I, например, производные камптотецина, например, камптотецин, иринотекан (СРТ11), SN-38 или топотекан,
(iv) Ингибиторы топоизомеразы II, например, противоопухолевые производные эпидофиллотоксинов или подофиллоытоксинов, например, этопозид, или тенипозид,
(v) Алкалоиды барвинка, например, винбластин, винкристин, липосомальный винкристин (Onco-TCS), винорелбин, виндезин, винфлунин или винвесир, (vi) Производные нукпеозидов, например, 5-фторурацил (5-FU, необязательно в комбинации с лейковорином), гемцитабин, капецитабин, тегафур, UFT, S1, кладрибин, цитарабин (Ara-С, цитазин-арабинозид), флударабин, кпофарабин или неларабин,
(vii) антиметаболиты, например, кпофарабин, аминоптерин, или метотрексат, азацитидин, цитарабин, флоксиридин, пентостатин, тиогуанин, тиопурин, 6-меркаптопурин, или гидроксимочевина (гидроксикарбамид) или трфлуридин (необязательно в комбинации с типирацилом),
(viii) Алкилирующие агенты, такие как азотистый иприт или нитрозомочевина, например, цикпофосфамид, хлорамбуцил, кармустин (BCNU), бендамустин, тиотепа, мелфалан, треосульфан, ломустин (CCNU), алтретамин, бусульфан, декарбазин, эстрамустин, фотемустин, ифосфамид (необязательно в комбинации с месной), пипроброман, прокарбазин, стрептозоцин, темозоломид, урацил, мехлорэтамин, метилцикпогексилхлорэтилнитрозомочевина или нимустин (ACNU),
(ix) Антрациклины, антрацендионы и родственные лекарственные средства, например, таунорубицин, доксорубицин (необязательно в комбинации с дексразоксаном), липосомные составы доксорубицина (например, Caelyx™, Myocet™, Doxil™), идарубицин, митоксантрон, эпирубицин, амскарин или валрубицин,
(x) эпотилоны, например, иксабепилон, патупилон, BMS-310705, KOS-862 и ZK-EFO, эпотилон А, эпотилон В, дезоксиэпотилон В (также известный как эпотилон D или KOS-862), азаэпотилон В (также известный как BMS-247550), аулималид, изоаулималид, или лейтеробин,
(xi) Ингибиторы ДНК-метил-трансферазы, например, тамозоломид, азацитидин, децитабин или гуадецитабин (SGI-110),
(xii) антифолаты, например, метотрексат, пеметрексед динатрия или ралитрексид,
(xiii) Цитотоксические антибиотики, например, антиномицин D, блеомицин, митомицин С, дактиномицин, карминомицин, дауномицин, левамизол, пликамицин или митрамицин,
(xiv) Тубулин-связывающие агенты, например, комбрестатин, колхицины или нокодазол,
(xv) Ингибиторы передачи сигнала, такие как ингибиторы киназ например, ингибиторы тирозинкиназных рецепторов (например, ингибиторы EGFR (рецептора эпителиального фактора роста), ингибиторы VEGFR (рецептора фактора роста эндотелия сосудов), ингибиторы PDGFR (рецептора тромбоцитарного фактора роста), ингибиторы Axl, MTKI (многоцелевые ингибиторы киназ), ингибиторы Raf, ингибиторы ROCK, ингибиторы mTOR, ингибиторы МЕК или ингибиторы PI3K) например, иматиниб мезилат, эрлотиниб, гефитиниб, дазатиниб, лапатиниб, довотиниб, акситиниб, нилотиниб, вандетаниб, ваталиниб, пазопаниб, сорафениб, сунитиниб, темсиролимус, эверолимус (RAD 001), вемурафениб (PLX4032 или RG7204), дабрафениб, энкорафениб, селуметиниб (AZD6244), траметиниб (GSK121120212), дактолисиб (BEZ235), бупарлисиб (ВКМ-120, NVP-BKM-120), BYL719, копанлисиб (BAY-80-6946), ZSTK-474, CUDC-907, апитолисиб (GDC-0980, RG-7422), пиктилисиб (пиктрелисиб, GDC-0941, RG-7321), GDC-0032, GDC-0068, GSK-2636771, иделалисиб (ранее CAL-101, GS 1101, GS-1101), MLN1117 (INK1117), MLN0128 (INK128), IPI-145 (INK1197), LY-3023414, ипатесертиб, афуресертиб, МК-2206, МК-8156, LY-3023414, LY294002, SF1126 или PI-103, сонолисиб (РХ-866) или АТ13148.
(xvi) Ингибиторы киназы Aurora, например, АТ9283, барасертиб (AZD1152), ТАК-901, МК0457 (VX680), сенисертиб (R-763), данусертиб (РНА-739358), алисертиб (MLN-8237) или МР-470,
(xvii) ингибиторы CDK, например, АТ7519, росковитин, селициклиб, альвоцидиб (флавопиридол), динациклиб (SCH-727965), 7-гидроксистауроспорин (UCN-01), JNJ-7706621, BMS-387032 (также известный как. SNS-032), РНА533533, ZK-304709 или AZD-5438, и включая ингибиторы CDK4, такие как палбоциклиб (PD332991) и рибоциклиб (LEE-011),
(xviii) ингибиторы пути РКА/В и РКВ (akt), например, АТ13148, AZD5363, Semaphore, SF1126 и ингибиторы MTOR, такие как аналоги рапамицина, АР23841 и АР23573, ингибиторы кальмодулина (ингибиторы транслокации фактора forkhead), API-2/TCN (трицирибин), RX-0201, энзастаурин HCI (LY317615), NL-71-101, SR-13668, РХ-316 или KRX-0401 (перифозин/ NSC 639966),
(xix) ингибиторы Hsp90, например, оналеспиб (АТ13387), гербимицин, гелданамицин (GA), 17-аллиламино-17-десметоксигелданамицин (17-AAG) например, NSC-330507, Kos-953 и CNF-1010, 17-диметиламиноэтиламино-17-деметоксигелданамицин гидрохлорид (17-DMAG) например, NSC-707545 и Kos-1022, NVP-AUY922 (VER-52296), NVP-BEP800, CNF-2024 (BIIB-021 - пурин для перорального введения), ганетеспиб (STA-9090), SNX-5422 (SC-102112), или IPI-504 или TAS-116, (xx) Моноклональные антитела (неконъюгированные или конъюгированные с радиоизотопами, токсинами или другими агентами), производные антител и родственные агенты, такие как анти-CD, анти-VEGFR, анти-HER2 или анти-EGFR антитела, например, ритуксимаб (CD20), офатумумаб (CD20), иритумомабтиуксетан (CD20), GA101 (CD20), тозитомумаб (CD20), эпратузумаб (CD22), линтузумаб (CD33), гемтузумаб озогамицин (CD33), алемтузумаб (CD52), галаксимаб (CD80), трастузумаб (антитело к HER2), пертузумаб (HER2), трастузумаб-DMI (HER2), эртумаксомаб (HER2 и CD3), цетуксимаб (рецептор эпидермального фактора роста), панитумумаб (рецептор эпидермального фактора роста), нецитумумаб (рецептор эпидермального фактора роста), нимотузумаб (рецептор эпидермального фактора роста), бевацизумаб (VEGF), катумаксумаб (ЕрСАМ и CD3), абаговомаб (СА125), фарлетузумаб (фолатный рецептор), элотузумаб (CS1), деносумаб (лиганд RANK), фигитумамаб (IGF1R), СР751.871 (IGF1R), мапатумумаб (рецептор TRAIL), metMAB (met), мутимомаб (ганглиозид GD3), наптумомаб эстафенатокс (5Т4), или силтуксимаб (IL6) или иммуномодилирующие агенты, такие как антитела, блокирующие CTLA-4, и/или антитела против PD-1 и PD-L1 и/или PD-L2 например, ипилимумаб (CTLA4), МК-3475 (пембролизумаб, ранее ламбролизумаб, анти-PD-1), ниволумаб (анти-PD-1), BMS-936559 (анти-PD-L1), MPDL320A, АМР-514 или MEDI4736 (анти-PD-U), или тремелимумаб (ранее тецилимумаб, СР-675,206, анти-CTLA-4),
(xxi) антагонисты рецептора эстрогена или селективные модуляторы рецептора эстрогена (SERM) или ингибиторы синтеза эстрогена, например, тамоксифен, фульвестрант, торемифен, дролоксифен, фаслодекс, или ралоксифен,
(xxii) ингибиторы ароматазы и родственные лекарственные средства, такие как эксеместан, анастрозол, летразол, тестолактон аминоглютенимид, митотан и ворозол,
(xxiii) антиандрогены (т.е., антагонисты андрогенового) и родственные агенты например, бикалютамид, нилутамид, флутамид, ципротерон или кетоконазол,
(xxiv) гормоны и их аналоги, такие как медроксипрогестерон, диэтилсильбэстрол (также известный как диэтилстильбэстрол) илиокреотид,
(xxv) Стероиды например, дромостанолон пропионат, мегестрол ацетат, нандролон (деканоат, фенпропионат), флуоксиместрон или госсипол,
(xxvi) Ингибитор стероидного цитохрома Р450 17альфа-гидроксиллазы-17,20-лиазы (CYP17), например, абиратерон, (xxvii) агонисты или антагонисты гонадоропин-высвобождающего гормона (GnRA) например, абареликс, гозерелин ацетат, гистрелин ацетат, лейпролид ацетат, трипторелина, бусерелин, или деслорелин,
(xxviii) глюкокортикоиды, например, преднизон, преднизолон, дексаметазон,
(xxix) Агенты, стимулирующие дифференцировку, такие как ретиноиды, рексиноиды, витамин D или ретиноевая кислота и агенты, блокирующие метаболизм ретиноевой кислоты (RAMBA), например, аккутан, алитретиноин, бексаротин или третиноин,
(xxx) Ингибиторы фарнезилтрансферазы например, типифарниб,
(xxxi) Терапевтические средства, нацеленные на хроматин, такие как ингибиторы гистондеацетилазы (HDAC) например, бутират натрия, субероиланилидгидроксамовая кислота (SAHA), депсипептид (FR 901228), дациностат (NVP-LAQ824), R306465 / JNJ-16241199, JNJ-26481585, трихостатинА, вориностат, хламидоцин, А-173, JNJ-MGCD-0103, PXD-101, или апицидин,
(xxxii) Лекарственные средства, нацеленные на убиквитин-протеасоомный путь, включая ингибиторы протеасом например, ботрезомиб, карфилзомиб, СЕР-18770, MLN-970 или ONX-0912, ингибиторы NEDD8, антагонист HDM2 и деубиквитиназы (DUB),
(xxxiii) Фотодинамические лекарственные средства например, порфимер натрия или темопорфин,
(xxxiv) Противораковые агенты, выделеные из морских организмов, такие как трабектидин,
(xxxv) Радиомеченные лекарственные средства для радиоиммунотерапии, например, с меченные изотопом, испускающим бета-частицы (например, йод -131, иттрий-90), или изотопом, испускающим альфа-частицы (например, висмут-213 или актиний-225) например, ибритумумаб, Йод-тозитомумаб или альфа-радий,
(xxxvi) ингибиторы теломеразы например, теломестатин,
(xxxvii) ингибиторы металлопротеиназы матрикса, например, батимастат, маримастат, приностат или метастат,
(xxxviii) рекомбинантные интерфероны (такие как интерферон-γ и интерферон α) и интерлейкины (например, интерлейкин 2), например, альдеслейкин, денилейкин дифтитокс, интерферон альфа 2а, интерферон альфа 2b или пегинтерферон альфа, (xxxix) Селективные модуляторы иммунных реакций, например, талидомид, или леналидомид,
(xl) терапевтические вакцины, такие как си пулей цел-Т (Provenge) или OncoVex,
(xli) цитокин-активирующие агенты включают Picibanil (пицибанил), Romurtide (ромуртид), Sizofiran (сиофиран), Virulizin (вирулизин) или тимозин,
(xlii) триоксид мышьяка,
(xliii) Ингибиторы рецепторов, сопряженных cG-белком (GPCR) например, атрасентан,
(xliv) Ферменты, такие как L-аспарагиназа, пегаспаргаза, расбуриказа или пегадемаза,
(xlv) ингибиторы репарации ДНК, такие как PARP ингибиторы например, олапариб, велапариб, инипариб, INO-1001, AG-014699 или ONO-2231,
(xlvi) агонисты клеточной смерти (например, рецептор родственного ФНО апоптоз-индуцирующего лиганда (TRAIL)), такие как мапатумумаб (ранее HGS-ETR1), конатумумаб (ранее AMG 655), PRO95780, лексатумумаб, дуланермин, CS-1008, апомаб или рекомбинантные лиганды TRAIL, такие как человеческий TRAIL/Apo2 лиганд,
(xlvii) Иммуно-терапевтические средства, такие как ингибиторы контрольных точек иммунитеты, противораковые вакцины и клеточная терапия CAR-T,
(xlviii) Регуляторы гибели клеток (апоптоза), включая антагонисты Bcl-2 (В-клеточноы лимфомы 2), такие как венетоклакс (АВТ-199 или GDC-0199), АВТ-737, АВТ-263, TW-37, сабутоклакс, обатоклакс, и антагонисты MIM1 и IAP, включая LCL-161 (Novartis), Debio-1143 (Debiopharma / Ascenta), AZD5582, биринапант / TL-32711 (TetraLogic), CUDC-427 / GDC-0917 / RG-7459 (Genentech), JP1201 (Joyant), T-3256336 (Takeda), GDC-0152 (Genentech), HGS-1029 / AEG-40826 (HGS/ Aegera) или ASTX-660,
(xlix) Профилактические агенты (вспомогательные), т.е., агенты снижают или облагчают побочные эффекты, связанные с химиотерапевтическим агентами,
- противорвотные средства,
- агенты, которые предотвращают или уменьшают длительность связанной с химиотерапией нейтропении и предотвращают осложнения, возникающие из-за снижения уровней тромбоцитов, эритроцитов или лейкоцитов, например, интерлейкин-11 (например, опрелвекин), эритропоэтин (ЕРО) и его аналоги (например, дарбепоэтин альфа), аналоги колониестимулирующего фактора, такие как колониестимулирующий фактор гранулоцитов-макрофагов (GM-CSF) (например, саргамостим), и колониестимулирующий фактор гранулоцитов (G-CSF) и его аналоги (например, филграстим, пегфилграстим),
- агенты, которые ингибируют резорбцию костей, такие как деносумаб или бисфосфонаты например, золедронат, золедроновая кислота, памидронат и ибандронат,
- агенты, которые подавляют воспалительные ответы, такие как дексаметазон, преднизон и преднизолон,
- агенты, применяемые для снижения в крови уровней гормона роста и инсулиноподобного фактора роста-1 (и других гормонов) у пациентов с акромегалией или другими редкими гормон-продуцирующими опухолями, такие как синтетические формы гормона соматостатина, например, октреотида ацетат,
- антидоты к лекарственным средствам, которые снижают уровни фолиевой кислоты, такие как лейковорин или фолиновая кислота,
- агенты против боли например, опиаты, такие как морфин, диаморфин и фентанил,
- нестероидные противовосплительные лекарственные средства (НСПВП), такие как ингибиторы СОХ-2 например, целекоксиб, эторикоксиб и лумиракоксиб,
- агенты против мукозита например, палифермин,
- агенты для лечения побочных эффектов, включая анорексию, кахексию, эдему, эпизоды тромбоэмболии, такие как мегастрол ацетат, и
(I) радиотерапия для радикальных, паллиативных или профилактических целей (для адъювантных или неоадъювантных целей).
Каждое соединение, присутствующее в комбинации согласно настоящему изобретению, можно давать с индивидуально варьируемым графиком введения и различными путями. Соответственно, каждый из двух или более агентов может отличаться своей схемой дозирования: каждый можно вводить в одно и то же время или в разные моменты времени. Специалист в данной области на основании своих общих знаний поймет, какие схемы введения и варианты комбинированной терапии можно применять. Например, соединение настоящего изобретения можно применять в комбинации с одним или более агентами, которые вводят в соответствии с существующими схемами комбинированной терапии для них. Примеры стандартных комбинированных режимов приведены ниже.
Соединения класса таксанов вводят преимущественно в дозировке от 50 до 400 мг на квадратный метр (мг/м2) поверхности тела, например, от 75 до 250 мг/м2, в частности, паклитаксел в дозировке приблизительно от 175 до 250 мг/м2 и доцетаксел в дозировки приблизительно от 75 до 150 мг/м2 на курс лечения.
Соединения класса кампотецинов вводят преимущественно в дозировке от 0,1 до 400 мг на квадратный метр (мг/м2) поверхности тела, например, от 1 до 300 мг/м2, в частности, иринотекан в дозировке приблизительно от 100 до 350 мг/м2, а топотекан в дозировке приблизительно от 1 до 2 мг/м2 на курс лечения.
Противоопухолевые производные подофиллотоксина вводят преимущественно в дозировке от 30 до 300 мг на квадратный метр (мг/м2) поверхности тела, например, от 50 до 250 мг/м2, в частности, этопозид в дозировке приблизительно от 35 до 100 мг/м2, и тенипозид в дозировке от 50 до 250 мг/м2 на курс лечения.
Противоопухолевые алкалоиды барвинка вводят преимущественно в дозировке от 2 до 30 мг на квадратный метр (мг/м2) поверхности тела, в частности, винбластин в дозировке приблизительно от 3 до 12 мг/м2, винкристин в дозировке приблизительно от 1 до 2 мг/м2, и винорелбин в дозировке от приблизительно 10 до 30 мг/м2 на курс лечения.
Противоопухолевые производные нуклеозидов вводят преимущественно в дозировке от 200 до 2500 мг на квадратный метр (мг/м2) поверхности тела, например, от 700 до 1500 мг/м2, в частности, в частности, 5-FU в дозировке от 200 до 500 мг/м2, гемцитабин в дозировке приблизительно от 800 до 1200 мг/м2 и капецитабин в дозировк приблизительно от 1000 до 2500 мг/м2 на курс лечения.
Алкилирующие агенты, такие как азотистый иприт или нитрозомочевина, вводят преимущественно в дозировке от 100 до 500 мг на квадратный метр (мг/м2) поверхности тела, например, от 120 до 200 мг/м2, в частности, циклофосфамид в дозировке приблизительно от 100 до 500 мг/м2, хлорамбуцил в дозировке приблизительно от 0,1 до 0,2 мг/кг, кармустин в дозировке приблизительно от 150 до 200 мг/м2, и ломустин в дозировке приблизительно от 100 до 150 мг/м2 на курс лечения.
Противоопухолевое производное антрациклина вводят преимущественно в дозировке от 10 до 75 мг на квадратный метр (мг/м2) поверхности тела, например, от 15 до 60 мг/м2, в частности, доксорубицин в дозировке приблизительно от 40 до 75 мг/м2, даунорубицин в дозировке приблизительно от 25 до 45 мг/м2, и иридарубицин в дозировке приблизительно от 10 до 15 мг/м2 на курс лечения.
Антиэстрогеновый агент вводят преимущественно в дозировке приблизительно 1-100 мг ежедневно в зависимости от конкретного агента и состояния, которое лечат. Тамоксифен преимущественно вводят перорально в дозировке от 5 до 50 мг, в частности, от 10 до 20 мг дважды в день, продолжая терапию в течение времени, достаточного для того, чтобы достичь и поддерживать терапевтический эффект. Торемифен преимущественно вводят перорально в дозировке приблизительно 60 мг один раз в день, продолжая терапию в течение времени, достаточного для того, чтобы достичь и поддерживать терапевтический эффект. Анастрозол преимущественно вводят перорально в дозировке приблизительно 1 мг один раз в день. Дролоксифен преимущественно вводят перорально в дозировке приблизительно 20-100 мг один раз в день. Ралоксифен преимущественно вводят перорально в дозировке приблизительно 60 мг один раз в день. Эксеместан преимущественно вводят перорально в дозировке приблизительно 25 мг один раз в день.
Антитела преимущественно вводят в дозировке приблизительно от 1 до 5 мг на квадратный метр (мг/м2) поверхности тела, или в другой дозировке, известной в данной области. Трастузумаб вводят преимущественно в дозировке от 1 до 5 мг на квадратный метр (мг/м2) поверхности тела, в частности, от 2 до 4 мг/м2, на курс лечения.
Когда соединение формулы (1) вводят в комбинированной терапии с одним, двумя, тремя, четырьмя или более другими терапевтическими агентами (в частности, с одним или двумя, более конкретно, с одним), указанные соединения можно вводить одновременно или последовательно. В последнем случае два или более соединений будут вводиться в течение периода и в количестве, и образом, которые достаточны для обеспечения достижения полезного или синергетического эффекта. При последовательном введении их можно вводить с близко расположенными интервалами (например, в течение периода 5-10 минут) или с более длительными интервалами (например, с интервалом в 1, 2, 3, 4 или более часов, или даже с более длительными интервалами друг от друга, где это необходимо), точный режим дозирования соразмерен свойствам терапевтического агента (агентов). Эти дозы могут быть введены, например, один раз, два или более за курс лечения, который может повторяться, например, каждые 7, 14, 21 или 28 дней.
Понятно, что конкретный способ и порядок введения и соответствующие размеры доз и режимы для каждого компонента комбинации будут зависеть от конкретного другого медицинского агента, и вводимого соединения согласно настоящему изобретению, пути из введения, конкретной опухоли, которую лечат и конкретного хозяина, которого лечат. Специалист в данной области сможет легко определить оптимальный способ и порядок введения, а также размеры доз и режим, с применением обычных способов с учетом информации, приведенной в настоящем документе.
Массовое отношение соединения в соответствии с настоящим изобретением к одному или более другим противораковым агентам, если они даны в виде комбинации, может быть определено специалистом в данной области техники. Отношение, точная дозировка и частота введения будут зависеть от конкретного соединения согласно настоящему изобретению и другого используемого противоракового агента (агентов), конкретного подвергаемого лечению состояния, тяжести подвергаемого лечению состояния, возраста, массы, пола, диеты, времени введения и общего физического состояния конкретного пациента, способа введения, а также других лекарств, которые может принимать индивидуум, как хорошо известно специалистам в данной области техники. Кроме того, очевидно, что эффективное суточное количество может быть уменьшено или увеличено в зависимости от реакции подвергаемого лечению субъекта и/или в зависимости от заключения врача, назначающего соединение согласно настоящему изобретению. Конкретное массовое отношение настоящего соединения формулы (1) к другому противораковому агенту может находиться в диапазоне от 1/10 до 10/1, более конкретно, от 1/5 до 5/1, еще более конкретно, от 1/3 до 3/1.
Соединение согласно настоящему изобретению также можно вводить в сочетании с нехимиотерапевтическими разновидностями лечения, такими как лучевая терапия, фотодинамическая терапия, генная терапия; хирургия и контролируемые диеты.
Соединение согласно настоящему изобретению также может иметь терапевтические применения при сенсибилизации опухолевых клеток для лучевой терапии и химиотерапии. Следовательно, соединение согласно настоящему изобретению можно применять в качестве «радиосенсибилизатора» и/или «хемосенсибилизатора» или его можно давать в комбинации с другим «радиосенсибилизатором» и/или «хемосенсибилизатором». Водном из вариантов реализаци соединение согласно настоящему изобретению предназначено для применения в качестве хемосенсибилизатора.
Термин «радиосенсибилизатор» определяют как молекулу, которую вводят пациентам в терапевтически эффективных количествах для повышения чувствительности клеток к ионизирующему излучению и/или для стимуляции лечения заболеваний, которые можно лечить иоизирующим излучением.
Термин «хемосенсибилизатор» определяют как молекулу, которую вводят пациентам в терапевтически эффективных количествах для повышения чувствительности клеток химиотерапии и/или стимуляции лечения заболеваний, которые можно лечить с применением химиотерапевтических средств.
В многих современных протоколах лечения рака применяют радиосенсибилизаторы в комбинации с рентгеновским излучением. Примеры активируемых рентгеновским излучением радиосенсибилизаторов включают следующие, но не ограничиваются ими: метронидазол, мезинидазол, десметилмедизидазол, пимонидазол, этанидазол, ниморазол, митомицин С, RSU 1069, SR 4233, ЕО9, RB 6145, никотинамид, 5-бромдезоксиуридин (BUdR), 5-йододезоксиуридин (IUdR), бромдезоксицитидин, фтордезоксиуридин (FudR), гидроксимочевину, цисплатин, и их терапевтически эффективные аналоги и производные.
В фотодинамической терапии (PDT) рака используют видимый свет в качестве активатора сенсибилизирующего средства. Примеры фотодинамических радиосенсибилизаторов включают следующие, но не ограничиваются ими: производные гематопорфирина, фотофрин, производные бензопорфирина, этиопорфирин олова, феоборбид-а, бактериохлорофилл-а, нафталоцианины, фталоцианины, фталоцианин цинка и их терапевтически эффективные аналоги и производные.
Радиосенсибилизаторы можно вводить в сочетании с терапевтически эффективным количеством одного или более соединениями, включая следующие, но не ограничиваясь ими: соединения, которые способствуют включению радиосенсибилизаторов в клетки-мишени, соединения, которые регулируют потоки терапевтических веществ, питательных веществ и/или кислорода в клетках-мишенях, химиотерапевтические средства, которые действуют на опухоль с дополнительным облучением или без него, другие терапевтически эффективные соединения для лечения рака или других заболеваний.
Хемосенсибилизаторы можно вводить в сочетании с терапевтически эффективным количеством одного или более соединений, включая следующие, но не ограничиваясь ими: соединения, которые способствуют включению хемосенсибилизаторов в клетки-мишени, соединения, которые регулируют потоки терапевтических веществ, питательных веществ и/или кислорода в клетках-мишенях, химиотерапевтические средства, которые действуют на опухоль или другие терапевтически эффективные соединения для лечения рака или других заболеваний. Антагонисты кальция, например, верапамил, применяются в комбинации с антинеопластическими агентами, обеспечивая хамочувствительность опухолевых клеток, устойчивых к принятым химиотерапевтическим агентам и потенцируя действия таких соединения на чувствительные к лекарственной терапии злокачественные новообразования.
Для применения в комбинированной терапии с другим химиотерапевтическим агентом соединение формулы (1) и один, два, три, четыре или более других терапевтических агентов могут быть, например, введены в состав вместе в лекарственной форме, содержащей два, три, четыре или больше терапевтических агентов, т.е. в унитарной фармацевтической композиции, содержащей все компоненты. В качестве альтернативы, индивидуальные терапевтические агенты могут быть введены в раздельные составы и представлены вместе в форме набора, необязательно с инструкциями по их применению. Из вышеизложенного будет понятно, что в другом варианте реализации изобретения предложена комбинация соединения формулы (1) и другого терапевтического агента, например другого терапевтического агента, как определено выше.
В другом варианте реализации настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (1) вместе с фармацевтически приемлемой переносящей средой и одним или более терапевтическими агентами, определенными выше.
В дополнительных вариантах реализации настоящего изобретения предложены:
• Комбинация, как определено в настоящем описании, для применения для лечения (для применения для ослабления или уменьшения частоты возникновения) заболевания или состояния, как описано в настоящем описании, в частности рака.
• Применение комбинации, как определено в настоящем описании, для получения лекарственного средства для лечения (для применения для ослабления или уменьшения частоты возникновения) заболевания или состояния, как описано в настоящем описании, в частности рака.
• Способ предотвращения или лечения (для применения для ослабления или уменьшения частоты возникновения) заболевания или состояния, как описано в настоящем описании, в частности рака, у субъекта (например, субъекта-млекопитающего, такого как человек, нуждающегося в этом), включающий введение субъекту терапевтически эффективного количества комбинации, как определено в настоящем описании.
• Комбинация, как определено в настоящем описании, для применения для ингибирования роста опухолевых клеток (например, у пациента).
• Применение комбинации, как определено в настоящем описании, для получения лекарственного средства для ингибирования роста опухолевых клеток у пациента.
• Способ ингибирования роста опухолевых клеток (например, у пациента), включающий контактирование опухолевых клеток с соединением формулы (1) или комбинацией, как определено в настоящем описании.
В каждом из вышеприведенных вариантов реализации указанное соединение формулы (1) и один или несколько других терапевтических агентов, по меньшей мере один из которых представляет собой противораковый агент, можно вводить одновременно, раздельно или последовательно при лечении пациентов, страдающих раком.
В другом варианте реализации настоящего изобретения предложен способ профилактики или лечения (или ослабления или уменьшения частоты возникновения) рака, включающий введение пациенту соединения формулы (1) в комбинации с радиотерапией или химиотерапией.
В другом варианте реализации настоящего изобретения предложено соединение формулы (1) для применения для профилактики или лечения (или ослабления или уменьшения частоты возникновения) рака в комбинации с радиотерапией или химиотерапией.
ПРИМЕРЫ
Ниже настоящее изобретение будет проиллюстрировано, но не ограничено, со ссылкой на конкретные варианты реализации, описанные в приведенных ниже примерах. Названия соединениям присвоены с применением автоматизированного пакета для генерирования названий, такого как AutoNom (MDL) или ChemAxon Structure to Name, или взяты их документации поставщика реагентов. В примерах приведены следующие сокращения.
Приведенные ниже соединения получали описанными способами и/или способами, аналогичными описанным.
Описанные ниже процедуры синтеза приведены для иллюстрации применяемых способов; для данного этапа получения применяемый предшественник необязательно должен происходить из индивидуальной партии, синтезированной в соответствии с указанным этапов в данном описании.
В тех случаях, когда соединения описаны как смесь двух диастереоизомеров / эпимеров, конфигурация стереоцентра не указана и представлена прямыми линиями.
Специалисту в данной области понятно, что соединения, синтезированные с применением указанных протоколов, могут существовать в форме сольвата, например, гидрата, и/или содержать остаточный растворитель или минорные примеси. Соединения, выделяемые в в форме соли, могут характеризоваться целочисленными стехиометрическими индексами, т.е., представлять собой моно- или ди-соли, или иметь промежуточную стехиометрию.
Некоторые из приведенных ниже соединению выделяют в форме солей, например, в зависимости от кислоты, применяемой в методе очистки. Некоторые соединения выделяют в форме свободных оснований.



BINAP - 2,2'-бис(дифенилфосфино)-1,1'-бинафталин; CDI - 1,1'-карбонилдиимидазол; DCE, ДХЭ - 1,2-дихлорэтан; ДХМ - дихлорметан; DIPEA - диизопропилэтиламин; ДМСО -диметилсульфоксид; ДМФА- N,N-диметилформамид; DMAP-диметиламинопиридин; EtOAc - этилацетат; ч - часы; HATU - N,N,N',N'-третраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат; HBTU - 3-[бис(диметиламино)метилийил]-3Н-бензотриазол-1-оксид гексафторфосфат; HCI - соляная кислота; ВЭЖХ - высокоэффективная жидкостная хроматография (жидкостная хроматография высокого давления); ЖХ-МС - хидкостная хроматография-масс-спектрометрия; LiHMDS - бис(триметилсилил)лития амид; мин - минуты; MeCN -ацетонитрил; МС - масс-спектрометрия; NBS - N-бромсукцинимид; ЯМР - ядерная магнито-резонансная спектроскопия; PdCl2(dppf)2 - (1,1'-бис(дифенилфосфино)-ферроцен)палладий(II) дихлорид; Pd2(dba)3 - трис(дибензилидинацетон)палладий (0); петролейный эфир - фракция петролейного эфира с температурой кипения в диапазоне 40-60°С; РуВОР - бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфат; КТ - комнатная температура; насыщ. - насыщенный; SCX -твердофазная катионообменная смола; SPhos - 2-дициклогексилфосфино-2',6'-диметоксибифенил; S-Phos Pd G3 - (2-дициклогексилфосфино-2',6'-диметоксибифенил) [2-(2'-амино-1,1'-бифенил)]палладий(II) метансульфонат; TBDMSCI - трет-бутилдиметилсилилхлорид; TBTU - O-(бензотриазол-1-ил)-N,N,N',N'-третраметилурония тетрафторборат; ТФУК - трифторуксусная кислота; ТГФ - тетрагидрофуран; XPhos - 2-дицикпогексилфосфино-2',4',6'-триизопропилбифенил; XantPhos - 4,5-Бис(дифенилфосфино)-9,9-диметилксантен.
Когда количества приведены в массовых эквивалентах (масс.) и объемных эквивалентах (об.), 1 об. равен 1 мл на грамм исходного материала (который определяют как имеющий значение эквивалентной массы 1 масс.). Например, при применении 50 мг (0,05 г) исходного материала (определяемого как имеющий массовый эквивалент 1 масс.), количество 20 об. составляет 1 мл (0,05 × 20=1).
Способы синтеза
Все исходные материалы и растворители либо получали из коммерческих источников, либо готовили в соответствии с цитируемыми литературными источниками. Если не указано иное, все реакционные смеси перемешивали. Органические растворы обычным образом сушили над безводным сульфатом магния. Гидрогенирование осуществляли на гидрогенаторе парровского типа, в поточном реакторе Thales H-cube в указанных условиях или под давлением водорода из баллона. Реакции с микроволновой обработкой проводили в микроволновом реакторе СЕМ Discover и Smithcreator, нагревание до постоянной температуры осуществляли с использованием микроволнового излучения переменной мощности. Хроматографию на колонке с нормальными фазами осуществляли обычным образом на автоматизированной хроматографической системе, такой как CombiFlash Companion или CombiFlash RF с применением картриджей, заполненных силикагелем (230-400 меш, 40-63 мкм). Твердофазную катионообменную смолу приобретали в Supelco и обрабатывали 1М соляной кислотой перед применением. Если не указано иное, очищаемую реакционную смесь сначала разбавляли метанолом (МеОН) и придавали ей кислотность несколькими каплями АсОН. Этот раствор загружали на твердофазную катионообменную смолу и промывали метанолом (МеОН). Затем целевой материал элюировали, промывая 1% NH3 в МеОН.
В тех случаях, когда соединения описаны как смесь двух диастереоизомеров / эпимеров, конфигурация стереоцентра не указана и представлена прямыми линиями.
Данные Я MP
1Н-ЯМР-спектры регистрировали на спектрометре Bruker Avance III при 400 МГц. В качестве эталонов использовали либо центральные пики хлороформа-d, диметилсульфоксида-d6, либо внутренний стандарт третраметилсилана. Для ЯМР, где определенное число протонов меньше теоретического числа протонов в молекуле, предполагалось, что отсутствующий сигнал (сигналы) перекрывается пиками растворителя и/или воды. Дополнительно, в тех случаях, когда спектры получают в протонных растворителях для ЯМР, происходит обмен протонами между NH и/или ОН и растворителем и, соответственно, такие сигналы обычно не наблюдаются.
Краткое описание чертежей
На фигуре 1 изображена дифрактограмма порошковой рентгеновской дифракции одной кристаллической формы («Форма А») (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида.
На фигуре 2 изображена дифрактограмма порошковой рентгеновской дифракции другой кристаллической формы («Форма В») (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида.
На фигуре 3 изображен скан дифференциальной сканирующей калориметрии (ДСК) кристаллической формы В (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида.
На фигуре 4 изображен профиль потери массы, полученный при термогравиметрическом анализе кристаллической формы В (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида.
На фигуре 5 изображен профиль массы кристаллической формы В (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида при различной влажности, полученный при динамическим анализе сорбции пара.
На фигуре 6 изображен 1Н-ЯМР спектр аморфной гидрохлоридной соли соединения формулы (1), полученной в примере 2.
На фигуре 7 изображен 1Н-ЯМР спектр аморфной сульфатной соли соединения формулы (1), полученной в примере 2.
На фигуре 8 изображен спектр 1Н-ЯМР аморфной нападизилатной соли соединения формулы (1), полученной в примере 2.
На фигуре 9 изображен 1Н-ЯМР спектр аморфной эдисилатной соли соединения формулы (1), полученной в примере 2.
На фигуре 10 изображен 1Н-ЯМР спектр аморфной тозилатной соли соединения формулы (1), полученной в примере 2.
На фигуре 11 изображен 1Н-ЯМР-спектр аморфной мезилатной соли соединения формулы (1), полученной в примере 2.
На фигуре 12 изображен 1Н-ЯМР спектр аморфной напсилатной соли соединения формулы (1), полученной в примере 2.
На фигуре 13 изображен 1Н-ЯМР спектр аморфной безилатной соли соединения формулы (1), полученной в примере 2.
На фигуре 14 изображен 1Н-ЯМР спектр аморфной изетионатной соли соединения формулы (1), полученной в примере 2.
На фигуре 15 изображен 1Н-ЯМР спектр аморфной эзилатной соли соединения формулы (1), полученной в примере 2.
На фигуре 16 изображен 1Н-ЯМР спектр аморфной гидробромидной соли соединения формулы (1), полученной в примере 2.
ПРИМЕР 1
Синтез аморфного (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино1пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида
Стадия 1: метил-5-бром-2-(бромметил)бензоат
В пятигорлую колбу на 10 л, снабженную конденсатором, магнитной мешалкой, впускным отверстием для N2 и барботером, к перемешивающемуся раствору 1,2-дихлорэтана (1,9 л) добавляли метил-5-бром-2-метилбензоат (500,0 г, 2,18 моль, 1,0 экв.) и N-бромсукцинимид (Fluorochem, 388,5 г, 2,18 моль, 1,0 экв.). Смесь нагревали до 90°С (масляная баня). Азобисизобутиронитрил (5,0 г, 0,03 моль, 0,014 экв.) растворяли в 1,2-дихлорэтане (ДХЭ) (100 мл) и 20 мл вносили через капельную воронку. Добавление в воронку осуществляли медленно, когда температура реакционной смеси достигла 85°С. Когда бурный обратный сток из конденсатора и пенообразование прекратились, к реакционной смеси добавляли оставшиеся 80 мл ДХЭ одной порцией и оставляли перемешиваться при 90°С в течение 1 часа. ЯМР показал, что реакция завершилась, при этом оставалось еще около 11% исходного материала. Затем реакционную смесь охлаждали до комнатной температуры с применением сухого льда в масляной бане и после того, как внутренняя температура снизилась до ~30°С, реакционную смесь гасили водой (2,0 л). Через 5 минут две идентичные реакционные смеси объединяли, переносили в делительную воронку и собирали органический слой. Водный слой снова подвергали экстракции, используя ДХМ (2 × 2,0 л). Все органические слои объединяли, промывали водой (2 л) и солевым раствором (2 л), сушили над MgSO4, отфильтровали и концентрировали в вакууме, получая оранжевую жидкость (1,397 кг, 104%, из 2×500 г опытов).
Стадия 2: трет-бутил-(2R)-2-(6-бром-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)пропаноат
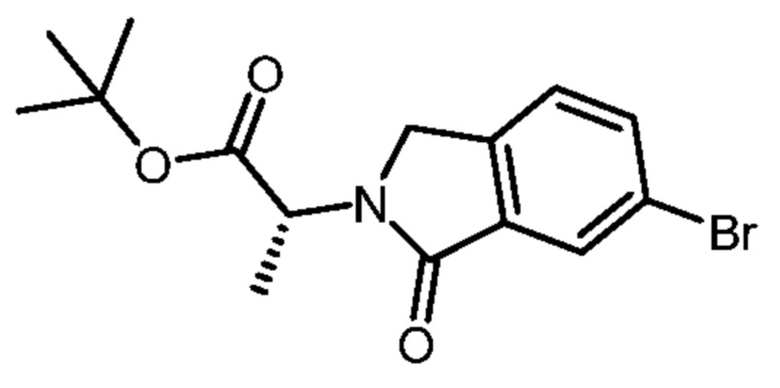
В пятигорлую колбу на 10 л, снабженную конденсатором, магнитной мешалкой, впускным отверстием для N2 и барботером, вносили в перемешиваемый раствор ТГФ (5,0 л) метил 5-бром-2-(бромметил)бензоат (со стадии 1) (660 г, 2,14 моль, 1,0 экв.), трет-бутил-(2R)-2-аминопропаноат⋅HCl (467 г, 2,57 моль, 1,2 экв.) и диизопропилэтиламин (1,0 л, 6,42 моль, 3,0 экв., d=0,742). Смесь нагревали до 80°С на масляной бане в течение ночи. Затем реакционную смесь охлаждали до комнатной температуры с применением сухого льда, а затем две большие идентичные реакционные смеси выливали в большую делительную воронку. Добавляли насыщенный водный раствор NaHCO3 (5,6 л) и смесь перемешивали в течение 5 минут. Органический слой собирали и водную фазу подвергали экстракции, используя этилацетат (2×4 л). Органические фазы объединяли и промывали солевым раствором (5,6 л), перемешивали в течение 3 минут, сушили над MgSO4, фильтровали и концентрировали под вакуумом, получая остаточное оранжево-коричневое клейкое твердое вещество, которое помещали на ночь в вакуумную печь при 40°С. Твердое вещество (1,4 кг) суспендировали в петролейном эфире 40-60 (2,5 л) и энергично перемешивали в течение ночи, а затем фильтровали и промывали петролейным эфиром (2×300 мл), получая желтое твердое вещество (480 г).
Стадия 3: трет-бутил-(2R)-2-[1-оксо-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3-дигидро-1Н-изоиндол-2-ил]пропаноат

В пятигорлую колбу на 10 л, снабженную верхнеприводной мешалкой, конденсатором, термозондом и впускным отверстием для N2, в перемешиваемый раствор диоксана (4 л) добавляли трет-бутил-(2R)-2-(6-бром-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)пропаноат (со стадии 2) (500 г, 1,469 моль, 1,0 экв.), бис(пинаколато)диборон (446,3 г, 1,764 моль, 1,2 экв.) и безводный ацетат калия (433,9 г, 4,409 моль, 3 экв.). Затем его дегазировали с помощью N2 в течение 30 минут перед добавлением Pd(dppf)Cl2 (21,5 г, 0,029 моль, 0,02 экв.) и реакционную смесь дополнительно дегазировали в течение 10 минут, и после этого нагревали до 90°С в масляной бане. Эту смесь оставляли перемешиваться в течение ночи. (Примечание: через 2-3 часа при 90°С реакция разогревается от 88°С до 104°С с большим обратным стоком из конденсатора, и раствор превращается из оранжево-красного в темно-коричневый в течение ~30 минут перед охлаждением обратно до 90°С). Реакционные смеси охлаждали до комнатной температуры с применением сухого льда перед объединением и фильтровали через слой целита (агломерат 4 л, слой толщиной ~ 2 дюйма). Слой промывали диоксаном (1 л) до полного отмывания цвета.
Стадия 4: трет-бутил-(2R)-2-[6-(2,5-дихлорпиримидин-4-ил)-1-оксо-2,3-дигидро-1Н-изоиндолин-2-ил]пропаноат

Затем раствор, полученный на стадии 3, поделили пополам и каждую половину поместили в пятигорлую колбу, такую же, как на предыдущем этапе. В эту пятигорлую колбу добавляли 2,4,5-трихлорпиримидин (404,4 г, 252,8 мл, 2,205 моль, 1,5 экв., D=1,6), карбонат калия (609,4 г, 4,409 моль, 3 экв.) и затем дегазировали при помощи N2 в течение 30 минут перед тем, как добавляли Pd(dppf)Cl2 (21,5 г, 0,029 моль, 0,02 экв.), с дальнейшим дегазированием в течение 10 минут. Реакционную смесь нагревали до 65°С на масляной бане, а затем в течение 5 минут через капельную воронку добавляли воду (500 мл) (Примечание: имеет место экзотермический эффект от 62°С до 72°С через несколько минут после первоначального добавления воды). Реакция была оставлена на ночь. Реакционные смеси охлаждали до комнатной температуры с применением сухого льда и отфильтровали через слой целита (агломерат 4 л, слой толщиной ~ 3 дюйма), промывая слой ДХМ (2 л). Затем фильтрат концентрировали почти до сухого состояния, получая неочищенную черно-коричневую смолу (1909 г).
Неочищенный материал разделяли на две части, вносили в сухом виде на колонку с диоксидом кремния и элюировали 6×4 л смесью 40% этилацетат/бензин. (Примечание: все фракции смешивали и таким образом объединяли, после чего концентрировали почти до сухого состояния: ТСХ продукта в смеси 30% этилацетат/бензин, Rf=~ 0,5). Оставшуюся коричневую смесь охлаждали до комнатной температуры, добавляли бензин (4 л) и перемешивали на ротационном смесителе в течение 2 часов для кристаллизации продукта. Его отфильтровали и осадок промывали бензином (2×2 л), получая белое твердое вещество (548 г).
В качестве альтернативы, трет-бутил-(2R)-2-[6-(2,5-дихлорпиримидин-4-ил)-1-оксо-2,3-дигидро-1Н-изоиндолин-2-ил]пропаноат (промежуточное соединение (4)) может быть синтезирован, как описано в PCT/IB2016/001507 (международная публикация №WO2017/068412) - см. примеры получения 76, 89 и 94 в ней.
Аморфный (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид (соединение 1) синтезировали изтрет-бутил-(2R)-2-[6-(2,5)-дихлорпиримидин-4-ил)-1-оксо-2,3-дигидро-1Н-изоиндолин-2-ил]пропаноата (со стадии 4) в соответствии со схемой синтеза, показанной ниже:
Стадия 5: (2R)-2-(6-{5-хлор-2-[(оксан-4-ил}амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид
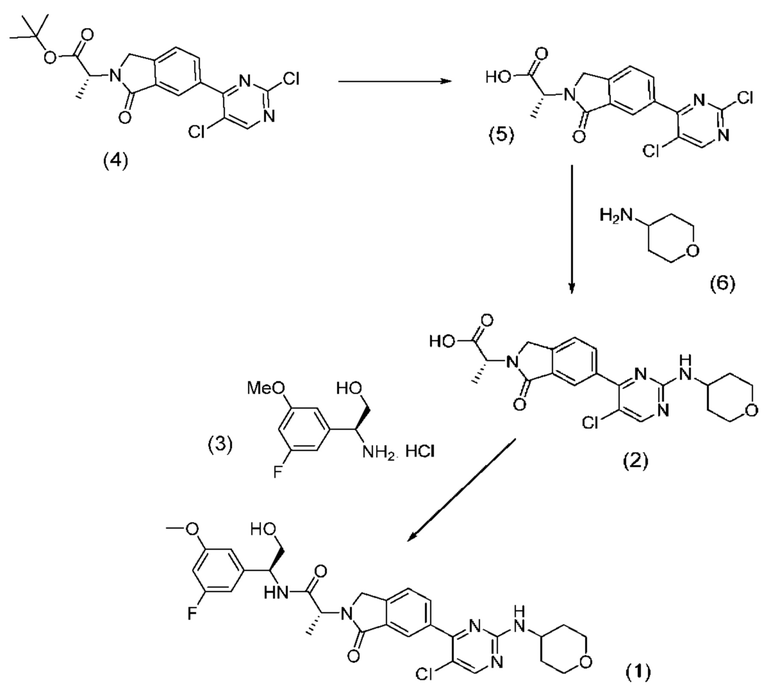
Стадия 1: Снятие Вос-защиты с (4)

К раствору (4) (поставляемому Manchester Organics) (1,0 масс.) в толуоле (13 об.) при 15-25°С добавляли концентрированную соляную кислоту (1 об.) с последующим промыванием линии толуолом (1 об.). Смесь нагревали до 35-40°С и перемешивали при этой температуре до достижения момента завершения реакции (критерий прохождения: ≤2,0% площади (4), ожидаемое время реакции от 16 до 24 часов). Суспензию концентрировали при пониженном давлении при температуре до 50°С с получением влажного твердого вещества. Вносили толуол (3×10 об.) с концентрированием при пониженном давлении при температуре до 50°С так, чтобы получить влажное твердое вещество после каждой последующей загрузки. Вносили толуол (4 об.) и суспензию вращали на роторном испарителе при атмосферном давлении при температуре до 50°С в течение от 10 до 20 минут. Колбу снимали с роторного испарителя и содержимое выдерживали по меньшей мере 1 ч при 15-25°С. Твердые вещества собирали фильтрацией, промывали толуолом (2×2 об.) и высушивали в атмосфере азота до тех пор, пока содержание толуола не составило ≤6,0% масс./масс. и содержание воды не составило ≤2,0% масс./масс. Промежуточное соединение (5) выделили в виде твердого вещества от почти белого до бледно-бежевого цвета (от 74 до 95% теор., от 64 до 82% масс./масс.).
Стадия 2. Сочетание промежуточного соединения (5) с 4-аминотетрагидропираном

К раствору (5) (1,0 масс, корр., 1,0 мол. экв.) в 1-метил-2-пирролидиноне (НМП) (9,5 об.) при 15-25°С добавляли карбонат калия (0,86 масс., 2,2 мол. экв.) с последующим добавлением 4-аминотетрагидропирана (6) (0,4 об., 1,3 мол. экв.) при температуре от 15 до 40°С и промывкой линии НМП (0,5 об.). Смесь нагревали до 80-95°С и перемешивали при этой температуре до достижения момента завершения реакции (критерий прохождения: ≤1,0 мол.% промежуточного соединения (5), время реакции от 4 до 6 часов). Смесь охлаждали до 15-25°С и вносили 3М соляную кислоту (10 об.), поддерживая температуру от 15 до 30°С. Добавляли дихлорметан (ДХМ) (10 об.) и фазы разделили. Кислотную водную фазу подвергли обратной экстракции ДХМ (5 об.) и объединенные органические фазы промывали очищенной водой (8×10 об.) до тех пор, пока содержание НМП не было отрегулировано до ≤15,0% масс./масс. Органическую фазу промывали 13%-ным масс./масс. раствором NaCl (10 об.), обработали активированным углем (0,3 масс.) и сушили над сульфатом магния (1,0 масс). Смесь отфильтровали для удаления осушающего агента, промывали ДХМ (2×2 об.) и объединенные фильтраты концентрировали при пониженном давлении и температуре до 35°С, получая (2) в виде светло-коричневой пены (от 74 до 90% теор., от 88 до 107% масс./масс.).
Стадия 3: получение соединения 1
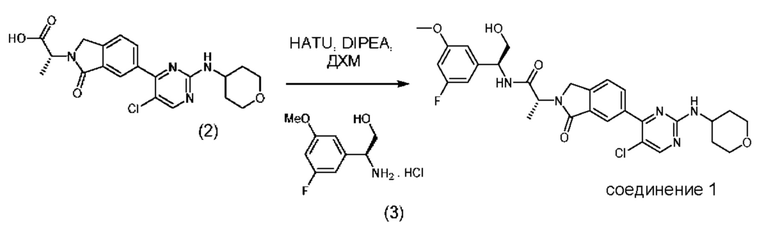
К раствору (2) (1,0 масс., 1,0 мол. экв.) в ДХМ (12,5 об.) добавляли амин (3) (0,68 масс., 1,27 мол. экв.). Суспензию охлаждали до 10-15°С и вносили DIPEA (1,67 об., 4,0 мол. экв.). Полученный раствор перемешивали в течение 5-10 минут перед частичным добавлением HATU (1,15 масс., 1,27 мол. экв.), поддерживая температуру реакцинной смеси <25°С. Смесь перемешивали при 15-25°С до тех пор, пока не сочли законченной по данным ВЭЖХ (площадь (2) <0,5%, обычно 1 час). После завершения реакционную смесь концентрировали при пониженном давлении и температуре до 38°С, получая густое подвижное оранжевое масло. Остаток растворяли в EtOAc (10 об.) и промывали очищенной водой (10 об.), 25% масс./масс. раствором хлорида аммония (2×10 об.), 8% масс./масс. раствором NaHCO3 (2×10 об.) и 13 масс./масс. раствором NaCl (6×10 об.), затем сушили над MgSO4 (1,0 масс.). Твердое вещество фильтровали и осадок на фильтре промывали этилацетатом (2×2 об.). Фильтраты концентрировали на роторном испарителе при температуре до 40°С с получением неочищенного соединения 1 в виде бледно-желтой пены. Неочищенное соединение 1 очищали сухой флэш-хроматографией.
Полное превращение (2) в соединение 1 также наблюдалось при воздействии вышеуказанных условий, но где 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCI) использовали в качестве агента сочетания вместо HATU, 4-диметиламинопиридин (4-DMAP) использовали в качестве основания вместо DIPEA, а диметилформамид (ДМФА) использовали в качестве растворителя вместо ДХМ.
Методика проведения хроматографии:
Диоксид кремния (20 масс.экв.) вносили в сухую флэш-колонку, и указанный диоксид кремния промывали и упаковывали, элюируя через него этилацетат (обычно 2×20 об.). Неочищенное соединение 1 (1 масс, нескорр.) растворяли в ДХМ (4 об.) и осторожно наносили на колонку. Затем указанную колонку элюировали следующим образом:
Все собранные фракции анализировали с помощью ВЭЖХ. Продукт обычно элюировался во фракциях с 12 по 45. Только те фракции, которые оказались наиболее чистыми и наиболее интенсивными по ТСХ (например, фракции с 15 по 25 в указанном выше диапазоне элюирования продукта) группировали вместе в отсутствие данных ВЭЖХ. Фракции, содержащие вышедший продукт, анализировали с помощью ВЭЖХ, чтобы определить, имеют ли они чистоту, подходящую для объединения с основными фракциями продукта. После того как все фракции были объединены и сконцентрированы либо до пены, либо до небольшого объема, продукт разбавляли этилацетатом (например, 10 об.), перемешивали с получением раствора и затем осветляли через фильтровальную бумагу из стекловолокна. Затем фильтраты концентрировали, получая аморфное соединение 1 в виде пены от грязно-белого до бледно-желтого цвета (от 65 до 85% теор., от 91 до 119% масс./масс.).
ПРИМЕР 2
Получение аморфных солей соединения 1
Готовили базовые растворы различных противоионов в 2-пропаноле. Соответствующий базовый раствор (500 мкл, 10 об.) вносили в раствор соединения 1 (полученного в соответствии с примером 1) в изопропилацетате (2,5 мл, 50 об.) и перемешивали. Твердые вещества, которые не кристаллизовались или осаждались слабо, охлаждали, концентрировали путем упаривания или дополнительно обрабатывали трет-бутилметиловым эфиром (ТБМЭ).
Твердые вещества отделяли фильтрованием, сушили в токе азота в течение 96 часов, выгружали и анализировали с помощью 1Н ЯМР и ПРД (см. следующую таблицу).
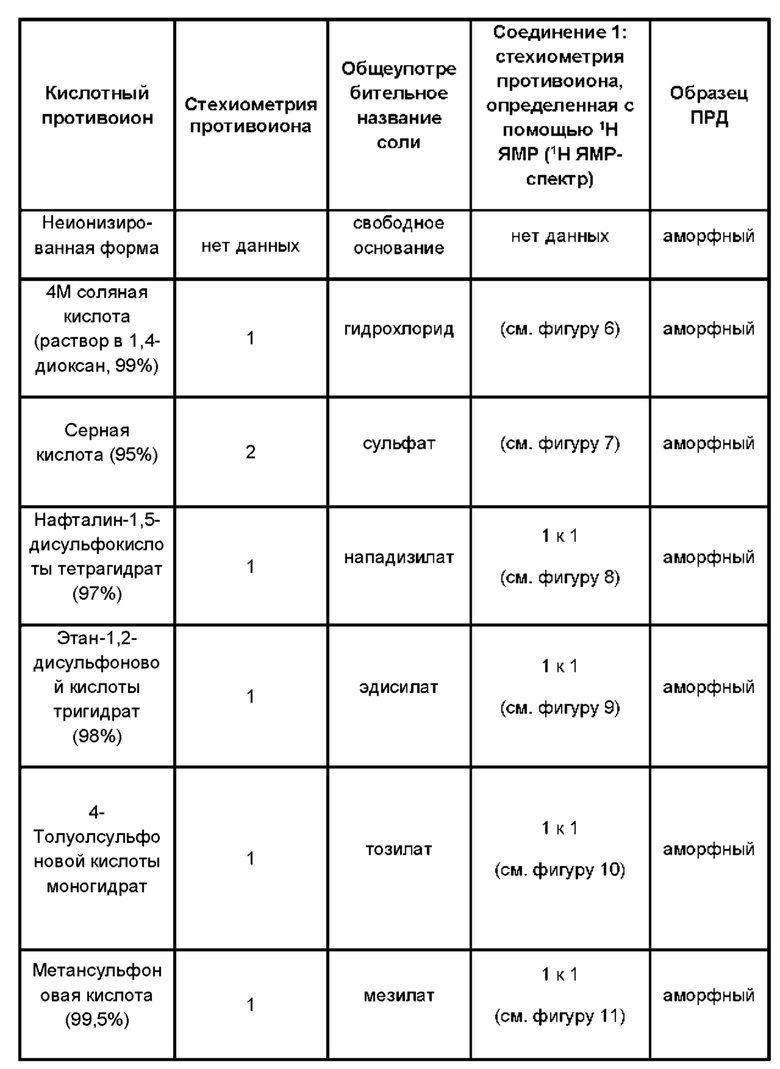
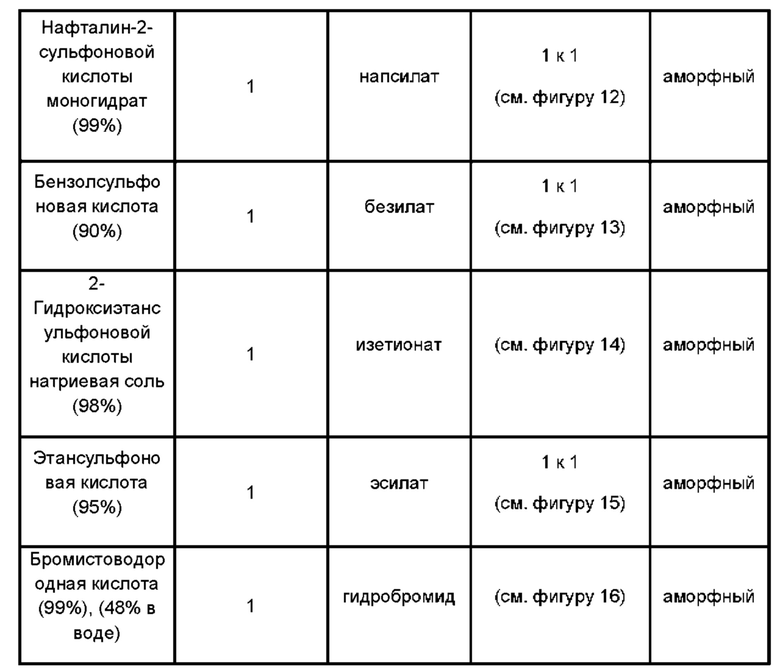
ПРИМЕР 3
Получение кристаллических форм соединения 1
Пример 3А - Получение кристаллической Формы А
Гидрохлоридную соль (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида (полученную в соответствии с примером 2) суспендировали в очищенной воде (35 объемов) при 70°С в течение 96 часов. Твердое вещество отделяли фильтрацией, сушили в токе азота в течение 96 часов, выгружали и анализировали методом ПРД (см. фигуру 1).
Пример 3В - Получение кристаллической Формы В (способ I)
Гидрохлоридную соль (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида (полученную в соответствии с примером 2) суспендировали в очищенной воде (2 мл, 20 об.) при 18-23°С. Растворы перемешивали при 45-50°С в течение 20 часов. Затем температуру понижали до 30-35°С и раствор перемешивали еще 96 часов. Показано, что ПРД полученного твердого вещества соответствовала образцу ПРД на фигуре 2.
Пример 3С - Получение кристаллической Формы В (способ II)
Гидрохлоридную соль (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида (полученную в соответствии с примером 2) суспендировали в очищенной воде (20 об.) и смесь перемешивали при 40°С в атмосфере азота в течение 20 часов. Вносили 2-пропанол (7,6 об.) и смесь перемешивали при 40°С в течение 20 часов. Ход конверсии контролировали по ПРД. Показано, что ПРД полученного твердого вещества соответствовала образцу ПРД на фигуре 2.
Пример 3D - Получение кристаллической Формы В (способ III)
(2R)-2-(6{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид (полученный в соответствии с примером 1) (100 мг, 1,0 масс.) вносили в сосуд с последующим добавлением либо этилацетата, либо толуола (15 об.) и трет-бутил метило во го эфира (15 об.). Суспензию перемешивали в течение 7 дней при 30°С. По истечении этого времени продукт отделяли фильтрацией, промывали ре циркулирующим растворителем для созревания, высушили в потоке азота при 18-23°С и проанализировали методом ПРД для подтверждения кристаллизации. Образец ПРД полученного твердого вещества соответствовал образцу ПРД на фигуре 2.
ПРИМЕР 4
Дальнейшая характеристика кристаллической Формы В соединения 1
Кристаллическая Форма В соединения 1, полученная в примерах 3В, 3С и 3D, была исследована с применением порошковой рентгеновской дифракции, дифференциальной сканирующей калориметрии, термогравиметрического анализа и динамической сорбции пара.
Пример 4А - Порошковая рентгеновская дифракция
Кристаллы кристаллической Формы В соединения 1 получали в соответствии со способом из примера 3. Анализ методом порошковой рентгеновской дифракции (ПРД) проводили с применением порошкового дифрактометра Bruker D2 Phaser, оборудованного детектором LynxEye. Образцы подвергали минимальной подготовке, но при необходимости их слегка мололи в ступке с пестиком перед сбором данных. Образцы располагали в центре кремниевого держателя образца внутри кармана длиной 5 мм (прибл. от 5 до 10 мг). Во время сбора данных образцы непрерывно вращали и сканировали с шагом в 0,02° два тета (29) в диапазоне от 4° до 40° два тета и с временем шага 34,5 секунды. Данные обрабатывали с применением Bruker Diffrac. Suite. Дифрактограмма порошковой рентгеновской дифракции кристаллической Формы В соединения показана на фигуре 2, а 2θ углы дифракции и интенсивности, связанные с каждым пиком, показаны в таблице ниже.

Пример 4В- Дифференциальная сканирующая калориметрия (ДСК)
Кристаллы кристаллической Формы В соединения 1 получали в соответствии со способом из примера 3. Для термического анализа использовали прибор Mettler Toledo DSC 821 с программным обеспечением STAReTM. Анализ проводили в открытых алюминиевых чашках на 40 мкл, в атмосфере азота, и размеры образцов находились в диапазоне от 1 до 10 мг. Как правило, анализ проводили в диапазоне температур от 20 до 250°С при градиенте температуры 10°С/мин. Скан ДСК кристаллического соединения изображен на фигуре 3.
Пример 4С - Термогравиметрический анализ (ТГА)
Кристаллы кристаллической Формы В соединения 1 получали в соответствии со способом примера 3. Приблизительно 7 мг образца помещали в платиновую НТ TGA чашку, которую очищали с применением бутановой паяльной горелки и тарировали с применением автоматической функции тары прибора. Образцы нагревали в атмосфере азота от 25 до 800°С со скоростью 10°С/мин. Профиль потери массы кристаллического соединения изображен на фигуре 4.
Пример 4D - Динамический анализ сорбции пара
Кристаллы кристаллической Формы В соединения 1 получали в соответствии со способом из примера 3. Приблизительно 20 мг образца взвешивали в алюминиевой чашке и вносили в прибор DVS Intrinsic, выдерживаемый при 25°С. Образцу давали уравновеситься в течение трех часов при относительной влажности 0%, затем повышали влажность от 0 до 30% относительной влажности с 5%-м приращением. За этим следовало линейное изменение с 10%-м приращением до относительной влажности 90%. Аналогичный профиль линейного изменения величины был применен для фазы десорбции. На каждой стадии в качестве параметра равновесия была установлена скорость изменения массы в единицу времени (dt), составляющая 0,002%/мин. Профиль сорбции/десорбции паров соединением показан на фигуре 5.
Пример 4Е - исследования рентгеновской дифракции на монокристалле
Рентгеновская структура монокристалла Формы В соединения формулы (1) была определена при 100К с применением кристалла, полученного из изопропилацетата медленным испарением.
Данные получали на дифрактометре Rigaku Oxford Diffraction Supernova Dual Source, Cu at Zero, Atlas CCD, оборудованном охлаждающим устройством Oxford Cryosystems Cobra. Данные получали с применением Cu Кα. Структуры определяли и уточняли с применением пакета Bruker AXS SHELXTL. Полную информацию можно найти в таблице ниже. Если не указано иное, атомы водорода, присоединенные к углероду, были расположены геометрически и позволили произвести уточнение с помощью параметра изотропного смещения во время движения. Атомы водорода, связанные с гетероатомом, были расположены в соответствии с разностным синтезом Фурье и позволили произвести уточнение с помощью параметра изотропного смещения без ограничений. Эталонную дифрактограмму для кристаллической структуры получали с использованием публикации Mercury (C.F.A. Macrae, «Mercury: visualization and analysis of crystal structures», J. Appl. Cryst., Vol.39, pp.453-457, 2006).
Сбор данных и уточнение структуры
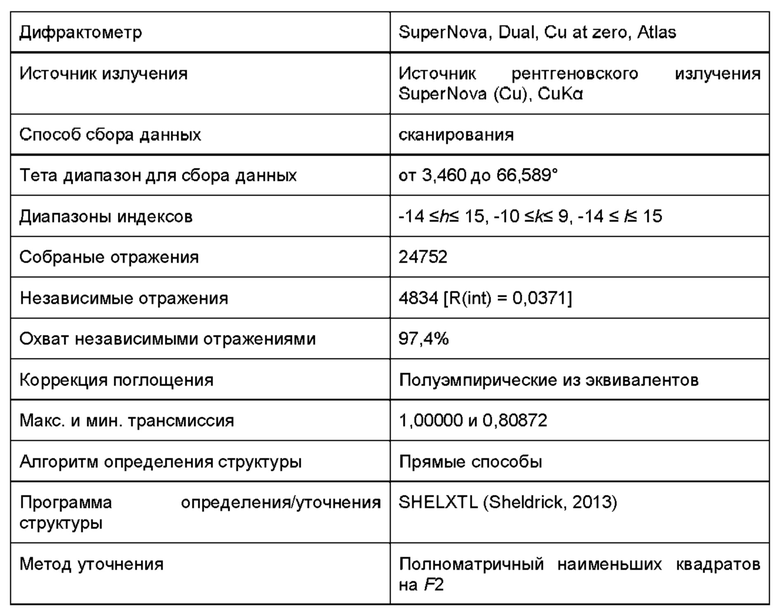
Данные о кристалле
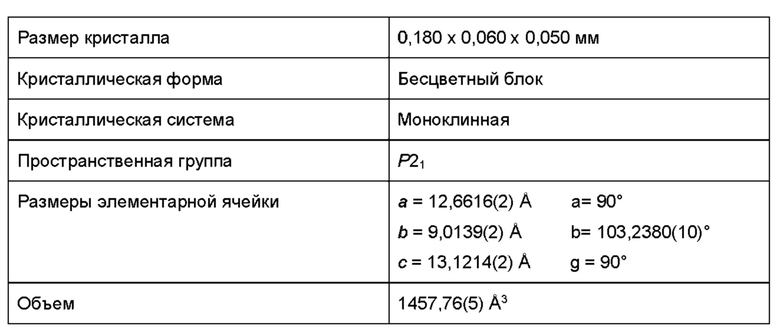
Было обнаружено, что кристаллы являются моноклинными, пространственная группа P21 с конечной R1=[l>2s(l)]=2,93%.
Абсолютную стереохимию соединения определяли с применением данных дифракции, и было подтверждено, что она соответствует приведенной здесь формуле (1) с параметром Флэка (Flack)=-0,004 (7).
В асимметричной единице имеется одна молекула соединения формулы (1) и одна молекула воды, обе полностью упорядочены. Эти данные подтверждают, что кристаллическая Форма В представляет собой моногидрат соединения формулы (1).
ПРИМЕР 5
Определение растворимости кристаллической Формы В соединения (1) в водных растворителях
Растворимость кристаллической Формы В и аморфной формы соединения формулы (1) определяли в очищенной воде и буферах при той же концентрации и перемешивании при 18-23°С до 24 ч по времени. рН и температуры растворов контролировали, чтобы подтвердить, что к концу эксперимента не произошло сдвига рН.
Общая методика
Каждый из образцов соединения формулы (1) (50 мг) суспендировали в очищенной воде (2 мл) или в подходящем буфере и перемешивали при температуре окружающей среды (18-23°С) в течение от 20 до 24 часов. По истечении этого времени суспензии центрифугировали, при необходимости разбавляли разбавителем ВЭЖХ образца (ацетонитрил/вода, 1/1, об./об.) и анализировали по площади пика ВЭЖХ. Измеренные значения сравнивали с соответствующей калибровочной кривой для соединения формулы (1). Рассчитывали приблизительные растворимости и использовали поправочные коэффициенты для соответствующих коэффициентов разбавления и % масс/масс, анализов. Коэффициент разбавления гарантировал, что значения площади пика попадали в требуемые диапазоны калибровочной кривой.
Слой твердого вещества, оставшийся в центрифужной пробирке, сушили в вакуумной печи и анализировали методом ПРД. Если образц демонстрировал какие-либо признаки кристалличности, отличные от ожидаемой дифракционной картины для кристаллической формы соединения (1), то для подтверждения химической идентичности проводили дальнейший анализ с помощью 1Н ЯМР.
Препараты буферов, примененных в исследовании, описаны ниже:
рН 1,2 - смесь 50 мл 0,2М раствора хлорида калия с 85 мл 0,2М раствора соляной кислоты.
рН 2 - смесь 50 мл 0,2М раствора хлорида калия с 13 мл 0,2М раствора соляной кислоты.
рН 3 - смесь 100 мл 0,1 М гидрофталата калия с 44,6 мл 0,1 М раствора соляной кислоты.
рН 4 - смесь 100 мл 0,1 М гидрофталата калия с 0,2 мл 0,1 М раствора соляной кислоты.
рН 5 - смесь 100 мл 0,1 М гидрофталата калия с 45,2 мл 0,1 М раствора гидроксида натрия.
рН 6 - смесь 100 мл 0,1 М дигидроортофосфата калия с 11,2 мл 0,1 М раствора гидроксида натрия.
рН 7 - смесь 100 мл 0,1 М дигидроортофосфата калия с 58,2 мл 0,1 М раствора гидроксида натрия.
рН 8 - смесь 100 мл 0,1 М дигидроортофосфата калия с 93,4 мл 0,1 М раствора гидроксида натрия.
Результаты
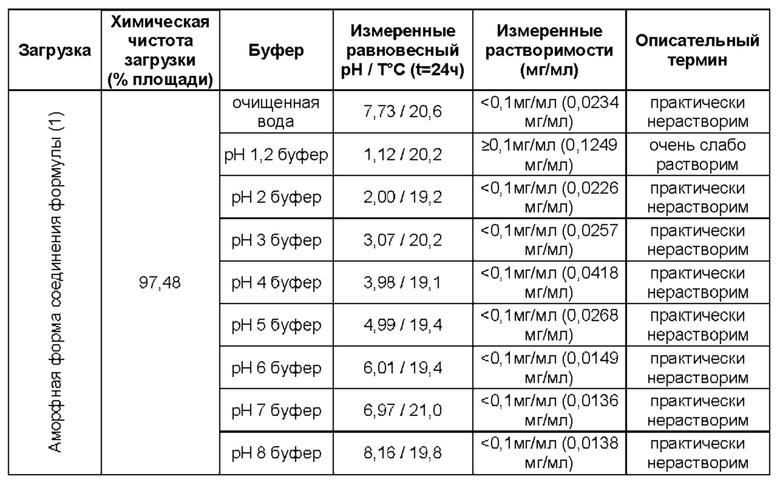
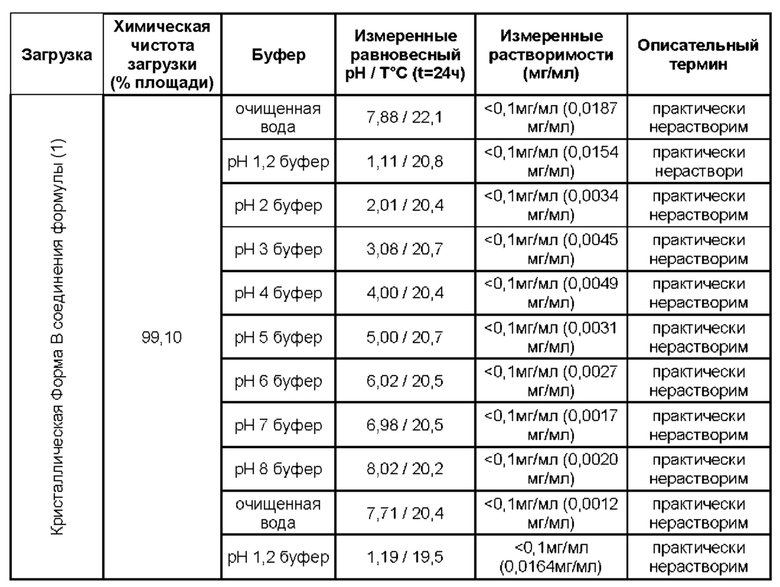
Заключения и наблюдения
Как кристаллическая, так и аморфная формы соединения формулы (1) были практически нерастворимы в буферах при рН от 1,2 до 8,0 (прибл. <0,1 мг/мл).
Определение растворимости кристаллической Формы В соединения (1) в неводных растворителях
Методика
Форму В соединения (1), полученную, какописано в примере 3, вносили в соответствующий растворитель (2 мл) с интервалами приблизительно 1 час. Указанные растворы целенаправленно насыщали и оставляли перемешиваться в течение 72 часов при температуре от 18 до 23°С. По истечении этого времени суспензии центрифугировали, отбирали их надосадочную жидкость, соответственно разбавляли и площади пиков соответствующих аналитов измеряли ВЭЖХ и сравнивали с пиками стандартных калибрантов. Рассчитывали приблизительные растворимости и физически проверяли указанные растворимости на безводной основе без растворителя, чтобы подтвердить, что результаты согласуются.
Наблюдения и результаты
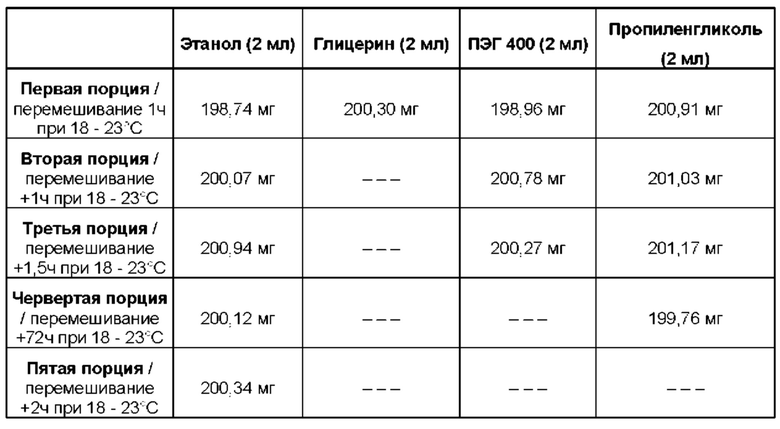
Насыщенные растворы центрифугировали и 100 мкл осветленной надосадочной жидкости вносили в мерную колбу на 250 мл (этанол) или в мерную колбу на 100 мл (остальные) и доводили до объема разбавителем образца. Разбавленные образцы затем анализировали по площади пика ВЭЖХ и получали расчетные максимальные растворимости, они представлены в таблице ниже.

Для подтверждения того, что расчетные растворимости были правильного порядка величины, готовили растворы с нужным порядком величины до предполагаемых максимальных измеряемых концентраций и перемешивали полученные смеси в течение ночи в тех же условиях для подтверждения растворения.
Заключение
Было показано, что соединение формулы (1) обладает значительно большей растворимостью в этаноле, ПЭГ 400 и пропиленгликоле, чем в воде.
ПРИМЕР 6
Жидкие составы -1
Растворители
а. Пропиленгликоль - Р4347 SIGMA-ALDRICH (соответствует требованиям испытаний Фармакопеи США (USP))
b. Этанол - 29221 SIGMA-ALDRICH (испытан в соответствии с Европейской Фармакопеей (Ph.Eur.))
c. Super Refined™ ПЭГ 400 - Croda (Японская Фармакопея (JP), Фармакопея США -Национальный формуляр (USP-NF), Европейская Фармакопея (PhEur))
d. Комбинация пропиленгликоля и этанола, 75:25 (% масс./масс.)
e. Комбинация пропиленгликоля и этанола, 85:15 (% масс./масс.)
Протокол
Соединение формулы (1) инкубируют в каждом из растворителей при 500 мг/мл в стеклянных виалах (при 25°С с применением нагревательного блока RS9000) и оставляют при постоянном взмучивании. При Т=1, 3, 7, 24 и 48 часов.
Полученную в результате суспензию подвергают ультрацентрифугированию (то есть от 500 до 1000 мкл образца) в течение 15 минут при 13000 об/мин и проводят визуальный осмотр для подтверждения осветления. Если удовлетворительное осветление не достигнуто, то процедуру центрифугирования повторяют для осаждения любого оставшегося нерастворенного соединения. Надосадочную жидкость отбирают в качестве релевантной (например, от 100 до 1000 мкл), разбавляют соответствующим образом и анализируют с помощью ВЭЖХ-УФ, чтобы зафиксировать фактическую концентрацию.
ПРИМЕР 7
Жидкие составы - II
Были приготовлены следующие жидкие составы.
7-1. Соединение формулы (1) растворяли в 5 г TPSG и 5 г пропиленгликоля.
7-2. Соединение формулы (1) растворяли в 6 г TPSG и 6 г пропиленгликоля.
7-3. Соединение формулы (1) растворяли в 2,5 г TPSG и 7,5 г пропиленгликоля.
7-4. Соединение формулы (1) растворяли в TPGS (20% масс./масс.), этаноле (15% масс/масс.) и пропиленгликоле (65% масс./масс.).
7-5. Соединение формулы (1) растворяли в пропиленгликоле (100% масс./масс.).
7-6. Соединение формулы (1) растворяли в TPGS (10% масс./масс.), этаноле (10% масс./масс.) и пропиленгликоле (80% масс./масс.).
Концентрация соединения формулы (1) в каждом составе составляла 100 мг/мл.
Для каждого состава проводили исследования растворения в модельной желудочной жидкости и модельной кишечной жидкости способами, описанными ниже. В указанных испытаниях концентрации соединения формулы (1) определяли с применением следующего метода ВЭЖХ-УФ.
Параметры метода жидкостной хроматографии
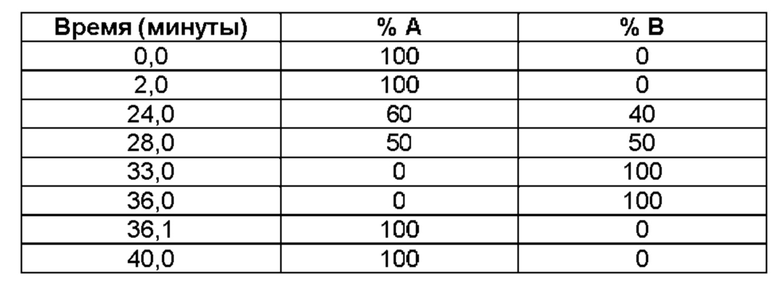
Исследование разведения
Каждый состав вносили в 50 мл FaSSGF (приготовленного в соответствии с способом, описанным в http://biorelenant. com/fassif-fessif-fassgf/how-to-make/) и отбирали образцы в три момента времени (Т=5 минут, 15 минут и 30 минут) с отбором по меньшей мере 2 мл на образец.
Затем смесь дополнительно разбавляли 44 мл среды, имитирующей кишечный сок натощак, FaSSIF (приготовленной в соответствии с способом, описанным в http://biorelevant.com/fassif-fessif-fassgf/how-to-make/) и отбирали образцы в восемь моментов времени (Т=5 минут, 15 минут, 30 минут, 45 минут, 1 час, 3 часа, 5 часов и 7 часов). Образцы анализировали на концентрацию соединения формулы (1) с помощью ВЭЖХ-УФ.
Для составов 7-1 и 7-2 около 65% соединения формулы (1) оставалось солюбилизированным после 30-минутного разведения в FaSSGF. На следующем этапе разведения при переходе на FaSSIF количество солюбилизированного соединения оставалось около 50% вплоть до 3 часов, а затем постепенно уменьшалось. Экстраполируя результаты этого исследования разведения на «сценарий разведения у человека», ожидалось, что >60% 1000 мг дозы соединения формулы (1) растворится в составе 7-1 или составе 7-2 (т.е. около 600 мг) и останется в растворе при поступлении в желудок.
Для состава 7-3 после 30 минут разведения в FaSSGF около 33% лекарственного средства еще оставалось солюбилизированным. На следующем этапе разведения при переходе на FaSSIF количество солюбилизированного лекарственного средства оставалось около 27% вплоть до 3 часов, а затем постепенно уменьшалось. Экстраполируя результаты этого исследования разведения на «сценарий разведения у человека», ожидается, что >30% от 1000 мг дозы растворится в составе 7-3 (т.е. около 300 мг) и останется в растворе при поступлении в желудок.
Для состава 7-4 после 30 минут разведения в FaSSGF около 30% лекарственного средства еще оставалось солюбилизированным. На следующем этапе разведения при переходе на FaSSIF количество солюбилизированного лекарственного средства оставалось около 24% вплоть до 3 часов, а затем постепенно уменьшалось. Экстраполируя результаты этого исследования разведения на «сценарий разведения у человека», ожидается, что >30% от дозы 1000 мг растворятся в составе 7-4 (то есть около 300 мг) и останется в растворе при поступлении в желудок.
Для состава 7-5 после 30 минут разведения в FaSSGF около 2% лекарственного средства еще оставалось солюбилизированным. На следующем этапе разведения при переходе на FaSSIF количество солюбилизированного лекарственного средства оставалось около 2%.
Для состава 7-6 после 30 минут разведения в FaSSGF около 17% лекарственного средства еще оставалось солюбилизированным. На следующем этапе разведения при переходе на FaSSIF количество солюбилизированного лекарственного средства оставалось около 11%.
Было обнаружено, что состав 7-6 особенно предпочтителен. Версия плацебо (т.е. один носитель и никакого соединения формулы (1)) этой композиции первоначально образовывала прозрачную жидкость, содержащую некоторое количество отстаивающихся частиц, но он был повторно растворен при нагревании до 50°С, и жидкость оставалась прозрачной без отстаивающихся частиц или мутности после 24 и 96 часов. Активный вариант состава 7-6, содержащий 100 мг/мл соединения формулы (1), образовывал прозрачный раствор, который оставался в виде прозрачной жидкости без осадка или мутности через 24 и 192 часов при комнатной температуре.
ПРИМЕР 8
Биологическая активность
Пример 8А- анализ ингибирования ERK2 in vitro
Ингибирующую активность соединения согласно настоящему изобретению определяли с применением изложенного ниже протокола.
Активность фермента ERK2 (Life Technologies) определяли с применением формата флуоресценции с временным разрешением, согласно которому определяют фосфорилирование усеченной версии активирующего транскрипционного фактора 2, меченного зеленым флуоресцентным белком (ATF2-GFP) (Life Technologies). Анализируемые реакции, при которых в смеси содержалось 50 мМ Трис рН 7,5, 10 мМ MgCl2, 1 мМ EGTA (этиленгликольтетрауксусная кислота), 0,01% Тритон Х-100, 1 мМ DTT (дитиотреитол), 2,5% ДМСО, 0,4 мкМ ATF2-GFP, 20 мкМ АТР (аденозинтрифосфорная кислота) и 0,25 нМ ERK2, были поставлены в присутствии соединения и проходили в течение 30 мин при комнатной температуре. Затем реакции были останавлены с использованием буфера для разведения TR-FRET (Life Technologies), 25 мМ ЭДТА и 2 нМ Tb-Anti-pATF2 (Thr71) (Life Technologies). После дополнительного периода инкубации по меньшей мере 30 минут определяли флуоресценцию на считывателе Pherastar (оптический модуль Lanthascreen; возбуждение 340 нм, излучение 520 нм (канал А), 495 нм (канал В)). Отношение между числом импульсов А и В использовали для расчета сигнала. Значения IC50 рассчитывали, используя сигмоидальное уравнение доза-ответ (программное обеспечение Prism GraphPad, La Jolla, Калифорния, США).
В анализах с применением ERK2 соединение формулы (1) имеет значение IC50 0,0027 мкМ.
Пример 8 В - Антипролиферативная активность
Антипролиферативные активности соединения согласно настоящему изобретению определяли путем измерения способности соединения формулы (1) ингибировать рост в клеточной линии меланомы человека А375.
Пролиферацию клеток определяли путем измерения превращения резазурина (Alamar Blue) в резоруфин в ответ на митохондриальную активность (Nociari, М.М, Shalev, А., Benias, P., Russo, С.Journal of Immunological Methods 1998, 213, 157-167). Клетки A375 (Американская коллекция типовых культур, Теддингтон, Великобритания) выращивали в модифицированной по Дульбекко среде Игла+10% FBS (фетальной бычьей сыворотки). В каждую лунку черного 96-луночного планшета с плоским дном высевали 2×103 клеток в 200 мкл полной культуральной среды за один день до обработки соединением. Клетки инкубировали с соединением в 0,1% (об./об.) диметилсульфоксиде (ДМСО) в течение 4 дней перед добавлением 20 мкл Alamar blue. После дальнейшей 6-часовой инкубации при 37°С планшет считывали на ридере Spectramax Gemini (Molecular Devices; возбуждение 535 нм, излучение 590 нм). Значения GI50 рассчитывали, с применением сигмоидального уравнения доза-ответ (программное обеспечение Prism GraphPad, La Jolla, Калифорния, США).
В анализах с применением клеток А375 соединение формулы (1) имеет значение GI50 0,0034 мкМ.
Комбинированный протокол для пролиферации клеток
Эффект соединения формулы (1) (Соединение I) в комбинации с противораковым агентом (Соединение II) можно оценить, используя следующую методику. Линии раковых клеток человека (например, А375) высевали на 96-луночные планшеты для тканевых культур в концентрации 2×104 - 4×103 клеток/на лунку. Клеткам давали восстановиться в течение 16-24 часов, после чего добавляли соединение (соединения) или контроль в виде ДМСО (0,1-0,5% ДМСО). Клетки инкубировали с соединением в 0,1%-0,5% (об./об.) диметилсульфоксиде (ДМСО) в течение 72-96 часов, после чего добавляли 20 мкл Alamar blue. После дальнейшей 6-часовой инкубации при 37°С данные в отношении планшета считывали на ридере Spectramax Gemini (Molecular Devices; возбуждение 535 нм, излучение 590 нм). Значения GI50 рассчитывали с применением сигмоидального уравнения доза-ответ (программное обеспечение Prism GraphPad, La Jolla, Калифорния, США). Определяли GI50 для Соединения II в присутствии различных доз Соединения I. Синергизм определяли при сдвиге GI50 вниз в присутствии субэффективных доз Соединения I. Аддитивность устанавливали, если отклик на Соединение II и Соединение I вместе приводил к эффекту, эквивалентному сумме двух соединений по отдельности. Антагонистические эффекты были охарактеризованы как те, которые вызывают сдвиг GI50 вверх, то есть те, где реакция на два соединения была меньше, чем сумма эффектов двух соединений.
ПРИМЕР 9
Фармацевтические составы
(i) Состав в виде таблетки
Композицию в виде таблетки, содержащую соединение формулы (1), получают путем смешивания подходящего количества соединения (например, 50-250 мг) с подходящим разбавителем, разрыхлителем, прессующим агентом и/или скользящим веществом. Одна возможная таблетка содержит 50 мг соединения с 197 мг лактозы (BP) в качестве разбавителя и 3 мг стеарата магния в качестве смазывающего вещества и прессуется с образованием таблетки известным способом. Прессованная таблетка может быть необязательно покрыта пленкой.
(ii) Состав в виде капсулы
Состав в виде капсулы получают путем смешивания 100-250 мг (например, 100 мг) соединения формулы (1) с эквивалентным количеством лактозы (например, 100 мг) и заполнения полученной смесью стандартных непрозрачных твердых желатиновых капсул.
По мере необходимости может быть включен подходящий разрыхлитель и/или скользящее вещество.
(iii) Инъекционный состав I
Парентеральную композицию для введения путем инъекции можно получить путем растворения соединения формулы (1) (например, в солевой форме) в воде, содержащей 10% пропиленгликоля, с получением концентрации активного соединения 1,5 масс. %. Затем указанный раствор стерилизуют фильтрацией, заполняют им ампулы и герметизируют. Факультативно перед стерилизацией раствор можно сделать изотоническим.
(iv) Инъекционный состав II
Парентеральную композицию для инъекций получают путем растворения в воде соединения формулы (1) (например, в солевой форме) (2 мг/мл) и маннита (50 мг/мл), стерильной фильтрации раствора и заполнения им герметизируемых виал на 1 мл или ампул или преднаполненного шприца.
(v) Инъекционный состав III
Состав для в/в (внутривенной) доставки путем инъекции или инфузии может быть получен путем растворения соединения формулы (1) (например, в солевой форме) в воде при 20 мг/мл и, необязательно, затем скорректирован по изотоничности. Виалу затем герметизируют и стерилизуют в автоклаве. В качестве альтернативы он может быть внесен в ампулу, или виалу, или преднаполненный шприц, простерелизован путем фильтрации и герметизирован.
(vi) Инъекционный состав IV
Состав для в/в доставки путем инъекции или инфузии может быть получен путем растворения соединения формулы (1) (например, в солевой форме) в воде, содержащей буфер (например, 0,2 М ацетатный буфер, рН 4,6) при 20 мг/мл. Виалу затем герметизируют и стерилизуют в автоклаве. В качестве альтернативы преднаполненный шприц затем герметизируют и стерилизуют в автоклаве или стерилизуют фильтрацией и герметизируют.
(vii) Состав для подкожных или внутримышечных инъекций
Композицию для подкожного (или внутримышечного) введения получают путем смешивания соединения формулы (1) с кукурузным маслом фармацевтического качества с получением концентрации 5-50 мг/мл (например, 5 мг/мл). Композицию стерилизуют и вносят в подходящий контейнер.
(viii) Лиофилизированный состав
Аликвоты введенного в состав соединения формулы (1) помещают в виалы на 50 мл и лиофилизируют. Во время лиофилизации композиции замораживают, используя одностадийный протокол замораживания при (-45°С). Температуру повышают до -10°С для закрепления, затем понижают до замерзания при -45°С, после чего проводят первичную сушку при +25°С в течение приблизительно 3400 минут, а затем вторичную сушку с постепенным увеличением температуры до 50°С. Давление при первичной и вторичной сушке устанавливается на уровне 80 миллиторр (примерно 11 Па).
(ix) Лиофилизированный состав II
Аликвоты введенного в состав соединения формулы (1) или его соли, как определено в настоящем описании, помещают в виалы на 50 мл и лиофилизируют. Во время лиофилизации композиции замораживают, используя одностадийный протокол замораживания при (-45°С). Температуру повышают до -10°С для закрепления, затем понижают до замерзания при -45°С, после чего проводят первичную сушку при +25°С в течение приблизительно 3400 минут, а затем вторичную сушку с постепенным увеличением температуры до 50°С. Давление при первичной и вторичной сушке устанавливается на уровне 80 миллиторр (примерно 11 Па).
(x) Лиофилизированный состав III для применения с целью внутривенного введения
Обработанный буфером водный раствор готовят путем растворения соединения формулы (1) в буфере. Указанный обработанный буфером раствор вносят в контейнер (такой как стеклянная виала типа 1) с фильтрацией для удаления механических включений и затем контейнер частично герметизируют (например, с помощью пробки Fluorotec). Если соединение и состав являются достаточно стабильными, указанный состав стерилизуют автоклавированием при 121°С в течение подходящего периода времени. Если состав не устойчив к автокпавированию, его можно стерилизовать с применением подходящего фильтра и внести в стерильных условиях в стерильные виалы. Раствор лиофилизируют с применением подходящего цикла. По завершении цикла сублимационной сушки виалы снова заполняют азотом до атмосферного давления, закупоривают и закрепляют (например, алюминиевым обжимом). Для внутривенного введения указанное лиофилизированное твердое вещество может быть восстановлено фармацевтически приемлемым разбавителем, таким как 0,9% солевой раствор или 5% декстроза. Раствор может быть дозирован в неизменном виде или может быть дополнительно перед введением разведен в инфузионный пакет (содержащий фармацевтически приемлемый разбавитель, такой как 0,9% солевой раствор или 5% декстрозу).
(xii) Порошок во флаконе
Композицию для перорального введения готовят путем внесения во флакон или виалу соединения формулы (1). Затем композицию восстанавливают подходящим разбавителем, например водой, фруктовым соком или коммерчески доступным носителем, таким как OraSweet или Syrspend. Восстановленный раствор может быть распределен по дозирующим чашкам или пероральным шприцам для введения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНИРОВАННАЯ ТЕРАПИЯ PAC-1 | 2016 |
|
RU2720509C2 |
| КОМПЛЕКСНАЯ ТЕРАПИЯ | 2013 |
|
RU2677245C2 |
| ХИНАЗОЛИНОВЫЕ ИНГИБИТОРЫ АКТИВИРУЮЩИХ МУТИРОВАННЫХ ФОРМ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА | 2014 |
|
RU2656597C2 |
| НОВЫЕ МАЛЫЕ МОЛЕКУЛЫ ДЛЯ ЦЕЛЕНАПРАВЛЕННОГО РАСЩЕПЛЕНИЯ НЕ ПОДДАЮЩЕГОСЯ ЦЕЛЕНАПРАВЛЕННОМУ ВОЗДЕЙСТВИЮ KRAS В ТЕРАПИИ РАКА | 2021 |
|
RU2839932C1 |
| СОЛЕВАЯ ФОРМА ИНГИБИТОРА ГИСТОН-МЕТИЛТРАНСФЕРАЗЫ EZH2 ЧЕЛОВЕКА | 2013 |
|
RU2658911C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2826182C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ И СОЛЬВАТЫ АВТ-263 ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С БЕЛКОМ BCL-2 | 2010 |
|
RU2551376C2 |
| КОМПОЗИЦИИ НА ОСНОВЕ ИНГИБИТОРА SHP2 И СПОСОБЫ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2805355C2 |
| АГЕНТ, ИНДУЦИРУЮЩИЙ КЛЕТОЧНУЮ ГИБЕЛЬ, ДЛЯ КЛЕТОК, ИМЕЮЩИХ МУТАЦИИ ГЕНА BRAF, АГЕНТ, ПОДАВЛЯЮЩИЙ РОСТ ТАКИХ КЛЕТОК, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ТЕРАПИИ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ ДЕФЕКТОМ РОСТА ТАКИХ КЛЕТОК | 2016 |
|
RU2760835C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ | 2018 |
|
RU2815400C2 |
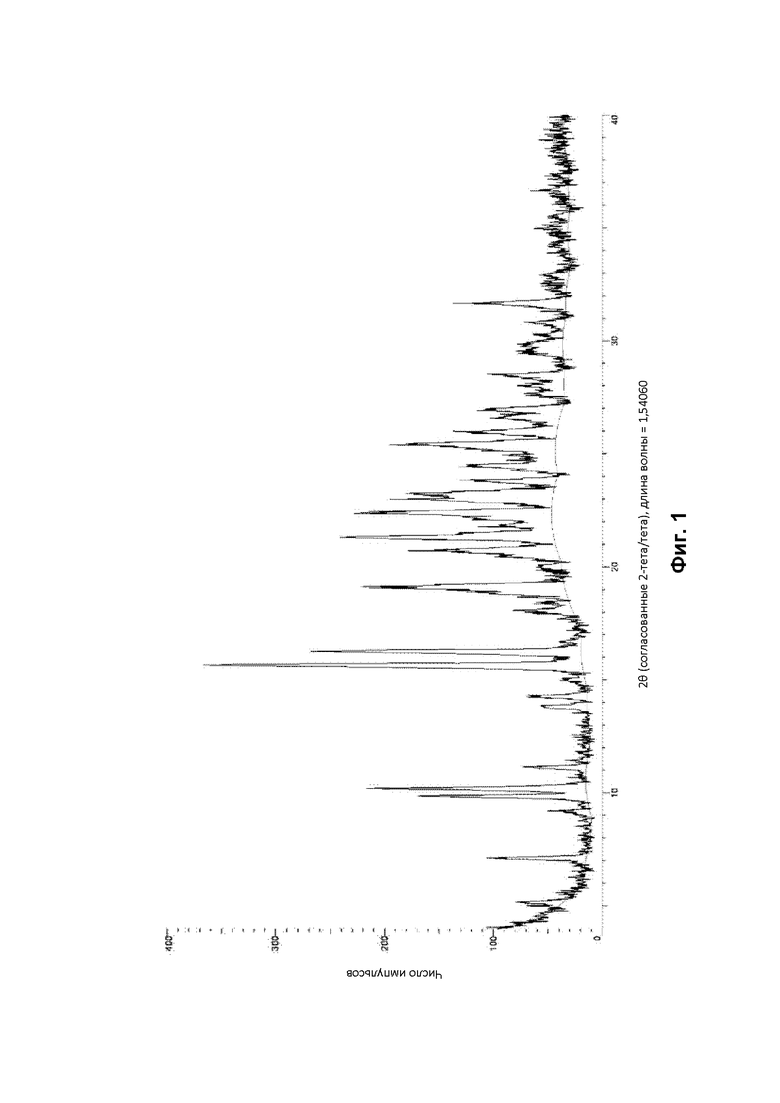

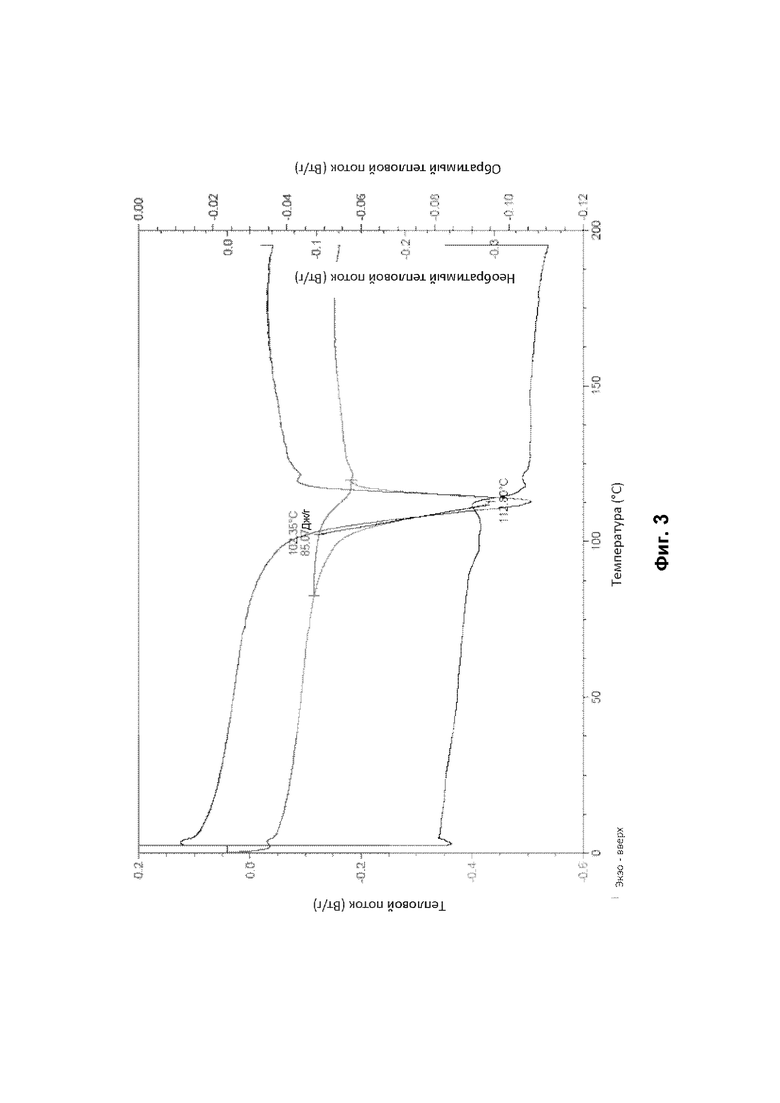
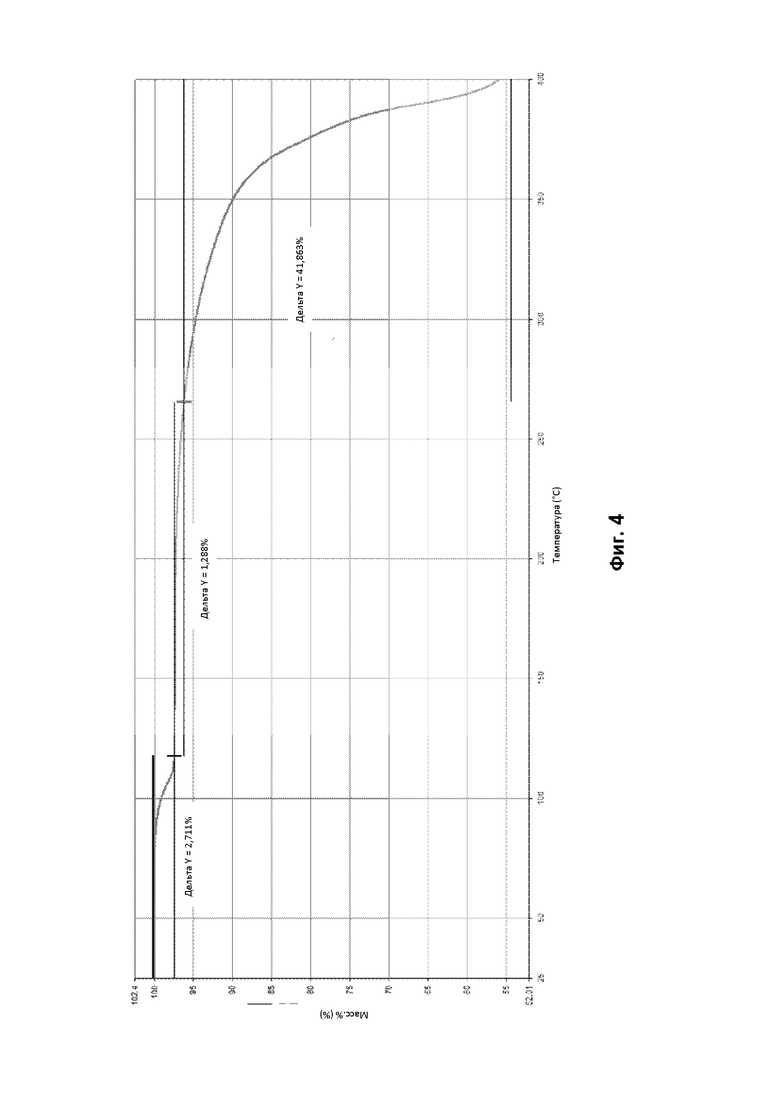
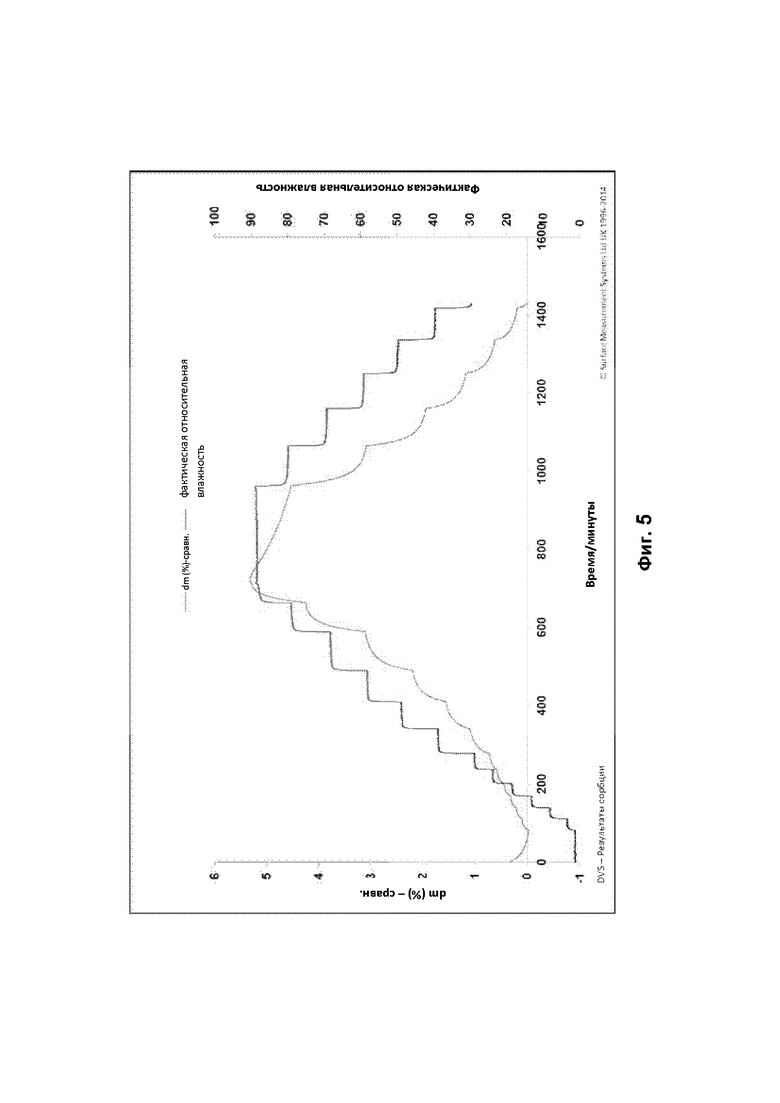




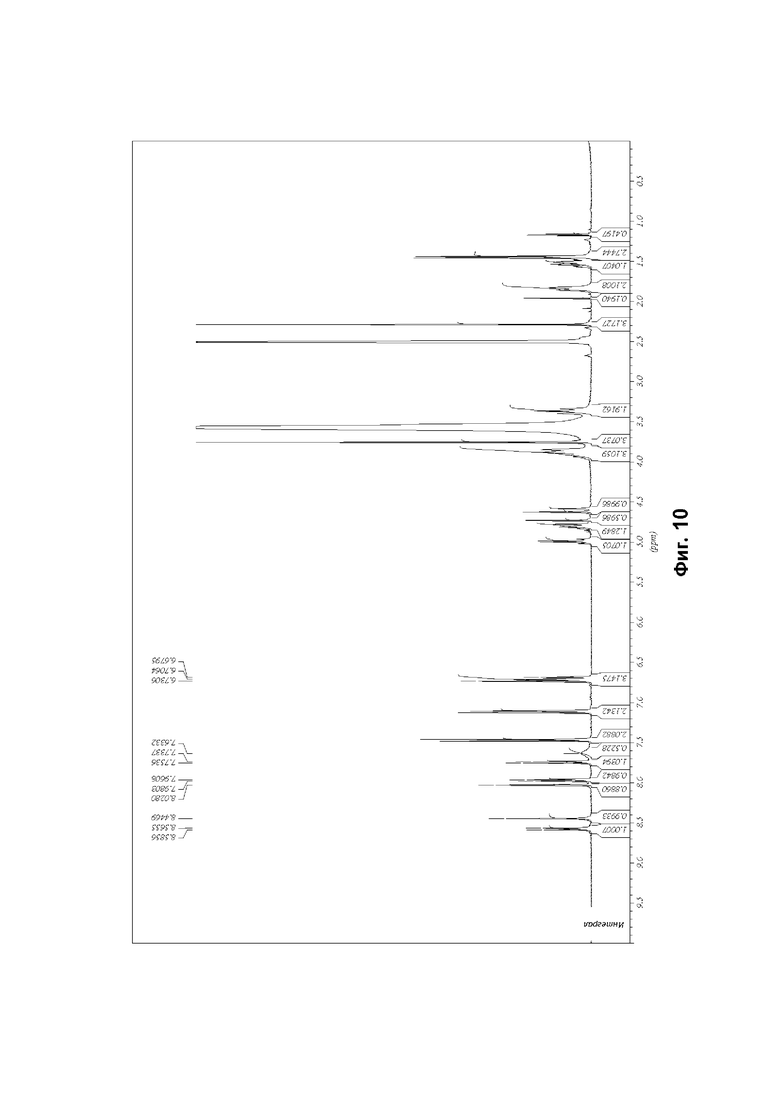

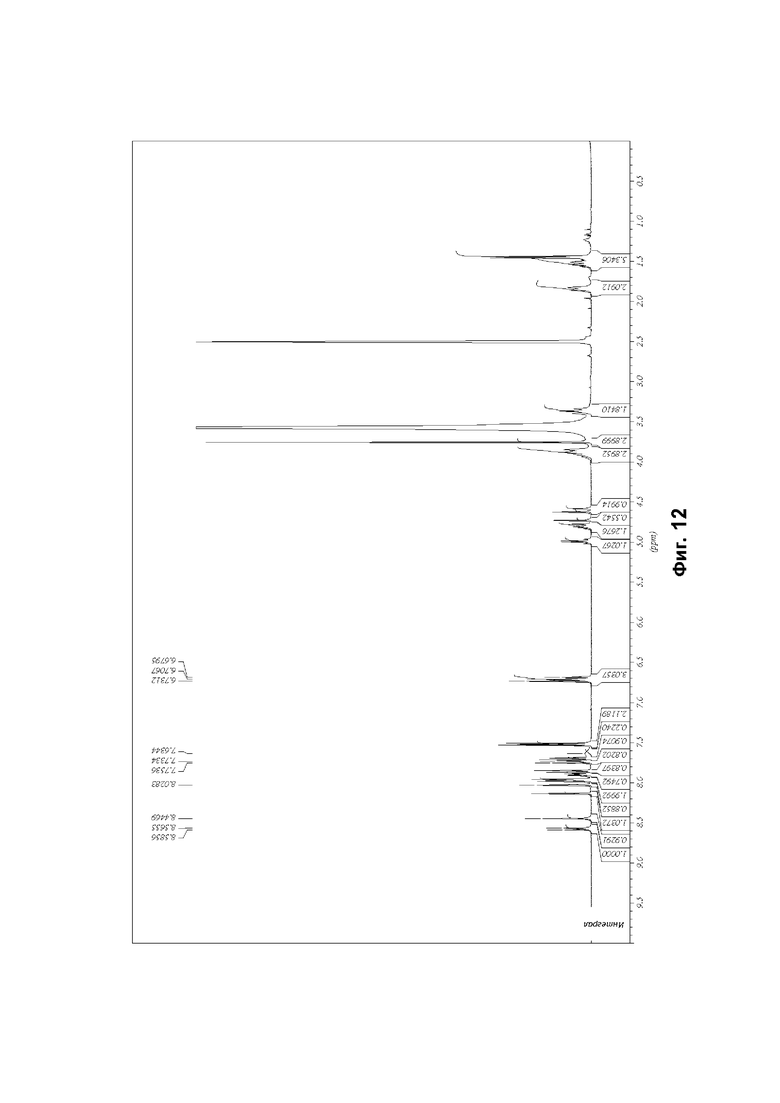
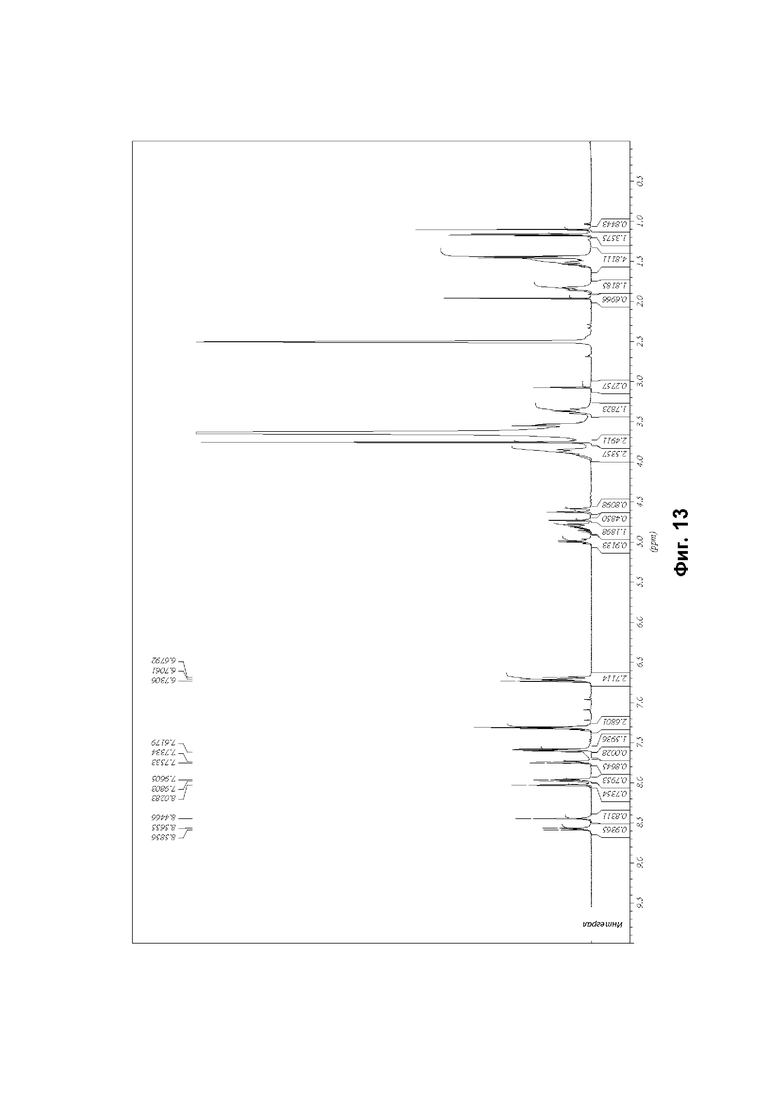


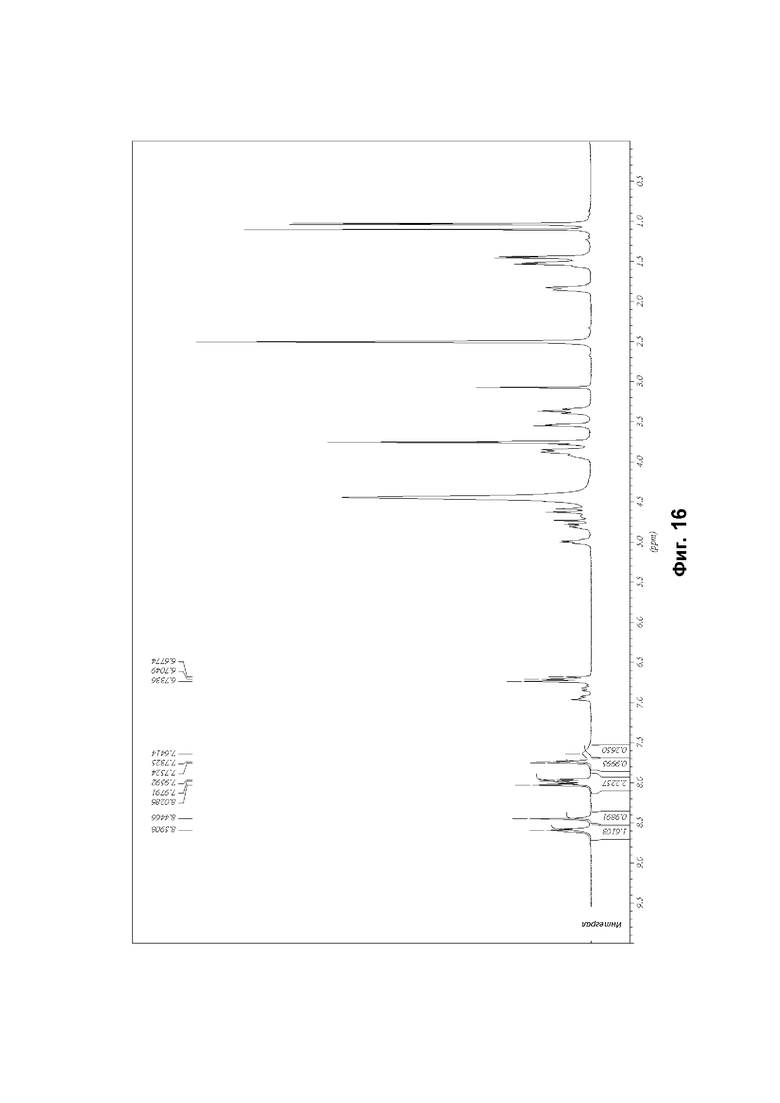
Настоящее изобретение относится к области органической химии и фармацевтики, а именно к ингибиторам ERK2. Раскрывается (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид, имеющий формулу (1) в кристаллической форме. Кроме того, раскрывается способ получения кристаллической формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, аморфные соли (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида и способ получения (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида. Также описана фармацевтическая композиция для профилактики или лечения заболеваний, опосредованных ERK1/2, содержащая эффективное количество (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида и носитель, описаны, кроме того, частицы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, имеющие среднемассовый диаметр от 1 мкм до 100 мкм. Помимо прочего, раскрыты применение указанных выше форм и солей (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида для получения лекарственного средства для профилактики или лечения заболевания или состояния, опосредованного ERK1/2, и способ профилактики или лечения заболевания или состояния, опосредованного ERK1/2, с использованием данных форм. Группа изобретений обеспечивает эффективное ингибирование ERK2. 33 н. и 157 з.п. ф-лы, 16 ил., 11 табл., 9 пр.
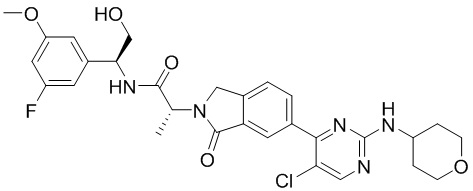 (1)
(1)
1. (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид, имеющий формулу (1):
 (1)
(1)
в кристаллической форме.
2. (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамид по п. 1, который является кристаллическим по меньшей мере на 55%, или по меньшей мере на 60%, или по меньшей мере на 70%, или по меньшей мере на 80%, или по меньшей мере на 90%, или по меньшей мере на 95%, или по меньшей мере на 98%, или по меньшей мере на 99%, или по меньшей мере на 99,5%, или по меньшей мере на 99,9 %.
3. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, имеющая дифрактограмму порошковой рентгеновской дифракции, характеризующуюся наличием основных пиков при углах дифракции 14,0°, и/или 20,6°, и/или 24,0°, и/или 24,2° (± 0,2°).
4. Кристаллическая форма (2R)-2-(6-{5-хлор-2[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, имеющая дифрактограмму порошковой рентгеновской дифракции, характеризующуюся наличием основных пиков при углах дифракции и межплоскостных расстояниях, представленных в таблице A:
5. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по п. 4, для которой указанная дифрактограмма порошковой рентгеновской дифракции дополнительно характеризуется присутствием одного или более дополнительных пиков при углах дифракции и межплоскостных расстояниях, представленных в таблице B.
6. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, демонстрирующая пики при по существу тех же углах дифракции, что и на дифрактограмме порошковой рентгеновской дифракции, изображенной на фигуре 2.
7. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, имеющая дифрактограмму порошковой рентгеновской дифракции, по существу такую же, как изображена на фиг. 2.
8. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-7, демонстрирующая при проведении дифференциальной сканирующей калориметрии эндотермический эффект с начальной температурой от 100°С до 110°С.
9. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-6, демонстрирующая при проведении дифференциальной сканирующей калориметрии эндотермический эффект с начальной температурой от 101°C до 108°C.
10. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-7, демонстрирующая при проведении дифференциальной сканирующей калориметрии эндотермический эффект с пиком между 110°С и 125°С.
11. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-6, демонстрирующая при проведении дифференциальной сканирующей калориметрии эндотермический эффект с пиком между 111°C и 113°C.
12. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-7, демонстрирующая при проведении термогравиметрического анализа потерю массы между 85°С и 130°С.
13. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1H-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-6, демонстрирующая при проведении термогравиметрического анализа потерю массы между 90°С и 120°С.
14. Кристаллическая форма по любому из пп. 1-13, которая представляет собой гидрат.
15. Кристаллическая форма по любому из пп. 1-13, которая представляет собой моногидрат.
16. Способ получения кристаллической формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15, включающий:
(i) получение водной суспензии кислотно-аддитивной соли (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида и перемешивание указанной суспензии при температуре от 25 до 75°С в течение периода времени, достаточного для того, чтобы обеспечить диспропорционирование указанной кислотно-аддитивной соли, и получение кристаллической формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, и после этого выделение кристаллической формы; или
(ii) получение водной суспензии аморфной формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, причем указанная водная суспензия не обработана буфером или обработана буфером до рН от 1,75 до 7,25, и перемешивание указанной водной суспензии при температуре от 25 до 55°С в течение периода времени, достаточного для того, чтобы обеспечить превращение аморфной формы (2R)-2-(6-{5-хлор-2)-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида в кристаллическую форму (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, и после этого выделение кристаллической формы.
17. Аморфный гидрохлорид, сульфат, нападизилат (нафталин-1,5-дисульфонат), эдисилат (этандисульфонат), тозилат (п-толуолсульфонат), мезилат (метансульфонат), напсилат (2-нафталинсульфонат), безилат (бензолсульфонат), изетионат (2-гидроксиэтансульфонат), эзилат (этансульфонат) или гидробромидная соль (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида.
18. Аморфная гидрохлоридная, сульфатная, гидробромидная или нападизилатная соль (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида.
19. Способ получения (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, включающий проведение реакции соединения формулы (2) с соединением формулы (3):
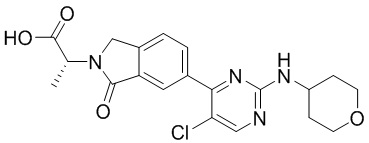 (2)
(2) 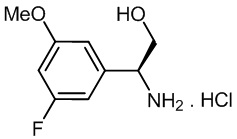 (3)
(3)
в апротонном растворителе в присутствии третичного аминного основания и промотирующего образование амидной связи агента, где указанный промотирующий образование амидной связи агент выбран из N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфата (HATU) и 1-этил-3-(3-диметиламинопропил)карбодиимида (EDCI).
20. Способ по п. 19, в котором указанное третичное аминное основание представляет собой диизопропилэтиламин (DIPEA).
21. Способ получения (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, включающий:
a) проведение реакции соединения формулы (5):
 (5)
(5)
с соединением формулы (6)
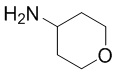 (6)
(6)
с получением соединения формулы (2):
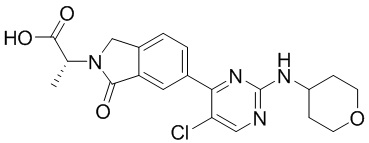 (2)
(2)
и
b) проведение реакции соединения формулы (2) с соединением формулы (3):
 (3)
(3)
с получением соединения формулы (1).
22. Способ по п. 21, который дополнительно включает получение соли или кристаллической формы соединения формулы (1).
23. Фармацевтическая композиция для профилактики или лечения заболеваний, опосредованных ERK1/2, содержащая эффективное количество (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида и носитель, выбранный из:
- C2-4 спирта;
- простого полиэфирного соединения;
- сложных моноэфиров жирных кислот с длиной цепи от C8 до C18 с глицерином или пропиленгликолем;
- ди- или триглицеридов жирных кислот с длиной цепи от C8 до C10;
и их смесей.
24. Фармацевтическая композиция по п. 23, дополнительно содержащая неионное поверхностно-активное вещество.
25. Частицы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида, имеющие среднемассовый диаметр от 1 мкм до 100 мкм.
26. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15 для применения в профилактике или лечении заболевания или состояния, опосредованного ERK1/2.
27. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15 для применения в предотвращении или лечении заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK.
28. Кристаллическая форма для применения по п. 27, в котором заболевание или состояние выбрано из: меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
29. Кристаллическая форма (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15 для применения в профилактике или лечении ракового заболевания.
30. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
31. Кристаллическая форма для применения по п. 29, в котором кристаллическую форму применяют в комбинации с одним или более других соединений или других видов терапии.
32. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
33. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из лейкозов и лимфом.
34. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
35. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
36. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
37. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
38. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из аденомы и карциномы.
39. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
40. Кристаллическая форма для применения по п. 29, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
41. Аморфная соль по п. 17 или 18 для применения в профилактике или лечении заболевания или состояния, опосредованного ERK1/2.
42. Аморфная соль по п. 17 или 18 для применения в предотвращении или лечении заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK.
43. Аморфная соль для применения по п. 42, в котором заболевание или состояние выбрано из меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
44. Аморфная соль по п. 17 или 18 для применения в профилактике или лечении ракового заболевания.
45. Аморфная соль для применения по п. 44, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
46. Аморфная соль для применения по п. 44, в котором аморфную соль применяют в комбинации с одним или более других соединений или других видов терапии.
47. Аморфная соль для применения по п. 44, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
48. Аморфная соль для применения по п. 44, в котором раковое заболевание выбрано из лейкозов и лимфом.
49. Аморфная соль для применения по п. 44, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
50. Аморфная соль для применения по п. 44, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
51. Аморфная соль для применения по п. 44, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
52. Aморфная соль для применения по п. 44, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
53. Аморфная соль для применения по п. 44, в котором раковое заболевание выбрано из аденомы и карциномы.
54. Аморфная соль для применения по п. 44, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
55. Аморфная соль для применения по п. 44, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
56. Фармацевтическая композиция по п. 23 или 24 для применения в профилактике или лечении заболевания или состояния, опосредованного ERK1/2.
57. Фармацевтическая композиция по п. 23 или 24 для применения в предотвращении или лечении заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK.
58. Фармацевтическая композиция для применения по п.57, в котором заболевание или состояние выбрано из: меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
59. Фармацевтическая композиция по п. 23 или 24 для применения в профилактике или лечении ракового заболевания.
60. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
61. Фармацевтическая композиция для применения по п. 59, в котором композицию применяют в комбинации с одним или более других соединений или других видов терапии.
62. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
63. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из лейкозов и лимфом.
64. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
65. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
66. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
67. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
68. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из аденомы и карциномы.
69. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
70. Фармацевтическая композиция для применения по п. 59, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
71. Частицы по п. 25 для применения в профилактике или лечении заболевания или состояния, опосредованного ERK1/2.
72. Частицы по п. 25 для применения в предотвращении или лечении заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK.
73. Частицы для применения по п. 72, в котором заболевание или состояние выбрано из: меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
74. Частицы по п. 25 для применения в профилактике или лечении ракового заболевания.
75. Частицы для применения по п. 74, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
76. Частицы для применения по п. 74, в котором частицы применяют в комбинации с одним или более других соединений или других видов терапии.
77. Частицы для применения по п. 74, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
78. Частицы для применения по п. 74, в котором раковое заболевание выбрано из лейкозов и лимфом.
79. Частицы для применения по п. 74, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
80. Частицы для применения по п. 74, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
81. Частицы для применения по п. 74, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
82. Частицы для применения по п. 74, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
83. Частицы для применения по п. 74, в котором раковое заболевание выбрано из аденомы и карциномы.
84. Частицы для применения по п. 74, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
85. Частицы для применения по п. 74, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
86. Применение кристаллической формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15 для получения лекарственного средства для профилактики или лечения заболевания или состояния, опосредованного ERK1/2.
87. Применение кристаллической формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15 для получения лекарственного средства для предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK.
88. Применение по п. 87, в котором заболевание или состояние выбрано из: меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
89. Применение кристаллической формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15 для получения лекарственного средства для профилактики или лечения ракового заболевания.
90. Применение по п. 89, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
91. Применение по п. 89, в котором кристаллическую форму применяют в комбинации с одним или более других соединений или других видов терапии.
92. Применение по п. 89, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
93. Применение по п. 89, в котором раковое заболевание выбрано из лейкозов и лимфом.
94. Применение по п. 89, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
95. Применение по п. 89, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
96. Применение по п. 89, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
97. Применение по п. 89, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
98. Применение по п. 89, в котором раковое заболевание выбрано из аденомы и карциномы.
99. Применение по п. 89, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
100. Применение по п. 89, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
101. Применение аморфной соли по п. 17 или 18 для получения лекарственного средства для профилактики или лечения заболевания или состояния, опосредованного ERK1/2.
102. Применение аморфной соли по п. 17 или 18 для получения лекарственного средства для предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK.
103. Применение по п. 102, в котором заболевание или состояние выбрано из: меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
104. Применение аморфной соли по п. 17 или 18 для получения лекарственного средства для профилактики или лечения ракового заболевания.
105. Применение по п. 104, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
106. Применение по п. 104, в котором аморфную соль применяют в комбинации с одним или более других соединений или других видов терапии.
107. Применение по п. 104, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
108. Применение по п. 104, в котором раковое заболевание выбрано из лейкозов и лимфом.
109. Применение по п. 104, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
110. Применение по п. 104, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
111. Применение по п. 104, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
112. Применение по п. 104, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
113. Применение по п. 104, в котором раковое заболевание выбрано из аденомы и карциномы.
114. Применение по п. 104, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
115. Применение по п. 104, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
116. Применение частиц по п. 25 для получения лекарственного средства для профилактики или лечения заболевания или состояния, опосредованного ERK1/2.
117. Применение частиц по п. 25 для получения лекарственного средства для предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK.
118. Применение по п. 117, в котором заболевание или состояние выбрано из: меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
119. Применение частиц по п. 25 для получения лекарственного средства для профилактики или лечения ракового заболевания.
120. Применение по п. 119, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
121. Применение по п. 119, в котором частицы применяют в комбинации с одним или более других соединений или других видов терапии.
122. Применение по п. 119, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
123. Применение по п. 119, в котором раковое заболевание выбрано из лейкозов и лимфом.
124. Применение по п. 119, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
125. Применение по п. 119, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
126. Применение по п. 119, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
127. Применение по п. 119, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
128. Применение по п. 119, в котором раковое заболевание выбрано из аденомы и карциномы.
129. Применение по п. 119, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
130. Применение по п. 119, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
131. Способ профилактики или лечения заболевания или состояния, опосредованного ERK1/2, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества кристаллической формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15.
132. Способ предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества кристаллической формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15.
133. Способ по п. 132, в котором заболевание или состояние выбрано из: меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
134. Способ профилактики или лечения ракового заболевания, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества кристаллической формы (2R)-2-(6-{5-хлор-2-[(оксан-4-ил)амино]пиримидин-4-ил}-1-оксо-2,3-дигидро-1Н-изоиндол-2-ил)-N-[(1S)-1-(3-фтор-5-метоксифенил)-2-гидроксиэтил]пропанамида по любому из пп. 1-15.
135. Способ по п. 134, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
136. Способ по п. 134, в котором кристаллическую форму применяют в комбинации с одним или более других соединений или других видов терапии.
137. Способ по п. 134, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
138. Способ по п. 134, в котором раковое заболевание выбрано из лейкозов и лимфом.
139. Способ по п. 134, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
140. Способ по п. 134, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
141. Способ по п. 134, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
142. Способ по п. 134, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
143. Способ по п. 134, в котором раковое заболевание выбрано из аденомы и карциномы.
144. Способ по п. 134, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
145. Способ по п. 134, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
146. Способ профилактики или лечения заболевания или состояния, опосредованного ERK1/2, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества аморфной соли по п. 17 или 18.
147. Способ предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества аморфной соли по п. 17 или 18.
148. Способ по п. 147, в котором заболевание или состояние выбрано из меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
149. Способ профилактики или лечения ракового заболевания, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества аморфной соли по п. 17 или 18.
150. Способ по п. 149, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
151. Способ по п. 149, в котором аморфную соль применяют в комбинации с одним или более других соединений или других видов терапии.
152. Способ по п. 149, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
153. Способ по п. 149, в котором раковое заболевание выбрано из лейкозов и лимфом.
154. Способ по п. 149, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
155. Способ по п. 149, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
156. Способ по п. 149, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
157. Способ по п. 149, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
158. Способ по п. 149, в котором раковое заболевание выбрано из аденомы и карциномы.
159. Способ по п. 149, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
160. Способ по п. 149, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
161. Способ профилактики или лечения заболевания или состояния, опосредованного ERK1/2, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества фармацевтической композиции по п. 23 или 24.
162. Способ предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества фармацевтической композиции по п. 23 или 24.
163. Способ по п. 162, в котором заболевание или состояние выбрано из: меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
164. Способ профилактики или лечения ракового заболевания, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества фармацевтической композиции по п. 23 или 24.
165. Способ по п. 164, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
166. Способ по п. 164, в котором фармацевтическую композицию применяют в комбинации с одним или более других соединений или других видов терапии.
167. Способ по п. 164, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
168. Способ по п. 164, в котором раковое заболевание выбрано из лейкозов и лимфом.
169. Способ по п. 164, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
170. Способ по п. 164, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
171. Способ по п. 164, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
172. Способ по п. 164, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
173. Способ по п. 164, в котором раковое заболевание выбрано из аденомы и карциномы.
174. Способ по п. 164, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
175. Способ по п. 164, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
176. Способ профилактики или лечения заболевания или состояния, опосредованного ERK1/2, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества частиц по п. 25.
177. Способ предотвращения или лечения заболевания или состояния с мутантной Ras, мутантной BRAF или мутантной MEK, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества частиц по п. 25.
178. Способ по п. 177, в котором заболевание или состояние выбрано из: меланомы, характеризующейся NRas, или острого миелогенного лейкоза (ОМЛ), характеризующегося NRas, рака легких, характеризующегося KRas, рака поджелудочной железы, характеризующегося KRas, и рака ободочной и прямой кишки (CRC), характеризующегося KRas, рака ободочной и прямой кишки (CRC), характеризующегося BRAF, рака легких, характеризующегося BRAF, или меланомы, характеризующейся BRAF.
179. Способ профилактики или лечения ракового заболевания, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества частиц по п. 25.
180. Способ по п. 179, в котором раковое заболевание выбрано из гепатоцеллюлярной карциномы, меланомы, раковых заболеваний пищевода, почек, толстого кишечника, ободочной и прямой кишки, легкого, мезотелиомы, аденокарциномы легкого, раковых заболеваний молочной железы, мочевого пузыря, желудочно-кишечного тракта, яичника и предстательной железы.
181. Способ по п. 179, в котором частицы применяют в комбинации с одним или более других соединений или других видов терапии.
182. Способ по п. 179, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы.
183. Способ по п. 179, в котором раковое заболевание выбрано из лейкозов и лимфом.
184. Способ по п. 179, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний лимфоидной линии.
185. Способ по п. 179, в котором раковое заболевание выбрано из острой лимфоцитарной лейкемии (ALL), хронической лимфоцитарной лейкемии (ХЛЛ), B-клеточных лимфом, таких как В-крупноклеточная диффузная лимфома (DLBCL, ВКДЛ), фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, T-клеточные лимфомы и лейкозы, лимфомы из клеток-природных киллеров (NK), ходжкинские лимфомы, волосатоклеточных лейкозов, моноклональной гаммапатии неясного значения, плазмацитомы, множественной миеломы или посттрансплантационных лимфопролиферативных нарушений.
186. Способ по п. 179, в котором раковое заболевание выбрано из злокачественных заболеваний кровеносной системы и родственных состояний миелоидной линии.
187. Способ по п. 179, в котором раковое заболевание выбрано из острого миелогенного лейкоза (ОМЛ), хронического миелогенного лейкоза (ХМЛ), хронического миеломоноцитарного лейкоза (CMML, ХММЛ), гиперэозинофильного синдрома, миелопролиферативных заболеваний, таких как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативного синдрома, миелодиспластического синдрома или промиелоцитарного лейкоза.
188. Способ по п. 179, в котором раковое заболевание выбрано из аденомы и карциномы.
189. Способ по п. 179, в котором раковое заболевание выбрано из опухолей эпителиального происхождения; злокачественных заболеваний кровеносной системы и предзлокачественных нарушений кровеносной системы и нарушений, пограничных со злокачественными; опухолей мезенхимального происхождения; опухолей из клеток, происходящих из нервного валика; опухолей центральной и периферической нервной системы; опухолей эндокринной системы; опухолей глазного яблока и придаточного аппарата глаза; опухолей половых клеток и трофбластических опухолей; и педиатрических и эмбриональных опухолей; или синдромов, врождённых или других, которые обуславливают подверженность субъекта злокачественным новообразованиям.
190. Способ по п. 179, в котором раковое заболевание выбрано из раковых заболеваний поджелудочной железы.
| WO 2015108861 А1, 23.07.2015 | |||
| WO 2016025649 A1, 18.02.2016 | |||
| ЛЕЧЕНИЕ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ С ИСПОЛЬЗОВАНИЕМ МОДУЛЯТОРОВ ИЗОФОРМ PI3-КИНАЗЫ | 2013 |
|
RU2702908C2 |

