Область техники, к которой относится изобретение
Изобретение относится к области производства карбоксилатов металлов, в частности, к кристаллической форме цис-2,3-эпоксисукцината кальция, характеризующейся модифицированной кристаллической решеткой. Полученную кристаллическую форму цис-2,3-эпоксисукцинат кальция используют, например, в производстве винной кислоты, поликарбоксилатов и т.д.
Уровень техники
L-(+)-винная кислота широко применяется в пищевой промышленности, медицине и фармакологии, аналитической химии, производстве гипсовых изделий и сухих строительных смесей и т.д.
Одним из способов получения L-(+)-винной кислоты является многостадийный процесс, включающий следующие стадии:
˗ получение малеата щелочного или щелочно-земельного металла;
˗ эпоксидирование малеата пероксидом в присутствии катализаторов эпоксидирования - молибдатов или вольфраматов щелочных или щелочно-земельных металлов с получением эпоксисукцинатов щелочного или щелочно-земельного металла. Для облегчения выделения эпоксисукцинатов из реакционной массы в качестве щелочного и щелочно-земельного металла, как правило, используют кальций или барий, соли которых малорастворимы или нерастворимы. Предпочтительно используют нетоксичные соли, т.е. соли кальция;
- ферментативный гидролиз эпоксисукцинатов с получением L-(+)-винной кислоты.
Из документа GB1423028 (MITSUBISHI GAS CHEMICAL COMPANY, INC., опубл. 28.01.1976) известен способ получения цис-2,3-эпоксисукцината кальция дигидрата заключающийся во взаимодействии кислого малеата кальция и перекиси водорода в присутствии водорастворимого катализатора эпоксидирования - одной или нескольких солей вольфрамовой и/или молибденовой кислоты. По окончании процесса эпоксидирования реакционную массу охлаждают до 25оС, цис-2,3-эпоксисукцинат кальция кристаллизуют и отфильтровывают от жидкой фазы. Недостатком данного способа является низкий выход соли, составляющий 64-75,9%.
Из документа GB1534195 (Takeda Chemical Industries, опубл. 29.11.1978) известен способ получения кристаллов эпоксисукцината кальция размером 100 мкм и менее, предпочтительно 70 мкм и менее, выбранный в качестве прототипа. Цис-2,3-эпоксисукцинат кальция получают в две стадии - первую стадию эпоксидирования с вольфраматом натрия проводят, используя кислый малеат кальция (при нейтрализации малеиновой кислоты 0,4-0,6 эквивалентами карбоната кальция), а вторую стадию кристаллизации цис-2,3-эпоксисукцинат кальций (пентагидрата) проводят при температуре не выше 70°С. Однако данный способ характеризуется недостаточно крупными кристаллами, что может приводить к замедлению растворения соли из-за низкой площади поверхности, и, следовательно, увеличенному времени ферментации. Так же, как будет далее проиллюстрировано в примерах, заявленный способ характеризуется образованием большого количества мелких кристаллов и гелеобразованием, что в конечном итоге приводит к сложному и долгому процессу фильтрации кристаллов цис-2,3-эпоксисукцината кальция.
Таким образом, существует необходимость в разработке улучшенных кристаллических форм цис-2,3-эпоксисукцината кальция и способов их получения, а также приготовления хорошо фильтрующихся суспензий цис-2,3-эпоксисукцината кальция для повышения эффективности стадии эпоксидирования и последующей стадии ферментации.
Сущность изобретения
Задачей настоящего изобретения является разработка новой кристаллической формы цис-2,3-эпоксисукцината кальция, позволяющей более эффективно проводить процесс ферментативного получения L-(+)-винной кислоты.
Технический результат заключается в получении новой кристаллической формы цис-2,3-эпоксисукцината кальция, где кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22 °С составляют a=15.1916(2) Å, b=8.9121(1) Å, c=7.4724(1) Å, beta=103.309(1)°, характеризующейся размером частиц от 60 до 115 мкм, предпочтительно 100 мкм.
Также техническим результатом является получение однородной и хорошо фильтрующейся суспензии кристаллической формы цис-2,3-эпоксисукцината кальция с размером кристаллов от 60 до 115 мкм, предпочтительно 100 мкм.
Дополнительным техническим результатом является отсутствие влияния температуры на процесс образования новой кристаллической формы цис-2,3-эпоксисукцината кальция.
Данная техническая задача решается, и достижение технического результата обеспечивается за счет получения новой кристаллической формы цис-2,3-эпоксисукцината кальция при выдерживании нестабильной кристаллической формы цис-2,3-эпоксисукцината кальция в течение от 24 до 240 часов, а также за счет дальнейшего использования новой кристаллической формы цис-2,3-эпоксисукцината кальция в качестве затравочных кристаллов при кристаллизации (затравки).
Здесь и далее под различными «кристаллическими формами вещества» понимают видоизменения кристаллической структуры, происходящие из-за способности одного вещества существовать в различных кристаллических формах, или структурах, называемых полиморфными модификациями.
Авторами настоящего изобретения было неожиданно обнаружено, что при выдерживании первоначальной трудно фильтрующейся кристаллической формы цис-2,3-эпоксисукцината кальция, образуются кристаллы новой кристаллической модификации. Дальнейшее использование новой кристаллической формы в качестве затравки позволяет получить хорошо фильтрующуюся суспензию цис-2,3-эпоксисукцината кальция со средним размером частиц 100 мкм.
Неожиданно было обнаружено, что при кристаллизации с затравкой, распределение кристаллов по размерам практически не зависит от температуры кристаллизации, максимум распределения размеров кристаллов составляет 100 мкм.
Настоящее изобретение также относится к способу получения кристаллической формы цис-2,3-эпоксисукцината кальция согласно настоящему изобретению, включающему следующие стадии:
a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция;
b) эпоксидирование кислого малеата кальция пероксидом водорода в присутствии катализаторов эпоксидирования с получением кислого цис-2,3-эпоксисукцината кальция;
c) нейтрализация кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция;
d) выдерживание цис-2,3-эпоксисукцината кальция в течение от 24 до 240 ч с получением кристаллической формы цис-2,3-эпоксисукцината кальция
е) выделение кристаллической формы цис-2,3-эпоксисукцината кальция.
Настоящее изобретение также относится к однородной суспензии кристаллической формы цис-2,3-эпоксисукцината кальция согласно настоящему изобретению, где кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22 °С составляют a=15.1916(2) Å, b=8.9121(1) Å, c=7.4724(1) Å, beta=103.309(1)°.
Также настоящее изобретение относится к применению кристаллической формы цис-2,3-эпоксисукцината кальция согласно настоящему изобретению в качестве затравки в способе получения новых порций кристаллической формы цис-2,3-эпоксисукцината кальция.
Настоящее изобретение также относится к способу получения кристаллической формы цис-2,3-эпоксисукцината кальция, включающему следующие стадии:
a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция;
b) эпоксидирование кислого малеата кальция пероксидом водорода в присутствии катализаторов эпоксидирования с получением кислого эпоксисукцината кальция;
c) добавление от 0,1 до 20 мас.% затравки;
d) нейтрализация кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция;
е) выделение кристаллической формы цис-2,3-эпоксисукцината кальция;
отличающийся тем, что в качестве затравки используют кристаллическую форму цис-2,3-эпоксисукцината кальция согласно настоящему изобретению.
Также настоящее изобретение относится к способу получения L-(+)-винной кислоты, включающему стадии:
a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция;
b) эпоксидирование кислого малеата кальция пероксидом водорода в присутствии катализаторов эпоксидирования с получением кислого цис-2,3-эпоксисукцината кальция;
c) нейтрализация кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция;
d) выдерживание цис-2,3-эпоксисукцината кальция в течение от 24 до 240 ч с получением кристаллической формы цис-2,3-эпоксисукцината кальция, где кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22°С составляют a=15.1916(2) Å, b=8.9121(1) Å, c=7.4724(1) Å, beta=103.309(1)°;
e) ферментативный гидролиз полученной кристаллической формы цис-2,3-эпоксисукцината кальция.
Настоящее изобретение также относится к способу получения L-(+)-винной кислоты, включающему стадии:
a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция;
b) эпоксидирование кислого малеата кальция пероксидом водорода в присутствии катализаторов эпоксидирования с получением кислого эпоксисукцината кальция;
c) добавление от 0,1 до 20 мас.% затравки;
d) нейтрализация кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция;
e) ферментативный гидролиз полученной кристаллической формы цис-2,3-эпоксисукцината кальция,
отличающийся тем, что в качестве затравки используют кристаллическую форму цис-2,3-эпоксисукцината кальция, где кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22°С составляют a=15.1916(2) Å, b=8.9121(1) Å, c=7.4724(1) Å, beta=103.309(1)°.
Описание фигур
Для пояснения технических решений, раскрывающих суть настоящего изобретения, представлены Фиг. 1 -7.
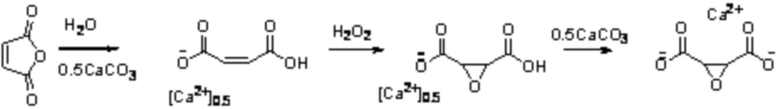
На Фиг. 1 представлена дифрактограмма цис-2,3-эпоксисукцината кальция, полученного по прототипу.
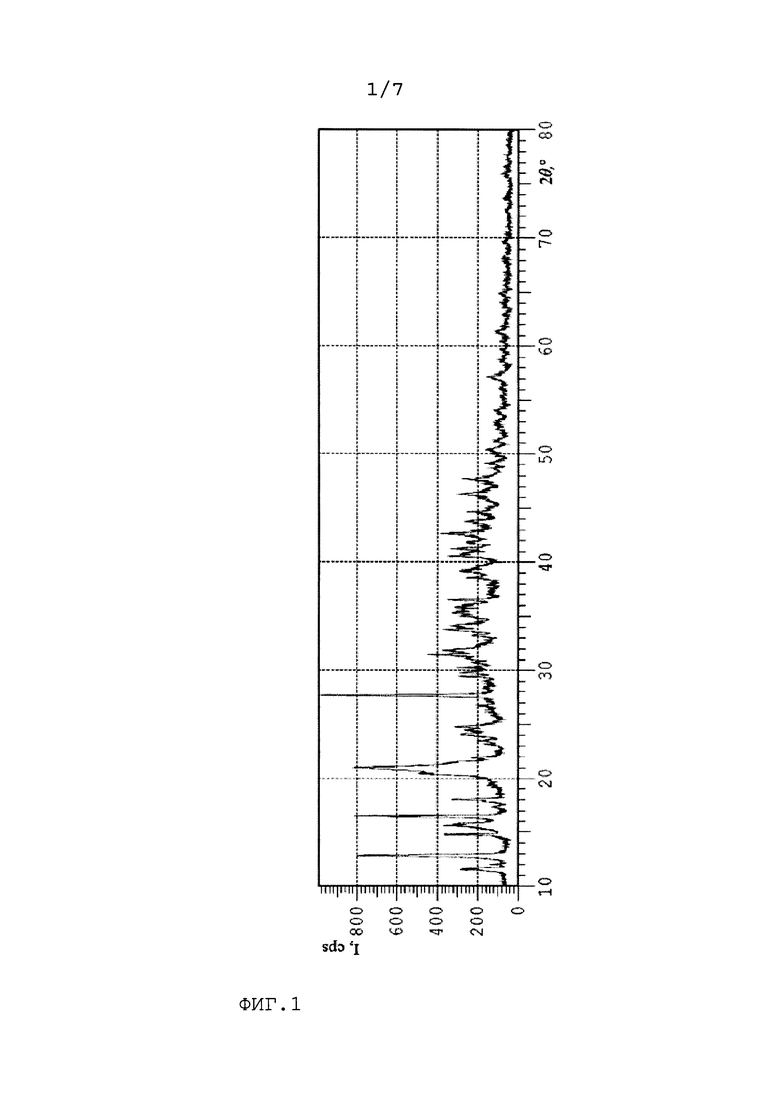
На Фиг. 2 представлена дифрактограмма цис-2,3-эпоксисукцината кальция, полученного по изобретению.
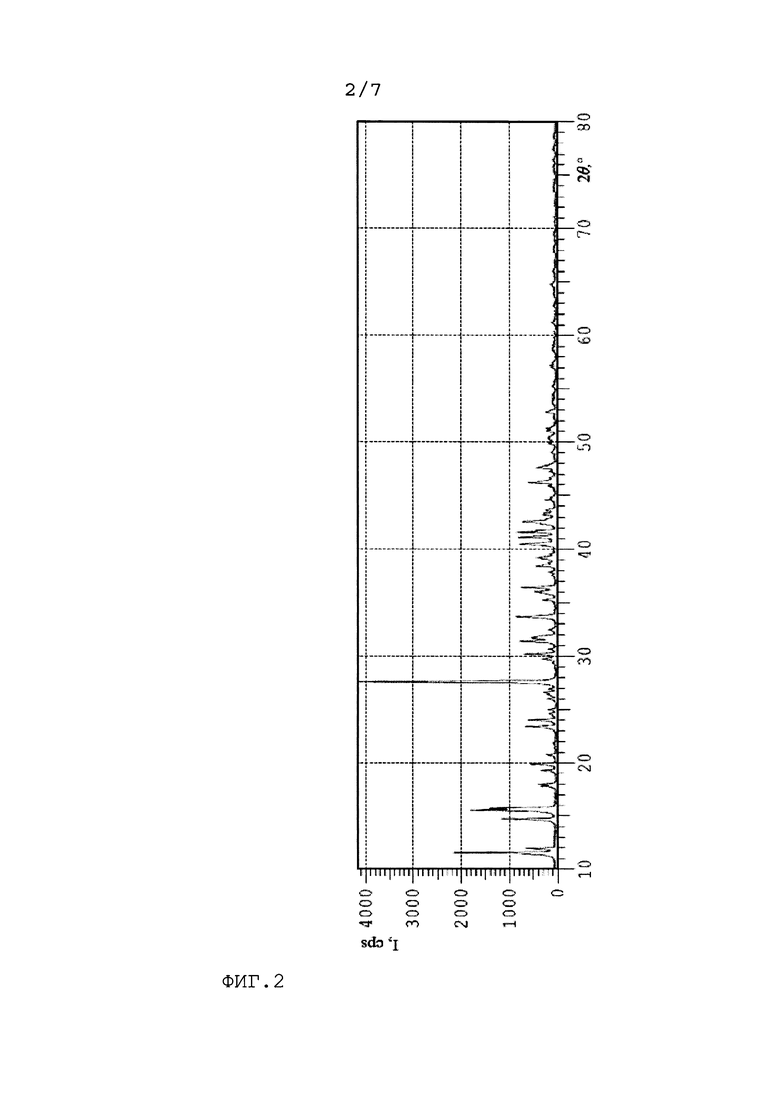
На Фиг. 3 представлено распределение по размерам частиц цис-2,3-эпоксисукцината кальция, полученного по изобретению при кристаллизации при 50°С.
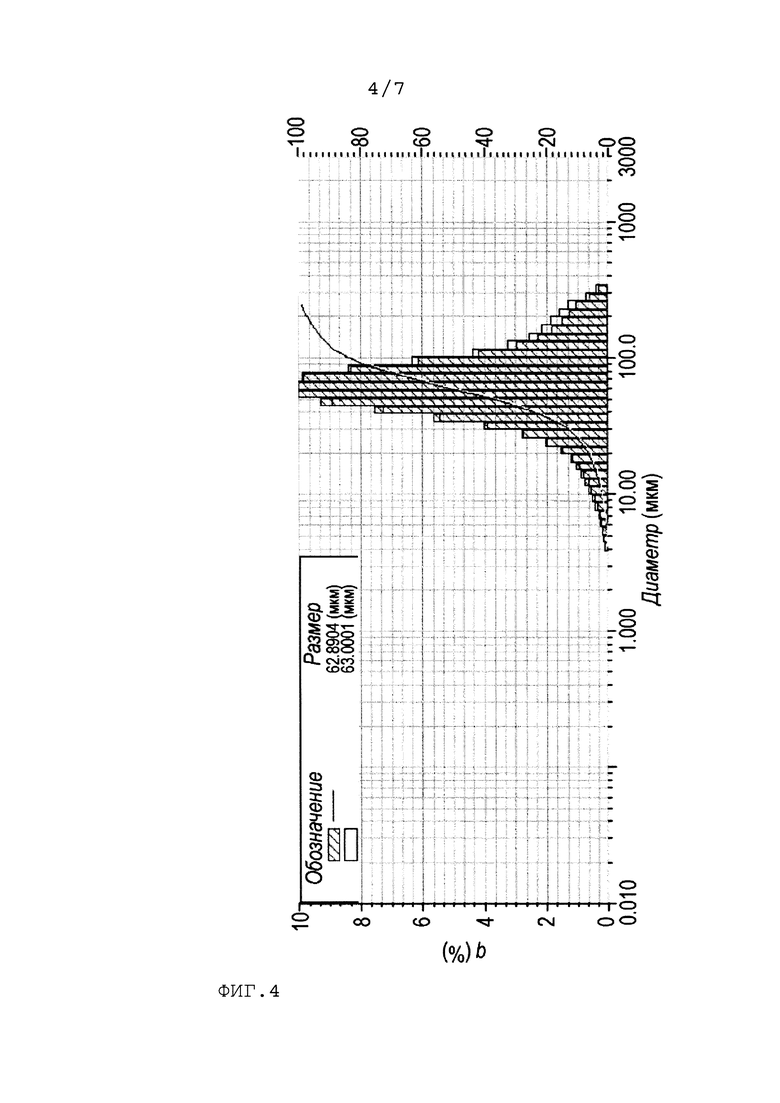
На Фиг. 4 представлено распределение по размерам частиц цис-2,3-эпоксисукцината кальция, полученного по изобретению при кристаллизации при 30°С.
На Фиг. 5 представлен ИК-спектр цис-2,3-эпоксисукцината кальция, полученного по изобретению, где сплошной линией представлен спектр цис-2,3-эпоксисукцината кальция, полученного по Примеру 3, пунктирной линией - спектр цис-2,3-эпоксисукцината кальция, полученного по Примеру 4.
На Фиг. 6 приведены результаты полнопрофильного уточнения дифрактограммы цис-2,3-эпоксисукцината кальция, полученного по изобретению, по методу ЛеБеля.
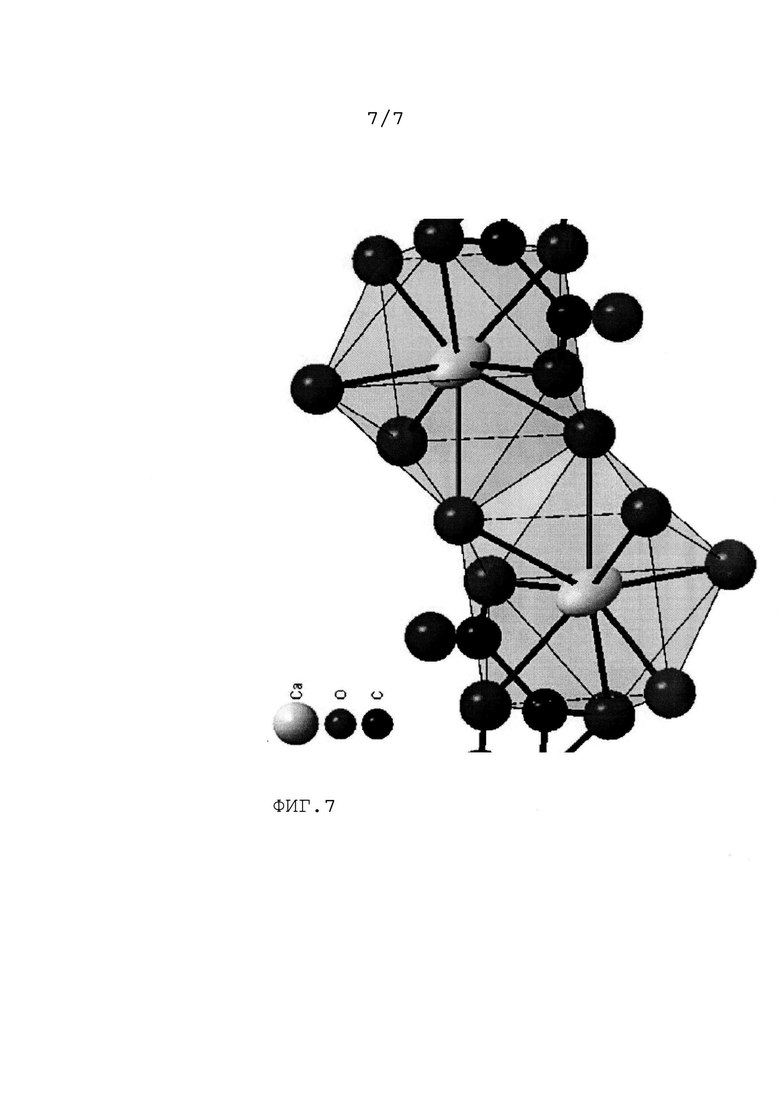
На Фиг.7 приведена проекция координационных полиэдров ионов кальция в кристаллической структуре цис-2,3-эпоксисукцината кальция, полученного по изобретению.
Подробное описание изобретения
Далее приводится описание различных аспектов реализации настоящего изобретения.
Настоящее изобретение относится к кристаллической форме цис-2,3-эпоксисукцината кальция, где ее кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22°С составляют a=15.1916(2) Å, b=8. 9121(1) Å, c=7.4724(1) Å, beta=103.309(1)°.
В одном варианте осуществления изобретения, раскрывается способ получения кристаллической модификации цис-2,3-эпоксисукцината кальция по настоящему изобретению, где кристаллическую форму получают следующим способом:
a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция;
b) эпоксидирование кислого малеата кальция пероксидом водорода в присутствии катализаторов эпоксидирования с получением кислого цис-2,3-эпоксисукцината кальция;
c) нейтрализация кислого цис-2,3-эпоксисукцината кальция добавлением соединения кальция;
d) выдерживание суспензии цис-2,3-эпоксисукцината кальция в течение от 24 до 240 ч с получением кристаллической формы цис-2,3-эпоксисукцината кальция;
e) выделение кристаллической формы цис-2,3-эпоксисукцината кальция.
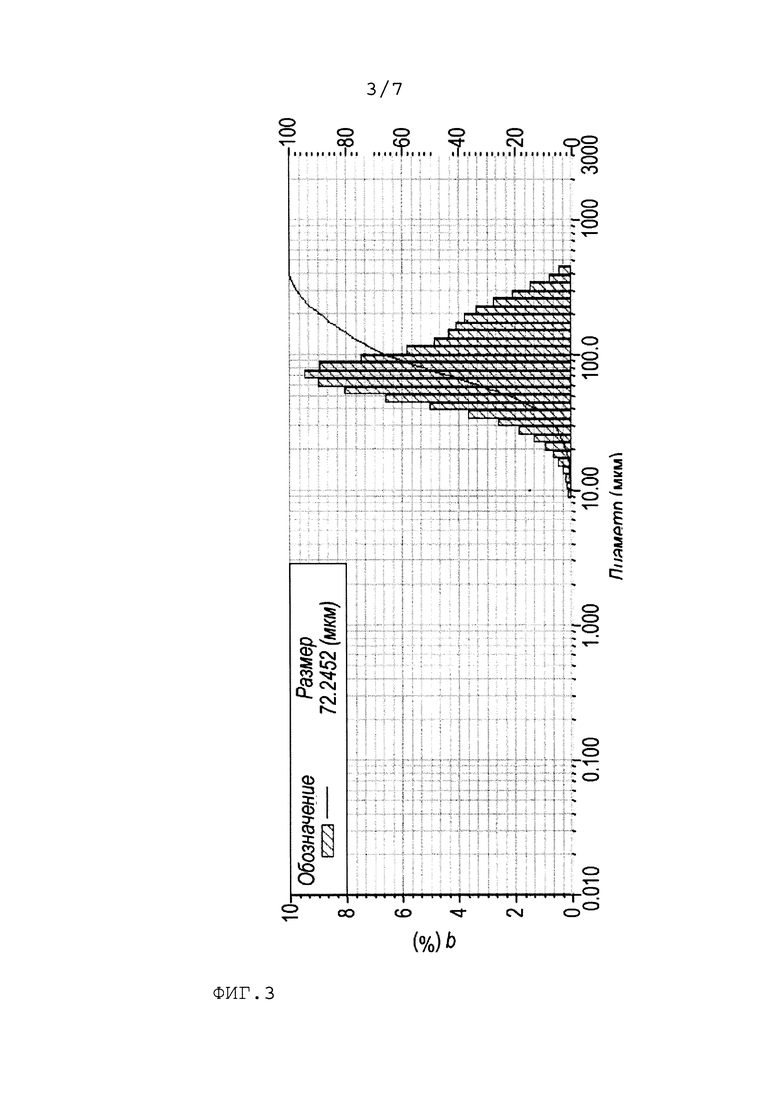
Общая схема получения цис-2,3-эпоксисукцината кальция представлена следующим образом:
Стадия а) получение кислого малеата кальция.
Кислый малеат кальция получают путем взаимодействия малеинового ангидрида или малеиновой кислоты с соединением кальция. В качестве соединения кальция используют карбонат кальция. Соотношение малеиновой кислоты или малеинового ангидрида к соединению кальция меньше эквимолярного (т.е. менее 1:1) и составляет предпочтительно 1:0,5.
Предпочтительно используют исходные вещества со степенью чистоты не менее 90%, предпочтительно не менее 95%, наиболее предпочтительно не менее 98% и выше. Предпочтительно реакцию проводят в водной среде, предварительно растворив малеиновую кислоту или малеиновый ангидрид в воде и добавляя в полученному раствору или суспензии соединения кальция в сухом виде.
Реакцию можно проводить при перемешивании для упрощения отведения диоксида углерода и ускорения растворения соединения кальция. Реакцию предпочтительно проводят в течение от 10 мин до 10 ч, предпочтительно от 20 мин до 3 ч, наиболее предпочтительно от 30 мин до 2 ч.
Температура реакции составляет от 0 до 100°С, предпочтительно от 15 до 70°С, наиболее предпочтительно от 20 до 60°С.
По окончании стадии получения кислого малеата кальция реакционная масса содержит, по существу, раствор кислого малеата кальция в воде или суспензию кислого малеата кальция в водном растворе.
Стадия b) получение кислого цис-2,3-эпоксисукцината кальция.
Кислый цис-2,3-эпоксисукцинат кальция получают взаимодействием в водном растворе кислого малеата кальция, полученного на стадии а) с пероксидом водорода в присутствии катализатора эпоксидирования. Массовая концентрация раствора или массовая доля суспензии кислого малеата кальция в воде может составлять от 5 до 50%, предпочтительно от 10 до 30%, наиболее предпочтительно от 15 до 25%. Массовая концентрация катализатора может составлять от 0,001 до 5%, предпочтительно от 0,1 до 0,5%.
Пероксид водорода вводят в реакционную смесь в виде раствора в воде. Концентрация раствора может составлять любую удобную концентрацию от 5 до 100%. Наиболее предпочтительно использовать товарные формы пероксида водорода с концентрацией от 20 до 40%. Пероксид водорода можно добавлять единовременно или по частям. Наиболее предпочтительно, с целью контроля температуры реакционной среды, добавлять пероксид водорода постепенно в течение от 5 мин до 3 ч.
В качестве катализатора используют любой известный из уровня техники водорастворимый катализатор эпоксидирования. В частности, используют вольфрамовую и/или молибденовую кислоты, гетерополикислоты вольфрама и молибдена, соли вольфрамовой и молибденовой кислот, например: вольфрамат натрия, вольфрамат калия, молибдат натрия, фосфорвольфрамовую кислоту, фосфомолибденовую кислоту и кремнийвольфрамовую кислоту. Предпочтительно используют вольфрамат натрия и вольфрамат калия.
Реакцию проводят в течение от 0,5 до 24 ч, предпочтительно от 1 до 6 ч, наиболее предпочтительно от 2 до 4 ч.
Температура реакции может составлять от 40 до 100°С, предпочтительно от 50 до 80°С, наиболее предпочтительно от 55 до 65°С. При более высокой температуре возможен гидролиз эпоксисукцината в D-тартрат, тогда как при более низкой температуре реакция может идти слишком медленно.
Стадия с) нейтрализация кислого цис-2,3-эпоксисукцината кальция.
По окончании реакции эпоксидирования проводят нейтрализацию кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция, описанных ранее на стадии а), в количестве 0,4-0,6 экв. для нейтрализации кислых солей и доведения значения pH до 5-8.
Стадия d) выдерживание цис-2,3-эпоксисукцината кальция.
Выдерживание можно проводить при температуре от 10 до 60°С, предпочтительно от 20 до 40°С.
Для выделения цис-2,3-эпоксисукцината кальция суспензию выдерживают в течение от 24 до 240 ч, предпочтительно от 32 до 120 ч, наиболее предпочтительно от 48 до 96 ч.
Стадия е) выделение кристаллической формы цис-2,3-эпоксисукцината кальция.
По окончании выдерживания визуально заметно образование двух кристаллических форм цис-2,3-эпоксисукцината кальция: мелких кристаллов, соответствующих по своей структуре кристаллам цис-2,3-эпоксисукцината кальция, известным из уровня техники, и крупных кристаллов, представляющих собой цис-2,3-эпоксисукцинат кальция по изобретению. Разделение кристаллических форм осуществляют, например, путем просеивания.
После разделения крупные кристаллы цис-2,3-эпоксисукцината кальция по изобретению размалываются. Размолотые кристаллы могут быть использованы как в качестве исходного компонента в органическом синтезе, так и в качестве затравки в процессе получения новых порций цис-2,3-эпоксисукцината кальция, как будет описано ниже.
В другом варианте осуществления изобретения, раскрывается способ получения кристаллической формы цис-2,3-эпоксисукцината кальция согласно настоящему изобретению, включающий следующие стадии:
a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция;
b) эпоксидирование кислого малеата пероксидом водорода в присутствии катализаторов с получением кислого цис-2,3-эпоксисукцината кальция;
c) добавление от 0,1 до 20 мас.% затравки;
d) нейтрализация кислых солей с одновременной кристаллизацией цис-2,3-эпоксисукцината кальция;
e) выделение кристаллической формы цис-2,3-эпоксисукцината кальция;
отличающийся тем, что в качестве затравки используют кристаллическую форму цис-2,3-эпоксисукцината кальция по настоящему изобретению.
Стадии а) получения малеата кальция и b) кислого цис-2,3-эпоксисукцината кальция идентичны стадиям а) и b) описанного выше способа.
Стадия с) добавление затравки.
Затравку, представляющую собой кристаллическую форму цис-2,3-эпоксисукцината кальция, добавляют в количестве от 0,1 до 20 мас.%, предпочтительно от 0,2 до 5 мас.%, наиболее предпочтительно от 0,5 до 2% от массы выпадающего цис-2,3-эпоксисукцината кальция.
При введении затравки в кристаллизатор, предпочтительно проводить интенсивное перемешивание для обеспечения образования достаточного количества зародышей новой кристаллической фазы. При использовании кристаллизатора с перемешивающим устройством, необходимо обеспечить достаточную скорость вращения, предпочтительно от 100 до 500 об/мин.
Стадия d) нейтрализация кислых солей.
Поcле введения в реакционную массу затравки проводят нейтрализацию кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция в количестве 0,4-0,6 экв. для нейтрализации кислых солей и доведения значения pH до 5-8. Наблюдается выпадение в осадок кристаллического цис-2,3-эпоксисукцината кальция.
Кристаллическая форма цис-2,3-эпоксисукцината кальция может образовываться в виде кристаллов различного размера. Однако, предпочтительно, кристаллическая форма образуется в виде мелких кристаллов со средним размером частиц от 65 до 115 мкм, предпочтительно 100 мкм, где кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22 °С составляют a=15.1916(2) Å, b=8.9121(1) Å, c=7.4724(1) Å, beta=103.309(1)°, которые удобно использовать в биотехнологической стадии раскрытия эпоксидного цикла.
В другом варианте осуществления изобретения раскрывается однородная водная суспензия кристаллической формы цис-2,3-эпоксисукцината кальция, где кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22°С составляют a=15.1916(2) Å, b=8.9121(1) Å, c=7.4724(1) Å, beta=103.309(1)°.
В другом варианте осуществления изобретения раскрывается применение кристаллической формы цис-2,3-эпоксисукцината кальция согласно настоящему изобретению в качестве затравки в способе получения новых порций кристаллической формы цис-2,3-эпоксисукцината кальция.
Кристаллическая форма цис-2,3-эпоксисукцината кальция согласно настоящему изобретению может быть использована для получения винной кислоты путем ферментативного гидролиза указанной кристаллической формы цис-2,3-эпоксисукцината кальция.
Также, однородная суспензия кристаллической формы цис-2,3-эпоксисукцината кальция может быть использована как подходящее сырье для получения (со)полимерных карбоксилатов, например, 2,3-полиэпоксисукцината натрия (PESA), используемых в синтетических моющих средствах, а также в качестве ингибиторов солеотложения в технике.
Осуществление изобретения
Методы исследования цис-2,3-эпоксисукцината кальция.
Элементный анализ проводили при помощи анализатора элементного состава «Elementar Vario MACRO CHNS».
Порошковая рентгенография
Первичный рентгенофазовый анализ образцов проводился на рентгеновском дифрактометре XRD-7000S (Shimadzu, Япония) на медном излучении CuKα (λ=1,5418 Ǻ) с вторичным графитовым монохроматором в геометрии Брегга-Брентано в режиме «на отражение» с использованием сцинтилляционного детектора с шагом сканирования 0,02° в диапазоне углов от 10 до 80° и временем экспозиции в каждой точке 0,6 с.
В дальнейшем, регистрация прецизионной дифрактограммы порошка цис-2,3-эпоксисукцината кальция была выполнена на дифрактометре STOE STADI-P (управляющее ПО WinXPow), излучение Co (кобальт) Kα1, с первичным Ge (111) монохроматором, изогнутым по Иогансону, в геометрии Брегга-Брентано в режиме «на просвет» (симметричное сканирование ω - 2θ) с использованием линейного газонаполненного позиционно-чувствительного детектора. Для регистрации дифрактограммы образец перетирали в агатовой ступке и наносили на рентгенаморфную PET-пленку, заранее смазанную тонким слоем вакуумной смазки. Толщину слоя образца подбирали эмпирически по интенсивности сигнала и соотношению «сигнал/фон». Сверху образец закрывали аналогичной майларовой пленкой и помещали в кольцевой держатель.
Инфракрасная спектроскопия (ИК)
ИК спектры регистрировали на ИК-Фурье спектрометре Varian Excalibur HE 3600 (Австралия) на приставке НПВО с кристаллом ZnSe/алмаз в области частот 400-4000 см-1.
Распределение размеров кристаллов
Определение размеров частиц проводили методом лазерной гранулометрии с помощью прибора Horiba LA-950V2 при суспендировании порошка эпоксисукцината кальция в деионизованной воде. Для расчетов использовали показатели преломления тартрата кальция (1,535) и деионизованной воды (1,333).
Пример 1 (сравнительный по прототипу). Получение кристаллов цис-эпоксисукцината кальция.
98 г малеинового ангидрида растворяли в 400 г воды в трехгорлой колбе. Затем добавляли 50 г карбоната кальция (0,5 моль карбоната кальция на 1 моль малеинового ангидрида). Перемешивали верхнеприводной мешалкой со скоростью 400 об/мин. После того как весь карбонат кальция прореагировал, добавляли 6,6 г вольфрамата натрия. Нагревали реакционную массу до 60°С и дозировали 102 г 35 мас.% раствора перекиси водорода через капельную воронку в течение 1 часа. По окончании дозирования всей перекиси водорода перемешивание продолжали в течение 7 часов. После этого охлаждали реакционную массу до 30°С и постепенно добавляли 49 г карбоната кальция. После реакционную массу охлаждали до 15-20°С. Полученные кристаллы цис-2,3-эпоксисукцината кальция образовывали плотную массу, не отделяющуюся от водной фазы. К реакционной массе добавляли 500 мл промывочной воды, при добавлении промывочной воды, кристаллы равномерно распределялись по объему водной фазы. Затем отделяли кристаллическую фазу от водной при помощи вакуум-фильтрования на фильтре с размером пор 3-5 мкм с разрежением 100 мбар. Фильтрование проводили в течение не менее 60 минут из-за медленного разделения жидкой и твердой фаз. Получившийся осадок представлял собой мелкодисперсные (размер около 10-100 мкм) кристаллы. Кристаллы сушили в течение 72 ч на воздухе, масса составила 88 г (выход 36%). Рентгенограмма полученных кристаллов представлена на Фиг.1.
Пример 2. Получение кристаллической формы цис-2,3-эпоксисукцината кальция.
98 г малеинового ангидрида растворяли в 400 г воды в трехгорлой колбе. Затем добавляли 50 г карбоната кальция (0,5 моль карбоната кальция на 1 моль малеинового ангидрида). После того как весь карбонат кальция прореагировал, добавляли 6,6 г вольфрамата натрия. Нагревали реакционную массу до 60°С и дозировали 102 г 35 мас.% раствора перекиси водорода через капельную воронку в течение 1 часа. По окончании дозирования всей перекиси водорода реакцию проводили в течение 7 часов. После этого охлаждали реакционную массу до 30°С и постепенно добавляли 49 г карбоната кальция. После реакционную массу охлаждали до 15-20°С. Полученную суспензию цис-2,3-эпоксисукцината кальций выдерживали в течение 72 ч. При этом происходило образование крупных сростков кристаллов (до 150 мкм) в смеси с первоначальными мелкодисперсными кристаллами. При этом водная фаза хорошо отделялась. Смесь фильтровали и сушили на воздухе. Смесь кристаллов просеивали и отделяли крупные кристаллы (более 50 мкм). Получали 100 г крупных кристаллов и 30 г мелких кристаллов. Крупные кристаллы размалывали в агатовой ступке и анализировали. Порошковая рентгенограмма полученной кристаллической формы представлена на Фиг.2.
Пример 3. Получение кристаллической формы цис-2,3-эпоксисукцината кальция (кристаллизация при 50°С с применением затравки).
98 г малеинового ангидрида растворяли в 400 г воды в трехгорлой колбе. Затем добавляли 50 г карбоната кальция (0,5 моль карбоната кальция на 1 моль малеинового ангидрида). После того как весь карбонат кальция прореагировал, добавляли 6,6 г вольфрамата натрия. Нагревали реакционную массу до 60°С и дозировали 102 г 35 мас.% раствора перекиси водорода через капельную воронку в течение 1 часа. По окончании дозирования перекиси водорода реакцию проводили в течение 7 часов. После этого охлаждали реакционную массу до 50°С и постепенно добавляли 4,9 г карбоната кальция и затравку в количестве молотого 2,5 г цис-2,3-эпоксисукцината кальция, полученного по Примеру 2. Затем постепенно добавляли оставшиеся 44,1 г карбоната кальция и перемешивали реакционную массу до окончания выделения газа. После реакционную массу охлаждали до 15-20°С, при этом происходило разделение реакционной массы на водную фазу и осадок. После этого отделяли кристаллическую фазу от водной при помощи вакуум-фильтрования на фильтре с размером пор 3-5 мкм с разрежением в приемнике 100 мбар. Промывали 500 мл воды. Фильтрование занимало в среднем 5 минут. Продукт представлял собой мелкодисперсные кристаллы (распределение по размерам представлено на Фиг.3). Масса после сушки составила 218,2 г (выход 90%).
Пример 4. Получение кристаллической формы цис-2,3-эпоксисукцината кальция (кристаллизация при 30°С с применением затравки).
98 г малеинового ангидрида растворяли в 400 г воды в трехгорлой колбе. Затем добавляли 50 г карбоната кальция (0,5 моль карбоната кальция на 1 моль малеинового ангидрида). После того как весь карбонат кальция прореагировал, добавляли 6,6 г вольфрамата натрия. Нагревали реакционную массу до 60°С и дозировали 102 г 35 мас.% раствора перекиси водорода через капельную воронку в течение 1 часа. По окончании дозирования всей перекиси водорода реакцию проводили в течение 7 часов. После этого охлаждали реакционную массу до 30°С и постепенно добавляли 4,9 г карбоната кальция и затравку в количестве 2,5 г цис-2,3-эпоксисукцината кальция по Примеру 2. Затем постепенно добавляли оставшиеся 44,1 г карбоната кальция и перемешивали реакционную массу до окончания выделения газа. После реакционную массу охлаждали до 15-20°С, при этом реакционная масса разделилась на водную фазу и осадок. После этого отделяли кристаллическую фазу от водной при помощи вакуум-фильтрования на фильтре с размером пор 3-5 мкм с разрежением в приемнике 100 мбар. Промывали 500 мл воды. Фильтрование занимало в среднем 5 минут. Продукт представлял собой мелкодисперсные кристаллы (распределение по размерам представлено на Фиг.4). Масса после осушки составила 225,5 г (93%).
Результаты рентгенофазового анализа кристаллов цис-2,3-эпоксисукцината кальция, полученных по Примеру 1 (сравнительный по прототипу) и Примеру 2, приведенные на Фиг.1 и Фиг.2, соответственно, указывают на разную кристаллическую форму, а именно, разное кристаллическое строение цис-2,3-эпоксисукцината кальция, получаемого в данных примерах. Таким образом, кристаллы цис-2,3-эпоксисукцината кальция по Примеру 2, полученные длительной выдержкой реакционной массы, характеризуются новой модификацией кристаллического цис-2,3-эпоксисукцината кальция пентагидрата.
Данные Примера 1 и Примера 4 по времени фильтрования показывают, что скорость фильтрования кристаллов цис-2,3-эпоксисукцината кальция при применении новой кристаллической модификации в качестве затравки, гораздо выше (Пример 4, время фильтрования 5 мин), чем в случае ее отсутствия (Пример 1, время фильтрования 60 мин).
По данным, представленным на Фиг.3 и Фиг. 4, можно сделать вывод, что температура кристаллизации не влияет на размер кристаллов суспензии соли, который составляет в среднем около 10 мкм.
ИК-спектры продуктов, полученных по Примеру 3 и Примеру 4, приведены на Фиг.5. Высокое сходство спектров указывает, что состав продукта, при разных температурах осаждения, также остается постоянным.
Таким образом, температура кристаллизации не оказывает влияния на процесс образования новой кристаллической модификации цис-2,3-эпоксисукцината кальция.
По данным анализа рентгенограммы продукта по Примеру 2 и повторных рентгенограмм (порошка по Примеру 4) проводили Рентгенофазовый и рентгеноструктурный анализ.
Качественный рентгенофазовый анализ проводили с использованием БД ICDD PDF-2 (2003 г.в.) и поисковой системы Crystallographica Search-Match 3.0. Полученные результаты не позволили провести отнесение рефлексов к известным кристаллическим фазам. Профильный анализ рефлексов выполняли в ПО WinXPow. Для моделирования рефлексов использовали функцию pseudo-Voigt с постоянным по всему угловому диапазону значением параметра η. Профильный анализ проводили в диапазоне 8-52 °2θ. Для моделирования зависимости полуширин рефлексов от угла использовали функцию Кальотти с варьируемым параметром W (V и U принимали равными нулю в связи с работой в узком угловом диапазоне). Индицирование дифрактограммы проводили в ПО STOE WinXPow с использованием ПО DICVOL. Полученные параметры были дополнительно уточнены с учетом возможного сдвига нуля (линейное приближение). Полученные в результате индицирования параметры приведены в Таблице 1.
На основании полученных при индицировании данных дифрактограмма была уточнена методом ЛеБеля в ПО Jana2006. Исходное уточнение проводили для пространственной группы P2/m (максимальной симморфной для моноклинной сингонии), затем - для группы P21/n.
Таблица 1. Параметры элементарной ячейки и критерий качества индицирования
Решение кристаллической структуры осуществляли методами прямого пространства в ПО FOX (Free Object for Xtallograhy). Решение проводили методом Монте-Карло в приближении постоянной заселенности позиций и штрафа за сближение атомов на расстояние менее 0.8 Å. В результате решения и уточнения структуры получили результаты, приведенные в Таблице 2. Уточнение кристаллической структуры позволило достичь высококачественного соответствия между теоретической и экспериментальной дифрактограммами. Результаты полнопрофильного уточнения приведены на Фиг.6.
Координационные полиэдры кальция представлены на Фиг.7. Ионы кальция имеют восемь лигандов каждый, с двумя мостиковыми молекулами воды. Каждый эпоксисукцинат-анион является тридентатным лигандом и соединяется только с одним ионом кальция. Следует отметить наличие в кристаллической структуре не связанной с кальцием молекулы H2O.
Таблица 2 - Результаты рентгеноструктурного анализа
По данным элементного анализа, кристаллы второй кристаллической модификации содержат С - 17,69% Н - 4,62%. По формуле пентагидрата цис-эпоксисукцината кальция C4H12O10Ca теоретически С 18,46%, H - 4,62%.
Пример 5. Получение поликарбоксилата - полимера цис-эпокси-1,4-бутандиовой кислоты (PESA).
Кристаллы цис-2,3- эпокисукцината кальция из примера 4 (200 г, 0,826 моль), суспендированные в 500 мл H2O, смешивали с водным раствором сульфата натрия (118 г, 0,831 моль, в 500 мл H2O) при 30°С. Смесь интенсивно перемешивали в течение 2 ч. Затем отфильтровывали осадок сульфата кальция дигидрата, осадок промывали 100 мл воды. Фильтрат представлял собой водный раствор динатрия цис-2,3-эпоксисукцината. К фильтрату добавляли 25 г (0,103 моль) кальция цис-2,3-эпокисукцината, полученного по Примеру 4, и 16,5 г (0,206 моль) 50% раствора NaOH. Смесь нагревали при перемешивании до 100°С и выдерживали 2 ч с обратным холодильником. Затем раствор подкисляли до pH 2 соляной кислотой и отфильтровывали осадок полимера. Полимер суспендировали в 200 мл H2O и подщелачивали 50% раствором NaOH до значения pH равного 6. Молекулярная масса поликарбоксилата Mw по данным ГПХ составляла 1083 г/моль.
Пример 6. Получение L-тартрата кальция и L-(+)-винной кислоты.
Штамм Acinetobacter tartarogenes KB-111 вносили в ферментер емкостью 2 л, содержащий 500 мл жидкой культуральной среды, состоящей из цис-2,3-эпоксисукцината кальция (0,6%), сульфата аммония (0,5%), дикалийфосфата (0,1%), сульфата магния (0,05%), с pH 7,0. Вышеуказанную смесь подвергали культивированию с возвратно-поступательным движением при 30°C в течение 24 часов и получали 500 мл культурального бульона. Этот культуральный бульон переносили в резервуар емкостью 50 л, который содержал 30 л жидкой культуральной среды, состоящей из пептона (0,1%), сульфата аммония (0,5%), дикалийфосфата (0,1%), сульфата магния (0,05%), сульфата железа (0,001%), глюкозы (0,2%), с pH 7.0; в то же время добавляли в резервуар 180 г цис-2,3-эпоксисукцината кальция по Примеру 4. Полученный продукт культивировали при 30°C с перемешиванием и аэрацией, при этом добавляли в культуру 20 г Terravis K1-2 (торговая марка Sasol) в качестве пеногасителя. По истечении 24 часов после начала культивирования добавляют 6 кг цис-2,3-эпоксисукцината кальция пентагидрата с последующей культивацией в течение 24 часов. Примерно 30 л полученного культурального бульона фильтровали с помощью фильтровального пресса и полученный осадок промывали водой. Продукт суспендировали в 10 л воды, центрифугировали, осадок вновь отделяли и промывали водой. Получали кристаллы L-тартрата кальция тетрагидрата массой 5,8 кг, выход 94%.
L-(+)-винную кислоту получали гидролизом L-тартрата кальция тетрагидрата путем пропускания полученных кристаллов L-тартрата кальция тетрагидрата через ионообменную колонку.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ ЦИС-2,3-ЭПОКСИСУКЦИНАТА КАЛЬЦИЯ | 2020 |
|
RU2763329C2 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ ЦИС-2,3-ЭПОКСИСУКЦИНАТА КАЛЬЦИЯ | 2020 |
|
RU2741870C1 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ ЦИС-2,3-ЭПОКСИСУКЦИНАТА КАЛЬЦИЯ | 2020 |
|
RU2756956C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕЛКОКРИСТАЛЛИЧЕСКОЙ ФОРМЫ ЦИС-2,3-ЭПОКСИСУКЦИНАТА КАЛЬЦИЯ | 2020 |
|
RU2762326C2 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ ЦИС-2,3-ЭПОКСИСУКЦИНАТА КАЛЬЦИЯ | 2020 |
|
RU2757039C2 |
| ПОЛУЧЕНИЕ КИСЛЫХ ПРОПИОНАТОВ | 2006 |
|
RU2454396C9 |
| СПОСОБ СТЕРЕОИЗБИРАТЕЛЬНОГО ПОЛУЧЕНИЯ Z-1,2-ДИАРИЛАЛЛИЛХЛОРИДОВ | 1990 |
|
RU2014317C1 |
| МНОГОКОМПОНЕНТНАЯ КРИСТАЛЛИЧЕСКАЯ СИСТЕМА, СОДЕРЖАЩАЯ НИЛОТИНИБ И ВЫБРАННЫЕ СОКРИСТАЛЛООБРАЗОВАТЕЛИ | 2013 |
|
RU2652121C2 |
| СПОСОБ ПОЛУЧЕНИЯ УЛИПРИСТАЛА АЦЕТАТА И ЕГО ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ | 2012 |
|
RU2624007C2 |
| КРИСТАЛЛЫ ГИДРОБРОМАТА ПРАСУГРЕЛЯ | 2010 |
|
RU2484094C1 |


Изобретение относится к области производства карбоксилатов металлов, в частности к кристаллической форме цис-2,3-эпоксисукцината кальция, характеризующейся тем, что кристаллы принадлежат к пространственной группе P21/n, где параметры кристаллической решетки при 22°С составляют a=15.1916(2)  , b=8.9121(1) , c=7.4724(1) , beta=103.309(1)°. Способ получения кристаллической формы цис-2,3-эпоксисукцината кальция осуществляют путем следующих стадий: a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция; b) эпоксидирование кислого малеата кальция пероксидом водорода в присутствии катализаторов эпоксидирования с получением кислого цис-2,3-эпоксисукцината кальция; c) нейтрализация кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция; d) выдерживание цис-2,3-эпоксисукцината кальция в течение от 24 до 240 ч с получением кристаллической формы цис-2,3-эпоксисукцината кальция и е) выделение кристаллов размером более 50 мкм, представляющих собой кристаллическую форму цис-2,3-эпоксисукцината кальция по изобретению. Изобретение также относится к способу получения L-(+)-винной кислоты, включающему применение в качестве затравки кристаллической формы цис-2,3-эпоксисукцината кальция, где кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22°С составляют a=15.1916(2) , b=8.9121(1) , c=7.4724(1) , beta=103.309(1)°, с последующей ферментацией полученной кристаллической формы цис-2,3-эпоксисукцината кальция с получением L-тартрата кальция и гидролиза L-тартрата кальция с получением L-винной кислоты. Технический результат – кристаллическая форма цис-2,3-эпоксисукцината кальция, позволяющая эффективно проводить процесс ферментативного получения L-(+)-винной кислоты. 3 н. и 26 з.п. ф-лы, 3 табл., 7 ил., 6 пр.
, b=8.9121(1) , c=7.4724(1) , beta=103.309(1)°. Способ получения кристаллической формы цис-2,3-эпоксисукцината кальция осуществляют путем следующих стадий: a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция; b) эпоксидирование кислого малеата кальция пероксидом водорода в присутствии катализаторов эпоксидирования с получением кислого цис-2,3-эпоксисукцината кальция; c) нейтрализация кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция; d) выдерживание цис-2,3-эпоксисукцината кальция в течение от 24 до 240 ч с получением кристаллической формы цис-2,3-эпоксисукцината кальция и е) выделение кристаллов размером более 50 мкм, представляющих собой кристаллическую форму цис-2,3-эпоксисукцината кальция по изобретению. Изобретение также относится к способу получения L-(+)-винной кислоты, включающему применение в качестве затравки кристаллической формы цис-2,3-эпоксисукцината кальция, где кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22°С составляют a=15.1916(2) , b=8.9121(1) , c=7.4724(1) , beta=103.309(1)°, с последующей ферментацией полученной кристаллической формы цис-2,3-эпоксисукцината кальция с получением L-тартрата кальция и гидролиза L-тартрата кальция с получением L-винной кислоты. Технический результат – кристаллическая форма цис-2,3-эпоксисукцината кальция, позволяющая эффективно проводить процесс ферментативного получения L-(+)-винной кислоты. 3 н. и 26 з.п. ф-лы, 3 табл., 7 ил., 6 пр.
1. Кристаллическая форма цис-2,3-эпоксисукцината кальция, отличающаяся тем, что кристаллы принадлежат к пространственной группе P21/n, где параметры кристаллической решетки при 22°С составляют a=15.1916(2)  , b=8.9121(1) , c=7.4724(1) , beta=103.309(1)°.
, b=8.9121(1) , c=7.4724(1) , beta=103.309(1)°.
2. Кристаллическая форма по п.1, где размер кристаллов составляет от 60 до 115 мкм.
3. Кристаллическая форма по п.2, где размер кристаллов составляет 100 мкм.
4. Способ получения кристаллической формы цис-2,3-эпоксисукцината кальция по п.1, включающий следующие стадии:
a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция;
b) эпоксидирование кислого малеата кальция пероксидом водорода в присутствии катализаторов эпоксидирования с получением кислого цис-2,3-эпоксисукцината кальция;
c) нейтрализация кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция;
d) выдерживание цис-2,3-эпоксисукцината кальция в течение от 24 до 240 ч с получением кристаллической формы цис-2,3-эпоксисукцината кальция;
е) выделение кристаллов размером более 50 мкм, представляющих собой кристаллическую форму цис-2,3-эпоксисукцината кальция по п.1.
5. Способ по п.4, где на стадии а) соотношение малеиновой кислоты или малеинового ангидрида к соединению кальция составляет 1:0,5.
6. Способ по п.4, где на стадии а) реакцию проводят в течение от 10 мин до 10 ч.
7. Способ по п.6, где на стадии а) реакцию проводят в течение от 20 мин до 3 ч.
8. Способ по п.7, где на стадии а) реакцию проводят в течение от 30 мин до 2 ч.
9. Способ по п.4, где на стадии а) температура реакции составляет от 0 до 100°С.
10. Способ по п.9, где на стадии а) температура реакции составляет от 15 до 70°С.
11. Способ по п.10, где на стадии а) температура реакции составляет от 20 до 60°С.
12. Способ по п.4, где пероксид водорода вводят в реакционную смесь в виде раствора в воде.
13. Способ по п.12, где используют раствор пероксида водорода в воде с концентрацией от 5 до 100%.
14. Способ по п.13, где используют раствор пероксида водорода в воде с концентрацией от 20 до 40%.
15. Способ по п.4, где пероксид водорода вводят единовременно.
16. Способ по п.4, где пероксид водород вводят по частям в течение от 5 мин до 3 ч.
17. Способ по п.4, где катализатор эпоксидирования представляет собой вольфрамовую и/или молибденовую кислоты, гетерополикислоты вольфрама и молибдена, соли вольфрамовой и молибденовой кислот.
18. Способ по п.17, где катализатор эпоксидирования представляет собой вольфрамат натрия, вольфрамат калия, молибдат натрия, фосфорвольфрамовую кислоту, фосфомолибденовую кислоту и кремнийвольфрамовую кислоту.
19. Способ по п.4, где на стадии b) температура реакции составляет от 40 до 100°С.
20. Способ по п.19, где на стадии b) температура реакции составляет от 50 до 80°С.
21. Способ по п.20, где на стадии b) температура реакции составляет от 55 до 65°С.
22. Способ по п.4, где на стадии b) реакцию проводят в течение от 0,5 до 24 ч.
23. Способ по п.22, где на стадии b) реакцию проводят в течение от 1 до 6 ч.
24. Способ по п.23, где на стадии b) реакцию проводят в течение от 2 до 4 ч.
25. Способ по п.4, где на стадии d) выдержку осуществляют от 32 до 120 ч.
26. Способ по п.25, где на стадии d) выдержку осуществляют от 48 до 96 ч.
27. Способ по п.4, где на стадии d) выдержку осуществляют при температуре от 10 до 60°С.
28. Способ по п.27, где на стадии d) выдержку осуществляют при температуре от 20 до 40°С.
29. Способ получения L-(+)-винной кислоты, включающий следующие стадии:
a) взаимодействие малеинового ангидрида или малеиновой кислоты с соединением кальция с получением кислого малеата кальция;
b) эпоксидирование кислого малеата кальция пероксидом водорода в присутствии катализаторов эпоксидирования с получением кислого цис-2,3-эпоксисукцината кальция;
c) нейтрализация кислого цис-2,3-эпоксисукцината кальция путем добавления соединения кальция;
d) выдерживание цис-2,3-эпоксисукцината кальция в течение от 24 до 240 ч с получением кристаллической формы цис-2,3-эпоксисукцината кальция;
e) выделение кристаллов размером более 50 мкм в качестве затравки, представляющих собой кристаллическую форму цис-2,3-эпоксисукцината кальция, где кристаллы принадлежат к пространственной группе P21/n, причем параметры кристаллической решетки при 22°С составляют a=15.1916(2) , b=8.9121(1) , c=7.4724(1) , beta=103.309(1)°;
f) использование выделенных кристаллов со стадии (е) в качестве затравки при получении новых порций кристаллической формы цис-эпоксисукцината кальция в соответствии со стадиями а)-с) на стадии (с), и осуществление стадий:
g) ферментации полученной кристаллической формы цис-2,3-эпоксисукцината кальция с получением L-тартрата кальция;
h) гидролиза L-тартрата кальция с получением L-винной кислоты.
| Двигатель внутреннего сгорания | 1988 |
|
SU1534195A1 |
| Устройство контроля качества внесения удобрений центробежным разбрасывателем | 1986 |
|
SU1423028A1 |
| US 9109084 B1, 18.08.2015 | |||
| WO 9417049 A1, 04.08.1994 | |||
| CN 101602852 B, 06.04.2011 | |||
| CN 105753818 A, 13.07.2016 | |||
| СПОСОБ ПОЛУЧЕНИЯ L-ВИННОЙ КИСЛОТЫ | 0 |
|
SU408943A1 |

