Ссылка на родственные заявки
Согласно настоящей заявке испрашивается приоритет в соответствии с предварительными заявками на выдачу патентов США с серийными №№62/453437, поданной 1 февраля 2017 года; 62/469912, поданной 10 марта 2017 года; 62/488366, поданной 21 апреля 2017 года, и 62/575248, поданной 20 октября 2017 года. Полное содержание данных заявок включено в настоящий документ посредством ссылки.
Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к гемисульфатной соли выбранного нуклеотидного соединения, которая обладает неожиданными терапевтическими свойствами для лечения хозяина, инфицированного гепатитом С, а также к фармацевтическим композициям и их лекарственным формам.
Предшествующий уровень техники настоящего изобретения
Вирус гепатита С (HCV) является содержащим однонитевую РНК вирусом и представителем рода Hepacivirus. По оценкам, 75% всех случаев заболеваний печени вызваны HCV. Инфекция HCV может привести к циррозу и раку печени, а при прогрессировании - к печеночной недостаточности, для которой может потребоваться пересадка печени. Приблизительно 71 миллион человек во всем мире живут с хроническими инфекциями HCV, и приблизительно 399000 человек ежегодно умирают от HCV, в основном от цирроза и печеночнотоклеточной карциномы.
РНК-полимераза является основной целью при разработке лекарственного средства против содержащих однонитевую РНК вирусов. РНК-зависимая РНК-полимераза - неструктурный белок NS5B HCV - является ключевым ферментом, ответственным за инициацию и катализ синтеза вирусной РНК. Существует два основных подкласса ингибиторов NS5B: нуклеозидные аналоги и ненуклеозидные ингибиторы (NNI). Нуклеозидные аналоги анаболизируются до активных трифосфатов, которые действуют как альтернативные субстраты для полимеразных и ненуклеозидных ингибиторов (NNI), связывающихся с аллостерическими областями в белке. Нуклеозидные или нуклеотидные ингибиторы имитируют природные субстраты полимеразы и действуют как терминаторы цепи. Они ингибируют инициацию транскрипции РНК и удлинение возникающей цепи РНК.
В дополнение к нацеливанию на РНК-полимеразу комбинированные терапевтические средства также могут быть нацелены на другие белки РНК-вирусов. Например, белками HCV, которые являются дополнительными целями для терапевтических подходов, являются NS3/4A (серинпротеаза) и NS5A (неструктурный белок, который является важным компонентом репликазы HCV и оказывает ряд эффектов на клеточные пути).
В декабре 2013 года был одобрен первый нуклеозидный ингибитор NS5B полимеразы софосбувир (Sovaldi®, Gilead Sciences). Sovaldi® является уридинфосфорамидатным пролекарством, которое поглощается гепатоцитами и подвергается внутриклеточной активации с образованием активного метаболита 2'-дезокси-2'-α-фтор-β-С-метилуридин-5'-трифосфата.
Sovaldi®

2'-дезокси-2'-α-фтор-β-С-метилуридин-5'-трифосфат
Sovaldi® является первым лекарственным средством, которое продемонстрировало безопасность и эффективность при лечении некоторых типов инфекции HCV без необходимости совместного введения интерферона. Sovaldi® является третьим лекарственным средством со статусом принципиально нового лекарственного средства, получившим одобрение FDA.
В 2014 году FDA США одобрило использование Harvoni® (ледиспасвир, ингибитор NS5A, и софосбувир) для лечения хронической инфекции вируса гепатита С генотипа 1. Harvoni® - первая комбинированная таблетка, одобренная для лечения хронической инфекции HCV генотипа 1. Это также первый одобренный режим, который не требует введения с интерфероном или рибавирином. Кроме того, FDA одобрило симепревир (Olysio™) в комбинации с софосбувиром (Sovaldi®) в качестве перорального лечения один раз в сутки без интерферона и рибавирина для взрослых с инфекцией HCV генотипа 1.
Также в 2014 году FDA США одобрило VIEKIRA Pak™ компании AbbVie, упаковку из нескольких таблеток, содержащую дасабувир (ненуклеозидный ингибитор NS5B полимеразы), омбитасвир (ингибитор NS5A), паритапревир (ингибитор NS3/4A) и ритонавир. VIEKIRA Pak™ можно использовать с рибавирином или без него для лечения инфицированных HCV генотипа 1 больных, в том числе больных с компенсированным циррозом печени. VIEKIRA Pak™ не требует совместной терапии с интерфероном.
В июле 2015 года FDA США одобрило Technivie™ и Daklinza™ для лечения заболеваний, вызванных HCV генотипа 4 и HCV генотипа 3, соответственно. Technivie™ (омбитасвир/паритапревир/ритонавир) был одобрен для использования в комбинации с рибавирином для лечения инфицированных HCV-4 больных без рубцов и цирроза и является первым вариантом для инфицированных HCV-4 больных, которым не требуется совместное введение с интерфероном. Daklinza™ был одобрен для применения с Sovaldi® для лечения инфекций HCV генотипа 3. Daklinza™ является первым лекарственным средством, которое продемонстрировало безопасность и эффективность при лечении заболеваний, вызванных HCV генотипа 3, без необходимости совместного введения интерферона или рибавирина.
В октябре 2015 года FDA США предупредило о том, что лечение заболеваний, вызванных HCV, с помощью Viekira Pak и Technivie может вызвать серьезное повреждение печени, в первую очередь у больных с основным прогрессирующим заболеванием печени, и потребовало добавить дополнительную информацию о безопасности на этикетке.
Другие одобренные в настоящее время терапевтические средства против HCV включают в себя интерферон альфа-2b или пегилированный интерферон альфа-2b (Pegintron®), который можно вводить с рибавирином (Rebetol®), телапревиром NS3/4A (Incivek®, Vertex и Johnson & Johnson), боцепревиром (Victrelis™, Merck), симепревиром (Olysio™, Johnson & Johnson), паритапревиром (AbbVie), омбитасвиром (AbbVie), NNI дасабувиром (ABT-333) и Zepatier™ компании Merck (однотаблеточная комбинация из двух лекарственных средств гразопревир и элбасвир).
Дополнительные ингибиторы NS5B полимеразы в настоящее время находятся в стадии разработки. Merck разрабатывает пролекарство уридинового нуклеотида MK-3682 (ранее Idenix IDX21437), и на данный момент это лекарственное средство находится на стадии комбинированных испытаний фазы II.
Патенты Соединенных Штатов Америки и международные заявки WO, в которых описываются нуклеозидные ингибиторы полимеразы для лечения заболеваний, вызванных Flaviviridae, в том числе HCV, включают в себя поданные Idenix Pharmaceuticals (патенты США №№6812219; 6914054; 7105493; 7138376; 7148206; 7157441; 7163929; 7169766; 7192936; 7365057; 7384924; 7456155; 7547704; 7582618; 7608597; 7608600; 7625875; 7635689; 7662798; 7824851; 7902202; 7932240; 7951789; 8193372; 8299038; 8343937; 8362068; 8507460; 8637475; 8674085; 8680071; 8691788, 8742101, 8951985; 9109001; 9243025; заявки на выдачу патентов США №№ US 2016/0002281; US 2013/0064794; международные заявки WO №№ WO 2015/095305; WO 2015/081133; WO 2015/061683; WO 2013/177219; WO 2013/039920; WO 2014/137930; WO 2014/052638; WO 2012/154321); Merck (патенты США №№6777395; 7105499; 7125855; 7202224; 7323449; 7339054; 7534767; 7632821; 7879815; 8071568; 8148349; 8470834; 8481712; 8541434; 8697694; 8715638, 9061041; 9156872 и международная заявка WO № WO 2013/009737); Emory University (патенты США №№6348587; 6911424; 7307065; 7495006; 7662938; 7772208; 8114994; 8168583; 8609627; заявка на выдачу патента США № US 2014/0212382 и международная заявка WO № WO 2014/1244430); Gilead Sciences/Pharmasset Inc. (патенты США №№7842672; 7973013; 8008264; 8012941; 8012942; 8318682; 8324179; 8415308; 8455451; 8563530; 8841275; 8853171; 8871785; 8877733; 8889159; 8906880; 8912321; 8957045; 8957046; 9045520; 9085573; 9090642 и 9139604) и (патенты США №№6908924; 6949522; 7094770; 7211570; 7429572; 7601820; 7638502; 7718790; 7772208; RE42015; 7919247; 7964580; 8093380; 8114997; 8173621; 8334270; 8415322; 8481713; 8492539; 8551973; 8580765; 8618076; 8629263; 8633309; 8642756; 8716262; 8716263; 8735345; 8735372; 8735569; 8759510 и 8765710); Hoffman La-Roche (патент США №6660721), Roche (патенты США №№6784166; 7608599, 7608601 и 8071567); Alios BioPharma Inc. (патенты США №№8895723; 8877731; 8871737, 8846896, 8772474; 8980865; 9012427; заявки на выдачу патентов США №№ US 2015/0105341; US 2015/0011497; US 2010/0249068; US 2012/0070411; международные заявки WO №№ WO 2015/054465; WO 2014/209979; WO 2014/100505; WO 2014/100498; WO 2013/142159; WO 2013/142157; WO 2013/096680; WO 2013/088155; WO 2010/108135), Enanta Pharmaceuticals (патенты США №№8575119; 8846638; 9085599; международные заявки WO №№ WO 2013/044030; WO 2012/125900), Biota (патенты США №№7268119; 7285658; 7713941; 8119607; 8415309; 8501699 и 8802840), Biocryst Pharmaceuticals (патенты США №№7388002; 7429571; 7514410; 7560434; 7994139; 8133870; 8163703; 8242085 и 8440813), Alia Chem, LLC (патент США №8889701 и международная заявка WO № WO 2015/053662), Inhibitex (патент США №8759318 и международная заявка WO № WO 2012/092484), Janssen Продукте (патенты США №№8399429; 8431588, 8481510, 8552021, 8933052; 900629 и 9012428), University of Georgia Foundation (патенты США №№6348587; 7307065; 7662938; 8168583; 8673926, 8816074; 8921384 и 8946244), RFS Pharma, LLC (патенты США №№8895531; 8859595; 8815829; 8609627; 7560550; заявки на выдачу патентов США №№ US 2014/0066395; US 2014/0235566; US 2010/0279969; международные заявки WO №№ WO 2010/091386 и WO 2012/158811) University College Cardiff Consultants Limited (международные заявки WO №№ WO 2014/076490, WO 2010/081082; WO 2008/062206), Achillion Pharmaceuticals, Inc. (международные заявки WO №№ WO 2014/169278 и WO 2014/169280), Cocrystal Pharma, Inc. (патент США №9173893), Katholieke Universiteit Leuven (международная заявка WO № WO 2015/158913), Catabasis (международная заявка WO № WO 2013/090420) и Regents of the University of Minnesota (международная заявка WO № WO 2006/004637).
Atea Pharmaceuticals, Inc. раскрыла β-D-2'-дезокси-2'-α-фтор-2'-β-С-замещенные-2-модифицированные-N6-(моно- и диметил)пуриновые нуклеотиды для лечение заболевания, вызванного HCV, в патенте США №9828410 и в поданной по процедуре РСТ заявке №WO2016/144918. Atea также раскрыла β-D-2'-дезокси-2'-замещенные-4'-замещенные-2-N6-замещенные-6-аминопуриновые нуклеотиды для лечения парамиксовирусных и ортомиксовирусных инфекций в заявке на выдачу патента США № US 2018/0009836 и в международной заявке WO № WO 2018/009623.
В медицине по-прежнему сохраняется острая потребность в разработке терапевтических средств против HCV, которые были бы безопасными, эффективными и хорошо переносимыми. Необходимость заостряется ожиданием устойчивости к лекарственному средству. Более мощные противовирусные препараты прямого действия могут значительно сократить продолжительность лечения и улучшить податливость и частоту SVR (устойчивый вирусный ответ) для больных, инфицированных всеми генотипами HCV.
Следовательно, цель настоящего изобретения заключается в обеспечении соединений, фармацевтических композиций, способов и лекарственных форм для лечения и/или предупреждения инфекций HCV.
Краткое раскрытие настоящего изобретения
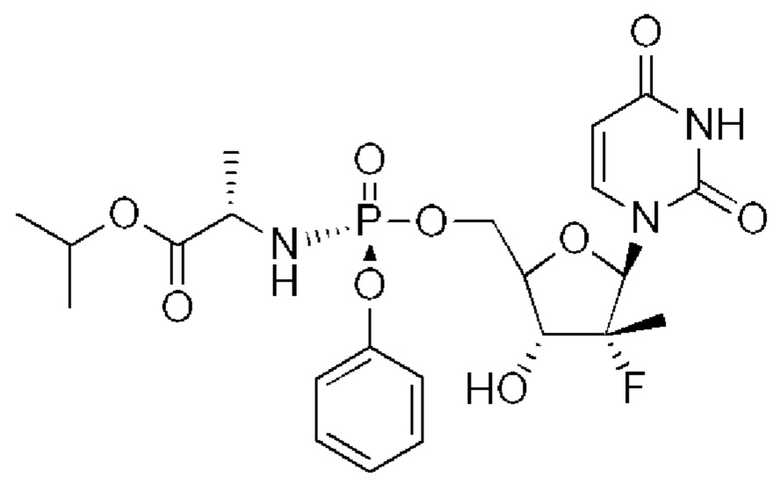
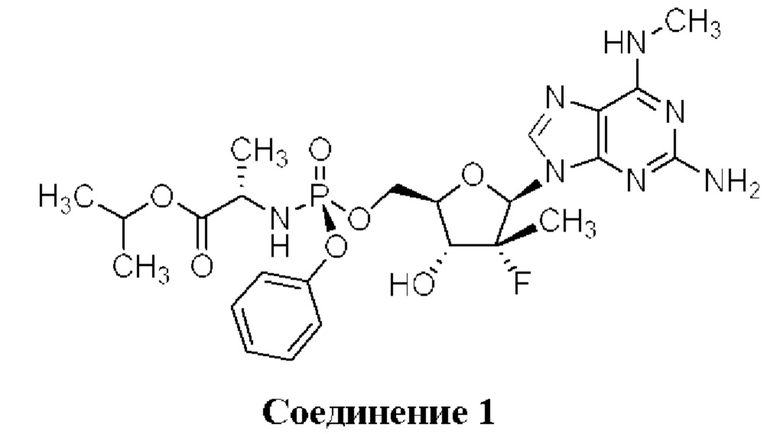
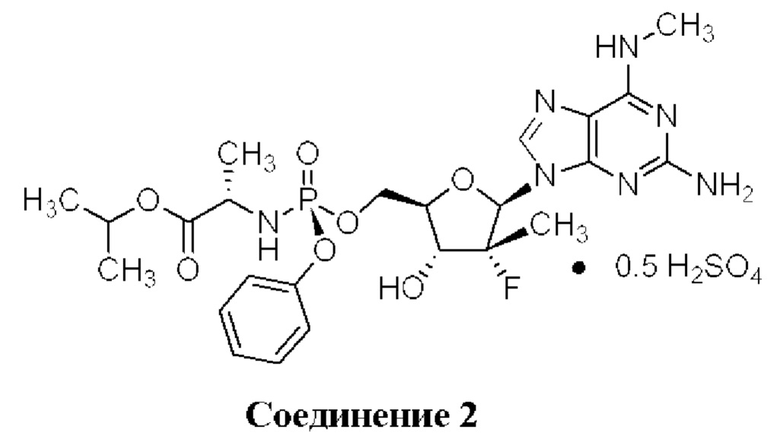
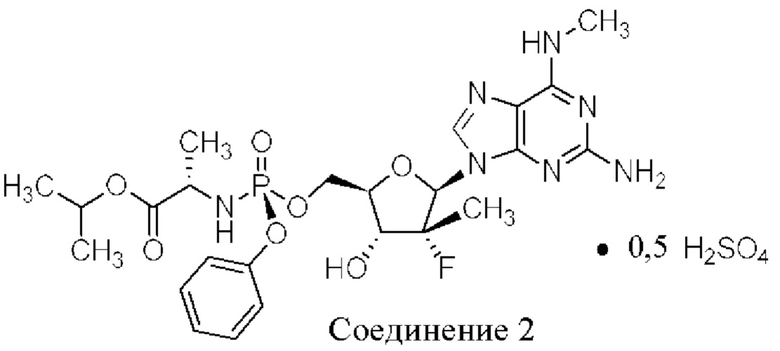
Неожиданно выяснили, что гемисульфатная соль соединения 1, которая представлена ниже как соединение 2, демонстрирует неожиданные благоприятные терапевтические свойства, в том числе повышенную биодоступность и селективность в отношении целевых органов, по сравнению с его свободным основанием (соединением 1). Эти неожиданные преимущества не могли быть предсказаны заранее. Соединение 2, таким образом, представляет собой терапевтически превосходную композицию вещества для введения в эффективном количестве хозяину, нуждающемуся в этом, как правило, человеку, для лечения гепатита С. Соединение 2 называют гемисульфатной солью изопропил-((S)-(((2R,3R,4R,5R)-5-(2-амино-6-(метиламино)-9H-пурин-9-ил)-4-фтор-3-гидрокси-4-метилтетрагидрофуран-2-ил)метокси)(фенокси)фосфорил)-L-аланината.
Соединение 1 раскрывается в патенте США №9828410.
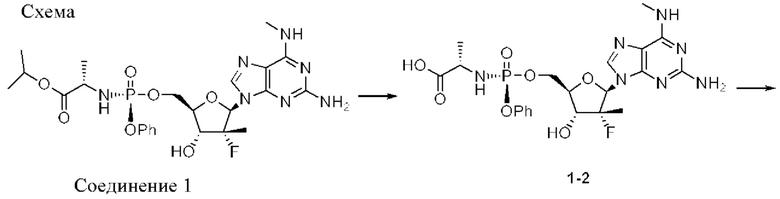
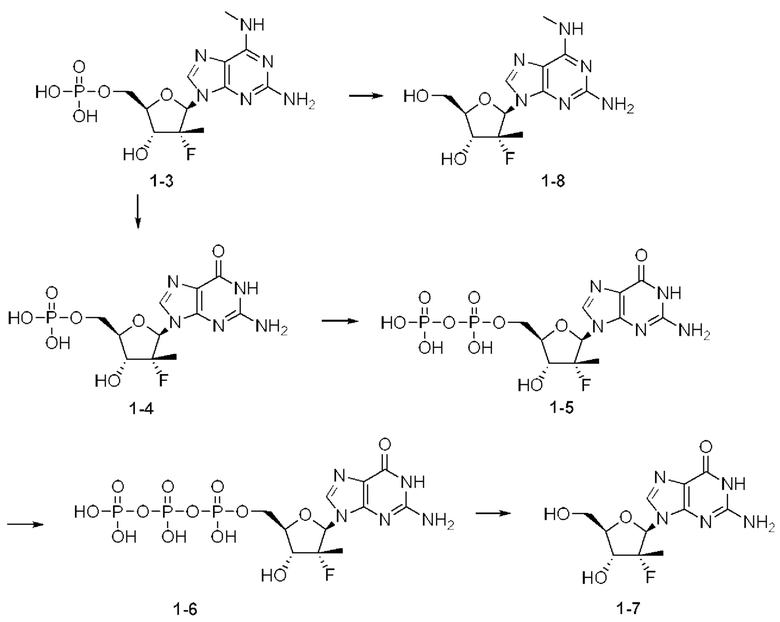
Соединение 2, как соединение 1, превращается в клетке в соответствующий ему трифосфат нуклеотида (соединение 1-6), который является активным метаболитом и ингибитором РНК-полимеразы (см. приведенную ниже схему 1). Поскольку соединение 1-6 продуцируется в клетке и не покидает клетку, оно не измеряется в плазме. Однако 5'-ОН метаболит соединение 1-7 (см. схему 1) экспортируется из клетки и, следовательно, измеряется в плазме и действует как заменитель концентрации внутриклеточного активного метаболита соединения 1-6.
Выяснили, что концентрация in vivo заменяющего соединения 1-7 в плазме, и таким образом внутриклеточного соединения 1-6 значительно выше, если вводят соединение 2 in vivo, чем если вводят соединение 1 in vivo. При прямом сравнении собак, получивших дозу соединения 1 и соединения 2 (пример 19, таблица 28), введение дозы соединения 2 достигало AUC(0-4 часа) конечного метаболита гуанинового 5'-ОН нуклеозида (1-7), которая в два раза превышала AUC после введения дозы соединения 1. Не ожидали, что нековалентная соль обладает таким эффектом на концентрацию в плазме исходного лекарственного средства (соединения 1).
Кроме того, соединение 2 селективно распределяется in vivo в печени, а не сердце (пример 19, таблица 29), что полезно, поскольку печень является больным органом у хозяев, инфицированных HCV. Собакам вводили дозу соединения 1 или соединения 2 и измеряли концентрацию активного трифосфата (1-6) в печени и сердце. Отношение концентраций активного трифосфата печень/сердце было выше после введения дозы соединения 2 по сравнению с соединением 1, как показано в таблице 29. В частности, отношение распределения печень/сердце для соединения 2 составляет 20 по сравнению с отношением распределения печень/сердце 3,1 для соединения 1. Неожиданно эти данные указывают на то, что введение соединения 2 дает в результате предпочтительное распределение активного трифосфата гуанина (соединения 1-6) в печени, превышающее таковое в сердце, по сравнении с соединением 1, что снижает потенциальные нецелевые эффекты. Неожиданным было то, что введение соединения 2 могло существенно снижать нежелательное нецелевое распределение. Это позволяет вводить соединения 2 при более высокой дозе, чем соединение 1, если это одобряет лечащий врач.
Кроме того, содержания в ткани печени и сердца активного трифосфатгуанинового производного соединения 2 (метаболита 1-6) измеряли после пероральных доз соединения 2 у крыс и обезьян (пример 20). Высокое содержание активного трифосфата гуанина (1-6) измеряли в печени всех тестируемых видов. Важно отметить, что не поддающееся количественному определению содержание трифосфата гуанина (1-6) измеряли в сердцах обезьян, и это указывает на специфическое для печени образование активного трифосфата. Таким образом, обнаружили, что по сравнению с введением дозы соединения 1 введение дозы соединения 2 улучшает распределение трифосфата гуанина (1-6).
При введении здоровым пациентам и больным, инфицированным гепатитом С, соединение 2 хорошо переносилось после однократной пероральной дозы, и фармакокинетические параметры Cmax, Tmax и AUCtot были сопоставимы в обеих группах (таблицы 34 и 35), как описано в примере 24, однократная доза соединения 2 у инфицированных HCV больных приводила в результате к значительной противовирусной активности. Содержание в плазме метаболита 1-7 было в основном пропорционально дозе в исследуемом диапазоне.
Индивидуальные фармакокинетические/фармакодинамические анализы больных, получивших дозу соединения 2, показали, что вирусологический ответ коррелировал с содержанием в плазме метаболита 1-7 соединения 2 (пример 24, фиг. 23А-23F), что указывает на то, что выраженные вирусологические ответы достижимы при повышенных дозах соединения 2.
Пример 24 подтверждает, что в качестве неограничивающих вариантов осуществления однократные пероральные дозы 300 мг, 400 мг и 600 мг приводят в результате к значительной противовирусной активности у людей. Остаточная концентрация в плазме С24 метаболита 1-7 после дозы 600 мг соединения 2 удваивалась по сравнению с остаточной концентрацией в плазме С24 метаболита 1-7 после дозы 300 мг соединения 2.
На фиг. 24 и в примере 25 показано неожиданное изобретение, заключающееся в соединении 2 для лечения гепатита С. Как показано на фиг. 24, прогнозировали равновесные остаточные содержания в плазме (C24,ss) метаболита 1-7 после введения дозы соединения 2 у людей (600 мг QD (550 мг эквивалента свободного основания) и 450 мг QD (400 мг эквивалента свободного основания)) и сравнивали с ЕС95 соединения 1 in vitro для ряда клинических изолятов HCV, чтобы определить, является ли равновесная концентрация в плазме стабильно выше, чем ЕС95, что может привести в результате к высокой эффективности против множества клинических изолятов in vivo. ЕС95 для соединения 1 является такой же, как ЕС95 соединения 2. Для того чтобы соединение 2 было эффективным, равновесное остаточное содержание в плазме метаболита 1-7 должно превышать ЕС95.
Как показано на фиг. 24, ЕС95 соединения 2 против всех тестируемых клинических изолятов, варьировала от приблизительно 18 нМ до 24 нМ.
Как показано на фиг. 24, соединение 2 при дозе 450 мг QD (400 мг эквивалента свободного основания) у людей обеспечивает прогнозируемую равновесную остаточную концентрацию в плазме (C24,ss) приблизительно 40 нг/мл. Соединение 2 при дозе 600 мг QD (550 мг эквивалента свободного основания) у людей обеспечивает прогнозируемую равновесную остаточную концентрацию в плазме (C24,ss) приблизительно 50 нг/мл.
Следовательно, прогнозируемая равновесная концентрация в плазме заменителя метаболита 1-7 почти вдвое превышает ЕС95 против всех тестируемых клинических изолятов (даже против трудно поддающегося лечению GT3a), что указывает на превосходную эффективность.
В отличие от этого, ЕС95 стандартного медицинского нуклеотида софосбувира (Sovaldi) варьирует от 50 нМ до 265 нМ для всех тестируемых клинических изолятов HCV, при этом ЕС95 меньше прогнозируемой равновесной концентрации при коммерческой дозировке 400 мг только для двух изолятов - GT2a и GT2b. ЕС95 при коммерческой дозировке 400 мг софосбувира больше, чем прогнозируемая равновесная концентрация для других клинических изолятов - GT1a, GT1b, GT3a, GT4a и GT4d.
Сравнение данных эффективности и фармакокинетических равновесных параметров на фиг. 24 четко демонстрирует неожиданное терапевтическое значение соединения 2 для лечения гепатита С. Действительно, прогнозируемое равновесное содержание в плазме (C24,ss) после введения соединения 2, как прогнозируют, по меньшей мере в 2 раза выше ЕС95 для всех тестируемых генотипов и в 3-5 раз эффективнее против GT2. Эти данные указывают на то, что соединение 2 обладает мощной активностью против вирусов всех генотипов у людей. Как показано на фиг. 24, ЕС95 софосбувира против GT1, GT3 и GT4 превышает 100 нг/мл. Таким образом, удивительно, что соединение 2 является активным против HCV в лекарственной форме, которая доставляет более низкую равновесную остаточную концентрацию (40-50 нг/мл), чем равновесная остаточная концентрация (приблизительно 100 нг/мл), достигаемая эквивалентной лекарственной формой софосбувира.
Поэтому, согласно одному варианту осуществления настоящее изобретение включает в себя лекарственную форму соединения 2, которая обеспечивает равновесную остаточную концентрацию в плазме (C24,ss) метаболита 1-7 от приблизительно 15 до 75 нг/мл, например, 20-60 нг/мл, 25-50 нг/мл, 40-60 нг/мл или даже 40-50 нг/мл. Это неожиданно в свете того факта, что равновесная концентрация эквивалентного метаболита софосбувира составляет приблизительно 100 нг/мл.
Кроме того, выяснили, что соединение 2 является необычайно стабильной, высоко растворимой негигроскопичной солью с активностью против HCV. Это удивительно, поскольку ряд солей соединения 1, отличных от гемисульфатной соли (соединения 2), в том числе моносульфатной соли (соединения 3), не являются физически стабильными, но вместо этого переходят в жидкое состояние или становятся смолистыми твердыми веществами (пример 4) и, таким образом, не подходят для стабильных твердых фармацевтических лекарственных форм. Удивительно, что, хотя соединение 2 не становится смолистыми, оно в 43 раза более растворимо в воде по сравнению с соединением 1 и более чем в 6 раз более растворимо, чем соединение 1, в условиях имитации желудочного сока (SGF) (пример 15).
Как обсуждается в примере 16, соединение 2 остается белым твердым веществом при IR, что соответствует эталонному стандарту, в течение 6 месяцев в условиях «ускоренного старения» (40°С/75% RH). Соединение 2 является стабильным в течение 9 месяцев при окружающих условиях (25°С/60% RH) и в условиях хранения в холодильнике (5°С).
Твердые лекарственные формы (таблетки по 50 мг и 100 мг) соединения 2 также являются химически стабильными в условиях «ускоренного старения» (40°С/75% RH) и в условиях хранения в холодильнике (5°С) в течение 6 месяцев (пример 26). Соединение 2 является стабильным при окружающих условиях (25°С/60% RH) в твердой лекарственной форме по меньшей мере в течение 9 месяцев.
Схема 1 обеспечивает метаболический путь соединения 1 и соединения 2, который включает в себя начальную деэстерификацию фосфорамидата (метаболита 1-1) с образованием метаболита 1-2. Затем метаболит 1-2 превращается в N6-метил-2,6-диаминопурин-5'-монофосфатное производное (метаболит 1-3), который, в свою очередь, метаболизируется до свободного 5'-гидроксил-N6-метил-2,6-диаминопуринового нуклеозида (метаболита 1-8) и ((2R,3R,4R,5R)-5-(2-амино-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-4-фтор-3-гидрокси-4-метилтетрагидрофуран-2-ил)метил-дигидрофосфата в виде 5'-монофосфата (метаболита 1-4). Метаболит 1-4 анаболизируется до соответствующего дифосфата (метаболита 1-5), а затем активного трифосфатного производного (метаболита 1-6). 5'-Трифосфат может быть далее метаболизирован с образованием 2-амино-9-((2R,3R,4R,5R)-3-фтор-4-гидрокси-5-(гидроксиметил)-3-метилтетрагидрофуран-2-ил)-1,9-дигидро-6Н-пурин-6-она (1-7). Метаболит 1-7 измеряется в плазме и, следовательно, является заменителем активного трифосфата (1-6), который не измеряется в плазме.
Согласно одному варианту осуществления настоящее изобретение относится к соединению 2 и к его применению для лечения гепатита С (HCV) у хозяина, нуждающегося в этом, необязательно в фармацевтически приемлемом носителе. Согласно одному аспекту соединение 2 используют в виде аморфного твердого вещества. Согласно другому аспекту соединение 2 используют в виде кристаллического твердого вещества.
Настоящее изобретение, кроме того, включает в себя типичный неограничивающий способ получения соединения 2, который включает
(i) первую стадию растворения соединения 1 в органическом растворителе, например, в ацетоне, этилацетате, метаноле, ацетонитриле, или эфире, или подобном, в колбе или контейнере;
(ii) загрузку второй колбы или контейнера вторым органическим растворителем, который может быть может быть таким же или отличаться от органического растворителя со стадии (i), необязательно охлаждение второго растворителя до 0-10 градусов Цельсия и добавление каплями H2SO4 во второй органический растворитель для создания смеси H2SO4/органический растворитель; и при этом растворителем может быть, например, метанол;
(iii) добавление каплями смеси H2SO4/растворитель при молярном отношении 0,5/1,0 со стадии (ii) в раствор соединения 1 со стадии (i) при окружающей или слегка повышенной или пониженной температуре (например, 23-35 градусов Цельсия);
(iv) перемешивание реакционной смеси со стадии (iii) до образования осадка соединения 2, например, при окружающей или слегка повышенной, или пониженной температуре;
(v) необязательно фильтрование полученного в результате осадка со стадии (iv) и промывания органическим растворителем; и
(vi) необязательно сушку полученного в результате соединения 2 в вакууме, необязательно при повышенной температуре, например, 55, 56, 57, 58, 59 или 60°С.
Согласно одному варианту осуществления органическим растворителем стадии (i) является 3-метил-2-пентанон. Согласно одному варианту осуществления органическим растворителем стадии (i) является этилизопропиловый кетон. Согласно одному варианту осуществления органическим растворителем стадии (i) является метилпропионат. Согласно одному варианту осуществления органическим растворителем стадии (i) является этилбутират.
Несмотря на объем литературы и заявок на выдачу патентов, касающихся противовирусных нуклеозидов, соединение 2 конкретно не было раскрыто. Следовательно, настоящее изобретение включает в себя соединение 2, или фармацевтически приемлемую композицию, или ее лекарственную форму, описываемые в настоящем документе.
Представлены соединения, способы, лекарственные формы и композиции для лечения хозяина, инфицированного вирусом HCV, путем введения эффективного количества соединения 2. Согласно некоторым вариантам осуществления соединение 2 вводят при дозе по меньшей мере приблизительно 100, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700,750, 800, 850, 900, 950 или 1000 мг. Согласно некоторым вариантам осуществления соединение 2 вводят сроком до 12 недель, сроком до 10 недель, сроком до 8 недель, сроком до 6 недель или сроком до 4 недель. Согласно альтернативным вариантам осуществления соединение 2 вводят в течение по меньшей мере 4 недель, в течение по меньшей мере 6 недель, в течение по меньшей мере 8 недель, в течение по меньшей мере 10 недель или в течение по меньшей мере 12 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки или через сутки. Согласно некоторым вариантам осуществления соединение 2 вводят в лекарственной форме, которая достигает равновесного остаточного содержания в плазме (C24,ss) метаболита 1-7 от приблизительно 15 до 75 нг/мл. Согласно одному варианту осуществления соединение 2 вводят в лекарственной форме, которая достигает равновесного остаточного содержания в плазме (C24,ss) метаболита 1-7 от приблизительно 20 до 60 нг/мл. Согласно некоторым вариантам осуществления соединение 2 вводят в лекарственной форме, которая достигает AUC метаболита 1-7 от приблизительно 1200 нг*час/мл до 3000 нг*час/мл. Согласно одному варианту осуществления соединение 2 вводят в лекарственной форме, которая достигает AUC метаболита 1-7 от приблизительно 1500 до 2100 нг*час/мл.
Соединения, композиции и лекарственные формы также могут быть использованы для лечения связанных состояний, таких как состояния с наличием антител против HCV или наличием антигенов, вирусное хроническое воспаление печени, рак печени в результате запущенного гепатита С (печеночноклеточная карцинома (НСС)), цирроз, хронический или острый гепатит С, молниеносный гепатит С, хронический персистирующий гепатит С и утомление, вызванное образованием антител против HCV. Соединение или составы, которые включают в себя соединения, также могут быть использованы профилактически для предупреждения или ограничения прогрессирования клинической болезни у индивидуумов с наличием антител против HCV или наличием антигенов, или которые подверглись воздействию вируса гепатита С.
Настоящее изобретение, таким образом, включает в себя следующие признаки:
(a) соединение 2, описываемое в настоящем документе;
(b) пролекарства соединения 2;
(c) применение соединения 2 в изготовлении медикамента для лечения инфекции вируса гепатита С;
(d) соединение 2 для применения при лечении гепатита С, необязательно в фармацевтически приемлемом носителе;
(e) способ изготовления медикамента, предназначенного для терапевтического применения для лечения инфекции вируса гепатита С, характеризующийся тем, что соединение 2 или фармацевтически приемлемую соль, описываемые в настоящем документе, используют в изготовлении;
(e) фармацевтический состав, содержащий эффективное для лечения хозяина количество соединения 2 с фармацевтически приемлемым носителем или разбавителем;
(f) способы получения терапевтических продуктов, которые содержат эффективное количество соединения 2;
(g) твердые лекарственные формы, в том числе те, которые обеспечивают благоприятный фармакокинетический профиль; и
(h) способы изготовления соединения 2, описываемого в настоящем документе.
Краткое описание графических материалов
На фиг. 1А представлено совмещение дифрактограмм XRPD образцов 1-1 (аморфного соединения 1), 1-2 (кристаллического соединения 1), и 1-3 (аморфного соединения 2) до исследований стабильности с целью характеристики, как описано в примере 2 и примере 5. По оси x показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 1В представлена хроматограмма HPLC аморфного соединения 1 (образца 1-1) для определения чистоты, как описано в примере 2. Чистота образца составляла 98,7%. По оси x показано время, измеренное в минутах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 2А представлена хроматограмма HPLC кристаллического соединения 1 (образца 1-2) для определения чистоты, как описано в примере 2. Чистота образца составляла 99,11%. По оси x показано время, измеренное в минутах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 2В представлен график DSC и TGA кристаллического соединения 1 (образца 1-2) перед любым из исследований стабильности с целью характеристики, как описано в примере 2. По оси x показана температура, измеряемая в °С, по левой оси у показан тепловой поток, измеряемый в Вт/г, а по правой оси у показана масса, измеряемая в процентах.
На фиг. 3 показано изображение рентгеновской кристаллографии соединения 1, демонстрирующее абсолютную стереохимию, описываемую в примере 2.
На фиг. 4А представлено совмещение дифрактограмм XRPD образцов 1-1 (аморфного соединения 1), 1-2 (кристаллического соединения 1) и 1-3 (аморфного соединения 2) после хранения при 25°С и 60% относительной влажности в течение 14 суток, как описано в примере 2. По оси x показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 4В представлено совмещение дифрактограмм XRPD образцов 1-4, 1-5, 1-6, 1-7 и 1-9 после хранения при 25°С и 60% относительной влажности в течение 7 суток, как описано в примере 4. По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 5А представлено совмещение дифрактограмм XRPD образцов 1-4, 1-6, 1-7 и 1-9 после хранения при 25°С и 60% относительной влажности в течение 14 суток, как описано в примере 4. По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 5В показан паттерн XRPD аморфного соединения 2 (образец 1-3), как описано в примере 5. По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 6А представлена хроматограмма HPLC аморфного соединения 2 (образца 1-3) для определения чистоты, как описано в примере 5. Чистота образца составляла 99,6%. По оси х показано время, измеренное в минутах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 6В представлен график DSC и TGA для аморфного соединения 2 (образца 1-3) перед любым из исследований стабильности с целью характеристики, как описано в примере 5. По оси х показана температура, измеряемая в °С, по левой оси у показан тепловой поток, измеряемый в Вт/г, а по правой оси у показана масса, измеряемая в процентах.
На фиг. 7А представлено совмещение дифрактограмм XRPD кристаллических образцов (образцов 2-2, 2-6 и 2-7) и слабо кристаллических образцов (образцов 2-3, 2-4, 2-5 и 2-8), идентифицированных при кристаллизациях соединения 2 (пример 6). По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 7В представлено совмещение дифрактограмм XRPD аморфных образцов (образцов 2-9, 2-10 и 2-11), идентифицированных при кристаллизациях соединения 2 (пример 6). По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 8А представлено совмещение дифрактограмм XRPD образцов (образцов 2-2, 2-3, 2-4, 2-5, 2-6, 2-7 и 2-8) после 6 суток хранения при 25°С и 60% относительной влажности (пример 6). По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 8В представлен график DSC и TGA для образца 2-2 (пример 6). По оси х показана температура, измеряемая в °С, по левой оси у показан тепловой поток, измеряемый в Вт/г, а по правой оси у показана масса, измеряемая в процентах. Экспериментальные процедуры для коллекции DSC и TGA приведены в примере 2.
На фиг. 9А представлен график DSC и TGA для образца 2-3 (пример 6). По оси х показана температура, измеряемая в °С, по левой оси у показан тепловой поток, измеряемый в Вт/г, а по правой оси у показана масса, измеряемая в процентах. Экспериментальные процедуры для коллекции DSC и TGA приведены в примере 2.
На фиг. 9В представлен график DSC и TGA для образца 2-4 (пример 6). По оси х показана температура, измеряемая в °С, по левой оси у показан тепловой поток, измеряемый в Вт/г, а по правой оси у показана масса, измеряемая в процентах. Экспериментальные процедуры для коллекции DSC и TGA приведены в примере 2.
На фиг. 10А представлен график DSC и TGA для образца 2-5 (пример 6). По оси х показана температура, измеряемая в °С, по левой оси у показан тепловой поток, измеряемый в Вт/г, а по правой оси у показана масса, измеряемая в процентах. Экспериментальные процедуры для коллекции DSC и TGA приведены в примере 2.
На фиг. 10В представлен график DSC и TGA для образца 2-6 (пример 6). По оси х показана температура, измеряемая в °С, по левой оси у показан тепловой поток, измеряемый в Вт/г, а по правой оси у показана масса, измеряемая в процентах. Экспериментальные процедуры для коллекции DSC и TGA приведены в примере 2.
На фиг. 11А представлен график DSC и TGA для образца 2-7 (пример 6). По оси х показана температура, измеряемая в °С, по левой оси у показан тепловой поток, измеряемый в Вт/г, а по правой оси у показана масса, измеряемая в процентах. Экспериментальные процедуры для коллекции DSC и TGA приведены в примере 2.
На фиг. 11В представлен график DSC и TGA для образца 2-8 (пример 6). По оси х показана температура, измеряемая в °С, по левой оси у показан тепловой поток, измеряемый в Вт/г, а по правой оси у показана масса, измеряемая в процентах. Экспериментальные процедуры для коллекции DSC и TGA приведены в примере 2.
На фиг. 12А показан паттерн XRPD аморфного соединения 4 (образца 3-12), обсуждаемого в примере 7. По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах. Кристаллизацию малонатной соли не наблюдали независимо от используемого растворителя.
На фиг. 12В представлено совмещение дифрактограмм XRPD аморфных образцов (образцов 3-6, 3-10, 3-11 и 3-12), идентифицированных при попытке кристаллизации соединения 1 с малонатной солью (пример 7). По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 13А представлена хроматограмма HPLC образца 3-12 при попытке кристаллизации соединения 1 с малонатной солью, как описано в примере 7. Образец характеризовался чистотой 99,2%. По оси х показано время, измеренное в минутах, а по оси у показана интенсивность, измеренная в мAu.
На фиг. 13В представлено совмещение дифрактограмм XRPD твердых образцов, полученных при кристаллизации с использованием LAG (образцов 4-13, 4-12, 4-9, 4-3 и 4-1), по сравнению с соединением 1 (образцом 1-2), как описано в примере 8. Все XRDP соответствуют паттернам противоиона кристаллической кислоты без дополнительных пиков. По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 14А представлено совмещение дифрактограмм XRPD образцов, полученных при использовании этилацетата в качестве кристаллизационного растворителя (образцов 6-13, 6-12, 6-11, 6-10, 6-8, 6-7, 6-6, 6-5, 6-4 и 6-2), по сравнению с кристаллическим соединением 1 (образцом 1-2), как описано в примере 10. Выяснили, что паттерны XRPD обычно соответствуют паттерну соединения 1 за исключением образцов 6-2, 6-4 и 6-5, которые демонстрируют небольшие отличия. По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 14В представлено совмещение дифрактограммы XRPD образца 5-1 после второго растворения в MEK и добавления антирастворителя циклогексана и памоевой кислоты, как описано в примере 9. Образец 5-1, кристаллизируемый в памоевой кислоте, был твердым после отстаивания, но паттерн XRPD соответствовал паттерну памоевой кислоты.
На фиг. 15А представлено совмещение дифрактограмм XRPD образцов, полученных при использовании этилацетата в качестве кристаллизационного растворителя (образцов 6-5, 6-4 и 6-2), по сравнению с кристаллическим соединением 1 (образцом 1-2), как описано в примере 10. Выяснили, что паттерны XRPD обычно соответствуют паттерну соединения 1 за исключением образцов 6-2, 6-4 и 6-5, которые демонстрируют небольшие отличия. По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах и помеченная кислотой, используемой при кристаллизации.
На фиг. 15В показан паттерн XRPD для соединения 2, как описано в примере 14. По оси х показан 2 Тета, измеренный в градусах, а по оси у показана интенсивность, измеренная в импульсах.
На фиг. 16А показан график уровней концентрации активного TP (метаболита 1-6) в печени и сердце крыс, собак и обезьян (пример 18). По оси х отмечена дозировка, измеряемая в мг/кг, для каждого вида, а по оси у отмечена концентрация активного TP, измеряемая в нг/г.
На фиг. 16В показан график уровней концентрации активного TP (метаболита 1-6) в печени и сердце собак (n=2), измеряемых через 4 часа после однократной пероральной дозы соединения 1 или соединения 2 (пример 19). По оси х отмечена дозировка каждого соединения, измеряемая в мг/кг, а по оси у отмечена концентрация активного TP, измеряемая в нг/г.
На фиг. 17 показан профиль в плазме соединения 1 и метаболита 1-7 у крыс, получивших однократную пероральную дозу 500 мг/кг соединения 2 (пример 20), измеряемый через 72 час после получения дозы. По оси х отмечено время, измеряемое в часах, по оси у отмечена концентрация в плазме, измеряемая в нг/мл.
На фиг. 18 показан профиль в плазме соединения 1 и метаболита 1-7 у обезьян, получивших однократные пероральные дозы 30 мг, 100 мг или 300 мг соединения 2 (пример 20), измеренный через 72 час после получения дозы. По оси х отмечено время, измеряемое в часах, а по оси у отмечена концентрация в плазме, измеряемая в нг/мл.
На фиг. 19 показан график ЕС95, измеряемой в нМ, для софосбувира и соединения 1 против клинических изолятов HCV. Значения ЕС95 для соединения 1 в 7-33 раза ниже, чем у софосбувира (пример 22). По оси х отмечен генотип, а по оси у показана ЕС95, измеряемая в нМ.
На фиг. 20 показан график ЕС50, измеряемой в нМ, для софосбувира и соединения 1 против лабораторных штаммов генотипов 1a, 1b, 2а, 3а, 4а и 5а HCV. Соединение 1 в приблизительно 6-11 раз эффективнее софосбувира в отношении генотипов 1-5 (пример 22). По оси х отмечен генотип, а по оси у показана ЕС50, измеряемая в нМ.
На фиг. 21 показан график профиля средней концентрации в плазме в зависимости от времени соединения 1 после введения однократной дозы соединения 2 во всех когортах части В исследования, как описано в примере 24. Соединение 1 быстро абсорбировалось и быстро метаболизировалось в пределах приблизительно 8 часов во всех когортах части В. По оси х отмечено время, измеряемое в часах, а по оси у отмечено геометрическое среднее концентрации в плазме, измеряемое в нг/мл.
На фиг. 22 показан график профиля средней концентрации в плазме в зависимости от времени метаболита 1-7 после введения однократной дозы соединения 2 во всех когортах части В исследования, как описано в примере 24. Метаболит 1-7 демонстрировал устойчивую концентрацию в плазме во всех когортах части В. По оси х отмечено время, измеряемое в часах, а по оси у отмечено геометрическое среднее концентрации в плазме, измеряемое в нг/мл.
На фиг. 23А показан индивидуальный фармакокинетический/фармакодинамический анализ субъекта, включенного в когорту 1b, как описано в примере 24. На графике показано воздействие метаболита 1-7 в плазме и снижение уровней РНК HCV. Пунктирная линия представляет минимальную концентрацию метаболита 1-7, необходимую для поддержания вирусологического ответа, превышающую значение ЕС95, против GT1b. По оси х отмечено время, измеряемое в часах. По левой оси у показана концентрация метаболита 1-7 в плазме, измеряемая в нг/мл, а по правой оси у показано снижение РНК HCV, измеряемое в log10 МЕ/мл.
На фиг. 23В показан индивидуальный фармакокинетический/фармакодинамический анализ субъекта, включенного в когорту 1b, как описано в примере 24. На графике показано воздействие метаболита 1-7 в плазме и снижение уровней РНК HCV. Пунктирная линия представляет минимальную концентрацию метаболита 1-7, необходимую для поддержания вирусологического ответа, превышающую значение ЕС95, против GT1b. По оси х отмечено время, измеряемое в часах. По левой оси у показана концентрация метаболита 1-7 в плазме, измеряемая в нг/мл, а по правой оси у показано снижение РНК HCV, измеряемое в log10 МЕ/мл.
На фиг. 23С показан индивидуальный фармакокинетический/фармакодинамический анализ субъекта, включенного в когорту 1b, как описано в примере 24. На графике показано воздействие метаболита 1-7 в плазме и снижение уровней РНК HCV. Пунктирная линия представляет минимальную концентрацию метаболита 1-7, необходимую для поддержания вирусологического ответа, превышающую значение ЕС95, против GT1b. По оси х отмечено время, измеряемое в часах. По левой оси у показана концентрация метаболита 1-7 в плазме, измеряемая в нг/мл, а по правой оси у показано снижение РНК HCV, измеряемое в log10 МЕ/мл.
На фиг. 23D показан индивидуальный фармакокинетический/фармакодинамический анализ субъекта, включенного в когорту 3b, как описано в примере 24. На каждом графике показано воздействие метаболита 1-7 в плазме и снижение уровней РНК HCV. Пунктирная линия представляет минимальную концентрацию метаболита 1-7, необходимую для поддержания вирусологического ответа, превышающую значение ЕС95, против GT1b. По оси х отмечено время, измеряемое в часах. По левой оси у показана концентрация метаболита 1-7 в плазме, измеряемая в нг/мл, а по правой оси у показано снижение РНК HCV, измеряемое в log10 МЕ/мл.
На фиг. 23Е показан индивидуальный фармакокинетический/фармакодинамический анализ субъекта, включенного в когорту 3b, как описано в примере 24. На каждом графике показано воздействие метаболита 1-7 в плазме и снижение уровней РНК HCV. Пунктирная линия представляет минимальную концентрацию метаболита 1-7, необходимую для поддержания вирусологического ответа, превышающую значение ЕС95, против GT1b. По оси х отмечено время, измеряемое в часах. По левой оси у показана концентрация метаболита 1-7 в плазме, измеряемая в нг/мл, а по правой оси у показано снижение РНК HCV, измеряемое в log10 МЕ/мл.
На фиг. 23F показан индивидуальный фармакокинетический/фармакодинамический анализ субъекта, включенного в когорту 3b, как описано в примере 24. На каждом графике показано воздействие метаболита 1-7 в плазме и снижение уровней РНК HCV. Пунктирная линия представляет минимальную концентрацию метаболита 1-7, необходимую для поддержания вирусологического ответа, превышающую значение ЕС95, против GT1b. По оси х отмечено время, измеряемое в часах. По левой оси у показана концентрация метаболита 1-7 в плазме, измеряемая в нг/мл, а по правой оси у показано снижение РНК HCV, измеряемое в log10 МЕ/мл.
На фиг. 24 показан график значений ЕС95 соединения 1 и софосбувира в отношении больных, инфицированных клиническими изолятами GT1, GT2, GT3 и GT4 HCV. Горизонтальная пунктирная линия ( ) представляет равновесную остаточную концентрацию (C24,ss) нуклеозида софосбувира после дозы 400 мг QD софосбувира. Сплошная горизонтальная линия (
) представляет равновесную остаточную концентрацию (C24,ss) нуклеозида софосбувира после дозы 400 мг QD софосбувира. Сплошная горизонтальная линия ( ) представляет равновесную остаточную концентрацию (C24,ss) метаболита 1-7 после 600 мг соединения 2 (что эквивалентно 550 мг соединения 1). Горизонтальная точечная линия (
) представляет равновесную остаточную концентрацию (C24,ss) метаболита 1-7 после 600 мг соединения 2 (что эквивалентно 550 мг соединения 1). Горизонтальная точечная линия ( ) представляет равновесную остаточную концентрацию (C24,ss) метаболита 1-7 после 450 мг соединения 2 (что эквивалентно 400 мг соединения 1). Как обсуждается в примере 25, прогнозируемое равновесное остаточное содержание в плазме (C24,ss) метаболита 1-7 после 600 мг и 450 мг соединения 2 превышает in vitro ЕС95 соединения 1 против всех тестируемых клинических изолятов. Равновесное остаточное содержание в плазме (C24,ss) софосбувира превышает только ЕС95 при клинических изолятах GT2. По оси х отмечены клинические изоляты, а в таблице под осью х приводятся значения ЕС95 для соединения 1 и софосбувира. По оси у показана ЕС95 против клинических изолятов, измеряемая в нг/мл. ЕС95 выражается в нуклеозидном эквиваленте. Софосбувир и соединение 2 вводили ежесуточно (QD).
) представляет равновесную остаточную концентрацию (C24,ss) метаболита 1-7 после 450 мг соединения 2 (что эквивалентно 400 мг соединения 1). Как обсуждается в примере 25, прогнозируемое равновесное остаточное содержание в плазме (C24,ss) метаболита 1-7 после 600 мг и 450 мг соединения 2 превышает in vitro ЕС95 соединения 1 против всех тестируемых клинических изолятов. Равновесное остаточное содержание в плазме (C24,ss) софосбувира превышает только ЕС95 при клинических изолятах GT2. По оси х отмечены клинические изоляты, а в таблице под осью х приводятся значения ЕС95 для соединения 1 и софосбувира. По оси у показана ЕС95 против клинических изолятов, измеряемая в нг/мл. ЕС95 выражается в нуклеозидном эквиваленте. Софосбувир и соединение 2 вводили ежесуточно (QD).
На фиг. 25 показана блок-схема, демонстрирующая способ изготовления 50-мг и 100-мг таблеток соединения 2, как описано в примере 26. На стадии 1 микрокристаллическую целлюлозу, соединение 2, моногидрат лактозы и кроскармеллозу натрия фильтруют через экран 600 мкМ. На стадии 2 содержимое со стадии 1 загружают в V-образный смеситель и смешивают в течение 5 минут при 25 оборотах в минуту. На стадии 3 стеарат магния фильтруют через экран 600 мкМ. На стадии 4 стеарат магния загружают в V-образный смеситель, содержащий содержимое со стадии 2 (микрокристаллическую целлюлозу, соединение 2, моногидрат лактозы и кроскармеллозу натрия) и смешивают в течение 2 минут при 25 оборотах в минуту. Затем всю смесь делят для получения 50-мг таблеток и 100-мг таблеток. Для получения 50-мг таблеток смесь со стадии 4 прессуют с помощью 6-мм круглого стандартного двояковогнутого инструмента. Для получения 100-мг таблеток смесь со стадии 4 прессуют с помощью 8-мм круглого стандартного двояковогнутого инструмента. Затем таблетки пакуют в HDPE бутылки, индукционно запечатанные РР колпачком с влагопоглотителем.
На фиг. 26 показана гемисульфатная соль, которая демонстрирует благоприятные фармакологические свойства по сравнению с соответствующим ей свободным основанием для лечения заболевания, вызванного вирусом HCV.
Подробное раскрытие настоящего изобретения
Настоящее изобретение, раскрываемое в настоящем документе, относится к соединению, способу, композиции и твердой лекарственной форме для лечения инфекций или воздействия на людей и других животных-хозяев вируса HCV, которые включают введение эффективного количества гемисульфатной соли изопропил-((S)-(((2R,3R,4R,5R)-5-(2-амино-6-(метиламино)-9H-пурин-9-ил)-4-фтор-3-гидрокси-4-метилтетрагидрофуран-2-ил)метокси)(фенокси)фосфорил)-L-аланината (соединения 2), описываемого в настоящем документе, необязательно в фармацевтически приемлемом носителе. Согласно одному варианту осуществления соединение 2 представляет собой аморфное твердое вещество. Согласно еще одному варианту осуществления соединение 2 представляет собой кристаллическое твердое вещество.
Соединение, композиции и лекарственные формы также могут быть использованы для лечения состояний, связанных с воздействием вируса HCV или возникающих в результате такового. Например, активное соединение может быть использовано для лечения состояний, которые сопровождаются наличием антител против HCV и антигенов, вызванных вирусом хронического воспаления печени, рака печени, вызванного запущенным гепатитом С (например, печеночноклеточной карциномы), цирроза, хронического или острого гепатита С, молниеносного гепатита С, хронического персистирующего гепатита С и утомления, вызванного образованием антител против HCV.
Активные соединения и композиции также могут быть использованы для лечения заболеваний, вызванных рядом генотипов HCV. В мире идентифицировано по меньшей мере шесть разных генотипов HCV, каждый из которых имеет несколько подтипов. Генотипы 1-3 являются преобладающими в мире, а генотипы 4, 5 и 6 географически более ограничены. Генотип 4 является обычным для Среднего Востока и Африки. Генотип 5 чаще всего встречается в Южной Африке. Генотип 6 преимущественно существует в Юго-Восточной Азии. Хотя наиболее распространенным генотипом в Соединенных Штатах Америки является генотип 1, определение генотипа и подтипа может помочь в определении типа и продолжительности лечения. Например, разные генотипы по-разному реагируют на различные лекарства, и оптимальное время лечения варьирует в зависимости от генотипа инфекции. Внутри генотипов подтипы, такие как генотип 1а и генотип 1b, также по-разному реагируют на лечение. Заражение одним типом генотипа не исключает последующего заражения другим генотипом.
Как описано в примере 22, соединение 2 является активным против ряд генотипов HCV, в том числе генотипов 1-5. Согласно одному варианту осуществления соединение 2 используют для лечения заболевания, вызванного HCV генотипа 1, HCV генотипа 2, HCV генотипа 3, HCV генотипа 4, HCV генотипа 5 или HCV генотипа 6. Согласно одному варианту осуществления соединение 2 используют для лечения заболевания, вызванного HCV генотипа 1а. Согласно одному варианту осуществления соединение 2 используют для лечения заболевания, вызванного HCV генотипа 1b. Согласно одному варианту осуществления соединение 2 используют для лечения заболевания, вызванного HCV генотипа 2а. Согласно одному варианту осуществления соединение 2 используют для лечения заболевания, вызванного HCV генотипа 2b. Согласно одному варианту осуществления соединение 2 используют для лечения заболевания, вызванного HCV генотипа 3а. Согласно одному варианту осуществления соединение 2 используют для лечения заболевания, вызванного HCV генотипа 4а. Согласно одному варианту осуществления соединение 2 используют для лечения заболевания, вызванного HCV генотипа 4d.
Согласно одному варианту осуществления соединение 1 или соединение 2 используют для лечения заболевания, вызванного HCV генотипа 5а. Согласно одному варианту осуществления соединение 1 или соединение 2 используют для лечения заболевания, вызванного HCV генотипа 6а. Согласно одному варианту осуществления соединение 1 или соединение 2 используют для лечения заболевания, вызванного HCV генотипов 6b, 6с, 6d, 6е, 6f, 6g, 6h, 6i, 6j, 6k, 6l, 6m, 6n, 6o, 6p, 6q, 6r, 6s, 6t или 6u.
Как обсуждается в примере 25 и показано на фиг. 24, прогнозируемая равновесная остаточная концентрация (C24,ss) метаболита 1-7 после дозы 450 мг (400 мг свободного основания) и дозы 600 мг (550 мг свободного основания) соединения 2 составляет от приблизительно 40 нг/мл до 50 нг/мл. Такой уровень C24,ss превышал ЕС95 соединения 1 при HCV генотипов 1a, 1b, 2а, 2b, 3а, 4а и 4d. Эти данные подтверждают, что соединение 2 обладает мощной активностью в отношении всех генотипов. Это удивительно, поскольку соединение 2 достигает более низкой равновесной остаточной концентрации (C24,ss), чем равновесная остаточная концентрация (C24,ss) нуклеозидного метаболита софосбувира после ведения эквивалентной дозы софосбувира. Равновесная остаточная концентрация (C24,ss) соответствующего нуклеозидного метаболита софосбувира составляет приблизительно 100 нг/мл, но этот уровень превышает только ЕС95 софосбувира против клинических изолятов GT2 (фиг. 24). Соединение 2 является более эффективным, чем софосбувир, против GT1, GT2, GT3 и GT4 и, следовательно, допускает лекарственную форму, доставляющую более низкую равновесную остаточную концентрацию этого метаболита, которая, тем не менее, эффективна против всех тестируемых генотипов HCV. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7 от приблизительно 15 до 75 нг/мл. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7 от приблизительно 20 до 60 нг/мл, от 20 до 50 нг/мл или от 20 до 40 нг/мл.
Согласно одному варианту осуществления соединение, составы или твердые лекарственные формы, которые включают в себя соединение, также могут быть использованы профилактически для предупреждения или ограничения прогрессирования клинической болезни у индивидуумов с наличием антител против HCV или наличием антигенов, или подвергнувшихся воздействию вируса гепатита С.
В частности, выяснили, что соединение 2 является активным против HCV и демонстрирует превосходные лекарственные и фармакологические свойства по сравнению с его свободным основанием (соединением 1). Удивительно, что соединение 2 является более биодоступным и достигает более высокой AUC, чем соединение 1 (пример 19), и соединение 2 является более селективным по отношению к целевому органу -печени, чем соединение 1 (пример 19).
Соединение 2 также превосходит соединение 1 в отношении растворимости и химической стабильности. Это удивительно, поскольку моносульфатная соль изопропил-((S)-(((2R,3R,4R,5R)-5-(2-амино-6-(метиламино)-9H-пурин-9-ил)-4-фтор-3-гидрокси-4-метилтетрагидрофуран-2-ил)метокси)(фенокси)фосфорил)-L-аланината (соединение 3) является нестабильной и имеет внешний вид липкой смолы, тогда как соединение 2, гемисульфатная соль, является стабильным белым твердым веществом. Гемисульфатная соль, как в виде твердого вещества, так и в твердой лекарственной форме, является очень стабильной на протяжении 9 месяцев и является негигроскопичной.

Несмотря на объем литературы по противовирусным нуклеозидам и поданных патентных документов соединение 2 не было конкретно раскрыто.
Соединение 2 имеет S-стереохимию по атому фосфора, что было подтверждено рентгеновской кристаллографией (фиг. 3, пример 2). Согласно альтернативным вариантам осуществления соединение 2 может быть использовано в форме фосфорных R- и S-энантиомеров в любом желаемом отношении, вплоть до чистых энантиомеров. Согласно некоторым вариантам осуществления соединение 2 используют в форме, которая по меньшей мере на 90% свободна от противоположного энантиомера и может быть по меньшей мере на 98%, 99% или даже 100% свободна от противоположного энантиомера. Если не описывается иное, энантиомерно обогащенное соединение 2 по меньшей мере на 90% свободно от противоположного энантиомера. Кроме того, согласно альтернативному варианту осуществления аминокислота фосфорамидата может иметь D- или L-конфигурацию или их смесь, в том числе рацемическую смесь.
Если не указано иное, соединения, описываемые в настоящем документе, представлены в β-D-конфигурации. Согласно альтернативному варианту осуществления соединения могут быть представлены в β-L-конфигурации. Подобным образом, любая группа заместителей, которая обладает хиральностью, может быть представлена в рацемической, энантиомерной, диастереомерной форме или любой их смеси. Если фосфорамидат обладает хиральностью, то он может быть обеспечен в виде R или S хирального фосфорного производного или их смеси, в том числе рацемической смеси. Все комбинации таких стереоконфигураций представляют собой альтернативные варианты осуществления настоящего изобретения, описываемого в настоящем документе. Согласно другому варианту осуществления по меньшей мере один из водородов соединения 2 (нуклеотида или гемисульфатной соли) может быть замещен дейтерием.
Такие альтернативные конфигурации включают в себя без ограничения
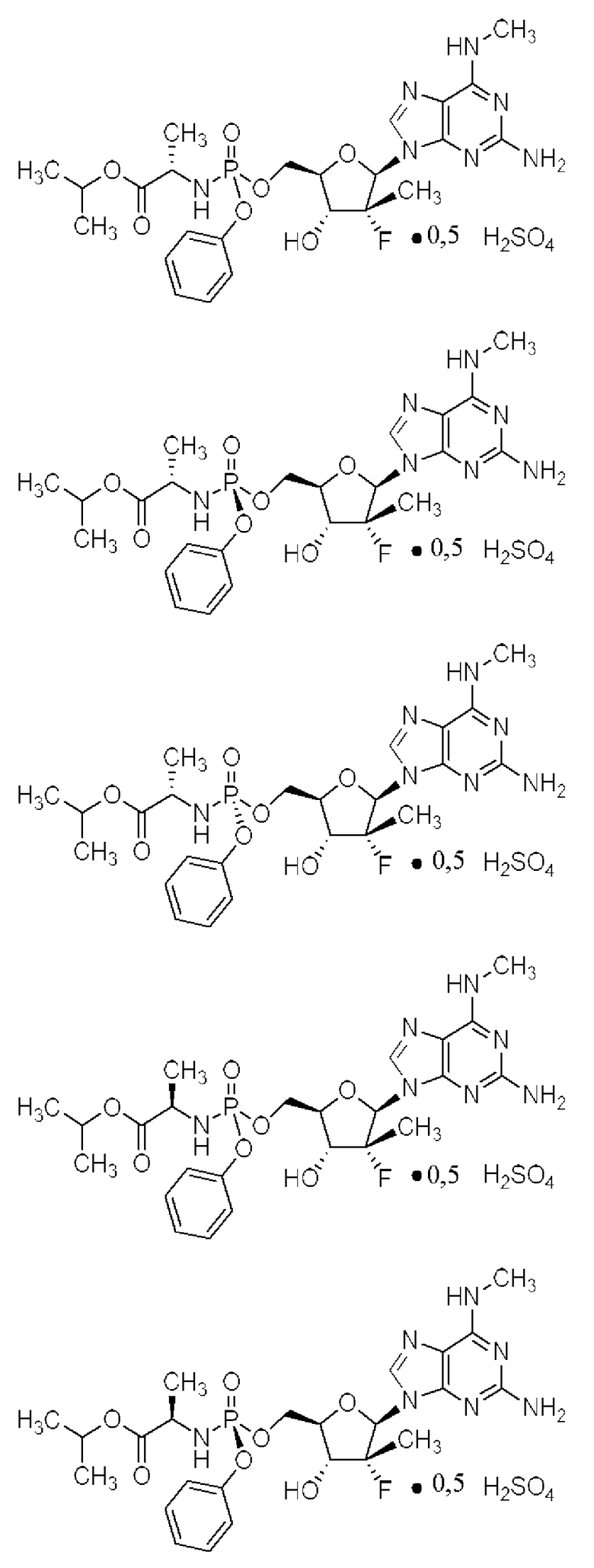
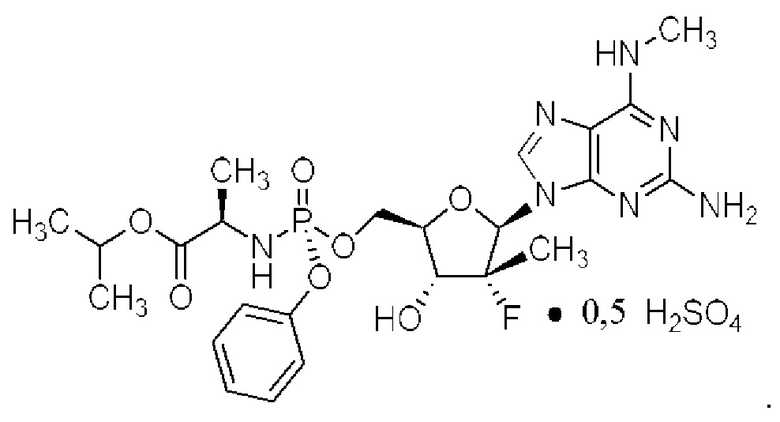
I. Гемисульфатная соль изопропил-((S)-(((2R,3R,4R,5R)-5-(2-амино-6-(метиламино)-9H-пурин-9-ил)-4-фтор-3-гидрокси-4-метилтетрагидрофуран-2-ил)метокси)(фенокси)фосфорил)-L-аланината (соединение 2)
Активным соединением в соответствии с настоящим изобретением является соединение 2, которое может быть обеспечено в фармацевтически приемлемой композиции или в его твердой лекарственной форме. Согласно одному варианту осуществления соединение 2 представляет собой аморфное твердое вещество. Согласно следующему варианту осуществления соединение 2 представляет собой кристаллическое твердое вещество.
Синтез соединения 2
Настоящее изобретение, кроме того, включает в себя типичный неограничивающий иллюстративный способ получения соединения 2, который включает
(i) первую стадию растворения соединения 1 в органическом растворителе, например, в ацетоне, этилацетате, метаноле, ацетонитриле, или эфире, или подобном, в колбе или контейнере;
(ii) загрузку второй колбы или контейнера вторым органическим растворителем, который может быть может быть таким же или отличаться от органического растворителя со стадии (i), необязательно охлаждение второго растворителя до 0-10 градусов Цельсия и добавление каплями H2SO4 во второй органический растворитель для создания смеси H2SO4/органический растворитель; и при этом растворителем может быть, например, метанол;
(iii) добавление каплями смеси H2SO4/растворитель при молярном отношении 0,5/1,0 со стадии (ii) в раствор соединения 1 со стадии (i) при окружающей или слегка повышенной или пониженной температуре (например, 23-35 градусов Цельсия);
(iv) перемешивание реакционной смеси со стадии (iii) до образования осадка соединения 2, например, при окружающей или слегка повышенной, или пониженной температуре;
(v) необязательно фильтрование полученного в результате осадка со стадии (iv) и промывания органическим растворителем; и
(vi) необязательно сушку полученного в результате соединения 2 в вакууме, необязательно при повышенной температуре, например, 55, 56, 57, 58, 59 или 60°С.
Согласно некоторым вариантам осуществления вышеупомянутую стадию (i) выполняют в ацетоне. Кроме того, вторым органическим растворителем на стадии (ii) может быть, например, метанол, а смесью органических растворителей на стадии (v) является метанол/ацетон.
Согласно одному варианту осуществления соединение 1 растворяют в этилацетате на стадии (i). Согласно одному варианту осуществления соединение 1 растворяют в тетрагидрофуране на стадии (i). Согласно одному варианту осуществления соединение 1 растворяют в ацетонитриле на стадии (i). Согласно дополнительному варианту осуществления соединение 1 растворяют в диметилформамиде на стадии (i).
Согласно одному варианту осуществления вторым органическим растворителем на стадии (ii) является этанол. Согласно одному варианту осуществления вторым органическим растворителем на стадии (ii) является изопропанол. Согласно одному варианту осуществления вторым органическим растворителем на стадии (ii) является н-бутанол.
Согласно одному варианту осуществления смесь растворителей используют для промывания на стадии (v), например, этанол/ацетон. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является изопропанол/ацетон. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является н-бутанол/ацетон. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является этанол/этил ацетат. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является изопропанол/этилацетат. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является н-бутанол/этилацетат. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является этанол/тетрагидрофуран. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является изопропанол/тетрагидрофуран. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является н-бутанол/тетрагидрофуран. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является этанол/ацетонитрил. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является изопропанол/ацетонитрил. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является н-бутанол/ацетонитрил. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является этанол/диметилформамид. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является изопропанол/диметилформамид. Согласно одному варианту осуществления смесью растворителей для промывания на стадии (v) является н-бутанол/диметилформамид.
II. Метаболизм изопропил-((S)-(((2R,3R,4R,5R)-5-(2-амино-6-(метиламино)-9H-пурин-9-ил)-4-фтор-3-гидрокси-4-метилтетрагидрофуран-2-ил)метокси)(фенокси)фосфорил)-L-аланината (соединения 2)
Метаболизм соединения 1 и соединения 2 предусматривает продуцирование 5'-монофосфата и последующий анаболизм N6-метил-2,6-диаминопуринового основания (1-3) с образованием ((2R,3R,4R,5R)-5-(2-амино-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-4-фтор-3-гидрокси-4-метилтетрагидрофуран-2-ил)метила дигидрофосфата (1-4) в виде 5'-монофосфата. Затем монофосфат далее анаболизируется до активного трифосфатного соединения - 5'-трифосфата (1-6). 5'-Трифосфат может быть далее метаболизирован с образованием 2-амино-9-((2R,3R,4R,5R)-3-фтор-4-гидрокси-5-(гидроксиметил)-3-метилтетрагидрофуран-2-ил)-1,9-дигидро-6Н-пурин-6-она (1-7). В качестве альтернативы, 5'-монофосфат 1-2 может быть метаболизирован с образованием пуринового основания 1-8. Метаболический путь для изопропил-((S)-(((2R,3R,4R,5R)-5-(2-амино-6-(метиламино)-9H-пурин-9-ил)-4-фтор-3-гидрокси-4-метилтетрагидрофуран-2-ил)метокси)(фенокси)фосфорил)-L-аланината показан на схеме 1 (показанной выше).
III. Дополнительные соли соединения 1
Согласно альтернативным вариантам осуществления настоящее изобретение обеспечивает соединение 1 в виде оксалатной соли (соединения 4) или соли HCl (соединения 5).
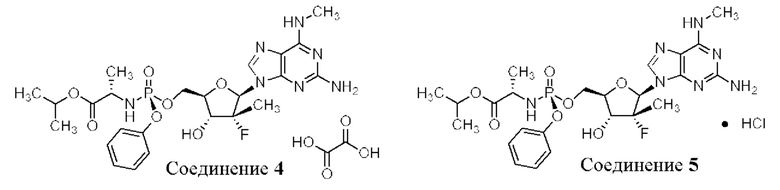
Как 1:1 оксалатная соль, так и 1:1 соль HCl образуют твердые вещества с приемлемыми свойствами для твердых лекарственных форм для лечения хозяина, такого как человек, с гепатитом С. Однако, оксалатная соль может быть менее желательной и, возможно, может не подходить, если больной восприимчив к образованию камней в почках. Соль HCl является более гигроскопичной, чем гемисульфатная соль. Таким образом, гемисульфатная соль остается наиболее желательной солевой формой соединения 1 с неожиданными свойствами.
IV. Определения
Используемый в контексте настоящего изобретения термин «D-конфигурация» относится к основной конфигурации, которая имитирует природную конфигурацию сахарных фрагментов по сравнению с не встречающимися в природе нуклеозидами или «L» конфигурацией. Термин «β» или «β-аномер» используют для обозначения нуклеозидных аналогов, в которых нуклеозидное основание сконфигурировано (расположено) над плоскостью фуранозного фрагмента в нуклеозидном аналоге.
Термины «совместно вводить» и «совместное введение» или комбинированная терапия используют для описания введения соединения 2 в соответствии с настоящим изобретением в комбинации по меньшей мере с одним другим активным средством, например, в случае необходимости, по меньшей мере с одним дополнительным средством против HCV. Время осуществления совместного введения лучше всего определять медицинскому специалисту, лечащему больного. Иногда предпочтительно вводить средства в одно и то же время. В качестве альтернативы, лекарственные средства, выбранные для комбинированной терапии, могут быть введены больному в разные моменты времени. Конечно, при наличии более чем одной вирусной или иной инфекции или другого состояния соединения в соответствии с настоящим изобретением при необходимости могут быть объединены с другими средствами для лечения этих других инфекции или состояния.
Используемый в настоящем документе термин «хозяин» относится к одноклеточному или многоклеточному организму, в котором вирус HCV может реплицироваться, в том числе к клеточным линиям, животным и, как правило, к человеку. Термин «хозяин», в частности, относится к инфицированным клеткам, клеткам, трансфицированным полным геномом HCV или его частью, и животным, в частности, приматам (в том числе шимпанзе) и людям. При большинстве применений для животных в соответствии с настоящим изобретением хозяином является больной человек. Тем не менее применения в ветеринарии, при определенных показаниях, явно предусматриваются настоящим изобретением (например, для шимпанзе). Хозяином могут быть, например, бычьи, лошадиные, птицы, собачьи, кошачьи и т.д.
Изотопное замещение
Настоящее изобретение относится к соединениям и к применению соединения 2 с требуемыми изотопными замещениями атомов в количествах, которые превышают распространенность изотопа в природе, т.е., к обогащенным. Изотопы представляют собой атомы с одинаковым атомным числом, но с разными массовыми числами, т.е. с одинаковым числом протонов, но разным числом нейтронов. В качестве общего примера и без ограничения, изотопы водорода, например, дейтерий (2Н) и тритий (3Н), могут быть использованы в любом месте в описанных структурах. Альтернативно или в дополнение, могут быть использованы изотопы углерода, например, 13С и 14С. Предпочтительным изотопным замещением является дейтерий для водорода в одном или нескольких местах молекулы для улучшения характеристики лекарственного средства. Дейтерий может быть связан в месте разрыва связи во время метаболизма (кинетический изотопный эффект α-дейтерия) или рядом с или возле места разрыва связи (кинетический изотопный эффект β-дейтерия). Компания Achillion Pharmaceuticals, Inc. (в международных заявках WO №№ WO 2014/169278 и WO 2014/169280) описывает дейтерирование нуклеотидов для улучшения их фармакокинетических или фармакодинамических показателей, включая 5-положение в молекуле.
Замещение изотопами, такими как дейтерий, может давать определенные терапевтические преимущества, возникающие вследствие большей метаболической стабильности, такие как, например, увеличенный in vivo период полувыведения или сниженные размеры дозировки. Замещение дейтерием водорода по участку метаболического расщепления может уменьшать скорость или элиминировать метаболизм по такой связи. В любом положении соединения, в котором может присутствовать атом водорода, атом водорода может быть любым изотопом водорода, в том числе протаем (1Н), дейтерием (2Н) и тритием (3Н). Таким образом, упоминание в настоящем изобретении соединения охватывает все возможные изотопные формы, если в контексте четко не указано иное.
Термин «изотопно меченный» аналог относится к аналогу, который представляет собой «дейтерированный аналог», «13С-меченный аналог» или «дейтерированный/13С-меченный аналог». Термин «дейтерированный аналог» означает описываемое в настоящем изобретении соединение, в соответствии с которым Н-изотоп, т.е. водород/протий (1Н), замещен Н-изотопом, т.е. дейтерием (2Н). Замещение дейтерием может быть частичным или полным. Частичное замещение дейтерием означает, что по меньшей мере один атом водорода замещен по меньшей мере одним дейтерием. Согласно определенным вариантам осуществления изотоп на 90, 95 или 99%, или более обогащен изотопом в любом представляющем интерес месте. Согласно некоторым вариантам осуществления он представляет собой дейтерий, который на 90, 95 или 99% обогащен в желаемом положении. Если не отмечено иное, дейтерирование составляет по меньшей мере 80% в выбранном положении. Дейтерирование нуклеозида может происходить по любому заменяемому водороду, который обеспечивает желаемые результаты.
V. Способы лечения или профилактики
Используемый в настоящем документе термин «лечение» относится к введению соединения 2 хозяину, например, человеку, который инфицирован вирусом HCV или может стать инфицированным вирусом HCV.
Используемый в настоящем документе термин «профилактическое» или «предупреждающее» относится к введению соединения 2 для предупреждения или снижения вероятности возникновения вирусного нарушения. Настоящее изобретение относится как к лечебным, так и к профилактическим или предупреждающим терапевтическим средствам. Согласно одному варианту осуществления соединение 2 вводят хозяину, который подвергся инфекции вируса гепатита С и, таким образом, рискует стать инфицированным таковым.
Настоящее изобретение относится к способу лечения или профилактики заболеваний, вызванных вирусом гепатита С, в том числе устойчивыми к лекарственному средству и устойчивыми к нескольким лекарственным средствам формами HCV, а также родственных болезненных состояний, состояний или осложнений инфекции HCV, в том числе цирроза и родственных гепатотоксичностей, а также других состояний, являющихся вторичными по отношению к инфекции HCV, таких как наряду с прочими слабость, потеря аппетита, потеря веса, увеличение молочной железы (особенно у мужчин), сыпь (особенно на ладонях), затруднение свертывания крови, звездчатые сосуды на коже, спутанность сознания, кома (энцефалопатия), накопление жидкости в брюшной полости (асциты), расширение вен пищевода, портальная гипертензия, почечная недостаточность, увеличение селезенки, уменьшение кровяных клеток, анемия, тромбоцитопения, желтуха и печеночноклеточный рак. Способ включает введение хозяину, нуждающемуся в этом, как правило, человеку, эффективного количества соединения 2, описываемого в настоящем документе, необязательно в комбинации по меньшей мере с одним дополнительным биоактивным средством, например, дополнительным средством против HCV, дополнительно в комбинации с фармацевтически приемлемыми носителем, добавкой и/или вспомогательным веществом.
Согласно другому аспекту настоящее изобретение относится к способу предупреждения или профилактики инфекции HCV, или болезненного состояния, или родственного, или последующего болезненного состояния, состояния или осложнения инфекции HCV, в том числе цирроза и родственных гепатотоксичностей, также других состояний, являющихся вторичными по отношению к инфекции HCV, таких как наряду с прочими слабость, потеря аппетита, потеря веса, увеличение молочной железы (особенно у мужчин), сыпь (особенно на ладонях), затруднение свертывания крови, звездчатые сосуды на коже, спутанность сознания, кома (энцефалопатия), накопление жидкости в брюшной полости (асциты), расширение вен пищевода, портальная гипертензия, почечная недостаточность, увеличение селезенки, уменьшение кровяных клеток, анемия, тромбоцитопения, желтуха и печеночноклеточный рак (печени), при этом указанный способ включает введение больному с риском эффективного количества соединения 2, как описывается выше, в комбинации с фармацевтически приемлемыми носителем, добавкой или вспомогательным веществом, необязательно в комбинации с другим средством против HCV. Согласно другому варианту осуществления активные соединения в соответствии с настоящим изобретением могут быть введены больному после связанной с гепатитом трансплантации печени для защиты нового органа.
Согласно альтернативному варианту осуществления соединение 2 представлено в виде гемисульфатной соли фосфорамидата соединения 1, отличного от специфического фосфорамидата, описанного в иллюстрации соединения. Специалистам в данной области известен широкий ряд фосфорамидатов, который включает в себя различные сложные эфиры и сложные фосфоэфиры, любая комбинация которых может быть использована для обеспечения активного соединения, описываемого в настоящем документе, в форме гемисульфатной соли.
VI. Фармацевтические композиции и лекарственные формы
Согласно аспекту настоящего изобретения фармацевтические композиции в соответствии с настоящим изобретением содержат эффективное против вируса HCV количество соединения 2, описываемого в настоящем документе, необязательно в комбинации с фармацевтически приемлемыми носителем, добавкой или вспомогательным средством, кроме того, необязательно в комбинации или при чередовании по меньшей мере с одним другим активным соединением. Согласно одному варианту осуществления настоящее изобретение включает в себя твердую лекарственную форму соединения 2 в фармацевтически приемлемом носителе.
Согласно аспекту настоящего изобретения фармацевтические композиции в соответствии с настоящим изобретением содержат эффективное против вируса HCV количество соединения 2, описываемого в настоящем документе, необязательно в комбинации с фармацевтически приемлемыми носителем, добавкой или вспомогательным веществом, кроме того, необязательно в комбинации по меньшей мере с одним другим противовирусным средством, таким как средство против HCV.
Настоящее изобретение относится к фармацевтическим композициям, которые включают в себя эффективное для лечения инфекции вируса гепатита С количество соединения 2 в соответствии с настоящим изобретением или пролекарства в фармацевтически приемлемом носителе или вспомогательном средстве. Согласно альтернативному варианту осуществления настоящее изобретение относится к фармацевтическим композициям, которые включают в себя эффективное для лечения инфекции вируса гепатита С количество соединения 2 в соответствии с настоящим изобретением или пролекарства в фармацевтически приемлемом носителе или вспомогательном средстве.
Специалисту в данной области будет очевидно, что терапевтически эффективное количество будет варьировать в зависимости от инфекции или состояния, подлежащих лечению, их тяжести, применяемого режима лечения, фармакокинетических показателей используемого средства, а также больного или субъекта (животного или человека), подлежащего лечению, и такое терапевтическое количество может быть определено лечащим врачом или специалистом.
Соединение 2 в соответствии с настоящим изобретением может быть составлено в смеси с фармацевтически приемлемым носителем. Как правило, предпочтительно вводить фармацевтическую композицию в перорально вводимой форме, в частности, в твердой лекарственной форме, такой как пилюля или таблетка. Некоторые составы можно вводить парентеральным, внутривенным, внутримышечным, местным, чрескожным, буккальным, подкожным, с помощью суппозитория или другим путем, в том числе интраназальным распылением. Внутривенные и внутримышечные составы часто вводят в стерильном солевом растворе. Специалист в данной области может модифицировать составы, чтобы сделать их более растворимыми в воде или другой среде-носителе, например, это можно легко осуществить с помощью небольших модификаций (солеобразования, эстерификации и т.д.), которые хорошо известны специалисту в данной области. Также специалисту в данной области хорошо известно, как модифицировать путь введения и режим лекарствения соединения 2 для регулирования фармакокинетических показателей соединений в соответствии с настоящим изобретением для максимально полезного эффекта для больных, как подробно описывается в настоящем документе.
В некоторых фармацевтических лекарственных формах для достижения желаемого эффекта может быть использована пролекарственная форма соединений, в том числе ацилированные (ацетилированные или иные) и эфирные (алкиловые и родственные) производные, фосфатные сложные эфиры, тиофосфорамидаты, фосфорамидаты и различные солевые формы соединений в соответствии с настоящим изобретением. Специалисту в данной области будет понятно, как легко модифицировать соединения в соответствии с настоящим изобретением в пролекарственные формы с целью облегчения доставки активных соединений в целевой участок организма хозяина или больного. Специалист также сможет использовать благоприятные фармакокинетические параметры пролекарственных форм при применении в доставке соединений в соответствии с настоящим изобретением в целевой участок организма хозяина или больного для максимизации предполагаемого эффекта соединения.
Количество соединения 2, включенного в терапевтически активные составы в соответствии с настоящим изобретением, представляет собой количество, эффективное для достижения желаемого результата в соответствии с настоящим изобретением, например, для лечения инфекции HCV, снижения вероятности возникновения инфекции HCV или ингибирования, снижения и/или устранения HCV или его вторичных эффектов, в том числе болезненных состояний, состояний и/или осложнений, которые являются вторичными по отношению к инфекции HCV. Как правило, терапевтически эффективное количество соединения в соответствии с настоящим изобретением в фармацевтической лекарственной форме обычно варьирует от приблизительно 0,001 мг/кг до приблизительно 100 мг/кг в сутки или больше, чаще, от чуть менее приблизительно 0,1 мг/кг до более чем приблизительно 25 мг/кг массы тела больного в сутки или существенно больше, в зависимости от используемого соединения, подлежащих лечению состояния или инфекции и пути введения. Соединение 2 часто вводят в количествах, варьирующих от приблизительно 0,1 мг/кг до приблизительно 15 мг/кг массы тела больного в сутки, в зависимости от фармакокинетических показателей средства у больного. Данный диапазон дозировки, как правило, обеспечивает эффективные уровни концентраций активного соединения в крови, которые могут варьировать от приблизительно 0,001 до приблизительно 100, от приблизительно 0,05 до приблизительно 100 микрограммов/см3 крови у больного.
Как правило, для лечения, предупреждения или замедления начала этих инфекций и/или для снижения вероятности инфекции вируса HCV или вторичного болезненного состояния, состояния или осложнения инфекции HCV соединение 2 будут вводить в твердой лекарственной форме в количестве, варьирующем от приблизительно 250 микрограммов до приблизительно 800 миллиграммов или больше, по меньшей мере один раз в сутки, например, по меньшей мере приблизительно 5, 10, 20, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750 или 800 миллиграммов или больше один раз, два раза, три раза или до четырех раз в сутки в соответствии с указанием медицинского работника. Соединение 2 часто вводится перорально, но может быть введено парентерально, местным путем или в форме суппозитория, а также интраназально, в виде назального распыления, или иным описываемым в настоящем документе способом. Чаще соединение 2 может быть введено таблеткой, капсулой, инъекцией, внутривенным составом, суспензией, жидкостью, эмульсией, имплантом, частицей, сферой, кремом, мазью, суппозиторием, ингаляционной формой, чрескожной формой, буккально, подъязычно, местным путем, гелем, через слизистую и т.п.
Если лекарственная форма в настоящем документе относится к миллиграммовой массовой дозе, то она относится к количеству соединения 2 (т.е. к массе гемисульфатной соли), если не указано иное.
Согласно некоторым вариантам осуществления фармацевтическая композиция находится в лекарственной форме, которая содержит от приблизительно 1 мг до приблизительно 2000 мг, от приблизительно 10 мг до приблизительно 1000 мг, от приблизительно 100 мг до приблизительно 800 мг, от приблизительно 200 мг до приблизительно 600 мг, от приблизительно 300 мг до приблизительно 500 мг или от приблизительно 400 мг до приблизительно 450 мг соединения 2 в единичной лекарственной форме. Согласно некоторым вариантам осуществления фармацевтическая композиция находится в лекарственной форме, например в твердой лекарственной форме, которая содержит до приблизительно 10, приблизительно 50, приблизительно 100, приблизительно 125, приблизительно 150, приблизительно 175, приблизительно 200, приблизительно 225, приблизительно 250, приблизительно 275, приблизительно 300, приблизительно 325, приблизительно 350, приблизительно 375, приблизительно 400, приблизительно 425, приблизительно 450, приблизительно 475, приблизительно 500, приблизительно 525, приблизительно 550, приблизительно 575, приблизительно 600, приблизительно 625, приблизительно 650, приблизительно 675, приблизительно 700, приблизительно 725, приблизительно 750, приблизительно 775, приблизительно 800, приблизительно 825, приблизительно 850, приблизительно 875, приблизительно 900, приблизительно 925, приблизительно 950, приблизительно 975 или приблизительно 1000 мг или больше соединения 2 в единичной лекарственной форме. Согласно одному варианту осуществления соединение 2 вводят в лекарственной форме, которая доставляет по меньшей мере приблизительно 300 мг. Согласно одному варианту осуществления соединение 2 вводят в лекарственной форме, которая доставляет по меньшей мере приблизительно 400 мг. Согласно одному варианту осуществления соединение 2 вводят в лекарственной форме, которая доставляет по меньшей мере приблизительно 500 мг. Согласно одному варианту осуществления соединение 2 вводят в лекарственной форме, которая доставляет по меньшей мере приблизительно 600 мг. Согласно одному варианту осуществления соединение 2 вводят в лекарственной форме, которая доставляет по меньшей мере приблизительно 700 мг. Согласно одному варианту осуществления соединение 2 вводят в лекарственной форме, которая доставляет по меньшей мере приблизительно 800 мг. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки сроком до 12 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки сроком до 10 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки сроком до 8 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки сроком до 6 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки сроком до 4 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки в течение по меньшей мере 4 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки в течение по меньшей мере 6 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки в течение по меньшей мере 8 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки в течение по меньшей мере 10 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере один раз в сутки в течение по меньшей мере 12 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере через сутки сроком до 12 недель, до 10 недель, до 8 недель, до 6 недель или до 4 недель. Согласно некоторым вариантам осуществления соединение 2 вводят по меньшей мере через сутки в течение по меньшей мере 4 недель, по меньшей мере 6 недель, по меньшей мере 8 недель, по меньшей мере 10 недель или по меньшей мере 12 недель. Согласно одному варианту осуществления по меньшей мере приблизительно 600 мг соединения 2 вводят по меньшей мере один раз в сутки сроком до 6 недель. Согласно одному варианту осуществления по меньшей мере приблизительно 500 мг соединения 2 вводят по меньшей мере один раз в сутки сроком до 6 недель. Согласно одному варианту осуществления по меньшей мере приблизительно 400 мг соединения 2 вводят по меньшей мере один раз в сутки сроком до 6 недель. Согласно одному варианту осуществления по меньшей мере 300 мг соединения 2 вводят по меньшей мере один раз в сутки сроком до 6 недель. Согласно одному варианту осуществления по меньшей мере 200 мг соединения 2 вводят по меньшей мере один раз в сутки сроком до 6 недель. Согласно одному варианту осуществления по меньшей мере 100 мг соединения 2 вводят по меньшей мере один раз в сутки сроком до 6 недель.
Метаболит 1-6 представляет собой активный трифосфат соединения 2, но метаболит 1-6 не измеряется в плазме. Заменителем метаболита 1-6 является метаболит 1-7. Метаболит 1-7 является нуклеозидным метаболитом, измеряемым в плазме, и, следовательно, является показателем внутриклеточных концентраций метаболита 1-6. Для максимальной активности против вируса HCV лекарственная форма соединения 2 должна достигать равновесной остаточной концентрации (C24,ss) метаболита 1-7, превышающей значение ЕС95 соединения 2. Как показано на фиг. 24, ЕС95 соединения 1 против клинических изолятов GT1, GT2, GT3 и GT4 составляет менее 25 нг/мл (значения ЕС95 соединения 1 и ЕС95 соединения 2 являются одинаковыми). Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7, составляющей от приблизительно 15 до 75 нг/мл. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7, составляющей от приблизительно 20 до 60 нг/мл. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7, составляющей от приблизительно 30 до 60 нг/мл. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7, составляющей от приблизительно 20 до 50 нг/мл. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7, составляющей от приблизительно 30 до 50 нг/мл. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7, составляющей от приблизительно 20 до 45 нг/мл. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7, составляющей от приблизительно 20 до 30 нг/мл. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7, составляющей от приблизительно 20 до 35 нг/мл. Согласно одному варианту осуществления доставляется лекарственная форма соединения 2, которая достигает равновесной остаточной концентрации (C24,ss) метаболита 1-7, составляющей от приблизительно 20 до 25 нг/мл. Аппроксимативные лекарственные формы составляют ±10% от равновесной остаточной концентрации.
Согласно одному варианту осуществления соединение 2 вводят дозой в количестве, которое достигает AUC (площади под кривой) метаболита 1-7 от приблизительно 1200 до 3000 нг/мл. Согласно одному варианту осуществления соединение 2 вводят дозой в количестве, которое достигает AUC метаболита 1-7 от приблизительно 1500 до 3000 нг/мл. Согласно одному варианту осуществления соединение 2 вводят дозой в количестве, которое достигает AUC метаболита 1-7 от приблизительно 1800 до 3000 нг/мл. Согласно одному варианту осуществления соединение 2 вводят дозой в количестве, которое достигает AUC метаболита 1-7 от приблизительно 2100 до 3000 нг/мл. Согласно предпочтительному варианту осуществления соединение 2 вводят дозой в количестве, которое достигает AUC метаболита 1-7 приблизительно 2200 нг*час/мл. Аппроксимативные лекарственные формы составляют ±10% от AUC.
В случае совместного введения соединения 2 в комбинации с другим соединением против HCV, иным образом описываемым в настоящем документе, количество соединения 2 в соответствии с настоящим изобретением, подлежащее введению, варьирует от приблизительно 0,01 мг/кг массы тела больного до приблизительно 800 мг/кг массы тела больного или больше или значительно больше, в зависимости от второго средства, подлежащего совместному введению, и его эффективности против вируса, состояния больного и тяжести заболевания или инфекции, подлежащих лечению, а также от пути введения. Другое средство против HCV может быть введено, например, в количествах, варьирующих от приблизительно 0,01 мг/кг до приблизительно 800 мг/кг. Примерами размеров дозировки второго активного средства являются количества, варьирующие от приблизительно 250 микрограммов до приблизительно 750 мг или больше по меньшей мере один раз в сутки, например, по меньшей мере приблизительно 5, 10, 20, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 600, 700 или 800 миллиграммов или больше, до четырех раз в сутки. Согласно некоторым предпочтительным вариантам осуществления соединение 2 можно часто вводить в количестве, варьирующем от приблизительно 0,5 мг/кг до приблизительно 50 мг/кг или больше (обычно до приблизительно 100 мг/кг), как правило, в зависимости от фармакокинетических показателей двух средств у больного. Такие диапазоны дозировки, как правило, обеспечивают эффективные уровни концентраций активного соединения в крови у больного.
Для целей настоящего изобретения профилактическое или превентивное эффективное количество композиций в соответствии с настоящим изобретением попадает в тот же диапазон концентраций, что изложен выше для терапевтически эффективного количества, и обычно является таким же, как и терапевтически эффективное количество.
Введение соединения 2 может варьировать от непрерывного (внутривенного капельного введения) до нескольких пероральных или интраназальных введений в сутки (например, Q.I.D.) или чрескожного введения и может предусматривать пероральное, местное, парентеральное, внутримышечное, внутривенное, подкожное, чрескожное (которое может предусматривать усиливающее проницаемость средство), буккальное введение и введение с помощью суппозитория, среди прочих путей введения. Покрытые энтеросолюбильной оболочкой пероральные таблетки также могут быть использованы для усиления биодоступности соединений для перорального пути введения. Наиболее эффективная лекарственная форма будет зависеть от биодоступности/фармакокинетических показателей конкретного выбранного средства, а также от тяжести заболевания у больного. Пероральные лекарственные формы являются особенно предпочтительными из-за простоты введения и предполагаемого благоприятного соблюдения больным режима лечения.
Для получения фармацевтических композиций в соответствии с настоящим изобретением терапевтически эффективное количество соединения 2 в соответствии с настоящим изобретением зачастую тщательно смешивают с фармацевтически приемлемым носителем согласно традиционным методикам фармацевтического составления для получения дозы. Носитель может принимать разнообразные формы в зависимости от формы препарата, желаемой для введения, например, пероральной или парентеральной. При получении фармацевтических композиций в пероральной лекарственной форме может быть использована любая из обычных фармацевтических сред. Таким образом, для жидких пероральных препаратов, таких как суспензии, эликсиры и растворы, могут быть использованы приемлемые носители и добавки, в том числе вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и т.п. Для твердых пероральных препаратов, таких как порошки, таблетки, капсулы, и для твердых препаратов, таких как суппозитории, могут быть использованы приемлемые носители и добавки, в том числе крахмалы, сахарные носители, такие как декстроза, manifold, лактоза, а также родственные носители, разбавители, гранулирующие средства, смазки, связующие, разрыхлители и т.п. При необходимости таблетки или капсулы могут быть покрыты энтеросолюбильным покрытием или обеспечены с замедленным высвобождением с помощью стандартных методик. Применение таких лекарственных форм может существенно улучшить биодоступность соединений у больного.
В парентеральных составах носитель, как правило, будет содержать стерильную воду или водный раствор натрия хлорида, хотя также могут быть включены другие ингредиенты, в том числе те, которые обеспечивают дисперсию. Конечно, если должна быть использована стерильная вода при сохранении ее стерильности, композиции и носители также должны быть стерилизованными. Также могут быть получены инъекционные суспензии, и в этом случае могут быть использованы подходящие жидкие носители, суспендирующие средства и т.п.
Липосомные суспензии (в том числе липосомы, нацеленные на вирусные антигены) также могут быть получены традиционными способами получения фармацевтически приемлемых носителей. Они могут подходить для доставки свободных нуклеозидов, ацил/алкилнуклеозидов или пролекарственных форм фосфатов сложных эфиров нуклеозидных соединений в соответствии с настоящим изобретением.
Согласно типичным вариантам осуществления настоящего изобретения соединение 2 и описываемые композиции используют для лечения, предупреждения или задержки развития инфекции HCV или вторичного связанного с HCV болезненного состояния, состояния или осложнения.
VII. Комбинированная и чередующаяся терапия
Хорошо известно, что устойчивые к лекарственному средству варианты вирусов могут появляться после длительного лечения противовирусным средством. Иногда устойчивость к лекарственному средству возникает из-за мутации гена, который кодирует фермент, используемый в вирусной репликации. Эффективность лекарственного средства против инфекции HCV может быть пролонгирована, усилена или восстановлена путем введения соединения в комбинации или при чередовании с другим, а возможно даже с двумя или тремя другими противовирусными соединениями, которые индуцируют другую мутацию или действуют через другой путь, по сравнению с основным лекарственным средством. В качестве альтернативы, фармакокинетические показатели, биораспределение, период полувыведения или другой параметр лекарственного средства могут быть изменены такой комбинированной терапией (которая может включать в себя чередующуюся терапию, если считается совместимой). Поскольку раскрываемое соединение 2 является ингибиторами NS5B полимеразы, может быть полезно вводить соединение хозяину в комбинации, например, с
(1) ингибитором протеазы, таким как ингибитор NS3/4A протеазы;
(2) ингибитором NS5A;
(3) другим ингибитором NS5B полимеразы;
(4) несубстратным ингибитором NS5B;
(5) интерфероном альфа-2а, который может быть пегилированным или иным образом модифицированным, и/или рибавирином;
(6) не субстратным ингибитором;
(7) ингибитором геликазы;
(8) антисмысловым олигодезоксинуклеотидом (S-ODN);
(9) аптамером;
(10) устойчивым к нуклеазе рибозимом;
(11) iRNA, в том числе microRNA и SiRNA;
(12) антителом, частичным антителом или доменным антителом против вируса
или
(13) вирусным антигеном или частичным антигеном, который индуцирует образование антител у хозяина.
Неограничивающими примерами средств против HCV, которые можно вводить в комбинации с соединением 2 в соответствии с настоящим изобретением отдельно или с несколькими лекарственными средствами из данного перечня, являются
(i) ингибиторы протеазы, такие как телапревир (mcivek®), боцепревир (Victrelis™), симепревир (Olysio™), паритапревир (АВТ-450), глекапревир (АВТ-493), ритонавир (Norvir), АСН-2684, AZD-7295, BMS-791325, данопревир, филибувир, GS-9256, GS-9451, MK-5172, сетробувир, совапревир, тегобувир, VX-135, VX-222 и ALS-220;
(ii) ингибитор NS5A, такой как АСН-2928, АСН-3102, IDX-719, даклатасвир, ледиспасвир, велпатасвир (Epclusa), элбасвир (MK-8742), гразопревир (MK-5172) и омбитасвир (АВТ-267);
(iii) ингибиторы NS5B, такие как AZD-7295, клемизол, дасабувир (Exviera), ITX-5061, PPI-461, PPI-688, софосбувир (Sovaldi®), MK-3682 и мерицитабин;
(iv) ингибиторы NS5B, такие как АВТ-333 и МВХ-700;
(v) антитело, такое как GS-6624;
(vi) комбинация лекарственных средств, таких как Harvoni (ледипасвир/софосбувир); Viekira Pak (омбитасвир/паритапревир/ритонавир/дасабувир); Viekirax (омбитасвир/паритапревир/ритонавир); G/P (паритапревир и глекапревир); Technivie (омбитасвир/паритапревир/ритонавир) и Epclusa (софосбувир/велпатасвир) и Zepatier (элбасвир и гразопревир).
Если соединение 2 вводят для лечения запущенного вирусного гепатита С, приводящего к раку печени или циррозу, согласно одному варианту осуществления соединение может быть введено в комбинации или при чередовании с другим лекарственным средством, которое, как правило, используют для лечения гепатоклеточной карциномы (НСС), например, как описано Andrew Zhu в "New Agents on the Horizon in Hepatocellular Carcinoma" Therapeutic Advances in Medical Oncology, V 5(1), January 2013, 41-50. Примеры приемлемых соединений для комбинированной терапии, если хозяин страдает НСС или рискует заболеть НСС, включают в себя противоангиогенные средства, сунитиниб, бриваниб, линифаниб, рамуцирумаб, бевацизумаб, цедираниб, пазопаниб, TSU-68, ленватиниб, антитела против EGFR, ингибиторы mTor, ингибиторы MEK и ингибиторы гистондеацетилазы.
Примеры
Общие способы
Спектры 1H, 19F и 31Р ЯМР регистрировали при 400 МГц на спектрометре с преобразованием Фурье производства  Спектры получали в DMSO-d6, если не указано иное. Спиновые мультиплетности обозначали символами s (синглет), d (дублет), t (триплет), m (мультиплет) и br (уширенный). Константы взаимодействия (J) выражали в Гц. Реакции, как правило, проводили в атмосфере осушенного азота с использованием безводных растворителей производства Sigma-Aldrich. Все обычные химические вещества приобретали из коммерческих источников.
Спектры получали в DMSO-d6, если не указано иное. Спиновые мультиплетности обозначали символами s (синглет), d (дублет), t (триплет), m (мультиплет) и br (уширенный). Константы взаимодействия (J) выражали в Гц. Реакции, как правило, проводили в атмосфере осушенного азота с использованием безводных растворителей производства Sigma-Aldrich. Все обычные химические вещества приобретали из коммерческих источников.
В примерах используют следующие сокращения.
AUC: площадь под кривой.
С24: концентрация лекарственного средства в плазме через 24 часа.
C24,ss: концентрация через 24 часа после введения дозы в равновесном состоянии.
Cmax: максимальная концентрация лекарственного средства, достигнутая в плазме.
DCM: дихлорметан.
EtOAc: этилацетат.
EtOH: этанол.
HPLC: жидкостная хроматография высокого давления.
NaOH: гидроксид натрия.
Na2SO4: сульфат натрия (безводный).
MeCN: ацетонитрил.
MeNH2: метиламин.
МеОН: метанол.
Na2SO4: сульфат натрия.
NaHCO3: бикарбонат натрия.
NH4Cl: хлорид аммония.
NH4OH: гидроксид аммония.
РЕ: петролейный эфир.
Ph3P: трифенилфосфин.
RH: относительная влажность.
Силикагель (230-400 меш, сорбент).
t-BuMgC1: трет-бутилмагния хлорид.
Tmax: время, за которое достигается Cmax.
THF: тетрагидрофуран (THF), безводный.
TP: трифосфат.
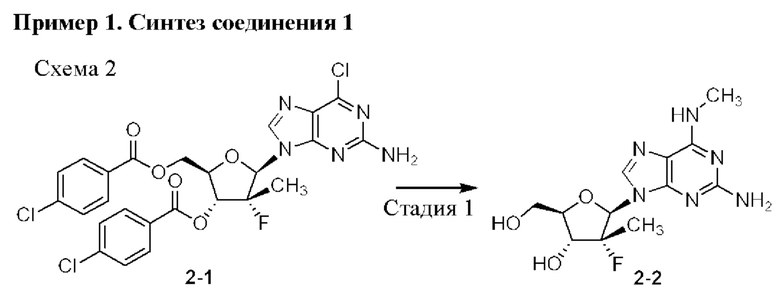
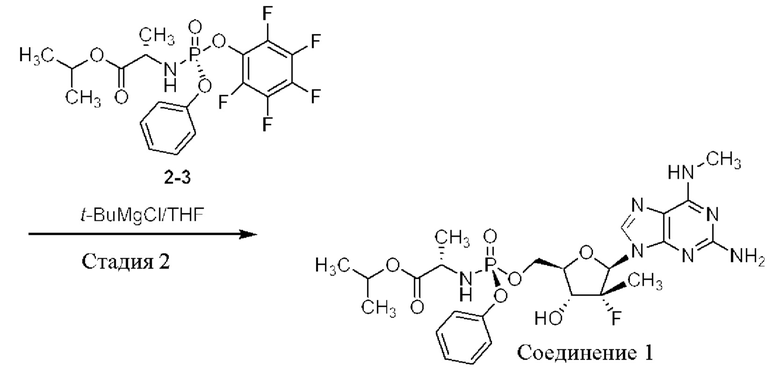
Стадия 1. Синтез (2R,3R,4R,5R)-5-(2-амино-6-(метиламино)-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)-4-метилтетрагидрофуран-3-ола (2-2)
В 50-л колбу загружали метанол (30 л) и взбалтывали при 10±5°С. NH2CH3 (3,95 кг) медленно вентилировали в реактор при 10±5°С. Соединение 2-1 (3,77 кг) добавляли партиями при 20±5°С и взбалтывали в течение 1 часа с получением прозрачного раствора. Реакционную смесь взбалтывали еще 6-8 часов, в этот момент HPLC показывала, что промежуточное соединение составило менее 0,1% раствора. Реактор загружали твердым NaOH (254 г), взбалтывали в течение 30 минут и концентрировали при 50±5°С (степень вакуума -0,095). В полученный в результате остаток загружали EtOH (40 л) и повторно суспендировали в течение 1 часа при 60°С. Смесь затем фильтровали через целит и осадок на фильтре повторно суспендировали с EtOH (15 л) в течение 1 часа при 60°С. Фильтрат еще раз фильтровали, объединяли с фильтратом из предыдущей фильтрации, а затем концентрировали при 50±5°С (степень вакуума -0,095). Осаждалось большое количество твердого вещества. EtOAc (6 л) добавляли к твердому остатку и смесь концентрировали при 50±5°С (степень вакуума -0,095). DCM затем добавляли к остатку и смесь повторно суспендировали с обратным холодильником в течение 1 часа, охлаждали до комнатной температуры, фильтровали и сушили при 50±5°С в вакуумной печи с получением соединения 2-2 в виде грязно-белого твердого вещества (1,89 кг, 95,3%, чистота 99,2%).
Аналитический способ для соединения 2-2. Чистоту соединения 2-2 (15 мг) достигали с использованием HPLC системы Agilent 1100 с колонкой Agilent Poroshell 120 ЕС-С18 4,6*150 мм 4 микрон при следующих условиях: скорость потока 1 мл/минута, считывание при 254 нм, температура колонки 30°С, объем введения 15 мкл и время выполнения 31 минута. Образец растворяли в ацетонитриле - воде (20:80) (объем/объем). Градиентный метод показан ниже.
Стадия 2: Синтез изопропил-((S)-(((2R,3R,4R,5R)-5-(2-амино-6-(метиламино)-9H-пурин-9-ил)-4-фтор-3-гидрокси-4-метилтетрагидрофуран-2-ил)метокси)(фенокси)фосфорил)-L-аланината (соединения 1)
Соединение 2-2 и соединение 2-3 (изопропил-(перфторфенокси)(фенокси)фосфорил)-1,-аланинат) растворяли в THF (1 л) и взбалтывали под азотом. Затем суспензию охлаждали до температуры ниже -5°С и медленно добавляли 1,7 М раствор t-BuMgCl (384 мл) за 1,5 часа, при этом поддерживали температуру 5-10°С. В суспензию добавляли раствор NH4Cl (2 л) и воду (8 л) при комнатной температуре, а затем DCM. Смесь взбалтывали в течение 5 минут перед добавлением 5% водного раствора K2CO3 (10 л) и смесь взбалтывали еще 5 минут перед фильтрованием через диатомит (500 г). Диатомит промывали с помощью DCM и фильтрат отделяли. Органическую фазу промывали с помощью 5% водного раствора K2CO3 (10 л × 2), солевого раствора (10 л × 3) и сушили над Na2SO4 (500 г) в течение приблизительно 1 часа. При этом данный полный процесс повторяли 7 раз параллельно и 8 партий объединяли. Органические фазы фильтровали и концентрировали при 45±5°С (степень вакуума 0,09 МПа). Добавляли EtOAc и смесь взбалтывали в течение 1 часа при 60°С, а затем при комнатной температуре в течение 18 часов. Смесь затем фильтровали и промывали с помощью EtOAc (2 л) с получением неочищенного соединения 1. Неочищенный материал растворяли в DCM (12 л), добавляли гептан (18 л) при 10-20°С и обеспечивали перемешивание смеси в течение 30 минут при этой температуре. Смесь фильтровали, промывали с помощью гептана (5 л) и сушили при 50±5°C с получением чистого соединения 1 (1650 г, 60%).
Аналитический способ для соединения 1. Чистоту соединения 1 (25 мг) достигали с использованием HPLC системы Agilent 1100 с колонкой Waters XTerra Phenyl 5 мкм 4,6*250 мм при следующих условиях: скорость потока 1 мл/минута, считывание при 254 нм, температура колонки 30°С, объем введения 15 мкл и время выполнения 25 минут. Образец растворяли в ацетонитриле - воде (50:50) (объем/объем). Градиентный метод показан ниже.

Пример 2. Характеристика аморфного и кристаллического соединения 1
Аморфное соединение 1 и кристаллическое соединение 1 сначала анализировали с помощью XRPD, 1Н ЯМР и HPLC. Паттерны XRPD для обоих соединений показаны на фиг. 1А, а выявленные с помощью HPLC следовые количества для определения чистоты показаны на фиг. 1В и 2А, соответсвенно. В таблице 1 приводится перечень пиковых значений от XRPD кристаллического соединения 1, а в таблице 2 приводится перечень относительного времени удержания (RTT) из выявленных с помощью HPLC следовых количеств. Чистота аморфного соединения 1 составляла 98,61%, а чистота кристаллического соединения 1 составляла 99,11%. Оба соединения представляли собой белое твердое вещество. На фиг. 2В представлены графики TGA и DSC для кристаллического соединения 1. Для кристаллического соединения 1 эндотерму наблюдали при 88,6°С, а также наблюдали потерю 7,8% массы от 80 до 110°С.
Образец соединения 1 перекристаллизовывали из EtOAc/гексана и рисовали с помощью ORTEP. Абсолютную структуру соединения 1 подтверждали путем перекристаллизации одного кристалла. На фиг. 3 представлен полученный с помощью ORTEP рисунок соединения 1. Кристаллографические данные и данные измерений показаны в таблице 3. Ниже показана абсолютная стереохимия соединения 1 на основании рентгеновской кристаллографии:
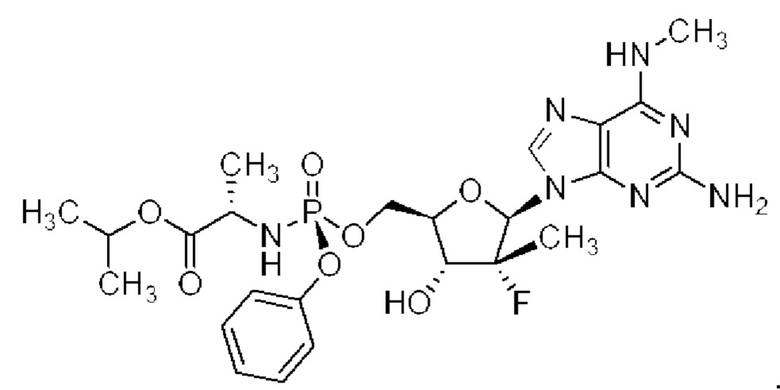
Данные DSC собирали с помощью ТА Instruments Q2000, оснащенного 50-позиционным автоматическим пробоотборником. Калибровку по теплоемкости выполняли с использованием сапфира, а калибровку по энергии и температуре выполняли с использованием сертифицированного индия. Как правило, приблизительно 3 мг каждого образца в алюминиевой чаше с игольчатым отверстием нагревали при 10°С/минута от 25°С до 200°С. Над образцом поддерживали продувку сухим азотом при 50 мл/минута. Программным обеспечением для контроля за прибором служили Advantage для Q Series v2.8.0.394 и Thermal Advantage v5.5.3, и данные анализировали с использованием Universal Analysis v4.5A.
Данные TGA собирали на ТА Instruments Q500 TGA, оснащенном 16-позиционным автоматическим пробоотборником. Прибор калибровали по температуре с использованием сертифицированного Alumel и никеля. Как правило, 5-10 мг каждого образца загружали в предварительно тарированную алюминиевую чашу DSC и нагревали при 10°С/минута от окружающей температуры до 350°С. Над образцом поддерживали продувку азотом при 60 мл/минута. Программным обеспечением для контроля за прибором служили Advantage для Q Series v2.5.0.256 и Thermal Advantage v5.5.3, и данные анализировали с использованием Universal Analysis v4.5.
Аморфное соединение 1 (1-1)
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,01-1,15 (m, 9Н), 1,21 (d, J=7,20 Гц, 3H), 2,75-3,08 (m, 3Н), 3,71-3,87 (m, 1Н), 4,02-4,13 (m, 1Н), 4,22-4,53 (m, 3Н), 4,81 (s, 1Н), 5,69-5,86 (m, 1Н), 6,04 (br d, J=19,33 Гц, 4H), 7,12-7,27 (m, 3H), 7,27-7,44 (m, 3H), 7,81 (s, 1H).
Кристаллическое соединение 1 (1-2)
1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,97-1,16 (m, 16Н), 1,21 (d, J=7,07 Гц, 3H), 2,87 (br s, 3H), 3,08 (s, 2H), 3,79 (br d, J=7,07 Гц, 1H), 4,08 (br d, J=7,58 Гц, 1H), 4,17-4,55 (m, 3H), 4,81 (quin, J=6,25 Гц, 1H), 5,78 (br s, 1 H), 5,91-6,15 (m, 4H), 7,10-7,26 (m, 3H), 7,26-7,44 (m, 3H), 7,81 (s, 1H).
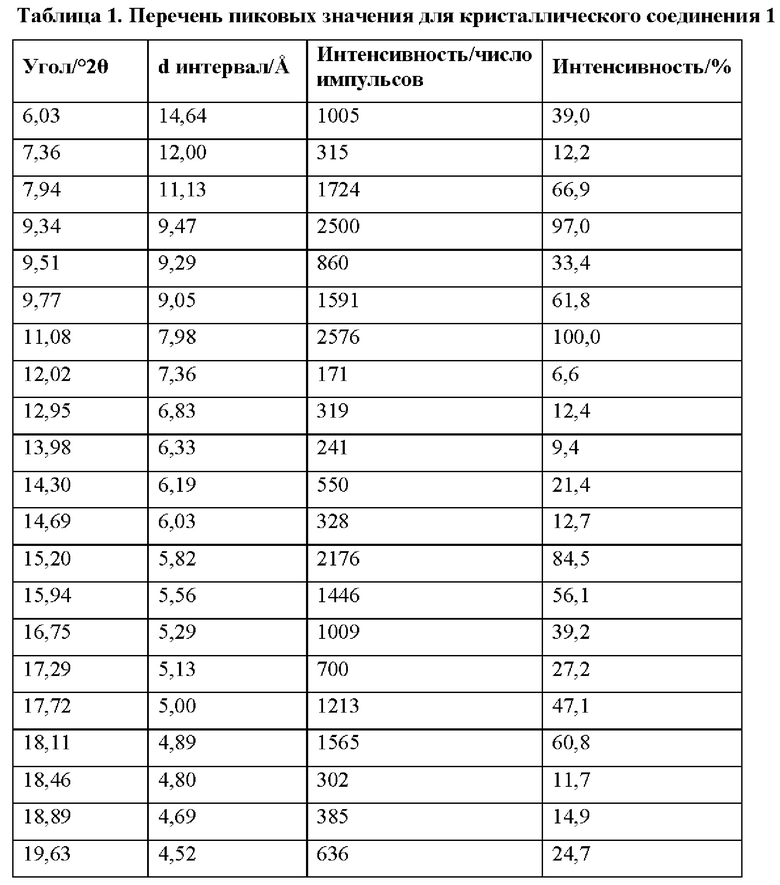
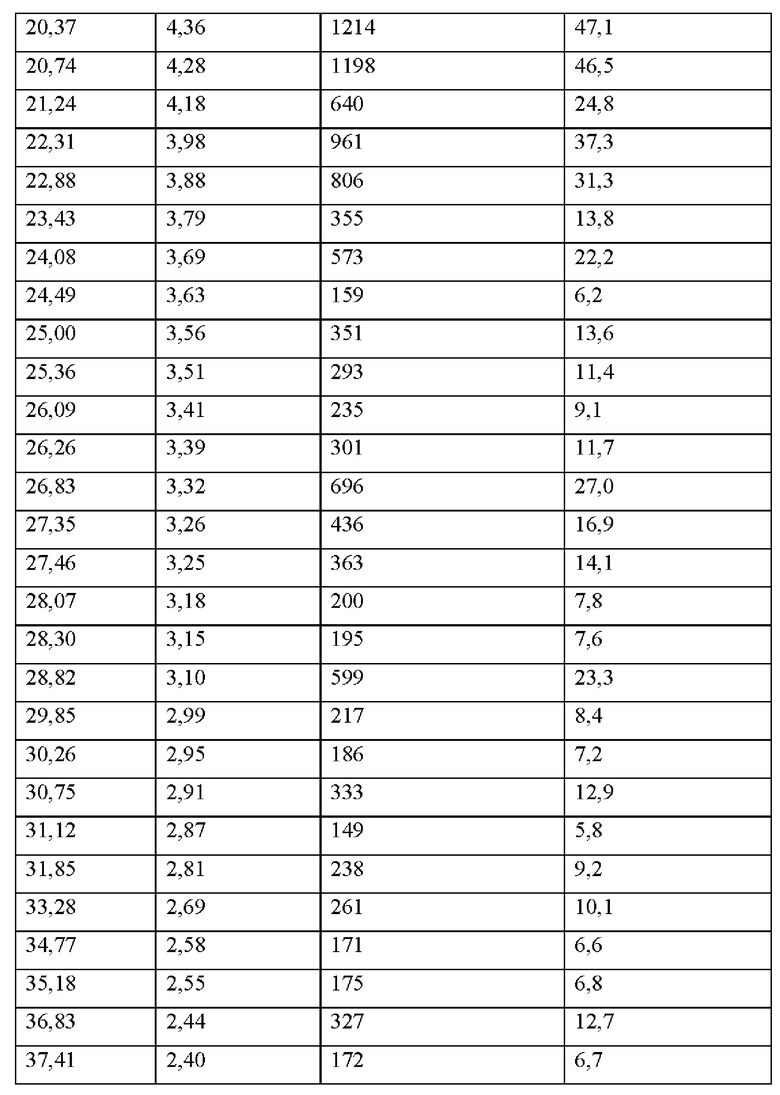
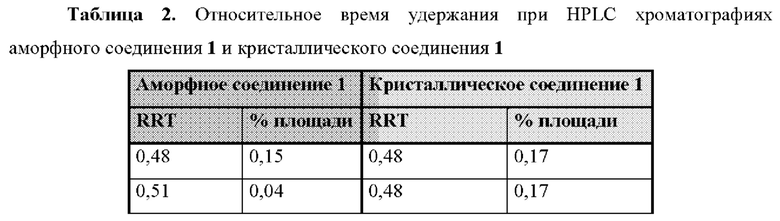
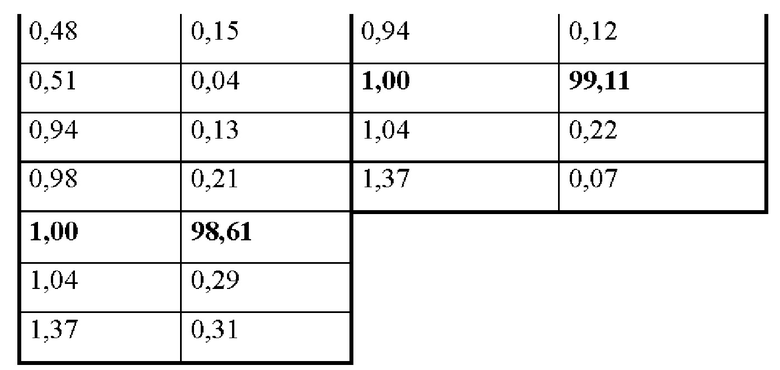
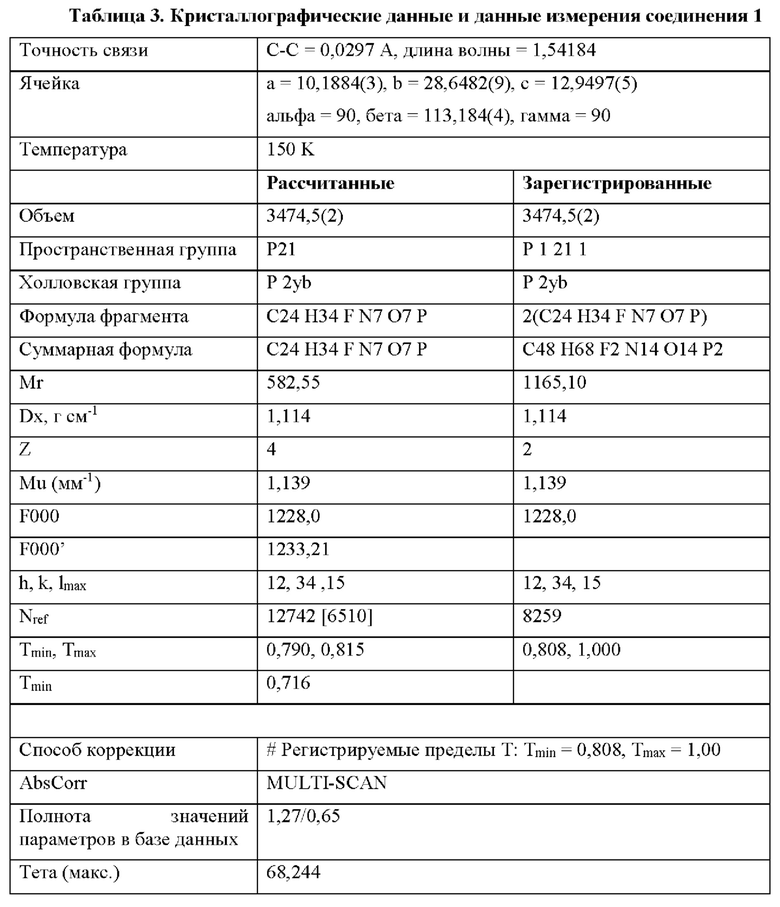

После этой изначальной характеристики следовало хранение при 25°С/60% относительной влажности (RH) в течение 14 суток с анализом с помощью HPLC и XRPD через 7 и 14 суток. На фиг. 4А показана XRPD после 14 суток при 25°С/60% (RH). Аморфное соединение 1 (образец 1-1) оставалось слабокристаллическим, тогда как кристаллическое соединение 1 (образец 1-2) сохраняло свою кристалличность, но оба соединения были стабильными после 14 суток при 25°С/60% (RH).
Пример 3. Состав оксалатной соли соединения 4
Сначала получали оксалатную соль соединения 1 - соединение 4 - путем смешивания оксалатной соли с растворителем (5 объемов, 100 мкл) и обеспечивали выпаривание любого раствора при комнатной температуре. Любую суспензию выдерживали (комнатная температура - 50°С) в течение 3 часов и оценивали кристалличность.
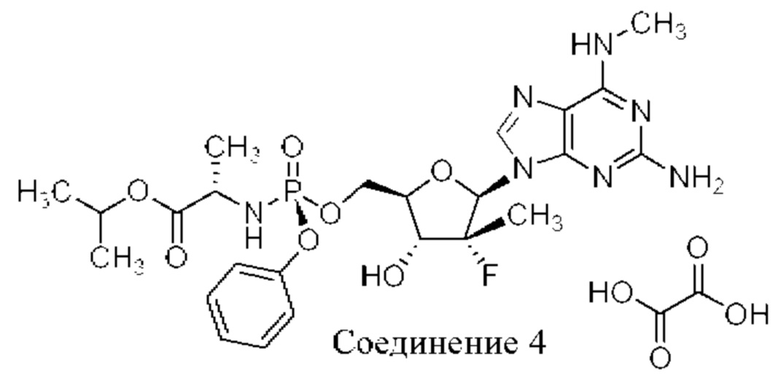
В таблице 4 показаны различные растворители, используемые при получении соединения 4. Все растворители, за исключением двух (циклогексана и н-гептана), давали кристаллические продукты. Несмотря на высокую кристалличность и растворимость соединения 4 оксалатные соли не являются приемлемыми для клинической разработки из-за потенциального формирования камней в почках, и изучали другие соли соединения 1.

Пример 4. Солевые соединения аморфного соединения 1
Поскольку оксалатное соединение 4 (пример 3) не могло быть передано на клинические испытания из-за его способности формировать камни в почках, получали аморфные соли соединения 1 с противоионами, приведенными в таблице 5. Соединение 1 растворяли в трет-бутаноле (20 объемов, 6 мл) и раствор обрабатывали кислотными противоионами (1 эквивалент для каждого образца, за исключением образца 1-9, который имел 0,5 эквивалента сульфата). Затем образцы замораживали при удалении растворителя лиофилизацией. Остаточное твердое вещество в образцах 1-4, 1-5, 1-6, 1-7, 1-8 и 1-9 сначала анализировали с помощью XRPD и HPLC.
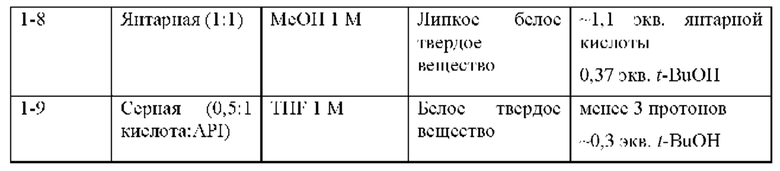
1Н ЯМР спектры получали для всех образцов.
Образец 1-4, соль HCl (1:1)
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 0,93-1,39 (m, 16 Н), 2,97 (br s, 2 H), 3,70-3,88 (m, 1 H), 4,10 (br s, 1 H), 4,18-4,49 (m, 3 H), 4,70-4,88 (m, 1 H), 5,71-5,94 (m, 1 H), 6,07 (br d, J=19,07 Гц, 2 H), 7,14-7,27 (m, 3 H), 7,29-7,44 (m, 2 H), 7,83-8,19 (m, 1 H).
Образец 1-5, соль серной кислоты (1:1)
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 0,97-1,38 (m, 15 Н), 2,96 (br s, 2 H), 4,06-4,18 (m, 1 H), 4,19-4,49 (m, 3 H), 4,66-4,91 (m, 1 H), 5,70-5,95 (m, 1 H), 5,96-6,16 (m, 2 H), 7,10-7,27 (m, 3 H), 7,30-7,43 (m, 2 H), 7,88-8,19 (m, 1 H).
Образец 1-6, соль фумаровой кислоты (1:1)
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 0,95-1,31 (m, 21 Н), 2,87 (br s, 3 H), 3,79 (br d, J=7,20 Гц, 1 H), 4,01-4,13 (m, 1 H), 4,16-4,23 (m, 1 H), 4,16-4,24 (m, 1 H), 4,20 (s, 1 H), 4,18-4,23 (m, 1 H), 4,24-4,52 (m, 1 H), 4,24-4,52 (m, 1 H), 4,24-4,49 (m, 1 H), 4,72-4,88 (m, 1 H), 5,68-5,86 (m, 1 H), 6,04 (br d, J=19,33 Гц, 4 H), 6,63 (s, 1 H), 6,61-6,66 (m, 1 H), 7,12-7,27 (m, 3 H), 7,27-7,45 (m, 3 H), 7,81 (s, 1 H), 13,16 (br s, 2 H).
Образец 1-7, соль бензойной кислоты (1:1)
1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,96-1,30 (m, 15 H), 2,87 (br s, 3 H), 3,79 (br d, J=7,07 Гц, 1 H), 4,07 (br s, 1 H), 4,20 (s, 1 H), 4,25-4,52 (m, 3 H), 4,81 (s, 1 H), 5.71-5,85 (m, 1 H), 6,04 (br d, J=19,33 Гц, 4 H), 7,08-7,27 (m, 3 H), 7,27-7,43 (m, 3 H), 7,45-7,57 (m, 2 H), 7,63 (s, 1 H), 7,81 (s, 1 H), 7,95 (dd, J=8,27, 1,33 Гц, 2 H), 12,98 (brs, 1 H).
Образен 1-8, соль янтарной кислоты (1:1)
1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,98-1,28 (m, 15 H), 2,42 (s, 5 H), 2,87 (br s, 3 H), 3,57-3,62 (m, 1 H), 3,70-3,86 (m, 1 H), 4,02-4,14 (m, 1 H), 4,20 (s, 1 H), 4,24-4,51 (m, 3 H), 4,70-4,88 (m, 1 H), 5,69-5,86 (m, 1 H), 6,04 (br d, J=19,33 Гц, 4 H), 7,12-7,27 (m, 3 H), 7,27-7,44 (m, 3 H), 7,81 (s, 1 H), 11,95-12,58 (m, 2 H).
Образец 1-9, соль серной кислоты (0,5:1)
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,02-1,31 (m, 15 Н), 2,94 (br s, 3 H), 3,79 (br d, J=7,20 Гц, 2 H), 4,09 (br s, 1 H), 4,22-4,48 (m, 3 H), 4,72-4,90 (m, 1 H), 5,71-5,92 (m, 1 H), 6,07 (br d, J=19,07 Гц, 2 H), 7,12-7,28 (m, 3 H), 7,31-7,44 (m, 2 H), 7,75-8,19 (m, 1 H).
Затем образцы подвергали хранению при 25°С/60% относительной влажности (RH) в течение 14 суток с анализом с помощью HPLC и XRPD через 7 (фиг. 4В) и 14 суток (фиг. 5А). Все полученные соли оставались аморфными, и наблюдения показаны в таблице 6. Выяснили, что моносульфатная (образец 1-5) и сукцинатная соли (образец 1-8) были физически нестабильными и переходили в жидкое состояние или превращались в смолу в ходе исследования. Выяснили, что и фумаратная (образец 1-6), и бензоатная соли (образец 1-7) представляли собой стекловидные твердые вещества. Выяснили, что соль HCl (образец 1-4) сохраняла свои физические свойства. Удивительно, что гемисульфатная соль (образец 1-9) также сохраняла свои физические свойства в виде белого твердого вещества в отличие от моносульфатного соединения (образец 1-5), которое представляло собой липкую смолу. Результаты показаны в таблице 6. Выяснили, что моно-HCl соль (образец 1-4) и гемисульфатная соль (образец 1-9) были физически и химически стабильным после 2 недель хранения при 25°С/60% относительной влажности (RH). Хотя обе соли были стабильными на протяжении двух недель, гемисульфатная соль превосходила соль HCl, поскольку соль HCl была гигроскопичной, что по сравнению с гемисульфатной солью делает ее менее применимой для длительного хранения или применения.
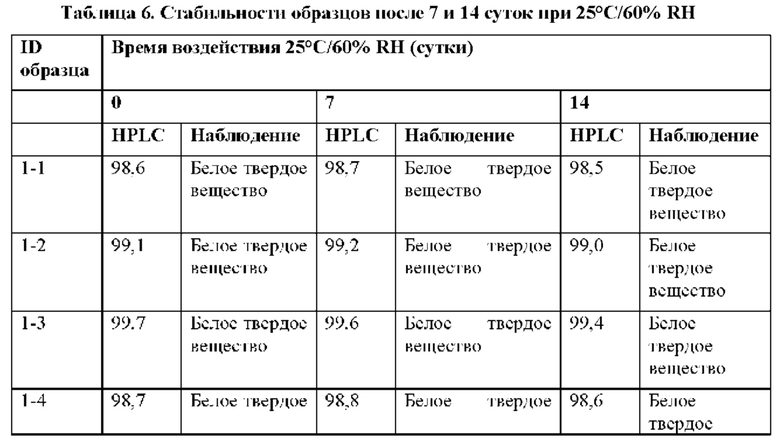
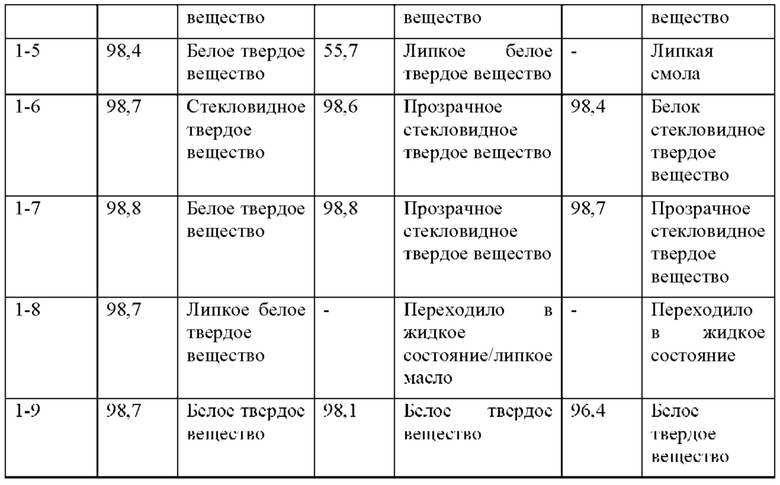
Пример 5. Характеристика аморфного соединения 2
Аморфное соединения 2 сначала анализировали с помощью XRPD, 1Н ЯМР, DSC, TGA и HPLC. Паттерн XRPD для аморфного соединения 2, совмещенный с таковым аморфного соединения 1 и кристаллического соединения 1, показан на фиг. 1А, а паттерн XRPD аморфного соединения 2 отдельно показан на фиг. 5В. В таблице 7 приводится перечень пиковых значения из паттерна XRPD, показанного на фиг. 5В. Выявленное с помощью HPLC следовое количество для определения чистоты показано на фиг. 6А. В таблице 8 приводится перечень относительного времени удержания (RTT) от выявленного с помощью HPLC следового количества, показанного на фиг. 6А. Чистота аморфного соединения 2 составляла 99,68%. На фиг. 6В представлен график TGA и DSC аморфного соединения 2. Экспериментальные подробности для экспериментов TGA и DSC приведены в примере 2.

Аморфное соединение 2
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 0,93-1,29 (m, 13 Н), 2,94 (br s, 3 H), 3,79 (td, J=10,04, 7,07 Гц, 2 H), 4,05-4,19 (m, 1 Н), 4,19-4,50 (m, 3 Н), 4,81 (quin, J=6,25 Гц, 1 H), 5,71-5,94 (m, 1 Н), 5,97-6,16 (m, 2 Н), 7,14-7,28 (m, 3 Н), 7,31-7,44 (m, 2 Н), 7,82-8,09 (m, 1 Н).
Пример 6. Кристаллизация аморфного соединения 2
Поскольку выяснили, что гемисульфатная соль остается твердой после 14-суточного исследования стабильности, как показано в таблице 6, проводили предварительные испытания, исследующие условия кристаллизации, с использованием 11 разных растворителей. Аморфное соединение 2 суспендировали в 5 объемах растворителя при 25°С (образцы 2-1, 2-2, 2-3, 2-4, 2-5, 2-6, 2-7, 2-8, 2-9, 2-10 и 2-11). К этим образцам, которые не были свободно текущими (2-1, 2-2, 2-3, 2-4, 2-5, 2-6, 2-7, 2-8 и 2-10), добавляли дополнительно 5 объемов растворителя. Затем образцы отстаивали при 25-50°С (1°С/минута между температурами и 4 часа при каждой температуре) в течение 6 суток, за исключением образца 2-1, который, как наблюдали, представлял собой прозрачный раствор через 1 сутки, и обеспечивали выпаривание при окружающих условиях. Результаты показаны в таблице 9. Кристаллические паттерны получали в результате при кристаллизации с изобутанолом (образец 2-1), ацетоном (образец 2-2), EtOAc (образец 2-6) и iPrOAc (образец 2-7). Два слабокристаллических образца также идентифицировали при кристаллизации с MEK (образец 2-4) и MIBK (образец 2-5). Паттерны XRPD показаны на фиг. 7А.

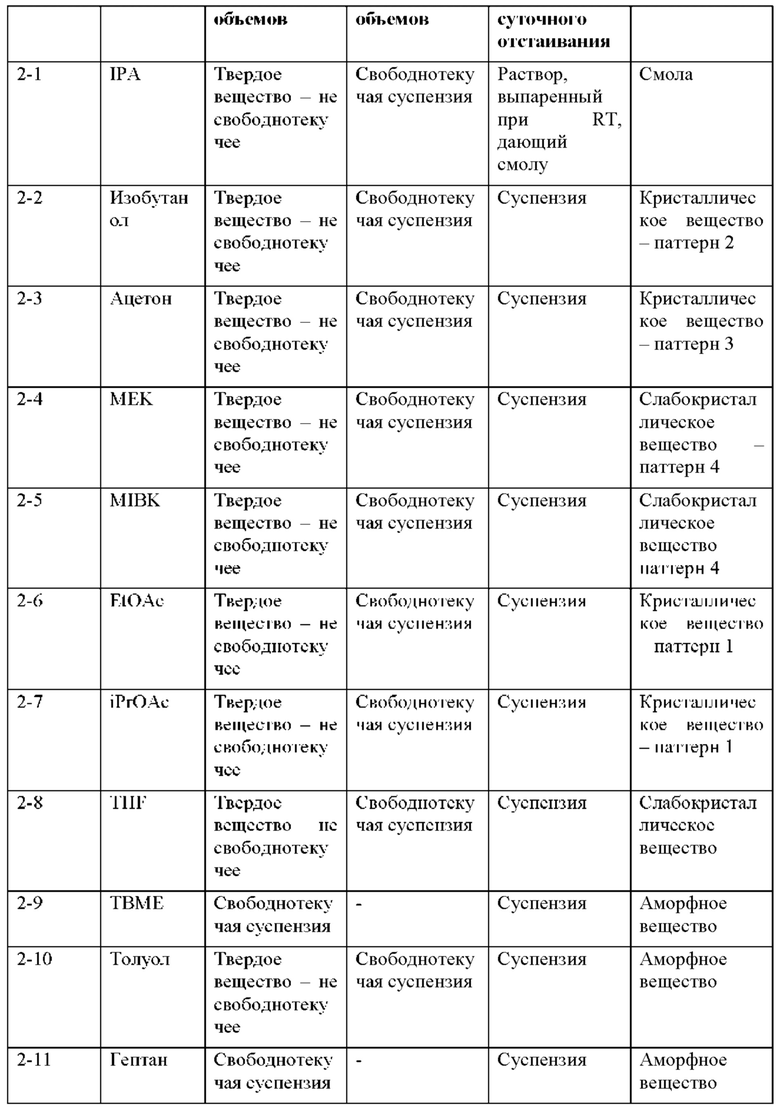
Семь образцов (образцы 2-2, 2-3, 2-4, 2-5, 2-6, 2-7 и 2-8) анализировали с помощью DSC, TGA, 1Н ЯМР и IC (таблица 10, фиг. 8А, фиг. 8В, фиг. 9А, фиг. 9В, фиг. 10А, фиг. 10В, фиг. 11А и фиг. 11В), а также с помощью XRPD после 6 суток хранения при 25°С/60% относительной влажности (RH) (все образцы оставались кристаллическими/слабокристаллическими после состояния равновесия). Все образцы сохраняли примерно половину эквивалента сульфата, но содержали относительно большое количество остаточного растворителя. Наложение рентгеновских дифрактограмм аморфных образцов 2-9, 2-10 и 2-11 показано на фиг. 7В.

1Н ЯМР спектры были получены для всех образцов и приведены ниже.
Образец 2-2
1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,83 (d, J=6,69 Гц, 7 H), 0,99-1,26 (m, 14 Н), 1,61 (dt, J=13,26, 6,63 Гц, 1 H), 3,73-3,87 (m, 2 Н), 4,03-4,18 (m, 1 Н), 4,18-4,51 (m, 4 Н), 4,66-4,92 (m, 1 Н), 4,70-4,90 (m, 1 Н), 4,72-4,88 (m, 1 Н), 5,81 (br s, 1 H), 5,93-6,11 (m, 2 H), 7,10-7,26 (m, 3 H), 7,14-7,26 (m, 1 H), 7,30-7,41 (m, 2 H), 7,94 (br s, 1 H).
Образец 2-3
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,00-1,26 (m, 13 H), 2,09 (s, 3 H), 3,74-3,87 (m, 2 H), 4,10 (br d, J=7,70 Гц, 1 H), 4,22-4,50 (m, 3 H), 4,81 (quin, J=6,28 Гц, 1 H), 5,71-5,90 (m, 1 H), 5,96-6,15 (m, 2 H), 7,12-7,26 (m, 3 H), 7,31-7,41 (m, 2 H), 7,79-8,07 (m, 1 H).
Образец 2-4
1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,91 (t, J=7,33 Гц, 3 H), 1,01-1,28 (m, 13 H), 2,08 (s, 2 H), 3,72-3,89 (m, 2 H), 4,10 (br d, J=8,08 Гц, 1 H), 4,23-4,47 (m, 3 H), 4,81 (quin, J=6,25 Гц, 1 H), 5,69-5,89 (m, 1 H), 5,94-6,13 (m, 2 H), 7,14-7,25 (m, 3 H), 7,32-7,41 (m, 2 H), 7,79-8,11 (m, 1 H).
Образец 2-5
1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,86 (d, J=6,69 Гц, 1 H), 0,98-1,33 (m, 13 H), 2,02-2,09 (m, 1 H), 4,03-4,17 (m, 1 H), 4,22-4,50 (m, 3 H), 4,81 (quin, J=6,25 Гц, 1 H), 5,81 (br s, 1 H), 5,93-6,15 (m, 2 H), 7,11-7,27 (m, 3 H), 7,31-7,41 (m, 2 H), 7,77-8,21 (m, 1 H).
Образец 2-6
1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,98-1,28 (m, 15 H), 2,00 (s, 3 H), 3,99-4,14 (m, 3 H), 4,21-4,49 (m, 3 H), 4,81 (quin, J=6,22 Гц, 1 H), 5,82 (br s, 1 H), 5,93-6,14 (m, 2 H), 7,11-7,26 (m, 3 H), 7,29-7,42 (m, 2 H), 7,79-8,17 (m, 1 H).
Образец 2-7
1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,92-1,28 (m, 17 H), 1,97 (s, 2 H), 4,04-4,16 (m, 1 H), 4,20-4,51 (m, 3 H), 4,71-4,93 (m, 2 H), 5,82 (br s, 1 H), 5,95-6,14 (m, 2 H), 7,11-7,28 (m, 3 H), 7,31-7,43 (m, 2 H), 7,75-8,21 (m, 1 H).
Образец 2-8
1H ЯМР (400 МГц, DMSO-d6) δ ppm 0,81-1,11 (m, 13 H), 1,19 (s, 1 H), 1,53-1,66 (m, 1 H), 3,87-4,01 (m, 1 H), 4,06-4,32 (m, 3 H), 4,64 (quin, J=6,25 Гц, 1 H), 5,55-5,75 (m, 1 H), 5,77-5,97 (m, 2 H), 6,94-7,10 (m, 3 H), 7,13-7,26 (m, 2 H), 7,66-7,96 (m, 1 H).
Пример 7. Невозможность кристаллизации аморфной малонатной соли (соединения 4)
Как показано в примере 3, кристаллическую оксалатную соль идентифицировали при определении подходящих солей для соединения 1, но оксалатная соль - соединение 4 - не могла быть передана на клинические испытания из-за ее способности формировать камни в почках. Следовательно, предпринимали попытку кристаллизации химически родственной малонатной соли (соединения 5) с использованием тех же 11 растворителей, что и для гемисульфатной соли. Соединение 1 (12 × 50 мг, образцы 3-1, 3-2, 3-3, 3-4, 3-5, 3-6, 3-7, 3-8, 3-9, 3-10, 3-11 и 3-12) растворяли в трет-бутаноле (20 объемов), а затем растворы обрабатывали 1 эквивалентом стокового раствора малоновой кислоты (1M в THF). Затем образцы замораживали при удалении растворителя путем лиофилизации. К образцам 3-1, 3-2, 3-3, 3-4, 3-5, 3-6, 3-7, 3-8, 3-9, 3-10 и 3-11 добавляли соответствующий растворитель (5 объемов) при комнатной температуре. Обеспечивали выпаривание любым получившимся в результате растворам при окружающих условиях, при этом смолы или твердые вещества отстаивали при 25-50°С (1°С/минута между температурами и 4 часа при каждой температуре) в течение 5 суток. Твердые вещества анализировали с помощью XRPD (фиг. 12В), но выяснили, что все образцы либо образовывали смолу, либо становились аморфными (фиг. 12В). Результаты показаны в таблице 11. Один твердый (аморфный) образец (3-12) анализировали с помощью 1Н ЯМР и HPLC и обнаружили, что он содержит около 1 эквивалента малоновой кислоты (пики перекрываются), а также 0,6 экв. t-BuOH. Чистота соединения составляла 99,2% (фиг. 13А). На фиг. 12А представлена XRDP образца 3-12. а на фиг. 13А представлена хроматограмма HPLC образца 3-12.
Образец 3-12
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 0,81-1,11 (m, 13 Н), 1,19 (s, 1 Н), 1,53-1,66 (m, 1 Н), 3,87-4,01 (m, 1 Н), 4,06-4,32 (m, 3 Н), 4,64 (quin, J=6,25 Гц, 1 H), 5,55-5,75 (m, 1 Н), 5,77-5,97 (m, 2 Н), 6,94-7,10 (m, 3 Н), 7,13-7,26 (m, 2 Н), 7,66-7,96 (m, 1 Н).

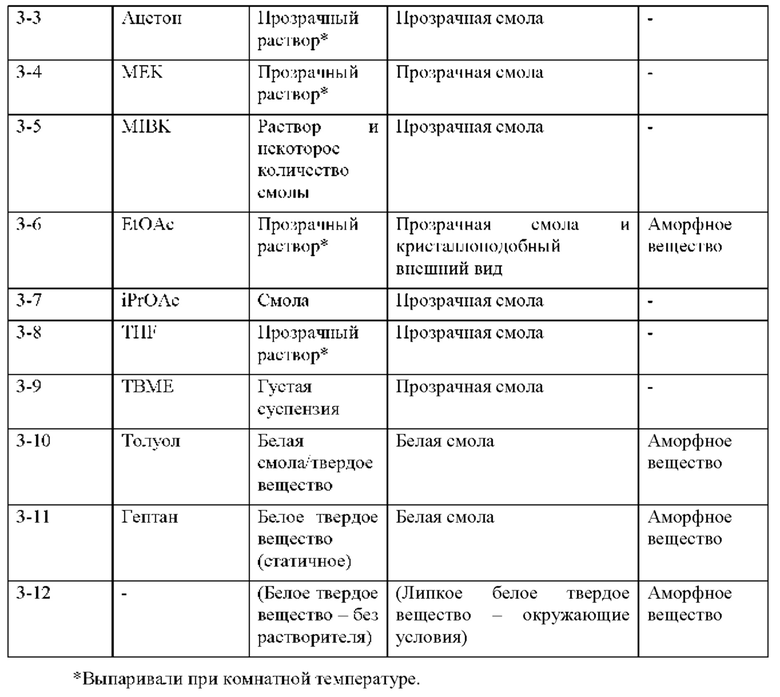
Пример 8. Невозможность надлежащего солеобразования с использованием измельчения в присутствии жидкости (LAG)
Выполняли исследование с измельчением в присутствии жидкости (LAG) для определения соответствующих солей, отличных от гемисульфата с использованием 14 кислотных противоионов из таблицы 12.

Соединение 1 (30 мг) помещали во флаконы для HPLC с двумя 3-мм шариками. Материалы смачивали растворителем (15 мкл этанола, образцы 4-1, 4-2, 4-3, 4-4, 4-5, 4-6, 4-7, 4-8, 4-9, 4-10, 4-11, 4-12, 4-13 и 4-14) и добавляли 1 эквивалент кислотного противоиона. Затем образцы измельчали в течение 2 часов при 650 оборотах в минуту с использованием системы помола Fritsch с адаптером Automaxion. Обнаружили, что большинство образцов после помола представляли собой прозрачные смолы и их далее не анализировали (таблица 13). Те, которые, как было обнаружено, содержали твердое вещество, анализировали с помощью XRPD, и во всех случаях, как выяснили, полученные паттерны соответствовали таковым противоиона кристаллической кислоты без дополнительных пиков (фиг. 13В).
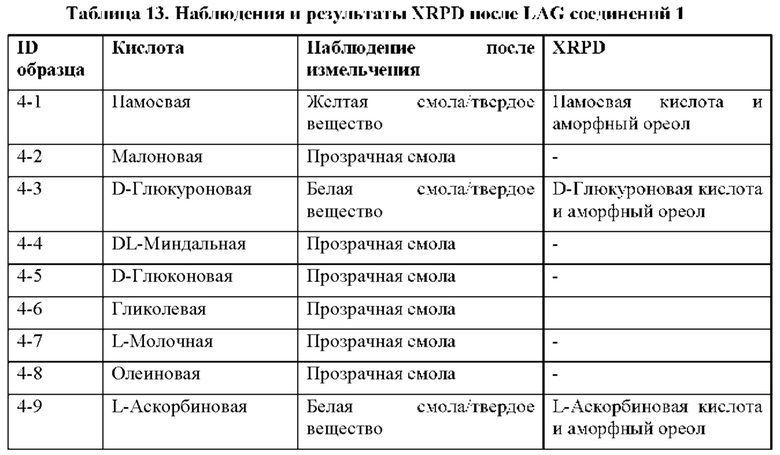
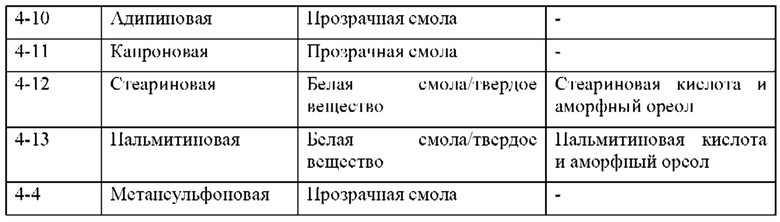
Пример 9. Невозможность получения надлежащего солеобразования с использованием метилэтилкетона (MEK)
Далее использовали метилэтилкетон (MEK) в качестве растворителя для исследования соответствующих солей, отличных от гемисульфатной соли. С использованием 14 кислотных противоионов в таблице 12, исследование выполняли путем растворения соединения 1 (50 мг) в MEK (20 объемов) при комнатной температуре. Растворы обрабатывали 1 эквивалентом выбранных противоионов (таблица 12). Затем образцы охлаждали до 5°С при 0,1°С/минута и взбалтывали при этой температуре на протяжении ночи. Обеспечивали выпаривание всех образцов при окружающих условиях и любые наблюдаемые твердые вещества анализировали с помощью XRPD. Такое выпаривание в основном давало смолы, за исключением образцов со стеариновой кислотой (образец 4-12) и пальмитиновой кислотой (образец 5-13), которые давали стекловидные растворители. Эти твердые вещества были аморфными по данным XRPD, и кристаллические формы соли не были получены. Результаты показаны в таблице 14. (фиг. 13А).
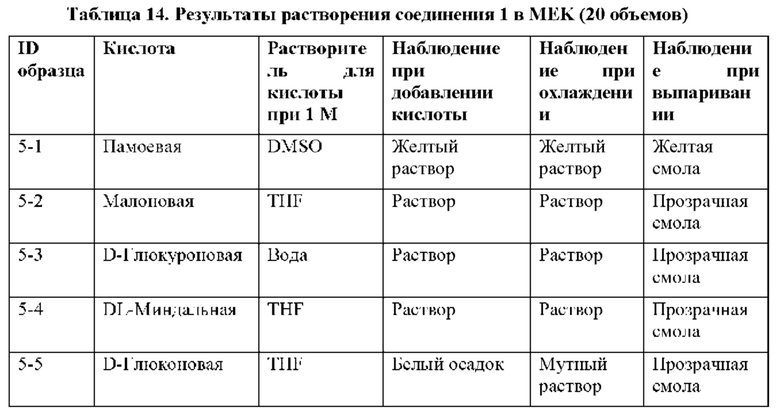
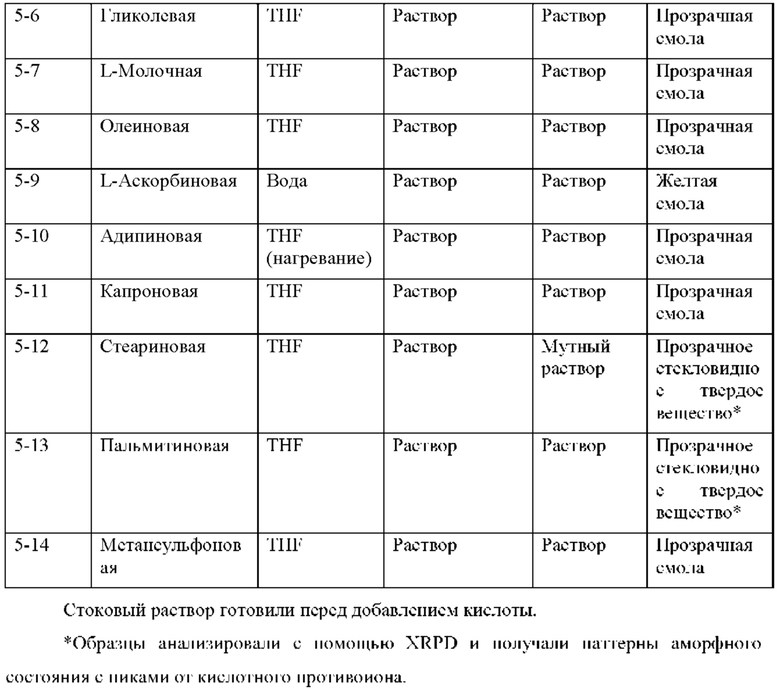
Поскольку все образцы были аморфными, их повторно растворяли в MEK (5 объемов) и добавляли циклогексан (20 объемов антирастворителя) при комнатной температуре, а затем 1 час перемешивали при 25°С. Затем образцы отстаивали при 50-5°С (1°С/минута между температурами, 4 часа при каждой температуре) в течение 2 суток, а затем цикл меняли на 50-25°С в течение следующих 4 суток. Образцы визуально осматривали после отстаивания. Результаты показаны в таблице 15. После отстаивания выяснили, что все образцы, за исключением 5-1 (с памоевой кислотой), представляли собой смолы. Образец 5-1 - желтое твердое вещество - анализировали с помощью XRPD, выяснили, что паттерн соответствует известной форме памоевой кислоты (фиг. 14В), и, следовательно, кристаллические формы соли не наблюдали.
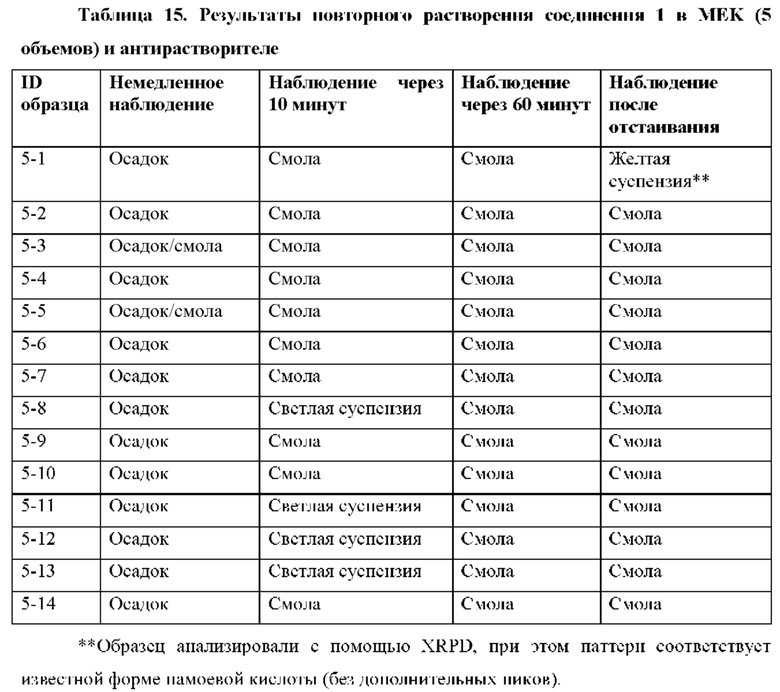
Пример 10. Невозможность получения надлежащего солеобразования с использованием этилацетата
Далее использовали этилацетат для исследования соответствующих солей, отличных от гемисульфатной соли. С использованием 14 кислотных противоионов в таблице 12 исследование выполняли путем растворения соединения 1 (50 мг) в этилацетате (20 объемов) при 50°С. Растворы обрабатывали 1 эквивалентом выбранных противоионов (таблица 12). Затем образцы охлаждали до 5°С при 0,1°С/минута и взбалтывали при этой температуре в течение 4 суток. Обеспечивали выпаривание всех образцов при окружающих условиях и любые наблюдаемые твердые вещества анализировали с помощью XRPD. Результаты кристаллизации с использованием этилацетата приводятся в таблице 16. В отличие от примера 8, в котором растворителем был MEK, наблюдали, что большинство образцов представляли собой суспензии после охлаждения смеси кислота: соединение (тем, которые были растворами, обеспечивали выпаривание при окружающих условиях). Однако, как обычно обнаруживали, дифрактограммы XRPD соответствовали кристаллическому соединению 1. Образцы 6-2, 6-4 и 6-5 имели некоторые небольшие различия (фиг. 14А и фиг. 15А). Кристаллические формы соли не были получены.

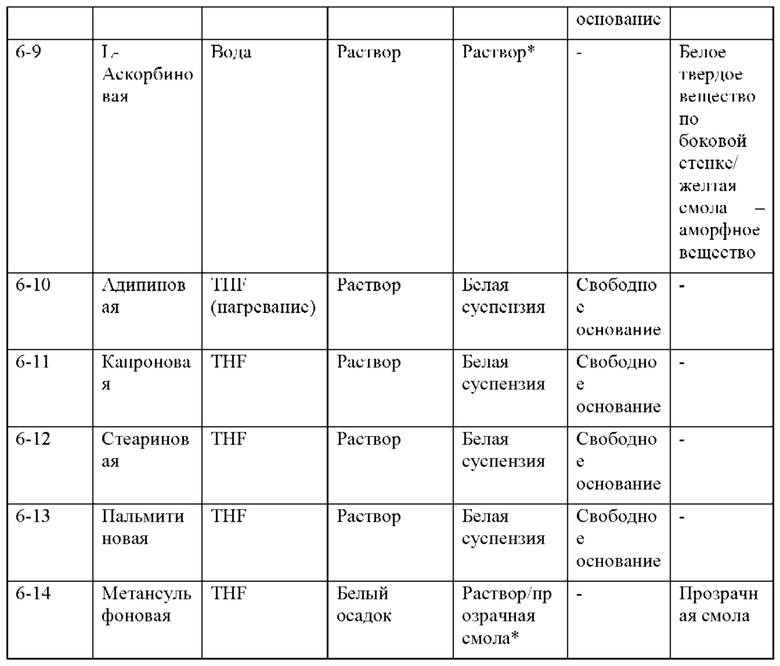
Пример 11. Определение химической чистоты с помощью HPLC
Анализ чистоты в примере 2 и примере 4 выполняли на последовательной системе Agilent HP1100, оснащенной детектором на диодной матрице, и с использованием программного обеспечения ChemStation vB.04.03 с помощью способа, показанного в таблице 17.
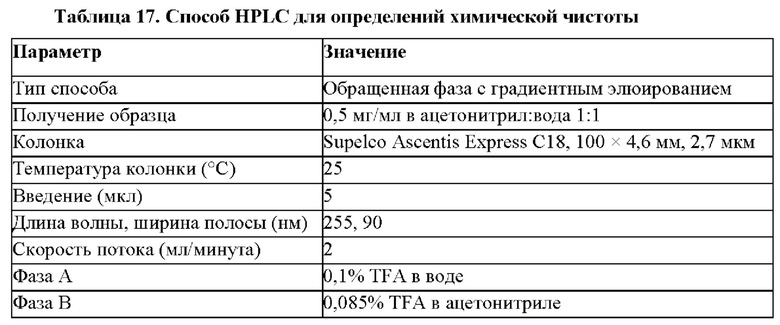
Пример 12. Методики порошковой рентгеновской дифракции (XRPD)
Паттерны XRPD в примерах 2, 3, 4, 5, 6, 7, 8, и 9 собирали на дифрактометре PANalytical Empyrean с использованием излучения Cu Kα (45 кВ, 40 мА) в геометрии пропускания. Для падающего луча использовали щели 0,5°, экрана 4 мм и щелей Соллера 0,4 рад с фокусирующим зеркалом. Детектор PIXcel3D, размещенный на дифрагированном пучке, был снабжен приемной щелью и щелями Соллера 0,04 рад. Технические характеристики прибора еженедельно проверяли с использованием кремниевого порошка. Программным обеспечением, используемым для сбора данных, являлось X'Pert Data Collector v. 5.3, данные анализировали и представляли с использованием Diffrac Plus EVA v. 15.0.0.0 или Highscore Plus v. 4.5. Образцы готовили и анализировали на металлическом планшете или на 96-луночном планшете Millipore в режиме пропускания. Рентгенопрозрачную пленку использовали между металлическими листами на планшете с металлическими лунками и порошки (приблизительно 1-2 мг) использовали в том виде, в котором они были получены. Планшет Millipore использовали для выделения и анализа твердых веществ из суспензий путем добавления небольшого количества суспензии непосредственно в планшет перед фильтрацией в легком вакууме.
В режиме сканирования для металлического планшета использовали ось сканирования gonio, тогда как для планшета Millipore использовали сканирование 2θ. Проверку технических характеристик проводили с использованием кремниевого порошка (планшет с металлическими лунками). Детали сбора данных представляли собой: угловой диапазон от 2,5 до 32,0°2θ, размер шага 0,0130°2θ и суммарное время сбора 2,07 минуты.
Образцы также собирали на дифрактометре Bruker D8 с использованием излучения Cu Kα (40 кВ, 40 мА), 0-2θ гониометра, расходимости V4 и приемных щелей, Ge монохроматора и детектора Lynxeye. Технические характеристики прибора проверяли с использованием сертифицированного стандарта Corundum (NIST 1976). Программным обеспечением для сбора данных являлось DiffracPlus XRD Commander v. 2.6.1, и данные анализировали и представляли с использованием Diffrac Plus EVA v. 15.0.0.0.
Образцы обрабатывали при окружающих условиях в виде плоских образцов с использованием порошка в том виде, в котором он был получен. Образец аккуратно упаковывали в полость, вырезанную в полированной кремниевой пластине с нулевым фоном (510). Образец вращали в его плоскости во время анализа. Детали сбора данных представляли собой: угловой диапазон от 2 до 42°2θ, размер шага 0,05°2θ и время сбора 0,5 секунды/шаг.

В 250-мл колбу загружали МеОН (151 мл) и раствор охлаждали до 0-5°С. Концентрированный раствор H2SO4 добавляли каплями за 10 минут. В отдельную колбу загружали соединение 1 (151 г) и ацетон (910 мл) и добавляли каплями раствор H2SO4/MeOH при 25-30°С за 2,5 часа. Осаждалось большое количество твердого вещества. После взбалтывания раствора в течение 12-15 часов при 25-30°С смесь фильтровали, промывали с помощью МеОН/ацетон (25 мл/150 мл) и сушили при 55-60°С в вакууме с получением соединения 2 (121 г, 74%).
Аналитический способ для соединения 2: Чистоты соединения 2 достигали с использованием HPLC системы Agilent 1100 с колонкой Waters XTerra Phenyl 5 мкм 4,6*250 мм при следующих условиях: скорость потока 1 мл/минута, считывание при 254 нм, температура колонки 30°С, объем введения 10 мкл и время выполнения 30 минут. Образец растворяли в ACN:вода (90:10, объем/объем). Градиентный метод для разделения представлен ниже. Rt (минуты) соединения 2 составляло приблизительно 12,0 минуты.

1H ЯМР: (400 МГц, DMSO-d6): δ 8,41 (br, 1H), 7,97 (s, 1H), 7,36 (t, J=8,0 Гц, 2Н), 7,22 (d, J=8,0 Гц, 2Н), 7,17 (t, J=8,0 Гц, 1H), 6,73 (s, 2H), 6,07 (d, J=8,0 Гц, 1H), 6,00 (dd, J=12,0, 8,0 Гц, 1H), 5,81 (br, 1H), 4,84-4,73 (m, 1H), 4,44-4,28 (m, 3H), 4,10 (t, J=8,0 Гц, 2H), 3,85-3,74 (m, 1H), 2,95 (s, 3H), 1,21 (s, J=4,0 Гц, 3H), 1,15-1,10 (m, 9H).
Пример 14. Характеристика соединения 2
Соединение 2 далее характеризовали визуально, с помощью 1Н ЯМР, 13С ЯМР, 19F ЯМР, MS, HPLC и XRPD (фиг. 15В). Остаточный растворитель измеряли с помощью GC. Содержание воды измеряли путем титрования по методу Карла Фишера, и содержание воды составляло всего лишь 0,70%. Данные кратко описываются в таблице 18.
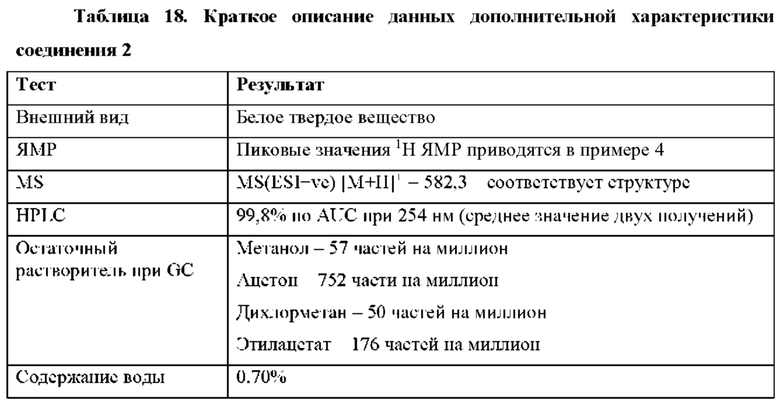
Пример 15. Растворимости соединения 1 и соединения 2
Соединение 1 и соединение 2 тестировали на предмет растворимости в биоадекватных для тестирования средах, в том числе в имитированном желудочном соке (SGF), имитированном желудочном соке натощак (FaSSIF) и в желудочном соке после приема пищи (FeSSIF). Результаты для соединения 1 показаны в таблице 19, а результаты для соединения 2 показаны в таблице 20. Образцы взбалтывали при комнатной температуре (20-25°С). Соединение 2 было более чем в 40 раз более растворимым, чем соединение 1, в воде через 2 часа и более чем в 25 раз более растворимым через 24 часа. При условиях SGF соединение 2 характеризовалось растворимостью 84,2 мг/мл через 24 часа по сравнению с растворимостью 15,6 мг/мл соединения 1 в тот же момент времени. Соединение 2 также было более растворимым через 2 часа при условиях SGF, чем соединение 1, и достаточно растворимым для обеспечения тестирования даже через 48 часов, тогда как тестирование соединения 1 через 48 часов не проводилось.
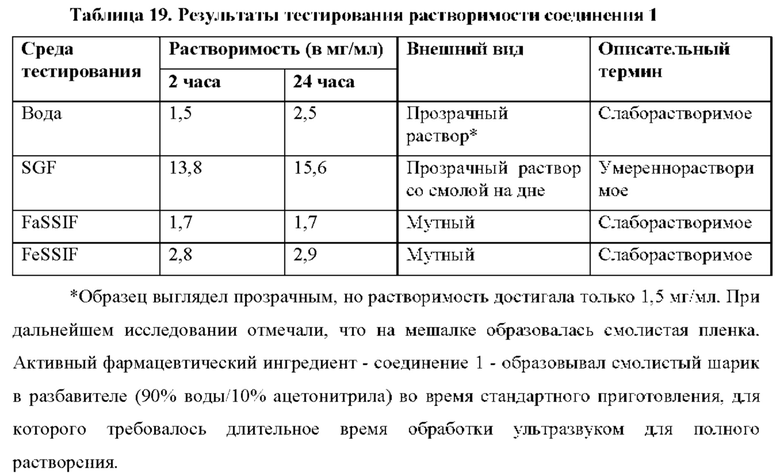
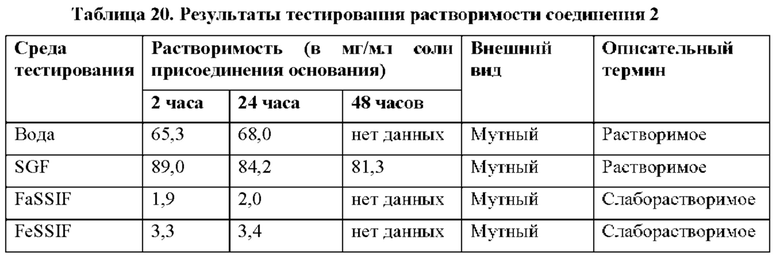
Пример 16. Химическая стабильность соединения 2
Соединение 2 тестировали на предмет химической стабильности при 25 и 40°С за период времени 6 месяцев путем мониторинга органической чистоты, содержания воды, 1H ЯМР, DSC и рамановского IR. Система укупоренного контейнера для исследования представляла собой комбинацию медицинского пакета с клапаном с фармацевтической ламинированной пленкой поверх пакета и осушающим силикагелем между двумя слоями. Соединение 2 (1 г) измеряли в каждом контейнере. Затем пакеты хранили при 25°С/60% RH (относительной влажности) и 40°С/75% RH (относительной влажности). Органическую чистоту, содержание воды, 1Н ЯМР, DSC и рамановское IR измеряли в момент времени 0, месяц 1, месяц 2, месяц 3 и месяц 6.
Значение чистоты соединения 2 получали с использованием системы Shimadzu LC-20AD с колонкой Waters XTerra Phenyl 5 мкм, 4,6 × 250 мм при следующих условиях: скорость потока 1 мл/минута, считывание при 254 нм, температура колонки 35°С и объем введения 10 мкл. Образец растворяли в ацетонитриле - воде (90:10) (объем/объем). Градиентный метод показан ниже.

Содержание воды соединения 2 (250 мг) определяли с помощью аппарата для титрования воды путем титрования по методу Карла Фишера.
Результаты показаны в таблице 21 и таблице 22. При хранении соединения 2 в течение 6 месяцев при 25 и 40°С скорость разложения была минимальной. Через 3 месяца соединение 2 было чистым на 99,75% процентов в условиях 25°С и чистым на 99,58% в условиях 40°С. Через 6 месяцев соединение 2 было все еще чистым на 99,74% в условиях 25°С и чистым на 99,30% в условиях 40°С. При 25°С процент разложения продукта повышался от 0,03% в сутки 0 до 0,08% через 6 месяцев. При 40°С процент разложения продукта повышался от 0,03% в сутки 0 до 0,39%. На протяжении 6 месяцев процент воды повышался приблизительно на 0,6% при 25°С и повышался на приблизительно 0,7% при 40°С.
Характеристика с помощью 1Н ЯМР, рамановской спектроскопии и DSC соединения 2 через 1, 2, 3 и 6 месяцев была такой же, как и характеристика соединения 2 в сутки 0 в условиях обеих температур (таблица 22), что подчеркивает длительную стабильность соединения 2.


Дополнительные исследования химической стабильности соединения 2 проводили для определения содержаний примеси и воды. Тестировали три условия: стабильность в условиях «ускоренного старения» (40±2°С/75±5% RH) за 6-месячный период времени, стабильность в окружающих условиях (25±2°С/60±5% RH) за 9-месячный период времени и стабильность в условиях хранения в холодильнике (5±3°С) за 9-месячный период времени. Результаты для стабильности в условиях «ускоренного старения», стабильности в окружающих условиях и в условиях хранения в холодильнике показаны в таблице 23, таблице 24 и таблице 25, соответсвенно. Исходя из результатов данных исследований соединение 2 является химически очень стабильным.
В исследовании стабильности в условиях «ускоренного старения» (таблица 23) в каждый момент времени (1ый месяц, 3ий месяц и 6ой месяц) измерения соединения 2 по внешнему виду соединение 2 всегда представляло собой белое твердое вещество, и IR соответствовал эталонному стандарту. Через шесть месяцев суммарные примеси родственного вещества 1 составляли всего лишь 0,08%, и не выявляли родственное вещество 2 и изомеры.

N.D.: не выявляли.
В исследовании стабильности в окружающих условиях при измерении внешнего вида, IR, содержаний воды и примеси в течение девяти месяцев соединение 2 по внешнему виду всегда представляло собой белое твердое вещество, a IR всегда соответствовал эталонному образцу. Результаты (таблица 24) подчеркивают химическую стабильность соединения 2. Через 9 месяцев процентное отношение воды в образце составляло всего лишь 0,20%, и суммарные примеси родственного вещества 1 составляли всего лишь 0,02%. Подобно исследованиям стабильности в условиях «ускоренного старения» родственное вещество 2 и какие-либо изомеры соединения 2 не выявляли.
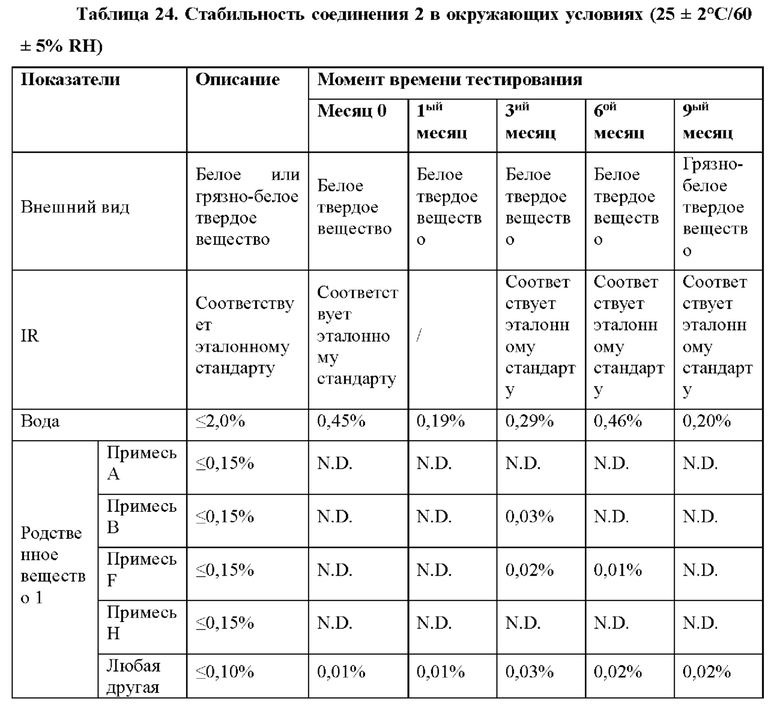
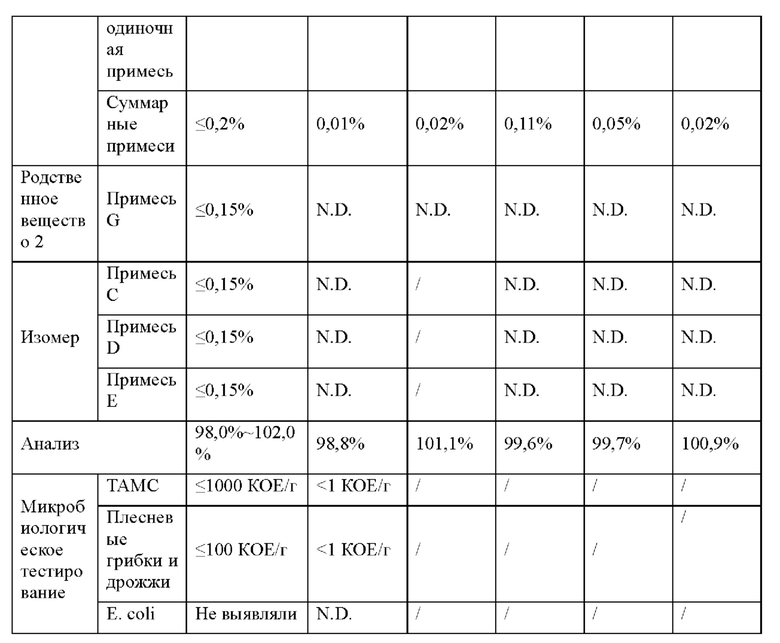
N.D.: не выявляли.
Результаты измерения стабильности в условиях хранения в холодильнике показаны в таблице 25. Единственные примеси, обнаруженные только через 9 месяцев, были связаны с родственным веществом 1 и водой. Содержание воды через 9 месяцев составляло 0,32%, а суммарные примеси родственного вещества 1 составляли всего лишь 0,01% образца. Соединение 2 является химически стабильным в высокой степени в условиях хранения в холодильнике.

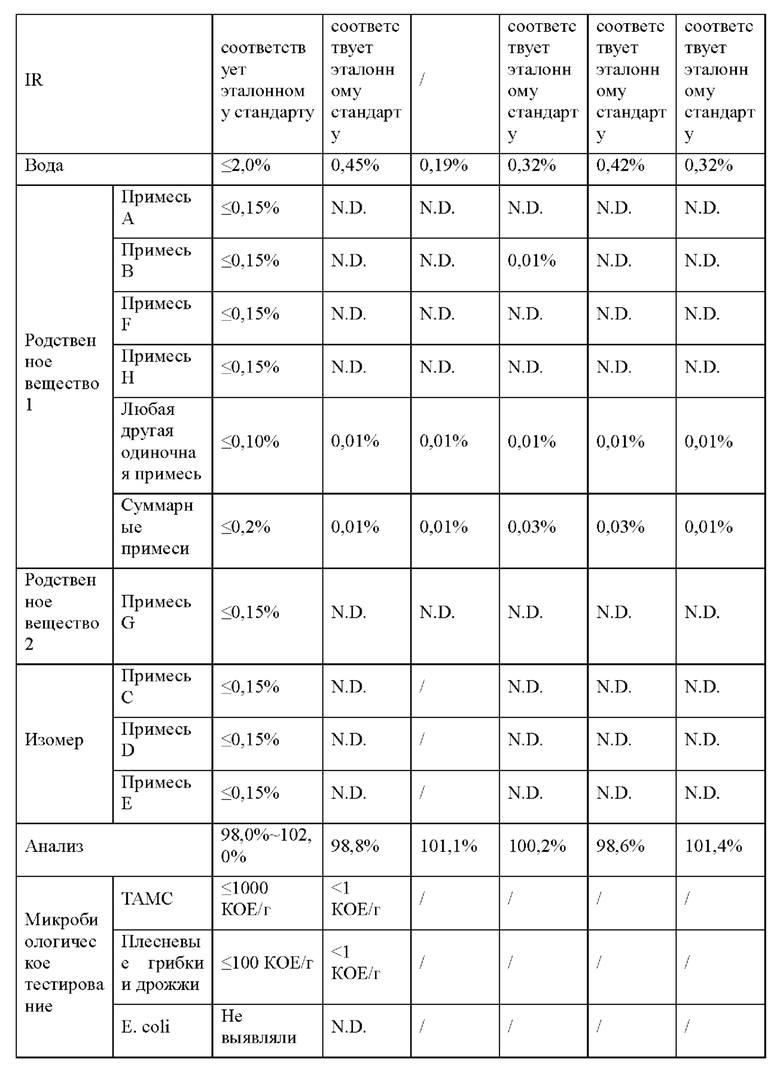
N.D.: не выявляли.
Пример 17. Содержания в плазме метаболитов после однократных пероральных доз соединения 2
Однократную пероральную дозу соединения 2 вводили крысам, собакам и обезьянам и измеряли содержания в плазме некоторых метаболитов, показанных на схеме 1.
Превращение соединения 2 в соединение 1 и метаболит 1-7 показаны в таблице 26, а результаты для метаболита 1-8 и метаболита 1-2 показаны в таблице 27. К крыс наблюдали низкие уровни воздействия соединения 1, но наблюдали высокие уровни воздействия метаболита 1-7 - нуклеозидного метаболита активного трифосфата (метаболита 1-6). У обезьян измеряли почти пропорциональное дозе воздействие соединения 1. У собак измеряли сверхпропорциональное воздействие соединение 1, что указывает на метаболический клиренс первого прохождения в печени. На протяжении всего исследования наблюдали значительно больше случаев рвоты у собак (5/5 в группе высокой дозы), чем у обезьян (1/5 в группе высокой дозы).
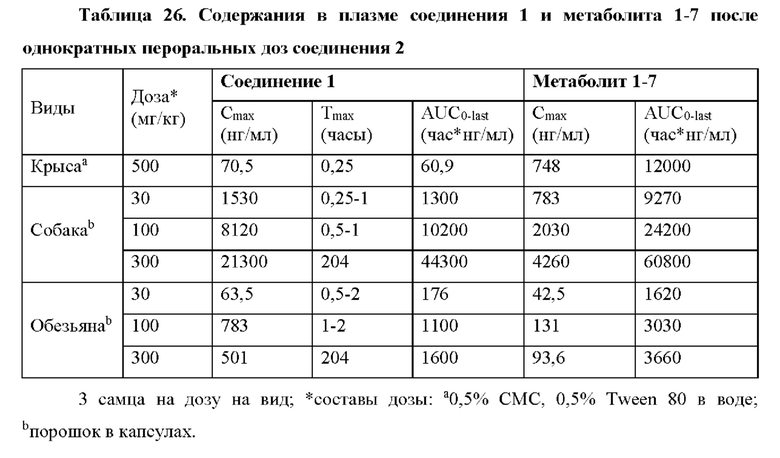
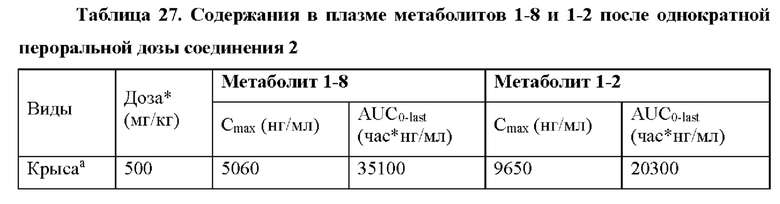
Пример 18. Воздействие на ткань активного трифосфата после пероральной дозы соединения 2
Измеряли содержания в ткани сердца и печени активного трифосфата (TP) соединения 2 (метаболита 1-6) через 4 часа после пероральных доз соединения 2. Образцы печени и сердца получали через 4 часа после однократной дозы соединения 2, мгновенно замораживали, гомогенизировали и анализировали с помощью LC-MS/MS на предмет внутриклеточных содержаний активного ТР. Содержания в тканях измеряли у крыс, собак и обезьян, как показано на фиг. 16А. Высокие содержания активного TP измеряли в печени всех тестируемых видов. Относительно низкие содержания активного TP измеряли в сердцах собак из-за насыщения почечного метаболизма первого прохождения, и не поддающиеся количественному определению содержания TP измеряли в сердцах крыс и обезьян, что указывает на специфический в отношении печени состав активного ТР. Хотя это не показано, по сравнению с введением дозы соединения 1 введение дозы соединения 2 улучшало распределение ТР.
Пример 19. Фармакологическое сравнение соединения 1 и соединения 2 у собак
Проводили прямое сравнение собак, получивших дозу соединения 1 и соединения 2. В ходе исследования измеряли содержания в плазме соединения 1 и метаболита 1-7 (со схемы 1) через 4 часа после введения дозы соединения 1 (25 мг/кг) и соединения 2 (30 мг/кг) (таблица 28), и AUC(0-4 часа) метаболита 1-7 была вдвое выше у соединения 2 по сравнению с соединением 1. Нормализованные по дозе воздействия соединения 1 и метаболита 1-7 показаны в таблице 28. Значения AUC(0-4hr) для соединения 1, метаболита 1-7 и сумма соединения 1 + метаболита 1-7 были выше после введения дозы соединения 2.
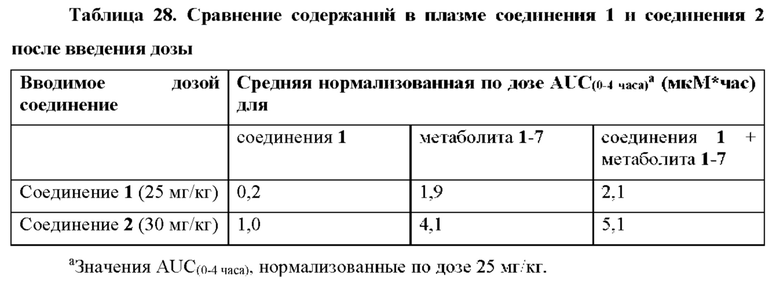
Отношение печень/сердце для концентраций трифосфата указывает на то, что введение дозы соединения 2 по сравнению с соединением 1 повышает селективную доставку трифосфата в печень, как показано в таблице 29. AUC(0-4 часа) активного гуанинового метаболита (1-6) после введения соединения 1, измеряемая в сердце, составляла 174 мкМ*час, тогда как AUC(0-4 часа) активного гуанинового метаболита (1-6) после введения соединения 2, измеряемая в сердце, составляла 28 мкМ*час. Отношение печень/сердце для соединения 2 составляло 20 по сравнению с отношением печень/сердце 3,1 для соединения 1.

Эффект повышенной селективности в отношении печени по сравнению с сердцем при введении соединения 2 по сравнению с соединением 1 также показан на фиг. 16В. Содержания в тканях сердца и печени активного трифосфата после введения дозы соединения 2 (30 мг/кг) сравнивали с содержаниями в тканях активного трифосфата после введения дозы соединения 1 (25 мг/кг). Концентрация активного TP была выше в печени, чем в сердце для соединения 1 и соединения 2, но активный TP был более селективным в отношении печени, чем в отношении сердца при введении дозы соединения 2 по сравнению с соединением 1.
Пример 20. Профили в плазме метаболитов соединения 2 у крыс и обезьян
Самцам крыс Sprague-Dawley и яванским макакам (3 животных на получавшую дозу группу) давали однократные пероральные дозы соединения 2. Аликвоты плазмы, полученной из образцов крови, обработанной с помощью Dichlorvos, анализировали с помощью LC-MS/MS на предмет концентраций соединения 1 и метаболита 1-7 (нуклеозидного метаболита активного трифосфата соединения 2, показанного на схеме 1), и фармакокинетические параметры определяли с использованием WinNonlin. Результаты для однократной дозы 500 мг/кг у крыс показаны на фиг. 17, а результаты для однократной дозы 30, 100 или 300 мг/кг у обезьян показаны на фиг. 18. Результаты также кратко описываются в таблице 30.
Высокие содержания в плазме метаболита 1-7 - нуклеозидного метаболита активного трифосфата (TP) соединения 2 указывают на образование высоких содержаний TP, даже у крыс, у которых наблюдали очень низкие содержания в плазме исходного нуклеотидного пролекарства, из-за короткого периода полувыведения соединения 1 в крови крыс (<2 минут). Персистирующие содержания в плазме метаболита 1-7 отражают длинный период полувыведения ТР.
У обезьян содержания в плазме (AUC) соединения 1 были почти пропорциональны дозе, тогда как воздействия метаболита 1-7 были несколько менее пропорциональны дозе, хотя значения AUC и для исходного лекарственного средства, и для нуклеозидного метаболита активного TP продолжала повышаться до самой высокой тестируемой дозы (300 мг/кг).
Пероральное введение соединения 2 крысам и обезьянам давало высокие и зависимые от дозы содержания в плазме метаболита 1-7 (нуклеозидного метаболита внутриклеточного активного трифосфата соединения 2); воздействия метаболита 1-7 продолжали усиливаться до самой высокой тестируемой дозы, отражая существенно образование активного TP у данных видов.
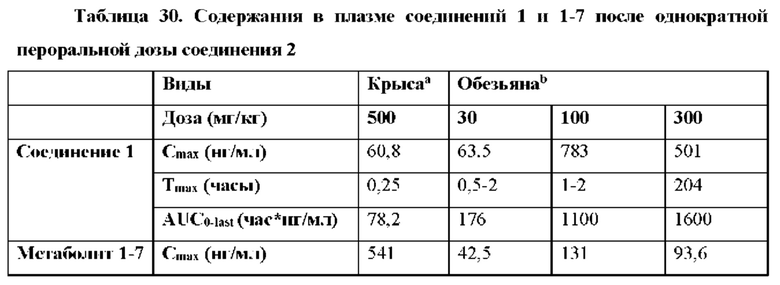
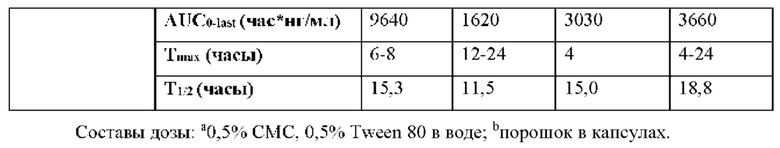
Пример 21. Эффект активного трифосфата соединения 1 и соединения 2 в отношении митохондриальной целостности
Относительную эффективность включения активного трифосфата (TP) соединения 1 и соединения 2, метаболита 1-6 (схема 1), человеческой митохондриальной РНК-полимеразой сравнивали с относительной эффективностью активного TP софосбувира и активного TP INX-189. Соединение 1 и соединение 2, по-видимому, не влияют на митохондриальную целостность, поскольку их активный трифосфат плохо включается человеческой митохондриальной РНК-полимеразой с эффективностью, подобной таковой трифосфата софосбувира; при этом относительная эффективность включения трифосфата INX-189 была до 55 раз выше. Результаты показаны в таблице 31. Включение этих аналогов человеческой митохондриальной РНК-зависимой РНК-полимеразой (POLRMT) определяли согласно Arnold et al. (Sensitivity of Mitochondrial Transcription and Resistance of RNA Polymerase II Dependent Nuclear Transcription to Antiviral Ribonucleotides. PLoS Pathog., 2012, 8, е1003030).
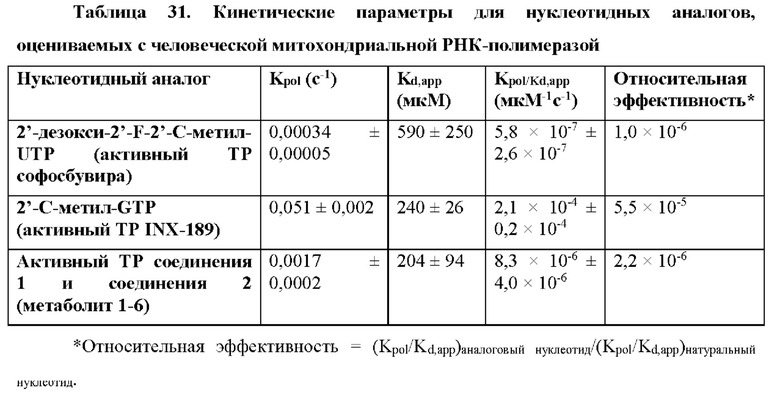
Пример 22. Активность соединения 1 против репликонов, содержащих последовательность NS5B
Панель репликонов, содержащих последовательности NS5B от различных генотипов HCV, полученных из 6 лабораторных эталонных штаммов (GT1a, 1b, 2а, 3а, 4а и 5а) (фиг. 19) и из 8 образцов плазмы больных HCV (GT1a, 1b, 2а, 2b, 3а-1, 3а-2, 4а и 4d) (фиг. 20), использовали для определения эффективности соединения 1 и софосбувира.
Соединение 1 было более эффективным, чем софосбувир против клинических и лабораторных штаммов HCV. Соединение 1 демонстрировало мощную активность против вирусов всех генотипов in vitro в отношении клинических изолятов дикого типа с ЕС95<80 нМ, которая была в 4-14 раз выше таковой у софосбувира. Как показано на фиг. 20, значения ЕС95 для соединения 1 были в 7-33 раза ниже, чем у софосбувира, против клинических изолятов всех тестируемых генотипов HCV. Значения ЕС50 для соединения 1 были в 6-11 ниже, чем у софосбувира, против лабораторных штаммов HCV генотипов 1-5 (фиг. 19).
Пример 23. Исследование однократной нарастающей дозы (SAD) соединения 2 на здоровых волонтерах (часть А) и инфицированных GT1-HCV больных (часть В)
Соединение 2 тестировали в исследовании однократной нарастающей дозы (SAD) для измерения его безопасности, переносимости и фармакокинетических показателей у здоровых субъектов (часть А). Часть А представляла собой рандомизованное, двойное слепое, с контролем по плацебо исследование SAD. Здоровые субъекты в части А получали однократную дозу соединения 2 или плацебо натощак. Субъектов помещали в клинику от суток -1 до суток 6.
Введение дозы в каждой когорте распределяли так, что 2 субъектов (1 получавший активное соединение: 1 получавший плацебо) оценивали в течение 48 часов после введения дозы до того, как получала дозу остальная часть когорты. Каждая когорта получала соединение 2 в порядке возрастания. Введение дозы следующим когортам происходило на основании анализа доступных данных по безопасности (до суток 5) и фармакокинетических данных плазмы (до 24 часов) предыдущей когорты.
Повышение дозы продолжали после удовлетворительного анализа этих данных. Поскольку фармакокинетические показатели и данные по безопасности получали от предыдущих когорт, дозы, оцененные в когортах 3а-4а, регулировали с шагом не более 100 мг. Суммарная максимальная доза, оцениваемая в части А, не превышала 800 мг. Режим введение дозы для части А показан в таблице 32.
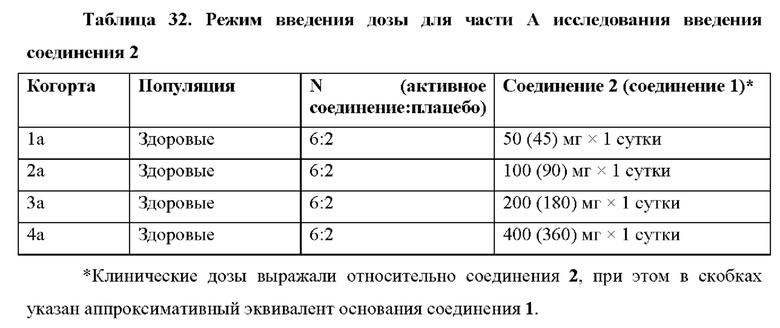
Здоровыми волонтерами в части А исследования были мужчины и женщины в возрасте от 18 до 65 лет. Реципиентов активного соединения и плацебо объединяли в каждую когорту части А когорта для сохранения анонимности исследования.
Соединение 2 также тестировали в исследовании однократной нарастающей дозы (SAD) для измерения его безопасности, переносимости, фармакокинетических показателей и противовирусной активности у инфицированных GT1-HCV больных (часть В). Субъекты в части В получали однократную дозу соединения 2 натощак. Больных помещали в клинику от суток -1 до суток 6.
Часть В начинали после анализа данных по безопасности (до суток 5) и фармакокинетических данных плазмы (до 24 часов) от когорты 3а в части А. Доступные данные по безопасности (до суток 5) и фармакокинетические данные (до 24 часов) анализировали для первой когорты в части В (когорты 1b) перед включением следующих когорт части В. Следующие когорты части В получали дозу только после анализа доступных данных по безопасности и фармакокинетических данных от соответствующих доз в части А, а также доступных данных по безопасности (до суток 5) от предыдущих когорт части В.
Повышение дозы до 600 мг у инфицированных HCV больных продолжали после удовлетворительного анализа этих данных. Режим введение дозы для части В показан в таблице 33.
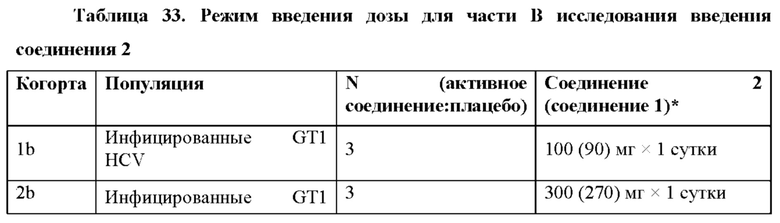

Больные, инфицированные HCV, были не получавшими лечения, нецирротическими инфицированными GT1 субъектами с вирусной нагрузкой ≥5 log10 МЕ/мл.
Никаких серьезных нежелательных явлений зарегистрировано не было, и не требовалось преждевременного прекращения приема средства ни в части А, ни в части В. Все побочные эффекты были от слабых до умеренных по интенсивности, и не было выявлено никаких зависимых от дозы паттернов, в том числе лабораторных параметров, показателей жизнедеятельности и ECG.
Пример 24. Результаты исследования однократной нарастающей дозы (SAD) соединения 2
Фармакокинетические параметры соединения 1 и нуклеозидного метаболита 1-7 измеряли после однократной дозы соединения 2. Остаточные концентрации в плазме С24 (C24h) метаболита 1-7 у инфицированных HCV больных после дозы 600 мг соединения 2 составляли 25,8 нг/мл, что более чем в два раза превышало концентрацию в плазме после дозы 300 мг соединения 2. Метаболит 1-7 (показан на схеме 1) мог быть образован только путем дефосфорилирования внутриклеточного фосфата метаболита 1-4, метаболита 1-5 и метаболита 1-6, которые являются активными соединениями. Следовательно, метаболит 1-7 можно считать заменителем активного соединения. Фармакокинетические данные для всех когорт показаны в таблице 34 и таблице 35. Значения регистрировали как среднее ± SD, за исключением Tmax, для которой регистрировали медиану (диапазон). Фармакокинетические параметры были сопоставимыми у здоровых субъектов и инфицированных HCV больных.
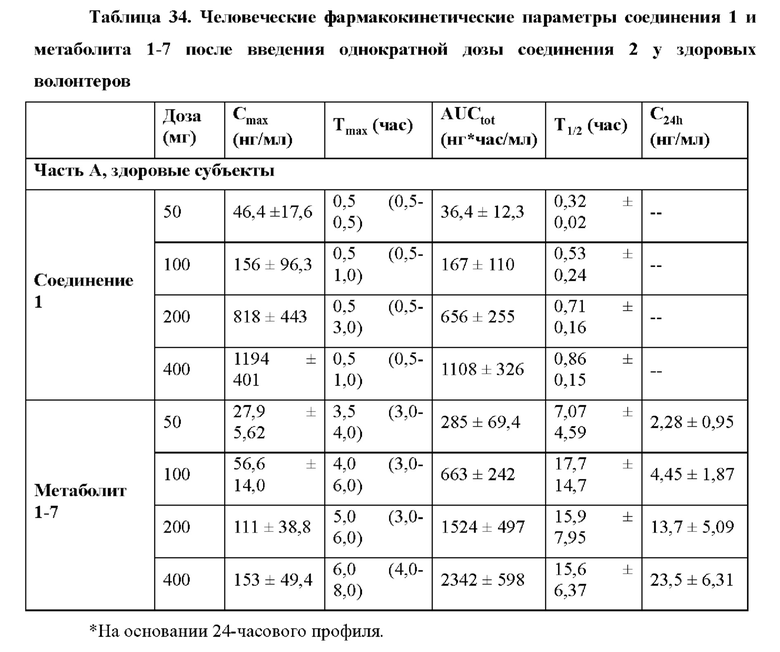
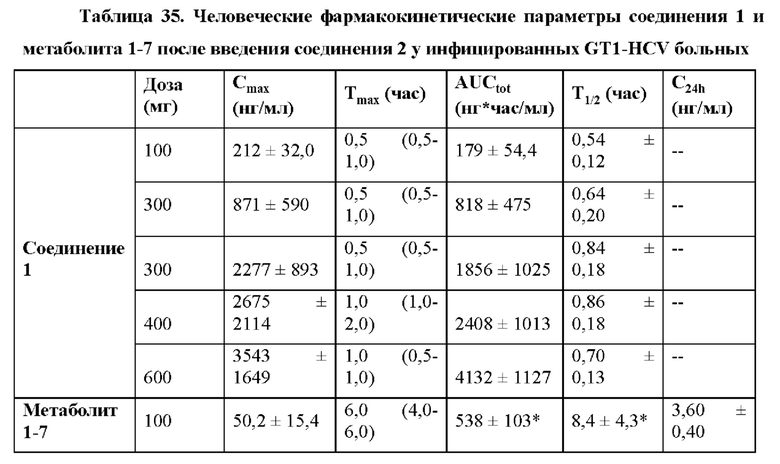
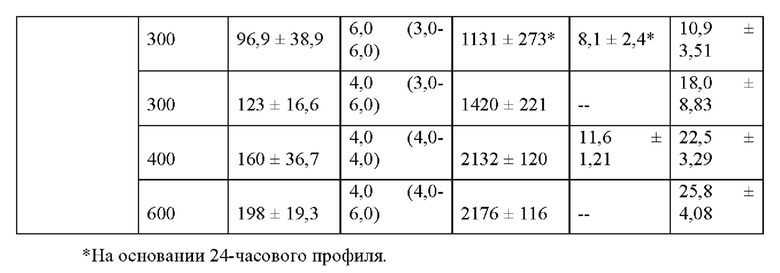
Также вычисляли профили средней концентрации в плазме в зависимости от времени соединения 1 и метаболита 1-7 для всех когорт части А и части В исследования. На фиг. 21 представлена средняя концентрация в плазме соединения 1 после однократной дозы соединения 2, а на фиг. 22 представлена средняя концентрация в плазме метаболита 1-7 после однократной дозы соединения 2. Как показано на фиг. 21, соединение 1 быстро абсорбировалось и быстро/активно метаболизировалось во всех когортах из части В. Как показано на фиг. 22, метаболит 1-7 был основным метаболитом и демонстрировал устойчивые концентрации в плазме. Содержание в плазме соединения 1 зависело от дозы, при этом воздействие метаболита 1-7 было пропорционально дозе.
Для инфицированных HCV субъектов части В измерения количественного определения РНК HCV выполняли до, во время и после введения соединения 2. Определения РНК HCV в плазме выполняли при применении валидированных коммерческих анализов. Исходный уровень определяли как среднее показателей в сутки - 1 и сутки 1 (перед введением дозы). Однократная доза 300 мг соединения 2 (что эквивалентно 270 мг соединения 1) приводила в результате к значительной противовирусной активности у инфицированных GT1b-HCV субъектов. Среднее максимальное снижение РНК HCV через 24 часа после введения однократной дозы 300 мг составляло 1,7 log10 МЕ/мл, и это сопоставимо со снижением -2 log10 МЕ/мл через 1 сутки после 400 мг монотерапии софосбувира у инфицированных GT1a HCV субъектов. Среднее максимальное снижение РНК HCV через 24 часа после введения однократной дозы 100 мг составляло 0,8 log10 МЕ/мл. Среднее максимальное снижение РНК HCV составляло 2,2 log10 МЕ/мл после однократной дозы 400 мг. Индивидуальные фармакокинетические/фармакодинамические анализы для отдельных субъектов из части В исследования показаны на фиг. 23А - 23F. Концентрацию метаболита 1-7 наносили на график против снижения концентрации РНК HCV, и как показано на фиг. 23А - 23F, снижение РНК HCV в плазме коррелирует с воздействием метаболита 1-7 в плазме. Вирусологический ответ являлся устойчивым с концентрациями в плазме метаболита 1-7, которые превышали значение ЕС95 в отношении GT1b. Корреляция между концентрацией в плазме и уровнями снижения РНК HCV указывает на то, что более выраженный ответ будет достигнут с более высокими дозами соединения 2.
Пример 25. Прогнозируемые равновесные содержания метаболита 1-7 превышают значения ЕС95 соединения 1 в отношении клинических изолятов GT 1-4 HCV
Как показано на фиг. 24, равновесные остаточные содержания в плазме (C24,ss) метаболита 1-7 после введения дозы соединения 2 у людей (600 мг QD (550 мг эквивалент свободного основания) и 450 мг QD (400 мг эквивалент свободного основания)) прогнозировали и сравнивали с ЕС95 соединения 1 in vitro по всем тестируемым клиническим изолятам, чтобы определить, является ли равновесная концентрация в плазме стабильно более высокой, чем ЕС95, что приводило бы в результате к высокой эффективности против любых или всех тестируемых клинических изолятов in vivo. ЕС95 для соединения 1 является такой же, что и ЕС95 соединения 2. Для того, чтобы соединение 2 было эффективным, равновесное остаточное содержание в плазме метаболита 1-7 должно превышать ЕС95.
Как показано на фиг. 24, ЕС95 соединения 2 против всех тестируемых клинических изолятов варьировала от приблизительно 18 до 24 нМ.
Как показано на фиг. 24, соединение 2 при дозе 450 мг QD (400 мг эквивалент свободного основания) у людей обеспечивает прогнозируемую равновесную остаточную концентрацию в плазме (C24,ss) приблизительно 40 нг/мл. Соединение 2 при дозе 600 мг QD (550 мг эквивалент свободного основания) у людей обеспечивает прогнозируемую равновесную остаточную концентрацию в плазме (C24,ss) приблизительно 50 нг/мл.
Следовательно, прогнозируемая равновесная концентрация в плазме заменителя метаболита 1-7 почти вдвое превышает ЕС95 в отношении всех тестируемых клинических изолятов (даже трудно поддающегося лечению GT3a), что указывает на превосходную эффективность.
В отличие от этого, ЕС95 стандарта лечения - нуклеотида софосбувира - варьирует от 50 до 265 нМ в отношении всех тестируемых клинических изолятов HCV, при этом ЕС95 меньше прогнозируемой равновесной концентрации при коммерческой дозировке 400 мг только для двух изолятов GT2a и GT2b. ЕС95 для коммерческой дозировки 400 мг софосбувира превышает прогнозируемую равновесную концентрацию для других клинических изолятов GT1a, GT1b, GT3a, GT4a и GT4d.
Равновесную остаточную концентрацию в плазме (C24,ss) 450 мг соединения 2 прогнозировали с использованием равновесной остаточной концентрации в плазме (C24,ss) 300 мг. Средняя равновесная остаточная концентрация в плазме (C24,ss) при 300 мг составляла 26,4 нг/мл, и, следовательно, расчет был следующим 26,4*450/300=39,6 нг/мл.
Равновесную остаточную концентрацию в плазме (C24,ss) 600 мг прогнозировали с использованием трех подходов: 1) средняя С24 при 600 мг в сутки 1 составляла 25,8 нг/мл, и предполагали 60% повышение для достижения равновесия. Следовательно, расчет был следующим 25,8*1,6=41,3 нг/мл; 2) средняя С24 при 400 мг в сутки 1 составляла 22,5 нг/мл, и предполагали 60% повышение для достижения равновесия. С учетом пропорциональной дозе PK расчет был следующим 22,5*1,6*600/400=54 нг/мл; и 3) равновесная остаточная концентрация в плазме (C24,ss) при 300 мг составляла 26,4 нг/мл, и предполагалась пропорциональная PK. Следовательно, расчет был следующим 26,4*2=52,8 нг/мл. Равновесная остаточная концентрация в плазме (C24,ss) при 600 мг является средним значением 3 точек данных ((41,3+54+52,8)/3=49,3 нг/мл). Как правило, при равновесном состоянии С24 увеличивается на приблизительно 60% по сравнению с С24 после однократной дозы.
Сравнение данных эффективности и фармакокинетических равновесных параметров на фиг. 24 четко демонстрирует неожиданную терапевтическую важность соединения 2 для лечения гепатита С. Действительно, прогнозируемое равновесное содержание в плазме после введения соединения 2, как предполагали, по меньшей мере в 2 раза превышает ЕС95 в отношении всех тестируемых генотипов и в 3-5 раз эффективнее против GT2. Эти данные указывают на то, что соединение 2 обладает мощной активностью против вирусов всех генотипов у людей. Как показано на фиг. 24, ЕС95 софосбувира в отношении GT1, GT3 и GT4 превышает 100 нг/мл. Таким образом удивительно, что соединение 2 является активным против HCV в лекарственной форме, которая обеспечивает более низкую равновесную остаточную концентрацию (40-50 нг/мл), чем равновесная остаточная концентрация (приблизительно 100 нг/мл), обеспечиваемая подобной лекарственной формой софосбувира.
Пример 26. Описание состава и изготовление соединения 2
Типичный неограничивающий состав партии для таблеток соединения 2 (50 мг и 100 мг) представлен в таблице 36. Таблетки получали из общей смеси с использованием процесса прямого прессования, как показано на фиг. 25. Активный фармацевтический ингредиент (API) регулировали на основании анализа без пересчета на сухое вещество с регулировкой процентного содержания микрокристаллической целлюлозы. API и вспомогательные средства (микрокристаллическую целлюлозу, моногидрат лактозы и кроскармеллозу натрия) просеивали, помещали в V-образный смеситель (PK Blendmaster, емкость 0,5 л) и смешивали в течение 5 минут при 25 оборотах в минуту. Затем стеарат магния просеивали, добавляли и смесь перемешивали еще 2 минуты. Общую смесь делили для применения в получении таблеток 50 мг и 100 мг. Затем смазанную смесь прессовали при скорости 10 таблеток/минута с использованием однопуансонного исследовательского таблеточного пресса (Korsch ХР1) и порошкового питателя гравитационного типа. 50-мг таблетки получали с использованием круглого стандартного вогнутого 6-мм инструмента и усилий 3,5 кН. 100-мг таблетки получали с использованием круглого стандартного вогнутого 8-мм инструмента и усилий 3,9-4,2 кН.
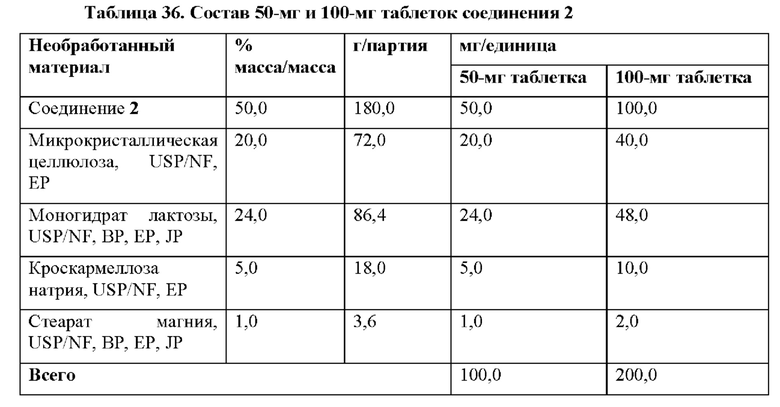
Соединение 2 регулировали на основании анализа без пересчета на сухое вещество с регулировкой процентного содержания микрокристаллической целлюлозы. Соединение 2 и вспомогательные средства (микрокристаллическую целлюлозу, моногидрат лактозы и кроскармеллозу натрия) просеивали, помещали в V-образный смеситель (PK Blendmaster, емкость 0,5 л) и смешивали в течение 5 минут при 25 оборотах в минуту. Затем стеарат магния просеивали, добавляли и смесь перемешивали еще 2 минуты. Общую смесь делили для применения в получении таблеток 50 мг и 100 мг. Затем смазанную смесь прессовали при скорости 10 таблеток/минута с использованием однопуансонного исследовательского таблеточного пресса (Korsch ХР1) и порошкового питателя гравитационного типа. 50-мг таблетки получали с использованием круглого стандартного вогнутого 6-мм инструмента и усилий 3,5 кН. 100-мг таблетки получали с использованием круглого стандартного вогнутого 8-мм инструмента и усилий 3,9-4,2 кН. Описания 50-мг и 100-мг таблеток представлены в таблице 37.

50-мг и 100-мг таблетки, полученные, как описано выше, подвергали 6-месячным исследованиям стабильности при трех условиях: 5°С (условия в холодильнике), 25°С/60% RH (окружающие условия) и 40°С/75% RH (условия «ускоренного старения»). И 50-мг, и 100-мг таблетки были химически стабильными при всех трех тестируемых условиях.
В условиях хранения в холодильнике (5°С) и 50-мг, и 100-мг таблетки оставались белыми твердыми, и их внешний вид не менялся от Т=0 до Т=6 месяцев. На протяжении 6-месячного исследования не регистрировали никаких примесей, превышающих 0,05%, на для 50-мг таблеток, ни для 100-мг таблеток. Содержание воды через 6 месяцев также составляло менее 3,0% масса/масса для обеих таблеток. Подобные результаты регистрировали, когда таблетки подвергали окружающим условиям (25°С/60% RH); никаких примесей, превышающих 0,05%, не регистрировали на протяжении 6 месяцев для обеих таблеток, и содержание воды не превышало 3,0% масса/масса на 6-месячной отметке. Когда таблетки подвергали условиям «ускоренного старения» (40°С/75% RH), внешний вид 50-мг и 100-мг таблеток не отличался от белой, круглой таблетки. Одну примесь зарегистрировали через 3 месяца, но примесь составляла всего лишь 0,09%. Вторую примесь зарегистрировали через 6 месяцев, но суммарное процентное содержание примеси составляло всего лишь 0,21% как для 50-мг, так и для 100-мг таблеток. Содержание воды составляло 3,4% масса/масса через 6 месяцев для 50-мг таблеток и 3,2% масса/масса для 100-мг таблеток.
В отдельном исследовании измеряли стабильность 50-мг и 100-мг таблеток соединения 2 при окружающих условиях (25°С/60% RH) через 9 месяцев. Внешний вид 50-мг и 100-мг таблеток не отличался от белой, круглой таблетки на протяжении 9 месяцев. Примеси в 50-мг таблетке составляли менее 0,10% через 9 месяцев, а примеси в 100-мг таблетке составляли менее 0,05%. Содержание воды в 50-мг таблетке и 100-мг таблетке через 9 месяцев составляло всего лишь 2,7% масса/масса и 2,6% масса/масса, соответсвенно.
Настоящее описание описывается со ссылкой на варианты осуществления настоящего изобретения. Однако специалисту в данной области будет понятно, что различные модификации и изменения могут быть выполнены без отступления от объема настоящего изобретения, изложенного в приведенной ниже формуле изобретения. Следовательно, настоящее описание следует рассматривать как иллюстративное, а не ограничивающее, и все такие модификации предназначены для включения в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2009 |
|
RU2519947C2 |
| Противовирусная композиция и способ ее применения | 2017 |
|
RU2650610C1 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ | 2005 |
|
RU2393863C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ОТ HELICOBACTER PYLORI | 2014 |
|
RU2671400C2 |
| КИШЕЧНОРАСТВОРИМЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2018 |
|
RU2812901C2 |
| СПОСОБЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ИНФЕКЦИИ ГЕПАТИТА С | 2006 |
|
RU2440822C2 |
| Пролекарство ингибитора NS5B HCV полимеразы, способ его получения и применения | 2017 |
|
RU2644156C1 |
| ПТЕРИДИНЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ HCV, И СПОСОБЫ ПОЛУЧЕНИЯ ПТЕРИДИНОВ | 2006 |
|
RU2447074C2 |
| ПРОЛЕКАРСТВО НА ОСНОВЕ УРИДИНФОСФОРАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЯ В МЕДИЦИНЕ | 2017 |
|
RU2740760C2 |
| ЛЕЧЕНИЕ ГИПЕРКИНЕТИЧЕСКИХ ДВИГАТЕЛЬНЫХ РАССТРОЙСТВ | 2015 |
|
RU2753740C2 |
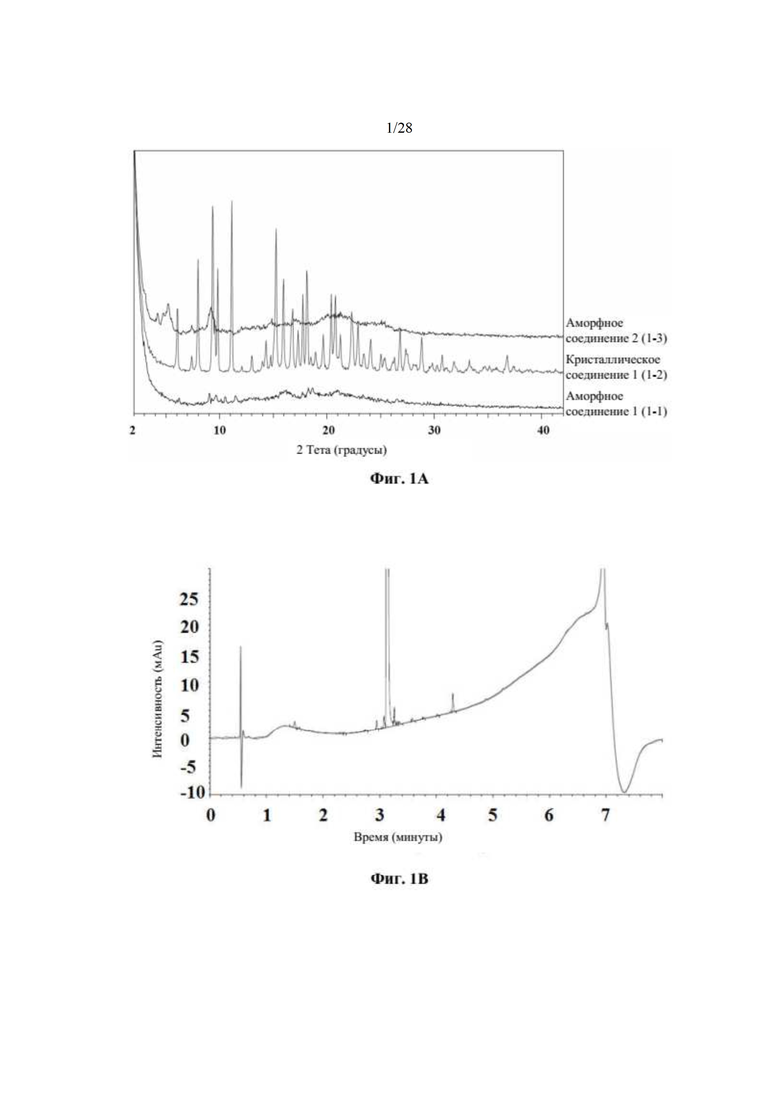
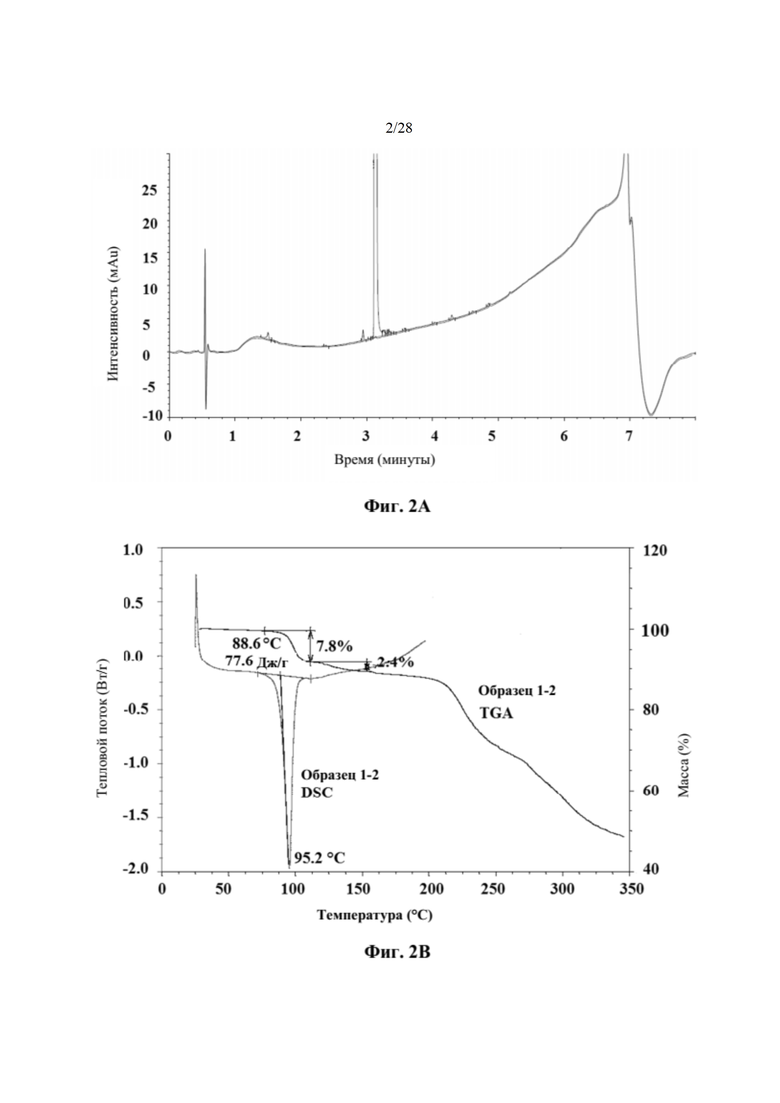
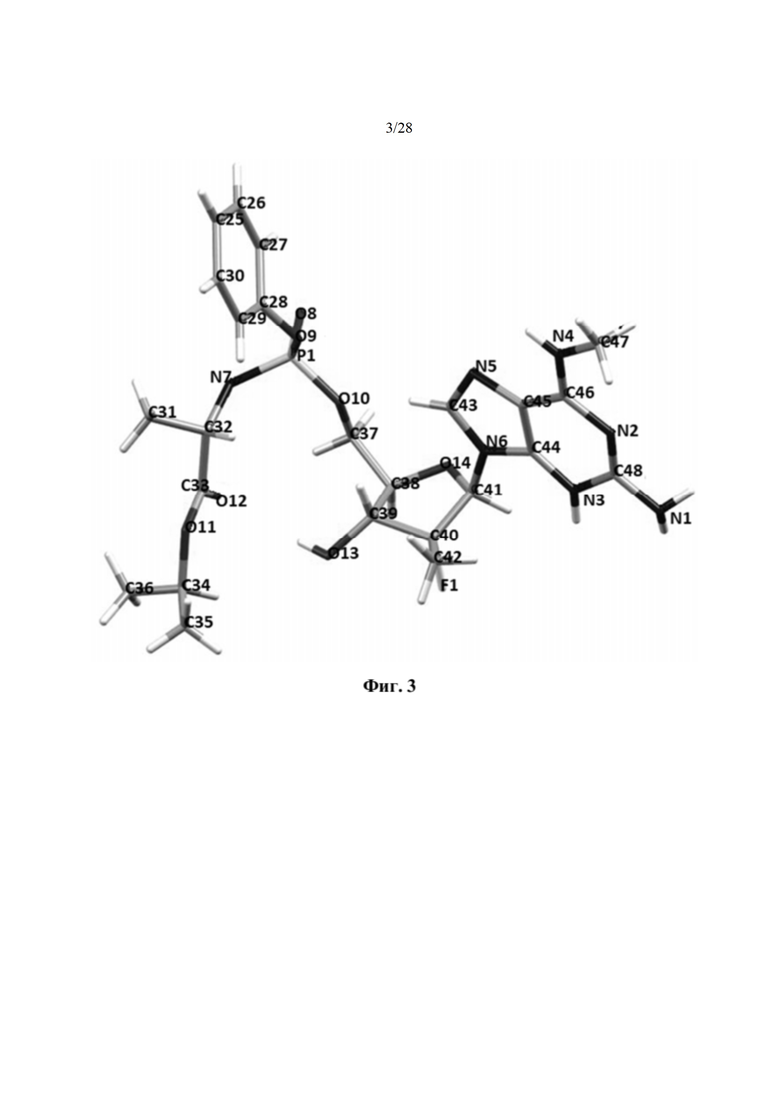

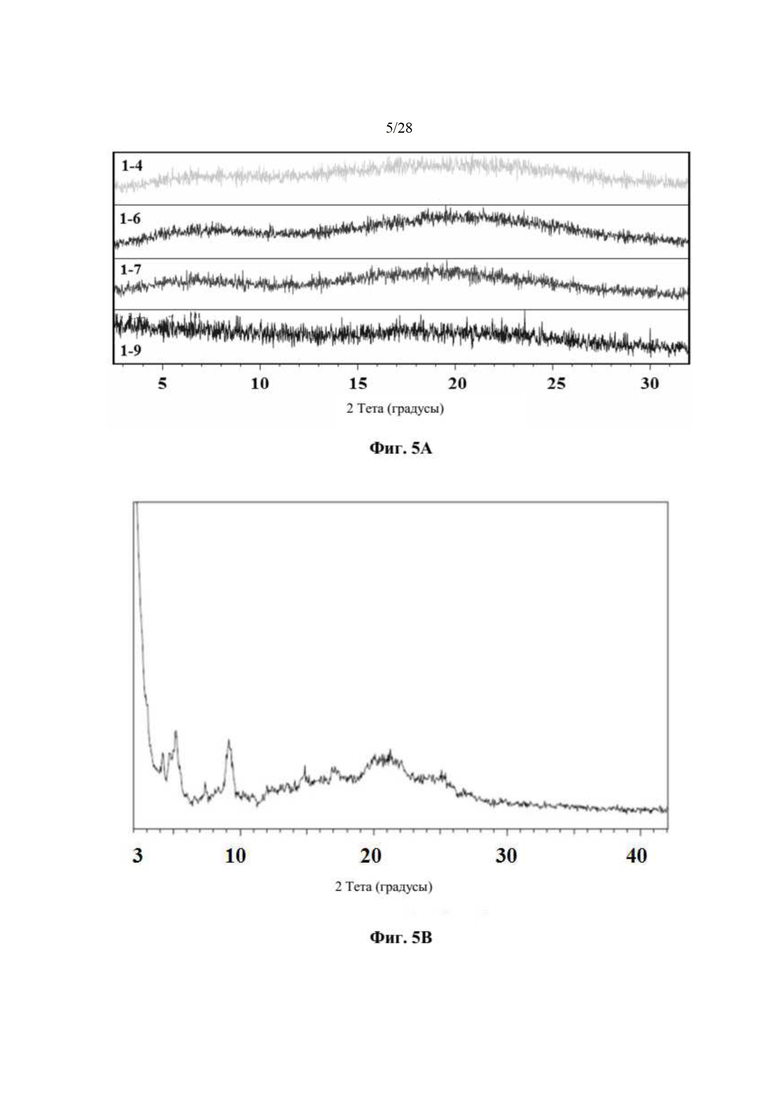
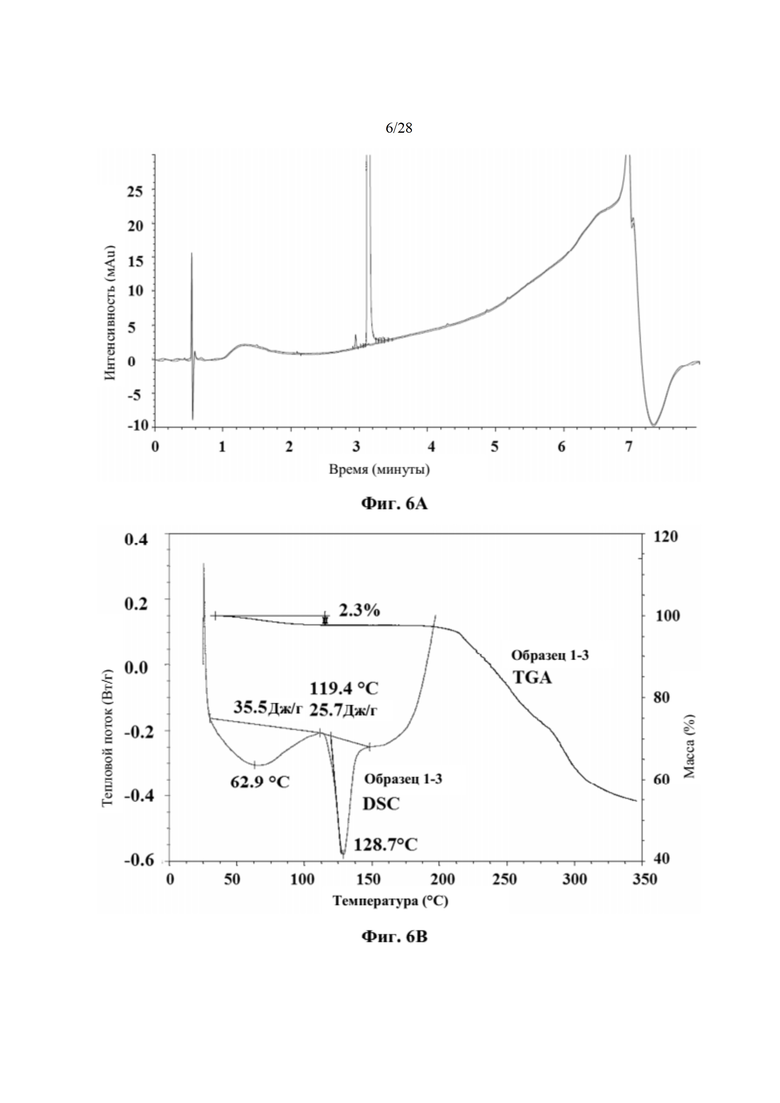
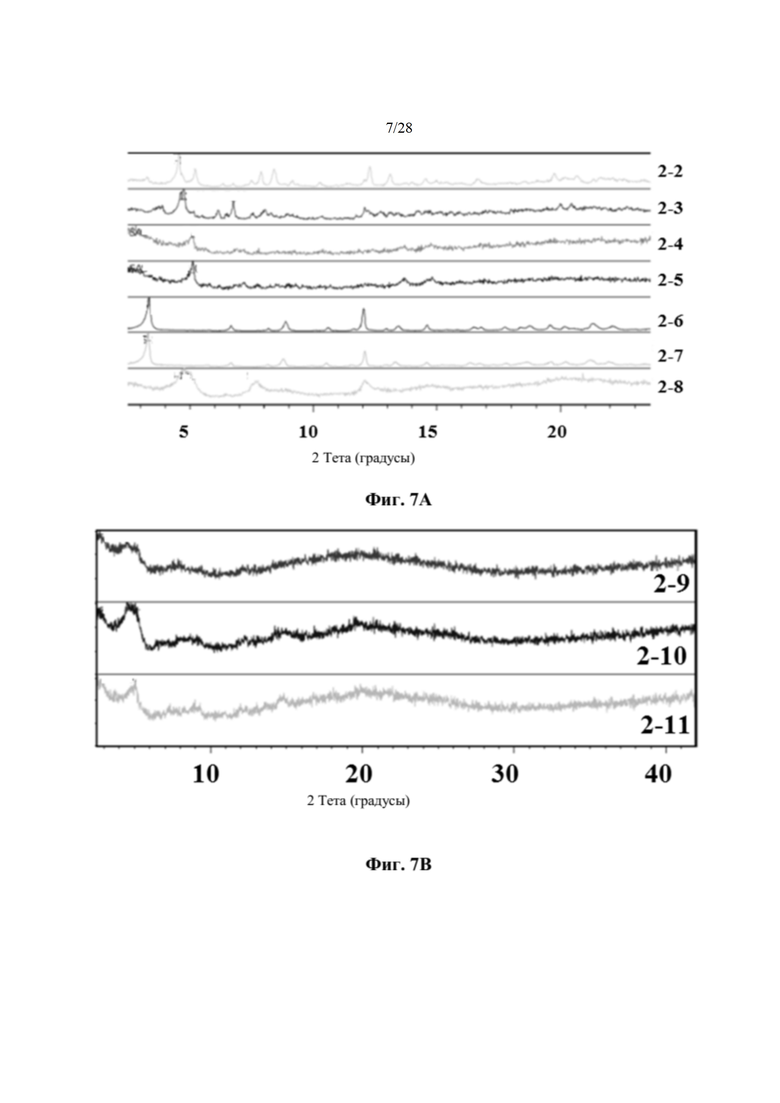
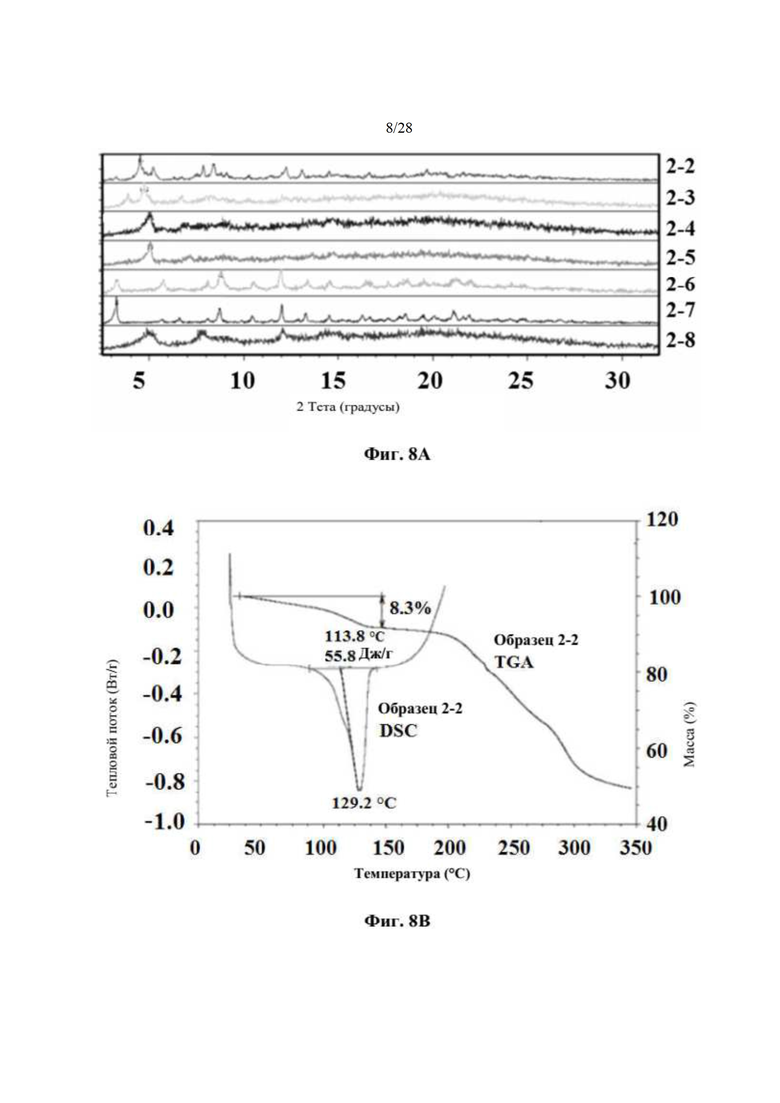
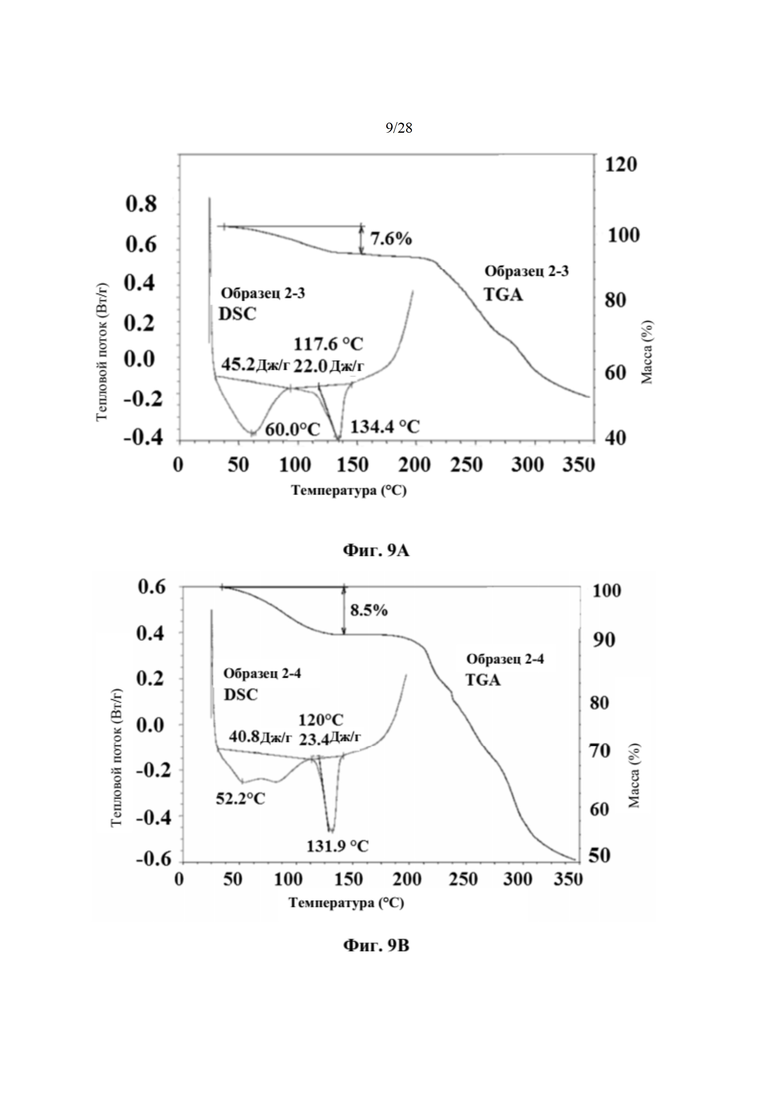
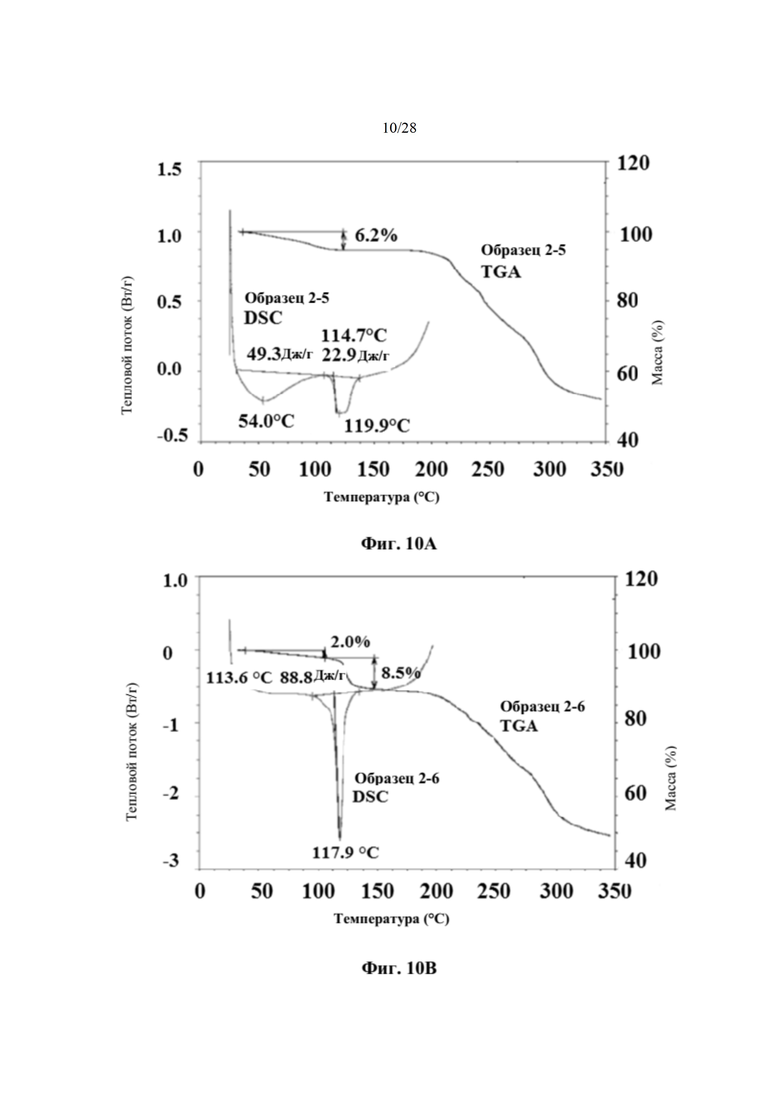
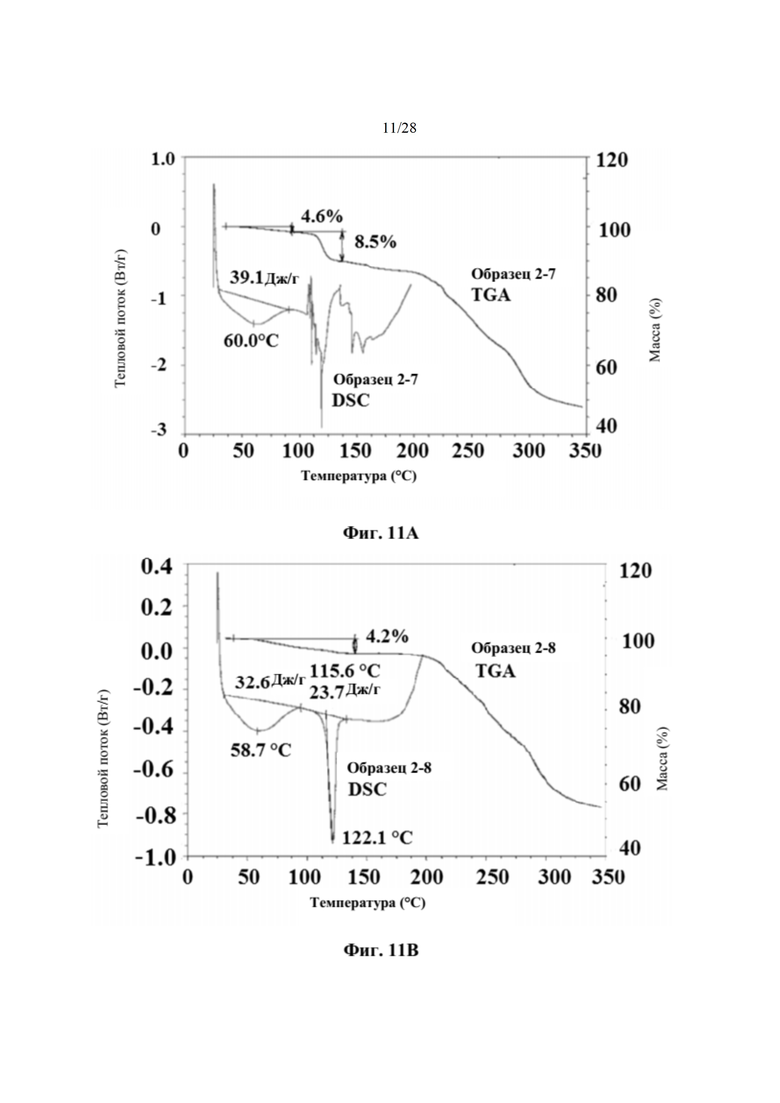

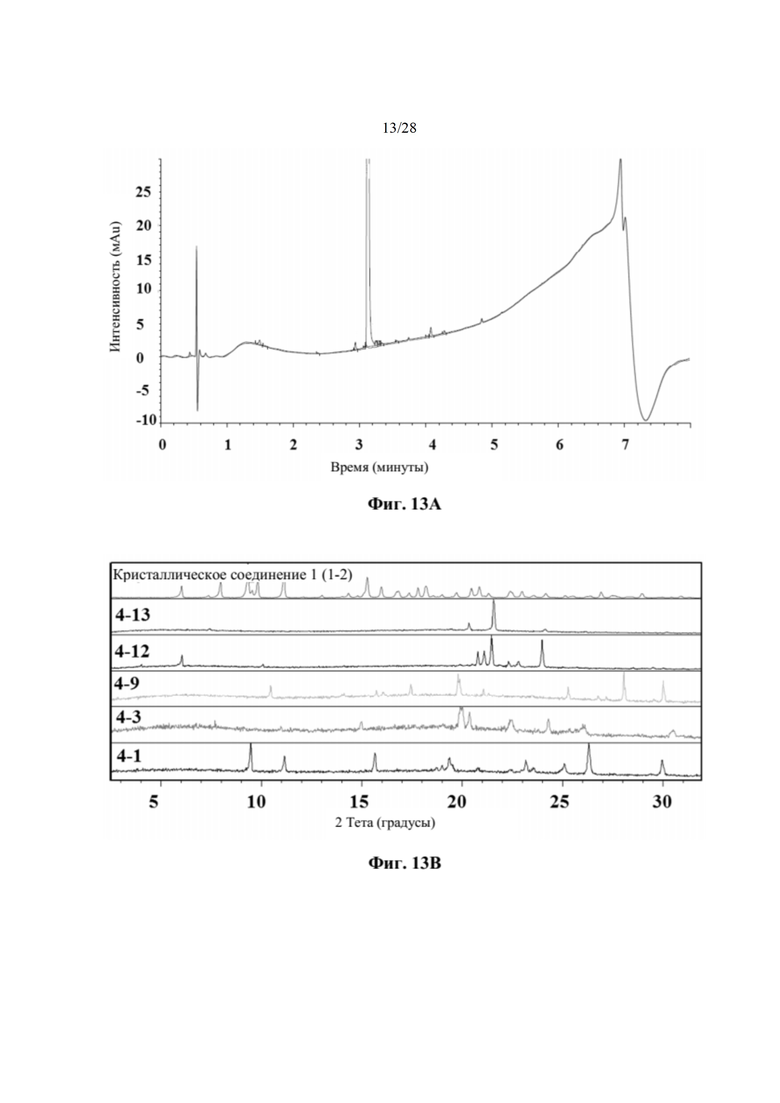
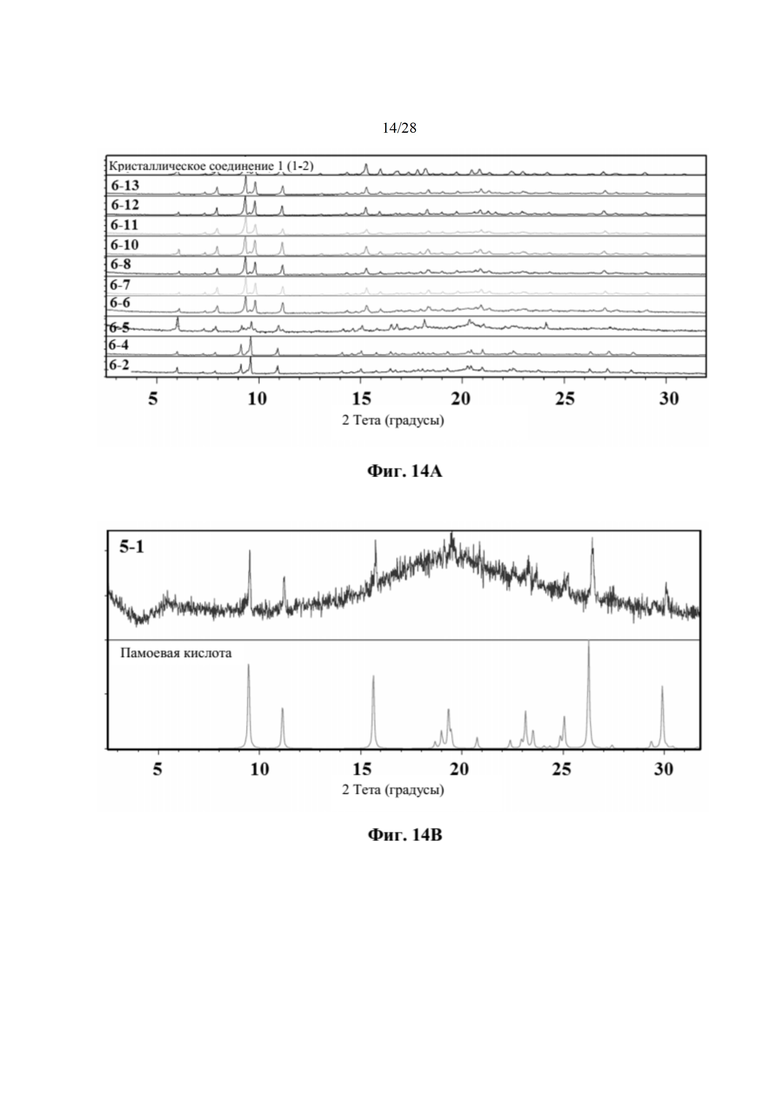
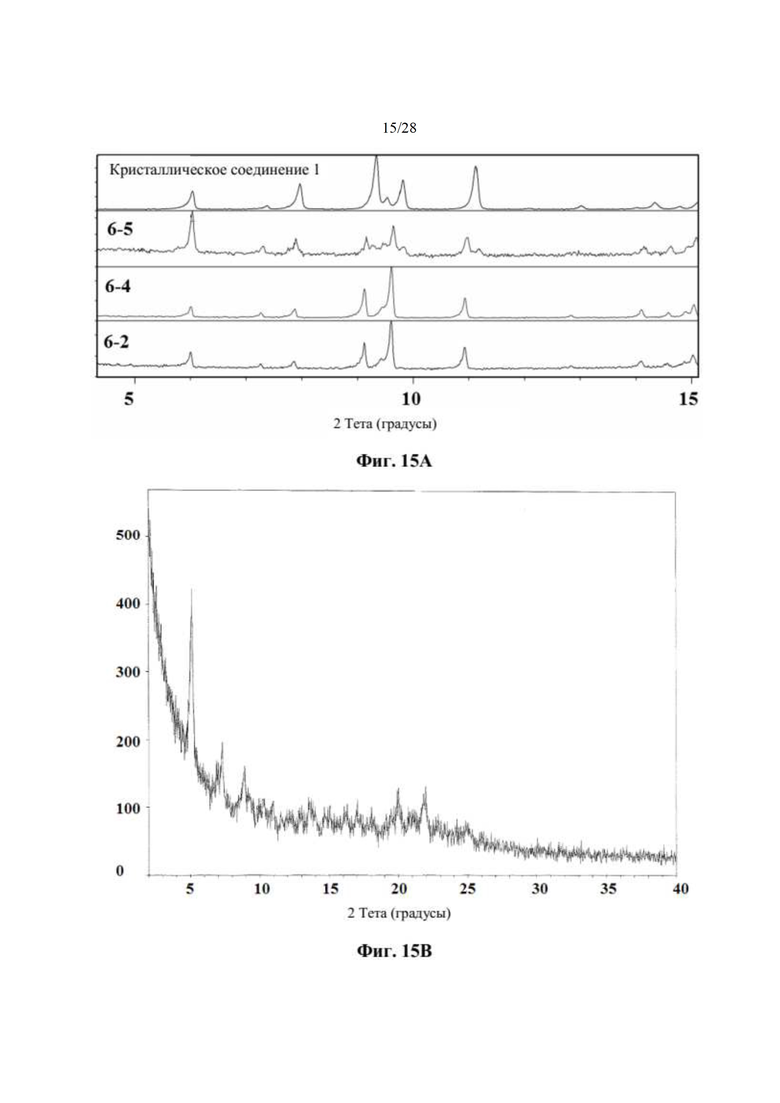
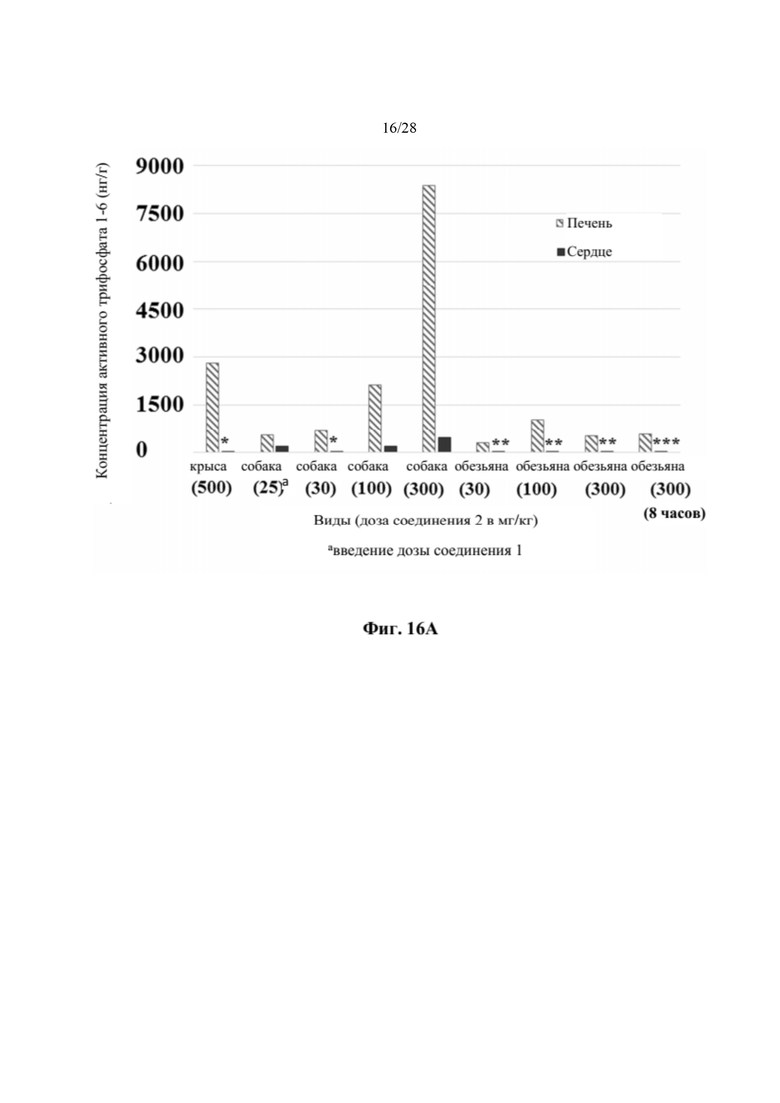
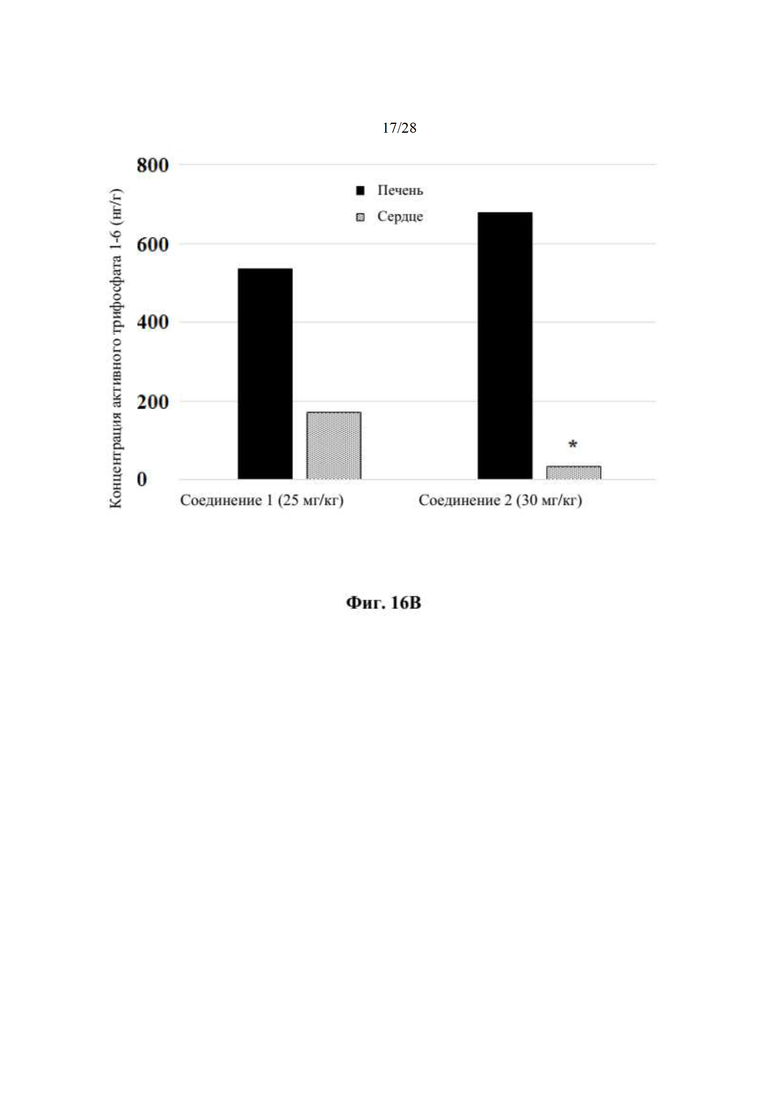
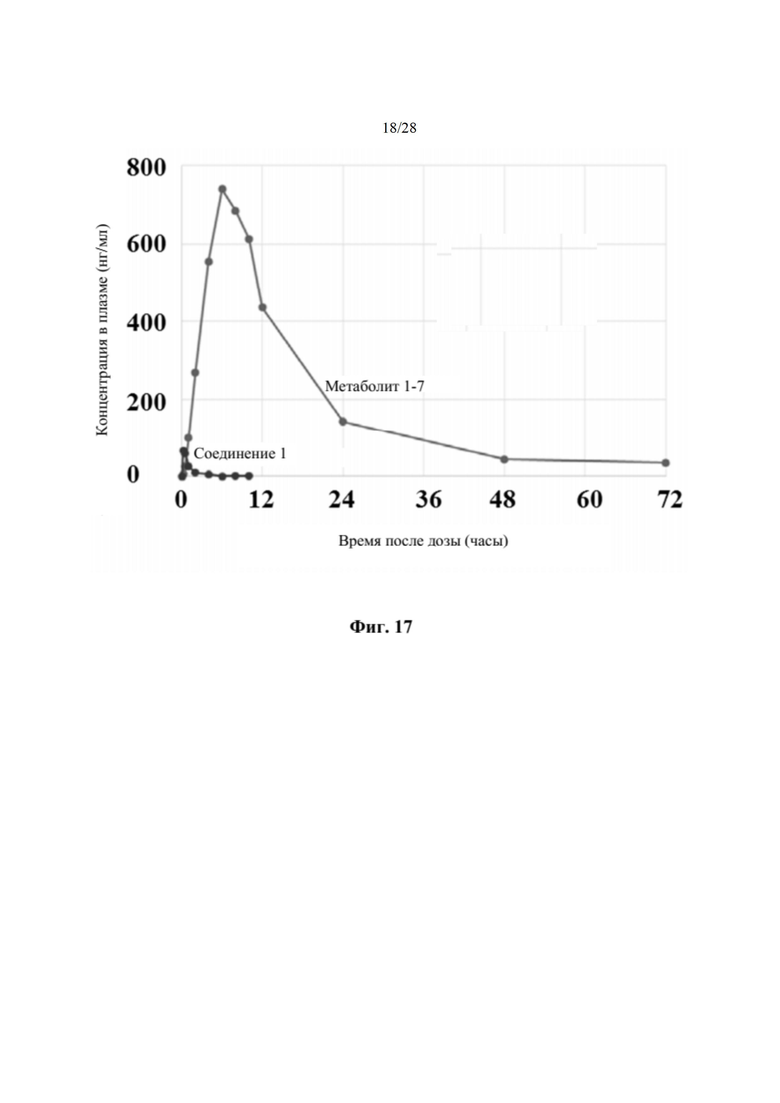
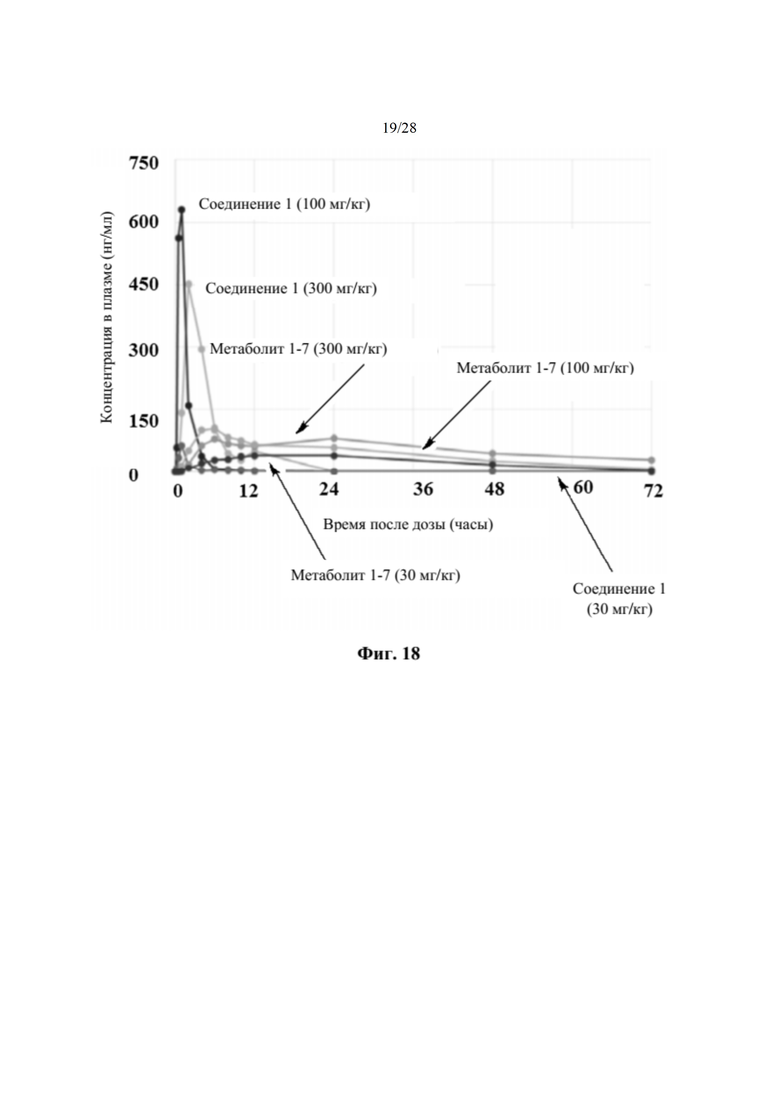

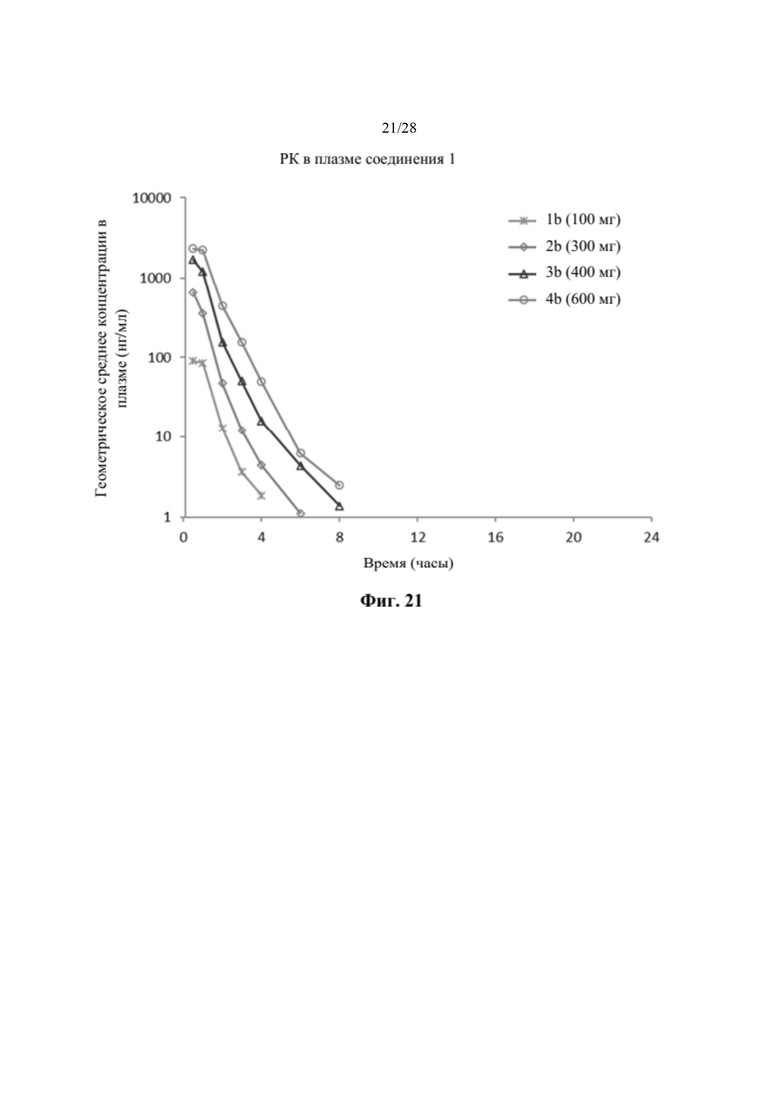
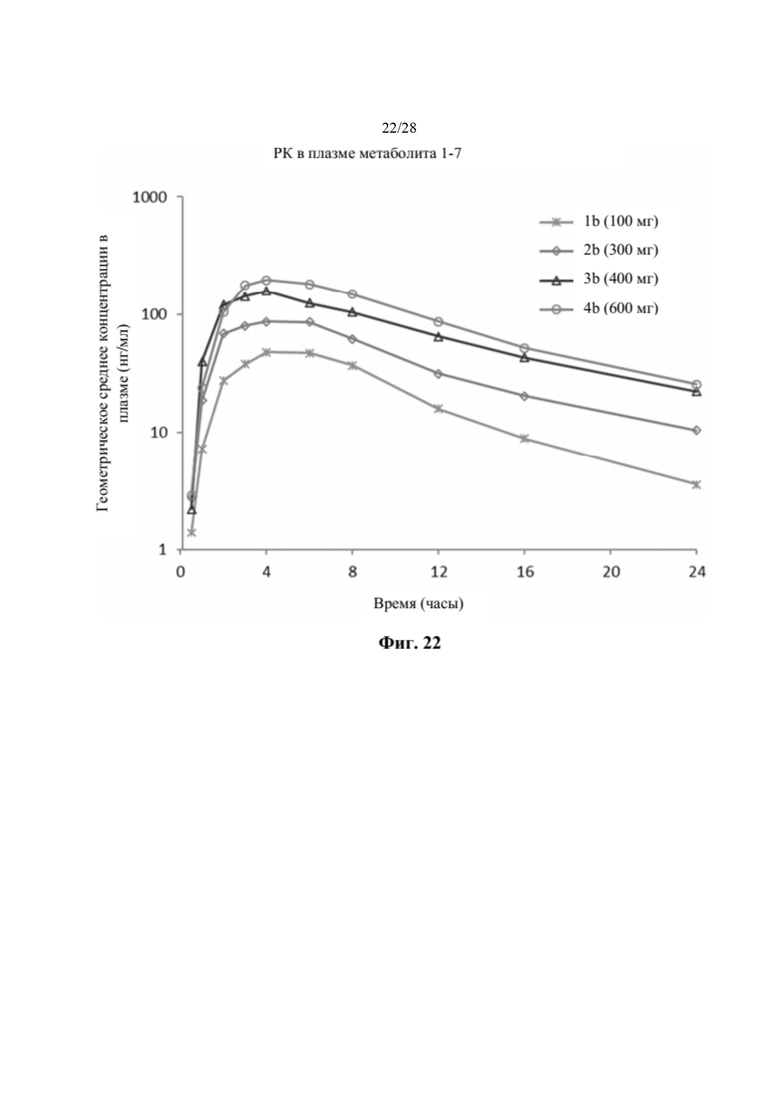

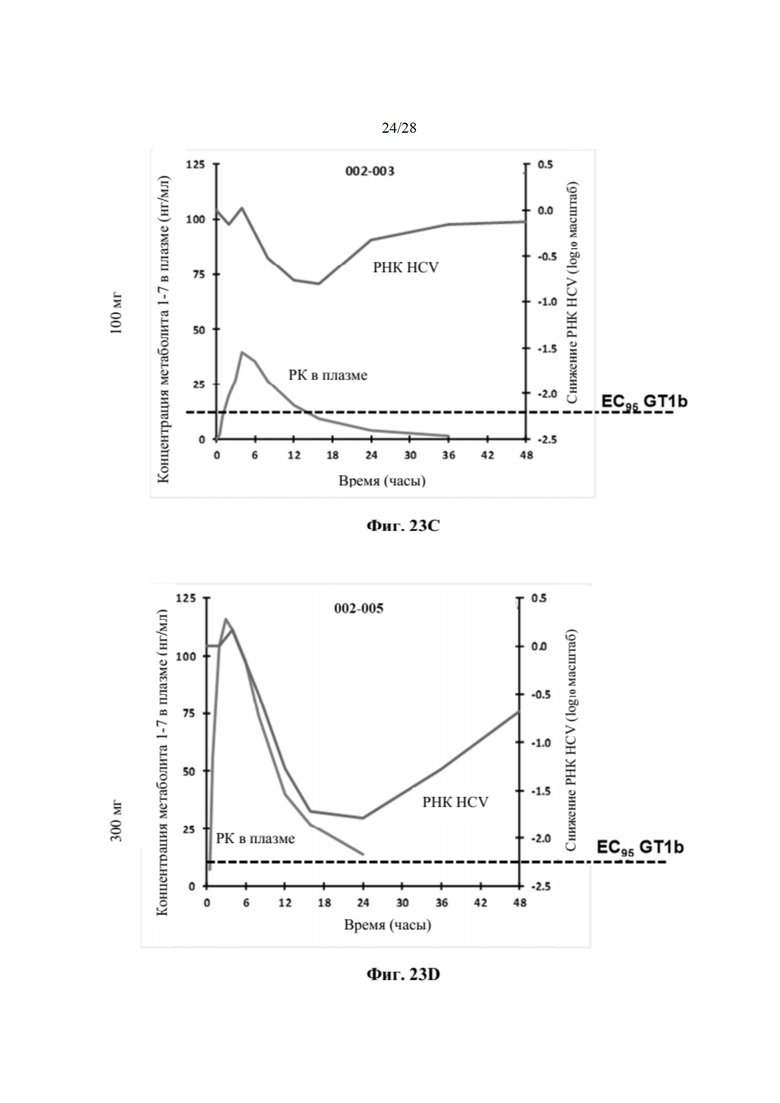
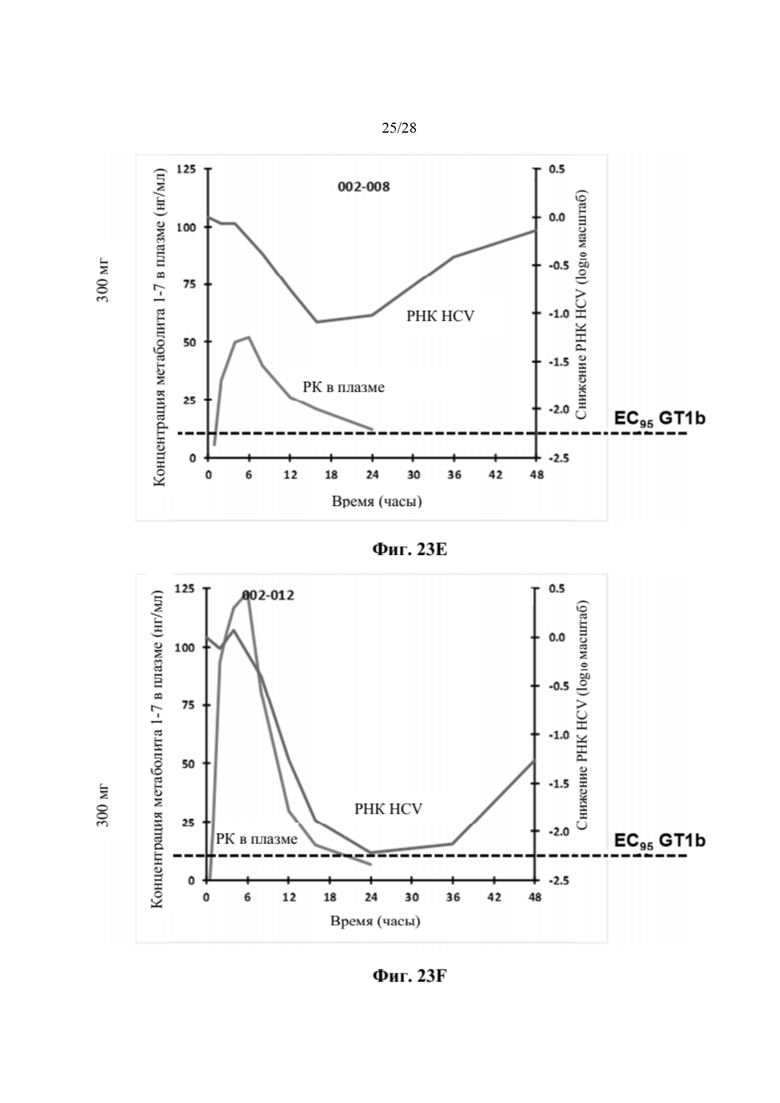
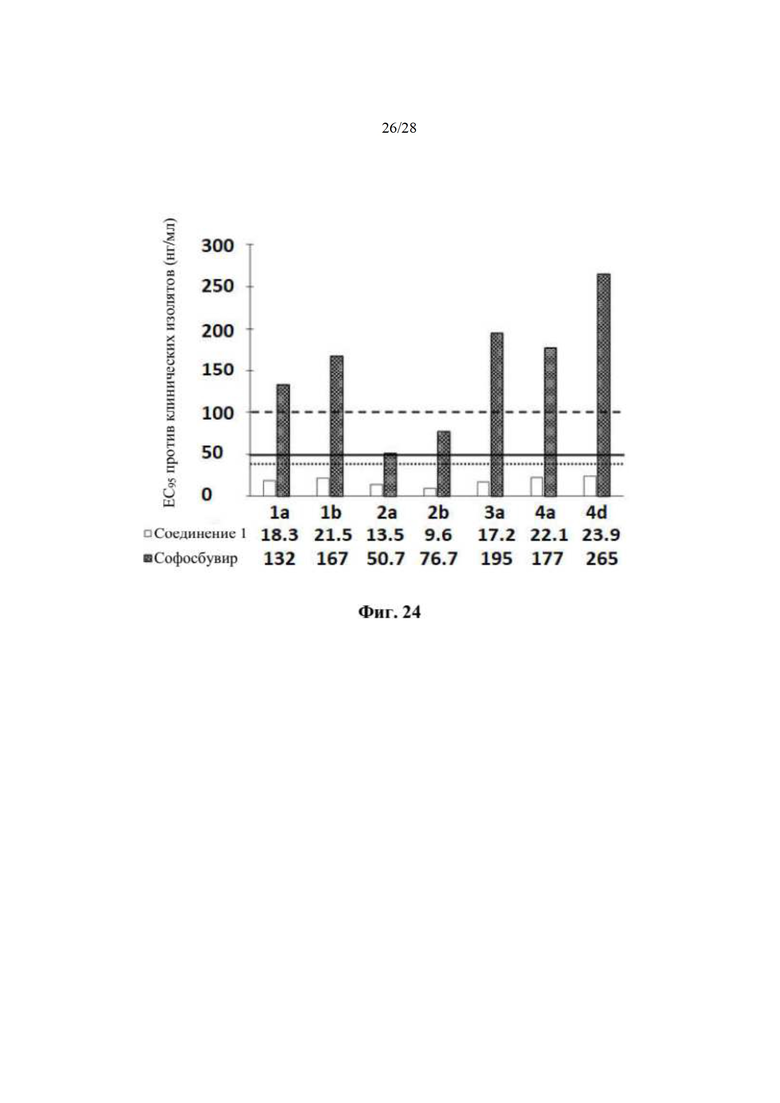

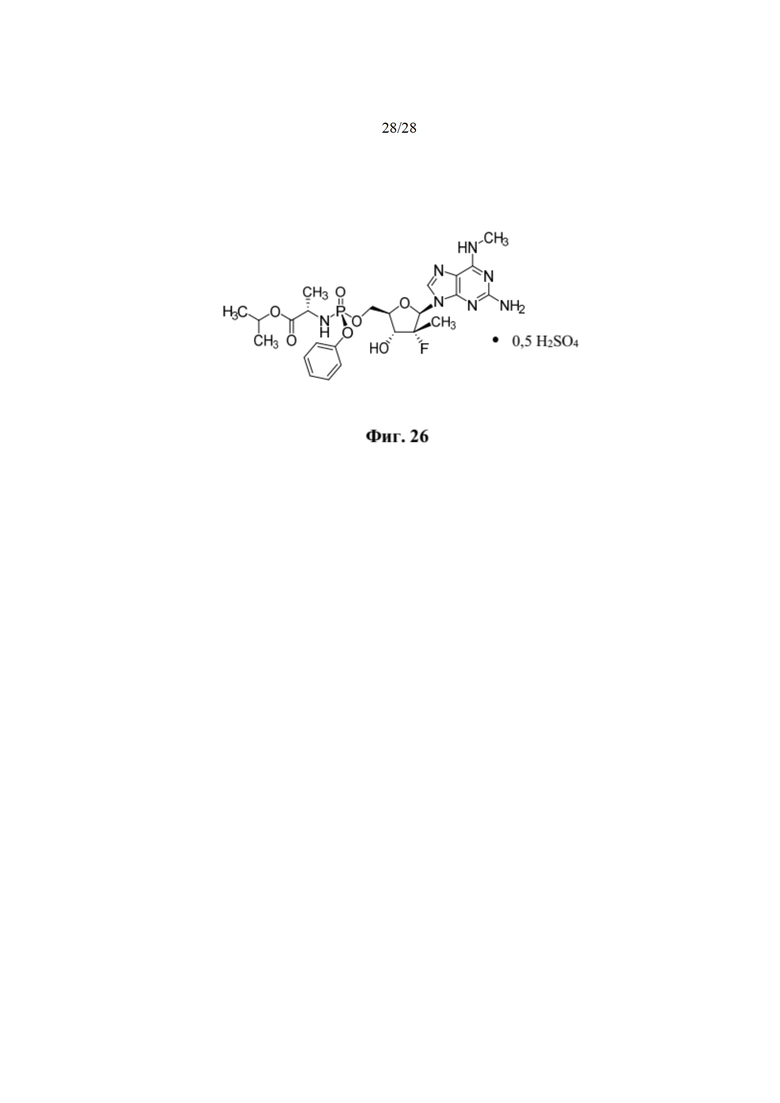
Группа изобретений относится к области органической химии и фармацевтики и направлена на лечение гепатита С. Раскрывается соединение представленной формулы. Кроме того, раскрыты фармацевтическая композиция для лечения или профилактики HCV, содержащая эффективное количество указанного соединения в фармацевтически приемлемом носителе, а также применение соединения по п. 1 для лечения вируса гепатита С (HCV) у человека. Группа изобретений обеспечивает эффективное лечение гепатита С. 4 н. и 28 з.п. ф-лы, 26 ил., 37 табл., 26 пр.

1. Соединение формулы:

2. Соединение по п. 1, причем соединение по меньшей мере на 90% свободно от противоположного R-энантиомера по фосфору.
3. Соединение по п. 1, причем соединение по меньшей мере на 98% свободно от противоположного R-энантиомера по фосфору.
4. Соединение по п. 1, причем соединение по меньшей мере на 99% свободно от противоположного R-энантиомера по фосфору.
5. Фармацевтическая композиция для лечения и/или профилактики HCV, содержащая эффективное количество соединения по любому из пп. 1-4 в фармацевтически приемлемом носителе.
6. Фармацевтическая композиция по п. 5 в пероральной лекарственной форме.
7. Фармацевтическая композиция по п. 6, причем пероральная лекарственная форма представляет собой твердую лекарственную форму.
8. Фармацевтическая композиция по п. 7, причем твердая лекарственная форма представляет собой таблетку.
9. Фармацевтическая композиция по п. 7, причем твердая лекарственная форма представляет собой капсулу.
10. Фармацевтическая композиция по п. 6, причем пероральная лекарственная форма представляет собой жидкую лекарственную форму.
11. Фармацевтическая композиция по п. 10, причем жидкая лекарственная форма представляет собой суспензию или раствор.
12. Фармацевтическая композиция по п. 5 во внутривенном составе.
13. Фармацевтическая композиция по п. 5 в парентеральном составе.
14. Фармацевтическая композиция по любому из пп. 5-13, которая доставляет по меньшей мере 500 мг соединения.
15. Фармацевтическая композиция по любому из пп. 5-13, которая доставляет по меньшей мере 600 мг соединения.
16. Фармацевтическая композиция по любому из пп. 5-13, которая доставляет по меньшей мере 700 мг соединения.
17. Применение соединения по п. 1, необязательно в фармацевтически приемлемом носителе, для лечения вируса гепатита С (HCV) у человека.
18. Применение соединения по любому из пп. 2-4, необязательно в фармацевтически приемлемом носителе, для лечения вируса гепатита С (HCV) у человека.
19. Применение по п. 17 или 18, в котором соединение вводится перорально.
20. Применение по п. 17 или 18, в котором соединение вводится внутривенно.
21. Применение по п. 17 или 18, в котором соединение вводится парентерально.
22. Применение по любому из пп. 19-21, в котором вводится по меньшей мере 500 мг соединения.
23. Применение по любому из пп. 19-21, в котором вводится по меньшей мере 600 мг соединения.
24. Применение по любому из пп. 19-21, в котором вводится по меньшей мере 700 мг соединения.
25. Применение по любому из пп. 19-24, в котором соединение вводится в течение вплоть до 6 недель.
26. Применение по любому из пп. 19-25, в котором соединение вводится четыре раза в сутки.
27. Применение по любому из пп. 19-25, в котором соединение вводится два раза в сутки.
28. Применение по любому из пп. 19-27, в котором вирус гепатита С относится к генотипу 1а или 1b.
29. Применение по любому из пп. 19-27, в котором вирус гепатита С относится к генотипу 2а или 2b.
30. Применение по любому из пп. 19-27, в котором вирус гепатита С относится к генотипу За.
31. Применение по любому из пп. 19-27, в котором вирус гепатита С относится к генотипу 4а или 4d.
32. Применение по любому из пп. 19-27, в котором вирус гепатита С относится к генотипу 5а.
| WO 2016144918 A1, 15.09.2016 | |||
| US 2010286083 A1, 11.11.2010 | |||
| WO 2010068708 A1, 17.06.2010 | |||
| RU 2013125713 A2, 22.11.2012. |

