ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к соединениям, которые являются ингибиторами киназной активности, фармацевтическим составам, содержащим эти соединения, и вариантам их применения в лечении и предотвращении вирусных инфекций и нарушений, вызываемых или усугубляемых такой вирусной инфекцией.
УРОВЕНЬ ТЕХНИКИ
Род Enterovirus (энтеровирусы) семейства Picornaviridae одноцепочечных вирусов, содержащих одноцепочечную РНК с положительной смысловой цепью, включает ряд патогенов человека, которые могут вызывать очень тяжелые болезни. Примеры включают вирус полиомиелита, вирус Коксаки В (асептический менингит, миокардит, панкректит и неспецифическая фибрильная болезнь), энтеровирус-А71 (асептический менингит, энцефалит и паралич, сходный с полиомиелитом), энтеровирус-D68 (острый вялый миелит) и парэховирус (миокардит и энцефалит). Энтеровирусные инфекции (вариант А24 вируса Коксаки и тип 70 энтеровируса) являются причиной большинства случаев острого геморрагического конъюнктивита (ОГК, АНС). Многочисленные эпидемии и три пандемии ОГК имели место начиная с 1969 (Yin-Murphy et al., British Journal of Ophthalmology, 1986, 70, 869; Nilsson et al., Journal of Virology, 2008 82, 3061). Однако у большинства людей с энтеровирусной инфекцией развиваются намного менее серьезные болезни. Простуда является одной из наиболее распространенных болезней человека, и она чаще всего связана с другим видом энтеровируса - риновирусом человека (HRV, РВЧ), который вызывает 30-50% случаев простуды.
Оптимальная температура для репликации HRV составляет 33-35°С, что способствует развитию инфекции верхних дыхательных путей (ИВДП) и болезни, которая чаще всего является мягкой и разрешается без медицинского вмешательства. Однако ИВДП могут иметь осложнения, и HRV был определен в среднем ухе у ~40% детей в возрасте до 7, страдающих эксудативным средним отитом, включая хронические случаи (Papadopoulus et al., Paediatric Allergy and Immunology, 2006: 17: 514), в мокроте у 26% из 291 пациента, страдающего острым бронхитом (Park et al. Plos One, 2016, 11, e0165553), в аспиратах и соскобах из верхнечелюстной пазухи у 15 из 34 пациентов, страдающих от острого синусита,  et al., Journal of Clinical Microbiology, 1997, 35, 1791 и Clinical Infectious Diseases, 2001 33, 909) и у 29% пациентов, проходящих функциональную эндоскопическую синусохирургию по поводу хронического риносинусита (Abshirini et al., Jundishapur Journal of Microbiology, 2015, 8, e20068).
et al., Journal of Clinical Microbiology, 1997, 35, 1791 и Clinical Infectious Diseases, 2001 33, 909) и у 29% пациентов, проходящих функциональную эндоскопическую синусохирургию по поводу хронического риносинусита (Abshirini et al., Jundishapur Journal of Microbiology, 2015, 8, e20068).
Температура в дыхательных путях крупного и среднего размера в легких также должна допускать репликацию HRV (McFadden ER Jr, et al., J. Appl. Physiol., 1985, 58, 564), и у некоторых групп пациентов инфекция может вызывать очень тяжелую болезнь. У детей в возрасте до 5 риновирусная инфекция часто приводит к госпитализации (4,8 случаев/1000 детей: Miller et al., Journal of Infectious Diseases, 2007; 195. 773), при этом тяжесть болезни близка к тяжести болезни, обусловленной инфекцией респираторным синцитиальным вирусом (RSV) (McMillan et al., Pediatric Infectious Disease Journal, 1993, 12, 321), и часто приводит к бронхиолиту и пневмонии (Kellner et al., Acta Paediatrica Scandinavica, 1989, 78, 390; McMillan et al., Pediatric Infectious Disease Journal, 1993, 12, 321; El-Sahly et al., Clinical infectious Diseases, 2000, 31, 96; Jartti and Korppi, Pediatric Allergy and Immunology, 2011, 22, 350). В исследовании, выполненном Asner с соавт.(Influenza and Other espiratory Viruses, 2014, 8, 436), большинство детей с инфекцией риновирусом человека/ энтеровирусом имели фоновую иммуносупрессию или сопутствующие кардио-респираторные заболевания; и хорошо известно, что последствия инфекции риновирусом человека могут быть особенно тяжелыми для пациентов с этими состояниями (Anzueto et al., Chest, 2003, 123. 1664; Rotbart, Antivir. Res. 2002, 53, 83). Например, у 7 из 22 реципиентов трансплантатов с миелосупрессией, инфицированных риновирусом, развилась пневмония, которая привела к смерти (Ghosh et al., Clinical infectious Diseases, 1999, 29, 528).
Энтеровирусные инфекции также часто связаны с кожными высыпаниями (пузырчатка полости рта и конечностей, экзема, связанная с вирусом Коксаки, и друие атипичные экзантемы: Hubisch et al., Pediatric Infectious Disease Journal, 2014, 33, e92; Korman et al., Journal of the American Academy of Dermatology, 2017, 76, 538; Drago et al., Future Microbiology, 2017 12, 171).
HRV представляет собой вирус, чаще всего связанный с обострением астмы (приблизительно 25% обострений у взрослых и 50% у детей, Nicholson et al., BMJ, 1993, 307. 982; Johnston et al., BMJ., 199, 310.1225) и хронической обструктивной болезни легких (ХОБЛ: 20-26% Seemungal et al., Am. J. Respir. Crit. Care Med., 2001, 164. 1618; Papi et al., Am. J. Respir. Crit. Care Med., 2006, 173. 1114), и в обоих случаях было показано, что экспериментальное заражение риновирусом приводит к обострению (усугублению) заболевания (Zambrano et al., J Аллергия Clin Immunol., 2003, 111, 1008; Mallia et al., Am. J. Respir. Crit. Care Med., 2011, 183. 734). Риновирусные инфекции также часто связаны с обострением бронхоэктаза (16 - 25%: Kapur et al. Arch Dis Child 2014, 99, 749; Gao et al Chest 2015, 147, 1635) и муковисцидоза (CF, MB) (Etherington, J. Cystic Fibrosis 2014, 13, 49; Flight et al. Thorax, 2014, 69. 247). В случае ХОБЛ, MB и бронхоэктаза обострения являются более тяжелыми, когда они связаны с вирусной инфекцией (Papi et al. Am. J. Respir. Crit. Care Med., 2006, 173. 1114; Etherington, J. Cystic Fibrosis, 2014, 13, 49; Kapur et al., Arch Dis Child, 2014, 99, 749), и в каждом случае обострения способствуют прогрессированию заболевания с снижению выживаемости (Liou et al., Am J Epidemiol., 2001, 153, 345; Soler-Cataluna et al., Thorax, 2005, 60. 925; Roberts et al., Intern Med J., 2012 42, 129). Большинство вызванных риновирусом обострений ХОБЛ сопровождаются последующей вторичной бактериальной инфекцией (Mallia et al., Am. J. Respir. Crit. Care Med., 2012, 186, 1117; George et al., Eur Respir J., 2014;. 87). Кроме того, инфекция риновирусом человека является одним из факторов, которые могут направлять развитие астматического фенотипа иммунной системы у младенца (D.J. Jackson et al., Am. J. Respir. Crit. Care Med., 2008, 178, 667).
Социоэкономическое влияние риновируса человека огромно, и лечение часто включает неоправданное использование антибиотиков. По оценкам, простуда является причиной по меньшей мере двадцати пяти миллионов случаев отсутствия на рабочем месте и примерно такого же количества случаев отсутствия в школе каждый год в США (Rotbart, Antivir. Res., 2002 53, 83). Прямой и непрямой ущерб от простуды и связанных с ней осложнений только у астматиков в США оценивается в сорок миллиардов долларов ежегодно (А. М. Fendrick et al., Arch. Intern. Med., 2003, 163, 487.
Существует три вида HRV (А, В и С), включающие более 150 генотипов. HRV-A и -С выявляются чаще всего, и последний по-видимому представляет более патогенную группу, по меньшей мере в популяции детей с астмой (Piralla et al., Journal of Clinical Virology, 2009, 45, 311; Bizzintino et al., Eur. Respir. J., 2011, 37, 1037). Риновирусы также делятся на 3 обширные группы на основании клеточного рецептора, опосредующего их проникновение в клетку. Основная группа риновирусов человека (приблизительно 90% веротипов HRV-A и В) проникают в клетку путем взаимодействия с человеческой молекулой межклеточной адгезии (ICAM-1). Остальные ~10% вирусов HRV-A и В составляют менее многочисленную группу и используют для проникновения в клетку рецептор липопротеинов низкой плотности. Обнаруженные позднее виды HRV-C связываются с членом 3 родственного кадгерину семейства (CDHR3) чтобы осуществить проникновение. Риновирусы человека проникают в клетку, запуская рецептор-опосредованный эндоцитоз, а разборка оболочки происходит в эндосомах. Различия между серотипами не только препятствуют развитию перекрестного иммунитета в организме, но и в значительной степени затрудняют разработку вакцин и других вирус-специфичных способов предотвращения и лечения.
РНК-геном риновируса (HRV) человека без белков (~8 к.б.) окружен капсидом, состоящим из шестидесяти копий каждого из четырех структурных белков, обозначаемых VP1 - VP4, в икосаэдорической конфигурации, образующих вирусную частицу с диаметром ~30 нм. Для репликации HRV необходима РНК-зависимая РНК-полимераза, зависимая от вирусной РНК, а также ряд свпомогательных белков вируса и клетки-хозяина. Геном HRV транслируется в форме единого полипротеина, который после трансляции расщепляется кодируемыми вирусом протеазами на три белка, которые в свою очередь расщепляются в образованием по меньшей мере одиннадцати белков. Репликация вирусного генома может начаться всего через час после инфицирования, и всего через четыре часа после проникновения вируса в клетки может высвободиться почти миллион полностью собранных клеточных частиц, что сопровождается гибелью клеток.
В настоящее время не существует одобренного лекарства для применения у человека, которое излечивало бы инфекцию, вызываемую HRV. Несколько попыток воздействовать напрямую на HRV-вирусы показали некоторые положительные результаты. 4-[2-[1-(6-метил-3-пиридазинил)-4-пиперидинил]-этокси]бензоат, известный также как "пиродавир" способен действовать как капсид-связывающий ингибитор, но проблемы с растворимостью, эндогенным расщеплением, и стоимость ограничили возможности его применение против HRV. Плеконарил (ПИКОВИР) продемонстрировал эффективность в ингибировании репликации, но не был одобрен Управление по контролю за продуктами и лекарствами США (FDA) со ссылкой на значительные сомнения в его безопасности. Рупинтривир, ингибитор вирусной протеазы 3С, был эффективен в исследованиях на людях с экспериментальным заражением HRV, но оказался неэффективным против инфекций природным HRV (Bauer et al., Current Opinion in Virology, 2017, 24, 1). Были высказаны предположения, что определенные имидазопиразины могут быть эффективными противовирусными средствами против HRV и других вирусов; механизм из действия неясен, хотя считается, что он не связан с циклин-зависимыми киназами (Патентная публикация США 2011/0166147, Macleod et al.).
В свете вышесказанного сохраняется потребность в новых терапевтических средствах против риновирусов человека и энтеровирусов.
Несмотря на другие различия, репликация генома РНК-вирусов с положительной цепью зависят от единственной фундаментальной стадии РНК-зависимого синтеза РНК. Эта стадия важна для жизненного цикла вируса, и известно, что эти вирусы нуждаются во многих белках хозяина для запуска и поддержания активности РНК-зависимой РНК-полимеразы. Без взаимодействия с факторами хозяина вирусы не смогли бы реплицироваться/выжить. Следовательно, возможным терапевтическим вмешательством для подавления вирусных инфекций этого класса является блокирование взаимодействия вируса с хозяином, особенно в отношении ингибирования репликации вирусного генома. Манипуляции с факторами хозяина, существенными для вируса, но не существенными для хозяина, могут позволить добиться значительного ингибирования распространения вируса. Кроме того, избыточные факторы хозяина могут представлять собой многообещающие мишени для вмешательства. Это особенно справедливо для случая, когда большие классы вирусов развили способность взаимодействовать только с одним из ряда избыточных факторов хозяина. Одним набором белков-хозяев, которые считаются потенциальными мишенями для ингибирования репликации вирусов, являются фосфатидилинозитол-4-киназы.
Фосфатидилинозитол-4-киназы (PI4K) участвуют в нескольких видах клеточной активности, включая слияние мембран, везикулярный транспорт и клеточную сигнализацию, за счет катализирования фосфорилирования фосфатидилинозитора с образованием фосфатидилинозитол-4 фосфата (PI4P). Существует несколько известных изоформ PI4K, которые различаются по нескольким характеристикам, включая последовательность, размер, локализацию в тканях и клетках и функцию.
Считается, что один тип PI4K, фосфатидилинозитол (типа III-4-киназы, бета-полипептид (PI4KIIIβ) (также известный в литературе как фосфатидилинозитол 4-киназа (III) β, Ptdlns 4-киназа (III) β, PI4Kβ, Pi4kcb and PI4K92) имеет важное значения для контроля локальных популяций PI4P, в первую очередь, в аппарате Гольджи, где он необходим для поддержания структурной целостности органеллы. Этот фермент также был обнаружен в ядре. В недавних исследованиях было высказано предположение, что PI4KIIIβ участвует в репликации генома нескольких РНК-вирусов, включая риновирус человека, энтеровирусы 68 и 71, вирус полиомиелита, вирус Коксаки, вирус гепатита С, кобувирус крупного рогатого скора, Айчивирус, вирус бешенства и другие (см., например, van der Schaar et al., Antimicrobial Agents and Chemotherapy, 57, 4971; Roulin et al., Cell Host & Microbe, 2014, 16, 677; Mello et al., Antimicrobial Agents and Chemotherapy 2014, 58, 1546; Jun Sasaki et al., EMBO J. 2011, 31 754; Hsu et al., Cell 2010, 141. 799; Borawski, J. Virology 2009, 83, 10058; Altan-Bonnet et al., TIBS 2012, 37, 293). Кроме того, было показано, что каталитическая активность PI4KB необходима для опосредуемого шиловидным белком проникновения в клетку коронавируса SARS - вируса, вызывающего тяжелый острый респираторный синдром (Yang et al., J. Biol. Chem., 2012, 287, 8457). SARS является причиной эпидемии, начавшейся в Южном Китае, которая насчитывала 8448 случаев и 774 смерти в 37 странах с ноября 2002 г. по июль 2003 г. Макроэкономический ущерб от этой вспышки оценивали в 30 - 100 миллиардов долларов США (Smith, Social Science & Medicine, 2006, 63, 3113). Общепризнано, что ингибирование PI4KB может значительно снизить репликацию вируса у многих РНК-вирусов и, в частности, вирусов с положительной РНК-цепью, что делает PI4KB потенциальной мишенью для разработки противовирусных агентов широкого спектра. Дополнительно, PI4-киназы вовлечены в проникновение и репликацию бактерий, и есть предположение, что PI4KB участвуют в развитии инфекции, вызываемой Legionella pneumophila, за счет роли PI4P в заякоривании бактериальных белков к внутриклеточным вакуолям, содержащим легионеллы (Clayton et al. Pu Progress in Lipid Research 2013, 52 294). Соответственно, ингибитор PI4KB может быть эффективен для противодействия острому поражению легких или острому респираторному дистресс-синдрому, ассоциированным с инфекциями, вызываемыми легионеллами, и, возможно, в качестве средства лечения других бактериальных инфекций.
Е.Р. Keaney с соавт.(Bioorg. Med. Chem. Lett., 24 (2014) 3714-3718) описывает 2-производные алкилоксазола в качестве ингибторов PI4KIIIβ для потенциального лечения инфекций, обуслоалвенных вирусом гепатита С.
I. Medrova с соавт. (J. Med. Chem., 2017, 60(1), 100-118) описывают ряд производных имидазо[1,2-b]пиридазина в качестве ингибиторов PI4KIIIβ для возможного лечения вирусных инфекций.
J.В. Shotwell, в слайдовой презентации, озаглавленной tled "Chemical Optimization of Novel Inhibitor Classes for PI4KIIIβ: A Critical Host Factor for Enterovirus Replication", поданной для участия в "The 27th International Conference on Antiviral Research" (проводимой, в г. Роли, шт. Северная Каролина, США) и представленной 12.05.2014 описал ряд соединений, проявляющих активность на рецептора PI4KB. Слайды в основном относились к пероральному введению включают слайд, озаглавленный "Existing Chemotyopes were optimized for IN Delivery" (Существующие хемотипы были оптимизированы для интраназальной доставки), на которой было показано влияние на легкие интраназального введения соединения GSK3180404A и соединения GSK3159043A в моделях на крысах. На этом слайде показано значительное накопление соединения в такни леких для обоих соединений. Следующий слайд, озаглавленный "Nasal Epithelian Findings Observed following IN dose" (Находки в эпителии носа при после интраназального введения), содержит ряд гистологических изображений, демонстрирующих изъязвление в носовой полости у крыс и гиперплазию эпителия бронхов в легких крыс в результате интраназального введения GSK3159043.

Существует потребность в соединениях, которые являются мощными ингибиторами PI4KIIIβ. Также существует потребность в соединениях, которые также можно применять в качестве селективных ингибиторов PI4KIIIβ.
В частности, существует представляет собой потребность в соединениях, которые являются мощными ингибиторами PI4KIIIβ, которые не накапливаются в значительных количествах в тканях организма, например, в тканях легкого, особенно при введении с использованием ингаляционного или интраназального путей. Такие соединения могут применяться в лечении вирусных инфекций и нарушений, вызываемых или усугубляемых такой вирусной инфекцией, в частности, инфекций, вызываемых риновирусом человека (HRV).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям Формулы (I) или к их фармацевтически приемлемым солям,

где R1, R2, R3, R4, R4b, R4c, R5, W, X, Y и Z определены в настоящем документе.
Было показано, что соединения Формулы (I) являются селективными ингибиторами PI4KIIIβ и могут применяться в лечении или предотвращении вирусных инфекций и нарушений, вызываемых или усугубляемых такой вирусной инфекцией. Нарушения, вызываемые или усугубляемые вирусными инфекциями, включают, в частности ХОБЛ, астму, муковисцидоз, бронхоэктаз и застойную сердечную недостаточность. Дополнительно, нарушения, вызываемые или усугубляемые риновирусными инфекциями, включают бронхиолит, средний отит, синусит и острый бронхит. Также риновирусные инфекции могут приводить к вторичным бактериальным инфекциям у детей, пожилых людей и людей с иммуносупрессией. Такие вторичные бактериальные инфекции могут вызывать пневмонию.
Соответственно, настоящее изобретение дополнительно относится к способам лечения или предотвращения вирусных инфекций и нарушений, вызываемых или усугубляемых такой вирусной инфекцией, причем указанные способы включают введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы (I) или его фармацевтически приемлемая соль.
Настоящее изобретение также относится к фармацевтическим составам, содержащим терапевтически эффективное количество соединения Формулы (I) или его фармацевтически приемлемой соли и одно или более фармацевтически приемлемых вспомогательных веществ.
Настоящее изобретение также относится к соединениям Формулы (I) или их фармацевтически приемлемым солям для применения в терапии
Настоящее изобретение также относится к применению соединений Формулы (I) или их фармацевтически приемлемых солей в изготовлении лекарственного средства для лечения или предотвращения вирусных инфекций и нарушений, вызываемых или усугубляемых такой вирусной инфекцией.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с первым аспектом настоящего изобретения предложено соединение общей Формулы (I) или его фармацевтически приемлемая соль,
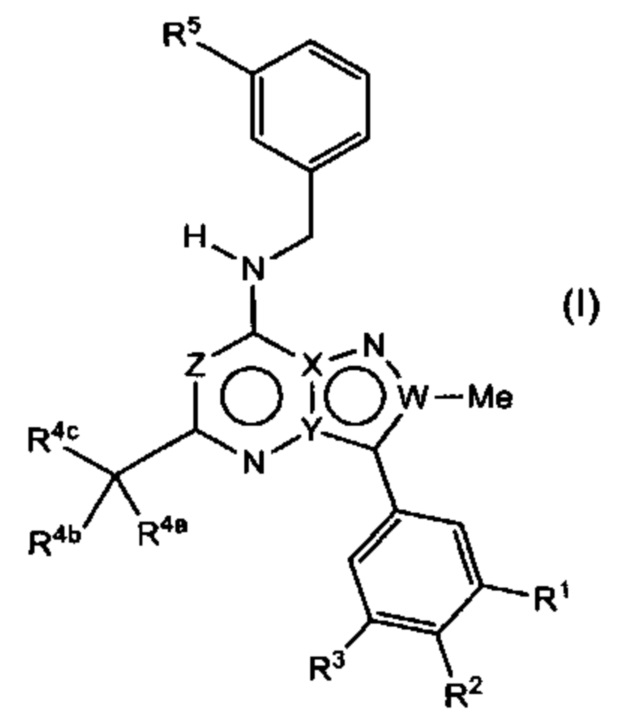
где
W представляет собой С, X представляет собой С, Y представляет собой N, и Z представляет собой С;
W представляет собой С, X представляет собой N, Y представляет собой С, и Z представляет собой С;
W представляет собой С, X представляет собой N, Y представляет собой С, и Z представляет собой N;
W представляет собой N, X представляет собой С, Y представляет собой С, и Z представляет собой N; или
W представляет собой N, X представляет собой С, Y представляет собой С, и Z представляет собой С;
R1 представляет собой С1-4 алкокси, -C(=O)N(R1aR1b), -S(=O)2-N(R1aR1b), -S(=O)2-R1c или -S(=O)-R1c, причем
R1a представляет собой С1-3алкил, галоС1-3алкил, гидроксиС1-3алкил, С1-3алкоксиС1-3алкил, тетрагидропиранил или тетрагидрофуранил; R1b представляет собой Н или С1-3алкил, или R1a и R1b, вместе с азотом, к которому они присоединены, образуют 4-7- членное кольцо, причем указанное кольцо содержит атомы углерода в кольце и необязательно один атом кислорода в кольце, при этом указанное кольцо а) необязательно замещено одной или двумя группами, выбранными из С1-3алкила, галогена, С1-3алкокси, гидрокси, гидроксиС1-3алкила и оксо, которые могут быть одинаковыми или разными или b) орто- или спиро-конденсировано с незамещенным 4-6-членным циклоалкановым кольцом или незамещенным 4-6-членным насыщенным гетероциклическим кольцом; и
R1c представляет собой С1-3алкил, С1-3алкокси, гидрокси, гидроксиС1-3алкил или С1-3алкоксиС1-3алкил;
R2 представляет собой Н, С1-3алкил, гало или -O-R2a, причем R2a представляет собой Н или незамещенную линейную С1-3апкильную цепь, причем один или два атома углерода в цепи необязательно заменены атомами кислорода;
R3 представляет собой Н или галоген;
и при этом либо
i) R4a представляет собой Н, С1-3алкил или гало, R4b представляет собой С1-3алкил, циклопропил или гидроксиС1-2алкил; или R4a и R4b вместе с атомом углерода, к которому они присоединены, образуют 3-6-членное насыщенное кольцо, содержащее атомы углерода в кольце и необязательно атом кислорода в кольце, причем указанное кольцо необязательно замещено одной С1-3алкильной группой или одной гидроксиС1-2алкильной группой; и R4c представляет собой ОН, гидроксиметил или гидроксиэтил;
ii) R4a Н, С1-3алкил, гало или ОН; R4b представляет собой Н, С1-3алкил или гало, R4c представляет собой незамещенное кольцо, выбранное из списка, состоящего из оксетанила, тетрагидрофуранила и тетрагидропиранила; или
iii) R4a представляет собой Н, и R4b и R4c вместе с атомом углерода, к которому они присоединены, образуют незамещенное кольцо, выбранное из списка, состоящего из оксетана, тетрагидрофурана или тетрагидропирана; и
R5 представляет собой
a) имидазол-2-ил, необязательно замещенный С1-3алкильной группой в положении 1 и необязательно замещенный метильной группой в положении 5; или
b) пиразол-1-ил, необязательно замещенный С1-3алкильной группой в положении 5 и необязательно замещенный метильной группой в положении 4.
В одном из вариантов реализации W представляет собой С, X представляет собой N,
Z представляет собой С, и Y представляет собой С.
В еще одном варианте реализации R4c представляет собой ОН.
В одном из вариантов реализации R2 представляет собой Н, С1-3алкил, хлор или -O-R2a, где R2a представляет собой Н или незамещенную линейную С1-3алкильную цепь, причем один или два атома углерода в цепи необязательно заменены атомами кислорода; и R3 представляет собой Н или фтор;
В другом варианте реализации i) R4a представляет собой Н, С1-3алкил или фтор; R4b представляет собой С1-3алкил, циклопропил или гидроксиС1-2алкил; или R4a и R4b вместе с атомом углерода, к которому они присоединены, образуют 3-6-членное насыщенное кольцо, содержащее атомы углерода в кольце и необязательно атом кислорода в кольце, причем указанное кольцо необязательно замещено одной С1-3алкильной группой или одной гидроксиС1-2алкильной группой; и R4c представляет собой ОН, гидроксиметил или гидроксиэтил;
ii) R4a Н, С1-3алкил, фтор или ОН; R4b представляет собой Н, С1-3алкил или фтор; R4c представляет собой незамещенное кольцо, выбранное из списка, состоящего из оксетанила, тетрагидрофуранила и тетрагидропиранила; или
iii) R4a представляет собой Н, и R4b и R4c вместе с атомом углерода, к которому они присоединены, образуют незамещенное кольцо, выбранное из списка, состоящего из оксетана, тетрагидрофурана или тетрагидропирана;
В одном из вариантов реализации настоящего изобретения предложены соединения Формулы (Ia) или их фармацевтически приемлемые соли:

где
X представляет собой N или С, Y представляет собой N или С, и Z представляет собой N или С; причем X и Y не могут оба быть N или оба быть С; и при этом если Z представляет собой N, X представляет собой N, и Y представляет собой С;
R1 представляет собой С1-4алкокси, -C(=O)N(R1aR1b), -S(=O)2-N(R1aR1b), -S(=O)2-R1c или -S(=O)-R1c, где
R1a представляет собой С1-3алкил, галоС1-3алкил, гидроксиС1-3алкил или С1-3алкоксиС1-3алкил; R1b представляет собой H или С1-3алкил, или R1a и R1b, вместе с азотом, к которому они присоединены, образуют 4-7-членное кольцо, причем указанное кольцо содержит атомы углерода в кольце и необязательно один атом кислорода в кольце, при этом указанное кольцо а) необязательно замещено одной или двумя группами, выбранными из С1-3алкила, галогена, С1-3апкокси, гидрокси и оксо, которые могут быть одинаковыми или разными или b) орто- или спиро-конденсировано с незамещенным 4-6-членным циклоалкановым кольцом или незамещенным 4-6-членным насыщенным гетероциклическим кольцом; и
R1c представляет собой С1-3алкил, С1-3алкокси, гидрокси, гидроксиС1-3алкил или С1-3алкоксиС1-3алкил;
R2 представляет собой H, С1-3алкил, хлор или -O-R2a, где R2a представляет собой н или незамещенную линейную С1-3алкильную цепь, причем один или два атома углерода в цепи необязательно заменены атомами кислорода;
R3 представляет собой H или фтор;
R4a представляет собой H или метил;
R4b представляет собой С1-3алкил или гидроксиС1-2алкил; и
R5 представляет собой
a) имидазол-2-ил, необязательно замещенный С1-3алкильной группой в положении 1 и необязательно замещенный метильной группой в положении 5; или
b) пиразол-1-ил, необязательно замещенный С1-3алкильной группой в положении 5 и необязательно замещенный метильной группой в положении 4.
В одном из вариантов реализации X представляет собой N, Z представляет собой С, и Y представляет собой С.
В одном из вариантов реализации соединение представляет собой соединение Формулы (Ib):
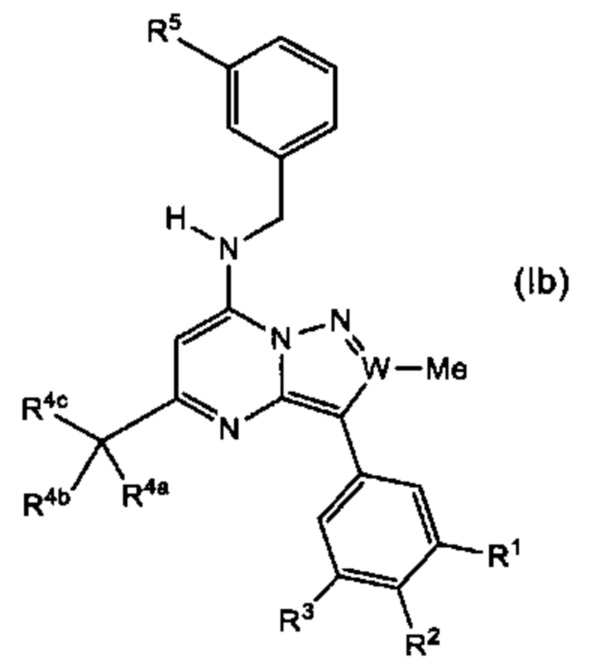
В другом варианте реализации соединение представляет собой соединение Формулы (Ic):

В одном из вариантов реализации представляет собой -C(=O)N(R1aR1b) или -S(=O)2-R1c. В еще одном варианте реализации R1 представляет собой -C(=O)N(R1aR1b).
В одном из вариантов реализации R1a представляет собой гидроксиС1-3алкил или тетрагидропиранил. В одном из вариантов реализации R1a представляет собой гидроксиС1-3алкил. В еще одном варианте реализации R1a представляет собой 3-гидрокси-1-пропил, 2-гидрокси-1-этил, 3-гидрокси-2-пропил или 4-тетрагидропиранил.
В одном из вариантов реализации R1b представляет собой С1-3алкил. В еще одном варианте реализации R1b представляет собой метил или этил.
В еще одном варианте реализации R1a представляет собой гидроксиС1-3алкил и R1b представляет собой С1-3алкил. В еще одном варианте реализации R1a представляет собой 3-гидрокси-1-пропил, и R1b представляет собой С1-3алкил. В еще одном варианте реализации R1a представляет собой 3-гидрокси-2-пропил и R1b представляет собой C1-3алкил. В еще одном варианте реализации R1a представляет собой 3-гидрокси-1 -пропил, и R1b представляет собой метил. В еще одном варианте реализации R1a представляет собой 3-гидрокси-2-пропил, и R1b представляет собой метил. В еще одном варианте реализации R1a представляет собой 2-гидрокси-1-этил, и R1b представляет собой этил. В еще одном варианте реализации R1a представляет собой 3-гидрокси-2-пропил, и R1b представляет собой метил. В еще одном варианте реализации R1a представляет собой 4-тетрагидропиранил, и R1b представляет собой метил.
В одном из вариантов реализации R1a и R1b, вместе с азотом, к которому они присоединены, образуют 4- 7-членное насыщенное кольцо, причем указанное кольцо содержит атомы углерода в кольце и необязательно один атом кислорода в кольце, при этом указанное кольцо а) необязательно замещено одной или двумя группами, выбранными из С1-3алкила, галогена, С1-3алкокси, гидрокси, гидроксиС1-3алкила и оксо, которые могут быть одинаковыми или разными или b) орто- или спиро-конденсировано с незамещенным 4-6-членным циклоалкановым кольцом или незамещенным 4-6-членным насыщенным гетероциклическим кольцом.
В одном из вариантов реализации R1a и R1b, вместе с азотом, к которому они присоединены, образуют необязательно замещенное пирролидиновое кольцо. В еще одном варианте реализации пирролидиновое кольцо замещено С1-3 алкилом, гидрокси или гидроксиС1-3алкилом.
В одном из вариантов реализации R1c представляет собой гидроксиС1-3алкил. В еще одном варианте реализации R1c представляет собой 2-гидрокси-1-этил.
В одном из вариантов реализации R2 представляет собой С1-3алкил, хлор или -O-R2a. В еще одном варианте реализации R2 представляет собой С1-3алкил, хлор или метокси.
В одном из вариантов реализации R3 представляет собой Н или фтор; В другом варианте реализации R3 представляет собой Н. В одном из вариантов реализации R4a представляет собой метил.
В одном из вариантов реализации R4b представляет собой С1-3алкил. В другом варианте реализации R4b представляет собой метил или этил. В еще одном варианте реализации R4b представляет собой метил.
В одном из вариантов реализации R4a представляет собой метил, и R4b представляет собой метил.
В одном из вариантов реализации R4a представляет собой С1-3алкил, R4b представляет собой С1-3алкил, и R4c представляет собой ОН. В другом варианте реализации R4a представляет собой метил, R4b представляет собой метил, и R4c представляет собой ОН.
В одном из вариантов реализации R5 представляет собой имидазол-2-ил, необязательно замещенный С1-3алкильной группой в положении 1 и необязательно замещенный метильной группой в положении 5. В еще одном варианте реализации R5 представляет собой 1-метил-1Н-имидазол-2-ил.
В одном из вариантов реализации указанное соединение представляет собой соединение, соответствующее формуле (I) и:
W представляет собой С, X представляет собой N, Z представляет собой С, и Y представляет собой С;
R1 представляет собой -C(=O)N(R1aR1b) или -S(=O)2-R1c, где R1a представляет собой гидроксиС1-3алкил, и R1b представляет собой С1-3алкил; или R1a и R1b, вместе с азотом, к которому они присоединены, образуют необязательно замещенное пирролидиновое кольцо; и при этом R1c представляет собой гидроксиС1-3алкил;
R2 представляет собой С1-3алкил, хлор или -O-R2a;
R3 представляет собой Н;
R4a представляет собой метил;
R4b представляет собой С1-3алкил;
R4c представляет собой ОН; и
R5 представляет собой имидазол-2-ил, необязательно замещенный С1-3алкильной группой в положении 1 и необязательно замещенный метильной группой в положении 5.
В одном из вариантов реализации указанное соединение представляет собой соединение, соответствующее формуле (I), и:
W представляет собой С, X. представляет собой N, Z представляет собой С, и Y представляет собой С;
R1 представляет собой -C(=O)N(R1aR1b), где R1a представляет собой гидроксиС1-3алкил, и R1b представляет собой С1-3алкил; или R1a и вместе с азотом, к которому они присоединены, образуют необязательно замещенное пирролидиновое кольцо, замещенное С1-3алкилом, гидрокси, гидроксиС1-3алкилом;
R2 представляет собой С1-3алкил, хлор или метокси;
R3 представляет собой Н;
R4a представляет собой метил;
R4b представляет собой метил;
R4c представляет собой ОН; и
R5 представляет 1-метил-1Н-имидазол-2-ил.
В одном из вариантов реализации соединение Формулы (I) выбрано из группы, состоящей из следующих соединений:
2-хлор-N-этил-N-(2-гидроксиэтил)-5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)бензамид (соединение 15);
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид (соединение 17);
N-этил-N-(2-гидроксиэтил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксибензамид (соединение 19);
(S)-N-(1-гидроксипропан-2-ил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метокси-N-метилбензамид (соединение 20);
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метокси-N-метил-N-(тетрагидро-2Н-пиран-4-ил)бензамид (соединение 21);
(S)-N-(1-гидроксипропан-2-ил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-[N,2-диметилбензамид (соединение 22);
(R)-(2-(гидроксиметил)пирролидин-1-ил)(5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метилфенил)метанон (соединение 25);
(S)-(5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метилфенил)(3-гидроксипирролидин-1-ил)метанон (соединение 29);
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-2-метокси-N-метилбензамид (соединение 32); и
(R)-(2-(гидроксиметил)пирролидин-1-ил)(5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксифенил)метанон (соединение 36);
N-этил-N-(2-гидроксиэтил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метилбензамид (соединение 38);
N-(2-гидроксиэтил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-a]пиримидин-3-ил)-N,2-диметилбензамид (соединение 43);
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N,2-диметил-N-(тетрагидрофуран-3-ил)бензамид (соединение 59);
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(2-гидроксипропил)-N,2-диметилбензамид, изомер 1 (соединение 62);
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(2-гидроксипропил)-N,2-диметилбензамид, изомер 2 (соединение 63);
2-Хлор-N-(2-гидроксиэтил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-метилбензамид (соединение 37);
N-((S)-1-гидроксипропан-2-ил)-5-(5-(1-гидроксипропил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин3-ил)-N,2-диметилбензамид, изомер 1 (соединение 80);
N-((S)-1-гидроксипропан-2-ил)-5-(5-(1-гидроксипропил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин3-ил)-N,2-диметилбензамид, изомер 2 (соединение 81);
5-(5-(1-гидроксибутен-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид, изомер 1 (соединение 66); и
5-(5-(1-гидроксибутен-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид, изомер 2 (соединение 67);
или фармацевтически приемлемых солей любого из приведенных выше соединений.
В одном из вариантов реализации соединение Формулы (I) представляет собой 5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид (соединение 17)

или его фармацевтически приемлемую соль.
В одном из вариантов реализации соединение Формулы (I) представляет собой N-этил-N-(2-гидроксиэтил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксибензамид (соединение 19)
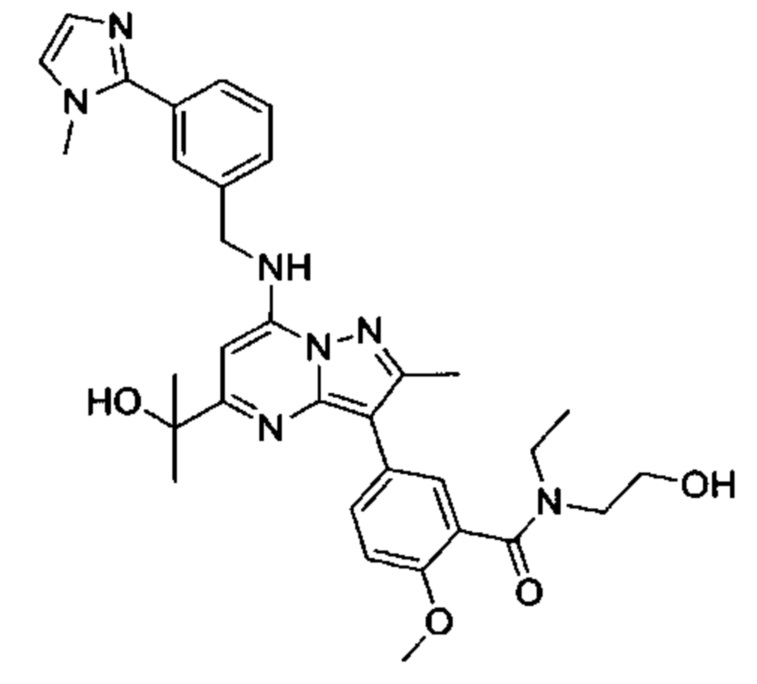
или его фармацевтически приемлемую соль.
В одном из вариантов реализации соединение Формулы (I) представляет собой 5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метокси-N-метил-N-(тетрагидро-2H-пиран-4-ил)бензамид (соединение 21)

или его фармацевтически приемлемую соль.
В одном из вариантов реализации соединение Формулы (I) представляет собой (S)-N-(1-гидроксипропан-2-ил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N,2-диметилбензамид (соединение 22)

или его фармацевтически приемлемую соль.
Термины и определения
Алкил представляет собой одновалентный радикал, образованный удалением атома водорода из ациклического алкана. Например, а С1-4алкил представляет собой алкил, содержащий от 1 до 4 атомов углерода. Алкил может быть линейным или разветвленным. Примерами С1-4алкила являются метил, этил, н-пропил, н-бутил, изопропил, изобутил, сек-бутил трет-бутил.
Алкокси представляет собой группу Формулы "-O-R", где R представляет собой алкил (в соответствии с определением выше). Например, С1-4алкокси представляет собой алкокси, состоящий из 1 - 4 атомов углерода. Примерами С1-4алкокси являются метокси, этокси, н-пропокси, н-бутокси, изо-пропокси, изо-бутокси, сек-бутокси и трет-бутокси.
Гало (галоген) относится к гапогеновому радикалу, т.е., фтор-, хлор-, брому- или йод-.
Галогеналкил представляет собой алкил (в соответствии с определением выше), замещенный одним или большим числом галогенов («гало» в соответствии с определением выше), причем галогены могут быть одинаковыми или разными. Например, галоС1-3алкил представляет собой галогеналкил, состоящий из 1 - 3 атомов углерода. Примерами галоС1-3алкила являются монофторметил, дифторметил, трифторметил и 1-хлор-2-фторэтил.
Гидроксиалкил представляет собой алкил (в соответствии с определением выше), замещенный одним или более гидрокси-заместителями. Например, гидроксиС3алкил имеет формулу -(СН2)3ОН (где "-" показывает, какой атом присоединен к соединению Формулы (I)).
Алкоксиалкил представляет собой алкил (в соответствии с определением выше), замещенный одним или более алкокси-заместителями. Например, С3алкоксиС2алкил имеет формулу -(СН2)2O(СН2)2СН3 (где "-" показывает, какой атом присоединен к соединению Формулы (I)).
Оксо представляет собой двухвалентный радикал формулы =O.
4-6-членное насыщенное гетероциклическое кольцо является моноциклическим и состоит из атомов углерода в кольце и гетероатомов в кольце, выбранных из группы, состоящей из азота, кислорода и серы. В одном из вариантов реализации гетероциклическое кольцо содержит 1 или 2 гетероатома в кольце. Примерами являются пирролидин, диоксолан, имидазолидин, пиразолидин, пиперидин, диоксан, морфолин, дитиан, тиоморфолин и пиперазин.
4-6-членное циклоалкановое кольцо не содержит ни одного гетероатома в кольце и является насыщенным и моноциклическим. Примерами являются циклобутан, циклопентан и циклогексан.
"Орто-сопряженная кольцевая система" содержит два кольца, содержащие только два общих атома и одну общую связь, например:

"Спиро-сопряженная кольцевая система" содержит два кольца, соединенные одним атомом углерода, например,

"Замещенный" применительно к группе указывает на то, что атом водорода, связанный с атомом-членом группы, заменен. Понятно, что термин «замещенный» подразумевает, что такое замещение соответствует допустимой валентности атома, содержащего заместитель, и заместителя и что замещение дает стабильное соединение включает (т.е., соединение, которое не претерпевает спонтанные трансформации, такие как перестройка, циклизация или отщепление). В некоторых вариантах реализации один атом может быть замещен более, чем одним заместителем, при условии, что такое замещение соответствует допустимой валентности этого атома. Подходящие заместители определены в настоящем документе для каждой замещенной или возможно замещенной группы.
"Фармацевтически приемлемый" относится к соединениям, материалам, составам и лекарственным формам, которые, в рамках обоснованного медицинского суждения, включены в объем настоящего изобретения, подходят для применения в контакте с тканями человека без чрезмерной токсичности, раздражения или других проблем или осложнений, и при разумном отношении риск/польза.
Во всем описании и следующей за ним формуле изобретения, если контекст не требует иного, слово «содержать» и его варианты, такие как «содержит» и «содержащий», будут пониматься как подразумевающие включение указанного объекта числа, этапа или группы объектов, но не исключение любого другого объекта, этапа или группы объектов или этапов.
Соединения Формулы (I) и формулы (Ia) и их фармацевтически приемлемые соли могут существовать в твердой или жидкой форме. В твердом состоянии они могут существовать в кристаллической или некристаллической форме, или в виде их смесей. В случае кристаллической форму, квалифицированный специалист поймет, что могут образовываться фармацевтически приемлемые сольваты, в которых молекулы растворителя внедряются в кристаллическую решетку в процессе кристаллизации. Сольваты включать неводные растворители, такие как этанол, изо-пропиловый спирт, N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО), уксусная кислота, этаноламин и этилацетат, или они могут включать воду в качестве растворителя, внедренного в кристаллическую решетку. Сольваты, в которых вода является растворителем, внедренным в кристаллческую решетку, обычно называются "гидратами". Гидраты включают стехиометрические гидраты, а также композиции, содержащие различные количества воды.
Соединения Формулы (I) и формулы (Ia) и их фармацевтически приемлемые соли, которые существуют в кристаллической форме, включая различные сольваты таких соединений, могут демонстрировать полиморфизм (т.е., способность существовать в виде различных кристаллических структур). Эти различные кристаллические формы обычно известны как "полиморфы". Настоящее изобретение включает все такие полиморфы. Полиморфы имеют одинаковый химический состав, но различаются по упаковке, геометрическому расположению и другим свойствам, описывающим твердое кристаллическое состояние. Соответственно, полиморфы могут иметь различные физические свойства, такие как форма, плотность, твердость, деформируемость, стабильность и свойства растворения, ИК-спектра и рентгеновская порошковая дифрактограмма, которые могут использоваться для идентификации. Понятно, что различные полиморфы могут быть получены, например, путем изменения или корректировки условий реакции или реагентов, применяемых при получении соединения. Например, изменения температуры, давления или растворителя могут дать полиморфы. Дополнительно, один полиморф может спонтанно превращаться в другой полиморф в определенных условиях.
Изобретение также включает меченные изотопами соединения, которые идентичны соединениям формулы (I) и формулы (Ia) и их фармацевтически приемлемым солям, за исключением того, что один или более атомов заменена атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, наиболее распространенных в природе. Примеры изотопов, которые могут быть включены в соединения согласно настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода и фтора, такие как 3Н, 11С, 14С и 18F.
Соединения, соответствующие формуле (I) и формуле (Ia) могут содержать один или более центров асимметрии (называемых также хиральными центрами) и могут, соответственно, существовать в виде отдельных энантиомеров, диастереоизомеров или других стереоизомерных формы или в виде их смесей. Хиральные центры, такие как атомы углерода, могут также присутствовать в заместителе, таком как алкильная группа. В тех случаях, когда стереохимия хирального центра, присутствующего в формуле (I) или в любой другой приведенной здесь химической структуре, не указана, предполагается, что эта структура охватывает любые стереоизомеры и все их смеси. Таким образом, соединения, соответствующие формуле (I), содержащие один или более хиральных центров, могут использоваться в виде рацемических смесей и рацематов, энантиомерно-обогащенных смесей или энантиомерно чистых отдельных стереоизомеров.
Индивидуальные стереоизомеры соединений формулы (I) и формулы (Ia), которые содержат один или несколько асимметричных центров, могут быть разделены способами, известными специалистам в данной области. Например, такое разделение может осуществлено (1) путем образования диастереоизомерных солей, комплексов или других производных; (2) путем селективной реакции со стереоизомер-специфическим реагентом, например, путем ферментативного окисления или восстановления; или (3) газожидкостной или жидкостной хроматографией в хиральной среде, например, на хиральном носителе, таком как диоксид кремния, со связанным хиральным лигандом или в присутствии хирального растворителя. Понятно, что когда желаемый стереоизомер преобразуют в другое химическое соединение с использованием одной из процедур разделения, описанных выше, необходим дополнительный этап для высвобождения желаемой формы. В качестве альтернативы, некоторые стереоизомеры можно синтезировать путем асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей, или путем преобразования одного энантиомера в другой путем асимметричного превращения.
Понятно, что когда желаемый стереоизомер преобразуют в другое химическое соединение с использованием одной из процедур разделения, описанных выше, для необходим дополнительный этап для высвобождения желаемой формы. В качестве альтернативы, конкретные стереоизомеры можно синтезировать путем асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей, или путем преобразования одного энантиомера в другой путем асимметричного превращения.
Следует понимать, указание в данном документе на соединение Формулы (I) и формулы (Ia) или его фармацевтически приемлемую соль включают соединение Формулы (I) и формулы (Ia), соответственно, в виде свободного основания или в виде его фармацевтически приемлемой соли. Таким образом, в одном варианте реализации изобретение относится к соединению Формулы (I). В другом варианте реализации изобретение относится к фармацевтически приемлемой соли соединения Формулы (I).
Фармацевтически приемлемые соли включают, среди прочего, соли, описанные в Berge, J. Pharm. Sci., 1977, 66, 1-19, или перечисленные в Р Н Stahl, С G Wermuth, editors, Handbook of Pharmaceutical Salts; Properties, Selection and Use, Second Edition Stahl/Wermuth: Wiley- VCH/VHCA, 2011
Соли, не являющиеся фармацевтически приемлемыми, можно применять, например, в качестве промежуточных соединений для получения соединения Формулы (I) или формулы (Ia) или его фармацевтически приемлемой соли. В качестве альтернативы, не являющиеся фармацевтически приемлемыми соли Формулы (I) и формулы (Ia) включены в настоящий документ.
Подходящие фармацевтически приемлемые соли могут включать соли присоединения кислоты.
Такие соли присоединения кислоты могут быть образованы в результате реакции соединения Формулы (I) или формулы (Ia) (которое, например, содержит основной амин или другую основную функциональную группу) с соответствующей кислотой, необязательно в подходящем растворителе, таком как органический растворитель, В результате чего получают соль, которую можно выделить различными методами, включая кристаллизацию и фильтрацию.
Соли могут быть получены in situ во время окончательного выделения и очистки соединения Формулы (I) или формулы (Ia). Если основное соединение Формулы (I) или формулы (Ia) выделяют в виде соли, соответствующая форма свободного основания этого соединения может быть получена любым подходящим способом, известным в данной области техники, включая обработку соли неорганическим или органическим основанием.
Будет понятно, что если соединение Формулы (I) или формулы (Ia) содержит два или более основных фрагмента, стехиометрия образования соли может включать 1, 2 или более эквивалентов кислоты. Такие соли могут содержать 1, 2 или более противоионов кислоты, например, соль дигидрохлорид. Стехиометрические и нестехиометрические формы фармацевтически приемлемой соли соединения Формулы (I) или формулы (Ia) включены в объем изобретения, включая субстехиометрические соли, например, где противоион содержит более одного кислотного протона.
Типичные фармацевтически приемлемые соли присоединения включают, но не ограничиваются перечисленными: 4-ацетамидобензоат, ацетат, адипат, альгинат, аскорбат, аспартат, бензолсульфонат (безилат), бензоат, бисульфат, биатртрат, бутират, эдетат кальция, камфорат, камфорсульфонат (камсилат), капрат (деканоат), капроат (гексаноат), каприлат (октаноат), циннамат, цитрат, цикламат, диглюконат, 2,5-дигидроксибензоат, дисукцинат, додецил сульфат (эстолат), эдетат (этилендиаминтетраацетат), эстолат (лаурилсульфат), этан-1,2-дисульфонат (эдисилат), этансульфонат (эзилат), формиат, фумарат, галактарат (мукат), гентизат (2,5-дигидроксибензоат), глюкогептонат (глюцептат), глюконат, глюкуронат, глутамат, глутарат, глицерофосфат, гликолат, гексилрезорцинат, гиппурат, гидрабамин (N,N'-ди(дегидроабиетил)-этилендиамин), гидробромид, гидрохлорид, гидройодид, гидроксинафтоат, изобутират, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, метансульфонат (мезилат), метилсульфат, мукат, нафталин-1,5-дисульфонат (нападизилат), нафталин-2-сульфонат (напсилат), никотинат, нитрат, олеат, пальмитат, п-аминобензолсульфонат, п-аминосалицилат, памоат (эмбонат), пантотеат, пектинат, персульфат, фенилацетат, фенилэтилбарбитурат, фосфат, полигалактоуронат, пропионат, п-толуолсульфонат (тозилат), пироглутамат, пируват, салицилат, себакат, стеарат, субацетат, сукцинат, сульфамат, сульфат, таннат, тартрат, теоклат (8-хлоротеофиллинат), тиоцианат, триэтиодид, ундеканоат, ундециленат и валерат.
Соединения Формулы (I) или формулы (Ia), а также их соли и фармацевтически приемлемые соли включают сольваты (включая гидраты), комплексы, полиморфы, пролекарства, меченные радиоактивными метками производные и стереоизомеры соединений Формулы (I) или формулы (Ia) и их фармацевтически приемлемых солей, здесь и далее называются "соединения согласно настоящему изобретению".
Общие пути
Соединения согласно настоящему изобретению могут быть получены различными способами. В следующих схемах реакций и далее, если не указано иное, R1 - R5, R4a, R4b, R4c, R5, W, X, Y и Z соответствуют определениям для первого аспекта. По всему тексту заявки общие формулы обозначаются римскими цифрами (I), (II), (III), (IV) и т.д..
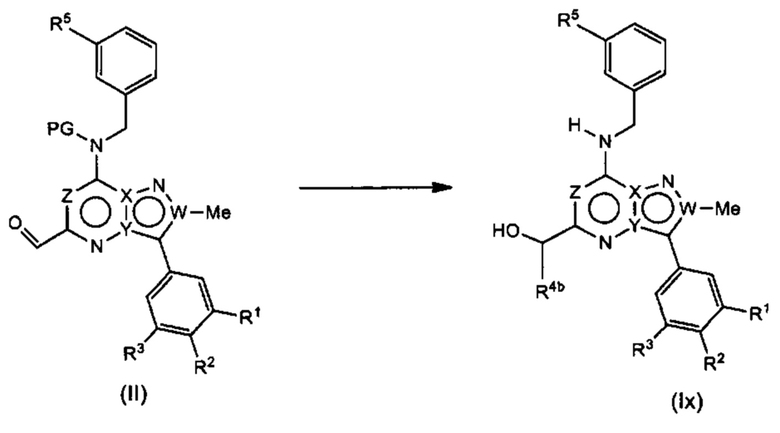
Общий путь получения формулы (I)
Соединения формулы (Ix), т.е., соединения Формулы (I), в которых R4a представляет собой Н, и R4c представляет собой ОН, могут быть получены в соответствии со схемой реакции 1 путем обработки (II) подходящим реагентом Гриньяра (таким как метилмагния бромид) в растворителе (например, ТГФ) с последующим удалением защиты подходящей кислотой (например, 4 М HCl в 1,4-диоксане) в растворителе (например, метаноле).
Схема 1
Соединения Формулы (II) могут быть получены в соответствии со схемой реакции 2 путем обработки соединений Формулы (III) бороновым эфиром (например, 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этан-1-олом) в растворителе (например, 1,4-диоксане и воде) в присутствии катализатора [например, PdCl2(dppf)] и основания (например, фторида калия).
Схема 2
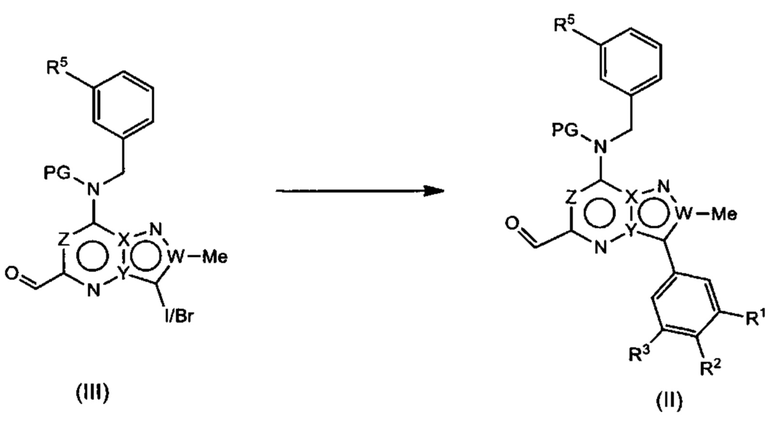
Соединения Формулы (III) могут быть получены в соответствии со схемой реакции 3 из соединений Формулы (IV) путем обработки (IV) подходящим окислителем (например, DMP) в растворителе (например, ДХМ).
Схема 3

Соединения Формулы (IV) могут быть получены в соответствии со схемой реакции 4 из соединений Формулы (V). Сначала, амин защищают подходящей амин-защитной группой (например, трет-бутилкарбаматом путем обработки подходящими реагентами например, ди-трет-бутилдикарбонатом, DIPEA м DMAP в ДХМ). Затем сложный эфир восстанавливают до первичного спирта подходящим восстановителем (например, борогидридом натрия) в растворителе (например, этанол).
Схеме 4
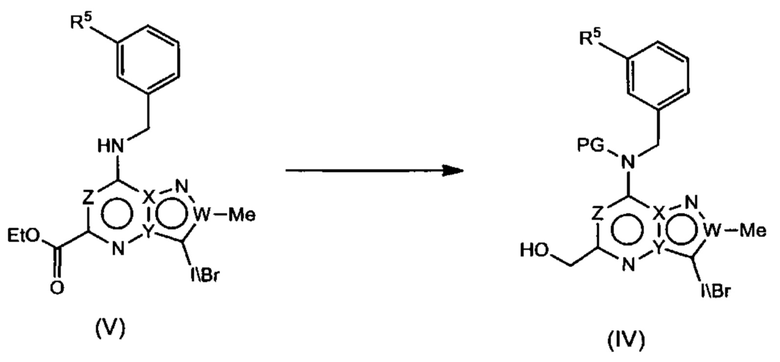
Соединения Формулы (V) могут быть получены в соответствии со схемой реакции 5 из соединений Формулы (VI) (где L представляет собой Cl или Br) или смесь соединений Формулы (VI), где L представляет собой Cl и Br) путем обработки амином (например, (3-(1-метил-1H-имидазол-2-ил)фенил)метиламином) и основанием (например, DIPEA) в растворителе (например, ДМСО).
Схема 5
Соединения Формулы (Ix'), т.е., соединения Формулы (I), где R4a и R4b представляют собой С1-3алкил и R4c, представляет собой ОН, могут быть получены в соответствии со схемой реакции 6 путем обработки (VII) бороновым эфиром (например, N-(3-гидроксипропил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамидом) в присутствии катализатора [например, PdCl2(dppf)] в растворителе (например, 1,4-диоксан и вода) в основанием (например, карбонат натрия).
Схема 6

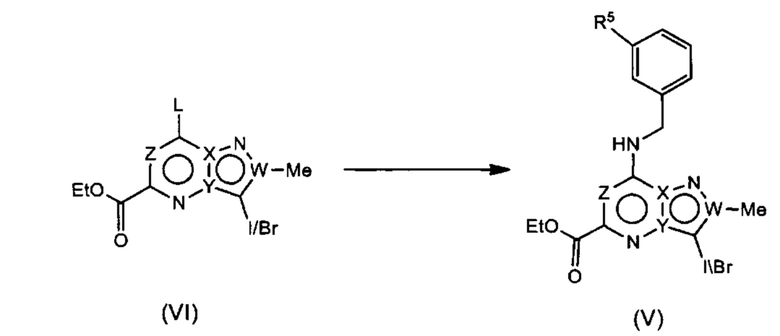
Соединения Формулы (VII) могут быть получены из соединений Формулы (V) в соответствии со схемой реакции 7 путем обработки (V) реагентом Гриньяра (например, метилмагния бромидом) в растворителе (например, ДХМ). Получение (V) показано на Схеме 5.
Схема 7

Соединения Формулы (VIa), т.е., соединения Формулы (VI) (см. Схему 5), в которых W представляет собой С, X представляет собой N, Y представляет собой С, Z представляет собой С, и L представляет собой Cl, могут быть получены в соответствии со схемой реакции 8. Сначала могут быть получены соединения Формулы (VIII) путем конденсации сложных эфиров (например, натриевой соли диэтилоксалацетата) и соединений Формулы (IX) с использованием кислоты (такой как HCl) в растворителе (таком как этанол) при нагревании (например, 85°С), в результате чего получали соединения формулы (VIII). Затем, обработка (VIII) хлорирующим реагентом (например, POCl3) при нагревании (например, при 90°С) дает соединения Формулы (X). Обработка соединений Формулы (X) источником йода или брома [например, N-йодосукцинимидом (N-йодосукцинимидом) или N-бромсукцинимидом (БСИ)] в растворителе (например, ДХМ) дает соединения Формулы (VIa).
Схема 8

Смесь соединений Формулы (VIb), т.е., соединений Формулы (VI) (см. схему 5), в которых W представляет собой С, X представляет собой С, Y представляет собой N, Z представляет собой С, и L представляет собой Cl и Br, может быть получена в соответствии со схемой реакции 9 в несколько этапов из соединений (XI). Сначала соединения Формулы (XI) обрабатывают бромирующим агентом например, N-бромсукцинимидом и бикарбонатом натрия в растворителе (например, метанол) с получением соединений формулы (XII). Затем обработка соединений Формулы (XII) 1-хлорпропан-2-оном при повышенной температуре (например, при 90°С) дает смесь соединений Формулы (XIII). Йодирование или бромирование смеси соединений Формулы (XIII), например, N-йодосукцинимидом или N-бромсукцинимидом, в растворителе (например, ДМФА) дает смесь соединений Формулы (VIb).
Схема 9

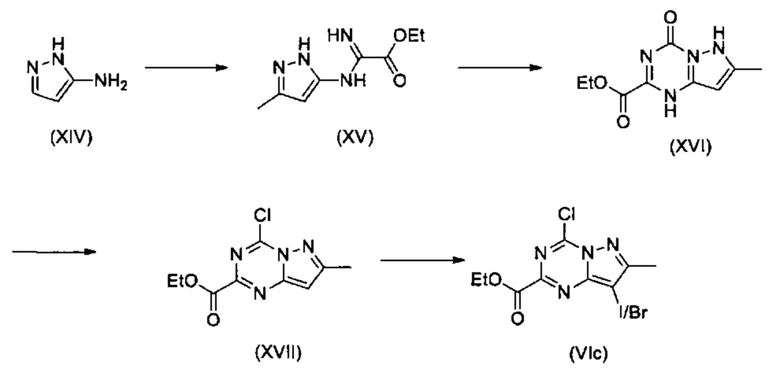
Соединения Формулы (VIc), т.е., соединения Формулы (VI) (см. Схему 5), где W представляет собой С, X представляет собой N, Y представляет собой С, Z представляет собой N, и L представляет собой Cl, могут быть получены в несколько этапов из соединений Формулы (XIV) в соответствии со схемой реакции 10. Сначала может быть проведена реакция соединений Формулы (XIV) с метилцианоформиатом с получением соединений (XV). Затем соединения Формулы (XV) можно циклизировать с использованием карбонилдиимидазола в растворителе (например, ДМСО) или, в качестве альтернативы, с использованием диэтилкарбоната в этоксиде натрия и этаноле с получением соединений формулы (XVI). Затем соединения Формулы (XVI) можно хлорировать с использованием, например, POCl3, при нагревании (например, при 90°С) с получением соединений формулы (XVII). Йодирование (с использованием например, N-йодосукцинимида) или бромирование (с использованием, например, N-бромсукцинимида), в растворителе (например, ДМФА) дает соединения Формулы (VIc).
Схема 10
Соединения Формулы (Ix''), т.е., соединения Формулы (I), в которых R4c представляет собой гидроксиметил, могут быть получены в соответствии со схемой реакции 11 из соединений Формулы (XVIII).
Схема 11

Соединения Формулы (XVIIIa), т.е., соединения Формулы (XVIII), в которых W представляет собой С, X представляет собой N, Y представляет собой С, и Z представляет собой С, могут быть получены в соответствии со схемой реакции 12.
Схема 12
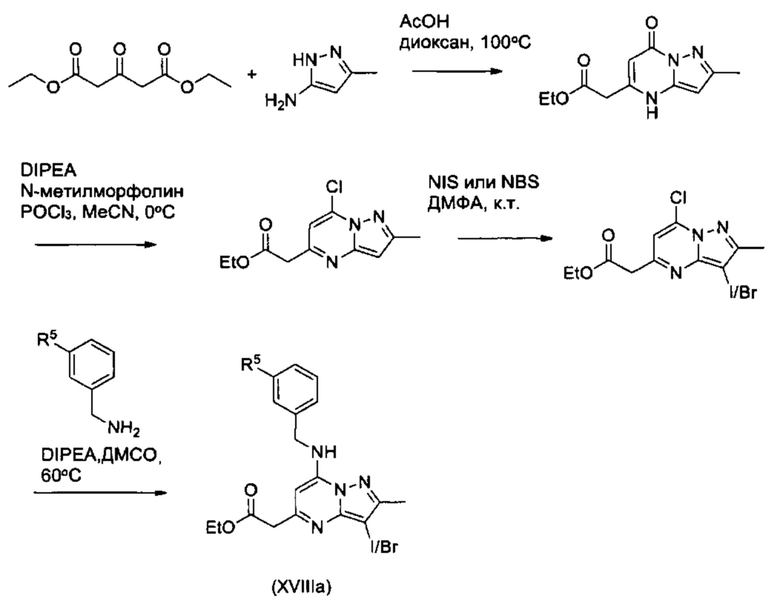
Соединения Формулы (XVIIIb), т.е., соединения Формулы (XVIII) (из схемы 11), в которых W представляет собой N, X представляет собой С, и Y представляет собой С, могут быть получены из соединений Формулы (XIX) в соответствии со схемой реакции 13.
Схема 13
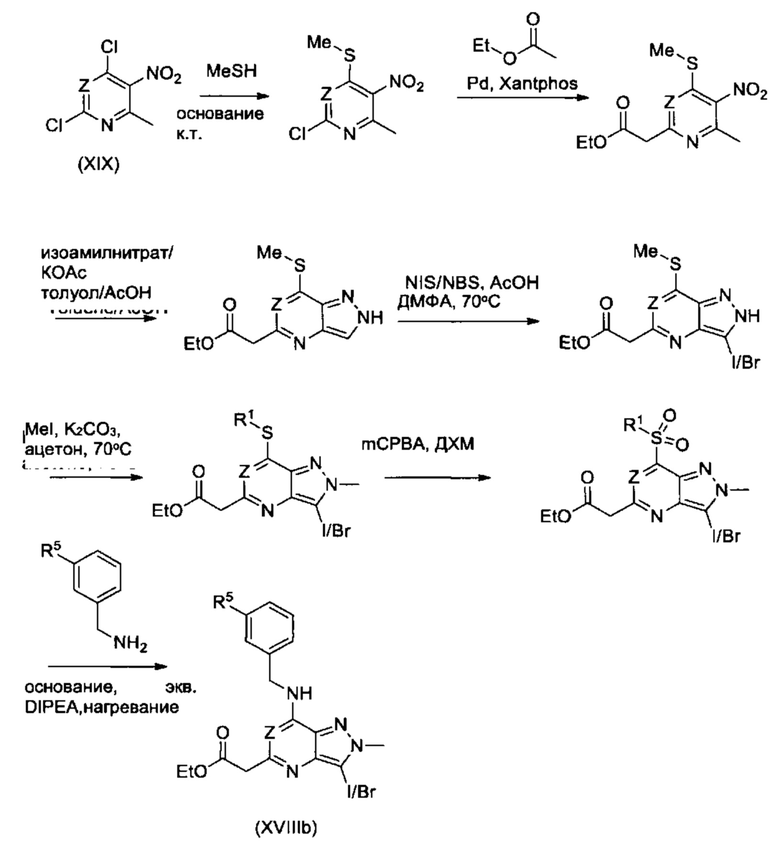
В качестве альтернативы, соединения Формулы (Ia) (см. Схему 1) могут быть получены из соединений Формулы (V) (см. Схему 4) в соответствии со схемой реакции 14. Соединения Формулы (Ia) могут быть разделены на отдельные энантиомеры с использованием методик, знакомых опытному химику, например, хиральной ВЭЖХ.
Схема 14

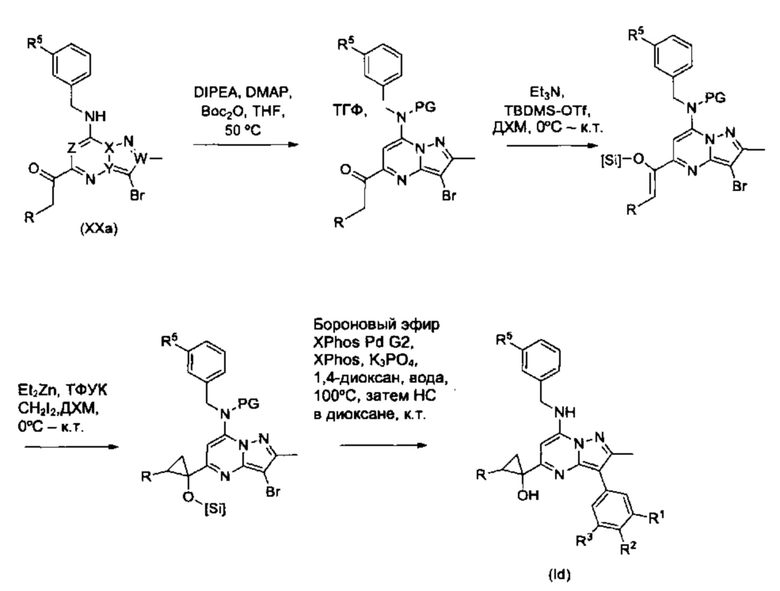
Соединения Формулы (Id), т.е., соединения общей формулы (I), в которой R4a и R4b вместе с атомом углерода, к которому они присоединены, образуют циклопропановое кольцо, замещенное группой R, которая представляет собой С1-3алкильную группу, могут быть получены в соответствии со схемой реакции 15 из соединений Формулы (ХХа). В схеме реакции 15, PG представляет собой защитную группу, такую как трет-бутилоксикарбонил, и [Si] представляет собой защитную группу на основе кремния для спиртовой группы, такую как TBDMS.
Схема 15
Соединения Формулы (Ie), т.е., соединения Формулы (I), в которых R4a представляет собой Н, и R4d и R4c вместе с атомом углерода, к которому они присоединены, образуют незамещенное тетрагидропирановое кольцо, могут быть получены из соединений Формулы (XXI) в соответствии со схемой реакции 15.
Схема 15
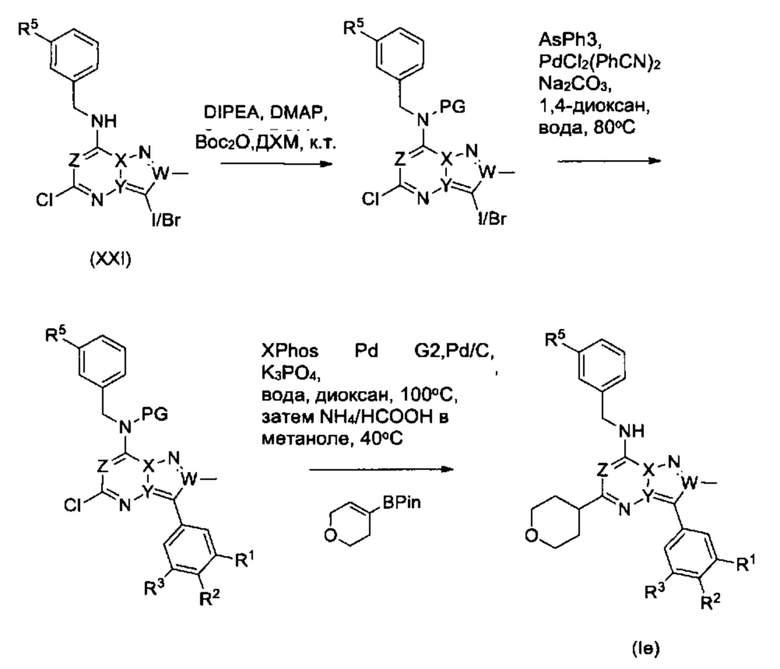
Соединения Формулы (XXI) могут быть получен из соединений Формулы (XXII) в соответствии со схемой реакции (16).
Схема 16
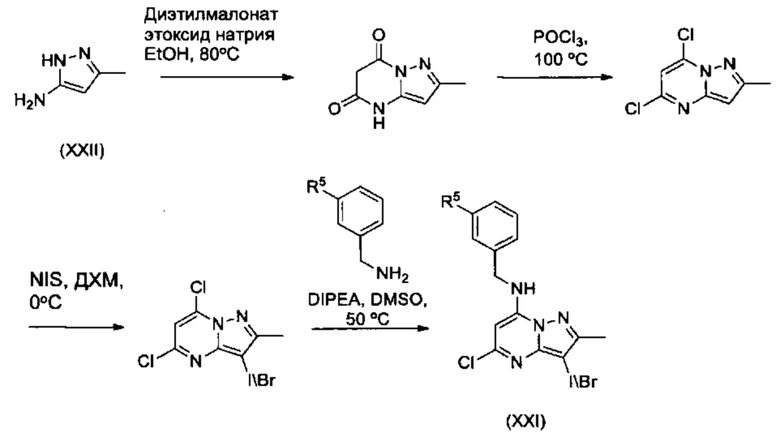
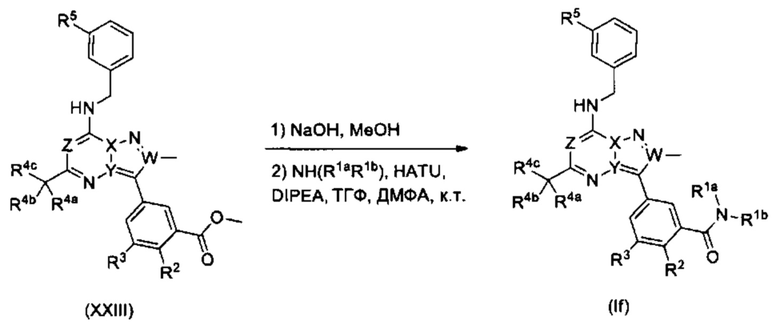
Для опытного химика понятно, что соединения Формулы (I) также можно получить путем преобразования в положении R1 на последнем этапе. В схеме реакции 17, соединения Формулы (If) т.е., соединения Формулы (I), в которых R1 представляет собой -C(=O)N(R1aR1b), могут быть получены из соединений Формулы (XXIII).
Схема 17
Для опытного химика понятно, что соединения Формулы (I) могут быть преобразованы в другие соединения Формулы (I) способами, известными в данной области. Дополнительно, промежуточные соединения, описанные в схемах реакций выше, могут быть преобразованы в другие промежуточные соединения, а затем преобразованы с использованием описанных способов в соединения Формулы (I). Также понятно, что соединения Формулы (I) могут быть получены с использованием другой последовательности преобразований, описанных в схемах реакций, включая соответствующие этапы введения/удаления защиты.
Общий путь Формулы (Ia)
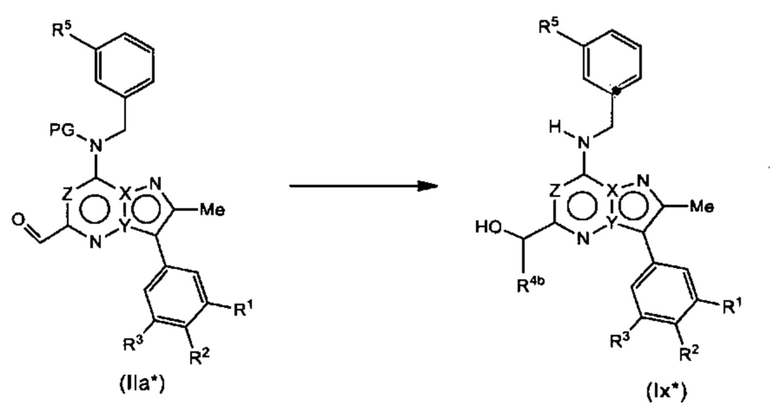
Соединения Формулы (Ix*), т.е., соединения Формулы (Ia), в которых R4a представляет собой Н, могут быть получены в соответствии со схемой реакции 1* путем обработки (IIa*) подходящим реагентом Гриньяра (таким как метилмагния бромид) в растворителе (например, ТГФ) с последующим удалением защиты подходящей кислотой (например, 4 М HCl в 1,4-диоксане) в растворителе (например, метанол).
Схема 1*
Соединения Формулы (IIa*) могут быть получены в соответствии со схемой реакции 2а* путем обработки соединений Формулы (IIIa*) бороновым эфиром (например, 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этан-1-олом) в растворителе (например, 1,4-диоксан и вода) в присутствии катализатора (например, PdCl2(dppf)) и основания (например, фторида калия).
Схема 2а*
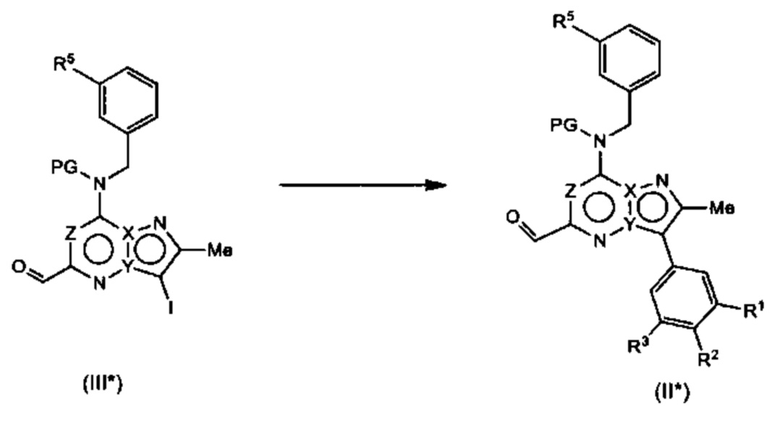
Соединения Формулы (III*) могут быть получены в соответствии со схемой реакции 3* из соединений Формулы (IV*) путем обработки (IV*) подходящим окислителем (например, DMP) в растворителе (например, ДХМ).
Схема 3*

Соединения Формулы (IV*) могут быть получены в соответствии со схемой реакции 4* из соединений Формулы (V*). Сначала амин защищают подходящей амин-защитной группой (например, трет-бутилкарбаматом, путем обработки подходящими реагентами например, ди-трет-бутил дикарбонатом, DIPEA м DMAP в ДХМ). Затем сложный эфир восстанавливают до первичного спирта подходящим восстанавливающим агентом (например, борогидридом натрия) в растворителе (например, этанол).
Схема 4*

Соединения Формулы (V*) могут быть получены в соответствии со схемой реакции 5* из соединений Формулы (VI*) (где L представляет собой Cl или Вr) или смеси соединений Формулы (VI), где L представляет собой Cl и Вr) путем обработки амином (например, (3-(1-метил-1Н-имидазол-2-ил)фенил)метиламином) и основанием (например, DIPEA) в растворителе (например, ДМСО).
Схема 5*
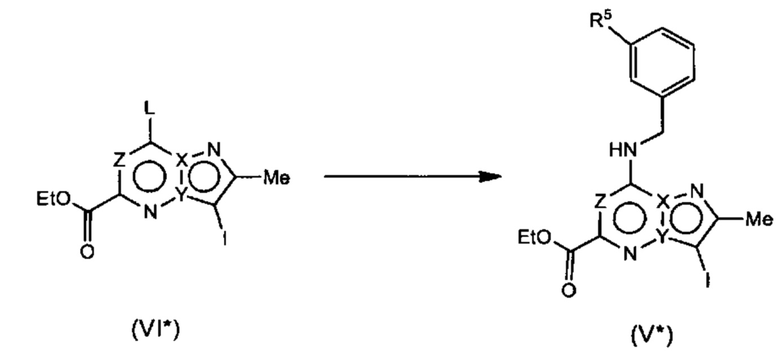
Соединения Формулы (Ib*), т.е., соединения Формулы (I*), где R4a и R4b представляют собой метил, могут быть получены в соответствии со схемой реакции 6 путем обработки (VII*) бороновым эфиром (например, N-(3-гидроксипропил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамидом) в присутствии катализатора (например, PdCl2(dppf)) в растворителе (например, 1,4-диоксан и вода) с основанием (например, карбонат натрия).
Схема 6*

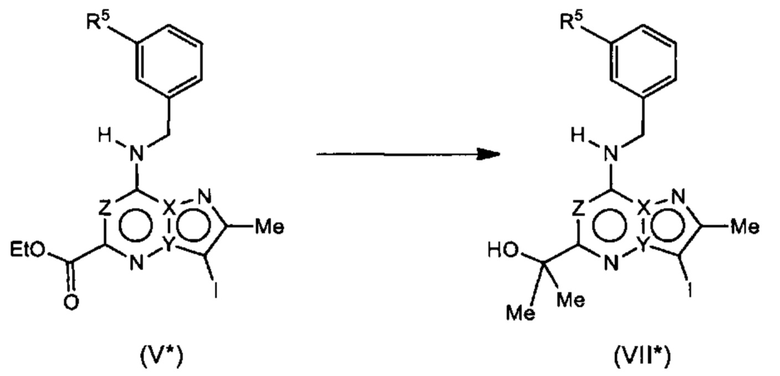
Соединения Формулы (VII*) могут быть получены из соединений Формулы (V*) в соответствии со схемой реакции 7* путем обработки (V*) реагентом Гриньяра (например, метилмагния бромидом) в растворителе (например, ДХМ).
Схема 7*
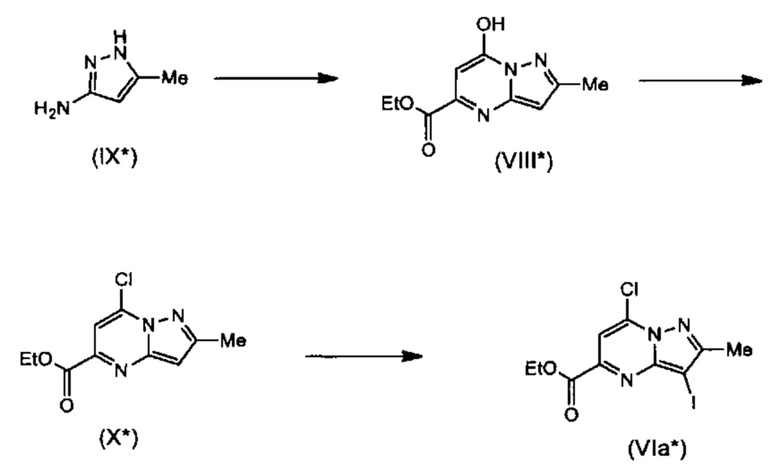
Соединения Формулы (Via*), т.е., соединения Формулы (VI*) (см. Схему 5*), где X представляет собой N, Y представляет собой С, Z представляет собой С, и L представляет собой Cl, могут быть получены в соответствии со схемой реакции 8*. Сначала соединения Формулы (VIII*) могут быть получены путем конденсации сложных эфиров (например, натриевой соли диэтилоксалацетата) и соединений Формулы (IX*) с использованием кислоты (такой как HCl) в растворителе (таком как этанол) при нагревании (например, 85°С) с получением соединения формулы VIII*). Затем обработка (VIII*) хлорирующим реагентом (например, РОСI3) при нагревании (например, при 90°С) дает соединения Формулы (X*). Обработка соединений Формулы (X*) источником йода (например, N-йодосукцинимидом) в растворителе (например, ДХМ) дает соединения Формулы (Via*).
Схема 8*
Смесь соединений Формулы (VIb*), т.е., соединений Формулы (VI*) (см. Схему 5*), в которых X представляет собой С, Y представляет собой N, Z представляет собой С, и L представляет собой Cl и Вr, может быть получена в соответствии со схемой реакции 9 в несколько этапов из соединений (ХI*). Сначала соединения Формулы (ХI*) обрабатывают бромирующим агентом, например, N-бромсукцинимид и бикарбонатом натрия в растворителе (например, метанол), в результате чего получают соединения формулы (XII*). Затем обработка соединений Формулы (ХII*) 1-хлорпропан-2-оном при повышенной температуре (например, при 90°С) дает смесь соединений Формулы (XIII*). Йодирование смеси соединений Формулы (XIII*), например, N-йодосукцинимидом, в растворителе (например, ДМФА) дает смесь соединений Формулы (VIb*).
Схема 9*

Соединения Формулы (Vic*), т.е., соединения Формулы (VI*) (см. схему 5*), в которых X представляет собой N, Y представляет собой С, Z представляет собой N, и L представляет собой CI, могут быть получены в несколько этапов из соединений Формулы (XIV*) в соответствии со схемой реакции 10*. Сначала может быть осуществлена реакция соединений Формулы (XIV*) с метилцианоформиатом с получением соединений (XV*). Затем соединения Формулы (XV*) могут быть циклизованы с использованием карбонилдиимидазола в растворителе (например, ДМСО) или, в качестве альтернативы, с использованием диэтилкарбоната в этоксида натрия и этаноле, с получением соединений (XVI*). Соединения Формулы (XVI*) можно затем хлорировать с использованием, например, POCl3, при нагревании (например, при 90°С) с получением соединений формулы (XVII*). Иодирование с использованием, например, N-йодосукцинимида, в растворителе (например, ДМФА) дает соединения Формулы (Vic*).
Схема 10*

Для квалифицированного химика понятно, что соединения Формулы (Ia) могут быть преобразованы в другие соединения Формулы (Ia) способами, известными в данной области. Дополнительно промежуточные соединения, описанные в приведенных выше схемах реакций, могут быть преобразованы в другие промежуточные соединения, а затем могут быть преобразованы, с использованием описанных способов, в соединения Формулы (Ia). Также понятно, что соединения Формулы (Ia) могут быть получены с использованием другой последовательности преобразований, описанных в схемах реакций, включая соответствующие этапы введения/удаления защиты. Способы применения
Было показано, что соединения согласно настоящему изобретению являются мощными ингибиторами PI4KIIIβ. Кроме того, соединения согласно настоящему изобретению являются селективными ингибиторами PI4KIIIβ. Соединения согласно настоящему изобретению могут применяться в лечении или предотвращении вирусных инфекций и нарушений, вызываемых или усугубляемых такой вирусной инфекцией. В частности, заболевания, вызываемые или усугубляемые вирусными инфекциями, включают ХОБЛ, астму, муковисцидоз, бронхоэктаз, застойную сердечную недостаточность, острый респираторный дистресс-синдром и острое поражение легких. Кроме того, заболевания, вызываемые или усугубляемые риновирусными инфекциями, включают бронхиолит, средний отит, синусит и острый бронхит. Также риновирусные инфекции могут вызывать вторичные бактериальные инфекции у детей, пожилых, и людей с подавленным иммунитетом. Такая вторичная бактериальная инфекция может вызвать пневмония.
В настоящем тексте «лечить», «лечение» или «средство лечения» применительно к нарушением обозначают: (1) облегчать нарушение или одно или более биологических проявлений нарушения; (2) показывать противодействие (а) одному или более элементам биологического каскада, который приводит к или отвечает за нарушение, или (b) одному или более биологическим проявлениям нарушения; (3) облегчать один или более симптомов или эффектов, связанных с нарушением; или (4) замедлять прогрессирование нарушения или одного или более биологических проявлений нарушения.
В настоящем тексте «пациент» относится к человеку (включая взрослых и детей) или другому животному. В одном варианте реализации «пациент» относится к человеку.
Предусмотрено, что соединения согласно настоящему изобретению можно вводить топически, например, путем ингаляции или интраназально. Ингаляция относится к введению в легкие пациента путем вдыхания через рот или через носовые ходы. В одном варианте реализации соединения Формулы (I) их фармацевтически приемлемые соли можно вводить топически. В другом варианте реализации соединения Формулы (I) или их фармацевтически приемлемые соли можно вводить путем ингаляции. В другом варианте реализации соединения Формулы (I) или их фармацевтически приемлемые соли можно вводить интраназально. В другом варианте реализации соединения Формулы (I) или их фармацевтически приемлемые соли можно вводить в глаз. В другом варианте реализации соединения Формулы (I) или их фармацевтически приемлемые соли можно вводить в ухо.
Соединения согласно настоящему изобретению можно вводить один раз в день или в соответствии со схемой дозирования, в которой несколько доз вводят через различные интервалы времени в течение определенного периода времени. Например, дозы можно вводить один, два, три или четыре раза в день. В одном варианте реализации дозу вводят один раз в день. В другом варианте реализации дозу вводят дважды в день. Дозу можно вводить до достижения желаемого терапевтического эффекта или неограниченное время для поддержания желаемого терапевтического эффекта. Подходящие схемы введения для соединения согласно настоящему изобретению зависят от фармакокинетических свойств соединения, таких как всасывание, распределение и время полужизни, которые могут быть определены квалифицированным специалистом. Дополнительно подходящие схемы введения, включая период, в течение которого такие схемы применяют, для соединения согласно настоящему изобретению зависят от заболевания, которое лечат, тяжести заболевания, которое лечат, возраста и физического состояния пациента, которого лечат, истории болезни пациента, которого лечат, природы сопутствующей терапии, желаемого терапевтического эффекта, и подобных факторов, находящихся в пределах знаний и опыта квалифицированного специалиста. Далее, таким квалифицированным специалистам будет понятно, что подходящие схемы дозирования могут потребовать корректировки в соответствии с индивидуальным ответом пациента на данную схему дозирования или с течением времени в зависимости от изменений потребностей конкретного пациента.
Типичные суточные дозировки для введения путем ингаляции лежат в диапазоне от 0.2 мкг до 0.02 мг на общей массы тела, например, от 0.5 мкг до 0.01 мг на общей массы тела. Например, суточные дозировки для введения путем ингаляции могут составлять от 20 мкг до 2.0 мг на пациента, например, от 50 мкг до 1.0 мг на пациента.
Дополнительно, соединения согласно настоящему изобретению можно вводить в виде пролекарств. В настоящем тексте «пролекарство» соединения согласно изобретению представляет собой функциональное производное, которое, при введении пациенту, высвобождает изобретение согласно настоящему изобретению in vivo. Введение соединения согласно настоящему изобретению в виде пролекарства может позволить квалифицированному специалисту сделать одно из следующего: (а) модифицировать начало проявления активности соединения in vivo; (b) модифицировать продолжительность действия соединения in vivo; (с) модифицировать транспорт или распределение соединения in vivo; (d) изменить растворимость соединения in vivo; и (е) преодолеть побочные эффекты или другие трудности, связанные с соединением. Типичные функциональные производные, применяемые для получения пролекарств, включают модификации соединения, которые могут подвергаться химическому или ферментативному расщеплению in vivo. Такие модификации, которые включают получение фосфатов, амидов, сложных эфиров, сложных тиоэфиро и карбаматов, хорошо известны квалифицированным специалистам.
Без привязки к конкретной теории, считается, что соединения согласно настоящему изобретению способны ингибировать активность клеточного фермента хозяина - РI4KIIIβ и за счет этого снижают способность вируса к репликации к клетке-хозяине. Многие вирусы используют PI4K для генерирования мембран, обогащенных фосфатидилинозитол-4-фосфатом (PI4P), которые могут использоваться как платформы для репликации. Механизм репликации вируса собирается на этих платформах в виде надмолекулярного комплекса, и липиды PI4P способствуют обеспечению синтеза вирусной РНК. Такие внутриклеточные липидные платформы создают более благоприятную среду для более эффективной репликации вируса. Нарушение способности вируса использовать молекулы PI4K и PI4KIIIβ, в частности, для создания этих липидных платформ и обеспечения вирусной репликации, может дать возможность лечения и/или предотвращения вирусных инфекций.
Соответственно, согласно еще одному аспекту настоящего изобретения предложен способ лечения вирусной инфекции, включающий введение соединения Формулы (I), или его фармацевтически приемлемой соли, нуждающемуся в этом пациенту.
Согласно другому аспекту настоящего изобретения предложен способ лечения нарушения, вызываемого или усугубляемого вирусной инфекцией, включающий введение соединения Формулы (I), или его фармацевтически приемлемой соли, нуждающемуся в этом пациенту.
Согласно другому аспекту настоящего изобретения предложен способ лечения вторичной бактериальной инфекцией, вызванной вирусной инфекцией, включающий введение соединения Формулы (I), или его фармацевтически приемлемой соли, нуждающемуся в этом пациенту.
Согласно другому аспекту настоящего изобретения предложено соединение Формулы (I), или его фармацевтически приемлемая соль, для применения в терапии.
Согласно другому аспекту настоящего изобретения предложено соединение Формулы (I), или его фармацевтически приемлемая соль для применения в лечении вирусной инфекции.
Согласно другому аспекту настоящего изобретения предложено соединение Формулы (I), или его фармацевтически приемлемая соль для применения в лечении нарушения, вызываемого или усугубляемого вирусной инфекцией. В одном из вариантов реализации нарушение представляет собой ХОБЛ, муковисцидоз, бронхоэктаз, астму или застойную сердечную недостаточность. В другом варианте реализации нарушение представляет собой ХОБЛ или астму. В еще одном варианте осуществления нарушение представляет собой ХОБЛ.
Согласно другому аспекту настоящего изобретения предложено соединение Формулы (I), или его фармацевтически приемлемая соль для применения в лечении вторичной бактериальной инфекции, вызванной бактериальной инфекцией.
Согласно другому аспекту настоящего изобретения предложено применение соединения Формулы (I), или его фармацевтически приемлемой соли, в изготовлении лекарственного средства для лечения вирусной инфекции.
Согласно другому аспекту настоящего изобретения предложено применение соединения Формулы (I), или его фармацевтически приемлемой соли, в изготовлении лекарственного средства для лечения нарушения, вызываемого или усугубляемого вирусной инфекцией.
Согласно другому аспекту настоящего изобретения предложено применение соединения Формулы (I), или его фармацевтически приемлемой соли, в изготовлении лекарственного средства для лечения вторичной бактериальной инфекции, вызванной вирусной инфекцией.
Для некоторых пациентов, как, например, у пациентов с ослабленной иммунной системой или кардио-легочными сопутствующими заболеваниями, у которых повышен риск развития тяжелой болезни после вирусной инфекции, предусмотрено профилактическое введение соединений согласно настоящему изобретению для того чтобы избежать усугубления нарушения, например, ХОБЛ, муковисцидоза, бронхоэктаза, астмы или застойной сердечной недостаточности. Понятно, что "предотвращение" не является абсолютным термином. В медицине под «предотвращением» понимают профилактическое введение лекарственного средства для сначительного снижения вероятности или тяжести нарушения или для отсрочки начала такого заболевания. Профилактическое введение может быть особенно полезным, в случаях, когда существует повышенный риск инфекции, например, в случае HRV - в течение зимних месяцев.
Согласно другому аспекту настоящего изобретения предложен способ предотвращения вирусной инфекции, включающий введение соединения Формулы (I), или его фармацевтически приемлемой соли, нуждающемуся в этом пациенту.
Согласно другому аспекту настоящего изобретения предложен способ предотвращения нарушения, вызываемого или усугубляемого вирусной инфекцией, включающий введение соединения Формулы (I), или его фармацевтически приемлемой соли, нуждающемуся в этом пациенту. В одном из вариантов реализации нарушение представляет собой ХОБЛ, муковисцидоз, бронхоэктаз, астму или застойную сердечную недостаточность. В другом варианте реализации нарушение представляет собой ХОБЛ или астму. В еще одном варианте осуществления нарушение представляет собой ХОБЛ.
Согласно другому аспекту настоящего изобретения предложено соединение Формулы (I), или его фармацевтически приемлемая соль для предотвращения вирусной инфекции.
Согласно другому аспекту настоящего изобретения предложено соединение Формулы (I), или его фармацевтически приемлемая соль для предотвращения нарушения, вызываемого или усугубляемого вирусной инфекцией. В одном из вариантов реализации нарушение представляет собой ХОБЛ, муковисцидоз, бронхоэктаз, астму или застойную сердечную недостаточность. В другом варианте реализации нарушение представляет собой ХОБЛ или астму. В еще одном варианте осуществления нарушение представляет собой ХОБЛ.
Согласно другому аспекту настоящего изобретения предложено применение соединения Формулы (I), или его фармацевтически приемлемой соли, в изготовлении лекарственного средства для предотвращения вирусной инфекции.
Согласно другому аспекту настоящего изобретения предложено применение соединения Формулы (I), или его фармацевтически приемлемая соль, в изготовлении лекарственного средства для предотвращения нарушения, вызываемого или усугубляемого вирусной инфекцией. В одном из вариантов реализации нарушение представляет собой ХОБЛ, муковисцидоз, бронхоэктаз, астму или застойную сердечную недостаточность. В другом варианте реализации нарушение представляет собой ХОБЛ или астму. В еще одном варианте осуществления нарушение представляет собой ХОБЛ.
Дополнительно, возможно профилактическое введение соединения согласно настоящему изобретению здоровым людям, если ожидается, что они подвергнутся повышенному риску вирусной инфекции, например, во время вспышки SARS или в условиях социального или медицинского учреждения, несколько резидентов которого имели контакт с HRV-инфекцией. Соответственно, согласно еще одному аспекту настоящего изобретения предложен способ предотвращения вирусной инфекции, включающий введение соединения Формулы (I), или его фармацевтически приемлемой соли, нуждающемуся в этом человеку.
Описанные ниже варианты реализации относятся к каждому из приведенных выше аспектов, связанных с медицинским применением. В одном из вариантов реализации вирус представляет собой одноцепочечный РНК-вирус. В еще одном варианте реализации вирус представляет собой одноцепочечный РНК-вирус с положительной смысловой цепью. В одном из вариантов реализации вирусная инфекция представляет собой РВЧ (HRV-риновирус человека), т.е. является РВЧ-инфекцией. В еще одном варианте реализации вирусная инфекция представляет собой РВЧ, и при этом нарушение, вызываемое вирусом, представляет собой простуду. В еще одном варианте реализации вирусная инфекция представляет собой РВЧ, причем нарушение, вызываемое вирусом, представляет собой бронхиолит, пневмонию, средний отит, синусит или острый бронхит. В еще одном варианте реализации вторичная бактериальная инфекция вызывает пневмонию. В другом варианте реализации нарушения, усугубляемые вирусом, представляют собой ХОБЛ, муковисцидоз, бронхоэктаз, астму или застойную сердечную недостаточность. В еще одном варианте реализации РВЧ предтсавляет собой РВЧ-А. В еще одном варианте реализации РВЧ представляет собой РВЧ-В. В еще одном варианте реализации РВЧ представляет сбой РВЧ-С. В другом варианте реализации соединение Формулы (I) вводят при начале назальных симптомов риновируса человека чтобы предотвратить легочную риновирусную инфекцию, и таким образом снизить частоту и тяжесть обострений астмы. В другом варианте реализации соединение Формулы (I) вводят при начале назальных симптомов риновируса человека чтобы предотвратить легочную риновирусную инфекцию, и таким образом снизить частоту и тяжесть обострений ХОБЛ. В другом варианте реализации соединение Формулы (I) вводят при начале назальных симптомов риновируса человека чтобы предотвратить легочную риновирусную инфекцию, и таким образом снизить частоту и тяжесть обострений муковисцидоза. В другом варианте реализации соединение Формулы (I) вводят при начале назальных симптомов риновируса человека чтобы предотвратить легочную риновирусную инфекцию, и таким образом снизить частоту и тяжесть обострений застойной сердечной недостаточности.
В одном из вариантов реализации вирусная инфекция является коронавирусной инфекцией, и при этом заболевание или состояние представляет собой тяжелый острый респираторный синдром (SARS или ТОРС).
Составы
Соединения согласно настоящему изобретению обычно, но не обязательно, будут готовиться в виде фармацевтических составов перед введением пациенту. Согласно другому аспекту настоящего изобретения предложен фармацевтический состав, содержащий соединение Формулы (I), или его фармацевтически приемлемую соль, и фармацевтически приемлемое вспомогательное вещество. Согласно другому аспекту настоящего изобретения предложен фармацевтический состав для лечения вирусной инфекции, содержащий Формулы (I) или его фармацевтически приемлемую соль. Согласно другому аспекту настоящего изобретения предложен фармацевтический состав для лечения нарушения, вызываемого или усугубляемого вирусной инфекцией, содержащий соединение Формулы (I) или его фармацевтически приемлемую соль. Согласно другому аспекту настоящего изобретения предложен фармацевтический состав для лечения вторичной бактериальной инфекции, вызванной вирусной инфекцией, содержащий соединение Формулы (I) или его фармацевтически приемлемая соль. Согласно другому аспекту настоящего изобретения предложен фармацевтический состав для предотвращения вирусной инфекции, содержащий соединение Формулы (I) или его фармацевтически приемлемую соль. Согласно другому аспекту настоящего изобретения предложен фармацевтический состав для предотвращения нарушения, вызываемого или усугубляемого вирусной инфекцией, содержащий Формулы (I) или его фармацевтически приемлемую соль.
В настоящем тексте «фармацевтически приемлемое вспомогательное вещество» обозначает фармацевтически приемлемый материал, композицию или основу, участвующие в придании формы или консистенции фармацевтическому составу. Каждое вспомогательное вещество должно быть совместимо с другими ингредиентами фармацевтического состава при объединении, чтобы избежать взаимодействий, которые существенно снизили бы эффективность соединений согласно настоящему изобретению при введении пациенту, и взаимодействий, которые могли бы привести к образованию фармацевтических составов, которые не являются фармацевтически приемлемыми. Кроме того, каждое вспомогательное вещество, конечно, должно быть фармацевтически приемлемым, например, достаточно высокой чистоты.
Предусмотрено, что соединения согласно настоящему изобретению можно вводить топически, например, путем ингаляции, интраназально, трансдермапьно, в глаз или в ухо.
Согласно другому аспекту настоящее изобретение относится к лекарственной форме, выполненной с возможностью введения пациенту путем ингаляции, например, в виде состава в форме сухого порошка, аэрозоля, суспензии или раствора.
Составы в форме сухого порошка для доставки в легкие путем ингаляции обычно содержат соединение согласно настоящему изобретению в виде тонкоизмельченного порошка совместно с одним или более фармацевтически приемлемыми вспомогательными веществами в виде тонкоизмельченных порошков. Фармацевтически приемлемые вспомогательные вещества, особенно подходящие для применения в сухом порошке, известны квалифицированных специалистам и включают лактозу, крахмал, маннитол, и моно-, ди-, и полисахариды. Тонкоизмельченный порошок может быть получен путем, например, микронизации и перемолки. Обычно, измельченное (например, микронизированное) соединение может характеризоваться значением D50 от примерно 1 до примерно 10 микрон (например, при измерении методом лазерной дифракции).
Сухой порошок можно вводить пациенту при помощи ингалятора сухого порошка с резервуаром (RDPI), имеющего резервуар, подходящий для хранения множества (неотмеренных доз) лекарственного средства в форме сухого порошка. RDPI обычно содержат для дозирования каждой дозы лекарственного средства из резервуара в место доставки. Например, средство дозирования может включать дозирующую чашку, которая выполнена с возможностью перемещения из первого положения, где чашка может быть заполнена лекарственным средством из резервуара, во второе положение, где отмеренная доза лекарственного средства становится доступной пациенту для ингаляции.
Составы в форме сухого порошка для применения в соответствии с настоящим изобретением можно вводить при помощи устройств для ингаляций. В качестве примера, такие устройства могут включать капсулы и картриджи из, например, желатина, или блистеры из, например, ламинированной алюминиевой фольги. В различных вариантах реализации каждая капсула, картридж или блистер может содержать дозы состава согласно настоящему описанию. Примеры устройств для ингаляции включают устройства, предназначенные для дозированной или многодозовой доставки состава, включая все устройства, представленные в настоящем документе. В качестве примера, в случае многодозовой доставки, составы могут быть заранее отмерены (например, как в DISKUS, см. GB2242134, патенты США №№6,032,666, 5,860,419, 5,873,360, 5,590,645, 6,378,519 и 6,536,427 или Diskhaler, см. GB 2178965, 2129691 и 2169265, патенты США №№4,778,054, 4,811,731, 5,035,237) или могут отмеряться при применении (например, как в устройстве Turbuhaler, см. ЕР 69715, или в устройствах, описанных в патенте США №6,321,747). Примером однодозового устройства является ROTAHALER (см. GB 2064336). В одном варианте реализации устройство для ингаляции DISKUS содержит продолговатую полоску, выполненную из основаного листа, содержащего множество углублений, расположенных на расстоянии по его длине, и покровного листа, который соединен к нему с возможности отслаивания, за счет чего образуется множество контейнеров, причем каждый контейнер содержит в чебе ингаляционный состав, содержащий соединения, необязательно с другими вспомогательными веществами и добавками, описанными в настоящем документе. Соединение с возможностью отслаивания представляет собой инженерное соединение и, в одном варианте реализации, это инженерное соединение представляет собой герметичное соединение. Предпочтительно, полоска является достаточно гибкой для сматывания в рулон. Покровный лист и лист основания предпочтительно имеют части ведущего конца, которые не соединяются друг с другом, и по меньшей мере одна из частей ведущего конца выполнена с возможностью присоединения к средству намотки. Также в предпочтительном случае инженерное соединение между основным и покровным листами проходит по всей ширине. Покровный лист предпочтительно может отслаиваться от основного листа в продольном направлении от первого конца основного листа.
Составы в форме сухого порошка также могут быть представлены в устройстве для ингаляции, которое выполнено с возможностью раздельного размещения двух разных компонентов состава. Таким образом, например, эти компоненты вводятся одновременно, но хранятся раздельно, например, в отдельных фармацевтических составах, например, как описано в например, WO 03/061743 А1 WO 2007/012871 А1, WO2007/068896, а также в патентах США №№8,113,199, 8,161,968, 8,511,304, 8,534,281, 8,746,242 и 9,333,310.
В одном варианте реализации ингаляционное устройство, выполненное с возможностью раздельного размещения компонентов, представляет собой ингалятор, имеющий две отслаиваемые блистерные полоски, каждая из которых содержит предварительно отмеренные дозы в блистерных карманах, расположенных вдоль ее длины, например, несколько контейнеров внутри каждой блистерной полоски, например, ELLIPTA. Такое устройство имеет внутренний механизм индексации, который при каждом приведении в действие устройства отслаивает, открывает карман каждой полоски и размещает блистеры так, чтобы каждая вновь экспонированная доза каждой полоски находилась рядом с коллектором, который сообщается с мундштуком устройства. Когда пациент вдыхает через мундштук, каждая доза одновременно вытягивается из соответствующего кармана в коллектор и увлекается через мундштук в дыхательные пути пациента. Еще одно устройство, выполненное с возможностью раздельного размещения различных компонентов - это DUOHALER от Innovata. Кроме того, различные конструкции ингаляционных устройств обеспечивают последовательную или раздельную доставку фармацевтического состава (ов) из устройства в дополнение к одновременной доставке.
В качестве альтернативы сухой порошок может быть представлен в капсулах (например, желатиновых или пластиковых), картриджах или блистерных упаковках для применения в многодозовом ингаляторе сухого порошка (MDPI). MDPI представляют собой ингаляторы, в которых лекарственное средство содержится в многодозовой упаковке, содержащей (или иным образом несущей) несколько определенных доз (или их частей) лекарственного средства. Когда сухой порошок представлен в виде блистерной упаковки, она содержит множество блистеров для хранения лекарственного средства в форме сухого порошка. Блистеры обычно расположены регулярным образом для облегчения высвобождения из них лекарственного средства. Например, блистеры могут быть расположены по существу по кругу на блистерной упаковке в форме диска, или блистеры могут быть удлиненными по форме, например, содержать полоску или ленту. Каждая капсула, картридж или блистер может, например, содержать от 200 мкг до 10 мг соединения Формулы (I) или его фармацевтически приемлемой соли.
Аэрозоли могут быть получены путем суспендирования или растворения соединения согласно настоящему изобретению в сжиженном пропелленте. Подходящие пропелленты включают галоуглероды, углеводороды и другие сжиженные газы. Типичные примеры пропеллентов включают: трихлорфторметан (пропеллент 11), дихлорфторметан (пропеллент 12), дихлортетрафторэтан (пропеллент 114), тетрафторэтан (HFA-134a), 1,1-дифторэтан (HFA-152a), дифторметан (HFA-32), пентафторэтан (HFA-12), гептафторпропан (HFA-227a), перфторпропан, перфторбутан, перфторпентан, бутан, изобутан и пентан. Аэрозоли, содержащие соединение согласно настоящему изобретению, обычно будут вводить пациенту при попощи дозирующего ингалятора (MDI). Такие устройства известны опытным специалистам.
Аэрозоль может содержать дополнительные фармацевтически приемлемые вспомогательные вещества, обычно применяемые в комбинации с MDI, такие как поверхностно-активные вещества, смазывающие вещества, сорастворители и другие вспомогательные вещества для улучшения физической стабильности состава, для улучшения работы клапана, для улучшения растворимости, для улучшения вкуса.
Согласно другому аспекту предложен фармацевтический аэрозольный состав, содержащий соединение Формулы (I) или его фармацевтически приемлемую соль и фторуглерод или хлорфторуглерод в качестве пропеллента, необязательно в комбинации с поверхностно-активным веществом и/или со-растворителем.
В соответствии с одним из вариантов осуществления пропеллент выбран из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3-гептафтор-н-пропана и их смесей.
Составы согласно настоящему изобретению могут быть забуферены путем добавления подходящих буферных веществ.
Капсулы и картриджи для применения в ингаляторе или инсуффляторе из, например, желатина могут быть выполнены содержащими порошковую смесь для ингаляции соединения согласно настоящему изобретению и порошковое основание, такое как лактоза или крахмал. Обычно каждая капсула или картридж могут содержать от 200 мкг до 10 мг соединения согласно настоящему изобретению. В качестве альтернативы соединение согласно настоящему изобретению может быть представлено без вспомогательных веществ, таких как лактоза.
Доля соединения согласно настоящему изобретению в составах для местного введения согласно настоящему изобретению зависит от точного типа состава, который нудно получить, но в целом будет находиться в диапазоне от 0.01 до 10% по массе. Обычно, для большинства типов препаратов, эта доля будет лежать в диапазоне от 0.05 до 1%, например, от 0.1 до 0.5%.
Аэрозольные составы предпочтительно выполняют таким образом, что каждая отмеренная доза или «затяжка» аэрозоля содержит от 20 мкг до 10 мг, предпочтительно от 20 мкг до 5 мг, более предпочтительно, от примерно 20 мкг до 0.5 мг соединения согласно настоящему изобретению. Введение можно осуществлять один раз в день или несколько раз в день, например, 2, 3, 4 или 8 раз, с введением например, 1, 2 или 3 доз каждый раз. Общая суточная доза при использовании аэрозоля будет лежать в диапазоне от 20 мкг до 2.0 мг, например, от 50 мкг до 1.0 мг. Общая суточная доза и отмеренные дозы, доставляемые посредством капсул и картриджей в ингаляторе инсуффляторе обычно будут в два раза больше доз, доставляемых в аэрозольных составах.
В случае суспензионных аэрозольных составов размер частиц лекарственного средства в виде частиц (например, микронизированного) должен быть таким чтобы обеспечивать возможность ингаляции по существу всего лекарственного средства в легкие при введении аэрозольного состава и, соответственно, будет меньше 100 микрон, желательно, меньше 20 микрон и, в частности, в диапазоне от 1 до 10 микрон, как, например, от 1 до 5 микрон, более предпочтительно от 2 до 3 микрон.
Составы согласно настоящему изобретению могут быть получены путем диспергирования или растворения медикамента и соединения согласно настоящему изобретению в выбранном пропелленте в подходящем контейнере, например, с помощью ультразвука или смесителя с высоким усилием сдвига. Указанный процесс желательно проводить в условиях контролируемой влажности.
Химическая и физическая стабильность, а также фармацевтическая приемлемость аэрозольных составов в соответствии с настоящим изобретением могут быть определены с применением методик, хорошо известных специалистам в данной области техники. Соответственно, например, химическая стабильность указанных компонентов может быть определена с применением ВЭЖХ-анализа, например, после продолжительного хранения продукта. Данные о физической стабильности могут быть получены с применением других стандартных методов анализа, таких как, например, испытания на отсутствие утечки, анализ поступления через клапан (средние массы впрыска на одно срабатывание), анализ воспроизводимости дозы (количество активного ингредиента на одно срабатывание) и анализ распределения спрея. Стабильность суспензионных аэрозольных составов в соответствии с настоящим изобретением может быть измерена с помощью стандартных методик, например, путем измерения гранулометрического состава флокулянта с применением инструмента для обратного рассеяния света; или путем измерения гранулометрического состава частиц методом каскадного импактора или аналитическим методом «двойного импинджера». В настоящем документе анализ с «двойным импинджером» означает «определение отложения испускаемой дозы при ингаляции под давлением с применением аппарата А» согласно определению в Британской фармакопее 1988 года, стр. А204-207, приложение XVII С.Такие методики позволяют рассчитать величину «вдыхаемой фракции» аэрозольных составов. Один способ, используемый для расчета «вдыхаемой фракции», основан на «фракции мелких частиц», которая представляет собой количество активного ингредиента, собираемое в нижней камере импинджера за одно срабатывание, выраженное в процентах от общего количества активного ингредиента, доставляемого за одно срабатывание, при использовании метода двойного импинджера, описанного выше.
Термин «дозирующий ингалятор», или MDI означает устройство, содержащее емкость, зафиксированную крышку, закрывающую емкость, и клапан дозирования состава, расположенный в крышке. Система MDI включает подходящее канализирующее устройство. Подходящие канализирующие устройства содержат, например, исполнительный клапан и цилиндрический или конусообразный канал, через который медикамент может быть доставлен из заполненного баллончика с помощью дозирующего клапана в нос или рот пациента, такой как клапан с мундштуком.
Баллончики MDI обычно включают контейнер, способный выдерживать давление пара используемого пропеллента, например, бутыль из пластика или покрытого пластиком стекла или, предпочтительно, металлическую емкость, например, из алюминия или сплава алюминия, который может необязательно быть анодирован, покрыт лаком и/или пластиком (например, согласно включенной в настоящий документ посредством ссылки WO 96/32099 все или некоторые внутренние поверхности покрыты одним или более фторуглеродными полимерами, необязательно в комбинации с одним или более не-фторуглеродными полимерами), и указанный контейнер закрыт дозирующим клапаном. Крышка может быть закреплена на емкости с помощью ультразвуковой сварки, резьбового соединения или обжатия. MDI, описанные в настоящем документе, могут быть получены с применением способов, известных в данной области техники (например см. Byron, выше, и WO 96/32099). Предпочтительно баллончик оборудован крышкой в сборе, причем в указанной крышке расположен дозирующий лекарство клапан, и она зафиксирована на месте путем обжатия.
В одном варианте реализации настоящего изобретения металлическая внутренняя поверхность емкости покрыта фторполимером, предпочтительнее -смешанную с не-фторполимером. Согласно другому варианту реализации настоящего изобретения металлическая внутренняя поверхность емкости покрыта смесью полимеров политетрафторэтилена (PTFE) и простого полиэфирсульфона (PES). В дополнительном варианте реализации согласно настоящему изобретению металлическая внутренняя поверхность емкости полностью покрыта смесью полимеров политетрафторэтилена (PTFE) и простого полиэфирсульфона (PES).
Дозирующие клапаны разработаны таким образом, чтобы доставлять отмеренное количество состава при каждом срабатывании и включают уплотнитель для предотвращения утечки пропеллента через клапан. Указанный уплотнитель может содержать любой подходящий эластичный материал, такой как, например, полиэтилен низкой плотности, хлорбутил, бромбутил, СКЭПТ, черный и белый бутадиен-акрилонитриловые каучуки, бутил каучук и неопрен. Подходящие клапаны коммерчески доступны у производителей, хорошо известных в аэрозольной промышленности, например, Valois, Франция (например, DF10, DF30, DF60), Bespak Pic, Великобритания (например, ВK300, ВK357), и 3M-Neotechnic Ltd, Великобритания (например, SPRAYMISER).
Согласно различным вариантам реализации указанные MDI могут также применяться в сочетании с другими структурами, такими как, без ограничения, внешняя упаковка для хранения, вмещающая MDI, в том числе описанные в патентах США №6119853; №6179118; №6315112; №6352152; №6390291; и №6679374, а также устройства для подсчета доз, такие как, но не ограничиваясь перечисленными, описанные в патентах США №6360739 и №6431168.
Обычные способы и оборудование для массового производства, хорошо известные специалистам в области приготовления фармацевтических аэрозолей, могут быть использованы для получения крупных партий для коммерческого производства заполненных баллончиков. Соответственно, например, согласно одному способу массового производства для получения суспензионных аэрозольных составов дозирующий клапан обжимают на алюминиевой емкости, получая пустой баллончик. Состоящий из частиц медикамент добавляют в сосуд для заправки и сжиженным пропеллентом совместно с необязательными вспомогательными веществами под давлением наполняют производственный сосуд через сосуд для заправки. Суспензию лекарственного средства перемешивают перед возвращением в наполняющий аппарат, после чего аликвотой суспензии лекарственного средства заполняют баллончик через дозирующий клапан. В одном примере способа массового производства для получения аэрозольных составов из растворов дозирующий клапан обжимают на алюминиевой емкости, получая пустой баллончик. Сжиженным пропеллентом вместе с необязательными вспомогательными веществами и растворенным медикаментом под давлением наполняют производственный сосуд через сосуд для заправки.
В альтернативном варианте процесса аликвоту сжиженного состава добавляют в открытый баллончик в условиях достаточно низких температур для того, чтобы состав не испарялся, а затем обжимают дозирующий клапан на баллончике.
Как правило, в партиях, подготовленных для фармацевтического применения, производят контрольное взвешивание каждого заполненного баллончика, наносят код номера партии и упаковывают в поддон для хранения перед тестированием готовой продукции.
Суспензии и растворы, содержащие соединение согласно настоящему изобретению, могут также вводиться пациенту с помощью небулайзера. Растворитель или суспензионный агент, используемый для небулизации, может представлять собой любую фармацевтически приемлемую жидкость, такую как вода, водный солевой раствор, спирты или гликоли, например, этанол, изопропиловый спирт, глицерин, пропиленгликоль, полиэтиленгликоль и т.п., или их смеси. Для солевых растворов используют соли, которые проявляют незначительную фармакологическую активность или не проявляют такой активности после введения. С указанной целью могут применяться как неорганические соли, такие как соли щелочных металлов или галоидные соли аммония, например, хлорид натрия, хлорид калия, так и органические соли, такие как калиевые, натриевые и аммониевые соли органических кислот, например, аскорбиновой кислоты, лимонной кислоты, уксусно кислоты, винной кислоты и т.п.
В суспензию или раствор могут быть добавлены другие фармацевтически приемлемые вспомогательные вещества. Соединение согласно настоящему изобретению может быть стабилизировано добавлением неорганической кислоты, например, соляной кислоты, азотной кислоты, серной кислоты и/или фосфорной кислоты; органической кислоты, например, аскорбиновой кислоты, лимонной кислоты, уксусной кислоты и винной кислоты, и т.п., комплексообразующего агента, такого как ЭДТК или лимонная кислота и их соли; или антиоксиданта, такого как антиоксидант витамин Е или аскорбиновая кислота. Указанные агенты могут применяться по отдельности или совместно для стабилизации соединения формулы (I) или его фармацевтически приемлемой соли. Могут быть добавлены консерванты, такие как бензалкония хлорид или бензойная кислота и ее соли. Может быть добавлено поверхностно-активное вещество, в частности, для улучшения физической стабильности суспензий. Указанные ПАВ включают лецитин, диоктилсульфосукцинат динатрия, олеиновую кислоту и сложные эфиры сорбита.
Составы для введения в нос могут включать аэрозольные составы под давлением и водные составы, которые вводятся в нос с помощью насоса под давлением. В частности, интерес представляют составы, не находящиеся под давлением и адаптированные для введения местно в носовую полость. Подходящие составы содержат воду в качестве разбавителя или носителя для указанной цели. Водные составы для введения в легкие или нос могут предоставляться со стандартными вспомогательными веществами, такими как буферные агенты, модифицирующие тоничность агенты и т.п. Водные составы могут также вводиться в нос путем небулизации.
Соединения согласно настоящему изобретению могут быть введены в жидкий состав для доставки из дозатора жидкости, например, дозатора жидкости с дозирующим соплом или дозирующим отверстием, через которое поступает отмеренная доза жидкого состава при приложении силы пользователем к механизму насоса указанного дозатора жидкости. Такие дозаторы жидкости обычно снабжены резервуаром с несколькими отмеренными дозами жидкого состава, которые могут быть выданы при последовательных приведениях насоса в действие. Дозирующее сопло или отверстие может быть выполнено с возможностью помещения в ноздри пользователя для распределения спрея жидкого состава в носовой полости. Дозатор жидкости вышеупомянутого типа описан и проиллюстрирован в WO 05/044354, все содержание которого полностью включено в настоящий документ посредством ссылки. Дозатор включает корпус, внутри которого размещено устройство для нагнетания жидкости, содержащее компрессионный насос, установленный на контейнере для жидкого состава. Корпус имеет по меньшей мере один боковой рычаг, управляемый пальцами, который способен смещаться в направлении внутрь относительно корпуса, отводя контейнер внутри корпуса вверх, в результате чего насос сжимается и выкачивает отмеренную дозу состава из штока насоса через назальную насадку на корпусе. В одном варианте реализации указанный дозатор жидкости представлен общим типом, показанным на фиг. 30-40 WO 05/044354.
Фармацевтические составы, адаптированные для интраназального введения, где носитель представляет собой твердое вещество, включают крупнодисперсный порошок с размерами частиц, например, в диапазоне 20-500 мкм, который вводят путем быстрого вдыхания через носовой канал из контейнера с порошком, удерживаемого возле носа. Подходящие составы, где носитель представляет собой жидкость, для введения в виде назального спрея или назальных капель включают водные или масляные растворы соединения согласно настоящему изобретению.
Некоторые из расстройств, вызываемых или усугубляемых вирусными инфекциями, могут приводить к проблемам с кожей, например, к кожной сыпи. Фармацевтические составы, адаптированные для чрескожного введения, могут быть представлены отдельными пластырями, предназначенными для близкого контакта с эпидермисом пациента в течение продолжительного периода времени. Например, активный ингредиент может доставляться из пластыря путем ионтофореза, как в общем описано в Pharmaceutical Research, 3(6), 318 (1986).
Фармацевтические составы, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел.
Мази, кремы и гели, могут, например, быть введены в состав с водной или масляной основой с добавлением подходящего загущающего и/или гелеобразующего агента и/или растворителей. Такие основы могут, соответственно, например, включать воду и/или масло, такое как жидкий парафин или растительное масло, такие как арахисовое масло или касторовое масло, или растворитель, такой как полиэтиленгликоль. Загущающие агенты и гелеобразующие агенты, которые могут применяться в зависимости от природы основы, включают мягкий парафин, стеарат алюминия, цетостеариловый спирт, полиэтиленгликоли, шерстяной жир, пчелиный воск, карбоксиполиметилен и производные целлюлозы, и/или глицерилмоностеарат и/или неионогенные эмульгирующие агенты.
Лосьоны могут быть введены в состав с водной или масляной основой и, в общем случае, также содержат один или более эмульгирующих агентов, стабилизирующих агентов, диспергирующих агентов, суспендирующих агентов или загущающих агентов.
Порошки для наружного применения могут быть получены с использованием любой подходящей порошковой основы, например, талька, лактозы или крахмала. Капли могут быть приготовлены с водной или неводной основой, также содержащей один или более диспергирующих агентов, солюбилизирующих агентов, суспендирующих агентов или консервантов.
Составы для местного применения могут вводиться путем одного или более нанесений в сутки на пораженный участок. На участках кожи предпочтительно использовать окклюзионные повязки. Непрерывная или продолжительная доставка может быть достигнута за счет применения адгезивной резервуарной системы.
Для интраокулярного или ушного лечения составы могут применяться в виде мази или крема для местного применения. При введении в состав мази соединение формулы (I) или его фармацевтически приемлемая соль могут применяться либо с парафиновой, либо с смешиваемой с водой основой для мази. Как вариант, соединение формулы (I) или его фармацевтически приемлемая соль могут быть введены в состав крема с основой для крема типа «масло в воде» или основой типа «вода в масле».
Фармацевтические составы, адаптированные для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатические средства и растворенные вещества, которые делают состав изотоническим относительно крови предполагаемого реципиента; а также водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загущающие агенты. Указанные составы могут быть предоставлены в порционных или многодозовых контейнерах, например, герметизированные ампулы и флаконы, и могут храниться в высушенном замораживанием (лиофилизированном) состоянии, требуя только добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед применением. Экстемпоральные инъецируемые растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток.
Соединения или фармацевтические составы согласно настоящему изобретению могут вводиться совместно с противовоспалительным агентом, таким как кортикостероид, или содержащим его фармацевтическим составом для лечения астмы, усугубленной вирусными инфекциями, в частности, инфекциями HRV. Например, соединения согласно настоящему изобретению могут быть введены совместно с противовоспалительным агентом, таким как кортикостероид, в один состав, такой как сухой порошковый состав для ингаляции. Как вариант, фармацевтический состав, содержащий соединение согласно настоящему изобретению, может вводиться в сочетании с фармацевтическим составом, содержащим противовоспалительный агент, такой как кортикостероид, либо одновременно, либо последовательно. Например, оба фармацевтических состава - фармацевтический состав, содержащий соединение согласно настоящему изобретению и дополнительный фармацевтический состав, содержащий противовоспалительный агент, такой как кортикостероид - могут находиться в устройстве, подходящем для одновременного введения обоих составов посредством ингаляции.
Подходящие кортикостероиды для введения совместно с соединениями согласно настоящему изобретению включают флутиказона фуроат, флутиказона пропионат, беклометазона дипропионат, будесонид, циклесонид, мометазона фуроат, триамцинолон, флунизолид и преднизолон. Подходящие кортикостероиды для введения совместно с соединениями согласно настоящему изобретению посредством ингаляции включают флутиказона фуроат, флутиказона пропионат, беклометазона дипропионат, будесонид, циклесонид, мометазона фуроат и флунизолид.
Соответственно, в соответствии с дополнительным аспектом согласно настоящему изобретению предложен фармацевтический состав, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, и один или более противовоспалительных агентов, такие как кортикостероид или ингибитор фосфатидилинозитол-4,5-бифосфат-3-киназы-дельта (PI3Kδ).
В соответствии с дополнительным аспектом согласно настоящему изобретению предложен способ лечения астмы, усугубленной вирусной инфекцией, например, HRV, который включает введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, и одного или более противовоспалительных агентов, таких как кортикостероид.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль, и один или более противовоспалительных агентов, таких как кортикостероид, для применения при лечении астмы, усугубленной вирусной инфекцией.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, и одного или более противовоспалительных агентов, таких как кортикостероид, при изготовлении медикамента для лечения астмы, усугубленной вирусной инфекцией.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложен способ лечения кистозного фиброза, усугубленного вирусной инфекцией, например, HRV, который включает введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, и одного или более противовоспалительных агентов, таких как кортикостероид.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль, и один или более противовоспалительных агентов, таких как кортикостероид, для применения при лечении кистозного фиброза, усугубленного вирусной инфекцией.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложено применение соединения формулы (I) или его фармацевтически приемлемая соль, и один или более противовоспалительных агентов, таких как кортикостероид, при изготовлении медикамента для лечения кистозного фиброза, усугубленного вирусной инфекцией.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложен способ лечения застойной сердечной недостаточности, усугубленной вирусной инфекцией, например, HRV, который включает введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, и одного или более противовоспалительных агентов, таких как кортикостероид.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль, и один или более противовоспалительных агентов, таких как кортикостероид, для применения при лечении застойной сердечной недостаточности, усугубленной вирусной инфекцией.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, и одного или более противовоспалительных агентов, таких как кортикостероид, при изготовлении медикамента для лечения застойной сердечной недостаточности, усугубленной вирусной инфекцией.
Приведенные ниже варианты реализации применимы к любому из вышеописанных аспектов, относящихся к комбинациям с одним или более противовоспалительными агентами.
Во варианте реализации указанный фармацевтический состав содержит один противовоспалительный агент.
В одном варианте реализации указанный противовоспалительный агент представляет собой кортикостероид.
В одном варианте реализации указанный кортикостероид представляет собой флутиказона фуроат.
В одном варианте реализации указанный кортикостероид представляет собой флутиказона пропионат.
В одном варианте реализации указанный противовоспалительный агент представляет собой ингибитор фосфатидилинозитол-4,5-бифосфат-3-киназы-дельта (PI3Kδ), такой как немиралисиб (см. Sriskantharajah et al., Annals of the New York Academy of Sciences, (2013), 1280, 35; Cahn et al., Pulmonary Pharmacology and Therapeutics, (2017), 46, 69; и Stark et al., Current Opinion in Pharmacology, (2015), 23, 82).
Соединения или фармацевтические составы согласно настоящему изобретению могут вводиться совместно с одним или более бронходилататорами, или содержащими их фармацевтическими составами, для лечения ХОБЛ, усугубленной вирусной инфекцией. Например, соединения согласно настоящему изобретению могут быть введены совместно с одним или более бронходилататорами в один состав, такой как сухой порошковый состав для ингаляции. Как вариант, фармацевтический состав, содержащий соединение согласно настоящему изобретению, может вводиться в сочетании с фармацевтическим составом, содержащим один или более бронходилататоров, либо одновременно, либо последовательно. В дополнительном альтернативном варианте реализации состав, содержащий соединение согласно настоящему изобретению и бронходилататор, может вводиться в сочетании с фармацевтическим составом, содержащим дополнительный бронходилататор. Например, каждый из составов - фармацевтический состав, содержащий соединение согласно настоящему изобретению, и дополнительный фармацевтический состав, содержащий один или более бронходилататоров - может находиться в устройстве, подходящем для одновременного введения обоих составов путем ингаляции.
Подходящие бронходилататоры для введения совместно с соединениями согласно настоящему изобретению включают агонисты β2-адренорецепторов и антихолинергические агенты. Примеры агонистов β2-адренорецепторов включают, например, вилантерол, салметерол, сальбутамол, формотерол, салмефамол, фенотерол, кармотерол, этантерол, наминтерол, кленбутерол, пирбутерол, флербутерол, репротерол, бамбутерол, индакатерол, тербуталин и их соли, например, ксинафоатная (1-гидрокси-2-нафталинкарбоксилат) соль салметерола, сульфатная соль сальбутамола или фумаратная соль формотерола. Примеры антихолинергических агентов включают умеклидиний (например, в виде бромида), ипратропий (например, в виде бромида), окситропий (например, в виде бромида) и тиотропий (например, в виде бромида). В одном варианте реализации соединение согласно настоящему изобретению может вводиться совместно с агонистом β2-адренорецепторов, таким как вилантерол, и антихолинергическим агентом, таким как умеклидиний.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложен фармацевтический состав, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, и один или более бронходилататоров.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложен способ лечения ХОБЛ, усугубленной вирусной инфекцией, который включает введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, и одного или более бронходилататоров.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль, и один или более бронходилататоров для применения при лечении ХОБЛ, усугубленной вирусной инфекцией.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, и одного или более бронходилататоров при изготовлении медикамента для лечения ХОБЛ, усугубленной вирусной инфекцией.
Приведенные ниже варианты реализации применимы к любому из вышеописанных аспектов, относящихся к комбинациям с одним или более бронходилататорами.
Во варианте реализации указанные один или более бронходилататоров содержат один или более агонистов β2-адренорецепторов.
Во варианте реализации указанные один или более бронходилататоров содержат один или более антихолинергических агентов.
Во варианте реализации указанные один или более бронходилататоров содержат один или более агонистов β2-адренорецепторов и один или более антихолинергических агентов.
Во варианте реализации указанные один или более бронходилататоров содержат агонист β2-адренорецепторов и антихолинергический агент.
Во варианте реализации указанные один или более бронходилататоров содержат один бронходилататор, который представляет собой агонист β2-адренорецепторов.
Во варианте реализации указанные один или более бронходилататоров содержат один бронходилататор, который представляет собой антихолинергический агент.
Во варианте реализации указанный агонист β2-адренорецепторов представляет собой вилантерол.
Во варианте реализации указанный антихолинергический агент представляет собой умеклидиний. В дополнительном варианте реализации указанный антихолинергический агент представляет собой умеклидиния бромид.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложен фармацевтический состав, содержащий а) соединение формулы (I) или его фармацевтически приемлемую соль, b) один или более бронходилататоров; и с) один или более противовоспалительных агентов.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложен способ лечения ХОБЛ, усугубленной вирусной инфекцией, который включает введение нуждающемуся в этом субъекту терапевтически эффективного количества а) соединения формулы (I) или его фармацевтически приемлемой соли, b) одного или более бронходилататоров; и с) одного или более противовоспалительных агентов.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложены а) соединение формулы (I) или его фармацевтически приемлемая соль, b) один или более бронходилататоров; и с) один или более противовоспалительных агентов для применения при лечении ХОБЛ, усугубленной вирусной инфекцией.
В соответствии с дополнительным аспектом согласно настоящему изобретению предложено применение а) соединения формулы (I) или его фармацевтически приемлемой соли, b) одного или более бронходилататоров; и с) одного или более противовоспалительных агентов при изготовлении медикамента для лечения ХОБЛ, усугубленной вирусной инфекцией.
Приведенные ниже варианты реализации применимы к любому из вышеописанных аспектов, относящихся к комбинациям с одним или более бронходилататорами и одним или более противовоспалительными агентами.
Во варианте реализации указанные один или более бронходилататоров содержат один или более агонистов β2-адренорецепторов.
Во варианте реализации указанные один или более бронходилататоров содержат один или более антихолинергических агентов.
Во варианте реализации указанные один или более бронходилататоров содержат один или более агонистов β2-адренорецепторов и один или более антихолинергических агентов.
Во варианте реализации указанные один или более бронходилататоров содержат агонист Рг-адренорецепторов и антихолинергический агент.
Во варианте реализации указанные один или более бронходилататоров содержат один бронходилататор, который представляет собой агонист β2-адренорецепторов.
Во варианте реализации указанные один или более бронходилататоров содержат один бронходилататор, который представляет собой антихолинергический агент.
Во варианте реализации указанный агонист β2-адренорецепторов представляет собой вилантерол.
Во варианте реализации указанный антихолинергический агент представляет собой умеклидиний. В дополнительном варианте реализации указанный антихолинергический агент представляет собой умеклидиния бромид.
Во варианте реализации указанные один или более противовоспалительных агентов представляют собой кортикостероид.
В дополнительном варианте реализации указанный кортикостероид представляет собой флутиказона фуроат.
В дополнительном варианте реализации указанный кортикостероид представляет собой флутиказона пропионат.
В дополнительном варианте реализации указанные один или более бронходилататоров представляют собой вилантерол и умеклидиний.
В дополнительном варианте реализации указанные один или более бронходилататоров представляют собой вилантерол и умеклидиний, а указанные один или более противовоспалительных агентов представляют собой флутиказона фуроат.
В дополнительном варианте реализации указанные один или более бронходилататоров представляют собой вилантерол и умеклидиний, а указанные один или более противовоспалительных агентов представляют собой флутиказона пропионат.
Соединения согласно настоящему изобретению могут иметь улучшенный профиль по сравнению с известными ингибиторами PI4KIIIβ, например, по сравнению с известными ингибиторами PI4KIIIβ определенные соединения согласно настоящему изобретению могут обладать одним или более из следующих свойств:
(i) более мощная ингибиторная активность в отношении PI4KIIIβ;
(ii) улучшенная селективность в отношении PI4KIIIβ;
(iii) увеличение T1/2 фермента;
(iv) улучшенная эффективность в клетках;
(v) Улучшенное удержание в легких
(vi) улучшенная растворимость; и/или
(vii) более низкие уровни накопления соединения в ткани организма.
Как указано выше, в ткани организма после введения могут накапливаться уровни соединений согласно настоящему изобретению, более низкие по сравнению с уровнями известных ингибиторов PI4KIIIβ. В частности, соединения согласно настоящему изобретению могут характеризоваться более низкими уровнями накопления в селезенке. Предыдущие исследования показали, что накопление ингибиторов PI4KIIIB в селезенке может быть неблагоприятным, например, может вызывать апоптоз. Подтверждающие соединения
Приведенные ниже ниже подтверждающие соединения иллюстрируют изобретение в качестве руководства по получению и применению соединений, составам и способам по изобретению для квалифицированного специалиста. Хотя описаны конкретные варианты осуществления изобретения, квалифицированный специалист поймет, что могут быть сделаны различные изменения и модификации. Указания процедур получения, осуществляемых аналогично другим процедурам получения или по общему способу, могут охватывать изменения в стандартных параметрах, таких как время, температура, условия обработки, изменения количеств реагентов и т.д.
Реакции с использованием гидридов металлов (включая гидрид натрия) и металлоорганических реагентов проводят в атмосфере аргона или азота, если не указано иное.
В описанных далее промежуточных соединениях и подтверждающий соединениях, для которых была идентифицирована относительная стереохимия соединения, это указано как в названии, так и в структуре соединения.
Для некоторых из описанных далее промежуточных соединений и подтверждающих соединений исходные материалы идентифицируются ссылкой на другие номера промежуточных соединений или соединений по изобретению. Это не означает, что фактический материал (или «партия»), полученный из какого-либо конкретного промежуточного соединения или подтверждающего соединения, обязательно использовался на последующем этапе, приведенном в настоящем изобретении в качестве примера.
Если не указано иное, исходные материалы были доступны для приобретения. Все растворители и товарные реагенты были лабораторного качества и использовались в неизмененном виде.
Если абсолютная стереохимия известна и соединение представляет собой один энантиомер, при необходимости используются жирные или заштрихованные клинья ( ). Там, где абсолютная стереохимия неизвестна, но известно, что это единственный энантиомер, используется звездочка (*).
). Там, где абсолютная стереохимия неизвестна, но известно, что это единственный энантиомер, используется звездочка (*).
Названия промежуточных соединений и подтверждающих соединений были получены с использованием программы генерирования названий в «ChemBioDraw Ultra v12» или «ACD Name Pro 6.02».
Используемые в настоящем документе символы и условные обозначения, используемые в этих процессах, схемах и примерах, соответствуют тем, которые используются в современной научной литературе, например, в Журнале Американского химического общества. Сокращения
В списке ниже приведены определения некоторых сокращений и символов, используемых в данном документе. Следует понимать, что список не является исчерпывающим, но значение тех сокращений и символов, которые не определены ниже в данном документе, понятно специалистам в данной области техники. В описании изобретения химические элементы идентифицированы в соответствии с периодической таблицей элементов.
Вu - бутил
m-СРВА - мета-хлорпероксибензойная кислота
CV - Объем(ы) колонки
ДХМ - Дихлорметан
DIBAL-H - Диизобутилалюминия гидрид
DIPEA - N,N-диизопропилэтиламин
DMAP - 4-диметиламинопиридин
DMP - периодинан Десс-Мартина
ДМФА - N,N-диметилформамид
ДМСО - Диметилсульфоксид
Dppf - 1,1'бис(дифенилфосфино)ферроцен
HATU - 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния 3-оксид гексафторфосфат
HCI - Соляная кислота
IPA - Изопропанол
2-метилтетрагидрофуран - 2-метилтетрагидрофуран
БСИ - N-бромсукцинимид
N-йодосукцинимид - N-йодосукцинимид
rt(вр.уд.) - Время удерживания
TFA - Трифторуксусная кислоты
ТГФ - Тетрагидрофуран
ВЭЖХ - Высокоэффективная жидкостная хроматография
MDAP - Масс-направленная автопрепаративная ВЭЖХ
XPhos - дициклогексил(2',4',6'-триизопропил-[1,1'-бифенил]-3-ил)фосфан
XPhos Pd G2 - хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(II)
Методы ЖХМС
Метод А
Колонка: Acquity ВЕН C18 (50 мм × 2.1 мм, 1.7 мкм). Подвижная фаза: А: 0.1% муравьиная кислота в воде; В: 0.1% муравьиная кислота в ацетонитриле. Время (мин)/% В: 0 / 3, 0.4 / 3, 2.0 / 98, 3.4 / 98, 3.5 / 3, 4.0 / 3. Температура колонки: 35°С, Скорость потока: 0.6 мл/мин.
Метод В
Колонка: XBridge ВЕН C18 (50 мм × 4.6 мм, 2.5 μМ). Подвижная фаза: А: 5 мМ бикарбонат аммония; В: ацетонитрил. Время (мин)/% В: 0 / 5, 0.5 / 5, 1 / 15, 3.3 / 98, 5.2 / 98, 5.5 / 5, 6.0 / 5. Температура колонки: 35°С, Скорость потока: 1.3 мл/мин.
Метод С
Колонка: Acquity ВЕН C18 (50 мм × 2.1 мм, 1.7 мкм). Подвижная фаза: А: 0.1% муравьиная кислота в воде; В: 0.1% муравьиная кислота в ацетонитриле. Время (мин)/% В: 0 / 3, 0.4 / 3, 2.5 / 98, 3.4 / 98, 3.5 / 3, 4.0 / 3. Температура колонки: 35°С, Скорость потока: 0.6 мл/мин.
Метод D
Колонка: XBridge ВЕН C18 (50 мм × 4.6 мм, 2.5 мкм). Подвижная фаза: А: 5 мМ бикарбонат аммония; В: ацетонитрил. Время (мин)/% В: 0 / 5, 1.5 / 15, 7 / 98, 9 / 98, 9.5 / 5, 10/5. Температура колонки: 35°С, Скорость потока: 1.3 мл/мин.
Метод Е
Колонка: Acquity ВЕН C18 (50 мм × 2.1 мм, 1.7 мкм). Подвижная фаза: А: 0.05% муравьиная кислота в воде; В: 0.05% муравьиная кислота в ацетонитриле. Время (мин)/% В: 0 / 3, 0.4 / 3, 3.2 / 98, 3.8 / 98, 4.2 / 3, 4.5 / 3. Температура колонки: 35°С, Скорость потока: 0.6 мл/мин.
Метод F
Колонка: Колонка Acquity UPLC CSH C18 (50 мм × 2.1 мм, внутр. диам 1.7 мкм) при 40°С. Использовали следующие растворители: А=10 мМ бикарбонат аммония в воде, доведенный до рН 10 раствором аммиака. В = лцетонитрил. Время (мин)/% В: 0 / 3, 0.05 /3, 1.5/ 95, 1.9 / 95, 2.0 / 3. Скорость потока: 1 мл/мин. МС: Waters ZQ. редим ионизации: Сканирующий режим положительного / отрицательного электроспрея
Метод G
Колонка: Acquity ВЕН C18 (50 мм × 2.1 мм, 1.7 мкм). Подвижная фаза: А: 0.1% муравьиная кислота в воде; В: 0.1% муравьиная кислота в ацетонитриле. Время (мин)/% В: 0 / 3, 0.4 / 3, 3.2 / 98, 3.8 / 98, 4.2 / 3, 4.5 / 3. Температура колонки: 35°С, Скорость потока: 0.6 мл/мин.
Метод Н
Колонка: Acquity ВЕН C18 (50 мм × 2.1 мм, 1.7 мкм). Подвижная фаза: А: 5 мМ бикарбонат аммонияв воде (рН 10); В: ацетонитрил. Время (мин)/% В: 0 / 3, 0.4 / 3, 2.5 / 98, 3.4 / 98, 3.5 / 3, 4.0 / 3. Температура колонки: 35°С, Скорость потока: 0.6 мл/мин.
Метод I
Колонка: Acquity ВЕН C18 (100 мм × 2.1 мм, 1.7 мкм). Подвижная фаза: А: 0.05% ТФУК в воде; В: ацетонитрил. Время (мин)/% В: 0 / 3, 0.4 / 3, 3.5 / 98, 4.5 / 98, 5.0 / 3, 5.5 / 3. Температура колонки: 35°С, Скорость потока: 0.45 мл/мин.
Метод J
Колонка: Acquity UPLC CSH C18 (50 мм × 2.1 мм, внутр. диам 1.7 мкм) при 40°С. Использовали следующие растворители: А=0.1% об./об. раствор муравьиной кислоты в воде. В=0.1% об./об. раствор муравьиной кислоты в ацетонитриле. Время (мин)/% В: 0 / 3, 1.5 / 95, 1.9 / 95, 2.0 / 3. Скорость потока: 1 мл/мин. МС: Waters ZQ. Режим ионизации: Сканирующий режим положительного / отрицательного электроспрея.
Метод K
Колонка: XSelect CSH C18(150 мм × 3.0 мм, 2.5 мкм). Подвижная фаза: А: 0.05% ТФУК в воде; В: 100% ацетонитрил. Время (мин)/% В: 0 / 3, 1 / 3, 8 / 98, 11 / 98, 11.1 / 3, 12 / 3. Темп. колонки: 35°С, Скорость потока: 0.7 мл/мин.
Метод L
Колонка: ВЕН C18 (100 мм × 2.1 мм, 1.7 мкм). Подвижная фаза: А: 0.1% ТФУК в воде, B: O.I % ТФУК в ацетонитриле. Время (мин)/% В: 0 / 3, 8.5 / 100, 9.0 / 100, 9.5 / 3, 10 / 3. Темп, колонки: 50°С, Скорость потока: 0.55 мл/мин.
Метод М
Колонка: CSH С18 (100 мм × 2.1 мм., внутр. диам. 1.7 мкм). Подвижная фаза: А: 0.1% об./об. раствор муравьиной кислоты в воде; В: 0.1% об./об. раствор муравьиной кислоты в ацетонитриле. Время (мин)/% В: 0 / 3, 8.5 / 99.9, 9 / 99.9, 9.5/3, 10 / 3. Темп, колонки: 50°С. Скорость потока: 0.8 мл/мин.
Масс-направленная автоматизированная препаративная ВЭЖХ (MDAP)
Методы масс-направленной автоматизированной препаративной ВЭЖХ, используемой для очистки соединений, описаны ниже. Использовали градиенты растворителей для элюирования от 0 до 99% Растворителя В в Растворителе А в течение периода времени до 25 мин. Для всех методов (если не указано иное):
Детектирование при помощи диодно-матричного детектроа осуществляли на длине волны от 210 нм до 350 нм. МС Условия: МС: Waters ZQ
Режим ионизации: Сканирующий режим положительного / отрицательного электроспрея Диапазон сканирования: 100-1000 а.е.м. Время сканирования: 0.2 с или 0.50 с. Промежуток между измерениями: 0.1 с или 0.2 с
Объем впрыска: 1 мл или 3 мл
Метод А
Колонка: Xselect CSH C18 (150 мм × 30 мм внутр. диам. 5 мкм заполняемый диаметр) при обычной температуре. Использовали следующие растворители: А=10 мМ бикарбонат аммония, доведенный до рН 10 аммиаком в воде. В = ацетонитрил. Скорость потока: 40 мл/мин.
Метод В
Колонка: колонка Xselect CSH C18 (150 мм × 30 мм, внутр. диам. 5 мкм- диаметр заполнения) при обычной температуре. Использовали следующие растворители:
А=0.1% об./об. раствор муравьиной кислоты в воде
В=0.1% об./об. раствор муравьиной кислоты в ацетонитриле.
Скорость потока: 40 мл/мин.
Промежуточное соединение 1
2-бром-1-метил-1Н-имидазол

1-Метил-1Н-имидазол (20 г, 244 ммоль) растворяли в безводном ТГФ (200 мл) в атмосфере азота. Раствор перемешивали при -78°С и добавляли по каплям n-BuLi (167 мл, 268 ммоль) медленно при -78°С. Через 1 ч добавляли раствор СВr4 (97 г, 292 ммоль) в безводном ТГФ (200 мл). Раствор перемешивали в течение 2 ч при -78°С и в течение 1 ч при комнатной температуре. Реакционную смесь гасили насыщенным водным раствором хлорида аммония (300 мл), экстрагировали этилацетатом (2 × 200 мл), сушили с использованием безводного сульфата натрия, фильтровали и выпаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя для элюирования 60% этилацетат в петролейном эфире, в результате чего получали указанное в заголовке соединение. ЖХМС (метод В): время удерживания =1.89, [М+Н]+=161.
Промежуточное соединение 2
3-(1-Метил-1H-имидазол-2-ил)бензонитрил
Смесь (3-цианофенил)бороновой кислоты (16 г, 109 ммоль), 2-бром-1-метил-1Н-имидазола (промежуточное соединение 1, 15.8 г, 98 ммоль) а карбоната натрия (46.2 г, 436 ммоль) в изопропиловом спирте (120 мл) и воде (120 мл) перемешивали и дегазировали азотом в течение 20 мин, а затем добавляли аддукт PdCl2(dppf)-ДXM (4.45 г, 5.44 ммоль) и перемешивали при 130°С в течение 18 ч в запаянной пробирке. После охлаждения реакционную смесь разбавляли этилацетатом (300 мл), промывали водой (300 мл), сушили с использованием безводного сульфата натрия, фильтровали и выпаривали при пониженном давлении. Неочищенное соединение очищали хроматографией на силикагеле, используя для элюирования 70% этилацетат в петролейном эфире, в результате чего получали указанное в заголовке соединение. ЖХМС (метод С): время удерживания =0.57, [М+Н]+=184.
Промежуточное соединение 3
(3-(1-метил-1Н-имидазол-2-ил)фенил)метанамин

К смеси 3-(1-метил-1Н-имидазол-2-ил)бензонитрила (промежуточное соединение 2, 12 г, 65.5 ммоль) в 7 М растворе аммония в метаноле (150 мл) добавляли никель Ренея (5 г, 65.5 ммоль) при 0°С. Реакционную смесь перемешивали под давлением водорода (60 фунтов/кв. дюйм при комнатной темп, в течение 24 ч. Реакционную смесь фильтровали через слой целита, промывали метанолом (300 мл) и концентрировали фильтрат при пониженном давлении. Затем реакцию повторяли в том же масштабе. Две партии неочищенного материала объединяли и очищали хроматографией на нейтральном оксиде алюминия, в результате чего получали указанное в заголовке соединение. ЖХМС (метод D): время удерживания =2.91, [М+Н]+=188.
Промежуточное соединение 4
Этил 7-гидрокси-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилат
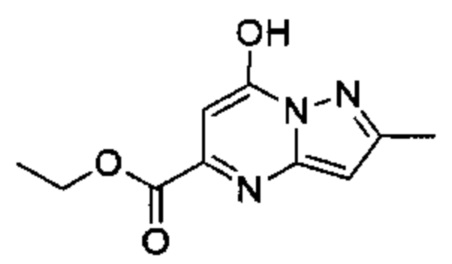
К раствору (Z)-1,4-диэтокси-1,4-диоксобут-2-ен-2-олата натрия (21.64 г, 103 ммоль) в этаноле (100 мл) добавляли 4 М HCI в 1,4 диоксане (28.3 мл), а затем 5-метил-1Н-пиразол-3-амин (10 г, 103 ммоль) при 0°С. Реакционную смесь нагревали до 85°С и перемешивали в течение 3 ч. Реакционную смесь охлаждали до комнатной темп, и концентрировали. Остаток растворяли в 10% метанол в ДХМ (200 мл) и промывали насыщенным раствором бикарбоната натрия (100 мл). Органический слой промывали солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и выпаривали досуха при пониженном давлении. Остаток растирали с диэтиловым эфиром, в результате чего получали указанное в заголовке соединение. ЖХМС (метод А): время удерживания =1.53, [М+Н]+=222.
Промежуточное соединение 5
Этил 7-хлор-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилат

К этил 7-гидрокси-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилату (промежуточное соединение 4, 10 г, 45.2 ммоль) добавляли оксихлорид фосфора (50 мл, 536 ммоль) и перемешивали реакционную смесь в течение 24 ч при 90°С. Реакционную смесь охлаждали до комнатной темп. и концентрировали при пониженном давлении. Остаток растворяли в ДХМ (100 мл) и доводили рН до нейтрального при помощи насыщенного раствора бикарбоната натрия (60 мл). Органический слой промывали водой (100 мл), солевым раствором (100 мл), сушили с использованием сульфата натрия, фильтровали и концентрировали. Остаток растворяли 10% метаноле в ДХМ (100 мл) и адсорбировали на силикагеле (14 г), затем очищали хроматографией на силикагеле (50 г), используя для элюирования 10% этилацетат в петролейном эфире. Содержащие продукт фракции объединяли и выпаривали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод В): время удерживания =3.13, [М+Н]+=240.
Промежуточное соединение 6
Этил 7-хлор-3-йодо-2-метилпиразоло[1.5-а]пиримидин-5-карбоксилат

К раствору этил 7-хлор-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилата (промежуточное соединение 5, 15 г, 62.6 ммоль) в ДХМ (220 мл) добавляли уксусную кислоту (25 мл) и N-йодосукцинимид (14.79 г, 65.7 ммоль). Реакционную смесь перемешивали в течение 6 ч при 28°С. Реакционную смесь разбавляли дихлорметаном (150 мл), промывали насыщенным тиосульфатом натрия (2 × 90 мл), солевым раствором (60 мл), сушили с использованием сульфата натрия, фильтровали и концентрировали, в результате чего получали указанное в заголовке соединение. ЖХМС (метод А): время удерживания =2.24, [М+Н]+=366.
Промежуточное соединение 7
Этил 3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат

К раствору DIPEA (14.33 мл, 82 ммоль) в ДМСО (50 мл) добавляли (3-(1-метил-1Н-имидазол-2-ил)фенил)метанамин (промежуточное соединение 3, 5.38 г, 28.7 ммоль) и этил 7-хлор-3-йодо-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 6, 10 г, 27.4 ммоль) и перемешивали реакционную смесь в течение 16 ч. Реакционную смесь вливали в ледяной воде (200 мл), и твердое вещество фильтровали, промывали водой (50 мл) и сушили под вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод А): время удерживания =1.79, [М+Н]+=517.
Промежуточное соединение 8
4-бром-1-метокси-2-(метилсульфонил)бензол

К перемешиваемому раствору сульфита натрия (16.55 г, 131 ммоль) и бикарбоната натрия (11.03 г, 131 ммоль) в воде (160 мл) добавляли 5-бром-2-метоксибензол-1-сульфонилхлорид (25 г, 88 ммоль) в 1,4-диоксане (160 мл) при 70°С. Реакционную смесь перемешивали при 70°С в течение 1 ч. Реакционную смесь охлаждали до комнатной темп. и удаляли растворитель при пониженном давлении, в результате чего получали указанное неочищенное промежуточное соединение в форме белого твердого вещества. Неочищенный продукт растворяли в ДМФА (300 мл) и добавляли метилйодид (10.95 мл, 175 ммоль) при комнатной темп., и перемешивали смесь при комнатной темп. в течение 2 ч. Реакционную смесь вливали в ледяную воду, экстрагировали этилацетатом (300 мл), сушили с использованием безводного сульфата натрия, фильтровали и сушили при пониженном давлении. Неочищенное соединение промывали н-пентаном (100 мл), в результате чего получали указанное в заголовке соединение. ЖХМС (метод А): время удерживания =1.84, [М+Н]+=265.
Промежуточное соединение 9
2-(4-метокси-3-(метилсульфонил)фенил)-4,4,5,5-третраметил-1,3,2-диоксаборолан

К перемешиваемому раствору 4-бром-1-метокси-2-(метилсульфонил)бензола (промежуточное соединение 8, 15 г, 56.6 ммоль) и 4,4,4',4',5,5,5',5-октаметил-2,2'-би(1,3,2-диоксаборолана) (21.55 г, 85 ммоль) в 1,4-диоксане (150 мл) добавляли ацетат калия (8.33 г, 85 ммоль) при комнатной темп. Реакционную смесь дегазировали аргоном в течение 15 мин, затем к реакционной смеси добавляли аддукт PdCI2(dppf)-ДXM (2.31 г, 2.83 ммоль) при комнатной темп. и снова дегазировали в течение 15 мин в атмосфере аргона. Реакционную смесь перемешивали при 100°С в течение 3 ч. После охлаждения реакционную смесь фильтровали через слой целита, промывали метанолом (50 мл) и концентрировали фильтрат при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле, используя для элюирования 20% этилацетат в гексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод Е): время удерживания =2.16, [М+Н]+=313.
Промежуточное соединение 10
Этил 3-(4-метокси-3-(метилсульфонил)фенил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат

2-(4-метокси-3-(метилсульфонил)фенил)-4,4,5,5-третраметил-1,3,2-диоксаборолан (промежуточное соединение 9, 680 мг, 2.179 ммоль), этил 3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 7, 750 мг, 1.453 ммоль), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(II) (57.1 мг, 0.073 ммоль) и фторид калия (253 мг, 4.36 ммоль) растворяли в 1,4-диоксане (10 мл) и воде (5 мл) в сосуде для микроволновой обработки, запаивали сосуд и дегазировали азотом. Реакционную смесь перемешивали при 80°С в течение 16 ч. Затем добавляли хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(Н) (57.1 мг, 0.073 ммоль), фторид калия (253 мг, 4.36 ммоль) и 2-(4-метокси-3-(метилсульфонил)фенил)-4,4,5,5-третраметил-1,3,2-диоксаборолан (промежуточное соединение 9, 680 мг, 2.179 ммоль) и перемешивали реакционную смесь в течение еще 8 ч при 80°С. Затем добавляли хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий (II) (57.1 мг, 0.073 ммоль), фторид калия (253 мг, 4.36 ммоль) и 2-(4-метокси-3-(метилсульфонил)фенил)-4,4,5,5-третраметил-1,3,2-диоксаборолан (промежуточное соединение 9, 680 мг, 2.179 ммоль) и перемешивали реакционную смесь в течение еще 16 ч при 80°С. Реакционную смесь охлаждали до комнатной темп., фильтровали через целит (промывая 3 × 20 мл этилацетата) и удаляли растворитель под вакуумом. Реакционную смесь разделяли между этилацетатом (50 мл) и водой (20 мл), и органическую фазу отделяли, сушили при помощи гидрофобного фильтра и удаляли растворитель под вакуумом. Очистка хроматографией на силикагеле (120 г) с использованием для элюирования 0-100% этилацетата в циклогексане (более 20 объемов колонки) давала указанное в заголовке соединение. ЖХМС (метод F): время удерживания =1.04, [М+Н]+=515.
Промежуточное соединение 11
(3-(4-метокси-3-(метилсульфонил)фенил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)метанол

Этил 3-(4-метокси-3-(метилсульфонил)фенил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 10, 180 мг, 0.313 ммоль) растворяли в ТГФ (5 мл) в атмосфере азота при 0°С и добавляли алюмогидрид лития (1 M в ТГФ) (0.626 мл, 0.626 ммоль) добавляли, зачем реакционную смесь перемешивали в течение еще 3 ч. Реакцию гасили путем добавления 2М раствора гидроксид натрия (5 мл) и перемешивали в течение еще 1 ч. Органическую фазу экстрагировали этилацетатом (3 × 10 мл), органическую фазу сушили при помощи гидрофобного фильтра и удаляли растворитель под вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания =0.89, [М+Н]+=533.
Промежуточное соединение 12
3-(4-метокси-3-(метилсульфонил)фенил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбальдегид

(3-(4-метокси-3-(метилсульфонил)фенил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)метанол (промежуточное соединение 11, 112 мг, 0.21 ммоль) растворяли в ДХМ (2.5 мл), добавляли диоксид марганца (32 мг, 0.368 ммоль) и перемешивали реакционную смесь при комнатной темп. в течение 4 ч. Затем добавляли диоксид марганца (183 мг, 2.103 ммоль) и перемешивали реакционную смесь при комнатной темп. в течение еще 16 ч. Реакционную смесь фильтровали через картридж с целитом, промывая дихлорметаном (3×5 мл) и удаляли растворитель под вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания =1.0, [М+Н]+=531.
Промежуточное соединение 13
Этил 7-((трет-бутоксикарбонил)(3-(1-метил-1Н-имидазол-2-ил)бензил)амино)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилат
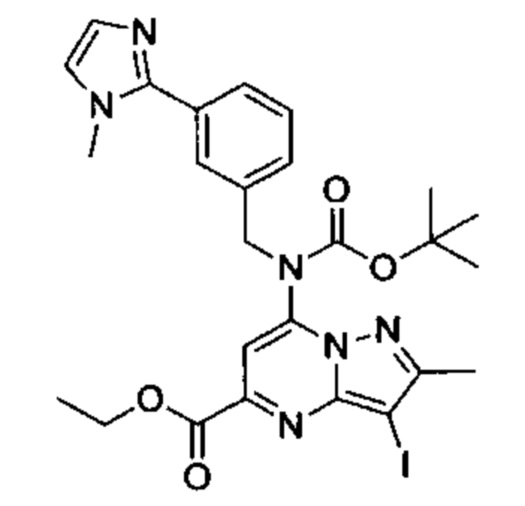
К раствору этил 2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилата (промежуточное соединение 7, 3.8 г, 9.73 ммоль) в ДХМ (25 мл) добавляли DIPEA (5.1 мл, 29.2 ммоль), DMAP (1.189 г, 9.73 ммоль) и Вос-ангидрид (3.39 мл, 14.6 ммоль) и перемешивали реакционную смесь в течение 16 ч при комнатной температуре. Реакционную смесь разбавляли дихлорметаном (50 мл), промывали водой (25 мл), сушили с использованием сульфата натрия, фильтровали и концентрировали. Остаток растворяли в ДХМ (30 мл), абсорбировали на силикагеле (5 г) и очищали хроматографией на силикагеле (15 г), используя для элюирования этилацетат в петролейном эфире, в результате чего получали указанное в заголовке соединение. ЖХМС (метод г): время удерживания = 2.14, [М+Н]+=617.
Промежуточное соединение 14
Трет-бутил(5-(гидроксиметил)-3-йодо-2-метилпиразоло[1,5-a]пиримидин-7-ил)(3-(1-метил-1H-имидазол-2-ил)бензил)карбамат
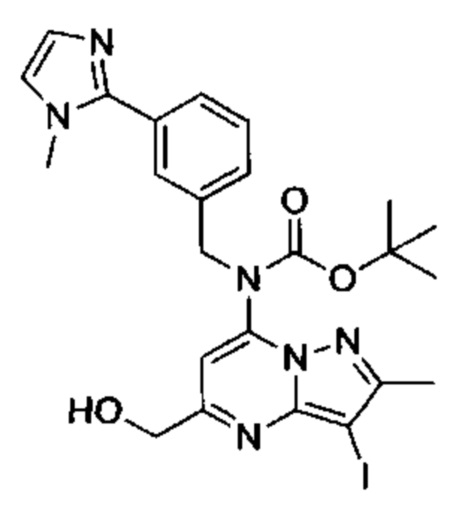
К раствору этил 7-((трет-бутоксикарбонил)(3-(1-метил-1Н-имидазол-2-ил)бензил)амино)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилата (промежуточное соединение 13, 500 мг, 0.811 ммоль) в этаноле (10 мл) добавляли борогидрид натрия (92 мг, 2.433 ммоль) и перемешивали реакционную смесь в течение 3 ч при 10°С. Реакционную смесь гасили с использованием 1 М HCl (5 мл), рН доводили до 6, затем концентрировали. Остаток растворяли в этилацетате (25 мл), промывали водой (20 мл), сушили с использованием сульфата натрия, фильтровали и концентрировали, в результате чего получали указанное в заголовке соединение. ЖХМС (метод Н): время удерживания=2.01, [М+Н]+=573.
Промежуточное соединение 15
Трет-бутил(5-формил-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат

DMP (1752 мг, 4.13 ммоль) добавляли одной порцией к перемешиваемому раствору Трет-бутил(5-(гидроксиметил)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамата (промежуточное соединение 14, 1306 мг, 2.274 ммоль) в ДХМ (7.5 мл). Реакционную смесь оставляли для перемешивания в течение 18 ч. Добавляли насыщенный водный раствор сульфита натрия (10 мл) и перемешивали реакционную смесь в течение 2 ч при комнатной темп. Реакционную смесь разделяли между ДХМ (150 мл) и водой (150 мл). Органический слой отделяли, а водный экстрагировали дополнительным количеством ДХМ (2 × 50 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (100 мл), водой (100 мл), пропускали через гидрофобный спеченный фильтр и выпаривали под вакуумом. Остаток растворяли в минимальном количестве ДХМ и очищали хроматографией на силикагеле (120 г), используя для элюирования 0 - 50% этилацетат: этанол (3:1, об./об.) в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.27, [М+Н]+=573.
Промежуточное соединение 16
1-метокси-2-(метилсульфонил)бензол

К перемешиваемому раствору (2-метоксифенил)(метил)сульфана (50 г, 324 ммоль) в ДХМ (1 л) при 0°С в атмосфере азота добавляли m-СРВА (140 г, 810 ммоль). Реакционной смеси давали перемешиваться при комнатной темп, в течение 2 ч. Реакционную смесь разбавляли дихлорметаном (300 мл), промывали насыщенным водным раствором карбоната натрия (2 × 500 мл), водную фазу экстрагировали дихлорметаном (200 мл). Объединенные органические вещества сушили с использованием безводного сульфата натрия, фильтровали и выпаривали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод I): время удерживания=2.28, [М+Н]+=187.
Промежуточное соединение 17
2-((2-метоксифенил)сульфонил)этанол
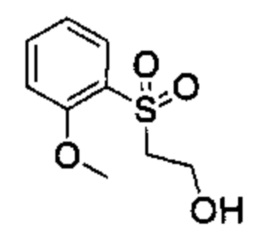
К перемешиваемому раствору 1-метокси-2-(метилсульфонил)бензола (промежуточное соединение 16, 25 г, 123 ммоль) в ТГФ (250 мл) при -78°С в атмосфере азота по каплям добавляли н-бутиллитий (115 мл, 184 ммоль) и перемешивали реакционную смесь при -78°С в течение 30 мин, затем охлаждали до 0°С. Добавляли порциями параформальдегид (73.6 г, 2450 ммоль), затем реакционную смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь гасили раствором хлорида аммония (120 мл), экстрагировали этилацетатом (2 × 200 мл). Объединенные органические вещества промывали солевым раствором (150 мл) и сушили с использованием безводного сульфата натрия, фильтровали и выпаривали при пониженном давлении. Неочищенное соединение предварительно абсорбировали на силикагеле (4 г) и очищали хроматографией на силикагеле (10 г колонка, 80% этилацетат в гексане), в результате чего получали указанное в заголовке соединение. ЖХМС (метод С): время удерживания=1.16, [М+Н]+=217.
Промежуточное соединение 18
2-((5-бром-2-метоксифенил)сульфонил)этанол

К перемешиваемому раствору 2-((2-метоксифенил)сульфонил)этанола (промежуточное соединение 17, 10 г, 46.2 ммоль) в ДМФА (100 мл) в атмосфере азота при комнатной темп, добавляли перекристаллизованный БСИ (16.46 г, 92 ммоль). Реакционную смесь нагревали до 50°С в течение 18 ч. Реакционную смесь охлаждали до комнатной темп, и разбавляли этилацетатом (300 мл) и промывали ледяной водой (2 × 500 мл). Органический слой отделяли, и водный слой снова экстрагировали этилацетатом (300 мл). Объединенные органические слои промывали солевым раствором (300 мл) и сушили с использованием сульфата натрия, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение предварительно абсорбировали на силикагеле (10 г) и очищали хроматографией на силикагеле (90 г колонка, 40% этилацетат в гексане), в результате чего получали указанное в заголовке соединение. ЖХМС (метод С): время удерживания=1.85, [М+Н]+=295.
Промежуточное соединение 19
2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этанол

К перемешиваемому раствору 2-((5-бром-2-метоксифенил)сульфонил)этанола (промежуточное соединение 18, 10 г, 33.9 ммоль) в 1,4-диоксане (150 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (17.2 г, 67.8 ммоль) и ацетат калия (9.98 г, 102 ммоль) при комнатной темп., затем реакционную смесь дегазировали азотом в течение 15 мин. Добавляли аддукт PdCl2(dppf)-ДХМ (2.77 г, 3.39 ммоль) и нагревали реакционную смесь при 100°С в течение 3 ч в запаянной пробирке. Реакционную смесь фильтровали на слое целита и промывали целит этилацетатом (2 × 100 мл). Объединенные органические вещества концентрировали при пониженном давлении. Затем реакцию повторяли в том же масштабе. Две партии неочищенного продукта смешивали и растворяли в ДХМ (100 мл). Смесь обрабатывали углем (5 г) и нагревали с обратным холодильником в течение 10 мин, после чего фильтровали на слое целита. Целит промывали дихлорметаном (2 × 100 мл). Объединенные органические вещества концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле (этилацетат в петролейном эфире), в результате чего получали указанное в заголовке соединение. ЖХМС (метод С): время удерживания=2.17, [М+Н]+=343.
Промежуточное соединение 20
Трет-бутил(5-формил-3-(3-((2-гидроксиэтил)сульфонил)-4-метоксифенил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1H-имидазол-2-ил)бензил)карбамат
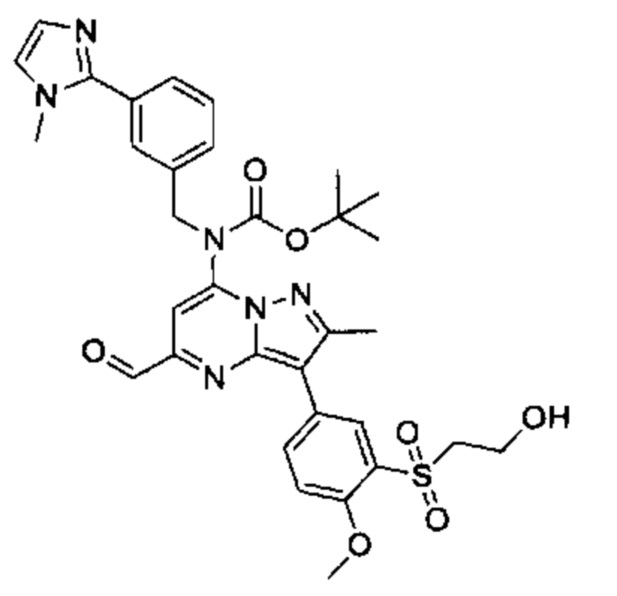
В сосуд для микроволновой обработки загружали 7рет-бутил(5-формил-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1H-имидазол-2-ил)бензил)карбамат (промежуточное соединение 15, 487 мг, 0.851 ммоль), 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этан-1-ол (промежуточное соединение 19, 415 мг, 1.03 ммоль), PdCl2(dppf) (64 мг, 0.087 ммоль) и фторид калия (151 мг, 2.6 ммоль) в 1,4-диоксане (2 мл) и воду (1 мл). Реакционный сосуд запаивали и нагревали в микроволновом реакторе при 100°С в течение в целом 1.5 ч. Реакционную смесь пропускали через целит, промывали метанолом (50 мл) и выпаривали под вакуумом. Остаток растворяли в ДХМ (30 мл) и разделяли с водой (15 мл). Органический слой отделяли и водную фазу экстрагировали дополнительным количеством ДХМ (2 × 20 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и выпаривали под вакуумом. Остаток очищали хроматографией на силикагеле (80 г), используя для элюирования 50-100% смесь этилацетат: этанол (3:1, об./об., содержащую 1% триэтиламина) в циклогексане (20 объемов колонки), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.06, [М+Н]+=661.
Промежуточное соединение 21
2-(3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а1пиримидин-5-ил)пропан-2-ол
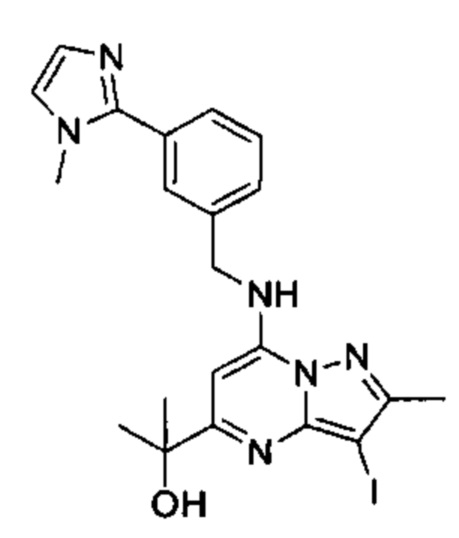
Этил-3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 7, 275 мг, 0.533 ммоль) перемешивали в ТГФ (5 мл) при 0°С в атмосфере азота. В течение 2 минут добавляли Метилмагния бромид (1 M в дибутиловом эфире) (1.75 мл, 1.75 ммоль) и давали смеси нагреться до комнатной темп., после чего оставляли на 4.5 с. Реакционную смесь гасили водой (2 мл) затем разделяли дихлорметаном (50 мл) и водой (50 мл). Органическую фазу собирали, а водную промывали дихлорметаном (50 мл), а затем этилацетатом (50 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и выпаривали досуха. Остаток загружали в ДХМ (2 мл) на колонку с силикагелем (40 г) и элюировали 10-60% смесью этилацетат: этанол (3:1, об./об., содержащей 1% триэтиламин) в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.7, [М+Н]+=503.
Промежуточное соединение 22
5-бром-2-хпор-N-(3-гидроксипропил)-N-метилбензамид
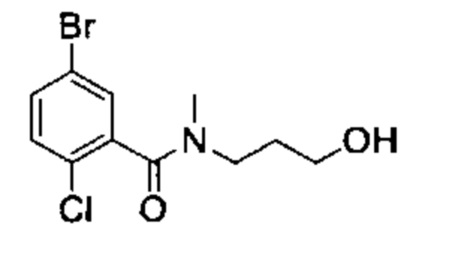
К перемешиваемому раствору 5-бром-2-хлорбензойной кислоты (12.5 г, 53.1 ммоль) в ТГФ (100 мл) добавляли 3-(метиламино)пропан-1-ол (5.21 г, 58.4 ммоль), DIPEA (27.8 мл, 159 ммоль) и HATU (22.2 г, 58.4 ммоль) и перемешивали реакционную смесь при комнатной темп, в течение 18 ч. Реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (200 мл). Органический слой сушили безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт растворяли в ДХМ (50 мл), предварительно абсорбировали на силикагеле (50 г) и очищали хроматографией на силикагеле (150 г, 100% этилацетат), в результате чего получали указанное в заголовке соединение. ЖХМС (метод К): время удерживания=5.30, [М+Н]+=306.
Промежуточное соединение 23
2-Хлор-N-(3-гидроксипропил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид

К перемешиваемому раствору 5-бром-2-хлор-N-(3-гидроксипропил)-N-метилбензамида (промежуточное соединение 22, 10 г, 32.6 ммоль) в 1,4-диоксане (100 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (9.69 г, 38.2 ммоль), ацетат калия (9.60 г, 98 ммоль) и дегазировали реакционную смесь аргоном в течение 30 мин. Добавляли аддукт PdCl2(dppf)-ДХМ (0.266 г, 0.326 ммоль) и дегазировали смесь в течение 20 мин. Реакционную смесь нагревали до 100°С в течение 18 ч. Реакционную смесь охлаждали до комнатной темп, и фильтровали через слой целита и промывали 10% метанолом в ДХМ, затем концентрировали при пониженном давлении. Неочищенный продукт растворяли в ДХМ (50 мл), предварительно абсорбировали на флоризиле (50 г) и очищали хроматографией на силикагеле (250 г, 0 - 100% этилацетат в петролейном эфире), в результате чего получали указанное в заголовке соединение. ЖХМС (метод L): время удерживания=2.45, [М+Н]+=354.
Промежуточное соединение 24
(S)-(5-бром-2-метоксифенил)(3-гидроксипирролидин-1-ил)метанон

DIPEA (1.134 мл, 6.49 ммоль) добавляли к раствору 5-бром-2-метоксибензойной кислоты (500 мг, 2.164 ммоль) и HATU (987 мг, 2.6 ммоль) в 2-метилтетрагидрофуране (10 мл) и оставляли реакционную смесь перемешиваться при комнатной темп, в течение 30 мин в атмосфере азота. Затем добавляли (S)-пирролидин-3-ол (226 мг, 2.6 ммоль) и оставляли реакционную смесь перемешиваться при комнатной темп, в течение ночи с добавлением диметилформамида (5 мл). Реакционную смесь концентрировали при пониженном давлении, разбавляли насыщенным водным раствором бикабоната натрия (50 мл) и 5% раствором хлорида лития (50 мл) и экстрагировали этилацетатом (2 × 100 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр, концентрировали при пониженном давлении, а затем сушили под потоком азота в течение ночи. Остаток загружали в ДХМ и очищали хроматографией на силикагеле (80 г), используя для элюирования смесь этилацетат: этанол (3:1, об./об., содержащую 1% триэтиламина) в этилацетате (0%, 2 объема колонки; 0-100%, 12 объемов колонки), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.73, [М+Н]+=300.
Промежуточное соединение 25
(S)-(3-гидроксипирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон
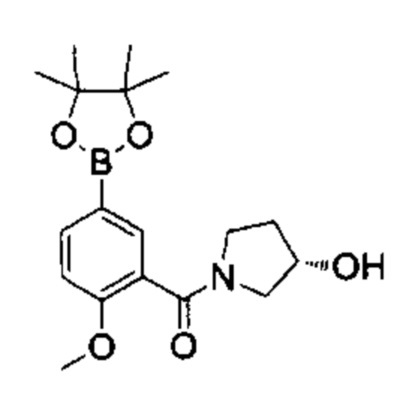
(S)-(5-бром-2-метоксифенил)(3-гидроксипирролидин-1-ил)метанон (промежуточное соединение 24, 750 мг, 2.249 ммоль), 4,4,4,4',5,5,5',5'-октаметил-2,2,-би(1,3,2-диоксаборолан) (628 мг, 2.474 ммоль), ацетат калия (662 мг, 6.75 ммоль), PdCl2(dppf) (165 мг, 0.225 ммоль) и 1,4-диоксан (6 мл) объединяли и помещали в атмосферу азота путем откачки и перезаполнения. Смесь нагревали в микроволновом реакторе при 100°С в течение 1 ч. Удаляли растворитель из реакционной смеси при пониженном давлении. Остаток суспендировали в этилацетате (50 мл) и воде (30 мл) и фильтровали через целит, промывая дополнительным количеством этилацетата (50 мл). Раствор разбавляли солевым раствором (20 мл) и промывали водный слой дополнительным количеством этилацетат (100 мл),объединенные органические слои пропускали через гидрофобный спеченный фильтр и удаляли растворитель при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.85, [М+Н]+=348.
Промежуточное соединение 26
Трет-бутил(5-(1-гидроксиэтил)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат
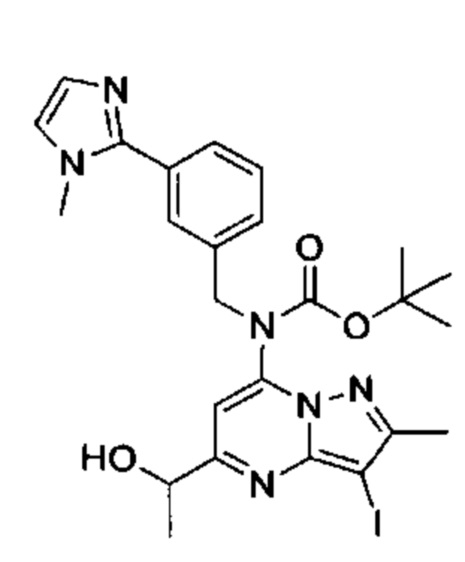
Трет-бутил(5-формил-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1H-имидазол-2-ил)бензил)карбамат (промежуточное соединение 15, 465 мг, 0.812 ммоль) растворяли в ТГФ (10 мл) и охлаждали до -78°С. По каплям в течение 2 мин добавляли метилмагния бромид (1 M в дибутиловом эфире) (0.85 мл, 0.85 ммоль). Через 1 ч 45 мин добавляли метилмагния бромид (1 M в дибутиловом эфире) (0.203 мл, 0.203 ммоль) и перемешивали смесь при -78°С в течение еще 3 ч. Добавляли насыщенный водный хлорид аммония (5 мл) и воду (10 мл), и перемешивали смесь в течение 10 мин, нагревая до комнатной темп. Добавляли ДХМ (20 мл), переносили смесь и собирали органический слой. Затем водный слой промывали дополнительным количеством дихлорметана (2 × 20 мл), пропускали объединенные органические слои через гидрофобный спеченный фильтр и выпаривали досуха. Остаток снова загружали влажным способом в ДХМ (1.5 мл) на колонку с силикагелем и элюировали 50-60% этилацетатом в циклогексане (30 объемов колонки), в затем 60-85% этилацетатом в циклогексане (10 объемов колонки). Содержащие продукт фракции объединяли и выпаривали досуха, затем растирали с диэтиловым эфиром, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.2, [М+Н]+=589.
Промежуточное соединение 27
5-бром-N-(3-гидроксипропил)-2-метокси-N-метилбензамид

HATU (458.3 мг, 1.205 ммоль) и DIPEA (0.655 мл, 3.75 ммоль) добавляли к перемешиваемому раствору 5-бром-2-метоксибензойной кислоты (288.9 мг, 1.250 ммоль) в ТГФ (10 мл) при комнатной темп. Через 10 добавляли мин 3-(метиламино)пропан-1-ол (0.146 мл, 1.500 ммоль). Через 2 ч удаляли растворитель под вакуумом, а полученный остаток разбавляли этилацетатом (10 мл) и водой (10 мл). Отделенную водную фазу далее экстрагировали этилацетатом (2 × 10 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.78, [М+Н]+=303.
Промежуточное соединение 28
N-(3-гидроксипропил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид
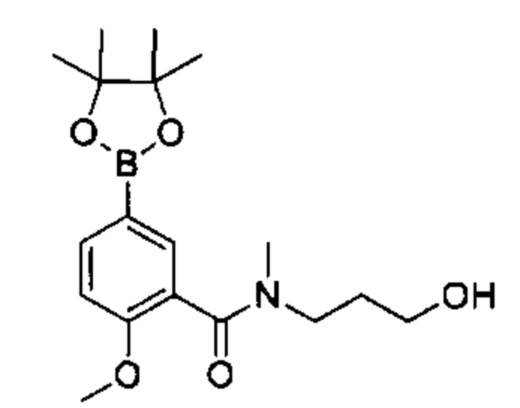
4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1.114 г, 4.39 ммоль), ацетат калия (1.077 г, 10.97 ммоль), аддукт PdCl2(dppf).ДХМ (0.299 г, 0.366 ммоль) и 1,4-диоксан (7 мл) объединяли и помещали в атмосферу азота (3 цикла вакуум/азот). Добавляли раствор 5-бром-N-(3-гидроксипропил)-2-метокси-N-метилбензамида (промежуточное соединение 27, 1.7 г, 3.66 ммоль) в 1,4-диоксане (7 мл) и обрабатывали смесь 3 циклами вакуум/азот. Затем реакционную смесь нагревали при 100°С в микроволновом реакторе в течение 90 мин. После охлаждения смесь фильтровали через целит, промывали дихлорметаном (50 мл) и удаляли растворитель под вакуумом. Полученный остаток растворяли в этилацетате (60 мл), воде (60 мл) и солевом растворе (20 мл). Отделенный водный слой повторно экстрагировали этилацетатом (2 × 30 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом. Неочищенный продукт очищали колоночной хроматографией на силикагеле, используя для элюирования 50 - 100% этилацетат в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.89, [М+Н]+=350.
Промежуточное соединение 29
5-бром-N-этил-N-(2-гидроксиэтил)-2-метоксибензамид

DIPEA (1.2 мл, 6.87 ммоль) HATU (980 мг, 2.58 ммоль) добавляли к перемешиваемому раствору 5-бром-2-метоксибензойной кислоты (500 мг, 2.164 ммоль) и оставляли реакционную смесь для перемешивания в течение ночи. Смесь разделяли между этилацетатом (50 мл) и насыщенным водным раствором бикарбоната натрия (50 мл). Водную фазу далее экстрагировали этилацетатом (50 мл). Объединенную органическую фазу промывали насыщенным водным раствором бикарбоната натрия (50 мл) и удаляли растворитель при пониженном давлении. Остаток очищали хроматографией на силикагеле, используя для элюирования 0 - 100% смесь этилацетат: этанол (3:1, содержащую 1% триэтиламина) в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.8, [М+Н]+=302.
Промежуточное соединение 30
N-этил-N-(2-гидроксиэтил)-2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид
5-бром-N-этил-N-(2-гидроксиэтил)-2-метоксибензамид (промежуточное соединение 29, 1.2 г, 3.97 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2,-би(1,3,2-диоксаборолан) (1.18 г, 4.65 ммоль), PdCl2(dppf) (0.301 г, 0.411 ммоль), ацетат калия (1.17 г, 11.92 ммоль) и 1,4-диоксан (8 мл) объединяли и помещали в атмосферу азота путем откачки и перезаполнения. Смесь нагревали при 100°С в микроволновом реакторе в течение 1.5 ч. После охлаждения реакционную смесь фильтровали через целит, промывали метанолом (250 мл) и удаляли растворитель при пониженном давлении. Остаток снова растворяли в ДХМ (30 мл) и разделяли водой (30 мл). Водный слой повторно экстрагировали дихлорметаном (3 × 30 мл), объединенные органические экстракты пропускали через гидрофобный спеченный фильтр и выпаривали досуха, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): rt 0.92, [М+Н]+=350.
Промежуточное соединение 31
5-бром-2-хлор-N-этил-N-(2-гидроксиэтил)бензамид

DIPEA (1.1 мл, 6.30 ммоль) и HATU (1.05 г, 2.76 ммоль) добавляли к перемешиваемому раствору 5-бром-2-хлорбензойной кислоты (509 мг, 2.162 ммоль) в 2-метилтетрагидрофуране (10 мл) и оставляли реакционную смесь перемешиваться при комнатной темп, в течение 15 мин. Добавляли 2-(этиламино)этан-1-ол (0.25 мл, 2.56 ммоль) и перемешивали реакционную смесь на протяжении выходных. Смесь разделяли между этилацетатом (50 мл) и насыщенным водным раствором бикарбоната натрия (50 мл). Затем водную фазу промывали этилацетатом (50 мл), а объединенные органические фазу промывали насыщенным водным раствором бикарбоната натрия (50 мл) и удаляли растворитель при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле, используя для элюирования 20 - 100% этилацетат в циклогексане. Содержащие продукт фракции объединяли и выпаривали досуха. Затем остаток разделяли обращенно-фазовой колоночной хроматографией, используя для элюирования 20 - 60% ацетонитрил (содержащий 0.01% аммиака) в 10 мМ аммония бикарбонате в воде, доведенной до рН 10 раствором аммиака, в результате чего получали указанное в заголовке соединение. ЖХМС (метод Е): время удерживания=0.85, [М+Н]+=306.
Промежуточное соединение 32
2-хлор-N-этил-N-(2-гидроксиэтил)-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид
4,4,4',4',5,5,5',5'-октаметил-2,2,-би(1,3,2-диоксаборолан) (462 мг, 1.819 ммоль), 5-бром-2-хлор-N-этил-N-(2-гидроксиэтил)бензамид (промежуточное соединение 31, 453 мг, 1.478 ммоль), PdCl2(dppf) (111 мг, 0.152 ммоль) и ацетат калия (445 мг, 4.53 ммоль) объединяли в 1,4-диоксане (4 мл). Смесь дегазировали и нагревали при 100°С в микроволновом реакторе в течение 1 ч, три раза. После охлаждения смесь фильтровали через целит, промывали метанолом (100 мл) и удаляли растворитель под вакуумом. Остаток разделяли между ДХМ (20 мл) и водой (10 мл). Затем отделенный водный слой промывали дихлорметаном (2 × 10 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод D): время удерживания=1.05, [М+Н]+=354.
Промежуточное соединение 33
5-бром-2-метокси-N-метил-N-(тетрагидро-2H-пиран-4-ил)бензамид
HATU (1.391 г, 3.66 ммоль) и DIPEA (1.72 мл, 9.85 ммоль) добавляли к перемешиваемой суспензии 5-бром-2-метоксибензойной кислоты (0.752 г, 3.25 ммоль) в ТГФ (10 мл) и перемешивали в течение 20 мин. Добавляли N-метилтетрагидро-2Н-пиран-4-амин (0.382 г, 3.32 ммоль) и оставляли реакционную смесь для перемешивания в течение 18 ч. Растворитель удаляли под вакуумом. Реакционную смесь разделяли между ДХМ (50 мл) и водой (50 мл), затем водную фазу экстрагировали дихлорметаном (2 × 20 мл), а органическую фазу фильтровали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом. Остаток растворяли в минимальном количестве ДХМ и очищали хроматографией на силикагеле (120 г), используя для элюирования 0 - 100% этилацетат: этанол (3:1, об./об.) в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.89, [М+Н]+=328.
Промежуточное соединение 34
2-метокси-N-метил-N-(тетрагидро-2Н-пиран-4-ил)-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид
5-бром-2-метокси-N-метил-N-(тетрагидро-2Н-пиран-4-ил)бензамид (промежуточное соединение 33, 746 мг, 2.273 ммоль), 4,4,4',4',5,5,5,,5,-октаметил-2,2,-би(1,3,2-диоксаборолан) (692 мг, 2.73 ммоль), PdCl2(dppf).ДХМ (194 мг, 0.238 ммоль), ацетат калия (664 мг, 6.77 ммоль) и 1,4-диоксан (10 мл) объединяли и нагревали в микроволновом реакторе при 100°С в течение 1 ч. Реакционную смесь фильтровали через целит и удаляли растворитель под вакуумом. Остаток разделяли между водой (25 мл) и ДХМ (25 мл), затем водную фазу экстрагировали дихлорметаном (2 × 15 мл), а органическую фазу фильтровали через гидрофобный спеченный фильтр, затем выпаривали досуха, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.01, [М+Н]+=376.
Промежуточное соединение 35
5-бром-N-(3-гидроксипропил)-N,2-диметилбензамид

5-бром-2-метилбензойную кислоту (504 мг, 2.344 ммоль), HATU (1053 мг, 2.77 ммоль) и DIPEA (0.5 мл, 2.86 ммоль) в ДМФА (4 мл) перемешивали при комнатной темп, в течение 30 мин, после чего добавляли 3-(метиламино)пропан-1-ол (0.275 мл, 2.83 ммоль). Через 3 ч смесь разделяли между этилацетатом (20 мл) и водой (15 мл). Отделенный водный слой далее экстрагировали этилацетатом (15 мл). Объединенные органические слои промывали 5% водным раствором хлорида лития (2×10 мл), водой (10 мл) и солевым раствором (10 мл). Органический слой пропускали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом. Неочищенный продукт очищали колоночной хроматографией на силикагеле, используя для элюирования 50 - 100% этилацетат в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.80, [М+Н]+=286.
Промежуточное соединение 36
N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид

Ацетат калия (636 мг, 6.48 ммоль), 5-бром-N-(3-гидроксипропил)-N,2-диметилбензамид (промежуточное соединение 35, 685 мг, 2.154 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (663 мг, 2.61 ммоль) и PdCl2(dppf) (131 мг, 0.179 ммоль) объединяли в 1,4-диоксане (5 мл). Смесь помещали в атмосферу азота путем осуществления пяти циклов вакуум/азот и нагревали в микроволновом реакторе при 100°С в течение 1 ч. После охлаждения удаляли растворитель под вакуумом и разделяли остаток между ДХМ (30 мл) и водой (20 мл). Затем отделенный водный слой промывали дихлорметаном (2 × 20 мл), а объединенные органические слои пропускали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.97, [М+Н]+=334.
Промежуточное соединение 37
(S)-(5-бром-2-хлорфенил)(3-гидроксипирролидин-1-ил)метанон
Получали аналогично промежуточному соединению 33, с использованием 5-бром-2-хлорбензойной кислоты (480 мг, 2.039 ммоль) и (S)-пирролидин-3-ола (178 мг, 2.039 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.73, [М+Н]+=303.
Промежуточное соединение 38
(S)-(2-хлор-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)(3-гидроксипиррол идин-1-ил)метанон

Получали аналогично промежуточному соединению 32 с использованием (S)-(5-бром-2-хлорфенил)(3-гидроксипирролидин-1-ил)метанона (промежуточное соединение 37), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.72, [М+Н]+=352.
Промежуточное соединение 39
(S)-5-бром-N-1(-гидроксипропан-2-ил)-2-метокси-N-метилбензамид
Получали аналогично промежуточному соединению 35, используя 5-бром-2-метоксибензойной кислоты (0.747 г, 3.23 ммоль) и (S)-2-(метиламино)пропан-1-ол (0.321 г, 3.6 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.77, [М+Н]+=302.
Промежуточное соединение 40
(S)-N-(1-гидроксипропан-2-ил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид

Получали аналогично промежуточному соединению 32, используя (S)-5-бром-N-(1-гидроксипропан-2-ил)-2-метокси-N-метилбензамид (промежуточное соединение 39), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.89, [М+Н]+=350.
Промежуточное соединение 41
Этил 3-бром-7-хлор-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилат
Получали аналогично промежуточному соединению 6, используя БСИ, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=1.11, [М+Н]+=320.
Промежуточное соединение 42
Этил 3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат
Этил-3-бром-7-хлор-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 41, 685 мг, 2.15 ммоль), (3-(1-метил-1H-имидазол-2-ил)фенил)метанамин (промежуточное соединение 3, 443 мг, 2.365 ммоль) и DIPEA (0.751 мл, 4.3 ммоль) перемешивали в ДМСО (5 мл) при 80°С в атмосфере азота в течение 3.5 ч. После охлаждения смесь вливали в ледяную воду и собирали преципитат фильтрацией. Остаток на фильтре растворяли в ДХМ, сушили при пониженном давлении и растирали с диэтиловым эфиром, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.06, [М+Н]+=469.
Альтернативный способ получения промежуточного соединения 42
Этил-3-бром-7-хлор-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 41, 50 г, 157 ммоль), (3-(1 -метил-1H-имидазол-2-ил)фенил)метанамин (промежуточное соединение 3, 35.3 г, 188 ммоль) и DIPEA (54.8 мл, 314 ммоль) перемешивали в ДМСО (500 мл) при 80°С в течение 3 ч. После охлаждения реакционную смесь вливали в ледяной воде и собирали преципитат фильтрацией. Полученное твердое вещество растирали с диэтиловым эфиром (500 мл), фильтровали и сушили, в результате чего получали указанное в заголовке соединение. ЖХМС (метод С): время удерживания=1.44, [М+Н]+=469.
Промежуточное соединение 43
2-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-2-ол
Этил 3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 42, 1.76 г, 3.75 ммоль) перемешивали в ТГФ (35 мл) при 0°С в атмосфере азота. На протяжении 10 минут добавляли метилмагния хлорид (3 М в ТГФ) (5.6 мл, 16.8 ммоль) и перемешивали смесь при 0°С в течение 30 мин. Смесь нагревали до комнатной темп, и перемешивали в течение ночи. Реакционную смесь гасили насыщенным водным раствором хлорида аммония (5 мл), затем разделяли дихлорметаном (100 мл) и насыщенным водным раствором хлорида аммония (100 мл). Органическую фазу собирали, а водную промывали дихлорметаном (2 × 25 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и выпаривали досуха. Остаток перемешивали в ТГФ (35 мл) в атмосфере азота при 0°С. На протяжении 10 минут добавляли метилмагния хлорид (3 М в ТГФ) (3.75 мл, 11.25 ммоль) и перемешивали смесь при 0°С в течение 40 мин, после чего ее перемешивали при комнатной темп, в течение 20 мин. Реакционную смесь гасили насыщенным водным раствором хлорида аммония (5 мл), затем разделяли дихлорметаном (100 мл) и насыщенным водным раствором хлорида аммония (100 мл). Органическую фазу собирали, а водную промывали дихлорметаном (2 × 25 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр, концентрировали при пониженном давлении и сушили в течение 4 дней в высоковакуумной установке, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.65, [М+Н]+=455.
Альтернативный способ получения промежуточного соединения 43
Этил-3-бром-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 42, 81 г, 173 ммоль) перемешивали в ДХМ (300 мл) и ТГФ (100 мл) при 5°С в атмосфере азота. По каплям добавляли метилмагния хлорид (3 М в ТГФ) (230 мл, 690 ммоль) на протяжении 45 мин, поддерживая внутреннюю температуру ниже 15°С, и перемешивали в течение еще 10 мин. Смесь гасили насыщенным водным раствором хлорида аммония (400 мл) и разделяли между ДХМ (2 л) и водой (2 л). Органическую фазу сушили при помощи сульфата магния и концентрировали при пониженном давлении. Полученный остаток растворяли в ДХМ (500 мл) и ТГФ (250 мл) и охлаждали до 0°С в атмосфере азота. Добавляли по каплям метилмагния хлорид (3 М в ТГФ) (173 мл, 518 ммоль) в течение 1 ч, поддерживая внутреннюю температуру ниже 10°С. После добавления смесь перемешивали при 5°С в течение 10 мин. Смесь гасили насыщенным водным раствором хлорида аммония (400 мл) и разделяли между ДХМ (2 л) и водой (2 л). Органическую фазу сушили при помощи сульфата магния и концентрировали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.60, [М+Н]+=455.
Промежуточное соединение 44
(S)-5-бром-N-(1-гидроксипропан-2-ил)-N,2-диметилбензамид
Получали аналогично промежуточному соединению 33, используя 5-бром-2-метилбензойную кислоту (800 мг, 3.72 ммоль) and (S)-2-(метиламино)пропан-1-ол (332 мг, 3.72 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.82, [М+Н]+=286.
Промежуточное соединение 45
(S)-N-(1-гидроксипропан-2-ил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид

Получали аналогично промежуточному соединению 32, используя (S)-5-бром-N-(1-гидроксипропан-2-ил)-N,2-диметилбензамид (промежуточное соединение 44), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.92, [М+Н]+=334.
Промежуточное соединение 46
5-бром-2-метокси-N,N-диметилбензамид

Получали аналогично промежуточному соединению 33, используя 5-бром-2-метоксибензойную кислоту (483 мг, 2.091 ммоль) и диметиламин (2 M в ТГФ) (1.254 мл, 2.509 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.85, [М+Н]+=258.
Промежуточное соединение 47
2-метокси-N,N-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид
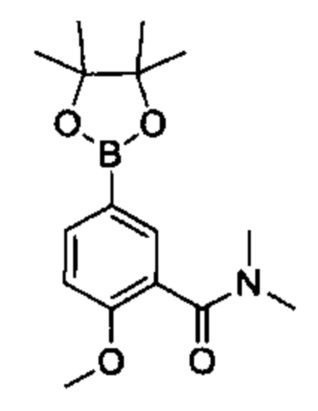
Получали аналогично промежуточному соединению 32, используя 5-бром-2-метокси-N,N-диметилбензамид (промежуточное соединение 46), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.01, [М+Н]+=306.
Промежуточное соединение 48
(R)-(5-бром-2-хлорфенил)(2-(гидроксиметил)пирролидин-1-ил)метанон
Получали аналогично промежуточному соединению 33, используя 5-бром-2-хлорбензойную кислоту (503 мг, 2.136 ммоль) и (R)-пирролидин-2-илметанол, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.89, [М+Н]+=320.
Промежуточное соединение 49
(R)-(2-хлор-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)(2-(гидроксиметил)пирролидин-1-ил)метанон
Получали аналогично промежуточному соединению 32, используя (R)-(5-бром-2-хлорфенил)(2-(гидроксиметил)пирролидин-1-ил)метанон (промежуточное соединение 48), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.94, [М+Н]+=366.
Промежуточное соединение 50
(R)-(5-бром-2-метилФенил)(2-(гидроксиметил)пирролидин-1-ил)метанон

5-бром-2-метилбензойную кислоту (534 мг, 2.483 ммоль), HATU (1074 мг, 2.82 ммоль) и DIPEA (0.520 мл, 2.98 ммоль) в ДМФА (1 мл) и ТГФ (5 мл) перемешивали при комнатной темп, в течение 30 мин, затем добавляли одной порцией (R)-пирролидин-2-илметанол (0.3 мл, 3.04 ммоль). Реакционную смесь перемешивали в течение 18 ч. Растворитель удаляли под вакуумом. Остаток растворяли в этилацетате (40 мл) и разделяли водой (20 мл). Органический слой отделяли и промывали 5% водным раствором хлорида лития (2×10 мл), водой (10 мл) и солевым раствором (10 мл). Затем органический слой пропускали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом. Остаток растворяли в минимальном количестве ДХМ и загружали на колонку с силикагелем (80 г) и элюировали 12 объемами колонки смеси 0 - 100% этилацетат: этанол (3:1, об./об., содержащую 1% триэтиламина) в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.87, [М+Н]+=298.
Промежуточное соединение 51
(R)-(2-(гидроксиметил)пирролидин-1-ил)(2-метил-5-(4,4,5,,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон

Получали аналогично промежуточному соединению 36, используя (R)-(5-бром-2-метилфенил)(2-(гидроксиметил)пирролидин-1-ил)метанон (промежуточное соединение 50), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.02, [М+Н]+=346.
Промежуточное соединение 52
5-бром-N-(2-гидроксиэтил)-2-метокси-N-метилбензамид

Получали аналогично промежуточному соединению 33, используя 5-бром-2-метоксибензойную кислоту (400 мг, 1.731 ммоль) и 2-(метиламино)этан-1-ол (0.167 мл, 2.078 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.72, [М+Н]+=288.
Промежуточное соединение 53
N-(2-гидроксиэтил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид

Получали аналогично промежуточному соединению 32, используя 5-бром-N-(2-гидроксиэтил)-2-метокси-N-метилбензамид (промежуточное соединение 52), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.85, [М+Н]+=336.
Промежуточное соединение 54
(5-бром-2-метоксифенил)(3-гидрокси-3-метилпирролидин-1-ил)метанон

Получали аналогично промежуточному соединению 33, используя 5-бром-2-метоксибензойную кислоту (503 мг, 2.177 ммоль) и 3-метилпирролидин-3-ол (244 мг, 2.412 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.76, [М+Н]+=314.
Промежуточное соединение 55
(3-гидрокси-3-метилпирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон

Получали аналогично промежуточному соединению 32, используя (5-бром-2-метоксифенил)(3-гидрокси-3-метилпирролидин-1-ил)метанон (промежуточное
соединение 54), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.91, [М+Н]+=362.
Промежуточное соединение 56
(5-бром-2-метоксиФенил)(1,4-оксазепан-4-ил)метанон

Получали аналогично промежуточному соединению 33, используя 5-бром-2-метоксибензойную кислоту (528 мг, 2.285 ммоль) и 1,4-оксазепан (272 мг, 2.69 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.87, [М+Н]+=316.
Промежуточное соединение 57
(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)(1,4-оксазепан-4-ил)метанон

Получали аналогично промежуточному соединению 32, используя (5-бром-2-метоксифенил)(1,4-оксазепан-4-ил)метанон (промежуточное соединение 56), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.0, [М+Н]+=362.
Промежуточное соединение 58
(S)-(5-бром-2-метилФенил)(3-гидроксипирролидин-1-ил)метанон
Получали аналогично промежуточному соединению 29, используя 5-бром-2-метилбензойную кислоту (493 мг, 2.293 ммоль) и (S)-пирролидин-3-ол (0.23 мл, 2.77 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.75, [М+Н]+=284.
Промежуточное соединение 59
(S)-(3-гидроксипирролидин-1-ил)(2-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон
Получали аналогично промежуточному соединению 32, используя (S)-(5-бром-2-метилфенил)(3-гидроксипирролидин-1-ил)метанон (промежуточное соединение 58), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.87, [М+Н]+=332.
Промежуточное соединение 60
(S)-(5-бром-2-метоксифенил)(3-метилморфолино)метанон
Получали аналогично промежуточному соединению 50, используя 5-бром-2-метоксибензойную кислоту (505 мг, 2.186 ммоль) и (S)-3-метилморфолин (251 мг, 2.481 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.92, [М+Н]+=314.
Промежуточное соединение 61
(S)-(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)(3-метилморфолино)метанон

Получали аналогично промежуточному соединению 32, используя (S)-(5-бром-2-метоксифенил)(3-метилморфолино)метанон (промежуточное соединение 60), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.04, [М+Н]+=362.
Промежуточное соединение 62
(5-бром-2-метилФенил)(3-гидрокси-3-метилпирролидин-1-ил)метанон
Получали аналогично промежуточному соединению 33, используя 5-бром-2-метилбензойную кислоту (394 мг, 1.832 ммоль) и 3-метилпирролидин-3-ол (222 мг, 2.199 ммоль), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.81, [М+Н]+=298.
Промежуточное соединение 63
(3-гидрокси-3-метилпирролидин-1-ил)(2-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон

Получали аналогично промежуточному соединению 32, используя (5-бром-2-метилфенил)(3-гидрокси-3-метилпирролидин-1-ил)метанон (промежуточное соединение 62), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.91, [М+Н]+=346.
Промежуточное соединение 64
Этил 7-((3-(1Н-пиразол-1-ил)бензил)амино)-3-бром-2-метилпиразолох[1,5-а]пиримидин-5-карбоксилат

Получали аналогично промежуточному соединению 42, используя (3-(1H-пиразол-1-ил)фенил)метанамин (можно приобрести в Sigma-Aldrich Inc.), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.21, [М+Н]+=455.
Промежуточное соединение 65
Этил-7-((3-(1Н-пиразол-1-ил)бензил)амино)-3-бром-2-метилпиразолоИ,5-а1пиримидин-5-карбоксилат
Получали аналогично промежуточному соединению 43, используя этил-7-((3-(1H-пиразол-1-ил)бензил)амино)-3-бром-2-метилпиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 64), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.20, [М+Н]+=441.
Промежуточное соединение 66
(R)-(5-бром-2-метоксифенил)(2-(гидроксиметил)пирролидин-1-ил)метанон
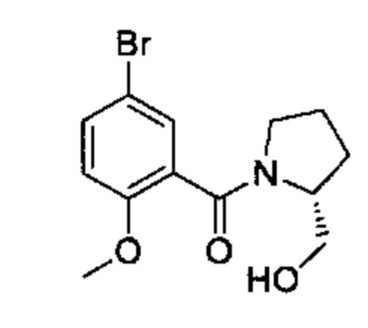
Получали аналогично промежуточному соединению 35, используя 5-бром-2-метоксибензойную кислоту и (R)-пирролидин-2-илметанол, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.83, [М+Н]+=314.
Промежуточное соединение 67
(R)-(2-(Гидроксиметил)пирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон
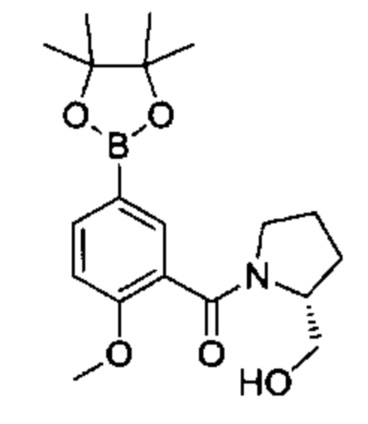
Получали аналогично промежуточному соединению 36, используя (R)-(5-бром-2-метоксифенил)(2-(гидроксиметил)пирролидин-1-ил)метанон (промежуточное соединение 66), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.02, [М+Н]+=362.
Промежуточное соединение 68
5-бром-2-хлор-N-(2-гидроксиэтил)-N-метилбензамид

Получали аналогично промежуточному соединению 29, используя 5-бром-2-хлорбензойную кислоту и 2-(метиламино)этан-1-ол в ТГФ, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.77, [М+Н]+=294.
Промежуточное соединение 69
2-хлор-N-(2-гидроксиэтил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид
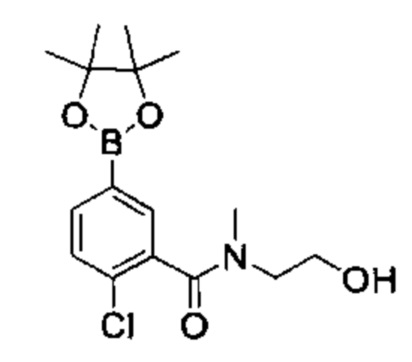
Получали аналогично промежуточному соединению 36, используя промежуточное соединение 68, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.75, [М+Н]+=340.
Промежуточное соединение 70
5-бром-N-(2-гидроксиэтил)-N,2-диметилбензамид

Получали аналогично промежуточному соединению 29, используя 5-бром-2-метилбензойную кислоту и 2-(метиламино)этан-1-ол, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.76, [М+Н]+=274.
Промежуточное соединение 71
N-(2-гидроксиэтил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид
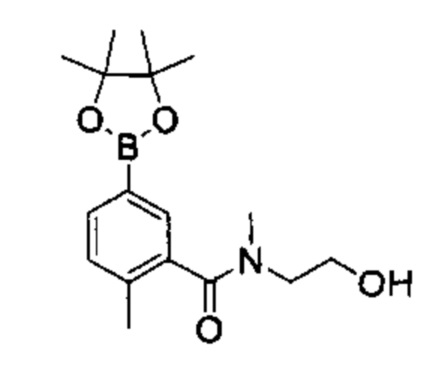
Получали аналогично промежуточному соединению 36, используя промежуточное соединение 70, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.85, [М+Н]+=320.
Промежуточное соединение 72
5-бром-N-(2-гидроксиэтил)-N,2-диметилбензамид
Получали аналогично промежуточному соединению 29, используя 5-бром-2-метилбензойную кислоту и 2-(этиламино)этан-1-ол, в ТГФ, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.84, [М+Н]+=286.
Промежуточное соединение 73 N-Этил-N-(2-гидроксиэтил)-2-метил-5-(4,4,5,5-третраметил1,3,2-диоксаборолан-2-ил)бензамид

Получали аналогично промежуточному соединению 36, используя промежуточное соединение 72, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.95, [М+Н]+=334.
Промежуточное соединение 74
Этил-2-(2-метил-7-оксо-4,7-дигидропиразоло[1,5-а]пиримидин-5-ил)ацетат
К перемешиваемому раствору 5-метил-1Н-пиразол-3-амина (25 г, 257 ммоль) в 1,4-диоксане (150 мл), добавляли диэтил-3-оксопентандиоат (56.8 мл, 309 ммоль), а затем уксусную кислоту (7.37 мл, 129 ммоль) при комнатной темп. Реакционную смесь перемешивали при 100°С в течение 6 ч. Смесь давали остыть до комнатной темп. Твердые вещества собирали фильтрацией, промывали этиловым эфиром и сушили, в результате чего получали указанное в заголовке соединение. ЖХМС (метод Н): время удерживания=1.55, [М+Н]+=236.
Промежуточное соединение 75
Этил 2-(7-хлор-2-метилпиразоло[1,5-а]пиримидин-5-ил)ацетат
К перемешиваемому раствору этил-2-(2-метил-7-оксо-4,7-дигидропиразоло[1,5-а]пиримидин-5-ил)ацетата (промежуточное соединение 74, 22.5 г, 96 ммоль) в ацетонитриле (250 мл), добавляли DIPEA (33.4 мл, 191 ммоль), N-метилморфолин (0.105 мл, 0.956 ммоль) и POCl3 (17.83 мл, 191 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной темп, в течение 24 ч. После завершения реакции смесь концентрировали, а затем выливали на смесь воды со людом. Смесь нейтрализовали бикарбонатом натрия и экстрагировали этилацетатом (2 × 300 мл). Объединенные органические слои промывали солевым раствором, сушили с использованием сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (0 - 20% этилацетат в гексанах), в результате чего получали указанное в заголовке соединение. ЖХМС (метод Н): время удерживания=2.05, [М+Н]+=254.
Промежуточное соединение 76
Этил 2-(7-хлор-3-йодо-2-метилпиразоло[1,5-а]пиримидин-5-ил)ацетат
К перемешиваемому раствору этил-2-(7-хлор-2-метилпиразоло[1,5-а]пиримидин-5-ил)ацетата (промежуточное соединение 75, 8 г, 31.5 ммоль) в ДМФА (160 мл) добавляли N-йодосукцинимид (7.09 г, 31.5 ммоль) при 0°С. Полученную реакционную смесь перемешивали при комнатной темп, в течение 1 ч. Реакционную смесь выливали на охлажденную льдом воду и собирали осажденное твердое вещество фильтрацией. Остаток очищали хроматографией на силикагеле (0 - 10% этилацетат в гексанах), в результате чего получали указанное в заголовке соединение. ЖХМС (метод Н): время удерживания=2.43, [М+Н]+=380.
Промежуточное соединение 77
Этил 2-(3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)ацетат

Смесь этил 2-(7-хлор-3-йодо-2-метилпиразоло[1,5-а]пиримидин-5-ил)ацетат (промежуточное соединение 76, 6 г, 15.81 ммоль), (3-(1-метил-1Н-имидазол-2-ил)фенил)метанамин (промежуточное соединение 3, 3.55 г, 18.97 ммоль) и DIPEA (5.52 мл, 31.6 ммоль) в ДМСО (50 мл) перемешивали и нагревали при 60°С в течение 2 ч. После охлаждения реакционную смесь выливали на охлажденную льдом воду (300 мл) и собирали осажденное твердое вещество фильтрацией, затем сушили при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод Н): время удерживания=1.74, [М+Н]+=531.
Промежуточное соединение 78
Метил 2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензоат

Раствор/суспензию метил-5-йодо-2-метоксибензоата (20 г, 68.5 ммоль), бис(пинаколато)дибора (17.39 г, 68.5 ммоль), ацетата калия (20.16 г, 205 ммоль) и бис(трифенилфосфин)палладия (II) хлорида (3.85 г, 5.48 ммоль) в 1,4-диоксане (200 мл) нагревали до 100°С в атмосфере азота. Реакционную смесь перемешивали при 100°С в течение 20 ч, а затем давали остыть. Реакционную смесь фильтровали через слой целита. Слой целита промывали этилацетатом (250 мл). К фильтрату добавляли 1 М соляной кислоты (250 мл). Органическую фазу промывали солевым раствором (100 мл), а затем сушили при помощи сульфата магния. Растворитель удаляли под вакуумом. Это растворяли в ДХМ и наносили на картридж с силикагелем (750 г) и элюировали градиентом 0 - 50% этилацетата в циклогексане с использованием 9 объемов колонки. Необходимые фракции объединяли и выпаривали под вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=1.16, [М+Н]+=293.
Промежуточное соединение 79 и Промежуточное соединение 80
Этил-2-(3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропаноат и этил-2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)-2-метилпропаноат

В высушенном сосуде этил-2-(3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)ацетат (промежуточное соединение 77, 400 мг, 0.754 ммоль) перемешивали в ТГФ (4.0 мл) в атмосфере азота при 0°С. Добавляли метилйодид (0.05 мл, 0.8 ммоль) и перемешивали смесь при 0°С в течение 1 мин, после чего добавляли LiHMDS (1 M в ТГФ) (1.6 мл, 1.6 ммоль). Реакционную смесь перемешивали при 0°С в течение 5 мин, затем давали нагреться до комнатной температуры. Через 15 минут реакционную смесь разбавляли насыщенным вод. NH4Cl (2 мл) и перемешивали в течение 5 мин. Смесь переносили в делительную воронку с дихлорметаном (20 мл) и водой (20 мл). Разделяли фазы и собирали органический слой. Затем водный слой промывали дополнительным количеством дихлорметана (2×10 мл), а объединенные органические слои промывали солевым раствором (20 мл), фильтровали через гидрофобный спеченный фильтр и выпаривали досуха. Неочищенный продукт загружали влажным методом из ДХМ (1 мл) на 28 г KP-NH колонку и элюировали 10-50% смесью этилацетат: этанол (3:1, содержащей 1% триэтиламина) в циклогексане. Затем пробу очищали обращенно-фазовой хроматографией, используя для элюирования 40-90% ацетонитрил в забуференной до рH10 карбонатом аммония воде. Фракции, содержащие каждый продукт, объединяли отдельно, концентрировали при пониженном давлении и сушили в вакуумной печи на протяжении выходных, в результате чего получали целевые указанные в заголовке соединения. Промежуточное соединение 79: ЖХМС (метод F): время удерживания=1.16, [М+Н]+=545. Промежуточное соединение 80: ЖХМС (метод F): время удерживания=1.27, [М+Н]+=559.
Промежуточное соединение 81
2-(3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол
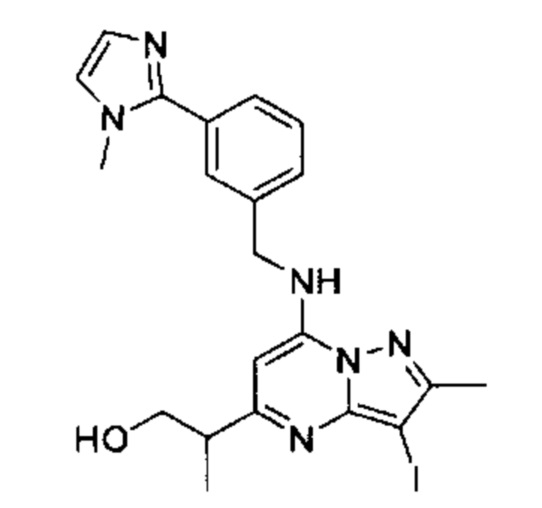
Этил-2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропаноат (промежуточное соединение 79, 87 мг, 0.160 ммоль) перемешивали в ТГФ (1.5 мл) при 0°С в атмосфере азота. Добавляли по каплям DIBAL-Н (1 М в ТГФ) (0.5 мл, 0.5 ммоль) и перемешивали смесь в течение 45 мин. Смесь нейтрализовали водным 1 М раствором соли Рошеля и интенсивно перемешивали в течение 30 мин. Смесь разделяли дихлорметаном (10 мл) и водой (10 мл). Отделенную водную фазу промывали дополнительным количеством дихлорметана (2 × 10 мл), а объединенные органические фазы пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.99, [М+Н]+=503.
Промежуточное соединение 82
2-(3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)-2-метилпропан-1-ол

Получали аналогично промежуточному соединению 81, используя этил-2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)-2-метилпропаноат (промежуточное соединение 80), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.12, [М+Н]+=517.
Промежуточное соединение 83
(5-бром-2-метоксифенил)(2-(гидроксиметил)пирролидин-1-ил)метанон

Получали аналогично промежуточному соединению 29, используя 5-бром-2-метоксибензойную кислоту и пирролидин-2-илметанол в ТГФ, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.84, [М+Н]+=314.
Промежуточное соединение 84
(2-(гидроксиметил)пирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон

Получали аналогично промежуточному соединению 36, используя промежуточное соединение 83, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.96, [М+Н]+=362.
Промежуточное соединение 85
(R)-5-бром-N-(1-гидроксипропан-2-ил)-N,2-диметилбензамид

Получали аналогично промежуточному соединению 29, с использованием 5-бром-2-метоксибензойной кислоты и (R)-2-(метиламино)пропан-1-ола в ТГФ, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.82, [М+Н]+=286.
Промежуточное соединение 86
(R)-N-(1-гидроксипропан-2-ил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид
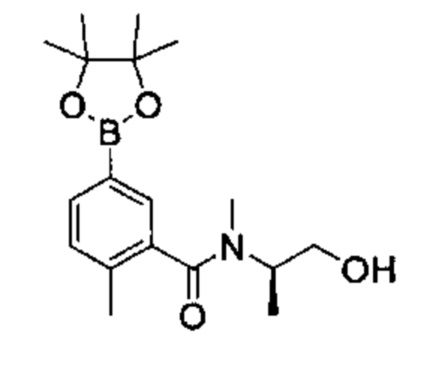
Получали аналогично промежуточному соединению 36, используя промежуточное соединение 85, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.91, [М+Н]+=334.
Промежуточное соединение 87
5-бром-N-(1-гидроксипропан-2-ил)-N,2-диметилбензамид

Получали аналогично промежуточному соединению 29, используя 5-бром-2-метилбензойную кислоту и 2-(метиламино)пропан-1-ол в ТГФ, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.82, [М+Н]+=286.
Промежуточное соединение 88 N-(1-гидроксипропан-2-ил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид

Получали аналогично промежуточному соединению 36, используя 5-бром-N-(1-гидроксипропан-2-ил)-N,2-диметилбензамид (промежуточное соединение 87), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.91, [М+Н]+=334.
Промежуточное соединение 89
(5-бром-2-метилфенил)(3-гидроксипирролидин-1-ил)метанон

Получали аналогично промежуточному соединению 29, используя 5-бром-2-метилбензойную кислоту и пирролидин-3-ол в ТГФ, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.75, [М+Н]+=284.
Промежуточное соединение 90
(3-гидроксипирролидин1-ил)(2-метил-5-(4,4,5,5-третраметил1,3,2-диоксаборолан-2-ил)фенил)метанон
Получали аналогично промежуточному соединению 36, используя (5-бром-2-метилфенил)(3-гидроксипирролидин-1-ил)метанон (промежуточное соединение 89), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.84, [М+Н]+=332.
Промежуточное соединение 91
5-бром-N-(1-гидроксипропан-2-ил)-2-метокси-N-метилбензамид

Получали аналогично промежуточному соединению 29, используя 5-бром-2-метоксибензойную кислоту и 2-(метиламино)пропан-1-ол в ТГФ, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.76, [М+Н]+=302.
Промежуточное соединение 92
N-(1-гидроксипропан2-ил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид
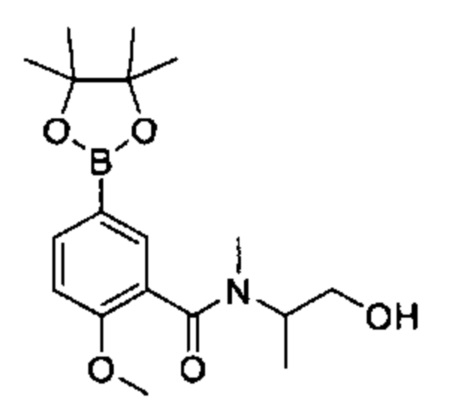
Получали аналогично промежуточному соединению 36, используя 5-бром-N-(1-гидроксипропан-2-ил)-2-метокси-N-метилбензамид (промежуточное соединение 91), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.89, [М+Н]+=350.
Промежуточное соединение 93 and Промежуточное соединение 94
Трет-бутил(5-(1-гидроксиэтил)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1H-имидазол-2-ил)бензил)карбамат, изомер 1 и изомер 2

Трет-бутил(5-(1-гидроксиэтил)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1H-имидазол-2-ил)бензил)карбамат (промежуточное соединение 26, 60 мг, 0.123 ммоль) раззделяли, используя колонку Chiralpak AD-H (30 мм × 250 мм, 5 мкм) и систему растворителей: 30% этанола (содержащий 0.2% изопропиламина)/гептан (содержащий 0.2% изопропиламина) при скорости 30 мл/мин, в результате чего получали хиральные соединения, указанные в заголовке, изомер 1: ЖХМС (метод F): время удерживания=0.97, [М+Н]+=489. Хиральная ВЭЖХ: время удерживания 10.73, 99.7%. изомер 2: ЖХМС (метод F): время удерживания=0.97, [М+Н]+=489. Хиральная ВЭЖХ: время удерживания 7.53, 100%.
Промежуточное соединение 95
5-бром-N,2-диметил-N-(тетрагидро-2Н-пиран-4-ил)бензамид
Получали аналогично промежуточному соединению 33, используя 5-бром-2-метилбензойную кислоту и N-метилтетрагидро-2H-пиран-4-амин в ТГФ, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.96, [М+Н]+=312.
Промежуточное соединение 96
N,2-диметил-N-(тетрагидро-2Н-пиран-4-ил)-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид

Получали аналогично промежуточному соединению 32, используя 5-бром-N,2-диметил-N-(тетрагидро-2H-пиран-4-ил)бензамид (промежуточное соединение 95), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.06, [М+Н]+=360.
Промежуточное соединение 97
2-метилпиразоло[1,5-а]пиримидин-5,7(4Н,6H)-дион

5-метил-1Н-пиразол-3-амин (50 г, 515 ммоль) растворяли в этаноле (500 мл) в атмосфере азота. Добавляли этанолат натрия (334 г, 1030 ммоль) в этаноле, а затем диэтилмалонат (86 мл, 566 ммоль). Реакционную смесь перемешивали при 80°С в течение 4 ч. Реакционной смеси давали остыть до комнатной темп, и собирали осажденное твердое вещество фильтрацией, промывали избытком этанола (1000 мл) и сушили, в результате чего получали указанное в заголовке соединение. ЖХМС (метод В): время удерживания=0.48, [М+Н]+=166.
Промежуточное соединение 98
5,7-дихлор-2-метилпиразоло[1,5-а]пиримидин
2-метилпиразоло[1,5-а]пиримидин-5,7(4Н,6Н)-дион (промежуточное соединение 97, 35 г, 212 ммоль) добавляли к оксихлориду фосфора (395 мл, 4239 ммоль) и нагревали смесь при 100°С в течение 24 ч. Смесь охлаждали до комнатной темп, и удаляли избыток оксихлорида фосфора при пониженном давлении. Смесь дистиллировали совместно с толуолом (2 × 200 мл). Неочищенный продукт очищали колоночной хроматографией на силикагеле (20% этилацетат в петролейном эфире), в результате чего получали указанное в заголовке соединение. ЖХМС (метод В): время удерживания=3.21, [М+Н]+=201.
Промежуточное соединение 99
5,7-дихлор-3-йодо-2-метилпиразоло[1,5-a]пиримидин

К перемешиваемому раствору 5,7-дихлор-2-метилпиразоло[1,5-а7пиримидина (промежуточное соединение 98, 15 г, 74.2 ммоль) в ДХМ (200 мл) при 0°С добавляли уксусную кислоту (29.8 мл, 520 ммоль), а затем порциями N-йодосукцинимид (16.70 г, 74.2 ммоль). Полученную реакционную смесь перемешивали при 0°С в течение 1 ч. Добавляли 10% Водный раствор сульфита натрия (200 мл) и интенсивно перемешивали при комнатной темп, в течение 30 мин. Слой в ДХМ отделяли и промывали насыщенным тиосульфатом натрия (300 мл), солевым раствором (200 мл), а затем водой (100 мл), а органический слой сушили при помощи безводного сульфата натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод Е): время удерживания=2.37, [М+Н]+=328.
Промежуточное соединение 100
5-хлор-3-йодо-2-метил-N-(3-(1-метил-1Н-имидазол-2-ил)бензил)пиразоло[1,5-а]пиримидин-7-амин

Смесь (3-(1-метил-1Н-имидазол-2-ил)фенил)метиламина (промежуточное соединение 3, 31 г, 122 ммоль), 5,7-дихлор-3-йодо-2-метилпиразоло[1,5-а]пиримидина (промежуточное соединение 99, 39.9 г, 122 ммоль) и DIPEA (63.8 мл, 365 ммоль) в ДМСО (500 мл) перемешивали и нагревали при 50°С в течение 18 ч. После охлаждения реакционную смесь выливали на охлажденную льдом воду (1500 мл). Осажденное твердое вещество собирали фильтрацией, сушили при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод С): время удерживания=2.19, [М+Н]+=479.
Промежуточное соединение 101
Трет-бутил(5-хлор-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат
К перемешиваемому раствору 5-хлор-3-йодо-2-метил-N-(3-(1-метил-1H-имидазол-2-ил)бензил)пиразоло[1,5-а]пиримидин-7-амина (промежуточное соединение 100, 50 г, 96 ммоль) в ДХМ (1000 мл) добавляли DIPEA (25.03 мл, 143 ммоль), DMAP (1.167 г, 9.55 ммоль) и ди-трет-бутил дикарбонат (33.3 мл, 143 ммоль). Реакционную смесь перемешивали при комнатной темп, в течение 3 ч. Реакционную смесь разбавляли дихлорметаном (1000 мл), промывали водой (1000 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле (80% этилацетат в петролейном эфире), в результате чего получали указанное в заголовке соединение. ЖХМС (метод D): время удерживания = 6.33, [М+Н]+ = 579.
Промежуточное соединение 102
Трет-бутил(5-хлор-3-(3-((2-гидроксиэтил)сульфонил)-4-метоксифенил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1H-имидазол-2-ил)бензил)карбамат
Трет-бутил(5-хлор-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат (промежуточное соединение 101, 506 мг, 0.874 ммоль), 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этан-1-ол (промежуточное соединение 19, 268 мг, 0.783 ммоль), трифениларсин (13 мг, 0.042 ммоль), PdCl2(PhCN)2 (18 мг, 0.047 ммоль) и карбонат натрия (193 мг, 1.821 ммоль) объединяли в 1,4-диоксане (6 мл) и воде (1.5 мл). Смесь барботировали азотом в течение 1 мин и нагревали до 80°С в течение 4 ч. Добавляли 2-((2-метокси-5-(4,4,5,5-третраметил1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этан-1-ол (промежуточное соединение 19, 113 мг, 0.330 ммоль), трифениларсин (13 мг, 0.042 ммоль) и PdCl2(PhCN)2 (18 мг, 0.047 ммоль). Смесь барботировали азотом в течение 1 мин, а затем нагревали до 80°С в течение ночи. Смесь давали остыть и концентрировали при пониженном давлении. Остаток суспендировали в этилацетате (20 мл) и фильтровали через целит, промывая этилацетатом (40 мл), а затем ДХМ (40 мл). Объединенный фильтрат концентрировали при пониженном давлении для удаления ДХМ, а затем разделяли водой (40 мл). Органическую фазу отделяли, пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток растворяли в смеси ДМСО: метанол (6 мл, 1:1) и очищали MDAP-хроматографией (метод А). Фракции, содержащие продукт, объединяли и концентрировали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 1.12, [М+Н]+ = 667.
Промежуточное соединение 103
Этил 2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)бутаноат
Получали аналогично промежуточному соединению 79, используя иодоэтан, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 1.24, [М+Н]+ = 559.
Промежуточное соединение 104
2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)бутен-1-ол
Получали аналогично промежуточному соединению 81, используя этил-2-(3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-a]пиримидин-5-ил)бутаноат (промежуточное соединение 103), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 1.05, [М+Н]+ = 517.
Промежуточное соединение 105
3-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пентан-3-ол
Этил-3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 42, 253 мг, 0.539 ммоль) перемешивали в ТГФ (5 мл) при 0°С в атмосфере азота. Этилмагния бромид (1 M в ТГФ) (2.4 мл, 2.4 ммоль) добавляли на протяжении 5 мин и перемешивали смесь при 0°С в течение 30 мин, затем нагревали до комнатной темп. Через 2 ч реакционную смесь гасили насыщенным водным раствором хлорида аммония (2 мл) и разделяли дихлорметаном (10 мл) и насыщенным водным раствором хлорида аммония (10 мл). Органическую фазу собирали, а водную промывали дихлорметаном (2×5 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и выпаривали досуха. Остаток перемешивали в ТГФ (5 мл) в атмосфере азота при 0°С. Добавляли этилмагния бромид (1 M в ТГФ) (1.6 мл, 1.6 ммоль) на протяжении 5 мин и перемешивали смесь при 0°С в течение 30 мин. Смесь гасили насыщенным водным раствором хлорида аммония (2 мл) и разделяли дихлорметаном (10 мл) и насыщенным водным раствором хлорида аммония (10 мл). Органическую фазу собирали, а водную промывали дихлорметаном (2×5 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток растворяли в смеси метанол: ДМСО (2 мл, 1:1) и очищали MDAP-хроматографией (метод А). Содержащие продукт фракции объединяли, выпаривали досуха, а затем сушили в вакуумной печи в течение ночи, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.80, [М+Н]+ = 483.
Промежуточное соединение 106
3-бром-N-метокси-N,2-диметил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксамид
Суспензию N,О-диметилгидроксиламина гидрохлорида (1.62 г, 16.61 ммоль) в ТГФ (10 мл) охлаждали до -10°С). По каплям добавляли н-бутиллитий (2.5 М в гексанах) (13 мл, 32.5 ммоль) на протяжении 40 мин, поддерживая температуру реакции ниже 5°С. Затем смесь перемешивали в ледяной ванне в течение 15 мин. К этому раствору медленно добавляли этил-3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксилат (промежуточное соединение 42, 1.5 г, 3.2 ммоль) в ТГФ (10 мл). В процессе добавления поддерживали температуру смеси ниже 0°С. После завершения добавления смесь перемешивали в ванне со льдом (at -8°С) в течение 20 мин, а затем давали нагреться до комнатной темп, и перемешивали в течение 1 ч. Добавляли насыщенный вод. раствор хлорида аммония (20 мл) и перемешивали смесь в течение 30 мин. Добавляли ДХМ (50 мл). Раствор обрабатывали ультразвуком и переносили в делительную воронку. Оставшееся твердое вещество суспендировали в смеси воды (50 мл) и ДХМ (50 мл) и обрабатывали ультразвуком. Смесь добавляли в делительную воронку. Органическую фазу отделяли, а водную далее экстрагировали дихлорметаном (100 мл × 2). Объединенные органические вещества пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток растворяли в ДХМ и очищали хроматографией на силикагеле, используя для элюирования смесь 30-100% этилацетата: этанол (3:1, об./об., содержащую 1% триэтиламина) в циклогексане (10 объемов колонки). Содержащие продукт фракции объединяли и концентрировали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.63, [М+Н]+ = 484.
Промежуточное соединение 107
1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-он

3-бром-N-метокси-N,2-диметил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбоксамид (промежуточное соединение 106, 530 мг, 1.094 ммоль) растворяли в ТГФ (20 мл) и охлаждали до 0°С. Медленно добавляли этилмагния бромид (1 M в ТГФ) (2.7 мл, 2.7 ммоль). Через 20 мин смесь гасили насыщенным водным раствором хлорида аммония (5 мл) и перемешивали интенсивно в течение 10 мин. Суспензию разделяли между водой (50 мл) и ДХМ (50 мл). Отделенную водную фазу промывали дополнительным количеством ДХМ (2×50 мл), а объединенные органические фазы пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток очищали обращенно-фазовой хроматографией (C18, 60 г), используя для элюирования 30-85% ацетонитрил в 10 мМ растворе бикарбоната аммония в воде (доведенным до рН 10 раствором аммиака) (25 объемов колонки). Фракции, содержащие целевой продукт, объединяли и концентрировали при пониженном давлении. Остаток очищали обращенно-фазовой хроматографией (C18, 40 г), используя для элюирования 20-50% ацетонитрил в воде (соедржащей 0.1% муравьиной кислоты). Фракции, содержащие целевой продукт, объединяли и нейтрализовали с использованием насыщенного вод. бикарбоната натрия, затем экстрагировали дихлорметаном (2×50 мл). Объединенные органические вещества пропускали через гидрофобный спеченный фильтр и сушили при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.84, [М+Н]+ = 453.
Промежуточные соединения 108 и 109
1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 1 и изомер 2.
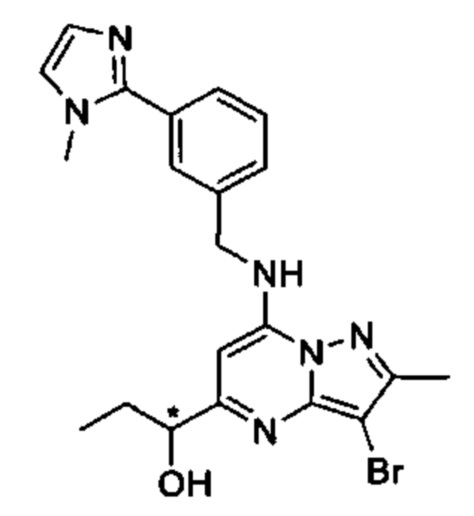
1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-он (промежуточное соединение 107, 164 мг, 0.362 ммоль) перемешивали в ТГФ (3 мл) при 0°С в атмосфере азота. Добавляли по каплям DIBAL-H (1 M в ТГФ) (0.75 мл, 0.75 ммоль) и перемешивали смесь в течение 90 мин. Смесь гасили водным раствором тартрата натрия-калия (1 М) и перемешивали интенсивно в течение 1 ч. Смесь отделяли дихлорметаном (20 мл) и водой (20 мл). Отделенную водную фазу промывали дополнительным количеством дихлорметана (2×20 мл), а объединенные органические вещества пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток очищали обращенно-фазовой хроматографией (C18, 30 г), используя для элюирования 10-40% ацетонитрил (содержащий 0.1% муравьиной кислоты) в воде (содержащей 0.1% муравьиной кислоты)(10 объемов колонки). Фракции, содержащие целевой продукт, объединяли и повышали основность с использованием насыщенного водного бикарбоната натрия, затем экстрагировали с использованием ДХМ (2×50 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток очищали с использованием колонки Chiralpak AD-H (30 мм × 250 мм, 5 мкм), используя для элюирования 30% этанол (содержащий 0.2% изопропиламина) в гексане (содержащем 0.2% изопропиламин), в результате чего получали указанные в заголовке соединения. Изомер 1: ЖХМС (метод J): время удерживания = 0.61, [М+Н]+ = 455. Хиральная ВЭЖХ, время удерживания 5.95, 100%. Изомер 2: ЖХМС (метод J): время удерживания = 0.61, [М+Н]+ = 455. Хиральная ВЭЖХ, время удерживания 9.02, 100%.
Промежуточное соединение 110
1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)этан-1-он

Получали аналогично промежуточному соединению 107, используя метил магния бромид, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.73, [М+Н]+ = 439.
Промежуточные соединения 111 и 112
1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)этан-1-ол, изомер 1 и изомер 2

Получали аналогично промежуточному соединению 108 и 109, в результате чего получали указанные в заголовке соединения. Изомер 1: ЖХМС (метод F): время удерживания = 0.95, [М+Н]+ = 441. Хиральная ВЭЖХ, время удерживания 7.37, 100%. Изомер 2: ЖХМС (метод F): время удерживания = 0.95, [М+Н]+ = 441. Хиральная ВЭЖХ, время удерживания 11.07, 99.4%.
Промежуточное соединение 113
Трет-бутил(5-ацетил-3-бром-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат

1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)этан-1-он (промежуточное соединение 110, 358 мг, 0.815 ммоль) суспендировали в ТГФ (10 мл). Добавляли DIPEA (0.21 мл, 1.202 ммоль) и DMAP (10 мг, 0.082 ммоль), а затем Вос-ангидрид (0.28 мл, 1.206 ммоль). Смесь перемешивали и нагревали до 50°С в течение 1.5 ч. Реакционную смесь охлаждали до температуры окружающей среды, разбавляли водой (25 мл) и этилацетатом (25 мл). Органическую фазу собирали, а водную дополнительно промывали этилацетатом (2×25 мл). Объединенные органические вещества пропускали через гидрофобный спеченный фильтр и выпаривали досуха. Остаток растворяли в минимальном количестве ДХМ (1.5 мл) и загружали влажным методом на 40 г силикагелевую колонку. Продукт элюировали с использованием смеси 0 - 50% этанол: этилацетат (3:1, об./об., содержащей 1% триэтиламина) в циклогексане. Фракции, содержащие продукт, объединяли и выпаривали досуха, затем сушили в высоком вакууме в течение 2 дней, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 1.35, [М+Н]+ = 539.
Промежуточное соединение 114
Трет-бутил(3-бром-5-(1-((трет-бутилдиметилсилил)окси)винил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-И-метил-1Н-имидазол-2-ил)бензил)карбамат

Трет-бутил(5-ацетил-3-бром-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат (промежуточное соединение 113, 456 мг, 0.676 ммоль) перемешивали в ДХМ (5 мл) и триэтиламине (0.32 мл, 2.296 ммоль) при 0°С в атмосфере азота. Добавляли TBS-OTf (0.21 мл, 0.914 ммоль) и давали смеси нагреться до комнатной темп., при которой перемешивали в течение 4 ч. Добавляли еще триэтиламин (0.3 мл, 2.152 ммоль), а затем TBS-OTf (0.25 мл, 1.089 ммоль) и оставляли смесь на ночь. Смесь разделяли между ДХМ (20 мл) и водой (20 мл). Отделенную водную фазу промывали дихлорметаном (2×10 мл), а объединенные органические фазы пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле, используя для элюирования 0-50% этилацетат в циклогексане. Остаток растирали с дихлорметаном и сушили в высоком вакууме в течение 2 дней, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 1.70, [М+Н]+ = 653.
Промежуточное соединение 115
Трет-бутил(3-бром-5-(1-((трет-бутилдиметилсилил)окси)циклопропил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат
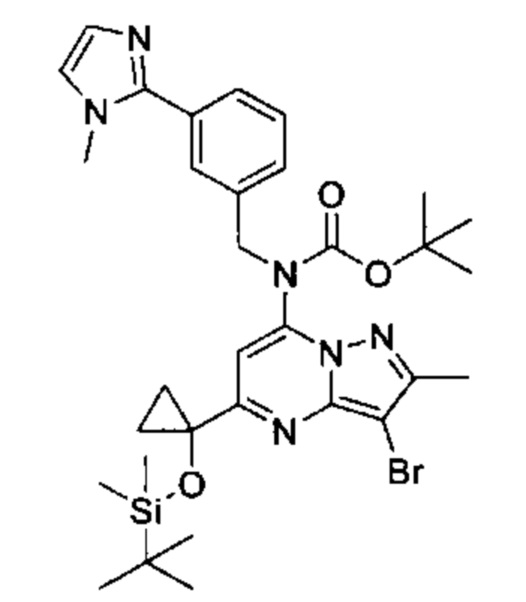
Диэтилцинк (1 M в гептане) (0.795 мл, 0.795 ммоль) перемешивали в ДХМ (0.5 мл) при 0°С в атмосфере азота. Очень медленно по каплям добавляли ТФУК (0.061 мл, 0.795 ммоль) в ДХМ (0.5 мл) и перемешивали смесь в течение 10 мин. Добавляли по каплям дийодометан (0.064 мл, 0.795 ммоль) в ДХМ (0.5 мл). Через 5 минут перемешивания добавляли трет-бутил(3-бром-5-(1-((трет-бутилдиметилсилил)окси)винил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат (промежуточное соединение 114, 260 мг, 0.398 ммоль) в ДХМ (2 мл). Через 1 ч смеси давали нагреться до темп, окружающей среды. Через 6 ч добавляли насыщенный вод. раствор хлорида аммония (1 мл) и перемешивали смесь интенсивно в течение 30 мин. Добавляли насыщ. вод. бикарбонат натрия (2 мл) и перемешивали смесь в течение еще 30 мин. Смесь разделяли между ДХМ (20 мл) и насыщенным водным раствором хлорида аммония (20 мл). Отделенную водную фазу промывали дихлорметаном (2×10 мл), а объединенные органические фазы пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Неочищенный материал объединяли с неочищенным материалом из аналогичной реакции с использованием трет-бутил(3-бром-5-(1-((трет-бутилдиметилсилил)окси)винил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамата (промежуточное соединение 114, 51 мг, 0.078 ммоль) и очищали MDAP-хроматографией (метод А). Содержащие продукт фракции объединяли, концентрировали при пониженном давлении и сушили в высоком вакууме, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 1.72, [М+Н]+ = 667.
Промежуточное соединение 116
Метил 2-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензоат
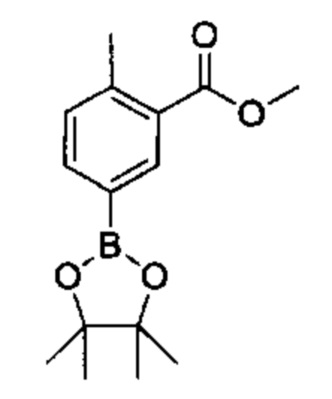
Метил 5-бром-2-метилбензоат (50 г, 218 ммоль), бис(пинаколато)дибор (55.4 г, 218 ммоль), ацетат калия (64.3 г, 655 ммоль) и бис(трифенилфосфин)палладия (II) хлорид (12.26 г, 17.46 ммоль) в 1,4-диоксане (500 мл) нагревали до 100°С в атмосфере азота в течение 2 ч. После охлаждения смесь фильтровали через целит и промывали этилацетатом (500 мл). К фильтрату добавляли 1 М водный раствор соляной кислоты (500 мл) и разделяли фазы. Органическую фазу промывали 1 М водным раствором соляной кислоты (250 мл), солевым раствором (250 мл), сушили при помощи сульфата магния и удаляли растворитель под вакуумом. Неочищенный продукт очищали хроматографией на колонке с силикагелем, используя для элюирования 0-50% этилацетат в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 1.34, [М+Н]+ = 277.
Промежуточное соединение 117
Метил 5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метилбензоат
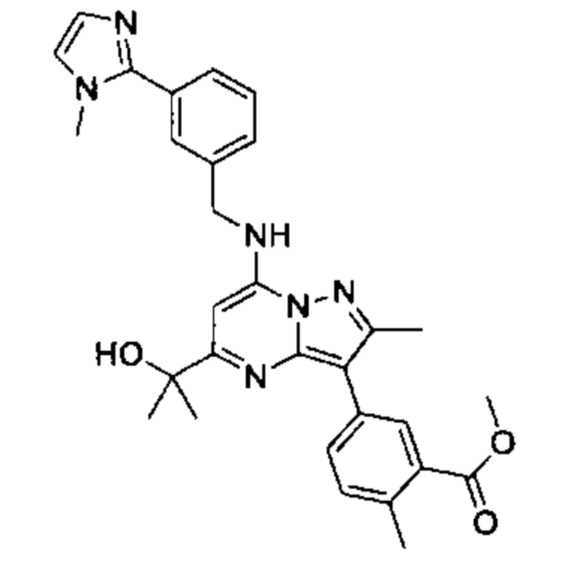
2-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-2-ол (промежуточное соединение 43, 25.77 г, 56.6 ммоль), метил-2-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензоат (промежуточное соединение 116, 23.44 г, 85 ммоль), фосфат калия (18.02 г, 85 ммоль), XPhos (2.70 г, 5.66 ммоль) и XPhos Pd G2 (4.45 г, 5.66 ммоль) объединяли в 1,4-диоксане (300 мл) и воде (100 мл). Смесь обрабатывали тремя циклами вакуум/азот, а затем перемешивали и нагревали до 100°С в течение 2 ч. После охлаждения смесь разделяли между этилацетатом (250 мл) и 2 М водным раствором соляной кислоты (250 мл). Отделенную органическую фазу промывали 2 М водным раствором соляной кислоты (250 мл). Основность объединенных водных фаз повышали 1 М водным раствором гидроксида натрия до ~рН 10 и экстрагировали этилацетатом (2×250 мл). Органические вещества промывали солевым раствором (100 мл), сушили при помощи сульфата магния и концентрировали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.78, [М+Н]+ = 525.
Промежуточное соединение 118
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метилбензойная кислота

Метил 5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метилбензоат (промежуточное соединение 117, 32.66 г, 62.3 ммоль) перемешивали в метаноле (300 мл). Добавляли 10 М водный раствор гидроксида натрия (31.1 мл, 311 ммоль) и перемешивали реакционную смесь при 50°С в течение 6 ч. После охлаждения добавляли этилацетат (500 мл) и воду (500 мл) и разделяли фазы. Органическую фазу промывали 1 М водным раствором гидроксида натрия (2×100 мл). Объединенные водные фазы нейтрализовали до ~рН 5-6 с использованием 2 М водного раствора соляной кислоты и экстрагировали этилацетатом (4×500 мл). Объединенные органические слои концентрировали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.65, [М+Н]+ = 511.
Промежуточное соединение 119
Метил-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксибензоат
2-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-2-ол (промежуточное соединение 43,10 г, 21.96 ммоль), метил 2-метокси-5-(4,4,5,5-третраметил1,3,2-диоксаборолан-2-ил)бензоат (промежуточное соединение 78, 9.62 г, 32.9 ммоль), фосфат калия (6.99 г, 32.9 ммоль), XPhos (1.047 г, 2.196 ммоль) и XPhos Pd G2 (1.728 г, 2.196 ммоль) в 1,4-диоксане (100 мл) и воду (33.3 мл) дегазировали (вакуум/азот×3). Реакционную смесь перемешивали в атмосфере азота при 100°С в течение 2 ч. Реакционной смеси давали остыть и разделяли между этилацетатом (250 мл) м 2 М соляной кислотой (200 мл). Органическую фазу промывали 2 М соляной кислотой (100 мл). Основность объединенных водных фаз повышали 1 М водным раствором гидроксида натрия до рН ~10 и экстрагировали этилацетатом (250 мл). Органическую фазу промывали солевым раствором и сушили при помощи сульфата магния. Растворитель выпаривали под вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.63, [М+Н]+ = 541.
Промежуточное соединение 120
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксибензойная кислота
К раствору метил-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксибензоата (промежуточное соединение 119, 13.55 г, 25.06 ммоль) в метаноле (100 мл) добавляли 1 М водный раствор гидроксида натрия (12.53 мл, 125 ммоль) и перемешивали реакционную смесь при 50°С в течение 6 ч. Реакционной смеси давали остыть, затем разбавляли этилацетатом (200 мл) и водой (200 мл), а затем фильтровали. Разделяли фазы. Водную фазу нейтрализовали до рН ~5-6 с использованием 2 М соляной кислоты, а затем экстрагировали с использованием этилацетата (3 раза). К водной фазе добавляли твердый хлорид натрия. Эту смесь экстрагировали этилацетатом (2×150 мл). Объединенные продукты экстракции этилацетатом сушили при помощи сульфата магния. Растворитель выпаривали под вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.57, [М+Н]+ = 527.
Промежуточное соединение 121
Этил 2-(3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)-2-(тетрагидро-2Н-пиран-4-ил)ацетат

В высушенном сосуде перемешивали этил 2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)ацетат (промежуточное соединение 77, 255 мг, 0.481 ммоль) в ТГФ (2 мл) в атмосфере азота при 0°С. Добавляли 4-йодотетрагидро-2Н-пиран (0.06 мл, 0.503 ммоль) и перемешивали смесь при 0°С в течение 1 мин, после чего добавляли LiHMDS (1Mb ТГФ) (1 мл, 1 ммоль). Реакционную смесь перемешивали при 0°С в течение 10 мин, затем давали нагреться до комнатной температуры, а затем нагревали до 40°С в течение 2 дней. Реакционную смесь разбавляли насыщенным водным раствором хлорида аммония (3 мл) и перемешивали в течение 5 мин. Суспензию переносили в делительную воронку с дихлорметаном (20 мл) и водой (20 мл). Разделяли фазы и собирали органический слой. Затем водный слой промывали дополнительным количеством дихлорметана (20 мл), а объединенные органические слои промывали солевым раствором (20 мл), затем фильтровали через гидрофобный спеченный фильтр и выпаривали досуха. Остаток загружали из ДХМ (1 мл) на колонку с силикагелем (40 г) и элюировали смесью 10-60% этанол: этилацетат (3:1, об./об., содержащей 1% триэтиламина) в циклогексане. Содержащие продукт фракции объединяли и выпаривали досуха, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 1.14, [М+Н]+ = 615.
Промежуточное соединение 122
3-йодо-2-метил-N-(3-(1-метил-1Н-имидазол-2-ил)бензил)-5-((тетрагидро-2H-пиран-4-ил)метил)пиразоло[1,5-а]пиримидин-7-амин
Этил2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)-2-(тетрагидро-2Н-пиран-4-ил)ацетат (промежуточное соединение 121, 158 мг, 0.188 ммоль) растворяли в ТГФ (2 мл) и 2 М гидроксиде натрия (вод.) (1 мл, 2 ммоль) и нагревали до 110°С в запаянной пробирке в течение 6 ч. После охлаждения добавляли ДХМ (20 мл) и воду (20 мл), и водную фазу нейтрализовали до рН 7 при помощи 2 М вод. раствора HCl. Разделяли фазы и собирали органический слой. Затем водный слой промывали дополнительным количеством дихлорметана (2×10 мл), а объединенные органические слои пропускали через гидрофобный спеченный фильтр и выпаривали досуха, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 1.07, [М+Н]+ = 543.
Промежуточное соединение 123
N-(3-гидроксипропил)-N-метил-3-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид

3-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензойную кислоту (245 мг, 0.988 ммоль) и HATU (413 мг, 1.086 ммоль) и DIPEA (0.35 мл, 2.004 ммоль) перемешивали в ТГФ (5 мл) в воздушной атмосфере при температуре окружающей среды. Через 10 минут добавляли 3-(метиламино)пропан-1-ол (0.12 мл, 1.234 ммоль) и перемешивали смесь в течение ночи. Смесь разделяли между насыщенным водным раствором бикарбоната натрия (30 мл) и этилацетатом (20 мл). Отделенную водную фазу промывали дополнительным количеством этилацетата (2×20 мл), а объединенные органические вещества промывали солевым раствором (20 мл). Органический слой пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле, используя для элюирования смесь 40-100% этилацетат: этанол (3:1 содержащую 1% триэтиламина) в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.93, [М+Н]+ = 320.
Примеры соединений
Соединение 1
1-(3-(4-метокси-3-(метилсульфонил)фенил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)этанол

3-(4-метокси-3-(метилсульфонил)фенил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-карбальдегид (промежуточное соединение 12, 30 мг, 0.057 ммоль) растворяли в ТГФ (2.5 мл) и охлаждали до 0°С.Добавляли по каплям метилмагния бромид (3 М в диэтиловом эфире) (0.1 мл, 0.3 ммоль) и давали реакционной смеси нагреться до комнатной темп, и перемешивали в течение еще 3 ч. Добавляли дополнительное количество метилмагния бромида (3 М в диэтиловом эфире) (0.1 мл, 0.3 ммоль) и перемешивали реакционную смесь в течение еще 20 ч. Реакцию гасили путем добавления 1 М HCl (10 мл), а затем экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы сушили через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом. Остаток очищали MDAP-хроматографией (метод А), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 0.94, [М+Н]+ = 547.
Соединение 2
2-((5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксифенил)сульфонил)этан-1-ол
Трет-бутил(5-формил-3-(3-((2-гидроксиэтил)сульфонил)-4-метоксифенил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат (промежуточное соединение 20, 100 мг, 0.151 ммоль) в ТГФ (3 мл) перемешивали при 0°С в атмосфере азота, затем одной порцией добавляли 3.4 М метилмагния бромид в 2-метилтетрагидрофуране (0.1 мл). Реакционной смеси давали остыть до комнатной темп, и перемешивали реакционную смесь в атмосфере азота в течение 1 ч. Реакционную смесь гачили водой (10 мл) и перемешивали в атмосфере азота в течение 5 мин. Растворитель удаляли под вакуумом. Неочищенный материал растворяли в ДХМ (20 мл) и разделяли водой (15 мл). Органический слой отделяли, а водную фазу экстрагировали дополнительным количеством ДХМ (2×20 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом. Неочищенный материал растворяли в метаноле (3 мл) и добавляли 4 М HCl в 1,4-диоксане (2 мл, 8 ммоль). Реакционную смесь перемешивали в атмосфере азота в течение 5 ч. Растворитель удаляли под вакуумом. Неочищенный материал растворяли в смеси метанол : ДМСО (2×1 мл, 1:1, об./об.) и очищали MDAP-хроматографией (метод А), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 0.86, [М+Н]+ = 577.
Приведенные ниже соединения получали способом, аналогичным получению соединения 2 с использованием следующих реагентов Гриньяра:
1 М Этилмагния бромид в ТГФ,
0.5 М циклопропилмагния бромид в ТГФ,
1 М изопропилмагния бромид в ТГФ.
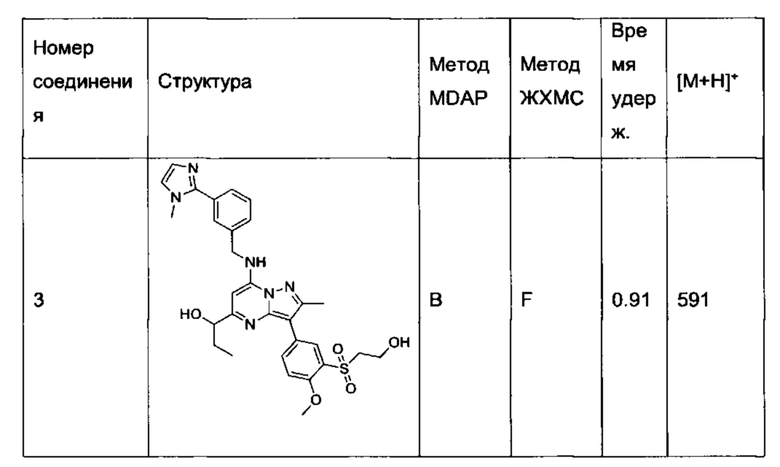

Соединение 6
2-(3-(3-((2-гидроксиэтил)сульфонил)-4-метоксифенил)-2-метил-7-((3-(1-1иетил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-2-ол
2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этан-1-ол (промежуточное соединение 19, 121 мг, 0.354 ммоль), карбонат натрия (87 мг, 0.821 ммоль), PdCl2(dppf) (19 мг, 0.026 ммоль), воду (1 мл) и 2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-2-ол (промежуточное соединение 21, 136 мг, 0.271 ммоль) в изопропиловом спирте (1 мл) объединяли и нагревали при 120°С в микроволновом реакторе в течение 1.5 ч. Реакционную смесь фильтровали через целит (промывая дихлорметаном) и выпаривали досуха. Затем фильтрат разделяли между водой (20 мл) и ДХМ (20 мл). Органическую фазу собирали, а водную промывали дихлорметаном (2×10 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и выпаривали досуха. Остаток растворяли в смеси ДМСО : метанол (2 мл) и очищали MDAP-хроматографией (метод А), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 0.91, [М+Н]+ = 591.
Соединение 7
2-хлор-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N-метилбензамид

2-хлор-N-(3-гидроксипропил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 23, 105 мг, 0.297 ммоль), карбонат натрия (69.6 мг, 0.657 ммоль), PdCl2(dppf)-ДХМ (12.5 мг, 0.015 ммоль), воду (1 мл) и 2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-2-ол (промежуточное соединение 21, 110 мг, 0.219 ммоль) в изопропиловом спирте (1 мл) объединяли и нагревали при 120°С в микроволновом реакторе в течение 2 ч. Добавляли еще 2-хлор-N-(3-гидроксипропил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 23, 105 мг, 0.297 ммоль) и нагревали реакционную смесь при 120°С в микроволновом реакторе в течение 2 ч. Реакционную смесь фильтровали через целит использованием ДХМ (20 мл). Фильтрат промывали водой (20 мл) и экстрагировали водную фазу дополнительным количеством ДХМ (2×20 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и удаляли растворитель при пониженном давлении. Остаток растворяли в смеси метанол : ДМСО (0.6 мл, 1:1, об./об.) и очищали MDAP-хроматографией (метод А). Остаток загружали в ДХМ (2 мл) и пропускали через картридж с силикагелем (1 г), используя для элюирования смесь этилацетат: этанол (3:1, об./об., содержащую 1% триэтиламина). Остаток загружали в ДХМ (2 мл) и очищали хроматографией на силикагеле (4 г), используя для элюирования смесь этилацетат: этанол (3:1, об./об., содержащую 1% триэтиламина) в циклогексане (0%, 2 объема колонки; 0-100%, 5 объемов колонки; 100%, 7 объемов колонки), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 0.98, [М+Н]+ = 602.
Соединение 8
(S)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксифенил)3-гидроксипирролидин-1-ил)метанон

Получали способом, аналогичным получению Соединения 7, используя (S)-(3-гидроксипирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 25), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 0.88, [М+Н]+ = 596.
Соединение 9
5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-2-метокси-N-метилбензамид

N-(3-гидроксипропил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 28, 80 мг, 0.160 ммоль) в изопропиловом спирте (0.5 мл), трет-бутил(5-(1-гидроксиэтил)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1H-имидазол-2-ил)бензил)карбамат (промежуточное соединение 26, 80 мг, 0.136 ммоль), PdCl2(dppf) (9 мг, 0.012 ммоль), карбонат натрия (45 мг, 0.425 ммоль) и воду (0.5 мл) объединяли и нагревали при 120°С в течение 1.5 ч в микроволновом реакторе. Реакционную смесь разбавляли в ДХМ (10 мл) и воде (10 мл) и разделяли фазы. Органический слой собирали, а водный дополнительно промывали дихлорметаном (2×10 мл), и объединенные органические вещества пропускали через гидрофобный спеченный фильтр и выпаривали досуха. Остаток растворяли в смеси ДМСО : метанол (1:1, об./об., 1 мл) и очищали MDAP-хроматографией (метод А). Содержащие продукт фракции объединяли и выпаривали досуха. Остаток очищали колоночной хроматографией на силикагеле, используя для элюирования 50-100% смесь этилацетат: этанол (3:1, об./об., содержащую 1% триэтиламина) в циклогексане, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 0.87, [М+Н]+ = 584.
Следующие соединения получали способом, аналогичным получению соединения 9, используя следующие сложные эфиры бороновой кислоты:
2-хлор-N-(3-гидроксипропил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 23), N-этил-N-(2-гидроксиэтил)-2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 30),

Соединение 12
5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1Н-имидаэол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метокси-N-метил-N-(тетрагидро-2H-пиран-4-ил)бензамид
Трет-бутил(5-(1-гидроксиэтил)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат (промежуточное соединение 26, 70 мг, 0.119 ммоль), 2-метокси-N-метил-N-(тетрагидро-2Н-пиран-4-ил)-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 34, 77 мг, 0.143 ммоль), PdCl2(dppf) (9 мг, 0.012 ммоль), карбонат натрия (41 мг, 0.387 ммоль), изопропиловый спирт (2 мл) и воду (1 мл) объединяли и нагревали в микроволновом реакторе при 120°С в течение 1.5 ч. Реакционную смесь фильтровали с использованием целита, промывали метанолом (100 мл) и удаляли растворитель под вакуумом. Остаток растворяли в ДХМ (20 мл) и разделяли водой (10 мл). Органический слой отделяли, а водную фазу экстрагировали дополнительным количеством ДХМ (2×10 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом. Остаток растворяли в метаноле (2 мл) и добавляли 4 М HCl в 1,4-диоксане (2 мл). Реакционную смесь перемешивали в атмосфере азота при комнатной темп, в течение 5 ч. Растворитель удаляли под вакуумом. Остаток растворяли в воде (15 мл), нейтрализовали 2 М гидроксидом натрия и разделяли дихлорметаном (20 мл). Органический слой отделяли, и водную фазу экстрагировали дополнительным количеством ДХМ (2×20 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и удаляли растворитель под вакуумом. Образец растворяли в смеси метанол : ДМСО (1 мл, 1:1, об./об.) и очищали MDAP-хроматографией (метод В). Растворитель удаляли под вакуумом. Остаток растирали с этилацетатом, затем с диэтиловым эфиром, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 0.94, [М+Н]+ = 610.
Приведенные ниже соединения получали способом, аналогичным получению соединения 12, варьируя органический реакционный растворитель между изопропиловым эфиром 1,4-диоксан, и используя сложные эфиры бороновой кислоты:
N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36),
(S)-(2-хлор-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)(3-гидроксипирролидин-1-ил)метанон (промежуточное соединение 38),
2-хлор-N-этил-N-(2-гидроксиэтил)-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 32),
(S)-N-(1-гидроксипропан-2-ил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 40),

Соединение 17
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид
N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36, 542 мг, 0.813 ммоль), 2-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-2-ол (промежуточное соединение 43, 333 мг, 0.622 ммоль), PdCl2(dppf) (47 мг, 0.064 ммоль), карбонат натрия (198 мг, 1.865 ммоль), 1,4-диоксан (3 мл) и воду (1 мл) объединяли и нагревали при 120°С в течение 1.5 ч в микроволновом реакторе. Добавляли N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36, 262 мг, 0.393 ммоль) и PdCl2(dppf) (20 мг, 0.027 ммоль) и нагревали реакционную смесь при 120°С в течение 1.5 ч в микроволновом реакторе. Реакционную смесь разбавляли в ДХМ (20 мл) и воде (20 мл) и разделяли фазы. Органический слой собирали, а водный дополнительно промывали дихлорметаном (2×20 мл), и пропускали объединенные органические вещества через гидрофобный спеченный фильтр, затем выпаривали досуха. Остаток растворяли в смеси ДМСО : метанол (4 мл) и очищали MDAP-хроматографией (метод А). Содержащие продукт фракции объединяли и выпаривали досуха. Остаток очищали колоночной хроматографией на силикагеле, используя для элюирования 20-100% смесь этилацетат: этанол (3:1, об./об., содержащую 1% триэтиламина) в циклогексане. Содержащие продукт фракции объединяли, выпаривали досуха, и затем сушили на высоковакуумной линии на протяжении 3 дней, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 0.96, [М+Н]+ = 582.
Следующие соединения получали способом, аналогичным получению соединения 17, используя либо 1,4-диоксан, либо изопропиловый спирт в качестве реакционного растворителя, и следующие сложные эфиры бороновой кислоты:
(S)-(2-хлор-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)(3-гидроксипирролидин-1-ил)метанон (промежуточное соединение 38),
N-этил-N-(2-гидроксиэтил)-2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 30),
(S)-N-(1-гидроксипропан-2-ил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 40),
N-(тетрагидро-2Н-пиран-4-ил)-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 34),
(S)-N-(1-гидроксипропан-2-ил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 45),
2-метокси-N,N-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 47),
(R)-(2-хлор-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)(2-(гидроксиметил)пирролидин-1-ил)метанон (промежуточное соединение 49),
(R)-(2-(гидроксиметил)пирролидин-1-ил)(2-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 51),
N-(2-гидроксиэтил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 53),
(3-гидрокси-3-метилпирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 55),
(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)(1,4-оксазепан-4-ил)метанон (промежуточное соединение 57),
(S)-(3-гидроксипирролидин-1-ил)(2-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 59),
(S)-(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)(3-метилморфолино)метанон (промежуточное соединение 61),
(3-гидрокси-3-метилпирролидин-1-ил)(2-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 63),
N-(3-гидроксипропил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 28)
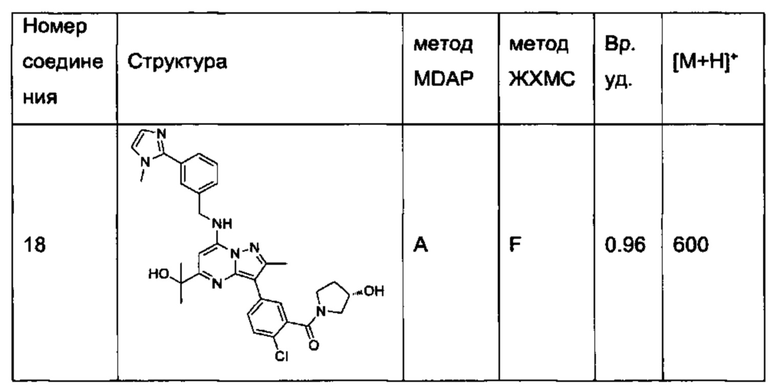

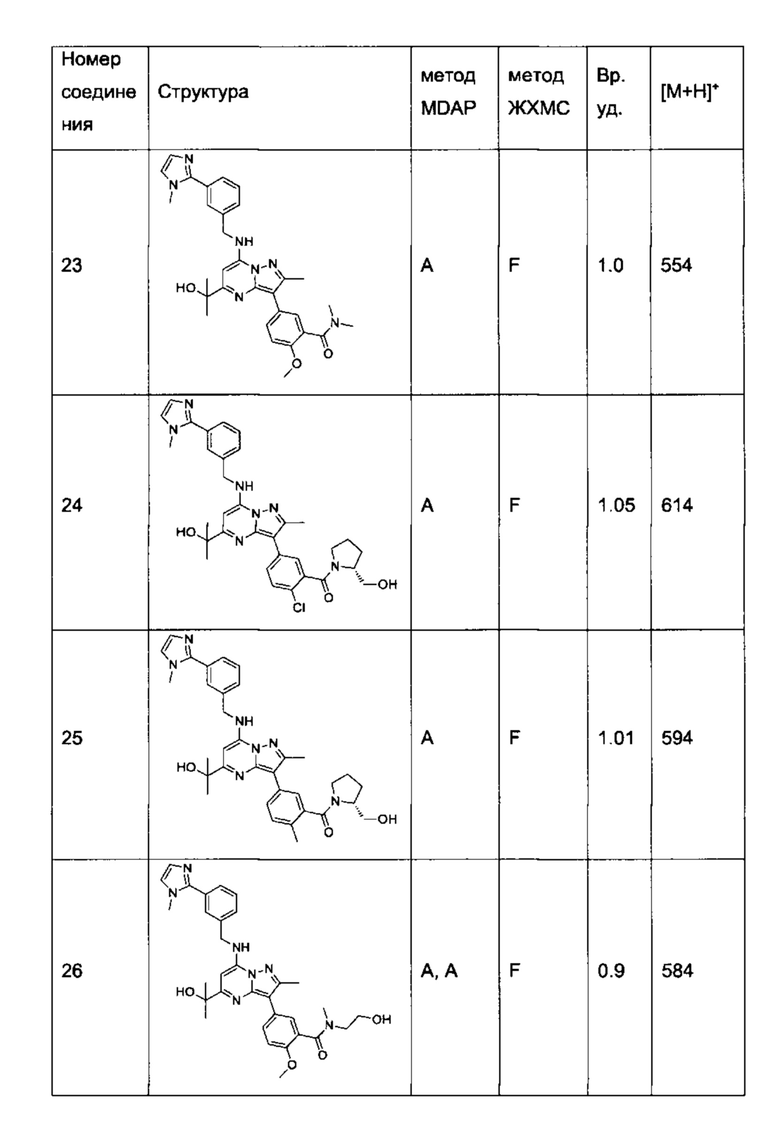
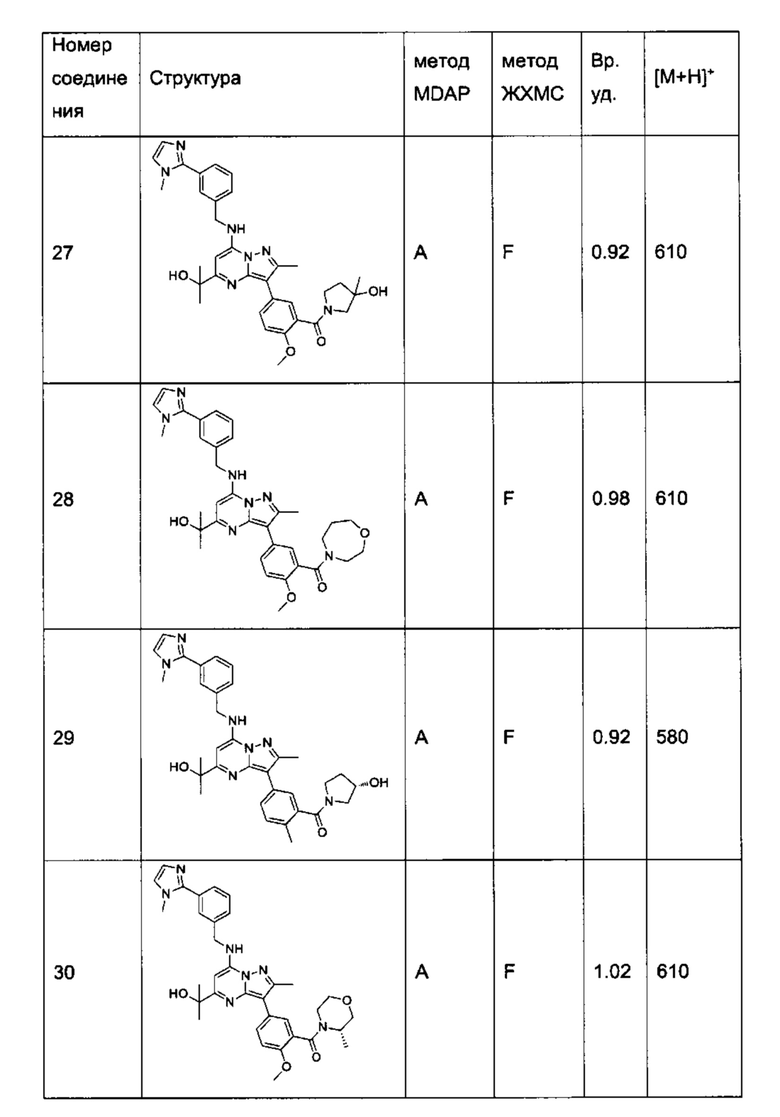

Следующие соединения получали способом, аналогичным получению соединения 17, используя промежуточное соединение 65, и либо 1,4-диоксан, либо изопропиловый спирт в качестве реакционного растворителя, и температуру между 100°С и 120°С, и следующие сложные эфиры бороновой кислоты:
N-(3-гидроксипропил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 28),
N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36),
(R)-(2-(гидроксиметил)пирролидин-1-ил)(2-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 51).
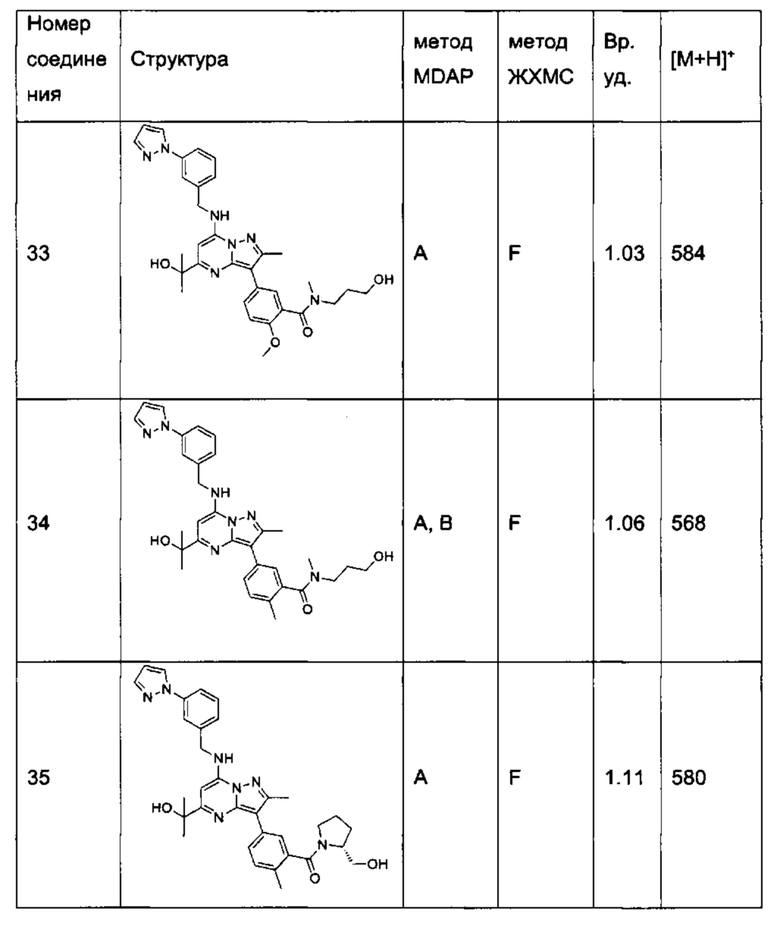
Следующие соединения получали способом, аналогичным получению соединения 17, используя следующие эфиры бороновой кислоты:
(R)-(2-(гидроксиметил)пирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 67).
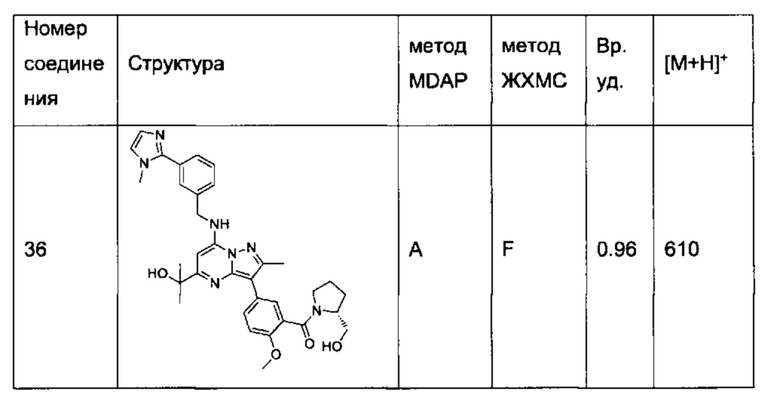
Альтернативный способ получения соединения 17 - 5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамида

5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метилбензойную кислоту (промежуточное соединение 118, 12 г, 23.5 ммоль), 3-(метиламино)пропан-1-ол (2.81 мл, 29.4 ммоль) и DIPEA (8.21 мл, 47.0 ммоль) перемешивали в ТГФ (100 мл). Добавляли HATU (11.17 г, 29.4 ммоль) и ДМФА (10 мл) и перемешивали смесь в атмосфере азота при комнатной температуре в течение 3 ч. Смесь разделяли между этилацетатом (400 мл) и насыщенным водным раствором бикарбоната натрия (400 мл). Водную фазу снова экстрагировали этилацетатом (200 мл). Объединенные органические фазы промывали солевым раствором (200 мл), сушили при помощи сульфата магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле, используя для элюирования 0-25% этанол в этилацетате (5 объемов колонки) и 25% этанол в этилацетате (7 объемов колонки). Фракции, содержащие продукт, объединяли, выпаривали под вакуумом и очищали тем же хроматографическим методом до достижения содержания материала >97.5% (согласно ВЭЖХ). Остаток растирали с диэтиловым эфиром и выпаривали растворитель под вакуумом. Добавляли этилацетат (40 мл) и перемешивали смесь при комнатной температуре в течение 5 дней.
Полученное белое твердое вещество собирали фильтрацией, промывали этилацетатом (25 мл) и сушили под вакуумом при 40°С, в результате чего получали указанное в заголовке соединение. ЖХМС (метод М): время удерживания = 2.49, [М+Н]+ = 582. δн (400 МГц, d6-ДМСО) (Смесь ротаметров) 8.53 (t, J=6.5 Гц, 1Н), 7.73 (s, 1Н), 7.71-7.67 (m, 0.75Н), 7.65 (s, 0.75Н), 7.61 (d, J=1.3 Гц, 0.5Н), 7.59-7.54 (m, 1Н), 7.48-7.42 (m, 2Н), 7.33-7.27 (m, 1Н), 7.22 (d, J=1.0 Гц, 1Н), 6.94 (d, J=1.0 Гц, 1Н), 6.45 (s, 1Н), 5.18 (s, 1Н), 4.70 (d, J=6.6 Гц, 2Н), 4.47 (t, J=5.3 Гц, 0.5Н), 4.34 (t, J=4.9 Гц, 0.5Н), 3.70 (s, 3Н), 3.56-3.46 (m, 2Н), 3.28-3.20 (m, 2Н), 3.00 (s, 1.5Н), 2.82 (s, 1.5Н), 2.58 (s, 3Н), 2.21 (арр. d, J=5.1 Гц, 3Н), 1.79-1.70 (m, 1Н), 1.66-1.57 (m, 1Н), 1.39 (s, 6Н) ppm. δc (151 МГц, d6-ДМСО) (Смесь ротамеров) 170.3, 170.2, 169.0, 150.3, 150.2, 146.39, 146.36, 146.3, 145.4, 138.6, 137.0, 136.9, 130.8, 130.14, 130.05, 130.0, 128.6, 127.5, 127.4, 126.89, 126.86, 126.81, 124.84, 124.79, 123.4, 104.72, 104.67, 81.55, 81.53, 72.39, 72.38, 58.4, 58.0, 47.4, 44.4, 43.5, 36.3, 34.3, 31.8, 31.0, 30.1, 30.0, 18.2, 18.1, 14.7, 14.6 ppm. Масс-спектроскопия высокого разрешения (ионизация электроспреем) расч. для C33H39N7O3+Н+ 582.3193, эксп. 582.3190 [М+Н]+.
Альтернативный способ получения соединения 22
(S)-N-(1-гидроксипропан-2-ил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N,2-диметилбензамид
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метилбензойную кислоту (промежуточное соединение 118, 7.55 г, 14.79 ммоль), (S)-2-(метиламино)пропан-1-ол (1.977 г, 22.18 ммоль) и DIPEA (5.17 мл, 29.6 ммоль) перемешивали в ТГФ (50 мл). Добавляли ДМФА (2.5 мл) и HATU (7.03 г, 18.48 ммоль) и перемешивали в атмосфере азота при комнатной температуре в течение 20 ч. Смесь разделяли между этилацетатом (200 мл) и насыщенным водным раствором бикарбоната натрия (200 мл). Водную фазу экстрагировали с использованием этилацетата (100 мл). Объединенные органические фазы промывали солевым раствором (100 мл), сушили при помощи сульфата магния и удаляли растворитель под вакуумом. Остаток подвергали хроматографии на колонке с силикагелем с концевым кэпированием амином (KP-NH), используя для элюирования 0-100% этилацетат в циклогексана. Затем смесь очищали обращенно-фазовой хроматографией, используя для элюирования 5-50% ацетонитрил (содержащий аммиак) в воде (10 мМ бикарбонат аммония/аммиак). Полученный остаток растирали в метаноле, а затем диэтиловом эфире (50 мл), в котором смесь быстро перемешивали в течение 30 минут. Растворитель удаляли под вакуумом, растворяли остаток в этилацетате (70 мл) и перемешивали с высокой скоростью в течение 24 ч. Твердое вещество собирали фильтрацией, промывали этилацетатом, сушили под вакуумом при 40°С. ЖХМС (метод М): время удерживания = 2.56, [М+Н]+ = 582. δн (400 МГц, d6-ДМСО) (Смесь ротамеров) 8.58-8.49 (m, 1Н), 7.81 (d, J=8.1 Гц, 0.3Н), 7.73 (s, 1Н), 7.71-7.62 (m, 1Н), 7.59-7.51 (m, 1.7Н), 7.49-7.42 (m, 2Н), 7.32-7.25 (m, 1Н), 7.22 (s, 1Н), 6.97-6.92 (m, 1Н), 6.49-6.42 (m, 1Н), 5.17 (s, 1Н), 4.80 (t, J=5.3 Гц, 0.7Н), 4.76 (t, J=5.5 Гц, 0.3Н), 4.70 (d, J=6.2 Гц, 2Н), 3.74-3.64 (m, 3.5Н), 3.64-3.57 (m, 0. 5Н), 3.53-3.39 (m, 1.3Н), 3.29-3.23 (m, 0.7Н), 2.90-2.84 (m, 2Н), 2.68 (s, 1Н), 2.60-2.56 (m, 3Н), 2.25-2.18 (m, 3Н), 1.41-1.35 (m, 6Н), 1.10 (d, J=7.0 Гц, 1Н), 1.06-1.00 (m, 2Н) ррт. δс (176 МГц, d6-ДМСО, 120°С) 170.2, 168.3, 149.8, 146.1, 146.0, 145.0, 137.9, 137.2, 130.7, 130.2, 129.3, 128.0, 127.1, 126.6, 126.50, 126.47, 124.5, 122.6, 104.8, 81.2, 71.8, 62.0, 44.5, 33.5, 29.5, 17.5, 13.8 ppm. МС высокого разрешения (ионизация электрораспылением), расч. для C33H39N7O3+Н+ 582.3193, эксп.582.3195 [М+Н]+.
Альтернативный способ получения соединения 36
(R)-(2-(Гидроксиметил)пирролидин-1-ил)(5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксифенил)метанон
К раствору/суспензии 5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксибензойной кислоты (промежуточное соединение 120, 3.0 г, 5.7 ммоль), (R)-пирролидин-2-илметанола (0.576 г, 5.7 ммоль) и DIPEA (1.990 мл, 11.39 ммоль) в ТГФ (100 мл) добавляли HATU (2.71 г, 7.12 ммоль). Реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 20 ч. Реакционную смесь разделяли между этилацетатом (200 мл) и насыщенным раствором бикарбоната натрия (200 мл). Органическую фазу промывали солевым раствором (100 мл) и сушили при помощи сульфата магния.
Растворитель удаляли под вакуумом. Остаток растворяли в ДХМ, наносили на картридж с силикагелем (120 г) и элюировали 0 - 25% этанолом в этилацетате (5 объемов колонки) и 25% этанолом в этилацетате (6 объемов колонки). Нужные фракции объединяли и выпаривали под вакуумом. Остаток растворяли в смеси ДМСО: метанол (1:1 об./об.) и наносили на картридж C18 (120 г), пре-кондиционированный до 5% ацетонитрила (содержащего аммиак) в воде (10 мМ бикарбоната аммония, содержащего аммиак). Затем осуществляли элюирование градиентом 5-40% ацетонитрила (содержащего аммиак) в воде (10 мМ бикарбонат аммония, содержащий аммиак). Градиент поддерживали с использованием 29% ацетонитрила (содержащего аммиак) в воде (10 мМ бикарбонат аммония, содержащий аммиак), пока элюировали продукт. Нужные фракции объединяли и выпаривали под вакуумом. Остаток растирали с диэтиловым эфиром и выпаривали растворитель. К остатку добавляли этилацетат (25 мл) и метанол (1 мл). Смесь подвергали воздействию температурных циклов от комнатной температуры до 45°С и обратно 6 раз, а затем оставляли при перемешивании при комнатной темп, на 18 ч. Полученное белое твердое вещество собирали фильтрацией и промывали этилацетатом (25 мл). Твердое вещество сушили под вакуумом при 40°С в течение 20 ч, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.59, [М+Н]+ = 610. δн (700 МГц, ДМСО-d6, 120°С) 7.94 (br s, 1Н), 7.75 (s, 1Н), 7.75-7.72 (m, 1Н), 7.65 (br s, 1Н), 7.58 (brd, J=7.3 Гц, 1Н), 7.50-7.47 (m, 1Н), 7.47-7.42 (m, 1Н), 7.16-7.14 (m, 1Н), 7.16-7.13 (m, 1Н), 6.95 (s, 1Н), 6.44 (s, 1Н), 4.74 (s, 2Н), 4.18 (br s, 1Н), 3.85 (s, 3Н), 3.75-3.71 (m, 1Н), 3.70 (s, 3Н), 3.51-3.42 (m, 1Н), 3.39-3.15 (m, 2Н), 2.56 (s, 3Н), 1.96-1.71 (m, 2Н), 2.04-1.68 (m, 2Н), 1.44 (s, 6Н) ppm. δC-ЯМР (176 МГц, ДМСО-d6, 120°С) 168.11, 166.71, 152.36, 149.48, 146.05, 146.01, 144.77, 137.90, 130.71, 128.70, 127.90, 127.35, 127.05, 126.65, 126.56, 126.44, 126.43, 125.52, 122.51, 111.87, 104.61, 81.02, 71.72, 61.77, 58.17, 55.49, 47.62, 44.49, 33.51, 29.45, 26.86, 23.05, 13.61 ppm. МС высокого разрешения (ионизация электрораспылением), расч. для C34H39N7O4+Н+ 610.3063, эксп. 610.3141 [М+Н]+.
Альтернативный способ получения соединения 21 5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метокси-N-метил-N-(тетрагидро-2Н-пиран-4-ил)бензамид
К раствору/суспензии 5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксибензойной кислоты (промежуточное соединение 120, 2.35 г, 4.46 ммоль) и DIPEA (1.6 мл, 9.16 ммоль) в ТГФ (50 мл) добавляли HATU (2.35 г, 6.18 ммоль). Смесь перемешивали при комнатной температуре в течение 10 мин и добавляли N-метилтетрагидро-2Н-пиран-4-амин (0.514 г, 4.46 ммоль). Смесь перемешивали в атмосфере азота при комнатной темп, в течение 16 ч. Реакционную смесь разделяли между этилацетатом (100 мл) нас. вод. раствором бикарбоната натрия (100 мл). Водную фазу экстрагировали второй раз этилацетатом (100 мл). Объединенные органические фазы промывали солевым раствором (100 мл) и пропускали через гидрофобный спеченный фильтр. Растворитель удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (340 г). Это соединение растворяли в минимальном количества ДХМ (+ несколько капель метанола), загружали поверх колонки путем инъекции, затем элюировали с использованием 0-25% этанола в этилацетате (6 объемов колонки), а затем обрабатывали 25% этанолом в этилацетате (6 объемов колонки). Фракции, содержащие целевой продукт, объединяли и концентрировали при пониженном давлении. Приблизительно половину материала растворяли в смеси ДМСО: метанол (1:1 об./об.) и наносили на картридж C18 (120 г), пре-кондиционированный до 5% ацетонитрила (содержащего аммиак) в воде (10 мМ бикарбонат аммония, содержащий аммиак), затем элюировали градиентом 5-20% ацетонитрила (с аммиаком) в воде (10 мМ бикарбонат аммония с аммиаком) (6 объемов колонки), а затем градиентом 20-60% ацетонитрила (с аммиаком) в воде (10 мМ бикарбонат аммония с аммиаком) (6 объемов колонки). Затем удерживали градиент при 47.5% ацетонитрил (с аммиаком) в воде (10 мМ бикарбонат аммония с аммиаком) пока элюировался продукт. Остальной материал очищали аналогичным образом, используя градиент 25-55% ацетонитрила (с аммиаком) в воде (10 мМ бикарбонат аммония с аммиаком), бикарбонат/NH3). Градиент удерживали на уровне 45% ацетонитрила (с аммиаком) в воде (10 мМ бикарбонат аммония с аммиаком) пока элюировался продукт. Фракции, содержащие чистый продукт, от обеих процедур очистки объединяли, концентрировали при пониженном давлении, затем сушили под вакуумом в течение 24 ч, в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания = 0.65, [М+Н]+ = 624. δн (600 МГц, d6-ДМСО) (Смесь ротамеров) 8.54-8.49 (m, 1Н), 7.77-7.72 (m, 2Н), 7.63 (d, J=1.8 Гц, 0.5Н), 7.59-7.54 (m, 1Н), 7.53 (d, J=2.2 Гц, 0.5Н), 7.47-7.43 (m, 2Н), 7.22 (s, 1Н), 7.18-7.13 (m, 1Н), 6.94 (s, 1Н), 6.43 (s, 1Н), 5.17 (s, 1Н), 4.69 (d, J=6.2 Гц, 2Н), 4.64-4.58 (m, 0.5Н), 3.98-3.91 (m, 1Н), 3.90-3.85 (m, 0.5Н), 3.82-3.75 (m, 3.5Н), 3.70 (s, 3Н), 3.57-3.50 (m, 0.5Н), 3.43 (t, J=11.2 Гц, 1Н), 3.12 (t, J=11.2 Гц, 0.5Н), 3.00 (t, J=11.6 Гц, 0.5Н), 2.88 (s, 1.5Н), 2.69 (s, 1.5H), 2.56 (ар. d, J=15.8 Гц, 3Н), 1.89-1.72 (m, 2H), 1.59-1.51 (m, 1.5H), 1.42 (d, J=11.4 Гц, 0.5H), 1.39-1.33 (m, 6H) ppm. δс (151 МГц, d6-ДМСО) (Смесь ротамеров) 168.80, 168.77, 168.1, 167.9, 152.50, 152.47, 150.0, 149.9, 146.4, 146.34, 146.26, 145.18, 145.16, 138.6, 130.8, 129.2, 128.9, 128.6, 127.5,126.89, 126.87, 126.80, 126.7, 126.6, 126.5, 125.9, 123.4, 111.6, 111.4, 104.7, 104.5, 81.4, 81.3, 72.4, 72.3, 66.5, 66.3, 66.2, 55.7, 55.4, 54.9, 54.8, 49.4, 44.4, 34.3, 30.5, 30.3, 30.09, 30.07, 30.0, 29.2, 26.6, 14.5, 14.4 ppm. MC высокого разрешения (ионизация электрораспылением), расч. для C35H41N7O4+H+ 624.3220, эксп. 624.3294 [М+Н]+.
Соединение 37
2-Хлор-N-(2-гидроксиэтил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-метилбензамид
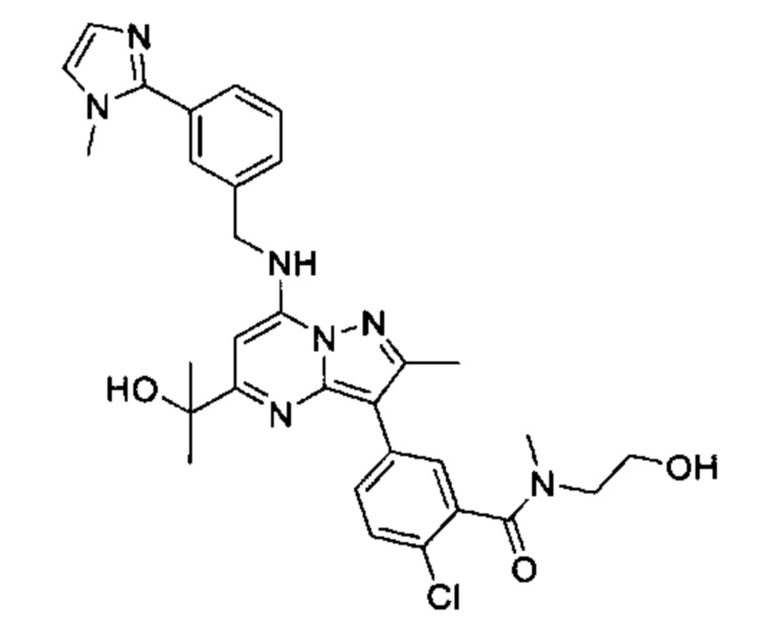
В сосуд для микроволновой обработки загружали 2-(3-бром-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-2-ол (промежуточное соединение 43, 305 мг, 0.636 ммоль), XPhos (32 мг, 0.067 ммоль), XPhos Pd G2 (53 мг, 0.067 ммоль) и трикалийфосфат (443 мг, 2.087 ммоль). 2-хлор-N-(2-гидроксиэтил)-N-метип-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 69), 446 мг, 0.788 ммоль) растворяли в 1,4-диоксане (7.5 мл) и добавляли в сосуд для микроволновой обработки, после чего добавляли воду (2.5 мл). Сосуд запечатывали и нагревали при микроволновой обработке при 100°С в течение 1.5 ч. Реакционную смесь разбавляли водой (25 мл) и разделяли дихлорметаном (25 мл). Органическую фазу собирали, а водную фазу промывали дихлорметаном (4×25 мл). Органические фазы объединяли и пропускали растворитель через гидрофобный спеченный фильтр, а затем через второй гидрофобный спеченный фильтр со слоем флоризила (глубиной 2 см). Слой флоризила промывали смесью этилацетатом: этанол (3:1, содержащую 1% триэтиламина) (25 мл). Фильтрат выпаривали досуха. Остаток растворяли в минимальном количестве смеси ДМСО: метанол (1:1, об./об.) и очищали MDAP-хроматографией (метод А). Нужные фракции объединяли и удаляли растворитель. Остаток очищали колоночной хроматографией на силикагеле, используя для элюирования 30-100 градиент этилацетат: этанол (3:1, содержащую 1% триэтиламина) в циклогексане. Нужные фракции объединяли и удаляли растворитель. Остаток растирали с минимальным количеством диэтилового эфира, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания = 0.96, [М+Н]+ = 588.
Следующие соединения получали способом, аналогичным получению соединения 37, используя следующие бороновые эфиры:
N-Этил-N-(2-гидроксиэтил)-2-метил-5-(4,4,5,5-третраметил1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 73),
(R)-N-(1-гидроксипропан-2-ил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 86),
N-(1-гидроксипропан-2-ил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 88),
(3-гидроксипирролидин1-ил)(2-метил-5-(4,4,5,5-третраметил1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 90),
N-(1-гидроксипропан2-ил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 92)
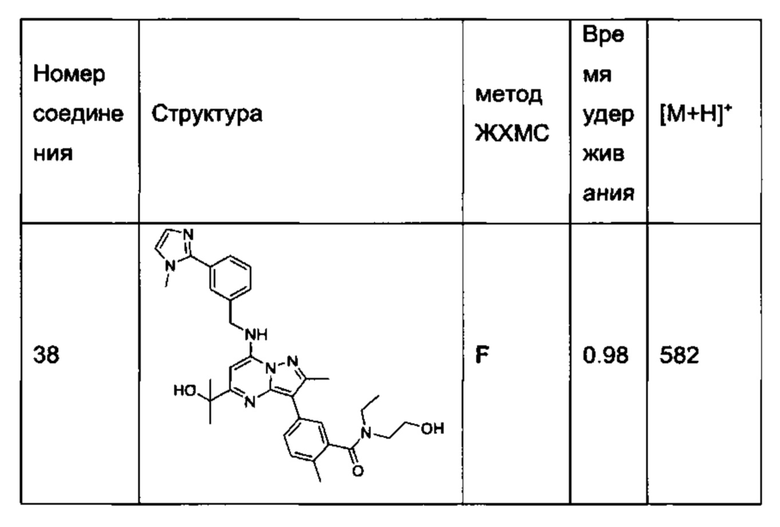
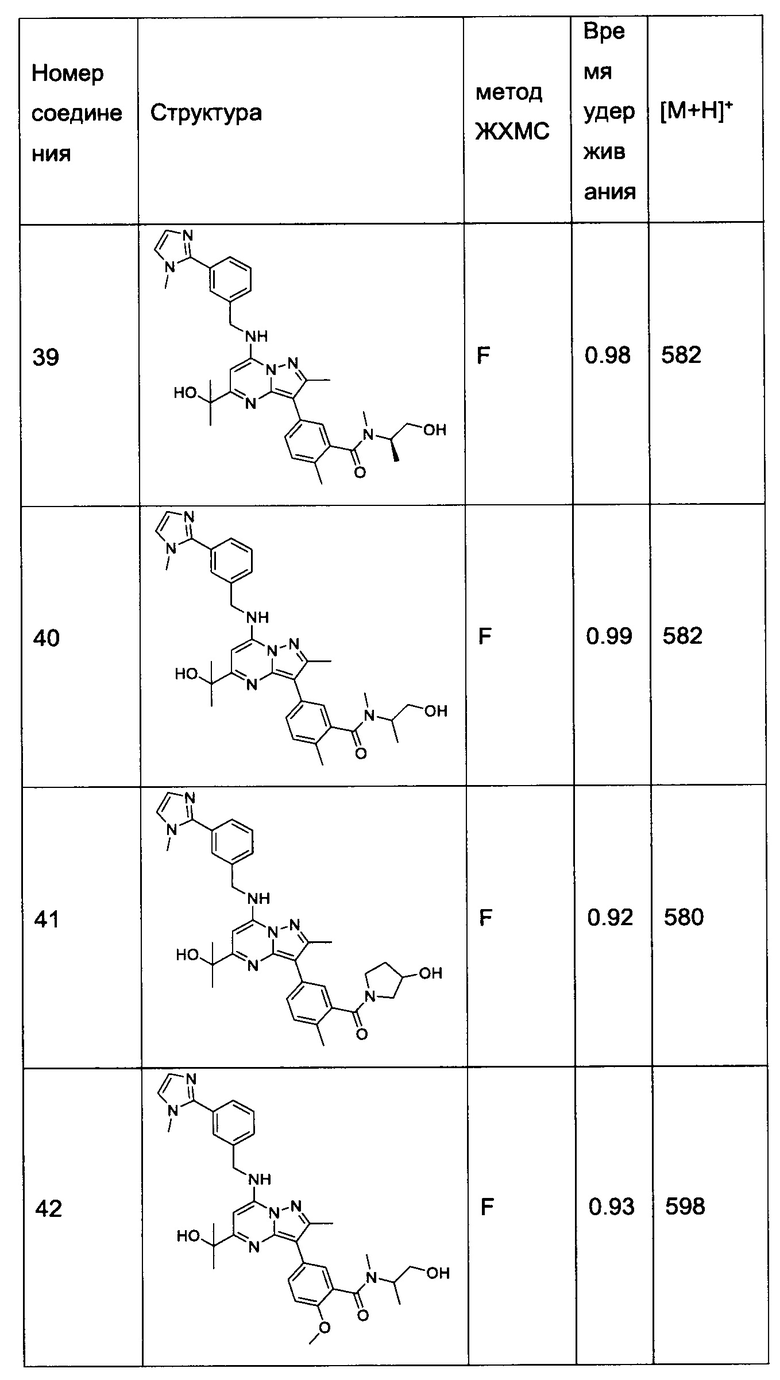
Следующие соединения получали способом, аналогичным получению соединения 17, используя либо 1,4-диоксан, либо изопропиловый спирт в качестве реакционного растворителя и температуру от 100°С до 140°С, и следующие бороновые эфиры:
N-(2-гидроксиэтил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 71),
(2-(Гидроксиметил)пирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 84),
N,2-Диметил-N-(тетрагидро-2Н-пиран-4-ил)-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 96),
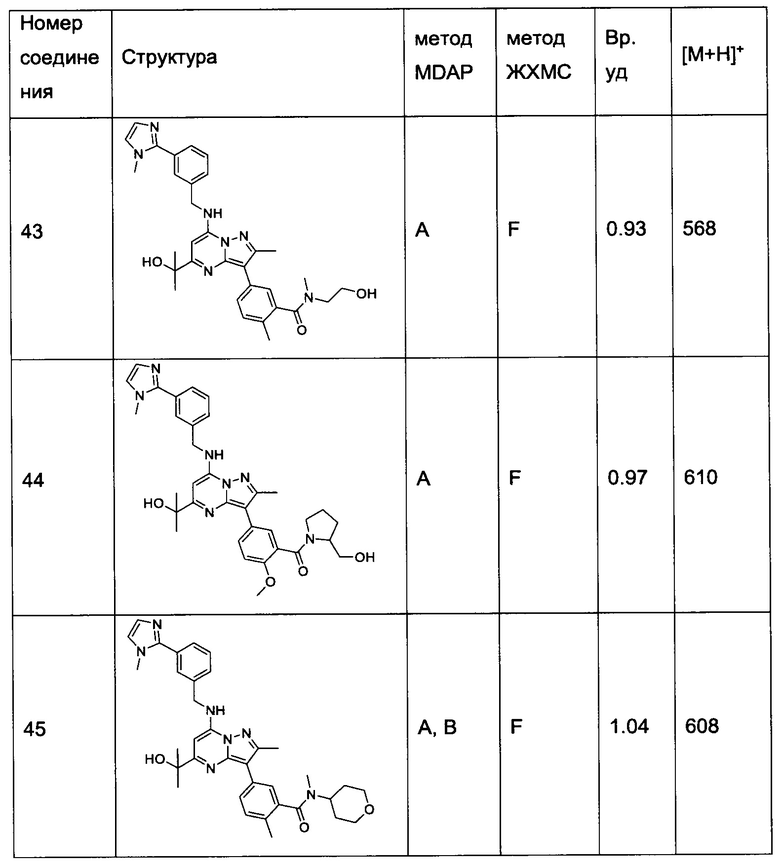
Следующие соединения получали способом, аналогичным получению соединения 17, используя промежуточное соединение 65, и либо 1,4-диоксан, либо изопропиловый спирт в качестве реакционного растворителя, и температуру от 100°С до 140°С, и следующие бороновые эфиры:
N-(2-гидроксиэтил)-2-метокси-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 53),
N-(2-гидроксиэтил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 71),
2-хлор-N-(2-гидроксиэтил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 69),
(R)-(2-(гидроксиметил)пирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 67),
(S)-(3-гидроксипирролидин-1-ил)(2-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 59),
(3-гидрокси-3-метилпирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 55),
(S)-(3-гидроксипирролидин-1-ил)(2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)метанон (промежуточное соединение 25),
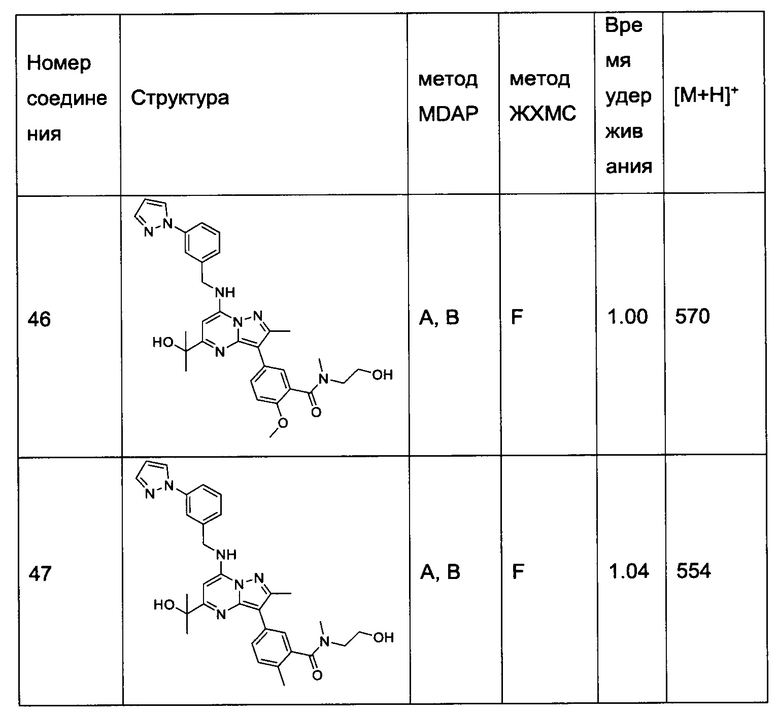
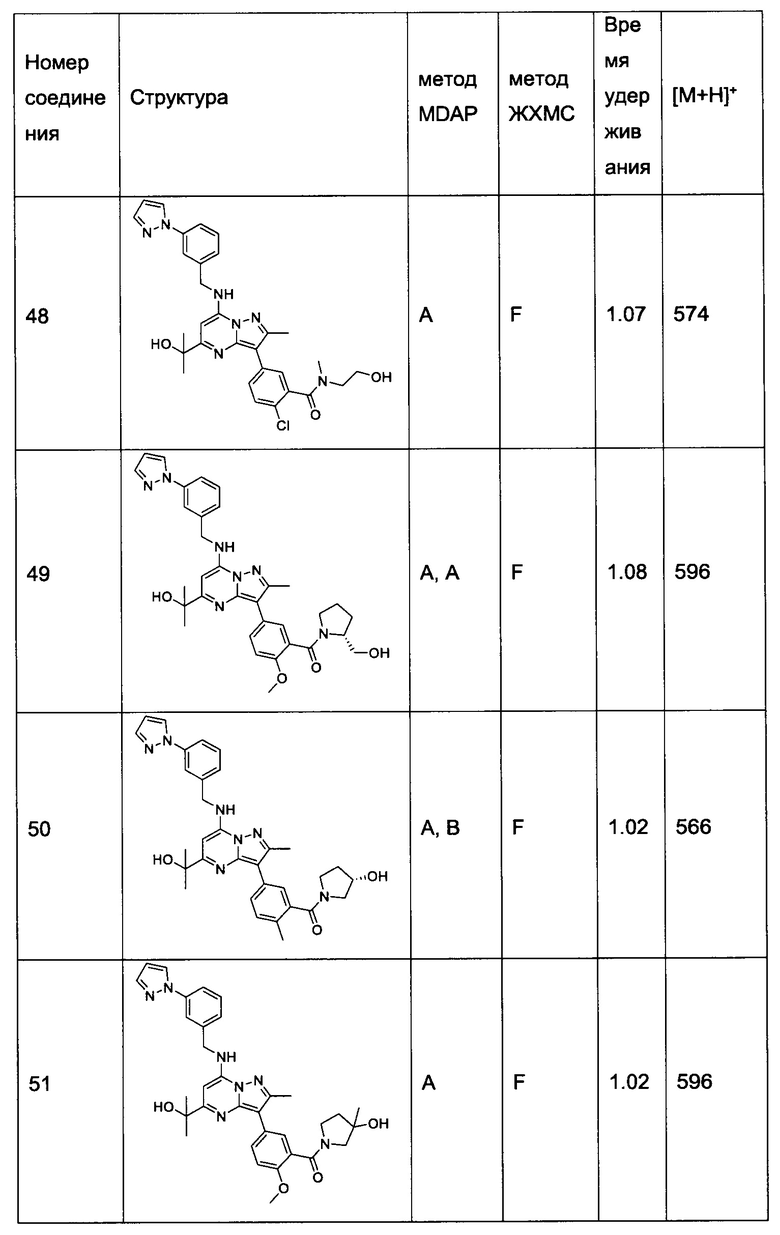
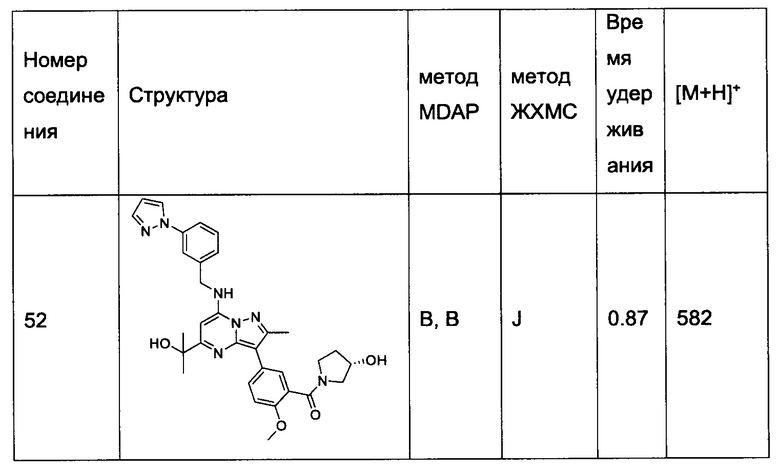
Соединение 53
2-(3-(3-((2-гидроксиэтил)сульфонил)-4-метоксифенил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол
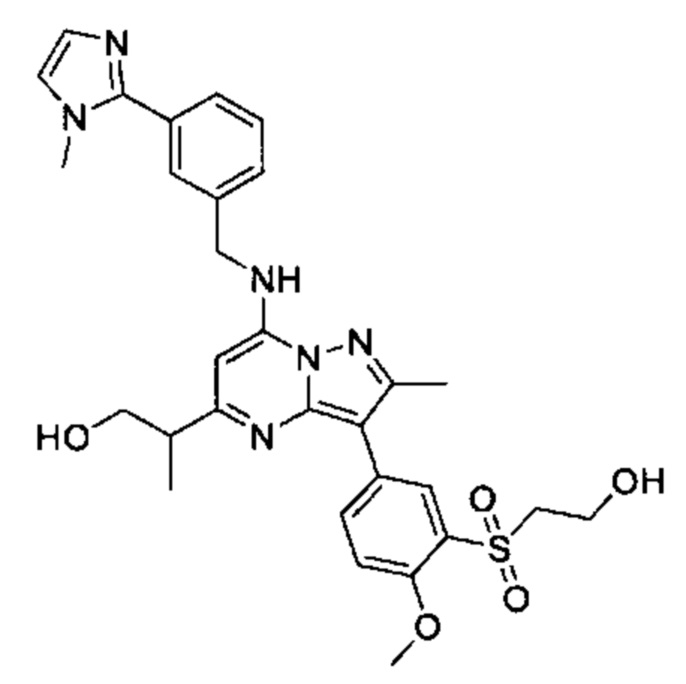
Получали аналогично соединению 37, используя 2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол (промежуточное соединение 81) и 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этанол (промежуточное соединение 19), в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.53, [М+Н]+=591.
Соединение 54
5-(5-(1-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид

Получали способом, аналогичным получению Соединения 17, используя 2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол (промежуточное соединение 81) и N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.92, [М+Н]+=582.
Соединение 55
5-(5-(1-гидрокси-2-метилпропан-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид
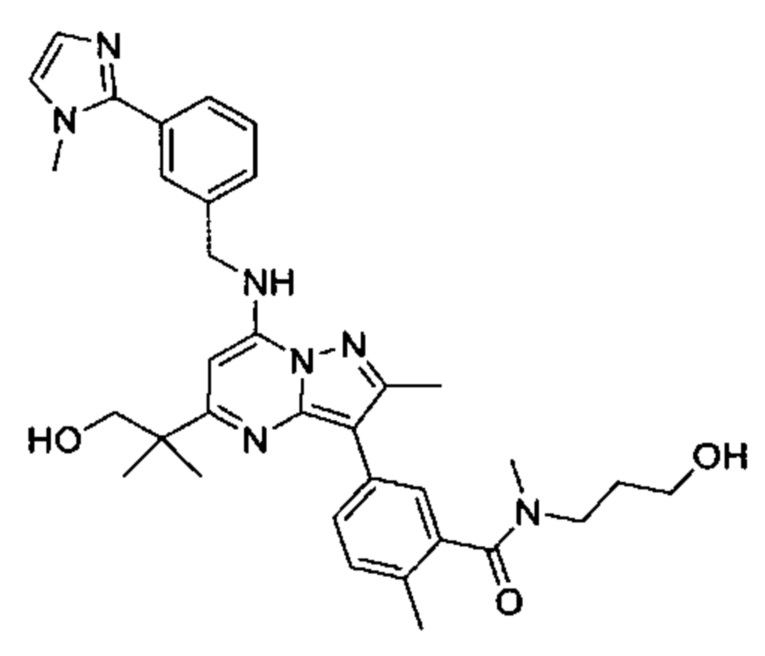
Получали способом, аналогичным получению Соединения 17, используя 2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)-2-метилпропан-1-ол (промежуточное соединение 82) и N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.99, [М+Н]+=596.
Соединение 56
2-(3-(3-((2-гидроксиэтил)сульФонил)-4-метоксисЬенил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)-2-метилпропан-1-ол
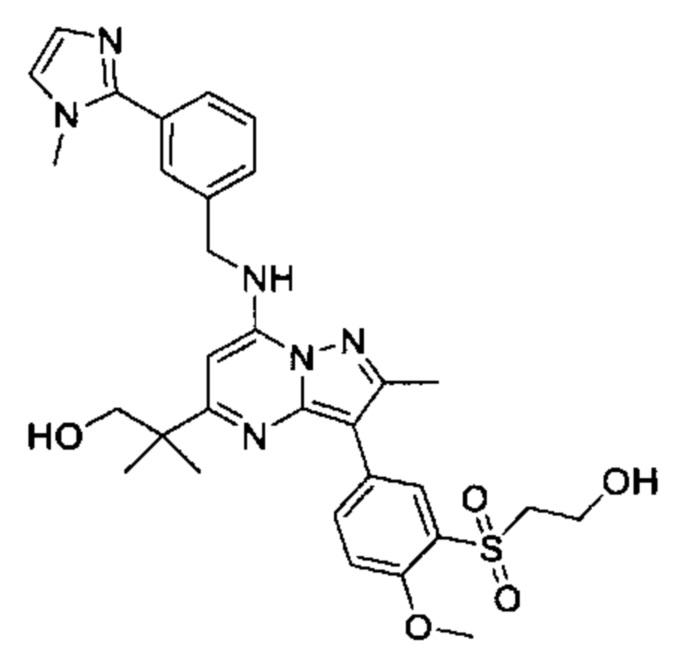
Получали способом, аналогичным получению Соединения 17, используя 2-(3-йодо-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)-2-метилпропан-1-ол (промежуточное соединение 82) и 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этанол (промежуточное соединение 19), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.95, [М+Н]+=605.
Соединение 57
2-хлор-5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N-метилбензамид, изомер 1
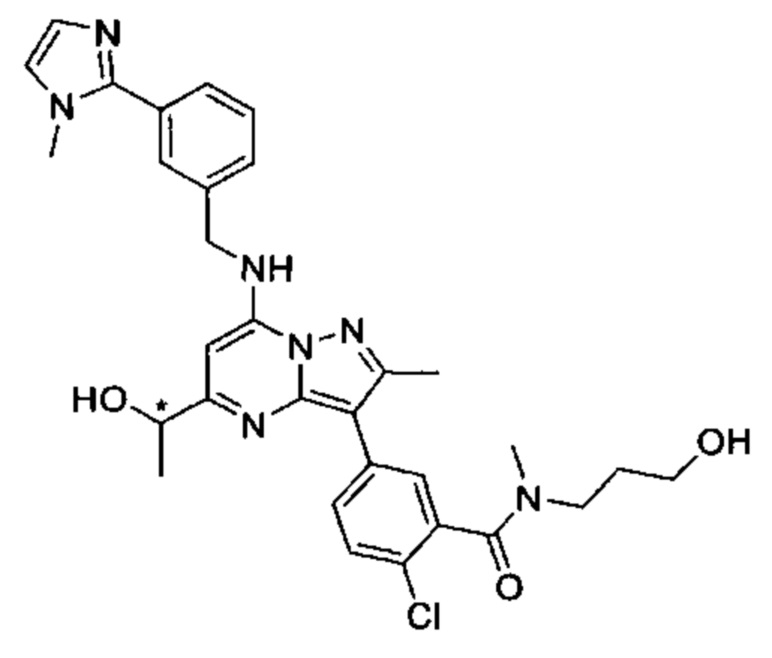
Получали способом, аналогичным получению Соединения 37, используя трет-бутил(5-(1-гидроксиэтил)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат, изомер 1 (промежуточное соединение 93) и 2-хлор-N-(3-гидроксипропил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 23), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.95, [М+Н]+=588.
Соединение 58
2-хлор-5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N-метилбензамид, изомер 2

Получали аналогично соединению 37, используя трет-бутил(5-(1-гидроксиэтил)-3-йодо-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат, изомер 2 (промежуточное соединение 94) и 2-хлор-N-(3-гидроксипропил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 23), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.95, [М+Н]+=588.
Соединение 59
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N,2-диметил-N-(тетрагидрофуран-3-ил)бензамид
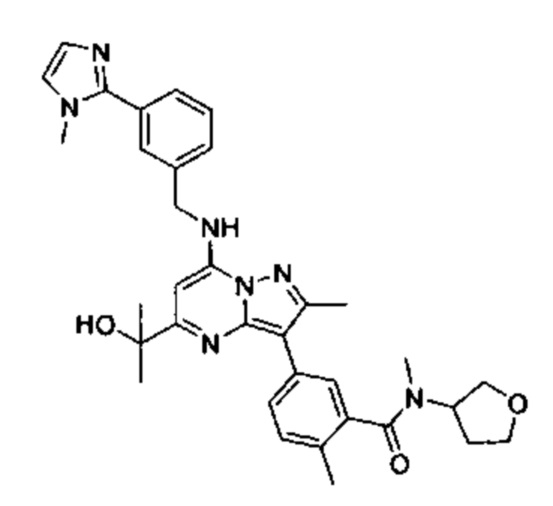
Раствор 5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метилбензойной кислоты (промежуточное соединение 118, 49 мг, 0.096 ммоль), HATU (40.1 мг, 0.106 ммоль) и DIPEA (0.034 мл, 0.192 ммоль) в ДМФА (0.4 мл) перемешивали в течение 20 мин при комнатной темп. К реакционной смеси добавляли N-Метилтетрагидрофуран-3-амин (0.013 мл, 0.115 ммоль) и перемешивали в течение 1 ч при комнатной температуре, затем оставляли на ночь. Реакционную смесь разбавляли метанолом (0.6 мл) и очищали MDAP-хроматографией (метод А). Нужные фракции объединяли, концентрировали, растирали с диэтиловым эфиром, затем сушили в высоком вакууме, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=1.04, [М+Н]+=594.
Следующие соединения получали способом, аналогичным получению Соединения 59, используя следующие амины:
3-Ааминопропан-1-ол,
1-(метиламино)пропан-2-ол
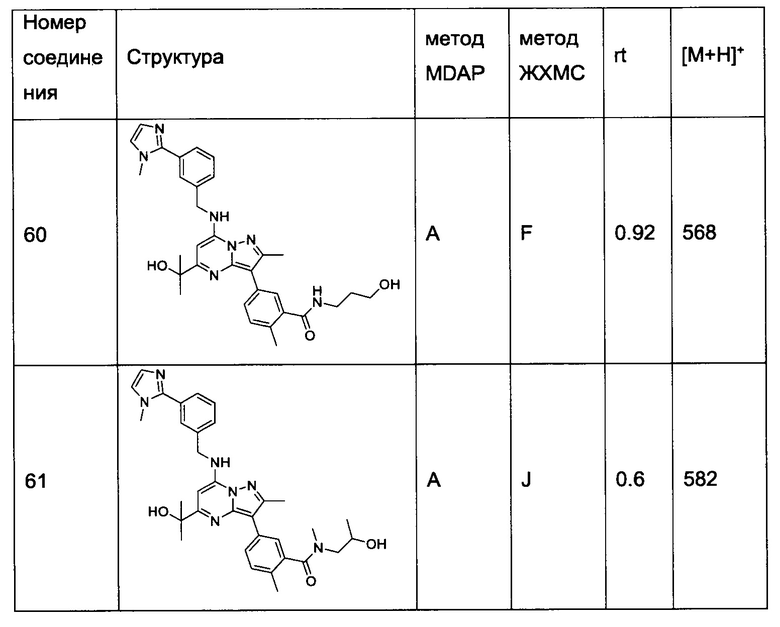
Соединение 62 и Соединение 63
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(2-гидроксипропил)-N,2-диметилбензамид, изомер 1 (соединение 62) и изомер 2 (соединение 63)
Соединение 61 очищали с использованием колонки Chiralpak AS-H (30 мм × 250 мм, 5 мкм), используя для элюирования ацетонитрил, содержащий 0.2% изопропиламин, в результате чего получали указанные в заголовке соединения. Изомер 1: ЖХМС (метод F): время удерживания=0.98, [М+Н]+=582. Хиральная ВЭЖХ: время удерживания 6.85, 100%. Изомер 2: ЖХМС (метод F): время удерживания=0.98, [М+Н]+=582. Хиральная ВЭЖХ: время удерживания 8.36, 98.5%.
Соединение 64
2-((2-метокси-5-(2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)-5-(тетрагидро-2Н-пиран-4-ил)пиразоло[1,5-а]пиримидин-3-ил)Фенил)сульфонил)этан-1-ол
В высушенный сосуд добавляли 2-(3,6-дигидро-2Н-пиран-4-ил)-4,4,5,5-третраметил-1,3,2-диоксаборолан (18 мг, 0.086 ммоль), Pd XPhos G2 (6 мг, 7.63 мкмоль), Pd/C (51 мг, 0.048 ммоль), трикалийфосфат (51 мг, 0.240 ммоль) и трет-бутил(5-хлор-3-(3-((2-гидроксиэтил)сульфонил)-4-метоксифенил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1H-имидазол-2-ил)бензил)карбамат (промежуточное соединение 102, 55 мг, 0.082 ммоль). Сосуд закрывали крышкой и барботировали азотом. Добавляли воду (0.2 мл) и 1,4-диоксан (0.8 мл) и нагревали смесь до 100°С в течение 45 мин. Смеси давали остыть до комнатной темп. Добавляли формиат аммония (1.25 М в метаноле) (0.659 мл, 0.824 ммоль) и перемешивали смесь в течение ночи. Смесь нагревали до 40°С в течение 6 ч. Сосуд продували азотом и фильтровали содержимое через целит, промывали дихлорметаном (10 мл). Фильтрат разделяли водой (10 мл) и отделенную водную фазу промывали дихлорметаном (2×10 мл). Объединенные органические слои пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток объединяли с остатком от аналогичной реакции, проведенной с трет-бутил-5-хлор-3-(3-((2-гидроксиэтил)сульфонил)-4-метоксифенил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбаматом (промежуточное соединение 102, 20 мг, 0.03 ммоль), растворяли в метаноле (1.4 мл) и добавляли соляную кислоту (3 М в СРМЕ) (0.35 мл, 1.05 ммоль). Смесь перемешивали в течение ночи при комнатной темп., затем при 40°С в течение 7 ч. Смеси давали выстояться при комнатной темп, на протяжении выходных. Добавляли насыщенный вод. бикарбонат натрия (1 мл) и перемешивали смесь в течение 5 мин. Добавляли ДХМ (2 мл) и разделяли смесь. Отделенную водную фазу промывали дихлорметаном (2×1 мл), а объединенные органические фазы пропускали через гидрофобный спеченный фильтр и концентрировали под потоком инертного газа. Остаток растворяли в смеси ДМСО: метанол (0.5 мл, 1:1) и очищали MDAP-хроматографией (метод А). Содержащие продукт фракции объединяли, концентрировали при пониженном давлении и сушили в установке с высоким вакуумом, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.95, [М+Н]+=617.
Соединение 65
5-(5-(1-гидроксибутен-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид

Получали способом, аналогичным получению Соединения 37, используя 2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)бутен-1-ол (промежуточное соединение 104) и N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.97, [М+Н]+=596.
Соединения 66 и 67
5-(5-(1-гидроксибутен-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид, изомер 1 (соединение 66) и изомер 2 (соединение 67)

Соединение 65 очищали с использованием колонки Chiralpak AD-H (30 мм × 250 мм, 5 мкм), используя для элюирования 40% этанол (содержащий 0.2% изопропиламина) в гептане (содержащим 0.2% изопропиламина), в результате чего получали указанные в заголовке соединения, изомер 1: ЖХМС (метод J): время удерживания=0.58, [М+Н]+=596. Хиральная ВЭЖХ: время удерживания 9.41, 100%, изомер 2: ЖХМС (метод J): время удерживания=0.58, [М+Н]+=596. Хиральная ВЭЖХ: время удерживания 14.57, 99.5%.
Соединение 68
5-(5-(3-гидроксипентан-3-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид

Получали способом, аналогичным получению Соединения 37, используя 3-(3-бром-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пентан-3-ол (промежуточное соединение 105) и N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36), в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.71, [М+Н]+=610.
Соединение 69
N-(3-гидроксипропил)-5-(5-(1-гидроксипропил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N,2-диметилбензамид, изомер 1
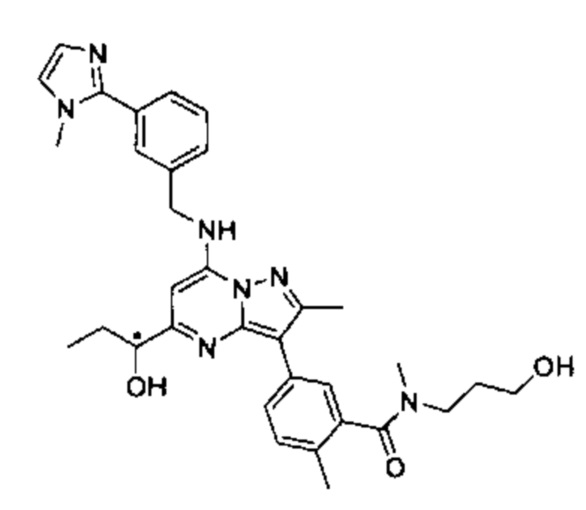
Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 1 (промежуточное соединение 108) и N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36), в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.65, [М+Н]+=582.
Соединение 70
N-(3-гидроксипропил)-5-(5-(1-гидроксипропил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N,2-диметилбензамид, изомер 2

Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1 Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 2 (промежуточное соединение 109) и N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36), в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.61, [М+Н]+=582.
Соединение 71
2-((5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксифенил)сульфонил)этан-1-ол, изомер 1
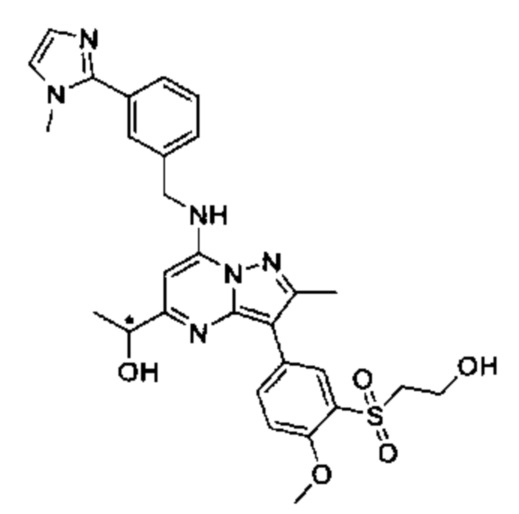
Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)этан-1-ол, изомер 1 (промежуточное соединение 111) и 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этанол (промежуточное соединение 19), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.87, [М+Н]+=577.
Соединение 72
2-((5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксиФенил)сульФонил)этан-1-ол, изомер 2

Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)этан-1-ол, изомер 2 (промежуточное соединение 112) и 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этанол (промежуточное соединение 19), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.86, [М+Н]+=577.
Соединение 73
2-Хлор-5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N-метилбензамид, изомер 1
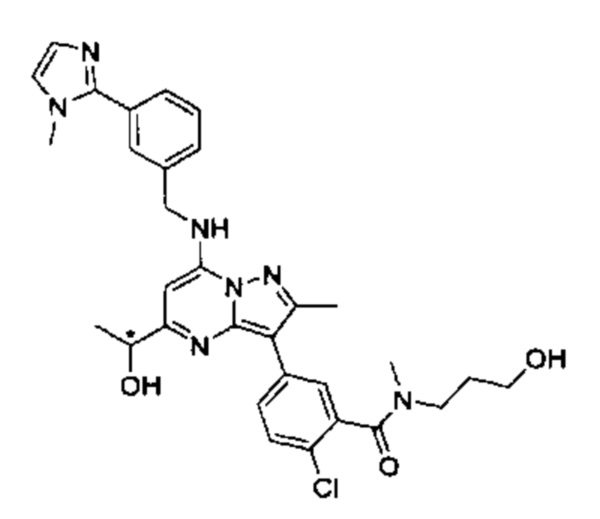
Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)этан-1-ол, изомер 1 (промежуточное соединение 111) и 2-хлор-N-(3-гидроксипропил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 23), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.93, [М+Н]+=588.
Соединение 74
2-Хлор-5-(5-(1-гидроксиэтил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N-метилбензамид, изомер 2
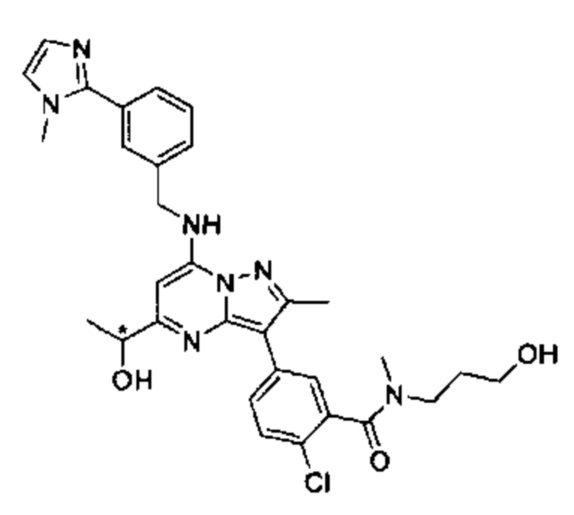
Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)этан-1-ол, изомер 2 (промежуточное соединение 112) и 2-хлор-N-(3-гидроксипропил)-N-метил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 23), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.93, [М+Н]+=588.
Соединение 75
1-(3-(3-((2-гидроксиэтил)сульфонил)-4-метоксиФенил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 1

Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 1 (промежуточное соединение 108) и 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этанол (промежуточное соединение 19), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.91, [М+Н]+=591.
Соединение 76
1-(3-(3-((2-гидроксиэтил)сульфонил)-4-метоксифенил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 2
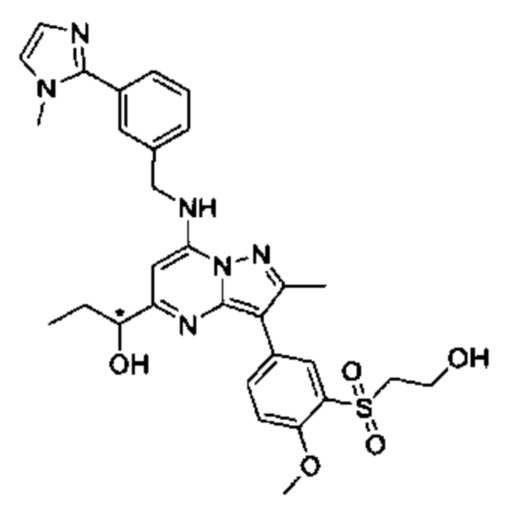
Получали способом, аналогичным получению Соединения 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 2 (промежуточное соединение 109), и 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этанол (промежуточное соединение 19), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.91, [М+Н]+=591.
Соединение 77
5-(5-(1-гидроксипропил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метокси-N-метил-N-(тетрагидро-2Н-пиран-4-ил)бензамид, изомер 1
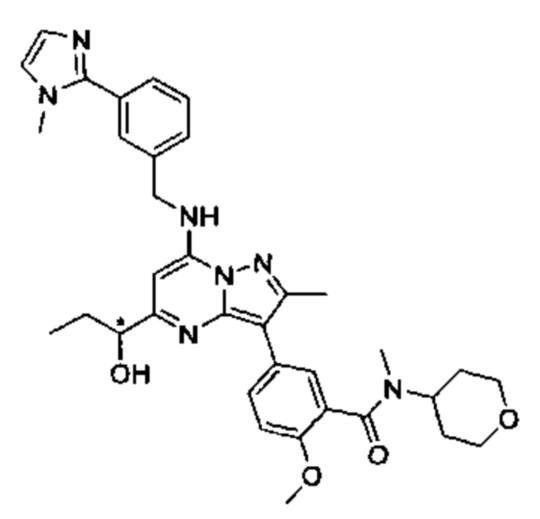
Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 1 (промежуточное соединение 108), и 2-метокси-N-метил-N-(тетрагидро-2Н-пиран-4-ил)-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 34), в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.63, [М+Н]+=624.
Соединение 78
5-(5-(1-гидроксипропил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метокси-N-метил-N-(тетрагидро-2Н-пиран-4-ил)бензамид, изомер 2

Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 2 (промежуточное соединение 109), и 2-метокси-N-метил-N-(тетрагидро-2H-пиран-4-ил)-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 34), в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.64, [М+Н]+=624.
Соединение 79
5-(5-(1-гидроксициклопропил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N,2-диметилбензамид
Трет-бутил(3-бром-5-(1-((трет-бутилдиметилсилил)окси)циклопропил)-2-метилпиразоло[1,5-а]пиримидин-7-ил)(3-(1-метил-1Н-имидазол-2-ил)бензил)карбамат (промежуточное соединение 115, 26 мг, 0.039 ммоль), фосфат калия (24 мг, 0.113 ммоль), XPhos (2 мг, 4.20 мкмоль), XPhos Pd G2 (3 мг, 3.81 мкмоль) и N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36, 25 мг, 0.053 ммоль) объединяли в 1,4-диоксане (0.4 мл) и воде (0.133 мл). Смесь нагревали до 60°С в запаянном сосуде в течение 1 ч 45 мин. XPhos Pd G2 (3 мг, 3.81 мкмоль) и добавляли N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36, 25 мг, 0.053 ммоль). Через 1 ч температуру реакции повышали до 80°С. Через 2 ч добавляли XPhos Pd G2 (3 мг, 3.81 мкмоль) и N-(3-гидроксипропил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 36, 25 мг, 0.053 ммоль) и нагревали смесь до 100°С в течение 1 ч. Смесь разделяли между водой (10 мл) и этилацетатом (10 мл). Отделенную водную фазу промывали этилацетатом (2×10 мл), а объединенные органические фазы пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток растворяли в 1,4-диоксане (1.0 мл), а затем добавляли HCl (4M в 1,4-диоксане) (0.1 мл). Смесь перемешивали при комнатной температуре в течение 4 дней Добавляли насыщенный водный раствор бикарбоната натрия (3 мл) и перемешивали смесь в течение 10 мин. Добавляли воду (10 мл) и ДХМ (10 мл) и разделяли фазы. Отделенную водную фазу промывали дихлорметаном (2×5 мл), а объединенные органические слои пропускали через гидрофобный спеченный фильтр и концентрировали при пониженном давлении. Остаток очищали MDAP-хроматографией (метод А). Фракции, содержащие нужный продукт, объединяли и концентрировали при пониженном давлении, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.97, [М+Н]+=580.
Соединение 80
N-((S)-1-гидроксипропан-2-ил)-5-(5-(1-гидроксипропил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин3-ил)-N,2-диметилбензамид, изомер 1

Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 1 (промежуточное соединение 108), и (S)-N-(1-гидроксипропан-2-ил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 45), в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.62, [М+Н]+=582.
Соединение 81
N-((S)-1-гидроксипропан-2-ил)-5-(5-(1-гидроксипропил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин3-ил)-N,2-диметилбензамид, изомер 2

Получали аналогично соединению 37, используя 1-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-1-ол, изомер 2 (промежуточное соединение 109), и (5)-N-(1-гидроксипропан-2-ил)-N,2-диметил-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 45), в результате чего получали указанное в заголовке соединение. ЖХМС (метод J): время удерживания=0.61, [М+Н]+=582.
Соединение 82
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1H-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(2-гидроксипропил)-2-метокси-N-метилбензамид

5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-2-метоксибензойную кислоту (промежуточное соединение 120, 65 мг, 0.101 ммоль), HATU (47 мг, 0.124 ммоль) и DIPEA (0.055 мл, 0.315 ммоль) перемешивали в ТГФ (1 мл) при комнатной температуре в течение 5 мин, после чего добавляли 1-(метиламино)пропан-2-ол (13 мг, 0.146 ммоль). Через 2 ч добавляли еще HATU (12 мг, 0.032 ммоль) и DIPEA (0.018 мл, 0.101 ммоль) добавляли. Через 1.5 ч реакционную смесь очищали MDAP-хроматографией (метод А). Содержащие продукт фракции объединяли и сушили под потоком инертного газа, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.96, [М+Н]+=598.
Соединения 83 и 84
5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(2-гидроксипропил)-2-метокси-N-метилбензамид, изомер 1 (соединение 83), и изомер 2 (соединение 84)

Соединение 82 очищали на колонке Chiralpak IC (30 мм × 250 мм, 5 мкм), используя для элюирования 30% этанол (содержащий 0.2% изопропиламин) в гептане (содержащем 0.2% изопропиламин), в результате чего получали указанные в заголовке соединения, изомер 1: ЖХМС (метод F): время удерживания=0.94, [М+Н]+=598. Хиральная ВЭЖХ: время удерживания 28.8, 100%. Изомер 2: ЖХМС (метод F): время удерживания=0.94, [М+Н]+=598. Хиральная ВЭЖХ: время удерживания 33.68, 96.6%.
Соединение 85
2-((2-метокси-5-(2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)-5-((тетрагидро-2Н-пиран-4-ил)метил)пиразоло[1,5-а]пиримидин-3-ил)фенил)сульфонил)этан-1-ол
Получали аналогично соединению 9, используя 3-йодо-2-метил-N-(3-(1-метил-1Н-имидазол-2-ил)бензил)-5-((тетрагидро-2Н-пиран-4-ил)метил)пиразоло[1,5-а]пиримидин-7-амин (промежуточное соединение 122) и 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этанол (промежуточное соединение 19), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.94, [М+Н]+=631. Соединение 86
2-(3-(3-((2-гидроксизтил)сульфонил)-4-метоксифенил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)бутен-1-ол
Получали аналогично соединению 37, используя 2-(3-йодо-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)бутен-1-ол (промежуточное соединение 104) и 2-((2-метокси-5-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)фенил)сульфонил)этанол (промежуточное соединение 19), в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.92, [М+Н]+=605.
Соединение 87
3-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N-(3-гидроксипропил)-N-метилбензамид
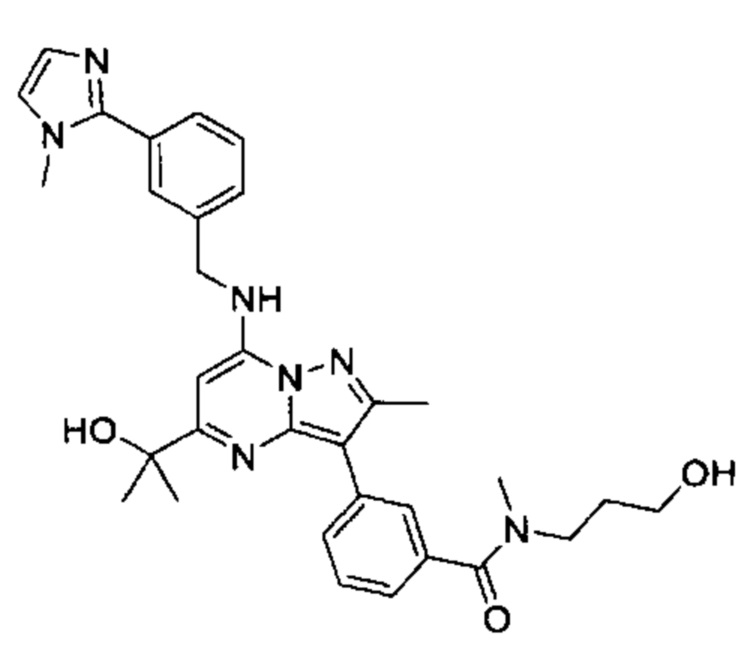
2-(3-бром-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-5-ил)пропан-2-ол (промежуточное соединение 43, 150 мг, 0.313 ммоль), фосфат калия (165 мг, 0.777 ммоль), дициклогексил(2',4',6'-триизопропил-[1,1'-бифенил]-2-ил)фосфан (12 мг, 0.025 ммоль), XPhos Pd G2 (22 мг, 0.028 ммоль) и N-(3-гидроксипропил)-N-метил-3-(4,4,5,5-третраметил-1,3,2-диоксаборолан-2-ил)бензамид (промежуточное соединение 123, 219 мг, 0.412 ммоль) объединяли в сосуде для микроволновой обработки. Добавляли 1,4-диоксан (1.8 мл) и воду (0.6 мл), запаивали сосуд и нагревали смесь до 100°С в микроволновом реакторе в течение 1 ч. После охлаждения органический слой двухфазной реакционной смеси фильтровали через вату и непосредственно очищали MDAP-хроматографией (метод А). Полученный остаток разделяли между насыщенным водным раствором бикарбоната натрия (10 мл) и ДХМ (5 мл). Отделенную водную фазу промывали дополнительным количеством ДХМ (2×5 мл). Органические вещества объединяли, пропускали через гидрофобный спеченный фильтр, концентрировали под потоком инертного газа и сушили в высоком вакууме при 40°С в течение 1 дня, в результате чего получали указанное в заголовке соединение. ЖХМС (метод F): время удерживания=0.94, [М+Н]+=568.
Биологические анализы
а) Анализ активности PI4KB
Ингибирование активности PI4KIII-бета 13-828 (D316-330) человека in vitro определяли, используя набор для анализа активности киназы ADP-Glo (ADP-Glo Kinase assay) от Promega. Ингибиторы растворяли в 100% ДМСО в концентрации 1 мМ. Разведения в 100% ДМСО получали путем пошаговых серийных разведений 1/3. Пробу объемом 60 нл, полученную при 11-точечном титровании, переносили в белый 384-луночный аналитический планшет Грейнера малого объема, добиваясь конечной концентрации ДМСО 1% во всем планшете и наивысшей итоговой концентрации ингибитора 10 мкМ. Анализ на PI4KIII-бета включал 25 мМ Hepes рН 7,5 (NaOH), 10 мМ MgCl2, 0,5 мМ EGTA, 0,1% Triton X-100, 2 мМ TCEP и 0,1 мг/мл БСА, 1 мМ АТФ, 60 мкМ фосфатидилинозитол и 1,5 нМ PI4K-бета 13-828 (D316-330) человека в общем объеме 6 мкл. Начало анализа инициировали добавлением фермента, накрывали и инкубировали при комнатной температуре в течение 3 часов. Анализы останавливали добавлением 6 мкл реагента ADP-GLO, содержащего 0,1% CHAPS для истощения непотребленного АТФ. Планшеты инкубировали при комнатной температуре в течение 60 минут, а затем добавляли 12 мкл реагента для детекции киназы, содержащего 0,1% CHAPS, для преобразования АДФ в АТФ, и вводили люциферазу и люциферин. После 40 минут инкубации при комнатной температуре люминесцентный сигнал считывали на BMG Labtech PHERAstar FS, используя следующие настройки; коэффициент усиления 3600, высота фокуса 13,7 мм, интервал между измерениями 1 с, время установления 0,2 с. Ингибиторный эффект соединений на активность PI4K-бета оценивали по IC50 ответа относительно значений для контроля с высокими значениями, без соединения, и контроля с низкими значениями, без фермента, путем аппроксимации четырехпараметрическим уравнением «доза - ответ».
При тестировании в указанном анализе: все соединения давали среднее значение pIC50, превышающее или равное 8,1; соединения 1-7, 9-11, 14, 15, 17-20, 22-29 и 31-87 давали среднее значение pIC50, превышающее или равное 8,4; соединения 17 и 19 давали среднее значение pIC50 8,6; соединение 21 давало среднее значение pIC50 8,2; и соединение 22 давала среднее значение pIC50 8,5.
b) Анализ времени удерживания
Подготовительный анализ Caliper
Разведения соединения получали в 100% ДМСО путем пошаговых серийных разведений 1/3 при наивысшей концентрации 1 мМ. Пробу объемом 200 нл, полученную при 12-точечном титровании, переносили в черный 384-луночный аналитический планшет Грейнера, добиваясь итоговой концентрации ДМСО 1% во всем планшете и наивысшей итоговой концентрации ингибитора 10 мкМ в итоговом объеме для анализа 20 мкл. В лунке 384-луночного аналитического планшета предварительно инкубировали 10 мкл 2Х ферментного буфера с соединением в течение 1 часа. Буфер без фермента использовали в качестве контроля со 100% ингибированием. Добавляли 10 мкл 2Х раствора субстрата для инициации начала анализа, так, чтобы итоговые концентрации составляли 2 мМ АТФ и 1 мкМ Bodipy-PI, и планшет инкубировали при 25°С в течение 3 часов. Ферментативную реакцию прекращали путем добавления 40 мкл останавливающего буфера; субстрат (PI) и продукт (PIP), присутствующие в каждом образце, разделяли электрофоретически с помощью инструмента для капиллярного электрофореза LabChip 3000, CALIPER LABCHIP 3000 Drug Discovery System, и детектировали с использованием голубого лазера (480 нм) для возбуждения и CCD в зеленом диапазоне (520 нм) для детекции (CCD2). Образцы для отрицательного контроля (0% - ингибирование в отсутствие ингибитора) и образцы для положительного контроля (100% - ингибирование в отсутствие фермента) собирали в 48 повторностях (по 4 повторности на сиппер-шприц Caliper) и использовали для расчета процентных значений ингибирования в каждой тестовой лунке. Процент ингибирования (Pinh) определяли, используя следующее уравнение:
Pinh=(PSR0% - PSRinh)/(PSR0% - PSR100%) × 100
где PSRinh - отношение количества продукта к суммарному количеству в присутствии ингибитора, PSR0% - среднее отношение количества продукта к суммарному количеству в отсутствие ингибитора, a PSR100% - среднее отношение количества продукта к суммарному количеству в контрольных образцах со100% ингибированием; значения IC50 для ингибиторов определяют путем аппроксимации кривых ингибирования (зависимость Pinh от концентрации ингибитора) 4-параметрической сигмоидальной моделью зависимости «доза - ответ» с помощью программного обеспечения XLfit 4 (IBDS).
Разведение с быстрым измерением концентрации ингибитора В небольшой пробирке Eppendorf 5 мкл 2х раствора фермента (0,4 мкМ - 0,8 мкМ) смешивали с 5 мкл 2х раствора соединения с 40-120-кратным превышением значений IC50, определенных с 2 мМ АТФ согласно описанию выше, и инкубировали в течение 1 часа при комнатной температуре. Начало анализа инициировали, быстро смешивая 2 мкл предварительно инкубированной ферментной смеси с 800 мкл смеси субстрата, содержащей 2 мМ АТФ и 1 мкМ Bodipy-PI. 60 мкл каждого образца переносили в 384-луночный планшет Грейнера малого объема; субстрат (PI) и продукт (PIP), присутствующие в каждом образце, разделяли электрофоретически с помощью инструмента для капиллярного электрофореза LabChip 3000, CALIPER LABCHIP 3000 Drug Discovery System, и детектировали с использованием голубого лазера (480 нм) для возбуждения и CCD в зеленом диапазоне (520 нм) для детекции (CCD2) на протяжении приблизительно 8 часов (по 200 измерений для каждой лунки). Кривые процесса, отражающие % преобразования от времени в присутствии соединения, сравнивают с кривыми для контрольных образцов без соединения. Кривые аппроксимировали уравнением кривой процесса, используя программное обеспечение XLfit:

где Vi - начальная скорость работы фермента, Vs - скорость работы в равновесном состоянии в присутствии разведенного ингибитора, и t - время после разведения.
Наблюдаемые значения Vi, Vs и Kobs зависят от природы ингибитора (т.е. быстрое достижение равновесия или прочное связывание). В зависимости от начальных условий анализа (относительной концентрации соединения и фермента) показатели Vi и Vs могут быть заранее установлены и/или фиксированы. Для медленно диссоциирующих соединений > [Е] начальная скорость работы фермента (после быстрого разведения) обычно не фиксирована; скорость работы в равновесном состоянии может быть фиксирована на уровне скорости в присутствии остаточного соединения (определенной по кривой IC50). Для некоторых протестированных быстро достигающих равновесия соединений (1/2 времени удерживания<разрешения анализа) > [Е] начальная скорость работы может быть фиксирована на уровне теоретического % активности, определенного по кривым IC50 при соответствующих концентрациях соединения, а скорость работы в равновесном состоянии может быть заранее установлена на уровне контроля, но не фиксирована. Для остальных быстро достигающих равновесия или медленно диссоциирующих соединений может быть использован альтернативный способ предварительного расчета Vi и Vs, не фиксированных на уровне теоретических скоростей, определенных по кривым IC50 при соответствующих концентрациях соединения, например, в тех случаях, когда вышеописанные стратегии не позволяют получить удовлетворительные результаты аппроксимации.
В указанном анализе соединение 17 продемонстрировало среднее время удерживания 542 минуты; соединение 19 продемонстрировало среднее время удерживания 997 минут; соединение 21 продемонстрировало среднее время удерживания 528 минут; и соединение 22 продемонстрировало среднее время удерживания 1666 минут.
с) Протокол анализа цитопатического эффекта (СРЕ)
Ингибирование активности PI4KB in vitro определяли путем анализа ингибирования истощения АТФ в клетках HeLa Ohio в ответ на инфекцию штаммом 16 риновируса человека типа A (HRVA16). Уровни АТФ определяли с применением реагента CellTiter-GLO от Promega. Соединения, которые ингибируют PI4KB, также являются мощными ингибиторами риновирусов человека и способны защищать клетки HeLa Ohio от CPE, индуцированного вирусной инфекцией и репликацией. Защищенные клетки сохраняют жизнеспособность после вирусной инфекции и, соответственно, уровни АТФ в них выше. Соединения растворяли в 100% ДМСО до концентрации 3 мМ с последующим пошаговым серийным разведением 1:3 в 100% ДМСО. Пробу объемом 0,5 мкл, полученную при 10-точечном титровании, переносили в белый 96-луночный плоскодонный планшет Грейнера для тканевых культур (655083). Пробу объемом 0,5 мкл 100% ДМСО вносили в вертикальные ряды планшета 11 и 12, с получением итоговой концентрации ДМСО во всем планшете, равной 0,33%, и наивысшей итоговой концентрации соединения в анализе 10 мкМ.
Клетки HeLa Ohio культивировали при 37°С, 5% CO2 в среде (DMEM с добавлением 10% фетальной бычьей сыворотки австралийского происхождения и 2 мМ глютамакса). Клетки пересевали после достижения >80% конфлюентности. Для проведения анализа клетки культивировали до 80-90% конфлюентности до рассоединения.
Клетки рассоединяли для проведения анализа, промывая в ФСБ и рассоединяя с использованием 3 мл TrypLE Express для 5 мл при 37°С. Рассоединенные клетки смешивали с 7 мл среды и центрифугировали при 300 g в течение 5 минут. Клеточный осадок ресуспендировали в 50 мл среды и подсчитывали количество клеток с помощью Beckman Coulter ViCell. Клетки разводили до плотности 6,6×104 клеток/мл и некоторую часть объема с клетками переносили в новую пробирку для использования в качестве контроля для 12 вертикального ряда. Стоковый материал HRVA16 добавляли в оставшуюся суспензию клеток, используя подходящее разведение вирусного стокового материала для достижения MOI, равной 1.
150 мкл суспензии с клетками и вирусом добавляли в каждую лунку вертикальных рядов 1-11, используя Multidrop Combi. Добавляли по 150 мкл отдельной суспензии клеток в каждую лунку 12 вертикального ряда, используя многоканальную пипетку. Аналитические планшеты герметизировали и инкубировали при 33°С, 5% CO2 на протяжении двух дней. Через 2 дня аналитические планшеты извлекали из инкубатора и оставляли для уравновешивания до комнатной температуры. Во все лунки добавляли по 60 мкл реагента CellTiter-Glo, используя Thermo Scientific Multidrop Combi. Планшеты инкубировали при комнатной температуре в течение 20 минут, после чего считывали на Perkin Elmer Envision (настройки: высота захвата 2,5, фиксированная высота измерения (мм) 6,5, расстояние между планшетом и детектором 0, время измерения (с) 0,1, поправочный коэффициент свечения (ct2) (0%)).
Ингибиторный эффект соединений на активность PI4KB оценивали по IC50 ответа относительно контролей с высоким уровнем (без вируса, вертикальный ряд 12) и низким уровнем (вирус + отсутствие ингибитора, вертикальный ряд 11) путем аппроксимации четырехпараметрическим уравнением зависимости «доза - ответ».
При тестировании в указанном анализе: все соединения давали среднее значение pIC50, равное или превышающее 7,2; соединений 1-8, 10-15 и 17-87 давали среднее значение pIC50, превышающее или равное 7,6; соединения 1, 3, 5, 6, 7, 10-12, 14, 15, 17-25, 29, 31, 33-37, 39-53, 55-59, 61-68, 70, 76, 80-82, 84 и 86 давали среднее значение pIC50, превышающее или равное 8,4; соединения 17 и 19 давали среднее значение pIC50 8,9; соединение 21 давало среднее значение pIC50 8,8; и соединение 22 давало среднее значение pIC50, равное по меньшей мере 9,1.
d) Анализ метаболической стабильности в микросомах человека
Краткое описание протокола
Тестируемое соединение (0,5 мкМ) инкубировали с объединенными микросомами печени. Тестируемое соединение инкубировали на протяжении 45-минутного эксперимента и анализировали с помощью ЖХ-МС/МС.
Экспериментальная процедура Объединенные микросомы печени человека приобретали у надежного коммерческого поставщика, например, Corning Life Sciences. Микросомы (итоговая концентрация белка 0,5 мг/мл), 50 мМ фосфатный буфер рН7,4 и НАДФН (итоговая концентрация=1 мМ) предварительно инкубировали при 37°С до добавления тестируемого соединения (итоговая концентрация субстрата=0,5 мкМ; итоговая концентрация ДМСО=0,25%) для инициации реакции. Итоговый инкубируемый объем составлял 500 мкл. Для каждого протестированного соединения включали инкубацию контроля, при этом добавляли 50 мМ фосфатный буфер, рН7,4, вместо НАДФН (минус НАДФН). Включали два контрольных соединения для каждого вида. Все инкубации проводят индивидуально для каждого тестируемого соединения.
Каждое соединение инкубировали в течение 45 минут и отбирали инкубированные образцы (50 мкл) в моменты времени через 0, 5, 15, 30 и 45 минут. Образец контроля (минус НАДФН) отбирали только в момент времени через 45 минут. Реакции останавливали путем добавления 100 мкл ацетонитрила, содержащего внутренний стандарт, в образец. После прекращения реакции образцы центрифугировали на 2500 об/мин в течение 20 минут при 4°С для осаждения белка.
Количественный анализ
После осаждения белка образцы анализировали с помощью ЖХ-МС/МС в стандартных условиях.
Анализ данных
По графику зависимости отношения площади пика (площадь пика соединения / площадь пика внутреннего стандарта) от времени определяли уклон кривой. Затем рассчитывали собственный клиренс, используя приведенные ниже уравнения, после чего преобразовывали в мл/мин/г:
Константа скорости элиминации (k)=(- уклон)
Время полужизни (t1/2) (мин)=0,693+k
Собственный клиренс (CLint) (мкл/мин/мг белка)=(V × 0,693)+t1/2
где V=Инкубируемый объем, мкл/мг микросомального белка.
При тестировании в указанном анализе собственный клиренс соединения 17 составил 28,9 мл/мин/г; собственный клиренс соединения 19 составил 11,6 мл/мин/г; собственный клиренс соединения 21 составил 34,0 мл/мин/г; и собственный клиренс соединения 22 составил 33,7 мл/мин/г.
е) Анализ концентрации в селезенке
Соединения 17, 18, 19, 22 и 23 тестировали в указанном анализе накопления в селезенке. Соединения вводили внутривенно крысам для определения уровня соединения в селезенке при полной абсорбции поглощенной дозы (перорально или через легкие) и введении в системный кровоток полной дозы.
Соединения 17, 19, 22 и 23 были введены в состав 2% ДМСО в клептозе (водн., 10% масса/объем). Соединение 18 было введено в состав с ДМСО:РЕС200:водой в соотношении 5:45:50. Составы вводили в виде внутривенных инфузий (1 мг/кг на протяжении 1 часа) одному самцу крысы Wistar Han.
Через 12 часов после начала дозирования крыс умерщвляли и собирали селезенки. Отбирали образцы селезенок массой приблизительно 0,5 г и гомогенизировали в 4 мл воды. Получали по 100 мкл итоговых гомогенатов, проводя осаждение белка с 300 мкл ацетонитрила, содержащего внутренний стандарт, и анализировали (наряду с калибровочными стандартами, приготовленными таким же образом) с применением обращенно-фазовой жидкостной хроматографии - масс-спектрометрии, используя подогреваемый источник электрораспыления и мониторинг множественных реакций в режиме положительных ионов. В качестве колонки для жидкостной хроматографии использовали Waters Cortecs С18 с размером частиц 2,7 мкм, размер колонки 50×2,1 мм, при 60°С; в качестве подвижной фазы, которая функционирует в качестве градиента, использовали (А) 0,1% муравьиную кислоту (водн.) и (В) 0,1% муравьиную кислоту в ацетонитриле, при работе со скоростью потока 1 мл/мин.
Концентрация всех соединений в селезенке составляла менее 30 нг/г. Концентрация соединений 17, 19, 22 и 23 в селезенке составляла менее 7 нг/г. В частности, концентрация соединения 17 в селезенке составляла 1,76 нг/г, а концентрация соединения 22 в селезенке составляла 5,54 нг/г..
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРРОЛОПИРИМИДИНЫ В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ МВТР | 2017 |
|
RU2757457C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ПРОИЗВОДНЫЕ НАФТИРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ АЛЬФА V БЕТА 6 ИНТЕГРИНА ДЛЯ ЛЕЧЕНИЯ, В ЧАСТНОСТИ, ФИБРОЗНЫХ ЗАБОЛЕВАНИЙ | 2015 |
|
RU2692775C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2687093C1 |
| ПРОИЗВОДНЫЕ 2-АМИНОПИРАЗИНА В КАЧЕСТВЕ ИНГИБИТОРОВ CSF-1R КИНАЗЫ | 2013 |
|
RU2642777C2 |
| ТИАЗОЛОПИРИМИДИНОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ РЕЦЕПТОРОВ NMDA | 2014 |
|
RU2703273C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2015 |
|
RU2691389C2 |
| ПИРАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ СОЕДИНЕНИЙ-АНТАГОНИСТОВ H4 | 2019 |
|
RU2821516C2 |
| ЗАМЕЩЕННЫЙ 2-АМИНОПИРИДИН В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2671212C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
Группа изобретений относится к области медицины, а именно к терапии, и предназначена для лечения риновирусной инфекции или нарушения, вызываемого или усугубляемого риновирусной инфекцией. Представлено соединение (S)-N-(1-гидроксипропан-2-ил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N,2-диметилбензамид. В другом воплощении обеспечивается фармацевтический состав, содержащий данное соединение. Также обеспечиваются способы лечения риновирусной инфекции или лечения нарушения, вызываемого или усугубляемого риновирусной инфекцией, включающие введение указанного соединения. Группа изобретений обеспечивает новое соединение (S)-N-(1-гидроксипропан-2-ил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N,2-диметилбензамид, а также фармацевтический состав, содержащий данное соединение, которое является мощным ингибитором PI4KIIIβ и применяется для лечения риновирусной инфекции и нарушений, вызываемых или усугубляемых такой вирусной инфекцией. 4 н. и 4 з.п. ф-лы.
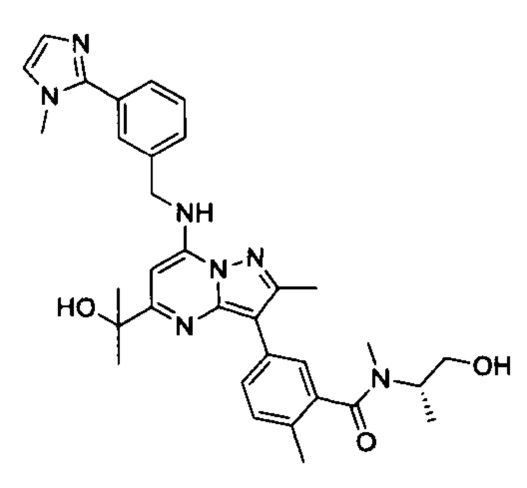
1. Соединение, которое представляет собой (S)-N-(1-гидроксипропан-2-ил)-5-(5-(2-гидроксипропан-2-ил)-2-метил-7-((3-(1-метил-1Н-имидазол-2-ил)бензил)амино)пиразоло[1,5-а]пиримидин-3-ил)-N,2-диметилбензамид (соединение 22)
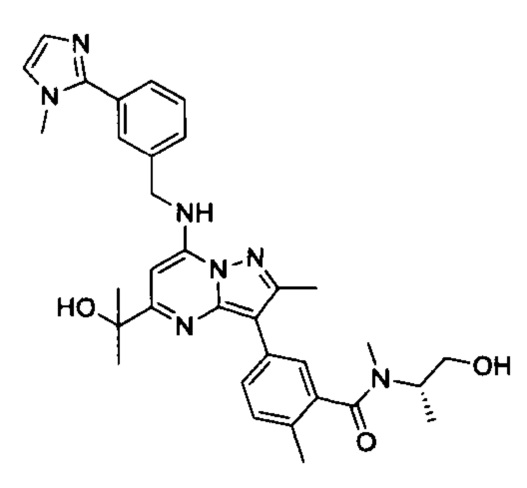
или его фармацевтически приемлемая соль.
3. Соединение по п. 1 или его фармацевтически приемлемая соль для применения в лечении риновирусной инфекции.
4. Соединение по п. 1 или его фармацевтически приемлемая соль для применения в лечении нарушения, вызываемого или усугубляемого риновирусной инфекцией.
5. Соединение по п. 1 или его фармацевтически приемлемая соль для применения по п. 4, отличающиеся тем, что нарушение представляет собой хроническую обструктивную болезнь легких (ХОБЛ).
6. Способ лечения риновирусной инфекции, включающий введение соединения по п. 1 или его фармацевтически приемлемой соли нуждающемуся в этом пациенту.
7. Способ лечения нарушения, вызываемого или усугубляемого риновирусной инфекцией, включающий введение соединения по п. 1 или его фармацевтически приемлемой соли нуждающемуся в этом пациенту.
8. Фармацевтический состав для лечения риновирусной инфекции или нарушения, вызываемого или усугубляемого риновирусной инфекцией, содержащий эффективное количество соединения по п. 1 или его фармацевтически приемлемой соли и фармацевтически приемлемое вспомогательное вещество.
| WO 2017055305 A1, 06.04.2017 | |||
| WO 2016206999 A1, 29.12.2016 | |||
| WO 2015110491 A2, 30.07.2015 | |||
| WO 2013128028 A1, 06.09.2013 | |||
| EA 201000085 A1, 29.10.2010 | |||
| БЕЛИКОВ В | |||
| Г | |||
| Фармацевтическая химия | |||
| Учебное пособие, М.: МЕДпресс-информ, 2007, С | |||
| Прибор с двумя призмами | 1917 |
|
SU27A1 |
| DELANG L | |||
| et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |

