Настоящее изобретение относится к атропизомерам производных триарилпиррола и их аналогов, способам их получения, содержащим их фармацевтическим композициям и их применению для лечения таких заболеваний, как рак.
Уровень техники
Белок, экспрессируемый нормальным геном KRAS, выполняет важную функцию при передаче сигналов в нормальной ткани. Мутация гена KRAS посредством замены одной аминокислоты и, в частности, замены одного нуклеотида ответственна за активирующую мутацию, которая является важным этапом в развитии многих раковых заболеваний. Предполагается, что образующийся мутированный белок причастен к развитию различных злокачественных новообразований, включая легочную аденокарциному, муцинозную аденому, протоковую карциному поджелудочной железы и колоректальную карциному. Как и другие члены семейства RAS, белок KRAS представляет собой гуанозинтрифосфатазу (GTP-азу) и участвует во многих путях передачи сигнала.
KRAS действует как молекулярный включатель/выключатель. Как только он включается, он рекрутирует и активирует белки, необходимые для распространения фактора роста и других рецепторных сигналов, таких как с-Raf и PI3 киназа. Нормальный KRAS связывается с GTP в активном состоянии и обладает собственной ферментативной активностью, за счет которой отщепляет концевой фосфат нуклеотида, превращая его в GDP. После преобразования GTP в GDP KRAS выключается. Скорость преобразования обычно низка, но может быть резко повышена с помощью акцессорного белка, относящегося к классу GTP-аза-активирующих белков (GAP), например, RasGAP. В свою очередь KRAS может связываться с белками, относящимися к классу факторов обмена гуаниновых нуклеотидов (GEF), например, SOS1, который ускоряет высвобождение связанного нуклеотида. Затем KRAS связывает GTP, присутствующий в цитозоле, a GEF высвобождается из ras-GTP. В мутантном гене KRAS его GTP-азная активность напрямую исключена, что переводит KRAS в постоянно активное состояние. Мутантный KRAS часто характеризуют по мутациям в кодонах 12, 13, 61 или их смесям.
Известно, что жизнеспособность раковых клеток, несущих мутантный KRAS, зависит от Polo-подобной Киназы 1 (PLK1), и было показано, что подавление экспрессии (сайленсинг) PLK1 приводит к гибели клеток, содержащих мутантный KRAS (см. Luo et al., Cell. 2009 May 29; 137(5): 835-848). Следовательно, соединения, ингибирующие PLK1, должны быть пригодны для лечения раковых заболеваний, возникающих в результате мутаций KRAS, но известные сегодня ингибиторы киназ, предназначенные для соединения с консервативным АТР-связующим доменом PLK1, могут быть слишком неизбирательными в отношении других киназ, чтобы перейти в предназначенный режим действия (см., например, Elsayed et al., Future Med Chem. (2019) 11(12), 1383-1386).
PLK1 представляет собой серин/треонин киназу, состоящую из 603 аминокислот и имеющую молекулярную массу 66 кДа, и является важным регулятором клеточного цикла. В частности, PLK1 важна для митоза и участвует в формировании и изменениях митотического веретена, а также в активации комплексов CDK/циклин во время М-фазы клеточного цикла.
Все Polo-подобные киназы содержат N-концевой серин/треонинкиназный каталитический домен и С-концевую область, содержащую один или два Polo-бокса (Lowery et al., Oncogene, (2005), 24, 248-259). Для Polo-подобных киназ 1, 2 и 3 вся С-концевая область, включающая оба Polo-бокса, функционирует как единый модульный фосфосерин/треонин-связующий домен, известный как Polo-Вох-домен (PBD). При отсутствии связанного субстрата PBD ингибирует базальную активность киназного домена. Фосфориляционно-зависимое связывание PBD с его лигандами высвобождает киназный домен, одновременно локализуя Polo-подобные киназы в специфических субклеточных структурах.
Было показано (Reindl et al., Chemistry & Biology, 15, 459-466, May 2008), что, поскольку PLKL1 локализуется на своих внутриклеточных якорных сайтах посредством своего Polo-бокс-домена, действие PLK1 может быть подавлено с помощью малых молекул, препятствующих его внутриклеточной локализации за счет подавления функции PBD.
Опухолевый белок р53 функционирует как опухолевый супрессор и играет свою роль в апоптозе, геномной стабильности и подавлении ангиогенеза. Известно, что опухоли с дефицитом р53 и высокой экспрессией PLK1 могут быть особенно чувствительны к ингибиторам PLK1 (Yim et al., Mutat Res Rev Mutat Res, (2014). 761, 31-39).
Таким образом, приведенные в литературе факты свидетельствуют о том, что малые молекулы, связывающиеся с PBD и подавляющие его функцию, должны быть эффективными ингибиторами PLK1-киназы и, следовательно, должны также быть пригодны для лечения раковых заболеваний, возникающих в результате мутаций KRAS и/или р53. В частности, поскольку домены PBD свойственны только PLK, возможность ингибиторов, предназначенных для этого домена, иметь большую селективность, чем предыдущие АТР-конкурентные ингибиторы, может обеспечить им большую способность избирательно воздействовать на раковые заболевания, связанные с мутациями KRAS и дефицитом р53.
Выявление и разработка лекарственных средств для лечения первичного рака головного мозга оказалась особенно сложной задачей. Терапевтические средства, избирательно направленные на раковые заболевания, и в частности терапевтические средства, в которых используют ингибиторы протеинкиназы, привлекли основное внимание фармацевтических и биотехнологических компаний (Nature Reviews Clinical Oncology 2016, 13, 209-227). Однако, хотя для применения в онкологии были одобрены более тридцати ингибиторов киназ, ни один из них не предназначен для лечения первичного рака головного мозга. Особая проблема заключалась в том, что большинство одобренных онкологических препаратов с ингибиторами киназ, не обладают качествами, необходимыми лекарственной субстанции, чтобы целенаправленно воздействовать на головной мозг в ситуациях, когда возникает потребность в их использовании при лечении рака головного мозга [JMC 2016, 59(22), 10030-10066].
Алкилирующий агент темозоломид (Temodar®, Temodal®) в настоящее время представляет собой терапевтическое средство первой линии для лечения мультиформной глиобластомы головного мозга, и его часто используют в комбинации с лучевой терапией. Однако серьезной проблемой при лечении глиобластомы является лекарственная резистентность, которая соответственно ограничивает применимость темозоломида. Поэтому в настоящее время злокачественная глиобластома остается неизлечимой.
Polo-подобная киназа 1 (PLK1) сверхэкспрессируется в ряде различных типов опухолей, включая и мультиформную глиобластому (Translational Oncology 2017, 10, 22-32). Кроме того, недавние исследования показали, что адаптацией контрольных точек и резистентностью к темозоломиду у мультиформной глиобластомы управляет PLK1 [Oncotarget 2017, 8, 15827-15837].
Эпендимомы представляют собой опухоли головного и спинного мозга, нынешний стандарт лечения которых ограничивается лишь хирургическим вмешательством и лучевой терапией. PLK1 также вовлечена в процессы развития эпендимом, и ингибиторы PLK1 соответственно активно действуют против клеточных линий эпидендимом [Gilbertson et. al., Cancer Cell (2011) 20, 384-399].
PLK1 также была исследована в качестве мишени при диффузной глиоме ствола головного мозга (DIPG), высокозлокачественной агрессивной детской опухоли головного мозга [Amani et al. ВМС Cancer (2016) 16, 647 и Cancer Biology and Therapy (2018) 19, 12, 1078-1087]
В частности, было показано, что ингибирование PLK1 повышает эффективность темозоломида при мутантных глиомах IDH1 [Oncotarget, (2017) 8, 9, 15827-15837] и подавляет рост опухоли в ксенотрансплантатной модели MMR-дефицитной и темозоломид-резистентной глиобластомы [Mol Cancer Ther; 17(12) December 2018]
В вышеуказанных случаях имеющиеся ныне ингибиторы не оказывают достаточного воздействия на мозг.
Можно ожидать, что соединения, ингибирующие PLK1, но не индуцирующие лекарственной резистентности, которые демонстрируют хорошее воздействие на мозг, будут пригодны для лечения мультиформной глиобластомы и других видов рака головного мозга.
PLK4 является членом семейства Poio-подобных киназ и относится к серин/треонин киназам, которые играют важнейшую роль в дупликации центросом, действуя в качестве центрального регулятора дупликации центриолей (Bettencourt-Dias, Curr Biol. 2005 15(24); 2199-207). PLK4-зависимые изменения в центросомах могут приводить к асимметричной сегрегации хромосом при митозе, что может спровоцировать гибель клеток при неправильной сегрегации хромосом и митотических дефектах.
PLK4 аберрантно экспрессируется при раке человека и вовлечена в процессы онкогенеза и метастазирования. Таким образом, PLK4 была намечена в качестве перспективной мишени при терапии рака (Zhao, J Canc Res Clin Oncol., 2019).
PLK4 сверхэкспрессируется при многих видах рака, включая рабдоидные опухоли, медуллобластому и другие эмбриональные опухоли головного мозга (Pediatr Biood Cancer. 2017), а также рак молочной железы, легких, меланому, рак желудка, колоректальный рак, рак поджелудочной железы и рак яичников. Повышенная или гиперактивированная PLK4 ассоциируется с низкой выживаемостью пациентов с онкологией, включая рак яичников, молочной железы и легких (Zhao, J Cane Res Clin Oncol., 2019).
Было изучено влияние ингибирования PLK4 при лечении мультиформной глиобластомы и показано, что PLK4 играет важнейшую роль в регуляции чувствительности к химиотерапии темозоломидом. Как было показано, комбинирование действия темозоломида с ингибированием PLK4 в PDX-моделях глиобластомы усиливает противоопухолевые эффекты по сравнению с действием одного только темозоломида (Cancer Letters, Vol 443, 2019, 91-107).
Сообщается, что при развитии рака PLK4 действует вкупе с инактивацией р53, и высказывается прогноз о том, что виды раковых заболеваний, характеризующиеся сверхэкспрессией PLK4 и дефицитом р53, склонны к формированию опухолей (Sercin, 2016; Nat Cell Biol 18:100-110). Следовательно, можно предполагать, что соединения, подавляющие активность PLK4, подходят для лечения раковых заболеваний, связанных с мутациями р53.
Ингибирование PLK4 при раке легких приводит к росту противоопухолевой активности, причем активность наблюдается при видах рака, несущих KRAS как дикого типа, так и мутантный (Kawakami, PNAS 2018, 115(8) 1913-18). Таким образом, можно ожидать, что соединения, подавляющие активность PLK4, будут пригодны для лечения раковых заболеваний, связанных с мутациями KRAS.
Известные ныне ингибиторы PLK4 действуют на активный центр киназы и не оптимальны для проникновения в головной мозг (Int. J. Mol. Sci. 2019, 20, 2112). Следовательно, можно предположить, что соединения, ингибирующие PLK4 PBD, но при этом демонстрирующие хорошее воздействие на головной мозг, будут полезны при лечении мультиформной глиобластомы и других раковых заболеваний головного мозга
В нашей более ранней заявке на международный патент WO 2018/197714 раскрыто соединение формулы (0):
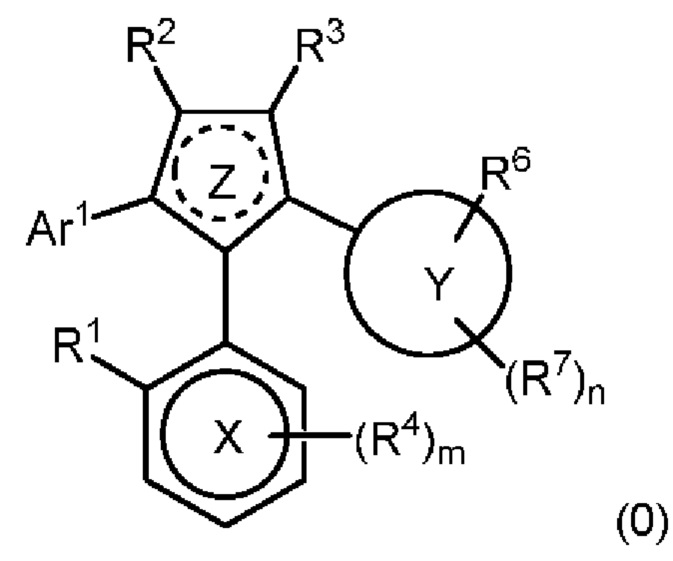
в котором кольцо X представляет собой бензольное или пиридиновое кольцо, кольцо Y представляет собой бензольное, пиридиновое, тиофеновое или фурановое кольцо, Ar1 представляет собой необязательно замещенное бензольное, пиридиновое, тиофеновое или фурановое кольцо, a R1-R4, R6, R7 представляют собой водород или различные заместители. Соединения описаны как вещества, обладающие противораковой активностью и хорошим воздействием на головной мозг после перорального введения, что делает их хорошими кандидатами в качестве средств для лечения различных видов рака головного мозга. Указанные соединения активны в отношении клеточных линий глиобластомы и, как полагают, действуют в качестве ингибиторов Polo-Box домена PLK1 киназы. Также раскрывается, что эти соединения активны в отношении клеточных линий раков с мутантными RAS (таких как НСТ116) и также должны быть пригодны для лечения раковых заболеваний, возникающих в результате мутаций KRAS.
Сущность изобретения
В настоящее время установлено, что соединения типа, раскрытого в нашей более ранней заявке, где R1 представляет собой заместитель, имеющий размер метильной группы или более и, в частности, трифторметильной группы, образуют атропизомеры. Атропизомеры представляют собой стереоизомеры, образующиеся в результате возникновения пространственных затруднений для вращения вокруг оси одинарной связи, причем энергетический барьер, препятствующий вращению, достаточно высок, чтобы можно было выделить отдельные вращательные (поворотные) изомеры (см. LaPlante et al., J. Med Chem., 54:7005-7022 (2011)).
Атропизомеры можно классифицировать по трем категориям, исходя из количества энергии, которую необходимо приложить к хиральной оси для рацемизации через вращение, и времени, необходимого для того, чтобы рацемизация произошла. Атропизомеры Класса 1 обладают энергетическими барьерами для вращения вокруг хиральной оси <84 кДж/моль (20 ккал/моль) и рацемизируются в течение периода времени, измеряемого минутами или менее при комнатной температуре, атропизомеры Класса 2 обладают энергетическим барьером для вращения в пределах от 84 до 117 кДж/моль (20-28 ккал/моль) и рацемизируются в течение периода времени, измеряемого часами и вплоть до месяцев при комнатной температуре, и атропизомеры Класса 3 обладают энергетически барьером для вращения >117 кДж/моль (28 ккал/моль) и рацемизируются в течение периода времени, измеряемого годами при комнатной температуре.
Атропизомеры могут быть классифицированы с использованием номенклатуры R и S Кана-Ингольда-Прелога, которая проиллюстрирована на примере (S)-6,6'-динитробифенил-2,2'-дикарбоновой кислоты на Фигуре 1.
В этой системе ближайшим заместителям по обе стороны от арил-арильной связи присваивают приоритет в порядке a-b-c-d. Поскольку заместители a, b и с располагаются по направлению против часовой стрелки, данный атропизомер представляет собой S-изомер. В соответствующем R-изомере заместители a, b и с расположены по часовой стрелке.
Атропизомерные соединения согласно настоящему изобретению достаточно стабильны, чтобы их можно было выделить и охарактеризовать, и они, как было установлено, не рацемизируются в сколь-нибудь значительной степени даже при нагревании до температур вплоть до 80°С в течение 10 дней. Следовательно, атропизомеры согласно настоящему изобретению можно классифицировать как атропизомеры Класса 3. Считается, что атропизомеризм возникает потому, что стерические взаимодействия между заместителем R1 и ароматическими кольцами Ar1 и Y препятствуют вращению вокруг связи, соединяющей кольца Z и X.
Было установлено, что отдельные атропизомеры данной пары обладают существенно разными биологическими свойствами. Так обычно один атропизомер в паре значительно более активен против определенных раковых мишеней, чем другой атропизомер в этой паре.
Согласно первому Варианту реализации (Варианту реализации 1.1) настоящего изобретения предложена:
(i) композиция веществ, состоящая по меньшей мере из 90 масс. % атропизомера формулы (1А) и 0-10 масс. % атропизомера формулы (1В); или
(ii) композиция веществ, состоящая по меньшей мере из 90 масс. % атропизомера формулы (1В) и 0-10 масс. % атропизомера формулы (1А);
где атропизомер формулы (1А) и атропизомер формулы (1В) представлены как:
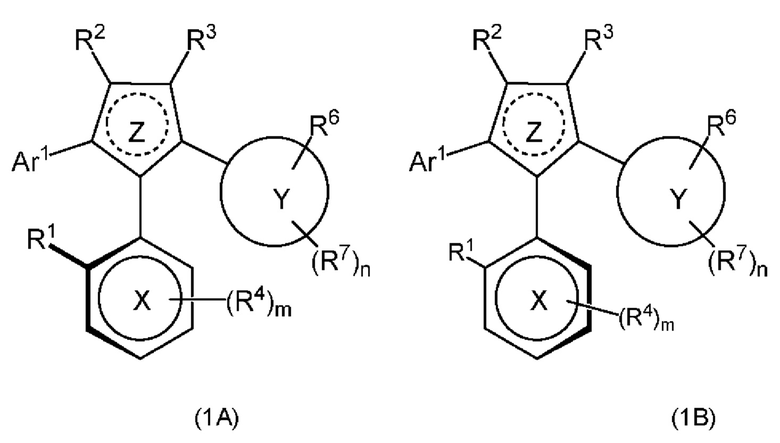
или их фармацевтически приемлемые соли или таутомеры, где: кольцо Z представляет собой 5-членное гетероарильное кольцо, содержащее один или два кольцевых атома азота и необязательно еще один кольцевой гетероатом, выбранный из N и О;
кольцо X представляет собой 6-членное карбоциклическое или гетероциклическое ароматическое кольцо, содержащее 0, 1 или 2 кольцевых гетероатома азота;
кольцо Y представляет собой 6-членное карбоциклическое кольцо или 5- или 6-членное гетероциклическое ароматическое кольцо, содержащее 1 или 2 кольцевых гетероатома, выбранных из N, О и S;
Ar1 представляет собой моноциклическое 5- или 6-членное ароматическое кольцо, необязательно содержащее 0, 1 или 2 гетероатома, выбранных из N, О и S, и необязательно содержит один или более заместителей R6;
m равен 0, 1 или 2;
n равен 0, 1 или 2;
R1 выбран из следующего:
- хлор;
- бром;
- гидроксил;
- цианогруппа;
- карбоксильная группа;
- С(O)O(Hyd1);
- CONH2;
- аминогруппа;
- -(Hyd2)NH;
- (Hyd2)2N и
- С1-5 углеводородная группа, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N, О и S, причем указанная углеводородная группа необязательно замещена одним или более атомов фтора;
Hyd1, Hyd1a, Hyd1d, Hyd2, Hyd2a, Hyd2b и Hyd2c могут быть одинаковыми или различными и представляют собой С1-4 углеводородные группы;
R2 выбран из водорода и С1-4 углеводородной группы;
R3 выбран из водорода и С1-4 углеводородной группы;
R4 выбран из следующего:
- фтор;
- хлор;
- бром;
- гидроксил;
- цианогруппа;
- карбоксильная группа;
- C(O)O(Hyd1a);
- CONH2;
- аминогруппа;
- -(Hyd2a)NH;
- (Hyd2a)2N и
- C1-5 углеводородная группа, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N, О и S, причем указанная углеводородная группа необязательно замещена одним или более атомов фтора;
R5 выбран из галогена; О-Ar2; цианогруппы, гидроксила; аминогруппы; Hyd1b-SO2- и неароматической С1-8 углеводородной группы, в которой 0, 1 или 2, но не все атомы углерода необязательно замещены гетероатомом, выбранным из N, О и S, причем углеводородная группа необязательно замещена одним или более атомами фтора;
Ar2 представляет собой фенил, пиридил или пиридоновую группу, необязательно замещенную 1 или 2 заместителями, выбранными из галогена, цианогруппы и С1-4 углеводородной группы, необязательно замещенной одним или более атомами фтора;
R6 выбран из галогена, цианогруппы, нитрогруппы и группы Q1-Ra-Rb;
Q1 отсутствует или представляет собой С1-6 насыщенный углеводородный линкер;
Ra отсутствует или выбран из О, С(О), С(O)O, CONRc, N(Rc)CO, N(Rc)CONRc, NRc, S, SO, SO2, SO2NRc и NRcSO2;
Rb выбран из следующего:
- водород;
- неароматическая C1-8 углеводородная группа, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N и О, причем неароматическая С1-8 углеводородная группа необязательно замещена одним или более заместителями, выбранными из фтора и группы Сус1, и
- группа Сус1;
При этом группа Сус1 представляет собой неароматическую 4-7-членную карбоциклическую или гетероциклическую кольцевую группу, содержащую 0, 1 или 2 кольцевых гетероатома, выбранных из N, О и S и необязательно замещенных одним или более заместителями, выбранными из гидроксила; аминогруппы; (Hyd2c)NH, (Hyd2c)2N и С1-5 углеводородной группы, в которой 0, 1 или 2 атомов углерода замещены гетероатомом, выбранным из N, О и S, причем углеводородная группа необязательно замещена одним или более атомами фтора или 5- или 6-членной гетероарильной группой, содержащей 1 или 2 кольцевых гетероатома, выбранных из N и О;
Rc выбран из водорода и неароматической С1-4 углеводородной группы и R7 независимо выбран из R4.
В формулах (1А) и (1В) кольцо Z представляет собой 5-членное гетероарильное кольцо, содержащее один или два кольцевых атома азота и необязательно еще один гетероатом, выбранный из N и О.
Следует понимать, что когда 5-членное гетероарильное кольцо Z содержит второй кольцевой гетероатом, например, когда оно представляет собой пиразол или изоксазол, один или оба из R2 и R3 будут отсутствовать. Соответственно, в каждом из указанных выше и последующих аспектов и вариантов реализации изобретения, в которых 5-членное гетероарильное кольцо не является пирролом, эти определения следует понимать как включающие в себя соединения, в которых отсутствует один или оба из R2 и R3.
Частные и предпочтительные аспекты и варианты реализации настоящего изобретения изложены ниже в Вариантах реализации 1.2-1.191.
1.2 Композиция веществ согласно Варианту реализации 1.1 при условии, что указанная композиция отличается от композиции, содержащей:
(i) атропизомер, в котором R1 представляет собой метил, a R4 представляет собой 4-цианогруппу или 4-карбамоильную группу;
(ii) атропизомер, в котором R6 представляет собой гидроксил, метоксиметил или незамещенную или фторзамещенную С1-8 алкоксигруппу (например, трифторметоксигруппу);
(iii) атропизомер, в котором кольцо Z представляет собой изоксазольное кольцо, Ar1 представляет собой незамещенную 4-пиридильную группу, присоединенную в положении 3 изоксазола, a R2 и R3 оба одновременно отсутствуют, или
(iv) атропизомер, в котором Z представляет собой изоксазольное кольцо, a R4 представляет собой азетидин-4-илоксигруппу.
1.3 Композиция веществ согласно Варианту реализации 1.1 или Варианту реализации 1.2, которые отличаются от пиррола, замещенного в каждом из его положений 1, 2 и 3 замещенным фенильным или пиридильным кольцом.
1.4 Композиция веществ согласно любому из Вариантов реализации 1.1-1.3, в которых кольцо Z не является 1,2,3-трехзамещенным пиррольным кольцом.
1.5 Композиция веществ согласно любому из Вариантов реализации 1.1-1.4, в которых кольцо Z не является имидазольным кольцом.
1.6 Композиция веществ согласно любому из Вариантов реализации 1.1-1.5, в которых кольцо Z не является 1,2,4-триазольным кольцом.
1.7 Композиция веществ согласно любому из Вариантов реализации 1.1-1.6, в которых кольцо Z представляет собой гетероарильное кольцо, содержащее кольцевой атом азота и необязательно еще один кольцевой гетероатом, выбранный из N и О, или кольцо Z представляет собой триазольное кольцо.
1.8 Композиция веществ согласно Варианту реализации 1.7, в которых кольцо Z выбрано из пиррольного, изоксазольного, имидазольного, пиразольного и триазольного колец.
1.9 Композиция веществ согласно Варианту реализации 1.8, в которых кольцо Z выбрано из пиррольного, пиразольного и изоксазольного колец.
1.10 Композиция веществ согласно варианту реализации 1.9, в которых кольцо Z представляет собой пиррольное кольцо.
1.11 Композиция веществ согласно Варианту реализации 1.10, в которых кольцо X присоединено к атому азота пиррольного кольца.
1.12 Композиция веществ согласно Варианту реализации 1.9, в которых кольцо Z представляет собой пиразольное кольцо.
1.13 Композиция веществ согласно Варианту реализации 1.12, в которых кольцо X присоединено к атому углерода пиразольного кольца.
1.14 Композиция веществ согласно Варианту реализации 1.12 или Варианту реализации 1.13, в которых кольцо Y присоединено к атому углерода пиразольного кольца.
1.15 Композиция веществ согласно Варианту реализации 1.12 или Варианту реализации 1.13, в которых кольцо Y присоединено к атому азота пиразольного кольца.
1.16 Композиция веществ согласно любому из Вариантов реализации 1.12-1.15, в которых заместитель Ar1 присоединен к атому углерода пиразольного кольца.
1.17 Композиция веществ согласно Варианту реализации 1.9, в которых кольцо Z представляет собой изоксазольное кольцо.
1.18 Композиция веществ согласно Варианту реализации 1.17, в которых кольцо X присоединено в положении 4 изоксазольного кольца.
1.19 Композиция веществ согласно Варианту реализации 1.17 или Варианту реализации 1.18, в которых кольцо Y присоединено в положении 5 изоксазольного кольца.
1.20 Композиция веществ согласно любому из Вариантов реализации 1.17-1.19, в которых заместитель Ar1 присоединен в положении 3 изоксазольного кольца.
1.21 Композиция веществ согласно любому из Вариантов реализации 1.1-1.20, в которых кольцо X представляет собой бензольное, пиридиновое или пиримидиновое кольцо.
1.22 Композиция веществ согласно Варианту реализации 1.21, в которых кольцо X представляет собой бензольное кольцо или пиридиновое кольцо.
1.23 Композиция веществ согласно Варианту реализации 1.22, в которых кольцо X представляет собой бензольное кольцо.
1.24 Композиция веществ согласно Варианту реализации 1.22, в которых кольцо X представляет собой пиридиновое кольцо.
1.25 Композиция веществ согласно любому из Вариантов реализации 1.21, 1.22 и 1.24, в которых пиридиновое кольцо представляет собой кольцо 2-пиридина.
1.26 Композиция веществ согласно любому из Вариантов реализации 1.21, 1.22 и 1.24, в которых пиридиновое кольцо представляет собой кольцо 3-пиридина.
1.27 Композиция веществ согласно любому из Вариантов реализации 1.21, 1.22 и 1.24, в которых пиридиновое кольцо представляет собой кольцо 4-пиридина.
1.28 Композиция веществ согласно любому из Вариантов реализации 1.21, 1.22 и 1.24, в которых пиридиновое кольцо представляет собой кольцо 2-пиридина или 3-пиридина.
1.29 Композиция веществ согласно любому из Вариантов реализации 1.1-1.28, в которых заместитель R1 выбран из следующего:
- хлор;
- бром;
- гидроксил;
- цианогруппа;
- карбоксильная группа;
- аминогруппа;
- -(Hyd2)NH;
- (Hyd2)2N;
- С1-5 углеводородная группа, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N, О и S, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора;
1.30 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из следующего:
- хлор;
- бром;
- гидроксил;
- карбоксильная группа;
- аминогруппа;
- метиламиногруппа;
- диметиламиногруппа;
- С1-5 углеводородная группа, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N, О и S, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора;
1.31 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из следующего:
- хлор;
- бром;
- гидроксил;
- аминогруппа;
- С1-5 углеводородная группа, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N, О и S, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора.
1.32 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель выбран из следующего:
- гидроксил;
- аминогруппа и
- С1-5 углеводородная группа, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N, О и S, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора;
1.33 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из следующего:
- гидроксил;
- аминогруппа и
- С1-4 углеводородная группа, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N и О, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора.
1.34 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из насыщенной С1-4 углеводородной группы, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N и О, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора.
1.35 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из насыщенной С1-4 углеводородной группы, в которой 0 или 1 атом углерода замещен гетероатомом, выбранным из N и О, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора.
1.36 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из С1-4 алкильной группы, в которой 0 или 1 атом углерода замещен гетероатомом, выбранным из N и О, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора.
1.37 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из гидроксила, карбоксила, аминогруппы, С1-4 алкильной группы, которая необязательно замещена одним или более атомами фтора, С1-3 алкоксигруппы, которая необязательно замещена одним или более атомами фтора, (диметиламино)метила и (метокси)метила.
1.38 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из гидроксила, карбоксила, аминогруппы, трифторметила, (диметиламино)метила и (метокси)метила.
1.39 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 представляет собой С1-4 алкильную группу, необязательно замещенную одним или более атомами фтора, или С1-3 алкоксигруппу, необязательно замещенную одним или более атомами фтора.
1.40 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 представляет собой С1-4 алкильную группу, замещенную одним или более атомами фтора.
1.41 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 представляет собой С1-2 алкильную группу, замещенную одним или более атомами фтора.
1.42 Композиция веществ согласно Варианту реализации 1.41, в которых заместитель R1 представляет собой метильную группу, замещенную двумя или тремя атомами фтора.
1.43 Композиция веществ согласно Варианту реализации 1.42, в которых заместитель R1 представляет собой трифторметил.
1.44 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из водорода, трифторметила, трифторметоксигруппы, дифторметила или дифторметоксигруппы, гидроксила, аминогруппы, карбоксила, (диметиламино)метила и (метокси)метила.
1.45 Композиция веществ согласно Варианту реализации 1.29, в которых заместитель R1 выбран из трифторметила, гидроксила, аминогруппы, (диметиламино)метила и (метокси)метила.
1.46 Композиция веществ согласно любому из Вариантов реализации 1.1-1.45, в которых m равен 0 или 1.
1.47 Композиция веществ согласно любому из Вариантов реализации 1.1-1.45, в которых m равен 0.
1.48 Композиция веществ согласно любому из Вариантов реализации 1.1-1.45, в которых m равен 1.
1.49 Композиция веществ согласно любому из Вариантов реализации 1.1-1.45, в которых m равен 2.
1.50 Композиция веществ согласно любому из Вариантов реализации 1.1-1.46, 1.48 и 1.49, в которых R4 выбран из следующего:
- фтор;
- хлор;
- бром;
- цианогруппа и
- С1-5 углеводородная группа, в которой 0, 1 или 2 атома углерода замещены гетероатомом, выбранным из N, О и S, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора.
1.51 Композиция веществ согласно Варианту реализации 1.50, в которых заместитель R4 выбран из следующего:
- фтор;
- хлор;
- бром;
- цианогруппа и
- С1-4 углеводородная группа, в которой 0 или 1 атомов углерода замещены гетероатомом, выбранным из N, О и S, причем указанная углеводородная группа необязательно замещена одним или более атомами фтора.
1.52 Композиция веществ согласно Варианту реализации 1.51, в которых заместитель R4 выбран из следующего:
- фтор;
- хлор;
- бром;
- цианогруппа и
- С1-4 алкильная группа, в которой 0 или 1 атомов углерода замещены гетероатомом, выбранным из N и О, причем указанная алкильная группа необязательно замещена одним или более атомами фтора.
1.53 Композиция веществ согласно Варианту реализации 1.52, в которых заместитель R4 выбран из следующего:
- фтор;
- хлор;
- бром и
- С1-4 алкильная группа, в которой 0 или 1 атомов углерода замещены гетероатомом О, причем указанная алкильная группа необязательно замещена одним или более атомами фтора.
1.54 Композиция веществ согласно Варианту реализации 1.53, в которых заместитель R4 выбран из фтора, хлора, брома и С1-4 алкила.
1.55 Композиция веществ согласно Варианту реализации 1.54, в которых заместитель R4 выбран из фтора, хлора и С1-4 алкила.
1.56 Композиция веществ согласно любому из Вариантов реализации 1.1-1.55, в которых заместитель R2 выбран из водорода и насыщенной С1-4 углеводородной группы.
1.57 Композиция веществ согласно Варианту реализации 1.56, в которых заместитель R2 выбран из водорода, С1-4 алкила, циклопропила и циклопропилметила.
1.58 Композиция веществ согласно Варианту реализации 1.57, в которых заместитель R2 выбран из водорода, С1-3 алкила и циклопропила.
1.59 Композиция веществ согласно Варианту реализации 1.58, в которых заместитель R2 выбран из водорода, метила и этила.
1.60 Композиция веществ согласно Варианту реализации 1.59, в которых заместитель R2 представляет собой водород или метил.
1.61 Композиция веществ согласно Варианту реализации 1.60, в которых заместитель R2 представляет собой водород.
1.62 Композиция веществ согласно любому из Вариантов реализации 1.1-1.61, в которых заместитель R3 выбран из водорода и насыщенной С1-4 углеводородной группы.
1.63 Композиция веществ согласно Варианту реализации 1.62, в которых заместитель R3 выбран из водорода, С1-4 алкила, циклопропила и циклопропилметила.
1.64 Композиция веществ согласно Варианту реализации 1.63, в которых заместитель R3 выбран из водорода, С1-3 алкила и циклопропила.
1.65 Композиция веществ согласно Варианту реализации 1.64, в которых заместитель R3 выбран из водорода, метила и этила.
1.66 Композиция веществ согласно Варианту реализации 1.65, в которых заместитель R3 представляет собой водород или метил.
1.67 Композиция веществ согласно Варианту реализации 1.66, в которых заместитель R3 представляет собой водород.
1.68 Композиция веществ согласно любому из Вариантов реализации 1.1-1.67, в которых заместитель Ar1 представляет собой моноциклическое ароматическое кольцо, выбранное из бензола, пиридина, пиримидина, тиофена и фурана, при этом каждое из моноциклических ароматических колец необязательно замещено одним или более заместителями R5.
1.69 Композиция веществ согласно Варианту реализации 1.68, в которых заместитель Ar1 представляет собой моноциклическое ароматическое кольцо, выбранное из бензола, пиридина и пиримидина, при этом каждое из моноциклических ароматических колец необязательно замещено одним или более заместителей R5.
1.70 Композиция веществ согласно Варианту реализации 1.69, в которых заместитель Ar1 представляет собой моноциклическое ароматическое кольцо, выбранное из бензола и пиридин, при этом каждое из моноциклических ароматических колец необязательно замещено одним или более заместителей R5.
1.71 Композиция веществ согласно Варианту реализации 1.70, в которых заместитель Ar1 представляет собой бензольное кольцо, необязательно замещенное одним или более заместителей R5.
1.72 Композиция веществ согласно Варианту реализации 1.70, в которых заместитель Ar1 представляет собой пиридиновое кольцо, необязательно замещенное одним или более заместителей R5.
1.73 Композиция веществ согласно любому из Вариантов реализации 1.1-1.72, в которых моноциклическое ароматическое кольцо Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5.
1.74 Композиция веществ согласно Варианту реализации 1.73, в которых моноциклическое ароматическое кольцо Ar1 является незамещенным или содержит 1 или 2 заместителя R5.
1.75 Композиция веществ согласно Варианту реализации 1.74, в которых моноциклическое ароматическое кольцо Ar1 является незамещенным или замещено 1 заместителем R5.
1.76 Композиция веществ согласно Варианту реализации 1.75, в которых моноциклическое ароматическое кольцо Ar1 замещено 1 заместителем R5.
1.77 Композиция веществ согласно варианту реализации 1.75, в которых моноциклическое ароматическое кольцо Ar1 является незамещенным.
1.78 Композиция веществ согласно варианту реализации 1,74, в которых моноциклическое ароматическое кольцо Ar1 замещено 2 заместителями R5.
1.79 Композиция веществ согласно любому из Вариантов реализации 1.1-1.76 и 1.78, в которых заместитель R5 выбран из галогена, О-Ar2, цианогруппы, Hyd1b-SO2- и C1-8 углеводородной группы, в которой 0, 1 или 2, но не все атомы углерода в углеводородной группе необязательно заменены на гетероатом, выбранный из N, О и S, и при этом углеводородная группа необязательно замещена одним или более атомами фтора.
1.80 Композиция веществ согласно любому из Вариантов реализации 1.1-1.76, 1.78 и 1.79, в которых заместитель Ar2 представляет собой фенильную или пиридильную группу, необязательно замещенную 1 или 2 заместителями, выбранными из фтора, хлора, цианогруппы и трифторметила.
1.81 Композиция веществ согласно Варианту реализации 1.80, в которых заместитель Ar2 представляет собой фенильную группу, необязательно замещенную 1 или 2 заместителями, выбранными из фтора, хлора, циано и трифторметила.
1.82 Композиция веществ согласно любому из Вариантов реализации 1.1-1.76 и 1.78-1.80, в которых заместитель Hyd1b представляет собой насыщенную углеводородную группу С1-4.
1.83 Композиция веществ согласно Варианту реализации 1.82, в которых заместитель Hyd1b выбран из С1-4 алкила, циклопропила и циклопропилметила.
1.84 Композиция веществ согласно Варианту реализации 1.78, в которых заместитель Hyd1b выбран из метила, этила, пропила, циклопропила и циклопропилметила.
1.85 Композиция веществ согласно Варианту реализации 1.79, в которых заместитель Hyd1b выбран из метила, этила, пропила и циклопропила.
1.86 Композиция веществ согласно Варианту реализации 1.80, в которых заместитель Hyd1b выбран из метила и этила.
1.87 Композиция веществ согласно Варианту реализации 1.81, в которых заместитель Hyd1b представляет собой метил.
1.88 Композиция веществ согласно любому из Вариантов реализации 1.1-1.76 и 1.78, в которых заместитель R5 выбран из брома, фтора, хлора, цианогруппы, феноксигруппы, С1-4 алкилсульфонила, С1-4 алкоксигруппы и С1-4 алкила, при этом каждая из С1-4 алкоксигруппы и С1-4 алкильной группы необязательно замещена одним или более атомами фтора.
1.89 Композиция веществ согласно Варианту реализации 1.88, в которых заместитель R5 выбран из брома, фтора, хлора, цианогруппы, феноксигруппы, метилсульфонила, метила, этила, изопропила, дифторметила, трифторметила, метоксигруппы, дифторметоксигруппы и трифторметоксигруппы.
1.90 Композиция веществ согласно Варианту реализации 1.89, в которых заместитель R5 выбран из брома, фтора, хлора, цианогруппы, феноксигруппы, метилсульфонила и изопропила.
1.90А Композиция веществ согласно любому из Вариантов реализации 1.1-1.90, в которых заместитель R5 находится в пара-положении на моноцикпическом ароматическом кольце Ar1.
1.91 Композиция веществ согласно Варианту реализации 1.90 или Варианту реализации 1.91, в которых заместитель R5 выбран из брома, фтора, хлора и цианогруппы.
1.92 Композиция веществ согласно Варианту реализации 1.91, в которых заместитель R5 выбран из фтора, хлора и цианогруппы.
1.93 Композиция веществ согласно Варианту реализации 1.92, в которых заместитель R5 представляет собой цианогруппу.
1.94 Композиция веществ согласно варианту реализации 1.93, в которых заместитель Ar1 представляет собой 4-цианофенил.
1.95 Композиция веществ согласно Варианту реализации 1.92, в которых заместитель R5 представляет собой хлор.
1.96 Композиция веществ согласно Варианту реализации 1.95, в которых заместитель Ar1 представляет собой 4-хлорфенил.
1.97 Композиция веществ согласно Варианту реализации 1.92, в которых заместитель R5 представляет собой фтор.
1.98 Композиция веществ согласно варианту реализации 1.97, в которых заместитель Ar1 представляет собой 4-фторфенил.
1.99 Композиция веществ согласно любому из Вариантов реализации 1.1-1.98, в которых кольцо Y представляет собой кольцо бензола, пиридина, пиримидина, фурана, тиофена или пиррола.
1.100 Композиция веществ согласно варианту реализации 1.99, в которых кольцо Y представляет собой (а) бензольное кольцо, (b) пиридиновое кольцо или (с) тиофеновое кольцо.
1.101 Композиция веществ согласно Варианту реализации 1.100, в которых кольцо Y представляет собой бензольное кольцо.
1.102 Композиция веществ согласно варианту реализации 1.100, в которых кольцо Y представляет собой пиридиновое кольцо.
1.103 Композиция веществ согласно любому из Вариантов реализации 1.1-1.102, в которых заместитель R6 представляет собой группу Q1-Ra-Rb.
1.104 Композиция веществ согласно любому из Вариантов реализации 1.1-1.103, в которых Q1 имеет формулу (CRpRq)r, где r равен 0, 1, 2, 3 или 4, a Rp и Rq независимо выбраны из водорода и метила, или Rp и Rq совместно с атомом углерода, к которому они присоединены, образуют 3- или 4-членное насыщенное циклическое углеводородное кольцо, при условии, что общее количество атомов углерода в Q1 не превышает 6.
1.105 Композиция веществ согласно любому из Вариантов реализации 1.1-1.104, в которых Q1 отсутствует или выбран из CH2, СН(СН3), С(СН3)2, циклопропан-1,1-диила и циклобутан-1,1-диила.
1.106 Композиция веществ согласно любому варианту реализации 1.105, в которых Q1 отсутствует.
1.107 Композиция веществ согласно варианту реализации 1.105, в которых Q1 представляет собой -СН2- группу.
1.108 Композиция веществ согласно любому из Вариантов реализации 1.1-1.107, в которых Ra отсутствует или выбран из О, С(О), С(O)O, CONRc, N(Rc)CO, N(Rc)CONRc, NRc и SO2.
1.109 Композиция веществ согласно Варианту реализации 1.108, в которых Ra отсутствует или выбран из О, CONRc, N(Rc)CO, N(Rc)CONRc, NRc и SO2.
1.110 Композиция веществ согласно Варианту реализации 1.108, в которых Ra представляет собой CONRc.
1.111 Композиция веществ согласно Варианту реализации 1.108, в которых Ra представляет собой N(Rc)CO.
1.112 Композиция веществ согласно Варианту реализации 1.108, в которых Ra представляет собой NRc.
1.113 Композиция веществ согласно Варианту реализации 1.108, в которых Ra отсутствует.
1.114 Композиция веществ согласно Варианту реализации 1.108, в которых Ra представляет собой О.
1.115 Композиция веществ согласно Варианту реализации 1.108, в которых Ra представляет собой С(О).
1.116 Композиция веществ согласно Варианту реализации 1.108, в которых Ra представляет собой С(O)O.
1.117 Композиция веществ согласно Варианту реализации 1.108, в которых Ra представляет собой SO2.
1.118 Композиция веществ согласно любому из Вариантов реализации 1.1-1.117, в которых Rb выбран из следующего:
- неароматическая C1-8 углеводородная группа, в которой 0, 1 или 2, но не все атомы углерода в углеводородной группе заменены на гетероатом, выбранный из N и О, причем неароматическая С1-8 углеводородная группа необязательно замещена одним или более заместителями, выбранными из фтора и группы Сус1, и
- группа Сус1;
при условии, что если Ra представляет собой С(O)O или CONRc, то Rb дополнительно выбран из водорода.
1.119 Композиция веществ согласно Варианту реализации 1.118, в которых Rb выбран из следующего:
- неароматическая C1-8 углеводородная группа, в которой 0, 1 или 2 атома углерода заменены на гетероатом, выбранный из N и О, причем неароматическая С1-8 углеводородная группа необязательно замещена одним или более заместителями, выбранными из фтора и группы Сус1, и
- группа Сус1.
1.120 Композиция веществ согласно Варианту реализации 1.119, в которых Rb выбран из следующего:
- неароматическая С1-8 углеводородная группа, в которой 0 или 1, но не все атомы углерода в углеводородной группе заменены на гетероатом, выбранный из N и О, причем неароматическая C1-8 углеводородная группа необязательно замещена одним или более заместителями, выбранными из фтора и группы Сус1, и
- группа Сус1.
1.121 Композиция веществ согласно Варианту реализации 1.120, в которых Rb выбран из следующего:
- неароматическая C1-8 углеводородная группа, в которой 0 или 1, но не все атомы углерода в углеводородной группе заменены на гетероатом, выбранный из N и О, причем неароматическая C1-8 углеводородная группа необязательно замещена группой Сус1, и
- группа Сус1.
1.122 Композиция веществ согласно Варианту реализации 1.121, в которых Rb выбран из следующего:
- неароматическая С1-8 углеводородная группа, в которой 1 из атомов углерода в углеводородной группе заменен на гетероатом, выбранный из N и О, и
- группа Сус1.
1.123 Композиция веществ согласно Варианту реализации 1.122, в которых Rb выбран из следующего:
- неароматическая C1-8 углеводородная группа, в которой 1 из атомов углерода в углеводородной группе заменен на гетероатом N, и
- группа Сус1.
1.124 Композиция веществ согласно любому из Вариантов реализации 1.1-1.117, в которых Rb представляет собой неароматическая C1-8 углеводородная группа, в которой 0, 1 или 2, но не все атомы углерода в углеводородной группе заменены на гетероатом, выбранный из N и О, причем неароматическая C1-8 углеводородная группа необязательно замещена одним или более заместителями, выбранными из фтора и группы Сус1.
1.125 Композиция веществ согласно Варианту реализации 1.124, в которых Rb выбран из следующего:
- неароматическая C1-8 углеводородная группа, в которой 0 или 1, но не все атомы углерода в углеводородной группе заменены на гетероатом, выбранный из N и О, причем неароматическая С1-8 углеводородная группа необязательно замещена одним или более заместителями, выбранными из фтора и группы Сус1.
1.126 Композиция веществ согласно Варианту реализации 1.125, в которых Rb выбран из следующего:
- неароматическая C1-8 углеводородная группа, в которой 0 или 1, но не все атомы углерода в углеводородной группе заменены на гетероатом, выбранный из N и О, причем неароматическая C1-8 углеводородная группа необязательно замещена группой Сус1.
1.127 Композиция веществ согласно Варианту реализации 1.126, в которых Rb выбран из следующего:
- неароматическая C1-8 углеводородная группа, в которой 1 из атомов углерода в углеводородной группе заменен на гетероатом, выбранный из N и О.
1.128 Композиция веществ согласно любому из Вариантов реализации 1.1-1.127, в которых Rb выбран из следующего:
- неароматическая С1-8 углеводородная группа, в которой 1 из атомов углерода в углеводородной группе заменен на гетероатом азота.
1.129 Композиция веществ согласно любому из Вариантов реализации 1.118
- 1.128, в которых Rb выбран из следующего:
- неароматическая C1-8 углеводородная группа, в которой атом углерода в углеводородной группе заменен гетероатомом азота, так чтобы образовать концевую диметиламиногруппу.
1.130 Композиция веществ согласно любому из Вариантов реализации 1.118-1.129, в которых неароматическая углеводородная группа является ациклической.
1.131 Композиция веществ согласно любому из Вариантов реализации 1.118-1.130, в которых неароматическая углеводородная группа является насыщенной.
1.132 Композиция веществ согласно любому из Вариантов реализации 1.118-1.131, в которых неароматическая углеводородная группа содержит от 1 до 6 атомов углерода.
1.133 Композиция веществ согласно любому из Вариантов реализации 1.118-1.132, в которых неароматическая углеводородная группа содержит от 1 до 5 атомов углерода.
1.134 Композиция веществ согласно любому из Вариантов реализации 1.118-1.133, в которых неароматическая углеводородная группа содержит от 3 до 5 атомов углерода.
1.135 Композиция веществ согласно любому из Вариантов реализации 1.1-1.126, в которых Rb представляет собой или содержит в себе группу Сус1.
1.136 Композиция веществ согласно варианту реализации 1.128, в которых Rb представляет собой группу Сус1.
1.137 Композиция веществ согласно Варианту реализации 1.136, в которых Сус1 представляет собой неароматическую 4-7-членную карбоциклическую или гетероциклическую кольцевую группу, содержащую 0, 1 или 2 кольцевых гетероатома, выбранных из N и О, и необязательно замещенную одним или более заместителями, выбранными из гидроксила, аминогруппы, моно-С1-4 алкиламиногруппы, ди-С1-4 алкиламиногруппы и насыщенной С1-5 углеводородной группы, в которой 0 или 1, но не все атомы углерода заменены на гетероатом, выбранный из N и О.
1.138 Композиция веществ согласно Варианту реализации 1.137, в которых Сус1 представляет собой неароматическую 4-7-членную гетероциклическую кольцевую группу, содержащую кольцевой гетероатом азота и необязательно второй кольцевой гетероатом, выбранный из N и О, при этом неароматическая 4-7-членная гетероциклическая кольцевая группа необязательно замещена одним или более заместителями, выбранными из гидроксила, аминогруппы, моно-C1-4 алкиламиногруппы, ди-С1-4 алкиламиногруппы и насыщенной С1-4 углеводородной группы, в которой 0 или 1, но не все атомы углерода заменены на гетероатом, выбранный из N и О.
1.139 Композиция веществ согласно Варианту реализации 1.138, в которых Сус1 представляет собой неароматическую 5-6-членную гетероциклическую кольцевую группу, содержащую кольцевой гетероатом азота и необязательно второй кольцевой гетероатом, выбранный из N и О, при этом неароматическая 5-6-членная гетероциклическая кольцевая группа необязательно замещена одним или более заместителями, выбранными из гидроксила, аминогруппы, моно-С1-4 алкиламиногруппы, ди-С1-4 алкиламиногруппы и насыщенной С1-4 углеводородной группы, в которой 0 или 1, но не все атомы углерода заменены на гетероатом, выбранный из N и О.
1.140 Композиция веществ согласно Варианту реализации 1.139, в которых Сус1 представляет собой неароматическую 5-6-членную гетероциклическую кольцевую группу, содержащую кольцевой гетероатом азота и необязательно второй кольцевой гетероатом, выбранный из N и О, при этом неароматическая 5-6-членная гетероциклическая кольцевая группа необязательно замещена одним или более заместителями, выбранными из гидроксила, аминогруппы, моно-С1-2 алкиламиногруппы, ди-С1-2 алкиламиногруппы и С1-4 алкильной группы, в которой 0 или 1, но не все атомы углерода заменены на гетероатом, выбранный из N и О.
1.141 Композиция веществ согласно любому из Вариантов реализации 1.1-1.126 и 1.135-1.140, в которых Сус1 представляет собой насыщенное кольцо.
1.142 Композиция веществ согласно Варианту реализации 1.141, в которых Сус1 выбран из пирролидина, пиперидина и пиперазина, каждый из которых необязательно замещен одним или более заместителями, выбранными из гидроксила, аминогруппы, моно-С1-2 алкиламиногруппы, ди-С1-2 алкиламиногруппы и С1-4 алкильной группы, в которой 0 или 1, но не все атомы углерода заменены на гетероатом, выбранный из N и О.
1.143 Композиция веществ согласно любому из Вариантов реализации 1.1-1.112 и 1.118-1.142, в которых R' выбран из водорода, метила, этила, пропила, изопропила, циклопропила, циклопропилметила, бутила, изобутила и циклобутила.
1.144 Композиция веществ согласно Варианту реализации 1.143, в которых Rc выбран из водорода и метила.
1.145 Композиция веществ согласно Варианту реализации 1.144, в которых Rc представляет собой водород.
1.146 Композиция веществ согласно варианту реализации 1.144, в которых Rc представляет собой метил.
1.147 Композиция веществ согласно любому из Вариантов реализации 1.1-1.103, в которых R6 выбран из групп А-AL, представленных в таблице ниже.
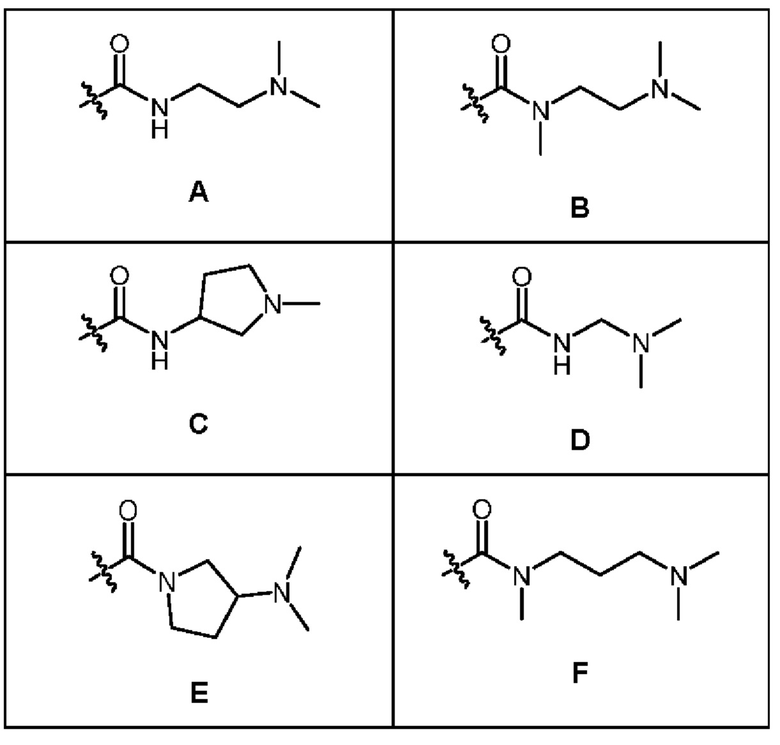
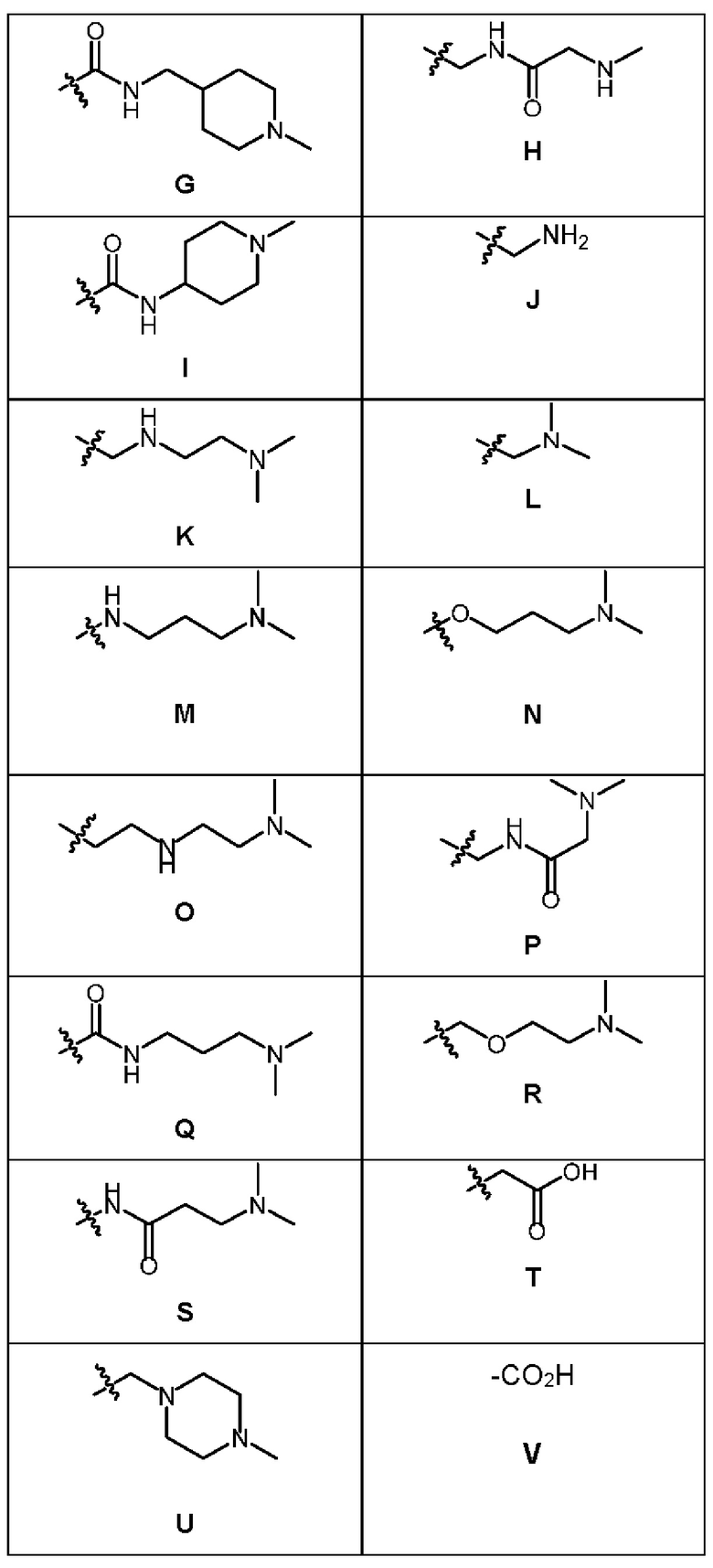
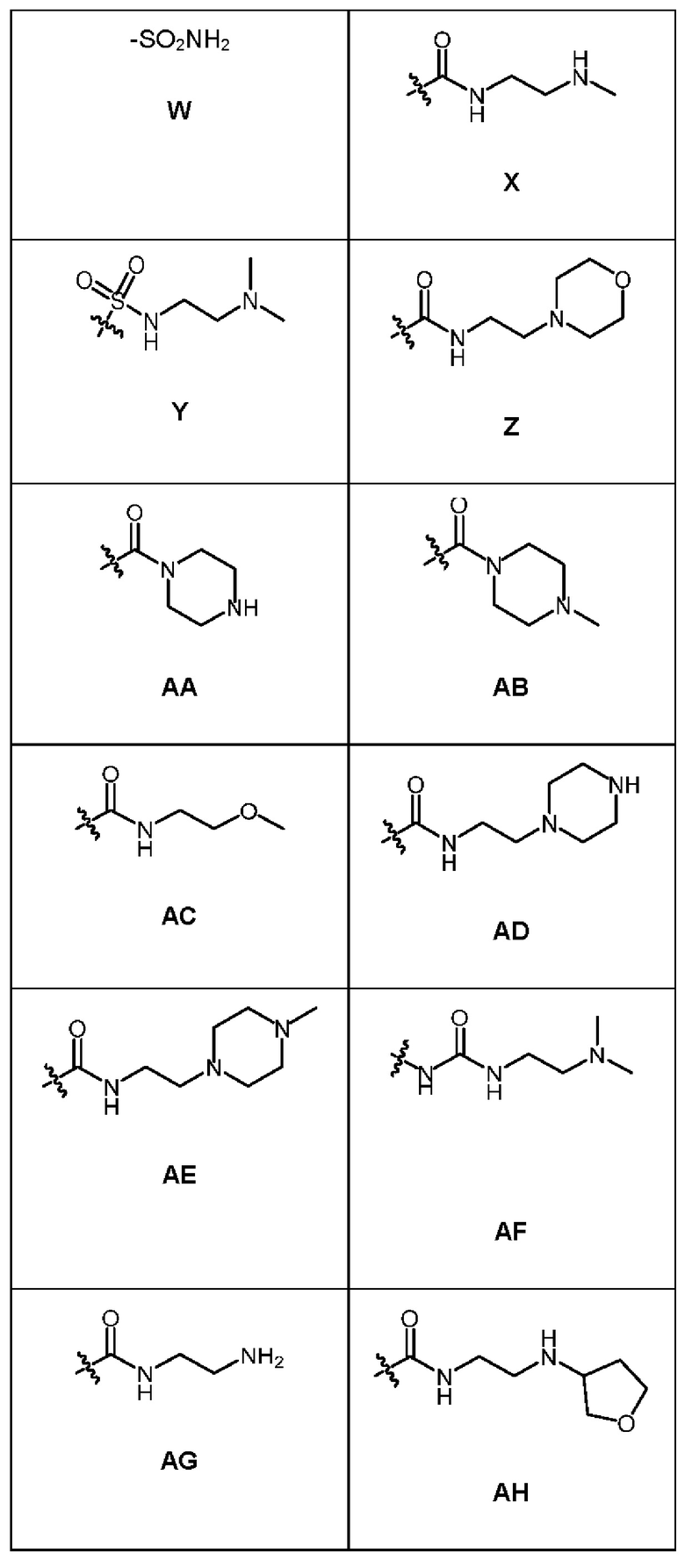
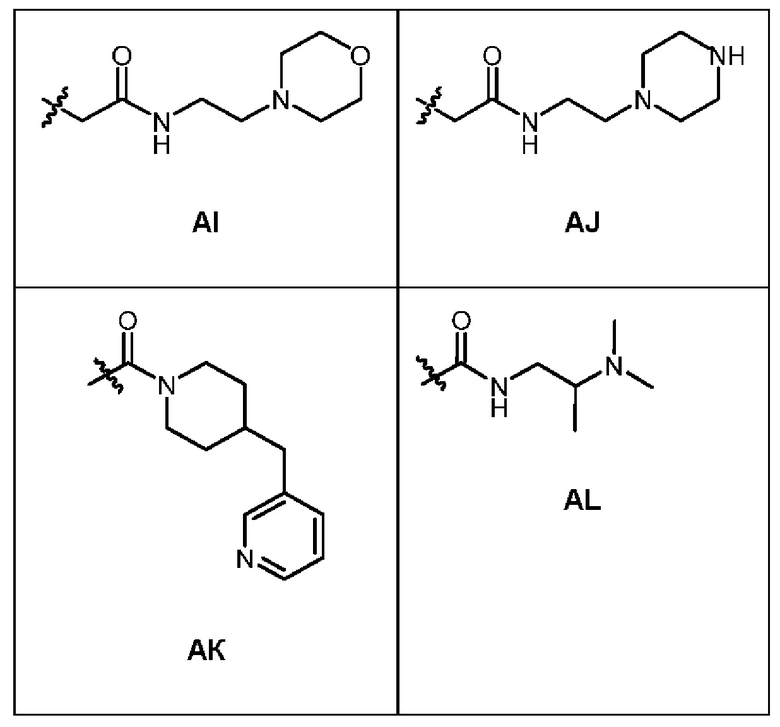
1.148 Композиция веществ согласно Варианту реализации 1.147, в которых R6 выбран из групп А и Q.
1.149 Композиция веществ согласно варианту реализации 1.148, в которых R6 представляет собой группу А.
1.149А Композиция веществ согласно любому из Вариантов реализации 1.1-1.149, в которых n равен 0 или 1.
1.149В Композиция веществ согласно вариантам реализации 1.149А, в которых n представляет собой 0.
1.149С Композиция веществ согласно Вариантам реализации 1.149А, в которых n представляет собой 1.
1.149D Композиция веществ согласно любому из Вариантов реализации 1.1-1.149А и 1.149С, в которых R7 выбран из фтора, хлора и метоксигруппы.
1.149Е Композиция веществ согласно Варианту реализации 1.149D, в которых R7 выбран из хлора и метоксигруппы.
1.149F Композиция веществ согласно варианту реализации 1.149D, в которых n равен 1, a R7 представляет собой хлор.
1.149G Композиция веществ согласно Варианту реализации 1.149D, в которых n равен 1, a R7 представляет собой метоксигруппу.
1.150 Композиция веществ согласно любому из Вариантов реализации 1.1-1.149, в которых: (i) если Y представляет собой шестичленное кольцо, то R6 присоединен в его мета- или пара-положении, или (ii) если Y представляет собой пятичленное кольцо, то R6 присоединен к кольцу Y в положении, которое не соседствует с членом кольца Y, к которому присоединено кольцо Z.
1.151 Композиция веществ согласно варианту реализации 1.150, в которых Y представляет собой шестичленное кольцо, a R6 присоединен в его мета- или пара-положении.
1.152 Композиция веществ согласно варианту реализации 1.151, в которых Y представляет собой шестичленное кольцо, a R6 присоединен в его мета-положении.
1.153 Композиция веществ согласно варианту реализации 1.151, в которых Y представляет собой шестичленное кольцо, a R6 присоединен в его пара-положении.
1.154 Композиция веществ, состоящая из по меньшей мере 90 масс. % атропизомера формулы (1А) и 0-10 масс. % атропизомера формулы (1В), причем атропизомер формулы (1А) и атропизомер формулы (1В) представлены как:
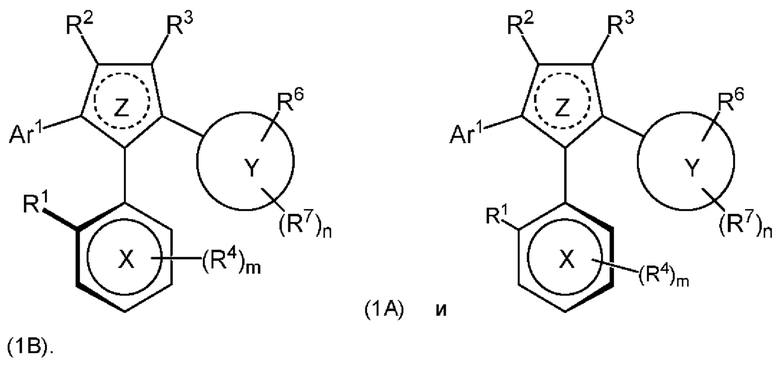
или их фармацевтически приемлемые соли или таутомеры, при этом R1-R7, Ar1, m, n, X, Y и Z являются такими, как это определено в любом из Вариантов реализации 1.1-1.153.
1.155 Композиция веществ согласно Варианту реализации 1.154, состоящая из по меньшей мере 95 масс. % атропизомера формулы (1А) или его соли или таутомера и 0-5 масс. % атропизомера формулы (1В) или его соли или таутомера.
1.156 Композиция веществ согласно Варианту реализации 1.154, состоящая из по меньшей мере 96 масс. % атропизомера формулы (1А) или его соли или таутомера и 0-4 масс. % атропизомера формулы (1В) или его соли или таутомера.
1.157 Композиция веществ согласно Варианту реализации 1.154, состоящая из по меньшей мере 97 масс. % атропизомера формулы (1А) или его соли или таутомера и 0-3 масс. % атропизомера формулы (1В) или его соли или таутомера.
1.158 Композиция веществ согласно Варианту реализации 1.154, состоящая из по меньшей мере 98 масс. % атропизомера формулы (1А) или его соли или таутомера и 0-2 масс. % атропизомера формулы (1В) или его соли или таутомера.
1.159 Композиция веществ согласно Варианту реализации 1.154, состоящая из по меньшей мере 99 масс. % атропизомера формулы (1А) или его соли или таутомера и 0-1 масс. % атропизомера формулы (1В) или его соли или таутомера.
1.160 Композиция веществ согласно Варианту реализации 1.154, состоящая из по меньшей мере 99,5 масс. % атропизомера формулы (1А) или его соли или таутомера и 0-0,5 масс. % атропизомера формулы (1В) или его соли или таутомера.
1.161 Композиция веществ, состоящая из по меньшей мере 90 масс. % атропизомера формулы (1В) и 0-10 масс. % атропизомера формулы (1А), причем атропизомер формулы (1А) и атропизомер формулы (1В) представлены как:
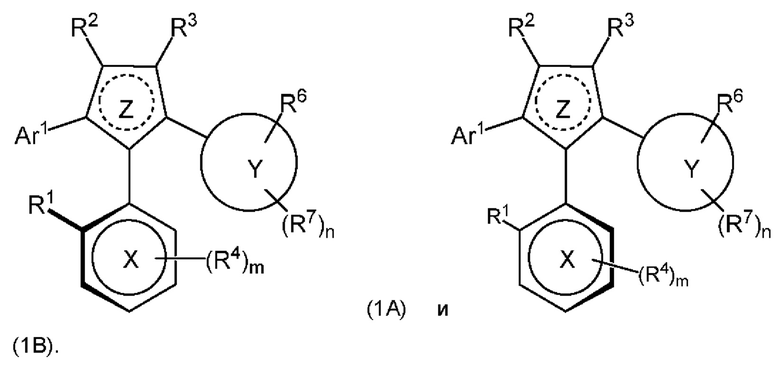
или их фармацевтически приемлемые соли или таутомеры, при этом R1-R7, Ar1, m, n, X, Y и Z являются такими, как это определено в любом из Вариантов реализации 1.1-1.153.
1.162 Композиция веществ согласно Варианту реализации 1.161, состоящая из по меньшей мере 95 масс. % атропизомера формулы (1В) или его соли или таутомера и 0-5 масс. % атропизомера формулы (1А) или его соли или таутомера.
1.163 Композиция веществ согласно Варианту реализации 1.161, состоящая из по меньшей мере 96 масс. % атропизомера формулы (1В) или его соли или таутомера и 0-4 масс. % атропизомера формулы (1А) или его соли или таутомера.
1.164 Композиция веществ согласно Варианту реализации 1.161, состоящая из по меньшей мере 97 масс. % атропизомера формулы (1В) или его соли или таутомера и 0-3 масс. % атропизомера формулы (1А) или его соли или таутомера.
1.165 Композиция веществ согласно Варианту реализации 1.161, состоящая из по меньшей мере 98 масс. % атропизомера формулы (1В) или его соли или таутомера и 0-2 масс. % атропизомера формулы (1А) или его соли или таутомера.
1.166 Композиция веществ согласно Варианту реализации 1.161, состоящая из по меньшей мере 99 масс. % атропизомера формулы (1В) или его соли или таутомера и 0-1 масс. % атропизомера формулы (1А) или его соли или таутомера.
1.167 Композиция веществ согласно Варианту реализации 1.161, состоящая из по меньшей мере 99,5 масс. % атропизомера формулы (1В) или его соли или таутомера и 0-0,5 масс. % атропизомера формулы (1А) или его соли или таутомера.
1.168 Композиция веществ:
(i) состоящая по меньшей мере из 90 масс. % атропизомера (2А) и 0-10 масс. % атропизомера формулы (2В), или
(ii) состоящая по меньшей мере из 90 масс. % атропизомера (2В) и 0-10 масс. % атропизомера формулы (2А),
в которой атропизомер формулы (2А) и атропизомер формулы (2В) представлены как:
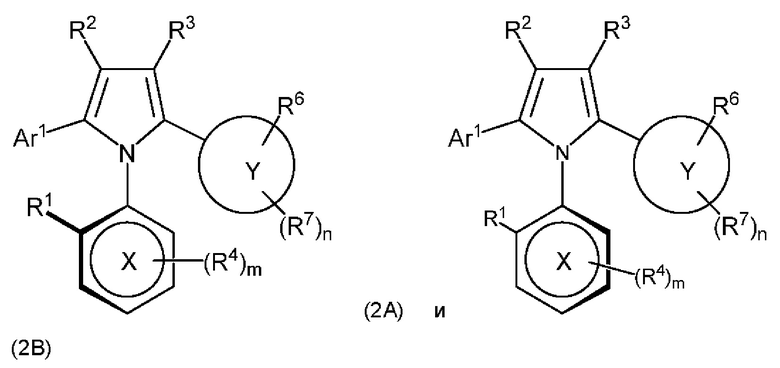
или представляют собой их фармацевтически приемлемые соли или таутомеры, при этом R1, R2, R3, R4, R6, R7, Ar1, X и Y являются такими, как это определено в любом из Вариантов реализации 1.1, 1.2, 1.8, 1.10, 1.11 и 1.21-1.153.
1.169 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 95 масс. % атропизомера формулы (2А) или его соли или таутомера и 0-5 масс. % атропизомера формулы (2В) или его соли или таутомера.
1.170 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 96 масс. % атропизомера формулы (2А) или его соли или таутомера и 0-4 масс. % атропизомера формулы (2В) или его соли или таутомера.
1.171 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 97 масс. % атропизомера формулы (2А) или его соли или таутомера и 0-3 масс. % атропизомера формулы (2В) или его соли или таутомера.
1.172 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 98 масс. % атропизомера формулы (2А) или его соли или таутомера и 0-2 масс. % атропизомера формулы (2В) или его соли или таутомера.
1.173 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 99 масс. % атропизомера формулы (2А) или его соли или таутомера и 0-1 масс. % атропизомера формулы (2В) или его соли или таутомера.
1.174 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 99,5 масс. % атропизомера формулы (2А) или его соли или таутомера и 0-0,5 масс. % атропизомера формулы (2В) или его соли или таутомера.
1.175 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 95 масс. % атропизомера формулы (2В) или его соли или таутомера и 0-5 масс. % атропизомера формулы (2А) или его соли или таутомера.
1.176 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 96 масс. % атропизомера формулы (2В) или его соли или таутомера и 0-4 масс. % атропизомера формулы (2А) или его соли или таутомера.
1.177 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 97 масс. % атропизомера формулы (2В) или его соли или таутомера и 0-3 масс. % атропизомера формулы (2А) или его соли или таутомера.
1.178 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 98 масс. % атропизомера формулы (2В) или его соли или таутомера и 0-2 масс. % атропизомера формулы (2А) или его соли или таутомера.
1.179 Композиция веществ согласно Варианту реализации 1.168, состоящая из по меньшей мере 99 масс. % атропизомера формулы (2В) или его соли или таутомера и 0-1 масс. % атропизомера формулы (2А) или его соли или таутомера.
1.180 Композиция веществ согласно любому из Вариантов реализации 1.168-1.179, в которых:
R1 выбран из трифторметила, гидроксила, аминогруппы, (диметиламино)метила и (метокси)метила;
R2 представляет собой водород;
R3 представляет собой водород;
R4 отсутствует или выбран из хлора, фтора и С1-4 алкила;
Ar1 представляет собой фенил или пиридил, необязательно замещенные одним или двумя заместителями R5, выбранными из брома, фтора, хлора, феноксигруппы, С1-4 алкила (например, изопропила), С1-4 алкилсульфонила (например, метилсульфонила) и цианогруппы;
X выбран из фенила и пиридила;
m равен 0 или 1;
Y выбран из фенила, пиридила и тиенила;
n равен 0 или 1;
R6 выбран из групп А-AM, представленных в Таблице 1 выше, и
R7 выбран из хлора, фтора и С1-4 алкоксигруппы (например, метоксигруппы).
1.181 Композиция веществ согласно Варианту реализации 1.180, в которых:
R1 представляет собой трифторметил;
R2 представляет собой водород;
R3 представляет собой водород;
Ar1 представляет собой фенил, замещенный заместителем R5, выбранным из фтора, хлора и цианогруппы;
X представляет собой фенил;
m равен 0;
Y представляет собой фенил или пиридил;
n равен 0 и
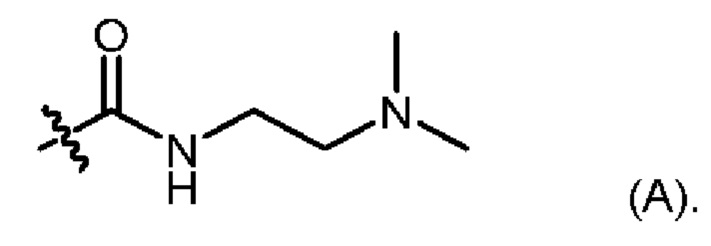
R6 представляет собой группу (А):
1.182 Композиция веществ согласно любому из Вариантов реализации 1.1 и 1.154-1.179, в которой атропизомер представляет собой атропизомер соединения согласно любому из Примеров А-1 - А-8 и В-2 - В-107.
1.183 Композиция веществ согласно любому из Вариантов реализации 1.1 и 1.154-1.179, в которой атропизомер представляет собой атропизомер соединения согласно любому из Примеров А-1 - А-8.
1.184 Композиция веществ, состоящая из 99,5-100 масс. % одинарного атропизомера, определенного в любом из Вариантов реализации 1.1-1.183.
1.185 Композиция веществ, состоящая из 99,9-100 масс. % одинарного атропизомера, определенного в любом из Вариантов реализации 1.1-1.183.
1.186 Одинарный атропизомер, имеющий химическую структуру, определенную в любом из Вариантов реализации 1.1-1.183, причем указанный одинарный атропизомер в композиции не сопровождается никаким другим атропизомером или сопровождается, но не более чем 0,5 масс. % любого другого атропизомера относительно массы указанного одинарного атропизомера.
1.187 Одинарный атропизомер, имеющий химическую структуру, определенную в любом из Вариантов реализации 1.1-1.183, причем указанный одинарный атропизомер в композиции не сопровождается никаким другим атропизомером или сопровождается, но не более чем 0,25 масс. % любого другого атропизомера относительно массы указанного одинарного атропизомера.
1.188 Одинарный атропизомер, имеющий химическую структуру, определенную в любом из Вариантов реализации 1.1-1.183, причем указанный одинарный атропизомер в композиции не сопровождается никаким другим атропизомером или сопровождается, но не более чем 0,1 масс. % любого другого атропизомера относительно массы указанного одинарного атропизомера.
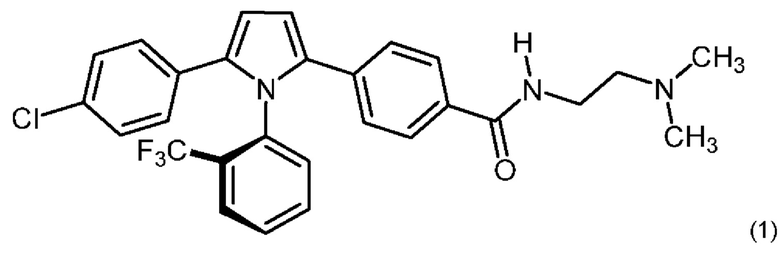
1.188А Одинарный атропизомер согласно Варианту реализации 1.188, который имеет конфигурацию R, представленную формулой (1), или его соль:
1.189 Композиция веществ, определенная в любом из Вариантов реализации 1.1-1.185, или отдельный одинарный атропизомер, определенный в любом из Вариантов реализации 1.186-1.188А, причем каждый атропизомер представлен в форме соли.
1.190 Композиция веществ, определенная в любом из Вариантов реализации 1.1-1.187, или отдельный одинарный атропизомер, определенный в Варианте реализации 1.188 или 1.188А, причем каждый атропизомер находится в форме кислотно-аддитивной соли.
1.191 Композиция веществ, определенная в любом из Вариантов реализации 1.1-1.187, или отдельный одинарный атропизомер, определенный в Варианте реализации 1.188 или 1.188А, причем каждый атропизомер находится в несолевой форме.
1.192 Композиция веществ, определенная в любом из Вариантов реализации 1.1-1.187, или отдельный одинарный атропизомер, определенный в Варианте реализации 1.188 или 1.188А, причем каждый атропизомер находится в форме кислотно-аддитивной соли (предпочтительно в соотношении с кислотой в соли приблизительно равном 1:1), образованной кислотой, выбранной из хлористоводородной, метансульфоновой, малеиновой, яблочной, винной, п-толуолсульфоновой, фосфорной и серной кислот.
Предпочтительной кислотно-аддитивной солью согласно настоящему изобретению является соль, образованная в соотношении 1:1 между одинарным атропизомером (Соединение (1)) согласно Варианту реализации изобретения 1.88А и (+)-L-винной кислотой.
(+)-L-винная кислота является особенно предпочтительной, поскольку она является высококристаллическим и стабильным твердым веществом, поглощающим только поверхностную влагу (<1% при 90% относительной влажности) с улучшенной водной растворимостью по сравнению со свободным основанием. Эти свойства делают ее особенно подходящей для фармацевтической разработки.
Соответственно, в дальнейших вариантах реализации (Варианты реализации 1.193-1.211) настоящего изобретения предложены:
1.193 Соль (R)-2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)-этил]бензамида и (+)-L-винной кислоты в мольном соотношение между кислотой и основанием равном приблизительно 1:1.
1.194 Соль (+)-L-винной кислоты и 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида, имеющая формулу (2):
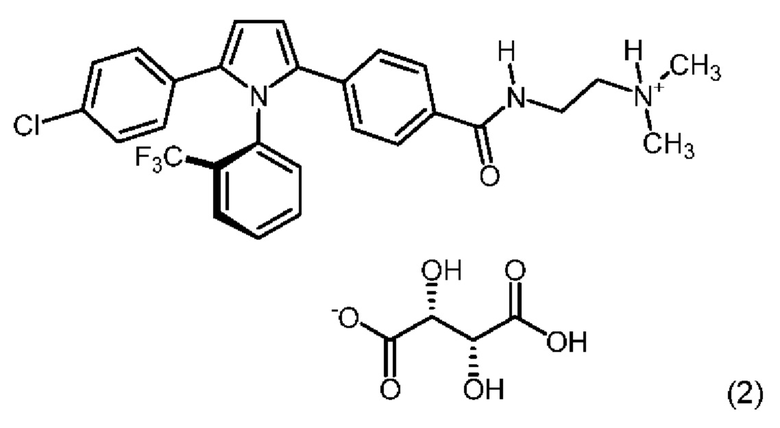
1.195 Соль (+)-L-винной кислоты и 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида, в которой мольное соотношение между кислотой и основанием составляет приблизительно 1:1 и где 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]-бензамид находится в виде единственного (одинарного) атропизомера.
1.196 Соль (+)-L-винной кислоты согласно Варианту реализации 1.195, в которой одинарный атропизомер представляет собой атропизомер формулы (1), определенный в Варианте реализации 1.188А.
1.197 Соль (+)-L-винной кислоты согласно Варианту реализации 1.195, в которой одинарный атропизомер представляет собой R-атропизомер 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида.
1.198 Соль(+)-L-винной кислоты согласно Варианту реализации 1.95, в которой одинарный атропизомер характеризуется любым одним или более из следующих параметров:
(i) рентгеновские кристаллографические данные по существу такие же, как это описано в Примере 3 настоящего документа;
(ii) время удерживания при определениях методом 1 хиральной ВЭЖХ составляет приблизительно 20 минут (например, приблизительно 20,5 минут), и
(iii) удельное оптическое вращение при измерениях методом, описанным в Примере 2 настоящего документа, составляет приблизительно -11,76°.
1.199 Соль (+)-L-винной кислоты согласно Варианту реализации 1.195, в которой одинарный атропизомер представляет собой атропизомер А-2 согласно описанию в Примерах, приведенных в настоящем документе.
1.200 Соль (+)-L-винной кислоты согласно Варианту реализации 1.195, в которой соль (+)-L-винной кислоты является такой, как это описано в Примерах, приведенных в настоящем документе.
1.201 Соль(+)-L-винной кислоты согласно любому из Вариантов реализации 1.193 - 1.200, которая находится в кристаллической форме.
1.202 Соль (+)-L-винной кислоты согласно Варианту реализации 1.201, которая является безводной (ангидратом).
1.203 Соль (+)-L-винной кислоты согласно Варианту реализации 1.202, которая является ангидратом, идентифицируемым в настоящем документе согласно Дифрактограмме В.
1.204 Соль (+)-L-винной кислоты согласно Варианту реализации 1.201, которая представляет собой сольват.
1.205 Соль(+)-L-винной кислоты согласно Варианту реализации 1.204, которая представляет собой сольват, идентифицируемый в настоящем документе согласно Дифрактограмме А.
1.206 Композиция веществ, содержащая соль(+)-L-винной кислоты согласно любому из Вариантов реализации 1.193-1.205, в которой либо (а) одинарный атропизомер является единственным атропизомером 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида, присутствующим в композиции, либо (b) имеется любой другой атропизомер 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида в количестве менее 10 мольных % относительно мольного количества указанного одинарного атропизомера.
1.207 Композиция веществ согласно Варианту реализации изобретения 1.206, в которой либо (а) одинарный атропизомер является единственным атропизомером 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида, присутствующим в композиции, либо (b) имеется любой другой атропизомер 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида в количестве менее 5 мольных % относительно мольного количества указанного одинарного атропизомера.
1.208 Композиция веществ согласно Варианту реализации изобретения 1.206, в которой либо (а) одинарный атропизомер является единственным атропизомером 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N,[2-(диметиламино)этил]бензамида, присутствующим в композиции, либо (b) имеется любой другой атропизомер 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида в количестве менее 2 мольных % относительно мольного количества указанного одинарного атропизомера.
1.209 Композиция веществ согласно Варианту реализации изобретения 1.206, в которой либо (а) одинарный атропизомер является единственным атропизомером 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида, присутствующем в композиции, либо (b) имеется любой другой атропизомер 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида в количестве менее 1,5 мольных % относительно мольного количества указанного одинарного атропизомера.
1.210 Композиция веществ согласно Варианту реализации изобретения 1.206, в которой либо (а) одинарный атропизомер является единственным атропизомером 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида, присутствующим в композиции, либо (b) имеется любой другой атропизомер 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида в количестве менее 1 мольного % относительно мольного количества указанного одинарного атропизомера.
1.211 Композиция веществ согласно Варианту реализации изобретения 1.206, в которой либо (а) одинарный атропизомер является единственным атропизомером 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида, присутствующим в композиции, либо (b) имеется любой другой атропизомер 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида в количестве менее 0,1 мольного % относительно мольного количества указанного одинарного атропизомера.
Определения
Термины «атропизомерное соединение (соединения)», «атропизомерное соединение (соединения) согласно настоящему изобретению», «соединение (соединения) формулы (1)», «соединение (соединения)» и «соединение (соединения) согласно настоящему изобретению» и подобные им термины могут быть использованы в настоящем документе для обозначения композиций и атропизомеров, определенных в любом из Вариантов реализации 1.1-1.211. Если в контексте не указано иное, такие термины могут быть восприняты как относящиеся к любому из атропизомеров согласно формулам (1А), (1В), (2А) и (2В) и всем подгруппам, предпочтениям, Вариантам реализации и Примерам, описанным в настоящем документе. Термин «соединение формулы (1)» может быть использован в настоящем документе в качестве родового понятия, охватывающего атропизомеры согласно формулам (1А), (1В), (2А) и (2В) и всем подгруппам, предпочтениям, Вариантам реализации и Примерам, а также смеси таких атропизомеров. Из контекста, в котором делается ссылка на соединение формулы (1), будет очевидно, относится ли оно к отдельным атропизомерам, композиции или смеси атропизомеров.
При использовании в настоящем документе термин «лекарственное средство» относится к фармацевтическому составу, который может быть использован для лечения, ухода или улучшения хода заболевания или для лечения, облегчения или смягчения симптомов заболевания. Фармацевтический состав содержит фармакологически активный ингредиент в форме, не вредной для субъекта, которому он вводится, и дополнительные компоненты, предназначенные для стабилизации активного ингредиента и влияющие на его поступление в кровеносную систему или целевую ткань.
Соли
Если атропизомеры, определенные в любом из Вариантов реализации 1.1-1.188А, содержат ионизируемые группы, они могут быть представлены в форме солей, как это определено в любом из Вариантов реализации 1.189, 1.190 и 1.192-1.211.
Например, если атропизомеры содержат основную (например, группу азотистого основания) группу или атом, атропизомеры могут быть представлены в форме кислотно-аддитивных солей.
Соли могут быть синтезированы из исходного соединения традиционными химическими способами, такими как способы, описанные в публикации Pharmaceutical Salts; Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camiile G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 pages, August 2002. Обычно такие соли могут быть получены путем проведения реакции соединения в форме свободного основания с раствором кислоты в воде или органическом растворителе, или в их смеси; обычно применяют неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Соли присоединения кислоты (как определено в Варианте реализации 1.190) могут быть образованы широким разнообразием кислот, как неорганических, так и органических. Примеры солей присоединения кислот включают соли, образованные кислотами, выбранными из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетамидобензойной, бутановой, (+)камфарной, камфарносульфоновой, (+)-(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламиновой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, бромистоводородной, хлористоводородной, иодистоводородной, изэтиновой, (+)-L-молочной, (±)-DL-молочной, лактобионовой, малеиновой, яблочной, (-)-L-оксиянтарной, малоновой, (±)-DL-миндальной, метансульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памоевой, фосфорной, пропионовой, L-пироглутаминовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, (+)-L-винной, тиоциановой, п-толуолсульфоновой, ундециленовой и валериановой кислот, а также ацилированных аминокислот и катионообменных смол.
Солевые формы композиций или атропизомеров согласно настоящему изобретению обычно представляют собой фармацевтически приемлемые соли, и примеры фармацевтически приемлемых солей обсуждаются в публикации Berge et al., 1977, "Pharmaceutical Acceptable Salts," J. Pnarm. Sci., Vol. 66, pp. 1-19. Однако, соли, которые не являются фармацевтически приемлемыми, также могут быть приготовлены в качестве некоторых промежуточных форм, которые затем могут быть преобразованы в фармацевтически приемлемые соли. Такие формы фармацевтически неприемлемых солей, которые могут быть полезны, например, при очистке или выделении композиций или атропизомеров согласно настоящему изобретению, также являются частью настоящего изобретения.
Геометрические изомеры и таутомеры
Помимо существующих в виде атропизомеров, композиции или атропизомеры согласно настоящему изобретению могут иметь и другие структурные особенности, которые обуславливают геометрический изомеризм и таутомеризм, и упоминание композиций или атропизомеров согласно определениям в Вариантах реализации 1.1-1.211, подразумевает все их геометрические изомерные и таутомерные формы. Во избежание неясности в случае, если атропизомер может существовать в одной из нескольких геометрических изомерных и таутомерных форм, а описана или показана только одна конкретная форма, то все остальные формы все равно считаются включенными в формулу (1А)-(1В) или ее подгруппы, подмножества, предпочтительные варианты и примеры.
Оптические изомеры
Если соединения согласно изобретению содержит один или более хиральных центров помимо структурных особенностей, приводящих к атропизомеризму, упоминание композиции или атропизомера включает все его оптически-изомерные формы (например, энантиомеры, эпимеры и диастереомеры), как в виде отдельных оптических изомеров, так и в виде их смесей (отличных от смесей атропизомеров), если из контекста не требуется иного.
Оптические изомеры могут быть охарактеризованы и идентифицированы по их оптической активности (т.е. как + и - изомеры, или d и l изомеры), или они могут быть охарактеризованы с точки зрения абсолютной стереохимии с использованием номенклатуры «R и S», разработанной Каном, Ингольдом и Прелогом, см. Advanced Organic Cnemistry by Jerry March, 4th Edition, John Wiley & Sons, New York, 1992, pages 109-114, и также см. Cahn, Ingold & Prelog, Angew. Chem. Inf. Ed. Engl., 1966, 5, 385-415.
Оптические изомеры можно разделить с использованием ряда методик, включая хиральную хроматографию (хроматографию на хиральном носителе), и такие методики хорошо известны специалисту в данной области техники.
В качестве альтернативы хиральной хроматографии оптические изомеры могут быть разделены путем образования диастереомерных солей с хиральными кислотами, такими как (+)-винная кислота, (-)-пироглутаминовая кислота, (-)-дитолуил-L-винная кислота, (+)-миндальная кислота, (-)-яблочная кислота и (-)-камфарсульфоновая кислота, разделения диастереомеров методами предпочтительной кристаллизации, после чего проводят диссоциацию солей и получают отдельные энантиомеры в форме свободных оснований.
Если соединения согласно настоящему изобретению существуют в виде двух или более оптических изомерных форм, один энантиомер в паре энантиомеров может показывать преимущества относительно другого энантиомера, например, с точки зрения биологической активности. Соответственно, в некоторых ситуациях может быть желательным использовать в качестве терапевтического агента только один из пары энантиомеров или только один из всего множества диастереоизомеров. Соответственно, в настоящем изобретении предложены композиции, содержащие атропизомер, имеющий один или более хиральных центров, причем по меньшей мере 55% (например, по меньшей мере 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95%) композиции или атропизомера формулы (1) присутствует в виде единственного оптического изомера (например, энантиомера или диастереоизомера). В общем варианте реализации изобретения 99% или более (например, практически все количество) от общего количества композиции или атропизомера формулы (1) может быть представлено в виде единственного оптического изомера (например, энантиомера или диастереоизомера).
Изотопы
Композиции или атропизомеры согласно настоящему изобретению, определенные в любом из Вариантов реализации 1.1-1.211, могут содержать один или более изотопных заместителей, и указание на какой-либо конкретный химический элемент включает в себя все множество изотопов этого элемента. Например, указание на водород включает в себя его изотопы 1Н, 2Н(D) и 3Н(Т). Аналогичным образом, указания на углерод и кислород включают в себя их изотопы 12С, 13С, 14С и 16О и 18О.
Изотопы могут быть радиоактивными и нерадиоактивными. Согласно одному варианту реализации изобретения композиции или атропизомеры не содержат радиоактивных изотопов. Такие соединения предпочтительны для терапевтического применения. Однако, согласно другому варианту реализации изобретения композиция или атропизомер может содержать один или более радиоизотопов. Соединения, содержащие такие радиоизотопы, могут быть пригодны в смысле диагностики.
Сольваты
Композиции или атропизомеры, определенные в любом из Вариантов реализации 1.1-1.211, могут образовывать сольваты и ангидраты.
Отдельные сольваты представляют собой сольваты, образованные путем включения в твердофазную структуру (например, кристаллическую структуру) композиций или атропизомеров согласно настоящему изобретению молекул нетоксичного и фармацевтически приемлемого растворителя (называемого далее сольватирующим растворителем). Примеры таких растворителей включают воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты могут быть получены путем перекристаллизации композиции или атропизомеров согласно настоящему изобретению в растворителе или смеси растворителей, содержащих сольватирующий растворитель. Образовался ли сольват в данном конкретном случае, можно определить, подвергнув кристаллы композиции или атропизомера анализу с помощью хорошо известных и стандартных методик, таких как термогравиметрический анализ (TGA), дифференциальная сканирующая калориметрия (DSC) и рентгеновская порошковая дифракция (XRPD).
Сольваты могут быть стехиометрическими или нестехиометрическими сольватами.
Особенно предпочтительными сольватами являются гидраты, и примеры гидратов включают полугидраты, моногидраты и дигидраты.
Более подробное обсуждение сольватов и способов их получения и определения характеристик см. в публикации Bryn et al., Solid-State Chemistry of Drugs, Second Edition, published by SSCl, Inc of West Lafayette, IN, USA, 1999, ISBN 0-967-06710-3.
Помимо сольватированных форм, композиции, соединения или соли, определенные в любом из Вариантов реализации 1.1-1.211, могут быть представлены в форме ангидратов (безводных форм). При использовании в настоящем документе термин «ангидрат» обозначает твердую дисперсную форму, которая не содержит воды (и предпочтительно не содержит никаких других растворителей) в своей трехмерной структуре (например, кристаллической форме), хотя частицы соли или соединения могут иметь молекулы воды, адсорбированные на их внешней поверхности.
Пролекарства
Соединения, соли, композиции или атропизомеры, определенные в любом из Вариантов реализации 1.1-1.211, могут быть представлены в форме пролекарства.
Термин «пролекарства» подразумевает, например, любое соединение, которое в условиях in vivo превращается в биологически активную композицию или атропизомер, определенные в любом из Вариантов реализации 1.1-1.211.
Например, некоторые пролекарства представляют собой сложные эфиры активного соединения (например, физиологически приемлемые, метаболически быстро разлагаемые эфиры). В процессе метаболизма сложноэфирная группа (-C(=O)OR) расщепляется с высвобождением активного лекарственного средства. Такие эфиры могут быть образованы путем эстерификации, например, любой гидроксильной группы, присутствующей в исходном соединении, с предварительной защитой (в соответствующих случаях) всех других реакционноспособных групп, присутствующих в исходном соединении, и последующим удалением защиты, если это будет необходимо.
Также некоторые пролекарства активируются под действием ферментов с образованием активного соединения или соединения, которое в результате дальнейшей химической реакции дает активное соединение (как, например, в концепциях ADEPT, GDEPT, UDEPT и т.д.). Например, пролекарство может быть производным сахара или другим конъюгатом гликозида, или может быть сложноэфирным производным аминокислоты.
Способы получения соединений согласно настоящему изобретению
Композиции веществ и атропизомеры согласно настоящему изобретению могут быть получены путем разделения смесей атропизомеров методами хиральной хроматографии и, в частности, хиральной ВЭЖХ.
Смеси атропизомеров формулы (1А), (1В), (2А), (2В) и их различных подгрупп могут быть приготовлены согласно синтетическим способам, хорошо известным специалисту в данной области техники. Если не указано иное, R1-R7, Ar1, X, Y и Z являются такими, как это определено выше. В последующих параграфах, относящихся к приготовлению смесей атропизомеров, эти смеси обычно называют соединениями формулы (1).
Соединения формулы (1), в которых Z представляет собой пиррольное кольцо, могут быть получены путем реакции 1,4-дикарбонильного соединения формулы (10) саминоарильным соединением формулы (11), как это показано на Схеме 1.
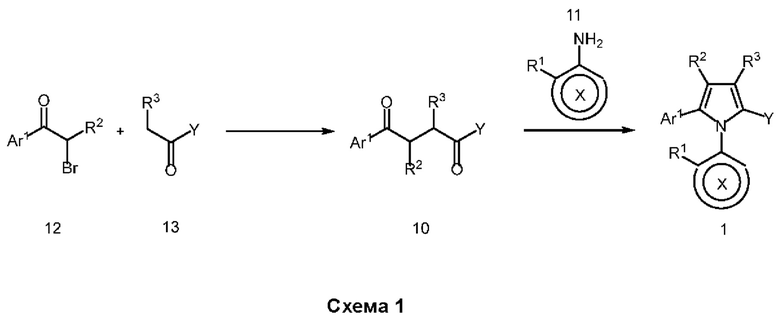
Исходным материалом для синтеза согласно пути, показанному на Схеме 1, является 1-арил-3-бромпропанон (12) с арилпропаноном (13), которые могут быть получены от коммерческих поставщиков.
1-Арил-2-бромэтанон (12) вступает в реакцию с арилпропаноном (13), в результате которой образуется 1,4-дикарбонильное соединение (10). Реакцию предпочтительно проводят в присутствии соли цинка (II) (например, хлорида цинка) в неполярном, апротонном растворителе (например, бензоле или толуоле). Предпочтительно также добавляют третичный спирт (например, трет-бутиловый) и третичный амин (например, триэтиламин). Реакцию можно проводить при комнатной температуре, например, в течение периода от 12 до 48 часов.
Затем 1,4-дикарбонильное соединение (10) может вступать в реакцию с аминоареном (11), в результате которой образуются тризамещенные пирролы согласно настоящему изобретению (1). Реакцию можно проводить в неполярном, апротонном растворителе (например, диоксане). Реакционную смесь можно подвергать нагреванию (например, до температур от 150 до 170°С) и/или микроволновому облучению. Реакцию можно проводить в течение от 1 до 12 часов, например, от 1 до 6 часов. Также в качестве катализатора можно добавлять сильную кислоту (например, п-толуолсульфоновую кислоту).
Альтернативно, соединения формулы (10), где R2 и R3 одновременно представляют собой водород, могут быть получены путем синтеза, показанным на Схеме 2.
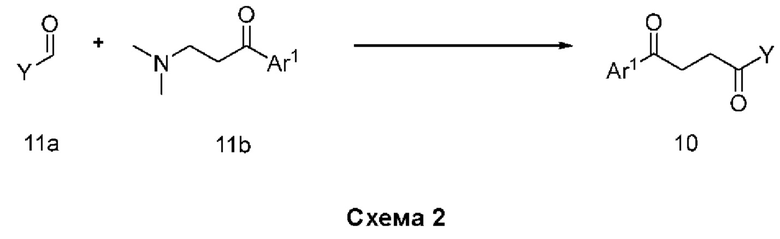
Исходный альдегид (11а) можно приготовить из соответствующей кислоты путем ее восстановления соответствующим восстановителем (например, NaBH4) с последующим окислением подходящим окислителем. Одним из таких примеров окислителя для приготовления альдегида без дальнейшего окисления до карбоновой кислоты является периодинан Десса-Мартина. Исходный амин (11b) может быть получен по реакции Манниха с гидрохлоридом диметиламина и формальдегидом в полярном, протонном растворителе (например, этаноле) в присутствии кислотного катализатора.
Соединения формулы (10) затем могут быть получены в результате реагирования соединений (11а) и (11b) в полярном апротонном растворителе (например, 1,2-диметоксиэтане) с подходящим катализатором. Одним из таких классов подходящих катализаторов являются соли тиазолия (например, бромид 3-этил-5-(2-гидроксиэтил)-4-метилтиазолия). Реакцию обычно проводят при повышенных температурах (например, при температурах от 80°С до 120°С) в течение от 1 до 24 часов, предпочтительнее в течение от 2 до 12 часов.
После получения соединения формулы (1) его можно преобразовать в другое соединение формулы (1) с помощью стандартных химических методик, хорошо известных в данной области техники. Примеры взаимопревращений функциональных групп рассмотрены, например, в публикациях March's Advanced Organic Chemistry, Michael В. Smith & Jerry March, 6th Edition, Wiley-Blackwell (ISBN: 0-471-72091-7), 2007 и Organic Syntheses, Volumes 1-9, John Wiley, edited by Jeremiah P. Freeman (ISBN: 0-471-12429), 1996.
Соединения формулы (1), в которых Y замещен заместителем R6, причем R6 представляет собой амидную группу формулы C(O)NHR8, где R8 представляет собой необязательно замещенную C1-8 углеводородную группу, могут быть получены согласно пути синтеза, показанному на Схеме 3.
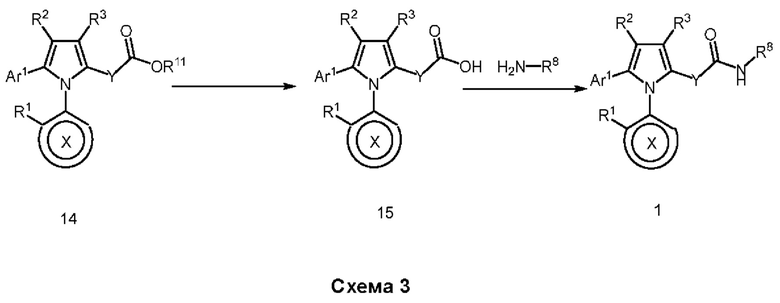
На Схеме 3 фрагмент Y представляет собой кольцо Y согласно определению в настоящем документе.
Соединение формулы (14) может быть получено в согласно пути синтеза, показанному на Схеме 1 выше, где R11 представляет собой C1-8 углеводородную группу или другую защитную группу для карбоновой кислоты. Сложный эфир (14) может быть гидролизован с получением карбоновой кислоты (15). Гидролиз предпочтительно проводят в смеси неполярного, апротонного растворителя (например, тетрагидрофурана) и полярного протонного растворителя (например, воды). Одной из таких подходящих систем растворителей является смесь тетрагидрофурана и воды в соотношении 1:1. В реакционную смесь добавляют сильное водорастворимое основание (например, гидроксид лития), после чего ее перемешивают при комнатной температуре в течение продолжительного периода времени, например, от 6 до 48 часов, обычно от 12 до 48 часов.
Затем кислотное соединение (15) можно ввести в реакцию с соответствующим амином (H2N-R8) в амид-образующих условиях, например, в присутствии реагента типа, обычно используемого с целью образования амидных связей, в результате чего получают соединение формулы (1), в котором R6 представляет собой амид. Примеры таких реагентов включают связующие агенты на основе карбодиимида, такие как 1,3-дициклогексилкарбодиимид (DCC) (Sheehan et al., J. Amer. Chem Soc. 1955, 77, 1067) и 1-этил-3-(3'-диметиламинопропил)-карбодиимид (обозначаемый в данном документе как EDC или EDCl) (Sheehan et al., J. Org. Chem., 7967, 26, 2525), которые обычно используют в комбинации с 1-гидрокси-7-азабензотриазолом (HOAt) (L.A. Carpino, J. Amer. Chem. Soc., 1993, 115, 4397) или 1-гидроксибензотриазолом (HOBt) (Konig et al, Chem. Ber., 103, 708, 2024-2034). Дополнительными примерами таких реагентов служат связующие агенты на основе урония, такие как гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU). Одним предпочтительным амидо-связующим агентом является HATU.
Реакцию связывания, как правило, проводят в неводном непротонном растворителе, таком как диметилформамид, при комнатной температуре в присутствии не конкурирующего основания, например, третичного амина, такого кактриэтиламин или N,N-диизопропилэтиламин.
Соединения формулы (15) в качестве альтернативы могут быть получены путем гидролиза соответствующего нитрила в соответствующих условиях для протекания гидролиза. Предпочтительно гидролиз проводят в присутствии сильного основания, например, гидроксида щелочного металла (например, гидроксида натрия) в полярном протонном растворителе или смеси полярных протонных растворителей. Одним таким примером подходящей системы растворителей служит смесь метанола и воды. Реакцию предпочтительно проводят при повышенной температуре в течение периода времени от 12 до 24 часов.
Соединения формулы (1), в котором Y замещен заместителем R6, где R6 представляет собой аминогруппу, имеющую формулу NHR9, могут быть получены согласно пути синтеза, показанному на Схеме 4.
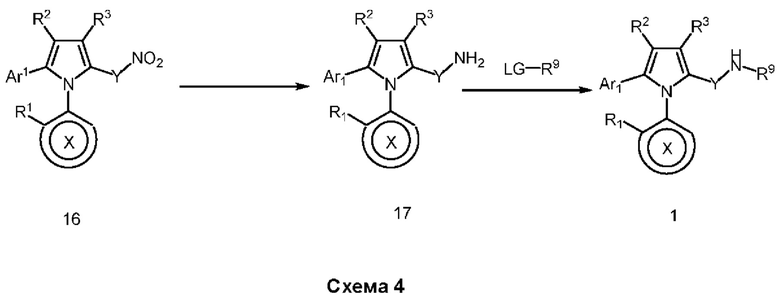
На Схеме 4 фрагмент Y представляет собой кольцо Y согласно определению в настоящем документе.
Соединение формулы (16) может быть получено путем синтеза, показанным на Схеме 1 выше. Соединение (16) затем можно восстановить до соединения (17) с помощью подходящего восстановителя (например, боргидрида натрия) и необязательно с каталитическими количествами соли меди (II) (например, ацетата меди (II)). Эту реакцию предпочтительно проводят в безводном, полярном, апротонном растворителе (например, метаноле).
Соединение (17) затем можно вводить в реакцию с соединением формулы LG-R9, где LG представляет собой подходящую уходящую группу (например, галоген, предпочтительнее хлор), a представляет собой необязательно замещенную неароматическую C1-8 углеводородную группу. Аминовое соединение (17) сначала обрабатывают подходящим основанием (например, гидридом натрия) в полярном апротонном растворителе (например, диметилформамиде), как правило, при комнатной температуре, а затем вводят в реакцию с соединением LG-R9, как правило, при повышенной температуре (например, в пределах от 60°С до 100°С).
Альтернативно, соединения формулы (1), где R6 представляет собой амид, в котором атом амидного азота связан с кольцом Y, могут быть получены, исходя из соединений формулы (17), способом, аналогичным показанному на Схеме 4, и карбоновых кислот или их активированных производных (таких как ацилхлориды или ангидриды кислот).
Альтернативно, соединения формулы (1), в котором R6 представляет собой амид формулы NHCOR10, где R10 представляет собой необязательно замещенную C1-8 углеводородную группу, могут быть получены, исходя из промежуточного соединения (17) в амид-образующих условиях, например, в присутствии реагента типа, обычно используемого с целью образования амидных связей, согласно Схеме 5.
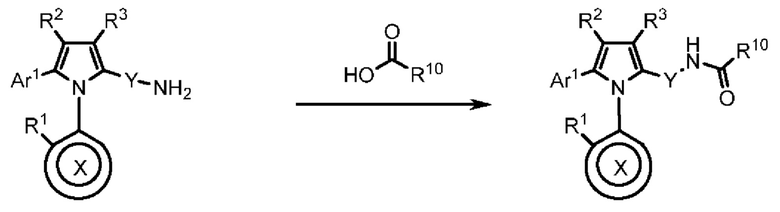

На Схеме 5 фрагмент Y представляет собой кольцо Y согласно определению в настоящем документе.
Примеры таких реагентов включают связующие агенты на основе карбодиимида, такие как 1,3-дициклогексилкарбодиимид (DCC) (Sheehan et al, J. Amer. Chem Soc. 1955, 77, 1067) и 1-этил-3-(3'-диметиламинопропил)-карбодиимид (обозначаемый в данном документе как EDC или EDCI) (Sheehan et al, J. Org. Chem., 1961, 26, 2525), которые обычно используют в комбинации с 1-гидрокси-7-азабензотриазолом (HOAt) (L.A. Carpino, J. Amer. Chem. Soc., 1993, 115, 4397) или 1-гидроксибензотриазолом (HOBt) (Konig et al, Chem. Ber., 103, 708, 2024-2034). Дополнительными примерами таких реагентов служат связующие агенты на основе урония, такие как гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU). Одним предпочтительным амидо-связующим агентом является HATU.
Реакцию связывания, как правило, проводят в неводном непротонном растворителе, таком как диметилформамид, при комнатной температуре в присутствии не конкурирующего основания, например, третичного амина, такого как триэтиламин или N,N-диизопропилэтиламин.
Соединения формулы (1), в которых Y замещен заместителем R6, причем R6 представляет собой любую группу, имеющую формулу OR12, где R12 представляет собой необязательно замещенную C1-8 углеводородную группу, могут быть получены согласно пути синтеза, показанному на Схеме 6.
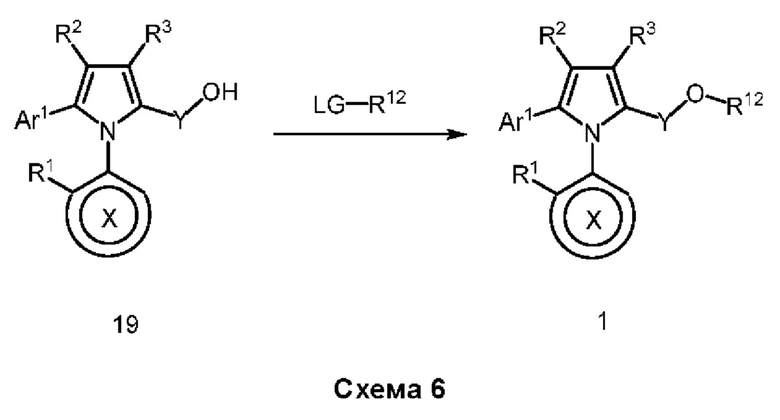
На Схеме 6 фрагмент Y представляет собой кольцо Y согласно определению в настоящем документе.
Соединение формулы (19) может быть получено путем синтеза, показанным на Схеме 1 выше. Соединение (19) затем можно вводить в реакцию с соединением формулы LG-R12, где LG представляет собой подходящую уходящую группу (например, галоген, предпочтительнее хлор), a R12 представляет собой необязательно замещенную неароматическую С1-8 углеводородную группу. Спиртовое соединение (19) сначала депротонируют подходящим основанием (например, гидридом натрия) в полярном апротонном растворителе (например, диметилформамиде). Эту реакцию можно проводить при комнатной температуре. Реакционную смесь затем обрабатывают соединением формулы LG-R12. Вторая стадия этой реакции может происходить при повышенных температурах, как правило, в пределах от 80°С до 100°С.
Соединения формулы (1), в которых R6 представляет собой Q1-Ra-Rb, a Q1 представляет собой метиленовую группу, могут быть получены согласно Схеме 7.
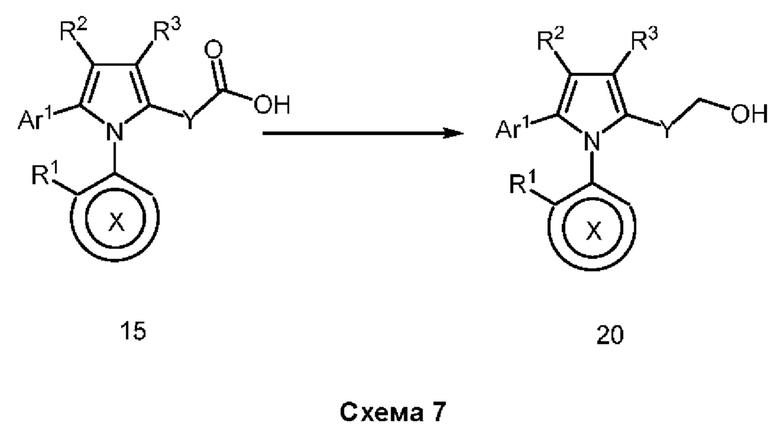
На Схеме 7 фрагмент Y представляет собой кольцо Y согласно определению в настоящем документе.
Соединение (15) (получаемое, как это описано на Схеме 3 выше) обрабатывают восстановителем (например, боргидридом натрия) в полярном апротонном растворителе, таком как тетрагидрофуран, в результате чего получают первичный спирт (20). Спирт (20) затем можно вводить в реакцию способом, описанным выше на Схеме 6, в результате чего могут быть получены дополнительные соединения формулы (1), в которых R6 представляет собой простой эфир.
Альтернативно, соединение (20) может претерпевать и другие обычные взаимопревращения функциональных групп, в результате чего могут быть получены дополнительные соединения формулы (1), например, посредством окисления до альдегида и восстановительного аминирования с получением амина. Амины, полученные этим способом, могут быть далее введены в реакцию с карбоновыми кислотами или производными кислот с получением амидных соединений формулы (1) посредством способа, описанного выше на Схеме 5.
Соединения формулы (1), в которых Z представляет собой 1,4,5-тризамещенный пиразол, могут быть получены по реакции арилгидразина (21) с α,β-ненасыщенным карбонильным соединением (22), как это показано на Схеме 8.
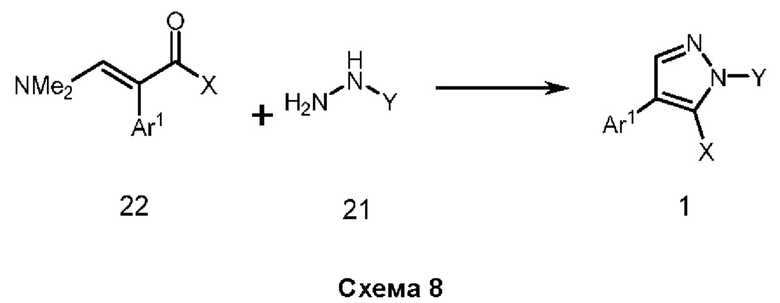
На Схеме 8 фрагменты X и Y представляют собой кольца X и Y соответственно, согласно определениям в настоящем документе.
Арилгидразин (21) и α,β-ненасыщенное карбонильное соединение (22) растворяют в подходящей полярной протонной системе растворителей (например, воды и метанола в соотношении 1:1) с подходящим основанием (например, карбонатом натрия). Смесь обычно перемешивают при комнатной или близкой к ней температуре (например, в течение примерно 15 минут), после чего добавляют слабую кислоту, например, уксусную кислоту. Полученную смесь затем нагревают (например, до температур от 100°С до 140°С) в течение продолжительного периода времени (например, от 6 до 12 часов), т.е. периода времени (например, 8 часов), достаточного для получения соединения формулы (1), в котором Z представляет собой 1,4,5-трехзамещенный пиразол.
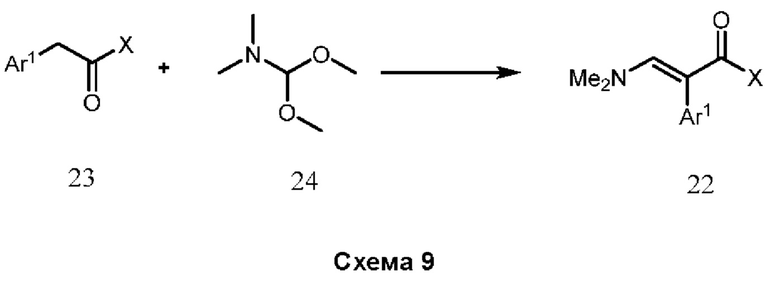
Исходное α,β-ненасыщенное карбонильное соединение (22) Схемы 8 может быть получено, исходя из соответствующего кетона (23) и диметилацеталя N,N-диметилформамида. К раствору кетона (23) добавляют раствор диметилацеталя N,N-диметилформамида в полярном апротонном растворителе, таком как ДМФА. Смесь, как правило, нагревают, например, до температур в пределах от 70°С до 110°С (например, приблизительно 90°С), в результате чего получают соединение (22). Соединение (23) может быть получено по реакции Гриньяра между Ar1CH2CHO и Br-Х с последующим окислением полученного спирта подходящим окислителем (например, периодинаном Десса-Мартина) в таком растворителе, как ДХМ, в результате чего получают кетон (23).
Как вариант, когда требуются альтернативные изомеры формулы (1), в которых Z представляет собой 3,4,5-трехзамещенный пиразол, они могут быть получены, как это описано на Схеме 10 ниже.
На Схеме 10 фрагменты X и Y представляют собой кольца X и Y соответственно, согласно определениям в настоящем документе.
Алкенилбромид (25) вводят во взаимодействие с диазосоединением (26) по реакции 1,3-диполярного циклоприсоединения путем смешивания двух этих соединений с сильным основанием (например, гидроксидом натрия) и нагревания (например, до температур приблизительно 70°С), в результате чего получают бромпиразол (27).
Бромпиразол (27) затем вводят в реакцию с бороновой кислотой, имеющей формулу Х-В(ОН)2 (где X представляет собой кольцо согласно определению в настоящем документе), в полярном растворителе, таком как диоксан, в присутствии палладиевого(0) катализатора, такого как бис(три-трет-бутилфосфин)палладий(0), и подходящего основания (такого как карбонат или фосфат цезия или калия) в условиях реакции Сузуки, в результате чего получают соединение формулы (1), в котором Z представляет собой пиразол или его защищенное производное. Бромпиразол (27) может находиться в защищенной форме. Например, в группе NH на пиразоле к атому азота может быть присоединена защитная группа, такая как группа Вое (трет-бутоксикарбонил), замещающая атом водорода. После реакции бороновой кислоты с пиразолом (27), может потребоваться стадия снятия защиты для получения соединения формулы (1). В случае защитной группы Вое она может быть удалена путем обработки кислотой, такой как хлористоводородная кислота.
Боронаты и бороновые кислоты широко доступны в продаже или могут быть получены, например, как описано в обзорной статье N. Miyaura and A. Suzuki, Chem. Rev. 1995, 95, 2457. Таким образом, боронаты могут быть получены путем проведения реакции соответствующего бромсодержащего соединения с алкиллитием, таким как бутиллитий, и последующего проведения реакции с эфиром борной кислоты. Полученное производное боронатного эфира при желании можно подвергнуть гидролизу с получением соответствующей бороновой кислоты.
Исходный материал (25) может быть получен путем обработки арилового альдегида тетрабромидом углерода и трифенилфосфином в растворителе, таком как ДХМ, при пониженной температуре (например, приблизительно при 0°С). Исходный материал (26) может быть получен, исходя из соответствующего арилальдегида, путем обработки гидразидом п-толуолсульфонила в полярном протонном растворителе, таком как метанол, и нагревания (например, до температуры примерно равной 60°С).
Соединения формулы (1), в которых Z представляет собой изоксазольную группу, могут быть получены согласно пути синтеза, показанного на Схеме 11.
На Схеме 11 фрагменты X и Y представляют собой кольца X и Y соответственно, согласно определениям в настоящем документе.
Промежуточное соединение (30) может быть получено путем взаимодействия алкина (28) с оксимом (29) путем их смешивания в полярном апротонном растворителе (таком как диэтиловый эфир) с основанием (таким как триэтиламин), например, при температурах вблизи комнатной температуры, в результате чего получают изоксазол (30). Затем изоксазол (30) можно бромировать подходящим бромирующим агентом, таким как N-бромсукцинимид в качестве источника брома, в результате чего получают бромоизоксазол (31). Реакцию обычно проводят в кислотном растворе (например, в уксусной кислоте) при повышенных температурах (например, в пределах от 90°С до 120°С).
Бромизоксазол (31) затем вводят в реакцию с бороновой кислотой, имеющей формулу Х-В(ОН)2 (где X представляет собой кольцо согласно определению в настоящем документе), в полярном растворителе, таком как диоксан, в присутствии палладиевого(0) катализатора, такого как бис(три-трет-бутилфосфин)палладий(0), и основания (например, карбоната или фосфата цезия или калия) в условиях реакции Сузуки, в результате чего получают соединение формулы (1), в котором Z представляет собой изоксазол или его защищенное производное. Бромоизоксазол (31) может находиться в защищенной форме. Например, в группе NH на группах Ar1 или Y к атому азота может быть присоединена защитная группа, такая как группа Boc (трет-бутоксикарбонил), замещающая атом водорода. После реакции бороновой кислоты с изоксазолом (31), может потребоваться стадия снятия защиты для получения соединения формулы (1). В случае защитной группы Вое она может быть удалена путем обработки кислотой, такой как хлористоводородная кислота.
Боронаты и бороновые кислоты широко доступны в продаже или могут быть получены, например, как описано в обзорной статье N. Miyaura and A. Suzuki, Chem. Rev. 1995, 95, 2457. Таким образом, боронаты могут быть получены путем проведения реакции соответствующего бромсодержащего соединения с алкиллитием, таким как бутиллитий, и последующего проведения реакции с эфиром борной кислоты. Полученное производное боронатного эфира при желании можно подвергнуть гидролизу с получением соответствующей бороновой кислоты.
Исходный материал (29) может быть получен, исходя из соответствующего арилового альдегида, в ходе двухстадийного процесса. Первая стадия заключается в обработке альдегида с помощью NH2OH и сильного основания (такого как гидроксид натрия) в системе полярных протонных растворителей (такой как смесь этанола и воды в соотношении 1:1), в результате чего получают арилоксим. Затем его можно хлорировать посредством смешивания с N-хлорсукцинимидом в диметилформамиде и перемешивания в течение 18 часов, в результате чего получают исходный материал (29).
Синтез соединений формулы (1) был проиллюстрирован выше на реакционных схемах для получения пирролов, изоксазолов и пиразолов. Однако легко понять, что аналогичные способы могут быть использованы и для получения соединений формулы (1), содержащих иные пятичленные гетероарильные кольца.
Конкретные пути синтеза для получения предпочтительного атропизомера, соединения (1), согласно настоящему изобретению показаны на Схеме 12 ниже.
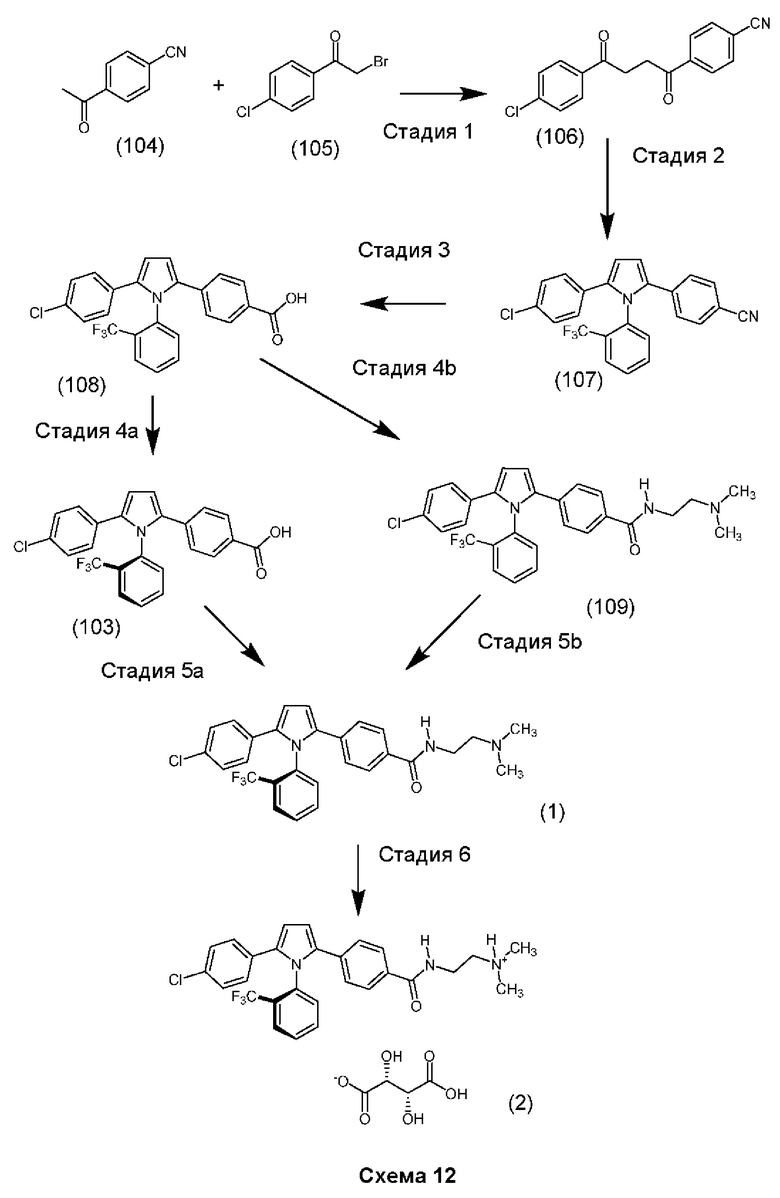
Исходными материалами для синтеза согласно пути, показанному на Схеме 1, являются 4-циано-ацетофенон (104) и 4-хлорфенацилбромид (105), которые оба легко доступны в продаже.
На Стадии 1 между собой реагируют 4-циано-ацетофенон (104) и 4-хлорфенацилбромид (105), в результате чего получают 4-[4-(4-хлорфенил)-4-оксо-бутаноил]бензонитрил (106). Реакцию обычно проводят в присутствии соли цинка (II) (например, хлорида цинка) в подходящем растворителе, например, смеси неполярного (например, углеводородного) растворителя, такого как бензол или толуол, и третичного спирта (например, трет-бутанола) в присутствии третичного амина, такого как триэтиламин. Реакцию можно проводить при комнатной или близкой к ней температуре, например, в течение периода от 12 до 60 часов.
На Стадии 2 полученный 4-[4-(4-хлорфенил)-4-оксо-бутаноил]бензонитрил (106) вводят в реакцию с 2-трифторметиланилином, в результате чего получают 4-(5-(4-хлорфенил)-1 -(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензонитрил (107). Реакцию, как правило, проводят в присутствии кислотного катализатора, такого как п-толуолсульфоновая кислота, в подходящем высококипящем растворителе (например, диоксане) при повышенной температуре (например, в диапазоне от 130 до 170°С) и/или микроволновом облучении. Реакцию можно проводить в течение от 1 до 12 часов, например, от 1 до 6 часов.
На Стадии 3 полученный 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензонитрил (107) подвергают щелочному гидролизу, в результате чего получают 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензойную кислоту (108). Реакцию гидролиза обычно проводят в водном растворителе, который может содержать спирт, такой как метанол, в присутствии гидроксида щелочного металла, такого как гидроксид натрия (обычно в избытке), и, как правило, при нагревании, например, до температур в пределах от 60-80°С или в течение периода времени вплоть до примерно 20 часов или более. После завершения гидролиза кислоту (8) обычно выделяют путем охлаждения и подкисления реакционной смеси.
После Стадии 3 можно использовать один из двух возможных путей к атропизомеру (1). В одном варианте, включающем Стадии 4b, 5b и 6, полученную 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензойную кислоту (108) подвергают реакции с N,N-диметилэтилендиамином в амид-образующих условиях, в результате чего получают рацемическую смесь атропизомеров 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)-N-(2-(диметиламино)этил)бензамида (109), которую затем разделяют на отдельные атропизомеры методами хирального разделения, в результате чего получают атропизомер (1).
В другом варианте рацемическую 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензойную кислоту (108) подвергают хиральному разделению, в результате чего получают атропизомер (103), который затем подвергают реакции с N,N-диметилэтилендиамином в амид-образующих условиях, в результате чего получают атропизомер (1).
Карбоновые кислоты (103) и (108) реагируют с N,N-диметилэтилендиамином в амид-образующих условиях в присутствии амидо-связующего реагента. Примеры таких амидо-связующих реагентов включают связующие реагенты на основе карбодиимида, такие как 1,3-дициклогексилкарбодиимид (DCC) (Sheehan et al, J. Amer. Chem Soc 1955, 77, 1067) и 1-этил-3-(3'-диметиламинопропил)-карбодиимид (обозначаемый в данном документе как EDC или EDCI) (Sheehan et al, J. Org. Chem., 7967, 26, 2525), которые обычно используют в комбинации с 1-гидрокси-7-азабензотриазолом (HOAt) (L.А. Carpino, J. Amer. Chem. Soc., 1993, 115, 4397) или 1-гидроксибензотриазолом (HOBt) (Konig et al, Chem. Ber., 103, 708, 2024-2034), связующие реагенты на основе урония, такие как гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) и ангидрид пропанофосфоновой кислоты (Т3Р) (см. A. Garcia, Synlett, 2007, №8, рр 1328-1329). Конкретными амидосвязующими реагентами для использования на Стадиях 5а и 5b процесса являются HATU и Т3Р.
Реакцию амидного связывания, как правило, проводят в неводном, полярном, непротонном растворителе, таком как тетрагидрофуран или диметилформамид, или их смеси при комнатной или близкой к ней температуре (например, при 18-30°С) в присутствии не конкурирующего основания, например, третичного амина, такого как триэтиламин или N,N-диизопропилэтиламин.
Определенные аспекты описанных выше процессов представляют собой дополнительные варианты реализации настоящего изобретения (Варианты реализации 2.1-2.8). Соответственно, в настоящем изобретении предложены:
2.1 Способ получения композиции веществ или одинарного атропизомера, определенных в любом из Вариантов реализации 1.1-1.211, включающий хиральное разделение смеси атропизомеров соединения формулы (0):
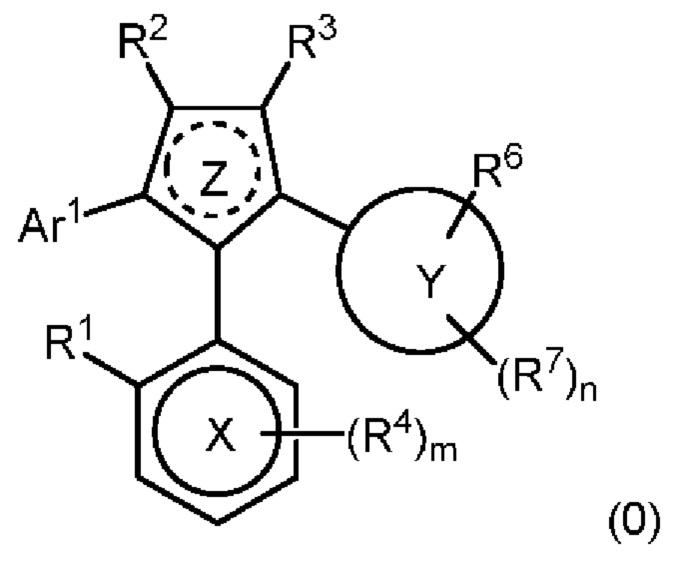
где кольцо X, кольцо Y, кольцо Z, Ar1, m, n и R1-R7 являются такими, как это определено в любом из Вариантов реализации 1.1-1.211.
2.2 Способ согласно Варианту реализации 2.1, отличающийся тем, что смесь атропизомеров соединения формулы (0) представляет собой рацемическую смесь.
2.3 Способ согласно Варианту реализации 2.1 или Варианту реализации 2.2, отличающийся тем, что хиральное разделение проводят путем:
(i) пропускания смеси атропизомеров через хиральную хроматографическую колонку, например, хиральную ВЭЖХ колонку, или
(ii) взаимодействия смеси атропизомеров соединения формулы (0) с хиральной кислотой с образованием солей обоих атропизомеров в смеси, разделения солей и их разложения, в результате чего получают соответствующие свободные основания каждого из атропизомеров.
2.4 Способ получения атропизомера (1), определенного в настоящем документе, включающий реакцию соединения формулы (103) с N,N-диметилэтилендиамином в амид-образующих условиях.
2.5 Способ согласно Варианту реализации 2.4, отличающийся тем, что амид-образующие условия включают присутствие амидо-связующего реагента, например, амидо-связующего агента согласно описанию в настоящем документе.
2.6 Способ согласно Варианту реализации 2.5, отличающийся тем, что амидо-связующий реагент представляет собой ангидрид пропанофосфоновой кислоты (Т3Р).
2.7 Способ получения соединения формулы (103) (см. Схему 12), включающий хиральное выделение соединения формулы (103) из смеси атропизомеров формулы (108), например, методами хиральной хроматографии или получения соли с хиральным основанием и перекристаллизации полученной хиральной соли.
2.8 Атропизомерное соединение, имеющее формулу (103), или его соль (например, соль с металлом, такая как соль со щелочным или щелочноземельным металлом или соль с аммонием или органическим амином).
Атропизомеры и композиции согласно настоящему изобретению могут быть предложены в солевых или в несолевых (например, в виде свободного основания) формах.
Кислотно-аддитивные соли основных атропизомеров согласно настоящему изобретению могут быть получены путем приведения атропизомера в виде свободного основания в контакт с подходящей солеобразующей кислотой в подходящем растворителе или смеси растворителей, как это описано в другом месте данного документа, и последующего выделения желаемой соли из растворителя или смеси растворителей.
Конкретной солью согласно настоящему изобретению является соль (+)-L-винной кислоты формулы (2), определенная в любом из Вариантов реализации 1.194-1.211.
Соль (+)-L-винной кислоты согласно настоящему изобретению может быть получена из атропизомера формулы (1) путем реакции с винной кислотой в растворителе или смеси растворителей с последующим выделением полученной соли винной кислоты (тартрата) из растворителя или смеси растворителей.
В одном из вариантов реализации изобретения (Вариант реализации 2.9) атропизомер формулы (1) можно растворить или суспендировать в одном растворителе с получением первой смеси, а (+)-L-винную кислоту растворить или суспендировать в том же или другом растворителе с получением второй смеси, после чего первую и вторую смеси объединяют и оставляют (например, при перемешивании) на определенный период времени, чтобы дать возможность образоваться соли, после чего выделяют соль(+)-L-винной кислоты.
При объединении первой и второй смесей предпочтительно, чтобы мольные количества атропизомера формулы (1) и(+)-L-винной кислоты были приблизительно эквивалентными, т.е. чтобы между атропизомером формулы (1) и (+)-L-винной кислотой предпочтительно было мольное соотношение 1:1.
Соль (+)-L-винной кислоты может быть выделена из объединенной смеси фильтрованием (в случае образования осадка) или путем выпаривания растворителей.
Таким образом, если в комбинированной смеси присутствует более одного растворителя, то могут быть выбраны различные растворители, чтобы действовать как сорастворители или как антирастворители.
Растворитель или смесь растворителей могут быть выбраны таким образом, чтобы они удерживали соль(+)-L-винной кислоты, хотя бы частично, в растворе при нагревании, но затем при охлаждении растворителя или смеси растворителей соль выпадала в осадок.
Растворитель, используемый для получения первой смеси (смеси, содержащей атропизомер формулы (1)), можно выбрать, например, из алифатических кетонов, алифатических сложных эфиров алифатических кислот, неароматических циклических эфиров и алифатических спиртов.
Конкретным примером алифатического кетона является ацетон.
Примеры алифатических сложных эфиров алифатических кислот включают С2-4 алкиловые эфиры уксусной кислоты, конкретным примером которых служит изопропилацетат.
Примеры неароматических циклических простых эфиров включают диоксан, 2-метилтетрагидрофуран и тетрагидрофуран, а конкретным примером служит 2-метилтетрагидрофуран.
Примерами алифатических спиртов служат С2-4 алифатические спирты и более конкретно С3-4 алканолы, такие как изопропиловый спирт и бутанол.
Растворитель, используемый для получения второй смеси (смеси, содержащей (+)-L-винную кислоту), может быть выбран из, например, воды, неароматических циклических эфиров и алифатических спиртов.
Конкретным примером растворителя алифатического спирта для второй смеси является этанол.
Конкретным примером неароматического циклического эфирного растворителя для второй смеси является тетрагидрофуран (ТГФ).
Другим конкретным примером растворителя для применения при получении второй смеси является вода.
Соль (+)-L-винной кислоты атропизомера формулы (1) может существовать в нескольких кристаллических формах, а именно в форме согласно Дифрактограмме А (являющейся сольватом) и в форме согласно Дифрактограмме В (являющейся ангидратом). Характеризующие подробности о различных кристаллических формах представлены в другом месте настоящего документа. Различные кристаллические формы могут быть получены путем подбора растворителей и теплового режима, используемого при получении солей.
Согласно одному способу (Вариант реализации 2.10) получения соли (+)-L-винной кислоты и атропизомера формулы (1), соответствующей Дифрактограмме А, раствор атропизомера в ацетоне смешивают с раствором (+)-L-винной кислоты в этаноле при температуре в диапазоне от 20°С до 30°С (например, приблизительно 25°С), полученную смесь перемешивают или как-либо иным образом взбалтывают в течение времени (например, 12-24 часов), достаточного для образования соли, после чего соль отделяют фильтрованием.
Согласно другому способу (Вариант реализации 2.11) получения соли (+)-L-винной кислоты и атропизомера формулы (1), соответствующей Дифрактограмме А, раствор атропизомера в изопропиловом спирте смешивают с раствором(+)-L-винной кислоты в этаноле при температуре от 35°С до 45°С (например, приблизительно 40°С), полученную смесь охлаждают до температур от 20°С до 30°С (например, приблизительно 25°С) в течение приблизительно 1-3 часов, после чего соль отделяют фильтрованием.
Согласно другому способу (Вариант реализации 2.12) получения соли (+)-L-винной кислоты и атропизомера формулы (1), соответствующей Дифрактограмме А, раствор атропизомера в 2-метилтетрагидрофуране смешивают с раствором (+)-L-винной кислоты в этаноле при температуре в диапазоне от 20°С до 30°С (например, приблизительно 25°С), полученную смесь перемешивают или как-либо иным образом взбалтывают в течение времени (например, 12-24 часов), достаточного для образования соли, после чего соль отделяют фильтрованием.
Согласно одному способу (Вариант реализации 2.13) получения соли (+)-L-винной кислоты и атропизомера формулы (1), соответствующей Дифрактограмме В, раствор атропизомера в изопропилацетате смешивают с раствором (+)-L-Винной кислоты в этаноле при температуре от 35°С до 45°С (например, приблизительно 40°С), полученную смесь охлаждают до температур от 20°С до 30°С (например, приблизительно 25°С) в течение приблизительно 1-3 часов, после чего соль отделяют фильтрованием.
Согласно другому способу (Вариант реализации 2.14) получения соли (+)-L-винной кислоты атропизомера формулы (1), соответствующей Дифрактограмме В, раствор атропизомера в изопропилацетате при температуре в диапазоне от 35°С до 45°С (например, приблизительно 40°С) смешивают (по частям или целиком в единой загрузке) с раствором (+)-L-винной кислоты в ТГФ, добавляют один или более затравочных кристаллов, соответствующих Дифрактограмме В, для инициирования осаждения осадка, смесь охлаждают до температур в пределах от 20°С до 30°С (например, приблизительно 25°С) и перемешивают или взбалтывают в течение периода времени (например, от 12 до 24 часов, в частности, приблизительно 20 часов), достаточного для того, чтобы дать осадку созреть до такого состояния, при котором его можно будет отделить фильтрованием.
Согласно другому способу (Вариант реализации 2.15) получения соли (+)-L-винной кислоты атропизомера формулы (1), соответствующей Дифрактограмме В, раствор атропизомера в бутаноле при высокой температуре в диапазоне от 70°С до 85°С (например, приблизительно 80°С) смешивают (по частям или целиком в единой загрузке) с раствором (+)-L-винной кислоты в воде, полученную смесь охлаждают до промежуточной температуры в диапазоне от 65°С до 70°С, после чего добавляют один или более затравочных кристаллов, соответствующих Дифрактограмме В и охлаждают смесь до низкой температуры в диапазоне от 3 до 10°С в течение периода времени в пределах от 8 до 15 часов, а затем перемешивают или как-либо иным образом взбалтывают полученную смесь при пониженной температуре в течение еще одного периода времени в пределах от 2 до 8 часов (например, приблизительно 6 часов), после чего отфильтровывают полученную таким образом соль, соответствующую Дифрактограмме В.
Защитные группы
Во многих описанных выше реакциях может потребоваться защита одной или более групп с целью исключения протекания реакции в нежелательных положениях в молекуле. Примеры защитных групп и способов защиты и снятия защиты функциональных групп можно найти в публикации Protective Groups in Organic Synthesis (Т. Green and P. Wuts; 3rd Edition; John Wiley and Sons, 1999).
Гидроксигруппу можно защитить, например, связав ее в виде простого эфира (-OR) или сложного эфира (-OC(=O)R), например, в виде трет-бутилового эфира, тетрагидропиранилового (ТНР) эфира, бензилового, бензгидрилового (дифенилметилового) или тритилового (трифенилметилового) эфира, триметилсилилового или трет-бутилдиметилсилилового эфира или ацетилового эфира (-ОС(=O)СН3, -ОАс).
Альдегидную или кетоновую группу можно защитить, например, связав ее в виде ацеталя (R-CH(OR)2) или кеталя (R2C(OR)2), соответственно, в котором карбонильная группа (>С=O) превращается в диэфир (>C(OR)2), путем реакции, например, с первичным спиртом. Альдегидную или кетоновую группу затем легко регенерировать путем гидролиза в большом избытке воды в присутствии кислоты.
Аминогруппу можно защитить, например, связав ее в виде амида (-NRCO-R) или уретана (-NRCO-OR), например, в виде метиламида (-NHCO-СН3), бензилоксиамида (-NHCO-OCH2C6H5, -NH-Cbz или NH-Z), в виде трет-бутокси амида (-NHCO-ОС(СН3)3, -NH-Boc), 2-бифенил-2-пропоксиамида (-NHCO-ОС(СН3)2С6Н4С6Н5, -NH-Bpoc), в виде 9-фторенилметоксиамида (-NH-Fmoc), в виде 6-нитровератрилоксиамида (-NH-Nvoc), в виде 2-триметилсилилэтилоксиамида (-NH-Teoc), в виде 2,2,2-трихлорэтилоксиамида (-NH-Troc), в виде аллилоксиамида (-NH-Alloc) или в виде 2(-фенилсульфонил)этилоксиамида (-NH-Psec).
Например, на Схеме 1 выше, когда фрагмент R3 в амине H2N-Y-R3 содержит вторую аминогруппу, такую как циклическая аминогруппа (например, пиперидиновая или пирролидиновая группа), втору аминогруппу можно защитить посредством защитной группы, определенной выше, при этом одна предпочтительная группа представляет собой трет-бутилоксикарбонильную (Boc) группу. Если последующая модификация второй аминогруппы не требуется, защитную группу можно провести через последовательность реакций, позволяющую получить N-защищенную форму соединения формулы (1), которую затем можно будет освободить от защиты стандартными способами (например, обработкой кислотой в случае группы Boc), в результате чего получают соединение формулы (1).
Другие защитные группы для таких аминов, как циклические амины и гетероциклические N-H группы, включают толуолсульфониловые (тозиловые) и метансульфониловые (мезиловые) группы, бензиловые группы, такие как пара-метоксибензиловая (ПМБ) группа и тетрагидропираниловая (ТГП) группа.
Группу карбоновой кислоты можно защитить, связав ее в виде сложного эфира, например, С1-7 алкилового эфира (например, метилового эфира, трет-бутилового эфира), С1-7 галогеналкилового эфира (например, С1-7 тригалогеналкилового эфира), триС1-7 алкилсилил-С1-7 алкилового эфира или С5-20 арил-С1-7 алкилового эфира (например, бензилового эфира, нитробензилового эфира), или в виде амида, например, в виде метиламида. Тиольную группу можно защитить, например, связав ее в виде тиоэфира (-SR), например, в виде бензилтиоэфира, ацетамидометилового эфира (-S-CH2NHC(=O)CH3).
Выделение и очистка соединений согласно настоящему изобретению
Соединения, полученные согласно вышеуказанным путям синтеза, могут быть выделены и частично очищены стандартными методами, хорошо известными специалисту в данной области техники, в результате чего получают смеси атропизомеров. Одним из способов, особенно полезных при очистке соединений, является препаративная жидкостная хроматография, использующая масс-спектрометрию в качестве средства детектирования очищенных соединений, выходящих из хроматографической колонки.
Препаративная ЖХ-МС представляет собой стандартный и эффективный способ, используемый для очистки малых органических молекул, таких как соединения, описанные в настоящем документе. Методы жидкостной хроматографии (ЖХ) и масс-спектрометрии (МС) можно варьировать с целью обеспечения наилучшего разделения неочищенных продуктов и более надежного детектирования образцов с помощью МС. Оптимизация метода препаративной градиентной ЖХ будет включать различные вариации с колонками, летучими элюентами и модификаторами, а также варьирование градиентов. Способы оптимизации методов препаративной ЖХ-МС и последующего их применения для очистки химических соединений хорошо известны в данной области техники. Такие способы описаны в публикациях Rosentreter U, Huber U.; Optimal fraction collecting in preparedative LC/MS; J Comb Chem.; 2004; 6(2), 159-64 и Leister W, Strauss K, Wisnoski D, Zhao Z, Lindsley C., Development of a custom high-throughputative preparedative liquid chromatography/mass spectrometer platform for the preparationative cleaning and analytical analysis of compound libraries; J Comb Cbem.; 2003; 5(3); 322-9.
Пример такой системы для очистки химических соединений методом препаративной ЖХ-МС описан ниже в разделе «Примеры» настоящей заявки (под заголовком «Система ЖХ-МС для очистки по показателям молекулярной массы»). Однако следует понимать, что могут быть использованы и другие системы и методы, альтернативные описанным. В частности, вместо описанных в данном документе методов препаративной ЖХ на основе обращенных фаз можно использовать методы с нормальными фазами. В большинстве препаративных ЖХ-МС систем используют обращенно-фазовую ЖХ и летучие кислотные модификаторы, поскольку данный метод очень эффективен для очистки малых молекул, а элюенты совместимы с системами масс-спектрометрии с электрораспылением положительных ионов. Для очистки соединений альтернативно можно использовать другие хроматографические решения, например, ЖХ с нормальными фазами, альтернативно забуференную подвижную фазу, основные модификаторы и т.д., как это описано в приведенных ниже аналитических методах.
После того, как смеси атропизомеров выделены и очищены до приемлемой степени, эти смеси можно затем подвергнуть процедурам разделения для выделения индивидуальных атропизомеров. Так, например, для выделения индивидуальных атропизомеров можно использовать хиральную хроматографию. Значения времени удерживания атропизомеров в процедурах хиральной хроматографии обеспечивают средства для дифференциации и характеристики индивидуальных атропизомеров, ЯМР и МС характеристики которых, как правило, одинаковы.
Колонки для хиральной хроматографии, которые можно использовать для выделения индивидуальных атропизомеров, содержат иммобилизованную хиральную неподвижную фазу (CSF), которая может быть получена, например, на основе функционализированной амилозы или целлюлозы. Примерами таких CSF являются амилоза и целлюлозы, функционализированные хлорзамещенными и/или метилзамещенными фенилкарбаматами. Конкретными примерами хиральных колонок, которые можно использовать для выделения индивидуальных атропизомеров согласно настоящему изобретению, являются колонки «CHIRALPAK IG», предлагаемые компанией Daicel Corporation.
Подвижные фазы, которые обычно могут быть использованы в сочетании с вышеуказанными хиральными колонками, включают смеси (А) жидких алканов, таких как н-гептан, содержащие небольшое количество (например, до 1% (об./об.), как правило, около 0,1% (об./об.)) алкиламинового основания, такого как диэтиламин, и (В) спиртов и их смесей, таких как смеси изопропилового спирта и метанола (например, смесь изопропанола и МеОН в соотношении 70:30). Например, подвижная фаза может содержать смесь А:В в диапазоне соотношений от 80:20 до 95:5, например, от примерно 85:15 до примерно 90:10. Подвижные фазы можно использовать в изократических или градиентных методах элюирования, но в одном варианте реализации изобретения их применяют в методе изократического элюирования.
Атропизомеры согласно настоящему изобретению также могут быть разделены методом хиральной ВЭЖХ в условиях сверхкритической жидкостной хроматографии (SFC). В сверхкритической жидкостной хроматографии подвижная фаза содержит сверхкритическую жидкость, такую как диоксид углерода, часто с сорастворителем, таким как спирт или смесь спиртов, например, метанола, этанола и изопропанола.
Упомянутые выше колонки CHIRALPAK IG можно использовать в хроматографических процедурах SFC с использованием смесей диоксида углерода, метанола и изопропанола в качестве подвижной фазы.
Другие комбинации хиральных колонок и сорастворителей для использования в SFC включают следующее:
Lux Cellulose 4 (МеОН, EtOH);
Lux Cellulose 2 (МеОН);
Lux Amylose 1 (МеОН, EtOH) и
YMC Amylose-SA (МеОН, EtOH)
Семейство Lux хиральных колонок доступно от компании Phenomenex, Inc.
Колонки YMC Amylose-SA доступны от компании YMC America, Inc.
Биологические свойства и терапевтическое применение
Свидетельства, приведенные в приведенных ниже примерах, указывают на то, что атропизомеры согласно настоящему изобретению, как это определено в настоящем документе, являются ингибиторами Polo-Box доменов (PBD) киназ PLK1 и PLK4, но не ингибируют каталитические домены киназ PLK1 и PLK4. Поскольку домены PBD присущи только киназам PLK, атропизомеры должны проявлять гораздо большую селективность (и, следовательно, меньше нежелательных побочных эффектов из-за нецелевого ингибирования киназ), чем соединения, являющиеся АТР-конкурентными ингибиторами киназ. Например, результаты, полученные в исследовании, описанном в Примере 11F ниже, в котором атропизомер формулы (1) прошел испытания в отношении ряда из девяносто семи киназ и продемонстрировал незначительную активность к другим (не-PLK) киназам, подтверждают, что атропизомер формулы (1) обладает высокой степенью селективности в отношении PLK1-PBD и PLK4-PBD по сравнению с другими структурно и функционально сходными киназами. Исходя из этих свидетельств, можно полагать, что и другие атропизомеры согласно настоящему изобретению, в частности, те, которые имеют такую же R-конфигурацию, что и атропизомер (1), должны обладать аналогичными преимуществами.
Еще одним преимуществом ингибирования домена PBD, а не каталитического домена, является то, что это может обеспечить пониженную тенденцию к возникновению лекарственной резистентности по сравнению с ингибиторами PLK1, подавляющими каталитический домен.
Активность атропизомеров согласно настоящему изобретению как ингибиторов домена PBD киназы PLK1 можно продемонстрировать с использованием метода флуоресцентного поляризационного (FP) анализа, описанного в публикации Narvaez et al., Cell Chemical Bioiogy, 24, 1017-1028, 2017, см. стр. 1018 и стр. 1026 (Подробное описание метода).
Считается, что соединения согласно настоящему изобретению могут эффективно использовать слабости в клеточных путях, возникающие в результате действия постоянно активирующих мутантов KRAS, и, следовательно, композиции или атропизомеры согласно настоящему изобретению могут быть пригодны для лечения заболеваний и состояний, опосредуемых модуляцией KRAS.
Мутация KRAS, возникающая в результате однонуклеотидного замещения, была ассоциирована с различными формами рака. В частности, мутации KRAS установлены во многих случаях лейкемии, рака толстой кишки, рака поджелудочной железы и рака легких.
Первичный скрининг на противораковую активность, в котором используют линию клеток рака (U87MG, головной мозг человека (глиобластома-астроцитома)), описан в Примере 11А ниже.
Кроме того, считается, что соединения согласно настоящему изобретению могут быть пригодны для лечения видов рака, характеризующихся дефицитом или мутацией р53 в гене ТР53. Считается, что PLK1 ингибирует р53 в раковых клетках. Следовательно, при лечении ингибиторами PLK1, фактор р53 в опухолевых клетках должен активироваться и, следовательно, начать индуцировать апоптоз.
Считается, что активность композиции или атропизомеров в отношении KRAS-мутантных и р53-дефицитных видов рака обуславливается, по меньшей мере частично, ингибированием киназы PLK1 и, в частности, С-концевого Polo-Box домена (PBD) киназы PLK1. Известно, что KRAS зависим от взаимодействия с PLK1.
Соединения согласно настоящему изобретению, которые ингибируют только домен PBD, а не N-концевой каталитический домен PLK1, являются более перспективными в том, что они селективны в отношении PLK1-PBD на фоне других структурно и функционально подобных киназ, в отношении которых они проявляют незначительную ингибиторную активность (см. Пример Е ниже).
Композиции или атропизомеры согласно настоящему изобретению индуцируют остановку митоза неконгрессированными хромосомами, т.е. проявляют свойство, которое, как полагают, возникает из активности по ингибированию PLK1-PBD и PLK4-PBD, свойственной указанным композициям или атропизомерам (см. Пример 11С ниже).
Атропизомеры индуцируют остановку митоза с фенотипом многополюсного веретена и вызывают амплификацию центриолей, хорошо описанного фенотипа ингибирования PLK4 (Lei 2018, Cell Death & Disease 9, 1066; Kawakami, PNAS 2018, 115(8) 1913-18). Считается, что эти фенотипы возникают в результате активности атропизомеров по ингибированию PLK4-PBD.
Еще одним преимуществом ингибирования домена PBD, а не каталитического домена, является то, что это может обеспечить пониженную тенденцию к возникновению лекарственной резистентности по сравнению с ингибиторами PLK1, подавляющими каталитический домен.
Активность соединений согласно настоящему изобретению как ингибиторов домена PBD киназы PLK1 можно продемонстрировать с использованием метода флуоресцентного поляризационного (FP) анализа, описанного в публикации Narvaez et al., Cell Chemical Biology, 24, 1017-1028, 2017, см. стр. 1018 и стр. 1026 (Подробное описание метода).
Соединения согласно настоящему изобретению обладают хорошей пероральной биодоступностью (см. Пример 11G ниже) и хорошей экспозицией в головном мозге при пероральном введении (см. Пример 11G ниже). Соответственно, композиция или атропизомеры согласно настоящему изобретению должны быть пригодными для лечения видов рака головного мозга, таких как глиомы и глиобластомы.
В дополнительных вариантах реализации (Вариантах реализации 3.1-3.27) изобретения предложены:
3.1. Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения в качестве ингибитора PLK1-PBD.
3.2. Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения в качестве ингибитора PLK4-PBD.
3.3. Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения в качестве ингибитора PLK1-PBD и PLK4-PBD.
3.4 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания, необязательно в комбинации с другим терапевтическим агентом или средством лечения (например, противораковым агентом или терапией), где раковое заболевание выбрано из опухолей эпителиального происхождения (аденомы и карциномы различных типов, включая аденокарциномы, плоскоклеточные карциномы, переход но-кпеточные карциномы и другие карциномы), таких как карциномы мочевого пузыря и мочевыводящих путей, молочной железы, желудочно-кишечного тракта (в том числе пищевода, желудка (гастрическая), тонкого кишечника, толстой кишки, прямой кишки и ануса), печени (гепатоцеллюлярная карцинома), желчного пузыря и желчевыводящей системы, экзокринной части поджелудочной железы, почек, легких (например, аденокарциномы, мелкоклеточные карциномы легкого, немелкоклеточные карциномы легких, бронхоальвеолярные карциномы и мезотелиомы), головы и шеи (например, раковые заболевания языка, щечной полости, гортани, глотки, носоглотки, миндалин, слюнных желез, носовой полости и придаточных пазух), яичников, фаллопиевых труб, брюшины, вагины, вульвы, пениса, шейки матки, миометрия, эндометрия, щитовидной железы (например, фолликулярная карцинома щитовидной железы), надпочечников, предстательной железы, кожи и придатков (например, меланома, базальноклеточная карцинома, плоскокпеточная карцинома, кератоантома, диспластический невус); гематологические злокачественные новообразования (т.е. лейкозы, лимфомы) и предраковые гематологические расстройства и нарушения пограничной злокачественности, включая гематологические злокачественные новообразования и связанные с ними состояния лимфоидного происхождения (например, острый лимфоцитарный лейкоз [ALL], хронический лимфоцитарный лейкоз [CLL], В-клеточные лимфомы, такие как диффузная В-крупнокпеточная лимфома [DLBCL], фолликулярная лимфома, лимфома Беркитта, мантийноклеточная лимфома, Т-клеточные лимфомы и лейкозы, лимфомы натуральных киллерных клеток [NK], лимфомы Ходжкина, волосатокпеточный лейкоз, моноклональная гаммопатия неопределенной значимости, плазмоцитома, множественная миелома и посттрансплантационные лимфопролиферативные расстройства), гематологические злокачественные новообразования и связанные с ними состояния миелоидного происхождения (например, острый миелоидный лейкоз [AML], хронический миелоидный лейкоз [CML], хронический миеломоноцитарный лейкоз [CMML], гиперэозинофильный синдром, миелопролиферативные нарушения, такие как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативный синдром, миелодиспластический синдром и промиелоцитарный лейкоз); опухоли мезенхимального происхождения, например, саркомы мягких тканей, костей или хрящей, такие как остеосаркомы, фибросаркомы, хондросаркомы, рабдомиосаркомы, лейомиосаркомы, липосаркомы, ангиосаркомы, саркомы Капоши, саркомы Юинга, синовиальные саркомы, эпителиоидные саркомы, желудочно-кишечные стромальные опухоли, доброкачественные и злокачественные гистиоцитомы и выбухающие дерматофибросаркомы; опухоли центральной или периферической нервной системы (например, астроцитомы, глиомы и глиобластомы, менингиомы, эпидендимомы, пинеальные опухоли и шванномы); эндокринные опухоли (например, опухоли гипофиза, опухоли надпочечников, опухоли островковых клеток, паращитовидные опухоли, карциноидные опухоли и медуллярная карцинома щитовидной железы), опухоли глаз и придатков (например, ретинобластома); зародышевые и трофобластные опухоли (например, тератомы, семиномы, дисгерминомы, хорионаденомы и хориокарциномы) и педиатрические и эмбриональные опухоли (например, медуллобластома, нейробластома, опухоль Вильмса и примитивные нейроэктодермальные опухоли) или синдромы, врожденные или иные, которые оставляют пациента восприимчивым к злокачественным новообразованиям (например, пигментная ксеродерма).
3.5 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания, необязательно в комбинации с другим терапевтическим агентом или средством лечениея (например, противораковым агентом или терапией), где раковое заболевание выбрано из рака поджелудочной железы, рака толстой кишки и прямой кишки, рака легких, рака головного мозга и нервов и рака крови, такого как лимфома и лейкоз.
3.6 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания, необязательно в комбинации с другим терапевтическим агентом или средством лечениея (например, противораковым агентом или терапией), где раковое заболевание выбрано из глиом и глиобластом (которые могут, например, быть выбраны из мультиформной глиобластомы, эпендимомы, диффузной глиомы ствола головного мозга, IDH1-мутантных глиом).
3.7 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания, необязательно в комбинации с другим терапевтическим агентом или средством лечениея (например, противораковым агентом или терапией), где раковое заболевание выбрано из рабдоидных опухолей, медуллобластомы и других эмбриональных опухолей головного мозга, рака молочной железы, рака легких, меланомы, рака желудка, рака толстой и прямой кишки, рака поджелудочной железы и рака яичников.
3.8 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания, необязательно в комбинации с другим терапевтическим агентом или средством лечениея (например, противораковым агентом или терапией), где раковое заболевание представляет собой рак, в котором участвует PLK1 (например, где имеет место сверхэкспрессия PLK1).
3.9 Композиция веществ, атропизомер или соль для применения согласно Варианту реализации 3.8, при этом рак является таким, как это определено в любом из вариантов реализации 3.4-3.7.
3.10 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания, необязательно в комбинации с другим терапевтическим агентом или средством лечениея (например, противораковым агентом или терапией), где раковое заболевание представляет собой рак, в котором участвует PLK4 (например, где имеет место сверхэкспрессия PLK4).
3.11 Композиция веществ, атропизомер или соль для применения согласно Варианту реализации 3.10, при этом рак является таким, как это определено в любом из вариантов реализации 3.4-3.7.
3.12 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания, необязательно в комбинации с другим терапевтическим агентом или средством лечениея (например, противораковым агентом или терапией), где раковое заболевание представляет собой рак, который характеризуется дефицитом р53 или мутацией в гене ТР53.
3.13 Композиция веществ, атропизомер или соль для применения согласно Варианту реализации 3.12, при этом рак является таким, как это определено в любом из вариантов реализации 3.4-3.7.
3.14 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания, где раковое заболевание представляет собой рак, характеризующийся наличием мутированной формы KRAS.
3.15 Композиция веществ, атропизомер или соль для применения согласно Варианту реализации 3.14, причем указанная мутированная форма KRAS имеет мутацию в аминокислоте белка, выбранной из глицина 12, глицина 13, глутамина 61 и их комбинаций.
3.16 Композиция веществ, атропизомер или соль для применения согласно Варианту реализации 3.14 или 3.15, при этом рак является таким, как это определено в любом из вариантов реализации 3.4-3.7.
3.17 Композиция веществ, атропизомер или соль, определенная в любом из Вариантов реализации 1.1-1.211 для применения в лекарственных препаратах или терапии, необязательно в комбинации с другим терапевтическим агентом или средством лечения (например, противораковым агентом или терапией).
3.18 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211 для применения, необязательно в комбинации с другим терапевтическим агентом или средством лечения (например, противораковым агентом или терапией), при профилактике или лечении заболеваний и состояний, характеризующихся аномальной экспрессией белка KRAS.
3.19 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения в качестве противоракового агента.
3.20 Способ лечения субъекта (например, млекопитающего субъекта, такого как человек), страдающего от ракового заболевания, определенного в любом из Вариантов реализации 3.4-3.16, включающий введение субъекту терапевтически эффективного количества композиции веществ, атропизомера или соли, определеных в любом из Вариантов реализации 1.1-1.211, необязательно в комбинации с другим терапевтическим агентом или средством лечения (например, противораковым агентом или терапией).
3.21 Применение композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, при получении лекарственного средства для применения согласно определению в любом из Вариантов реализации 3.1-3.19.
3.22 Способ подавления PLK1-PBD, включающий приведение в контакт с PLK1-PBD эффективного ингибирующего киназу количества композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211.
3.23 Способ подавления PLK1 киназы, включающий приведение в контакт с PLK1 киназой ингибирующего киназу количества композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211.
3.24 Способ подавления PLK4-PBD, включающий приведение в контакт с PLK4-PBD эффективного ингибирующего количества композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211.
3.25 Способ подавления PLK4 киназы, включающий приведение в контакт с PLK4 киназой ингибирующего киназу количества композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211.
3.26 Способ подавления PLK1-PBD и PLK4-PBD, включающий приведение в контакт с PLK1-PBD и PLK4-PBD эффективного ингибирующего количества композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211.
3.27 Способ согласно любому из Вариантов реализации 3.22-3.26, отличающийся тем, что эффективное ингибирующее количество композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, приводят в контакт с PLK1-PBD и/или PLK4-PBD в условиях in vivo, например, в организме млекопитающего субъекта, такого как человек.
Перед введением композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, пациент может пройти скрининг с целью определения, является ли раковое заболевание, от которого страдает (или возможно будет страдать) пациент, раковым заболеванием, характеризующимся повышенными уровнями PLK1 и/или PLK4 киназы, которое, следовательно, будет поддаваться лечению с применением соединения, обладающего активностью против PLK1 и/или PLK4 киназы.
Например, биологический образец, полученный от пациента, может быть проанализирован с целью определения, будет ли раковое заболевание, от которого страдает или возможно будет страдать пациент, раковым заболеванием, характеризующимся генетическим сбоем или аномальной экспрессией белка, которые приводят к повышающей регуляции киназы PLK1 и/или PLK4. Термин повышающая регуляция включает повышенную экспрессию или сверхэкспрессию, включая амплификацию (т.е. появление множественных копий) гена, его повышенную экспрессию посредством транскрипционного эффекта, гиперактивность и активацию, включая активацию посредством мутаций. Таким образом, может быть проведено диагностическое исследование пациента с целью обнаружения маркерного признака повышающей регуляции киназы PLK1 и/или PLK4. Термин «диагноз» включает скрининг. Термин «маркер» в настоящей заявке охватывает генетические маркеры, включающие, например, измерение состава ДНК с целью идентификации мутаций в PLK1 и/или PLK4 киназах. Термин «маркер» также включает маркеры, которые характеризуют повышающую регуляцию PLK1 и/или PLK4, включая активность ферментов, уровни ферментов, состояние ферментов (например, фосфорилированные или нет) и уровни мРНК вышеупомянутых белков.
Опухоли с повышающей регуляцией киназы PLK1 и/или PLK4 могут быть особенно восприимчивы к действию ингибиторов PLK1. Предпочтительно может быть проведен скрининг опухолей на наличие повышающей регуляции PLK1 и/или PLK4. Так может быть проведено диагностическое исследование пациента с целью обнаружения маркера, характеризующего повышающую регуляцию PLK1 и/или PLK4. Диагностические исследования обычно проводят на биологическом образце, выбранном из образцов биопсии опухолей, образцов крови (выделение и обогащение выделяющихся опухолевых клеток), биопсии кала, слюны, анализа хромосом, плевральной жидкости и перитонеальной жидкости.
Способы идентификации и анализа мутаций и повышающей регуляции белков хорошо известны специалистам в области техники. Способы скрининга могут включать, но не ограниваются ими, стандартные способы, такие как полимеразная цепная реакция с обратной транскриптазой (RT-PCR) или in-situ гибридизация.
При скрининге методом RT-PCR, проводят оценку уровня мРНК в опухоли путем создания кДНК копии мРНК с последующей амплификацией кДНК методом PCR. Методы PCR-амплификации, выбора праймеров и условий для амплификации хорошо известны специалисту в данной области техники. Манипулирование нуклеиновыми кислотами и PCR проводят с помощью стандартных методов, как это описано, например, в публикации Ausubel, F.M. et al., eds. Current Protocols in Molecular Biology, 2004, John Wiley & Sons Inc. или Innis, M.A. et-al., eds. PCR Protocols: a guide to methods and applications, 1990, Academic Press, San Diego. Реакции и манипуляции с нуклеиновокислотными методами также описаны в публикации Sambrook et al., 2001, 3rd Ed, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press. Как вариант можно использовать доступный в продаже набор для проведения RT-PCR (например, от компании Roche Molecular Biochemicals) или методики, изложенные в патентах США 4,666,828; 4,683,202; 4,801,531; 5,192,659, 5,272,057, 5,882,864 и 6,218,529, включенных в настоящую заявку посредством ссылки.
Примером метода гибридизации in-situ для оценки экспрессии мРНК может являться метод флуоресцентной in-situ гибридизации (FISH) (см., Angerer, 1987 Meth. Enzymol., 152: 649).
В целом, in situ гибридизация включает следующие основные стадии: (1) фиксацию ткани, подлежащей анализу, (2) предгибридизационную обработку образца для повышения доступности целевой нуклеиновой кислоты и для уменьшения неспецифического связывания, (3) гибридизацию смеси нуклеиновых кислот с нуклеиновой кислотой в биологической структуре или ткани, (4) постгибридизационную промывку для удаления фрагментов нуклеиновых кислот, не связанных при гибридизации, и (5) детектирование фрагментов гибридизованной нуклеиновой кислоты. Зонды, используемые для такого применения, обычно являются мечеными, например, радиоизотопами или флуоресцентными репортерами. Предпочтительные зонды являются достаточно длинными, например, приблизительно от 50, 100, или 200 нуклеотидов до приблизительно 1000 или более нуклеотидов, чтобы обеспечить возможность специфической гибридизации с целевой нуклеиновой кислотой (кислотами) в жестких условиях. Стандартные способы проведения FISH описаны в публикации Ausubel, F.M. et al., eds. Current Protocols in Molecular Biology, 2004, John Wiley & Sons Inc and Fluorescence In Situ Hybridization: Technical Overview by John M.S. Bartlett in Molecular Diagnosis of Cancer, Methods and Protocols, 2nd ed.; ISBN: 1-59259-760-2; March 2004, pps. 077-088; Series: Methods in Molecular Medicine.
Как вариант, белковые продукты, экспрессируемые из мРНК, можно анализировать методами иммуногистохимии образцов опухоли, твердофазного иммуноферментного анализа с микротитрационными планшетами, вестерн-блоттинга, 2-мерного электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия (SDS), ELISA, проточной цитометрии и другими методами, известными в данной области техники для обнаружения специфических белков. Методы детектирования включают применение сайт-специфических антител. Специалисту в области техники будет понятно, что в данном случае применимы все хорошо известные способы выявления повышающей регуляции киназ PLK1 и/или PLK4.
В качестве альтернативы или дополнения, перед введением композиции, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, пациент может быть подвергнут скринингу, чтобы определить, характеризуется ли раковое заболевание, от которого страдает (или возможно будет страдать) пациент, наличием мутированного KRAS, и, следовательно, будет ли оно восприимчиво к лечению с применением соединения, обладающего активностью против раковых клеток, несущих мутантный KRAS.
Например, взятый у пациента биологический образец, может быть проанализирован с целью определения того, характеризуется ли раковое заболевание, от которого страдает (или возможно будет страдать) пациент, наличием мутированного KRAS. Так, например, может быть проведено диагностическое исследование пациента с целью обнаружения мутаций в кодонах 12, 13, 61 (глицин 12, глицин 13 и глутамин 61) или их смесей в белке KRAS. Коммерчески доступные диагностические тесты для мутантного KRAS включают COBAS® KRAS Mutation Test от Roche Molecular Systems, inc и Therascreen KRAS RGQ PCR Kit от Qiagen Manchester, Ltd.
Опухоли с мутантным KRAS могут быть особенно восприимчивы к ингибиторам PLK1 и/или PLK4. Методы идентификации и анализа мутаций и повышающей регуляции белков хорошо известны специалистам в данной области техники. Методы скрининга могут включать, но не ограниваются ими, стандартные методы, такие как полимеразная цепная реакция с обратной транскриптазой (RT-PCR), или in-situ гибридизация, уже описанная выше.
Соответственно, в дальнейших вариантах реализации (Варианты реализации 3.28-3.38) настоящего изобретения предложены:
3.28 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания у субъекта (например, у человеческого субъекта), который прошел скрининг и был определен как страдающий от ракового заболевания, характеризующегося повышенными уровнями киназы PLK1 (например, сверхэкспрессией PLK1).
3.29 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания у субъекта (например, у человеческого субъекта), который прошел скрининг и был определен как страдающий от ракового заболевания, характеризующегося повышенными уровнями киназы PLK4 (например, сверхэкспрессией PLK4).
3.30 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания у субъекта (например, у человеческого субъекта), который прошел скрининг и был определен как страдающий от ракового заболевания, характеризующегося повышенными уровнями PLK1 киназы и PLK4 киназы (например, сверхэкспрессией PLK1 и PLK4 киназ).
3.31 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания у субъекта (например, у человеческого субъекта), который прошел скрининг и был определен как страдающий или подверженный риску заболевания или состояния, которое поддается лечению с применением соединения, обладающего активностью против KRAS.
3.32 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения при лечении ракового заболевания у субъекта (например, у человеческого субъекта), который прошел скрининг и был определен как страдающий от ракового заболевания, которое характеризуется наличием мутированного KRAS и которое будет восприимчиво к лечению с применением соединения, обладающего активностью против KRAS или против раковых клеток, несущих мутантный KRAS.
3.33 Композиция веществ, атропизомер или соль для применения согласно любому из Вариантов реализации 3.28-3.32, при этом раковое заболевание представляет собой рак, определенный в любом из Вариантов реализации 3.4-3.16.
3.34 Применение композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, для получения лекарственного средства для применения, определенного в любом из Вариантов реализации 3.28-3.33.
3.35 Способ диагностики и лечения заболевания или состояния (например, ракового заболевания, такого как рака, определенного в любом из Вариантов реализации 3.4-3.16), опосредованного KRAS или характеризующегося наличием мутированной формы KRAS, включающий (i) проведение скрининга субъекта (например, человеческого субъекта) с целью определения, будет ли заболевание или состояние, от которого страдает или возможно будет страдать субъект, поддаваться лечению с применением соединения, обладающего активностью против KRAS, и (ii) если будет показано, что заболевание или состояние, от которого страдает или возможно будет страдать субъект, восприимчиво к такому лечению, введение субъекту терапевтически эффективного количества композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211.
3.36 Способ лечения заболевания или состояния (например, ракового заболевания, такого как рака, определенного в любом из Вариантов реализации 3.4-3.16), характеризующегося наличием мутированной формы KRAS, включающий введение терапевтически эффективного количества композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, субъекту (например, человеческому субъекту), который прошел скрининг и был определен как страдающий или подверженный риску заболевания или состояния, которое будет восприимчиво к лечению с применением соединения, обладающего активностью против KRAS.
3.37 Способ диагностики и лечения ракового заболевания, характеризующегося повышенными уровнями PLK1 киназы, включающий (i) проведение скрининга пациента с целью определения, характеризуется ли раковое заболевание, от которого страдает пациент, повышенными уровнями PLK1 киназы и (ii) если отмечено, что раковое заболевание характеризуется повышенными уровнями PLK1 киназы, введение пациенту терапевтически эффективного количества композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211.
3.38 Применение композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, для получения лекарственного средства для лечения или профилактики заболевания или состояния у пациента, который прошел скрининг и был определен как страдающий или подверженный риску заболевания или состояния, которое поддается лечению с применением соединения, обладающего активностью против KRAS.
Лекарственные Формы
Композицию веществ или атропизомеры согласно настоящему изобретению обычно вводят пациентам в форме фармацевтической композиции. Соответственно, в другом варианте реализации изобретения (Вариант реализации 4.1) предложена фармацевтическая композиция, содержащая композицию веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, и фармацевтически приемлемое вспомогательное вещество.
Согласно дальнейшим вариантам реализации настоящего изобретения предложены:
4.2 Фармацевтическая композиция согласно Варианту реализации 4.1, которая содержит от приблизительно 1 масс. % до приблизительно 95 масс. % композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, и от 99 масс. % до 5 масс. % фармацевтически приемлемого вспомогательного вещества или комбинации вспомогательных веществ и необязательно одного или более дополнительных терапевтически активных ингредиентов.
4.3 Фармацевтическая композиция согласно Варианту реализации 4.2, которая содержит от приблизительно 5 масс. % до приблизительно 90 масс. % композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, и от 95 масс. % до 10 масс. % фармацевтически приемлемого вспомогательного вещества или комбинации вспомогательных веществ и необязательно одного или более дополнительных терапевтически активных ингредиентов.
4.4 Фармацевтическая композиция согласно Варианту реализации 4.3, которая содержит от приблизительно 10 масс. % до приблизительно 90 масс. % композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, и от 90 масс. % до 10 масс. % фармацевтически приемлемого вспомогательного вещества или комбинации вспомогательных веществ.
4.5 Фармацевтическая композиция согласно Варианту реализации 4.4, которая содержит от приблизительно 20 масс. % до приблизительно 90 масс. % композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, и от 80 масс. % до 10 масс. % фармацевтически приемлемого вспомогательного вещества или комбинации вспомогательных веществ.
4.6 Фармацевтическая композиция согласно Варианту реализации 4.5, которая содержит от приблизительно 25 масс. % до приблизительно 80 масс. % композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, и от 75 масс. % до 20 масс. % фармацевтически приемлемого вспомогательного вещества или комбинации вспомогательных веществ.
Фармацевтические композиции согласно настоящему изобретению могут быть представлены в любой форме, подходящей для перорального, парентерального, местного, интраназального, интрабронхиального, офтальмического, ушного, ректального, внутривагинального или трансдермального применения. В случае, когда композиции предназначены для парентерального введения, они могут быть представлены в виде лекарственной формы для внутривенного, внутримышечного, внутрибрюшинного, подкожного введения или для непосредственной доставки в целевой орган или ткань путем инъекции, инфузии или других средств доставки.
Фармацевтические дозированные формы, подходящие для перорального применения, включают таблетки, капсулы, каплеты, пилюли, таблетки для рассасывания, сиропы, растворы, спреи, порошки, гранулы, эликсиры и суспензии, подъязычные таблетки, спреи, капсулы-имплантаты или наружные и буккальные пластыри.
Соответственно, в дальнейших вариантах реализации изобретения предложено следующее:
4.7 Фармацевтическая композиция согласно любому из Вариантов реализации 4.1-4.6, подходящая для перорального введения.
4.8 Фармацевтическая композиция согласно Варианту реализации 4.7, которая выбрана из таблеток, капсул, каплет, пилюль, таблеток для рассасывания, сиропов, растворов, спреев, порошков, гранул, эликсиров и суспензий, подъязычных таблеток, спреев, капсул-имплантатов или наружных и буккальных пластырей.
4.9 Фармацевтическая композиция согласно Варианту реализации 4.8, которая выбрана из таблеток и капсул.
4.10 Фармацевтическая композиция согласно любому из Вариантов реализации 4.1-4.6, подходящая для парентерального введения.
4.11 Фармацевтическая композиция согласно Варианту реализации 4.10, которая сформулирована для внутривенного, внутримышечного, внутрибрюшинного, подкожного введения или для непосредственной доставки в целевой орган или ткань путем инъекции, инфузии или других средств доставки.
4.12 Фармацевтическая композиция согласно Варианту реализации 4.11, которая представляет собой раствор или суспензию для инъекций или инфузий.
Фармацевтические композиции (например, определенные в любом из Вариантов реализации 4.1-4.12), содержащие композицию, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211 настоящего изобретения, могут быть приготовлены в виде лекарственных форм в соответствии с известными методиками, см, например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, USA.
Таким образом, таблетированные композиции (как в Варианте реализации 4.9) могут содержать единичную дозировку активного соединения совместно с инертным разбавителем или носителем, таким как сахар или сахарный (многоатомный) спирт, например, лактоза, сахароза, сорбит или маннит, и/или разбавителем, не производным от Сахаров, таким как карбонат натрия, фосфат кальция, тальк, карбонат кальция или целлюлоза или ее производное, такое как метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза, и крахмалы, такие как кукурузный крахмал. Таблетки также могут содержать такие стандартные ингредиенты, как связующие и гранулирующие агенты, (например, поливинилпирролидон), разрыхлители (например, набухающие сшитые полимеры, такие как сшитая карбоксиметилцеллюлоза), смазывающие агенты (например, стеараты), консерванты (например, парабены), антиоксиданты (например, ВНТ), буферизующие агенты (например, фосфатный или цитратный буферы) и шипучие агенты, такие как смеси цитрат-бикарбонат.Такие вспомогательные вещества хорошо известны и не требуют подробного описании в настоящей заявке.
Лекарственные формы в виде капсул (как в Варианте реализации 4.9) могут представлять собой твердые желатиновые или мягкие желатиновые разновидности и содержать активный компонент в твердой, полутвердой или жидкой форме. Желатиновые капсулы могут быть сформированы из животного желатина или его эквивалентов синтетического или растительного происхождения.
Твердые дозировочные формы (например, таблетки, капсулы и т.д.) могут быть покрыты или не покрыты оболочкой, но обычно имеют покрытие, например, в виде защитной пленки (например, воск или глазурь) или покрытия, регулирующего высвобождение активного компонента. Покрытие (например, полимер типа Eudragit™) может быть разработано таким образом, чтобы высвобождать активное соединение в желаемом месте желудочно-кишечного тракта. Таким образом, покрытие может быть подобрано таким образом, чтобы разрушаться при определенных условиях рН внутри желудочно-кишечного тракта и посредством чего селективно высвобождать композицию или атропизомер в желудке, или подвздошной кишке, или двенадцатиперстной кишке.
Вместо покрытия или в дополнение к нему лекарственное средство может быть представлено в составе твердой матрицы, включающей агент, регулирующий высвобождение активного вещества, например, агент, замедляющий его высвобождение, который может быть адаптирован для селективного высвобождения композиции или атропизомера в определенных условиях кислотности или щелочности, варьирующихся в желудочно-кишечном тракте. Как вариант, материал матрицы или покрытия, замедляющий высвобождение активного вещества, может быть взят в виде распадающегося полимера (например, полимера малеинового ангидрида), который по существу непрерывно размывается по мере продвижения лекарственной формы по желудочно-кишечному тракту.
Композиции для местного применения включают мази, кремы, спреи, пластыри, гели, жидкие капли и вкладыши (например, внутриглазные пленки). Такие композиции могут быть приготовлены согласно известным способам.
Композиции для парентерального введения (как в Вариантах реализации 4.10-4.12) обычно представлены в виде стерильных водных или масляных растворов или мелкодисперсных суспензий, или могут быть представлены в форме мелкодисперсного стерильного порошка для приготовления раствора непосредственно перед применением с использованием стерильной воды для инъекций.
Примеры лекарственных форм для ректального или внутривагинального введения, включают пессарии и суппозитории, которые могут быть, например, сформованы из формуемого пластичного или воскообразного материала, содержащего активное соединение.
Композиции для ингаляционного введения могут быть представлены в форме вдыхаемых порошковых композиций, или жидких или порошковых спреев, и их можно вводить в стандартной форме с помощью порошковых ингаляторов или аэрозольных дозаторов. Такие устройства хорошо известны. В случае ингаляционного введения в порошковые формы обычно включают активное соединение совместно с инертным твердым порошкообразным разбавителем, таким как лактоза.
Композиции веществ или атропизомеры согласно настоящему изобретению в целом будут представлены в единичной дозировочной форме и соответственно будут, как правило, содержать достаточное количество соединения, чтобы получить желаемый уровень биологической активности. Например, согласно любому из Вариантов реализации 3.1-3.9), композиция, предназначенная для перорального введения, может содержать от 2 до 200 миллиграммов активного ингредиента, обычно от 10 до 100 миллиграммов, например, 12,5, 25 и 50 миллиграммов.
Дозировка
Активное соединение (композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211) может быть введено пациенту, нуждающемуся в нем (например, пациенту-человеку или пациенту-животному), в количестве, достаточном для достижения желаемого терапевтического эффекта, например, эффекта, указанного в приведенных выше Вариантах реализации 3.1-3.38.
Композицию веществ, атропизомер или соль обычно вводят субъекту, нуждающемуся в таком введении, например, пациенту-человеку или пациенту-животному, предпочтительно человеку.
Композицию веществ, атропизомер или соль, как правило, вводят в количествах, которые являются терапевтически или профилактически полезными и которые в целом нетоксичны. Однако в определенных ситуациях преимущества, достигаемые при введении композиции веществ, атропизомера или соли могут перевешивать недостатки, связанные с любыми токсическими или побочными эффектами, и в этом случае может оказаться желательным вводить соединения в количествах, которые подразумевают определенную степень токсичности.
Обычная суточная доза композиции веществ, атропизомера или соли может составлять величину в пределах от 0,025 до 5 миллиграмм на килограмм массы тела, например, до 3 миллиграмм на килограмм массы тела, но, как правило, от 0,15 до 5 миллиграмм на килограмм массы тела, хотя в случае необходимости могут быть введены и более высокие или низкие дозы.
К примеру начальную стартовую дозу 12,5 мг можно вводить от 2 до 3 раз в сутки. Дозировка может быть увеличена на 12,5 мг в сутки через каждые 3-5 дней, пока не будет достигнута определяемая лечащим врачом максимальная переносимая и эффективная доза для данного индивидуума. В конечном счете, количество вводимого соединения должно соответствовать природе заболевания или физиологического состояния, подлежащего лечению, терапевтическим преимуществам, наличию или отсутствию побочных эффектов, возникающих при данном режиме дозирования, и должно определяться по усмотрению лечащего врача.
Комбинированная терапия
Предполагается, что композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, будут пригодны в качестве химиотерапевтического агента как сами по себе, так и (более вероятно) в составе комбинированной терапии совместно с химиотерапевтическими агентами или лучевой терапией при профилактике или лечении ряда пролиферативных заболеваний или состояний. Примеры таких заболеваний и состояний изложены выше.
Конкретные примеры химиотерапевтических агентов или других лекарственных средств, которые могут быть введены совместно с композицией веществ, атропизомером или солью, определенными в любом из Вариантов реализации 1.1-1.211, включают следующее:
• Ингибиторы топоизомеразы I
• Антиметаболиты (например, цитарабин)
• Тубулин-направленные агенты
• ДНК-связывающие и ингибирующие топоизомеразу II агенты
• Ингибиторы EGFR (например, гефитиниб - см. Biochemical Pharmacology 78 2009 460-468)
• Ингибиторы mTOR (например, эверолимус)
• Ингибиторы пути PI3K (например, PI3K, PDK1)
• Ингибиторы Akt
• Алкилирующие агенты (например, темозоломид)
• Моноклональные антитела.
• Антигормоны
• Ингибиторы сигнальной трансдукции
• Ингибиторы протеасом
• Ингибиторы ДНК-метилтрансферазы
• Циокины и ретиноиды
• Инициируемые гипоксией ДНК-повреждающие агенты (например, тирапазамин)
• Ингибиторы ароматазы
• Антитела к HER2 (см, например, http://www.wipo.int/pctdb/en/wo.jsp?wo=2007056118)
• Антитела к CD20
• Ингибиторы ангиогенеза
• Ингибиторы HDAC
• Ингибиторы MEK
• Ингибиторы B-Raf
• Ингибиторы ERK
• Низкомолекулярные ингибиторы HER2 (например, лапатиниб)
• Ингибиторы Bcr-AbI-тирозинкиназы (например, иматиниб)
• Ингибитор CDK4/6 (например, ибранс)
• Ингибиторы Mps1/TTK
• Ингибиторы Aurora В
• Ингибиторы FLT3 киназы
• Ингибиторы IDH1 или IDH2
• Ингибиторы BRD4
• темозоломид
• Ингибиторы компонентов сигнализации блокады иммунных контрольных точек, включая PD1, PDL-1 и CTLA4, и
• лучевая терапия.
Соответственно, в дальнейших вариантах реализации изобретения предложено следующее:
5.1 Фармацевтическая комбинация, содержащая композицию веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, и другой терапевтически активный агент.
5.2 Фармацевтическая комбинация согласно Варианту реализации 5.1, в которой указанный другой терапевтический агент выбран из химиотерапевтических агентов, перечисленных выше.
5.3 Фармацевтическая комбинация согласно Варианту реализации 5.1, в которой указанный другой терапевтический агент представляет собой противораковый агент.
5.4 Фармацевтическая комбинация согласно любому из Вариантов реализации 5.1-5.3, в которой композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, и указанный другой терапевтически активный агент представлены в единой фармацевтической композиции или упаковке для пациента.
5.5 Фармацевтическая комбинация, содержащая композицию веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, другой терапевтически активный агент и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
5.6 Способ лечения субъекта, страдающего от ракового заболевания, включающий введение субъекту терапевтически эффективного количества фармацевтической комбинации согласно любому из Вариантов реализации 5.1-5.5.
5.7 Композиция веществ, атропизомер или соль, определенные в любом из Вариантов реализации 1.1-1.211, для применения с целью усиления терапевтического эффекта лучевой терапии или химиотерапии при лечении пролиферативного заболевания, такого как рак.
5.8 Применение композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, или их фармацевтически приемлемых солей для приготовления лекарственного средства, предназначенного для усиления терапевтического эффекта лучевой терапии или химиотерапии при лечении пролиферативного заболевания, такого как рак.
5.9 Способ профилактики или лечения пролиферативного заболевания, такого как рак, включающий введение пациенту в сочетании с лучевой терапией или химиотерапией композиции веществ, атропизомера или соли, определенных в любом из Вариантов реализации 1.1-1.211, или их фармацевтически приемлемых солей.
Краткое описание графических материалов
Фигура 1 представляет собой схематическую диаграмму, иллюстрирующую систему RIS классификации для атропизомеров.
Фигура 2 представляет собой изображение трехмерной структуры 2,4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)-этил]бензамида Атропизомера А-2, определенной с помощью монокристалльных рентгеновских кристаллографических исследований.
Фигура 3 представляет собой схематическую стереохимическую иллюстрацию двух атропизомеров, а именно А-1 (S) и А-2 (R), а также основание для назначения им данных стереохимических структур согласно правилам последовательности Кана-Ингольда-Прелога (CIP).
Фигура 4 представляет собой спектр рентгеновской порошковой дифракции для Атропизомера А-2 в форме свободного основания.
Фигура 5 представляет собой спектр рентгеновской порошковой дифракции для Атропизомера А-2 в форме виннокислой соли (тартрата) согласно Дифрактограмме А (нижняя линия) и виннокислой соли согласно Дифрактограмме В (верхняя и средняя линии).
Фигура 6 иллюстрирует температурный профиль Атропизомера А-2 в форме свободного основания и демонстрирует график дифференциальной сканирующей калориметрии (линия 6А) и график термогравиметрического анализа (линия 6В).
Фигура 7 иллюстрирует температурный профиль Атропизомера А-2 в форме виннокислой соли согласно Дифрактограмме А и демонстрирует график дифференциальной сканирующей калориметрии (линия 7А) и график термогравиметрического анализа (линия 7В).
Фигура 8 иллюстрирует температурный профиль Атропизомера А-2 в форме виннокислой соли согласно Дифрактограмме В и демонстрирует график дифференциальной сканирующей калориметрии (линия 8А) и график термогравиметрического анализа (линия 8В).
Фигура 9 представляет собой график изменения массы в зависимости от относительной влажности в исследованиях методом гравиметрической сорбции паров (GVS), проведенных на Атропизомере А-2 в форме виннокислой соли, соответствующей Дифрактограмме В.
Фигура 10 представляет собой гистограмму, показывающую количественные соотношения различных наблюдаемых митотических фенотипов (неконгрессированных хромосом, многополярных веретен/аномального цитокинез, монополярных веретен, нормальной прометафазы, нормальной метафазы), полученных после обработки клеток U87MG 0,03 мкМ концентрациями Атропизомера А-1 или Атропизомера А-2.
Фигура 11 представляет собой гистограмму, показывающую количество центриолей, присутствующих в клетках HeLa после обработки 0,02 мкМ концентрациями Атропизомера А-1 или Атропизомера А-2.
Фигура 12 представляет собой график временной зависимости концентрации в плазме крови после перорального и внутривенного введения мышам Атропизомера А-2. Нижняя линия протяженностью вплоть до 24 часов является линией для дозы 2 мг/кг (внутривенно). Другая линия соответствует дозе 10 мг/кг (перорально).
Фигура 13 представляет собой график временной зависимости концентрации Атропизомера А-3 в плазме крови после его перорального и внутривенного введения мышам. Нижняя линия протяженностью вплоть до 24 часов соответствует внутривенной дозе. Другая линия соответствует пероральной дозе.
Фигура 14 представляет собой график временной зависимости концентрации Атропизомера А-2 в плазме крови и головном мозге после его перорального введения (10 мг/кг) мышам. Верхняя линия показывает концентрации в головном мозге, а нижняя - концентрации в плазме крови.
Фигура 15 представляет собой график временной зависимости концентраций Атропизомера А-3 в плазме крови и головном мозге после его перорального введения мышам. Верхняя линия показывает концентрации в головном мозге, а нижняя - концентрации в плазме крови.
Фигура 16 представляет собой график временной зависимости объема опухоли у самцов бестимусных мышей на модели подкожного ксенотрансплантата U87MG после введения Атропизомера А-2.
Фигура 17 представляет собой графическое сравнение биолюминесцентного сигнала, привязанного к росту опухоли у самцов бестимусных мышей в ортотопической модели ксенотрансплантата U87-Luc после введения Атропизомера А-2.
Фигура 18 представляет собой график временной зависимости объема опухоли у самцов бестимусных мышей на модели подкожного ксенотрансплантата НСТ116 после введения Атропизомера А-2.
Фигура 19 иллюстрирует графики XRPD для Атропизомера А-2 в формах гидрохлоридной соли, соответствующих Дифрактограммам А и В.
Фигура 20 иллюстрирует графики XRPD для Атропизомера А-2 в форме мезилатной соли.
Фигура 21 иллюстрирует графики XRPD для Атропизомера А-2 в формах малеиновокислой соли (малеата), соответствующих Дифрактограммам А и В.
Фигура 22 иллюстрирует графики XRPD для Атропизомера А-2 в формах яблочнокислой соли (малата), соответствующих Дифрактограммам А и В.
Фигура 23 иллюстрирует графики XRPD для Атропизомера А-2 в форме тозилатной соли, соответствующей Дифрактограмме А.
Фигура 24 иллюстрирует графики XRPD для Атропизомера А-2 в формах фосфорнокислой соли (фосфата), соответствующих Дифрактограммам А и В.
Фигура 25 иллюстрирует графики XRPD для Атропизомера А-2 в формах сернокислой соли (сульфата), соответствующих Дифрактограммам А и В.
ПРИМЕРЫ
Далее изобретение будет проиллюстрировано на приведенных ниже примерах, но не ограничиваясь ими, со ссылкой на описанные в них конкретные варианты реализации.
В примерах используются следующие аббревиатуры.
водн. - водный
CaCl2 - кальция хлорид
ДХМ - дихлорметан
ДЭА - диэтиламин
ДИПЭА - N,N-диизопропилэтиламин
ДМФА - диметилформамид
DMP - периодинатДесса-Мартина
ДМСО - диметилсульфоксид
Et2O - диэтиловый эфир
EtOAc - этилацетат
EtOH - этанол
ч - час(ы)
HATU - (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний-3-оксида гексафторфосфат)
HCl - хлористый водород
ВЭЖХ - высокоэффективная жидкостная хроматография
H2SO4 - серная кислота
ИПС - изопропиловый спирт
ЖХ - жидкостная хроматография
ЖХ-МС - жидкостная хроматография с масс-спектроскопией
LiOH - лития гидроксид
MeCN - ацетонитрил
МеОН - метанол
мин - минута(ы)
МТБЭ - метилтретбутиловый эфир
NaBH4 - натрия боргидрид
NaHCO3 - натрия гидрокарбонат
NaOH - натрия гидроксид
Na2SO4 - натрия сульфат
NH4Cl - аммония хлорид
ЯМР - ядерный магнитный резонанс
ПТСК - п-толуолсульфоновая кислота
ТЭА - триэтиламин
ТГФ – тетрагидрофуран.
Подробности о соединениях и экспериментальная часть
Атропизомеры А-1 - А-8

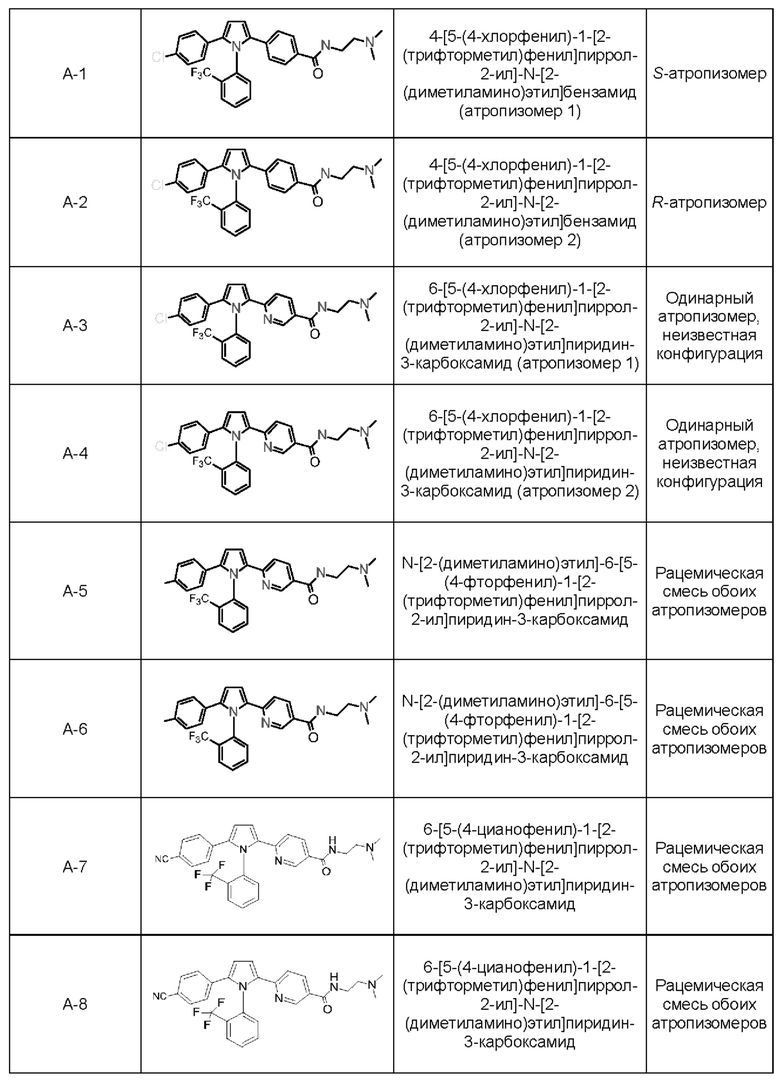
Спектры протонного магнитного резонанса (1H ЯМР) регистрировали на приборе Bruker 400, работающем при 400 МГц в DMSO-d6 или MeOH-d4 (где как указано) при 27°С, если не указано иное, и в отчете указывалось следующее: химический сдвиг δ/ppm (мультиплетность, где s = синглет, d = дублет, dd = двойной дублет, dt = двойной триплет, t = триплет, q = квартет, m = мультиплет, br = уширенный, количество протонов). Остаточный протонный растворитель использовали в качестве внутреннего эталона.
Анализ методом жидкостной хроматографии с масс-спектроскопией проводили, используя указанные ниже системы и условия хроматографирования. Если присутствуют атомы, имеющие разные изотопы, и указана единая масса, то заявленная масса соединения представляет собой моноизотопную массу (т.е. 35Cl, 79Br и т.д.)
УСЛОВИЯ ЖХ-МС ХРОМАТОГРАФИРОВАНИЯ
Данные ЖХ-МС, приведенные в следующих примерах, были получены с использованием одного из описанных ниже методов.
ЖХ-МС Метод 1
ЖХ-МС хроматографирование проводили на приборе UPLC AQUITY с фотодиодным матричным детектором PDA и масс-детектором QDa. Использовали колонку С18, 2,1×50 мм, 1,9 мкм. Поток через колонку составлял 1,2 мл/мин, и использовали следующую подвижную фазу: (А) 0,1% раствор муравьиной кислоты в воде MilliQ (рН=2,70), (В) 0,1% раствор муравьиной кислоты в воде + MeCN (10:90), объем вводимой пробы составлял от 4 до 7 мкл. Образец готовили в смеси МеОН и MeCN, так чтобы получить приблизительную концентрацию в 250 ppm.
Для элюирования использовали следующий градиент:
Параметры масс-спектрометрии
Зонд: ESI капилляр
Температура источника: 120°С
Температура зонда: 600°С
Напряжение на капилляре: 0,8 кВ (+Ve и -Ve)
Напряжение на конусе: 10 и 30 В
Режим ионизации: Положительный и отрицательный
ЖХ-МС Метод 2
ЖХ-МС хроматографирование проводили на приборе Agilent Infinity II G6125C LCMS. Использовали колонку XBridge С18, 50 × 4,6 мм, 3,5 мкм. Поток через колонку составлял 1,0 мл/мин, и использовали следующую подвижную фазу: (А) 5 мМ раствор бикарбоната аммония в воде Milli-Q и (В) МеОН. Объем вводимой пробы составлял 5 мкл. Образец готовили в смеси воды и MeCN, так чтобы получить приблизительную концентрацию в 250 ppm.
Для элюирования использовали следующий градиент.
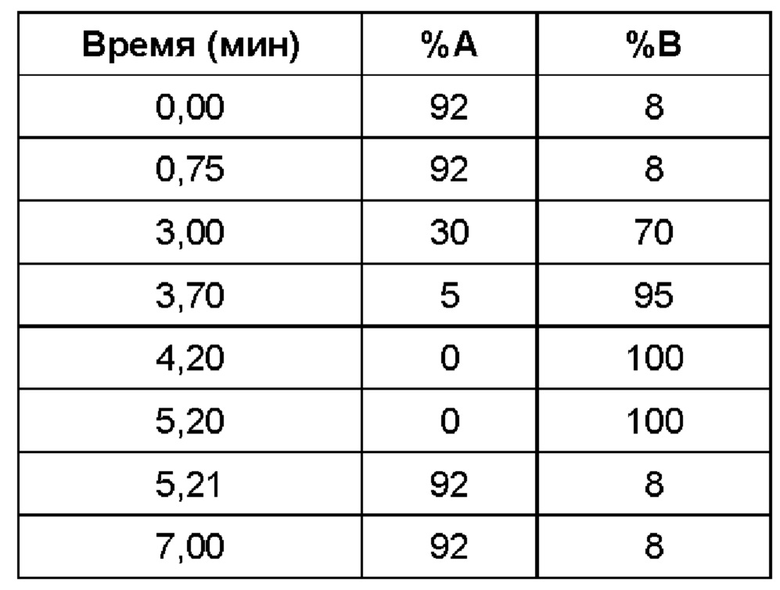
Параметры масс-спектрометрии
Источник ионов: MMI
Напряжение фрагментации: 70 В
Режим ионизации: Положительный и отрицательный
Температура газа: 250°С
Испаритель: 160°С
Поток газа: 10 л/мин
Давление в распылителе: 45 psi
ВЭЖХ Метод 1
ВЭЖХ-анализ проводили на ВЭЖХ системе Agilent Technologies 1100/1200 series. Использовали колонку АСЕ 3 С18; 150 × 4,6 мм, размер частиц 3,0 мкм (например, Hichrom, кат. номер: АСЕ-111-1546). Скорость пропускания (скорость потока) составляла 1,0 мл/мин. Подвижная фаза А представляла собой воду с трифторуксусной кислотой (100:0,1%), подвижная фаза В представляла собой ацетонитрил с трифторуксусной кислотой (100:0,1%). Объем вводимой пробы составлял 5 мкл, и использовали следующий градиент:
Анализ методами хиральной ВЭЖХ
Представленные данные хиральной ВЭЖХ были получены с использованием одного из описанных ниже методов.
Хиральная ВЭЖХ, Метод 1
Анализ методом хиральной ВЭЖХ проводили на системе Agilent Technologies 1200 series HPLC. Использовали колонку CHIRAL PAK IG, 250 × 4,6 мм, 5 мкм. Скорость потока в колонке составляла 1,0 мл/мин, а подвижная фаза представляла собой (А) 0,1% об./об. ДЭА в н-гептане и (В) смесь ИПС и МеОН (70:30). Объем вводимой пробы составлял 25 мкл. Образцы готовили в смеси ИПС и МеОН, так чтобы получить приблизительную концентрацию в 250 ppm, с использованием следующего изократического метода:

Хиральная ВЭЖХ. Метод 2
Анализ методом хиральной ВЭЖХ проводили на системе Agilent Technologies 1200 series HPLC. Использовали колонку CHIRALPAK IG SFC, 21 × 250 мм, 5 мкм. Скорость потока в колонке составляла 1,0 мл/мин, а подвижная фаза представляла собой (А) 0,1% об./об. ДЭА в н-гептане и (В) смесь ИПС и МеОН (70:30). Объем вводимой пробы составлял 20 мкл. Образцы готовили в смеси ИПС и МеОН, так чтобы получить приблизительную концентрацию в 250 ppm, с использованием следующего изократического метода:
Хиральная ВЭЖХ. Метод 3
Анализ методом хиральной ВЭЖХ проводили на системе Agilent Technologies 1200 series HPLC. Использовали колонку CHIRAL PAK IG, 250 × 4,6 мм, 5 мкм. Скорость потока в колонке составляла 1,0 мл/мин, а подвижная фаза представляла собой (А) 0,1% об./об. ДЭА в н-гептане и (В) смесь ИПС и МеОН (70:30). Объем вводимой пробы составлял 10 мкл. Образцы готовили в смеси ИПС и MeCN, так чтобы получить приблизительную концентрацию в 250 ppm, с использованием следующего изократического метода:

Хиральная ВЭЖХ, Метод 4
Условия идентичны хиральному Методу 3, за исключением того, что использовали следующий изократический метод:

Хиральная ВЭЖХ. Метод 5
Условия идентичны хиральному Методу 3, за исключением того, что использовали следующий изократический метод:

Хиральная ВЭЖХ, Метод 7
Анализ методом хиральной ВЭЖХ проводили на системе Agilent Technologies 1100/1200 series HPLC. Использовали колонку CHIRALPAK IA; 250 × 4,6 мм, 5,0 мкм. Скорость потока в колонке составлял 1,0 мл/мин, а подвижная фаза представляла собой смесь гексана, EtOH и этаноламина (90:10:0,1%). Объем вводимой пробы составлял 5 мкл.Образцы готовили в 100% EtOH, так чтобы получить приблизительную концентрацию в 0,5 мг/мл.
Методы препаративной ВЭЖХ:
Итоговые соединения очищали с использованием одного из следующих методов препаративной ВЭЖХ.
Препаративная ВЭЖХ, Метод 1
Препаративную ВЭЖХ проводили с использованием колонки SUNFIRE Prep С18 OBD, 19 × 250 мм, 5 мкм, с подвижной фазой (А) 0,05% HCl в воде и (В) 100% MeCN и скоростью пропускания 17 мл/мин, при этом для элюирования использовали следующую изократическую систему:
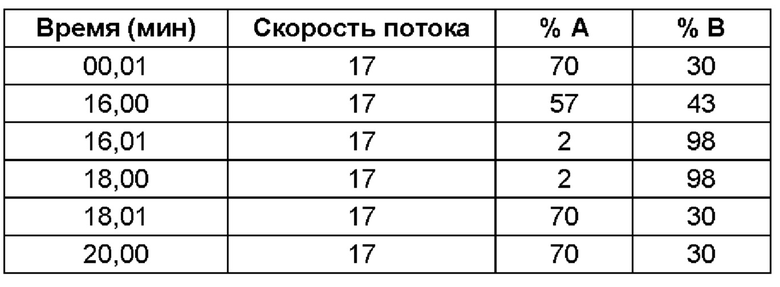
Препаративная ВЭЖХ, Метод 2
Препаративную ВЭЖХ проводили с использованием колонки X-bridge Prep, С18, 30 × 250 мм, 5 мкм, с подвижной фазой (А) 0,05% раствор HCl в воде и (В) 100% MeCN и скоростью пропускания 25 мл/мин, при этом для элюирования использовали следующую изократическую систему:
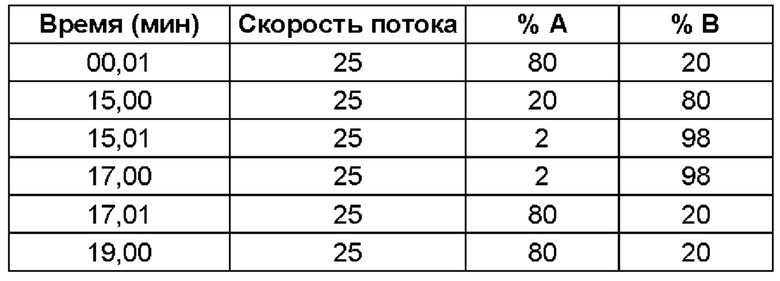
Методы препаративной хиральной ВЭЖХ:
Атропизомеры выделяли с использованием одного из следующих методов препаративной хиральной ВЭЖХ.
Препаративная хиральная ВЭЖХ. Метод 1
Препаративную хиральную ВЭЖХ проводили с использованием колонки CHIRALPAK IG SFC, 21 × 250 мм, 5 мкм, для элюирования использовали подвижную фазу (А) 0,1% ДЭА в гептане и (В) ИПС со скоростью потока 30 мл/мин и следующей изократической системой:
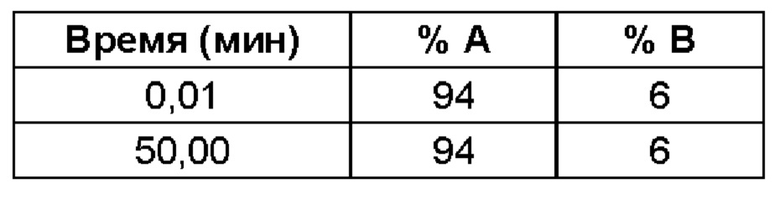
Препаративная хиральная ВЭЖХ, Метод 2
Препаративную хиральную ВЭЖХ проводили с использованием колонки CHIRALPAK IG SFC, 21 × 250 мм, 5 мкм, для элюирования использовали подвижную фазу (А) 0,1% ДЭА в гептане и (В) смесь ИПС с МеОН (90:10) со скоростью потока 22 мл/мин и следующей изократической системой:
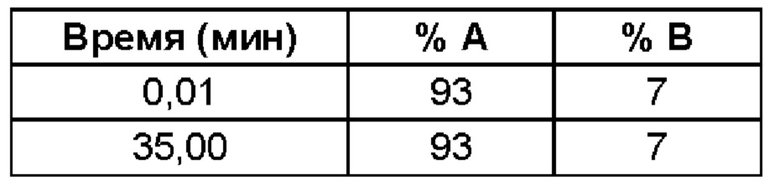
Протокол хирального анализа и определения величин удельного оптического вращения
Приборы и оборудование: Автоматический поляриметр Optical Activity АА-10
Automatic Polarimeter
Длина волны: 589 нм
Температура: 23°С
Длина оптического пути ячейки: 1 дм
Растворитель: Хлороформ (Fisher, класс чистоты «для ВЭЖХ»)
Концентрация: 1,0 г/100 мл
Метод отбора проб
Прибор включали и оставляли стабилизироваться в течение 30 минут перед проверкой калибровки с помощью образцовой кварцевой пластинки для контроля оптической активности Optical Activity Quartz Controi Plate (кат. №00049). Угловое вращение измеряли при 23°С на длине волны желтой D-линии натрия и получали величину 34,16° (после первоначальной установки прибора на ноль и без измерительной пробирки). Затем проверяли качество измерительной пробирки для образцов, для чего производили установку прибора на ноль, затем заполняли измерительную пробирку хлороформом и проверяли показания прибора, которые должны оставаться на 0,00 (±0,02). Прибор снова устанавливали на ноль с установленной в нем измерительной пробиркой с холостым образцом (хлороформом).
Образец растворяли в CHCl3 (2 мг в 2 мл), фильтровали, и в измерительную ячейку переносили пипеткой 2 мл для измерений величины α.
Удельное оптическое вращение рассчитывали согласно следующему уравнению: 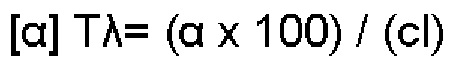
Синтез промежуточных соединений:
Промежуточное соединение А: 1-(4-хлорфенил)-3-(диметиламино)пропан-1-она гидрохлооид
К раствору 4'-хлорацетофенона (10 г, 65 ммоль) в абсолютном EtOH (50 мл) при комнатной температуре добавляли параформальдегид (1,94 г, 64 моль), N,N-диметиламина гидрохлорид (5,27 г, 64,68 ммоль) и конц. HCl (2 мл). Полученную реакционную смесь перемешивали при 80-90°С в течение 30 ч. Реакционную смесь концентрировали при пониженном давлении, и полученный остаток очищали методом колоночной хроматографии с силикагелем (60-120 меш), элюируя смесью 2% EtOAc/гексан, и растиранием с Et2O (100 мл), в результате чего получали указанное в заголовке соединение (10 г, 40 ммоль, выход 62%).
Промежуточное соединение В: 3-(диметиламино)-1-(4-фторфенил)пропан-1-она гидрохлорид
Промежуточное соединение В получали таким же способом, который был описан для получения Промежуточного соединения А, за исключением того, что использовали 4'-фторацетофенон (20 г, 144,87 ммоль), а полученный остаток очищали методом колоночной хроматографии с силикагелем (60-120 меш), элюируя смесью 4% МеОН/ДХМ), и последующим растиранием с Et2O (400 мл), в результате чего получали указанное в заголовке соединение (15 г, 77 ммоль, выход 53%).
Промежуточное соединение С: 4-(3-(диметиламино)поопанол)бензонитоила гидрохлорид
Промежуточное соединение С получали таким же способом, который был описан для получения Промежуточного соединения А, за исключением того, что использовали 4-ацетилбензонитрил (25 г, 172 ммоль), а полученный остаток очищали методом колоночной хроматографии с силикагелем (60-120 меш), элюируя смесью 5% МеОН/ДХМ, и последующим растиранием с Et2O (400 мл), в результате чего получали указанное в заголовке соединение (20 г, 99 ммоль, выход 57%).
ПРИМЕР 1
Получение Атропизомеров А-1 и А-2
Атропизомеры А-1 и А-2 могут быть получены согласно Пути синтеза А, как это показано ниже.
Путь синтеза А
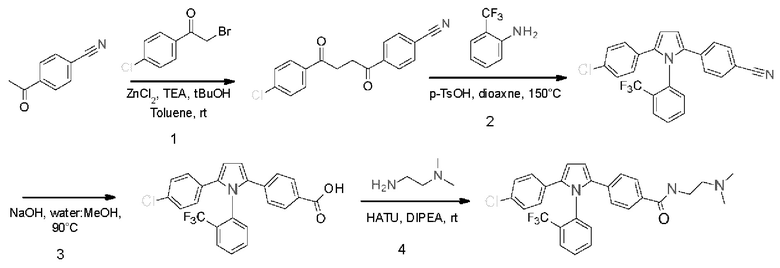
Стадия 1: 4-[4-(4-хлорфенил)-4-оксо-бутаноил]бензонитрил
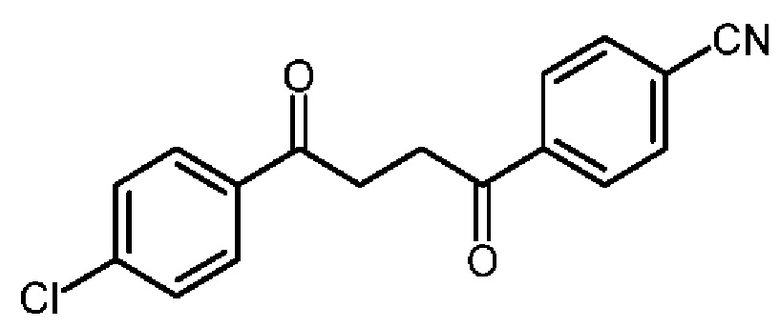
Хлорид цинка (30,5 г, 223 ммоль) нагревали до плавления под вакуумом и затем охлаждали до комнатной температуры. Затем добавляли толуол (100 мл), трет-бутанол (16,5 мл, 172 ммоль) и ТЭА (24 мл, 172 ммоль), и смесь перемешивали при комнатной температуре в атмосфере азота в течение 2 часов, и за это время хлорид цинка полностью растворился. Затем добавляли 4-цианоацетофенон (25 г, 172 ммоль) и 4-хлорфенацилбромид (40,2 г, 172 ммоль), и полученную реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Реакционную смесь разбавляли EtOAc (300 мл) и промывали водой (5 × 100 мл). Объединенные органические экстракты сушили (над Na2SO4) и упаривали при пониженном давлении. Полученный остаток очищали путем растирания с МТБЭ (400 мл), в результате чего получали указанное в заголовке соединение (30 г, 101 ммоль, выход 59%).
Стадия 2: 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензонитрил
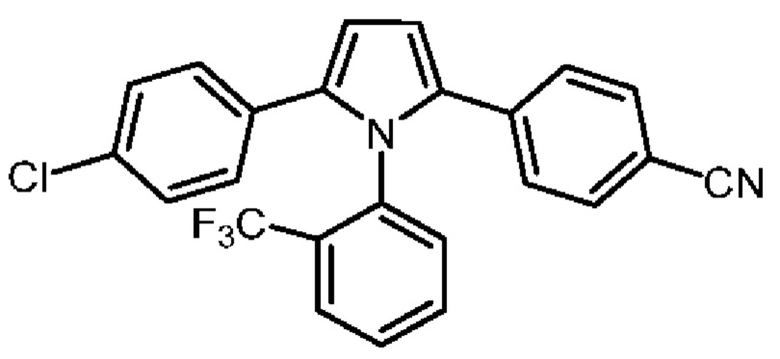
Перемешиваемый раствор 4-(4-(4-хлорфенил)-4-оксобутаноил)бензонитрила (30 г, 101 ммоль), 2-трифторметиланилина (48,79 г, 303 ммоль) и ПТСК (1,92 г, 10,099 ммоль) в диоксане (300 мл) нагревали при 150°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, и полученный остаток очищали методом колоночной хроматографии на силикагеле (60-120 меш), используя смесь 8% EtOAc/гексан в качестве элюента, в результате чего получали указанное в заголовке соединение (30 г, 71 ммоль, выход 70%).
Стадия 3: 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензойная кислота
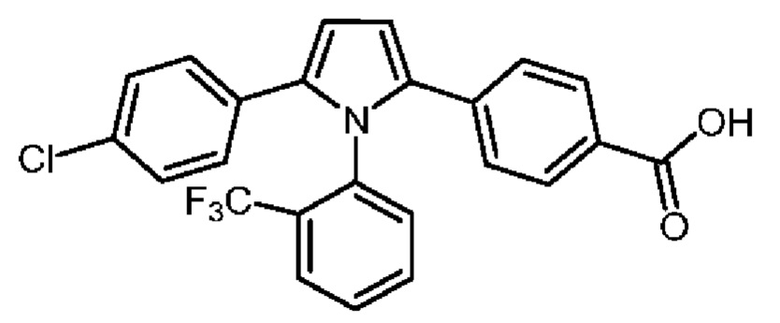
К раствору 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензонитрила (2 г, 4,739 ммоль) в МеОН (20 мл) добавляли NaOH (1,89 г, 47 ммоль) в воде (10 мл), и полученную смесь перемешивали при 90°С в течение 24 часов. Смесь концентрировали при пониженном давлении, и полученный остаток очищали растиранием с Et2O (10 мл), в результате чего получали указанное в заголовке соединение (1,8 г, 4,1 ммоль, выход 86%).
Стадия 4: 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)-N-(2-(диметиламино)этил)бензамид
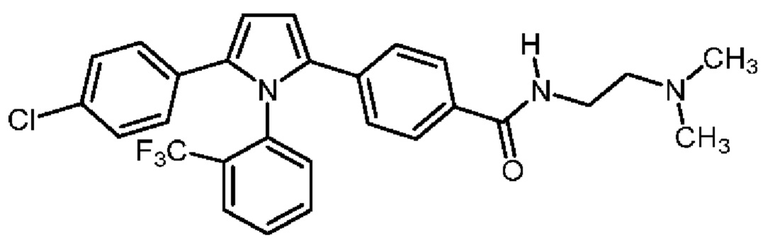
К раствору 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензойной кислоты (1,8 г, 4,0 ммоль) в ДМФА (12 мл) при постоянном перемешивании добавляли ДИПЭА (2,13 мл, 22 ммоль), а затем HATU (4,65 г, 12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, после чего к ней по каплям добавляли N,N-диметилэтилендиамин (1,08 г, 12 ммоль) и продолжали перемешивание при комнатной температуре в течение 4 ч. Затем смесь выливали в ледяную воду (150 мл) и экстрагировали EtOAc (3 × 100 мл). Объединенные органические слои сушили (над Na2SO4) и концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на нейтральном оксиде алюминия, элюируя смесью 6% МеОН/ДХМ, в результате чего получали указанное в заголовке соединение (1,2 г, 2,3 ммоль, выход 57%) в виде смеси атропизомеров.
Разделение атропизомеров
Атропизомеры (А-1 и А-2) 4-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида могут быть разделены с помощью хиральной ВЭЖХ с использованием Метода 1 препаративной хиральной ВЭЖХ.
Выделяли два пика:
Пик 1: Атропизомер А-1, 4-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2(диметиламино)этил]бензамид (атропизомер 1) (0,3 г, 0,58 ммоль, выход 38%, >99% э.и.), и
Пик 2: Атропизомер А-2, 4-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2(диметиламино)этил]бензамид (атропизомер 2) (0,31 г, 0,606 ммоль, выход 39%, 98% э.и.).
Соединения также могут быть выделены в виде их гидрохлоридных солей.
ПРИМЕР 2
Дальнейшая очистка и определение характеристик атропизомеров
Атропизомер А-1: 4-[5-(4-хлооФенил)-1-[2-(триФтоометил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида гидрохлоридная соль
Материал согласно Пику 1 (0,31 г, 0,606 ммоль) дополнительно очищали путем перемешивания в воде класса чистоты «для ВЭЖХ» (30 мл), после чего обрабатывали ультразвуком в течение 10 мин и экстрагировали EtOAc (3 × 30 мл). Объединенные органические слои сушили (над Na2SO4), фильтровали и концентрировали при пониженном давлении с последующей лиофилизацией, в результате чего получали аморфное твердое вещество (0,290 г, 0,567 ммоль, выход 94%), которое растворяли в ДХМ (7,12 мл). Полученный раствор охлаждали до 0°С, и к нему добавляли 4 н. раствор HCl в диоксане (1,42 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь концентрировали и сушили под глубоким вакуумом. Очистка растиранием с Et2O (10 мл) и последующей лиофилизацией давала указанное в заголовке соединение (0,3 г, 0,56 ммоль, выход 98%) в виде почти белого твердого вещества.
1H ЯМР (ДМСО-d6) δ 10,03, (brs, 1Н), 8,62 (s, 1Н), 7,81 - 7,68 (m, 6Н), 7,25 (d, J=8,4 Hz, 2Н), 7,10 - 7,03 (m, 4Н), 6,67 - 6,58 (m, 2Н), 3,56 - 3,54 (m, 2Н), 3,20 - 3,18 (m, 2Н), 2,76 (s, 6Н). ЖХ-МС (Метод 1): RT 2,54, МН+ 512,4
Атропизомер А-2: 4-[5-(4-хлорфенил)-1-[2-(триФторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида гидрохлоридная соль
Гидрохлоридную соль Атропизомера А-2 получали с использованием того же способа, что и для Атропизомера А-1, начиная с Пика 2, в результате чего получали указанное в заголовке соединение (0,31 г, 0,56 ммоль, выход 99%) в виде почти белого твердого вещества.
1H ЯМР (ДМСО-d6) δ 9,91 (brs, 1Н), 8,69 (s, 1Н), 7,81 - 7,68 (m, 6Н), 7,25 (d, J=8,0 Гц, 2Н), 7,10 - 7,03 (m, 4Н), 6,67 - 6,58 (m, 2Н), 3,56 - 3,54 (m, 2Н), 3,20 - 3,18 (m, 2Н), 2,77 (s, 6Н). ЖХ-МС (Метод 1): RT 2,56, МН+ 512,4
Монокристальный рентгено-кристаллографический анализ Атропизомера А-2 (см. Пример 3 ниже) показал, что Атропизомер А-2 представляет собой R-изомер (Соединение (1)), и, следовательно, Атропизомер А-1 должен представлять собой S-изомер.
Хиоальный анализ
Анализ хиральных свойств Атропизомеров А-1 и А-2 проводили путем измерения их величин оптического вращения и времени удерживания, полученных методом хиральной ВЭЖХ с использованием описанных выше способов, в результате чего получили данные, приведенные в таблице ниже.
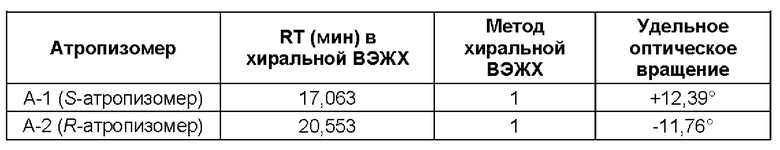
Классификация атропизомеров
Проводили исследования стабильности выделенных атропизомеров, т.е. Атропизомеров А-1 и А-2.
Для оценки процесса взаимопревращений между Атропизомером А-1 и Атропизомером А-2 проводили мониторинг их хиральной стабильности при 40°С и 80°С. Как видно из приведенных ниже результатов, при нагревании в течение 10 дней при любой температуре никаких взаимных превращений не наблюдалось.
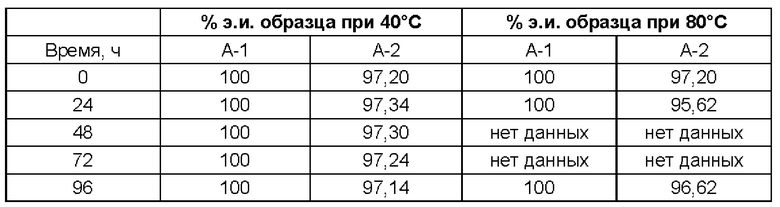

Протокол:
1. В закрытом мерном флаконе растворяли 2 × 1 мг чистого атропизомера в 1 мл EtOH.
2. Часть флаконов нагревали при 40°С, другую часть - при 80°С.
3. В определенные моменты времени отбирали аликвоту 20 мкл каждого исходного раствора (1 мл), которую быстро охлаждали, выливая во флакон для ВЭЖХ, содержащий 80 мкл смеси гексана и EtOH (80:20), в результате чего получали раствор с конечной концентрации 200 ppm, и образец такого раствора анализировали с помощью хиральной ВЭЖХ
4. Анализ проводили в следующих временных точках: 0 ч, 24 ч, 48 ч, 72 ч, 96 ч и 240 ч для образцов, хранившихся при 40°С и 24 ч, 96 ч и 240 ч для образцов, хранившихся при 80°С, с использованием Метода 5 хиральной ВЭЖХ.
Стабильность выделенных атропизомеров, т.е. Атропизомеров А-1 и А-2, показала, что они являются атропизомерами Класса 3 (LaPlante et al., J. Med. Chem., 54: 7005-7022 (2011)).
ПРИМЕР 3
Рентгеновский кристаллографический анализ Атропизомера А-2
Готовили Атропизомер А-2 в виде свободного основания, и его монокристалл подвергали рентгеновским кристаллографическим исследованиям согласно описанному ниже.
Экспериментальная часть:
Монокристаллы Атропизомера А-2 неопределенной морфологии получали перекристаллизацией из метилизобутилкетона (МИБК). Выбирали подходящий кристалл размерами 0,19 мм × 0,13 мм × 0,04 мм, который с помощью держателя MiTiGen MicroMount устанавливали на дифрактометр Rigaku XtaLAB Syngery-S, снабженный детектором HyPix-6000НЕ. В ходе сбора данных температуру кристалла стабильно поддерживали на уровне Т=123(2)K.
Данные получали с использованием излучения линии CuKα. Максимальное достигнутое разрешение составило Θ=74,263° (0,80 Å). Проводили обработку данных, масштабирование и корректировку величин поглощения. Окончательная полнота составила 100,00% из величин до 74,263° Θ. Коэффициент поглощения μ соединения определяли как 1,761 мм-1 при данной длине волны (λ=1,542Å).
Данные были собраны и обработаны с использованием программного обеспечения CrysAlisPro, и структура была определена с помощью программы определения структур SheIXT (Sheldrick, 2015) методом Intrinsic Phasing (внутренней фазировки) и с помощью графического интерфейса Olex2 (Dolomanov et al., 2009). Модель была доработана с использованием версии 2018/3 SheIXL-2018/3 (Sheidrick, 2018) и метода наименьших квадратов.
Было установлено, что кристаллическая структура является моноклинной, и ей была присвоена пространственная группа Р21 (#4).
Все неводородные атомы уточняли анизотропно. Положения атомов водорода рассчитывали геометрически и уточняли с помощью модели «наездника».
Справочная литература: O.V. Dolomanov and L.J. Bourhis and R.J. Gildea and J.A.K. Howard and H. Puschmann, Olex2: A complete structure solution, refinement and analysis program, J. Appl. Cryst., (2009), 42, 339-341.
Sheldrick, G.M., Crystal structure refinement with SheIXL, Acta Cryst., (2015), C71, 3-8.
Sheldrick, G.M., SheIXT-Integrated space-group and crystal-structure determination, Acta Cryst., (2015), A71, 3-8.
Результаты исследований приведены ниже в Таблицах 1-7.
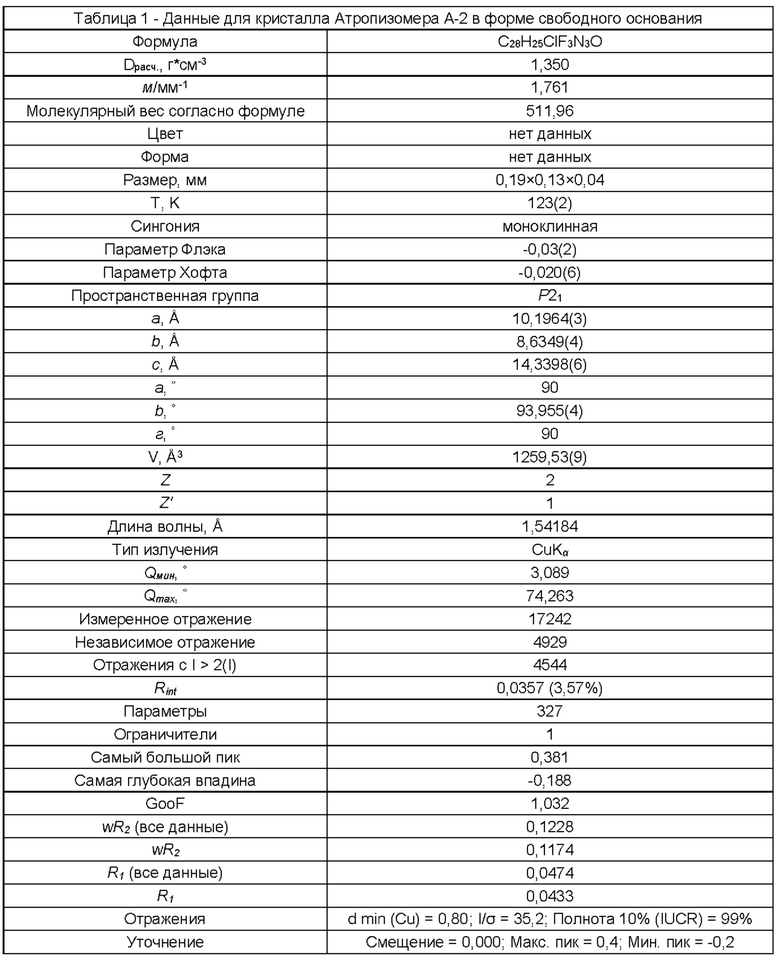
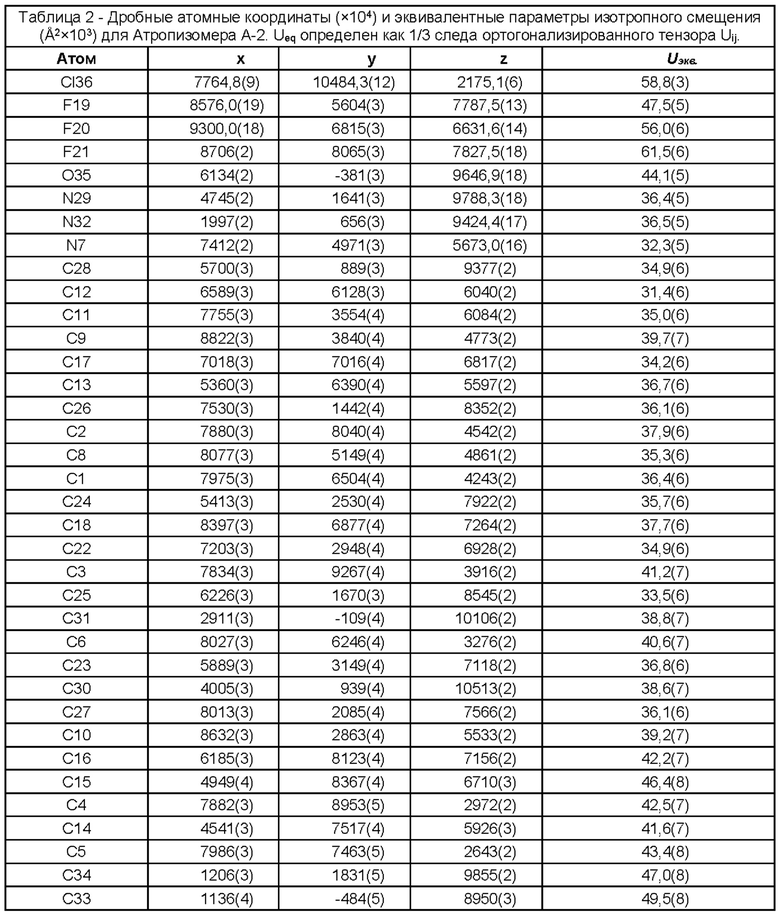
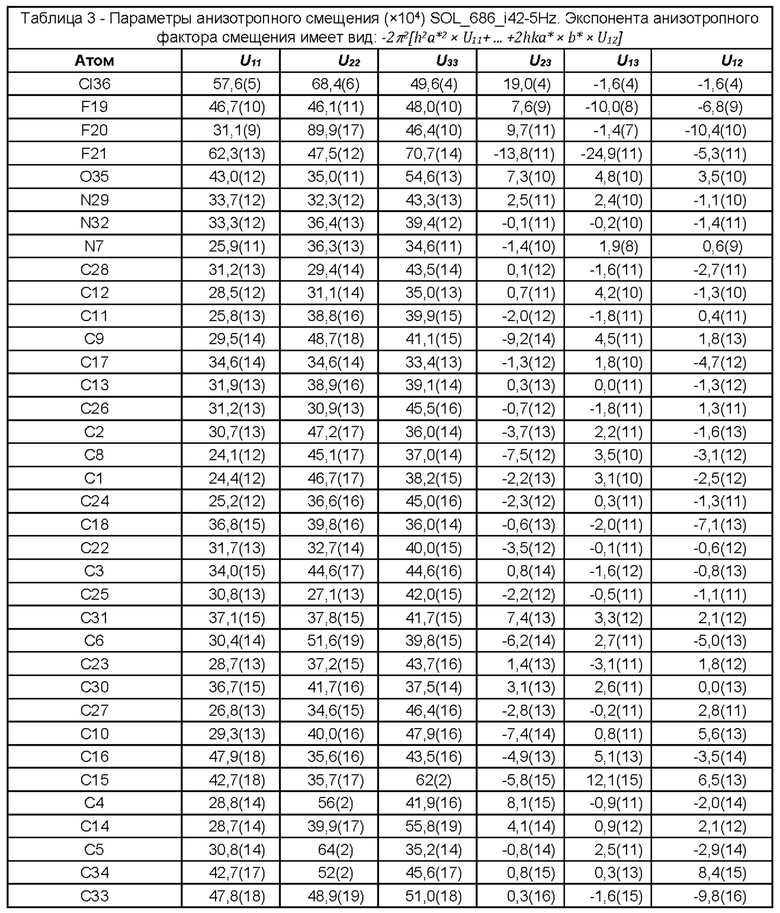
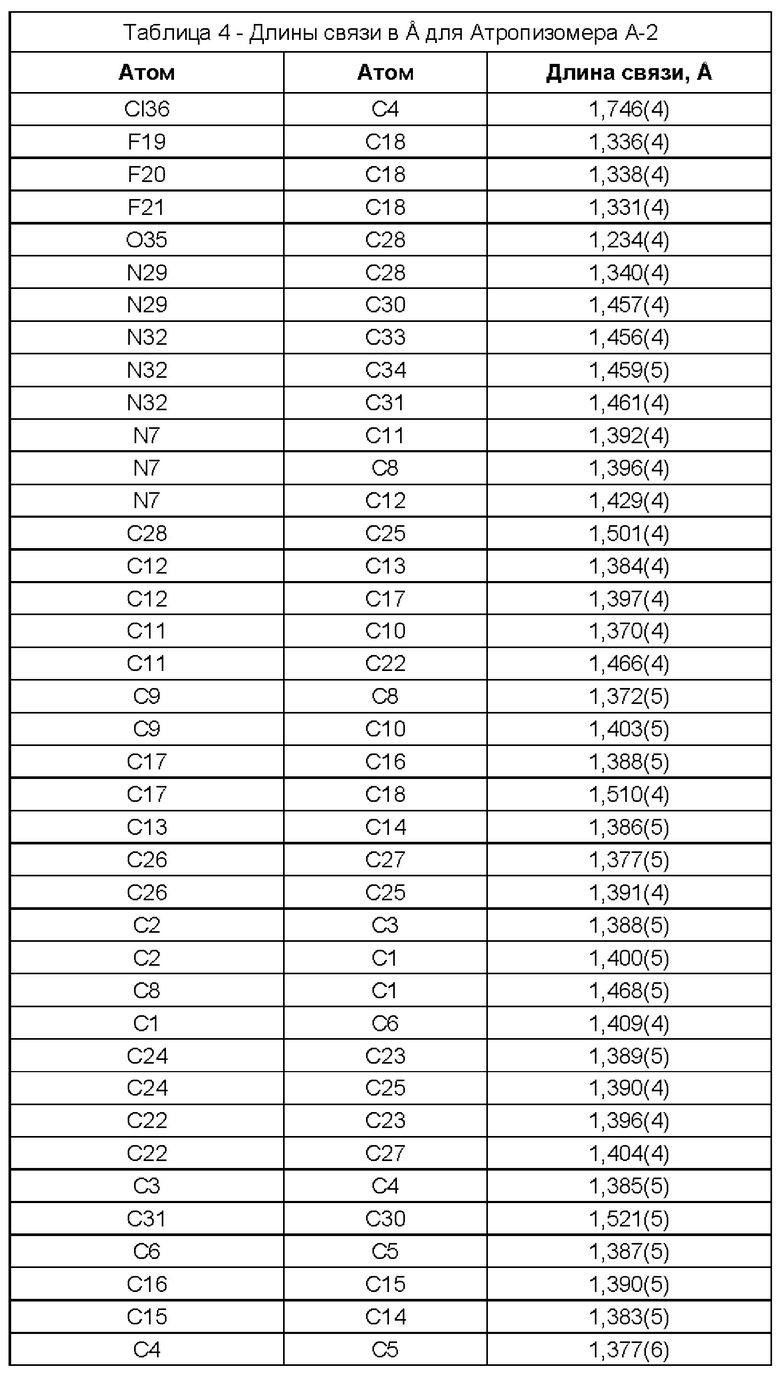
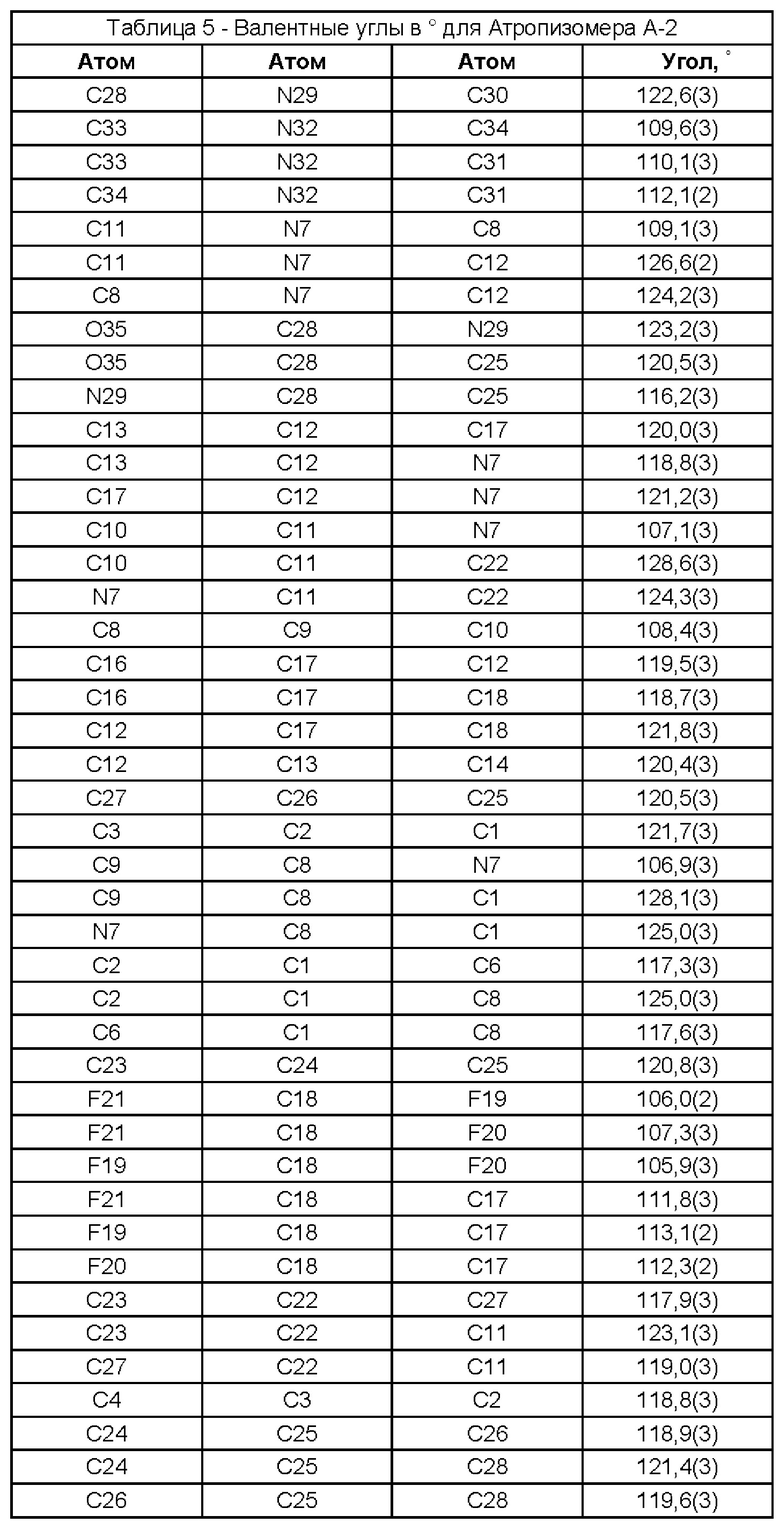
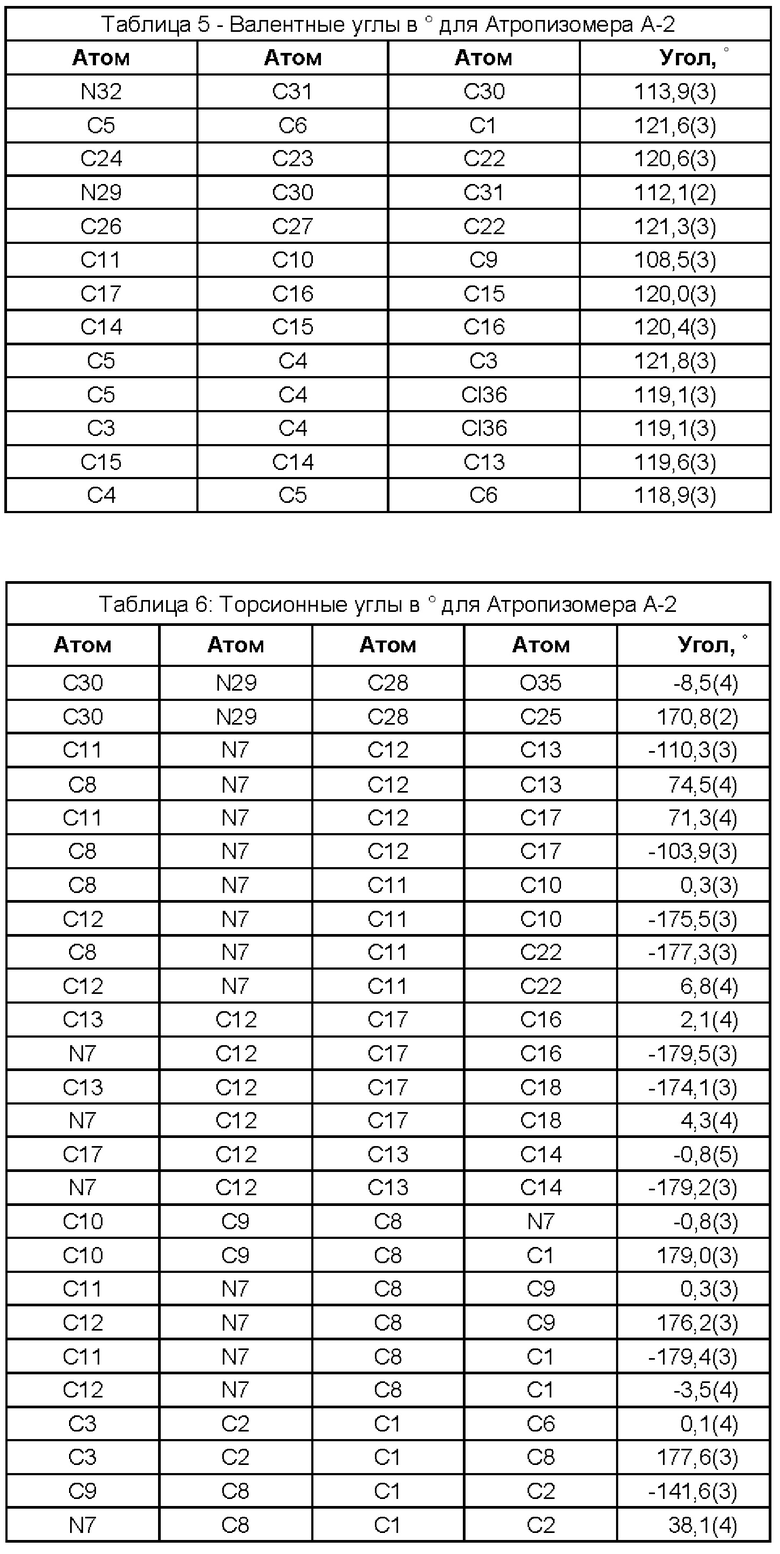
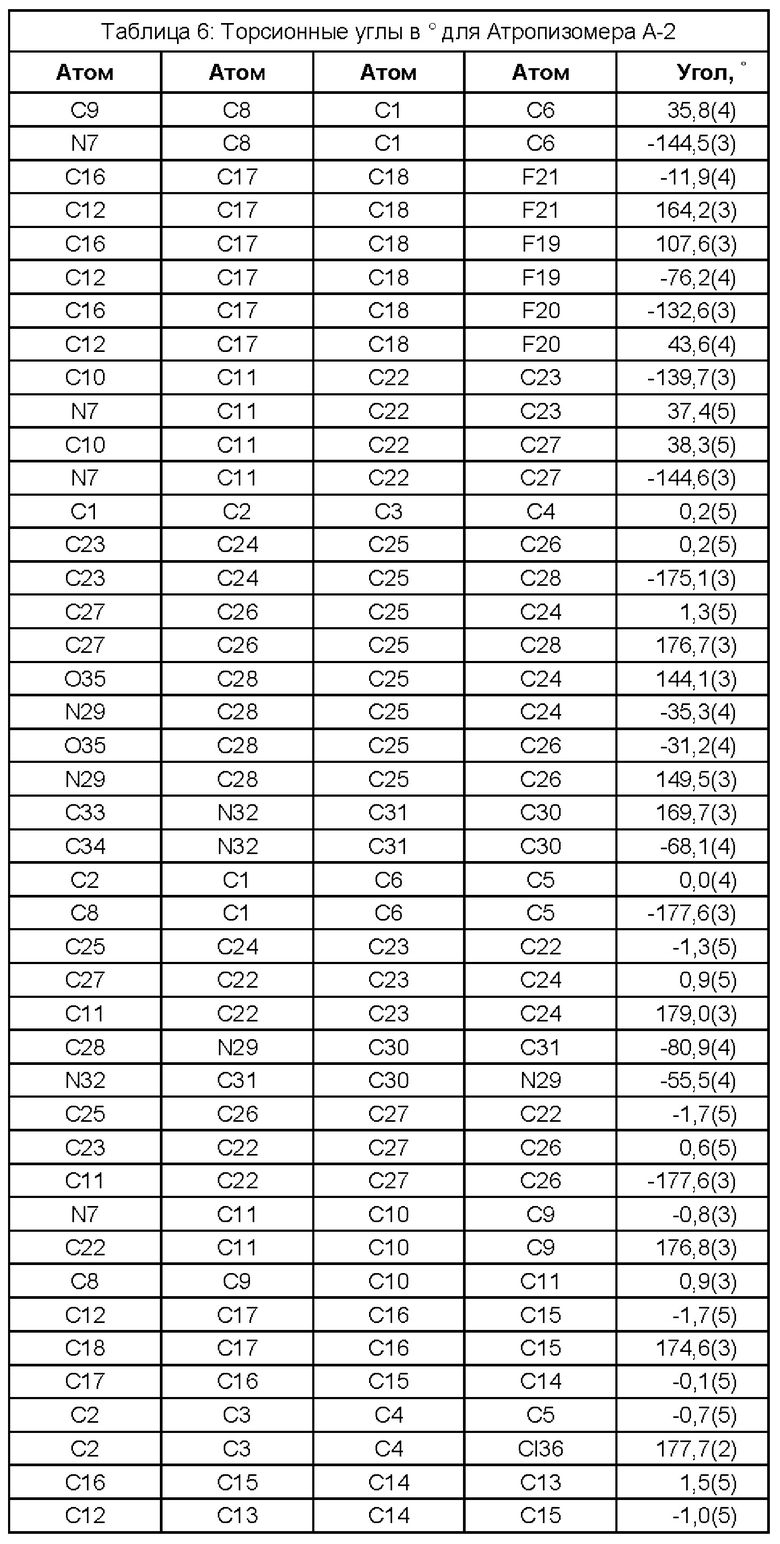
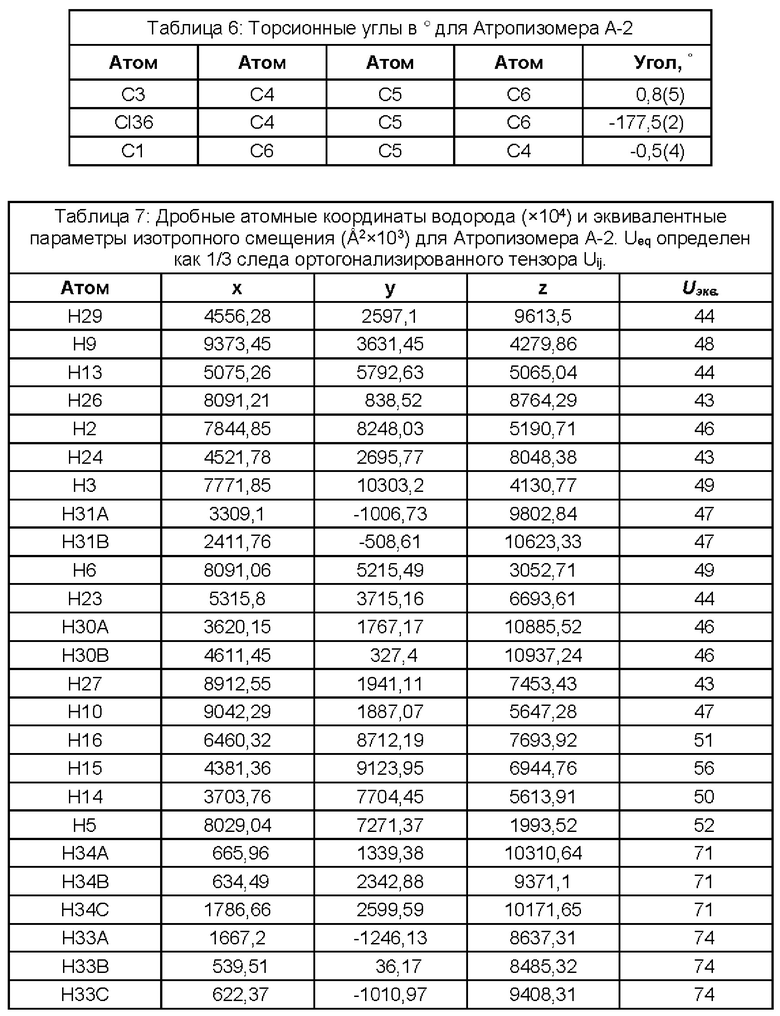
На основании данных, изложенных ниже, полагают, что Атропизомер А-2 имеет конфигурацию R, как это показано на Фиг. 2 и 3, и, следовательно, может быть назван как (R)-4-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамид.
ПРИМЕР 4
Получение Атропизомеров А-3 и А-4
Атропизомеры А-3 и А-4 получали, следуя Пути синтеза В, как это показано ниже.
Путь синтеза В
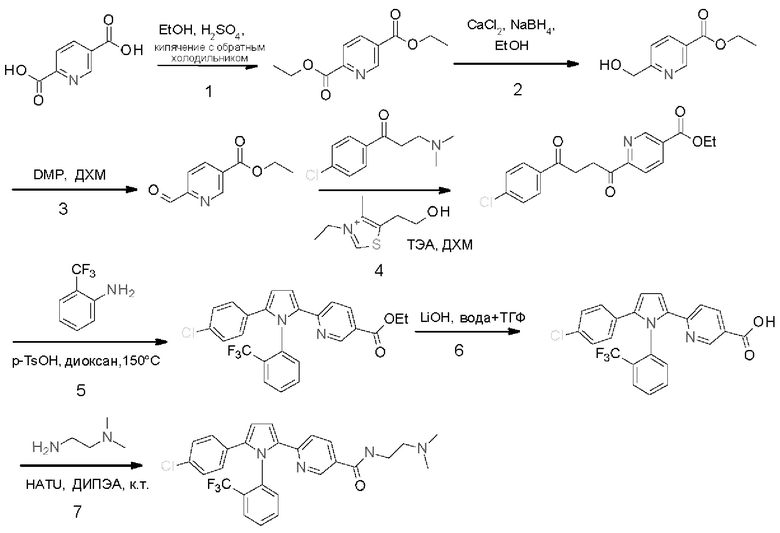
Стадия 1: Диэтилпиридин-2,5-дикарбоксилат
К суспензии 2,5-пиридиндикарбоновой кислоты (20 г, 120 ммоль) в абсолютном EtOH (120 мл) по каплям в течение 30 мин добавляли конц. H2SO4 (25,6 мл, 0,048 ммоль). Полученную реакционную смесь кипятили с обратным холодильником в течение 48 часов. Затем реакционную смесь концентрировали, и полученный остаток подщелачивали до уровня рН 8 (насыщенным водн. раствором NaHCO3). Полученный водный слой экстрагировали EtOAC (4 × 200 мл). Объединенные органические слои промывали солевым раствором, сушили (над Na2SO4) и концентрировали. Параллельно проводили аналогичную реакцию с четырьмя другими партиями по 20 г, и полученный в каждой реакции сырой материал объединяли и очищали методом колоночной хроматографии на силикагеле (60-120 меш), элюируя смесью 5% EtOAC/гексан, в результате чего получали указанное в заголовке соединение (65 г, 291 ммоль, выход 49%).
Стадия 2: Этил-6-(гидроксиметил)пиридин-3-карбоксилат
К охлажденному (на ледяной бане) раствору диэтилпиридин-2,5-дикарбоксилата (10 г, 45 ммоль) в смеси абсолютного EtOH (40 мл) и ТГФ (3,5 мл) в атмосфере азота по частям в течение 30 мин добавляли NaBH4 (4,26 г, 112 ммоль) и безводный CaCl2 (7,86 г, 71 ммоль). Полученную реакционную смесь перемешивали при 0°С в течение 5 ч. Затем реакционную смесь выливали в насыщенный водный раствор NH4Cl (150 мл) и экстрагировали EtOAc (4 × 150 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Параллельно проводили аналогичную реакцию с шестью другими партиями по 10 г и одной партией 5 г, и полученный в каждой реакции сырой материал объединяли и очищали методом колоночной хроматографии с силикагелем (60-120 меш) с элюированием смесью 20% EtOAc/гексан, в результате чего получали указанное в заголовке соединение (55 г, 320 ммоль, выход 100%).
Стадия 3: Этил-6-формилпиридин-3-карбоксилат
К охлажденному (на ледяной бане) раствору этил-6-(гидроксиметил)пиридин-3-карбоксилата (30 г, 166 ммоль) в ДХМ (360 мл) в атмосфере азота по частям в течение 20 мин добавляли DMP (84,32 г, 199 ммоль). Реакционную смесь перемешивали при к.т. в течение 3 ч. Реакционную смесь выливали в ледяную воду (1,5 л), и полученную смесь подщелачивали до уровня ~рН 8 (насыщенным водным раствором NHCO3) и экстрагировали EtOAc (4 × 1000 мл). Объединенные органические слои промывали солевым раствором, сушили (над Na2SO4) и концентрировали. Полученный остаток очищали методом колоночной хроматографии с силикагелем (60-120 меш), элюируя смесью 12% EtOAc/гексан, в результате чего получали указанное в заголовке соединение (19 г, 106 ммоль, выход 33%).
Стадия 4: Этил-6-[4-(4-хлорфенил)-4-оксо-бутаноил]пиридин-3-карбоксилат
К перемешиваемому раствору Промежуточного соединения А (1,17 г, 5,6 ммоль) и ТЭА (1,56 мл, 11,2 ммоль) в 1,2-диметоксиэтане (10 мл) при комнатной температуре добавляли этил-6-формилпиридин-3-карбоксилат (1 г, 5,6 ммоль) и 3-этил-5-(2-гидроксиэтил)-4-метилтиазол-3-ий бромид (0,28 г, 11,2 ммоль). Полученный раствор нагревали при 80-90°С в течение 5 ч. Реакционную смесь разбавляли ледяной водой (400 мл) и экстрагировали EtOAc (3 × 200 мл). Объединенные органические слои сушили (над Na2SO4) и концентрировали. Полученный остаток очищали методом колоночной хроматографии с силикагелем (60-120 меш), элюируя смесью 8% EtOAc/гексан, в результате чего получали указанное в заголовке соединение (5,5 г, 15,9 ммоль, выход 17%).
Стадия 5: Этил-6-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]пиридин-3-карбоксилат
К раствору этил-6-[4-(4-хлорфенил)-4-оксо-бутаноил]пиридин-3-карбоксилата (2,5 г, 7,2 ммоль) в 1,4-диоксане (25 мл) при комнатной температуре добавляли 2-аминобензотрифторид (3,5 г, 21,7 ммоль) и ПТСК (0,14 г, 0,72 ммоль). Полученный раствор нагревали при 150°С в течение 48 ч. Реакционную смесь концентрировали и очищали методом колоночной хроматографии с силикагелем (60-120 меш) с элюированием смесью 6% EtOAc/гексан, в результате чего получали указанное в заголовке соединение (2,5 г, 5,3 ммоль, выход 64%).
Стадия 6: 6-[5-(4-Хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]пиридин-3-карбоновая кислота
К раствору этил-6-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]пиридин-3-карбоксилата (2,2 г, 4,7 ммоль) в смеси ТГФ (10 мл) и воды (10 мл) при комнатной температуре добавляли LiOH (0,59 г, 14 ммоль). Полученный раствор перемешивали при 80°С в течение 16 ч. Реакционную смесь концентрировали, разбавляли водой (150 мл) и экстрагировали EtOAc (4 × 150 мл). Объединенные органические экстракты сушили (над Na2SO4) и концентрировали. Полученный материал растирали с н-пентаном (15 мл) и Et2O (15 мл), в результате чего получали указанное в заголовке соединение (2 г, 4,5 ммоль, выход 97%).
Стадия 7: 4-[5-(4-Хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамид
К раствору 6-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]пиридин-3-карбоновой кислоты (2,8 г, 6,33 моль) в ДМФА (20 мл) добавляли HATU (7,22 г, 19 моль), и реакционную смесь перемешивали при комнатной температуре в течение 20 мин. Затем добавляли unsym-N,N-диметилэтилендиамин (1,11 г, 12,7 моль) и ДИПЭА (3,31 мл, 19 моль), и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь разбавляли ледяной водой (200 мл) и экстрагировали EtOAc (4 × 100 мл). Объединенные органические экстракты сушили (над Na2SO4) и концентрировали. Полученный остаток очищали методом колоночной хроматографии с силикагелем (60-120 меш), элюируя смесью 30% EtOAc/гексан, в результате чего получали указанное в заголовке соединение (2,4 г, 4,7 ммоль, выход 74%).
Стадия 8: Разделение Атропизомеров А-3 и А-4
Атропизомеры 6-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]пиридин-3-карбоксамида могут быть разделены с помощью хиральной ВЭЖХ с использованием Метода 2 препаративной хиральной ВЭЖХ.
Выделяли два пика:
Пик 1: Атропизомер А-3, 6-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]пиридин-3-карбоксамид (атропизомер 1) (70 мг, 0,14 ммоль, выход 355%), твердое вещество коричневого цвета.
Пик 2: Атропизомер А-4, 6-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]пиридин-3-карбоксамид (атропизомер 2) (75 мг, 0,15 ммоль, выход 38%), твердое вещество коричневого цвета.
Материалы согласно обоим пикам дополнительно очищали с целью удаления алифатических примесей:
Пик 1: (А-3) (57 мг, 0,11 ммоль) разбавляли водой класса чистоты «для ВЭЖХ» (25 мл), затем обрабатывали ультразвуком в течение 10 минут и экстрагировали EtOAc (3 × 20 мл). Объединенные органические экстракты сушили (над Na2SO4), фильтровали, концентрировали и лиофилизировали, в результате чего получали Атропизомер А-3 (56 мг, 0,11 ммоль, выход 98%, >99% э.и.).
1H ЯМР (ДМСО-d6) δ 8,45 - 8,43 (m, 2Н), 8,01 (d, J=6,8 Гц, 1Н), 7,74 - 7,68 (m, 2Н), 7,65 - 7,60 (m, 3Н), 7,25 (d, J=8,4 Гц, 2Н), 7,11 - 7,04 (m, 3Н), 6,60 (d, J=4 Гц, 1Н), 3,32 (m, 2Н, перекрытый пиком остаточной воды), 2,30 (m, 2Н, перекрытый пиком остаточного растворителя), 2,19 (s, 6Н). ЖХ-МС (Метод 1): RT 2,41, МН+ 513,4
Пик 2: (А-4) (60 мг, 0,117 ммоль) разбавляли водой класса чистоты «для ВЭЖХ» (25 мл), затем обрабатывали ультразвуком в течение 10 минут и экстрагировали EtOAc (3 × 20 мл). Объединенные органические экстракты сушили (над Na2SO4), фильтровали, концентрировали и лиофилизировали, в результате чего получали Атропизомер А-4 (60 мг, 0,12 ммоль, выход 99%, 95% э.и.).
1H ЯМР (ДМСО-d6) δ 8,47 - 8,43 (m, 2Н), 8,02 (d, J=7,2 Гц, 1Н), 7,74 - 7,68 (m, 2Н), 7,65 - 7,60 (m, 3Н), 7,25 (d, J=8,4 Гц, 2Н), 7,11 - 7,04 (m, 3Н), 6,60 (d, J=4 Гц, 1Н), 3,32 (m, 2Н, перекрытый пиком остаточной воды), 2,30 (m, 2Н, перекрытый пиком остаточного растворителя), 2,20 (s, 6Н). ЖХ-МС (Метод 1): RT 2,41, МН+ 513,4
Хиральный анализ
Анализ хиральных свойств Атропизомеров А-3 и А-4 проводили путем измерения их величин оптического вращения и времени удерживания, полученных методом хиральной ВЭЖХ с использованием описанных выше способов, в результате чего получили данные, приведенные в таблице ниже.
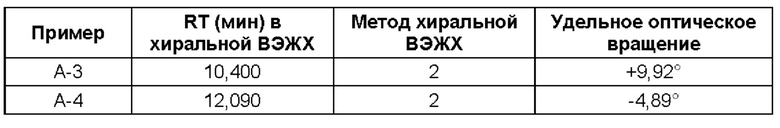
ПРИМЕР 5
Получение Атропизомеров А-5 и А-6: N-[2-(диметиламино)этил]-6-[5-(4-фторфенил)-1-[2-(триФтоометил)фенил]пиррол-2-ил]пиридин-3-карбоксамид
Атропизомеры А-5 и А-6 получали в виде рацемической смеси тем же способом, который был описан выше в Примере 4 для получения Атропизомеров А-3 и А-4 за исключением следующего: (а) на Стадии 4 использовали Промежуточное соединение В (3,23 г, 16,58 ммоль) и 3-бензил-5-(2-гидроксиэтил)-4-метилтиазол-3-ий бромид (0,678 г, 2,51 ммоль), а очистку проводили с использованием в качестве элюента смеси 10% EtOAc/гексан, (b) на Стадии 5 для очистки в качестве элюента использовали смесь 1,3% EtOAc/гексан, (с) на Стадии 6 вместо ТГФ использовали МеОН, а очистку проводили растиранием с Et2O, (d) на Стадии 7 выделенный остаток очищали методом хроматографии с гелем щелочного оксида алюминия, элюируя ДХМ с получением указанного в заголовке соединения (0,16 г, 0,32 ммоль, выход 55%), (е) очистку проводили Методом 1 препаративной ВЭЖХ, в результате чего получали указанное в заголовке соединение (61 мг, 0,12 ммоль, выход 38%) (рацемическую смесь атропизомеров) в виде гидрохлоридной соли, представляющей собой твердое вещество светло-желтого цвета.
1Н ЯМР (ДМСО-d6) δ 10,09 (bs, 1Н), 8,85 (m, 1Н), 8,49 (s, 1Н), 8,11 (d, J=8,0 Гц, 1Н), 7,73 - 7,70 (m, 2Н), 7,69 - 7,61 (m, 3Н), 7,13 - 7,10 (m, 3Н), 7,06- 7,02 (m, 2Н), 6,56 (d, J=4,0 Гц, 1Н), 3,56 (m, 2Н), 3,20 (m, 2Н), 2,76 (d, J=4,4 Гц, 6Н). ЖХ-МС (Метод 2): RT 5,06, МН+ 497,2
Хиральный ВЭЖХ анализ Методом 3 хиральной ВЭЖХ показал смесь атропизомеров, соответствующих Пику 1 с RT 9,95 мин и 49,8% площади (Атропизомер А-5) и Пику 2 с RT 11,52 мин и 50,2% площади (Атропизомер А-6).
ПРИМЕР 6
Получение Атропизомеров А-7 и А-8 6-[5-(4-цианофенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]пиридин-3-карбоксамида
Атропизомеры А-7 и А-8 получали в виде рацемической смеси тем же способом, который был описан выше в Примере 4 для получения Атропизомеров А-3 и А-4 за исключением следующего: (а) на Стадии 4 использовали Промежуточное соединение С (0,28 г, 1,39 ммоль) и 3-бензил-5-(2-гидроксиэтил)-4-метилтиазол-3-ий бромид (0,04 г, 0,14 ммоль), а очистку проводили с использованием в качестве элюента смеси 10% EtOAc/гексан, (р) на Стадии 5 для очистки в качестве элюента использовали смесь 7% EtOAc/гексан, (с) на Стадии 7 выделенный остаток очищали методом хроматографии с гелем щелочного оксида алюминия, элюируя смесью 10% EtOAc/гексан с получением указанного в заголовке соединения (0,13 г, 0,25 ммоль, выход 75%), (d) очистку проводили Методом 2 препаративной ВЭЖХ, в результате чего получали указанное в заголовке соединение (54 мг, 0,11 ммоль, выход 36%) в виде гидрохлоридной соли, представляющей собой твердое вещество светло-желтого цвета.
1H ЯМР (ДМСО-d6) δ 9,83 (brs, 1Н), 8,80 (t, J=5,2 Гц, 1Н), 8,50 (d, J=1,6 Гц, 1Н), 8,11 (dd, J=8,4, 2,0 Гц, 1Н), 7,79 - 7,64 (m, 6Н), 7,32 - 7,07 (m, 4Н), 6,83 (d, J=4,0 Гц, 1Н), 3,56 - 3,46 (m, 2Н), 3,22 - 3,18 (m, 2Н), 2,78 (d, J=4,8 Гц, 6Н). ЖХ-МС (Метод 1): RT 2,05, МН+ 504,1
Хиральный ВЭЖХ анализ Методом 4 хиральной ВЭЖХ показал смесь атропизомеров, соответствующих Пику 1 с RT 8,82 мин и 50,2% площади (Атропизомер А-7) и Пику 2 с RT 10,10 мин и 49,8% площади (Атропизомер А-8).
ПРИМЕР 7
Получение Соединений В-2 - В-107
Дополнительные примеры атропизомерных соединений согласно настоящему изобретению можно приготовить путем получения рацемических смесей соединений, показанных в таблице ниже, и последующего выделения отдельных атропизомеров с использованием методов хиральной ВЭЖХ, описанных выше, или сходных с ними методов. В данной таблице указанные номера соединений соответствуют номерам Примеров в нашей более ранней международной патентной заявке WO 2018/197714, но с добавлением префикса «В-». Таким образом, Соединение В-2 соответствует Примеру 2 в WO 2018/197714, Соединение В-3 соответствует Примеру 3 в WO 2018/197714 и так далее. Данные ЯМР, ЖХ-МС и другие характеристики рацемических соединений, а также данные по их биологической активности соответствуют данным, приведенным B WO 2018/197714.
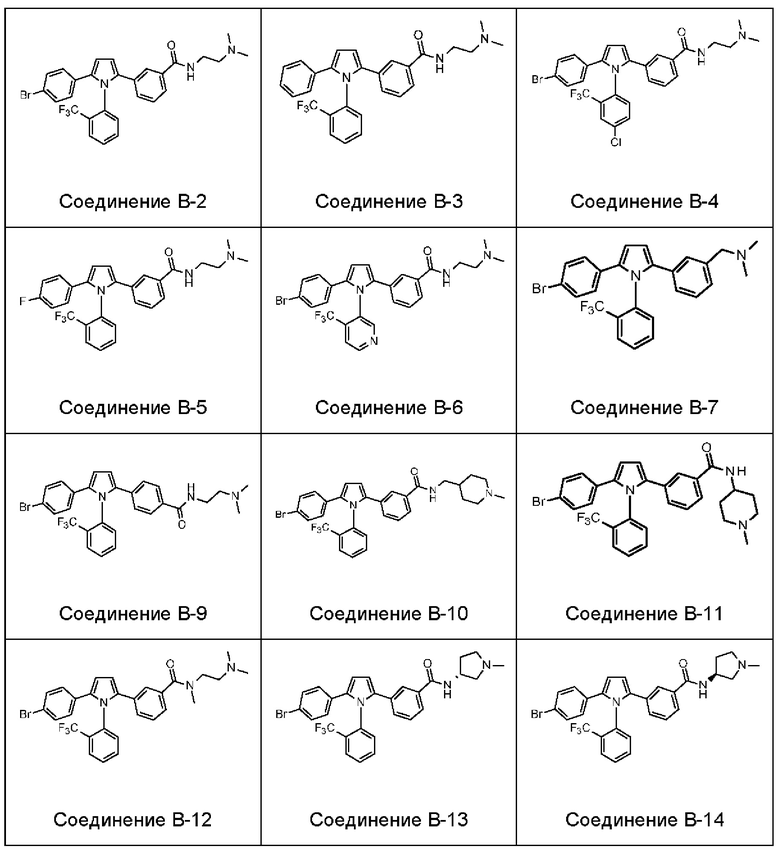
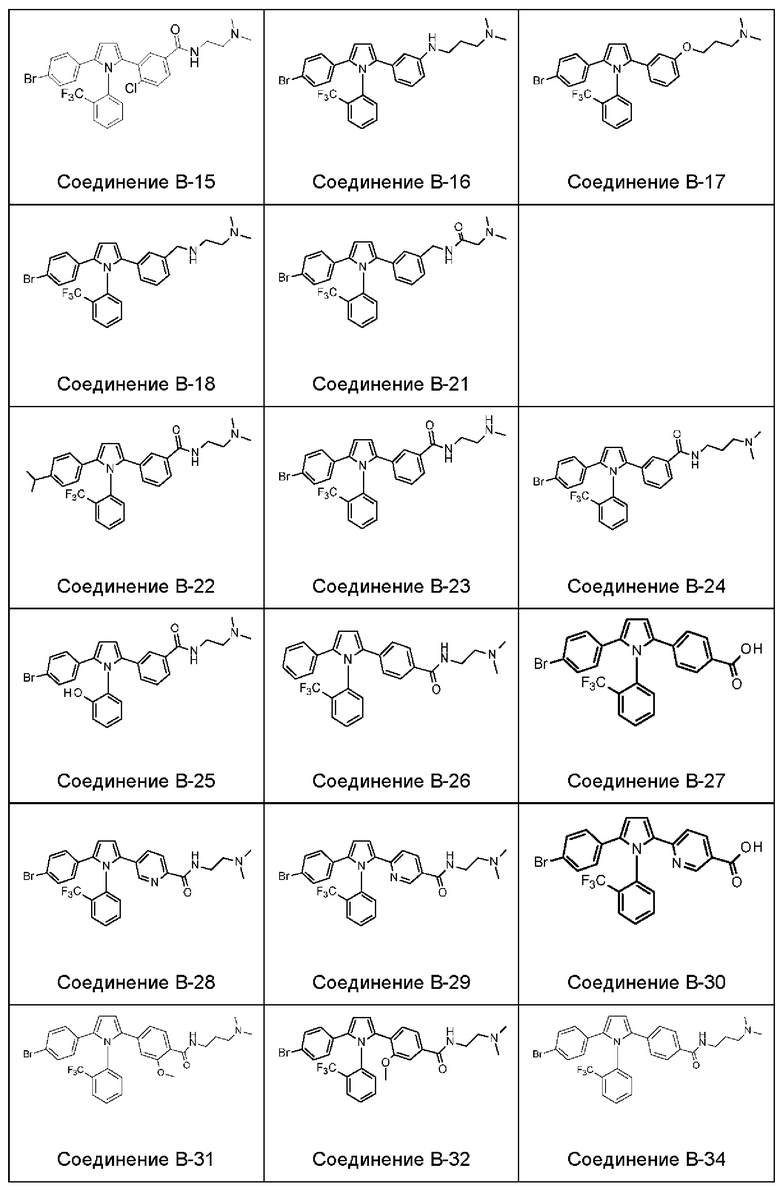
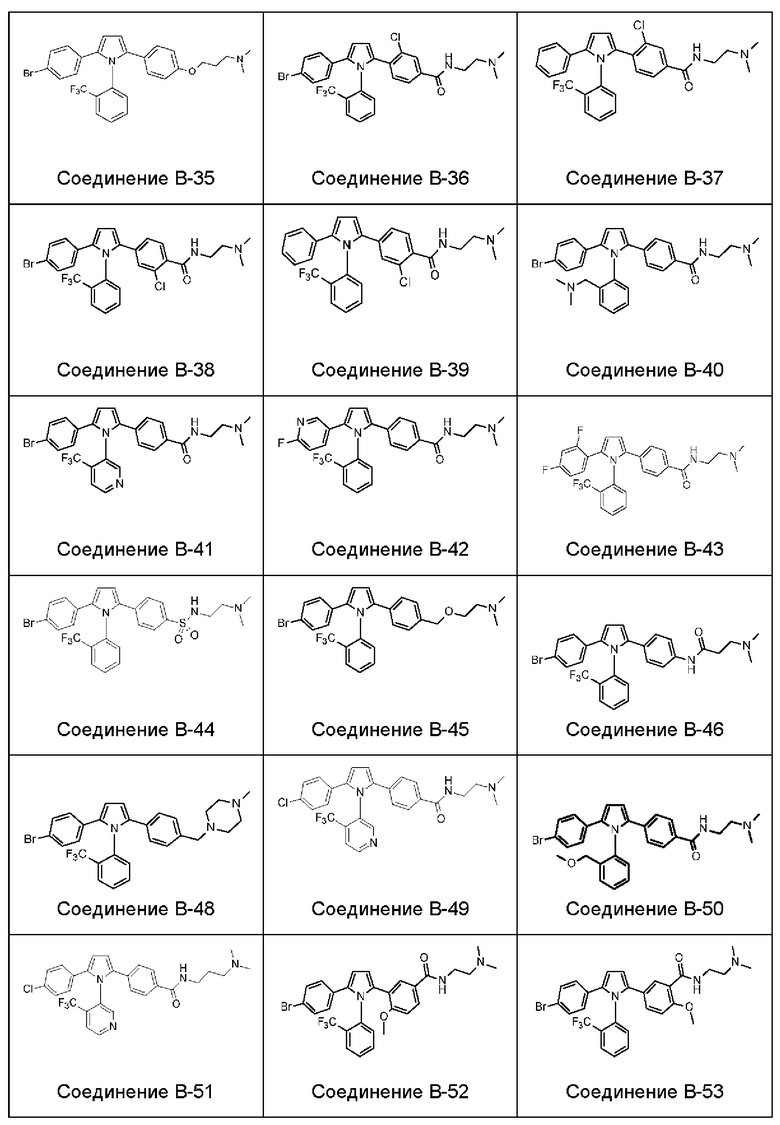
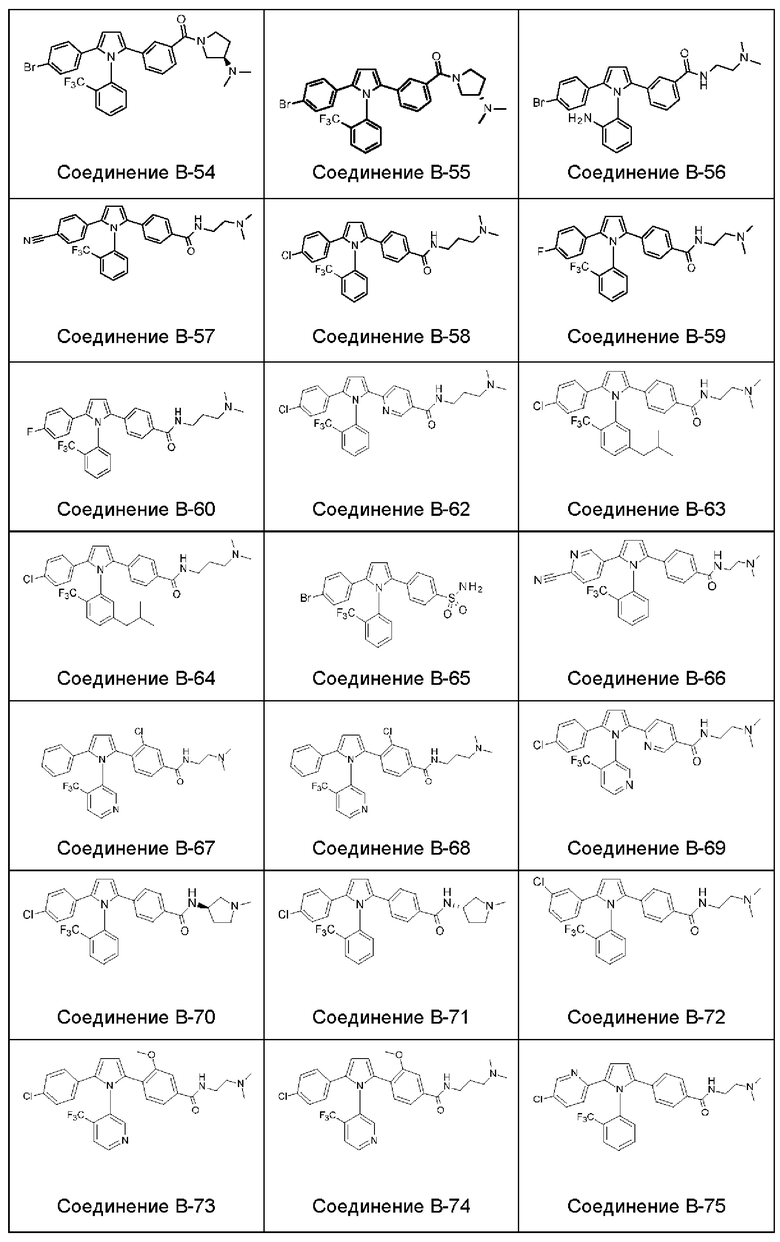
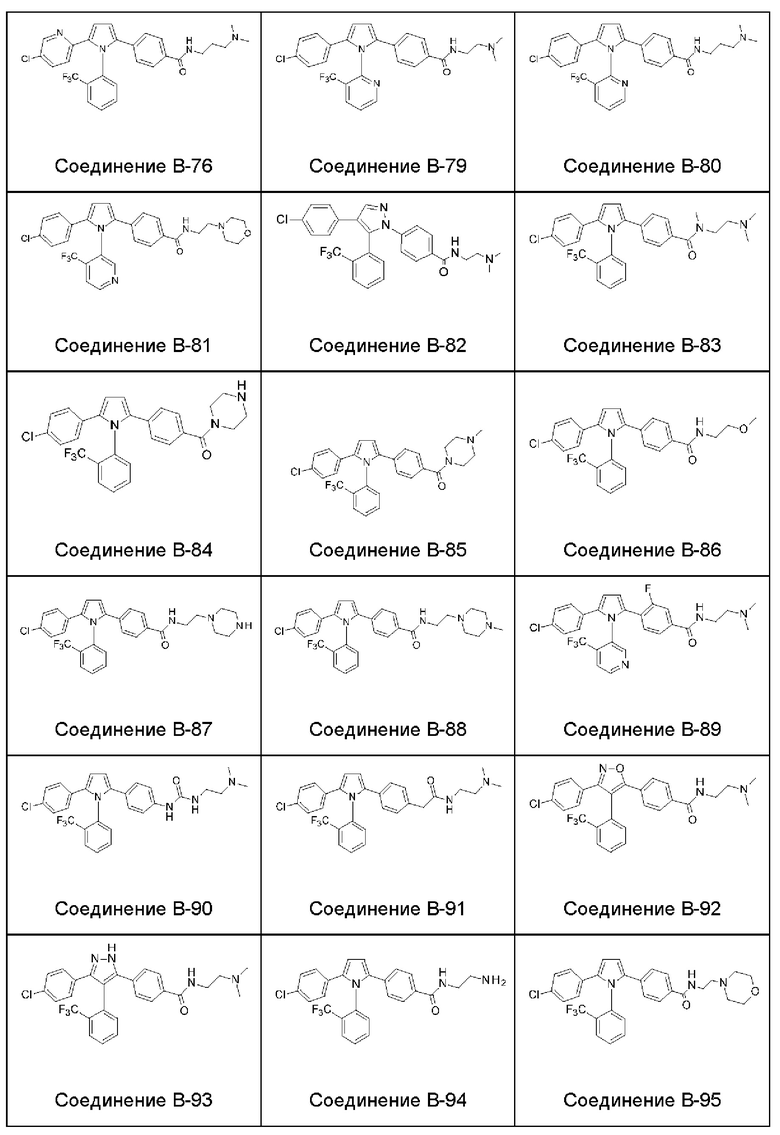
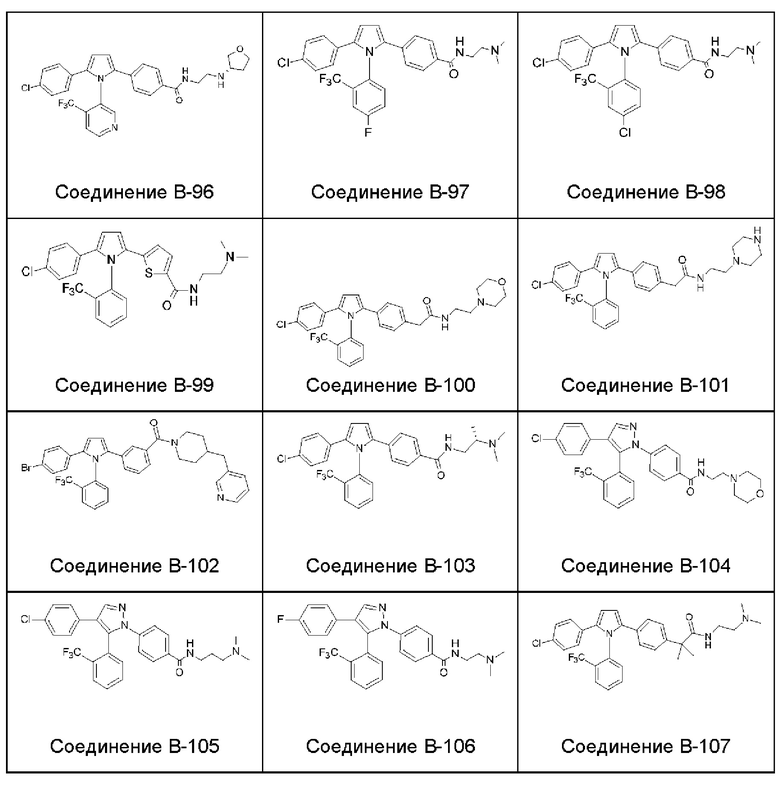
ПРИМЕР 8
Альтернативный способ получения (R)-4-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида (Атропизомера А-2)
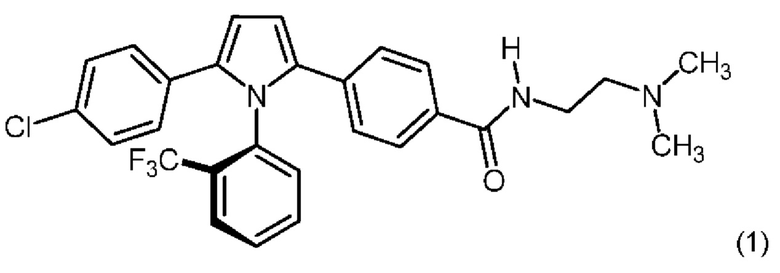
Указанное в заголовке соединение получали согласно Стадиям 1, 2, 3, 4а и 5а путей синтеза, показанных на Схеме 1 выше. Согласно этому пути хиральное разделение проводили на промежуточном соединении в форме карбоновой кислоты (8), а не на диметиламиноэтиламиде (9).
Стадия 1: 4-[4-(4-Хлорфенил)-4-оксо-бутаноил]бензонитрил (6)
В колбу заливали тетрагидрофуран (4 мл/г) и порциями добавляли хлорид цинка (1,222 г/г, 1,3 экв.), в результате чего получали белую подвижную суспензию, которую перемешивали в течение 15 мин. Затем добавляли трет-бутанол (0,66 мл/г, 1 экв), после чего порциями добавляли триэтиламин (0,96 мл/г, 1 экв.), поддерживая температуру ниже 40°С. Реакционную смесь перемешивали в течение 2 ч. Затем добавляли 4-цианоацетофенон (1 г/г, 1 экв.) и 4-хлорфенацилбромид (1,61 г/г, 1 экв), и реакционную смесь перемешивали при 20°С (±5) в течение 48 часов или до завершения реакции. Полученный продукт выделяли путем осаждения водным раствором НС! и суспендировали в смеси водной HCl и метанола. Полученное твердое вещество сушили под вакуумом (45°С), в результате чего получали указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета.
Стадия 2: 4-(5-(4-Хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензонитрил (7)
В колбу загружали 4-(4-(4-хлорфенил)-4-оксобутаноил)бензонитрил (1 г/г, 1 экв.) и добавляли диоксан (10 мл/г), и в результате получали желтую суспензию. Затем единой порцией добавляли 2-трифторметиланилин (1,269 мл/г, 3 экв), после чего добавляли п-толуолсульфоновую кислоту (0,06399 г/г, 0,1 экв.), и полученную реакционную смесь нагревали при 101°С в течение 40-72 часов (при необходимости через каждые 8 часов добавляли дополнительные порции п-толуолсульфоновой кислоты (0,1 экв.), так чтобы добиться завершения реакции). Реакционную смесь охлаждали до комнатной температуры и концентрировали под вакуумом. Полученный маслянистый остаток очищали путем суспендирования в метаноле (10 мл/г). Твердое вещество отделяли фильтрованием и сушили под вакуумом (45°С), в результате чего получали указанное в заголовке соединение в виде твердого вещества желтого цвета.
Стадия 3: 4-(5-(4-Хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензойная кислота (8)
К 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензонитрилу (1 г/г, 1 экв.) в метаноле (10,9 мл/г) по каплям в течение 15 минут добавляли гидроксид натрия (0,948 г/г, 10 экв.) в воде (5 мл/г), и полученную смесь перемешивали при 70-76°С в течение 18 часов или до полного завершения реакции. Реакционную смесь охлаждали до комнатной температуры, подкисляли, и полученный продукт отделяли фильтрованием, промывая водой (5 мл/г) и ацетонитрилом (3 мл/г). Затем продукт суспендировали в смеси ацетон/вода (20 объемов, 75:25) при 50-55°С и сушили под вакуумом (при 60°С), в результате чего получали указанное в заголовке соединение в виде твердого вещества желтого цвета.
Стадия 4а: (R)4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензойная кислота (3) путем хирального разрешения (8)
В колбу брали навеску 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензойной кислоты (1 г/г, 1 экв.) и добавляли тетрагидрофуран (2 мл/г) и ацетонитрил (0,75 мл/г). Затем по каплям в течение 5 мин добавляли (S)-1-(4-метоксифенил)этиламин (0,335 мл/г, 1 экв.), и полученную реакционную смесь перемешивали при 40-50°С в течение 15 мин, после чего охлаждали до комнатной температуры. Затем добавляли ацетонитрил (7,25 мл/г) и затравку для инициирования реакции (0,0001 г/г, 99% э.и., соль желаемого атропизомера с (S)-1-(4-метоксифенил)этиламином). Реакционную смесь перемешивали в течение 16 ч, и полученные твердые вещества отделяли фильтрованием, промывая ацетонитрилом. Горячая (75-80°С) суспензия в ацетонитриле давала хиральную соль в виде твердого вещества белого цвета (выход 40%, 98,16% э.и.). Разложение соли проводили в смеси ТГФ и воды (2:2 по объему) с помощью 1М HCl (2,2 экв.), в результате чего получали кислоту, которую далее очищали в виде суспензии в воде, в результате чего получали указанное в заголовке соединение (90,52 г, выход по реакции разложения соли 97%, общий выход 39%, 98,06% э.и.). 1H ЯМР (ДМСО-d6) δ 12,83 (brs, 1Н), 7,77 - 7,67 (m, 6Н), 7,23 - 7,10 (m, 2Н), 7,08 - 7,01 (m, 4Н), 6,68 (d, J=4,0 Гц, 1Н), 6,59 - 6,58 (d, J=4,0 Гц, 1Н). Хиральная ВЭЖХ, проведенная согласно Методу 6 хиральной ВЭЖХ показала наличие одинарного атропизомера, RT 6,083 мин, площадь пика 99,02% (небольшое количество другого атропизомера с RT 7,07 мин, площадь пика 0,98%).
Хиральное разделение также может быть достигнуто с помощью (S)-(-)-1-фенилэтиламина.
Стадия 5а: (R)-4-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамид (1)
В ТГФ (5 мл/г) растворяли 4-(5-(4-хлорфенил)-1-(2-(трифторметил)фенил)-1Н-пиррол-2-ил)бензойную кислоту (одинарный атропизомер) (1 г/г, 1 экв.) и добавляли сначала по каплям N,N-диметилэтилендиамин (0,75 мл/г, 3 экв.) и затем ДИПЭА (1,58 мл/г, 4 экв.). Затем по каплям добавляли раствор 50% ТЗР в ТГФ (2,72 мл/г, 2 экв), и реакционную смесь перемешивали при 20°С в течение 15 минут. Добавляли дополнительные порции раствора 50% ТЗР в ТГФ вплоть до завершения реакции. Реакционную смесь разбавляли 10% солевым раствором (2 мл/г) и раствором гидроксида натрия (2 мл/г) до уровня рН 8-10. Слои разделяли, и водный слой экстрагировали этилацетатом (2×5 мл/г). Объединенные органические слои промывали солевым раствором, сушили (над MgS04) и концентрировали, в результате чего получали указанное в заголовке соединение (80 г, 156 ммоль, выход 71%) в виде триболюминесцентного твердого вещества белого цвета. Хиральная ВЭЖХ, проведенная согласно Методу 7 хиральной ВЭЖХ показала наличие одинарного атропизомера, RT 12,62 мин, площадь пика 99,32% (небольшое количество другого атропизомера с RT 10,58 мин, площадь пика 0,67%).
ПРИМЕР 9
Получение и определение характеристик тартратной соли (R)-4-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида
Метод 1: Приготовление тартратной соли в небольших объемах
Атропизомера А-2 в форме свободного основания (904,2 мг) суспендировали в ацетоне (9,042 мл, 10 об.) и перемешивали при 25°С в течение 40 минут. После полного растворения всех видимых частиц раствор разделили на 12 равных аликвот (603 мкл), что давало приблизительное активное содержание порядка 60,3 мг на образец.
К аликвоте раствора свободного основания при 25°С добавляли аликвоту 247 мкл (1,05 экв.) 0,5M раствора винной кислоты в этаноле. Смесь перемешивали при 25°С в течение 18 часов, за это время образовывалась белая суспензия, и полученные твердые вещества отделяли фильтрованием (через фриттованный картридж, ПТФЭ 10 мкм) и сушили под вакуумом при 40°С в течение примерно 72 часов. Полученную соль помечали как Тартрат согласно Дифрактограмме А (сольват).
Метод 2: Получение тартратной соли с использованием изопропилацетатного раствора Атропизомера А-2
Атропизомер А-2 (749,8 мг) суспендировали в изопропилацетате (15 мл, 20 об.), и суспензию нагревали до 40°С при перемешивании. После полного растворения всех видимых частиц раствор разделили на 12 равных аликвот (1 мл), что давало приблизительное активное содержание порядка 50 мг на образец. К аликвоте раствора свободного основания при 40°С добавляли аликвоту 195,3 мкл 1М раствора Атропизомера А-2 в этаноле. Полученную смесь охлаждали до 25°С со скоростью охлаждения приблизительно 10°С/час. При этом образовывалась белая суспензия, и полученные твердые вещества выделяли из нее фильтрованием (через фриттованный картридж, ПТФЭ 10 мкм) и сушили под вакуумом при 40°С в течение примерно 18 часов. Полученную соль помечали как Тартрат согласно Дифрактограмме В.
Метод 3: Получение тартратной соли с использованием раствора Атропизомера А-2 в изопропиловом спирте
Тартратную соль, соответствующую Дифрактограмме А, готовили, следуя Методу 2, за исключением того, что Атропизомер А-2 (750,1 мг) сначала суспендировали в изопропиловом спирте (15 мл, 20 об.).
Метод 4: Получение тартратной соли с использованием раствора Атропизомера А-2 в 2-метилтетрагидрофуране
Повторяли процедуру согласно Методу 1, за исключением того, что Атропизомер А-2 (913,9 мг) сначала суспендировали в 2-метилтетрагидрофуране (15 мл, 20 об), (9,139 мл, 10 об.) и перемешивали при 25°С в течение примерно 40 минут, а затем к аликвоте раствора Атропизомер А-2 в виде свободного основания добавляли аликвоту 250 мкл (1,05 экв.) 1М раствора винной кислоты в этаноле, в результате чего получали соль Тартрата согласно Дифрактограмме А.
Метод 5: Приготовление Атропизомера А-2 как Тартрата согласно Дифрактограмме В в объеме 500 мг
В стеклянный флакон брали навеску Атропизомера А-2 в форме свободного основания (521,5 мг), и заливали в него изопропилацетат (20 об., 10,430 мл). Смесь нагревали до 40°С и перемешивали в течение 15 минут, в результате чего получали прозрачный раствор. В полученный раствор добавляли винную кислоту (1,05 экв., 162,5 мг), растворенную в 3 мл тетрагидрофурана. В полученную смесь добавляли затравку Атропизомера А-2 в форме Тартрата согласно Дифрактограмме В, что приводило к немедленному выпадению соли при 40°С с образованием подвижной суспензии. Смесь охлаждали до 25°С и перемешивали в течение 20 часов. Полученное твердое вещество отделяли фильтрованием и сушили при 40°С под вакуумом, в результате чего получали Атропизомер А-2 в форме Тартрата согласно Дифрактограмме В с выходом 84%.
Метод 6: Приготовление соли Атропизомера А-2 в форме Тартрата (безводная форма) согласно Дифрактограмме В в увеличенных объемах
В колбу Бюхи брали навеску Атропизомера А-2 в форме свободного основания (10,0497 мг), и заливали изопропилацетат (20 об., 200 мл). Смесь нагревали до 40°С, в результате чего получали прозрачный раствор, свободный от механических включений, который перемешивали в течение 30 минут.В этот раствор добавляли винную кислоту (3,1954 г, 1,08 экв), растворенную в тетрагидрофуране (50 мл), при этом кислоту добавляли порциями в следующем порядке: добавляли 15 мл при 40°С, вносили затравку Атропизомера А-2 в форме Тартрата согласно Дифрактограмме В, и перемешивали в течение 30 минут, добавляли 10 мл и перемешивали в течение 1 часа, добавляли 10 мл и перемешивали в течение 30 минут и добавляли 15 мл и перемешивали в течение 30 минут. Итоговую белую суспензию охлаждали до к.т. со скоростью охлаждения 10°С/ч и перемешивали в течение 18 часов. Полученное твердое вещество отделяли фильтрованием под вакуумом, промывали изопропилацетатом (2×2 объема) и высушивали под вакуумом при 40°С в течение 20 часов, в результате чего получали соль Атропизомера А-2 в форме Тартрата согласно Дифрактограмме В (безводного) с выходом 97%, чистотой по ВЭЖХ 99,74% (Метод 1 ВЭЖХ) и хиральной чистотой 99,27% (Метод 7 хиральной ВЭЖХ).
Метод 7: Альтернативное приготовление соли Атропизомера А-2 в форме Тартрата (безводная форма) согласно Дифрактограмме В в увеличенных объемах путем кристаллизации из смеси бутанола и воды (96:4) при охлаждении.
В колбе брали навеску Атропизомера А-2 в форме свободного основания (36,79 г) и добавляли бутанол (282,57 мл, 7,68 об.). Смесь нагревали до 80°С (получая бледно-желтый опалесцирующий раствор) и перемешивали в течение 30 минут, после чего осветляли в сосуде Муа*, предварительно нагретом до 80°С. Затем в раствор добавляли L-(+)-винную кислоту (1,023 экв., 11,0806 г) в виде раствора в воде (11,77 мл, 0,32 объема от исходной загрузки АФИ). Добавление осуществляли по каплям при 80°С, дожидаясь осветления раствора кислоты. Затем смесь охлаждали до 68°С в течение 30 минут, вносили затравку из 0,1% размолотых кристаллов соли Атропизомера А-2 в форме Тартрата согласно Дифрактограмме В (32,6 мг) и выдерживали в течение 1 часа. Затем смесь охлаждали до 5°С со скоростью охлаждения 5°С/час и перемешивали при 5°С в течение 6 часов, после чего отделяли твердую фазу. Твердое вещество отфильтровывали под вакуумом, дважды промывали бутанолом и высушивали в течение 15 минут сначала на фильтре и затем при 40°С в течение 20 часов, в результате чего получали соль Атропизомера А-2 в форме Тартрата согласно Дифрактограмме В (безводного) с выходом 83%, чистотой по ВЭЖХ 99,84% (Метод 1 ВЭЖХ) и хиральной чистотой 99,66% (Метод 7 хиральной ВЭЖХ).
*Примечание: В упомянутых выше процессах уравновешивания или кристаллизации, требующих контроля температуры и/или определенных профилей нагрева/охлаждения, использовали реакционную станцию Муа4 от компании Radley. Реакционная станция Муа4 компании Radley представляет собой 4-зонную реакционную станцию с магнитными и верхнеприводными мешалками, диапазоном температур от -30 до 180°С и объемами реакционных смесей от 2 до 400 мл. Требуемые условия реакции программировали на панели управления Муа 4 Control Pad.
Определение характеристик тартратных солей Атропизомера А-2
Идентичность солей как солей со стехиометрией 1:1 (мольное соотношение свободного основания и винной кислоты) была подтверждена на основании их спектров 1H ЯМР, которые получали с помощью спектрометра JEOL ЕСХ 400MHz, оборудованного автоматическим пробоотборником. Образцы растворяли в подходящем дейтерированном растворителе класса чистоты «для анализа». Данные получали с помощью программного обеспечения Delta NMR Processing and Control Software версии 4.3.
Характеристики тартратных солей получали с помощью методов рентгеновской порошковой дифракции (XRPD), дифференциальной сканирующей калориметрии (DSC), термогравиметрического анализа (TGA), гравиметрических испытаний на растворимость и испытаний на гравиметрическую сорбцию паров, используя описанные ниже процедуры.
Рентгеновская порошковая дифракция (XRPD)
Дифрактограммы рентгеновской порошковой дифракции получали на аналитическом дифрактометре PANalytical с использованием излучения линии CuKα (45 кВ, 40 мА), гониометра θ - θ, фокусирующего зеркала, щели расходимости (1/2''), щелей Соллера как на падающем, так и на расходящемся пучке (4 мм) и детектора PIXcel. Для сбора данных использовали программное обеспечение X'Pert Data Coliector версии 2.2f, а представление данных производили с помощью средства просмотра данных X'Pert Data Viewer, версии 1.2d. Дифрактограммы XRPD получали при окружающих условиях через стадию пропускания излучения сквозь тонкопленочный образец (полиимид Kapton, пленка толщиной 12,7 мкм) при окружающих условиях с использованием программного обеспечения PANaiyticai X'Pert PRO. Диапазон сбора данных составлял 2,994 - 35°2θ со скоростью непрерывного сканирования 0,202004°/сек.
Дифференциальная сканирующая калориметрия (DSC)
Данные DSC получали на приборе PerkinElmer Pyris 6000 DSC, оборудованном 45-позиционным держателем образцов. Прибор верифицировали на калибровку по энергии и температуре с использованием сертифицированного индия. Заданное количество образца (0,5-3,0 мг) помещали на пористый алюминиевый поддон и нагревали со скоростью 20°С/мин от 30 до 350°С или варьировали в зависимости от требований эксперимента. Образец постоянно обдували сухим азотом со скоростью 20 мл/мин. Управление прибором, сбор и анализ данных производили с помощью программного обеспечения Pyris Software v11.1.1 revision H.
Термогравиметрический анализ (TGA)
Данные TGA получали на приборе PerkinElmer Pyris 1 TGA, оборудованном 20-позиционным автоматическим пробоотборником. Прибор калибровали по температуре с помощью сертифицированного груза и сертифицированных эталонных сплавов алюмеля и перкаллоя. Заданное количество образца (1-5 мг) помещали в тарированный алюминиевый тигель и нагревали со скоростью 20°С/мин от окружающей температуры до 400°С. Образец постоянно обдували азотом со скоростью 20 мл/мин. Управление прибором, сбор и анализ данных производили с помощью программного обеспечения Pyris Software v11.1.1 revision H.
Гравиметрический анализ растворимости
Растворимость в воде солей измеряли с использованием протокола гравиметрического анализа растворимости.
В пробирки для кристаллизации добавляли по 1 мл воды. Навеску твердого образца брали в тарированном стеклянном флаконе, добавляли порциями в растворы, а флакон взвешивали после каждого добавления до тех пор, пока не наблюдалось помутнение раствора. Затем рассчитывали количество израсходованного образца в мг, чтобы получить растворимость в мг/мл.
Результаты, полученные в исследованиях по определению характеристик, приведены в Таблице 8 ниже.
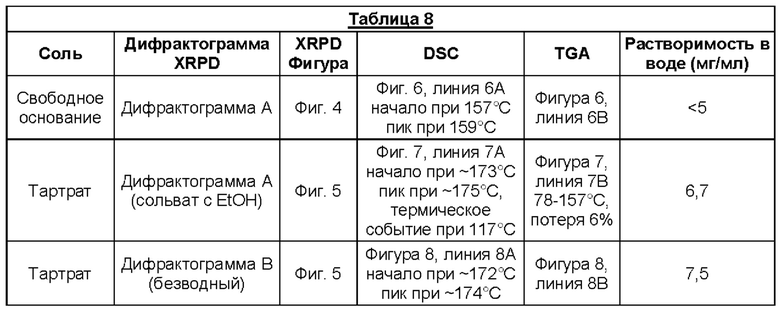
Гравиметрическая сорбция паров (GVS)
Исследования GVS были проведены на соли Атропизомера А-2 в форме Тартрата согласно Дифрактограмме В с использованием изложенного ниже протокола:
Изотермы сорбции были получены с помощью анализатора сорбции влаги Hiden Isochema (модель IGAsorp), управляемого посредством программного обеспечения IGAsorp Systems V6.50.48. Образец поддерживали при постоянной температуре (25°С) с помощью регуляторов прибора. Влажность контролировали путем смешивания потоков сухого и влажного азота с общей скоростью пропускания 250 мл/мин. Прибор верифицировали на определение относительной влажности путем измерения трех калиброванных солевых растворов Rotronic (10, 50 и 88%). Отслеживали изменение массы образца как функции от влажности посредством микрохимических весов (точность +/-0,005 мг). Определенное количество образца помещали в тарированную сетчатую корзинку из нержавеющей стали при окружающих условиях. Полный экспериментальный цикл обычно включал три сканирования (сорбция, десорбция и сорбция) при постоянной температуре (25°С) интервалами по 10% RH в диапазоне 0-90% (по 60 минут для каждого уровня влажности). Эксперимент такого типа должен продемонстрировать способность исследуемых образцов поглощать (или не поглощать) влагу в ряду четко определенных диапазонов влажности
GVS анализ (см. Фигуру 9) показал содержание влаги около 0,3% перед первой десорбцией. В диапазоне от 80 до 90% RH имеется несколько большее повышение влажности, при этом твердое вещество поглощает примерно 0,8% влаги. Второй цикл абсорбции/десорбции показывает, что поглощение влаги является полностью обратимым и возвращается к величине 0 масс. % при относительной влажности 0% RH. XRPD анализ после выполнения цикла GVS, проводимого при 0% RH и 90% RH в течение по меньшей мере 3 часов, показал безводный материал согласно Дифрактограмме В при обоих значениях относительной влажности.
Таким образом, можно сделать вывод, что соль Атропизомера А-2 в форме Тартрата согласно Дифрактограмме В существует в виде стабильного твердого вещества, поглощая только поверхностную влагу без изменения формы.
ПРИМЕР 10
Получение и определение характеристик других солей (R)-4-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида
Были получены и охарактеризованы гидрохлоридные, мезилатные, малеатные, малатные, тозилатные, сульфатные и фосфатные соли (R)-4-[5-(4-хлорфенил)-1-[2-(трифторметил)фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамида. Их дифрактограммы рентгеновской порошковой дифракции (XRPD), термические профили (DSC и TGA) и растворимость в воде приведены в таблице ниже.
Для всех солей ЯМР показал, что соотношение между свободным основанием и противоионом в них составляет 1:1.
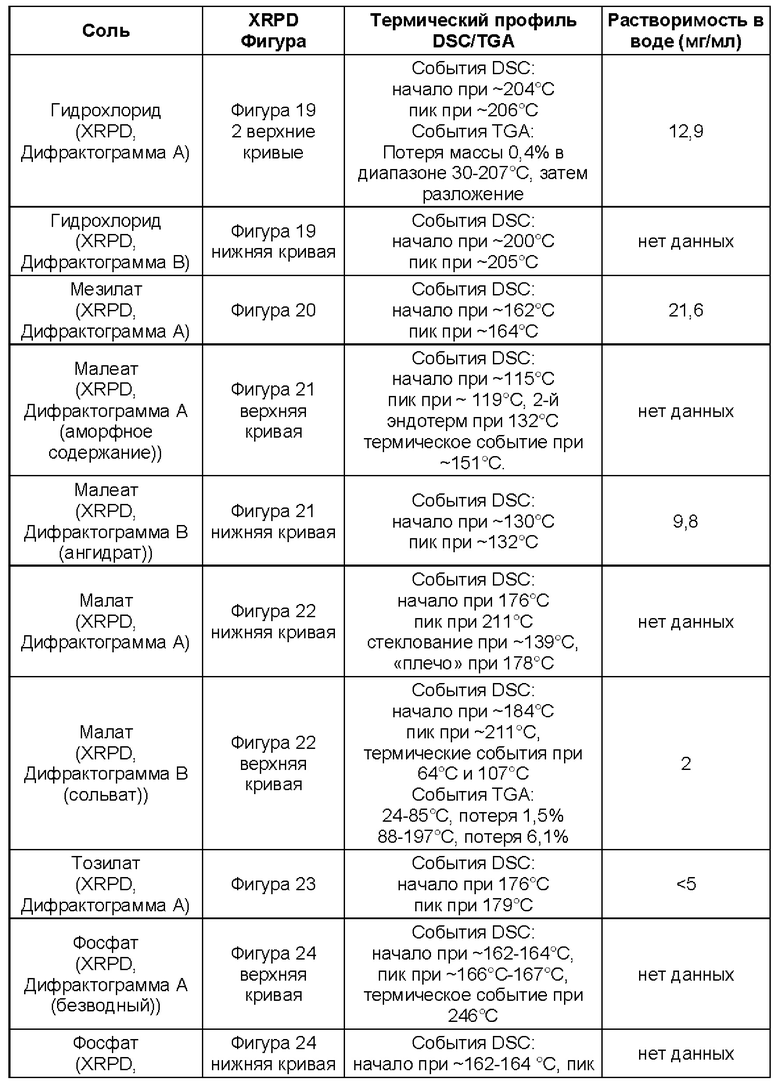
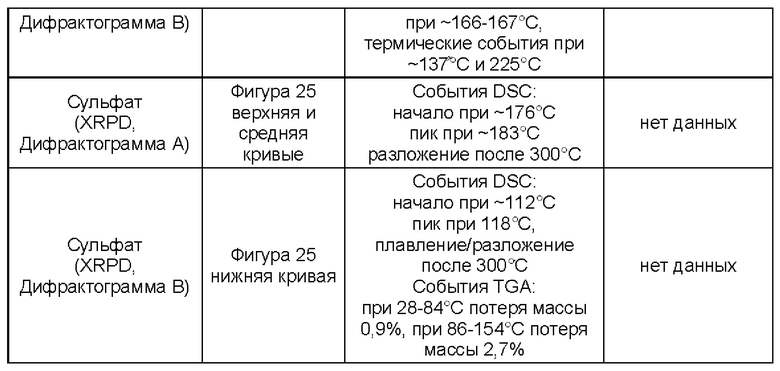
Растворимость в воде солей измеряли с использованием протокола гравиметрического анализа растворимости. Так, в пробирки для кристаллизации добавляли по 1 мл воды. Навеску твердого образца брали в тарированном стеклянном флаконе, добавляли порциями в растворы, а флакон взвешивали после каждого добавления до тех пор, пока не наблюдалось помутнение раствора. Затем определяли количество добавленного образца в мг, получая величину растворимости в мг/мл.
Протонная ЯМР спектроскопия
Спектры 1Н ЯМР получали с помощью спектрометра JEOL ЕСХ 400MHz, оборудованного автоматическим пробоотборником. Образцы растворяли в подходящем дейтерированном растворителе класса чистоты «для анализа». Данные получали с использованием программного обеспечения Delta NMR Processing and Control Software версии 4.3.
Приготовление солей
Получение солей Атропизомера А-2 в небольших объемах
Метод 1: В среде ацетона
Атропизомер А-2 в форме свободного основания (904,2 мг) суспендировали в ацетоне (9,042 мл, 10 об.) и перемешивали при 25°С в течение 40 минут. После полного растворения всех видимых частиц раствор разделили на 12 равных аликвот (603 мкл), что давало приблизительное активное содержание порядка 60,3 мг на образец.
В полученные растворы добавляли исходные 0,5М или 1М растворы кислот (247 мкл или 124 мкл, 1,05 экв.) в EtOH при 25°С. Смеси перемешивали при 25°С в течение 18 часов. При необходимости образцы подвергали дополнительным манипуляциям (например, растиранием твердых веществ и добавлением антирастворителя), чтобы извлечь для анализа твердые вещества, которые отделяли и сушили под вакуумом при 40°С в течение приблизительно 72 часов.
В таблице ниже приведены используемые количества кислот, применяемые антирастворители и получаемые кристаллические формы. Для выделения солей можно использовать и другие альтернативные методы.
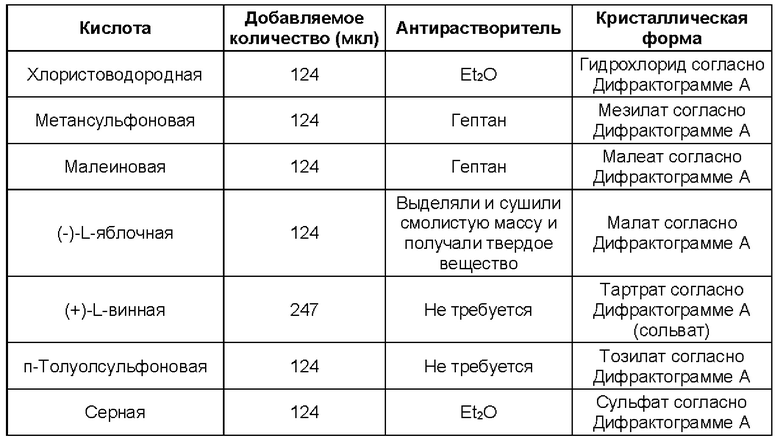
Метод 2: Вереде изопропилацетата
Атропизомер А-2 (749,8 мг) суспендировали в iPrOAc (15 мл, 20 об.), и суспензию нагревали до 40°С при перемешивании. После полного растворения всех видимых частиц раствор разделили на 12 равных аликвот (1 мл), что давало приблизительное активное содержание порядка 50 мг на образец. В полученные растворы добавляли исходные 0,5М или 1М растворы кислот (195,3 мкл или 97,7 мкл, 1 экв.) в EtOH при 40°С. Смеси охлаждали до 25°С со скоростью приблизительно 10°С/ч. При необходимости образцы подвергали дополнительным манипуляциям (например, растиранием твердых веществ и добавлением антирастворителя), чтобы извлечь для анализа твердые вещества, которые отделяли и сушили под вакуумом при 40°С в течение приблизительно 18 часов.
Методом 2 можно получать гидрохлорид согласно Дифрактограмме А (антирастворитель МТБЭ), тартрат согласно Дифрактограмме В (исходный 1М раствор кислоты (195,3 мкл) в EtOH), тозилат согласно Дифрактограмме А и фосфат согласно Дифрактограмме В.
Метод 3: Вереде ИПС
Метод идентичен Методу 2, за исключением того, что Атропизомер А-2 (750,1 мг) суспендировали в ИПС (15 мл, 20 об.).
Методом 3 можно получать гидрохлорид согласно Дифрактограмме А (антирастворитель МТБЭ), тартрат согласно Дифрактограмме В (исходный 1М раствор кислоты (195,3 мкл) в EtOH), тозилат согласно Дифрактограмме А и фосфат согласно Дифрактограмме В.
Метод 4: В среде 2-метилтетрагидрофурана
Метод идентичен Методу 1, за исключением того, что Атропизомер А-2 (913,9 мг) суспендировали в 2-метил-ТГФ (9,139 мл, 10 об.) и перемешивали при 25°С в течение примерно 40 мин, и использовали 0,5М или 1М исходные растворы кислот (250 мкл или 125 мкл, 1,05 экв.) в EtOH.
Методом 4 можно получать гидрохлорид согласно Дифрактограмме В (гептан в качестве антирастворителя), малеат согласно Дифрактограмме А (гептан в качестве антирастворителя), тартрат согласно Дифрактограмме А (1М раствор кислоты (250 мкл, 1,05 экв.) в EtOH) и тозилат согласно Дифрактограмме А.
Определенное подмножество солей получали в увеличенных объемах и характеризовали более подробно.
Получение солей Атропизомера А-2 в объемах порядка 500 мг
Гидрохлоридная соль
В стеклянный флакон брали навеску Атропизомера А-2 в форме свободного основания (524,9 мг), заливали в него ИПС (20 об., 10,498 мл) и нагревали до 40°С. Раствор перемешивали при 40°С в течение 40 мин и затем добавляли раствор HCl (4,4М в ИПС, 1,2 экв., 280 мкл). Затем в полученную смесь вносили затравку гидрохлоридной соли согласно Дифрактограмме В, и смесь перемешивали при 40°С в течение 15 минут, после чего охлаждали до 25°С. Смесь концентрировали под вакуумом и получали остаток в виде бледно-желтой маслянистой жидкости. Маслянистую жидкость суспендировали в 10 объемах МТБЭ и перемешивали при 25°С в течение 72 ч, получая белую суспензию. Из этой суспензии выделяли твердое вещество, которое высушивали при 40°С под вакуумом в течение 18 часов, в результате чего получали указанную в заголовке соль согласно Дифрактограмме А с выходом 73%.
Мезилатная соль
В стеклянный флакон брали навеску Атропизомера А-2 в форме свободного основания (503,9 мг), и заливали в него 2-метилТГФ (10 об., 5,039 мл). Смесь перемешивали при к.т. в течение 30 минут. Затем в раствор добавляли метансульфоновую кислоту (1М раствор в EtOH, 1,05 экв., 1,033 мл), вносили затравку мезилатной соли Атропизомера А-2 согласно Дифрактограмме А и перемешивали при 25°С в течение 30 минут. Смесь сначала превращалась в опалесцирующий раствор, и затем образовывалась белая суспензия, которую перемешивали при 25°С в течение 72 часов. Твердое вещество отделяли фильтрованием и сушили под вакуумом при 40°С в течение 18 часов, в результате чего получали указанную в заголовке соль с выходом 46%.
Тартратная соль
В стеклянный флакон брали навеску Атропизомера А-2 в форме свободного основания (521,5 мг), и заливали в него iPrOAc (20 об., 10,430 мл). Смесь нагревали до 40°С и перемешивали в течение 15 минут, в результате чего получали прозрачный раствор. В полученный раствор добавляли винную кислоту (1,05 экв., 162,5 мг), растворенную в 3 мл ТГФ. В полученную смесь добавляли затравку Атропизомера А-2 в форме Тартрата согласно Дифрактограмме В, что приводило к немедленному выпадению соли при 40°С с образованием подвижной суспензии. Смесь охлаждали до 25°С и перемешивали в течение 20 ч. Твердое вещество отделяли фильтрованием и сушили при 40°С под вакуумом, в результате чего получали указанную в заголовке соль согласно Дифрактограмме В с выходом 84%.
Тозилатная соль
В стеклянный флакон брали навеску Атропизомера А-2 в форме свободного основания (504,5 мг), заливали в него iPrOAc (20 об., 10,090 мл) и нагревали до 40°С. Раствор перемешивали при 40°С в течение 40 мин и затем добавляли раствор п-толуолсульфоновой кислоты (1М в EtOH, 1,05 экв., 1,04 мкл). Затем в полученную смесь вносили затравку небольшого количества тозилатной соли Атропизомера А-2 согласно Дифрактограмме А, и смесь перемешивали при 40°С в течение 15 минут, после чего охлаждали до 25°С. Смесь быстро превращалась в белую суспензию, и ее перемешивали при 25°С в течение 72 часов. Из этой суспензии выделяли твердое вещество, которое высушивали при 40°С под вакуумом в течение 18 часов, в результате чего получали указанную в заголовке соль согласно Дифрактограмме А с выходом 82%.
Малеатная соль
В стеклянный флакон брали навеску Атропизомера А-2 в форме свободного основания (523,9 мг), и заливали в него 2-метилТГФ (10 об., 5,239 мл). Смесь перемешивали при к.т.в течение 30 мин и получали прозрачный раствор. Затем в раствор добавляли малеиновую кислоту (0,5М раствор в ТГФ, 1,05 экв., 2,149 мл), вносили затравку малеатной соли Атропизомера А-2 согласно Дифрактограмме А, и раствор перемешивали при 25°С в течение 30 минут.Смесь упаривали под вакуумом с получением белой смолистой массы. Эту смолу суспендировали в 10 объемах гептана и перемешивали при 25°С в течение 72 ч. Твердое вещество отделяли и сушили под вакуумом при 40°С в течение 18 ч, в результате чего получали указанную в заголовке соль согласно Дифрактограмме В. Спектр 1Н ЯМР соответствует структуре, но показывает стехиометрию ~1:0,8.
Малатная соль
В стеклянный флакон брали навеску Атропизомера А-2 в форме свободного основания (524,9 мг), заливали в него ИПС (20 об., 10,618 мл) и нагревали до 40°С. Раствор перемешивали при 40°С в течение 40 мин и затем добавляли раствор яблочой кислоты (1М в EtOH, 1,05 экв., 1,09 мкл). Затем смесь перемешивали при 40°С в течение 15 минут, после чего охлаждали до 25°С. Смесь, которая оставалась в виде раствора при 25°С, упаривали под вакуумом, получая масляный остаток. Маслянистую жидкость суспендировали в 10 объемах гептана и перемешивали при 25°С в течение 70 ч, получая белую суспензию. Твердое вещество отделяли и сушили под вакуумом при 40°С в течение 18 ч, в результате чего получали указанную в заголовке соль согласно Дифрактограмме В.
Сульфатная соль
В стеклянный флакон брали навеску Атропизомера А-2 в форме свободного основания (520 мг), и заливали в него ацетон (10 об., 5,2 мл). Смесь перемешивали при к.т. в течение 30 мин и получали прозрачный раствор. Затем в раствор добавляли серную кислоту (1М раствор в EtOH, 1,05 экв., 1,066 мл), вносили затравку сульфатной соли Атропизомера А-2 согласно Дифрактограмме А, и раствор перемешивали при 25°С в течение 30 минут. Смесь оставалась в виде раствора, поэтому ее упаривали под вакуумом со слабым током азота, после чего оставалась белая смолистая масса.
Эту смолу суспендировали в 10 объемах диэтилового эфира, и суспензию перемешивали при 25°С в течение 70 ч. Твердое вещество отделяли и сушили под вакуумом при 40°С в течение 18 ч, в результате чего получали указанную в заголовке соль согласно Дифрактограмме A sim (аналогичную, но не идентичную ранее выделенной сульфатной соли согласно Дифрактограмме А).
ПРИМЕР 11
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
A. Анализ для измерения степени влияния соединений согласно настоящему изобретению на жизнеспособность раковых клеток глиобластомы человека U87MG
Для определения степени влияния соединений согласно настоящему изобретению на жизнеспособность клеток U87MG использовали следующий протокол.
Клетки U87MG выращивали в рекомендованных для них питательных средах/добавках (АТСС). Клетки высевали в концентрации 5000 клеток на лунку в 96-луночные планшеты и выдерживали в течение ночи при 37°С и 5% СО2. Затем клетки обрабатывали соответствующими концентрациями испытуемого соединения на протяжении 72 часов. После 72 часов инкубации жизнеспособность клеток определяли методом колориметрического анализа с сульфородамином В (SRB). Процент жизнеспособности рассчитывали относительно среднего значения обработанных ДМСО контрольных образцов, а значения IC50 для ингибирования роста клеток рассчитывали с использованием программного обеспечения GraphPad Prism методом нелинейной регрессии (через 4-параметрическое логистическое уравнение).
Исходя из результатов, полученных согласно вышеуказанному протоколу, определяли значения IC50 для атропизомеров согласно Примерам в отношении линии клеток U87MG, как это показано ниже в Таблице 9.
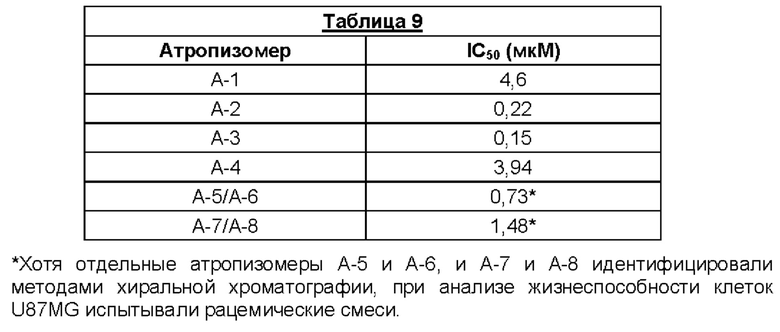
B. Анализ для определения степени влияния Атропизомеров А-1 и А-2 на жизнеспособность раковых клеток в составе панели различных линий раковых клеток
Для идентификации типов опухолей, проявляющих восприимчивость к Атропизомерам А-1 и А-2, проводили скрининг в отношении различных линий раковых клеток. Панель из 48 клеточных линий ракового происхождения подвергали скринингу в ходе высокопроизводительного анализа пролиферации с использованием различных разведений Атропизомеров А-1/А-2. Клеточные линии, подвергнутые скринингу, включали линии, представляющие рак поджелудочной железы, толстого кишечника/ободочной и прямой кишки, легких, мозга и нервов, а также клеточные линии лимфомы и лейкемии. Клеточные линии обрабатывали последовательными полулогарифмическими разведениями испытуемого соединения и анализировали через 72 часа на пролиферацию с использованием анализа CellTiter-Glo (Promega). Значения IC50 рассчитывали путем аппроксимации данных доза-ответ с использованием модели нелинейной регрессии. Значения IC50 (микромолярные концентрации) для Атропизомеров А-1 и А-2 приведены ниже в Таблице 10.
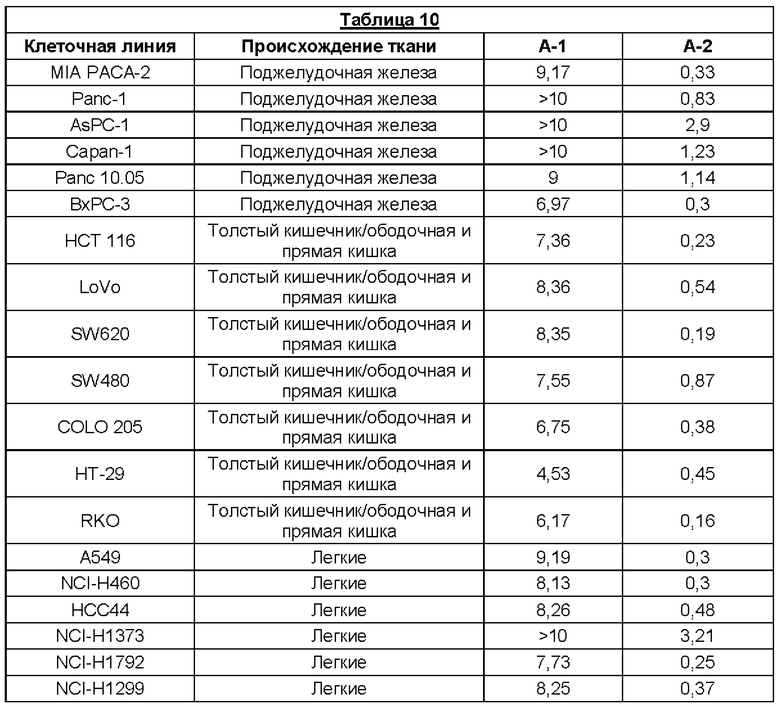
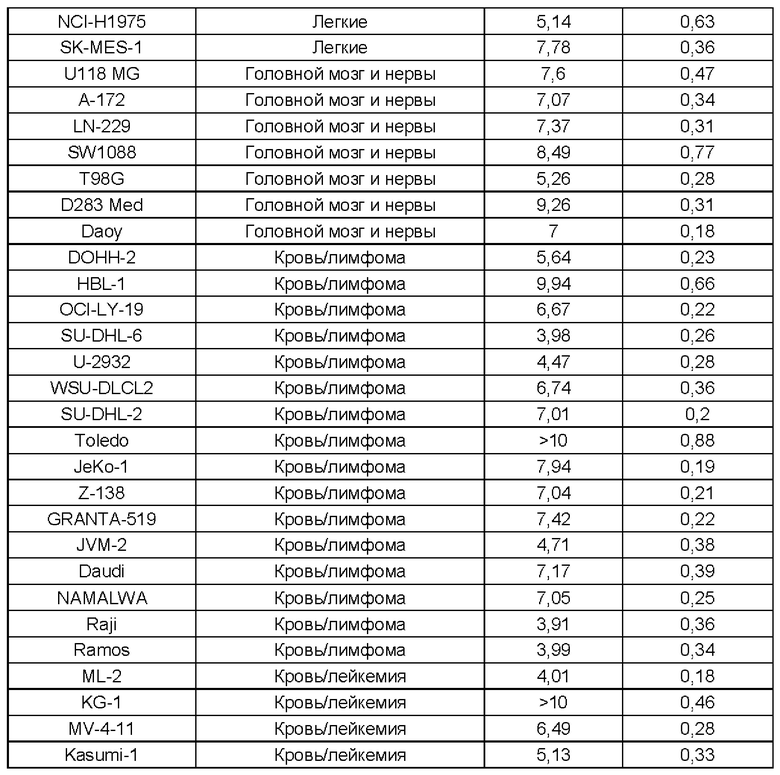
Как можно видеть из приведенных данных, Атропизомер А-2 был значительно более активным ингибитором роста клеток, чем Атропизомер А-1, в отношении всех клеточных линий.
С. Анализ для измерения степени влияния соединений согласно настоящему изобретению на делящиеся клетки
Известно, что ингибирование способности PLK1 и PLK4 привязываться к своим партнерам через их PBD-домены вызывает остановку митоза клеток. Экспериментально это может быть измерено путем оценки количества клеток, находящихся в митозе, в определенное время после их обработки испытуемым соединением, используя метод иммунофлуоресцентного обнаружения фосфорилированного гистона Н3 (рН3), метки, присутствующей только в делящихся (митотических) клетках. Ожидается, что ингибиторы PLK1/4-PBD будут вызывать дозозависимый прирост рН3-положительных клеток, отражаемый в отчетности митотическим индексом (MI) - процентом клеток, которые в данный момент времени положительны на наличие этой митотической метки.
После ингибирования PLK1 и PLK4 в клетках индуцируются отчетливо выраженные митотические фенотипы. Было продемонстрировано, что нарушение PBD-домена в PLK1 вызывает митотическую остановку с неконгрессирующими хромосомами, т.е. фенотипом, отличным от фенотипа монополярного веретена, индуцируемого АТФ-конкурентными ингибиторами PLK1 (Hanisch et al., 2006 Mol. Biol. Cell 17, 448-459). Сборку центриолей контролирует PLK4, при этом ингибиторы индуцируют фенотип многополярного веретена из-за дефектов центросомы, что приводит к аномальному цитокинезу (Wong et al., 2015. Science 348(6239); 1155-1160).
Для измерения степени влияния Атропизомера А-2 и Атропизомера А-3 на остановку митоза клеток применяли следующий протокол.
Клетки высевали в количестве 10000 на лунку в 96-луночные планшеты и инкубировали в течение ночи. На следующий день исходные растворы Атропизомера А-2 в ДМСО разбавляли в питательной среде и затем добавляли к клеткам с максимальной конечной концентрацией ДМСО на клетках 0,2%. Клетки инкубировали с исследуемым соединением в течение 24 часов и затем фиксировали в 3,7% растворе формальдегида. Клетки пермеабилизовали с помощью 0,1% Triton Х-100, затем инкубировали с антителом кфосфогистону Н3 (Ser10) (Abcam). Клетки промывали PBS, затем инкубировали с меченым красителем AlexaFluor 488 антителом козы к IgG кролика (Invitrogen) в присутствии 4 мкг/мл красителя Hoechst 33342 (Invitrogen). Клетки промывали в PBS и затем получали их изображение на приборе Arrayscan VTi HCS с использованием программного приложения Target Activation V4 Bioapplication. Для идентификации митотических клеток по интенсивности окрашивания фосфогистона НЗ применяли заданный пользователем порог.
Для построения графической зависимости % митотических клеток от концентрации соединения использовали программный продукт GraphPad Prism, для чего брали зависимость «log (ингибитора)-ответ» с изменяемым наклоном, аппроксимацией по методу наименьших квадратов и без ограничений.
Исходя из результатов, полученных согласно вышеуказанному протоколу, были получены значения ЕС50 и процент митотических клеток в отношении клеточных линий HeLa и U87MG для Атропизомера А-2 и Атропизомера А-3. Значения ЕС50 приведены ниже в Таблице 11.

Исследование фенотипов
В отдельном исследовании, проводимом согласно вышеуказанному протоколу и с использованием концентраций одинарных соединений 0,03 мкМ для Атропизомера А-1 и Атропизомера А-2, частоту наблюдаемых митотических фенотипов в клетках U87MG оценивали вручную и классифицировали по следующим фенотипам: неконгрессированные хромосомы, многополярные веретена/аномальный цитокинез, монополярные веретена, нормальная прометафаза, нормальная метафаза для каждого из А-1 и А-2 атропизомеров. Результаты показаны на Фигуре 10.
Результаты
Результаты, представленные на Фиг. 10, демонстрируют, что Атропизомер А-2 оказывает гораздо больший эффект нарушения нормального митоза, чем атропизомер А-1. Таким образом, в случае А-1 76% клеток демонстрировали нормальный митотический фенотип, сопоставимый с 77% клеток, обработанных ДМСО в контрольном опыте, и 24% демонстрировали аномальный цитокинез, что сравнимо с 23% клеток, обработанных ДМСО в контрольном опыте. Ни в клетках, обработанных ДМСО, ни в клетках, обработанных Атропизомером А-1, не было обнаружено признаков фенотипа неконгрессированных хромосом. И напротив, обработка клеток более активным Атропизомером А-2 приводила только к 17% клеток с нормальным митотическим фенотипом, 70% с аномальным цитокинезом и 13% с неконгрессированными хромосомами. Эти фенотипы согласуются с нарушением активности PLK1 и PLK4 во время митоза.
Р. Анализ для измерения степени влияния Атропизомера А-2 на центросомы
Приведенные выше результаты исследования С показывают, что Атропизомер А-2 вызывает митотические эффекты, которые характерны для разрегулированной функции центросомы. Поэтому дополнительно исследовали влияние атропизомеров А-1 и А-2 на функцию центросомы. Клетки HeLa, стабильно экспрессирующие гибридный белок Centrin1-GFP, высевали в 96-луночные планшеты и оставляли на ночь. Клетки обрабатывали Атропизомером А-1 или Атропизомером А-2 (в виде растворов концентрацией 0,02 мкМ в ДМСО) или просто ДМСО (в качестве контроля) в течение 72 часов, а затем получали их изображение с флуоресцентного микроскопа. Для каждого вида обработки делали снимки, захватывающие несколько полей клеток, а затем изображения анализировали вручную. Centrin1-GFP специфически маркирует центриоли в виде дискретных очагов, и поэтому может быть использован для количественного определения количества центриолей на клетку. Так, для каждого вида обработки анализировали по 100 клеток и регистрировали количество центриолей, присутствующих в каждой клетке. Затем данные были распределены по нескольким корзинам (без центриолей, с 1 центриолью, с 2 центриолями и с более чем 2 центриоли) и представлены на Фиг. 11.
Исходя из этих данных, можно заключить, что Атропизомер А-2 демонстрирует признаки фенотипов ингибирования PLK4 на клетках HeLa.
Е. Анализ для измерения степени влияния соединений согласно настоящему изобретению на жизнеспособность клеток дикого типа в сравнении с клетками KRAS HeLa
Атропизомеры А-1, А-2, А-3 и А-4 испытывали на клетках HeLa, сконструированных генной инженерией с целью индуцируемой экспрессии трансгенов дикого типа или онкогенных KRasG12V посредством системы FLP-in/T-Rex (invitrogen). Клетки высевали, а затем обрабатывали доксициклином или без него для индуцирования экспрессии трансгена, а затем обрабатывали последовательно разведенными ингибиторами PBD. Через 72 часа инкубации жизнеспособность клеток оценивали с помощью реагента Cell litre Blue (Promega) и планшет-ридера BMG Pherastar. Влияние PBD-ингибирования на жизнеспособность клеток дикого типа или онкогенного G12V KRAS типа оценивали с помощью программного продукта GraphPad Prism.
Исходя из результатов, полученных в соответствии с вышеуказанным протоколом, определяли значения GI50 каждого из атропизомеров в отношении линии клеток HeLa дикого типа и с KRAS G12V, которые представлены в Таблице 12.
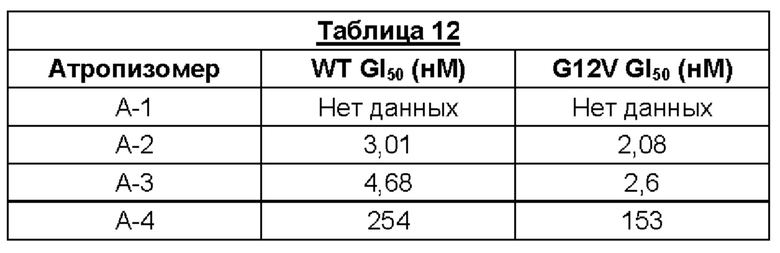
F. Анализ селективности киназы
Соединения согласно настоящему изобретению связываются с доменом PBD в PLK1 и PLK4, но не с каталитическими доменами PLK1 и PLK4, и должны проявлять хорошую селективность в присутствии других киназ. Атропизомер А-2 был испытан на нецелевую (побочную) активность в отношении панели из девяносто семи киназ, распределенных по киному в концентрации 3 мкМ, с использованием анализа DiscoverX KinomeScreen. Результаты показаны в Таблице 13 ниже.
Анализ DiscoverX KinomeScreen представляет собой сайт-специфический конкурентный анализ связывания, который измеряет аффинность связывания соединения с киназой путем использования твердого контрольного соединения на подложке, которое может связывать или захватывать киназы, находящиеся в растворе. В отсутствие испытуемого соединения, ингибирующего киназу, вся киназа будет связываться с твердой подложкой. Если к аналитической смеси добавляют испытуемое соединение, ингибирующее киназу, то количество киназы, связывающейся с твердой подложкой, будет снижаться, причем степень снижения зависит от эффективности испытуемого соединения как ингибитора киназы. Эффективность испытуемых соединений в отношении киназ может быть выражена как процент (относительно контроля) связывания киназы с твердой подложкой при данной концентрации испытуемого соединения, при этом чем ниже процент, тем более эффективна способность испытуемого соединения связывать киназу. Так, значение 100% относительно контроля указывает на то, что испытуемое соединение вообще не связывается с данной киназой, поскольку вся эта киназа связана с твердой подложкой. И наоборот, значение процента относительно контроля, равное 0%, указывает на то, что тестируемое соединение связывает всю киназу, поскольку никакая ее часть не связана с твердой подложкой.
Протокол:
Для большинства анализов штаммы Т7 фагов, меченные киназой, выращивали параллельно в 24-луночных блоках в хозяине Е. coli, произведенном из штамма BL21.
Е. coli выращивали до фазы логарифмического роста и инфицировали Т7 фагом из замороженного запаса (множественность заражения = 0,4) и инкубировали со встряхиванием при 32°С до лизиса (90-150 минут). Лизаты центрифугировали (6000 × g) и фильтровали (0,2 мкм) для удаления клеточного детрита. Оставшиеся киназы продуцировали в клетках HEK-293, а затем помечали ДНК для выявления методом кПЦР. Магнитные гранулы, покрытые стрептавидином, обрабатывали биотинилированными низкомолекулярными лигандами в течение 30 минут при комнатной температуре, чтобы выработать аффинные смолы для анализа киназ. Лигандированные гранулы блокировали избытком биотина и промывали блокирующим буфером (SeaBlock (Pierce), 1% BSA, 0,05% Tween 20, 1 мМ DTT) для удаления несвязанного лиганда и снижения неспецифического связывания фага. Реакции связывания собирали путем комбинирования киназ, лигандированных аффинных гранул и испытуемых соединений в 1х буфере для связывания (20% SeaBlock, 0,17х PBS, 0,05% Tween 20, 6 мМ DTT). Испытуемые соединения готовили в виде 40-кратных исходных растворов в 100% ДМСО и разбавляли непосредственно в ходе анализа. Все реакции проводили в полипропиленовых 384-луночных планшетах с конечным объемом 0,02 мл. Аналитические планшеты инкубировали при комнатной температуре со встряхиванием в течение 1 часа и аффинные гранулы промывали промывочным буфером (1х PBS, 0,05% Tween 20). Затем гранулы повторно суспендировали в элюирующем буфере (1х PBS, 0,05% Tween 20, 0,5 мкМ небиотинилированного аффинного лиганда) и инкубировали при комнатной температуре со встряхиванием в течение 30 минут.Концентрацию киназы в элюатах измеряли посредством кПЦР.
Соединения, связывающие активный сайт киназы и прямо (стерически) или косвенно (аллостерически) препятствующие связыванию киназы с иммобилизованным лигандом, уменьшают количество киназы, захваченной на твердой подложке. И наоборот, испытуемые молекулы, которые не связывают киназу, не влияют на количество киназы, захваченной на твердой подложке.
Сила связывания испытуемой молекулы с киназой может быть выражена как процент от контроля (% Ctrl)
Процент от контроля (% Ctrl)
Соединение (соединения) подвергали скринингу при концентрации 3000 нМ, и результаты первичного скрининга связующих взаимодействий представляли в отчете как «% Ctrl», где более низкие числа указывают на более сильные попадания в матрицу на следующей странице (страницах).
Расчет величины % Ctrl
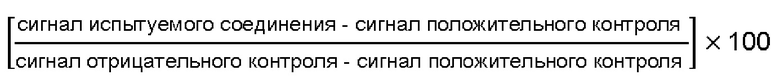
отрицательный контроль = ДМСО (100% Cfrl)
положительный контроль = контрольное соединение (0% Cfrl)
Значения % Ctrl для Атропизомера А-2 в отношении киназ, представленных в панели, приведены в Таблице 13 ниже.
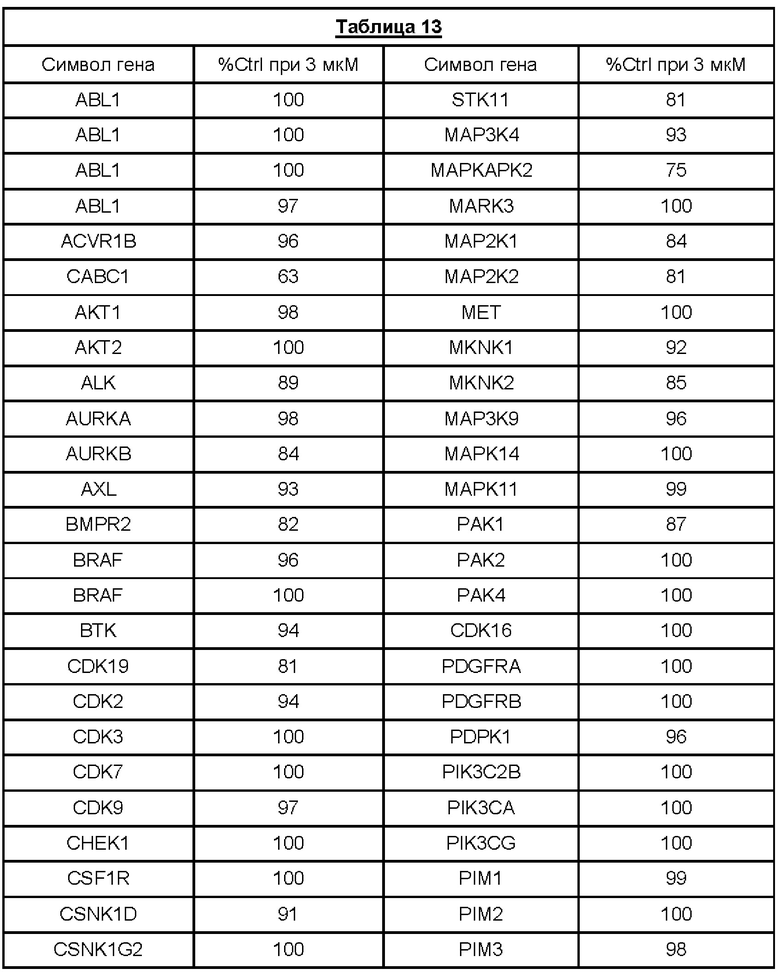
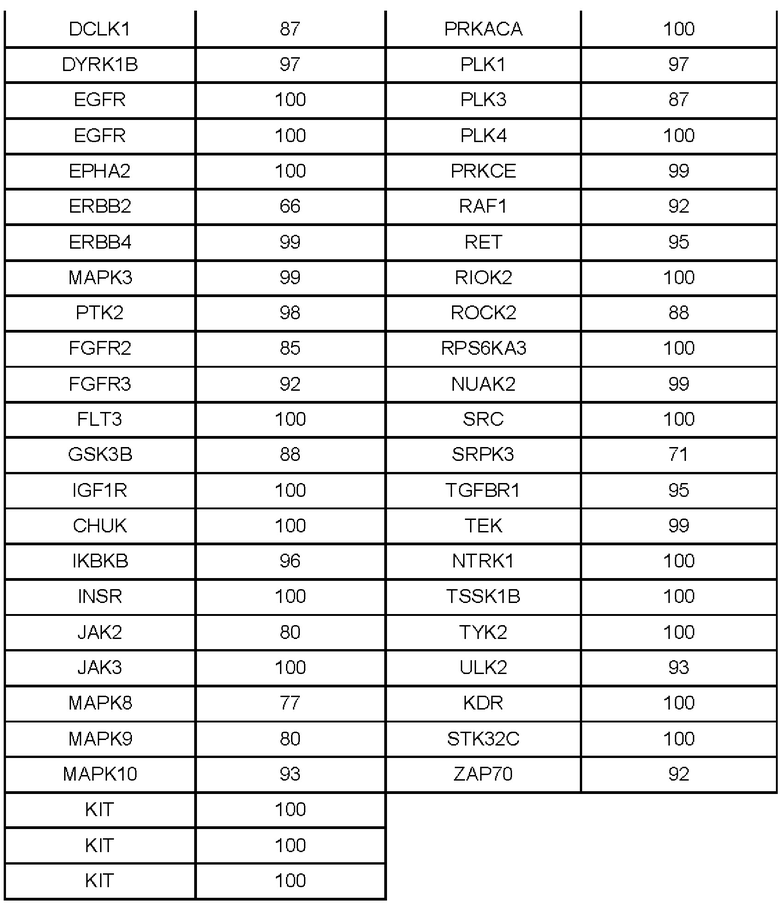
Результаты в отношении девяносто семи киназ показывают, что Атропизомер А-2 обладает плохой или отсутствующей связывающей активностью в отношении широкого диапазона киназ и, следовательно, вряд ли будет создавать проблемы, связанные с нецелевым (побочным) ингибированием киназ.
В случае PLK1 и PLK4 киназ Атропизомер А-2 показал незначительную или нулевую аффинность связывания с каталитическими доменами этих киназ (значения % Ctrl 97% и 100% соответственно). Поэтому делается вывод, что профили активности, указывающие на активность по ингибированию PLK1/PLK4, продемонстрированные в приведенных выше примерах, являются следствием связывания с некаталитическими Polo Box доменами в PLK1 и PLK4.
G. Определение пеоооальной биодоступности и воздействия на головной мозг согласно ФК у мышей
Атропизомеры А-2 и А-3 оценивали in vivo на мышиных моделях путем определения их концентраций в головном мозге и плазме крови после перорального и внутривенного введения.
При этом соблюдали следующий протокол:
Самцам мышей CD-1 вводили соединения согласно Примерам А-2 и А-3 путем либо внутривенного (2 мг/кг), либо перорального введения (10 мг/кг). Для анализа из лапки животного, получившего внутривенную дозу, отбирали восемь образцов через 2, 10, 30 мин, 1, 2, 4, 8 и 24 часа, и отбирали 9 образцов из лапки животного, получившего пероральную дозу, через 15, 30 мин, 1, 2, 4, 8, 24, 48 и 72 часа.
Оба соединения согласно Примерам А-2 и А-3 были приготовлены в форме раствора в смеси 10% DMSO/90% гидроксипропил-бета-циклодекстрин (20% масс./об. в воде), как для внутривенного, так и для перорального дозирования. N=3 мыши на временную точку.
У отдельных животных после введения доз отбирали терминальные образцы крови и помещали их в меченые полипропиленовые пробирки, содержащие антикоагулянт (ЭДТА). Образцы выдерживали на влажном льду в течение максимум 30 минут, в то время как отбор проб всех животных в когорте был завершен. Образцы крови центрифугировали для получения плазмы (при 4°С и 21100 g в течение 5 мин), и полученную плазму переносили в соответствующие маркированные пробирки. Терминальный головной мозг от каждого животного, получавшего пероральную дозу, иссекали, промывали солевым раствором и помещали в предварительно взвешенные меченые полипропиленовые пробирки, а образцы повторно взвешивали перед хранением.
Количественный биоанализ проводили методом жидкостной хроматографии - масс-спектроскопии. Полученные результаты представлены в Таблицах 14, 15 и 16 ниже и на Фиг. 12-15.
Пероральная биодоступность
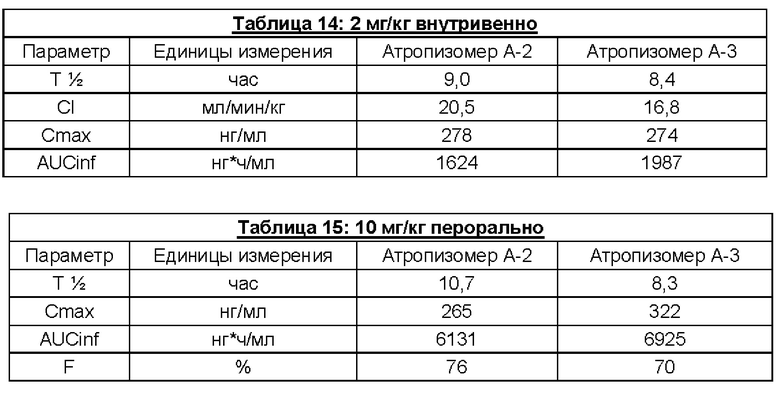
Эти результаты показывают, что Атропизомеры А-2 и А-3 имеют высокую степень абсорбции после их перорального введения доз мышам.
Воздействие на головной мозг
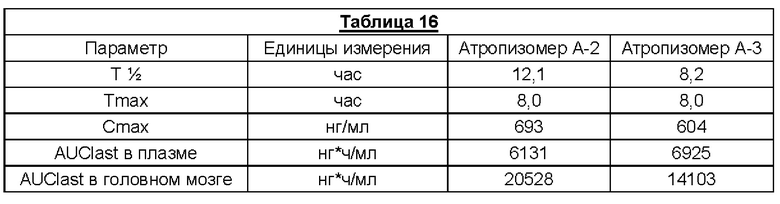
Результаты исследований по воздействию на головной мозг, представленные в Таблице 16, демонстрируют, что Атропизомеры А-2 и А-3 обладают высокой степенью воздействия на головной мозг.В случае Атропизомера А-2 результаты показывают, что Атропизомер А-2 имеет высокую экспозицию в головном мозге с соотношением AUC В:Р равным 3,3 после перорального введения его доз мышам.
Н. Эффективность in vivo
Атропизомер А-2 демонстрирует эффективность в мышиных моделях глиобластомы, когда опухоли имплантируют подкожно и ортотопически, как это указано в исследованиях, описанных ниже.
(i) Противораковая активность in vivo в модели подкожного ксенотрансплантата U87MG
Самцам бестимусных мышей, несущих опухоли U87MG, перорально вводили дозу 100 мг/кг Атропизомера А-2 в дни 1, 4 и 7, и объемы опухолей измеряли на протяжении 20 дней. Также измеряли объемы опухолей в контрольной группе опухоленесущих мышей, которые получали только носитель в те же моменты времени. Группа, получавшая лечение, показала значительное уменьшение объема опухоли по сравнению с контролем (3,85% Т/С в день 13), как это показано на Фигуре 16.
(ii) Противораковая активность in vivo в модели оототопического ксенотрансплантата U87-Luc
Клетки U87-Luc интрацеребрально имплантировали в мозг самцов бестимусных мышей, и рост опухоли контролировали с помощью биолюминесцентного сигнала. В группе лечения животным вводили пероральную дозу 100 мг/кг Атропизомера А-2 в 1, 4, 7, 10 и 13 дни. Животным контрольной группы вводили только носитель. Результаты, показанные на Фиг. 17, демонстрируют снижение сигнала опухоли у группы лечения относительно контрольной группы в День 15.
(iii) Противораковая активность in vivo у мышей, несущих опухоли НСТ116
Атропизомер А-2 показал эффективность в модели колоректального рака с мутацией KRAS, как это описано ниже.
Самцам бестимусных мышей, несущих ксенотрансплантатные опухоли НСТ116, перорально вводили дозу 100 мг/кг Атропизомера А-2 в дни 1, 8 и 15, а объемы опухолей измеряли в течение 3 недель. Также измеряли объемы опухолей в контрольной группе опухоленесущих мышей, которые получали только носитель в те же моменты времени.
Результаты, показанные на Фиг. 18, демонстрируют выраженное влияние на рост опухоли на День 20 (TGI 60%).
ФАРМАЦЕВТИЧЕСКИЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ
(i) Лекарственная форма в виде таблетки
Таблеточную лекарственную форму, содержащую композицию или атропизомер согласно настоящему изобретению, получают путем смешивания 50 мг соединения со 197 мг лактозы (BP) в качестве разбавителя и 3 мг стеарата магния в качестве лубриканта и последующего прессования с получением таблетки известным способом.
(ii) Лекарственная форма в виде капсулы
Лекарственную форму в виде капсул получают путем смешивания 100 мг композиции или атропизомера согласно настоящему изобретению со 100 мг лактозы и наполнения полученной смесью стандартных непрозрачных твердых желатиновых капсул.
(iii) Инъекционная лекарственная Форма I
Лекарственная форма для парентерального введения путем инъекции может быть приготовлена растворением композиции или атропизомера согласно настоящему изобретению (например, в форме соли) в воде, содержащей 10% пропиленгликоля, так чтобы получить концентрацию активного вещества 1,5 масс. %. Затем раствор стерилизуют фильтрацией, разливают в ампулы и запаивают.
(iv) Инъекционная лекарственная форма II
Лекарственная форма для парентерального введения путем инъекции может быть приготовлена растворением в воде композиции или атропизомера согласно настоящему изобретению (например, в форме соли) (2 мг/мл) и маннита (50 мг/мл), после чего полученный раствор стерилизуют фильтрацией и разливают в плотно укупориваемые флаконы объемом 1 мл или ампулы.
(iv) Инъекционная лекарственная форма III
Лекарственная форма для внутривенной доставки путем инъекции или инфузии может быть приготовлена путем растворения композиции или атропизомера согласно настоящему изобретению (например, в форме соли) в воде, так чтобы получить концентрации 20 мг/мл. Затем флакон укупоривают и стерилизуют автоклавированием.
(vi) Инъекционная лекарственная форма IV
Лекарственная форма для внутривенной доставки путем инъекции или инфузии может быть приготовлена путем растворения композиции или атропизомера согласно настоящему изобретению (например, в форме соли) в воде, содержащей буфер (например, 0,2М ацетатный с рН 4,6), так чтобы получить концентрацию 20 мг/мл. Затем флакон укупоривают и стерилизуют автоклавированием.
(vii) Лекарственная формула для подкожного введения
Лекарственную форму для подкожного введения получают смешиванием композиции или атропизомера согласно настоящему изобретению с кукурузным маслом фармацевтической степени чистоты, так чтобы получить концентрацию 5 мг/мл. Композицию стерилизуют и помещают в подходящую емкость.
(viii) Лиофилизиоованная лекарственная форма
Аликвоты композиции или атропизомера согласно настоящему изобретению в готовой смеси помещают во флаконы объемом 50 мл и лиофилизируют. Во время лиофилизации композиции замораживают с соблюдением одноступенчатого протокола замораживания при (-45°С). Температуру поднимают до -10°С для отпуска напряжений, затем снижают до -45°С для замораживания, после чего производят первичную сушку при +25°С в течение приблизительно 3400 минут и затем вторичную сушку с поэтапными повышениями температуры до 50°С. Давление в процессе первичной и вторичной сушки устанавливают на 80 миллиТорр.
Эквиваленты
Приведенные выше примеры представлены лишь с целью иллюстрации изобретения и их не следует рассматривать как ограничивающие объем настоящего изобретения. Должно быть очевидно, что в описанные выше и проиллюстрированные в примерах конкретные варианты реализации изобретения, не отступая от принципов, лежащих в основе настоящего изобретения, могут быть внесены самые разнообразные модификации и изменения. Предполагается, что настоящее изобретение охватывает любые подобные модификации и изменения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКОЕ СОЕДИНЕНИЕ | 2018 |
|
RU2786994C2 |
| НОВЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ (ВАРИАНТЫ) И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2550346C2 |
| ПРОИЗВОДНЫЕ 2-АМИНОХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ P70S6 КИНАЗЫ | 2016 |
|
RU2736123C1 |
| СОЕДИНЕНИЯ И СПОСОБЫ МОДУЛИРОВАНИЯ КИНАЗ И ПОКАЗАНИЯ К ПРИМЕНЕНИЮ УКАЗАННЫХ СОЕДИНЕНИЙ И СПОСОБОВ | 2008 |
|
RU2487121C2 |
| 4, 5-ДИГИДРО-[1, 2, 4]ТРИАЗОЛО[4, 3-F]ПТЕРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ Plk1 ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2441006C2 |
| ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ КАК МОДУЛЯТОРЫ ФОСФОИНОЗИТИД-3-КИНАЗЫ | 2014 |
|
RU2672910C9 |
| СТЕРЕОИЗОМЕРНО ОБОГАЩЕННЫЕ 3-АМИНОКАРБОНИЛЬНЫЕ БИЦИКЛОГЕПТЕНОВЫЕ ПИРИМИДИНДИАМИНЫ И ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2416604C2 |
| КОМПЛЕКСНАЯ ТЕРАПИЯ | 2013 |
|
RU2677245C2 |
| АМИНОПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2012 |
|
RU2674017C2 |
| КОНФОРМАЦИОННО ОГРАНИЧЕННЫЕ ИНГИБИТОРЫ PI3K И mTOR | 2014 |
|
RU2669696C2 |
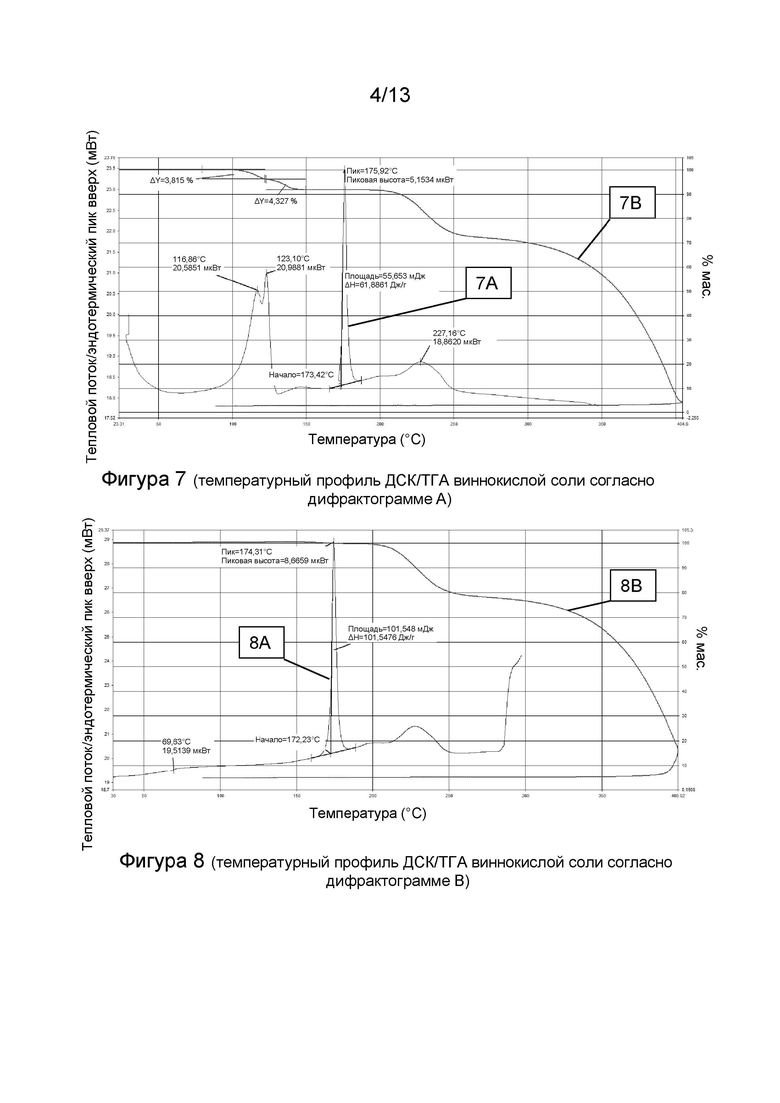
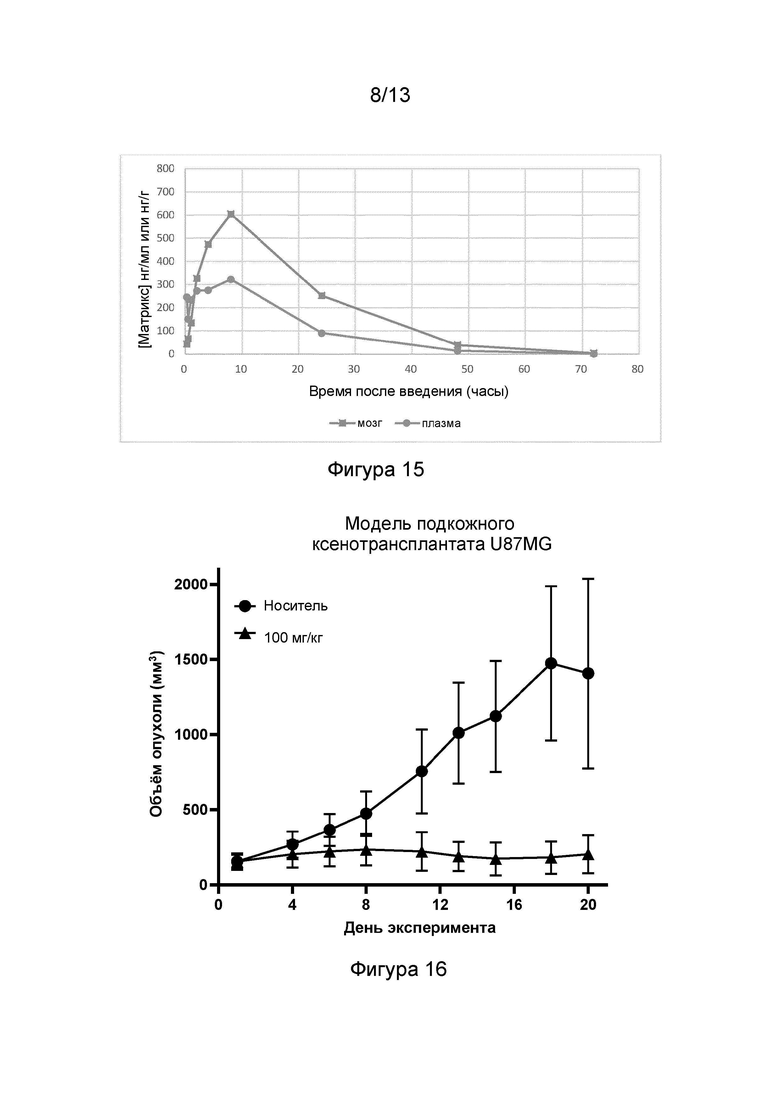
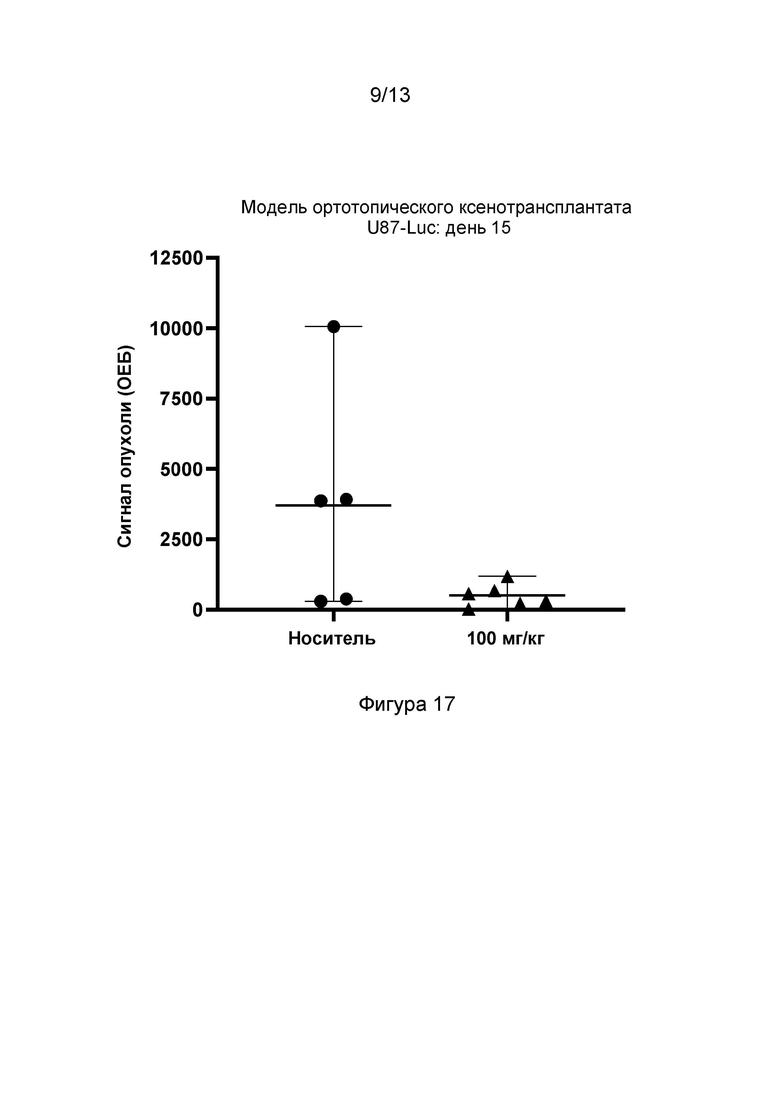
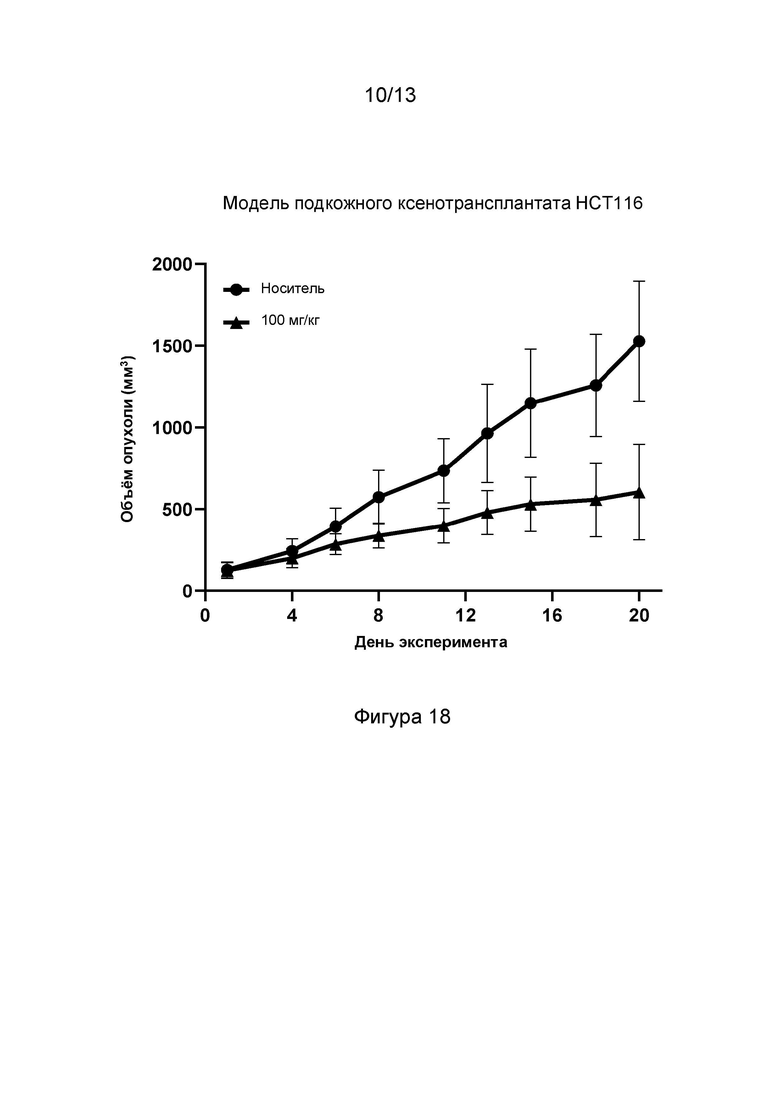
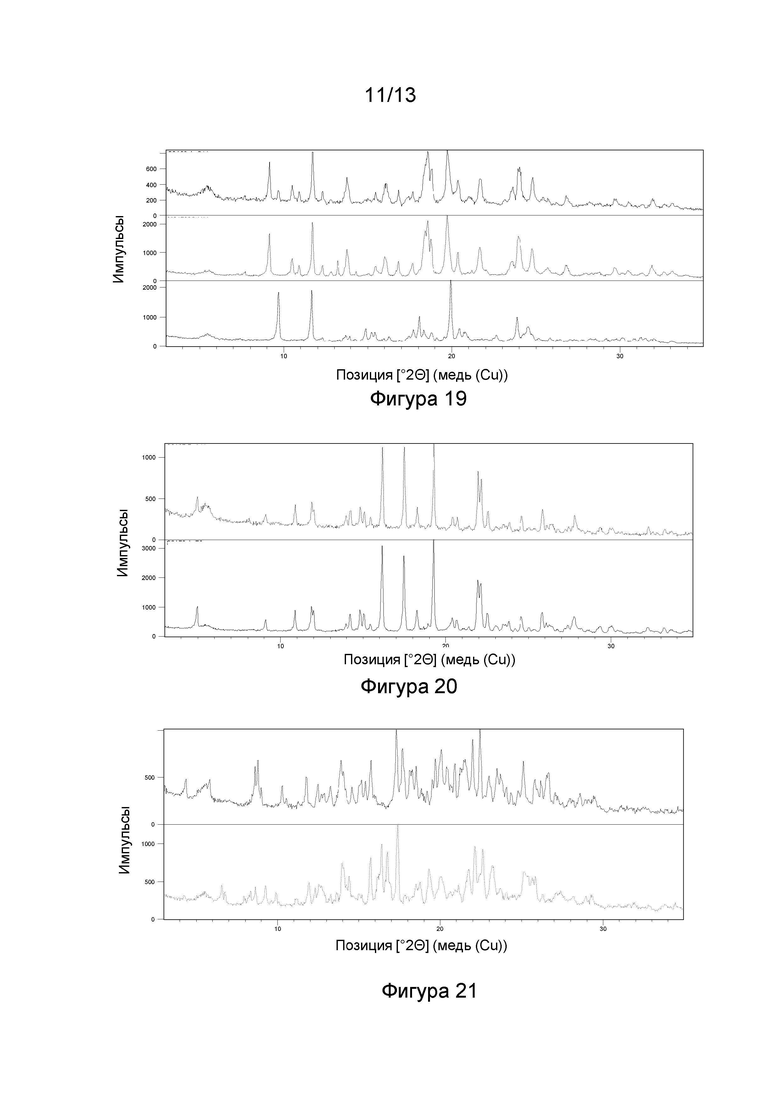
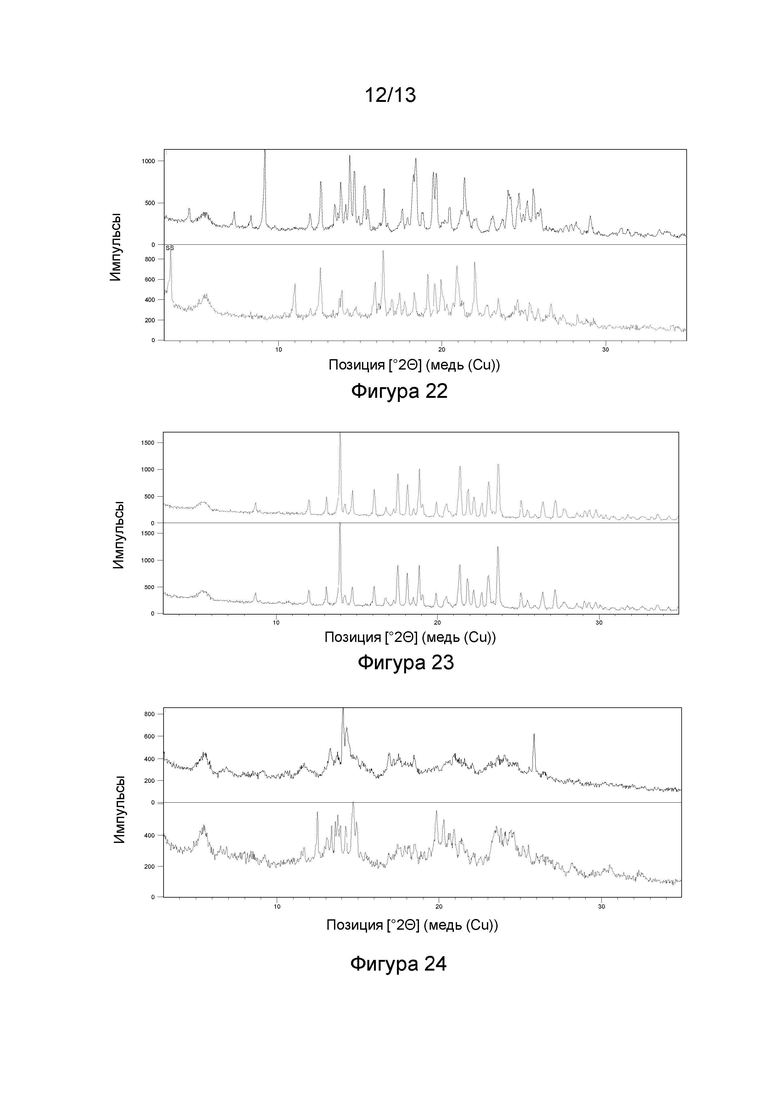
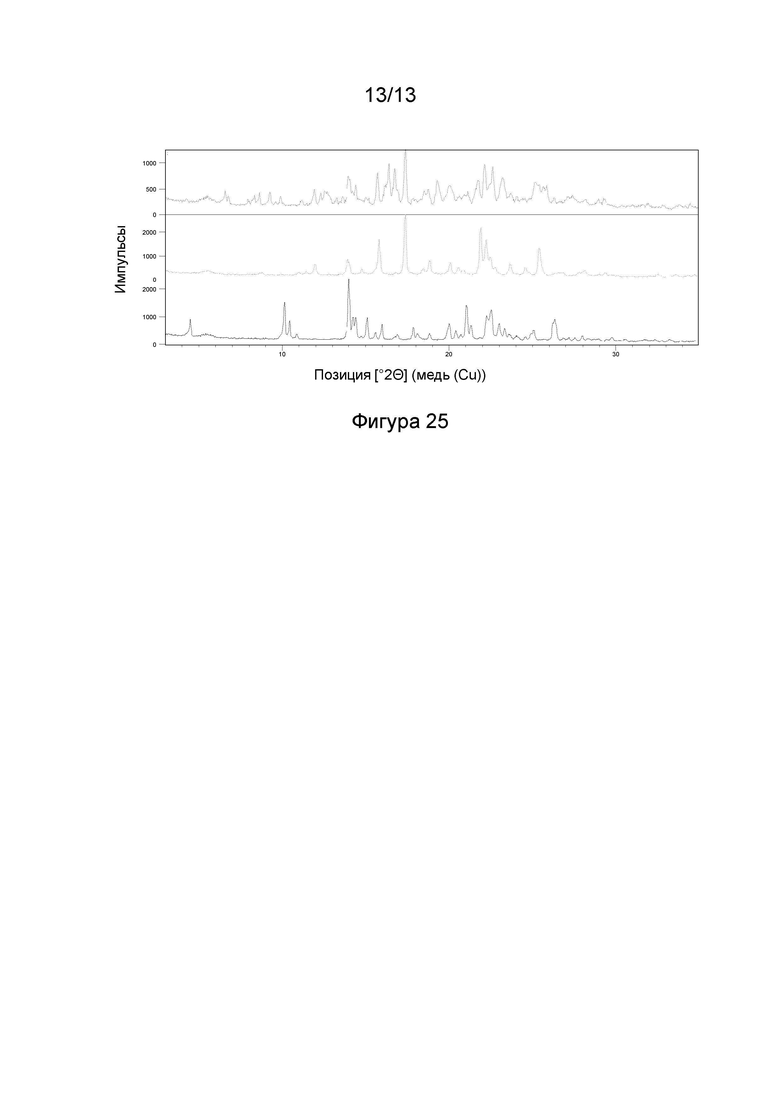
Группа изобретений относится к лечению рака. Композиция для лечения ракового заболевания состоит из по меньшей мере на 90 масс. % из атропизомера формулы (1) или его фармацевтически приемлемой соли или его таутомера; на 0-10 масс. % из атропизомера формулы (2) или его фармацевтически приемлемой соли или его таутомера:
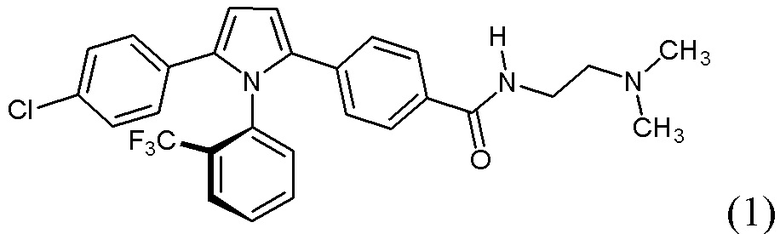
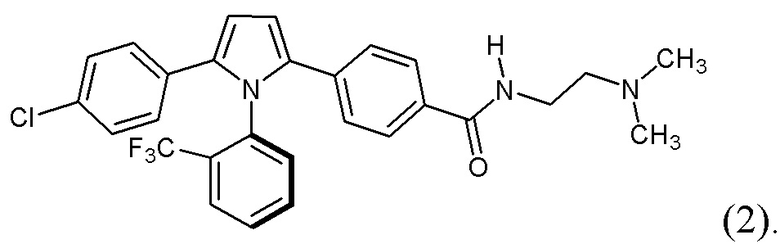
Также раскрыты одинарный атропизомер, фармацевтическая композиция для лечения ракового заболевания, их применение для лечения ракового заболевания, в процесс развития которого вовлечена PLK1 и/или PLK4, применение в качестве ингибитора PLK1-PBD и/или PLK4-PBD, применение при получении лекарственного средства для лечения ракового заболевания, способ лечения ракового заболевания, способ получения композиции или одинарного атропизомера. Группа изобретений обеспечивает эффективное лечение рака. 9 н. и 9 з.п. ф-лы, 13 ил., 16 табл., 11 пр.
1. Композиция для лечения ракового заболевания, состоящая из
(i) по меньшей мере на 90 масс. % из атропизомера формулы (1)
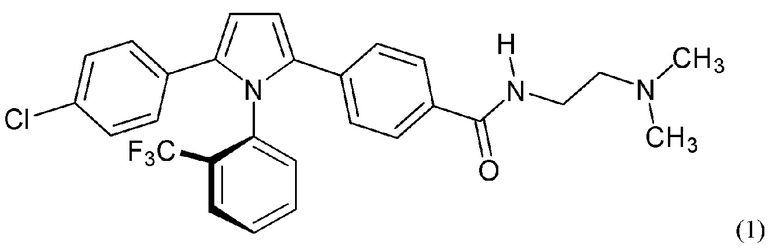
или его фармацевтически приемлемой соли или его таутомера; и
(ii) на 0-10 масс. % из атропизомера формулы (2)
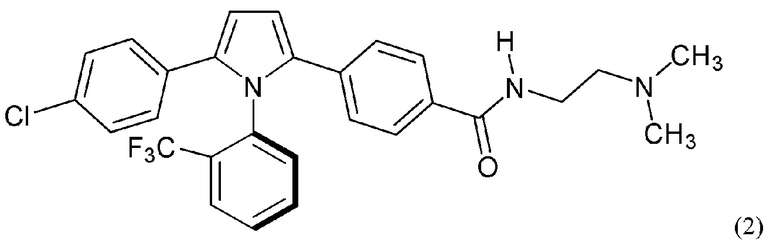
или его фармацевтически приемлемой соли или его таутомера.
2. Композиция по п. 1, состоящая из по меньшей мере на 99,5 масс. % из атропизомера формулы (1) или его фармацевтически приемлемой соли или его таутомера; и не более чем на 0,5 масс. % из атропизомера формулы (2) или его фармацевтически приемлемой соли или его таутомера.
3. Одинарный атропизомер, представляющий собой (R)-4-[5-(4-хлорфенил)-1-[2-(трифторметил)-фенил]пиррол-2-ил]-N-[2-(диметиламино)этил]бензамид, или его фармацевтически приемлемая соль или его таутомер.
4. Фармацевтическая композиция для лечения ракового заболевания, содержащая композицию по любому из пп. 1-2 или одинарный атропизомер по п. 3 и фармацевтически приемлемое вспомогательное вещество.
5. Применение композиции по любому из пп. 1, 2, 4, или одинарного атропизомера по п. 3 для лечения ракового заболевания, в процесс развития которого вовлечена PLK1 и/или PLK4.
6. Применение композиции по любому из пп. 1, 2, 4, или одинарного атропизомера по п. 3 в качестве ингибитора PLK1-PBD и/или PLK4-PBD.
7. Применение композиции по любому из пп. 1, 2, 4, или одинарного атропизомера по п. 3 для лечения ракового заболевания, выбранного из:
a) рака поджелудочной железы, рака толстой кишки и прямой кишки, рака легких, рака молочной железы, рака яичников, рака простаты и рака головного мозга и нервов;
b) рака крови, такого как лимфома и лейкоз, и миелопролиферативные нарушения, включая острый миелоидный лейкоз (AML), острый лимфоцитарный лейкоз (ALL), B- и Т-клеточные лимфомы;
c) глиомы и глиобластомы;
d) рабдоидных опухолей, медуллобластомы и других эмбриональных опухолей головного мозга; рака молочной железы, рака легких, меланомы, рака желудка, рака толстой и прямой кишки, рака поджелудочной железы и рака яичников;
e) сарком, включая рабдомиосаркому, остеосаркому;
f) рака, в котором участвует PLK1;
g) рака, в котором участвует PLK4;
h) рака, который характеризуется дефицитом p53 или мутацией в гене TP53; и
i) рака, характеризующегося наличием мутированной формы KRAS.
8. Применение композиции по любому из пп. 1, 2, 4, или одинарного атропизомера по п. 3 при получении лекарственного средства для лечения ракового заболевания, где раковое заболевание выбрано из:
a) рака поджелудочной железы, рака толстой кишки и прямой кишки, рака легких, рака молочной железы, рака яичников, рака простаты и рака головного мозга и нервов;
b) рака крови, такого как лимфома и лейкоз, и миелопролиферативные нарушения, включая острый миелоидный лейкоз (AML), острый лимфоцитарный лейкоз (ALL), B- и Т-клеточные лимфомы;
c) глиомы и глиобластомы;
d) рабдоидных опухолей, медуллобластомы и других эмбриональных опухолей головного мозга; рака молочной железы, рака легких, меланомы, рака желудка, рака толстой и прямой кишки, рака поджелудочной железы и рака яичников;
e) сарком, включая рабдомиосаркому, остеосаркому;
f) рака, в котором участвует PLK1;
g) рака, в котором участвует PLK4;
h) рака, который характеризуется дефицитом p53 или мутацией в гене TP53; и
i) рака, характеризующегося наличием мутированной формы KRAS.
9. Способ лечения ракового заболевания, выбранного из:
a) рака поджелудочной железы, рака толстой кишки и прямой кишки, рака легких, рака молочной железы, рака яичников, рака простаты и рака головного мозга и нервов;
b) рака крови, такого как лимфома и лейкоз, и миелопролиферативные нарушения, включая острый миелоидный лейкоз (AML), острый лимфоцитарный лейкоз (ALL), B- и Т-клеточные лимфомы;
c) глиомы и глиобластомы;
d) рабдоидных опухолей, медуллобластомы и других эмбриональных опухолей головного мозга; рака молочной железы, рака легких, меланомы, рака желудка, рака толстой и прямой кишки, рака поджелудочной железы и рака яичников;
e) сарком, включая рабдомиосаркому, остеосаркому;
f) рака, в котором участвует PLK1;
g) рака, в котором участвует PLK4;
h) рака, который характеризуется дефицитом p53 или мутацией в гене TP53; и
i) рака, характеризующегося наличием мутированной формы KRAS;
где способ включает введение эффективного количества композиции по любому из пп. 1, 2, 4, или одинарного атропизомера по п. 3.
10. Применение по п. 8, где раковое заболевание выбрано из мультиформной глиобластомы, эпендимомы, диффузной глиомы ствола головного мозга, IDH1-мутантных глиом.
11. Применение по п. 8, где раковое заболевание представляет собой раковое заболевание, при котором имеет место сверхэкспрессия PLK1.
12. Применение по п. 8, где раковое заболевание представляет собой раковое заболевание, при котором имеет место сверхэкспрессия PLK4.
13. Способ по п. 9, где раковое заболевание выбрано из мультиформной глиобластомы, эпендимомы, диффузной глиомы ствола головного мозга, IDH1-мутантных глиом.
14. Способ по п. 9, где раковое заболевание представляет собой раковое заболевание, при котором имеет место сверхэкспрессия PLK1.
15. Способ по п. 9, где раковое заболевание представляет собой раковое заболевание, при котором имеет место сверхэкспрессия PLK4.
16. Способ получения композиции по любому из пп. 1, 2, 4, или одинарного атропизомера по п. 3, включающий хиральное разделение смеси атропизомеров соединения формулы (0):
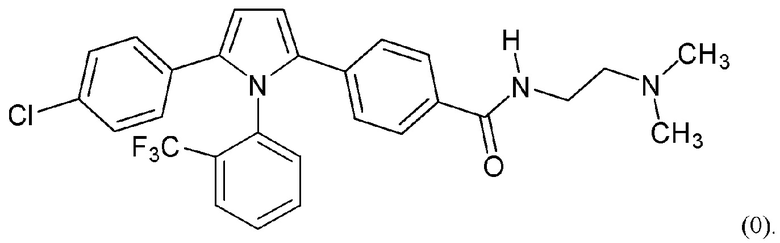
17. Способ по п. 16, где смесь атропизомеров соединения формулы (0) представляет собой рацемическую смесь.
18. Способ по п. 16 или 17, где хиральное разделение проводят путем:
(i) пропускания смеси атропизомеров через хиральную хроматографическую колонку, или
(ii) взаимодействия смеси атропизомеров соединения формулы (0) с хиральной кислотой с образованием солей обоих атропизомеров в смеси, разделения солей и их разложения, в результате чего получают соответствующие свободные основания каждого из атропизомеров.
| WO 2018197714 A1, 01.11.2018 | |||
| ЗАМЕЩЕННЫЕ ПОЛИЦИКЛИЧЕСКИЕ ПИРАЗОЛЬНЫЕ ИНГИБИТОРЫ КИНАЗНОЙ АКТИВНОСТИ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2655921C2 |
| ХАРКЕВИЧ Д.А | |||
| Фармакология: Учебник, 2010, 10-е издание, стр.72-82 | |||
| ДУРНОВ Л.А., ГОЛДОБЕНКО Г.В | |||
| Детская онкология, Москва, "Медицина", 2002, стр.139; Малая медицинская энциклопедия, т.5, Москва, "Медицина", 1996, стр | |||
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |

